Job Results:
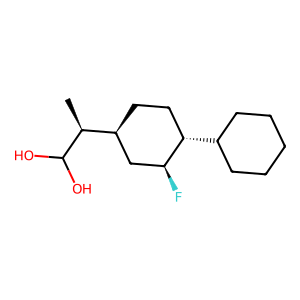
Ligand
Structure
Job ID
6a37619723fa756e073677fca12f4afa
Job name
NA
Time
2024-07-14 16:31:23
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Multidrug resistance protein 3 (ABCB4) | 6S7P | 7.30 | |
Target general information Gen name ABCB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PGY3; MDR3; ATP-binding cassette sub-family B member 4; ABCB4 Protein family ABC transporter superfamily, ABCB family, Multidrug resistance exporter (TC 3.A.1.201) subfamily Biochemical class Acid anhydrides hydrolase Function Mediates ATP-dependent export of organic anions and drugs from the cytoplasm. Hydrolyzes ATP with low efficiency. Not capable of conferring drug resistance. Mediates the translocation of phosphatidylcholine across the canalicular membrane of the hepatocyte. Related diseases Cholestasis, progressive familial intrahepatic, 3 (PFIC3) [MIM:602347]: A disorder characterized by early onset of cholestasis that progresses to hepatic fibrosis, cirrhosis, and end-stage liver disease before adulthood. PFIC3 inheritance is autosomal recessive. {ECO:0000269|PubMed:11313315, ECO:0000269|PubMed:12671900, ECO:0000269|PubMed:17726488, ECO:0000269|PubMed:21119540, ECO:0000269|PubMed:24045840, ECO:0000269|PubMed:24594635, ECO:0000269|PubMed:24806754, ECO:0000269|PubMed:28012258, ECO:0000269|PubMed:9419367}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cholestasis of pregnancy, intrahepatic 3 (ICP3) [MIM:614972]: A liver disorder of pregnancy. It presents during the second or, more commonly, the third trimester of pregnancy with intense pruritus which becomes more severe with advancing gestation and cholestasis. It causes fetal distress, spontaneous premature delivery and intrauterine death. Patients have spontaneous and progressive disappearance of cholestasis after delivery. Cholestasis results from abnormal biliary transport from the liver into the small intestine. {ECO:0000269|PubMed:10767346, ECO:0000269|PubMed:12746424, ECO:0000269|PubMed:15077010}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gallbladder disease 1 (GBD1) [MIM:600803]: One of the major digestive diseases. Gallstones composed of cholesterol (cholelithiasis) are the common manifestations in western countries. Most people with gallstones, however, remain asymptomatic through their lifetimes. {ECO:0000269|PubMed:11313316, ECO:0000269|PubMed:12891548, ECO:0000269|PubMed:22331132, ECO:0000269|PubMed:23533021, ECO:0000269|PubMed:24723470, ECO:0000269|PubMed:28012258, ECO:0000269|PubMed:28587926, ECO:0000269|Ref.2}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06414; DB06207 Interacts with NA EC number EC 7.6.2.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cytoplasm; Cytoplasmic vesicle; Disease variant; Glycoprotein; Intrahepatic cholestasis; Lipid transport; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 123919 Length 1128 Aromaticity 0.1 Instability index 29.44 Isoelectric point 8.78 Charge (pH=7) 11.08 2D Binding mode Binding energy (Kcal/mol) -8.01 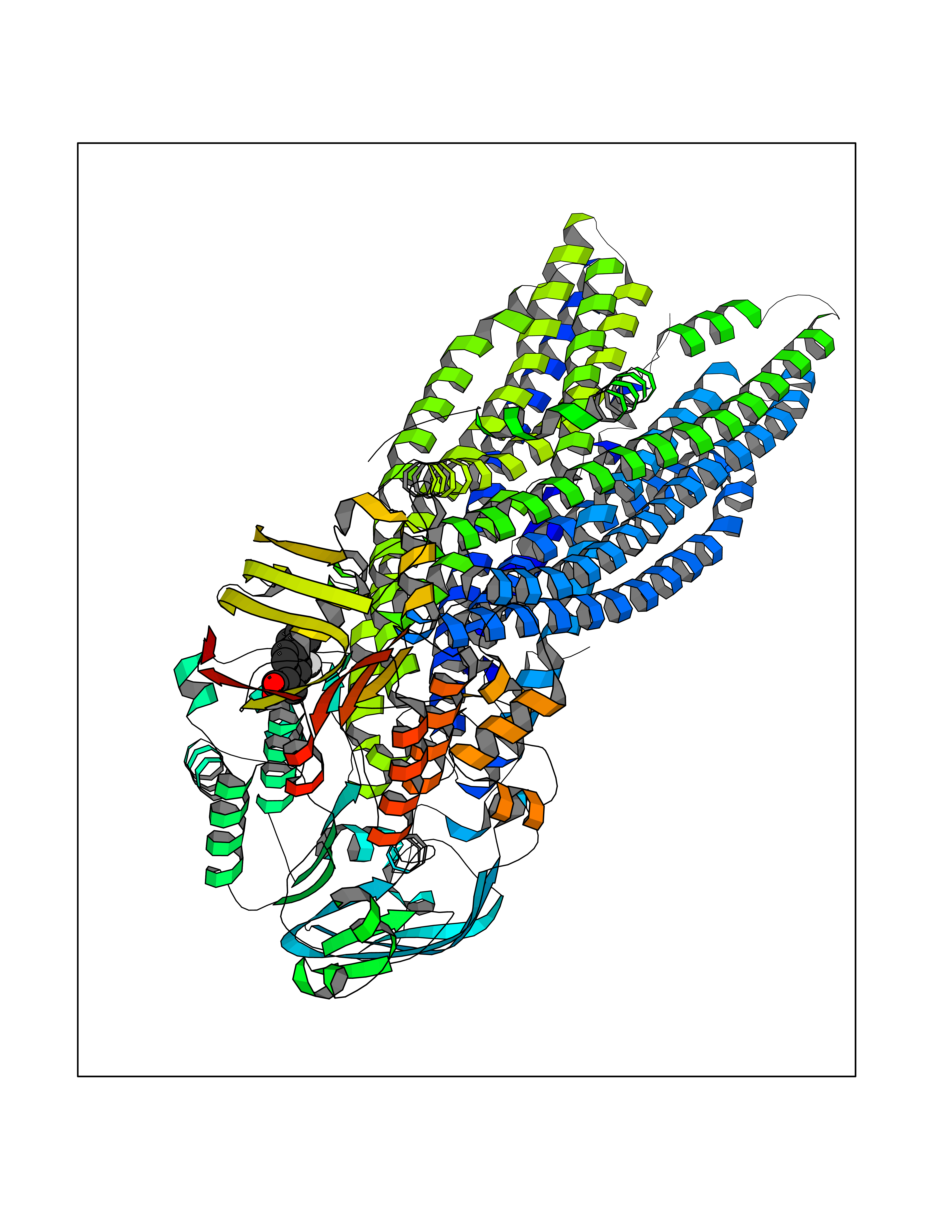 Molscript Map 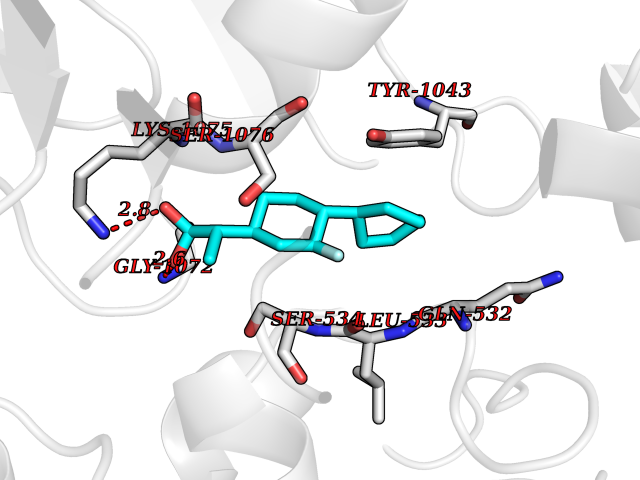 Pymol Map 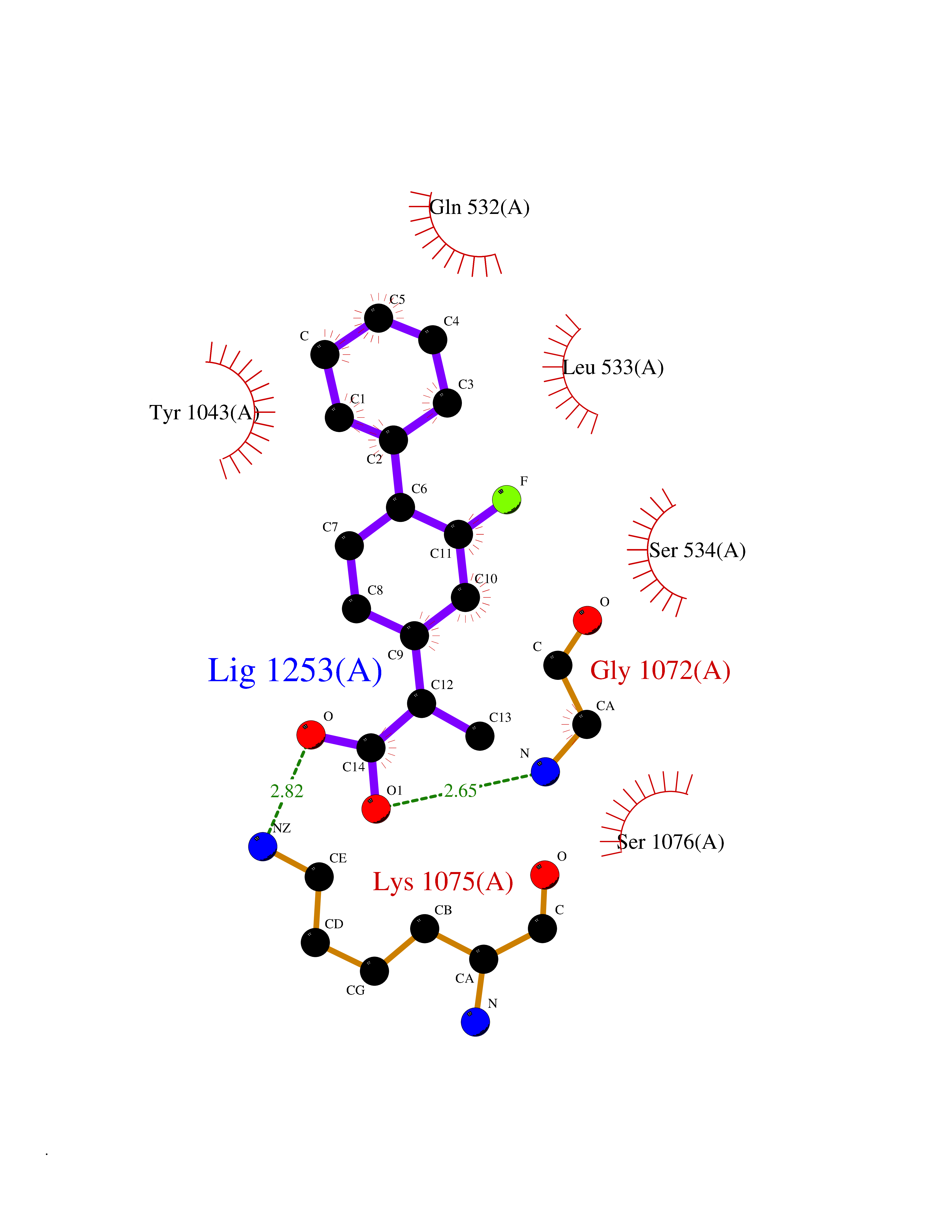 Ligplot Map 3D Binding mode Sequence VLTLFRYSDWQDKLFMSLGTIMAIAHGSGLPLMMIVFGEMTDKPGKILEEEMTRYAYYYSGLGAGVLVAAYIQVSFWTLAAGRQIRKIRQKFFHAILRQEIGWFDINDTTELNTRLTDDISKISEGIGDKVGMFFQAVATFFAGFIVGFIRGWKLTLVIMAISPILGLSAAVWAKILSAFSDKELAAYAKAGAVAEEALGAIRTVIAFGGQNKELERYQKHLENAKEIGIKKAISANISMGIAFLLIYASYALAFWYGSTLVISKEYTIGNAMTVFFSILIGAFSVGQAAPCIDAFANARGAAYVIFDIIDNNPKIDSFSERGHKPDSIKGNLEFNDVHFSYPSRANVKILKGLNLKVQSGQTVALVGSSGCGKSTTVQLIQRLYDPDEGTINIDGQDIRNFNVNYLREIIGVVSQEPVLFSTTIAENICYGRGNVTMDEIKKAVKEANAYEFIMKLPQKFDTLVGERGAQLSGGQKQRIAIARALVRNPKILLLDQATSALDTESEAEVQAALDKAREGRTTIVIAHRLSTVRNADVIAGFEDGVIVEQGSHSELMKKEGVYFKLVNVPPVSFLKVLKLNKTEWPYFVVGTVCAIANGGLQPAFSVIFSEIIAIFGPGDDAVKQQKCNIFSLIFLFLGIISFFTFFLQGFTFGKAGEILTRRLRSMAFKAMLRQDMSWFDDHKNSTGALSTRLATDAAQVQGATGTRLALIAQNIANLGTGIIISFIYGWQLTLLLLAVVPIIAVSGIVEMKLLAGNAKRDKKELEAAGKIATEAIENIRTVVSLTQERKFESMYVEKLYGPYRNSVQKAHIYGITFSISQAFMYFSYAGCFRFGAYLIVNGHMRFRDVILVFSAIVFGAVALGHASSFAPDYAKAKLSAAHLFMLFERQPLIDSYSEEGLKPDKFEGNITFNEVVFNYPTRANVPVLQGLSLEVKKGQTLALVGSSGCGKSTVVQLLERFYDPLAGTVLLDGQEAKKLNVQWLRAQLGIVSQEPILFDCSIAENIAYGDNSRVVSQDEIVSAAKAANIHPFIETLPHKYETRVGDKGTQLSGGQKQRIAIARALIRQPQILLLDQATSALDTESEKVVQEALDKAREGRTCIVIAHRLSTIQNADLIVVFQNGRVK Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Histone acetyltransferase p300 (EP300) | 5LKX | 7.30 | |
Target general information Gen name EP300 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p300 HAT; Protein propionyltransferase p300; P300; Histone crotonyltransferase p300; Histone butyryltransferase p300; E1Aassociated protein p300; E1A-associated protein p300 Protein family NA Biochemical class Acyltransferase Function Acetylates all four core histones in nucleosomes. Histone acetylation gives an epigenetic tag for transcriptional activation. Mediates cAMP-gene regulation by binding specifically to phosphorylated CREB protein. Mediates acetylation of histone H3 at 'Lys-122' (H3K122ac), a modification that localizes at the surface of the histone octamer and stimulates transcription, possibly by promoting nucleosome instability. Mediates acetylation of histone H3 at 'Lys-27' (H3K27ac). Also functions as acetyltransferase for non-histone targets, such as ALX1, HDAC1, PRMT1 or SIRT2. Acetylates 'Lys-131' of ALX1 and acts as its coactivator. Acetylates SIRT2 and is proposed to indirectly increase the transcriptional activity of TP53 through acetylation and subsequent attenuation of SIRT2 deacetylase function. Acetylates HDAC1 leading to its inactivation and modulation of transcription. Acts as a TFAP2A-mediated transcriptional coactivator in presence of CITED2. Plays a role as a coactivator of NEUROD1-dependent transcription of the secretin and p21 genes and controls terminal differentiation of cells in the intestinal epithelium. Promotes cardiac myocyte enlargement. Can also mediate transcriptional repression. Acetylates FOXO1 and enhances its transcriptional activity. Acetylates BCL6 wich disrupts its ability to recruit histone deacetylases and hinders its transcriptional repressor activity. Participates in CLOCK or NPAS2-regulated rhythmic gene transcription; exhibits a circadian association with CLOCK or NPAS2, correlating with increase in PER1/2 mRNA and histone H3 acetylation on the PER1/2 promoter. Acetylates MTA1 at 'Lys-626' which is essential for its transcriptional coactivator activity. Acetylates XBP1 isoform 2; acetylation increases protein stability of XBP1 isoform 2 and enhances its transcriptional activity. Acetylates PCNA; acetylation promotes removal of chromatin-bound PCNA and its degradation during nucleotide excision repair (NER). Acetylates MEF2D. Acetylates and stabilizes ZBTB7B protein by antagonizing ubiquitin conjugation and degragation, this mechanism may be involved in CD4/CD8 lineage differentiation. In addition to protein acetyltransferase, can use different acyl-CoA substrates, such as (2E)-butenoyl-CoA (crotonyl-CoA), butanoyl-CoA (butyryl-CoA) or propanoyl-CoA (propionyl-CoA), and is able to mediate protein crotonylation, butyrylation or propionylation, respectively. Acts as a histone crotonyltransferase; crotonylation marks active promoters and enhancers and confers resistance to transcriptional repressors. Histone crotonyltransferase activity is dependent on the concentration of (2E)-butenoyl-CoA (crotonyl-CoA) substrate and such activity is weak when (E)-but-2-enoyl-CoA (crotonyl-CoA) concentration is low. Also acts as a histone butyryltransferase; butyrylation marks active promoters. Functions as a transcriptional coactivator for SMAD4 in the TGF-beta signaling pathway. Acetylates PCK1 and promotes PCK1 anaplerotic activity. Functions as histone acetyltransferase and regulates transcription via chromatin remodeling. Related diseases Defects in EP300 may play a role in epithelial cancer.; DISEASE: Chromosomal aberrations involving EP300 may be a cause of acute myeloid leukemias. Translocation t(8;22)(p11;q13) with KAT6A.; DISEASE: Rubinstein-Taybi syndrome 2 (RSTS2) [MIM:613684]: A disorder characterized by craniofacial abnormalities, postnatal growth deficiency, broad thumbs, broad big toes, intellectual disability and a propensity for development of malignancies. Some individuals with RSTS2 have less severe mental impairment, more severe microcephaly, and a greater degree of changes in facial bone structure than RSTS1 patients. {ECO:0000269|PubMed:15706485}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Menke-Hennekam syndrome 2 (MKHK2) [MIM:618333]: A form of Menke-Hennekam syndrome, a congenital autosomal dominant disease characterized by developmental delay, growth retardation, and craniofacial dysmorphism. Patients have intellectual disability of variable severity, speech delay, autistic behavior, short stature and microcephaly. Main facial characteristics include short palpebral fissures, telecanthi, depressed nasal ridge, short nose, anteverted nares, short columella and long philtrum. {ECO:0000269|PubMed:29460469}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NXW9; P27695; Q9UBL3; Q8WXX7; Q9NPI1; P24941; Q99967; P61201; P16220-1; P17844; Q01844; P35637; Q00403; Q16665; Q9H2X6; Q92831; P55209; O60934; P20265; Q96KQ4; Q8WUF5; Q13761; Q96EB6; Q13309; O95863; P42226; Q9UL17; P56279; P05549; P04637; Q13625; O15350; P11473; P67809; K4P3M7; P03122; P06422; P06790; Q61221; Q9QXM1; P04608; P03070; P03255; P03255-2; P03259 EC number EC 2.3.1.48 Uniprot keywords 3D-structure; Acetylation; Acyltransferase; Biological rhythms; Bromodomain; Cell cycle; Chromosomal rearrangement; Chromosome; Citrullination; Cytoplasm; Direct protein sequencing; Disease variant; Host-virus interaction; Intellectual disability; Isopeptide bond; Metal-binding; Methylation; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 64477.2 Length 554 Aromaticity 0.12 Instability index 45.78 Isoelectric point 7.01 Charge (pH=7) 0.05 2D Binding mode Binding energy (Kcal/mol) -8.31 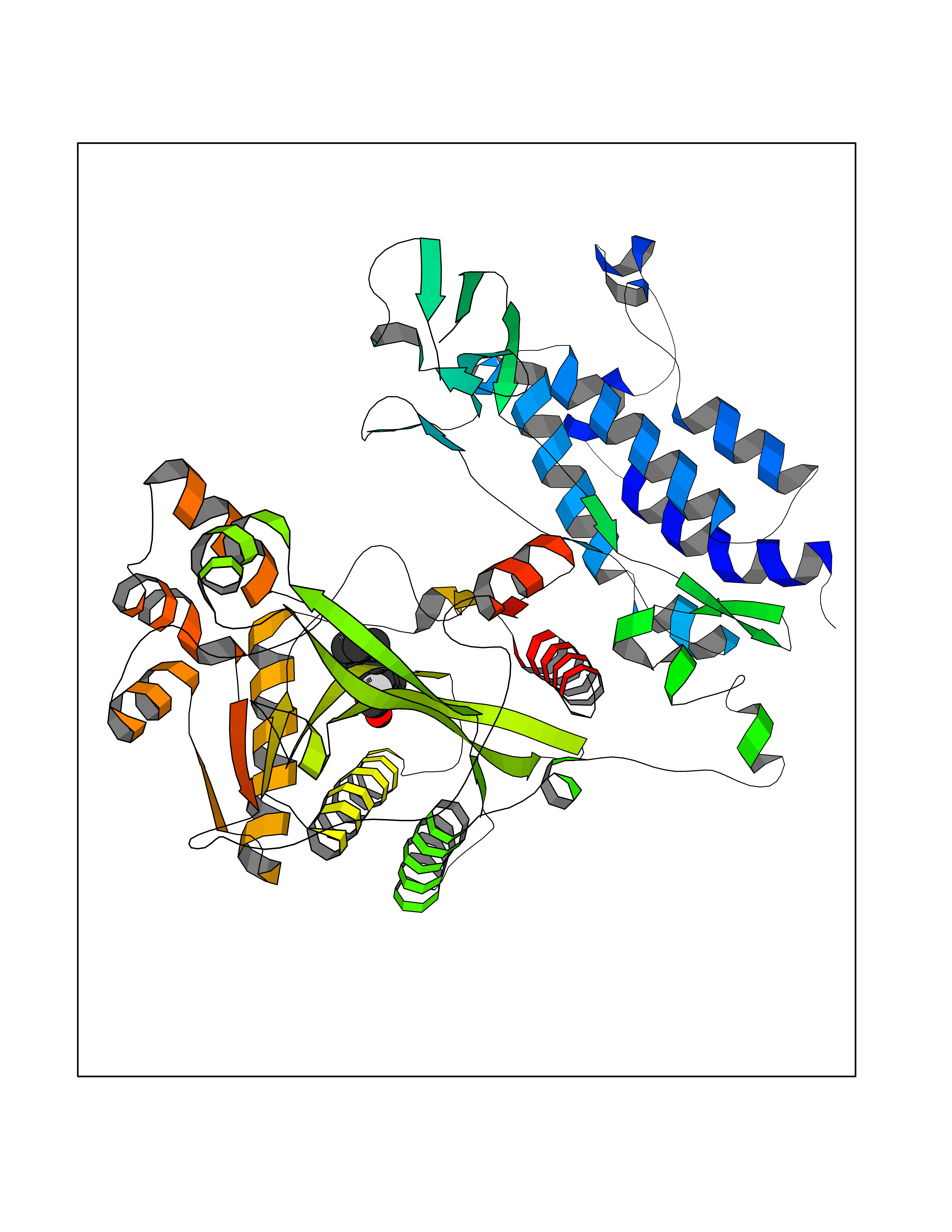 Molscript Map 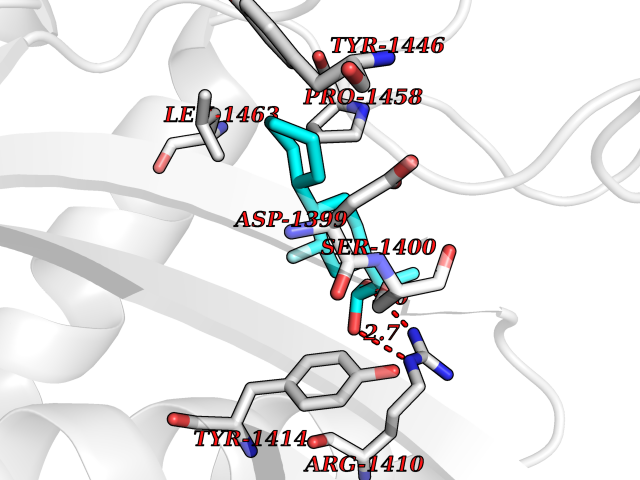 Pymol Map 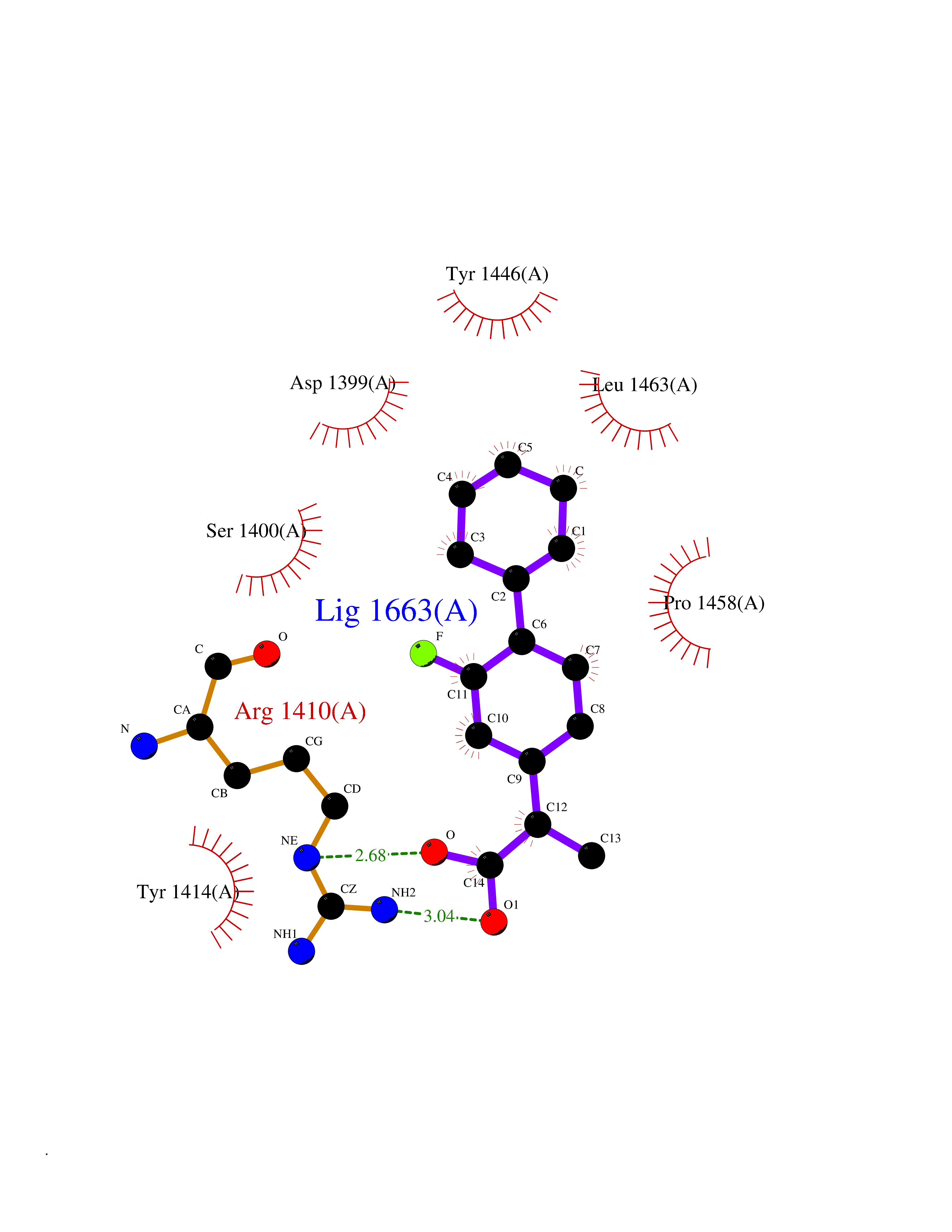 Ligplot Map 3D Binding mode Sequence KKIFKPEELRQALMPTLEALYRQDPESLPFRQPVDPQLLGIPDYFDIVKSPMDLSTIKRKLDTGQYQEPWQYVDDIWLMFNNAWLYNRKTSRVYKYCSKLSEVFEQEIDPVMQSLGYCCGRKLEFSPQTLCCYGKQLCTIPRDATYYSYQNRYHFCEKCFNEIQGESVSLGQTTINKEQFSKRKNDTLDPELFVECTECGRKMHQICVLHHEIIWPAGFVCDGCLKKSARTRKENKFSAKRLPSTRLGTFLENRVNDFLRRQNHPESGEVTVRVVHASDKTVEVKPGMKARFVDSGEMAESFPYRTKALFAFEEIDGVDLCFFGMHVQEYGSDCPPPNQRRVYISYLDSVHFFRPKCLRTAVYHEILIGYLEYVKKLGYTTGHIWACPPSEGDDYIFHCHPPDQKIPKPKRLQEWFKKMLDKAVSERIVHDYKDIFKQATEDRLTSAKELPYFEGDFWPNVLEESIKESGGSGSQKLYATMEKHKEVFFVIRLIAGPAANSLPPIVDPDPLIPCDLMDGRDAFLTLARDKHLEFSSLRRAQWSTMCMLVELHTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Protein arginine methyltransferase 5 (PRMT5) | 7MXC | 7.29 | |
Target general information Gen name PRMT5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Shk1 kinase-binding protein 1 homolog; SKB1Hs; SKB1 homolog; SKB1; Protein arginine N-methyltransferase 5; Jak-binding protein 1; JBP1; IBP72; Histone-arginine N-methyltransferase PRMT5; HRMT1L5; 72 k Protein family Class I-like SAM-binding methyltransferase superfamily, Protein arginine N-methyltransferase family Biochemical class Methyltransferase Function Specifically mediates the symmetrical dimethylation of arginine residues in the small nuclear ribonucleoproteins Sm D1 (SNRPD1) and Sm D3 (SNRPD3); such methylation being required for the assembly and biogenesis of snRNP core particles. Methylates SUPT5H and may regulate its transcriptional elongation properties. Mono- and dimethylates arginine residues of myelin basic protein (MBP) in vitro. May play a role in cytokine-activated transduction pathways. Negatively regulates cyclin E1 promoter activity and cellular proliferation. Methylates histone H2A and H4 'Arg-3' during germ cell development. Methylates histone H3 'Arg-8', which may repress transcription. Methylates the Piwi proteins (PIWIL1, PIWIL2 and PIWIL4), methylation of Piwi proteins being required for the interaction with Tudor domain-containing proteins and subsequent localization to the meiotic nuage. Methylates RPS10. Attenuates EGF signaling through the MAPK1/MAPK3 pathway acting at 2 levels. First, monomethylates EGFR; this enhances EGFR 'Tyr-1197' phosphorylation and PTPN6 recruitment, eventually leading to reduced SOS1 phosphorylation. Second, methylates RAF1 and probably BRAF, hence destabilizing these 2 signaling proteins and reducing their catalytic activity. Required for induction of E-selectin and VCAM-1, on the endothelial cells surface at sites of inflammation. Methylates HOXA9. Methylates and regulates SRGAP2 which is involved in cell migration and differentiation. Acts as a transcriptional corepressor in CRY1-mediated repression of the core circadian component PER1 by regulating the H4R3 dimethylation at the PER1 promoter. Methylates GM130/GOLGA2, regulating Golgi ribbon formation. Methylates H4R3 in genes involved in glioblastomagenesis in a CHTOP- and/or TET1-dependent manner. Symmetrically methylates POLR2A, a modification that allows the recruitment to POLR2A of proteins including SMN1/SMN2 and SETX. This is required for resolving RNA-DNA hybrids created by RNA polymerase II, that form R-loop in transcription terminal regions, an important step in proper transcription termination. Along with LYAR, binds the promoter of gamma-globin HBG1/HBG2 and represses its expression. Symmetrically methylates NCL. Methylates TP53; methylation might possibly affect TP53 target gene specificity. Arginine methyltransferase that can both catalyze the formation of omega-N monomethylarginine (MMA) and symmetrical dimethylarginine (sDMA), with a preference for the formation of MMA. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P01019; Q9NX04; Q8WUW1; Q08289; P78371; Q16543; Q8N8U2; P54105; P21964-2; Q9NQ92; Q16526; Q9Y6K1; Q01094; Q08426; P38919; Q14241; O15197-2; Q6ZV65; P01100; O95995; P62993; Q8TE85; Q9BX10; P62805; P31269; Q00613; Q63ZY3; P03952; Q8TBB1; P06858; Q86UQ8-1; Q96HA8; Q8WVJ2; P24928; O14744; Q86U06; Q9BRS2; O75044; Q96RU7; P31930; P40337-2; Q9BQA1; P63104; Q96E35; Q91XC0; P03418; Q6ZV65-2 EC number EC 2.1.1.320 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Chromosome; Cytoplasm; Direct protein sequencing; Golgi apparatus; Methyltransferase; Nucleus; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 71188.3 Length 621 Aromaticity 0.11 Instability index 44.6 Isoelectric point 5.95 Charge (pH=7) -9.68 2D Binding mode Binding energy (Kcal/mol) -8.48 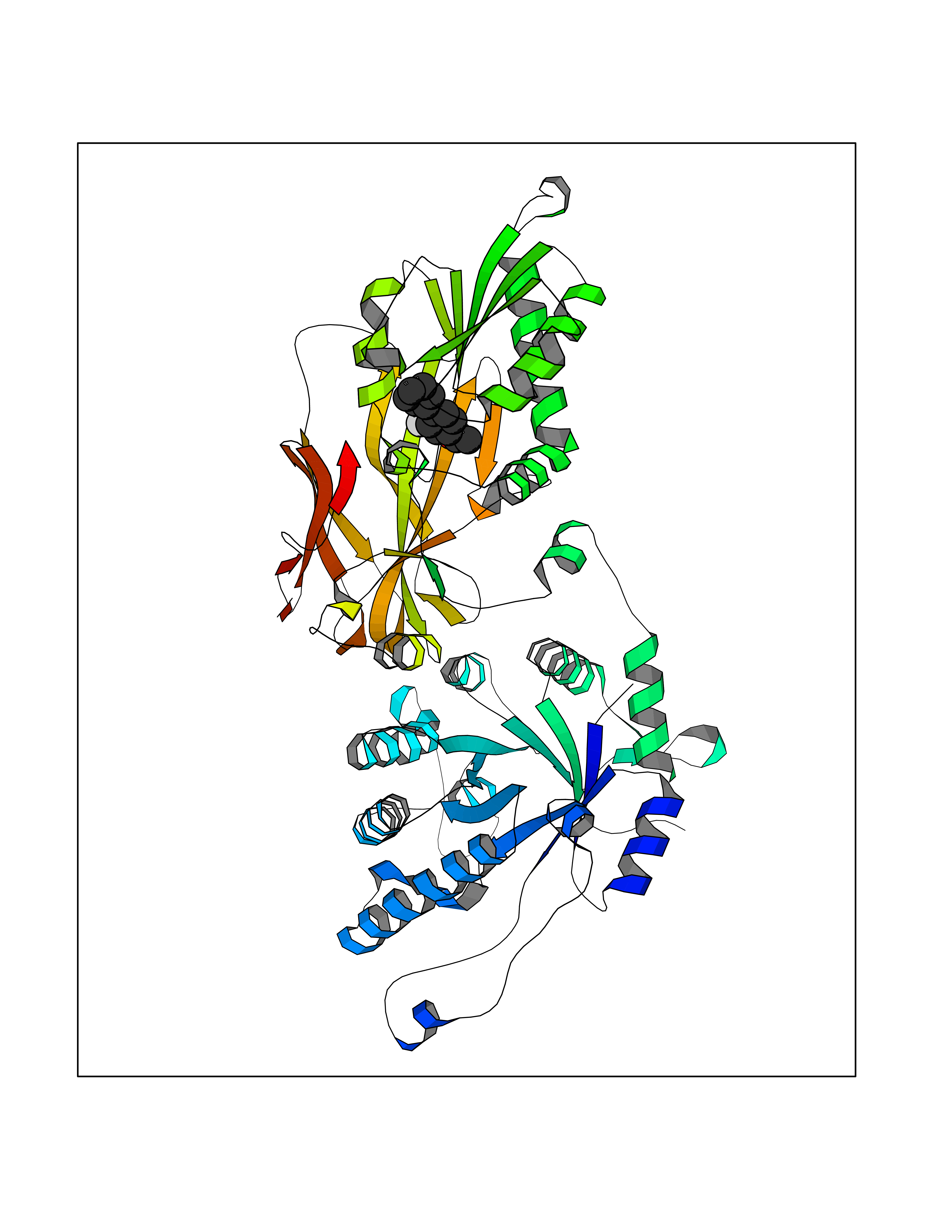 Molscript Map 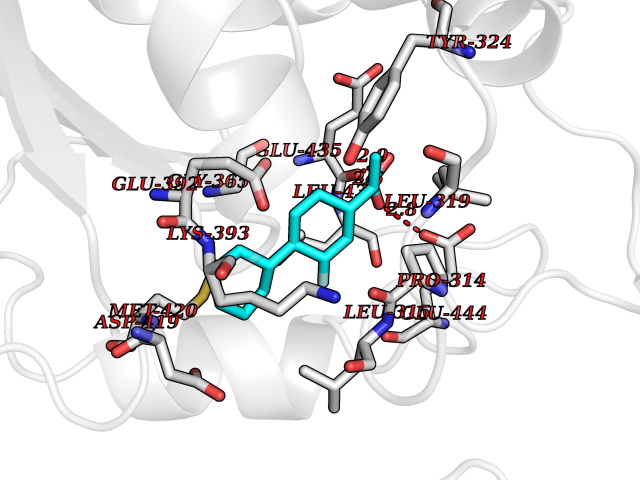 Pymol Map 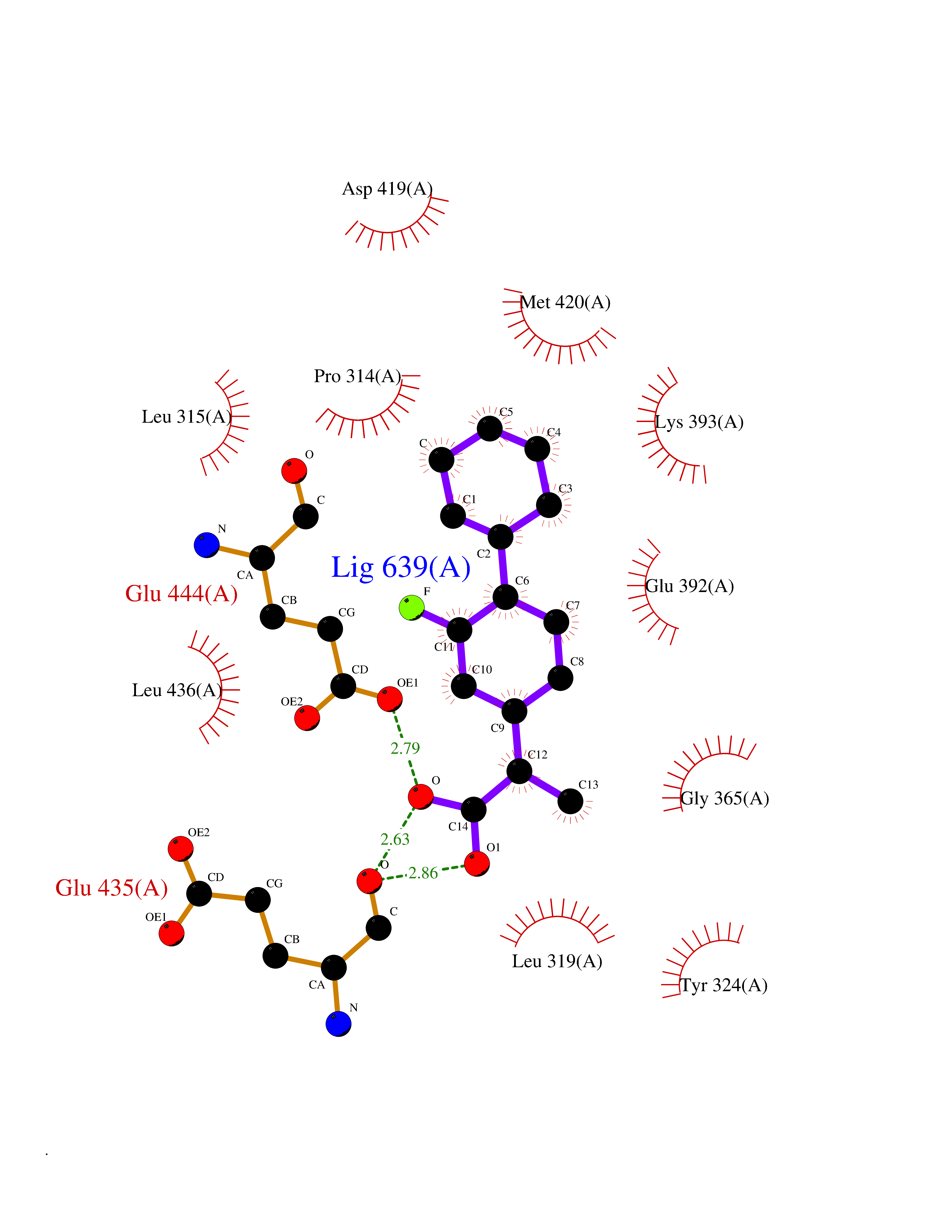 Ligplot Map 3D Binding mode Sequence RVSSGRDLNCVPEIADTLGAVAKQGFDFLCMPVFHPRFKREFIQEPAKNRPGPQTRSDLLLSGRDWNTLIVGKLSPWIRPDSKVEKIRRNSEAAMLQELNFGAYLGLPAFLLPLNQEDNTNLARVLTNHIHTGHHSSMFWMRVPLVAPEDLRDDIIENAPTTHTEEYSGEEKTWMWWHNFRTLCDYSKRIAVALEIGADLPSNHVIDRWLGEPIKAAILPTSIFLTNKKGFPVLSKMHQRLIFRLLKLEVQFIITGTNHHSCSYLQYLEYLSQNRPPPNAYELFAKGYEDYLQSPLQPLMDNLESQTYEVFEKDPIKYSQYQQAIYKCLLDRVPEEEKDTNVQVLMVLGAGRGPLVNASLRAAKQADRRIKLYAVEKNPNAVVTLENWQFEEWGSQVTVVSSDMREWVAPEKADIIVSELLGSFADNELSPECLDGAQHFLKDDGVSIPGEYTSFLAPISSSKLYNEVRACREKDRDPEAQFEMPYVVRLHNFHQLSAPQPCFTFSHPNRDPMIDNNRYCTLEFPVEVNTVLHGFAGYFETVLYQDITLSIRPETHSPGMFSWFPILFPIKQPITVREGQTICVRFWRCSNSKKVWYEWAVTAPVCSAIHNPTGRSYTIGL Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Glutamate receptor ionotropic kainate 1 (GRIK1) | 3FV2 | 7.29 | |
Target general information Gen name GRIK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glutamate receptor 5; GluR5 kainate receptor; GluR5; GluR-5; GRIK1; Excitatory amino acid receptor 3; EAA3 Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, GRIK1 subfamily Biochemical class Glutamate-gated ion channel Function Ionotropic glutamate receptor. L-glutamate acts as an excitatory neurotransmitter at many synapses in the central nervous system. Binding of the excitatory neurotransmitter L- glutamate induces a conformation change, leading tothe opening of the cation channel, and thereby converts the chemical signal to an electrical impulse. The receptor then desensitizes rapidly and enters a transient inactive state, characterized by the presence of bound agonist. May be involved in the transmission of light information from the retina to the hypothalamus. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB00237; DB00142; DB06354; DB00273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; RNA editing; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 29057.1 Length 256 Aromaticity 0.1 Instability index 24.38 Isoelectric point 8.3 Charge (pH=7) 1.97 2D Binding mode Binding energy (Kcal/mol) -8.22 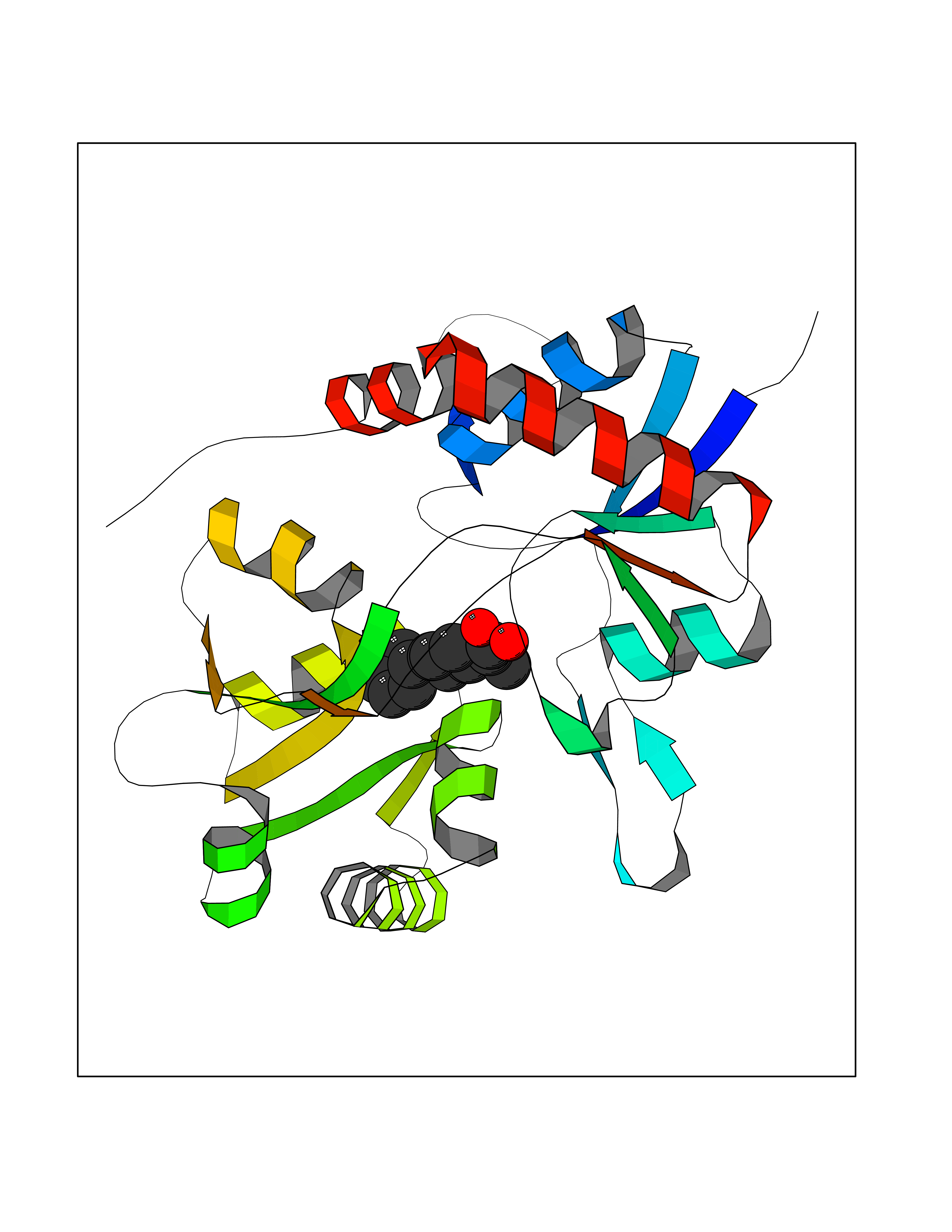 Molscript Map 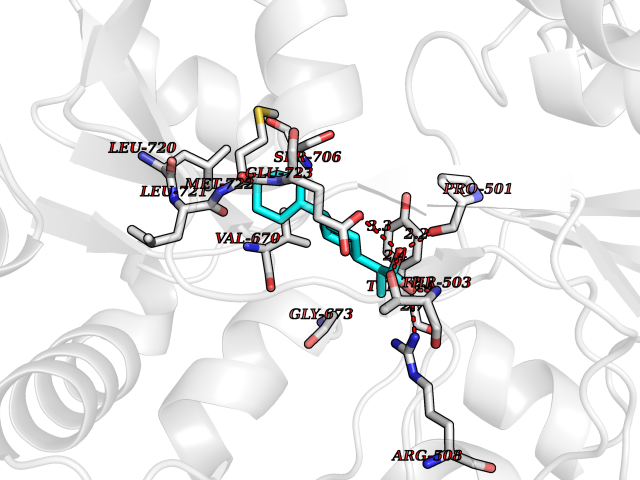 Pymol Map 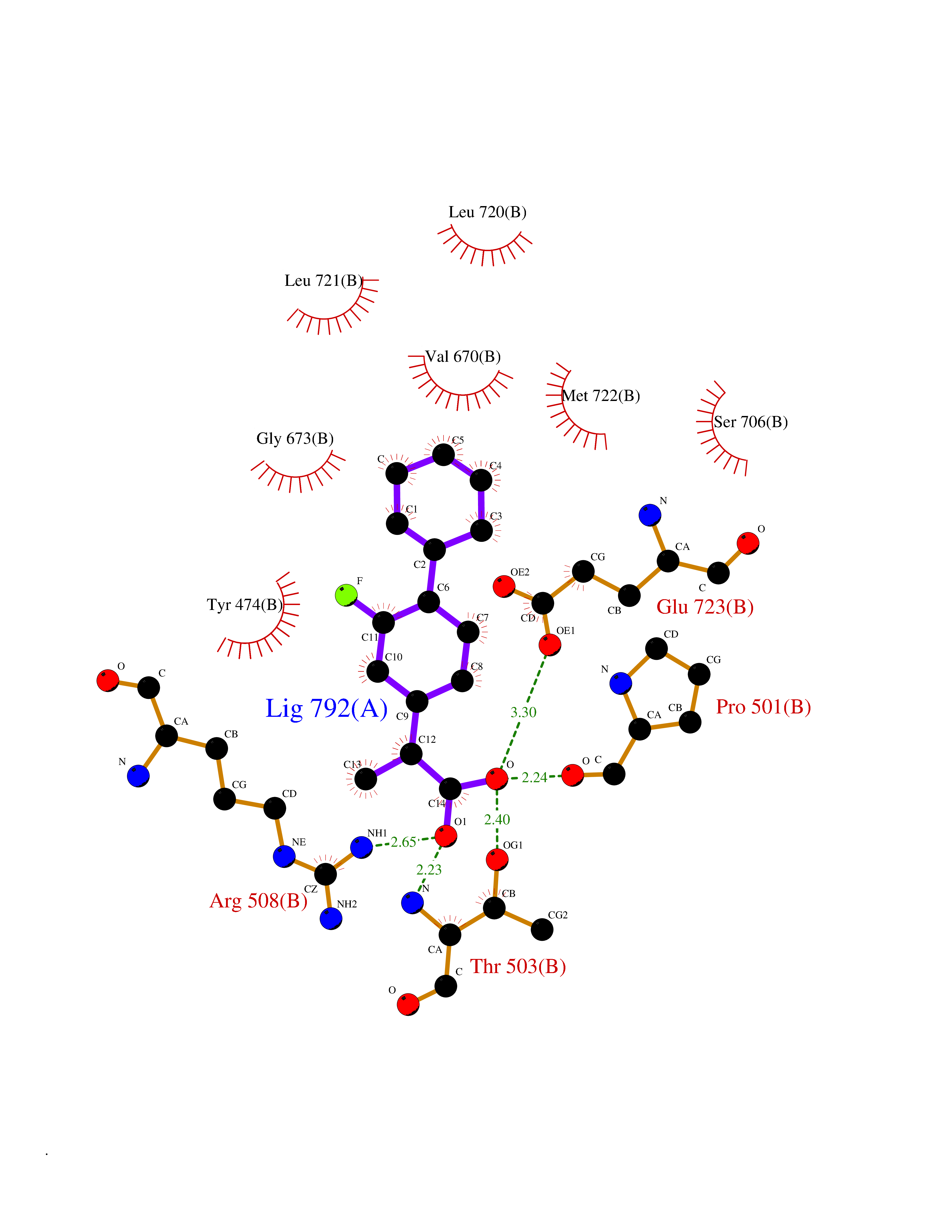 Ligplot Map 3D Binding mode Sequence ANRTLIVTTILEEPYVMYRKSDKPLYGNDRFEGYCLDLLKELSNILGFIYDVKLVPDGKYGAQNDKGEWNGMVKELIDHRADLAVAPLTITYVREKVIDFSKPFMTLGISILYRKGTPIDSADDLAKQTKIEYGAVRDGSTMTFFKKSKISTYEKMWAFMSSRQQTALVRNSDEGIQRVLTTDYALLMESTSIEYVTQRNCNLTQIGGLIDSKGYGVGTPIGSPYRDKITIAILQLQEEGKLHMMKEKWWRGNGCP Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Retinal rod rhodopsin-sensitive cGMP 3',5'-cyclic phosphodiesterase subunit gamma | 3JWR | 7.28 | |
Target general information Gen name PDE6G Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PDEG Protein family Rod/cone cGMP-PDE gamma subunit family Biochemical class Hydrolase Function 3',5'-cyclic-GMP phosphodiesterase activity.CGMP binding.Enzyme inhibitor activity.Spectrin binding. Related diseases Retinitis pigmentosa 57 (RP57) [MIM:613582]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:20655036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07954; DB00203; DB00820; DB00862 Interacts with O14503; Q96JM7; A0A6Q8PF08; O43741; Q8R511; P62994; Q9QY17; Q63787 EC number 3.1.4.35 Uniprot keywords 3D-structure; Acetylation; cGMP; Hydrolase; Reference proteome; Retinitis pigmentosa; Sensory transduction; Vision Protein physicochemical properties Chain ID C,D Molecular weight (Da) 40027.7 Length 345 Aromaticity 0.09 Instability index 37.44 Isoelectric point 6.02 Charge (pH=7) -6.88 2D Binding mode Binding energy (Kcal/mol) -8.19  Molscript Map 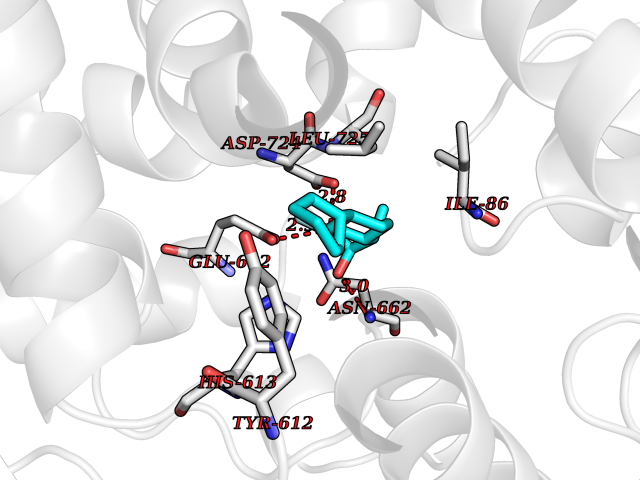 Pymol Map 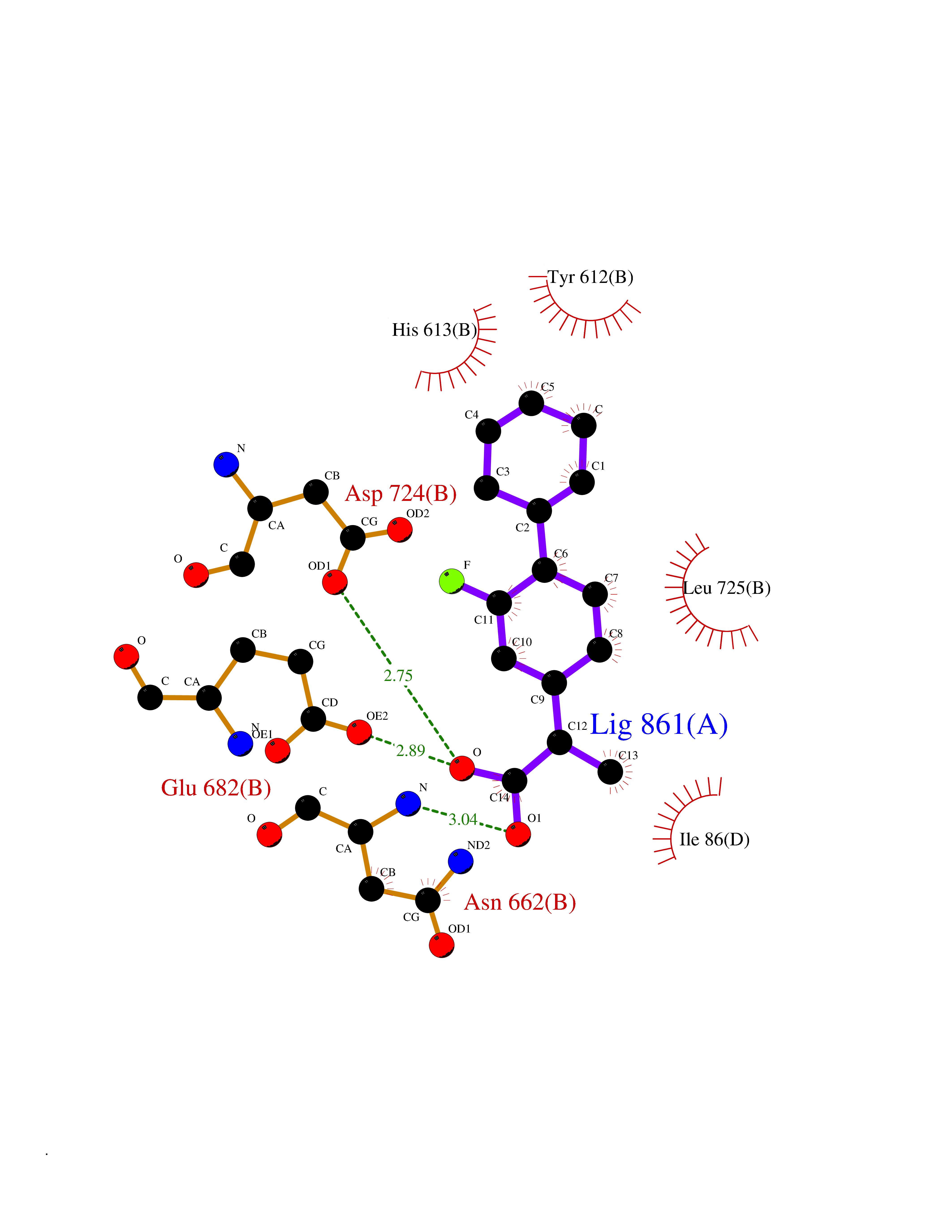 Ligplot Map 3D Binding mode Sequence EAFNHLELHELAQYGIISHMEETRELQSLAAAVVPSAQTLKITDFSFSDFELSDLETALCTIRMFTDLNLVQNFQMKHEVLCRWILSVKKNYRKNVAYHNWRHAFNTAQCMFAALKAGKIQNKLTDLEILALLIAALSHDLDHRGVNNSYIQRSEHPLAQLYCHSIMEHHHFDQCLMILNSPGNQILSGLSIEEYKTTLKIIKQAILATDLALYIKRRGEFFELIRKNQFNLEDPHQKELFLAMLMTACDLSAITKPWPIQQRIAELVATEFWEQGDLERTVLQQQPIPMMDRNKRDELPKLQVGFIDFVCTQLYEALTHVSEDCFPLLDGCRKNRQKWQALAEQ Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Phosphatidylinositol-4-kinase beta (PI4KB) | 4WAE | 7.27 | |
Target general information Gen name PI4KB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PtdIns 4-kinase beta; Phosphatidylinositol 4-kinase beta; PIK4CB; PI4Kbeta; PI4K92; PI4K-beta; NPIK Protein family PI3/PI4-kinase family, Type III PI4K subfamily Biochemical class NA Function Phosphorylates phosphatidylinositol (PI) in the first committed step in the production of the second messenger inositol-1,4,5,-trisphosphate (PIP). May regulate Golgi disintegration/reorganization during mitosis, possibly via its phosphorylation. Involved in Golgi-to-plasma membrane trafficking (By similarity). Related diseases Deafness, autosomal dominant, 87 (DFNA87) [MIM:620281]: A form of non-syndromic, sensorineural hearing loss. Sensorineural hearing loss results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. DFNA87 is characterized by prelingual, profound sensorineural hearing loss with inner ear anomalies, including cochlear maldevelopment, absence of the osseous spiral lamina, and/or an enlarged vestibular aqueduct. {ECO:0000269|PubMed:33358777}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with Q9H3P7; P27348; P03495; PRO_0000424692 [P03300]; P62491 EC number EC 2.7.1.67 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cytoplasm; Deafness; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Golgi apparatus; Host-virus interaction; Kinase; Lipid metabolism; Membrane; Mitochondrion; Mitochondrion outer membrane; Non-syndromic deafness; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 53130 Length 463 Aromaticity 0.1 Instability index 49.16 Isoelectric point 6.78 Charge (pH=7) -0.96 2D Binding mode Binding energy (Kcal/mol) -8.1  Molscript Map 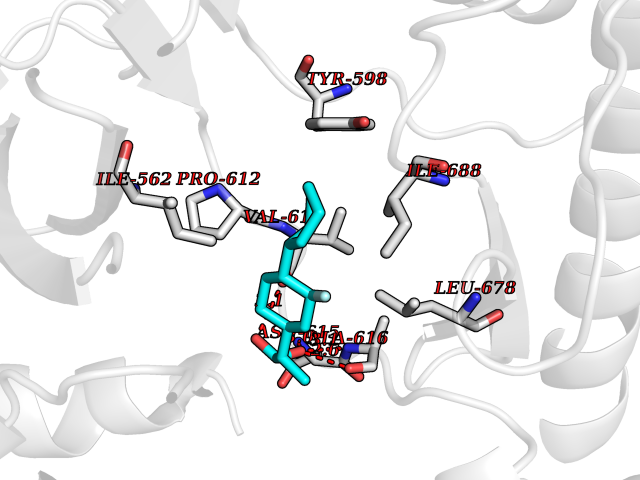 Pymol Map 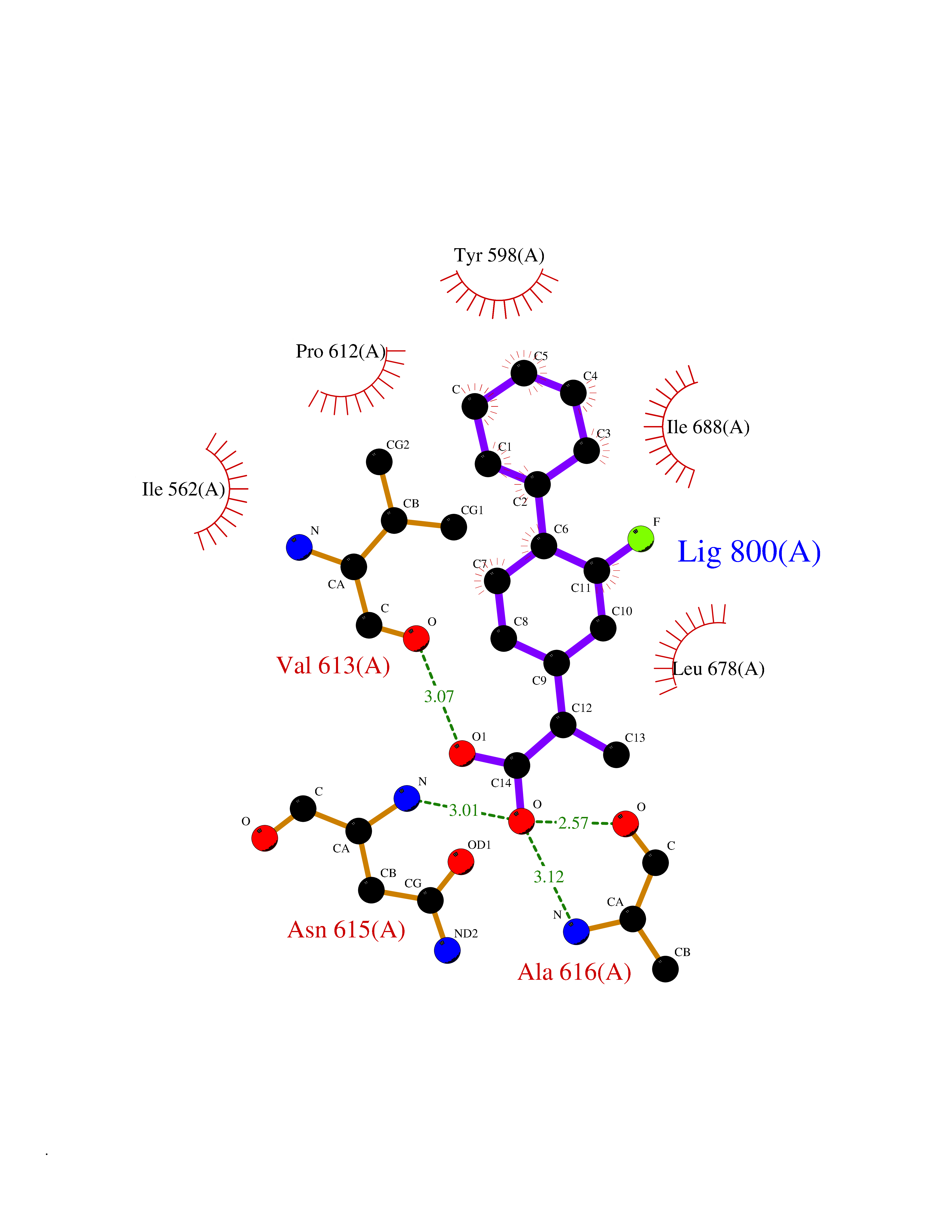 Ligplot Map 3D Binding mode Sequence LLRLFESKLFDISMAISYLYNSKEPGVQAYIGNRLFCFRNEDVDFYLPQLLNMYIHMDEDVGDAIKPYIVHRCRQSINFSLQCALLLGAYSSDMNSFSSPVRLAPEREFIKSLMAIGKRLATLPTKEQKTQRLISELSLLNHKLPARVWLPTAGFDHHVVRVPHTQAVVLNSKDKAPYLIYVEVLECENFDTTSVPARIPEWQEKVRRIREGSPYGHLPNWRLLSVIVKCGDDLRQELLAFQVLKQLQSIWEQERVPLWIKPYKILVISADSGMIEPVVNAVSIHQVKKQSQLSLLDYFLQEHGSYTTEAFLSAQRNFVQSCAGYCLVCYLLQVKDRHNGNILLDAEGHIIHIDFGFILSSSPRNLGFETSAFKLTTEFVDVMGGLDGDMFNYYKMLMLQGLIAARKHMDKVVQIVEIMQQGSQLPCFHGSSTIRNLKERFHMSMTEEQLQLLVEQMVDGSMR Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Acetylcholine receptor subunit alpha | 4ZJS | 7.27 | |
Target general information Gen name CHRNA1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACHRA;CHNRA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-1/CHRNA1 sub-subfamily Biochemical class Immune system Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ion channel activity.Ligand-gated ion channel activity. Related diseases Multiple pterygium syndrome, lethal type (LMPS) [MIM:253290]: Multiple pterygia are found infrequently in children with arthrogryposis and in fetuses with fetal akinesia syndrome. In lethal multiple pterygium syndrome there is intrauterine growth retardation, multiple pterygia, and flexion contractures causing severe arthrogryposis and fetal akinesia. Subcutaneous edema can be severe, causing fetal hydrops with cystic hygroma and lung hypoplasia. Oligohydramnios and facial anomalies are frequent. {ECO:0000269|PubMed:18252226}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: The alpha subunit is the main focus for antibody binding in myasthenia gravis. Myasthenia gravis is characterized by sporadic muscular fatigability and weakness, occurring chiefly in muscles innervated by cranial nerves, and characteristically improved by cholinesterase-inhibiting drugs.; DISEASE: Myasthenic syndrome, congenital, 1A, slow-channel (CMS1A) [MIM:601462]: A common congenital myasthenic syndrome. Congenital myasthenic syndromes are characterized by muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1A is a slow-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in prolonged AChR channel opening episodes, prolonged endplate currents, and depolarization block. This is associated with calcium overload, which may contribute to subsequent degeneration of the endplate and postsynaptic membrane. {ECO:0000269|PubMed:16685696, ECO:0000269|PubMed:7619526, ECO:0000269|PubMed:8872460, ECO:0000269|PubMed:9158151, ECO:0000269|PubMed:9221765}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 1B, fast-channel (CMS1B) [MIM:608930]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1B is a fast-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in brief opening and activity of the channel, with a rapid decay in endplate current, failure to achieve threshold depolarization of the endplate and consequent failure to fire an action potential. {ECO:0000269|PubMed:10195214, ECO:0000269|PubMed:12588888, ECO:0000269|PubMed:15079006}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08838; DB00565; DB00555 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46717.8 Length 411 Aromaticity 0.11 Instability index 38.02 Isoelectric point 4.77 Charge (pH=7) -22.31 2D Binding mode Binding energy (Kcal/mol) -9.19 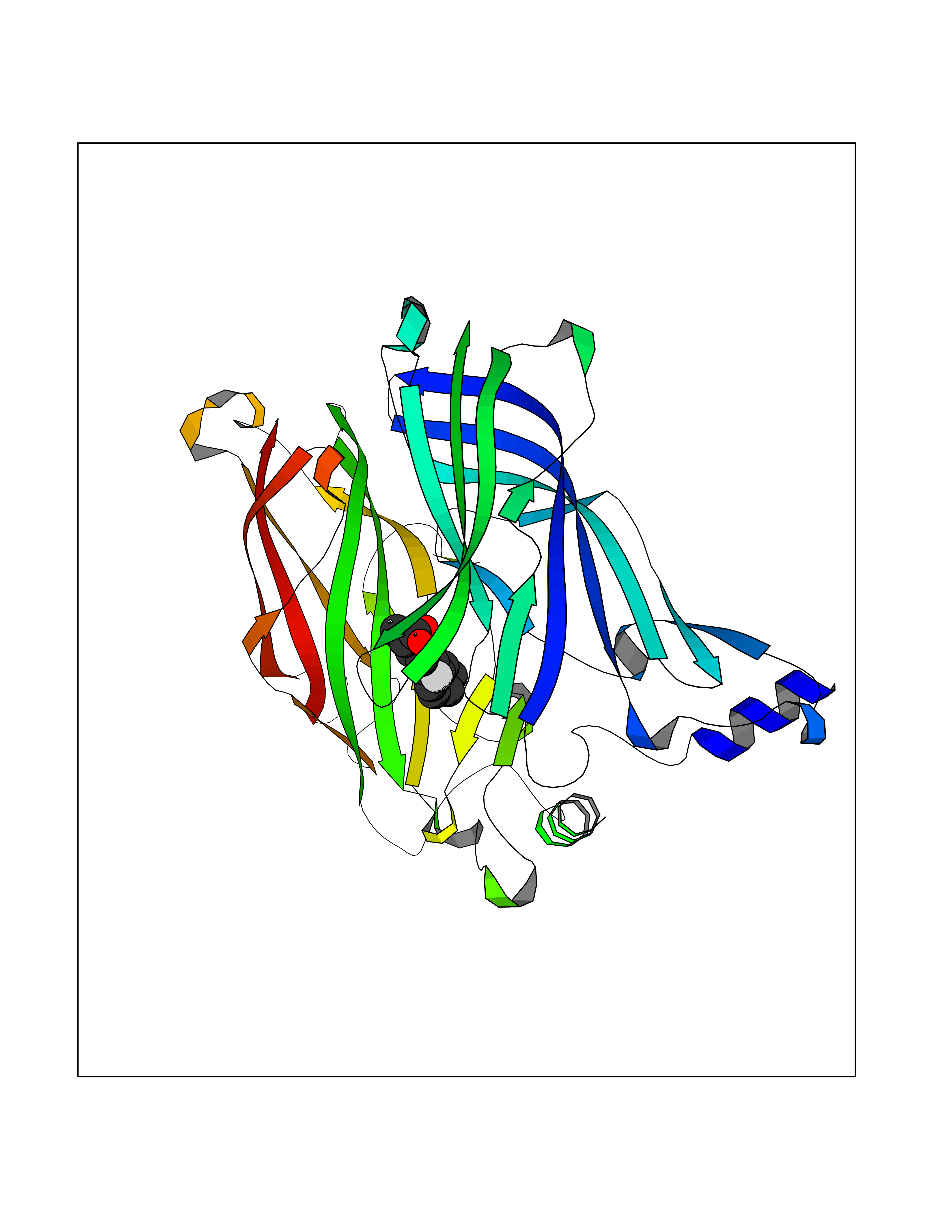 Molscript Map 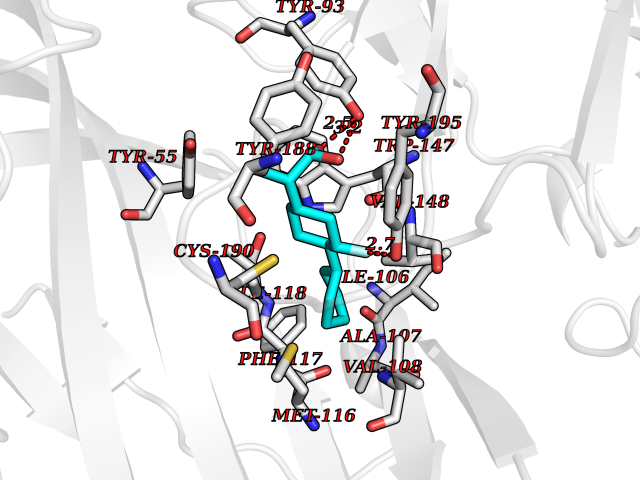 Pymol Map 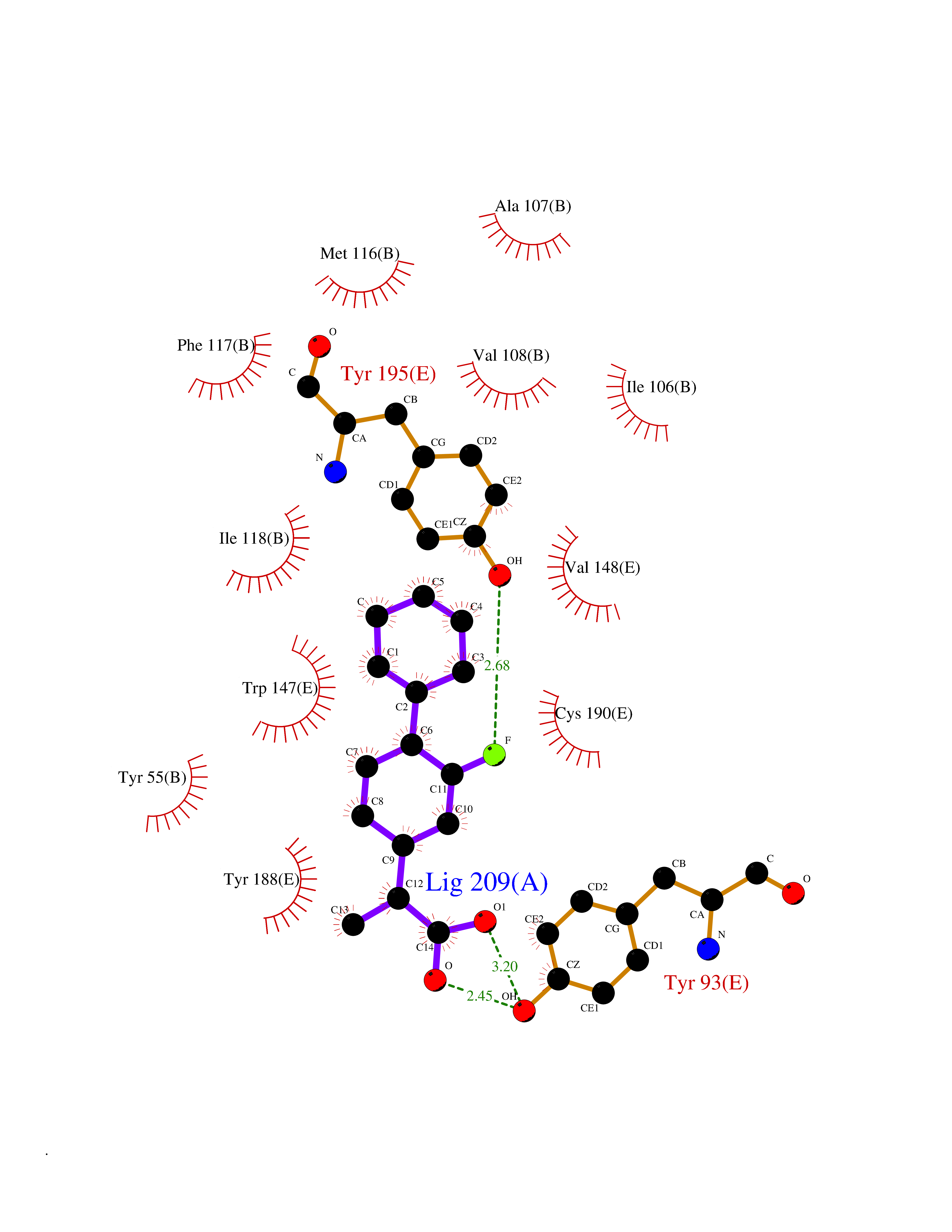 Ligplot Map 3D Binding mode Sequence EHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFREEHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFRER Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | GTPase HRas (HRAS) | 7L0F | 7.26 | |
Target general information Gen name HRAS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p21ras; cHras; c-H-ras; Transforming protein p21; HaRas; Ha-Ras; H-Ras-1; GTPase HRas, Nterminally processed Protein family Small GTPase superfamily, Ras family Biochemical class Small GTPase Function Ras proteins bind GDP/GTP and possess intrinsic GTPase activity. Involved in the activation of Ras protein signal transduction. Related diseases Costello syndrome (CSTLO) [MIM:218040]: A rare condition characterized by prenatally increased growth, postnatal growth deficiency, intellectual disability, distinctive facial appearance, cardiovascular abnormalities (typically pulmonic stenosis, hypertrophic cardiomyopathy and/or atrial tachycardia), tumor predisposition, skin and musculoskeletal abnormalities. {ECO:0000269|PubMed:16170316, ECO:0000269|PubMed:16329078, ECO:0000269|PubMed:16443854, ECO:0000269|PubMed:17054105, ECO:0000269|PubMed:18039947, ECO:0000269|PubMed:18247425, ECO:0000269|PubMed:19995790}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy with excess of muscle spindles (CMEMS) [MIM:218040]: Variant of Costello syndrome. {ECO:0000269|PubMed:17412879}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Mutations which change positions 12, 13 or 61 activate the potential of HRAS to transform cultured cells and are implicated in a variety of human tumors. {ECO:0000269|PubMed:3670300}.; DISEASE: Bladder cancer (BLC) [MIM:109800]: A malignancy originating in tissues of the urinary bladder. It often presents with multiple tumors appearing at different times and at different sites in the bladder. Most bladder cancers are transitional cell carcinomas that begin in cells that normally make up the inner lining of the bladder. Other types of bladder cancer include squamous cell carcinoma (cancer that begins in thin, flat cells) and adenocarcinoma (cancer that begins in cells that make and release mucus and other fluids). Bladder cancer is a complex disorder with both genetic and environmental influences. {ECO:0000269|PubMed:6298635, ECO:0000269|PubMed:6844927}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Schimmelpenning-Feuerstein-Mims syndrome (SFM) [MIM:163200]: A disease characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects. Many oral manifestations have been reported, not only including hypoplastic and malformed teeth, and mucosal papillomatosis, but also ankyloglossia, hemihyperplastic tongue, intraoral nevus, giant cell granuloma, ameloblastoma, bone cysts, follicular cysts, oligodontia, and odontodysplasia. Sebaceous nevi follow the lines of Blaschko and these can continue as linear intraoral lesions, as in mucosal papillomatosis. {ECO:0000269|PubMed:22683711}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04315; DB04137; DB02210; DB08751; DB03226; DB15588 Interacts with Q99996-3; P53677-2; P10398; Q9NXL2-1; Q9UII2; Q9H7T9; Q00994; Q9H2G9; P15056; Q7Z569; Q5PSV4; Q9ULD4-2; Q96LL4; Q96HB5; Q49A88-3; Q96GN5-2; P24941; O95674; Q9H3R5; Q9Y4F5-3; Q86XR8; Q494V2-2; Q8WUX9; Q14117; Q9Y6W6; O14641; A0AVK6; Q8NB25; Q8IZU1; O94868-3; P15407; P15408; P52655; Q96CS2; Q9BT25; Q8IV36; O43248; Q53GQ0; P10809; Q8NDH6-2; Q8IY31-2; Q8NA54; Q13352; P28290-2; Q9BVG8-5; Q2M2Z5; Q6P597; P57682; Q9UH77; P08727; Q14525; Q14847-2; Q96LR2; P27338; Q99558; Q96EZ8; Q8TAC0; Q5JXC2; Q8NEH6; Q9Y605; Q96HT8; Q9GZM8; P21359; Q8N5V2; Q6PHZ7; Q9BZ95-3; A5D8V7; O43482; Q9BR81; O15534; Q9BUL5; O00329; O00329-2; Q9UPR0; Q96I34; Q15435-3; P04049; P11233; Q15311; Q12967; Q9NS23-2; Q9NS23-4; Q8WWW0; Q8TBY0; Q9P2K3-2; Q9NZL6; O15211; Q8IXN7; Q13671; Q13671-1; Q8WVD3; Q9BY12-3; Q13435; Q12824; Q13573; Q07889; Q86W54-2; Q92783-2; O75886; Q13586; Q8N4C7; O75528; P54274-2; Q9BXU0; Q5T0J7-2; Q5T1C6; Q8IUR5-4; P36406; Q86WT6-2; Q99598; Q6PF05; Q9UGJ1-2; Q9Y5Z9; P22415; Q495M9; Q9H270; Q8NEZ2; P19544-6; O43829; Q9C0F3; Q7Z637; Q86V28; P42337; Q9Z0S9; Q9EQZ6; P27671; Q5EBH1; Q5EBH1-1; P52306-5 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Glycoprotein; Golgi apparatus; GTP-binding; Hydrolase; Isopeptide bond; Lipoprotein; Membrane; Methylation; Nucleotide-binding; Nucleus; Palmitate; Prenylation; Proteomics identification; Proto-oncogene; Reference proteome; S-nitrosylation; Ubl conjugation Protein physicochemical properties Chain ID E,F Molecular weight (Da) 28737.2 Length 259 Aromaticity 0.1 Instability index 30.69 Isoelectric point 5.64 Charge (pH=7) -4.15 2D Binding mode Binding energy (Kcal/mol) -8.69 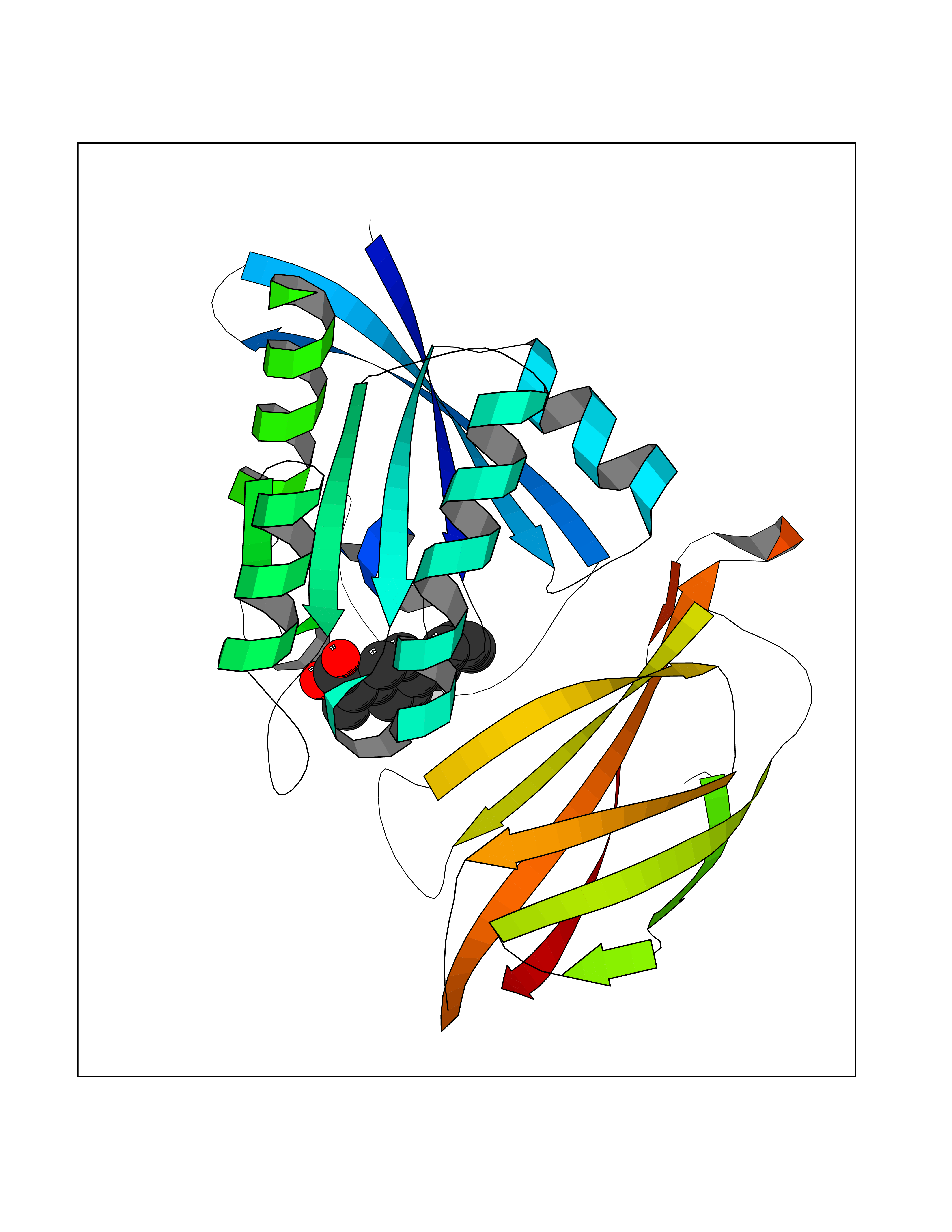 Molscript Map 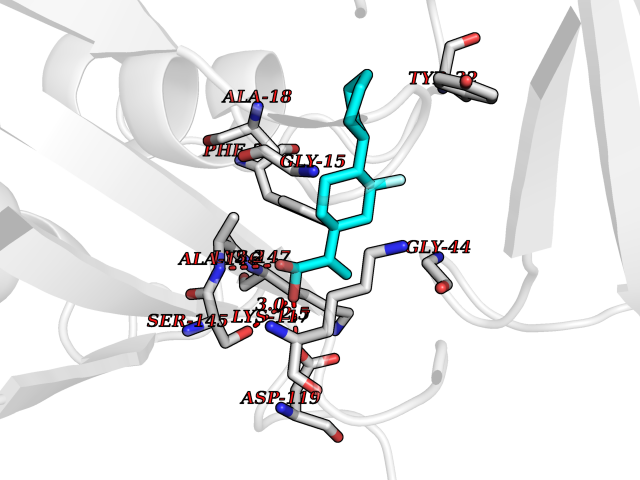 Pymol Map 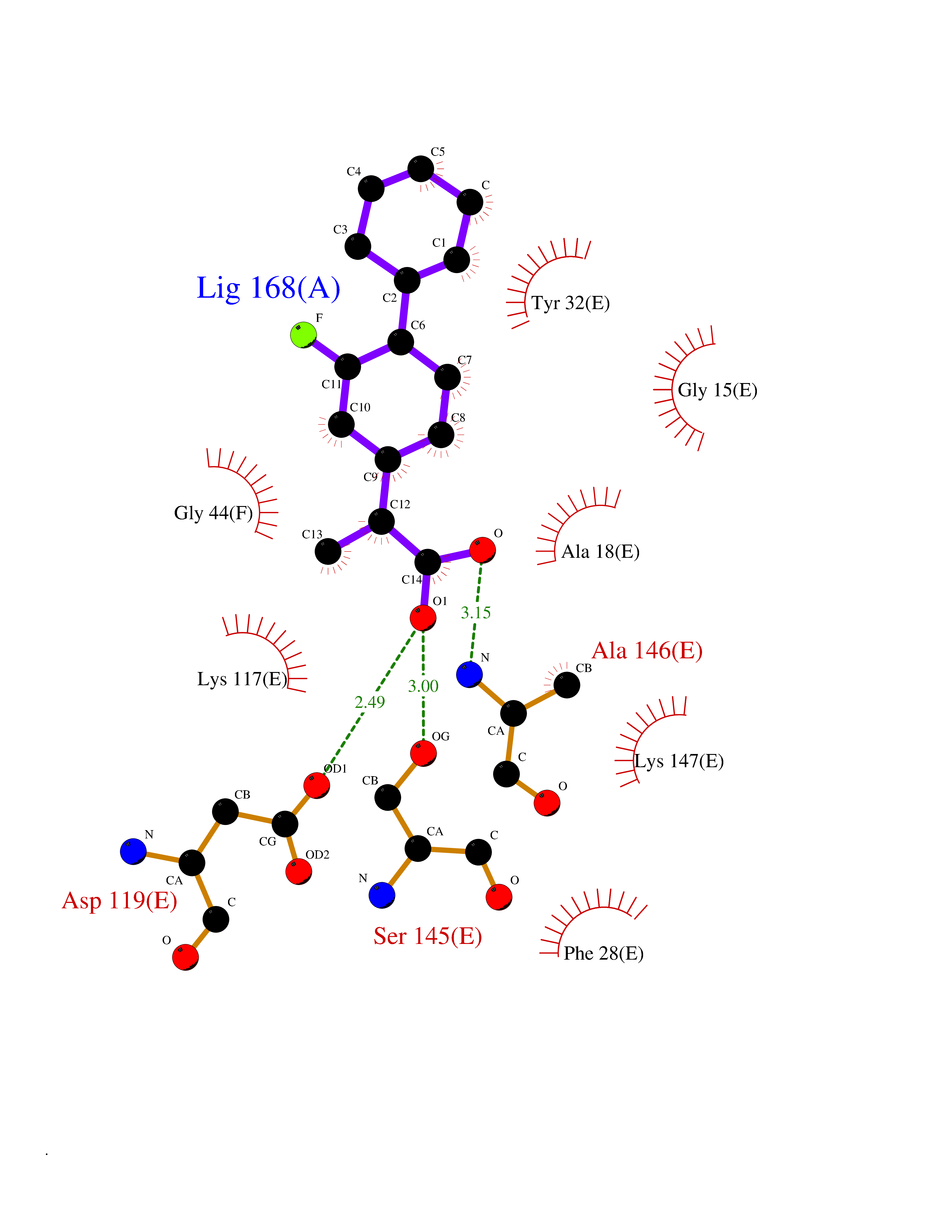 Ligplot Map 3D Binding mode Sequence MTEYKLVVVGAGGVGKSALTIQLIQNHFVDEYDPTIEDSYRKQVVIDGETCLLDILDTAGQEEYSAMRDQYMRTGEGFLCVFAINNTKSFEDIHQYREQIKRVKDSDDVPMVLVGNKCDLAARTVESRQAQDLARSYGIPYIETSAKTRQGVEDAFYTLVREIRQHSVPTKLEVVAATPTSLLISWDAPAVTVFFYIIAYGETGHGVGAFQAFRVPGSKSTATISGLKPGVDYTITVYARGYSKQGPYKPSPISINYRT Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Glucagon-like peptide 1 receptor (GLP1R) | 6X1A | 7.26 | |
Target general information Gen name GLP1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GLP-1R; GLP-1-R; GLP-1 receptor Protein family G-protein coupled receptor 2 family Biochemical class GPCR secretin Function Ligand binding triggers activation of a signaling cascade that leads to the activation of adenylyl cyclase and increased intracellular cAMP levels. Plays a role in regulating insulin secretion in response to GLP-1. G-protein coupled receptor for glucagon-like peptide 1 (GLP-1). Related diseases Lynch syndrome 2 (LYNCH2) [MIM:609310]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:10323887, ECO:0000269|PubMed:10375096, ECO:0000269|PubMed:10386556, ECO:0000269|PubMed:10413423, ECO:0000269|PubMed:10480359, ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10627141, ECO:0000269|PubMed:10660333, ECO:0000269|PubMed:10671064, ECO:0000269|PubMed:10713887, ECO:0000269|PubMed:10777691, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:11139242, ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:11726306, ECO:0000269|PubMed:11748856, ECO:0000269|PubMed:11754112, ECO:0000269|PubMed:11781295, ECO:0000269|PubMed:11793442, ECO:0000269|PubMed:11839723, ECO:0000269|PubMed:11870161, ECO:0000269|PubMed:12095971, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12200596, ECO:0000269|PubMed:12362047, ECO:0000269|PubMed:12373605, ECO:0000269|PubMed:12655562, ECO:0000269|PubMed:12658575, ECO:0000269|PubMed:14635101, ECO:0000269|PubMed:14961575, ECO:0000269|PubMed:15064764, ECO:0000269|PubMed:15139004, ECO:0000269|PubMed:15365995, ECO:0000269|PubMed:15365996, ECO:0000269|PubMed:16083711, ECO:0000269|PubMed:16451135, ECO:0000269|PubMed:17301300, ECO:0000269|PubMed:17510385, ECO:0000269|PubMed:18561205, ECO:0000269|PubMed:20020535, ECO:0000269|PubMed:21120944, ECO:0000269|PubMed:22753075, ECO:0000269|PubMed:7757073, ECO:0000269|PubMed:8566964, ECO:0000269|PubMed:8571956, ECO:0000269|PubMed:8574961, ECO:0000269|PubMed:8797773, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:8993976, ECO:0000269|PubMed:9048925, ECO:0000269|PubMed:9067757, ECO:0000269|PubMed:9218993, ECO:0000269|PubMed:9272156, ECO:0000269|PubMed:9298827, ECO:0000269|PubMed:9311737, ECO:0000269|PubMed:9326924, ECO:0000269|PubMed:9399661, ECO:0000269|PubMed:9559627, ECO:0000269|PubMed:9718327, ECO:0000269|PubMed:9833759, ECO:0000269|PubMed:9927034, ECO:0000269|Ref.5}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mismatch repair cancer syndrome 1 (MMRCS1) [MIM:276300]: An autosomal recessive form of mismatch repair cancer syndrome, a childhood cancer predisposition syndrome encompassing a broad tumor spectrum. This includes hematological malignancies, central nervous system tumors, Lynch syndrome-associated malignancies such as colorectal tumors as well as multiple intestinal polyps, embryonic tumors and rhabdomyosarcoma. Multiple cafe-au-lait macules, a feature reminiscent of neurofibromatosis type 1, are often found as first manifestation of the underlying cancer. {ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:17440981, ECO:0000269|PubMed:7661930}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muir-Torre syndrome (MRTES) [MIM:158320]: Rare autosomal dominant disorder characterized by sebaceous neoplasms and visceral malignancy. {ECO:0000269|PubMed:8751876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in MLH1 may contribute to lobular carcinoma in situ (LCIS), a non-invasive neoplastic disease of the breast.; DISEASE: Endometrial cancer (ENDMC) [MIM:608089]: A malignancy of endometrium, the mucous lining of the uterus. Most endometrial cancers are adenocarcinomas, cancers that begin in cells that make and release mucus and other fluids. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Some epigenetic changes can be transmitted unchanged through the germline (termed 'epigenetic inheritance'). Evidence that this mechanism occurs in humans is provided by the identification of individuals in whom 1 allele of the MLH1 gene is epigenetically silenced throughout the soma (implying a germline event). These individuals are affected by Lynch syndrome but does not have identifiable mutations in MLH1, even though it is silenced, which demonstrates that an epimutation can phenocopy a genetic disease.; DISEASE: Colorectal cancer (CRC) [MIM:114500]: A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history. {ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12655564, ECO:0000269|PubMed:14504054, ECO:0000269|PubMed:15184898, ECO:0000269|PubMed:18033691, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:9032648, ECO:0000269|PubMed:9087566, ECO:0000269|PubMed:9611074}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09043; DB09045; DB15650; DB01276; DB00040; DB16697; DB06655; DB09265; DB13928; DB14027; DB15171 Interacts with A8MQ03; Q07627; Q8IUG1; P60409; P60410; P60411; Q9BYP8; P26371; Q7Z3S9; P0DPK4 EC number NA Uniprot keywords 3D-structure; ADP-ribosylation; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 45579.6 Length 390 Aromaticity 0.16 Instability index 39.66 Isoelectric point 6.73 Charge (pH=7) -0.68 2D Binding mode Binding energy (Kcal/mol) -8.96 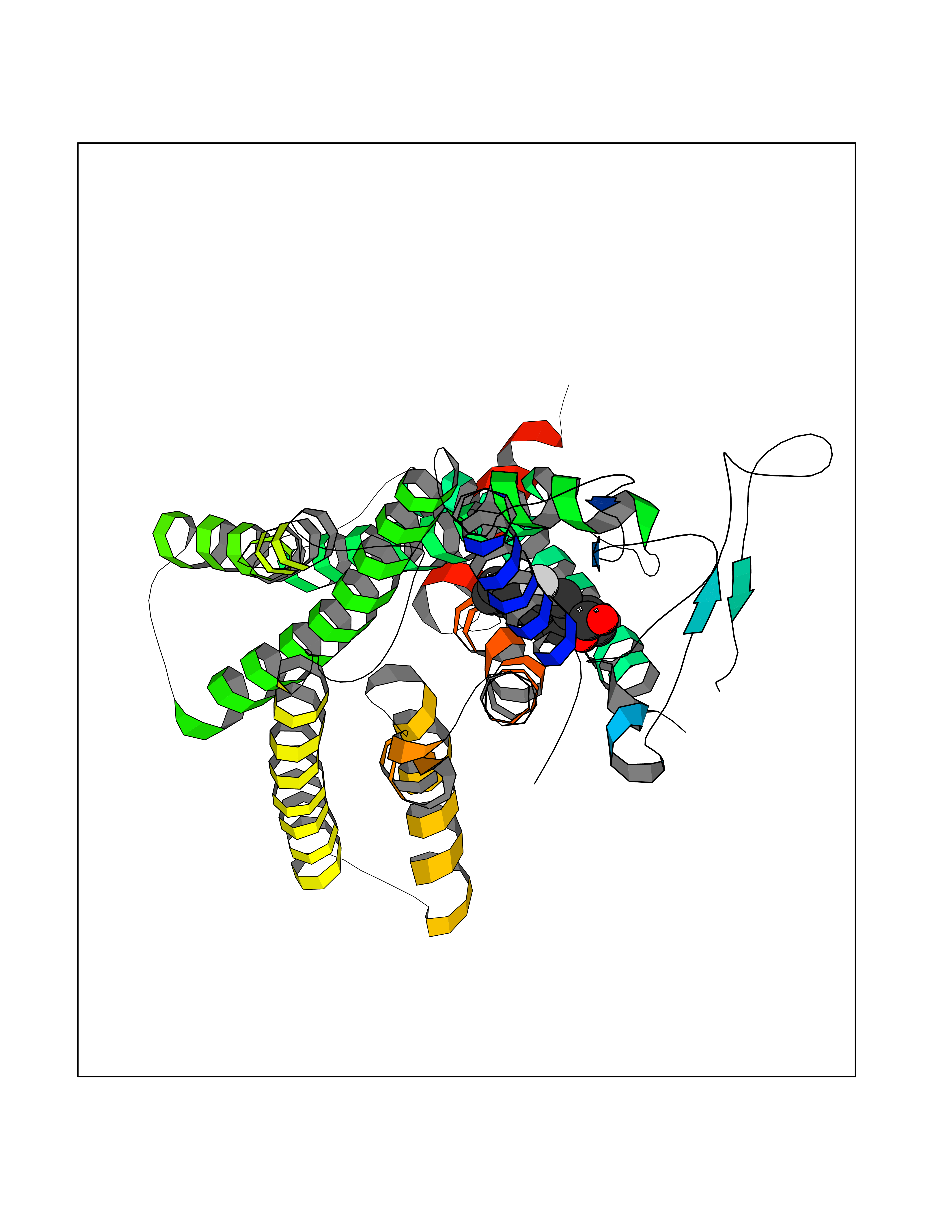 Molscript Map 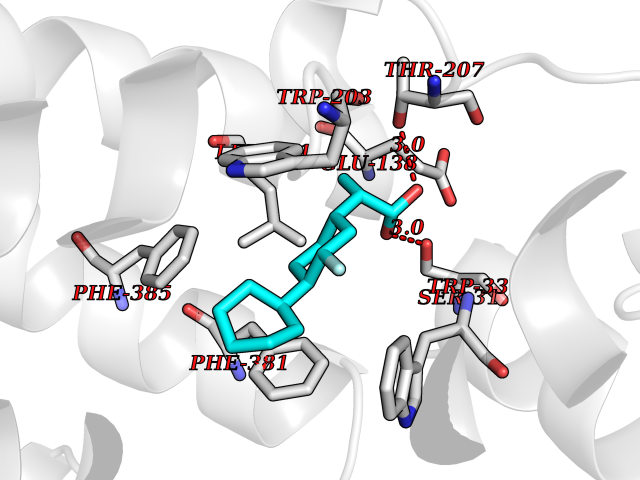 Pymol Map 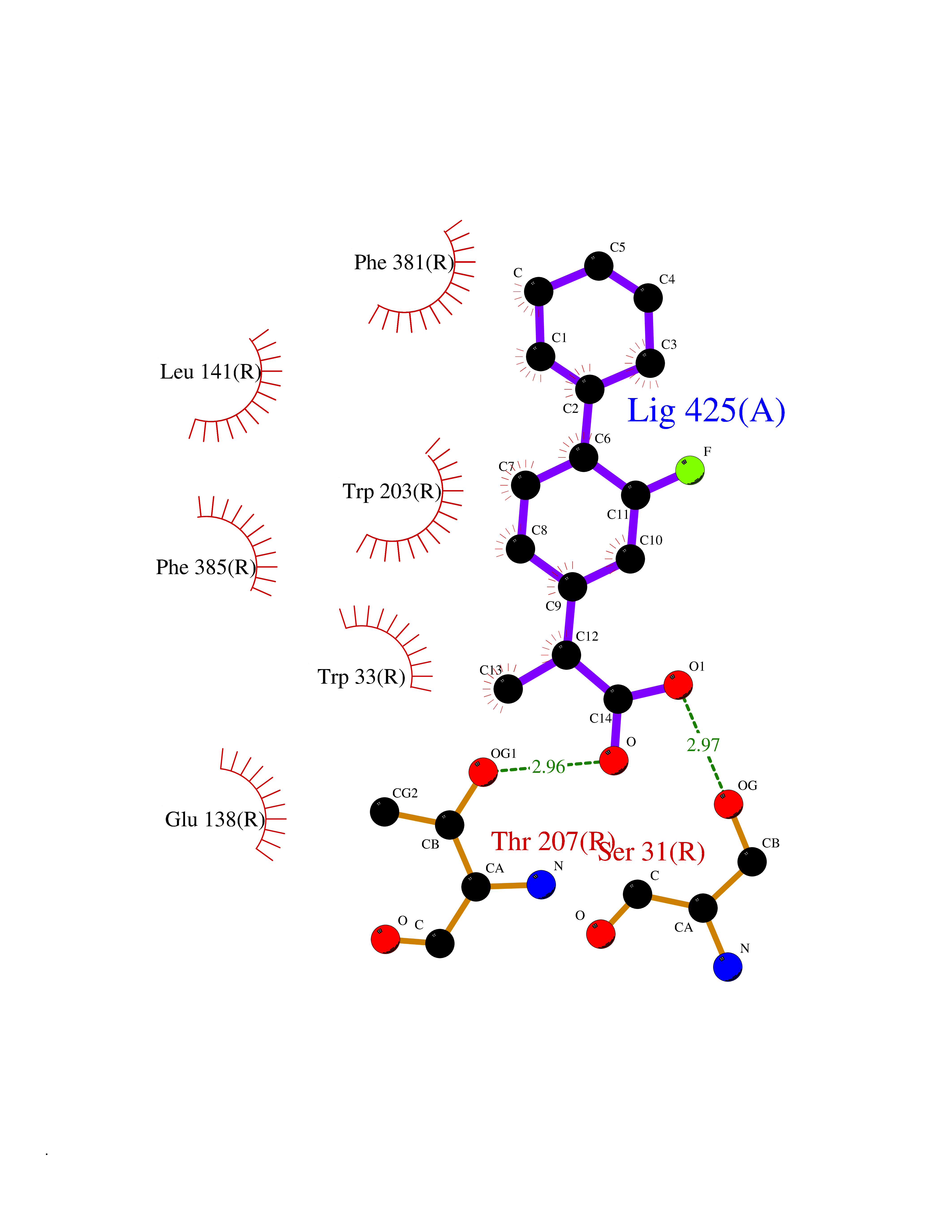 Ligplot Map 3D Binding mode Sequence ATVSLWETVQKWREYRRQCQRSLTEDPPPATDLFCNRTFDEYACWPDGEPGSFVNVSCPWYLPWASSVPQGHVYRFCTAEGLWLQKDNSSLPWRDLSECEESSPEEQLLFLYIIYTVGYALSFSALVIASAILLGFRHLHCTRNYIHLNLFASFILRALSVFIKDAALKWMYSTAAQQHQWDGLLSYQDSLSCRLVFLLMQYCVAANYYWLLVEGVYLYTLLAFSVFSEQWIFRLYVSIGWGVPLLFVVPWGIVKYLYEDEGCWTRNSNMNYWLIIRLPILFAIGVNFLIFVRVICIVVSKLKANLMCKTDIKCRLAKSTLTLIPLLGTHEVIFAFVMDEHARGTLRFIKLFTELSFTSFQGLMVAILYCFVNNEVQLEFRKSWERWRLE Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Gamma-aminobutyric acid type B receptor subunit 1 | 4MS4 | 7.25 | |
Target general information Gen name GABBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPRC3B;GPR51 Protein family G-protein coupled receptor 3 family, GABA-B receptor subfamily Biochemical class Signaling protein / antagonist Function G-protein coupled GABA receptor activity. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08891; DB08892; DB00181; DB00363; DB02530; DB05010; DB09072 Interacts with Q9UBS5; Q9UBS5-2; P46459; Q86UR5 EC number NA Uniprot keywords 3D-structure; Cell membrane; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Intellectual disability; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 46502.1 Length 408 Aromaticity 0.12 Instability index 50.05 Isoelectric point 5.78 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -9.16 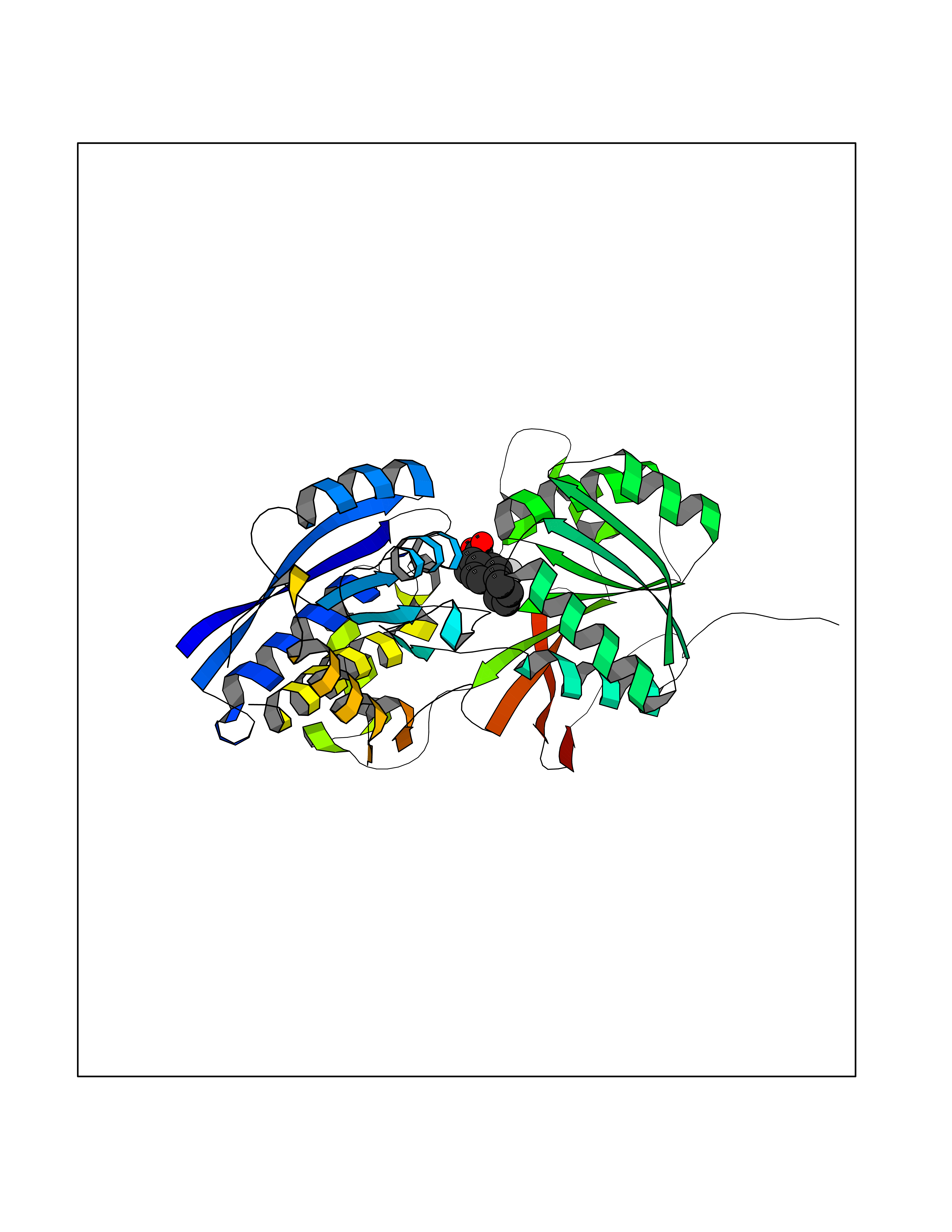 Molscript Map 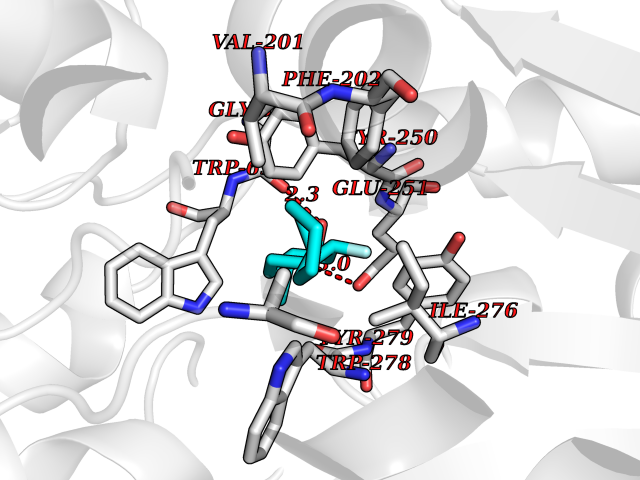 Pymol Map 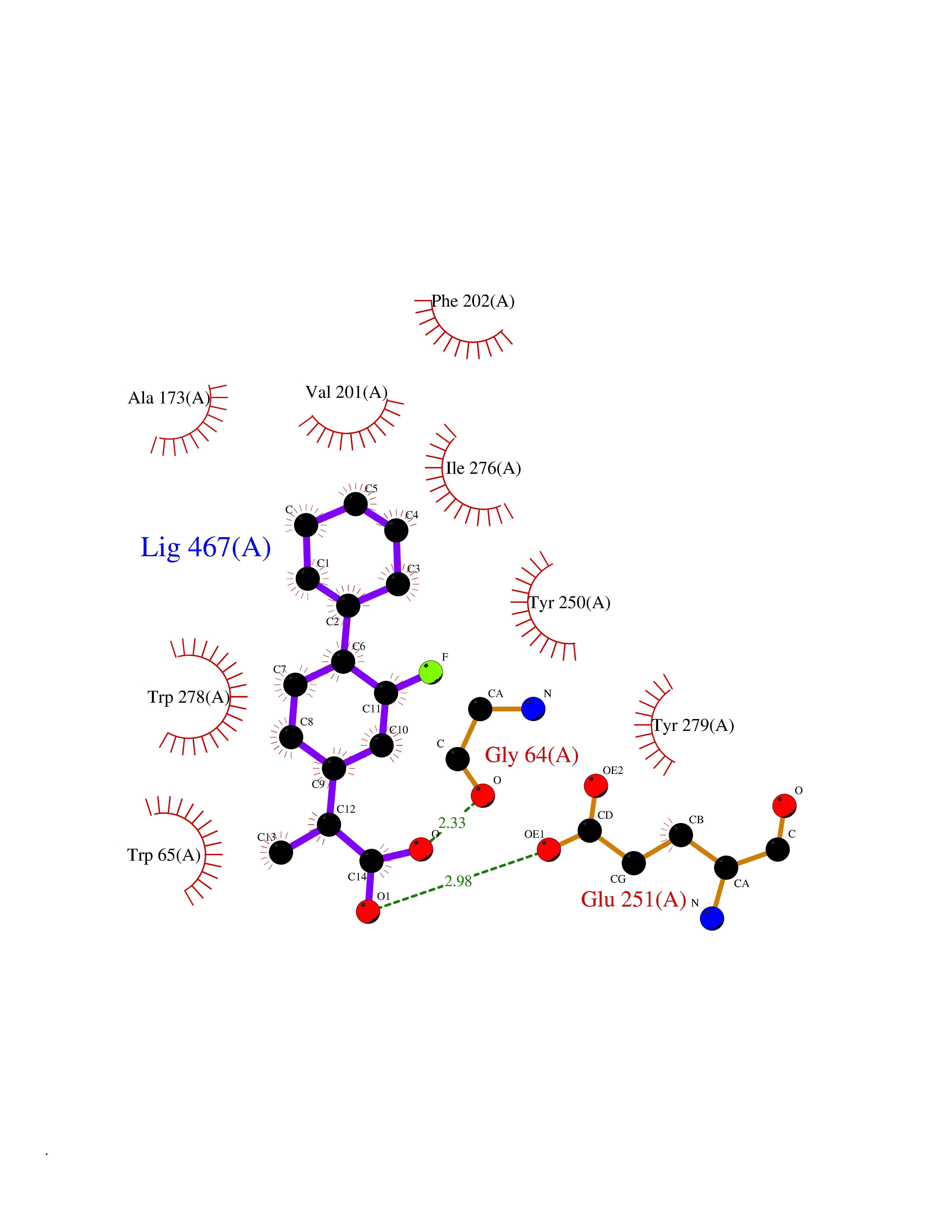 Ligplot Map 3D Binding mode Sequence RRAVYIGALFPMSGGWPGGQACQPAVEMALEDVNSRRDILPDYELKLIHHDSKCDPGQATKYLYELLYNDPIKIILMPGCSSVSTLVAEAARMWNLIVLSYGSSSPALSNRQRFPTFFRTHPSATLHNPTRVKLFEKWGWKKIATIQQTTEVFTSTLDDLEERVKEAGIEITFRQSFFSDPAVPVKNLKRQDARIIVGLFYETEARKVFCEVYKERLFGKKYVWFLIGWYADNWFKIYDPSINCTVDEMTEAVEGHITTEIVMLNPANTRSISNMTSQEFVEKLTKRLKRHPEETGGFQEAPLAYDAIWALALALNKTSRLEDFNYNNQTITDQIYRAMNSSSFEGVSGHVVFDASGSRMAWTLIEQLQGGSYKKIGYYDSTKDDLSWSKTDKWIGGSPPADDYKDDD Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Inosine-5'-monophosphate dehydrogenase 1 (IMPDH1) | 1JCN | 7.24 | |
Target general information Gen name IMPDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Superoxide-inducible protein 12; SOI12; Probable inosine-5'-monophosphate dehydrogenase IMD1; NAD-dependent inosine monophosphate dehydrogenase; Inosine dehydrogenase; IMPDH-I; IMPDH 1; IMPDH; IMPD1; Protein family IMPDH/GMPR family Biochemical class CH-OH donor oxidoreductase Function Could also have a single-stranded nucleic acid-binding activity and could play a role in RNA and/or DNA metabolism. It may also have a role in the development of malignancy and the growth progression of some tumors. Catalyzes the conversion of inosine 5'-phosphate (IMP) to xanthosine 5'-phosphate (XMP), the first committed and rate-limiting step in the de novo synthesis of guanine nucleotides, and therefore plays an important role in the regulation of cell growth. Related diseases Retinitis pigmentosa 10 (RP10) [MIM:180105]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:11875049, ECO:0000269|PubMed:11875050, ECO:0000269|PubMed:16384941}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leber congenital amaurosis 11 (LCA11) [MIM:613837]: A severe dystrophy of the retina, typically becoming evident in the first years of life. Visual function is usually poor and often accompanied by nystagmus, sluggish or near-absent pupillary responses, photophobia, high hyperopia and keratoconus. {ECO:0000269|PubMed:16384941}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03948; DB00993; DB01033; DB00688; DB01024; DB00157; DB00811; DB06408; DB06103 Interacts with Q96D03; P20839-3; P12268; O75928-2; Q9Y4B4; P78317; Q7KZS0 EC number EC 1.1.1.205 Uniprot keywords 3D-structure; Alternative splicing; CBS domain; Cytoplasm; Direct protein sequencing; Disease variant; DNA-binding; GMP biosynthesis; Leber congenital amaurosis; Metal-binding; Methylation; NAD; Nucleus; Oxidoreductase; Phosphoprotein; Potassium; Proteomics identification; Purine biosynthesis; Reference proteome; Repeat; Retinitis pigmentosa; RNA-binding Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42447.5 Length 395 Aromaticity 0.06 Instability index 33.17 Isoelectric point 5.61 Charge (pH=7) -6.1 2D Binding mode Binding energy (Kcal/mol) -7.79 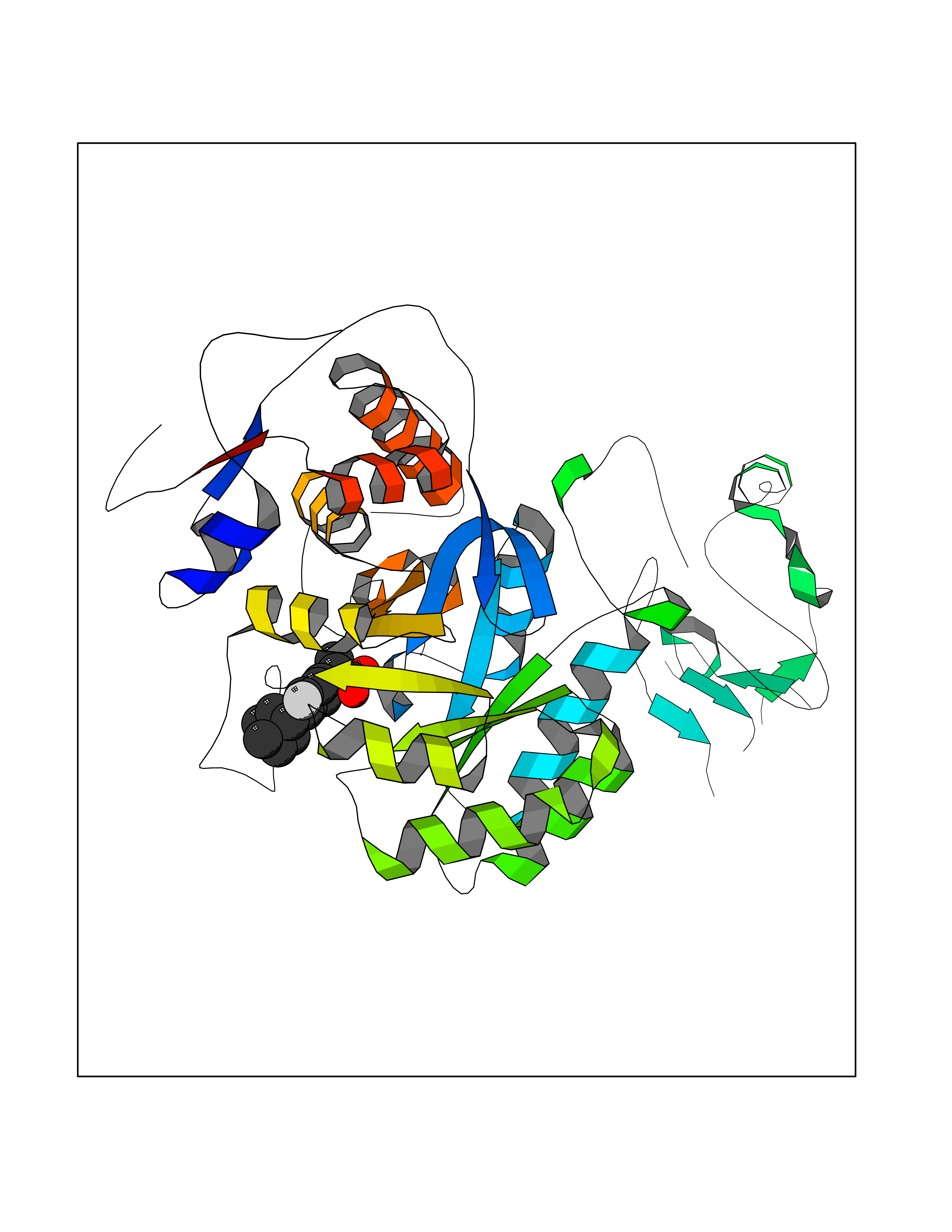 Molscript Map 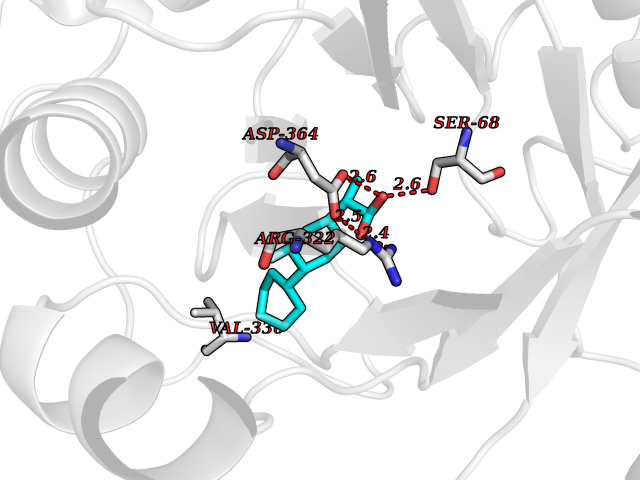 Pymol Map 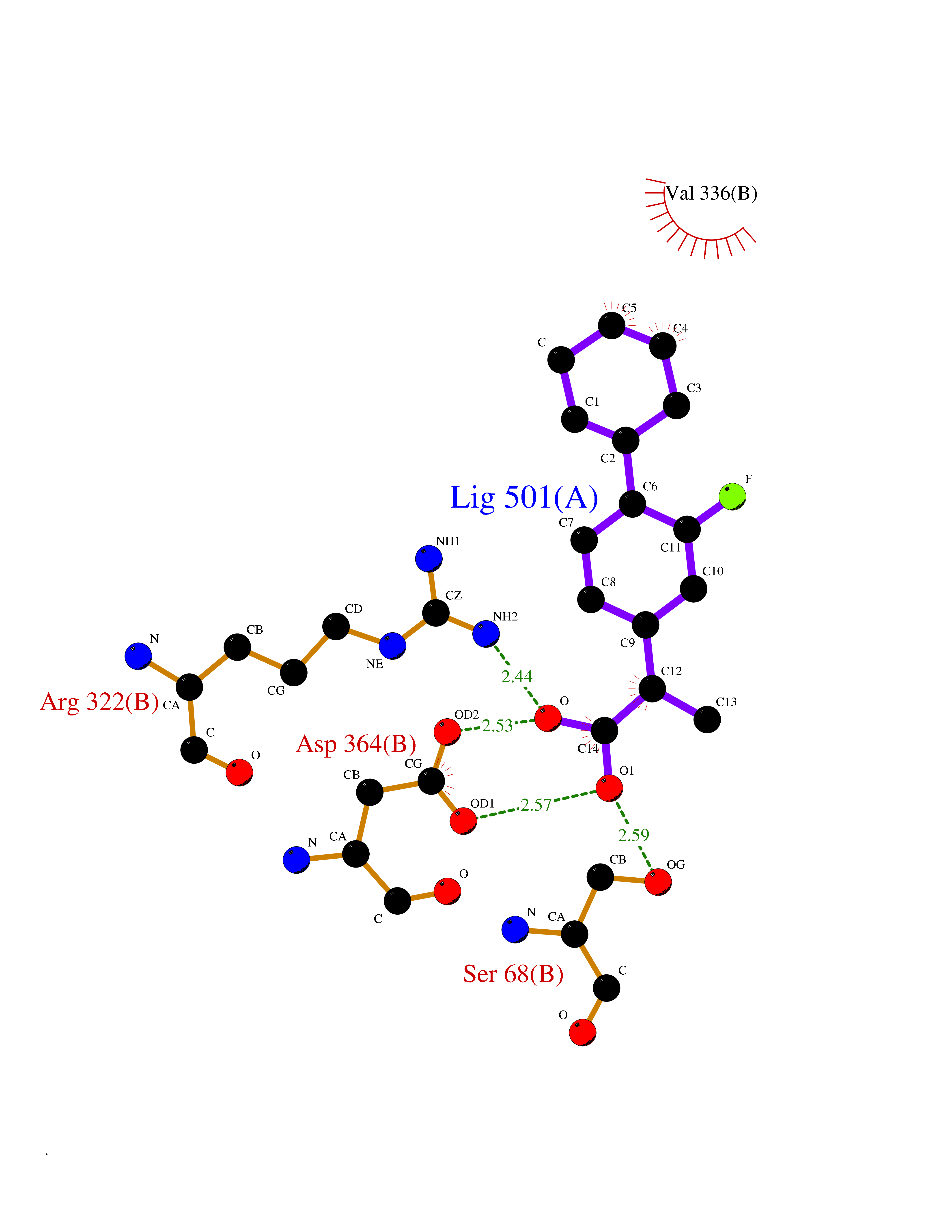 Ligplot Map 3D Binding mode Sequence TGYVPEDGLTAQQLFASADDLTYNDFLILPGFIDFIADEVDLTSALTRKITLKTPLISSPMDTVTEADMAIAMALMGGIGFIHHNCTPEFQANEVRKVKNFEQGFITDPVVLSPGIPITEVGIVTSRDIDPRIELVVAPAGVTLKEANEILQRSKKGKLPIVNDCDELVRTDLKKNRDYPLASKDSQKQLLCGAAVGTREDDKYRLDLLTQAGVDVIVLDSSQGNSVYQIAMVHYIKQKYPHLQVIGGNVVTAAQAKNLIDAGVDGLRVGMGCGSICITQEVMACGRPQGTAVYKVAEYARRFGVPIIADGGIQTVGHVVKALALGASTVMMGSLLAATTEAPGEKGSIQKFVPYLIAGIQHGCQDIGARSLSVLRSMMYSGELKFEKRTMSAQI Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Phosphodiesterase 9 (PDE9) | 4E90 | 7.24 | |
Target general information Gen name PDE9A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms High affinity cGMPspecific 3',5'cyclic phosphodiesterase 9A; High affinity cGMP-specific 3',5'-cyclic phosphodiesterase 9A Protein family Cyclic nucleotide phosphodiesterase family, PDE9 subfamily Biochemical class Phosphoric diester hydrolase Function Highly specific: compared to other members of the cyclic nucleotide phosphodiesterase family, has the highest affinity and selectivity for cGMP. Specifically regulates natriuretic-peptide-dependent cGMP signaling in heart, acting as a regulator of cardiac hypertrophy in myocytes and muscle. Does not regulate nitric oxide-dependent cGMP in heart. Additional experiments are required to confirm whether its ability to hydrolyze natriuretic-peptide-dependent cGMP is specific to heart or is a general feature of the protein. In brain, involved in cognitive function, such as learning and long-term memory. Specifically hydrolyzes the second messenger cGMP, which is a key regulator of many important physiological processes. Related diseases Macular degeneration, age-related, 7 (ARMD7) [MIM:610149]: A form of age-related macular degeneration, a multifactorial eye disease and the most common cause of irreversible vision loss in the developed world. In most patients, the disease is manifest as ophthalmoscopically visible yellowish accumulations of protein and lipid that lie beneath the retinal pigment epithelium and within an elastin-containing structure known as Bruch membrane. {ECO:0000269|PubMed:17053108, ECO:0000269|PubMed:17053109}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Cerebral arteriopathy, autosomal recessive, with subcortical infarcts and leukoencephalopathy (CARASIL) [MIM:600142]: A cerebrovascular disease characterized by non-hypertensive arteriopathy of cerebral small vessels with subcortical infarcts, alopecia, and spondylosis. Small cerebral arteries show arteriosclerotic changes, fibrous intimal proliferation, and hyaline degeneration with splitting of the intima and/or the internal elastic membrane. Neurologic features include progressive dementia, gait disturbances, extrapyramidal and pyramidal signs, and demyelination of the cerebral white matter with sparing of U fibers. {ECO:0000269|PubMed:19387015}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cerebral arteriopathy, autosomal dominant, with subcortical infarcts and leukoencephalopathy, 2 (CADASIL2) [MIM:616779]: A cerebrovascular disease characterized by multiple subcortical infarcts, pseudobulbar palsy, dementia, and the presence of granular deposits in small cerebral arteries producing ischemic stroke. {ECO:0000269|PubMed:26063658}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07954; DB00201; DB03597; DB09283 Interacts with O95817; P49759; Q49AN0; Q9H8Y8; P60410; Q9BYR5; Q9BYQ4; Q14657; Q8NAJ2; P25791; Q9BRA0; Q7Z3S9; O76083-2; Q96FC7-2; P49888; Q13049; Q9BRU9; Q9Y260; O95817; Q16543; P49759-3; Q49AN0; A8MQ03; Q15051-2; Q63ZY3; O76011; Q07627; Q8IUG1; P60409; P60410; P60411; P59991; P60328; P26371; Q14657; Q16649; O76083-2; Q96FC7; Q99633; O00560; P49888; Q13049; O15205; P61964; O76083-2; Q13049 EC number EC 3.1.4.35 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; cGMP; Cytoplasm; Endoplasmic reticulum; Golgi apparatus; Hydrolase; Magnesium; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 38689.4 Length 328 Aromaticity 0.11 Instability index 53.51 Isoelectric point 5.14 Charge (pH=7) -16.23 2D Binding mode Binding energy (Kcal/mol) -8.62 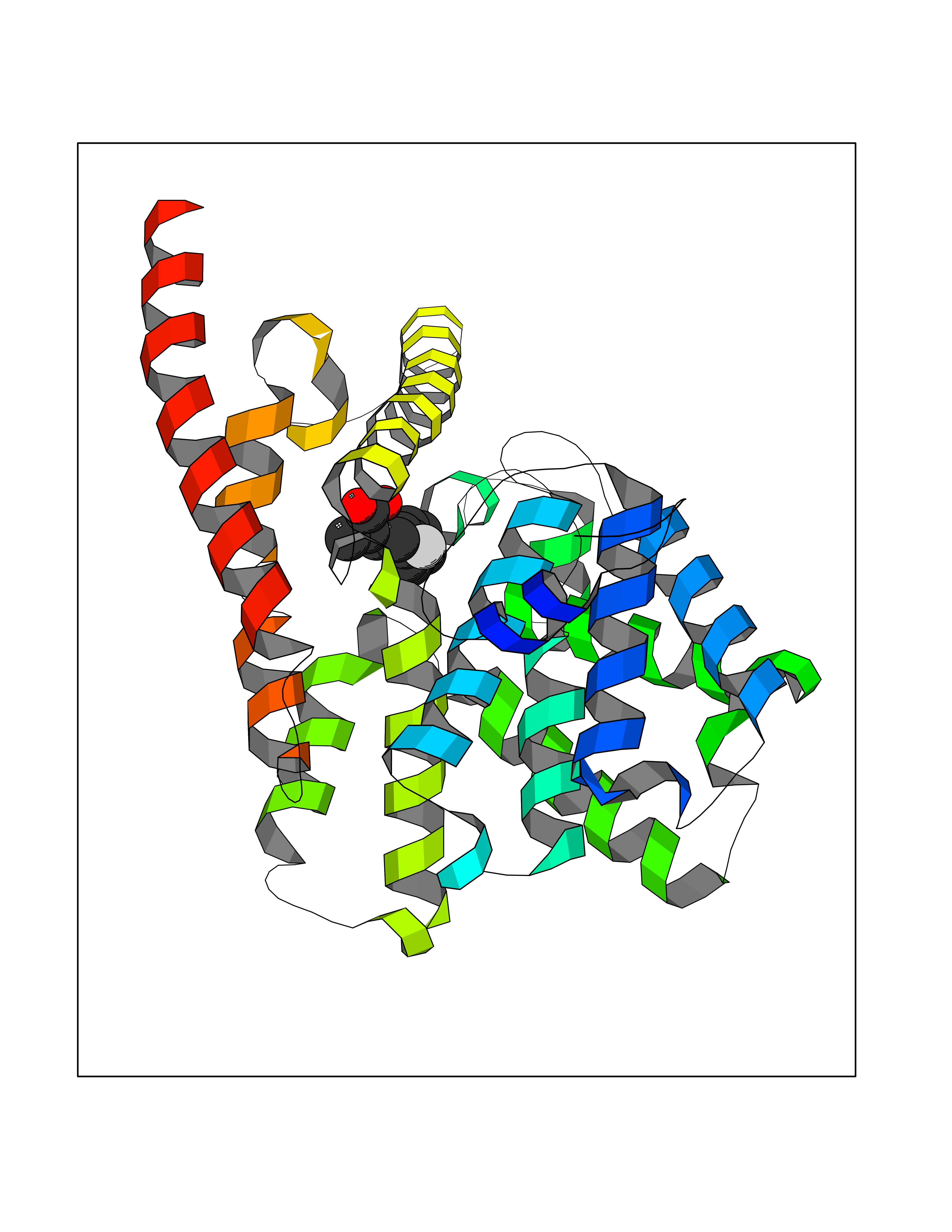 Molscript Map 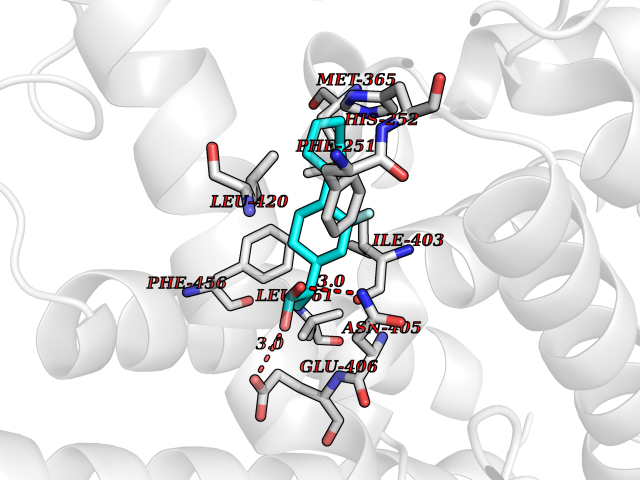 Pymol Map 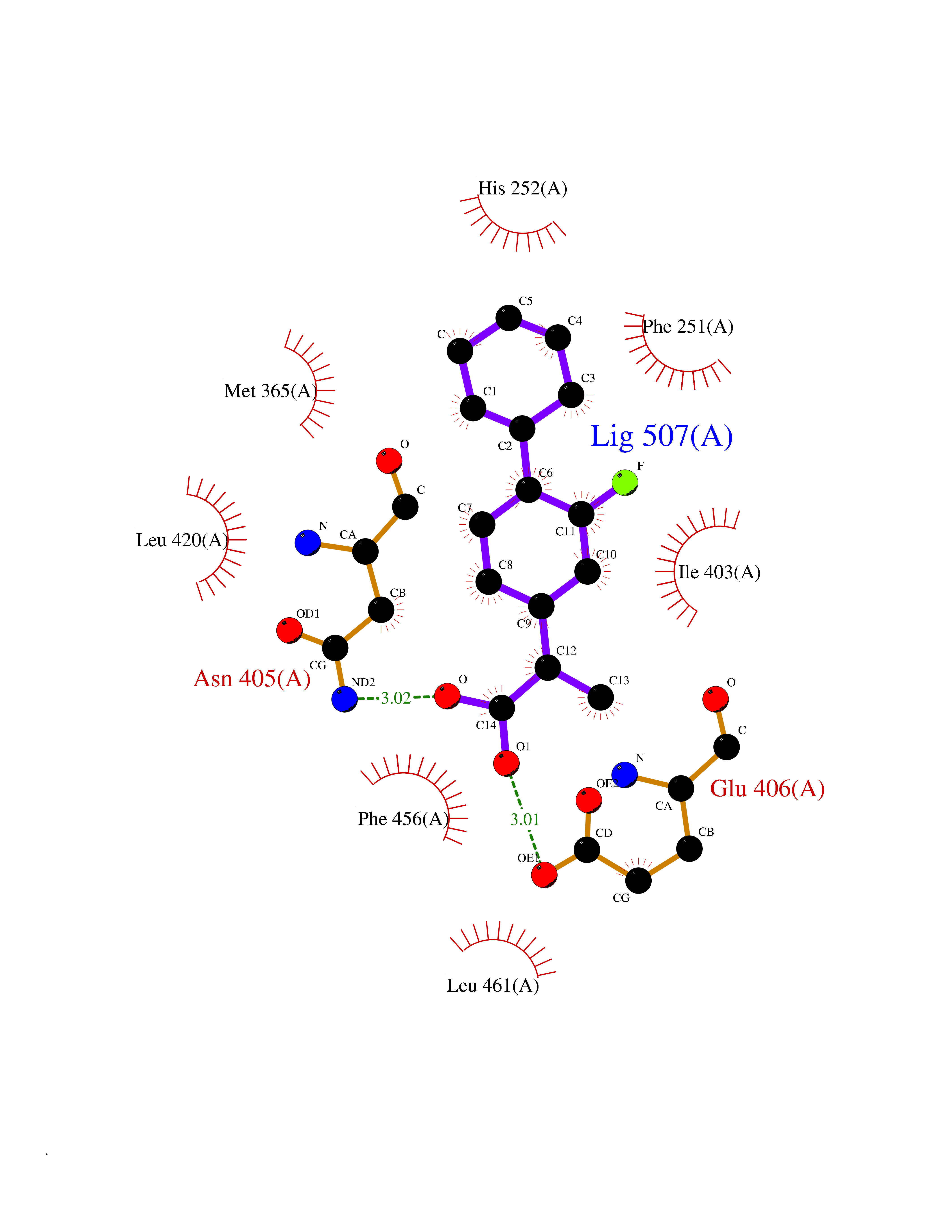 Ligplot Map 3D Binding mode Sequence GSHMTYPKYLLSPETIEALRKPTFDVWLWEPNEMLSCLEHMYHDLGLVRDFSINPVTLRRWLFCVHDNYRNNPFHNFRHCFCVAQMMYSMVWLCSLQEKFSQTDILILMTAAICHDLDHPGYNNTYQINARTELAVRYNDISPLENHHCAVAFQILAEPECNIFSNIPPDGFKQIRQGMITLILATDMARHAEIMDSFKEKMENFDYSNEEHMTLLKMILIKCCDISNEVRPMEVAEPWVDCLLEEYFMQSDREKSEGLPVAPFMDRDKVTKATAQIGFIKFVLIPMFETVTKLFPMVEEIMLQPLWESRDRYEELKRIDDAMKELQK Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Eukaryotic translation initiation factor 2-alpha kinase 4 (EIF2AK4) | 7QQ6 | 7.23 | |
Target general information Gen name EIF2AK4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms eIF-2-alpha kinase GCN2; KIAA1338; GCN2-like protein; GCN2 Protein family Protein kinase superfamily, Ser/Thr protein kinase family, GCN2 subfamily Biochemical class NA Function Metabolic-stress sensing protein kinase that phosphorylates the alpha subunit of eukaryotic translation initiation factor 2 (eIF-2-alpha/EIF2S1) on 'Ser-52' in response to low amino acid availability. Plays a role as an activator of the integrated stress response (ISR) required for adapatation to amino acid starvation. Converts phosphorylated eIF-2-alpha/EIF2S1 either to a competitive inhibitor of the translation initiation factor eIF-2B, leading to a global protein synthesis repression, and thus to a reduced overall utilization of amino acids, or to a translational initiation activation of specific mRNAs, such as the transcriptional activator ATF4, and hence allowing ATF4-mediated reprogramming of amino acid biosynthetic gene expression to alleviate nutrient depletion. Binds uncharged tRNAs (By similarity). Involved in cell cycle arrest by promoting cyclin D1 mRNA translation repression after the unfolded protein response pathway (UPR) activation or cell cycle inhibitor CDKN1A/p21 mRNA translation activation in response to amino acid deprivation. Plays a role in the consolidation of synaptic plasticity, learning as well as formation of long-term memory. Plays a role in neurite outgrowth inhibition. Plays a proapoptotic role in response to glucose deprivation. Promotes global cellular protein synthesis repression in response to UV irradiation independently of the stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) and p38 MAPK signaling pathways (By similarity). Plays a role in the antiviral response against alphavirus infection; impairs early viral mRNA translation of the incoming genomic virus RNA, thus preventing alphavirus replication (By similarity). Related diseases Pulmonary venoocclusive disease 2, autosomal recessive (PVOD2) [MIM:234810]: A disease characterized by widespread fibrous obstruction and intimal thickening of septal veins and preseptal venules, a low diffusing capacity for carbon monoxide, occult alveolar hemorrhage, and nodular ground-glass opacities, septal lines and lymph node enlargement showed by high-resolution computed tomography of the chest. It is frequently associated with pulmonary capillary dilatation and proliferation, and is a rare and devastating cause of pulmonary hypertension. {ECO:0000269|PubMed:24135949, ECO:0000269|PubMed:24292273}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with NA EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Activation of host autophagy by virus; Activator; Adaptive immunity; Alternative splicing; Antiviral defense; ATP-binding; Cell cycle; Coiled coil; Cytoplasm; Disease variant; Growth arrest; Host-virus interaction; Immunity; Kinase; Neurogenesis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; RNA-binding; Serine/threonine-protein kinase; Stress response; Transferase; Translation regulation; tRNA-binding Protein physicochemical properties Chain ID A Molecular weight (Da) 31090.3 Length 269 Aromaticity 0.1 Instability index 42.78 Isoelectric point 7.75 Charge (pH=7) 1.43 2D Binding mode Binding energy (Kcal/mol) -8.8  Molscript Map 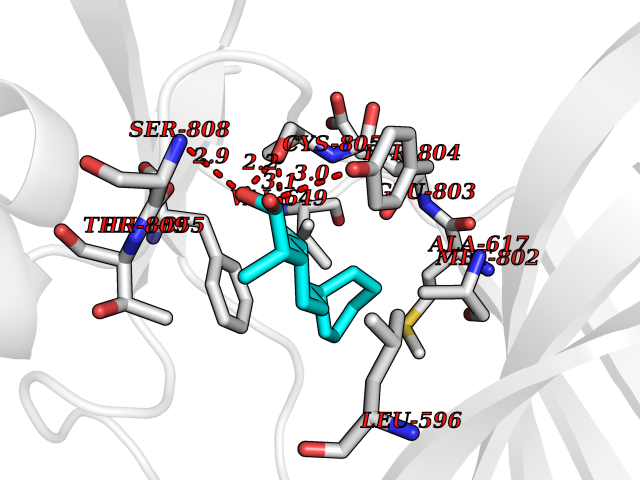 Pymol Map 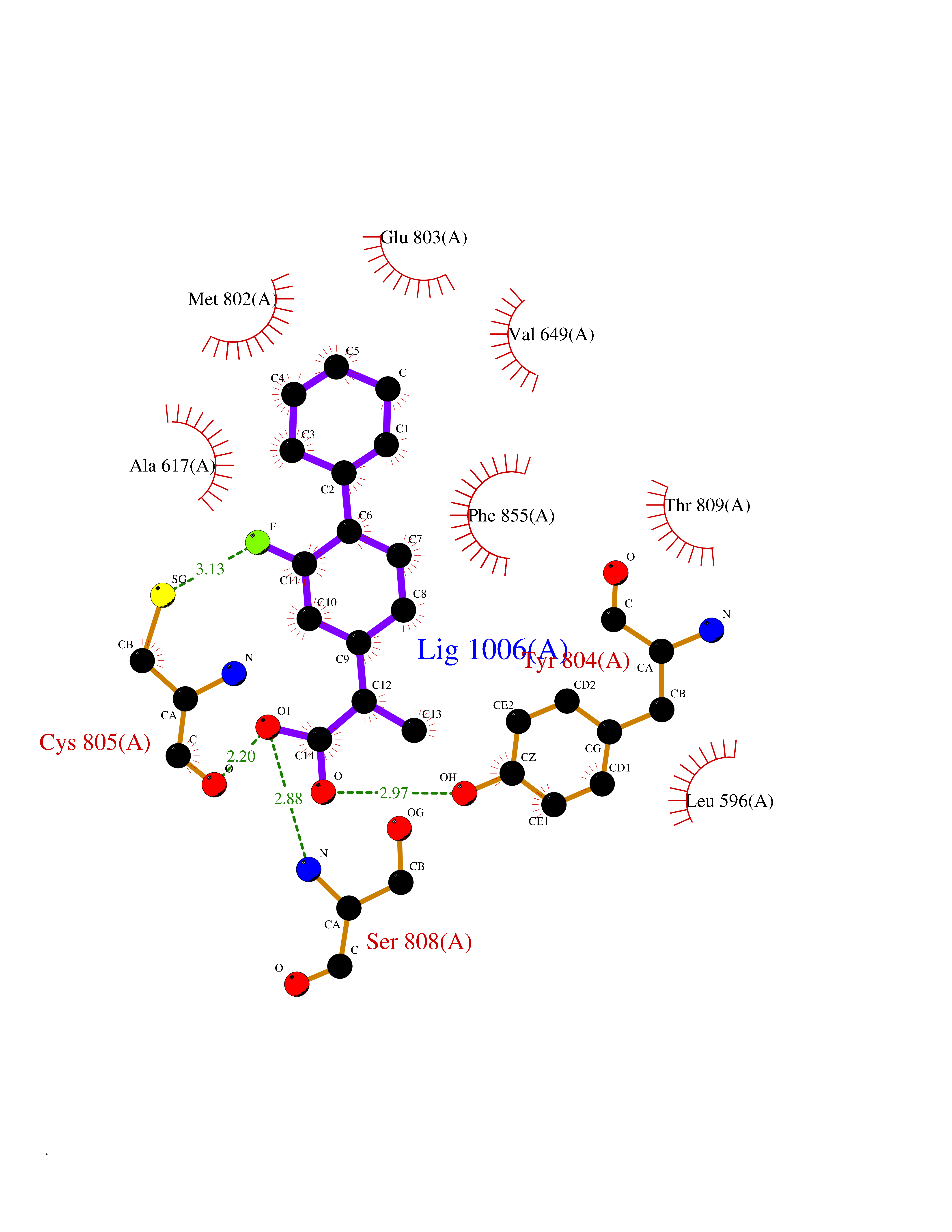 Ligplot Map 3D Binding mode Sequence SRYFIEFEELQLLGKGAFGAVIKVQNKLDGCCYAVKRIPINPASRQFRRIKGEVTLLSRLHHENIVRYYNAWIERHERPVHYLYIQMEYCEKSTLRDTIDQGLYRDTVRLWRLFREILDGLAYIHEKGMIHRNLKPVNIFLDSDDHVKIGDFGLIKSDPSGHLTGMVGTALYVSPEVQGSTKSAYNQKVDLFSLGIIFFEMSYHPMVTASERIFVLNQLRDPTSPKFPEDFDDGEHAKQKSVISWLLNHDPAKRPTATELLKSELLPPP Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Leukocyte immunoglobulin-like receptor B2 (LILRB2) | 6BCS | 7.23 | |
Target general information Gen name LILRB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LIR-2; Leukocyte immunoglobulin-like receptor 2; CD85 antigen-like family member D; Immunoglobulin-like transcript 4; ILT-4; Monocyte/macrophage immunoglobulin-like receptor 10; MIR-10; DE AltName: CD Protein family NA Biochemical class NA Function Receptor for class I MHC antigens. Recognizes a broad spectrum of HLA-A, HLA-B, HLA-C, HLA-G and HLA-F alleles. Involved in the down-regulation of the immune response and the development of tolerance. Recognizes HLA-G in complex with B2M/beta-2 microglobulin and a nonamer self-peptide (peptide-bound HLA-G-B2M) triggering differentiation of type 1 regulatory T cells and myeloid-derived suppressor cells, both of which actively maintain maternal-fetal tolerance. Competes with CD8A for binding to class I MHC antigens. Inhibits FCGR1A-mediated phosphorylation of cellular proteins and mobilization of intracellular calcium ions. Related diseases Cardiomyopathy, familial hypertrophic, 1 (CMH1) [MIM:192600]: A hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death. {ECO:0000269|PubMed:10065021, ECO:0000269|PubMed:10329202, ECO:0000269|PubMed:10521296, ECO:0000269|PubMed:10563488, ECO:0000269|PubMed:10679957, ECO:0000269|PubMed:10862102, ECO:0000269|PubMed:11113006, ECO:0000269|PubMed:11133230, ECO:0000269|PubMed:11214007, ECO:0000269|PubMed:11424919, ECO:0000269|PubMed:11733062, ECO:0000269|PubMed:11861413, ECO:0000269|PubMed:11968089, ECO:0000269|PubMed:12081993, ECO:0000269|PubMed:12566107, ECO:0000269|PubMed:12590187, ECO:0000269|PubMed:12707239, ECO:0000269|PubMed:12818575, ECO:0000269|PubMed:12820698, ECO:0000269|PubMed:12951062, ECO:0000269|PubMed:12974739, ECO:0000269|PubMed:12975413, ECO:0000269|PubMed:1417858, ECO:0000269|PubMed:15358028, ECO:0000269|PubMed:15483641, ECO:0000269|PubMed:1552912, ECO:0000269|PubMed:15563892, ECO:0000269|PubMed:15856146, ECO:0000269|PubMed:15858117, ECO:0000269|PubMed:16199542, ECO:0000269|PubMed:16267253, ECO:0000269|PubMed:1638703, ECO:0000269|PubMed:16650083, ECO:0000269|PubMed:16938236, ECO:0000269|PubMed:17095604, ECO:0000269|PubMed:17372140, ECO:0000269|PubMed:18175163, ECO:0000269|PubMed:18403758, ECO:0000269|PubMed:1975517, ECO:0000269|PubMed:25182012, ECO:0000269|PubMed:7581410, ECO:0000269|PubMed:7731997, ECO:0000269|PubMed:7848441, ECO:0000269|PubMed:7874131, ECO:0000269|PubMed:7909436, ECO:0000269|PubMed:8250038, ECO:0000269|PubMed:8254035, ECO:0000269|PubMed:8268932, ECO:0000269|PubMed:8282798, ECO:0000269|PubMed:8343162, ECO:0000269|PubMed:8435239, ECO:0000269|PubMed:8483915, ECO:0000269|PubMed:8533830, ECO:0000269|PubMed:8655135, ECO:0000269|PubMed:8899546, ECO:0000269|PubMed:9544842, ECO:0000269|PubMed:9822100, ECO:0000269|PubMed:9829907}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 7A, myosin storage, autosomal dominant (CMYO7A) [MIM:608358]: A skeletal muscle disorder characterized by prominent axial and proximal weakening, spinal stiffness, severe scoliosis, with or without respiratory and cardiac involvement. The age at symptom onset can range from early childhood to late adulthood, and disease severity ranges from asymptomatic to severe muscular weakness and respiratory insufficiency. Histopathological examination shows variable findings including subsarcolemmal hyaline bodies in type 1 fibers. {ECO:0000269|PubMed:14520662, ECO:0000269|PubMed:15136674, ECO:0000269|PubMed:16684601, ECO:0000269|PubMed:17336526}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cardiomyopathy, dilated, 1S (CMD1S) [MIM:613426]: A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. {ECO:0000269|PubMed:11106718, ECO:0000269|PubMed:12379228, ECO:0000269|PubMed:15769782, ECO:0000269|PubMed:18506004, ECO:0000269|PubMed:21127202, ECO:0000269|PubMed:21846512}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, distal, 1 (MPD1) [MIM:160500]: A muscular disorder characterized by early-onset selective weakness of the great toe and ankle dorsiflexors, followed by weakness of the finger extensors. Mild proximal weakness occasionally develops years later after the onset of the disease. {ECO:0000269|PubMed:15322983, ECO:0000269|PubMed:17548557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 7B, myosin storage, autosomal recessive (CMYO7B) [MIM:255160]: A skeletal muscle disorder characterized by the onset of scapuloperoneal muscle weakness in early childhood or young adulthood. Affected individuals have difficulty walking, steppage gait, and scapular winging due to shoulder girdle involvement. The severity and progression of the disorder is highly variable. Most patients develop respiratory insufficiency and restrictive lung disease. Some develop hypertrophic cardiomyopathy. Histopathological examination shows variable findings including subsarcolemmal hyaline bodies in type 1 fibers. {ECO:0000269|PubMed:25666907}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Left ventricular non-compaction 5 (LVNC5) [MIM:613426]: A form of left ventricular non-compaction, a cardiomyopathy due to myocardial morphogenesis arrest and characterized by a hypertrophic left ventricle, a severely thickened 2-layered myocardium, numerous prominent trabeculations, deep intertrabecular recesses, and poor systolic function. Clinical manifestations are variable. Some affected individuals experience no symptoms at all, others develop heart failure. In some cases, left ventricular non-compaction is associated with other congenital heart anomalies. LVNC5 is an autosomal dominant condition. {ECO:0000269|PubMed:18506004}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UKU9; Q86XS5; PRO_0000000092 [P05067]; P30447; Q95J06; P01889; P10321; P17693; P17693-2; P17693-5; P17693-6; P46531; P29350 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative initiation; Alternative promoter usage; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 21853.4 Length 196 Aromaticity 0.1 Instability index 50.2 Isoelectric point 8.21 Charge (pH=7) 1.8 2D Binding mode Binding energy (Kcal/mol) -7.26  Molscript Map 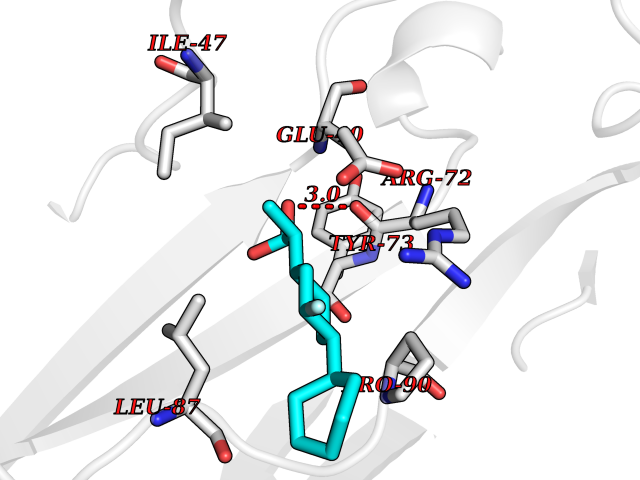 Pymol Map 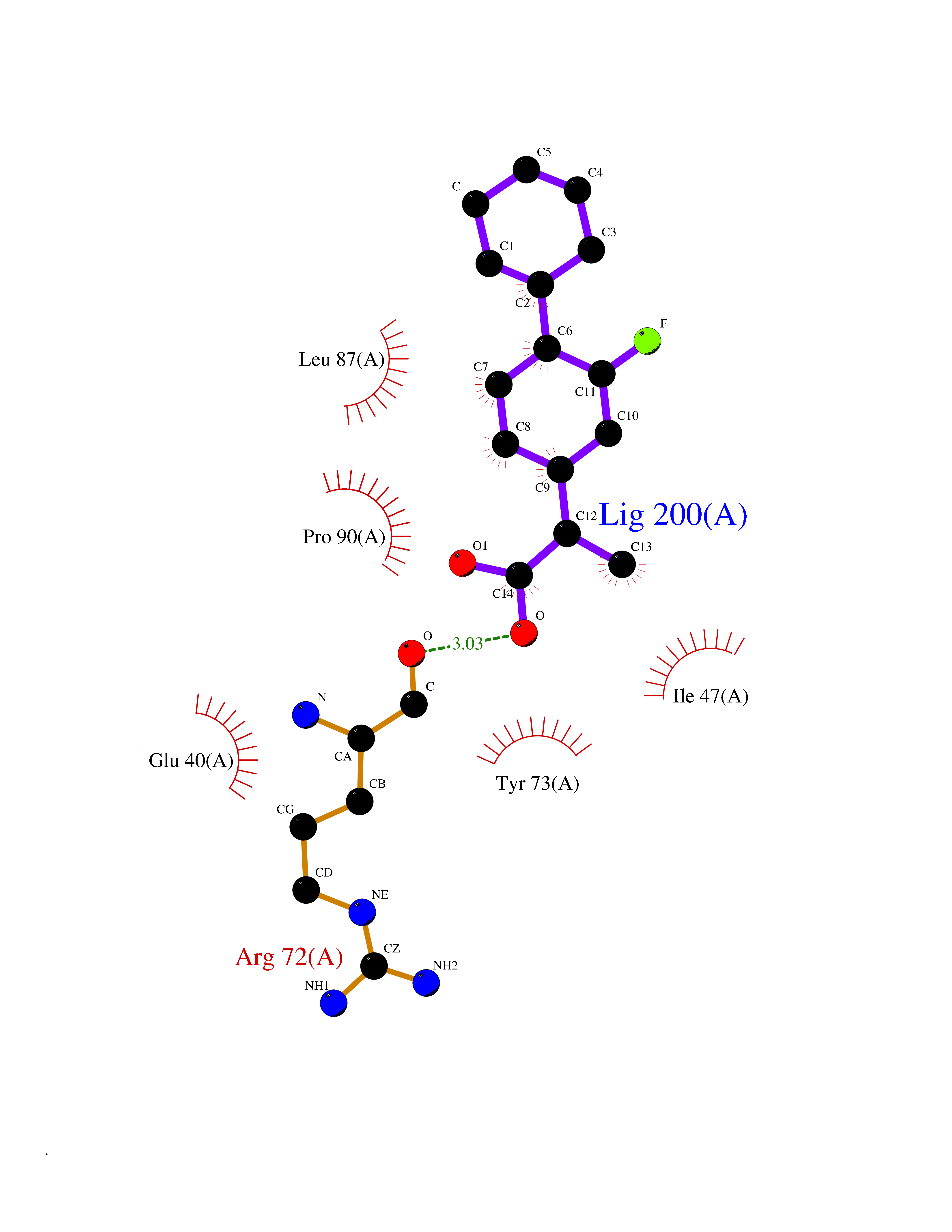 Ligplot Map 3D Binding mode Sequence TIPKPTLWAEPDSVITQGSPVTLSCQGSLEAQEYRLYREKKSASWITRIRPELVKNGQFHIPSITWEHTGRYGCQYYSRARWSELSDPLVLVMTGAYPKPTLSAQPSPVVTSGGRVTLQCESQVAFGGFILCKEGEDEHPQCLNSQPHARGSSRAIFSVGPVSPNRRWSHRCYGYDLNSPYVWSSPSDLLELLVPG Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Kidney-type arginase (ARG2) | 4HZE | 7.22 | |
Target general information Gen name ARG2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Non-hepatic arginase; Human penile arginase; Arginase II; ARG2 Protein family Arginase family Biochemical class Carbon-nitrogen hydrolase Function May play a role in the regulation of extra-urea cycle arginine metabolism and also in down-regulation of nitric oxide synthesis. Extrahepatic arginase functions to regulate L-arginine bioavailability to NO synthase. Since NO synthase is found in the penile corpus cavernosum smooth muscle, the clitoral corpus cavernosum and the vagina, arginase II plays a role in both male and female sexual arousal. It is therefore a potential target for the treatment of male and female sexual arousal disorders. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00125; DB00129; DB03731 Interacts with Q3SYB3 EC number EC 3.5.3.1 Uniprot keywords 3D-structure; Adaptive immunity; Arginine metabolism; Hydrolase; Immunity; Innate immunity; Manganese; Metal-binding; Mitochondrion; Proteomics identification; Reference proteome; Transit peptide; Urea cycle Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 33194.3 Length 306 Aromaticity 0.07 Instability index 33.35 Isoelectric point 5.52 Charge (pH=7) -9.28 2D Binding mode Binding energy (Kcal/mol) -7.94 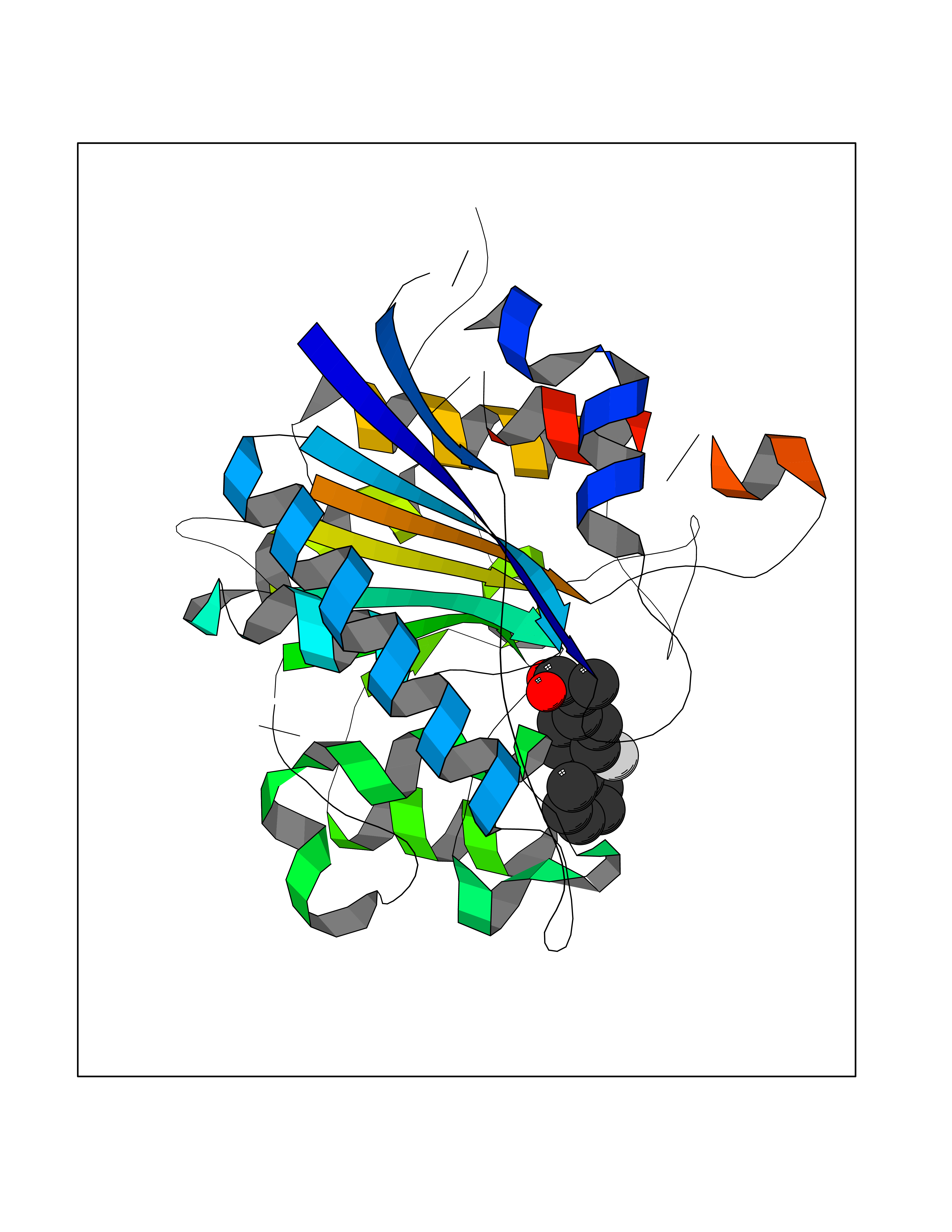 Molscript Map 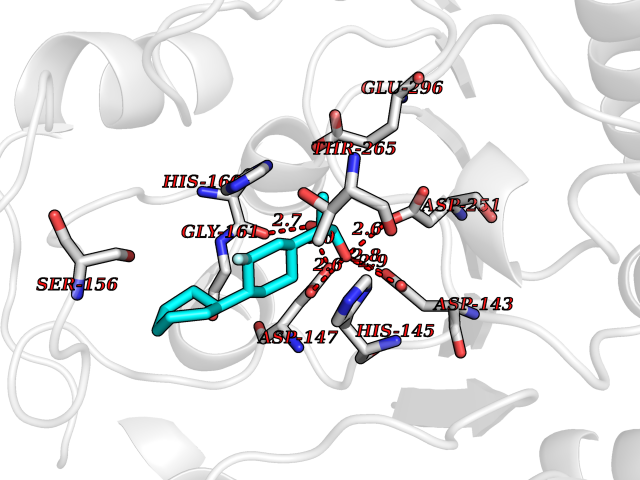 Pymol Map 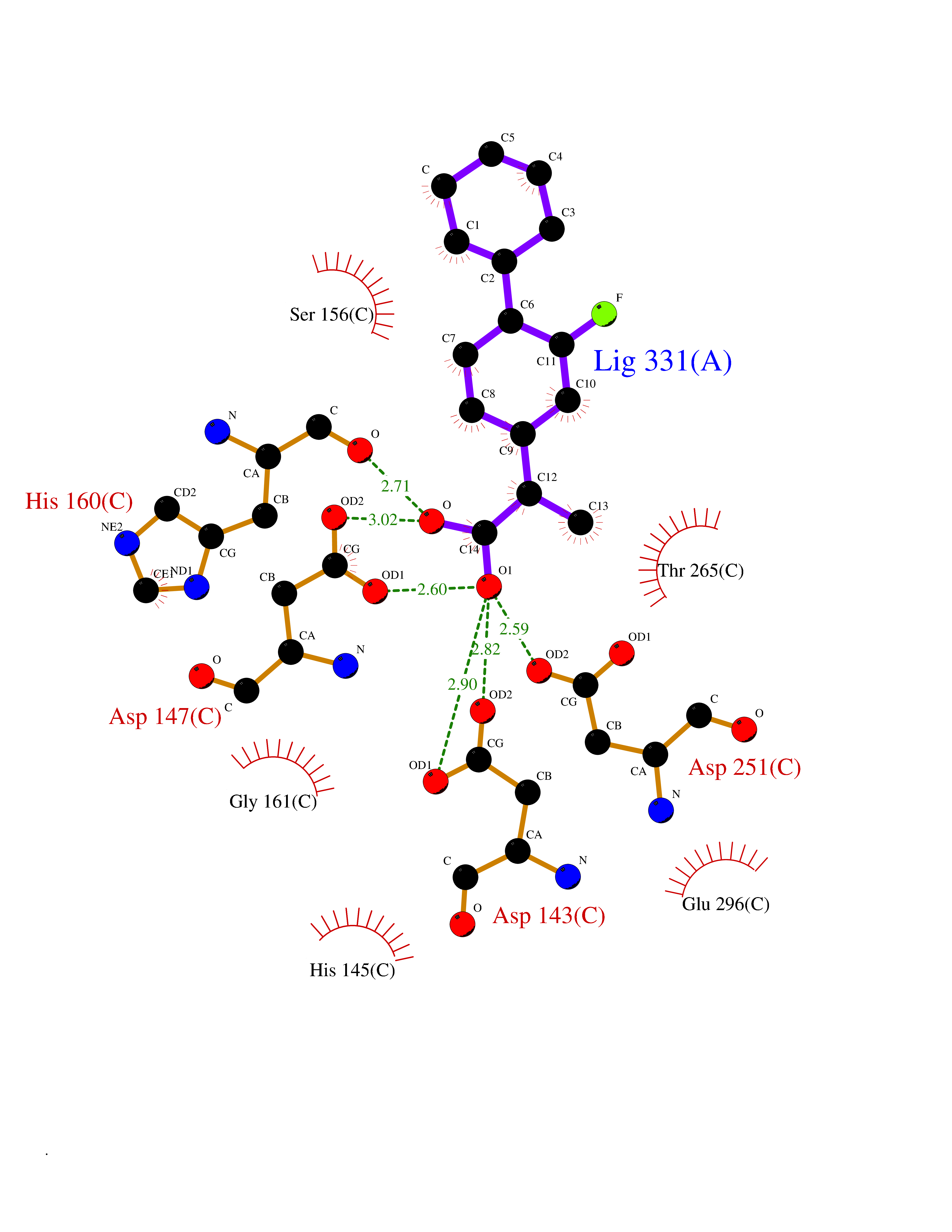 Ligplot Map 3D Binding mode Sequence HSVAVIGAPFSQGQKRKGVEHGPAAIREAGLMKRLSSLGCHLKDFGDLSFTPVPKDDLYNNLIVNPRSVGLANQELAEVVSRAVSDGYSCVTLGGDHSLAIGTISGHARHCPDLCVVWVDAHADINTPLTTSSGNLHGQPVSFLLRELQDKVPQLPGFSWIKPCISSASIVYIGLRDVDPPEHFILKNYDIQYFSMRDIDRLGIQKVMERTFDLLIGKRQRPIHLSFDIDAFDPTLAPATGTPVVGGLTYREGMYIAEEIHNTGLLSALDLVEVNPQLATSEEEAKTTANLAVDVIASSFGQTREG Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Aldo-keto reductase family 1 member C3 | 1S1P | 7.22 | |
Target general information Gen name AKR1C3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PGFS;DDH1;HSD17B5;KIAA0119 Protein family Aldo/keto reductase family Biochemical class Oxidoreductase Function 15-hydroxyprostaglandin-D dehydrogenase (NADP+) activity.Alditol:NADP+ 1-oxidoreductase activity.Aldo-keto reductase (NADP) activity.Androsterone dehydrogenase activity.Delta4-3-oxosteroid 5beta-reductase activity.Dihydrotestosterone 17-beta-dehydrogenase activity.Geranylgeranyl reductase activity.Indanol dehydrogenase activity.Ketoreductase activity.Ketosteroid monooxygenase activity.NADP-retinol dehydrogenase activity.Oxidoreductase activity, acting on NAD(P)H, quinone or similar compound as acceptor.Phenanthrene 9,10-monooxygenase activity.Prostaglandin D2 11-ketoreductase activity.Prostaglandin-F synthase activity.Prostaglandin H2 endoperoxidase reductase activity.Retinal dehydrogenase activity.Retinol dehydrogenase activity.Testosterone 17-beta-dehydrogenase (NADP+) activity.Testosterone dehydrogenase (NAD+) activity.Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase activity. Related diseases Neurodevelopmental disorder with language impairment and behavioral abnormalities (NEDLIB) [MIM:618917]: A neurodevelopmental disorder characterized by global developmental delay, impaired intellectual development, poor or absent speech, and behavioral abnormalities, such as autism spectrum disorder, repetitive behaviors, and hyperactivity. Some patients develop seizures and manifest developmental regression. {ECO:0000269|PubMed:31300657}. The disease is caused by variants affecting the gene represented in this entry. The genetic variation producing the missense variant p.Q607E, associated with NEDLIB, is predicted to deeply affect RNA editing. In a physiological context, the adenosine (A) residue of the original glutamine (Q) codon CAG is post-transcriptionaly edited to inosine (I) by ADAR2, leading to a codon recognized by the ribosome as arginine (R). The glutamate (E) codon GAG, resulting from the genetic variation, is predicted to be edited 90% less than the normal CAG codon. If edited, the codon GIG would be translated as p.Q607G. {ECO:0000305|PubMed:31300657}. Drugs (DrugBank ID) DB07700; DB01561; DB01536; DB00997; DB01039; DB02266; DB13751; DB00328; DB06077; DB00959; DB00157; DB03461; DB09074; DB00776; DB02056; DB01698; DB02901 Interacts with P17516 EC number 1.1.1.-; 1.1.1.188; 1.1.1.210; 1.1.1.239; 1.1.1.357; 1.1.1.53; 1.1.1.62; 1.1.1.64 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Lipid metabolism; NAD; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35846.8 Length 315 Aromaticity 0.09 Instability index 47.59 Isoelectric point 8.54 Charge (pH=7) 4.51 2D Binding mode Binding energy (Kcal/mol) -8.75  Molscript Map 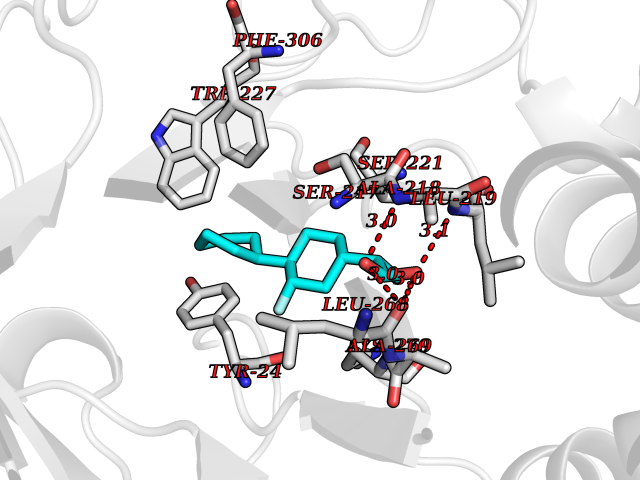 Pymol Map 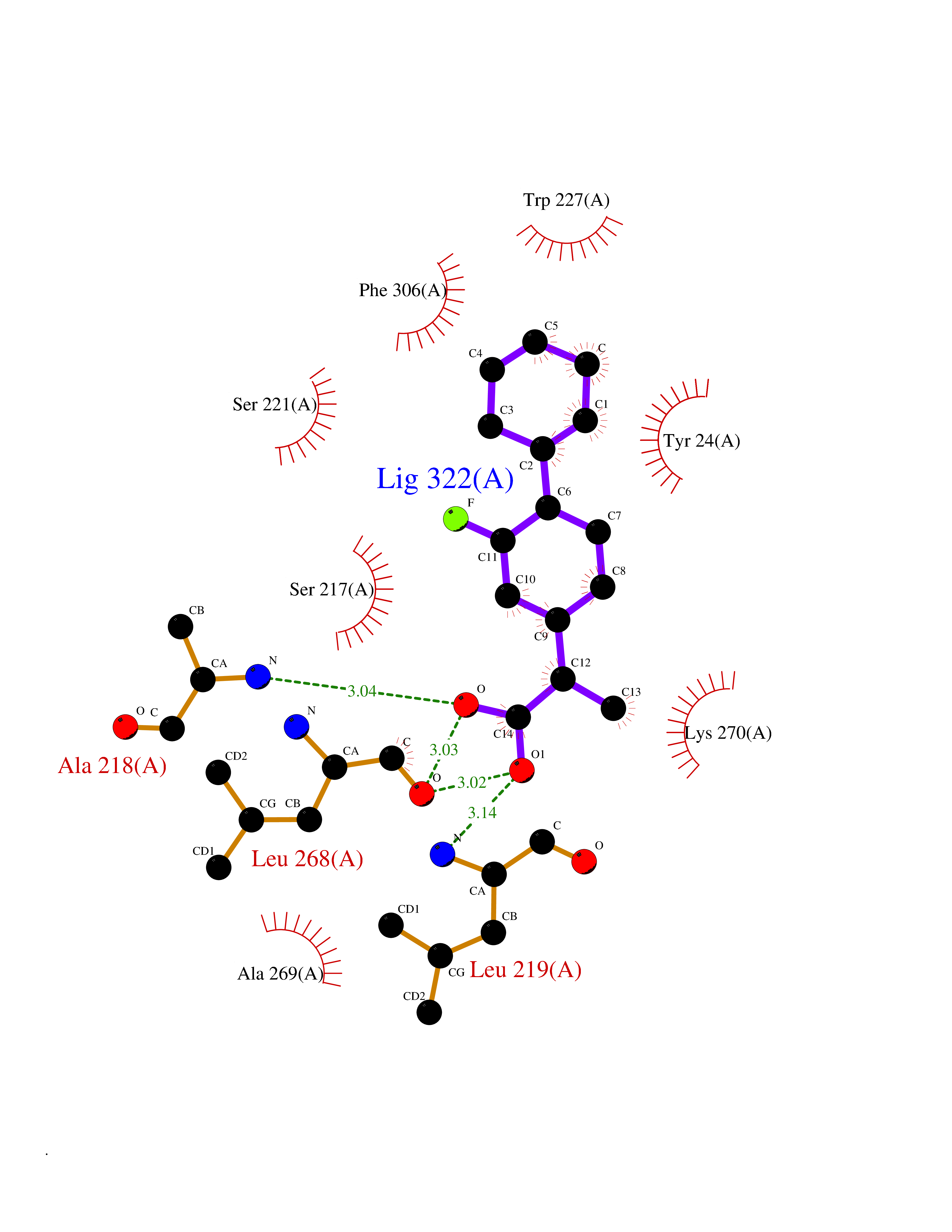 Ligplot Map 3D Binding mode Sequence QCVKLNDGHFMPVLGFGTYAPPEVPRSKALEVTKLAIEAGFRHIDSAHLYNNEEQVGLAIRSKIADGSVKREDIFYTSKLWSTFHRPELVRPALENSLKKAQLDYVDLYLIHSPMSLKPGEELSPTDENGKVIFDIVDLCTTWEAMEKCKDAGLAKSIGVSNFNRRQLEMILNKPGLKYKPVCNQVECHPYFNRSKLLDFCKSKDIVLVAYSALGSQRDKRWVDPNSPVLLEDPVLCALAKKHKRTPALIALRYQLQRGVVVLAKSYNEQRIRQNVQVFEFQLTAEDMKAIDGLDRNLHYFNSDSFASHPNYPYS Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Cytoplasmic thioredoxin reductase (TXNRD1) | 3QFA | 7.21 | |
Target general information Gen name TXNRD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Thioredoxin reductase TR1; Thioredoxin reductase 1, cytoplasmic; KM-102-derived reductase-like factor; KDRF; Gene associated with retinoic and interferon-induced mortality 12 protein; Gene associated Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Sulfur donor oxidoreductase Function Isoform 1 may possess glutaredoxin activity as well as thioredoxin reductase activity and induces actin and tubulin polymerization, leading to formation of cell membrane protrusions. Isoform 4 enhances the transcriptional activity of estrogen receptors alpha and beta while isoform 5 enhances the transcriptional activity of the beta receptor only. Isoform 5 also mediates cell death induced by a combination of interferon-beta and retinoic acid. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB01169; DB00126; DB03147; DB04106; DB05428; DB02338; DB05448; DB11127; DB11135; DB03566 Interacts with Q03135; P03372; Q92731 EC number EC 1.8.1.9 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Disulfide bond; Electron transport; FAD; Flavoprotein; NADP; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Redox-active center; Reference proteome; Selenocysteine; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 54161.4 Length 494 Aromaticity 0.08 Instability index 34.36 Isoelectric point 6.22 Charge (pH=7) -3.51 2D Binding mode Binding energy (Kcal/mol) -8.57 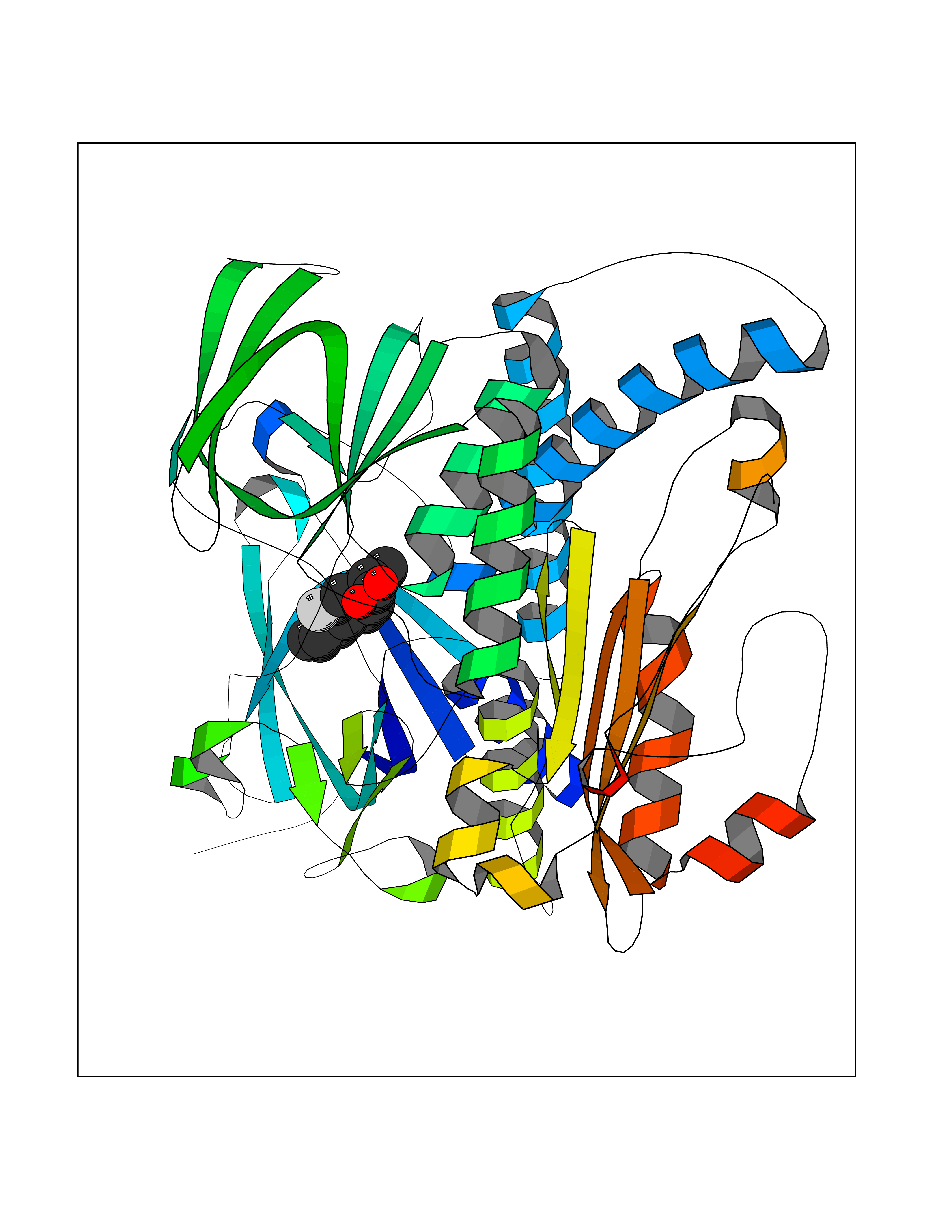 Molscript Map 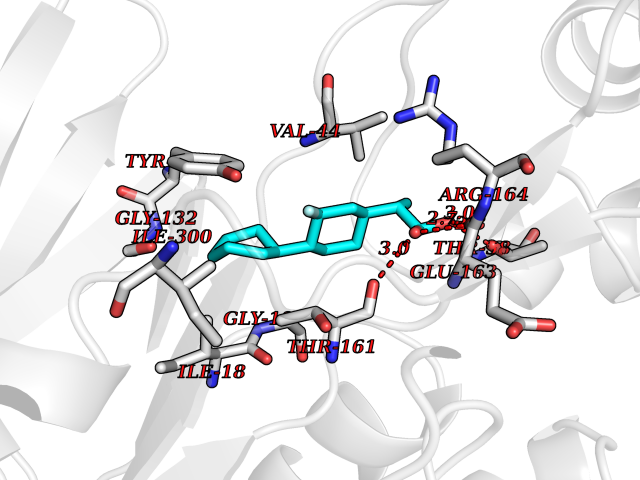 Pymol Map 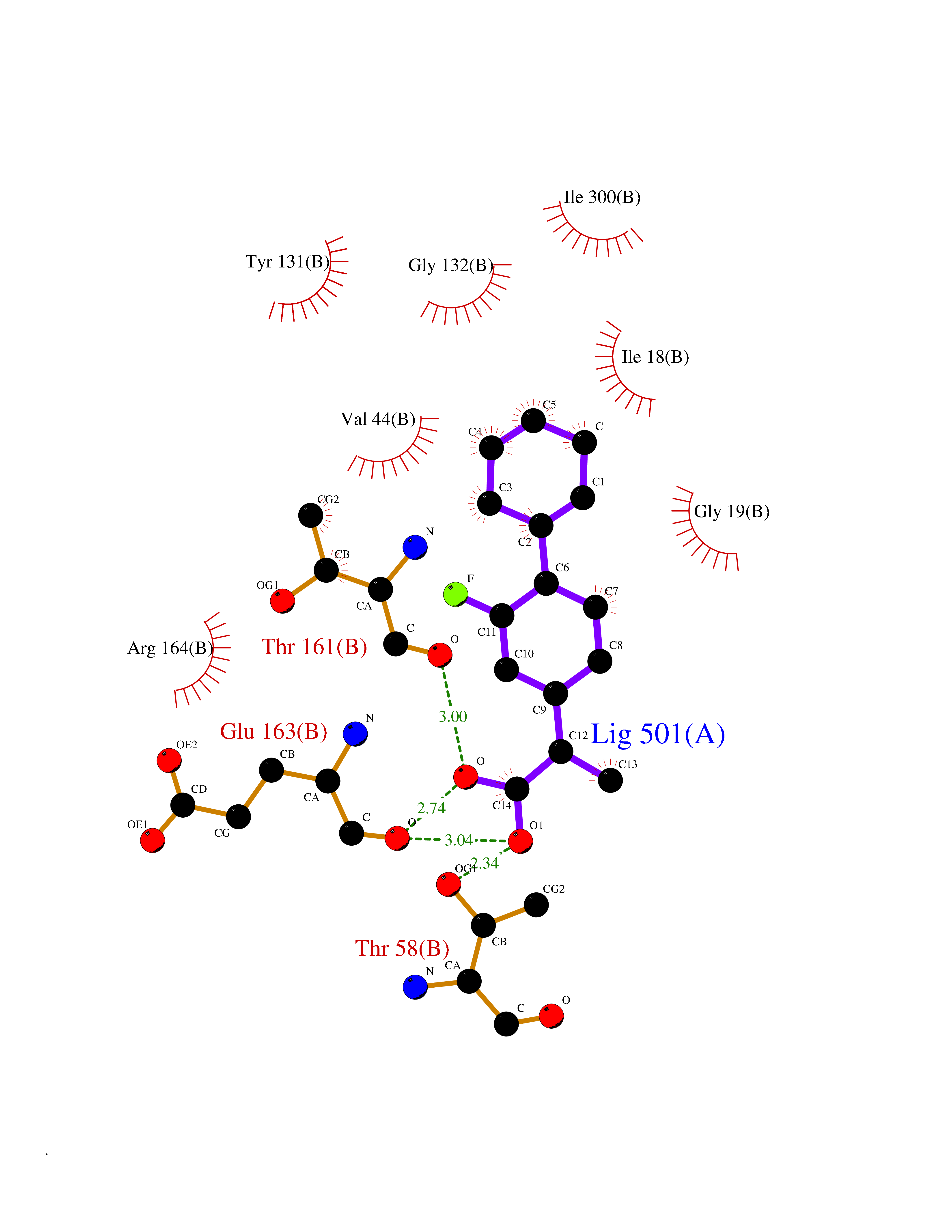 Ligplot Map 3D Binding mode Sequence DLPKSYDYDLIIIGGGSGGLAAAKEAAQYGKKVMVLDFVTPTPLGTRWGLGGTCVNVGCIPKKLMHQAALLGQALQDSRNYGWKVEETVKHDWDRMIEAVQNHIGSLNWGYRVALREKKVVYENAYGQFIGPHRIKATNNKGKEKIYSAERFLIATGERPRYLGIPGDKEYCISSDDLFSLPYCPGKTLVVGASYVALECAGFLAGIGLDVTVMVRSILLRGFDQDMANKIGEHMEEHGIKFIRQFVPIKVEQIEAGTPGRLRVVAQSTNSEEIIEGEYNTVMLAIGRDACTRKIGLETVGVKINEKTGKIPVTDEEQTNVPYIYAIGDILEDKVELTPVAIQAGRLLAQRLYAGSTVKCDYENVPTTVFTPLEYGACGLSEEKAVEKFGEENIEVYHSYFWPLEWTIPSRDNNKCYAKIICNTKDNERVVGFHVLGPNAGEVTQGFAAALKCGLTKKQLDSTIGIHPVCAEVFTTLSVTKRSGASILQAGSCG Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 7.20 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -8.99 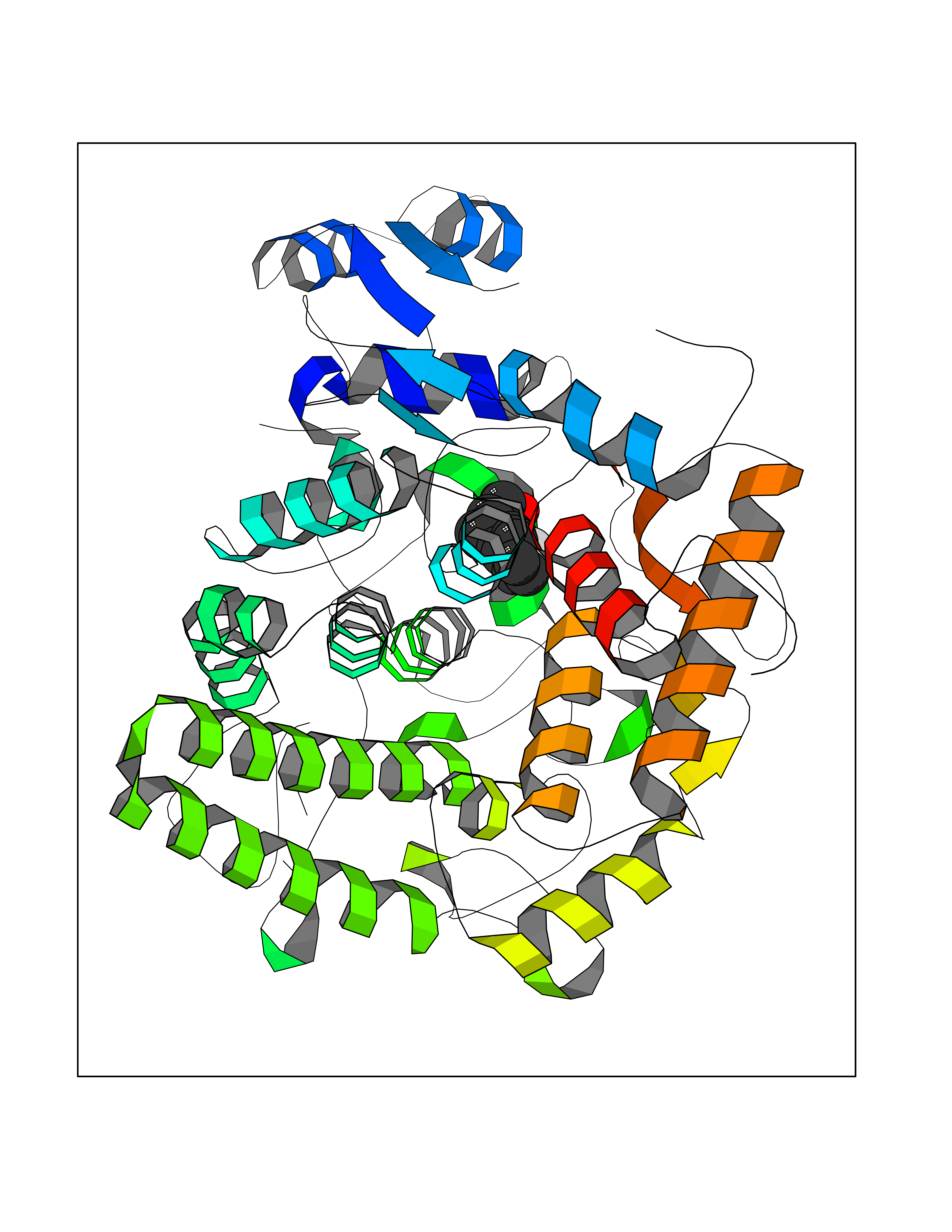 Molscript Map 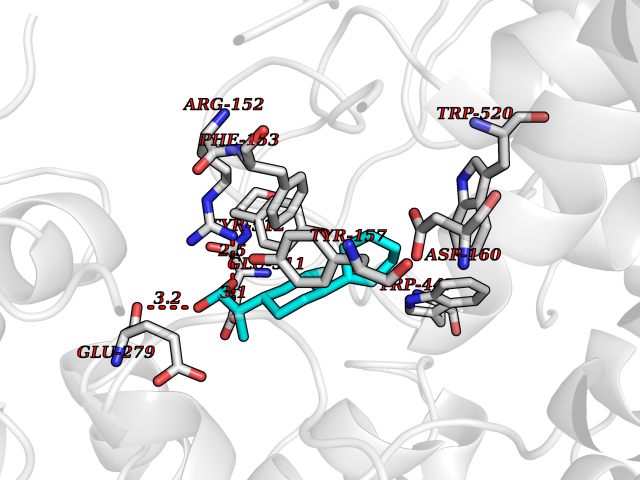 Pymol Map 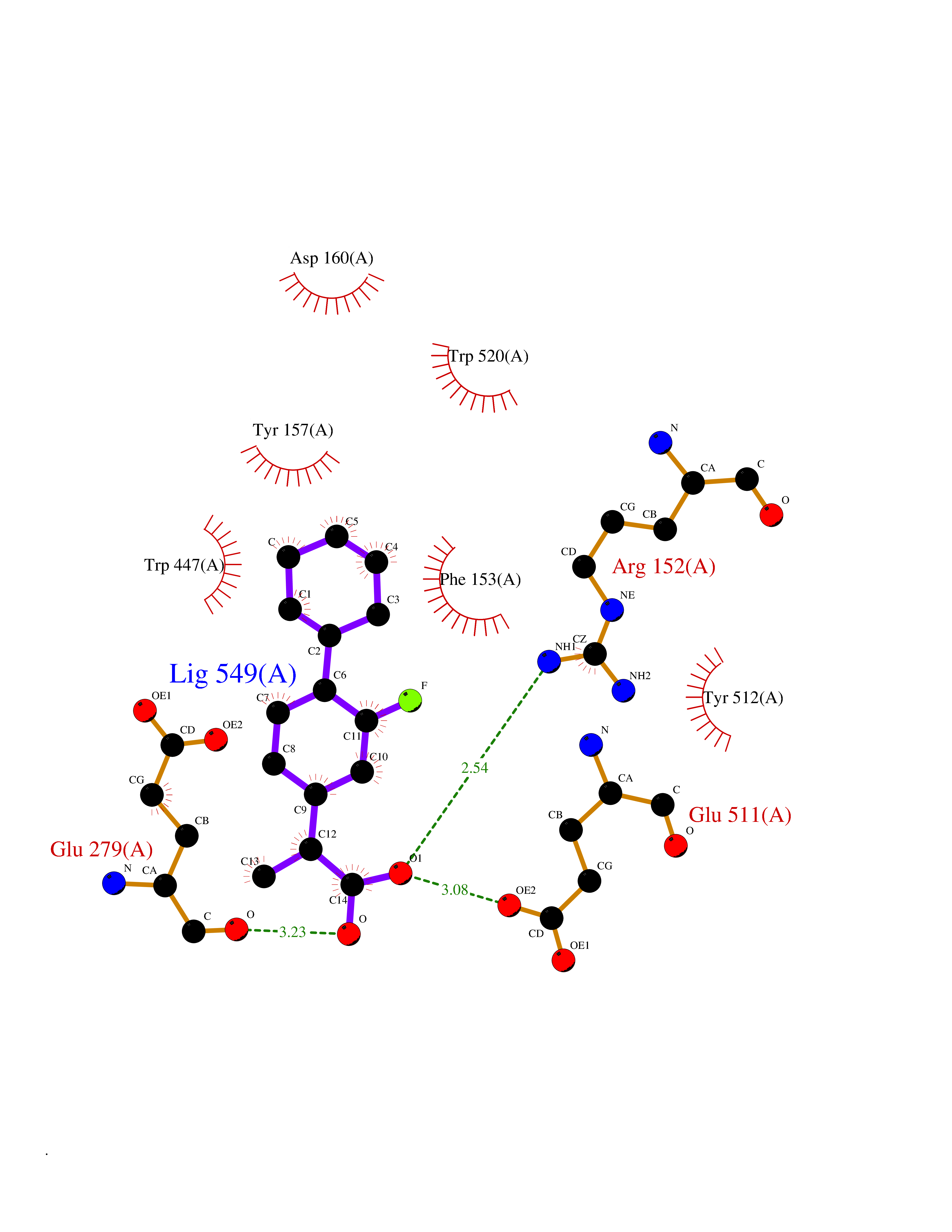 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Carbonic anhydrase I (CA-I) | 5E2M | 7.20 | |
Target general information Gen name CA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase B; Carbonic anhydrase 1; Carbonate dehydratase I; CAB Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Can hydrates cyanamide to urea. Reversible hydration of carbon dioxide. Related diseases Severe combined immunodeficiency autosomal recessive T-cell-negative/B-cell-negative/NK-cell-negative due to adenosine deaminase deficiency (ADASCID) [MIM:102700]: An autosomal recessive disorder accounting for about 50% of non-X-linked SCIDs. SCID refers to a genetically and clinically heterogeneous group of rare congenital disorders characterized by impairment of both humoral and cell-mediated immunity, leukopenia, and low or absent antibody levels. Patients with SCID present in infancy with recurrent, persistent infections by opportunistic organisms. The common characteristic of all types of SCID is absence of T-cell-mediated cellular immunity due to a defect in T-cell development. ADA deficiency has been diagnosed in chronically ill teenagers and adults (late or adult onset). Population and newborn screening programs have also identified several healthy individuals with normal immunity who have partial ADA deficiency. {ECO:0000269|PubMed:10200056, ECO:0000269|PubMed:1284479, ECO:0000269|PubMed:2166947, ECO:0000269|PubMed:2783588, ECO:0000269|PubMed:3182793, ECO:0000269|PubMed:3839802, ECO:0000269|PubMed:6208479, ECO:0000269|PubMed:7599635, ECO:0000269|PubMed:8227344, ECO:0000269|PubMed:8299233, ECO:0000269|PubMed:9361033}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08156; DB00819; DB00381; DB00436; DB00562; DB01194; DB00880; DB00310; DB00606; DB01144; DB00869; DB08846; DB01031; DB00311; DB08157; DB00774; DB00703; DB00423; DB00232; DB08155; DB01325; DB09460; DB09472; DB00273; DB01021; DB01593; DB14487; DB00909 Interacts with Q12800 EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lyase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 28483.4 Length 257 Aromaticity 0.1 Instability index 24.13 Isoelectric point 6.63 Charge (pH=7) -1.26 2D Binding mode Binding energy (Kcal/mol) -8.1  Molscript Map 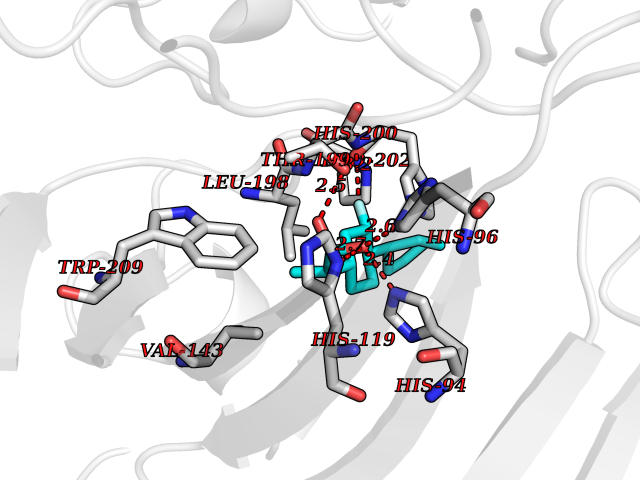 Pymol Map 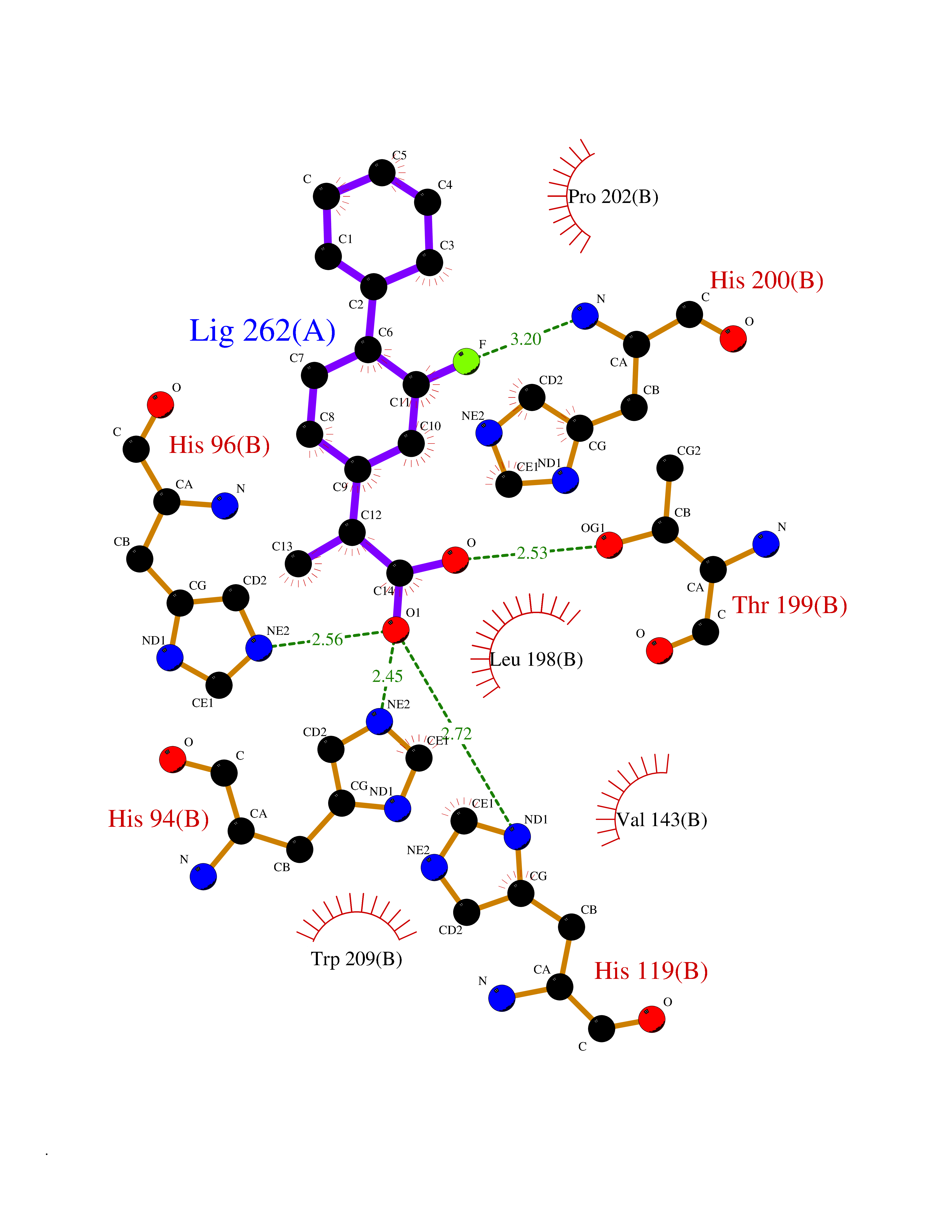 Ligplot Map 3D Binding mode Sequence DWGYDDKNGPEQWSKLYPIANGNNQSPVDIKTSETKHDTSLKPISVSYNPATAKEIINVGHSFHVNFEDNDNRSVLKGGPFSDSYRLFQFHFHWGSTNEHGSEHTVDGVKYSAELHVAHWNSAKYSSLAEAASKADGLAVIGVLMKVGEANPKLQKVLDALQAIKTKGKRAPFTNFDPSTLLPSSLDFWTYPGSLTHPPLYESVTWIICKESISVSSEQLAQFRSLLSNVEGDNAVPMQHNNRPTQPLKGRTVRASF Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Multidrug resistance protein 3 (ABCB4) | 6S7P | 7.20 | |
Target general information Gen name ABCB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PGY3; MDR3; ATP-binding cassette sub-family B member 4; ABCB4 Protein family ABC transporter superfamily, ABCB family, Multidrug resistance exporter (TC 3.A.1.201) subfamily Biochemical class Acid anhydrides hydrolase Function Mediates ATP-dependent export of organic anions and drugs from the cytoplasm. Hydrolyzes ATP with low efficiency. Not capable of conferring drug resistance. Mediates the translocation of phosphatidylcholine across the canalicular membrane of the hepatocyte. Related diseases Cholestasis, progressive familial intrahepatic, 3 (PFIC3) [MIM:602347]: A disorder characterized by early onset of cholestasis that progresses to hepatic fibrosis, cirrhosis, and end-stage liver disease before adulthood. PFIC3 inheritance is autosomal recessive. {ECO:0000269|PubMed:11313315, ECO:0000269|PubMed:12671900, ECO:0000269|PubMed:17726488, ECO:0000269|PubMed:21119540, ECO:0000269|PubMed:24045840, ECO:0000269|PubMed:24594635, ECO:0000269|PubMed:24806754, ECO:0000269|PubMed:28012258, ECO:0000269|PubMed:9419367}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cholestasis of pregnancy, intrahepatic 3 (ICP3) [MIM:614972]: A liver disorder of pregnancy. It presents during the second or, more commonly, the third trimester of pregnancy with intense pruritus which becomes more severe with advancing gestation and cholestasis. It causes fetal distress, spontaneous premature delivery and intrauterine death. Patients have spontaneous and progressive disappearance of cholestasis after delivery. Cholestasis results from abnormal biliary transport from the liver into the small intestine. {ECO:0000269|PubMed:10767346, ECO:0000269|PubMed:12746424, ECO:0000269|PubMed:15077010}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gallbladder disease 1 (GBD1) [MIM:600803]: One of the major digestive diseases. Gallstones composed of cholesterol (cholelithiasis) are the common manifestations in western countries. Most people with gallstones, however, remain asymptomatic through their lifetimes. {ECO:0000269|PubMed:11313316, ECO:0000269|PubMed:12891548, ECO:0000269|PubMed:22331132, ECO:0000269|PubMed:23533021, ECO:0000269|PubMed:24723470, ECO:0000269|PubMed:28012258, ECO:0000269|PubMed:28587926, ECO:0000269|Ref.2}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06414; DB06207 Interacts with NA EC number EC 7.6.2.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cytoplasm; Cytoplasmic vesicle; Disease variant; Glycoprotein; Intrahepatic cholestasis; Lipid transport; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 123919 Length 1128 Aromaticity 0.1 Instability index 29.44 Isoelectric point 8.78 Charge (pH=7) 11.08 2D Binding mode Binding energy (Kcal/mol) -7.86 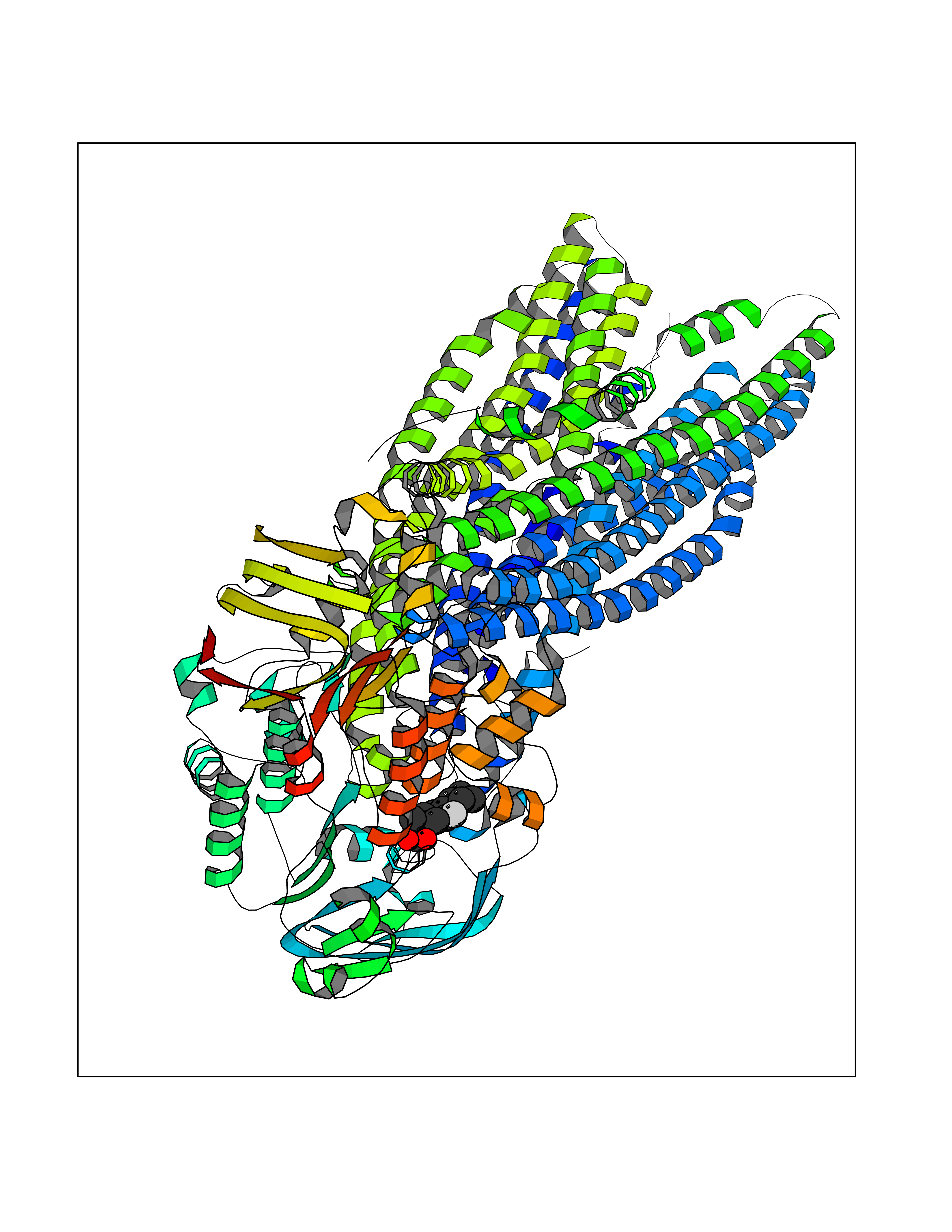 Molscript Map 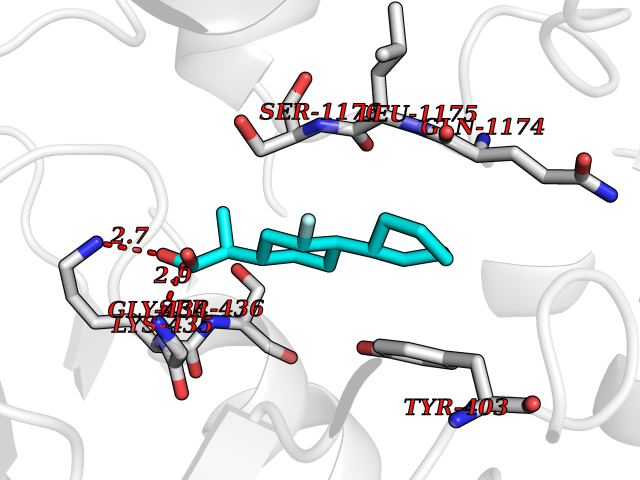 Pymol Map 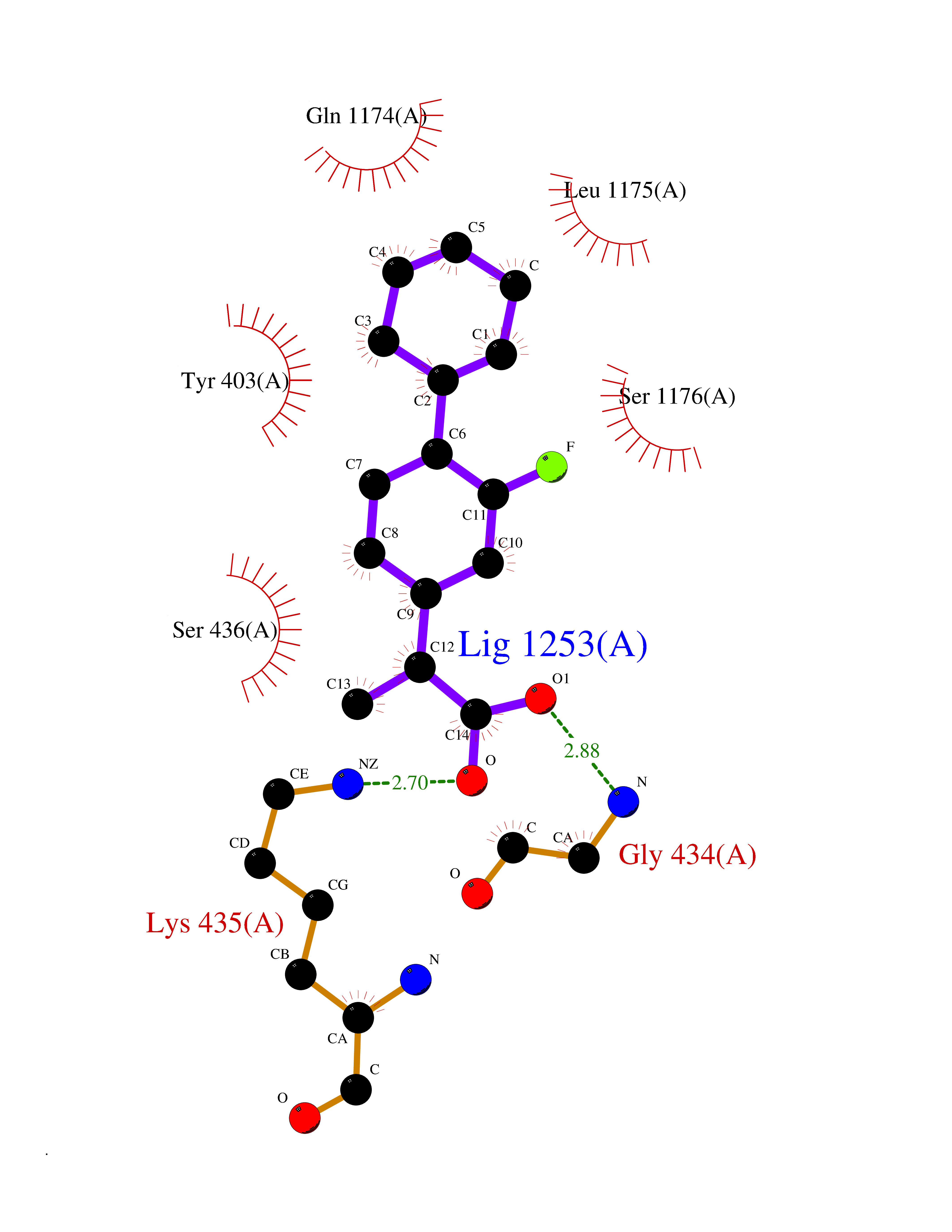 Ligplot Map 3D Binding mode Sequence VLTLFRYSDWQDKLFMSLGTIMAIAHGSGLPLMMIVFGEMTDKPGKILEEEMTRYAYYYSGLGAGVLVAAYIQVSFWTLAAGRQIRKIRQKFFHAILRQEIGWFDINDTTELNTRLTDDISKISEGIGDKVGMFFQAVATFFAGFIVGFIRGWKLTLVIMAISPILGLSAAVWAKILSAFSDKELAAYAKAGAVAEEALGAIRTVIAFGGQNKELERYQKHLENAKEIGIKKAISANISMGIAFLLIYASYALAFWYGSTLVISKEYTIGNAMTVFFSILIGAFSVGQAAPCIDAFANARGAAYVIFDIIDNNPKIDSFSERGHKPDSIKGNLEFNDVHFSYPSRANVKILKGLNLKVQSGQTVALVGSSGCGKSTTVQLIQRLYDPDEGTINIDGQDIRNFNVNYLREIIGVVSQEPVLFSTTIAENICYGRGNVTMDEIKKAVKEANAYEFIMKLPQKFDTLVGERGAQLSGGQKQRIAIARALVRNPKILLLDQATSALDTESEAEVQAALDKAREGRTTIVIAHRLSTVRNADVIAGFEDGVIVEQGSHSELMKKEGVYFKLVNVPPVSFLKVLKLNKTEWPYFVVGTVCAIANGGLQPAFSVIFSEIIAIFGPGDDAVKQQKCNIFSLIFLFLGIISFFTFFLQGFTFGKAGEILTRRLRSMAFKAMLRQDMSWFDDHKNSTGALSTRLATDAAQVQGATGTRLALIAQNIANLGTGIIISFIYGWQLTLLLLAVVPIIAVSGIVEMKLLAGNAKRDKKELEAAGKIATEAIENIRTVVSLTQERKFESMYVEKLYGPYRNSVQKAHIYGITFSISQAFMYFSYAGCFRFGAYLIVNGHMRFRDVILVFSAIVFGAVALGHASSFAPDYAKAKLSAAHLFMLFERQPLIDSYSEEGLKPDKFEGNITFNEVVFNYPTRANVPVLQGLSLEVKKGQTLALVGSSGCGKSTVVQLLERFYDPLAGTVLLDGQEAKKLNVQWLRAQLGIVSQEPILFDCSIAENIAYGDNSRVVSQDEIVSAAKAANIHPFIETLPHKYETRVGDKGTQLSGGQKQRIAIARALIRQPQILLLDQATSALDTESEKVVQEALDKAREGRTCIVIAHRLSTIQNADLIVVFQNGRVK Hydrogen bonds contact Hydrophobic contact | ||||