Job Results:
Ligand
Structure
Job ID
85eee3bcbedbd74b81bc48f797dc6af1
Job name
NA
Time
2026-02-27 13:44:11
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Staphylococcus Enoyl ACP reductase (Stap-coc fabI) | 6YUR | 5.11 | |
Target general information Gen name Stap-coc fabI Organism Staphylococcus aureus (strain NCTC 8325 / PS 47) Uniprot ID TTD ID Synonyms NADPHdependent enoylACP reductase; Enoyl[acylcarrierprotein] reductase [NADPH] FabI Protein family Short-chain dehydrogenases/reductases (SDR) family, FabI subfamily Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the reduction of a carbon-carbon double bond in an enoyl moiety that is covalently linked to an acyl carrier protein (ACP). It has a preference for a long chain (C12) substrate compared to the shorter (C4) acyl group. Involved in the elongation cycle of fatty acid which are used in the lipid metabolism. Related diseases Amyloidosis, hereditary systemic 4, Finnish type (AMYLD4) [MIM:105120]: A form of hereditary systemic amyloidosis, a disorder characterized by amyloid deposition in multiple tissues resulting in a wide clinical spectrum. AMYLD4 is due to gelsolin amyloid deposition and is typically characterized by cranial neuropathy and lattice corneal dystrophy. Most patients have modest involvement of internal organs, but severe systemic disease can develop in some individuals causing peripheral polyneuropathy, amyloid cardiomyopathy, and nephrotic syndrome leading to renal failure. AMYLD4 is usually inherited in an autosomal dominant pattern. However, homozygotes with a more severe phenotype have also been reported. {ECO:0000269|PubMed:1338910, ECO:0000269|PubMed:19666512, ECO:0000269|PubMed:2176481}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; NAD; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,C Molecular weight (Da) 55674.6 Length 510 Aromaticity 0.07 Instability index 33.86 Isoelectric point 5.64 Charge (pH=7) -9.3 2D Binding mode Binding energy (Kcal/mol) -6.97 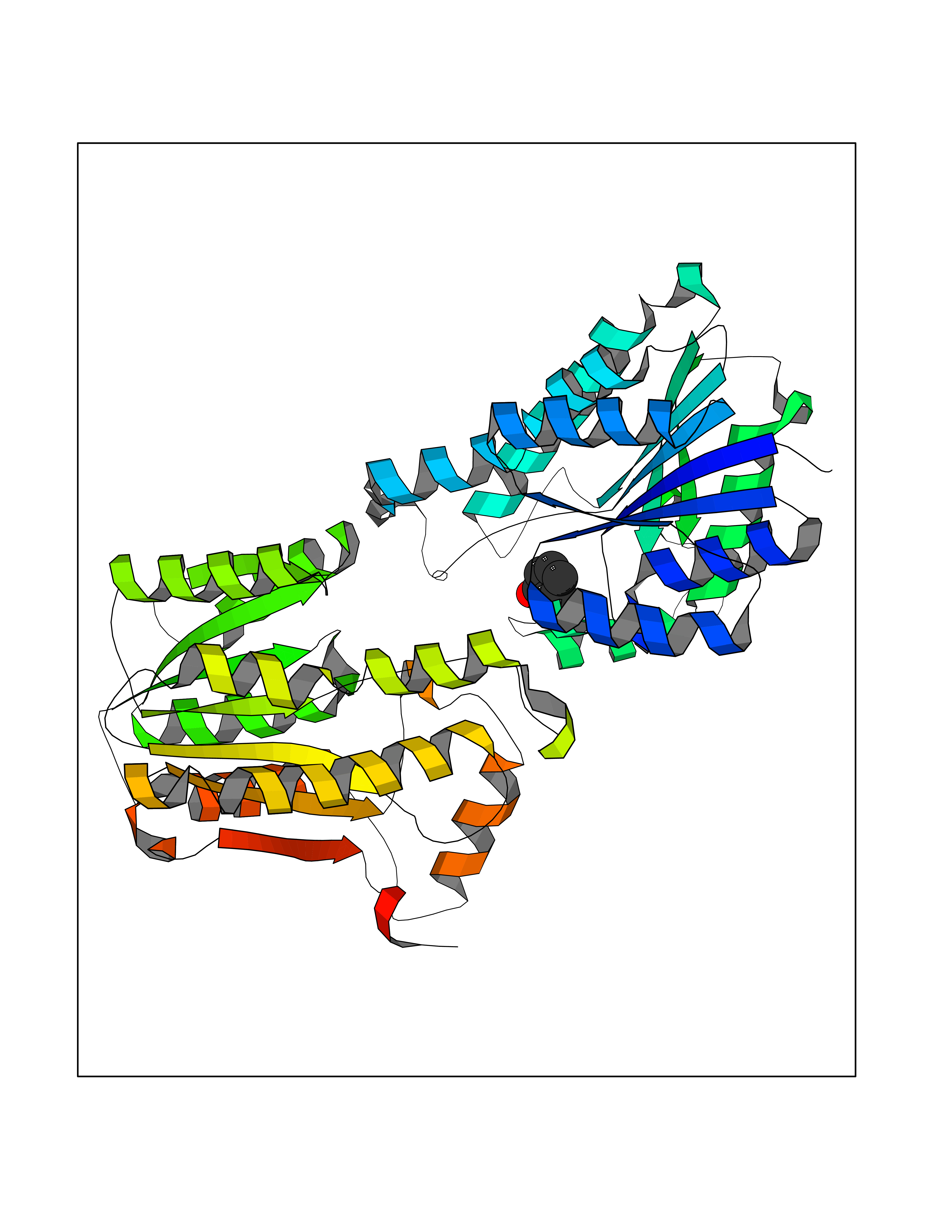 Molscript Map 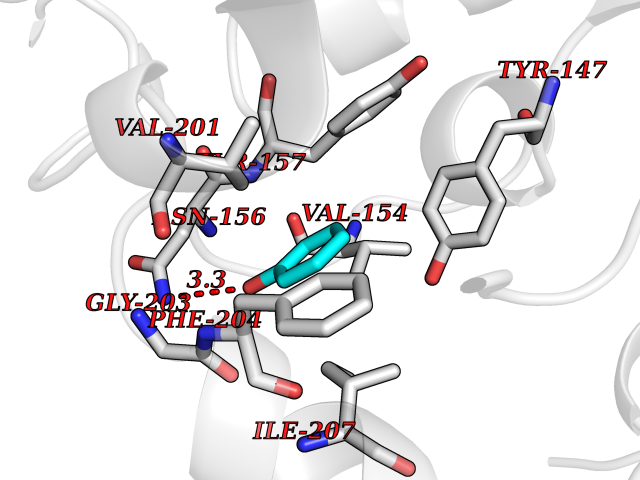 Pymol Map 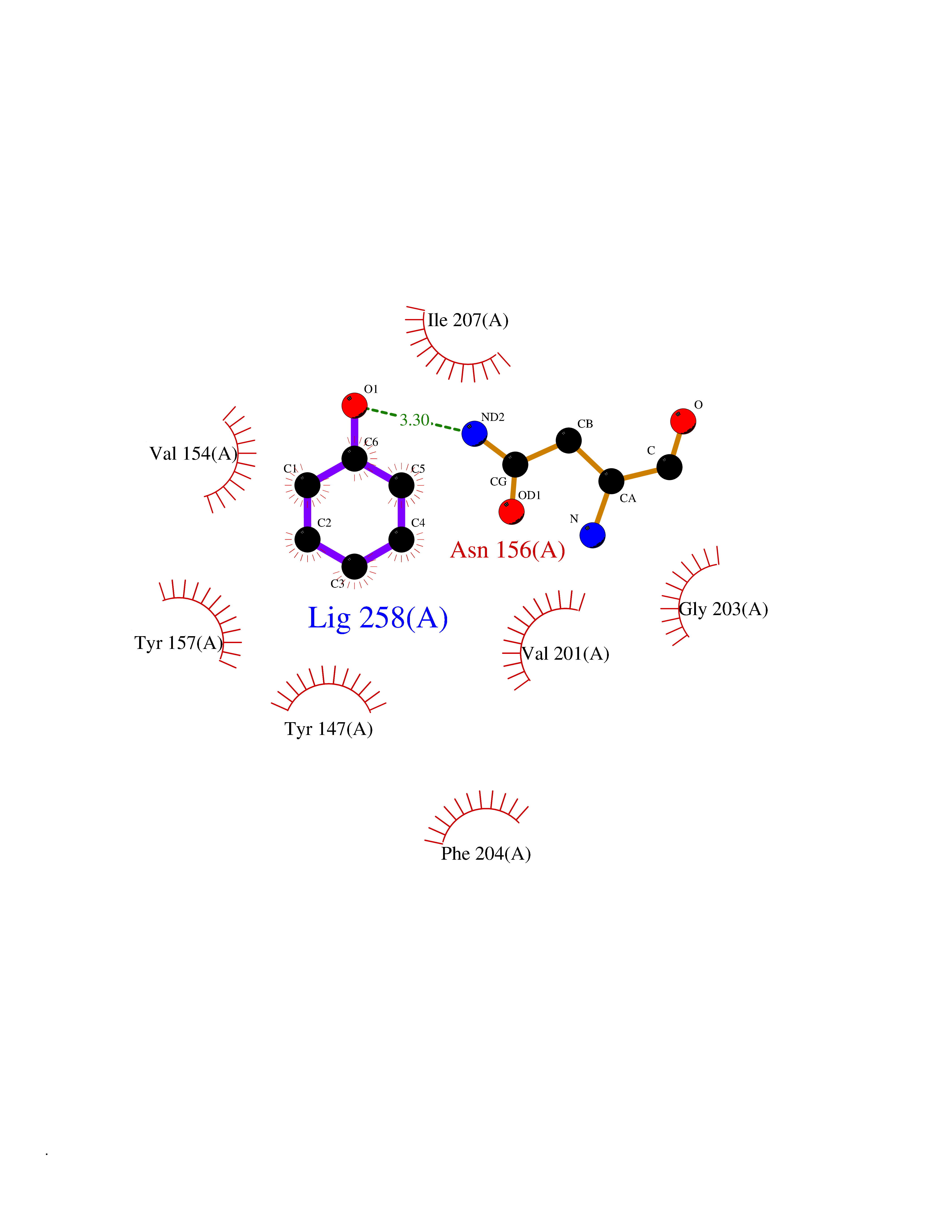 Ligplot Map 3D Binding mode Sequence VNLENKTYVIMGIANKRSIAFGVAKVLDQLGAKLVFTYRKERSRKELEKLLEQLNQPEAHLYQIDVQSDEEVINGFEQIGKDVGNIDGVYHSIAFANMEDLRGRFSETSREGFLLAQDISSYSLTIVAHEAKKLMPEGGSIVATTYLGGEFAVQNYNVMGVAKASLEANVKYLALDLGPDNIRVNAISAGPIRTLSAKGVGGFNTILKEIEERAPLKRNVDQVEVGKTAAYLLSDLSSGVTGENIHVDSGFHAIKVNLENKTYVIMGIANKRSIAFGVAKVLDQLGAKLVFTYRKERSRKELEKLLEQLNQPEAHLYQIDVQSDEEVINGFEQIGKDVGNIDGVYHSIAFANMEDLRGRFSETSREGFLLAQDISSYSLTIVAHEAKKLMPEGGSIVATTYLGGEFAVQNYNVMGVAKASLEANVKYLALDLGPDNIRVNAISAGPIRTLSAKGVGGFNTILKEIEERAPLKRNVDQVEVGKTAAYLLSDLSSGVTGENIHVDSGFHAIK Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Somatostatin receptor type 4 (SSTR4) | 7XMT | 5.11 | |
Target general information Gen name SSTR4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SSTR4; SS4R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for somatostatin-14. The activity of this receptor is mediated by G proteins which inhibits adenylyl cyclase. It is functionally coupled not only to inhibition of adenylate cyclase, but also to activation of both arachidonate release and mitogen-activated protein (MAP) kinase cascade. Mediates antiproliferative action of somatostatin in tumor cells. Related diseases Oocyte/zygote/embryo maturation arrest 21 (OZEMA21) [MIM:620610]: An autosomal dominant, female infertility disorder characterized by zygote development arrest due to failure of pronuclei fusion. {ECO:0000269|PubMed:33948904, ECO:0000269|PubMed:33953335}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB13985; DB09099 Interacts with P35346 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 29548.3 Length 265 Aromaticity 0.12 Instability index 45.65 Isoelectric point 9.78 Charge (pH=7) 14.03 2D Binding mode Binding energy (Kcal/mol) -6.96 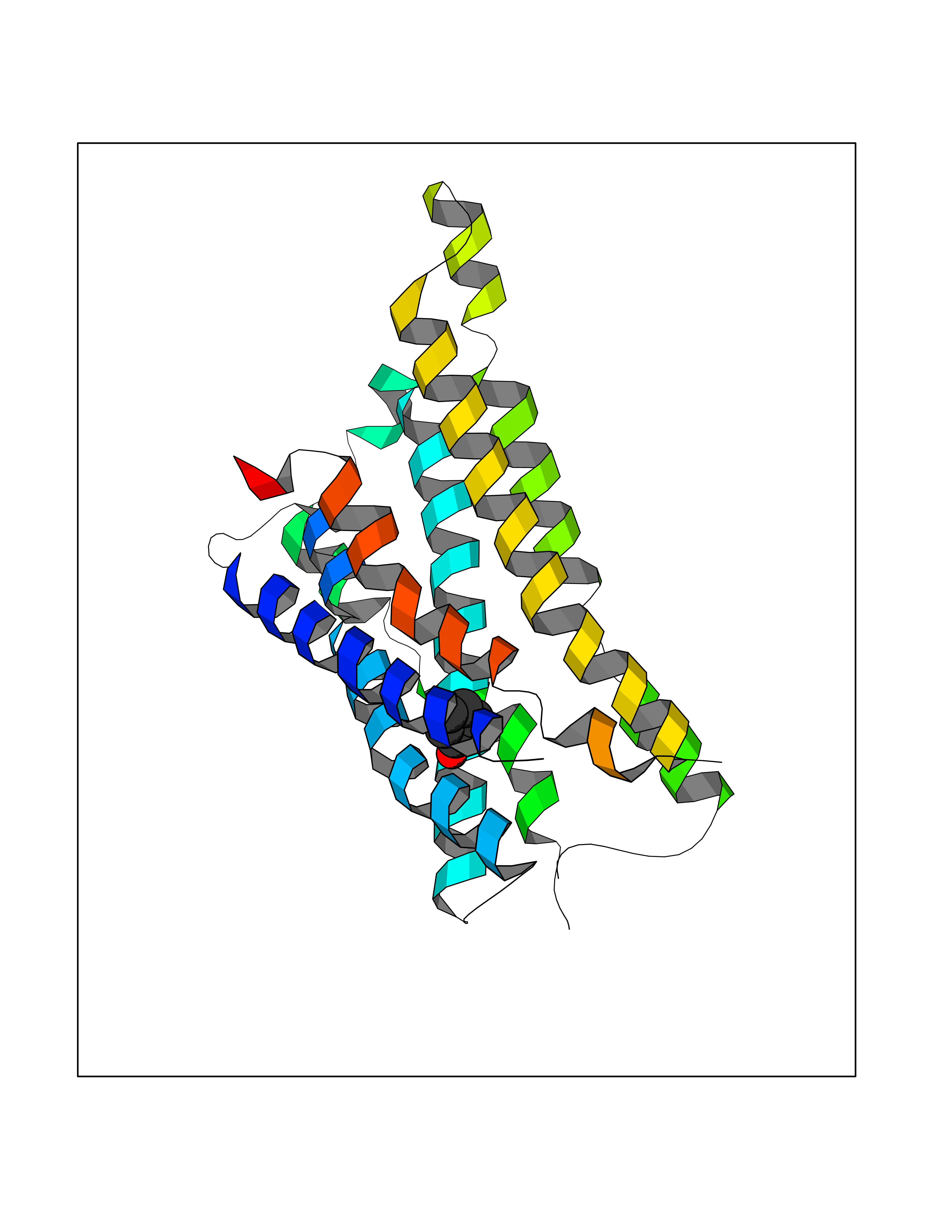 Molscript Map 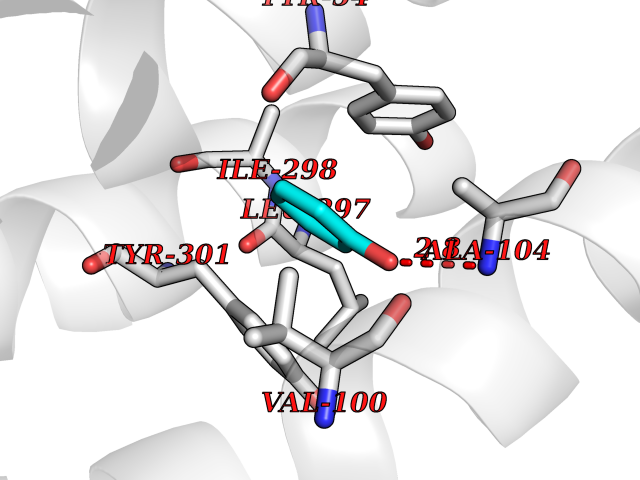 Pymol Map 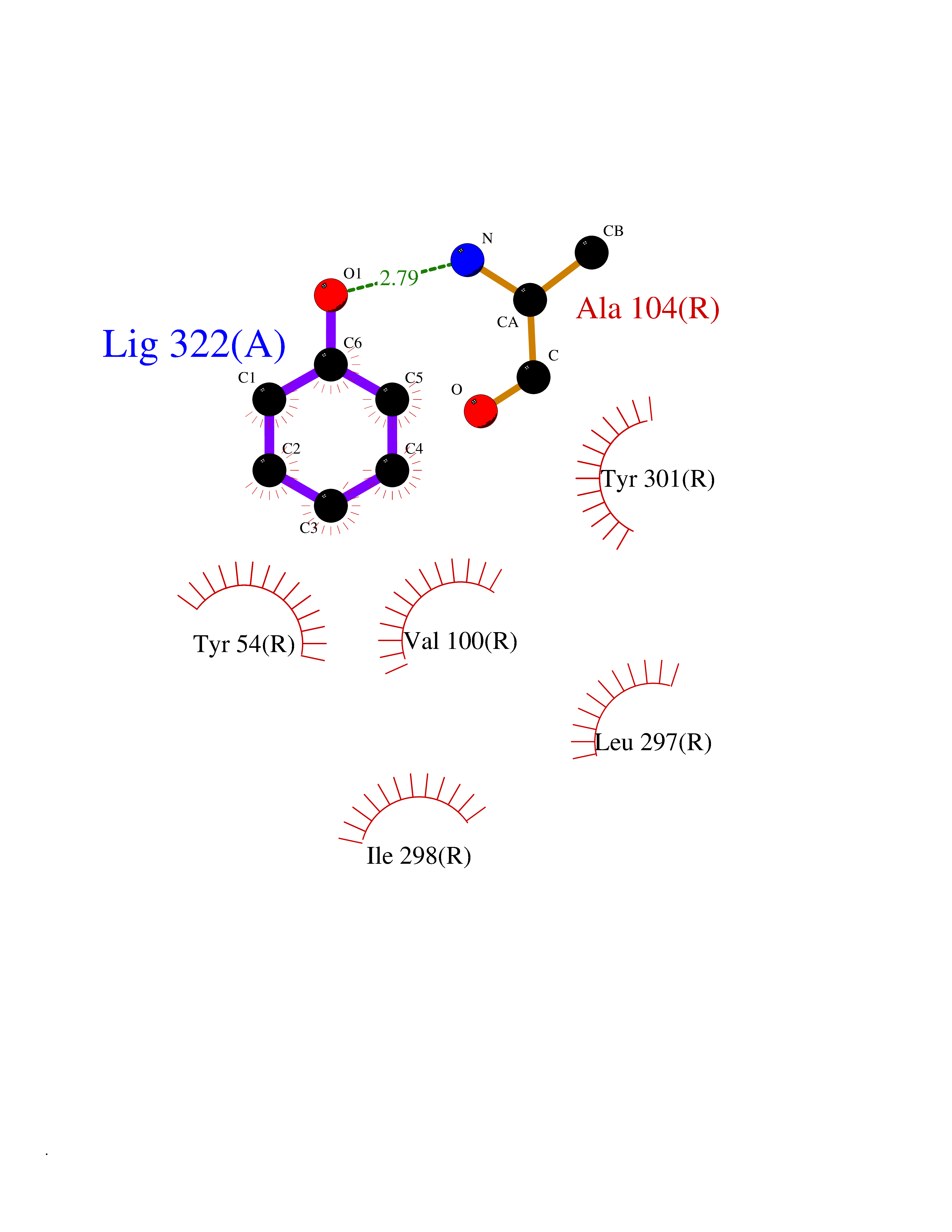 Ligplot Map 3D Binding mode Sequence GMVAIQCIYALVCLVGLVGNALVIFVILRYAKMKTATNIYLLNLAVADELFMLSVPFVASSAALRHWPFGSVLCRAVLSVDGLNMFTSVFCLTVLSVDRYVAVVHPLRAATYRRPSVAKLINLGVWLASLLVTLPIAIFADTRPACNLQWPHPAWSAVFVVYTFLLGFLLPVLAIGLCYLLIVGKMRAVALRAGWQQRRRSEKKITRLVLMFVVVFVLCWMPFYVVQLLNLFLDATVNHVSLILSYANSCANPILYGFLSDNFRR Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | 4-hydroxyphenylpyruvate dioxygenase | 3ISQ | 5.10 | |
Target general information Gen name HPD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PPD Protein family 4HPPD family Biochemical class Oxidoreductase Function 4-hydroxyphenylpyruvate dioxygenase activity.Metal ion binding. Related diseases Tyrosinemia 3 (TYRSN3) [MIM:276710]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, seizures and mild intellectual disability. {ECO:0000269|PubMed:10942115, ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hawkinsinuria (HWKS) [MIM:140350]: An inborn error of tyrosine metabolism characterized by failure to thrive, persistent metabolic acidosis, fine and sparse hair, and excretion of the unusual cyclic amino acid metabolite, hawkinsin, in the urine. {ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02850; DB00348 Interacts with NA EC number 1.13.11.27 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Dioxygenase; Disease variant; Endoplasmic reticulum; Golgi apparatus; Intellectual disability; Iron; Membrane; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 43164.8 Length 376 Aromaticity 0.11 Instability index 32.38 Isoelectric point 6.73 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -6.96 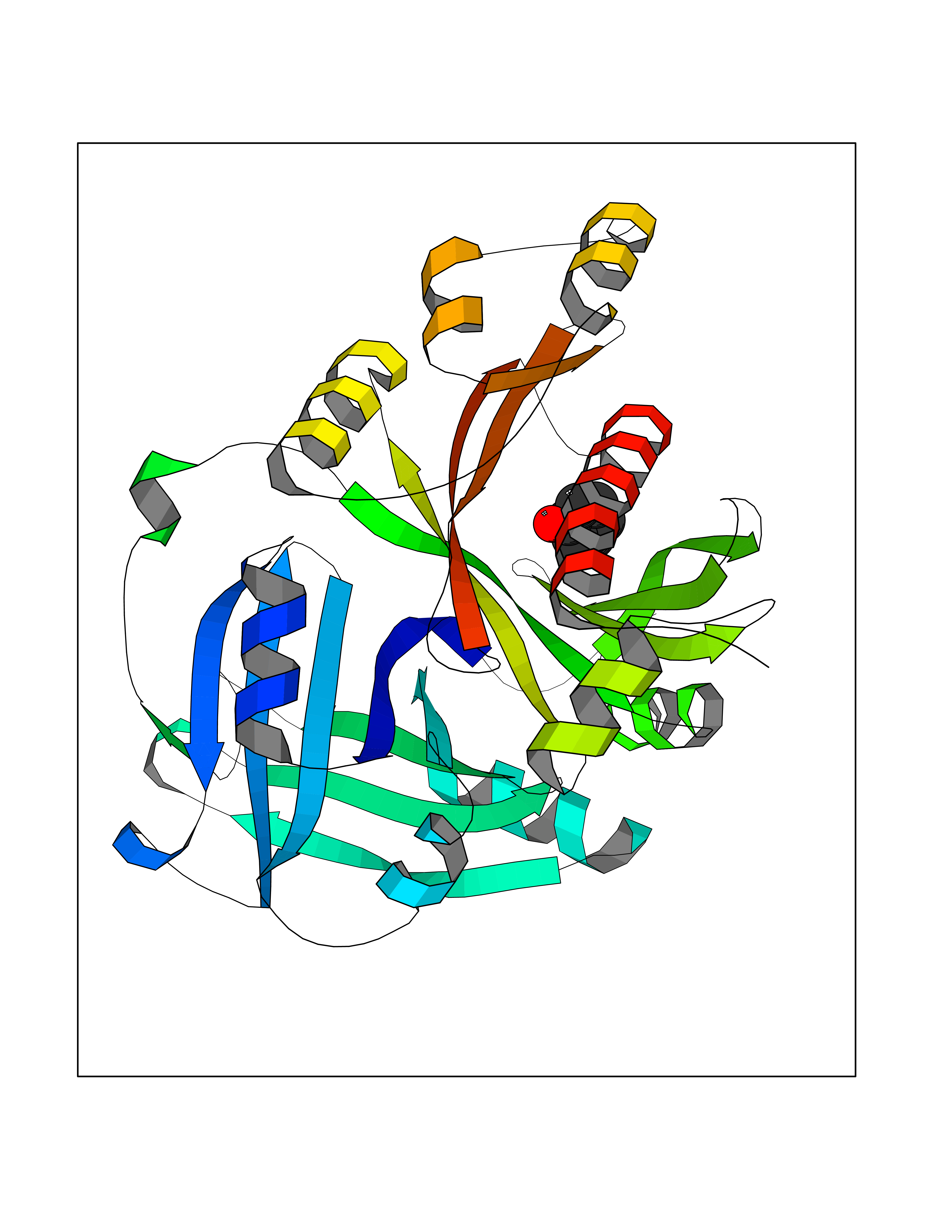 Molscript Map 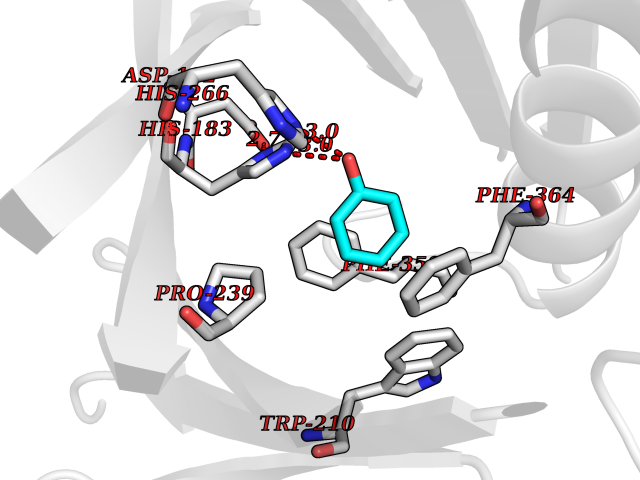 Pymol Map 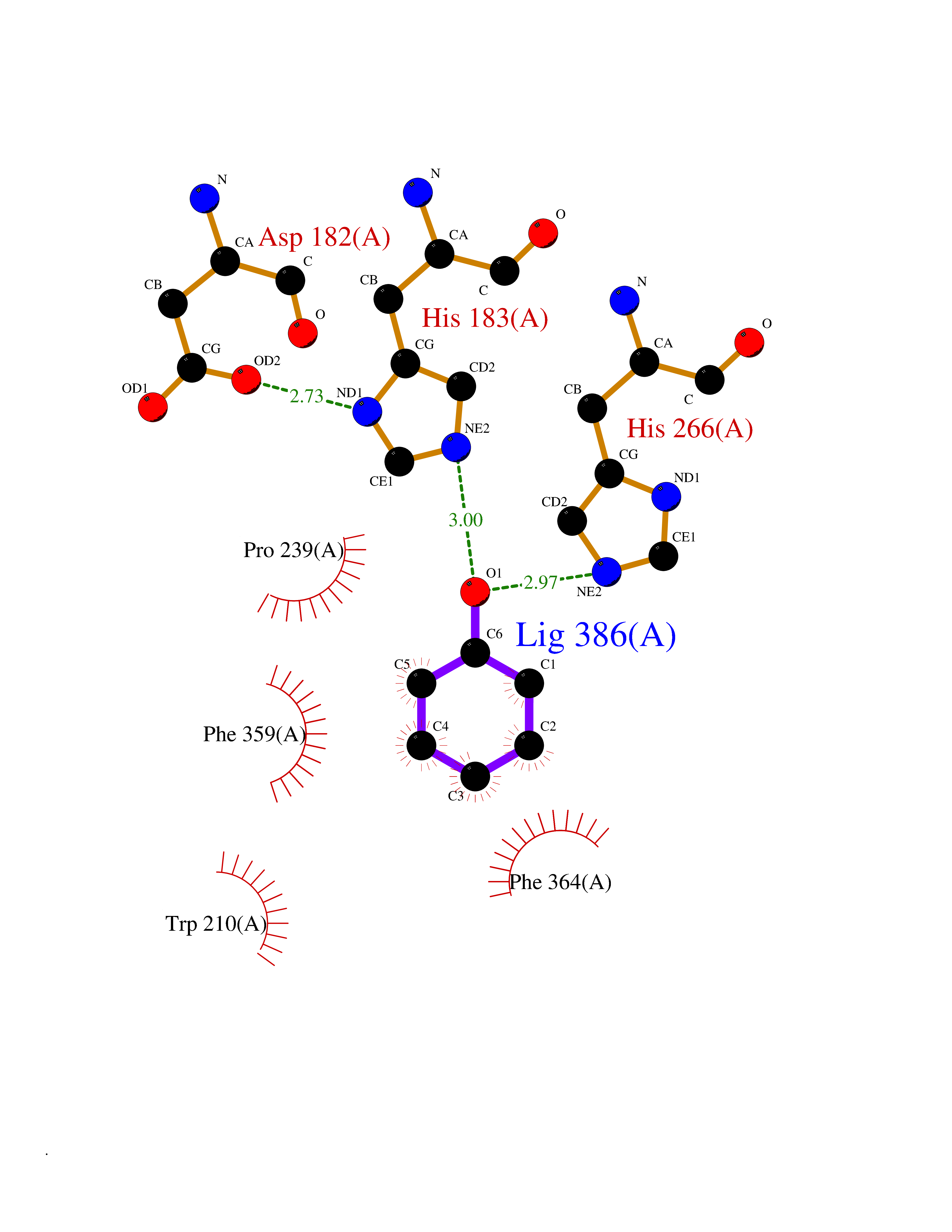 Ligplot Map 3D Binding mode Sequence AKPERGRFLHFHSVTFWVGNAKQAASFYCSKMGFEPLAYRGLETGSREVVSHVIKQGKIVFVLSSALNPWNKEMGDHLVKHGDGVKDIAFEVEDCDYIVQKARERGAKIMREPWVEQDKFGKVKFAVLQTYGDTTHTLVEKMNYIGQFLPGYEAPAFMDPLLPKLPKCSLEMIDHIVGNQPDQEMVSASEWYLKNLQFHRFWSVDDTQVHTEYSSLRSIVVANYEESIKMPINEPAPGKKKSQIQEYVDYNGGAGVQHIALKTEDIITAIRHLRERGLEFLSVPSTYYKQLREKLKTAKIKVKENIDALEELKILVDYDEKGYLLQIFTKPVQDRPTLFLEVIQRHNHQGFGAGNFNSLFKAFEEEQNLRGNLTNM Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Aspartate beta-hydroxylase (ASPH) | 5APA | 5.10 | |
Target general information Gen name ASPH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peptideaspartate betadioxygenase; Aspartyl/asparaginyl betahydroxylase; ASPH; ASP betahydroxylase Protein family Aspartyl/asparaginyl beta-hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Isoform 8: membrane-bound Ca(2+)-sensing protein, which is a structural component of the ER-plasma membrane junctions. Isoform 8 regulates the activity of Ca(+2) released-activated Ca(+2) (CRAC) channels in T-cells. {ECO:0000269|PubMed:22586105}. Related diseases Facial dysmorphism, lens dislocation, anterior segment abnormalities, and spontaneous filtering blebs (FDLAB) [MIM:601552]: A syndrome characterized by dislocated crystalline lenses and anterior segment abnormalities in association with a distinctive facies involving flat cheeks and a beaked nose. Some affected individuals develop highly unusual non-traumatic conjunctival cysts (filtering blebs). {ECO:0000269|PubMed:24768550}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00128; DB00139 Interacts with O43681; O95994; P02818; P01037; Q5JRM2; P81605; P60022; Q8N5I4; Q14204; Q6UWR7; Q8NFU4; Q14512; Q9Y680; P22466; P01350; O43681; Q8NBJ4; Q8TDV5; Q96SL4; Q8TED1; O15499; Q02747; P42858; Q9HBE4; Q9Y5Q6; Q9NZI2-2; Q9Y2W7; Q6PIL6; Q92876; P80188; P30533; P01374; Q9NX40; Q6UW60; P04085-2; Q9NWW9; Q9H8W4; Q59EV6; Q96NZ9; A5D903; P02812; P01270; P21246; Q15293; Q8NC24; O60930; P0DMR2; P34741; Q15428; Q96EQ0; Q3SXP7; Q86UW2; P58511; Q96E16; P08294; P00995; Q9NRX6; Q9Y320; O14763; A0A384ME17; Q96J42; O75310; Q8WWY7; Q8TCV5; O60844; A0A087WZY1; A0A1U9X8X8; P42858 EC number EC 1.14.11.16 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Dioxygenase; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Iron; Membrane; Metal-binding; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Sarcoplasmic reticulum; Signal-anchor; TPR repeat; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 22760.8 Length 196 Aromaticity 0.1 Instability index 41.41 Isoelectric point 8.51 Charge (pH=7) 2.96 2D Binding mode Binding energy (Kcal/mol) -6.95 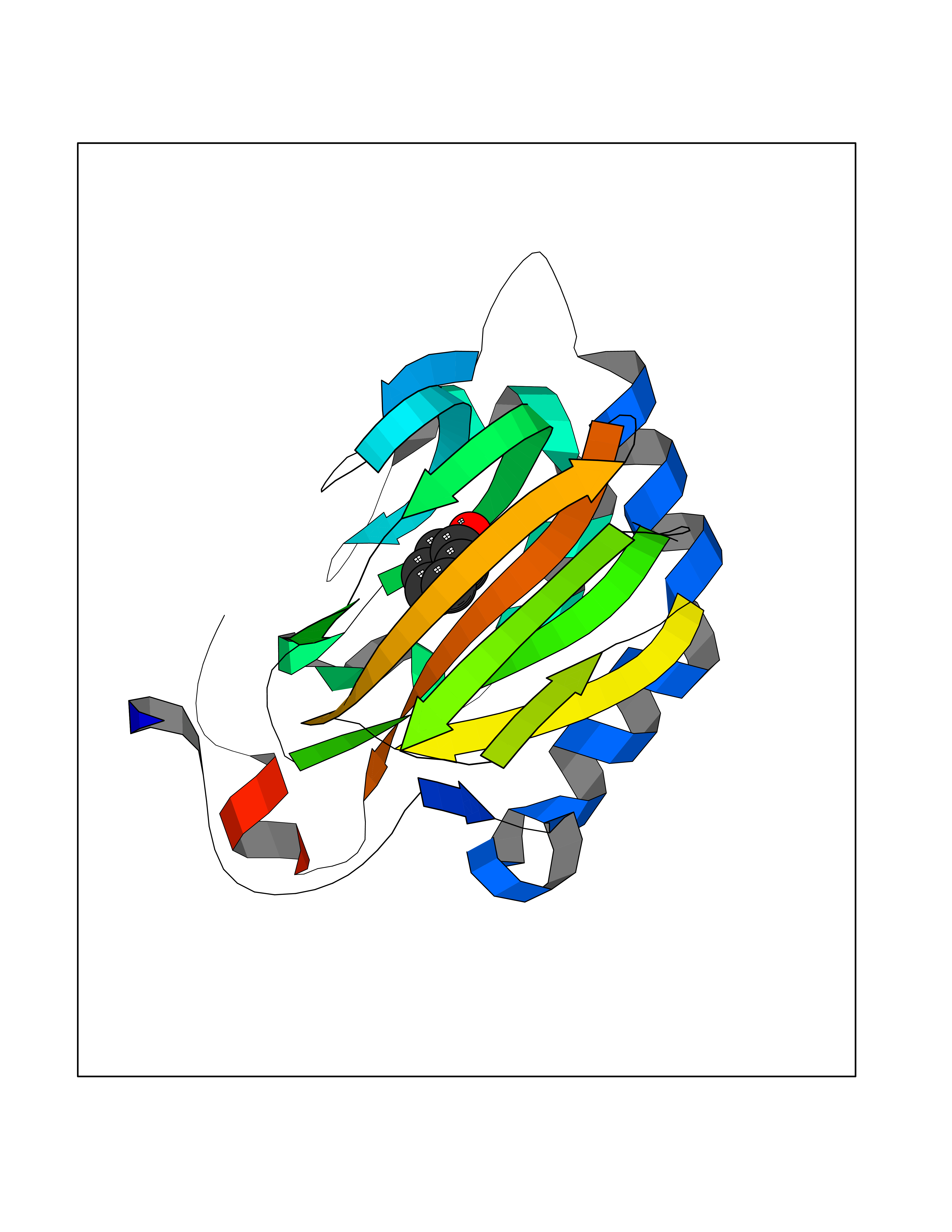 Molscript Map 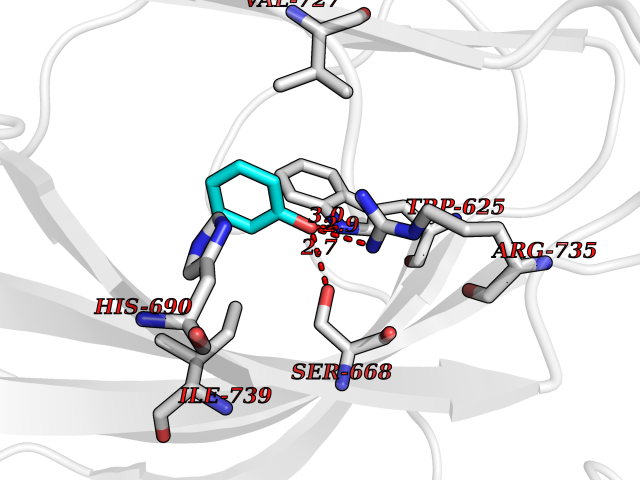 Pymol Map 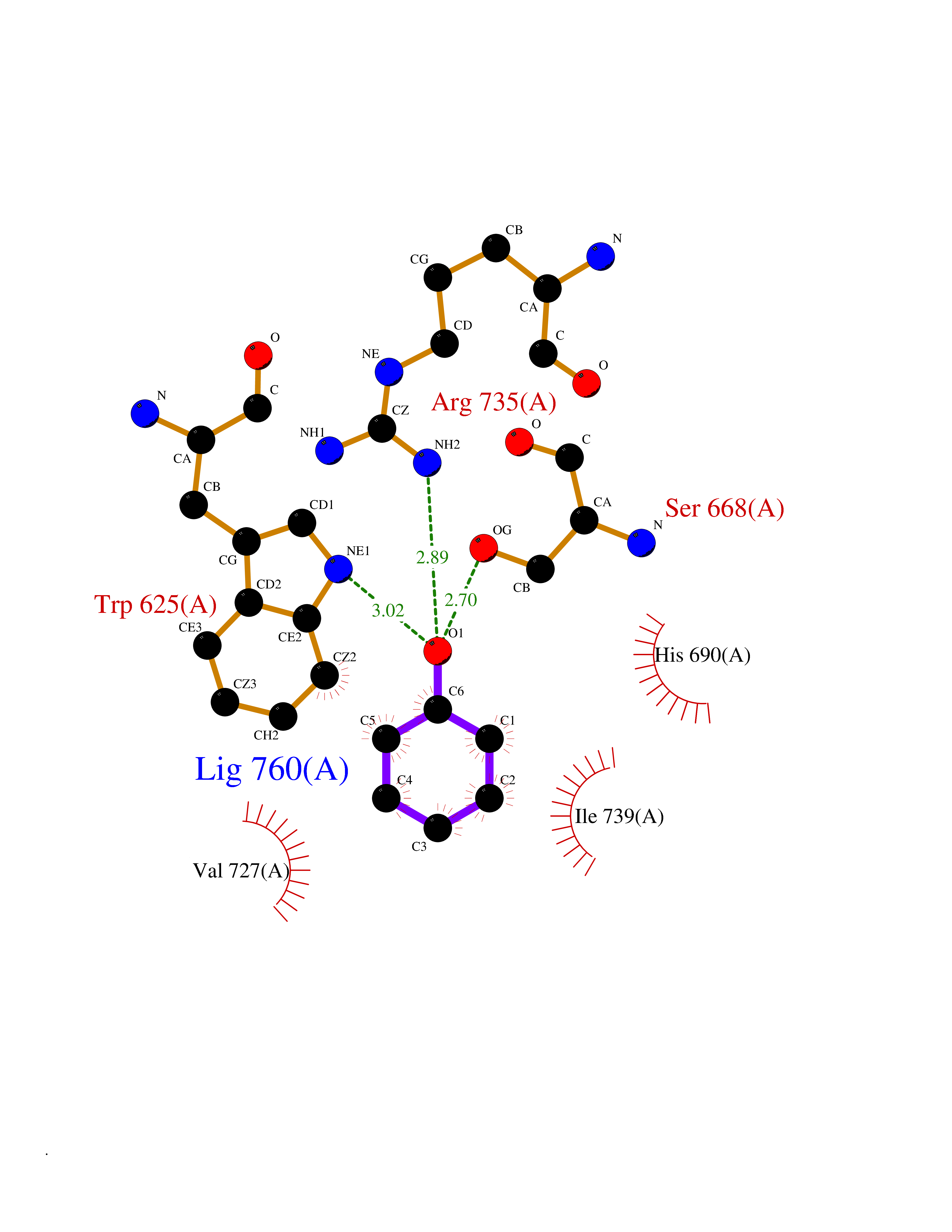 Ligplot Map 3D Binding mode Sequence SLYNVNGLKAQPWWTPKETGYTELVKSLERNWKLIRDEGLAVMDKAKGLFLPEDENLREKGDWSQFTLWQQGRRNENACKGAPKTCTLLEKFPETTGCRRGQIKYSIMHPGTHVWPHTGPTNCRLRMHLGLVIPKEGCKIRCANETKTWEEGKVLIFDDSFEHEVWQDASSFRLIFIVDVWHPELTPQQRRSLPAI Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 5.10 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -6.95 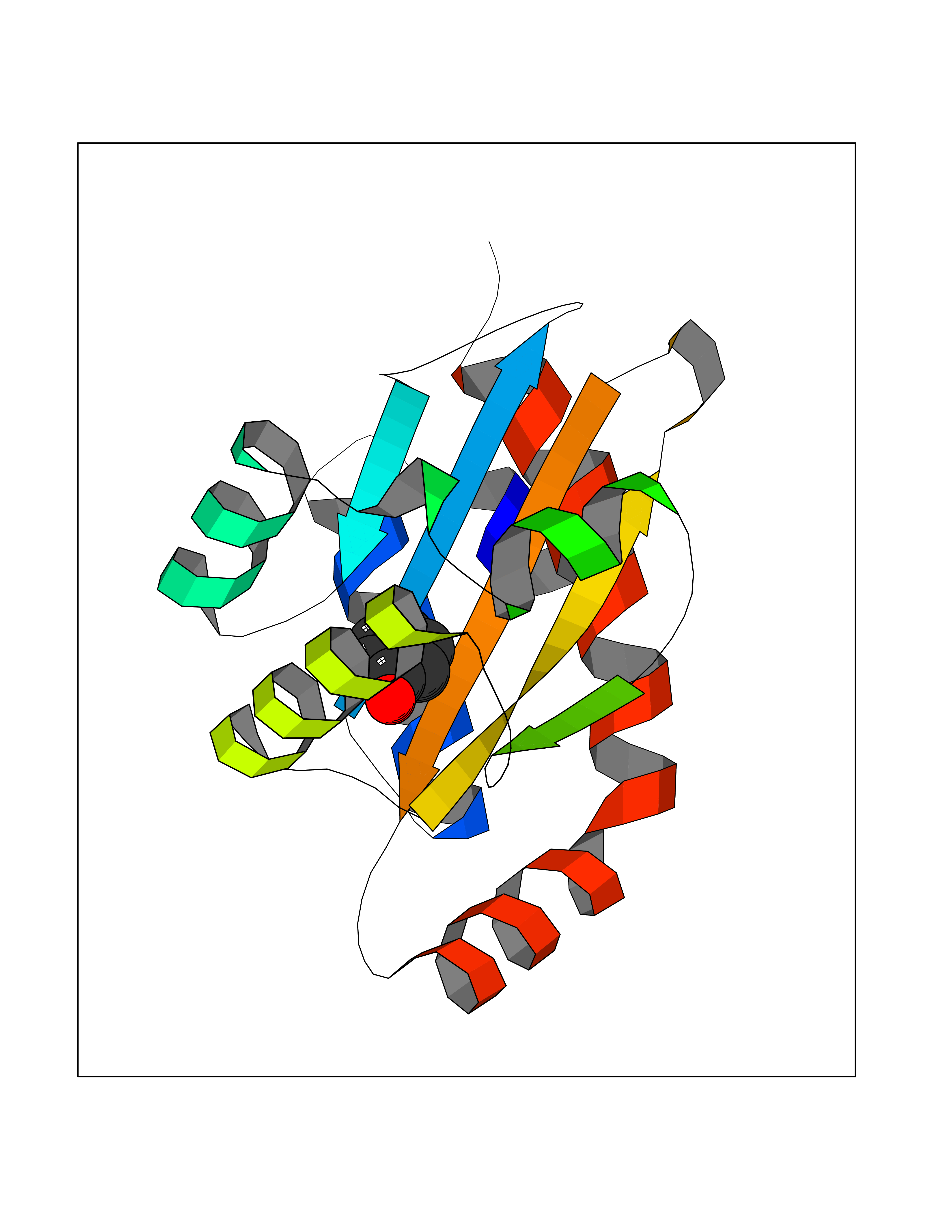 Molscript Map 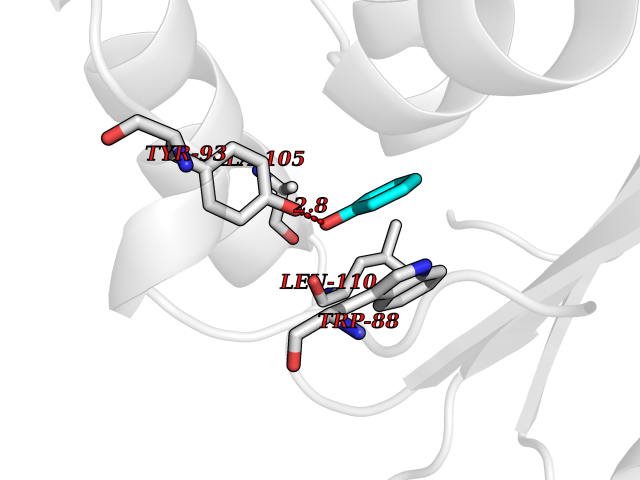 Pymol Map 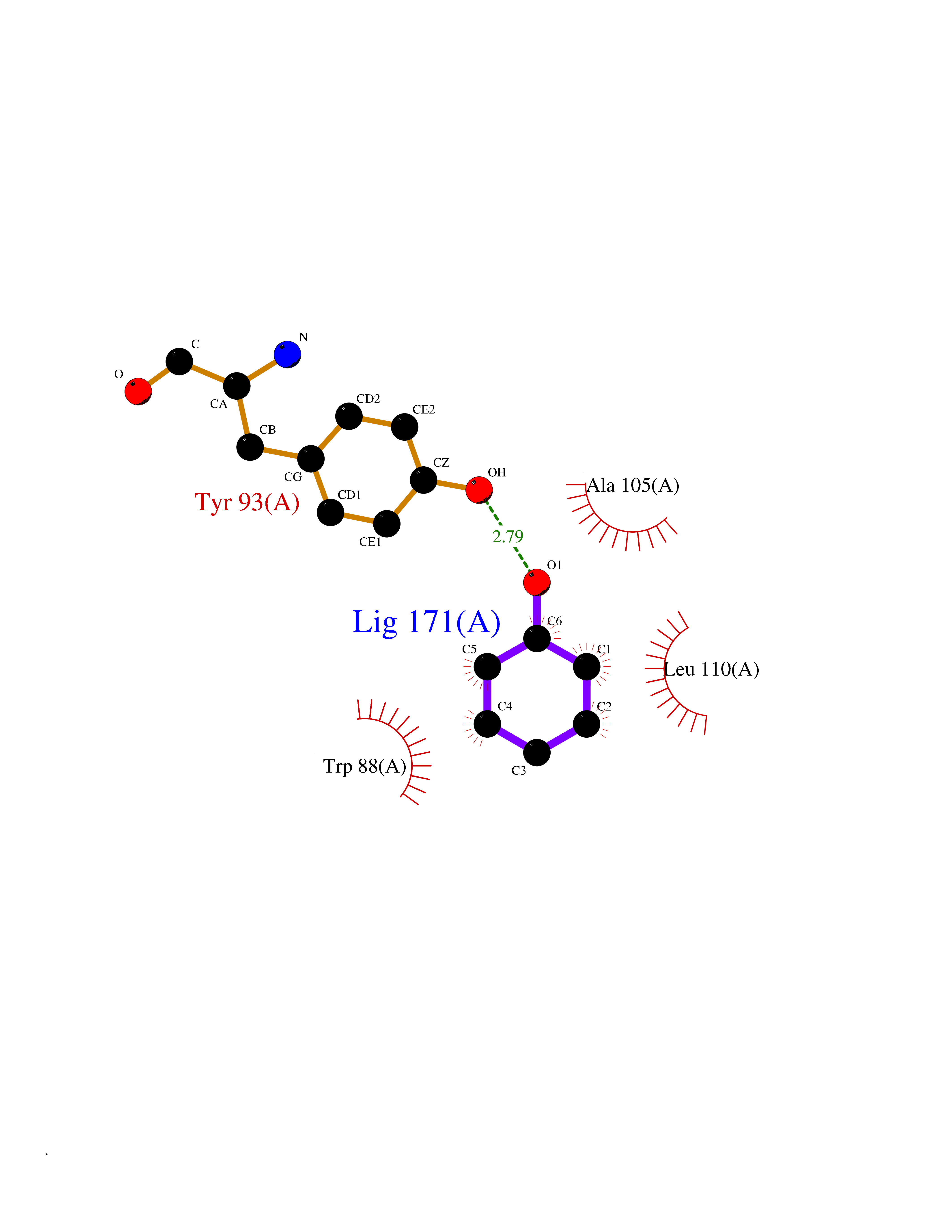 Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | ERK activator kinase 1 (MEK1) | 7M0U | 5.10 | |
Target general information Gen name MAP2K1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PRKMK1; Mitogen-activated protein kinase kinase 1; MKK1; MEK 1; MAPKK 1; MAPK/ERKkinase 1; MAPK/ERK kinase 1; MAP kinase kinase 1; Dual specificity mitogen-activated protein kinase kinase 1 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase subfamily Biochemical class Kinase Function Binding of extracellular ligands such as growth factors, cytokines and hormones to their cell-surface receptors activates RAS and this initiates RAF1 activation. RAF1 then further activates the dual-specificity protein kinases MAP2K1/MEK1 and MAP2K2/MEK2. Both MAP2K1/MEK1 and MAP2K2/MEK2 function specifically in the MAPK/ERK cascade, and catalyze the concomitant phosphorylation of a threonine and a tyrosine residue in a Thr-Glu-Tyr sequence located in the extracellular signal-regulated kinases MAPK3/ERK1 and MAPK1/ERK2, leading to their activation and further transduction of the signal within the MAPK/ERK cascade. Depending on the cellular context, this pathway mediates diverse biological functions such as cell growth, adhesion, survival and differentiation, predominantly through the regulation of transcription, metabolism and cytoskeletal rearrangements. One target of the MAPK/ERK cascade is peroxisome proliferator-activated receptor gamma (PPARG), a nuclear receptor that promotes differentiation and apoptosis. MAP2K1/MEK1 has been shown to export PPARG from the nucleus. The MAPK/ERK cascade is also involved in the regulation of endosomal dynamics, including lysosome processing and endosome cycling through the perinuclear recycling compartment (PNRC), as well as in the fragmentation of the Golgi apparatus during mitosis. Dual specificity protein kinase which acts as an essential component of the MAP kinase signal transduction pathway. Related diseases Cardiofaciocutaneous syndrome 3 (CFC3) [MIM:615279]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. Distinctive features of CFC3 include macrostomia and horizontal shape of palpebral fissures. {ECO:0000269|PubMed:16439621, ECO:0000269|PubMed:18042262}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melorheostosis, isolated (MEL) [MIM:155950]: A sclerosing bone disorder characterized by hyperostosis of the cortex of tubular bones, frequently involving one limb. The lesions may be accompanied by abnormalities of adjacent soft tissue, joint contractures, sclerodermatous skin lesions, muscle atrophy, or hemangioma. {ECO:0000269|PubMed:29643386}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06892; DB07046; DB08208; DB03115; DB11967; DB06616; DB05239; DB02152; DB07101; DB08130; DB14904; DB11689; DB08911 Interacts with Q8N9N5; Q8N9N5-2; Q9NR09; P15056; Q9Y297; O15519-1; P28482; P27361; Q13526; Q9H8W4; P04049; Q8WWU5-7; Q86Y07; Q86Y07-1; P46937 EC number EC 2.7.12.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cardiomyopathy; Cytoplasm; Cytoskeleton; Direct protein sequencing; Disease variant; Ectodermal dysplasia; Intellectual disability; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID B Molecular weight (Da) 34785.9 Length 311 Aromaticity 0.07 Instability index 46.58 Isoelectric point 6.29 Charge (pH=7) -2.54 2D Binding mode Binding energy (Kcal/mol) -6.96 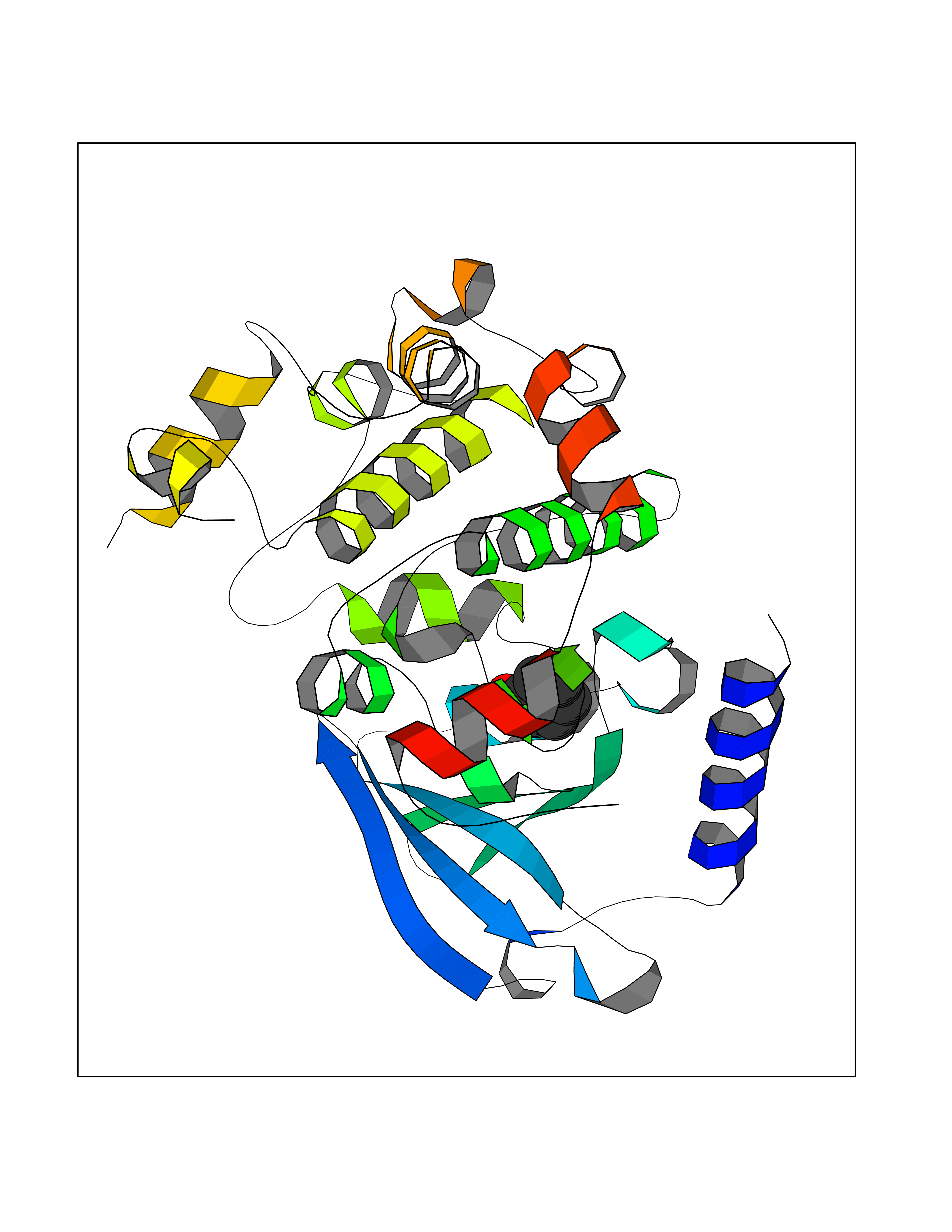 Molscript Map 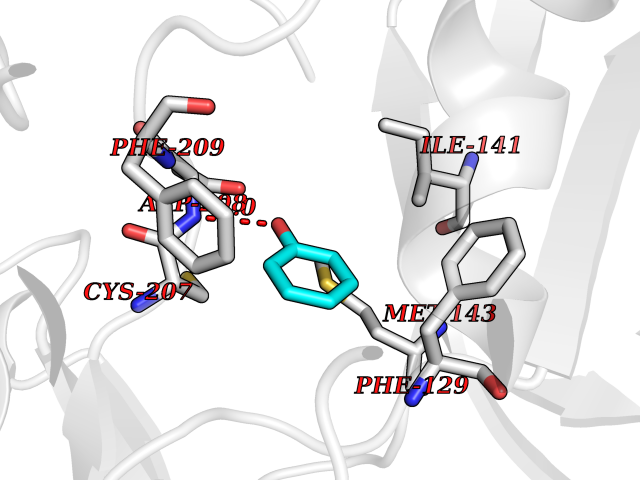 Pymol Map 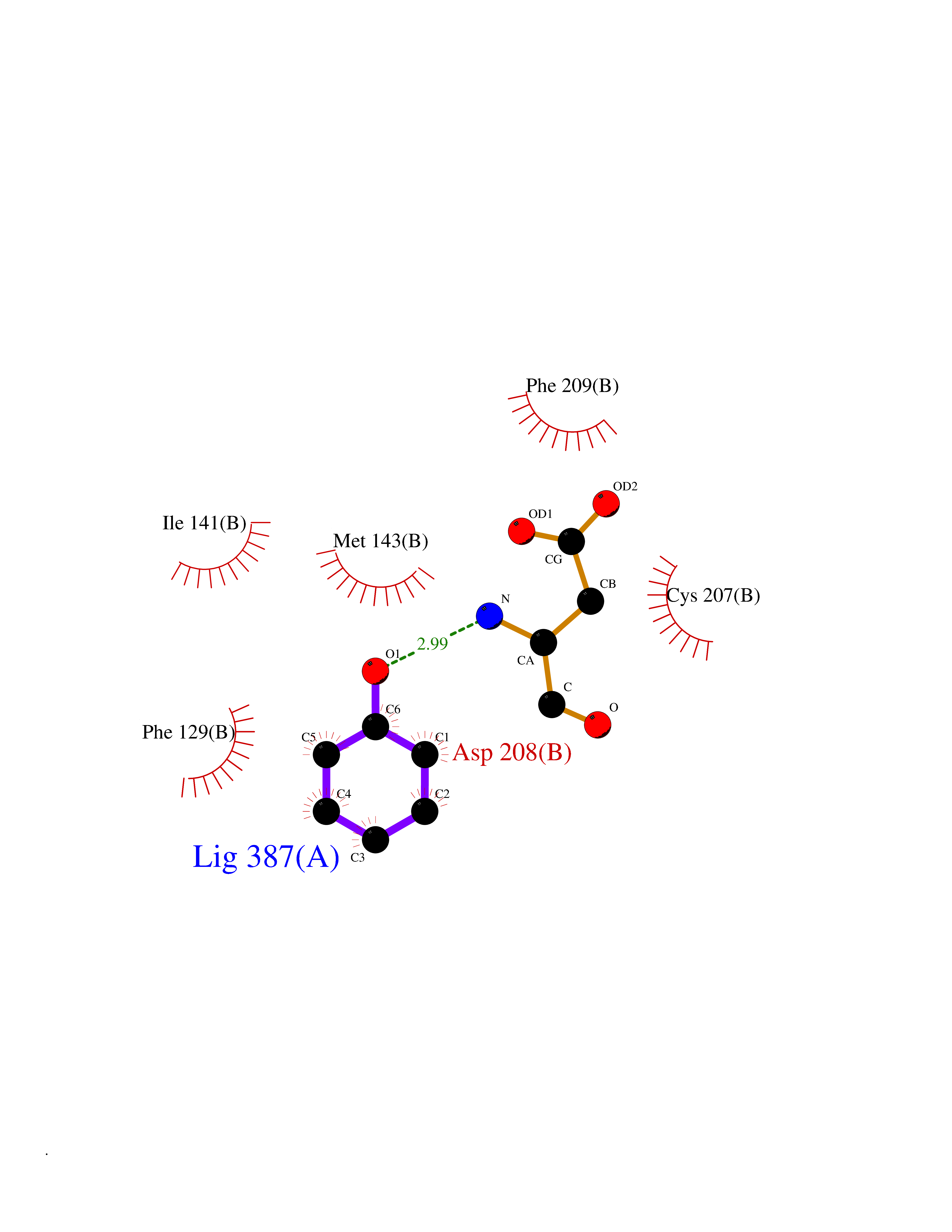 Ligplot Map 3D Binding mode Sequence DEQQRKRLEAFLTQKQKVGELKDDDFEKISELGAGNGGVVFKVSHKPSGLVMARKLIHLEIKPAIRNQIIRELQVLHECNSPYIVGFYGAFYSDGEISICMEHMDGGSLDQVLKKAGRIPEQILGKVSIAVIKGLTYLREKHKIMHRDVKPSNILVNSRGEIKLCDFGVSGQLIDAMANAFVGTRSYMSPERLQGTHYSVQSDIWSMGLSLVEMAVGRYPIPPPDAKELELMPMAIFELLDYIVNEPPPKLPSGVFSLEFQDFVNKCLIKNPAERADLKQLMVHAFIKRSDAEEVDFAGWLCSTIGLNQPS Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | DNA mismatch repair protein MSH2 (MSH2) | 3THX | 5.10 | |
Target general information Gen name MSH2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hMSH2; MutS protein homolog 2; Mismatch repair gene Msh2 Protein family DNA mismatch repair MutS family Biochemical class NA Function Forms two different heterodimers: MutS alpha (MSH2-MSH6 heterodimer) and MutS beta (MSH2-MSH3 heterodimer) which binds to DNA mismatches thereby initiating DNA repair. When bound, heterodimers bend the DNA helix and shields approximately 20 base pairs. MutS alpha recognizes single base mismatches and dinucleotide insertion-deletion loops (IDL) in the DNA. MutS beta recognizes larger insertion-deletion loops up to 13 nucleotides long. After mismatch binding, MutS alpha or beta forms a ternary complex with the MutL alpha heterodimer, which is thought to be responsible for directing the downstream MMR events, including strand discrimination, excision, and resynthesis. Recruits DNA helicase MCM9 to chromatin which unwinds the mismatch containg DNA strand. ATP binding and hydrolysis play a pivotal role in mismatch repair functions. The ATPase activity associated with MutS alpha regulates binding similar to a molecular switch: mismatched DNA provokes ADP-->ATP exchange, resulting in a discernible conformational transition that converts MutS alpha into a sliding clamp capable of hydrolysis-independent diffusion along the DNA backbone. This transition is crucial for mismatch repair. MutS alpha may also play a role in DNA homologous recombination repair. In melanocytes may modulate both UV-B-induced cell cycle regulation and apoptosis. Component of the post-replicative DNA mismatch repair system (MMR). Related diseases Lynch syndrome 1 (LYNCH1) [MIM:120435]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:10375096, ECO:0000269|PubMed:10386556, ECO:0000269|PubMed:10528862, ECO:0000269|PubMed:10573010, ECO:0000269|PubMed:10612836, ECO:0000269|PubMed:10777691, ECO:0000269|PubMed:10829038, ECO:0000269|PubMed:11726306, ECO:0000269|PubMed:11870161, ECO:0000269|PubMed:11920458, ECO:0000269|PubMed:12112654, ECO:0000269|PubMed:12124176, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12200596, ECO:0000269|PubMed:12362047, ECO:0000269|PubMed:12373605, ECO:0000269|PubMed:12655564, ECO:0000269|PubMed:12655568, ECO:0000269|PubMed:12658575, ECO:0000269|PubMed:14635101, ECO:0000269|PubMed:15046096, ECO:0000269|PubMed:15300854, ECO:0000269|PubMed:15342696, ECO:0000269|PubMed:15365995, ECO:0000269|PubMed:15613555, ECO:0000269|PubMed:15870828, ECO:0000269|PubMed:15896463, ECO:0000269|PubMed:15991316, ECO:0000269|PubMed:15996210, ECO:0000269|PubMed:16451135, ECO:0000269|PubMed:17101317, ECO:0000269|PubMed:17128465, ECO:0000269|PubMed:18561205, ECO:0000269|PubMed:18625694, ECO:0000269|PubMed:18781619, ECO:0000269|PubMed:18822302, ECO:0000269|PubMed:18951462, ECO:0000269|PubMed:21120944, ECO:0000269|PubMed:22102614, ECO:0000269|PubMed:22371642, ECO:0000269|PubMed:7874129, ECO:0000269|PubMed:8261515, ECO:0000269|PubMed:8700523, ECO:0000269|PubMed:8797773, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:9048925, ECO:0000269|PubMed:9240418, ECO:0000269|PubMed:9298827, ECO:0000269|PubMed:9311737, ECO:0000269|PubMed:9419403, ECO:0000269|PubMed:9559627, ECO:0000269|PubMed:9621522, ECO:0000269|PubMed:9718327, ECO:0000269|PubMed:9889267}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muir-Torre syndrome (MRTES) [MIM:158320]: Rare autosomal dominant disorder characterized by sebaceous neoplasms and visceral malignancy. {ECO:0000269|PubMed:7713503}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Endometrial cancer (ENDMC) [MIM:608089]: A malignancy of endometrium, the mucous lining of the uterus. Most endometrial cancers are adenocarcinomas, cancers that begin in cells that make and release mucus and other fluids. {ECO:0000305|PubMed:11306449, ECO:0000305|PubMed:21642682}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Mismatch repair cancer syndrome 2 (MMRCS2) [MIM:619096]: An autosomal recessive form of mismatch repair cancer syndrome, a childhood cancer predisposition syndrome encompassing a broad tumor spectrum. This includes hematological malignancies, central nervous system tumors, Lynch syndrome-associated malignancies such as colorectal tumors as well as multiple intestinal polyps, embryonic tumors and rhabdomyosarcoma. Multiple cafe-au-lait macules, a feature reminiscent of neurofibromatosis type 1, are often found as first manifestation of the underlying cancer. {ECO:0000269|PubMed:12549480, ECO:0000269|PubMed:16372347}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Colorectal cancer (CRC) [MIM:114500]: A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history. {ECO:0000269|PubMed:12792735, ECO:0000269|PubMed:14504054, ECO:0000269|PubMed:15996210, ECO:0000269|PubMed:9559627}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q92624; Q9UQ84-1; P09429; P20585; P52701; Q8IY92; P39875 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Chromosome; Disease variant; DNA damage; DNA repair; DNA-binding; Hereditary nonpolyposis colorectal cancer; Isopeptide bond; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Tumor suppressor; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 97883.4 Length 868 Aromaticity 0.08 Instability index 36.78 Isoelectric point 5.79 Charge (pH=7) -10.09 2D Binding mode Binding energy (Kcal/mol) -6.96 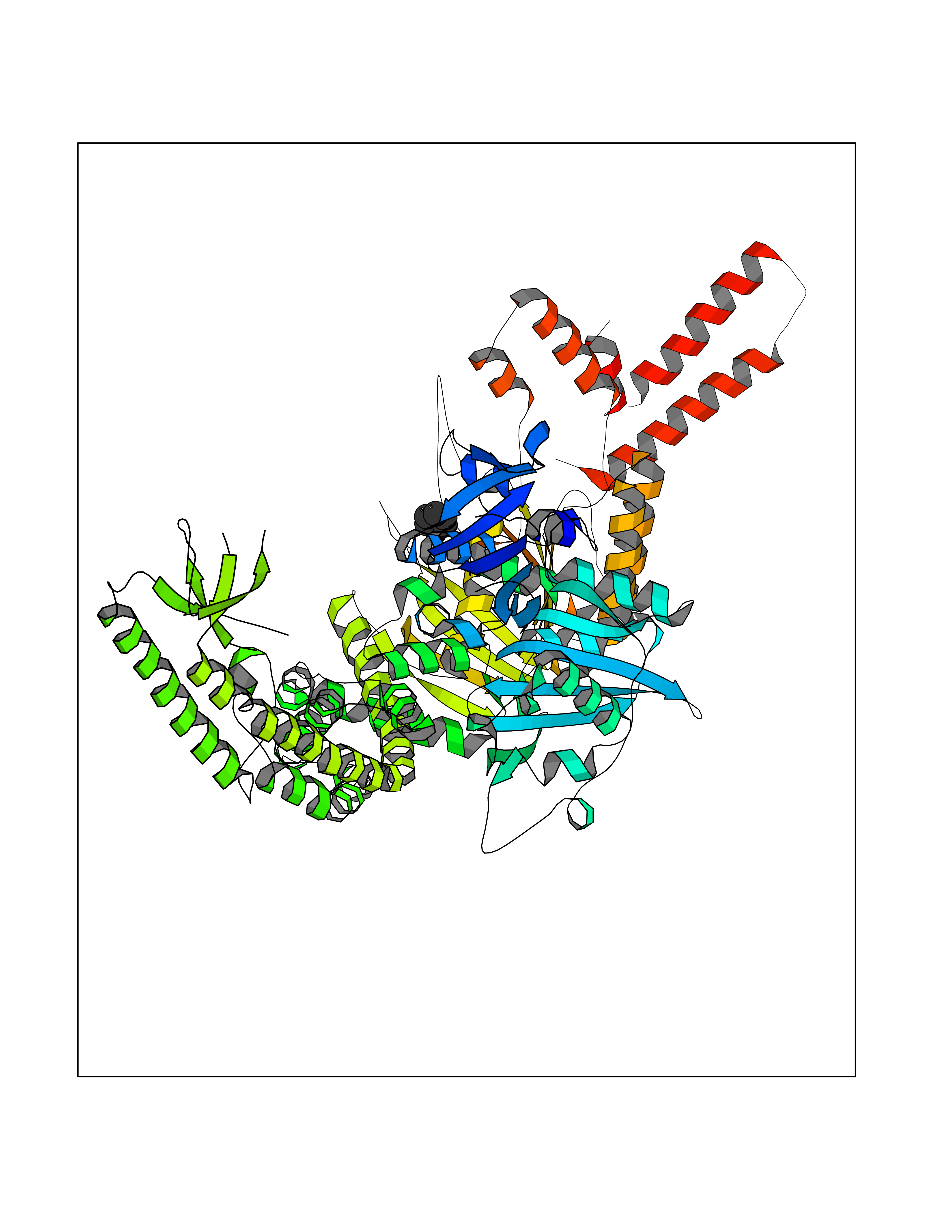 Molscript Map 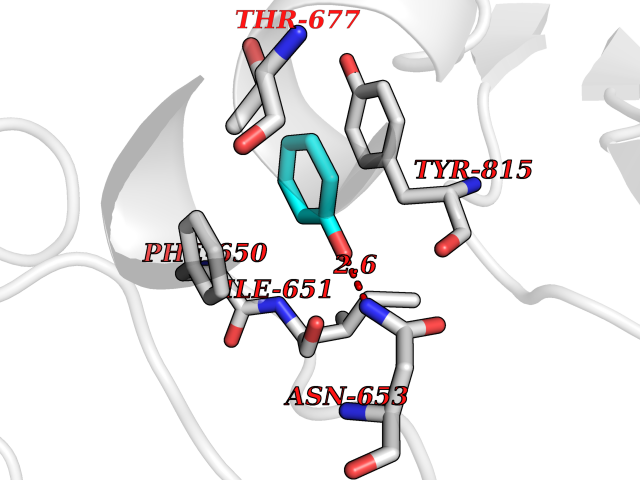 Pymol Map 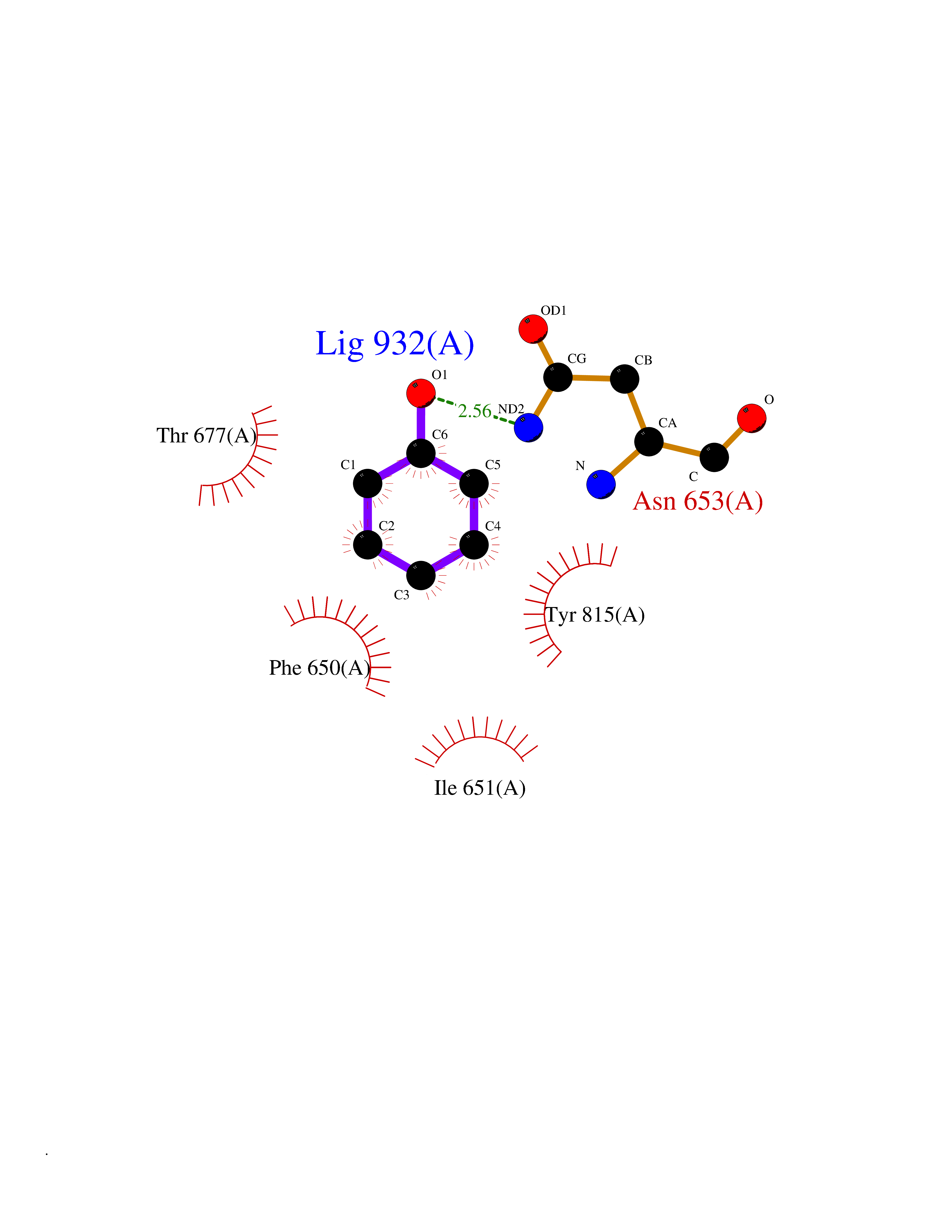 Ligplot Map 3D Binding mode Sequence ESAAEVGFVRFFQGMPEKPTTTVRLFDRGDFYTAHGEDALLAAREVFKTQGVIKYMGPAGAKNLQSVVLSKMNFESFVKDLLLVRQYRVEVYKNRASKENDWYLAYKASPGNLSQFEDILFIGVVGVKMSAVDGQRQVGVGYVDSIQRKLGLCEFPDNDQFSNLEALLIQIGPKECVLPGGETAGDMGKLRQIIQRGGILITERKKADFSTKDIYQDLNRLLKGKKGEQMNSAVLPEMENQVAVSSLSAVIKFLELLSDDSNFGQFELTTFDFSQYMKLDIAAVRALNLFQQSLAALLNKCKTPQGQRLVNQWIKQPLMDKNRIEERLNLVEAFVEDAELRQTLQEDLLRRFPDLNRLAKKFQRQAANLQDCYRLYQGINQLPNVIQALEKHEGKHQKLLLAVFVTPLTDLRSDFSKFQEMIETTLDMDQVENHEFLVKPSFDPNLSELREIMNDLEKKMQSTLISAARDLGLDPGKQIKLDSSAGYYFRVTCKEEKVLRNNKNFSTVDIQGVKFTNSKLTSLNEEYTKNKTEYEEAQDAIVKEIVNISSGYVEPMQTLNDVLAQLDAVVSFAHVSNGAPVPYVRPAILEKGQGRIILKASRHACVEVQIAFIPNDVYFEKDKQMFHIITGPNMGGKSTYIRQTGVIVLMAQIGCFVPCESAEVSIVDCILARVGSTFMAEMLETASILRSATKDSLIIIDELGRGTSTYDGFGLAWAISEYIATKIGAFCMFATHFHELTALANQIPTVNNLHVTALTTEETLTMLYQVKKGVCDQSFGIHVAELANFPKHVIECAKQKALELEEFQYKCYLEREQGEKIIQEFLSKVKQMPFTEMSEENITIKLKQLKAEVIAKNNSFVNEIISRI Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | MAPK signal-integrating kinase 1 (MKNK1) | 5WVD | 5.10 | |
Target general information Gen name MKNK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mnk1; MAP kinase signal-integrating kinase 1 Protein family Protein kinase superfamily, CAMK Ser/Thr protein kinase family Biochemical class Protein kinase superfamily. CAMK Ser/Thr protein kinase family Function May play a role in the response to environmental stress and cytokines. Appears to regulate translation by phosphorylating EIF4E, thus increasing the affinity of this protein for the 7-methylguanosine-containing mRNA cap. Related diseases Defects in MELK are associated with some cancers, such as brain or breast cancers. Expression is dramatically increased in aggressive undifferentiated tumors, correlating with poor patient outcome in breast and brain cancers, suggesting a role in tumor-initiating cells and proliferation via its function in cell proliferation regulation. Drugs (DrugBank ID) DB12010 Interacts with P54253; Q03060-25; P42858; P28482; Q16539; Q96CV9 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Translation regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 27536.2 Length 241 Aromaticity 0.11 Instability index 50.42 Isoelectric point 6.02 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -6.96  Molscript Map 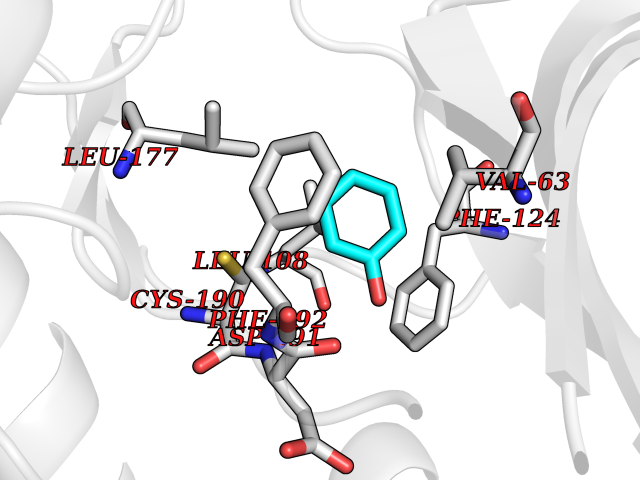 Pymol Map 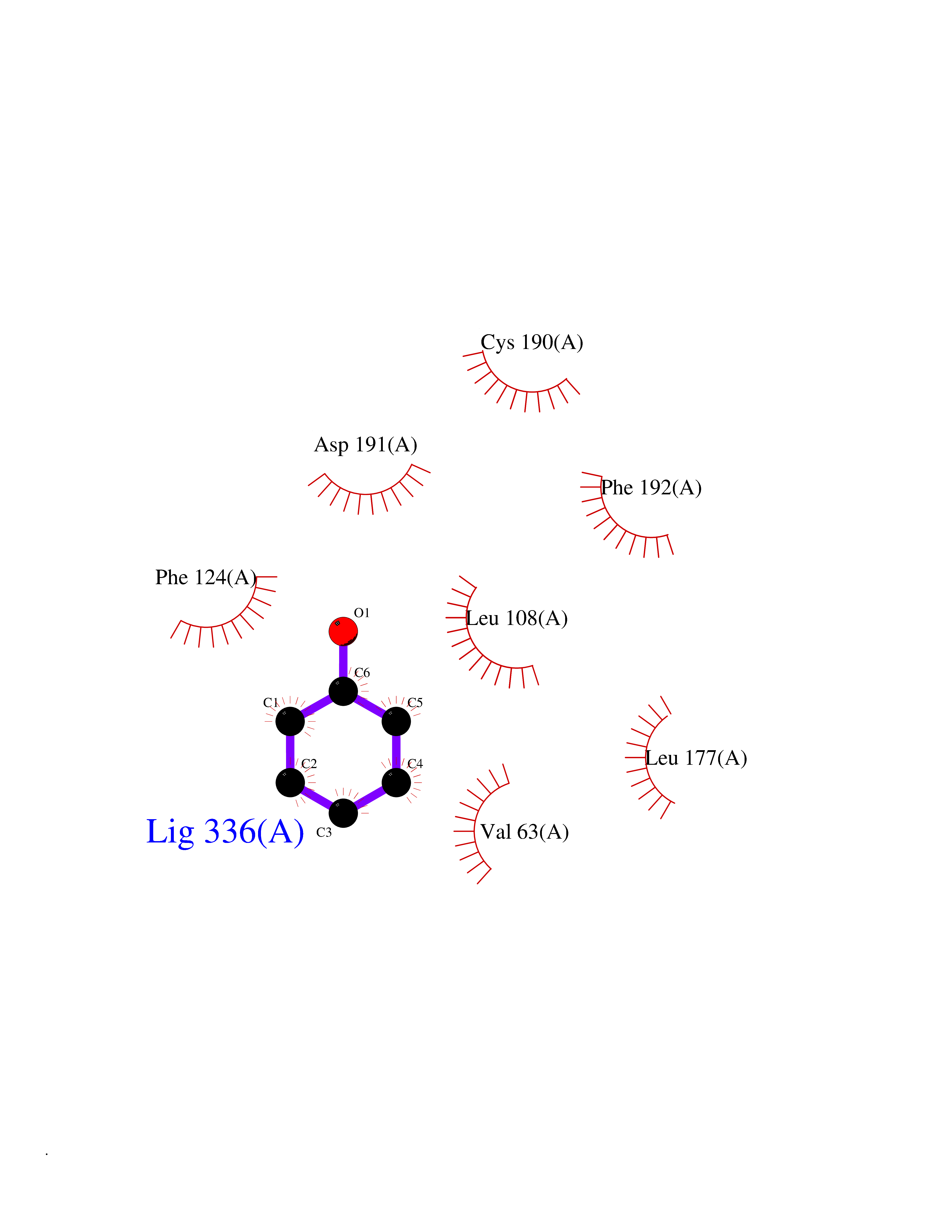 Ligplot Map 3D Binding mode Sequence PGKFEDMYKLTSELLGEGAYAKVQGAVSLQNGKEYAVKIIEKQAGHSRSRVFREVETLYQCQGNKNILELIEFFEDDTRFYLVFEKLQGGSILAHIQKQKHFNEREASRVVRDVAAALDFLHTKGIAHRDLKPENILCESPEKVSPVKICDFDLGSGYMAPEVVEVFTDQATFYDKRCDLWSLGVVLYIMLSGYPPFKYEFPDKDWAHISSEAKDLISKLLVRDAKQRLSAAQVLQHPWVQ Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Fatty acid-binding protein, intestinal | 3AKM | 5.09 | |
Target general information Gen name FABP2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FABPI Protein family Calycin superfamily, Fatty-acid binding protein (FABP) family Biochemical class Transport protein Function Fatty acid binding.Transporter activity. Related diseases Usher syndrome 3B (USH3B) [MIM:614504]: A syndrome characterized by progressive vision and hearing loss during early childhood. Some patients have the so-called 'Charles Bonnet syndrome,' involving decreased visual acuity and vivid visual hallucinations. USH is a genetically heterogeneous condition characterized by the association of retinitis pigmentosa with sensorineural deafness. Age at onset and differences in auditory and vestibular function distinguish Usher syndrome type 1 (USH1), Usher syndrome type 2 (USH2) and Usher syndrome type 3 (USH3). USH3 is characterized by postlingual, progressive hearing loss, variable vestibular dysfunction, and onset of retinitis pigmentosa symptoms, including nyctalopia, constriction of the visual fields, and loss of central visual acuity, usually by the second decade of life. {ECO:0000269|PubMed:22279524}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2W (CMT2W) [MIM:616625]: An autosomal dominant, axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2W patients manifest a peripheral neuropathy mainly affecting the lower limbs and resulting in gait difficulties and distal sensory impairment. Most patients also have upper limb involvement. {ECO:0000269|PubMed:22930593, ECO:0000269|PubMed:26072516, ECO:0000269|PubMed:29235198}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04557; DB09213; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB01050; DB08231; DB03796; DB01138 Interacts with O95994; Q9NYB0 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 15075.9 Length 131 Aromaticity 0.11 Instability index 32.01 Isoelectric point 6.88 Charge (pH=7) -0.09 2D Binding mode Binding energy (Kcal/mol) -6.94 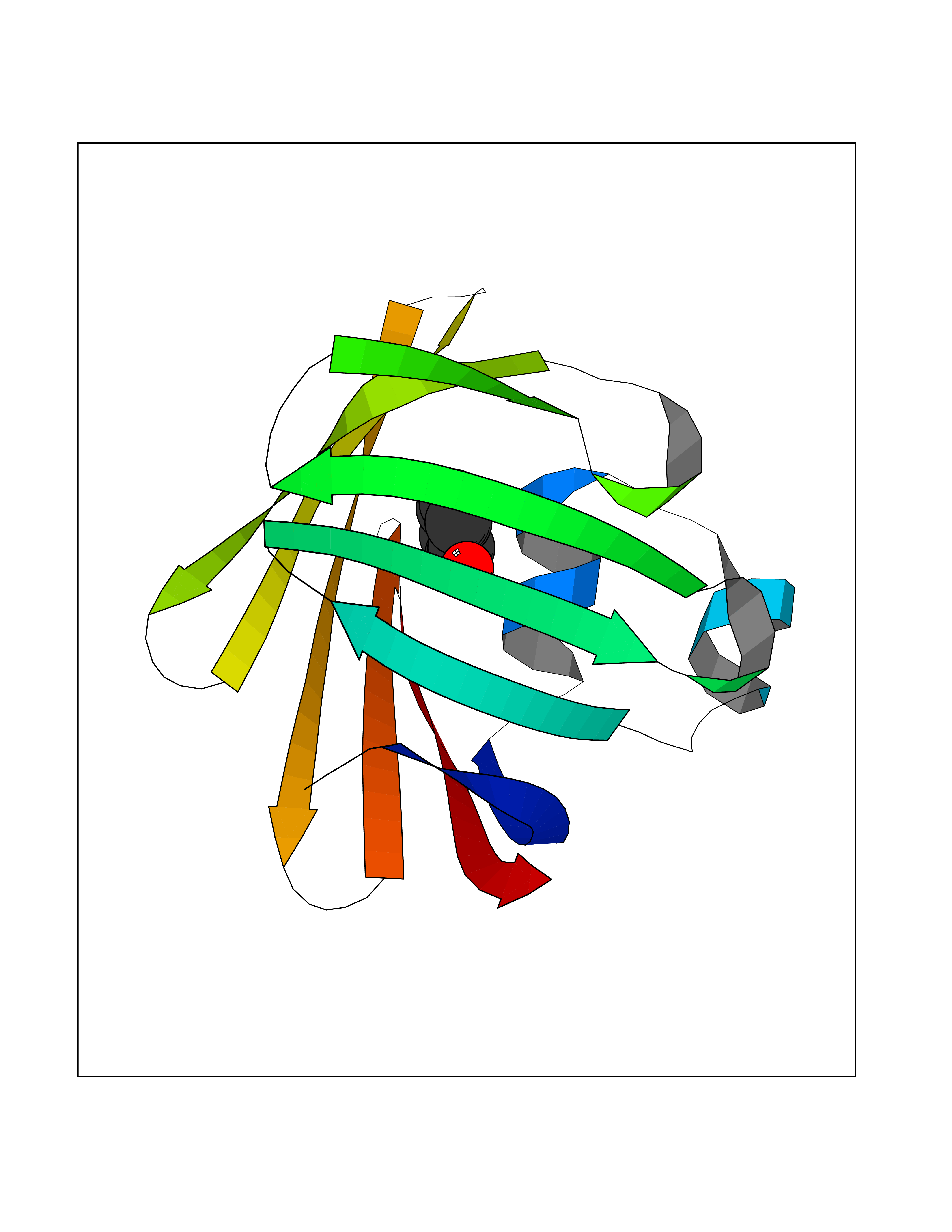 Molscript Map 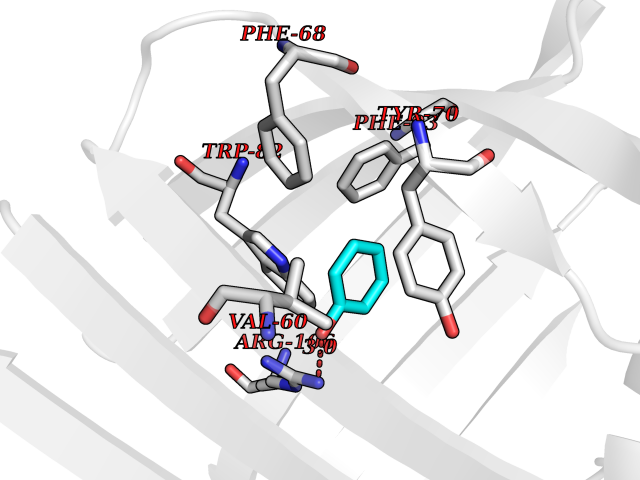 Pymol Map 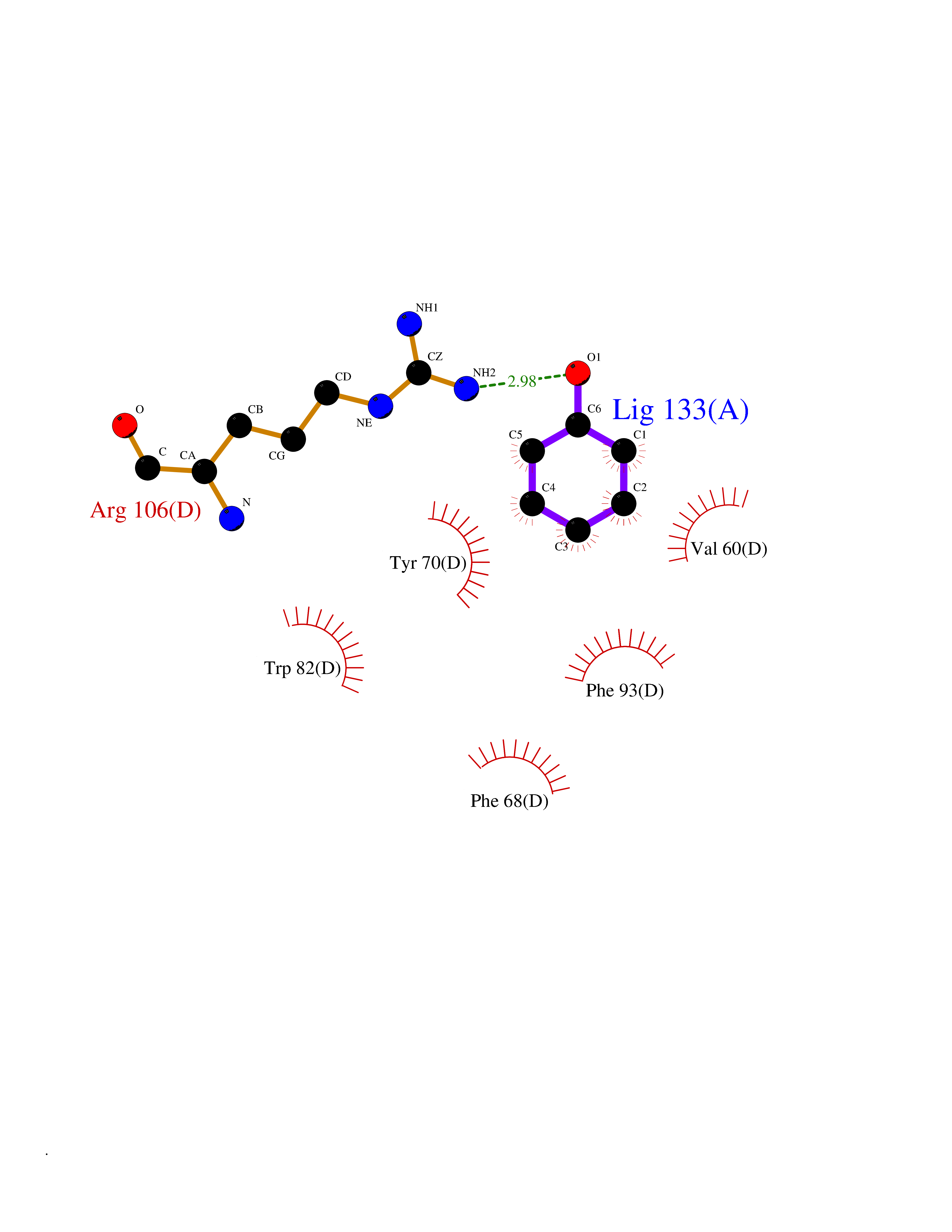 Ligplot Map 3D Binding mode Sequence AFDSTWKVDRSENYDKFMEKMGVNIVKRKLAAHDNLKLTITQEGNKFTVKESSAFRNIEVVFELGVTFNYNLADGTELRGTWSLEGNKLIGKFKRTDNGNELNTVREIIGDELVQTYVYEGVEAKRIFKKD Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 5.09 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -6.95  Molscript Map 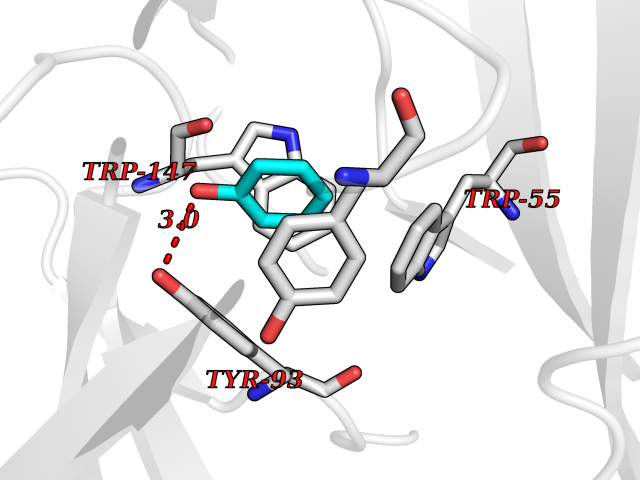 Pymol Map 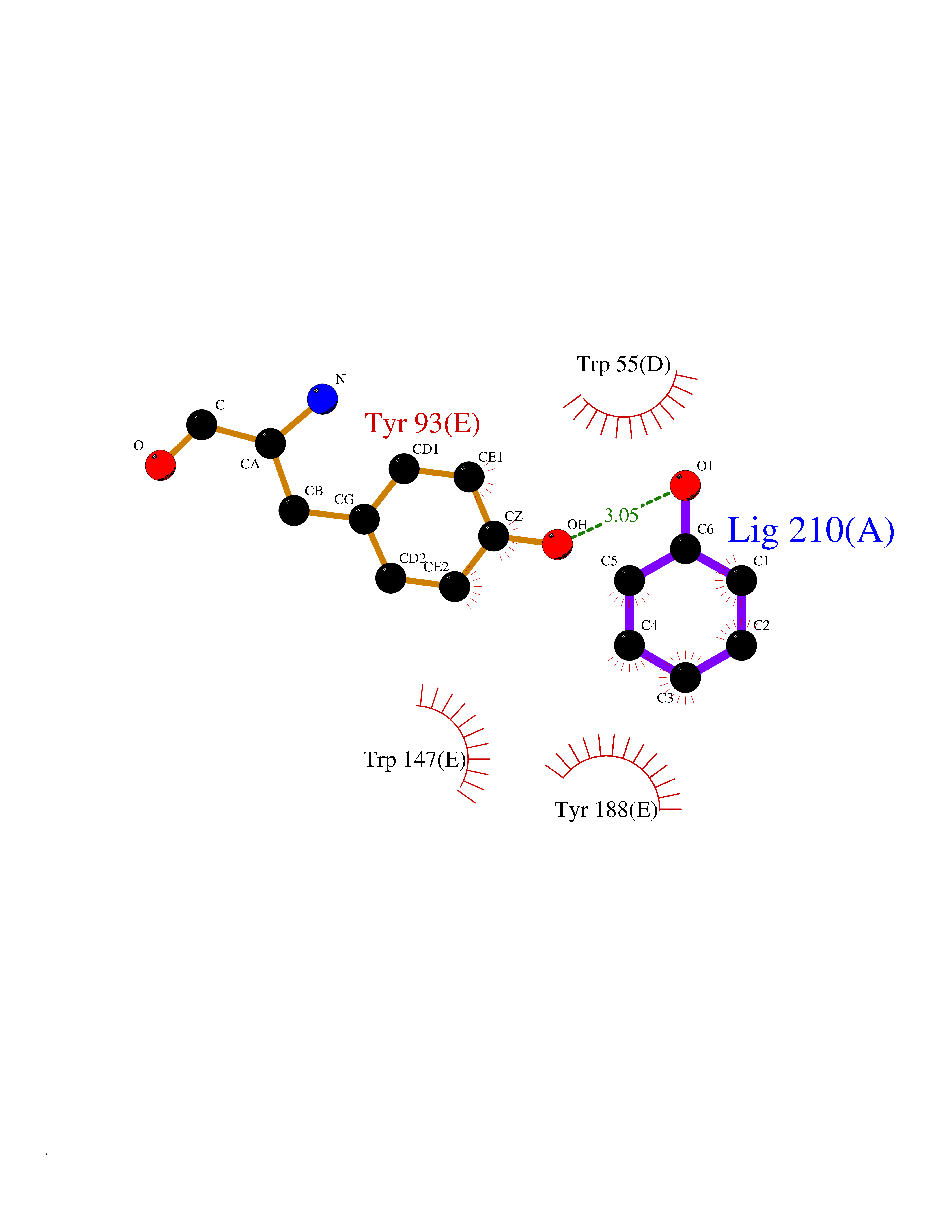 Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 5.09 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -6.94  Molscript Map 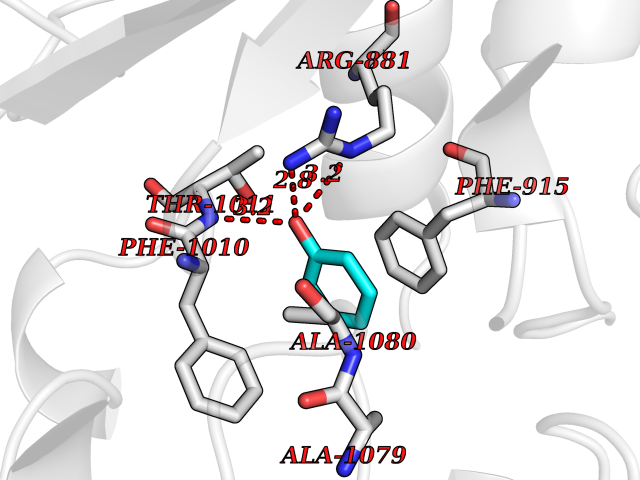 Pymol Map 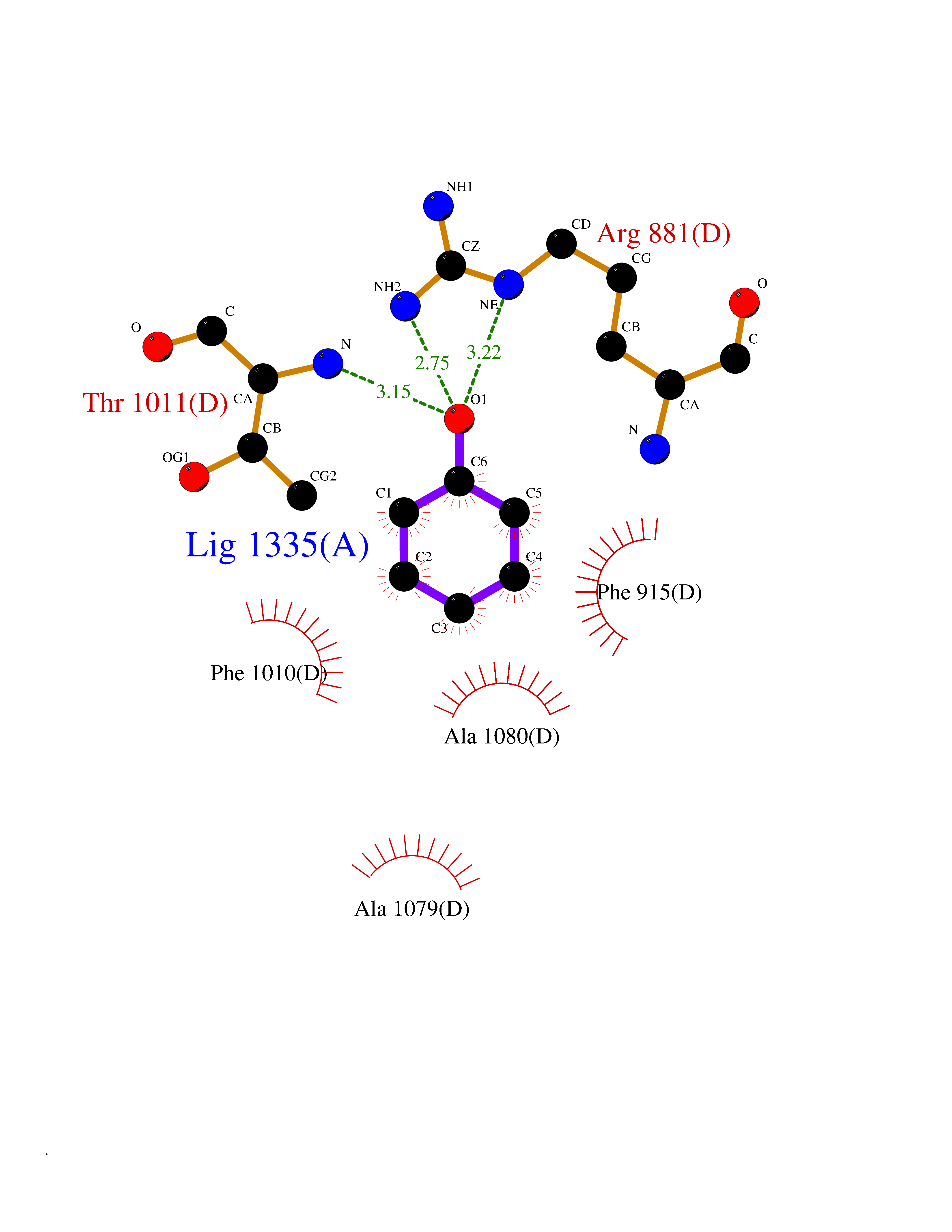 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Branched-chain-amino-acid transaminase 1 (BCAT1) | 2COI | 5.09 | |
Target general information Gen name BCAT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein ECA39; ECA39; Branched-chain-amino-acid aminotransferase, cytosolic; BCT1; BCAT(c) Protein family Class-IV pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Catalyzes the first reaction in the catabolism of the essential branched chain amino acids leucine, isoleucine, and valine. Related diseases Charcot-Marie-Tooth disease, dominant intermediate C (CMTDIC) [MIM:608323]: A form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. The dominant intermediate type C is characterized by clinical and pathologic features intermediate between demyelinating and axonal peripheral neuropathies, and motor median nerve conduction velocities ranging from 25 to 45 m/sec. {ECO:0000269|PubMed:16429158}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurologic, endocrine, and pancreatic disease, multisystem, infantile-onset 2 (IMNEPD2) [MIM:619418]: An autosomal recessive disorder with variable clinical manifestations and severity. Main features include cholestatic hepatitis, poor feeding, poor overall growth, and hypoglycemia apparent from infancy. Most patients have variable global developmental delay, sensorineural deafness, retinal abnormalities with visual defects, and hypotonia. Some patients have endocrine abnormalities. Brain imaging often shows dysmyelination, thin corpus callosum, cerebral atrophy, and white matter abnormalities. Death in early childhood may occur. {ECO:0000269|PubMed:27633801, ECO:0000269|PubMed:29232904, ECO:0000269|PubMed:30304524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in YARS1 may be the cause of proximal-predominant motor neuropathy. Affected individuals may develop tremors, cramping of hands, asymmetric weakness in the upper and lower extremities, and present with elevated creatine kinase levels. {ECO:0000269|PubMed:36307205}. Drugs (DrugBank ID) DB00996; DB00142; DB00167; DB00149; DB07544; DB00114; DB00161 Interacts with P55212; O75190-2; O14645; P22607; P06396; O14901; P13473-2; O75400-2 EC number EC 2.6.1.42 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Amino-acid biosynthesis; Aminotransferase; Branched-chain amino acid biosynthesis; Cytoplasm; Lipid metabolism; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40123.7 Length 356 Aromaticity 0.1 Instability index 32.34 Isoelectric point 5.39 Charge (pH=7) -9 2D Binding mode Binding energy (Kcal/mol) -6.94  Molscript Map 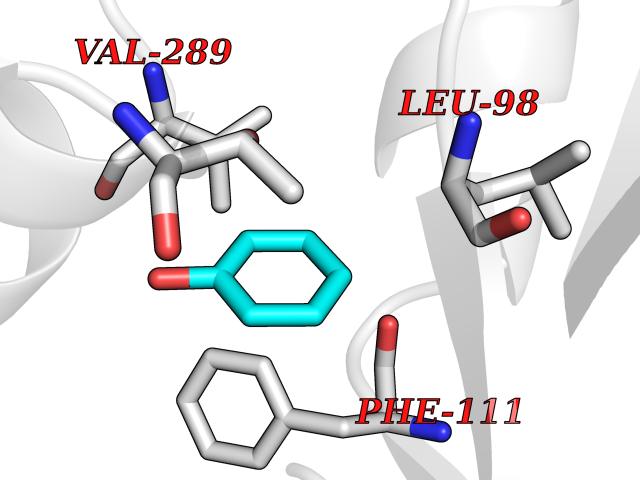 Pymol Map 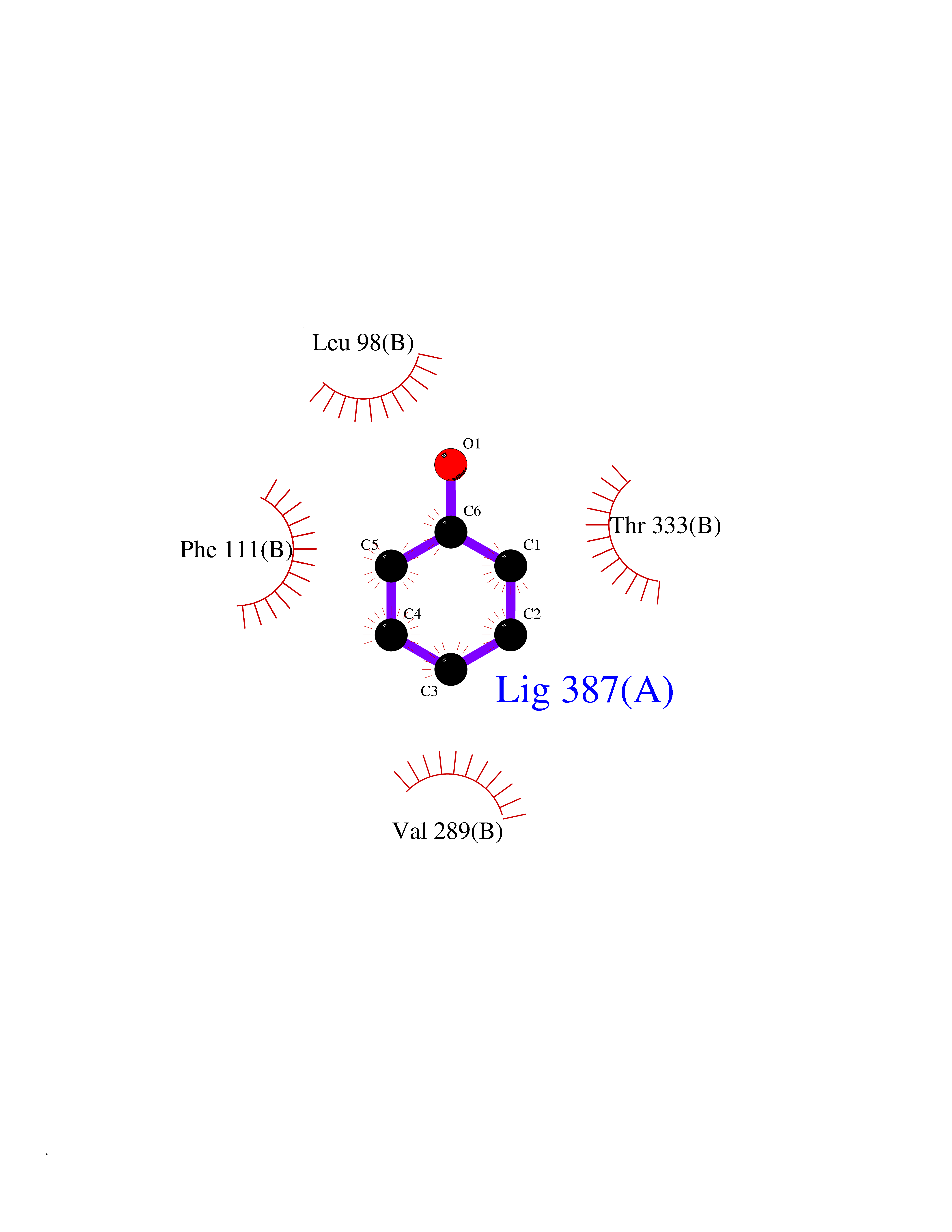 Ligplot Map 3D Binding mode Sequence TFKAKDLIVTPATILKEKPDPNLVFGTVFTDHMLTVEWSSEFGWEKPHIKPLQNLSLHPGSSALHYAVELFEGLKAFRGVDNKIRLFQPNLNMDRMYRSAVRATLPVFDKEELLECIQQLVKLDQEWVPYSTSASLYIRPTFIGTEPSLGVKKPTKALLFVLLSPVGPYFNPVSLWANPKYVRAWKGGTGDCKMGGNYGSSLFAQCEAVDNGCQQVLWLYGEDHQITEVGTMNLFLYWINEDGEEELATPPLDGIILPGVTRRCILDLAHQWGEFKVSERYLTMDDLTTALEGNRVREMFGSGTACVVCPVSDILYKGETIHIPTMENGPKLASRILSKLTDIQYGREERDWTIVL Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | 2-oxoglutarate dehydrogenase, mitochondrial | 3ERY | 5.09 | |
Target general information Gen name OGDH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Alpha-ketoglutarate dehydrogenase family Biochemical class Immune system Function Chaperone binding.Heat shock protein binding.Metal ion binding.Oxoglutarate dehydrogenase (NAD+) activity.Oxoglutarate dehydrogenase (succinyl-transferring) activity.Thiamine pyrophosphate binding. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00157; DB00313; DB09092 Interacts with P54253; P42858 EC number 1.2.4.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Calcium; Glycolysis; Isopeptide bond; Magnesium; Metal-binding; Mitochondrion; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Thiamine pyrophosphate; Transit peptide; Ubl conjugation Protein physicochemical properties Chain ID P,Q Molecular weight (Da) 18311.1 Length 158 Aromaticity 0.15 Instability index 37.26 Isoelectric point 5.64 Charge (pH=7) -2.98 2D Binding mode Binding energy (Kcal/mol) -6.94 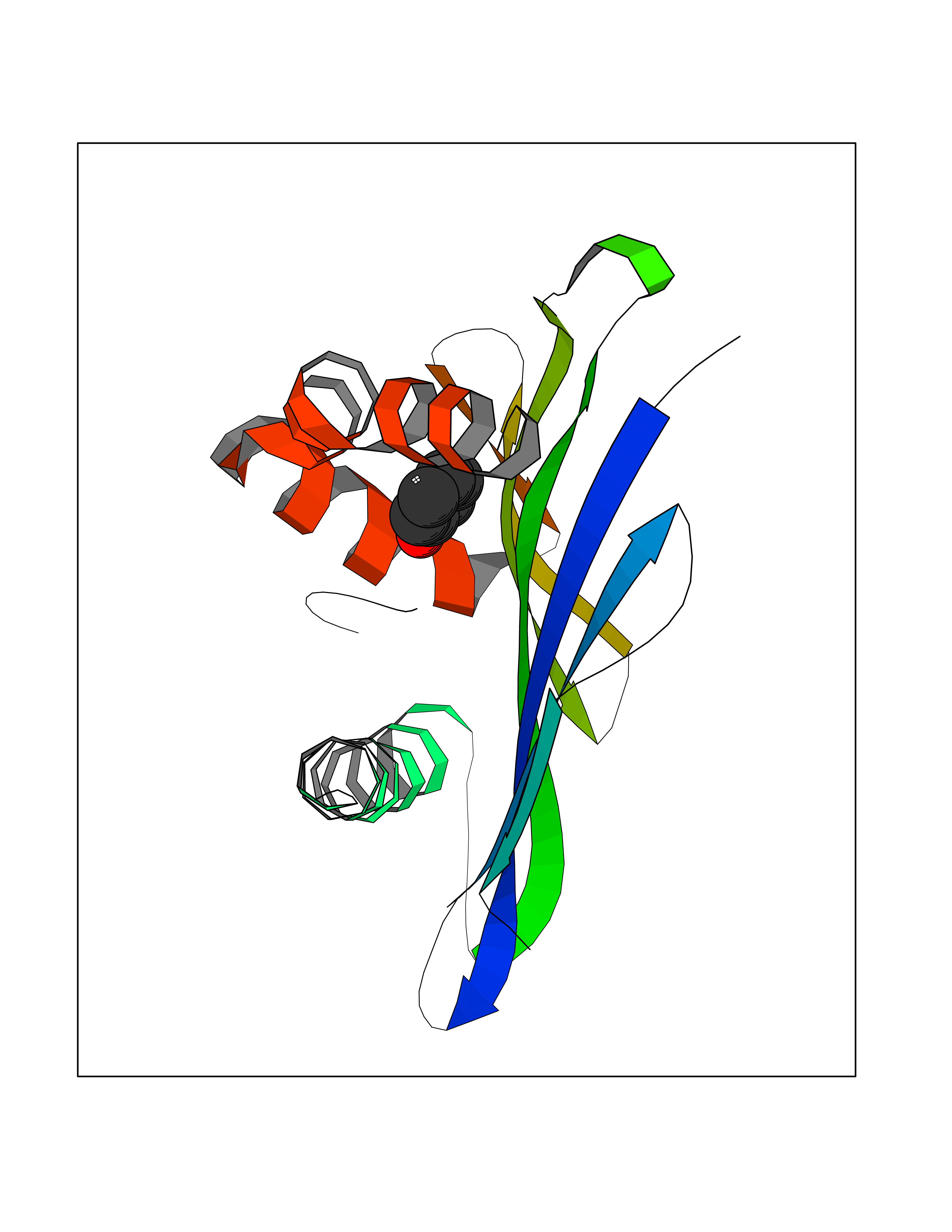 Molscript Map 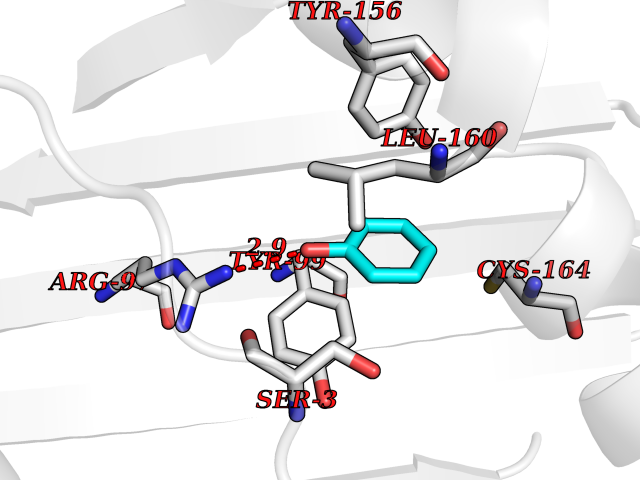 Pymol Map 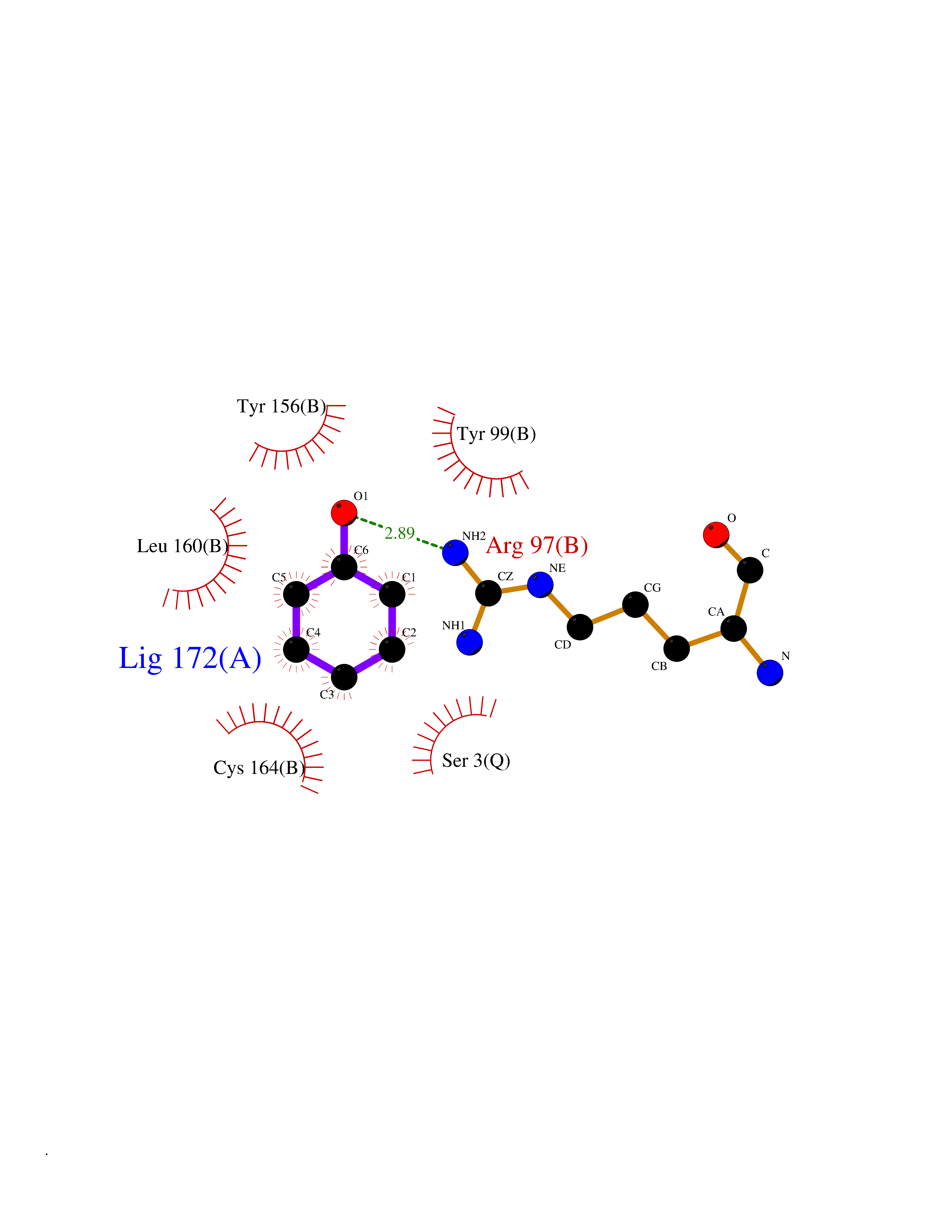 Ligplot Map 3D Binding mode Sequence QLSPFPFDLGPHSMRYYETATSRRGLGEPRYTSVGYVDDKEFVRFDSDARITQVAKGQEQWFRVNLRTLLGYYNQSAGGTHTLQRMYGCDVGSDGRLLRGYEQFAYDGCDYIALNEDLRTWTAADMAAQITRRKWEQAGAAEYYRAYLEGECVEWLHR Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | T-cell-specific kinase (ITK) | 4HCU | 5.09 | |
Target general information Gen name ITK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine kinase ITK; Inducible T cell kinase; EMT Protein family Protein kinase superfamily, Tyr protein kinase family, TEC subfamily Biochemical class Kinase Function Regulates the development, function and differentiation of conventional T-cells and nonconventional NKT-cells. When antigen presenting cells (APC) activate T-cell receptor (TCR), a series of phosphorylation lead to the recruitment of ITK to the cell membrane, in the vicinity of the stimulated TCR receptor, where it is phosphorylated by LCK. Phosphorylation leads to ITK autophosphorylation and full activation. Once activated, phosphorylates PLCG1, leading to the activation of this lipase and subsequent cleavage of its substrates. In turn, the endoplasmic reticulum releases calcium in the cytoplasm and the nuclear activator of activated T-cells (NFAT) translocates into the nucleus to perform its transcriptional duty. Phosphorylates 2 essential adapter proteins: the linker for activation of T-cells/LAT protein and LCP2. Then, a large number of signaling molecules such as VAV1 are recruited and ultimately lead to lymphokine production, T-cell proliferation and differentiation. Phosphorylates TBX21 at 'Tyr-530' and mediates its interaction with GATA3. Tyrosine kinase that plays an essential role in regulation of the adaptive immune response. Related diseases Lymphoproliferative syndrome 1 (LPFS1) [MIM:613011]: A rare immunodeficiency characterized by extreme susceptibility to infection with Epstein-Barr virus (EBV). Inadequate immune response to EBV can have a fatal outcome. Clinical features include splenomegaly, lymphadenopathy, anemia, thrombocytopenia, pancytopenia, recurrent infections. There is an increased risk for lymphoma. {ECO:0000269|PubMed:19425169}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010; DB06589; DB14924; DB02010; DB15035 Interacts with P04626; P48023; P08238; Q13094; P31947; P62258; P10686 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Adaptive immunity; ATP-binding; Cytoplasm; Direct protein sequencing; Disease variant; Immunity; Kinase; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; SH2 domain; SH3 domain; Transferase; Tyrosine-protein kinase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 30116.1 Length 263 Aromaticity 0.11 Instability index 37.47 Isoelectric point 5.03 Charge (pH=7) -11.73 2D Binding mode Binding energy (Kcal/mol) -6.94  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence WVIDPSELTFVQEIGSGQFGLVHLGYWLNKDKVAIKTIREGAMSEEDFIEEAEVMMKLSHPKLVQLYGVCLEQAPICLVFEFMEHGCLSDYLRTQRGLFAAETLLGMCLDVCEGMAYLEEACVIHRDLAARNCLVGENQVIKVSDFGMTRFVLDDQYTSSTGTKFPVKWASPEVFSFSRYSSKSDVWSFGVLMWEVFSEGKIPYENRSNSEVVEDISTGFRLYKPRLASTHVYQIMNHCWRERPEDRPAFSRLLRQLAEIAES Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 5.09 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -6.94  Molscript Map 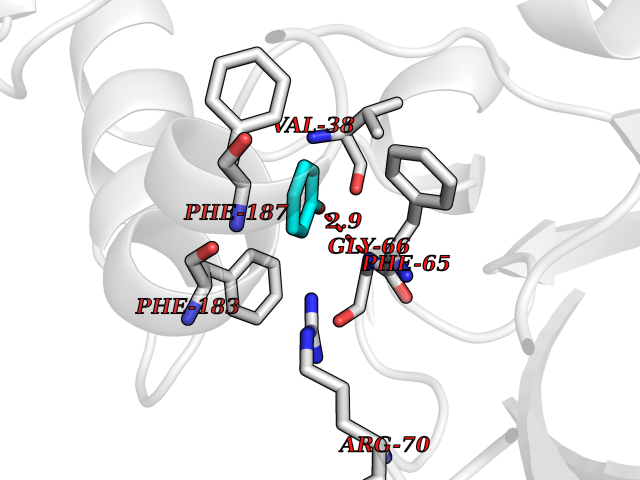 Pymol Map 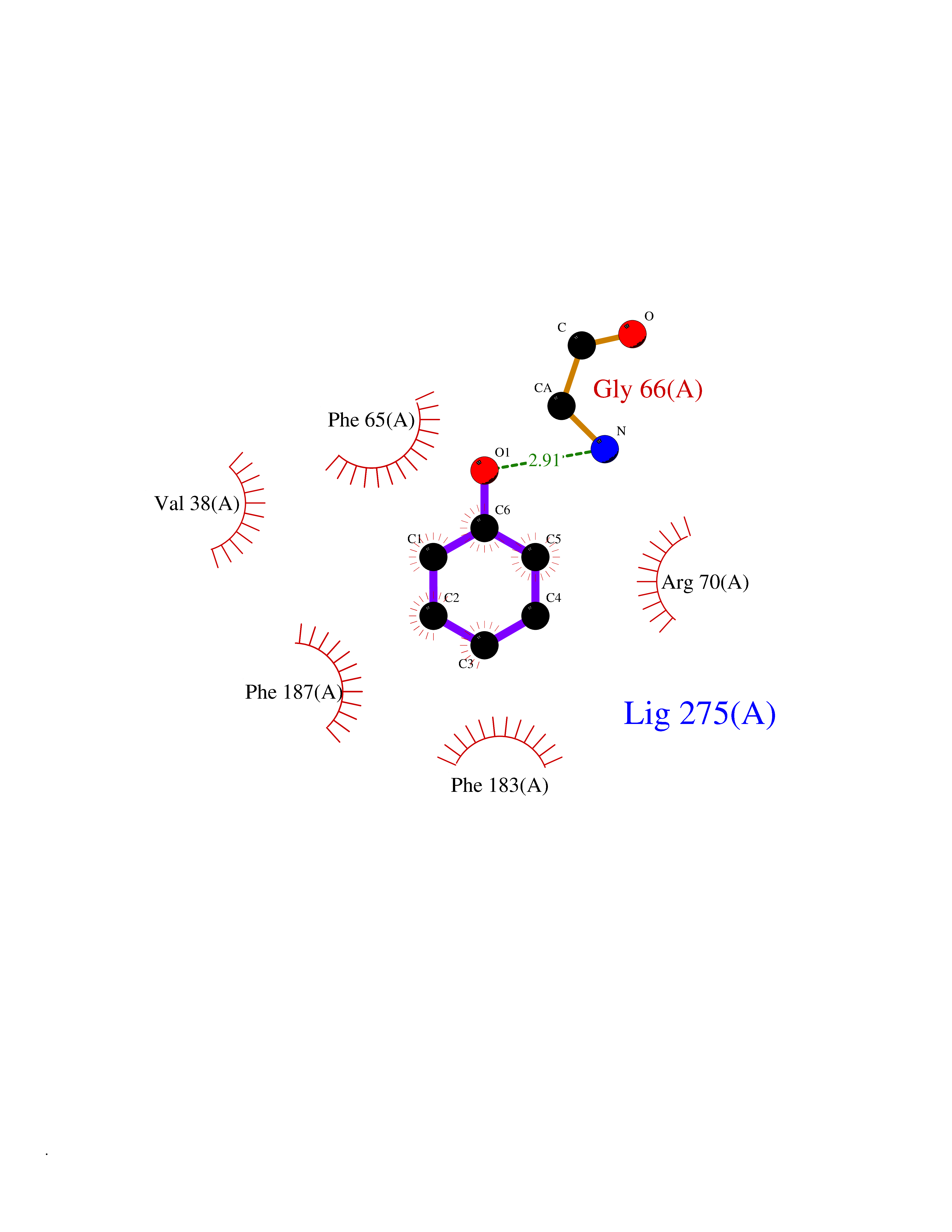 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Adrenergic receptor alpha-2A (ADRA2A) | 7EJ8 | 5.09 | |
Target general information Gen name ADRA2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Alpha-2AAR; Alpha-2A adrenoreceptor; Alpha-2A adrenoceptor; Alpha-2A adrenergic receptor; Alpha-2 adrenergic receptor subtype C10; ADRAR; ADRA2R Protein family G-protein coupled receptor 1 family, Adrenergic receptor subfamily, ADRA2A sub-subfamily Biochemical class GPCR rhodopsin Function The rank order of potency for agonists of this receptor is oxymetazoline > clonidine > epinephrine > norepinephrine > phenylephrine > dopamine > p-synephrine > p-tyramine > serotonin = p-octopamine. For antagonists, the rank order is yohimbine > phentolamine = mianserine > chlorpromazine = spiperone = prazosin > propanolol > alprenolol = pindolol. Alpha-2 adrenergic receptors mediate the catecholamine-induced inhibition of adenylate cyclase through the action of G proteins. Related diseases Lipodystrophy, familial partial, 8 (FPLD8) [MIM:620679]: An autosomal dominant form of partial lipodystrophy, a disorder characterized by abnormal subcutaneous fat distribution. FPLD8 patients show selective loss of subcutaneous adipose tissue from the limbs, beginning around 13 to 15 years of age, and abnormal accumulation of subcutaneous adipose tissue in the dorsal neck and face, as well as in the posterior thoracic and abdominal regions. The disorder is associated with metabolic abnormalities, including diabetes mellitus and hyperlipidemia. {ECO:0000269|PubMed:27376152}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01472; DB00321; DB00543; DB00182; DB00714; DB00964; DB09229; DB01238; DB14185; DB06216; DB00865; DB00217; DB00484; DB01200; DB00248; DB01136; DB04846; DB00477; DB09202; DB00575; DB00363; DB01151; DB00633; DB01576; DB11273; DB13345; DB00320; DB00449; DB11278; DB09167; DB04855; DB06262; DB01363; DB05492; DB00751; DB00668; DB01049; DB00696; DB01175; DB06678; DB09194; DB00800; DB06623; DB00629; DB01018; DB00502; DB11577; DB00555; DB06707; DB00589; DB04948; DB09195; DB00408; DB08815; DB00934; DB01365; DB01577; DB01403; DB00968; DB06148; DB00370; DB09205; DB09242; DB06711; DB01149; DB00368; DB00540; DB06229; DB00935; DB01267; DB00715; DB01186; DB01608; DB00925; DB00692; DB00397; DB09286; DB09244; DB06153; DB00413; DB00457; DB00433; DB01069; DB00852; DB01224; DB11124; DB11738; DB00268; DB09304; DB06764; DB13025; DB00697; DB00797; DB00193; DB00656; DB00726; DB11477; DB06694; DB01392; DB00246; DB01624 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Methylation; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 30303.9 Length 263 Aromaticity 0.16 Instability index 35.08 Isoelectric point 9.66 Charge (pH=7) 16.82 2D Binding mode Binding energy (Kcal/mol) -6.94 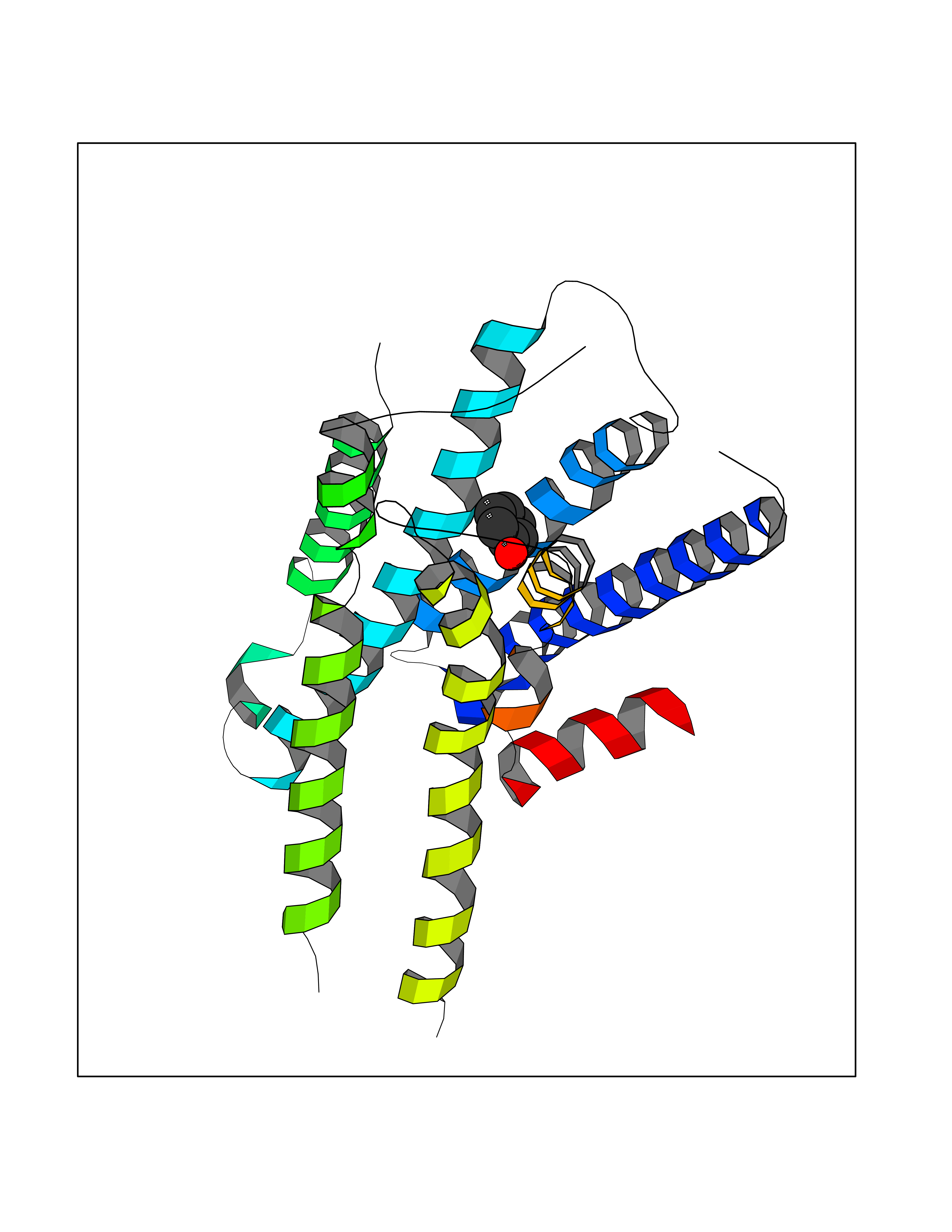 Molscript Map 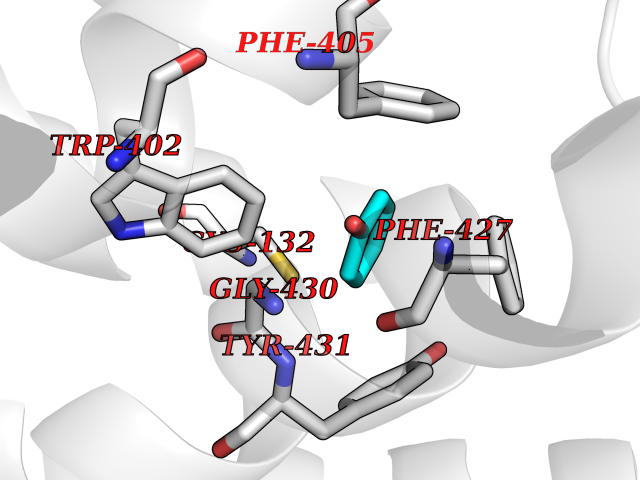 Pymol Map 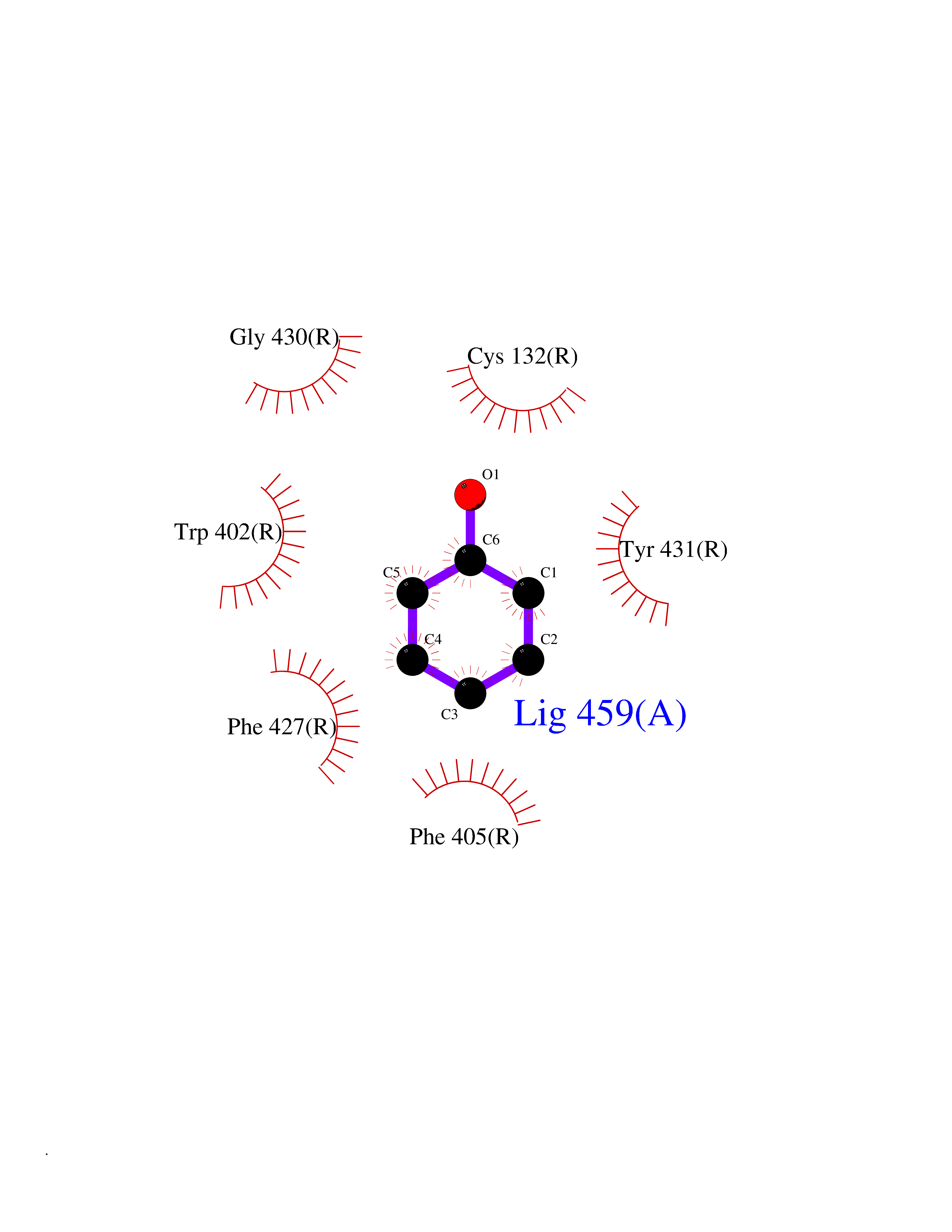 Ligplot Map 3D Binding mode Sequence YSLQVTLTLVCLAGLLMLLTVFGNVLVIIAVFTSRALKAPQNLFLVSLASADILVATLVIPFSLANEVMGYWYFGKAWCEIYLALDVLFCTSSIVHLCAISLDRYWSITQAIEYNLKRTPRRIKAIIITVWVISAVISFPPRCEINDQKWYVISSCIGSFFAPCLIMILVYVRIYQIAKRRTRRGRQNREKRFTFVLAVVIGVFVVCWFPFFFTYTLTAVGCSVPRTLFKFFFWFGYCNSSLNPVIYTIFNHDFRRAFKKILC Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Zinc finger-containing ubiquitin peptidase 1 (ZUP1) | 6EI1 | 5.09 | |
Target general information Gen name ZUP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Zinc finger with UFM1-specific peptidase domain protein; ZUFSP; Lys-63-specific deubiquitinase ZUFSP; DUB; C6orf113 Protein family Peptidase C78 family, ZUFSP subfamily Biochemical class Peptidase Function Shows only weak activity against 'Lys-11' and 'Lys-48'-linked chains. Plays an important role in genome stability pathways, functioning to prevent spontaneous DNA damage and also promote cellular survival in response to exogenous DNA damage. Modulates the ubiquitination status of replication protein A (RPA) complex proteins in response to replication stress. Deubiquitinase with endodeubiquitinase activity that specifically interacts with and cleaves 'Lys-63'-linked long polyubiquitin chains. Related diseases WHIM syndrome 2 (WHIMS2) [MIM:619407]: An autosomal recessive form of WHIM syndrome, a primary immunodeficiency disorder characterized by warts, hypogammaglobulinemia, infections, and myelokathexis. Myelokathexis is a unique form of non-cyclic severe congenital neutropenia caused by accumulation of mature and degenerating neutrophils in the bone marrow. Monocytopenia and lymphopenia, especially B lymphopenia, also commonly occur. There is significant phenotypic variation among patients, such that some individuals may have an incomplete form of the disorder in which one or more of the classic tetrad features are not present. {ECO:0000269|PubMed:24777453}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q92619; P50281; Q8WVC2 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Hydrolase; Metal-binding; Nucleus; Proteomics identification; Reference proteome; Repeat; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 46930.3 Length 410 Aromaticity 0.07 Instability index 58.67 Isoelectric point 9 Charge (pH=7) 9.7 2D Binding mode Binding energy (Kcal/mol) -6.95 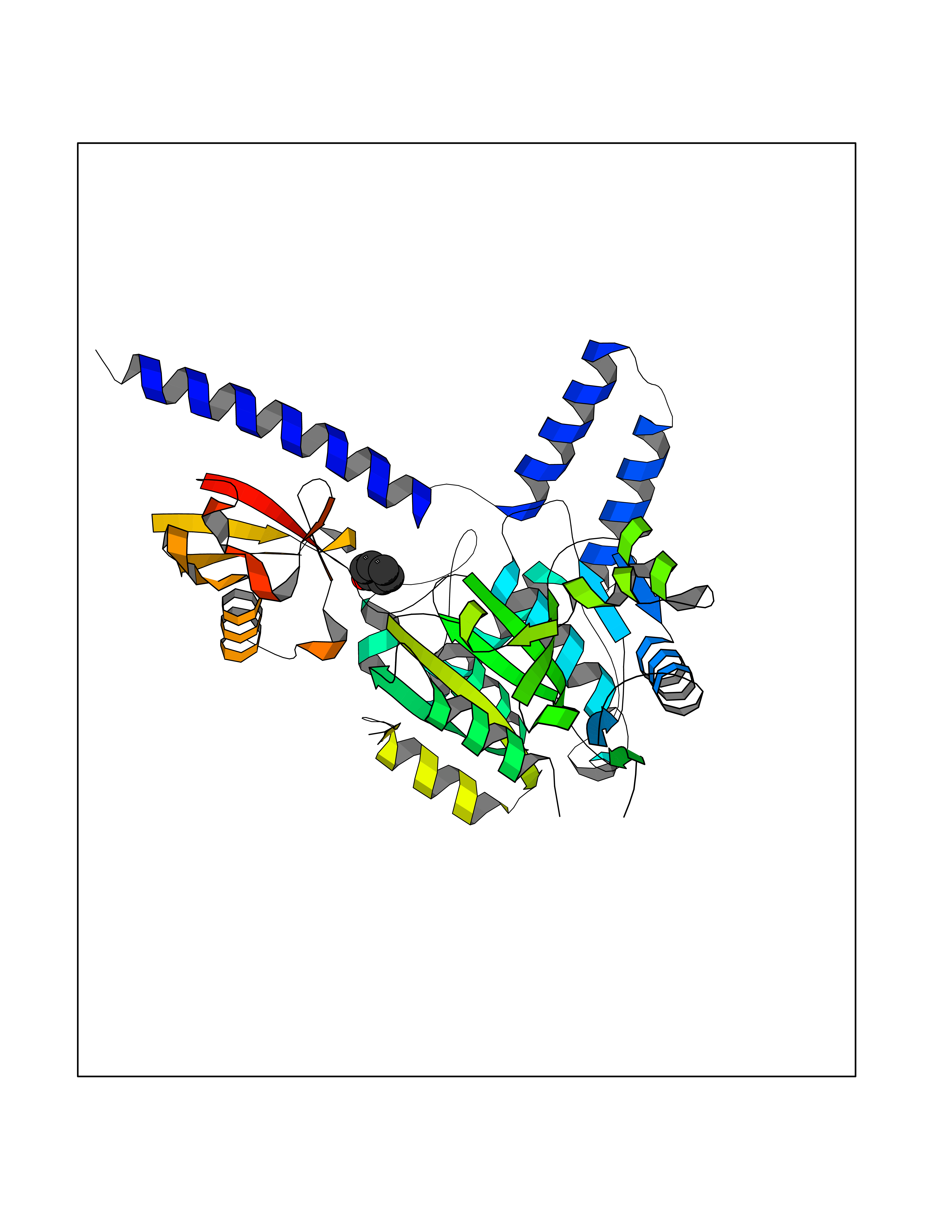 Molscript Map 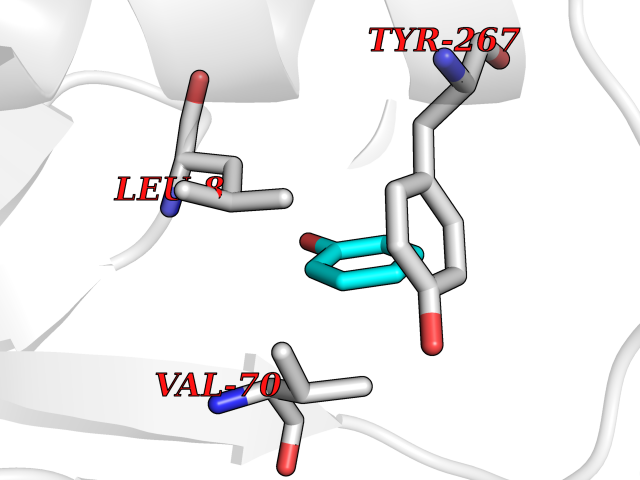 Pymol Map 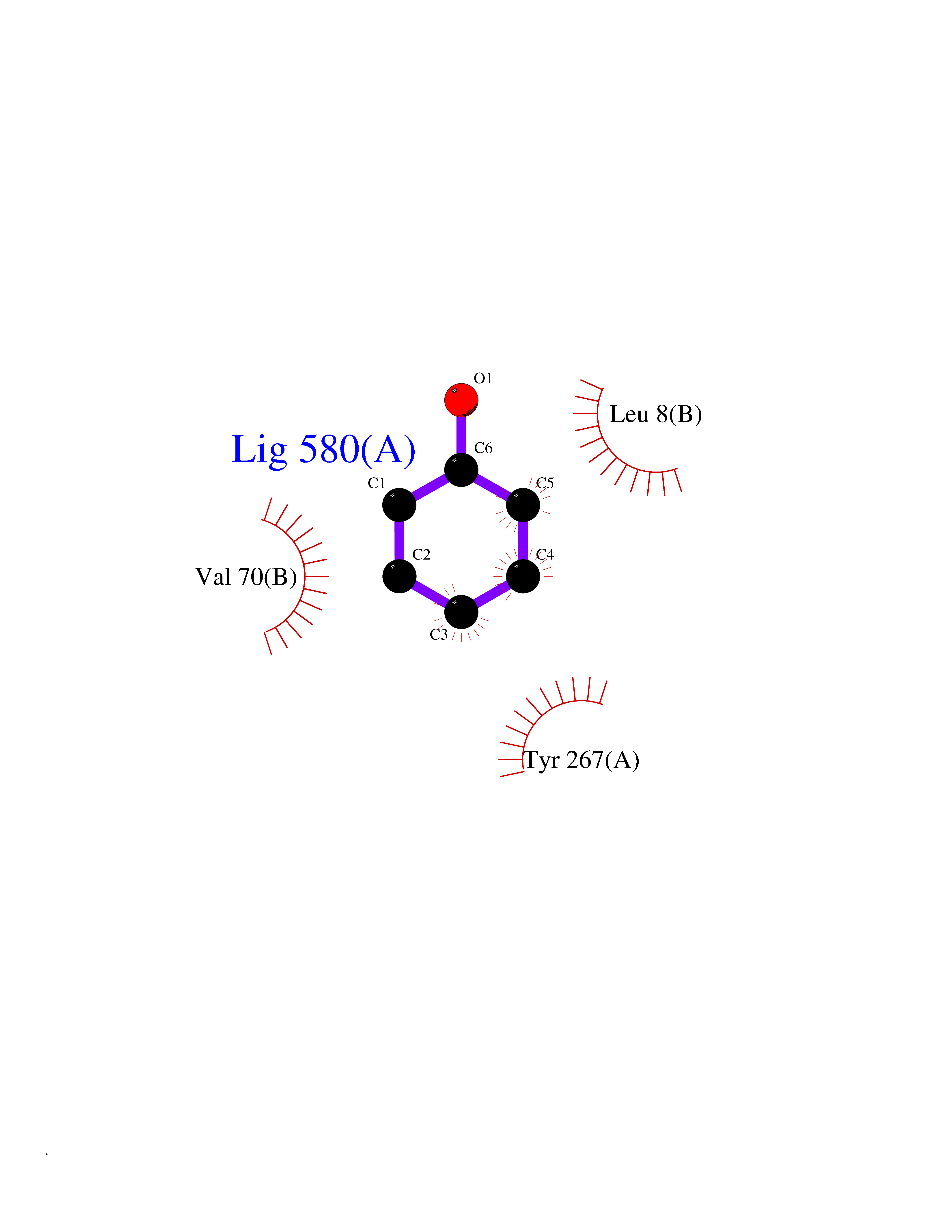 Ligplot Map 3D Binding mode Sequence LQQEEDRKRRSEESRQEIEEFQKLQRQYGLDNSGGYKQQQLRNMEIEVNRGRMPPSEFHRRKADMMESLALGFDDGKTKTSGIIEALHRYYQNAATDVRRVWLSSVVDHFHSSLGDKGWGCGYRNFQMLLSSLLQNDAYNDCLKGMLIPCIPKIQSMIEDAWKEGFDPQGASQLNNRLQGTKAWIGACEVYILLTSLRVKCHIVDFHKSTGPLGTHPRLFEWILNYYSSSPKVVCTSKPPIYLQHQGHSRTVIGIEEKKNRTLCLLILDPGCPSREMQKLLKQDIEASSLKQLRKSMGNLKHKQYQILAVEGALSLEEKLARRQASQVFTAEKIPMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRG Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Ephrin type-B receptor 3 (EPHB3) | 5L6O | 5.09 | |
Target general information Gen name EPHB3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hEK2; Tyrosine-protein kinase TYRO6; TYRO6; Embryonic kinase 2; ETK2; EPH-like tyrosine kinase 2; EPH-like kinase 2; EK2 Protein family Protein kinase superfamily, Tyr protein kinase family, Ephrin receptor subfamily Biochemical class Kinase Function The signaling pathway downstream of the receptor is referred to as forward signaling while the signaling pathway downstream of the ephrin ligand is referred to as reverse signaling. Generally has an overlapping and redundant function with EPHB2. Like EPHB2, functions in axon guidance during development regulating for instance the neurons forming the corpus callosum and the anterior commissure, 2 major interhemispheric connections between the temporal lobes of the cerebral cortex. In addition to its role in axon guidance plays also an important redundant role with other ephrin-B receptors in development and maturation of dendritic spines and the formation of excitatory synapses. Controls other aspects of development through regulation of cell migration and positioning. This includes angiogenesis, palate development and thymic epithelium development for instance. Forward and reverse signaling through the EFNB2/EPHB3 complex also regulate migration and adhesion of cells that tubularize the urethra and septate the cloaca. Finally, plays an important role in intestinal epithelium differentiation segregating progenitor from differentiated cells in the crypt. Receptor tyrosine kinase which binds promiscuously transmembrane ephrin-B family ligands residing on adjacent cells, leading to contact-dependent bidirectional signaling into neighboring cells. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) NA Interacts with P37235; O75031 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Angiogenesis; ATP-binding; Cell membrane; Cell projection; Developmental protein; Disulfide bond; Glycoprotein; Kinase; Membrane; Neurogenesis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30412.9 Length 267 Aromaticity 0.09 Instability index 37.42 Isoelectric point 7.74 Charge (pH=7) 1 2D Binding mode Binding energy (Kcal/mol) -6.94 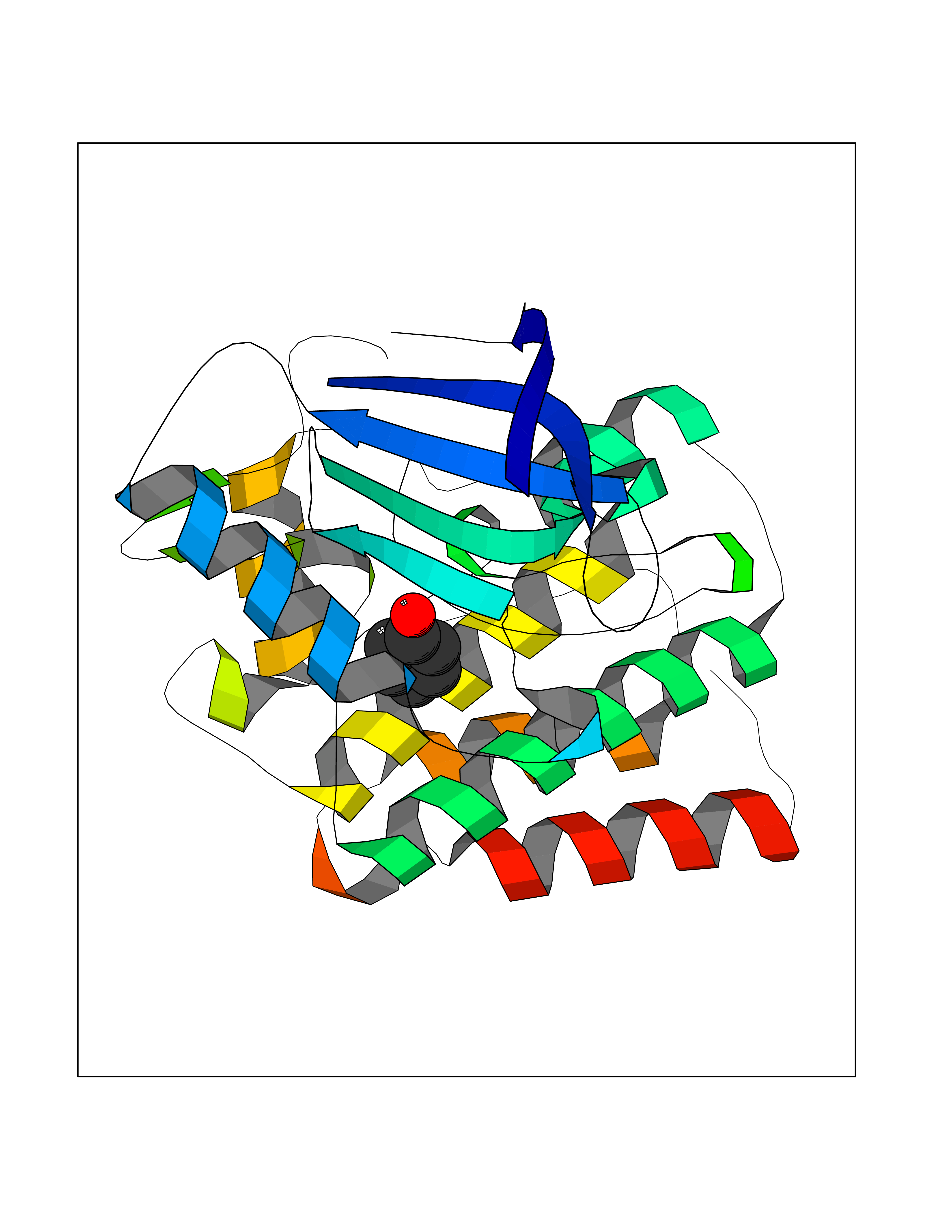 Molscript Map 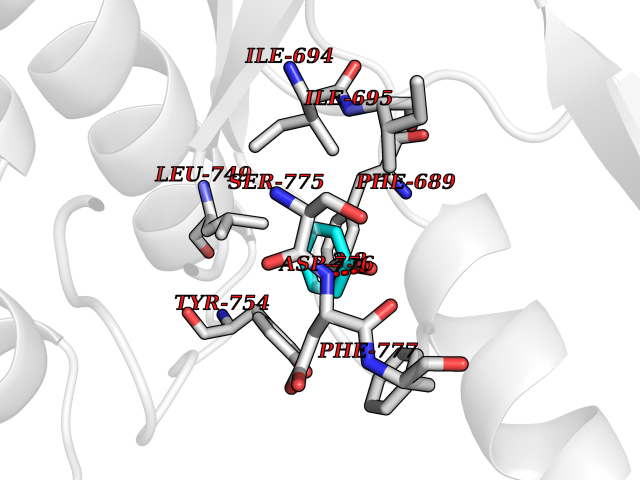 Pymol Map 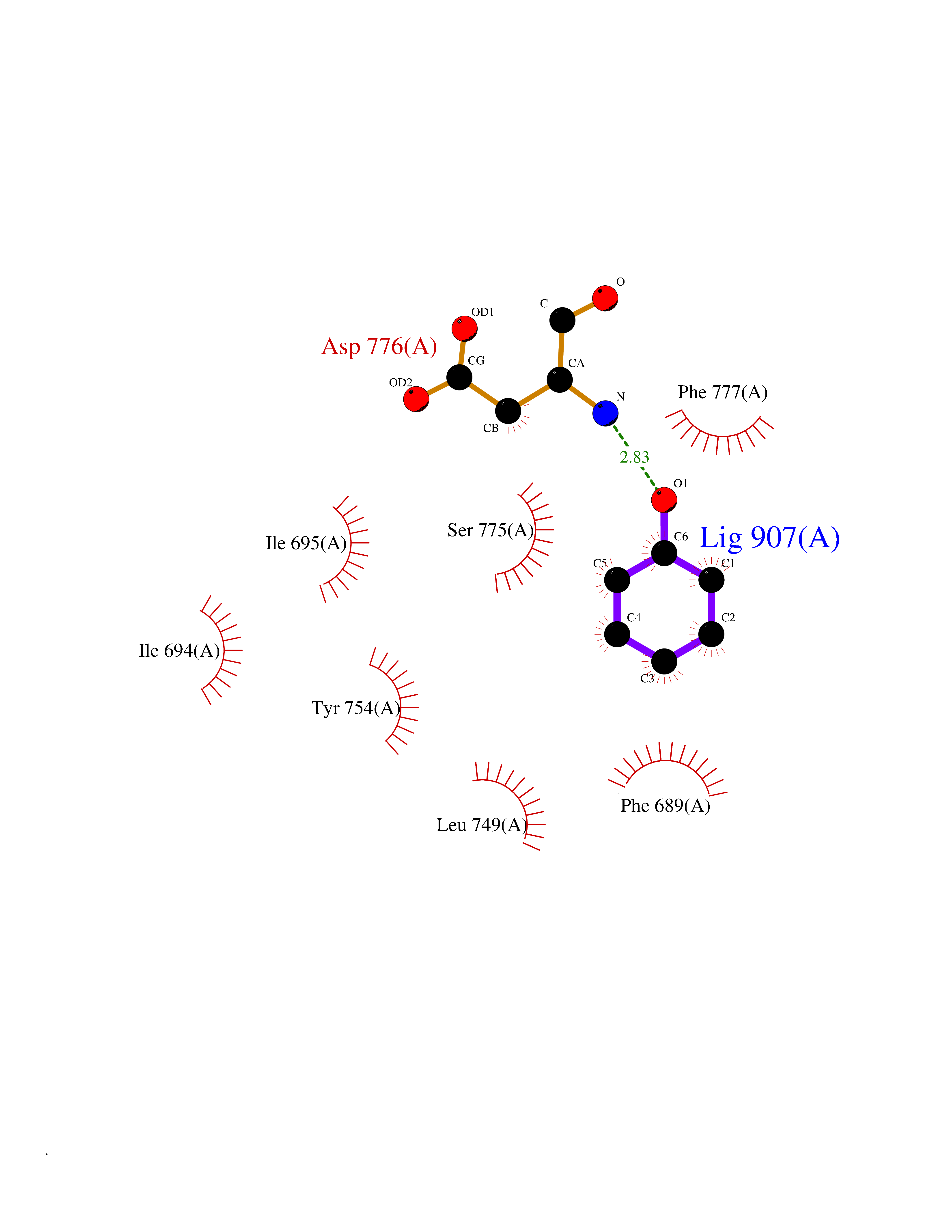 Ligplot Map 3D Binding mode Sequence CVKIEEVIGAGEVCRGRLKQPGRREVFVAIKTLKVGYTERQRRDFLSEASIMGQFDHPNIIRLEGVVTKSRPVMILTEFMENCALDSFLRLNDGQFTVIQLVGMLRGIAAGMKYLSEMNYVHRDLAARNILVNSNLVCKVSDFGLEDDPSDPTYTSSLGGKIPIRWTAPEAIAYRKFTSASDVWSYGIVMWEVMSYGERPYWDMSNQDVINAVEQDYRLPPPMDCPTALHQLMLDCWVRDRNLRPKFSQIVNTLDKLIRNPASLKVI Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Cytosolic 10-formyltetrahydrofolate dehydrogenase | 2CFI | 5.08 | |
Target general information Gen name ALDH1L1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FTHFD Protein family GART family; Aldehyde dehydrogenase family, ALDH1L subfamily Biochemical class Oxidoreductase Function Aldehyde dehydrogenase (NAD) activity.Catalytic activity.Formyltetrahydrofolate dehydrogenase activity.Hydroxymethyl-, formyl- and related transferase activity. Related diseases Developmental and epileptic encephalopathy 39 with leukodystrophy (DEE39) [MIM:612949]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE39 is characterized by global hypomyelination of the central nervous system, with the gray matter appearing relatively unaffected. Inheritance is autosomal recessive. {ECO:0000269|PubMed:19641205, ECO:0000269|PubMed:24515575}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00116 Interacts with Q3SY69; Q92624 EC number 1.5.1.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; NADP; One-carbon metabolism; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33869.5 Length 308 Aromaticity 0.08 Instability index 30.42 Isoelectric point 6.09 Charge (pH=7) -3.83 2D Binding mode Binding energy (Kcal/mol) -6.93 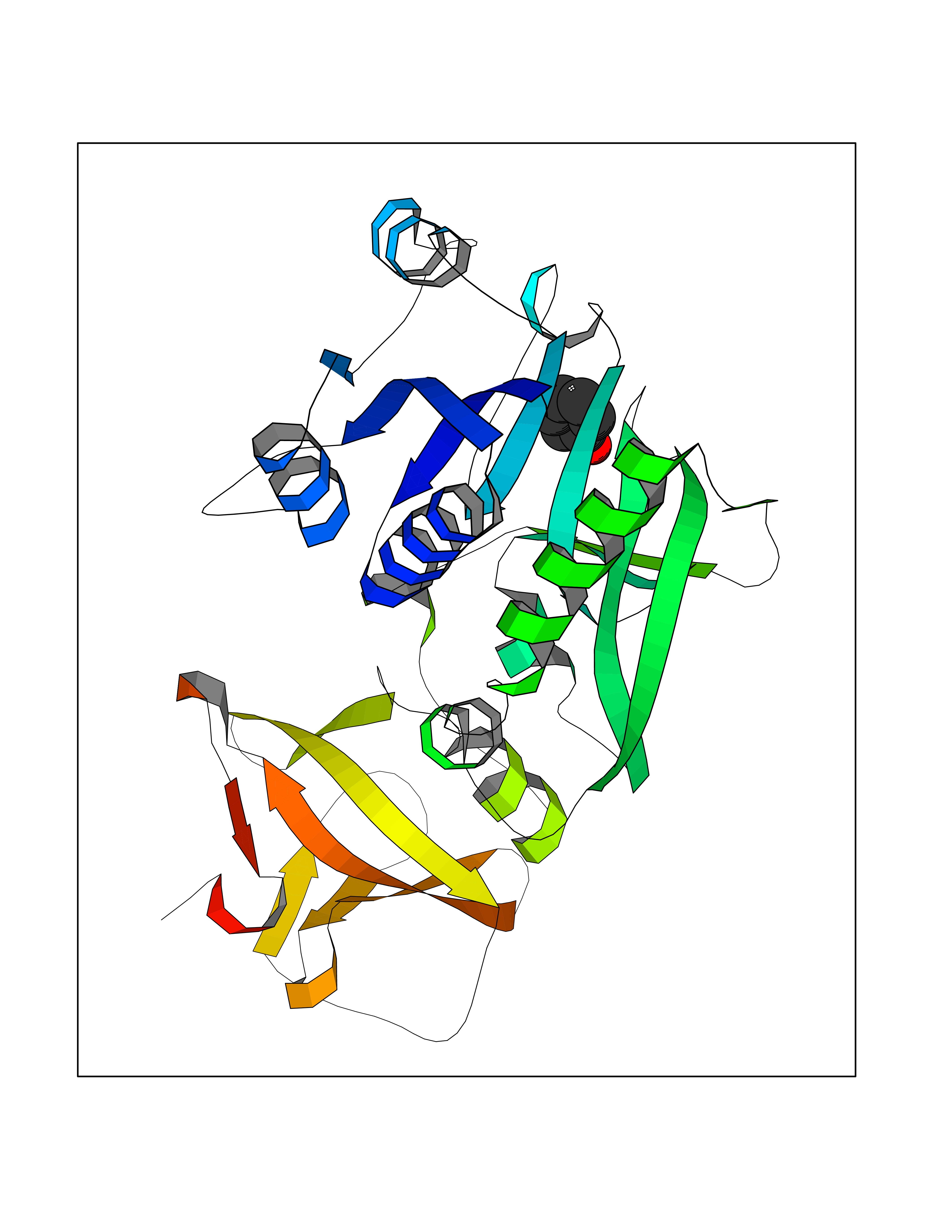 Molscript Map 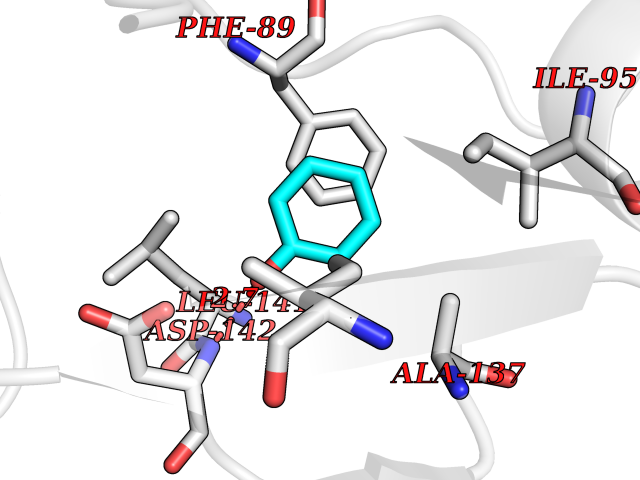 Pymol Map 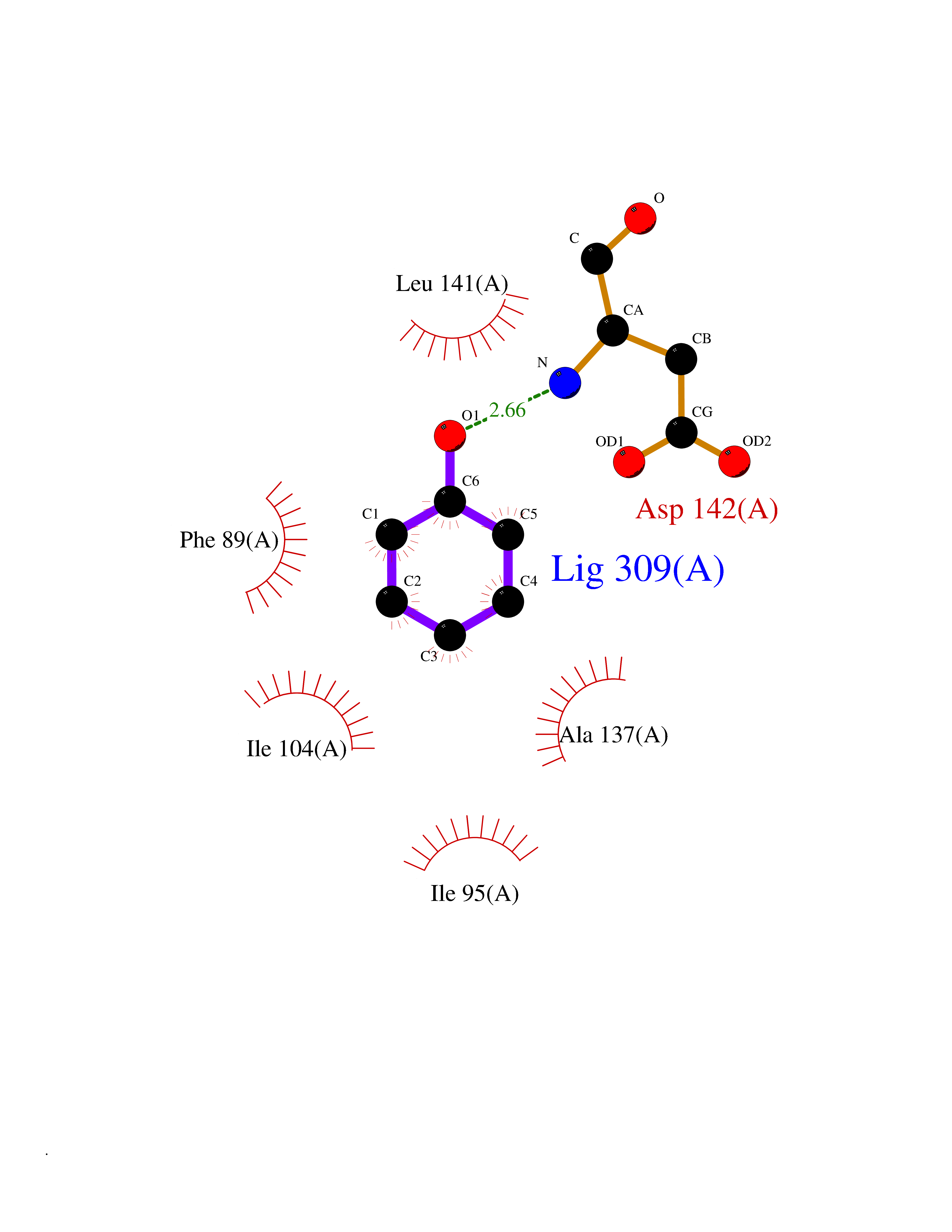 Ligplot Map 3D Binding mode Sequence SMKIAVIGQSLFGQEVYCHLRKEGHEVVGVFTVPDKDGKADPLGLEAEKDGVPVFKYSRWRAKGQALPDVVAKYQALGAELNVLPFCSQFIPMEIISAPRHGSIIYHPSLLPRHRGASAINWTLIHGDKKGGFSIFWADDGLDTGDLLLQKECEVLPDDTVSTLYNRFLFPEGIKGMVQAVRLIAEGKAPRLPQPEEGATYEGIQKKETAKINWDQPAEAIHNWIRGNDKVPGAWTEACEQKLTFFNSTLNTSGLVPEGDALPIPGAHRPGVVTKAGLILFGNDDKMLLVKNIQLEDGKMILASNFFK Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Inositol monophosphatase 1 | 1IMB | 5.08 | |
Target general information Gen name IMPA1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms IMPA Protein family Inositol monophosphatase superfamily Biochemical class Hydrolase Function Identical protein binding.Inositol monophosphate 1-phosphatase activity.Inositol monophosphate 3-phosphatase activity.Inositol monophosphate 4-phosphatase activity.Inositol monophosphate phosphatase activity.Lithium ion binding.Magnesium ion binding.Manganese ion binding.Protein homodimerization activity. Related diseases Intellectual developmental disorder, autosomal recessive 59 (MRT59) [MIM:617323]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:26416544}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03542; DB14509; DB01356; DB14507; DB14508 Interacts with P29218; O14732; P54253; G5E9A7; P42858; P29218-3; O14732; Q7Z699 EC number 3.1.3.25; 3.1.3.94 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Hydrolase; Intellectual disability; Lithium; Magnesium; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 59299.9 Length 544 Aromaticity 0.07 Instability index 37.53 Isoelectric point 5.38 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -6.93 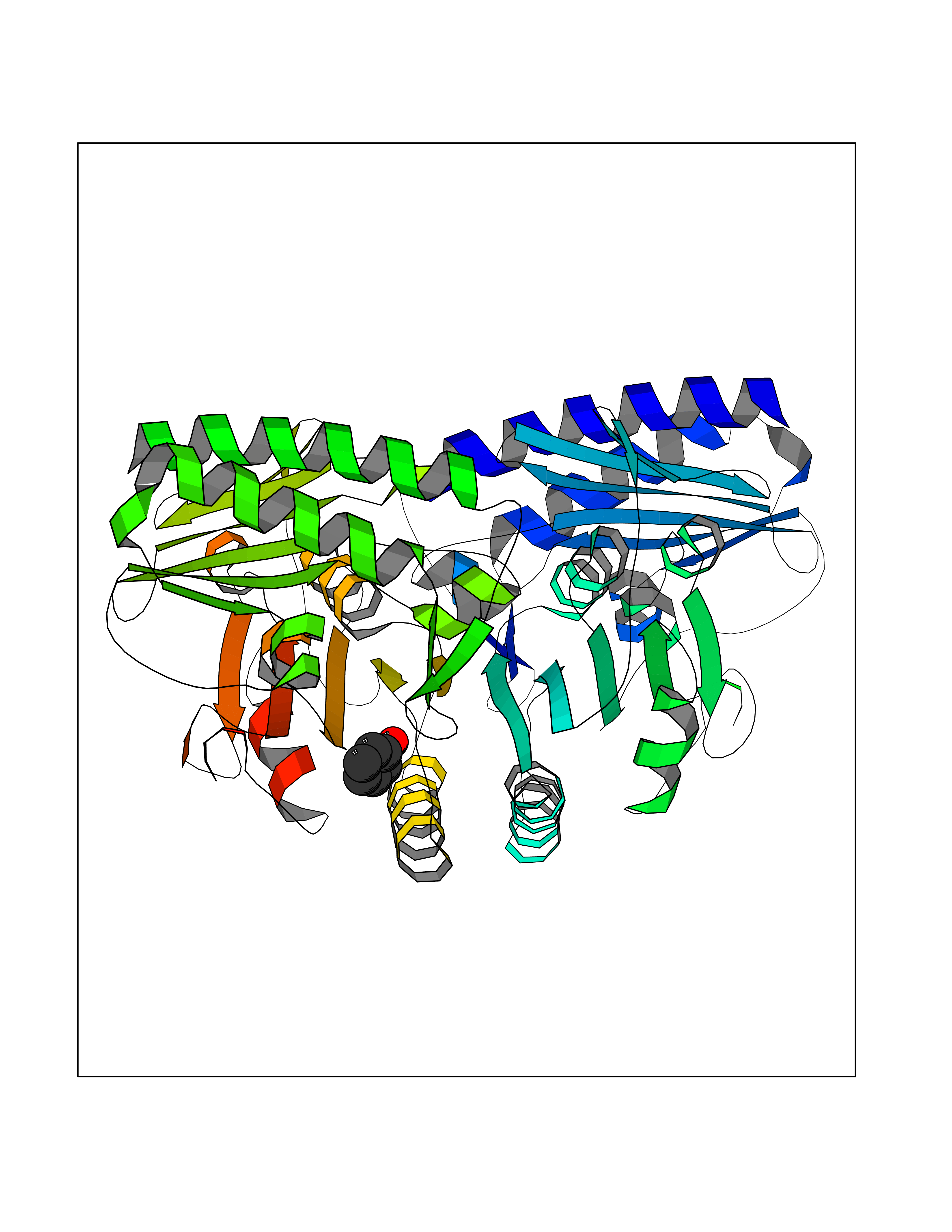 Molscript Map 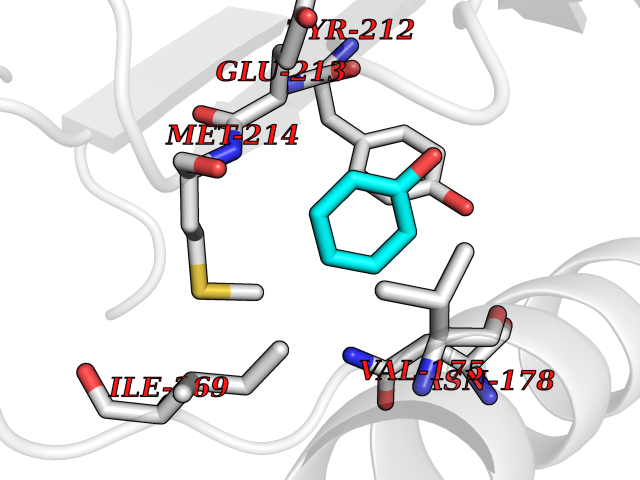 Pymol Map 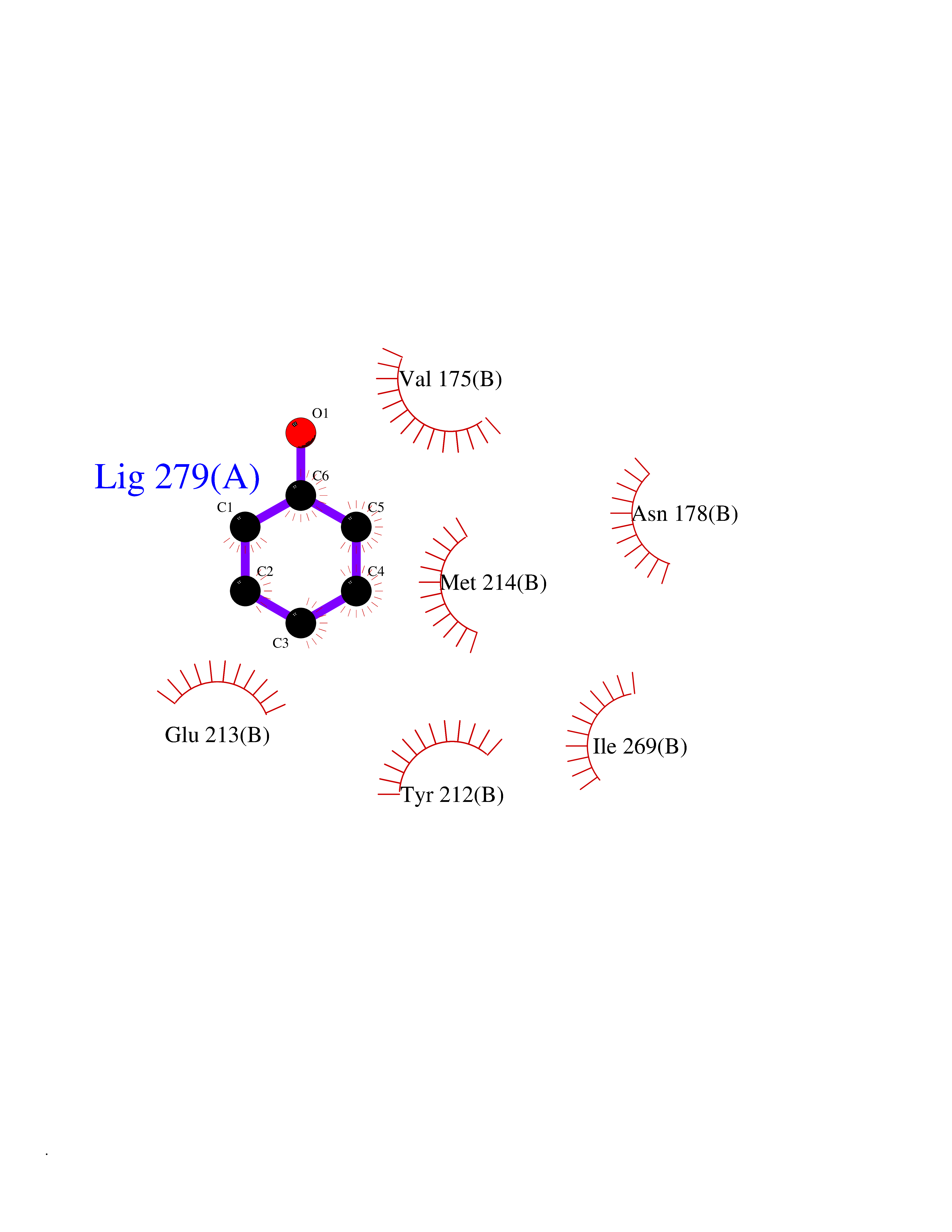 Ligplot Map 3D Binding mode Sequence WQECMDYAVTLARQAGEVVCEAIKNEMNVMLKSSPVDLVTATDQKVEKMLISSIKEKYPSHSFIGEESVAAGEKSILTDNPTWIIDPIDGTTNFVHRFPFVAVSIGFAVNKKIEFGVVYSCVEGKMYTARKGKGAFCNGQKLQVSQQEDITKSLLVTELGSSRTPETVRMVLSNMEKLFCIPVHGIRSVGTAAVNMCLVATGGADAYYEMGIHCWDVAGAGIIVTEAGGVLMDVTGGPFDLMSRRVIAANNRILAERIAKEIQVIPLQRDDEWQECMDYAVTLARQAGEVVCEAIKNEMNVMLKSSPVDLVTATDQKVEKMLISSIKEKYPSHSFIGEESVAAGEKSILTDNPTWIIDPIDGTTNFVHRFPFVAVSIGFAVNKKIEFGVVYSCVEGKMYTARKGKGAFCNGQKLQVSQQEDITKSLLVTELGSSRTPETVRMVLSNMEKLFCIPVHGIRSVGTAAVNMCLVATGGADAYYEMGIHCWDVAGAGIIVTEAGGVLMDVTGGPFDLMSRRVIAANNRILAERIAKEIQVIPLQRDDE Hydrogen bonds contact Hydrophobic contact | ||||