Job Results:
Ligand
Structure
Job ID
5c75d0793a5c7fa3ef7fd4eba45db829
Job name
NA
Time
2026-02-27 11:58:42
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Angiopoietin-related protein 4 (ANGPTL4) | 6U73 | 5.06 | |
Target general information Gen name ANGPTL4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ171/PRO197; PSEC0166; PP1158; PGAR; Hepatic fibrinogen/angiopoietin-related protein; HFARP; Angiopoietin-like protein PP1158; Angiopoietin-like protein 4; Angiopoietin-like 4; ARP4 Protein family NA Biochemical class Fibrinogen protein Function May also play a role in regulating glucose homeostasis and insulin sensitivity. Inhibits proliferation, migration, and tubule formation of endothelial cells and reduces vascular leakage. Upon heterologous expression, inhibits the adhesion of endothelial cell to the extracellular matrix (ECM), and inhibits the reorganization of the actin cytoskeleton, formation of actin stress fibers and focal adhesions in endothelial cells that have adhered to ANGPTL4-containing ECM (in vitro). Depending on context, may modulate tumor-related angiogenesis. Mediates inactivation of the lipoprotein lipase LPL, and thereby plays a role in the regulation of triglyceride clearance from the blood serum and in lipid metabolism. Related diseases Mucopolysaccharidosis 1H (MPS1H) [MIM:607014]: A severe form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1H usually present, within the first year of life, a combination of hepatosplenomegaly, skeletal deformities, corneal clouding and severe intellectual disability. Obstructive airways disease, respiratory infection and cardiac complications usually result in death before 10 years of age. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:1301941, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:24036510, ECO:0000269|PubMed:31194252, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:7951228, ECO:0000269|PubMed:8019563, ECO:0000269|PubMed:8328452, ECO:0000269|PubMed:8401515, ECO:0000269|Ref.20}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1H/S (MPS1H/S) [MIM:607015]: A form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. MPS1H/S represents an intermediate phenotype of the MPS1 clinical spectrum. It is characterized by relatively little neurological involvement, but most of the somatic symptoms described for severe MPS1 develop in the early to mid-teens, causing considerable loss of mobility. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8401515}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1S (MPS1S) [MIM:607016]: A mild form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1S may have little or no neurological involvement, normal stature and life span, but present development of joints stiffness, mild hepatosplenomegaly, aortic valve disease and corneal clouding. {ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:25256405, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8213840}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9BY76; P05556; P18084 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; Coiled coil; Direct protein sequencing; Disulfide bond; Extracellular matrix; Glycoprotein; Lipid metabolism; Proteomics identification; Reference proteome; Secreted; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 24429.1 Length 216 Aromaticity 0.12 Instability index 40.6 Isoelectric point 8.51 Charge (pH=7) 2.46 2D Binding mode Binding energy (Kcal/mol) -6.9 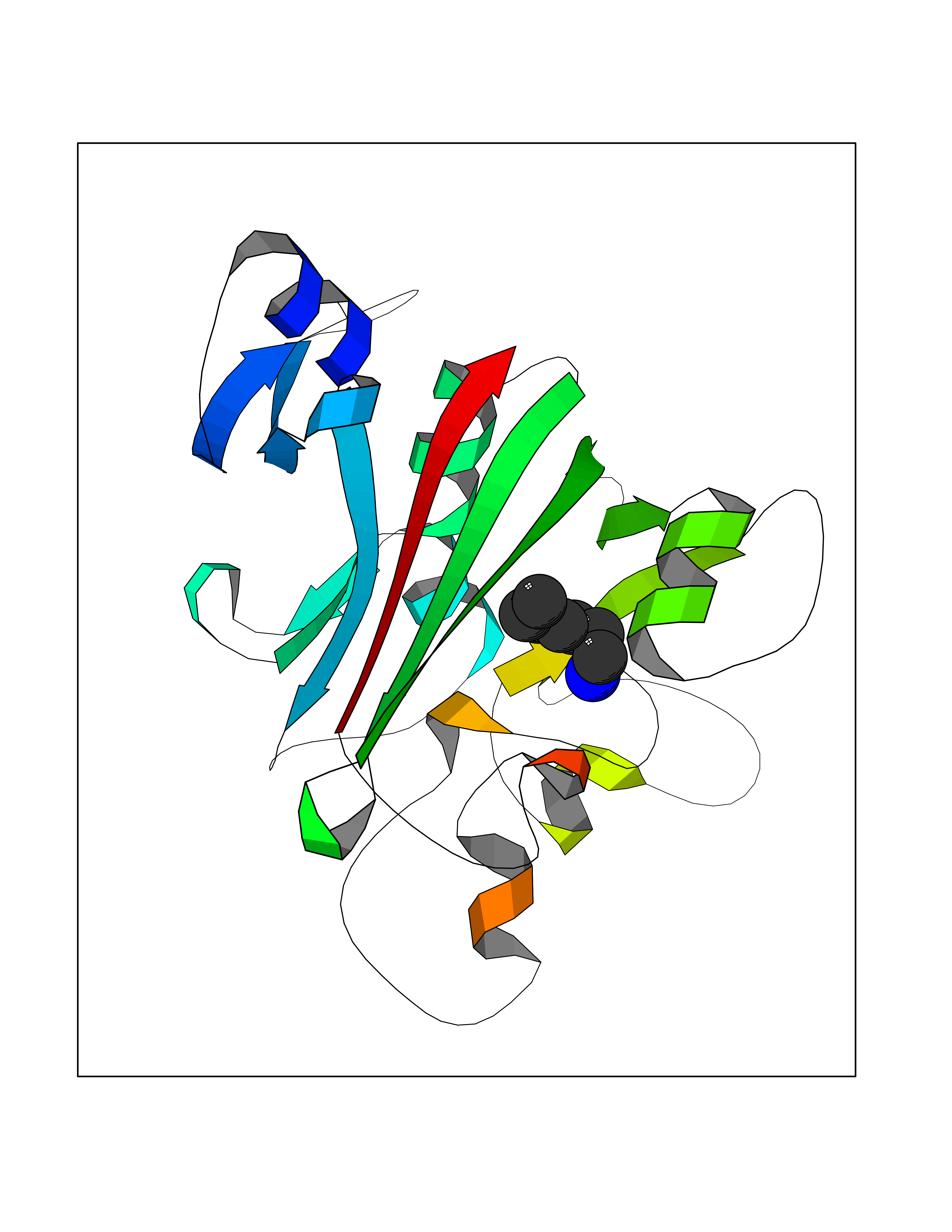 Molscript Map 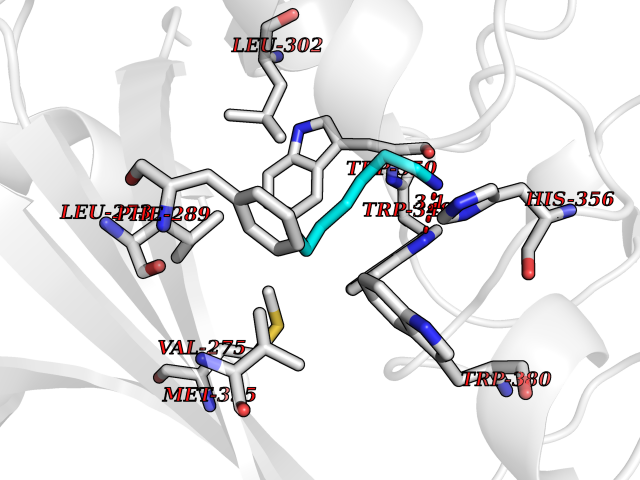 Pymol Map 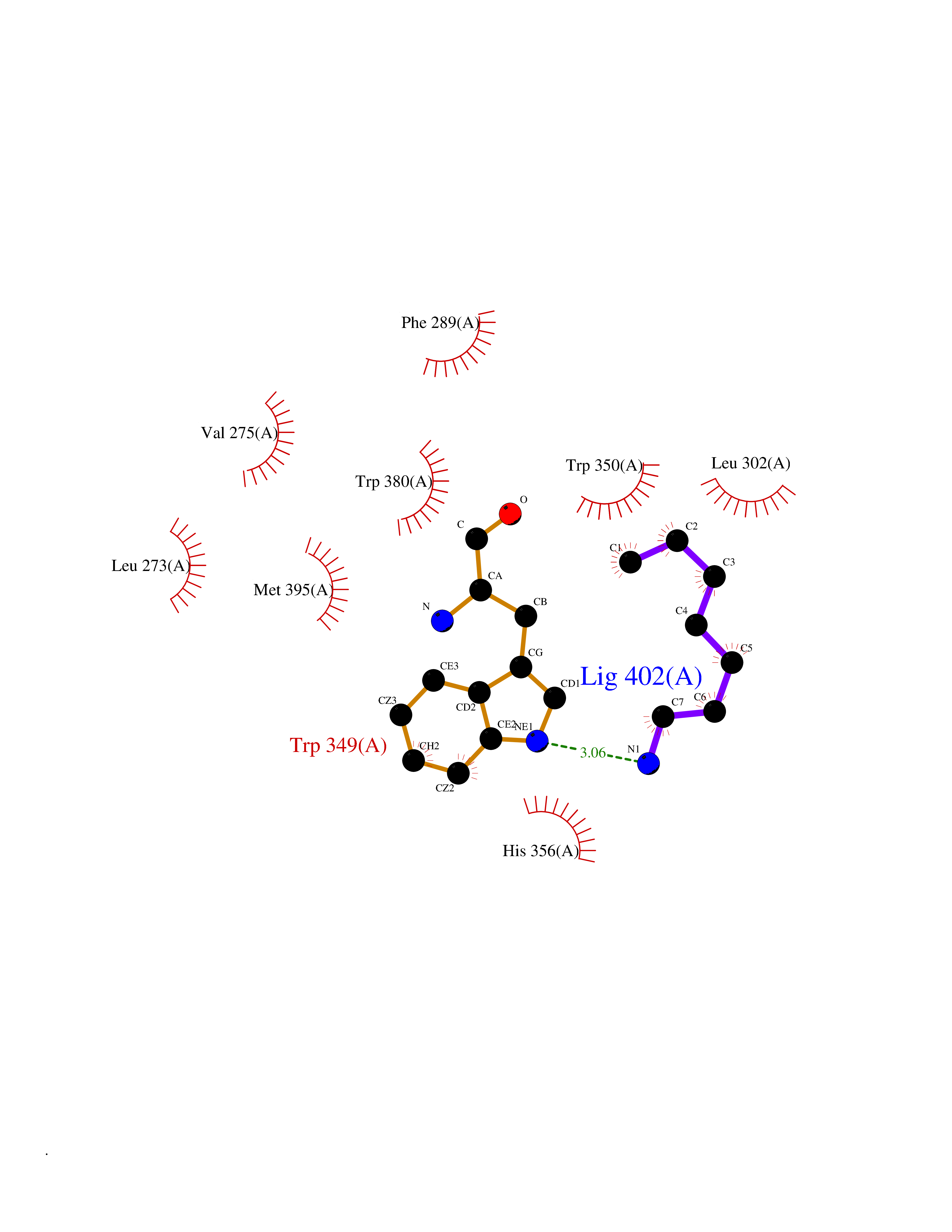 Ligplot Map 3D Binding mode Sequence PRDCQELFQVGERQSGLFEIQPQGSPPFLVNCKMTSDGGWTVIQRRHDGSVDFNRPWEAYKAGFGDPHGEFWLGLEKVHSITGDRNSRLAVQLRDWDGNAELLQFSVHLGGEDTAYSLQLTAPVAGQLGATTVPPSGLSVPFSTWDQDHDLRRDKNCAKSLSGGWWFGTCSHSNLNGQYFRSIPQQRQKLKKGIFWKTWRGRYYPLQATTMLIQPM Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Cytochrome P450 1A2 | 2HI4 | 5.05 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -6.89 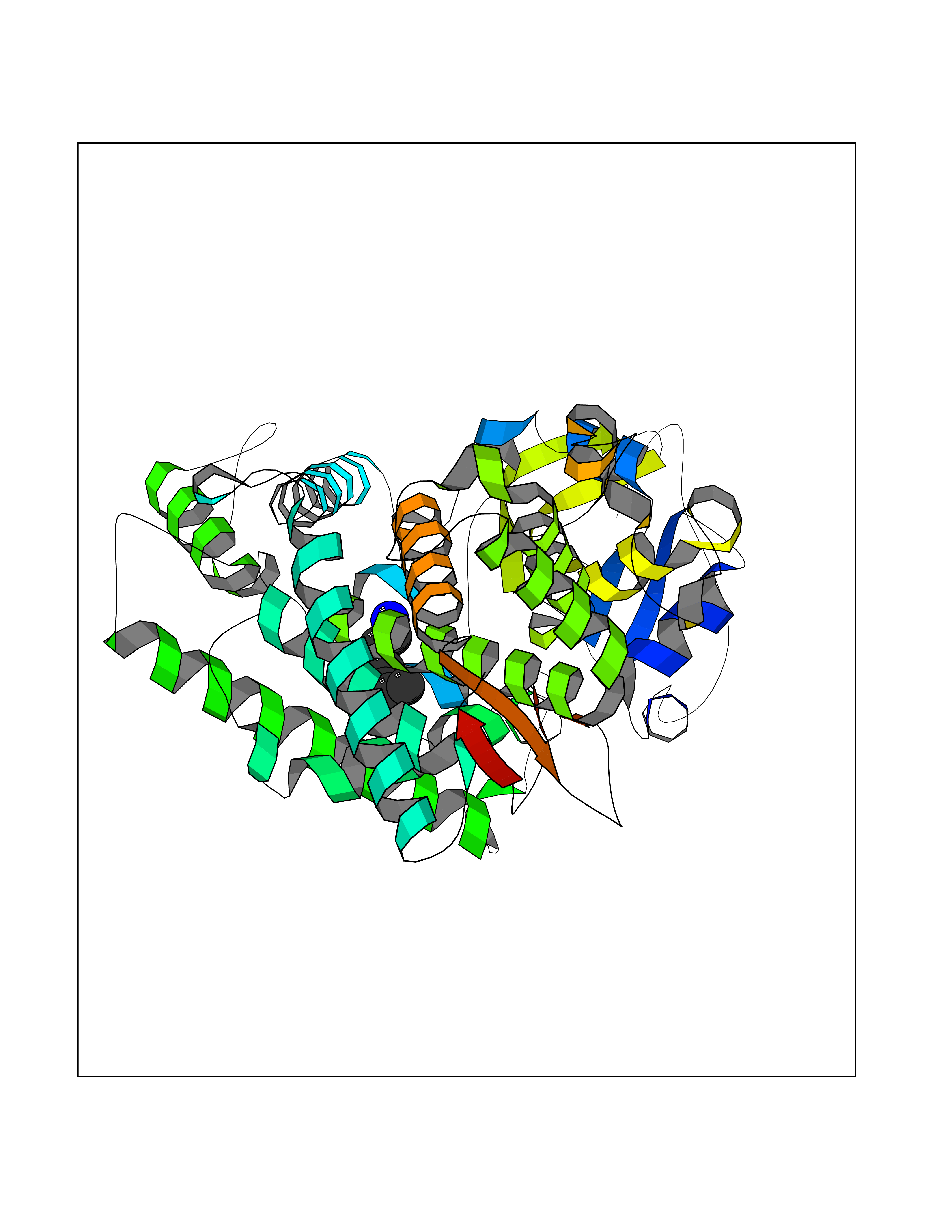 Molscript Map 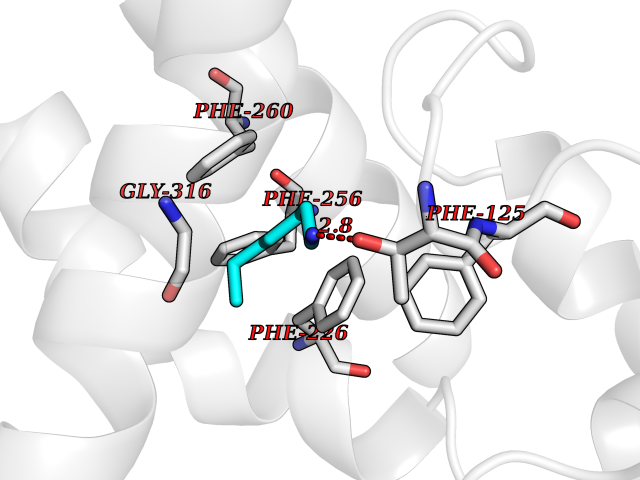 Pymol Map 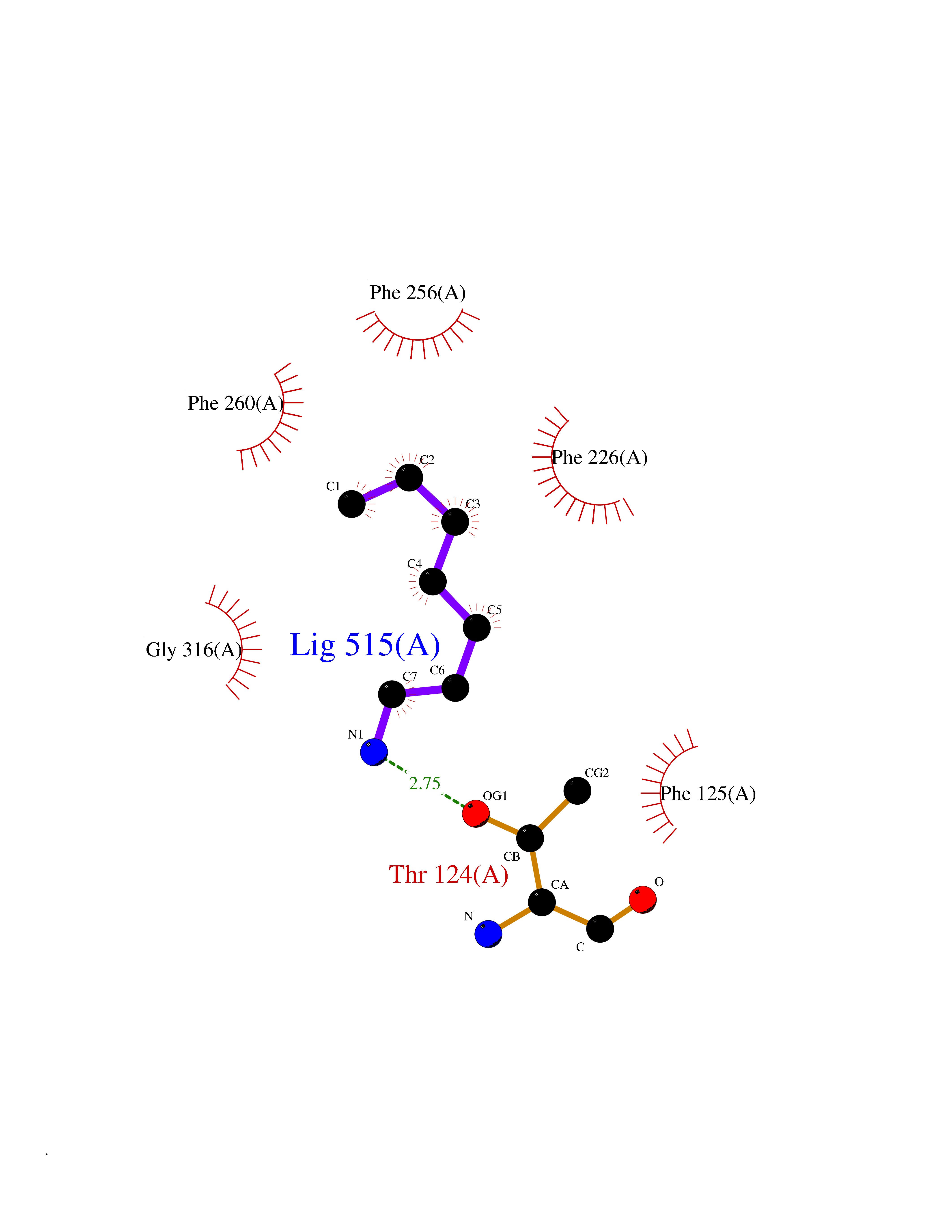 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | 5,10-methylenetetrahydrofolate reductase | 3FST | 5.05 | |
Target general information Gen name metF Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3941;JW3913 Protein family Methylenetetrahydrofolate reductase family Biochemical class Oxidoreductase Function FAD binding.Methylenetetrahydrofolate reductase (NAD(P)H) activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.5.1.54 Uniprot keywords 3D-structure; Amino-acid biosynthesis; FAD; Flavoprotein; Methionine biosynthesis; NAD; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,C,E Molecular weight (Da) 30855.9 Length 274 Aromaticity 0.09 Instability index 27.54 Isoelectric point 5.84 Charge (pH=7) -4.61 2D Binding mode Binding energy (Kcal/mol) -6.89 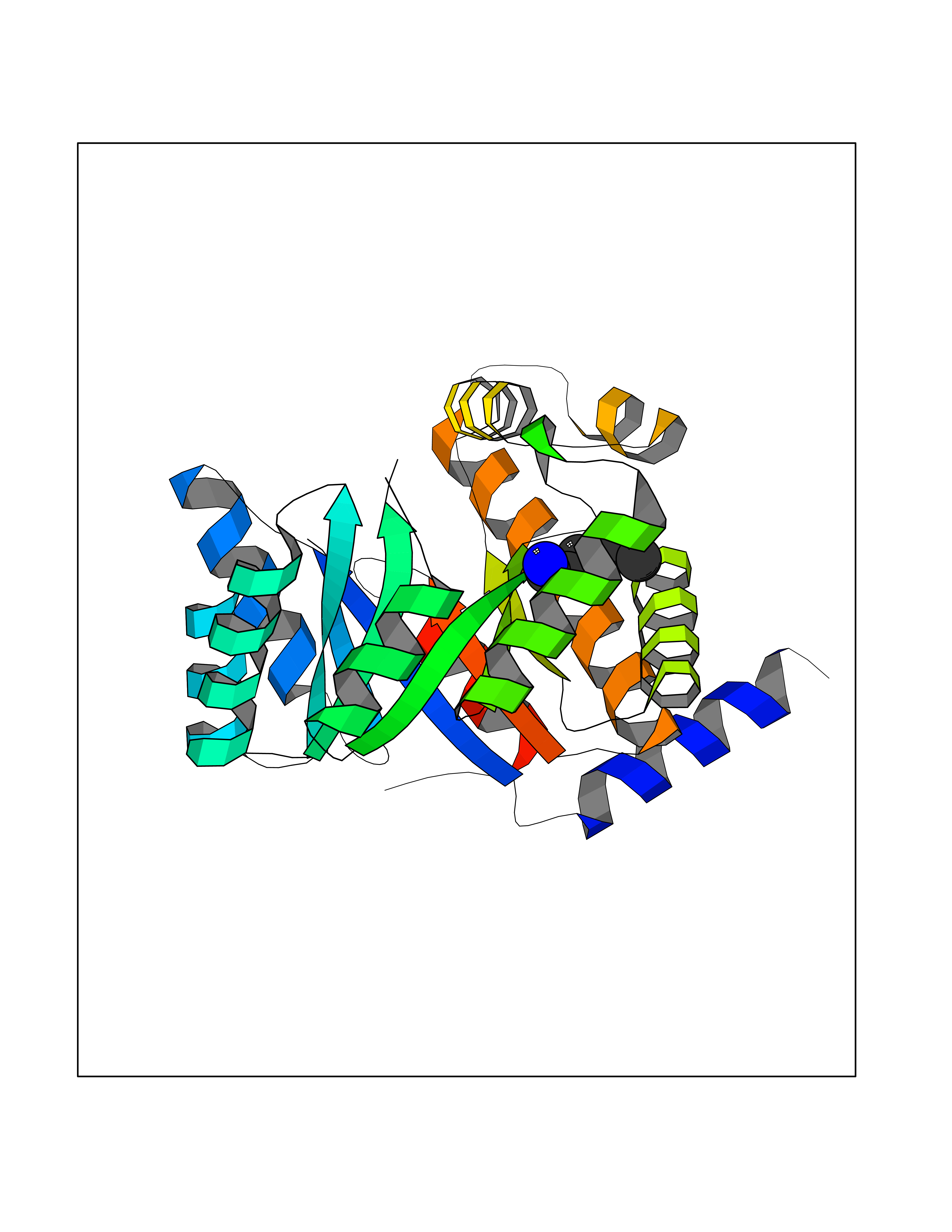 Molscript Map 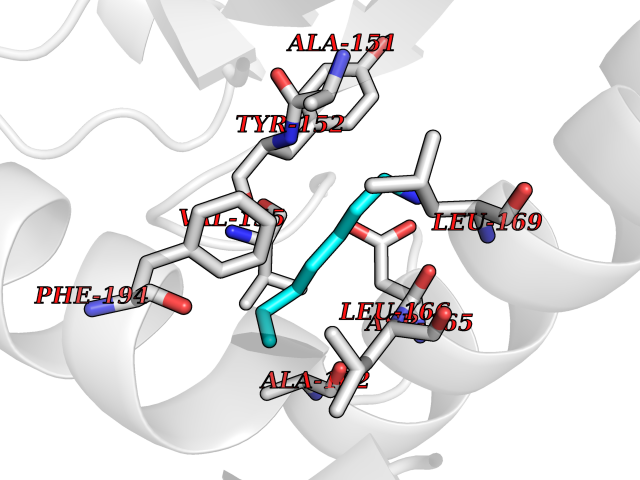 Pymol Map 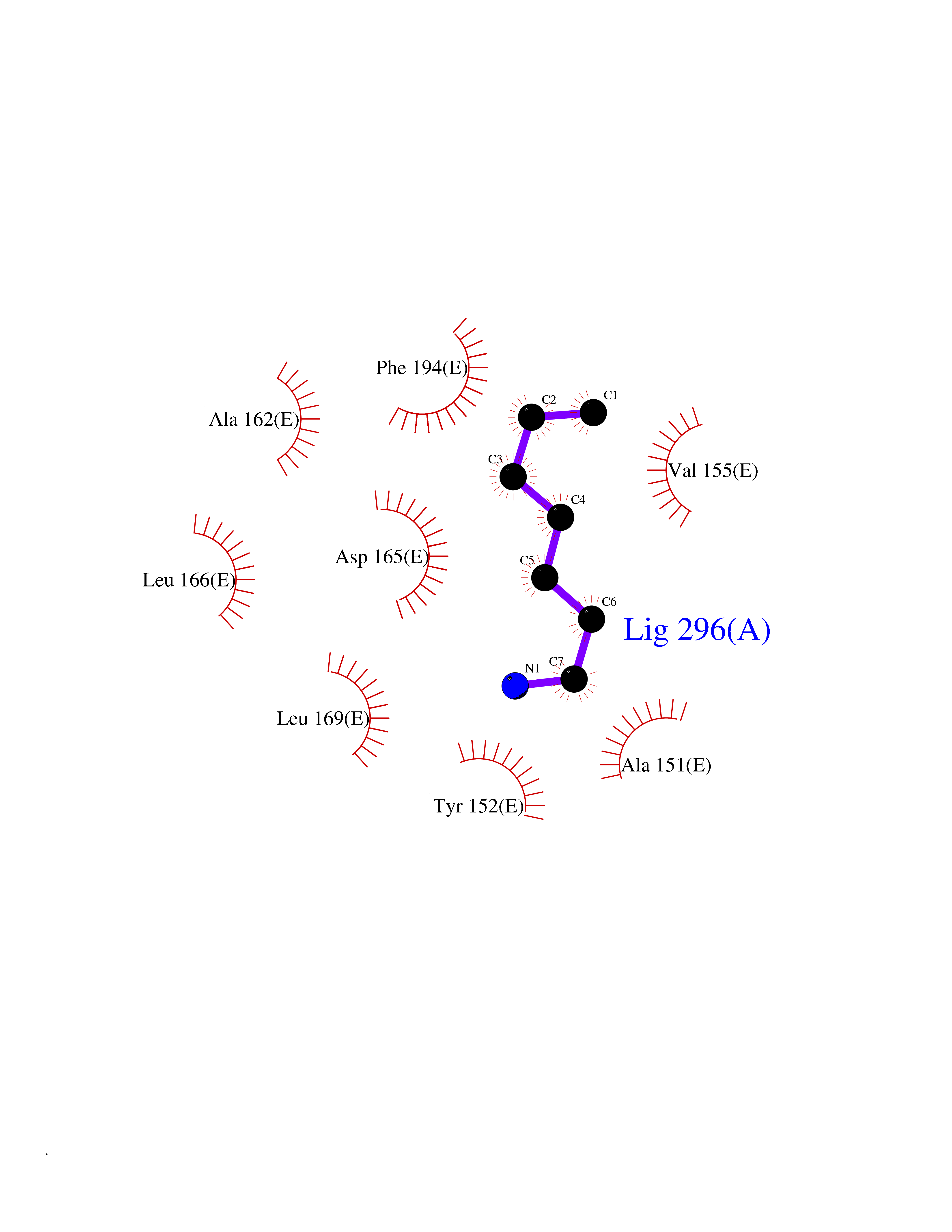 Ligplot Map 3D Binding mode Sequence FHASQRDALNQSLAEVQGQINVSFEFFPPRTSEMEQTLWNSIDRLSSLKPKFVSVTYTHSIIKGIKDRTGLEAAPHLTCIDATPDELRTIARDYWNNGIRHIVALRGDEMYASDLVTLLKEVADFDISVAAYPEVHPEAKSAQADLLNLKRKVDAGANRAITQFFFDVESYLRFRDRCVSAGIDVEIIPGILPVSNFKQAKKLADMTNVRIPAWMAQMFDGLDDDAETRKLVGANIAMDMVKILSREGVKDFHFYTLNRAEMSYAICHTLGVRP Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Retinoic acid receptor gamma (RARG) | 1FCY | 5.05 | |
Target general information Gen name RARG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-gamma; Nuclear receptor subfamily 1 group B member 3; NR1B3 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Receptor for retinoic acid. Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. Required for limb bud development. In concert with RARA or RARB, required for skeletal growth, matrix homeostasis and growth plate function (By similarity). Related diseases Cystic fibrosis (CF) [MIM:219700]: A common generalized disorder of the exocrine glands which impairs clearance of secretions in a variety of organs. It is characterized by the triad of chronic bronchopulmonary disease (with recurrent respiratory infections), pancreatic insufficiency (which leads to malabsorption and growth retardation) and elevated sweat electrolytes. It is the most common genetic disease in Caucasians, with a prevalence of about 1 in 2'000 live births. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10094564, ECO:0000269|PubMed:10869121, ECO:0000269|PubMed:10923036, ECO:0000269|PubMed:11242048, ECO:0000269|PubMed:12167682, ECO:0000269|PubMed:12394343, ECO:0000269|PubMed:12529365, ECO:0000269|PubMed:1284466, ECO:0000269|PubMed:1284468, ECO:0000269|PubMed:1284529, ECO:0000269|PubMed:1284530, ECO:0000269|PubMed:1284548, ECO:0000269|PubMed:1379210, ECO:0000269|PubMed:15528182, ECO:0000269|PubMed:15716351, ECO:0000269|PubMed:16822950, ECO:0000269|PubMed:1695717, ECO:0000269|PubMed:1699669, ECO:0000269|PubMed:17098864, ECO:0000269|PubMed:1710600, ECO:0000269|PubMed:1712898, ECO:0000269|PubMed:17182731, ECO:0000269|PubMed:20008117, ECO:0000269|PubMed:20150177, ECO:0000269|PubMed:20691141, ECO:0000269|PubMed:21884936, ECO:0000269|PubMed:2236053, ECO:0000269|PubMed:23818989, ECO:0000269|PubMed:25330774, ECO:0000269|PubMed:26846474, ECO:0000269|PubMed:27241308, ECO:0000269|PubMed:28001373, ECO:0000269|PubMed:28067262, ECO:0000269|PubMed:28087700, ECO:0000269|PubMed:32026723, ECO:0000269|PubMed:33572515, ECO:0000269|PubMed:7504969, ECO:0000269|PubMed:7505694, ECO:0000269|PubMed:7505767, ECO:0000269|PubMed:7508414, ECO:0000269|PubMed:7513296, ECO:0000269|PubMed:7517264, ECO:0000269|PubMed:7520022, ECO:0000269|PubMed:7522211, ECO:0000269|PubMed:7524909, ECO:0000269|PubMed:7524913, ECO:0000269|PubMed:7525450, ECO:0000269|PubMed:7537150, ECO:0000269|PubMed:7541273, ECO:0000269|PubMed:7541510, ECO:0000269|PubMed:7543567, ECO:0000269|PubMed:7544319, ECO:0000269|PubMed:7581407, ECO:0000269|PubMed:7606851, ECO:0000269|PubMed:7680525, ECO:0000269|PubMed:7683628, ECO:0000269|PubMed:7683954, ECO:0000269|PubMed:8081395, ECO:0000269|PubMed:8406518, ECO:0000269|PubMed:8522333, ECO:0000269|PubMed:8723693, ECO:0000269|PubMed:8723695, ECO:0000269|PubMed:8800923, ECO:0000269|PubMed:8829633, ECO:0000269|PubMed:8910473, ECO:0000269|PubMed:8956039, ECO:0000269|PubMed:9101301, ECO:0000269|PubMed:9222768, ECO:0000269|PubMed:9375855, ECO:0000269|PubMed:9401006, ECO:0000269|PubMed:9443874, ECO:0000269|PubMed:9452048, ECO:0000269|PubMed:9452054, ECO:0000269|PubMed:9452073, ECO:0000269|PubMed:9482579, ECO:0000269|PubMed:9507391, ECO:0000269|PubMed:9521595, ECO:0000269|PubMed:9554753, ECO:0000269|PubMed:9736778, ECO:0000269|PubMed:9804160, ECO:0000269|PubMed:9921909}. The disease is caused by variants affecting the gene represented in this entry. There is some evidence that the functional defect caused by the most common variant Phe-508 DEL can be corrected by the binding to the snake phospholipase A2 crotoxin basic subunit CB. This toxin both disrupts the Phe-508 DEL-cytokeratin 8 complex, allowing for the escape from degradation, and increases the chloride channel current (PubMed:27241308). {ECO:0000269|PubMed:27241308}.; DISEASE: Congenital bilateral absence of the vas deferens (CBAVD) [MIM:277180]: An autosomal recessive disease characterized by vas deferens aplasia resulting in azoospermia and male infertility. CBAVD may occur in isolation or as a manifestation of cystic fibrosis. {ECO:0000269|PubMed:10066035, ECO:0000269|PubMed:10651488, ECO:0000269|PubMed:17329263, ECO:0000269|PubMed:7529962, ECO:0000269|PubMed:7539342, ECO:0000269|PubMed:9067761, ECO:0000269|PubMed:9736778, ECO:0000269|Ref.117}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07294; DB07031; DB00459; DB00210; DB00523; DB02466; DB03466; DB02741; DB03279; DB00926; DB00982; DB05785; DB05467; DB02258; DB00799; DB00755; DB12808 Interacts with Q96RK4; P13349; P31321; P28702; P48443; O60504-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 26574.9 Length 236 Aromaticity 0.06 Instability index 49.98 Isoelectric point 5.76 Charge (pH=7) -2.95 2D Binding mode Binding energy (Kcal/mol) -6.89 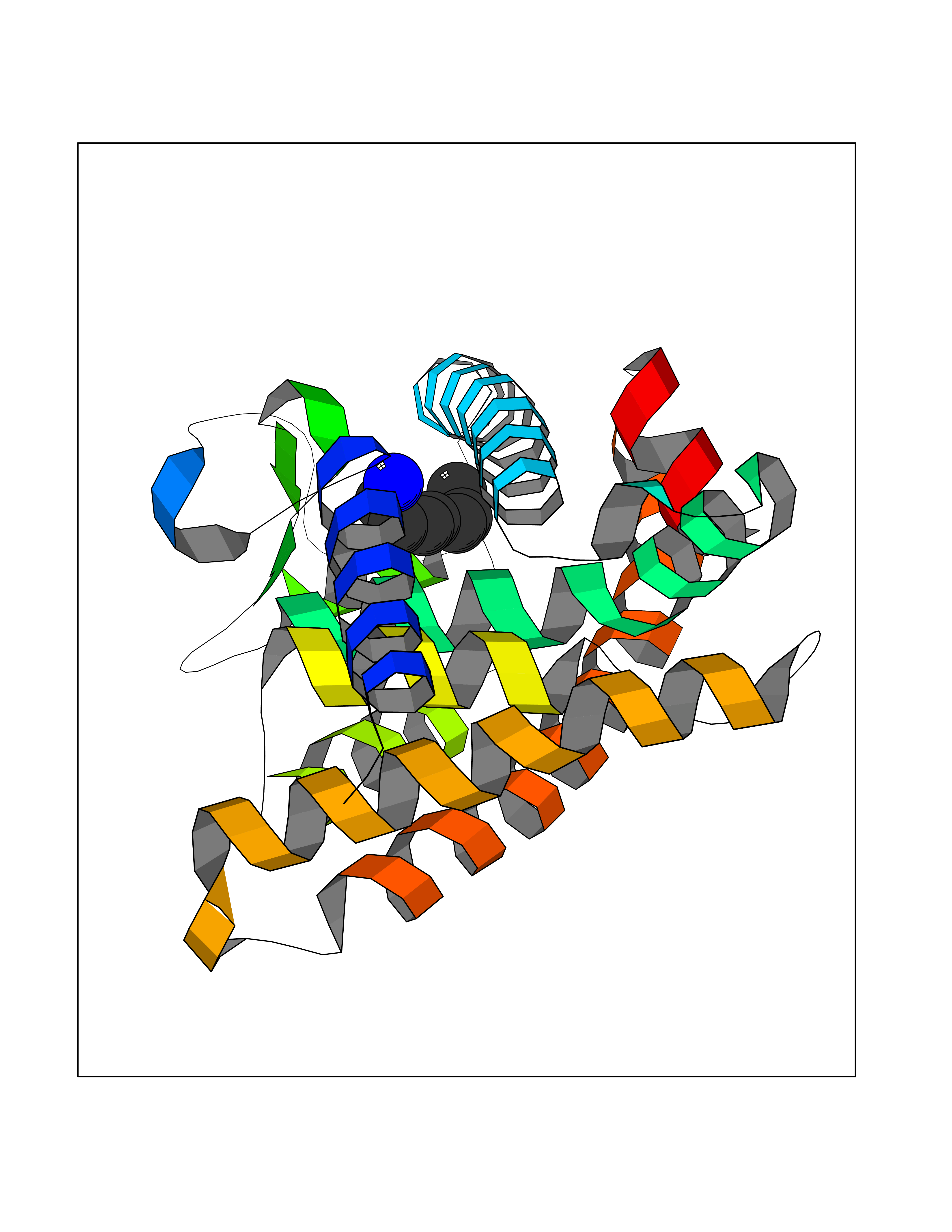 Molscript Map 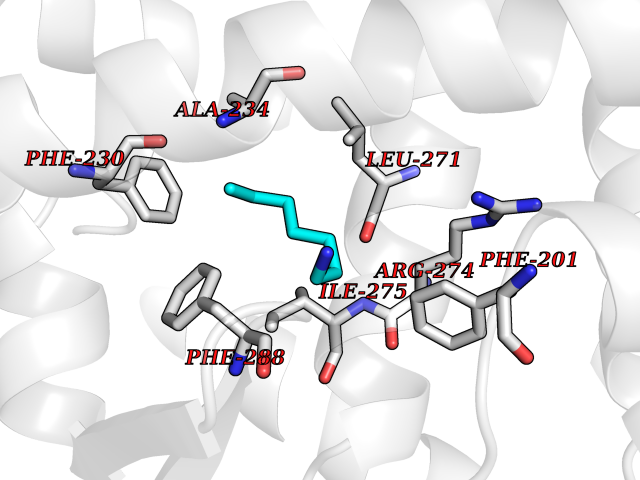 Pymol Map 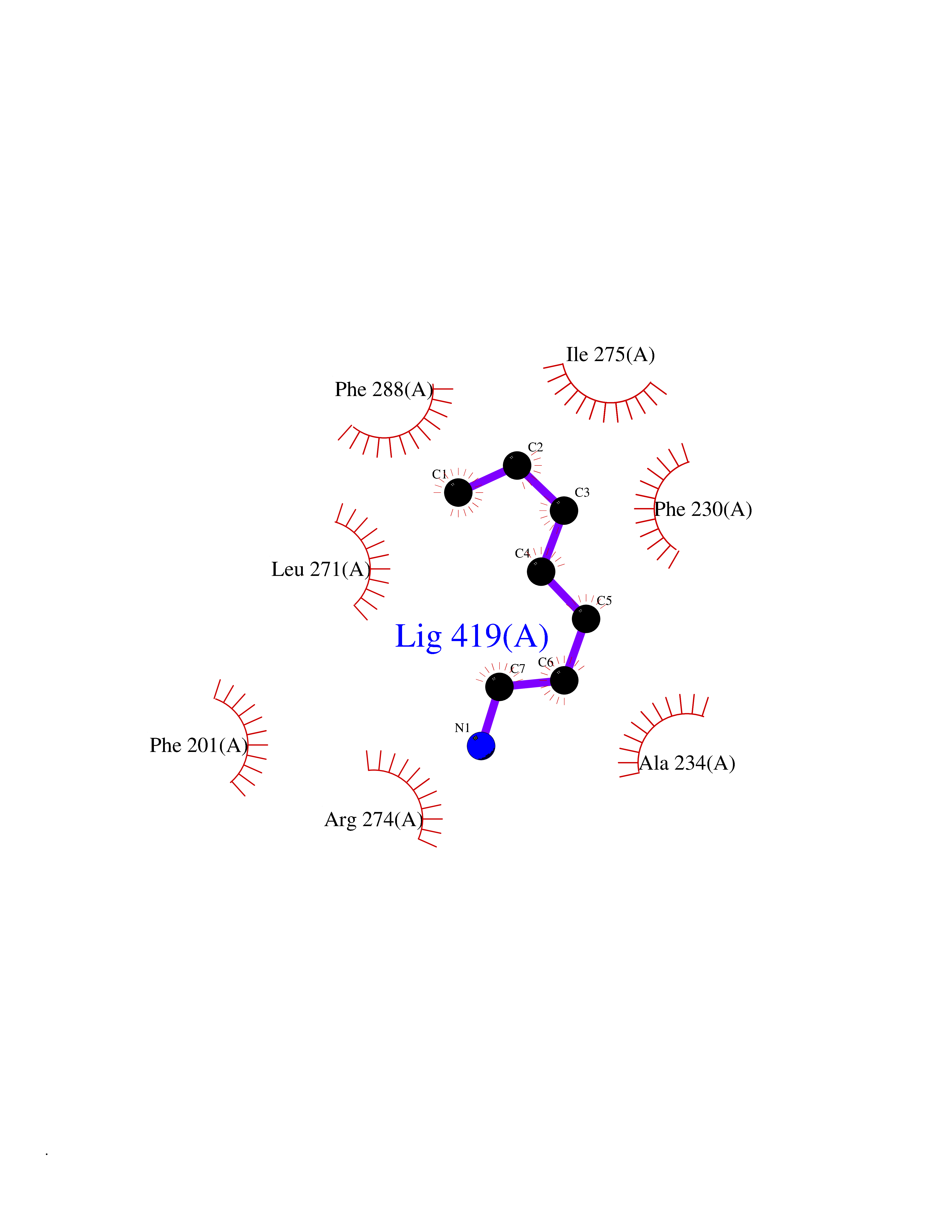 Ligplot Map 3D Binding mode Sequence ASPQLEELITKVSKAHQETFPSLCQLGKYTTNSSADHRVQLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLSIADQITLLKAACLDILMLRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFAGQLLPLEMDDTETGLLSAICLICGDRMDLEEPEKVDKLQEPLLEALRLYARRRRPSQPYMFPRMLMKITDLRGISTKGAERAITLKMEIPGPMPPLIREMLE Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Peroxisome proliferator-activated receptor delta (PPARD) | 3TKM | 5.05 | |
Target general information Gen name PPARD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peroxisomeproliferator-activated receptor beta; Peroxisomeproliferator activated receptor beta/delta; Peroxisome proliferator-activated receptor beta; PPARdelta; PPARB; PPAR-delta; PPAR-beta; Nuclear Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Receptor that binds peroxisome proliferators such as hypolipidemic drugs and fatty acids. Has a preference for poly-unsaturated fatty acids, such as gamma-linoleic acid and eicosapentanoic acid. Once activated by a ligand, the receptor binds to promoter elements of target genes. Regulates the peroxisomal beta-oxidation pathway of fatty acids. Functions as transcription activator for the acyl-CoA oxidase gene. Decreases expression of NPC1L1 once activated by a ligand. Ligand-activated transcription factor. Related diseases Aminoacylase-1 deficiency (ACY1D) [MIM:609924]: An enzymatic deficiency resulting in encephalopathy, unspecific psychomotor delay, psychomotor delay with atrophy of the vermis and syringomyelia, marked muscular hypotonia or normal clinical features. Epileptic seizures are a frequent feature. All affected individuals exhibit markedly increased urinary excretion of several N-acetylated amino acids. {ECO:0000269|PubMed:16274666, ECO:0000269|PubMed:16465618, ECO:0000269|PubMed:17562838, ECO:0000269|PubMed:21414403}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07070; DB07691; DB00132; DB01393; DB05416; DB04801; DB09006; DB05187; DB13873; DB13961; DB09462; DB03338; DB00159; DB07724; DB05188; DB04224; DB02746; DB00412; DB00605; DB00374; DB00197; DB00313; DB08078 Interacts with O60341; P55055-1; Q13133; P42858; P60409; P02545; Q7Z699; Q96EG3 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 30805.5 Length 270 Aromaticity 0.1 Instability index 37.2 Isoelectric point 7.88 Charge (pH=7) 1.64 2D Binding mode Binding energy (Kcal/mol) -6.88  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GSHMQVADLKAFSKHIYNAYLKNFNMTKKKARSILTGKASHTAPFVIHDIETLWQAEKGLVWGLPPYKEISVHVFYRCQCTTVETVRELTEFAKSIPSFSSLFLNDQVTLLKYGVHEAIFAMLASIVNKDGLLVANGSGFVTREFLRSLRKPFSDIIEPKFEFAVKFNALELDDSDLALFIAAIILCGDRPGLMNVPRVEAIQDTILRALEFHLQANHPDAQYLFPKLLQKMADLRQLVTEHAQMMQRIKKTETETSLHPLLQEIYKDMY Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | TRANSPORT INHIBITOR RESPONSE 1 protein | 2P1Q | 5.05 | |
Target general information Gen name IAA7 Organism Arabidopsis thaliana (Mouse-ear cress) Uniprot ID TTD ID NA Synonyms AXR2;At3g23050;MXC7.8 Protein family Aux/IAA family Biochemical class Signaling protein Function DNA binding transcription factor activity. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) NA Interacts with Q9LW29; Q9C5W9; Q8RYC8; Q94AH6; Q9ZR12; P49677; Q38828; Q38829; Q38830; Q38831; O24407; O24408; O24409; P49678; O24410; Q8LAL2; Q9XFM0; Q38822; Q9M1R4; Q9C5X0; Q9C8Y3; Q39255; Q570C0 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Auxin signaling pathway; Nucleus; Reference proteome; Repressor; Transcription; Transcription regulation Protein physicochemical properties Chain ID B,C Molecular weight (Da) 65385.2 Length 581 Aromaticity 0.09 Instability index 47.83 Isoelectric point 7.46 Charge (pH=7) 1.23 2D Binding mode Binding energy (Kcal/mol) -6.89  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence QVVGWPPVRNYRKFPEEVLEHVFSFIQLDKDRNSVSLVCKSWYEIERWCRRKVFIGNCYAVSPATVIRRFPKVRSVELKGKPHFADFNLVPDGWGGYVYPWIEAMSSSYTWLEEIRLKRMVVTDDCLELIAKSFKNFKVLVLSSCEGFSTDGLAAIAATCRNLKELDLRESDVDDVSGHWLSHFPDTYTSLVSLNISCLASEVSFSALERLVTRCPNLKSLKLNRAVPLEKLATLLQRAPQLEELGTGGYTAEVRPDVYSGLSVALSGCKELRCLSGFWDAVPAYLPAVYSVCSRLTTLNLSYATVQSYDLVKLLCQCPKLQRLWVLDYIEDAGLEVLASTCKDLRELRVFPSEPFVMEPNVALTEQGLVSVSMGCPKLESVLYFCRQMTNAALITIARNRPNMTRFRLCIIEPKAPDYLTLEPLDIGFGAIVEHCKDLRRLSLSGLLTDKVFEYIGTYAKKMEMLSVAFAGDSDLGMHHVLSGCDSLRKLEIRDCPFGDKALLANASKLETMRSLWMSSCSVSFGACKLLGQKMPKLNVEVIDERGAPDSRPESCPVERVFIYRTVAGPRFDMPGFVWNM Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Histone-lysine N-methyltransferase KMT5B (KMT5B) | 3S8P | 5.05 | |
Target general information Gen name KMT5B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 5B; Lysine-specific methyltransferase 5B; Suppressor of variegation 4-20 homolog 1; Su(var)4-20 homolog 1; Suv4-20h1; [histone H4]-N-methyl-L-lysine20 N-methyltransferase KM Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar4-20 subfamily Biochemical class NA Function Histone methyltransferase that specifically methylates monomethylated 'Lys-20' (H4K20me1) and dimethylated 'Lys-20' (H4K20me2) of histone H4 to produce respectively dimethylated 'Lys-20' (H4K20me2) and trimethylated 'Lys-20' (H4K20me3) and thus regulates transcription and maintenance of genome integrity. In vitro also methylates unmodified 'Lys-20' (H4K20me0) of histone H4 and nucleosomes. H4 'Lys-20' trimethylation represents a specific tag for epigenetic transcriptional repression. Mainly functions in pericentric heterochromatin regions, thereby playing a central role in the establishment of constitutive heterochromatin in these regions. KMT5B is targeted to histone H3 via its interaction with RB1 family proteins (RB1, RBL1 and RBL2) (By similarity). Plays a role in myogenesis by regulating the expression of target genes, such as EID3. Facilitates TP53BP1 foci formation upon DNA damage and proficient non-homologous end-joining (NHEJ)-directed DNA repair by catalyzing the di- and trimethylation of 'Lys-20' of histone H4. May play a role in class switch reconbination by catalyzing the di- and trimethylation of 'Lys-20' of histone H4 (By similarity). Related diseases Intellectual developmental disorder, autosomal dominant 51 (MRD51) [MIM:617788]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:28191889, ECO:0000269|PubMed:29276005}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H2G4; Q61026 EC number EC 2.1.1.361 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Disease variant; Intellectual disability; Isopeptide bond; Metal-binding; Methyltransferase; Myogenesis; Nucleus; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 26230.2 Length 233 Aromaticity 0.11 Instability index 42.29 Isoelectric point 5.64 Charge (pH=7) -6.07 2D Binding mode Binding energy (Kcal/mol) -6.89  Molscript Map  Pymol Map 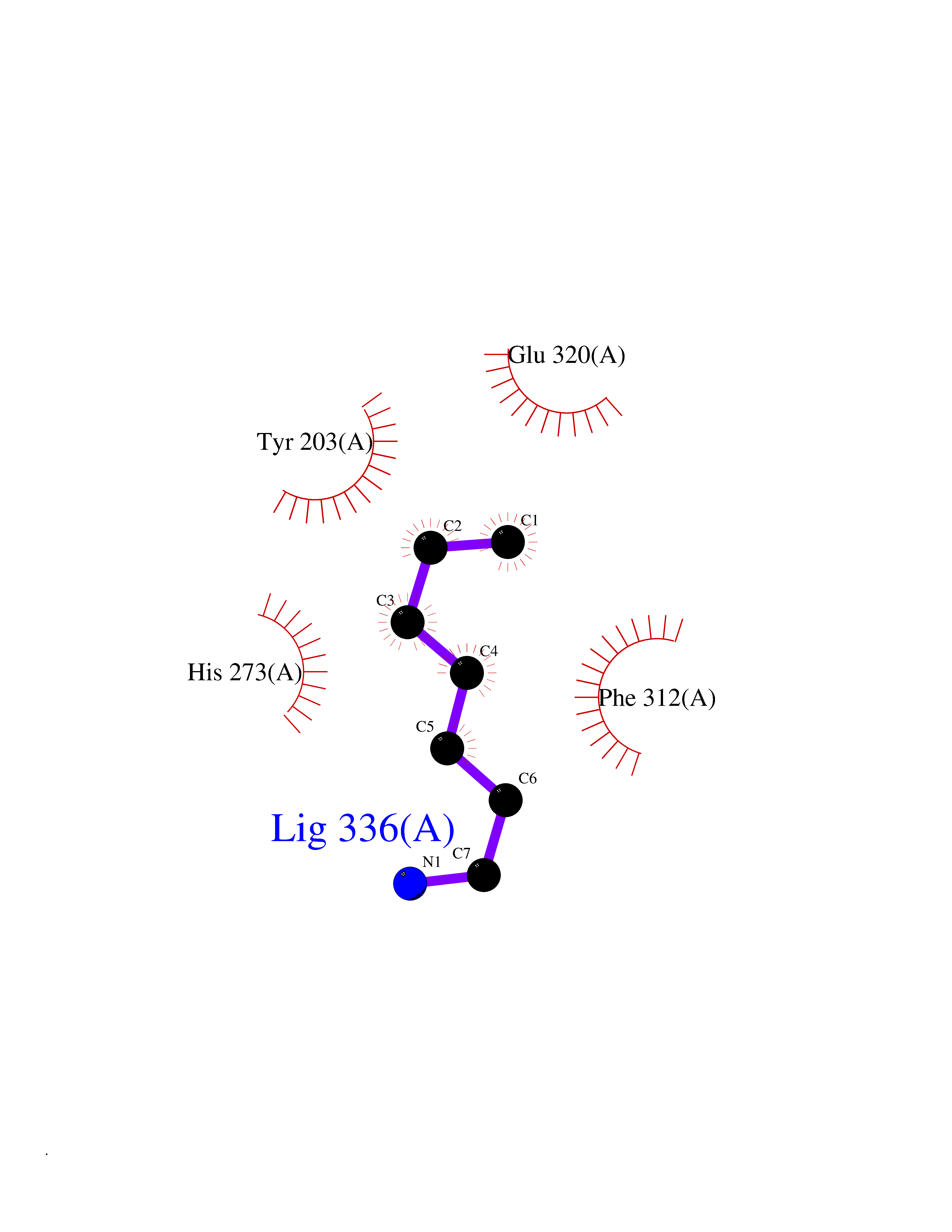 Ligplot Map 3D Binding mode Sequence XSAKELCENDDLATSLVLDPYLGFQTHKXNTRQEELKEVIERFKKDEHLEKAFKCLTSGEWARHYFLNKNKXQEKLFKEHVFIYLRXFATDSGFEILPCNRYSSEQNGAKIVATKEWKRNDKIELLVGCIAELSEIEENXLLRHGENDFSVXYSTRKNCAQLWLGPAAFINHDCRPNCKFVSTGRDTACVKALRDIEPGEEISCYYGDGFFGENNEFCECYTCERRGTGAFKS Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Ubiquitin carboxyl-terminal hydrolase 14 (USP14) | 6IIK | 5.05 | |
Target general information Gen name USP14 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin-specific-processing protease 14; Ubiquitin thioesterase 14; TGT; Deubiquitinating enzyme 14 Protein family Peptidase C19 family, USP14/UBP6 subfamily Biochemical class Peptidase Function Ensures the regeneration of ubiquitin at the proteasome. Is a reversibly associated subunit of the proteasome and a large fraction of proteasome-free protein exists within the cell. Required for the degradation of the chemokine receptor CXCR4 which is critical for CXCL12-induced cell chemotaxis. Serves also as a physiological inhibitor of endoplasmic reticulum-associated degradation (ERAD) under the non-stressed condition by inhibiting the degradation of unfolded endoplasmic reticulum proteins via interaction with ERN1. Indispensable for synaptic development and function at neuromuscular junctions (NMJs). Plays a role in the innate immune defense against viruses by stabilizing the viral DNA sensor CGAS and thus inhibiting its autophagic degradation. Proteasome-associated deubiquitinase which releases ubiquitin from the proteasome targeted ubiquitinated proteins. Related diseases Hypophosphatemic rickets, autosomal dominant (ADHR) [MIM:193100]: A disease characterized by isolated renal phosphate wasting, hypophosphatemia, and inappropriately normal 1,25-dihydroxyvitamin D3 (calcitriol) levels. Patients frequently present with bone pain, rickets, and tooth abscesses. {ECO:0000269|PubMed:11062477, ECO:0000269|PubMed:11409890, ECO:0000269|PubMed:16638743}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Tumoral calcinosis, hyperphosphatemic, familial, 2 (HFTC2) [MIM:617993]: A form of hyperphosphatemic tumoral calcinosis, a rare autosomal recessive metabolic disorder that manifests with hyperphosphatemia and massive calcium deposits in the skin and subcutaneous tissues. Some patients have recurrent, transient, painful swellings of the long bones associated with the radiographic findings of periosteal reaction and cortical hyperostosis and absence of skin involvement. {ECO:0000269|PubMed:15590700, ECO:0000269|PubMed:16030159, ECO:0000269|PubMed:16151858, ECO:0000269|PubMed:24680727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12695 Interacts with Q08209 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell membrane; Cytoplasm; Hydrolase; Immunity; Innate immunity; Membrane; Phosphoprotein; Protease; Proteasome; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 38476.7 Length 335 Aromaticity 0.1 Instability index 61.05 Isoelectric point 5.6 Charge (pH=7) -4.84 2D Binding mode Binding energy (Kcal/mol) -6.88 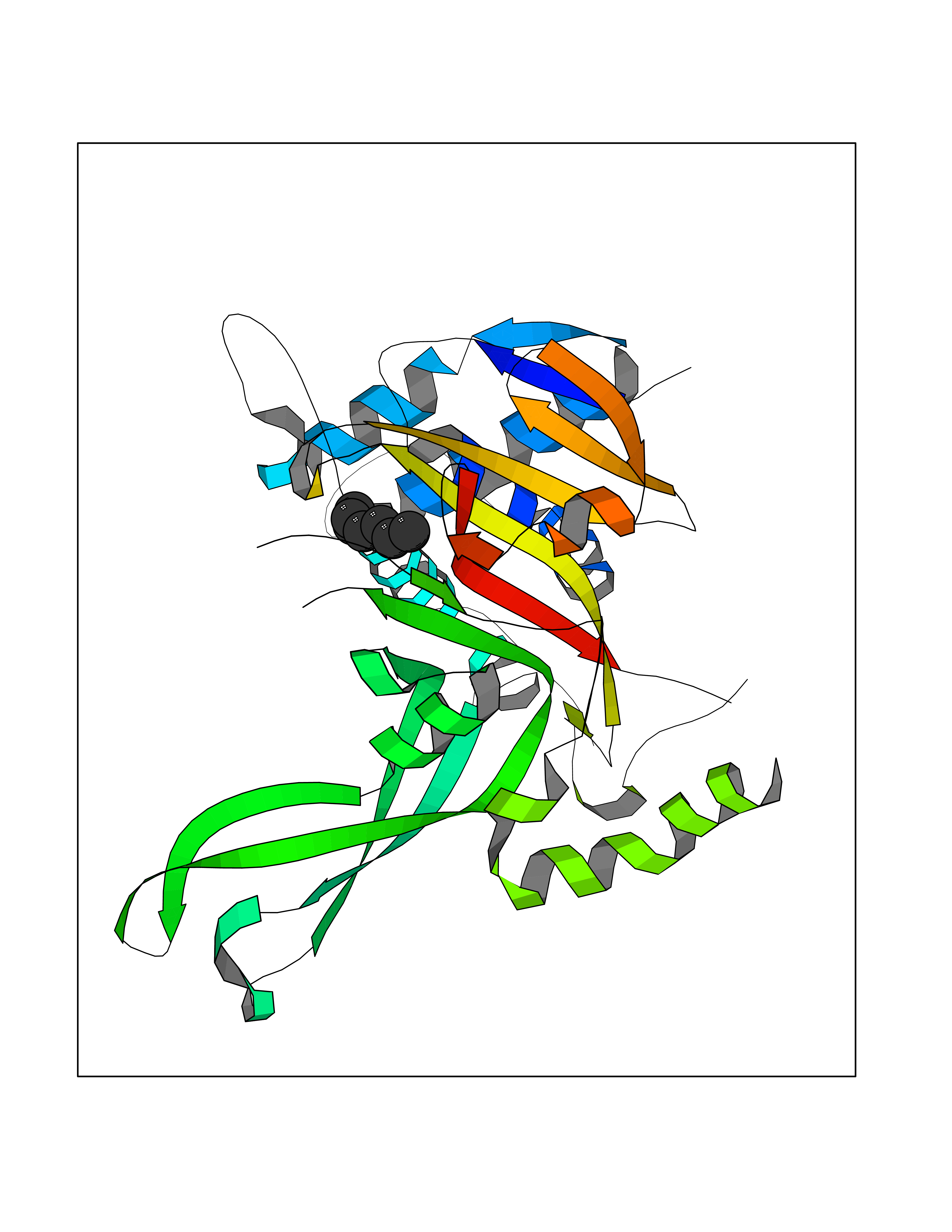 Molscript Map 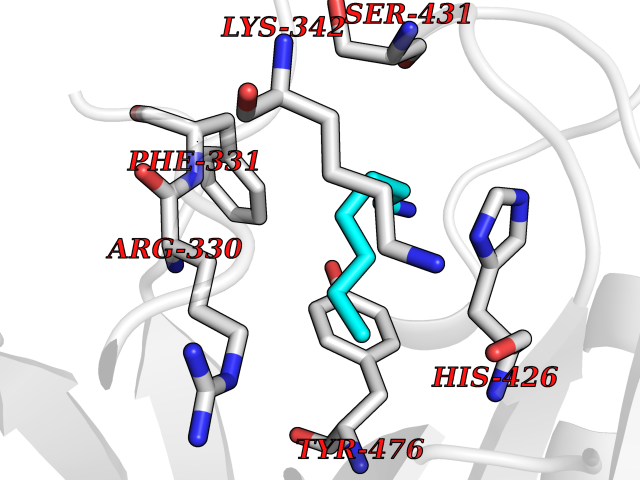 Pymol Map 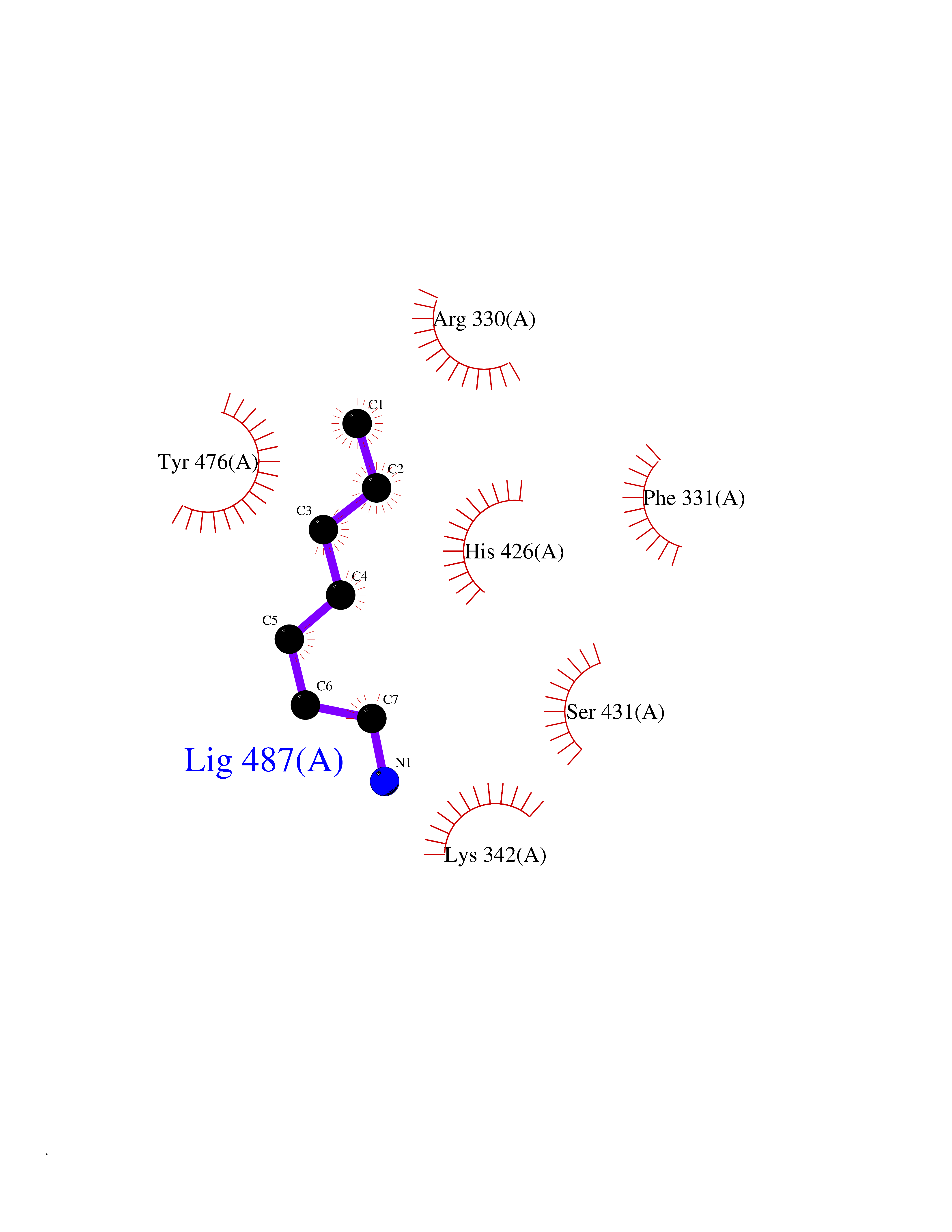 Ligplot Map 3D Binding mode Sequence ELPCGLTNLGNTCYMNATVQCIRSVPELKDALKRYAGALRASGEMASAQYITAALRDLFDSMDKTSSSIPPIILLQFLHMAFPQFAEKGEQGQYLQQDANECWIQMMRVLQQKLEAIEDKSLIDQFFGVEFETTMKCTESEEEEVTKGKENQLQLSCFINQEVKYLFTGLKLRLQEEITKQSPTLQRNALYIKSSKISRLPAYLTIQMVRFFNAKVLKDVKFPLMLDMYELCTPELQEKMVSFRSKFKDLYEPFSFADDIGSNNCGYYDLQAVLTHQGRSSSSGHYVSWVKRKQDEWIKFDDDKVSIVTPEDILRLSGGGDWHIAYVLLYGPRRV Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Cerebron E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase) | 4CI1 | 5.05 | |
Target general information Gen name CUL4A/CUL4B-DDB1-CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NA Protein family Cullin family Biochemical class NA Function NA Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; Q86VP6; Q16531; Q92466; P08238; O94888; P55072 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; DNA damage; DNA repair; Host-virus interaction; Isopeptide bond; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID B Molecular weight (Da) 42669.7 Length 368 Aromaticity 0.1 Instability index 44.94 Isoelectric point 8.72 Charge (pH=7) 6.58 2D Binding mode Binding energy (Kcal/mol) -6.89  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MINFDTSLPTSHMYLGSDMEEFHGRTLHDDDSCQVIPVLPHVMVMLIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVREREAHFGTTAEIYAYREEQEYGIETVKVKAIGRQRFKVLEIRTQSDGIQQAKVQILPERVLPSTMSAVQLQSLSRRHIRAFRQWWQKYQKRKFHCASLTSWPPWLYSLYDAETLMERVKRQLHEWDENLKDESLPTNPIDFSYRVAACLPIDDALRIQLLKIGSAIQRLRELDIMNKTSLCCKQCQDTEITTKNEIFSLSLCGPMAAYVNPHGYIHETLTVYKACNLNLSGRPSTEHSWFPGYAWTIAQCRICGNHMGWKFTATKKDMSPQKFWGLTRSALLPR Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Pyruvate dehydrogenase kinase 1 (PDHK1) | 2Q8G | 5.05 | |
Target general information Gen name PDK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Pyruvate dehydrogenase kinase isoform 1; Pyruvate dehydrogenase (acetyl-transferring) kinase isozyme 1, mitochondrial; PDHK1; PDH kinase 1 Protein family PDK/BCKDK protein kinase family Biochemical class Kinase Function Kinase that plays a key role in regulation of glucose and fatty acid metabolism and homeostasis via phosphorylation of the pyruvate dehydrogenase subunits PDHA1 and PDHA2. This inhibits pyruvate dehydrogenase activity, and thereby regulates metabolite flux through the tricarboxylic acid cycle, down-regulates aerobic respiration and inhibits the formation of acetyl-coenzyme A from pyruvate. Plays an important role in cellular responses to hypoxia and is important for cell proliferation under hypoxia. Protects cells against apoptosis in response to hypoxia and oxidative stress. Related diseases TP53 is found in increased amounts in a wide variety of transformed cells. TP53 is frequently mutated or inactivated in about 60% of cancers. TP53 defects are found in Barrett metaplasia a condition in which the normally stratified squamous epithelium of the lower esophagus is replaced by a metaplastic columnar epithelium. The condition develops as a complication in approximately 10% of patients with chronic gastroesophageal reflux disease and predisposes to the development of esophageal adenocarcinoma.; DISEASE: Esophageal cancer (ESCR) [MIM:133239]: A malignancy of the esophagus. The most common types are esophageal squamous cell carcinoma and adenocarcinoma. Cancer of the esophagus remains a devastating disease because it is usually not detected until it has progressed to an advanced incurable stage. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Li-Fraumeni syndrome (LFS) [MIM:151623]: An autosomal dominant familial cancer syndrome that in its classic form is defined by the existence of a proband affected by a sarcoma before 45 years with a first degree relative affected by any tumor before 45 years and another first degree relative with any tumor before 45 years or a sarcoma at any age. Other clinical definitions for LFS have been proposed and called Li-Fraumeni like syndrome (LFL). In these families affected relatives develop a diverse set of malignancies at unusually early ages. Four types of cancers account for 80% of tumors occurring in TP53 germline mutation carriers: breast cancers, soft tissue and bone sarcomas, brain tumors (astrocytomas) and adrenocortical carcinomas. Less frequent tumors include choroid plexus carcinoma or papilloma before the age of 15, rhabdomyosarcoma before the age of 5, leukemia, Wilms tumor, malignant phyllodes tumor, colorectal and gastric cancers. {ECO:0000269|PubMed:10484981, ECO:0000269|PubMed:1565144, ECO:0000269|PubMed:1737852, ECO:0000269|PubMed:1933902, ECO:0000269|PubMed:1978757, ECO:0000269|PubMed:2259385, ECO:0000269|PubMed:36108750, ECO:0000269|PubMed:7887414, ECO:0000269|PubMed:8825920, ECO:0000269|PubMed:9452042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Squamous cell carcinoma of the head and neck (HNSCC) [MIM:275355]: A non-melanoma skin cancer affecting the head and neck. The hallmark of cutaneous SCC is malignant transformation of normal epidermal keratinocytes. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Lung cancer (LNCR) [MIM:211980]: A common malignancy affecting tissues of the lung. The most common form of lung cancer is non-small cell lung cancer (NSCLC) that can be divided into 3 major histologic subtypes: squamous cell carcinoma, adenocarcinoma, and large cell lung cancer. NSCLC is often diagnosed at an advanced stage and has a poor prognosis. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Papilloma of choroid plexus (CPP) [MIM:260500]: A benign tumor of neuroectodermal origin that generally occurs in childhood, but has also been reported in adults. Although generally found within the ventricular system, choroid plexus papillomas can arise ectopically in the brain parenchyma or disseminate throughout the neuraxis. Patients present with signs and symptoms of increased intracranial pressure including headache, hydrocephalus, papilledema, nausea, vomiting, cranial nerve deficits, gait impairment, and seizures. {ECO:0000269|PubMed:12085209}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Adrenocortical carcinoma (ADCC) [MIM:202300]: A malignant neoplasm of the adrenal cortex and a rare childhood tumor. It occurs with increased frequency in patients with Beckwith-Wiedemann syndrome and Li-Fraumeni syndrome. {ECO:0000269|PubMed:11481490}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Basal cell carcinoma 7 (BCC7) [MIM:614740]: A common malignant skin neoplasm that typically appears on hair-bearing skin, most commonly on sun-exposed areas. It is slow growing and rarely metastasizes, but has potentialities for local invasion and destruction. It usually develops as a flat, firm, pale area that is small, raised, pink or red, translucent, shiny, and waxy, and the area may bleed following minor injury. Tumor size can vary from a few millimeters to several centimeters in diameter. {ECO:0000269|PubMed:21946351}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Bone marrow failure syndrome 5 (BMFS5) [MIM:618165]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFS5 is an autosomal dominant form characterized by infantile onset of severe red cell anemia requiring transfusion. Additional features include hypogammaglobulinemia, poor growth with microcephaly, developmental delay, and seizures. {ECO:0000269|PubMed:30146126}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07403; DB08809 Interacts with P05067; P08559; Q16513; P31749-1; P31751-1 EC number EC 2.7.11.2 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Kinase; Mitochondrion; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 42249.9 Length 368 Aromaticity 0.11 Instability index 49.91 Isoelectric point 6.83 Charge (pH=7) -0.46 2D Binding mode Binding energy (Kcal/mol) -6.89  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GVPGQVDFYARFSPSPLSMKQFLDFGSVNACEKTSFMFLRQELPVRLANIMKEISLLPDNLLRTPSVQLVQSWYIQSLQELLDFKDKSAEDAKAIYDFTDTVIRIRNRHNDVIPTMAQGVIEYKESFDPVTSQNVQYFLDRFYMSRISIRMLLNQHSLLFGKHIGSINPNCNVLEVIKDGYENARRLCDLYYINSPELELEELNAKSPGQPIQVVYVPSHLYHMVFELFKNAMRATMEHHANRGVYPPIQVHVTLGNEDLTVKMSDRGGGVPLRKIDRLFNYMYSTAPRPRVETSRAVPLAGFGYGLPISRLYAQYFQGDLKLYSLEGYGTDAVIYIKALSTDSIERLPVYNKAAWKHYNTNDDWCVP Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Staphylococcus Enoyl ACP reductase (Stap-coc fabI) | 6YUR | 5.05 | |
Target general information Gen name Stap-coc fabI Organism Staphylococcus aureus (strain NCTC 8325 / PS 47) Uniprot ID TTD ID Synonyms NADPHdependent enoylACP reductase; Enoyl[acylcarrierprotein] reductase [NADPH] FabI Protein family Short-chain dehydrogenases/reductases (SDR) family, FabI subfamily Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the reduction of a carbon-carbon double bond in an enoyl moiety that is covalently linked to an acyl carrier protein (ACP). It has a preference for a long chain (C12) substrate compared to the shorter (C4) acyl group. Involved in the elongation cycle of fatty acid which are used in the lipid metabolism. Related diseases Amyloidosis, hereditary systemic 4, Finnish type (AMYLD4) [MIM:105120]: A form of hereditary systemic amyloidosis, a disorder characterized by amyloid deposition in multiple tissues resulting in a wide clinical spectrum. AMYLD4 is due to gelsolin amyloid deposition and is typically characterized by cranial neuropathy and lattice corneal dystrophy. Most patients have modest involvement of internal organs, but severe systemic disease can develop in some individuals causing peripheral polyneuropathy, amyloid cardiomyopathy, and nephrotic syndrome leading to renal failure. AMYLD4 is usually inherited in an autosomal dominant pattern. However, homozygotes with a more severe phenotype have also been reported. {ECO:0000269|PubMed:1338910, ECO:0000269|PubMed:19666512, ECO:0000269|PubMed:2176481}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; NAD; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,C Molecular weight (Da) 55674.6 Length 510 Aromaticity 0.07 Instability index 33.86 Isoelectric point 5.64 Charge (pH=7) -9.3 2D Binding mode Binding energy (Kcal/mol) -6.89  Molscript Map  Pymol Map 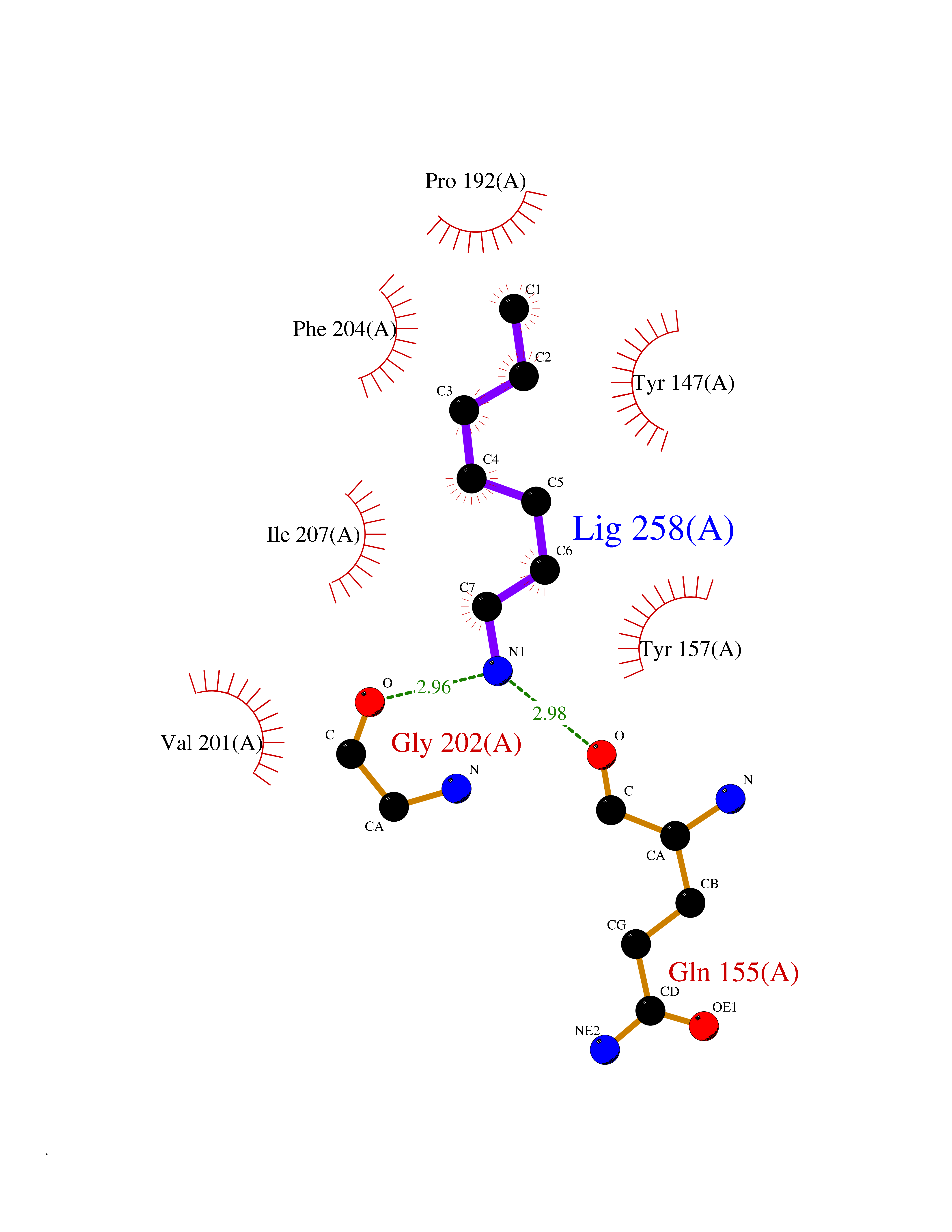 Ligplot Map 3D Binding mode Sequence VNLENKTYVIMGIANKRSIAFGVAKVLDQLGAKLVFTYRKERSRKELEKLLEQLNQPEAHLYQIDVQSDEEVINGFEQIGKDVGNIDGVYHSIAFANMEDLRGRFSETSREGFLLAQDISSYSLTIVAHEAKKLMPEGGSIVATTYLGGEFAVQNYNVMGVAKASLEANVKYLALDLGPDNIRVNAISAGPIRTLSAKGVGGFNTILKEIEERAPLKRNVDQVEVGKTAAYLLSDLSSGVTGENIHVDSGFHAIKVNLENKTYVIMGIANKRSIAFGVAKVLDQLGAKLVFTYRKERSRKELEKLLEQLNQPEAHLYQIDVQSDEEVINGFEQIGKDVGNIDGVYHSIAFANMEDLRGRFSETSREGFLLAQDISSYSLTIVAHEAKKLMPEGGSIVATTYLGGEFAVQNYNVMGVAKASLEANVKYLALDLGPDNIRVNAISAGPIRTLSAKGVGGFNTILKEIEERAPLKRNVDQVEVGKTAAYLLSDLSSGVTGENIHVDSGFHAIK Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | ERK activator kinase 1 (MEK1) | 7M0U | 5.05 | |
Target general information Gen name MAP2K1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PRKMK1; Mitogen-activated protein kinase kinase 1; MKK1; MEK 1; MAPKK 1; MAPK/ERKkinase 1; MAPK/ERK kinase 1; MAP kinase kinase 1; Dual specificity mitogen-activated protein kinase kinase 1 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase subfamily Biochemical class Kinase Function Binding of extracellular ligands such as growth factors, cytokines and hormones to their cell-surface receptors activates RAS and this initiates RAF1 activation. RAF1 then further activates the dual-specificity protein kinases MAP2K1/MEK1 and MAP2K2/MEK2. Both MAP2K1/MEK1 and MAP2K2/MEK2 function specifically in the MAPK/ERK cascade, and catalyze the concomitant phosphorylation of a threonine and a tyrosine residue in a Thr-Glu-Tyr sequence located in the extracellular signal-regulated kinases MAPK3/ERK1 and MAPK1/ERK2, leading to their activation and further transduction of the signal within the MAPK/ERK cascade. Depending on the cellular context, this pathway mediates diverse biological functions such as cell growth, adhesion, survival and differentiation, predominantly through the regulation of transcription, metabolism and cytoskeletal rearrangements. One target of the MAPK/ERK cascade is peroxisome proliferator-activated receptor gamma (PPARG), a nuclear receptor that promotes differentiation and apoptosis. MAP2K1/MEK1 has been shown to export PPARG from the nucleus. The MAPK/ERK cascade is also involved in the regulation of endosomal dynamics, including lysosome processing and endosome cycling through the perinuclear recycling compartment (PNRC), as well as in the fragmentation of the Golgi apparatus during mitosis. Dual specificity protein kinase which acts as an essential component of the MAP kinase signal transduction pathway. Related diseases Cardiofaciocutaneous syndrome 3 (CFC3) [MIM:615279]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. Distinctive features of CFC3 include macrostomia and horizontal shape of palpebral fissures. {ECO:0000269|PubMed:16439621, ECO:0000269|PubMed:18042262}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melorheostosis, isolated (MEL) [MIM:155950]: A sclerosing bone disorder characterized by hyperostosis of the cortex of tubular bones, frequently involving one limb. The lesions may be accompanied by abnormalities of adjacent soft tissue, joint contractures, sclerodermatous skin lesions, muscle atrophy, or hemangioma. {ECO:0000269|PubMed:29643386}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06892; DB07046; DB08208; DB03115; DB11967; DB06616; DB05239; DB02152; DB07101; DB08130; DB14904; DB11689; DB08911 Interacts with Q8N9N5; Q8N9N5-2; Q9NR09; P15056; Q9Y297; O15519-1; P28482; P27361; Q13526; Q9H8W4; P04049; Q8WWU5-7; Q86Y07; Q86Y07-1; P46937 EC number EC 2.7.12.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cardiomyopathy; Cytoplasm; Cytoskeleton; Direct protein sequencing; Disease variant; Ectodermal dysplasia; Intellectual disability; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID B Molecular weight (Da) 34785.9 Length 311 Aromaticity 0.07 Instability index 46.58 Isoelectric point 6.29 Charge (pH=7) -2.54 2D Binding mode Binding energy (Kcal/mol) -6.88 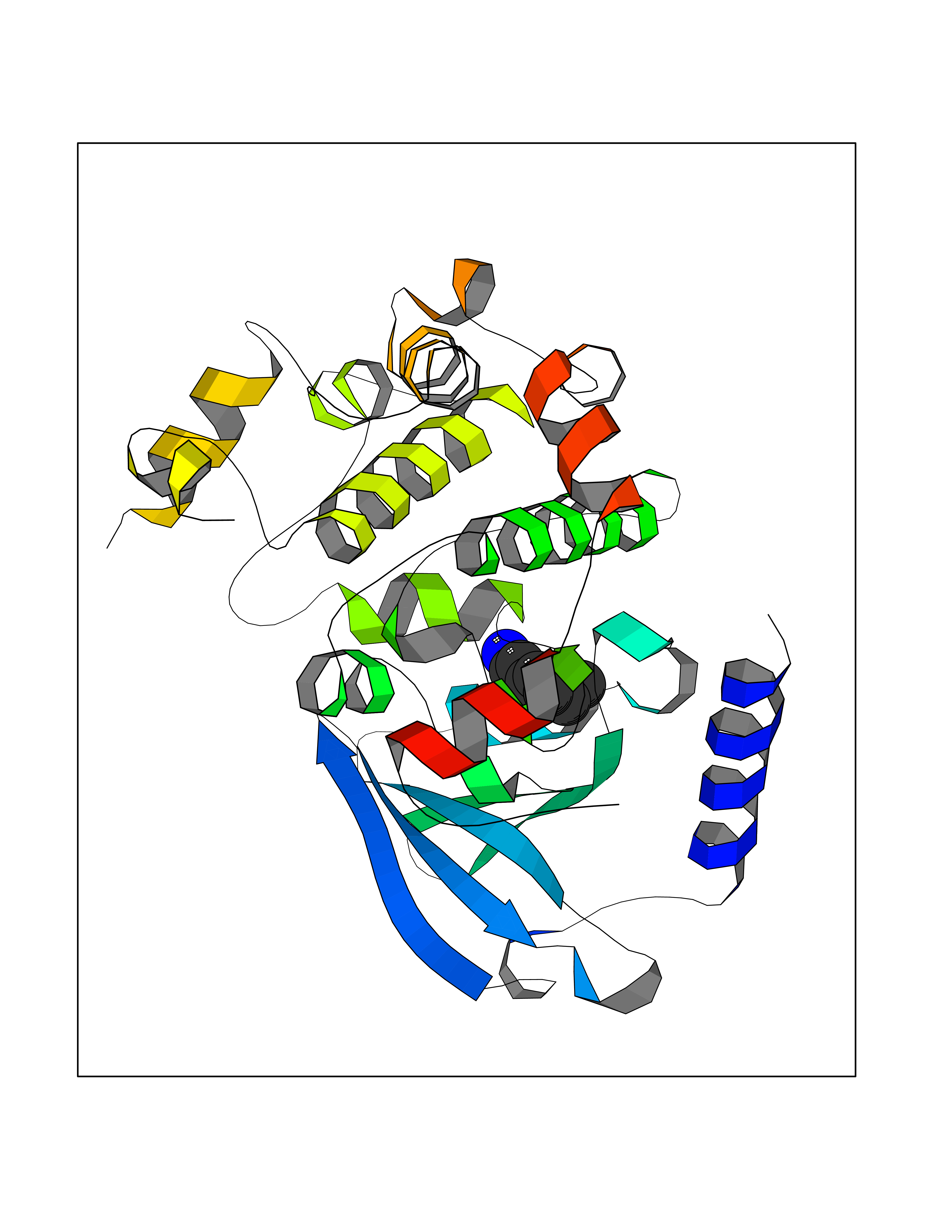 Molscript Map 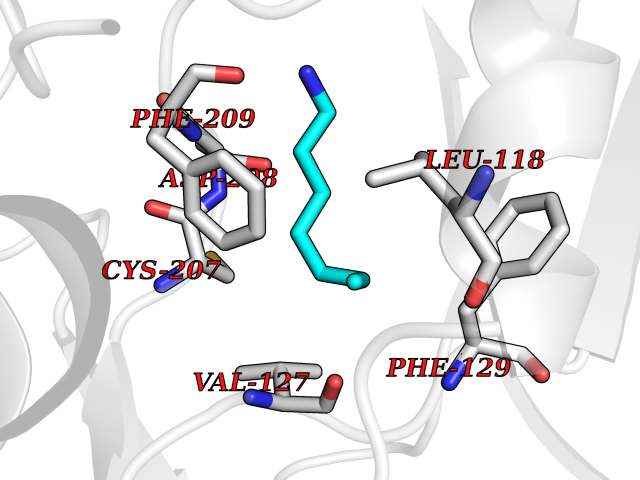 Pymol Map 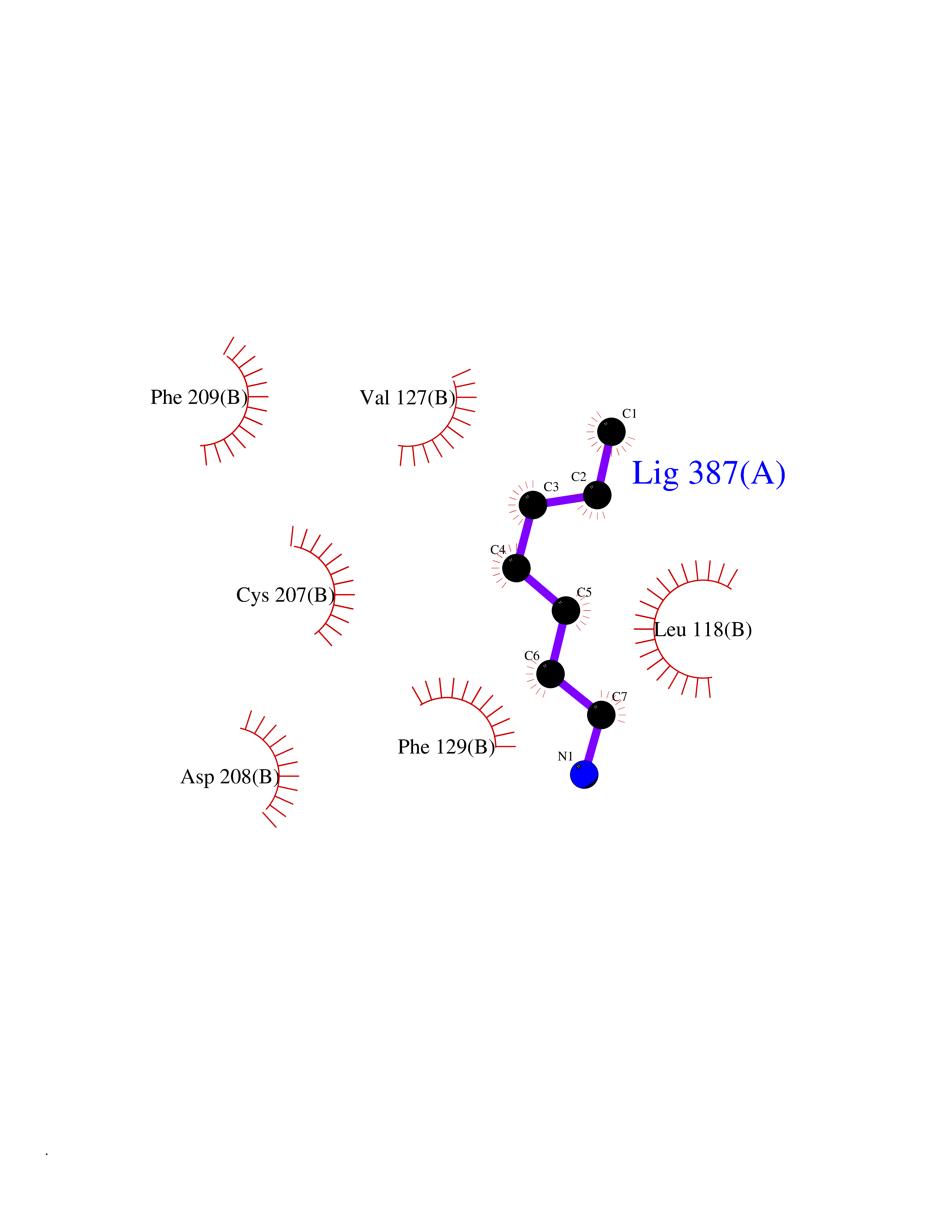 Ligplot Map 3D Binding mode Sequence DEQQRKRLEAFLTQKQKVGELKDDDFEKISELGAGNGGVVFKVSHKPSGLVMARKLIHLEIKPAIRNQIIRELQVLHECNSPYIVGFYGAFYSDGEISICMEHMDGGSLDQVLKKAGRIPEQILGKVSIAVIKGLTYLREKHKIMHRDVKPSNILVNSRGEIKLCDFGVSGQLIDAMANAFVGTRSYMSPERLQGTHYSVQSDIWSMGLSLVEMAVGRYPIPPPDAKELELMPMAIFELLDYIVNEPPPKLPSGVFSLEFQDFVNKCLIKNPAERADLKQLMVHAFIKRSDAEEVDFAGWLCSTIGLNQPS Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Bromodomain-containing protein 9 (BRD9) | 6V0X | 5.05 | |
Target general information Gen name BRD9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Rhabdomyosarcoma antigen MU-RMS-40.8 Protein family NA Biochemical class Bromodomain Function Plays a role in chromatin remodeling and regulation of transcription. Acts as a chromatin reader that recognizes and binds acylated histones: binds histones that are acetylated and/or butyrylated. Component of SWI/SNF chromatin remodeling subcomplex GBAF that carries out key enzymatic activities, changing chromatin structure by altering DNA-histone contacts within a nucleosome in an ATP-dependent manner. Related diseases Major depressive disorder (MDD) [MIM:608516]: A common psychiatric disorder. It is a complex trait characterized by one or more major depressive episodes without a history of manic, mixed, or hypomanic episodes. A major depressive episode is characterized by at least 2 weeks during which there is a new onset or clear worsening of either depressed mood or loss of interest or pleasure in nearly all activities. Four additional symptoms must also be present including changes in appetite, weight, sleep, and psychomotor activity; decreased energy; feelings of worthlessness or guilt; difficulty thinking, concentrating, or making decisions; or recurrent thoughts of death or suicidal ideation, plans, or attempts. The episode must be accompanied by distress or impairment in social, occupational, or other important areas of functioning. {ECO:0000269|PubMed:15229186}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q7Z7H3; Q7Z7H3 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Bromodomain; Chromatin regulator; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 11491.3 Length 99 Aromaticity 0.13 Instability index 13.58 Isoelectric point 9.24 Charge (pH=7) 3.8 2D Binding mode Binding energy (Kcal/mol) -6.88  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence STPIQQLLEHFLRQLQRKDPHGFFAFPVTDAIAPGYSMIIKHPMDFGTMKDKIVANEYKSVTEFKADFKLMCDNAMTYNRPDTVYYKLAKKILHAGFKM Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Nicotinamide phosphoribosyltransferase (NAMPT) | 2E5D | 5.05 | |
Target general information Gen name NAMPT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Visfatin; PreBcell colonyenhancing factor 1; PreB cellenhancing factor; Pre-B-cell colony-enhancing factor 1; Pre-B cell-enhancing factor; PBEF1; PBEF; Nampt; NAmPRTase Protein family NAPRTase family Biochemical class Glycosyltransferases Function It is the rate limiting component in the mammalian NAD biosynthesis pathway. The secreted form behaves both as a cytokine with immunomodulating properties and an adipokine with anti-diabetic properties, it has no enzymatic activity, partly because of lack of activation by ATP, which has a low level in extracellular space and plasma. Plays a role in the modulation of circadian clock function. NAMPT-dependent oscillatory production of NAD regulates oscillation of clock target gene expression by releasing the core clock component: CLOCK-ARNTL/BMAL1 heterodimer from NAD-dependent SIRT1-mediated suppression. Catalyzes the condensation of nicotinamide with 5-phosphoribosyl-1-pyrophosphate to yield nicotinamide mononucleotide, an intermediate in the biosynthesis of NAD. Related diseases Hemolytic anemia, non-spherocytic, due to glucose phosphate isomerase deficiency (HA-GPID) [MIM:613470]: A form of anemia in which there is no abnormal hemoglobin or spherocytosis. It is caused by glucose phosphate isomerase deficiency. {ECO:0000269|PubMed:28803808, ECO:0000269|PubMed:7989588, ECO:0000269|PubMed:8499925, ECO:0000269|PubMed:8822952, ECO:0000269|PubMed:8822954, ECO:0000269|PubMed:9446754, ECO:0000269|PubMed:9856489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12980; DB12731; DB05217 Interacts with P02792; Q01628; P03886; P43490; Q70CQ1-2 EC number EC 2.4.2.12 Uniprot keywords 3D-structure; Acetylation; Biological rhythms; Cytokine; Cytoplasm; Glycosyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Pyridine nucleotide biosynthesis; Reference proteome; Secreted; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 105483 Length 932 Aromaticity 0.11 Instability index 34.4 Isoelectric point 6.68 Charge (pH=7) -2.24 2D Binding mode Binding energy (Kcal/mol) -6.89  Molscript Map 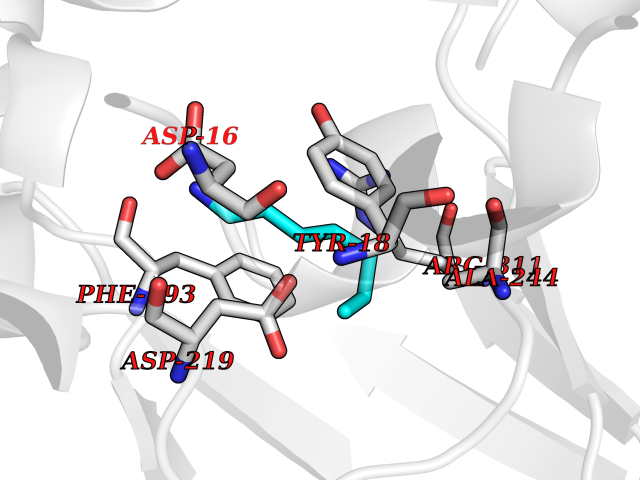 Pymol Map 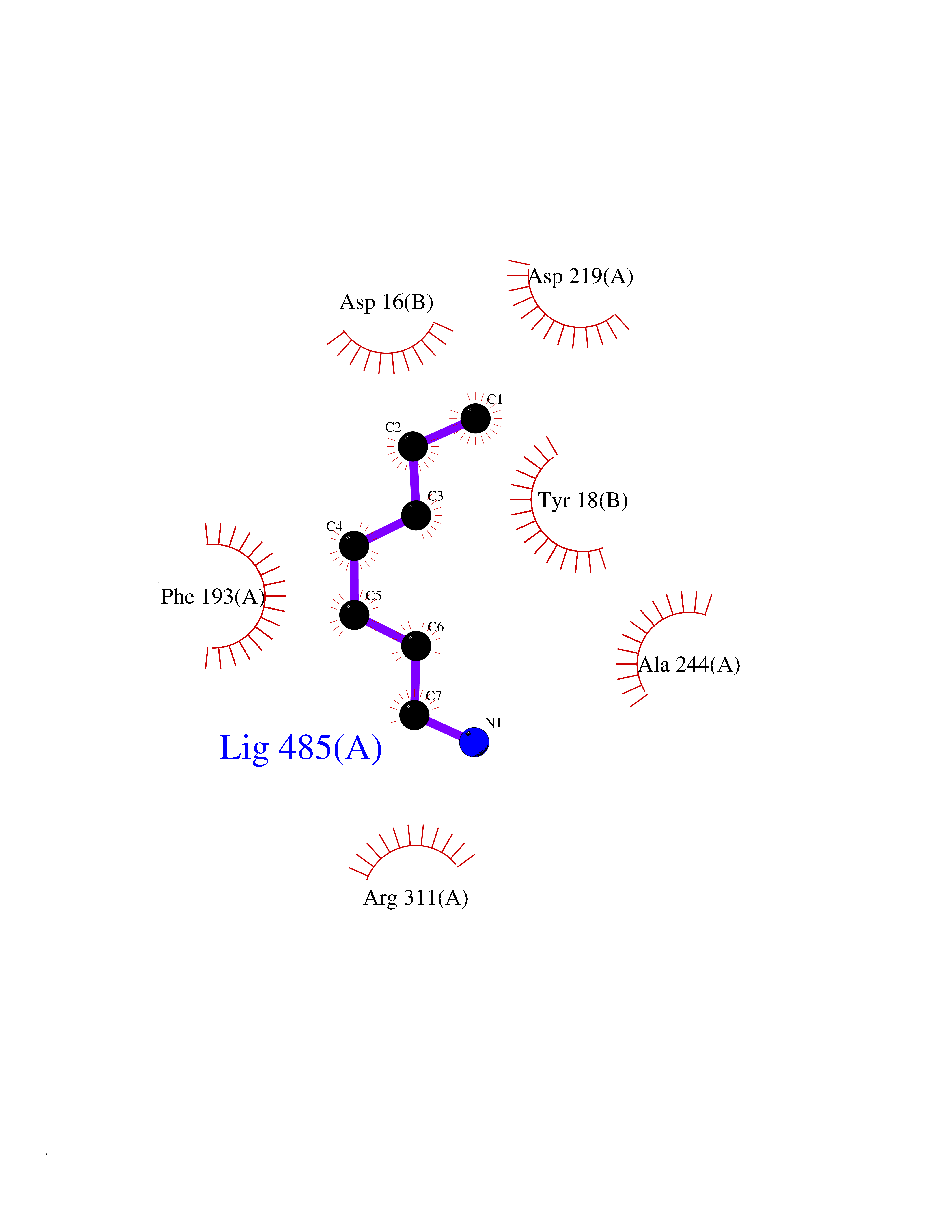 Ligplot Map 3D Binding mode Sequence EFNILLATDSYKVTHYKQYPPNTSKVYSYFECREKKYEETVFYGLQYILNKYLKGKVVTKEKIQEAKDVYKEHFQDDVFNEKGWNYILEKYDGHLPIEIKAVPEGFVIPRGNVLFTVENTDPECYWLTNWIETILVQSWYPITVATNSREQKKILAKYLLETSGNLDGLEYKLHDFGYRGVSSQETAGIGASAHLVNFKGTDTVAGLALIKKYYGTKDPVPGYSVPAAEHSTITAWGKDHEKDAFEHIVTQFSSVPVSVVSDSYDIYNACEKIWGEDLRHLIVSRSTQAPLIIRPDSGNPLDTVLKVLEILGKKFPVTENSKGYKLLPPYLRVIQGDGVDINTLQEIVEGMKQKMWSIENIAFGSGGGLLQKLTRDLLNCSFKCSYVVTNGLGINVFKDPVADPNKRSKKGRLSLHRTPAGNFVTLEEGKGDLEEYGQDLLHTVFKNGKVTKSYSFDEIRKNAQLNEFNILLATDSYKVTHYKQYPPNTSKVYSYFECREKKYEETVFYGLQYILNKYLKGKVVTKEKIQEAKDVYKEHFQDDVFNEKGWNYILEKYDGHLPIEIKAVPEGFVIPRGNVLFTVENTDPECYWLTNWIETILVQSWYPITVATNSREQKKILAKYLLETSGNLDGLEYKLHDFGYRGVSSQETAGIGASAHLVNFKGTDTVAGLALIKKYYGTKDPVPGYSVPAAEHSTITAWGKDHEKDAFEHIVTQFSSVPVSVVSDSYDIYNACEKIWGEDLRHLIVSRSTQAPLIIRPDSGNPLDTVLKVLEILGKKFPVTENSKGYKLLPPYLRVIQGDGVDINTLQEIVEGMKQKMWSIENIAFGSGGGLLQKLTRDLLNCSFKCSYVVTNGLGINVFKDPVADPNKRSKKGRLSLHRTPAGNFVTLEEGKGDLEEYGQDLLHTVFKNGKVTKSYSFDEIRKNAQLN Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Programmed cell death 1 ligand 1 (PD-L1) | 5J89 | 5.05 | |
Target general information Gen name CD274 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hPD-L1; Programmed death ligand 1; PDL1; PDCD1LG1; PDCD1L1; PDCD1 ligand 1; B7H1; B7-H1; B7 homolog 1 Protein family Immunoglobulin superfamily, BTN/MOG family Biochemical class Immunoglobulin Function As a ligand for the inhibitory receptor PDCD1/PD-1, modulates the activation threshold of T-cells and limits T-cell effector response. Through a yet unknown activating receptor, may costimulate T-cell subsets that predominantly produce interleukin-10 (IL10). Plays a critical role in induction and maintenance of immune tolerance to self. Related diseases Truncation of the 3'-untranslated (3'-UTR) region of CD274 transcripts leads to elevated expression of CD274 in multiple cancers including T-cell leukemia, diffuse large B-cell lymphoma and stomach adenocarcinoma (PubMed:27281199). Disruption of 3'-UTR region is caused by structural variants that stabilize CD274 transcripts, leading to overexpression (PubMed:27281199). Increased expression in tumors promotes immune evasion and tumor cell growth by allowing malignant cells to escape destruction by the immune system (PubMed:27281199). {ECO:0000269|PubMed:27281199}. Drugs (DrugBank ID) DB15773; DB11595; DB15771; DB11945; DB15772; DB14776; DB15770; DB11714; DB15769; DB09035; DB09037; DB00203; DB00313 Interacts with P33681; Q8IZR5; Q9NX76; Q15116; Q15116 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Endosome; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Nucleus; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID C,D Molecular weight (Da) 28335.2 Length 249 Aromaticity 0.1 Instability index 35.39 Isoelectric point 6.15 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -6.89 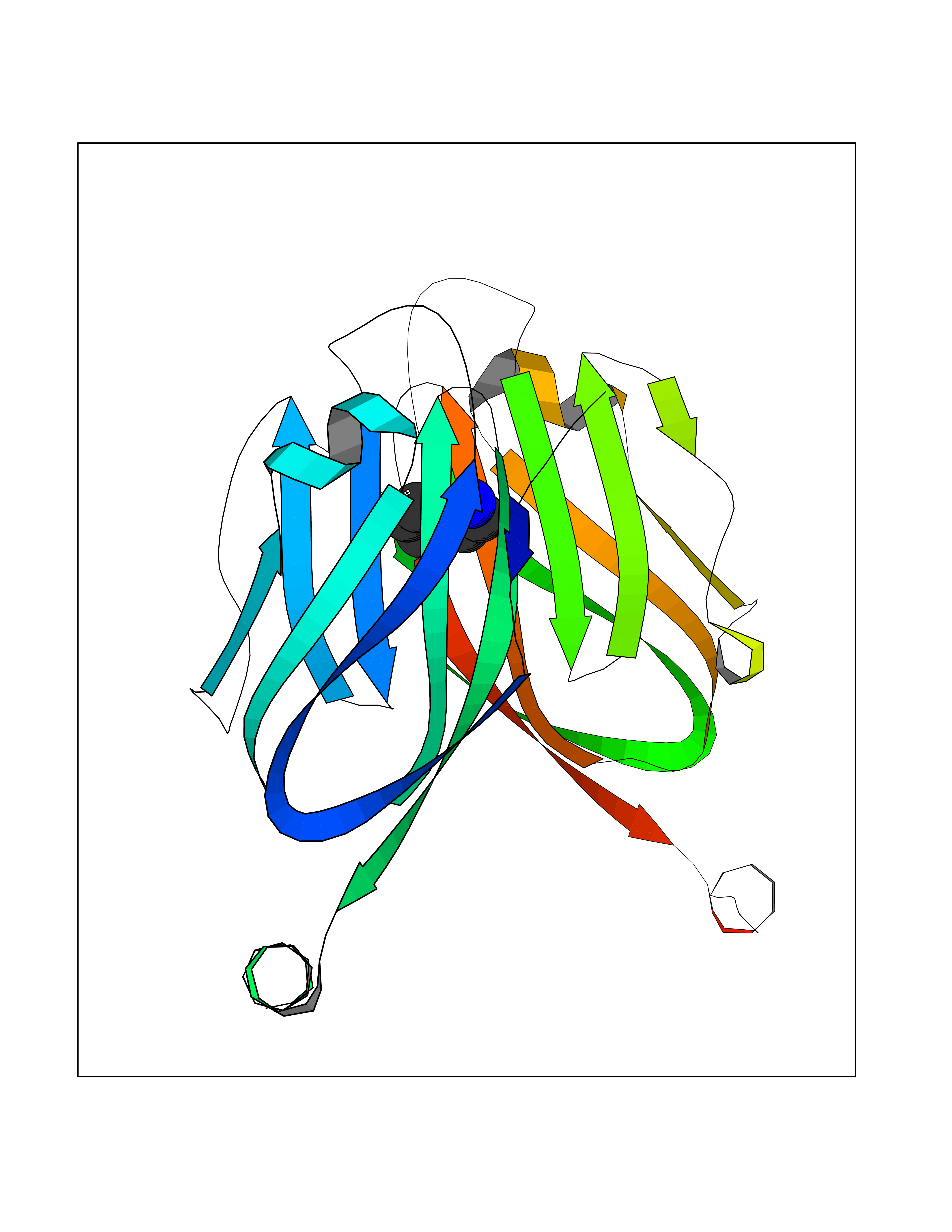 Molscript Map 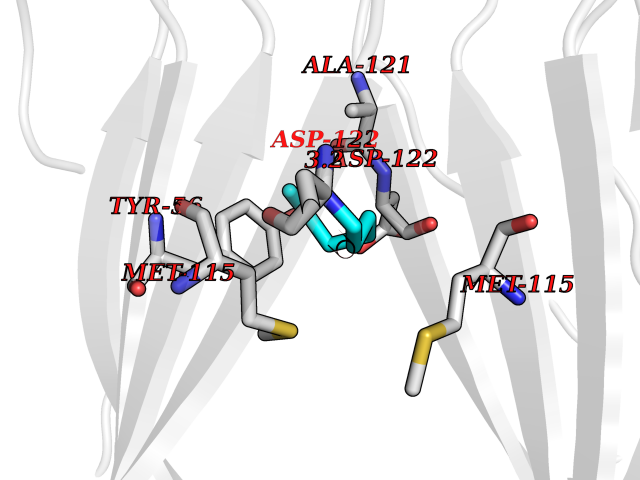 Pymol Map 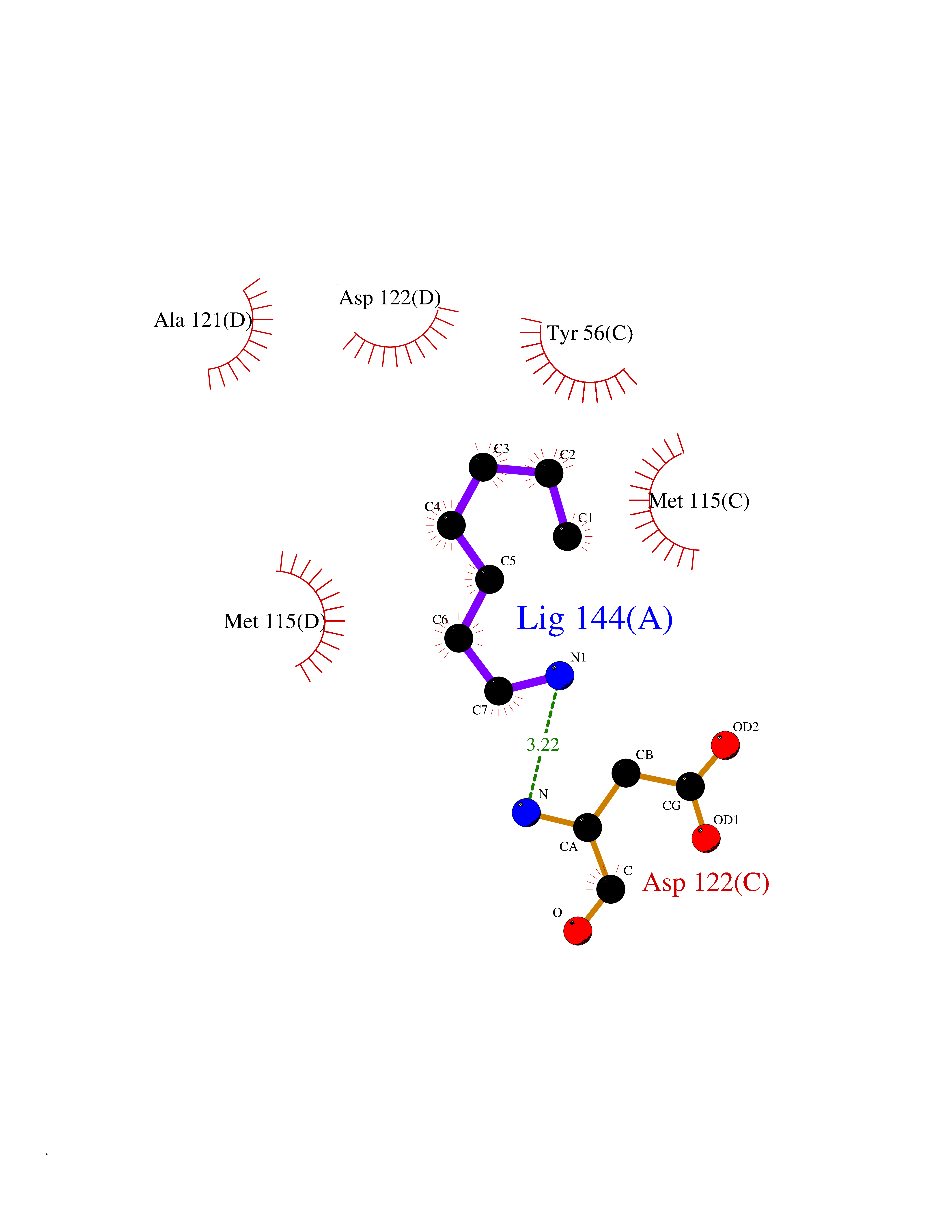 Ligplot Map 3D Binding mode Sequence AFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHHHAFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHH Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Intestinal maltase-glucoamylase (MGAM) | 3L4Y | 5.04 | |
Target general information Gen name MGAM Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MGAM Protein family Glycosyl hydrolase 31 family Biochemical class Glycosylase Function May serve as an alternate pathway for starch digestion when luminal alpha-amylase activity is reduced because of immaturity or malnutrition. May play a unique role in the digestion of malted dietary oligosaccharides used in food manufacturing. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB00284; DB00491; DB04878 Interacts with Q13520; Q7Z7G2; Q96BA8; O15529; P14410; P54219-3; Q9NUH8 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; Glycosidase; Hydrolase; Membrane; Multifunctional enzyme; Proteomics identification; Reference proteome; Repeat; Signal-anchor; Sulfation; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 97779.4 Length 863 Aromaticity 0.12 Instability index 32.47 Isoelectric point 5.2 Charge (pH=7) -28.27 2D Binding mode Binding energy (Kcal/mol) -6.88  Molscript Map 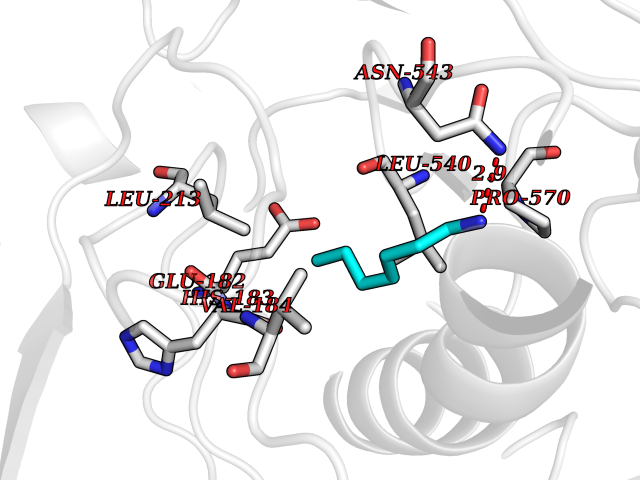 Pymol Map 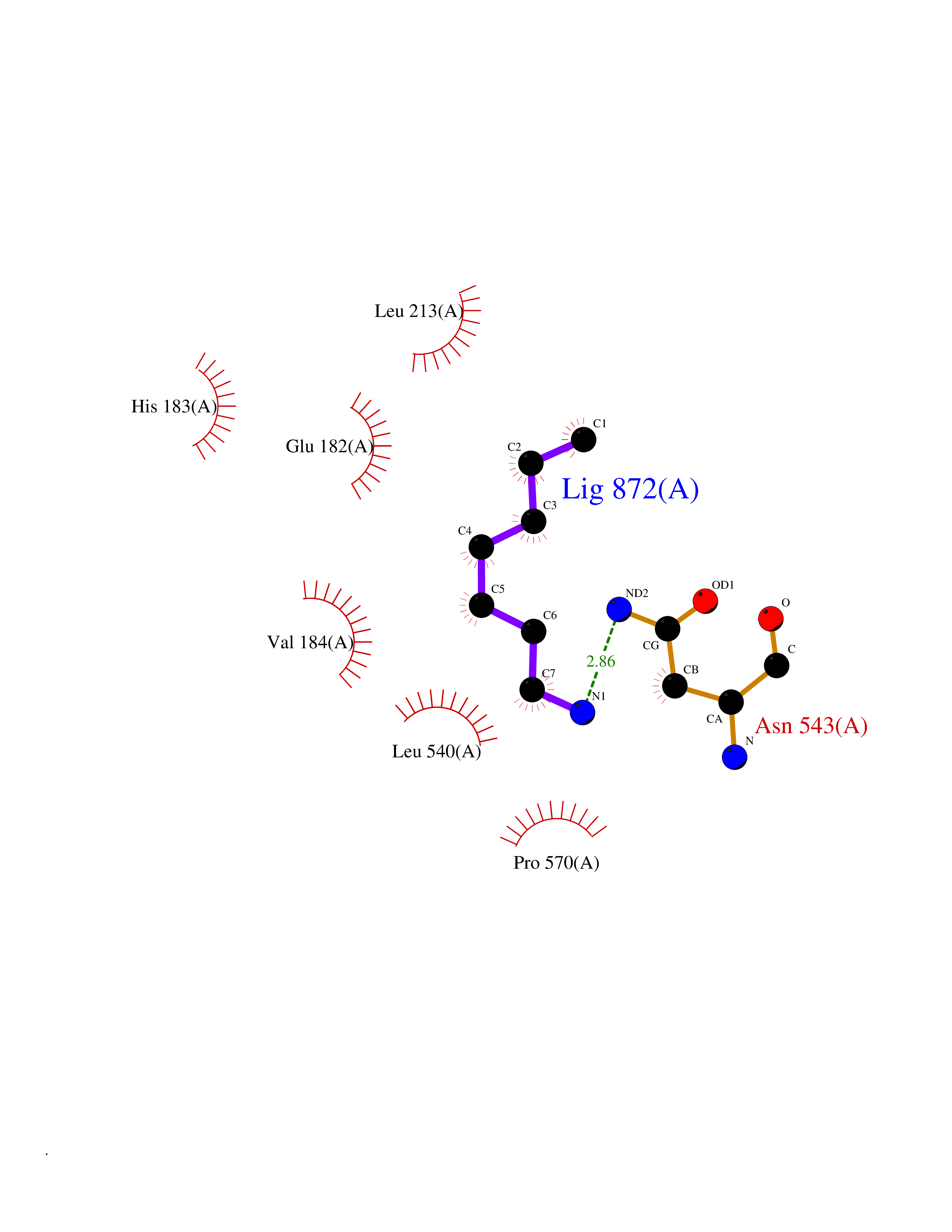 Ligplot Map 3D Binding mode Sequence VNELERINCIPDQPPTKATCDQRGCCWNPQGAVSVPWCYYSKNHSYHVEGNLVNTNAGFTARLKNLPSSPVFGSNVDNVLLTAEYQTSNRFHFKLTDQTNNRFEVPHEHVQSFSGNAAASLTYQVEISRQPFSIKVTRRSNNRVLFDSSIGPLLFADQFLQLSTRLPSTNVYGLGEHVHQQYRHDMNWKTWPIFNRDTTPNGNGTNLYGAQTFFLCLEDASGLSFGVFLMNSNAMEVVLQPAPAITYRTIGGILDFYVFLGNTPEQVVQEYLELIGRPALPSYWALGFHLSRYEYGTLDNMREVVERNRAAQLPYDVQHADIDYMDERRDFTYDSVDFKGFPEFVNELHNNGQKLVIIVDPAISNNSSSSKPYGPYDRGSDMKIWVNSSDGVTPLIGEVWPGQTVFPDYTNPNCAVWWTKEFELFHNQVEFDGIWIDMNEVSNFVDGSVSGCSTNNLNNPPFTPRILDGYLFCKTLCMDAVQHWGKQYDIHNLYGYSMAVATAEAAKTVFPNKRSFILTRSTFAGSGKFAAHWLGDNTATWDDLRWSIPGVLEFNLFGIPMVGPDICGFALDTPEELCRRWMQLGAFYPFSRNHNGQGYKDQDPASFGADSLLLNSSRHYLNIRYTLLPYLYTLFFRAHSRGDTVARPLLHEFYEDNSTWDVHQQFLWGPGLLITPVLDEGAEKVMAYVPDAVWYDYETGSQVRWRKQKVEMELPGDKIGLHLRGGYIFPTQQPNTTTLASRKNPLGLIIALDENKEAKGELFWDDGETKDTVANKVYLLCEFSVTQNRLEVNISQSTYKDPNNLAFNEIKILGTEEPSNVTVKHNGVPSTSPTVTYDSNLKVAIITDIDLLLGEAYTVEWAH Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Fatty acid-binding protein, intestinal | 3AKM | 5.04 | |
Target general information Gen name FABP2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FABPI Protein family Calycin superfamily, Fatty-acid binding protein (FABP) family Biochemical class Transport protein Function Fatty acid binding.Transporter activity. Related diseases Usher syndrome 3B (USH3B) [MIM:614504]: A syndrome characterized by progressive vision and hearing loss during early childhood. Some patients have the so-called 'Charles Bonnet syndrome,' involving decreased visual acuity and vivid visual hallucinations. USH is a genetically heterogeneous condition characterized by the association of retinitis pigmentosa with sensorineural deafness. Age at onset and differences in auditory and vestibular function distinguish Usher syndrome type 1 (USH1), Usher syndrome type 2 (USH2) and Usher syndrome type 3 (USH3). USH3 is characterized by postlingual, progressive hearing loss, variable vestibular dysfunction, and onset of retinitis pigmentosa symptoms, including nyctalopia, constriction of the visual fields, and loss of central visual acuity, usually by the second decade of life. {ECO:0000269|PubMed:22279524}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2W (CMT2W) [MIM:616625]: An autosomal dominant, axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2W patients manifest a peripheral neuropathy mainly affecting the lower limbs and resulting in gait difficulties and distal sensory impairment. Most patients also have upper limb involvement. {ECO:0000269|PubMed:22930593, ECO:0000269|PubMed:26072516, ECO:0000269|PubMed:29235198}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04557; DB09213; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB01050; DB08231; DB03796; DB01138 Interacts with O95994; Q9NYB0 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 15075.9 Length 131 Aromaticity 0.11 Instability index 32.01 Isoelectric point 6.88 Charge (pH=7) -0.09 2D Binding mode Binding energy (Kcal/mol) -6.88 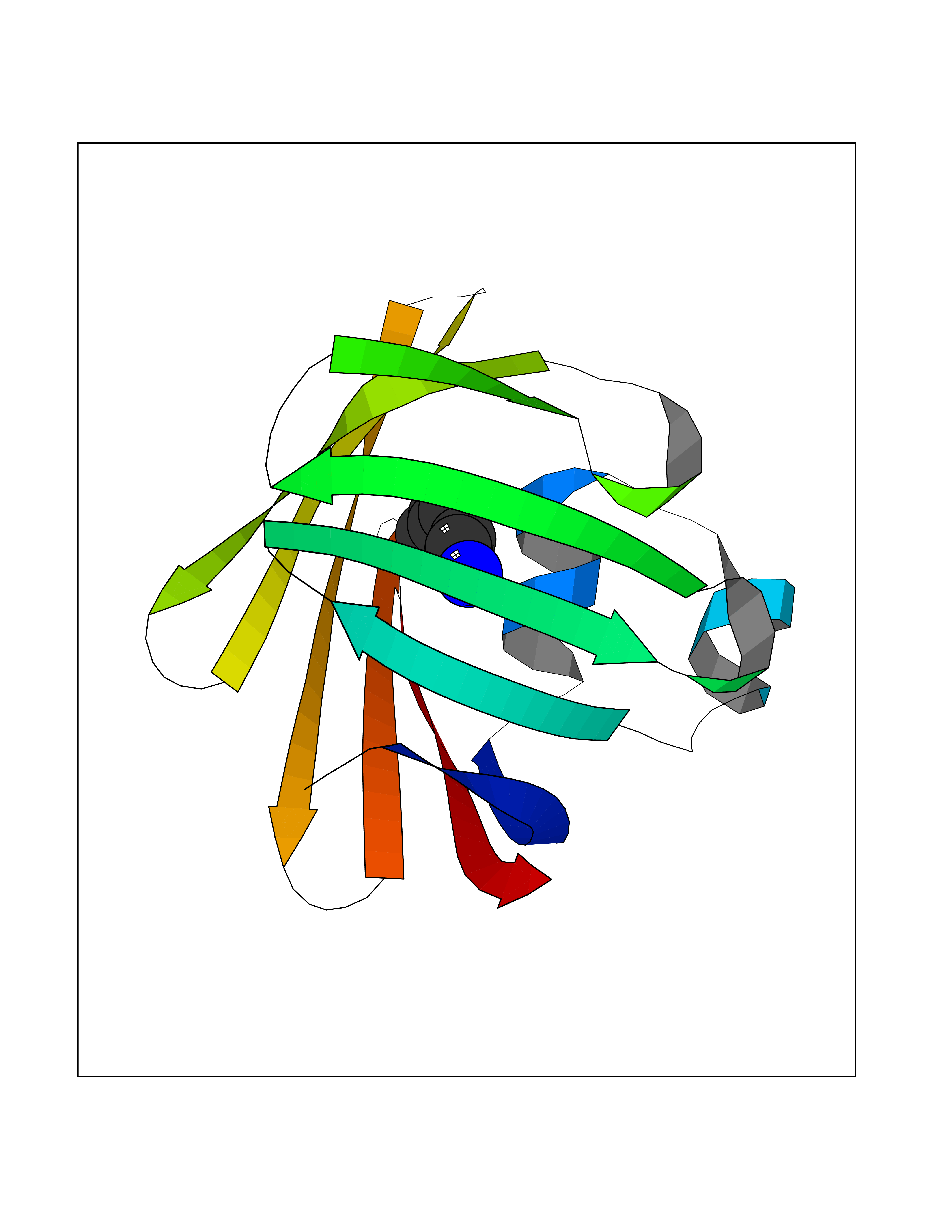 Molscript Map 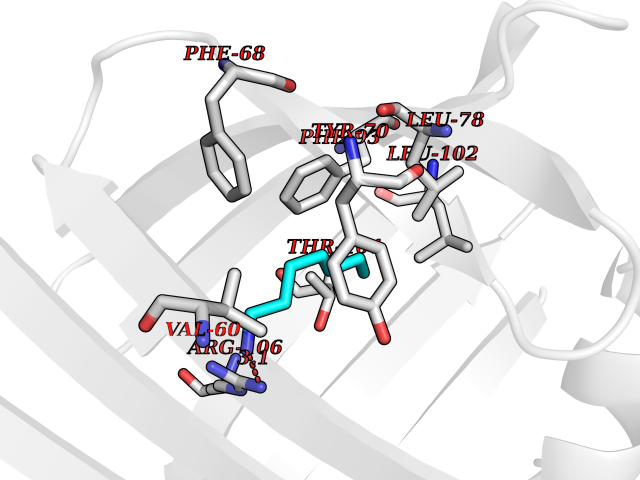 Pymol Map 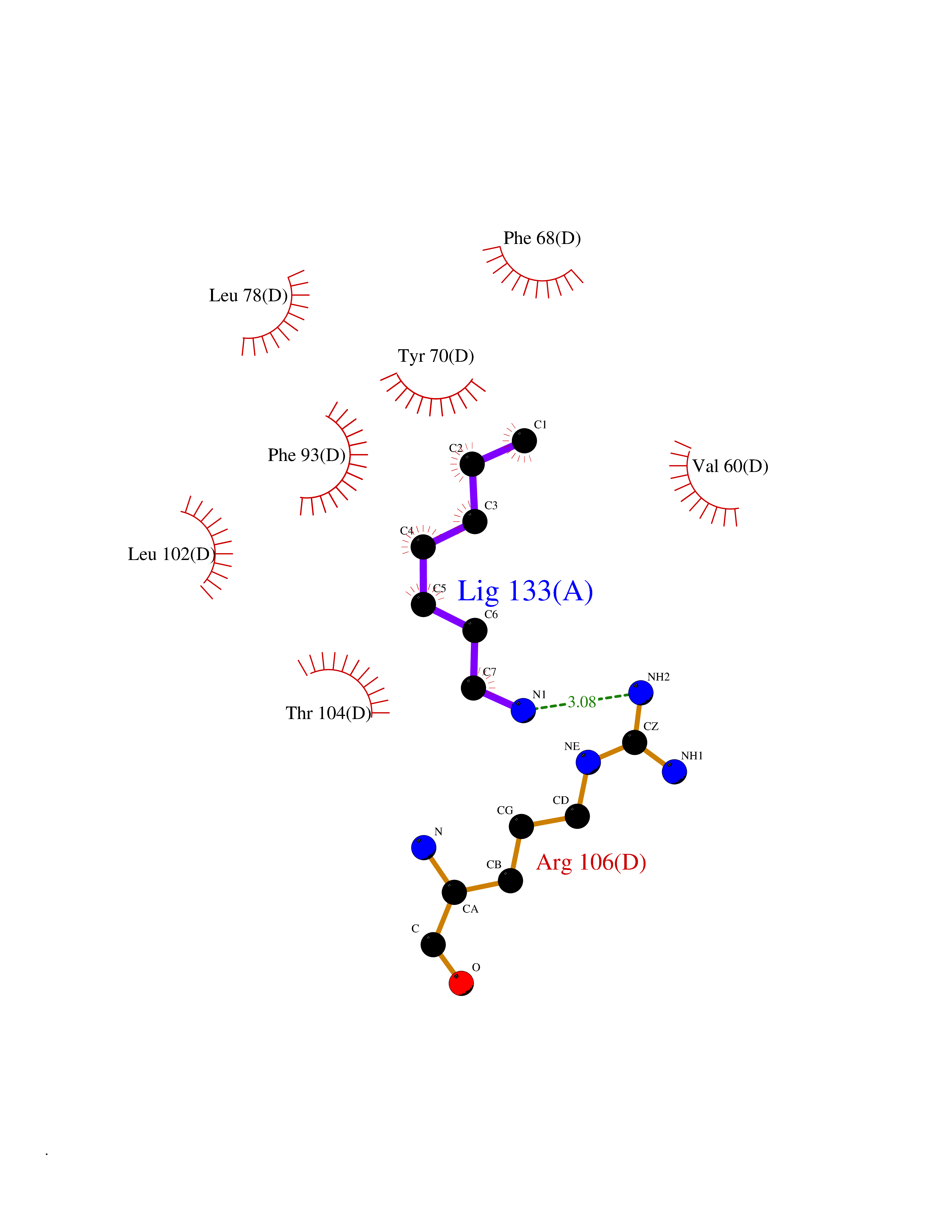 Ligplot Map 3D Binding mode Sequence AFDSTWKVDRSENYDKFMEKMGVNIVKRKLAAHDNLKLTITQEGNKFTVKESSAFRNIEVVFELGVTFNYNLADGTELRGTWSLEGNKLIGKFKRTDNGNELNTVREIIGDELVQTYVYEGVEAKRIFKKD Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Retinaldehyde-binding protein 1 | 3HX3 | 5.04 | |
Target general information Gen name RLBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CRALBP Protein family NA Biochemical class Transport protein Function 11-cis retinal binding.Retinol binding.Transporter activity. Related diseases Bothnia retinal dystrophy (BRD) [MIM:607475]: A type of retinitis punctata albescens. Affected individuals show night blindness from early childhood with features consistent with retinitis punctata albescens and macular degeneration. {ECO:0000269|PubMed:10102298}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rod-cone dystrophy Newfoundland (NFRCD) [MIM:607476]: A rod-cone dystrophy reminiscent of retinitis punctata albescens but with a substantially lower age at onset and more-rapid and distinctive progression. Rod-cone dystrophies results from initial loss of rod photoreceptors, later followed by cone photoreceptors loss. {ECO:0000269|PubMed:11868161}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Retinitis punctata albescens (RPA) [MIM:136880]: A form of fleck retina disease characterized by aggregation of white flecks posteriorly in the retina, causing night blindness and delayed dark adaptation. It differs from fundus albipunctatus in being progressive and evolving to generalized atrophy of the retina. {ECO:0000269|PubMed:10102299, ECO:0000269|PubMed:11453974, ECO:0000269|PubMed:9326942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00162 Interacts with Q9P2G9-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Disease variant; Proteomics identification; Reference proteome; Retinol-binding; Sensory transduction; Transport; Vision Protein physicochemical properties Chain ID A Molecular weight (Da) 28328.6 Length 250 Aromaticity 0.14 Instability index 52.64 Isoelectric point 4.96 Charge (pH=7) -9.87 2D Binding mode Binding energy (Kcal/mol) -6.88 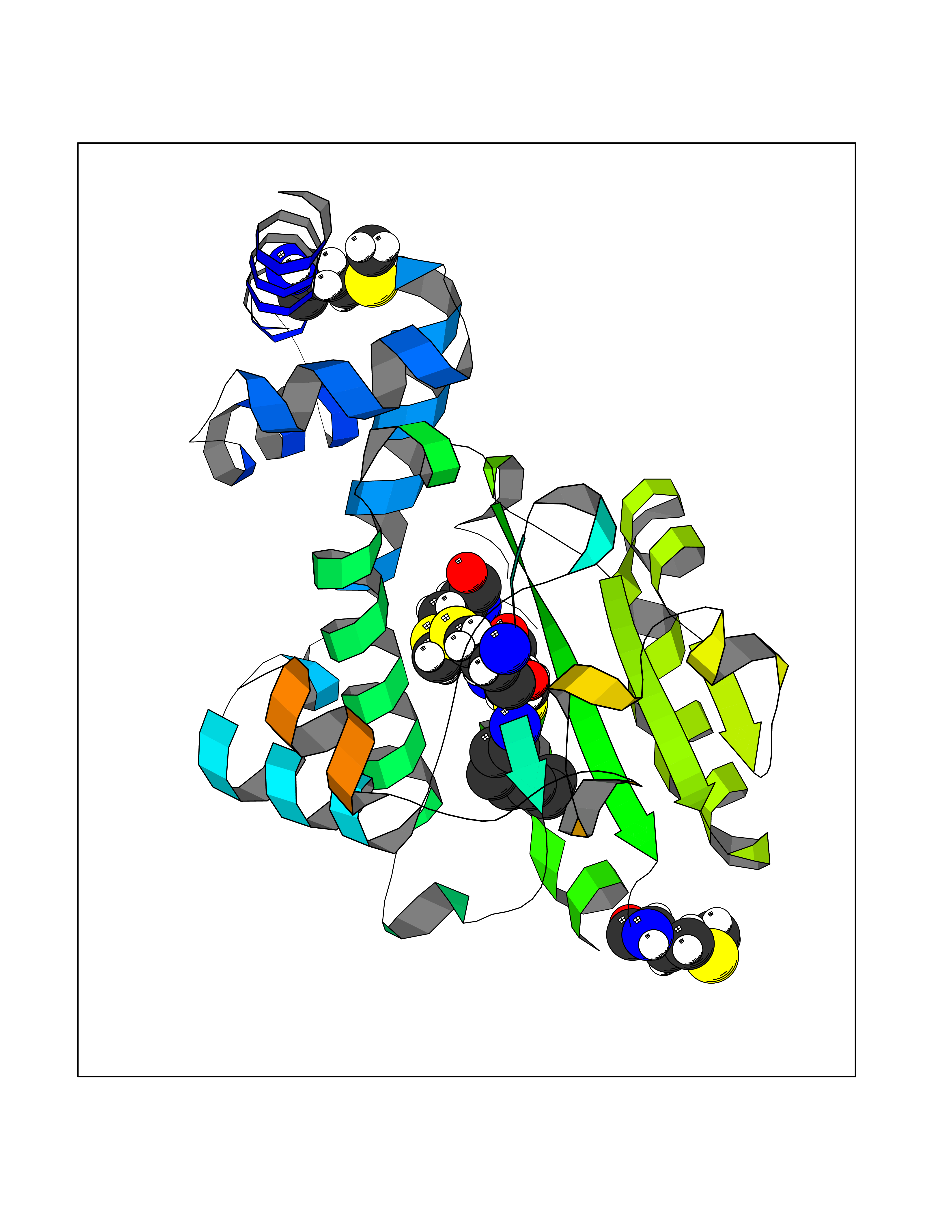 Molscript Map 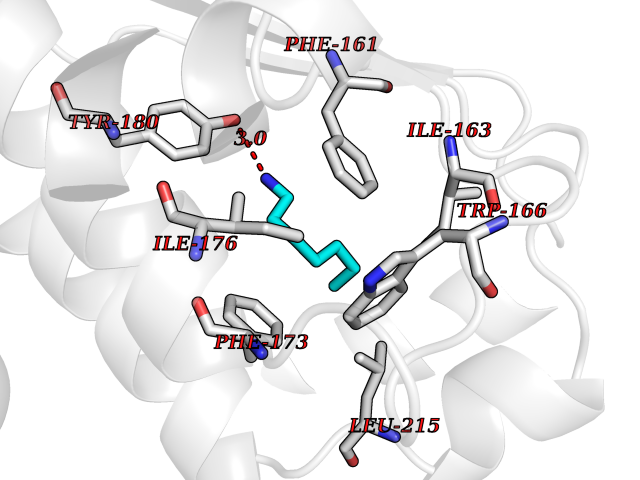 Pymol Map 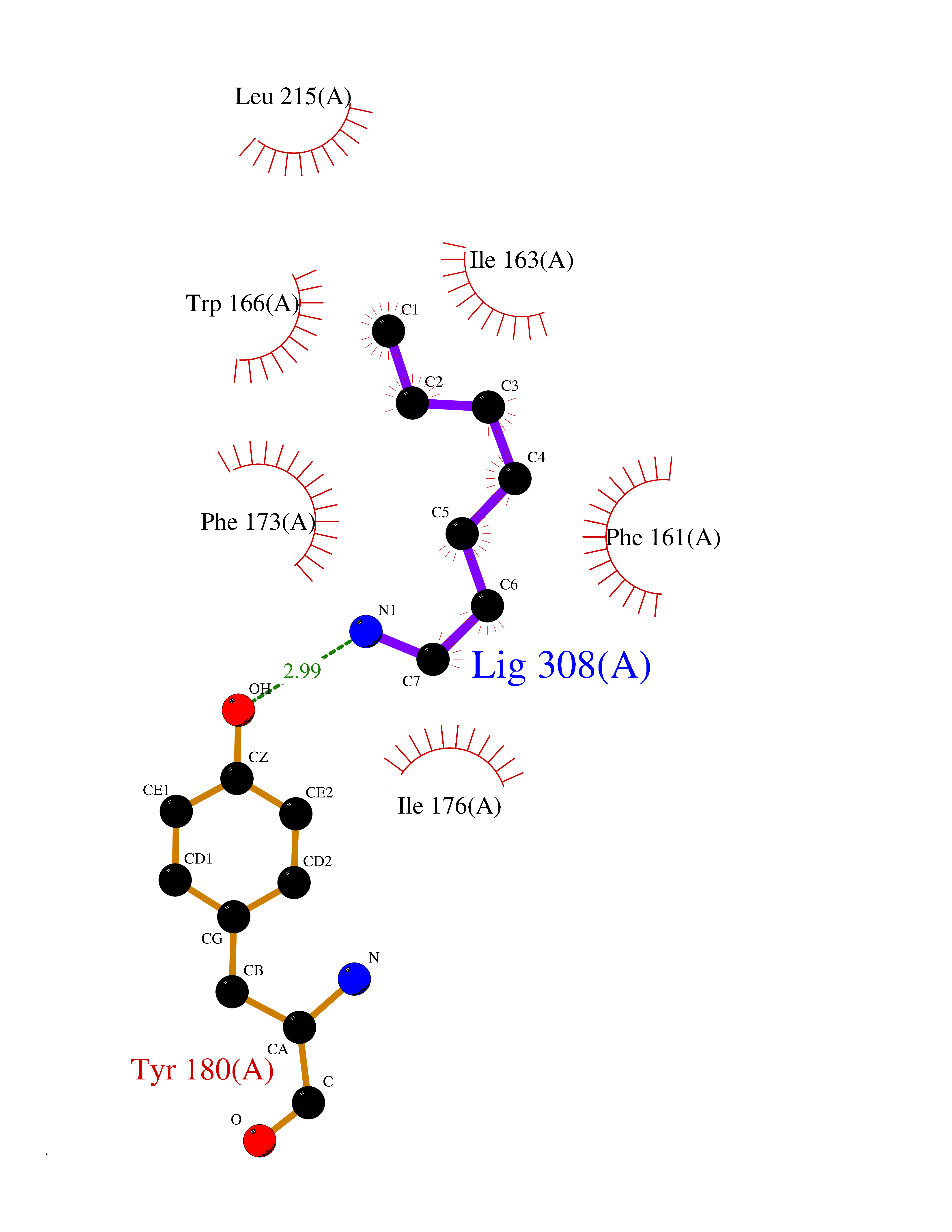 Ligplot Map 3D Binding mode Sequence ETREEAVRELQEXVQAQAASGEELAVAVAERVQEKDSGFFLRFIRARKFNVGRAYELLRGYVNFRLQYPELFDSLSPEAVRCTIEAGYPGVLSSRDKYGRVVXLFNIENWQSQEITFDEILQAYCFILEKLLENEETQINGFCIIENFKGFTXQQAASLRTSDLRKXVDXLQDSFPAWFKAIHFIHQPWYFTTTYNVVKPFLKSKLLERVFVHGDDLSGFYQEIDENILPSDFGGTLPKYDGKAVAEQLF Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | 5'-methylthioadenosine/S-adenosylhomocysteine nucleosidase | 4WKC | 5.04 | |
Target general information Gen name mtnN Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms pfs;b0159;yadA;JW0155;mtn Protein family PNP/UDP phosphorylase family, MtnN subfamily Biochemical class hydrolase / hydrolase inhibitor Function Adenosylhomocysteine nucleosidase activity.Identical protein binding.Methylthioadenosine nucleosidase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02158; DB08606; DB02933; DB00173; DB02281 Interacts with P0AF12 EC number 3.2.2.9 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Hydrolase; Methionine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 24353.7 Length 232 Aromaticity 0.05 Instability index 22.1 Isoelectric point 5.09 Charge (pH=7) -9.9 2D Binding mode Binding energy (Kcal/mol) -6.87 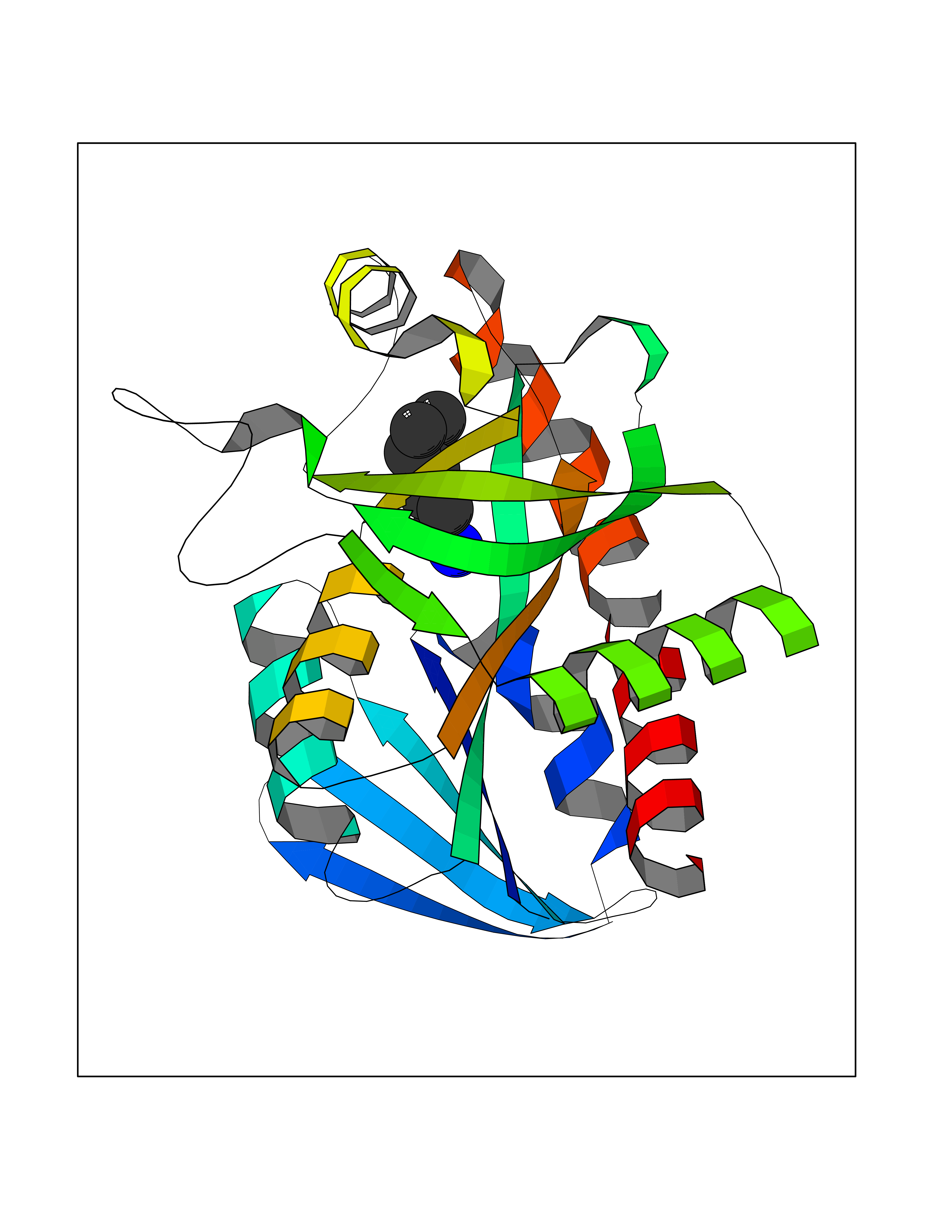 Molscript Map 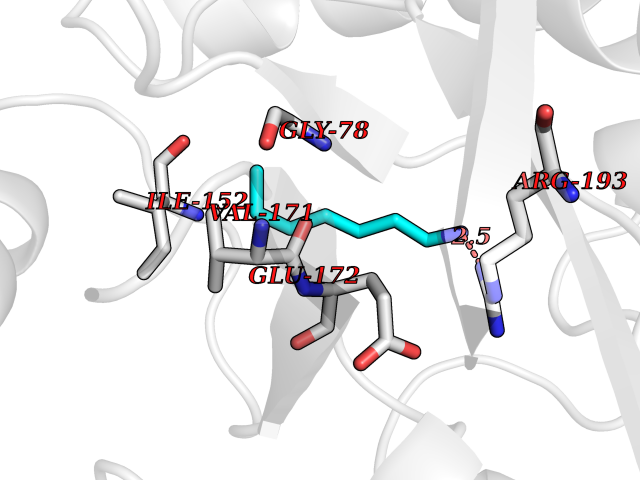 Pymol Map 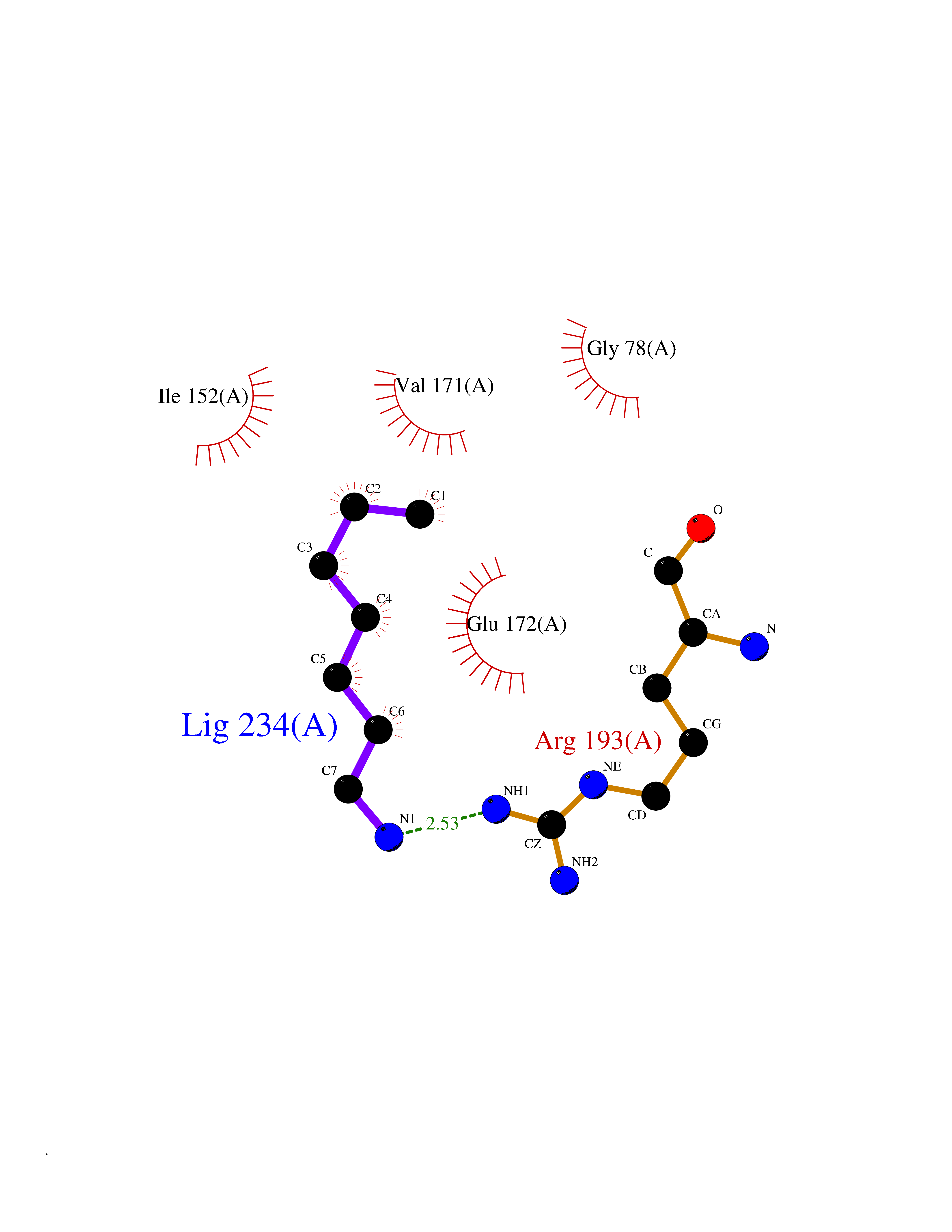 Ligplot Map 3D Binding mode Sequence MKIGIIGAMEEEVTLLRDKIENRQTISLGGCEIYTGQLNGTEVALLKSGIGKVAAALGATLLLEHCKPDVIINTGSAGGLAPTLKVGDIVVSDEARYHDADVTAFGYEYGQLPGCPAGFKADDKLIAAAEACIAELNLNAVRGLIVSGDAFINGSVGLAKIRHNFPQAIAVEMEATAIAHVCHNFNVPFVVVRAISDVADQQSHLSFDEFLAVAAKQSSLMVESLVQKLAHG Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Pyruvate synthase | 2C42 | 5.04 | |
Target general information Gen name por Organism Desulfocurvibacter africanus (Desulfovibrio africanus) Uniprot ID TTD ID NA Synonyms NA Protein family Pyruvate:ferredoxin/flavodoxin oxidoreductase family Biochemical class Oxidoreductase Function 4 iron, 4 sulfur cluster binding.Iron ion binding.Pyruvate synthase activity.Thiamine pyrophosphate binding. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02410; DB01987; DB00507 Interacts with NA EC number 1.2.7.1 Uniprot keywords 3D-structure; 4Fe-4S; Calcium; Cytoplasm; Direct protein sequencing; Disulfide bond; Electron transport; Iron; Iron-sulfur; Magnesium; Metal-binding; Oxidoreductase; Pyruvate; Thiamine pyrophosphate; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 115569 Length 1065 Aromaticity 0.09 Instability index 31.51 Isoelectric point 6.32 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -6.87  Molscript Map 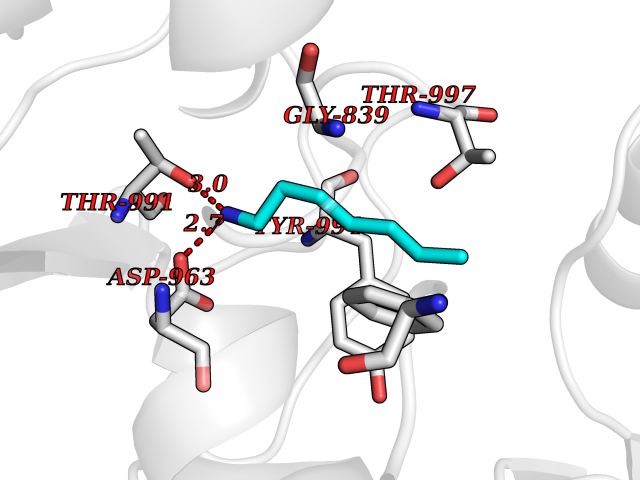 Pymol Map 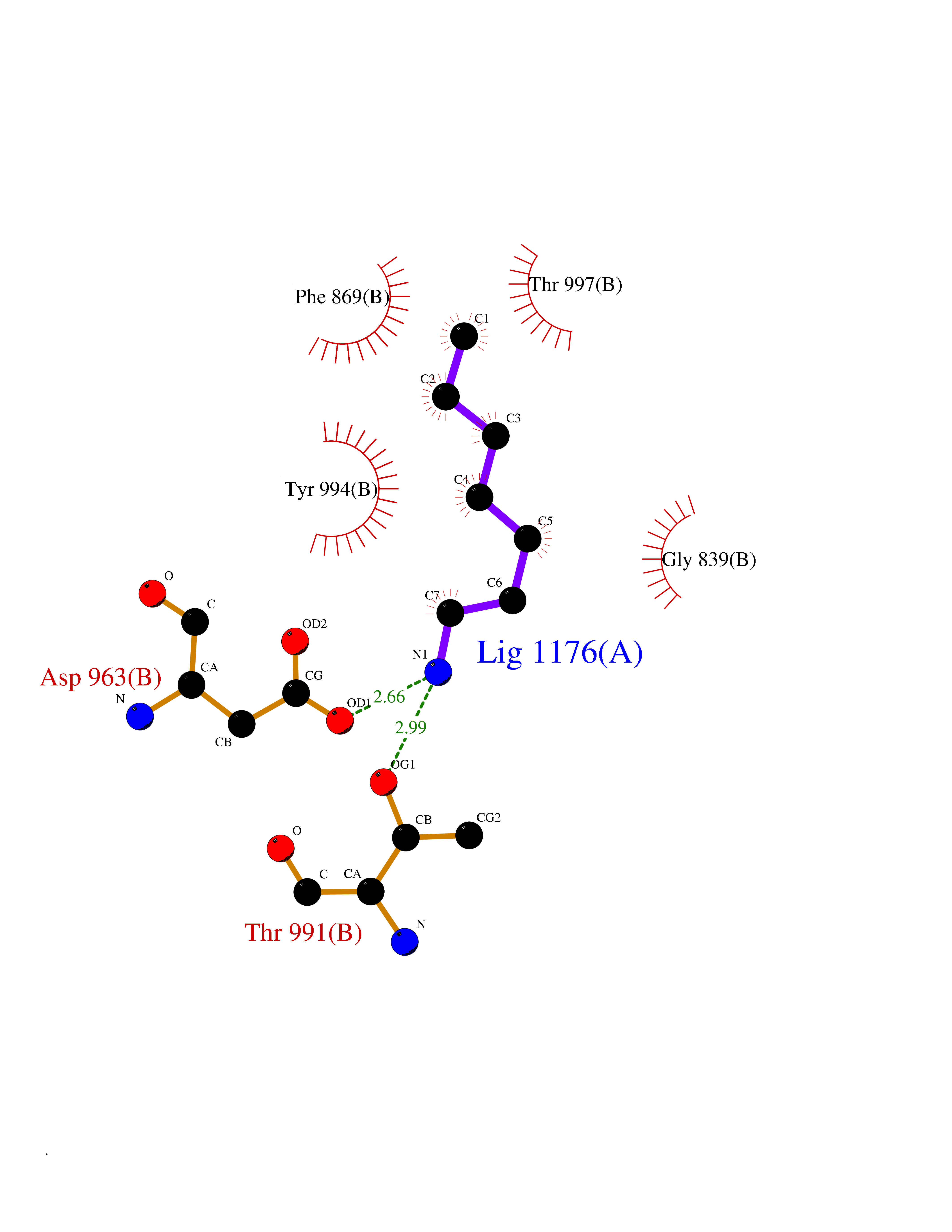 Ligplot Map 3D Binding mode Sequence GKKMMTTDGNTATAHVAYAMSEVAAIYPITPSSTMGEEADDWAAQGRKNIFGQTLTIREMQSEAGAAGAVHGALAAGALTTTFTASQGLLLMIPNMYKISGELLPGVFHVTARAIAAHALSIFGDHQDIYAARQTGFAMLASSSVQEAHDMALVAHLAAIESNVPFMHFFDGFRTSHEIQKIEVLDYADMASLVNQKALAEFRAKSPGIVAEYMQKVASLTGRSYKLFDYVGAPDAERVIVSMGSSCETIEEVINHLAAKGEKIGLIKVRLYRPFVSEAFFAALPASAKVITVLDRTKEPGAPGDPLYLDVCSAFVERGEAMPKILAGRYGLGSKEFSPAMVKSVYDNMSGAKKNHFTVGIEDDVTGTSLPVDNAFADTTPKGTIQCQFWGLGADGTVGANKQAIKIIGDNTDLFAQGYFSYDSKKSGGITISHLRFGEKPIQSTYLVNRADYVACHNPAYVGIYDILEGIKDGGTFVLNSPWSSLEDMDKHLPSGIKRTIANKKLKFYNIDAVKIATDVGLGGRINMIMQTAFFKLAGVLPFEKAVDLLKKSIHKAYGKKGEKIVKMNTDAVDQAVTSLQEFKYPDSWKDAPAETKAEPMTNEFFKNVVKPILTQQGDKLPVSAFEADGRFPLGTSQFEKRGVAINVPQWVPENCIQCNQCAFVCPHSAILPVLAKEEELVGAPANFTALEAKGKELKGYKFRIQINTLDCMGCGNCADICPPKEKALVMQPLDTQRDAQVPNLEYAARIPVKSEVLPRDSLKGSQFQEPLMEFSGACSGCGETPYVRVITQLFGERMFIANATGCSSIWGASAPSMPYKTNRLGQGPAWGNSLFEDAAEYGFGMSVWIFGGDGWAYDIGYGGLDHVLASGEDVNVFVMDTEVYSNTGGQSSKATPTGAVAKFAAAGKRTGKKDLARMVMTYGYVYVATVSMGYSKQQFLKVLKEAESFPGPSLVIAYATCINQGLRKGMGKSQDVMNTAVKSGYWPLFRYDPRLAAQGKNPFQLDSKAPDGSVEEFLMAQNRFAVLDRSFPEDAKRLRAQVAHELDVRFKELEHMAATNIFES Hydrogen bonds contact Hydrophobic contact | ||||