Job Results:
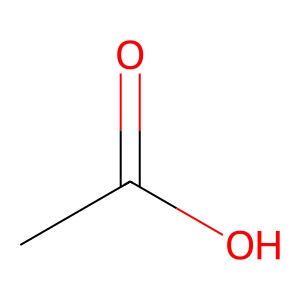
Ligand
Structure
Job ID
6365174e5cc200e6a930804dda23a0ba
Job name
NA
Time
2026-02-27 16:33:59
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | cAMP-dependent protein kinase A type I (PRKAR1A) | 5KJZ | 4.40 | |
Target general information Gen name PRKAR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-dependent protein kinase type I-alpha regulatory subunit; Tissue-specific extinguisher 1; TSE1; PRKAR1; PKR1 Protein family CAMP-dependent kinase regulatory chain family Biochemical class Kinase Function Regulatory subunit of the cAMP-dependent protein kinases involved in cAMP signaling in cells. Related diseases Carney complex 1 (CNC1) [MIM:160980]: CNC is a multiple neoplasia syndrome characterized by spotty skin pigmentation, cardiac and other myxomas, endocrine tumors, and psammomatous melanotic schwannomas. {ECO:0000269|PubMed:15371594, ECO:0000269|PubMed:18241045, ECO:0000269|PubMed:22785148, ECO:0000269|PubMed:23323113, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Intracardiac myxoma (INTMYX) [MIM:255960]: Inheritance is autosomal recessive. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Primary pigmented nodular adrenocortical disease 1 (PPNAD1) [MIM:610489]: A rare bilateral adrenal defect causing ACTH-independent Cushing syndrome. Macroscopic appearance of the adrenals is characteristic with small pigmented micronodules observed in the cortex. Clinical manifestations of Cushing syndrome include facial and truncal obesity, abdominal striae, muscular weakness, osteoporosis, arterial hypertension, diabetes. PPNAD1 is most often diagnosed in patients with Carney complex, a multiple neoplasia syndrome. However it can also be observed in patients without other manifestations. {ECO:0000269|PubMed:12213893}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Acrodysostosis 1, with or without hormone resistance (ACRDYS1) [MIM:101800]: A form of skeletal dysplasia characterized by short stature, severe brachydactyly, facial dysostosis, and nasal hypoplasia. Affected individuals often have advanced bone age and obesity. Laboratory studies show resistance to multiple hormones, including parathyroid, thyrotropin, calcitonin, growth hormone-releasing hormone, and gonadotropin. However, not all patients show endocrine abnormalities. {ECO:0000269|PubMed:21651393, ECO:0000269|PubMed:22464250, ECO:0000269|PubMed:22464252, ECO:0000269|PubMed:22723333, ECO:0000269|PubMed:23043190, ECO:0000269|PubMed:23425300, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01790; DB02527; DB02315; DB05798 Interacts with Q9GZX7; P24588; O43687-2; Q9BSF0; Q9H6J7-2; Q86Y01; P0C7A2-2; Q9H0R8; Q9H8W4; P17612; P31321; P51817; P35250; Q86UC2; Q01105; Q8N0X7; O96006; P03259-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; cAMP; cAMP-binding; Cell membrane; Cushing syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat Protein physicochemical properties Chain ID A Molecular weight (Da) 17007.4 Length 149 Aromaticity 0.08 Instability index 49.36 Isoelectric point 6.36 Charge (pH=7) -0.52 2D Binding mode Binding energy (Kcal/mol) -6 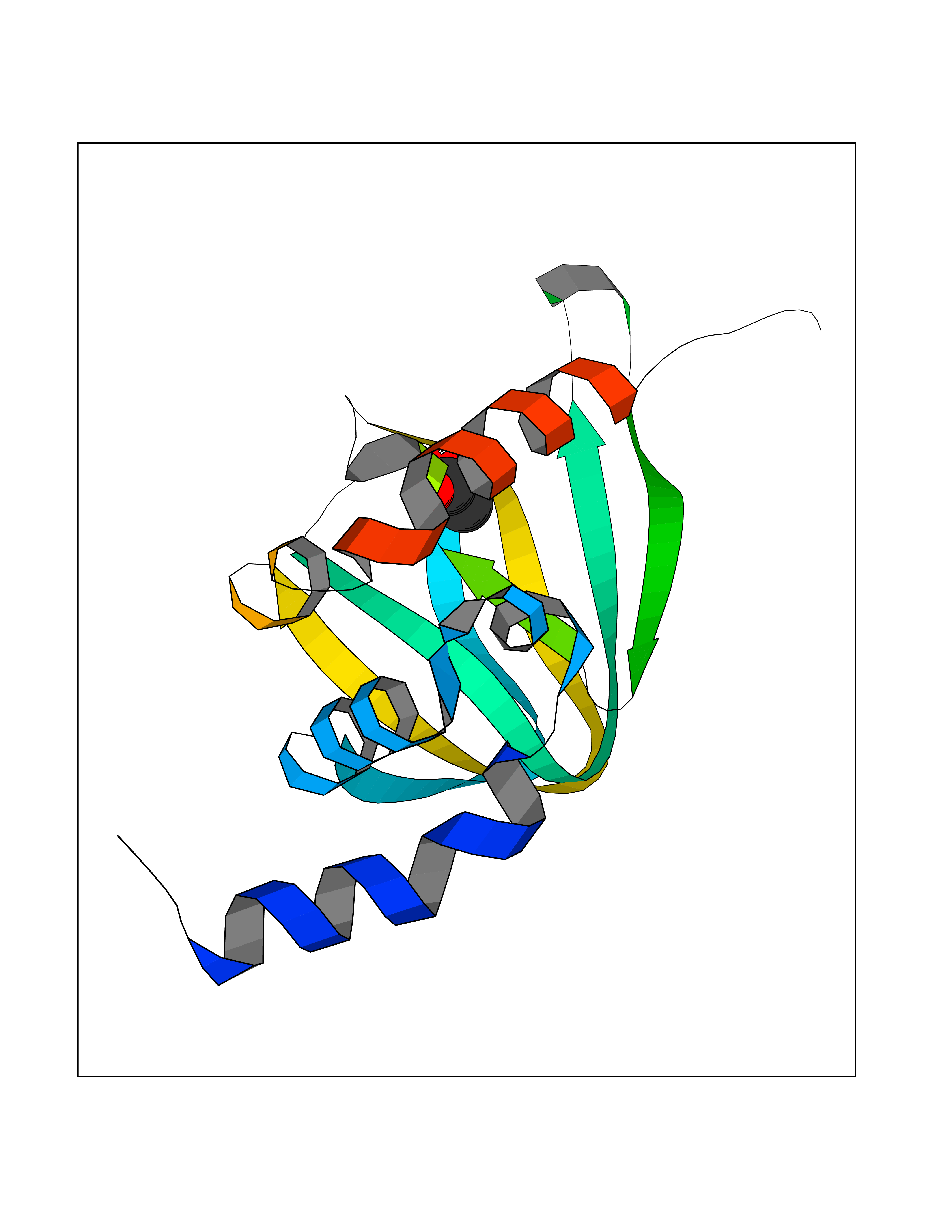 Molscript Map 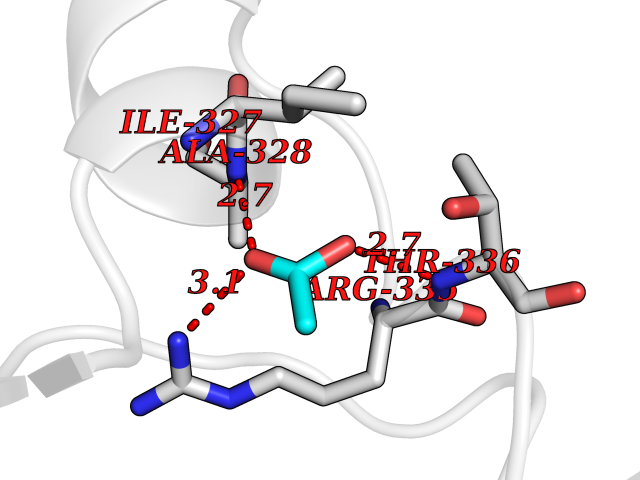 Pymol Map 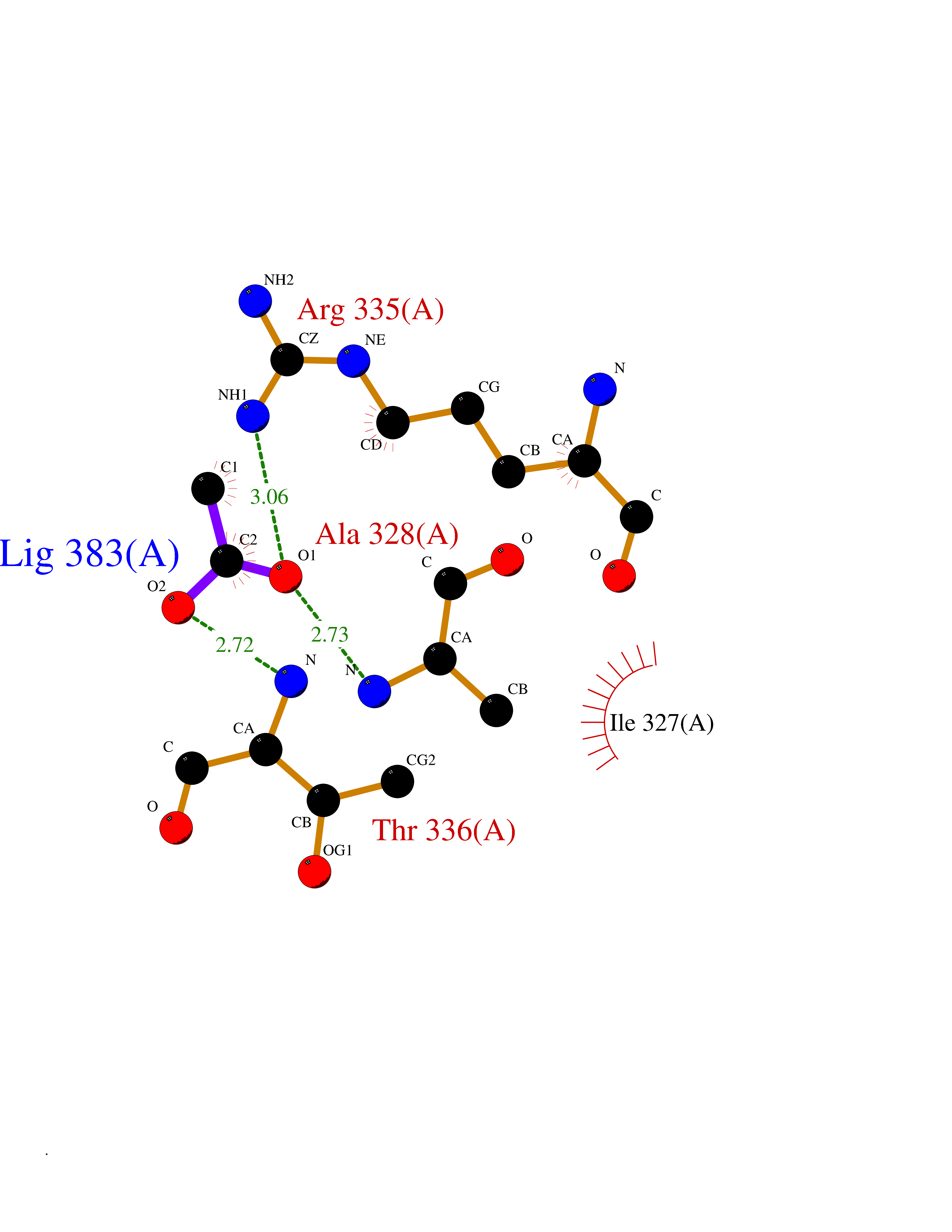 Ligplot Map 3D Binding mode Sequence SILMGSTLRKRKMYEEFLSKVSILESLDKWERLTVADALEPVQFEDGQKIVVQGEPGDEFFIILEGSAAVLQRRSENEEFVEVRRLGPSDYFGEIALLMNRPRTATVVARGPLKCVKLDRPRFERVLGPCSDILKRNIQQYNSFVSLSV Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | mRNA-capping enzyme | 2C46 | 4.39 | |
Target general information Gen name RNGTT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CAP1A Protein family Non-receptor class of the protein-tyrosine phosphatase family; Eukaryotic GTase family Biochemical class Transferase Function GTP binding.MRNA guanylyltransferase activity.Polynucleotide 5'-phosphatase activity.Protein tyrosine/serine/threonine phosphatase activity.Protein tyrosine phosphatase activity.RNA guanylyltransferase activity.Triphosphatase activity. Related diseases Atrial fibrillation, familial, 14 (ATFB14) [MIM:615378]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:19808477}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Genetic variations in SCN2B may be involved in Brugada syndrome (PubMed:23559163). This tachyarrhythmia is characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:23559163}. Drugs (DrugBank ID) NA Interacts with Q92624; P16333-1 EC number 2.7.7.50; 3.6.1.74 Uniprot keywords 3D-structure; Alternative splicing; GTP-binding; Host-virus interaction; Hydrolase; mRNA capping; mRNA processing; Multifunctional enzyme; Nucleotide-binding; Nucleotidyltransferase; Nucleus; Protein phosphatase; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 21849.8 Length 189 Aromaticity 0.11 Instability index 53.71 Isoelectric point 5.89 Charge (pH=7) -2.91 2D Binding mode Binding energy (Kcal/mol) -5.99 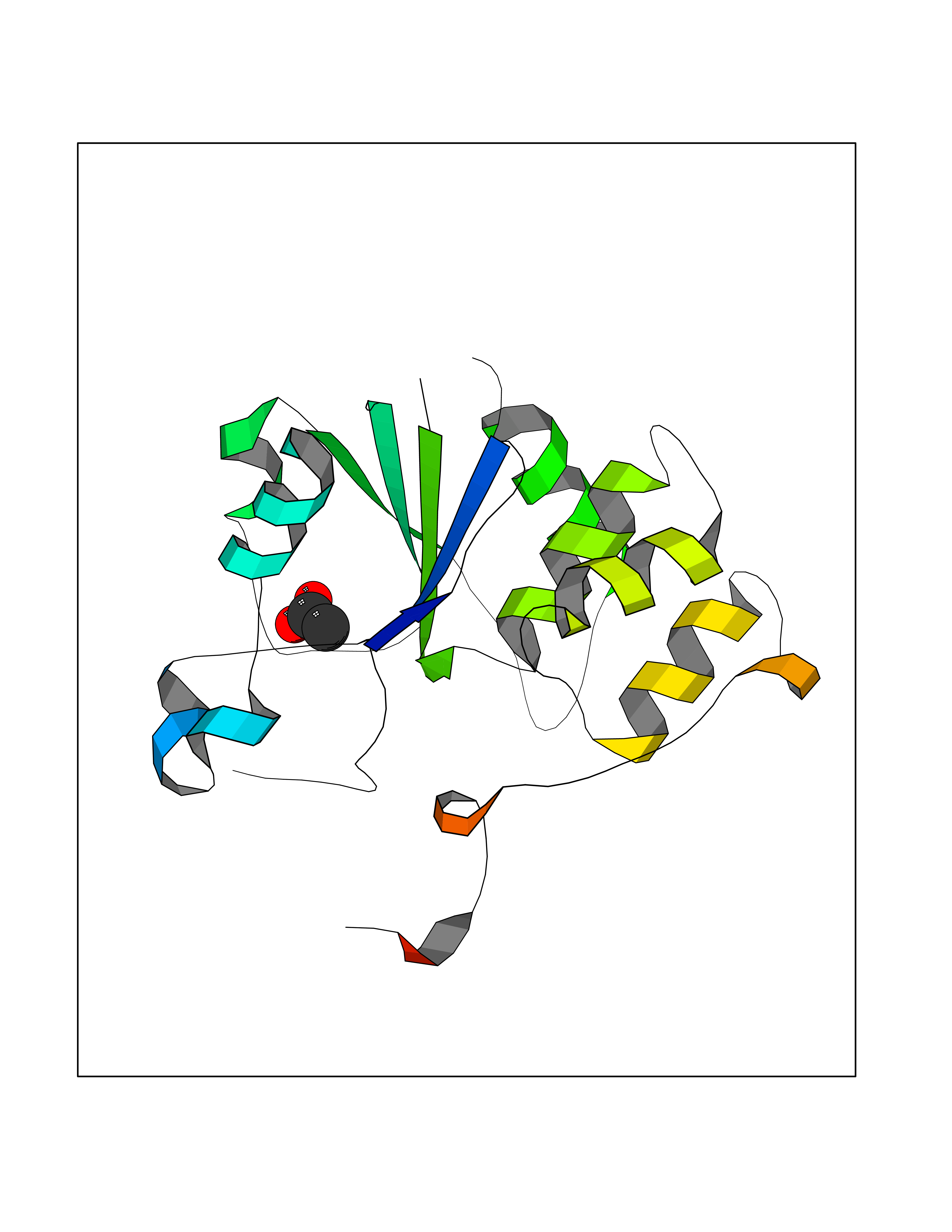 Molscript Map 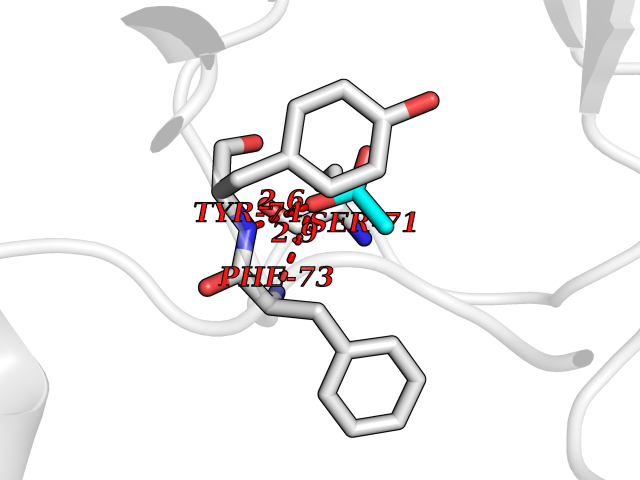 Pymol Map 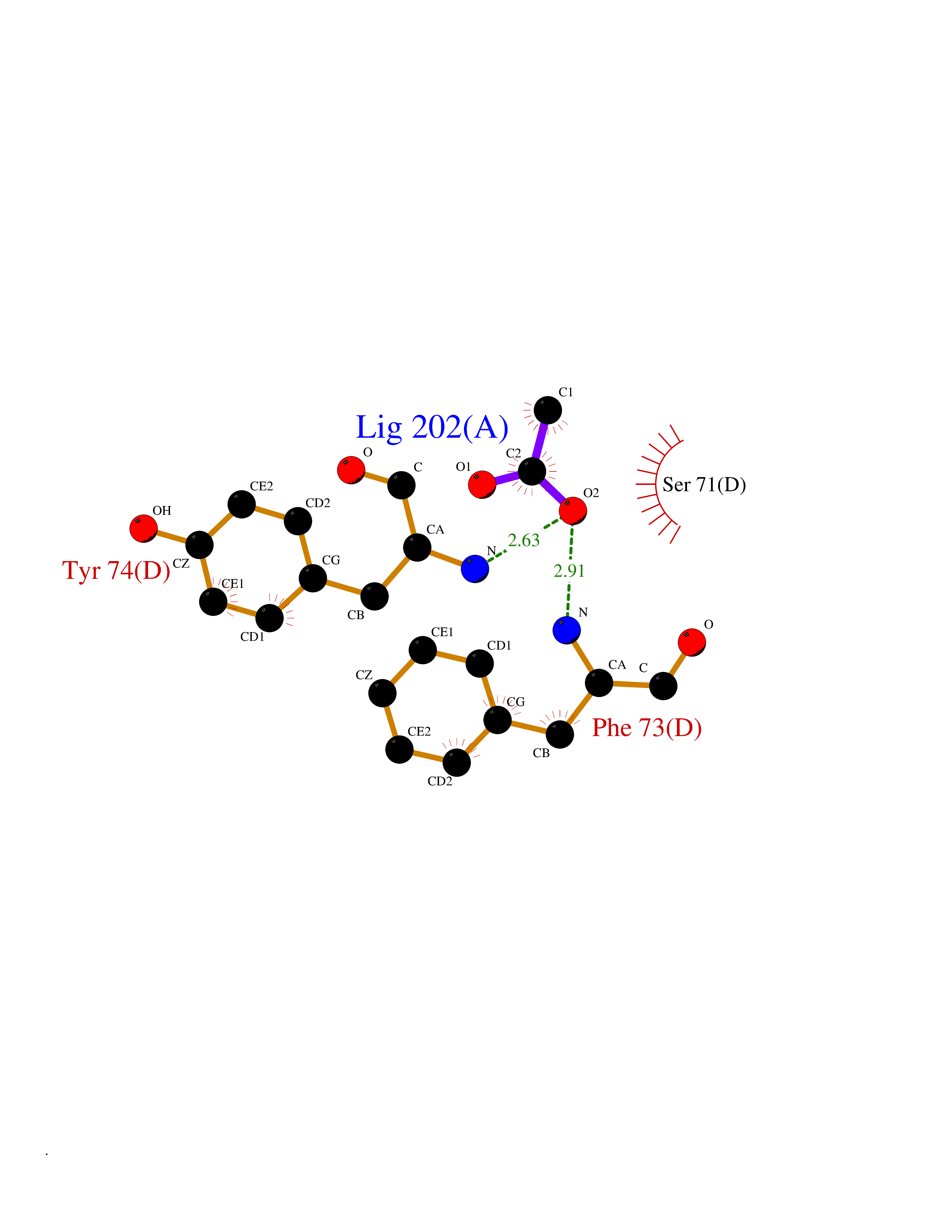 Ligplot Map 3D Binding mode Sequence NKIPPRWLNCPRRGQPVAGRFLPLKTMLGPRYDSQVAEENRFHPSMLSNYLKSVKMGLLVDLTNTSRFYDRNDIEKEGIKYIKLQCKGHGECPTTENTETFIRLCERFELIGVHCTHGFNRTGFLICAFLVEKMDWSIEAAVATFAQARPPGIYKGDYLKELFRRYGDIEEAPPPPLLPDWCFEDDEDE Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Tyrosine 3-monooxygenase (TH) | 2XSN | 4.39 | |
Target general information Gen name TH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine 3-hydroxylase; TH Protein family Biopterin-dependent aromatic amino acid hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Plays an important role in the physiology of adrenergic neurones. Related diseases Segawa syndrome autosomal recessive (ARSEGS) [MIM:605407]: A form of DOPA-responsive dystonia presenting in infancy or early childhood. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Some cases present with parkinsonian symptoms in infancy. Unlike all other forms of dystonia, it is an eminently treatable condition, due to a favorable response to L-DOPA. {ECO:0000269|PubMed:10585338, ECO:0000269|PubMed:11196107, ECO:0000269|PubMed:11246459, ECO:0000269|PubMed:15505183, ECO:0000269|PubMed:15747353, ECO:0000269|PubMed:16049992, ECO:0000269|PubMed:17696123, ECO:0000269|PubMed:18058633, ECO:0000269|PubMed:18554280, ECO:0000269|PubMed:19491146, ECO:0000269|PubMed:20056467, ECO:0000269|PubMed:20430833, ECO:0000269|PubMed:21940685, ECO:0000269|PubMed:22264700, ECO:0000269|PubMed:22815559, ECO:0000269|PubMed:23762320, ECO:0000269|PubMed:23939262, ECO:0000269|PubMed:24753243, ECO:0000269|PubMed:7814018, ECO:0000269|PubMed:8528210, ECO:0000269|PubMed:8817341, ECO:0000269|PubMed:9613851, ECO:0000269|PubMed:9703425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: May play a role in the pathogenesis of Parkinson disease (PD). A genome-wide copy number variation analysis has identified a 34 kilobase deletion over the TH gene in a PD patient but not in any controls. {ECO:0000269|PubMed:20809526}. Drugs (DrugBank ID) DB03552; DB04400; DB00765; DB00120; DB00360; DB00135 Interacts with P29762; P61978-2; Q99750; P08651-5; O75928-2; Q9UHX1-2; P0DJD3-2; P07101-3; Q9UJ04; C9J7I0; Q5MCW4 EC number EC 1.14.16.2 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Cell projection; Cytoplasm; Cytoplasmic vesicle; Disease variant; Dystonia; Iron; Metal-binding; Monooxygenase; Neurotransmitter biosynthesis; Nucleus; Oxidoreductase; Parkinson disease; Parkinsonism; Phosphoprotein; Proteomics identification; Reference proteome; Synapse Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 34997 Length 306 Aromaticity 0.12 Instability index 42.59 Isoelectric point 5.32 Charge (pH=7) -12.31 2D Binding mode Binding energy (Kcal/mol) -5.99 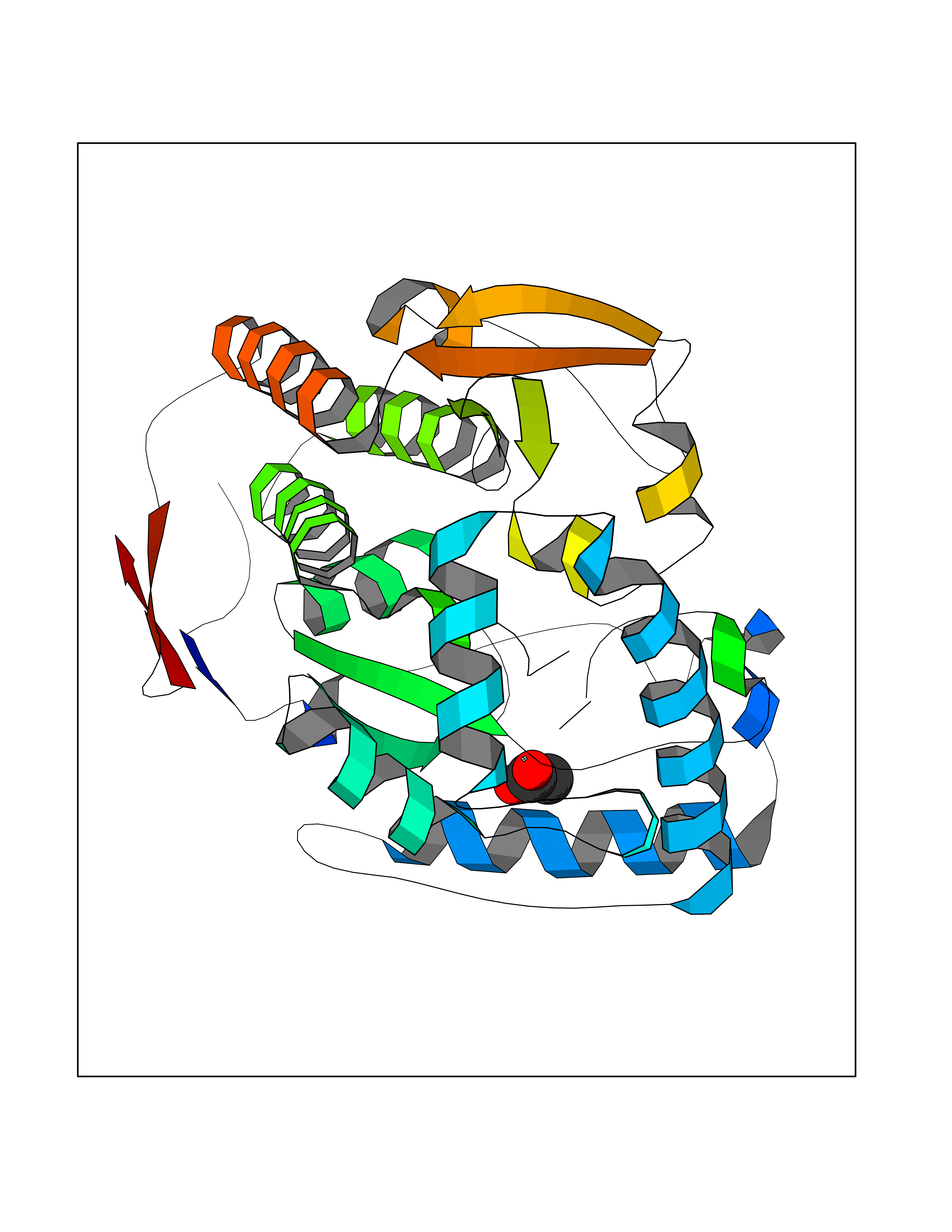 Molscript Map 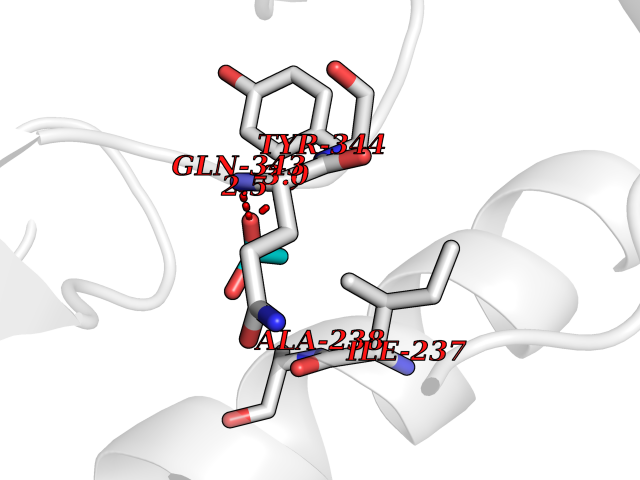 Pymol Map 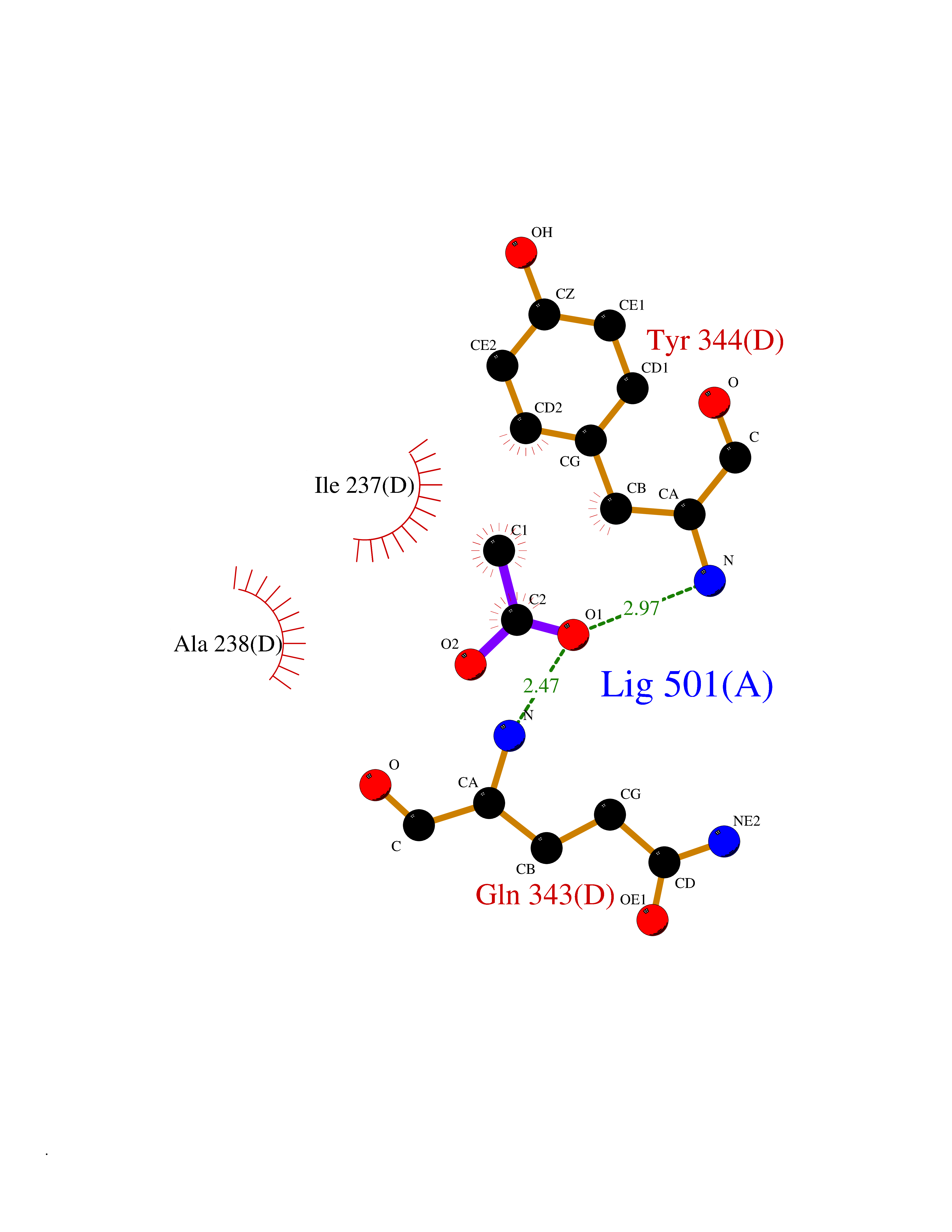 Ligplot Map 3D Binding mode Sequence VPWFPRKVSELDKCHHLVTKFDPDLDLDHPGFSDQVYRQRRKLIAEIAFQYRHGDPIPRVEYTAEEIATWKEVYTTLKGLYATHACGEHLEAFALLERFSGYREDNIPQLEDVSRFLKERTGFQLRPVAGLLSARDFLASLAFRVFQCTQYIRHASSPMHSPEPDCCHELLGHVPMLADRTFAQFSQDIGLASLGASDEEIEKLSTLYWFTVEFGLCKQNGEVKAYGAGLLSSYGELLHCLSEEPEIRAFDPEAAAVQPYQDQTYQSVYFVSESFSDAKDKLRSYASRIQRPFSVKFDPYTLAIDV Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Acetylcholinesterase (AChE) (EC 3.1.1.7) | 1GPK | 4.39 | |
Target general information Gen name ache Organism Tetronarce californica (Pacific electric ray) (Torpedo californica) Uniprot ID TTD ID NA Synonyms NA Protein family Type-B carboxylesterase/lipase family Biochemical class NA Function Terminates signal transduction at the neuromuscular junction by rapid hydrolysis of the acetylcholine released into the synaptic cleft. May be involved in cell-cell interactions. Related diseases Noonan syndrome 5 (NS5) [MIM:611553]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:17603482, ECO:0000269|PubMed:17603483, ECO:0000269|PubMed:20683980}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: LEOPARD syndrome 2 (LPRD2) [MIM:611554]: A disorder characterized by lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonic stenosis, abnormalities of genitalia, retardation of growth, and sensorineural deafness. {ECO:0000269|PubMed:17603483}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cardiomyopathy, dilated, 1NN (CMD1NN) [MIM:615916]: A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. {ECO:0000269|PubMed:24777450}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.1.1.7 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; Neurotransmitter degradation; Serine esterase; Signal; Synapse Protein physicochemical properties Chain ID A Molecular weight (Da) 59779.2 Length 529 Aromaticity 0.12 Instability index 48.49 Isoelectric point 5.8 Charge (pH=7) -8.48 2D Binding mode Binding energy (Kcal/mol) -5.99  Molscript Map 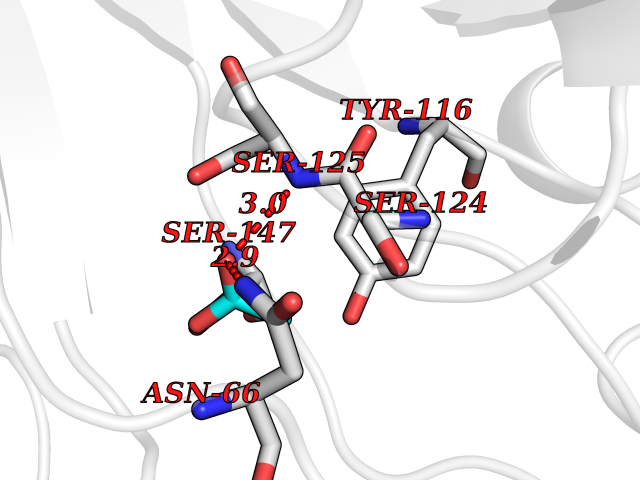 Pymol Map 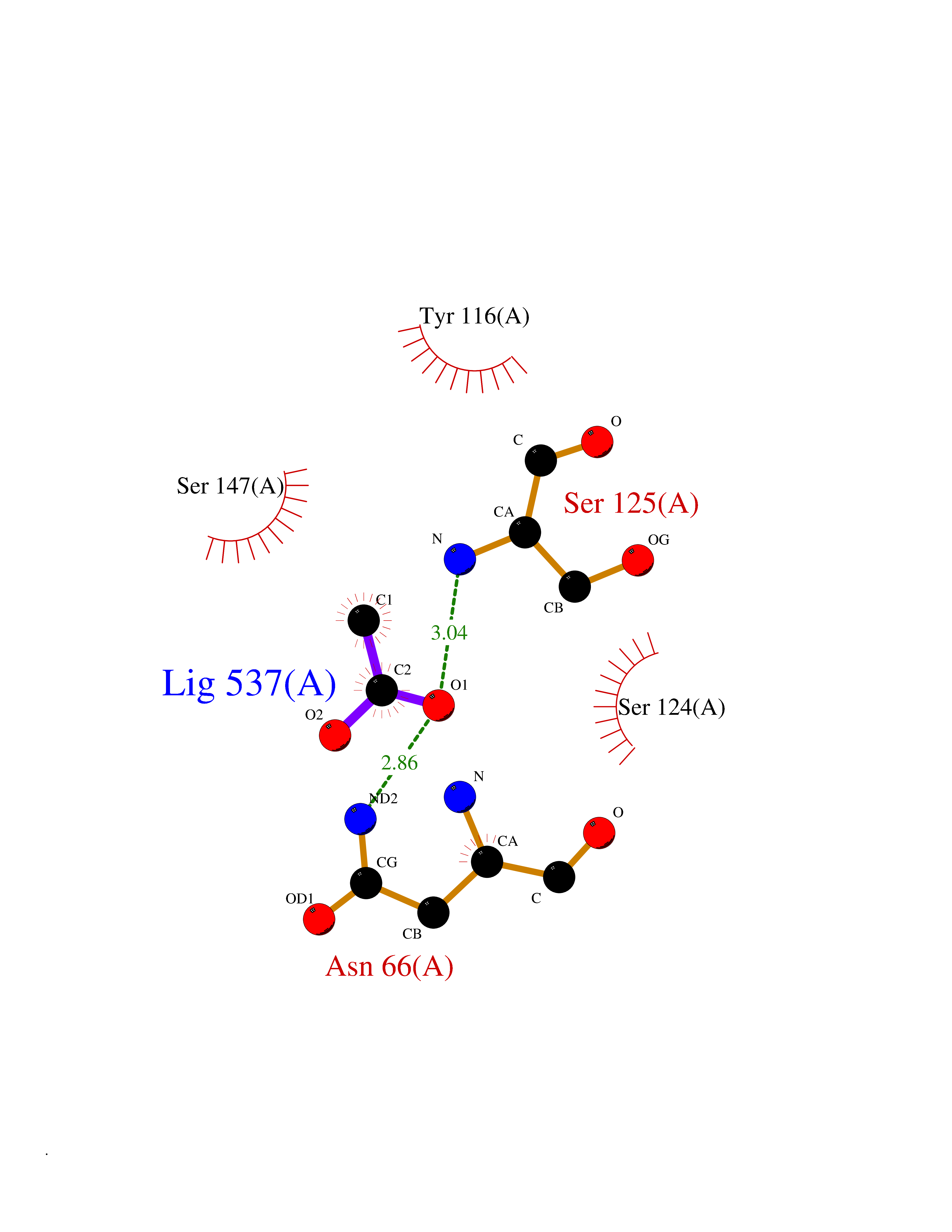 Ligplot Map 3D Binding mode Sequence SELLVNTKSGKVMGTRVPVLSSHISAFLGIPFAEPPVGNMRFRRPEPKKPWSGVWNASTYPNNCQQYVDEQFPGFSGSEMWNPNREMSEDCLYLNIWVPSPRPKSTTVMVWIYGGGFYSGSSTLDVYNGKYLAYTEEVVLVSLSYRVGAFGFLALHGSQEAPGNVGLLDQRMALQWVHDNIQFFGGDPKTVTIFGESAGGASVGMHILSPGSRDLFRRAILQSGSPNCPWASVSVAEGRRRAVELGRNLNCNLNSDEELIHCLREKKPQELIDVEWNVLPFDSIFRFSFVPVIDGEFFPTSLESMLNSGNFKKTQILLGVNKDEGSFFLLYGAPGFSKDSESKISREDFMSGVKLSVPHANDLGLDAVTLQYTDWMDDNNGIKNRDGLDDIVGDHNVICPLMHFVNKYTKFGNGTYLYFFNHRASNLVWPEWMGVIHGYEIEFVFGLPLVKELNYTAEEEALSRRIMHYWATFAKTGNPNEPESKWPLFTTKEQKFIDLNTEPMKVHQRLRVQMCVFWNQFLPKLLNAT Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Xylose isomerase | 1XIM | 4.39 | |
Target general information Gen name xylA Organism Actinoplanes missouriensis (strain ATCC 14538 / DSM 43046 / CBS 188.64 / JCM 3121 / NBRC 102363 / NCIMB 12654 / NRRL B-3342 / UNCC 431) Uniprot ID TTD ID NA Synonyms AMIS_10350;XI Protein family Xylose isomerase family Biochemical class Isomerase(intramolecular oxidoreductase) Function Metal ion binding.Xylose isomerase activity. Related diseases Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Renal tubular dysgenesis (RTD) [MIM:267430]: Autosomal recessive severe disorder of renal tubular development characterized by persistent fetal anuria and perinatal death, probably due to pulmonary hypoplasia from early-onset oligohydramnios (the Potter phenotype). {ECO:0000269|PubMed:16116425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Microvascular complications of diabetes 3 (MVCD3) [MIM:612624]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. {ECO:0000269|PubMed:10099885}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Intracerebral hemorrhage (ICH) [MIM:614519]: A pathological condition characterized by bleeding into one or both cerebral hemispheres including the basal ganglia and the cerebral cortex. It is often associated with hypertension and craniocerebral trauma. Intracerebral bleeding is a common cause of stroke. {ECO:0000269|PubMed:15277638}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11195 Interacts with NA EC number 5.3.1.5 Uniprot keywords 3D-structure; Carbohydrate metabolism; Cytoplasm; Isomerase; Magnesium; Metal-binding; Reference proteome; Xylose metabolism Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 35820.8 Length 322 Aromaticity 0.1 Instability index 32.29 Isoelectric point 5.32 Charge (pH=7) -11.33 2D Binding mode Binding energy (Kcal/mol) -5.99  Molscript Map  Pymol Map 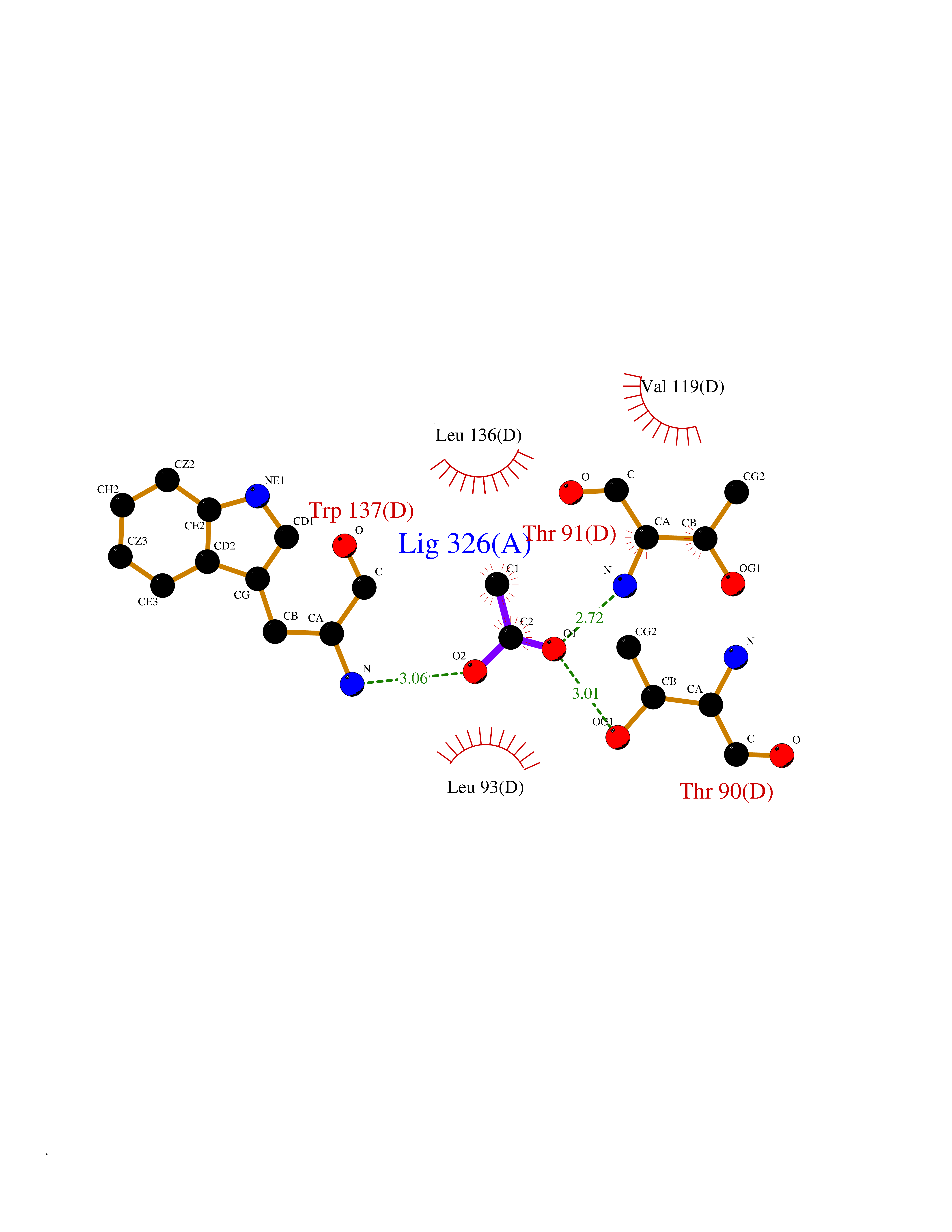 Ligplot Map 3D Binding mode Sequence VQATREDKFSFGLWTVGWQARDAFGDATRTALDPVEAVHKLAEIGAYGITFHDDDLVPFGSDAQTRDGIIAGFKKALDETGLIVPMVTTNLFTHPVFKDGGFTSNDRSVRRYAIRKVLRQMDLGAELGAKTLVLWGGREGAEYDSAKDVSAALDRYREALNLLAQYSEDRGYGLRFAIEPKPNEPRGDILLPTAGHAIAFVQELERPELFGINPETGHEQMSNLNFTQGIAQALWHKKLFHIDLNGQHGPKFDQDLVFGHGDLLNAFSLVDLLENGPDGAPAYDGPRHFDYKPSRTEDYDGVWESAKANIRMYLLLKERAKA Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Protein-tyrosine phosphatase 1B (PTP1B) | 2F71 | 4.39 | |
Target general information Gen name PTPN1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein phosphatase non-receptor type 1; PTP-1B Protein family Protein-tyrosine phosphatase family, Non-receptor class 1 subfamily Biochemical class Phosphoric monoester hydrolase Function Mediates dephosphorylation of EIF2AK3/PERK; inactivating the protein kinase activity of EIF2AK3/PERK. May play an important role in CKII- and p60c-src-induced signal transduction cascades. May regulate the EFNA5-EPHA3 signaling pathway which modulates cell reorganization and cell-cell repulsion. May also regulate the hepatocyte growth factor receptor signaling pathway through dephosphorylation of MET. Tyrosine-protein phosphatase which acts as a regulator of endoplasmic reticulum unfolded protein response. Related diseases Can contribute to cancer cell survival, proliferation, migration, and invasion, and tumor angiogenesis and metastasis. May contribute to cancer pathogenesis by promoting inflammatory responses and recruitment of tumor-infiltrating macrophages.; DISEASE: Abnormally high expression of soluble isoforms (isoform 2, isoform 3 or isoform 4) may be a cause of preeclampsia. Drugs (DrugBank ID) DB08549; DB08783; DB03483; DB08593; DB04800; DB03670; DB03102; DB02072; DB02622; DB07295; DB04088; DB01820; DB02259; DB03311; DB04142; DB02620; DB07298; DB01734; DB08147; DB03557; DB07197; DB06829; DB07130; DB03714; DB07480; DB02014; DB07730; DB08001; DB07134; DB08591; DB07289; DB08397; DB04001; DB02827; DB07719; DB06887; DB04204; DB02420; DB07263; DB02615; DB03982; DB06521; DB05506; DB08003; DB03661; DB04525; DB02784; DB07651; DB02662; DB08371; DB02977; DB06333; DB02436; DB04285; DB02651; DB03154 Interacts with Q13520; P56945; P11274-1; P07384; Q03135; Q14247; P00533; Q9GZR5; P19235; P10912; P62993; P08069; P06213; P06213-1; P05556; P05106; O60674; O43561; P08581; P04629; Q16288; P09619; P57054; P08922; P12931; P40763; P42229; Q9NPL8; Q96HV5; Q8N661; Q9H1D0; P10599; Q61140; Q63767; P15116; Q63768; P62994; P35570; P05622; P10686; P34152; Q8VI36; Q9WUD9; P63166 EC number EC 3.1.3.48 Uniprot keywords 3D-structure; Acetylation; Direct protein sequencing; Endoplasmic reticulum; Hydrolase; Membrane; Oxidation; Phosphoprotein; Protein phosphatase; Proteomics identification; Reference proteome; S-nitrosylation Protein physicochemical properties Chain ID A Molecular weight (Da) 34541 Length 297 Aromaticity 0.1 Instability index 35.91 Isoelectric point 5.91 Charge (pH=7) -5.37 2D Binding mode Binding energy (Kcal/mol) -5.99 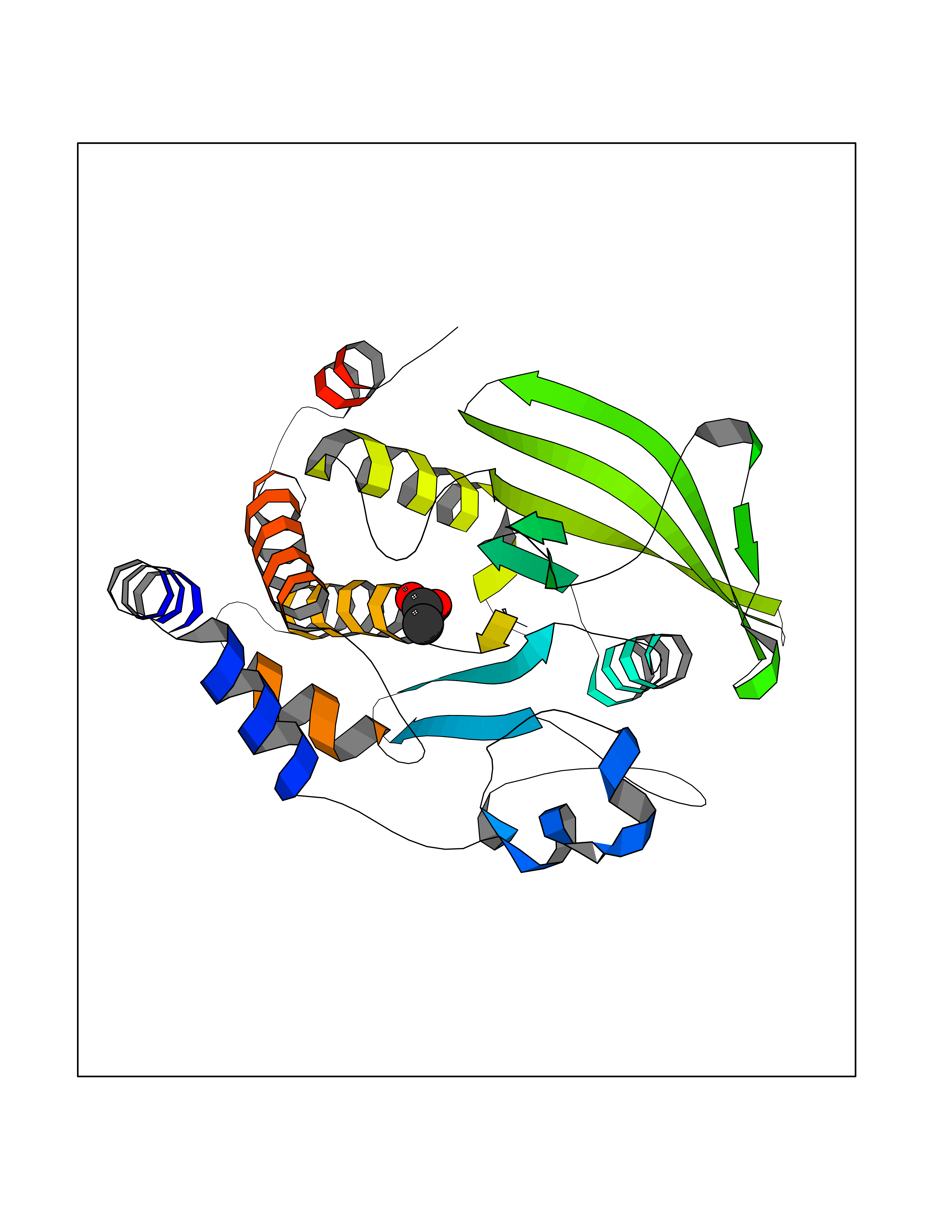 Molscript Map 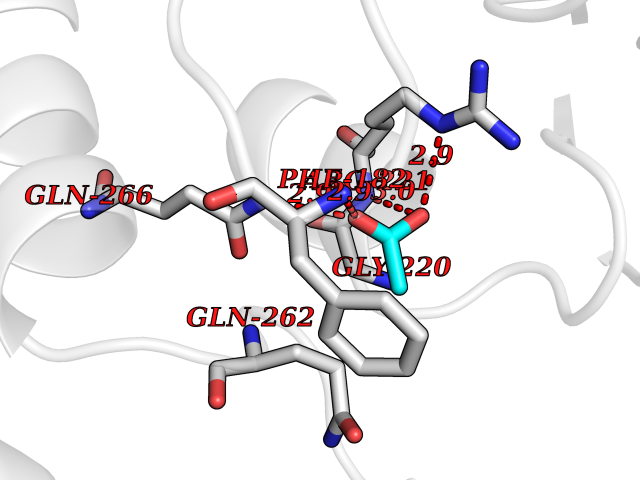 Pymol Map 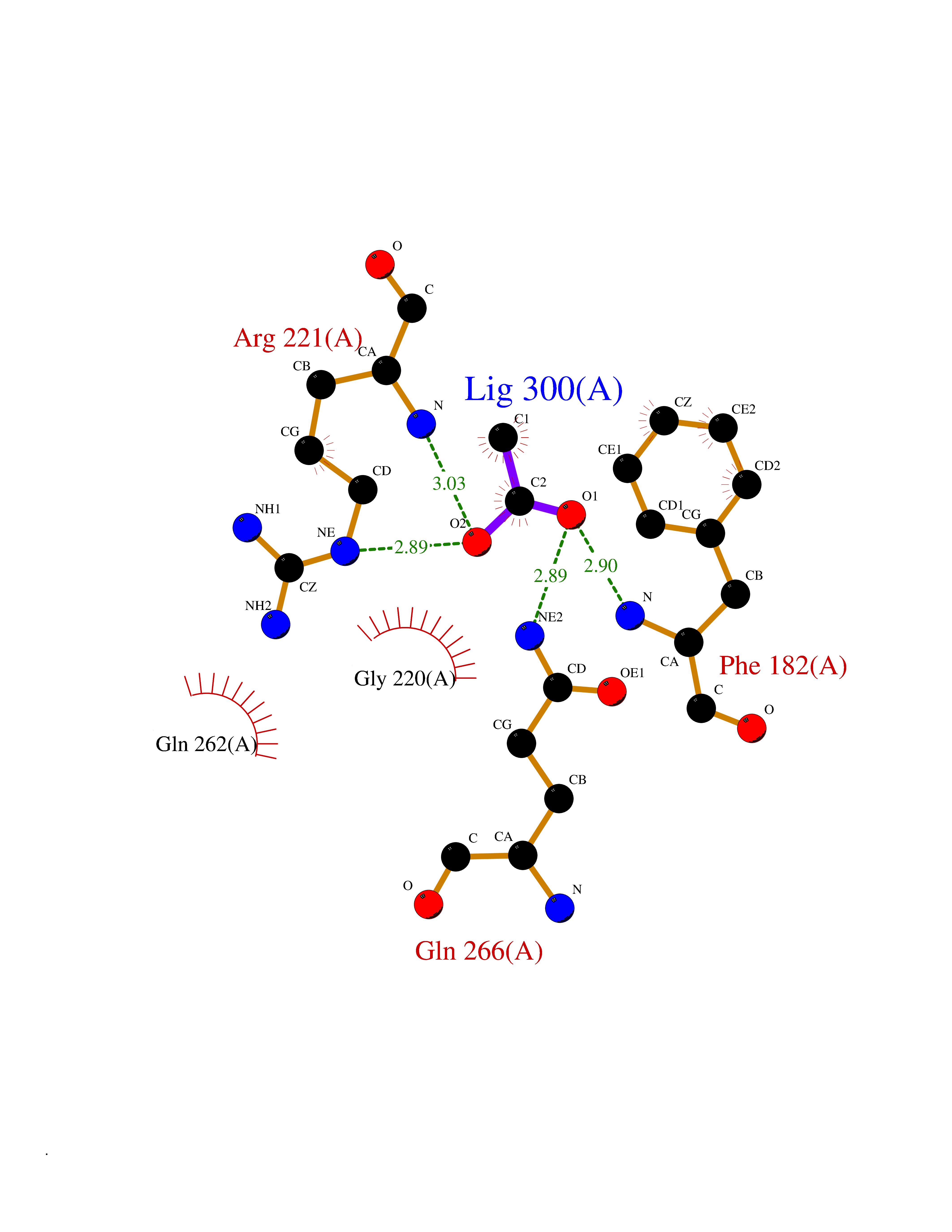 Ligplot Map 3D Binding mode Sequence EMEKEFEQIDKSGSWAAIYQDIRHEASDFPCRVAKLPKNKNRNRYRDVSPFDHSRIKLHQEDNDYINASLIKMEEAQRSYILTQGPLPNTCGHFWEMVWEQKSRGVVMLNRVMEKGSLKCAQYWPQKEEKEMIFEDTNLKLTLISEDIKSYYTVRQLELENLTTQETREILHFHYTTWPDFGVPESPASFLNFLFKVRESGSLSPEHGPVVVHCSAGIGRSGTFCLADTCLLLMDKRKDPSSVDIKKVLLEMRKFRMGLIQTADQLRFSYLAVIEGAKFIMGDSSVQDQWKELSHED Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Beta-glucosidase A | 1E4I | 4.39 | |
Target general information Gen name bglA Organism Paenibacillus polymyxa (Bacillus polymyxa) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 1 family Biochemical class Hydrolase Function Beta-glucosidase activity.Scopolin beta-glucosidase activity. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02658; DB04282; DB04304 Interacts with NA EC number 3.2.1.21 Uniprot keywords 3D-structure; Carbohydrate metabolism; Cellulose degradation; Glycosidase; Hydrolase; Polysaccharide degradation Protein physicochemical properties Chain ID A Molecular weight (Da) 51515.2 Length 447 Aromaticity 0.14 Instability index 38.44 Isoelectric point 5.28 Charge (pH=7) -18.1 2D Binding mode Binding energy (Kcal/mol) -5.99 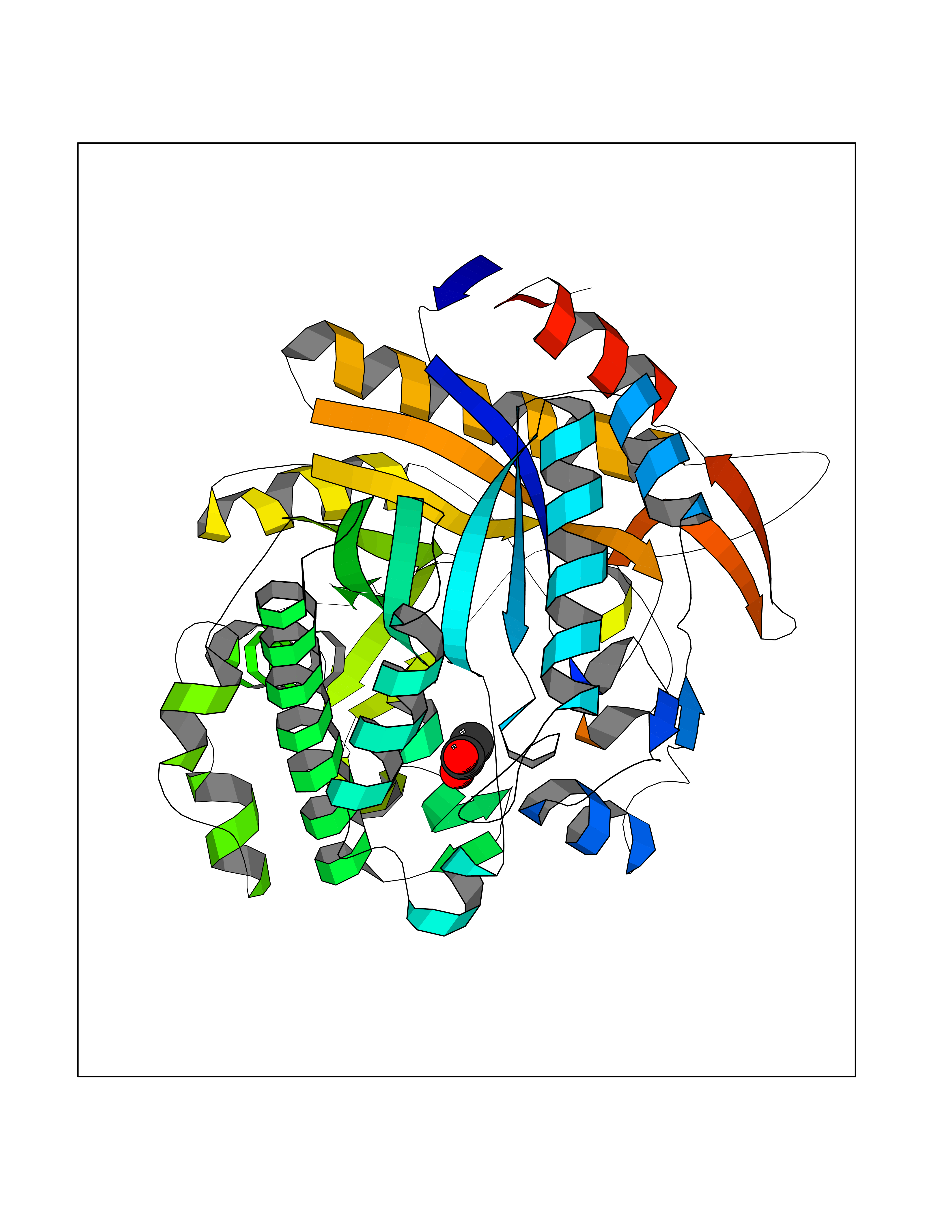 Molscript Map 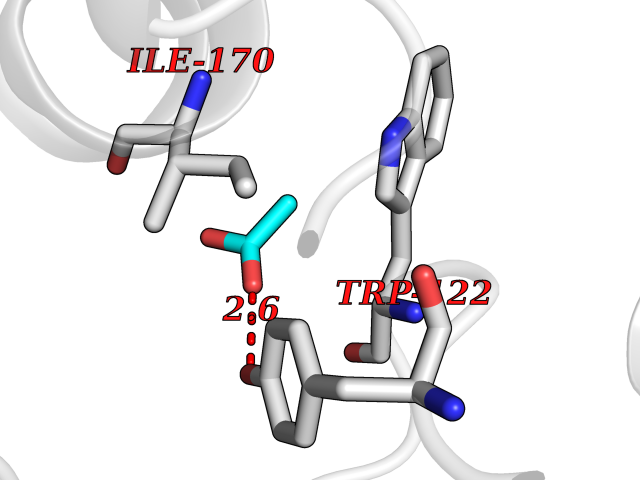 Pymol Map 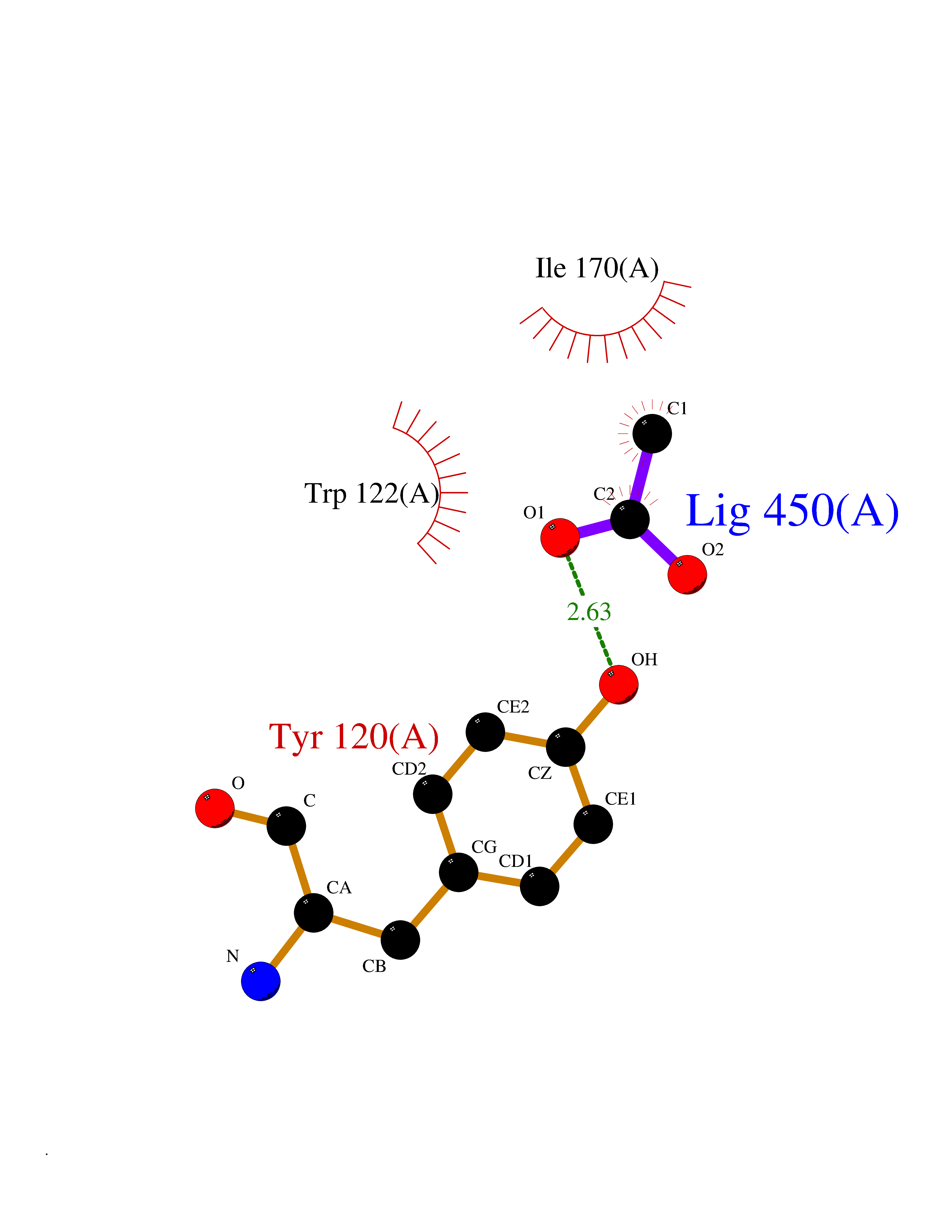 Ligplot Map 3D Binding mode Sequence TIFQFPQDFMWGTATAAYQIEGAYQEDGRGLSIWDTFAHTPGKVFNGDNGNVACDSYHRYEEDIRLMKELGIRTYRFSVSWPRIFPNGDGEVNQKGLDYYHRVVDLLNDNGIEPFCTLYHWDLPQALQDAGGWGNRRTIQAFVQFAETMFREFHGKIQHWLTFNEPWCIAFLSNMLGVHAPGLTNLQTAIDVGHHLLVAHGLSVRRFRELGTSGQIGIAPNVSWAVPYSTSEEDKAACARTISLHSDWFLQPIYQGSYPQFLVDWFAEQGATVPIQDGDMDIIGEPIDMIGINYYSMSVNRFNPEAGFLQSEEINMGLPVTDIGWPVESRGLYEVLHYLQKYGNIDIYITENGACINDEVVNGKVQDDRRISYMQQHLVQVHRTIHDGLHVKGYMAWSLLDNFEWAEGYNMRFGMIHVDFRTQVRTPKQSYYWYRNVVSNNWLETRR Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Vascular endothelial growth factor receptor 3 | 4BSJ | 4.39 | |
Target general information Gen name FLT4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms VEGFR3 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Transferase Function ATP binding.Growth factor binding.Protein homodimerization activity.Protein phosphatase binding.Transmembrane receptor protein tyrosine kinase activity.Vascular endothelial growth factor-activated receptor activity.VEGF-C-activated receptor activity. Related diseases Lymphatic malformation 1 (LMPHM1) [MIM:153100]: A form of primary lymphedema, a disease characterized by swelling of body parts due to developmental anomalies and functional defects of the lymphatic system. Patients with lymphedema may suffer from recurrent local infections. LMPHM1 is an autosomal dominant form with variable expression and severity. Onset is usually at birth or in early childhood but can occur later. Affected individuals manifest lymphedema, predominantly in the lower limbs, and hypoplasia of lymphatic vessels. Additional features are hemangioma and nail dysplasia or papillomatosis. {ECO:0000269|PubMed:10835628, ECO:0000269|PubMed:10856194, ECO:0000269|PubMed:12881528, ECO:0000269|PubMed:15102829, ECO:0000269|PubMed:16924388, ECO:0000269|PubMed:16965327, ECO:0000269|PubMed:17458866, ECO:0000269|PubMed:19289394, ECO:0000269|PubMed:26091405, ECO:0000269|PubMed:9817924}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hemangioma, capillary infantile (HCI) [MIM:602089]: A condition characterized by dull red, firm, dome-shaped hemangiomas, sharply demarcated from surrounding skin, usually presenting at birth or occurring within the first two or three months of life. They result from highly proliferative, localized growth of capillary endothelium and generally undergo regression and involution without scarring. {ECO:0000269|PubMed:11807987}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Plays an important role in tumor lymphangiogenesis, in cancer cell survival, migration, and formation of metastases.; DISEASE: Congenital heart defects, multiple types, 7 (CHTD7) [MIM:618780]: An autosomal dominant disorder with incomplete penetrance characterized by congenital developmental abnormalities involving structures of the heart. Common defects include tetralogy of Fallot, pulmonary stenosis or atresia, absent pulmonary valve, right aortic arch, double aortic arch, and major aortopulmonary collateral arteries. {ECO:0000269|PubMed:28991257, ECO:0000269|PubMed:30232381}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06626; DB05932; DB12010; DB11679; DB06101; DB09078; DB06080; DB09079; DB06589; DB08896; DB15685; DB00398; DB01268; DB05075; DB11800; DB04879 Interacts with P08238; P35968; P49767 EC number 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 24029.2 Length 213 Aromaticity 0.1 Instability index 47.75 Isoelectric point 8.34 Charge (pH=7) 2.35 2D Binding mode Binding energy (Kcal/mol) -5.99 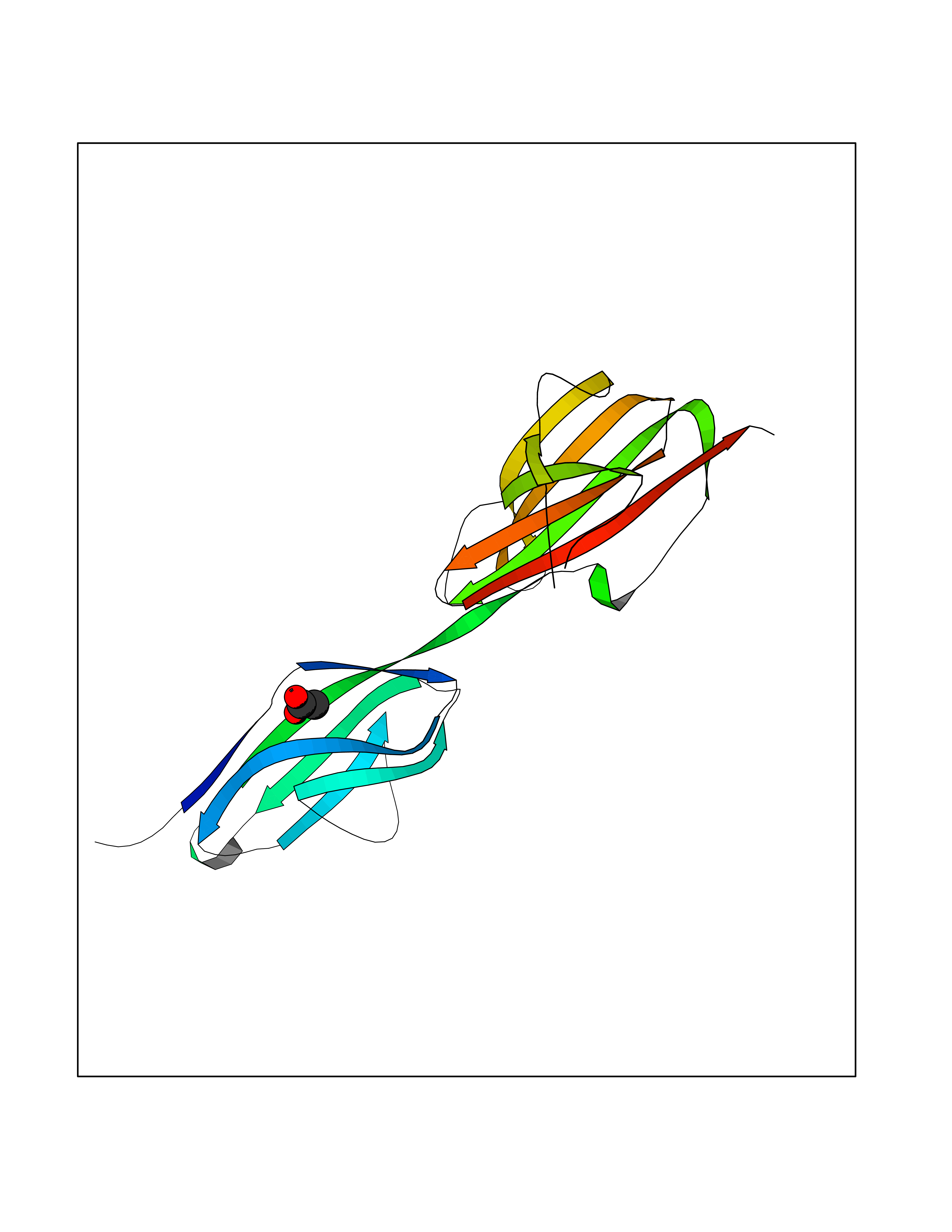 Molscript Map 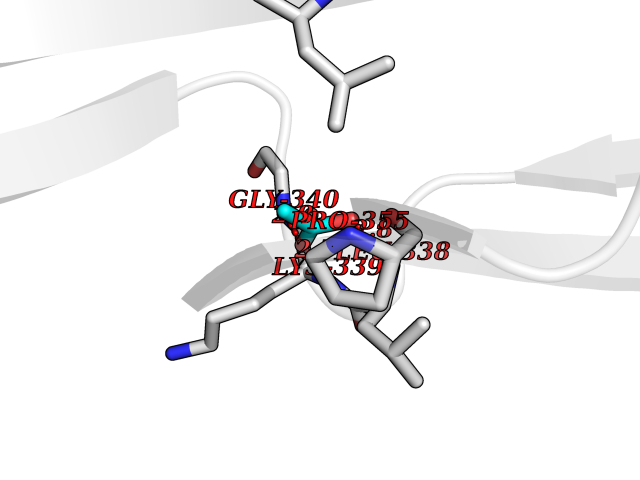 Pymol Map 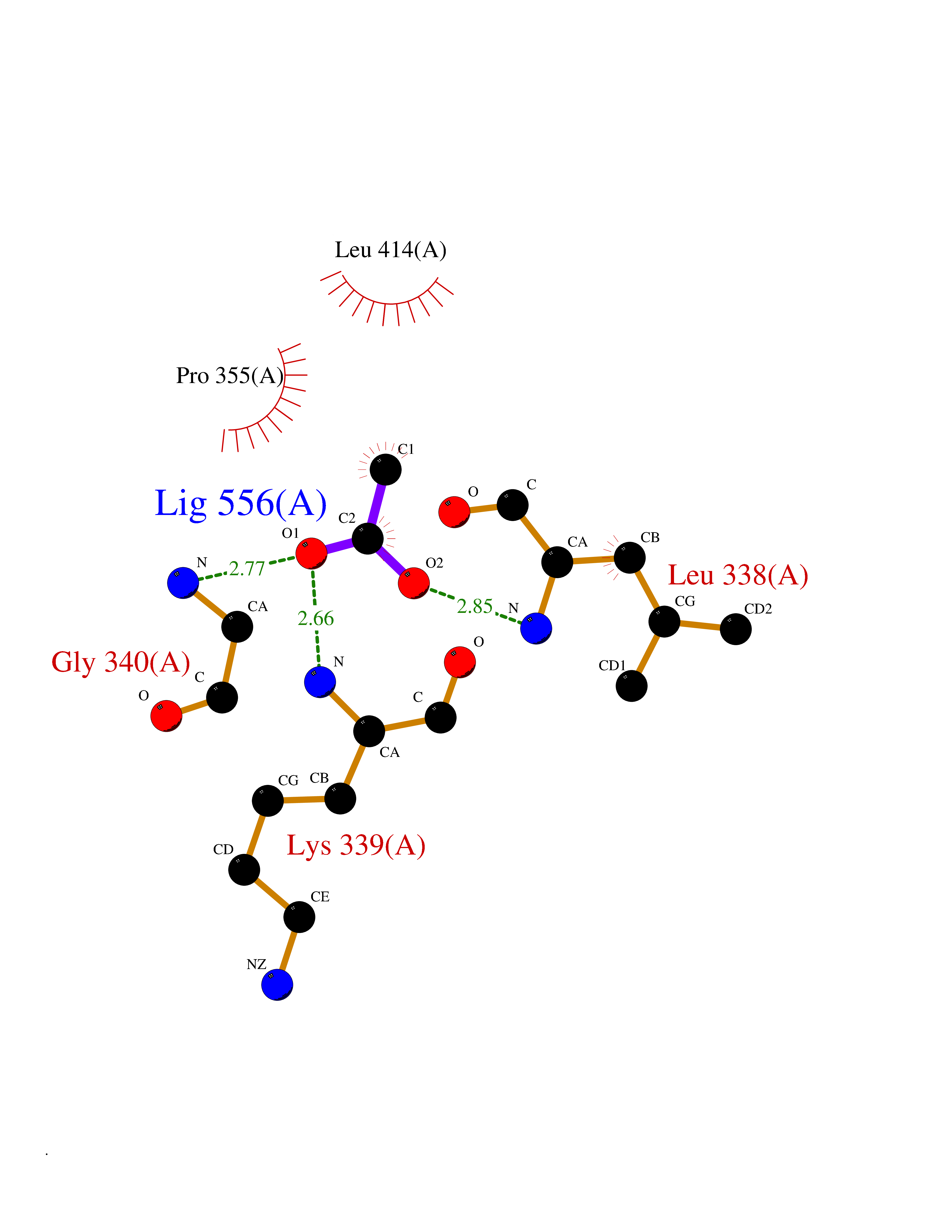 Ligplot Map 3D Binding mode Sequence DHNPFISVEWLKGPILEATAGDELVKLPVKLAAYPPPEFQWYKDGKALSGRHSPHALVLKEVTEASTGTYTLALWNSAAGLRRNISLELVVNVPPQIHEKEASSPSIYSRHSRQALTCTAYGVPLPLSIQWHWRPWTPCKMFPQCRDWRAVTTQDAVNPIESLDTWTEFVEGKNKTVSKLVIQNANVSAMYKCVVSNKVGQDERLIYFYVTTH Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | 2,4-dienoyl-CoA reductase | 1PS9 | 4.39 | |
Target general information Gen name fadH Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3081;ygjL;JW3052 Protein family NADH:flavin oxidoreductase/NADH oxidase family Biochemical class Oxidoreductase Function 2,4-dienoyl-CoA reductase (NADPH) activity.4 iron, 4 sulfur cluster binding.FAD binding.FMN binding.Metal ion binding. Related diseases Smith-Kingsmore syndrome (SKS) [MIM:616638]: An autosomal dominant syndrome characterized by intellectual disability, macrocephaly, seizures, umbilical hernia, and facial dysmorphic features. {ECO:0000269|PubMed:25851998, ECO:0000269|PubMed:26542245, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Focal cortical dysplasia 2 (FCORD2) [MIM:607341]: A form of focal cortical dysplasia, a malformation of cortical development that results in medically refractory epilepsy in the pediatric population and in adults. FCORD2 is a severe form, with onset usually in childhood, characterized by disrupted cortical lamination and specific cytological abnormalities. It is classified in 2 subtypes: type IIA characterized by dysmorphic neurons and lack of balloon cells; type IIB with dysmorphic neurons and balloon cells. {ECO:0000269|PubMed:25799227, ECO:0000269|PubMed:25878179, ECO:0000269|PubMed:26018084, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03698; DB03147; DB03247; DB03461 Interacts with P11349; P19318 EC number 1.3.1.34 Uniprot keywords 3D-structure; 4Fe-4S; Direct protein sequencing; FAD; Flavoprotein; FMN; Iron; Iron-sulfur; Metal-binding; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 40003.1 Length 366 Aromaticity 0.07 Instability index 33.42 Isoelectric point 5.65 Charge (pH=7) -9.48 2D Binding mode Binding energy (Kcal/mol) -5.98 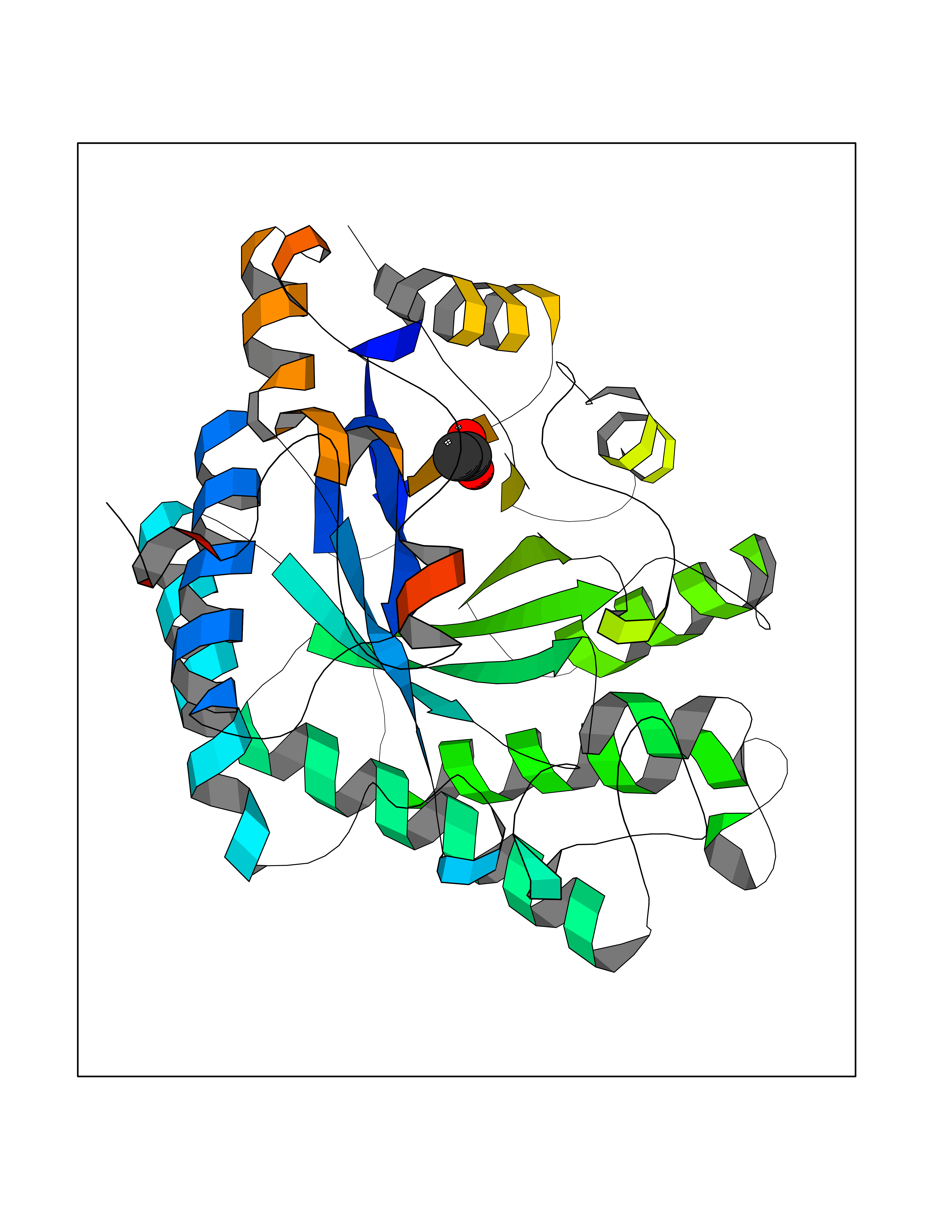 Molscript Map  Pymol Map 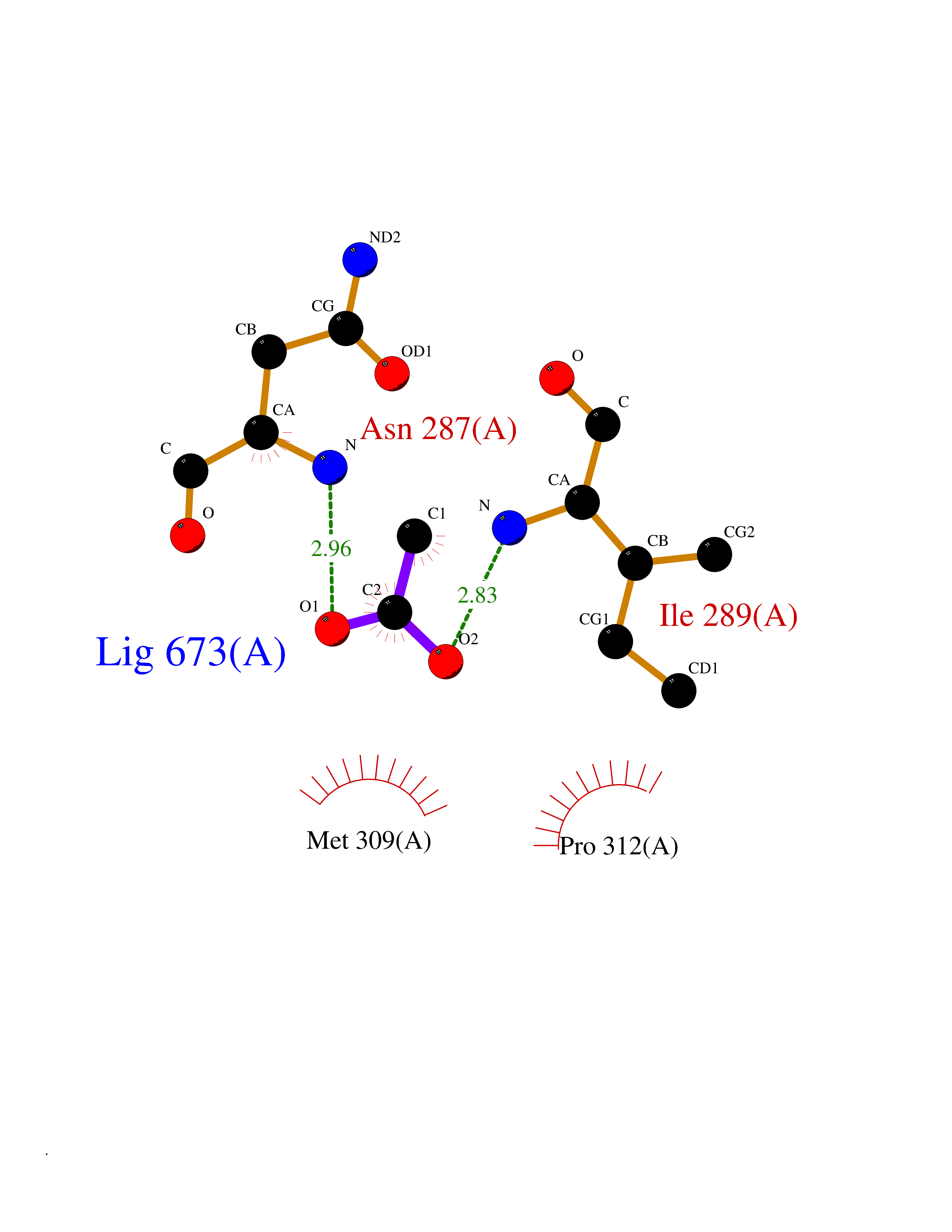 Ligplot Map 3D Binding mode Sequence SYPSLFAPLDLGFTTLKNRVLMGSMHTGLEEYPDGAERLAAFYAERARHGVALIVSGGIAPDLTGVGMEGGAMLNDASQIPHHRTITEAVHQEGGKIALQILHTGRYSYQPHLVAPSALQAPINRFVPHELSHEEILQLIDNFARCAQLAREAGYDGVEVMGSEGYLINEFLTLRTNQRSDQWGGDYRNRMRFAVEVVRAVRERVGNDFIIIYRLSMLDLVEDGGTFAETVELAQAIEAAGATIINTGIGWHEARIPTIATPVPRGAFSWVTRKLKGHVSLPLVTTNRINDPQVADDILSRGDADMVSMARPFLADAELLSKAQSGRADEINTCIGCNQACLDQIFVGKVTSCLVNPRACHETKMP Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 4.39 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -5.99 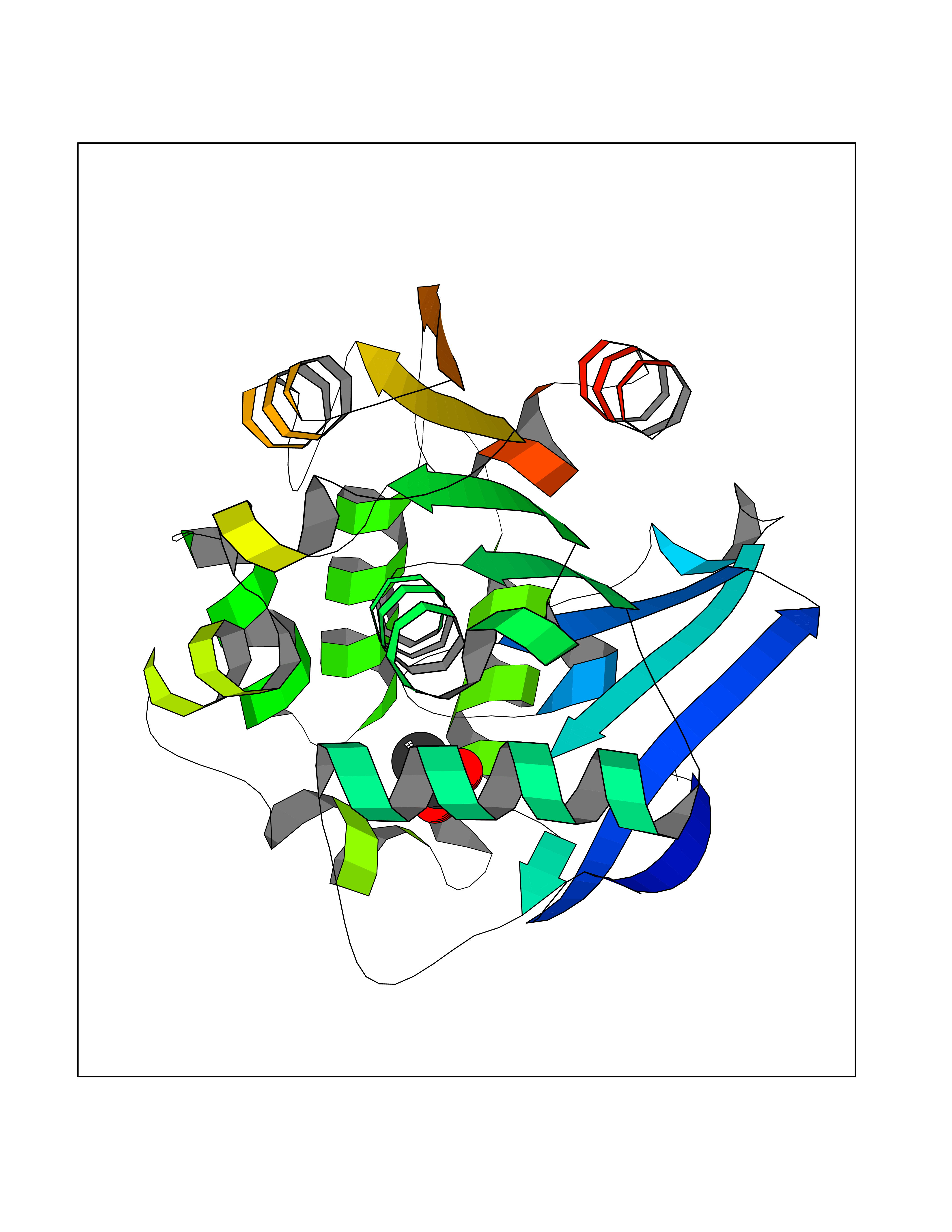 Molscript Map 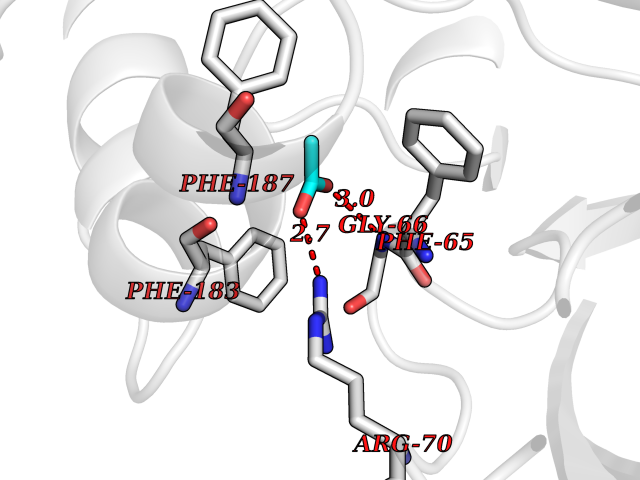 Pymol Map 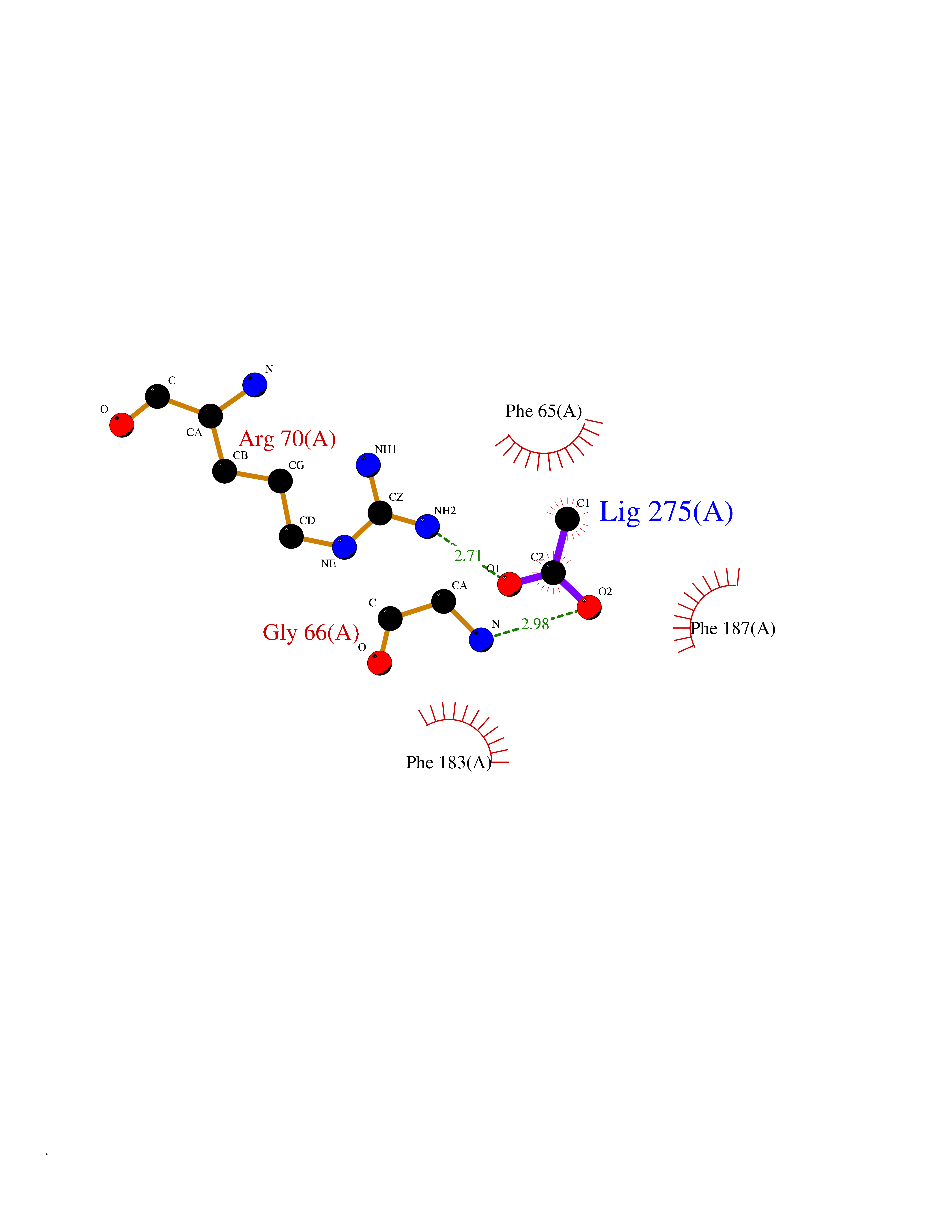 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Protein cereblon (CRBN) | 5FQD | 4.39 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -5.99 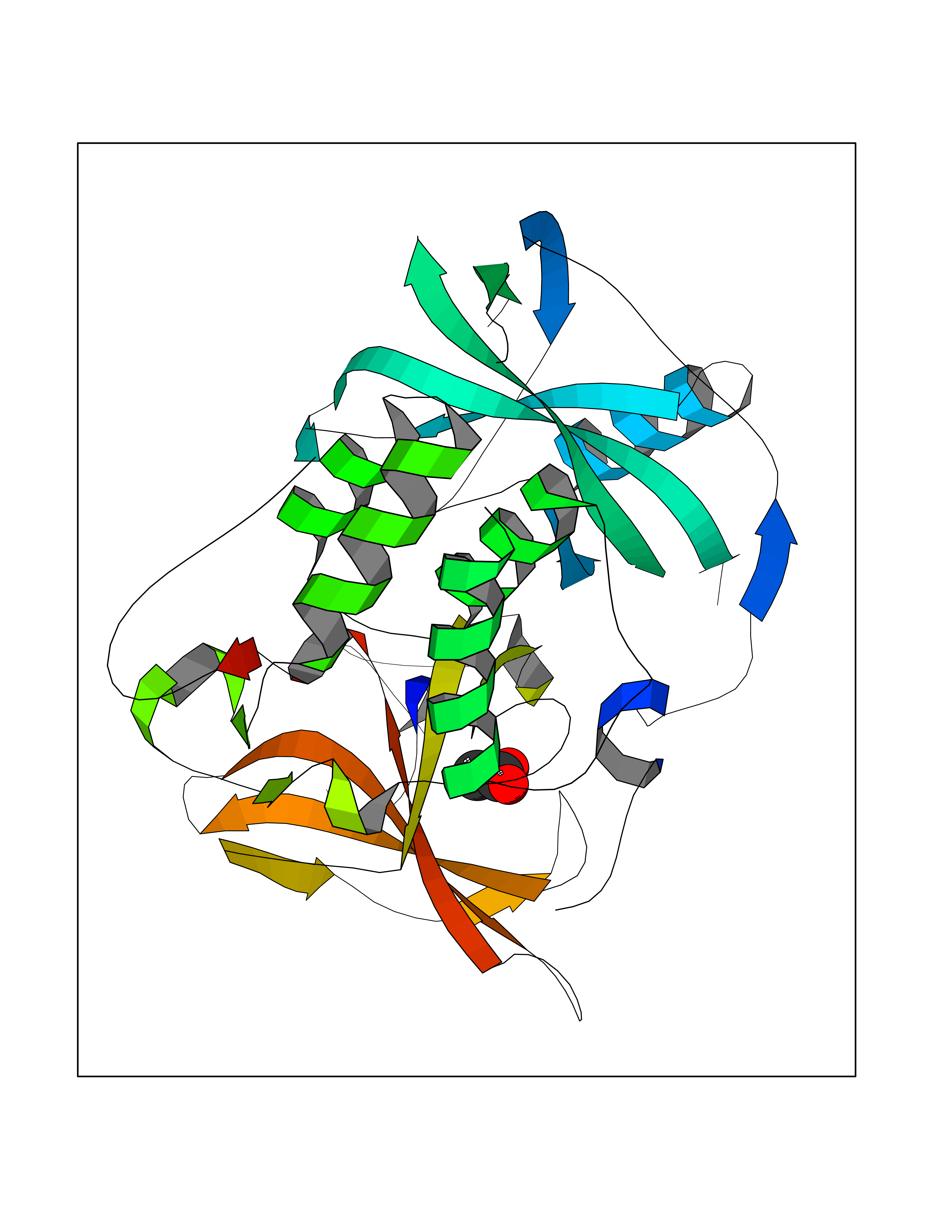 Molscript Map 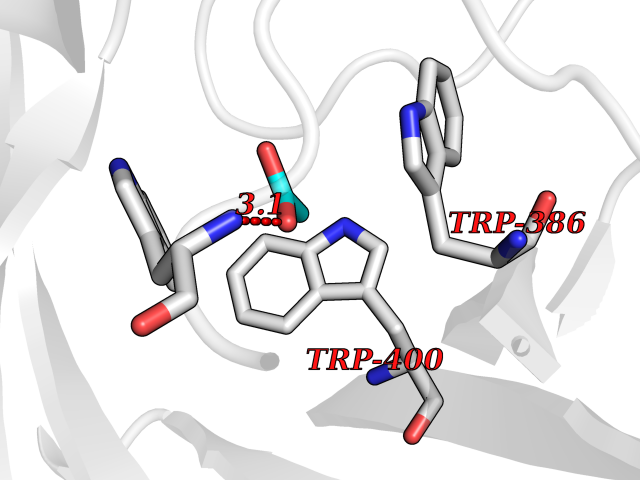 Pymol Map 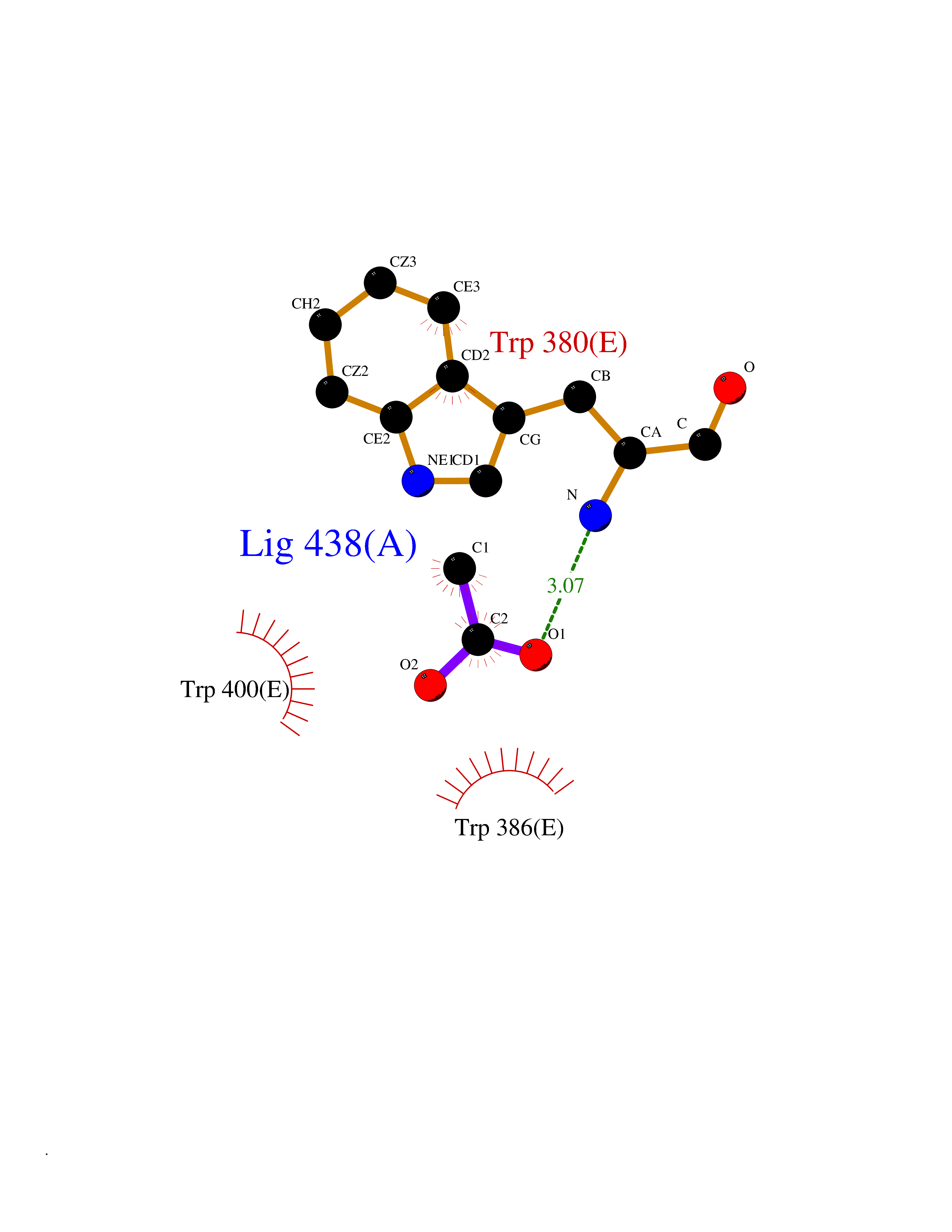 Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 4.39 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -5.98 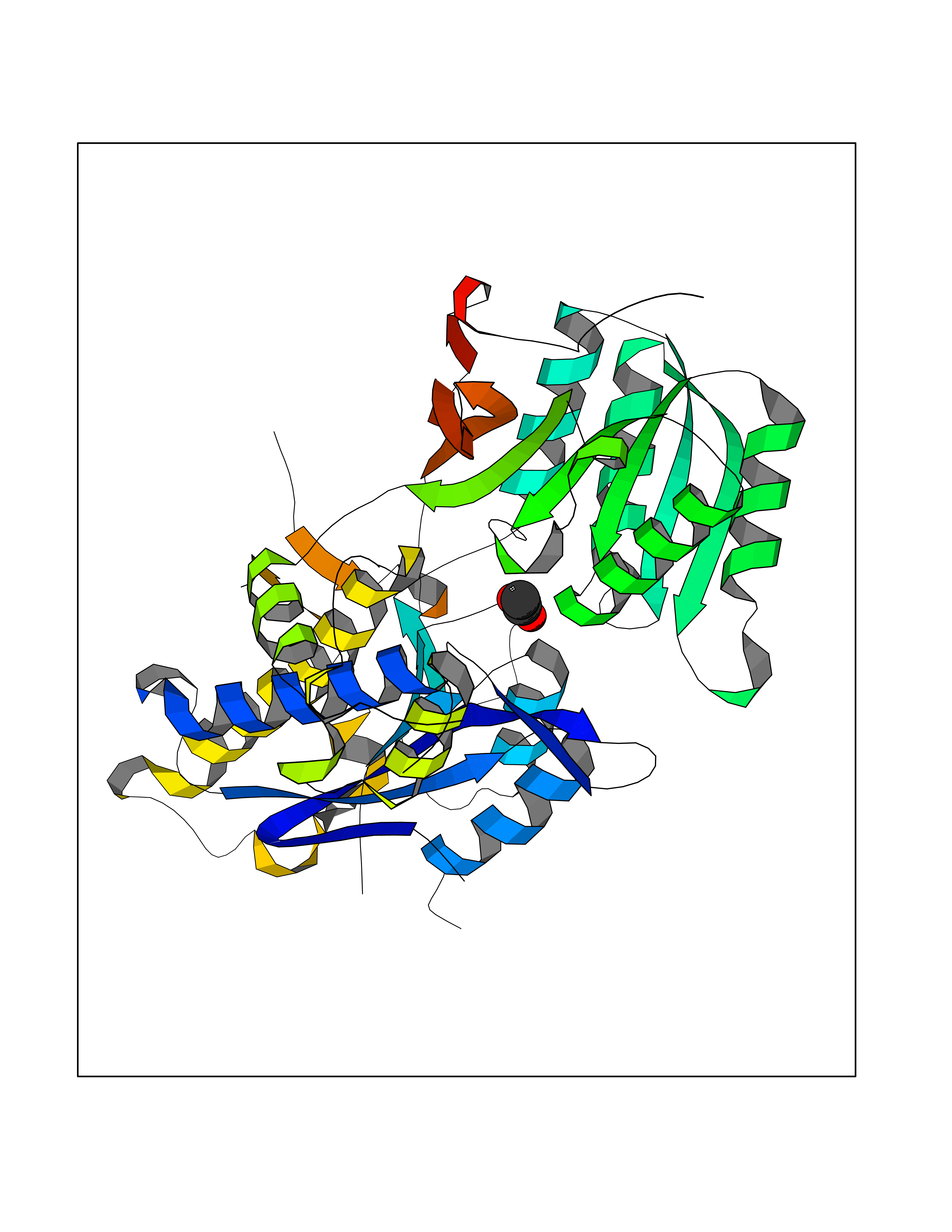 Molscript Map 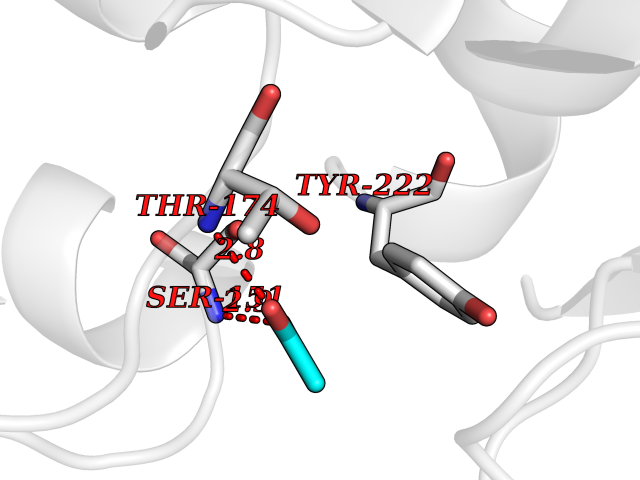 Pymol Map 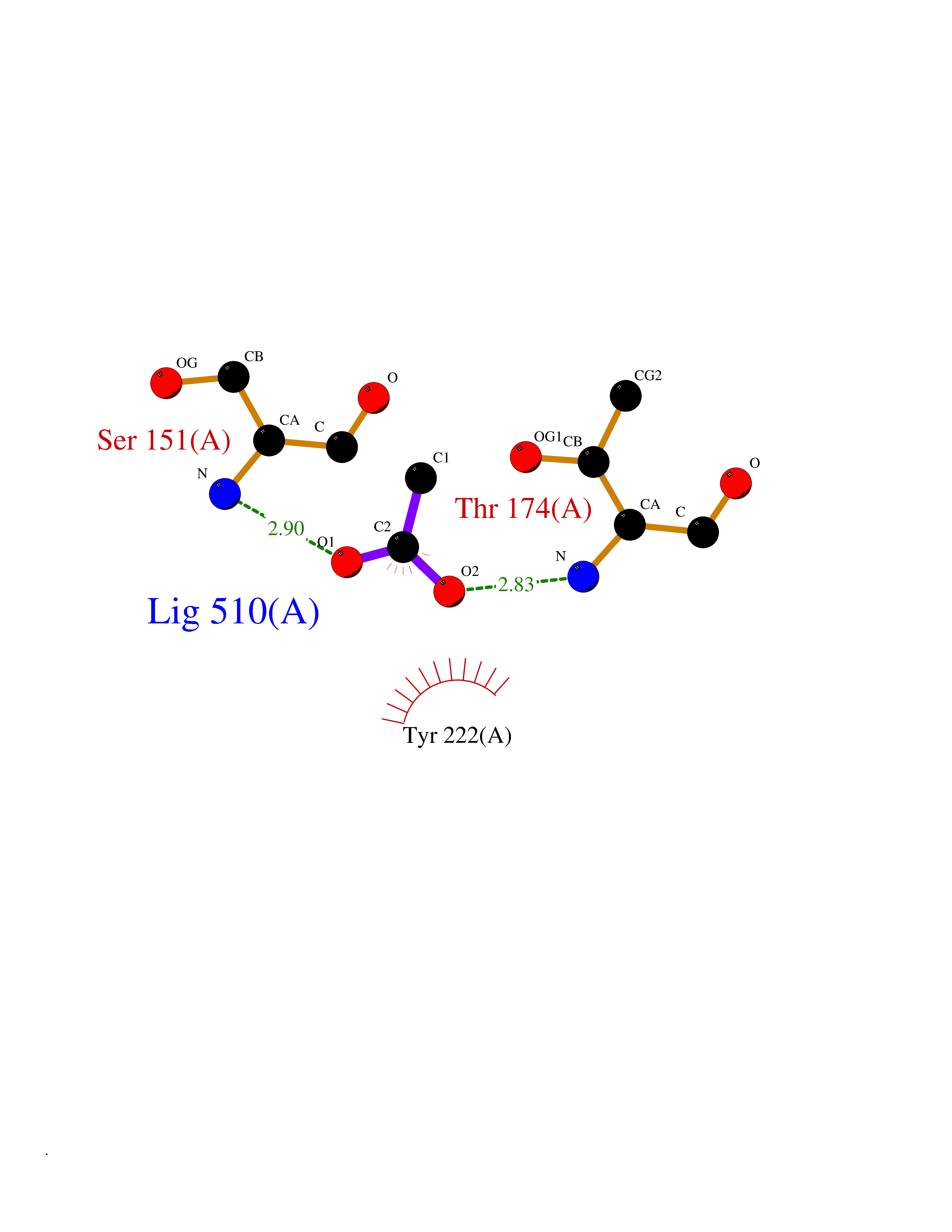 Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Pyridoxal phosphate phosphatase (PDXP) | 2CFT | 4.39 | |
Target general information Gen name PDXP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PLPP; PLP phosphatase; PDXP Protein family HAD-like hydrolase superfamily Biochemical class Phosphoric monoester hydrolase Function Protein serine phosphatase that dephosphorylates 'Ser-3' in cofilin and probably also dephosphorylates phospho-serine residues in DSTN. Regulates cofilin-dependent actin cytoskeleton reorganization. Required for normal progress through mitosis and normal cytokinesis. Does not dephosphorylate phospho-threonines in LIMK1. Does not dephosphorylate peptides containing phospho- tyrosine. Pyridoxal phosphate phosphatase. Has some activity towards pyridoxal 5'-phosphate (PLP), pyridoxine 5'-phosphate (PMP) and pyridoxine 5'-phosphate (PNP), with a highest activity with PLP followed by PNP. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00114; DB00165 Interacts with P29066 EC number EC 3.1.3.3 Uniprot keywords 3D-structure; Alternative initiation; Alternative splicing; Cell membrane; Cell projection; Cytoplasm; Cytoskeleton; Direct protein sequencing; Hydrolase; Magnesium; Membrane; Metal-binding; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 31285.4 Length 292 Aromaticity 0.06 Instability index 41.9 Isoelectric point 6.61 Charge (pH=7) -0.98 2D Binding mode Binding energy (Kcal/mol) -5.98 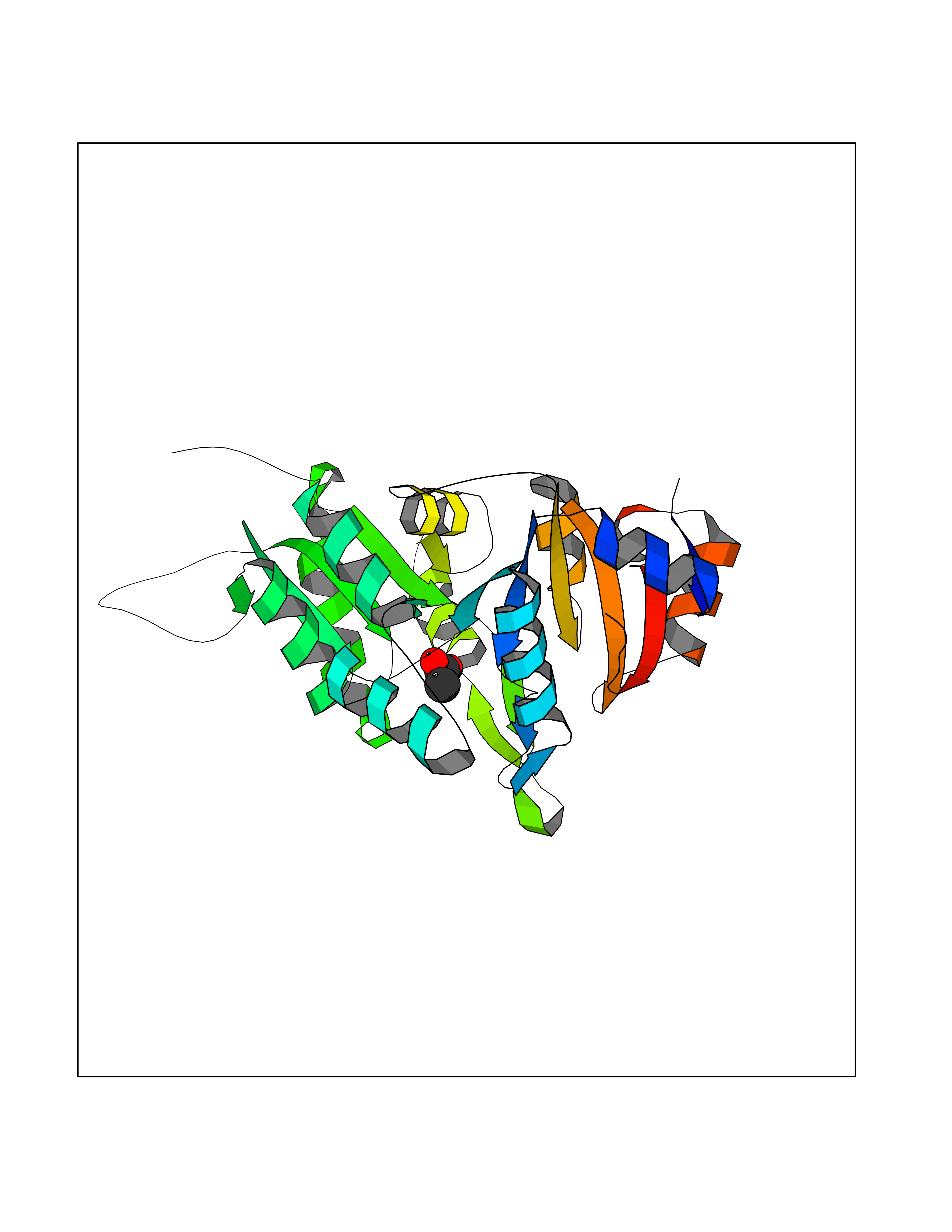 Molscript Map 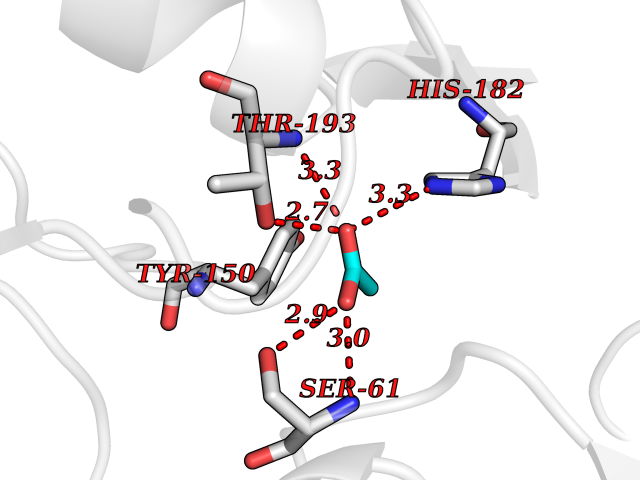 Pymol Map 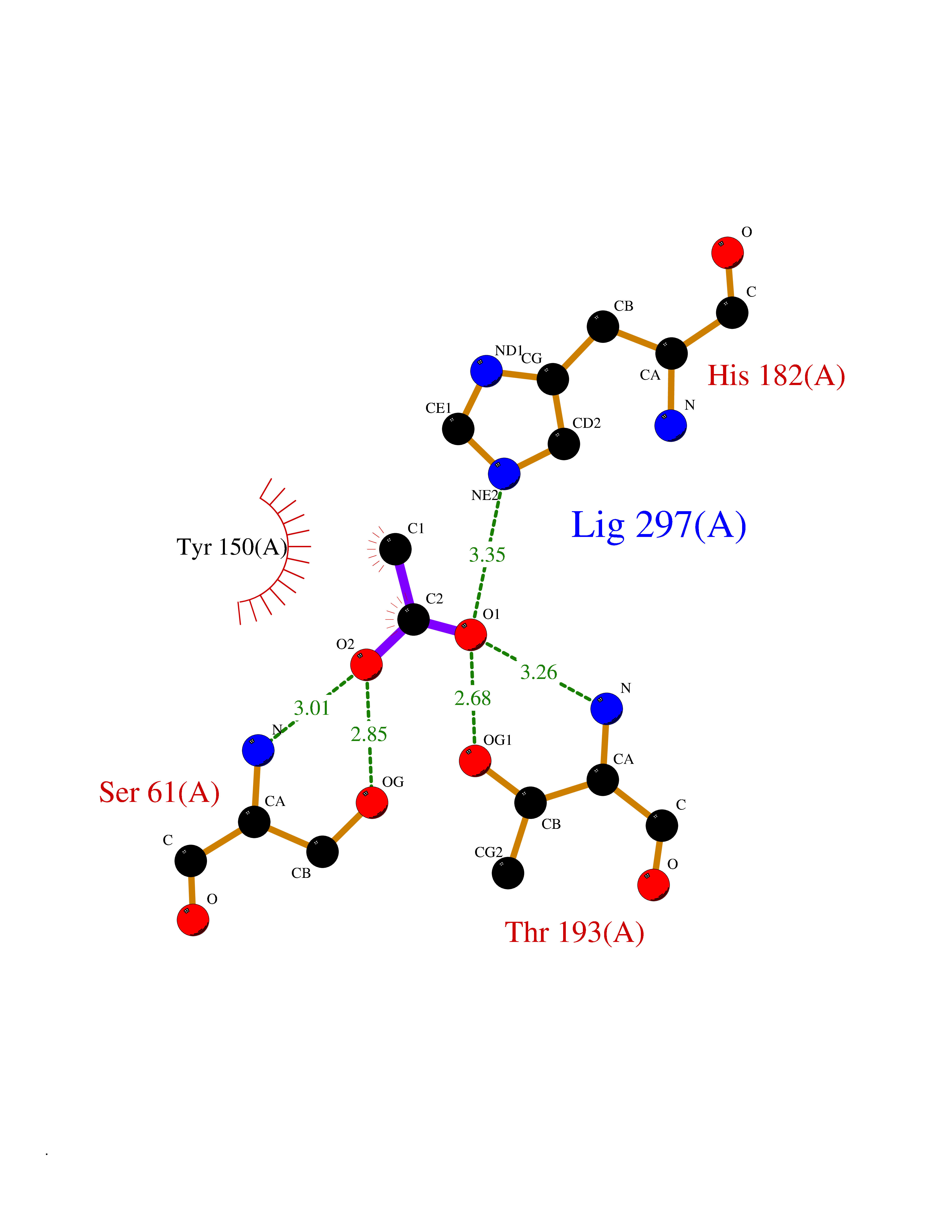 Ligplot Map 3D Binding mode Sequence MARCERLRGAALRDVLGRAQGVLFDCDGVLWNGERAVPGAPELLERLARAGKAALFVSNNSRRARPELALRFARLGFGGLRAEQLFSSALCAARLLRQRLPGPPDGAVFVLGGEGLRAELRAAGLRLAGDPSAGDGAAPRVRAVLVGYDEHFSFAKLREACAHLRDPECLLVATDRDPWHPLSDGSRTPGTGSLAAAVETASGRQALVVGKPSPYMFECITENFSIDPARTLMVGDRLETDILFGHRCGMTTVLTLTGVSRLEEAQAYLAAGQHDLVPHYYVESIADLTEGL Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Chymase (CYM) | 4K69 | 4.39 | |
Target general information Gen name CMA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mast cell protease I; CYH; Alpha-chymase Protein family Peptidase S1 family, Granzyme subfamily Biochemical class Peptidase Function Major secreted protease of mast cells with suspected roles in vasoactive peptide generation, extracellular matrix degradation, and regulation of gland secretion. Related diseases Weaver syndrome (WVS) [MIM:277590]: A syndrome of accelerated growth and osseous maturation, unusual craniofacial appearance, hoarse and low-pitched cry, and hypertonia with camptodactyly. Distinguishing features of Weaver syndrome include broad forehead and face, ocular hypertelorism, prominent wide philtrum, micrognathia, deep horizontal chin groove, and deep-set nails. In addition, carpal bone development is advanced over the rest of the hand. {ECO:0000269|PubMed:22177091, ECO:0000269|PubMed:22190405, ECO:0000269|PubMed:23239504, ECO:0000269|PubMed:26694085, ECO:0000269|PubMed:28229514}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03814; DB04016; DB07680; DB03297 Interacts with NA EC number EC 3.4.21.39 Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 23715.1 Length 215 Aromaticity 0.07 Instability index 37.79 Isoelectric point 9.51 Charge (pH=7) 11.49 2D Binding mode Binding energy (Kcal/mol) -5.99 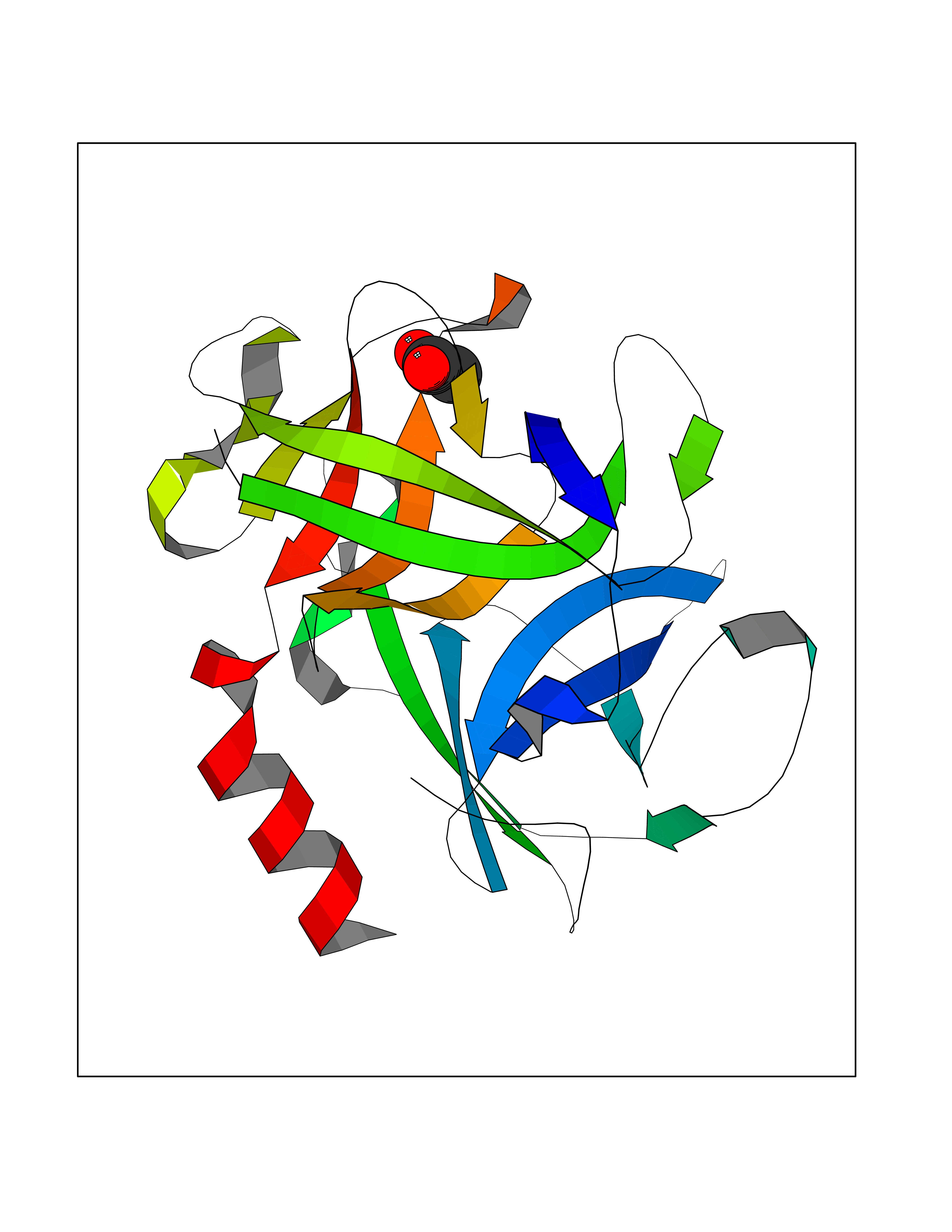 Molscript Map 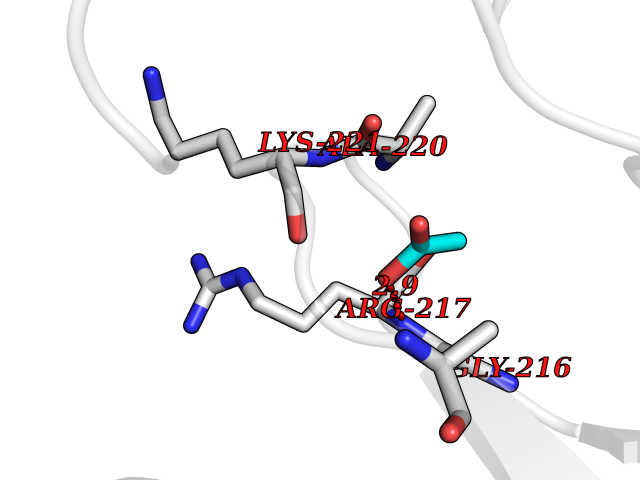 Pymol Map 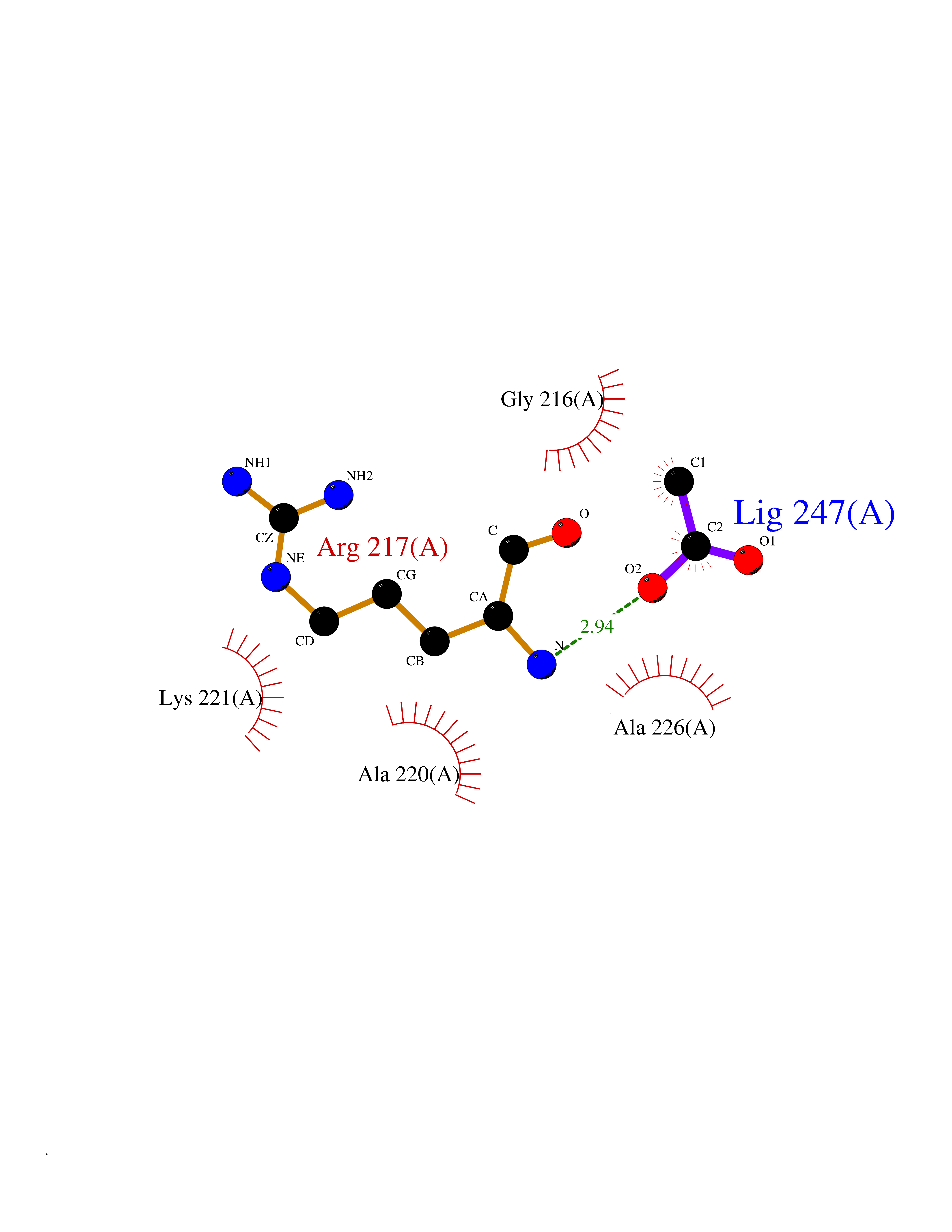 Ligplot Map 3D Binding mode Sequence IIGGTECKPHSRPYMAYLEIVTSNGPSKFCGGFLIRRNFVLTAAHCAGRSITVTLGAHNITEEEDTWQKLEVIKQFRHPKYNTSTLHHDIMLLKLKEKASLTLAVGTLGRMCRVAGWGRTGVLKPGSDTLQEVKLRLMDPQACSHFRDFDHNLQLCVGNPRKTKSAFKGDSGGPLLCAGAAQGIVSYGRSDAKPPAVFTRISHYQPWINQILQAN Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Myosin-7 (MYH7) | 4DB1 | 4.39 | |
Target general information Gen name MYH7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Myosin heavy chain, cardiac muscle beta isoform; Myosin heavy chain slow isoform; Myosin heavy chain 7; MyHCslow; MyHCbeta; MYH7 Protein family TRAFAC class myosin-kinesin ATPase superfamily, Myosin family Biochemical class TRAFAC class myosin-kinesin ATPase Function Muscle contraction. Related diseases Cardiomyopathy, familial hypertrophic, 1 (CMH1) [MIM:192600]: A hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death. {ECO:0000269|PubMed:10065021, ECO:0000269|PubMed:10329202, ECO:0000269|PubMed:10521296, ECO:0000269|PubMed:10563488, ECO:0000269|PubMed:10679957, ECO:0000269|PubMed:10862102, ECO:0000269|PubMed:11113006, ECO:0000269|PubMed:11133230, ECO:0000269|PubMed:11214007, ECO:0000269|PubMed:11424919, ECO:0000269|PubMed:11733062, ECO:0000269|PubMed:11861413, ECO:0000269|PubMed:11968089, ECO:0000269|PubMed:12081993, ECO:0000269|PubMed:12566107, ECO:0000269|PubMed:12590187, ECO:0000269|PubMed:12707239, ECO:0000269|PubMed:12818575, ECO:0000269|PubMed:12820698, ECO:0000269|PubMed:12951062, ECO:0000269|PubMed:12974739, ECO:0000269|PubMed:12975413, ECO:0000269|PubMed:1417858, ECO:0000269|PubMed:15358028, ECO:0000269|PubMed:15483641, ECO:0000269|PubMed:1552912, ECO:0000269|PubMed:15563892, ECO:0000269|PubMed:15856146, ECO:0000269|PubMed:15858117, ECO:0000269|PubMed:16199542, ECO:0000269|PubMed:16267253, ECO:0000269|PubMed:1638703, ECO:0000269|PubMed:16650083, ECO:0000269|PubMed:16938236, ECO:0000269|PubMed:17095604, ECO:0000269|PubMed:17372140, ECO:0000269|PubMed:18175163, ECO:0000269|PubMed:18403758, ECO:0000269|PubMed:1975517, ECO:0000269|PubMed:25182012, ECO:0000269|PubMed:7581410, ECO:0000269|PubMed:7731997, ECO:0000269|PubMed:7848441, ECO:0000269|PubMed:7874131, ECO:0000269|PubMed:7909436, ECO:0000269|PubMed:8250038, ECO:0000269|PubMed:8254035, ECO:0000269|PubMed:8268932, ECO:0000269|PubMed:8282798, ECO:0000269|PubMed:8343162, ECO:0000269|PubMed:8435239, ECO:0000269|PubMed:8483915, ECO:0000269|PubMed:8533830, ECO:0000269|PubMed:8655135, ECO:0000269|PubMed:8899546, ECO:0000269|PubMed:9544842, ECO:0000269|PubMed:9822100, ECO:0000269|PubMed:9829907}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 7A, myosin storage, autosomal dominant (CMYO7A) [MIM:608358]: A skeletal muscle disorder characterized by prominent axial and proximal weakening, spinal stiffness, severe scoliosis, with or without respiratory and cardiac involvement. The age at symptom onset can range from early childhood to late adulthood, and disease severity ranges from asymptomatic to severe muscular weakness and respiratory insufficiency. Histopathological examination shows variable findings including subsarcolemmal hyaline bodies in type 1 fibers. {ECO:0000269|PubMed:14520662, ECO:0000269|PubMed:15136674, ECO:0000269|PubMed:16684601, ECO:0000269|PubMed:17336526}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cardiomyopathy, dilated, 1S (CMD1S) [MIM:613426]: A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. {ECO:0000269|PubMed:11106718, ECO:0000269|PubMed:12379228, ECO:0000269|PubMed:15769782, ECO:0000269|PubMed:18506004, ECO:0000269|PubMed:21127202, ECO:0000269|PubMed:21846512}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, distal, 1 (MPD1) [MIM:160500]: A muscular disorder characterized by early-onset selective weakness of the great toe and ankle dorsiflexors, followed by weakness of the finger extensors. Mild proximal weakness occasionally develops years later after the onset of the disease. {ECO:0000269|PubMed:15322983, ECO:0000269|PubMed:17548557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 7B, myosin storage, autosomal recessive (CMYO7B) [MIM:255160]: A skeletal muscle disorder characterized by the onset of scapuloperoneal muscle weakness in early childhood or young adulthood. Affected individuals have difficulty walking, steppage gait, and scapular winging due to shoulder girdle involvement. The severity and progression of the disorder is highly variable. Most patients develop respiratory insufficiency and restrictive lung disease. Some develop hypertrophic cardiomyopathy. Histopathological examination shows variable findings including subsarcolemmal hyaline bodies in type 1 fibers. {ECO:0000269|PubMed:25666907}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Left ventricular non-compaction 5 (LVNC5) [MIM:613426]: A form of left ventricular non-compaction, a cardiomyopathy due to myocardial morphogenesis arrest and characterized by a hypertrophic left ventricle, a severely thickened 2-layered myocardium, numerous prominent trabeculations, deep intertrabecular recesses, and poor systolic function. Clinical manifestations are variable. Some affected individuals experience no symptoms at all, others develop heart failure. In some cases, left ventricular non-compaction is associated with other congenital heart anomalies. LVNC5 is an autosomal dominant condition. {ECO:0000269|PubMed:18506004}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. Drugs (DrugBank ID) DB08378; DB14921 Interacts with Q5SYC1; Q9BQD3; Q96DD0 EC number NA Uniprot keywords 3D-structure; Actin-binding; ATP-binding; Calmodulin-binding; Cardiomyopathy; Coiled coil; Cytoplasm; Disease variant; Methylation; Motor protein; Muscle protein; Myosin; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Thick filament Protein physicochemical properties Chain ID A Molecular weight (Da) 81702.7 Length 716 Aromaticity 0.11 Instability index 36.28 Isoelectric point 6.43 Charge (pH=7) -3.73 2D Binding mode Binding energy (Kcal/mol) -5.99 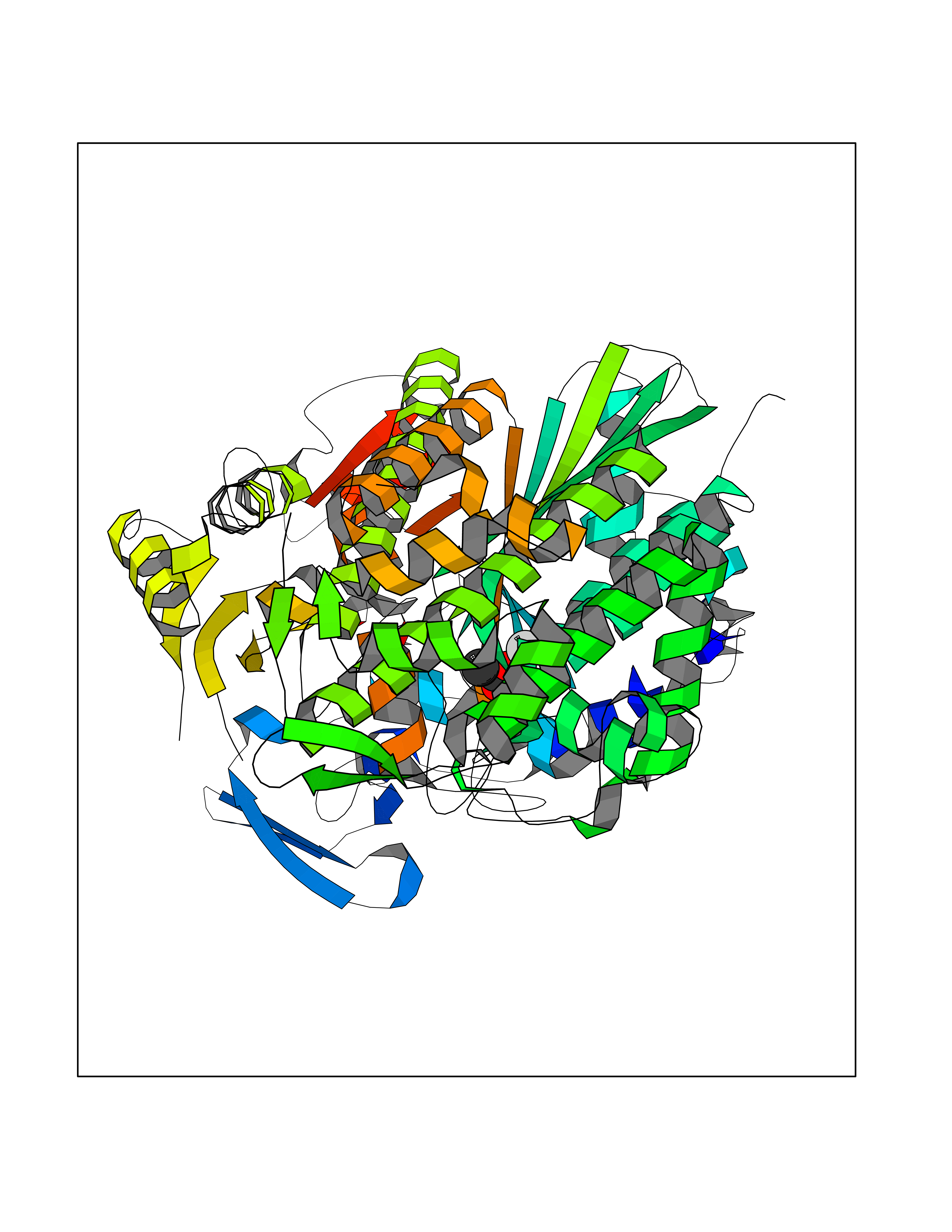 Molscript Map 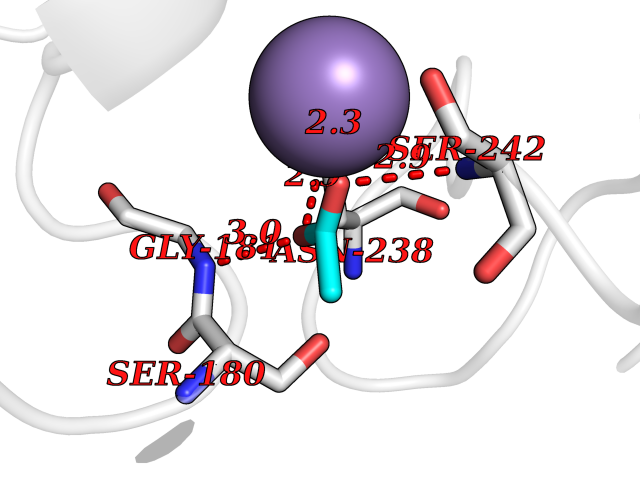 Pymol Map 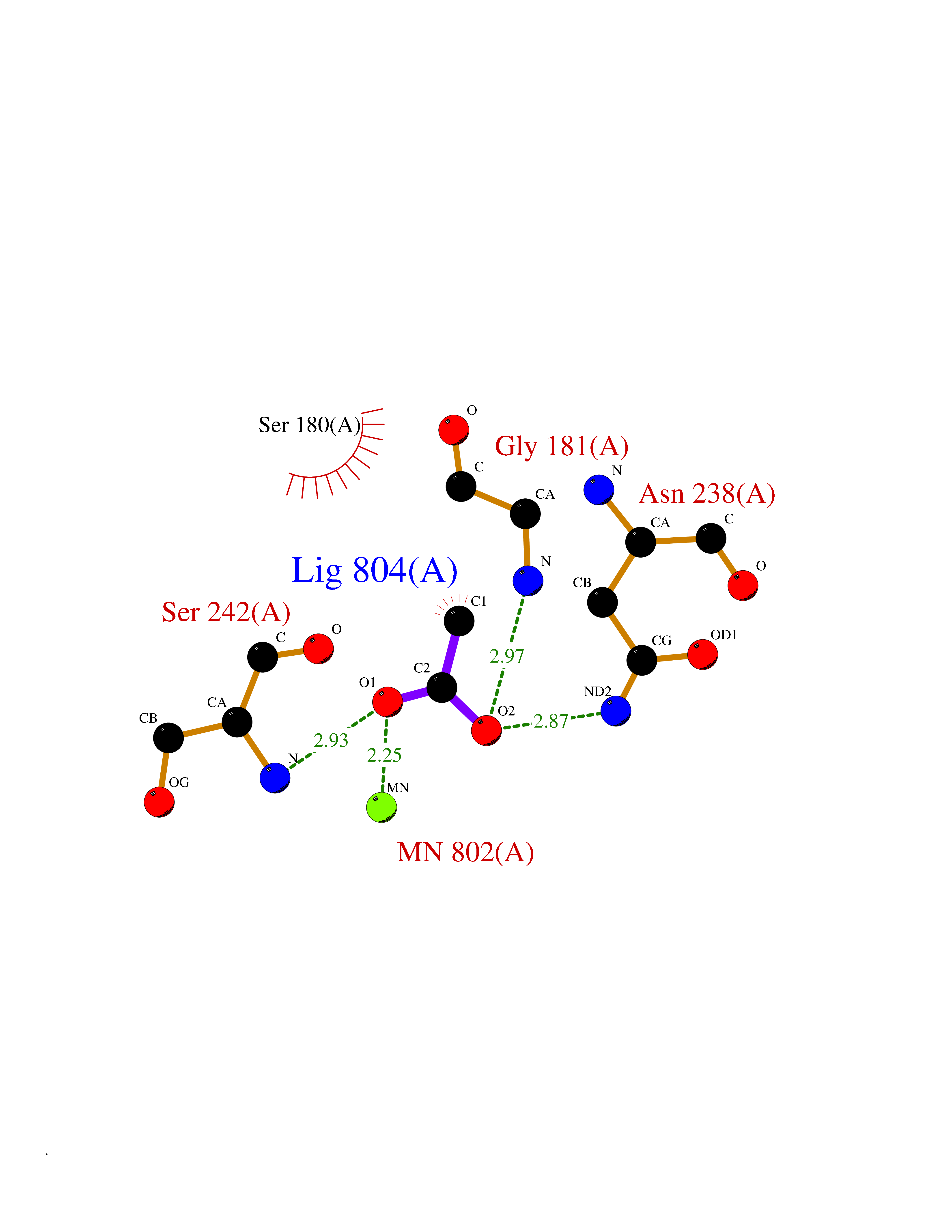 Ligplot Map 3D Binding mode Sequence GGDSEMAVFGAAAPYLRKSEKERLEAQTRPFDLKKDVFVPDDKQEFVKAKIVSREGGKVTAETEYGKTVTVKEDQVMQQNPPKFDKIEDMAMLTFLHEPAVLYNLKDRYGSWMIYTYSGLFCVTVNPYKWLPVYTPEVVAAYRGKKRSEAPPHIFSISDNAYQYMLTDRENQSILITGESGAGKTVNTKRVIQYFAVIAAIGDRGKGTLEDQIIQANPALEAFGNAKTVRNDNSSRFGKFIRIHFGATGKLASADIETYLLEKSRVIFQLKAERDYHIFYQILSNKKPELLDMLLITNNPYDYAFISQGETTVASIDDAEELMATDNAFDVLGFTSEEKNSMYKLTGAIMHFGNMKFKLKQREEQAEPDGTEEADKSAYLMGLNSADLLKGLCHPRYVTKGQNVQQVIYATGALAKAVYERMFNWMVTRINATLETKQPRQYFIGVLDIAGFEIFDFNSFEQLCINFTNEKLQQFFNHHMFVLEQEEYKKEGIEWTFIDFGMDLQACIDLIEKPMGIMSILEEECMFPKATDMTFKAKLFDNHLGKSANFQKPRNPEAHFSLIHYAGIVDYNIIGWLQKNKDPLNETVVGLYQKSSLKLLSTLFANYAFQTVSALHRENLNKLMTNLRSTHPHFVRCIIPNETKSPGVMDNPLVMHQLRCNGVLEGIRICRKGFPNRILYAEKLLSSLDIDHNQYKFGHTKVFFKAGLLGLLEEMR Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Oxidative stress responsive 1 (OXSR1) | 2VWI | 4.39 | |
Target general information Gen name OXSR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein kinase OSR1; Oxidative stress-responsive 1 protein; KIAA1101 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, STE20 subfamily Biochemical class NA Function Phosphorylates RELL1, RELL2 and RELT. Phosphorylates PAK1. Phosphorylates PLSCR1 in the presence of RELT. Related diseases Hyperparathyroidism, transient neonatal (HRPTTN) [MIM:618188]: An autosomal recessive disease characterized by impaired transplacental maternal-fetal transport of calcium, high serum PTH levels and signs of metabolic bone disease in the neonatal period. Skeletal anomalies include generalized osteopenia, narrow chest, short ribs with multiple healing fractures, and bowing or fractures of long bones. Affected individuals experience postnatal respiratory and feeding difficulties. The condition improves within a short time after birth once calcium is provided orally. {ECO:0000269|PubMed:29861107, ECO:0000269|PubMed:30820485}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with Q9Y376; O95747; Q8NC24; P55011; Q9H4A3; Q9Y3S1; Q96J92 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cytoplasm; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 26440.4 Length 236 Aromaticity 0.08 Instability index 44.04 Isoelectric point 6.48 Charge (pH=7) -1.55 2D Binding mode Binding energy (Kcal/mol) -5.98 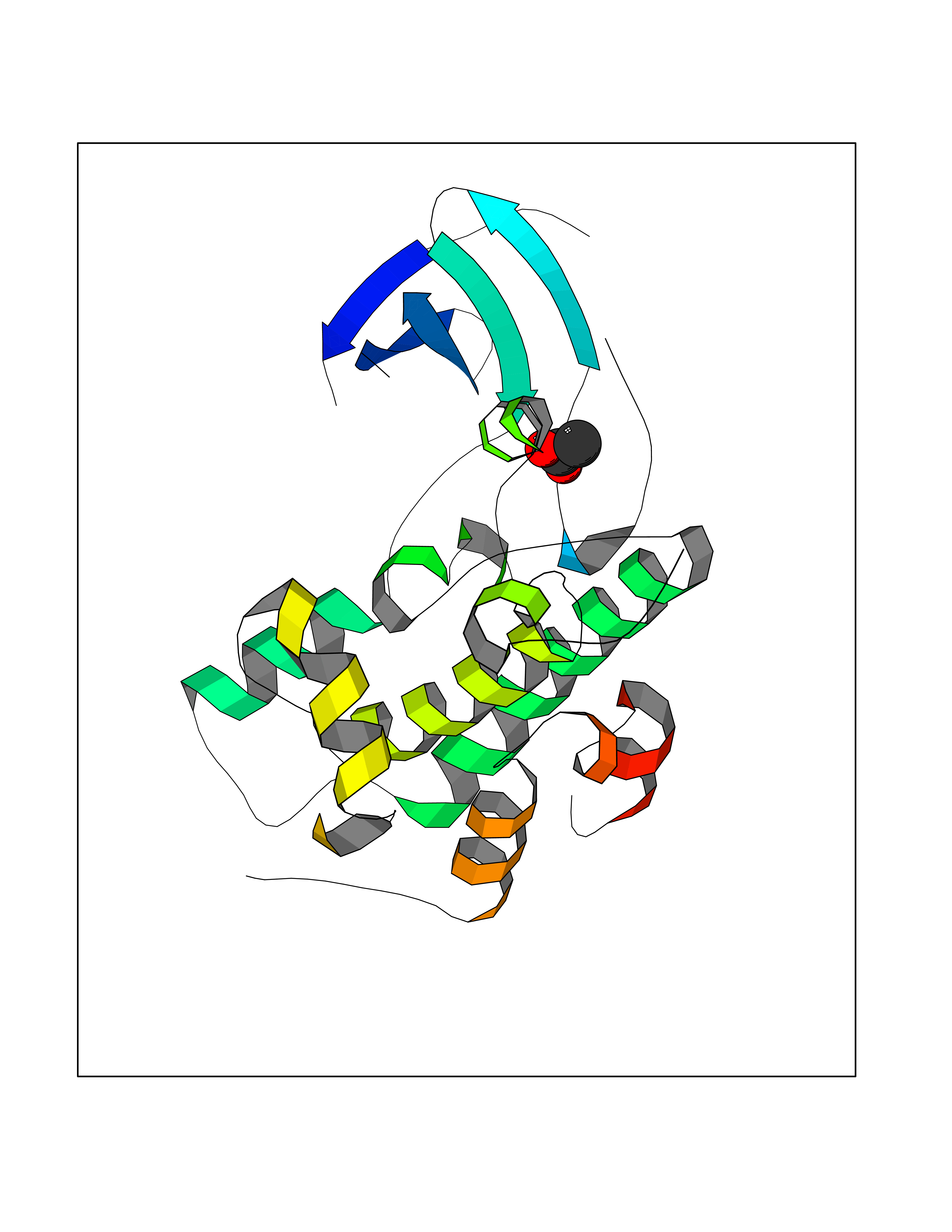 Molscript Map  Pymol Map 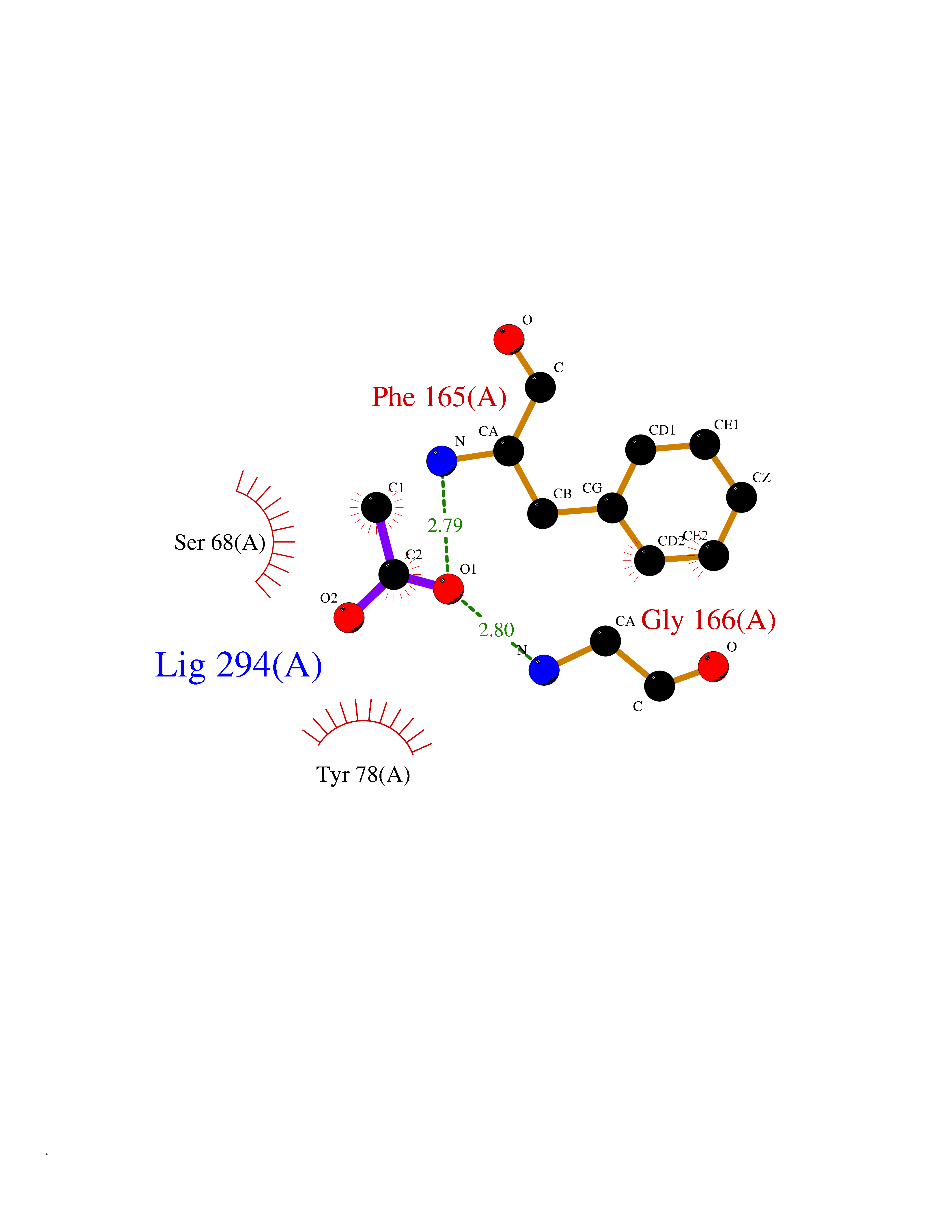 Ligplot Map 3D Binding mode Sequence NRDDYELQEVIGTAVVQAAYCEKVAIKRINAMSQCHHPNIVSYYTSFVVKDELWLVMKLLSGGSVLDIIKHIVAKGEHKSGVLDESTIATILREVLEGLEYLHKNGQIHRDVKAGNILLGEDGSVQIADFGVSAFLAGTPCWMAPEVMEQVRGYDFKADIWSFGITAIELATGAAPYHKYPPMKVLMLTLQNDPPSLETEMLKKYGKSFRKMISLCLQKDPEKRPTAAELLRHKFF Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | C5a anaphylatoxin chemotactic receptor (C5AR1) | 5O9H | 4.39 | |
Target general information Gen name C5AR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CD88; C5aR; C5a-R; C5a anaphylatoxin chemotactic receptor 1; C5R1 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function The ligand interacts with at least two sites on the receptor: a high-affinity site on the extracellular N-terminus, and a second site in the transmembrane region which activates downstream signaling events. Receptor activation stimulates chemotaxis, granule enzyme release, intracellular calcium release and superoxide anion production. Receptor for the chemotactic and inflammatory peptide anaphylatoxin C5a. Related diseases Acrokeratosis verruciformis (AKV) [MIM:101900]: A localized disorder of keratinization, which is inherited as an autosomal dominant trait. Its onset is early in life with multiple flat-topped, flesh-colored papules on the hands and feet, punctate keratoses on the palms and soles, with varying degrees of nail involvement. The histopathology shows a distinctive pattern of epidermal features with hyperkeratosis, hypergranulosis and acanthosis together with papillomatosis. These changes are frequently associated with circumscribed elevations of the epidermis that are said to resemble church spires. There are no features of dyskeratosis or acantholysis, the typical findings in lesions of Darier disease. {ECO:0000269|PubMed:12542527}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Darier disease (DD) [MIM:124200]: A skin disorder characterized by warty papules and plaques in seborrheic areas (central trunk, flexures, scalp and forehead), palmoplantar pits and distinctive nail abnormalities. It is due to loss of adhesion between epidermal cells (acantholysis) and abnormal keratinization. Patients with mild disease may have no more than a few scattered keratotic papules or subtle nail changes, whereas those with severe disease are handicapped by widespread malodorous keratotic plaques. Some patients present with hemorrhage into acantholytic vesicles on the palms and dorsal aspects of the fingers which gives rise to black macules. In a few families affected by Darier disease, neuropsychiatric abnormalities such as mild intellectual disability, schizophrenia, bipolar disorder and epilepsy have been reported. Stress, UV exposure, heat, sweat, friction and oral contraception exacerbate disease symptoms. Clinical variants of Darier disease include hypertrophic, vesicobullous, hypopigmented, cornifying, zosteriform or linear, acute and comedonal subtypes. Comedonal Darier disease is characterized by the coexistence of acne-like comedonal lesions with typical Darier hyperkeratotic papules on light-exposed areas. At histopathologic level, comedonal Darier disease differs from classic Darier disease in the prominent follicular involvement and the presence of greatly elongated dermal villi. {ECO:0000269|PubMed:10080178, ECO:0000269|PubMed:10441323, ECO:0000269|PubMed:10441324, ECO:0000269|PubMed:10441325, ECO:0000269|PubMed:19995371, ECO:0000269|PubMed:28035777}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15011 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Chemotaxis; Cytoplasmic vesicle; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Sulfation; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 65967.4 Length 589 Aromaticity 0.12 Instability index 35.93 Isoelectric point 9.27 Charge (pH=7) 18.32 2D Binding mode Binding energy (Kcal/mol) -5.98  Molscript Map 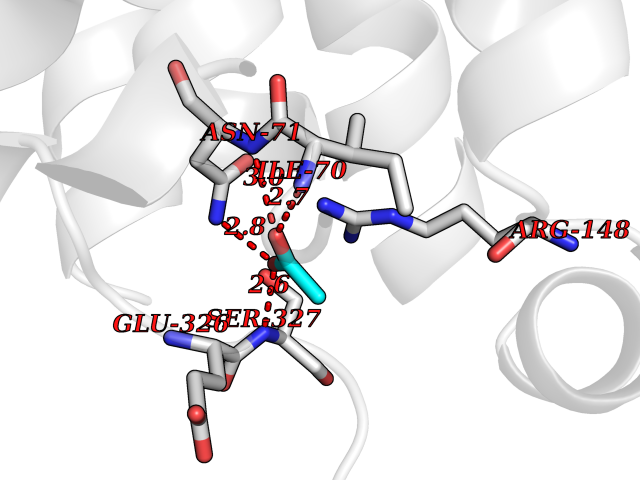 Pymol Map 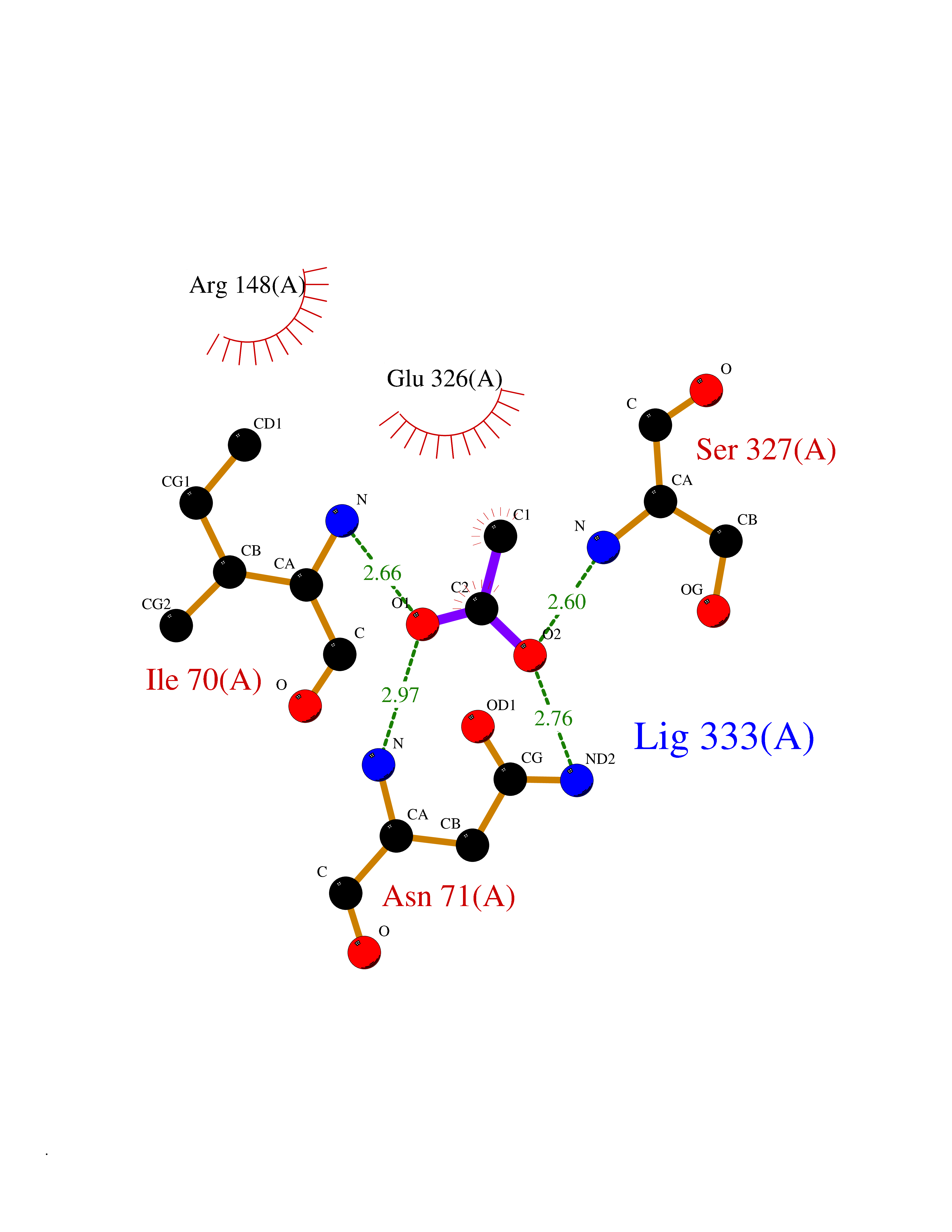 Ligplot Map 3D Binding mode Sequence NTLRVPDILALVIFAVVFLVGVLGNALVVWVTAFEAKRTINAIWFLNLAVADFLACLALPALFTSIVQHHHWPFGGAACSILPSLILLNMYASILLLATISADRFLLVFKPAWCQRFRGAGLAWILCAVAWGLALLLTIPSALYRVVREEYFPPKVLCGVDHDKRRERAVAIVRLVLGFLWPLLTLTICYTFILLRTWSARETRSTKTLKVVVAVVASFFIFWLPYQVTGIMMSFLEPSSPTFLLLKKLDSLCVSFAYINCCINPIIYVVAGQGFQKSLPELLREVLTEESVVRNTLRVPDILALVIFAVVFLVGVLGNALVVWVTAFEAKRTINAIWFLNLAVADFLACLALPALFTSIVQHHHWPFGGAACSILPSLILLNMYASILLLATISADRFLLVFKPAWCQRFRGAGLAWILCAVAWGLALLLTIPSALYRVVREEYFPPKVLCGVDYSHDKRRERAVAIVRLVLGFLWPLLTLTICYTFILLRTWSARETRSTKTLKVVVAVVASFFIFWLPYQVTGIMMSFLEPSSPTFLLLKKLDSLCVSFAYINCCINPIIYVVAGQRKSLPELLREVLTEESVVRE Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | 6YND | 4.39 | |
Target general information Gen name GAPDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peptidyl-cysteine S-nitrosylase GAPDH; OK/SW-cl.12; GAPD; D-glyceraldehyde-3-phosphate dehydrogenase; CDABP0047 Protein family Glyceraldehyde-3-phosphate dehydrogenase family Biochemical class Aldehyde/oxo donor oxidoreductase Function Participates in nuclear events including transcription, RNA transport, DNA replication and apoptosis. Nuclear functions are probably due to the nitrosylase activity that mediates cysteine S-nitrosylation of nuclear target proteins such as SIRT1, HDAC2 and PRKDC. Modulates the organization and assembly of the cytoskeleton. Facilitates the CHP1-dependent microtubule and membrane associations through its ability to stimulate the binding of CHP1 to microtubules. Glyceraldehyde-3-phosphate dehydrogenase is a key enzyme in glycolysis that catalyzes the first step of the pathway by converting D-glyceraldehyde 3-phosphate (G3P) into 3-phospho-D-glyceroyl phosphate. Component of the GAIT (gamma interferon-activated inhibitor of translation) complex which mediates interferon-gamma-induced transcript-selective translation inhibition in inflammation processes. Upon interferon-gamma treatment assembles into the GAIT complex which binds to stem loop-containing GAIT elements in the 3'-UTR of diverse inflammatory mRNAs (such as ceruplasmin) and suppresses their translation. Has both glyceraldehyde-3-phosphate dehydrogenase and nitrosylase activities, thereby playing a role in glycolysis and nuclear functions, respectively. Related diseases Bone marrow failure and diabetes mellitus syndrome (BMFDMS) [MIM:620044]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFDMS is an autosomal recessive form characterized by various degrees of bone marrow failure, ranging from dyserythropoiesis to bone marrow aplasia, with onset in infancy or early childhood, and non-autoimmune insulin-dependent diabetes mellitus appearing in the first or second decades. Many patients show pigmentary skin abnormalities and short stature. {ECO:0000269|PubMed:28073829, ECO:0000269|PubMed:35611808, ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07347; DB02059; DB11638; DB09130; DB00157; DB03893; DB09092 Interacts with Q6UY14-3; Q9UIJ7; P05067; Q9UQM7; Q14194; P35222; Q9BPW9-4; P00533; O00471; O75344; P06241; P04406; O14556; Q8NEA9; P42858; Q92993-2; P42695; P35228; P12004; P00558; P48147; P17612; Q8WUY3; Q9UHX1-2; P15927; P05109; Q96GZ6; P00441; Q9BSI4; P10599 EC number EC 1.2.1.12 Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Alternative splicing; Apoptosis; Cytoplasm; Cytoskeleton; Direct protein sequencing; Glycolysis; Glycoprotein; Immunity; Innate immunity; Isopeptide bond; Membrane; Methylation; NAD; Nucleus; Oxidation; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; S-nitrosylation; Transferase; Translation regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35818.4 Length 333 Aromaticity 0.08 Instability index 13.69 Isoelectric point 8.64 Charge (pH=7) 3.64 2D Binding mode Binding energy (Kcal/mol) -5.98 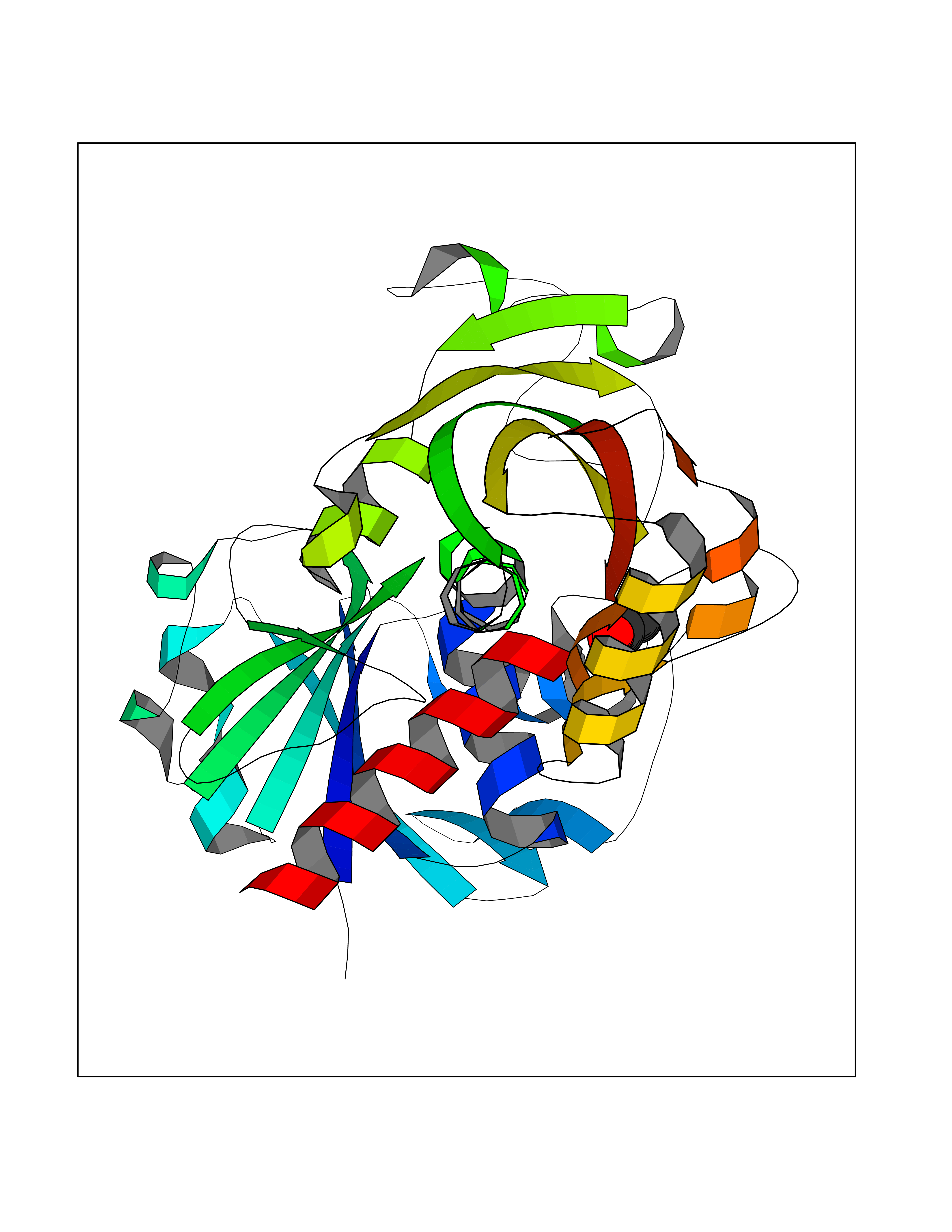 Molscript Map 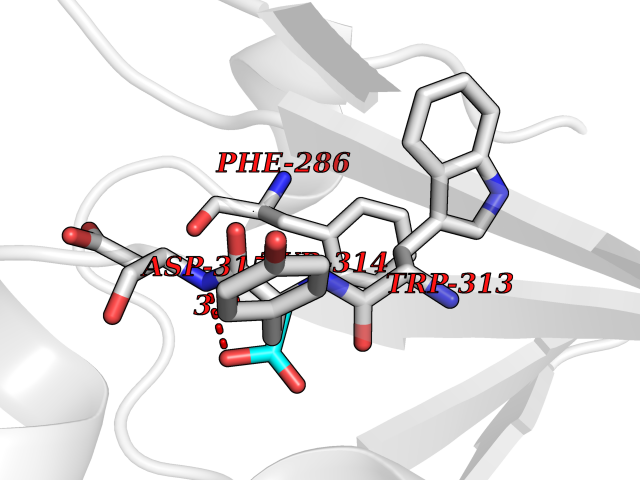 Pymol Map 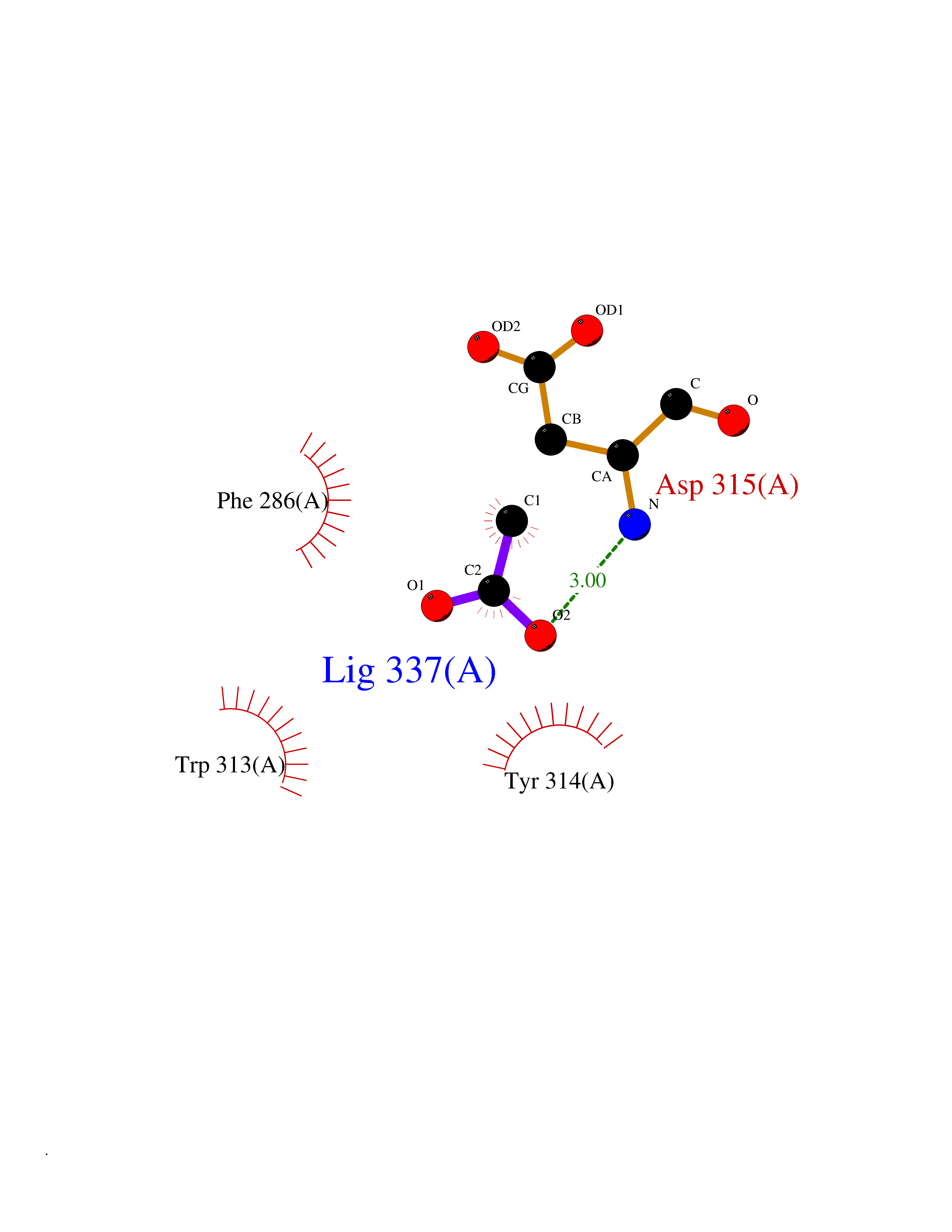 Ligplot Map 3D Binding mode Sequence GKVKVGVNGFGRIGRLVTRAAFNSGKVDIVAINDPFIDLNYMVYMFQYDSTHGKFHGTVKAENGKLVINGNPITIFQERDPSKIKWGDAGAEYVVESTGVFTTMEKAGAHLQGGAKRVIISAPSADAPMFVMGVNHEKYDNSLKIISNASTTNCLAPLAKVIHDNFGIVEGLMTTVHAITATQKTVDGPSGKLWRDGRGALQNIIPASTGAAKAVGKVIPELNGKLTGMAFRVPTANVSVVDLTCRLEKPAKYDDIKKVVKQASEGPLKGILGYTEHQVVSSDFNSDTHSSTFDAGAGIALNDHFVKLISWYDNEFGYSNRVVDLMAHMASKE Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Kallikrein-6 (KLK6) | 1LO6 | 4.39 | |
Target general information Gen name KLK6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Zyme; Serine protease 9; Serine protease 18; SP59; Protease M; PRSS9; PRSS18; Neurosin; MSP; K6 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function Shows activity against amyloid precursor protein, myelin basic protein, gelatin, casein and extracellular matrix proteins such as fibronectin, laminin, vitronectin and collagen. Degrades alpha-synuclein and prevents its polymerization, indicating that it may be involved in the pathogenesis of Parkinson disease and other synucleinopathies. May be involved in regulation of axon outgrowth following spinal cord injury. Tumor cells treated with a neutralizing KLK6 antibody migrate less than control cells, suggesting a role in invasion and metastasis. Serine protease which exhibits a preference for Arg over Lys in the substrate P1 position and for Ser or Pro in the P2 position. Related diseases Prieto syndrome (PRS) [MIM:309610]: An X-linked recessive disorder characterized by impaired intellectual development, developmental delay, autism spectrum disorder, variable epilepsy, craniofacial dysmorphism, and structural brain abnormalities including polymicrogyria and cerebral atrophy. {ECO:0000269|PubMed:35678782}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03127 Interacts with Q8NC06-3; P11117; Q53FZ2-2; Q96BT7-2; P05067; Q9NP61; Q5H9R4-2; Q8WXK3-2; Q12797-6; Q9Y6H3; P27449; O95817; Q8TBE0; Q9UQB8-3; Q9UQB8-6; P51572; O15155-2; Q9BXY8; Q7L1Q6-2; Q9H0W9-3; Q9H257-2; Q86XM0; O75309; P49336-2; Q9Y281; Q8NE62; O75508; Q9BT09; P20849; Q86WV2; P68400; P09668; P09172; P61962; Q9BTE7; Q6ZPD9-2; P63167; Q13144; P23588; P16452; Q5RHP9-3; Q6NXG1; Q6NXG1-3; Q7L5A8; Q6NZ36-4; Q14296; P62861; P31994; Q7L622; P24522; P15976-2; Q9NXC2; B2RAF7; P52790; Q9P0W2; Q4VB01; Q7LGA3-3; Q96D96-2; Q8IYA8; Q14005-2; Q0VD86; Q9BT40; Q9Y283-3; P57682; Q9Y2M5; O60259; P08727; Q14533; Q3LI72; Q3SYF9; Q8IUC2; Q92615; Q14847-2; Q6DKI2; Q8TE12-2; O95332; Q96JB6; Q99683; Q15759; P42679; Q8N6R0; Q14728; A0A0A0MR05; Q9BRA0; Q92886; P48645; Q9Y239; P06748; Q6P4D5-2; Q8WW12; Q16549; Q13371; Q9NZ53-2; Q9GZS1; P19388; Q6ZMI0-5; Q96QH2; Q86UA1; P61289; P21246; P53801; Q9NWB1-5; Q9BWF3; P52756; P47804-3; Q9H0X6; Q969K3; Q9BY12-3; Q86SQ7-2; Q9NTN9-3; Q8IUQ4-2; Q9H2B4-2; Q99717; P37840; Q5T0L3; Q496A3; Q9BUD6; Q9C004; Q99469; O75558; Q9UMX1; O43463; O60506-4; Q8TDR4; Q96A09; Q01664; Q6YHU6; Q9H808; Q8IU80-2; Q8IUR5-4; Q8WVP5; Q96KP6; O14787-2; O94900; P06753-2; Q9NX07; O60636; Q86UF1; Q5VYS8-5; Q9GZX9; Q13404; Q9H9P5-5; Q9NVA1; P61964; Q9NZC7-5; O00308; Q9HAV4; Q8N0Y2-2; Q7Z783; Q96EJ4 EC number EC 3.4.21.- Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Cleavage on pair of basic residues; Cytoplasm; Direct protein sequencing; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Hydrolase; Microsome; Mitochondrion; Nucleus; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 24300.3 Length 221 Aromaticity 0.06 Instability index 37.16 Isoelectric point 6.65 Charge (pH=7) -1.29 2D Binding mode Binding energy (Kcal/mol) -5.98  Molscript Map 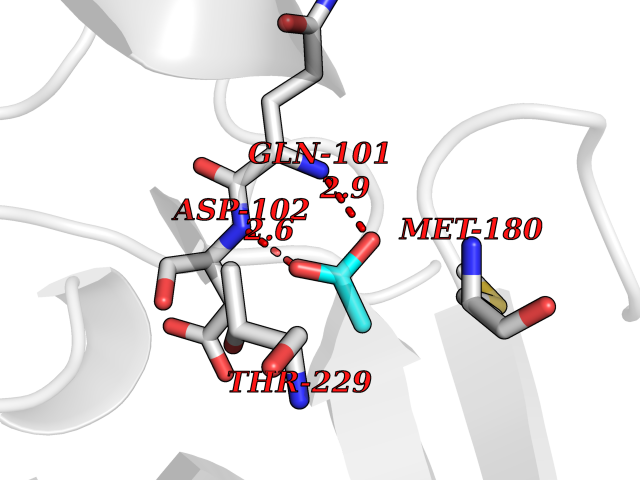 Pymol Map 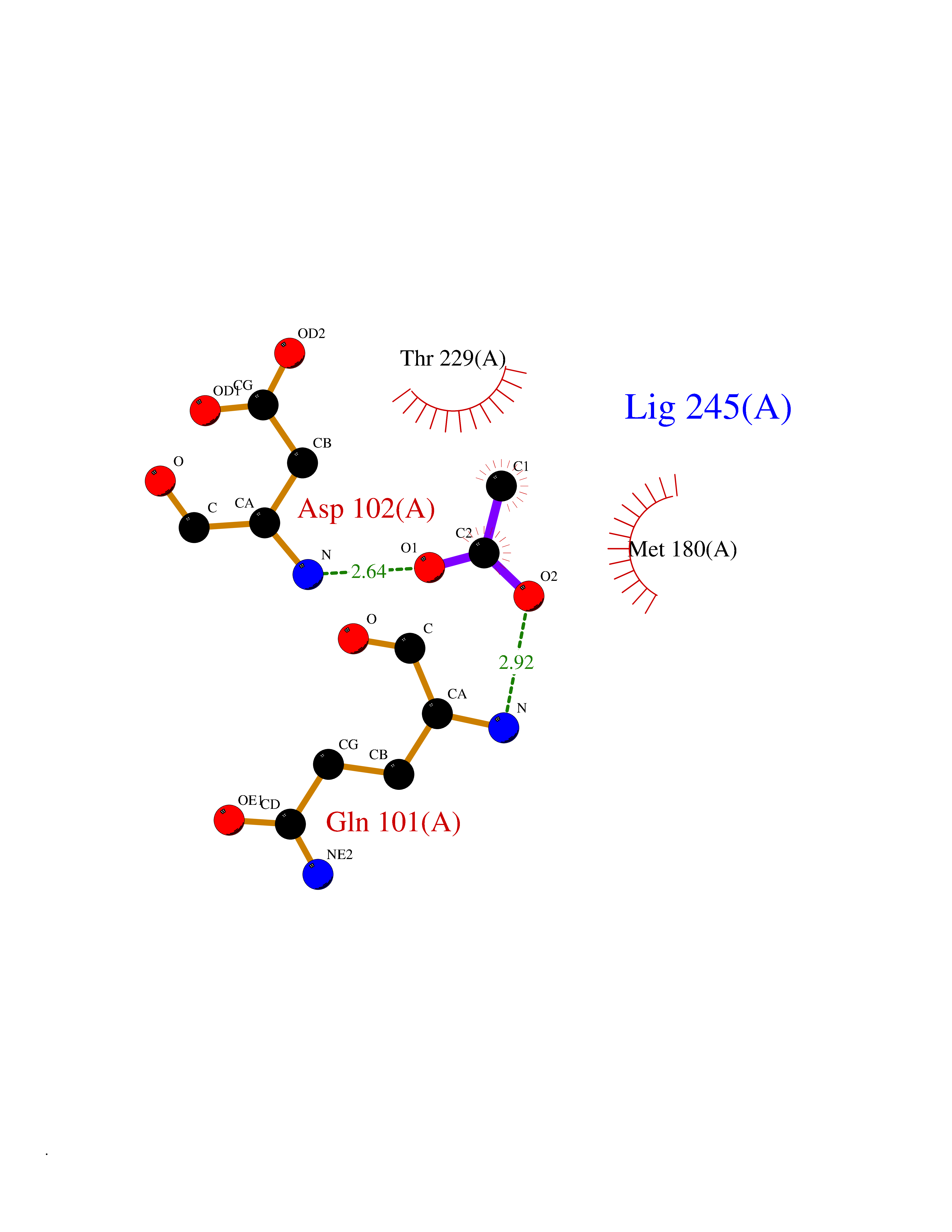 Ligplot Map 3D Binding mode Sequence LVHGGPCDKTSHPYQAALYTSGHLLCGGVLIHPLWVLTAAHCKKPNLQVFLGKHNLRQRESSQEQSSVVRAVIHPDYDAASHDQDIMLLRLARPAKLSELIQPLPLERDCSANTTSCHILGWGKTADGDFPDTIQCAYIHLVSREECEHAYPGQITQNMLCAGDEKYGKDSCQGDSGGPLVCGDHLRGLVSWGNIPCGSKEKPGVYTNVCRYTNWIQKTIQ Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Deubiquitinating enzyme 1 (USP1) | 7ZH4 | 4.39 | |
Target general information Gen name USP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hUBP; Ubiquitin-specific-processing protease 1; Ubiquitin thioesterase 1; Ubiquitin carboxyl-terminal hydrolase 1 Protein family Peptidase C19 family Biochemical class Peptidase Function Involved in PCNA-mediated translesion synthesis (TLS) by deubiquitinating monoubiquitinated PCNA. Has almost no deubiquitinating activity by itself and requires the interaction with WDR48 to have a high activity. Negative regulator of DNA damage repair which specifically deubiquitinates monoubiquitinated FANCD2. Related diseases Brachydactyly A2 (BDA2) [MIM:112600]: A form of brachydactyly. Brachydactyly defines a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. In brachydactyly type A2 shortening of the middle phalanges is confined to the index finger and the second toe, all other digits being more or less normal. Because of a rhomboid or triangular shape of the affected middle phalanx, the end of the second finger usually deviates radially. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}. The gene represented in this entry is involved in disease pathogenesis. Duplications of a cis-regulatory element located approximately 110 kb downstream of BMP2 have been found in BDA2 families. They likely cause altered BMP2 expression with pathological consequences. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}.; DISEASE: Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 1 (SSFSC1) [MIM:617877]: An autosomal dominant disorder characterized by short stature, facial dysmorphism, skeletal anomalies, and variable cardiac defects. Distinctive facial features include midface retrusion, short upturned nose, long philtrum, high-arched or cleft palate, and variable degrees of micrognathia and dental crowding. Skeletal anomalies include patterning defects of the axial skeleton, characterized by 11 pairs of ribs and brachydactyly of the fifth ray. Congenital heart defects are variably observed and appear to involve primarily the cardiac outflow tract. {ECO:0000269|PubMed:29198724}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q8TAF3; Q8TAF3-1 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Autocatalytic cleavage; DNA damage; DNA repair; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID D Molecular weight (Da) 32426 Length 285 Aromaticity 0.1 Instability index 50.73 Isoelectric point 5.85 Charge (pH=7) -4.67 2D Binding mode Binding energy (Kcal/mol) -5.99 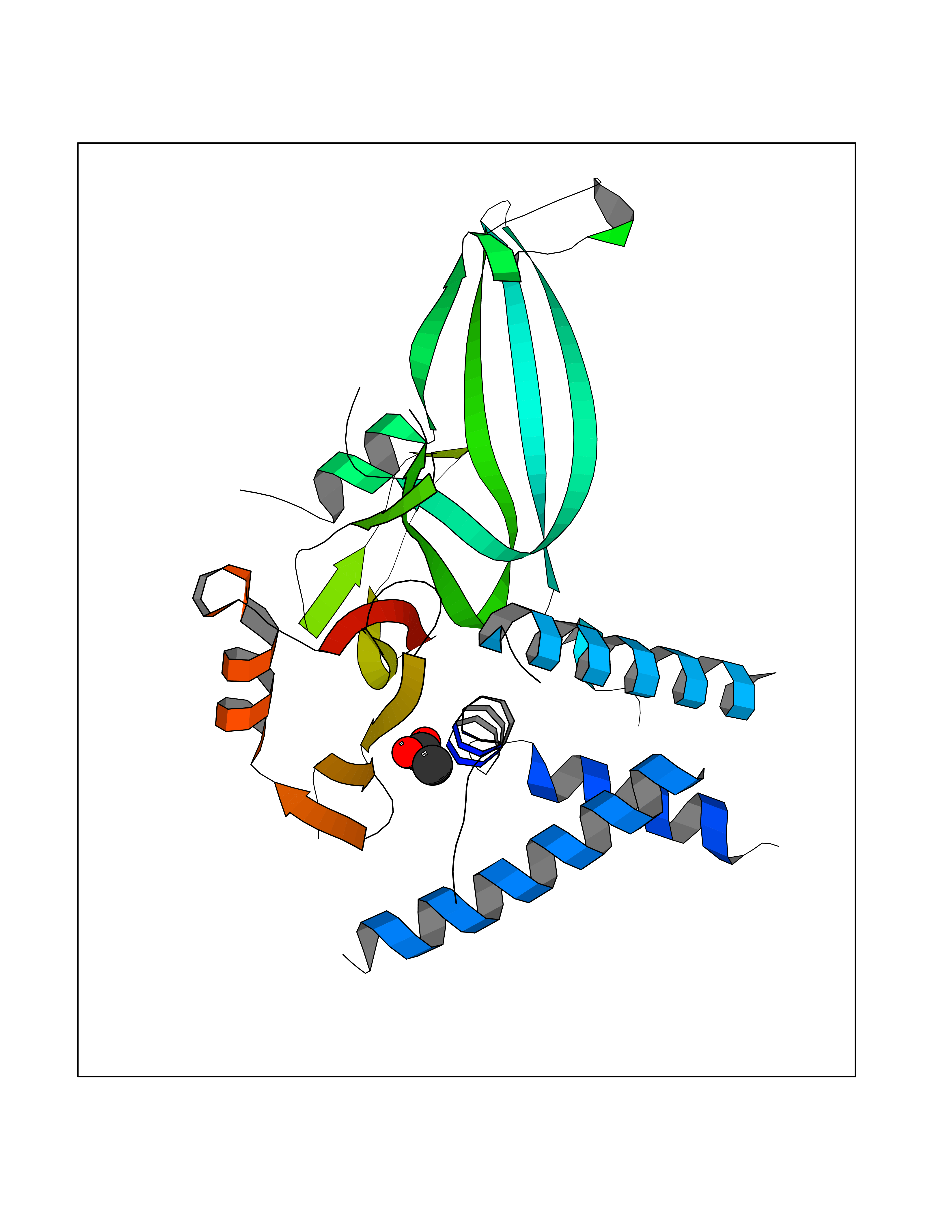 Molscript Map  Pymol Map 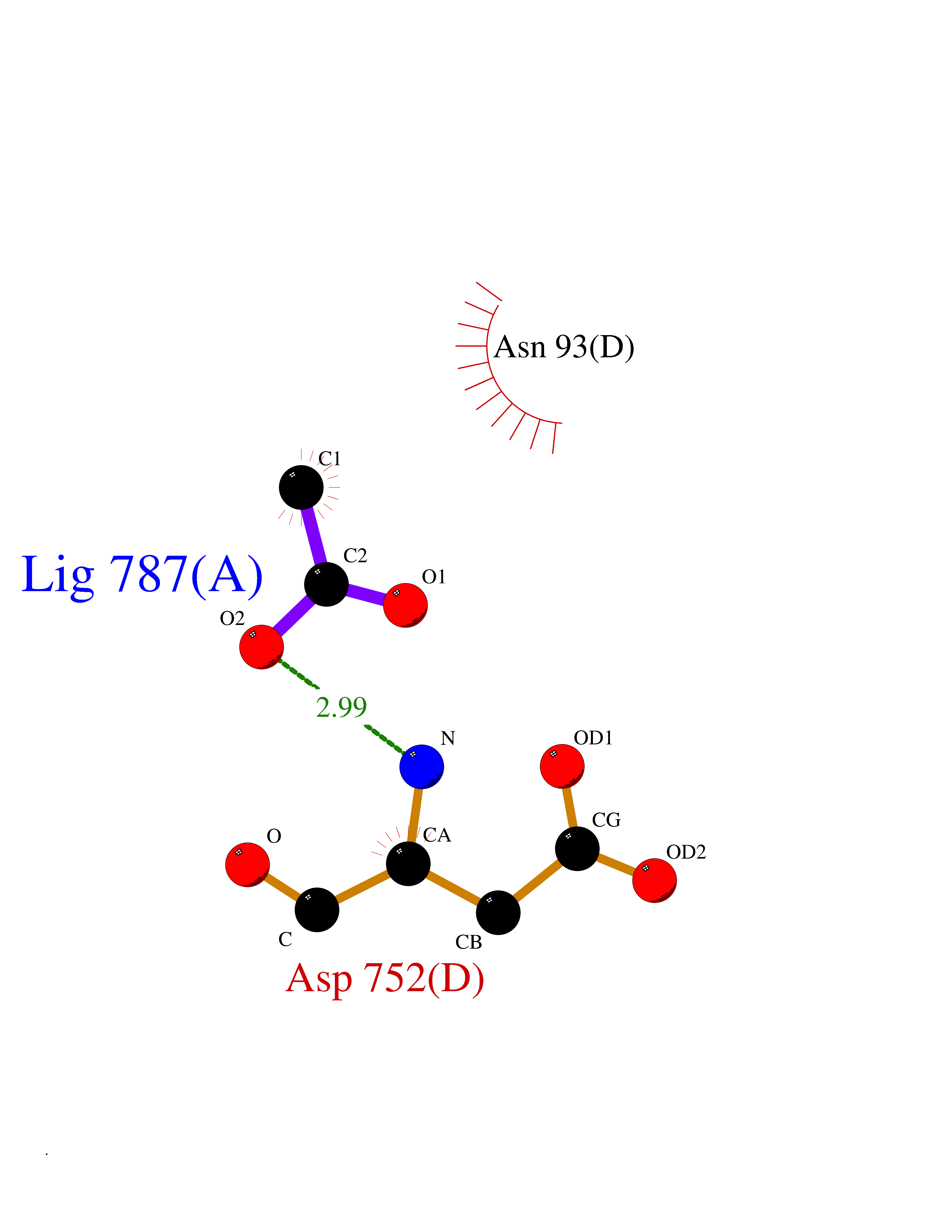 Ligplot Map 3D Binding mode Sequence GLNNLGNTSYLNSILQVLYFCPGFKSGVKHLFNIISRKKYELICSLQSLIISVEQLQASFLLNPLQHDAQEVLQCILGNIQETCQLLKKGFELVEKLFQGQLVLRTRCLECESLTERREDFQDISVPVQEDMKTLRWAISQFASVERIVGEDKYFCENCHHYTEAERSLLFDKMPEVITIHLKCFAASGLSKINTPLLTPLKLSLEEWSTKPTNDSYGLFAVVMHSGITISSGHYTASVKVTYEGKWLLFDDSEVKVTEEKDFLNSLSPSTSPTSTPYLLFYKKL Hydrogen bonds contact Hydrophobic contact | ||||