Job Results:
Ligand
Structure
Job ID
8e57bce38c3d7c3304510f6b58eb7b68
Job name
NA
Time
2026-02-27 11:59:36
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Serine--pyruvate aminotransferase | 5HHY | 4.77 | |
Target general information Gen name AGXT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AGT1;SPAT Protein family Class-V pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transferase Function Alanine-glyoxylate transaminase activity.Amino acid binding.Identical protein binding.Protein homodimerization activity.Protein self-association.Pyridoxal phosphate binding.Receptor binding.Serine-pyruvate transaminase activity.Transaminase activity. Related diseases Hyperoxaluria primary 1 (HP1) [MIM:259900]: An inborn error of glyoxylate metabolism characterized by increased excretion of oxalate and glycolate, and progressive tissue accumulation of insoluble calcium oxalate. Affected individuals are at risk for nephrolithiasis, nephrocalcinosis and early onset end-stage renal disease. {ECO:0000269|PubMed:10394939, ECO:0000269|PubMed:10453743, ECO:0000269|PubMed:10541294, ECO:0000269|PubMed:10862087, ECO:0000269|PubMed:10960483, ECO:0000269|PubMed:12559847, ECO:0000269|PubMed:12777626, ECO:0000269|PubMed:1301173, ECO:0000269|PubMed:1349575, ECO:0000269|PubMed:15253729, ECO:0000269|PubMed:15849466, ECO:0000269|PubMed:15961946, ECO:0000269|PubMed:15963748, ECO:0000269|PubMed:16971151, ECO:0000269|PubMed:1703535, ECO:0000269|PubMed:17495019, ECO:0000269|PubMed:2039493, ECO:0000269|PubMed:23229545, ECO:0000269|PubMed:24055001, ECO:0000269|PubMed:24934730, ECO:0000269|PubMed:26149463, ECO:0000269|PubMed:8101040, ECO:0000269|PubMed:9192270, ECO:0000269|PubMed:9604803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08060; DB00160; DB02079; DB00145; DB04083; DB00114; DB00133 Interacts with Q5BKX5-3; P21549; P0C7T5; Q9NR55; Q9H5F2; Q9UKJ5; A8MQ03; O43281-2; A1KXE4-2; Q5TD97; P49356; P53539; P49639; Q15323; O76011; O76014; Q6A162; Q07627; P60328; Q9BYR8; Q3LI67; Q8IUC2; Q9BYQ4; O60711; Q99750; Q5VZ52; P0DPK4; O43482; P50542-1; O15496; Q6ZR37; Q9NQX0; Q8HWS3; Q5W111-2; Q8WVR3; Q6DKK2 EC number 2.6.1.44; 2.6.1.51 Uniprot keywords 3D-structure; Acetylation; Aminotransferase; Disease variant; Peroxisome; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84537 Length 771 Aromaticity 0.07 Instability index 36.81 Isoelectric point 8.42 Charge (pH=7) 6.81 2D Binding mode Binding energy (Kcal/mol) -6.51  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKLLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKK Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Monoamine oxidase type B (MAO-B) | 2V5Z | 4.77 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -6.5 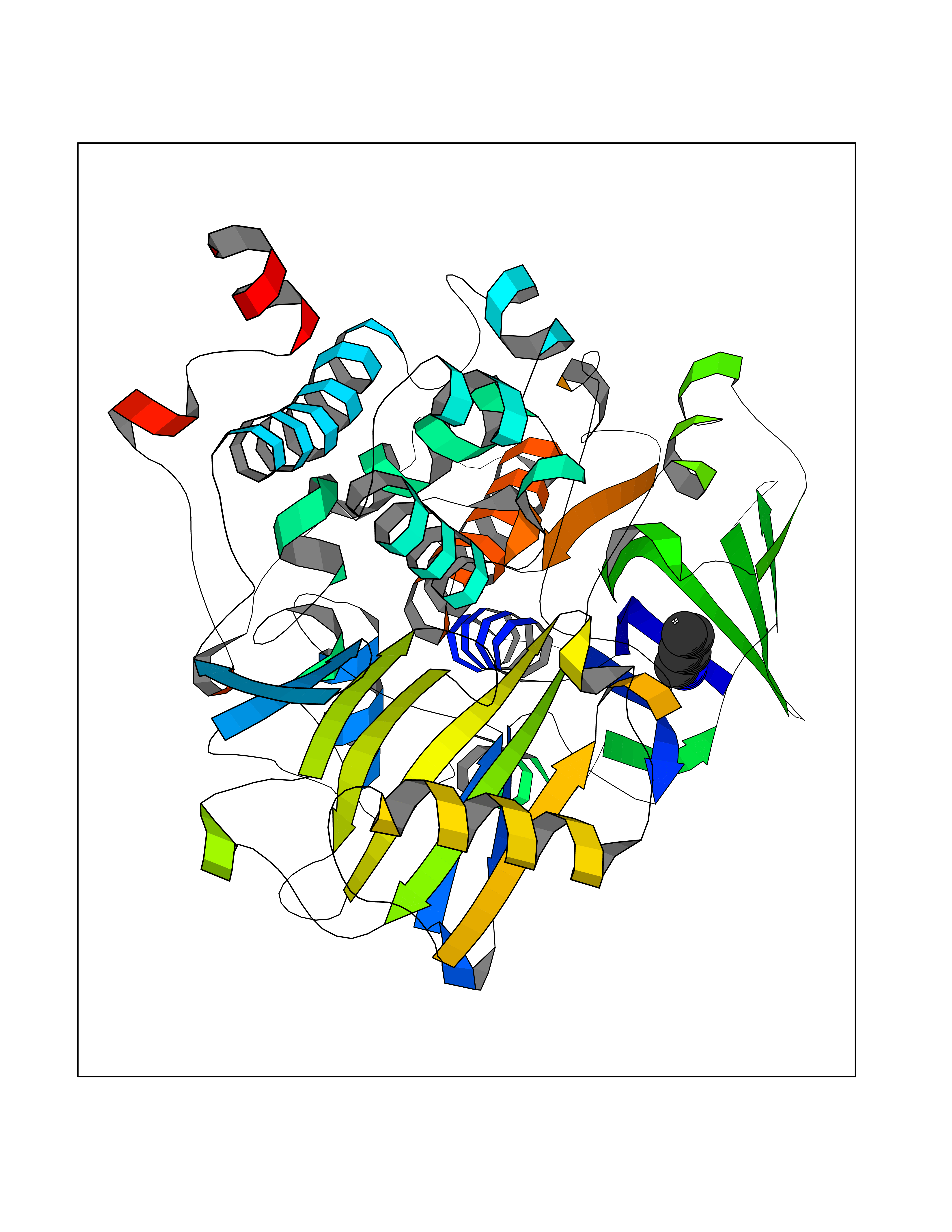 Molscript Map 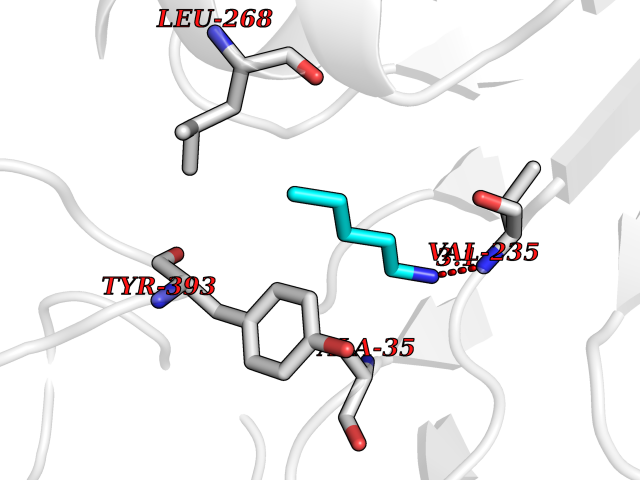 Pymol Map 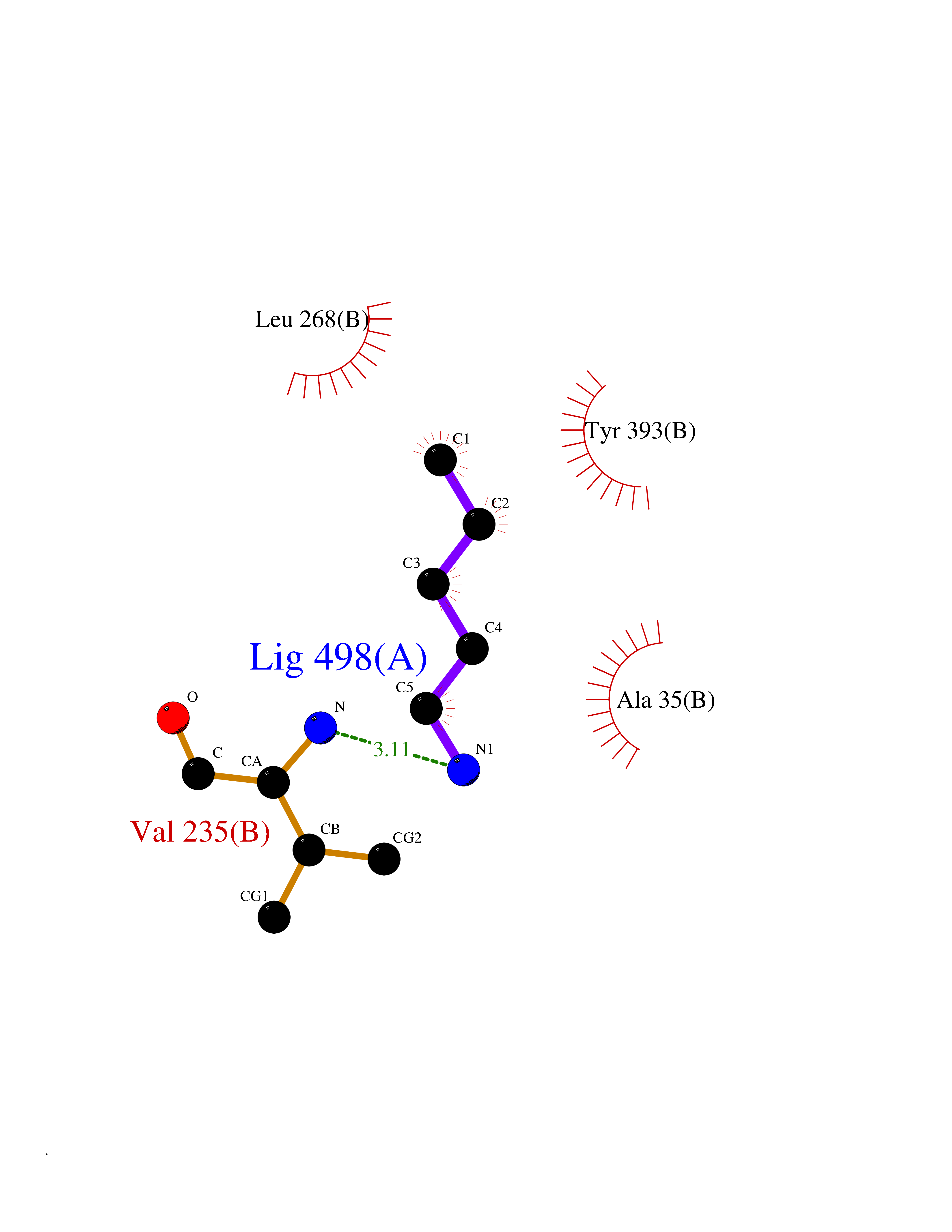 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Serine hydroxymethyltransferase, mitochondrial | 3OU5 | 4.77 | |
Target general information Gen name SHMT2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SHMT family Biochemical class Transferase Function Amino acid binding.Chromatin binding.Glycine hydroxymethyltransferase activity.Identical protein binding.L-allo-threonine aldolase activity.Pyridoxal phosphate binding. Related diseases Neurodevelopmental disorder with cardiomyopathy, spasticity, and brain abnormalities (NEDCASB) [MIM:619121]: An autosomal recessive neurodevelopmental disorder characterized by global developmental delay, moderate to severe intellectual disability, spastic paraparesis, ataxia, and/or peripheral neuropathy. Patients also exhibit dysmorphic features and congenital microcephaly. Most affected individuals develop progressive hypertrophic cardiomyopathy in childhood or have cardiac developmental anomalies. Brain imaging shows corpus callosum abnormalities in all patients, and perisylvian polymicrogyria-like pattern in some individuals. {ECO:0000269|PubMed:33015733}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11638; DB00145; DB00114; DB00116 Interacts with Q15041; P46736; Q9Y376; Q96DZ9; Q96DZ9-2; Q8IZU9; Q969L2; P34897 EC number 2.1.2.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cardiomyopathy; Cytoplasm; Disease variant; Intellectual disability; Membrane; Mitochondrion; Mitochondrion inner membrane; Mitochondrion nucleoid; Nucleus; One-carbon metabolism; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 46797.9 Length 422 Aromaticity 0.07 Instability index 38.89 Isoelectric point 7.24 Charge (pH=7) 0.55 2D Binding mode Binding energy (Kcal/mol) -6.5 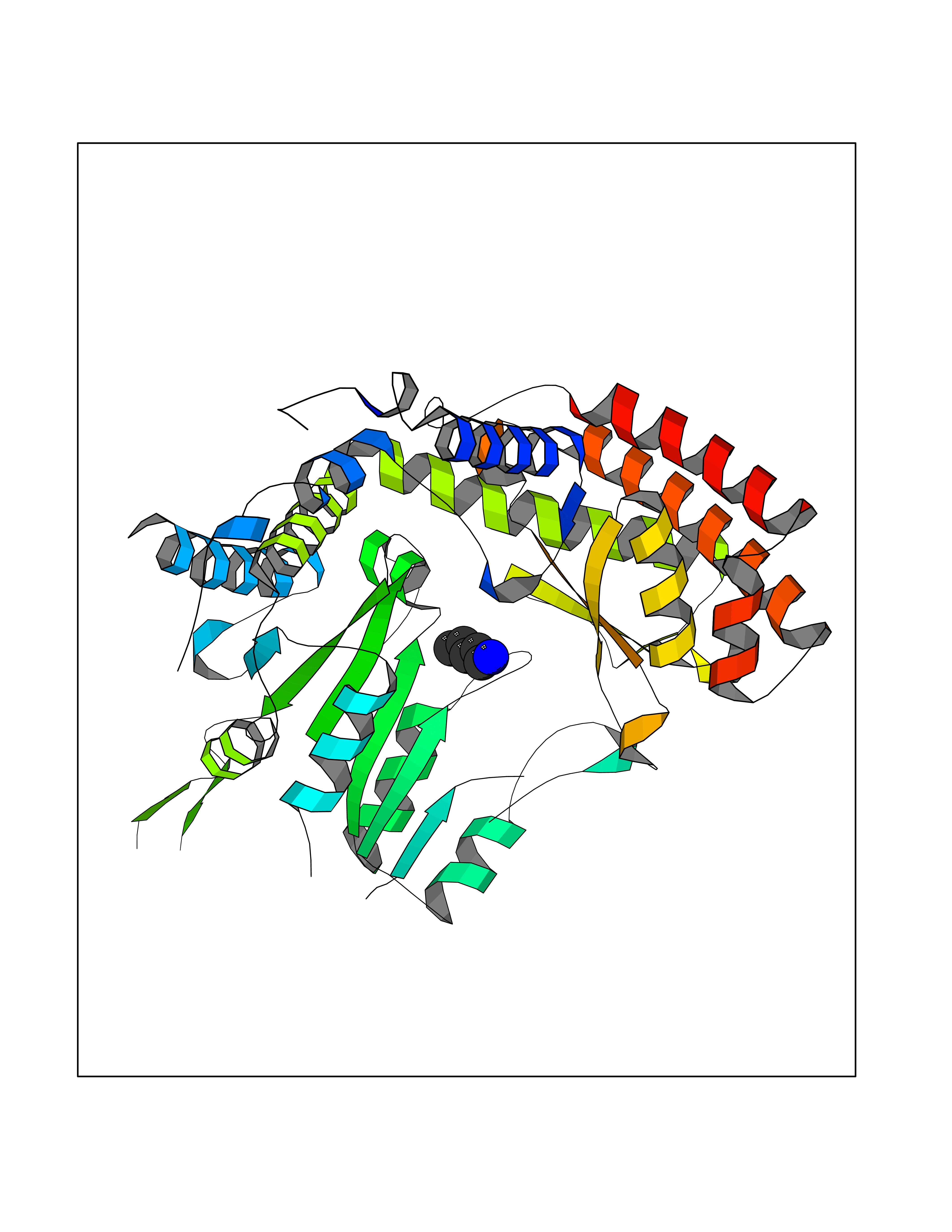 Molscript Map 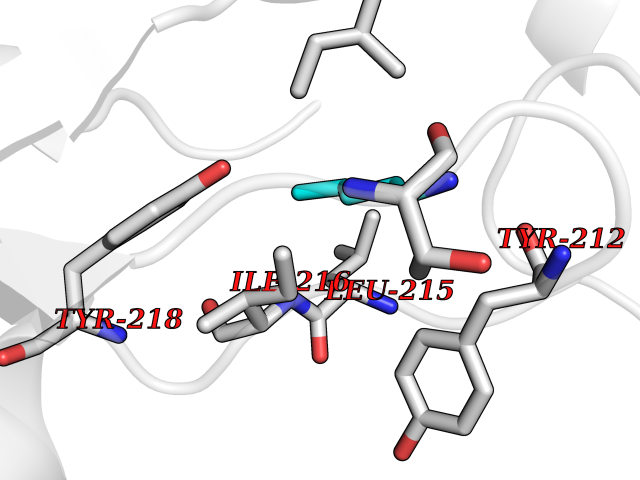 Pymol Map 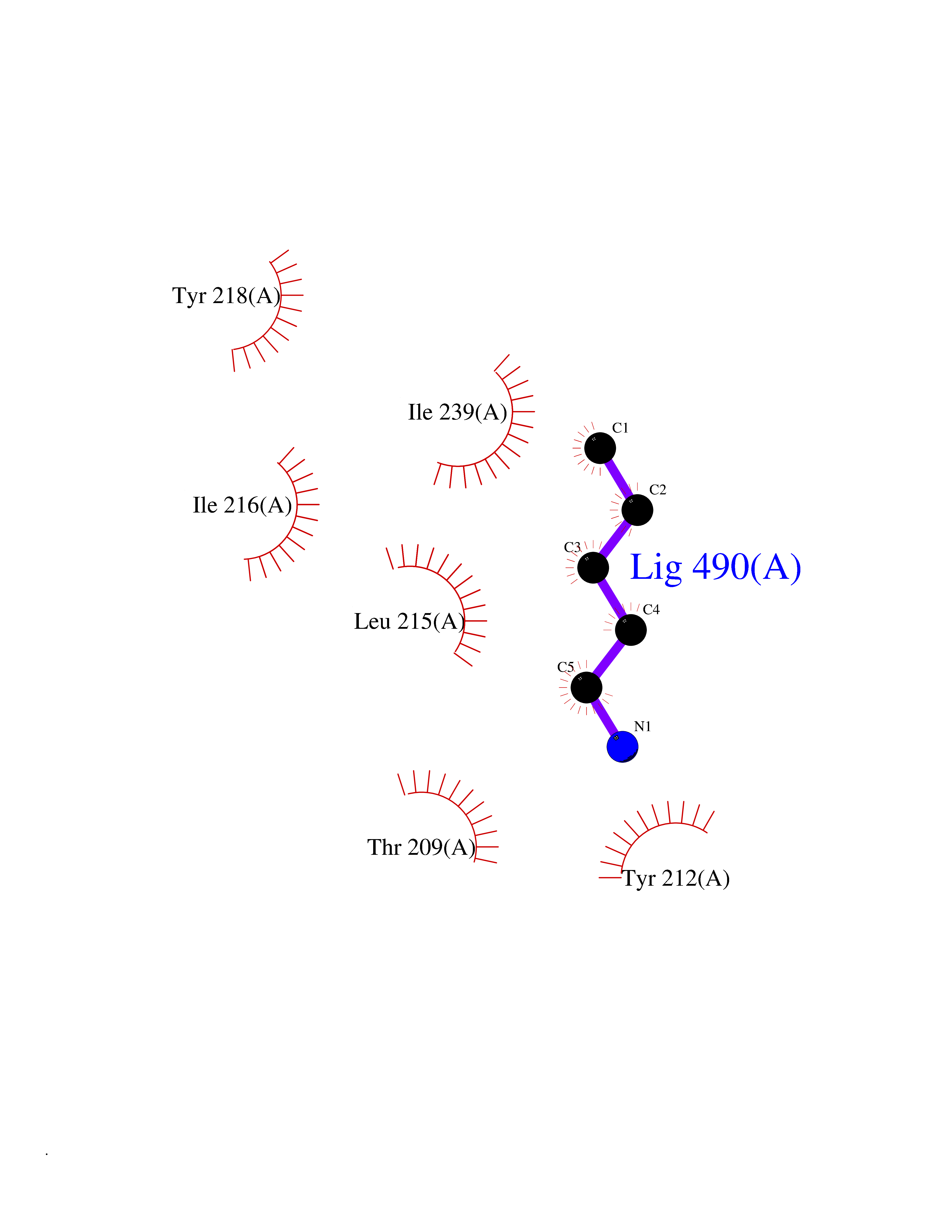 Ligplot Map 3D Binding mode Sequence GWTGQESLSDSDPEMWELLQREKDRQCRGLELIASENFCSRAALEALGSCLNNKYSEGEVVDEIELLCQRRALEAFDLDPAQWGVNVQPYSGSPANLAVYTALLQHDRIMGLDPYKLNPKTGLIDYNQLALTARLFRPRLIIAGTSAYARLIDYARMREVCDEVKAHLLADMAHISGLVAAKVIPSPFKHADIVTTTTHKTLRGARSGLIFYRKGVKAVDPGREIPYTFEDRINFAVFPSLQGGPHNHAIAAVAVALKQACTPMFREYSLQVLKNARAMADALLERGYSLVSGGTDNHLVLVDLRPKGLDGARAERVLELVSITANKNTCPGDRSAITPGGLRLGAPALTSRQFREDDFRRVVDFIDEGVNIGLEVKSKTAKLQDFKSFLLKDSETSQRLANLRQRVEQFARAFPMPGFDEH Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Carbonic anhydrase 13 | 4KNN | 4.77 | |
Target general information Gen name CA13 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Alpha-carbonic anhydrase family Biochemical class Lyase / lyase inhibitor Function Carbonate dehydratase activity.Zinc ion binding. Related diseases Long QT syndrome 10 (LQT10) [MIM:611819]: A heart disorder characterized by a prolonged QT interval on the ECG and polymorphic ventricular arrhythmias. They cause syncope and sudden death in response to exercise or emotional stress, and can present with a sentinel event of sudden cardiac death in infancy. {ECO:0000269|PubMed:17592081}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Atrial fibrillation, familial, 17 (ATFB17) [MIM:611819]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:23604097}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00562; DB00606; DB07115; DB00909 Interacts with Q8N4Y2-3; Q6UUV7 EC number 4.2.1.1 Uniprot keywords 3D-structure; Lyase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 28955.1 Length 258 Aromaticity 0.1 Instability index 40.81 Isoelectric point 6.3 Charge (pH=7) -3.47 2D Binding mode Binding energy (Kcal/mol) -6.5 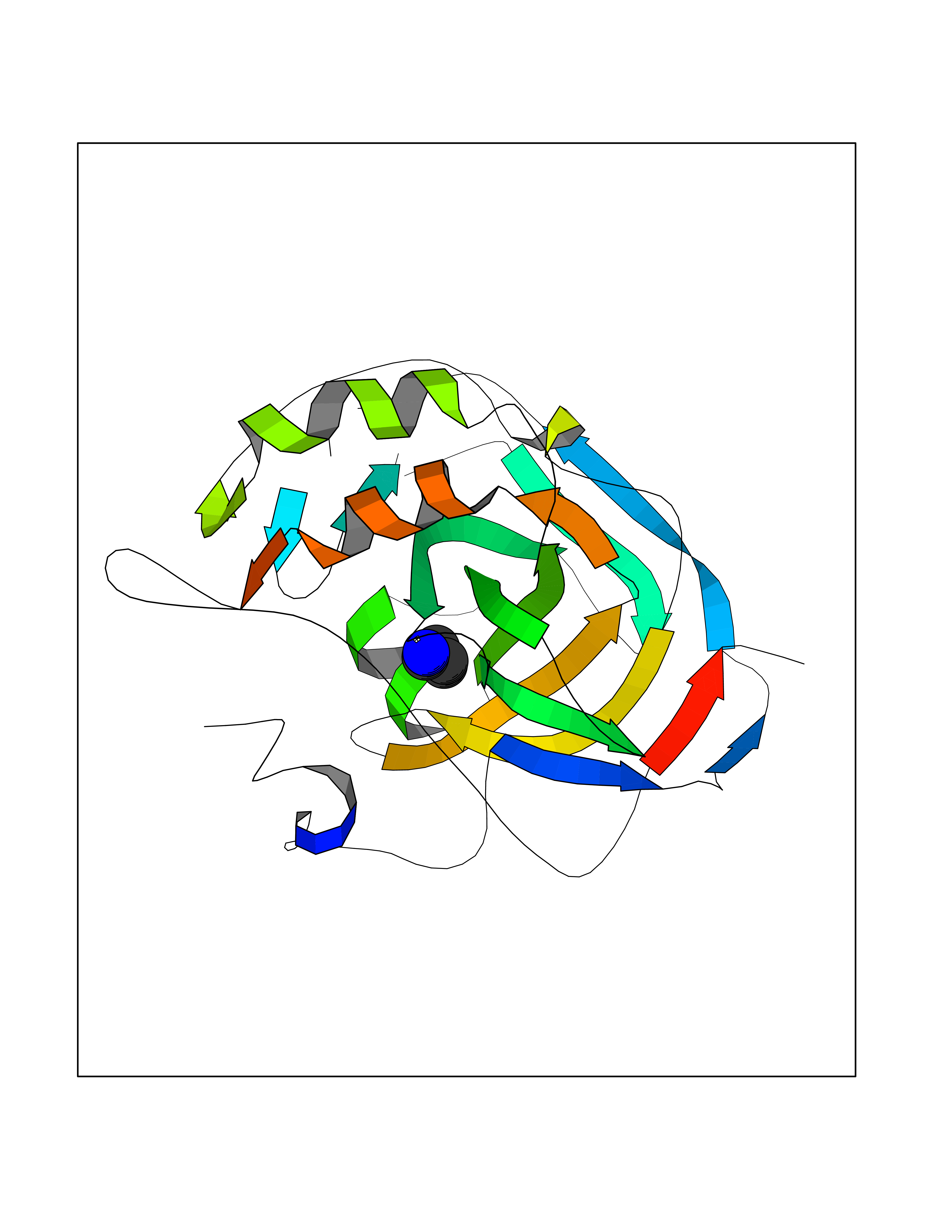 Molscript Map  Pymol Map 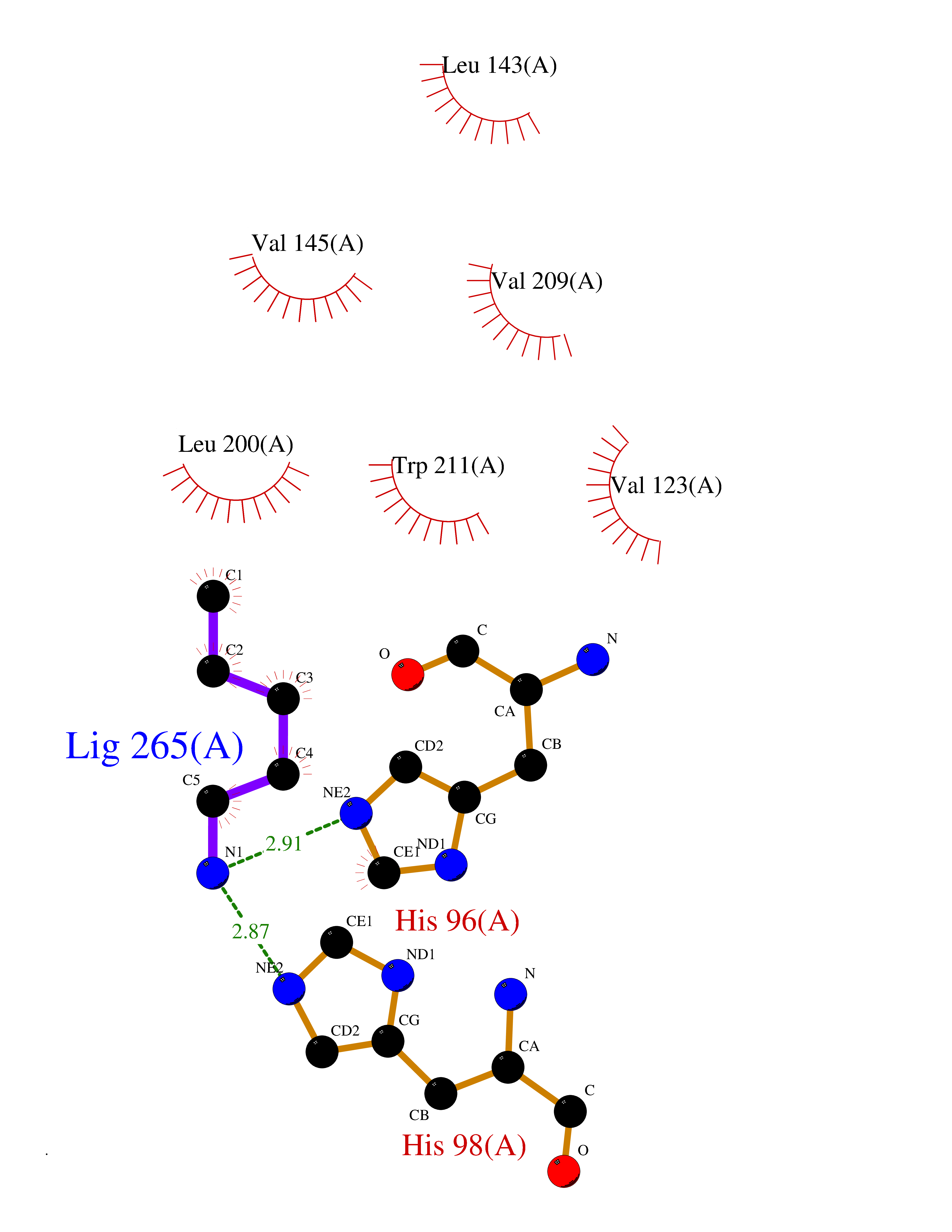 Ligplot Map 3D Binding mode Sequence SWGYREHNGPIHWKEFFPIADGDQQSPIEIKTKEVKYDSSLRPLSIKYDPSSAKIISNSGHSFNVDFDDTENKSVLRGGPLTGSYRLRQVHLHWGSADDHGSEHIVDGVSYAAELHVVHWNSDKYPSFVEAAHEPDGLAVLGVFLQIGEPNSQLQKITDTLDSIKEKGKQTRFTNFDLLSLLPPSWDYWTYPGSLTVPPLLESVTWIVLKQPINISSQQLAKFRSLLCTAEGEAAAFLVSNHRPPQPLKGRKVRASFH Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 4.77 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -6.5  Molscript Map  Pymol Map 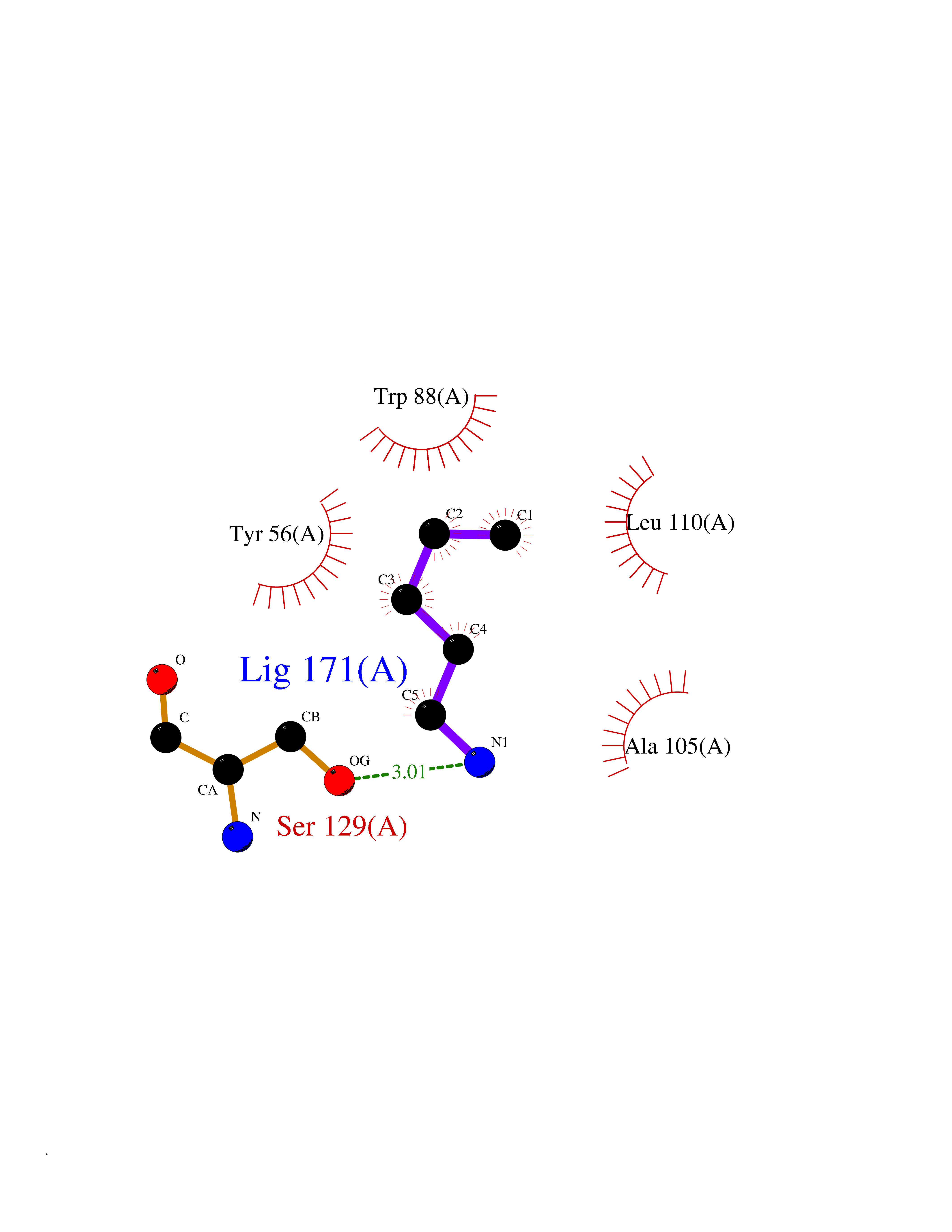 Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Neuronal acetylcholine receptor beta-4 (CHRNB4) | 6PV7 | 4.77 | |
Target general information Gen name CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNB4; Beta-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-4/CHRNB4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. {ECO:0000269|PubMed:20054297, ECO:0000269|PubMed:23622243, ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}. The disease may be caused by variants affecting the gene represented in this entry. Defects of SETD2 are associated with loss of DNA methylation at non-promoter regions (PubMed:23792563). SETD2 defects lead to aberrant and reduced nucleosome compaction and chromatin association of key replication proteins, such as MCM7 and DNA polymerase delta, leading to hinder replication fork progression and prevent loading of RAD51 homologous recombination repair factor at DNA breaks (PubMed:25728682). {ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}.; DISEASE: Luscan-Lumish syndrome (LLS) [MIM:616831]: An autosomal dominant syndrome with a variable phenotype. Clinical features include macrocephaly, distinctive facial appearance, postnatal overgrowth, various degrees of learning difficulties, autism spectrum disorder, and intellectual disability. {ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24852293, ECO:0000269|PubMed:26084711, ECO:0000269|PubMed:27317772}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute lymphoblastic (ALL) [MIM:613065]: A subtype of acute leukemia, a cancer of the white blood cells. ALL is a malignant disease of bone marrow and the most common malignancy diagnosed in children. The malignant cells are lymphoid precursor cells (lymphoblasts) that are arrested in an early stage of development. The lymphoblasts replace the normal marrow elements, resulting in a marked decrease in the production of normal blood cells. Consequently, anemia, thrombocytopenia, and neutropenia occur to varying degrees. The lymphoblasts also proliferate in organs other than the marrow, particularly the liver, spleen, and lymphnodes. {ECO:0000269|PubMed:24509477, ECO:0000269|PubMed:24662245}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16314571, ECO:0000269|PubMed:24509477}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Intellectual developmental disorder, autosomal dominant 70 (MRD70) [MIM:620157]: An autosomal dominant disorder characterized by mild global developmental delay, moderately impaired intellectual disability with speech difficulties, and behavioral abnormalities. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rabin-Pappas syndrome (RAPAS) [MIM:620155]: An autosomal dominant neurodevelopmental disorder characterized by severely impaired global development, intellectual disability, microcephaly, facial dysmorphism, and variable congenital anomalies affecting the skeletal, genitourinary, cardiac, and other organ systems. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00237; DB00565; DB00514; DB07720; DB00898; DB00472; DB01227; DB00184; DB01090; DB00202 Interacts with Q6FHY5 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -6.5  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Non-heme chloroperoxidase | 1A8U | 4.76 | |
Target general information Gen name cpo Organism Kitasatospora aureofaciens (Streptomyces aureofaciens) Uniprot ID TTD ID NA Synonyms cpoT Protein family AB hydrolase superfamily, Bacterial non-heme haloperoxidase / perhydrolase family Biochemical class Haloperoxidase Function Chloride peroxidase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB03793 Interacts with NA EC number 1.11.1.- Uniprot keywords 3D-structure; Chloride; Oxidoreductase; Peroxidase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60428.4 Length 554 Aromaticity 0.13 Instability index 31.26 Isoelectric point 4.65 Charge (pH=7) -32.72 2D Binding mode Binding energy (Kcal/mol) -6.49  Molscript Map 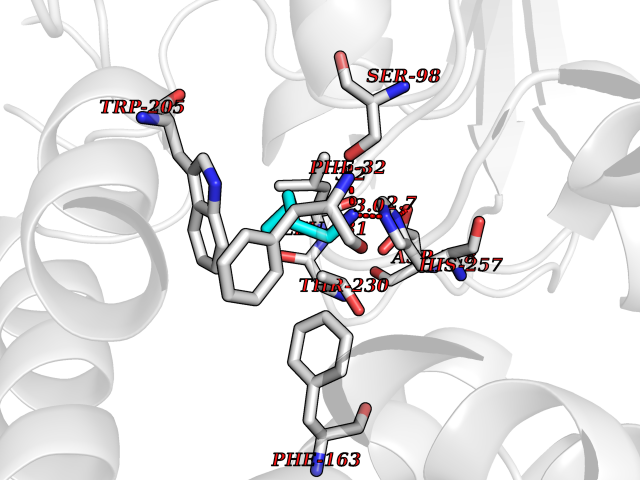 Pymol Map 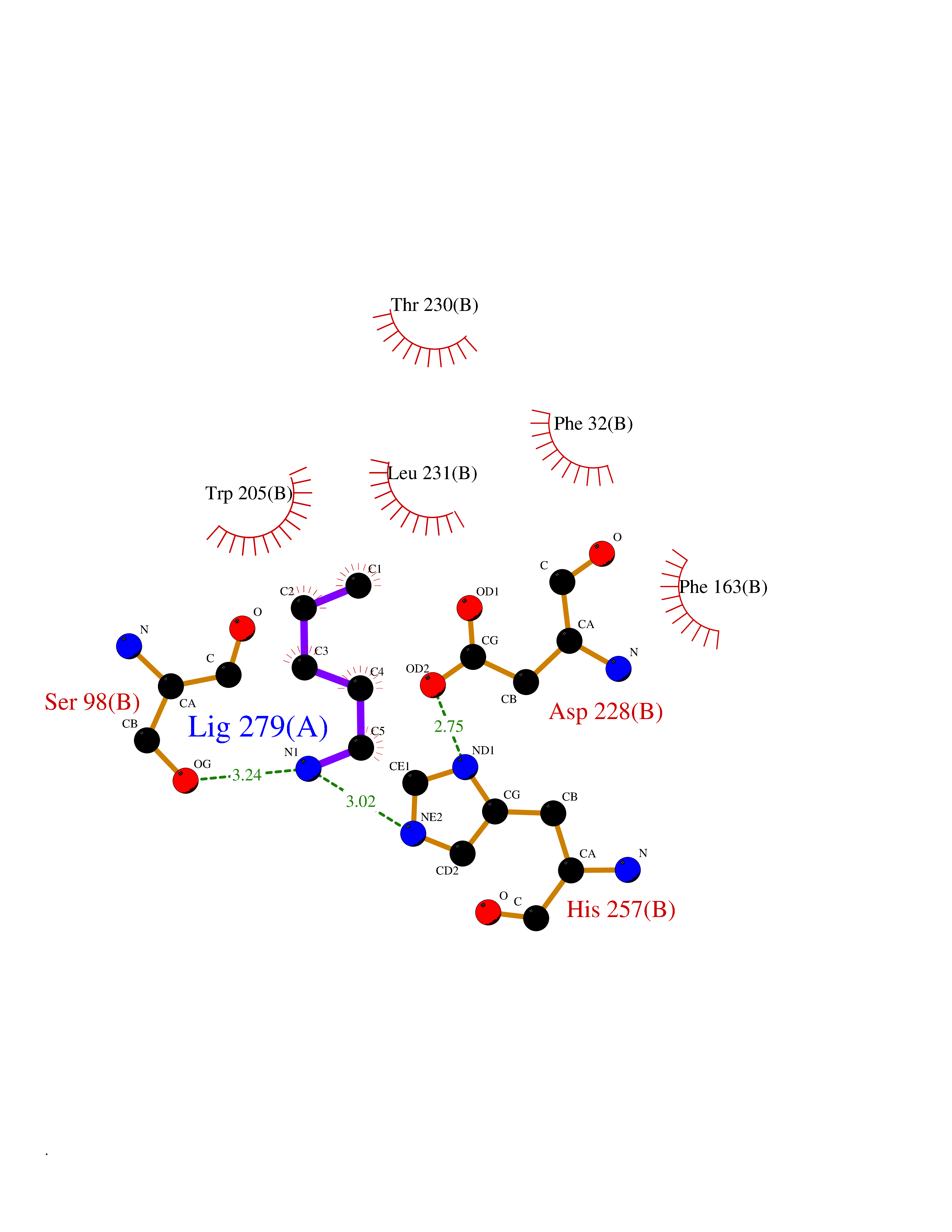 Ligplot Map 3D Binding mode Sequence PFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAKPFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | 72 kDa type IV collagenase | 3AYU | 4.76 | |
Target general information Gen name MMP2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CLG4A Protein family Peptidase M10A family Biochemical class Hydrolase / hydrolase inhibitor Function Metalloendopeptidase activity.Metallopeptidase activity.Serine-type endopeptidase activity.Zinc ion binding. Related diseases Multicentric osteolysis, nodulosis, and arthropathy (MONA) [MIM:259600]: An autosomal recessive syndrome characterized by severe multicentric osteolysis with predominant involvement of the hands and feet. Additional features include coarse face, corneal opacities, patches of thickened, hyperpigmented skin, hypertrichosis and gum hypertrophy. {ECO:0000269|PubMed:11431697, ECO:0000269|PubMed:15691365, ECO:0000269|PubMed:16542393}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01197; DB06423; DB04866; DB00786; DB05387; DB12843; DB01630 Interacts with P05067; PRO_0000000092 [P05067]; PRO_0000000093 [P05067]; P20908; Q8NBP7; Q04864-2; Q8IX30; P16035 EC number 3.4.24.24 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; Autocatalytic cleavage; Calcium; Collagen degradation; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Mitochondrion; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 19623.6 Length 176 Aromaticity 0.14 Instability index 15.48 Isoelectric point 4.84 Charge (pH=7) -10.61 2D Binding mode Binding energy (Kcal/mol) -6.49  Molscript Map 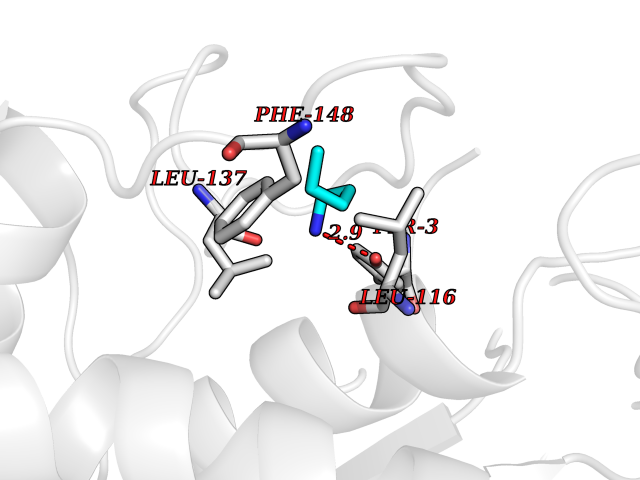 Pymol Map 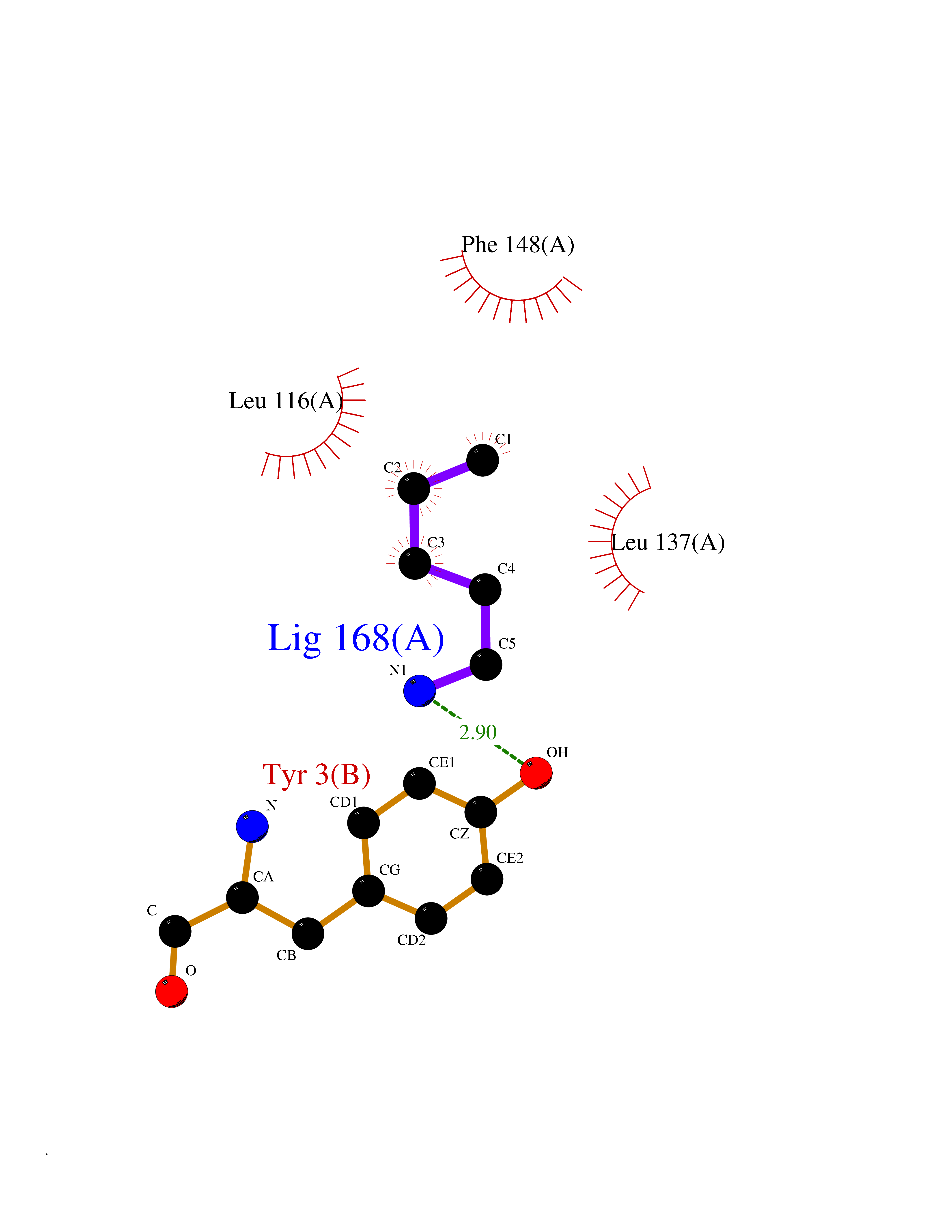 Ligplot Map 3D Binding mode Sequence YNFFPRKPKWDKNQITYRIIGYTPDLDPETVDDAFARAFQVWSDVTPLRFSRIHDGEADIMINFGRWEHGDGYPFDGKDGLLAHAFAPGTGVGGDSHFDDDELWTLGKGVGYSLFLVAAHAFGHAMGLEHSQDPGALMAPIYTYTKNFRLSQDDIKGIQELYGASPISYGNDALMP Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Cytochrome b (Complex III subunit 3) (Complex III subunit III) (Cytochrome b-c1 complex subunit 3) (Ubiquinol-cytochrome-c reductase complex cytochrome b subunit) | 1SQB | 4.76 | |
Target general information Gen name MT-CYB Organism Bos taurus (Bovine) Uniprot ID TTD ID NA Synonyms CYTB;MTCYB;COB Protein family Cytochrome b family Biochemical class NA Function Component of the ubiquinol-cytochrome c reductase complex (complex III or cytochrome b-c1 complex) that is part of the mitochondrial respiratory chain. The b-c1 complex mediates electron transfer from ubiquinol to cytochrome c. Contributes to the generation of a proton gradient across the mitochondrial membrane that is then used for ATP synthesis. {ECO:0000269|PubMed:1327781, ECO:0000269|PubMed:20025846, ECO:0000269|PubMed:9485330, ECO:0000305|PubMed:189810}." Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Electron transport; Heme; Iron; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Reference proteome; Respiratory chain; Transmembrane; Transmembrane helix; Transport; Ubiquinone Protein physicochemical properties Chain ID C,D,F,G,H Molecular weight (Da) 99281.2 Length 866 Aromaticity 0.12 Instability index 43.81 Isoelectric point 8.32 Charge (pH=7) 7.12 2D Binding mode Binding energy (Kcal/mol) -6.49 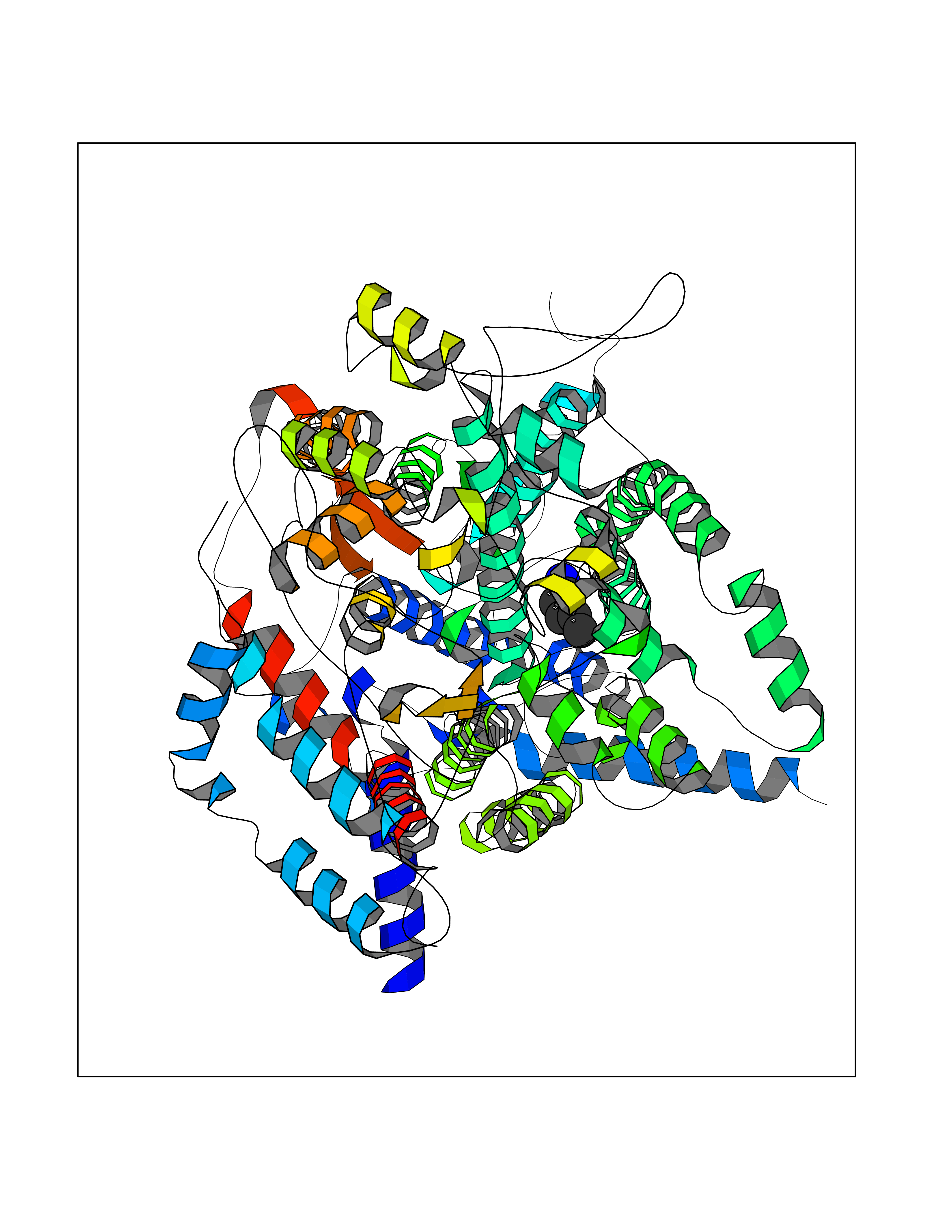 Molscript Map 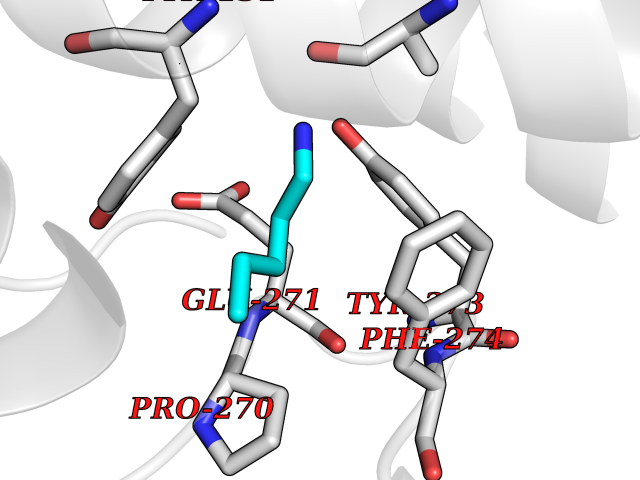 Pymol Map 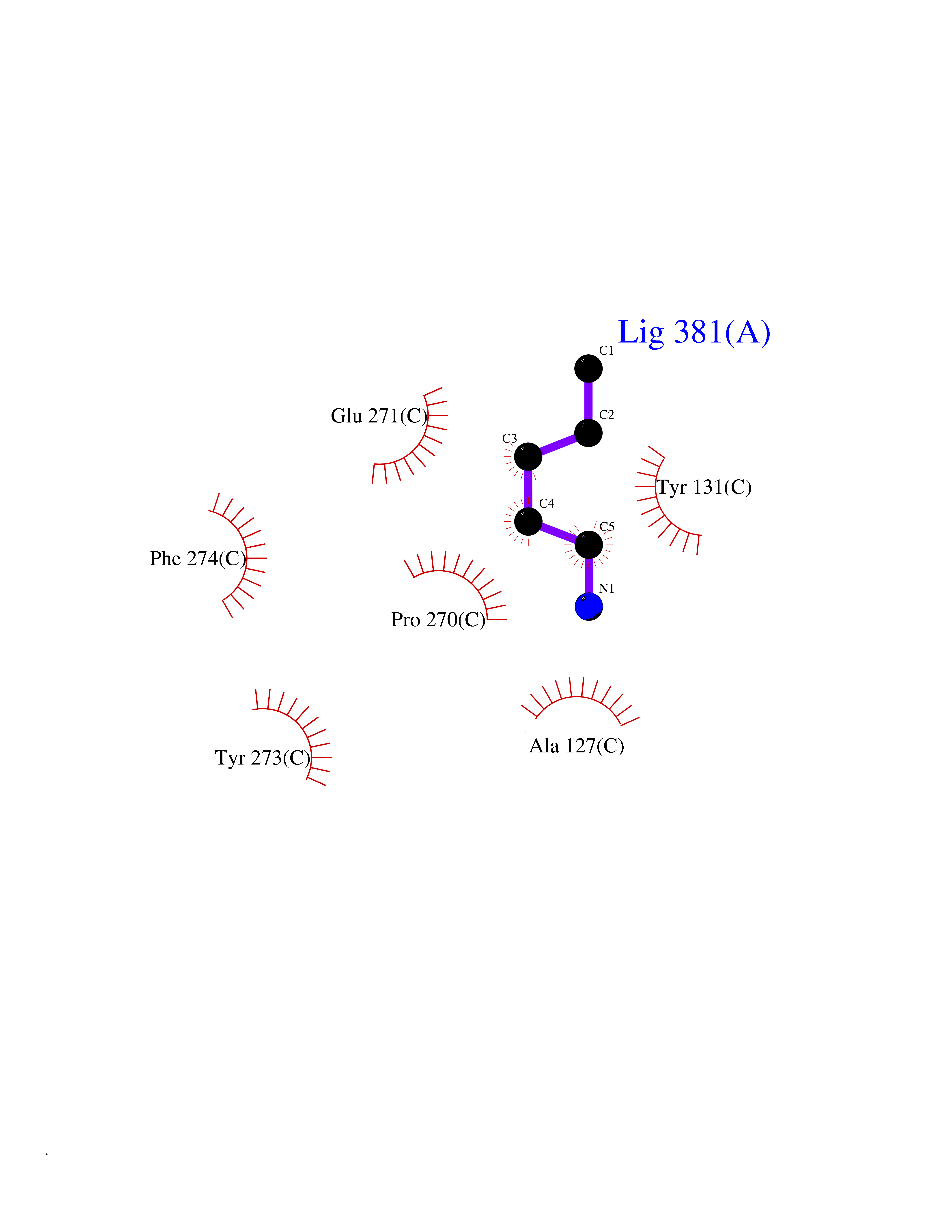 Ligplot Map 3D Binding mode Sequence VSASSRWLEGIRKWYYNAAGFNKLGLMRDDTIHENDDVKEAIRRLPENLYDDRVFRIKRALDLSMRQQILPKEQWTKYEEDKSYLEPYLKEVIRERKEREEWAKKELVDPLTTVREQCEQLEKCVKARERLELCDERVSSRSQTEEDCTEELLDFLHARDHCVAHKLFNSLKTNIRKSHPLMKIVNNAFIDLPAPSNISSWWNFGSLLGICLILQILTGLFLAMHYTSDTTTAFSSVTHICRDVNYGWIIRYMHANGASMFFICLYMHVGRGLYYGSYTFLETWNIGVILLLTVMATAFMGYVLPWGQMSFWGATVITNLLSAIPYIGTNLVEWIWGGFSVDKATLTRFFAFHFILPFIIMAIAMVHLLFLHETGSNNPTGISSDVDKIPFHPYYTIKDILGALLLILALMLLVLFAPDLLGDPDNYTPANPLNTPPHIKPEWYFLFAYAILRSIPNKLGGVLALAFSILILALIPLLHTSKQRSMMFRPLSQCLFWALVADLLTLTWIGGQPVEHPYITIGQLASVLYFLLILVLMPTAGTIENKLLKWSDLELHPPSYPWSHRGLLSSLDHTSIRRGFQVYKQVCSSCHSMDYVAYRHLVGVCYTEDEAKALAEEVEVQDGPNEDGEMFMRPGKLSDYFPKPYPNPEAARAANNGALPPDLSYIVRARHGGEDYVFSLLTGYCEPPTGVSLREGLYFNPYFPGQAIGMAPPIYNEVLEFDDGTPATMSQVAKDVCTFLRWAAEPEHDHRKRMGLKMLLMMGLLLPLVYAMKRHKWSVLKSRKLAYRPPKGRQFGHLTRVRHVITYSLSPFEQRAFPHYFSKGIPNVLRRTRACILRVAPPFVAFYLVYTWGTQEFEKSKRKNPA Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Dopamine beta-hydroxylase | 4ZEL | 4.76 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -6.49 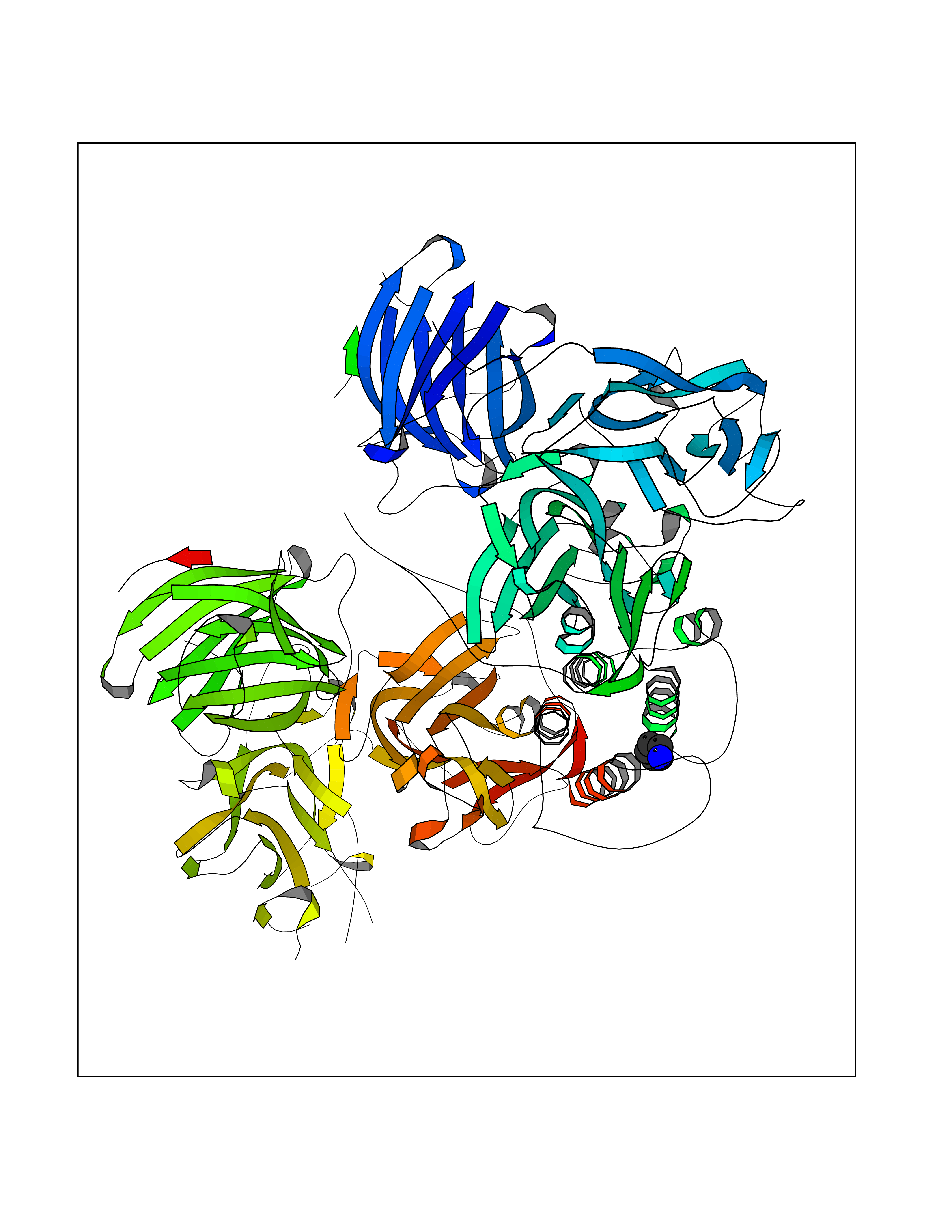 Molscript Map 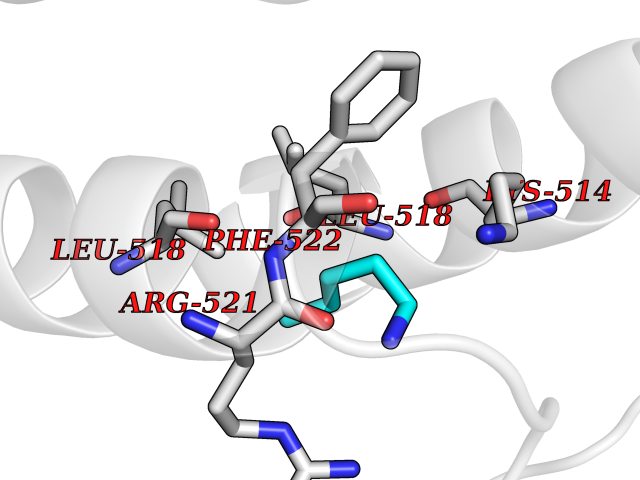 Pymol Map 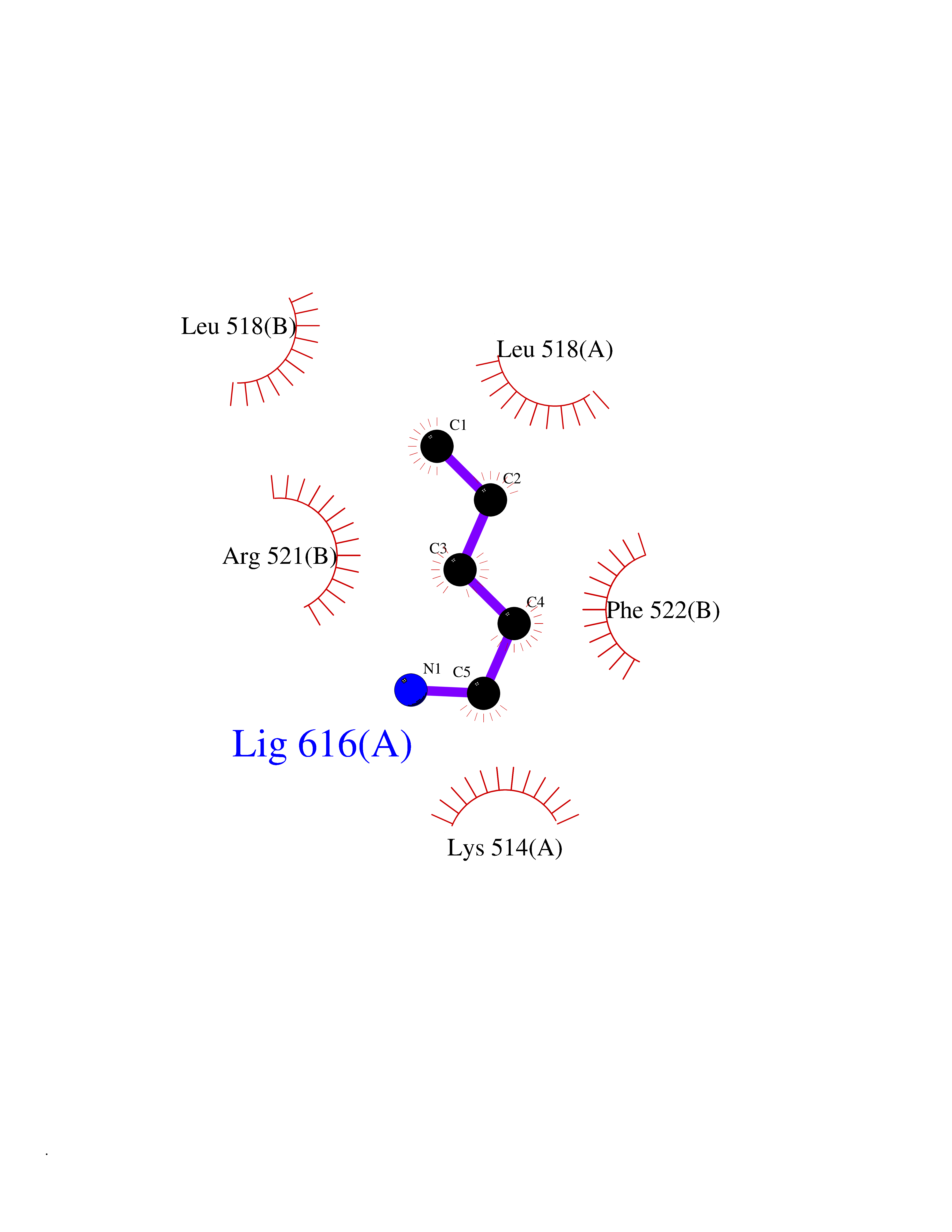 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Oxygen-insensitive NADPH nitroreductase | 1F5V | 4.76 | |
Target general information Gen name nfsA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms mda18;b0851;mdaA;ybjB;JW0835 Protein family Flavin oxidoreductase frp family Biochemical class Oxidoreductase Function Chromate reductase activity.FMN binding.Oxidoreductase activity, acting on NAD(P)H, nitrogenous group as acceptor. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03247; DB00698 Interacts with P28630 EC number 1.-.-.- Uniprot keywords 3D-structure; Flavoprotein; FMN; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 53582.7 Length 480 Aromaticity 0.07 Instability index 45.43 Isoelectric point 6.47 Charge (pH=7) -3.13 2D Binding mode Binding energy (Kcal/mol) -6.5 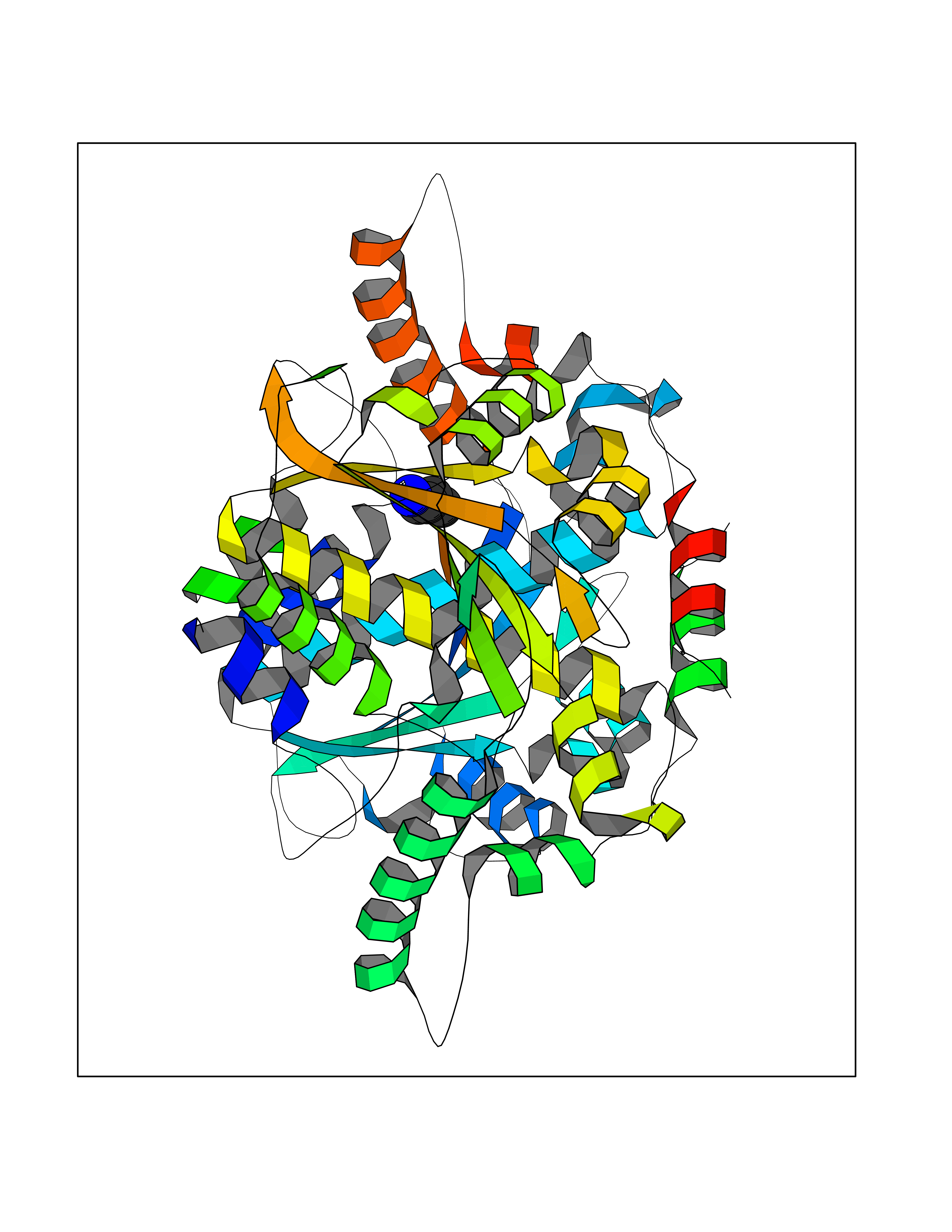 Molscript Map 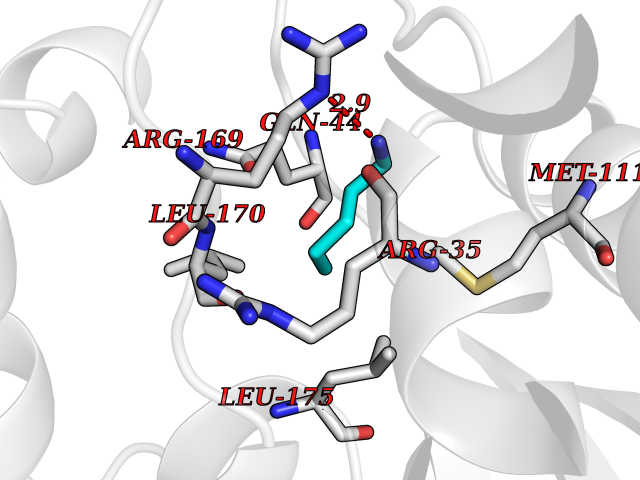 Pymol Map 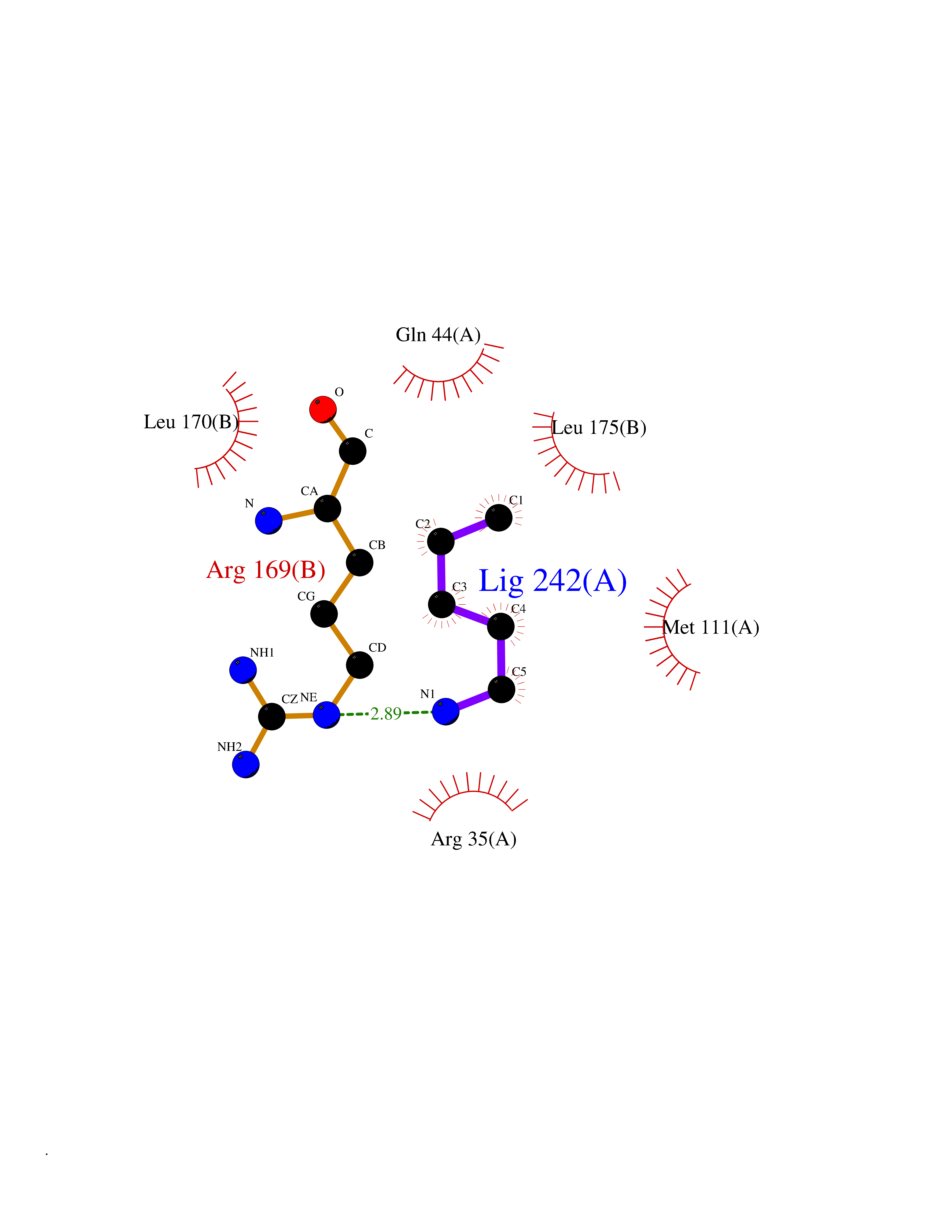 Ligplot Map 3D Binding mode Sequence MTPTIELICGHRSIRHFTDEPISEAQREAIINSARATSSSSFLQCSSIIRITDKALREELVTLTGGQKHVAQAAEFWVFCADFNRHLQICPDAQLGLAEQLLLGVVDTAMMAQNALIAAESLGLGGVYIGGLRNNIEAVTKLLKLPQHVLPLFGLCLGWPADNPDLKPRLPASILVHENSYQPLDKGALAQYDEQLAEYYLTRGSNNRRDTWSDHIRRTIIKESRPFILDYLHKQGWATRMTPTIELICGHRSIRHFTDEPISEAQREAIINSARATSSSSFLQCSSIIRITDKALREELVTLTGGQKHVAQAAEFWVFCADFNRHLQICPDAQLGLAEQLLLGVVDTAMMAQNALIAAESLGLGGVYIGGLRNNIEAVTKLLKLPQHVLPLFGLCLGWPADNPDLKPRLPASILVHENSYQPLDKGALAQYDEQLAEYYLTRGSNNRRDTWSDHIRRTIIKESRPFILDYLHKQGWATR Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Cyclin-dependent kinase 4 | 2W96 | 4.76 | |
Target general information Gen name CDK4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Cell cycle Function ATP binding.Cyclin binding.Cyclin-dependent protein serine/threonine kinase activity.Cyclin-dependent protein serine/threonine kinase regulator activity.Protein complex binding. Related diseases Melanoma, cutaneous malignant 3 (CMM3) [MIM:609048]: A malignant neoplasm of melanocytes, arising de novo or from a pre-existing benign nevus, which occurs most often in the skin but may also involve other sites. {ECO:0000269|PubMed:7652577, ECO:0000269|PubMed:8528263, ECO:0000269|PubMed:9311594, ECO:0000269|PubMed:9425228}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12001; DB03496; DB12010; DB09073; DB02733; DB11730; DB15442 Interacts with Q9UH17; P24385; P30279; P30281; Q16543; P50613; P38936; P46527; P49918; P42771; P42772; P42773; P55273; Q9UJC3; P08238; Q9UKT9; Q0VD86; P01106; Q9ULD0; P28749; Q08999; P09936; Q8N720 EC number 2.7.11.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Cell division; Cytoplasm; Disease variant; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 30138.4 Length 267 Aromaticity 0.09 Instability index 36.2 Isoelectric point 5.78 Charge (pH=7) -5.83 2D Binding mode Binding energy (Kcal/mol) -6.49 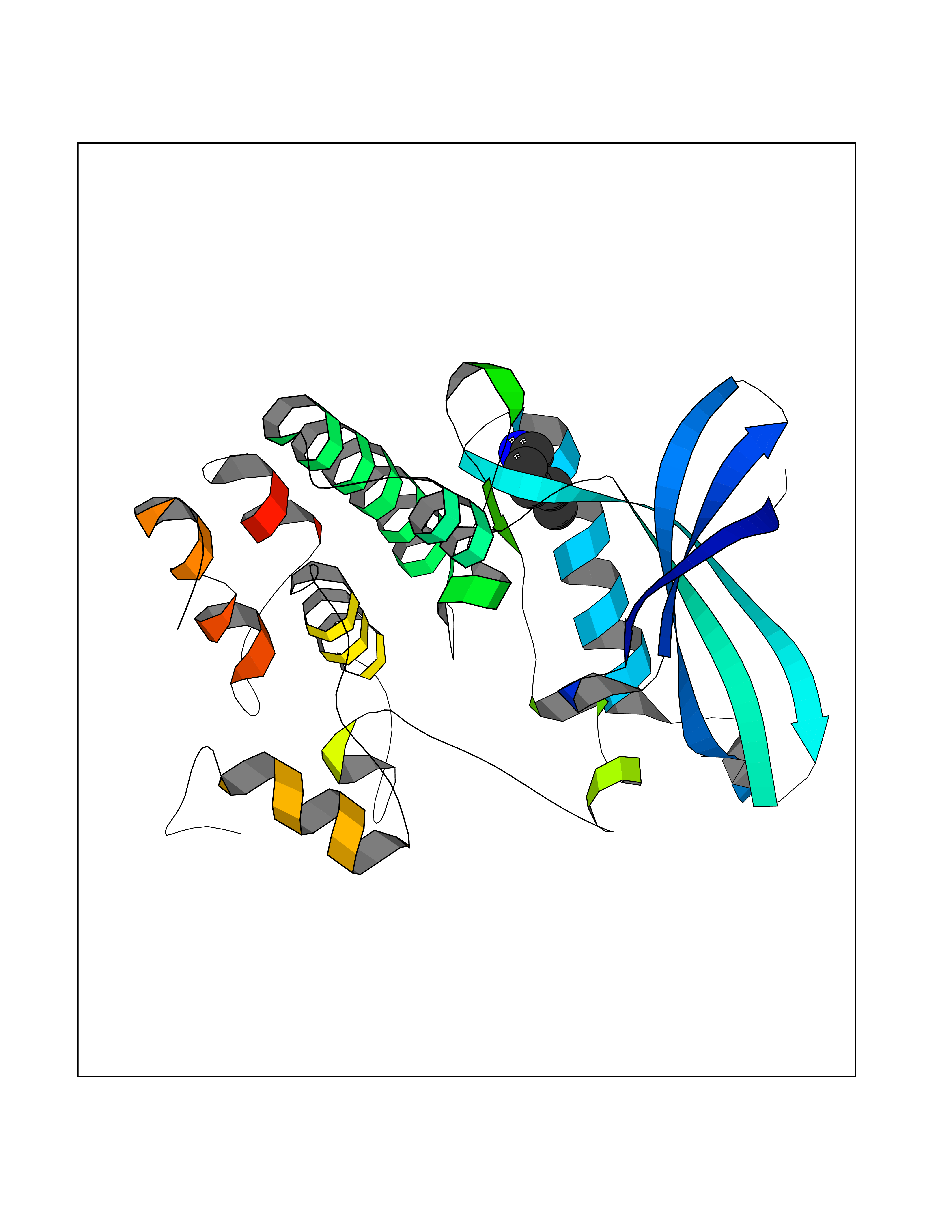 Molscript Map 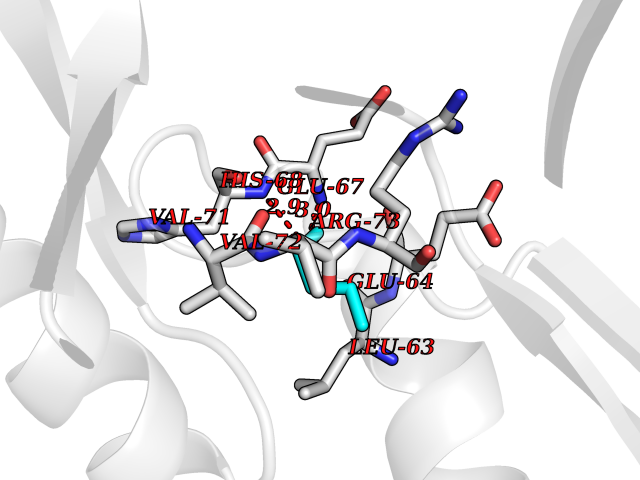 Pymol Map 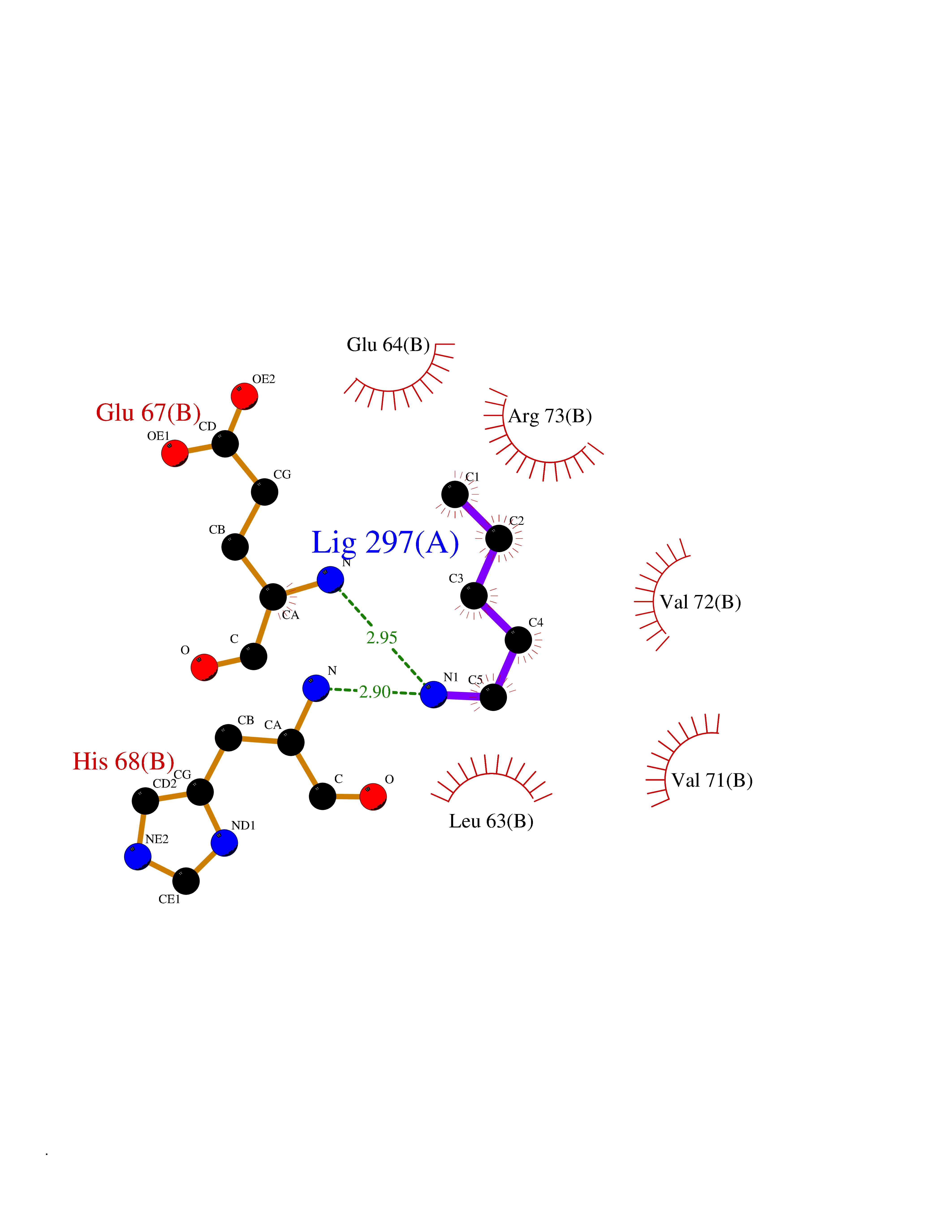 Ligplot Map 3D Binding mode Sequence SRYEPVAEIGVGAYGTVYKARDPHSGHFVALKSVRVPNGEEGLPISTVREVALLRRLEAFEHPNVVRLMDVCATSRTDREIKVTLVFEHVDQDLRTYLDKAPPPGLPAETIKDLMRQFLRGLDFLHANCIVHRDLKPENILVTSGGTVKLADFGLARIYSYQMALDPVVVTLWYRAPEVLLQSTYATPVDMWSVGCIFAEMFRRKPLFCGNSEADQLGKIFDLIGLPPEDDWVPEMEESGAQLLLEMLTFNPHKRISAFRALQHSYL Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Diamine oxidase (AOC1) | 3HIG | 4.76 | |
Target general information Gen name AOC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Kidney amine oxidase; KAO; Histaminase; Amiloride-binding protein; AOC1; ABP Protein family Copper/topaquinone oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the degradation of compounds such as putrescine, histamine, spermine, and spermidine, substances involved in allergic and immune responses, cell proliferation, tissue differentiation, tumor formation, and possibly apoptosis. Placental DAO is thought to play a role in the regulation of the female reproductive function. Related diseases Lichtenstein-Knorr syndrome (LIKNS) [MIM:616291]: An autosomal recessive neurologic disorder characterized by progressive cerebellar ataxia and severe progressive sensorineural hearing loss. {ECO:0000269|PubMed:25205112, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00594; DB01373; DB09130; DB03608; DB05383 Interacts with Q15038; O75593; Q8IUC2; Q96HA8; Q7Z3K3; Q6ZRY4; Q01085-2; O43711; Q96K80 EC number EC 1.4.3.22 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Copper; Direct protein sequencing; Disulfide bond; Glycoprotein; Heparin-binding; Membrane; Metal-binding; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal; TPQ Protein physicochemical properties Chain ID A,B Molecular weight (Da) 69037.3 Length 607 Aromaticity 0.13 Instability index 43.33 Isoelectric point 6.52 Charge (pH=7) -4.61 2D Binding mode Binding energy (Kcal/mol) -6.49  Molscript Map 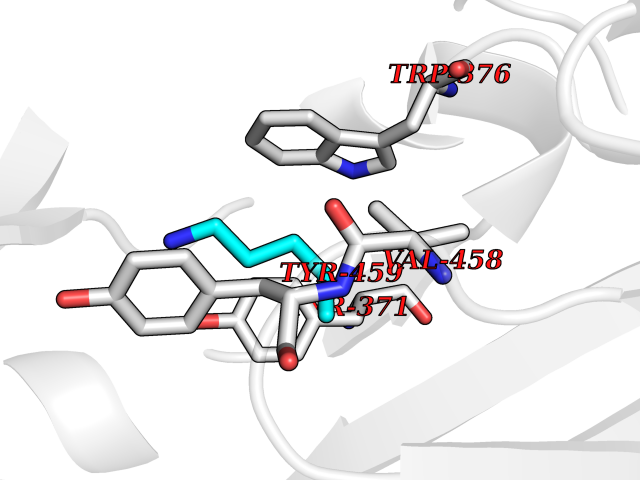 Pymol Map 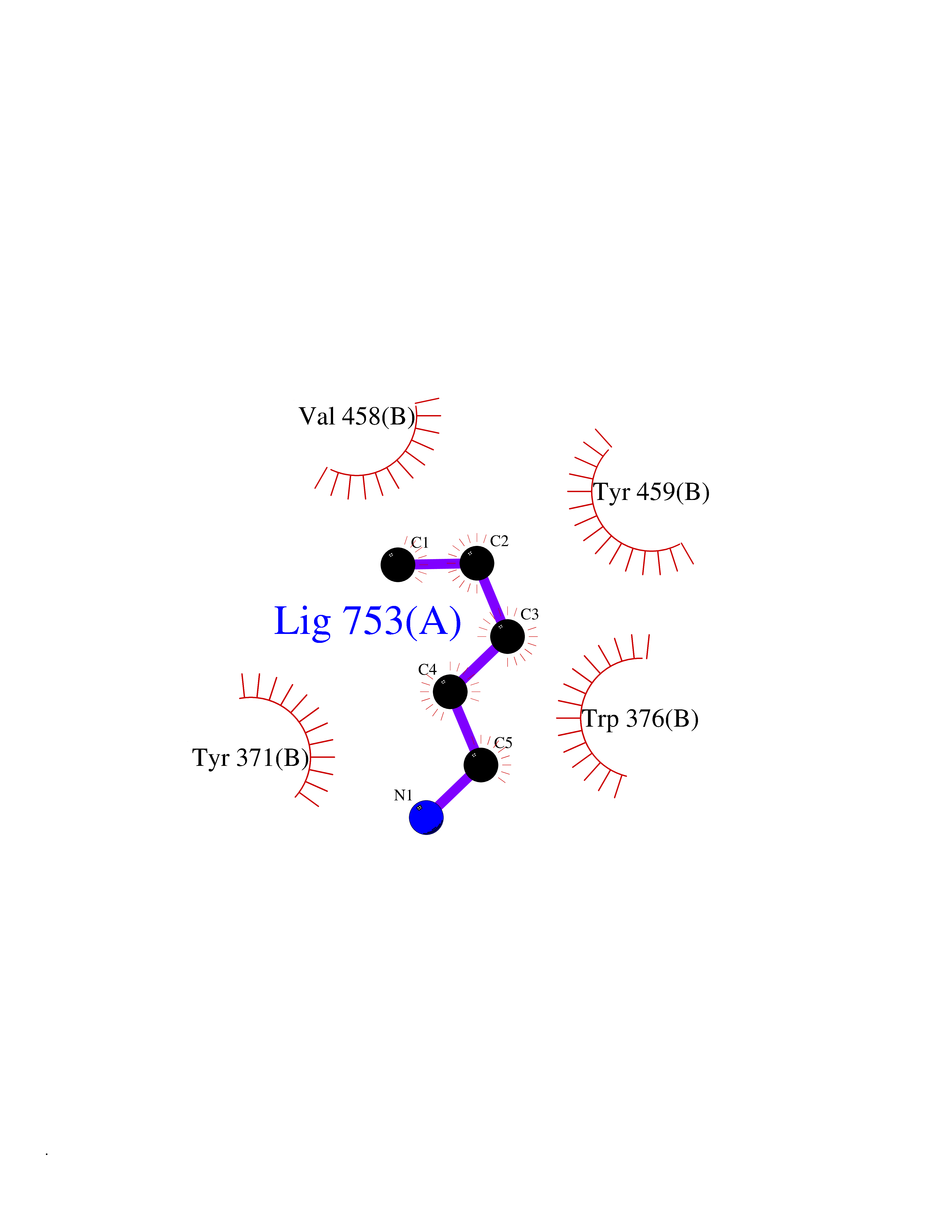 Ligplot Map 3D Binding mode Sequence RKAGVFSDLSNQELKAVHSFLWSKKELRLQPSSTTTMAKNTVFLIEMLLPKKYHVLRFLDKGERHPVREARAVIFFGDQEHPNVTEFAVGPLPGPCYMRALSPRPGYQSSWASRPISTAEYALLYHTLQEATKPLHQFFLNTTGFSFQDCHDRCLAFTDVAPRGVASGQRRSWLIIQRYVEGYFLHPTGLELLVDHGSTDAGHWAVEQVWYNGKFYGSPEELARKYADGEVDVVVLEPPLFSSLVQPHGPRFRLEGNAVLYGGWSFAFRLRSSSGLQVLNVHFGGERIAYEVSVQEAVALYGGHTPAGMQTKYLDVGWGLGSVTHELAPGIDCPETATFLDTFHYYDADDPVHYPRALCLFEMPTGVPLKGQVLVLRTTSTVYNXDYIWDFIFYPNGVMEAKMHATGYVHATFYTPEGLRHGTRLHTHLIGNIHTHLVHYRVDLDVAGTKNSFQTLQYSWERQAAFRFKRKLPKYLLFTSPQENPWGHKRSYRLQIHSMADQVLPPGWQEEQAITWARYPLAVTKYRESELCSSSIYHQNDPWDPPVVFEQFLHNNENIENEDLVAWVTVGFLHIPHSEDIPNTATPGNSVGFLLRPFNFNGTYRPV Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | S-adenosylmethionine decarboxylase proenzyme (AMD1) | 1JL0 | 4.76 | |
Target general information Gen name AMD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SamDC; S-adenosylmethioninedecarboxylase; AdoMetDC; AMD Protein family Eukaryotic AdoMetDC family Biochemical class Carbon-carbon lyase Function Promotes maintenance and self-renewal of embryonic stem cells, by maintaining spermine levels. Essential for biosynthesis of the polyamines spermidine and spermine. Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08163; DB00118; DB01917 Interacts with P17707; Q96A98; Q8WY91 EC number EC 4.1.1.50 Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Decarboxylase; Direct protein sequencing; Lyase; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Pyruvate; Reference proteome; S-adenosyl-L-methionine; Schiff base; Spermidine biosynthesis; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 35790.5 Length 311 Aromaticity 0.14 Instability index 39.47 Isoelectric point 6.03 Charge (pH=7) -2.01 2D Binding mode Binding energy (Kcal/mol) -6.49 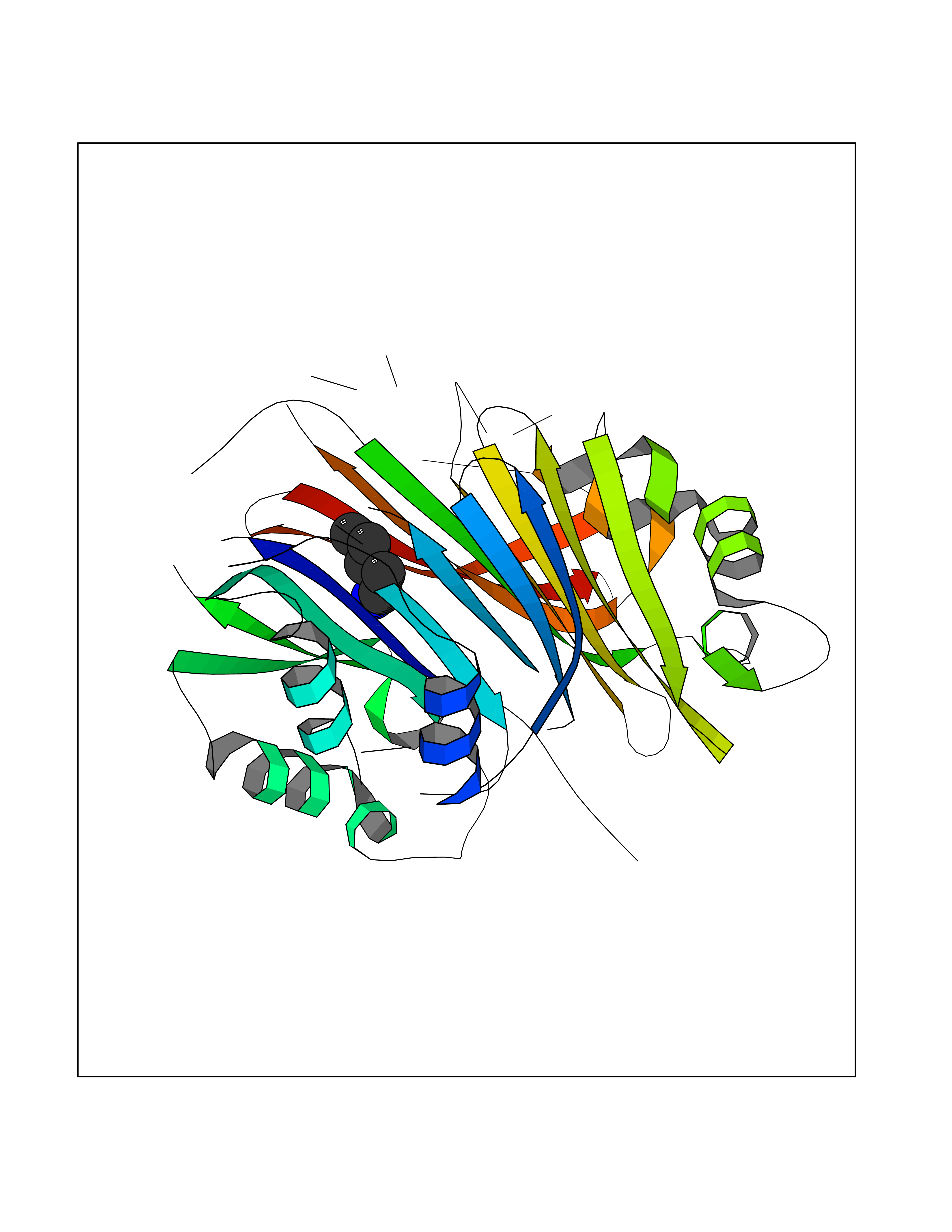 Molscript Map 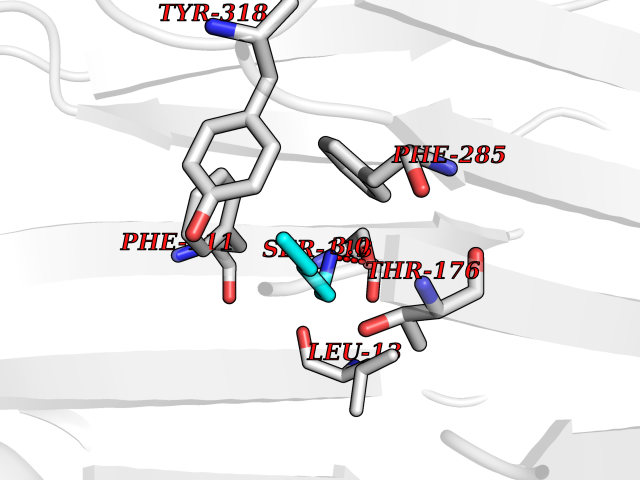 Pymol Map 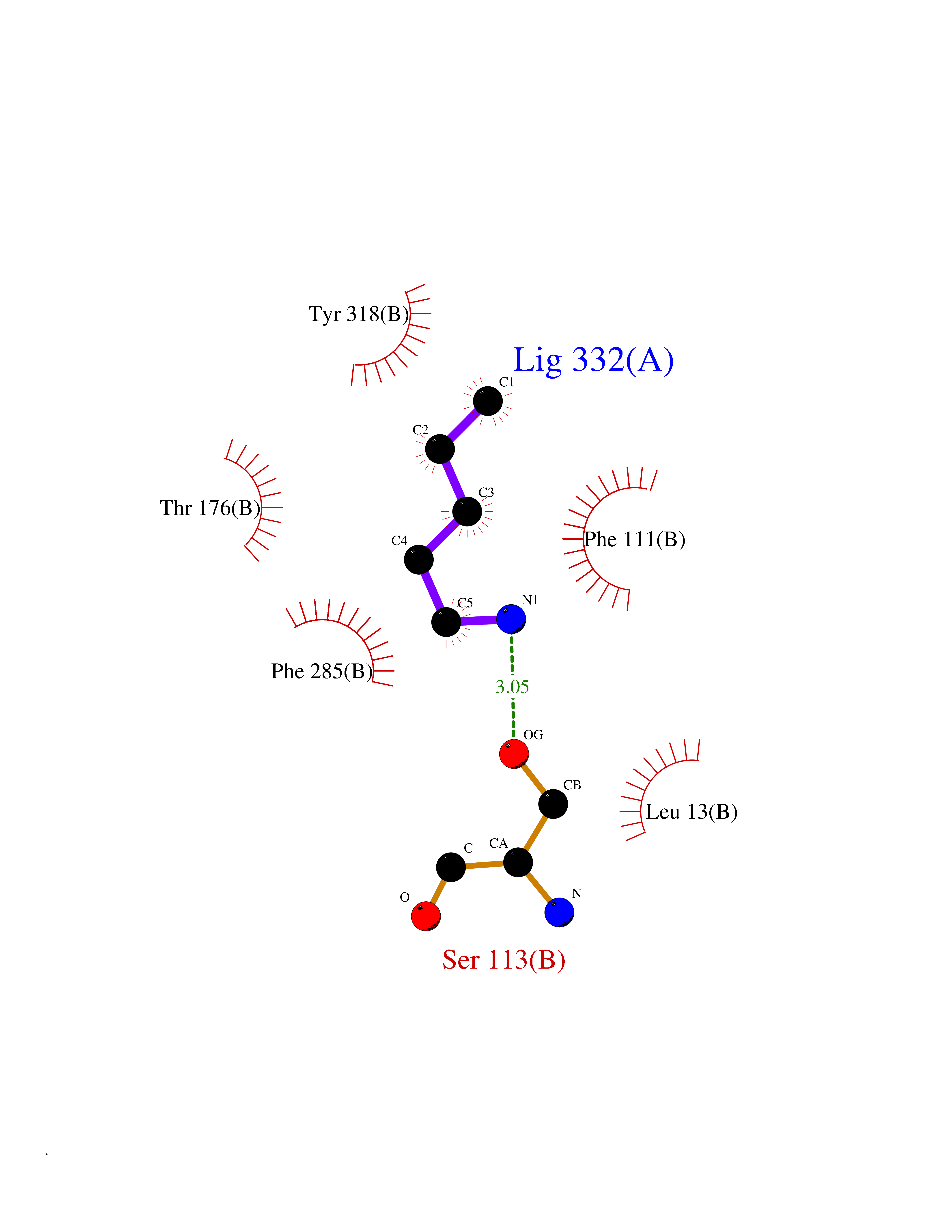 Ligplot Map 3D Binding mode Sequence HFFEGTEKLLEVWFSRQGSGDLRTIPRSEWDILLKDVQCSIISVTKTDKQEAYVLSESSMFVSKRRFILKTCGTTLLLKALVPLLKLARDYSGFDSIQSFFYSRKNFMKPSHQGYPHRNFQEEIEFLNAIFPNGAGYCMGRMNSDCWYLYTLDFRVISQPDQTLEILMSELDPAVMDQFYMKDGVTAKDVTRESGIRDLIPGSVIDATMFNPCGYSMNGMKSDGTYWTIAITPEPEFSYVSFETNLSQTSYDDLIRKVVEVFKPGKFVTTLFVNQSSKCPQKIEGFKRLDCQSAMFNDYNFVFTSFAKKQQ Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 4.76 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -6.49 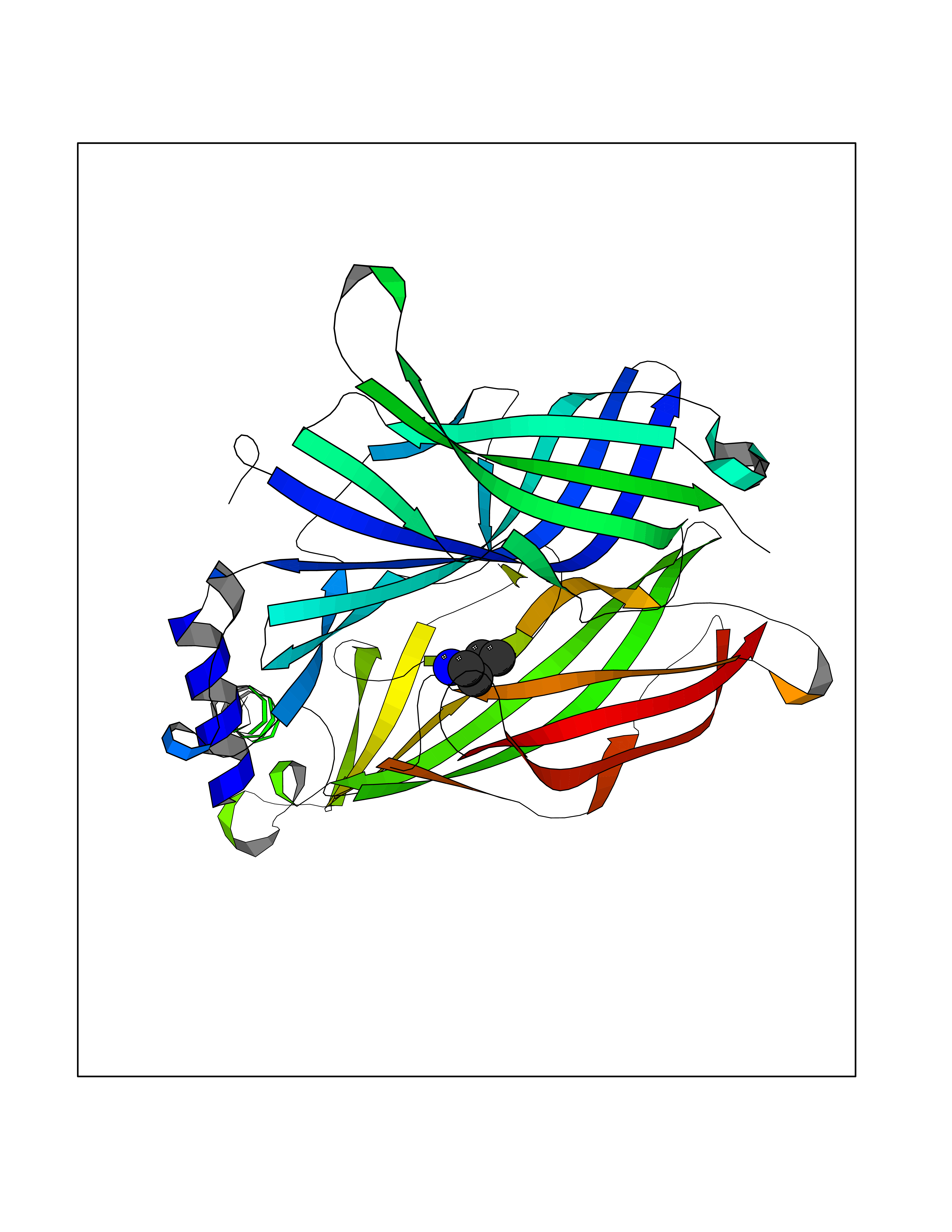 Molscript Map 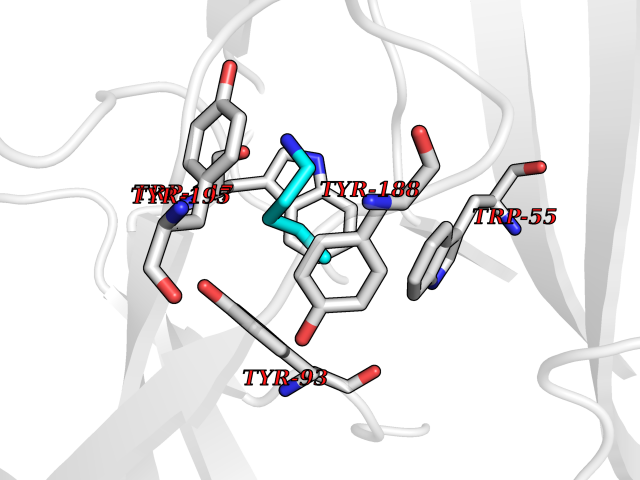 Pymol Map 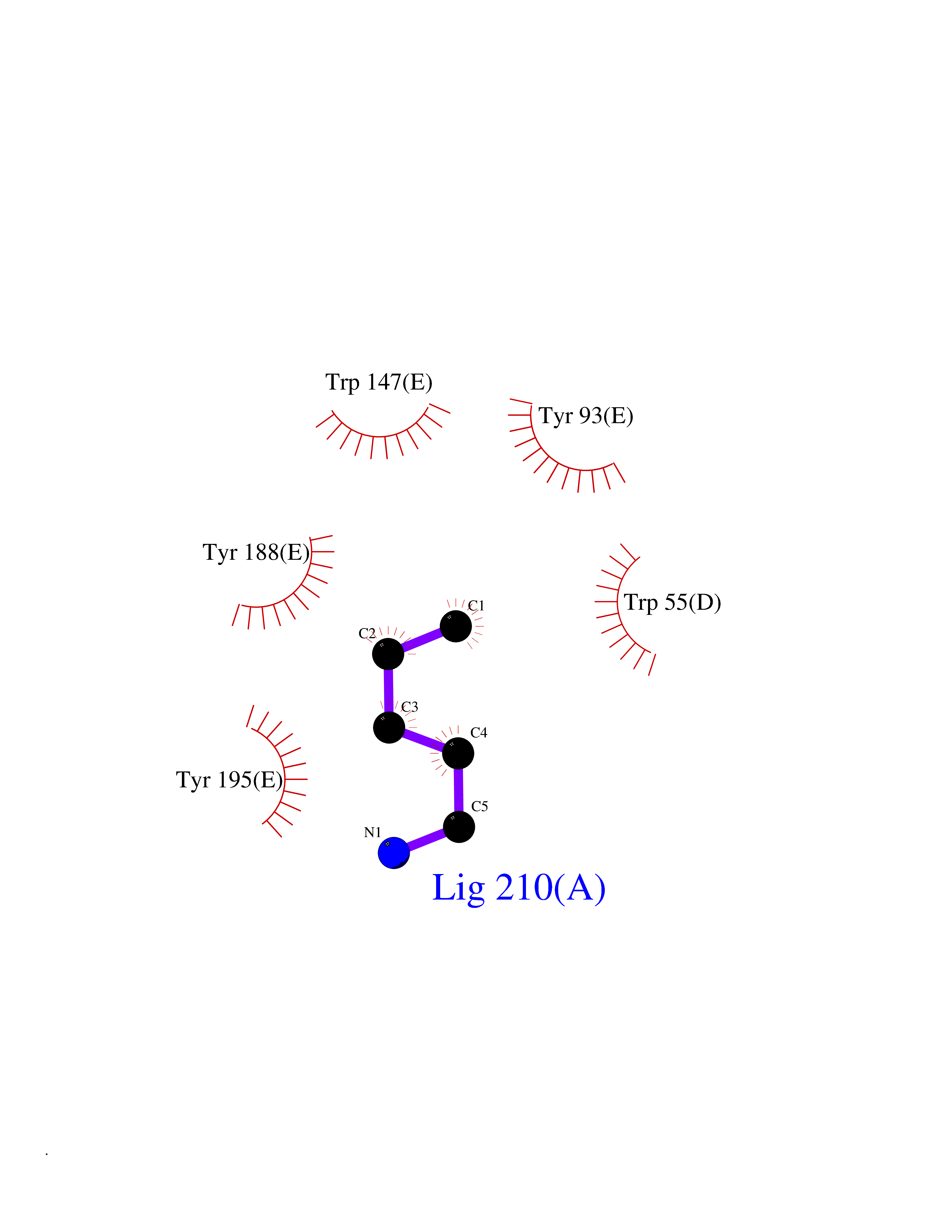 Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Fms-like tyrosine kinase 3 (FLT-3) | 1RJB | 4.76 | |
Target general information Gen name FLT3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Stem cell tyrosine kinase 1; STK1; STK-1; Receptor-type tyrosine-protein kinase FLT3; Fetal liver kinase-2; FLT-3; FLK2; FLK-2; FL cytokine receptor; CD135 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Tyrosine-protein kinase that acts as cell-surface receptor for the cytokine FLT3LG and regulates differentiation, proliferation and survival of hematopoietic progenitor cells and of dendritic cells. Promotes phosphorylation of SHC1 and AKT1, and activation of the downstream effector MTOR. Promotes activation of RAS signaling and phosphorylation of downstream kinases, including MAPK1/ERK2 and/or MAPK3/ERK1. Promotes phosphorylation of FES, FER, PTPN6/SHP, PTPN11/SHP-2, PLCG1, and STAT5A and/or STAT5B. Activation of wild-type FLT3 causes only marginal activation of STAT5A or STAT5B. Mutations that cause constitutive kinase activity promote cell proliferation and resistance to apoptosis via the activation of multiple signaling pathways. Related diseases Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:11090077, ECO:0000269|PubMed:11290608, ECO:0000269|PubMed:11442493, ECO:0000269|PubMed:14504097, ECO:0000269|PubMed:16266983, ECO:0000269|PubMed:18305215, ECO:0000269|PubMed:8946930, ECO:0000269|PubMed:9737679}. The gene represented in this entry may be involved in disease pathogenesis. Somatic mutations that lead to constitutive activation of FLT3 are frequent in AML patients. These mutations fall into two classes, the most common being in-frame internal tandem duplications of variable length in the juxtamembrane region that disrupt the normal regulation of the kinase activity. Likewise, point mutations in the activation loop of the kinase domain can result in a constitutively activated kinase. Drugs (DrugBank ID) DB12742; DB12267; DB12500; DB12010; DB12141; DB06469; DB06080; DB06595; DB11763; DB09079; DB11697; DB12978; DB08901; DB15822; DB12874; DB00398; DB01268; DB05465; DB11800; DB05014 Interacts with P00519; P42684; P46108; P46109; P06241; Q13322; Q9Y6K9; P06239; P27986; P20936; P43405; Q8R4L0 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 34209.2 Length 298 Aromaticity 0.14 Instability index 39.68 Isoelectric point 5.57 Charge (pH=7) -4.02 2D Binding mode Binding energy (Kcal/mol) -6.49 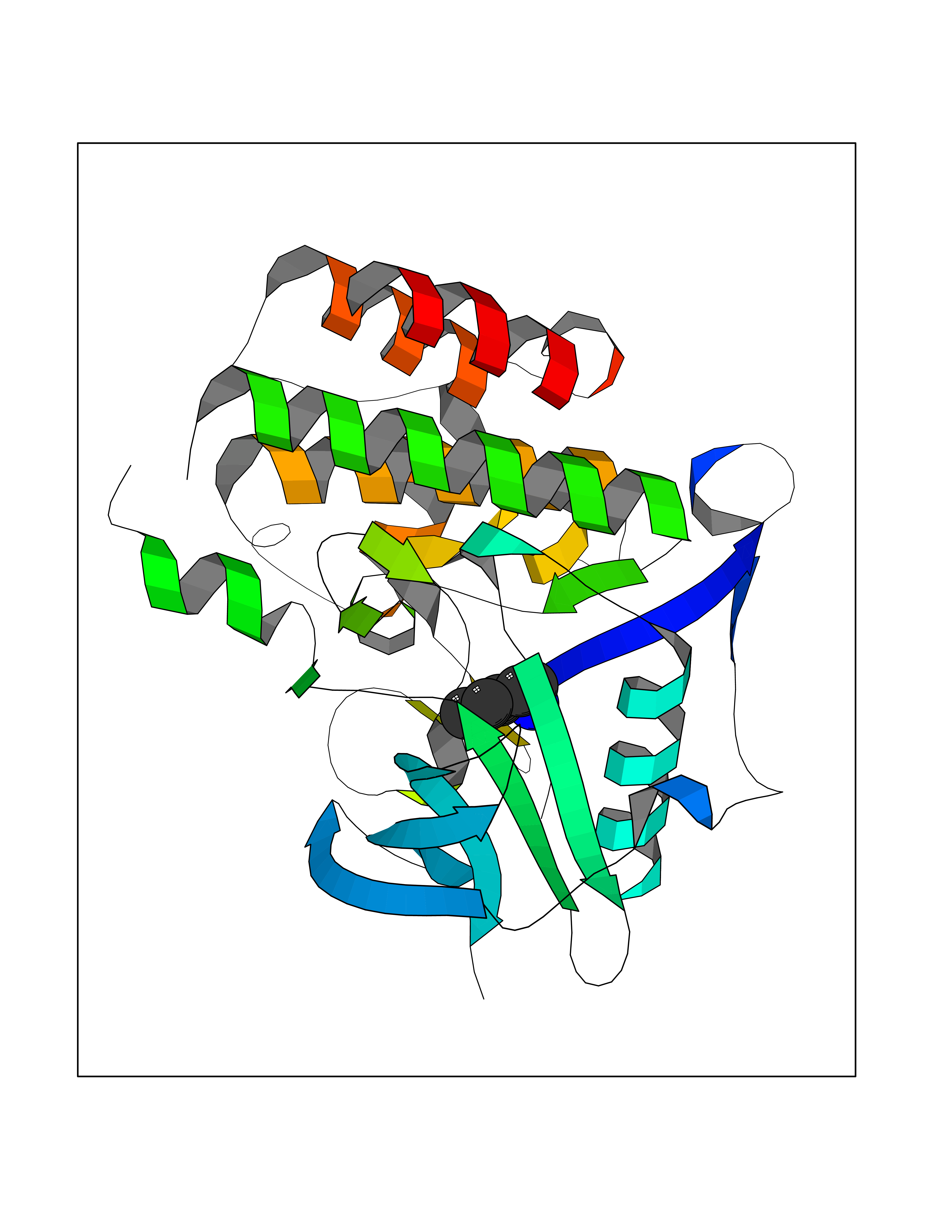 Molscript Map  Pymol Map 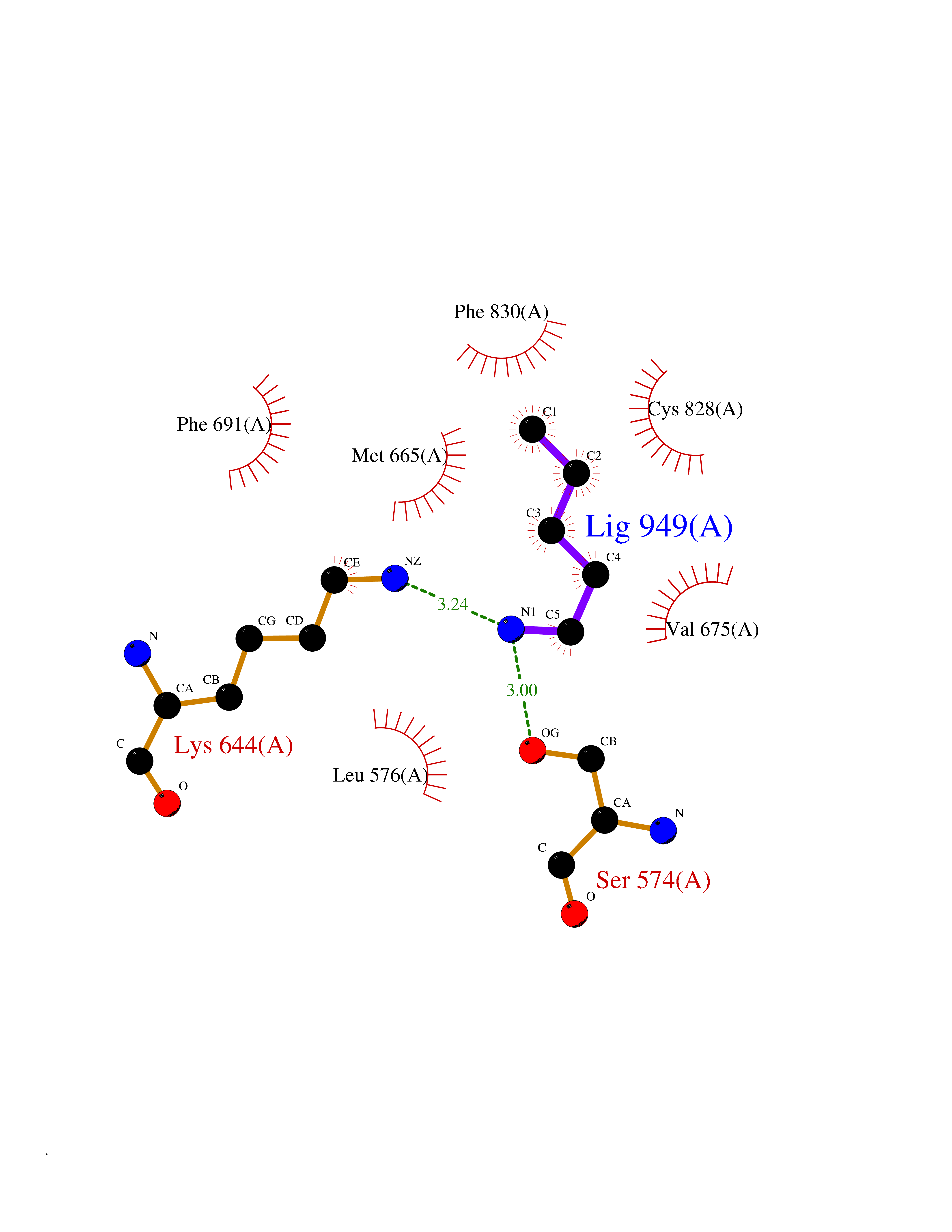 Ligplot Map 3D Binding mode Sequence YESQLQMVQVTGSSDNEYFYVDFREYEYDLKWEFPRENLEFGKVLGSGAFGKVMNATAYGISKTGVSIQVAVKMLKEREALMSELKMMTQLGSHENIVNLLGACTLSGPIYLIFEYCCYGDLLNYLRSKREKFLTFEDLLCFAYQVAKGMEFLEFKSCVHRDLAARNVLVTHGKVVKICDFGLARDIMSDSNYVVRGNARLPVKWMAPESLFEGIYTIKSDVWSYGILLWEIFSLGVNPYPGIPVDANFYKLIQNGFKMDQPFYATEEIYIIMQSCWAFDSRKRPSFPNLTSFLGCQL Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Monomeric sarcosine oxidase | 2GF3 | 4.76 | |
Target general information Gen name soxA Organism Bacillus sp. (strain B-0618) Uniprot ID TTD ID NA Synonyms sox Protein family MSOX/MTOX family, MSOX subfamily Biochemical class Oxidoreductase Function Sarcosine oxidase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB03098; DB01918; DB03517; DB03147; DB03366; DB02083; DB02543 Interacts with NA EC number 1.5.3.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42606.4 Length 385 Aromaticity 0.1 Instability index 26.97 Isoelectric point 5.27 Charge (pH=7) -17.18 2D Binding mode Binding energy (Kcal/mol) -6.49 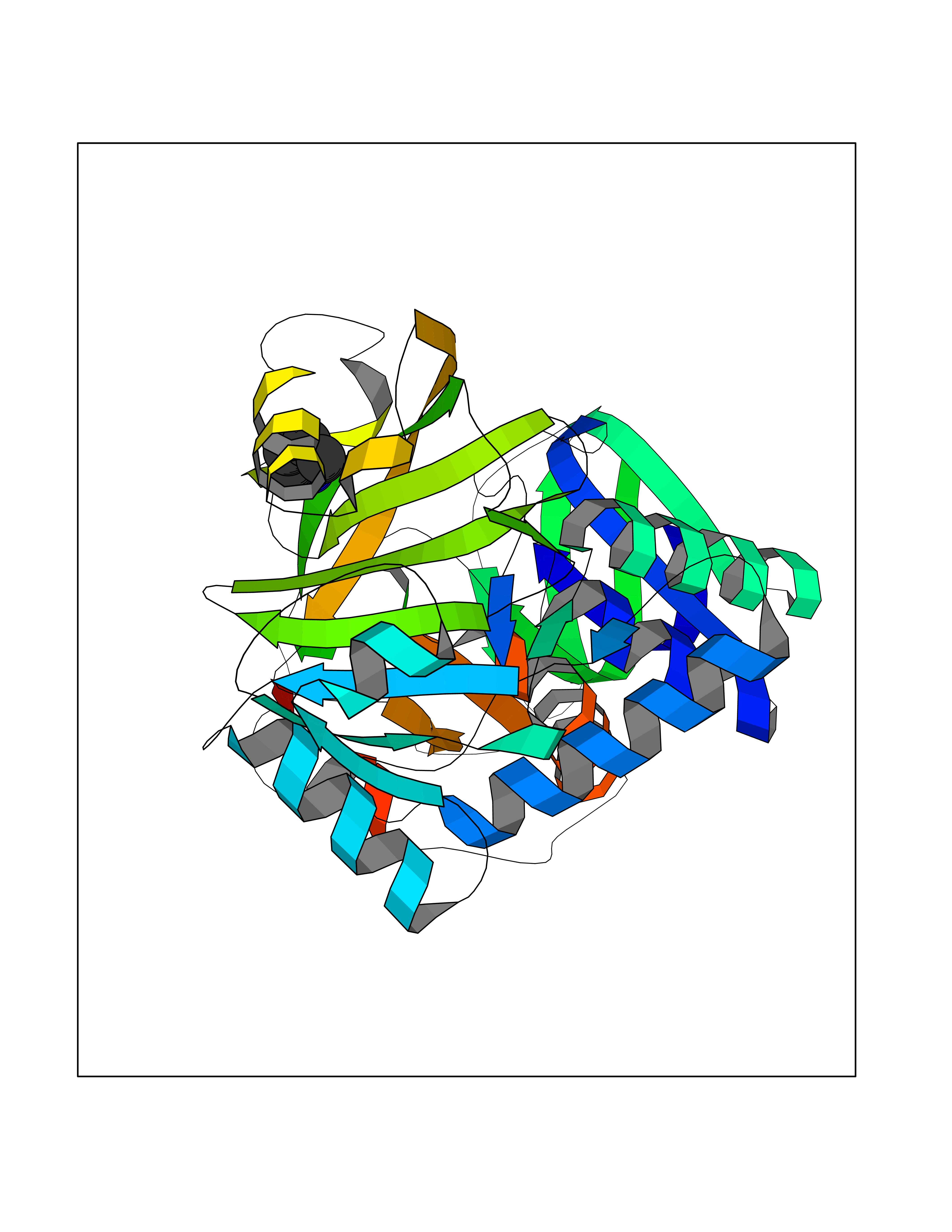 Molscript Map 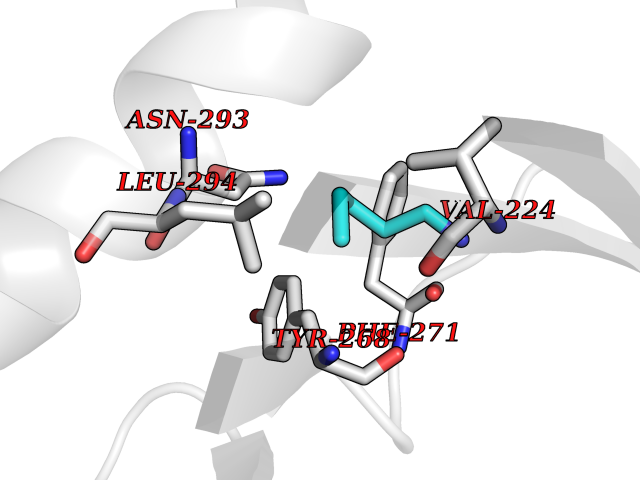 Pymol Map 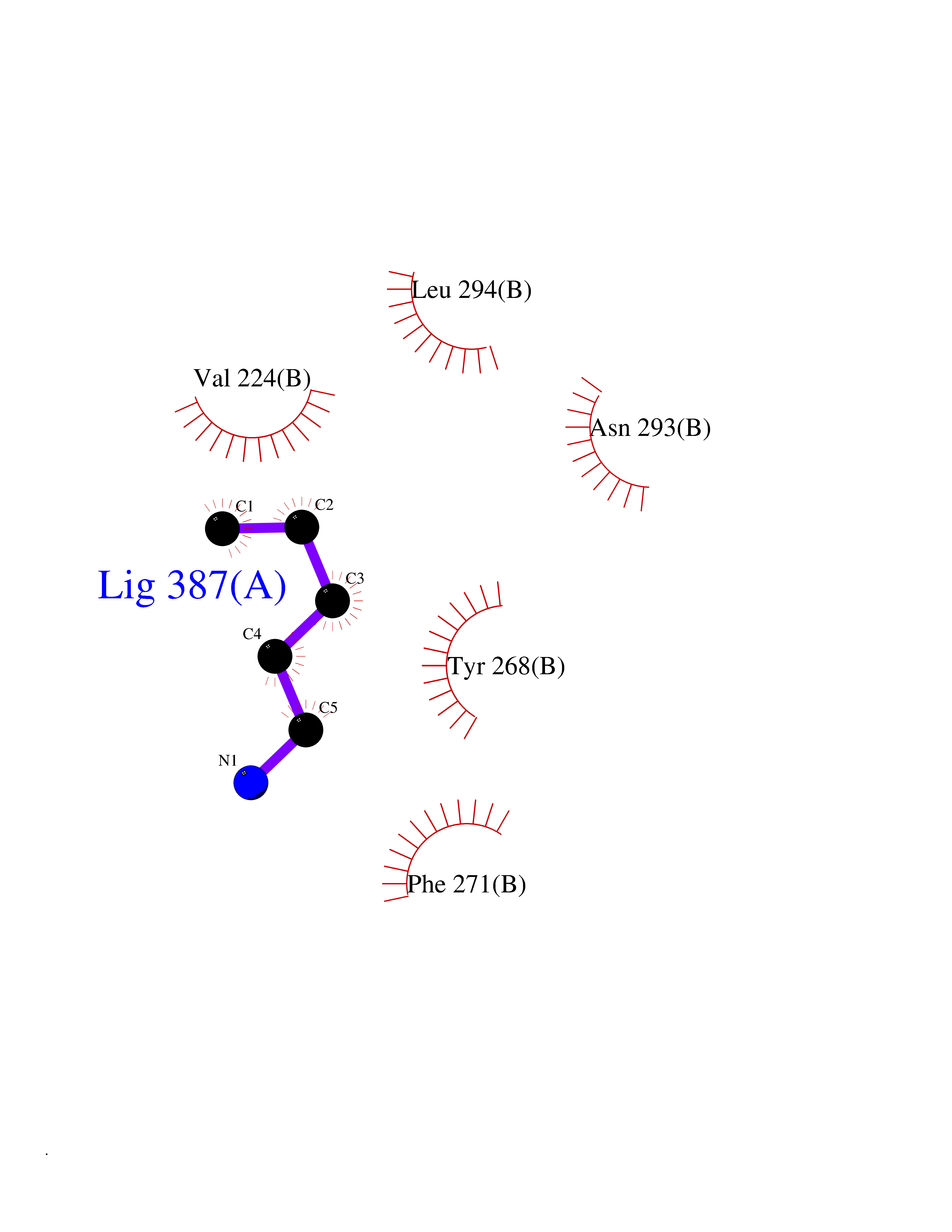 Ligplot Map 3D Binding mode Sequence STHFDVIVVGAGSMGMAAGYQLAKQGVKTLLVDAFDPPHTNGSHHGDTRIIRHAYGEGREYVPLALRSQELWYELEKETHHKIFTKTGVLVFGPKGESAFVAETMEAAKEHSLTVDLLEGDEINKRWPGITVPENYNAIFEPNSGVLFSENCIRAYRELAEARGAKVLTHTRVEDFDISPDSVKIETANGSYTADKLIVSMGAWNSKLLSKLNLDIPLQPYRQVVGFFESDESKYSNDIDFPGFMVEVPNGIYYGFPSFGGCGLKLGYHTFGQKIDPDTINREFGVYPEDESNLRAFLEEYMPGANGELKRGAVCMYTKTLDEHFIIDLHPEHSNVVIAAGFSGHGFKFSSGVGEVLSQLALTGKTEHDISIFSINRPALKESLQ Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Thymidine kinase 1 (TK1) | 1W4R | 4.76 | |
Target general information Gen name TK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Thymidine kinase, cytosolic Protein family Thymidine kinase family Biochemical class Kinase Function cytosol, identical protein binding, thymidine kinase activity, zinc ion binding, DNA metabolic process, nucleobase-containing compound metabolic process, protein homotetramerization, pyrimidine nucleoside salvage, thymidine metabolic process Related diseases Seizures, benign familial infantile, 3 (BFIS3) [MIM:607745]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS3 inheritance is autosomal dominant. {ECO:0000269|PubMed:11371648, ECO:0000269|PubMed:12243921, ECO:0000269|PubMed:15048894, ECO:0000269|PubMed:16417554, ECO:0000269|PubMed:17021166, ECO:0000269|PubMed:17386050, ECO:0000269|PubMed:18479388, ECO:0000269|PubMed:20371507, ECO:0000269|PubMed:22612257, ECO:0000269|PubMed:23360469, ECO:0000269|PubMed:23758435, ECO:0000269|PubMed:25982755, ECO:0000269|PubMed:26291284, ECO:0000269|PubMed:29844171, ECO:0000269|PubMed:30144217}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 11 (DEE11) [MIM:613721]: An autosomal dominant seizure disorder characterized by neonatal or infantile onset of refractory seizures with resultant delayed neurologic development and persistent neurologic abnormalities. Patients may progress to West syndrome, which is characterized by tonic spasms with clustering, arrest of psychomotor development, and hypsarrhythmia on EEG. {ECO:0000269|PubMed:19783390, ECO:0000269|PubMed:19786696, ECO:0000269|PubMed:20956790, ECO:0000269|PubMed:22677033, ECO:0000269|PubMed:23033978, ECO:0000269|PubMed:23195492, ECO:0000269|PubMed:23550958, ECO:0000269|PubMed:23662938, ECO:0000269|PubMed:23708187, ECO:0000269|PubMed:23935176, ECO:0000269|PubMed:23988467, ECO:0000269|PubMed:24463883, ECO:0000269|PubMed:24579881, ECO:0000269|PubMed:24659627, ECO:0000269|PubMed:24710820, ECO:0000269|PubMed:25457084, ECO:0000269|PubMed:25459969, ECO:0000269|PubMed:25772804, ECO:0000269|PubMed:25818041, ECO:0000269|PubMed:26138355, ECO:0000269|PubMed:26291284, ECO:0000269|PubMed:26993267, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:29625812, ECO:0000269|PubMed:29844171, ECO:0000269|PubMed:30144217, ECO:0000269|PubMed:30415926}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in SCN2A are associated with genetic epilepsy with febrile seizures plus (GEFS+), a familial autosomal dominant epilepsy syndrome, a clinical subset of febrile seizures, characterized by frequent episodes after 6 years of age and various types of subsequent epilepsy. {ECO:0000269|PubMed:29635106}.; DISEASE: Defects in SCN2A are associated with autism spectrum disorders (ASD). It seems that mutations resulting in sodium channel gain of function and increased neuron excitability lead to infantile seizures, whereas variants resulting in sodium channel loss of function and decrease neuron excitability are associated with ASD. {ECO:0000269|PubMed:28256214}.; DISEASE: Episodic ataxia 9 (EA9) [MIM:618924]: An autosomal dominant neurologic disorder characterized by episodic ataxia manifesting in the first years of life, early-onset seizures, difficulty walking, dizziness, slurred speech, headache, vomiting, and pain. The duration of ataxic episodes is heterogeneous. Most patients show episodes lasting minutes to maximum several hours, but periods lasting days up to weeks have been reported. Some patients have mildly delayed development with speech delay and/or autistic features or mildly impaired intellectual development. {ECO:0000269|PubMed:26645390, ECO:0000269|PubMed:27159988, ECO:0000269|PubMed:27328862, ECO:0000269|PubMed:28065826}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01692; DB04485; DB02452; DB00432; DB00495 Interacts with P05067; A0A087WZT3; Q92993; Q1RN33; P04183 EC number EC 2.7.1.21 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cytoplasm; DNA synthesis; Kinase; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 19373.5 Length 174 Aromaticity 0.09 Instability index 36.21 Isoelectric point 8.63 Charge (pH=7) 3.88 2D Binding mode Binding energy (Kcal/mol) -6.49 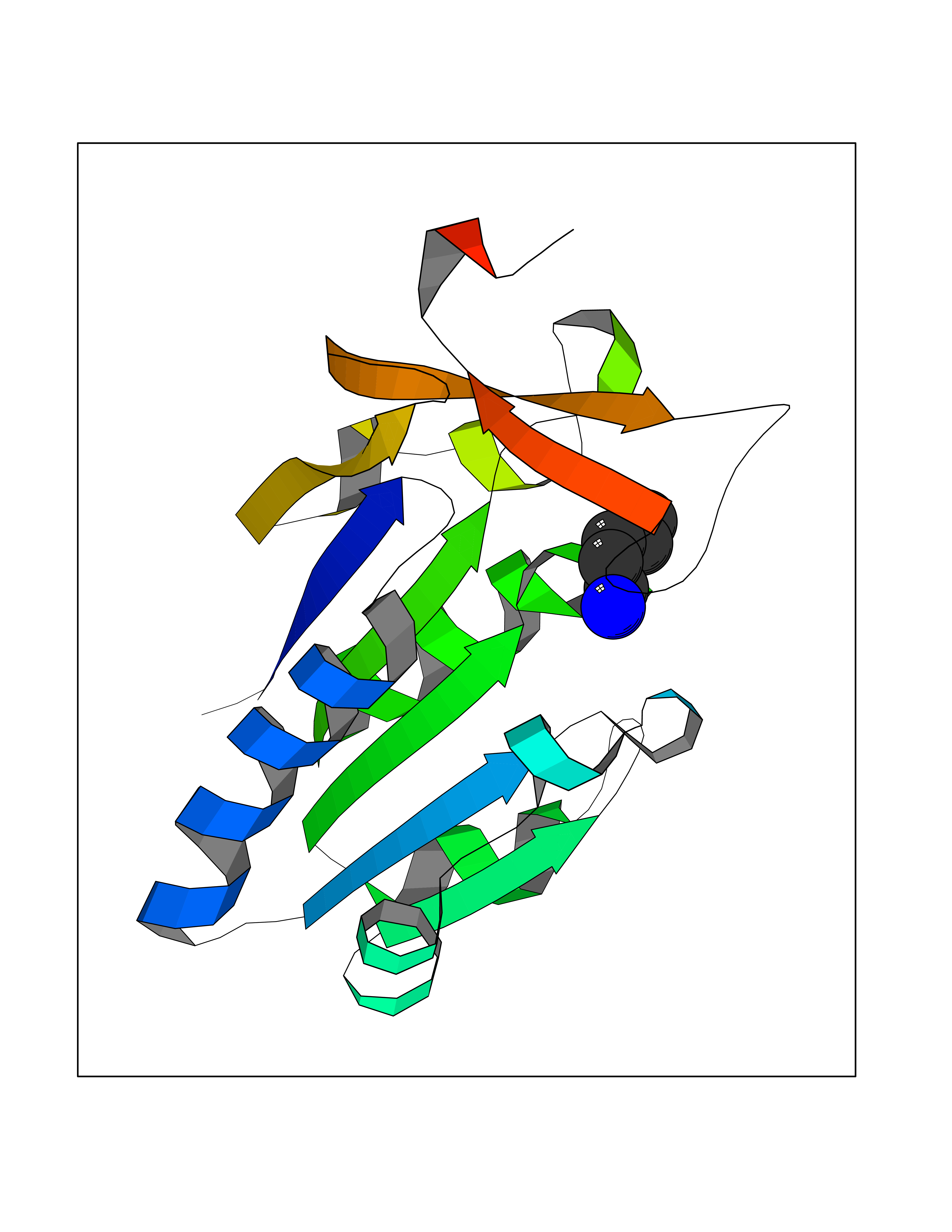 Molscript Map 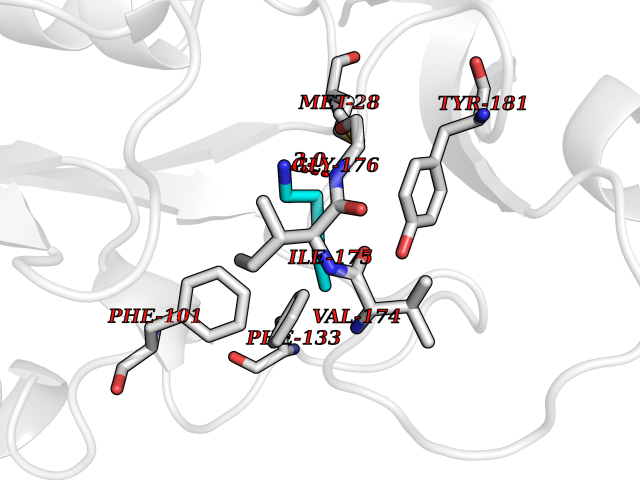 Pymol Map 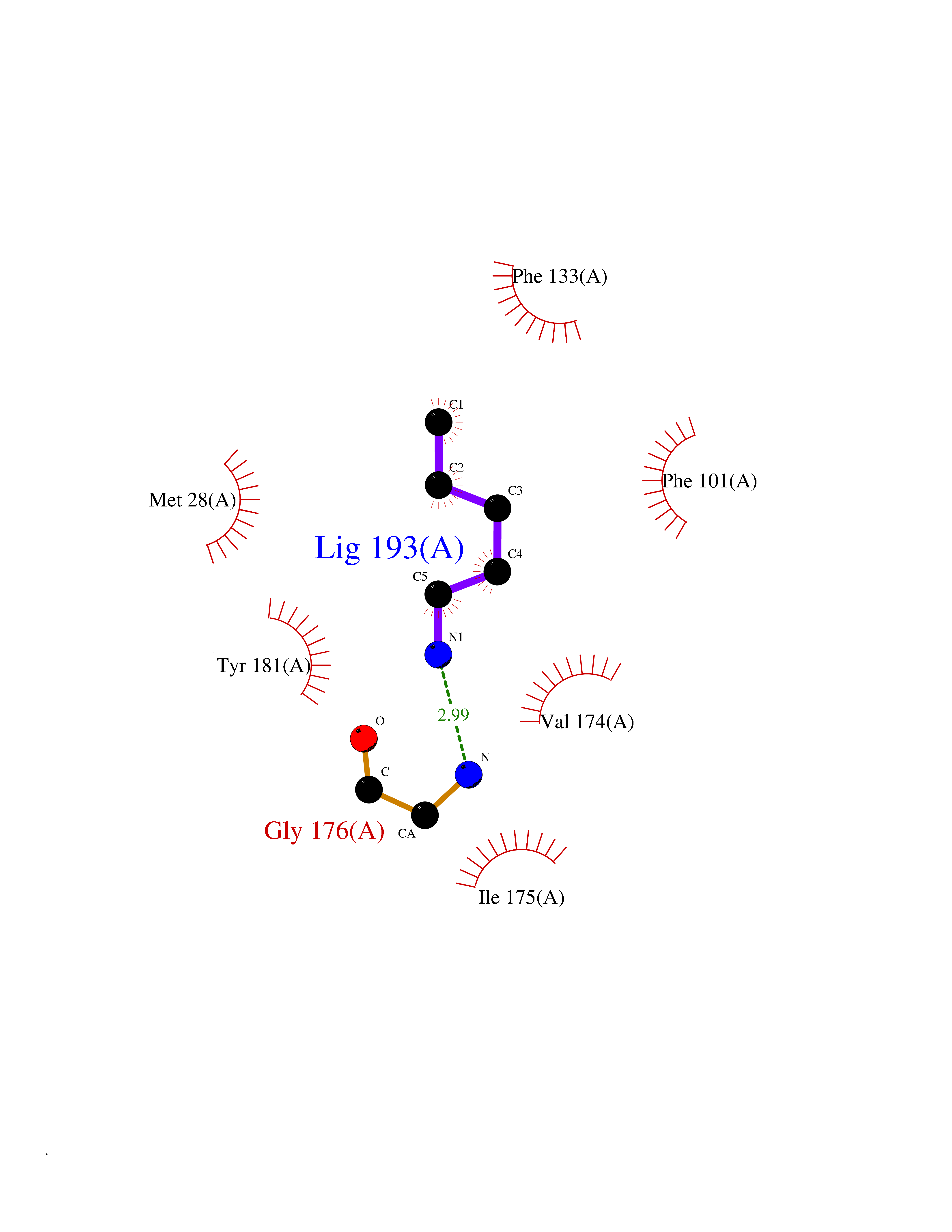 Ligplot Map 3D Binding mode Sequence RGQIQVILGPMFSGKSTELMRRVRRFQIAQYKCLVIKYAKDTRYSSSFCTHDRNTMEALPACLLRDVAQEALGVAVIGIDEGQFFPDIVEFCEAMANAGKTVIVAALDGTFQRKPFGAILNLVPLAESVVKLTAVCMECFREAAYTKRLGTEKEVEVIGGADKYHSVCRLCYFK Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Histone-lysine N-methyltransferase SMYD3 (SMYD3) | 6P7Z | 4.76 | |
Target general information Gen name SMYD3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SET and MYND domain-containing protein 3; Zinc finger MYND domain-containing protein 1 Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family Biochemical class NA Function Histone methyltransferase. Specifically methylates 'Lys-4' of histone H3, inducing di- and tri-methylation, but not monomethylation . Also methylates 'Lys-5' of histone H4. Plays an important role in transcriptional activation as a member of an RNA polymerase complex. Binds DNA containing 5'-CCCTCC-3' or 5'-GAGGGG-3' sequences. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H0L4; Q9H0I2; Q13064; Q7Z3B4; Q16512; Q92529; Q15915; Q9Y2U5 EC number EC 2.1.1.354 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Chromatin regulator; Cytoplasm; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 48694.1 Length 425 Aromaticity 0.08 Instability index 44.91 Isoelectric point 6.86 Charge (pH=7) -0.4 2D Binding mode Binding energy (Kcal/mol) -6.49 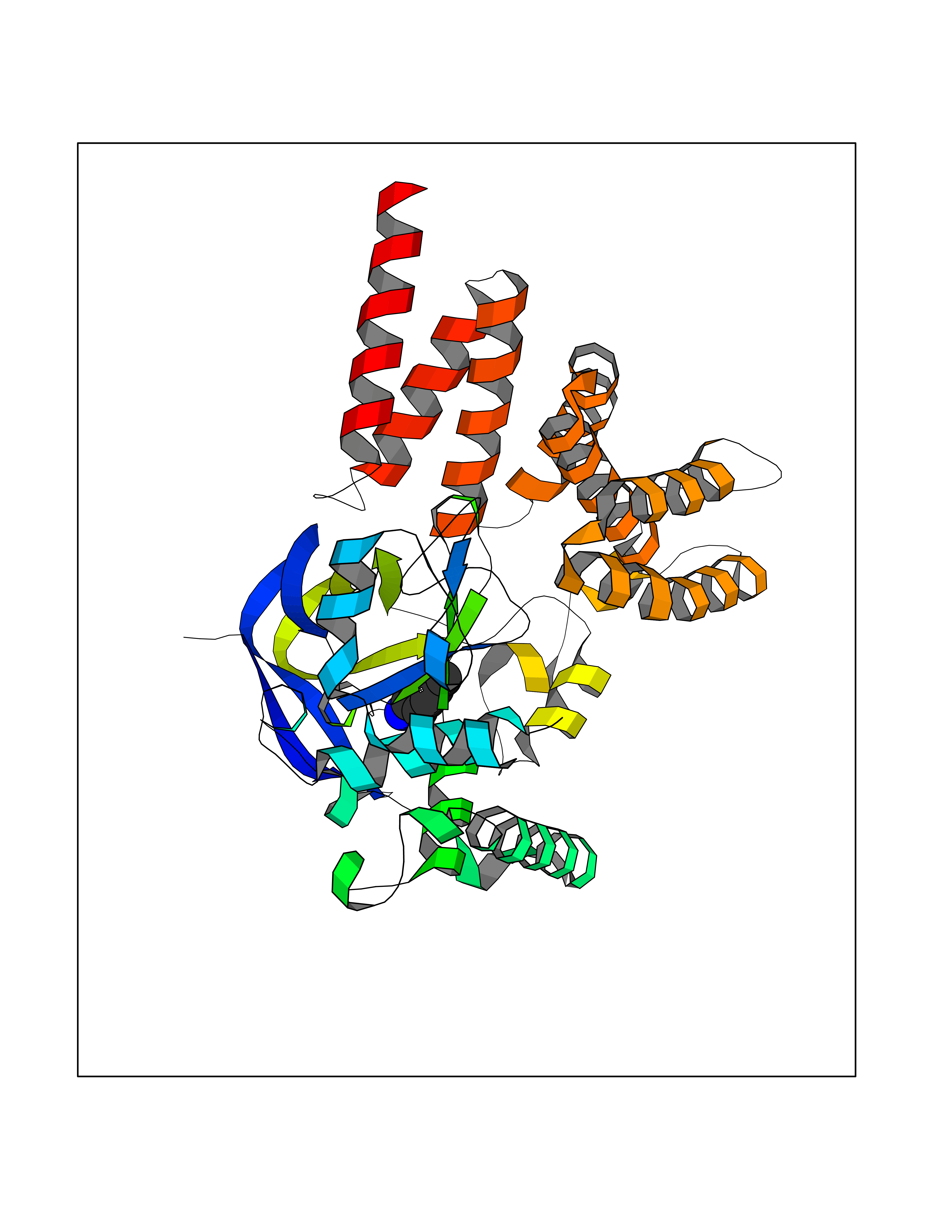 Molscript Map 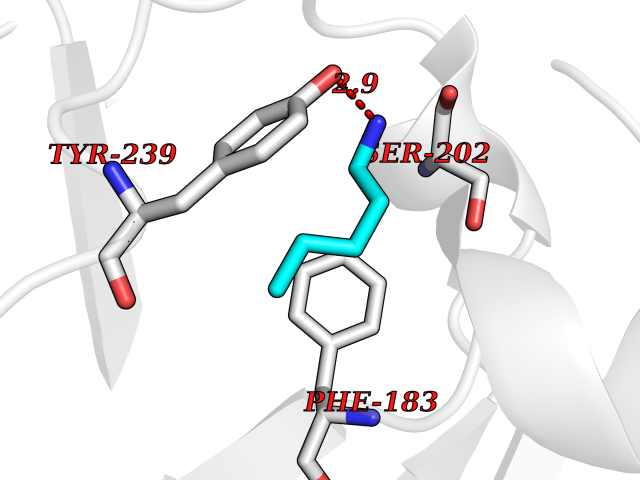 Pymol Map 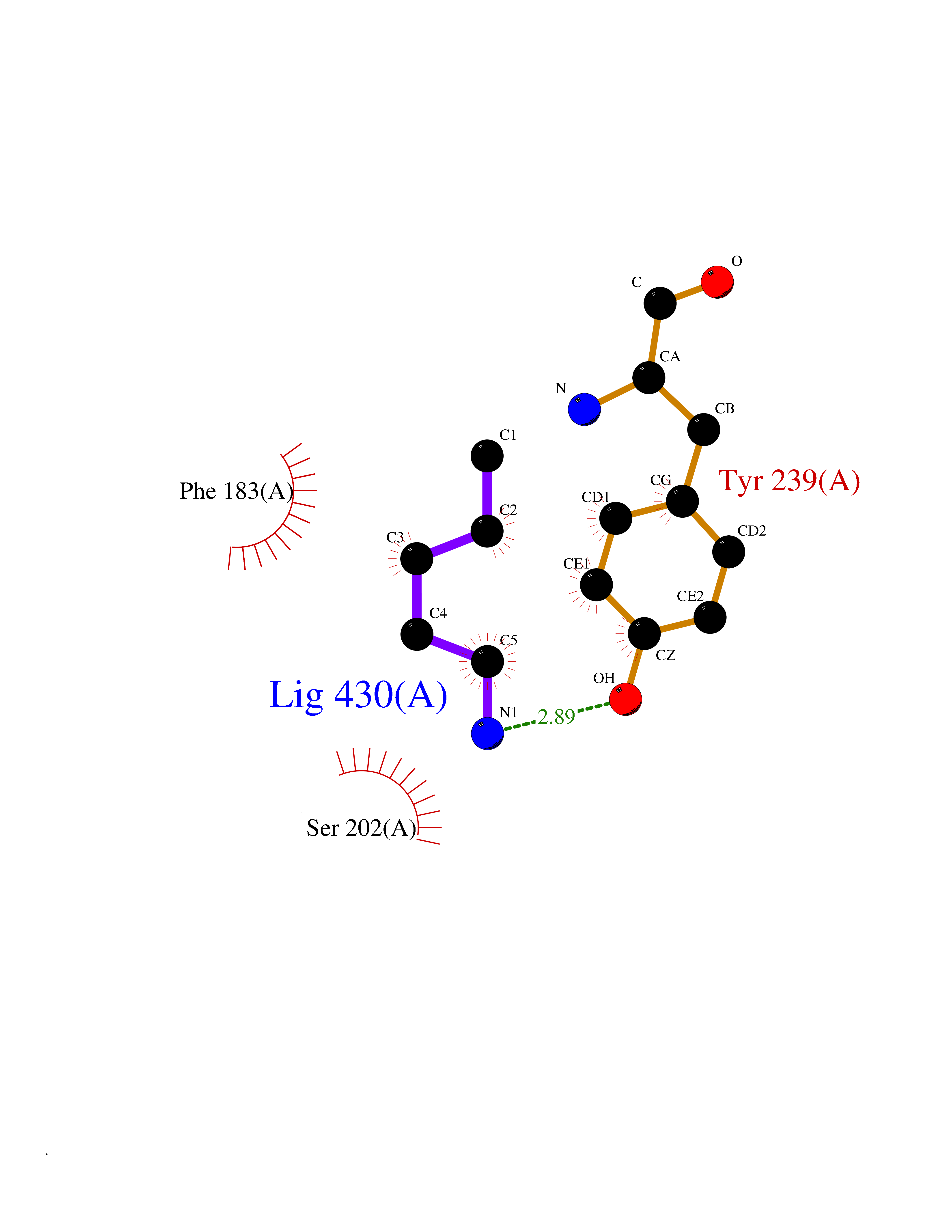 Ligplot Map 3D Binding mode Sequence PLKVEKFATANRGNGLRAVTPLRPGELLFRSDPLAYTVCKGSRGVVCDRCLLGKEKLMRCSQCRVAKYCSAKCQKKAWPDHKRECKCLKSCPRYPPDSVRLLGRVVFKLMDGAPSESEKLYSFYDLESNINKLTEDKKEGLRQLVMTFQHFMREEIQDASQLPPAFDLFEAFAKVICNSFTICNAEMQEVGVGLYPSISLLNHSCDPNCSIVFNGPHLLLRAVRDIEVGEELTICYLDMLMTSEERRKQLRDQYCFECDCFRCQTQDKDADMLTGDEQVWKEVQESLKKIEELKAHWKWEQVLAMCQAIISSNSERLPDINIYQLKVLDCAMDACINLGLLEEALFYGTRTMEPYRIFFPGSHPVRGVQVMKVGKLQLHQGMFPQAMKNLRLAFDIMRVTHGREHSLIEDLILLLEECDANIRAS Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Zinc finger-containing ubiquitin peptidase 1 (ZUP1) | 6EI1 | 4.76 | |
Target general information Gen name ZUP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Zinc finger with UFM1-specific peptidase domain protein; ZUFSP; Lys-63-specific deubiquitinase ZUFSP; DUB; C6orf113 Protein family Peptidase C78 family, ZUFSP subfamily Biochemical class Peptidase Function Shows only weak activity against 'Lys-11' and 'Lys-48'-linked chains. Plays an important role in genome stability pathways, functioning to prevent spontaneous DNA damage and also promote cellular survival in response to exogenous DNA damage. Modulates the ubiquitination status of replication protein A (RPA) complex proteins in response to replication stress. Deubiquitinase with endodeubiquitinase activity that specifically interacts with and cleaves 'Lys-63'-linked long polyubiquitin chains. Related diseases WHIM syndrome 2 (WHIMS2) [MIM:619407]: An autosomal recessive form of WHIM syndrome, a primary immunodeficiency disorder characterized by warts, hypogammaglobulinemia, infections, and myelokathexis. Myelokathexis is a unique form of non-cyclic severe congenital neutropenia caused by accumulation of mature and degenerating neutrophils in the bone marrow. Monocytopenia and lymphopenia, especially B lymphopenia, also commonly occur. There is significant phenotypic variation among patients, such that some individuals may have an incomplete form of the disorder in which one or more of the classic tetrad features are not present. {ECO:0000269|PubMed:24777453}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q92619; P50281; Q8WVC2 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Hydrolase; Metal-binding; Nucleus; Proteomics identification; Reference proteome; Repeat; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 46930.3 Length 410 Aromaticity 0.07 Instability index 58.67 Isoelectric point 9 Charge (pH=7) 9.7 2D Binding mode Binding energy (Kcal/mol) -6.49  Molscript Map 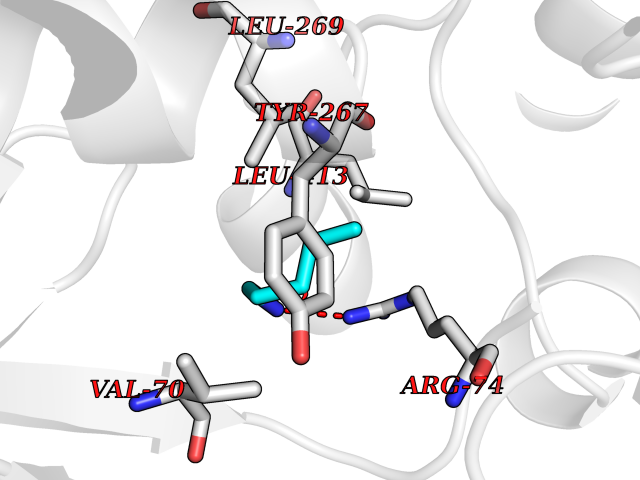 Pymol Map 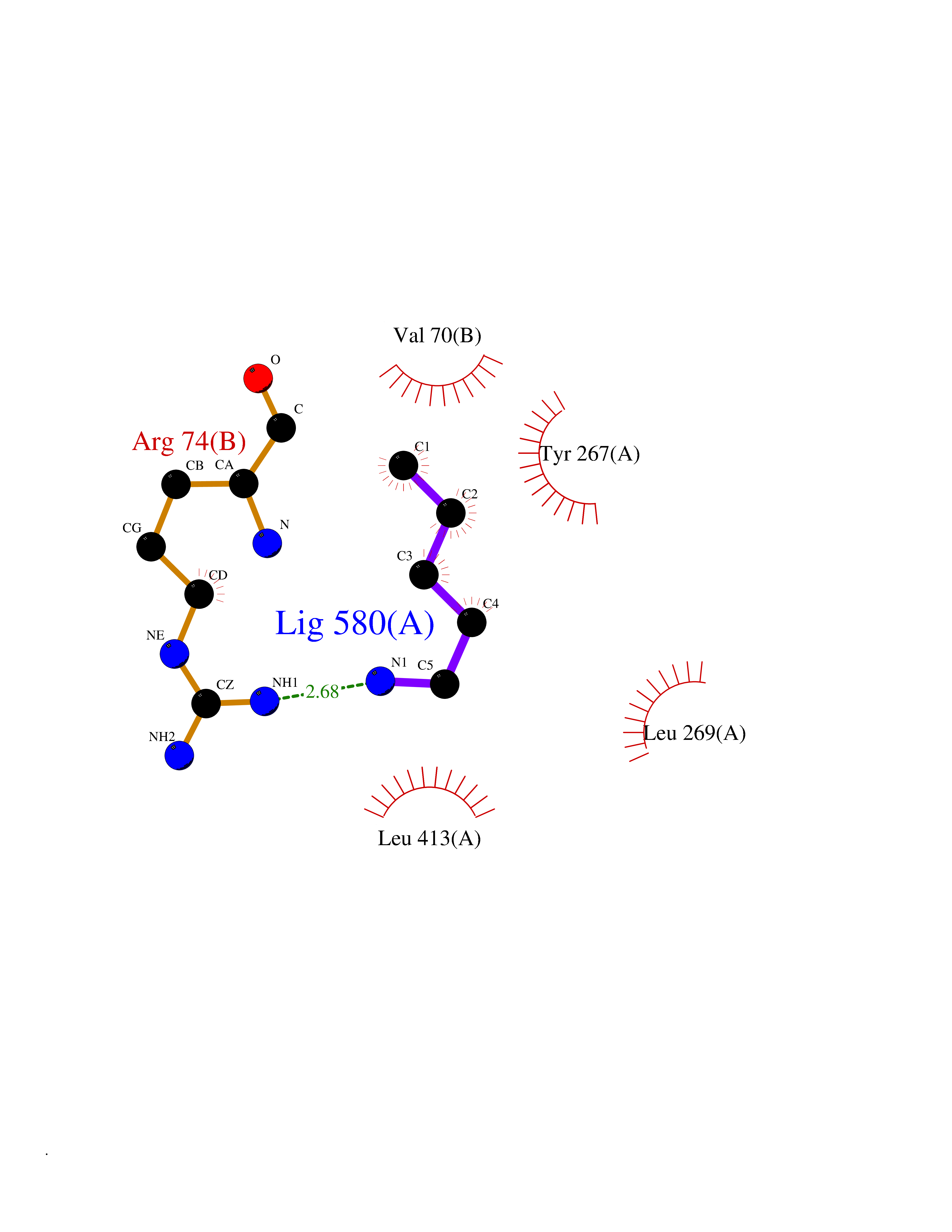 Ligplot Map 3D Binding mode Sequence LQQEEDRKRRSEESRQEIEEFQKLQRQYGLDNSGGYKQQQLRNMEIEVNRGRMPPSEFHRRKADMMESLALGFDDGKTKTSGIIEALHRYYQNAATDVRRVWLSSVVDHFHSSLGDKGWGCGYRNFQMLLSSLLQNDAYNDCLKGMLIPCIPKIQSMIEDAWKEGFDPQGASQLNNRLQGTKAWIGACEVYILLTSLRVKCHIVDFHKSTGPLGTHPRLFEWILNYYSSSPKVVCTSKPPIYLQHQGHSRTVIGIEEKKNRTLCLLILDPGCPSREMQKLLKQDIEASSLKQLRKSMGNLKHKQYQILAVEGALSLEEKLARRQASQVFTAEKIPMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRG Hydrogen bonds contact Hydrophobic contact | ||||