Job Results:
Ligand
Structure
Job ID
4d96340bab84003cd843e60272b3611c
Job name
NA
Time
2025-02-13 15:23:33
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Farnesyl pyrophosphate synthase | 4NUA | 4.00 | |
Target general information Gen name FDPS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA1293;FPS Protein family FPP/GGPP synthase family Biochemical class Transferase Function Dimethylallyltranstransferase activity.Geranyltranstransferase activity.Metal ion binding.RNA binding. Related diseases Porokeratosis 9, multiple types (POROK9) [MIM:616631]: A form of porokeratosis, a disorder of faulty keratinization characterized by one or more atrophic patches surrounded by a distinctive hyperkeratotic ridgelike border called the cornoid lamella. The keratotic lesions can progress to overt cutaneous neoplasms, typically squamous cell carcinomas. Multiple clinical variants of porokeratosis are recognized, including porokeratosis of Mibelli, linear porokeratosis, disseminated superficial actinic porokeratosis, palmoplantar porokeratosis, and punctate porokeratosis. Different clinical presentations can be observed among members of the same family. Individuals expressing more than one variant have also been reported. {ECO:0000269|PubMed:26202976}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00630; DB01785; DB07780; DB02552; DB07841; DB00710; DB06255; DB04714; DB02508; DB06548; DB00282; DB00884; DB00399 Interacts with O95870; P54253; Q6ZMZ0; Q9BRI3; Q8WWF3 EC number 2.5.1.1; 2.5.1.10 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Cytoplasm; Disease variant; Host-virus interaction; Hydroxylation; Isoprene biosynthesis; Lipid biosynthesis; Lipid metabolism; Magnesium; Metal-binding; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 38722.9 Length 338 Aromaticity 0.12 Instability index 45.6 Isoelectric point 5.1 Charge (pH=7) -9.86 2D Binding mode Binding energy (Kcal/mol) -5.45 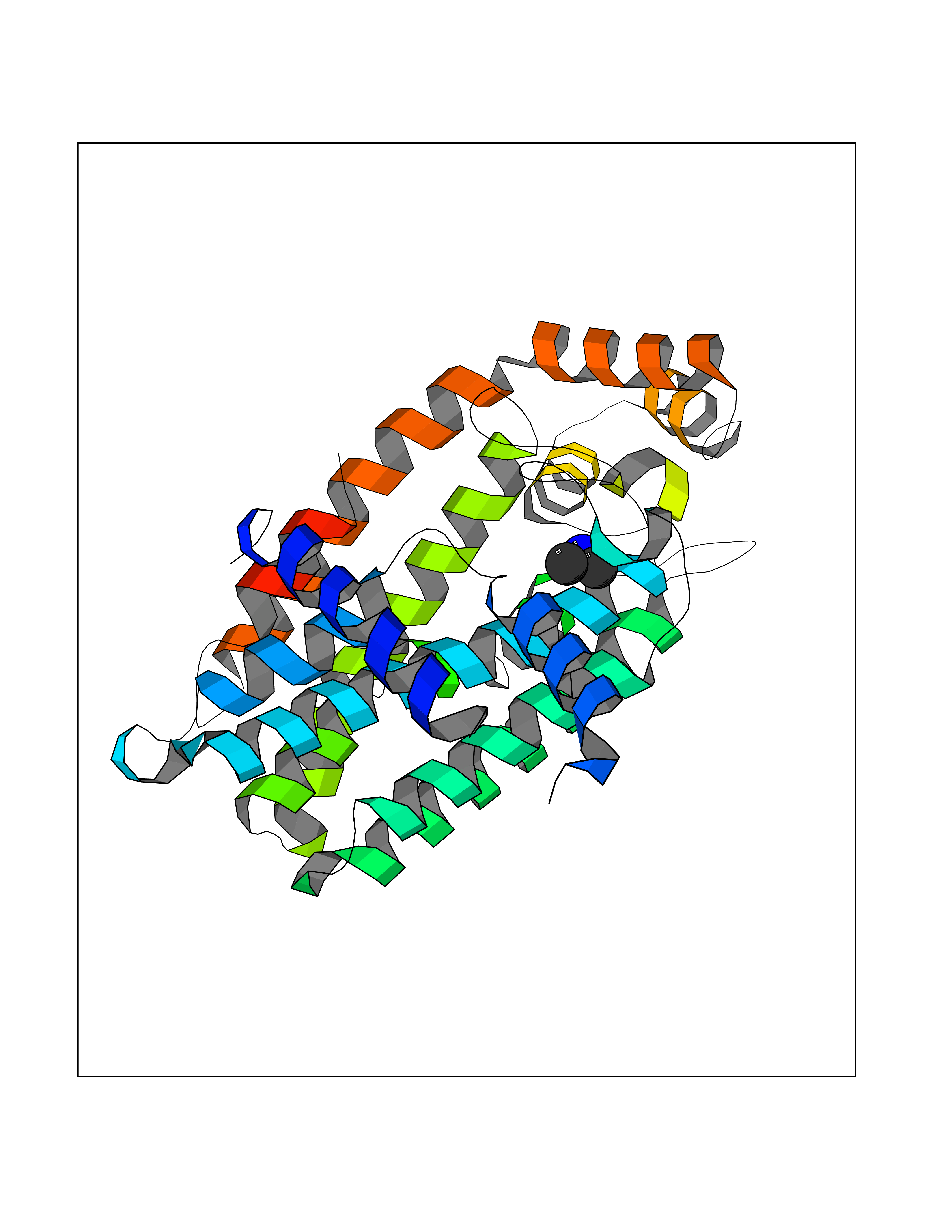 Molscript Map 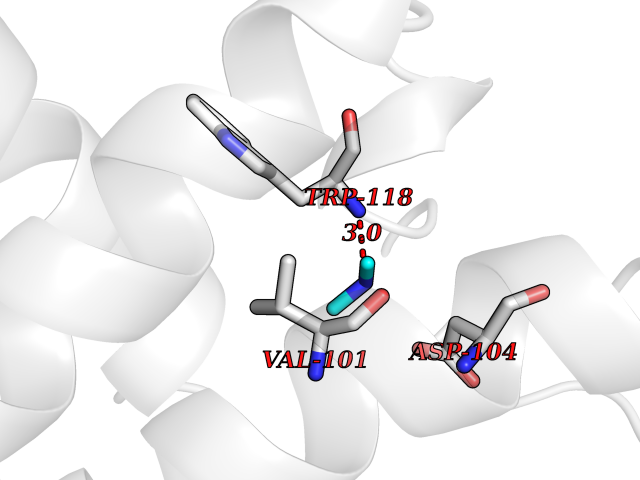 Pymol Map 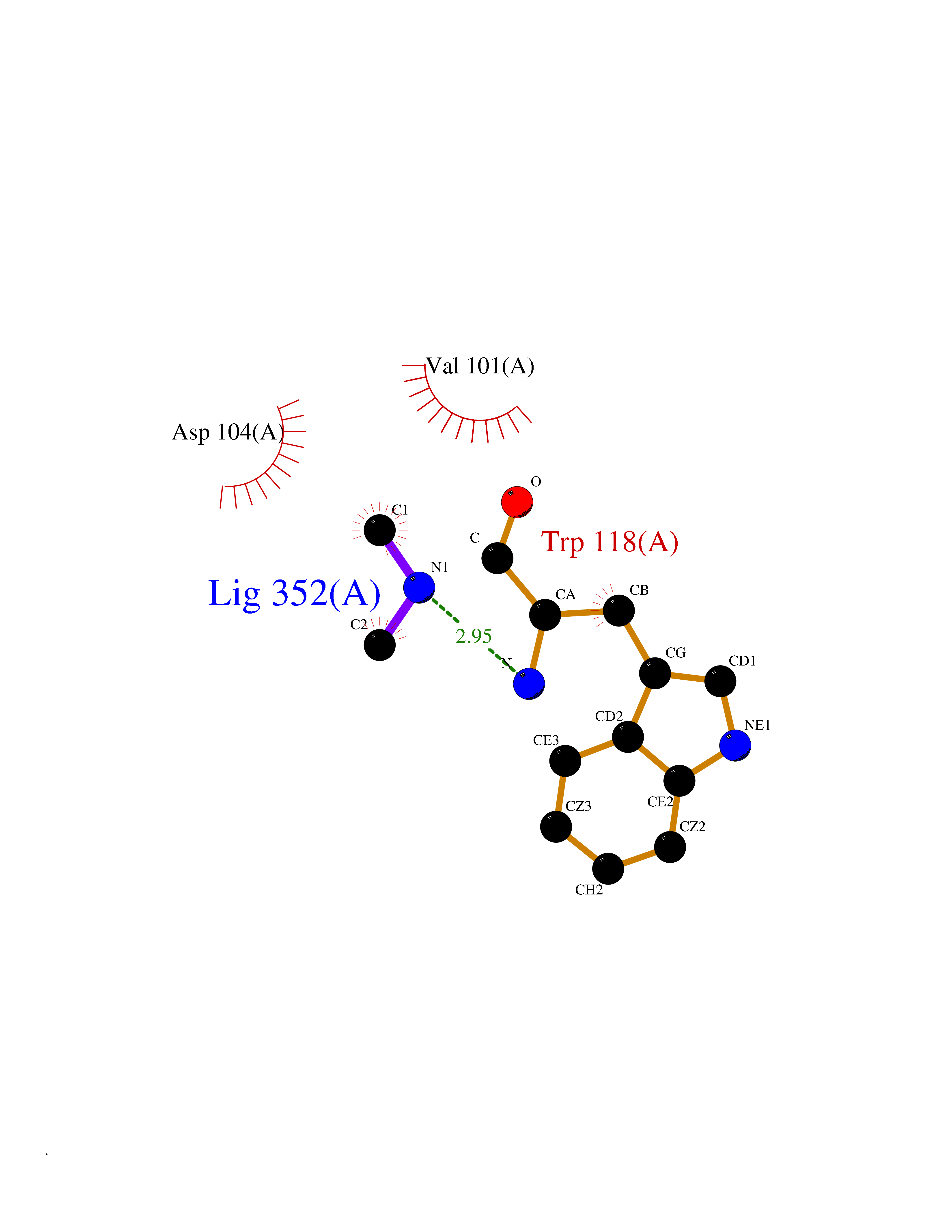 Ligplot Map 3D Binding mode Sequence AYAQEKQDFVQHFSQIVRVLTEHPEIGDAIARLKEVLEYNTIGGKYNRGLTVVVAFRELVEPRKQDADSLQRAWTVGWCVELLQAFFLVADDIMDSSLTRRGQICWYQKPGVGLDAINDANLLEACIYRLLKLYCREQPYYLNLIELFLQSSYQTEIGQTLDLLTAPQGNVDLVRFTEKRYKSIVKYKTAFYSFYLPIAAAMYMAGIDGEKEHANAKKILLEMGEFAQIQDDYLDLFGDPSVTGKIGTDIQDNKCSWLVVQCLQRATPEQYQILKENYGQKEAEKVARVKALYEELDLPAVFLQYEEDSYSHIMALIEQYAAPLPPAVFLGLARKIYK Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | S-adenosylmethionine decarboxylase proenzyme (AMD1) | 1JL0 | 4.00 | |
Target general information Gen name AMD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SamDC; S-adenosylmethioninedecarboxylase; AdoMetDC; AMD Protein family Eukaryotic AdoMetDC family Biochemical class Carbon-carbon lyase Function Promotes maintenance and self-renewal of embryonic stem cells, by maintaining spermine levels. Essential for biosynthesis of the polyamines spermidine and spermine. Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08163; DB00118; DB01917 Interacts with P17707; Q96A98; Q8WY91 EC number EC 4.1.1.50 Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Decarboxylase; Direct protein sequencing; Lyase; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Pyruvate; Reference proteome; S-adenosyl-L-methionine; Schiff base; Spermidine biosynthesis; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 35790.5 Length 311 Aromaticity 0.14 Instability index 39.47 Isoelectric point 6.03 Charge (pH=7) -2.01 2D Binding mode Binding energy (Kcal/mol) -5.45 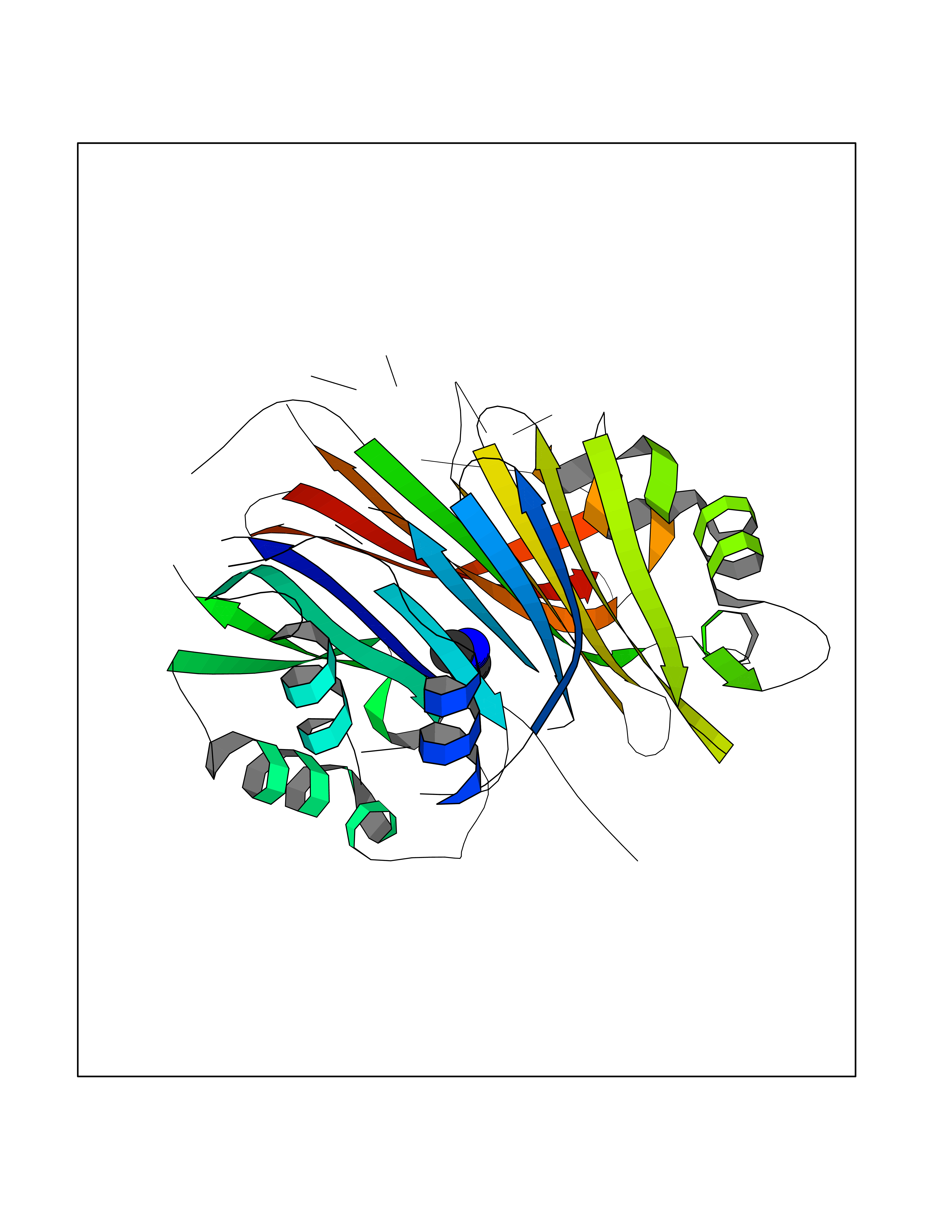 Molscript Map 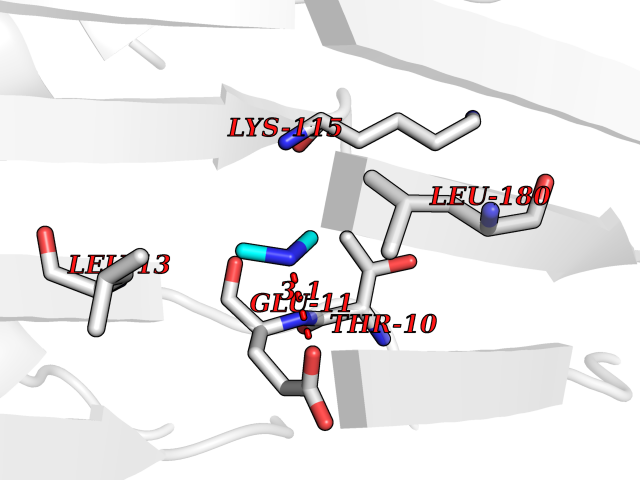 Pymol Map 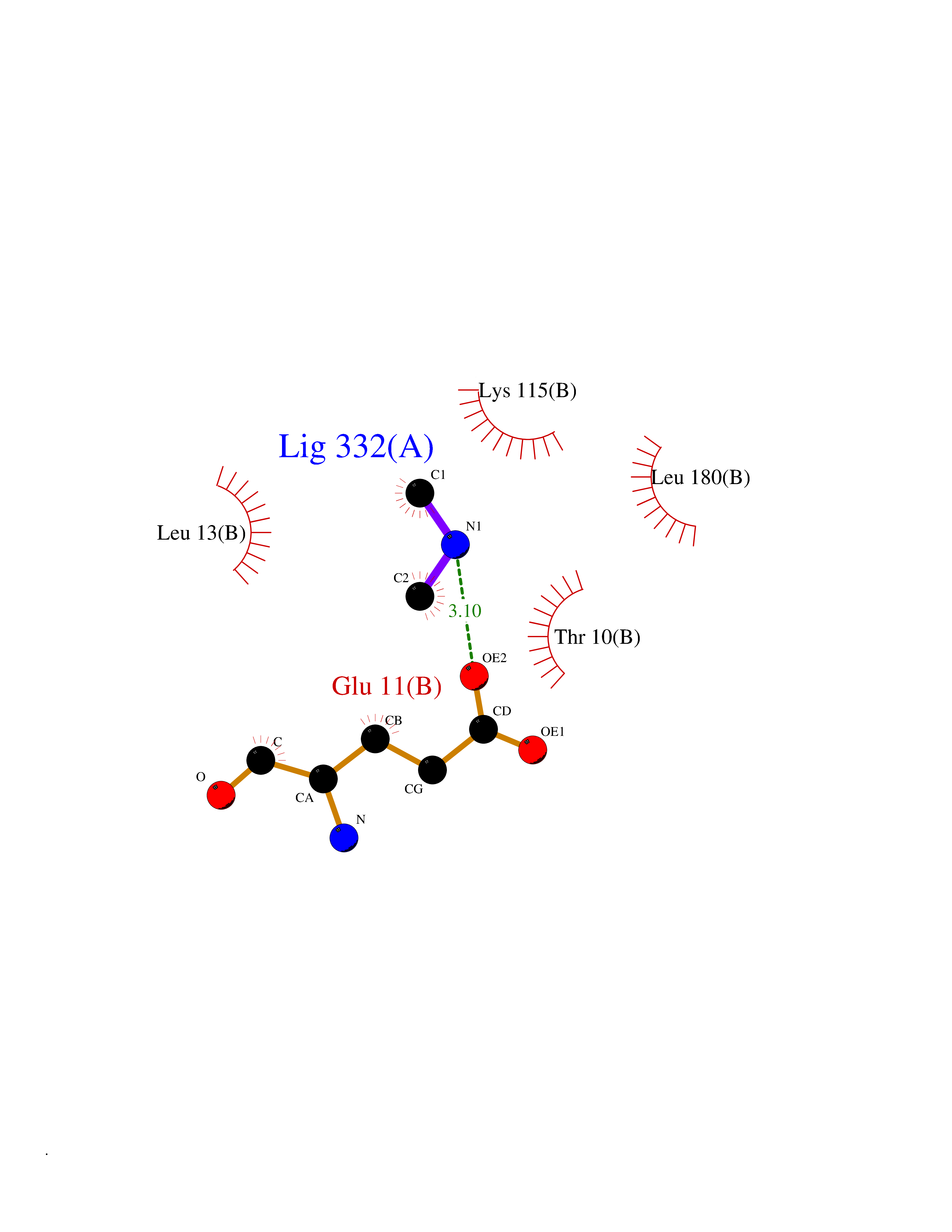 Ligplot Map 3D Binding mode Sequence HFFEGTEKLLEVWFSRQGSGDLRTIPRSEWDILLKDVQCSIISVTKTDKQEAYVLSESSMFVSKRRFILKTCGTTLLLKALVPLLKLARDYSGFDSIQSFFYSRKNFMKPSHQGYPHRNFQEEIEFLNAIFPNGAGYCMGRMNSDCWYLYTLDFRVISQPDQTLEILMSELDPAVMDQFYMKDGVTAKDVTRESGIRDLIPGSVIDATMFNPCGYSMNGMKSDGTYWTIAITPEPEFSYVSFETNLSQTSYDDLIRKVVEVFKPGKFVTTLFVNQSSKCPQKIEGFKRLDCQSAMFNDYNFVFTSFAKKQQ Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Cytoplasmic aspartate aminotransferase (GOT1) | 3II0 | 4.00 | |
Target general information Gen name GOT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glutamate oxaloacetate transaminase-1; GOT1 Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Biosynthesis of L-glutamate from L-aspartate or L- cysteine. Important regulator of levels of glutamate, the major excitatory neurotransmitter of the vertebrate central nervous system. Acts as a scavenger of glutamate in brain neuroprotection. The aspartate aminotransferase activity is involved in hepatic glucose synthesis during development and in adipocyte glyceroneogenesis. Using L-cysteine as substrate, regulates levels of mercaptopyruvate, an important source of hydrogen sulfide. Mercaptopyruvate is converted into H(2)S via the action of 3- mercaptopyruvate sulfurtransferase (3MST). Hydrogen sulfide is an important synaptic modulator and neuroprotectant in the brain. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00210; DB00128; DB09130; DB00151; DB00142; DB04299; DB00114 Interacts with NA EC number EC 2.6.1.1 Uniprot keywords 3D-structure; Alternative splicing; Amino-acid biosynthesis; Aminotransferase; Cytoplasm; Direct protein sequencing; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 51732.3 Length 462 Aromaticity 0.1 Instability index 31.83 Isoelectric point 7.48 Charge (pH=7) 0.86 2D Binding mode Binding energy (Kcal/mol) -5.46 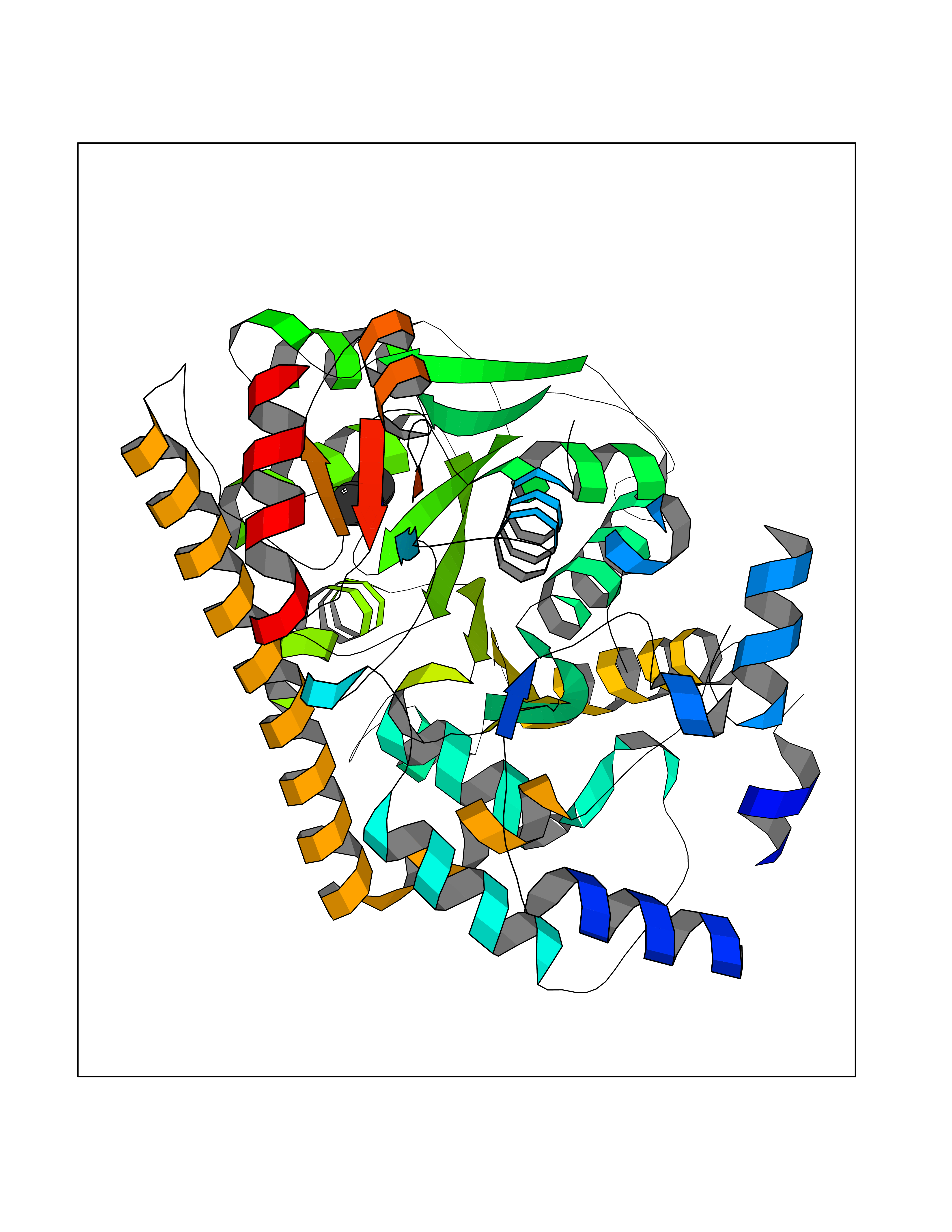 Molscript Map 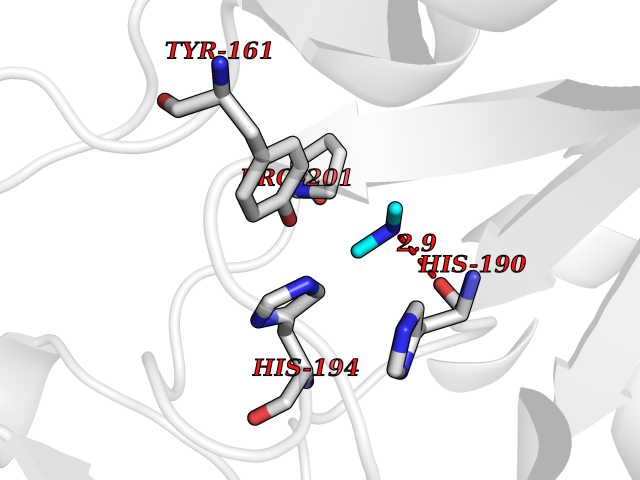 Pymol Map 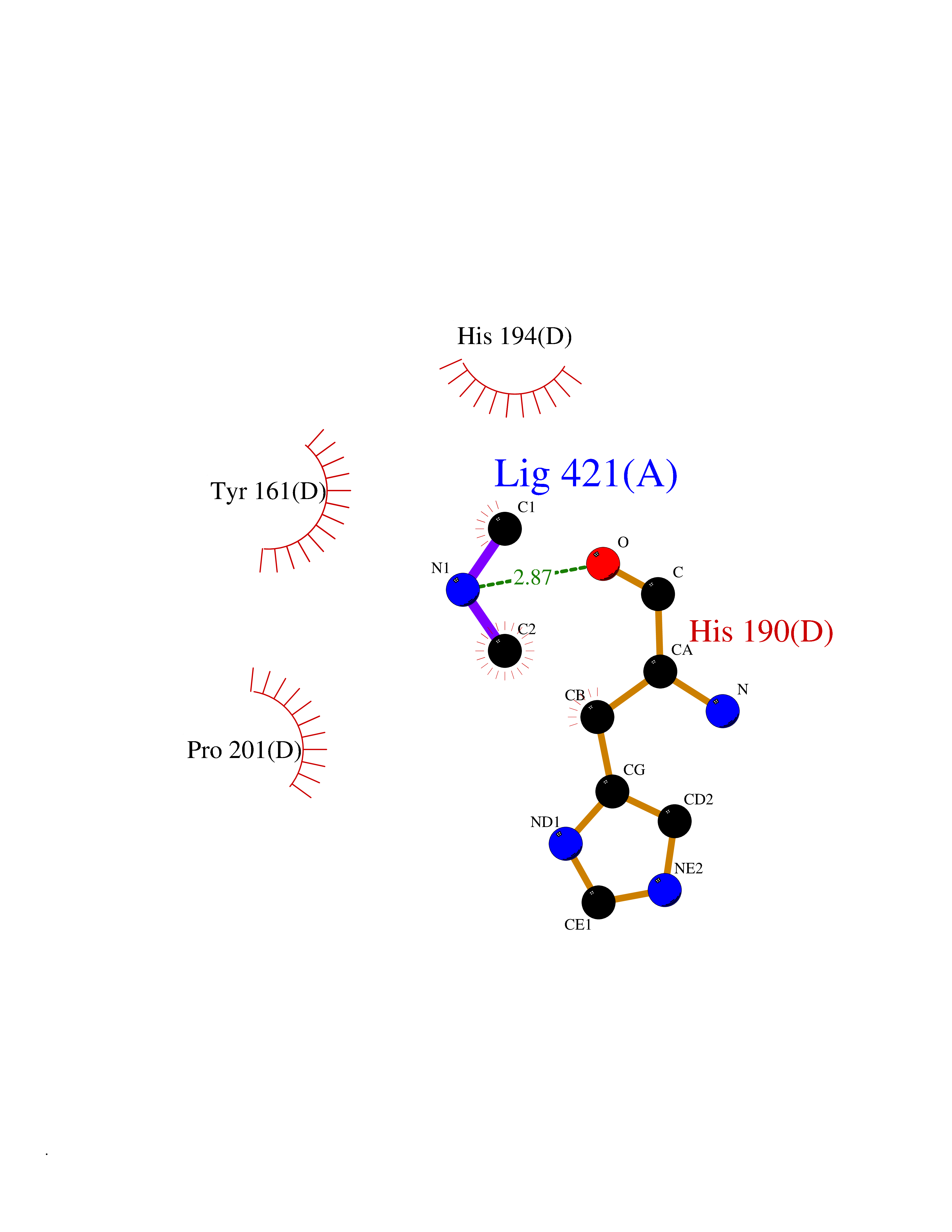 Ligplot Map 3D Binding mode Sequence PVLVFKLTVLPVVKKVEQKIANDNSLNHEYLPILGLAEFRVQSLGGTGALRIGADEKIVRITWSNPMQPVLVFKLTADFREDPDPRKVNLGVGAYRTDDCHPWVLPVVKKVEQKIANDNSLNHEYLPILGLAEFRSCASRLALGDDSPALKEKRVGGVQSLGGTGALRIGADFLARWYNGTNNKNTPVYVSSPTWENHNAVFSAAGFKDIRSYRYWDAEKRGLDLQGFLNDLENAPEFSIVVLHACAHNPTGIDPTPEQWKQIASVMKHRFLFPFFDSAYQGFASGNLERDAWAIRYFVSEGFEFFCAQSFSKNFGLYNERVGNLTVVGKEPESILQVLSQMEKIVRITWSNPPAQGARIVASTLSNPELFEEWTGNVKTMADRILTMRSELRARLEALKTPGTWNHITDQIGMFSFTGLNPKQVEYLVNEKHIYLLPSGRINVSGLTTKNLDYVATSIHEA Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Deoxycytidine kinase (DCK) | 1P5Z | 4.00 | |
Target general information Gen name DCK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms dCK Protein family DCK/DGK family Biochemical class Kinase Function Required for the phosphorylation of the deoxyribonucleosides deoxycytidine (dC), deoxyguanosine (dG) and deoxyadenosine (dA). Has broad substrate specificity, and does not display selectivity based on the chirality of the substrate. It is also an essential enzyme for the phosphorylation of numerous nucleoside analogs widely employed as antiviral and chemotherapeutic agents. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB02594; DB00242; DB00631; DB00987; DB01262; DB05494; DB00879; DB01073; DB00441; DB00709; DB01280; DB00642; DB04961; DB00943 Interacts with Q16854 EC number EC 2.7.1.74 Uniprot keywords 3D-structure; ATP-binding; Direct protein sequencing; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 27128.5 Length 229 Aromaticity 0.13 Instability index 52.5 Isoelectric point 5.26 Charge (pH=7) -7.8 2D Binding mode Binding energy (Kcal/mol) -5.46 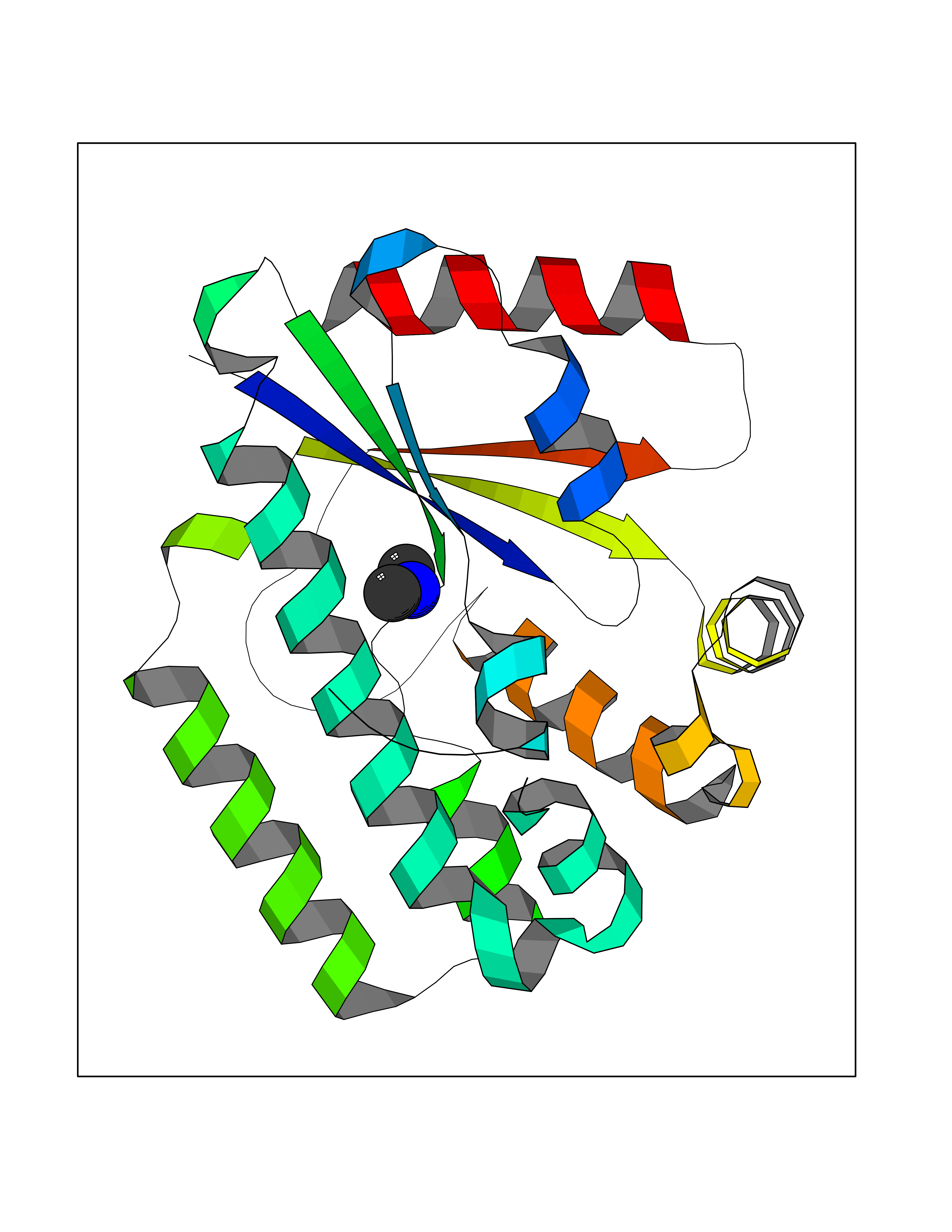 Molscript Map 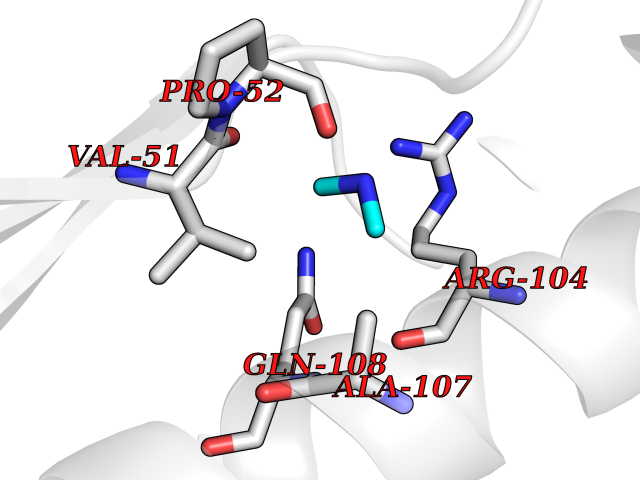 Pymol Map 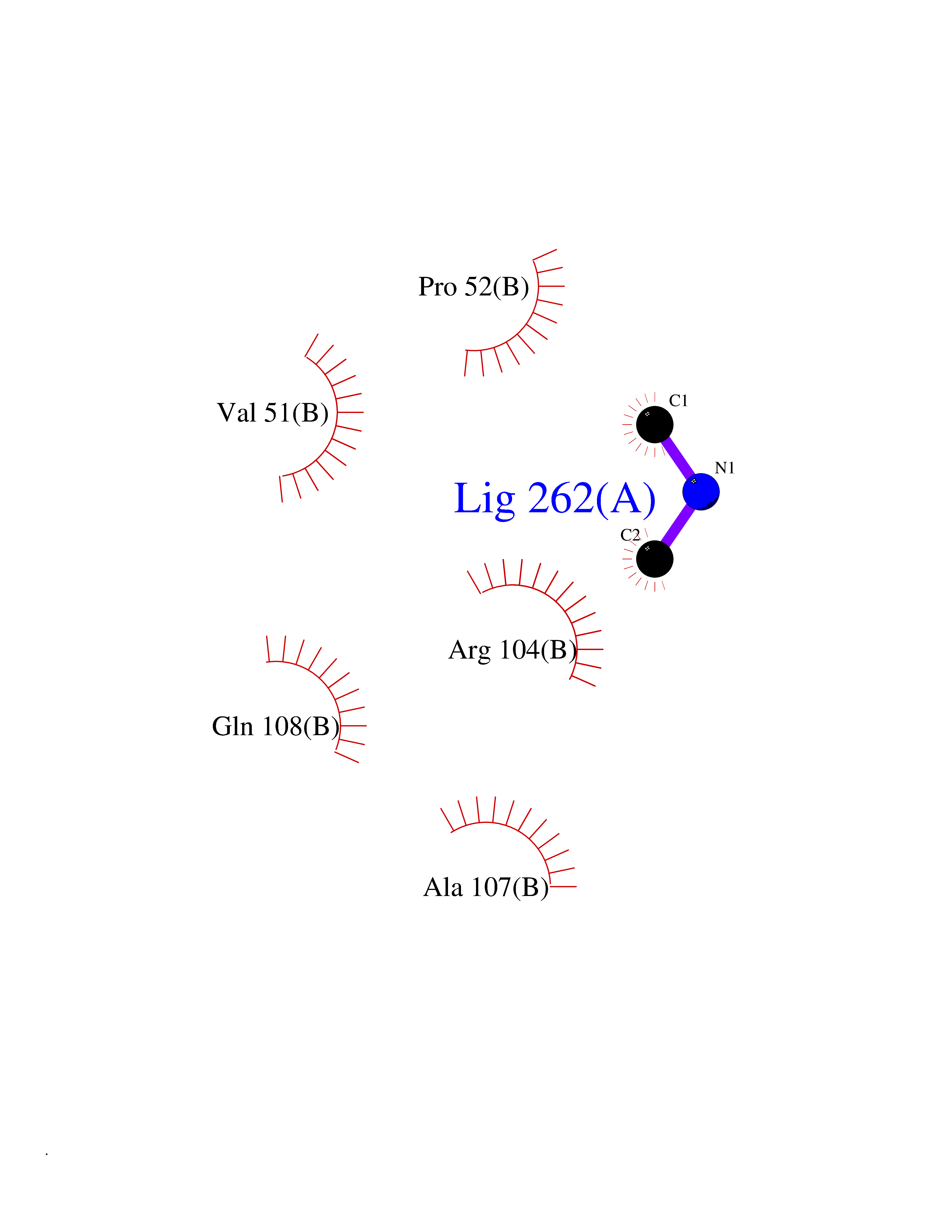 Ligplot Map 3D Binding mode Sequence RIKKISIEGNIAAGKSTFVNILKQLCEDWEVVPEPVARWCNVQSTNGGNVLQMMYEKPERWSFTFQTYACLSRIRAQLASLNGKLKDAEKPVLFFERSVYSDRYIFASNLYESECMNETEWTIYQDWHDWMNNQFGQSLELDGIIYLQATPETCLHRIYLRGRNEEQGIPLEYLEKLHYKHESWLLHRTLKTNFDYLQEVPILTLDVNEDFKDKYESLVEKVKEFLSTL Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Nitric-oxide synthase brain (NOS1) | 5ADF | 4.00 | |
Target general information Gen name NOS1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peptidyl-cysteine S-nitrosylase NOS1; Nitric oxide synthase, brain; Neuronal NOS; NOS, type I; NOS type I; NNOS; NC-NOS; N-NOS; BNOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function In the brain and peripheral nervous system, NO displays many properties of a neurotransmitter. Probably has nitrosylase activity and mediates cysteine S-nitrosylation of cytoplasmic target proteins such SRR. Produces nitric oxide (NO) which is a messenger molecule with diverse functions throughout the body. Related diseases Variation Asp-298 in NOS3 may be associated with susceptibility to coronary spasm. {ECO:0000269|PubMed:11740345, ECO:0000269|PubMed:9737779}. Drugs (DrugBank ID) DB02143; DB02727; DB01997; DB03892; DB02207; DB03710; DB00155; DB00843; DB00997; DB03147; DB03247; DB01942; DB01221; DB02077; DB01821; DB09241; DB03144; DB03449; DB02044; DB02644; DB08019; DB08018; DB02027; DB03461; DB04223; DB06096; DB02991; DB03707 Interacts with Q08AM6 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calmodulin-binding; Cell membrane; Cell projection; FAD; Flavoprotein; FMN; Heme; Iron; Membrane; Metal-binding; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Synapse; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34875.7 Length 299 Aromaticity 0.1 Instability index 42.94 Isoelectric point 5.96 Charge (pH=7) -6.25 2D Binding mode Binding energy (Kcal/mol) -5.45 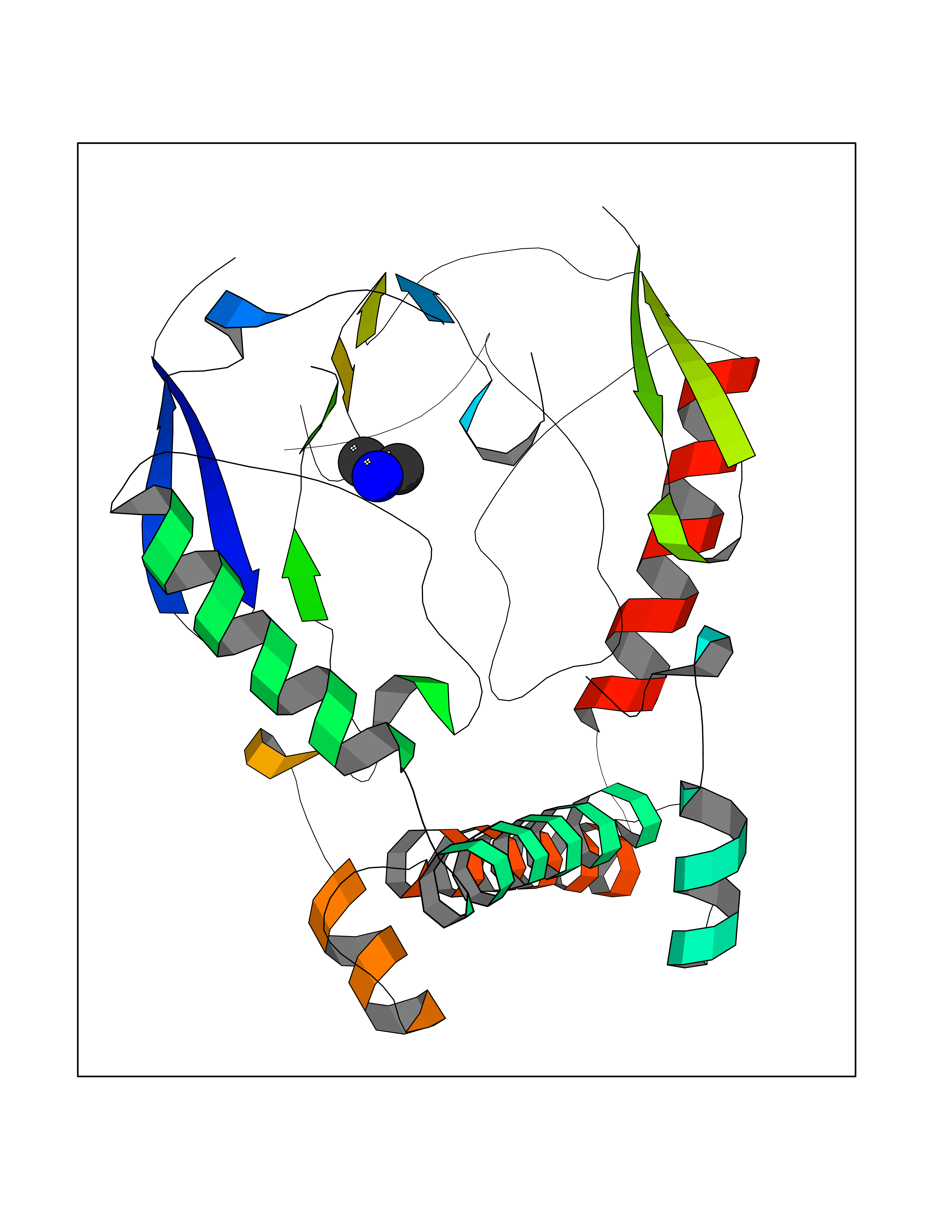 Molscript Map  Pymol Map 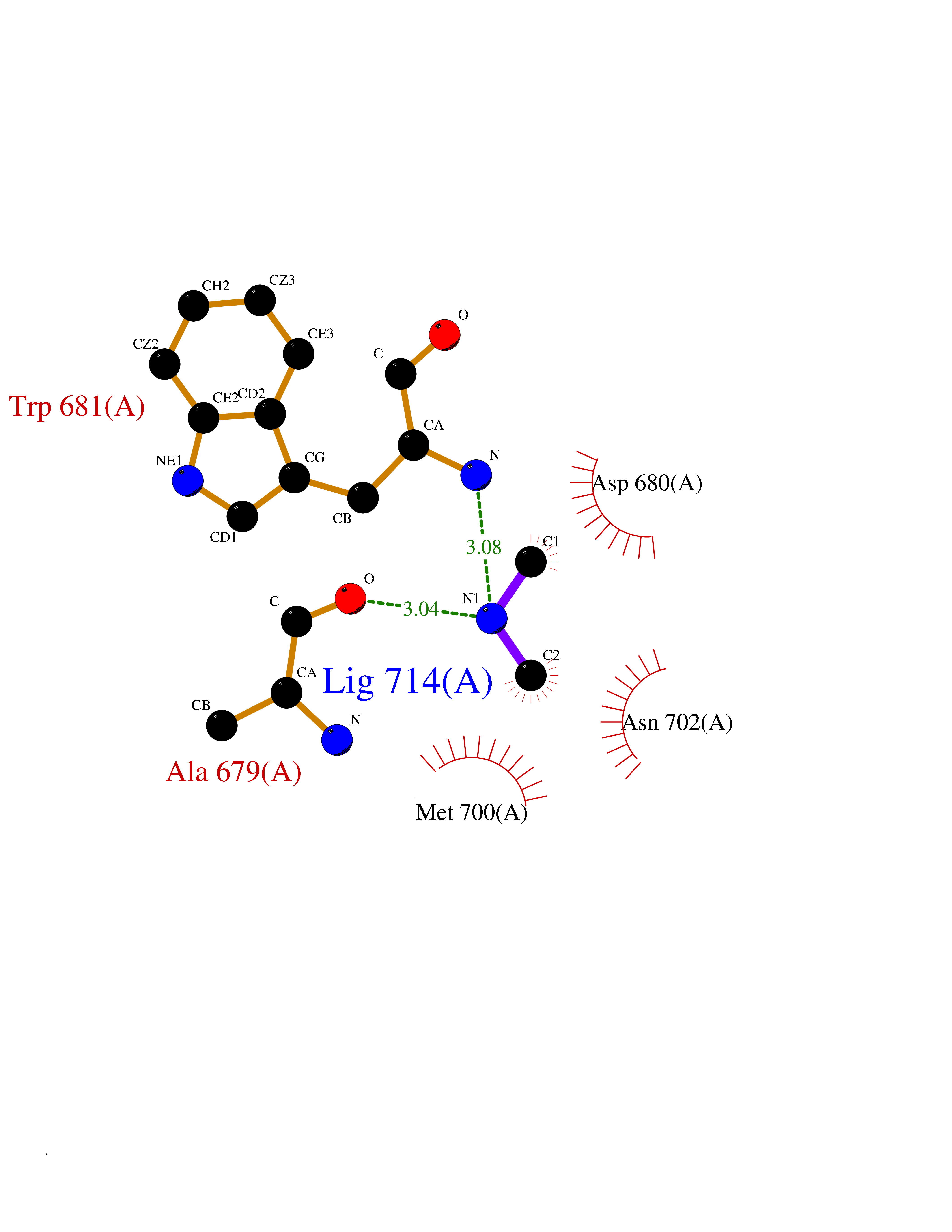 Ligplot Map 3D Binding mode Sequence CPRFLKVKNWETEVVLTDTLHLKSTLETGCTEYICMGSIMHPRDYCDNSRYNILEEVAKKMNLDMRKTSSLWKDQALVEINIAVLYSFQSDKVTIVDHHSATESFIKHMENEYRCRGGCPADWVWIVPPMSGSITPVFHQEMLNYRLRFLKVKNWETEVVLTDTLHLKSTLETGCTEYICMGSIMHPRDYCDNSRYNILEEVAKKMNLDMRKTSSLWKDQALVEINIAVLYSFQSDKVTIVDHHSATESFIKHMENEYRCRGGCPADWVWIVPPMSGSITPVFHQEMLNYRLTPSFEYQ Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Cytosolic purine 5'-nucleotidase | 2JC9 | 4.00 | |
Target general information Gen name NT5C2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PNT5;NT5B;NT5CP Protein family 5'(3')-deoxyribonucleotidase family Biochemical class Hydrolase Function 5'-nucleotidase activity.Metal ion binding.Nucleoside phosphotransferase activity.Nucleotide binding. Related diseases Spastic paraplegia 45, autosomal recessive (SPG45) [MIM:613162]: A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. Some SPG45 patients manifest intellectual disability, contractures and learning disability. {ECO:0000269|PubMed:24482476, ECO:0000269|PubMed:28884889}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00171; DB00811; DB06408 Interacts with P48047; P51116; Q8IVS8; Q7L9L4; Q86TA1; Q70IA8; Q9Y5B8; Q6ZVK8; O00560; Q9NRS6 EC number 2.7.1.77; 3.1.3.5; 3.1.3.99 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Cytoplasm; Disease variant; Hereditary spastic paraplegia; Hydrolase; Magnesium; Metal-binding; Neurodegeneration; Nucleotide metabolism; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 53978.4 Length 467 Aromaticity 0.14 Instability index 30.87 Isoelectric point 8.25 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -5.46 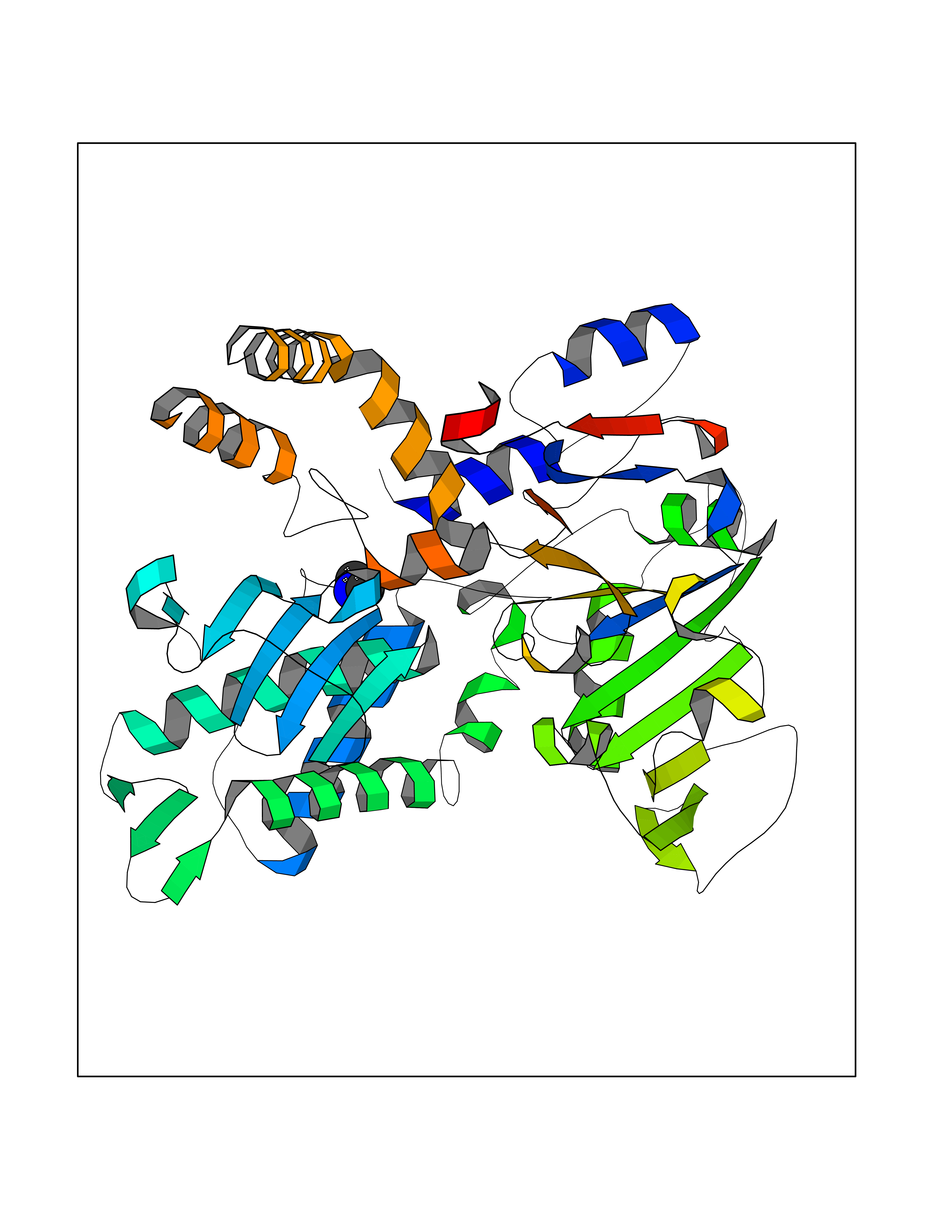 Molscript Map 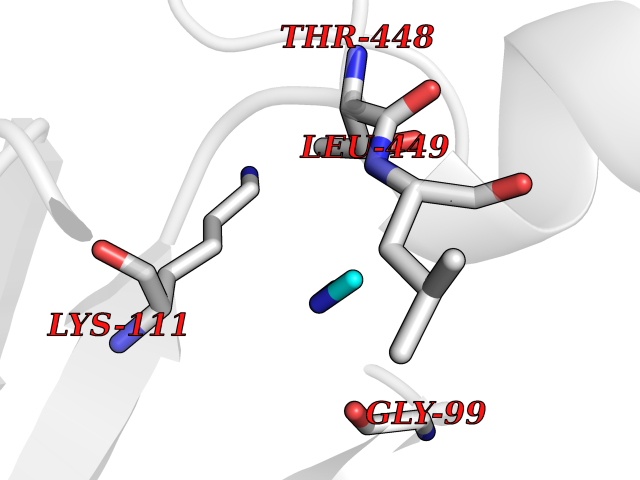 Pymol Map 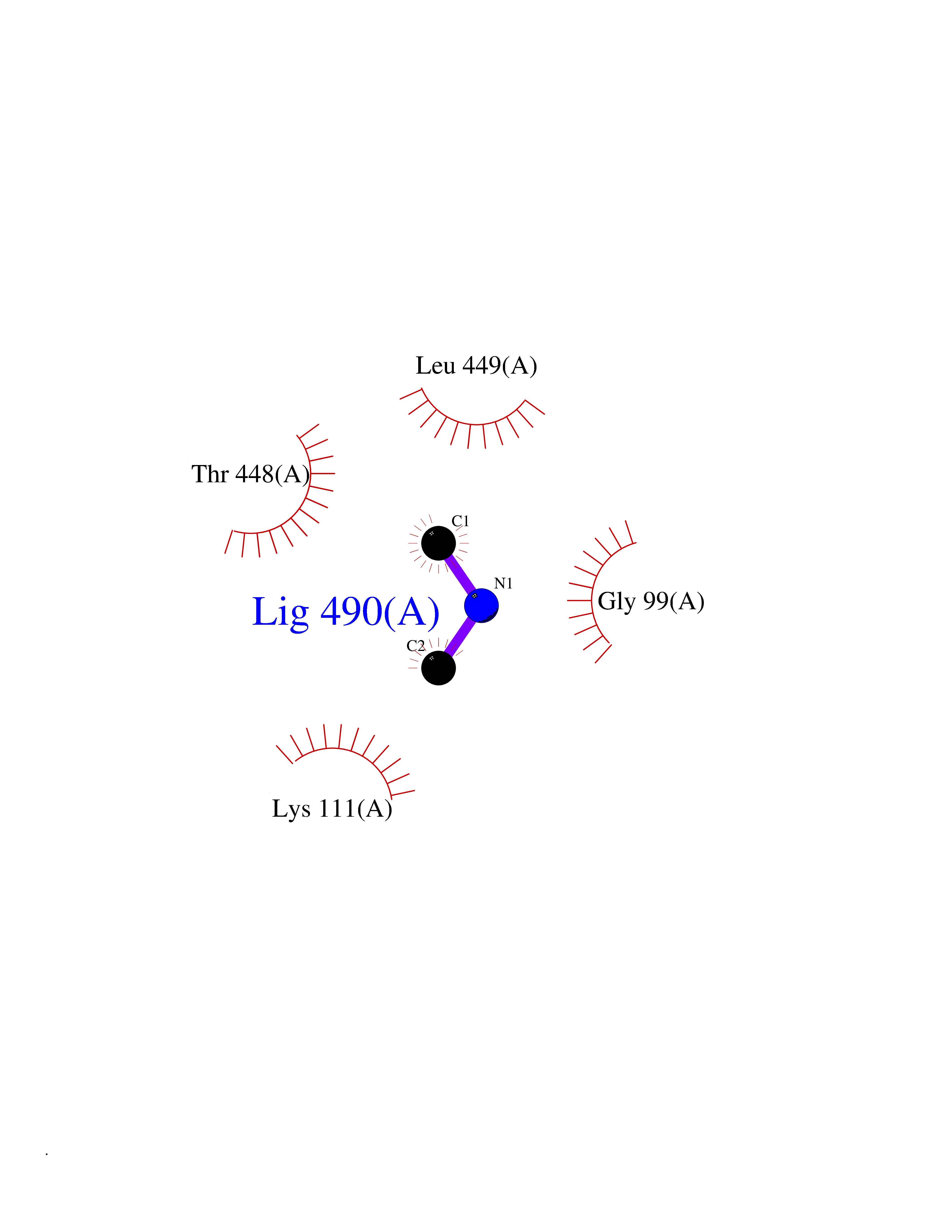 Ligplot Map 3D Binding mode Sequence TSWSDRLQNAADMPANMDKHALKKYRREAYHRVFVNRSLAMEKIKCFGFDMDYTLAVYKSPEYESLGFELTVERLVSIGYPQELLSFAYDSTFPTRGLVFDTLYGNLLKVDAYGNLLVCAHGFNFIRGPETREQYPNKFIQRDDTERFYILNTLFNLPETYLLACLVDFFTNCPRYTSCETGFKDGDLFMSYRSMFQDVRDAVDWVHYKGSLKEKTVENLEKYVVKDGKLPLLLSRMKEVGKVFLATNSDYKYTDKIMTYLFDFPHGPKPGSSHRPWQSYFDLILVDARKPLFFGEGTVLRQVDTKTGKLKIGTYTGPLQHGIVYSGGSSDTICDLLGAKGKDILYIGDHIFGDILKSKKRQGWRTFLVIPELAQELHVWTDKSSLFEELQSLDIFLAQRRIKKVTHDMDMCYGMMGSLFRSGSRQTLFASQVMRYADLYAASFINLLYYPFSYLFRAAHVLMPHES Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Oxygen-insensitive NAD(P)H nitroreductase | 1KQB | 4.00 | |
Target general information Gen name nfsB Organism Enterobacter cloacae Uniprot ID TTD ID NA Synonyms nfsI;nfnB Protein family Nitroreductase family Biochemical class Oxidoreductase Function Oxidoreductase activity. Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03793; DB03247 Interacts with NA EC number 1.-.-.- Uniprot keywords 3D-structure; Flavoprotein; FMN; NAD; NADP; Oxidoreductase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 47619.4 Length 432 Aromaticity 0.08 Instability index 38.43 Isoelectric point 5.52 Charge (pH=7) -12.98 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 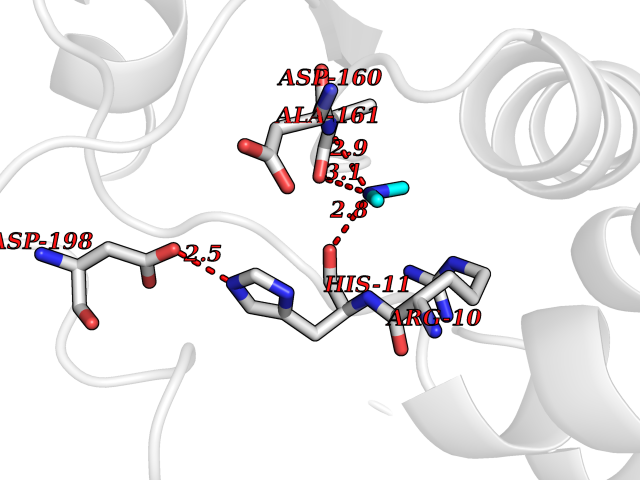 Pymol Map 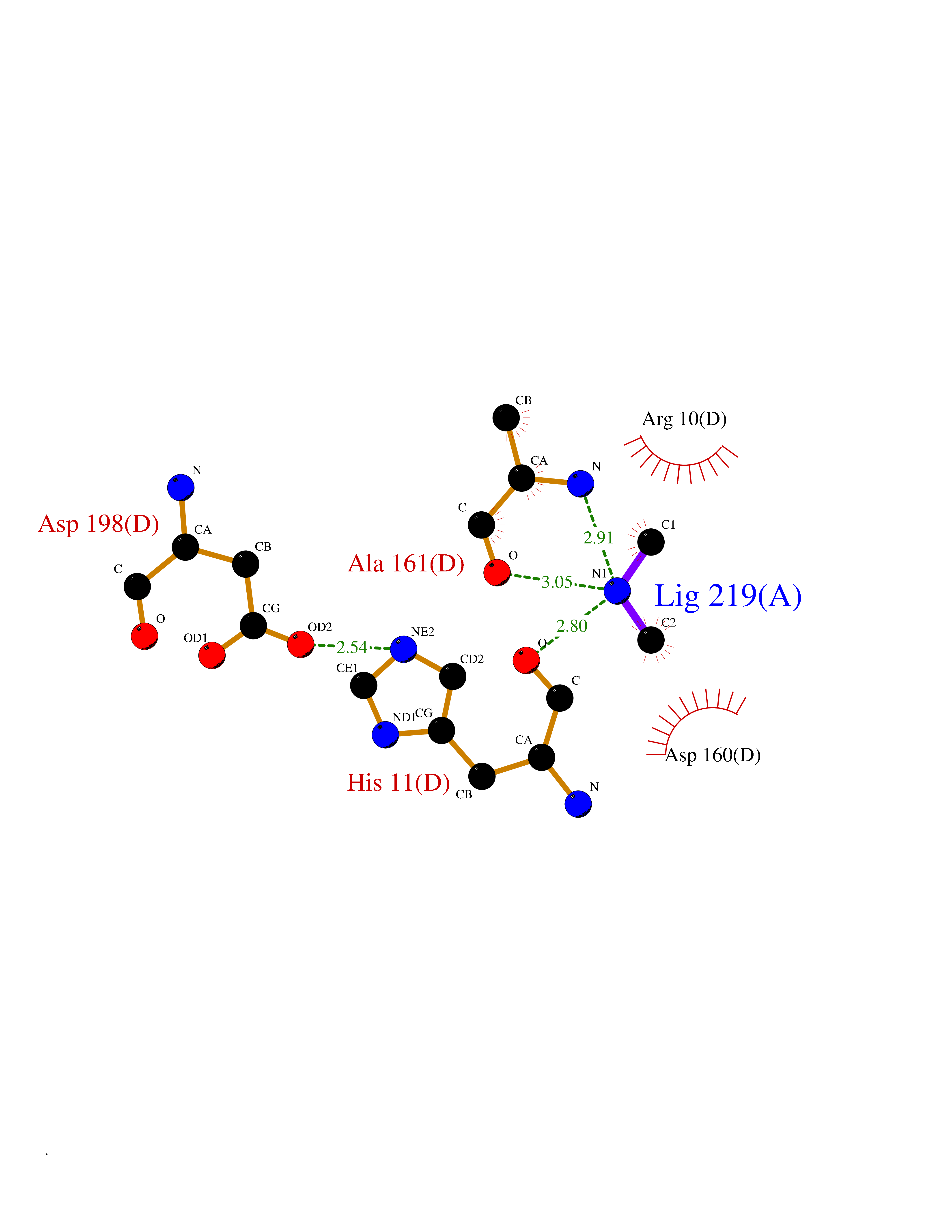 Ligplot Map 3D Binding mode Sequence DIISVALKRHSTKAFDASKKLTAEEAEKIKTLLQYSPSSTNSQPWHFIVASTEEGKARVAKSAAGTYVFNERKMLDASHVVVFCAKTAMDDAWLERVVDQEEADGRFNTPEAKAANHKGRTYFADMHRVDLKDDDQWMAKQVYLNVGNFLLGVGAMGLDAVPIEGFDAAILDEEFGLKEKGFTSLVVVPVGHHSVEDFNATLPKSRLPLSTIVTECDIISVALKRHSTKAFDASKKLTAEEAEKIKTLLQYSPSSTNSQPWHFIVASTEEGKARVAKSAAGTYVFNERKMLDASHVVVFCAKTAMDDAWLERVVDQEEADGRFNTPEAKAANHKGRTYFADMHRVDLKDDDQWMAKQVYLNVGNFLLGVGAMGLDAVPIEGFDAAILDEEFGLKEKGFTSLVVVPVGHHSVEDFNATLPKSRLPLSTIVTEC Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Aldehyde oxidoreductase | 4USA | 4.00 | |
Target general information Gen name mop Organism Megalodesulfovibrio gigas (Desulfovibrio gigas) Uniprot ID TTD ID NA Synonyms NA Protein family Xanthine dehydrogenase family Biochemical class Oxidoreductase Function 2 iron, 2 sulfur cluster binding.Aldehyde dehydrogenase (FAD-independent) activity.Electron carrier activity.Metal ion binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB02137 Interacts with NA EC number 1.2.99.7 Uniprot keywords 2Fe-2S; 3D-structure; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 96930.4 Length 907 Aromaticity 0.07 Instability index 29.17 Isoelectric point 5.69 Charge (pH=7) -17.56 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map  Pymol Map 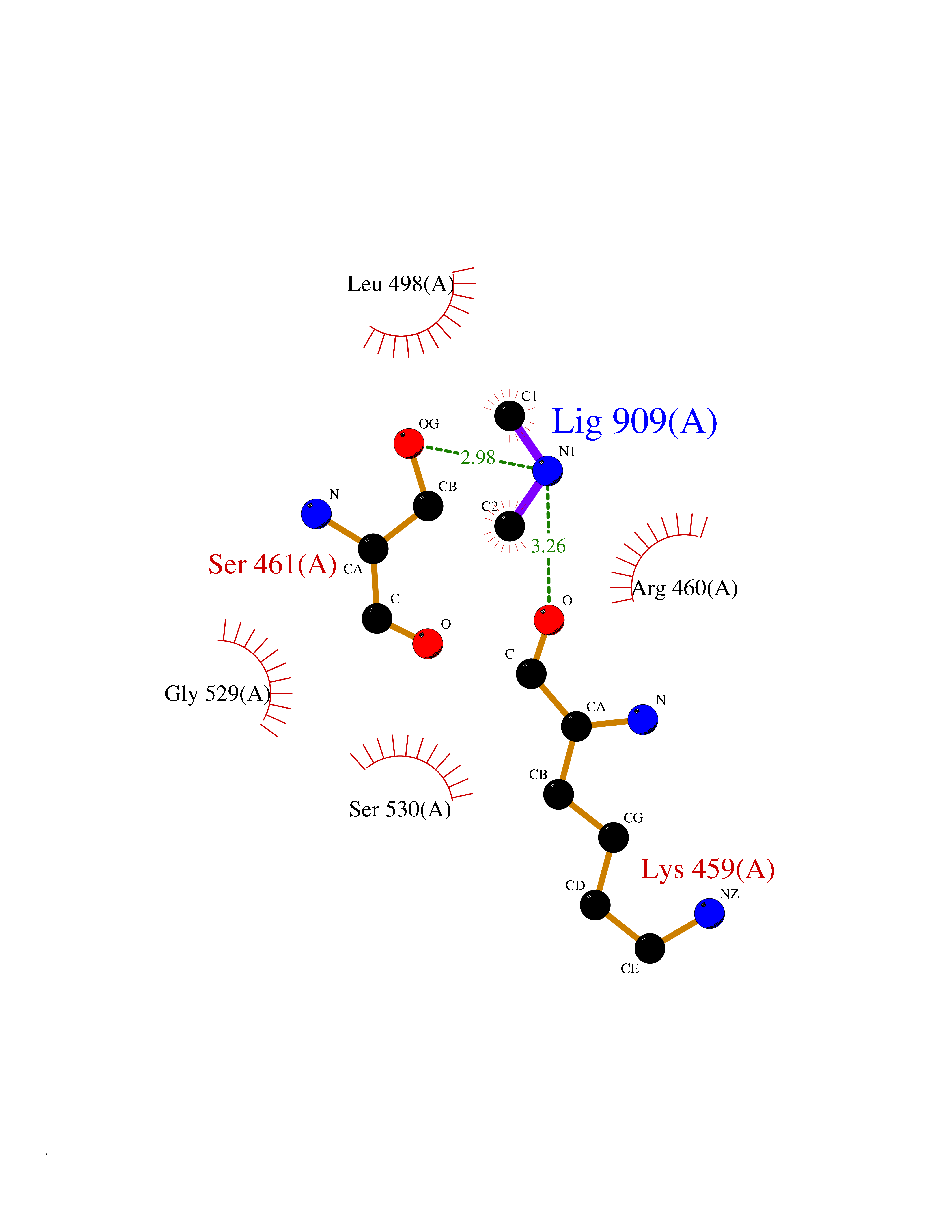 Ligplot Map 3D Binding mode Sequence MIQKVITVNGIEQNLFVDAEALLSDVLRQQLGLTGVKVGCEQGQCGACSVILDGKVVRACVTKMKRVADGAQITTIEGVGQPENLHPLQKAWVLHGGAQCGFCSPGFIVSAKGLLDTNADPSREDVRDWFQKHRNACRCTGYKPLVDAVMDAAAVINGKKPETDLEFKMPADGRIWGSKYPRPTAVAKVTGTLDYGADLGLKMPAGTLHLAMVQAKVSHANIKGIDTSEALTMPGVHSVITHKDVKGKNRITGLITFPTNKGDGWDRPILXDEKVFQYGDCIALVCADSEANARAAAEKVKVDLEELPAYMSGPAAAAEDAIEIHPGTPNVYFEQPIVKGEDTGPIFASADVTVEGDFYVGRQPHMPIEPDVAFAYMGDDGKCYIHSKSIGVHLHLYMIAPGVGLEPDQLVLVANPMGGTFGYKFSPTSEALVAVAAMATGRPVHLRYNYQQQQQYTGKRSPWEMNVKFAAKKDGTLLAMESDWLVDHGPYSEFGDLLTLRGAQFIGAGYNIPNIRGLGRTVATNHVWGSAFRGYGAPQSMFASECLMDMLAEKLGMDPLELRYKNAYRPGDTNPTGQEPEVFSLPDMIDQLRPKYQAALEKAQKESTATHKKGVGISIGVYGSGLDGPDASEAWAELNADGTITVHTAWEDHGQGADIGCVGTAHEALRPMGVAPEKIKFTWPNTATTPNSGPSGGSRQQVMTGNAIRVACENLLKACEKPGGGYYTYDELKAADKPTKITGNWTASGATHCDAVTGLGKPFVVYMYGVFMAEVTVDVATGQTTVDGMTLMADLGSLCNQLATDGQIYGGLAQGIGLALSEDFEDIKKHATLVGAGFPFIKQIPDKLDIVYVNHPRPDGPFGASGVGELPLTSPHAAIINAIKSATGVRIYRLPAYPEKVLEALKA Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Smoothened homolog (SMO) | 4JKV | 4.00 | |
Target general information Gen name SMO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Smo-D473H; SMOH; Protein Gx Protein family G-protein coupled receptor Fz/Smo family Biochemical class GPCR frizzled Function Binding of sonic hedgehog (SHH) to its receptor patched is thought to prevent normal inhibition by patched of smoothened (SMO). Required for the accumulation of KIF7, GLI2 and GLI3 in the cilia. Interacts with DLG5 at the ciliary base to induce the accumulation of KIF7 and GLI2 at the ciliary tip for GLI2 activation. G protein-coupled receptor that probably associates with the patched protein (PTCH) to transduce the hedgehog's proteins signal. Related diseases Curry-Jones syndrome (CRJS) [MIM:601707]: A multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. The disease is caused by variants affecting the gene represented in this entry. 8 individuals have been identified with the disease-causing mutation Phe-412 and all were mosaic. The mutation could not be reliably detected in blood, greatest success rates were obtained with affected tissues obtained by invasive procedures. It is thought that the mutation has arisen postzygotically early during embryonic development (PubMed:27236920). This mutation has also been identified in ameloblastoma, medulloblastoma, meningioma, and basal cell carcinoma, and has been reported as the oncogenic driver in some of these tumors (PubMed:24859340). {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. Drugs (DrugBank ID) DB01047; DB11978; DB06786; DB09143; DB08828 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; Developmental protein; Disease variant; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37420.1 Length 333 Aromaticity 0.16 Instability index 25.17 Isoelectric point 6.65 Charge (pH=7) -0.76 2D Binding mode Binding energy (Kcal/mol) -5.45 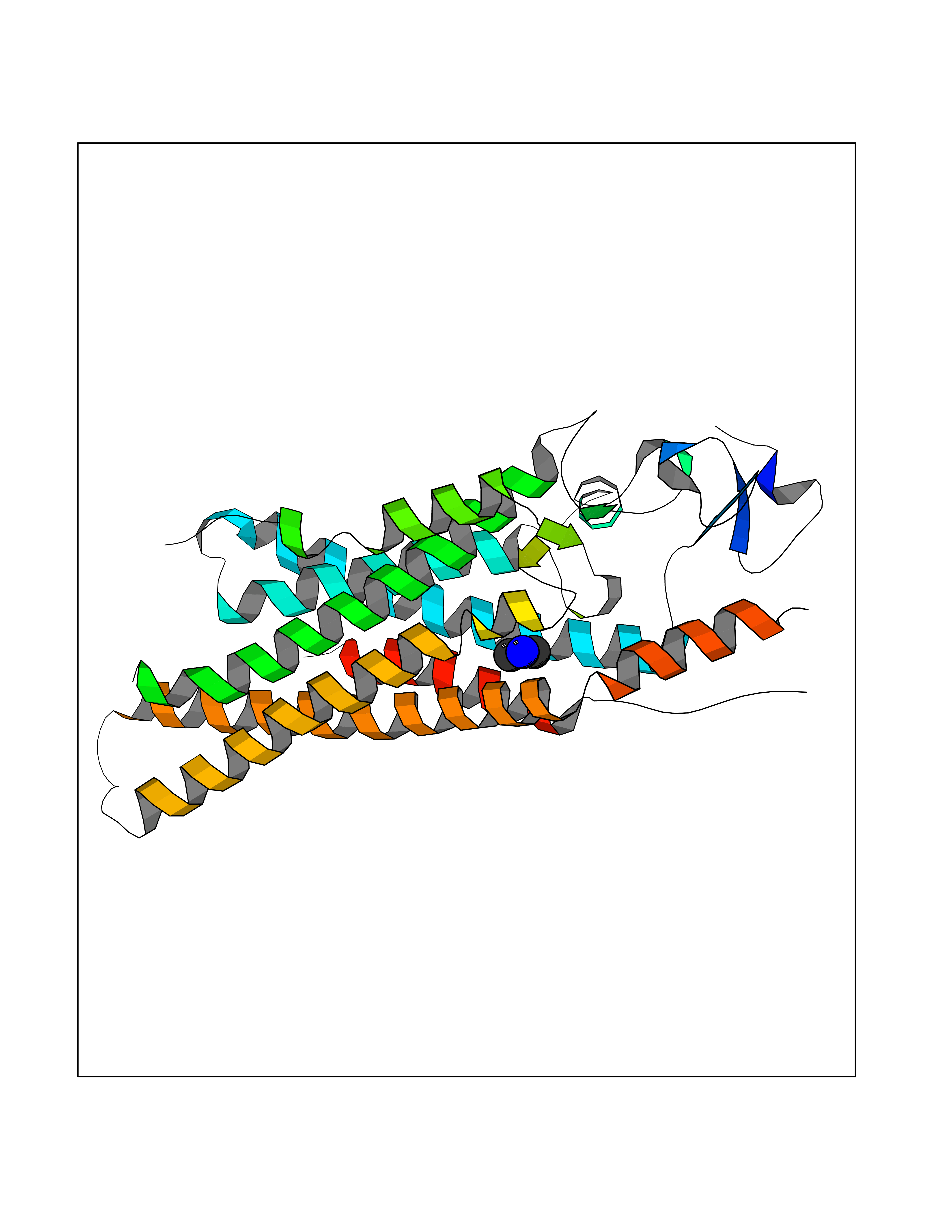 Molscript Map 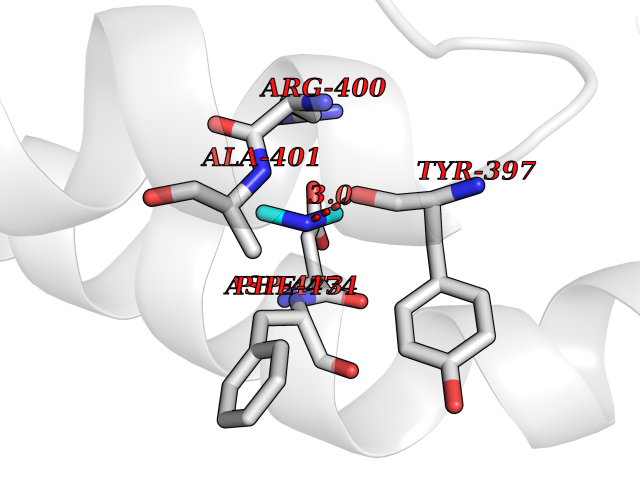 Pymol Map 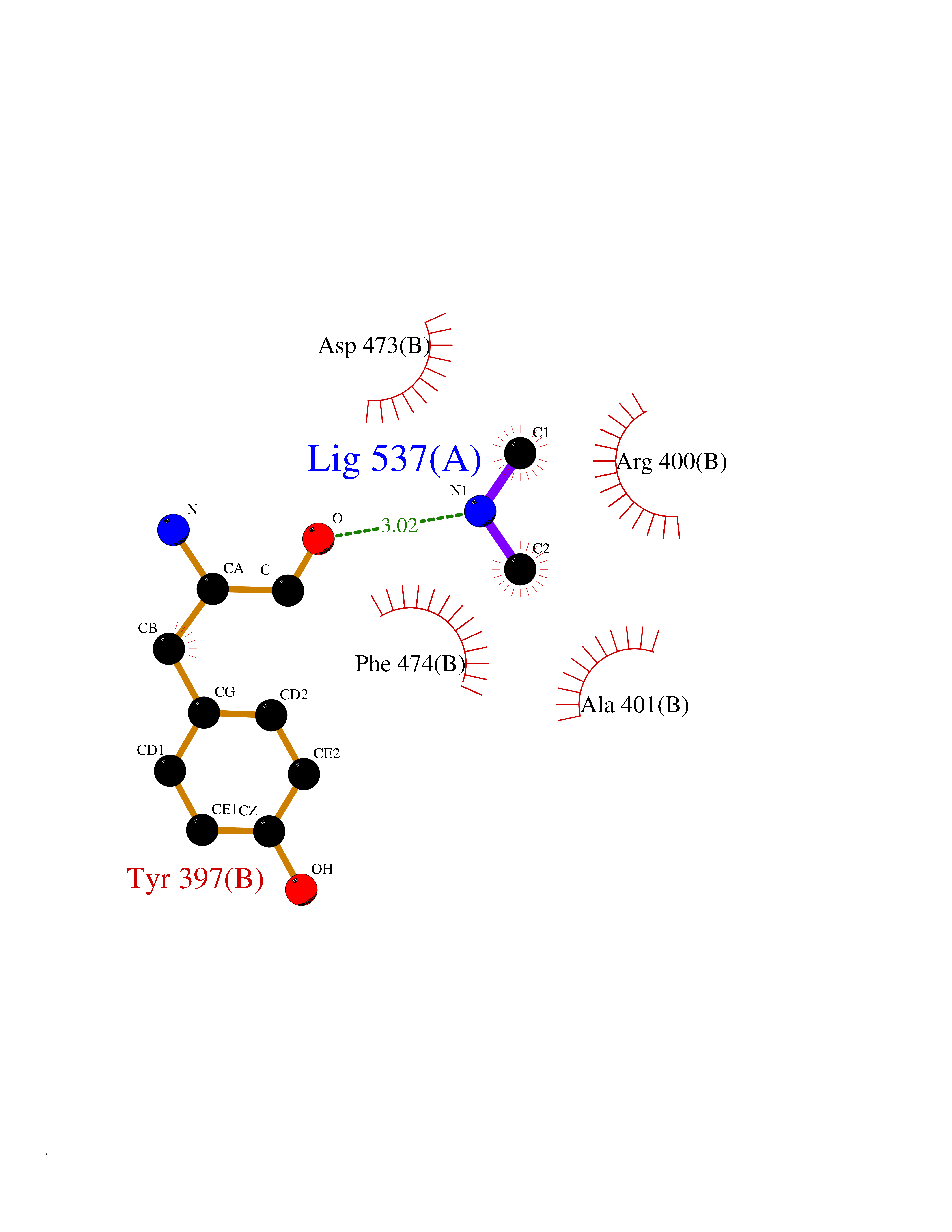 Ligplot Map 3D Binding mode Sequence AYIQKYLSGQCEVPLVRTDNPKSWYEDVEGCGIQCQNPLFTEAEHQDMHSYIAAFGAVTGLCTLFTLATFVADWRNSNRYPAVILFYVNACFFVGSIGWLAQFMDGARREIVCRADGTMRLGEPTSNETLSCVIIFVIVYYALMAGVVWFVVLTYAWHTSFKALGKTSYFHLLTWSLPFVLTVAILAVAQVDGDSVSGICFVGYKNYRYRAGFVLAPIGLVLIVGGYFLIRGVMTLFSIKSNHPGLLSEKAASKINETMLRLGIFGFLAFGFVLITFSCHFYDFFNQAEWERSFRDYVLCQANDCEIKNRPSLLVEKINLFAMFGTGIAMSTW Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Alanine aminotransferase 2 | 3IHJ | 4.00 | |
Target general information Gen name GPT2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AAT2;ALT2 Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family, Alanine aminotransferase subfamily Biochemical class Transferase Function L-alanine:2-oxoglutarate aminotransferase activity.Pyridoxal phosphate binding. Related diseases Neurodevelopmental disorder with spastic paraplegia and microcephaly (NEDSPM) [MIM:616281]: An autosomal recessive syndrome characterized by severe psychomotor developmental delay, dysarthria, walking difficulties, moderately to severely impaired intellectual development, poor or absent speech, and progressive microcephaly. {ECO:0000269|PubMed:25758935}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00160; DB00142; DB00780; DB00114 Interacts with NA EC number 2.6.1.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminotransferase; Disease variant; Intellectual disability; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 48717.7 Length 436 Aromaticity 0.09 Instability index 47.17 Isoelectric point 6.07 Charge (pH=7) -4.32 2D Binding mode Binding energy (Kcal/mol) -5.45 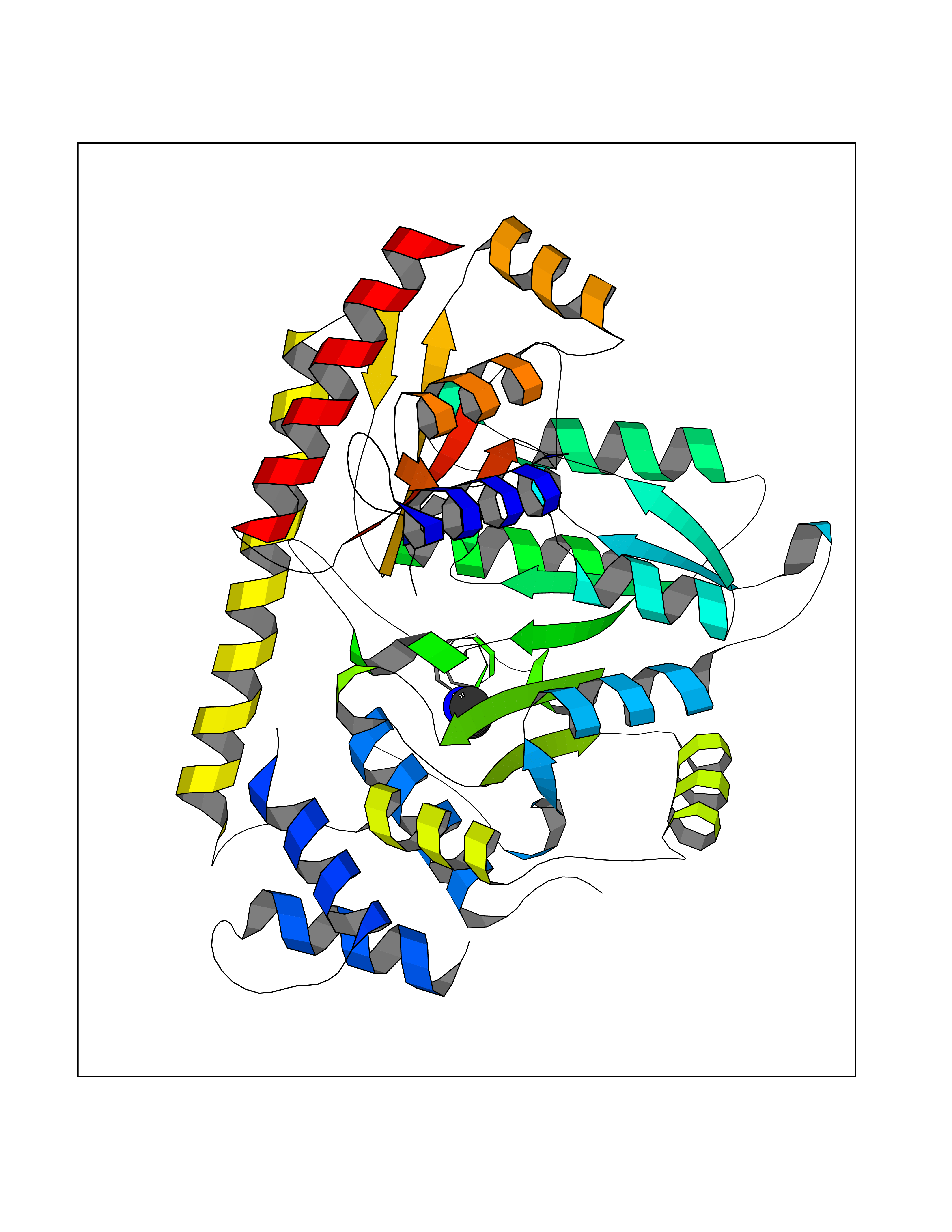 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PIVLKAGEIELELQRGIKKPFTEVIRANPITFLRQVMALCTYPNLLDSPSFPEDAKKRARRILQACSQGVNCIREDVAAYITRRDGGVPADPDNIYLTTGASDGISTILKILVSGGGKSRTGVMIPIPQYPLYSAVISELDAIQVNYYLDEENCWALNVNELRRAVQEAKDHCDPKVLCIINPGNPTGQVQSRKCIEDVIHFAWEEKLFLLADEVYQDNVYSPDCRFHSFKKVLYEMGPEYSSNVELASFHSTSKGYMGECGYRGGYMEVINLHPEIKGQLVKLLSVRLCPPVSGQAAMDIVVNPPVAGEESFEQFSREKESVLGNLAKKAKLTEDLFNQVPGIHCNPLQGAMYAFPRIFIPAKAVEAAQAHQMAPDMFYCMKLLEETGICVVPGSGFGQREGTYHFRMTILPPVEKLKTVLQKVKDFHINFLEKY Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | MAPK-activated protein kinase 2 (MAPKAPK2) | 1NXK | 4.00 | |
Target general information Gen name MAPKAPK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MK2; MK-2; MAPKactivated protein kinase 2; MAPKAPK-2; MAPKAP-K2; MAPKAP kinase 2; MAP kinaseactivated protein kinase 2; MAP kinase-activated protein kinase 2 Protein family Protein kinase superfamily, CAMK Ser/Thr protein kinase family Biochemical class Kinase Function Following stress, it is phosphorylated and activated by MAP kinase p38-alpha/MAPK14, leading to phosphorylation of substrates. Phosphorylates serine in the peptide sequence, Hyd-X-R-X(2)-S, where Hyd is a large hydrophobic residue. Phosphorylates ALOX5, CDC25B, CDC25C, CEP131, ELAVL1, HNRNPA0, HSP27/HSPB1, KRT18, KRT20, LIMK1, LSP1, PABPC1, PARN, PDE4A, RCSD1, RPS6KA3, TAB3 and TTP/ZFP36. Phosphorylates HSF1; leading to the interaction with HSP90 proteins and inhibiting HSF1 homotrimerization, DNA-binding and transactivation activities. Mediates phosphorylation of HSP27/HSPB1 in response to stress, leading to the dissociation of HSP27/HSPB1 from large small heat-shock protein (sHsps) oligomers and impairment of their chaperone activities and ability to protect against oxidative stress effectively. Involved in inflammatory response by regulating tumor necrosis factor (TNF) and IL6 production post-transcriptionally: acts by phosphorylating AU-rich elements (AREs)-binding proteins ELAVL1, HNRNPA0, PABPC1 and TTP/ZFP36, leading to the regulation of the stability and translation of TNF and IL6 mRNAs. Phosphorylation of TTP/ZFP36, a major post-transcriptional regulator of TNF, promotes its binding to 14-3-3 proteins and reduces its ARE mRNA affinity, leading to inhibition of dependent degradation of ARE-containing transcripts. Phosphorylates CEP131 in response to cellular stress induced by ultraviolet irradiation which promotes binding of CEP131 to 14-3-3 proteins and inhibits formation of novel centriolar satellites. Also involved in late G2/M checkpoint following DNA damage through a process of post-transcriptional mRNA stabilization: following DNA damage, relocalizes from nucleus to cytoplasm and phosphorylates HNRNPA0 and PARN, leading to stabilization of GADD45A mRNA. Involved in toll-like receptor signaling pathway (TLR) in dendritic cells: required for acute TLR-induced macropinocytosis by phosphorylating and activating RPS6KA3. Stress-activated serine/threonine-protein kinase involved in cytokine production, endocytosis, reorganization of the cytoskeleton, cell migration, cell cycle control, chromatin remodeling, DNA damage response and transcriptional regulation. Related diseases Erythrocytosis, familial, 4 (ECYT4) [MIM:611783]: An autosomal dominant disorder characterized by elevated serum hemoglobin and hematocrit, and normal platelet and leukocyte counts. {ECO:0000269|PubMed:18184961, ECO:0000269|PubMed:18378852, ECO:0000269|PubMed:19208626, ECO:0000269|PubMed:22367913}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07430; DB07431; DB07406; DB08358; DB07728; DB07234; DB02010 Interacts with Q00613; P04792; Q16539; Q9QWH1; P47811 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; DNA damage; Isopeptide bond; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32100.5 Length 290 Aromaticity 0.1 Instability index 43.53 Isoelectric point 8.76 Charge (pH=7) 5.44 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map  Pymol Map 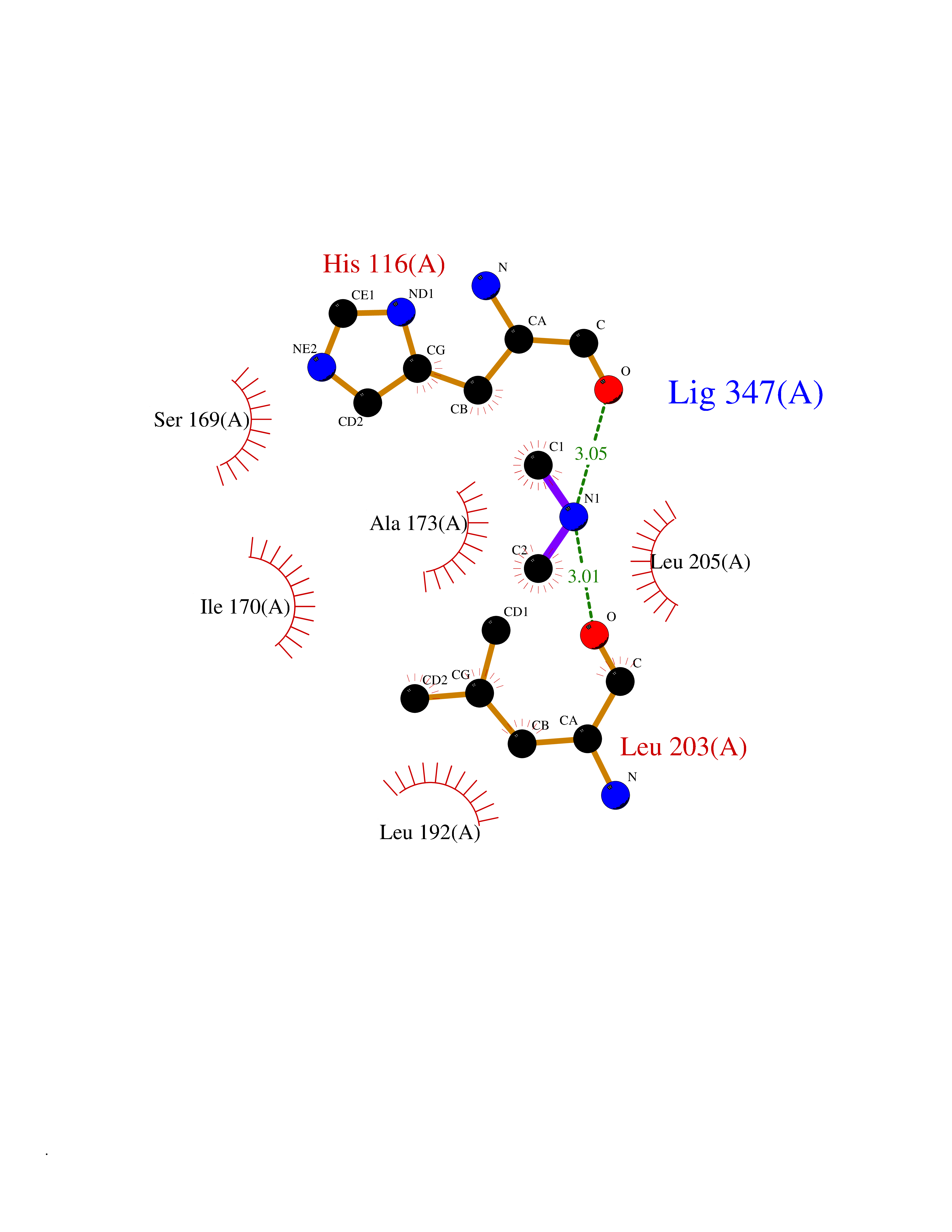 Ligplot Map 3D Binding mode Sequence QFPQFHVKSGLQIKKNAIIDDYKVTSQVLGLGINGKVLQIFNKRTQEKFALKXLQDCPKARREVELHWRASQCPHIVRIVDVYENLYAGRKCLLIVXECLDGGELFSRIQDRAFTEREASEIXKSIGEAIQYLHSINIAHRDVKPENLLYTSKRPNAILKLTDFGFAKETTPYYVAPEVLGPEKYDKSCDXWSLGVIXYILLCGYPPFYSNHGLAISPGXKTRIRXGQYEFPNPEWSEVSEEVKXLIRNLLKTEPTQRXTITEFXNHPWIXQSTKVPQTPLHTSRVLKED Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | N-acetylmannosamine kinase (GNE) | 4ZHT | 4.00 | |
Target general information Gen name GNE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UDPGlcNAc2epimerase/ManAc kinase; GNE; Bifunctional UDPNacetylglucosamine 2epimerase/Nacetylmannosamine kinase Protein family UDP-N-acetylglucosamine 2-epimerase family; ROK (NagC/XylR) family Biochemical class Kinase Function Regulates and initiates biosynthesis of N- acetylneuraminic acid (NeuAc), a precursor of sialic acids. Plays an essential role in early development. Required for normal sialylation in hematopoietic cells. Sialylation is implicated in cell adhesion, signal transduction, tumorigenicity and metastatic behavior of malignant cells. {ECO:0000250, ECO:0000269|PubMed:10334995}. Related diseases Sialuria (SIALURIA) [MIM:269921]: In sialuria, free sialic acid accumulates in the cytoplasm and gram quantities of neuraminic acid are secreted in the urine. The metabolic defect involves lack of feedback inhibition of UDP-GlcNAc 2-epimerase by CMP-Neu5Ac, resulting in constitutive overproduction of free Neu5Ac. Clinical features include variable degrees of developmental delay, coarse facial features and hepatomegaly. Sialuria inheritance is autosomal dominant. {ECO:0000269|PubMed:10330343, ECO:0000269|PubMed:10356312, ECO:0000269|PubMed:11326336, ECO:0000269|PubMed:2808337}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Nonaka myopathy (NM) [MIM:605820]: An autosomal recessive myopathy characterized by early adult onset and progressive distal muscle weakness that preferentially affects the anterior tibial muscles, usually sparing the quadriceps femoris. Some individuals may have involvement of the upper limbs or proximal muscles. Muscle biopsy reveals presence of rimmed vacuoles. {ECO:0000269|PubMed:11528398, ECO:0000269|PubMed:11916006, ECO:0000269|PubMed:12177386, ECO:0000269|PubMed:12325084, ECO:0000269|PubMed:12409274, ECO:0000269|PubMed:12473753, ECO:0000269|PubMed:12473769, ECO:0000269|PubMed:12473780, ECO:0000269|PubMed:12497639, ECO:0000269|PubMed:12811782, ECO:0000269|PubMed:12913203, ECO:0000269|PubMed:14707127, ECO:0000269|PubMed:15146476, ECO:0000269|PubMed:16503651}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombocytopenia 12 with or without myopathy (THC12) [MIM:620757]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC12 is an autosomal recessive form manifesting from infancy or early childhood with bleeding episodes. Clinical features include petechiae, easy bruising, epistaxis, hematomas, menorrhagia, and increased bleeding after trauma or surgery. Rare patients may have thrombocytopenia without bleeding. Some affected individuals have myopathic features, usually apparent in the second or third decades of life. {ECO:0000269|PubMed:25257349, ECO:0000269|PubMed:30171045, ECO:0000269|PubMed:33198675, ECO:0000269|PubMed:34788986, ECO:0000269|PubMed:34858435, ECO:0000269|PubMed:35052006, ECO:0000269|PubMed:38237079}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P12814; Q6UY14-3; Q969Y2; Q15323; P60370; P60409; P60410; P60411; Q9BQ66; P26371; Q9BYQ4; Q7Z3S9; O43597; Q6UY14-3; Q6P5X5; P27918; A8MQ03; Q16610; Q9UHF1; P28799; P49639; Q5T749; Q15323; O76011; Q6A162; P78385; P78386; O43790; Q07627; Q8IUG1; P60409; P60410; Q8IUC1; P60328; Q52LG2; Q3SY46; Q9BYP8; Q3LHN2; Q3SYF9; Q9BYR8; Q9BYR6; Q9BYQ7; Q9BYQ6; Q9BYR3; P26371; Q3LI64; Q3LI66; Q3LI67; Q9BYQ4; Q9BYQ3; Q9BYQ0; Q99750; Q8IV28; P0DPK4; O15496; O43609; O43610; P14373; Q8IWZ5; Q15654; O14817; Q2TAL6; Q9BRX9; O76024; Q9NZC7-5 EC number NA Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Cytoplasm; Disease variant; Hydrolase; Kinase; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 41589.2 Length 384 Aromaticity 0.07 Instability index 35.07 Isoelectric point 7.05 Charge (pH=7) 0.19 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map  Pymol Map 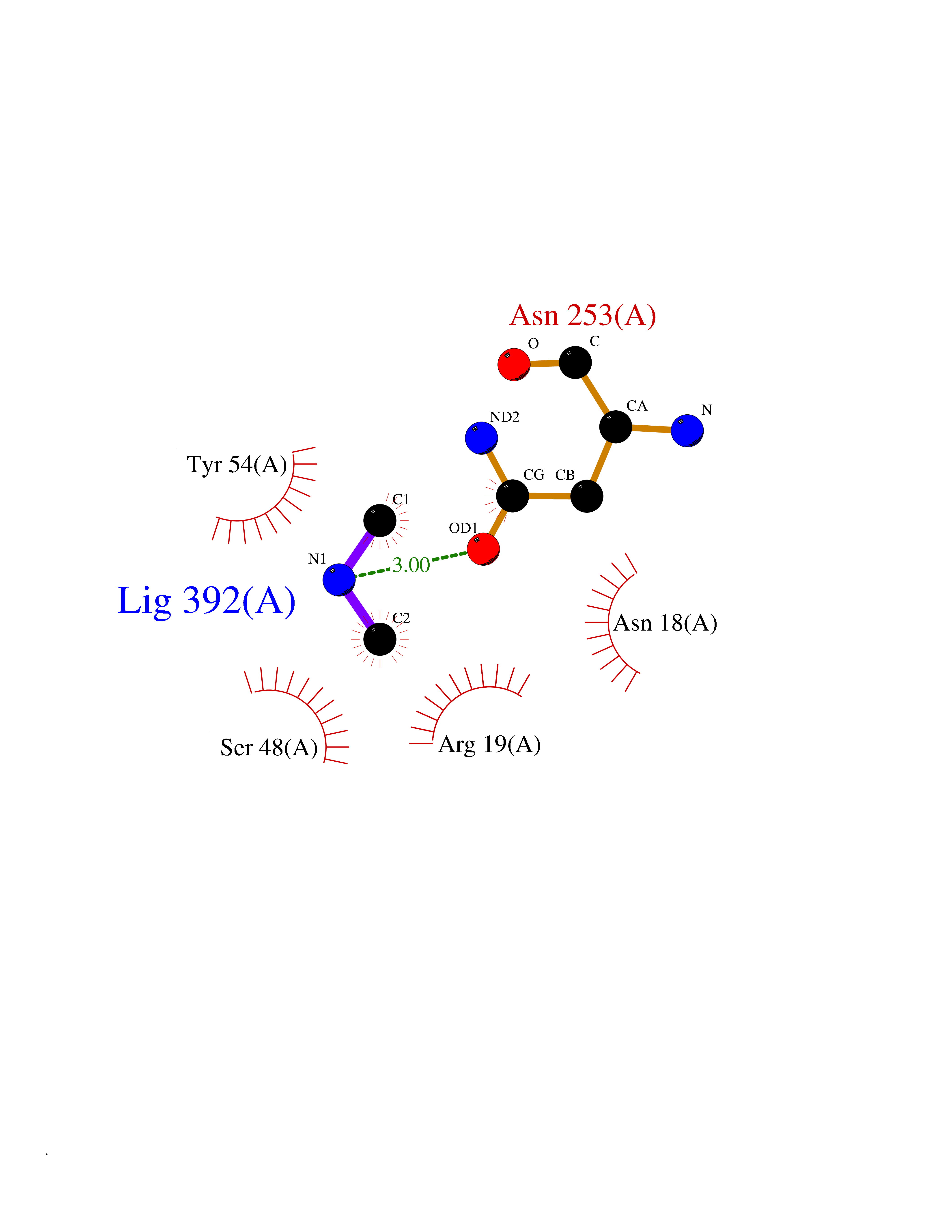 Ligplot Map 3D Binding mode Sequence NRKLRVCVATCNRADYSKLAPIXFGIKTEPEFFELDVVVLGSHLIDDYGNTYRXIEQDDFDINTRLHTIVRGEDEAAXVESVGLALVKLPDVLNRLKPDIXIVHGDRFDALALATSAALXNIRILHIEGGEVSGTIDDSIRHAITKLAHYHVCCTRSAEQHLISXCEDHDRILLAGCPSYDKLLSAKNKDYXSIIRXWLGDDVKSKDYIVALQHPVTTDIKHSIKXFELTLDALISFNKRTLVLFPNIDAGSKEXVRVXRKKGIEHHPNFRAVKHVPFDQFIQLVAHAGCXIGNSSCGVREVGAFGTPVINLGTRQIGRETGENVLHVRDADTQDKILQALHLQFGKQYPCSKIYGDGNAVPRILKFLKSIDLQEPLQKKFCFP Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Staphylococcus Peptide deformylase (Stap-coc def) | 1Q1Y | 4.00 | |
Target general information Gen name Stap-coc def Organism Staphylococcus aureus Uniprot ID TTD ID Synonyms def; Stap-coc Polypeptide deformylase Protein family Polypeptide deformylase family Biochemical class Carbon-nitrogen hydrolase Function Removes the formyl group from the N-terminal Met of newly synthesized proteins. Requires at least a dipeptide for an efficient rate of reaction. N-terminal L-methionine is a prerequisite for activity but the enzyme has broad specificity at other positions. Related diseases Immunodeficiency 43 (IMD43) [MIM:241600]: A disorder characterized by marked reduction in serum concentrations of immunoglobulins and albumin, and hypoproteinemia due to hypercatabolism. Patients may suffer from recurrent respiratory tract infections and severe skin disease. {ECO:0000269|PubMed:16549777}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Amyloidosis, hereditary systemic 6 (AMYLD6) [MIM:620659]: A form of hereditary systemic amyloidosis, a disorder characterized by amyloid deposition in multiple tissues resulting in a wide clinical spectrum. AMYLD6 is mainly characterized by gastrointestinal and cardiac symptoms. Neurologic involvement, sicca syndrome, and carpal tunnel syndrome may also be present. Inheritance is autosomal dominant. {ECO:0000269|PubMed:22693999}. The disease is caused by variants affecting the gene represented in this entry. Apart from the presence of causative mutations, beta-2-microglobulin may adopt the fibrillar configuration of amyloid, resulting in amyloidosis, when its serum levels are persistently high, as seen in patients on long-term hemodialysis (PubMed:7918443). In contrast to patients with dialysis-related amyloidosis, patients with hereditary amyloidosis have normal circulating concentrations of beta2-microglobulin (PubMed:22693999). {ECO:0000269|PubMed:22693999, ECO:0000269|PubMed:7918443}. Drugs (DrugBank ID) DB04310 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase; Iron; Metal-binding; Protein biosynthesis Protein physicochemical properties Chain ID A Molecular weight (Da) 20835.6 Length 186 Aromaticity 0.05 Instability index 42.9 Isoelectric point 5.65 Charge (pH=7) -7.59 2D Binding mode Binding energy (Kcal/mol) -5.45 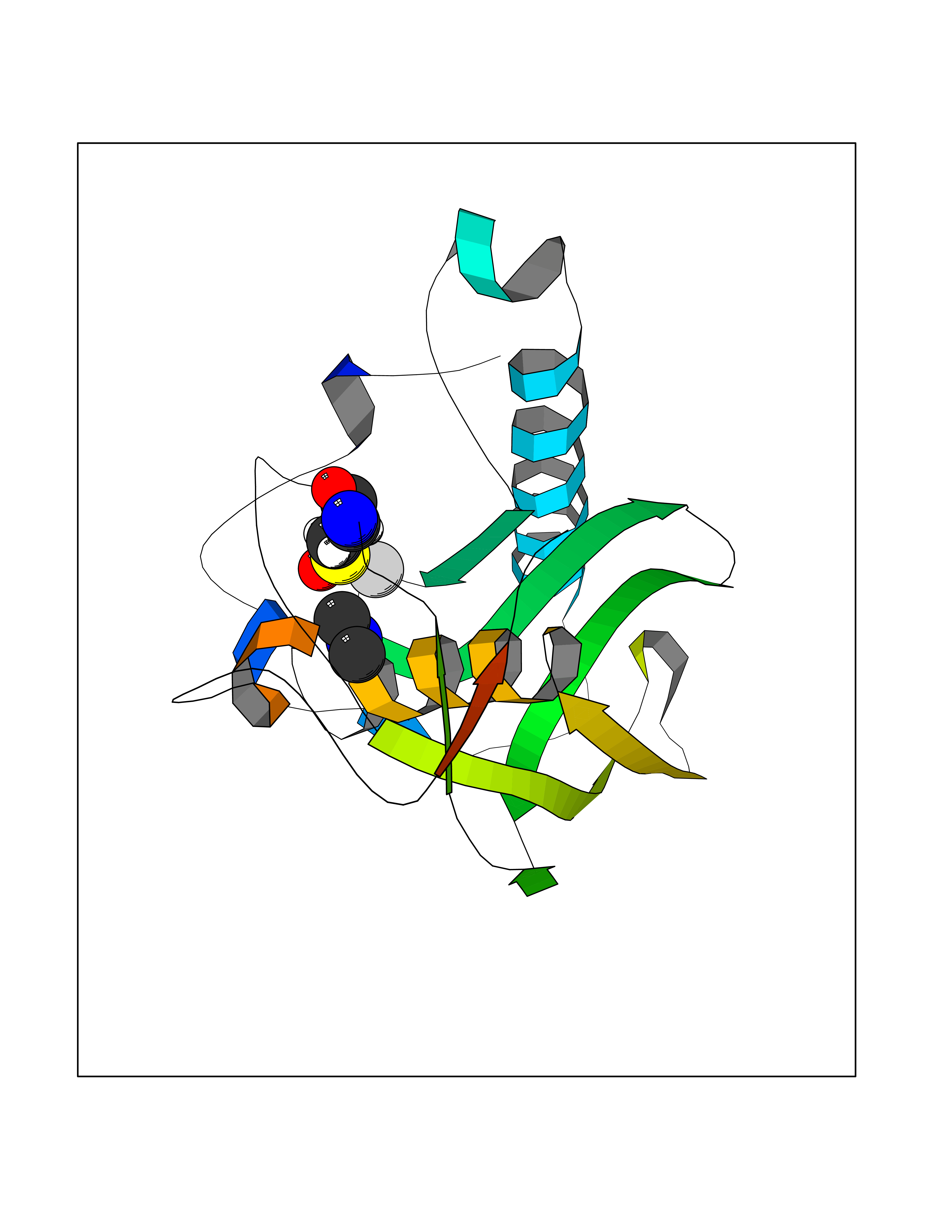 Molscript Map 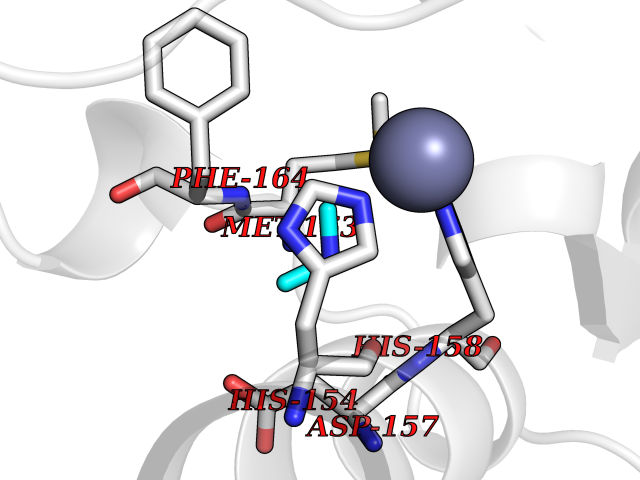 Pymol Map 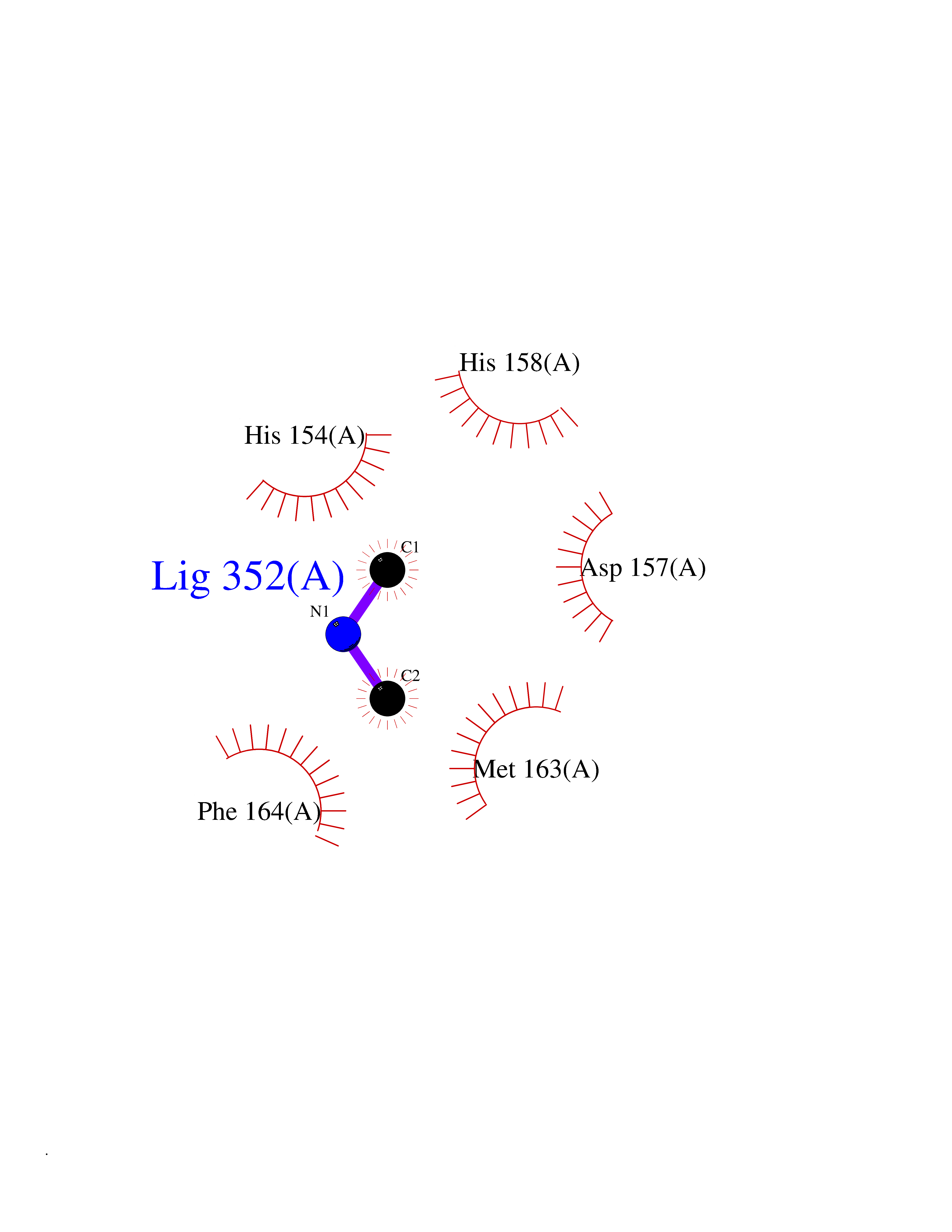 Ligplot Map 3D Binding mode Sequence MLTMKDIIRDGHPTLRQKAAELELPLTKEEKETLIAMREFLVNSQDEEIAKRYGLRSGVGLAAPQINISKRMIAVLIPDDGSGKSYDYMLVNPKIVSHSVQEAYLPTGEGXLSVDDNVAGLVHRHNRITIKAKDIEGNDIQLRLKGYPAIVFQHEIDHLNGVMFYDHIDKDHPLQPHTDAVEVLEH Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Tissue factor (F3) | 6R2W | 4.00 | |
Target general information Gen name F3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Thromboplastin; TF; F3; Coagulation factor III; CD142 antigen Protein family Tissue factor family Biochemical class NA Function Initiates blood coagulation by forming a complex with circulating factor VII or VIIa. The [TF:VIIa] complex activates factors IX or X by specific limited protolysis. TF plays a role in normal hemostasis by initiating the cell-surface assemblyand propagation of the coagulation protease cascade. Related diseases Intellectual developmental disorder, autosomal recessive 54 (MRT54) [MIM:617028]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT54 patients manifest intellectual disability, delayed speech and hyperactivity. {ECO:0000269|PubMed:27106596}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07207; DB07247; DB08232; DB06552; DB13150; DB00036; DB16732 Interacts with P55085; P08709; Q9UM19; Q92544 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Disulfide bond; Glycoprotein; Hemostasis; Lipoprotein; Membrane; Palmitate; Proteomics identification; Reference proteome; Secreted; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID H Molecular weight (Da) 27479.3 Length 249 Aromaticity 0.09 Instability index 32.92 Isoelectric point 7.22 Charge (pH=7) 0.48 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map 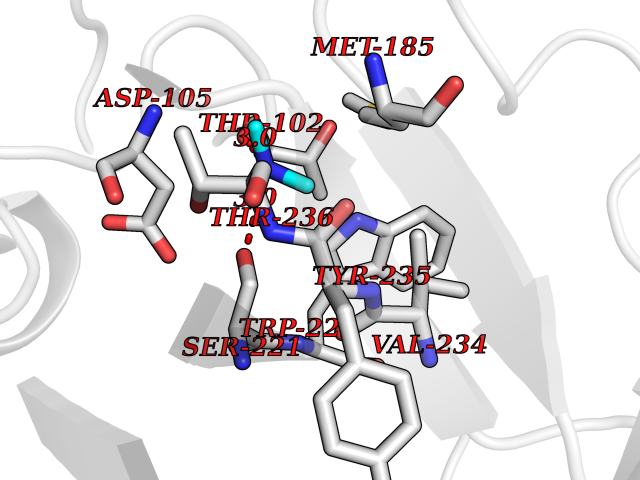 Pymol Map 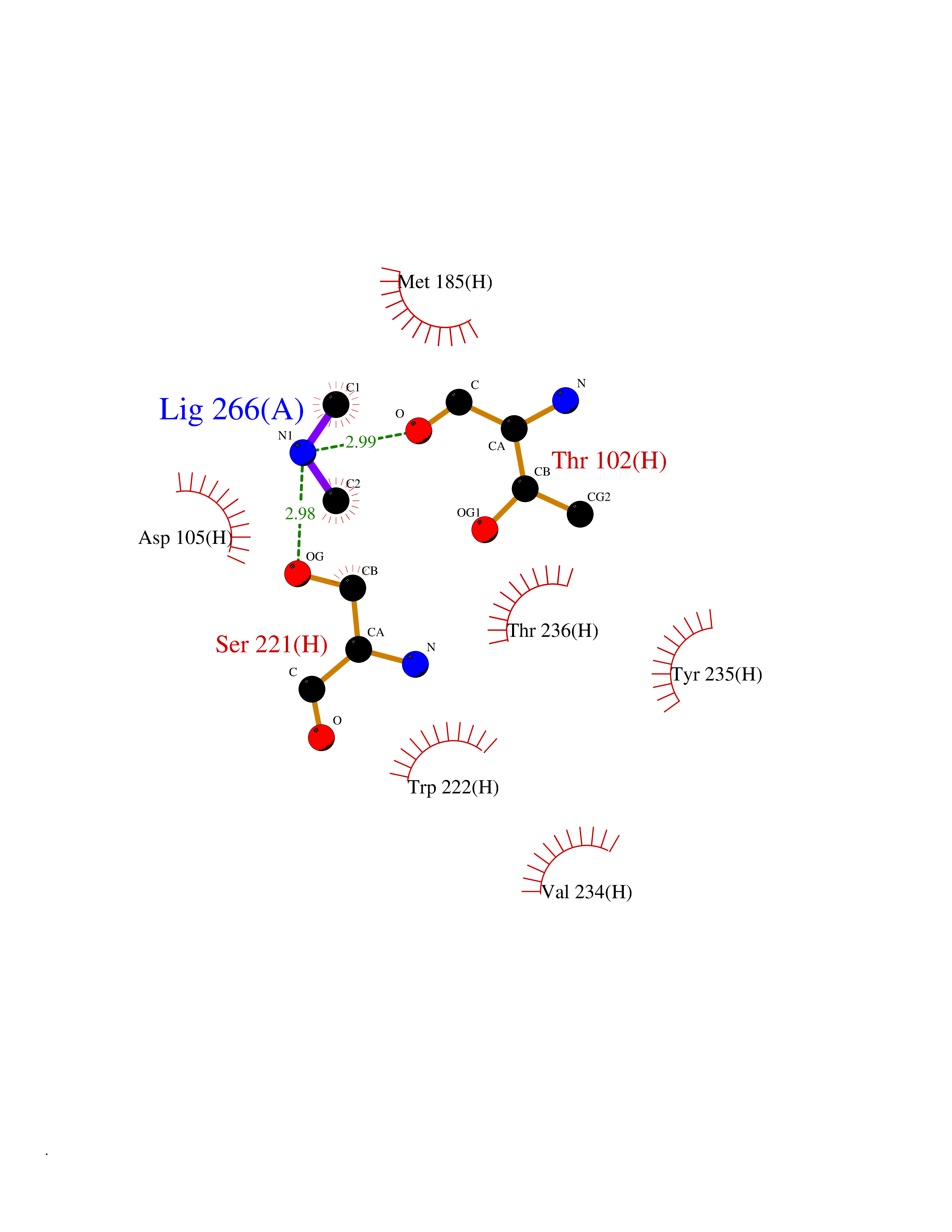 Ligplot Map 3D Binding mode Sequence IVGGKVCPKGECPWQVLLLVNGAQLCGGTLINTIWVVSAAHCFDKIKNWRNLIAVLGEHDLSEHDGDEQSRRVAQVIIPSTYVPGTTNHDIALLRLHQPVVLTDHVVPLCLPERTFSERTLAFVRFSLVSGWGQLLDRGATALELMVLNVPRVMTQDCEASYPGKITEYMFCAGYSDGSKDSCKGDSGGPHATHYRGTWYLTGIVSWGQGCATVGHFGVYTRVSQYIEWLQKLMRSEPRPGVLLRAPFP Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Zinc finger protein Helios (IKZF2) | 7LPS | 4.00 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -5.46 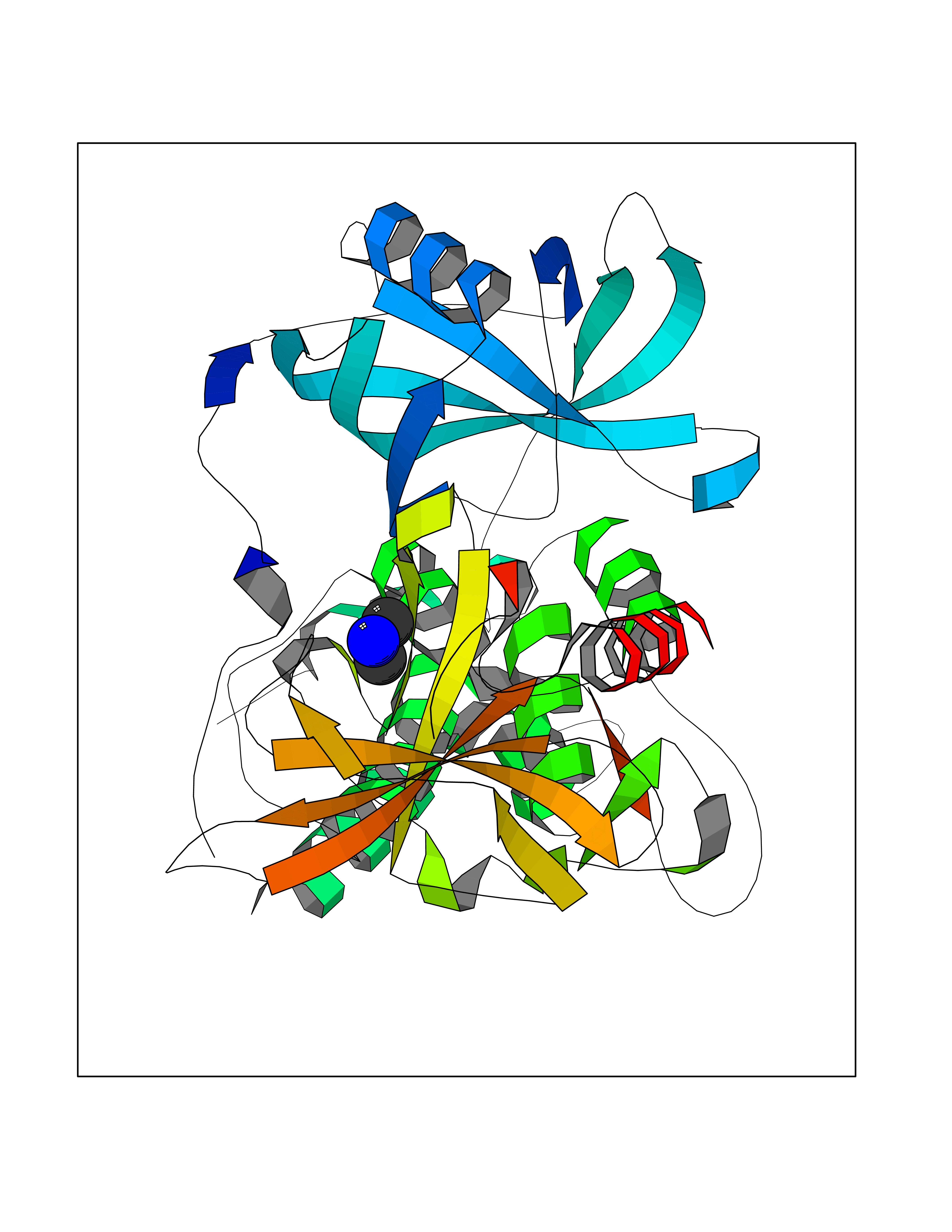 Molscript Map 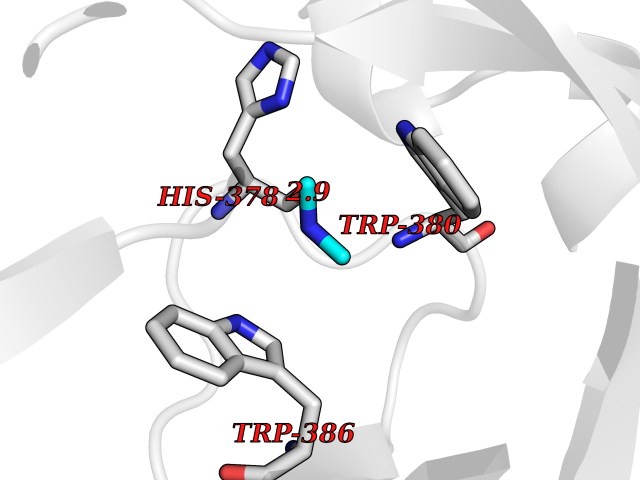 Pymol Map 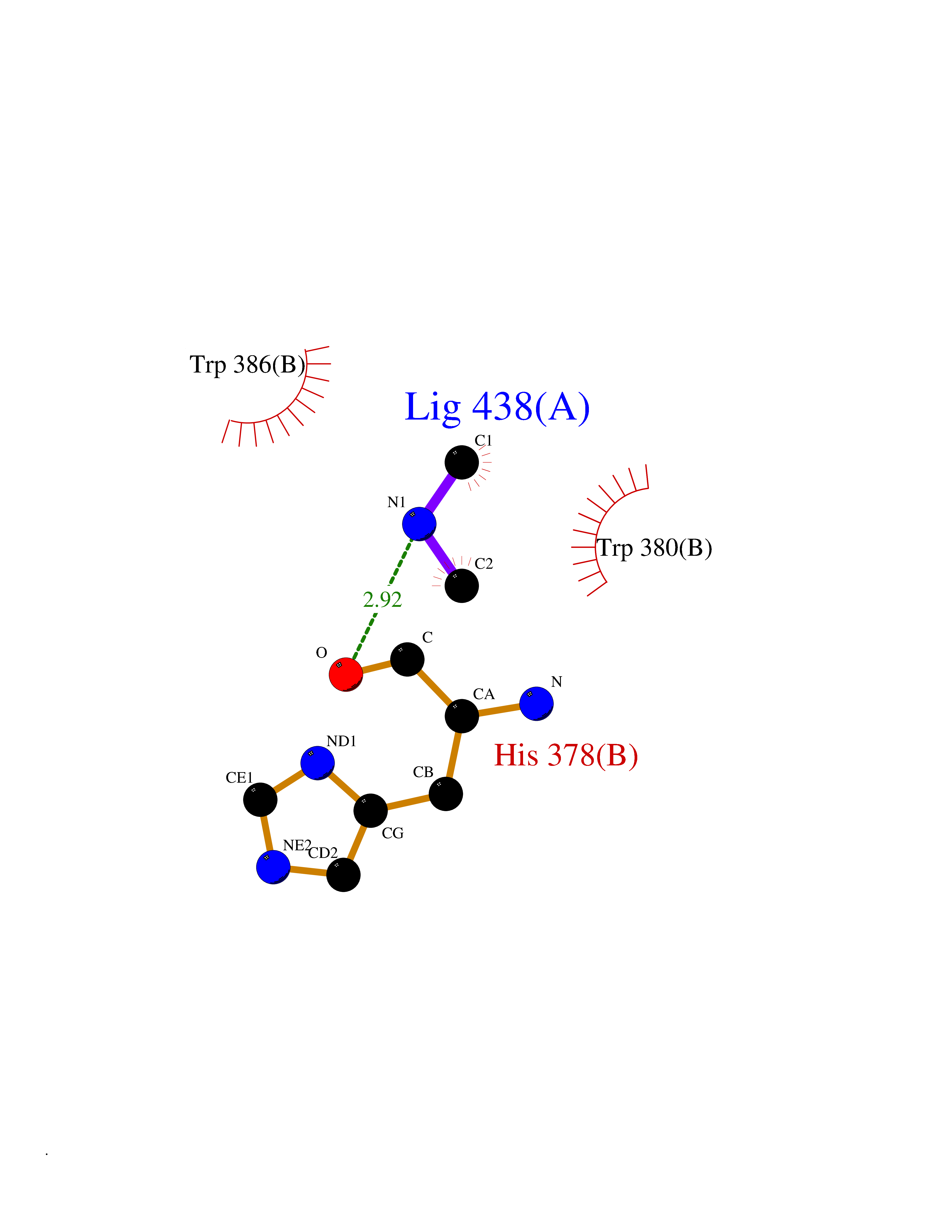 Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | N-glycosylase/DNA lyase (OGG1) | 1M3Q | 4.00 | |
Target general information Gen name OGG1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms OGH1; MUTM; MMH Protein family Type-1 OGG1 family Biochemical class Type-1 OGG1 family Function DNA repair enzyme that incises DNA at 8-oxoG residues. Excises 7,8-dihydro-8-oxoguanine and 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine (FAPY) from damaged DNA. Has a beta-lyase activity that nicks DNA 3' to the lesion. Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; DNA damage; DNA repair; Glycosidase; Hydrolase; Lyase; Mitochondrion; Multifunctional enzyme; Nucleus; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID C,A Molecular weight (Da) 36555.1 Length 329 Aromaticity 0.09 Instability index 53.82 Isoelectric point 6.66 Charge (pH=7) -1.28 2D Binding mode Binding energy (Kcal/mol) -5.45 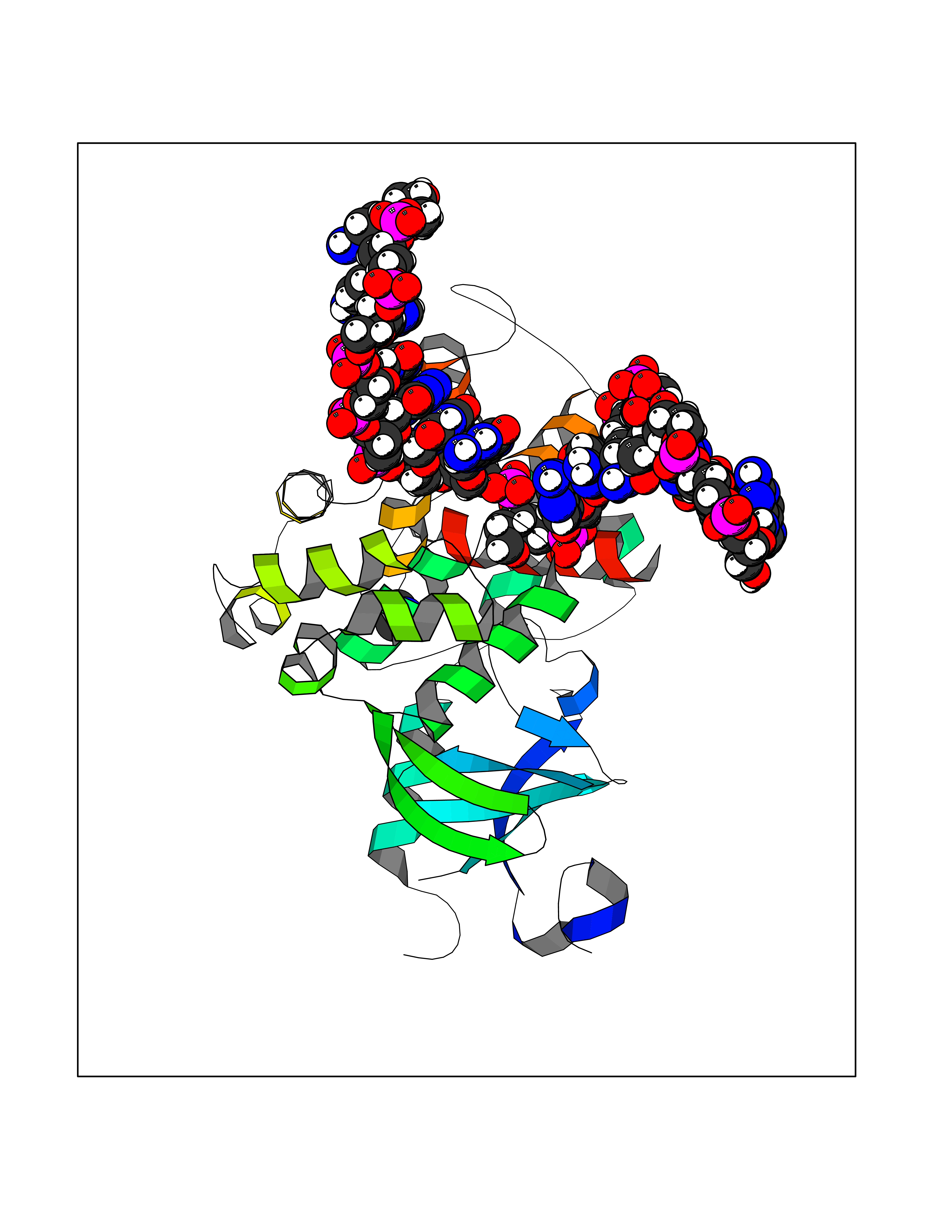 Molscript Map 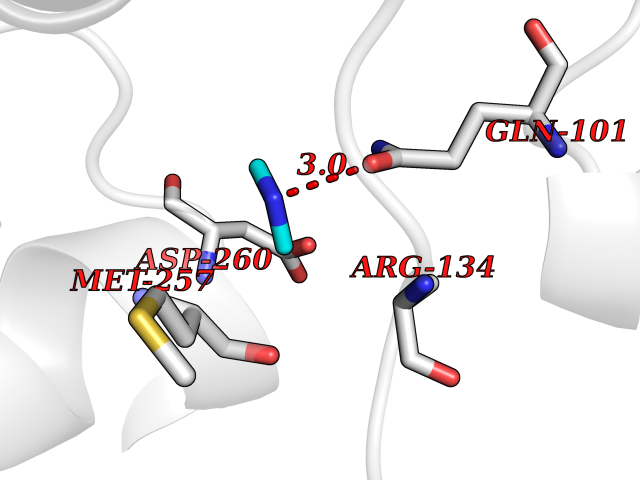 Pymol Map 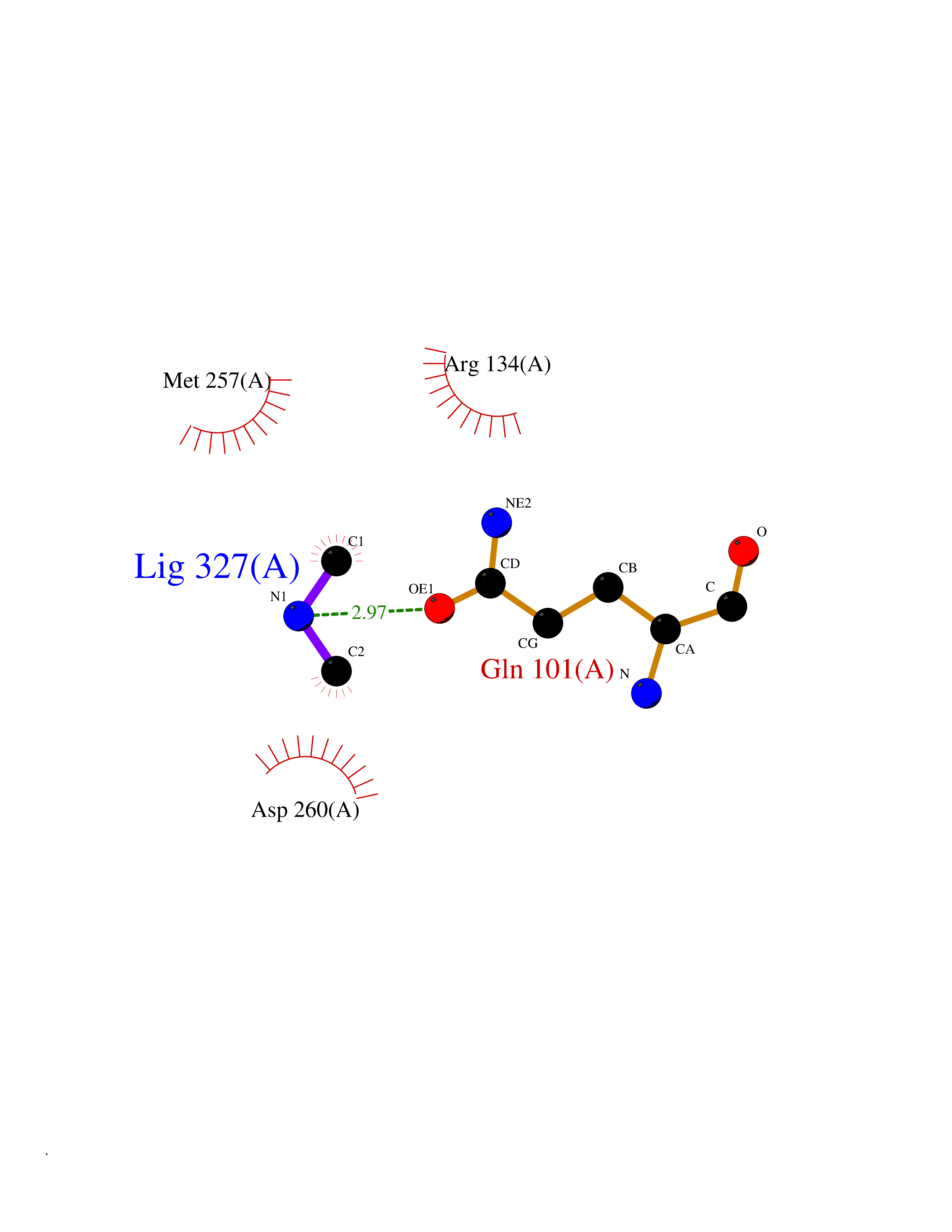 Ligplot Map 3D Binding mode Sequence GCGTCCAXGTCTACCGSEGHRTLASTPALWASIPCPRSELRLDLVLPSGQSFRWREQSPAHWSGVLADQVWTLTQTEEQLHCTVYRSQASRPTPDELEAVRKYFQLDVTLAQLYHHWGSVDSHFQEVAQKFQGVRLLRQDPIECLFSFICSSNNNIARITGMVERLCQAFGPRLIQLDDVTYHGFPSLQALAGPEVEAHLRKLGLGYRARYVSASARAILEEQGGLAWLQQLRESSYEEAHKALCILPGVGTKVADCICLMALDKPQAVPVEVHMWHIAQRDYSWHPTTSQAKGPSPQTNKELGNFFRSLWGPYAGWAQAVLFSADLRQ Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Aldehyde oxidase (AOX1) | 7OPN | 4.00 | |
Target general information Gen name AOX1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms AOX1 Protein family Xanthine dehydrogenase family Biochemical class Aldehyde/oxo donor oxidoreductase Function Oxidase with broad substrate specificity, oxidizing aromatic azaheterocycles, such as N1-methylnicotinamide and N- methylphthalazinium, as well as aldehydes, such as benzaldehyde, retinal, pyridoxal, and vanillin. Plays a key role in the metabolism of xenobiotics and drugs containing aromatic azaheterocyclic substituents. Participates in the bioactivation of prodrugs such as famciclovir, catalyzing the oxidation step from 6-deoxypenciclovir topenciclovir, which is a potent antiviral agent. Is probably involved in the regulation of reactive oxygen species homeostasis. May be a prominent source of superoxide generation via the one-electron reduction of molecular oxygen. Also may catalyze nitric oxide (NO) production via the reduction of nitrite to NO with NADH or aldehyde as electron donor. May play a role in adipogenesis. Related diseases Progressive familial heart block 1B (PFHB1B) [MIM:604559]: A cardiac bundle branch disorder characterized by progressive alteration of cardiac conduction through the His-Purkinje system, with a pattern of a right bundle-branch block and/or left anterior hemiblock occurring individually or together. It leads to complete atrio-ventricular block causing syncope and sudden death. {ECO:0000269|PubMed:19726882, ECO:0000269|PubMed:20562447, ECO:0000269|PubMed:21887725}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythrokeratodermia variabilis et progressiva 6 (EKVP6) [MIM:618531]: A form of erythrokeratodermia variabilis et progressiva, a genodermatosis characterized by the coexistence of two independent skin lesions: transient erythema and hyperkeratosis that is usually localized but occasionally occurs in its generalized form. Clinical presentation varies significantly within a family and from one family to another. Palmoplantar keratoderma is present in around 50% of cases. EKVP6 inheritance is autosomal dominant. {ECO:0000269|PubMed:30528822}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00437; DB00513; DB00484; DB11791; DB00215; DB00924; DB03516; DB01175; DB00426; DB12466; DB09054; DB09078; DB00170; DB01033; DB00563; DB08840; DB00157; DB00339; DB00481; DB04827; DB00962; DB00246; DB00909 Interacts with Q06278 EC number EC 1.2.3.1 Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; FAD; Flavoprotein; Iron; Iron-sulfur; Lipid metabolism; Metal-binding; Molybdenum; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 286845 Length 2590 Aromaticity 0.08 Instability index 42.7 Isoelectric point 6.92 Charge (pH=7) -1.62 2D Binding mode Binding energy (Kcal/mol) -5.46 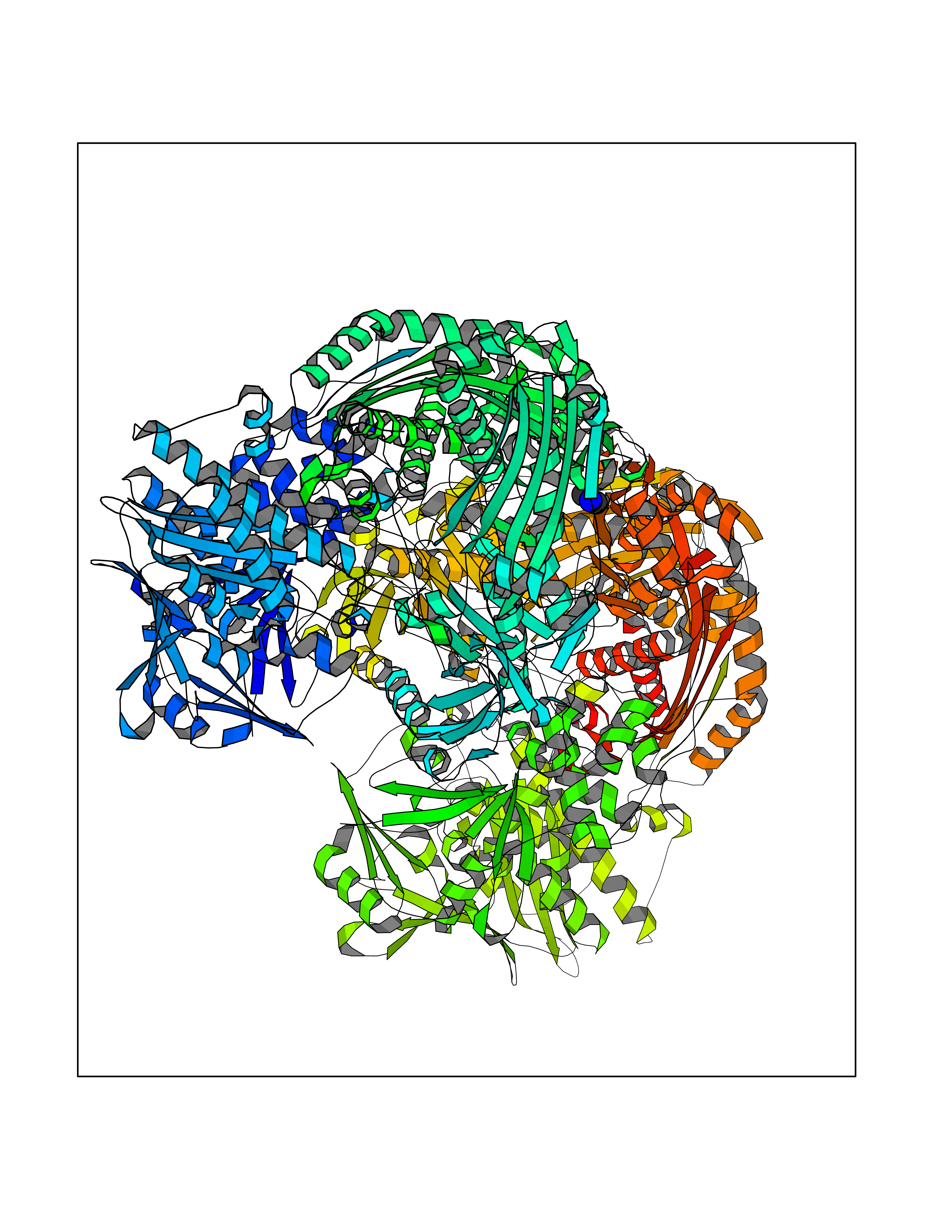 Molscript Map 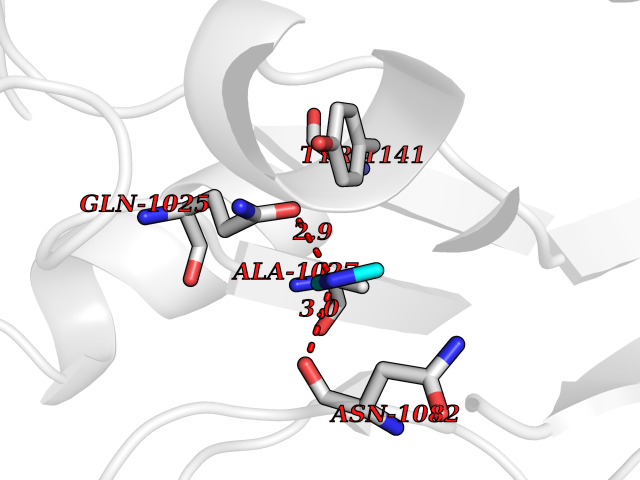 Pymol Map 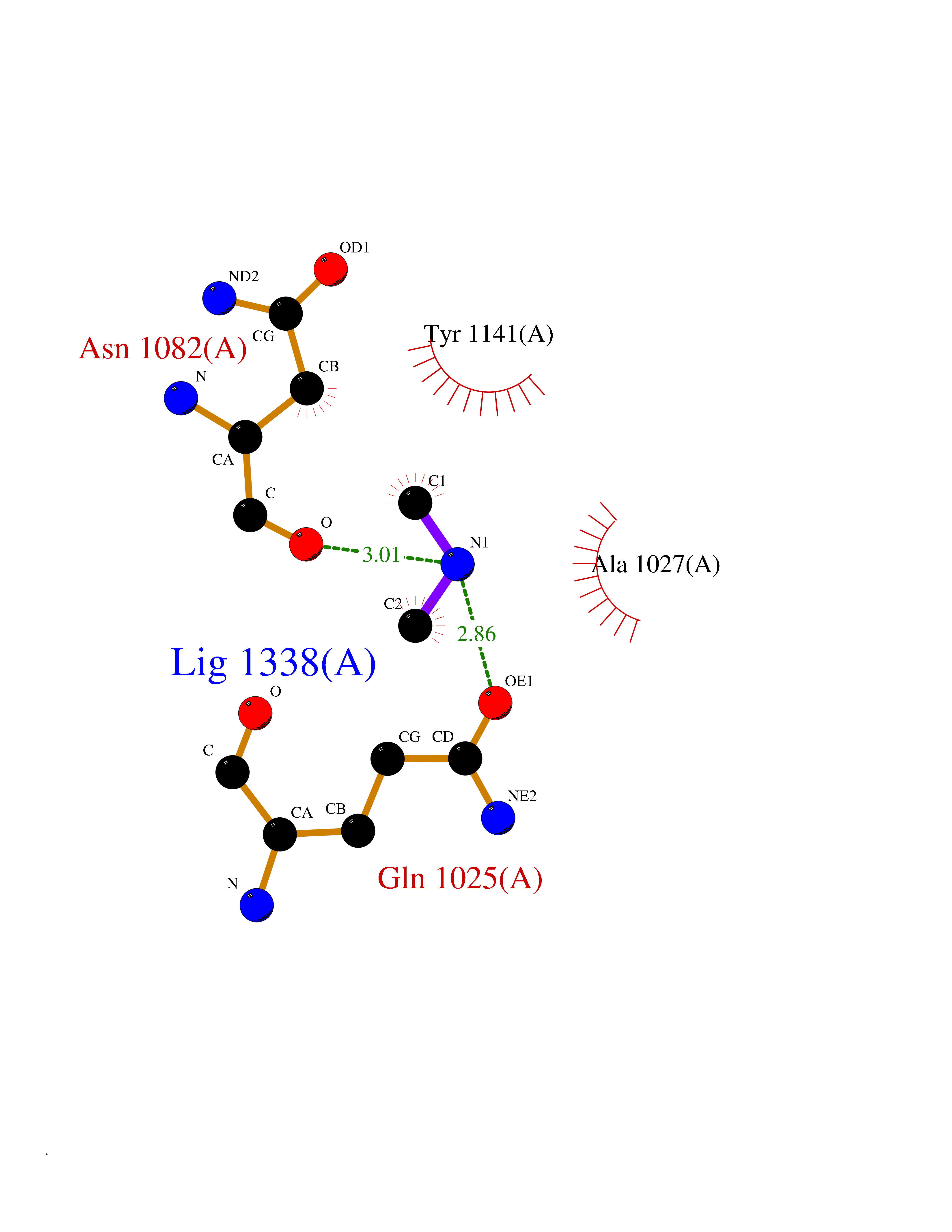 Ligplot Map 3D Binding mode Sequence ASELLFYVNGRKVIEKNVDPETMLLPYLRKKLRLTGTKYGCGGGGCGACTVMISRYNPITKRIRHHPANACLIPICSLYGAAVTTVEGIGSTHTRIHPVQERIAKCHGTQCGFCTPGMVMSIYTLLRNHPEPTLDQLTDALGGNLCRCTGYRPIIDACKTFCKPKLFAEEEFLPLDPTQELIFPPELMIMAEKQSQRTRVFGSERMMWFSPVTLKELLEFKFKYPQAPVIMGNTSVGPEVKFKGVFHPVIISPDRIEELSVVNHAYNGLTLGAGLSLAQVKDILADVVQKLPEEKTQMYHALLKHLGTLAGSQIRNMASLGGHIISRHPDSDLNPILAVGNCTLNLLSKEGKRQIPLNEQFLSKCPNADLKPQEILVSVNIPYSRKWEFVSAFRQAQRQENALAIVNSGMRVFFGEGDGIIRELCISYGGVGPATICAKNSCQKLIGRHWNEQMLDIACRLILNEVSLLGSAPGGKVEFKRTLIISFLFKFYLEVSQILKKMDPVHYPSLADKYESALEDLHSKHHCSTLKYQNQHPEDPIGHPIMHLSGVKHATGEAIYCDDMPLVDQELFLTFVTSSRAHAKIVSIDLSEALSMPGVVDIMTAEHLSDVNSFCFFTEAEKFLATDKVFCVGQLVCAVLADSEVQAKRAAKRVKIVYQDLEPLILTIEESIQHNSSFKPERKLEYGNVDEAFKVVDQILEGEIHMGGQEHFYMETQSMLVVPKGEDQEMDVYVSTQFPKYIQDIVASTLKLPANKVMCHVRRVGGAFGGKVLKTGIIAAVTAFAANKHGRAVRCVLERGEDMLITGGRHPYLGKYKAGFMNDGRILALDMEHYSNAGASLDESLFVIEMGLLKMDNAYKFPNLRCRGWACRTNLPSNTAFRGFGFPQAALITESCITEVAAKCGLSPEKVRIINMYKEIDQTPYKQEINAKNLIQCWRECMAMSSYSLRKVAVEKFNAENYWKKKGLAMVPLKFPVGLGSRAAGQAAALVHIYLDGSVLVTHGGIEMGQGVHTKMIQVVSRELRMPMSNVHLRGTSTETVPNANISGGSVVADLNGLAVKDACQTLLKRLEPIISKNPKGTWKDWAQTAFDESINLSAVGYFRGYESDMNWEKGEGQPFEYFVYGAACSEVEIDCLTGDHKNIRTDIVMDVGCSINPAIDIGQIEGAFIQGMGLYTIEELNYSPQGILHTHGPDQYKIPAICDMPTELHIALLPPSQNSNTLYSSKGLGESGVFLGCSVFFAIHDAVSAARQERGLHLTLNSPLTPEKIRMACEDKFTKMIPRDEPGSYVPWNVASELLFYVNGRKVIEKNVDPETMLLPYLRKKLRLTGTKYGCGGGGCGACTVMISRYNPITKRIRHHPANACLIPICSLYGAAVTTVEGIGSTHTRIHPVQERIAKCHGTQCGFCTPGMVMSIYTLLRNHPEPTLDQLTDALGGNLCRCTGYRPIIDACKTFCKPKLFAEEEFLPLDPTQELIFPPELMIMAEKQSQRTRVFGSERMMWFSPVTLKELLEFKFKYPQAPVIMGNTSVGPEVKFKGVFHPVIISPDRIEELSVVNHAYNGLTLGAGLSLAQVKDILADVVQKLPEEKTQMYHALLKHLGTLAGSQIRNMASLGGHIISRHPDSDLNPILAVGNCTLNLLSKEGKRQIPLNEQFLSKCPNADLKPQEILVSVNIPYSRKWEFVSAFRQAQRQENALAIVNSGMRVFFGEGDGIIRELCISYGGVGPATICAKNSCQKLIGRHWNEQMLDIACRLILNEVSLLGSAPGGKVEFKRTLIISFLFKFYLEVSQILKKMDPVHYPSLADKYESALEDLHSKHHCSTLKYQNQHPEDPIGHPIMHLSGVKHATGEAIYCDDMPLVDQELFLTFVTSSRAHAKIVSIDLSEALSMPGVVDIMTAEHLSDVNSFCFFTEAEKFLATDKVFCVGQLVCAVLADSEVQAKRAAKRVKIVYQDLEPLILTIEESIQHNSSFKPERKLEYGNVDEAFKVVDQILEGEIHMGGQEHFYMETQSMLVVPKGEDQEMDVYVSTQFPKYIQDIVASTLKLPANKVMCHVRRVGGAFGGKVLKTGIIAAVTAFAANKHGRAVRCVLERGEDMLITGGRHPYLGKYKAGFMNDGRILALDMEHYSNAGASLDESLFVIEMGLLKMDNAYKFPNLRCRGWACRTNLPSNTAFRGFGFPQAALITESCITEVAAKCGLSPEKVRIINMYKEIDQTPYKQEINAKNLIQCWRECMAMSSYSLRKVAVEKFNAENYWKKKGLAMVPLKFPVGLGSRAAGQAAALVHIYLDGSVLVTHGGIEMGQGVHTKMIQVVSRELRMPMSNVHLRGTSTETVPNANISGGSVVADLNGLAVKDACQTLLKRLEPIISKNPKGTWKDWAQTAFDESINLSAVGYFRGYESDMNWEKGEGQPFEYFVYGAACSEVEIDCLTGDHKNIRTDIVMDVGCSINPAIDIGQIEGAFIQGMGLYTIEELNYSPQGILHTHGPDQYKIPAICDMPTELHIALLPPSQNSNTLYSSKGLGESGVFLGCSVFFAIHDAVSAARQERGLHLTLNSPLTPEKIRMACEDKFTKMIPRDEPGSYVPWNV Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Ribosomal protein S6 kinase alpha-6 (RSK6) | 6G77 | 4.00 | |
Target general information Gen name RPS6KA6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms pp90RSK4; p90RSK6; p90-RSK 6; S6K-alpha-6; Ribosomal S6 kinase 4; RSK4; RSK-4; 90 kDa ribosomal protein S6 kinase 6 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, S6 kinase subfamily Biochemical class NA Function Constitutively active serine/threonine-protein kinase that exhibits growth-factor-independent kinase activity and that may participate in p53/TP53-dependent cell growth arrest signaling and play an inhibitory role during embryogenesis. Related diseases Symptomatic deficiency in lactate transport (SDLT) [MIM:245340]: Deficiency of lactate transporter may result in an acidic intracellular environment created by muscle activity with consequent degeneration of muscle and release of myoglobin and creatine kinase. This defect might compromise extreme performance in otherwise healthy individuals. {ECO:0000269|PubMed:10590411}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperinsulinemic hypoglycemia, familial, 7 (HHF7) [MIM:610021]: A form of hyperinsulinemic hypoglycemia, a clinically and genetically heterogeneous disorder characterized by inappropriate insulin secretion from the pancreatic beta-cells in the presence of low blood glucose levels. HHF7 features include exercise-induced hyperinsulinism, loss of consciousness due to hypoglycemia, and hypoglycemic seizures. HHF7 inheritance is autosomal dominant. {ECO:0000269|PubMed:17701893}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Monocarboxylate transporter 1 deficiency (MCT1D) [MIM:616095]: A metabolic disorder characterized by recurrent ketoacidosis, a pathologic state due to ketone formation exceeding ketone utilization. The clinical consequences of ketoacidosis are vomiting, osmotic diuresis, dehydration, and Kussmaul breathing. The condition may progress to decreased consciousness and, ultimately, death. {ECO:0000269|PubMed:25390740}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with Q7Z698 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 33406.4 Length 291 Aromaticity 0.11 Instability index 38.08 Isoelectric point 8.4 Charge (pH=7) 2.55 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map 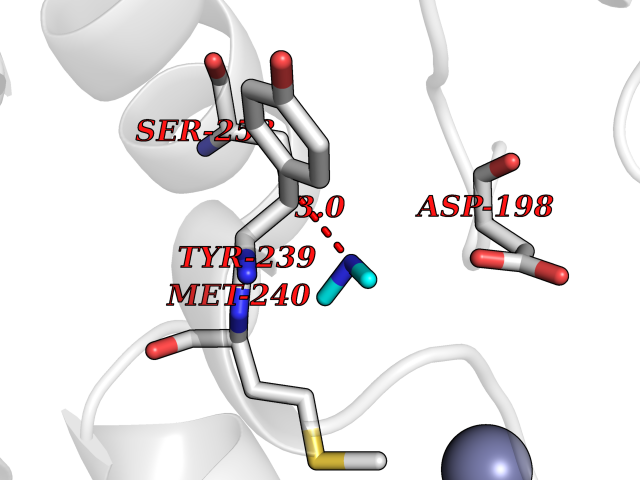 Pymol Map 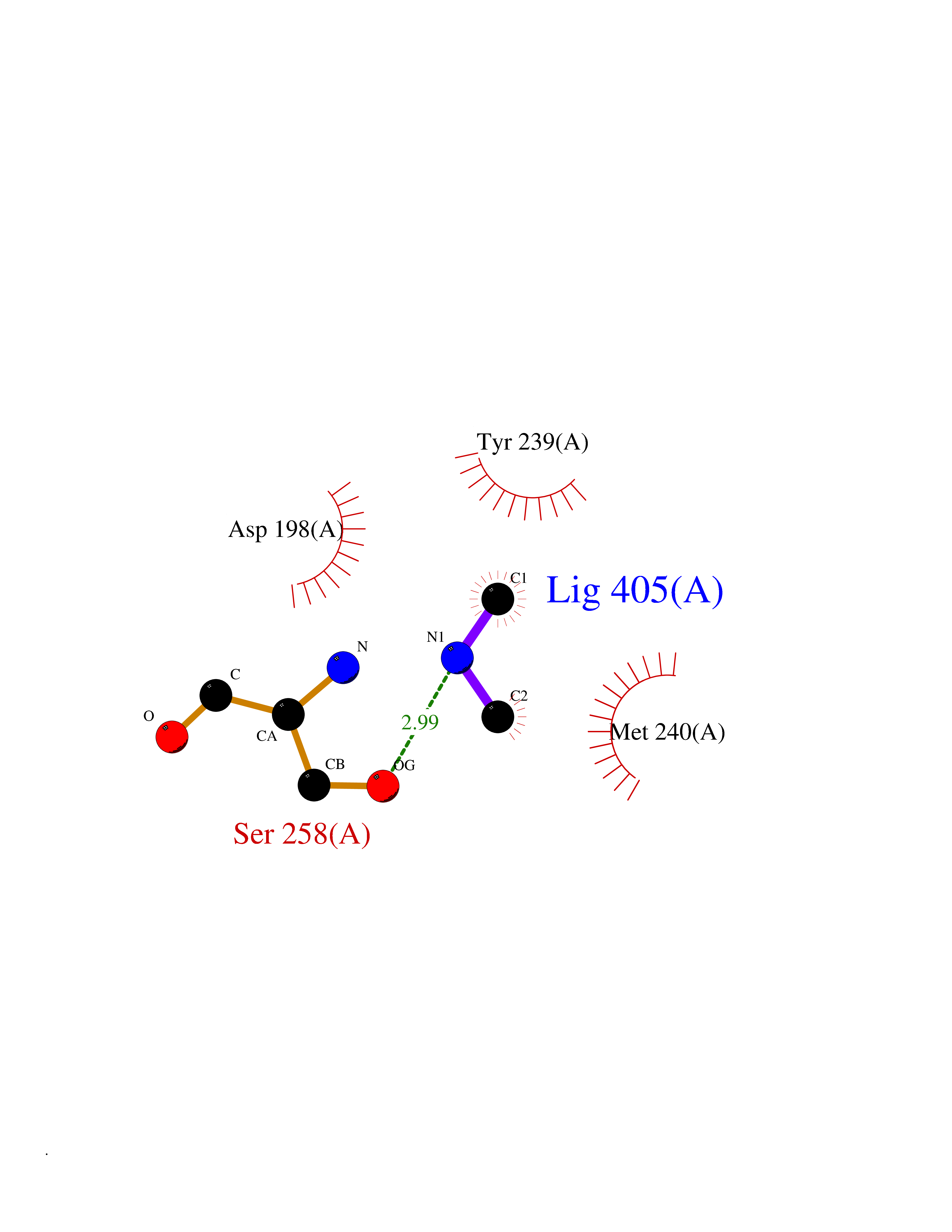 Ligplot Map 3D Binding mode Sequence VVKEIPITHHVKEGYEKADPAQFELLKVLGQGSFGKVFLVRKKTGPDAGQLYAMKVLKKASLKVRDDILVEVNHPFIVKLHYAFQTEGKLYLILDFLRGGDVFTRLSKEVLFTEEDVKFYLAELALALDHLHQLGIVYRDLKPENILLDEIGHIKLTDFGLSKESVDQEKKAYSFCGTVEYMAPEVVNRRGHSQSADWWSYGVLMFEMLTGTLPFQGKDRNETMNMILKAKLGMPQFLSAEAQSLLRMLFKRNPANRLGSEGVEEIKRHLFFANIDWDKLYKREVQPPFKP Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Glutathione S-transferase LANCL1 (LANCL1) | 3E73 | 4.00 | |
Target general information Gen name LANCL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p40; LanC-like protein 1; GPR69A; 40 kDa erythrocyte membrane protein Protein family LanC-like protein family Biochemical class NA Function Functions as glutathione transferase. Catalyzes conjugation of the glutathione (GSH) to artificial substrates 1-chloro-2,4-dinitrobenzene (CDNB) and p-nitrophenyl acetate. Mitigates neuronal oxidative stress during normal postnatal development and in response to oxidative stresses probably through GSH antioxidant defense mechanism (By similarity). May play a role in EPS8 signaling. Binds glutathione. Related diseases Spermatogenic failure 5 (SPGF5) [MIM:243060]: An infertility disorder caused by spermatogenesis defects. Semen from affected men show close to 100% morphologically abnormal multiflagellar spermatozoa with low motility, oversized irregular heads, and abnormal midpiece and acrosome. {ECO:0000269|PubMed:17435757, ECO:0000269|PubMed:21733974}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UHR4; P42858; Q08509 EC number EC 2.5.1.18 Uniprot keywords 3D-structure; Acetylation; Cell membrane; Cytoplasm; Direct protein sequencing; Membrane; Metal-binding; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 46005.4 Length 405 Aromaticity 0.14 Instability index 33.7 Isoelectric point 7.13 Charge (pH=7) 0.4 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 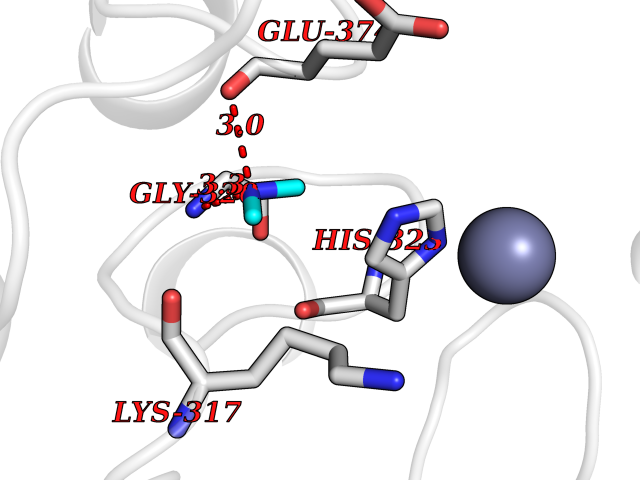 Pymol Map 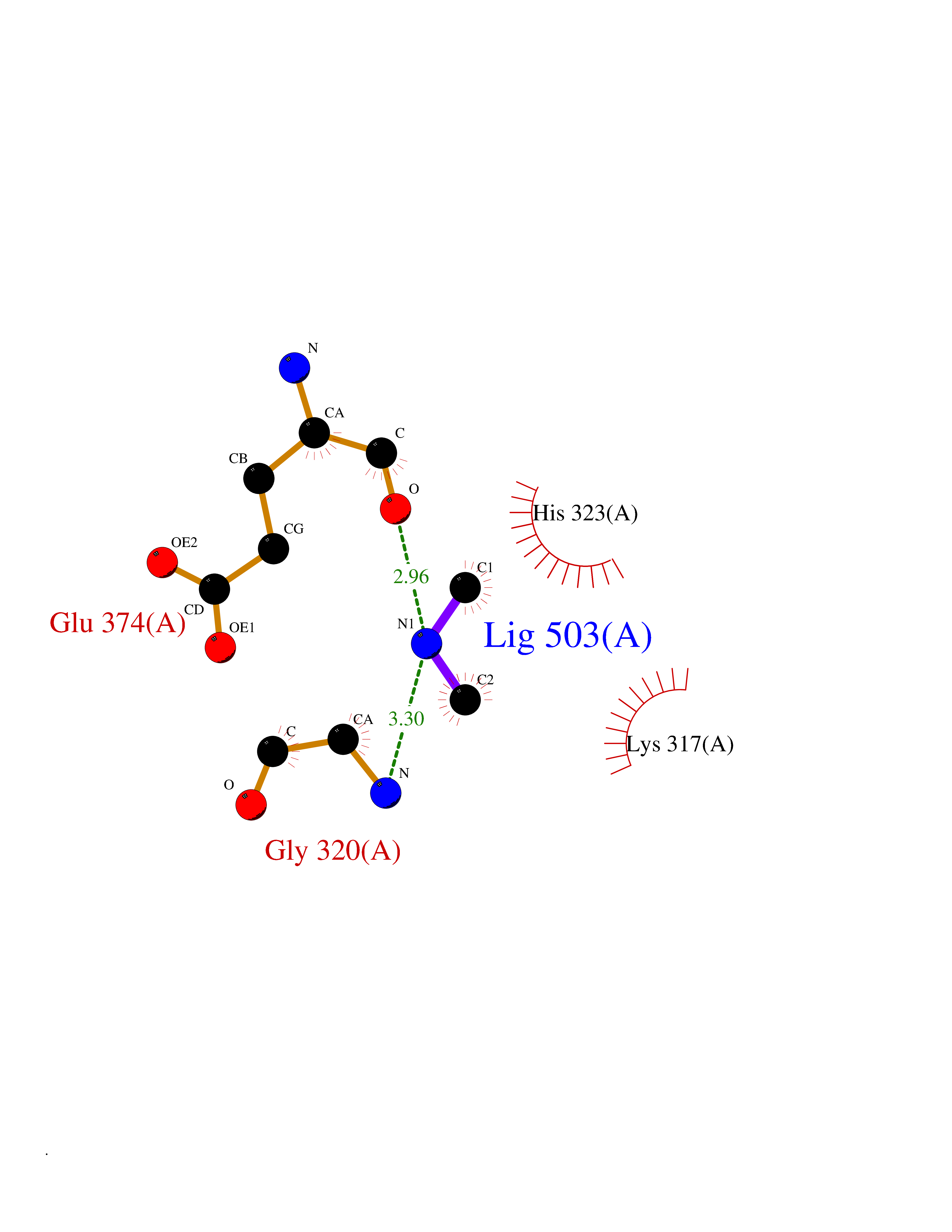 Ligplot Map 3D Binding mode Sequence SMDIEFMAQRAFPNPYADYNKSLAEGYFDAAGRLTPEFSQRLTNKIRELLQQMERGLKSADPRDGTGYTGWAGIAVLYLHLYDVFGDPAYLQLAHGYVKQSLNCLTKRSITFLCGDAGPLAVAAVLYHKMNNEKQAEDCITRLIHLNKIDPHAPNEMLYGRIGYIYALLFVNKNFGVEKIPQSHIQQICETILTSGENLARKRNFTAKSPLMYEWYQEYYVGAAHGLAGIYYYLMQPSLQVSQGKLHSLVKPSVDYVCQLKFPSGNYPPCIGDNRDLLVHWCHGAPGVIYMLIQAYKVFREEKYLCDAYQCADVIWQYGLLKKGYGLCHGSAGNAYAFLTLYNLTQDMKYLYRACKFAEWCLEYGEHGCRTPDTPFSLFEGMAGTIYFLADLLVPTKARFPAFEL Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Histone deacetylase 7 (HDAC7) | 3C0Z | 4.00 | |
Target general information Gen name HDAC7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histone deacetylase 7A; HDAC7A; HD7a; HD7 Protein family Histone deacetylase family, HD type 2 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Involved in muscle maturation by repressing transcription of myocyte enhancer factors such as MEF2A, MEF2B and MEF2C. During muscle differentiation, it shuttles into the cytoplasm, allowing the expression of myocyte enhancer factors. May be involved in Epstein-Barr virus (EBV) latency, possibly by repressing the viral BZLF1 gene. Positively regulates the transcriptional repressor activity of FOXP3. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4). Related diseases Mucopolysaccharidosis 1H (MPS1H) [MIM:607014]: A severe form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1H usually present, within the first year of life, a combination of hepatosplenomegaly, skeletal deformities, corneal clouding and severe intellectual disability. Obstructive airways disease, respiratory infection and cardiac complications usually result in death before 10 years of age. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:1301941, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:24036510, ECO:0000269|PubMed:31194252, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:7951228, ECO:0000269|PubMed:8019563, ECO:0000269|PubMed:8328452, ECO:0000269|PubMed:8401515, ECO:0000269|Ref.20}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1H/S (MPS1H/S) [MIM:607015]: A form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. MPS1H/S represents an intermediate phenotype of the MPS1 clinical spectrum. It is characterized by relatively little neurological involvement, but most of the somatic symptoms described for severe MPS1 develop in the early to mid-teens, causing considerable loss of mobility. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8401515}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1S (MPS1S) [MIM:607016]: A mild form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1S may have little or no neurological involvement, normal stature and life span, but present development of joints stiffness, mild hepatosplenomegaly, aortic valve disease and corneal clouding. {ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:25256405, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8213840}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12565; DB05015; DB01262; DB11841; DB12645; DB06603; DB06819; DB03766; DB12847; DB06176; DB04297; DB00313; DB02546 Interacts with P00533; Q9BZS1-1; Q9BZS1-2; Q9BZL6; P31947; P63104; P08393; Q8CFN5; Q13137; Q04864; Q0D2K3; Q8WXI4-2; Q9BQD7; Q03989; Q9NSI6-4; Q3SXR2; Q13137; P60953; Q7L2Z9; Q96D03; Q9NQ30; Q9UBI6; A6NEM1; A5PKX9; Q9BXK1; Q6ZNG9; O43679; Q6FHY5; Q9BRT3; O94964-4; O95411; O00746; Q9BQI9; B7ZLY0; Q96I34; P63000; P15153; P60763; Q04864-2; Q0D2K3; P62070; O15427; O95164 EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Cytoplasm; Hydrolase; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Repressor; Transcription; Transcription regulation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 41454.5 Length 383 Aromaticity 0.08 Instability index 38.49 Isoelectric point 6.26 Charge (pH=7) -5.18 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 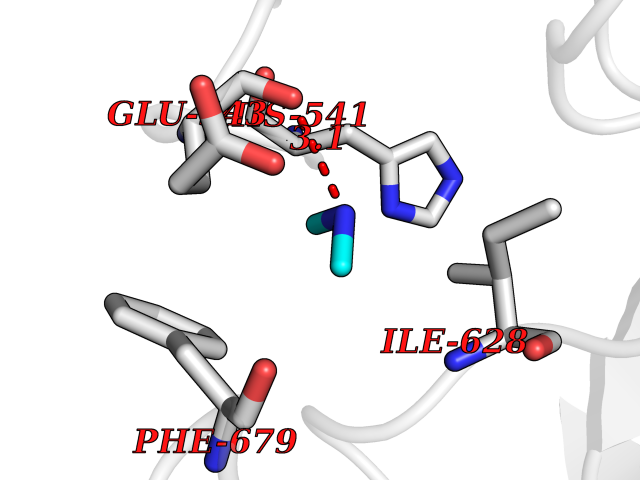 Pymol Map 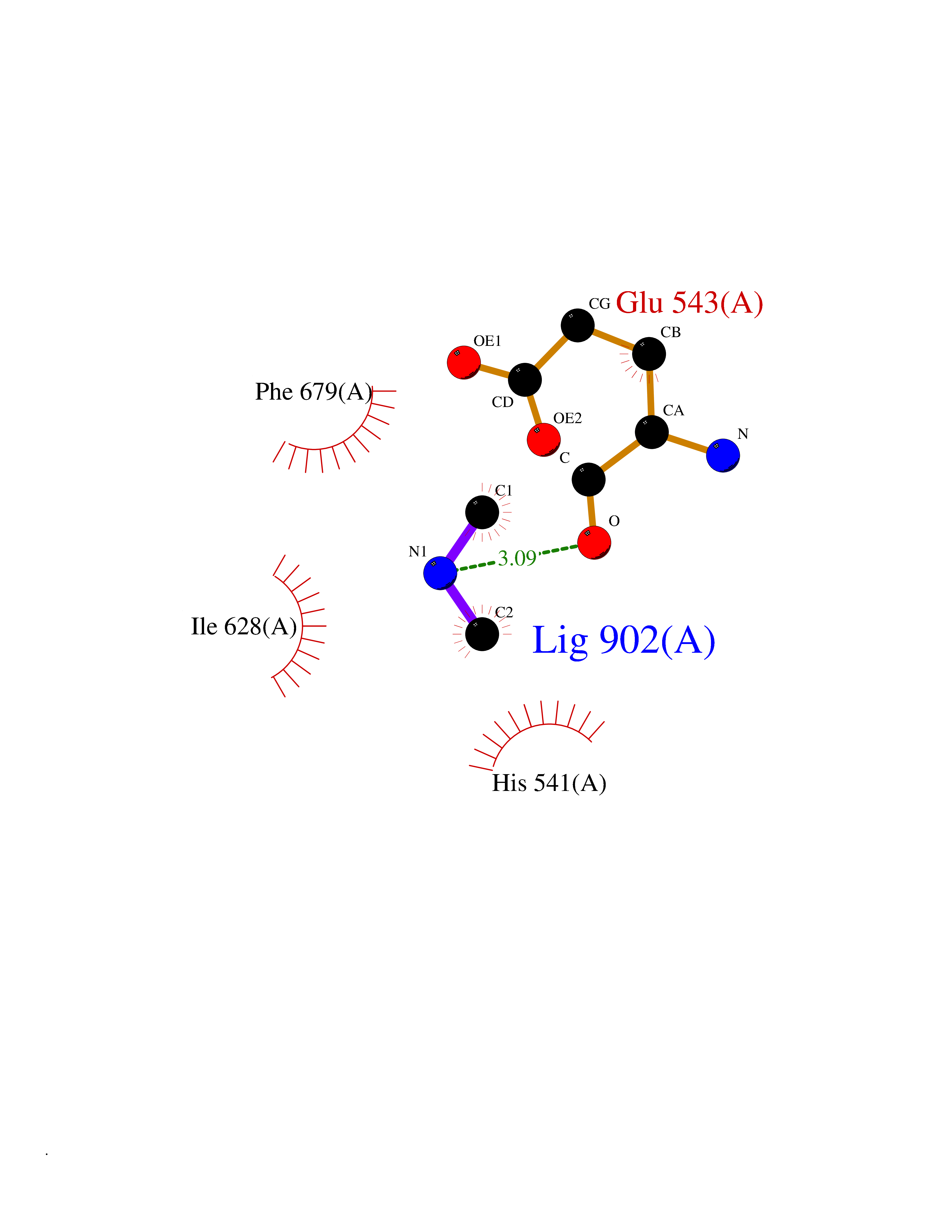 Ligplot Map 3D Binding mode Sequence TLPFTTGLIYDSVMLKHQCSCGDNSRHPEHAGRIQSIWSRLQERGLRSQCECLRGRKASLEELQSVHSERHVLLYGTNPLSRLKLDNGKLAGLLAQVMLPCGGVGVDTDTIWNELHSSNAARWAAGSVTDLAFKVASRELKNGFAVVRPPGHHADHSTAMGFCFFNSVAIACRQLQQQSKASKILIVDWDVHHGNGTQQTFYQDPSVLYISLHRHDDGNFFPGSGAVDEVGAGSGEGFNVNVAWAGGLDPPMGDPEYLAAFRIVVMPIAREFSPDLVLVSAGFDAAEGHPAPLGGYHVSAKCFGYMTQQLMNLAGGAVVLALEGGHDLTAICDASEACVAALLGNRVDPLSEEGWKQKPNLNAIRSLEAVIRVHSKYWGCMQR Hydrogen bonds contact Hydrophobic contact | ||||