Job Results:
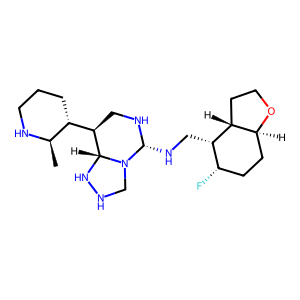
Ligand
Structure
Job ID
b66da62373d29d285a275497218b65f3
Job name
NA
Time
2024-11-27 22:32:54
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Phosphodiesterase 9 (PDE9) | 4E90 | 8.03 | |
Target general information Gen name PDE9A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms High affinity cGMPspecific 3',5'cyclic phosphodiesterase 9A; High affinity cGMP-specific 3',5'-cyclic phosphodiesterase 9A Protein family Cyclic nucleotide phosphodiesterase family, PDE9 subfamily Biochemical class Phosphoric diester hydrolase Function Highly specific: compared to other members of the cyclic nucleotide phosphodiesterase family, has the highest affinity and selectivity for cGMP. Specifically regulates natriuretic-peptide-dependent cGMP signaling in heart, acting as a regulator of cardiac hypertrophy in myocytes and muscle. Does not regulate nitric oxide-dependent cGMP in heart. Additional experiments are required to confirm whether its ability to hydrolyze natriuretic-peptide-dependent cGMP is specific to heart or is a general feature of the protein. In brain, involved in cognitive function, such as learning and long-term memory. Specifically hydrolyzes the second messenger cGMP, which is a key regulator of many important physiological processes. Related diseases Macular degeneration, age-related, 7 (ARMD7) [MIM:610149]: A form of age-related macular degeneration, a multifactorial eye disease and the most common cause of irreversible vision loss in the developed world. In most patients, the disease is manifest as ophthalmoscopically visible yellowish accumulations of protein and lipid that lie beneath the retinal pigment epithelium and within an elastin-containing structure known as Bruch membrane. {ECO:0000269|PubMed:17053108, ECO:0000269|PubMed:17053109}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Cerebral arteriopathy, autosomal recessive, with subcortical infarcts and leukoencephalopathy (CARASIL) [MIM:600142]: A cerebrovascular disease characterized by non-hypertensive arteriopathy of cerebral small vessels with subcortical infarcts, alopecia, and spondylosis. Small cerebral arteries show arteriosclerotic changes, fibrous intimal proliferation, and hyaline degeneration with splitting of the intima and/or the internal elastic membrane. Neurologic features include progressive dementia, gait disturbances, extrapyramidal and pyramidal signs, and demyelination of the cerebral white matter with sparing of U fibers. {ECO:0000269|PubMed:19387015}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cerebral arteriopathy, autosomal dominant, with subcortical infarcts and leukoencephalopathy, 2 (CADASIL2) [MIM:616779]: A cerebrovascular disease characterized by multiple subcortical infarcts, pseudobulbar palsy, dementia, and the presence of granular deposits in small cerebral arteries producing ischemic stroke. {ECO:0000269|PubMed:26063658}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07954; DB00201; DB03597; DB09283 Interacts with O95817; P49759; Q49AN0; Q9H8Y8; P60410; Q9BYR5; Q9BYQ4; Q14657; Q8NAJ2; P25791; Q9BRA0; Q7Z3S9; O76083-2; Q96FC7-2; P49888; Q13049; Q9BRU9; Q9Y260; O95817; Q16543; P49759-3; Q49AN0; A8MQ03; Q15051-2; Q63ZY3; O76011; Q07627; Q8IUG1; P60409; P60410; P60411; P59991; P60328; P26371; Q14657; Q16649; O76083-2; Q96FC7; Q99633; O00560; P49888; Q13049; O15205; P61964; O76083-2; Q13049 EC number EC 3.1.4.35 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; cGMP; Cytoplasm; Endoplasmic reticulum; Golgi apparatus; Hydrolase; Magnesium; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 38689.4 Length 328 Aromaticity 0.11 Instability index 53.51 Isoelectric point 5.14 Charge (pH=7) -16.23 2D Binding mode Binding energy (Kcal/mol) -8.75 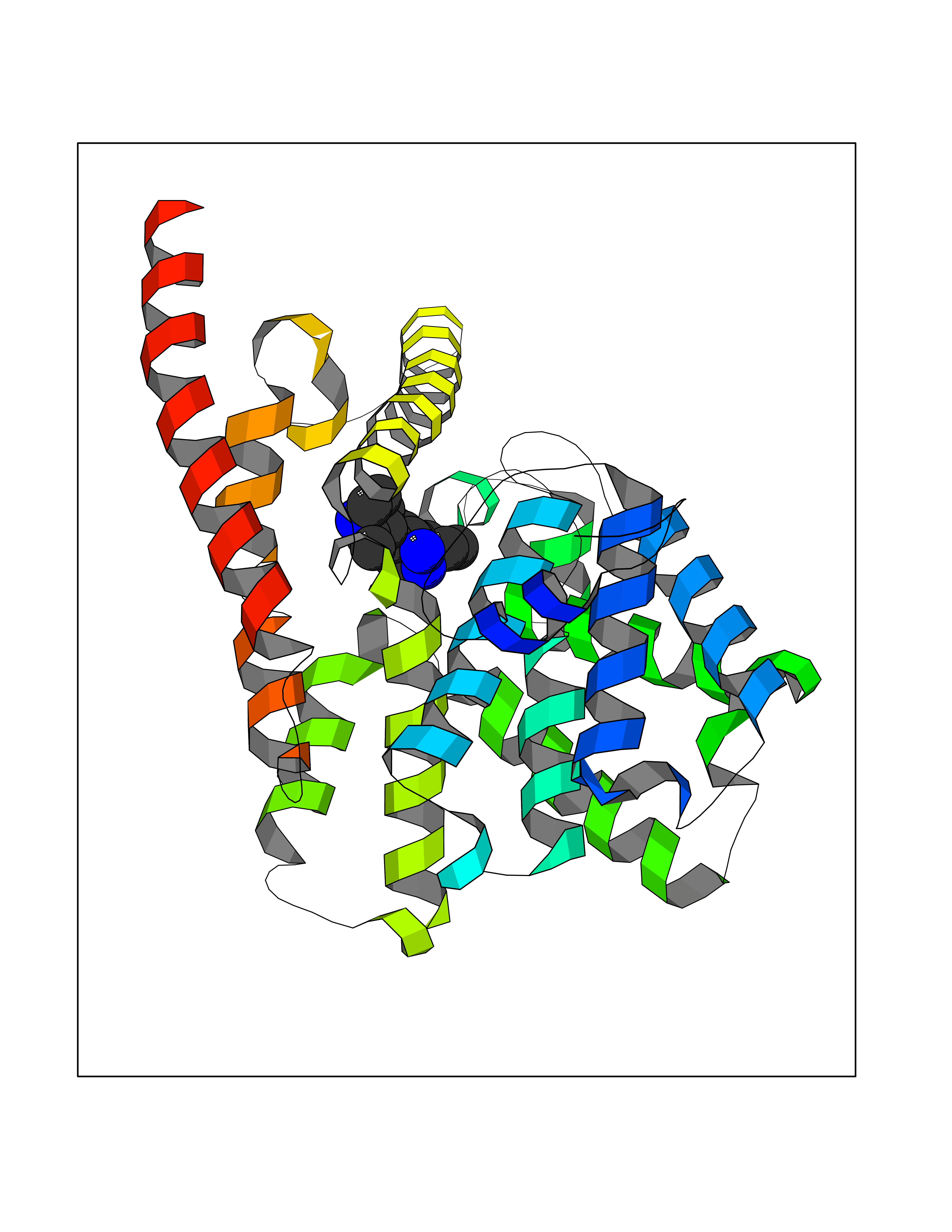 Molscript Map 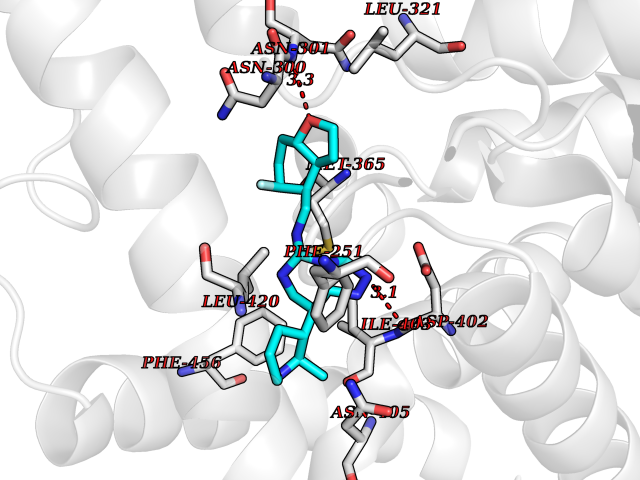 Pymol Map 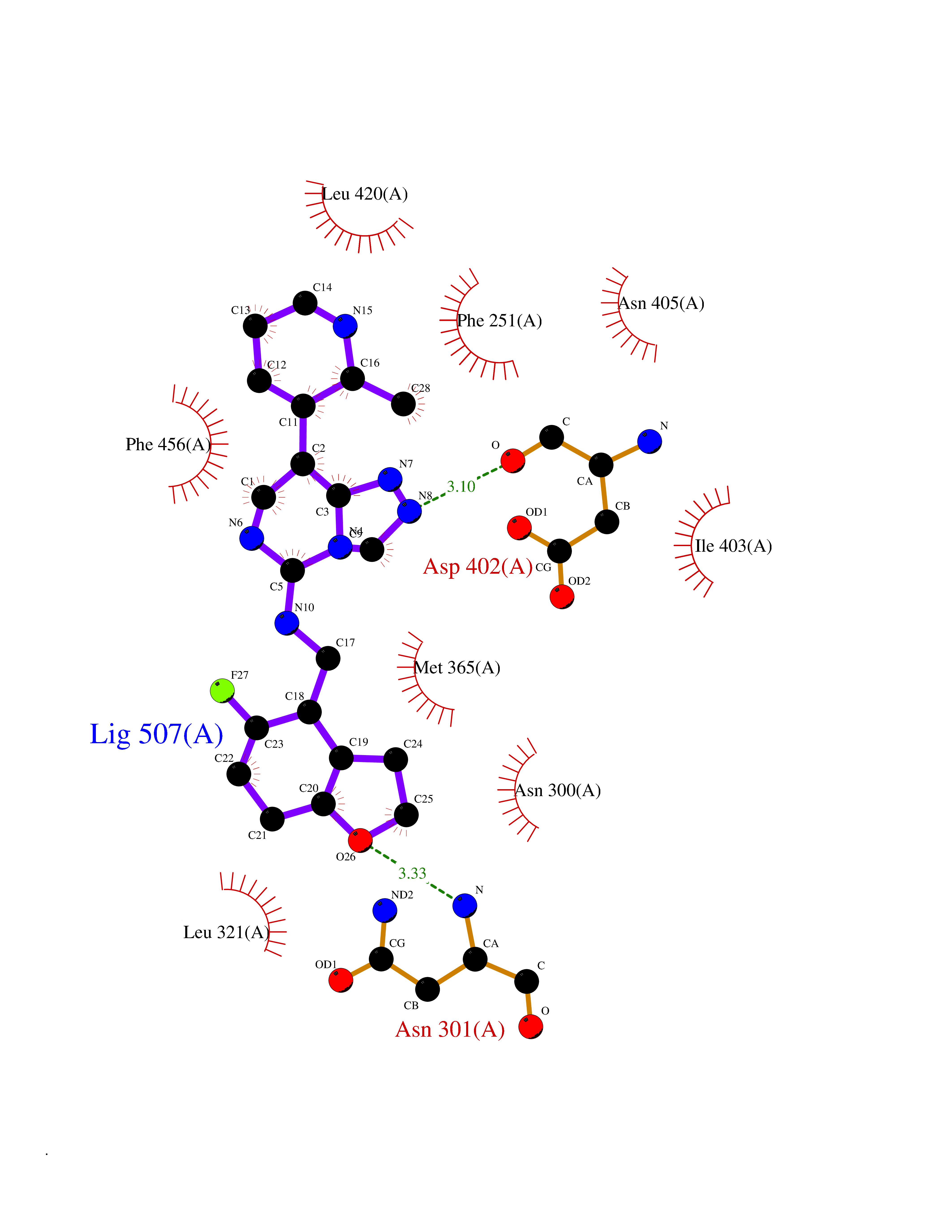 Ligplot Map 3D Binding mode Sequence GSHMTYPKYLLSPETIEALRKPTFDVWLWEPNEMLSCLEHMYHDLGLVRDFSINPVTLRRWLFCVHDNYRNNPFHNFRHCFCVAQMMYSMVWLCSLQEKFSQTDILILMTAAICHDLDHPGYNNTYQINARTELAVRYNDISPLENHHCAVAFQILAEPECNIFSNIPPDGFKQIRQGMITLILATDMARHAEIMDSFKEKMENFDYSNEEHMTLLKMILIKCCDISNEVRPMEVAEPWVDCLLEEYFMQSDREKSEGLPVAPFMDRDKVTKATAQIGFIKFVLIPMFETVTKLFPMVEEIMLQPLWESRDRYEELKRIDDAMKELQK Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Janus kinase 1 (JAK-1) | 4E5W | 8.02 | |
Target general information Gen name JAK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase JAK1; JAK1B; JAK1A Protein family Protein kinase superfamily, Tyr protein kinase family, JAK subfamily Biochemical class Kinase Function Kinase partner for the interleukin (IL)-2 receptor as well as interleukin (IL)-10 receptor. Tyrosine kinase of the non-receptor type, involved in the IFN-alpha/beta/gamma signal pathway. Related diseases Autoinflammation, immune dysregulation, and eosinophilia (AIIDE) [MIM:618999]: An autosomal dominant disorder characterized by immune dysregulation, severe atopic dermatitis, and chronic gastrointestinal inflammation. Additional features include asthma, food or environmental allergies, as well as poor overall growth with short stature. {ECO:0000269|PubMed:28111307, ECO:0000269|PubMed:32750333}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04716; DB14973; DB11817; DB12500; DB14845; DB12010; DB16756; DB11763; DB02375; DB15822; DB08877; DB08895; DB15091 Interacts with P04626; P48551; P40189-1; O60674; Q99650; P42224; P40763 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Disease variant; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; SH2 domain; Transferase; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32050.7 Length 280 Aromaticity 0.09 Instability index 44.51 Isoelectric point 8.07 Charge (pH=7) 2.39 2D Binding mode Binding energy (Kcal/mol) -8.86  Molscript Map 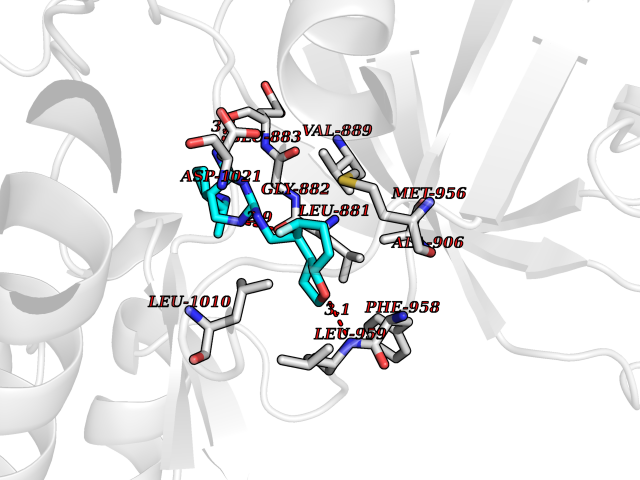 Pymol Map 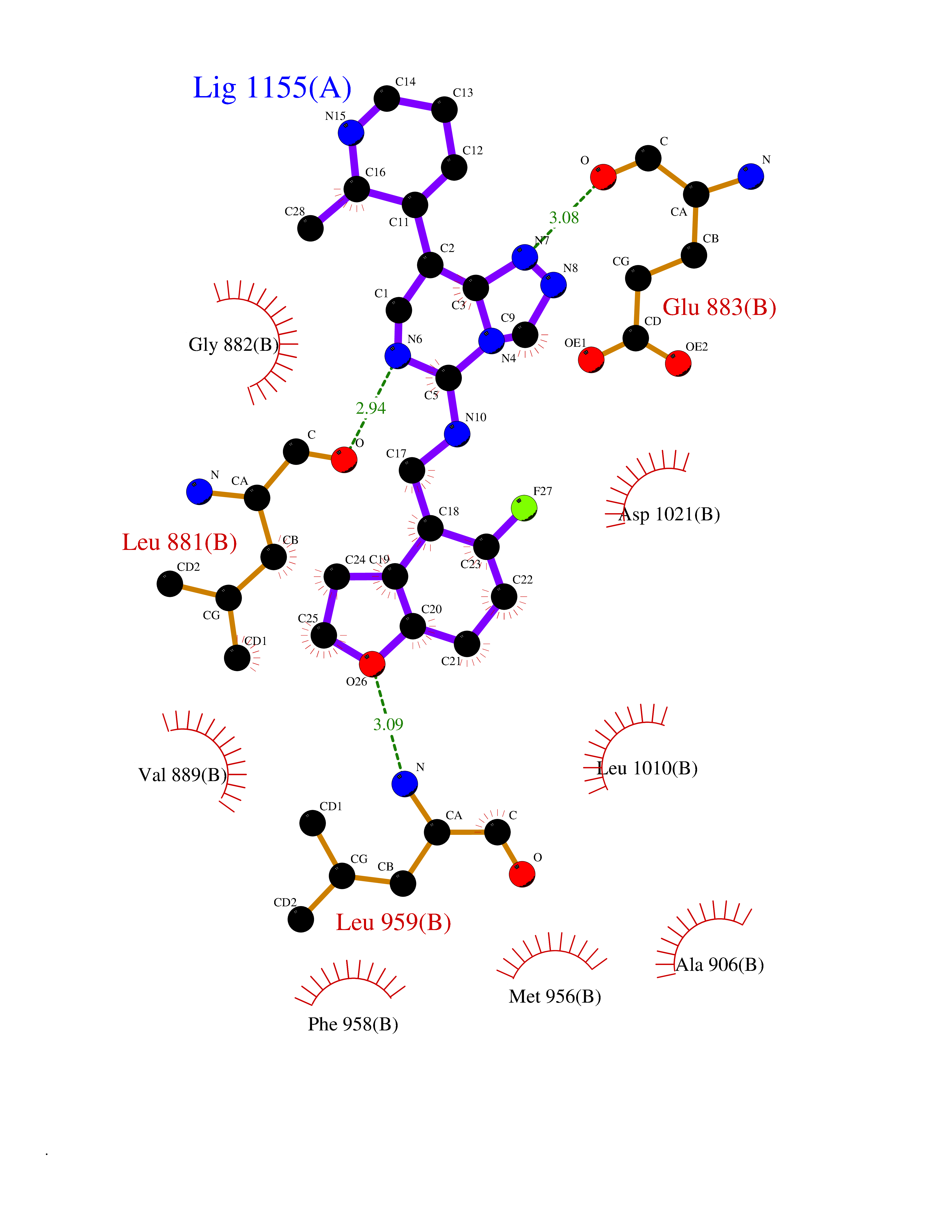 Ligplot Map 3D Binding mode Sequence VDPTHFEKRFLKRIRDLGEGHFGKVELCRYDPEGDNTGEQVAVKSLKPNHIADLKKEIEILRNLYHENIVKYKGICTENGIKLIMEFLPSGSLKEYLPKNKNKINLKQQLKYAVQICKGMDYLGSRQYVHRDLAARNVLVESEHQVKIGDFGLTKAIEKEXXTVKDDRDSPVFWYAPECLMQSKFYIASDVWSFGVTLHELLTYCDSDSSPMALFLKMIGPTHGQMTVTRLVNTLKEGKRLPCPPNCPDEVYQLMRKCWEFQPSNRTSFQNLIEGFEALL Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Dopamine beta-hydroxylase | 4ZEL | 8.02 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -9.07  Molscript Map 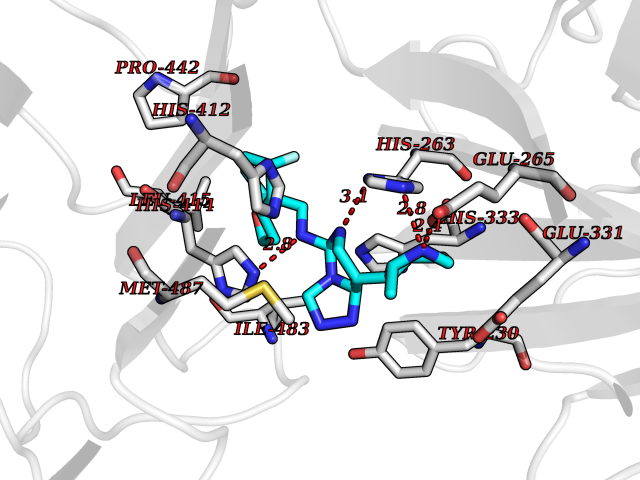 Pymol Map 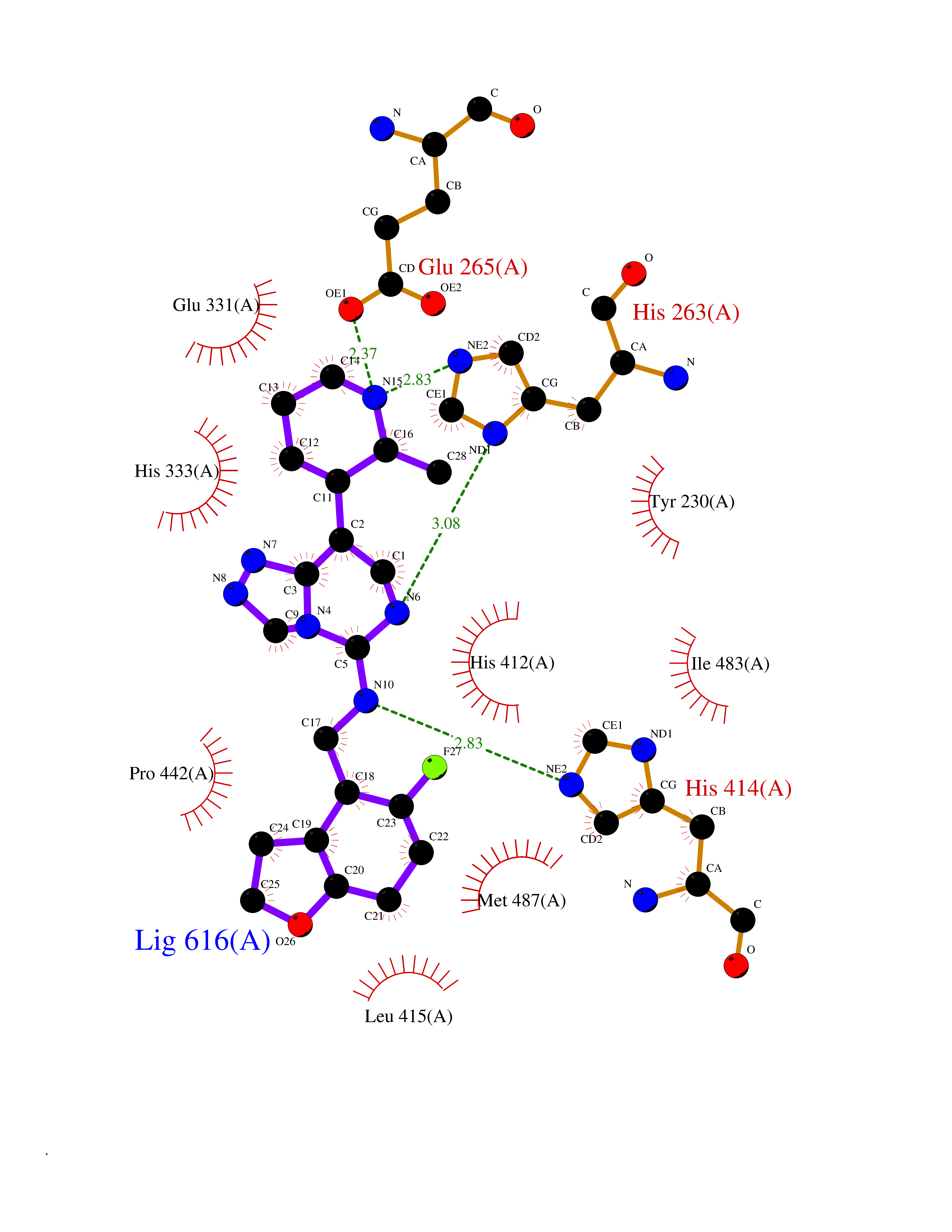 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Platelet glycoprotein VI (GP6) | 5OU7 | 8.02 | |
Target general information Gen name GP6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glycoprotein 6; GPVI Protein family NA Biochemical class NA Function Collagen receptor involved in collagen-induced platelet adhesion and activation. Plays a key role in platelet procoagulant activity and subsequent thrombin and fibrin formation. This procoagulant function may contribute to arterial and venous thrombus formation. The signaling pathway involves the FcR gamma-chain, the Src kinases (likely FYN or LYN) and SYK, the adapter protein LAT and leads to the activation of PLCG2. Related diseases Bleeding disorder, platelet-type, 11 (BDPLT11) [MIM:614201]: A mild to moderate bleeding disorder caused by defective platelet activation and aggregation in response to collagen. {ECO:0000269|PubMed:19549989, ECO:0000269|PubMed:19552682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P06241; P07948; P06241; P07948 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hemostasis; Immunoglobulin domain; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 19027.4 Length 173 Aromaticity 0.11 Instability index 37.14 Isoelectric point 8.68 Charge (pH=7) 2.52 2D Binding mode Binding energy (Kcal/mol) -7.13  Molscript Map 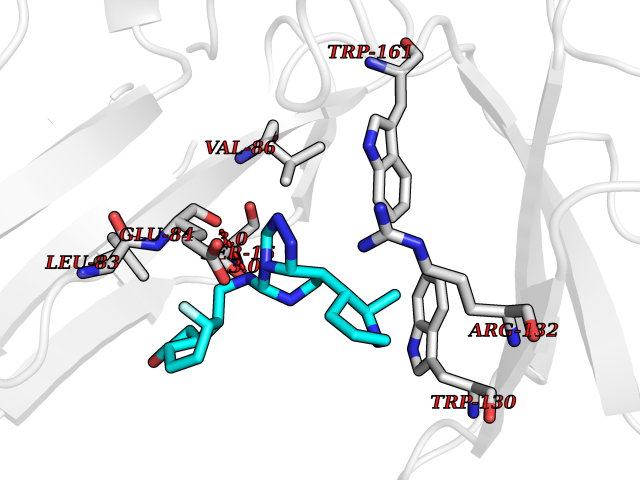 Pymol Map 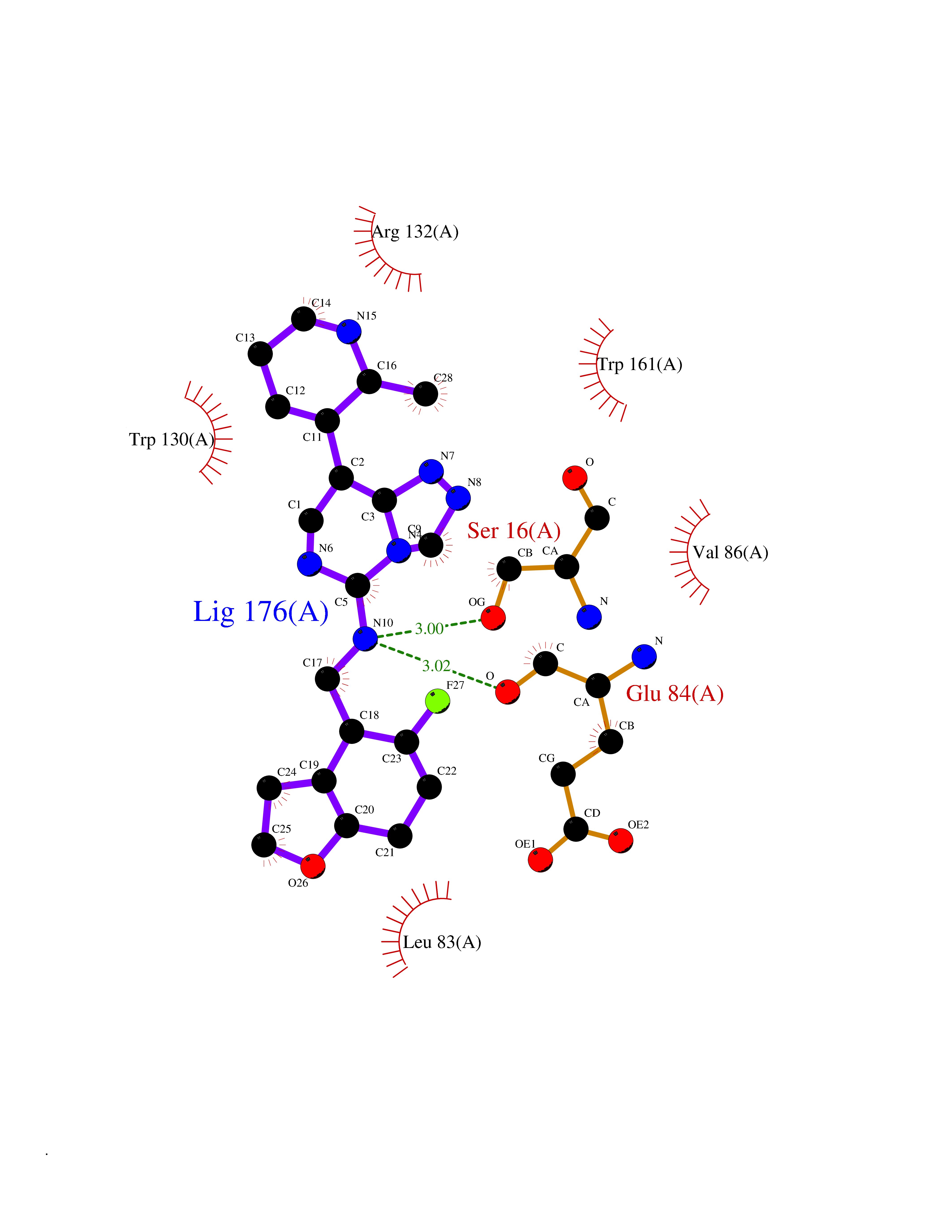 Ligplot Map 3D Binding mode Sequence SGPLPKPSLQALPSSLVPLEKPVTLRCQGPPGVDLYRLEKLSSSRYQDQAVLFIPAMKRSLAGRYRCSYQNGSLWSLPSDQLELVATGVFAKPSLSAQPGSGGDVTLQCQTRYGFDQFALYKEGDPERWYRASFPIITVTAAHSGTYRCYSFSSRDPYLWSAPSDPLELVVTG Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Kallikrein-5 (KLK5) | 6QFE | 8.01 | |
Target general information Gen name KLK5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ570/PRO1132; Stratum corneum tryptic enzyme; SCTE; Kallikrein-like protein 2; KLK-L2 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function May be involved in desquamation. Related diseases Lipodystrophy, familial partial, 8 (FPLD8) [MIM:620679]: An autosomal dominant form of partial lipodystrophy, a disorder characterized by abnormal subcutaneous fat distribution. FPLD8 patients show selective loss of subcutaneous adipose tissue from the limbs, beginning around 13 to 15 years of age, and abnormal accumulation of subcutaneous adipose tissue in the dorsal neck and face, as well as in the posterior thoracic and abdominal regions. The disorder is associated with metabolic abnormalities, including diabetes mellitus and hyperlipidemia. {ECO:0000269|PubMed:27376152}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P20930; Q9NQG1 EC number EC 3.4.21.- Uniprot keywords 3D-structure; Disulfide bond; Glycoprotein; Hydrolase; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50299.2 Length 454 Aromaticity 0.07 Instability index 40.74 Isoelectric point 9.25 Charge (pH=7) 23.09 2D Binding mode Binding energy (Kcal/mol) -8.43 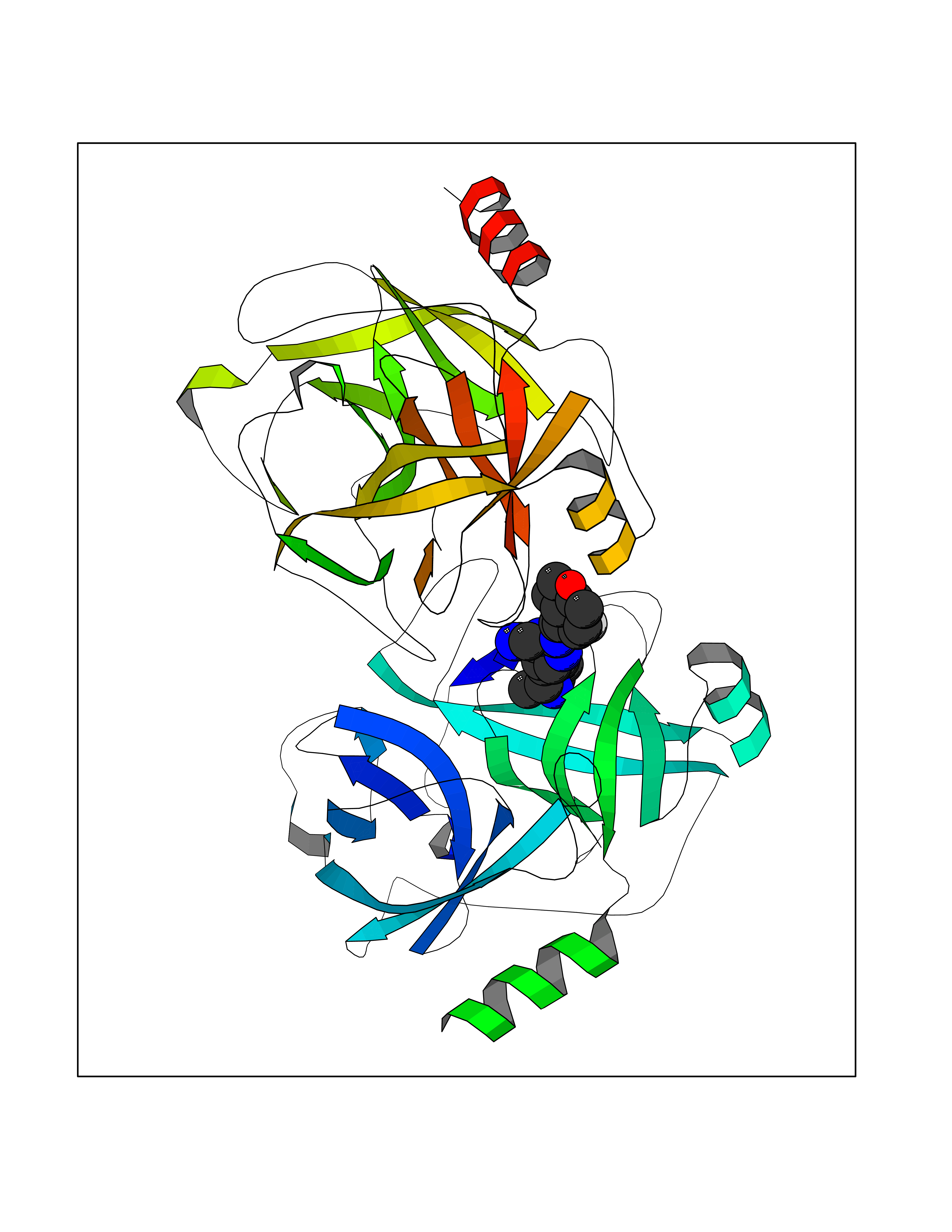 Molscript Map 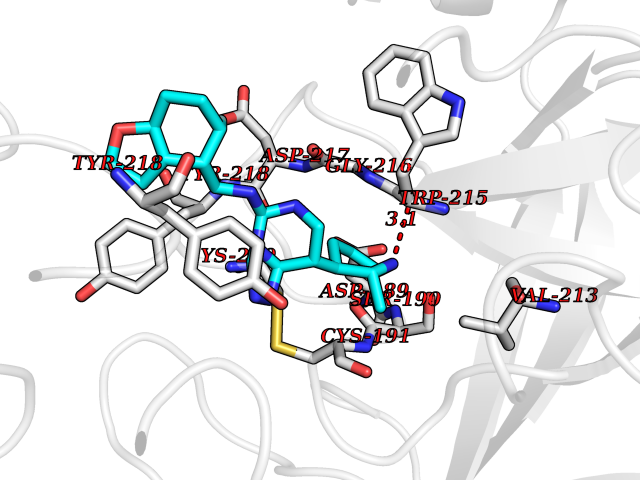 Pymol Map 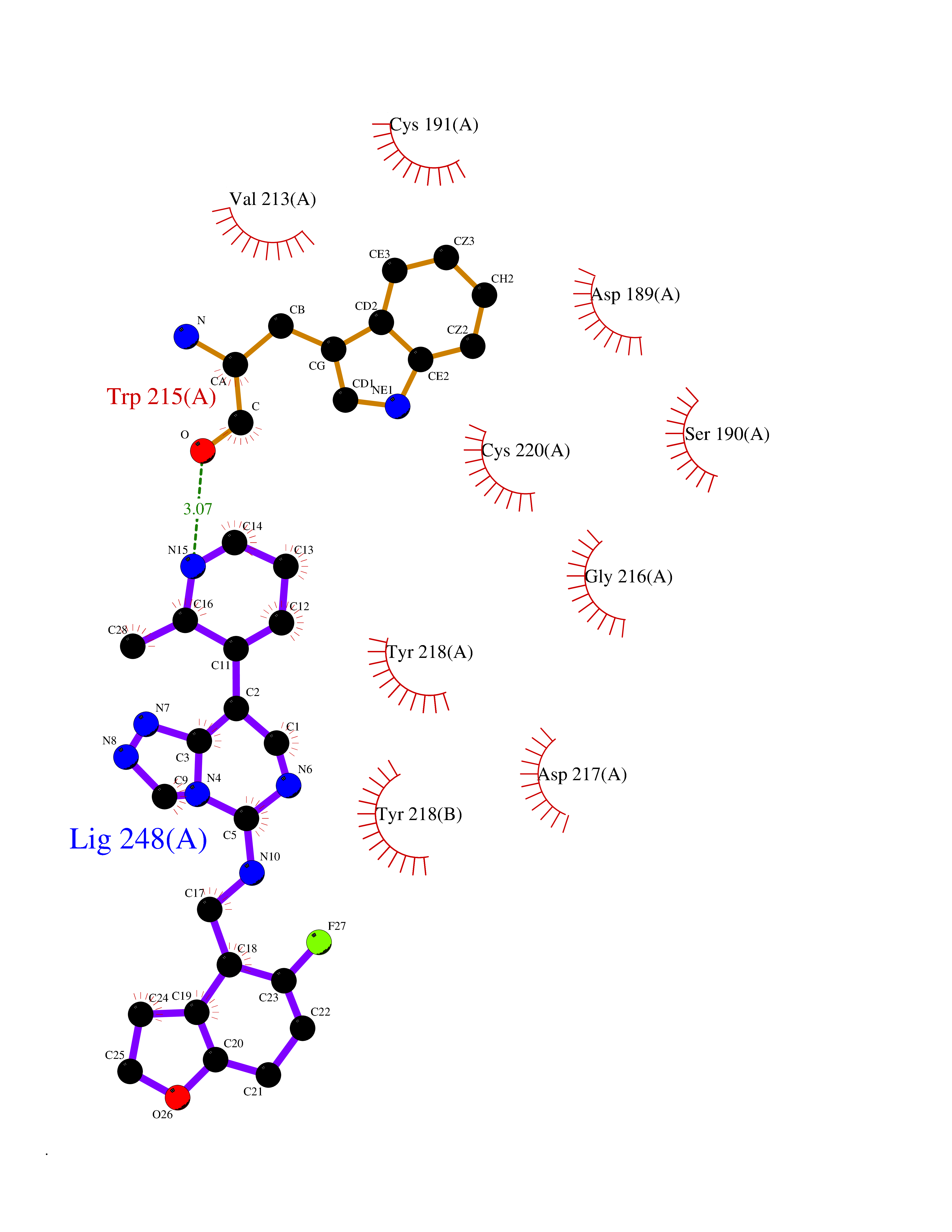 Ligplot Map 3D Binding mode Sequence IINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANSIINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANS Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 8.01 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -9.57 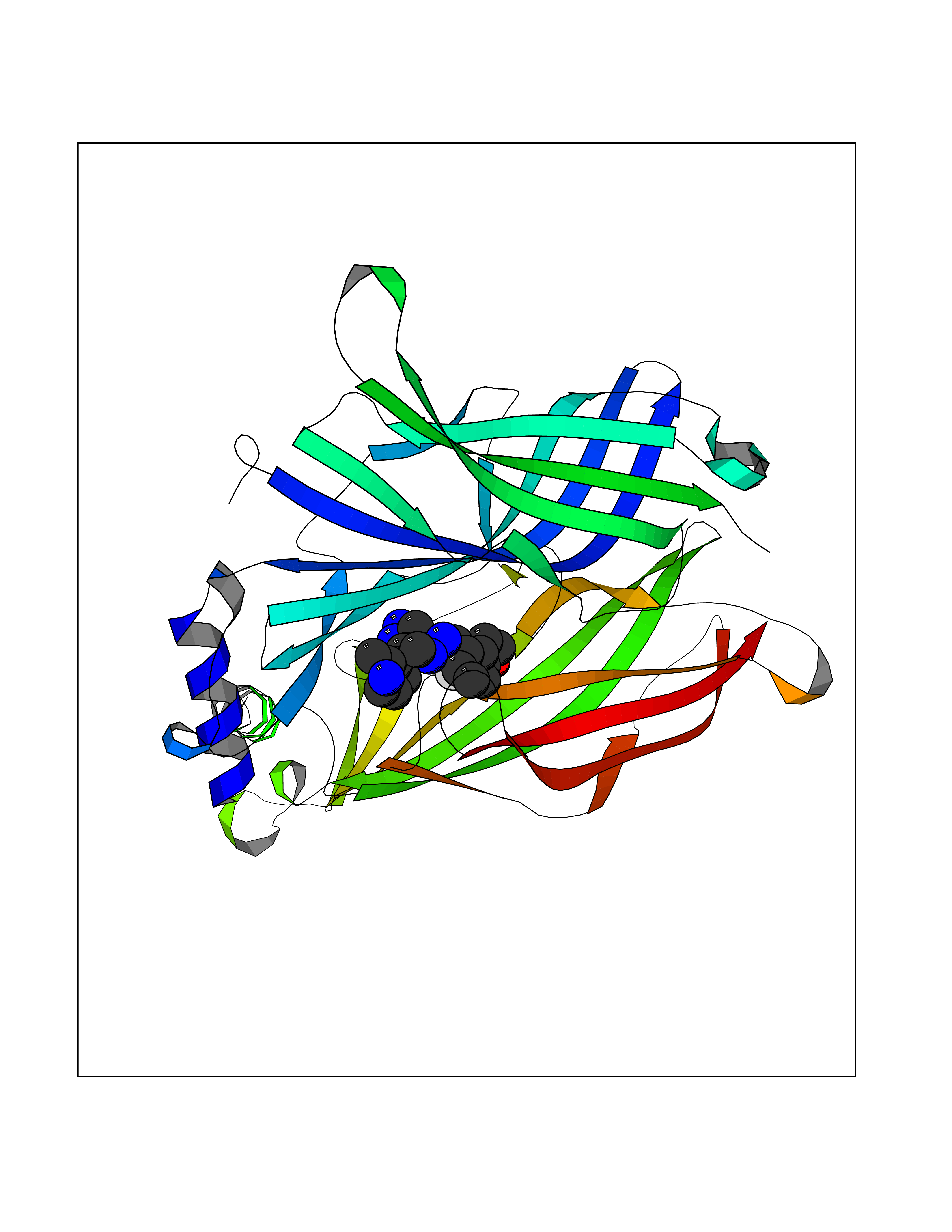 Molscript Map 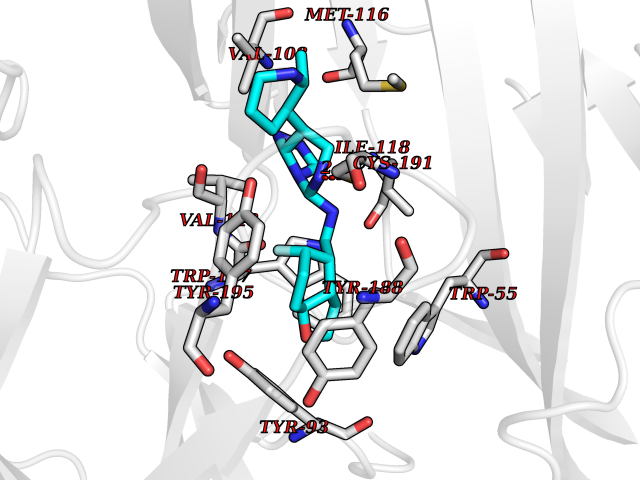 Pymol Map 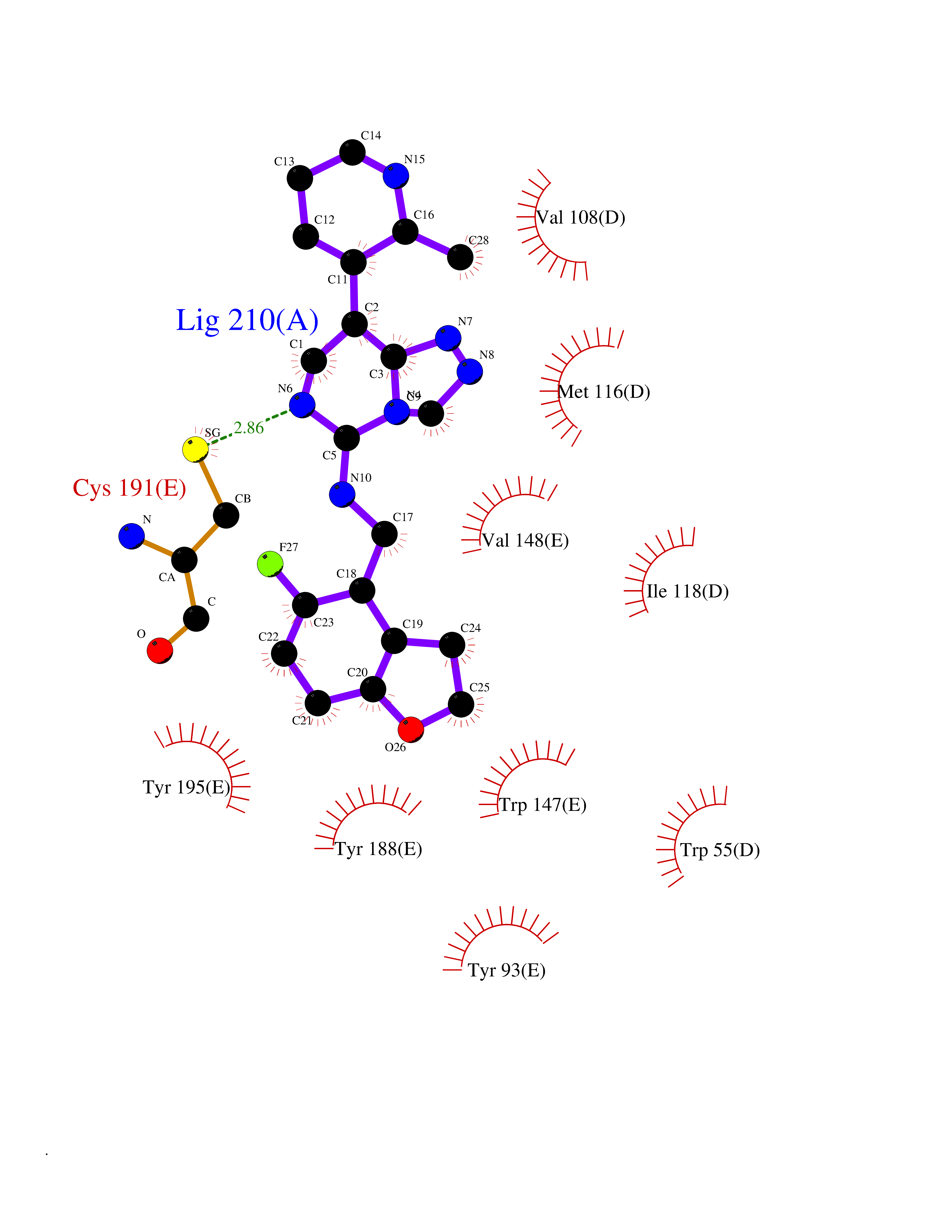 Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Soluble epoxide hydrolase (EPHX2) | 1ZD3 | 8.00 | |
Target general information Gen name EPHX2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Bifunctional epoxide hydrolase 2 Protein family AB hydrolase superfamily, Epoxide hydrolase family Biochemical class Ether bond hydrolase Function Bifunctional enzyme. The C-terminal domain has epoxide hydrolase activity and acts on epoxides (alkene oxides, oxiranes) and arene oxides. Plays a role in xenobiotic metabolism by degrading potentially toxic epoxides (By similarity). Also determines steady-state levels of physiological mediators. The N-terminal domain has lipid phosphatase activity, with the highest activity towards threo-9,10-phosphonooxy-hydroxy-octadecanoic acid, followed by erythro-9,10-phosphonooxy-hydroxy-octadecanoic acid, 12-phosphonooxy-octadec-9Z-enoic acid and 12-phosphonooxy-octadec-9E-enoic acid. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08257; DB08258; DB08259; DB06345; DB12610; DB08256; DB02029; DB04213; DB03677 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aromatic hydrocarbons catabolism; Cytoplasm; Detoxification; Direct protein sequencing; Hydrolase; Lipid metabolism; Lipoprotein; Magnesium; Metal-binding; Multifunctional enzyme; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 61744.9 Length 547 Aromaticity 0.09 Instability index 43.97 Isoelectric point 5.81 Charge (pH=7) -7.76 2D Binding mode Binding energy (Kcal/mol) -9.36  Molscript Map 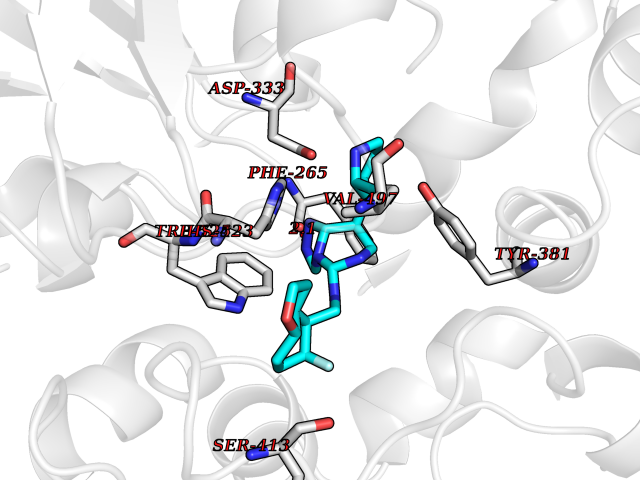 Pymol Map 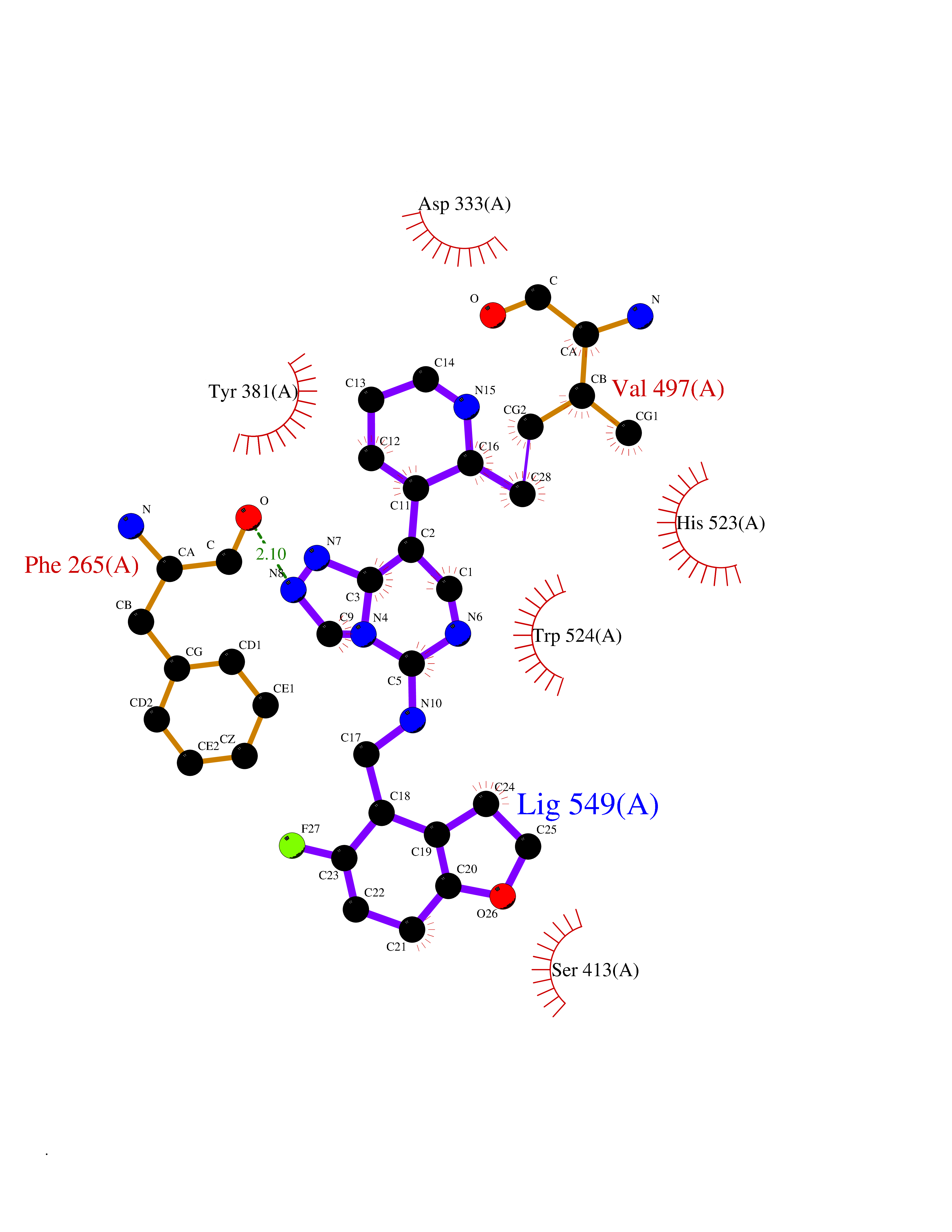 Ligplot Map 3D Binding mode Sequence TLRAAVFDLDGVLALPAVFGVLGRTEEALALPRGLLNDAFQKGGPEGATTRLMKGEITLSQWIPLMEENCRKCSETAKVCLPKNFSIKEIFDKAISARKINRPMLQAALMLRKKGFTTAILTNTWLDDRAERDGLAQLMCELKMHFDFLIESCQVGMVKPEPQIYKFLLDTLKASPSEVVFLDDIGANLKPARDLGMVTILVQDTDTALKELEKVTGIQLLNTPAPLPTSCNPSDMSHGYVTVKPRVRLHFVELGSGPAVCLCHGFPESWYSWRYQIPALAQAGYRVLAMDMKGYGESSAPPEIEEYCMEVLCKEMVTFLDKLGLSQAVFIGHDWGGMLVWYMALFYPERVRAVASLNTPFIPANPNMSPLESIKANPVFDYQLYFQEPGVAEAELEQNLSRTFKSLFRASDESVLSMHKVCEAGGLFVNSPEEPSLSRMVTEEEIQFYVQQFKKSGFRGPLNWYRNMERNWKWACKSLGRKILIPALMVTAEKDFVLVPQMSQHMEDWIPHLKRGHIEDCGHWTQMDKPTEVNQILIKWLDSDARN Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 8.00 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -9.65 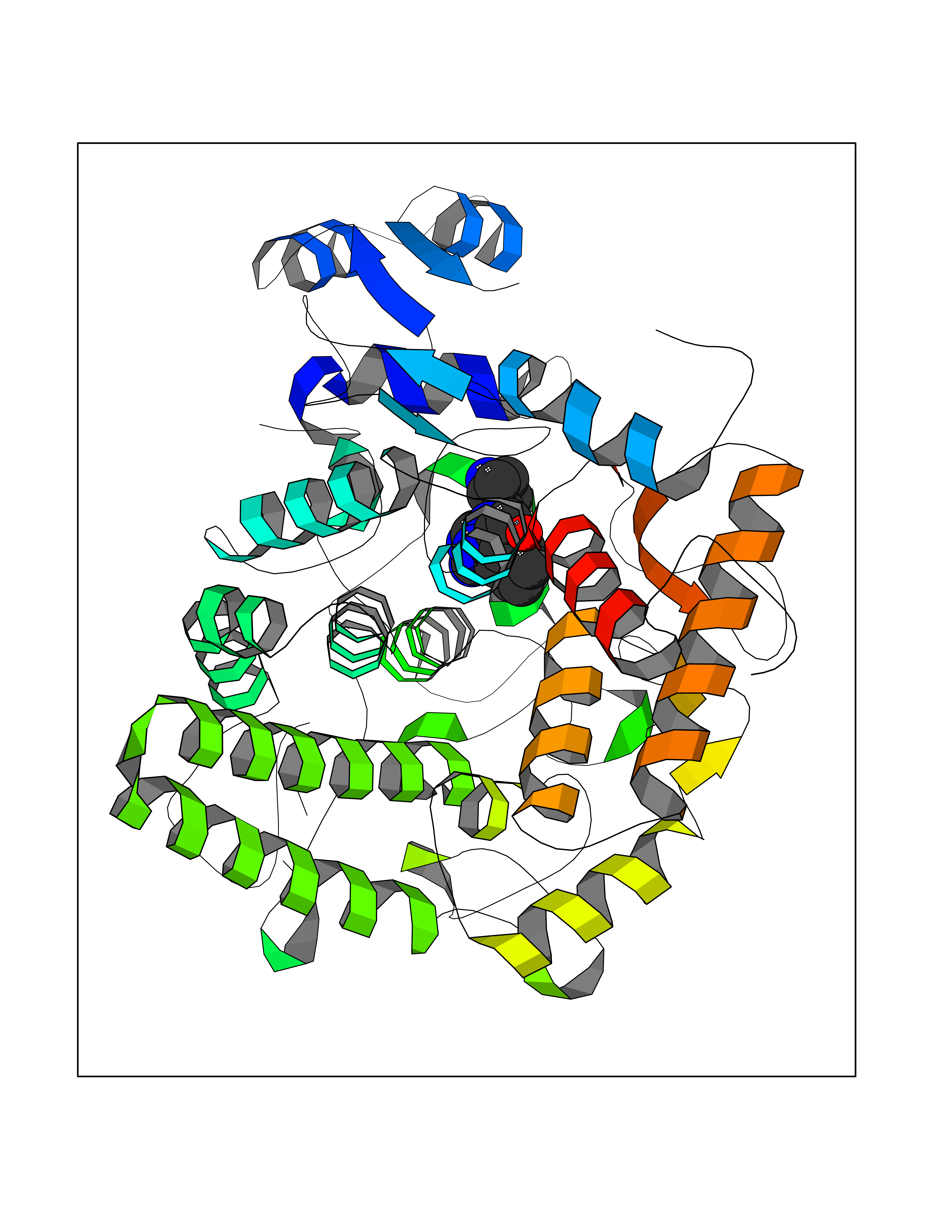 Molscript Map 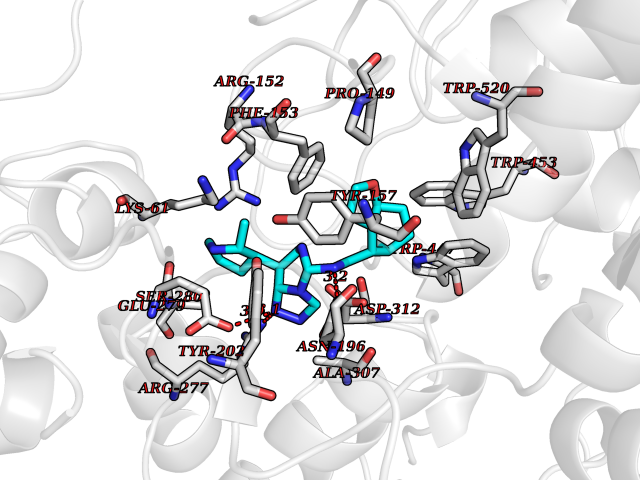 Pymol Map 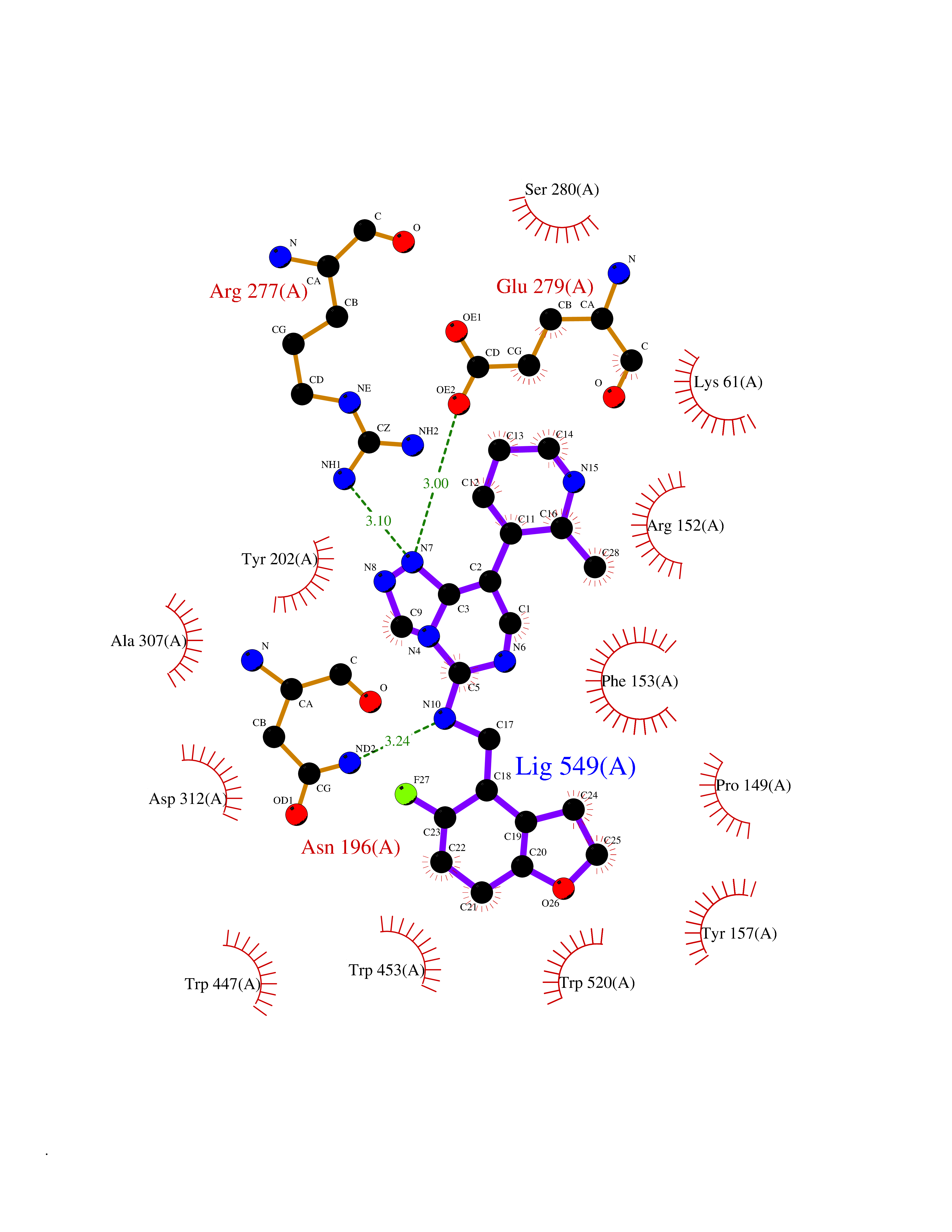 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Trypanosoma Dihydrolipoamide dehydrogenase (Trypano LPD) | 2QAE | 8.00 | |
Target general information Gen name Trypano LPD Organism Trypanosoma cruzi Uniprot ID TTD ID Synonyms Lipoamide dehydrogenase; LipDH; LPD; Glycine cleavage system L protein; Dihydrolipoamide dehydrogenase Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Sulfur donor oxidoreductase Function Lipoamide dehydrogenase is a component of the glycine cleavage system as well as of the alpha-ketoacid dehydrogenase complexes. Related diseases Focal segmental glomerulosclerosis 2 (FSGS2) [MIM:603965]: A renal pathology defined by the presence of segmental sclerosis in glomeruli and resulting in proteinuria, reduced glomerular filtration rate and progressive decline in renal function. Renal insufficiency often progresses to end-stage renal disease, a highly morbid state requiring either dialysis therapy or kidney transplantation. {ECO:0000269|PubMed:15879175, ECO:0000269|PubMed:15924139, ECO:0000269|PubMed:19458060, ECO:0000269|PubMed:19936226, ECO:0000269|PubMed:20798252, ECO:0000269|PubMed:21511817, ECO:0000269|PubMed:21734084, ECO:0000269|PubMed:22732337, ECO:0000269|PubMed:23014460, ECO:0000269|PubMed:23291369, ECO:0000269|PubMed:26892346}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Disulfide bond; FAD; Flavoprotein; NAD; Oxidoreductase; Redox-active center Protein physicochemical properties Chain ID A,B Molecular weight (Da) 98183.9 Length 930 Aromaticity 0.06 Instability index 29.02 Isoelectric point 6.32 Charge (pH=7) -6.37 2D Binding mode Binding energy (Kcal/mol) -9.39 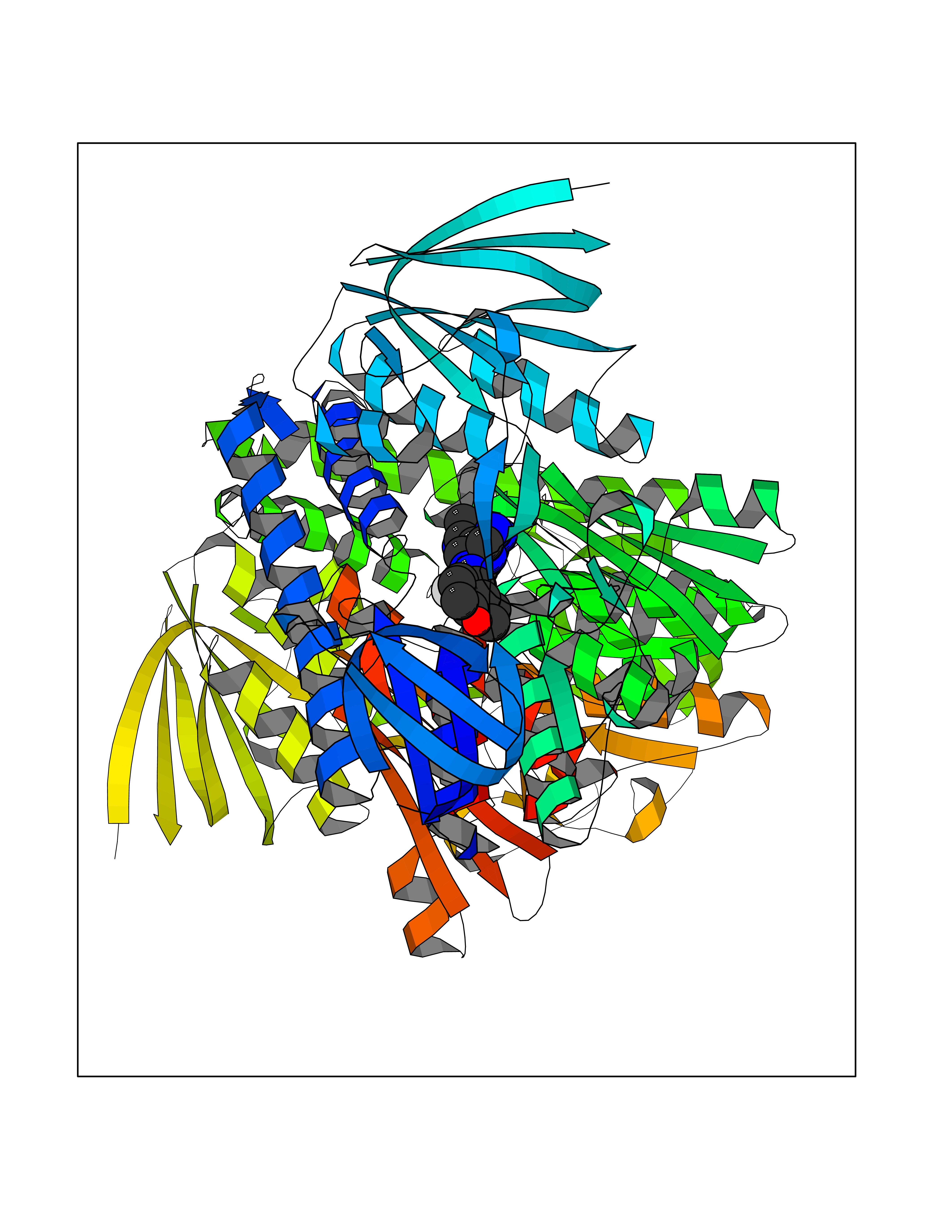 Molscript Map 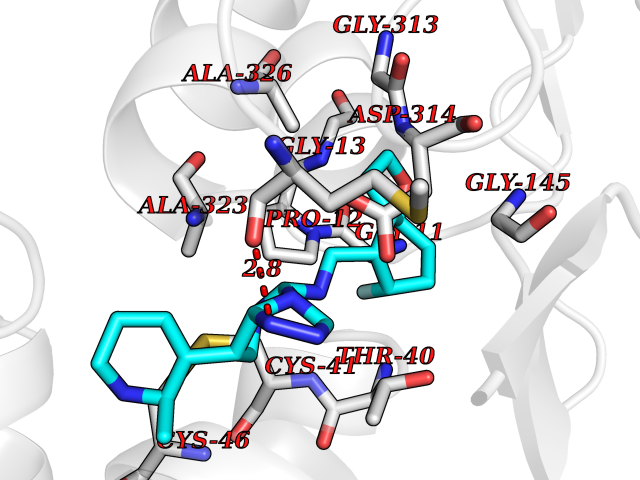 Pymol Map 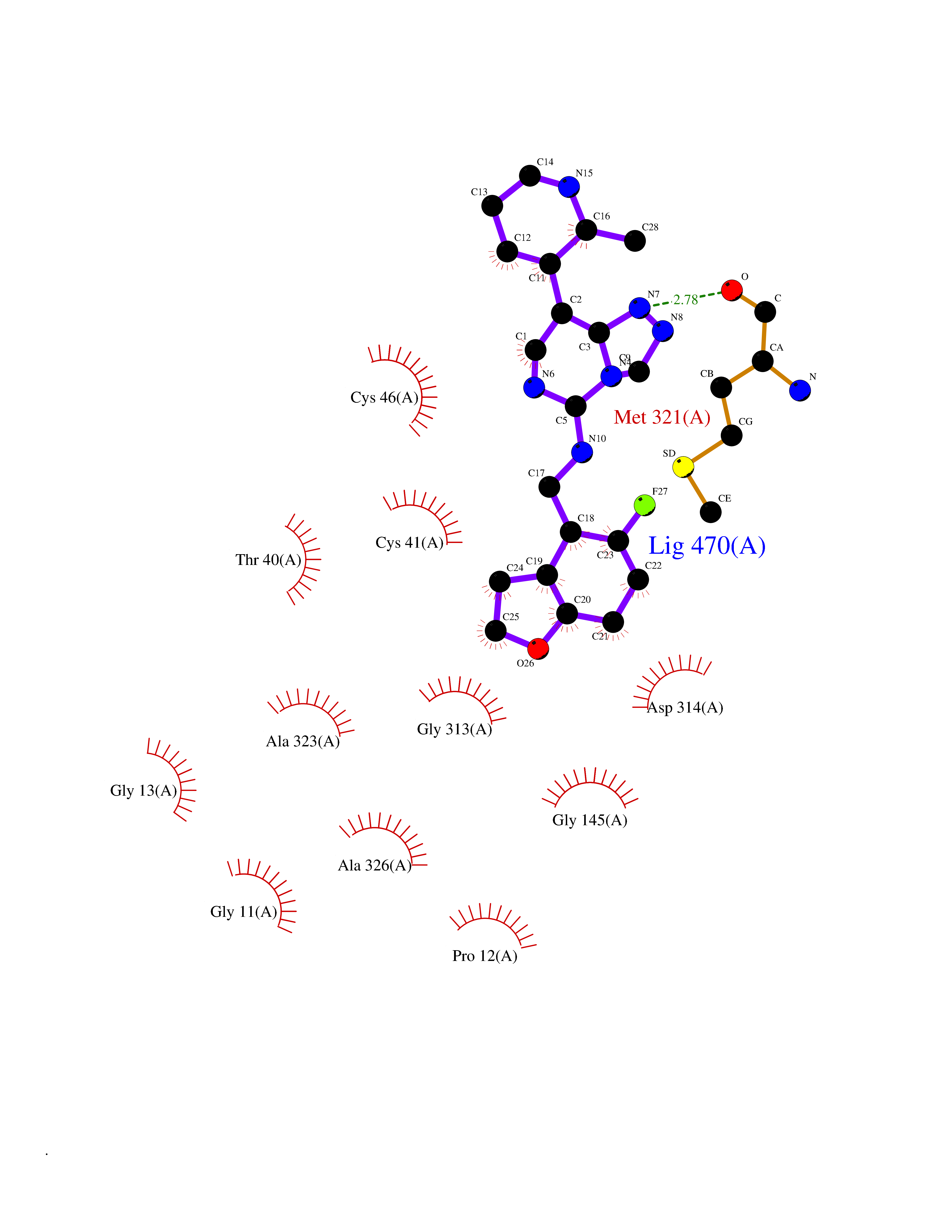 Ligplot Map 3D Binding mode Sequence NPYDVVVIGGGPGGYVASIKAAQLGMKTACVEKRGALGGTCLNVGCIPSKALLHATHLYHDAHANFARYGLMGGEGVTMDSAKMQQQKERAVKGLTGGVEYLFKKNKVTYYKGEGSFETAHSIRVNGLDGKQEMLETKKTIIATGSEPTELPFLPFDEKVVLSSTGALALPRVPKTMVVIGGGVIGLELGSVWARLGAEVTVVEFAPRCAPTLDEDVTNALVGALAKNEKMKFMTSTKVVGGTNNGDSVSLEVEGKRETVTCEALLVSVGRRPFTGGLGLDKINVAKNERGFVKIGDHFETSIPDVYAIGDVVDKGPMLAHKAEDEGVACAEILAGKPGHVNYGVIPAVIYTMPEVASVGKSEDELKKEGVAYKVGKFPFNANSRAKAVSTEDGFVKVLVDKATDRILGVHIVCTTAGELIGEACLAMEYGASSEDVGRTCHAHPTMSEALKEACMALFAKTINFNPYDVVVIGGGPGGYVASIKAAQLGMKTACVEKRGALGGTCLNVGCIPSKALLHATHLYHDAHANFARYGLMGGEGVTMDSAKMQQQKERAVKGLTGGVEYLFKKNKVTYYKGEGSFETAHSIRVNGLDGKQEMLETKKTIIATGSEPTELPFLPFDEKVVLSSTGALALPRVPKTMVVIGGGVIGLELGSVWARLGAEVTVVEFAPRCAPTLDEDVTNALVGALAKNEKMKFMTSTKVVGGTNNGDSVSLEVEGKRETVTCEALLVSVGRRPFTGGLGLDKINVAKNERGFVKIGDHFETSIPDVYAIGDVVDKGPMLAHKAEDEGVACAEILAGKPGHVNYGVIPAVIYTMPEVASVGKSEDELKKEGVAYKVGKFPFNANSRAKAVSTEDGFVKVLVDKATDRILGVHIVCTTAGELIGEACLAMEYGASSEDVGRTCHAHPTMSEALKEACMALFAKTINF Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Protein arginine methyltransferase 5 (PRMT5) | 7MXC | 7.99 | |
Target general information Gen name PRMT5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Shk1 kinase-binding protein 1 homolog; SKB1Hs; SKB1 homolog; SKB1; Protein arginine N-methyltransferase 5; Jak-binding protein 1; JBP1; IBP72; Histone-arginine N-methyltransferase PRMT5; HRMT1L5; 72 k Protein family Class I-like SAM-binding methyltransferase superfamily, Protein arginine N-methyltransferase family Biochemical class Methyltransferase Function Specifically mediates the symmetrical dimethylation of arginine residues in the small nuclear ribonucleoproteins Sm D1 (SNRPD1) and Sm D3 (SNRPD3); such methylation being required for the assembly and biogenesis of snRNP core particles. Methylates SUPT5H and may regulate its transcriptional elongation properties. Mono- and dimethylates arginine residues of myelin basic protein (MBP) in vitro. May play a role in cytokine-activated transduction pathways. Negatively regulates cyclin E1 promoter activity and cellular proliferation. Methylates histone H2A and H4 'Arg-3' during germ cell development. Methylates histone H3 'Arg-8', which may repress transcription. Methylates the Piwi proteins (PIWIL1, PIWIL2 and PIWIL4), methylation of Piwi proteins being required for the interaction with Tudor domain-containing proteins and subsequent localization to the meiotic nuage. Methylates RPS10. Attenuates EGF signaling through the MAPK1/MAPK3 pathway acting at 2 levels. First, monomethylates EGFR; this enhances EGFR 'Tyr-1197' phosphorylation and PTPN6 recruitment, eventually leading to reduced SOS1 phosphorylation. Second, methylates RAF1 and probably BRAF, hence destabilizing these 2 signaling proteins and reducing their catalytic activity. Required for induction of E-selectin and VCAM-1, on the endothelial cells surface at sites of inflammation. Methylates HOXA9. Methylates and regulates SRGAP2 which is involved in cell migration and differentiation. Acts as a transcriptional corepressor in CRY1-mediated repression of the core circadian component PER1 by regulating the H4R3 dimethylation at the PER1 promoter. Methylates GM130/GOLGA2, regulating Golgi ribbon formation. Methylates H4R3 in genes involved in glioblastomagenesis in a CHTOP- and/or TET1-dependent manner. Symmetrically methylates POLR2A, a modification that allows the recruitment to POLR2A of proteins including SMN1/SMN2 and SETX. This is required for resolving RNA-DNA hybrids created by RNA polymerase II, that form R-loop in transcription terminal regions, an important step in proper transcription termination. Along with LYAR, binds the promoter of gamma-globin HBG1/HBG2 and represses its expression. Symmetrically methylates NCL. Methylates TP53; methylation might possibly affect TP53 target gene specificity. Arginine methyltransferase that can both catalyze the formation of omega-N monomethylarginine (MMA) and symmetrical dimethylarginine (sDMA), with a preference for the formation of MMA. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P01019; Q9NX04; Q8WUW1; Q08289; P78371; Q16543; Q8N8U2; P54105; P21964-2; Q9NQ92; Q16526; Q9Y6K1; Q01094; Q08426; P38919; Q14241; O15197-2; Q6ZV65; P01100; O95995; P62993; Q8TE85; Q9BX10; P62805; P31269; Q00613; Q63ZY3; P03952; Q8TBB1; P06858; Q86UQ8-1; Q96HA8; Q8WVJ2; P24928; O14744; Q86U06; Q9BRS2; O75044; Q96RU7; P31930; P40337-2; Q9BQA1; P63104; Q96E35; Q91XC0; P03418; Q6ZV65-2 EC number EC 2.1.1.320 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Chromosome; Cytoplasm; Direct protein sequencing; Golgi apparatus; Methyltransferase; Nucleus; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 71188.3 Length 621 Aromaticity 0.11 Instability index 44.6 Isoelectric point 5.95 Charge (pH=7) -9.68 2D Binding mode Binding energy (Kcal/mol) -9.73 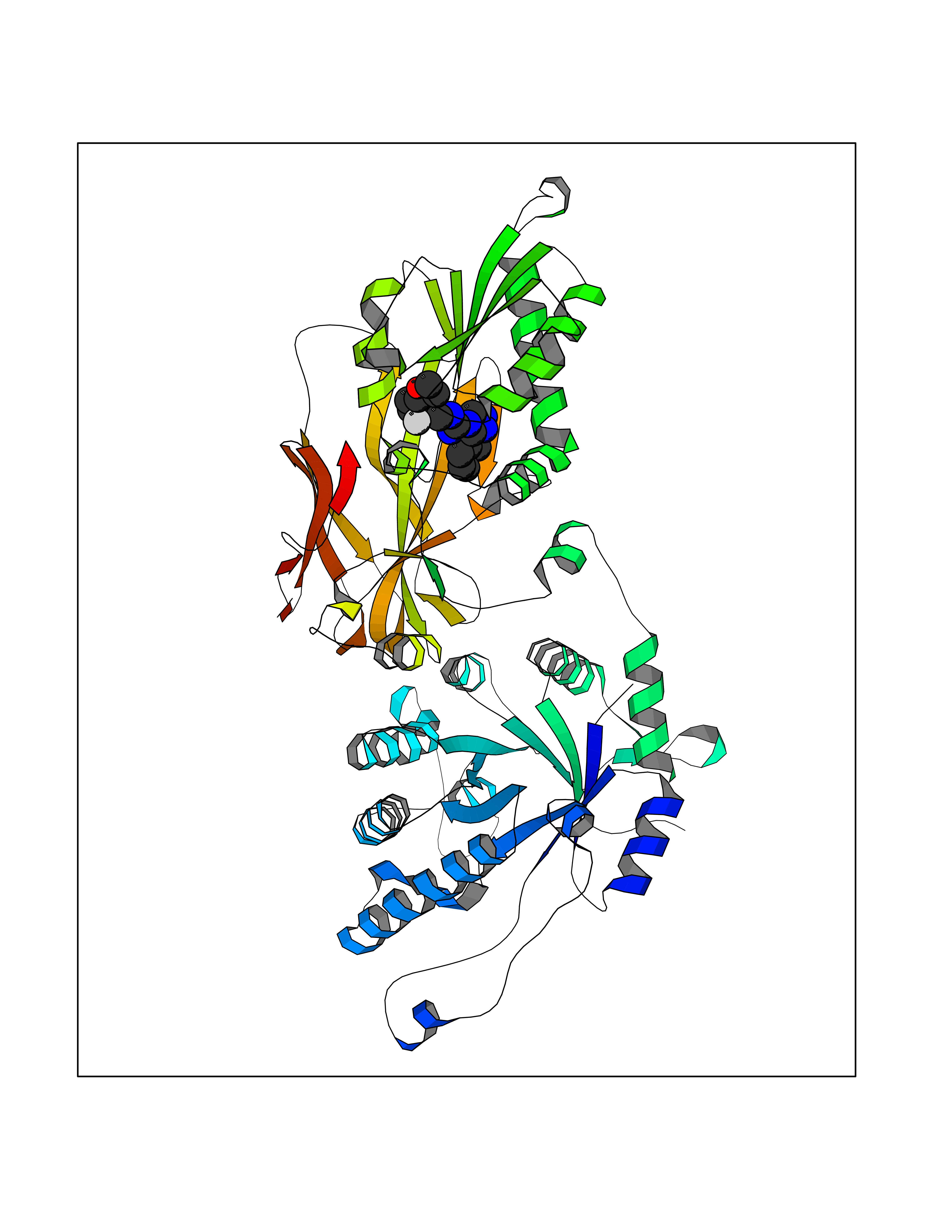 Molscript Map 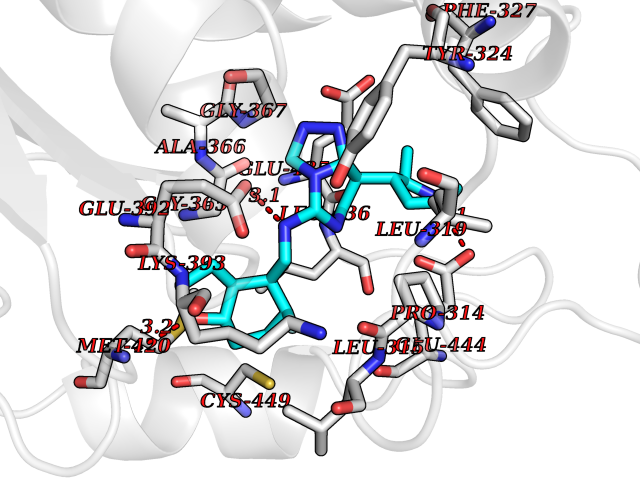 Pymol Map 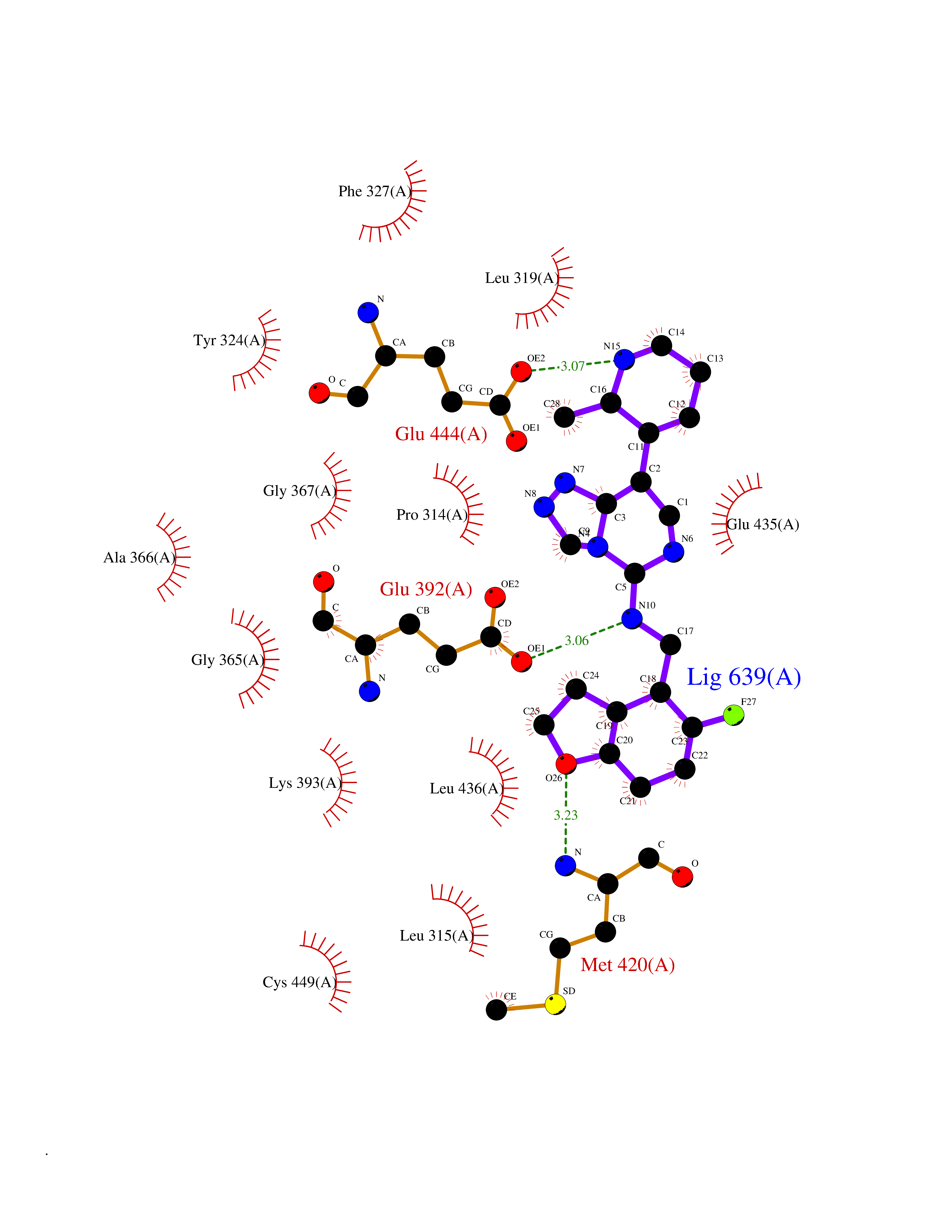 Ligplot Map 3D Binding mode Sequence RVSSGRDLNCVPEIADTLGAVAKQGFDFLCMPVFHPRFKREFIQEPAKNRPGPQTRSDLLLSGRDWNTLIVGKLSPWIRPDSKVEKIRRNSEAAMLQELNFGAYLGLPAFLLPLNQEDNTNLARVLTNHIHTGHHSSMFWMRVPLVAPEDLRDDIIENAPTTHTEEYSGEEKTWMWWHNFRTLCDYSKRIAVALEIGADLPSNHVIDRWLGEPIKAAILPTSIFLTNKKGFPVLSKMHQRLIFRLLKLEVQFIITGTNHHSCSYLQYLEYLSQNRPPPNAYELFAKGYEDYLQSPLQPLMDNLESQTYEVFEKDPIKYSQYQQAIYKCLLDRVPEEEKDTNVQVLMVLGAGRGPLVNASLRAAKQADRRIKLYAVEKNPNAVVTLENWQFEEWGSQVTVVSSDMREWVAPEKADIIVSELLGSFADNELSPECLDGAQHFLKDDGVSIPGEYTSFLAPISSSKLYNEVRACREKDRDPEAQFEMPYVVRLHNFHQLSAPQPCFTFSHPNRDPMIDNNRYCTLEFPVEVNTVLHGFAGYFETVLYQDITLSIRPETHSPGMFSWFPILFPIKQPITVREGQTICVRFWRCSNSKKVWYEWAVTAPVCSAIHNPTGRSYTIGL Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Cholesterol oxidase | 4REK | 7.99 | |
Target general information Gen name choA Organism Streptomyces sp. (strain SA-COO) Uniprot ID TTD ID NA Synonyms NA Protein family GMC oxidoreductase family Biochemical class Oxidoreductase Function Cholesterol oxidase activity.Flavin adenine dinucleotide binding.Steroid delta-isomerase activity. Related diseases Bothnia retinal dystrophy (BRD) [MIM:607475]: A type of retinitis punctata albescens. Affected individuals show night blindness from early childhood with features consistent with retinitis punctata albescens and macular degeneration. {ECO:0000269|PubMed:10102298}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rod-cone dystrophy Newfoundland (NFRCD) [MIM:607476]: A rod-cone dystrophy reminiscent of retinitis punctata albescens but with a substantially lower age at onset and more-rapid and distinctive progression. Rod-cone dystrophies results from initial loss of rod photoreceptors, later followed by cone photoreceptors loss. {ECO:0000269|PubMed:11868161}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Retinitis punctata albescens (RPA) [MIM:136880]: A form of fleck retina disease characterized by aggregation of white flecks posteriorly in the retina, causing night blindness and delayed dark adaptation. It differs from fundus albipunctatus in being progressive and evolving to generalized atrophy of the retina. {ECO:0000269|PubMed:10102299, ECO:0000269|PubMed:11453974, ECO:0000269|PubMed:9326942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB02332 Interacts with NA EC number 1.1.3.6; 5.3.3.1 Uniprot keywords 3D-structure; Cholesterol metabolism; Direct protein sequencing; FAD; Flavoprotein; Isomerase; Lipid metabolism; Oxidoreductase; Secreted; Signal; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54367.8 Length 498 Aromaticity 0.1 Instability index 30.62 Isoelectric point 6.69 Charge (pH=7) -0.71 2D Binding mode Binding energy (Kcal/mol) -9.36  Molscript Map  Pymol Map 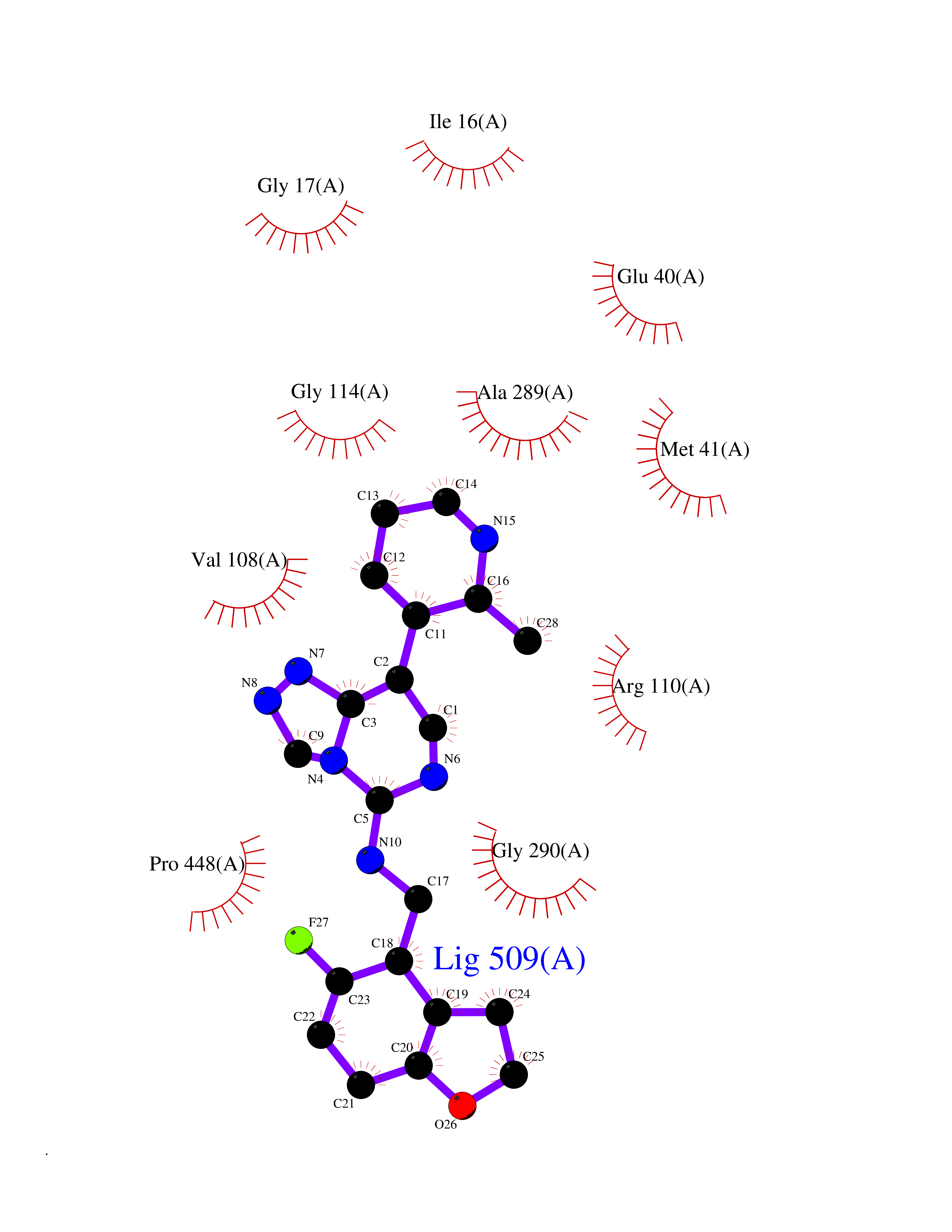 Ligplot Map 3D Binding mode Sequence GYVPAVVIGTGYGAAVSALRLGEAGVQTLMLEMGQLWNQPGPDGNIFCGMLNPDKRSSWFKNRTEAPLGSFLWLDVVNRNIDPYAGVLDRVNYDQMSVYVGRGVGGGSLVNGGMAVEPKRSYFEEILPRVDSSEMYDRYFPRANSMLRVNHIDTKWFEDTEWYKFARVSREQAGKAGLGTVFVPNVYDFGYMQREAAGEVPKSALATEVIYGNNHGKQSLDKTYLAAALGTGKVTIQTLHQVKTIRQTKDGGYALTVEQKDTDGKLLATKEISCRYLFLGAGSLGSTELLVRARDTGTLPNLNSEVGAGWGPNGNIMTARANHMWNPTGAHQSSIPALGIDAWDNSDSSVFAEIAPMPAGLETWVSLYLAITKNPQRGTFVYDAATDRAKLNWTRDQNAPAVNAAKALFDRINKANGTIYRYDLFGTQLKAFADDFCYHPLGGCVLGKATDDYGRVAGYKNLYVTDGSLIPGSVGVNPFVTITALAERNVERIIKQDV Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Ribosomal protein S6 kinase beta-1 (S6K1) | 7N93 | 7.99 | |
Target general information Gen name RPS6KB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p70-S6K 1; p70 ribosomal S6 kinase alpha; p70 S6KA; p70 S6K-alpha; p70 S6 kinase alpha; Serine/threonine-protein kinase 14A; STK14A; S6K-beta-1; S6K; Ribosomal protein S6 kinase I; P70S6K1; P70-S6K; 7 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, S6 kinase subfamily Biochemical class Kinase Function Regulates protein synthesis through phosphorylation of EIF4B, RPS6 and EEF2K, and contributes to cell survival by repressing the pro-apoptotic function of BAD. Under conditions of nutrient depletion, the inactive form associates with the EIF3 translation initiation complex. Upon mitogenic stimulation, phosphorylation by the mammalian target of rapamycin complex 1 (mTORC1) leads to dissociation from the EIF3 complex and activation. The active form then phosphorylates and activates several substrates in the pre-initiation complex, including the EIF2B complex and the cap-binding complex component EIF4B. Also controls translation initiation by phosphorylating a negative regulator of EIF4A, PDCD4, targeting it for ubiquitination and subsequent proteolysis. Promotes initiation of the pioneer round of protein synthesis by phosphorylating POLDIP3/SKAR. In response to IGF1, activates translation elongation by phosphorylating EEF2 kinase (EEF2K), which leads to its inhibition and thus activation of EEF2. Also plays a role in feedback regulation of mTORC2 by mTORC1 by phosphorylating RICTOR, resulting in the inhibition of mTORC2 and AKT1 signaling. Mediates cell survival by phosphorylating the pro-apoptotic protein BAD and suppressing its pro-apoptotic function. Phosphorylates mitochondrial URI1 leading to dissociation of a URI1-PPP1CC complex. The free mitochondrial PPP1CC can then dephosphorylate RPS6KB1 at Thr-412, which is proposed to be a negative feedback mechanism for the RPS6KB1 anti-apoptotic function. Mediates TNF-alpha-induced insulin resistance by phosphorylating IRS1 at multiple serine residues, resulting in accelerated degradation of IRS1. In cells lacking functional TSC1-2 complex, constitutively phosphorylates and inhibits GSK3B. May be involved in cytoskeletal rearrangement through binding to neurabin. Phosphorylates and activates the pyrimidine biosynthesis enzyme CAD, downstream of MTOR. Following activation by mTORC1, phosphorylates EPRS and thereby plays a key role in fatty acid uptake by adipocytes and also most probably in interferon-gamma-induced translation inhibition. Serine/threonine-protein kinase that acts downstream of mTOR signaling in response to growth factors and nutrients to promote cell proliferation, cell growth and cell cycle progression. Related diseases Progressive familial heart block 1B (PFHB1B) [MIM:604559]: A cardiac bundle branch disorder characterized by progressive alteration of cardiac conduction through the His-Purkinje system, with a pattern of a right bundle-branch block and/or left anterior hemiblock occurring individually or together. It leads to complete atrio-ventricular block causing syncope and sudden death. {ECO:0000269|PubMed:19726882, ECO:0000269|PubMed:20562447, ECO:0000269|PubMed:21887725}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythrokeratodermia variabilis et progressiva 6 (EKVP6) [MIM:618531]: A form of erythrokeratodermia variabilis et progressiva, a genodermatosis characterized by the coexistence of two independent skin lesions: transient erythema and hyperkeratosis that is usually localized but occasionally occurs in its generalized form. Clinical presentation varies significantly within a family and from one family to another. Palmoplantar keratoderma is present in around 50% of cases. EKVP6 inheritance is autosomal dominant. {ECO:0000269|PubMed:30528822}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P31749; Q06481-5; P55884; P08151; Q5VY09; Q00005; P13051-2; P08151; P05067; P27986-2 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Alternative initiation; Alternative splicing; Apoptosis; ATP-binding; Cell cycle; Cytoplasm; Kinase; Membrane; Mitochondrion; Mitochondrion outer membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Synapse; Synaptosome; Transferase; Translation regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 35187.6 Length 312 Aromaticity 0.1 Instability index 39.58 Isoelectric point 8.8 Charge (pH=7) 5.54 2D Binding mode Binding energy (Kcal/mol) -8.53  Molscript Map 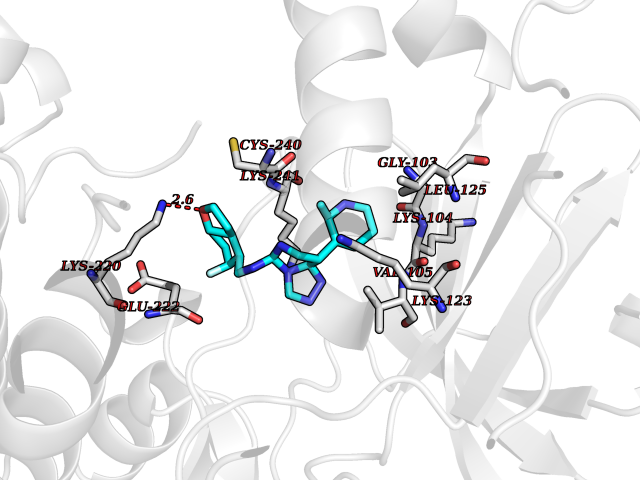 Pymol Map 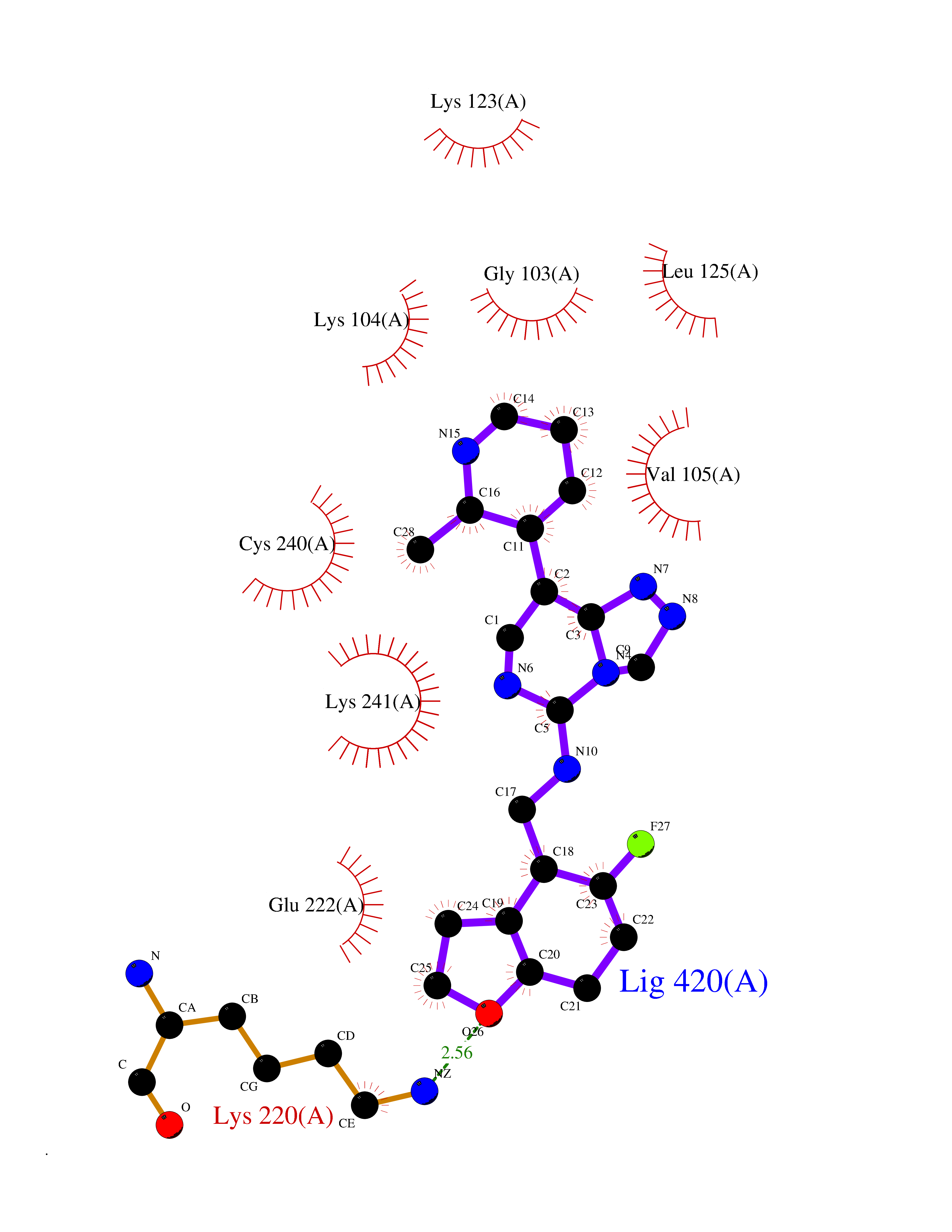 Ligplot Map 3D Binding mode Sequence RPECFELLRVLGKGGYGKVFQVRKVTGANTGKIFAMKVLKKAMIVRNAKDTAHTKAERNILEEVKHPFIVDLIYAFQTGGKLYLILEYLSGGELFMQLEREGIFMEDTACFYLAEISMALGHLHQKGIIYRDLKPENIMLNHQGHVKLTDFGLCKECGTIEYMAPEILMRSGHNRAVDWWSLGALMYDMLTGAPPFTGENRKKTIDKILKCKLNLPPYLTQEARDLLKKLLKRNAASRLGAGPGDAGEVQAHPFFRHINWEELLARKVEPPFKPLLQSEEDVSQFDSKFTRQXPVDXPNQVFLGFEYVAPSV Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Hydroxymethylglutaryl-CoA synthase 2 (HMGCS2) | 2WYA | 7.99 | |
Target general information Gen name HMGCS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HMGCS2; HMG-CoAsynthase; 3-hydroxy-3-methylglutaryl coenzyme A synthase 2 Protein family Thiolase-like superfamily, HMG-CoA synthase family Biochemical class Acyltransferase Function This enzyme condenses acetyl-CoA with acetoacetyl-CoA to form HMG-CoA, which is the substrate for HMG-CoA reductase. Related diseases 3-hydroxy-3-methylglutaryl-CoA synthase-2 deficiency (HMGCS2D) [MIM:605911]: A metabolic disorder characterized by severe hypoketotic hypoglycemia, encephalopathy, and hepatomegaly. {ECO:0000269|PubMed:11228257, ECO:0000269|PubMed:11479731, ECO:0000269|PubMed:12647205, ECO:0000269|PubMed:16601895, ECO:0000269|PubMed:23751782, ECO:0000269|PubMed:25511235, ECO:0000269|PubMed:29597274}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.3.3.10 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Disease variant; Lipid biosynthesis; Lipid metabolism; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase; Transit peptide Protein physicochemical properties Chain ID A,D Molecular weight (Da) 102499 Length 920 Aromaticity 0.1 Instability index 33.48 Isoelectric point 6.72 Charge (pH=7) -1.41 2D Binding mode Binding energy (Kcal/mol) -9.17 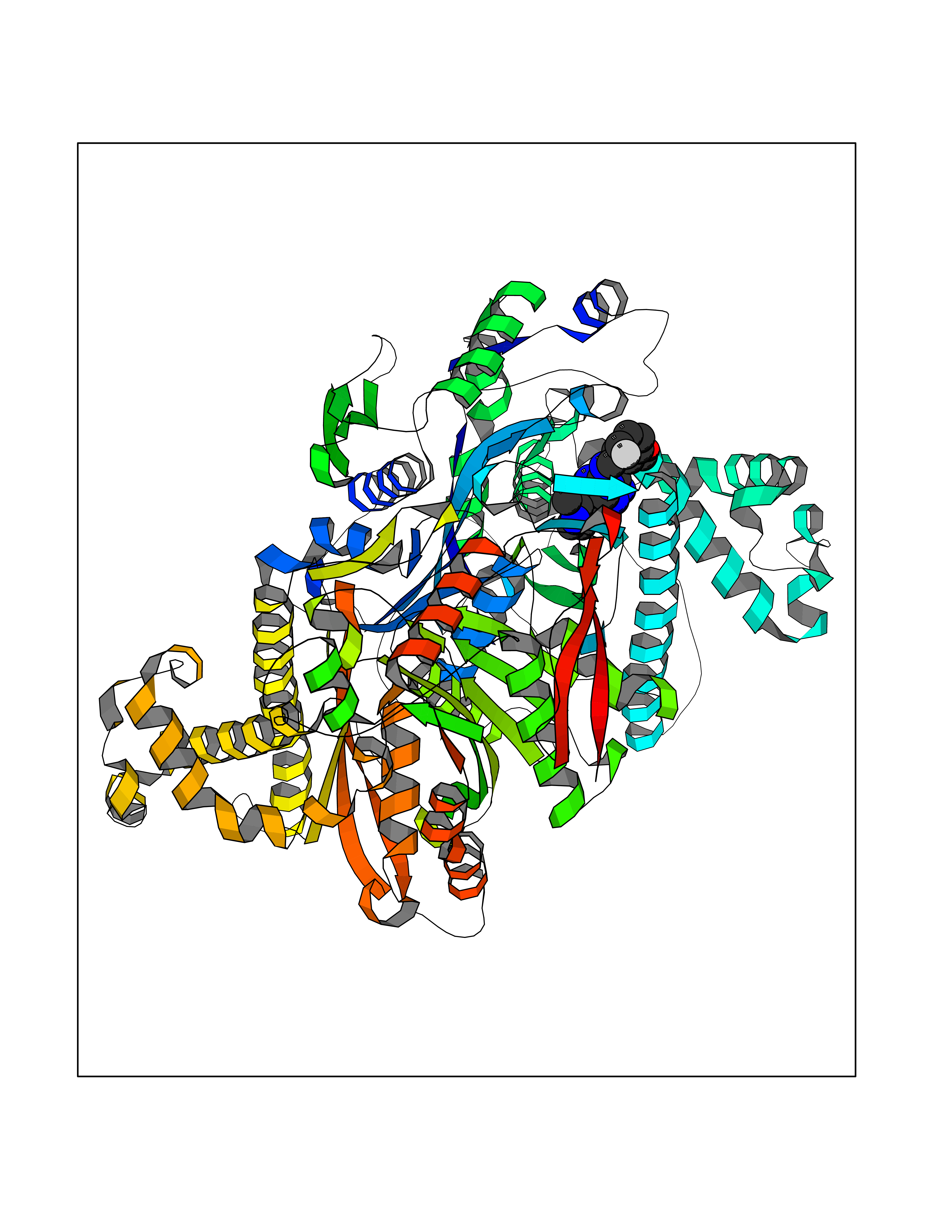 Molscript Map 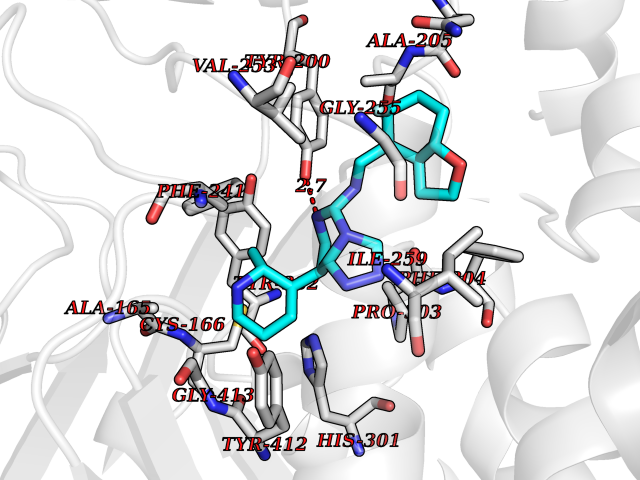 Pymol Map 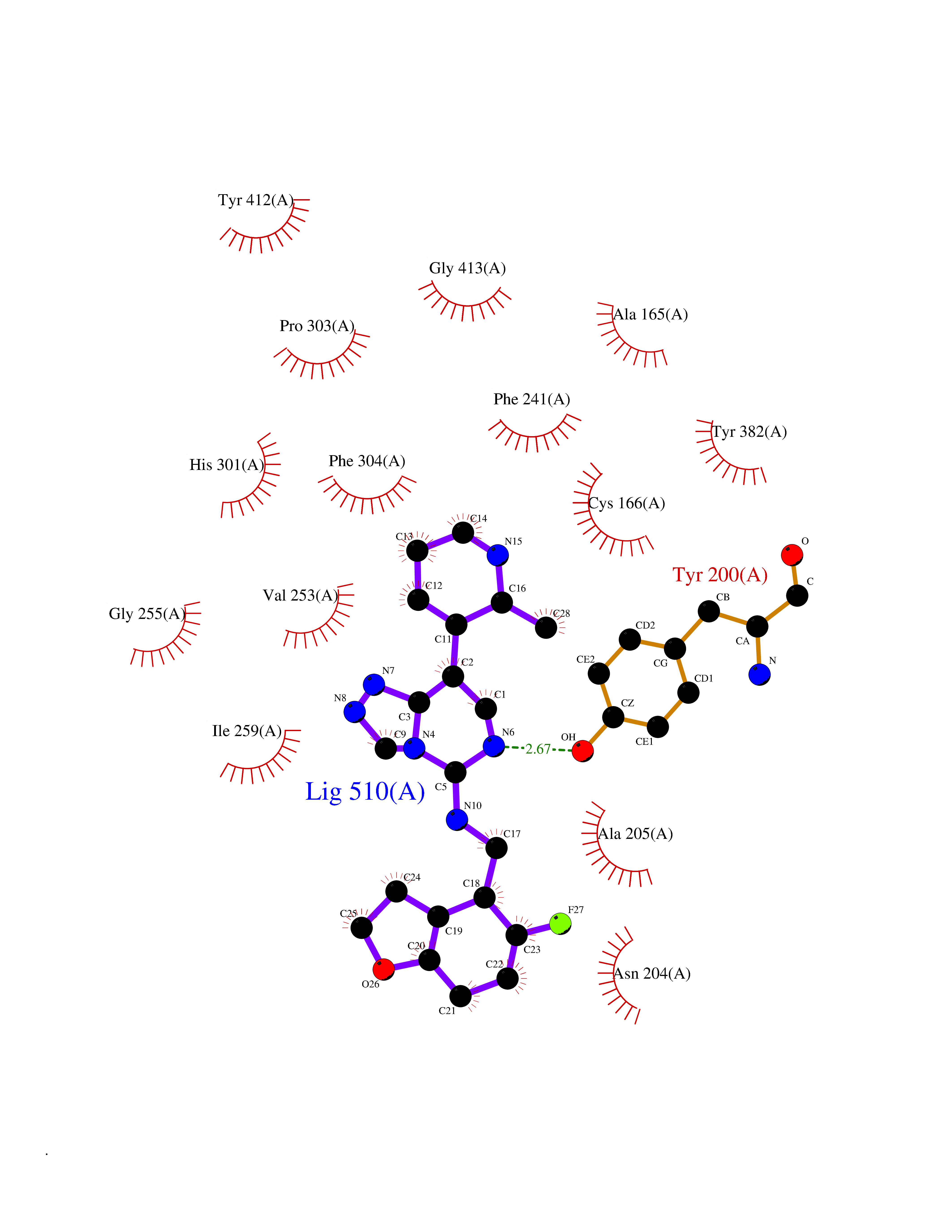 Ligplot Map 3D Binding mode Sequence SMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPVSMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPV Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Activin receptor type IIA (ACVR2A) | 3Q4T | 7.99 | |
Target general information Gen name ACVR2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Activin receptor type2A; Activin receptor type-2A; ACVR2; ACTRIIA; ACTR-IIA Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family, TGFB receptor subfamily Biochemical class Kinase Function Type II receptors phosphorylate and activate type I receptors which autophosphorylate, then bind and activate SMAD transcriptional regulators. Receptor for activin A, activin B and inhibin A. Mediates induction of adipogenesis by GDF6. On ligand binding, forms a receptor complex consisting of two type II and two type I transmembrane serine/threonine kinases. Related diseases Periventricular nodular heterotopia 8 (PVNH8) [MIM:618185]: A form of periventricular nodular heterotopia, a disorder resulting from a defect in the pattern of neuronal migration in which ectopic collections of neurons lie along the lateral ventricles of the brain or just beneath, contiguously or in isolated patches. PVNH8 is an autosomal dominant disease characterized by developmental disabilities, speech delay, seizures and attention deficit-hyperactivity disorder. {ECO:0000269|PubMed:28868155}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12118 Interacts with NA EC number EC 2.7.11.30 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Disulfide bond; Glycoprotein; Kinase; Magnesium; Manganese; Membrane; Metal-binding; Nucleotide-binding; Proteomics identification; Receptor; Reference proteome; Serine/threonine-protein kinase; Signal; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 34599.3 Length 305 Aromaticity 0.09 Instability index 44.48 Isoelectric point 5.7 Charge (pH=7) -8.29 2D Binding mode Binding energy (Kcal/mol) -8.43  Molscript Map 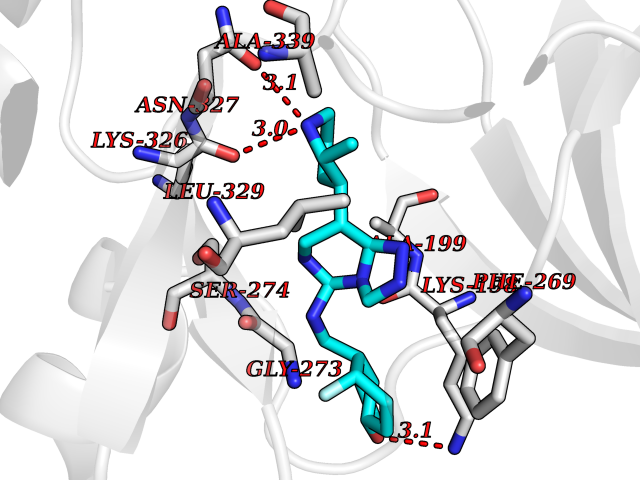 Pymol Map 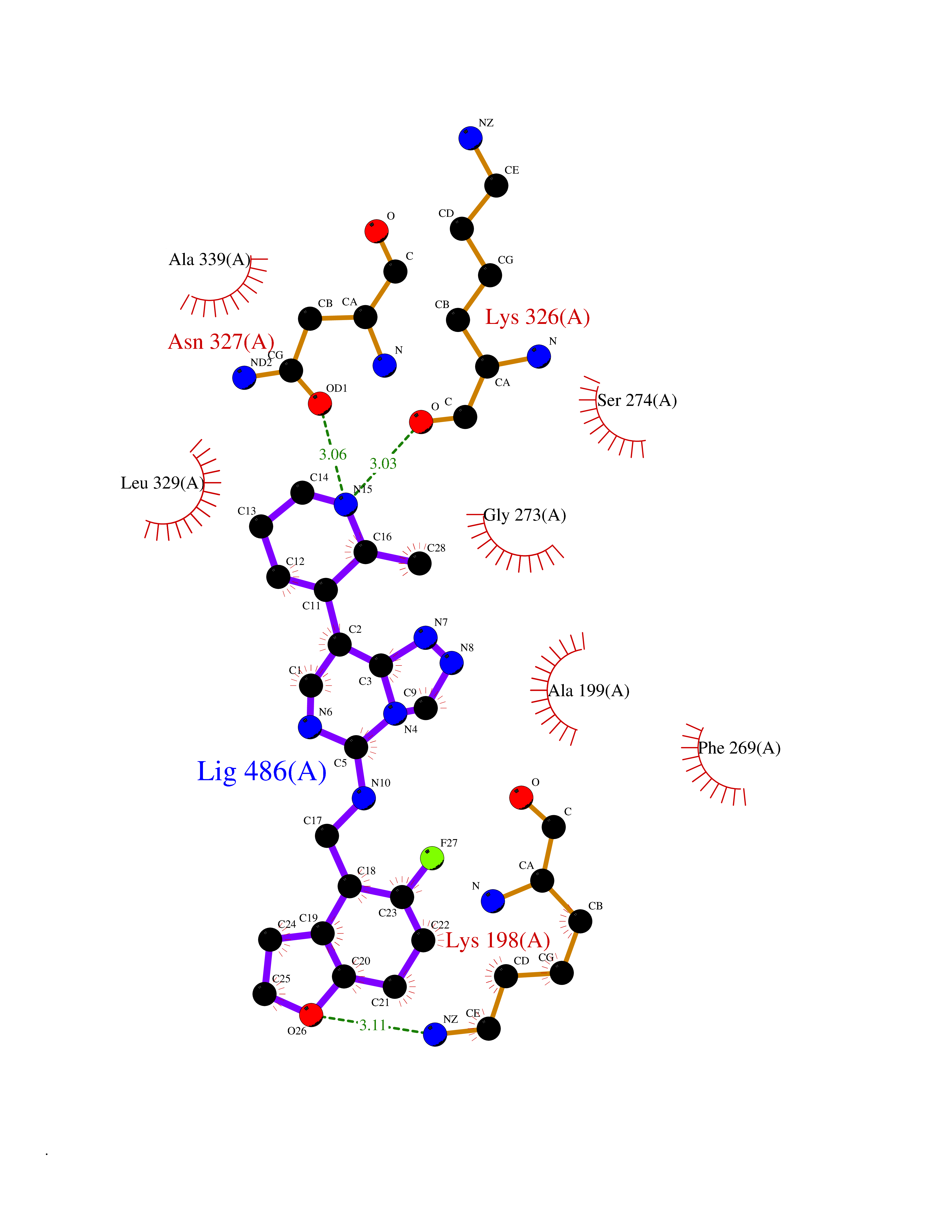 Ligplot Map 3D Binding mode Sequence LGTENLYFQSMPLQLLEVKARGRFGCVWKAQLLNEYVAVKIFPIQDKQSWQNEYEVYSLPGMKHENILQFIGAEKRGTSVDVDLWLITAFHEKGSLSDFLKANVVSWNELCHIAETMARGLAYLHEDIPGLKDGHKPAISHRDIKSKNVLLKNNLTACIADFGLALKFEAGKSAGDTHGQVGTRRYMAPEVLEGAINFQRDAFLRIDMYAMGLVLWELASRCTAADGPVDEYMLPFEEEIGQHPSLEDMQEVVVHKKKRPVLRDYWQKHAGMAMLCETIEECWDHDAEARLSAGCVGERITQMQR Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Phosphodiesterase 5A (PDE5A) | 1TBF | 7.99 | |
Target general information Gen name PDE5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-specific 3',5'-cyclic phosphodiesterase; PDE5A; CGMP-binding cGMP-specific phosphodiesterase; CGB-PDE Protein family Cyclic nucleotide phosphodiesterase family Biochemical class Phosphoric diester hydrolase Function Plays a role in signal transduction by regulating the intracellular concentration of cyclic nucleotides. This phosphodiesterase catalyzes the specific hydrolysis of cGMP to 5'-GMP. Specifically regulates nitric-oxide-generated cGMP. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07954; DB08729; DB06237; DB00201; DB00975; DB06246; DB12010; DB03597; DB01972; DB09282; DB05415; DB01113; DB00203; DB00820; DB00277; DB09283; DB06267; DB00862 Interacts with Q13976; Q9NZD8 EC number EC 3.1.4.35 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; cGMP; cGMP-binding; Hydrolase; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 37400.7 Length 325 Aromaticity 0.08 Instability index 36.9 Isoelectric point 5.64 Charge (pH=7) -8.3 2D Binding mode Binding energy (Kcal/mol) -9  Molscript Map 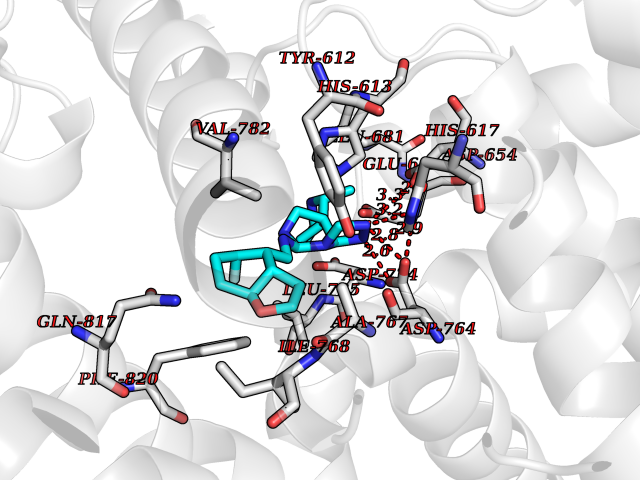 Pymol Map 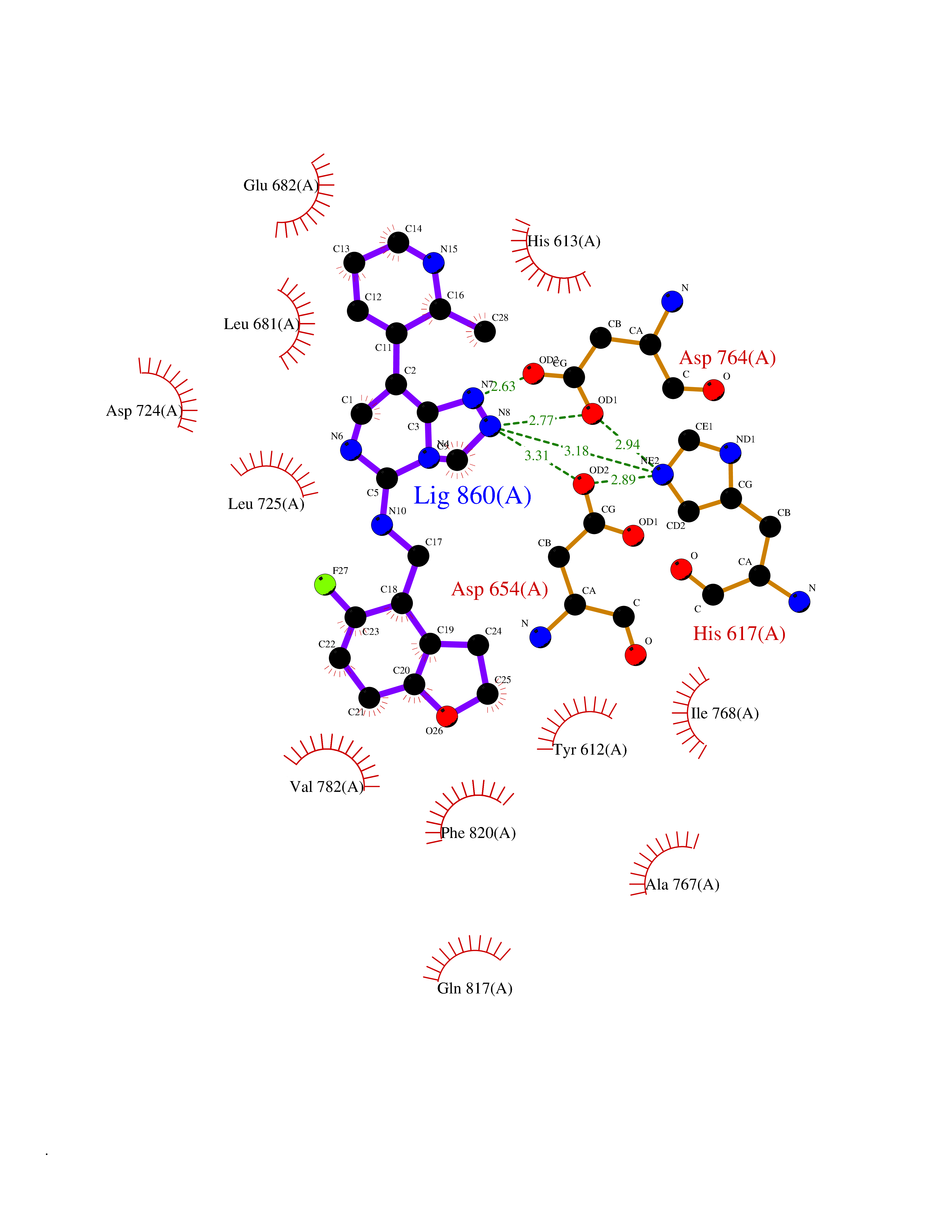 Ligplot Map 3D Binding mode Sequence EEETRELQSLAAAVVPSAQTLKITDFSFSDFELSDLETALCTIRMFTDLNLVQNFQMKHEVLCRWILSVKKNYRKNVAYHNWRHAFNTAQCMFAALKAGKIQNKLTDLEILALLIAALSHDLDHPGVSNQFLINTNSELALMNDESVLEHHHFDQCLMILNSPGNQILSGLSIEEYKTTLKIIKQAILATDLALYIKRRGEFFELIRKNQFNLEDPHQKELFLAMLMTACDLSAITKPWPIQQRIAELVATEFFDQGDRERKELNIEPTDLMNREKKNKIPSMQVGFIDAICLQLYEALTHVSEDCFPLLDGCRKNRQKWQALAE Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | PRKR-like endoplasmic reticulum kinase (PERK) | 4G31 | 7.99 | |
Target general information Gen name EIF2AK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PEK Protein family Protein kinase superfamily, Ser/Thr protein kinase family, GCN2 subfamily Biochemical class Kinase Function Converts phosphorylated eIF-2-alpha/EIF2S1 either in a global protein synthesis inhibitor, leading to a reduced overall utilization of amino acids, or to a translation initiation activator of specific mRNAs, such as the transcriptional activator ATF4, and hence allowing ATF4-mediated reprogramming of amino acid biosynthetic gene expression to alleviate nutrient depletion. Serves as a critical effector of unfolded protein response (UPR)-induced G1 growth arrest due to the loss of cyclin-D1 (CCND1). Involved in control of mitochondrial morphology and function. Metabolic-stress sensing protein kinase that phosphorylates the alpha subunit of eukaryotic translation initiation factor 2 (eIF-2-alpha/EIF2S1) on 'Ser-52' during the unfolded protein response (UPR) and in response to low amino acid availability. Related diseases Wolcott-Rallison syndrome (WRS) [MIM:226980]: A rare autosomal recessive disorder, characterized by permanent neonatal or early infancy insulin-dependent diabetes and, at a later age, epiphyseal dysplasia, osteoporosis, growth retardation and other multisystem manifestations, such as hepatic and renal dysfunctions, intellectual disability and cardiovascular abnormalities. {ECO:0000269|PubMed:10932183, ECO:0000269|PubMed:12086964, ECO:0000269|PubMed:12960215, ECO:0000269|PubMed:16813601, ECO:0000269|PubMed:24168455, ECO:0000269|PubMed:24194294, ECO:0000269|PubMed:27145240, ECO:0000269|PubMed:28220546, ECO:0000269|PubMed:30906465, ECO:0000269|PubMed:30922274, ECO:0000269|PubMed:32216767, ECO:0000269|PubMed:34123975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NZJ5; P11021 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; ADP-ribosylation; ATP-binding; Diabetes mellitus; Disease variant; Endoplasmic reticulum; Glycoprotein; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Signal; Stress response; Transferase; Translation regulation; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Unfolded protein response Protein physicochemical properties Chain ID A Molecular weight (Da) 29033.5 Length 248 Aromaticity 0.1 Instability index 43.71 Isoelectric point 7.75 Charge (pH=7) 1.27 2D Binding mode Binding energy (Kcal/mol) -9.65 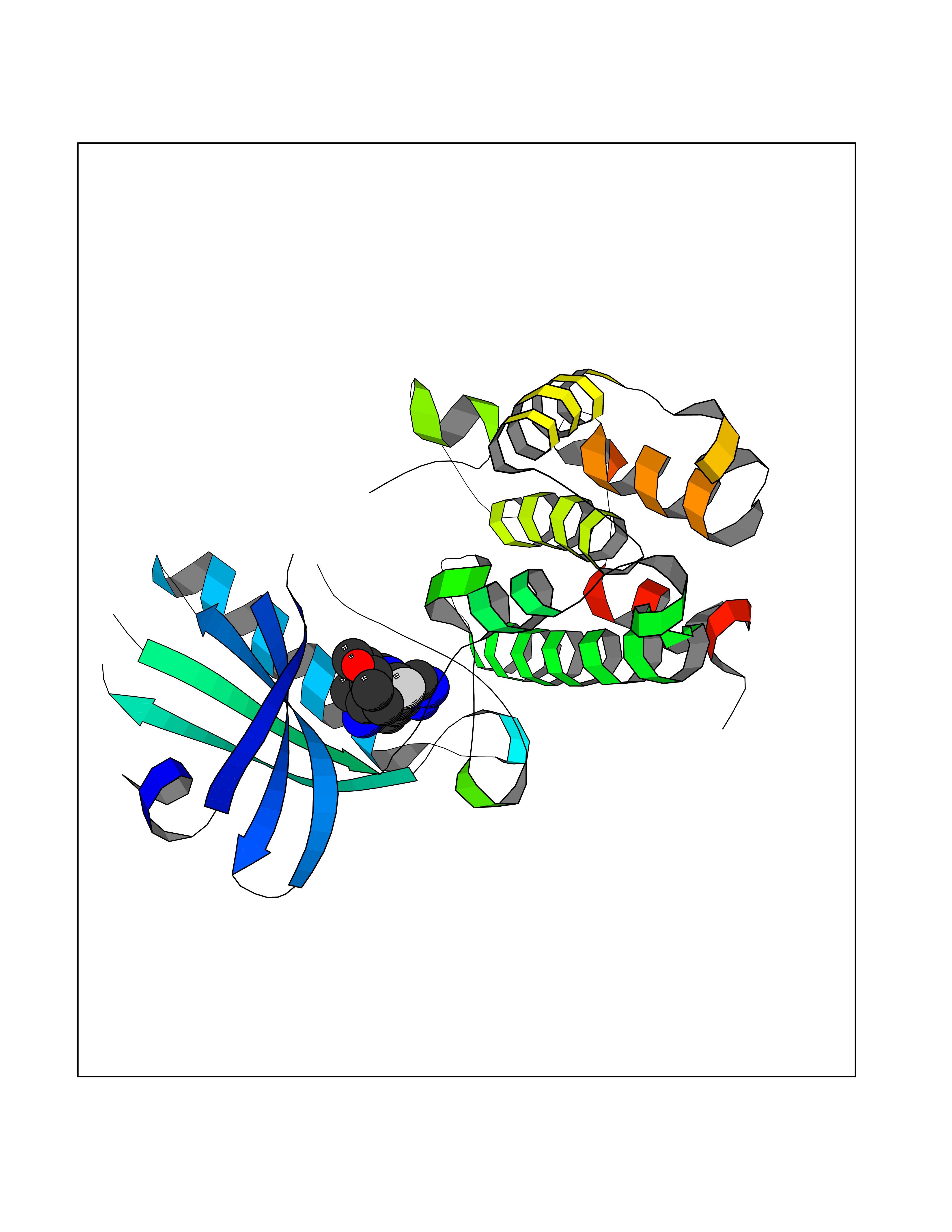 Molscript Map 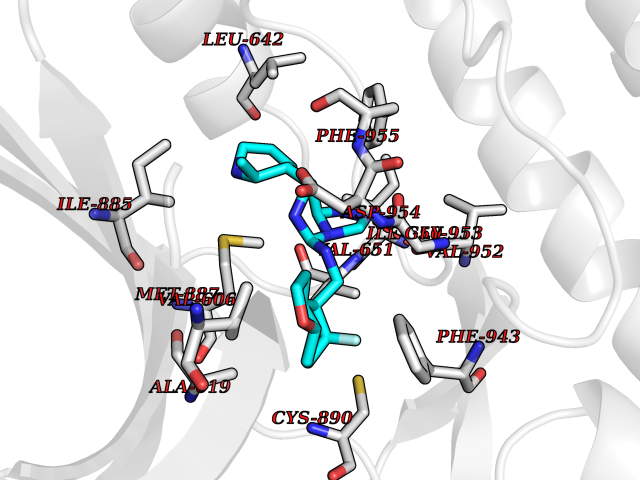 Pymol Map 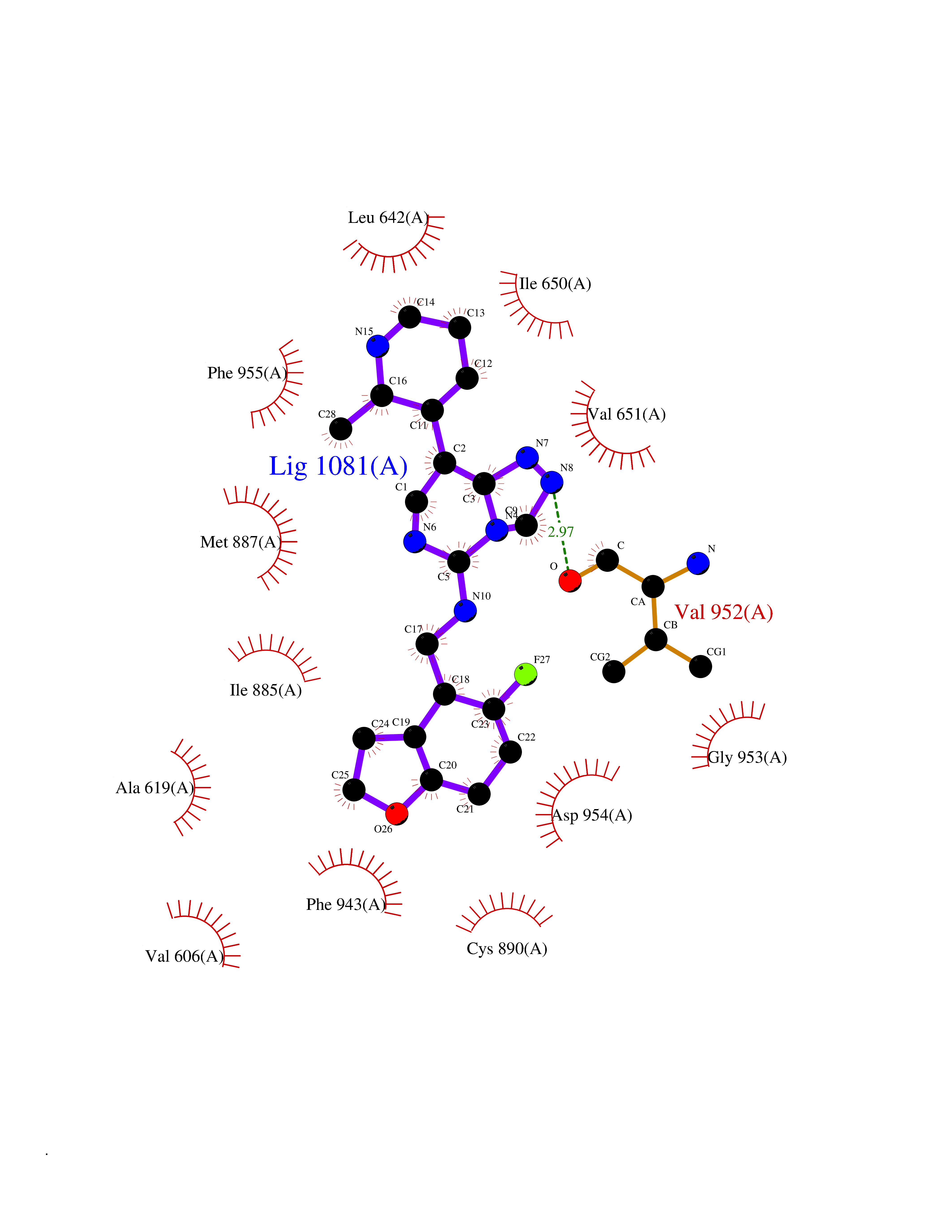 Ligplot Map 3D Binding mode Sequence GRYLTDFEPIQCLGRGGVVFEAKNKVDDCNYAIKRIRLPNRELAREKVMREVKALAKLEHPGIVRYFNAWLEKNKVYLYIQMQLCRKENLKDWMNGRCTIEERERSVCLHIFLQIAEAVEFLHSKGLMHRDLKPSNIFFTMDDVVKVGDFGLVGTKLYMSPEQIHGNSYSHKVDIFSLGLILFELLYPFSTQMERVRTLTDVRNLKFPPLFTQKYPCEYVMVQDMLSPSPMERPEAINIIENAVFEDL Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Phosphatidylinositol-4-kinase beta (PI4KB) | 4WAE | 7.98 | |
Target general information Gen name PI4KB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PtdIns 4-kinase beta; Phosphatidylinositol 4-kinase beta; PIK4CB; PI4Kbeta; PI4K92; PI4K-beta; NPIK Protein family PI3/PI4-kinase family, Type III PI4K subfamily Biochemical class NA Function Phosphorylates phosphatidylinositol (PI) in the first committed step in the production of the second messenger inositol-1,4,5,-trisphosphate (PIP). May regulate Golgi disintegration/reorganization during mitosis, possibly via its phosphorylation. Involved in Golgi-to-plasma membrane trafficking (By similarity). Related diseases Deafness, autosomal dominant, 87 (DFNA87) [MIM:620281]: A form of non-syndromic, sensorineural hearing loss. Sensorineural hearing loss results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. DFNA87 is characterized by prelingual, profound sensorineural hearing loss with inner ear anomalies, including cochlear maldevelopment, absence of the osseous spiral lamina, and/or an enlarged vestibular aqueduct. {ECO:0000269|PubMed:33358777}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with Q9H3P7; P27348; P03495; PRO_0000424692 [P03300]; P62491 EC number EC 2.7.1.67 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cytoplasm; Deafness; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Golgi apparatus; Host-virus interaction; Kinase; Lipid metabolism; Membrane; Mitochondrion; Mitochondrion outer membrane; Non-syndromic deafness; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 53130 Length 463 Aromaticity 0.1 Instability index 49.16 Isoelectric point 6.78 Charge (pH=7) -0.96 2D Binding mode Binding energy (Kcal/mol) -7.81 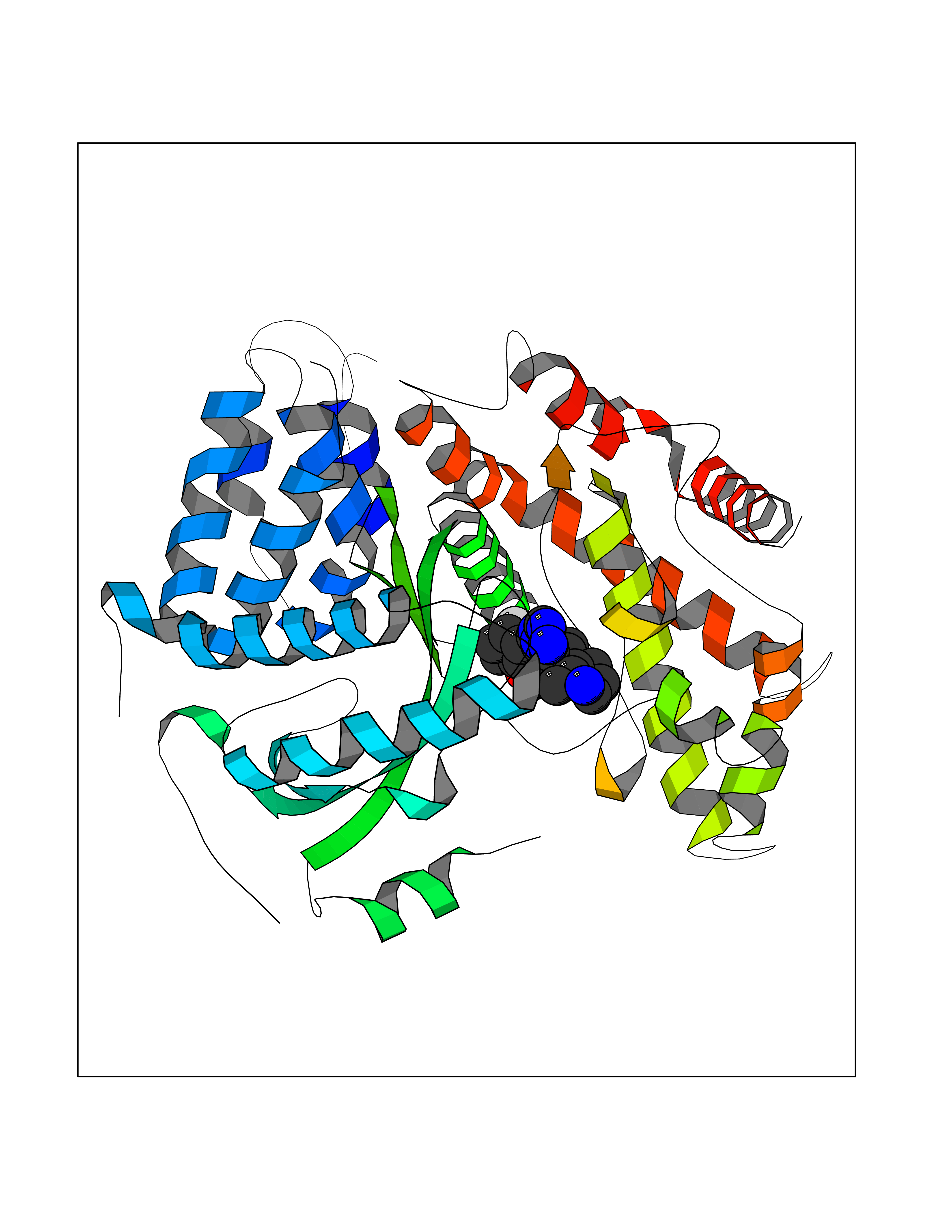 Molscript Map 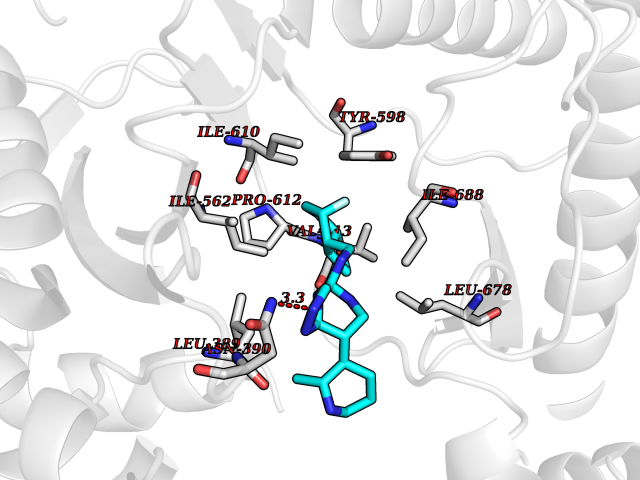 Pymol Map 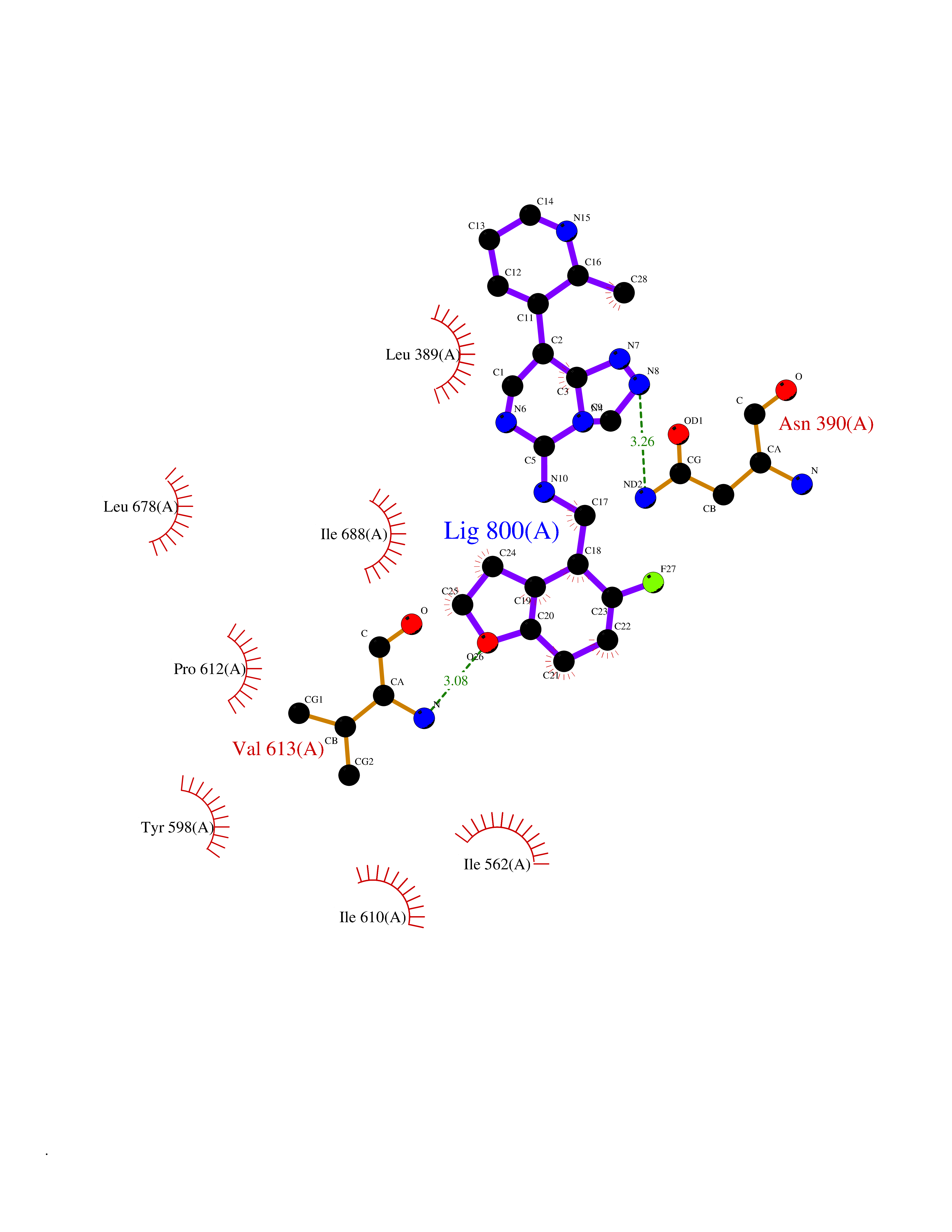 Ligplot Map 3D Binding mode Sequence LLRLFESKLFDISMAISYLYNSKEPGVQAYIGNRLFCFRNEDVDFYLPQLLNMYIHMDEDVGDAIKPYIVHRCRQSINFSLQCALLLGAYSSDMNSFSSPVRLAPEREFIKSLMAIGKRLATLPTKEQKTQRLISELSLLNHKLPARVWLPTAGFDHHVVRVPHTQAVVLNSKDKAPYLIYVEVLECENFDTTSVPARIPEWQEKVRRIREGSPYGHLPNWRLLSVIVKCGDDLRQELLAFQVLKQLQSIWEQERVPLWIKPYKILVISADSGMIEPVVNAVSIHQVKKQSQLSLLDYFLQEHGSYTTEAFLSAQRNFVQSCAGYCLVCYLLQVKDRHNGNILLDAEGHIIHIDFGFILSSSPRNLGFETSAFKLTTEFVDVMGGLDGDMFNYYKMLMLQGLIAARKHMDKVVQIVEIMQQGSQLPCFHGSSTIRNLKERFHMSMTEEQLQLLVEQMVDGSMR Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Coagulation factor Xa (F10) | 2JKH | 7.98 | |
Target general information Gen name F10 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Fxa; Factor Xa; F10; Activated coagulation factor X Protein family Peptidase S1 family Biochemical class Peptidase Function Factor Xa is avitamin K-dependent glycoprotein that converts prothrombin to thrombin in the presence of factor Va, calcium and phospholipid during blood clotting. Related diseases Factor X deficiency (FA10D) [MIM:227600]: A hemorrhagic disease with variable presentation. Affected individuals can manifest prolonged nasal and mucosal hemorrhage, menorrhagia, hematuria, and occasionally hemarthrosis. Some patients do not have clinical bleeding diathesis. {ECO:0000269|PubMed:10468877, ECO:0000269|PubMed:10739379, ECO:0000269|PubMed:10746568, ECO:0000269|PubMed:11248282, ECO:0000269|PubMed:11728527, ECO:0000269|PubMed:12574802, ECO:0000269|PubMed:12945883, ECO:0000269|PubMed:15075089, ECO:0000269|PubMed:15650540, ECO:0000269|PubMed:17393015, ECO:0000269|PubMed:19135706, ECO:0000269|PubMed:1973167, ECO:0000269|PubMed:1985698, ECO:0000269|PubMed:25313940, ECO:0000269|PubMed:26222694, ECO:0000269|PubMed:2790181, ECO:0000269|PubMed:7669671, ECO:0000269|PubMed:7860069, ECO:0000269|PubMed:8529633, ECO:0000269|PubMed:8845463, ECO:0000269|PubMed:8910490}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07211; DB08746; DB07974; DB07277; DB07605; DB08487; DB08495; DB04673; DB08745; DB08488; DB07804; DB08174; DB08173; DB07872; DB07843; DB07848; DB07875; DB08143; DB07847; DB07844; DB13884; DB06552; DB13151; DB13192; DB00025; DB11166; DB06605; DB09258; DB12364; DB00100; DB13152; DB13150; DB00036; DB09075; DB16662; DB13923; DB01225; DB06920; DB00569; DB03847; DB07278; DB01109; DB06406; DB09332; DB06245; DB13998; DB05713; DB13999; DB07630; DB07629; DB07973; DB07800; DB12598; DB13933; DB06635; DB09141; DB13149; DB11311; DB06228; DB05362; DB07261; DB08426; DB09109; DB14738 Interacts with P0DPK4; Q9UK55; Q9UHD9 EC number EC 3.4.21.6 Uniprot keywords 3D-structure; Blood coagulation; Calcium; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Gamma-carboxyglutamic acid; Glycoprotein; Hemostasis; Hydrolase; Hydroxylation; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 31315.2 Length 280 Aromaticity 0.09 Instability index 38.33 Isoelectric point 6.36 Charge (pH=7) -1.82 2D Binding mode Binding energy (Kcal/mol) -8.46 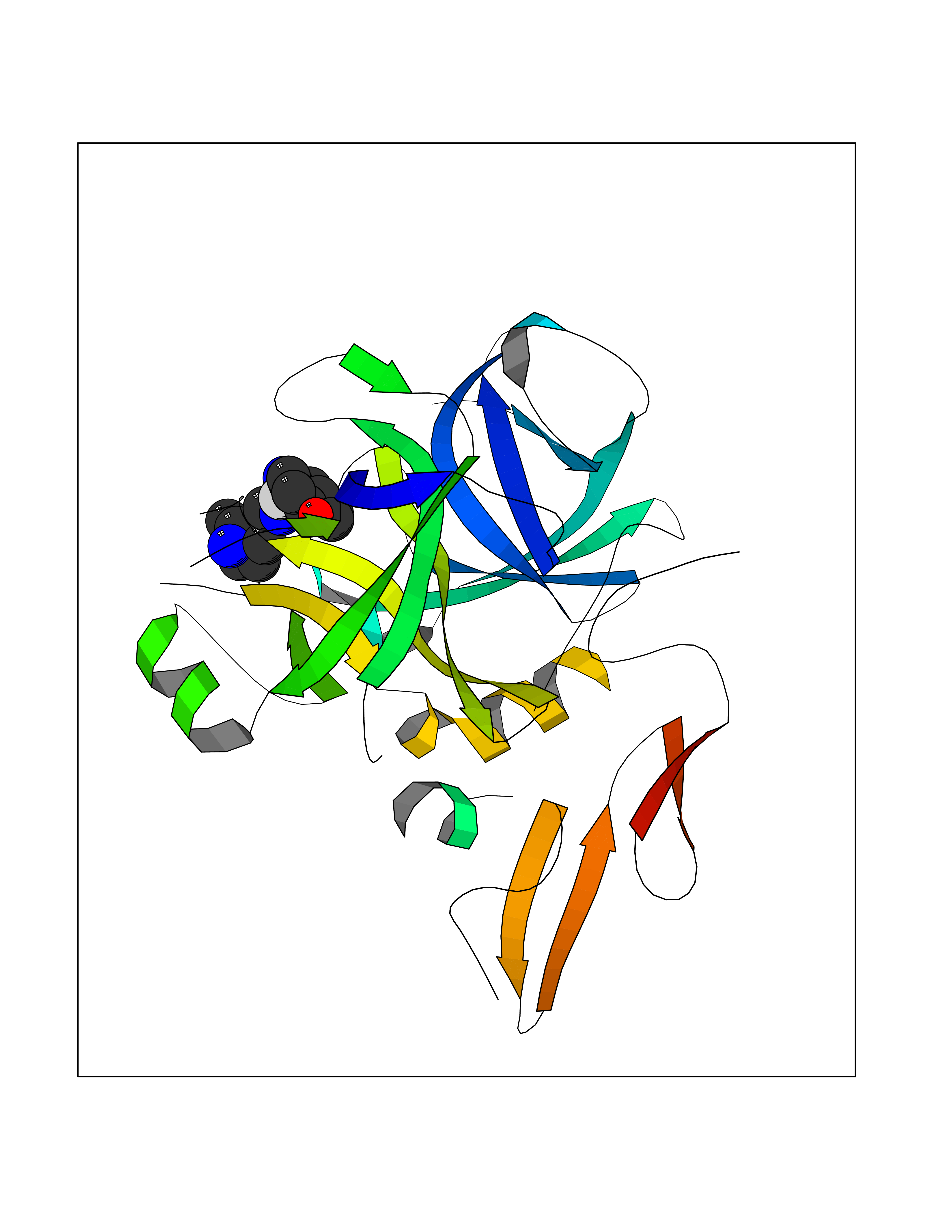 Molscript Map 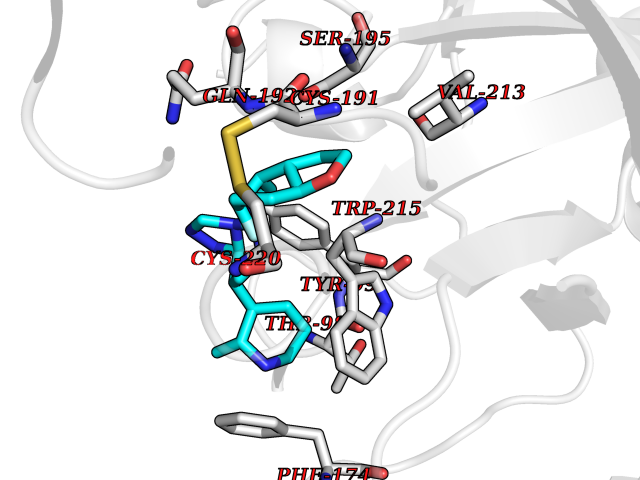 Pymol Map 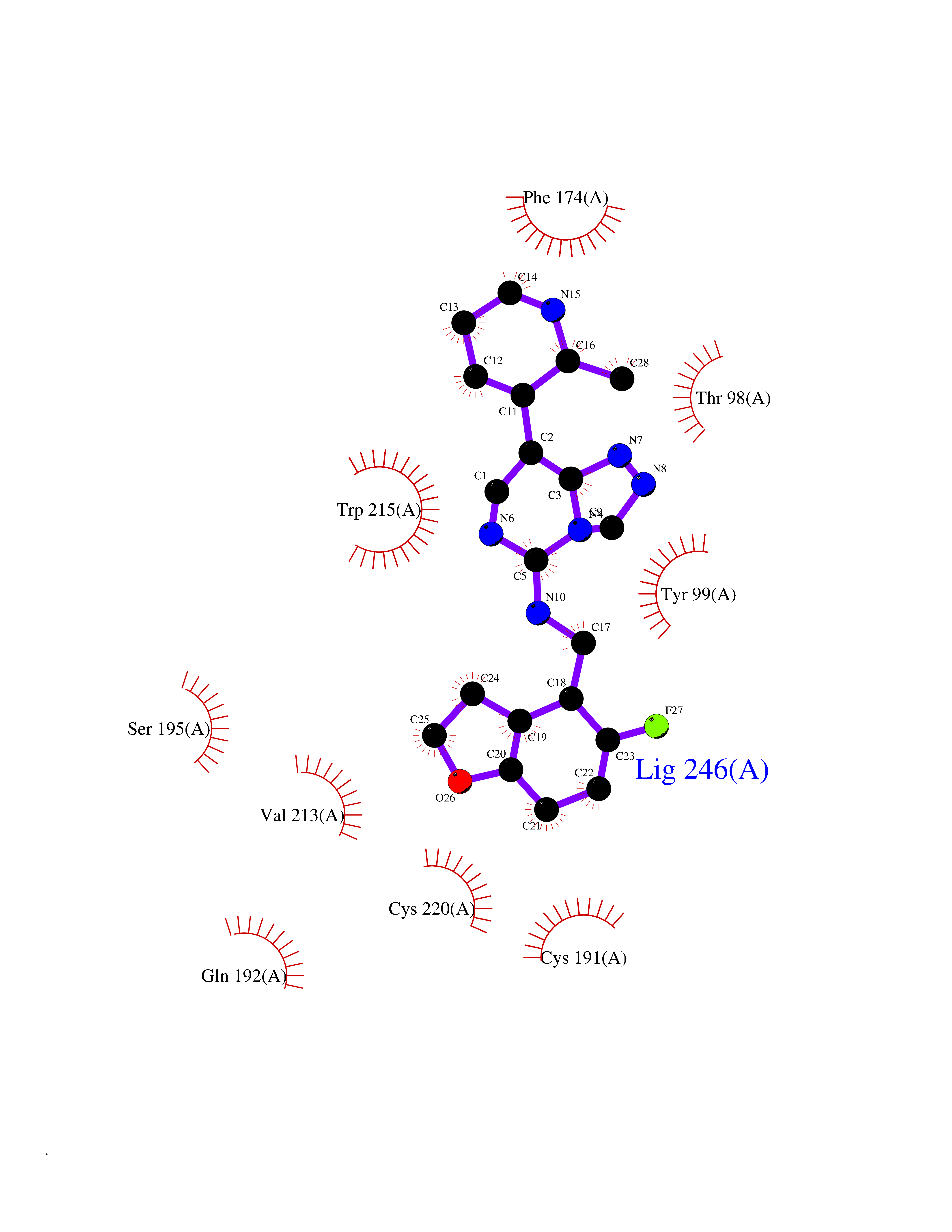 Ligplot Map 3D Binding mode Sequence IVGGQECKDGECPWQALLINEENEGFCGGTILSEFYILTAAHCLYAKRFKVRVGDRNTEQEEGGEAVHEVEVVIKHNRFTKETYDFDIAVLRLKTPITFRMNVAPACLERDWAESMTQKTGIVSGFGRTHEKGEQSTRLKMLEVPYVDRNSCKLSSSFIITQNMFCAGTKQEDACQGDSGGPHVTRFKDTYFVTGIVSWGEGCARGKYGIYTKVTAFLKWIDRSMKKLCSLDNGDCDQFCHEEQNSVVCSCARGYTLADNGKACIPTGPYPCGKQTLERR Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Lysine-specific demethylase 4C (KDM4C) | 4XDO | 7.97 | |
Target general information Gen name KDM4C Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms KIAA0780; Jumonji domain-containing protein 2C; JmjC domain-containing histone demethylation protein 3C; JMJD2C; JHDM3C; Gene amplified in squamous cell carcinoma 1 protein; GASC1; GASC-1 protein Protein family JHDM3 histone demethylase family Biochemical class Paired donor oxygen oxidoreductase Function Does not demethylate histone H3 'Lys-4', H3 'Lys-27' nor H4 'Lys-20'. Demethylates trimethylated H3 'Lys-9' and H3 'Lys-36' residue, while it has no activity on mono- and dimethylated residues. Demethylation of Lys residue generates formaldehyde and succinate. Histone demethylase that specifically demethylates 'Lys-9' and 'Lys-36' residues of histone H3, thereby playing a central role in histone code. Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. {ECO:0000269|PubMed:20054297, ECO:0000269|PubMed:23622243, ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}. The disease may be caused by variants affecting the gene represented in this entry. Defects of SETD2 are associated with loss of DNA methylation at non-promoter regions (PubMed:23792563). SETD2 defects lead to aberrant and reduced nucleosome compaction and chromatin association of key replication proteins, such as MCM7 and DNA polymerase delta, leading to hinder replication fork progression and prevent loading of RAD51 homologous recombination repair factor at DNA breaks (PubMed:25728682). {ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}.; DISEASE: Luscan-Lumish syndrome (LLS) [MIM:616831]: An autosomal dominant syndrome with a variable phenotype. Clinical features include macrocephaly, distinctive facial appearance, postnatal overgrowth, various degrees of learning difficulties, autism spectrum disorder, and intellectual disability. {ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24852293, ECO:0000269|PubMed:26084711, ECO:0000269|PubMed:27317772}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute lymphoblastic (ALL) [MIM:613065]: A subtype of acute leukemia, a cancer of the white blood cells. ALL is a malignant disease of bone marrow and the most common malignancy diagnosed in children. The malignant cells are lymphoid precursor cells (lymphoblasts) that are arrested in an early stage of development. The lymphoblasts replace the normal marrow elements, resulting in a marked decrease in the production of normal blood cells. Consequently, anemia, thrombocytopenia, and neutropenia occur to varying degrees. The lymphoblasts also proliferate in organs other than the marrow, particularly the liver, spleen, and lymphnodes. {ECO:0000269|PubMed:24509477, ECO:0000269|PubMed:24662245}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16314571, ECO:0000269|PubMed:24509477}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Intellectual developmental disorder, autosomal dominant 70 (MRD70) [MIM:620157]: An autosomal dominant disorder characterized by mild global developmental delay, moderately impaired intellectual disability with speech difficulties, and behavioral abnormalities. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rabin-Pappas syndrome (RAPAS) [MIM:620155]: An autosomal dominant neurodevelopmental disorder characterized by severely impaired global development, intellectual disability, microcephaly, facial dysmorphism, and variable congenital anomalies affecting the skeletal, genitourinary, cardiac, and other organ systems. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.14.11.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Dioxygenase; Iron; Metal-binding; Nucleus; Oxidoreductase; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 39355.6 Length 338 Aromaticity 0.14 Instability index 38.34 Isoelectric point 8.04 Charge (pH=7) 2.41 2D Binding mode Binding energy (Kcal/mol) -8.16 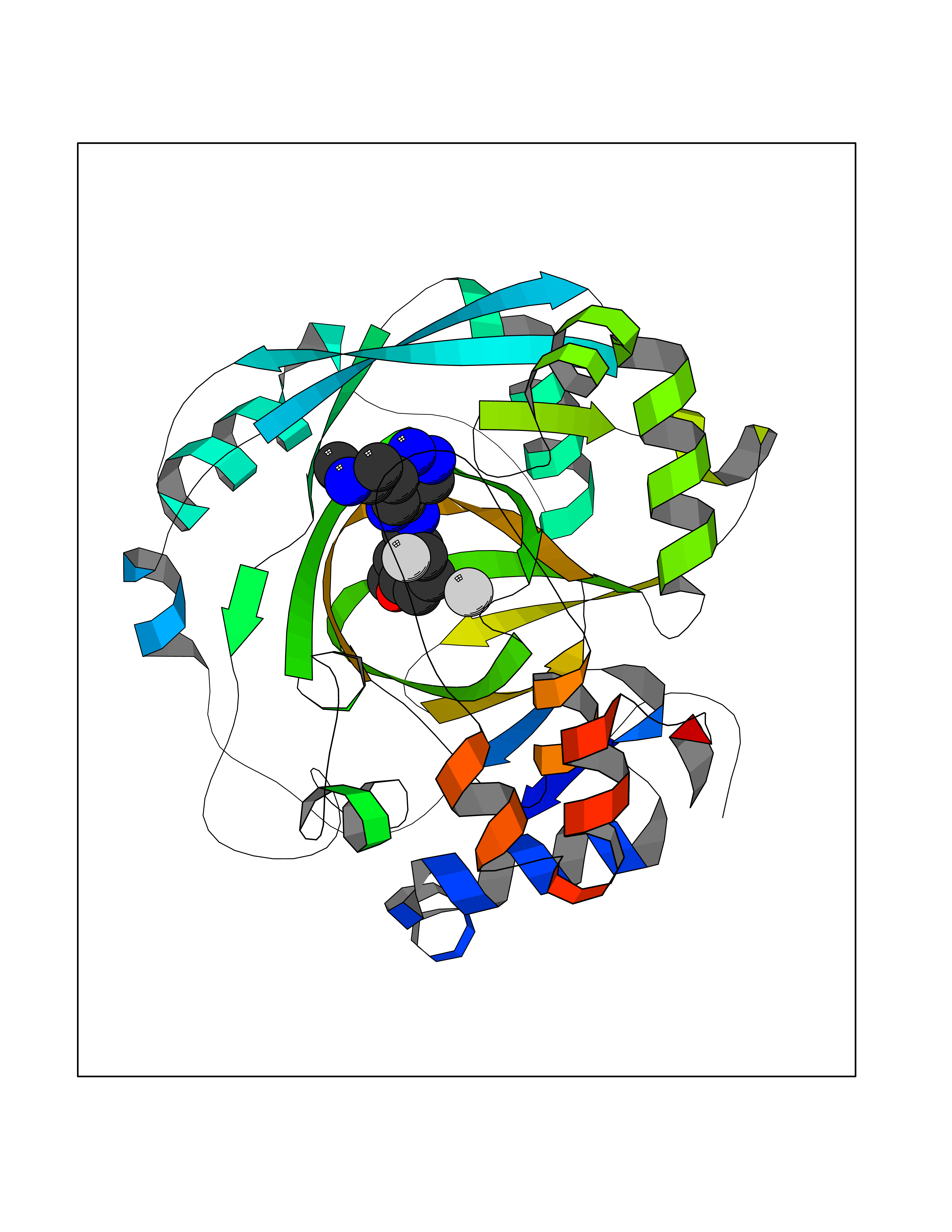 Molscript Map 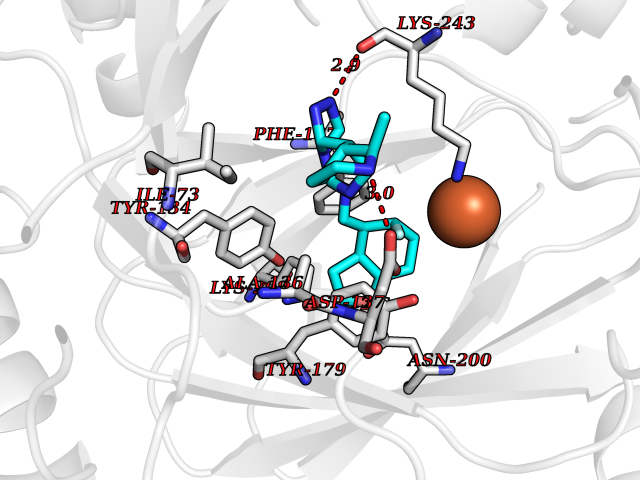 Pymol Map 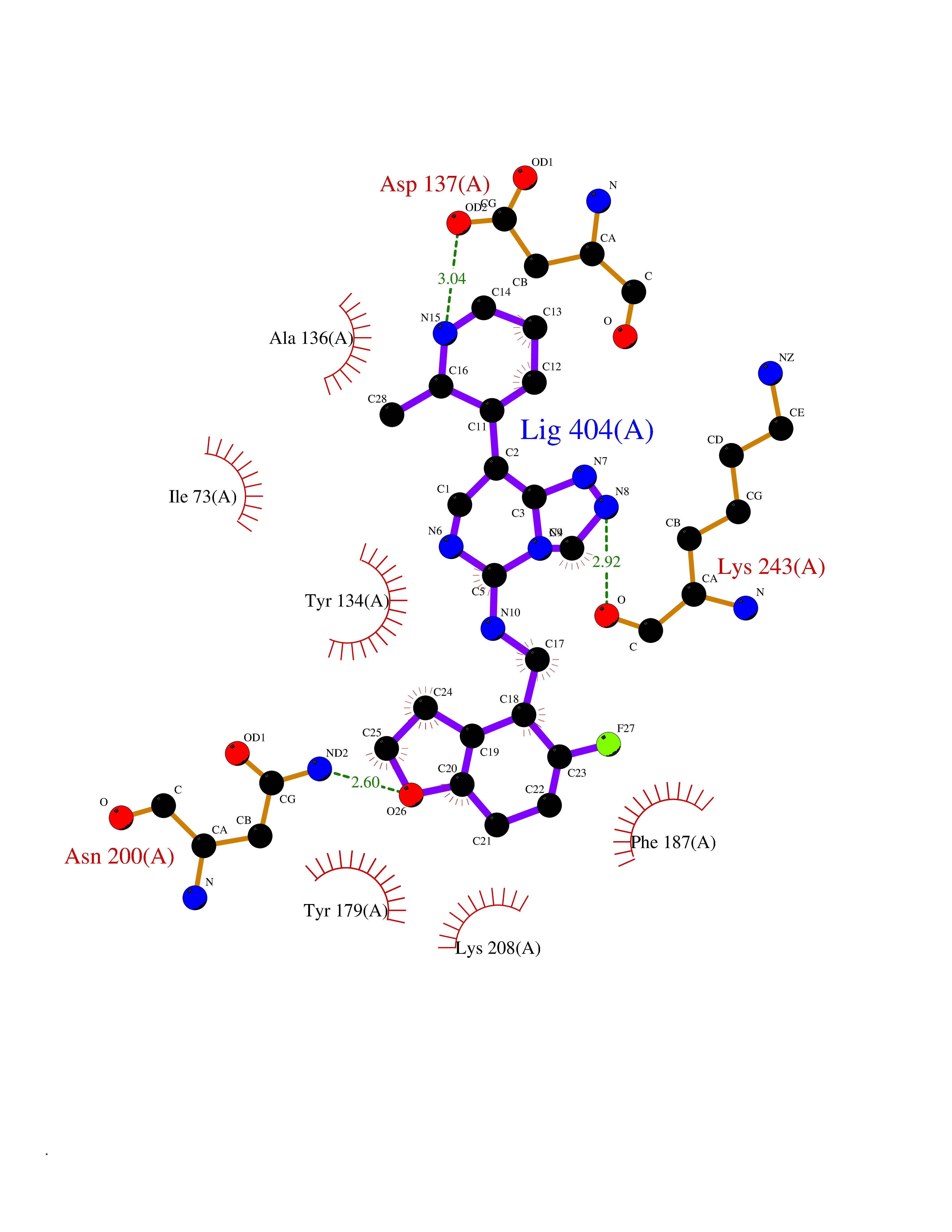 Ligplot Map 3D Binding mode Sequence LNPSCKIMTFRPSMEEFREFNKYLAYMESKGAHRAGLAKVIPPKEWKPRQCYDDIDNLLIPAPIQQMVTGQSGLFTQYNIQKKAMTVKEFRQLANSGKYCTPRYLDYEDLERKYWKNLTFVAPIYGADINGSIYDEGVDEWNIARLNTVLDVVEEECGISIEGVNTPYLYFGMWKTTFAWHTEDMDLYSINYLHFGEPKSWYAIPPEHGKRLERLAQGFFPSSSQGCDAFLRHKMTLISPSVLKKYGIPFDKITQEAGEFMITFPYGYHAGFNHGFNCAESTNFATVRWIDYGKVAKLCTCRKDMVKISMDIFVRKFQPDRYQLWKQGKDIYTIDHTK Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Bacterial DD-carboxypeptidase (Bact vanYB) | 5ZHW | 7.97 | |
Target general information Gen name Bact vanYB Organism Enterococcus faecalis (strain ATCC 700802 / V583) Uniprot ID TTD ID Synonyms vanYB; DD-peptidase; DD-carboxypeptidase; D-alanyl-D-alanine carboxypeptidase-transpeptidase Protein family Peptidase M15B family Biochemical class Peptidase Function Vancomycin-inducible, penicillin-resistant, DD- carboxypeptidase that hydrolyzes depsipeptide- and D-alanyl-D- alanine-containing peptidoglycan precursors. Insensitive to beta- lactams. Related diseases Brachyolmia 3 (BCYM3) [MIM:113500]: A form of brachyolmia, a clinically and genetically heterogeneous skeletal dysplasia primarily affecting the spine and characterized by a short trunk, short stature, and platyspondyly. BCYM3 is an autosomal dominant form with severe scoliosis with or without kyphosis, and flattened irregular cervical vertebrae. {ECO:0000269|PubMed:18587396}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondylometaphyseal dysplasia Kozlowski type (SMDK) [MIM:184252]: A form of spondylometaphyseal dysplasia, a group of short stature disorders distinguished by abnormalities in the vertebrae and the metaphyses of the tubular bones. It is characterized by postnatal dwarfism, significant scoliosis and mild metaphyseal abnormalities in the pelvis. The vertebrae exhibit platyspondyly and overfaced pedicles. {ECO:0000269|PubMed:19232556, ECO:0000269|PubMed:20577006, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metatropic dysplasia (MTD) [MIM:156530]: A severe spondyloepimetaphyseal dysplasia characterized by short limbs with limitation and enlargement of joints and usually severe kyphoscoliosis. Radiologic features include severe platyspondyly, severe metaphyseal enlargement and shortening of long bones. {ECO:0000269|PubMed:19232556, ECO:0000269|PubMed:20425821, ECO:0000269|PubMed:20577006, ECO:0000269|PubMed:22702953, ECO:0000269|PubMed:26249260, ECO:0000269|Ref.6}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 8 (HMND8) [MIM:600175]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:20037588, ECO:0000269|PubMed:22526352, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2C (CMT2C) [MIM:606071]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:20037586, ECO:0000269|PubMed:20037587, ECO:0000269|PubMed:20037588, ECO:0000269|PubMed:21115951, ECO:0000269|PubMed:21288981, ECO:0000269|PubMed:22702953, ECO:0000269|PubMed:25256292}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Scapuloperoneal spinal muscular atrophy (SPSMA) [MIM:181405]: A clinically variable neuromuscular disorder characterized by neurogenic scapuloperoneal amyotrophy, laryngeal palsy, congenital absence of muscles, progressive scapuloperoneal atrophy and progressive distal weakness and amyotrophy. {ECO:0000269|PubMed:20037587, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepiphyseal dysplasia Maroteaux type (SEDM) [MIM:184095]: A clinically variable spondyloepiphyseal dysplasia with manifestations limited to the musculoskeletal system. Clinical features include short stature, brachydactyly, platyspondyly, short and stubby hands and feet, epiphyseal hypoplasia of the large joints, and iliac hypoplasia. Intelligence is normal. {ECO:0000269|PubMed:20503319, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parastremmatic dwarfism (PSTD) [MIM:168400]: A bone dysplasia characterized by severe dwarfism, kyphoscoliosis, distortion and bowing of the extremities, and contractures of the large joints. Radiographically, the disease is characterized by a combination of decreased bone density, bowing of the long bones, platyspondyly and striking irregularities of endochondral ossification with areas of calcific stippling and streaking in radiolucent epiphyses, metaphyses and apophyses. {ECO:0000269|PubMed:20503319}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Digital arthropathy-brachydactyly, familial (FDAB) [MIM:606835]: A disorder characterized by irregularities in the proximal articular surfaces of the distal interphalangeal joints of the hand. Individuals appear normal at birth, with no clinical or radiographic evidence of a developmental skeletal dysplasia. The earliest changes appear during the first decade of life. By adulthood, all interphalangeal, metacarpophalangeal, and metatarsophalangeal joints are affected by a deforming, painful osteoarthritis. The remainder of the skeleton is clinically and radiographically unaffected. {ECO:0000269|PubMed:21964574}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Avascular necrosis of the femoral head, primary 2 (ANFH2) [MIM:617383]: A disease characterized by mechanical failure of the subchondral bone, and degeneration of the hip joint. It usually leads to destruction of the hip joint in the third to fifth decade of life. The clinical manifestations, such as pain on exertion, a limping gait, and a discrepancy in leg length, cause considerable disability. {ECO:0000269|PubMed:27330106}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.16.4 Uniprot keywords 3D-structure; Antibiotic resistance; Carboxypeptidase; Cell membrane; Cell shape; Cell wall biogenesis/degradation; Hydrolase; Membrane; Metal-binding; Peptidoglycan synthesis; Protease; Reference proteome; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 20547.5 Length 181 Aromaticity 0.11 Instability index 33.61 Isoelectric point 4.81 Charge (pH=7) -10.78 2D Binding mode Binding energy (Kcal/mol) -9.17 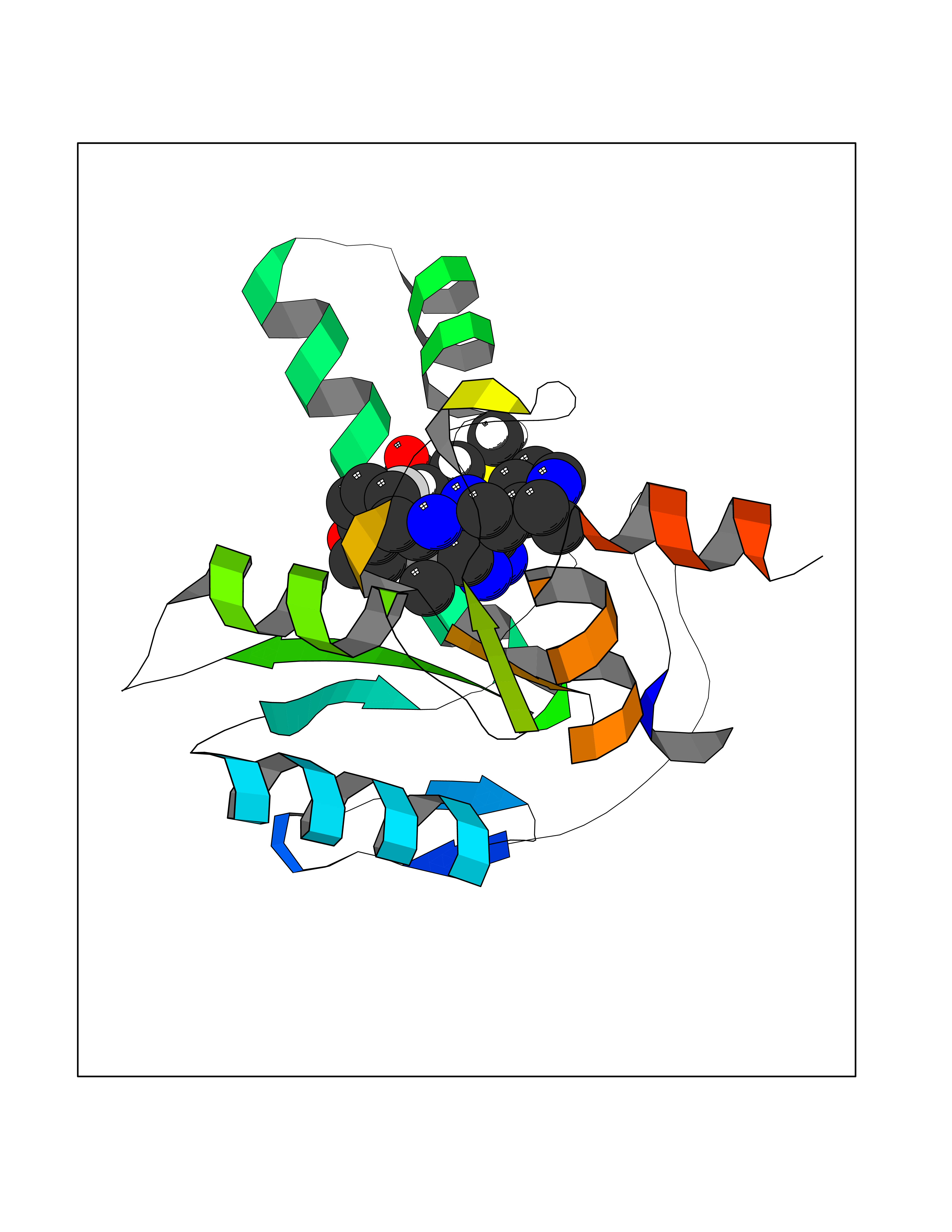 Molscript Map 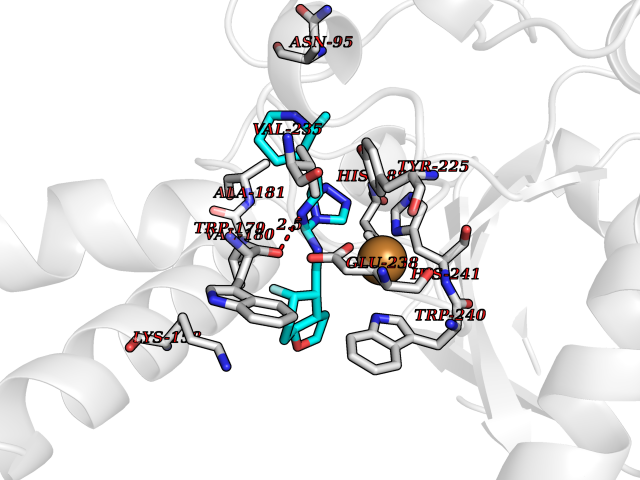 Pymol Map 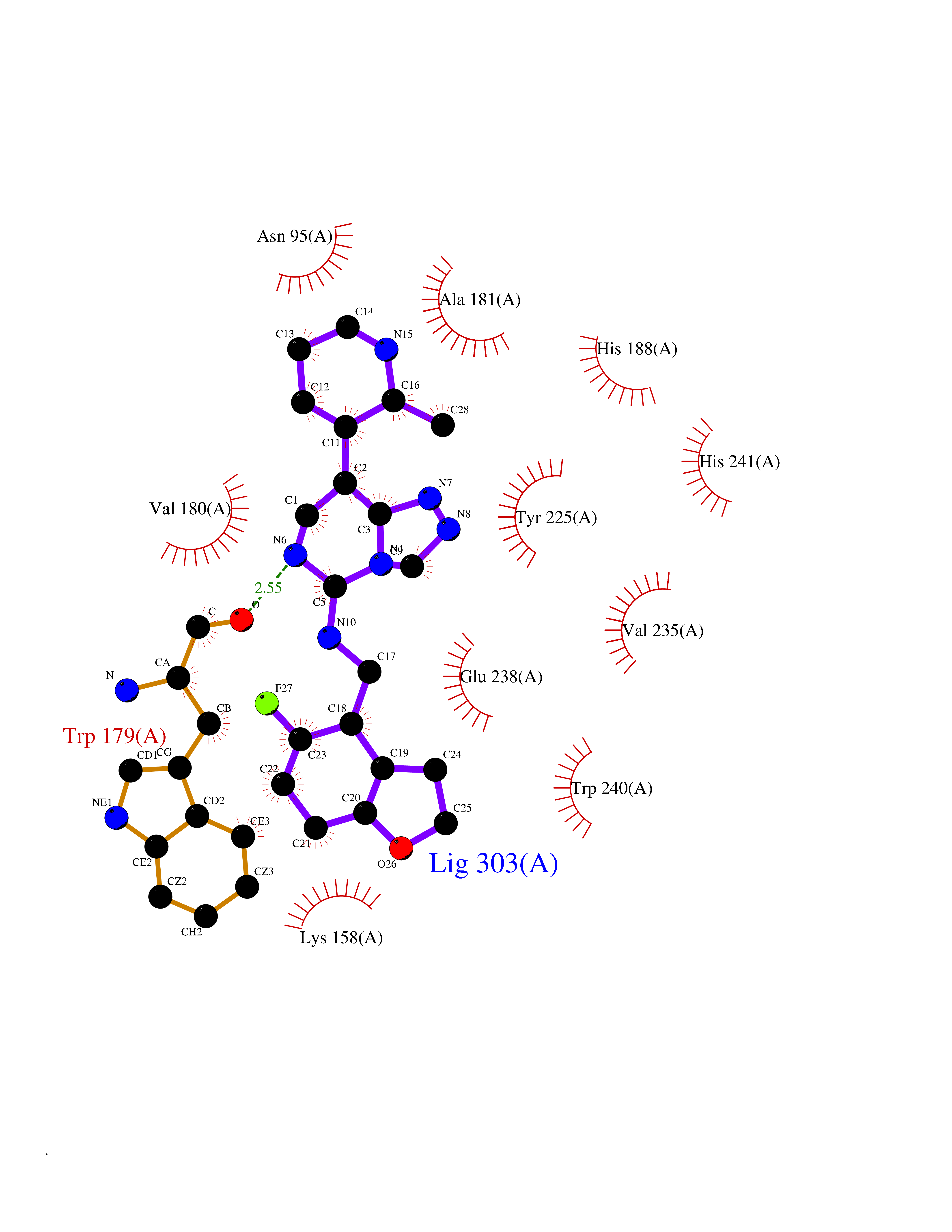 Ligplot Map 3D Binding mode Sequence EWSLILVNRQNPIPAQYDVELEQLSNGERIDIRISPYLQDLFDAARADGVYPIVASGYRTTEKQQEIXDEKVAEYKAKGYTSAQAKAEAETWVAVPGTSEHQLGLAVDINADGIHSTGNEVYRWLDENSYRFGFIRRYPPDKTEITGVSNEPWHYRYVGIEAATKIYHQGLCLEEYLNTEK Hydrogen bonds contact Hydrophobic contact | ||||