Job Results:
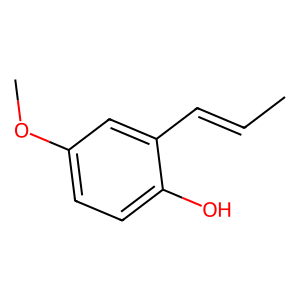
Ligand
Structure
Job ID
58e2f3b701186914fd3b121ac4df0edd
Job name
NA
Time
2026-02-27 13:43:39
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Guanidinoacetate N-methyltransferase | 3ORH | 5.64 | |
Target general information Gen name GAMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, RMT2 methyltransferase family Biochemical class Transferase Function Guanidinoacetate N-methyltransferase activity.Methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 2 (CCDS2) [MIM:612736]: An autosomal recessive disorder characterized by developmental delay and regression, intellectual disability, severe disturbance of expressive and cognitive speech, intractable seizures, movement disturbances, severe depletion of creatine and phosphocreatine in the brain, and accumulation of guanidinoacetic acid in brain and body fluids. {ECO:0000269|PubMed:12468279, ECO:0000269|PubMed:15108290, ECO:0000269|PubMed:15651030, ECO:0000269|PubMed:16293431, ECO:0000269|PubMed:16855203, ECO:0000269|PubMed:17101918, ECO:0000269|PubMed:17466557, ECO:0000269|PubMed:19388150, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:24415674, ECO:0000269|PubMed:8651275}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00148; DB02751; DB00536; DB13191; DB01752 Interacts with O95363; Q969Q5; Q9HCM9-2 EC number 2.1.1.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Disease variant; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 24656 Length 219 Aromaticity 0.11 Instability index 46.5 Isoelectric point 5.91 Charge (pH=7) -4.34 2D Binding mode Binding energy (Kcal/mol) -7.69  Molscript Map 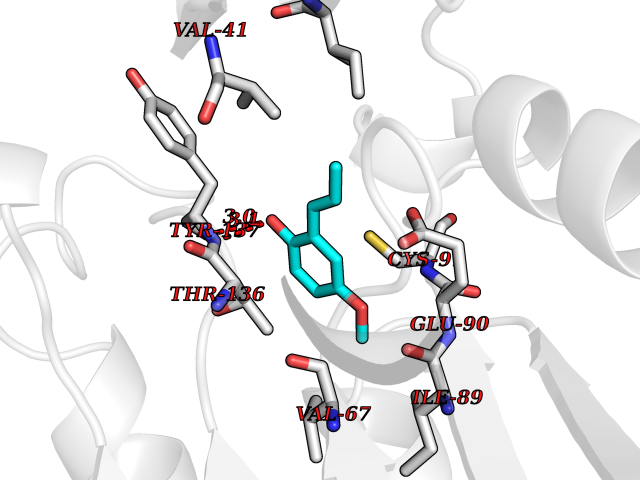 Pymol Map 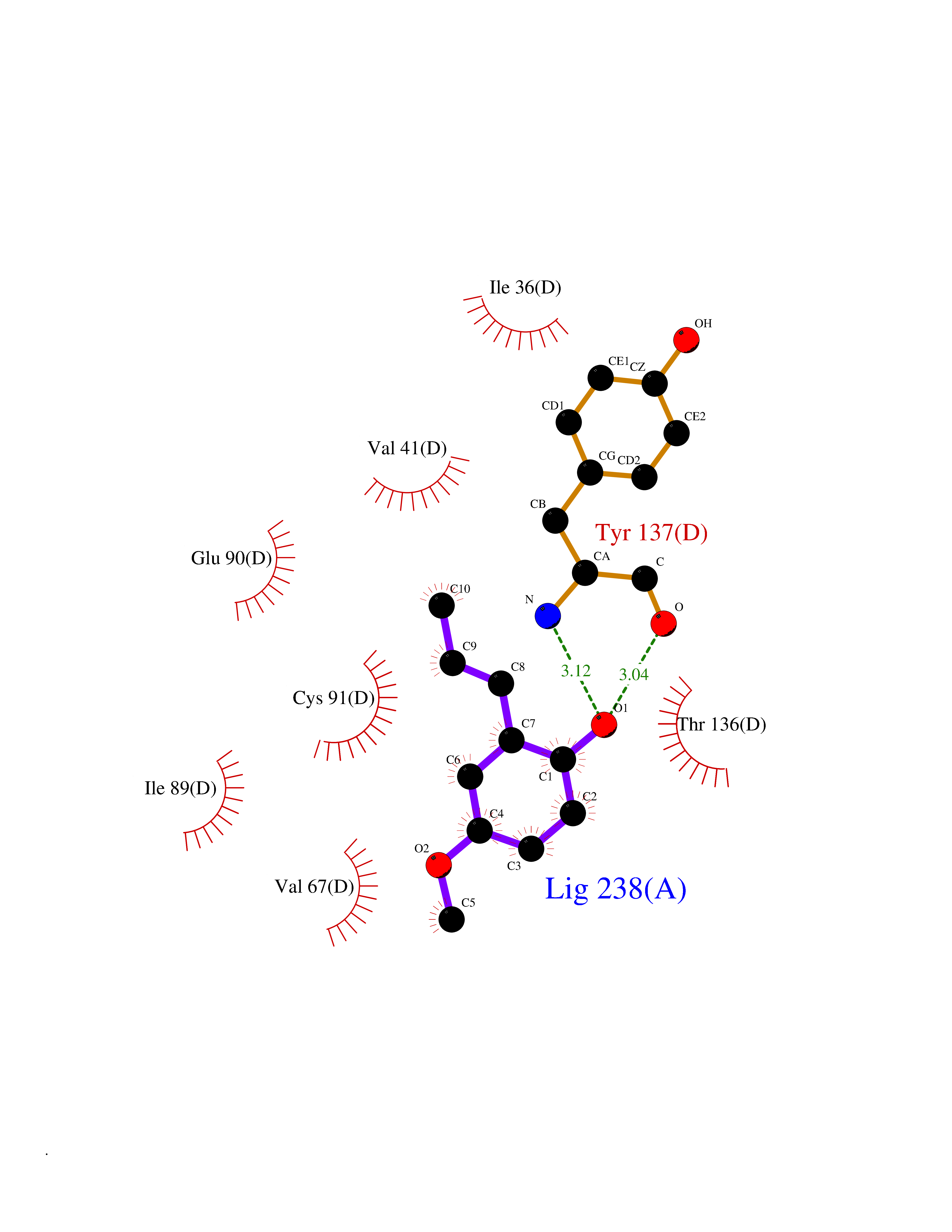 Ligplot Map 3D Binding mode Sequence PAWGAAPAAYDAADTHLRILGKPVMERWETPYMHALAAAASSKGGRVLEVGFGMAIAASKVQEAPIDEHWIIECNDGVFQRLRDWAPRQTHKVIPLKGLWEDVAPTLPDGHFDGILYDTYPLSEETWHTHQFNFIKNHAFRLLKPGGVLTYCNLTSWGELMKSKYSDITIMFEETQVPALLEAGFRRENIRTEVMALVPPADCRYYAFPQMITPLVTKG Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 5.64 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -7.7 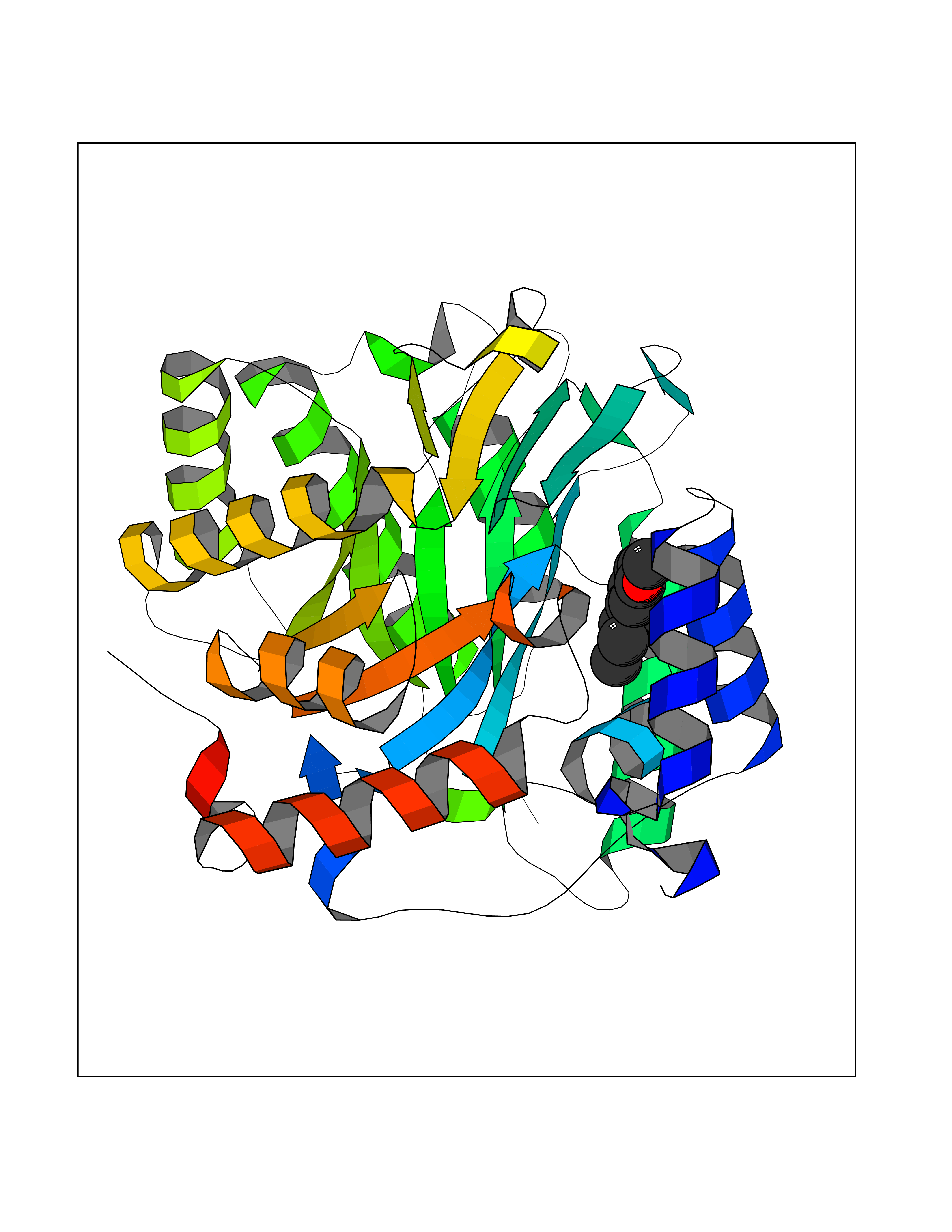 Molscript Map 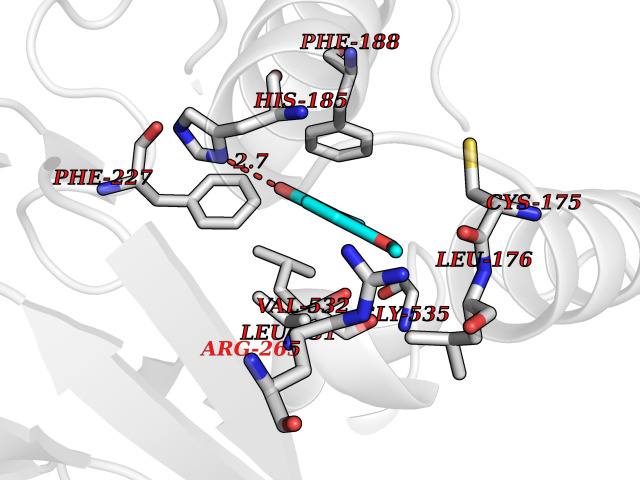 Pymol Map 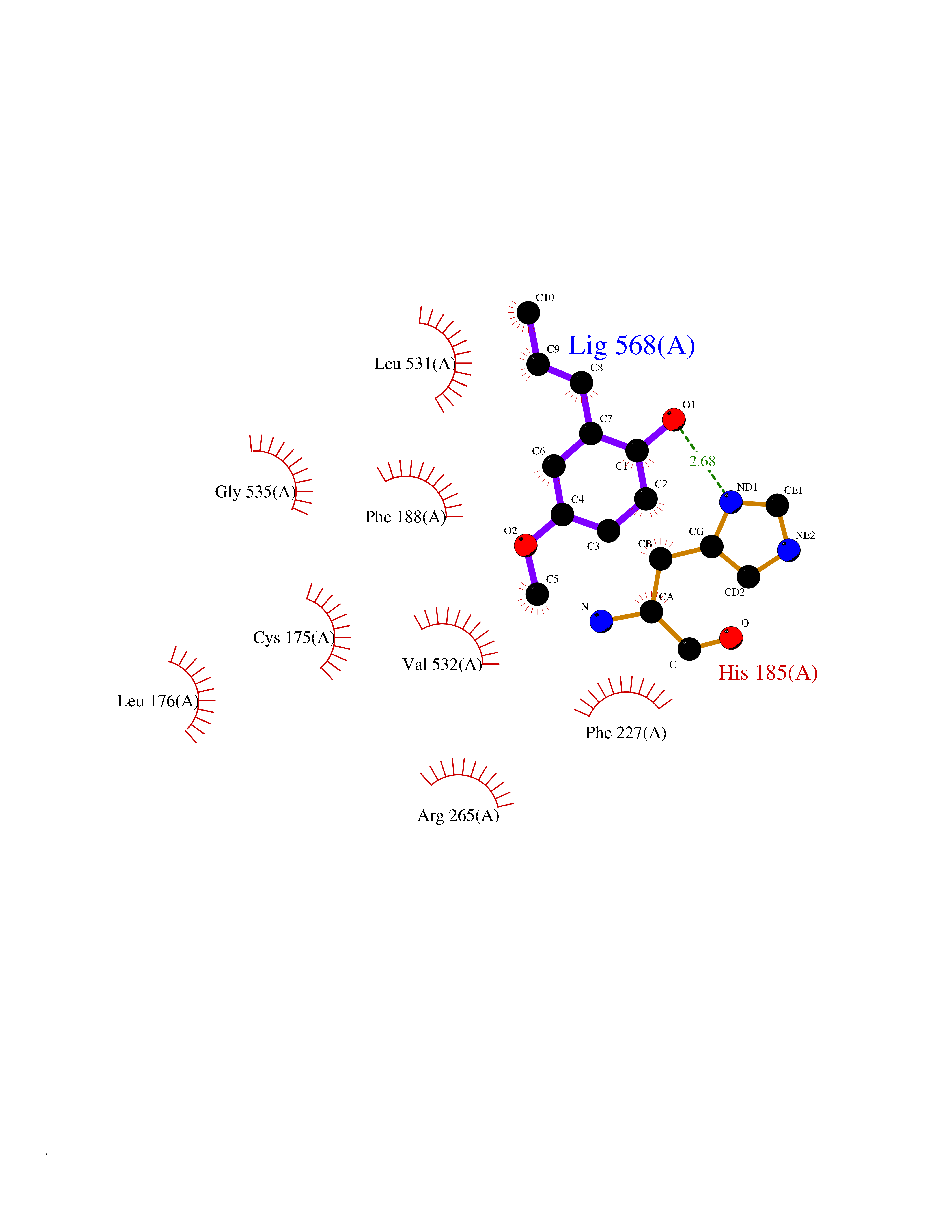 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Pseudomonas Methionine gamma-lyase (Pseudo mdeA) | 1PG8 | 5.64 | |
Target general information Gen name Pseudo mdeA Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID Synonyms Pseudo MGL; L-methionine gamma-lyase; L-methioninase; Homocysteine desulfhydrase Protein family Trans-sulfuration enzymes family, L-methionine gamma-lyase subfamily Biochemical class Carbon-sulfur lyases Function Catalyzes the alpha,gamma-elimination of L-methionine to produce methanethiol, 2-oxobutanoate and ammonia. Is involved in L-methionine catabolism. In fact, shows a multicatalytic function since it also catalyzes gamma-replacement of L-methionine with thiol compounds, alpha,gamma-elimination and gamma-replacement reactions of L-homocysteine and its S-substituted derivatives, O-substituted-L-homoserines and DL-selenomethionine, and, to a lesser extent, alpha,beta-elimination and beta-replacement reactions of L-cysteine, S-methyl-L-cysteine, and O-acetyl-L-serine. Also catalyzes deamination and gamma-addition reactions of L-vinylglycine. Thus, the enzyme is able to cleave C-S, C-Se, and C-O bonds of sulfur, selenium, and oxygen amino acids, respectively. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04083 Interacts with NA EC number EC 4.4.1.11 Uniprot keywords 3D-structure; Direct protein sequencing; Lyase; Pyridoxal phosphate Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85234.4 Length 796 Aromaticity 0.07 Instability index 37.1 Isoelectric point 6.21 Charge (pH=7) -11.34 2D Binding mode Binding energy (Kcal/mol) -7.69 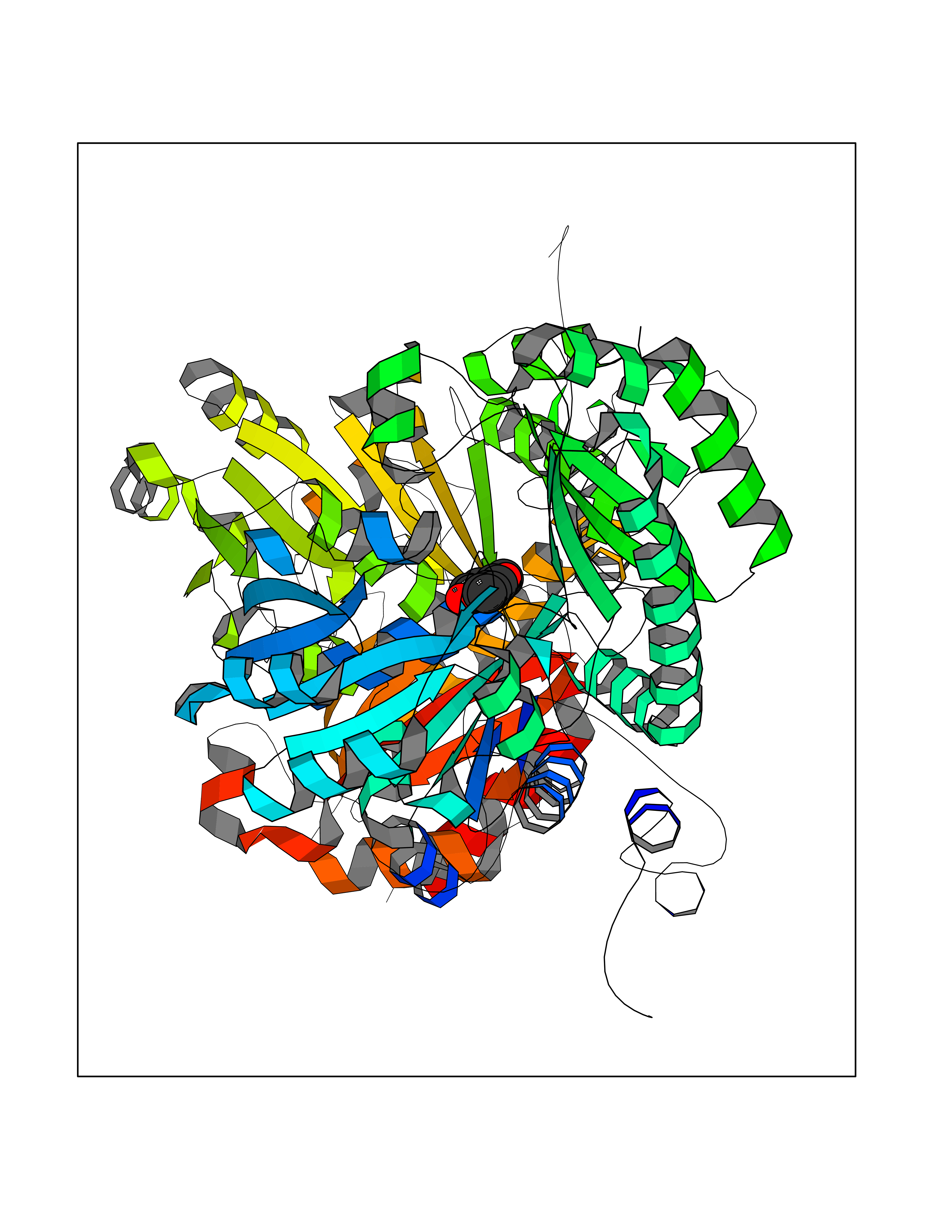 Molscript Map 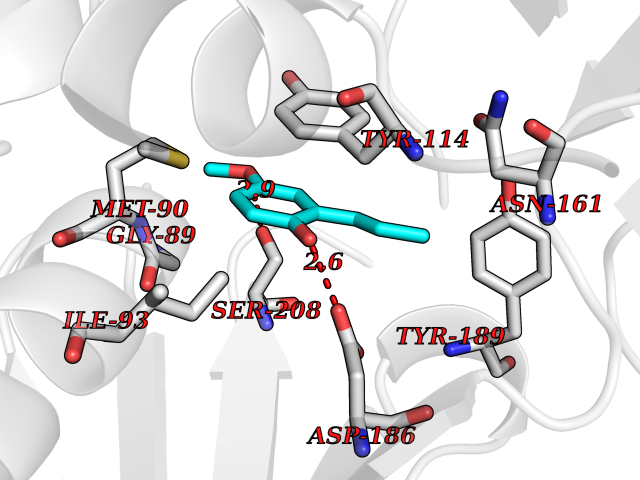 Pymol Map 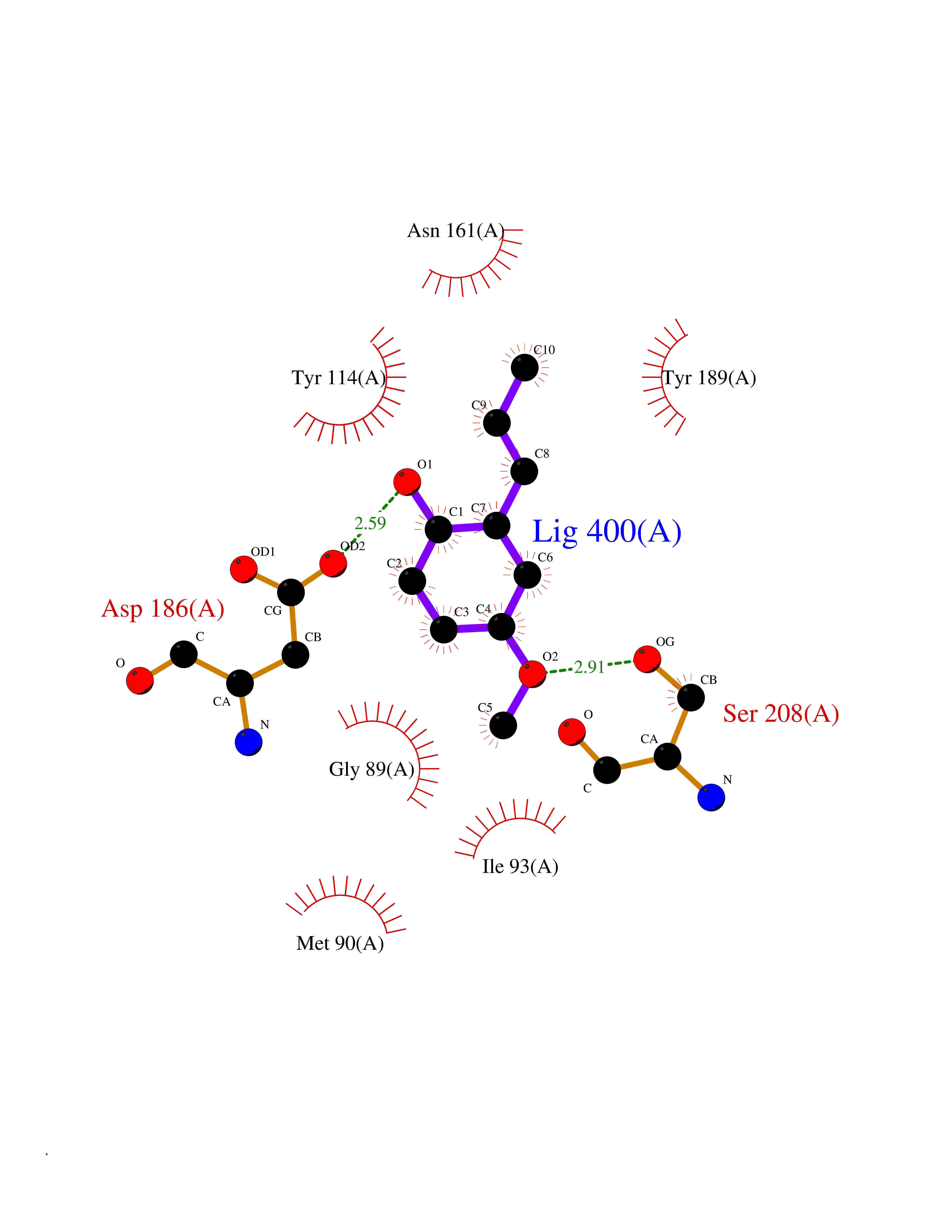 Ligplot Map 3D Binding mode Sequence MHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASAMHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASA Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Endothelin-converting enzyme 1 (ECE1) | 3DWB | 5.64 | |
Target general information Gen name ECE1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ECE-1 Protein family Peptidase M13 family Biochemical class Peptidase Function Converts big endothelin-1 to endothelin-1. Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07171 Interacts with P49760; A8MQ03; Q8IUG1; P60370; P60410 EC number EC 3.4.24.71 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Hirschsprung disease; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 75247.9 Length 660 Aromaticity 0.12 Instability index 46.29 Isoelectric point 5.33 Charge (pH=7) -18.3 2D Binding mode Binding energy (Kcal/mol) -7.69 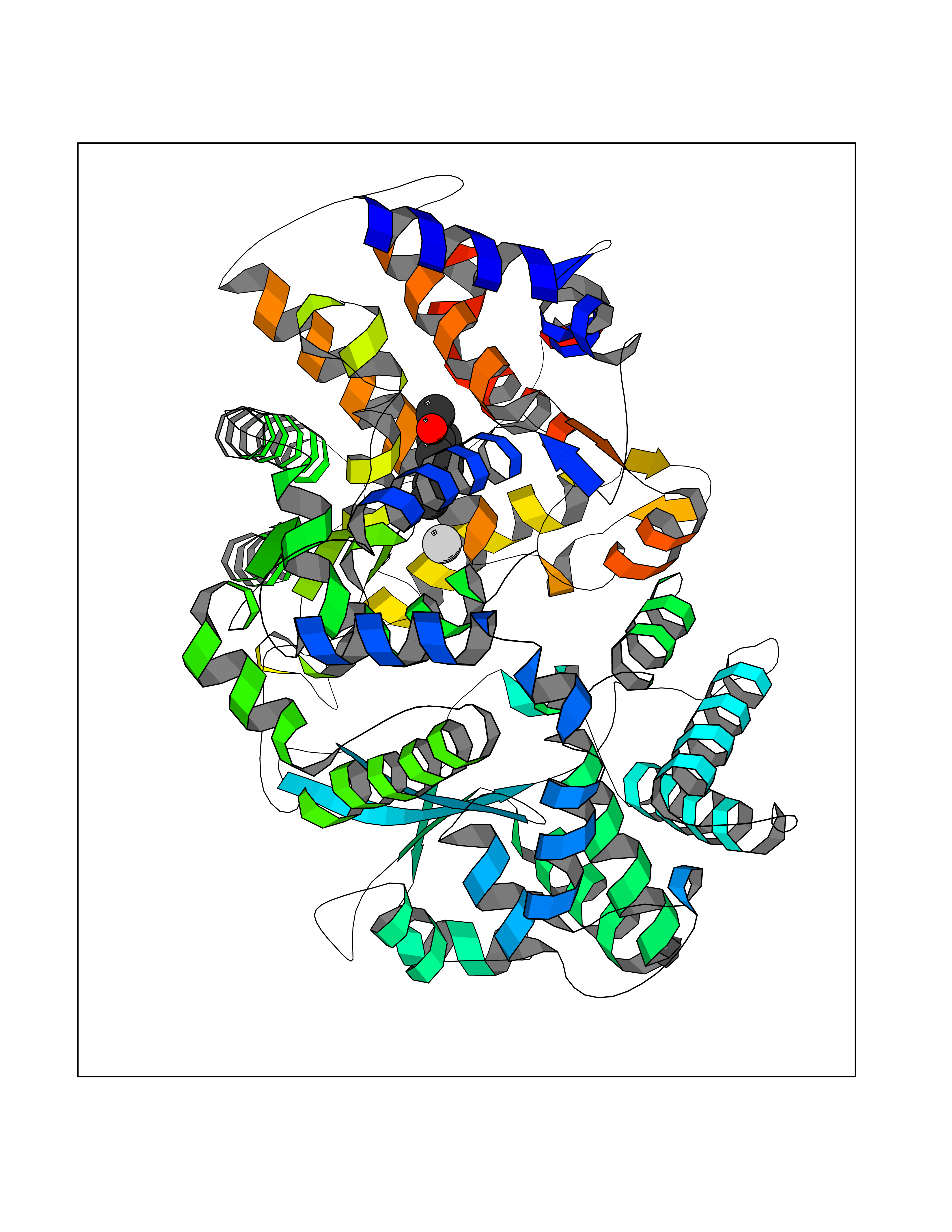 Molscript Map 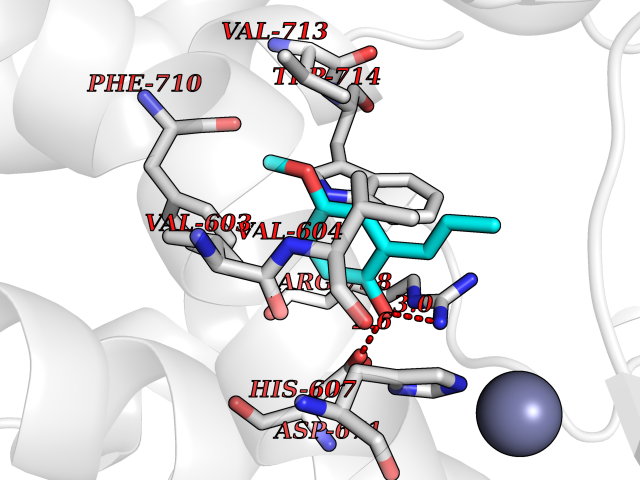 Pymol Map 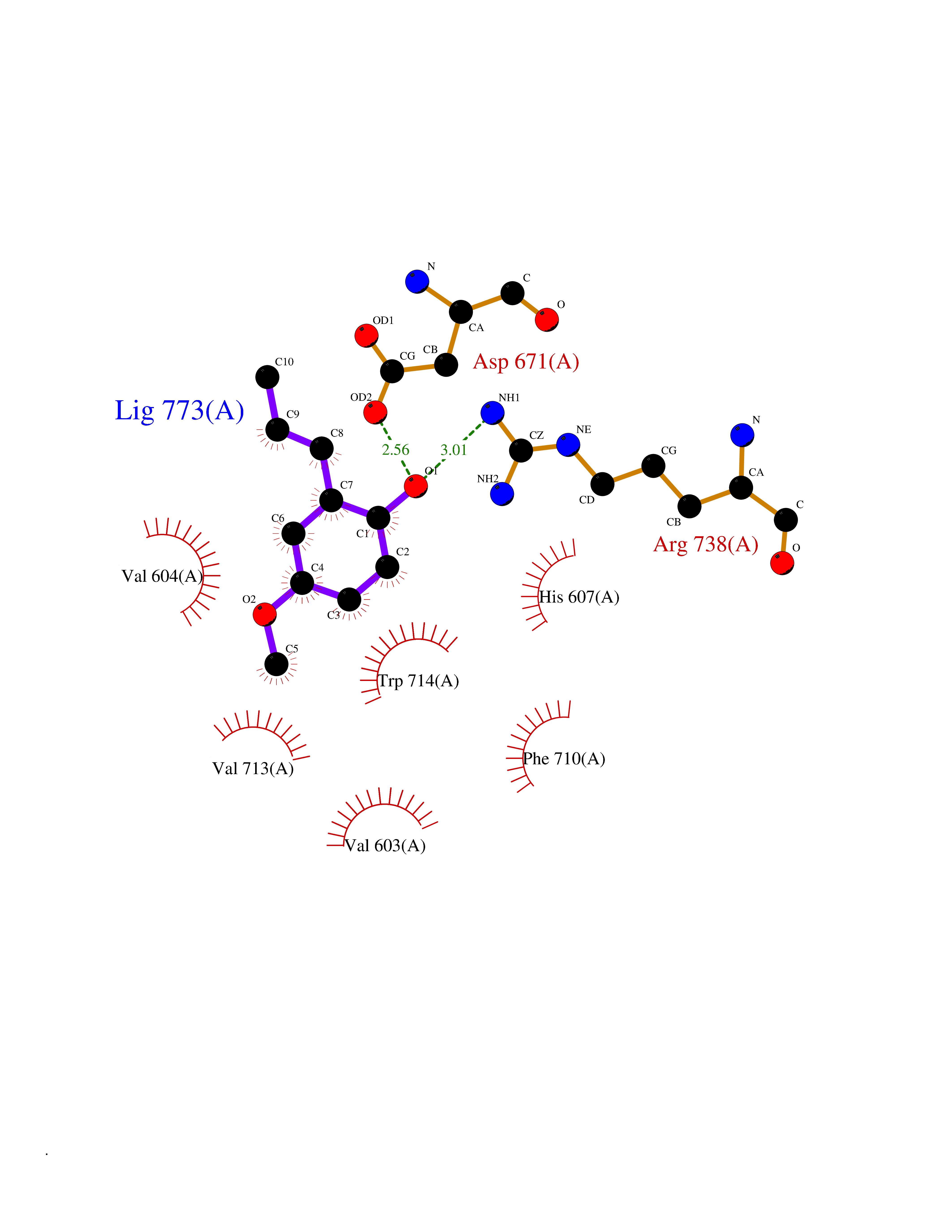 Ligplot Map 3D Binding mode Sequence SEACVSVTSSILSSMDPTVDPCHDFFSYACGGWIKANPVPDGHSRWGTFSNLWEHNQAIIKHLLENSTASVSEAERKAQVYYRACMNETRIEELRAKPLMELIERLGGWNITGPWAKDNFQDTLQVVTAHYRTSPFFSVYVSADSKNSNSNVIQVDQSGLGLPSRDYYLNKTENEKVLTGYLNYMVQLGKLLGGGDEEAIRPQMQQILDFETALANITIPQEKRRDEELIYHKVTAAELQTLAPAINWLPFLNTIFYPVEINESEPIVVYDKEYLEQISTLINTTDRCLLNNYMIWNLVRKTSSFLDQRFQDADEKFMEVMWKFCVSDTENNLGFALGPMFVKATFAEDSKSIATEIILEIKKAFEESLSTLKWMDEETRKSAKEKADAIYNMIGYPNFIMDPKELDKVFNDYTAVPDLYFENAMRFFNFSWRVTADQLRKAPNRDQWSMTPPMVNAYYSPTKNEIVFPAGILQAPFYTRSSPKALNFGGIGVVVGHELTHAFDDQGREYDKDGNLRPWWKNSSVEAFKRQTECMVEQYSNYSVNGEPVNGRHTLGENIADNGGLKAAYRAYQNWVKKNGAEHSLPTLGLTNNQLFFLGFAQVWCSVRTPESSHEGLITDPHSPSRFRVIGSLSNSKEFSEHFRCPPGSPMNPPHKCEVW Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Tumor-associated calcium signal transducer 1 (EPCAM) | 4MZV | 5.64 | |
Target general information Gen name EPCAM Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hEGP314; TROP1; TACSTD1; Major gastrointestinal tumor-associated protein GA733-2; MIC18; M4S1; M1S2; KSA; KS 1/4 antigen; Gastrointestinal carcinoma antigen GA733; GA733-2; Epithelial glycoprotein 314 Protein family EPCAM family Biochemical class NA Function Plays a role in embryonic stem cells proliferation and differentiation. Up-regulates the expression of FABP5, MYC and cyclins A and E. May act as a physical homophilic interaction molecule between intestinal epithelial cells (IECs) and intraepithelial lymphocytes (IELs) at the mucosal epithelium for providing immunological barrier as a first line of defense against mucosal infection. Related diseases Diarrhea 5, with tufting enteropathy, congenital (DIAR5) [MIM:613217]: An intractable diarrhea of infancy characterized by villous atrophy and absence of inflammation, with intestinal epithelial cell dysplasia manifesting as focal epithelial tufts in the duodenum and jejunum. {ECO:0000269|PubMed:18572020, ECO:0000269|PubMed:24142340}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Lynch syndrome 8 (LYNCH8) [MIM:613244]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:19098912}. The disease is caused by variants affecting the gene represented in this entry. LYNCH8 results from heterozygous deletion of 3-prime exons of EPCAM and intergenic regions directly upstream of MSH2, resulting in transcriptional read-through and epigenetic silencing of MSH2 in tissues expressing EPCAM. Drugs (DrugBank ID) DB06607; DB11075; DB05831; DB05319; DB09336 Interacts with P27797; P12830; Q15078; P36957; Q8TDX7 EC number NA Uniprot keywords 3D-structure; Cell junction; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hereditary nonpolyposis colorectal cancer; Membrane; Proteomics identification; Reference proteome; Repeat; Signal; Tight junction; Transmembrane; Transmembrane helix; Tumor antigen Protein physicochemical properties Chain ID A Molecular weight (Da) 27439.8 Length 243 Aromaticity 0.07 Instability index 33.77 Isoelectric point 6.03 Charge (pH=7) -1.99 2D Binding mode Binding energy (Kcal/mol) -7.69  Molscript Map 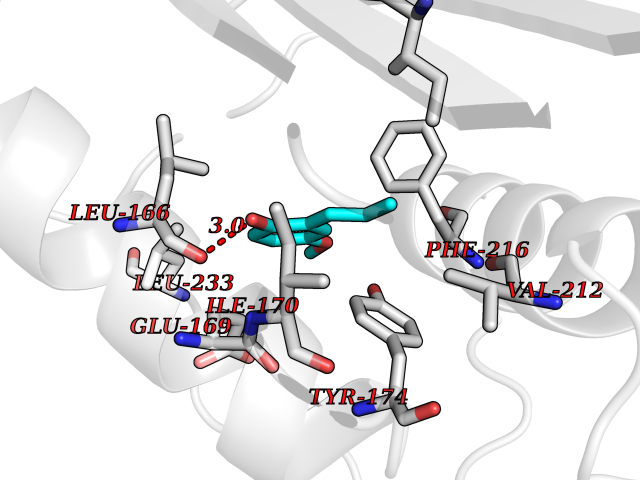 Pymol Map 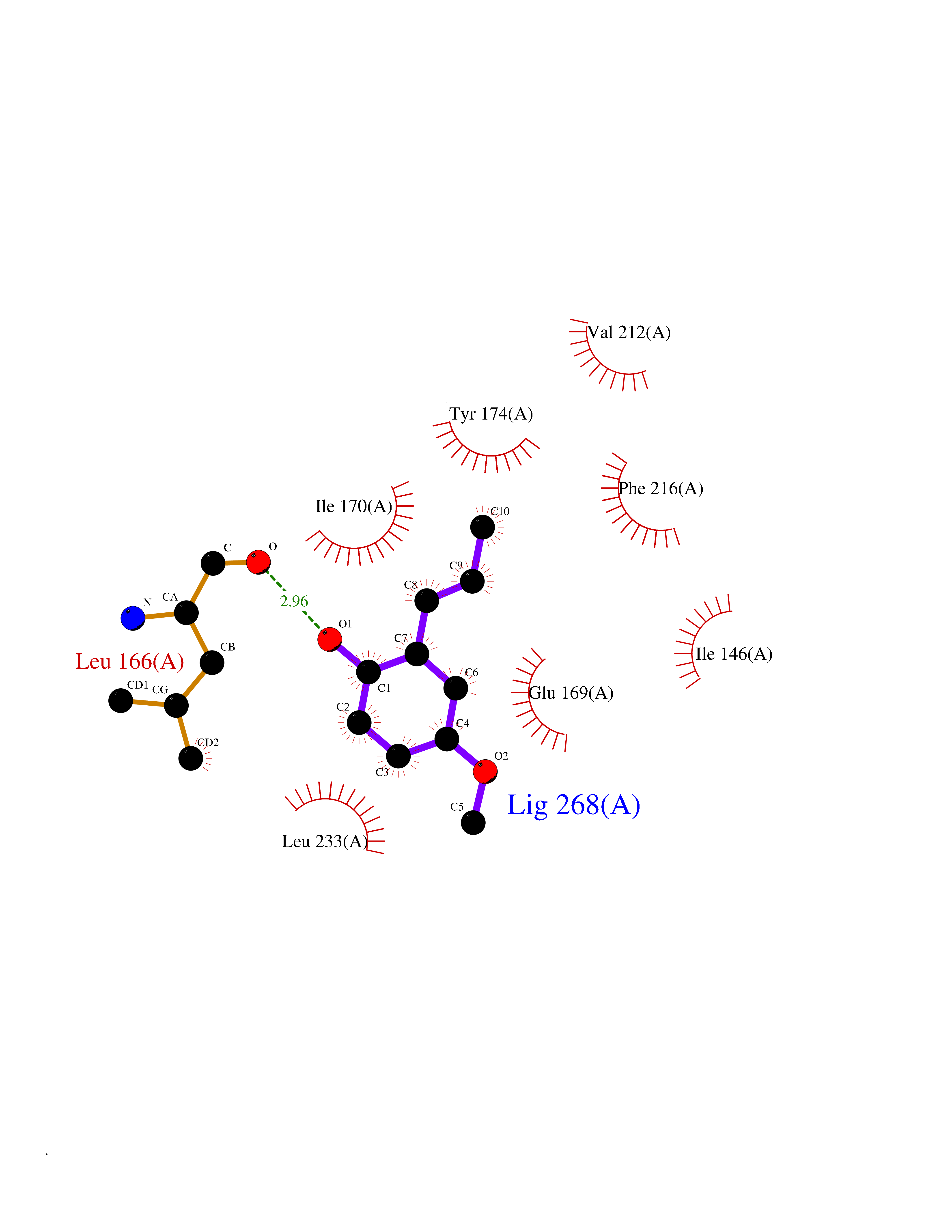 Ligplot Map 3D Binding mode Sequence XEECVCENYKLAVNCFVNNNRQCQCTSVGAQNTVICSKLAAKCLVMKAEMQGSKLGRRAKPEGALQNNDGLYDPDCDESGLFKAKQCQGTSTCWCVNTAGVRRTDKDTEITCSERVRTYWIIIELKHKAREKPYDSKSLRTALQKEITTRYQLDPKFITSILYENNVITIDLVQQSSQKTQNDVDIADVAYYFEKDVKGESLFHSKKMDLTVNGEQLDLDPGQTLIYYVDEKAPEFSMQGLKH Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Extracellular lysophospholipase D (E-NPP2) | 4ZGA | 5.64 | |
Target general information Gen name ENPP2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LysoPLD; Ectonucleotide pyrophosphatase/phosphodiesterase family member 2; E-NPP 2; Autotaxin; ATX Protein family Nucleotide pyrophosphatase/phosphodiesterase family Biochemical class Phosphoric diester hydrolase Function Hydrolyzes lysophospholipids to produce the signaling molecule lysophosphatidic acid (LPA) in extracellular fluids. Major substrate is lysophosphatidylcholine. Also can act on sphingosylphosphorylcholine producing sphingosine-1-phosphate, a modulator of cell motility. Can hydrolyze, in vitro, bis-pNPP, to some extent pNP-TMP, and barely ATP. Involved in several motility-related processes such as angiogenesis and neurite outgrowth. Acts as an angiogenic factor by stimulating migration of smooth muscle cells and microtubule formation. Stimulates migration of melanoma cells, probably via a pertussis toxin-sensitive G protein. May have a role in induction of parturition. Possible involvement in cell proliferation and adipose tissue development (Probable). Tumor cell motility-stimulating factor. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.1.4.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Chemotaxis; Cleavage on pair of basic residues; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lipid degradation; Lipid metabolism; Metal-binding; Obesity; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 85397.4 Length 740 Aromaticity 0.12 Instability index 58.63 Isoelectric point 7.16 Charge (pH=7) 1.02 2D Binding mode Binding energy (Kcal/mol) -7.69 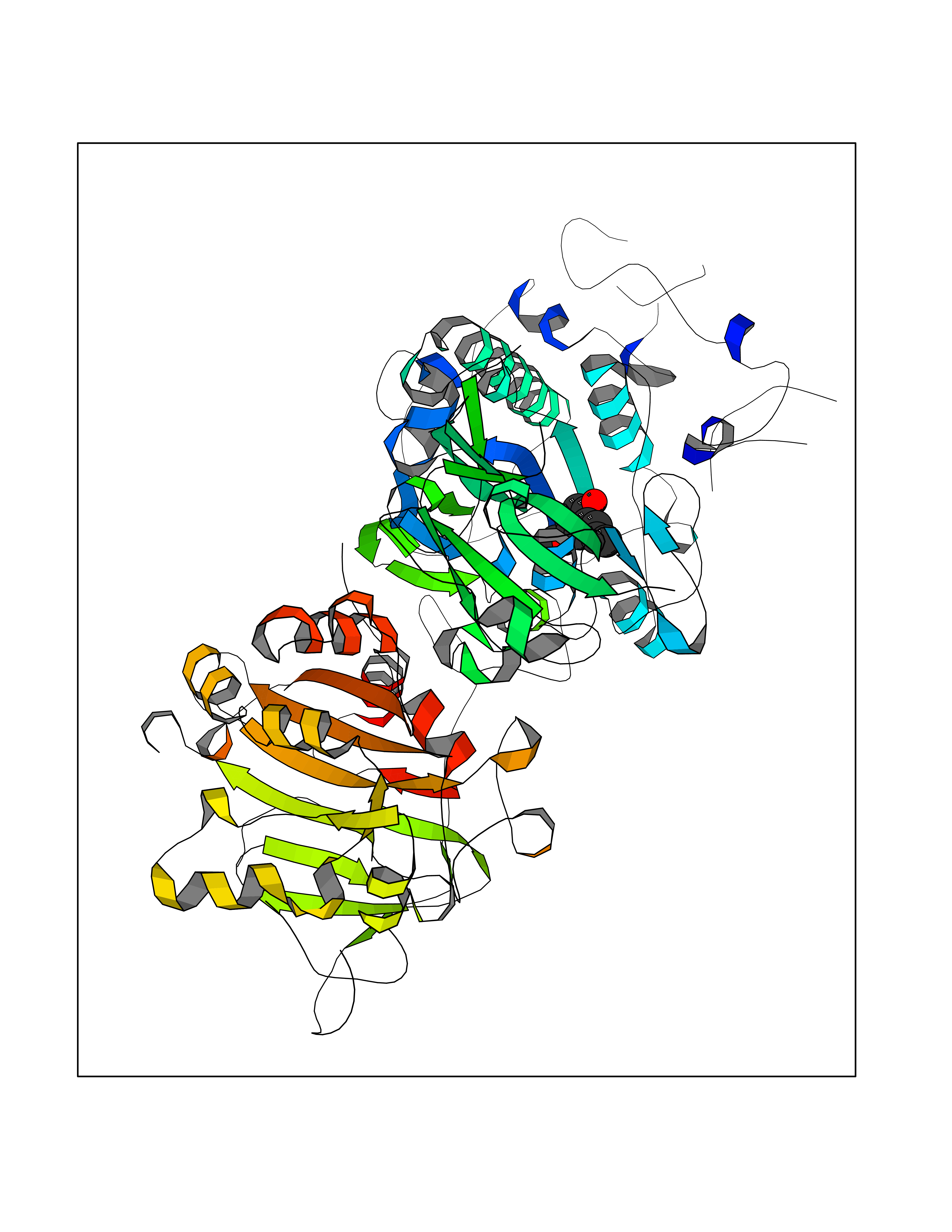 Molscript Map 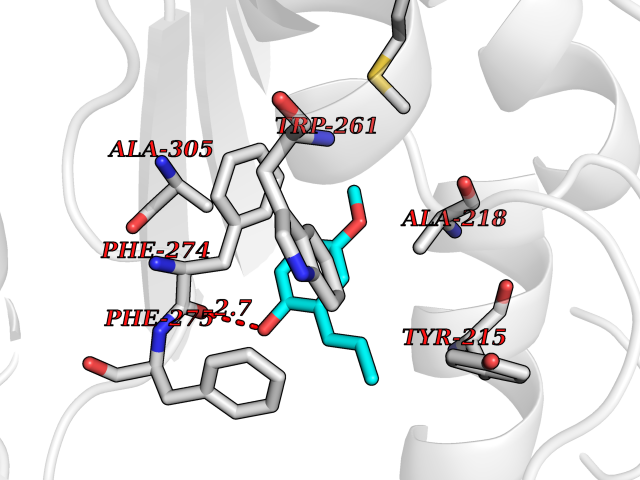 Pymol Map 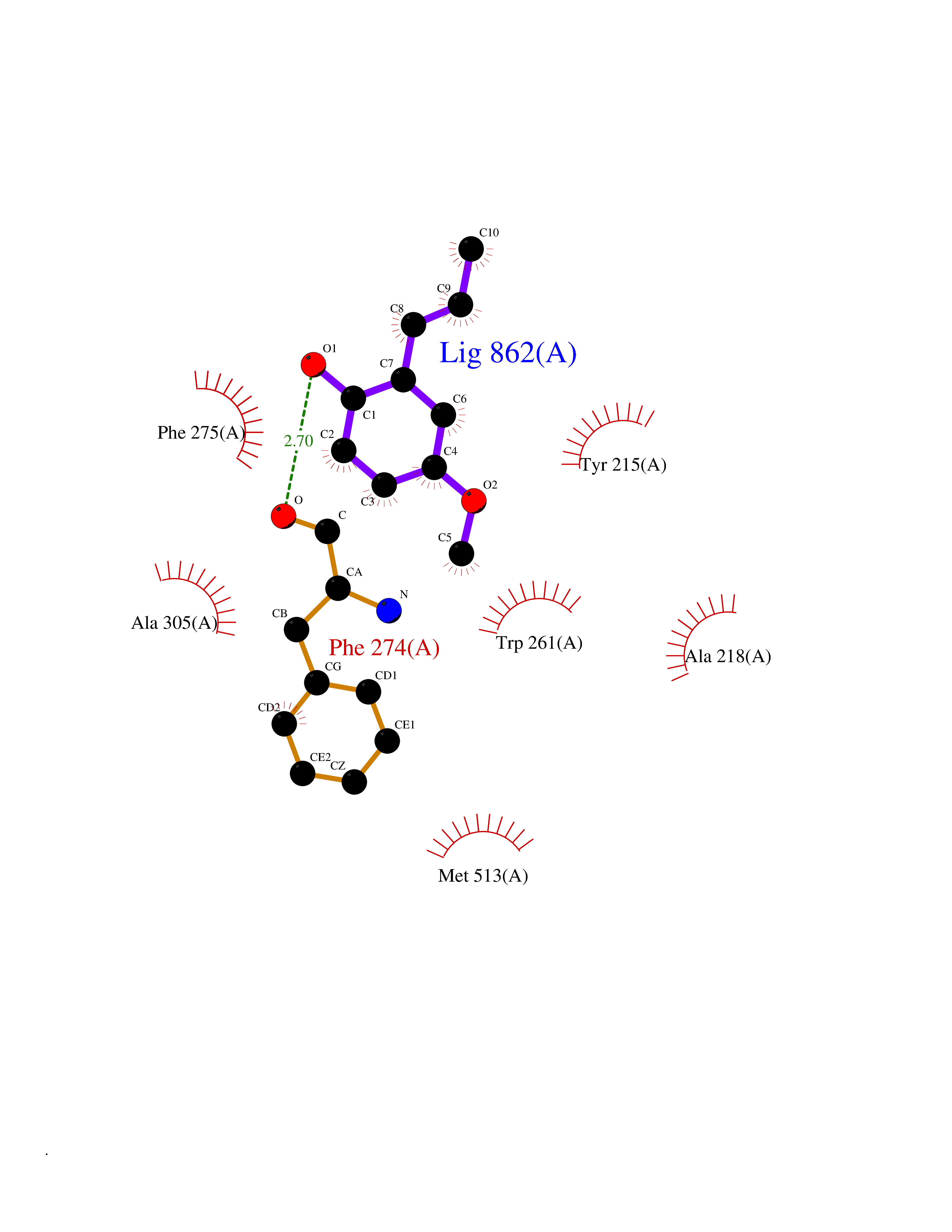 Ligplot Map 3D Binding mode Sequence GSCKGRCFELCRCDNLCKSYTSCCHDFDELCLKTARGWECTKDRCGNEENACHCEDCLARGDCCTNYQVVCKGESHWVDDDCEEIKAAECPAGFVRPPLIIFSVDGFRASYMKKGSKVMPNIEKLRSCGTHSPYMRPVYPTKTFPNLYTLATGLYPESHGIVGNSMYDPVFDATFHLRGREKFNHRWWGGQPLWITATKQGVKAGTFFWSVVIPHERRILTILQWLTLPDHERPSVYAFYSEQPDFSGHKYGPFGPEMTNPLREIDKIVGQLMDGLKQLKLHRCVNVIFVGDHGMEDVTCDRTEFLSNYLTNVDDITLVPGTLGRIRSKFDPKAIIANLTCKKPDQHFKPYLKQHLPKRLHYANNRRIEDIHLLVERRWHVARKPFFQGDHGFDNKVNSMQTVFVGYGSTFKYKTKVPPFENIELYNVMCDLLGLKPAPNNGTHGSLNHLLRTNTFRPTMPEEVTRPNYPGIMYLQSDFDLGTEERHLLYGRPAVLYRTRYDILYHTDFESGYSEIFLMPLWTSYTVSKQACVRPDVRVSPSFSQNCLAYKNDKQMSYGFLFPPYLSSSPEAKYDAFLVTNMVPMYPAFKRVWNYFQRVLVKKYASERNGVNVISGPIFDYDYDGLHDTEDKIKQYVEGSSIPVPTHYYSIITSCLDFTQPADKCDGPLSVSSFILPHRPDNEESCNSSEDESKWVEELMKMHTARVRDIEHLTSLDFFRKTSRSYPEILTLKTYLHTYE Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Cytochrome P450 1B1 (CYP1B1) | 3PM0 | 5.64 | |
Target general information Gen name CYP1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CYPIB1 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function In liver microsomes, this enzyme is involved in an NADPH-dependent electron transport pathway. It oxidizes a variety of structurally unrelated compounds, including steroids, fatty acids, retinoid and xenobiotics. Preferentially oxidizes 17beta-estradiol to the carcinogenic 4-hydroxy derivative, and a variety of procarcinogenic compounds to their activated forms, including polycyclic aromatic hydrocarbons. Promotes angiogenesis by removing cellular oxygenation products, thereby decreasing oxidative stress, release of antiangiogenic factor THBS2, then allowing endothelial cells migration, cell adhesion and capillary morphogenesis. These changes are concommitant with the endothelial nitric oxide synthase activity and nitric oxide synthesis. Plays an important role in the regulation of perivascular cell proliferation, migration, and survival through modulation of the intracellular oxidative state and NF-kappa-B expression and/or activity, during angiogenesis. Contributes to oxidative homeostasis and ultrastructural organization and function of trabecular meshwork tissue through modulation of POSTN expression. Cytochromes P450 are a group of heme-thiolate monooxygenases. Related diseases Anterior segment dysgenesis 6 (ASGD6) [MIM:617315]: A form of anterior segment dysgenesis, a group of defects affecting anterior structures of the eye including cornea, iris, lens, trabecular meshwork, and Schlemm canal. Anterior segment dysgeneses result from abnormal migration or differentiation of the neural crest derived mesenchymal cells that give rise to components of the anterior chamber during eye development. Different anterior segment anomalies may exist alone or in combination, including iris hypoplasia, enlarged or reduced corneal diameter, corneal vascularization and opacity, posterior embryotoxon, corectopia, polycoria, abnormal iridocorneal angle, ectopia lentis, and anterior synechiae between the iris and posterior corneal surface. Clinical conditions falling within the phenotypic spectrum of anterior segment dysgeneses include aniridia, Axenfeld anomaly, Reiger anomaly/syndrome, Peters anomaly, and iridogoniodysgenesis. ASGD6 patients predominantly manifest Peters anomaly. Peters anomaly consists of corneal leukoma, defects in the posterior structures of the cornea such as absence of the posterior corneal stroma and Descemet membrane, and a variable degree of iridocorneal and/or keratolenticular adhesions. Over 50% of patients develop glaucoma in childhood. {ECO:0000269|PubMed:11403040}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glaucoma 3, primary congenital, A (GLC3A) [MIM:231300]: An autosomal recessive form of primary congenital glaucoma (PCG). PCG is characterized by marked increase of intraocular pressure at birth or early childhood, large ocular globes (buphthalmos) and corneal edema. It results from developmental defects of the trabecular meshwork and anterior chamber angle of the eye that prevent adequate drainage of aqueous humor. {ECO:0000269|PubMed:10227395, ECO:0000269|PubMed:10655546, ECO:0000269|PubMed:11184479, ECO:0000269|PubMed:11527932, ECO:0000269|PubMed:11774072, ECO:0000269|PubMed:11980847, ECO:0000269|PubMed:12036985, ECO:0000269|PubMed:12525557, ECO:0000269|PubMed:14635112, ECO:0000269|PubMed:14640114, ECO:0000269|PubMed:15255109, ECO:0000269|PubMed:15342693, ECO:0000269|PubMed:15475877, ECO:0000269|PubMed:16490498, ECO:0000269|PubMed:16688110, ECO:0000269|PubMed:16735994, ECO:0000269|PubMed:16862072, ECO:0000269|PubMed:18470941, ECO:0000269|PubMed:9463332, ECO:0000269|PubMed:9497261}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Glaucoma 1, open angle, A (GLC1A) [MIM:137750]: A form of primary open angle glaucoma (POAG). POAG is characterized by a specific pattern of optic nerve and visual field defects. The angle of the anterior chamber of the eye is open, and usually the intraocular pressure is increased. However, glaucoma can occur at any intraocular pressure. The disease is generally asymptomatic until the late stages, by which time significant and irreversible optic nerve damage has already taken place. {ECO:0000269|PubMed:11774072}. The gene represented in this entry acts as a disease modifier. Digenic mutations in CYP1B1 and MYOC have been found in a family segregating both primary adult-onset and juvenile forms of open angle glaucoma (PubMed:11774072). All affected family members with mutations in both MYOC and CYP1B1 had juvenile glaucoma, whereas those with only the MYOC mutation had the adult-onset form (PubMed:11774072). {ECO:0000269|PubMed:11774072}. Drugs (DrugBank ID) DB02342; DB00613; DB06732; DB00443; DB00121; DB01222; DB00201; DB09061; DB14737; DB01254; DB00694; DB01248; DB00997; DB00470; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB07776; DB00499; DB01645; DB01381; DB00741; DB01064; DB01026; DB00448; DB14009; DB01065; DB00170; DB00959; DB01204; DB14011; DB03467; DB00338; DB01229; DB14631; DB00635; DB01087; DB00396; DB00818; DB04216; DB02709; DB00675; DB00624; DB13946; DB00277; DB12245; DB11155 Interacts with Q02763 EC number EC 1.14.14.- Uniprot keywords 3D-structure; Disease variant; Endoplasmic reticulum; Fatty acid metabolism; Glaucoma; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Mitochondrion; Monooxygenase; Oxidoreductase; Peters anomaly; Proteomics identification; Reference proteome; Steroid metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 51875.9 Length 459 Aromaticity 0.1 Instability index 34.16 Isoelectric point 8.64 Charge (pH=7) 4.89 2D Binding mode Binding energy (Kcal/mol) -7.69 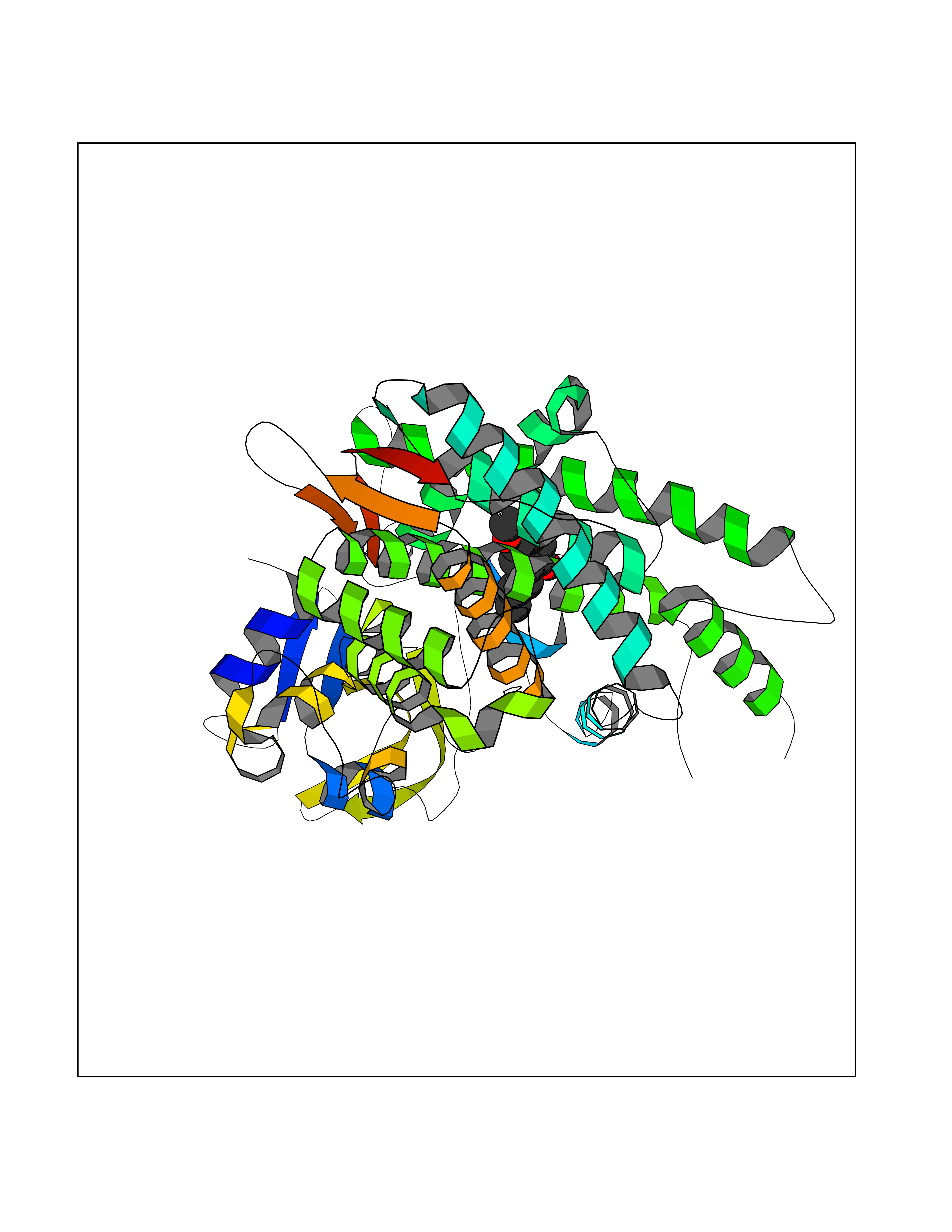 Molscript Map 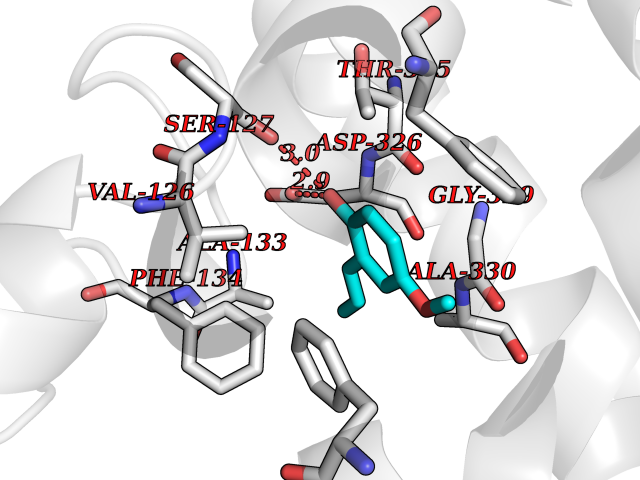 Pymol Map 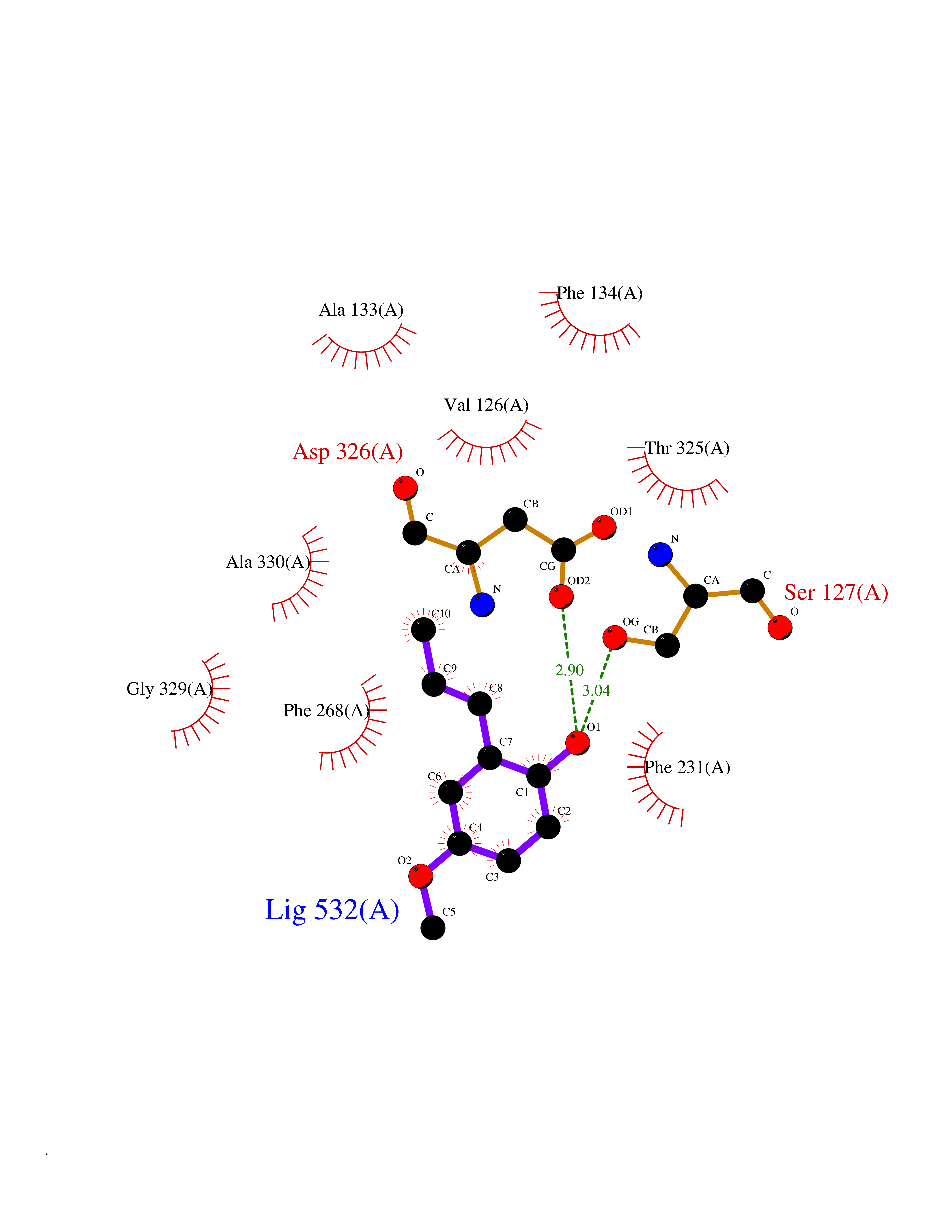 Ligplot Map 3D Binding mode Sequence QAAHLSFARLARRYGDVFQIRLGSCPIVVLNGERAIHQALVQQGSAFADRPSFASFRVVSGGRSMAFGHYSEHWKVQRRAAHSMMRNFFTRQPRSRQVLEGHVLSEARELVALLVRGSADGAFLDPRPLTVVAVANVMSAVCFGCRYSHDDPEFRELLSHNEEFGRTVGAGSLVDVMPWLQYFPNPVRTVFREFEQLNRNFSNFILDKFLRHCESLRPGAAPRDMMDAFILSAEKKAAGDGARLDLENVPATITDIFGASQDTLSTALQWLLLLFTRYPDVQTRVQAELDQVVGRDRLPCMGDQPNLPYVLAFLYEAMRFSSFVPVTIPHATTANTSVLGYHIPKDTVVFVNQWSVNHDPLKWPNPENFDPARFLDKDGLINKDLTSRVMIFSVGKRRCIGEELSKMQLFLFISILAHQCDFRANPNEPAKMNFSYGLTIKPKSFKVNVTLRESMELLD Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Receptor-interacting protein 1 (RIPK1) | 5TX5 | 5.64 | |
Target general information Gen name RIPK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Receptor-interacting serine/threonine-protein kinase 1; RIP1; RIP-1; RIP; Cell death protein RIP Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family Biochemical class Kinase Function Upon activation of TNFR1 by the TNF-alpha family cytokines, TRADD and TRAF2 are recruited to the receptor. Phosphorylates DAB2IP at 'Ser-728' in a TNF-alpha-dependent manner, and thereby activates the MAP3K5-JNK apoptotic cascade. Ubiquitination by TRAF2 via 'Lys-63'-link chains acts as a critical enhancer of communication with downstream signal transducers in the mitogen-activated protein kinase pathway and the NF-kappa-B pathway, which in turn mediate downstream events including the activation of genes encoding inflammatory molecules. Polyubiquitinated protein binds to IKBKG/NEMO, the regulatory subunit of the IKK complex, a critical event for NF-kappa-B activation. Interaction with other cellular RHIM-containing adapters initiates gene activation and cell death. RIPK1 and RIPK3 association, in particular, forms a necrosis-inducing complex. Serine-threonine kinase which transduces inflammatory and cell-death signals (programmed necrosis) following death receptors ligation, activation of pathogen recognition receptors (PRRs), and DNA damage. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with P04083; Q13490; Q13489; Q92851; Q14790; Q8IVM0; P48729; Q13158; Q9Y6K9; Q96AB6; Q9ULZ3; Q13546; Q9Y572; P19438; Q13077; Q12933; Q13114; Q13107; B7UI21; PRO_0000449629 [P0DTD1]; U5TQE9 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell membrane; Cytoplasm; Disease variant; Glycoprotein; Host-virus interaction; Inflammatory response; Isopeptide bond; Kinase; Membrane; Necrosis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 29554.2 Length 259 Aromaticity 0.08 Instability index 48.26 Isoelectric point 6.29 Charge (pH=7) -2.52 2D Binding mode Binding energy (Kcal/mol) -7.69 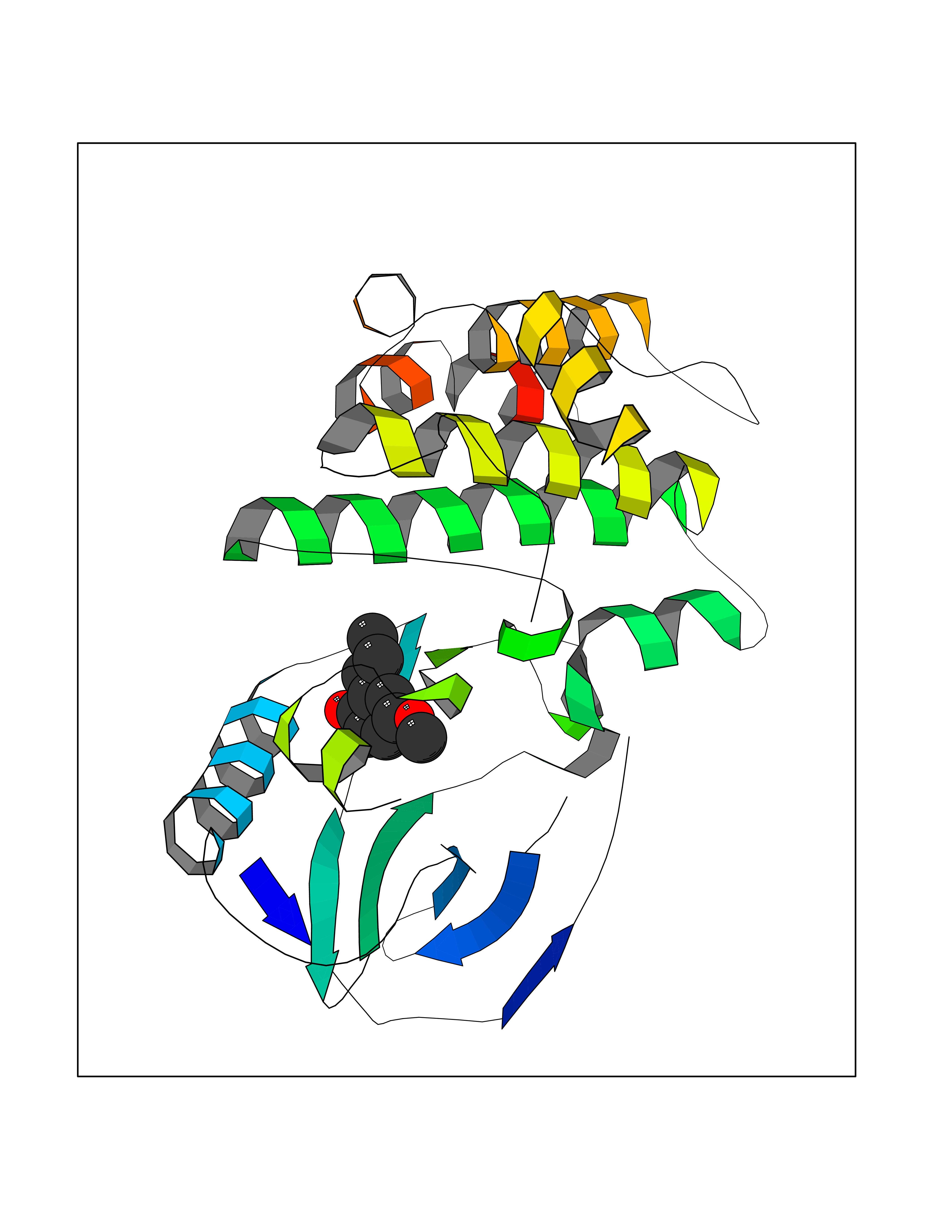 Molscript Map 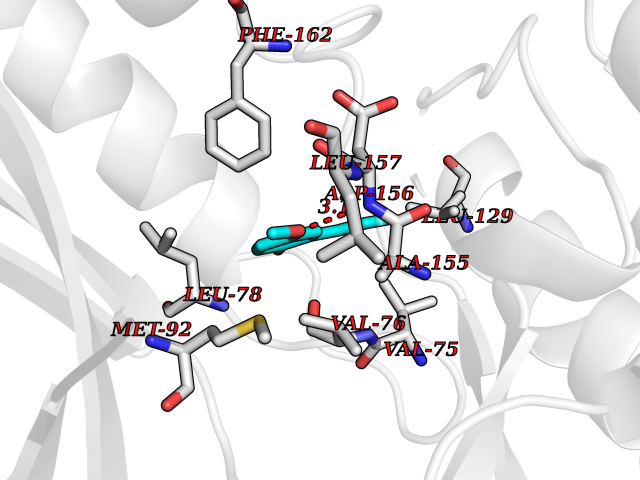 Pymol Map 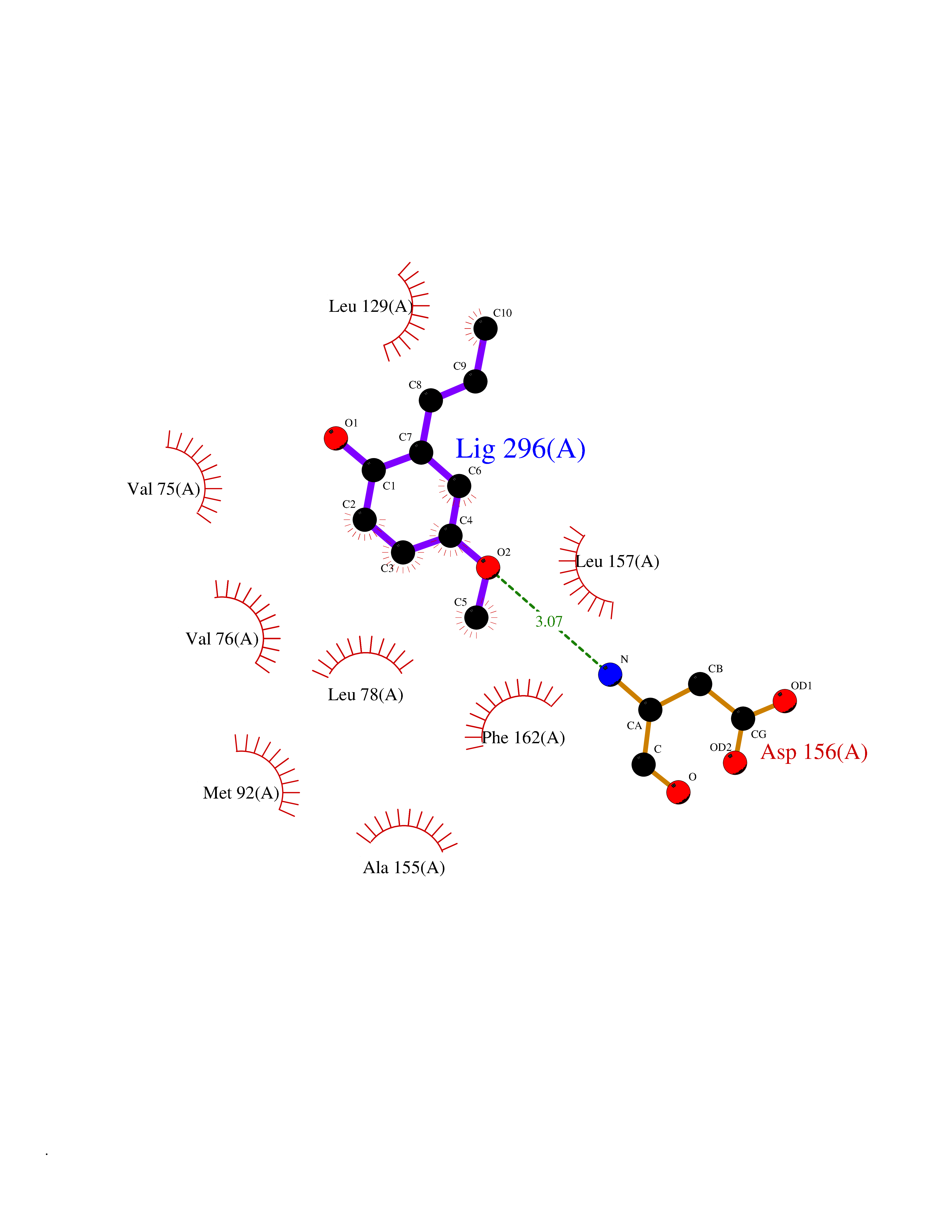 Ligplot Map 3D Binding mode Sequence IKMKSSDFLESAELDSGGKVSLAFHRTQGLMIMKTVYKGPNCIEHNEALLEEAKMMNRLRHSRVVKLLGVIIEEGKYSLVMEYMEKGNLMHVLKAEMSTPLSVKGRIILEIIEGMAYLHGKGVIHKDLKPENILVDNDFHIKIADLGLASFKMWSKLNGTLYYMAPEHLNDVNAKPTEKSDVYSFAVVLWAIFANKEPYQQLIMAIKSGNRPDVDDITEYCPREIISLMKLCWEANPEARPTFPGIEEKFRPFYLSQLE Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Thymidine kinase | 1E2K | 5.63 | |
Target general information Gen name TK Organism Human herpesvirus 1 (strain 17) (HHV-1) (Human herpes simplex virus 1) Uniprot ID TTD ID NA Synonyms UL23 Protein family Herpesviridae thymidine kinase family Biochemical class Transferase Function ATP binding.Thymidine kinase activity. Related diseases Atransferrinemia (ATRAF) [MIM:209300]: A rare autosomal recessive disorder characterized by abnormal synthesis of transferrin leading to iron overload and microcytic hypochromic anemia. {ECO:0000269|PubMed:11110675, ECO:0000269|PubMed:15466165}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 2.7.1.21 Uniprot keywords 3D-structure; ATP-binding; DNA synthesis; Early protein; Kinase; Nucleotide-binding; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 67808.8 Length 624 Aromaticity 0.07 Instability index 43.94 Isoelectric point 6.1 Charge (pH=7) -7.02 2D Binding mode Binding energy (Kcal/mol) -7.68  Molscript Map 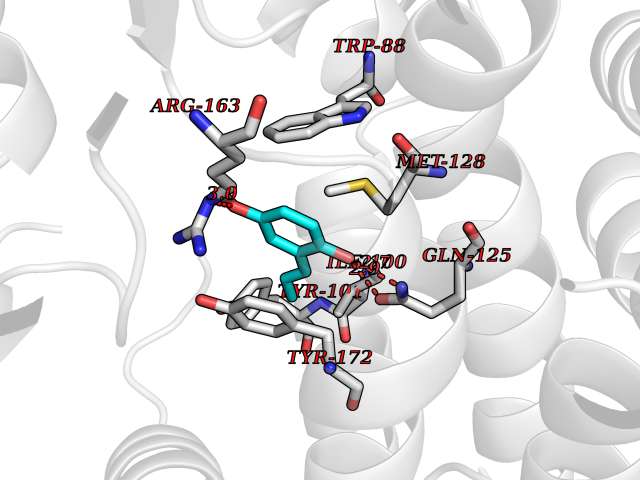 Pymol Map 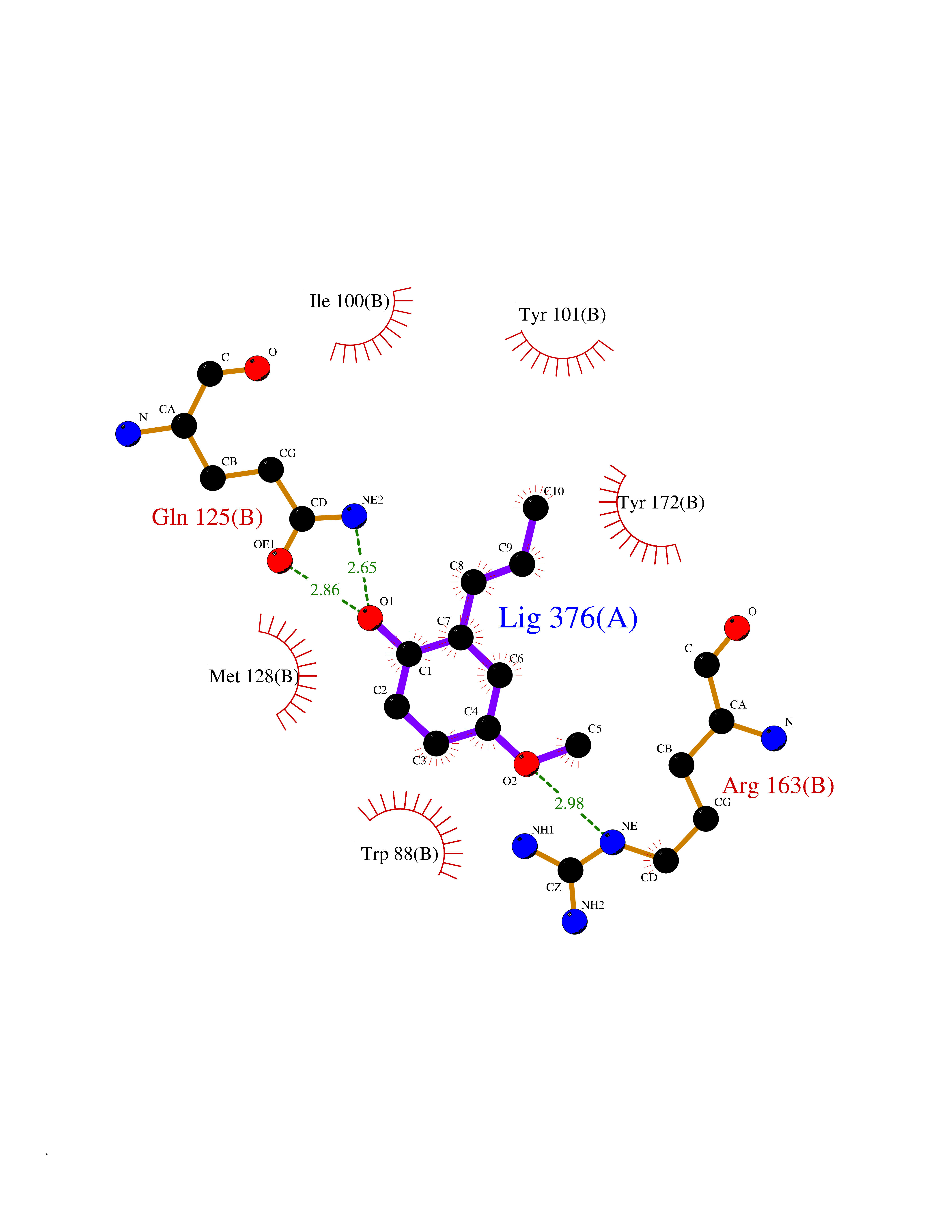 Ligplot Map 3D Binding mode Sequence MPTLLRVYIDGPHGMGKTTTTQLLVADDIVYVPEPMTYWRVLGASETIANIYTTQHRLDQGEISAGDAAVVMTSAQITMGMPYAVTDAVLAPHIGGEAGPPPALTLIFDRHPIAALLCYPAARYLMGSMTPQAVLAFVALIPPTLPGTNIVLGALPEDRHIDRLAKRQRPGERLDLAMLAAIRRVYGLLANTVRYLQCGGSWREDWGQLSGTGPRPHIGDTLFTLFRAPELLAPNGDLYNVFAWALDVLAKRLRSMHVFILDYDQSPAGCRDALLQLTSGMVQTHVTTPGSIPTICDLARTFAREMGEMPTLLRVYIDGPHGMGKTTTTQLLVALGSRDDIVYVPEPMTYWRVLGASETIANIYTTQHRLDQGEISAGDAAVVMTSAQITMGMPYAVTDAVLAPHIGGEAHAPPPALTLIFDRHPIAALLCYPAARYLMGSMTPQAVLAFVALIPPTLPGTNIVLGALPEDRHIDRLAKRERLDLAMLAAIRRVYGLLANTVRYLQCGGSWREDWGQLSGTAVPQSNAGPRPHIGDTLFTLFRAPELLAPNGDLYNVFAWALDVLAKRLRSMHVFILDYDQSPAGCRDALLQLTSGMVQTHVTTPGSIPTICDLARTFAREMGE Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Leukotriene A-4 hydrolase (LTA4H) | 3U9W | 5.63 | |
Target general information Gen name LTA4H Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Leukotriene A4 hydrolase; Leukotriene A(4)Leukotriene A-4 hydrolase hydrolase; Leukotriene A(4) hydrolase; LTA4; LTA-H; LTA-4hydrolase; LTA-4 hydrolase Protein family Peptidase M1 family Biochemical class Ether bond hydrolase Function Has also aminopeptidase activity. Epoxide hydrolase that catalyzes the final step in the biosynthesis of the proinflammatory mediator leukotriene B4. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07102; DB06917; DB07258; DB07094; DB07259; DB02352; DB07292; DB07104; DB06828; DB08466; DB01197; DB05177; DB03366; DB08040; DB06851; DB02062; DB07099; DB07260; DB07196; DB11781; DB03424; DB07237 Interacts with Q9BSI4 EC number EC 3.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Leukotriene biosynthesis; Lipid metabolism; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 68927 Length 608 Aromaticity 0.1 Instability index 38.84 Isoelectric point 5.87 Charge (pH=7) -9.86 2D Binding mode Binding energy (Kcal/mol) -7.67 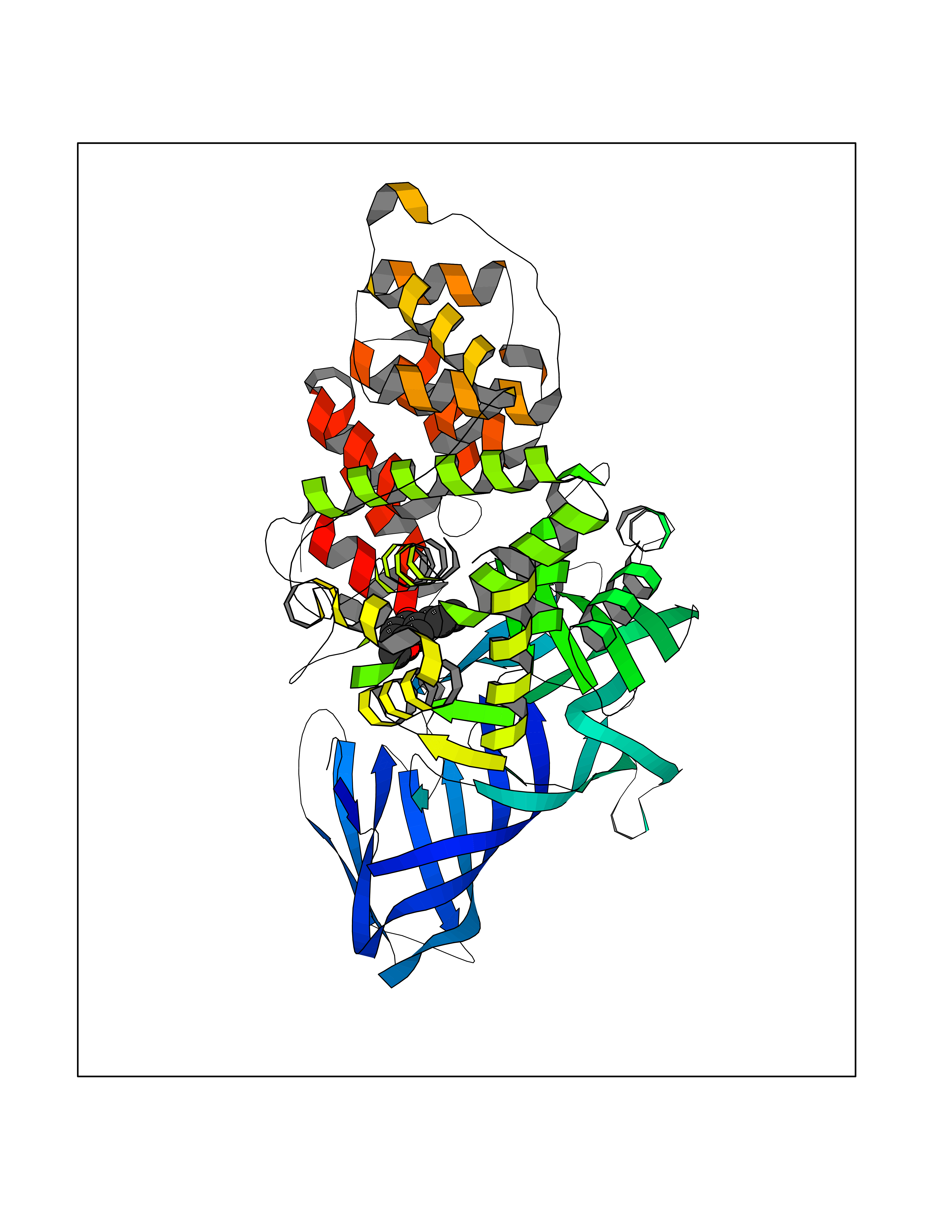 Molscript Map 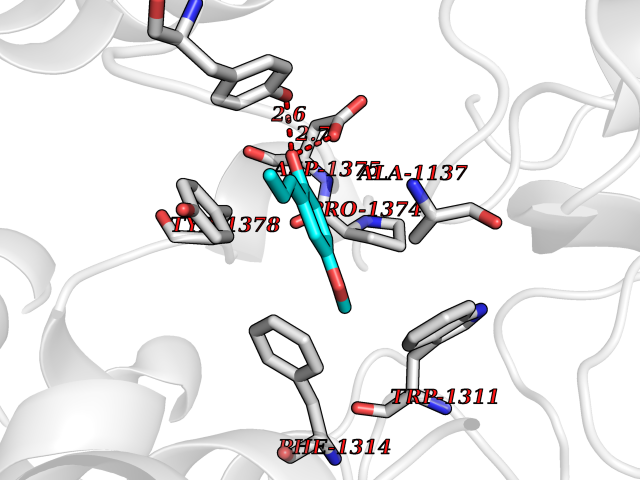 Pymol Map 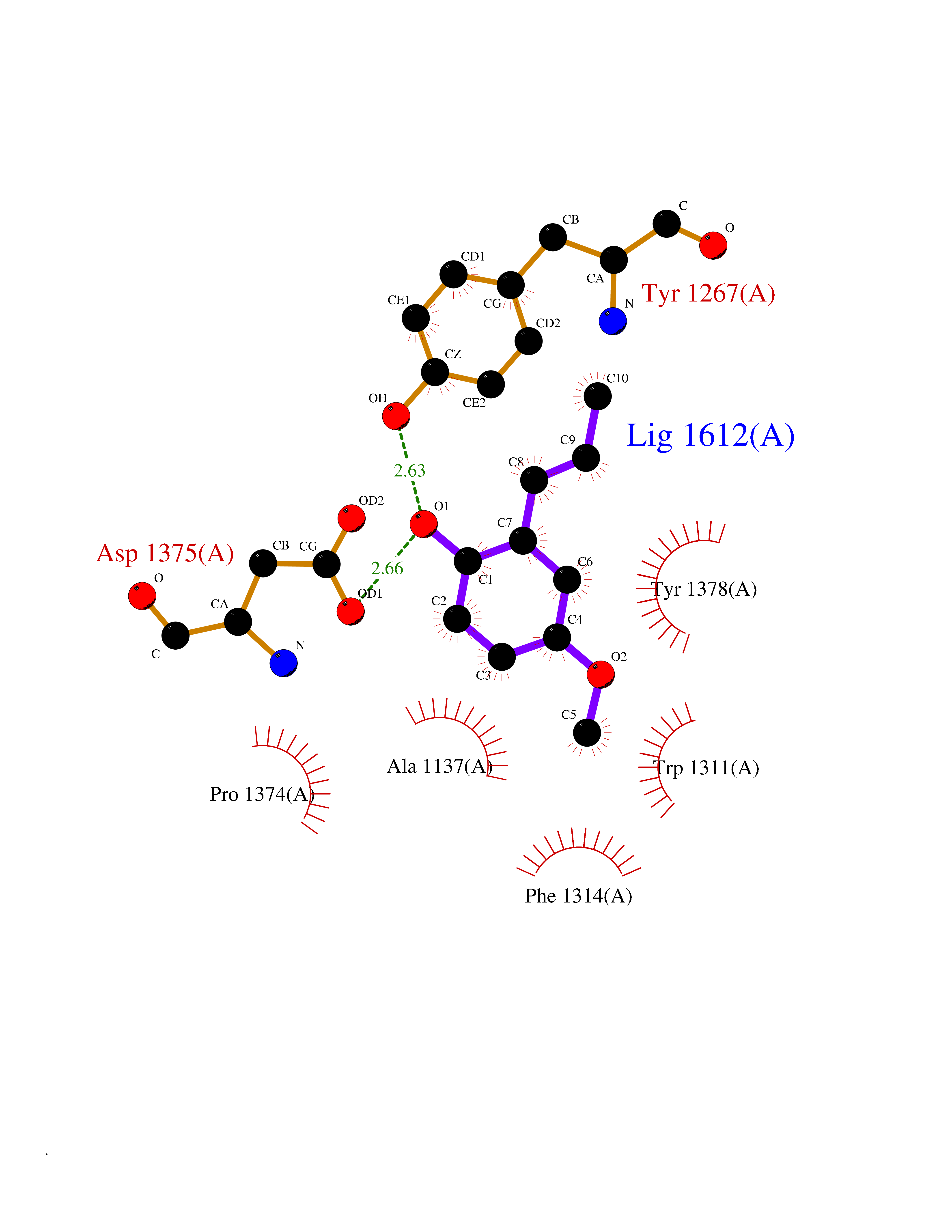 Ligplot Map 3D Binding mode Sequence IVDTCSLASPASVCRTKHLHLRCSVDFTRRTLTGTAALTVQSQEDNLRSLVLDTKDLTIEKVVINGQEVKYALGERQSYKGSPMEISLPIALSKNQEIVIEISFETSPKSSALQWLTPEQTSGKEHPYLFSQCQAIHCRAILPCQDTPSVKLTYTAEVSVPKELVALMSAIRDGETPDPEDPSRKIYKFIQKVPIPCYLIALVVGALESRQIGPRTLVWSEKEQVEKSAYEFSETESMLKIAEDLGGPYVWGQYDLLVLPPSFPYGGMENPCLTFVTPTLLAGDKSLSNVIAHEISHSWTGNLVTNKTWDHFWLNEGHTVYLERHICGRLFGEKFRHFNALGGWGELQNSVKTFGETHPFTKLVVDLTDIDPDVAYSSVPYEKGFALLFYLEQLLGGPEIFLGFLKAYVEKFSYKSITTDDWKDFLYSYFKDKVDVLNQVDWNAWLYSPGLPPIKPNYDMTLTNACIALSQRWITAKEDDLNSFNATDLKDLSSHQLNEFLAQTLQRAPLPLGHIKRMQEVYNFNAINNSEIRFRWLRLCIQSKWEDAIPLALKMATEQGRMKFTRPLFKDLAAFDKSHDQAVRTYQEHKASMHPVTAMLVGKDLKVD Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Free fatty acid receptor 1 | 4PHU | 5.63 | |
Target general information Gen name FFAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPR40 Protein family G-protein coupled receptor 1 family Biochemical class Fatty acid binding protein / hydrolase Function Bioactive lipid receptor activity.G-protein coupled receptor activity.Guanyl-nucleotide exchange factor activity.Lipid binding. Related diseases Refsum disease (RD) [MIM:266500]: A rare autosomal recessive peroxisomal disorder characterized by the accumulation of the branched-chain fatty acid, phytanic acid, in blood and tissues. Cardinal clinical features are retinitis pigmentosa, peripheral neuropathy, cerebellar ataxia, and elevated protein levels in the cerebrospinal fluid (CSF). Half of all patients exhibit generalized, mild to moderate ichthyosis resembling ichthyosis vulgaris. Less constant features are nerve deafness, anosmia, skeletal abnormalities, cataracts and cardiac impairment. {ECO:0000269|PubMed:10709665, ECO:0000269|PubMed:10767344, ECO:0000269|PubMed:14974078, ECO:0000269|PubMed:9326939, ECO:0000269|PubMed:9326940}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00159 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 28319.1 Length 272 Aromaticity 0.11 Instability index 27.3 Isoelectric point 9.07 Charge (pH=7) 6.85 2D Binding mode Binding energy (Kcal/mol) -7.68  Molscript Map 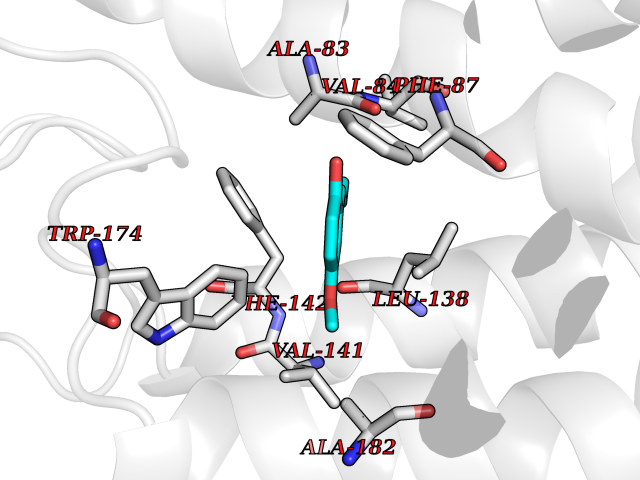 Pymol Map  Ligplot Map 3D Binding mode Sequence MDLPPQLSFGLYVAAFALGFPLNVLAIRGATAHARLRLTPSAVYALNLGCSDLLLTVSLPLKAVEALASGAWPLPASLCPVFAVAHFAPLYAGGGFLAALSAARYLGAAFPPCYSWGVCAAIWALVLCHLGLVFGLEAPGGWLDHSNTSLGINTPVNGSPVCLEAWDPASAGPARFSLSLLLFFLPLAITAFCFVGCLRALARGSLTHRRKLRAAWVAGGALLTLLLCVGPYNASNVASFLYPNLGGSWRKLGLITGAWSVVLNPLVTGYLG Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Retinoic acid receptor RXR-alpha (RXRA) | 2P1T | 5.63 | |
Target general information Gen name RXRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor alpha; RXRalpha; Nuclear receptor subfamily 2 group B member 1; NR2B1 Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. The high affinity ligand for RXRs is 9-cis retinoic acid. RXRA serves as a common heterodimeric partner for a number of nuclear receptors. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. The RXRA/PPARA heterodimer is required for PPARA transcriptional activity on fatty acid oxidation genes such as ACOX1 and the P450 system genes. Receptor for retinoic acid. Related diseases Lichtenstein-Knorr syndrome (LIKNS) [MIM:616291]: An autosomal recessive neurologic disorder characterized by progressive cerebellar ataxia and severe progressive sensorineural hearing loss. {ECO:0000269|PubMed:25205112, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08063; DB08402; DB07863; DB07557; DB00459; DB00210; DB01436; DB00523; DB00132; DB04557; DB00307; DB01393; DB03756; DB00749; DB00926; DB05956; DB04224; DB02746; DB00412; DB00755; DB08601 Interacts with O14503; P35637; Q15648; Q71SY5; Q15788; Q15596; P55055; P55055-1; Q13133; P27986; P37231; P37231-1; P10276; P42224; P11473; P97792-1; Q9JLI4; P04625; PRO_0000278730 [Q03463] EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; DNA-binding; Host-virus interaction; Isopeptide bond; Metal-binding; Mitochondrion; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 24958.9 Length 221 Aromaticity 0.07 Instability index 39.47 Isoelectric point 6.16 Charge (pH=7) -3.45 2D Binding mode Binding energy (Kcal/mol) -7.67  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DMPVERILEAELAVEDPVTNICQAADKQLFTLVEWAKRIPHFSELPLDDQVILLRAGWNELLIASFSHRSIAVKDGILLATGLHVHRNSAHSAGVGAIFDRVLTELVSKMRDMQMDKTELGCLRAIVLFNPDSKGLSNPAEVEALREKVYASLEAYCKHKYPEQPGRFAKLLLRLPALRSIGLKCLEHLFFFKLIGDTPIDTFLMEMLEAPHKILHRLLQD Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Peptostreptococcal albumin-binding protein | 2VDB | 5.63 | |
Target general information Gen name pab Organism Finegoldia magna (Peptostreptococcus magnus) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Protein binding Function Binds serum albumin. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03600; DB00788 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell wall; Peptidoglycan-anchor; Secreted; Signal Protein physicochemical properties Chain ID B Molecular weight (Da) 21751.6 Length 189 Aromaticity 0.1 Instability index 49.17 Isoelectric point 5.44 Charge (pH=7) -6.7 2D Binding mode Binding energy (Kcal/mol) -7.68 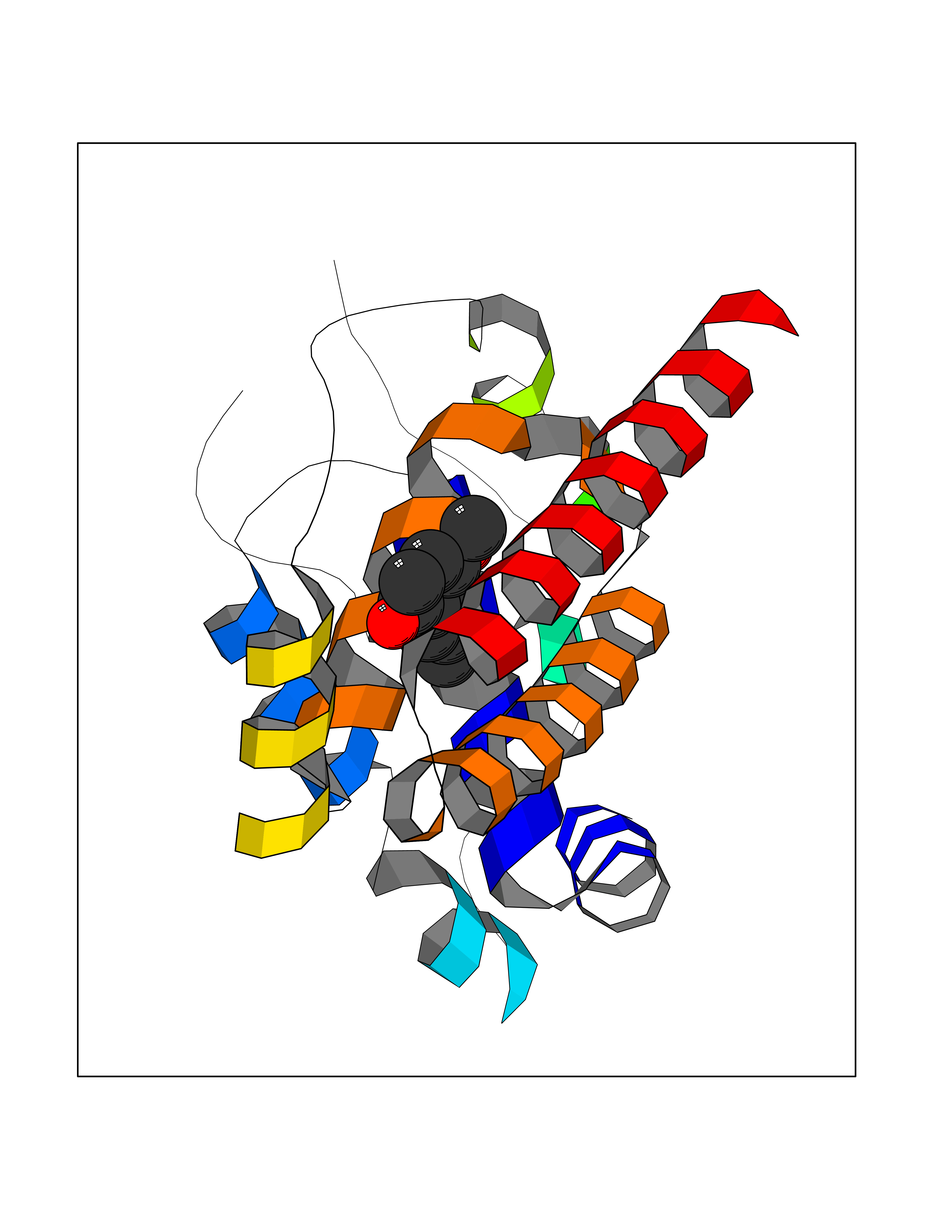 Molscript Map 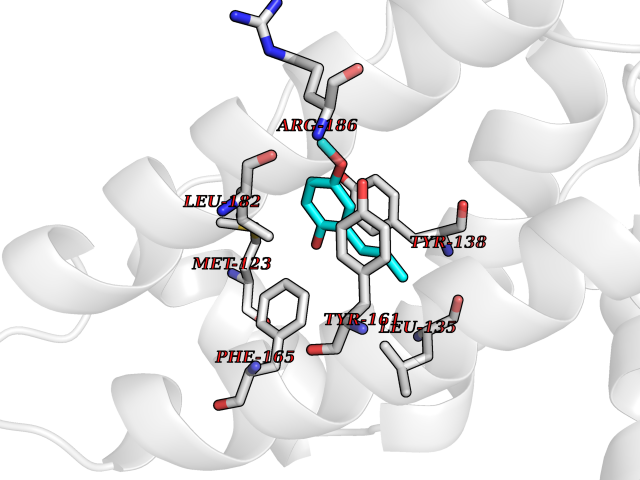 Pymol Map 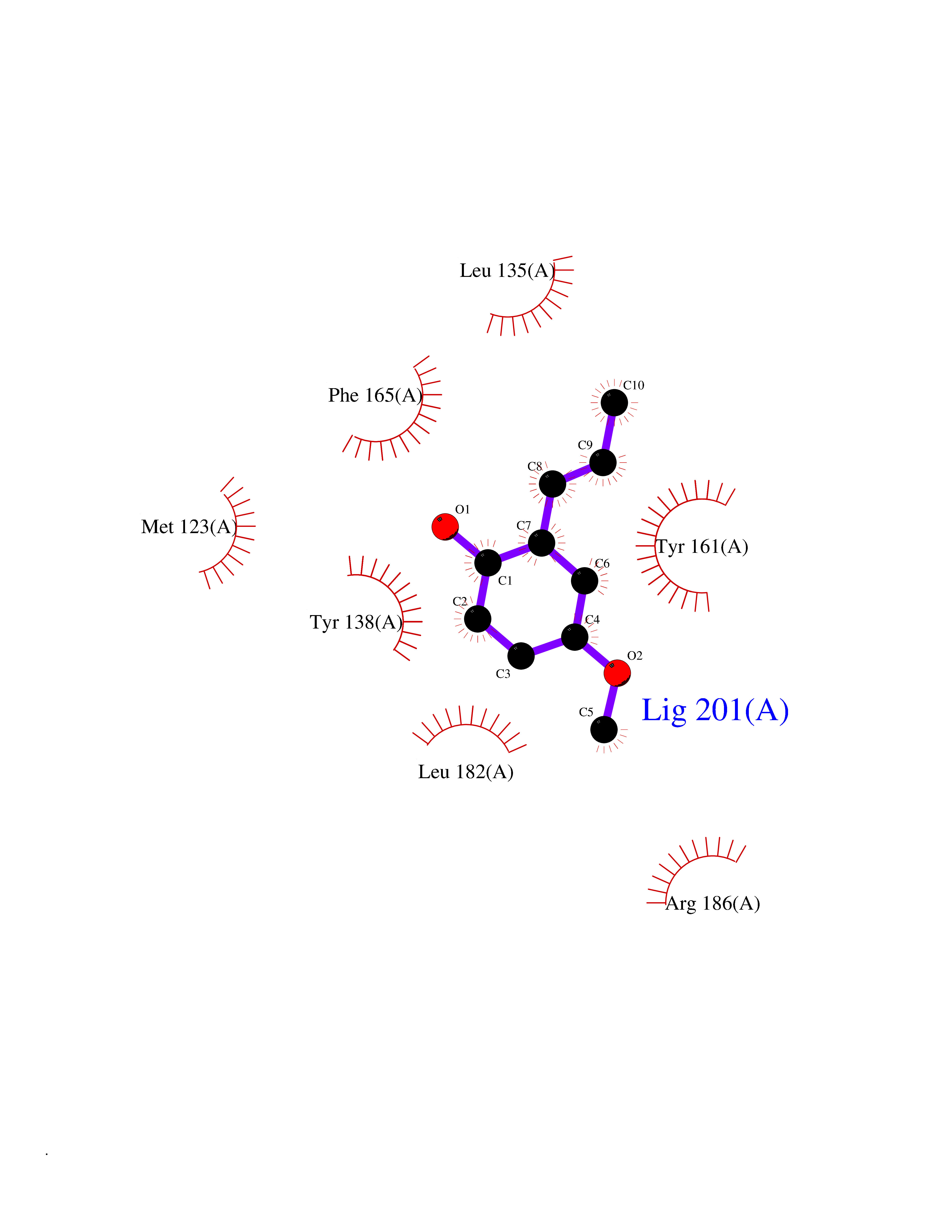 Ligplot Map 3D Binding mode Sequence EVAHRFKDLGEENFKALVLIAFAQYLQQCPFEDHVKLVNEVTEFAKTCVADESAENCDKSLHTLFGDKLCTVATLEMADCCAKQEPERNECFLQHKDDNPNLPRLVRPEVDVMCTAFHDNEETFLKKYLYEIARRHPYFYAPELLFFAKRYKAAFTECCQAADKAACLLPKLDELRDEGKASSAKQRLK Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Histone deacetylase 2 (HDAC2) | 4LY1 | 5.63 | |
Target general information Gen name HDAC2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HD2 Protein family Histone deacetylase family, HD type 1 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Forms transcriptional repressor complexes by associating with MAD, SIN3, YY1 and N-COR. Interacts in the late S-phase of DNA-replication with DNMT1 in the other transcriptional repressor complex composed of DNMT1, DMAP1, PCNA, CAF1. Deacetylates TSHZ3 and regulates its transcriptional repressor activity. Component of a RCOR/GFI/KDM1A/HDAC complex that suppresses, via histone deacetylase (HDAC) recruitment, a number of genes implicated in multilineage blood cell development. May be involved in the transcriptional repression of circadian target genes, such as PER1, mediated by CRY1 through histone deacetylation. Involved in MTA1-mediated transcriptional corepression of TFF1 and CDKN1A. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4). Related diseases Ventricular tachycardia, catecholaminergic polymorphic, 1, with or without atrial dysfunction and/or dilated cardiomyopathy (CPVT1) [MIM:604772]: An arrhythmogenic disorder characterized by stress-induced, bidirectional ventricular tachycardia that may degenerate into cardiac arrest and cause sudden death. Patients present with recurrent syncope, seizures, or sudden death after physical activity or emotional stress. CPVT1 inheritance is autosomal dominant. {ECO:0000269|PubMed:11157710, ECO:0000269|PubMed:11159936, ECO:0000269|PubMed:11208676, ECO:0000269|PubMed:12093772, ECO:0000269|PubMed:12106942, ECO:0000269|PubMed:14571276, ECO:0000269|PubMed:15046072, ECO:0000269|PubMed:15046073, ECO:0000269|PubMed:15466642, ECO:0000269|PubMed:15544015, ECO:0000269|PubMed:16188589, ECO:0000269|PubMed:24793461, ECO:0000269|PubMed:25372681, ECO:0000269|PubMed:27733687}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ventricular arrhythmias due to cardiac ryanodine receptor calcium release deficiency syndrome (VACRDS) [MIM:115000]: An autosomal dominant arrhythmogenic disorder characterized by syncope, cardiac arrest and/or sudden unexpected death, often in association with physical exertion or acute emotional stress. Patients who survive manifest polymorphic ventricular tachycardia and ventricular fibrillation. Unlike typical catecholaminergic ventricular tachycardia, arrhythmias are not reproducible on exercise stress testing or adrenaline challenge. {ECO:0000269|PubMed:12093772, ECO:0000269|PubMed:17984046, ECO:0000269|PubMed:33536282}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12565; DB01223; DB01076; DB05015; DB01262; DB11841; DB01095; DB12645; DB00227; DB11830; DB01303; DB06603; DB06819; DB05223; DB00175; DB03766; DB12847; DB06176; DB00641; DB00277; DB09091; DB00313; DB02546 Interacts with Q9C0K0; Q9HCU9; P68400; Q9UER7; P51610; Q13547; Q9UIS9; Q13330; P01106; P06748; P48382; Q96ST3; O95863; Q9HD15; O43463; Q9H3M7; Q92618; Q17R98; Q2HR82; PRO_0000449623 [P0DTD1] EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Cytoplasm; Hydrolase; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-nitrosylation; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 42020.5 Length 366 Aromaticity 0.13 Instability index 29.52 Isoelectric point 6.52 Charge (pH=7) -2.16 2D Binding mode Binding energy (Kcal/mol) -7.68 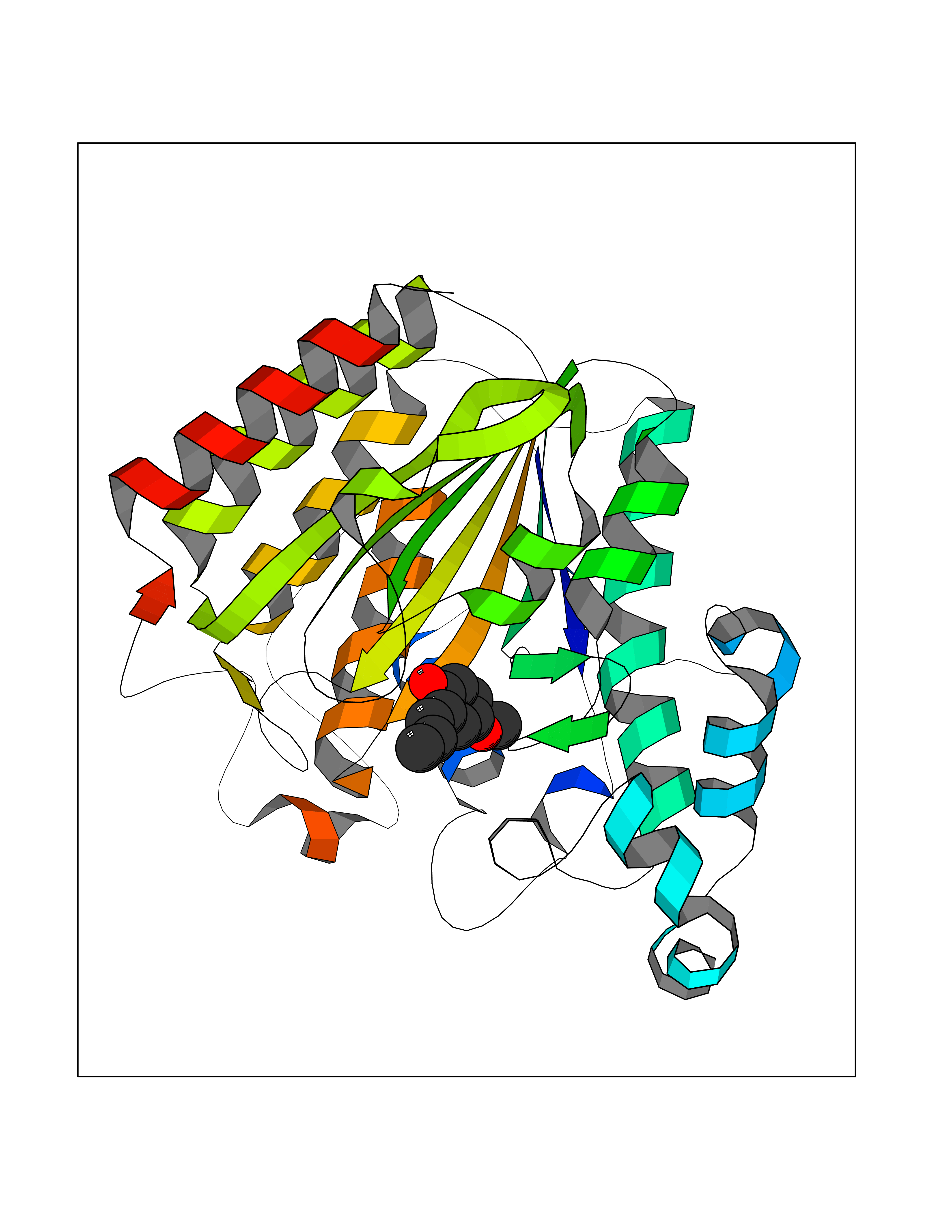 Molscript Map 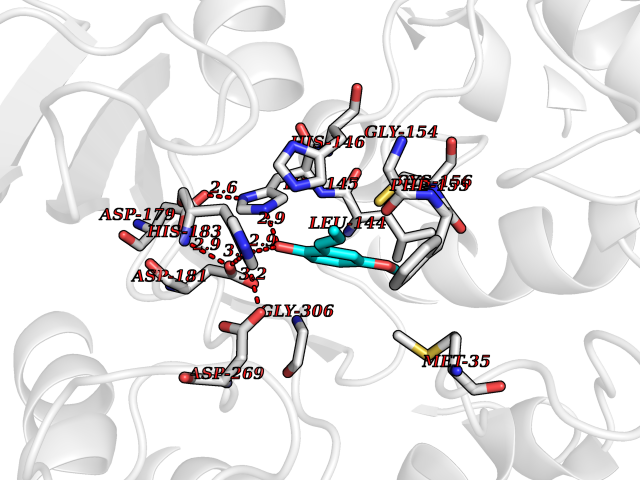 Pymol Map 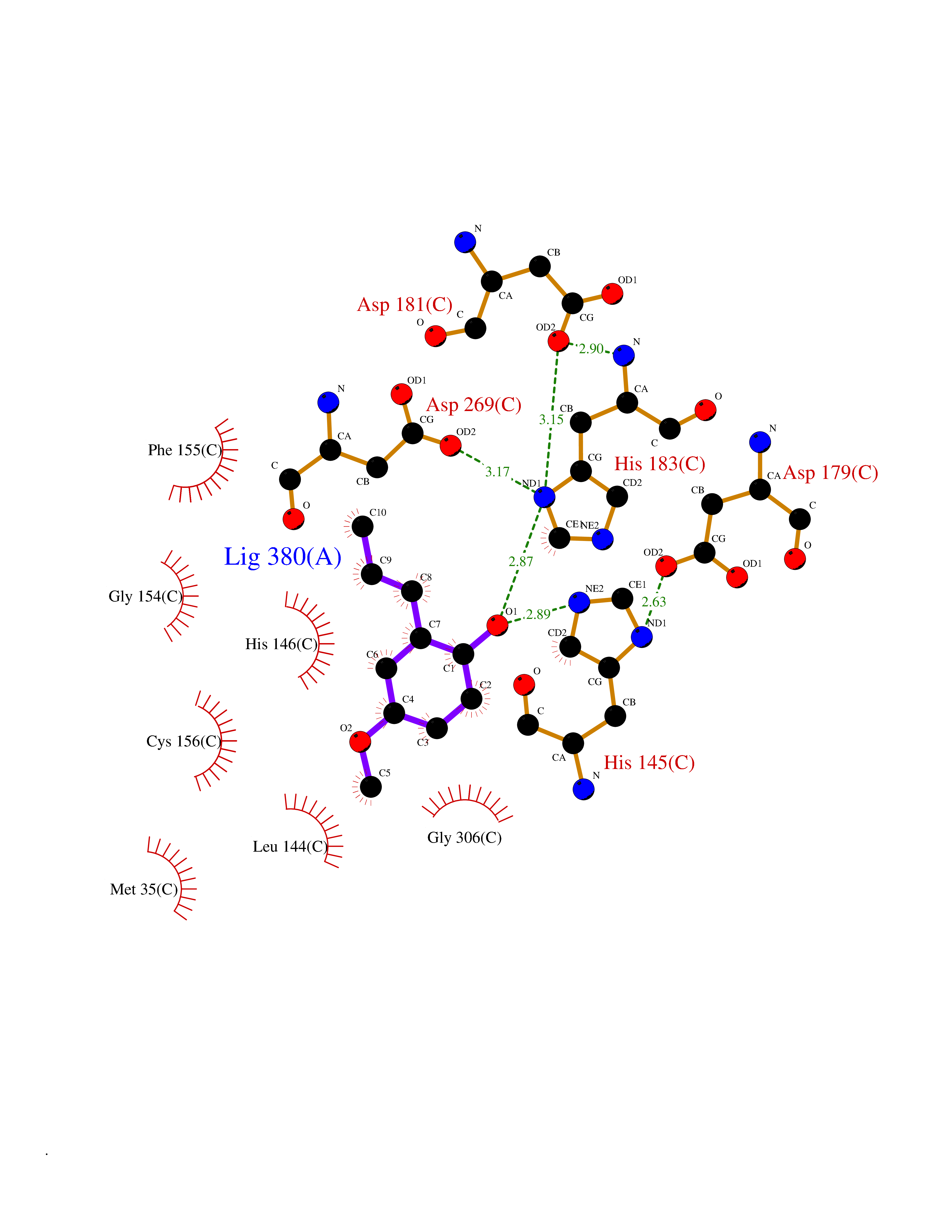 Ligplot Map 3D Binding mode Sequence KKKVCYYYDGDIGNYYYGQGHPMKPHRIRMTHNLLLNYGLYRKMEIYRPHKATAEEMTKYHSDEYIKFLRSIRPDNMSEYSKQMQRFNVGEDCPVFDGLFEFCQLSTGGSVAGAVKLNRQQTDMAVNWAGGLHHAKKSEASGFCYVNDIVLAILELLKYHQRVLYIDIDIHHGDGVEEAFYTTDRVMTVSFHKYGEYFPGTGDLRDIGAGKGKYYAVNFPMRDGIDDESYGQIFKPIISKVMEMYQPSAVVLQCGADSLSGDRLGCFNLTVKGHAKCVEVVKTFNLPLLMLGGGGYTIRNVARCWTYETAVALDCEIPNELPYNDYFEYFGPDFKLHISPSNMTNQNTPEYMEKIKQRLFENLRML Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | PRKR-like endoplasmic reticulum kinase (PERK) | 4G31 | 5.63 | |
Target general information Gen name EIF2AK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PEK Protein family Protein kinase superfamily, Ser/Thr protein kinase family, GCN2 subfamily Biochemical class Kinase Function Converts phosphorylated eIF-2-alpha/EIF2S1 either in a global protein synthesis inhibitor, leading to a reduced overall utilization of amino acids, or to a translation initiation activator of specific mRNAs, such as the transcriptional activator ATF4, and hence allowing ATF4-mediated reprogramming of amino acid biosynthetic gene expression to alleviate nutrient depletion. Serves as a critical effector of unfolded protein response (UPR)-induced G1 growth arrest due to the loss of cyclin-D1 (CCND1). Involved in control of mitochondrial morphology and function. Metabolic-stress sensing protein kinase that phosphorylates the alpha subunit of eukaryotic translation initiation factor 2 (eIF-2-alpha/EIF2S1) on 'Ser-52' during the unfolded protein response (UPR) and in response to low amino acid availability. Related diseases Wolcott-Rallison syndrome (WRS) [MIM:226980]: A rare autosomal recessive disorder, characterized by permanent neonatal or early infancy insulin-dependent diabetes and, at a later age, epiphyseal dysplasia, osteoporosis, growth retardation and other multisystem manifestations, such as hepatic and renal dysfunctions, intellectual disability and cardiovascular abnormalities. {ECO:0000269|PubMed:10932183, ECO:0000269|PubMed:12086964, ECO:0000269|PubMed:12960215, ECO:0000269|PubMed:16813601, ECO:0000269|PubMed:24168455, ECO:0000269|PubMed:24194294, ECO:0000269|PubMed:27145240, ECO:0000269|PubMed:28220546, ECO:0000269|PubMed:30906465, ECO:0000269|PubMed:30922274, ECO:0000269|PubMed:32216767, ECO:0000269|PubMed:34123975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NZJ5; P11021 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; ADP-ribosylation; ATP-binding; Diabetes mellitus; Disease variant; Endoplasmic reticulum; Glycoprotein; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Signal; Stress response; Transferase; Translation regulation; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Unfolded protein response Protein physicochemical properties Chain ID A Molecular weight (Da) 29033.5 Length 248 Aromaticity 0.1 Instability index 43.71 Isoelectric point 7.75 Charge (pH=7) 1.27 2D Binding mode Binding energy (Kcal/mol) -7.68  Molscript Map 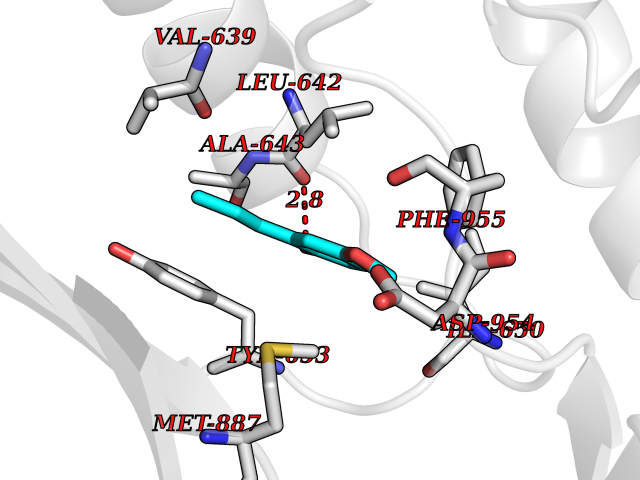 Pymol Map 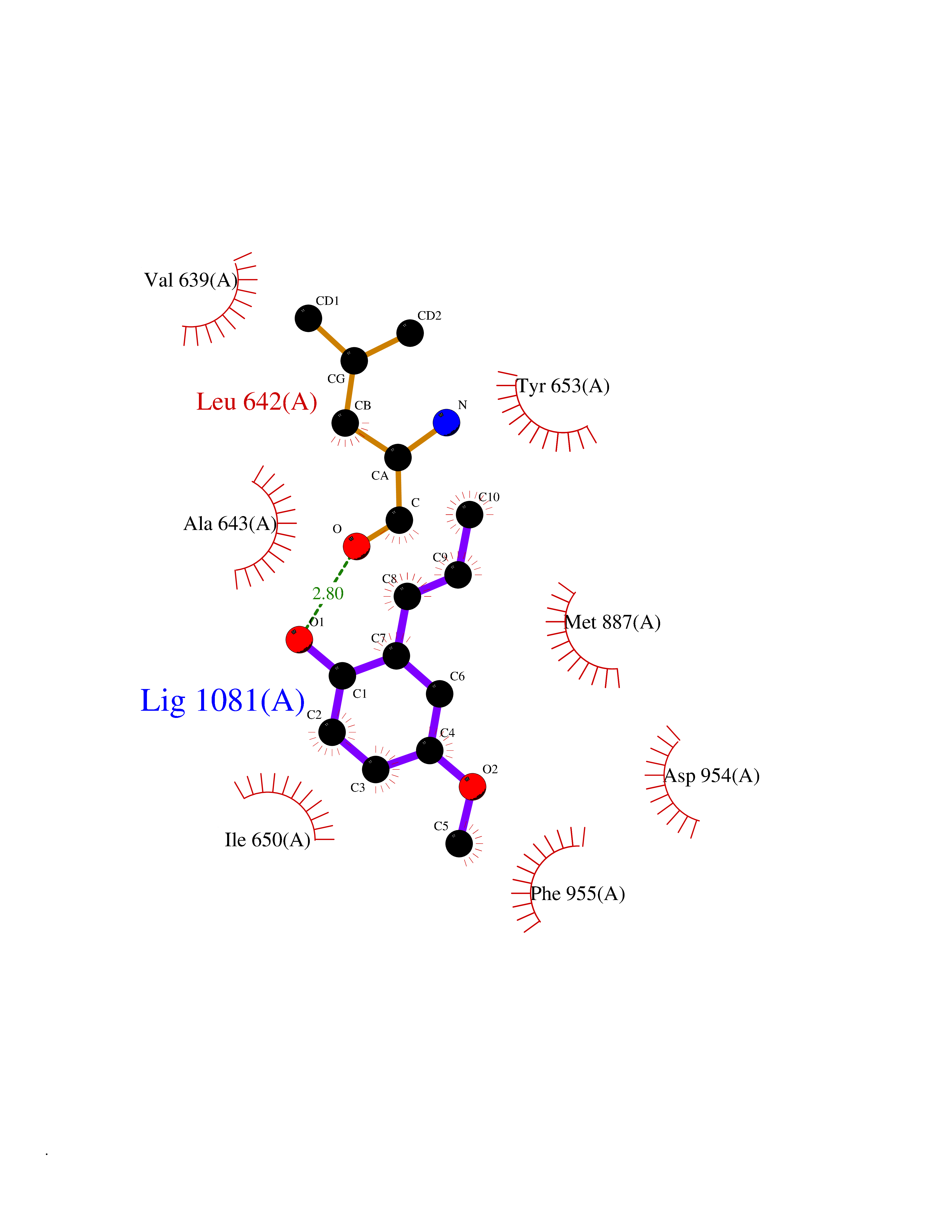 Ligplot Map 3D Binding mode Sequence GRYLTDFEPIQCLGRGGVVFEAKNKVDDCNYAIKRIRLPNRELAREKVMREVKALAKLEHPGIVRYFNAWLEKNKVYLYIQMQLCRKENLKDWMNGRCTIEERERSVCLHIFLQIAEAVEFLHSKGLMHRDLKPSNIFFTMDDVVKVGDFGLVGTKLYMSPEQIHGNSYSHKVDIFSLGLILFELLYPFSTQMERVRTLTDVRNLKFPPLFTQKYPCEYVMVQDMLSPSPMERPEAINIIENAVFEDL Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Eukaryotic translation initiation factor 2-alpha kinase 4 (EIF2AK4) | 7QQ6 | 5.63 | |
Target general information Gen name EIF2AK4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms eIF-2-alpha kinase GCN2; KIAA1338; GCN2-like protein; GCN2 Protein family Protein kinase superfamily, Ser/Thr protein kinase family, GCN2 subfamily Biochemical class NA Function Metabolic-stress sensing protein kinase that phosphorylates the alpha subunit of eukaryotic translation initiation factor 2 (eIF-2-alpha/EIF2S1) on 'Ser-52' in response to low amino acid availability. Plays a role as an activator of the integrated stress response (ISR) required for adapatation to amino acid starvation. Converts phosphorylated eIF-2-alpha/EIF2S1 either to a competitive inhibitor of the translation initiation factor eIF-2B, leading to a global protein synthesis repression, and thus to a reduced overall utilization of amino acids, or to a translational initiation activation of specific mRNAs, such as the transcriptional activator ATF4, and hence allowing ATF4-mediated reprogramming of amino acid biosynthetic gene expression to alleviate nutrient depletion. Binds uncharged tRNAs (By similarity). Involved in cell cycle arrest by promoting cyclin D1 mRNA translation repression after the unfolded protein response pathway (UPR) activation or cell cycle inhibitor CDKN1A/p21 mRNA translation activation in response to amino acid deprivation. Plays a role in the consolidation of synaptic plasticity, learning as well as formation of long-term memory. Plays a role in neurite outgrowth inhibition. Plays a proapoptotic role in response to glucose deprivation. Promotes global cellular protein synthesis repression in response to UV irradiation independently of the stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) and p38 MAPK signaling pathways (By similarity). Plays a role in the antiviral response against alphavirus infection; impairs early viral mRNA translation of the incoming genomic virus RNA, thus preventing alphavirus replication (By similarity). Related diseases Pulmonary venoocclusive disease 2, autosomal recessive (PVOD2) [MIM:234810]: A disease characterized by widespread fibrous obstruction and intimal thickening of septal veins and preseptal venules, a low diffusing capacity for carbon monoxide, occult alveolar hemorrhage, and nodular ground-glass opacities, septal lines and lymph node enlargement showed by high-resolution computed tomography of the chest. It is frequently associated with pulmonary capillary dilatation and proliferation, and is a rare and devastating cause of pulmonary hypertension. {ECO:0000269|PubMed:24135949, ECO:0000269|PubMed:24292273}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with NA EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Activation of host autophagy by virus; Activator; Adaptive immunity; Alternative splicing; Antiviral defense; ATP-binding; Cell cycle; Coiled coil; Cytoplasm; Disease variant; Growth arrest; Host-virus interaction; Immunity; Kinase; Neurogenesis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; RNA-binding; Serine/threonine-protein kinase; Stress response; Transferase; Translation regulation; tRNA-binding Protein physicochemical properties Chain ID A Molecular weight (Da) 31090.3 Length 269 Aromaticity 0.1 Instability index 42.78 Isoelectric point 7.75 Charge (pH=7) 1.43 2D Binding mode Binding energy (Kcal/mol) -7.67  Molscript Map 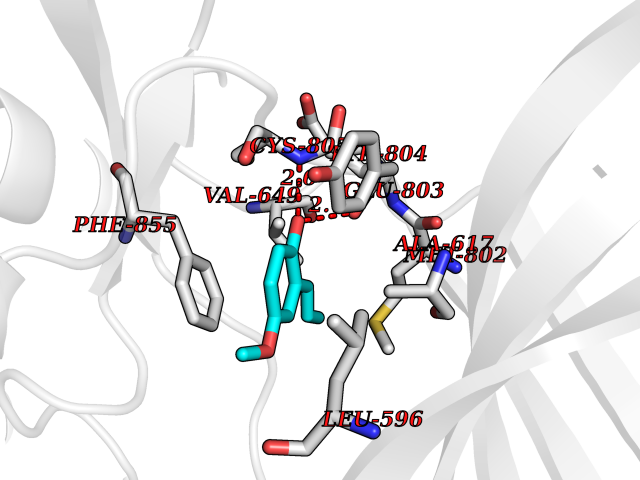 Pymol Map 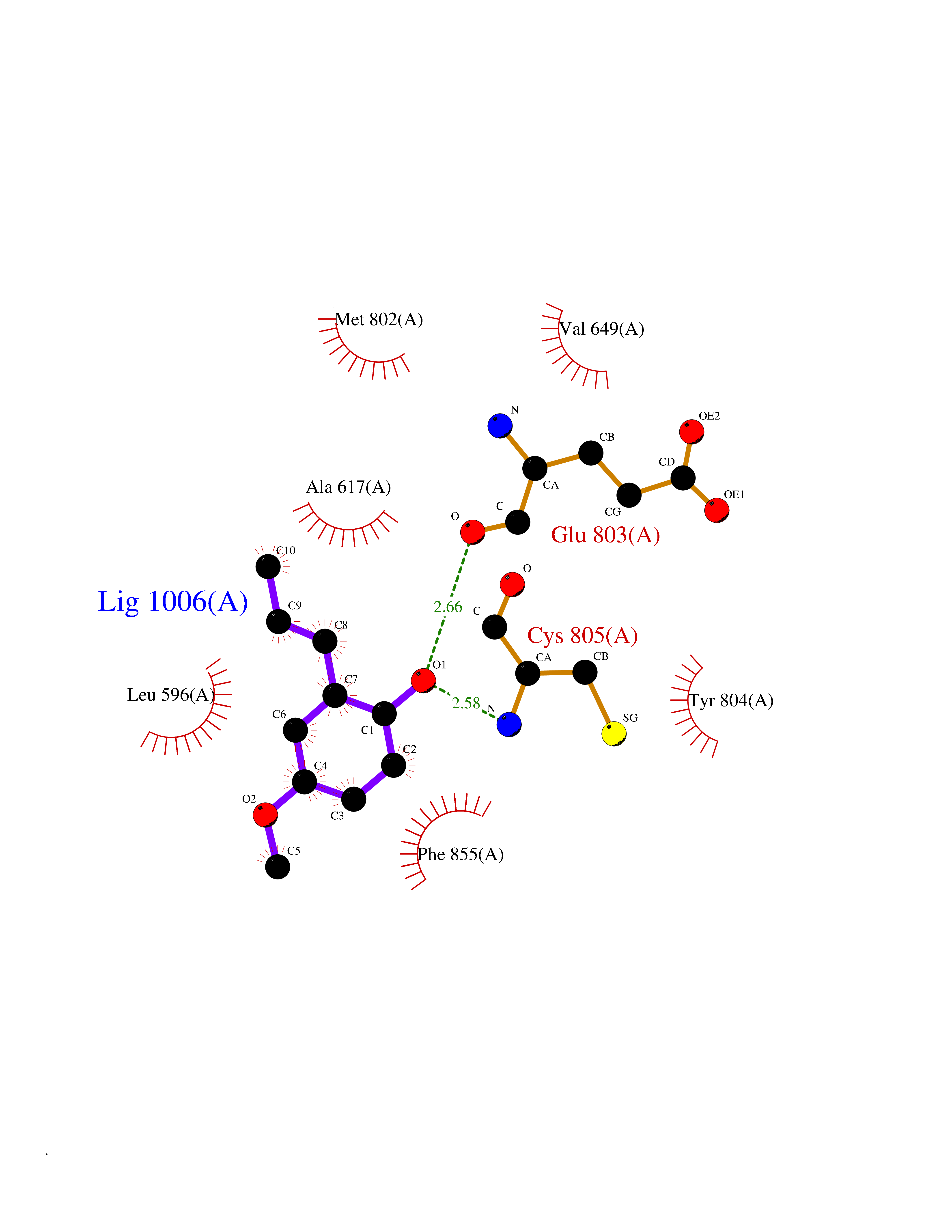 Ligplot Map 3D Binding mode Sequence SRYFIEFEELQLLGKGAFGAVIKVQNKLDGCCYAVKRIPINPASRQFRRIKGEVTLLSRLHHENIVRYYNAWIERHERPVHYLYIQMEYCEKSTLRDTIDQGLYRDTVRLWRLFREILDGLAYIHEKGMIHRNLKPVNIFLDSDDHVKIGDFGLIKSDPSGHLTGMVGTALYVSPEVQGSTKSAYNQKVDLFSLGIIFFEMSYHPMVTASERIFVLNQLRDPTSPKFPEDFDDGEHAKQKSVISWLLNHDPAKRPTATELLKSELLPPP Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Bacterial Flavohemoglobin (Bact hmp) | 1GVH | 5.62 | |
Target general information Gen name Bact hmp Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms Nitric oxide dioxygenase; NOD; NO oxygenase; Hemoglobin-like protein; HMP; Ferrisiderophore reductase B; Dihydropteridine reductase Protein family Globin family, Two-domain flavohemoproteins subfamily; Flavoprotein pyridine nucleotide cytochrome reductase family Biochemical class Paired donor oxygen oxidoreductase Function Various electron acceptors arealso reduced by HMP in vitro, including dihydropterine, ferrisiderophores, ferric citrate, cytochrome c, nitrite, S-nitrosoglutathione, and alkylhydroperoxides. However, it is unknown if these reactions are of any biological significance in vivo. Related diseases Ovarian dysgenesis 1 (ODG1) [MIM:233300]: An autosomal recessive disease characterized by primary amenorrhea, variable development of secondary sex characteristics, poorly developed streak ovaries, and high serum levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH). {ECO:0000269|PubMed:10551778, ECO:0000269|PubMed:11889179, ECO:0000269|PubMed:12571157, ECO:0000269|PubMed:12915623, ECO:0000269|PubMed:7553856, ECO:0000269|PubMed:9769327, ECO:0000269|PubMed:9851774}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ovarian hyperstimulation syndrome (OHSS) [MIM:608115]: Disorder which occurs either spontaneously or most often as an iatrogenic complication of ovarian stimulation treatments for in vitro fertilization. The clinical manifestations vary from abdominal distention and discomfort to potentially life-threatening, massive ovarian enlargement and capillary leak with fluid sequestration. Pathologic features of this syndrome include the presence of multiple serous and hemorrhagic follicular cysts lined by luteinized cells, a condition called hyperreactio luteinalis. {ECO:0000269|PubMed:12930927, ECO:0000269|PubMed:12930928, ECO:0000269|PubMed:15080154, ECO:0000269|PubMed:16278261, ECO:0000269|PubMed:17721928, ECO:0000269|PubMed:24058690, ECO:0000269|PubMed:25581598}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number EC 1.14.12.17 Uniprot keywords 3D-structure; Cytoplasm; Detoxification; Direct protein sequencing; FAD; Flavoprotein; Heme; Iron; Metal-binding; NAD; NADP; Oxidoreductase; Oxygen transport; Reference proteome; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 43867.1 Length 396 Aromaticity 0.1 Instability index 28.85 Isoelectric point 5.48 Charge (pH=7) -12.32 2D Binding mode Binding energy (Kcal/mol) -7.67 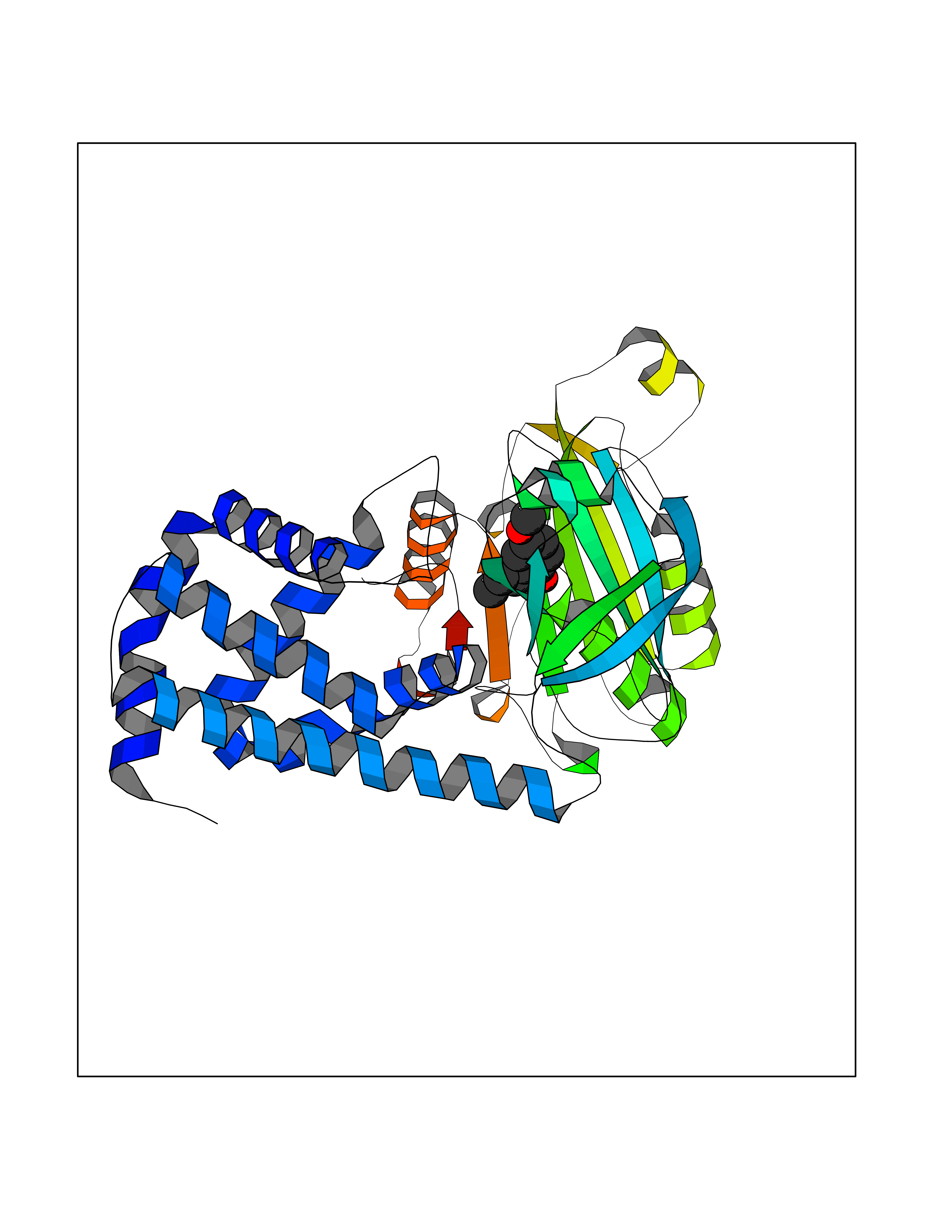 Molscript Map 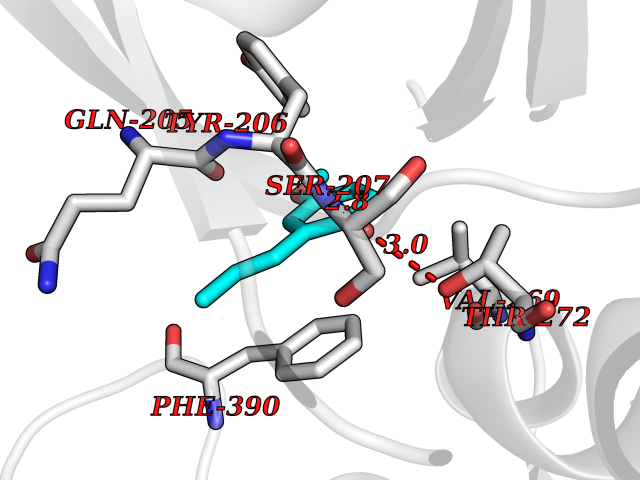 Pymol Map 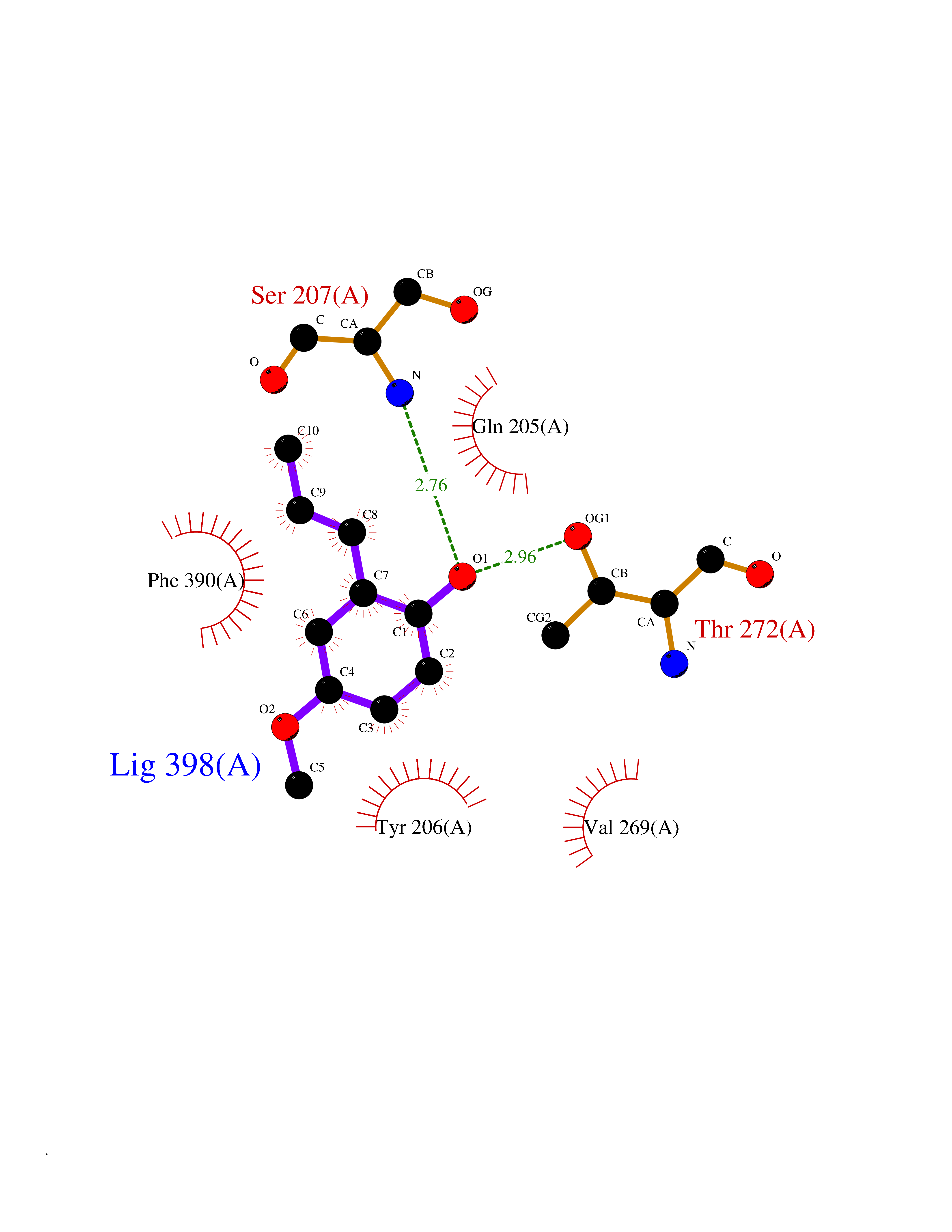 Ligplot Map 3D Binding mode Sequence MLDAQTIATVKATIPLLVETGPKLTAHFYDRMFTHNPELKEIFNMSNQRNGDQREALFNAIAAYASNIENLPALLPAVEKIAQKHTSFQIKPEQYNIVGEHLLATLDEMFSPGQEVLDAWGKAYGVLANVFINREAEIYNENASKAGGWEGTRDFRIVAKTPRSALITSFELEPVDGGAVAEYRPGQYLGVWLKPEGFPHQEIRQYSLTRKPDGKGYRIAVKREEGGQVSNWLHNHANVGDVVKLVAPAGDFFMAVADDTPVTLISAGVGQTPMLAMLDTLAKAGHTAQVNWFHAAENGDVHAFADEVKELGQSLPRFTAHTWYRQPSEADRAKGQFDSEGLMDLSKLEGAFSDPTMQFYLCGPVGFMQFTAKQLVDLGVKQENIHYECFGPHKVL Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Cytochrome P450 2C19 | 4GQS | 5.62 | |
Target general information Gen name CYP2C19 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function (R)-limonene 6-monooxygenase activity.(S)-limonene 6-monooxygenase activity.(S)-limonene 7-monooxygenase activity.Arachidonic acid epoxygenase activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxygen binding.Steroid hydroxylase activity. Related diseases Cystathioninuria (CSTNU) [MIM:219500]: Autosomal recessive phenotype characterized by abnormal accumulation of plasma cystathionine, leading to increased urinary excretion. {ECO:0000269|PubMed:12574942, ECO:0000269|PubMed:18476726}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB14055; DB05812; DB14973; DB01418; DB00945; DB00546; DB00518; DB00918; DB12015; DB06403; DB00357; DB01424; DB01118; DB00321; DB00381; DB00701; DB01435; DB11901; DB06605; DB00673; DB01274; DB06413; DB06697; DB11638; DB12597; DB11586; DB00289; DB01076; DB06442; DB06626; DB00972; DB01483; DB16703; DB15463; DB12319; DB01086; DB00443; DB01128; DB11967; DB13746; DB00188; DB12151; DB05541; DB01558; DB01222; DB00297; DB00921; DB09061; DB14737; DB08502; DB00564; DB06016; DB00395; DB14984; DB06119; DB00446; DB00672; DB01166; DB00501; DB00604; DB00215; DB12499; DB04920; DB14025; DB00349; DB06470; DB01242; DB00758; DB01559; DB00363; DB14635; DB00531; DB00091; DB08912; DB00250; DB00705; DB06700; DB01234; DB14649; DB09213; DB05351; DB13762; DB00514; DB00829; DB00586; DB00343; DB01093; DB01075; DB09167; DB05928; DB00590; DB01142; DB00470; DB00476; DB00625; DB00216; DB15444; DB13874; DB11718; DB00109; DB08899; DB01175; DB11823; DB14575; DB09119; DB00736; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00330; DB00898; DB00977; DB09166; DB01628; DB14766; DB06414; DB12500; DB00949; DB00574; DB01039; DB12265; DB15669; DB00196; DB01544; DB00472; DB01095; DB00176; DB00983; DB01320; DB11679; DB05087; DB00317; DB01241; DB06730; DB01120; DB01016; DB00986; DB01018; DB00502; DB01355; DB00956; DB00741; DB06789; DB01050; DB09054; DB01181; DB00619; DB00458; DB00328; DB11633; DB06636; DB00951; DB09570; DB06738; DB01026; DB00598; DB06218; DB00448; DB01259; DB09078; DB12070; DB01006; DB08918; DB09198; DB04948; DB06448; DB16220; DB01601; DB00455; DB04871; DB12130; DB00678; DB00227; DB08933; DB09280; DB01283; DB12474; DB08932; DB09238; DB14921; DB14009; DB01065; DB01043; DB00170; DB00454; DB00532; DB00333; DB00763; DB05246; DB09241; DB00849; DB00959; DB01110; DB06595; DB16236; DB01171; DB00745; DB11763; DB14011; DB09049; DB01183; DB04861; DB00220; DB00622; DB00184; DB00665; DB06712; DB12005; DB00717; DB00540; DB00334; DB14881; DB16267; DB00338; DB11632; DB04911; DB11837; DB04938; DB00776; DB00239; DB00935; DB11697; DB05467; DB00213; DB00715; DB00738; DB00312; DB00850; DB03783; DB00780; DB01174; DB00252; DB13941; DB01621; DB04951; DB17472; DB06209; DB01058; DB14631; DB00635; DB00794; DB00396; DB01131; DB00420; DB00818; DB00571; DB01589; DB04216; DB01224; DB00468; DB01129; DB00980; DB08896; DB16826; DB02709; DB00615; DB01045; DB11753; DB01201; DB01220; DB08864; DB00503; DB06176; DB05271; DB12332; DB11614; DB06654; DB12543; DB12834; DB00418; DB01037; DB11689; DB06731; DB06739; DB01104; DB00203; DB00641; DB06268; DB15093; DB00052; DB00398; DB12548; DB01323; DB09118; DB00675; DB06204; DB06083; DB12020; DB00966; DB12095; DB00444; DB00857; DB00624; DB13943; DB13944; DB13946; DB11712; DB01041; DB00599; DB00679; DB00208; DB00373; DB01007; DB00932; DB06137; DB08895; DB01124; DB01036; DB00273; DB01685; DB05109; DB00752; DB12245; DB00347; DB00726; DB00197; DB15328; DB00313; DB00862; DB00285; DB00661; DB16349; DB06684; DB08828; DB11739; DB00582; DB09068; DB14975; DB00682; DB00549; DB00425; DB00909; DB09120 Interacts with NA EC number 1.14.14.1; 1.14.14.51; 1.14.14.52; 1.14.14.53; 1.14.14.75 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 51680.3 Length 452 Aromaticity 0.1 Instability index 39.98 Isoelectric point 6.12 Charge (pH=7) -4.4 2D Binding mode Binding energy (Kcal/mol) -7.67 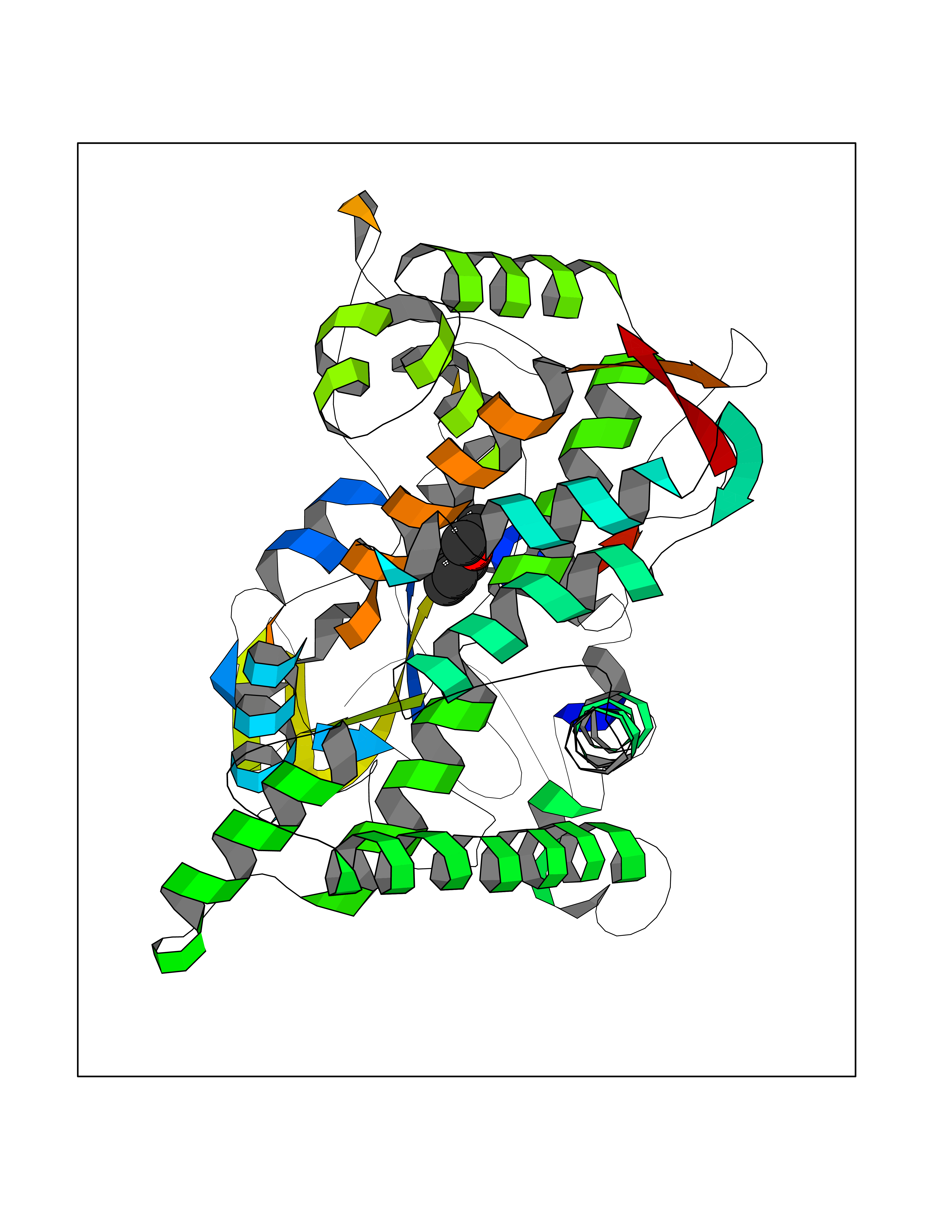 Molscript Map 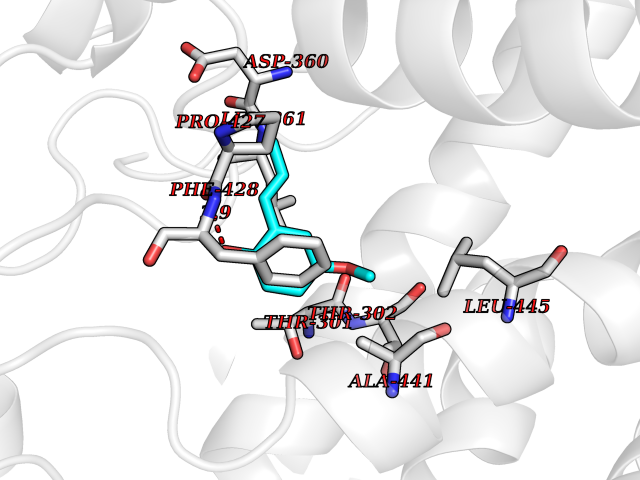 Pymol Map 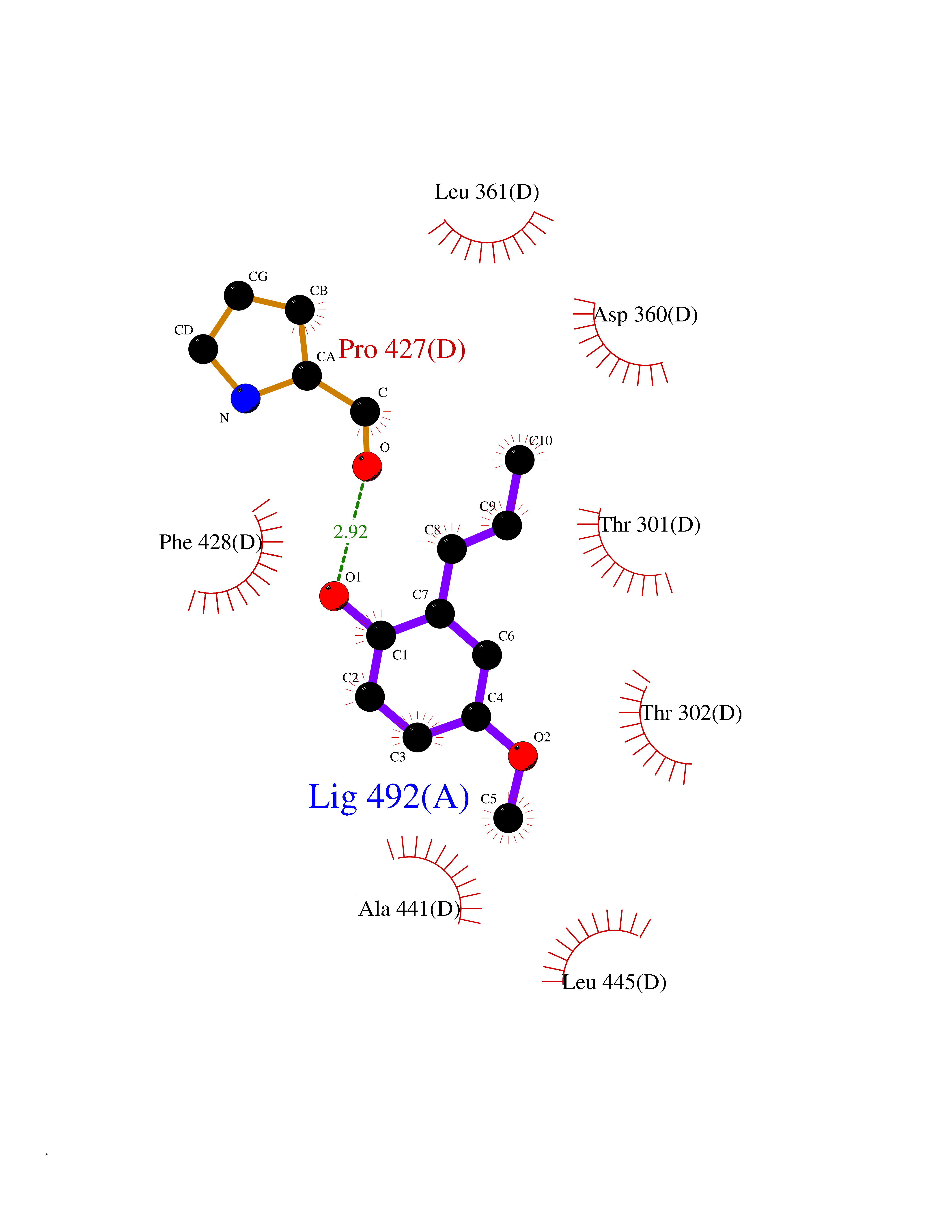 Ligplot Map 3D Binding mode Sequence LPPGPTPLPVIGNILQIDIKDVSKSLTNLSKIYGPVFTLYFGLERMVVLHGYEVVKEALIDLGEEFSGRGHFPLAERANRGFGIVFSNGKRWKEIRRFSLMTLEDRVQEEARCLVEELRKTKASPCDPTFILGCAPCNVICSIIFQKRFDYKDQQFLNLMEKLNENIRIVSTPWIQICNNFPTIIDYFPGTHNKLLKNLAFMESDILEKVKEHQESMDINNPRDFIDCFLIKMEKEKQNQQSEFTIENLVITAADLLGAGTETTSTTLRYALLLLLKHPEVTAKVQEEIERVVGRNRSPCMQDRGHMPYTDAVVHEVQRYIDLIPTSLPHAVTCDVKFRNYLIPKGTTILTSLTSVLHDNKEFPNPEMFDPRHFLDEGGNFKKSNYFMPFSAGKRICVGEGLARMELFLFLTFILQNFNLKSLIDPKDLDTTPVVNGFASVPPFYQLCFIPI Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Nitric-oxide synthase inducible (NOS2) | 3E7G | 5.62 | |
Target general information Gen name NOS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms iNOS; Peptidyl-cysteine S-nitrosylase NOS2; Nitric oxide synthase, inducible; NOS2A; NOS type II; Inducible NOS; Inducible NO synthase; Hepatocyte NOS; HEP-NOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function Produces nitric oxide (NO) which is a messenger molecule with diverse functions throughout the body. In macrophages, NO mediates tumoricidal and bactericidal actions. Also has nitrosylase activity and mediates cysteine S-nitrosylation of cytoplasmic target proteins such PTGS2/COX2 (By similarity). As component of the iNOS-S100A8/9 transnitrosylase complex involved in the selective inflammatory stimulus-dependent S-nitrosylation of GAPDH on 'Cys-247' implicated in regulation of the GAIT complex activity and probably multiple targets including ANXA5, EZR, MSN and VIM. Involved in inflammation, enhances the synthesis of proinflammatory mediators such as IL6 and IL8. Related diseases Cerebellar ataxia, impaired intellectual development, and dysequilibrium syndrome 3 (CAMRQ3) [MIM:613227]: An autosomal recessive, congenital cerebellar ataxia associated with dysarthia, quadrupedal gait and intellectual disability. {ECO:0000269|PubMed:19461874}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07003; DB07007; DB07011; DB07405; DB08750; DB01997; DB07029; DB07008; DB08214; DB07002; DB01835; DB06879; DB04534; DB03100; DB02207; DB00125; DB00155; DB01234; DB14649; DB11327; DB00997; DB07306; DB07388; DB05252; DB01381; DB03366; DB05214; DB04400; DB09237; DB00244; DB01110; DB01017; DB03144; DB01686; DB03449; DB06916; DB07318; DB07389; DB02044; DB02644; DB05383; DB02234; DB03953; DB02462; DB08814 Interacts with P04406 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calmodulin-binding; Cytoplasm; FAD; Flavoprotein; FMN; Heme; Iron; Metal-binding; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 48633 Length 421 Aromaticity 0.12 Instability index 46.5 Isoelectric point 6.75 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -7.66  Molscript Map 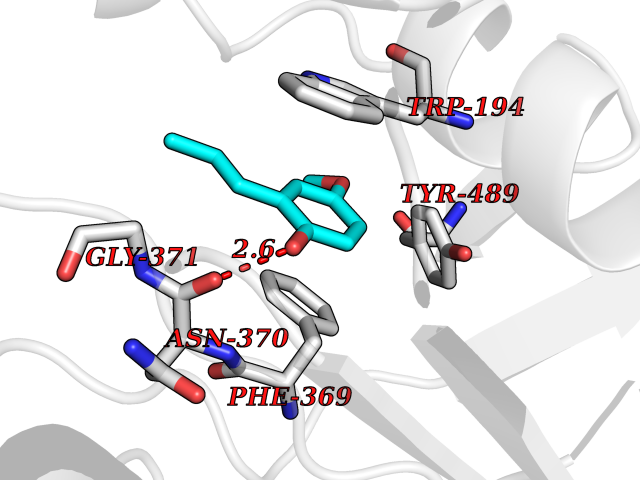 Pymol Map  Ligplot Map 3D Binding mode Sequence RHVRIKNWGSGMTFQDTLHHKAKGILTCRSKSCLGSIMTPKSLTRGPRDKPTPPDELLPQAIEFVNQYYGSFKEAKIEEHLARVEAVTKEIETTGTYQLTGDELIFATKQAWRNAPRCIGRIQWSNLQVFDARSCSTAREMFEHICRHVRYSTNNGNIRSAITVFPQRSDGKHDFRVWNAQLIRYAGYQMPDGSIRGDPANVEFTQLCIDLGWKPKYGRFDVVPLVLQANGRDPELFEIPPDLVLEVAMEHPKYEWFRELELKWYALPAVANMLLEVGGLEFPGCPFNGWYMGTEIGVRDFCDVQRYNILEEVGRRMGLETHKLASLWKDQAVVEINIAVLHSFQKQNVTIMDHHSAAESFMKYMQNEYRSRGGCPADWIWLVPPMSGSITPVFHQEMLNYVLSPFYYYQVEAWKTHVWQD Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 5.62 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -7.67  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||