Job Results:
Ligand
Structure
Job ID
634b273143b25dfaacd81a63c2250095
Job name
NA
Time
2026-02-27 17:35:01
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Zinc finger-containing ubiquitin peptidase 1 (ZUP1) | 6EI1 | 5.38 | |
Target general information Gen name ZUP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Zinc finger with UFM1-specific peptidase domain protein; ZUFSP; Lys-63-specific deubiquitinase ZUFSP; DUB; C6orf113 Protein family Peptidase C78 family, ZUFSP subfamily Biochemical class Peptidase Function Shows only weak activity against 'Lys-11' and 'Lys-48'-linked chains. Plays an important role in genome stability pathways, functioning to prevent spontaneous DNA damage and also promote cellular survival in response to exogenous DNA damage. Modulates the ubiquitination status of replication protein A (RPA) complex proteins in response to replication stress. Deubiquitinase with endodeubiquitinase activity that specifically interacts with and cleaves 'Lys-63'-linked long polyubiquitin chains. Related diseases WHIM syndrome 2 (WHIMS2) [MIM:619407]: An autosomal recessive form of WHIM syndrome, a primary immunodeficiency disorder characterized by warts, hypogammaglobulinemia, infections, and myelokathexis. Myelokathexis is a unique form of non-cyclic severe congenital neutropenia caused by accumulation of mature and degenerating neutrophils in the bone marrow. Monocytopenia and lymphopenia, especially B lymphopenia, also commonly occur. There is significant phenotypic variation among patients, such that some individuals may have an incomplete form of the disorder in which one or more of the classic tetrad features are not present. {ECO:0000269|PubMed:24777453}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q92619; P50281; Q8WVC2 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Hydrolase; Metal-binding; Nucleus; Proteomics identification; Reference proteome; Repeat; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 46930.3 Length 410 Aromaticity 0.07 Instability index 58.67 Isoelectric point 9 Charge (pH=7) 9.7 2D Binding mode Binding energy (Kcal/mol) -7.33  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LQQEEDRKRRSEESRQEIEEFQKLQRQYGLDNSGGYKQQQLRNMEIEVNRGRMPPSEFHRRKADMMESLALGFDDGKTKTSGIIEALHRYYQNAATDVRRVWLSSVVDHFHSSLGDKGWGCGYRNFQMLLSSLLQNDAYNDCLKGMLIPCIPKIQSMIEDAWKEGFDPQGASQLNNRLQGTKAWIGACEVYILLTSLRVKCHIVDFHKSTGPLGTHPRLFEWILNYYSSSPKVVCTSKPPIYLQHQGHSRTVIGIEEKKNRTLCLLILDPGCPSREMQKLLKQDIEASSLKQLRKSMGNLKHKQYQILAVEGALSLEEKLARRQASQVFTAEKIPMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRG Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Dipeptidyl peptidase 9 (DPP-9) | 6EOR | 5.38 | |
Target general information Gen name DPP9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dipeptidyl peptidase-like protein 9; Dipeptidyl peptidase IX; Dipeptidyl peptidase IV-related protein 2; DPRP2; DPRP-2; DPP IX; DPLP9; DP9 Protein family Peptidase S9B family, DPPIV subfamily Biochemical class Peptidase Function Dipeptidyl peptidase that cleaves off N-terminal dipeptides from proteins having a Pro or Ala residue at position 2. Related diseases Hatipoglu immunodeficiency syndrome (HATIS) [MIM:620331]: An autosomal recessive immunologic disorder manifesting in infancy or early childhood, and characterized by failure to thrive, short stature, skin pigmentation abnormalities, pancytopenia, and susceptibility to recurrent infections. {ECO:0000269|PubMed:36112693}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NXR5; Q86TI2; Q6NUP5; P46379-2; Q8WUW1; Q96A83-2; O75190-2; O14645; Q01658; P29692-2; Q06787-7; Q9Y5Q9; O14901; Q9BVL2; Q96CV9; Q06830; P14678-2; P49458; Q11203; Q13148; P14927 EC number EC 3.4.14.5 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminopeptidase; Cytoplasm; Disease variant; Hydrolase; Nucleus; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A Molecular weight (Da) 92797.4 Length 808 Aromaticity 0.12 Instability index 37.45 Isoelectric point 6.34 Charge (pH=7) -8.98 2D Binding mode Binding energy (Kcal/mol) -7.33  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence AARFQVQKHSWDGLRSIIHGSRKAPHDFQFVQKSGPHSHRLYYLGMPYRENSLLYSEIPKLLLSWKQMLDHFQATPHHGVYSREEELLRERKRLGVFGITSYDFHSESGLFLFQASNSLFHCRDGGKNGFMVSPMKPLEIKTQCSGPRMDPKICPADPAFFSFINNSDLWVANIETGEERRLTFCHQNVLDDPKSAGVATFVIQEEFDRFTGYWWCPTASWEGLKTLRILYEEVDESEVEVIHVPSPALEERKTDSYRYPRTGSKNPKIALKLAEFQTDSQGKIVSTQEKELVQPFSSLFPKVEYIARAGWTRDGKYAWAMFLDRPQQWLQLVLLPPALFIPSTENEEQRLASARAVPRNVQPYVVYEEVTNVWINVHDIFYPFPQLCFLRANECKTGFCHLYKVTAVLKSQGYDWSEPFSPGEDEFKCPIKEEIALTSGEWEVLARHGSKIWVNEETKLVYFQGTKDTPLEHHLYVVSYEAAGEIVRLTTPGFSHSCSMSQNFDMFVSHYSSVSTPPCVHVYKLSGPDDDPLHKQPRFWASMMEADYVPPEIFHFHTRSDVRLYGMIYKPHALQPGKKHPTVLFVYGGPQVQLVNNSFKGIKYLRLNTLASLGYAVVVIDGRGSCQRGLRFEGALKNQMGQVEIEDQVEGLQFVAEKYGFIDLSRVAIHGWSYGGFLSLMGLIHKPQVFKVAIAGAPVTVWMAYDTGYTERYMDVPENNQHGYEAGSVALHVEKLPNEPNRLLILHGFLDENVHFFHTNFLVSQLIRAGKPYQLQIYPNERHSIRCPESGEHYEVTLLHFLQEYLHH Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Neuronal acetylcholine receptor beta-4 (CHRNB4) | 6PV7 | 5.38 | |
Target general information Gen name CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNB4; Beta-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-4/CHRNB4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. {ECO:0000269|PubMed:20054297, ECO:0000269|PubMed:23622243, ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}. The disease may be caused by variants affecting the gene represented in this entry. Defects of SETD2 are associated with loss of DNA methylation at non-promoter regions (PubMed:23792563). SETD2 defects lead to aberrant and reduced nucleosome compaction and chromatin association of key replication proteins, such as MCM7 and DNA polymerase delta, leading to hinder replication fork progression and prevent loading of RAD51 homologous recombination repair factor at DNA breaks (PubMed:25728682). {ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}.; DISEASE: Luscan-Lumish syndrome (LLS) [MIM:616831]: An autosomal dominant syndrome with a variable phenotype. Clinical features include macrocephaly, distinctive facial appearance, postnatal overgrowth, various degrees of learning difficulties, autism spectrum disorder, and intellectual disability. {ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24852293, ECO:0000269|PubMed:26084711, ECO:0000269|PubMed:27317772}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute lymphoblastic (ALL) [MIM:613065]: A subtype of acute leukemia, a cancer of the white blood cells. ALL is a malignant disease of bone marrow and the most common malignancy diagnosed in children. The malignant cells are lymphoid precursor cells (lymphoblasts) that are arrested in an early stage of development. The lymphoblasts replace the normal marrow elements, resulting in a marked decrease in the production of normal blood cells. Consequently, anemia, thrombocytopenia, and neutropenia occur to varying degrees. The lymphoblasts also proliferate in organs other than the marrow, particularly the liver, spleen, and lymphnodes. {ECO:0000269|PubMed:24509477, ECO:0000269|PubMed:24662245}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16314571, ECO:0000269|PubMed:24509477}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Intellectual developmental disorder, autosomal dominant 70 (MRD70) [MIM:620157]: An autosomal dominant disorder characterized by mild global developmental delay, moderately impaired intellectual disability with speech difficulties, and behavioral abnormalities. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rabin-Pappas syndrome (RAPAS) [MIM:620155]: An autosomal dominant neurodevelopmental disorder characterized by severely impaired global development, intellectual disability, microcephaly, facial dysmorphism, and variable congenital anomalies affecting the skeletal, genitourinary, cardiac, and other organ systems. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00237; DB00565; DB00514; DB07720; DB00898; DB00472; DB01227; DB00184; DB01090; DB00202 Interacts with Q6FHY5 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -7.34  Molscript Map 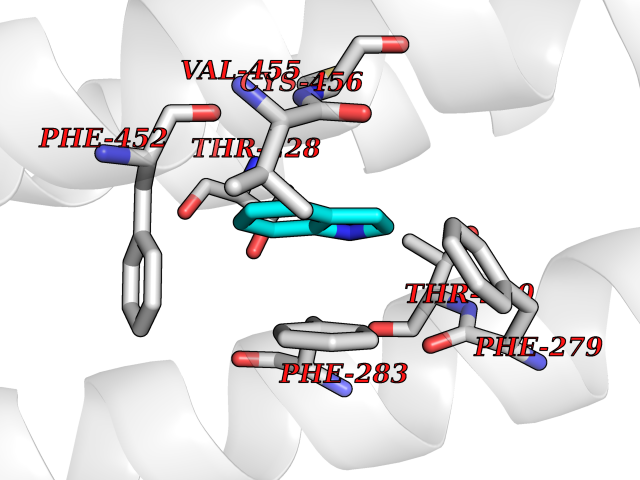 Pymol Map 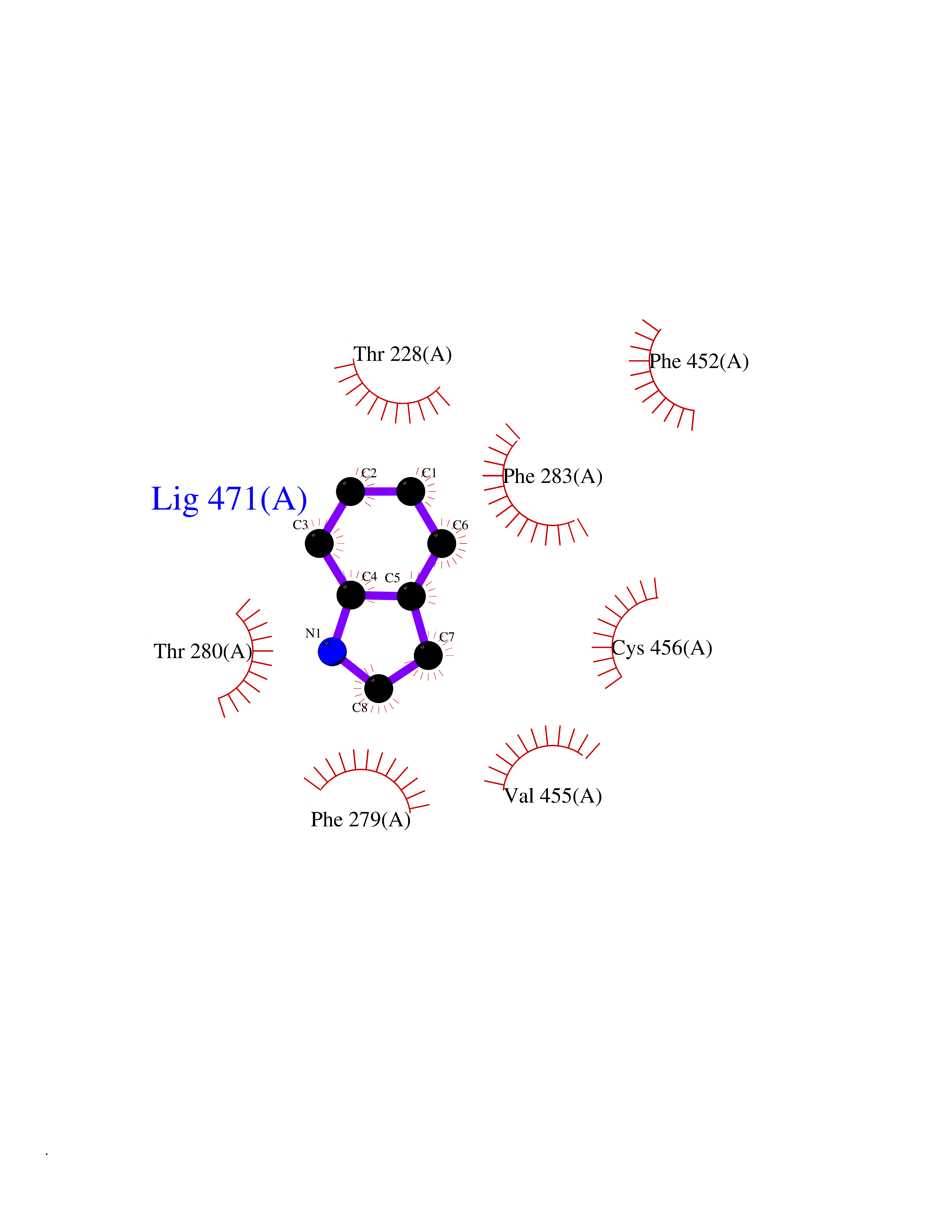 Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Bacterial Nicotinate-nucleotide adenylyltransferase (Bact nadD) | 1K4K | 5.38 | |
Target general information Gen name Bact nadD Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms nadD of Escherichia coli (strain K12); Nicotinate mononucleotide adenylyltransferase of Escherichia coli (strain K12); NaMN adenylyltransferase of Escherichia coli (strain K12); Deamido-NAD(+)Nicotina Protein family NadD family Biochemical class Kinase Function Catalyzes the reversible adenylation of nicotinate mononucleotide (namn) to nicotinic acid adenine dinucleotide (naad). Related diseases Asthma-related traits 5 (ASRT5) [MIM:611064]: Asthma-related traits include clinical symptoms of asthma, such as coughing, wheezing, dyspnea, bronchial hyperresponsiveness as assessed by methacholine challenge test, serum IgE levels, atopy and atopic dermatitis. {ECO:0000269|PubMed:17503328}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.7.7.18 Uniprot keywords 3D-structure; ATP-binding; NAD; Nucleotide-binding; Nucleotidyltransferase; Pyridine nucleotide biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 24527.6 Length 213 Aromaticity 0.09 Instability index 48.92 Isoelectric point 5.46 Charge (pH=7) -7.78 2D Binding mode Binding energy (Kcal/mol) -7.34  Molscript Map 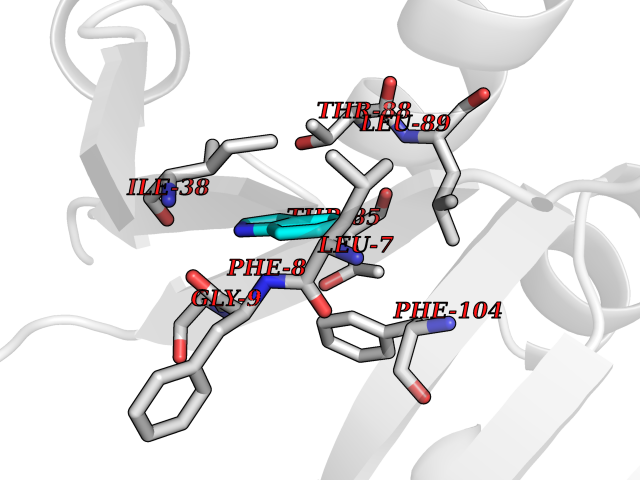 Pymol Map  Ligplot Map 3D Binding mode Sequence MKSLQALFGGTFDPVHYGHLKPVETLANLIGLTRVTIIPNNVPPHRPQPEANSVQRKHMLELAIADKPLFTLDERELKRNAPSYTAQTLKEWRQEQGPDVPLAFIIGQDSLLTFPTWYEYETILDNAHLIVCRRPGYPLEMAQPQYQQWLEDHLTHNPEDLHLQPAGKIYLAETPWFNISATIIRERLQNGESCEDLLPEPVLTYINQQGLYR Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Opioid receptor sigma 1 (OPRS1) | 5HK1 | 5.37 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 20805.3 Length 185 Aromaticity 0.14 Instability index 31.72 Isoelectric point 5.44 Charge (pH=7) -6.63 2D Binding mode Binding energy (Kcal/mol) -7.32  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence VFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLFG Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Leucine carboxyl methyltransferase 1 | 3IEI | 5.37 | |
Target general information Gen name LCMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CGI-68;LCMT Protein family Methyltransferase superfamily, LCMT family Biochemical class Transferase Function Protein C-terminal carboxyl O-methyltransferase activity.Protein C-terminal leucine carboxyl O-methyltransferase activity.S-adenosylmethionine-dependent methyltransferase activity. Related diseases Neurodevelopmental disorder, mitochondrial, with abnormal movements and lactic acidosis, with or without seizures (NEMMLAS) [MIM:617710]: An autosomal recessive, mitochondrial disorder with a broad phenotypic spectrum ranging from severe neonatal lactic acidosis, encephalomyopathy and early death to an attenuated course with milder manifestations. Clinical features include delayed psychomotor development, intellectual disability, hypotonia, dystonia, ataxia, and spasticity. Severe combined respiratory chain deficiency may be found in severely affected individuals. {ECO:0000269|PubMed:28236339, ECO:0000269|PubMed:28650581, ECO:0000269|PubMed:28905505, ECO:0000269|PubMed:30920170, ECO:0000269|PubMed:35074316}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parkinsonism-dystonia 3, childhood-onset (PKDYS3) [MIM:619738]: An autosomal recessive neurodegenerative disorder with onset in infancy or early childhood. Affected individuals present with progressive movement abnormalities, including parkinsonism with tremor, dystonia, myoclonus ataxia, and hyperkinetic movements such as ballismus. The parkinsonism features may be responsive to treatment with levodopa, although many patients develop levodopa-induced dyskinesia. Some patients may have mild cognitive impairment or psychiatric disturbances. {ECO:0000269|PubMed:29120065, ECO:0000269|PubMed:31970218, ECO:0000269|PubMed:34890876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00149 Interacts with P51116 EC number 2.1.1.233 Uniprot keywords 3D-structure; Alternative splicing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 35803 Length 310 Aromaticity 0.08 Instability index 42.77 Isoelectric point 6.13 Charge (pH=7) -3.58 2D Binding mode Binding energy (Kcal/mol) -7.32  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GVRGTCEDASLCKRFAVSIGYWHDPYIQHFVRLSKERKAPEINRGYFARVHGVSQLIKAFLRKTECHCQIVNLGAGMDTTFWRLKDEDLLSSKYFEVDFPMIVTRKLHSIKCKPPLSSPILELHSEDTLQMDGHILDSKRYAVIGADLRDLSELEEKLKKCNMNTQLPTLLIAECVLVYMTPEQSANLLKWAANSFERAMFINYEQVNMGDRFGQIMIENLRRRQCDLAGVETCKSLESQKERLLSNGWETASAVDMMELYNRLPRAEVSRIESLEFLDEMELLEQLMRHYCLCWATKGGNELGLKEITY Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Proto-oncogene c-Met (MET) | 3DKC | 5.37 | |
Target general information Gen name MET Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase Met; Scatter factor receptor; SF receptor; Met proto-oncogene tyrosine kinase; Hepatocyte growth factor receptor; HGF/SF receptor; HGF-SF receptor; HGF receptor; C-met; C-Met r Protein family Protein kinase superfamily, Tyr protein kinase family Biochemical class Kinase Function Regulates many physiological processes including proliferation, scattering, morphogenesis and survival. Ligand binding at the cell surface induces autophosphorylation of MET on its intracellular domain that provides docking sites for downstream signaling molecules. Following activation by ligand, interacts with the PI3-kinase subunit PIK3R1, PLCG1, SRC, GRB2, STAT3 or the adapter GAB1. Recruitment of these downstream effectors by MET leads to the activation of several signaling cascades including the RAS-ERK, PI3 kinase-AKT, or PLCgamma-PKC. The RAS-ERK activation is associated with the morphogenetic effects while PI3K/AKT coordinates prosurvival effects. During embryonic development, MET signaling plays a role in gastrulation, development and migration of muscles and neuronal precursors, angiogenesis and kidney formation. In adults, participates in wound healing as well as organ regeneration and tissue remodeling. Promotes also differentiation and proliferation of hematopoietic cells. May regulate cortical bone osteogenesis. Receptor tyrosine kinase that transduces signals from the extracellular matrix into the cytoplasm by binding to hepatocyte growth factor/HGF ligand. Related diseases Activation of MET after rearrangement with the TPR gene produces an oncogenic protein.; DISEASE: Defects in MET may be associated with gastric cancer.; DISEASE: Hepatocellular carcinoma (HCC) [MIM:114550]: A primary malignant neoplasm of epithelial liver cells. The major risk factors for HCC are chronic hepatitis B virus (HBV) infection, chronic hepatitis C virus (HCV) infection, prolonged dietary aflatoxin exposure, alcoholic cirrhosis, and cirrhosis due to other causes. {ECO:0000269|PubMed:9927037}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Renal cell carcinoma papillary (RCCP) [MIM:605074]: A subtype of renal cell carcinoma tending to show a tubulo-papillary architecture formed by numerous, irregular, finger-like projections of connective tissue. Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. {ECO:0000269|PubMed:10327054, ECO:0000269|PubMed:10417759, ECO:0000269|PubMed:10433944, ECO:0000269|PubMed:9140397, ECO:0000269|PubMed:9563489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A common allele in the promoter region of the MET shows genetic association with susceptibility to autism in some families. Functional assays indicate a decrease in MET promoter activity and altered binding of specific transcription factor complexes.; DISEASE: MET activating mutations may be involved in the development of a highly malignant, metastatic syndrome known as cancer of unknown primary origin (CUP) or primary occult malignancy. Systemic neoplastic spread is generally a late event in cancer progression. However, in some instances, distant dissemination arises at a very early stage, so that metastases reach clinical relevance before primary lesions. Sometimes, the primary lesions cannot be identified in spite of the progresses in the diagnosis of malignancies.; DISEASE: Deafness, autosomal recessive, 97 (DFNB97) [MIM:616705]: A form of non-syndromic sensorineural hearing loss with prelingual onset. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:25941349}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Osteofibrous dysplasia (OSFD) [MIM:607278]: A congenital disorder of osteogenesis characterized by non-neoplastic, radiolucent lesions that affect the cortical bone immediately under the periosteum. It usually manifests as a painless swelling or anterior bowing of the long bones, most commonly the tibia and fibula. {ECO:0000269|PubMed:26637977}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Disease-associated variants identified in 4 families cause the deletion of exon 14. This results in the exclusion of an ubiquitination target site within the cytoplasmic domain, hence in protein stabilization. The persistent presence of MET at the cell surface in conditions of ligand-dependent activation retards osteoblastic differentiation. {ECO:0000269|PubMed:26637977}.; DISEASE: Arthrogryposis, distal, 11 (DA11) [MIM:620019]: A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. DA11 is an autosomal dominant form characterized mainly by camptodactyly. Other features include absent flexion creases and limited forearm supination. {ECO:0000269|PubMed:30777867}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06896; DB08791; DB06997; DB07969; DB08079; DB16695; DB12742; DB12267; DB08875; DB11791; DB08865; DB12010; DB02152; DB07369; DB06995; DB06314; DB01268; DB15133; DB12200; DB11800 Interacts with P22681; Q96EY1; Q96EY1-2; P00533; P09769; P14210; P14210-6; O15357; P35968; P06239; P07948; P08581; P41218; P15941; P16333; O43639; Q16288; P27986; O00459; Q92569; P19174; O43157; O15031; Q9ULL4; Q8TCU6; P18031; Q06124; P23467; Q12913; Q16827; P20936; Q9UQQ2; O60880; O14796; Q9NP31; Q8N5H7; Q15464; P29353; P98077; Q6S5L8; Q96IW2; Q9H6Q3; O75159; O14544; P12931; Q9ULZ2; P43405; P42680; Q9HBL0; Q63HR2; Q68CZ2; Q9UKW4; P07947; P43403; Q08048; P0DQD2; P35918; Q00944 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromosomal rearrangement; Deafness; Disease variant; Disulfide bond; Glycoprotein; Kinase; Membrane; Non-syndromic deafness; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35229.5 Length 312 Aromaticity 0.1 Instability index 37.98 Isoelectric point 7.79 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -7.33  Molscript Map  Pymol Map 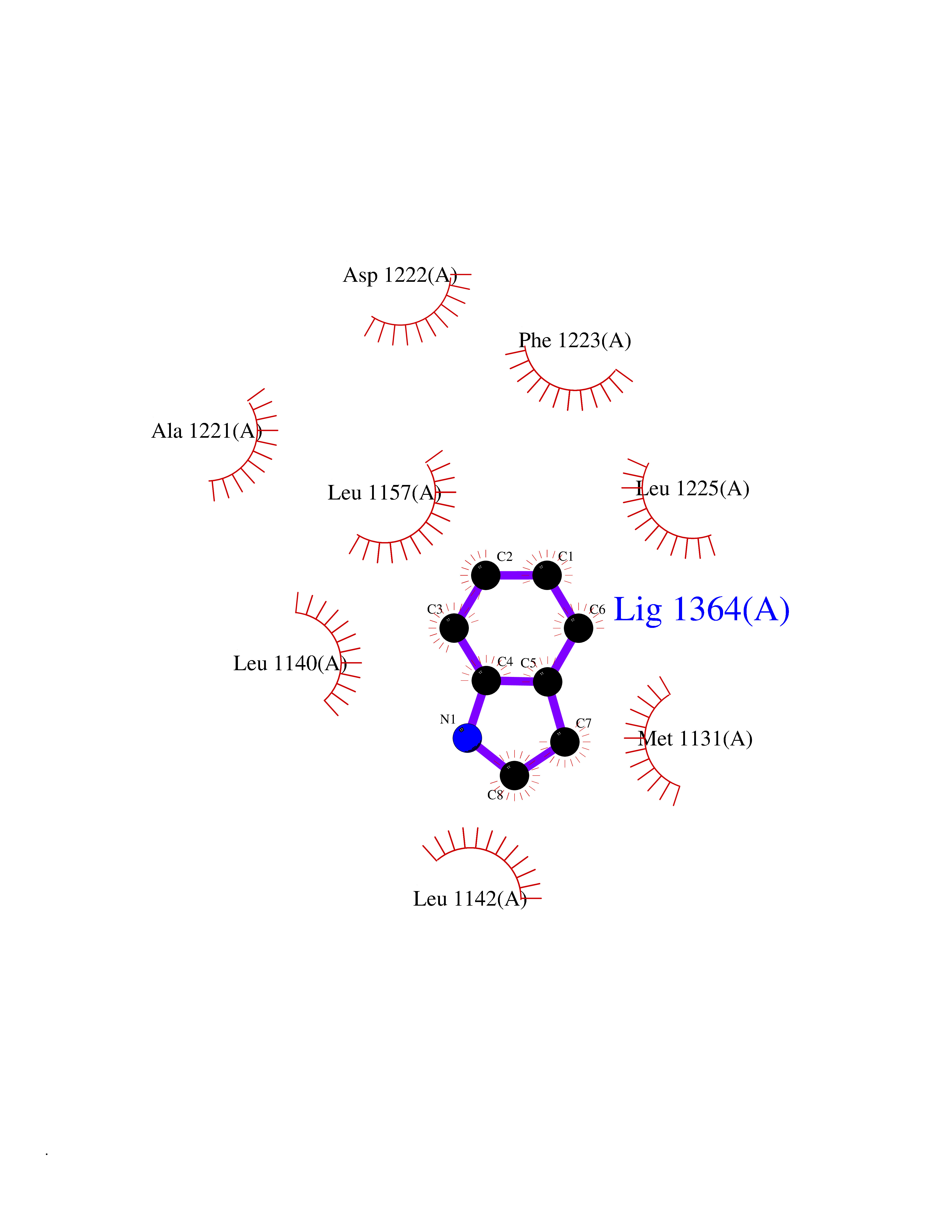 Ligplot Map 3D Binding mode Sequence VHIDLSALNPELVQAVQHVVIGPSSLIVHFNEVIGRGHFGCVYHGTLLDNDGKKIHCAVKSLNRITDIGEVSQFLTEGIIMKDFSHPNVLSLLGICLRSEGSPLVVLPYMKHGDLRNFIRNETHNPTVKDLIGFGLQVAKGMKFLASKKFVHRDLAARNCMLDEKFTVKVADFGLARDMYDKEFDSVHNKTGAKLPVKWMALESLQTQKFTTKSDVWSFGVLLWELMTRGAPPYPDVNTFDITVYLLQGRRLLQPEYCPDPLYEVMLKCWHPKAEMRPSFSELVSRISAIFSTFIGEHYVHVNATYVNVKEG Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Acyloxyacyl hydrolase (neutrophil) | 5W7C | 5.37 | |
Target general information Gen name AOAH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Acyloxyacyl hydrolase Protein family NA Biochemical class NA Function Removes the secondary (acyloxyacyl-linked) fatty acyl chains from the lipid A region of bacterial lipopolysaccharides. By breaking down LPS, terminates the host response to bacterial infection and prevents prolonged and damaging inflammatory responses (By similarity). In peritoneal macrophages, seems to be important for recovery from a state of immune tolerance following infection by Gram-negative bacteria (By similarity). Related diseases Major depressive disorder (MDD) [MIM:608516]: A common psychiatric disorder. It is a complex trait characterized by one or more major depressive episodes without a history of manic, mixed, or hypomanic episodes. A major depressive episode is characterized by at least 2 weeks during which there is a new onset or clear worsening of either depressed mood or loss of interest or pleasure in nearly all activities. Four additional symptoms must also be present including changes in appetite, weight, sleep, and psychomotor activity; decreased energy; feelings of worthlessness or guilt; difficulty thinking, concentrating, or making decisions; or recurrent thoughts of death or suicidal ideation, plans, or attempts. The episode must be accompanied by distress or impairment in social, occupational, or other important areas of functioning. {ECO:0000269|PubMed:15229186}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q15700 EC number EC 3.1.1.77 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lipid metabolism; Metal-binding; Proteomics identification; Reference proteome; Secreted; Signal; Zymogen Protein physicochemical properties Chain ID C Molecular weight (Da) 47779.7 Length 420 Aromaticity 0.1 Instability index 43.45 Isoelectric point 7.72 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -7.33 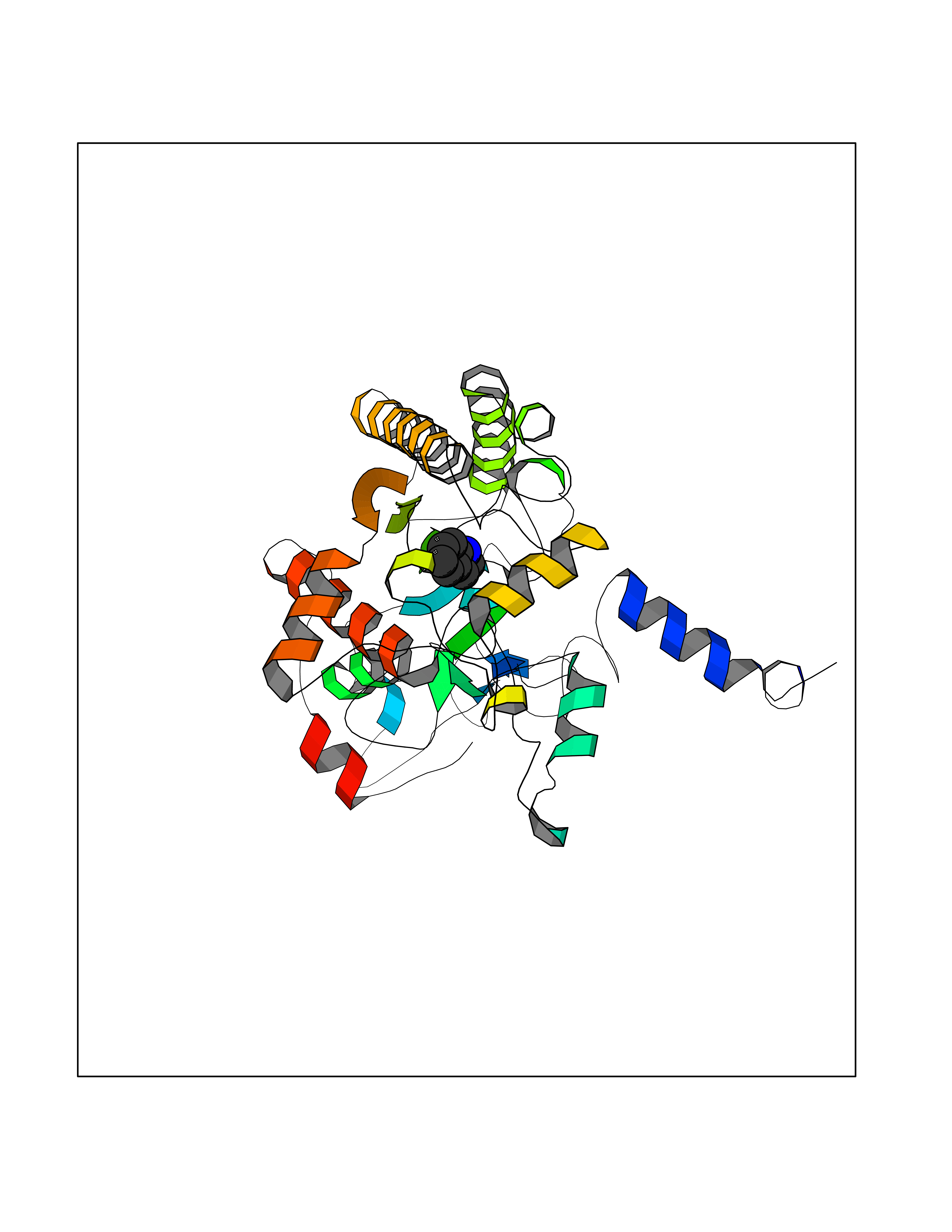 Molscript Map 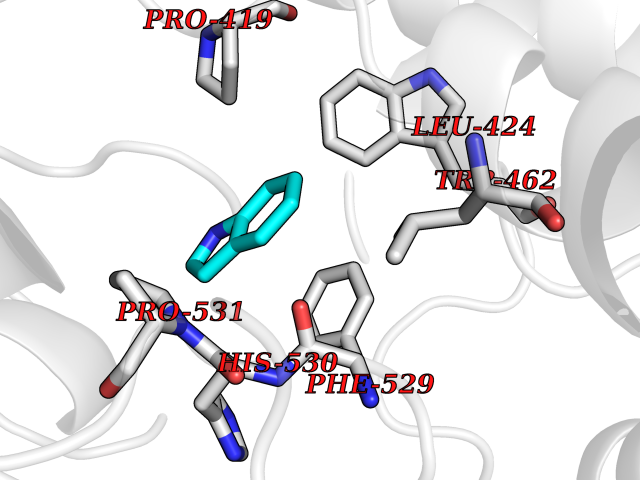 Pymol Map 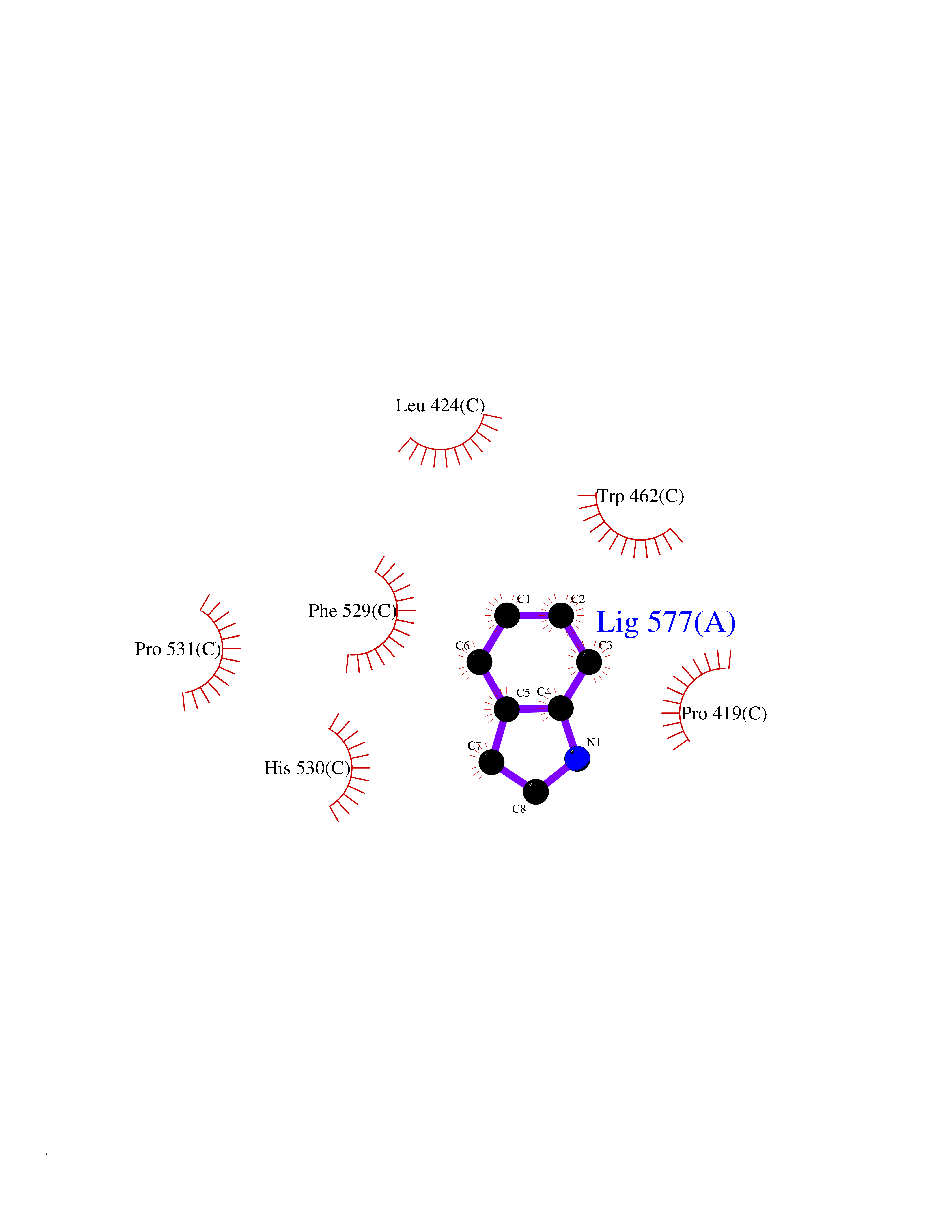 Ligplot Map 3D Binding mode Sequence GSDICSLPVLAKICQKIKLAMEQSVPFKDVDSDKYSVFPTLRGYHWRGRDCNDSDESVYPGRRPNNWDVHQDSNCNGIWGVDPKDGVPYEKKFCEGSQPRGIILLGDAAGAHFHISPEWITASQMSLNSFINLPTALTNELDWPQLSGATGFLDSTVGIKEKSIYLRLWKRNHCNHRDYQNISRNGASSRNLKKFIESLSRNKVLDYPAIVIYAMIGNDVCSGKSDPVPAMTTPEKLYSNVMQTLKHLNSHLPNGSHVILYGLPDGTFLWDNLHNRYHPLGQLNKDMTYAQLYSFLNCLQVSPCHGWMSSNKTLRTLTSERAEQLSNTLKKIAASEKFTNFNLFYMDFAFHEIIQEWQKRGGQPWQLIEPVDGFHPNEVALLLLADHFWKKVQLQWPQILGKENPFNPQIKQVFGDQGGH Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Mutated Histone H3.3 (H3F3A) | 4GUS | 5.37 | |
Target general information Gen name H3F3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PP781; Histone H3.3; H3F3; H3.3B; H3.3A Protein family Histone H3 family Biochemical class NA Function Variant histone H3 which replaces conventional H3 in a wide range of nucleosomes in active genes. Constitutes the predominant form of histone H3 in non-dividing cells and is incorporated into chromatin independently of DNA synthesis. Deposited at sites of nucleosomal displacement throughout transcribed genes, suggesting that it represents an epigenetic imprint of transcriptionally active chromatin. Nucleosomes wrap and compact DNA into chromatin, limiting DNA accessibility to the cellular machineries which require DNA as a template. Histones thereby play a central role in transcription regulation, DNA repair, DNA replication and chromosomal stability. DNA accessibility is regulated via a complex set of post-translational modifications of histones, also called histone code, and nucleosome remodeling. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) NA Interacts with Q9NVP2; P45973; Q13111; Q9UER7; Q9UER7-1; Q9Y6K1; P62805; P49321-2; Q8IZL8; Q5VWG9; Q9VK33; Q8R5C8 EC number NA Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Chromosome; Citrullination; Direct protein sequencing; Disease variant; DNA-binding; Hydroxylation; Intellectual disability; Lipoprotein; Methylation; Nucleosome core; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,C Molecular weight (Da) 86148.9 Length 766 Aromaticity 0.1 Instability index 37.57 Isoelectric point 8.25 Charge (pH=7) 7.16 2D Binding mode Binding energy (Kcal/mol) -7.32  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKPNTAIKPETSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQL Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 5.36 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -7.31  Molscript Map 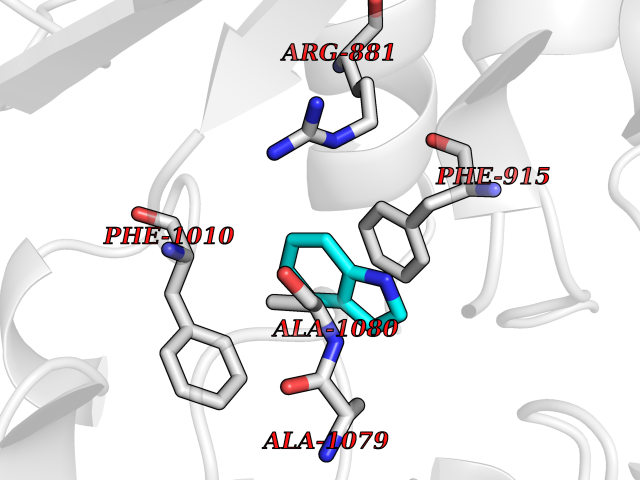 Pymol Map 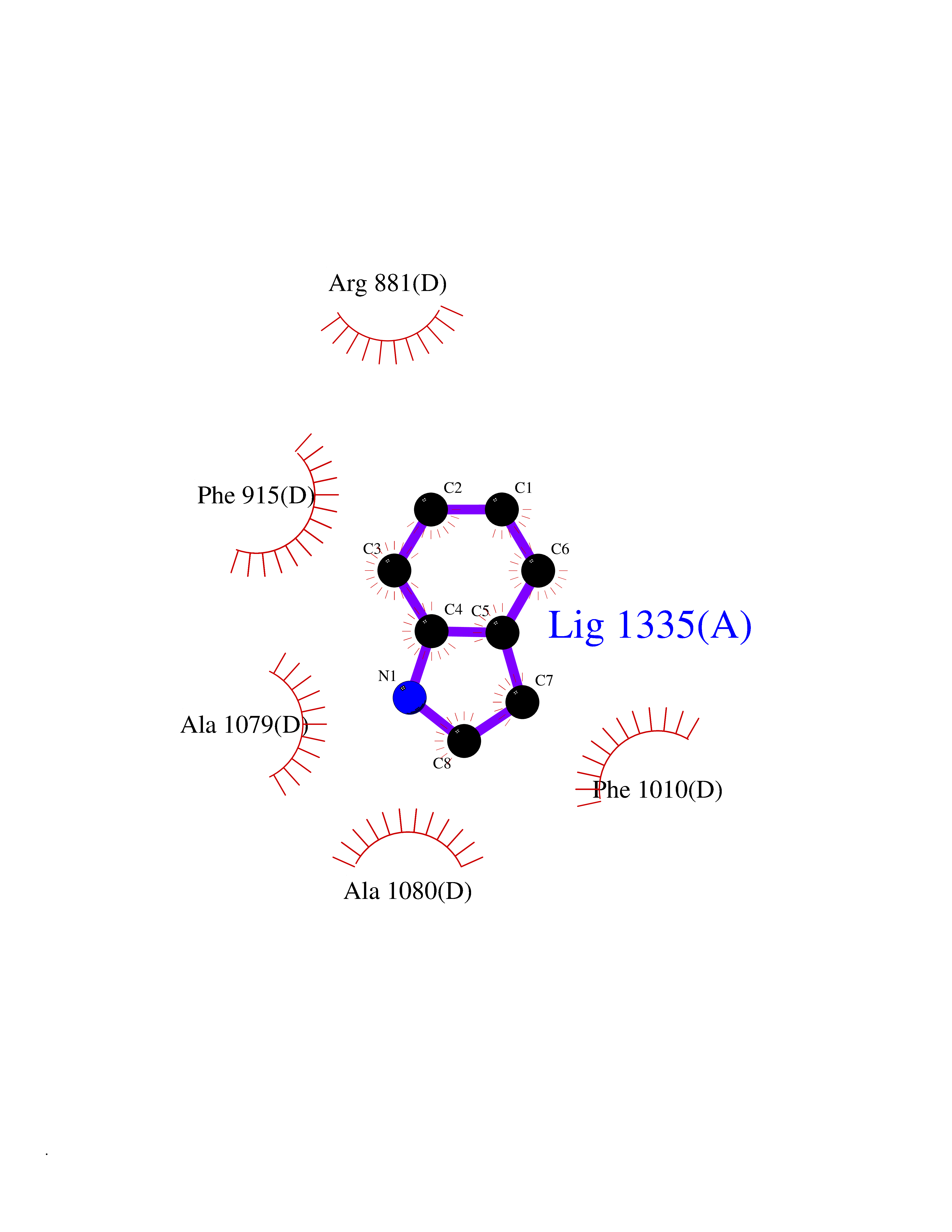 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Matrix metalloproteinase-16 (MMP-16) | 1RM8 | 5.36 | |
Target general information Gen name MMP16 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Membrane-type-3 matrix metalloproteinase; Membrane-type matrix metalloproteinase 3; MTMMP3; MT3MMP; MT3-MMP; MT-MMP 3; MMPX2; MMP-X2; C8orf57 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates progelatinase A. Involved in the matrix remodeling of blood vessels. Isoform short cleaves fibronectin and also collagen type III, but at lower rate. It has no effect on type I, II, IV and V collagen. However, upon interaction with CSPG4, it may be involved in degradation and invasion of type I collagen by melanoma cells. Endopeptidase that degrades various components of the extracellular matrix, such as collagen type III and fibronectin. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03880; DB00786 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Cleavage on pair of basic residues; Collagen degradation; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18853.6 Length 169 Aromaticity 0.12 Instability index 33.65 Isoelectric point 4.88 Charge (pH=7) -12.42 2D Binding mode Binding energy (Kcal/mol) -7.31  Molscript Map 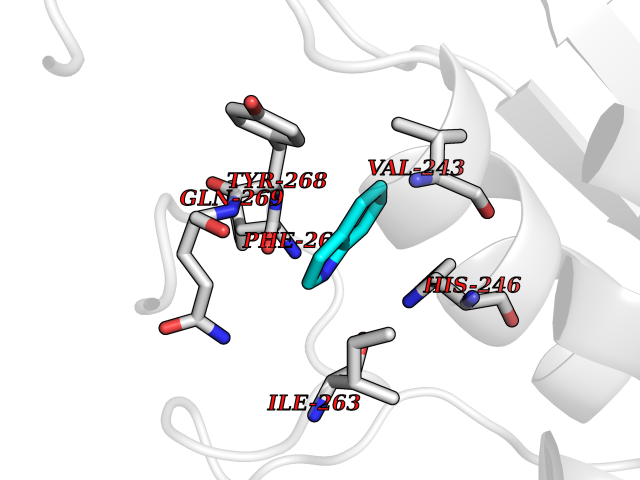 Pymol Map 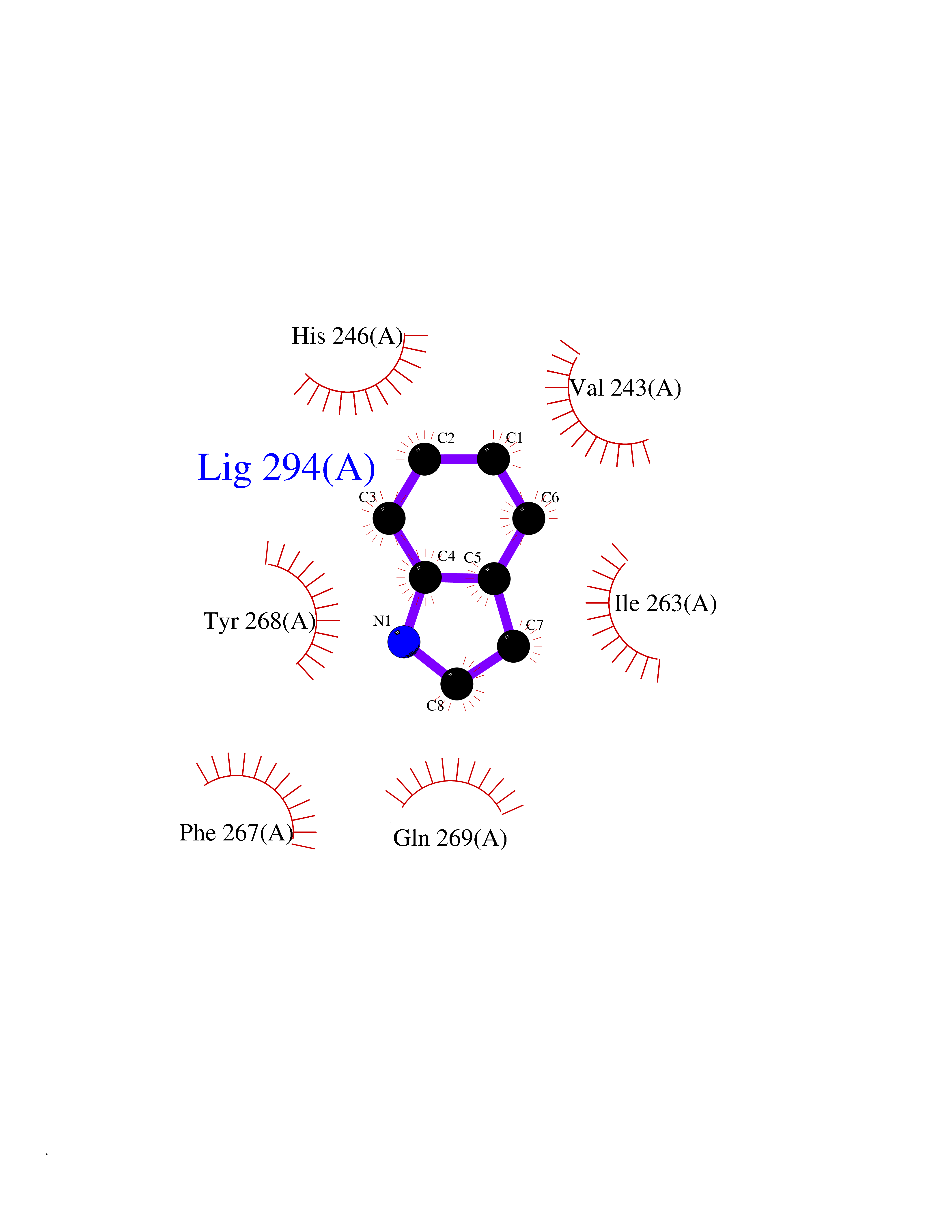 Ligplot Map 3D Binding mode Sequence GQKWQHKHITYSIKNVTPKVGDPETRKAIRRAFDVWQNVTPLTFEEVPYSELENGKRDVDITIIFASGFHGDSSPFDGEGGFLAHAYFPGPGIGGDTHFDSDEPWTLGNPNHDGNDLFLVAVHELGHALGLEHSNDPTAIMAPFYQYMETDNFKLPNDDLQGIQKIYGP Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 5.36 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -7.31 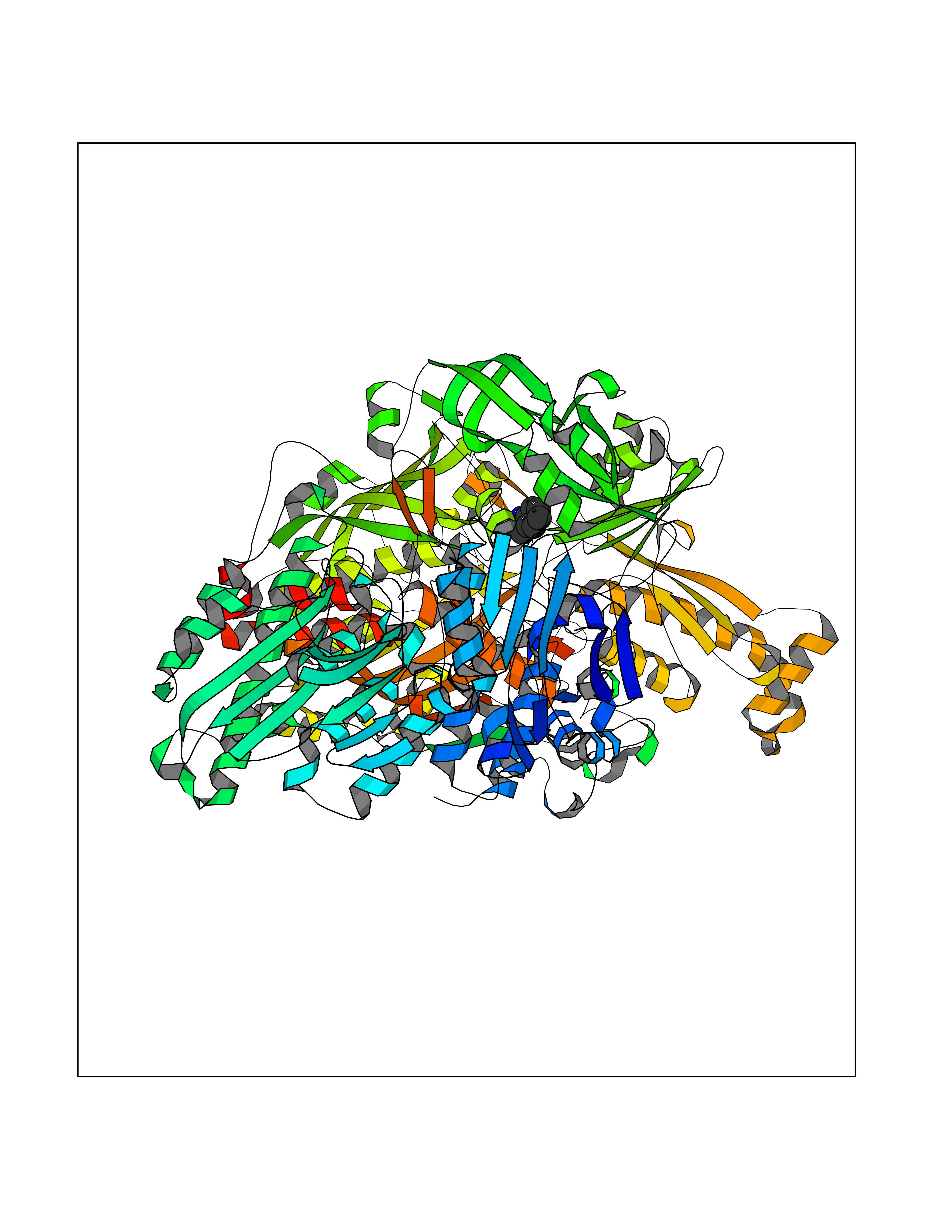 Molscript Map 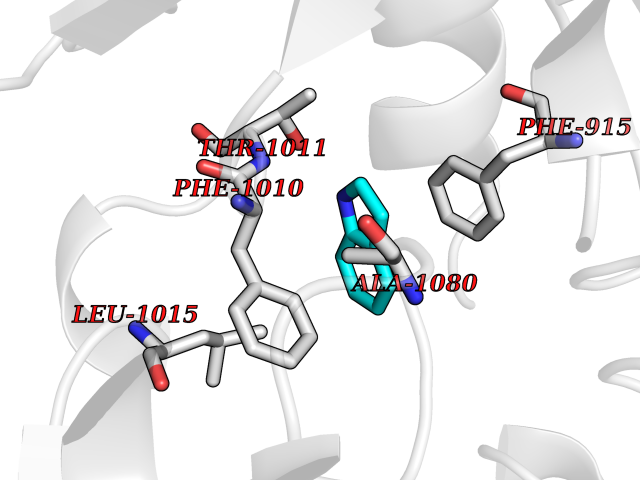 Pymol Map 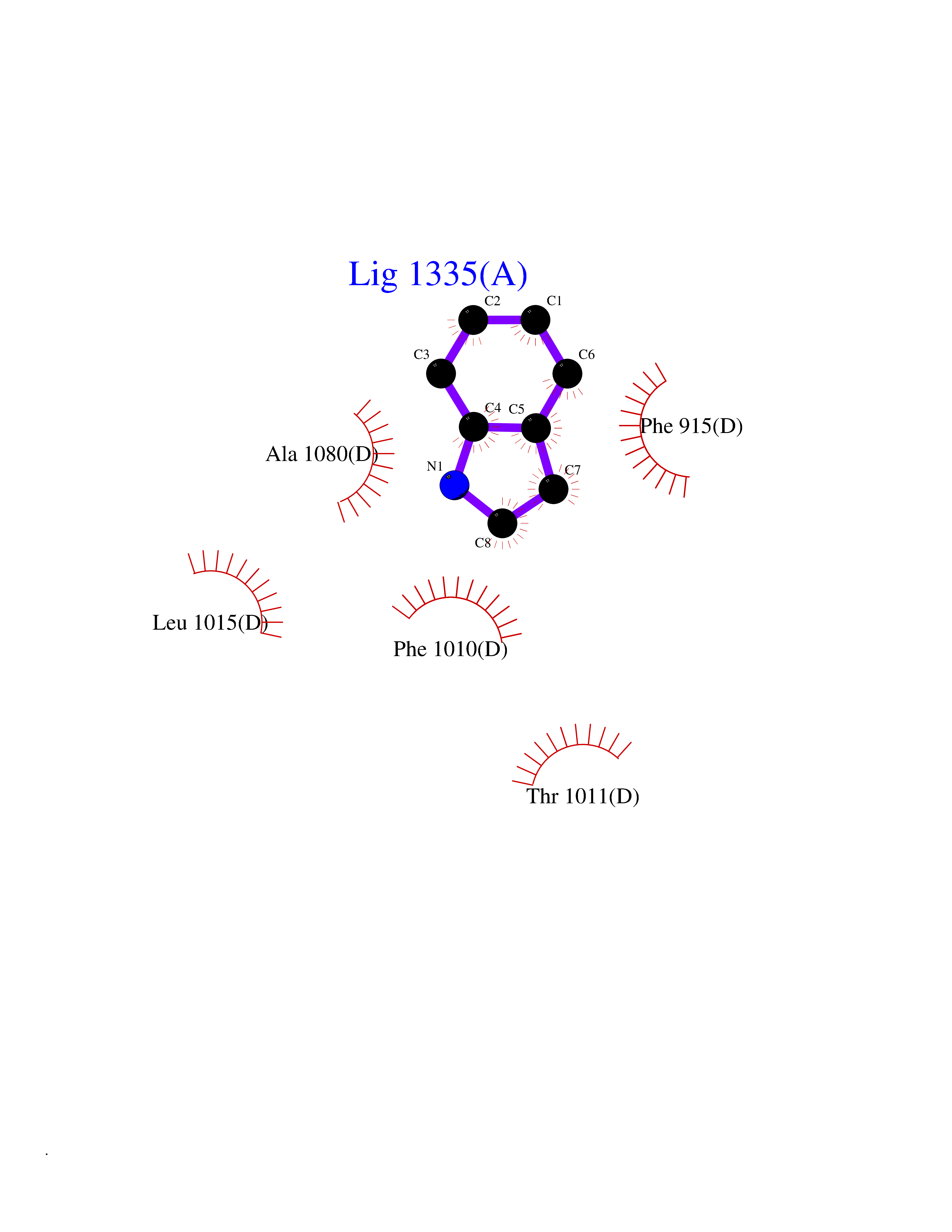 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Histone-lysine N-methyltransferase KMT5B (KMT5B) | 3S8P | 5.36 | |
Target general information Gen name KMT5B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 5B; Lysine-specific methyltransferase 5B; Suppressor of variegation 4-20 homolog 1; Su(var)4-20 homolog 1; Suv4-20h1; [histone H4]-N-methyl-L-lysine20 N-methyltransferase KM Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar4-20 subfamily Biochemical class NA Function Histone methyltransferase that specifically methylates monomethylated 'Lys-20' (H4K20me1) and dimethylated 'Lys-20' (H4K20me2) of histone H4 to produce respectively dimethylated 'Lys-20' (H4K20me2) and trimethylated 'Lys-20' (H4K20me3) and thus regulates transcription and maintenance of genome integrity. In vitro also methylates unmodified 'Lys-20' (H4K20me0) of histone H4 and nucleosomes. H4 'Lys-20' trimethylation represents a specific tag for epigenetic transcriptional repression. Mainly functions in pericentric heterochromatin regions, thereby playing a central role in the establishment of constitutive heterochromatin in these regions. KMT5B is targeted to histone H3 via its interaction with RB1 family proteins (RB1, RBL1 and RBL2) (By similarity). Plays a role in myogenesis by regulating the expression of target genes, such as EID3. Facilitates TP53BP1 foci formation upon DNA damage and proficient non-homologous end-joining (NHEJ)-directed DNA repair by catalyzing the di- and trimethylation of 'Lys-20' of histone H4. May play a role in class switch reconbination by catalyzing the di- and trimethylation of 'Lys-20' of histone H4 (By similarity). Related diseases Intellectual developmental disorder, autosomal dominant 51 (MRD51) [MIM:617788]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:28191889, ECO:0000269|PubMed:29276005}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H2G4; Q61026 EC number EC 2.1.1.361 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Disease variant; Intellectual disability; Isopeptide bond; Metal-binding; Methyltransferase; Myogenesis; Nucleus; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 26230.2 Length 233 Aromaticity 0.11 Instability index 42.29 Isoelectric point 5.64 Charge (pH=7) -6.07 2D Binding mode Binding energy (Kcal/mol) -7.31  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence XSAKELCENDDLATSLVLDPYLGFQTHKXNTRQEELKEVIERFKKDEHLEKAFKCLTSGEWARHYFLNKNKXQEKLFKEHVFIYLRXFATDSGFEILPCNRYSSEQNGAKIVATKEWKRNDKIELLVGCIAELSEIEENXLLRHGENDFSVXYSTRKNCAQLWLGPAAFINHDCRPNCKFVSTGRDTACVKALRDIEPGEEISCYYGDGFFGENNEFCECYTCERRGTGAFKS Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Oxysterols receptor LXR-beta (NR1H2) | 4RAK | 5.36 | |
Target general information Gen name NR1H2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitously-expressed nuclear receptor; Nuclear receptor subfamily 1 group H member 2; Nuclear receptor NER; Nuclear orphan receptor LXR-beta; NER; Liver X receptor beta; LXRbeta; LXRB Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Binds preferentially to double-stranded oligonucleotide direct repeats having the consensus half-site sequence 5'-AGGTCA-3' and 4-nt spacing (DR-4). Regulates cholesterol uptake through MYLIP-dependent ubiquitination of LDLR, VLDLR and LRP8; DLDLR and LRP8. Interplays functionally with RORA for the regulation of genes involved in liver metabolism. Plays an anti-inflammatory role during the hepatic acute phase response by acting as a corepressor: inhibits the hepatic acute phase response by preventing dissociation of the N-Cor corepressor complex. Nuclear receptor that exhibits a ligand-dependent transcriptional activation activity. Related diseases Wolcott-Rallison syndrome (WRS) [MIM:226980]: A rare autosomal recessive disorder, characterized by permanent neonatal or early infancy insulin-dependent diabetes and, at a later age, epiphyseal dysplasia, osteoporosis, growth retardation and other multisystem manifestations, such as hepatic and renal dysfunctions, intellectual disability and cardiovascular abnormalities. {ECO:0000269|PubMed:10932183, ECO:0000269|PubMed:12086964, ECO:0000269|PubMed:12960215, ECO:0000269|PubMed:16813601, ECO:0000269|PubMed:24168455, ECO:0000269|PubMed:24194294, ECO:0000269|PubMed:27145240, ECO:0000269|PubMed:28220546, ECO:0000269|PubMed:30906465, ECO:0000269|PubMed:30922274, ECO:0000269|PubMed:32216767, ECO:0000269|PubMed:34123975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07082; DB03848; DB11994; DB03791; DB13174; DB07080 Interacts with Q92828; Q99750; O75376; P19793; P48443; Q07869-1; Q03181; P37231; P19793 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 27215.1 Length 236 Aromaticity 0.09 Instability index 54.18 Isoelectric point 7.08 Charge (pH=7) 0.11 2D Binding mode Binding energy (Kcal/mol) -7.32  Molscript Map 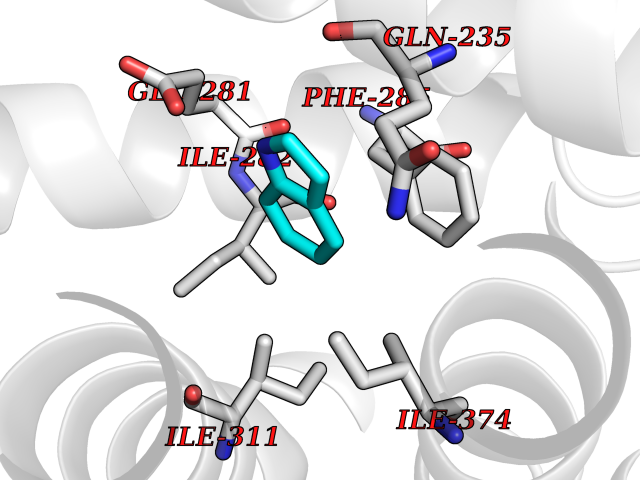 Pymol Map 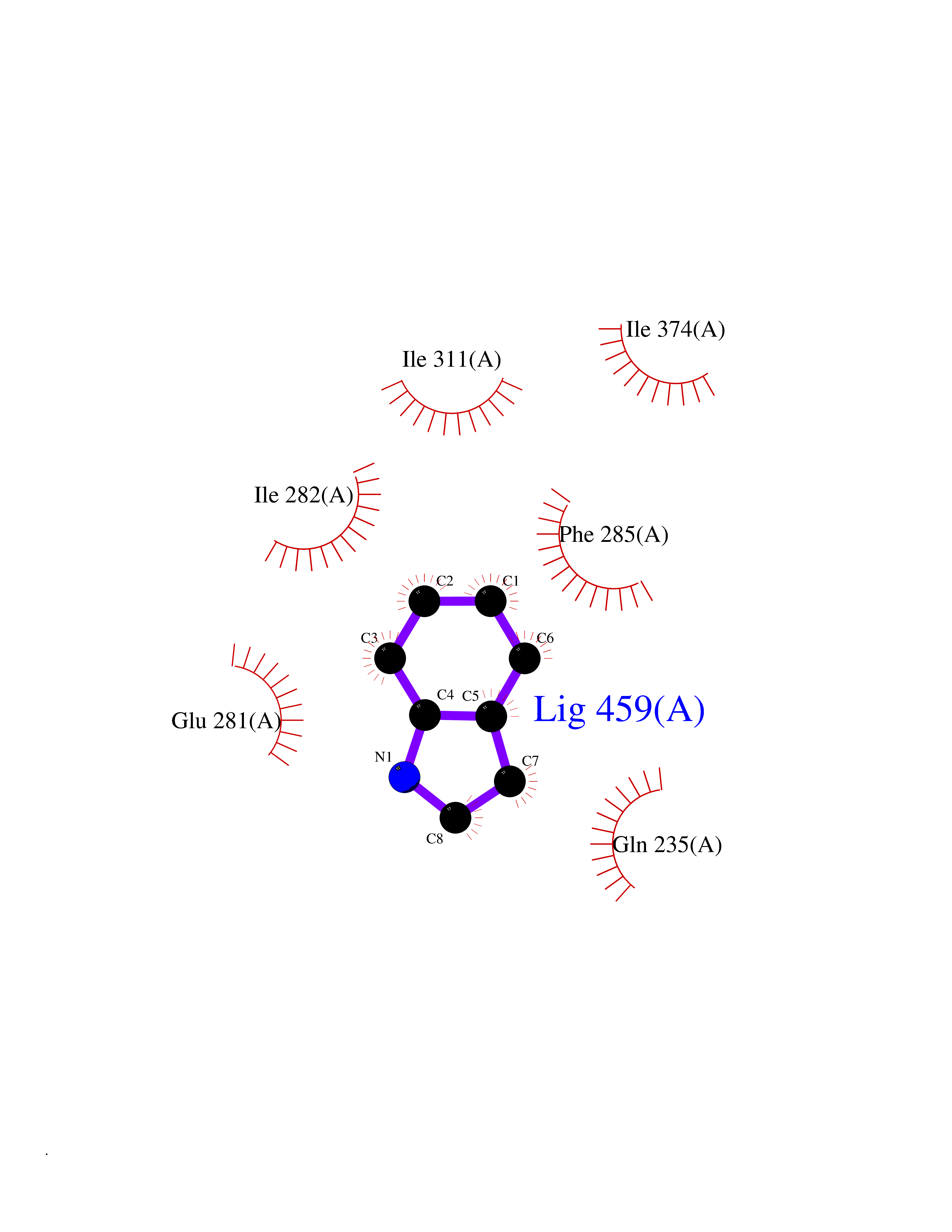 Ligplot Map 3D Binding mode Sequence VQLTAAQELMIQQLVAAQLQCNKRSFSDQPKVTPWPLGADPQSRDARQQRFAHFTELAIISVQEIVDFAKQVPGFLQLGREDQIALLKASTIEIMLLETARRYNHETECITFLKDFTYSKDDFHRAGLQVEFINPIFEFSRAMRRLGLDDAEYALLIAINIFSADRPNVQEPGRVEALQQPYVEALLSYTRIKRPQDQLRFPRMLMKLVSLRTLSSVHSEQVFALRKLPPLLSEIW Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Monoglyceride lipase (MAGL) | 3PE6 | 5.36 | |
Target general information Gen name MGLL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoacylglycerol lipase; MGL; Lysophospholipaselike; Lysophospholipase-like; Lysophospholipase homolog; HUK5; HU-K5 Protein family AB hydrolase superfamily, Monoacylglycerol lipase family Biochemical class Carboxylic ester hydrolase Function Hydrolyzes the endocannabinoid 2-arachidonoylglycerol, and thereby contributes to the regulation of endocannabinoid signaling, nociperception and perception of pain. Regulates the levels of fatty acids that serve as signaling molecules and promote cancer cell migration, invasion and tumor growth. Converts monoacylglycerides to free fatty acids and glycerol. Related diseases Systemic lupus erythematosus 9 (SLEB9) [MIM:610927]: A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. {ECO:0000269|PubMed:17360460}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency, common variable, 7 (CVID7) [MIM:614699]: A primary immunodeficiency characterized by antibody deficiency, hypogammaglobulinemia, recurrent bacterial infections and an inability to mount an antibody response to antigen. The defect results from a failure of B-cell differentiation and impaired secretion of immunoglobulins; the numbers of circulating B-cells is usually in the normal range, but can be low. {ECO:0000269|PubMed:22035880}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07550; P37235 EC number EC 3.1.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Lipid biosynthesis; Lipid degradation; Lipid metabolism; Membrane; Nitration; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 31808.4 Length 289 Aromaticity 0.08 Instability index 29.7 Isoelectric point 6.73 Charge (pH=7) -0.91 2D Binding mode Binding energy (Kcal/mol) -7.31 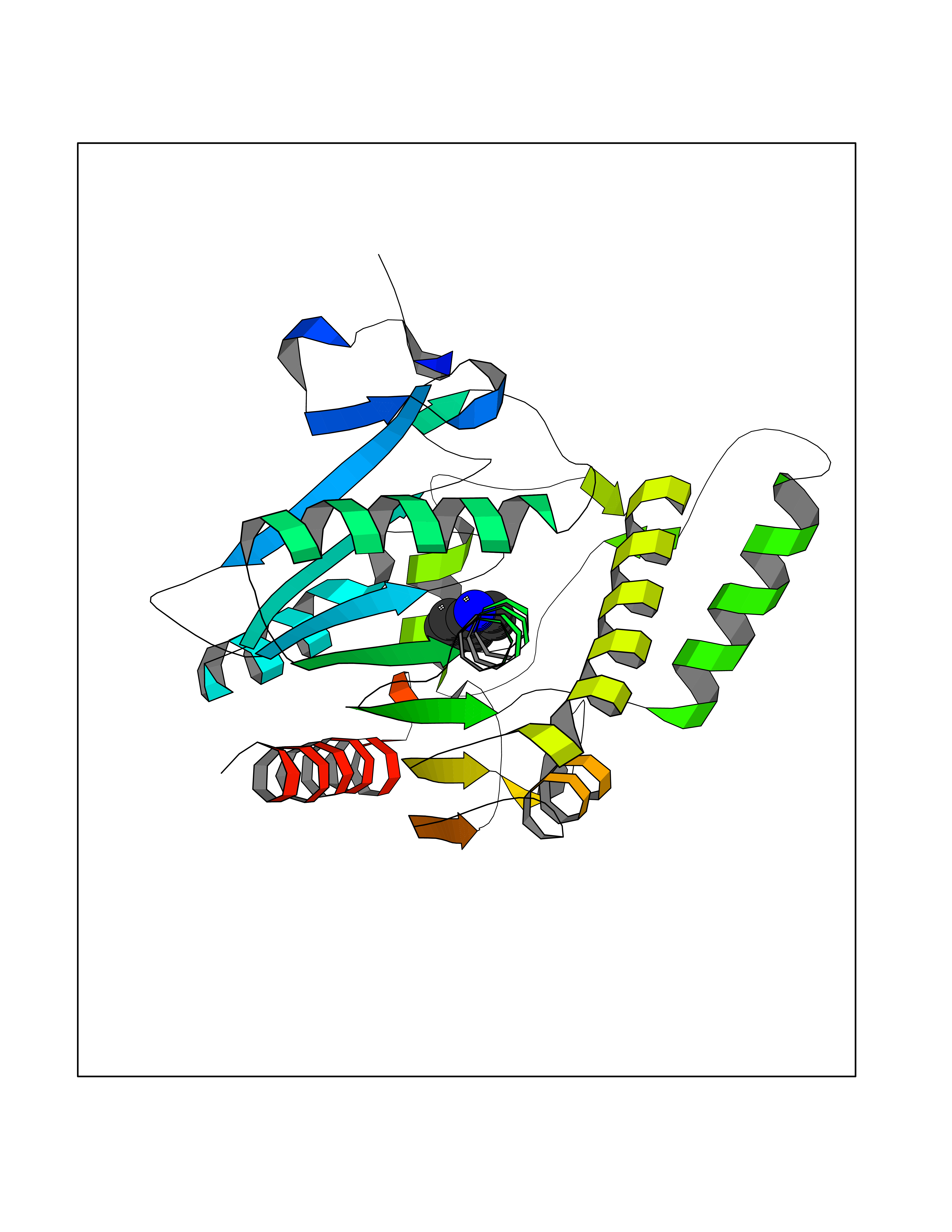 Molscript Map 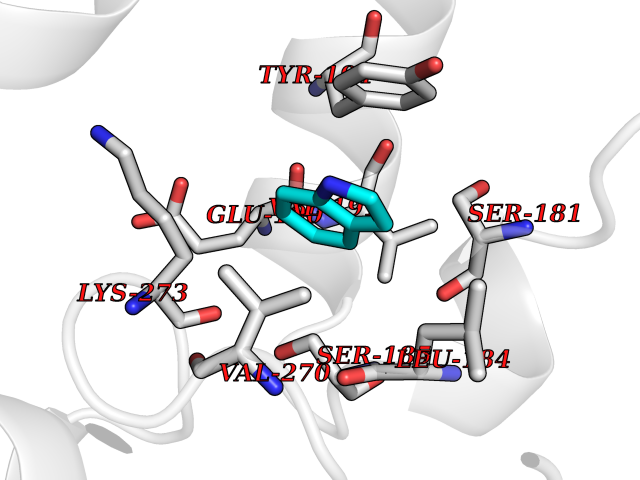 Pymol Map  Ligplot Map 3D Binding mode Sequence PRRTPQSIPYQDLPHLVNADGQYLFCRYWAPTGTPKALIFVSHGAGEHSGRYEELARMLMGLDLLVFAHDHVGHGQSEGERMVVSDFHVFVRDVLQHVDSMQKDYPGLPVFLLGHSMGGAIAILTAAERPGHFAGMVLISPLVLANPESATTFKVLAAKVLNSVLPNLSSGPIDSSVLSRNKTEVDIYNSDPLICRAGLKVCFGIQLLNAVSRVERALPKLTVPFLLLQGSADRLCDSKGAYLLMELAKSQDKTLKIYEGAYHVLHKELPEVTNSVFHEINMWVSQRTA Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Somatostatin receptor type 4 (SSTR4) | 7XMT | 5.36 | |
Target general information Gen name SSTR4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SSTR4; SS4R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for somatostatin-14. The activity of this receptor is mediated by G proteins which inhibits adenylyl cyclase. It is functionally coupled not only to inhibition of adenylate cyclase, but also to activation of both arachidonate release and mitogen-activated protein (MAP) kinase cascade. Mediates antiproliferative action of somatostatin in tumor cells. Related diseases Oocyte/zygote/embryo maturation arrest 21 (OZEMA21) [MIM:620610]: An autosomal dominant, female infertility disorder characterized by zygote development arrest due to failure of pronuclei fusion. {ECO:0000269|PubMed:33948904, ECO:0000269|PubMed:33953335}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB13985; DB09099 Interacts with P35346 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 29548.3 Length 265 Aromaticity 0.12 Instability index 45.65 Isoelectric point 9.78 Charge (pH=7) 14.03 2D Binding mode Binding energy (Kcal/mol) -7.32  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GMVAIQCIYALVCLVGLVGNALVIFVILRYAKMKTATNIYLLNLAVADELFMLSVPFVASSAALRHWPFGSVLCRAVLSVDGLNMFTSVFCLTVLSVDRYVAVVHPLRAATYRRPSVAKLINLGVWLASLLVTLPIAIFADTRPACNLQWPHPAWSAVFVVYTFLLGFLLPVLAIGLCYLLIVGKMRAVALRAGWQQRRRSEKKITRLVLMFVVVFVLCWMPFYVVQLLNLFLDATVNHVSLILSYANSCANPILYGFLSDNFRR Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Neuronal acetylcholine receptor alpha-3/beta-4 (CHRNA3/B4) | 6PV7 | 5.36 | |
Target general information Gen name CHRNA3-CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Neuronal acetylcholine receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Neurotransmitter receptor Function A type of nicotinic acetylcholine receptor, consisting of 3 and 4 subunits. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -7.31  Molscript Map 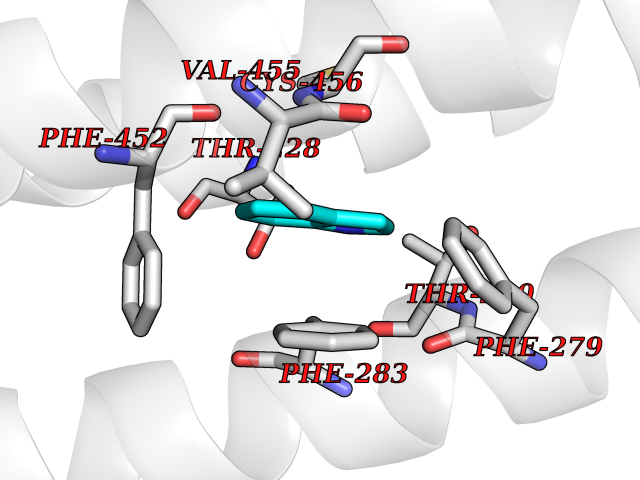 Pymol Map 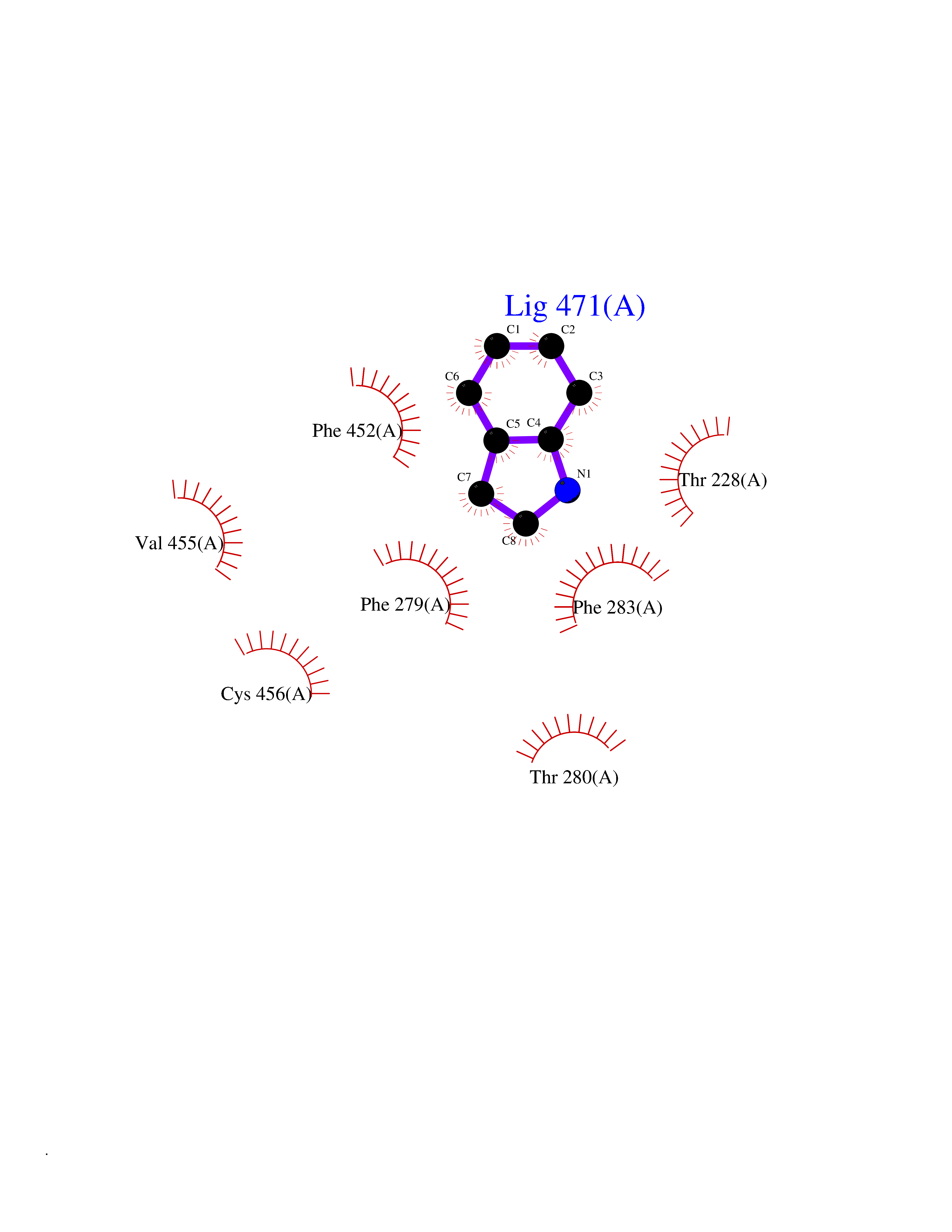 Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Receptor-type protein-tyrosine phosphatase zeta (PTPRZ1) | 5H08 | 5.36 | |
Target general information Gen name PTPRZ1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Receptor protein tyrosine phosphatase zeta; R-PTP-zeta; PTPRZ1 Protein family Protein-tyrosine phosphatase family, Receptor class 5 subfamily Biochemical class Phosphoric monoester hydrolase Function Protein tyrosine phosphatase that negatively regulates oligodendrocyte precursor proliferation in the embryonic spinal cord. Required for normal differentiation of the precursor cells into mature, fully myelinating oligodendrocytes. May play a role in protecting oligondendrocytes against apoptosis. May play a role in the establishment of contextual memory, probably via the dephosphorylation of proteins that are part of important signaling cascades. Related diseases Optic atrophy 1 (OPA1) [MIM:165500]: A condition that features progressive visual loss in association with optic atrophy. Atrophy of the optic disk indicates a deficiency in the number of nerve fibers which arise in the retina and converge to form the optic disk, optic nerve, optic chiasm and optic tracts. OPA1 is characterized by an insidious onset of visual impairment in early childhood with moderate to severe loss of visual acuity, temporal optic disk pallor, color vision deficits, and centrocecal scotoma of variable density. {ECO:0000269|PubMed:11017079, ECO:0000269|PubMed:11017080, ECO:0000269|PubMed:11440988, ECO:0000269|PubMed:11440989, ECO:0000269|PubMed:11810270, ECO:0000269|PubMed:12036970, ECO:0000269|PubMed:12566046, ECO:0000269|PubMed:14961560, ECO:0000269|PubMed:15948788, ECO:0000269|PubMed:16513463, ECO:0000269|PubMed:16617242, ECO:0000269|PubMed:18204809, ECO:0000269|PubMed:18360822, ECO:0000269|PubMed:19319978, ECO:0000269|PubMed:19325939, ECO:0000269|PubMed:19969356, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:22382025, ECO:0000269|PubMed:22857269, ECO:0000269|PubMed:23401657}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Dominant optic atrophy plus syndrome (DOA+) [MIM:125250]: A neurologic disorder characterized most commonly by an insidious onset of visual loss and sensorineural hearing loss in childhood with variable presentation of other clinical manifestations including progressive external ophthalmoplegia, muscle cramps, hyperreflexia, and ataxia. There appears to be a wide range of intermediate phenotypes. {ECO:0000269|PubMed:15531309, ECO:0000269|PubMed:16240368, ECO:0000269|PubMed:18065439, ECO:0000269|PubMed:18158317, ECO:0000269|PubMed:18195150, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:21112924, ECO:0000269|PubMed:23387428}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Behr syndrome (BEHRS) [MIM:210000]: An autosomal recessive syndrome characterized by optic atrophy beginning in early childhood associated with ataxia, pyramidal signs, spasticity, intellectual disability, and posterior column sensory loss. The ataxia, spasticity, and muscle contractures, mainly of the hip adductors, hamstrings, and soleus, are progressive and become more prominent in the second decade. {ECO:0000269|PubMed:21636302, ECO:0000269|PubMed:25012220, ECO:0000269|PubMed:25146916}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mitochondrial DNA depletion syndrome 14, cardioencephalomyopathic type (MTDPS14) [MIM:616896]: An autosomal recessive mitochondrial disorder characterized by lethal infantile encephalopathy, hypertrophic cardiomyopathy and optic atrophy. Skeletal muscle biopsies show significant mtDNA depletion and abnormal mitochondria. {ECO:0000269|PubMed:26561570}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UM73; Q12860 EC number EC 3.1.3.48 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Hydrolase; Membrane; Phosphoprotein; Protein phosphatase; Proteoglycan; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32208.1 Length 282 Aromaticity 0.11 Instability index 38.98 Isoelectric point 7.35 Charge (pH=7) 0.89 2D Binding mode Binding energy (Kcal/mol) -7.31  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GPAIPIKHFPKHVADLHASSGFTEEFEEVQSCTVDLGITADSSNHPDNKHKNRYINIVAYDHSRVKLAQLAEKDGKLTDYINANYVDGYNRPKAYIAAQGPLKSTAEDFWRMIWEHNVEVIVMITNLVEKGRRKCDQYWPADGSEEYGNFLVTQKSVQVLAYYTVRNFTLRNTKIRVVTQYHYTQWPDMGVPEYSLPVLTFVRKAAYAKRHAVGPVVVHCSAGVGRTGTYIVLDSMLQQIQHEGTVNIFGFLKHIRSQRNYLVQTEEQYVFIHDTLVEAILS Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Cytosolic 10-formyltetrahydrofolate dehydrogenase | 2CFI | 5.35 | |
Target general information Gen name ALDH1L1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FTHFD Protein family GART family; Aldehyde dehydrogenase family, ALDH1L subfamily Biochemical class Oxidoreductase Function Aldehyde dehydrogenase (NAD) activity.Catalytic activity.Formyltetrahydrofolate dehydrogenase activity.Hydroxymethyl-, formyl- and related transferase activity. Related diseases Developmental and epileptic encephalopathy 39 with leukodystrophy (DEE39) [MIM:612949]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE39 is characterized by global hypomyelination of the central nervous system, with the gray matter appearing relatively unaffected. Inheritance is autosomal recessive. {ECO:0000269|PubMed:19641205, ECO:0000269|PubMed:24515575}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00116 Interacts with Q3SY69; Q92624 EC number 1.5.1.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; NADP; One-carbon metabolism; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33869.5 Length 308 Aromaticity 0.08 Instability index 30.42 Isoelectric point 6.09 Charge (pH=7) -3.83 2D Binding mode Binding energy (Kcal/mol) -7.29  Molscript Map  Pymol Map 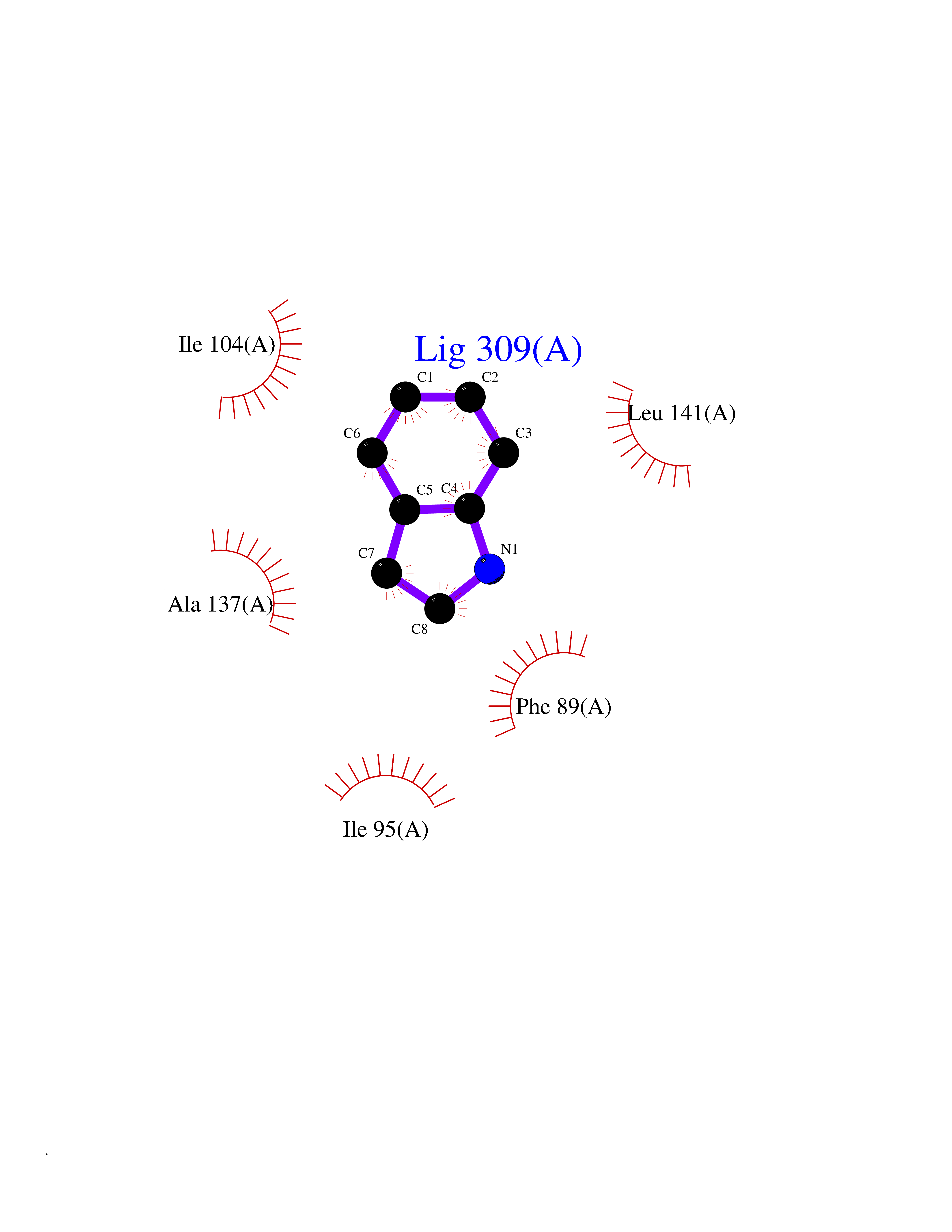 Ligplot Map 3D Binding mode Sequence SMKIAVIGQSLFGQEVYCHLRKEGHEVVGVFTVPDKDGKADPLGLEAEKDGVPVFKYSRWRAKGQALPDVVAKYQALGAELNVLPFCSQFIPMEIISAPRHGSIIYHPSLLPRHRGASAINWTLIHGDKKGGFSIFWADDGLDTGDLLLQKECEVLPDDTVSTLYNRFLFPEGIKGMVQAVRLIAEGKAPRLPQPEEGATYEGIQKKETAKINWDQPAEAIHNWIRGNDKVPGAWTEACEQKLTFFNSTLNTSGLVPEGDALPIPGAHRPGVVTKAGLILFGNDDKMLLVKNIQLEDGKMILASNFFK Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Natriuretic peptides B | 1YK1 | 5.35 | |
Target general information Gen name NPPB Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Natriuretic peptide family Biochemical class Hormone / growth factor receptor Function Diuretic hormone activity.Hormone activity.Peptide hormone receptor binding.Receptor binding. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01136; DB06412 Interacts with A8MQ03; P57678; Q6A162; P60411; Q7Z3S9; P25788; Q9UJW9 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disulfide bond; Glycoprotein; Hormone; Pharmaceutical; Proteoglycan; Proteomics identification; Reference proteome; Secreted; Signal; Vasoactive; Vasodilator Protein physicochemical properties Chain ID E Molecular weight (Da) 46353.1 Length 415 Aromaticity 0.1 Instability index 37.91 Isoelectric point 5.51 Charge (pH=7) -12.09 2D Binding mode Binding energy (Kcal/mol) -7.3 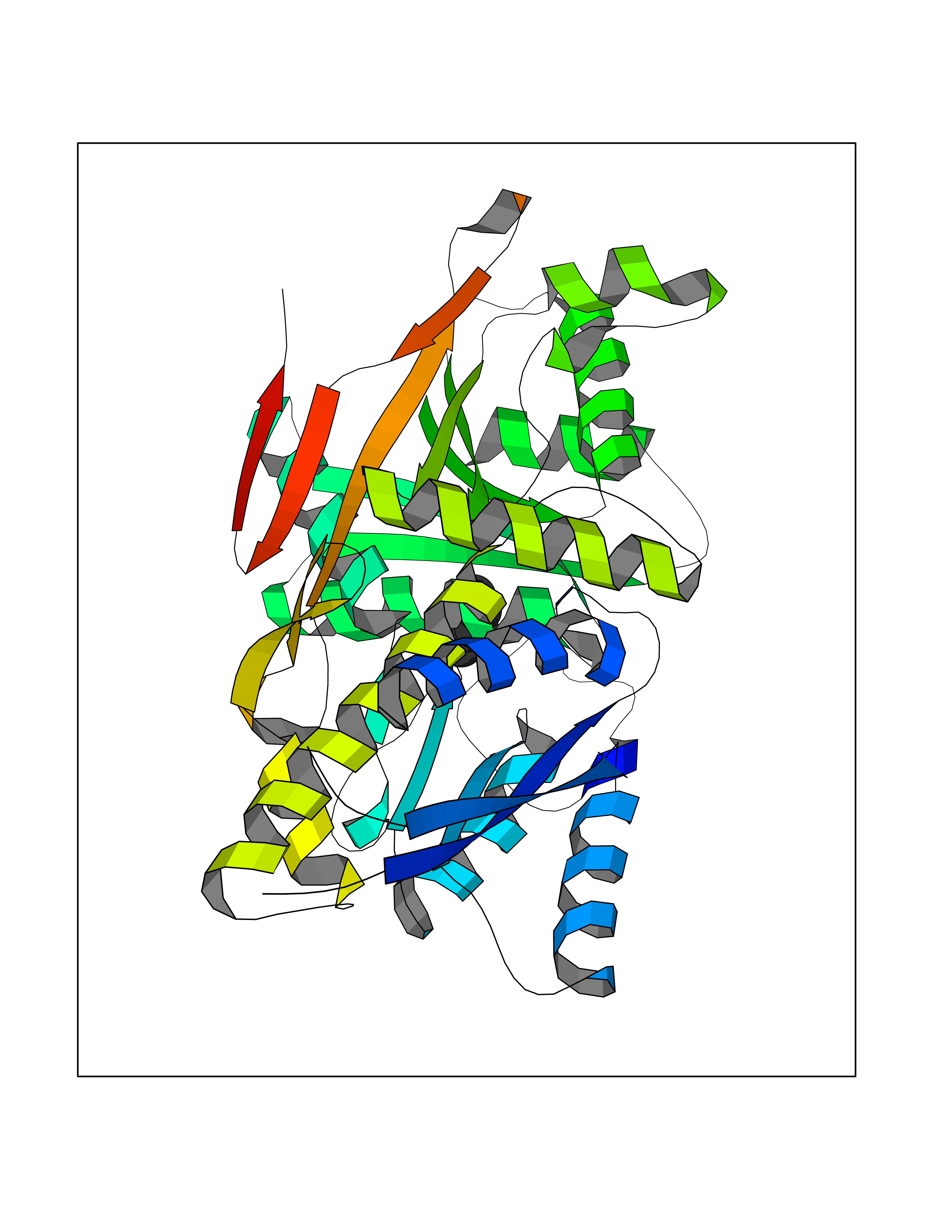 Molscript Map 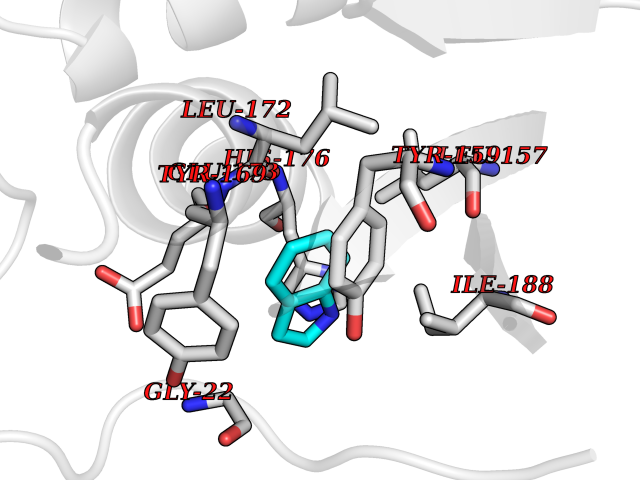 Pymol Map 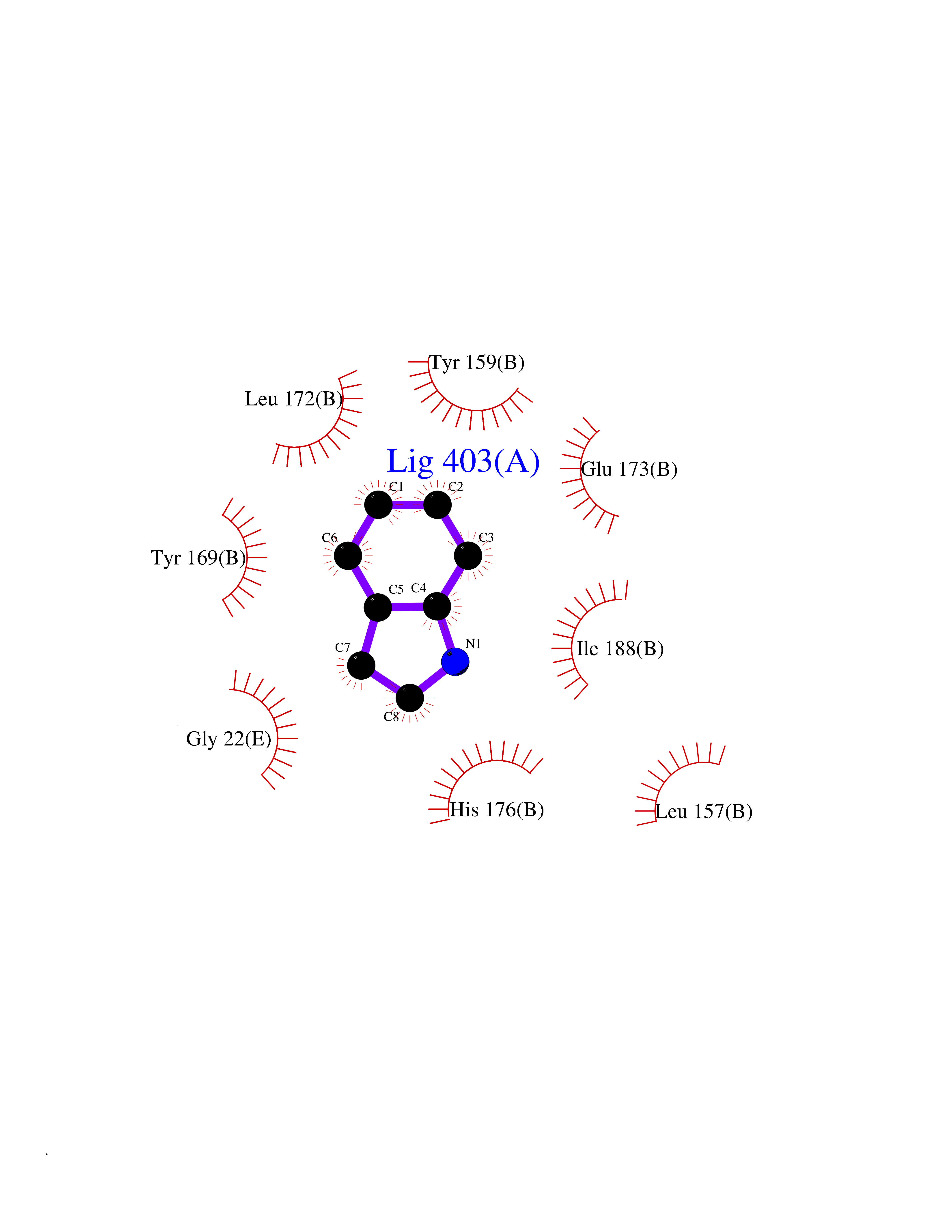 Ligplot Map 3D Binding mode Sequence GCFGRKMDRISSSSGLGCKVLALPPQKIEVLVLLPQDDSYLFSLTRVRPAIEYALRSVEGLLPPGTRFQVAYEDSDCGNRALFSLVDRVAAARGAKPDLILGPVCEYAAAPVARLASHWDLPMLSAGALAAGFQHKDSEYSHLTRVAPAYAKMGEMMLALFRHHHWSRAALVYSDDKLERNCYFTLEGVHEVFQEEGLHTSIYSFDETKDLDLEDIVRNIQASERVVIMCASSDTIRSIMLVAHRHGMTSGDYAFFNIELFNSSSYGDGSWKRGDKHDFEAKQAYSSLQTVTLLRTVKPEFEKFSMEVKSSVEKQGLNMEDYVNMFVEGFHDAILLYVLALHEVLRAGYSKKDGGKIIQQTWNRTFEGIAGQVSIDANGDRYGDFSVIAMTDVEAGTQEVIGDYFGKEGRFEMRP Hydrogen bonds contact Hydrophobic contact | ||||