Job Results:
Ligand
Structure
Job ID
40faeca68ee7fb64f0c588e9129de017
Job name
NA
Time
2026-02-27 13:42:04
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Gamma-butyrobetaine dioxygenase | 4C5W | 4.97 | |
Target general information Gen name BBOX1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms BBH;BBOX Protein family Gamma-BBH/TMLD family Biochemical class Oxidoreductase Function Gamma-butyrobetaine dioxygenase activity.Identical protein binding.Iron ion binding.Zinc ion binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126 Interacts with O75936; A0MZ66-7 EC number 1.14.11.1 Uniprot keywords 3D-structure; Carnitine biosynthesis; Cytoplasm; Dioxygenase; Iron; Metal-binding; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 31642.5 Length 275 Aromaticity 0.12 Instability index 35.68 Isoelectric point 6.33 Charge (pH=7) -2.46 2D Binding mode Binding energy (Kcal/mol) -6.77 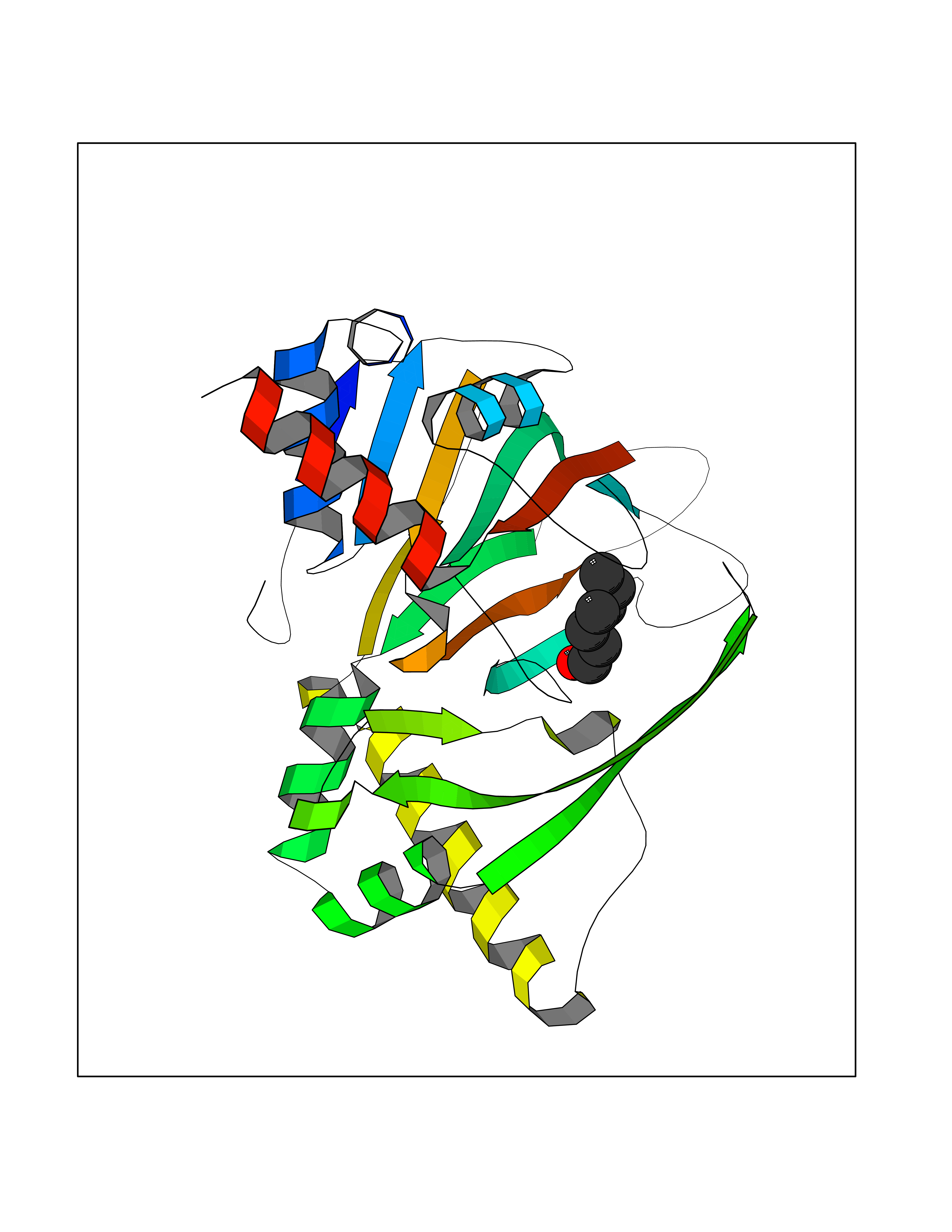 Molscript Map 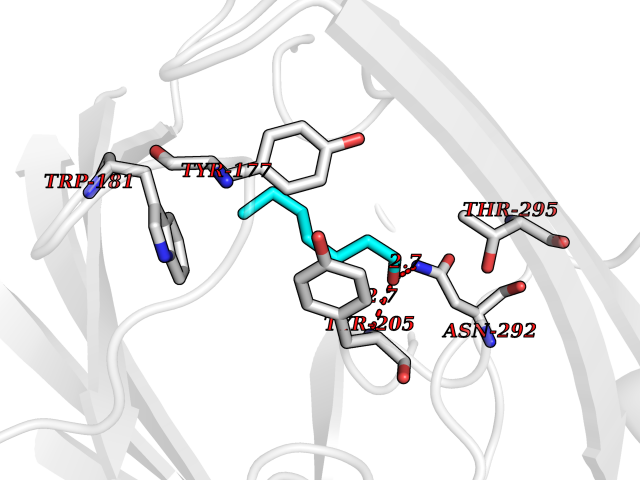 Pymol Map 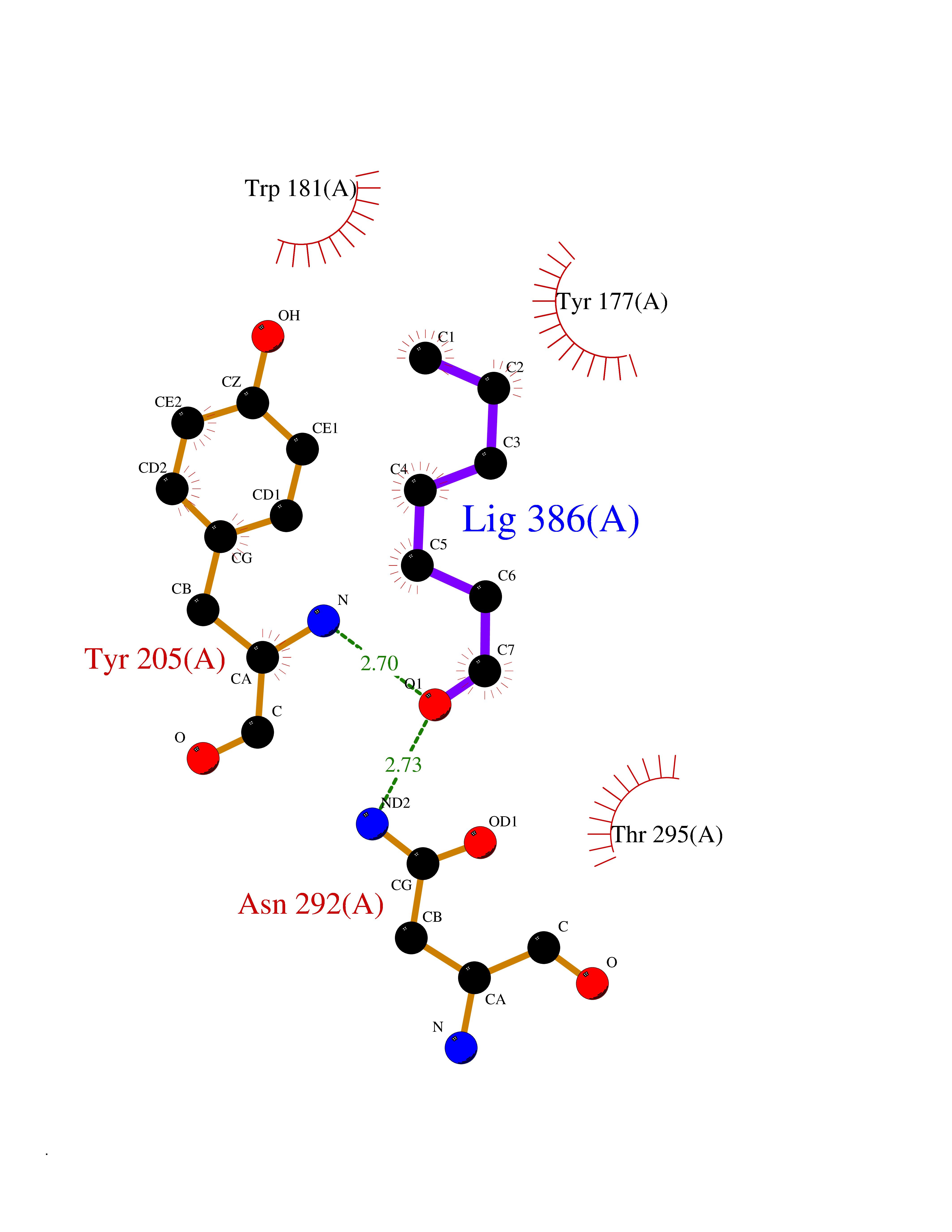 Ligplot Map 3D Binding mode Sequence FPECQYWGSELQLPTLDFEDVLRYDEHAYKWLSTLKKVGIVRLTGASDKPGEVSKLGKRMGFLYLTFYGHTWQVQDKIDANNVAYTTGKLSFHTDYPALHHPPGVQLLHCIKQTVTGGDSEIVDGFNVCQKLKKNNPQAFQILSSTFVDFTDIGVDYCDFSVQSKHKIIELDDKGQVVRINFNNATRDTIFDVPVERVQPFYAALKEFVDLMNSKESKFTFKMNPGDVITFDNWRLLHGRRSYEAGTEISRHLEGAYADWDVVMSRLRILRQRVE Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Aldose reductase (AKR1B1) | 1US0 | 4.97 | |
Target general information Gen name AKR1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde reductase; AKR1B1 Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies. Related diseases Glutamine deficiency, congenital (GLND) [MIM:610015]: An autosomal recessive disorder characterized by variable brain malformations, encephalopathy, severe developmental delay, seizures, and decreased glutamine levels in bodily fluids. Death in early infancy may occur. {ECO:0000269|PubMed:16267323, ECO:0000269|PubMed:26711351, ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 116 (DEE116) [MIM:620806]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE116 is autosomal dominant form characterized by severe developmental delay, seizures, and white matter abnormalities. {ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry. DEE116 is caused by variants that disrupt the canonical translation start codon in GLUL resulting in initiation of translation at Met-18 (PubMed:38579670). The resulting protein is enzymatically competent but insensitive to negative feedback regulation via glutamine-induced degradation. {ECO:0000269|PubMed:38579670}. Drugs (DrugBank ID) DB07028; DB07030; DB07450; DB02101; DB08449; DB08000; DB07139; DB07498; DB02007; DB02020; DB11859; DB02994; DB04272; DB07187; DB00694; DB00997; DB06246; DB01039; DB02021; DB16707; DB00143; DB02834; DB08084; DB01689; DB07063; DB06077; DB02518; DB00157; DB03461; DB05383; DB05533; DB05327; DB02712; DB00605; DB02383; DB02132; DB08772; DB07093; DB07999; DB08098 Interacts with Q9BUY7 EC number EC 1.1.1.300 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35447.6 Length 313 Aromaticity 0.09 Instability index 36.41 Isoelectric point 7.1 Charge (pH=7) 0.26 2D Binding mode Binding energy (Kcal/mol) -6.78  Molscript Map 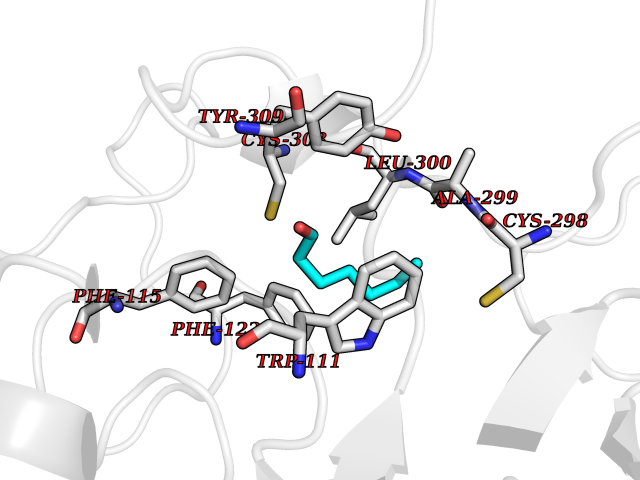 Pymol Map 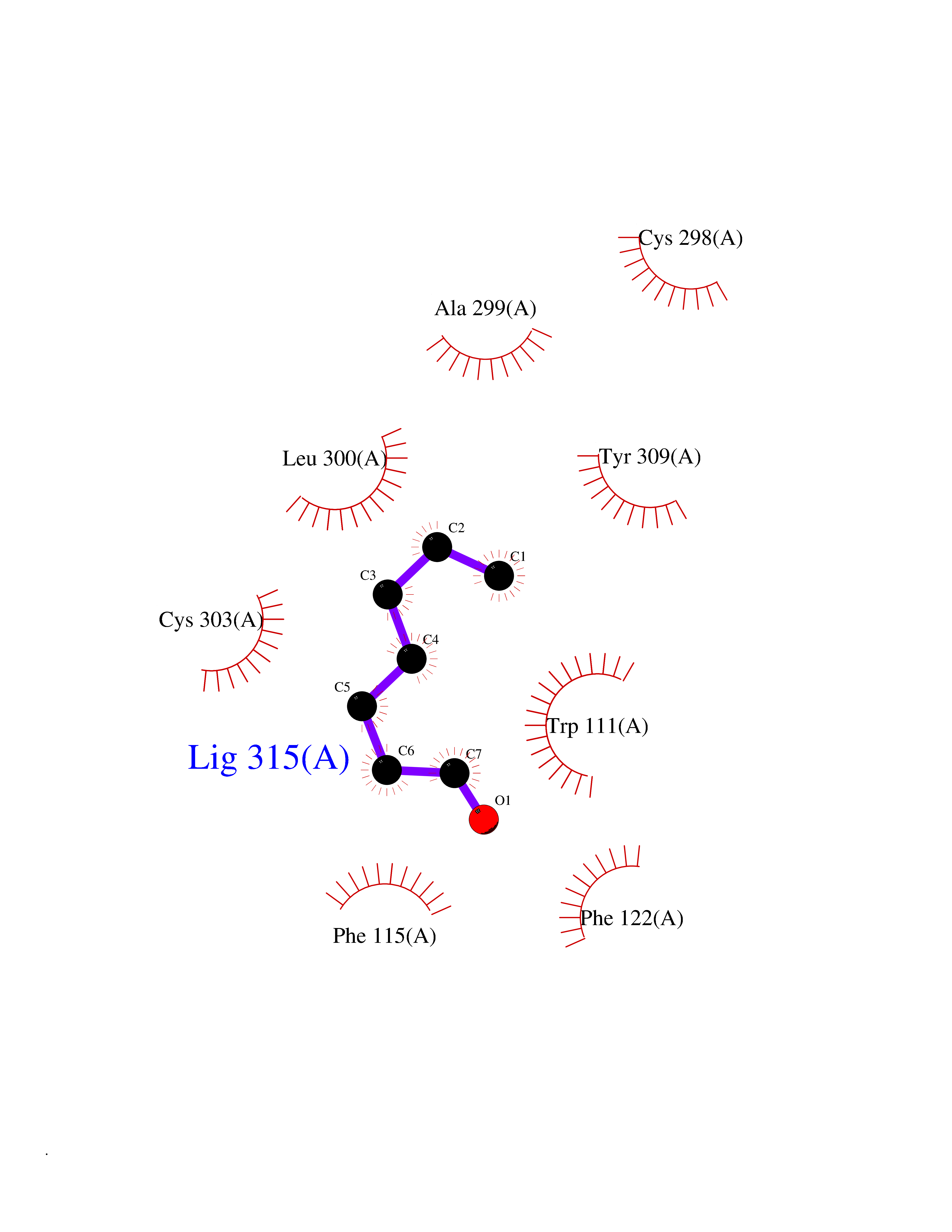 Ligplot Map 3D Binding mode Sequence MASRILLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEKLREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEFFPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAVNQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHNKTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRNWRVCALLSCTSHKDYPFH Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Leukotriene A-4 hydrolase (LTA4H) | 3U9W | 4.97 | |
Target general information Gen name LTA4H Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Leukotriene A4 hydrolase; Leukotriene A(4)Leukotriene A-4 hydrolase hydrolase; Leukotriene A(4) hydrolase; LTA4; LTA-H; LTA-4hydrolase; LTA-4 hydrolase Protein family Peptidase M1 family Biochemical class Ether bond hydrolase Function Has also aminopeptidase activity. Epoxide hydrolase that catalyzes the final step in the biosynthesis of the proinflammatory mediator leukotriene B4. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07102; DB06917; DB07258; DB07094; DB07259; DB02352; DB07292; DB07104; DB06828; DB08466; DB01197; DB05177; DB03366; DB08040; DB06851; DB02062; DB07099; DB07260; DB07196; DB11781; DB03424; DB07237 Interacts with Q9BSI4 EC number EC 3.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Leukotriene biosynthesis; Lipid metabolism; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 68927 Length 608 Aromaticity 0.1 Instability index 38.84 Isoelectric point 5.87 Charge (pH=7) -9.86 2D Binding mode Binding energy (Kcal/mol) -6.78  Molscript Map 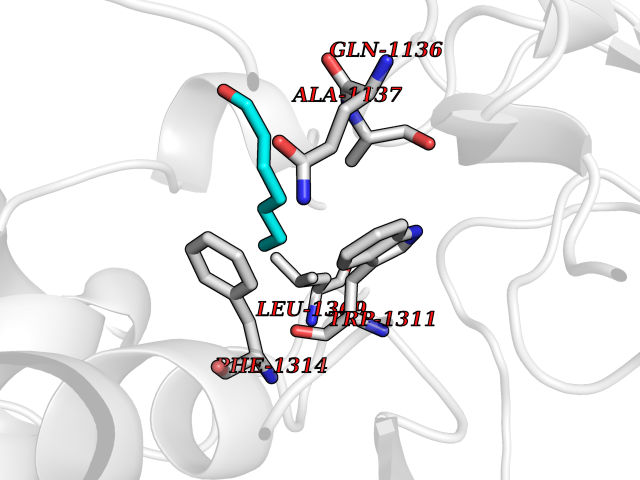 Pymol Map 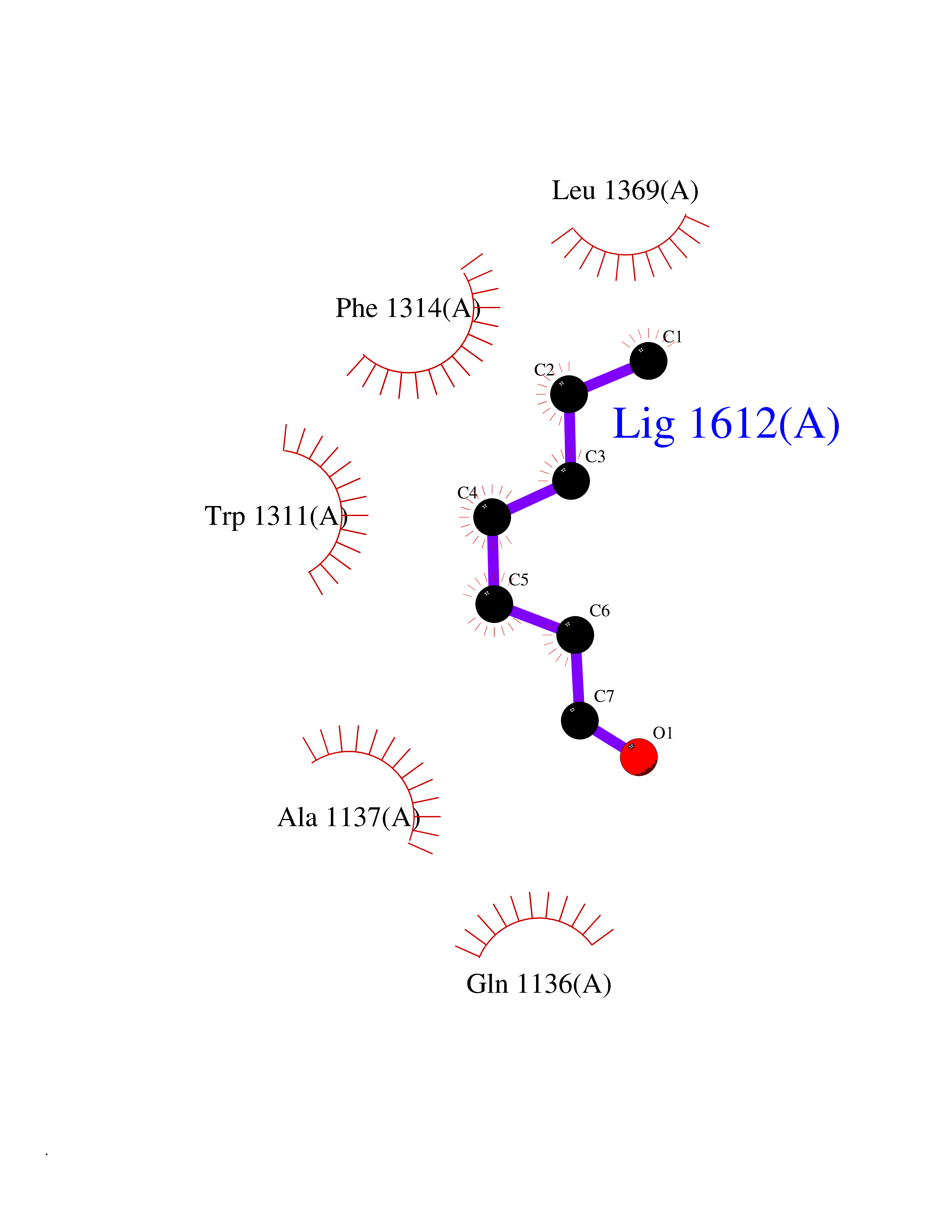 Ligplot Map 3D Binding mode Sequence IVDTCSLASPASVCRTKHLHLRCSVDFTRRTLTGTAALTVQSQEDNLRSLVLDTKDLTIEKVVINGQEVKYALGERQSYKGSPMEISLPIALSKNQEIVIEISFETSPKSSALQWLTPEQTSGKEHPYLFSQCQAIHCRAILPCQDTPSVKLTYTAEVSVPKELVALMSAIRDGETPDPEDPSRKIYKFIQKVPIPCYLIALVVGALESRQIGPRTLVWSEKEQVEKSAYEFSETESMLKIAEDLGGPYVWGQYDLLVLPPSFPYGGMENPCLTFVTPTLLAGDKSLSNVIAHEISHSWTGNLVTNKTWDHFWLNEGHTVYLERHICGRLFGEKFRHFNALGGWGELQNSVKTFGETHPFTKLVVDLTDIDPDVAYSSVPYEKGFALLFYLEQLLGGPEIFLGFLKAYVEKFSYKSITTDDWKDFLYSYFKDKVDVLNQVDWNAWLYSPGLPPIKPNYDMTLTNACIALSQRWITAKEDDLNSFNATDLKDLSSHQLNEFLAQTLQRAPLPLGHIKRMQEVYNFNAINNSEIRFRWLRLCIQSKWEDAIPLALKMATEQGRMKFTRPLFKDLAAFDKSHDQAVRTYQEHKASMHPVTAMLVGKDLKVD Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Medium-chain specific acyl-CoA dehydrogenase, mitochondrial | 4P13 | 4.97 | |
Target general information Gen name ACADM Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Acyl-CoA dehydrogenase activity.Flavin adenine dinucleotide binding.Identical protein binding.Medium-chain-acyl-CoA dehydrogenase activity. Related diseases Acyl-CoA dehydrogenase medium-chain deficiency (ACADMD) [MIM:201450]: An inborn error of mitochondrial fatty acid beta-oxidation which causes fasting hypoglycemia, hepatic dysfunction and encephalopathy, often resulting in death in infancy. {ECO:0000269|PubMed:10767181, ECO:0000269|PubMed:11349232, ECO:0000269|PubMed:11409868, ECO:0000269|PubMed:11486912, ECO:0000269|PubMed:1363805, ECO:0000269|PubMed:1671131, ECO:0000269|PubMed:1684086, ECO:0000269|PubMed:1902818, ECO:0000269|PubMed:2251268, ECO:0000269|PubMed:2393404, ECO:0000269|PubMed:2394825, ECO:0000269|PubMed:7603790, ECO:0000269|PubMed:7929823, ECO:0000269|PubMed:8198141, ECO:0000269|PubMed:9158144, ECO:0000269|PubMed:9882619}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03415; DB03147; DB02910 Interacts with PRO_0000000502 [P11310] EC number 1.3.8.7 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; FAD; Fatty acid metabolism; Flavoprotein; Lipid metabolism; Mitochondrion; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 85080.3 Length 773 Aromaticity 0.09 Instability index 30.55 Isoelectric point 5.71 Charge (pH=7) -7.7 2D Binding mode Binding energy (Kcal/mol) -6.78 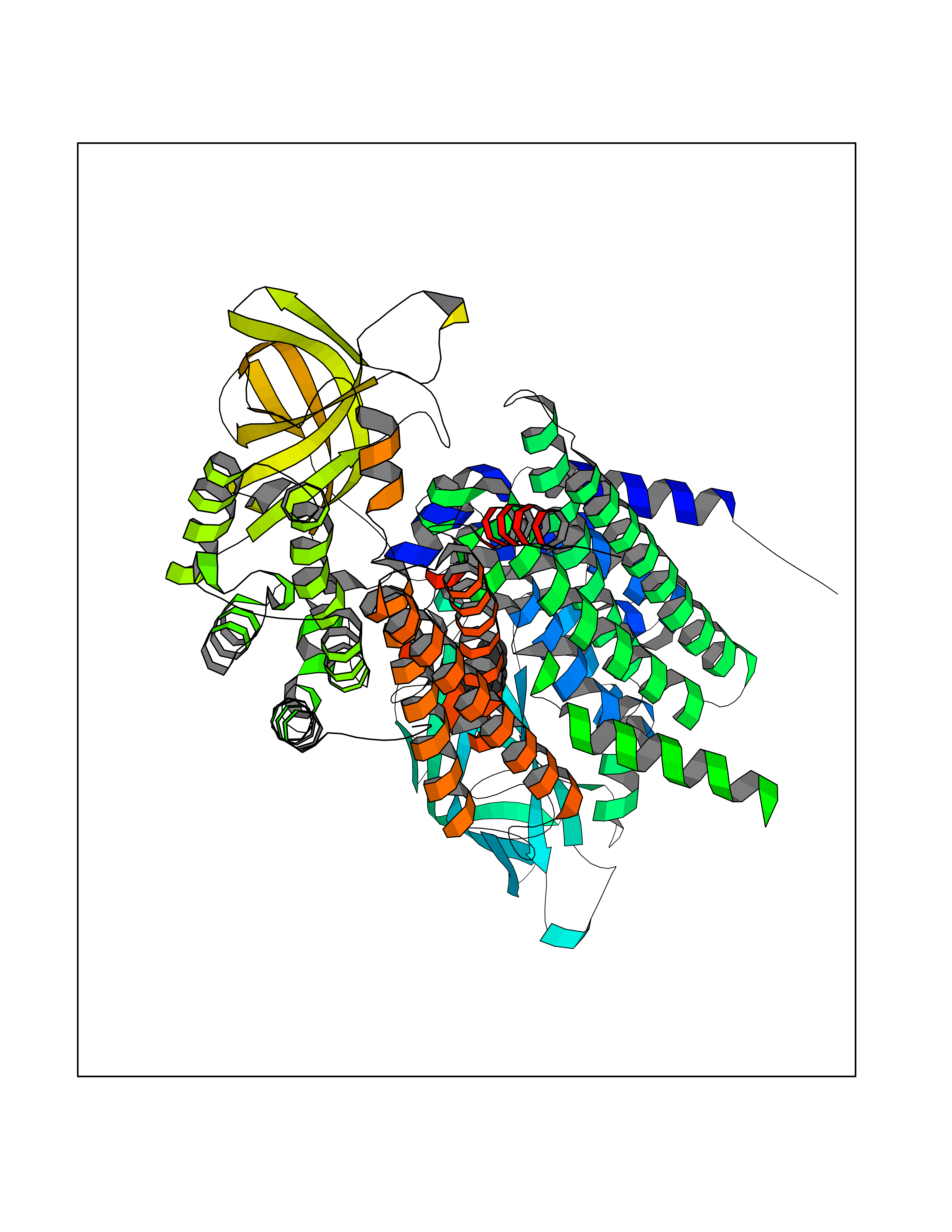 Molscript Map  Pymol Map 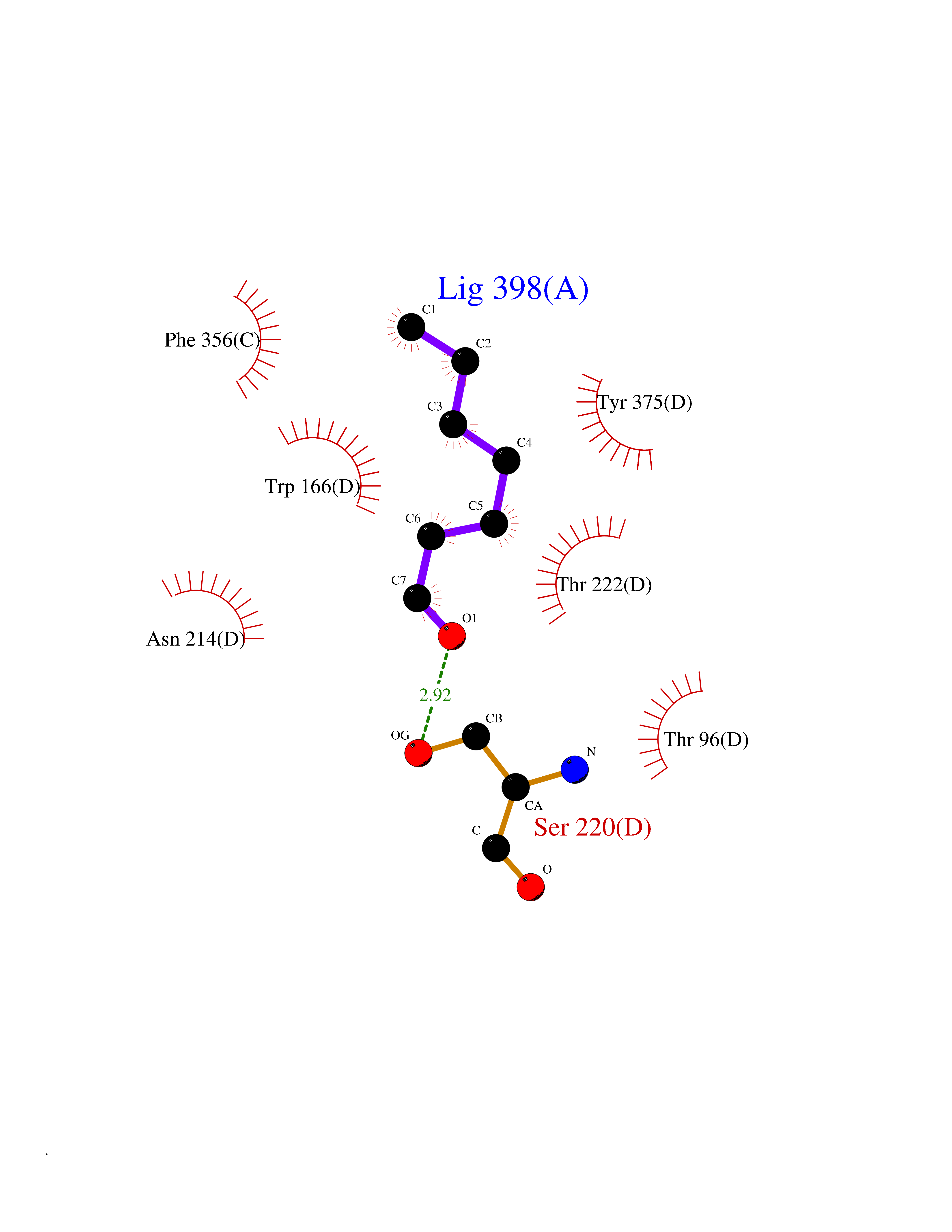 Ligplot Map 3D Binding mode Sequence LGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKLGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKN Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | S-adenosylmethionine decarboxylase proenzyme (AMD1) | 1JL0 | 4.97 | |
Target general information Gen name AMD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SamDC; S-adenosylmethioninedecarboxylase; AdoMetDC; AMD Protein family Eukaryotic AdoMetDC family Biochemical class Carbon-carbon lyase Function Promotes maintenance and self-renewal of embryonic stem cells, by maintaining spermine levels. Essential for biosynthesis of the polyamines spermidine and spermine. Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08163; DB00118; DB01917 Interacts with P17707; Q96A98; Q8WY91 EC number EC 4.1.1.50 Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Decarboxylase; Direct protein sequencing; Lyase; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Pyruvate; Reference proteome; S-adenosyl-L-methionine; Schiff base; Spermidine biosynthesis; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 35790.5 Length 311 Aromaticity 0.14 Instability index 39.47 Isoelectric point 6.03 Charge (pH=7) -2.01 2D Binding mode Binding energy (Kcal/mol) -6.77  Molscript Map 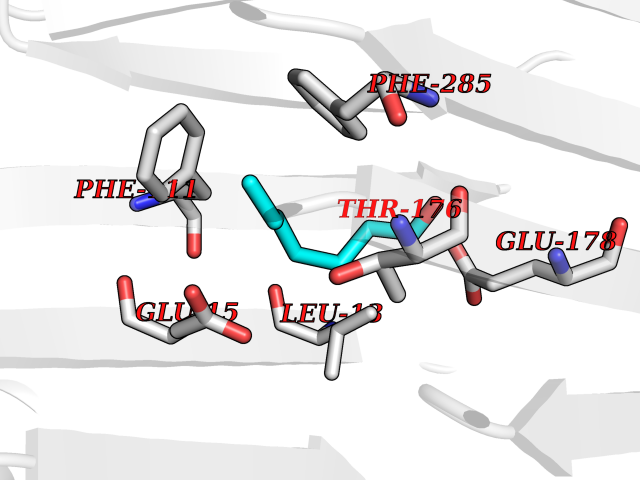 Pymol Map 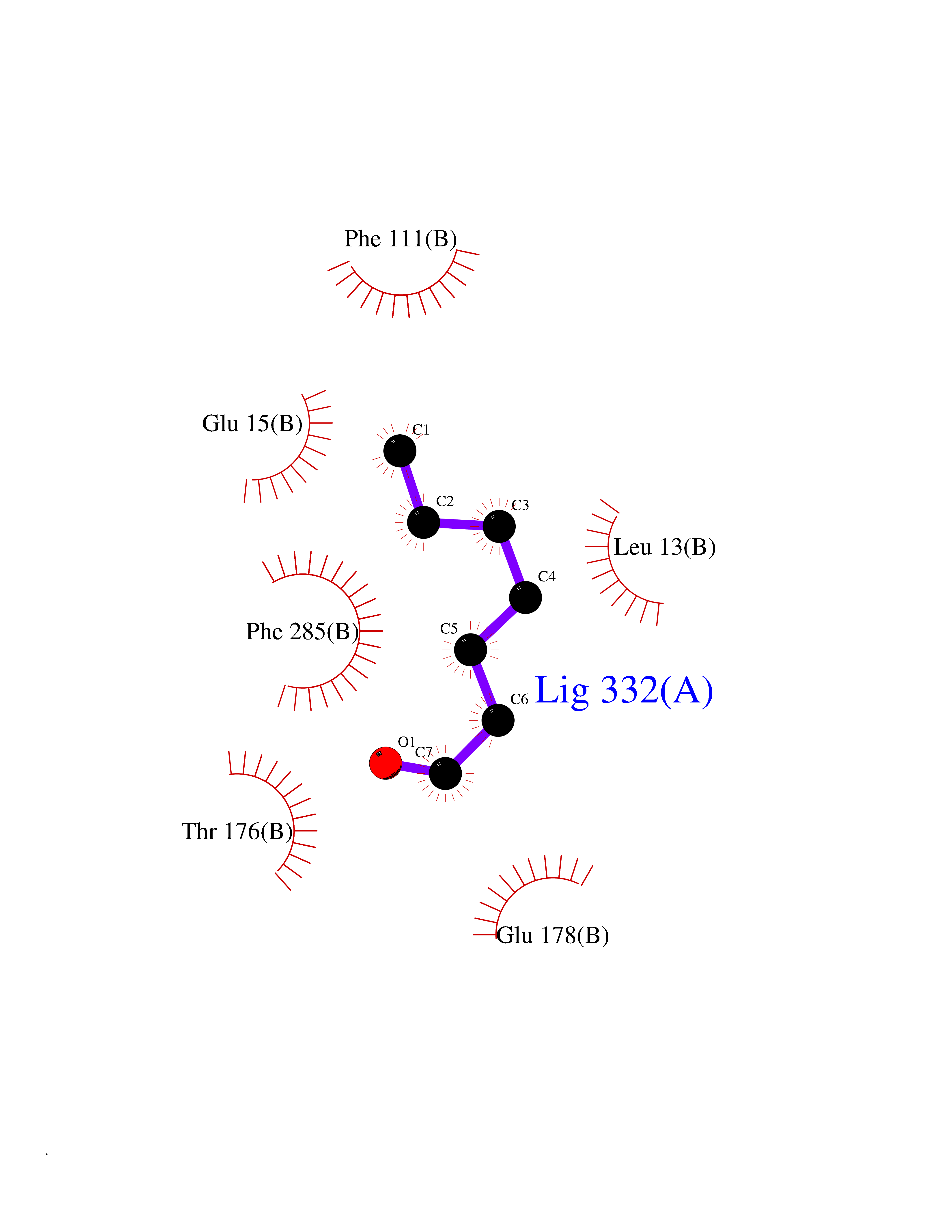 Ligplot Map 3D Binding mode Sequence HFFEGTEKLLEVWFSRQGSGDLRTIPRSEWDILLKDVQCSIISVTKTDKQEAYVLSESSMFVSKRRFILKTCGTTLLLKALVPLLKLARDYSGFDSIQSFFYSRKNFMKPSHQGYPHRNFQEEIEFLNAIFPNGAGYCMGRMNSDCWYLYTLDFRVISQPDQTLEILMSELDPAVMDQFYMKDGVTAKDVTRESGIRDLIPGSVIDATMFNPCGYSMNGMKSDGTYWTIAITPEPEFSYVSFETNLSQTSYDDLIRKVVEVFKPGKFVTTLFVNQSSKCPQKIEGFKRLDCQSAMFNDYNFVFTSFAKKQQ Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Matrix metalloproteinase-16 (MMP-16) | 1RM8 | 4.97 | |
Target general information Gen name MMP16 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Membrane-type-3 matrix metalloproteinase; Membrane-type matrix metalloproteinase 3; MTMMP3; MT3MMP; MT3-MMP; MT-MMP 3; MMPX2; MMP-X2; C8orf57 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates progelatinase A. Involved in the matrix remodeling of blood vessels. Isoform short cleaves fibronectin and also collagen type III, but at lower rate. It has no effect on type I, II, IV and V collagen. However, upon interaction with CSPG4, it may be involved in degradation and invasion of type I collagen by melanoma cells. Endopeptidase that degrades various components of the extracellular matrix, such as collagen type III and fibronectin. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03880; DB00786 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Cleavage on pair of basic residues; Collagen degradation; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18853.6 Length 169 Aromaticity 0.12 Instability index 33.65 Isoelectric point 4.88 Charge (pH=7) -12.42 2D Binding mode Binding energy (Kcal/mol) -6.78 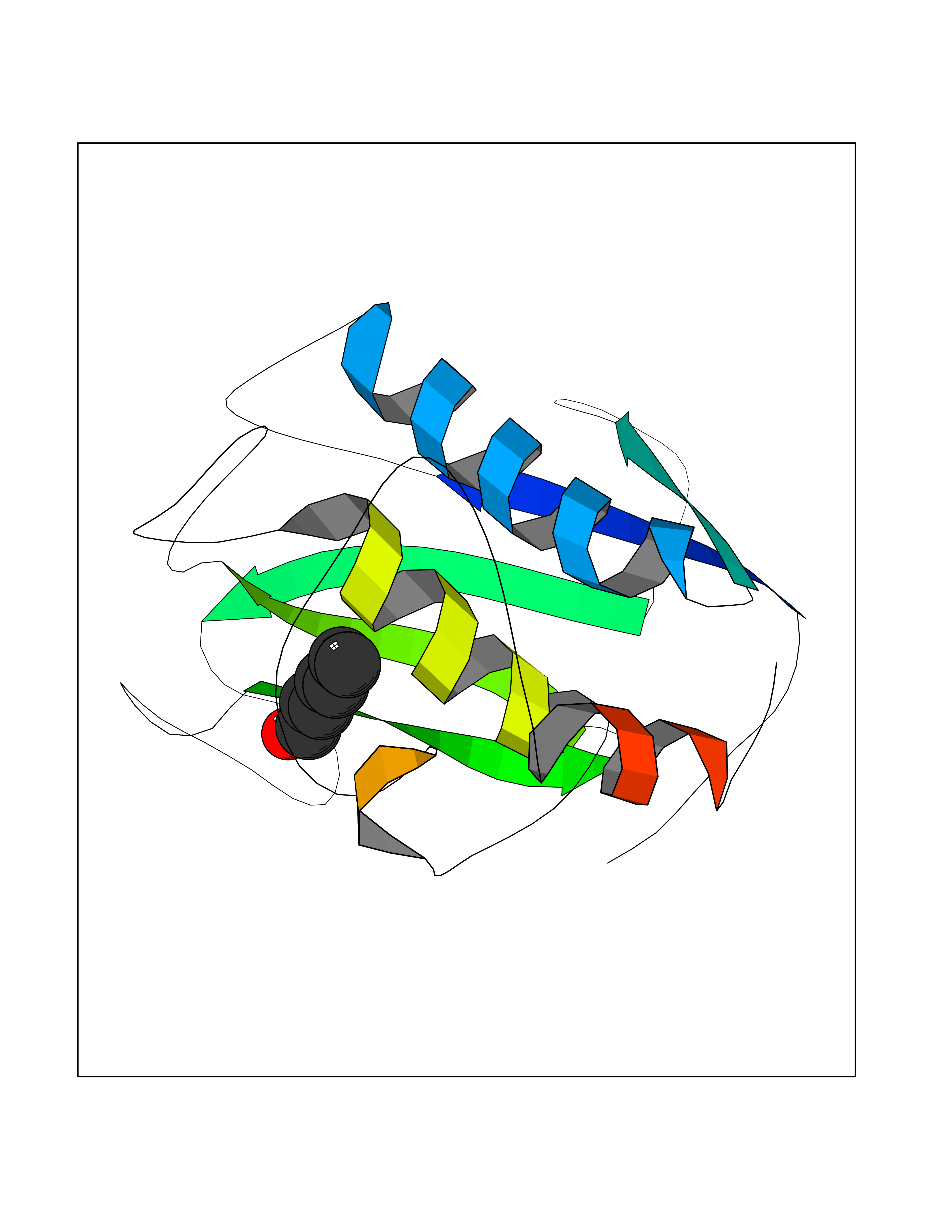 Molscript Map 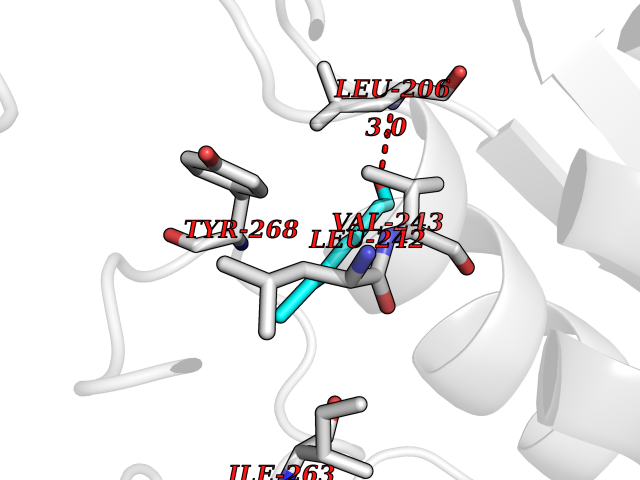 Pymol Map 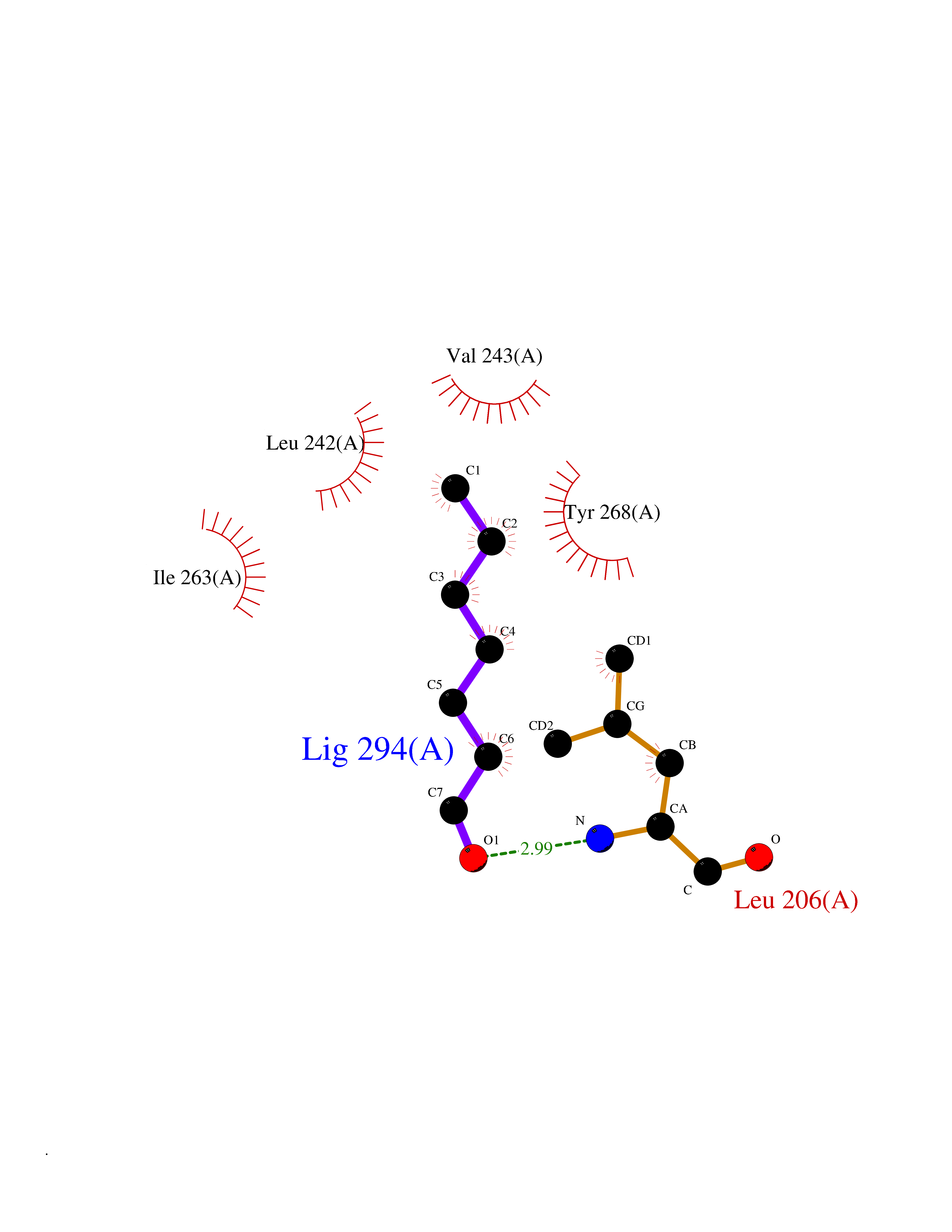 Ligplot Map 3D Binding mode Sequence GQKWQHKHITYSIKNVTPKVGDPETRKAIRRAFDVWQNVTPLTFEEVPYSELENGKRDVDITIIFASGFHGDSSPFDGEGGFLAHAYFPGPGIGGDTHFDSDEPWTLGNPNHDGNDLFLVAVHELGHALGLEHSNDPTAIMAPFYQYMETDNFKLPNDDLQGIQKIYGP Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Hyperpolarization cyclic nucleotide-gated channel 4 (HCN4) | 3OTF | 4.97 | |
Target general information Gen name HCN4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 4 Protein family Potassium channel HCN family Biochemical class Voltage-gated ion channel Function Contributes to the native pacemaker currents in heart (If) that regulate the rhythm of heart beat. May contribute to the native pacemaker currents in neurons (Ih). May mediate responses to sour stimuli. Hyperpolarization-activated ion channel with very slow activation and inactivation exhibiting weak selectivity for potassium over sodium ions. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60741; Q9Y3Q4 EC number NA Uniprot keywords 3D-structure; Brugada syndrome; cAMP; cAMP-binding; Cell membrane; Disease variant; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Nucleotide-binding; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 23211.2 Length 197 Aromaticity 0.12 Instability index 42.69 Isoelectric point 8.67 Charge (pH=7) 3.11 2D Binding mode Binding energy (Kcal/mol) -6.78 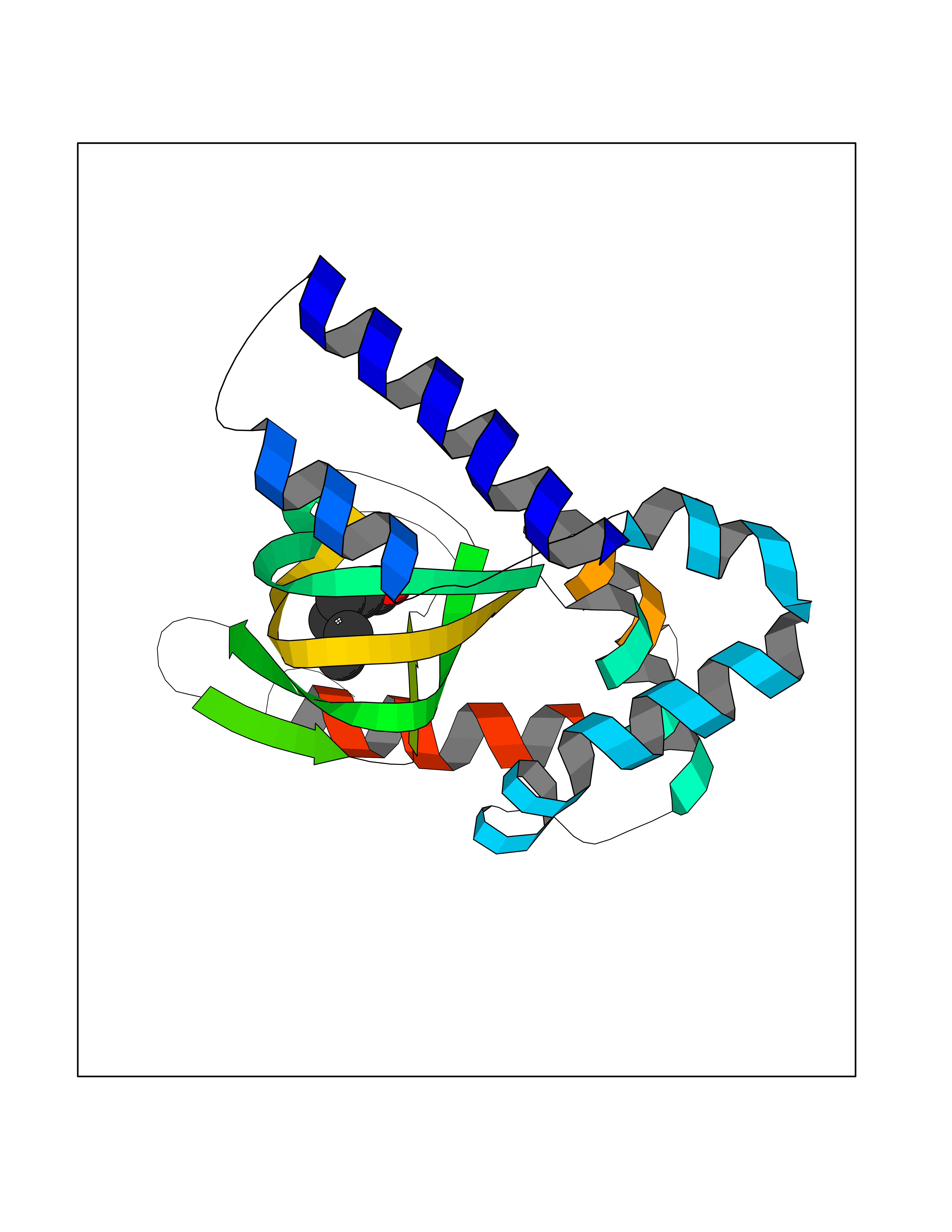 Molscript Map 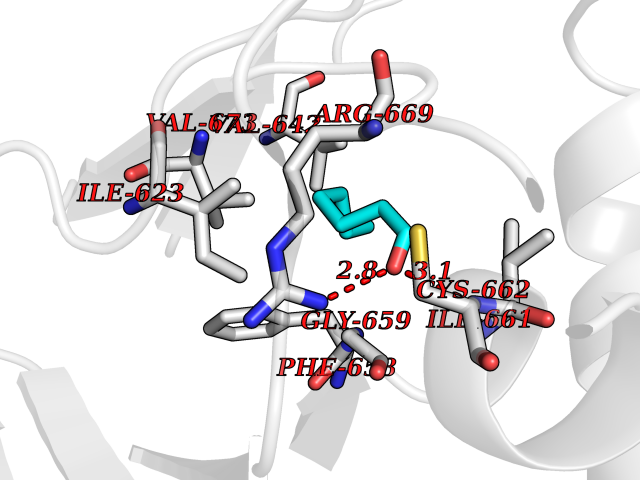 Pymol Map 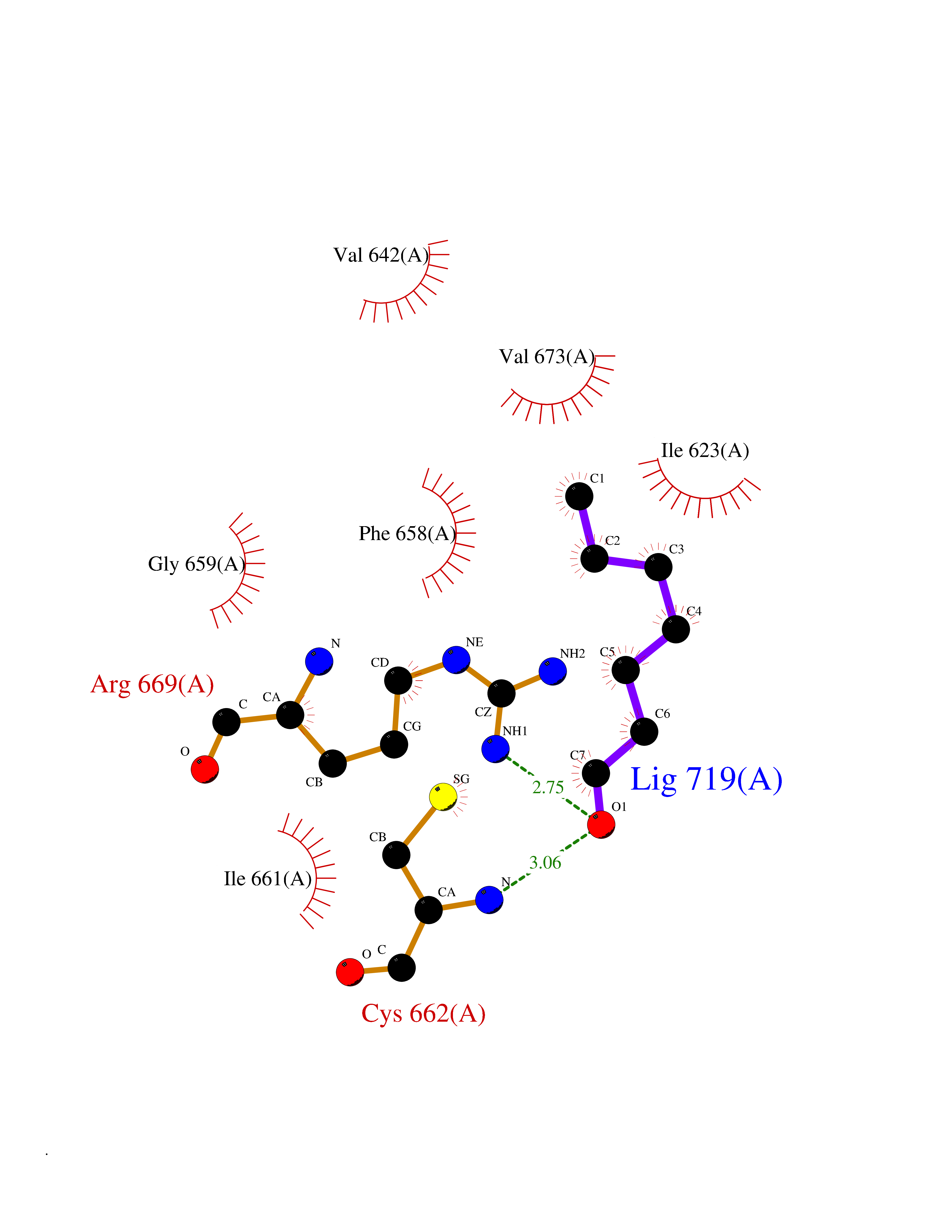 Ligplot Map 3D Binding mode Sequence DSSRRQYQEKYKQVEQYMSFHKLPPDTRQRIHDYYEHRYQGKMFDEESILGELSEPLREEIINFNCRKLVASMPLFANADPNFVTSMLTKLRFEVFQPGDYIIREGTIGKKMYFIQHGVVSVLTKGNKETKLADGSYFGEICLLTRGRRTASVRADTYCRLYSLSVDNFNEVLEEYPMMRRAFETVALDRLDRIGKK Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Lysine N-methyltransferase 3B (NSD1) | 3OOI | 4.97 | |
Target general information Gen name NSD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor-binding SET domain-containing protein 1; NR-binding SET domain-containing protein; NR-binding SET domain containing protein; KMT3B; Hypothetical protein FLJ22263 similar to nuclear re Protein family Class V-like SAM-binding methyltransferase superfamily Biochemical class Methyltransferase Function Preferentially methylates 'Lys-36' of histone H3 and 'Lys-20' of histone H4 (in vitro). Transcriptional intermediary factor capable of both negatively or positively influencing transcription, depending on the cellular context. Histone methyltransferase. Related diseases Sotos syndrome (SOTOS) [MIM:117550]: An autosomal dominant, childhood overgrowth syndrome characterized by pre- and postnatal overgrowth, developmental delay, intellectual disability, advanced bone age, and abnormal craniofacial morphology including macrodolichocephaly with frontal bossing, frontoparietal sparseness of hair, apparent hypertelorism, downslanting palpebral fissures, and facial flushing. Common oral findings include: premature eruption of teeth; high, arched palate; pointed chin and, more rarely, prognathism. {ECO:0000269|PubMed:11896389, ECO:0000269|PubMed:12464997, ECO:0000269|PubMed:12807965, ECO:0000269|PubMed:14997421}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Beckwith-Wiedemann syndrome (BWS) [MIM:130650]: A disorder characterized by anterior abdominal wall defects including exomphalos (omphalocele), pre- and postnatal overgrowth, and macroglossia. Additional less frequent complications include specific developmental defects and a predisposition to embryonal tumors. {ECO:0000269|PubMed:14997421}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A chromosomal aberration involving NSD1 is found in childhood acute myeloid leukemia. Translocation t(5;11)(q35;p15.5) with NUP98.; DISEASE: A chromosomal aberration involving NSD1 is found in an adult form of myelodysplastic syndrome (MDS). Insertion of NUP98 into NSD1 generates a NUP98-NSD1 fusion product. {ECO:0000269|PubMed:15382262}. Drugs (DrugBank ID) NA Interacts with Q13283; Q16778; Q04206; O95994; Q86Z20; A8MQ03; Q3LI66; Q99750 EC number EC 2.1.1.43 Uniprot keywords 3D-structure; Activator; Alternative splicing; Chromatin regulator; Chromosomal rearrangement; Chromosome; Disease variant; Isopeptide bond; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; Repeat; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 26487.9 Length 232 Aromaticity 0.08 Instability index 37.29 Isoelectric point 8.1 Charge (pH=7) 2.69 2D Binding mode Binding energy (Kcal/mol) -6.78 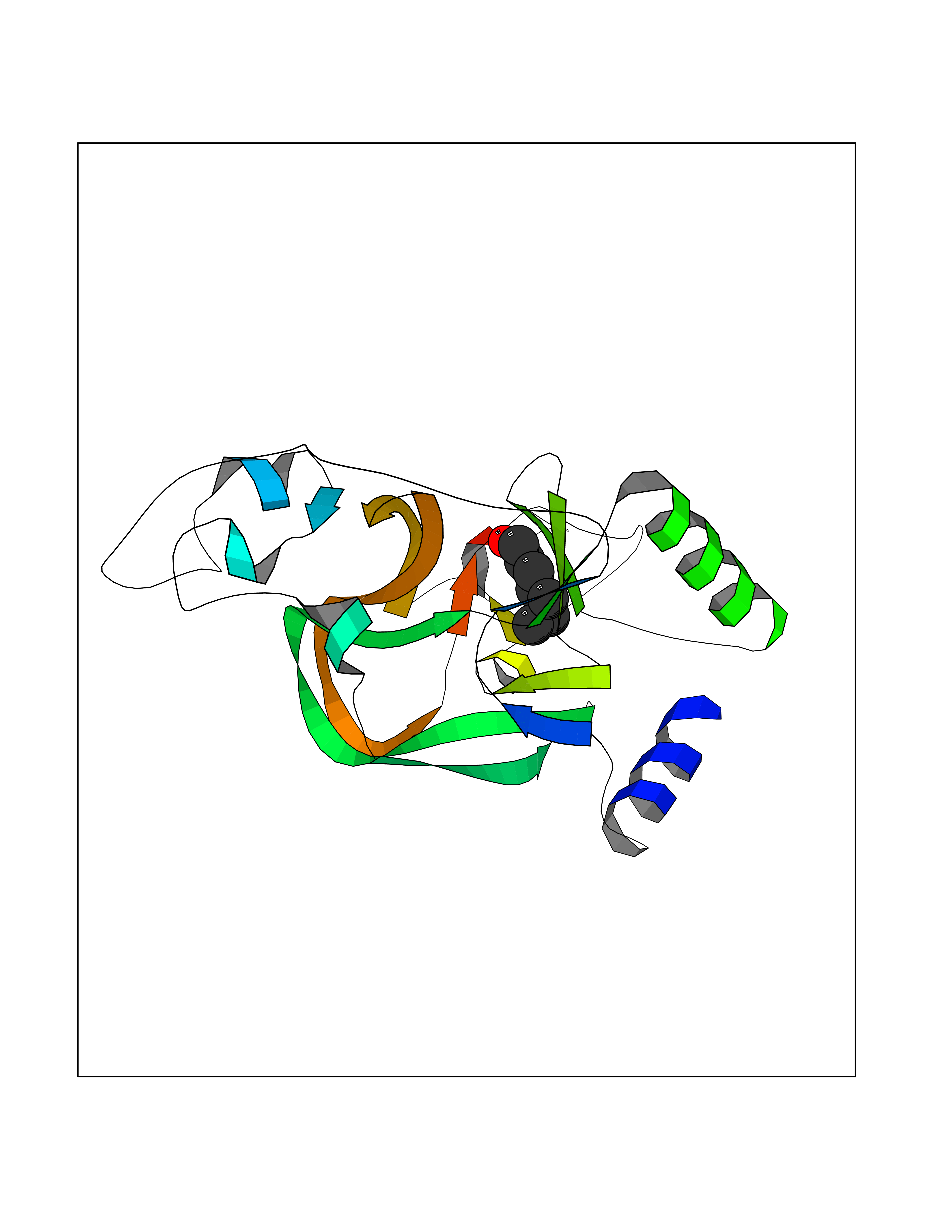 Molscript Map 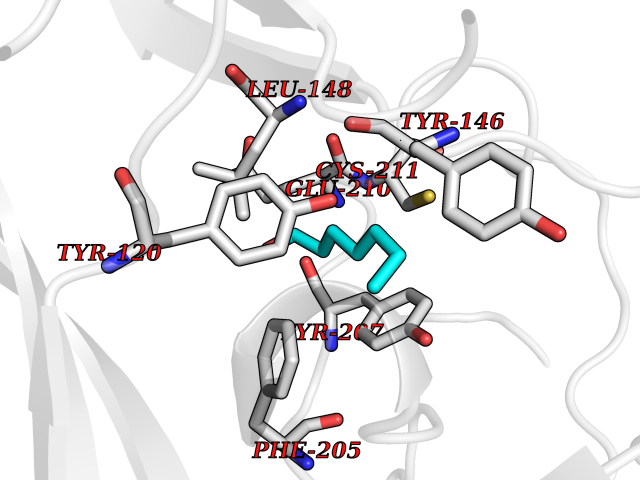 Pymol Map 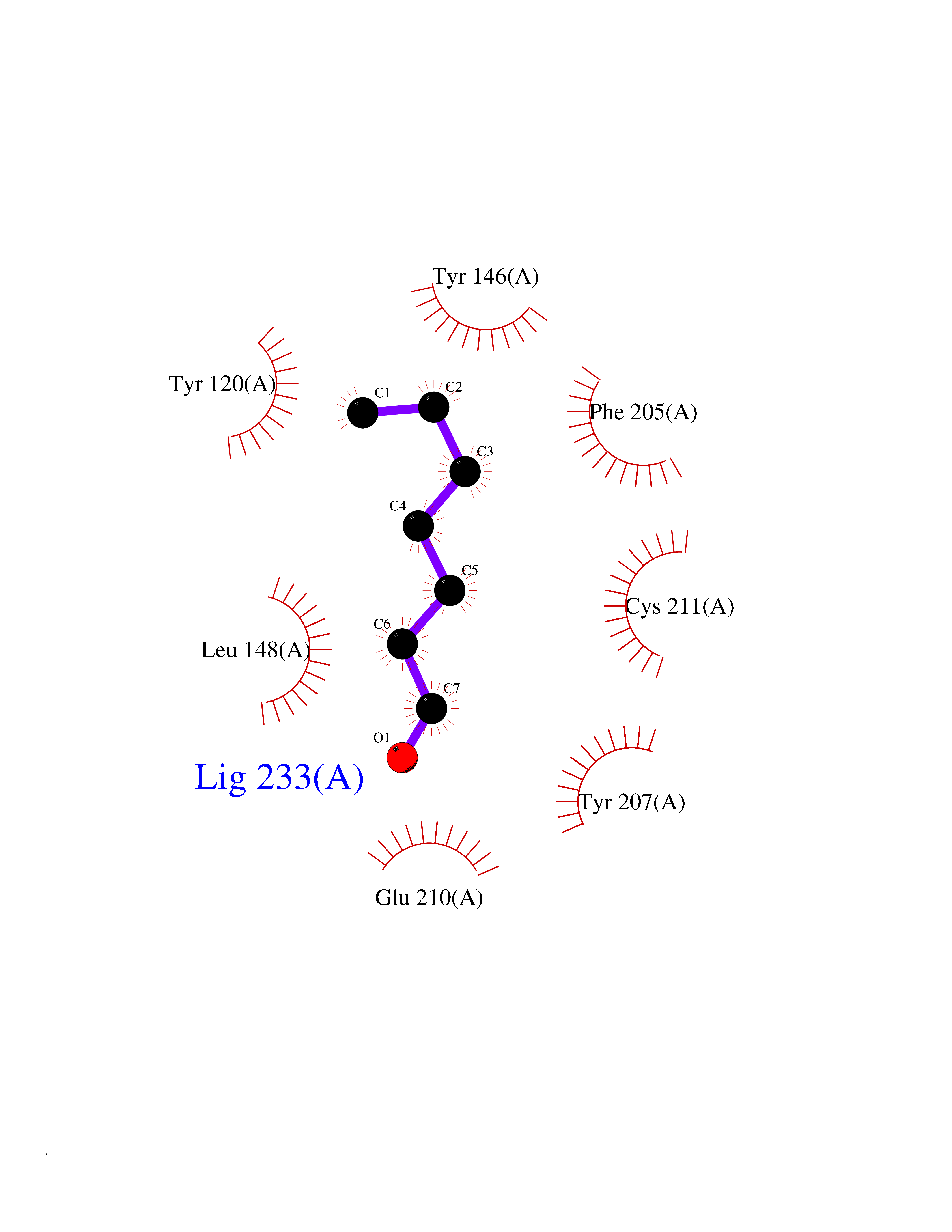 Ligplot Map 3D Binding mode Sequence SKELRQLQEDRKNDKKPPPYKHIKVNRPIGRVQIFTADLSEIPRCNCKATDENPCGIDSECINRMLLYECHPTVCPAGGRCQNQCFSKRQYPEVEIFRTLQRGWGLRTKTDIKKGEFVNEYVGELIDEEECRARIRYAQEHDITNFYMLTLDKDRIIDAGPKGNYARFMNHCCQPNCETQKWSVNGDTRVGLFALSDIKAGTELTFNYNLECLGNGKTVCKCGAPNCSGFLG Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Neprilysin | 1R1H | 4.96 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms EPN Protein family Peptidase M13 family Biochemical class Hydrolase Function Endopeptidase activity.Exopeptidase activity.Metalloendopeptidase activity.Metallopeptidase activity.Peptide binding.Zinc ion binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -6.76 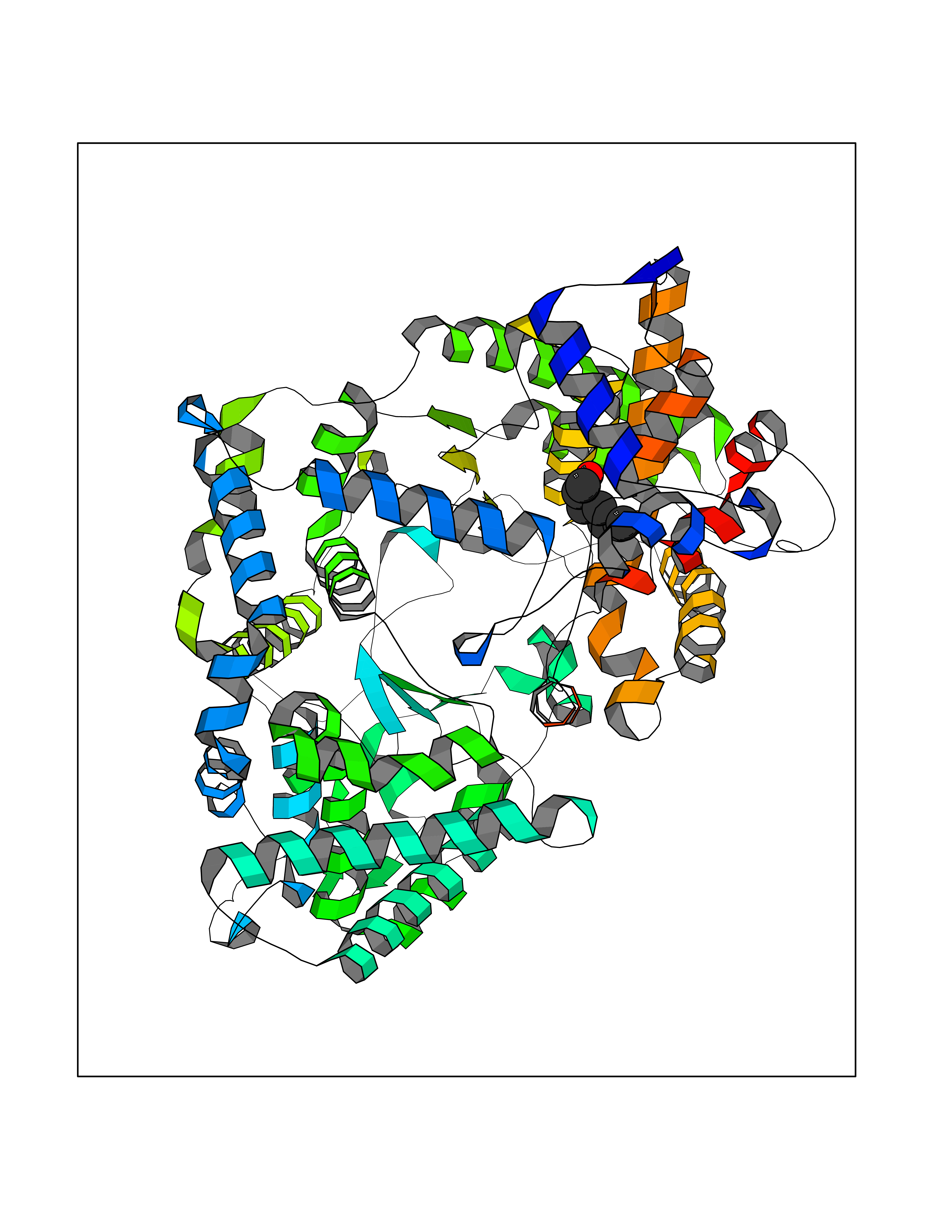 Molscript Map 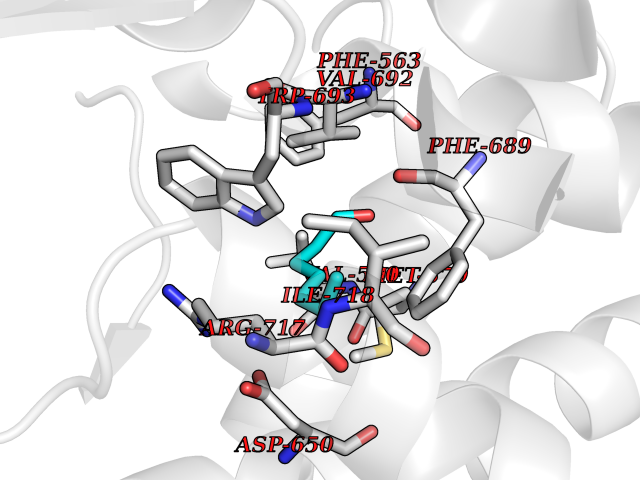 Pymol Map 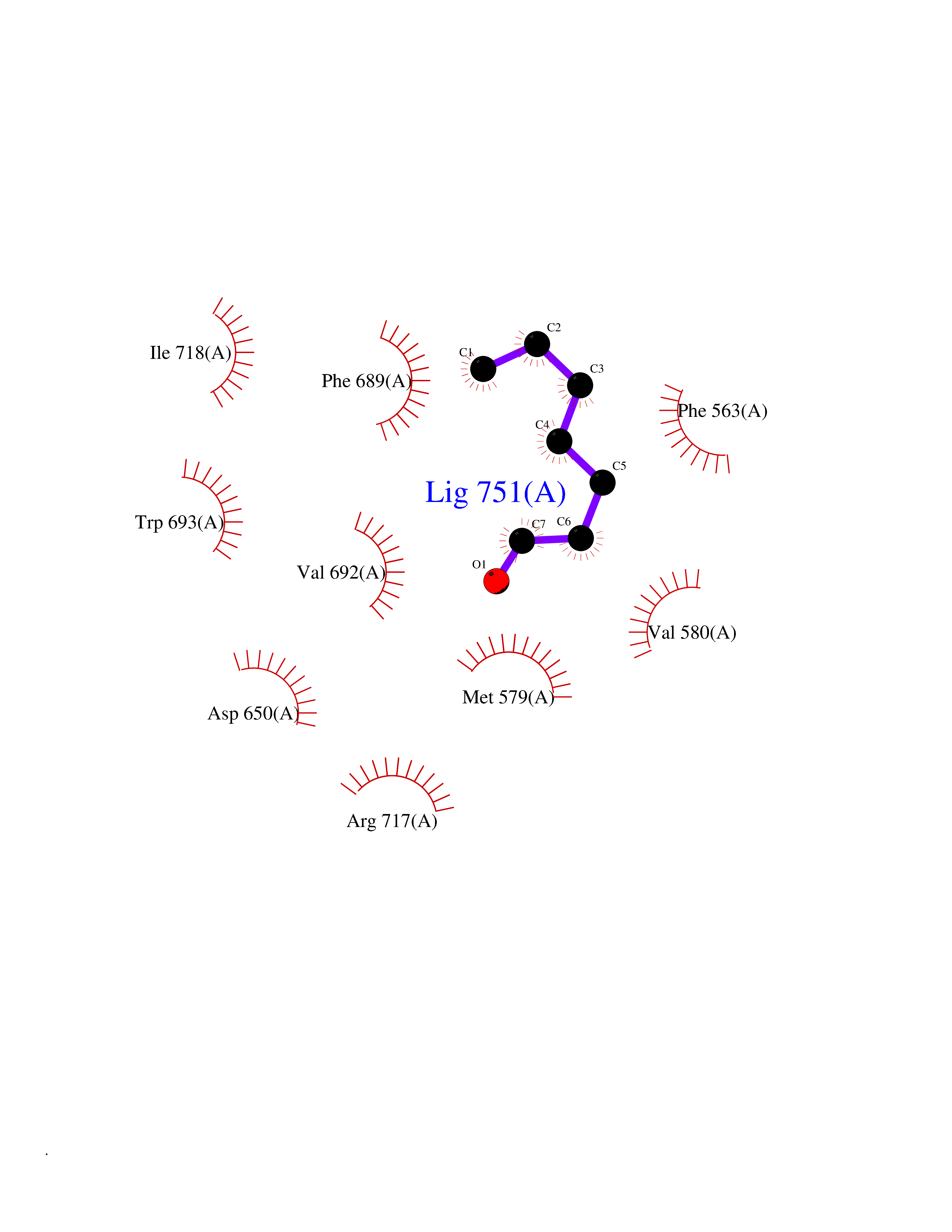 Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Cytochrome b (Complex III subunit 3) (Complex III subunit III) (Cytochrome b-c1 complex subunit 3) (Ubiquinol-cytochrome-c reductase complex cytochrome b subunit) | 1SQB | 4.96 | |
Target general information Gen name MT-CYB Organism Bos taurus (Bovine) Uniprot ID TTD ID NA Synonyms CYTB;MTCYB;COB Protein family Cytochrome b family Biochemical class NA Function Component of the ubiquinol-cytochrome c reductase complex (complex III or cytochrome b-c1 complex) that is part of the mitochondrial respiratory chain. The b-c1 complex mediates electron transfer from ubiquinol to cytochrome c. Contributes to the generation of a proton gradient across the mitochondrial membrane that is then used for ATP synthesis. {ECO:0000269|PubMed:1327781, ECO:0000269|PubMed:20025846, ECO:0000269|PubMed:9485330, ECO:0000305|PubMed:189810}." Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Electron transport; Heme; Iron; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Reference proteome; Respiratory chain; Transmembrane; Transmembrane helix; Transport; Ubiquinone Protein physicochemical properties Chain ID C,D,F,G,H Molecular weight (Da) 99281.2 Length 866 Aromaticity 0.12 Instability index 43.81 Isoelectric point 8.32 Charge (pH=7) 7.12 2D Binding mode Binding energy (Kcal/mol) -6.77  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence VSASSRWLEGIRKWYYNAAGFNKLGLMRDDTIHENDDVKEAIRRLPENLYDDRVFRIKRALDLSMRQQILPKEQWTKYEEDKSYLEPYLKEVIRERKEREEWAKKELVDPLTTVREQCEQLEKCVKARERLELCDERVSSRSQTEEDCTEELLDFLHARDHCVAHKLFNSLKTNIRKSHPLMKIVNNAFIDLPAPSNISSWWNFGSLLGICLILQILTGLFLAMHYTSDTTTAFSSVTHICRDVNYGWIIRYMHANGASMFFICLYMHVGRGLYYGSYTFLETWNIGVILLLTVMATAFMGYVLPWGQMSFWGATVITNLLSAIPYIGTNLVEWIWGGFSVDKATLTRFFAFHFILPFIIMAIAMVHLLFLHETGSNNPTGISSDVDKIPFHPYYTIKDILGALLLILALMLLVLFAPDLLGDPDNYTPANPLNTPPHIKPEWYFLFAYAILRSIPNKLGGVLALAFSILILALIPLLHTSKQRSMMFRPLSQCLFWALVADLLTLTWIGGQPVEHPYITIGQLASVLYFLLILVLMPTAGTIENKLLKWSDLELHPPSYPWSHRGLLSSLDHTSIRRGFQVYKQVCSSCHSMDYVAYRHLVGVCYTEDEAKALAEEVEVQDGPNEDGEMFMRPGKLSDYFPKPYPNPEAARAANNGALPPDLSYIVRARHGGEDYVFSLLTGYCEPPTGVSLREGLYFNPYFPGQAIGMAPPIYNEVLEFDDGTPATMSQVAKDVCTFLRWAAEPEHDHRKRMGLKMLLMMGLLLPLVYAMKRHKWSVLKSRKLAYRPPKGRQFGHLTRVRHVITYSLSPFEQRAFPHYFSKGIPNVLRRTRACILRVAPPFVAFYLVYTWGTQEFEKSKRKNPA Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | T-cell-specific kinase (ITK) | 4HCU | 4.96 | |
Target general information Gen name ITK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine kinase ITK; Inducible T cell kinase; EMT Protein family Protein kinase superfamily, Tyr protein kinase family, TEC subfamily Biochemical class Kinase Function Regulates the development, function and differentiation of conventional T-cells and nonconventional NKT-cells. When antigen presenting cells (APC) activate T-cell receptor (TCR), a series of phosphorylation lead to the recruitment of ITK to the cell membrane, in the vicinity of the stimulated TCR receptor, where it is phosphorylated by LCK. Phosphorylation leads to ITK autophosphorylation and full activation. Once activated, phosphorylates PLCG1, leading to the activation of this lipase and subsequent cleavage of its substrates. In turn, the endoplasmic reticulum releases calcium in the cytoplasm and the nuclear activator of activated T-cells (NFAT) translocates into the nucleus to perform its transcriptional duty. Phosphorylates 2 essential adapter proteins: the linker for activation of T-cells/LAT protein and LCP2. Then, a large number of signaling molecules such as VAV1 are recruited and ultimately lead to lymphokine production, T-cell proliferation and differentiation. Phosphorylates TBX21 at 'Tyr-530' and mediates its interaction with GATA3. Tyrosine kinase that plays an essential role in regulation of the adaptive immune response. Related diseases Lymphoproliferative syndrome 1 (LPFS1) [MIM:613011]: A rare immunodeficiency characterized by extreme susceptibility to infection with Epstein-Barr virus (EBV). Inadequate immune response to EBV can have a fatal outcome. Clinical features include splenomegaly, lymphadenopathy, anemia, thrombocytopenia, pancytopenia, recurrent infections. There is an increased risk for lymphoma. {ECO:0000269|PubMed:19425169}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010; DB06589; DB14924; DB02010; DB15035 Interacts with P04626; P48023; P08238; Q13094; P31947; P62258; P10686 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Adaptive immunity; ATP-binding; Cytoplasm; Direct protein sequencing; Disease variant; Immunity; Kinase; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; SH2 domain; SH3 domain; Transferase; Tyrosine-protein kinase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 30116.1 Length 263 Aromaticity 0.11 Instability index 37.47 Isoelectric point 5.03 Charge (pH=7) -11.73 2D Binding mode Binding energy (Kcal/mol) -6.76  Molscript Map 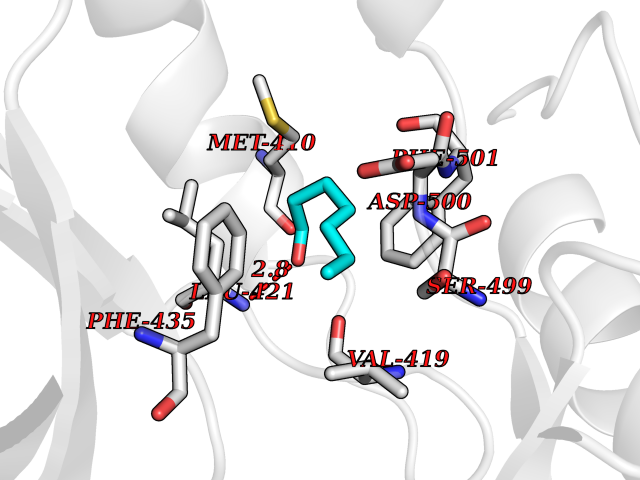 Pymol Map 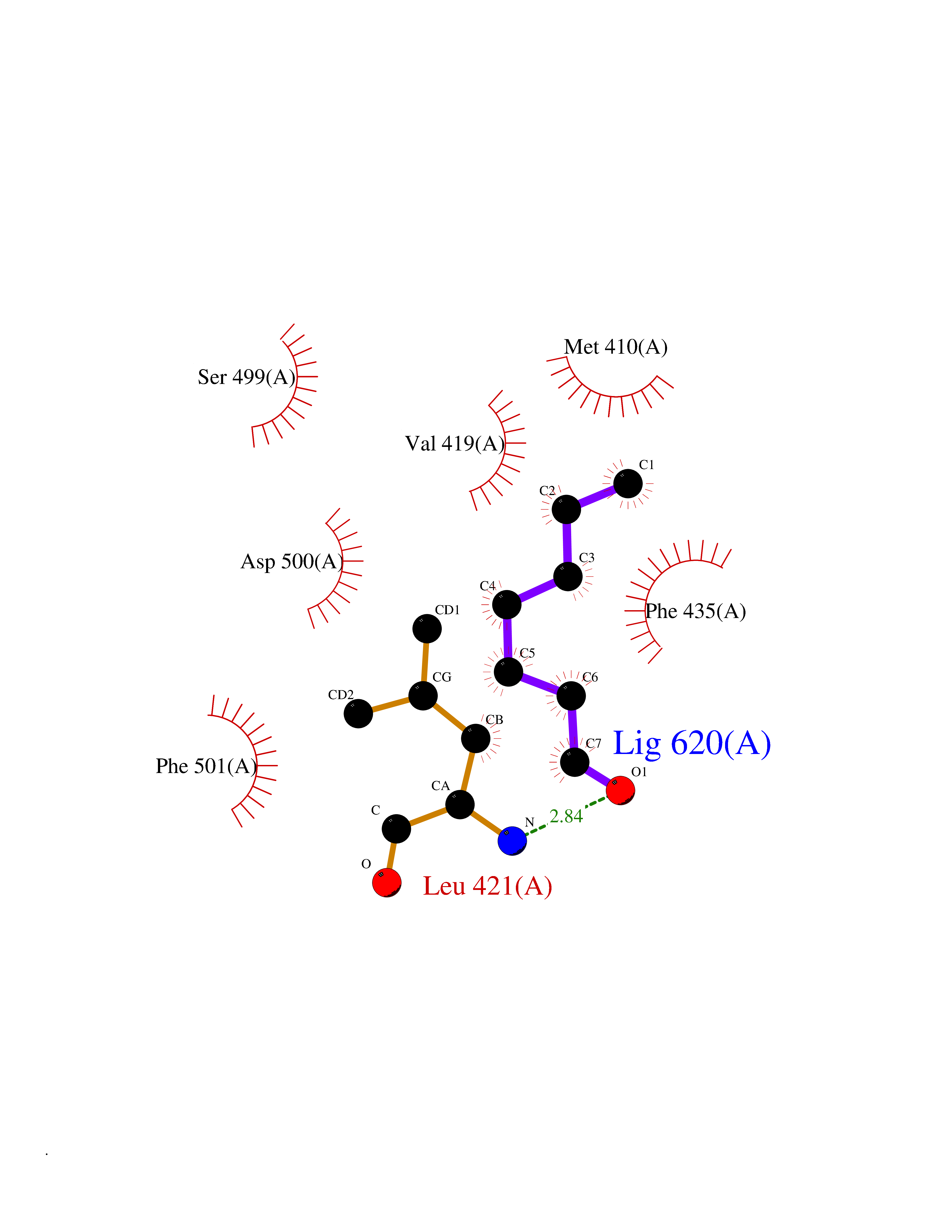 Ligplot Map 3D Binding mode Sequence WVIDPSELTFVQEIGSGQFGLVHLGYWLNKDKVAIKTIREGAMSEEDFIEEAEVMMKLSHPKLVQLYGVCLEQAPICLVFEFMEHGCLSDYLRTQRGLFAAETLLGMCLDVCEGMAYLEEACVIHRDLAARNCLVGENQVIKVSDFGMTRFVLDDQYTSSTGTKFPVKWASPEVFSFSRYSSKSDVWSFGVLMWEVFSEGKIPYENRSNSEVVEDISTGFRLYKPRLASTHVYQIMNHCWRERPEDRPAFSRLLRQLAEIAES Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | TRANSPORT INHIBITOR RESPONSE 1 protein | 2P1Q | 4.96 | |
Target general information Gen name IAA7 Organism Arabidopsis thaliana (Mouse-ear cress) Uniprot ID TTD ID NA Synonyms AXR2;At3g23050;MXC7.8 Protein family Aux/IAA family Biochemical class Signaling protein Function DNA binding transcription factor activity. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) NA Interacts with Q9LW29; Q9C5W9; Q8RYC8; Q94AH6; Q9ZR12; P49677; Q38828; Q38829; Q38830; Q38831; O24407; O24408; O24409; P49678; O24410; Q8LAL2; Q9XFM0; Q38822; Q9M1R4; Q9C5X0; Q9C8Y3; Q39255; Q570C0 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Auxin signaling pathway; Nucleus; Reference proteome; Repressor; Transcription; Transcription regulation Protein physicochemical properties Chain ID B,C Molecular weight (Da) 65385.2 Length 581 Aromaticity 0.09 Instability index 47.83 Isoelectric point 7.46 Charge (pH=7) 1.23 2D Binding mode Binding energy (Kcal/mol) -6.77 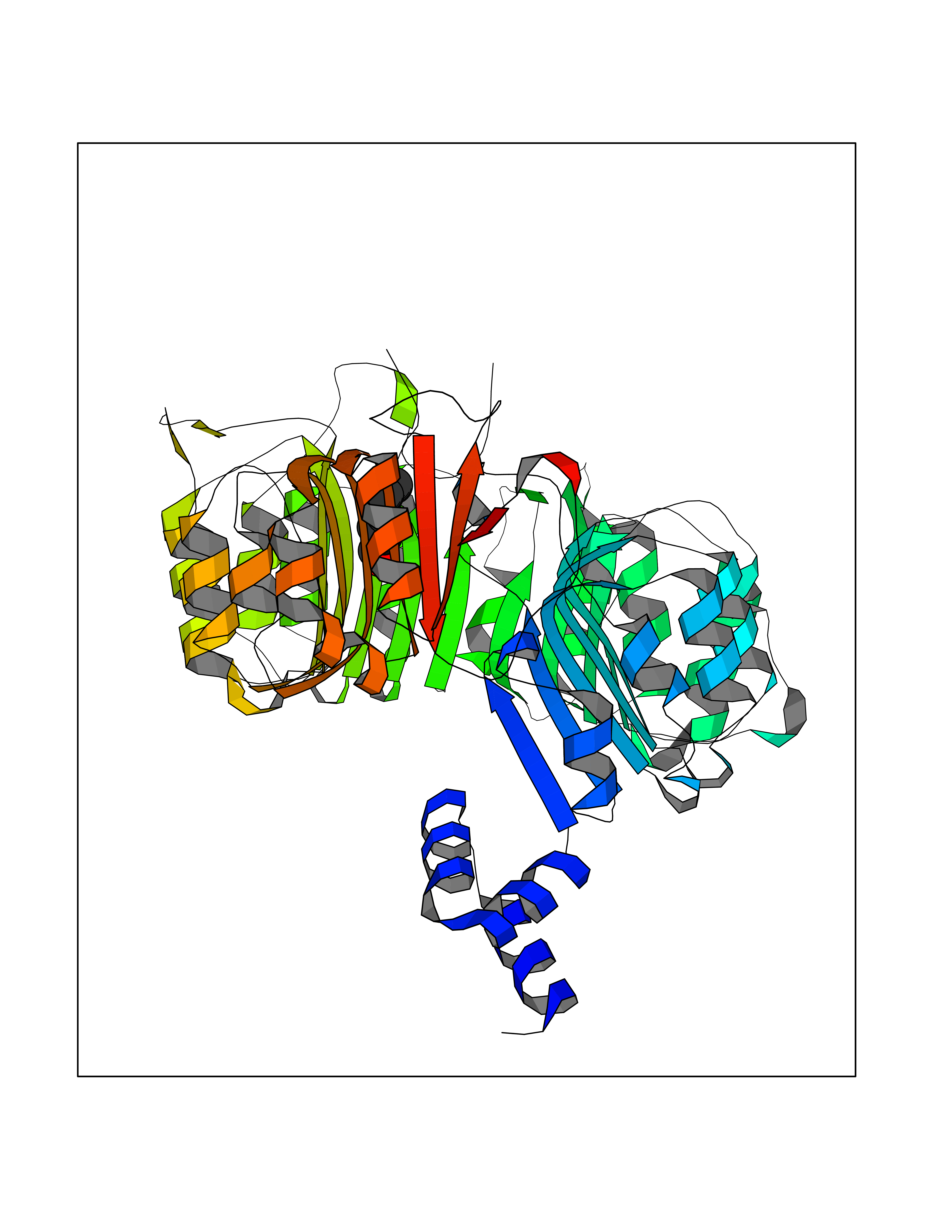 Molscript Map 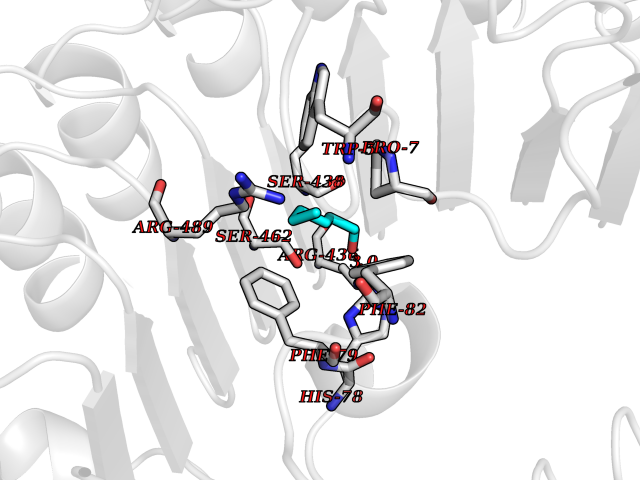 Pymol Map 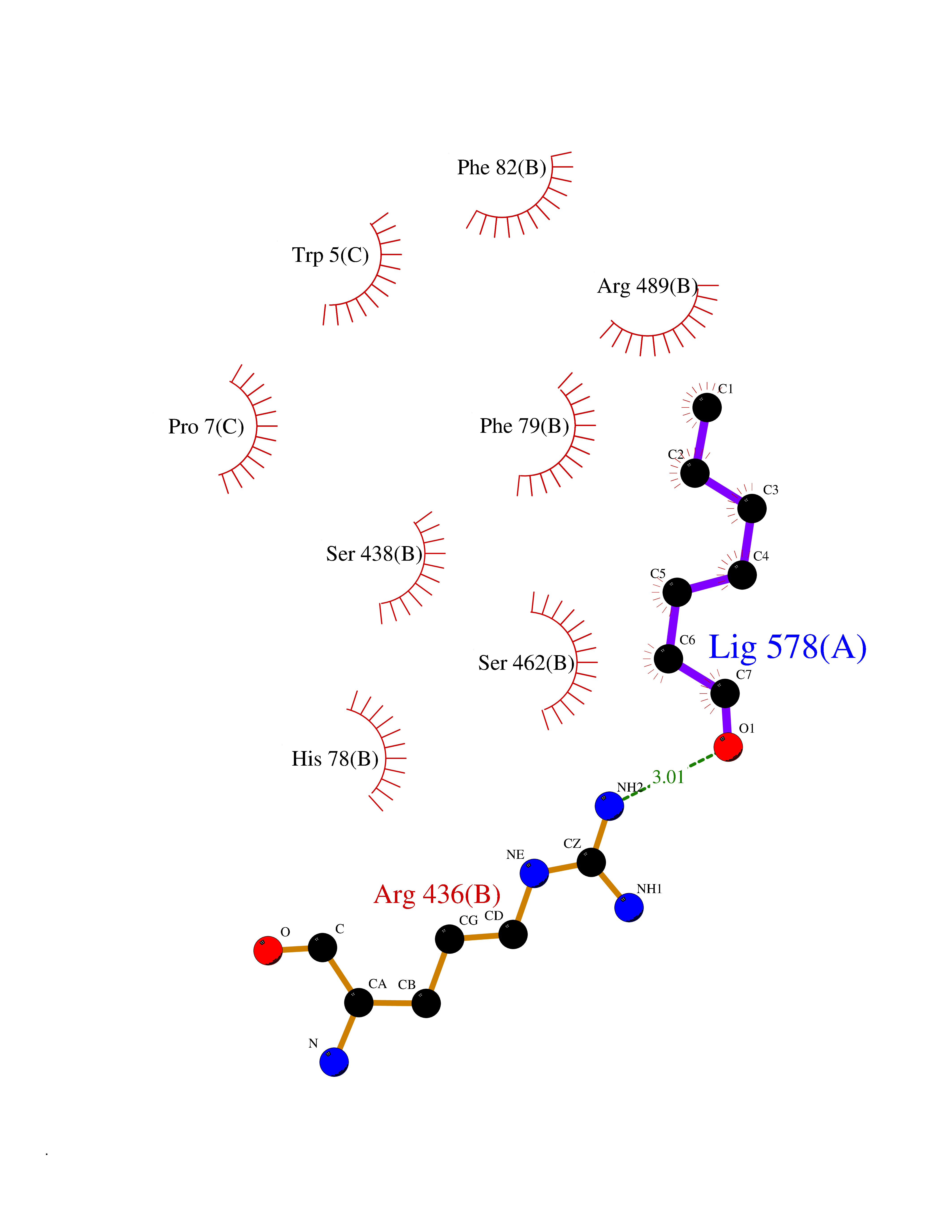 Ligplot Map 3D Binding mode Sequence QVVGWPPVRNYRKFPEEVLEHVFSFIQLDKDRNSVSLVCKSWYEIERWCRRKVFIGNCYAVSPATVIRRFPKVRSVELKGKPHFADFNLVPDGWGGYVYPWIEAMSSSYTWLEEIRLKRMVVTDDCLELIAKSFKNFKVLVLSSCEGFSTDGLAAIAATCRNLKELDLRESDVDDVSGHWLSHFPDTYTSLVSLNISCLASEVSFSALERLVTRCPNLKSLKLNRAVPLEKLATLLQRAPQLEELGTGGYTAEVRPDVYSGLSVALSGCKELRCLSGFWDAVPAYLPAVYSVCSRLTTLNLSYATVQSYDLVKLLCQCPKLQRLWVLDYIEDAGLEVLASTCKDLRELRVFPSEPFVMEPNVALTEQGLVSVSMGCPKLESVLYFCRQMTNAALITIARNRPNMTRFRLCIIEPKAPDYLTLEPLDIGFGAIVEHCKDLRRLSLSGLLTDKVFEYIGTYAKKMEMLSVAFAGDSDLGMHHVLSGCDSLRKLEIRDCPFGDKALLANASKLETMRSLWMSSCSVSFGACKLLGQKMPKLNVEVIDERGAPDSRPESCPVERVFIYRTVAGPRFDMPGFVWNM Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Protein-tyrosine phosphatase SHP-2 (PTPN11) | 2SHP | 4.96 | |
Target general information Gen name PTPN11 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein phosphatase non-receptor type 11; SHPTP2; SHP2; SHP-2; SH-PTP3; SH-PTP2; Protein-tyrosine phosphatase SHP2; Protein-tyrosine phosphatase 2C; Protein-tyrosine phosphatase 1D; PTP2C; PT Protein family Protein-tyrosine phosphatase family, Non-receptor class 2 subfamily Biochemical class Phosphoric monoester hydrolase Function Positively regulates MAPK signal transduction pathway. Dephosphorylates GAB1, ARHGAP35 and EGFR. Dephosphorylates ROCK2 at 'Tyr-722' resulting in stimulatation of its RhoA binding activity. Dephosphorylates CDC73. Acts downstream of various receptor and cytoplasmic protein tyrosine kinases to participate in the signal transduction from the cell surface to the nucleus. Related diseases LEOPARD syndrome 1 (LPRD1) [MIM:151100]: A disorder characterized by lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonic stenosis, abnormalities of genitalia, retardation of growth, and sensorineural deafness. {ECO:0000269|PubMed:12058348, ECO:0000269|PubMed:14961557, ECO:0000269|PubMed:15121796, ECO:0000269|PubMed:15389709, ECO:0000269|PubMed:15520399, ECO:0000269|PubMed:15690106, ECO:0000269|PubMed:16679933, ECO:0000269|PubMed:16733669, ECO:0000269|PubMed:24891296, ECO:0000269|PubMed:26742426}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 1 (NS1) [MIM:163950]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. Some patients with NS1 develop multiple giant cell lesions of the jaw or other bony or soft tissues, which are classified as pigmented villonodular synovitis (PVNS) when occurring in the jaw or joints. {ECO:0000269|PubMed:11704759, ECO:0000269|PubMed:11992261, ECO:0000269|PubMed:12161469, ECO:0000269|PubMed:12325025, ECO:0000269|PubMed:12529711, ECO:0000269|PubMed:12634870, ECO:0000269|PubMed:12717436, ECO:0000269|PubMed:12739139, ECO:0000269|PubMed:12960218, ECO:0000269|PubMed:15384080, ECO:0000269|PubMed:15889278, ECO:0000269|PubMed:15948193, ECO:0000269|PubMed:19020799, ECO:0000269|PubMed:24891296, ECO:0000269|PubMed:28074573}. The disease is caused by variants affecting the gene represented in this entry. Mutations in PTPN11 account for more than 50% of the cases.; DISEASE: Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:12717436, ECO:0000269|PubMed:26742426}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metachondromatosis (MC) [MIM:156250]: A skeletal disorder with radiologic features of both multiple exostoses and Ollier disease, characterized by the presence of exostoses, commonly of the bones of the hands and feet, and enchondromas of the metaphyses of long bones and iliac crest. {ECO:0000269|PubMed:20577567}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02779 Interacts with P10275; P32239; Q9BZW8; P20138; Q08345; P00533; P29317; P04626; Q8WU20; Q13480; Q9UQC2; P62993; P08069; P06213; P35568; P43628; P10721; P08581; O95297; Q15116; P09619; P16284; P49023; P49247; Q13049; P68105; Q71V39; P35570; P97710; Q6P1J9; Q13480; O75496; Q9UKI8 EC number EC 3.1.3.48 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Deafness; Disease variant; Hydrolase; Nucleus; Phosphoprotein; Protein phosphatase; Proteomics identification; Reference proteome; Repeat; SH2 domain Protein physicochemical properties Chain ID A Molecular weight (Da) 56341.2 Length 491 Aromaticity 0.09 Instability index 41.37 Isoelectric point 7.76 Charge (pH=7) 2.43 2D Binding mode Binding energy (Kcal/mol) -6.76  Molscript Map 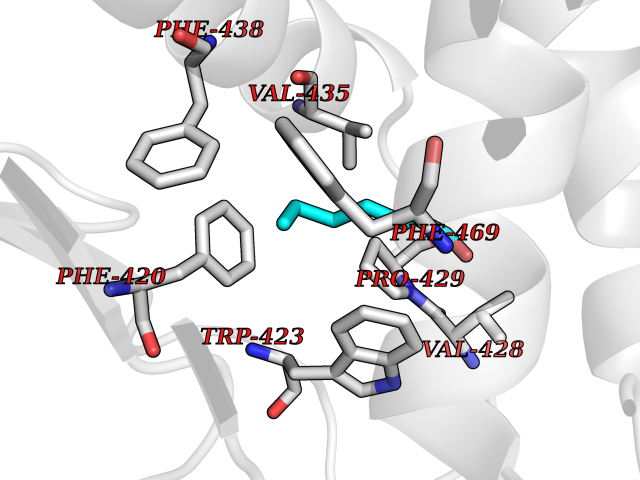 Pymol Map 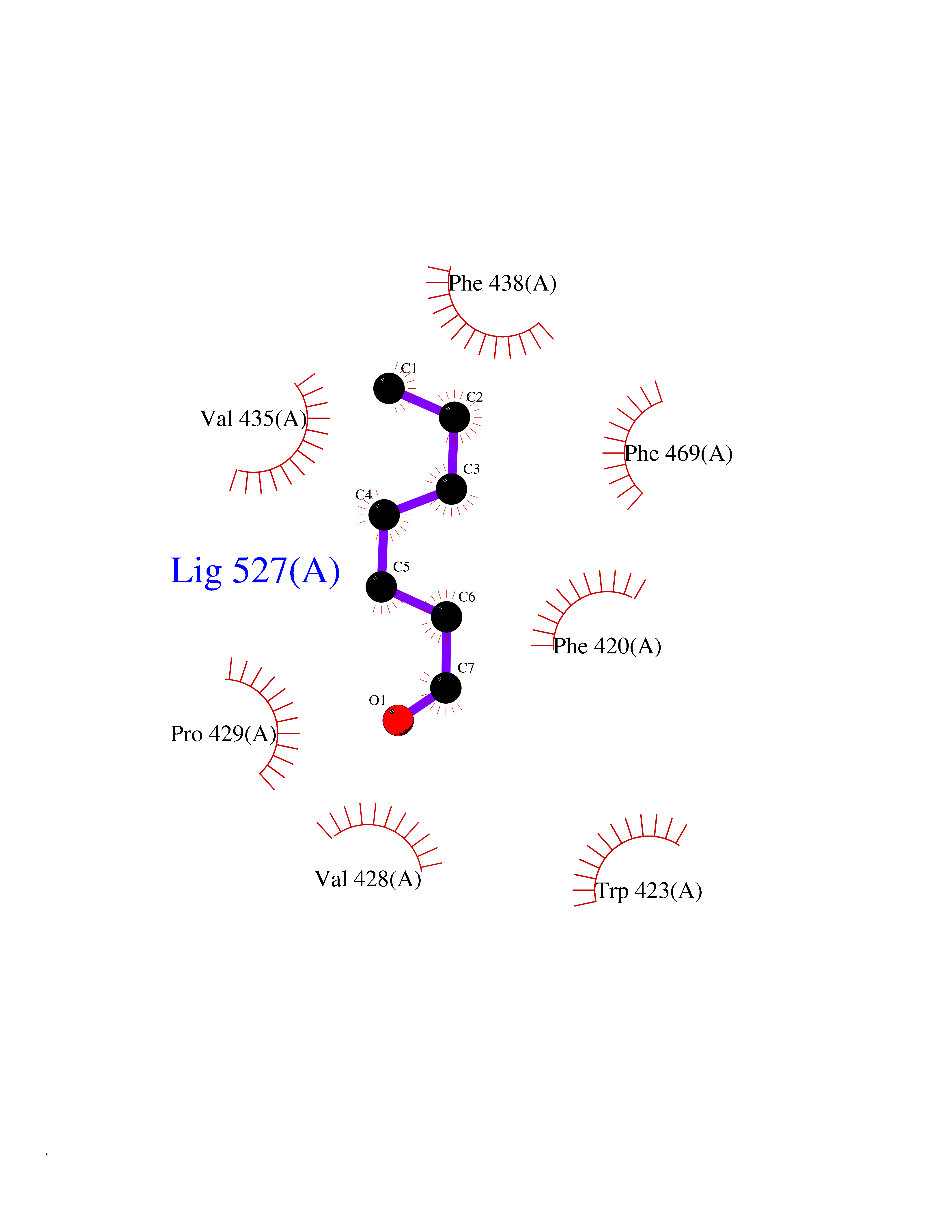 Ligplot Map 3D Binding mode Sequence KSRRWFHPNITGVEAENLLLTRGVDGSFLARPSKSNPGDLTLSVRRNGAVTHIKIQNTGDYYDLYGGEKFATLAELVQYYMEHHGQLKEKNGDVIELKYPLNCADPTSERWFHGHLSGKEAEKLLTEKGKHGSFLVRESQSHPGDFVLSVRTGDNDGKSKVTHVMIRCQELKYDVGGGERFDSLTDLVEHYKKNPMVETLGTVLQLKQPLNTTRINAAEIESRVRELSKGFWEEFETLQQQECKLLYSRKEGQRQENKNKNRYKNILPFDHTRVVLHDSDYINANIIMPKKSYIATQGCLQNTVNDFWRMVFQENSRVIVMTTKEVERGKSKCVKYWPDEYALKEYGVMRVRNVKESAAHDYTLRELKLSKVGQGNTERTVWQYHFRTWPDHGVPSDPGGVLDFLEEVHHKQESIMDAGPVVVHCSAGIGRTGTFIVIDILIDIIREKGVDCDIDVPKTIQMVRSQRSGMVQTEAQYRSIYMAVQHYIETL Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Ubiquitin carboxyl-terminal hydrolase 14 (USP14) | 6IIK | 4.96 | |
Target general information Gen name USP14 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin-specific-processing protease 14; Ubiquitin thioesterase 14; TGT; Deubiquitinating enzyme 14 Protein family Peptidase C19 family, USP14/UBP6 subfamily Biochemical class Peptidase Function Ensures the regeneration of ubiquitin at the proteasome. Is a reversibly associated subunit of the proteasome and a large fraction of proteasome-free protein exists within the cell. Required for the degradation of the chemokine receptor CXCR4 which is critical for CXCL12-induced cell chemotaxis. Serves also as a physiological inhibitor of endoplasmic reticulum-associated degradation (ERAD) under the non-stressed condition by inhibiting the degradation of unfolded endoplasmic reticulum proteins via interaction with ERN1. Indispensable for synaptic development and function at neuromuscular junctions (NMJs). Plays a role in the innate immune defense against viruses by stabilizing the viral DNA sensor CGAS and thus inhibiting its autophagic degradation. Proteasome-associated deubiquitinase which releases ubiquitin from the proteasome targeted ubiquitinated proteins. Related diseases Hypophosphatemic rickets, autosomal dominant (ADHR) [MIM:193100]: A disease characterized by isolated renal phosphate wasting, hypophosphatemia, and inappropriately normal 1,25-dihydroxyvitamin D3 (calcitriol) levels. Patients frequently present with bone pain, rickets, and tooth abscesses. {ECO:0000269|PubMed:11062477, ECO:0000269|PubMed:11409890, ECO:0000269|PubMed:16638743}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Tumoral calcinosis, hyperphosphatemic, familial, 2 (HFTC2) [MIM:617993]: A form of hyperphosphatemic tumoral calcinosis, a rare autosomal recessive metabolic disorder that manifests with hyperphosphatemia and massive calcium deposits in the skin and subcutaneous tissues. Some patients have recurrent, transient, painful swellings of the long bones associated with the radiographic findings of periosteal reaction and cortical hyperostosis and absence of skin involvement. {ECO:0000269|PubMed:15590700, ECO:0000269|PubMed:16030159, ECO:0000269|PubMed:16151858, ECO:0000269|PubMed:24680727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12695 Interacts with Q08209 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell membrane; Cytoplasm; Hydrolase; Immunity; Innate immunity; Membrane; Phosphoprotein; Protease; Proteasome; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 38476.7 Length 335 Aromaticity 0.1 Instability index 61.05 Isoelectric point 5.6 Charge (pH=7) -4.84 2D Binding mode Binding energy (Kcal/mol) -6.76  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ELPCGLTNLGNTCYMNATVQCIRSVPELKDALKRYAGALRASGEMASAQYITAALRDLFDSMDKTSSSIPPIILLQFLHMAFPQFAEKGEQGQYLQQDANECWIQMMRVLQQKLEAIEDKSLIDQFFGVEFETTMKCTESEEEEVTKGKENQLQLSCFINQEVKYLFTGLKLRLQEEITKQSPTLQRNALYIKSSKISRLPAYLTIQMVRFFNAKVLKDVKFPLMLDMYELCTPELQEKMVSFRSKFKDLYEPFSFADDIGSNNCGYYDLQAVLTHQGRSSSSGHYVSWVKRKQDEWIKFDDDKVSIVTPEDILRLSGGGDWHIAYVLLYGPRRV Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Haemophilus influenzae NadR protein (Hae-influ nadR) | 1LW7 | 4.96 | |
Target general information Gen name Hae-influ nadR Organism Haemophilus influenzae (strain ATCC 51907 / DSM 11121 / KW20 / Rd) Uniprot ID TTD ID Synonyms nadR; Transcriptional regulator nadR Protein family Bacterial NMN adenylyltransferase family; Bacterial RNK family Biochemical class Nicotinamide ribonucleoside uptake permease Function This enzyme has twoactivities: nicotinamide mononucleotide (NMN) adenylyltransferase and ribosylnicotinamide (RN) kinase. The RN kinase activity catalyzes the phosphorylation of RN to form nicotinamide ribonucleotide. The NMN adenylyltransferase activity catalyzes the transfer of the AMP moiety of ATP to nicotinamide ribonucleotide to form NAD(+). Related diseases Involved in the epigenetic regulation of ESR1 expression in breast cancer in a TFAP2C, IFI16 and HDAC4/5/6-dependent manner. {ECO:0000269|PubMed:24413532}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative initiation; ATP-binding; Cell membrane; Cytoplasm; Kinase; Membrane; Multifunctional enzyme; NAD; Nucleotide-binding; Pyridine nucleotide biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 39581.5 Length 344 Aromaticity 0.14 Instability index 41.39 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -6.77  Molscript Map 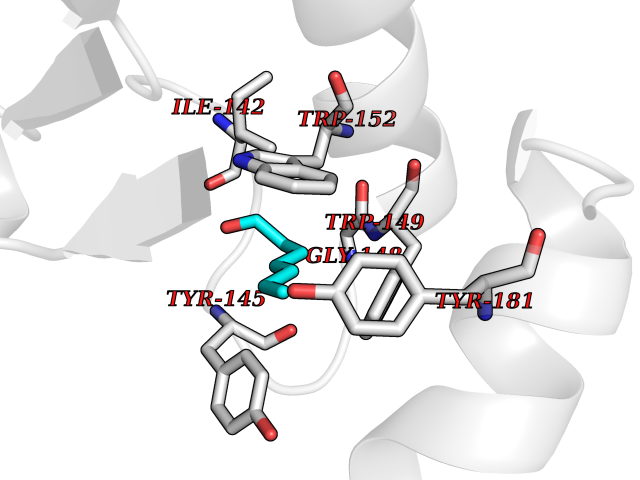 Pymol Map 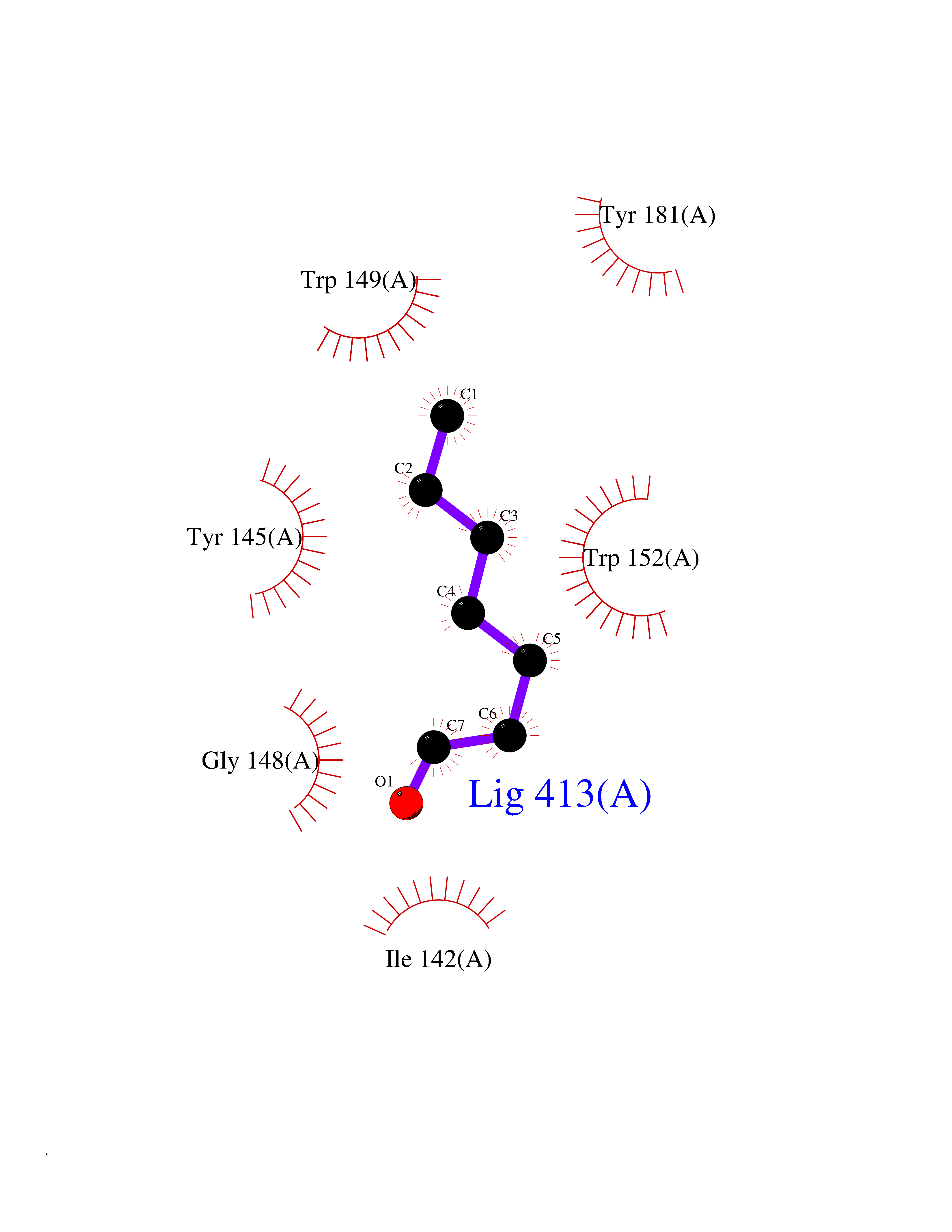 Ligplot Map 3D Binding mode Sequence EKKVGVIFGKFYPVHTGHINXIYEAFSKVDELHVIVCSDTVRDLKLFYDSKXKRXPTVQDRLRWXQQIFKYQKNQIFIHHLVEDGIPSYPNGWQSWSEAVKTLFHEKHFEPSIVFSSEPQDKAPYEKYLGLEVSLVDPDRTFFNVSATKIRTTPFQYWKFIPKEARPFFAKTVAILGGESSGKSVLVNKLAAVFNTTSAWEYGREFVFEKLGGDEQAMQYSDYPQXALGHQRYIDYAVRHSHKIAFIDTDFITTQAFCIQYEGKAHPFLDSXIKEYPFDVTILLKNNTEQKQRQQFQQLLKKLLDKYKVPYIEIESPSYLDRYNQVKAVIEKVLNEEEISELQN Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Debrisoquine 4-hydroxylase (CYP2D6) | 4WNV | 4.96 | |
Target general information Gen name CYP2D6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms P450-DB1; Cytochrome P450-DB1; Cytochrome P450 2D6; CYPIID6; CYP2DL1 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function It is involved in the metabolism of drugs such as antiarrhythmics, adrenoceptor antagonists, and tricyclic antidepressants. Responsible for the metabolism of many drugs and environmental chemicals that it oxidizes. Related diseases A chromosomal aberration involving BCL2 has been found in chronic lymphatic leukemia. Translocation t(14;18)(q32;q21) with immunoglobulin gene regions. BCL2 mutations found in non-Hodgkin lymphomas carrying the chromosomal translocation could be attributed to the Ig somatic hypermutation mechanism resulting in nucleotide transitions. {ECO:0000269|PubMed:2875799, ECO:0000269|PubMed:3285301}. Drugs (DrugBank ID) DB01562; DB01472; DB14010; DB12001; DB05812; DB01193; DB00316; DB15568; DB00918; DB06203; DB00866; DB01424; DB01118; DB00321; DB00381; DB00613; DB00543; DB00182; DB00701; DB11785; DB01435; DB01429; DB01274; DB01238; DB14185; DB09204; DB11638; DB06216; DB00637; DB11586; DB00335; DB00289; DB01076; DB00972; DB04957; DB09013; DB16703; DB01086; DB06770; DB01244; DB15982; DB00195; DB01295; DB12236; DB01128; DB04889; DB00810; DB13975; DB08807; DB00188; DB09128; DB12151; DB12752; DB06726; DB00297; DB08808; DB00921; DB01156; DB00490; DB09173; DB00201; DB09061; DB14737; DB06016; DB00521; DB01136; DB00482; DB04846; DB00439; DB00185; DB00608; DB01114; DB00477; DB00356; DB01410; DB01166; DB00501; DB01012; DB00568; DB00604; DB00215; DB12499; DB00283; DB04920; DB14025; DB00349; DB00845; DB01242; DB00575; DB13508; DB00257; DB00363; DB09065; DB05239; DB00907; DB00318; DB11672; DB14635; DB00924; DB00091; DB11963; DB06292; DB04884; DB00496; DB01264; DB09183; DB04840; DB00705; DB06512; DB01151; DB06700; DB16650; DB12161; DB13679; DB09555; DB01191; DB00633; DB01576; DB00514; DB00647; DB11994; DB01551; DB00343; DB01093; DB01075; DB00757; DB01184; DB00843; DB09167; DB00590; DB01142; DB00997; DB00470; DB04855; DB00476; DB00625; DB11979; DB00216; DB15444; DB09039; DB13874; DB01228; DB06735; DB11718; DB00494; DB13757; DB00751; DB00530; DB13443; DB01175; DB06678; DB00187; DB00330; DB01466; DB01628; DB01590; DB12500; DB01023; DB00574; DB06702; DB12265; DB01195; DB04841; DB00472; DB00623; DB01095; DB00176; DB00983; DB02703; DB15149; DB00674; DB05087; DB00317; DB08909; DB00986; DB01218; DB00502; DB00956; DB01611; DB00557; DB09053; DB01177; DB04946; DB00619; DB00458; DB08952; DB00224; DB06370; DB13293; DB04818; DB16200; DB11633; DB06636; DB00951; DB11757; DB00602; DB09570; DB01026; DB00598; DB12212; DB00448; DB11732; DB16217; DB09078; DB00528; DB12070; DB09351; DB01210; DB08918; DB00281; DB04948; DB01206; DB00836; DB01601; DB00455; DB04871; DB09195; DB06708; DB04829; DB09238; DB00934; DB14921; DB00737; DB14009; DB09224; DB00170; DB00454; DB00532; DB13530; DB06691; DB01071; DB00933; DB01577; DB00333; DB00763; DB01403; DB01028; DB09241; DB01214; DB01233; DB00264; DB00379; DB06148; DB01388; DB01110; DB00211; DB01454; DB06595; DB00834; DB00805; DB08893; DB00370; DB12523; DB01171; DB00745; DB14011; DB09049; DB00731; DB04861; DB01149; DB00220; DB09048; DB00238; DB00627; DB00622; DB00699; DB02701; DB00184; DB01115; DB04868; DB12005; DB00540; DB00334; DB14881; DB00338; DB00904; DB11130; DB04911; DB01173; DB11837; DB04938; DB01096; DB01580; DB01062; DB00497; DB06412; DB01192; DB01267; DB00377; DB06603; DB00715; DB06589; DB00022; DB01359; DB00738; DB01074; DB08922; DB00850; DB03783; DB00780; DB00914; DB00252; DB05316; DB01100; DB00960; DB00592; DB01621; DB04951; DB17472; DB11642; DB08901; DB01297; DB15822; DB01087; DB01035; DB00433; DB00396; DB01131; DB00420; DB01069; DB09288; DB01182; DB00571; DB04216; DB01224; DB00908; DB00468; DB01129; DB00863; DB00243; DB00234; DB14761; DB00409; DB06506; DB02709; DB11855; DB13174; DB11753; DB08864; DB14840; DB00734; DB12693; DB00503; DB00953; DB09291; DB15119; DB00412; DB05271; DB12332; DB11614; DB06654; DB01232; DB01037; DB06144; DB01104; DB00203; DB00641; DB01591; DB00398; DB12713; DB00489; DB06727; DB01323; DB09118; DB06820; DB06729; DB06608; DB11770; DB00675; DB00706; DB06204; DB06083; DB01079; DB12095; DB06287; DB00857; DB00342; DB13775; DB04905; DB04844; DB11712; DB00277; DB00679; DB01623; DB00208; DB00373; DB01409; DB00932; DB06137; DB01036; DB05109; DB00193; DB00752; DB00656; DB12245; DB00726; DB00792; DB00209; DB15328; DB09076; DB13609; DB15091; DB11915; DB00862; DB08881; DB00285; DB00661; DB06217; DB06684; DB09185; DB00570; DB00361; DB11739; DB09068; DB01392; DB00549; DB15688; DB00425; DB01624 Interacts with NA EC number EC 1.14.14.- Uniprot keywords 3D-structure; Alternative splicing; Cholesterol metabolism; Endoplasmic reticulum; Fatty acid metabolism; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 51898.1 Length 464 Aromaticity 0.09 Instability index 43.83 Isoelectric point 6.76 Charge (pH=7) -0.99 2D Binding mode Binding energy (Kcal/mol) -6.76  Molscript Map 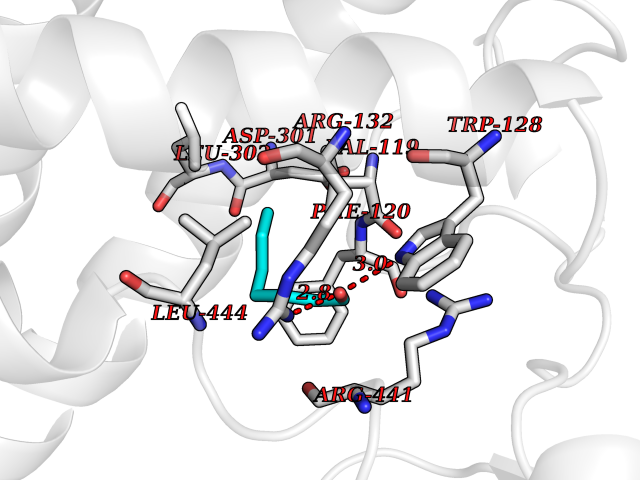 Pymol Map 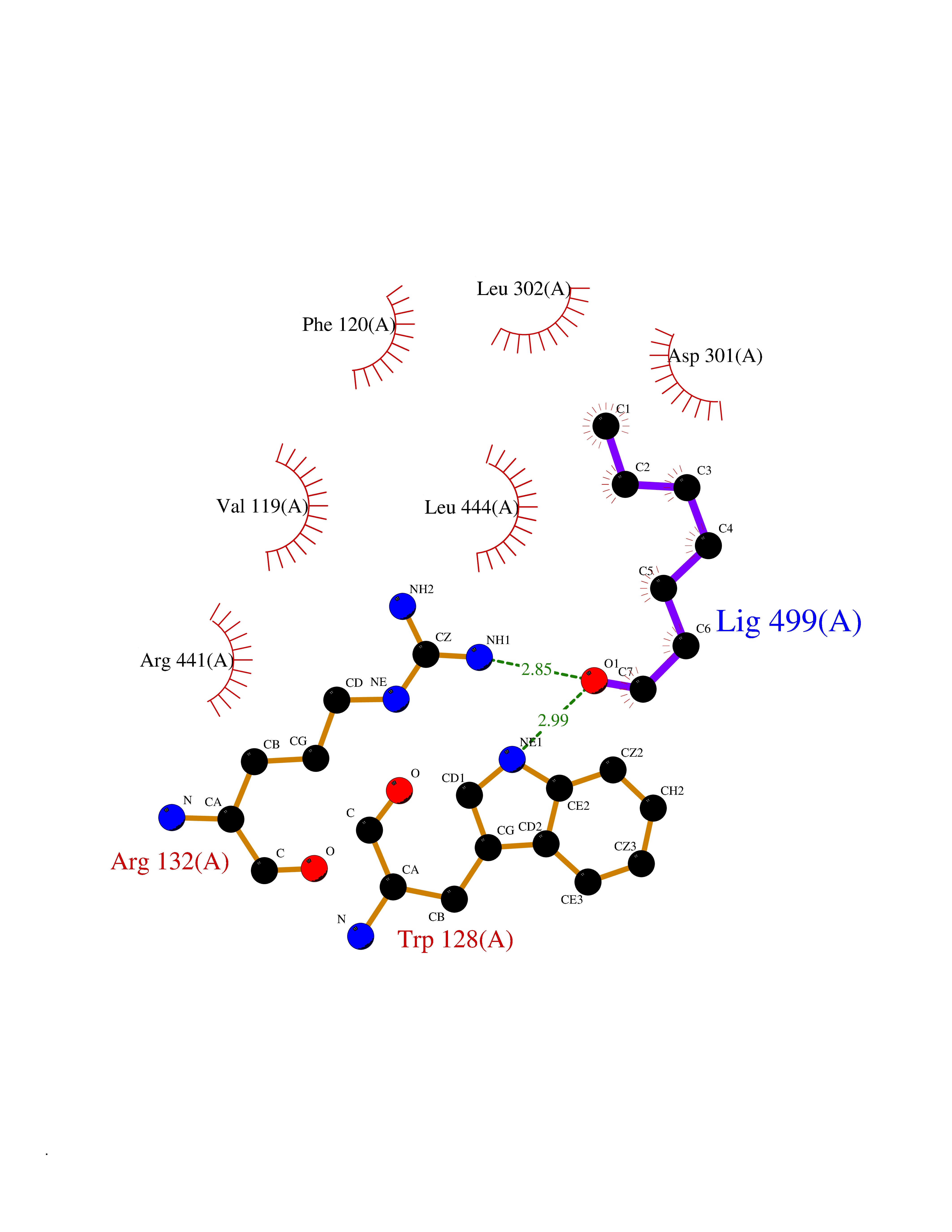 Ligplot Map 3D Binding mode Sequence GKLPPGPLPLPGLGNLLFQNTPYCFDQLRRRFGDVFSLQLAWTPVVVLNGLAAVREALVTHGEDTADRPPVPITQILGFGPRSQGVFLARYGPAWREQRRFSVSTLRNLGLGKKSLEQWVTEEAACLCAAFANHSGRPFRPNGLLDKAVSNVIASLTCGRRFEYDDPRFLRLLDLAQEGLKEESGFLREVLNAVPVLLHIPALAGKVLRFQKAFLTQLDELLTEHRMTWDPAQPPRDLTEAFLAEMEKAKGNPESSFNDENLRIVVADLFSAGMVTTSTTLAWGLLLMILHPDVQRRVQQEIDDVIGQVRRPEMGDQAHMPYTTAVIHEVQRFGDIVPLGVTHMTSRDIEVQGFRIPKGTTLITNLSSVLKDEAVWEKPFRFHPEHFLDAQGHFVKPEAFLPFSAGRRACLGEPLARMELFLFFTSLLQHFSFSVPTGQPRPSHHGVFAFLVSPSPYELCAVPR Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Ephrin type-B receptor 3 (EPHB3) | 5L6O | 4.96 | |
Target general information Gen name EPHB3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hEK2; Tyrosine-protein kinase TYRO6; TYRO6; Embryonic kinase 2; ETK2; EPH-like tyrosine kinase 2; EPH-like kinase 2; EK2 Protein family Protein kinase superfamily, Tyr protein kinase family, Ephrin receptor subfamily Biochemical class Kinase Function The signaling pathway downstream of the receptor is referred to as forward signaling while the signaling pathway downstream of the ephrin ligand is referred to as reverse signaling. Generally has an overlapping and redundant function with EPHB2. Like EPHB2, functions in axon guidance during development regulating for instance the neurons forming the corpus callosum and the anterior commissure, 2 major interhemispheric connections between the temporal lobes of the cerebral cortex. In addition to its role in axon guidance plays also an important redundant role with other ephrin-B receptors in development and maturation of dendritic spines and the formation of excitatory synapses. Controls other aspects of development through regulation of cell migration and positioning. This includes angiogenesis, palate development and thymic epithelium development for instance. Forward and reverse signaling through the EFNB2/EPHB3 complex also regulate migration and adhesion of cells that tubularize the urethra and septate the cloaca. Finally, plays an important role in intestinal epithelium differentiation segregating progenitor from differentiated cells in the crypt. Receptor tyrosine kinase which binds promiscuously transmembrane ephrin-B family ligands residing on adjacent cells, leading to contact-dependent bidirectional signaling into neighboring cells. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) NA Interacts with P37235; O75031 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Angiogenesis; ATP-binding; Cell membrane; Cell projection; Developmental protein; Disulfide bond; Glycoprotein; Kinase; Membrane; Neurogenesis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30412.9 Length 267 Aromaticity 0.09 Instability index 37.42 Isoelectric point 7.74 Charge (pH=7) 1 2D Binding mode Binding energy (Kcal/mol) -6.77 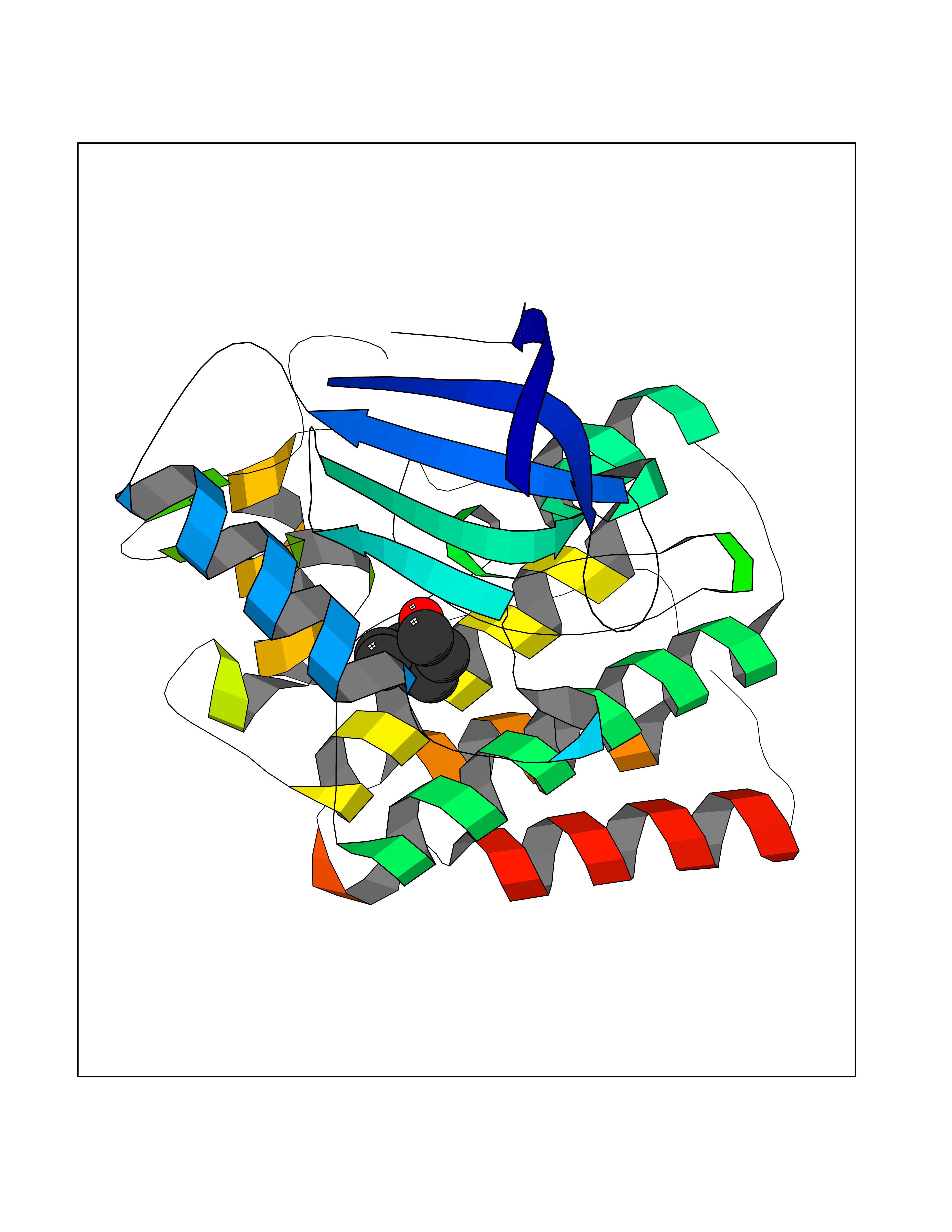 Molscript Map 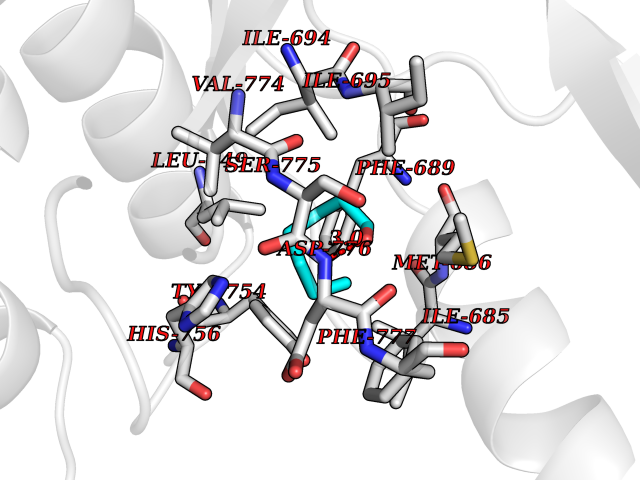 Pymol Map 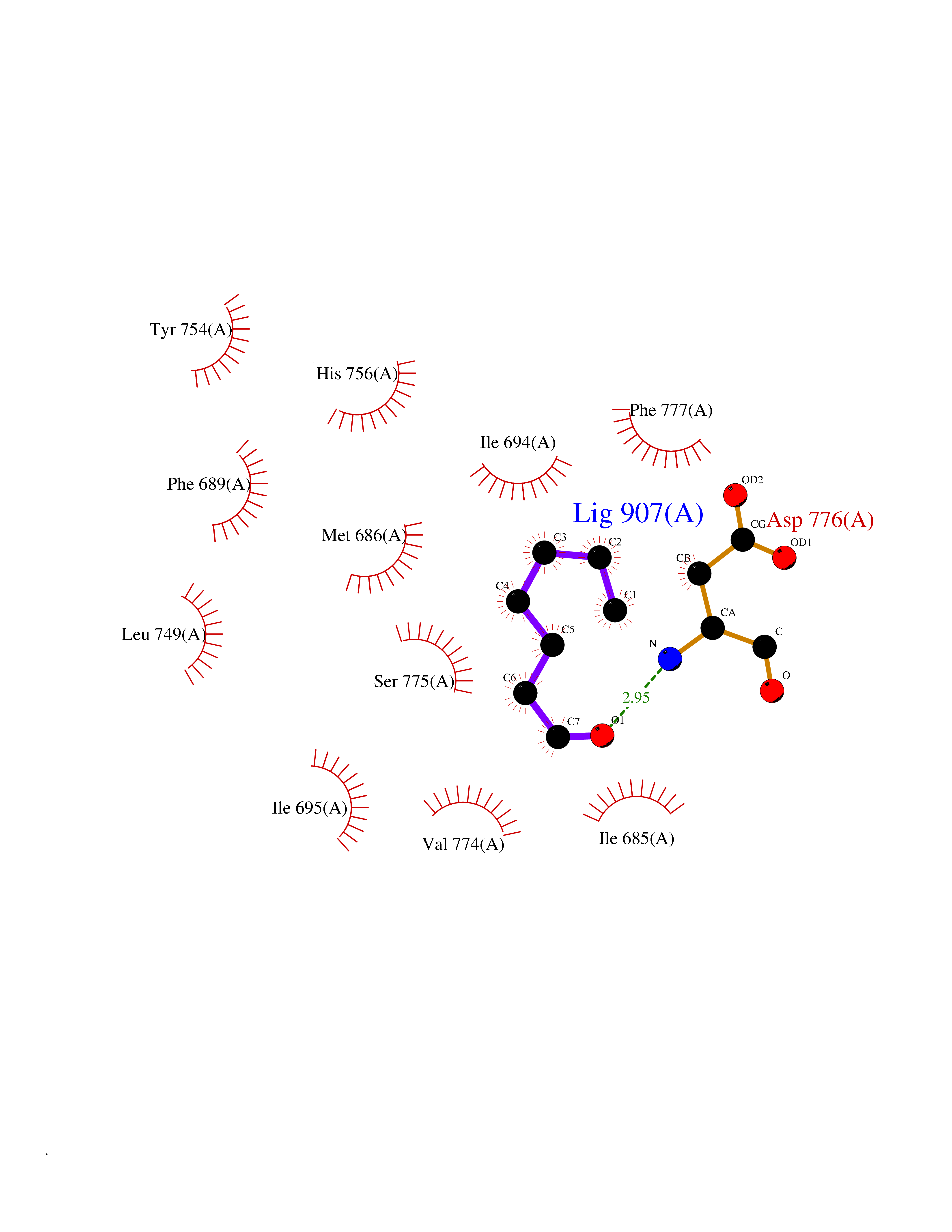 Ligplot Map 3D Binding mode Sequence CVKIEEVIGAGEVCRGRLKQPGRREVFVAIKTLKVGYTERQRRDFLSEASIMGQFDHPNIIRLEGVVTKSRPVMILTEFMENCALDSFLRLNDGQFTVIQLVGMLRGIAAGMKYLSEMNYVHRDLAARNILVNSNLVCKVSDFGLEDDPSDPTYTSSLGGKIPIRWTAPEAIAYRKFTSASDVWSYGIVMWEVMSYGERPYWDMSNQDVINAVEQDYRLPPPMDCPTALHQLMLDCWVRDRNLRPKFSQIVNTLDKLIRNPASLKVI Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Hyperpolarization cyclic nucleotide-gated channel 2 (HCN2) | 3U10 | 4.96 | |
Target general information Gen name HCN2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 2; Brain cyclic nucleotide-gated channel 2; BCNG2; BCNG-2 Protein family Potassium channel HCN family Biochemical class Voltage-gated ion channel Function Contributes to the native pacemaker currents in heart (If) and in neurons (Ih). Can also transport ammonium in the distal nephron. Produces a large instantaneous current. Modulated by intracellular chloride ions and pH; acidic pH shifts the activation to more negative voltages. Hyperpolarization-activated ion channel exhibiting weak selectivity for potassium over sodium ions. Related diseases Epilepsy, idiopathic generalized 17 (EIG17) [MIM:602477]: A form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. Both autosomal dominant and autosomal recessive EIG17 inheritance have been reported. {ECO:0000269|PubMed:22131395, ECO:0000269|PubMed:29064616}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Febrile seizures, familial, 2 (FEB2) [MIM:602477]: Seizures associated with febrile episodes in childhood without any evidence of intracranial infection or defined pathologic or traumatic cause. It is a common condition, affecting 2-5% of children aged 3 months to 5 years. The majority are simple febrile seizures (generally defined as generalized onset, single seizures with a duration of less than 30 minutes). Complex febrile seizures are characterized by focal onset, duration greater than 30 minutes, and/or more than one seizure in a 24 hour period. The likelihood of developing epilepsy following simple febrile seizures is low. Complex febrile seizures are associated with a moderately increased incidence of epilepsy. FEB2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:24324597}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02527; DB02315; DB09083 Interacts with Q9UL51; Q4ACU6-1 EC number NA Uniprot keywords 3D-structure; Ammonia transport; cAMP; cAMP-binding; Cell membrane; Disease variant; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Methylation; Nucleotide-binding; Palmitate; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 23672.9 Length 202 Aromaticity 0.12 Instability index 38.05 Isoelectric point 8.85 Charge (pH=7) 4.11 2D Binding mode Binding energy (Kcal/mol) -6.77 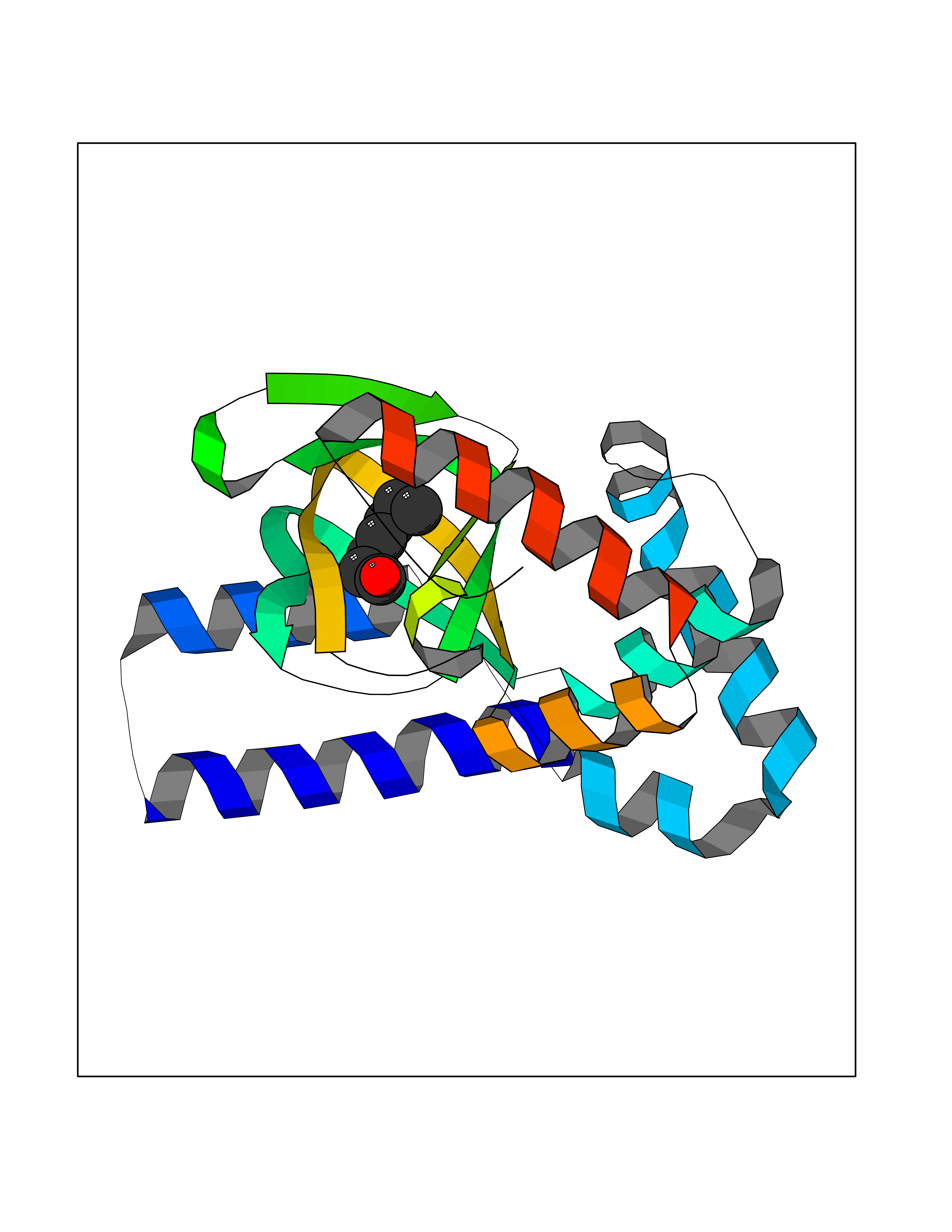 Molscript Map 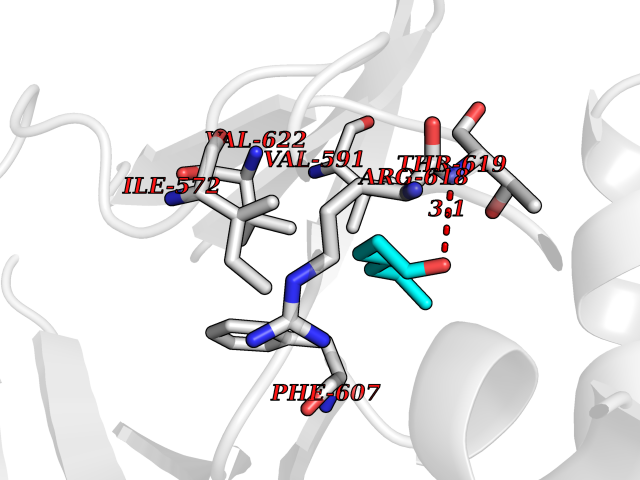 Pymol Map 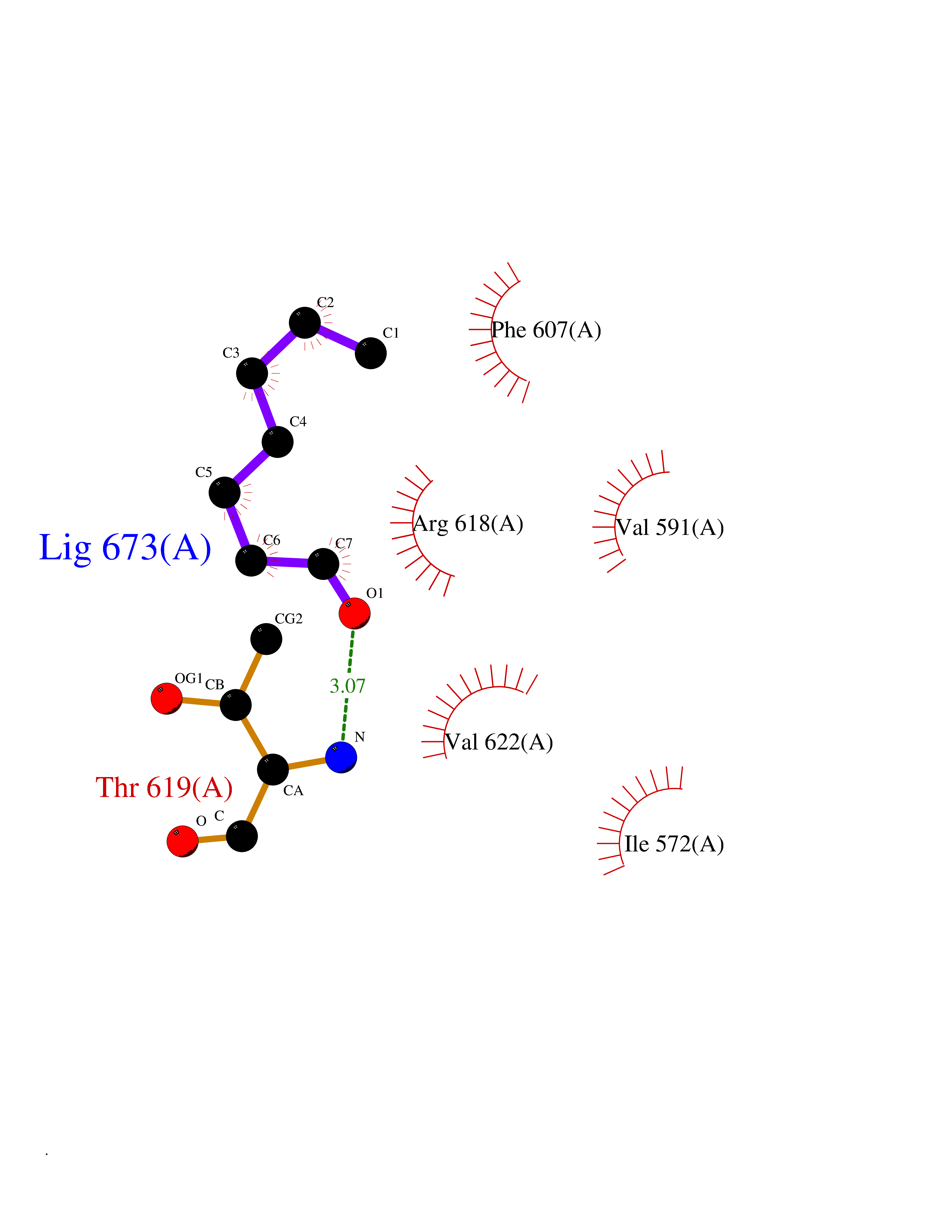 Ligplot Map 3D Binding mode Sequence DSSRRQYQEKYKQVEQYMSFHKLPADFRQKIHDYYEHRYQGKMFDEDSILGELNGPLREEIVNFNCRKLVASMPLFANADPNFVTAMLTKLKFEVFQPGDYIIREGTIGKKMYFIQHGVVSVLTKGNKEMKLSDGSYFGEICLLTRGRRTASVRADTYCRLYSLSVDNFNEVLEEYPMMRRAFETVAIDRLDRIGKKNSILL Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Peroxiredoxin-4 (PRDX4) | 4RQX | 4.96 | |
Target general information Gen name PRDX4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Thioredoxindependent peroxide reductase A0372; Thioredoxin peroxidase AO372; PrxIV; Peroxiredoxin4; Peroxiredoxin IV; PRDX4; Antioxidant enzyme AOE372; AOE372 Protein family Peroxiredoxin family, AhpC/Prx1 subfamily Biochemical class Peroxidases Function Probably involved in redox regulation of the cell. Regulates the activation of NF-kappa-B in the cytosol by a modulation of I-kappa-B-alpha phosphorylation. Related diseases May be involved in T-cell exhaustion associated with chronic viral infections such as with human immunodeficiency virus (HIV) and hepatitic C virus (HCV). {ECO:0000269|PubMed:19001139, ECO:0000269|PubMed:19587053}.; DISEASE: T-cell lymphoma, subcutaneous panniculitis-like (SPTCL) [MIM:618398]: An uncommon form of T-cell non-Hodgkin lymphoma, in which cytotoxic CD8+ T-cells infiltrate subcutaneous adipose tissue, and rimming adipocytes in a lace-like pattern. Affected individuals typically present with multiple subcutaneous nodules, systemic B-cell symptoms, and, in a subset of cases, autoimmune disorders, most commonly systemic lupus erythematosus. A subset of patients develop hemophagocytic lymphohistiocytosis. SPTCL transmission pattern is consistent with autosomal recessive inheritance with incomplete penetrance. {ECO:0000269|PubMed:30374066, ECO:0000269|PubMed:30792187, ECO:0000269|Ref.2}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; P18428; P07237; P30101; Q15084; Q06830; P21731; Q8NBS9 EC number EC 1.11.1.15 Uniprot keywords 3D-structure; Antioxidant; Cytoplasm; Direct protein sequencing; Disulfide bond; Endoplasmic reticulum; Oxidoreductase; Peroxidase; Proteomics identification; Redox-active center; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 18846.2 Length 166 Aromaticity 0.12 Instability index 30.43 Isoelectric point 5.54 Charge (pH=7) -3.89 2D Binding mode Binding energy (Kcal/mol) -6.77 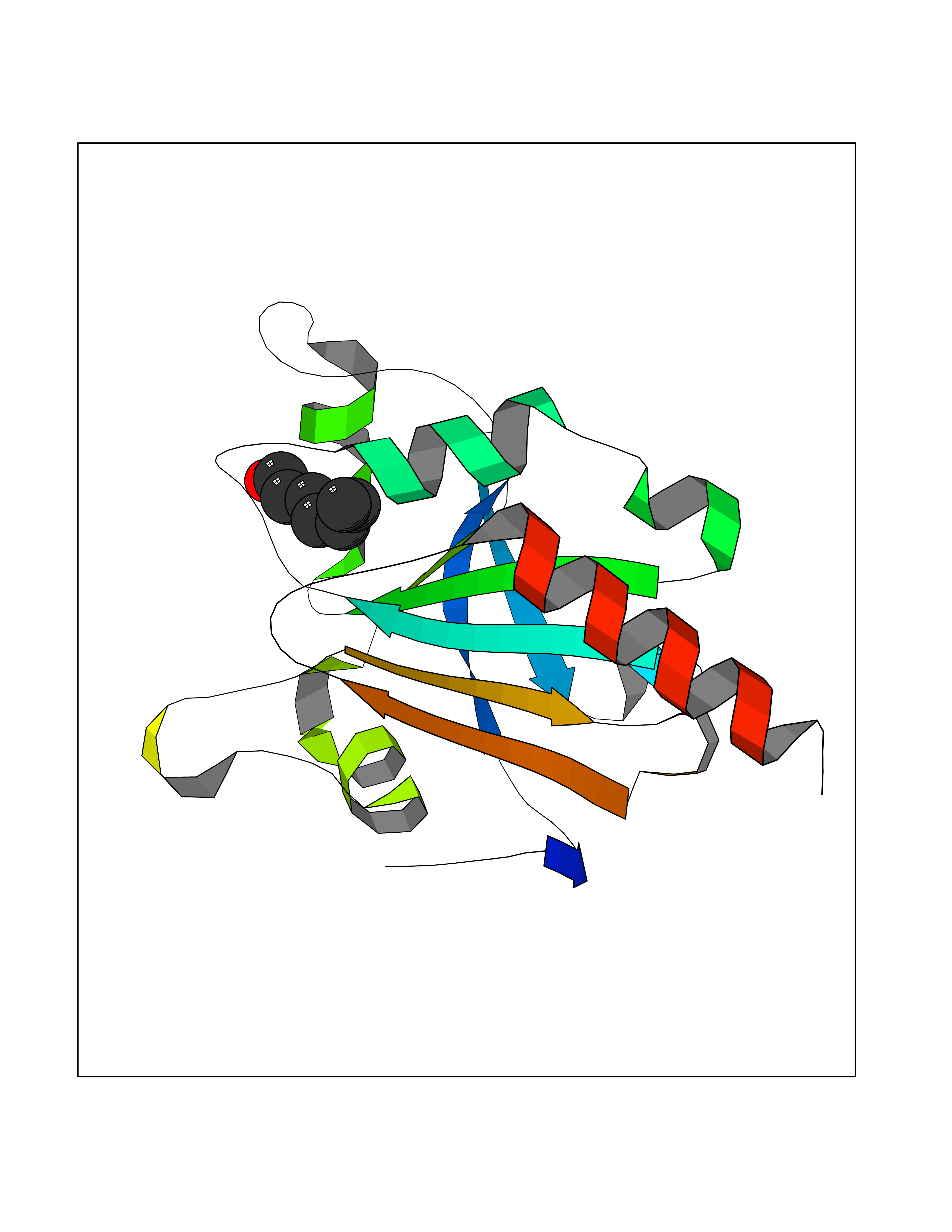 Molscript Map 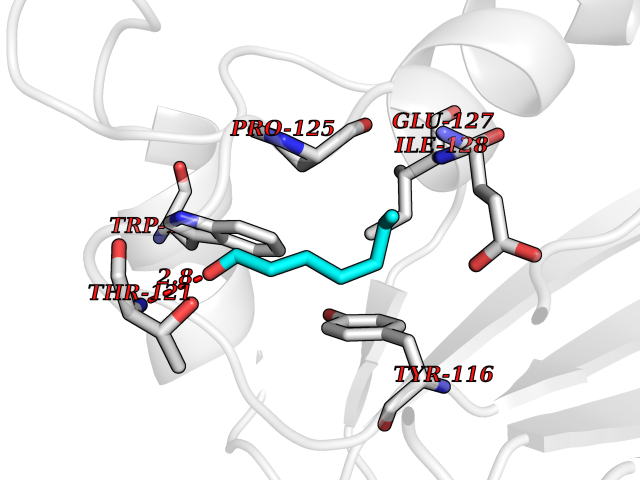 Pymol Map 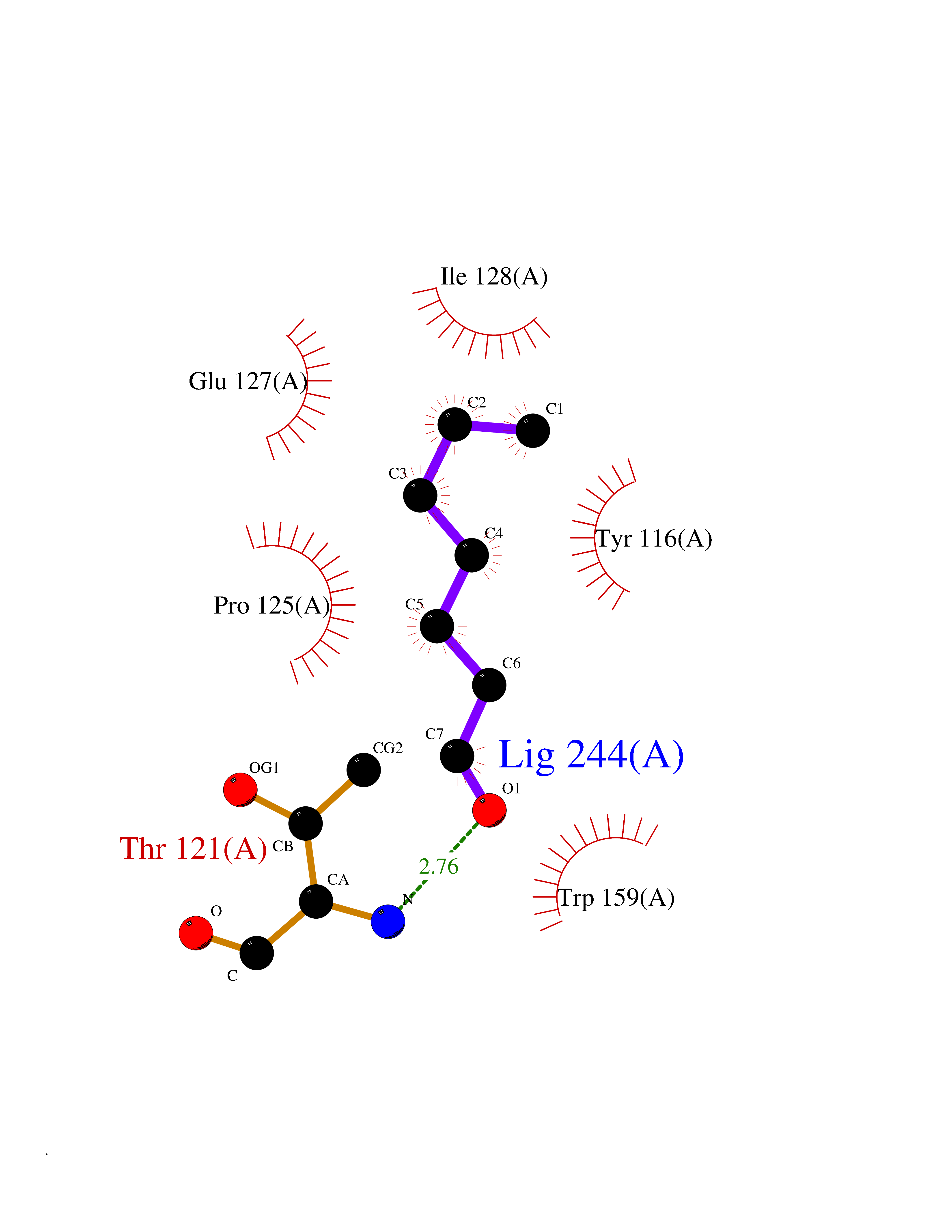 Ligplot Map 3D Binding mode Sequence GTAKISKPAPYWEGTAVIDGEFKELKLTDYRGKYLVFFFYPLDFTFVCPTEIIAFGDRLEEFRSINTEVVACSVDSQFTHLAWINTPRRQGGLGPIRIPLLSDLTHQISKDYGVYLEDSGHTLRGLFIIDDKGILRQITLNDLPVGRSVDETLRLVQAFQYTDKHG Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Histamine H1 receptor (H1R) | 7DFL | 4.96 | |
Target general information Gen name HRH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HH1R; H1R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function In peripheral tissues, the H1 subclass of histamine receptors mediates the contraction of smooth muscles, increase in capillary permeability due to contraction of terminal venules, and catecholamine release from adrenal medulla, as well as mediating neurotransmission in the central nervous system. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01615; DB09488; DB06766; DB01246; DB00321; DB00543; DB08799; DB01238; DB14185; DB06216; DB00637; DB00719; DB00972; DB00245; DB00767; DB04890; DB06698; DB11591; DB09128; DB01237; DB00835; DB00354; DB09016; DB00748; DB06016; DB00341; DB08936; DB08800; DB01114; DB00477; DB01239; DB00568; DB00215; DB00283; DB04837; DB00363; DB01176; DB00434; DB01151; DB00967; DB00405; DB09555; DB00985; DB08801; DB01075; DB01146; DB09167; DB01142; DB00366; DB01084; DB05492; DB00751; DB01175; DB06678; DB00950; DB04841; DB00502; DB05381; DB05079; DB00557; DB04946; DB00458; DB08802; DB00920; DB00555; DB01106; DB06282; DB00455; DB09195; DB00408; DB00934; DB00737; DB06691; DB01071; DB00902; DB01403; DB06148; DB00370; DB00540; DB05080; DB06229; DB00334; DB00768; DB01173; DB01267; DB00715; DB08922; DB01619; DB01620; DB06153; DB00433; DB00420; DB01069; DB00777; DB01224; DB00912; DB00734; DB11614; DB05345; DB00342; DB04905; DB11235; DB00797; DB00656; DB00726; DB00792; DB00427; DB09185; DB00246; DB01624 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32298.5 Length 275 Aromaticity 0.15 Instability index 36.59 Isoelectric point 9.54 Charge (pH=7) 16.01 2D Binding mode Binding energy (Kcal/mol) -6.77 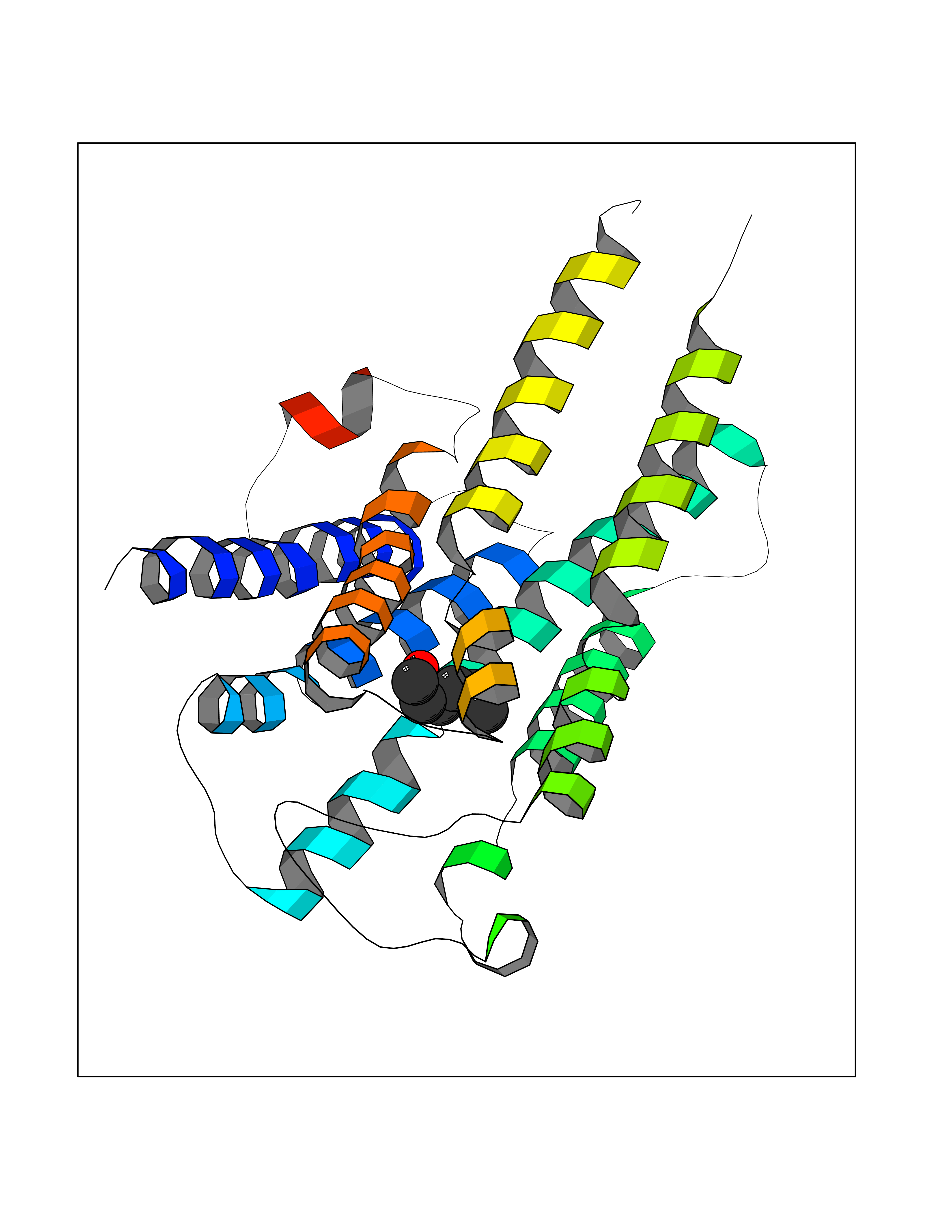 Molscript Map 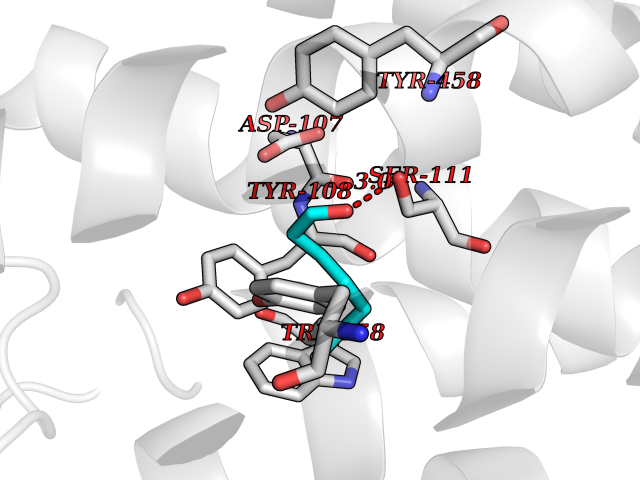 Pymol Map 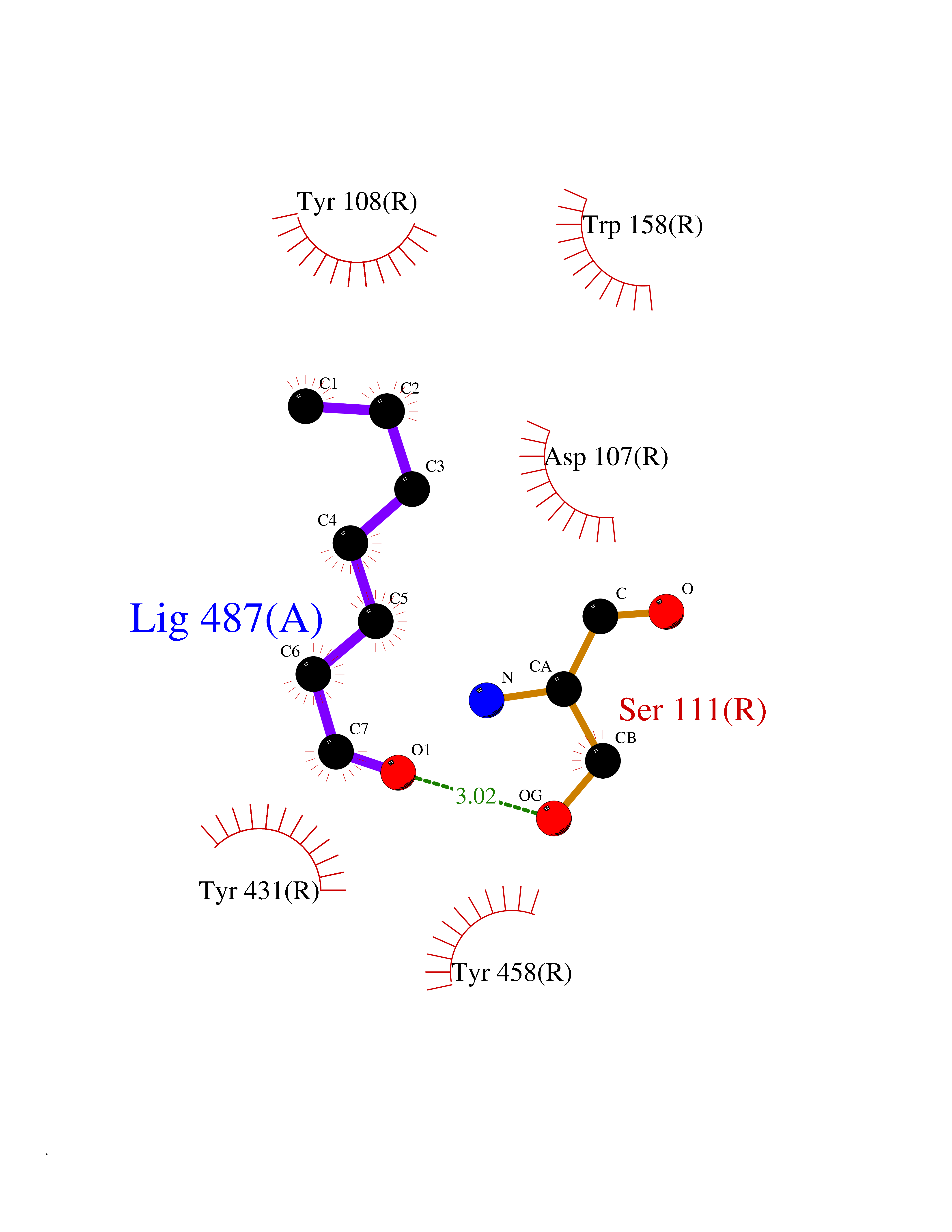 Ligplot Map 3D Binding mode Sequence MPLVVVLSTICLVTVGLNLLVLYAVRSERKLHTVGNLYIVSLSVADLIVGAVVMPMNILYLLMSKWSLGRPLCLFWLSMDYVASTASIFSVFILCIDRYRSVQQPLRYLKYRTKTRASATILGAWFLSFLWVIPILGWNHFMQQTSVRREDKCETDFYDVTWFKVMTAIINFYLPTLLMLWFYAKIYKAVRQHCLHMNRERKAAKQLGFIMAAFILCWIPYFIFFMVIAFCKNCCNEHLHMFTIWLGYINSTLNPLIYPLCNENFKKTFKRILHI Hydrogen bonds contact Hydrophobic contact | ||||