Job Results:
Ligand
Structure
Job ID
7f371ea108ffadeed56066fb8d4cae63
Job name
NA
Time
2024-12-09 11:06:55
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | 2-oxoglutarate dehydrogenase, mitochondrial | 3ERY | 4.74 | |
Target general information Gen name OGDH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Alpha-ketoglutarate dehydrogenase family Biochemical class Immune system Function Chaperone binding.Heat shock protein binding.Metal ion binding.Oxoglutarate dehydrogenase (NAD+) activity.Oxoglutarate dehydrogenase (succinyl-transferring) activity.Thiamine pyrophosphate binding. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00157; DB00313; DB09092 Interacts with P54253; P42858 EC number 1.2.4.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Calcium; Glycolysis; Isopeptide bond; Magnesium; Metal-binding; Mitochondrion; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Thiamine pyrophosphate; Transit peptide; Ubl conjugation Protein physicochemical properties Chain ID P,Q Molecular weight (Da) 18311.1 Length 158 Aromaticity 0.15 Instability index 37.26 Isoelectric point 5.64 Charge (pH=7) -2.98 2D Binding mode Binding energy (Kcal/mol) -6.47  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence QLSPFPFDLGPHSMRYYETATSRRGLGEPRYTSVGYVDDKEFVRFDSDARITQVAKGQEQWFRVNLRTLLGYYNQSAGGTHTLQRMYGCDVGSDGRLLRGYEQFAYDGCDYIALNEDLRTWTAADMAAQITRRKWEQAGAAEYYRAYLEGECVEWLHR Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | cAMP-specific 3',5'-cyclic phosphodiesterase 4C | 2QYM | 4.74 | |
Target general information Gen name PDE4C Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms DPDE1 Protein family Cyclic nucleotide phosphodiesterase family, PDE4 subfamily Biochemical class Hydrolase Function 3',5'-cyclic-AMP phosphodiesterase activity.Metal ion binding. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01427; DB00201; DB05219; DB00651; DB06246; DB05266; DB01088; DB01791; DB01656; DB01954; DB09283 Interacts with Q96D03; O15499; P26718; P50221; Q6FHY5; Q9UJX0; P26367; P30626; P59817; P30626 EC number 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cell projection; Cilium; Hydrolase; Manganese; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 33042.2 Length 291 Aromaticity 0.08 Instability index 23.45 Isoelectric point 4.91 Charge (pH=7) -18.1 2D Binding mode Binding energy (Kcal/mol) -6.47 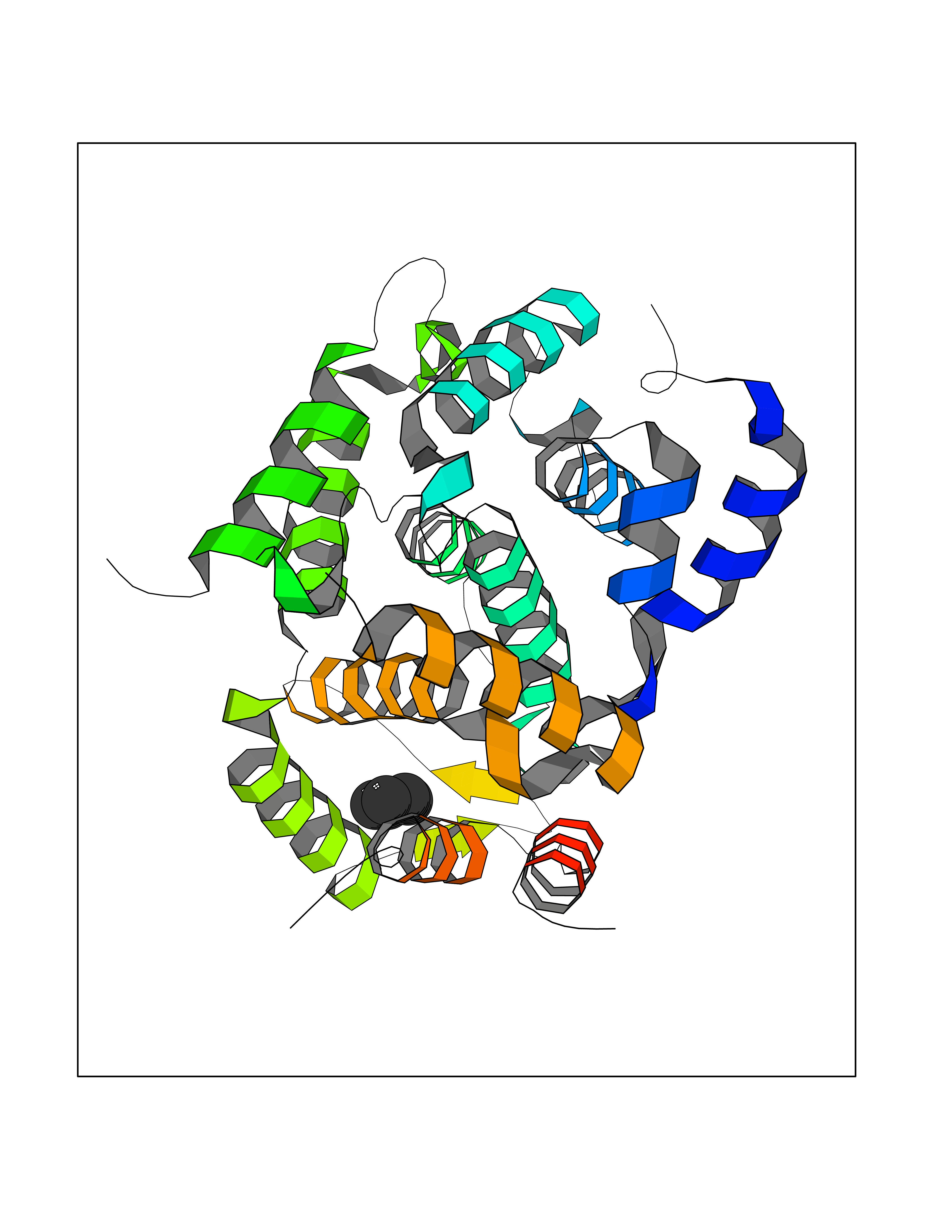 Molscript Map 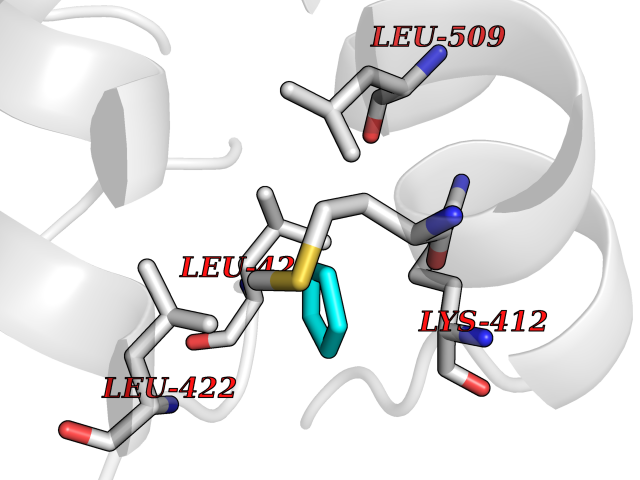 Pymol Map 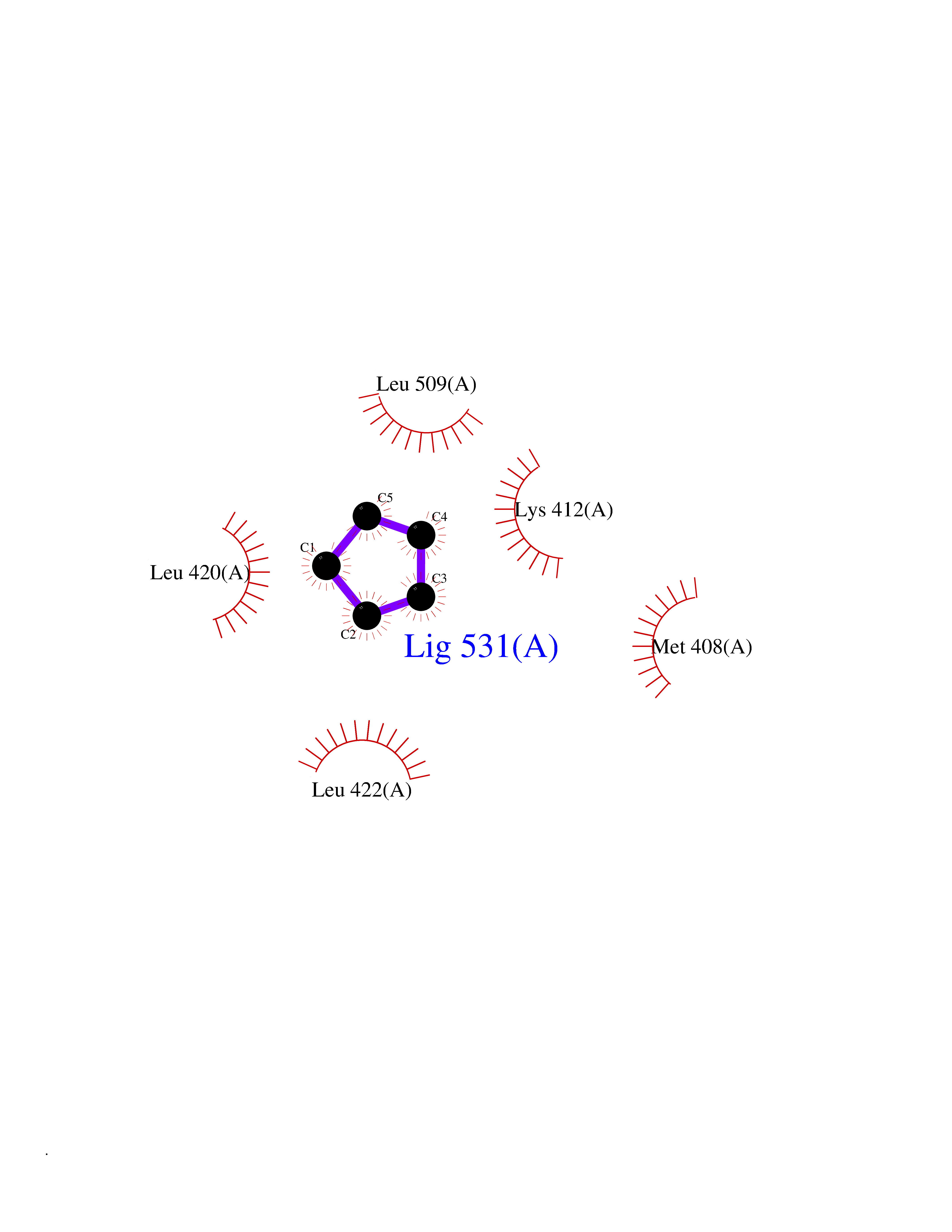 Ligplot Map 3D Binding mode Sequence VPRFGVQTDQEEQLAKELEDTNKWGLDVFKVAELSGNRPLTAIIFSIFQERDLLKTFQIPADTLATYLLMLEGHYHANVAYHNSLHAADVAQSTHVLLATPALEAVFTDLEILAALFASAIHDVDHPGVSNQNDASVLENHHLAVGFKLLQAENCDIFQNLSAKQRLSLRRMVIDMVLATDMSKHMNLLADLKTMVETKKVTSLGVLLLDNYSDRIQVLQNLVHCADLSNPTKPLPLYRQWTDRIMAEFFQQQVGFIDYIAHPLWETWADLVHPDAQDLLDTLEDNREWYQ Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Lecithin-cholesterol acyltransferase (LCAT) | 6MVD | 4.74 | |
Target general information Gen name LCAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phospholipidcholesterolacyltransferase; Phospholipid-cholesterol acyltransferase; Phosphatidylcholinesterol acyltransferase; Phosphatidylcholine-sterol acyltransferase Protein family AB hydrolase superfamily, Lipase family Biochemical class Acyltransferase Function Synthesized mainly in the liver and secreted into plasma where it converts cholesterol and phosphatidylcholines (lecithins) to cholesteryl esters and lysophosphatidylcholines on the surface of high and low density lipoproteins (HDLs and LDLs). The cholesterol ester is then transported back to the liver. Has a preference for plasma 16:0-18:2 or 18:O-18:2 phosphatidylcholines. Also produced in the brain by primary astrocytes, and esterifies free cholesterol on nascent APOE-containing lipoproteins secreted from glia and influences cerebral spinal fluid (CSF) APOE- and APOA1 levels. Together with APOE and the cholesterol transporter ABCA1, plays a key role in the maturation of glial-derived, nascent lipoproteins. Required for remodeling high-density lipoprotein particles into their spherical forms. Central enzyme in the extracellular metabolism of plasma lipoproteins. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P02647; O76024 EC number EC 2.3.1.43 Uniprot keywords 3D-structure; Acyltransferase; Cholesterol metabolism; Corneal dystrophy; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipid metabolism; Proteomics identification; Reference proteome; Secreted; Signal; Steroid metabolism; Sterol metabolism; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42715.4 Length 376 Aromaticity 0.12 Instability index 42.05 Isoelectric point 5.69 Charge (pH=7) -9.12 2D Binding mode Binding energy (Kcal/mol) -6.46  Molscript Map 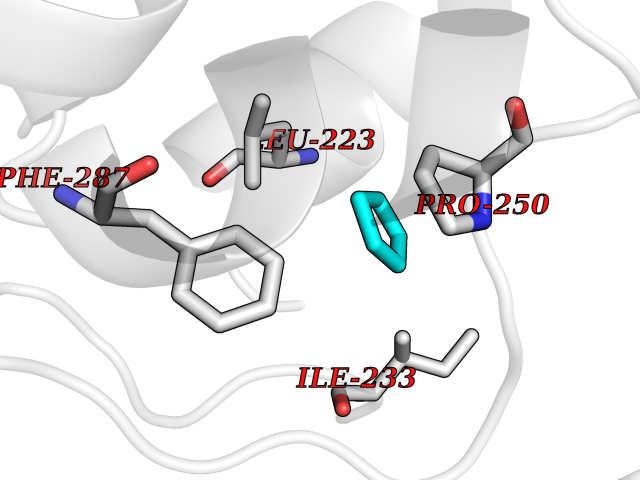 Pymol Map 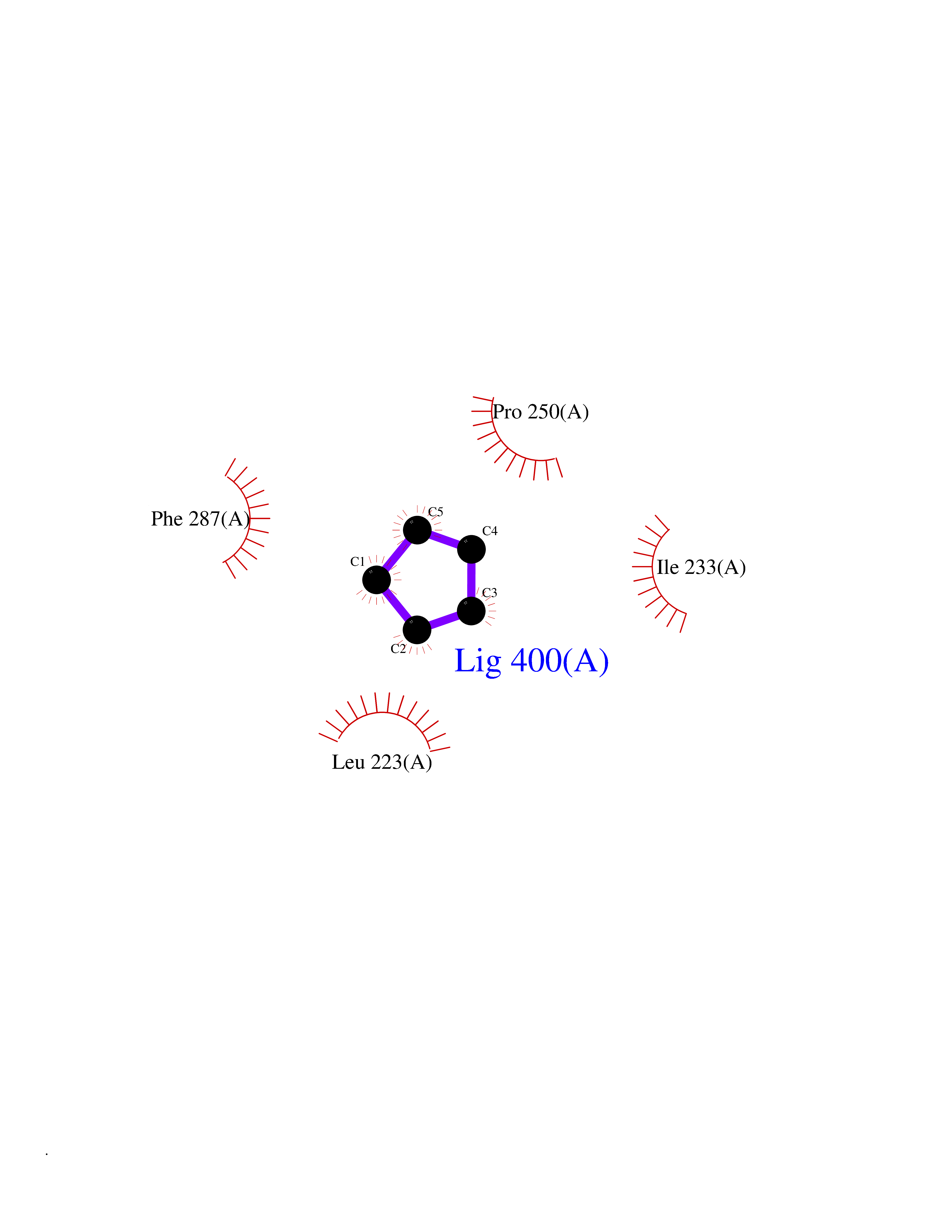 Ligplot Map 3D Binding mode Sequence HTRPVILVPGCLGNQLEAKLDKPDVVNWMCYRKTEDFFTIWLDLNMFLPLGVDCWIDNTRVVYNRSSGLVSNAPGVQIRVPGFGKTYSVEYLDSSKLAGYLHTLVQNLVNNGYVRDETVRAAPYDWRLEPGQQEEYYRKLAGLVEEMHAAYGKPVFLIGHSLGCLHLLYFLLRQPQAWKDRFIDGFISLGAPWGGSIKPMLVLASGDNQGIPIMSSIKEEQRITTTSPWMFPSRMAWPEDHVFISTPSFNYTGRDFQRFFADLHFEEGWYMWLQSRDLLAGLPAPGVEVYCLYGVGLPTPRTYIYDHGFPYTDPVGVLYEDGDDTVATRSTELCGLWQGRQPQPVHLLPLHGIQHLNMVFSNLTLEHINAILLGAH Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | AP endonuclease 1 (APEX1) | 5WN2 | 4.74 | |
Target general information Gen name APEX1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Redox factor-1; REF1; REF-1; HAP1; DNA-(apurinic or apyrimidinic site) lyase; Apurinic-apyrimidinic endonuclease 1; APX; APEX nuclease; APEX; APEN; APE1; APE-1; APE Protein family DNA repair enzymes AP/ExoA family Biochemical class Alpha-carbonic anhydrase Function Multifunctional protein that plays a central role in the cellular response to oxidative stress. The two major activities of APEX1 are DNA repair and redox regulation of transcriptional factors. Functions as a apurinic/apyrimidinic (AP) endodeoxyribonuclease in the DNA base excision repair (BER) pathway of DNA lesions induced by oxidative and alkylating agents. Initiates repair of AP sites in DNA by catalyzing hydrolytic incision of the phosphodiester backbone immediately adjacent to the damage, generating a single-strand break with 5'-deoxyribose phosphate and 3'-hydroxyl ends. Does also incise at AP sites in the DNA strand of DNA/RNA hybrids, single-stranded DNA regions of R-loop structures, and single-stranded RNA molecules. Has a 3'-5' exoribonuclease activity on mismatched deoxyribonucleotides at the 3' termini of nicked or gapped DNA molecules during short-patch BER. Possesses a DNA 3' phosphodiesterase activity capable of removing lesions (such as phosphoglycolate) blocking the 3' side of DNA strand breaks. May also play a role in the epigenetic regulation of gene expression by participating in DNA demethylation. Acts as a loading factor for POLB onto non-incised AP sites in DNA and stimulates the 5'-terminal deoxyribose 5'-phosphate (dRp) excision activity of POLB. Plays a role in the protection from granzymes-mediated cellular repair leading to cell death. Also involved in the DNA cleavage step of class switch recombination (CSR). On the other hand, APEX1 also exerts reversible nuclear redox activity to regulate DNA binding affinity and transcriptional activity of transcriptional factors by controlling the redox status of their DNA-binding domain, such as the FOS/JUN AP-1 complex after exposure to IR. Involved in calcium-dependent down-regulation of parathyroid hormone (PTH) expression by binding to negative calcium response elements (nCaREs). Together with HNRNPL or the dimer XRCC5/XRCC6, associates with nCaRE, acting as an activator of transcriptional repression. Stimulates the YBX1-mediated MDR1 promoter activity, when acetylated at Lys-6 and Lys-7, leading to drug resistance. Acts also as an endoribonuclease involved in the control of single-stranded RNA metabolism. Plays a role in regulating MYC mRNA turnover by preferentially cleaving in between UA and CA dinucleotides of the MYC coding region determinant (CRD). In association with NMD1, plays a role in the rRNA quality control process during cell cycle progression. Associates, together with YBX1, on the MDR1 promoter. Together with NPM1, associates with rRNA. Binds DNA and RNA. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB04967 Interacts with Q09472; Q8N4N3; Q16236; Q96EB6; O88846 EC number EC 3.1.-.- Uniprot keywords 3D-structure; Acetylation; Activator; Cleavage on pair of basic residues; Cytoplasm; Direct protein sequencing; Disulfide bond; DNA damage; DNA recombination; DNA repair; DNA-binding; Endonuclease; Endoplasmic reticulum; Exonuclease; Hydrolase; Magnesium; Metal-binding; Mitochondrion; Nuclease; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; RNA-binding; S-nitrosylation; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 31820.9 Length 285 Aromaticity 0.1 Instability index 42.58 Isoelectric point 8.35 Charge (pH=7) 3.27 2D Binding mode Binding energy (Kcal/mol) -6.47 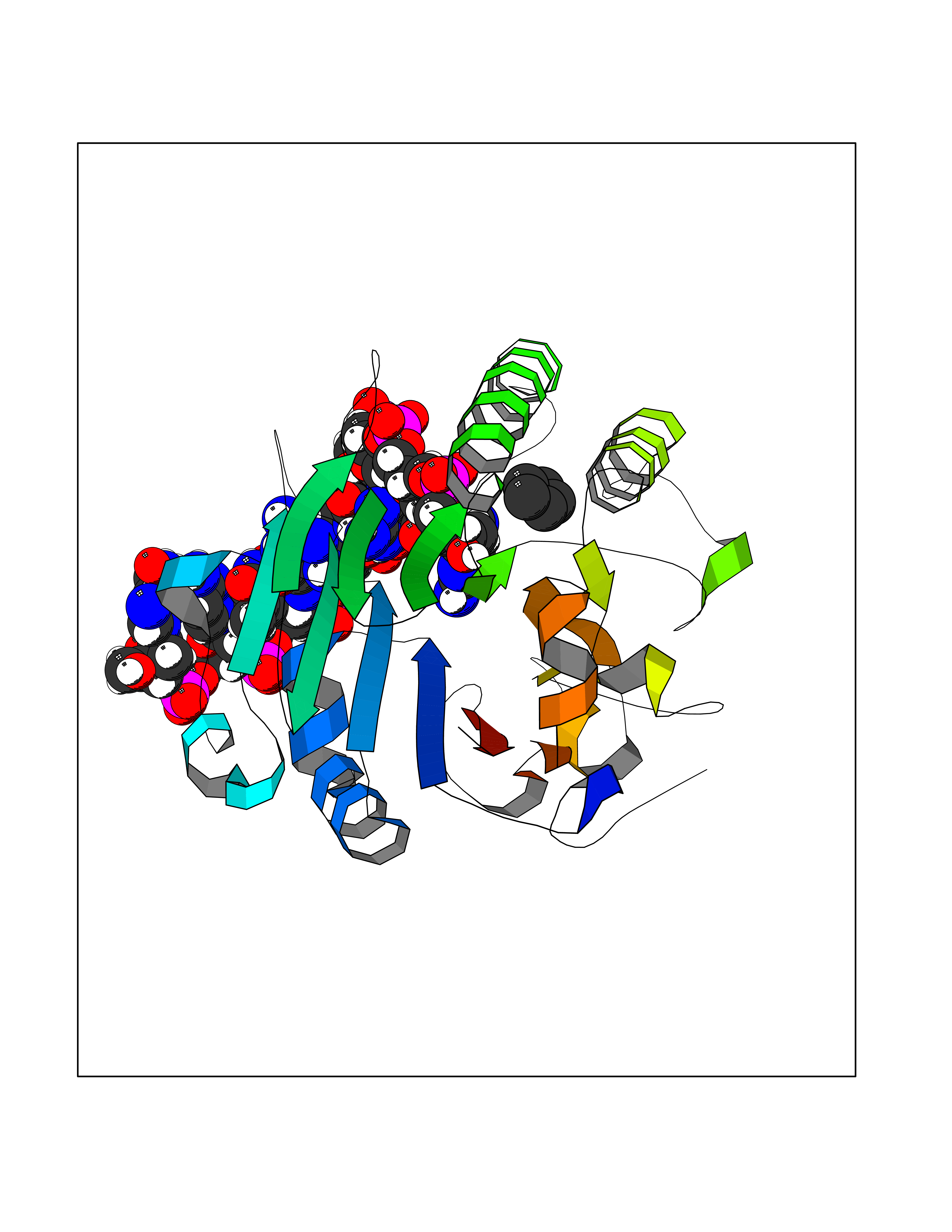 Molscript Map 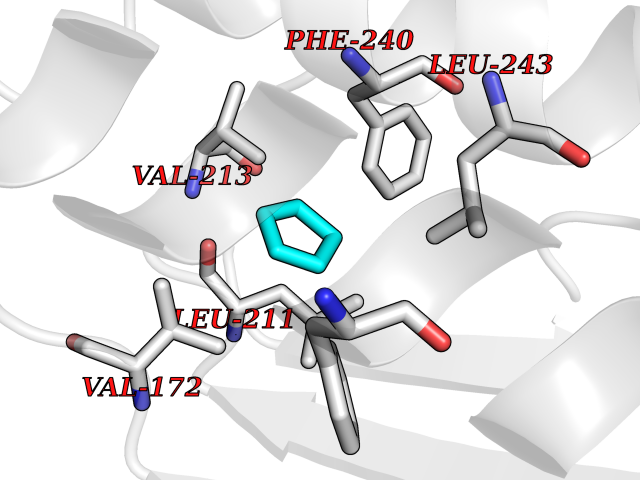 Pymol Map  Ligplot Map 3D Binding mode Sequence ALYEDPPDQKTSPSGKPATLKICSWNVDGLRAWIKKKGLDWVKEEAPDILCLQQTKCSENKLPAELQELPGLSHQYWSAPSDKEGYSGVGLLSRQAPLKVSYGIGDEEHDQEGRVIVAEFDSFVLVTAYVPNAGRGLVRLEYRQRWDEAFRKFLKGLASRKPLVLCGNLNVAHEEIDLRNPKGNKKNAGFTPQERQGFGELLQAVPLADSFRHLYPNTPYAYTFWTYMMNARSKNVGWRLDYFLLSHSLLPALCDSKIRSKALGSDHCPITLYLALGCTGATGCG Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Hepatocyte nuclear factor 4-alpha (HNF4A) | 6CHT | 4.74 | |
Target general information Gen name HNF4A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Transcription factor HNF-4; Transcription factor 14; TCF14; TCF-14; Nuclear receptor subfamily 2 group A member 1; NR2A1; HNF4; HNF-4-alpha Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Activates the transcription of CYP2C38. Represses the CLOCK-ARNTL/BMAL1 transcriptional activity and is essential for circadian rhythm maintenance and period regulation in the liver and colon cells. Transcriptional regulator which controls the expression of hepatic genes during the transition of endodermal cells to hepatic progenitor cells, facilitating the recruitment of RNA pol II to the promoters of target genes. Related diseases Maturity-onset diabetes of the young 1 (MODY1) [MIM:125850]: A form of diabetes that is characterized by an autosomal dominant mode of inheritance, onset in childhood or early adulthood (usually before 25 years of age), a primary defect in insulin secretion and frequent insulin-independence at the beginning of the disease. {ECO:0000269|PubMed:10389854, ECO:0000269|PubMed:17407387, ECO:0000269|PubMed:9243109, ECO:0000269|PubMed:9313765}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Type 2 diabetes mellitus (T2D) [MIM:125853]: A multifactorial disorder of glucose homeostasis caused by a lack of sensitivity to insulin. Affected individuals usually have an obese body habitus and manifestations of a metabolic syndrome characterized by diabetes, insulin resistance, hypertension and hypertriglyceridemia. The disease results in long-term complications that affect the eyes, kidneys, nerves, and blood vessels. {ECO:0000269|PubMed:9449683}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 4 with maturity-onset diabetes of the young (FRTS4) [MIM:616026]: An autosomal dominant disease characterized by Fanconi syndrome associated with a beta cell phenotype of neonatal hyperinsulinism with macrosomia and young onset diabetes. Fanconi syndrome is a proximal tubulopathy resulting in generalized aminoaciduria, low molecular weight proteinuria, glycosuria, hyperphosphaturia and hypouricemia. Some FRTS4 patients have nephrocalcinosis, renal impairment, hypercalciuria with relative hypocalcemia, and hypermagnesemia. {ECO:0000269|PubMed:22802087, ECO:0000269|PubMed:24285859}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05447; DB03017; DB08231 Interacts with Q99967; A8MYZ6; P04150; Q9UBK2; Q92786; P23246; Q12772; P04637; P11532; O14602; O43688 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative promoter usage; Alternative splicing; Biological rhythms; Diabetes mellitus; Disease variant; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repressor; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B Molecular weight (Da) 23446.1 Length 205 Aromaticity 0.08 Instability index 39.17 Isoelectric point 5.05 Charge (pH=7) -6.97 2D Binding mode Binding energy (Kcal/mol) -6.46  Molscript Map 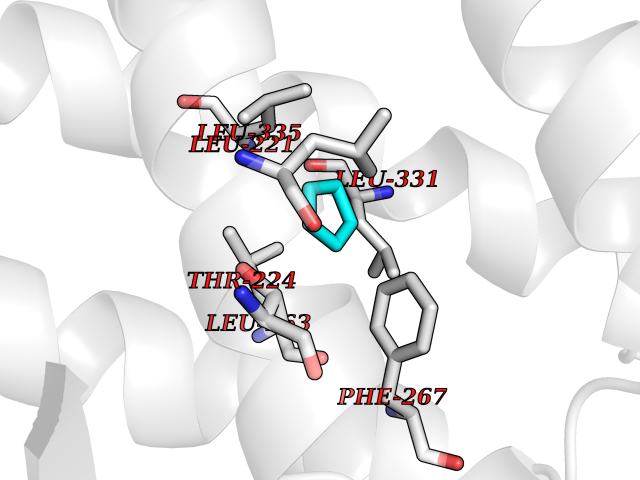 Pymol Map  Ligplot Map 3D Binding mode Sequence GINGDIRAKKIASIADVCESMKEQLLVLVEWAKYIPAFCELPLDDQVALLRAHAGEHLLLGATKRSMVFKDVLLLGNDYIVPRHCPELAEMSRVSIRILDELVLPFQELQIDDNEYAYLKAIIFFDPDAKGLSDPGKIKRLRSQVQVSLEDYINDRQYDSRGRFGELLLLLPTLQSITWQMIEQIQFIKLFGMAKIDNLLQEMLL Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Bacterial Botulinum toxin A (Bact botA) | 6XCF | 4.74 | |
Target general information Gen name Bact botA Organism Clostridium botulinum Uniprot ID TTD ID Synonyms botA Protein family Peptidase M27 family Biochemical class Peptidase Function Inhibits acetylcholine release. The botulinum toxin binds with high affinity to peripheral neuronal presynaptic membrane to the secretory vesicle protein SV2. It binds directly to the largest luminal loop of SV2A, SV2B and SV2C. It is then internalized by receptor-mediated endocytosis. The C-terminus of the heavy chain (H) is responsible for the adherence of the toxin to the cell surface while the N-terminus mediates transport of the light chain from the endocytic vesicle to the cytosol. After translocation, the light chain (L) hydrolyzes the 197-Gln-|-Arg- 198 bond in SNAP-25, thereby blocking neurotransmitter release. Inhibition of acetylcholine release results in flaccid paralysis, with frequent heart or respiratory failure. Related diseases Major depressive disorder (MDD) [MIM:608516]: A common psychiatric disorder. It is a complex trait characterized by one or more major depressive episodes without a history of manic, mixed, or hypomanic episodes. A major depressive episode is characterized by at least 2 weeks during which there is a new onset or clear worsening of either depressed mood or loss of interest or pleasure in nearly all activities. Four additional symptoms must also be present including changes in appetite, weight, sleep, and psychomotor activity; decreased energy; feelings of worthlessness or guilt; difficulty thinking, concentrating, or making decisions; or recurrent thoughts of death or suicidal ideation, plans, or attempts. The episode must be accompanied by distress or impairment in social, occupational, or other important areas of functioning. {ECO:0000269|PubMed:15229186}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q02563; Q496J9; Q9Z2I6 EC number NA Uniprot keywords 3D-structure; Cell wall; Direct protein sequencing; Disulfide bond; Host cell membrane; Host cytoplasm; Host cytoplasmic vesicle; Host membrane; Host synapse; Hydrolase; Lipid-binding; Membrane; Metal-binding; Metalloprotease; Neurotoxin; Pharmaceutical; Protease; Secreted; Toxin; Transmembrane; Transmembrane helix; Ubl conjugation; Virulence; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 47759.6 Length 417 Aromaticity 0.13 Instability index 21.7 Isoelectric point 6.36 Charge (pH=7) -1.96 2D Binding mode Binding energy (Kcal/mol) -6.47  Molscript Map  Pymol Map 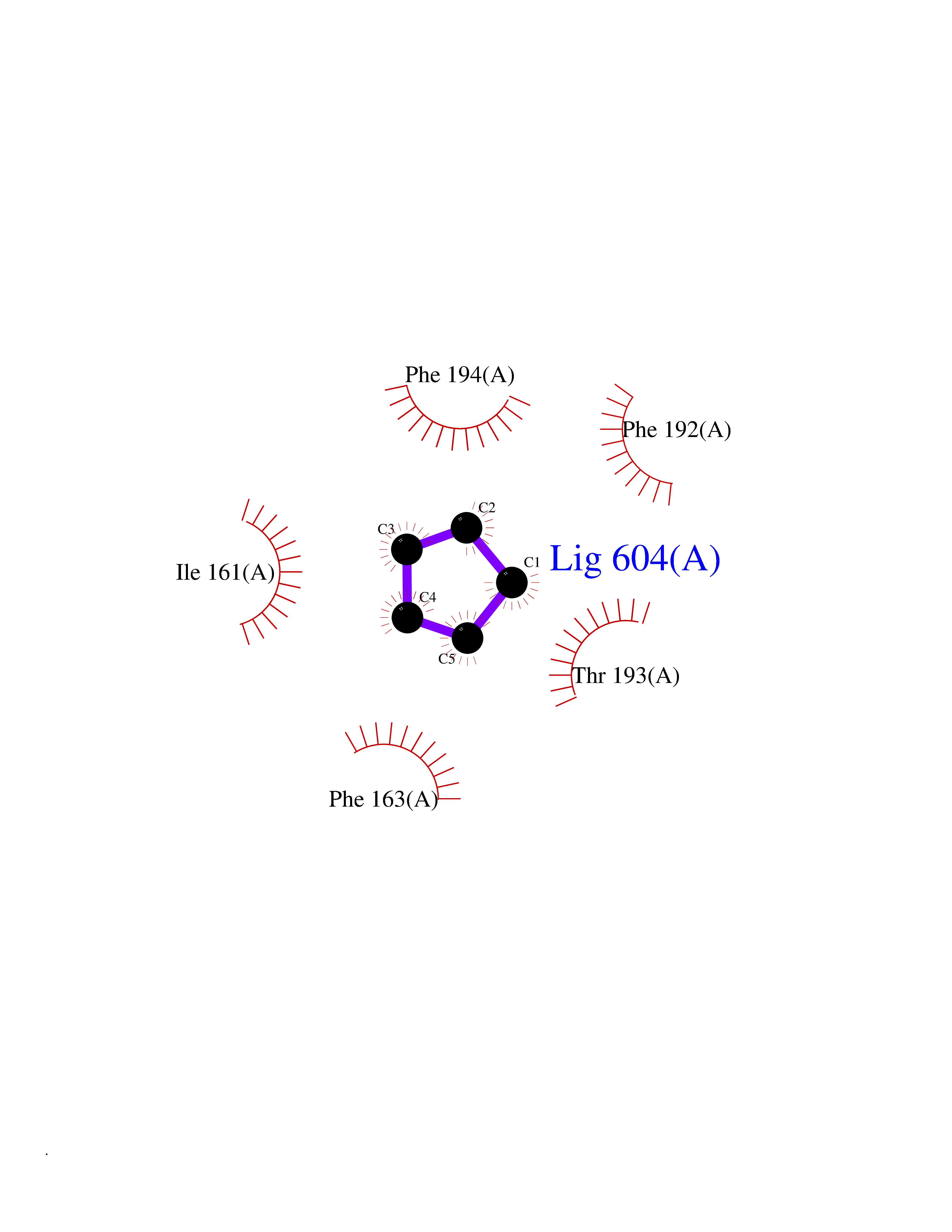 Ligplot Map 3D Binding mode Sequence MQFVNKQFNYKDPVNGVDIAYIKIPNVGQMQPVKAFKIHNKIWVIPERDTFTNPEEGDLNPPPPVSYYDSTYLSTDNEKDNYLKGVTKLFERIYSTDLGRMLLTSIVRGIPFWGGSTIDTELKVIDTNCINVIQPDGSYRSEELNLVIIGPSADIIQFECKSFGHEVLNLTRNGYGSTQYIRFSPDFTFGFEESLEVDTNPLLGAGKFATDPAVTLAHELIHAGHRLYGIAINPNRVFKVNTNAYYEMSGLEVSFEELRTFGGHDAKFIDSLQENEFRLYYYNKFKDIASTLNKAKSIVGTTASLQYMKNVFKEKYLLSEDTSGKFSVDKLKFDKLYKMLTEIYTEDNFVKFFKVLNRKTYLNFDKAVFKINIVPKVNYTIYDGFNLRNTNLAANFNGQNTEINNMNFTKLKNFTGL Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Neuronal acetylcholine receptor alpha-2 (CHRNA2) | 5FJV | 4.74 | |
Target general information Gen name CHRNA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNA2 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-2/CHRNA2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Epilepsy, nocturnal frontal lobe, 4 (ENFL4) [MIM:610353]: An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking. {ECO:0000269|PubMed:16826524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Seizures, benign familial infantile, 6 (BFIS6) [MIM:610353]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS6 inheritance is autosomal dominant. {ECO:0000269|PubMed:25847220}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00732; DB00237; DB00411; DB00565; DB01245; DB00514; DB01135; DB07720; DB00898; DB00472; DB00483; DB08960; DB00657; DB01336; DB00416; DB01226; DB00184; DB01337; DB01338; DB00721; DB00728; DB05740; DB00202; DB01199; DB01339 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 48323.4 Length 413 Aromaticity 0.15 Instability index 32 Isoelectric point 5.69 Charge (pH=7) -6.58 2D Binding mode Binding energy (Kcal/mol) -6.47  Molscript Map 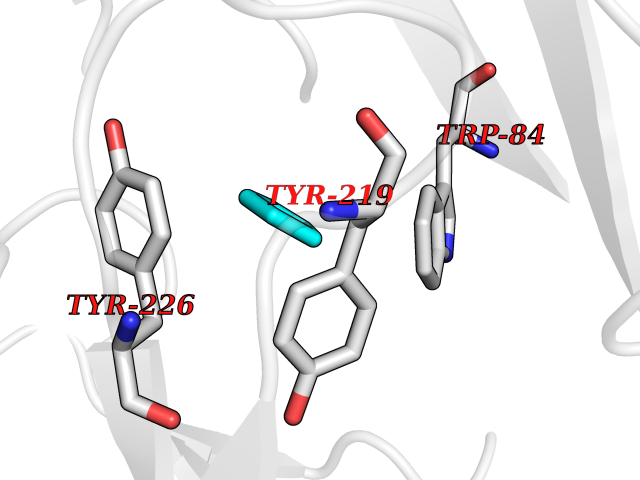 Pymol Map 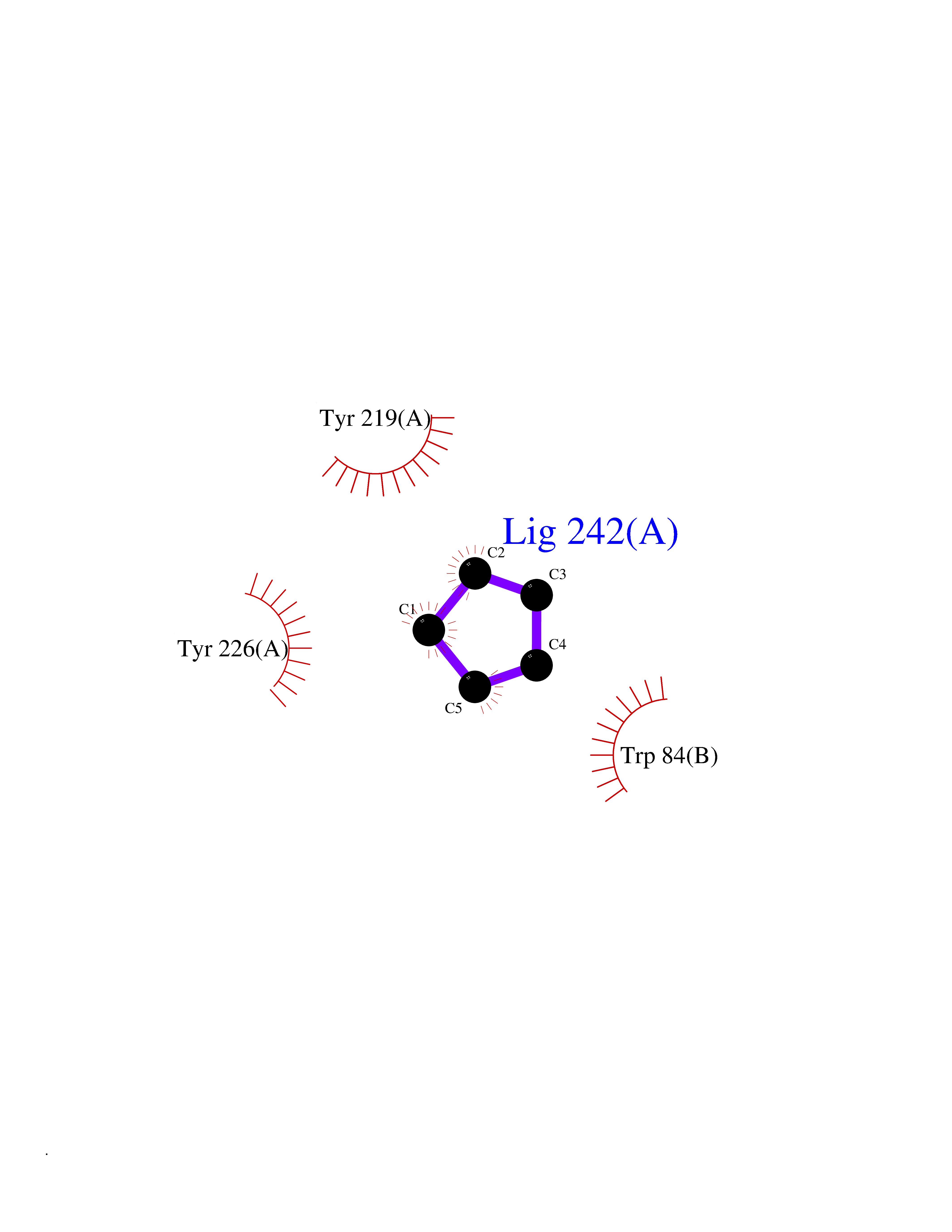 Ligplot Map 3D Binding mode Sequence DRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLP Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Ephrin type-B receptor 3 (EPHB3) | 5L6O | 4.74 | |
Target general information Gen name EPHB3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hEK2; Tyrosine-protein kinase TYRO6; TYRO6; Embryonic kinase 2; ETK2; EPH-like tyrosine kinase 2; EPH-like kinase 2; EK2 Protein family Protein kinase superfamily, Tyr protein kinase family, Ephrin receptor subfamily Biochemical class Kinase Function The signaling pathway downstream of the receptor is referred to as forward signaling while the signaling pathway downstream of the ephrin ligand is referred to as reverse signaling. Generally has an overlapping and redundant function with EPHB2. Like EPHB2, functions in axon guidance during development regulating for instance the neurons forming the corpus callosum and the anterior commissure, 2 major interhemispheric connections between the temporal lobes of the cerebral cortex. In addition to its role in axon guidance plays also an important redundant role with other ephrin-B receptors in development and maturation of dendritic spines and the formation of excitatory synapses. Controls other aspects of development through regulation of cell migration and positioning. This includes angiogenesis, palate development and thymic epithelium development for instance. Forward and reverse signaling through the EFNB2/EPHB3 complex also regulate migration and adhesion of cells that tubularize the urethra and septate the cloaca. Finally, plays an important role in intestinal epithelium differentiation segregating progenitor from differentiated cells in the crypt. Receptor tyrosine kinase which binds promiscuously transmembrane ephrin-B family ligands residing on adjacent cells, leading to contact-dependent bidirectional signaling into neighboring cells. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) NA Interacts with P37235; O75031 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Angiogenesis; ATP-binding; Cell membrane; Cell projection; Developmental protein; Disulfide bond; Glycoprotein; Kinase; Membrane; Neurogenesis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30412.9 Length 267 Aromaticity 0.09 Instability index 37.42 Isoelectric point 7.74 Charge (pH=7) 1 2D Binding mode Binding energy (Kcal/mol) -6.47  Molscript Map  Pymol Map 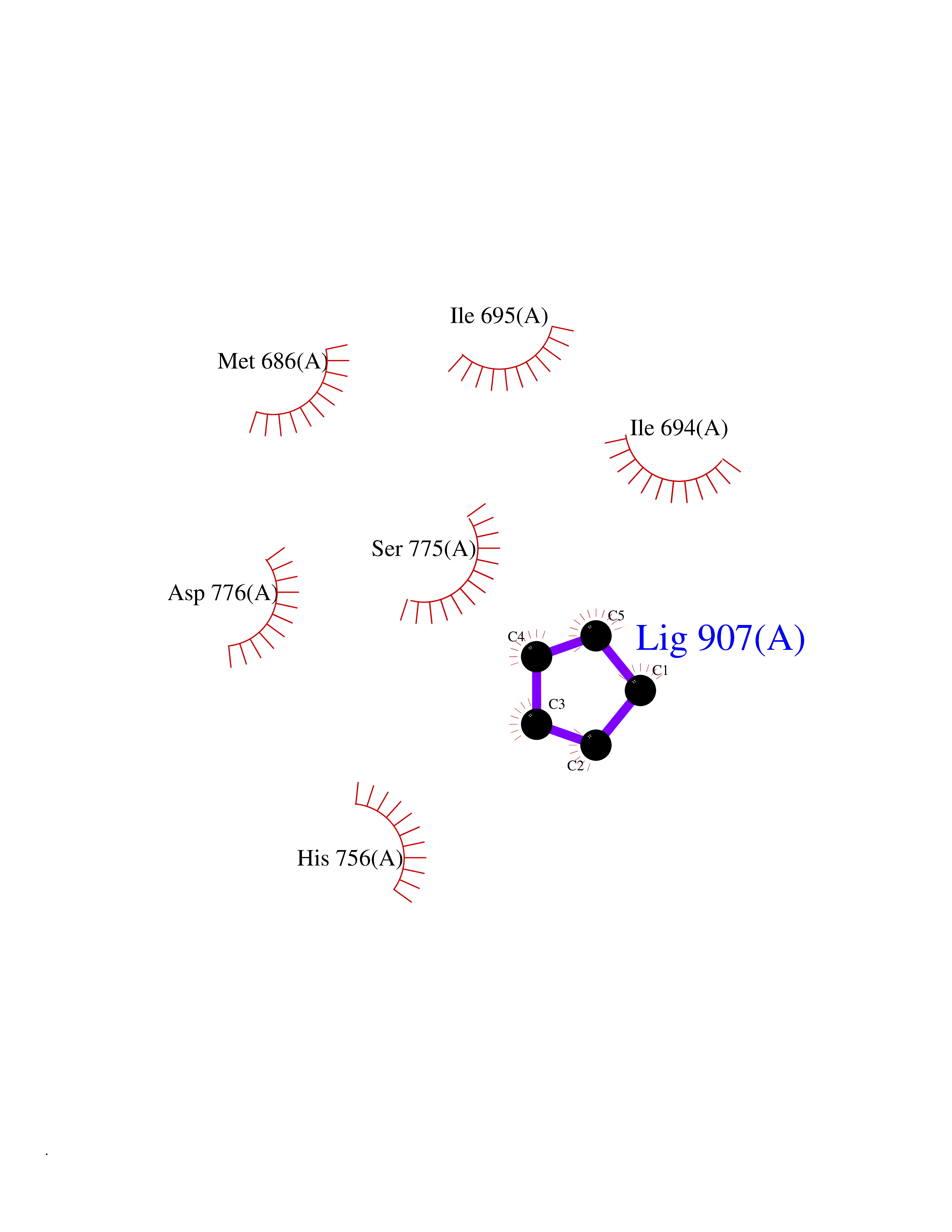 Ligplot Map 3D Binding mode Sequence CVKIEEVIGAGEVCRGRLKQPGRREVFVAIKTLKVGYTERQRRDFLSEASIMGQFDHPNIIRLEGVVTKSRPVMILTEFMENCALDSFLRLNDGQFTVIQLVGMLRGIAAGMKYLSEMNYVHRDLAARNILVNSNLVCKVSDFGLEDDPSDPTYTSSLGGKIPIRWTAPEAIAYRKFTSASDVWSYGIVMWEVMSYGERPYWDMSNQDVINAVEQDYRLPPPMDCPTALHQLMLDCWVRDRNLRPKFSQIVNTLDKLIRNPASLKVI Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Voltage-gated calcium channel alpha Cav3.1 (CACNA1G) | 6KZP | 4.74 | |
Target general information Gen name CACNA1G Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Voltage-gated calcium channel alpha subunit Cav3.1; Voltage-dependent T-type calcium channel; NBR13; Cav3.1c; CACNA1G Protein family Calcium channel alpha-1 subunit (TC 1.A.1.11) family, CACNA1G subfamily Biochemical class Voltage-gated ion channel Function Voltage-sensitive calcium channels (vscc) mediate the entry of calcium ions into excitable cells and are also involved in a variety of calcium-dependent processes, including muscle contraction, hormone or neurotransmitter release and gene expression. Related diseases Spinocerebellar ataxia 42 (SCA42) [MIM:616795]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA42 is a slowly progressive, autosomal dominant form with variable severity. {ECO:0000269|PubMed:26456284, ECO:0000269|PubMed:26715324}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 42, early-onset, severe, with neurodevelopmental deficits (SCA42ND) [MIM:618087]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA42ND is an early-onset, severe form associated with motor and cognitive impairment, cerebellar atrophy as well as variable features such as facial dysmorphisms, digital anomalies, microcephaly and epilepsy. SCA42ND inheritance is autosomal dominant. {ECO:0000269|PubMed:29878067}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09227; DB09231; DB13746; DB11148; DB11093; DB11348; DB14481; DB09061; DB00568; DB09235; DB00228; DB00153; DB00593; DB04841; DB09238; DB14009; DB05246; DB01388; DB14011; DB00622; DB01115; DB06712; DB09089; DB00347; DB00661; DB00909 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calcium channel; Calcium transport; Cell membrane; Cytoplasm; Disease variant; Glycoprotein; Ion channel; Ion transport; Membrane; Neurodegeneration; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 116116 Length 1014 Aromaticity 0.13 Instability index 33.06 Isoelectric point 6.61 Charge (pH=7) -2.04 2D Binding mode Binding energy (Kcal/mol) -6.46 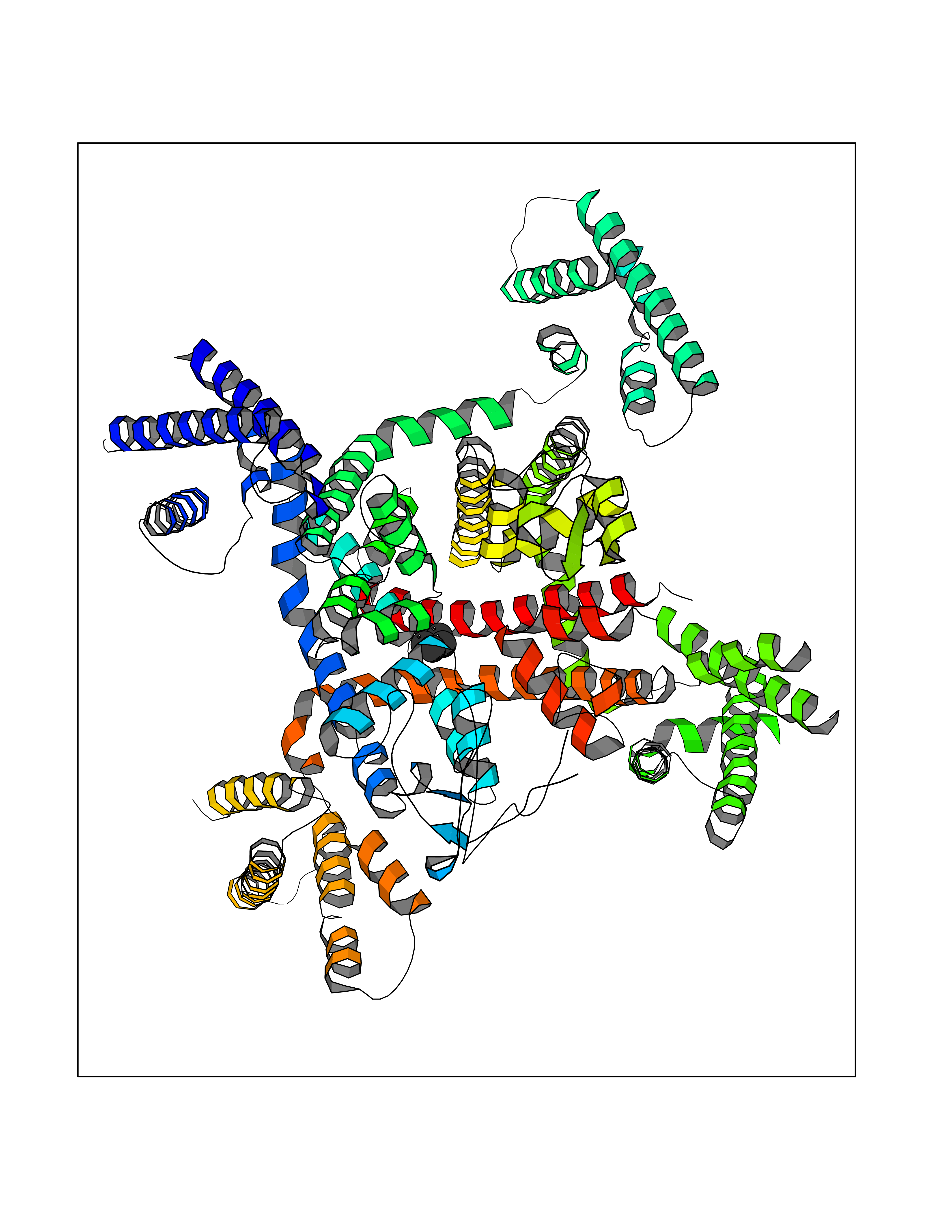 Molscript Map 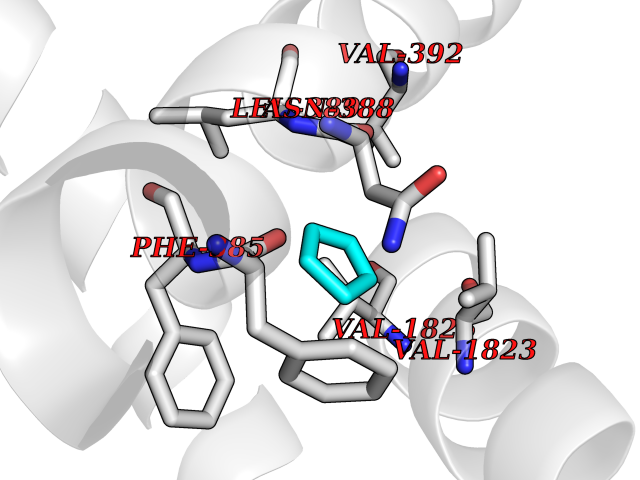 Pymol Map 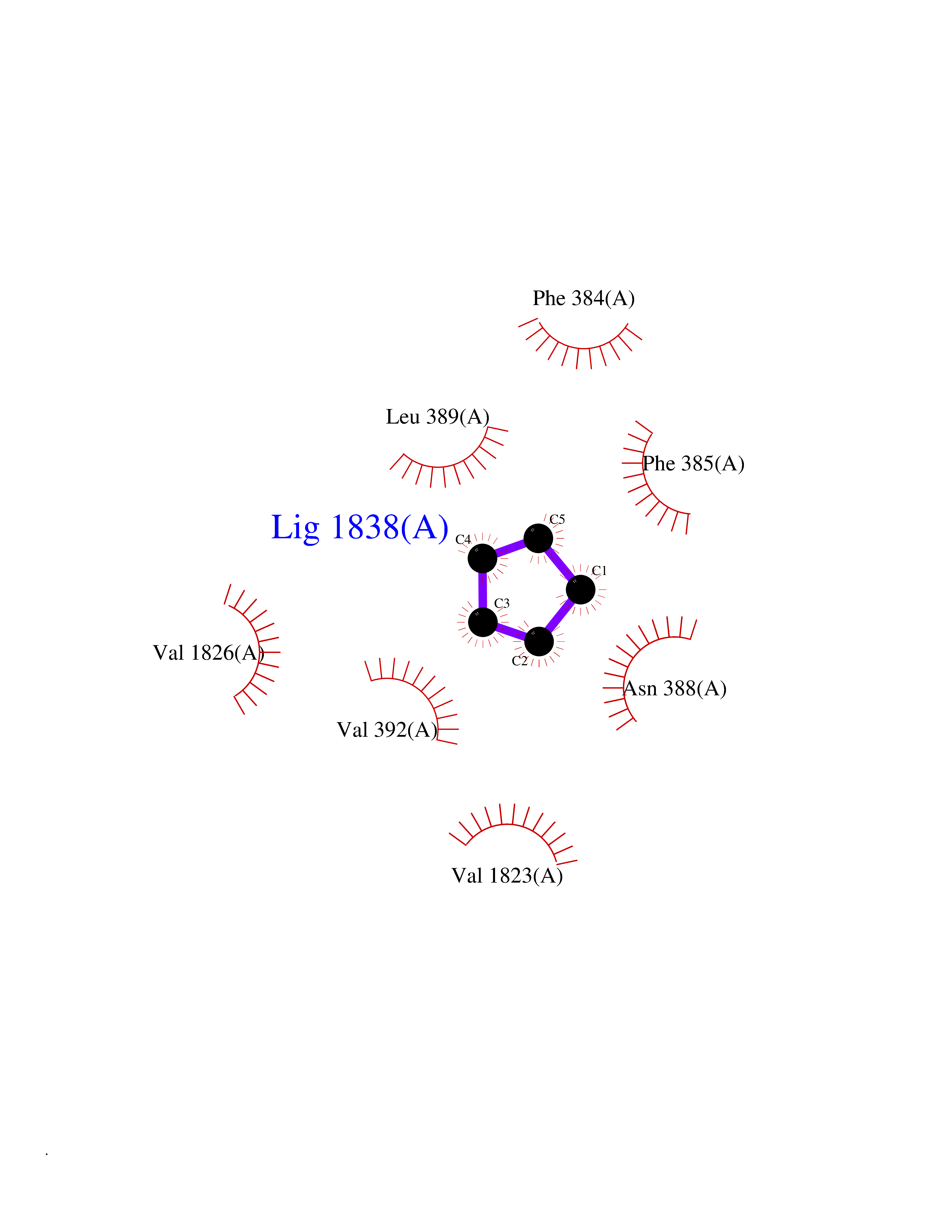 Ligplot Map 3D Binding mode Sequence NPWFERISMLVILLNCVTLGMFRPCEDIACDSQRCRILQAFDDFIFAFFAVEMVVKMVAGDTWNRLDFFIVIAGMLEYSLDLQNVSFSAVRTVRVLRPLRAINRVPSMRILVTLLLDTLPMLGNVLLLCFFVFFIFGIVGVQLWAGLLRNRCFLPENFSLPLSVDLERYYQTENEDESPFICSQPRENGMRSCRSVPTLRCVNWNQYYTNCSAGEHNPFKGAINFDNIGYAWIAIFQVITLEGWVDIMYFVMDAHSFYNFIYFILLIIVGSFFMINLCLVVIATQFSETKQREIVDSKYFGRGIMIAILVNTLSMGIEYHEQPEELTNALEISNIVFTSLFALEMLLKLLVYGPFGYIKNPYNIFDGVIVVISVWEIVSVLRTFRLMRVLKLVRFLPALQRQLVVLMKTMDNVATFCMLLMLFIFIFSILGMHLFGCKFASLPDRKNFDSLLWAIVTVFQILTQEDWNKVLYNGMASTSSWAALYFIALMTFGNYVLFNLLVAILVEGFQFRLLCHRIITHKMFDHVVLVIIFLNCITIAMERPKIDPHSAERIFLTLSNYIFTAVFLAEMTVKVVALGSSWNVLDGLLVLISVIDILVSMVSKILGMLRVLRLLRTLRPLRVISRAQGLKLVVETLMSSLKPIGNIVVICCAFFIIFGILGVQLFKGKFFVCQGEDTRNITNKSDCAEASYRWVRHKYNFDNLGQALMSLFVLASKDGWVDIMYDGLDAVGVDQQPIMNHNPWMLLYFISFLLIVAFFVLNMFVGVVVENFHYLDLFITGVIGLNVVTMAMEHYQQPQILDEALKICNYIFTVIFVLESVFKLVAFGFRRFFQDRWNQLDLAIVLLSIMGITLEEIEVNASLPINPTIIRIMRVLRIARVLKLLKMAVGMRALLDTVMQALPQVGNLGLLFMLLFFIFAALGVELFGDLECDETHPCEGLGRHATFRNFGMAFLTLFRVSTGDNWNGIMKDTLRDYNTVISPIYFVSFVLTAQFVLVNVVIAVLMKHLEESNK Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Neuronal acetylcholine receptor beta-4 (CHRNB4) | 6PV7 | 4.74 | |
Target general information Gen name CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNB4; Beta-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-4/CHRNB4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. {ECO:0000269|PubMed:20054297, ECO:0000269|PubMed:23622243, ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}. The disease may be caused by variants affecting the gene represented in this entry. Defects of SETD2 are associated with loss of DNA methylation at non-promoter regions (PubMed:23792563). SETD2 defects lead to aberrant and reduced nucleosome compaction and chromatin association of key replication proteins, such as MCM7 and DNA polymerase delta, leading to hinder replication fork progression and prevent loading of RAD51 homologous recombination repair factor at DNA breaks (PubMed:25728682). {ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}.; DISEASE: Luscan-Lumish syndrome (LLS) [MIM:616831]: An autosomal dominant syndrome with a variable phenotype. Clinical features include macrocephaly, distinctive facial appearance, postnatal overgrowth, various degrees of learning difficulties, autism spectrum disorder, and intellectual disability. {ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24852293, ECO:0000269|PubMed:26084711, ECO:0000269|PubMed:27317772}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute lymphoblastic (ALL) [MIM:613065]: A subtype of acute leukemia, a cancer of the white blood cells. ALL is a malignant disease of bone marrow and the most common malignancy diagnosed in children. The malignant cells are lymphoid precursor cells (lymphoblasts) that are arrested in an early stage of development. The lymphoblasts replace the normal marrow elements, resulting in a marked decrease in the production of normal blood cells. Consequently, anemia, thrombocytopenia, and neutropenia occur to varying degrees. The lymphoblasts also proliferate in organs other than the marrow, particularly the liver, spleen, and lymphnodes. {ECO:0000269|PubMed:24509477, ECO:0000269|PubMed:24662245}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16314571, ECO:0000269|PubMed:24509477}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Intellectual developmental disorder, autosomal dominant 70 (MRD70) [MIM:620157]: An autosomal dominant disorder characterized by mild global developmental delay, moderately impaired intellectual disability with speech difficulties, and behavioral abnormalities. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rabin-Pappas syndrome (RAPAS) [MIM:620155]: An autosomal dominant neurodevelopmental disorder characterized by severely impaired global development, intellectual disability, microcephaly, facial dysmorphism, and variable congenital anomalies affecting the skeletal, genitourinary, cardiac, and other organ systems. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00237; DB00565; DB00514; DB07720; DB00898; DB00472; DB01227; DB00184; DB01090; DB00202 Interacts with Q6FHY5 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -6.46  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | ERK activator kinase 2 (MEK2) | 1S9I | 4.74 | |
Target general information Gen name MAP2K2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PRKMK2; MKK2; MEK 2; MAPKK 2; MAPK/ERK kinase 2; MAP kinase kinase 2; Dual specificity mitogenactivated protein kinase kinase 2; Dual specificity mitogen-activated protein kinase kinase 2 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase subfamily Biochemical class Kinase Function Activates the ERK1 and ERK2 MAP kinases. Catalyzes the concomitant phosphorylation of a threonine and a tyrosine residue in a Thr-Glu-Tyr sequence located in MAP kinases. Related diseases Cardiofaciocutaneous syndrome 4 (CFC4) [MIM:615280]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. {ECO:0000269|PubMed:16439621, ECO:0000269|PubMed:18042262, ECO:0000269|PubMed:20358587}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11967; DB06616; DB12010; DB14904; DB11689; DB08911 Interacts with P05067; P10398; Q96II5; P15056; O95273; Q12959; P61978-2; Q8IVT5; P00540; P04049 EC number EC 2.7.12.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cardiomyopathy; Cytoplasm; Direct protein sequencing; Disease variant; Ectodermal dysplasia; Intellectual disability; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 33960.9 Length 303 Aromaticity 0.07 Instability index 45.61 Isoelectric point 6.29 Charge (pH=7) -2.53 2D Binding mode Binding energy (Kcal/mol) -6.46 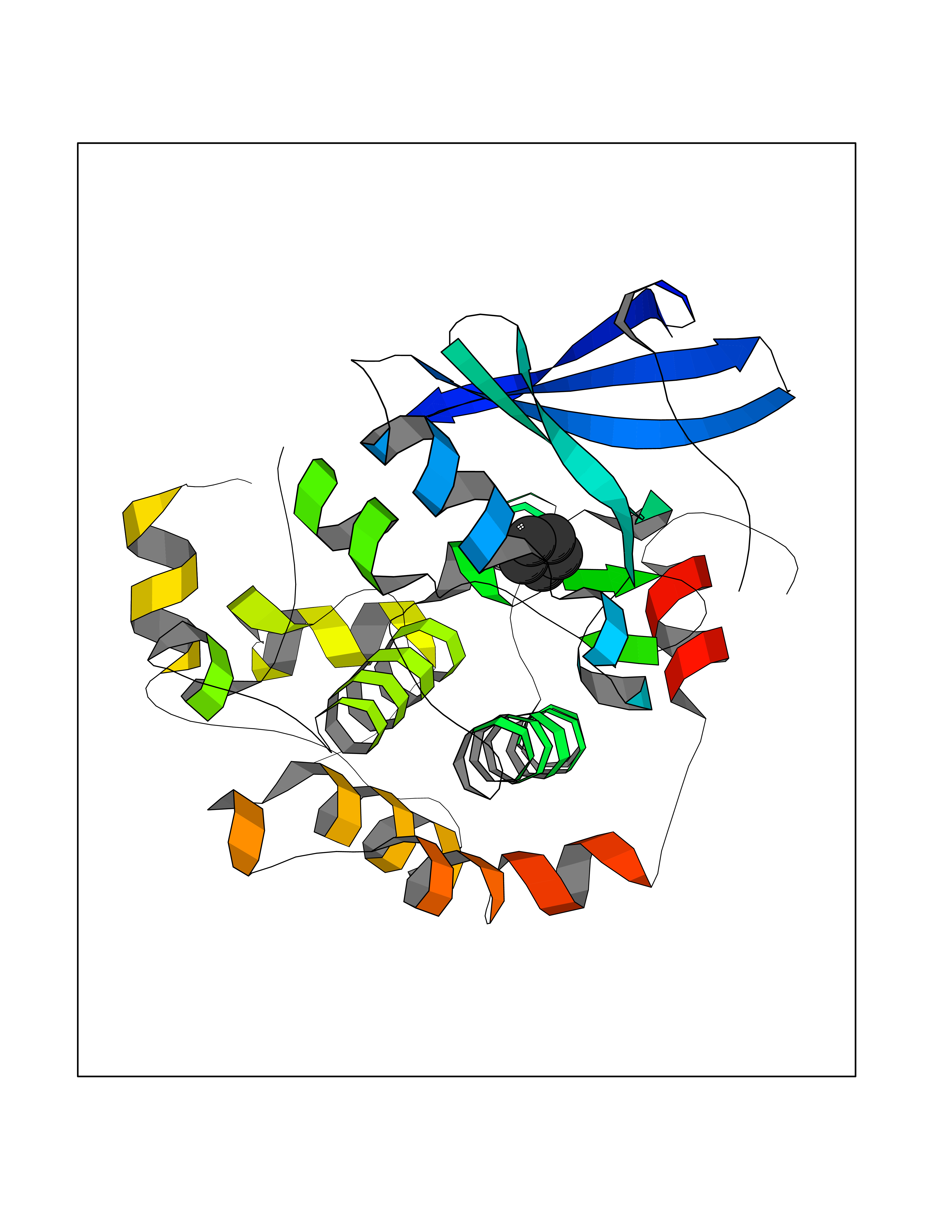 Molscript Map 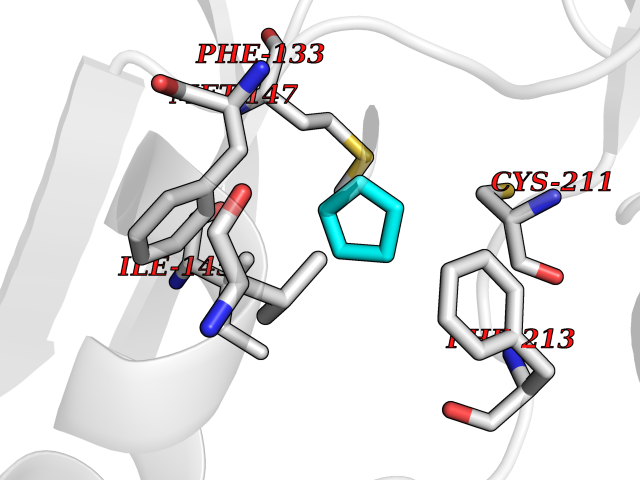 Pymol Map 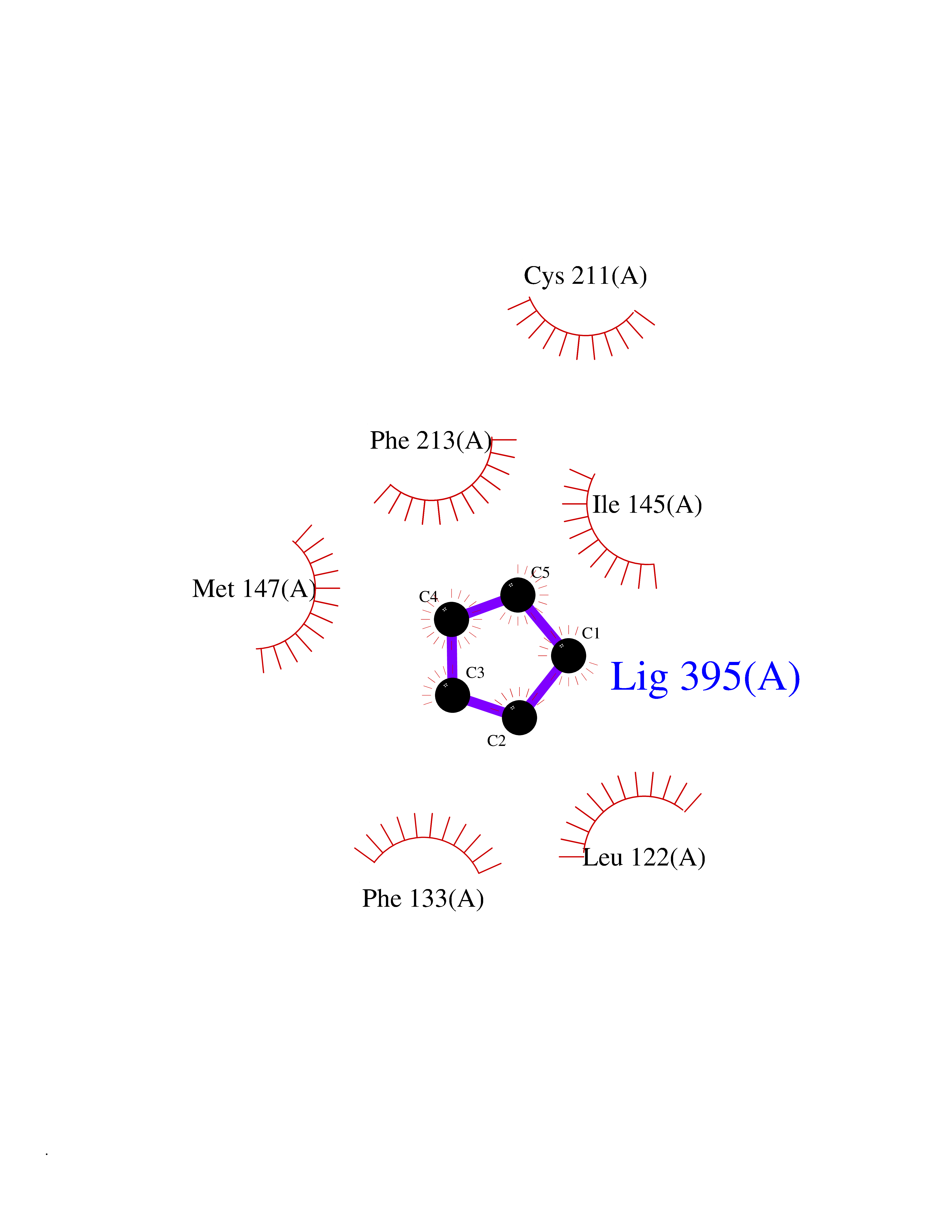 Ligplot Map 3D Binding mode Sequence QKAKVGELKDDDFERISELGAGNGGVVTKVQHRPSGLIMARKLIHLEIKPAIRNQIIRELQVLHECNSPYIVGFYGAFYSDGEISICMEHMDGGSLDQVLKEAKRIPEEILGKVSIAVLRGLAYLREKHQIMHRDVKPSNILVNSRGEIKLCDFGVSGQLIDSMVGTRSYMAPERLQGTHYSVQSDIWSMGLSLVELAVGRYPIPPPDAKELEAIFGRPVVDRPAMAIFELLDYIVNEPPPKLPNGVFTPDFQEFVNKCLIKNPAERADLKMLTNHTFIKRSEVEEVDFAGWLCKTLRLNQPG Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Programmed cell death 1 ligand 1 (PD-L1) | 5J89 | 4.74 | |
Target general information Gen name CD274 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hPD-L1; Programmed death ligand 1; PDL1; PDCD1LG1; PDCD1L1; PDCD1 ligand 1; B7H1; B7-H1; B7 homolog 1 Protein family Immunoglobulin superfamily, BTN/MOG family Biochemical class Immunoglobulin Function As a ligand for the inhibitory receptor PDCD1/PD-1, modulates the activation threshold of T-cells and limits T-cell effector response. Through a yet unknown activating receptor, may costimulate T-cell subsets that predominantly produce interleukin-10 (IL10). Plays a critical role in induction and maintenance of immune tolerance to self. Related diseases Truncation of the 3'-untranslated (3'-UTR) region of CD274 transcripts leads to elevated expression of CD274 in multiple cancers including T-cell leukemia, diffuse large B-cell lymphoma and stomach adenocarcinoma (PubMed:27281199). Disruption of 3'-UTR region is caused by structural variants that stabilize CD274 transcripts, leading to overexpression (PubMed:27281199). Increased expression in tumors promotes immune evasion and tumor cell growth by allowing malignant cells to escape destruction by the immune system (PubMed:27281199). {ECO:0000269|PubMed:27281199}. Drugs (DrugBank ID) DB15773; DB11595; DB15771; DB11945; DB15772; DB14776; DB15770; DB11714; DB15769; DB09035; DB09037; DB00203; DB00313 Interacts with P33681; Q8IZR5; Q9NX76; Q15116; Q15116 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Endosome; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Nucleus; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID C,D Molecular weight (Da) 28335.2 Length 249 Aromaticity 0.1 Instability index 35.39 Isoelectric point 6.15 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -6.46  Molscript Map 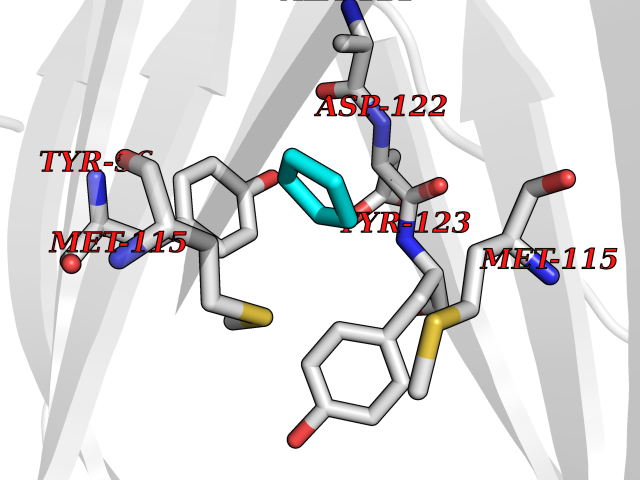 Pymol Map 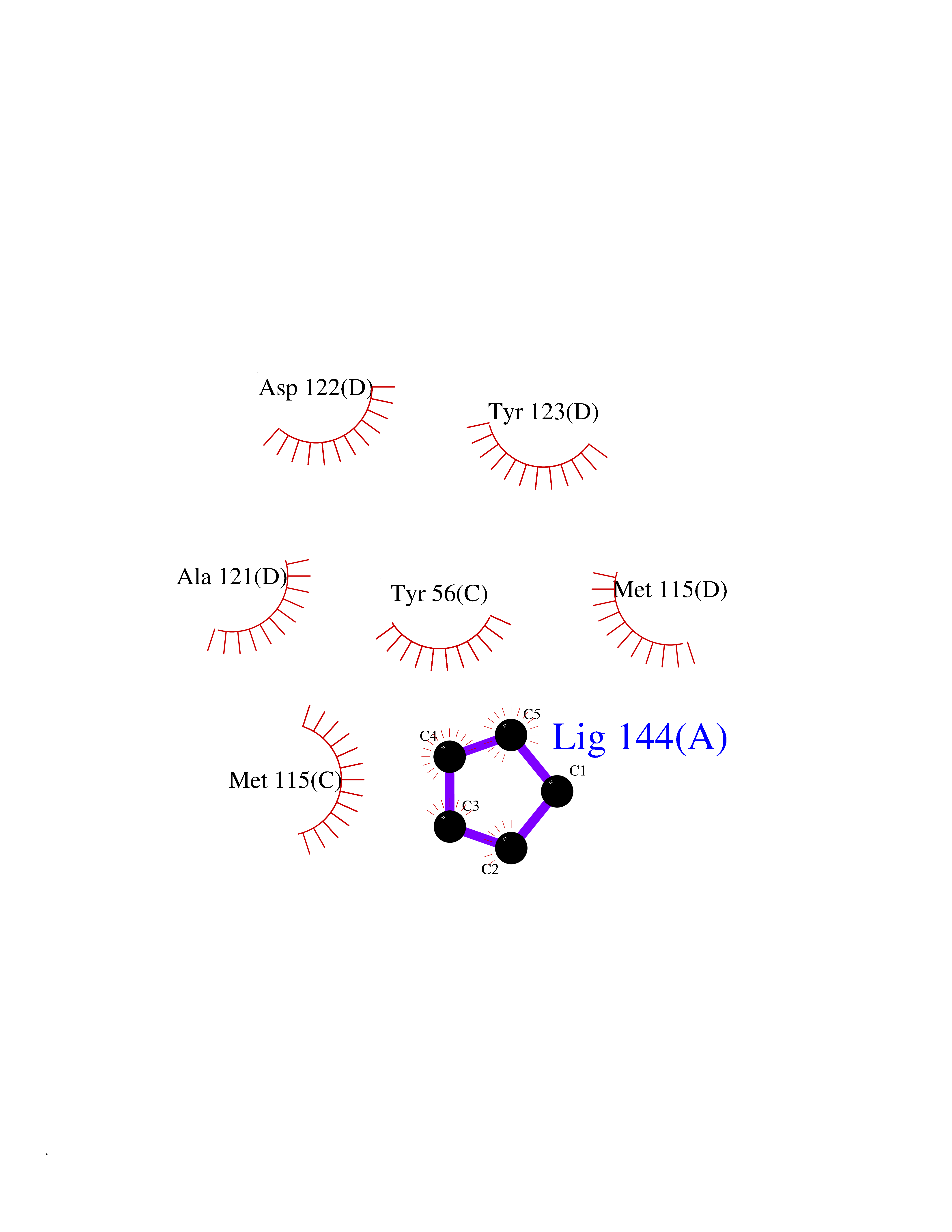 Ligplot Map 3D Binding mode Sequence AFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHHHAFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHH Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Endolysin | 1AM7 | 4.73 | |
Target general information Gen name R Organism Escherichia phage lambda (Bacteriophage lambda) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 24 family Biochemical class Glycosidase Function Lyase activity.Lysozyme activity.Lytic transglycosylase activity. Related diseases Estrogen resistance (ESTRR) [MIM:615363]: A disorder characterized by partial or complete resistance to estrogens, in the presence of elevated estrogen serum levels. Clinical features include absence of the pubertal growth spurt, delayed bone maturation, unfused epiphyses, reduced bone mineral density, osteoporosis, continued growth into adulthood and very tall adult stature. Glucose intolerance, hyperinsulinemia and lipid abnormalities may also be present. {ECO:0000269|PubMed:23841731, ECO:0000269|PubMed:27754803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04206 Interacts with NA EC number 4.2.2.n2 Uniprot keywords 3D-structure; Antimicrobial; Bacteriolytic enzyme; Cytolysis; Direct protein sequencing; Host cell lysis by virus; Host cytoplasm; Lyase; Reference proteome; Viral release from host cell Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 49834.9 Length 462 Aromaticity 0.07 Instability index 18.78 Isoelectric point 9.6 Charge (pH=7) 18.29 2D Binding mode Binding energy (Kcal/mol) -6.45 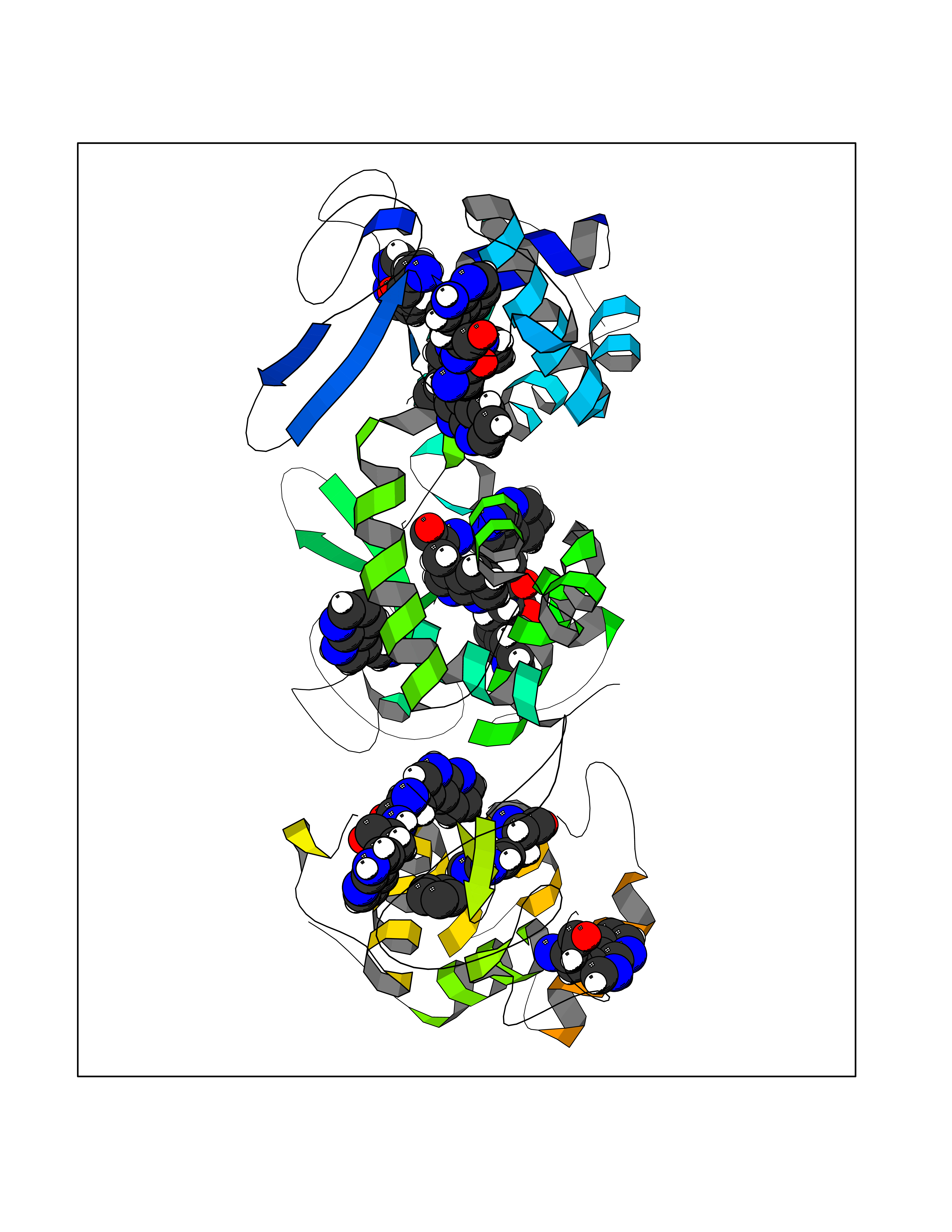 Molscript Map  Pymol Map 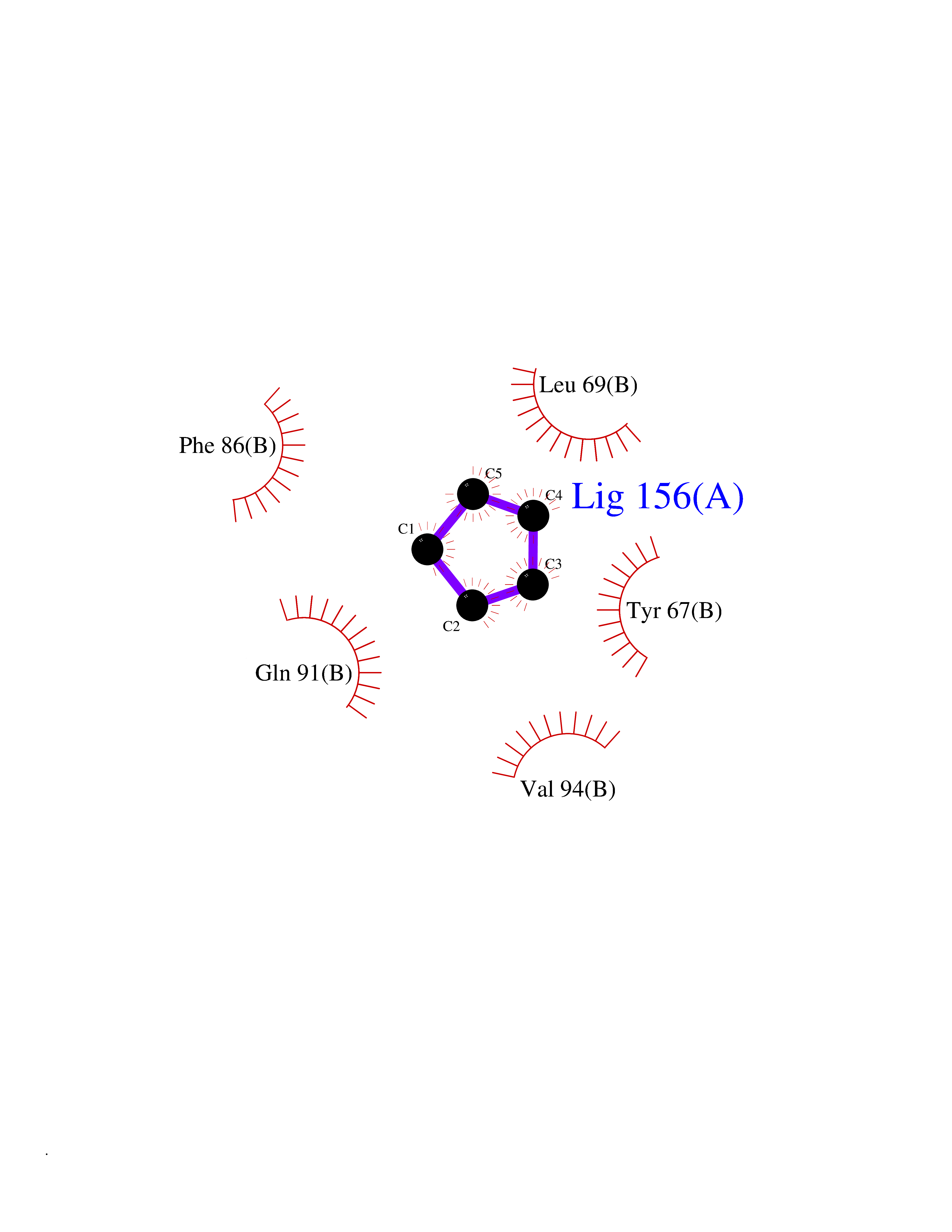 Ligplot Map 3D Binding mode Sequence MVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVR Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Natriuretic peptides B | 1YK1 | 4.73 | |
Target general information Gen name NPPB Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Natriuretic peptide family Biochemical class Hormone / growth factor receptor Function Diuretic hormone activity.Hormone activity.Peptide hormone receptor binding.Receptor binding. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01136; DB06412 Interacts with A8MQ03; P57678; Q6A162; P60411; Q7Z3S9; P25788; Q9UJW9 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disulfide bond; Glycoprotein; Hormone; Pharmaceutical; Proteoglycan; Proteomics identification; Reference proteome; Secreted; Signal; Vasoactive; Vasodilator Protein physicochemical properties Chain ID E Molecular weight (Da) 46353.1 Length 415 Aromaticity 0.1 Instability index 37.91 Isoelectric point 5.51 Charge (pH=7) -12.09 2D Binding mode Binding energy (Kcal/mol) -6.45 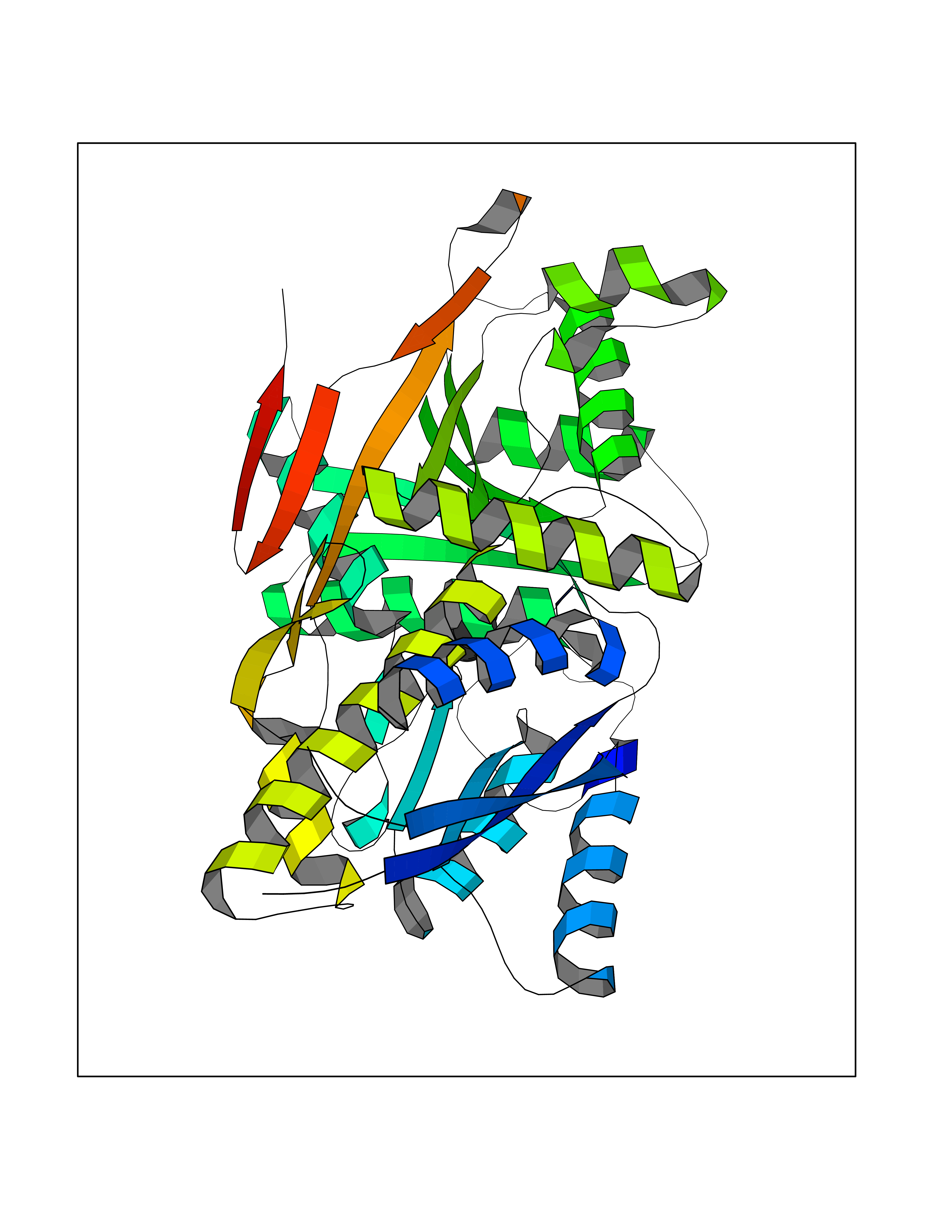 Molscript Map 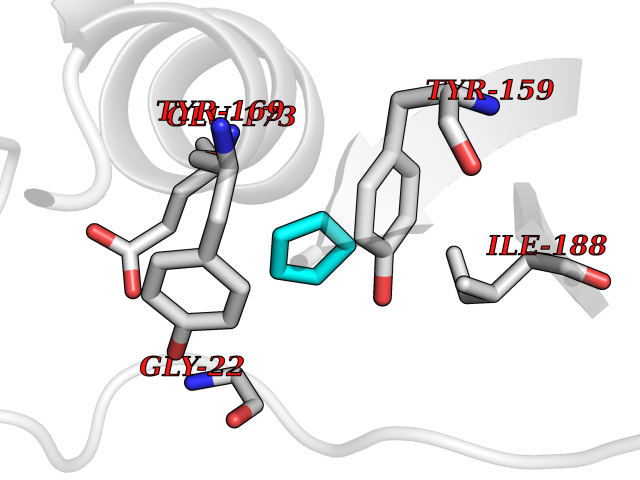 Pymol Map 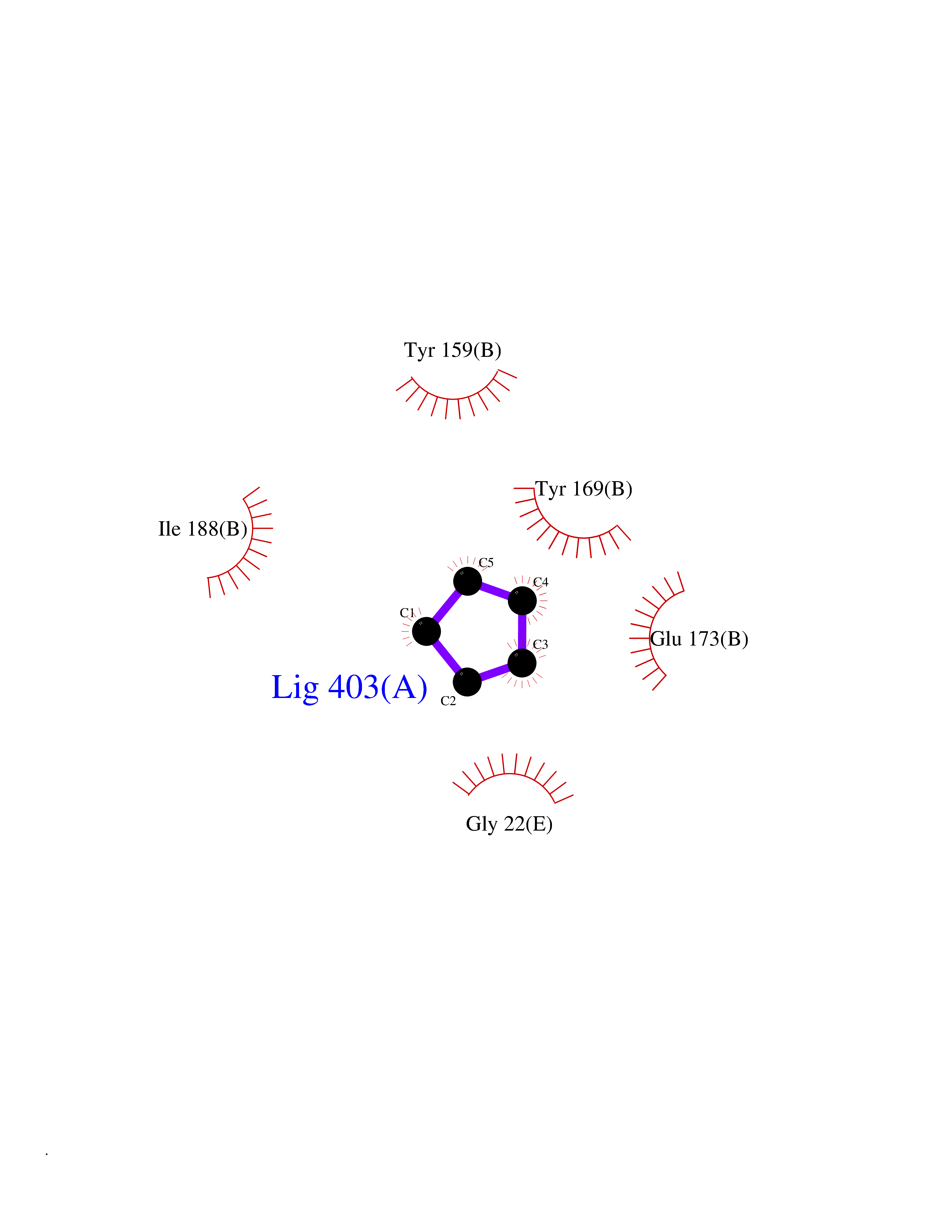 Ligplot Map 3D Binding mode Sequence GCFGRKMDRISSSSGLGCKVLALPPQKIEVLVLLPQDDSYLFSLTRVRPAIEYALRSVEGLLPPGTRFQVAYEDSDCGNRALFSLVDRVAAARGAKPDLILGPVCEYAAAPVARLASHWDLPMLSAGALAAGFQHKDSEYSHLTRVAPAYAKMGEMMLALFRHHHWSRAALVYSDDKLERNCYFTLEGVHEVFQEEGLHTSIYSFDETKDLDLEDIVRNIQASERVVIMCASSDTIRSIMLVAHRHGMTSGDYAFFNIELFNSSSYGDGSWKRGDKHDFEAKQAYSSLQTVTLLRTVKPEFEKFSMEVKSSVEKQGLNMEDYVNMFVEGFHDAILLYVLALHEVLRAGYSKKDGGKIIQQTWNRTFEGIAGQVSIDANGDRYGDFSVIAMTDVEAGTQEVIGDYFGKEGRFEMRP Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Chromodomain-helicase-DNA-binding protein 1 | 4O42 | 4.73 | |
Target general information Gen name CHD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SNF2/RAD54 helicase family Biochemical class Dna binding protein / viral protein Function ATP binding.ATP-dependent DNA helicase activity.DNA binding.Methylated histone binding. Related diseases Pilarowski-Bjornsson syndrome (PILBOS) [MIM:617682]: An autosomal dominant disorder characterized by developmental delay, speech apraxia, intellectual disability, autism, and facial dysmorphic features. Some patients may have seizures. {ECO:0000269|PubMed:28866611}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60341-1; B2BUF1; P28799; O76024 EC number 3.6.4.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromatin regulator; Cytoplasm; Disease variant; DNA-binding; Helicase; Hydrolase; Intellectual disability; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 20969.1 Length 180 Aromaticity 0.12 Instability index 46.35 Isoelectric point 5.88 Charge (pH=7) -2.83 2D Binding mode Binding energy (Kcal/mol) -6.45 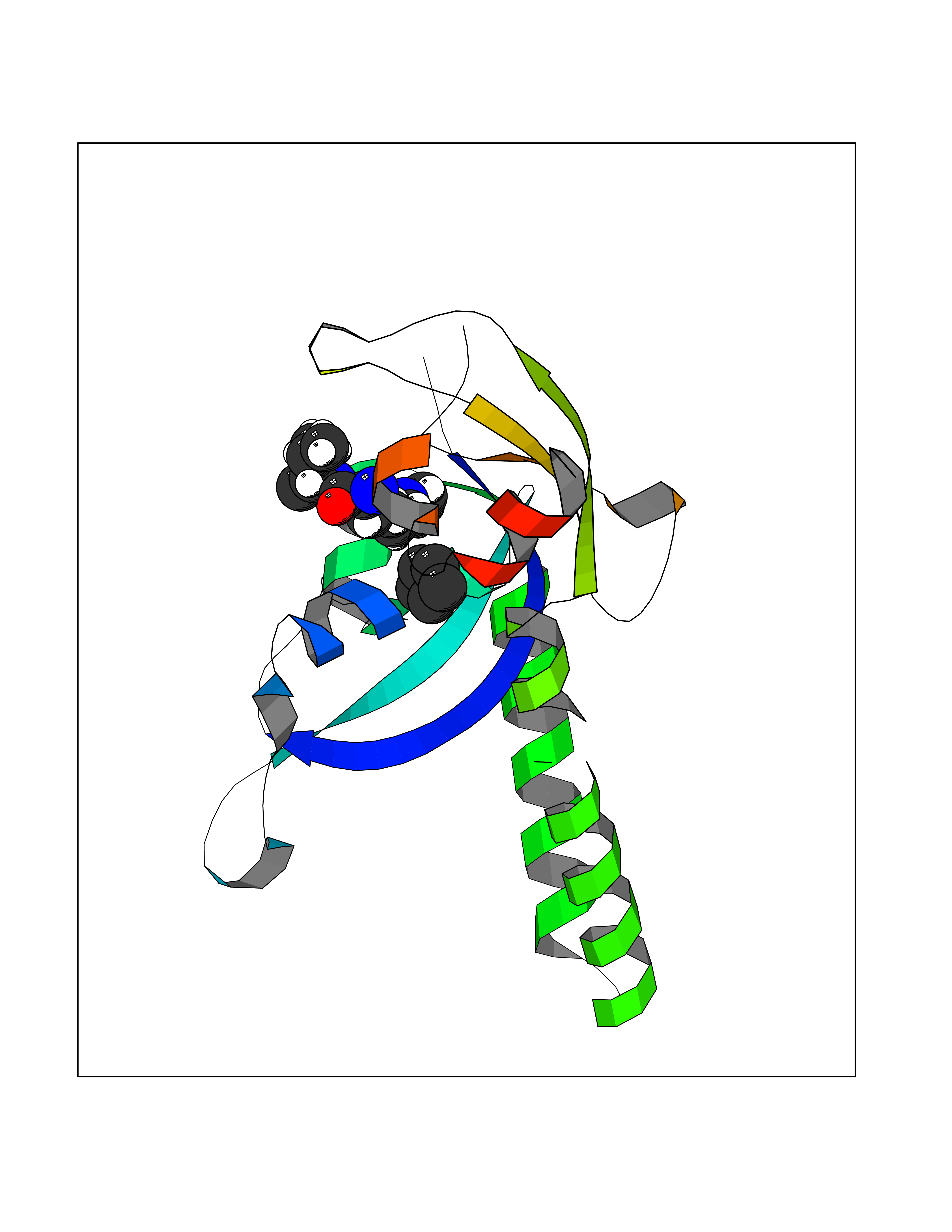 Molscript Map 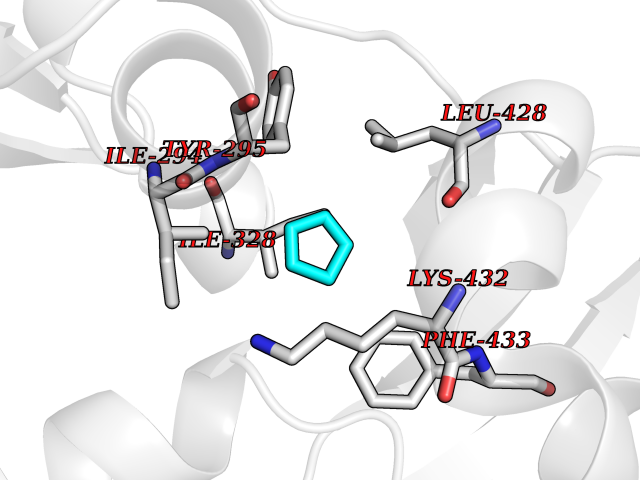 Pymol Map 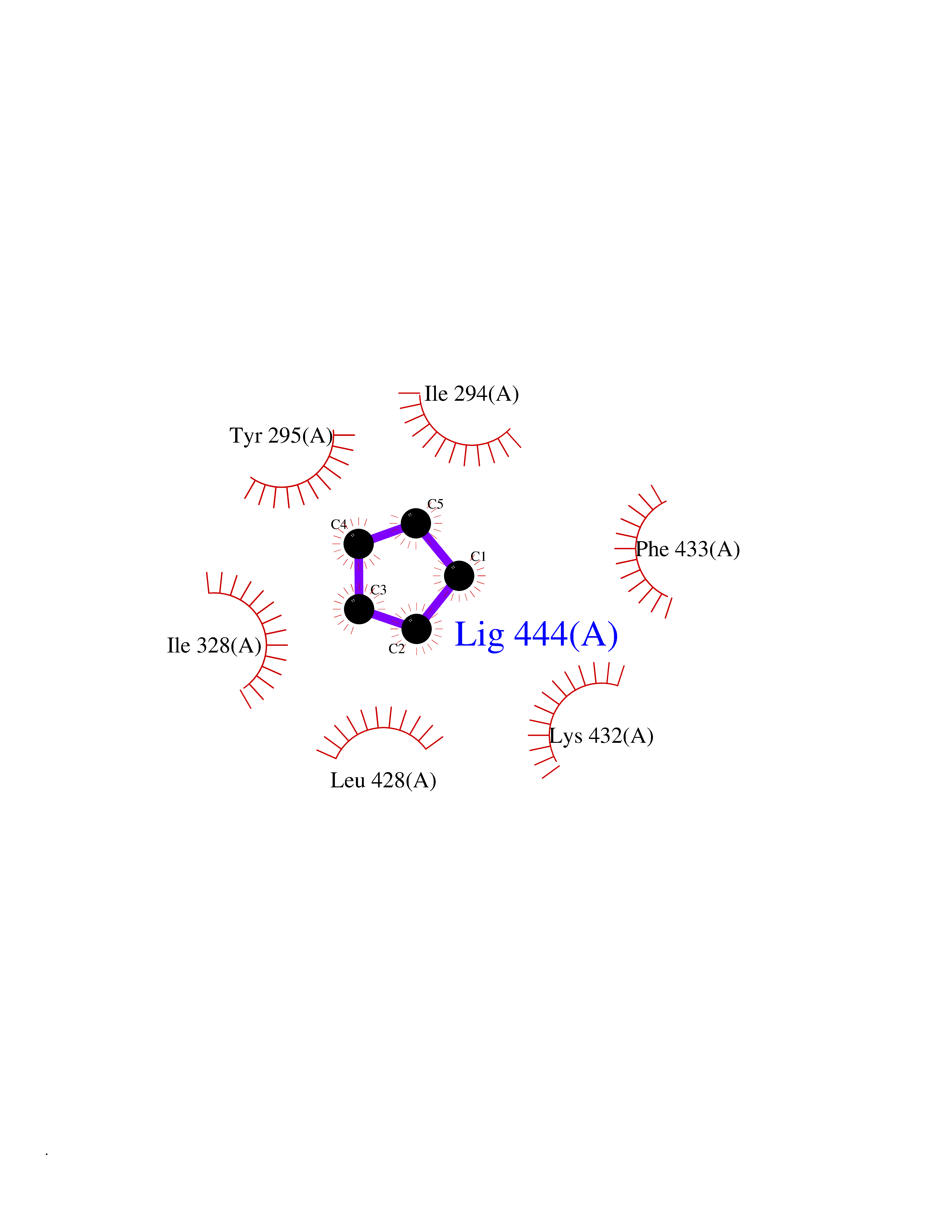 Ligplot Map 3D Binding mode Sequence EFETIERFMDCRIGRKGATGATTTIYAVEADGDPNAGFEKNKEPGEIQYLIKWKGWSHIHNTWETEETLKQQNVRGMKKLDNYKKKDQETKRWLKNASPEDVEYYNCQQELTDDLHKQYQIVERIIAHSNQKSAAGYPDYYCKWQGLPYSECSWEDGALISKKFQACIDEYFSRTARSXV Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Nitric-oxide synthase inducible (NOS2) | 3E7G | 4.73 | |
Target general information Gen name NOS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms iNOS; Peptidyl-cysteine S-nitrosylase NOS2; Nitric oxide synthase, inducible; NOS2A; NOS type II; Inducible NOS; Inducible NO synthase; Hepatocyte NOS; HEP-NOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function Produces nitric oxide (NO) which is a messenger molecule with diverse functions throughout the body. In macrophages, NO mediates tumoricidal and bactericidal actions. Also has nitrosylase activity and mediates cysteine S-nitrosylation of cytoplasmic target proteins such PTGS2/COX2 (By similarity). As component of the iNOS-S100A8/9 transnitrosylase complex involved in the selective inflammatory stimulus-dependent S-nitrosylation of GAPDH on 'Cys-247' implicated in regulation of the GAIT complex activity and probably multiple targets including ANXA5, EZR, MSN and VIM. Involved in inflammation, enhances the synthesis of proinflammatory mediators such as IL6 and IL8. Related diseases Cerebellar ataxia, impaired intellectual development, and dysequilibrium syndrome 3 (CAMRQ3) [MIM:613227]: An autosomal recessive, congenital cerebellar ataxia associated with dysarthia, quadrupedal gait and intellectual disability. {ECO:0000269|PubMed:19461874}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07003; DB07007; DB07011; DB07405; DB08750; DB01997; DB07029; DB07008; DB08214; DB07002; DB01835; DB06879; DB04534; DB03100; DB02207; DB00125; DB00155; DB01234; DB14649; DB11327; DB00997; DB07306; DB07388; DB05252; DB01381; DB03366; DB05214; DB04400; DB09237; DB00244; DB01110; DB01017; DB03144; DB01686; DB03449; DB06916; DB07318; DB07389; DB02044; DB02644; DB05383; DB02234; DB03953; DB02462; DB08814 Interacts with P04406 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calmodulin-binding; Cytoplasm; FAD; Flavoprotein; FMN; Heme; Iron; Metal-binding; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 48633 Length 421 Aromaticity 0.12 Instability index 46.5 Isoelectric point 6.75 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -6.45  Molscript Map 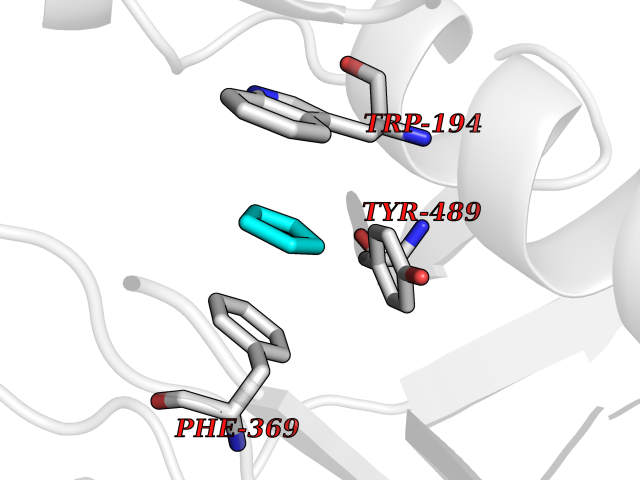 Pymol Map 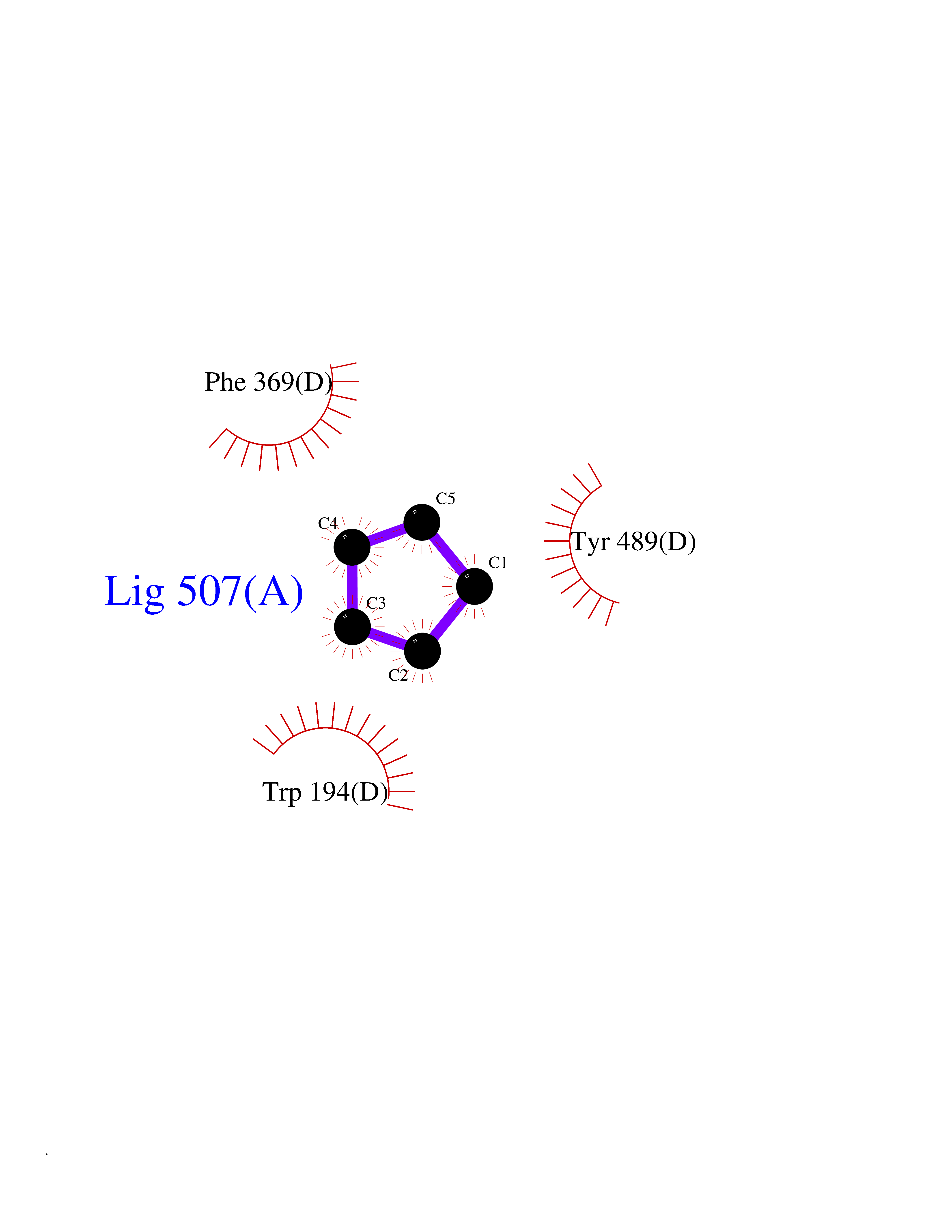 Ligplot Map 3D Binding mode Sequence RHVRIKNWGSGMTFQDTLHHKAKGILTCRSKSCLGSIMTPKSLTRGPRDKPTPPDELLPQAIEFVNQYYGSFKEAKIEEHLARVEAVTKEIETTGTYQLTGDELIFATKQAWRNAPRCIGRIQWSNLQVFDARSCSTAREMFEHICRHVRYSTNNGNIRSAITVFPQRSDGKHDFRVWNAQLIRYAGYQMPDGSIRGDPANVEFTQLCIDLGWKPKYGRFDVVPLVLQANGRDPELFEIPPDLVLEVAMEHPKYEWFRELELKWYALPAVANMLLEVGGLEFPGCPFNGWYMGTEIGVRDFCDVQRYNILEEVGRRMGLETHKLASLWKDQAVVEINIAVLHSFQKQNVTIMDHHSAAESFMKYMQNEYRSRGGCPADWIWLVPPMSGSITPVFHQEMLNYVLSPFYYYQVEAWKTHVWQD Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Tyrosine-protein kinase BTK (ATK) | 4RFZ | 4.73 | |
Target general information Gen name BTK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Bruton's tyrosine kinase; Bruton tyrosine kinase; BPK; B-cell progenitor kinase; B cell progenitor kinase; Agammaglobulinemia tyrosine kinase; Agammaglobulinaemia tyrosine kinase; AGMX1 Protein family Protein kinase superfamily, Tyr protein kinase family, TEC subfamily Biochemical class Kinase Function Binding of antigen to the B-cell antigen receptor (BCR) triggers signaling that ultimately leads to B-cell activation. After BCR engagement and activation at the plasma membrane, phosphorylates PLCG2 at several sites, igniting the downstream signaling pathway through calcium mobilization, followed by activation of the protein kinase C (PKC) family members. PLCG2 phosphorylation is performed in close cooperation with the adapter protein B-cell linker protein BLNK. BTK acts as a platform to bring together a diverse array of signaling proteins and is implicated in cytokine receptor signaling pathways. Plays an important role in the function of immune cells of innate as well as adaptive immunity, as a component of the Toll-like receptors (TLR) pathway. The TLR pathway acts as a primary surveillance system for the detection of pathogens and are crucial to the activation of host defense. Especially, is a critical molecule in regulating TLR9 activation in splenic B-cells. Within the TLR pathway, induces tyrosine phosphorylation of TIRAP which leads to TIRAP degradation. BTK plays also a critical role in transcription regulation. Induces the activity of NF-kappa-B, which is involved in regulating the expression of hundreds of genes. BTK is involved on the signaling pathway linking TLR8 and TLR9 to NF-kappa-B. Transiently phosphorylates transcription factor GTF2I on tyrosine residues in response to BCR. GTF2I then translocates to the nucleus to bind regulatory enhancer elements to modulate gene expression. ARID3A and NFAT are other transcriptional target of BTK. BTK is required for the formation of functional ARID3A DNA-binding complexes. There is however no evidence that BTK itself binds directly to DNA. BTK has a dual role in the regulation of apoptosis. Non-receptor tyrosine kinase indispensable for B lymphocyte development, differentiation and signaling. Related diseases X-linked agammaglobulinemia (XLA) [MIM:300755]: Humoral immunodeficiency disease which results in developmental defects in the maturation pathway of B-cells. Affected boys have normal levels of pre-B-cells in their bone marrow but virtually no circulating mature B-lymphocytes. This results in a lack of immunoglobulins of all classes and leads to recurrent bacterial infections like otitis, conjunctivitis, dermatitis, sinusitis in the first few years of life, or even some patients present overwhelming sepsis or meningitis, resulting in death in a few hours. Treatment in most cases is by infusion of intravenous immunoglobulin. {ECO:0000269|PubMed:10220140, ECO:0000269|PubMed:10612838, ECO:0000269|PubMed:10678660, ECO:0000269|PubMed:7627183, ECO:0000269|PubMed:7633420, ECO:0000269|PubMed:7633429, ECO:0000269|PubMed:7711734, ECO:0000269|PubMed:7809124, ECO:0000269|PubMed:7849006, ECO:0000269|PubMed:7849697, ECO:0000269|PubMed:7849721, ECO:0000269|PubMed:7880320, ECO:0000269|PubMed:7897635, ECO:0000269|PubMed:8013627, ECO:0000269|PubMed:8162018, ECO:0000269|PubMed:8162056, ECO:0000269|PubMed:8594569, ECO:0000269|PubMed:8634718, ECO:0000269|PubMed:8695804, ECO:0000269|PubMed:8723128, ECO:0000269|PubMed:8834236, ECO:0000269|PubMed:9016530, ECO:0000269|PubMed:9260159, ECO:0000269|PubMed:9280283, ECO:0000269|PubMed:9445504, ECO:0000269|PubMed:9545398}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 3, with agammaglobulinemia (IGHD3) [MIM:307200]: An X-linked recessive disorder characterized by growth hormone deficiency, short stature, delayed bone age, agammaglobulinemia with markedly reduced numbers of B cells, and good response to treatment with growth hormone. {ECO:0000269|PubMed:8013627}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15327; DB11703; DB15347; DB01254; DB15170; DB14785; DB12010; DB09053; DB01863; DB17472; DB14924; DB11764; DB15227; DB16657; DB05204; DB15035 Interacts with Q13444; Q99856; Q8WV28; Q06187; P78347; P08238; Q9BVA0; P21145; P50222; Q04759; O60239; P42768 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Acetylation; Adaptive immunity; Alternative promoter usage; Apoptosis; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Dwarfism; Immunity; Innate immunity; Kinase; Lipid-binding; Membrane; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; SH2 domain; SH3 domain; Transcription; Transcription regulation; Transferase; Tyrosine-protein kinase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 30491.7 Length 263 Aromaticity 0.12 Instability index 45.93 Isoelectric point 5.39 Charge (pH=7) -7.65 2D Binding mode Binding energy (Kcal/mol) -6.46 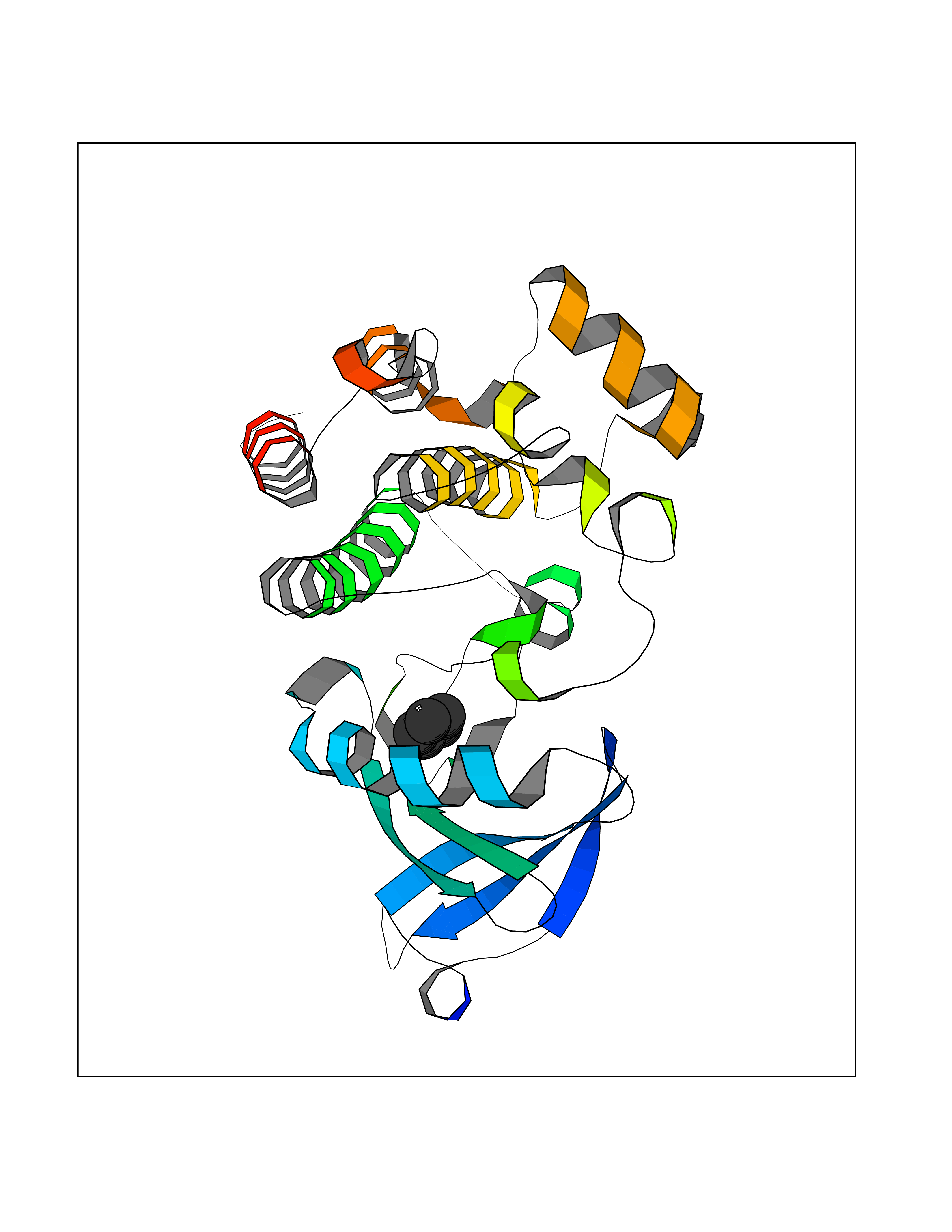 Molscript Map 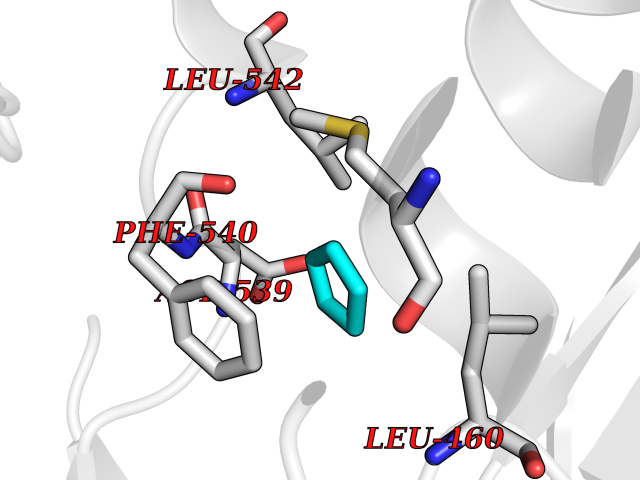 Pymol Map 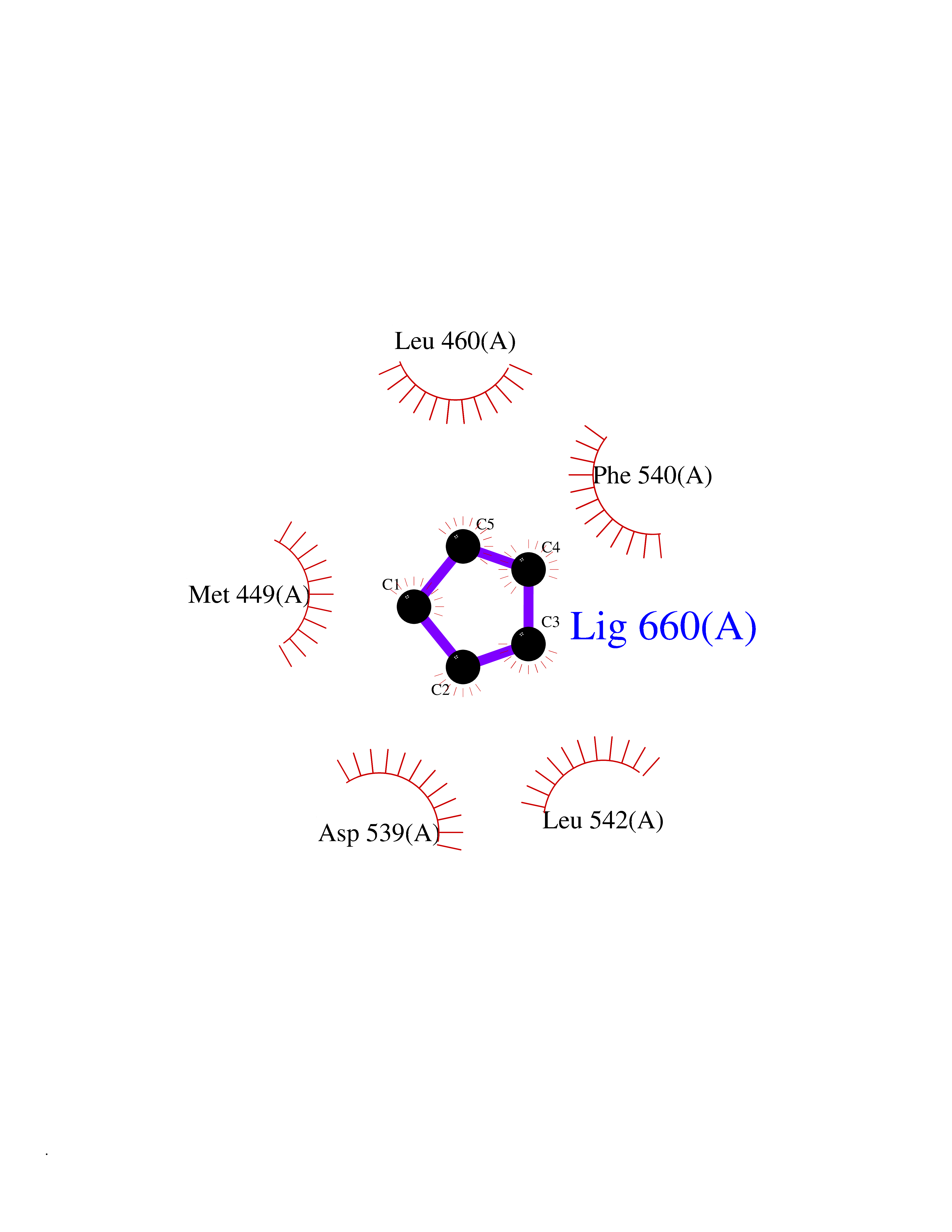 Ligplot Map 3D Binding mode Sequence EIDPKDLTFLKELGTGQFGVVKYGKWRGQYDVAIKMIKEGSMSEDEFIEEAKVMMNLSHEKLVQLYGVCTKQRPIFIITEYMANGCLLNYLREARHAFQTQQLLEMCKDVCEAMEYLESKQFLHRDLAARNCLVNDQGVVKVSDFGLSRYVLDDEYTSSVGSKFPVRWSPPEVLMYSKFSSKSDIWAFGVLMWEIYSLGKMPYERFTNSETAEHIAQGLRLYRPHLASAAVYTIMYSCWHEKADERPTFKILLSNILDVMDEE Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Calcium-binding mitochondrial carrier protein Aralar2 | 4P5W | 4.73 | |
Target general information Gen name SLC25A13 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Mitochondrial carrier (TC 2.A.29) family Biochemical class Transport protein Function Acidic amino acid transmembrane transporter activity.Calcium ion binding.L-aspartate transmembrane transporter activity.L-glutamate transmembrane transporter activity.Transporter activity. Related diseases Citrullinemia 2 (CTLN2) [MIM:603471]: A form of citrullinemia, an autosomal recessive disease characterized primarily by elevated serum and urine citrulline levels. Ammonia intoxication is another manifestation. Citrullinemia type 2 is characterized by neuropsychiatric symptoms including abnormal behaviors, loss of memory, seizures and coma. Death can result from brain edema. Onset is sudden and usually between the ages of 20 and 50 years. {ECO:0000269|PubMed:10369257, ECO:0000269|PubMed:10610724}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cholestasis, neonatal intrahepatic, caused by citrin deficiency (NICCD) [MIM:605814]: A form of citrullinemia type 2 with neonatal onset, characterized by suppression of the bile flow, hepatic fibrosis, low birth weight, growth retardation, hypoproteinemia, variable liver dysfunction. Neonatal intrahepatic cholestasis due to citrin deficiency is generally not severe and symptoms disappear by one year of age with an appropriate diet. Years or even decades later, however, some individuals develop the characteristic features of citrullinemia type 2 with neuropsychiatric symptoms. {ECO:0000269|PubMed:11793471}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00128 Interacts with O75746 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Calcium; Disease variant; Intrahepatic cholestasis; Membrane; Metal-binding; Methylation; Mitochondrion; Mitochondrion inner membrane; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34724.3 Length 306 Aromaticity 0.12 Instability index 28.83 Isoelectric point 5.55 Charge (pH=7) -7.5 2D Binding mode Binding energy (Kcal/mol) -6.45 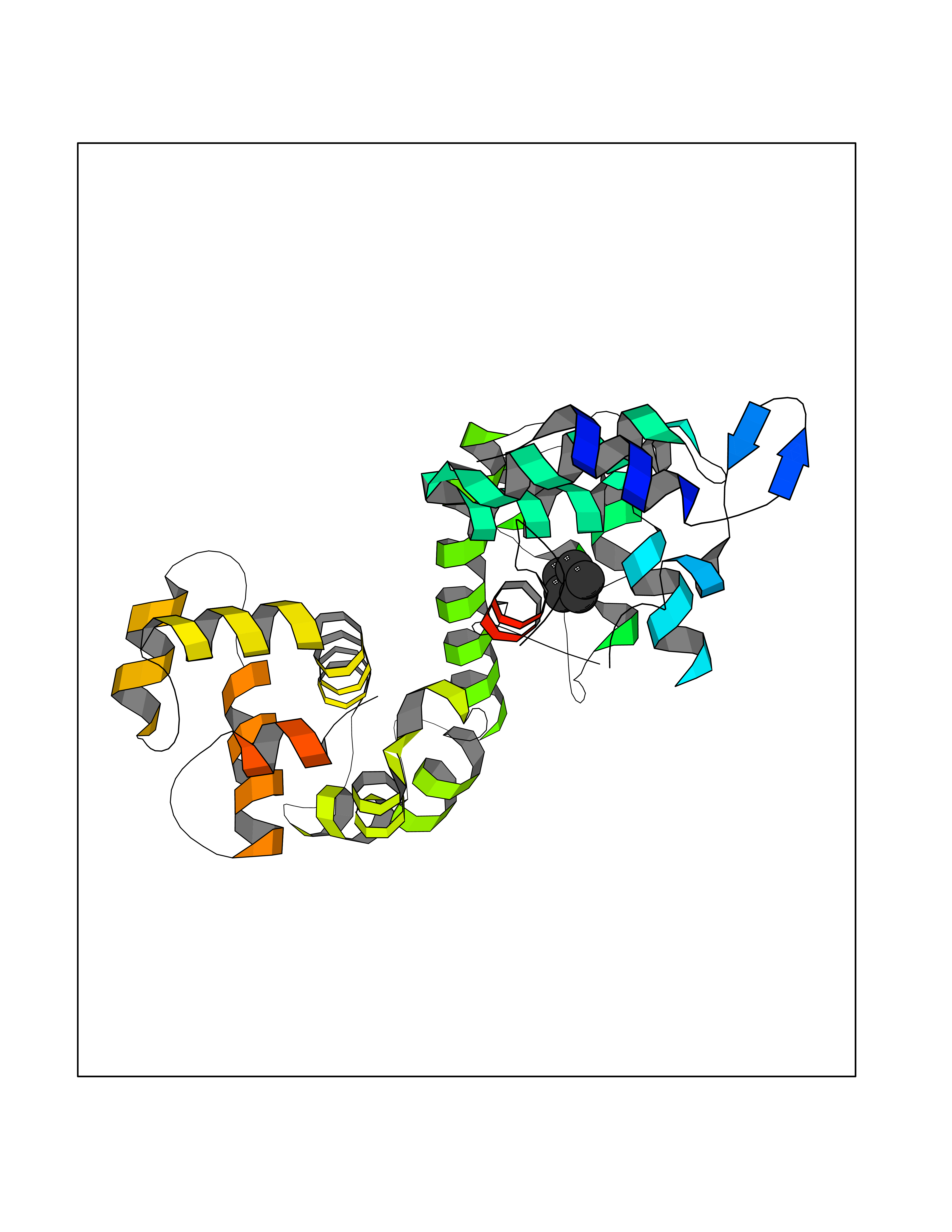 Molscript Map 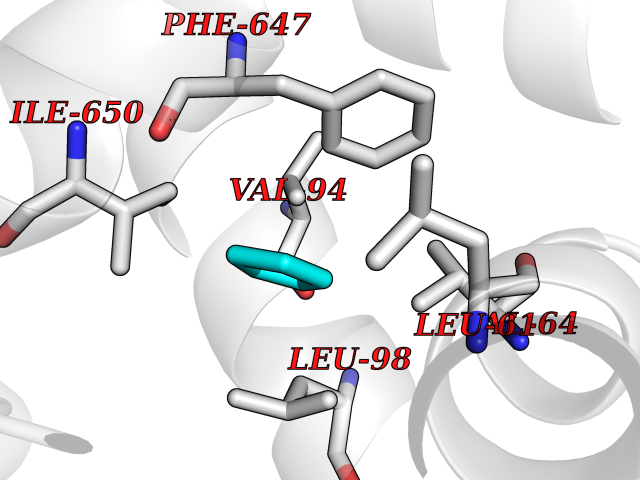 Pymol Map 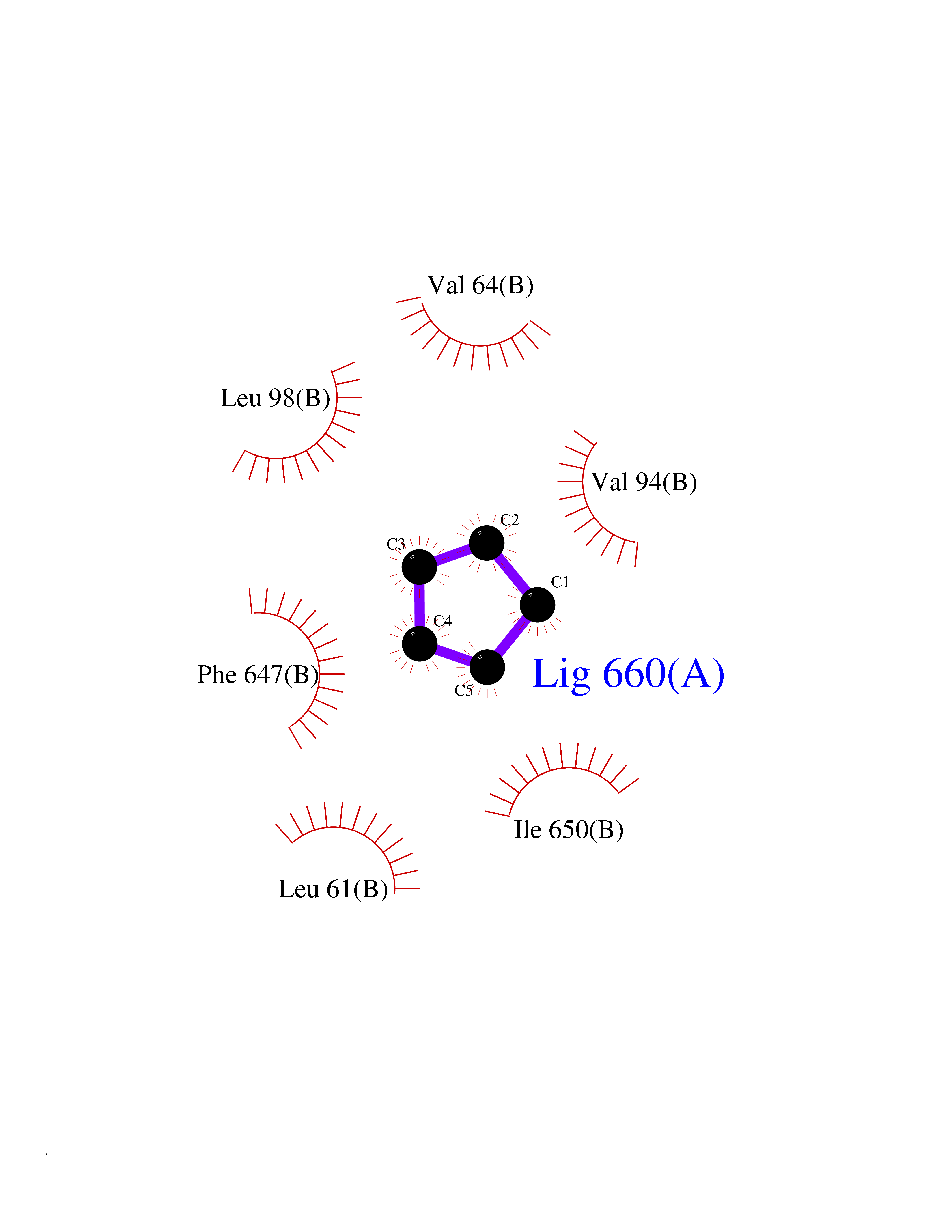 Ligplot Map 3D Binding mode Sequence RADPAELRTIFLKYASIEKNGEFFMSPNDFVTRYLNINPKTVELLSGVVDQTKDGLISFQEFVAFESVLCAPDALFMVAFQLFDKAGKGEVTFEDVKQVFGQTTIHQHIPFNWDSEFVQLHFGKERKRHLTYAEFTQFLLEIQLEHAKQAFVQRDNARTGRVTAIDFRDIMVTIRPHVLTPFVEECLVAAAGGTTSHQVSFSYFNGFNSLLNNMELIRKIYSTLAGTRKDVEVTKEEFVLAAQKFGQVTPMEVDILFQLADLYEPRGRMTLADIERIAPPNPDHVGGYKLAVATFAGIENKFGLYL Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Vitamin K epoxide reductase complex 1 (VKORC1) | 6WV3 | 4.73 | |
Target general information Gen name VKORC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin K1 2,3-epoxide reductase subunit 1; VKORC1; VKOR; UNQ308/PRO351; MSTP576; MSTP134 Protein family VKOR family Biochemical class Short-chain dehydrogenases reductase Function Involved invitamin K metabolism. Catalytic subunit of the vitamin K epoxide reductase (VKOR) complex which reduces inactive vitamin K 2,3-epoxide to active vitamin K. Vitamin K is required for the gamma-carboxylation of various proteins, including clotting factors, and is required for normal blood coagulation, but also for normal bone development. Related diseases Combined deficiency of vitamin K-dependent clotting factors 2 (VKCFD2) [MIM:607473]: VKCFD leads to a bleeding tendency that is usually reversed by oral administration of vitamin K. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:16270630}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Coumarin resistance (CMRES) [MIM:122700]: A condition characterized by partial or complete resistance to warfarin or other 4-hydroxycoumarin derivatives. These drugs are used as anti-coagulants for the prevention of thromboembolic diseases in subjects with deep vein thrombosis, atrial fibrillation, or mechanical heart valve replacement. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:20946155}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01418; DB00266; DB09332; DB00170; DB00498; DB00946; DB01022; DB00682 Interacts with Q13323; Q7Z7G2; Q96BA8; Q9Y282; Q5JX71; Q96KR6; Q5T7V8; Q8TDT2; Q9NQG1; P15941-11; Q96TC7; Q9NR31; A0A0S2Z4U3; Q8TBB6; O15393-2; Q19QW4 EC number EC 1.17.4.4 Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Disulfide bond; Endoplasmic reticulum; Membrane; Oxidoreductase; Proteomics identification; Quinone; Redox-active center; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 42656.4 Length 381 Aromaticity 0.1 Instability index 32.12 Isoelectric point 7.73 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -6.45  Molscript Map 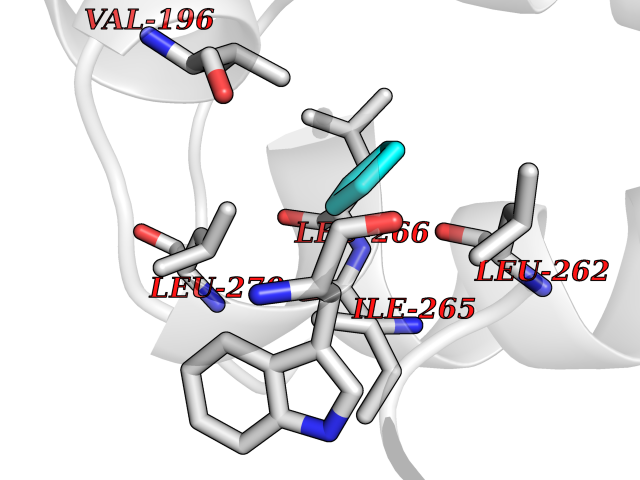 Pymol Map 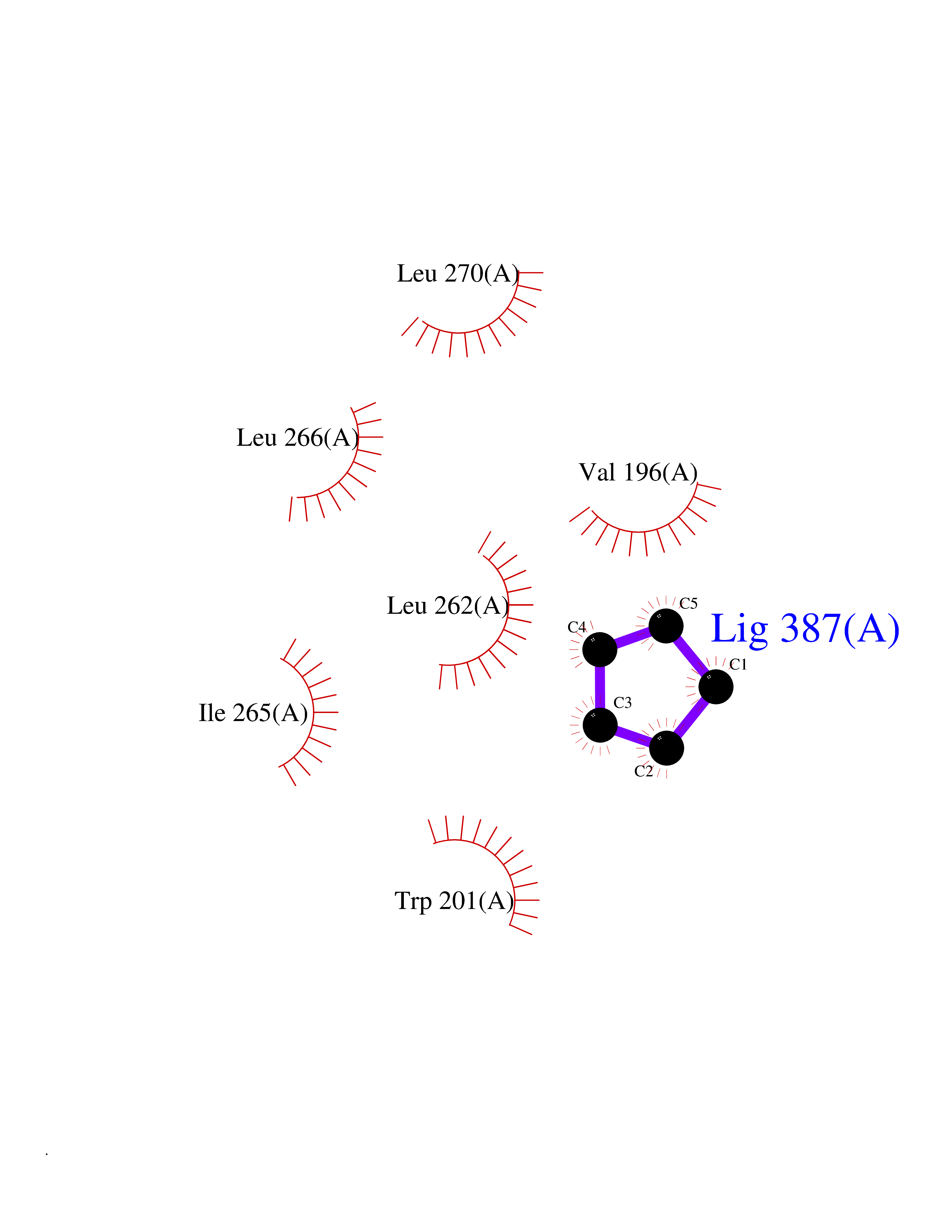 Ligplot Map 3D Binding mode Sequence KGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLXVQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNSTWGSPGWVRLALCLTGLVLSLYALHVKAARARDRDYRALCDVGTAISCSRVFSSRWGRGFGLVEHVLGQDSILNQSNSIFGCIFYTLQLLLGCLRTRWASVLMLLSSLVSLAGSVYLAWILFFVLYDFCIVCITTYAINVSLMWLSFRKVQENSHNVYITADKQKNGIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHHH Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Influenza Polymerase acidic endonuclease (Influ PA) | 4ZI0 | 4.73 | |
Target general information Gen name Influ PA Organism Influenza A virus (strain A/Puerto Rico/8/1934 H1N1) Uniprot ID TTD ID Synonyms RNA-directed RNA polymerase subunit P2; Polymerase acidic protein Protein family Influenza viruses PA family Biochemical class NA Function Plays an essential role in viral RNA transcription and replication by forming the heterotrimeric polymerase complex together with PB1 and PB2 subunits. The complex transcribes viral mRNAs by using a unique mechanism called cap-snatching. It consists in the hijacking and cleavage of host capped pre-mRNAs. These short capped RNAs are then used as primers for viral mRNAs. The PB2 subunit is responsible for the binding of the 5' cap of cellular pre-mRNAs which are subsequently cleaved after 10-13 nucleotides by the PA subunit that carries the endonuclease activity. Related diseases Lymphatic malformation 8 (LMPHM8) [MIM:618773]: A form of primary lymphedema, a disease characterized by swelling of body parts due to developmental anomalies and functional defects of the lymphatic system. Adult patients with lymphedema may suffer from recurrent local infections. Impaired lymphatic drainage in the fetus can develop into hydrops fetalis, a severe condition characterized by excessive fluid accumulation in more than two fetal extra-vascular compartments and body cavities, placental enlargement and edema, pericardial or pleural effusion, or ascites. LMPHM8 is an autosomal recessive form characterized by onset in utero and fetal death due to non-immune hydrops fetalis. {ECO:0000269|PubMed:30115739}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB13997 Interacts with P03485; P03466; P03431 EC number EC 3.1.-.- Uniprot keywords 3D-structure; Cap snatching; Endonuclease; Eukaryotic host gene expression shutoff by virus; Eukaryotic host transcription shutoff by virus; Host cytoplasm; Host gene expression shutoff by virus; Host nucleus; Host-virus interaction; Hydrolase; Inhibition of host RNA polymerase II by virus; Manganese; Metal-binding; Nuclease; Phosphoprotein; Reference proteome; Ribosomal frameshifting Protein physicochemical properties Chain ID A Molecular weight (Da) 21288.1 Length 181 Aromaticity 0.11 Instability index 49.23 Isoelectric point 6.27 Charge (pH=7) -1.8 2D Binding mode Binding energy (Kcal/mol) -6.45  Molscript Map  Pymol Map 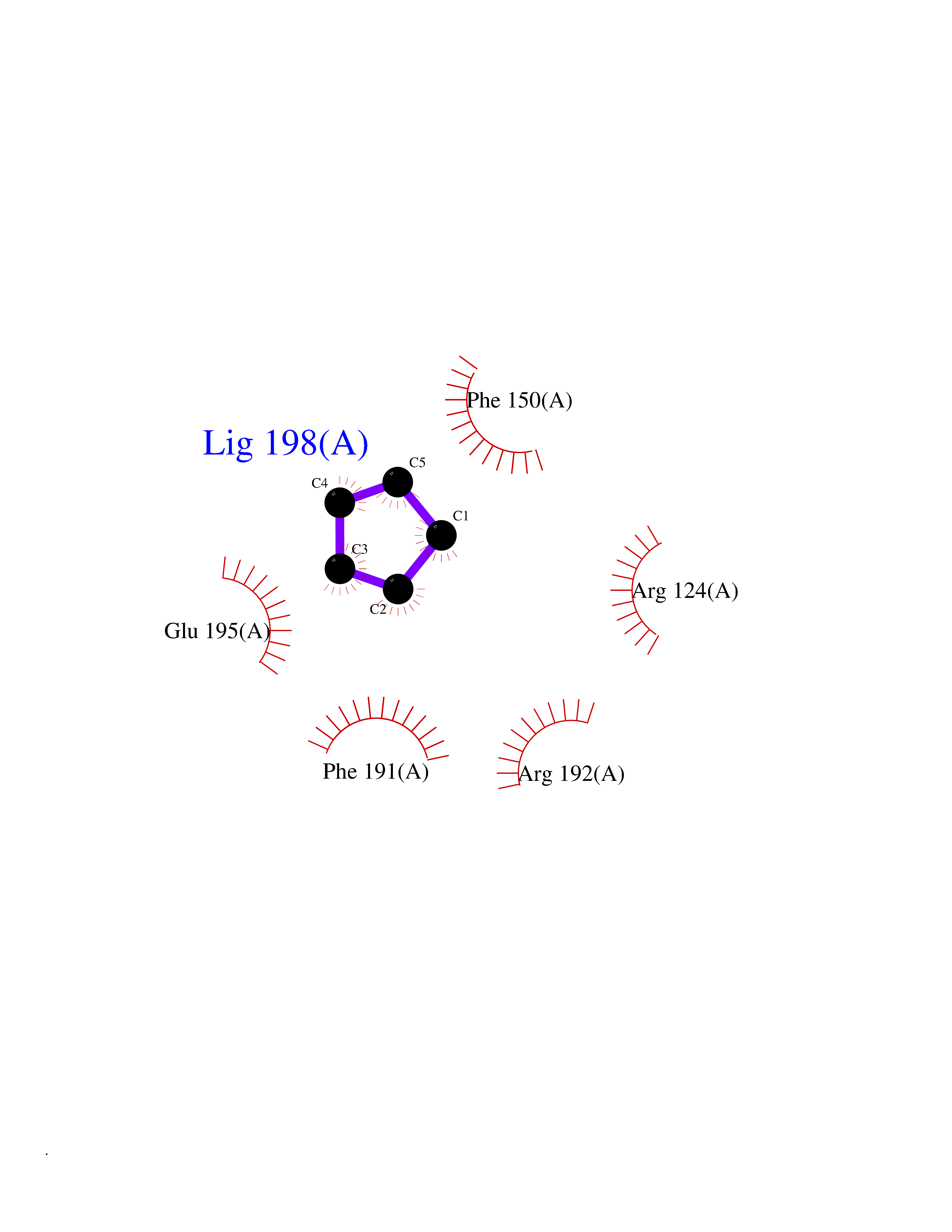 Ligplot Map 3D Binding mode Sequence GPLGSMEDFVRQCFNPMIVELAEKTMKEYGEDLKIETNKFAAICTHLEVCFMYSDASKHRFEIIEGRDRTMAWTVVNSICNTTGAEKPKFLPDLYDYKENRFIEIGVTRREVHIYYLEKANKIKSEKTHIHIFSFTGEEMATKADYTLDEESRARIKTRLFTIRQEMASRGLWDSFRQSER Hydrogen bonds contact Hydrophobic contact | ||||