Job Results:
Ligand
Structure
Job ID
0006599cba9dbe83f9144f4d36fa70dc
Job name
NA
Time
2024-05-29 06:52:44
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Janus kinase 1 (JAK-1) | 4E5W | 8.01 | |
Target general information Gen name JAK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase JAK1; JAK1B; JAK1A Protein family Protein kinase superfamily, Tyr protein kinase family, JAK subfamily Biochemical class Kinase Function Kinase partner for the interleukin (IL)-2 receptor as well as interleukin (IL)-10 receptor. Tyrosine kinase of the non-receptor type, involved in the IFN-alpha/beta/gamma signal pathway. Related diseases Autoinflammation, immune dysregulation, and eosinophilia (AIIDE) [MIM:618999]: An autosomal dominant disorder characterized by immune dysregulation, severe atopic dermatitis, and chronic gastrointestinal inflammation. Additional features include asthma, food or environmental allergies, as well as poor overall growth with short stature. {ECO:0000269|PubMed:28111307, ECO:0000269|PubMed:32750333}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04716; DB14973; DB11817; DB12500; DB14845; DB12010; DB16756; DB11763; DB02375; DB15822; DB08877; DB08895; DB15091 Interacts with P04626; P48551; P40189-1; O60674; Q99650; P42224; P40763 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Disease variant; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; SH2 domain; Transferase; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32050.7 Length 280 Aromaticity 0.09 Instability index 44.51 Isoelectric point 8.07 Charge (pH=7) 2.39 2D Binding mode Binding energy (Kcal/mol) -8.8  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence VDPTHFEKRFLKRIRDLGEGHFGKVELCRYDPEGDNTGEQVAVKSLKPNHIADLKKEIEILRNLYHENIVKYKGICTENGIKLIMEFLPSGSLKEYLPKNKNKINLKQQLKYAVQICKGMDYLGSRQYVHRDLAARNVLVESEHQVKIGDFGLTKAIEKEXXTVKDDRDSPVFWYAPECLMQSKFYIASDVWSFGVTLHELLTYCDSDSSPMALFLKMIGPTHGQMTVTRLVNTLKEGKRLPCPPNCPDEVYQLMRKCWEFQPSNRTSFQNLIEGFEALL Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Inosine-5'-monophosphate dehydrogenase 1 (IMPDH1) | 1JCN | 8.01 | |
Target general information Gen name IMPDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Superoxide-inducible protein 12; SOI12; Probable inosine-5'-monophosphate dehydrogenase IMD1; NAD-dependent inosine monophosphate dehydrogenase; Inosine dehydrogenase; IMPDH-I; IMPDH 1; IMPDH; IMPD1; Protein family IMPDH/GMPR family Biochemical class CH-OH donor oxidoreductase Function Could also have a single-stranded nucleic acid-binding activity and could play a role in RNA and/or DNA metabolism. It may also have a role in the development of malignancy and the growth progression of some tumors. Catalyzes the conversion of inosine 5'-phosphate (IMP) to xanthosine 5'-phosphate (XMP), the first committed and rate-limiting step in the de novo synthesis of guanine nucleotides, and therefore plays an important role in the regulation of cell growth. Related diseases Retinitis pigmentosa 10 (RP10) [MIM:180105]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:11875049, ECO:0000269|PubMed:11875050, ECO:0000269|PubMed:16384941}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leber congenital amaurosis 11 (LCA11) [MIM:613837]: A severe dystrophy of the retina, typically becoming evident in the first years of life. Visual function is usually poor and often accompanied by nystagmus, sluggish or near-absent pupillary responses, photophobia, high hyperopia and keratoconus. {ECO:0000269|PubMed:16384941}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03948; DB00993; DB01033; DB00688; DB01024; DB00157; DB00811; DB06408; DB06103 Interacts with Q96D03; P20839-3; P12268; O75928-2; Q9Y4B4; P78317; Q7KZS0 EC number EC 1.1.1.205 Uniprot keywords 3D-structure; Alternative splicing; CBS domain; Cytoplasm; Direct protein sequencing; Disease variant; DNA-binding; GMP biosynthesis; Leber congenital amaurosis; Metal-binding; Methylation; NAD; Nucleus; Oxidoreductase; Phosphoprotein; Potassium; Proteomics identification; Purine biosynthesis; Reference proteome; Repeat; Retinitis pigmentosa; RNA-binding Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42447.5 Length 395 Aromaticity 0.06 Instability index 33.17 Isoelectric point 5.61 Charge (pH=7) -6.1 2D Binding mode Binding energy (Kcal/mol) -8.6  Molscript Map 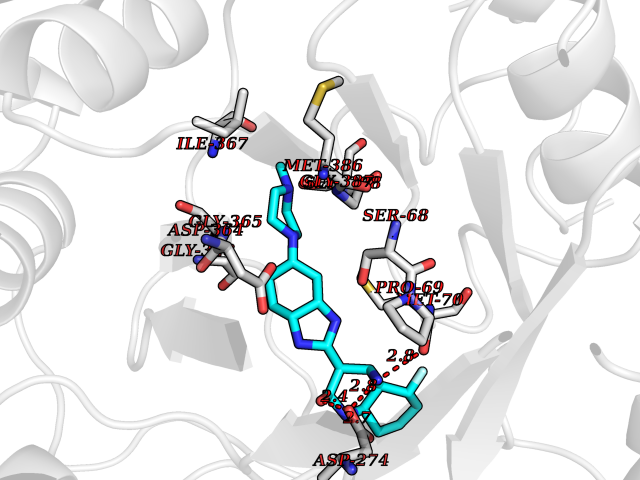 Pymol Map 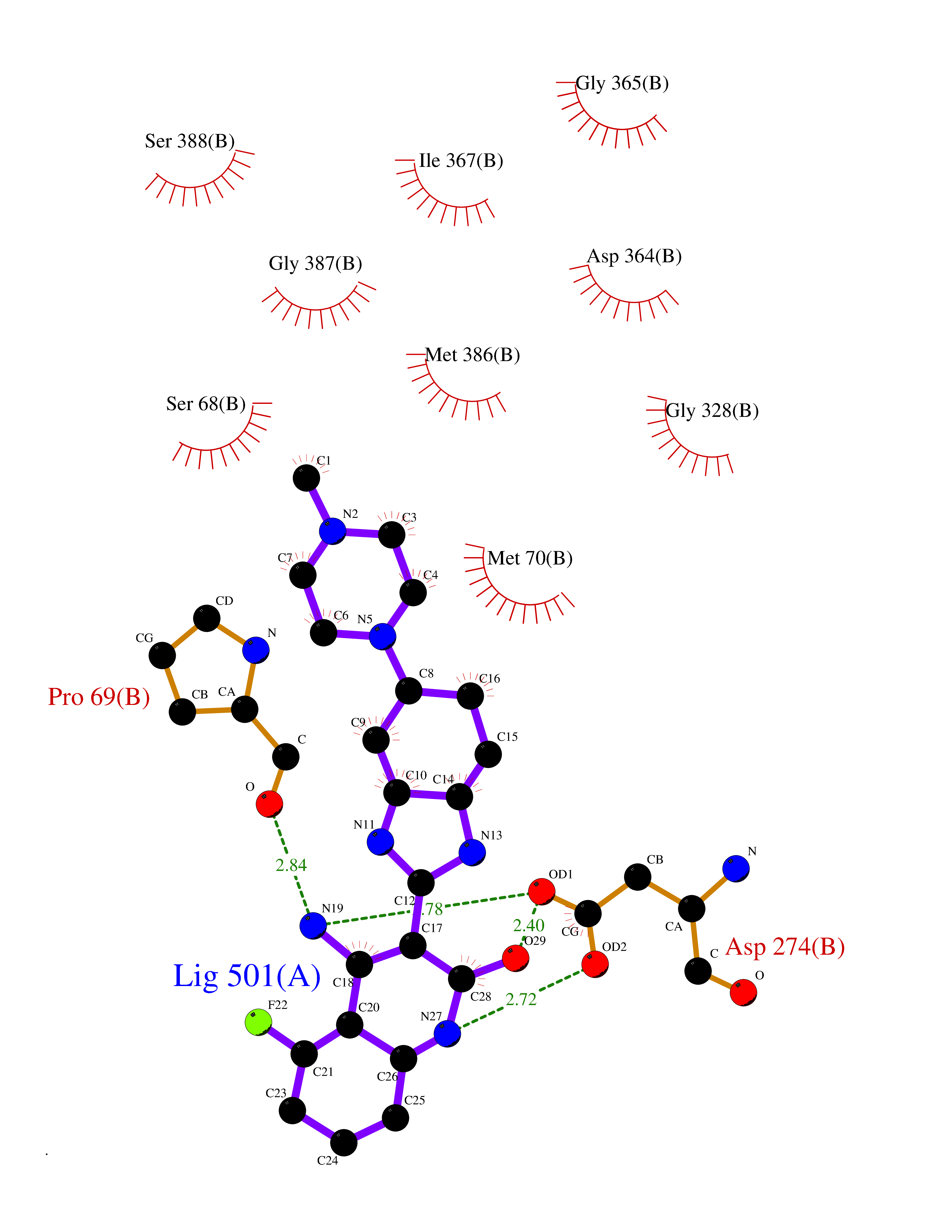 Ligplot Map 3D Binding mode Sequence TGYVPEDGLTAQQLFASADDLTYNDFLILPGFIDFIADEVDLTSALTRKITLKTPLISSPMDTVTEADMAIAMALMGGIGFIHHNCTPEFQANEVRKVKNFEQGFITDPVVLSPGIPITEVGIVTSRDIDPRIELVVAPAGVTLKEANEILQRSKKGKLPIVNDCDELVRTDLKKNRDYPLASKDSQKQLLCGAAVGTREDDKYRLDLLTQAGVDVIVLDSSQGNSVYQIAMVHYIKQKYPHLQVIGGNVVTAAQAKNLIDAGVDGLRVGMGCGSICITQEVMACGRPQGTAVYKVAEYARRFGVPIIADGGIQTVGHVVKALALGASTVMMGSLLAATTEAPGEKGSIQKFVPYLIAGIQHGCQDIGARSLSVLRSMMYSGELKFEKRTMSAQI Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Notch-1 receptor (NOTCH1) | 5L0R | 8.00 | |
Target general information Gen name NOTCH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hN1; Translocationassociated notch protein TAN1; Translocation-associated notch protein TAN-1; TAN1; Notch 1 intracellular domain; Notch 1; Neurogenic locus notch homolog protein 1; NICD Protein family NOTCH family Biochemical class Notch protein Function Upon ligand activation through the released notch intracellular domain (NICD) it forms a transcriptional activator complex with RBPJ/RBPSUH and activates genes of the enhancer of split locus. Affects the implementation of differentiation, proliferation and apoptotic programs. Involved in angiogenesis; negatively regulates endothelial cell proliferation and migration and angiogenic sprouting. Involved in the maturation of both CD4(+) and CD8(+) cells in the thymus. Important for follicular differentiation and possibly cell fate selection within the follicle. During cerebellar development, functions as a receptor for neuronal DNER and is involved in the differentiation of Bergmann glia. Represses neuronal and myogenic differentiation. May play an essential role in postimplantation development, probably in some aspect of cell specification and/or differentiation. May be involved in mesoderm development, somite formation and neurogenesis. May enhance HIF1A function by sequestering HIF1AN away from HIF1A. Required for the THBS4 function in regulating protective astrogenesis from the subventricular zone (SVZ) niche after injury. Involved in determination of left/right symmetry by modulating the balance between motile and immotile (sensory) cilia at the left-right organiser (LRO). Functions as a receptor for membrane-bound ligands Jagged-1 (JAG1), Jagged-2 (JAG2) and Delta-1 (DLL1) to regulate cell-fate determination. Related diseases Aortic valve disease 1 (AOVD1) [MIM:109730]: A common defect in the aortic valve in which two rather than three leaflets are present. It is often associated with aortic valve calcification, stenosis and insufficiency. In extreme cases, the blood flow may be so restricted that the left ventricle fails to grow, resulting in hypoplastic left heart syndrome. {ECO:0000269|PubMed:16025100}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Adams-Oliver syndrome 5 (AOS5) [MIM:616028]: A form of Adams-Oliver syndrome, a disorder characterized by the congenital absence of skin (aplasia cutis congenita) in combination with transverse limb defects. Aplasia cutis congenita can be located anywhere on the body, but in the vast majority of the cases, it is present on the posterior parietal region where it is often associated with an underlying defect of the parietal bones. Limb abnormalities are typically limb truncation defects affecting the distal phalanges or entire digits (true ectrodactyly). Only rarely, metatarsals/metacarpals or more proximal limb structures are also affected. Apart from transverse limb defects, syndactyly, most commonly of second and third toes, can also be observed. The clinical features are highly variable and can also include cardiovascular malformations, brain abnormalities and vascular defects such as cutis marmorata and dilated scalp veins. {ECO:0000269|PubMed:25132448}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q13315; Q969H0; Q16665; P78504; O60341; Q8N423; Q92585; P19838; P46531; Q13153; Q13526; Q06330; Q06330-6; Q13573; P98170; Q8IZL2; Q96JK9 EC number NA Uniprot keywords 3D-structure; Activator; Angiogenesis; ANK repeat; Calcium; Cell membrane; Developmental protein; Differentiation; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Endosome; Glycoprotein; Hydroxylation; Isopeptide bond; Membrane; Metal-binding; Notch signaling pathway; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transcription; Transcription regulation; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 46121.2 Length 394 Aromaticity 0.13 Instability index 39.95 Isoelectric point 7.22 Charge (pH=7) 0.63 2D Binding mode Binding energy (Kcal/mol) -9.49  Molscript Map 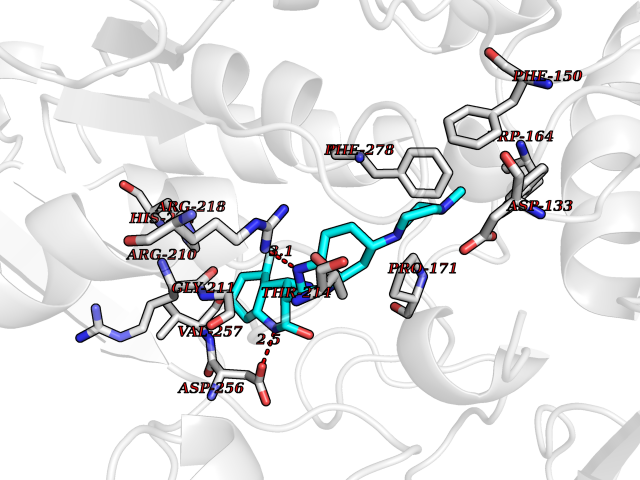 Pymol Map 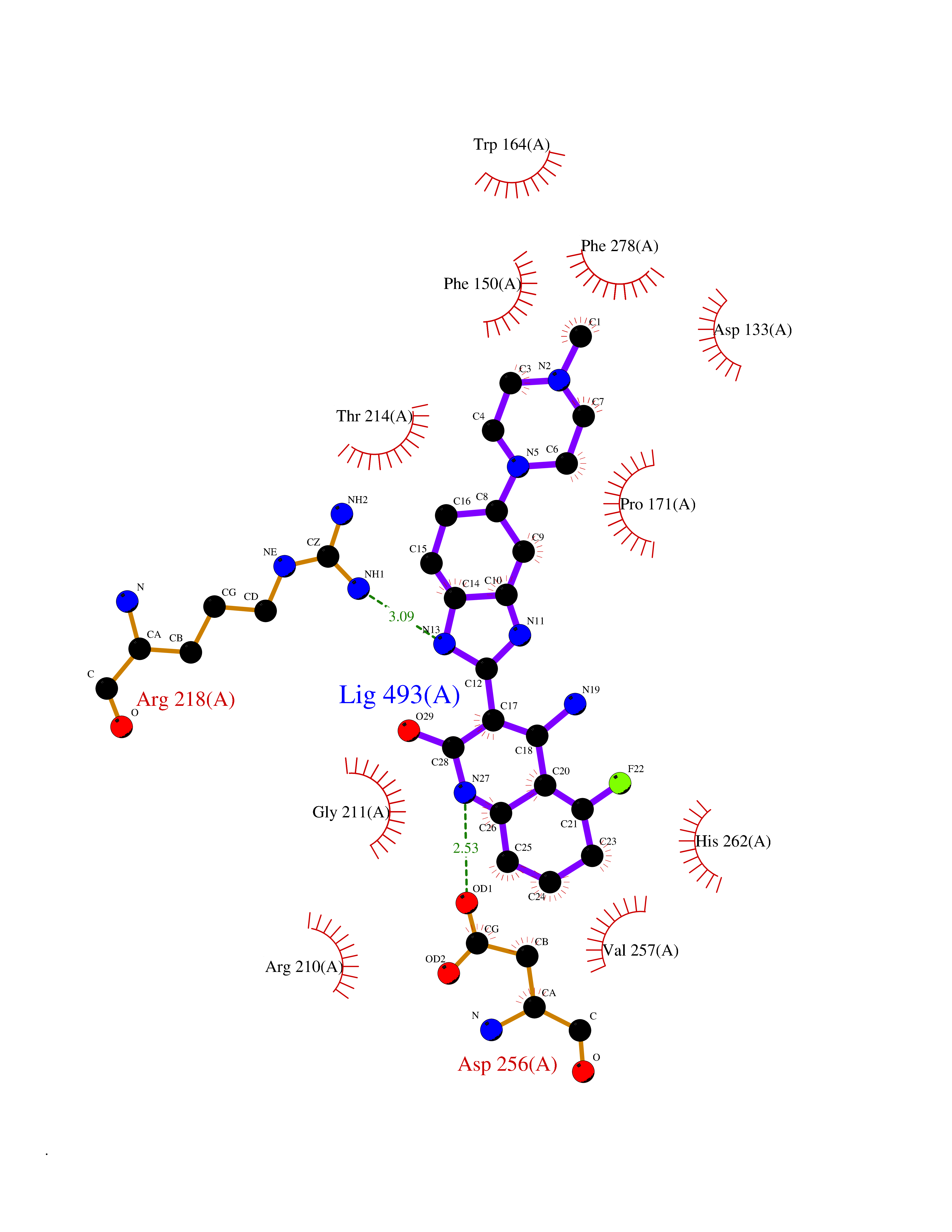 Ligplot Map 3D Binding mode Sequence KWKVFIDQINRSLENYEPCSSQNCSCYHGVIEEDLTPFRGGISRKMMAEVVRRKLGTHYQITKNRLYRENDCMFPSRCSGVEHFILEVIGRLPDMEMVINVRDYPQVPKWMEPAIPVFSFSKTSEYHDIMYPAWTFWEGGPAVWPIYPTGLGRWDLFREDLVRSAAQWPWKKKNSTAYFRGSRTSPERDPLILLSRKNPKLVDAEYTKNQAWKSMKDTLGKPAAKDVHLVDHCKYKYLFNFRGVAASFRFKHLFLCGSLVFHVGDEWLEFFYPQLKPWVHYIPVKTDLSNVQELLQFVKANDDVAQEIAERGSQFIRNHLQMDDITCYWENLLSEYSKFLSYNVTRRKGYDQIIPVNECVSNPCQNDATCLDQIGEFQCICMPGYEGVHCEVNT Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Iron hydrogenase 1 | 3C8Y | 8.00 | |
Target general information Gen name N/A Organism Clostridium pasteurianum Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Oxidoreductase Function 4 iron, 4 sulfur cluster binding.Electron carrier activity.Ferredoxin hydrogenase activity.Iron ion binding. Related diseases Noonan syndrome 13 (NS13) [MIM:619087]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. NS13 inheritance is autosomal dominant. There is considerable variability in severity. {ECO:0000269|PubMed:32721402}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 1.12.7.2 Uniprot keywords 3D-structure; 4Fe-4S; Direct protein sequencing; Iron; Iron-sulfur; Metal-binding; Oxidoreductase; Repeat Protein physicochemical properties Chain ID A Molecular weight (Da) 47536.9 Length 430 Aromaticity 0.08 Instability index 33.26 Isoelectric point 6.52 Charge (pH=7) -1.83 2D Binding mode Binding energy (Kcal/mol) -8.79  Molscript Map 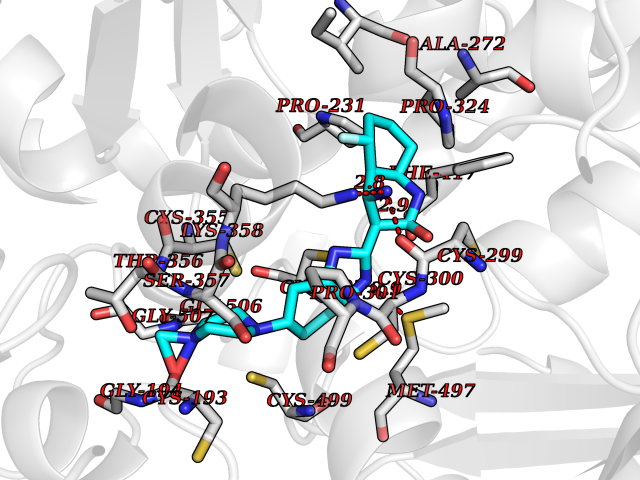 Pymol Map 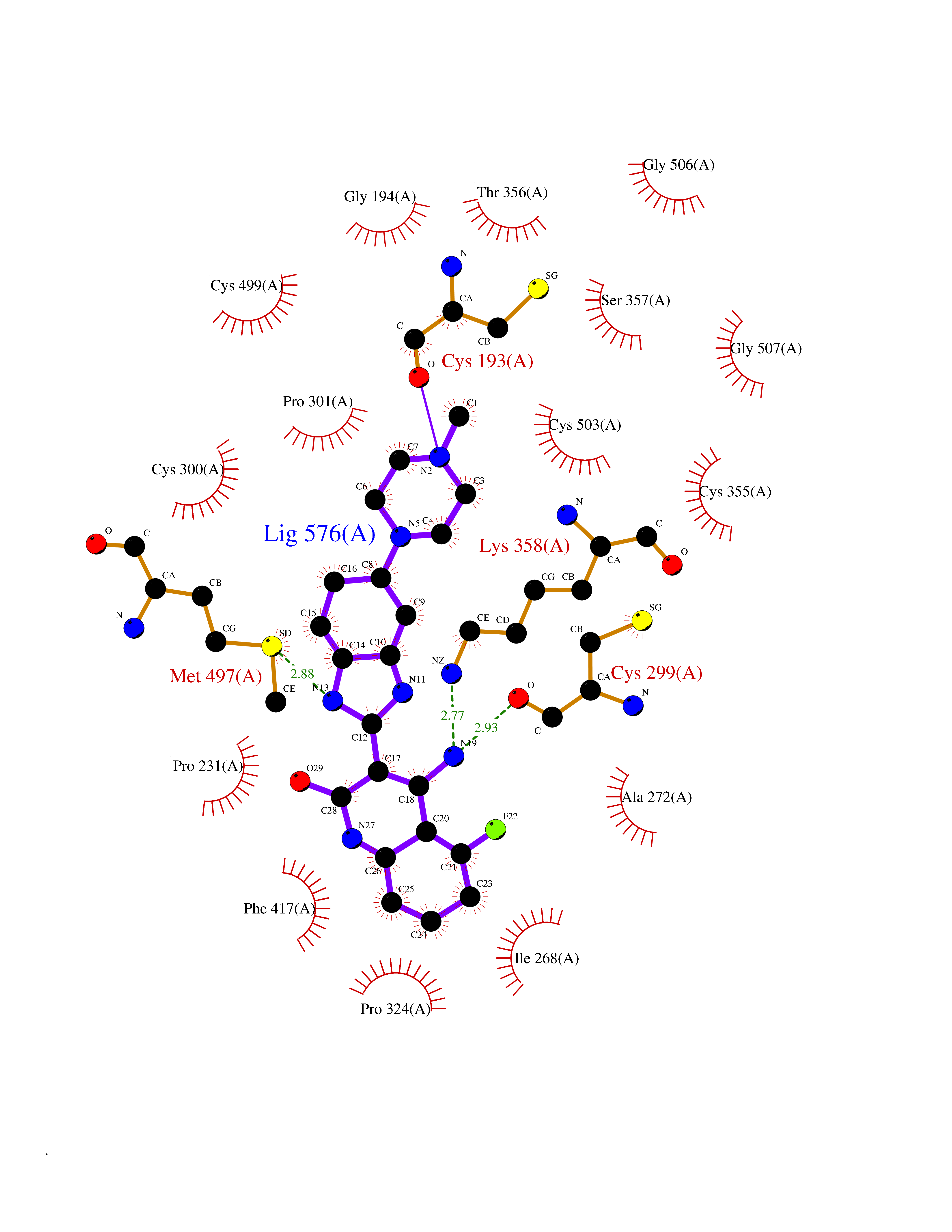 Ligplot Map 3D Binding mode Sequence SKSLTVDRTKCLLCGRCVNACGKNTETYAMKFIGAEDEKCFDDTNCLLCGQCIIACPVAALSEKSHMDRVKNALNAPEKHVIVAMAPSVRASIGELFNMGFGVDVTGKIYTALRQLGFDKIFDINFGADMTIMEEATELVQRIENNGPFPMFTSCCPGWVRQAENYYPELLNNLSSAKSPQQIFGTASKTYYPSISGLDPKNVFTVTVMPCTSKKFEADRPQMEKDGLRDIDAVITTRELAKMIKDAKIPFAKLEDSEADPAMGEYSGAGAIFGATGGVMEAALRSAKDFAENAELEDIEYKQVRGLNGIKEAEVEINNNKYNVAVINGASNLFKFMKSGMINEKQYHFIEVMACHGGCVNGGGQPHVNPKDLEKVDIKKVRASVLYNQDEHLSKRKSHENTALVKMYQNYFGKPGEGRAHEILHFKYKK Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | CAAX farnesyltransferase beta (FNTB) | 1SA4 | 7.99 | |
Target general information Gen name FNTB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAS proteins prenyltransferasebeta; FTase-beta; FNTB; CAAX farnesyltransferase beta subunit Protein family Protein prenyltransferase subunit beta family Biochemical class Alkyl aryl transferase Function Essential subunit of the farnesyltransferase complex. Catalyzes the transfer of a farnesyl moiety from farnesyl diphosphate to a cysteine at the fourth position from the C- terminus of several proteins having the C-terminal sequence Cys- aliphatic-aliphatic-X. Related diseases Lethal congenital contracture syndrome 2 (LCCS2) [MIM:607598]: A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS2 patients manifest craniofacial/ocular findings, lack of hydrops, multiple pterygia, and fractures, as well as a normal duration of pregnancy and a unique feature of a markedly distended urinary bladder (neurogenic bladder defect). The phenotype suggests a spinal cord neuropathic etiology. {ECO:0000269|PubMed:17701904}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythroleukemia, familial (FERLK) [MIM:133180]: An autosomal dominant myeloproliferative disorder characterized by neoplastic proliferation of erythroblastic and myeloblastic elements with atypical erythroblasts and myeloblasts in the peripheral blood. Disease penetrance is incomplete. {ECO:0000269|PubMed:27416908}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Visceral neuropathy, familial, 1, autosomal recessive (VSCN1) [MIM:243180]: An autosomal recessive disorder characterized by intestinal dysmotility due to aganglionosis (Hirschsprung disease), hypoganglionosis, and/or chronic intestinal pseudoobstruction. Additional variable features are progressive peripheral neuropathy, arthrogryposis, hypoplasia or aplasia of the olfactory bulb and of the external auditory canals, microtia or anotia, and facial dysmorphism. Some patients present structural cardiac anomalies and arthrogryposis with multiple pterygia. {ECO:0000269|PubMed:33497358}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07216; DB08674; DB08676; DB06953; DB07771; DB07895; DB04893; DB07780; DB07841; DB07227; DB06448; DB04960 Interacts with P21549; Q9BWW8; Q8N5M1; Q8N4L8; G5E9A7; P49354; P31273; A5PL33-2; Q9H0W8; Q7Z699; O43711 EC number EC 2.5.1.58 Uniprot keywords 3D-structure; Alternative splicing; Lipid metabolism; Metal-binding; Phosphoprotein; Prenyltransferase; Proteomics identification; Reference proteome; Repeat; Transferase; Zinc Protein physicochemical properties Chain ID B,A Molecular weight (Da) 83751.2 Length 725 Aromaticity 0.11 Instability index 46.44 Isoelectric point 5.61 Charge (pH=7) -20.1 2D Binding mode Binding energy (Kcal/mol) -8.69 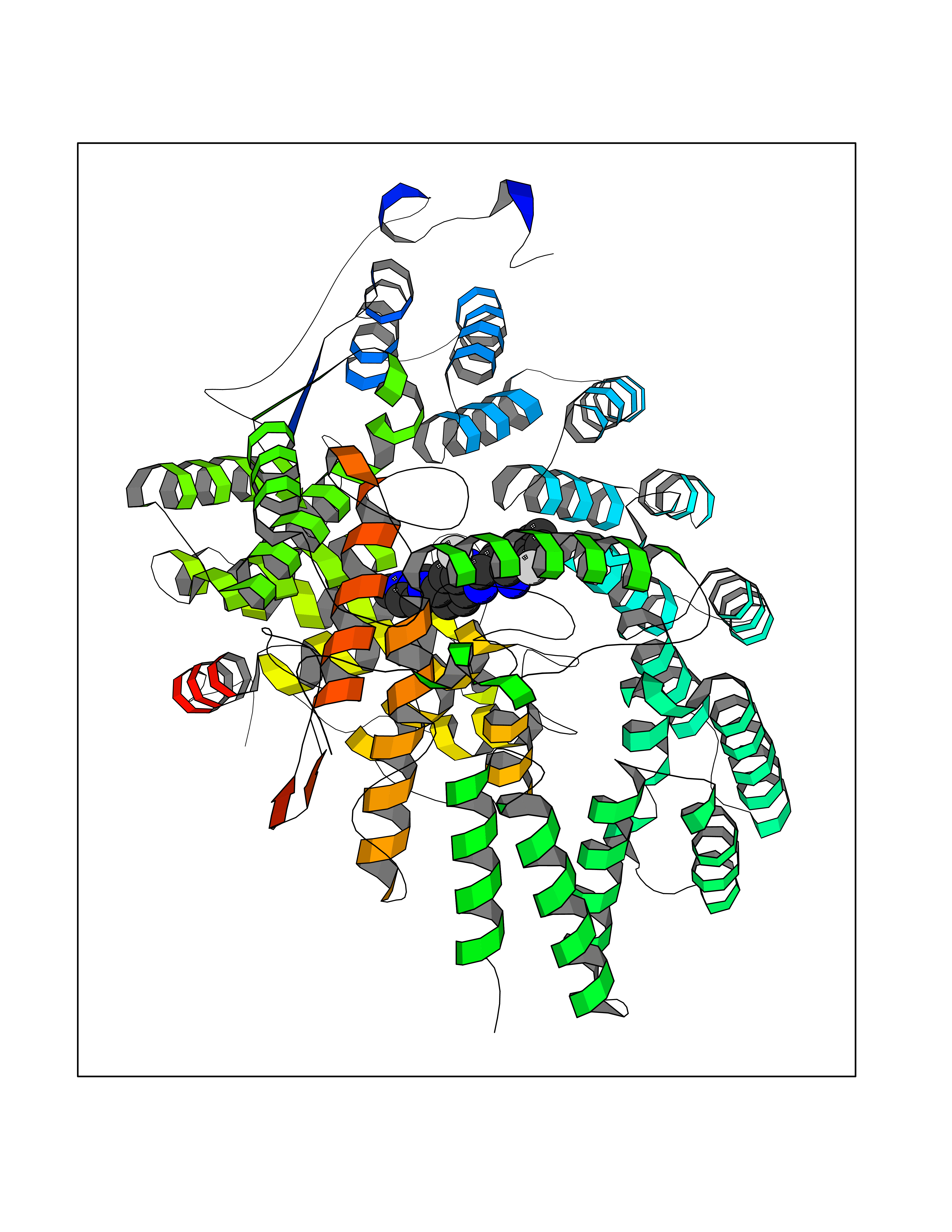 Molscript Map 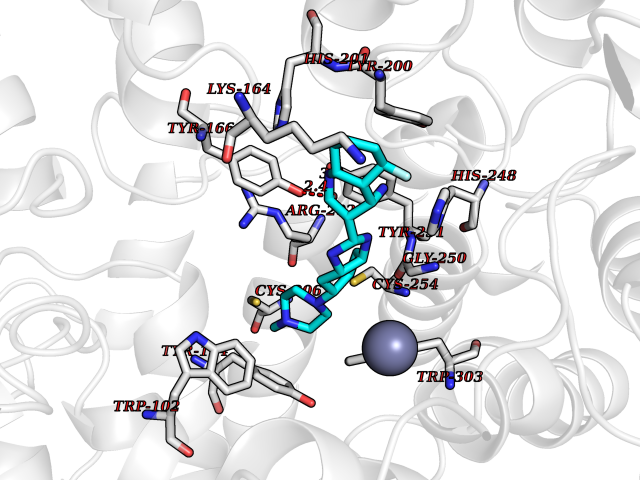 Pymol Map 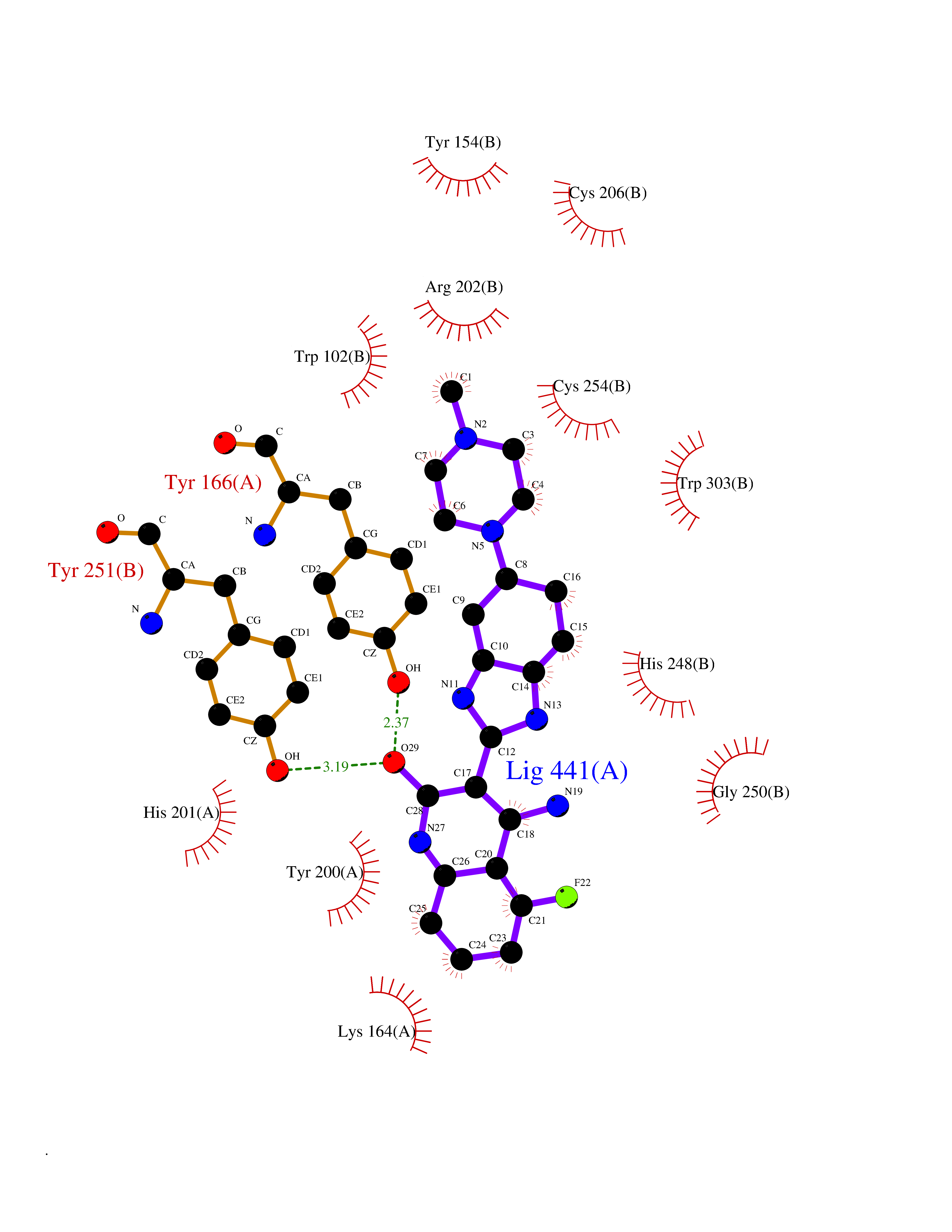 Ligplot Map 3D Binding mode Sequence FVSLDSPSYVLYRDRAEWADIDPVPQNDGPNPVVQIIYSDKFRDVYDYFRAVLQRDERSERAFKLTRDAIELNAANYTVWHFRRVLLKSLQKDLHEEMNYITAIIEEQPKNYQVWHHRRVLVEWLRDPSQELEFIADILNQDAKNYHAWQHRQWVIQEFKLWDNELQYVDQLLKEDVRNNSVWNQRYFVISNTTGYNDRAVLEREVQYTLEMIKLVPHNESAWNYLKGILQDRGLSKYPNLLNQLLDLQPSHSSPYLIAFLVDIYEDMLENQCDNKEDILNKALELCEILAKEKDTIRKEYWRYIGRSLQSKHSTSSPVWSEPLYSLRPEHARERLQDDSVETVTSIEQAKVEEKIQEVFSSYKFNHLVPRLVLQREKHFHYLKRGLRQLTDAYECLDASRPWLCYWILHSLELLDEPIPQIVATDVCQFLELCQSPEGGFGGGPGQYPHLAPTYAAVNALCIIGTEEAYDIINREKLLQYLYSLKQPDGSFLMHVGGEVDVRSAYCAASVASLTNIITPDLFEGTAEWIARCQNWEGGIGGVPGMEAHGGYTFCGLAALVILKRERSLNLKSLLQWVTSRQMRFEGGFQGRCNKLVDGCYSFWQAGLLPLLHRALHAQGDPALSMSHWMFHQQALQEYILMCCQCPAGGLLDKPGKSRDFYHTCYCLSGLSIAQHFGSGAMLHDVVLGVPENALQPTHPVYNIGPDKVIQATTYFLQKPVPGFE Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Thyroid hormone receptor beta (THRB) | 1N46 | 7.98 | |
Target general information Gen name THRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms c-erbA-beta; c-erbA-2; THR1; Nuclear receptor subfamily 1 group A member 2; NR1A2; ERBA2 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function High affinity receptor for thyroid hormones, including triiodothyronine and thyroxine. Nuclear hormone receptor that can act as a repressor or activator of transcription. Related diseases Thyroid hormone resistance, generalized, autosomal dominant (GRTHD) [MIM:188570]: An autosomal dominant disease characterized by high levels of circulating thyroid hormones (T3-T4), goiter, abnormal mental functions, increased susceptibility to infections, abnormal growth and bone maturation, tachycardia and deafness. Affected individuals may also have attention deficit-hyperactivity disorders (ADHD) and language difficulties. Patients have normal or slightly elevated thyroid stimulating hormone (TSH). {ECO:0000269|PubMed:10660344, ECO:0000269|PubMed:12511610, ECO:0000269|PubMed:12554782, ECO:0000269|PubMed:1314846, ECO:0000269|PubMed:1324420, ECO:0000269|PubMed:1563081, ECO:0000269|PubMed:1587388, ECO:0000269|PubMed:1619012, ECO:0000269|PubMed:1661299, ECO:0000269|PubMed:16804041, ECO:0000269|PubMed:1846005, ECO:0000269|PubMed:19268523, ECO:0000269|PubMed:2153155, ECO:0000269|PubMed:2510172, ECO:0000269|PubMed:7833659, ECO:0000269|PubMed:8175986, ECO:0000269|PubMed:8514853, ECO:0000269|PubMed:8664910, ECO:0000269|PubMed:8889584}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid hormone resistance, generalized, autosomal recessive (GRTHR) [MIM:274300]: An autosomal recessive disorder characterized by goiter, clinical euthyroidism, end-organ unresponsiveness to thyroid hormone, abnormal growth and bone maturation, and deafness. Patients also have high levels of circulating thyroid hormones, with elevated thyroid stimulating hormone. {ECO:0000269|PubMed:1653889}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Selective pituitary thyroid hormone resistance (PRTH) [MIM:145650]: Variant form of thyroid hormone resistance and is characterized by clinical hyperthyroidism, with elevated free thyroid hormones, but inappropriately normal serum TSH. Unlike GRTH, where the syndrome usually segregates with a dominant allele, the mode of inheritance in PRTH has not been established. {ECO:0000269|PubMed:7528740, ECO:0000269|PubMed:8381821}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08085; DB03181; DB02106; DB01118; DB00509; DB05035; DB03788; DB03176; DB00451; DB00279; DB01583; DB05192; DB07425; DB09100; DB03604 Interacts with Q60974; Q9Y618 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Deafness; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 27235.4 Length 239 Aromaticity 0.09 Instability index 43.29 Isoelectric point 5.42 Charge (pH=7) -8.55 2D Binding mode Binding energy (Kcal/mol) -9.37 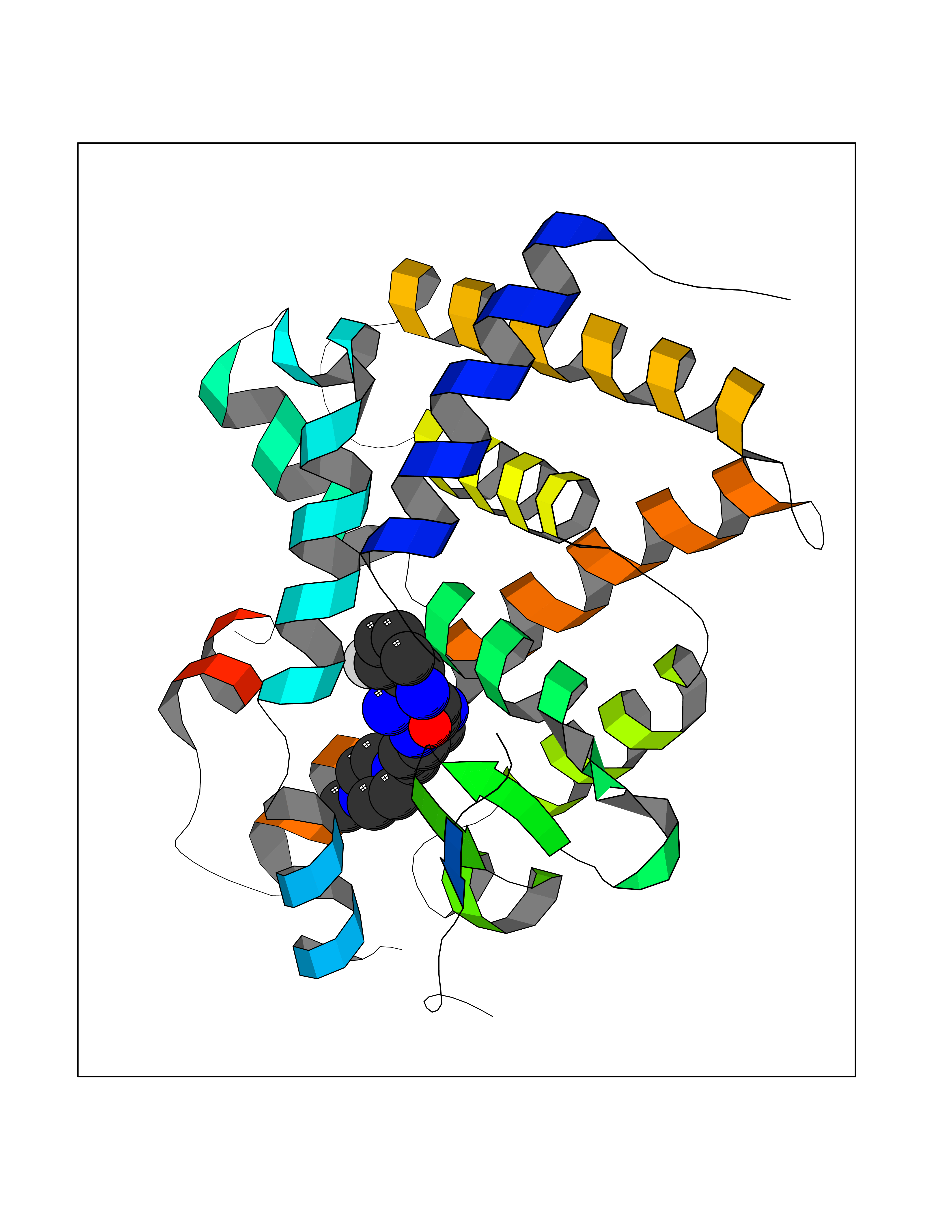 Molscript Map 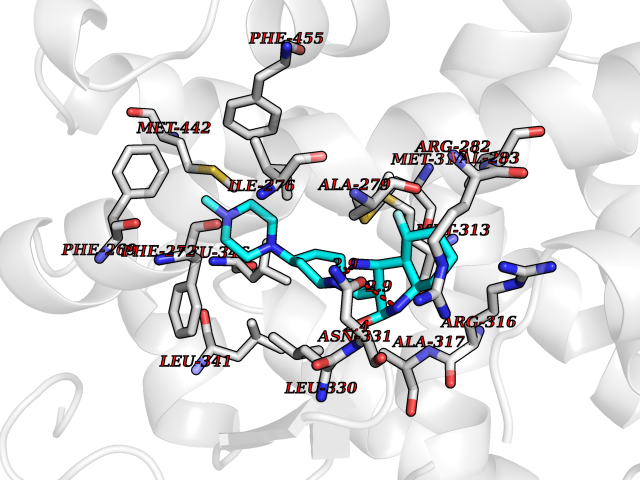 Pymol Map 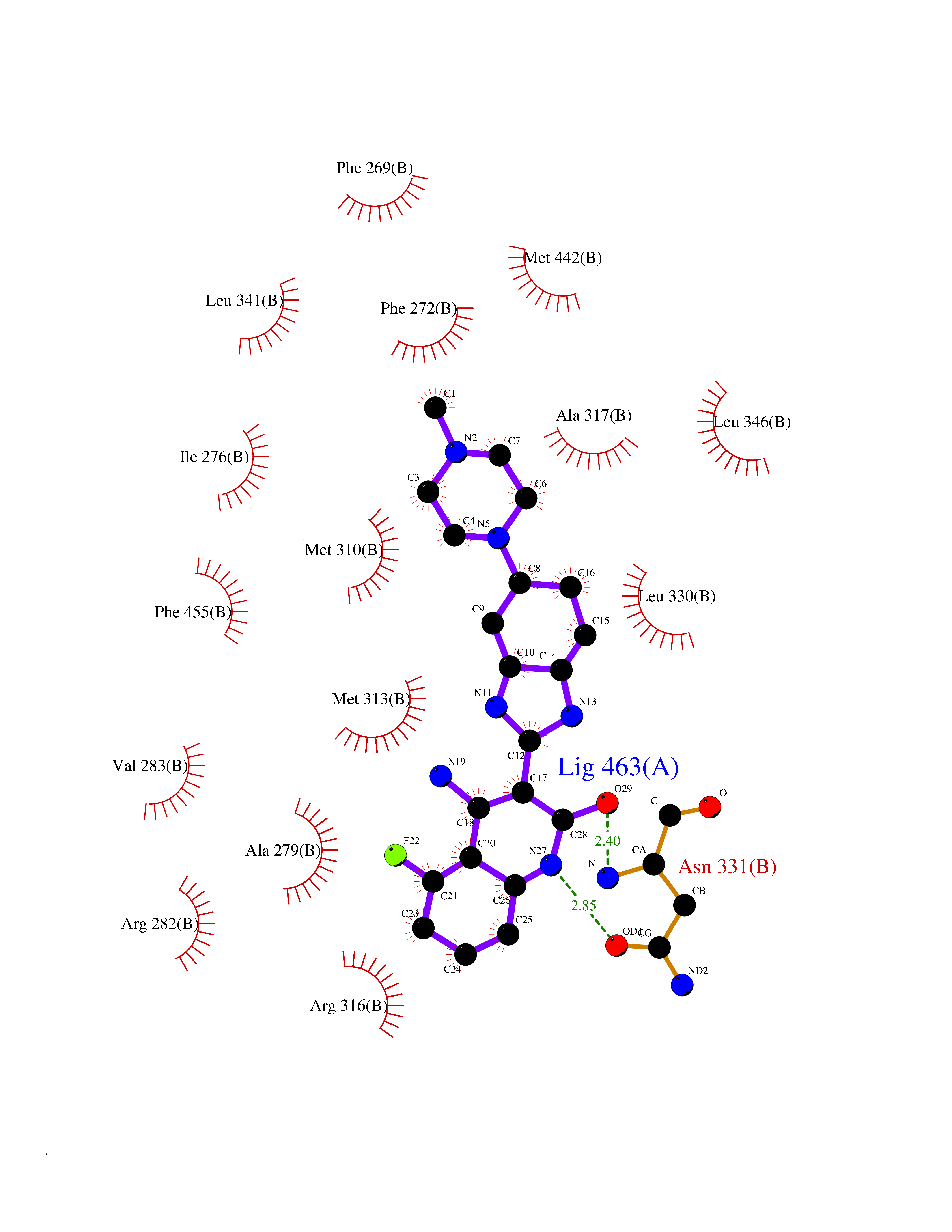 Ligplot Map 3D Binding mode Sequence KPEPTDEEWELIKTVTEAHVATNAQWKQKRKFLPEDIGQAKVDLEAFSHFTKIITPAITRVVDFAKKLPMFCELPCEDQIILLKGCCMEIMSLRAAVRYDPESETLTLNGEMAVTRGQLKNGGLGVVSDAIFDLGMSLSSFNLDDTEVALLQAVLLMSSDRPGLACVERIEKYQDSFLLAFEHYINYRKHHVTHFWPKLLMKVTDLRMIGACHASRFLHMKVECPTELFPPLFLEVFED Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Creatine kinase S-type, mitochondrial | 4Z9M | 7.98 | |
Target general information Gen name CKMT2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family ATP:guanido phosphotransferase family Biochemical class Transferase Function ATP binding.Creatine kinase activity. Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00787; DB00148; DB13191; DB00300 Interacts with O15182; P12532; Q9P2K6; Q16656-4; Q7Z4N8 EC number 2.7.3.2 Uniprot keywords 3D-structure; ATP-binding; Direct protein sequencing; Kinase; Membrane; Mitochondrion; Mitochondrion inner membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 41558.1 Length 365 Aromaticity 0.08 Instability index 40.19 Isoelectric point 7.23 Charge (pH=7) 0.53 2D Binding mode Binding energy (Kcal/mol) -8.95 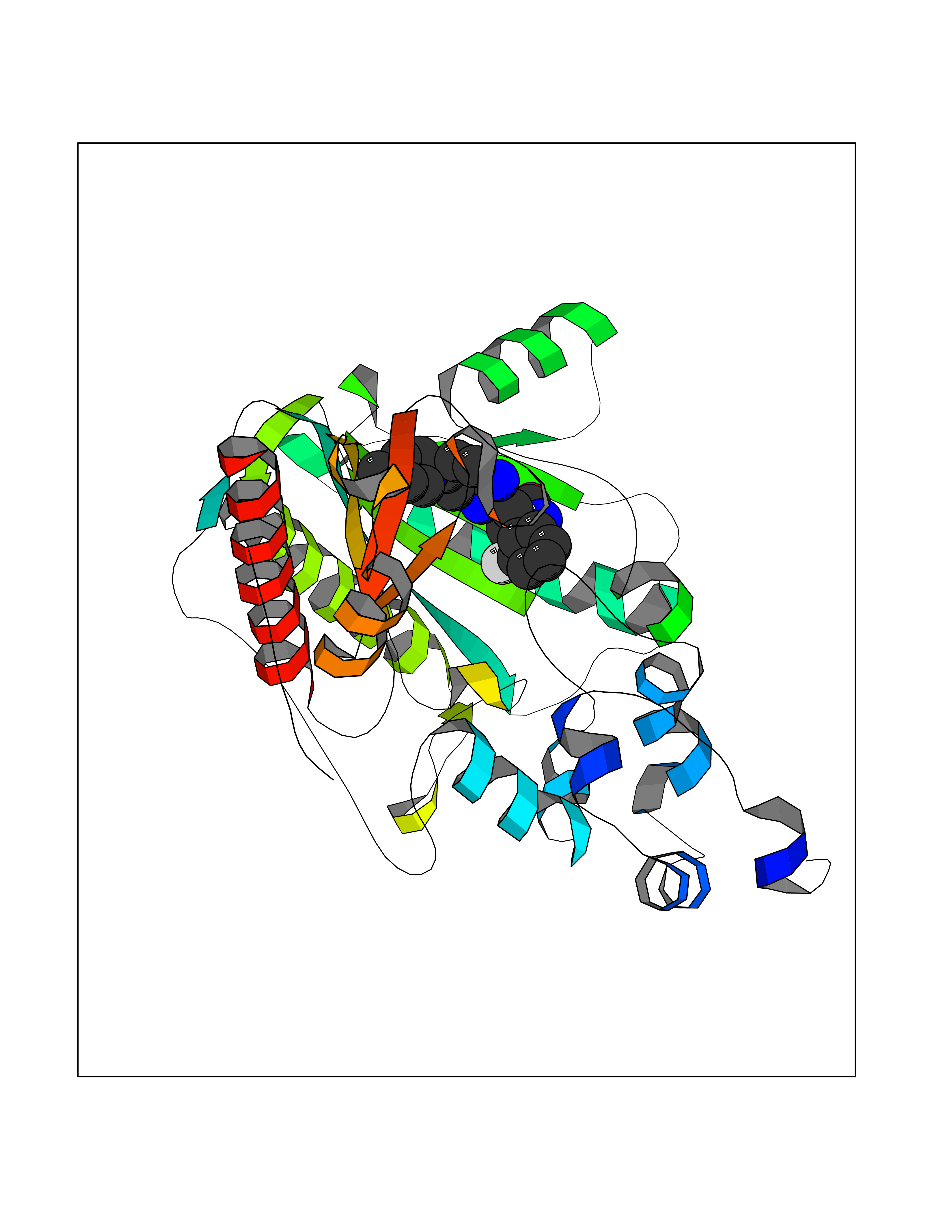 Molscript Map 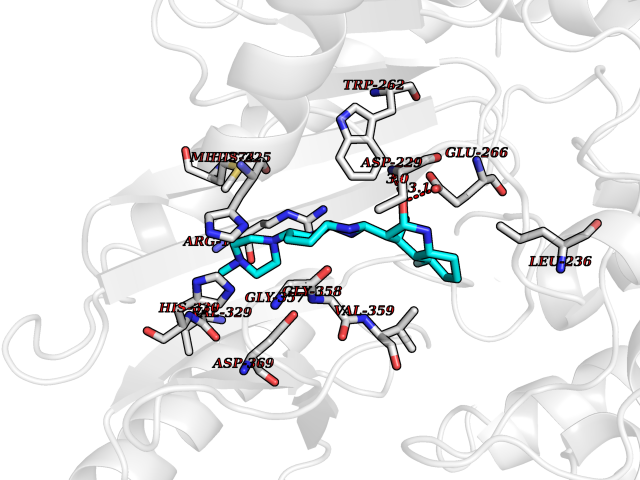 Pymol Map 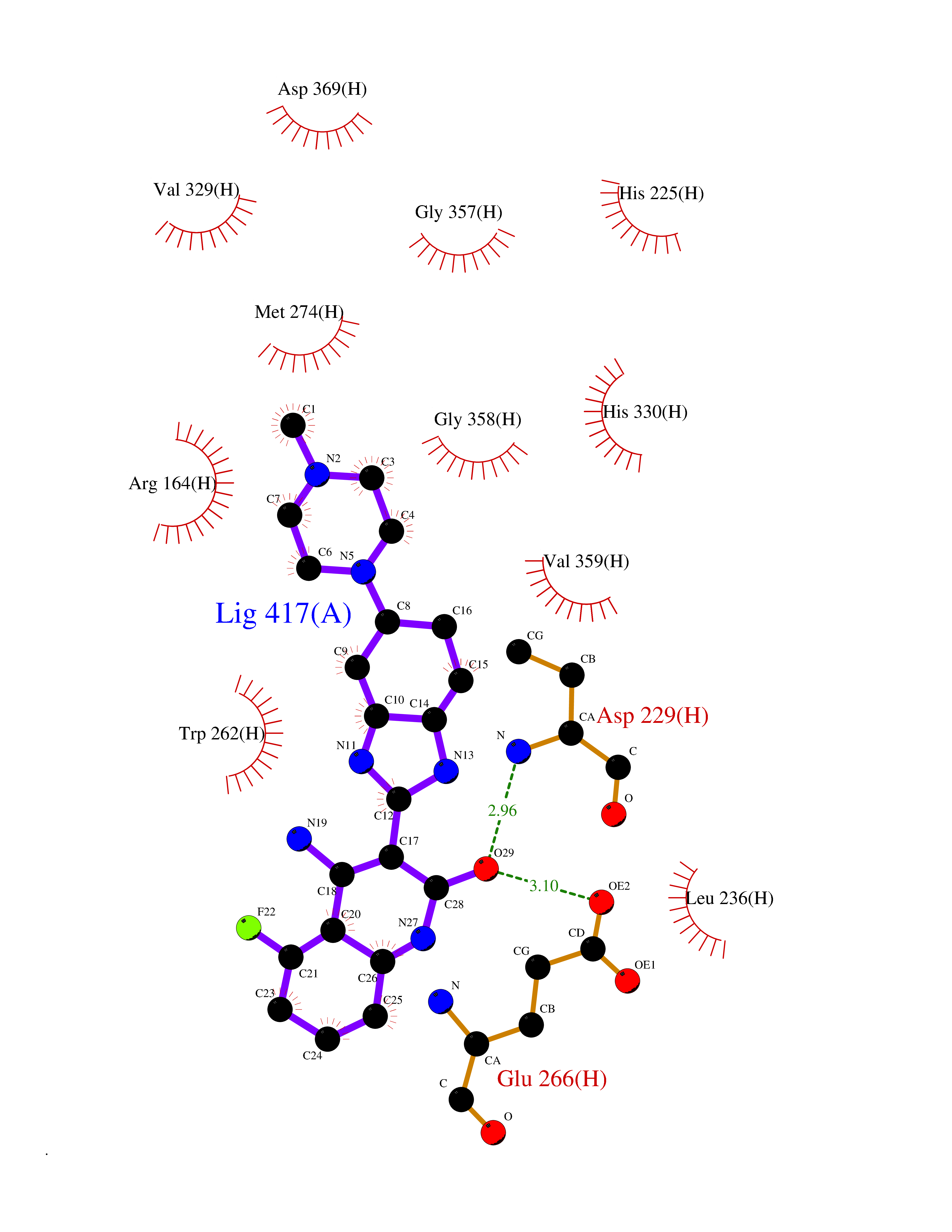 Ligplot Map 3D Binding mode Sequence RLFPPSADYPDLRKHNNCMAECLTPAIYAKLRNKVTPNGYTLDQCIQTGVDNPGHPFIKTVGMVAGDEESYEVFADLFDPVIKLRHNGYDPRVMKHTTDLDASKITQGQFDEHYVLSSRVRTGRSIRGLSLPPACTRAERREVENVAITALEGLKGDLAGRYYKLSEMTEQDQQRLIDDHFLFDKPVSPLLTCAGMARDWPDARGIWHNYDKTFLIWINEEDHTRVISMEKGGNMKRVFERFCRGLKEVERLIQERGWEFMWNERLGYILTCPSNLGTGLRAGVHVRIPKLSKDPRFSKILENLRLQKRGTGGVDTAAVADVYDISNIDRIGRSEVELVQIVIDGVNYLVDCEKKLIKVPPPLPQ Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | DNA polymerase alpha subunit B | 4Y97 | 7.97 | |
Target general information Gen name POLA2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family DNA polymerase alpha subunit B family Biochemical class Transferase Function DNA binding.DNA-directed DNA polymerase activity.Protein heterodimerization activity. Related diseases Timothy syndrome (TS) [MIM:601005]: Disorder characterized by multiorgan dysfunction including lethal arrhythmias, webbing of fingers and toes, congenital heart disease, immune deficiency, intermittent hypoglycemia, cognitive abnormalities and autism. {ECO:0000269|PubMed:15454078, ECO:0000269|PubMed:15863612, ECO:0000269|PubMed:25260352, ECO:0000269|PubMed:26253506, ECO:0000269|PubMed:30023270, ECO:0000269|PubMed:30172029, ECO:0000269|PubMed:31430211}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 3 (BRGDA3) [MIM:611875]: A heart disease characterized by the association of Brugada syndrome with shortened QT intervals. Brugada syndrome is a tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:17224476}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Long QT syndrome 8 (LQT8) [MIM:618447]: A form of long QT syndrome, a heart disorder characterized by a prolonged QT interval on the ECG and polymorphic ventricular arrhythmias. They cause syncope and sudden death in response to exercise or emotional stress, and can present with a sentinel event of sudden cardiac death in infancy. LQT8 transmission pattern is consistent with autosomal dominant inheritance with incomplete penetrance. {ECO:0000269|PubMed:23677916, ECO:0000269|PubMed:24728418, ECO:0000269|PubMed:25633834, ECO:0000269|PubMed:30345660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with hypotonia, language delay, and skeletal defects with or without seizures (NEDHLSS) [MIM:620029]: An autosomal dominant disorder characterized by global developmental delay apparent from infancy, intellectual disability, poor or absent speech, behavioral abnormalities, and hypotonia with delayed walking or inability to walk. Additional features include epilepsy, mild skeletal defects, and non-specific dysmorphic features. {ECO:0000269|PubMed:30513141, ECO:0000269|PubMed:34163037}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00851 Interacts with P55212; O75460-2; Q0VDC6; P54652; P13473-2; O75400-2; P62826; P49591; Q9Y371; P31948; PRO_0000449619 [P0DTD1] EC number NA Uniprot keywords 3D-structure; Alternative splicing; DNA replication; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,C,E,G Molecular weight (Da) 48300.5 Length 436 Aromaticity 0.08 Instability index 52 Isoelectric point 5.2 Charge (pH=7) -13.29 2D Binding mode Binding energy (Kcal/mol) -9.53  Molscript Map 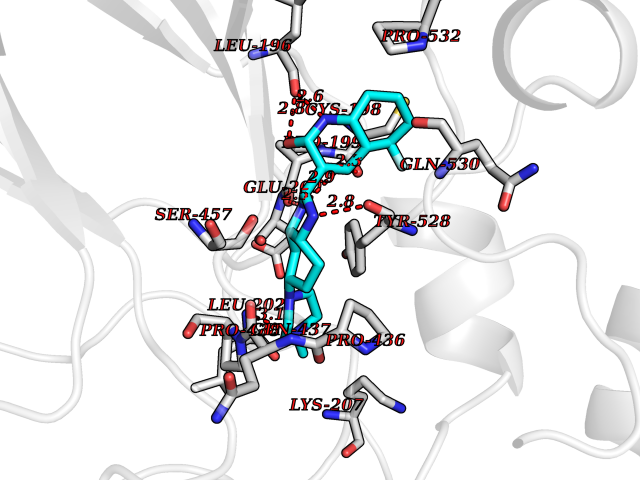 Pymol Map  Ligplot Map 3D Binding mode Sequence PSQKYNSRSNRGEVVTSFGLAQGVSWSGRGGAGNISLKVLGCPEALKSMFQKLPDIREVLTCKIEELGSELKEHYKIEAFTPLLAPAQEPVTLLGQIGCDSNGKLNNKSVILEGDREHSSGAQIPVDLSELKEYSLFPGQVVIMEGINTTGRKLVATKLYEGVPLPFYQPTEEDADFEQSMVLVACGPYTTSDSITYDPLLDLIAVINHDRPDVCILFGPFLDAKHEQVENCLLTSPFEDIFKQCLRTIIEGTRSSGSHLVFVPSLRDVHHEPVYPQPPFSYSDLSREDKKQVQFVSEPCSLSINGVIFGLTSTDLLFHLGAEEISSSTSDRFSRILKHILTQRSYYPLYPPQEDMAIDYESFYVYAQLPVTPDVLIIPSELRYFVKDVLGCVCVNPGRLTKGQVGGTFARLYLRRPAADGAERQSPCIAVQVVRI Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Voltage-gated sodium channel alpha Nav1.5 (SCN5A) | 6LQA | 7.97 | |
Target general information Gen name SCN5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Voltage-gated sodium channel subunit alpha Nav1.5; Sodium channel protein type V subunit alpha; Sodium channel protein type 5 subunit alpha; Sodium channel protein cardiac muscle subunit alpha; HH1 Protein family Sodium channel (TC 1.A.1.10) family, Nav1.5/SCN5A subfamily Biochemical class Voltage-gated ion channel Function Assuming opened or closed conformations in response to the voltage difference across the membrane, the protein forms a sodium-selective channel through which Na(+) ions may pass in accordance with their electrochemical gradient. It is a tetrodotoxin-resistant Na(+) channel isoform. This channel is responsible for the initial upstroke of the action potential. Channel inactivation is regulated by intracellular calcium levels. This protein mediates the voltage-dependent sodium ion permeability of excitable membranes. Related diseases Progressive familial heart block 1A (PFHB1A) [MIM:113900]: A cardiac bundle branch disorder characterized by progressive alteration of cardiac conduction through the His-Purkinje system, with a pattern of a right bundle-branch block and/or left anterior hemiblock occurring individually or together. It leads to complete atrio-ventricular block causing syncope and sudden death. {ECO:0000269|PubMed:11234013, ECO:0000269|PubMed:11804990, ECO:0000269|PubMed:12569159, ECO:0000269|PubMed:12574143, ECO:0000269|PubMed:19251209, ECO:0000269|PubMed:23420830}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Long QT syndrome 3 (LQT3) [MIM:603830]: A heart disorder characterized by a prolonged QT interval on the ECG and polymorphic ventricular arrhythmias. They cause syncope and sudden death in response to exercise or emotional stress, and can present with a sentinel event of sudden cardiac death in infancy. {ECO:0000269|PubMed:10377081, ECO:0000269|PubMed:10508990, ECO:0000269|PubMed:10590249, ECO:0000269|PubMed:10627139, ECO:0000269|PubMed:10911008, ECO:0000269|PubMed:10973849, ECO:0000269|PubMed:11304498, ECO:0000269|PubMed:11410597, ECO:0000269|PubMed:11710892, ECO:0000269|PubMed:11889015, ECO:0000269|PubMed:11997281, ECO:0000269|PubMed:12209021, ECO:0000269|PubMed:12454206, ECO:0000269|PubMed:12673799, ECO:0000269|PubMed:15840476, ECO:0000269|PubMed:16414944, ECO:0000269|PubMed:16922724, ECO:0000269|PubMed:18060054, ECO:0000269|PubMed:18378609, ECO:0000269|PubMed:18451998, ECO:0000269|PubMed:18708744, ECO:0000269|PubMed:18848812, ECO:0000269|PubMed:18929331, ECO:0000269|PubMed:19716085, ECO:0000269|PubMed:26392562, ECO:0000269|PubMed:7651517, ECO:0000269|PubMed:7889574, ECO:0000269|PubMed:8541846, ECO:0000269|PubMed:9506831, ECO:0000269|PubMed:9686753, ECO:0000269|Ref.36}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 1 (BRGDA1) [MIM:601144]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:10532948, ECO:0000269|PubMed:10618304, ECO:0000269|PubMed:10690282, ECO:0000269|PubMed:11410597, ECO:0000269|PubMed:11748104, ECO:0000269|PubMed:11823453, ECO:0000269|PubMed:11901046, ECO:0000269|PubMed:12051963, ECO:0000269|PubMed:12106943, ECO:0000269|PubMed:15023552, ECO:0000269|PubMed:15338453, ECO:0000269|PubMed:15579534, ECO:0000269|PubMed:15851320, ECO:0000269|PubMed:16266370, ECO:0000269|PubMed:16325048, ECO:0000269|PubMed:16616735, ECO:0000269|PubMed:17075016, ECO:0000269|PubMed:17081365, ECO:0000269|PubMed:17198989, ECO:0000269|PubMed:18252757, ECO:0000269|PubMed:18341814, ECO:0000269|PubMed:18451998, ECO:0000269|PubMed:18456723, ECO:0000269|PubMed:18616619, ECO:0000269|PubMed:19251209, ECO:0000269|PubMed:19272188, ECO:0000269|PubMed:20129283, ECO:0000269|PubMed:23085483, ECO:0000269|PubMed:23420830, ECO:0000269|PubMed:24167619, ECO:0000269|PubMed:26279430, ECO:0000269|PubMed:26776555, ECO:0000269|PubMed:32850980, ECO:0000269|PubMed:9521325}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Sick sinus syndrome 1 (SSS1) [MIM:608567]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS1 onset is in utero, infancy, or early childhood. {ECO:0000269|PubMed:11748104, ECO:0000269|PubMed:14523039, ECO:0000269|PubMed:22795782}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Familial paroxysmal ventricular fibrillation 1 (VF1) [MIM:603829]: A cardiac arrhythmia marked by fibrillary contractions of the ventricular muscle due to rapid repetitive excitation of myocardial fibers without coordinated contraction of the ventricle and by absence of atrial activity. {ECO:0000269|PubMed:10940383}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Sudden infant death syndrome (SIDS) [MIM:272120]: SIDS is the sudden death of an infant younger than 1 year that remains unexplained after a thorough case investigation, including performance of a complete autopsy, examination of the death scene, and review of clinical history. Pathophysiologic mechanisms for SIDS may include respiratory dysfunction, cardiac dysrhythmias, cardiorespiratory instability, and inborn errors of metabolism, but definitive pathogenic mechanisms precipitating an infant sudden death remain elusive. {ECO:0000269|PubMed:18596570, ECO:0000269|PubMed:19302788}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Atrial standstill 1 (ATRST1) [MIM:108770]: A rare arrhythmia characterized by the absence of electrical and mechanical activity in the atria. Electrocardiographically, it is characterized by bradycardia, the absence of P waves, and a junctional narrow complex escape rhythm. {ECO:0000269|PubMed:12522116, ECO:0000269|PubMed:23420830}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry. A mutation in SCN5A has been detected in combination with a rare GJA5 genotype in a large family with atrial standstill.; DISEASE: Cardiomyopathy, dilated, 1E (CMD1E) [MIM:601154]: A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. {ECO:0000269|PubMed:15466643, ECO:0000269|PubMed:23420830}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Atrial fibrillation, familial, 10 (ATFB10) [MIM:614022]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:18088563, ECO:0000269|PubMed:18378609}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01426; DB09088; DB01429; DB00868; DB13746; DB05541; DB00564; DB01161; DB00527; DB00907; DB13269; DB00280; DB04855; DB01228; DB00754; DB13961; DB01195; DB01320; DB00473; DB00192; DB11633; DB00555; DB00281; DB00532; DB00379; DB00680; DB00776; DB11186; DB00252; DB09345; DB00750; DB01035; DB01069; DB01182; DB09342; DB00908; DB01346; DB00243; DB00740; DB09085; DB01056; DB00273; DB00313; DB06217; DB00909 Interacts with P0DP25; Q13557; P61328-2; P26045; Q49AR9; Q8N9N5-2; Q9Y3B6; Q8WW24; Q96E35 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Atrial fibrillation; Brugada syndrome; Calmodulin-binding; Cardiomyopathy; Cell junction; Cell membrane; Cytoplasm; Disease variant; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Long QT syndrome; Membrane; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Ubl conjugation; Voltage-gated channel Protein physicochemical properties Chain ID B Molecular weight (Da) 131190 Length 1151 Aromaticity 0.14 Instability index 32.8 Isoelectric point 7.56 Charge (pH=7) 1.76 2D Binding mode Binding energy (Kcal/mol) -8.66  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PIRRAAVKILVHSLFNMLIMCTILTNCVFMAQHDPPPWTKYVEYTFTAIYTFESLVKILARGFCLHAFTFLRDPWNWLDFSVIIMAYTTEFVDLGNVSALRTFRVLRALKTISVISGLKTIVGALIQSVKKLADVMVLTVFCLSVFALIGLQLFMGNLRHKCVRNFTALNGTNGSVEADGLVWESLDLYLSDPENYLLKNGTSDVLLCGNSSDAGTCPEGYRCLKAGENPDHGYTSFDSFAWAFLALFRLMTQDCWERLYQQTLRSAGKIYMIFFMLVIFLGSFYLVNLILAVVAMAYEEQNQATIAETEECCPLWMSIKQGVKLVVMDPFTDLTITMCIVLNTLFMALEHYNMTSEFEEMLQVGNLVFTGIFTAEMTFKIIALDPYYYFQQGWNIFDSIIVILSLMELGLSRMSNLSVLRSFRLLRVFKLAKSWPTLNTLIKIIGNSVGALGNLTLVLAIIVFIFAVVGMQLFGKNYSELRDSDSGLLPRWHMMDFFHAFLIIFRILCGEWIETMWDCMEVSGQSLCLLVFLLVMVIGNLVVLNLFLALLLSSFSAGKVWWRLRKTCYHIVEHSWFETFIIFMILLSSGALAFEDIYLEERKTIKVLLEYADKMFTYVFVLEMLLKWVAYGFKKYFTNAWCWLDFLIVDVSLVSLVANTLGFAEMGPIKSLRTLRALRPLRALSRFEGMRVVVNALVGAIPSIMNVLLVCLIFWLIFSIMGVNLFAGKFGRCINQTEGDLPLNYTIVNNKSQCESLNLTGELYWTKVKVNFDNVGAGYLALLQVATFKGWMDIMYAAVDSRGYEEQPQWEYNLYMYIYFVIFIIFGSFFTLNLFIGVIIDNFNQQKKKLGGQDIFMTEEQKKYYNAMKKLGSKKPQKPIPRPLNKYQGFIFDIVTKQAFDVTIMFLICLNMVTMMVETDDQSPEKINILAKINLLFVAIFTGECIVKLAALRHYYFTNSWNIFDFVVVILSIVGTVLSDIIQKYFFSPTLFRVIRLARIGRILRLIRGAKGIRTLLFALMMSLPALFNIGLLLFLVMFIYSIFGMANFAYVKWEAGIDDMFNFQTFANSMLCLFQITTSAGWDGLLSPILNTGPPYCDPTLPNSNGSRGDCGSPAVGILFFTTYIIISFLIVVNMYIAIILENFSVATEE Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Phosphodiesterase 10A (PDE10) | 2OUR | 7.96 | |
Target general information Gen name PDE10A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP and cAMPinhibited cGMP 3',5'cyclic phosphodiesterase 10A; cAMP and cAMP-inhibited cGMP 3',5'-cyclic phosphodiesterase 10A Protein family Cyclic nucleotide phosphodiesterase family Biochemical class Phosphoric diester hydrolase Function Can hydrolyze both cAMP and cGMP, but has higher affinity for cAMP and is more efficient with cAMP as substrate. May play a critical role in regulating cAMP and cGMP levels in the striatum, a region of the brain that contributes to the control of movement and cognition. Plays a role in signal transduction by regulating the intracellular concentration of cyclic nucleotides. Related diseases Dyskinesia, limb and orofacial, infantile-onset (IOLOD) [MIM:616921]: An autosomal recessive, early-onset hyperkinetic movement disorder characterized by axial hypotonia, dyskinesia of the limbs and trunk, orofacial dyskinesia, drooling, and dysarthria. The severity of the hyperkinesis is variable. {ECO:0000269|PubMed:27058446}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Striatal degeneration, autosomal dominant 2 (ADSD2) [MIM:616922]: An autosomal dominant disorder characterized by striatal degeneration and dysfunction of basal ganglia, resulting in hyperkinesis. {ECO:0000269|PubMed:27058447}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08384; DB08386; DB08383; DB08389; DB00201; DB00975; DB08387; DB01113; DB08391; DB08811; DB09283; DB08814 Interacts with NA EC number EC 3.1.4.17 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative initiation; Alternative splicing; cAMP; cAMP-binding; cGMP; cGMP-binding; Cytoplasm; Disease variant; Hydrolase; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37469.9 Length 324 Aromaticity 0.1 Instability index 48.52 Isoelectric point 6.28 Charge (pH=7) -4.08 2D Binding mode Binding energy (Kcal/mol) -8.53 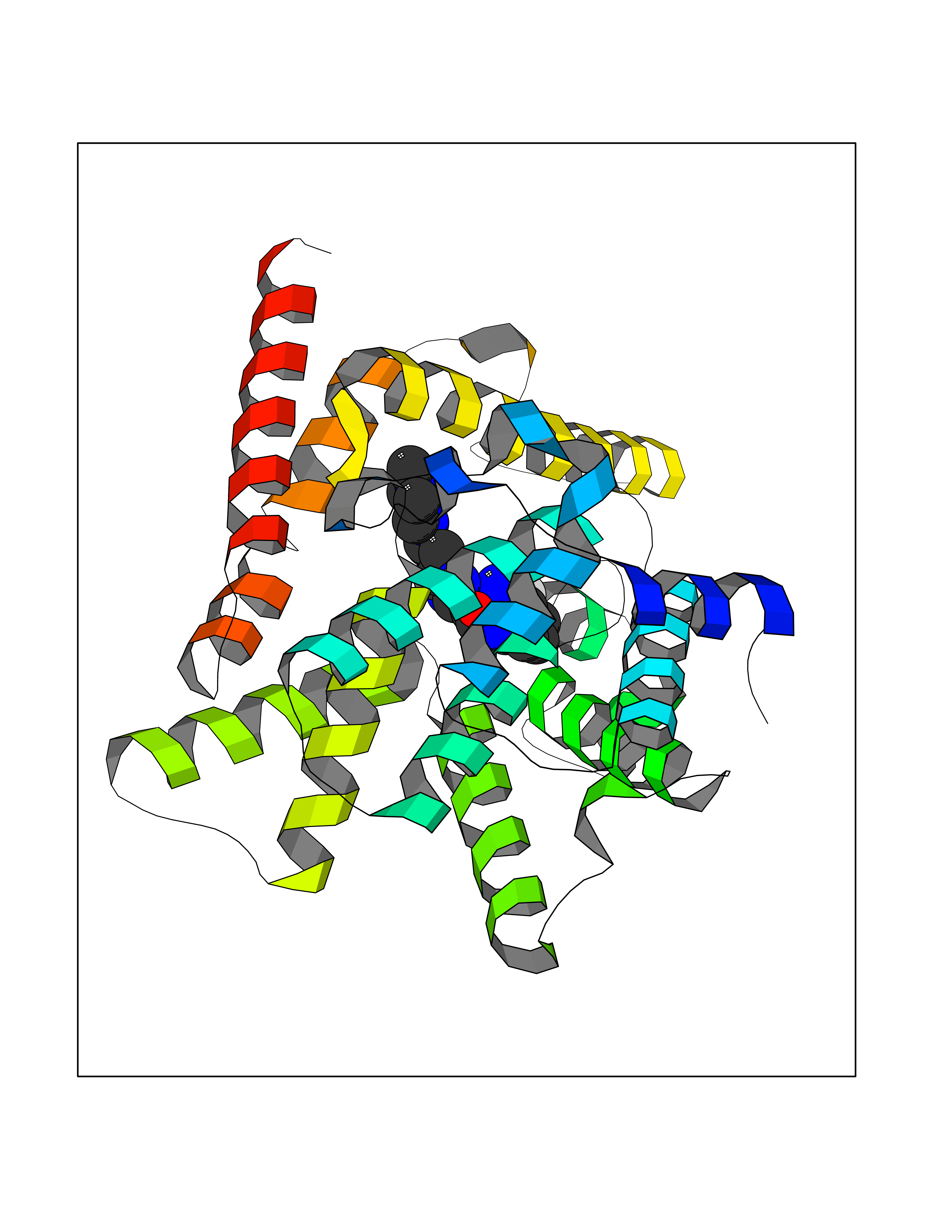 Molscript Map 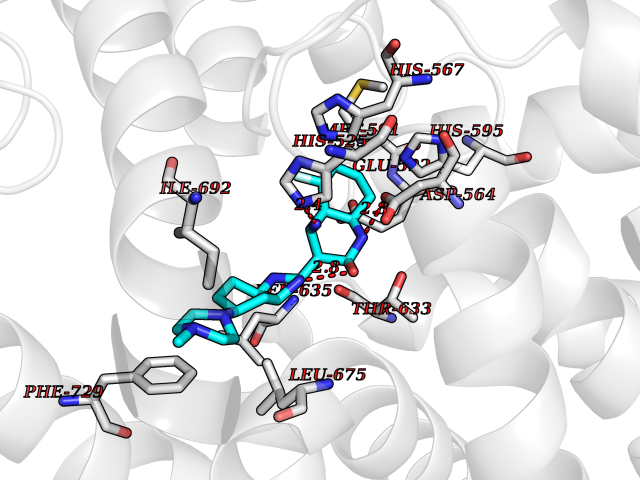 Pymol Map 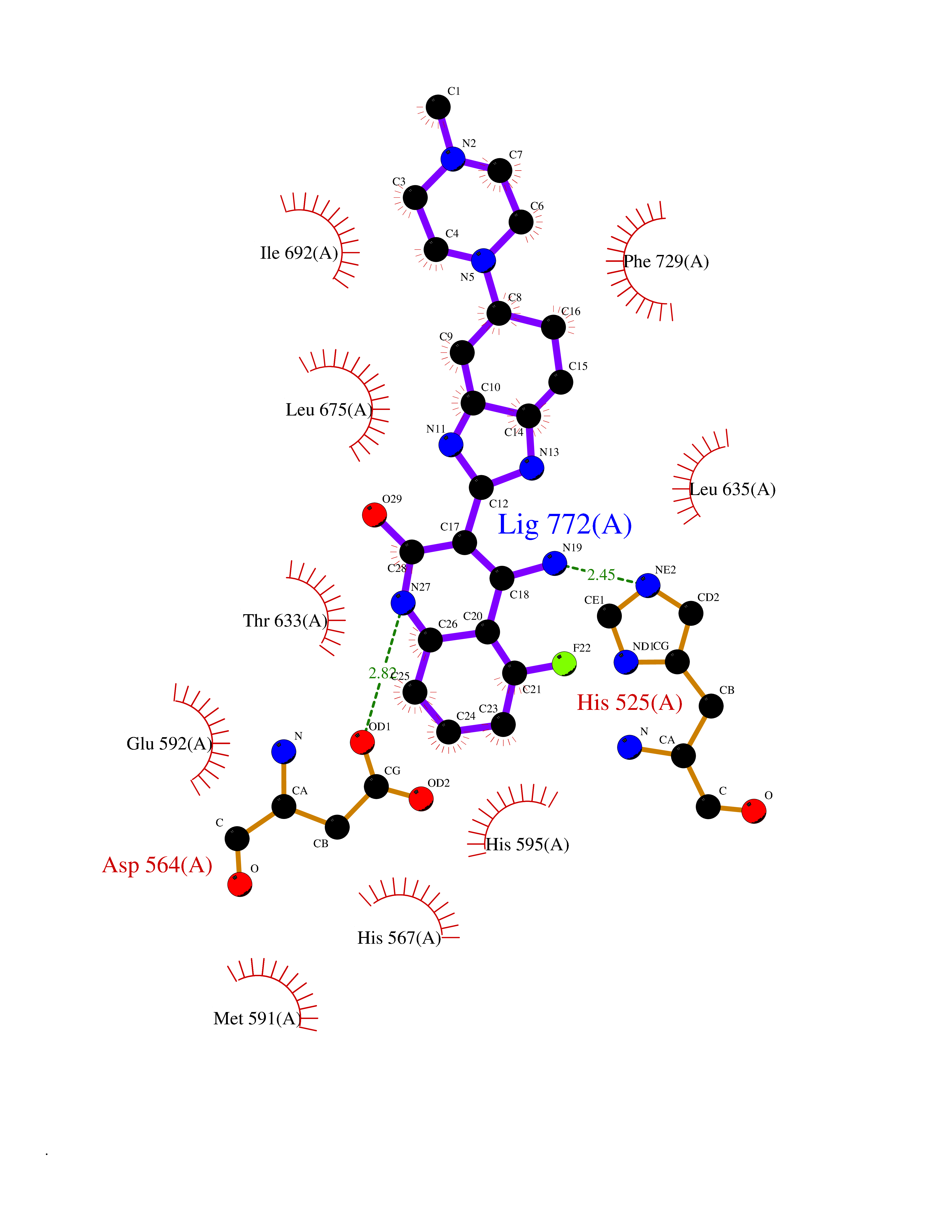 Ligplot Map 3D Binding mode Sequence HMSICTSEEWQGLMQFTLPVRLCKEIELFHFDIGPFENMWPGIFVYMVHRSCGTSCFELEKLCRFIMSVKKNYRRVPYHNWKHAVTVAHCMYAILQNNHTLFTDLERKGLLIACLCHDLDHRGFSNSYLQKFDHPLAALYSTSTMEQHHFSQTVSILQLEGHNIFSTLSSSEYEQVLEIIRKAIIATDLALYFGNRKQLEEMYQTGSLNLNNQSHRDRVIGLMMTACALCSVTKLWPVTKLTANDIYAEFWAEGDEMKKLGIQPIPMMDRDKKDEVPQGQLGFYNAVAIPCYTTLTQILPPTEPLLKACRDNLSQWEKVIRGEE Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Kallikrein-5 (KLK5) | 6QFE | 7.96 | |
Target general information Gen name KLK5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ570/PRO1132; Stratum corneum tryptic enzyme; SCTE; Kallikrein-like protein 2; KLK-L2 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function May be involved in desquamation. Related diseases Lipodystrophy, familial partial, 8 (FPLD8) [MIM:620679]: An autosomal dominant form of partial lipodystrophy, a disorder characterized by abnormal subcutaneous fat distribution. FPLD8 patients show selective loss of subcutaneous adipose tissue from the limbs, beginning around 13 to 15 years of age, and abnormal accumulation of subcutaneous adipose tissue in the dorsal neck and face, as well as in the posterior thoracic and abdominal regions. The disorder is associated with metabolic abnormalities, including diabetes mellitus and hyperlipidemia. {ECO:0000269|PubMed:27376152}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P20930; Q9NQG1 EC number EC 3.4.21.- Uniprot keywords 3D-structure; Disulfide bond; Glycoprotein; Hydrolase; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50299.2 Length 454 Aromaticity 0.07 Instability index 40.74 Isoelectric point 9.25 Charge (pH=7) 23.09 2D Binding mode Binding energy (Kcal/mol) -8.57 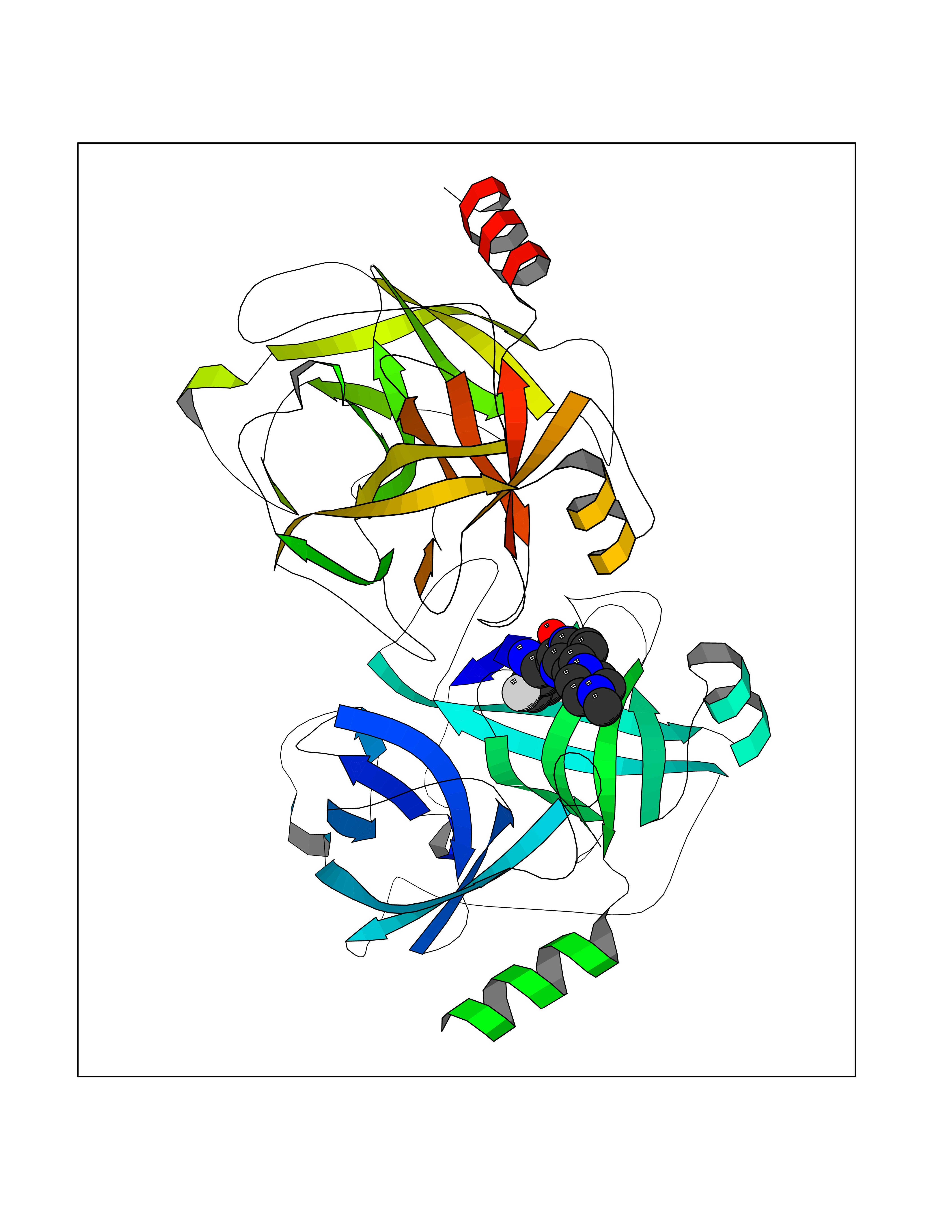 Molscript Map 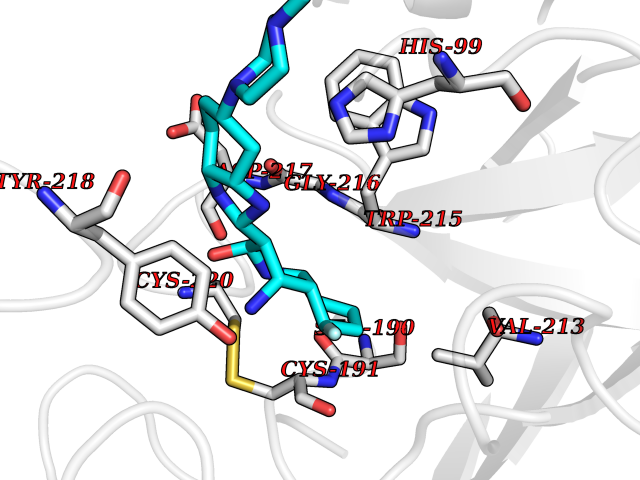 Pymol Map 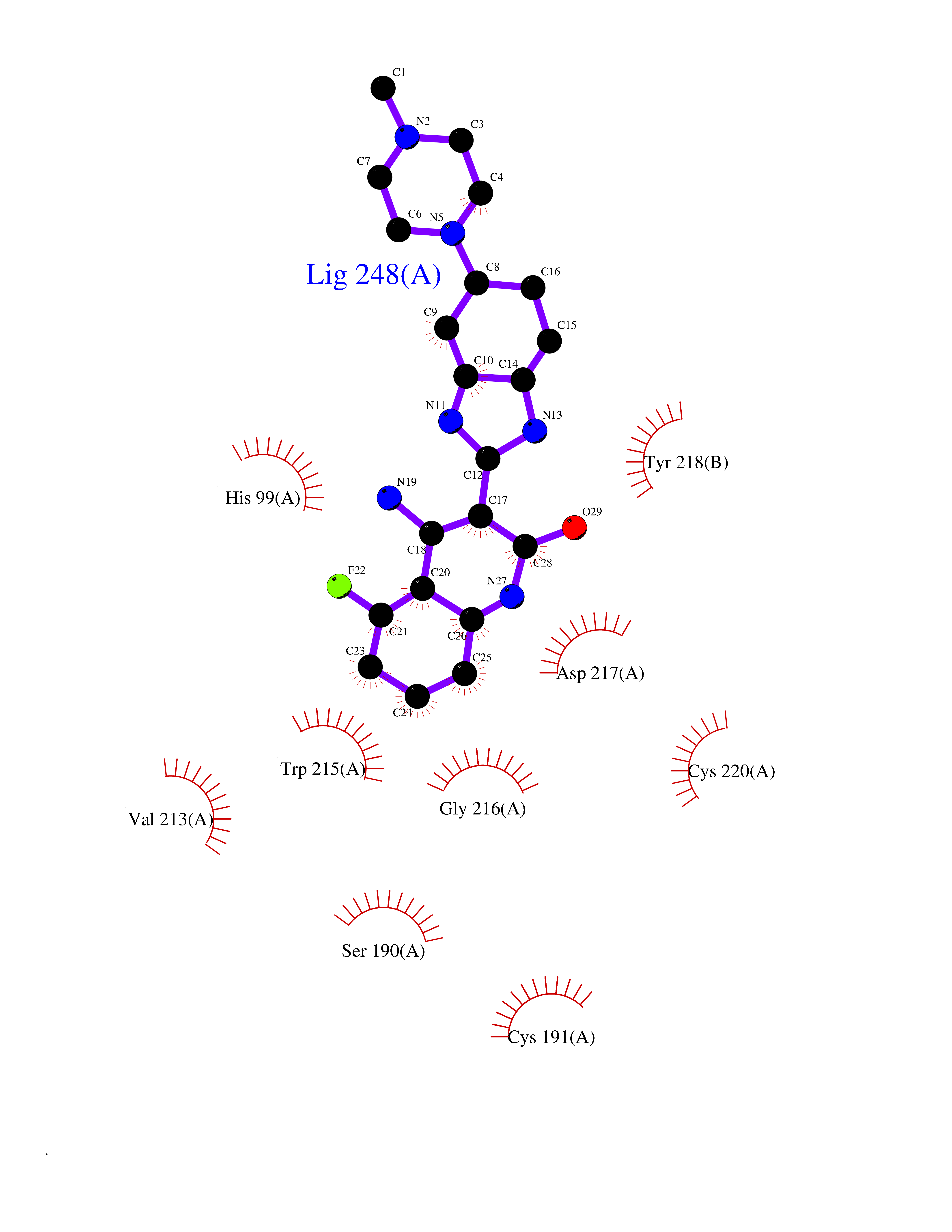 Ligplot Map 3D Binding mode Sequence IINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANSIINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANS Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Glutamate carboxypeptidase III (NAALAD2) | 3FED | 7.95 | |
Target general information Gen name NAALAD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NAALADase II; NAALAD2 Protein family Peptidase M28 family, M28B subfamily Biochemical class Peptidase Function Has N-acetylated-alpha-linked-acidic dipeptidase (NAALADase) activity. Also exhibits a dipeptidyl-peptidase IV type activity. Inactivate the peptide neurotransmitter N- acetylaspartylglutamate. Related diseases Dystonia 1, torsion, autosomal dominant (DYT1) [MIM:128100]: A primary torsion dystonia, and the most common and severe form. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Dystonia type 1 is characterized by involuntary, repetitive, sustained muscle contractions or postures involving one or more sites of the body, in the absence of other neurological symptoms. Typically, symptoms develop first in an arm or leg in middle to late childhood and progress in approximately 30% of patients to other body regions (generalized dystonia) within about five years. 'Torsion' refers to the twisting nature of body movements observed in DYT1, often affecting the trunk. Distribution and severity of symptoms vary widely between affected individuals, ranging from mild focal dystonia to severe generalized dystonia, even within families. {ECO:0000269|PubMed:14970196, ECO:0000269|PubMed:15505207, ECO:0000269|PubMed:16361107, ECO:0000269|PubMed:17428918, ECO:0000269|PubMed:18167355, ECO:0000269|PubMed:18477710, ECO:0000269|PubMed:18827015, ECO:0000269|PubMed:19955557, ECO:0000269|PubMed:20169475, ECO:0000269|PubMed:21102408, ECO:0000269|PubMed:24930953, ECO:0000269|PubMed:27490483, ECO:0000269|PubMed:9288096}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Arthrogryposis multiplex congenita 5 (AMC5) [MIM:618947]: A form of arthrogryposis multiplex congenita, a developmental condition characterized by multiple joint contractures resulting from reduced or absent fetal movements. AMC5 is an autosomal recessive form characterized by severe congenital contractures, developmental delay, strabismus and tremor. {ECO:0000269|PubMed:28516161, ECO:0000269|PubMed:29053766, ECO:0000269|PubMed:30244176}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UHD4; Q6NTF9-3; B2RUZ4; O76024 EC number EC 3.4.17.21 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Carboxypeptidase; Cell membrane; Dipeptidase; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Multifunctional enzyme; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 77761.6 Length 690 Aromaticity 0.12 Instability index 39.42 Isoelectric point 8.48 Charge (pH=7) 4.65 2D Binding mode Binding energy (Kcal/mol) -9.54 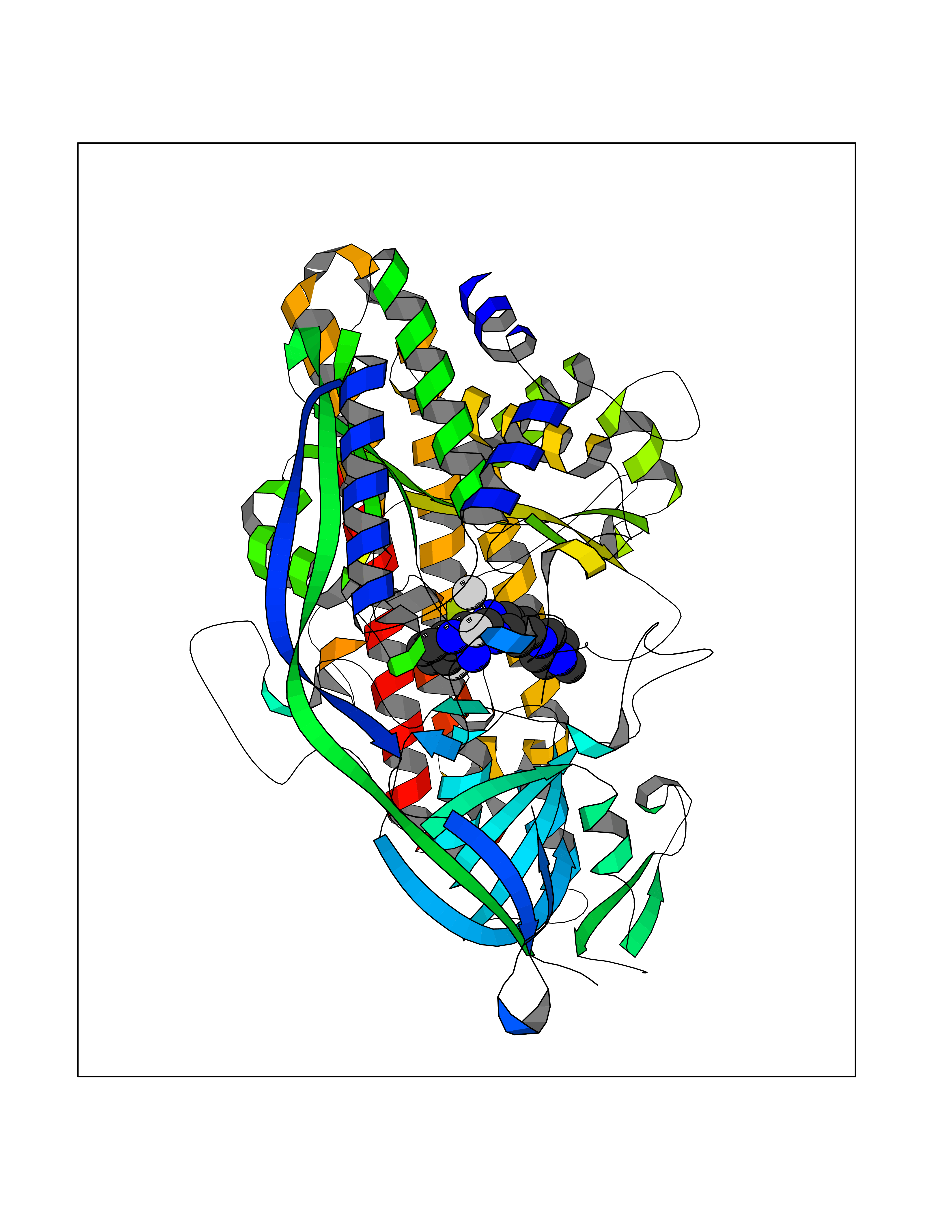 Molscript Map 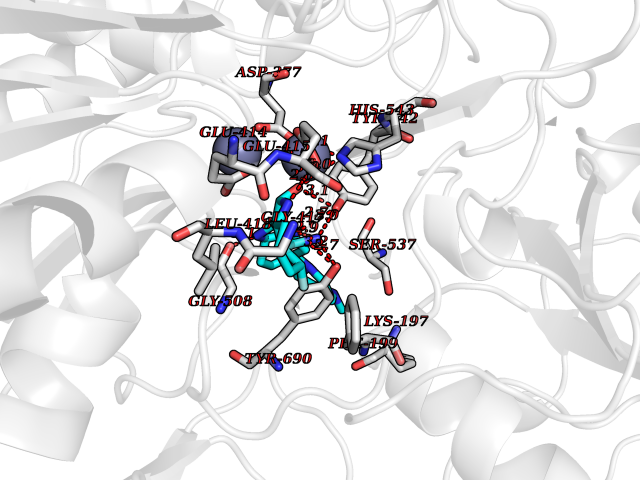 Pymol Map 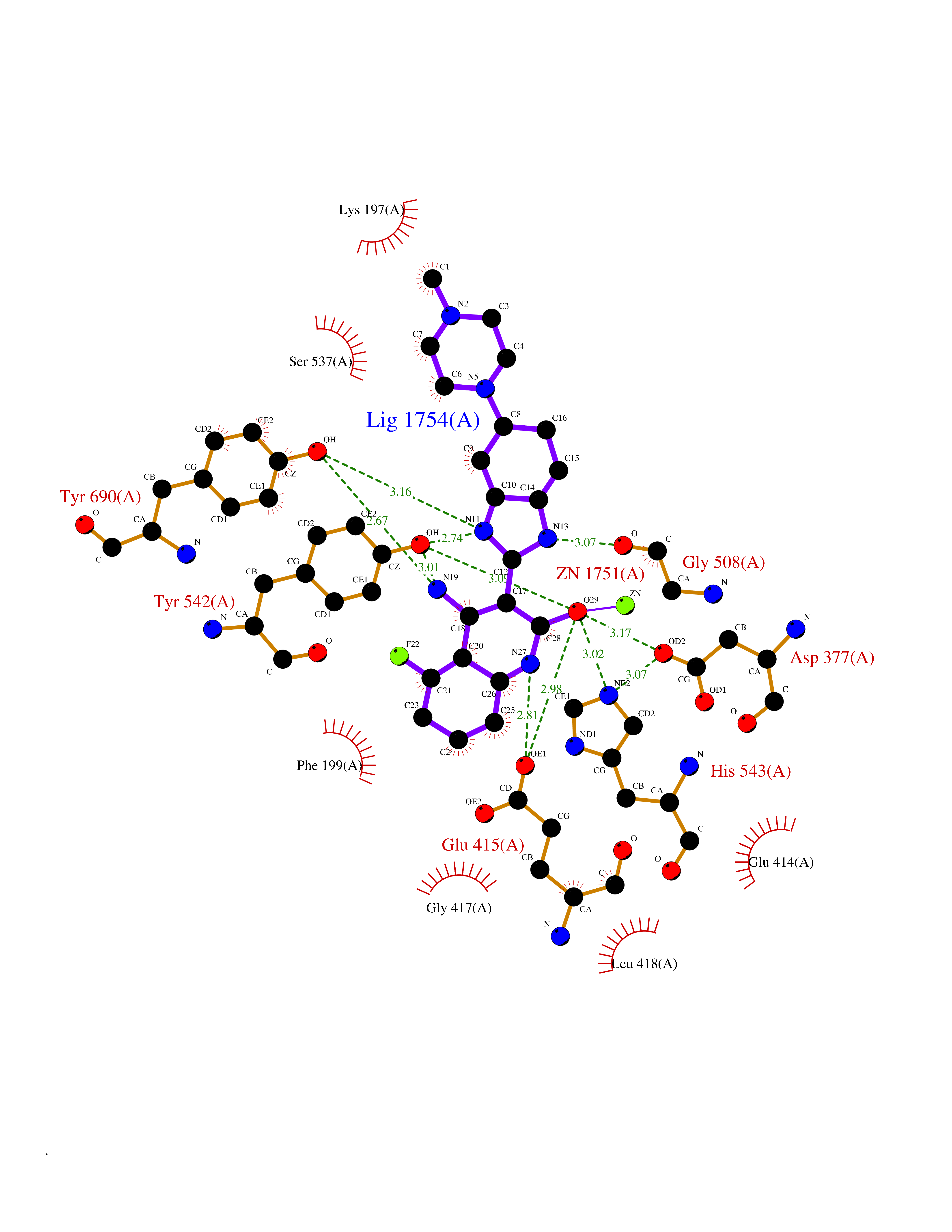 Ligplot Map 3D Binding mode Sequence SIRWKLVSEMKAENIKSFLRSFTKLPHLAGTEQNFLLAKKIQTQWKKFGLDSAKLVHYDVLLSYPNETNANYISIVDEHETEIFKTSPPPDGYENVTNIVPPYNAFSAQGMPEGDLVYVNYARTEDFFKLEREMGINCTGKIVIARYGKIFRGNKVKNAMLAGAIGIILYSDPADYFAPEVQPYPKGWNLPGTAAQRGNVLNLNGAGDPLTPGYPAKEYTFRLDVEEGVGIPRIPVHPIGYNDAEILLRYLGGIAPPDKSWKGALNVSYSIGPGFTGSSFRKVRMHVYNINKITRIYNVVGTIRGSVEPDRYVILGGHRDSWVFGAIDPTSGVAVLQEIARSFGKLMSKGWRPRRTIIFASWDAEEFGLLGSTEWAEENVKILQERSIAYINSDSSIEGNYTLRVDCTPLLYQLVYKLTKEIPSPDDGFESKSLYESWLEKDPSPENKNLPRINKLGSGSDFEAYFQRLGIASGRARYTKNKKTDKYSSYPVYHTIYETFELVEKFYDPTFKKQLSVAQLRGALVYELVDSKIIPFNIQDYAEALKNYAASIYNLSKKHDQQLTDHGVSFDSLFSAVKNFSEAASDFHKRLIQVDLNNPIAVRMMNDQLMLLERAFIDPLGLPGKLFYRHIIFAPSSHNKYAGESFPGIYDAIFDIENKANSRLAWKEVKKHISIAAFTIQAAAGTLKEV Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Melanoma derived growth regulator (MIA) | 5IXB | 7.94 | |
Target general information Gen name MIA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Melanoma-derived growth regulatory protein; Melanoma inhibitory activity protein Protein family MIA/OTOR family Biochemical class NA Function Elicits growth inhibition on melanoma cells in vitro as well as some other neuroectodermal tumors, including gliomas. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Disulfide bond; Growth factor; Proteomics identification; Reference proteome; Secreted; SH3 domain; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 11955.6 Length 105 Aromaticity 0.12 Instability index 26.45 Isoelectric point 8.7 Charge (pH=7) 2.64 2D Binding mode Binding energy (Kcal/mol) -8.17  Molscript Map 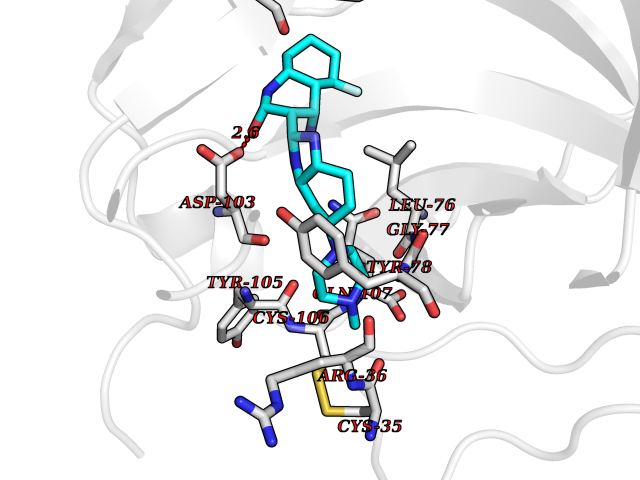 Pymol Map 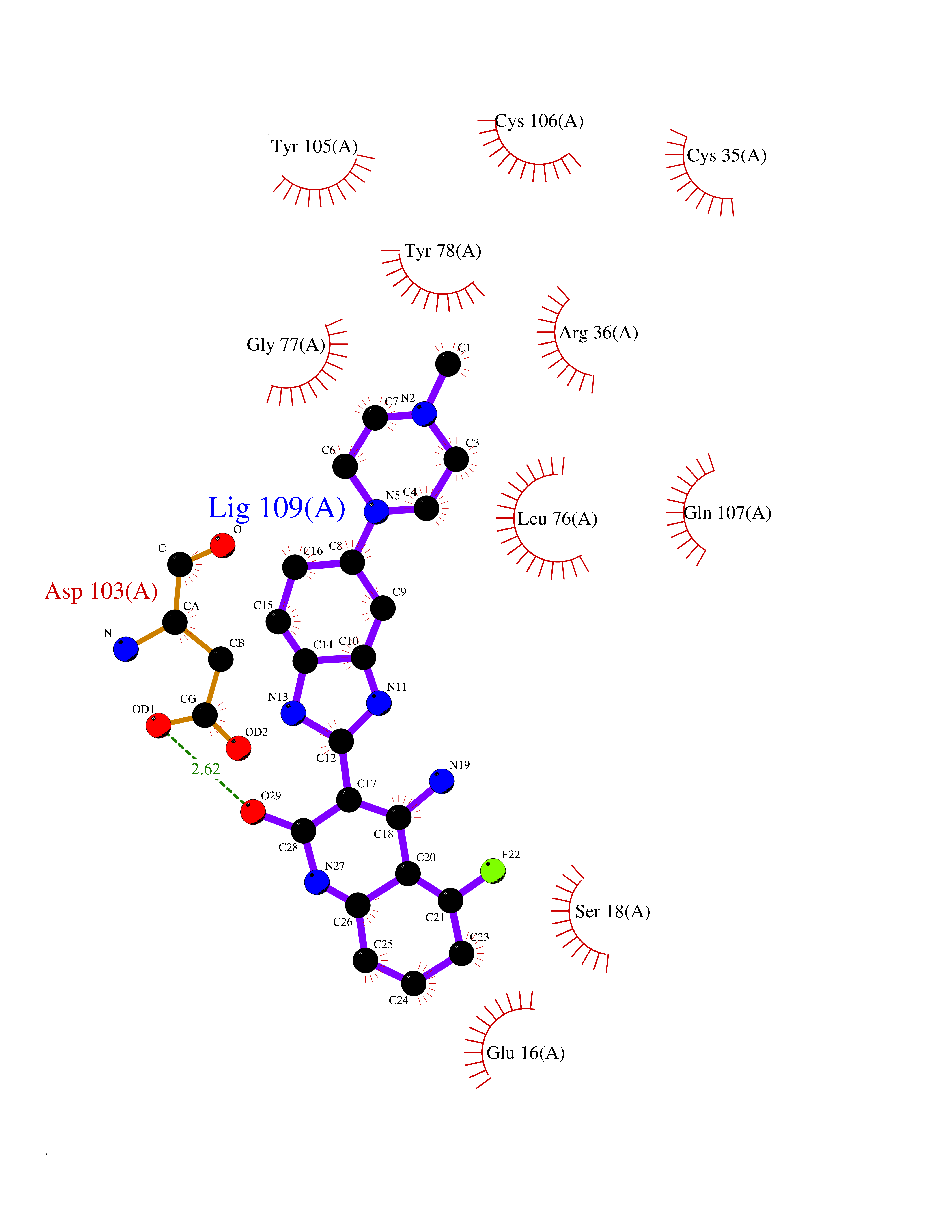 Ligplot Map 3D Binding mode Sequence MPKLADRKLCADQECSHPISMAVALQDYMAPDCRFLTIHRGQVVYVFSKLKGRGRLFWGGSVQGDYYGDLAARLGYFPSSIVREDQTLKPGKVDVKTDKWDFYCQ Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | 1-deoxy-D-xylulose-5-phosphate synthase (EC 2.2.1.7) (1-deoxyxylulose-5-phosphate synthase) (DXP synthase) (DXPS) | 2O1X | 7.93 | |
Target general information Gen name dxs Organism Deinococcus radiodurans (strain ATCC 13939 / DSM 20539 / JCM 16871 / CCUG 27074 / LMG 4051 / NBRC 15346 / NCIMB 9279 / VKM B-1422 / R1) Uniprot ID TTD ID NA Synonyms DR_1475 Protein family Transketolase family, DXPS subfamily Biochemical class NA Function Catalyzes the acyloin condensation reaction between C atoms 2 and 3 of pyruvate and glyceraldehyde 3-phosphate to yield 1-deoxy-D-xylulose-5-phosphate (DXP). {ECO:0000250}. Related diseases Brugada syndrome 7 (BRGDA7) [MIM:613120]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:20031595}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Atrial fibrillation, familial, 16 (ATFB16) [MIM:613120]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:20558140, ECO:0000269|PubMed:21051419}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 2.2.1.7 Uniprot keywords 3D-structure; Isoprene biosynthesis; Magnesium; Metal-binding; Reference proteome; Thiamine biosynthesis; Thiamine pyrophosphate; Transferase Protein physicochemical properties Chain ID C,D Molecular weight (Da) 114550 Length 1068 Aromaticity 0.06 Instability index 28.21 Isoelectric point 5.51 Charge (pH=7) -32.76 2D Binding mode Binding energy (Kcal/mol) -8.43  Molscript Map 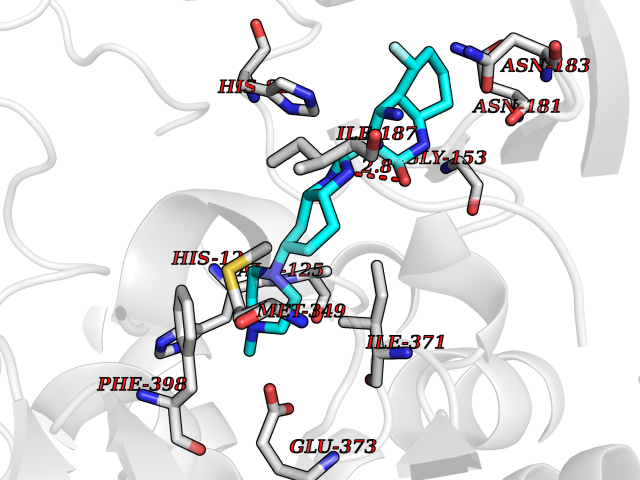 Pymol Map 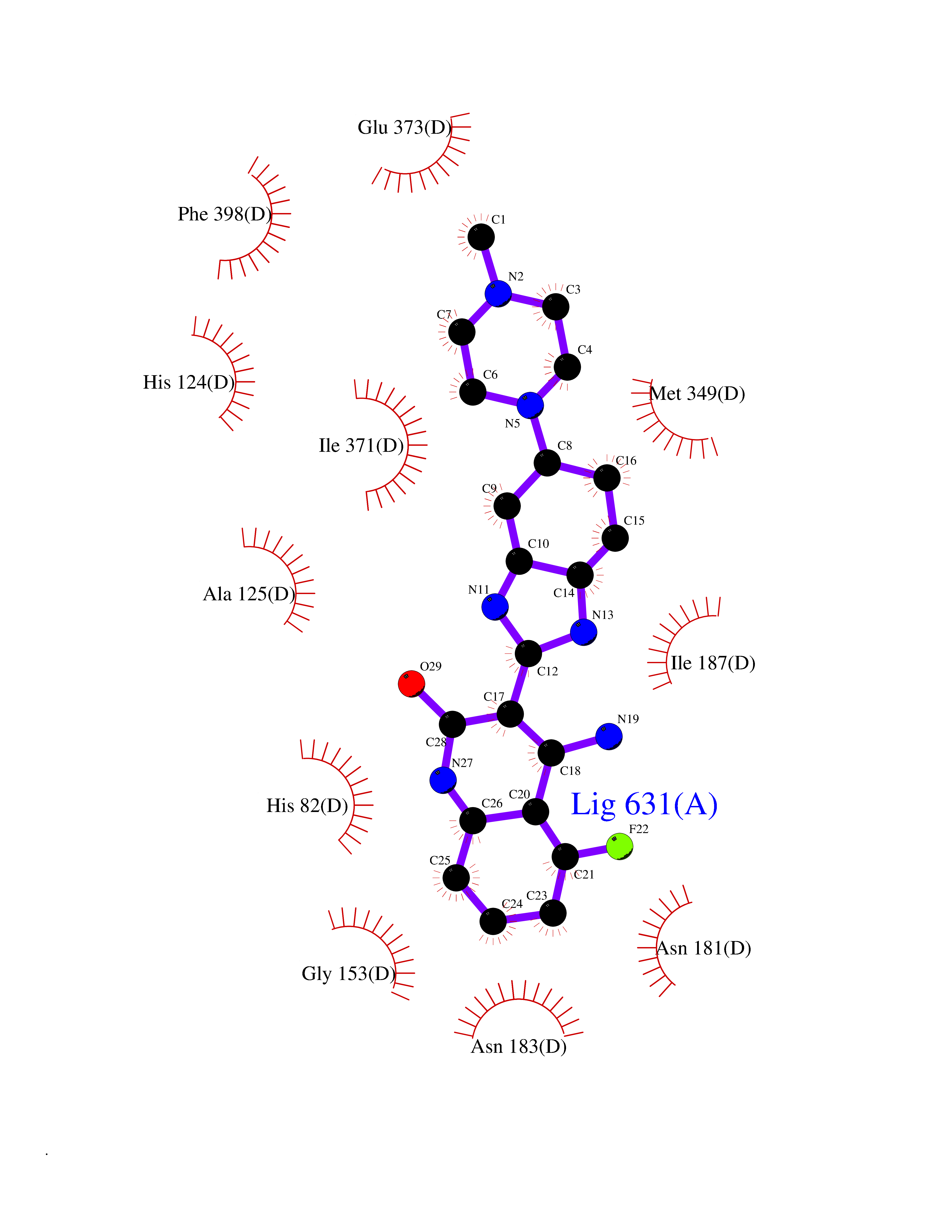 Ligplot Map 3D Binding mode Sequence HGPKDLKRLSREQLPALTEELRGEIVRVCSRGGLHLASSLGAVDIITALHYVLDSPRDRILFDVGHQAYAHKILTGRRDQMADIKKEGGISGFTKVSESEHDAITVGHASTSLTNALGMALARDAQGKDFHVAAVIGDGSLTGGMALAALNTIGDMGRKMLIVLNDNEMSISENVVNPFAAMGVRYVGPVDGHNVQELVWLLERLVDLDGPTILHIVTTKGAYSWSAAFGEAVTEWAKTDPRTFVVTPAMREGSGLVEFSRVHPHRYLDVGIAEEVAVTTAAGMALQGMRPVVAIYSTFLQRAYDQVLHDVAIEHLNVTFCIDRAGIVGADGATHNGVFDLSFLRSIPGVRIGLPKDAAELRGMLKYAQTHDGPFAIRYPRGNTAQVPAGTWPDLKWGEWERLKGGDDVVILAGGKALDYALKAAEDLPGVGVVNARFVKPLDEEMLREVGGRARALITVEDNTVVGGFGGAVLEALNSMNLHPTVRVLGIPDEFQEHATAESVHARAGIDAPAIRTVLAELGVDVPIEVTSDTPLLDQIHGPKDLKRLSREQLPALTEELRGEIVRVCSRGGLHLASSLGAVDIITALHYVLDSPRDRILFDVGHQAYAHKILTGRRDQMADIKKEGGISGFTKVSESEHDAITVGHASTSLTNALGMALARDAQGKDFHVAAVIGDGSLTGGMALAALNTIGDMGRKMLIVLNDNEMSISENVNPFAAMGVRYVGPVDGHNVQELVWLLERLVDLDGPTILHIVTTKGAYSWSAAFGEAVTEWAKTDPRTFVVTPAMREGSGLVEFSRVHPHRYLDVGIAEEVAVTTAAGMALQGMRPVVAIYSTFLQRAYDQVLHDVAIEHLNVTFCIDRAGIVGADGATHNGVFDLSFLRSIPGVRIGLPKDAAELRGMLKYAQTHDGPFAIRYPRGNTAQVPAGTWPDLKWGEWERLKGGDDVVILAGGKALDYALKAAEDLPGVGVVNARFVKPLDEEMLREVGGRARALITVEDNTVVGGFGGAVLEALNSMNLHPTVRVLGIPDEFQEHATAESVHARAGIDAPAIRTVLAELGVDVPIE Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Amylin receptor (IAPPR) | 6ZIS | 7.93 | |
Target general information Gen name CALCR-RAMP1/RAMP2/RAMP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complex of Calcitonin receptor and Receptor activity-modifying protein Protein family RAMP family Biochemical class NA Function Transports the calcitonin gene-related peptide type 1 receptor (CALCRL) to the plasma membrane. Acts as a receptor for calcitonin-gene-related peptide (CGRP) together with CALCRL. Related diseases Immunodeficiency 9 (IMD9) [MIM:612782]: An immune disorder characterized by recurrent infections, impaired activation and proliferative response of T-cells, decreased T-cell production of cytokines, and normal lymphocytes counts and serum immunoglobulin levels. In surviving patients ectodermal dysplasia with anhidrosis and non-progressive myopathy may be observed. {ECO:0000269|PubMed:16147976, ECO:0000269|PubMed:16582901}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, tubular aggregate, 2 (TAM2) [MIM:615883]: A rare congenital myopathy characterized by regular arrays of membrane tubules on muscle biopsies without additional histopathological hallmarks. Tubular aggregates in muscle are structures of variable appearance consisting of an outer tubule containing either one or more microtubule-like structures or amorphous material. TAM2 patients have myopathy and pupillary abnormalities. {ECO:0000269|PubMed:24591628, ECO:0000269|PubMed:28058752}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01278 Interacts with Q16602; P21145; Q5J8X5; Q16617 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 63832.6 Length 569 Aromaticity 0.12 Instability index 22.42 Isoelectric point 5.07 Charge (pH=7) -16.96 2D Binding mode Binding energy (Kcal/mol) -8.85  Molscript Map 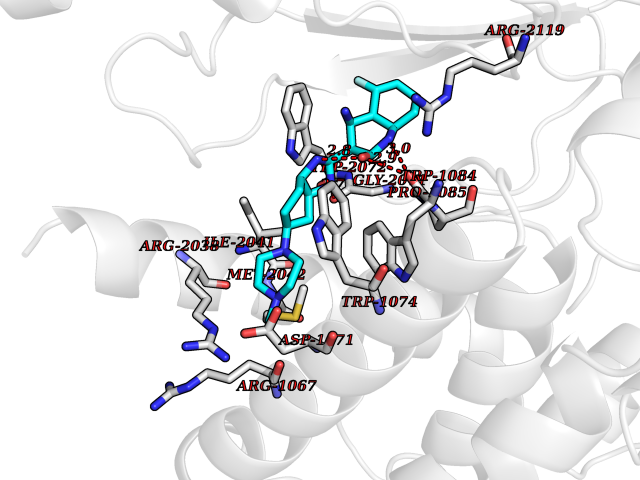 Pymol Map 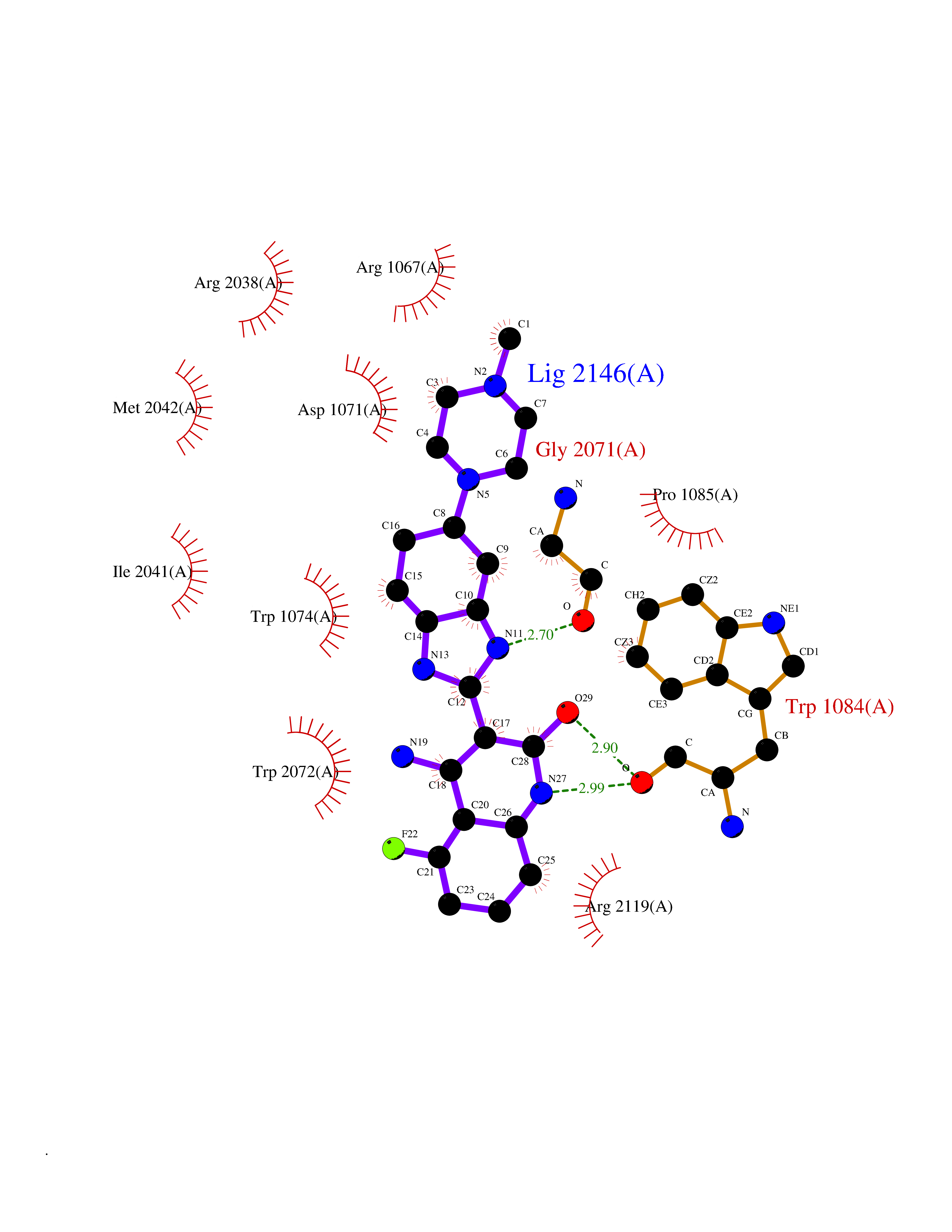 Ligplot Map 3D Binding mode Sequence SAKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQTNAAAEFTTACQEANYGALLRELCLTQFQVDMEAVGETLWCDWGRTIRSYRELADCTWHMAEKLGCFWPNAEVDRFFLAVHGRYFRSCPISIQLGVTRNKIMTAQYECYQKIMQDPIQQGVYCQRTWDGWLCWNDVAAGTESMQLCPDYFQDFDPSEKVTKICDQDGNWFRHPASQRTWTDYTQCNVNTHEKVKTALNLFYL Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Prostaglandin E2 receptor EP2 (PTGER2) | 7CX2 | 7.93 | |
Target general information Gen name PTGER2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Prostanoid EP2 receptor; Prostaglandin E2 receptor EP2 subtype; PGE2 receptor EP2 subtype; PGE receptor, EP2 subtype; PGE receptor EP2 subtype; EP2 receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function The activity of this receptor is mediated by G(s) proteins that stimulate adenylate cyclase. The subsequent raise in intracellular cAMP is responsible for the relaxing effect of this receptor on smooth muscle. Receptor for prostaglandin E2 (PGE2). Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00770; DB00917; DB08964; DB09211; DB00929; DB15071; DB16315; DB00374; DB04297 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 29928.4 Length 266 Aromaticity 0.12 Instability index 58.45 Isoelectric point 9.17 Charge (pH=7) 7.21 2D Binding mode Binding energy (Kcal/mol) -9.2 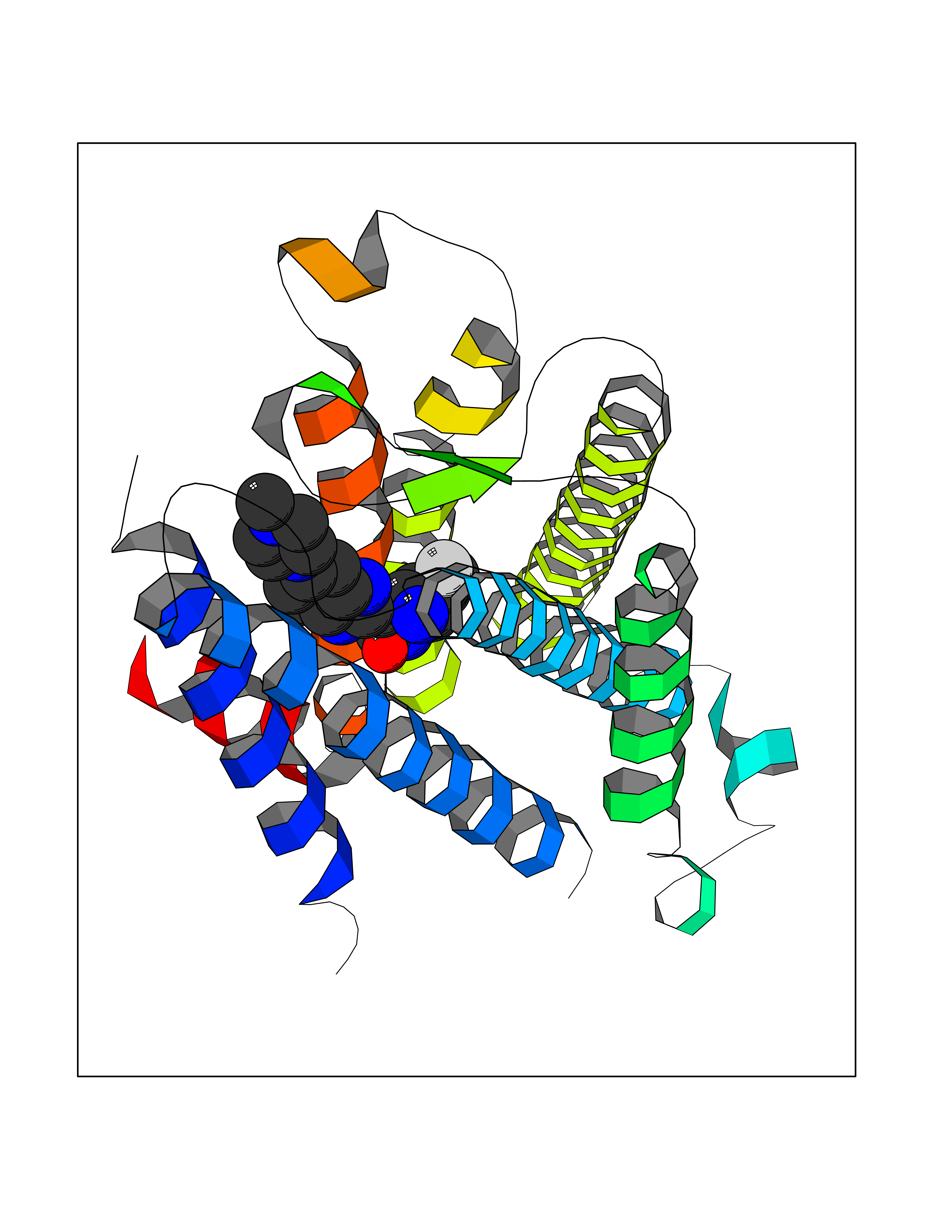 Molscript Map 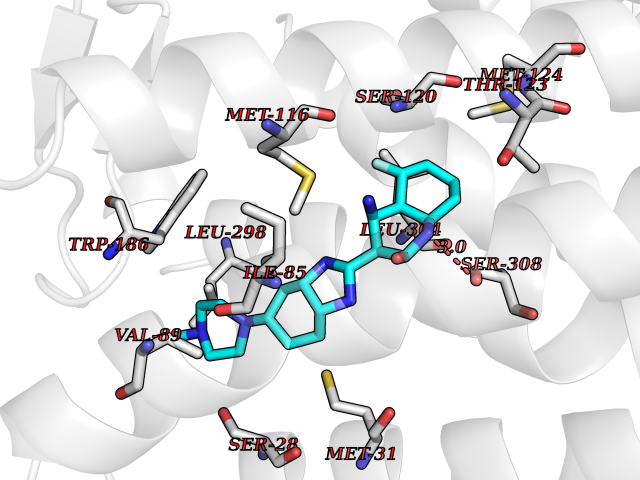 Pymol Map 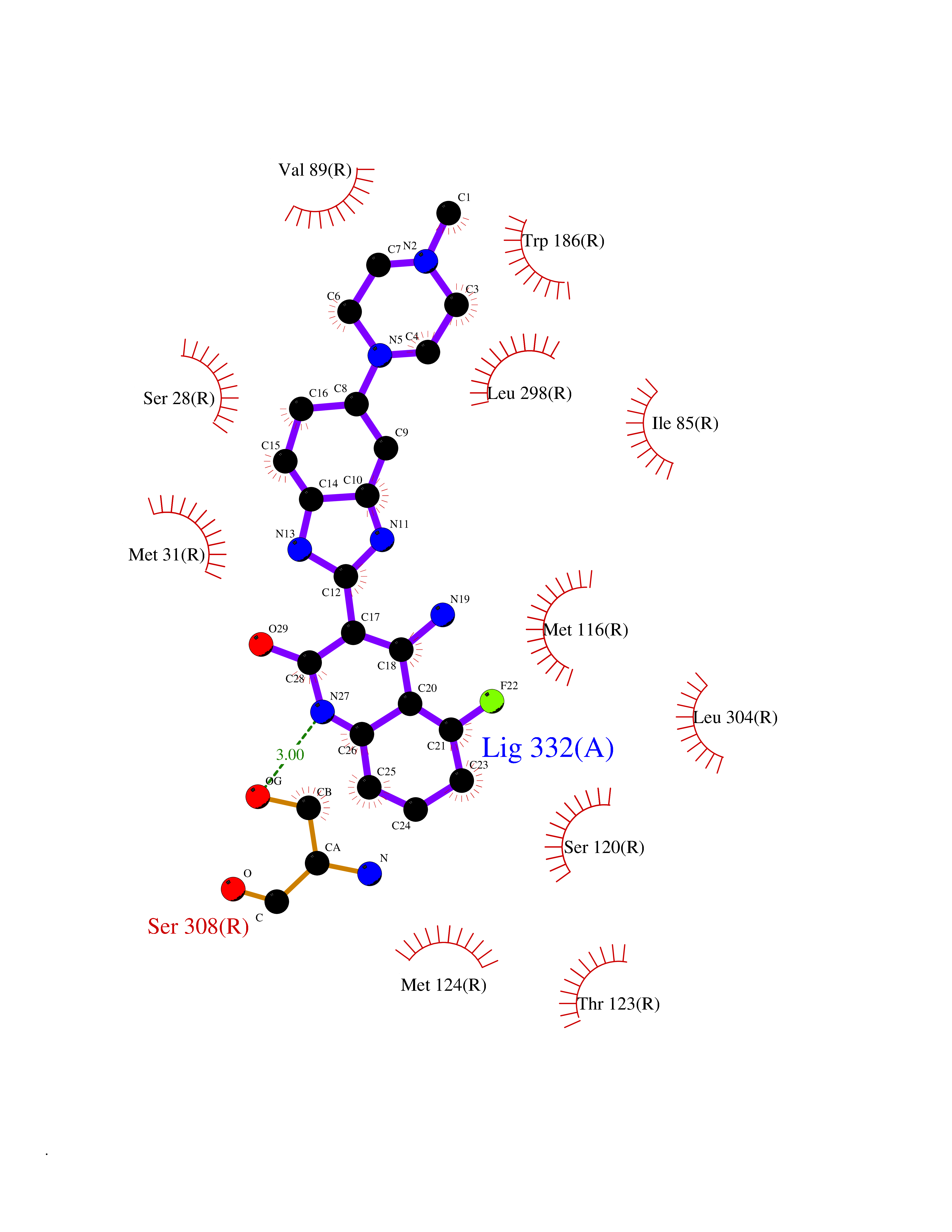 Ligplot Map 3D Binding mode Sequence ESPAISSVMFSAGVLGNLIALALLARSLFHVLVTELVFTDLLGTCLISPVVLASYARNQTLVALAPESRACTYFAFAMTFFSLATMLMLFAMALERYLSIGHPYFYQRRVSRSGGLAVLPVIYAVSLLFCSLPLLDYGQYVQYCPGTWCFIRHGRTAYLQLYATLLLLLIVSVLACNFSVILNLIRMHRRSRAEETDHLILLAIMTITFAVCSLPFTIFAYMNETSSRKEKWDLQALRFLSINSIIDPWVFAILRPPVLRLMRSVL Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Dopamine beta-hydroxylase | 4ZEL | 7.91 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -7.4  Molscript Map 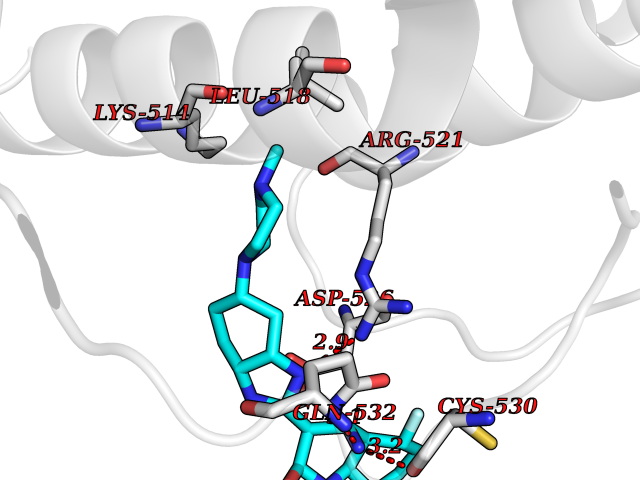 Pymol Map 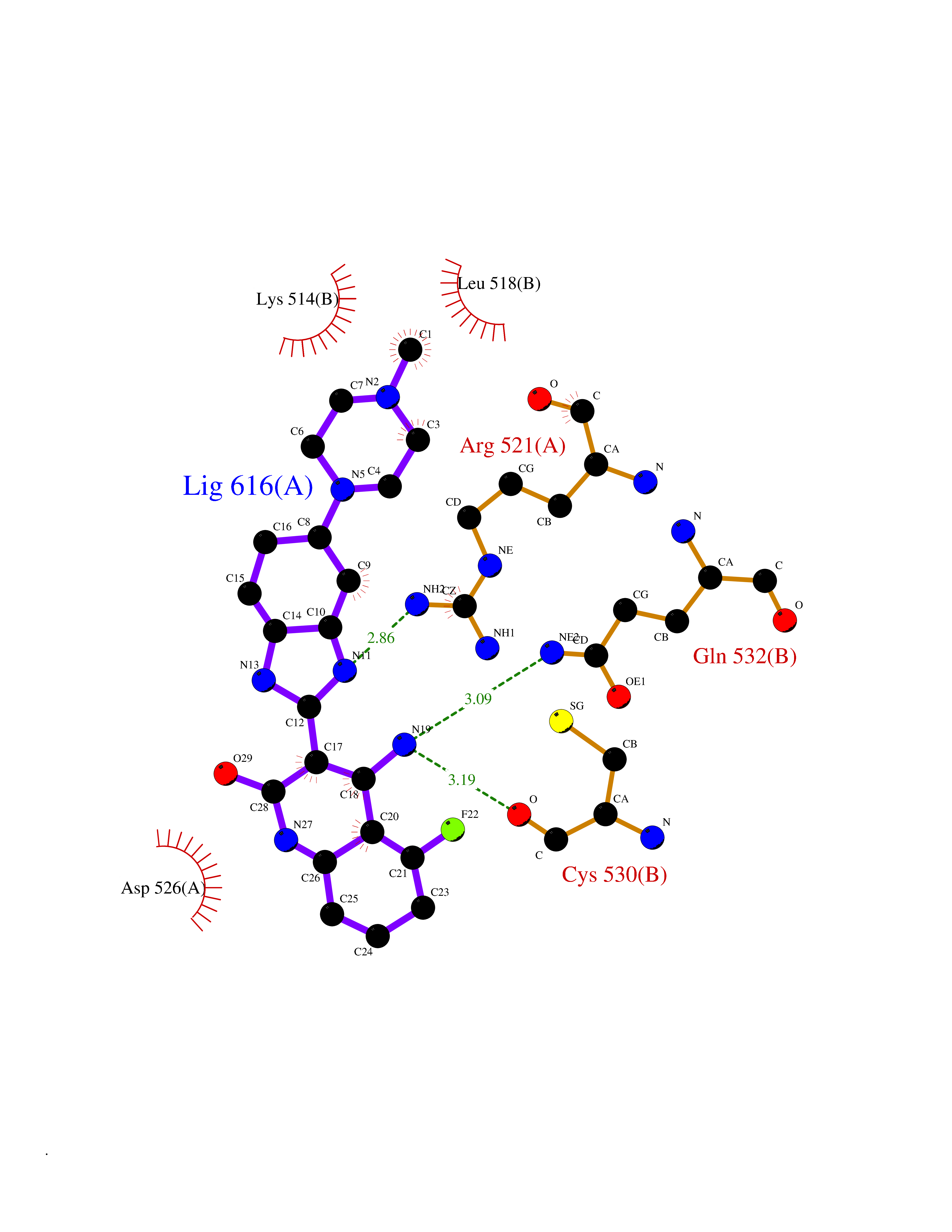 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 7.91 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -9.54  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Kallikrein-4 (KLK4) | 7JOW | 7.90 | |
Target general information Gen name KLK4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine protease 17; Prostase; PSTS; PRSS17; Kallikreinlike protein 1; Kallikrein4; Kallikrein-like protein 1; KLKL1; KLK-L1; Enamel matrix serine proteinase 1; EMSP1 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function Required during the maturation stage of tooth development for clearance of enamel proteins and normal structural patterning of the crystalline matrix. Has a major role in enamel formation. Related diseases Amelogenesis imperfecta, hypomaturation type, 2A1 (AI2A1) [MIM:204700]: A defect of enamel formation. The disorder involves both primary and secondary dentitions. The teeth have a shiny agar jelly appearance and the enamel is softer than normal. Brown pigment is present in middle layers of enamel. {ECO:0000269|PubMed:15235027}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P20155; Q06418 EC number EC 3.4.21.- Uniprot keywords 3D-structure; Alternative splicing; Amelogenesis imperfecta; Biomineralization; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Metal-binding; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID E,I Molecular weight (Da) 41763.9 Length 387 Aromaticity 0.07 Instability index 32.75 Isoelectric point 5.38 Charge (pH=7) -11.36 2D Binding mode Binding energy (Kcal/mol) -8.61  Molscript Map 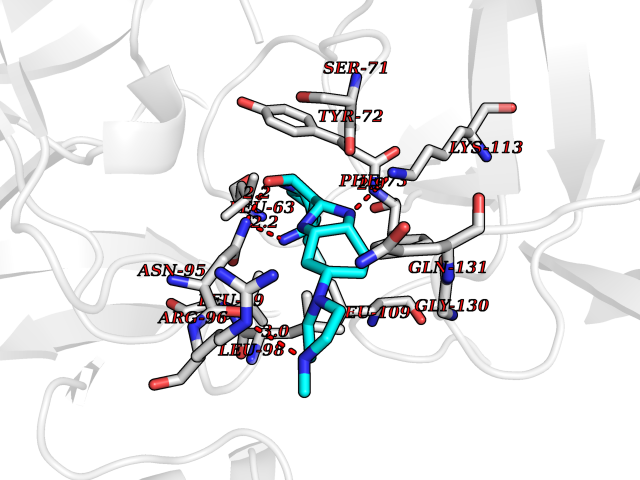 Pymol Map 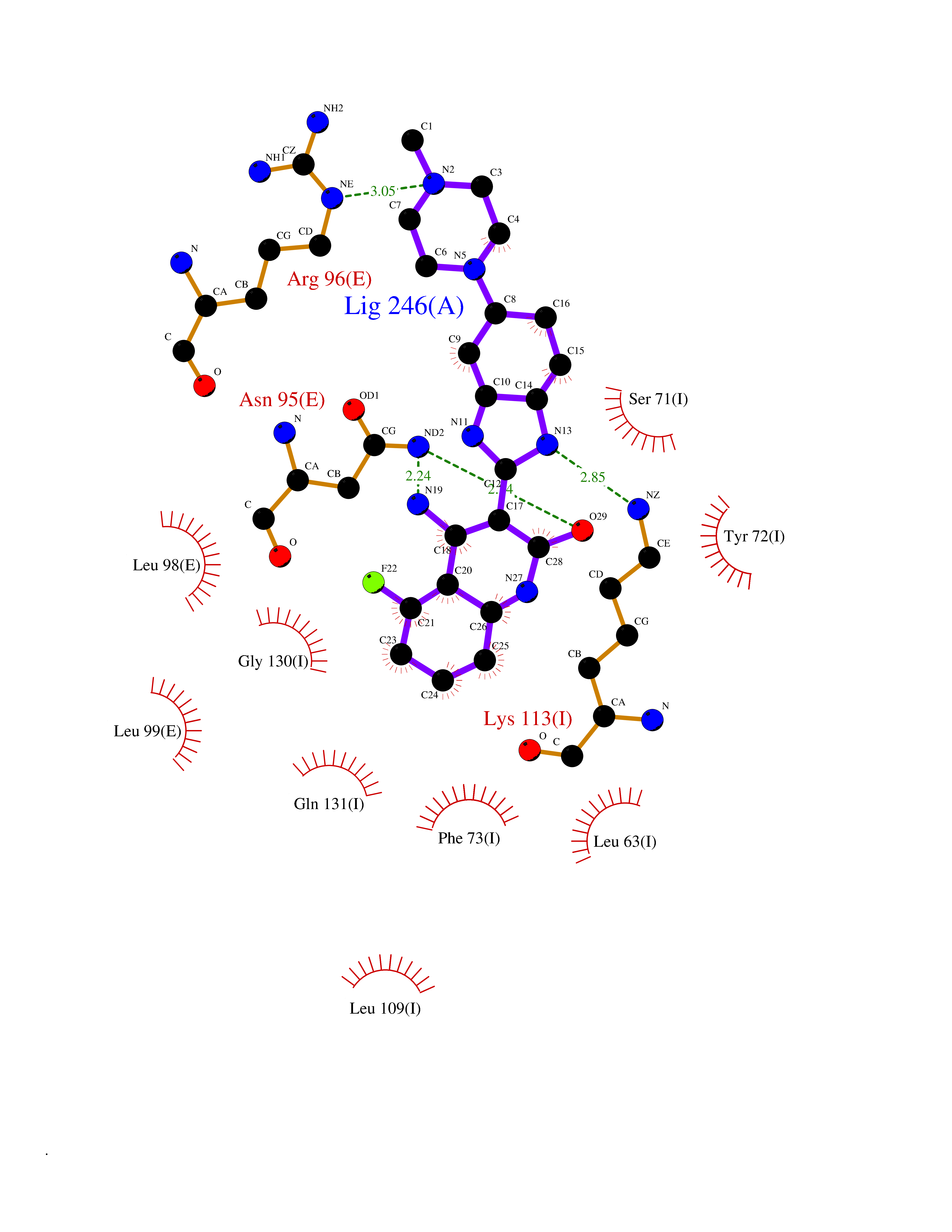 Ligplot Map 3D Binding mode Sequence IINGEDCSPHSQPWQAALVMENELFCSGVLVHPQWVLSAAHCFQNSYTIGLGLHSLEADQEPGSQMVEASLSVRHPEYNRPLLANDLMLIKLDESVSESDTIRSISIASQCPTAGNSCLVSGWGLLANGRMPTVLQCVNVSVVSEEVCSKLYDPLYHPSMFCAGGGQDQKDSCNGDSGGPLICNGYLQGLVSFGKAPCGQVGVPGVYTNLCKFTEWIEKTVQAGSSVVVDTNGQPVSNGADAYYLVPVSHGHAGLALAKIGNEAEPRAVVLDPHHRPGLPVRFESPLRINIIKESYFLNIKFGPSSSDSGVWDVIQQDPIGLAVKVTDTKSLLGPFKVEKEGEGYKIVYYPERGQTGLDIGLVHRNDKYYLAVKDGEPCVFKIRKAT Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Beta-arrestin-1 (ARRB1) | 6TKO | 7.90 | |
Target general information Gen name ARRB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Non-visual arrestin-2; Betaarrestin1; Arrestin beta1; Arrestin beta-1; ARR1 Protein family Arrestin family Biochemical class Arrestin protein Function During homologous desensitization, beta-arrestins bind to the GPRK-phosphorylated receptor and sterically preclude its coupling to the cognate G-protein; the binding appears to require additional receptor determinants exposed only in the active receptor conformation. The beta-arrestins target many receptors for internalization by acting as endocytic adapters (CLASPs, clathrin-associated sorting proteins) and recruiting the GPRCs to the adapter protein 2 complex 2 (AP-2) in clathrin-coated pits (CCPs). However, the extent of beta-arrestin involvement appears to vary significantly depending on the receptor, agonist and cell type. Internalized arrestin-receptor complexes traffic to intracellular endosomes, where they remain uncoupled from G-proteins. Two different modes of arrestin-mediated internalization occur. Class A receptors, like ADRB2, OPRM1, ENDRA, D1AR and ADRA1B dissociate from beta-arrestin at or near the plasma membrane and undergo rapid recycling. Class B receptors, like AVPR2, AGTR1, NTSR1, TRHR and TACR1 internalize as a complex with arrestin and traffic with it to endosomal vesicles, presumably as desensitized receptors, for extended periods of time. Receptor resensitization then requires that receptor-bound arrestin is removed so that the receptor can be dephosphorylated and returned to the plasma membrane. Involved in internalization of P2RY4 and UTP-stimulated internalization of P2RY2. Involved in phosphorylation-dependent internalization of OPRD1 ands subsequent recycling. Involved in the degradation of cAMP by recruiting cAMP phosphodiesterases to ligand-activated receptors. Beta-arrestins function as multivalent adapter proteins that can switch the GPCR from a G-protein signaling mode that transmits short-lived signals from the plasma membrane via small molecule second messengers and ion channels to a beta-arrestin signaling mode that transmits a distinct set of signals that are initiated as the receptor internalizes and transits the intracellular compartment. Acts as signaling scaffold for MAPK pathways such as MAPK1/3 (ERK1/2). ERK1/2 activated by the beta-arrestin scaffold is largely excluded from the nucleus and confined to cytoplasmic locations such as endocytic vesicles, also called beta-arrestin signalosomes. Recruits c-Src/SRC to ADRB2 resulting in ERK activation. GPCRs for which the beta-arrestin-mediated signaling relies on both ARRB1 and ARRB2 (codependent regulation) include ADRB2, F2RL1 and PTH1R. For some GPCRs the beta-arrestin-mediated signaling relies on either ARRB1 or ARRB2 and is inhibited by the other respective beta-arrestin form (reciprocal regulation). Inhibits ERK1/2 signaling in AGTR1- and AVPR2-mediated activation (reciprocal regulation). Is required for SP-stimulated endocytosis of NK1R and recruits c-Src/SRC to internalized NK1R resulting in ERK1/2 activation, which is required for the antiapoptotic effects of SP. Is involved in proteinase-activated F2RL1-mediated ERK activity. Acts as signaling scaffold for the AKT1 pathway. Is involved in alpha-thrombin-stimulated AKT1 signaling. Is involved in IGF1-stimulated AKT1 signaling leading to increased protection from apoptosis. Involved in activation of the p38 MAPK signaling pathway and in actin bundle formation. Involved in F2RL1-mediated cytoskeletal rearrangement and chemotaxis. Involved in AGTR1-mediated stress fiber formation by acting together with GNAQ to activate RHOA. Appears to function as signaling scaffold involved in regulation of MIP-1-beta-stimulated CCR5-dependent chemotaxis. Involved in attenuation of NF-kappa-B-dependent transcription in response to GPCR or cytokine stimulation by interacting with and stabilizing CHUK. May serve as nuclear messenger for GPCRs. Involved in OPRD1-stimulated transcriptional regulation by translocating to CDKN1B and FOS promoter regions and recruiting EP300 resulting in acetylation of histone H4. Involved in regulation of LEF1 transcriptional activity via interaction with DVL1 and/or DVL2 Also involved in regulation of receptors other than GPCRs. Involved in Toll-like receptor and IL-1 receptor signaling through the interaction with TRAF6 which prevents TRAF6 autoubiquitination and oligomerization required for activation of NF-kappa-B and JUN. Binds phosphoinositides. Binds inositolhexakisphosphate (InsP6). Involved in IL8-mediated granule release in neutrophils. Required for atypical chemokine receptor ACKR2-induced RAC1-LIMK1-PAK1-dependent phosphorylation of cofilin (CFL1) and for the up-regulation of ACKR2 from endosomal compartment to cell membrane, increasing its efficiency in chemokine uptake and degradation. Involved in the internalization of the atypical chemokine receptor ACKR3. Negatively regulates the NOTCH signaling pathway by mediating the ubiquitination and degradation of NOTCH1 by ITCH. Participates to the recruitment of the ubiquitin-protein ligase to the receptor. Functions in regulating agonist-mediated G-protein coupled receptor (GPCR) signaling by mediating both receptor desensitization and resensitization processes. Related diseases Intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) [MIM:617333]: An autosomal dominant neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, delayed language, and facial dysmorphisms, most notably ptosis. Additional features may include poor growth, hypotonia, and seizures. {ECO:0000269|PubMed:27939639, ECO:0000269|PubMed:27939640}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P63010-2; O15169; P0DP25; P20963; P25101; P50148; Q5JWF2; Q14749; P06396; Q16665; P11142; Q99683; P53779; P45984; Q00987; P19338; Q14978; P14618; P14859-6; P35813; O75688; Q13523; P06702; P12931; Q15208; Q13428; P04637; P27348; P25490; O43298; O95218; Q7DB77 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; Coated pit; Cytoplasm; Cytoplasmic vesicle; Membrane; Nucleus; Phosphoprotein; Protein transport; Proteomics identification; Reference proteome; Signal transduction inhibitor; Transcription; Transcription regulation; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32455.6 Length 293 Aromaticity 0.12 Instability index 28.99 Isoelectric point 9.12 Charge (pH=7) 9.86 2D Binding mode Binding energy (Kcal/mol) -9.64 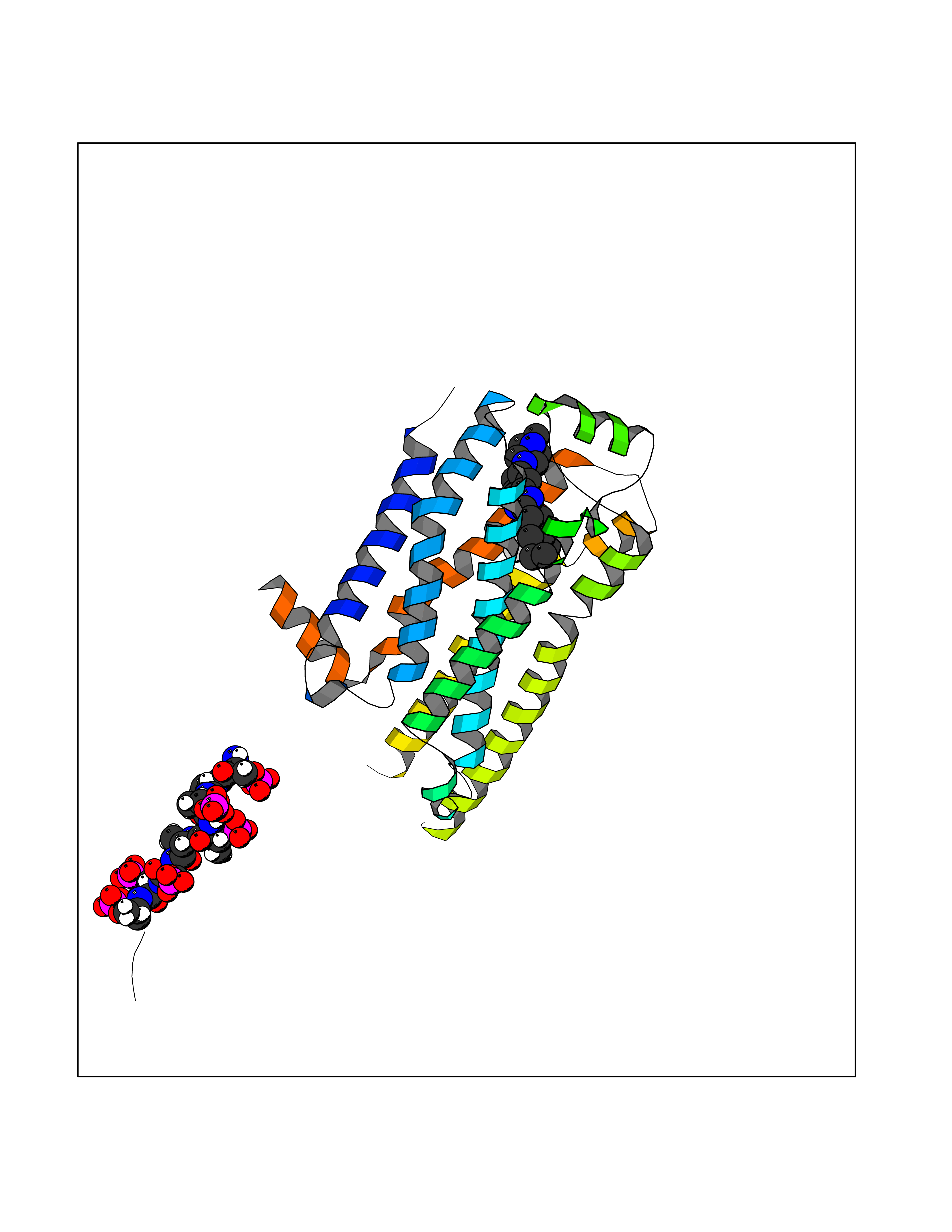 Molscript Map 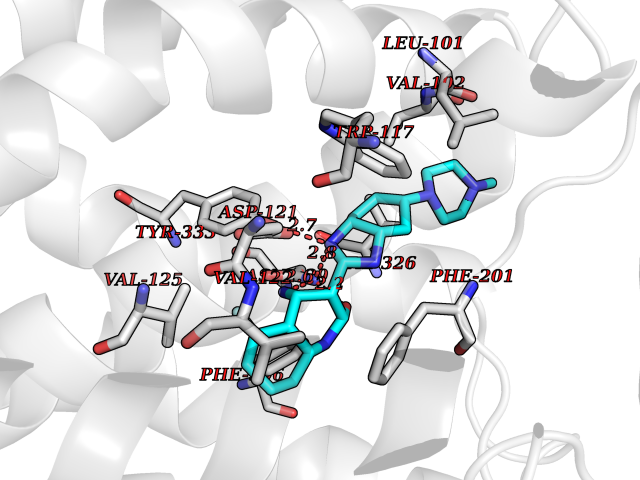 Pymol Map  Ligplot Map 3D Binding mode Sequence AGCSLLMALVVLLIVAGNVLVIAAIGRTQRLQTLTNLFITSLACADLVVGLLVVPFGATLVCRGTWLWGSFLCELWTSLDVLCVTASIWTLCVIAIDRYLAITSPFRYQSLMTRARAKVIICTVWAISALVSFLPIMMHWWRDEDPQALKCYQDPGCCDFVTNRAYAIASSIISFYIPLLIMIFVYLRVYREAKEQIRKIDVMAMREHKALKTLGIIMGVFTLCWLPFFLVNIVNVFNRDLVPKWLFVAFNWLGYANSAMNPIIYCRSPDFRKAFKRLLAEXAXXAXXXLAKD Hydrogen bonds contact Hydrophobic contact | ||||