Job Results:
Ligand
Structure
Job ID
d1681d1a563fba4481a5af742fb3bb0c
Job name
NA
Time
2026-02-27 11:58:22
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Ephrin type-B receptor 3 (EPHB3) | 5L6O | 4.62 | |
Target general information Gen name EPHB3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hEK2; Tyrosine-protein kinase TYRO6; TYRO6; Embryonic kinase 2; ETK2; EPH-like tyrosine kinase 2; EPH-like kinase 2; EK2 Protein family Protein kinase superfamily, Tyr protein kinase family, Ephrin receptor subfamily Biochemical class Kinase Function The signaling pathway downstream of the receptor is referred to as forward signaling while the signaling pathway downstream of the ephrin ligand is referred to as reverse signaling. Generally has an overlapping and redundant function with EPHB2. Like EPHB2, functions in axon guidance during development regulating for instance the neurons forming the corpus callosum and the anterior commissure, 2 major interhemispheric connections between the temporal lobes of the cerebral cortex. In addition to its role in axon guidance plays also an important redundant role with other ephrin-B receptors in development and maturation of dendritic spines and the formation of excitatory synapses. Controls other aspects of development through regulation of cell migration and positioning. This includes angiogenesis, palate development and thymic epithelium development for instance. Forward and reverse signaling through the EFNB2/EPHB3 complex also regulate migration and adhesion of cells that tubularize the urethra and septate the cloaca. Finally, plays an important role in intestinal epithelium differentiation segregating progenitor from differentiated cells in the crypt. Receptor tyrosine kinase which binds promiscuously transmembrane ephrin-B family ligands residing on adjacent cells, leading to contact-dependent bidirectional signaling into neighboring cells. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) NA Interacts with P37235; O75031 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Angiogenesis; ATP-binding; Cell membrane; Cell projection; Developmental protein; Disulfide bond; Glycoprotein; Kinase; Membrane; Neurogenesis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30412.9 Length 267 Aromaticity 0.09 Instability index 37.42 Isoelectric point 7.74 Charge (pH=7) 1 2D Binding mode Binding energy (Kcal/mol) -6.3  Molscript Map 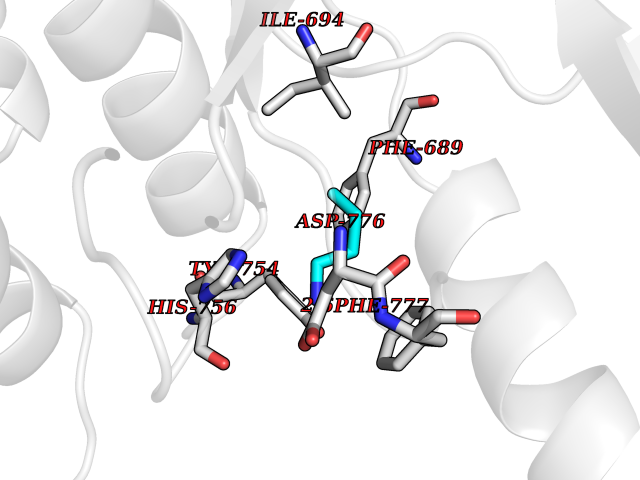 Pymol Map 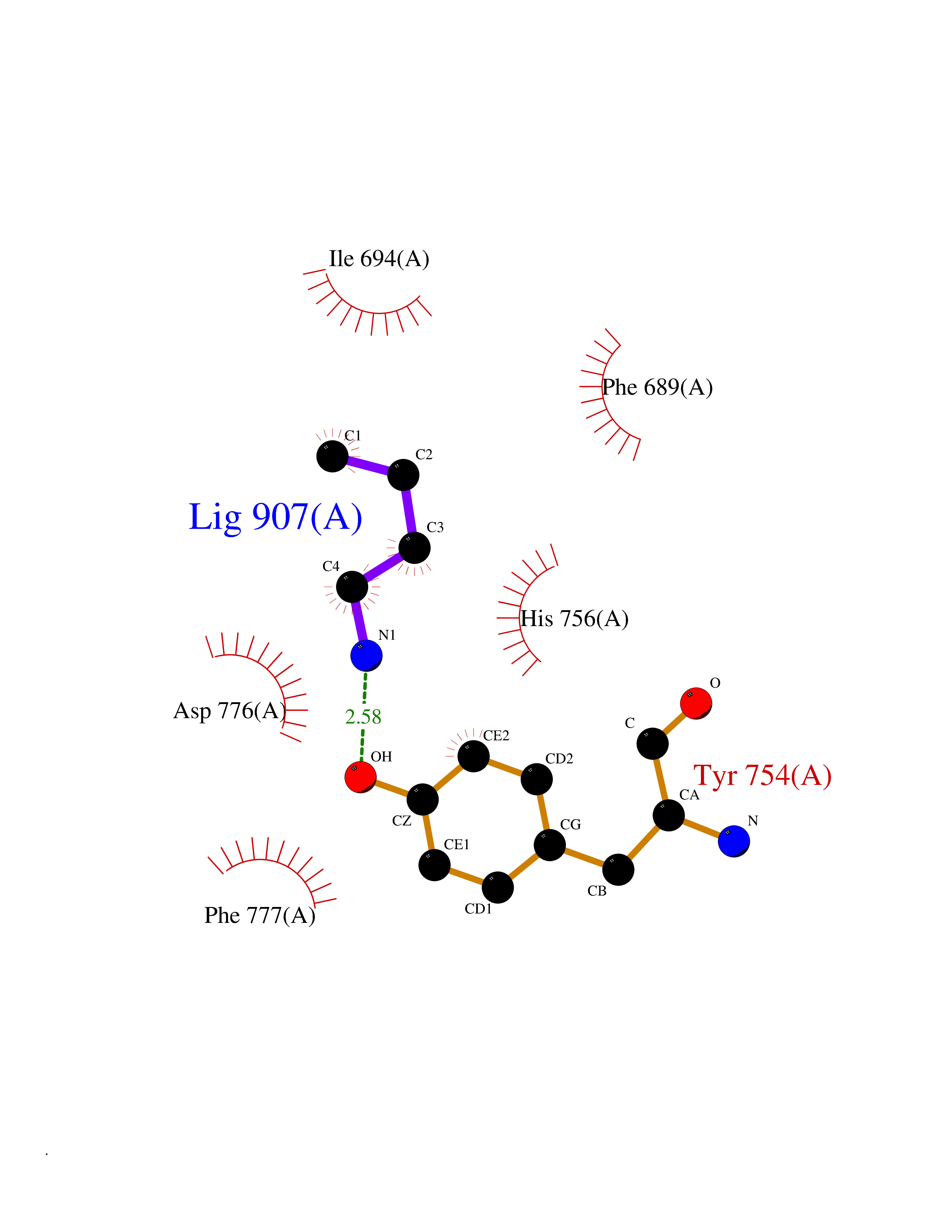 Ligplot Map 3D Binding mode Sequence CVKIEEVIGAGEVCRGRLKQPGRREVFVAIKTLKVGYTERQRRDFLSEASIMGQFDHPNIIRLEGVVTKSRPVMILTEFMENCALDSFLRLNDGQFTVIQLVGMLRGIAAGMKYLSEMNYVHRDLAARNILVNSNLVCKVSDFGLEDDPSDPTYTSSLGGKIPIRWTAPEAIAYRKFTSASDVWSYGIVMWEVMSYGERPYWDMSNQDVINAVEQDYRLPPPMDCPTALHQLMLDCWVRDRNLRPKFSQIVNTLDKLIRNPASLKVI Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Neuronal acetylcholine receptor alpha-3/beta-4 (CHRNA3/B4) | 6PV7 | 4.62 | |
Target general information Gen name CHRNA3-CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Neuronal acetylcholine receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Neurotransmitter receptor Function A type of nicotinic acetylcholine receptor, consisting of 3 and 4 subunits. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -6.3  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Intestinal maltase-glucoamylase (MGAM) | 3L4Y | 4.61 | |
Target general information Gen name MGAM Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MGAM Protein family Glycosyl hydrolase 31 family Biochemical class Glycosylase Function May serve as an alternate pathway for starch digestion when luminal alpha-amylase activity is reduced because of immaturity or malnutrition. May play a unique role in the digestion of malted dietary oligosaccharides used in food manufacturing. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB00284; DB00491; DB04878 Interacts with Q13520; Q7Z7G2; Q96BA8; O15529; P14410; P54219-3; Q9NUH8 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; Glycosidase; Hydrolase; Membrane; Multifunctional enzyme; Proteomics identification; Reference proteome; Repeat; Signal-anchor; Sulfation; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 97779.4 Length 863 Aromaticity 0.12 Instability index 32.47 Isoelectric point 5.2 Charge (pH=7) -28.27 2D Binding mode Binding energy (Kcal/mol) -6.29  Molscript Map 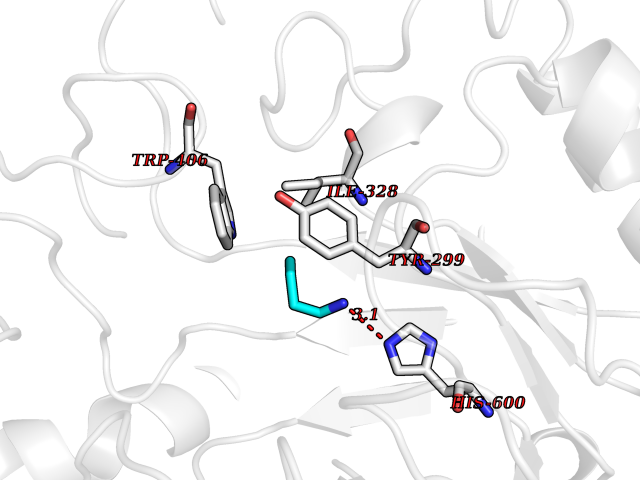 Pymol Map 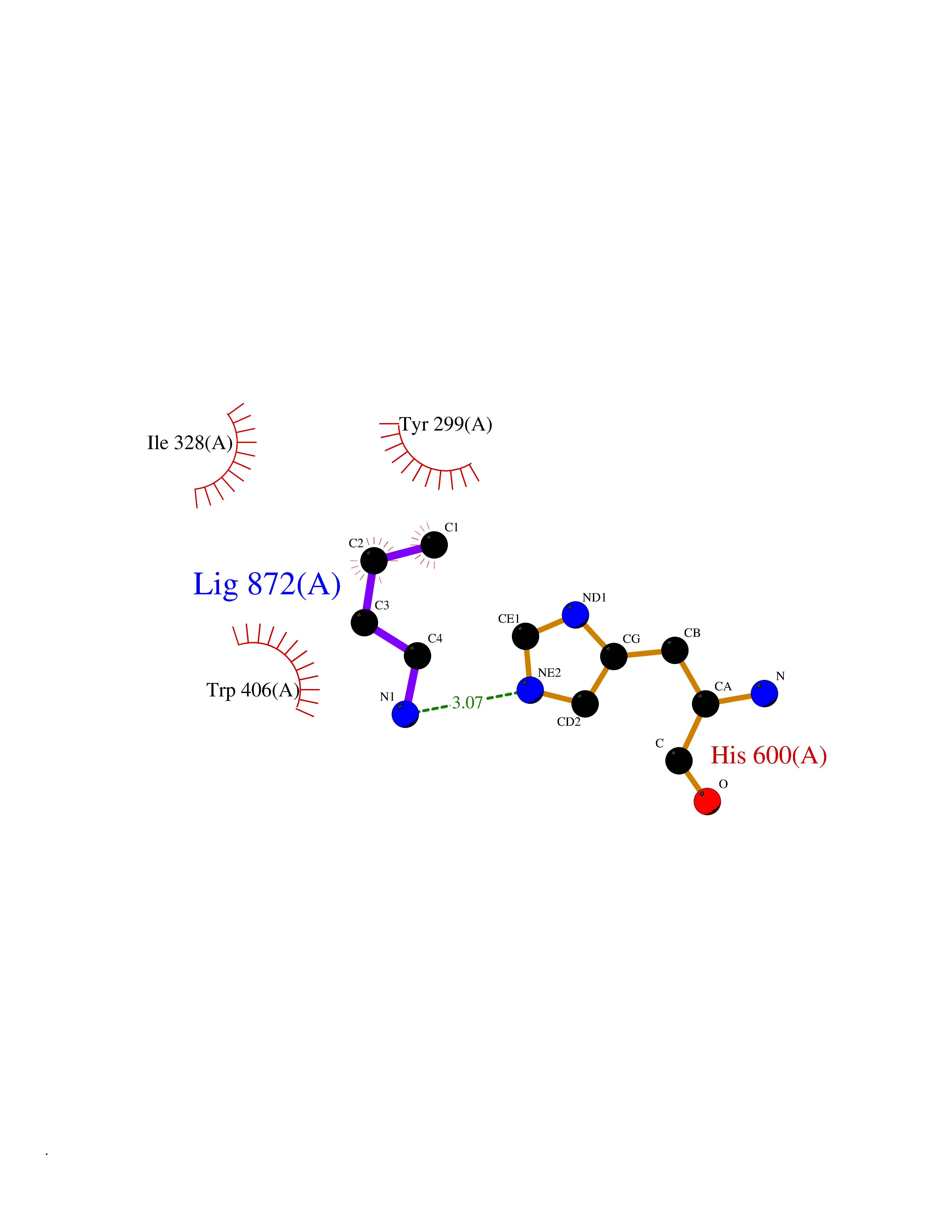 Ligplot Map 3D Binding mode Sequence VNELERINCIPDQPPTKATCDQRGCCWNPQGAVSVPWCYYSKNHSYHVEGNLVNTNAGFTARLKNLPSSPVFGSNVDNVLLTAEYQTSNRFHFKLTDQTNNRFEVPHEHVQSFSGNAAASLTYQVEISRQPFSIKVTRRSNNRVLFDSSIGPLLFADQFLQLSTRLPSTNVYGLGEHVHQQYRHDMNWKTWPIFNRDTTPNGNGTNLYGAQTFFLCLEDASGLSFGVFLMNSNAMEVVLQPAPAITYRTIGGILDFYVFLGNTPEQVVQEYLELIGRPALPSYWALGFHLSRYEYGTLDNMREVVERNRAAQLPYDVQHADIDYMDERRDFTYDSVDFKGFPEFVNELHNNGQKLVIIVDPAISNNSSSSKPYGPYDRGSDMKIWVNSSDGVTPLIGEVWPGQTVFPDYTNPNCAVWWTKEFELFHNQVEFDGIWIDMNEVSNFVDGSVSGCSTNNLNNPPFTPRILDGYLFCKTLCMDAVQHWGKQYDIHNLYGYSMAVATAEAAKTVFPNKRSFILTRSTFAGSGKFAAHWLGDNTATWDDLRWSIPGVLEFNLFGIPMVGPDICGFALDTPEELCRRWMQLGAFYPFSRNHNGQGYKDQDPASFGADSLLLNSSRHYLNIRYTLLPYLYTLFFRAHSRGDTVARPLLHEFYEDNSTWDVHQQFLWGPGLLITPVLDEGAEKVMAYVPDAVWYDYETGSQVRWRKQKVEMELPGDKIGLHLRGGYIFPTQQPNTTTLASRKNPLGLIIALDENKEAKGELFWDDGETKDTVANKVYLLCEFSVTQNRLEVNISQSTYKDPNNLAFNEIKILGTEEPSNVTVKHNGVPSTSPTVTYDSNLKVAIITDIDLLLGEAYTVEWAH Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | 72 kDa type IV collagenase | 3AYU | 4.61 | |
Target general information Gen name MMP2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CLG4A Protein family Peptidase M10A family Biochemical class Hydrolase / hydrolase inhibitor Function Metalloendopeptidase activity.Metallopeptidase activity.Serine-type endopeptidase activity.Zinc ion binding. Related diseases Multicentric osteolysis, nodulosis, and arthropathy (MONA) [MIM:259600]: An autosomal recessive syndrome characterized by severe multicentric osteolysis with predominant involvement of the hands and feet. Additional features include coarse face, corneal opacities, patches of thickened, hyperpigmented skin, hypertrichosis and gum hypertrophy. {ECO:0000269|PubMed:11431697, ECO:0000269|PubMed:15691365, ECO:0000269|PubMed:16542393}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01197; DB06423; DB04866; DB00786; DB05387; DB12843; DB01630 Interacts with P05067; PRO_0000000092 [P05067]; PRO_0000000093 [P05067]; P20908; Q8NBP7; Q04864-2; Q8IX30; P16035 EC number 3.4.24.24 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; Autocatalytic cleavage; Calcium; Collagen degradation; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Mitochondrion; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 19623.6 Length 176 Aromaticity 0.14 Instability index 15.48 Isoelectric point 4.84 Charge (pH=7) -10.61 2D Binding mode Binding energy (Kcal/mol) -6.28 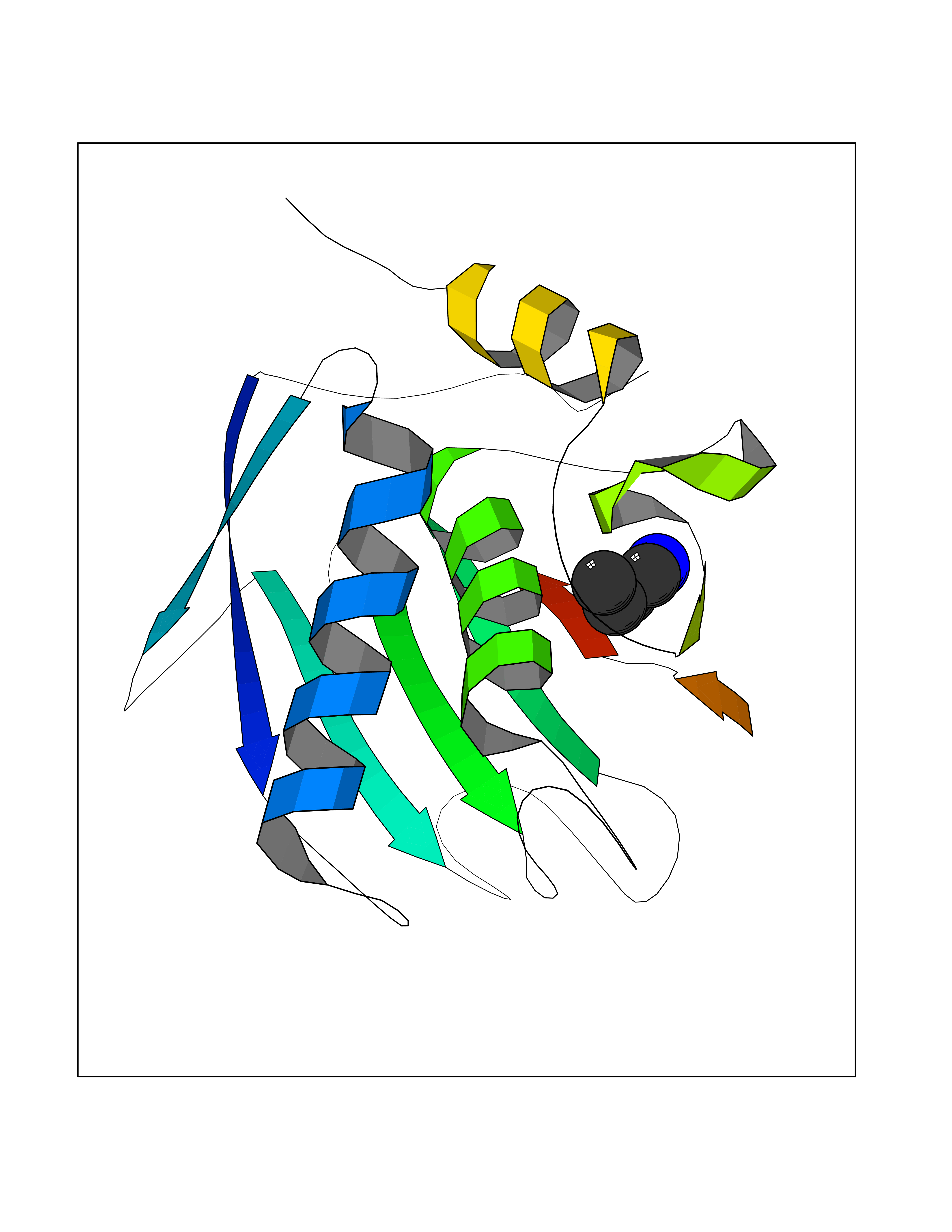 Molscript Map 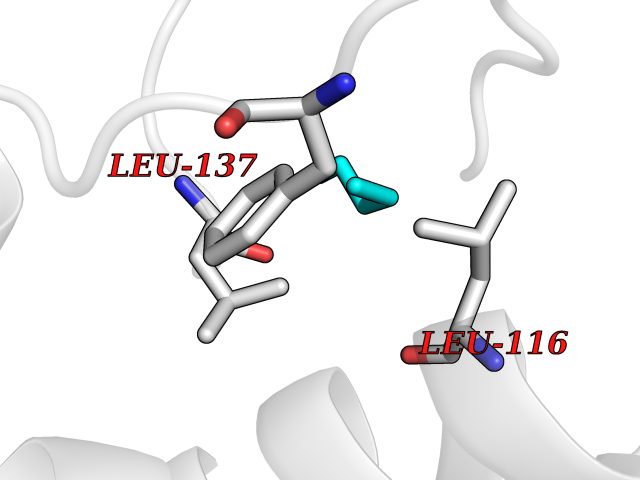 Pymol Map 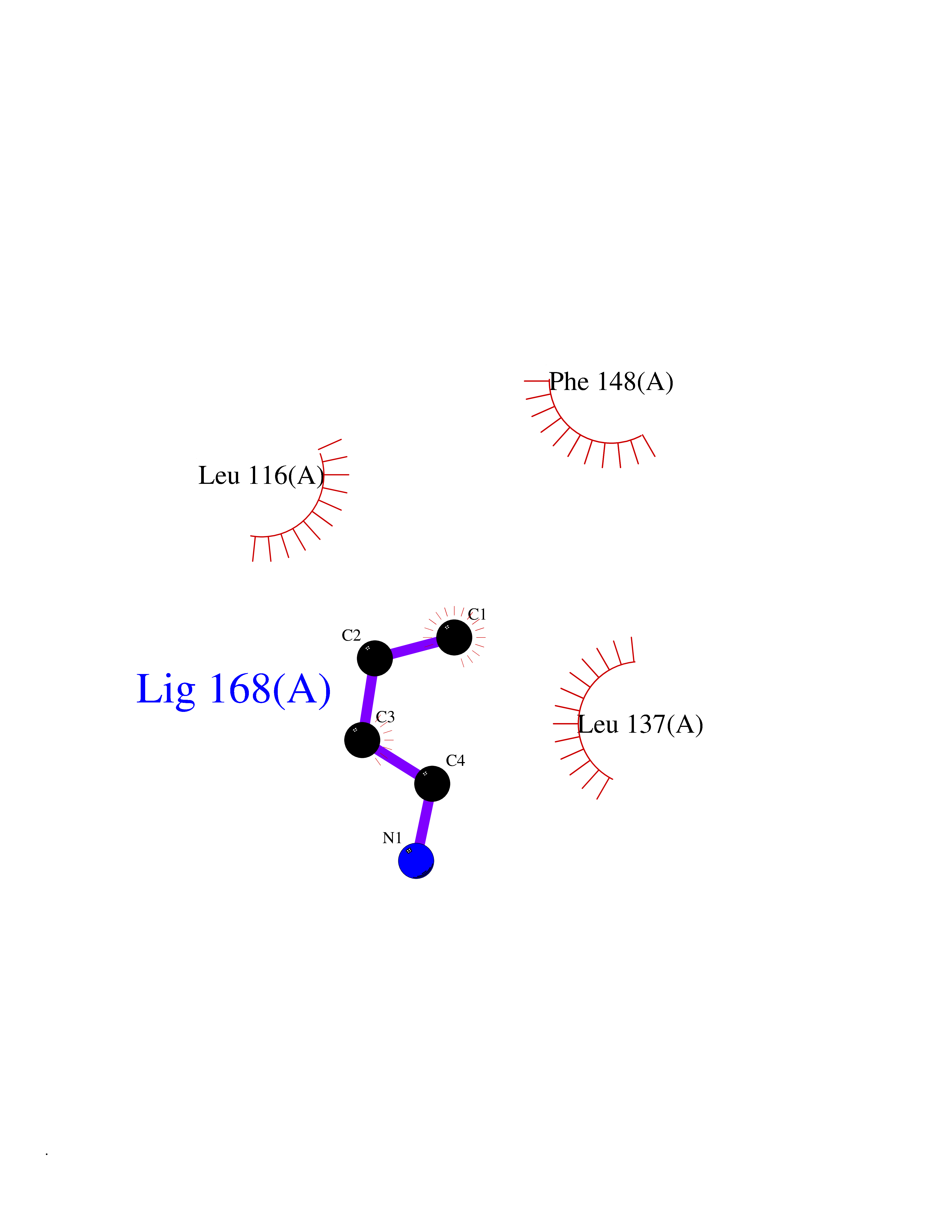 Ligplot Map 3D Binding mode Sequence YNFFPRKPKWDKNQITYRIIGYTPDLDPETVDDAFARAFQVWSDVTPLRFSRIHDGEADIMINFGRWEHGDGYPFDGKDGLLAHAFAPGTGVGGDSHFDDDELWTLGKGVGYSLFLVAAHAFGHAMGLEHSQDPGALMAPIYTYTKNFRLSQDDIKGIQELYGASPISYGNDALMP Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | DNA polymerase catalytic subunit | 1YYP | 4.61 | |
Target general information Gen name UL54 Organism Human cytomegalovirus (strain AD169) (HHV-5) (Human herpesvirus 5) Uniprot ID TTD ID NA Synonyms HFLF2 Protein family DNA polymerase type-B family Biochemical class Replication / transferase Function 3'-5' exonuclease activity.DNA binding.DNA-directed DNA polymerase activity.Nucleotide binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00369; DB00529 Interacts with NA EC number 2.7.7.7 Uniprot keywords 3D-structure; DNA replication; DNA-binding; DNA-directed DNA polymerase; Host nucleus; Nucleotidyltransferase; Reference proteome; Transferase; Viral DNA replication Protein physicochemical properties Chain ID B Molecular weight (Da) 30127.5 Length 269 Aromaticity 0.09 Instability index 36.39 Isoelectric point 8.41 Charge (pH=7) 3.48 2D Binding mode Binding energy (Kcal/mol) -6.29 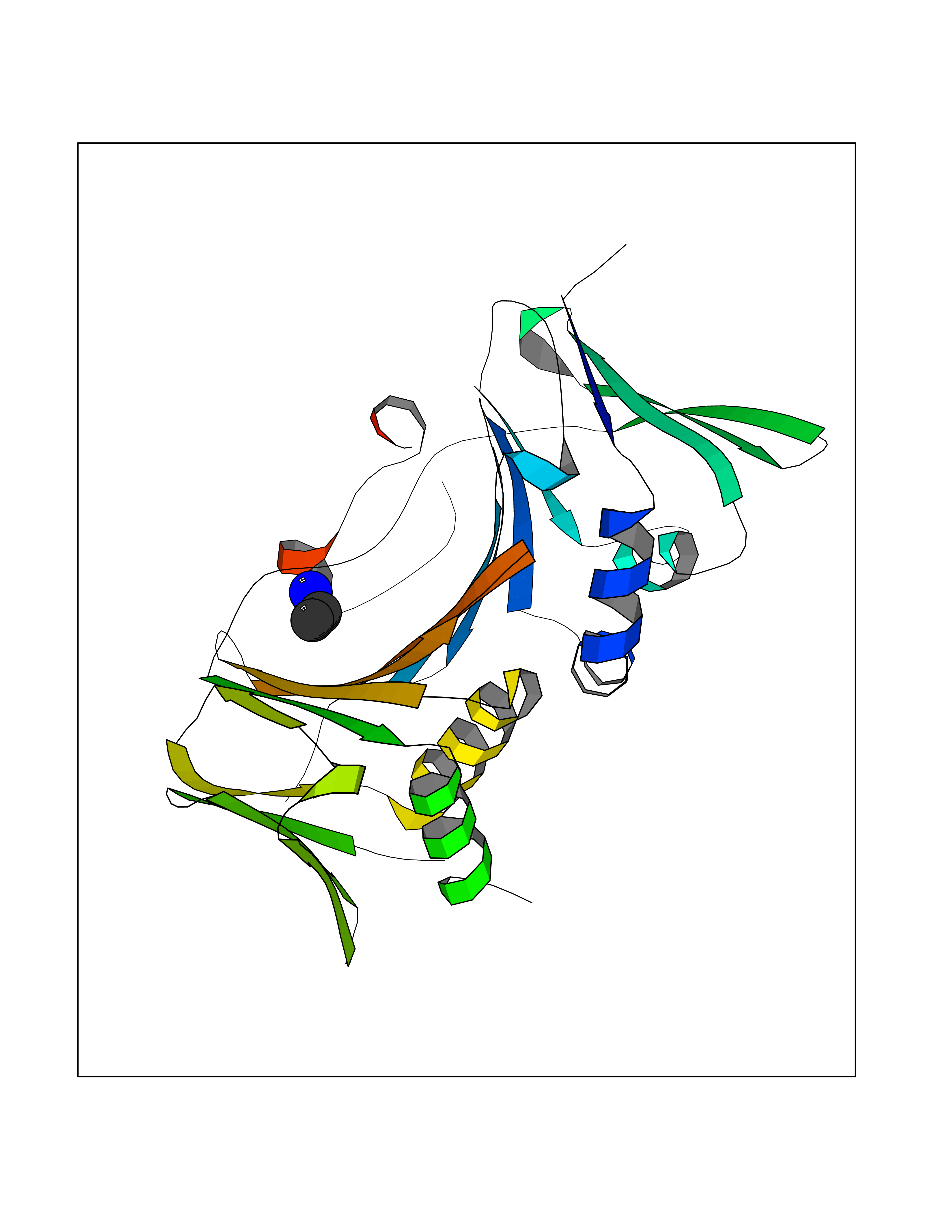 Molscript Map 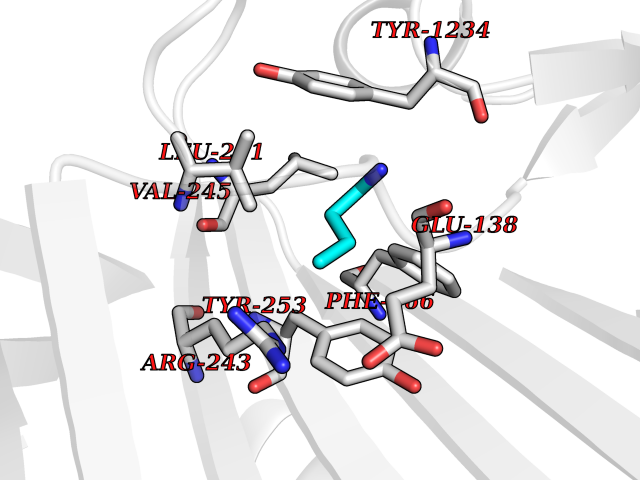 Pymol Map 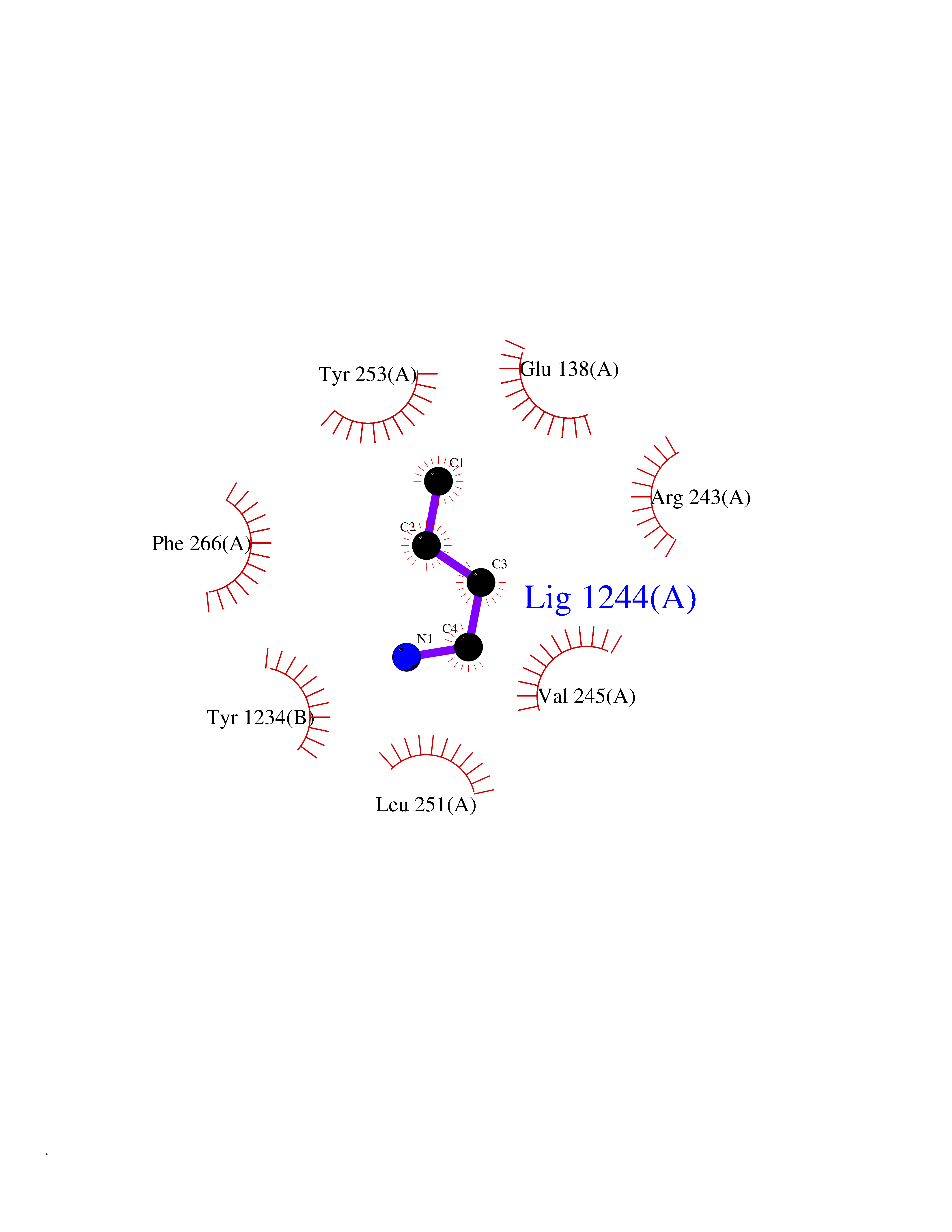 Ligplot Map 3D Binding mode Sequence EPPTLALRLKPYKTAIQQLRSVIRALKENTTVTFLPTPSLILQTVRSHCVSKITFNSSCLYITDKSFQPKTINNSTPLLGNFMYLTSSKDLTKFYVQDISDLSAKISMCAPDFNMEFSSACVHGQDIVRESENSAVHVDLDFGVVADLLKWIGPTGTVQILVHAGPPAIKFILTNGSELEFTSNNRVSFHGVKNMRINVQLKNFYQTLLNCAVTKLPCTLRIVTEHDTLLYVASRNGLFAVENFLTEEPRRLHLEPAFLPYSVKAHECC Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Dopamine D3 receptor (D3R) | 3PBL | 4.61 | |
Target general information Gen name DRD3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms D(3) dopamine receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Dopamine receptor whose activity is mediated by G proteins which inhibit adenylyl cyclase. Promotes cell proliferation. Related diseases Tremor, hereditary essential 1 (ETM1) [MIM:190300]: A common movement disorder mainly characterized by postural tremor of the arms. Head, legs, trunk, voice, jaw, and facial muscles may also be involved. The condition can be aggravated by emotions, hunger, fatigue and temperature extremes, and may cause a functional disability or even incapacitation. Inheritance is autosomal dominant. {ECO:0000269|PubMed:16650084, ECO:0000269|PubMed:16809426}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Glycine at position 9 results in gain of function and is associated with susceptibility to essential tremor. {ECO:0000269|PubMed:16650084, ECO:0000269|PubMed:16809426}.; DISEASE: Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:1362221, ECO:0000269|PubMed:9514583}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Glycine at position 9 results in gain of function and may be a risk factor for schizophrenia. {ECO:0000269|PubMed:1362221, ECO:0000269|PubMed:9514583}. Drugs (DrugBank ID) DB06288; DB00543; DB00714; DB01238; DB14185; DB09207; DB06216; DB09223; DB09128; DB01200; DB00490; DB00248; DB09014; DB06016; DB00477; DB01239; DB00363; DB11274; DB13345; DB00320; DB01184; DB00988; DB11275; DB01049; DB00875; DB00502; DB04946; DB01235; DB00589; DB00408; DB01403; DB06148; DB08804; DB05766; DB00334; DB01267; DB12061; DB01186; DB01100; DB09286; DB12478; DB00413; DB01224; DB00409; DB00268; DB05271; DB06454; DB00391; DB06477; DB09289; DB13025; DB01392; DB00246 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Receptor; Reference proteome; Schizophrenia; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 29257.6 Length 263 Aromaticity 0.13 Instability index 40.85 Isoelectric point 8.79 Charge (pH=7) 6.89 2D Binding mode Binding energy (Kcal/mol) -6.29  Molscript Map 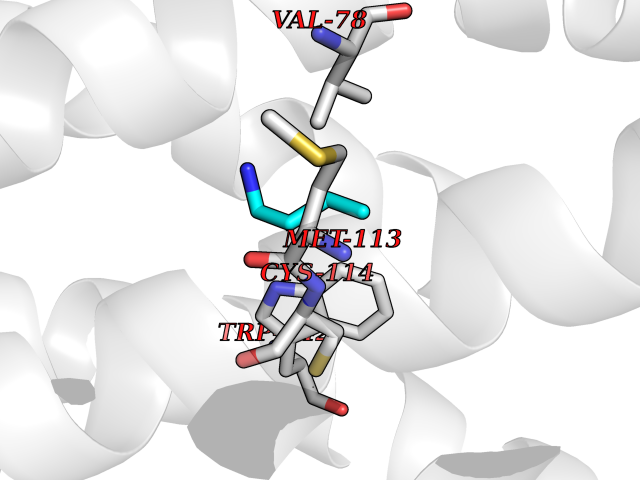 Pymol Map 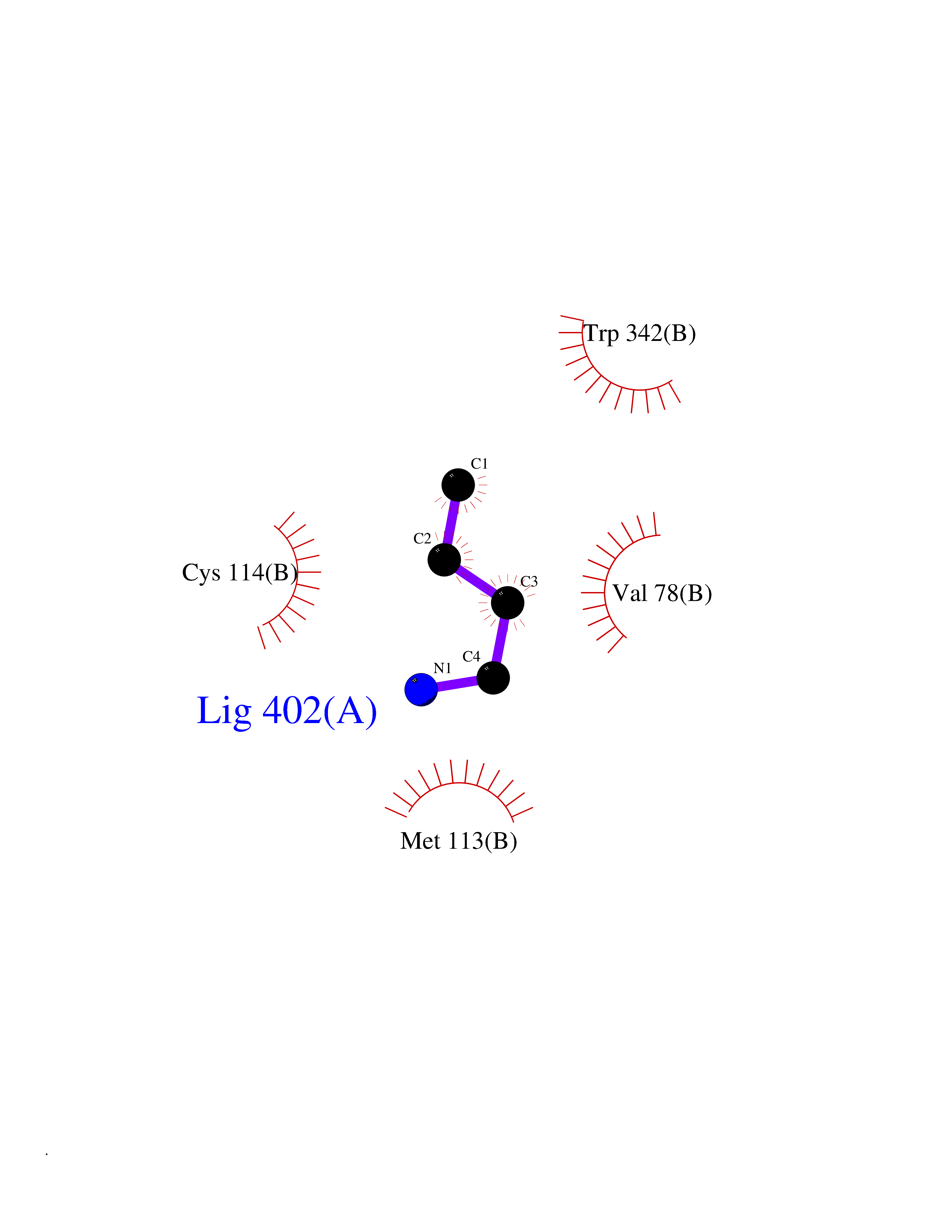 Ligplot Map 3D Binding mode Sequence YALSYCALILAIVFGNGLVCMAVLKERALQTTTNYLVVSLAVADLLVATLVMPWVVYLEVTGGVWNFSRICCDVFVTLDVMMCTASIWNLCAISIDRYTAVVMPSSCRRVALMITAVWVLAFAVSCPLLFGFNTTGDPTVCSISNPDFVIYSSVVSFYLPFGVTVLVYARIYVVLKQRRRKGVPLREKKATQMVAIVLGAFIVCWLPFFLTHVLNTHCQTCHVSPELYSATTWLGYVNSALNPVIYTTFNIEFRKAFLKILSC Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Plasmepsin-2 | 2BJU | 4.61 | |
Target general information Gen name N/A Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase A1 family Biochemical class Hydrolase Function Aspartic-type endopeptidase activity. Related diseases Short/branched-chain acyl-CoA dehydrogenase deficiency (SBCADD) [MIM:610006]: Autosomal recessive disorder and consists of a defect in catabolism of L-isoleucine which is characterized by an increase of 2-methylbutyrylglycine and 2-methylbutyrylcarnitine in blood and urine. Affected individuals have seizures and psychomotor delay as the main clinical features. {ECO:0000269|PubMed:10832746, ECO:0000269|PubMed:11013134, ECO:0000269|PubMed:16317551}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04378; DB04373; DB11638; DB01218; DB02505; DB03063 Interacts with NA EC number 3.4.23.39 Uniprot keywords 3D-structure; Aspartyl protease; Direct protein sequencing; Disulfide bond; Hydrolase; Membrane; Protease; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Vacuole; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 36923.5 Length 329 Aromaticity 0.13 Instability index 44.31 Isoelectric point 4.67 Charge (pH=7) -17.94 2D Binding mode Binding energy (Kcal/mol) -6.29  Molscript Map 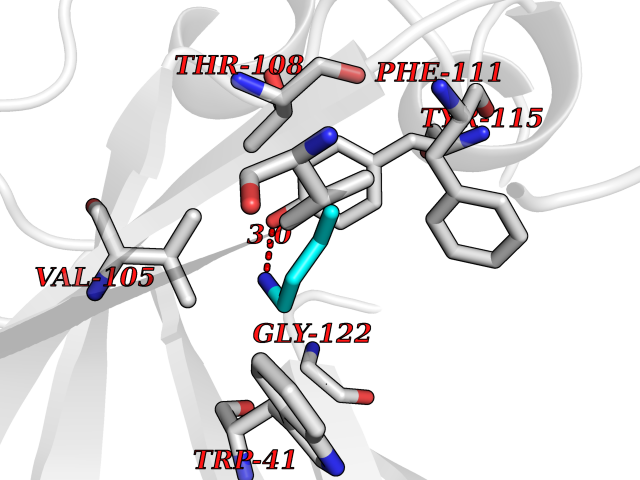 Pymol Map 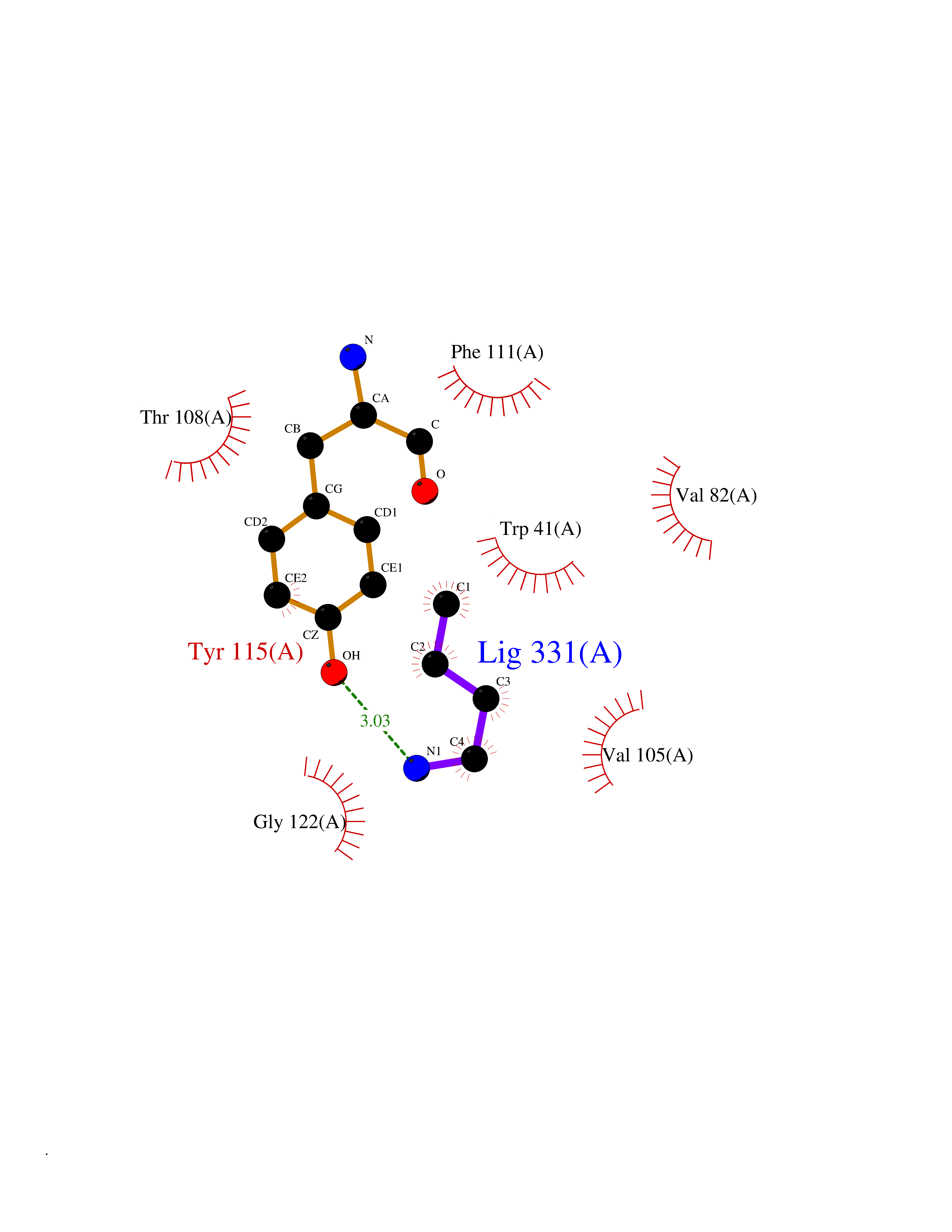 Ligplot Map 3D Binding mode Sequence SSNDNIELVDFQNIMFYGDAEVGDNQQPFTFILDTGSANLWVPSVKCTTAGCLTKHLYDSSKSRTYEKDGTKVEMNYVSGTVSGFFSKDLVTVGNLSLPYKFIEVIDTNGFEPTYTASTFDGILGLGWKDLSIGSVDPIVVELKNQNKIENALFTFYLPVHDKHTGFLTIGGIEERFYEGPLTYEKLNHDLYWQITLDAHVGNIMLEKANCIVDSGTSAITVPTDFLNKMLQNLDVIKVPFLPFYVTLCNNSKLPTFEFTSENGKYTLEPEYYLQHIEDVGPGLCMLNIIGLDFPVPTFILGDPFMRKYFTVFDYDNHSVGIALAKKNL Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Aminoacylase-1 | 1Q7L | 4.61 | |
Target general information Gen name ACY1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase M20A family Biochemical class Hydrolase Function Aminoacylase activity.Identical protein binding.Metal ion binding.Metallopeptidase activity. Related diseases Aminoacylase-1 deficiency (ACY1D) [MIM:609924]: An enzymatic deficiency resulting in encephalopathy, unspecific psychomotor delay, psychomotor delay with atrophy of the vermis and syringomyelia, marked muscular hypotonia or normal clinical features. Epileptic seizures are a frequent feature. All affected individuals exhibit markedly increased urinary excretion of several N-acetylated amino acids. {ECO:0000269|PubMed:16274666, ECO:0000269|PubMed:16465618, ECO:0000269|PubMed:17562838, ECO:0000269|PubMed:21414403}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06151; DB00128; DB09130 Interacts with Q03154; O75934; Q96HA8; P36639; P36639-2; Q8TCT1; P0CG20; Q96A09; P54274; O43711; Q9UPN9; Q9NZC7-5 EC number 3.5.1.14 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,C Molecular weight (Da) 31172.2 Length 275 Aromaticity 0.11 Instability index 36.46 Isoelectric point 6 Charge (pH=7) -5.26 2D Binding mode Binding energy (Kcal/mol) -6.28 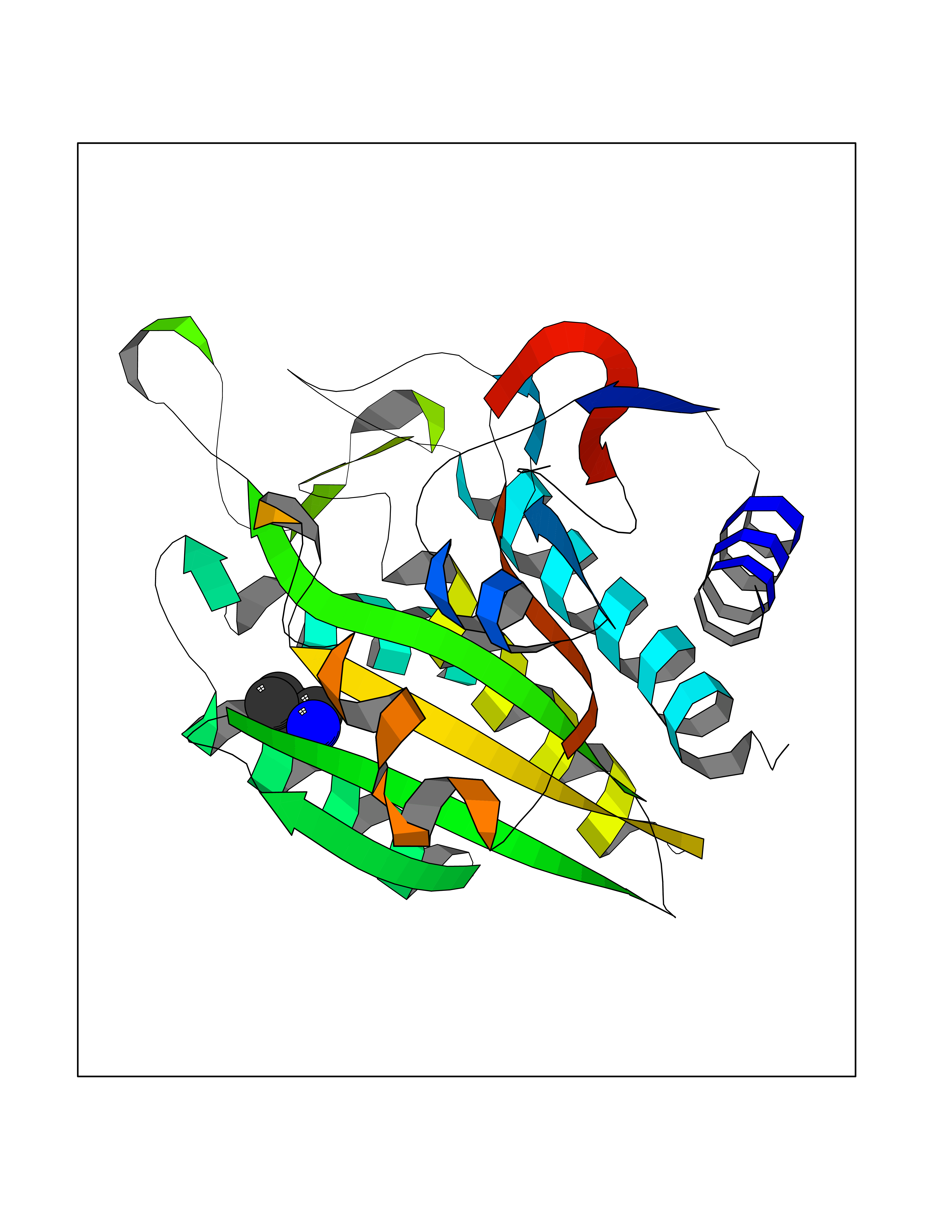 Molscript Map 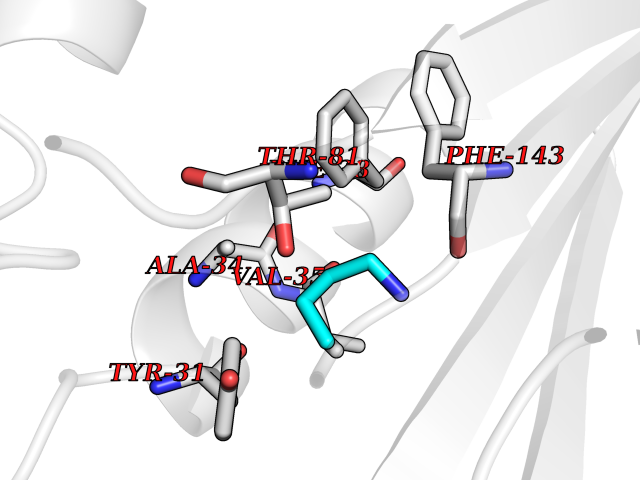 Pymol Map 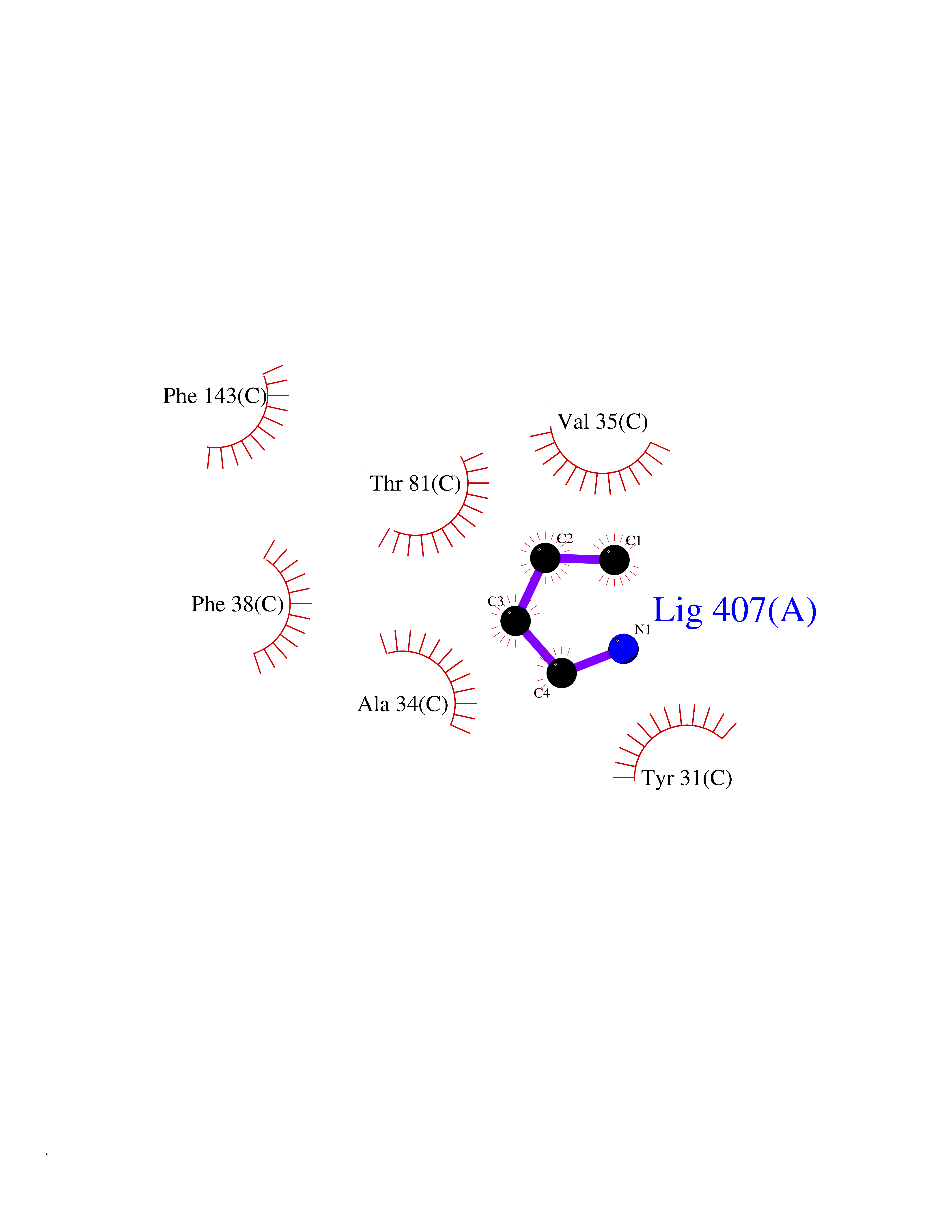 Ligplot Map 3D Binding mode Sequence NPWWAAFSRVCKDMNLTLEPEIMPAAGDNRYIRAVGVPALGFSPMNRTPVLLHDHDERLHEAVFLRGVDIYTRLLPALASVPALPEEHPSVTLFRQYLRIRTVQPKPDYGAAVAFFEETARQLGLGCQKVEVAPGYVVTVLTWPGTNPTLSSILLNSHTDVVPVFKEHWSHDPFEAFKDSEGYIYARGAQDMKCVSIQYLEAVRRLKVEGHRFPRTIHMTFVPDEEVGGHQGMELFVQRPEFHALRAGFALDEGIANPTDAFTVFYSERSPWWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Carbonic anhydrase 13 | 4KNN | 4.61 | |
Target general information Gen name CA13 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Alpha-carbonic anhydrase family Biochemical class Lyase / lyase inhibitor Function Carbonate dehydratase activity.Zinc ion binding. Related diseases Long QT syndrome 10 (LQT10) [MIM:611819]: A heart disorder characterized by a prolonged QT interval on the ECG and polymorphic ventricular arrhythmias. They cause syncope and sudden death in response to exercise or emotional stress, and can present with a sentinel event of sudden cardiac death in infancy. {ECO:0000269|PubMed:17592081}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Atrial fibrillation, familial, 17 (ATFB17) [MIM:611819]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:23604097}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00562; DB00606; DB07115; DB00909 Interacts with Q8N4Y2-3; Q6UUV7 EC number 4.2.1.1 Uniprot keywords 3D-structure; Lyase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 28955.1 Length 258 Aromaticity 0.1 Instability index 40.81 Isoelectric point 6.3 Charge (pH=7) -3.47 2D Binding mode Binding energy (Kcal/mol) -6.29  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SWGYREHNGPIHWKEFFPIADGDQQSPIEIKTKEVKYDSSLRPLSIKYDPSSAKIISNSGHSFNVDFDDTENKSVLRGGPLTGSYRLRQVHLHWGSADDHGSEHIVDGVSYAAELHVVHWNSDKYPSFVEAAHEPDGLAVLGVFLQIGEPNSQLQKITDTLDSIKEKGKQTRFTNFDLLSLLPPSWDYWTYPGSLTVPPLLESVTWIVLKQPINISSQQLAKFRSLLCTAEGEAAAFLVSNHRPPQPLKGRKVRASFH Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Lecithin-cholesterol acyltransferase (LCAT) | 6MVD | 4.61 | |
Target general information Gen name LCAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phospholipidcholesterolacyltransferase; Phospholipid-cholesterol acyltransferase; Phosphatidylcholinesterol acyltransferase; Phosphatidylcholine-sterol acyltransferase Protein family AB hydrolase superfamily, Lipase family Biochemical class Acyltransferase Function Synthesized mainly in the liver and secreted into plasma where it converts cholesterol and phosphatidylcholines (lecithins) to cholesteryl esters and lysophosphatidylcholines on the surface of high and low density lipoproteins (HDLs and LDLs). The cholesterol ester is then transported back to the liver. Has a preference for plasma 16:0-18:2 or 18:O-18:2 phosphatidylcholines. Also produced in the brain by primary astrocytes, and esterifies free cholesterol on nascent APOE-containing lipoproteins secreted from glia and influences cerebral spinal fluid (CSF) APOE- and APOA1 levels. Together with APOE and the cholesterol transporter ABCA1, plays a key role in the maturation of glial-derived, nascent lipoproteins. Required for remodeling high-density lipoprotein particles into their spherical forms. Central enzyme in the extracellular metabolism of plasma lipoproteins. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P02647; O76024 EC number EC 2.3.1.43 Uniprot keywords 3D-structure; Acyltransferase; Cholesterol metabolism; Corneal dystrophy; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipid metabolism; Proteomics identification; Reference proteome; Secreted; Signal; Steroid metabolism; Sterol metabolism; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42715.4 Length 376 Aromaticity 0.12 Instability index 42.05 Isoelectric point 5.69 Charge (pH=7) -9.12 2D Binding mode Binding energy (Kcal/mol) -6.29  Molscript Map 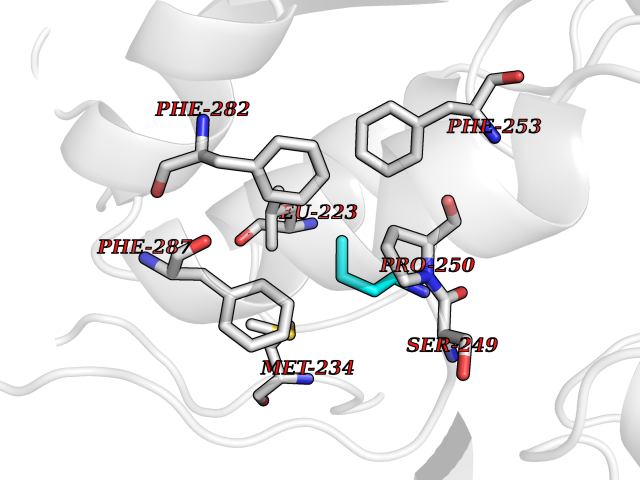 Pymol Map 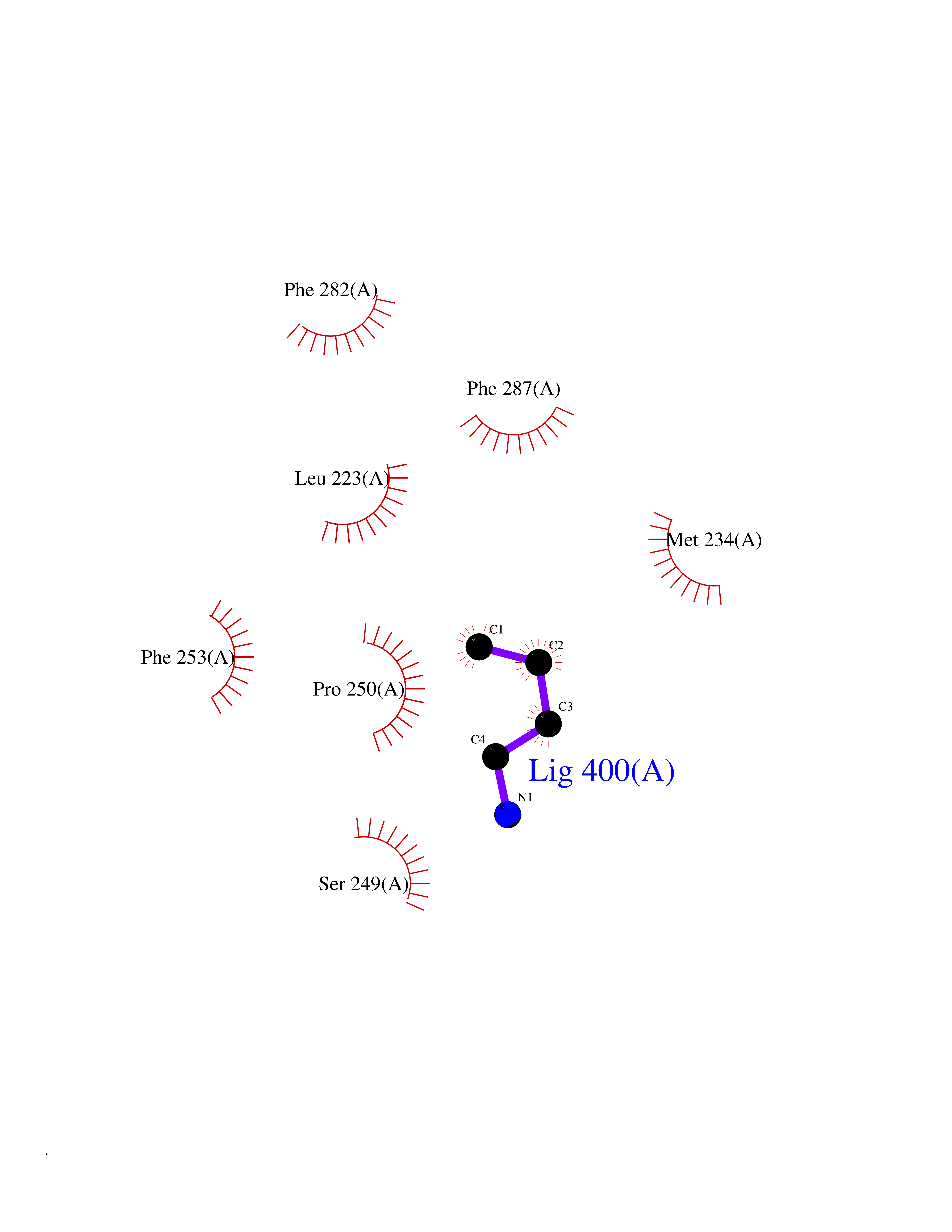 Ligplot Map 3D Binding mode Sequence HTRPVILVPGCLGNQLEAKLDKPDVVNWMCYRKTEDFFTIWLDLNMFLPLGVDCWIDNTRVVYNRSSGLVSNAPGVQIRVPGFGKTYSVEYLDSSKLAGYLHTLVQNLVNNGYVRDETVRAAPYDWRLEPGQQEEYYRKLAGLVEEMHAAYGKPVFLIGHSLGCLHLLYFLLRQPQAWKDRFIDGFISLGAPWGGSIKPMLVLASGDNQGIPIMSSIKEEQRITTTSPWMFPSRMAWPEDHVFISTPSFNYTGRDFQRFFADLHFEEGWYMWLQSRDLLAGLPAPGVEVYCLYGVGLPTPRTYIYDHGFPYTDPVGVLYEDGDDTVATRSTELCGLWQGRQPQPVHLLPLHGIQHLNMVFSNLTLEHINAILLGAH Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Aspartate carbamoyltransferase (CAD) | 6HFQ | 4.61 | |
Target general information Gen name CAD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CAD Protein family CarA family; CarB family; Metallo-dependent hydrolases superfamily, DHOase family, CAD subfamily; Aspartate/ornithine carbamoyltransferase superfamily, ATCase family Biochemical class Carbon-nitrogen ligase Function This protein is a "fusion" protein encoding four enzymatic activities of the pyrimidine pathway (GATase, CPSase, ATCase and DHOase). Related diseases Developmental and epileptic encephalopathy 50 (DEE50) [MIM:616457]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE50 is an autosomal recessive, progressive disease with onset in infancy and favorable response to treatment with oral uridine. {ECO:0000269|PubMed:25678555, ECO:0000269|PubMed:28087732}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00128; DB00130; DB03459 Interacts with P27708; Q8N137; P63104 EC number NA Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; ATP-binding; Congenital disorder of glycosylation; Cytoplasm; Direct protein sequencing; Disease variant; Epilepsy; Hydrolase; Ligase; Magnesium; Manganese; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Repeat; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 39410.8 Length 362 Aromaticity 0.06 Instability index 41.36 Isoelectric point 5.94 Charge (pH=7) -9.57 2D Binding mode Binding energy (Kcal/mol) -6.28 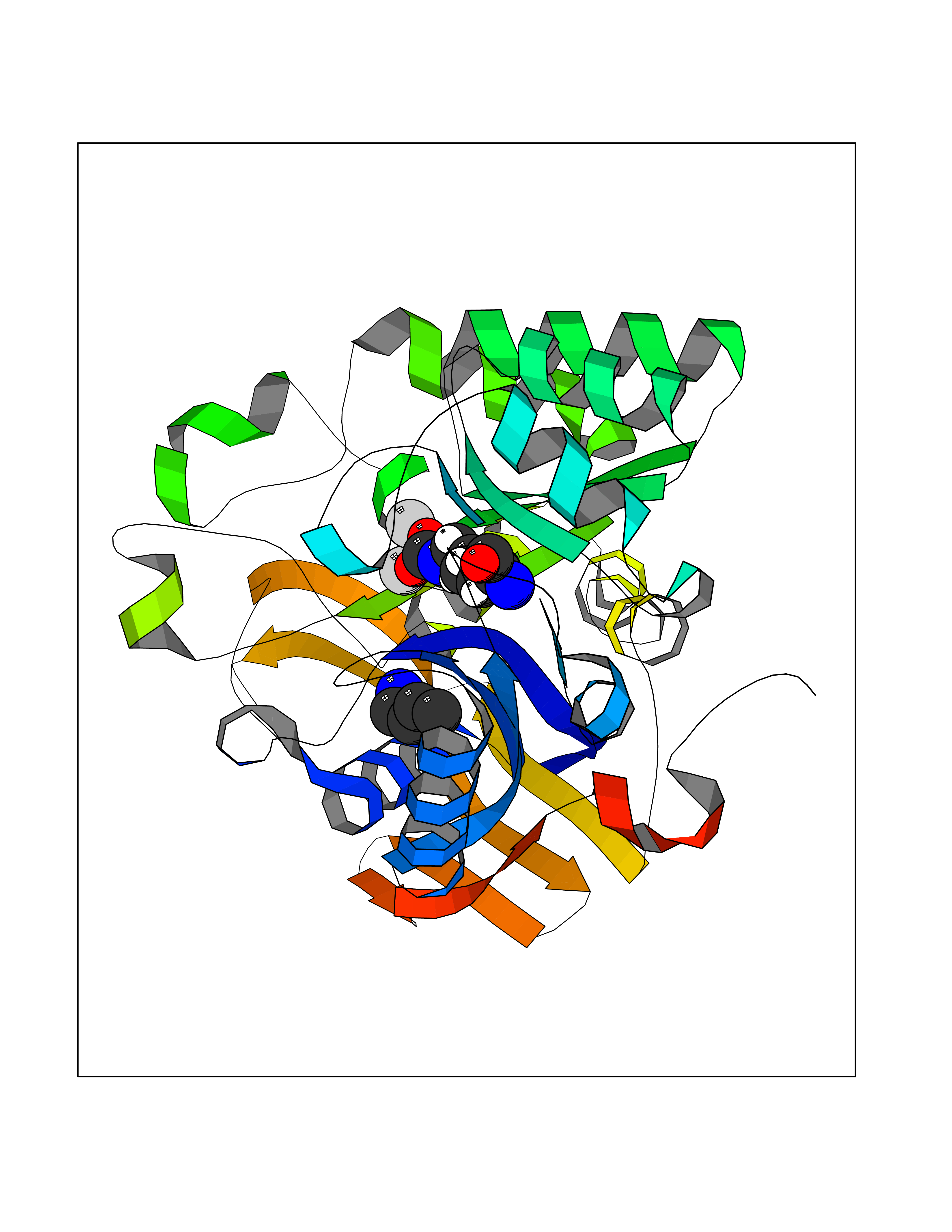 Molscript Map  Pymol Map 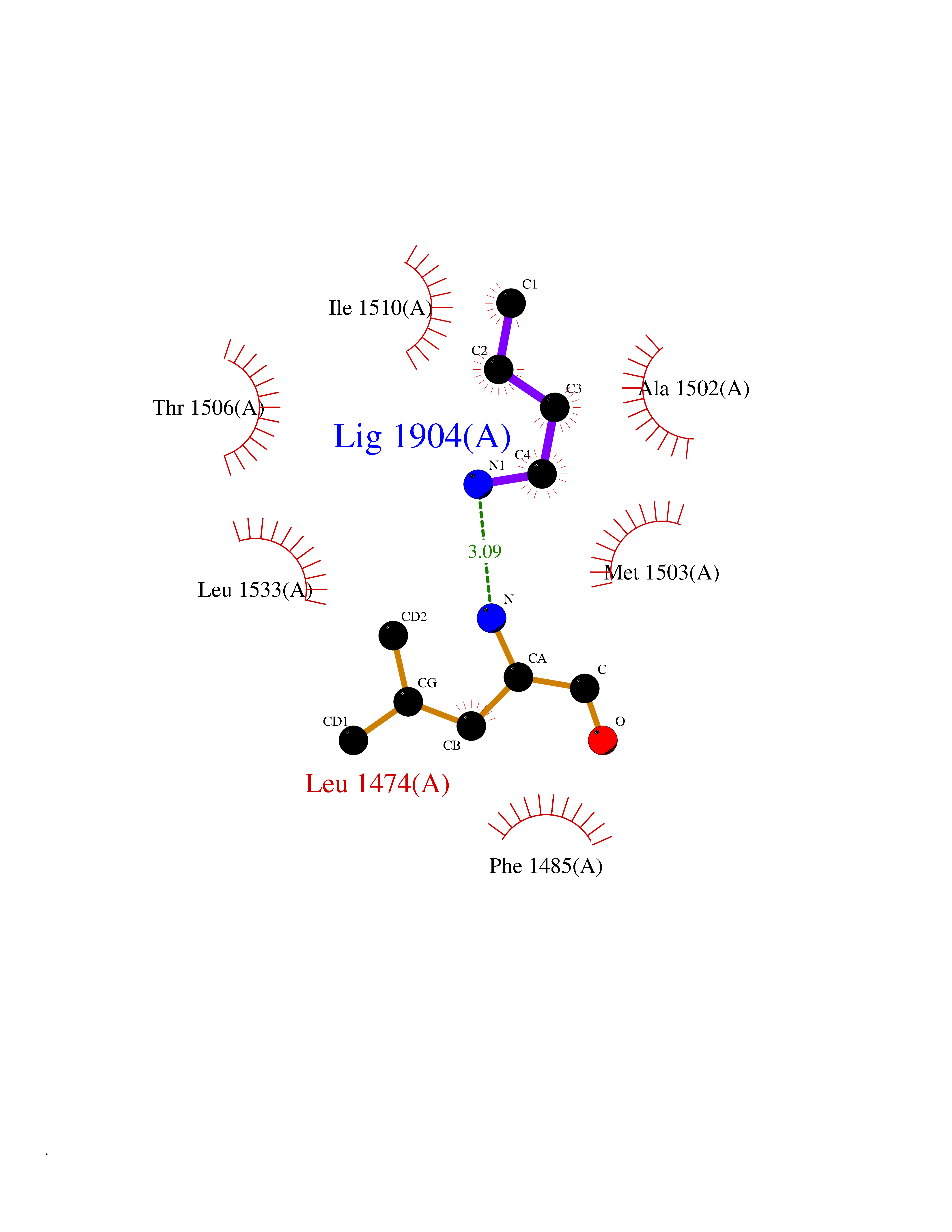 Ligplot Map 3D Binding mode Sequence KLVRLPGLIDVHVHLREPGGTHKEDFASGTAAALAGGITMVCAMPNTRPPIIDGPALALAQKLAEAGARCDFALFLGASSENAGTLGTVAGSAAGLXLYLNETTSELRLDSVVQWMEHFETWPSHLPIVAHAEQQTVAAVLMVAQLTQRSVHICHVARKEEILLIKAAKARGLPVTCEVAPHHLFLSHDDLERLGPGKGEVRPELGSRQDVEALWENMAVIDCFASDHAPHTLEEKCGSRPPPGFPGLETMLPLLLTAVSEGRLSLDDLLQRLHHNPRRIFHLPPQEDTYVEVDLEHEWTIPSHMPFSKAHWTPFEGQKVKGTVRRVVLRGEVAYIDGQVLVPPGYGQDVRKWPQGAVPQLP Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Voltage-gated potassium channel Kv7.1 (KCNQ1) | 6V01 | 4.61 | |
Target general information Gen name KCNQ1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Voltage-gated potassium channel subunit Kv7.1; Potassium voltage-gated channel subfamily KQT member 1; Kv7.1; KVLQT1; KQT-like 1; KCNQ1 channel; KCNA9; KCNA8; IKs producing slow voltage-gated potassiu Protein family Potassium channel family, KQT (TC 1.A.1.15) subfamily, Kv7.1/KCNQ1 sub-subfamily Biochemical class Voltage-gated ion channel Function Associates with KCNE beta subunits that modulates current kinetics. Induces a voltage-dependent by rapidly activating and slowly deactivating potassium-selective outward current. Promotes also a delayed voltage activated potassium current showing outward rectification characteristic. During beta-adrenergic receptor stimulation participates in cardiac repolarization by associating with KCNE1 to form the I(Ks) cardiac potassium current that increases the amplitude and slows down the activation kinetics of outward potassium current I(Ks). Muscarinic agonist oxotremorine-M strongly suppresses KCNQ1/KCNE1 current. When associated with KCNE3, forms the potassium channel that is important for cyclic AMP-stimulated intestinal secretion of chloride ions. This interaction with KCNE3 is reduced by 17beta-estradiol, resulting in the reduction of currents. During conditions of increased substrate load, maintains the driving force for proximal tubular and intestinal sodium ions absorption, gastric acid secretion, and cAMP-induced jejunal chloride ions secretion. Allows the provision of potassium ions to the luminal membrane of the secretory canaliculus in the resting state as well as during stimulated acid secretion. When associated with KCNE2, forms a heterooligomer complex leading to currents with an apparently instantaneous activation, a rapid deactivation process and a linear current-voltage relationship and decreases the amplitude of the outward current. When associated with KCNE4, inhibits voltage-gated potassium channel activity. When associated with KCNE5, this complex only conducts current upon strong and continued depolarization. Also forms a heterotetramer with KCNQ5; has a voltage-gated potassium channel activity. Binds with phosphatidylinositol 4,5-bisphosphate. Potassium channel that plays an important role in a number of tissues, including heart, inner ear, stomach and colon. Related diseases Long QT syndrome 1 (LQT1) [MIM:192500]: A heart disorder characterized by a prolonged QT interval on the ECG and polymorphic ventricular arrhythmias. They cause syncope and sudden death in response to exercise or emotional stress, and can present with a sentinel event of sudden cardiac death in infancy. {ECO:0000269|PubMed:10024302, ECO:0000269|PubMed:10220144, ECO:0000269|PubMed:10220146, ECO:0000269|PubMed:10367071, ECO:0000269|PubMed:10409658, ECO:0000269|PubMed:10482963, ECO:0000269|PubMed:10728423, ECO:0000269|PubMed:10973849, ECO:0000269|PubMed:11799244, ECO:0000269|PubMed:12442276, ECO:0000269|PubMed:15840476, ECO:0000269|PubMed:16414944, ECO:0000269|PubMed:16556865, ECO:0000269|PubMed:16922724, ECO:0000269|PubMed:18165683, ECO:0000269|PubMed:18400097, ECO:0000269|PubMed:19540844, ECO:0000269|PubMed:19716085, ECO:0000269|PubMed:19808498, ECO:0000269|PubMed:21241800, ECO:0000269|PubMed:24184248, ECO:0000269|PubMed:24269949, ECO:0000269|PubMed:24713462, ECO:0000269|PubMed:25037568, ECO:0000269|PubMed:25139741, ECO:0000269|PubMed:25705178, ECO:0000269|PubMed:28096388, ECO:0000269|PubMed:34398675, ECO:0000269|PubMed:8528244, ECO:0000269|PubMed:8818942, ECO:0000269|PubMed:8872472, ECO:0000269|PubMed:9024139, ECO:0000269|PubMed:9272155, ECO:0000269|PubMed:9302275, ECO:0000269|PubMed:9312006, ECO:0000269|PubMed:9323054, ECO:0000269|PubMed:9386136, ECO:0000269|PubMed:9482580, ECO:0000269|PubMed:9570196, ECO:0000269|PubMed:9641694, ECO:0000269|PubMed:9693036, ECO:0000269|PubMed:9702906, ECO:0000269|PubMed:9799083, ECO:0000269|PubMed:9927399, ECO:0000269|Ref.36}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Jervell and Lange-Nielsen syndrome 1 (JLNS1) [MIM:220400]: An autosomal recessive disorder characterized by congenital deafness, prolongation of the QT interval, syncopal attacks due to ventricular arrhythmias, and a high risk of sudden death. {ECO:0000269|PubMed:10090886, ECO:0000269|PubMed:10728423, ECO:0000269|PubMed:18400097, ECO:0000269|PubMed:18441444, ECO:0000269|PubMed:25705178, ECO:0000269|PubMed:9781056}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Atrial fibrillation, familial, 3 (ATFB3) [MIM:607554]: An autosomal dominant form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:12522251}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Short QT syndrome 2 (SQT2) [MIM:609621]: An autosomal dominant form of short QT syndrome, a heart disorder characterized by idiopathic persistently and uniformly short QT interval on ECG in the absence of structural heart disease in affected individuals. It can cause syncope and sudden death. {ECO:0000269|PubMed:15159330}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Type 2 diabetes mellitus (T2D) [MIM:125853]: A multifactorial disorder of glucose homeostasis caused by a lack of sensitivity to insulin. Affected individuals usually have an obese body habitus and manifestations of a metabolic syndrome characterized by diabetes, insulin resistance, hypertension and hypertriglyceridemia. The disease results in long-term complications that affect the eyes, kidneys, nerves, and blood vessels. {ECO:0000269|PubMed:18711366, ECO:0000269|PubMed:18711367, ECO:0000269|PubMed:24390345}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04957; DB01244; DB04855; DB00228; DB06089; DB11633; DB01110; DB01069 Interacts with P15382; P0DP25 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Atrial fibrillation; Calmodulin-binding; Cell membrane; Coiled coil; Cytoplasmic vesicle; Deafness; Diabetes mellitus; Disease variant; Endoplasmic reticulum; Endosome; Glycoprotein; Ion channel; Ion transport; Long QT syndrome; Membrane; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Short QT syndrome; Transmembrane; Transmembrane helix; Transport; Ubl conjugation; Voltage-gated channel Protein physicochemical properties Chain ID J,L,A Molecular weight (Da) 80494.8 Length 702 Aromaticity 0.14 Instability index 31.57 Isoelectric point 10.06 Charge (pH=7) 49.05 2D Binding mode Binding energy (Kcal/mol) -6.28 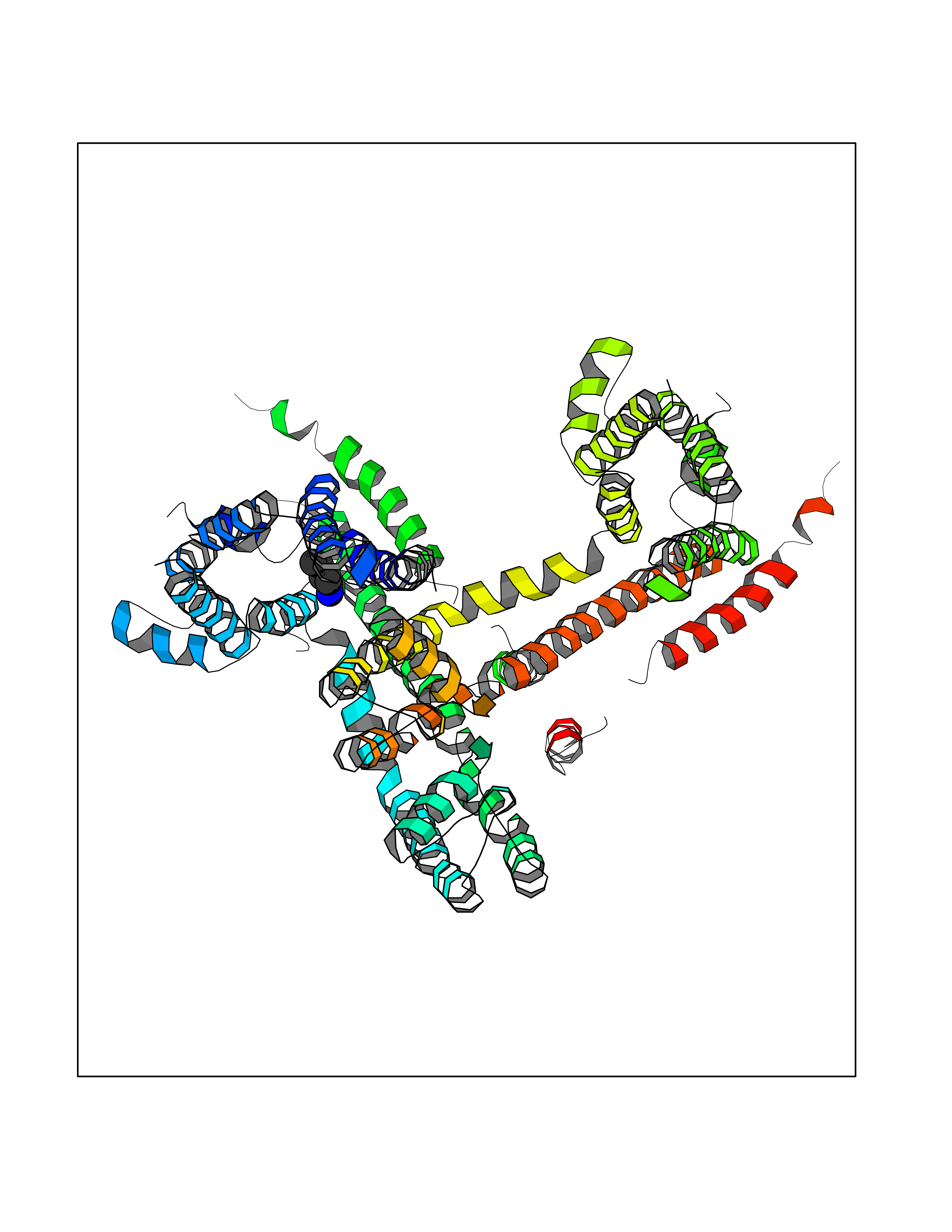 Molscript Map 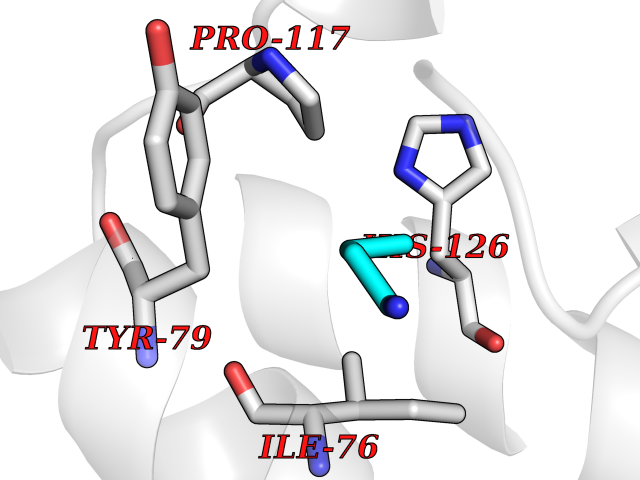 Pymol Map 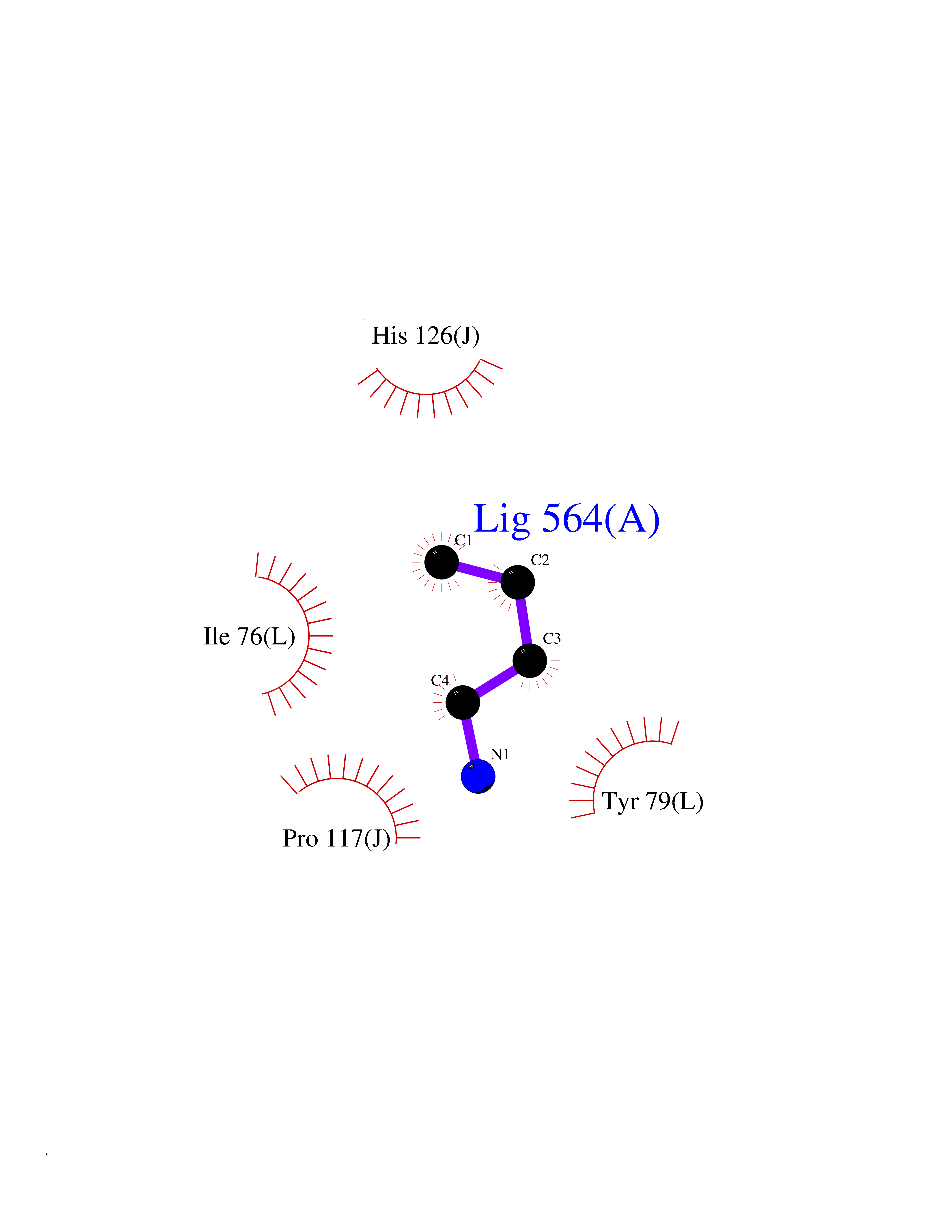 Ligplot Map 3D Binding mode Sequence RDDNSYMYILFVMFLFAVTVGSLILGYTRSRKVDKRSDPYTHVQGRVYNFLERPTGWKCFVYHFAVFLIVLVCLIFSVLSTIEQYAALATGTLFWMEIVLVVFFGTEYVVRLWSAGCRSKYVGLWGRLRFARKPISIIDLIVVVASMVVLCVGSKSAIRGIRFLQILRMLHVDRQGGTWRLLGSVVFIHRQELITTLYIGFLGLIFSSYFVYLAEKDAVNESGRVEFGSYADALWWGVVTVTTIGYGDKVPQTWVGKTIASCFSVFAISFFALPAGILGSGFALKVQQKQRQKHFNRQIPAAASLIQTAWRCYAAENPLREHHRATIKVIRRMQYFVAKKKFQQARKPYDIEQYSQGHLNLMVRIKELQRRTHVQGRVYNFLERPTGWKCFVYHFAVFLIVLVCLIFSVLSTIEQYAALATGTLFWMEIVLVVFFGTEYVVRLWSAGCRSKYVGLWGRLRFARKPISIIDLIVVVASMVVLCVGSKSAIRGIRFLQILRMLHVDRQGGTWRLLGSVVFIHRQELITTLYIGFLGLIFSSYFVYLAEKDAVNESGRVEFGSYADALWWGVVTVTTIGYGDKVPQTWVGKTIASCFSVFAISFFALPAGILGSGFALKVQQKQRQKHFNRQIPAAASLIQTAWRCYAAENPLREHHRATIKVIRRMQYFVAKKKFQQARKPYDIEQYSQGHLNLMVRIKELQRR Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Neuronal acetylcholine receptor alpha-3/beta-4 (CHRNA3/B4) | 6PV7 | 4.61 | |
Target general information Gen name CHRNA3-CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Neuronal acetylcholine receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Neurotransmitter receptor Function A type of nicotinic acetylcholine receptor, consisting of 3 and 4 subunits. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -6.29 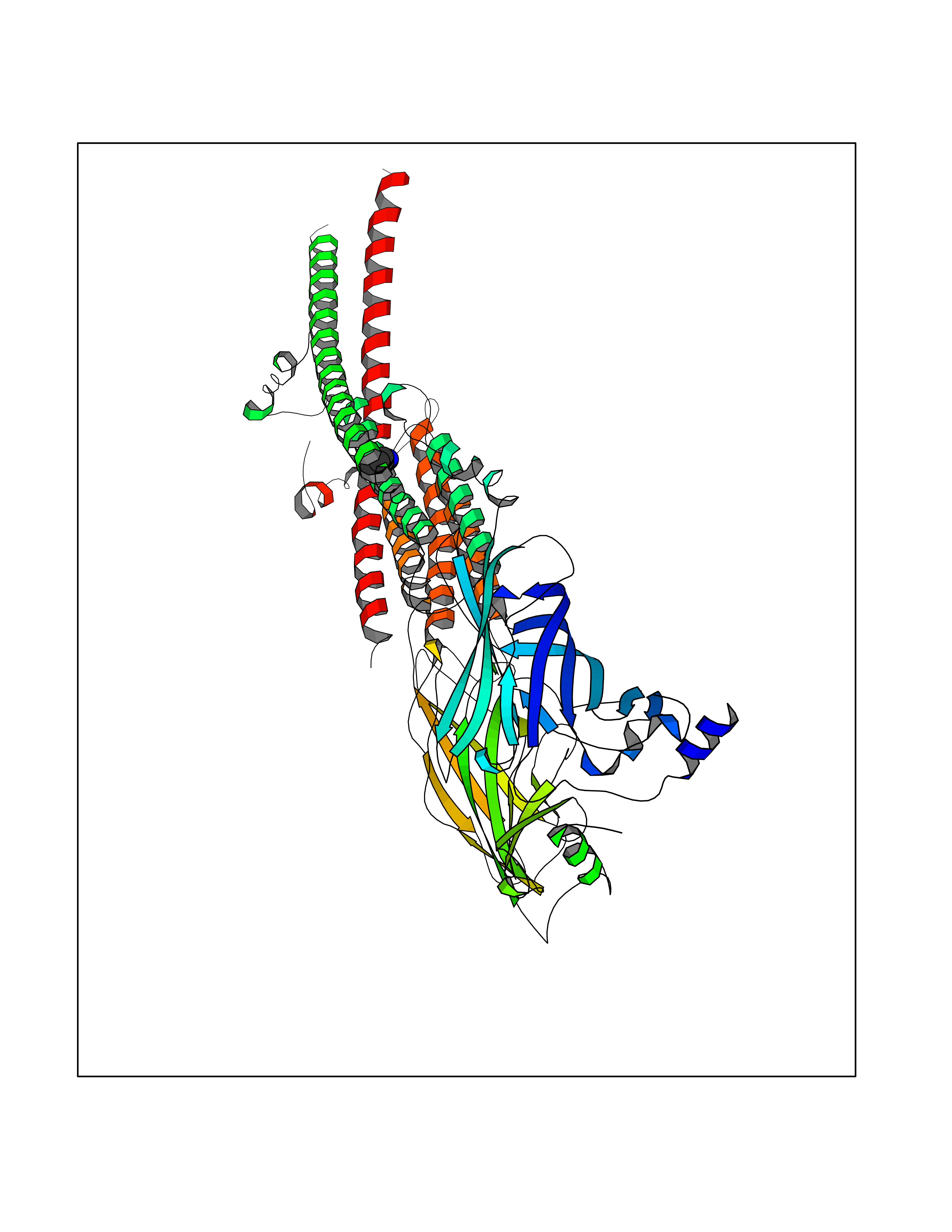 Molscript Map 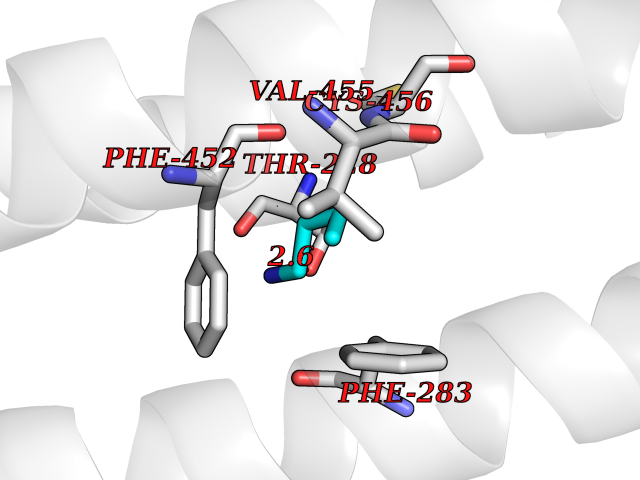 Pymol Map 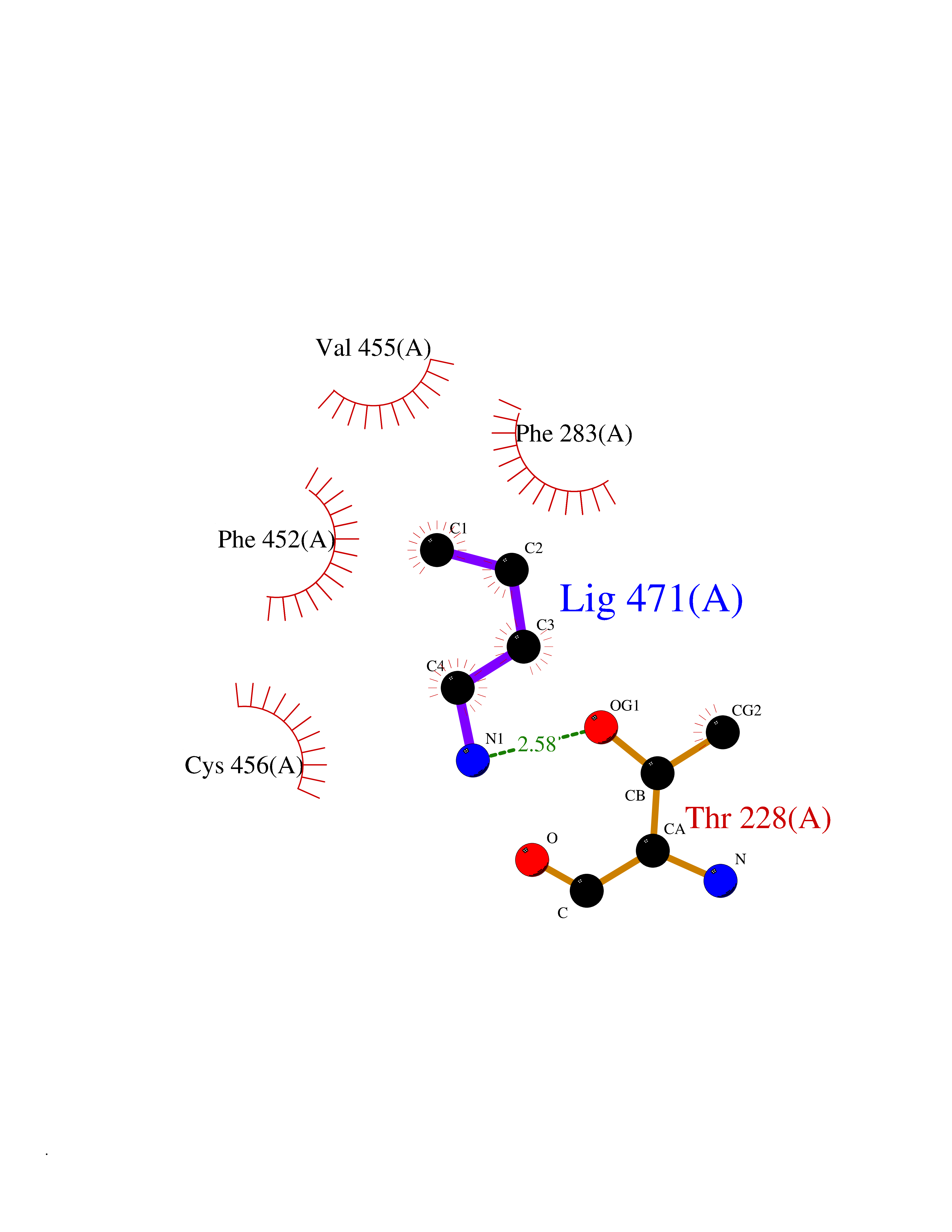 Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Neuronal acetylcholine receptor beta-4 (CHRNB4) | 6PV7 | 4.61 | |
Target general information Gen name CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNB4; Beta-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-4/CHRNB4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. {ECO:0000269|PubMed:20054297, ECO:0000269|PubMed:23622243, ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}. The disease may be caused by variants affecting the gene represented in this entry. Defects of SETD2 are associated with loss of DNA methylation at non-promoter regions (PubMed:23792563). SETD2 defects lead to aberrant and reduced nucleosome compaction and chromatin association of key replication proteins, such as MCM7 and DNA polymerase delta, leading to hinder replication fork progression and prevent loading of RAD51 homologous recombination repair factor at DNA breaks (PubMed:25728682). {ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}.; DISEASE: Luscan-Lumish syndrome (LLS) [MIM:616831]: An autosomal dominant syndrome with a variable phenotype. Clinical features include macrocephaly, distinctive facial appearance, postnatal overgrowth, various degrees of learning difficulties, autism spectrum disorder, and intellectual disability. {ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24852293, ECO:0000269|PubMed:26084711, ECO:0000269|PubMed:27317772}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute lymphoblastic (ALL) [MIM:613065]: A subtype of acute leukemia, a cancer of the white blood cells. ALL is a malignant disease of bone marrow and the most common malignancy diagnosed in children. The malignant cells are lymphoid precursor cells (lymphoblasts) that are arrested in an early stage of development. The lymphoblasts replace the normal marrow elements, resulting in a marked decrease in the production of normal blood cells. Consequently, anemia, thrombocytopenia, and neutropenia occur to varying degrees. The lymphoblasts also proliferate in organs other than the marrow, particularly the liver, spleen, and lymphnodes. {ECO:0000269|PubMed:24509477, ECO:0000269|PubMed:24662245}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16314571, ECO:0000269|PubMed:24509477}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Intellectual developmental disorder, autosomal dominant 70 (MRD70) [MIM:620157]: An autosomal dominant disorder characterized by mild global developmental delay, moderately impaired intellectual disability with speech difficulties, and behavioral abnormalities. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rabin-Pappas syndrome (RAPAS) [MIM:620155]: An autosomal dominant neurodevelopmental disorder characterized by severely impaired global development, intellectual disability, microcephaly, facial dysmorphism, and variable congenital anomalies affecting the skeletal, genitourinary, cardiac, and other organ systems. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00237; DB00565; DB00514; DB07720; DB00898; DB00472; DB01227; DB00184; DB01090; DB00202 Interacts with Q6FHY5 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -6.29 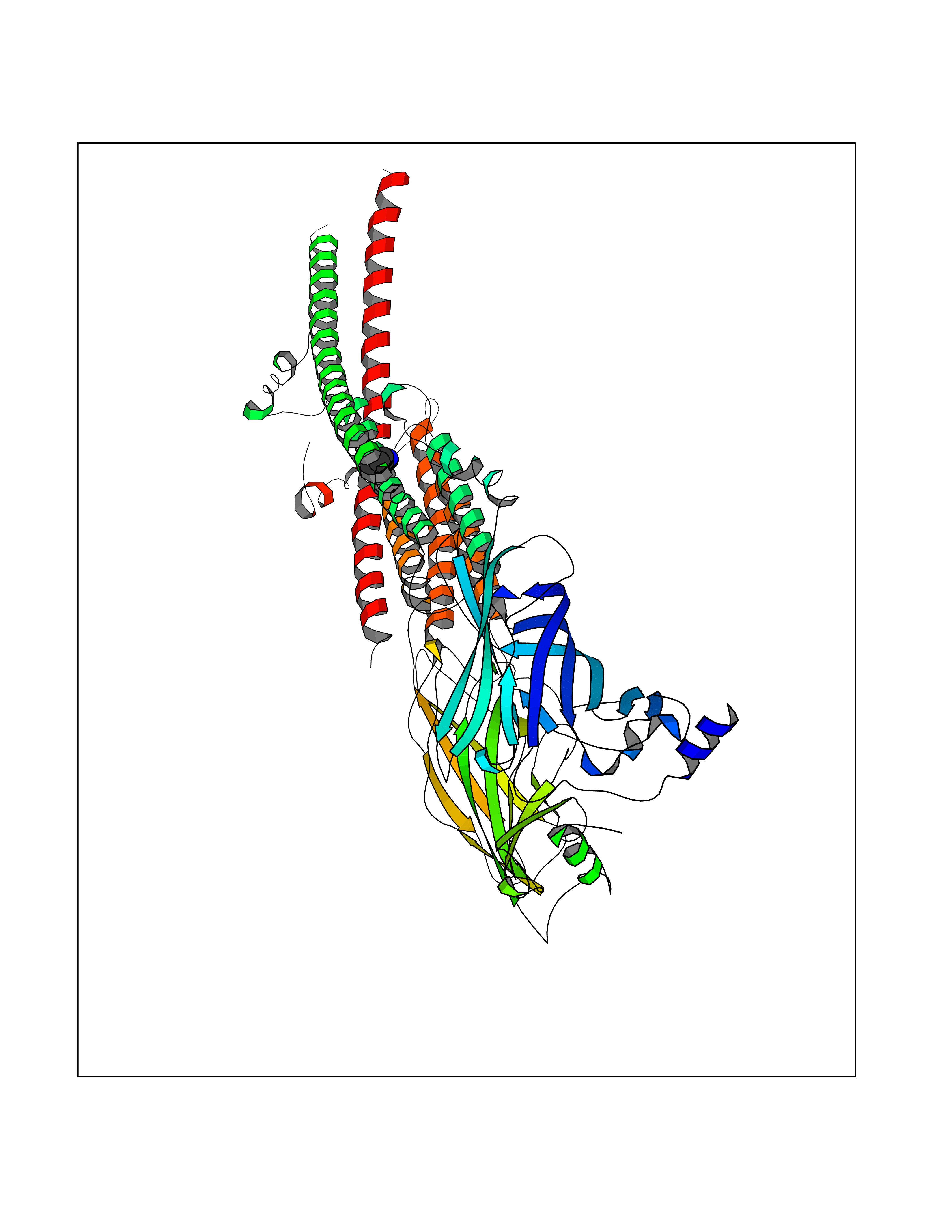 Molscript Map 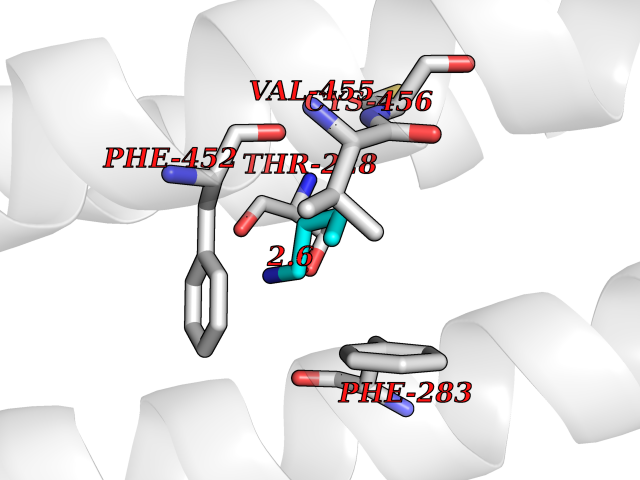 Pymol Map 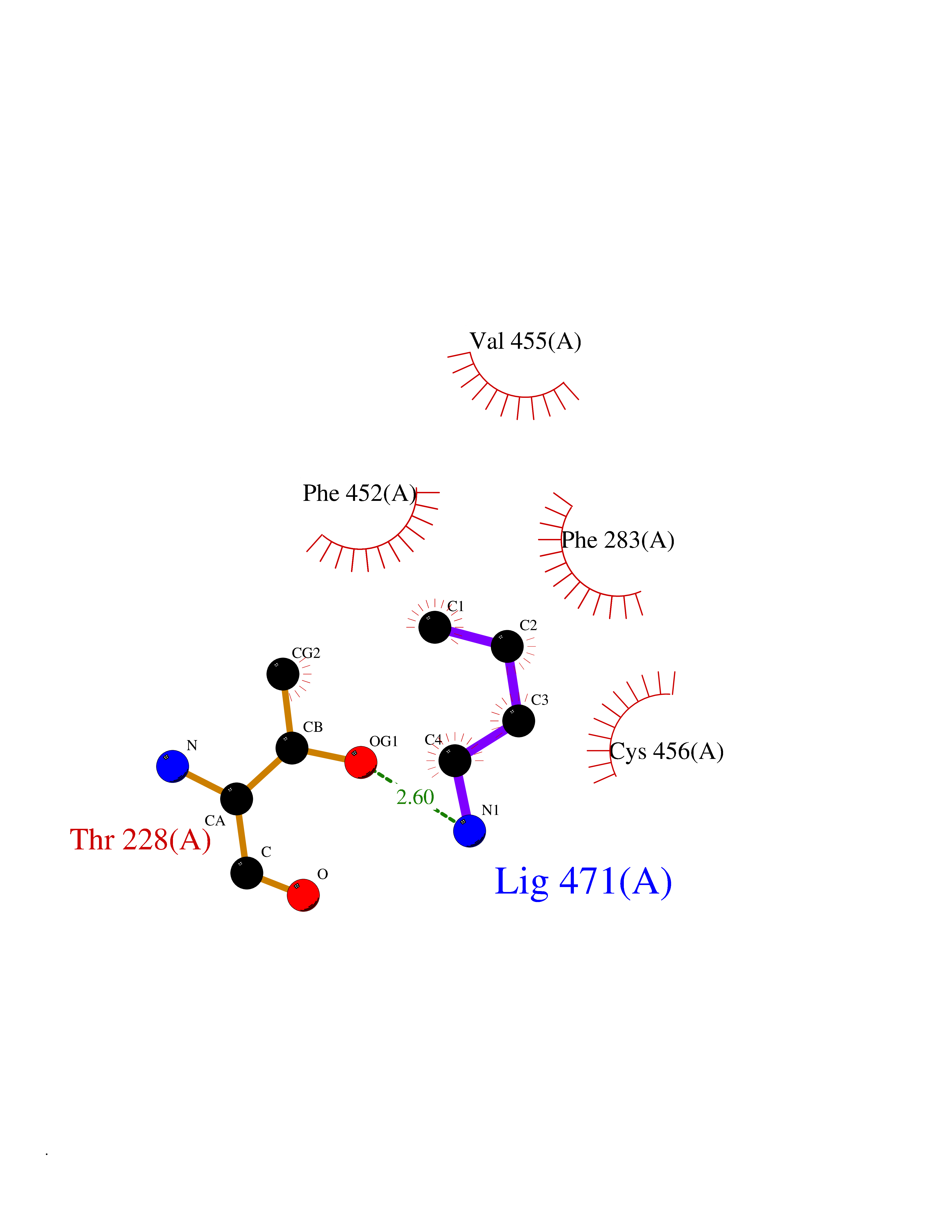 Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Folate receptor alpha (FOLR1) | 4LRH | 4.61 | |
Target general information Gen name FOLR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ovarian tumorassociated antigen MOv18; KB cells FBP; Folate receptor, adult; Folate receptor 1; FRalpha; FOLR1; Adult folatebinding protein Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pHafter receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Required for normal embryonic development and normal cell proliferation. Related diseases Neurodegeneration due to cerebral folate transport deficiency (NCFTD) [MIM:613068]: An autosomal recessive neurodegenerative disorder resulting from brain-specific folate deficiency early in life. Onset is apparent in late infancy with severe developmental regression, movement disturbances, epilepsy and leukodystrophy. {ECO:0000269|PubMed:19732866}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05595; DB00158; DB00563; DB12489; DB15413; DB05168 Interacts with Q8N357 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Endosome; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Neurodegeneration; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 24216 Length 207 Aromaticity 0.13 Instability index 49.36 Isoelectric point 8.14 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -6.28 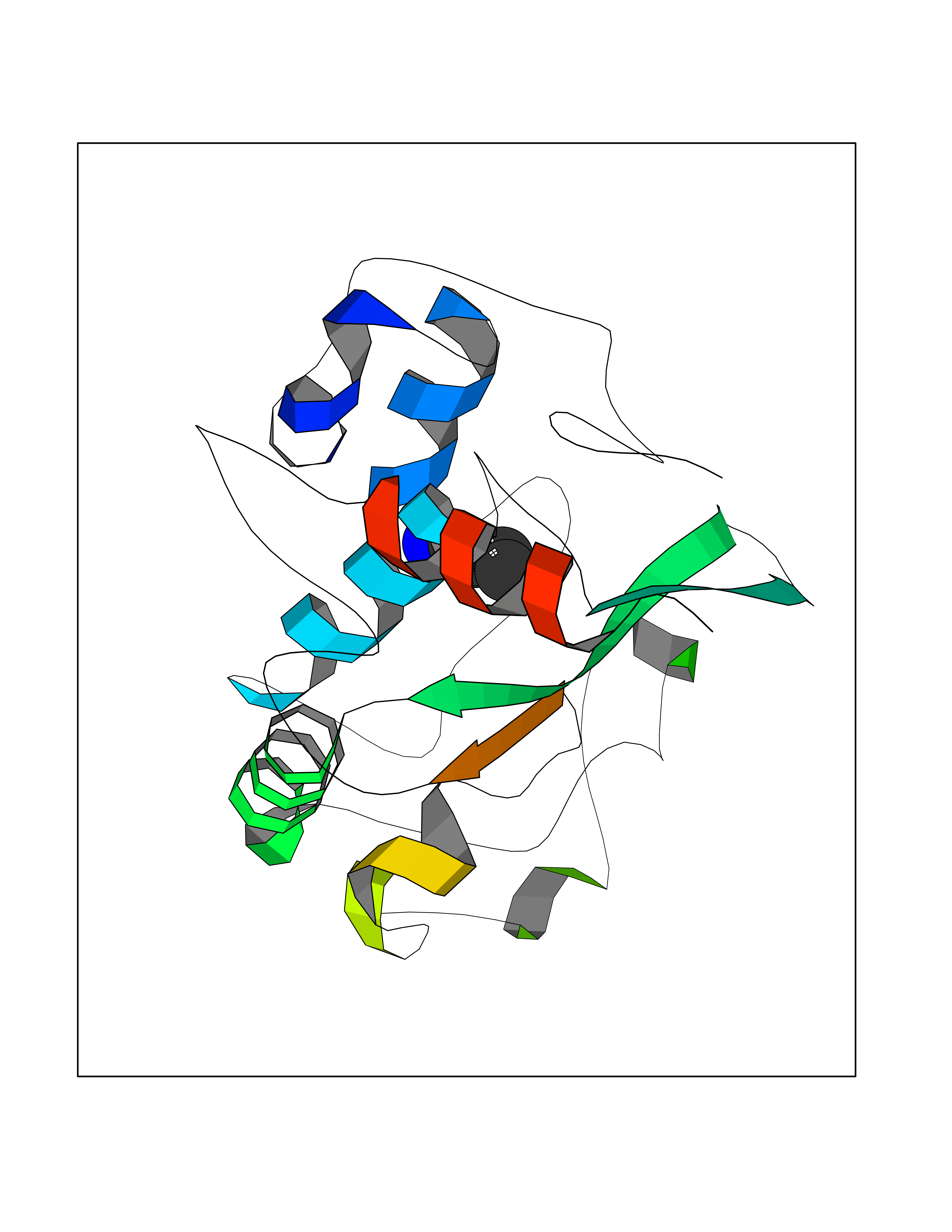 Molscript Map 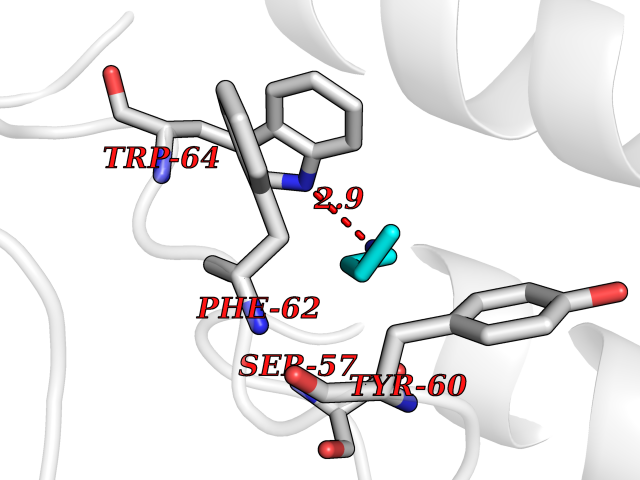 Pymol Map 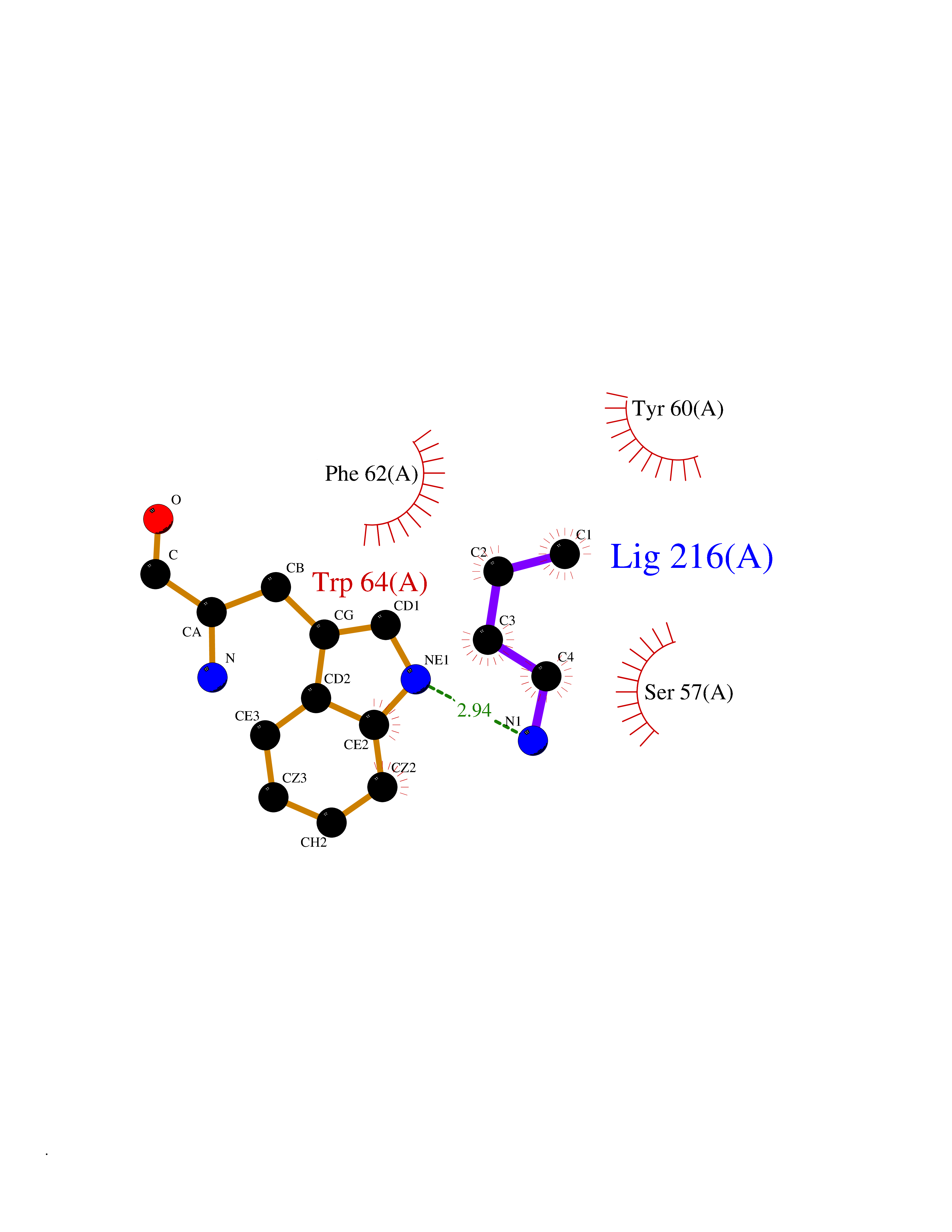 Ligplot Map 3D Binding mode Sequence RTELLNVCMNAKHHKEKPGPEDKLHEQCRPWRKNACCSTNTSQEAHKDVSYLYRFNWNHCGEMAPACKRHFIQDTCLYECSPNLGPWIQQVDQSWRKERVLNVPLCKEDCEQWWEDCRTSYTCKSNWHKGWNWTSGFNKCAVGAACQPFHFYFPTPTVLCNEIWTHSYKVSNYSRGSGRCIQMWFDPAQGNPNEEVARFYAAAMSGT Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Bacterial Nicotinate-nucleotide adenylyltransferase (Bact nadD) | 1K4K | 4.61 | |
Target general information Gen name Bact nadD Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms nadD of Escherichia coli (strain K12); Nicotinate mononucleotide adenylyltransferase of Escherichia coli (strain K12); NaMN adenylyltransferase of Escherichia coli (strain K12); Deamido-NAD(+)Nicotina Protein family NadD family Biochemical class Kinase Function Catalyzes the reversible adenylation of nicotinate mononucleotide (namn) to nicotinic acid adenine dinucleotide (naad). Related diseases Asthma-related traits 5 (ASRT5) [MIM:611064]: Asthma-related traits include clinical symptoms of asthma, such as coughing, wheezing, dyspnea, bronchial hyperresponsiveness as assessed by methacholine challenge test, serum IgE levels, atopy and atopic dermatitis. {ECO:0000269|PubMed:17503328}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.7.7.18 Uniprot keywords 3D-structure; ATP-binding; NAD; Nucleotide-binding; Nucleotidyltransferase; Pyridine nucleotide biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 24527.6 Length 213 Aromaticity 0.09 Instability index 48.92 Isoelectric point 5.46 Charge (pH=7) -7.78 2D Binding mode Binding energy (Kcal/mol) -6.29 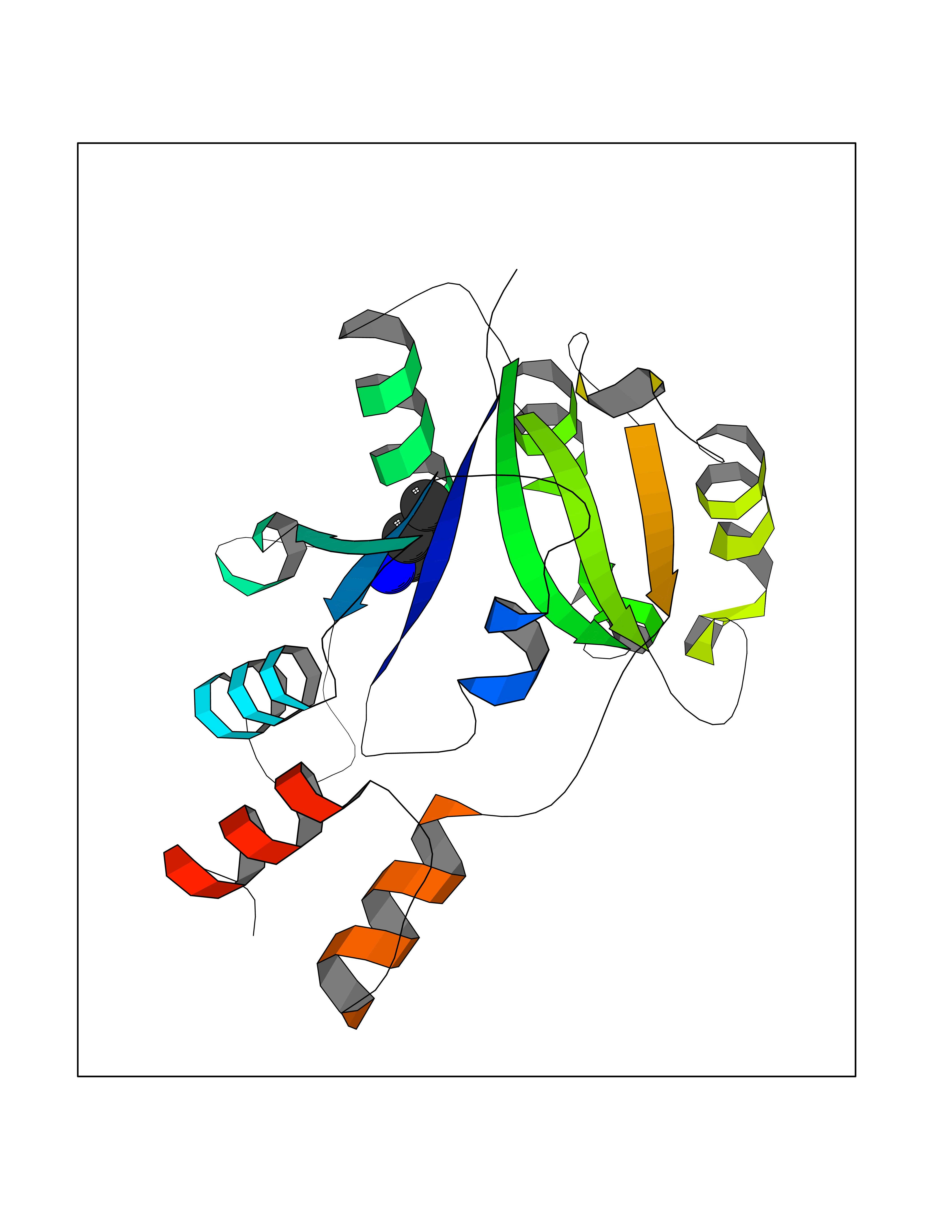 Molscript Map 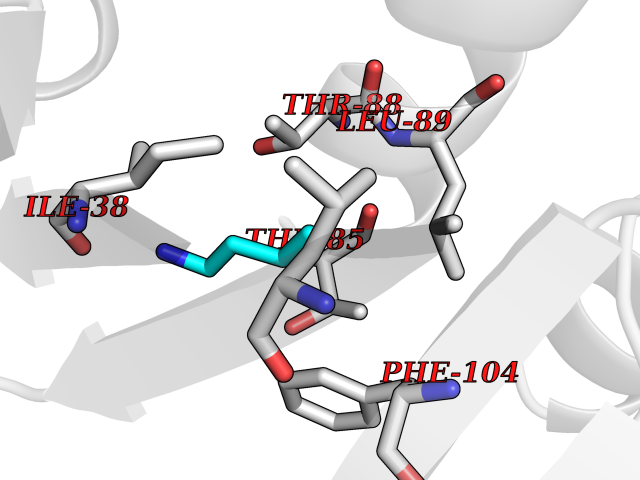 Pymol Map 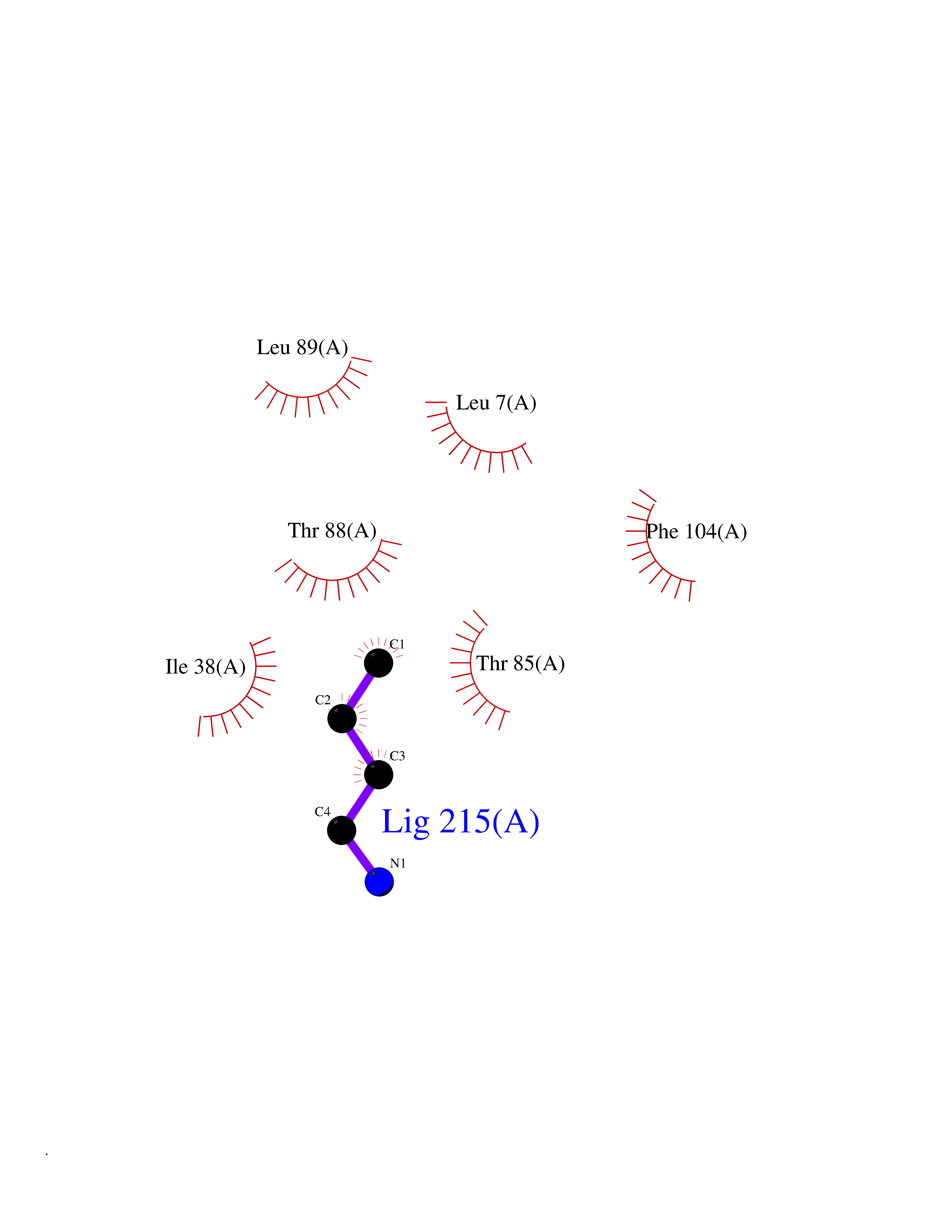 Ligplot Map 3D Binding mode Sequence MKSLQALFGGTFDPVHYGHLKPVETLANLIGLTRVTIIPNNVPPHRPQPEANSVQRKHMLELAIADKPLFTLDERELKRNAPSYTAQTLKEWRQEQGPDVPLAFIIGQDSLLTFPTWYEYETILDNAHLIVCRRPGYPLEMAQPQYQQWLEDHLTHNPEDLHLQPAGKIYLAETPWFNISATIIRERLQNGESCEDLLPEPVLTYINQQGLYR Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Histone-lysine N-methyltransferase (HLNM) | 3QOW | 4.61 | |
Target general information Gen name DOT1L Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 4; KMT4; KIAA1814; Histone-lysine N-methyltransferase, H3 lysine-79 specific; Histone H3-K79 methyltransferase; H3-K79-HMTase; DOT1-like protein Protein family Class I-like SAM-binding methyltransferase superfamily, DOT1 family Biochemical class Methyltransferase Function Histone methyltransferase. Methylates 'Lys-79' of histone H3. Nucleosomes are preferred as substrate compared to free histones. Binds to DNA. Related diseases Defects in DOTL1 are associated with an autosomal dominant form of global developmental delay and intellectual disability, with or without one or more major congenital anomalies (PubMed:37827158). The patient phenotypes are characterized by central nervous system (CNS) dysfunction, such as mild motor delay and significant speech and language delay, and a range of congenital anomalies, including brain structural anomalies, cardiac defects, varied urogenital features and growth restriction (PubMed:37827158). Variants may cause a gain-of-function effect leading to an increase in cellular H3K79 methylation levels (PubMed:37827158). {ECO:0000269|PubMed:37827158}. Drugs (DrugBank ID) NA Interacts with Q03111; P42568 EC number EC 2.1.1.43 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Disease variant; DNA-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 37367.1 Length 322 Aromaticity 0.11 Instability index 32.71 Isoelectric point 6.03 Charge (pH=7) -5.25 2D Binding mode Binding energy (Kcal/mol) -6.28 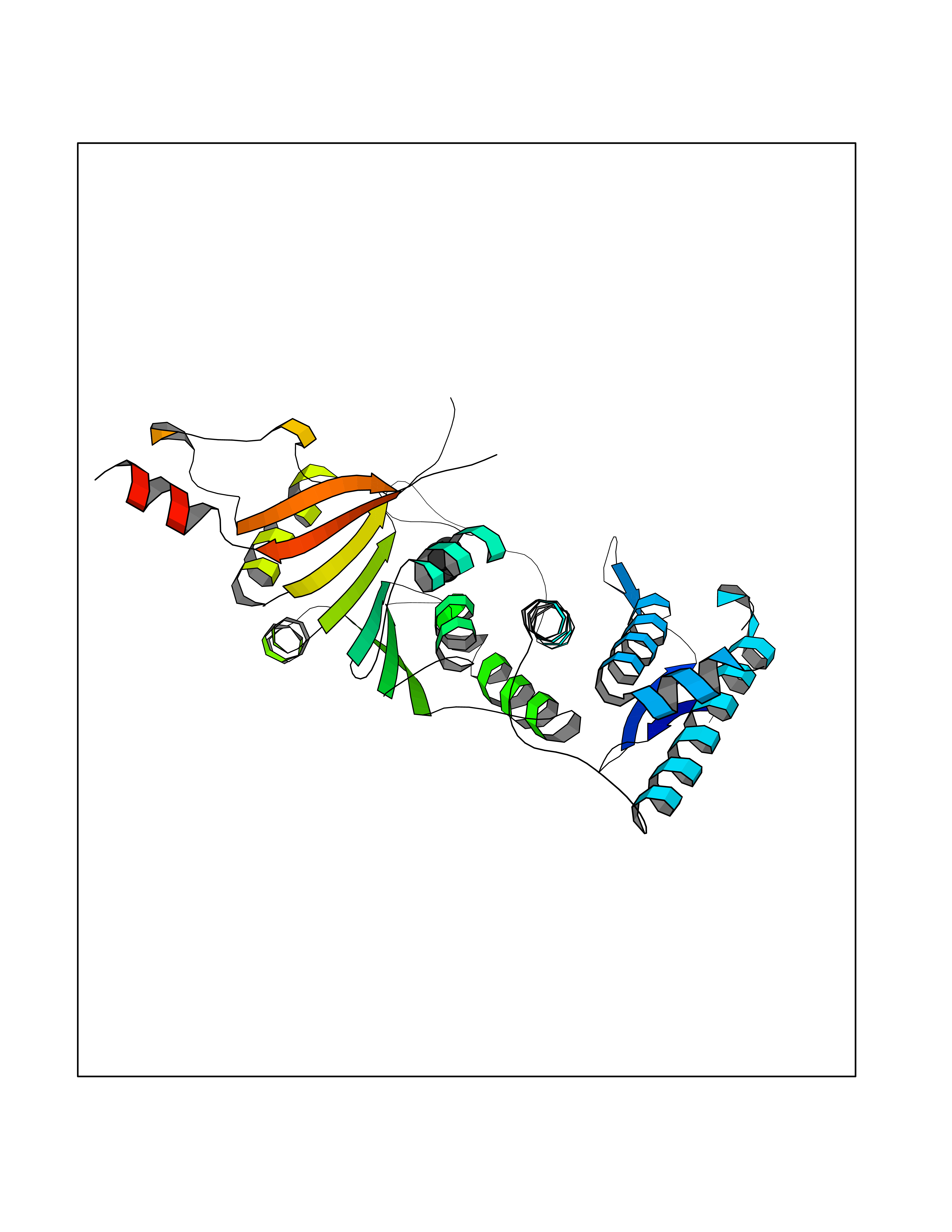 Molscript Map 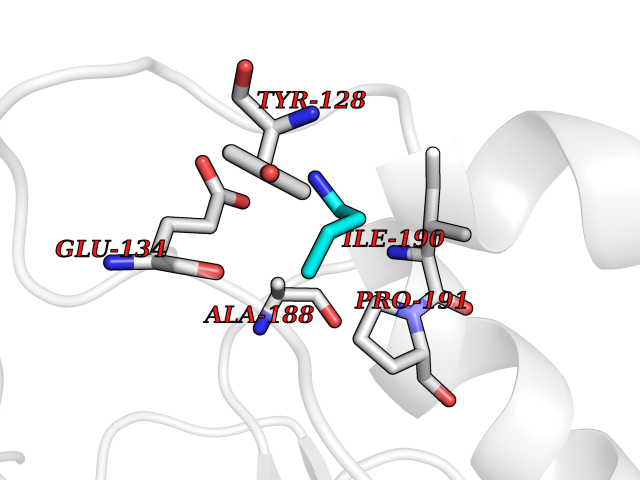 Pymol Map 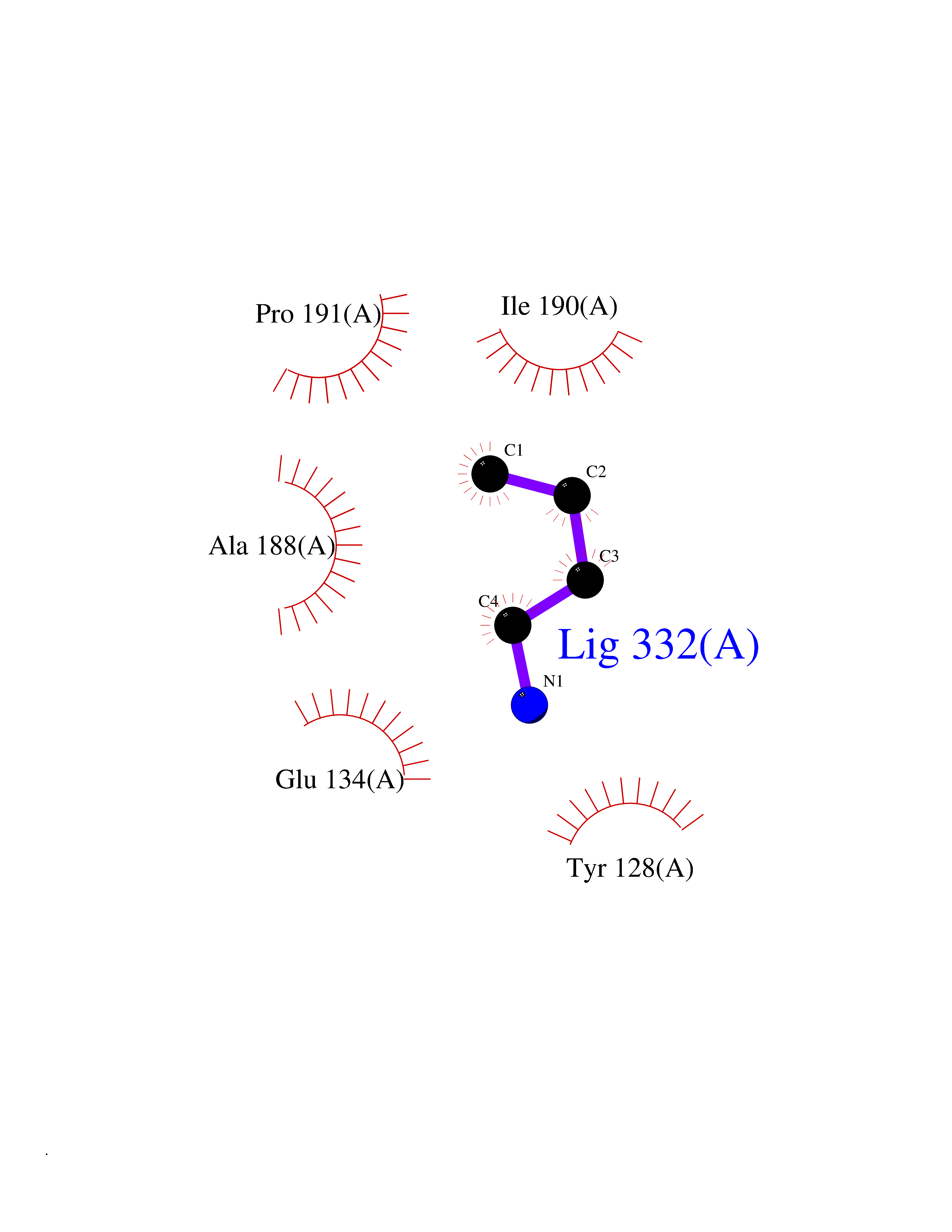 Ligplot Map 3D Binding mode Sequence KLELRLKSPVGAEPAVYPWPLPVYDKHHDAAHEIIETIRWVCEEIPDLKLAMENYLIDYDTKSFESMQRLCDKYNRAIDSIHQLWKGTTQPMKLNTRPSTGLLRHILQQVYNHSVTDPEKLNNYEPFSPEVYGETSFDLVAQMIDEIKMTDDDLFVDLGSGVGQVVLQVAAATNCKHHYGVEKADIPAKYAETMDREFRKWMKWYGKKHAEYTLERGDFLSEEWRERIANTSVIFVNNFAFGPEVDHQLKERFANMKEGGRIVSSKPFAPLNFRINSRNLSDIGTIMRVVELSPLKWTGKPVSYYLHTIDRTILENYFSSLK Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Helicobacter pylori Methylthioadenosine nucleosidase (HELPY mtnN) | 4BMZ | 4.61 | |
Target general information Gen name HELPY mtnN Organism Helicobacter pylori (strain ATCC 700392 / 26695) (Campylobacter pylori) Uniprot ID TTD ID Synonyms MTAN; MTA/SAH nucleosidase; Aminofutalosine nucleosidase; Aminodeoxyfutalosine nucleosidase; AFL nucleosidase; 6-amino-6-deoxyfutalosine N-ribosylhydrolase; 5'-methylthioadenosine/S-adenosylhomocystei Protein family PNP/UDP phosphorylase family Biochemical class NA Function Catalyzes the direct conversion of aminodeoxyfutalosine (AFL) into dehypoxanthine futalosine (DHFL) and adenine via the hydrolysis of the N-glycosidic bond; this reaction seems to represent an essential step in the menaquinone biosynthesis pathway in Helicobacter species. Can also probably catalyzes the hydrolysis of 5'-methylthioadenosine (MTA) and S-adenosylhomocysteine (SAH) to adenine and the corresponding thioribose, 5'-methylthioribose and S-ribosylhomocysteine, respectively. These other activities highlight the tremendous versatility of the enzyme, which also plays key roles in S-adenosylmethionine recycling and in the biosynthesis of the quorum-sensing molecule autoinducer-2. Does not act on futalosine (FL) as substrate. Related diseases Progressive familial heart block 1B (PFHB1B) [MIM:604559]: A cardiac bundle branch disorder characterized by progressive alteration of cardiac conduction through the His-Purkinje system, with a pattern of a right bundle-branch block and/or left anterior hemiblock occurring individually or together. It leads to complete atrio-ventricular block causing syncope and sudden death. {ECO:0000269|PubMed:19726882, ECO:0000269|PubMed:20562447, ECO:0000269|PubMed:21887725}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythrokeratodermia variabilis et progressiva 6 (EKVP6) [MIM:618531]: A form of erythrokeratodermia variabilis et progressiva, a genodermatosis characterized by the coexistence of two independent skin lesions: transient erythema and hyperkeratosis that is usually localized but occasionally occurs in its generalized form. Clinical presentation varies significantly within a family and from one family to another. Palmoplantar keratoderma is present in around 50% of cases. EKVP6 inheritance is autosomal dominant. {ECO:0000269|PubMed:30528822}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Amino-acid biosynthesis; Hydrolase; Menaquinone biosynthesis; Methionine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50547.6 Length 464 Aromaticity 0.08 Instability index 26.92 Isoelectric point 5.13 Charge (pH=7) -20.92 2D Binding mode Binding energy (Kcal/mol) -6.29 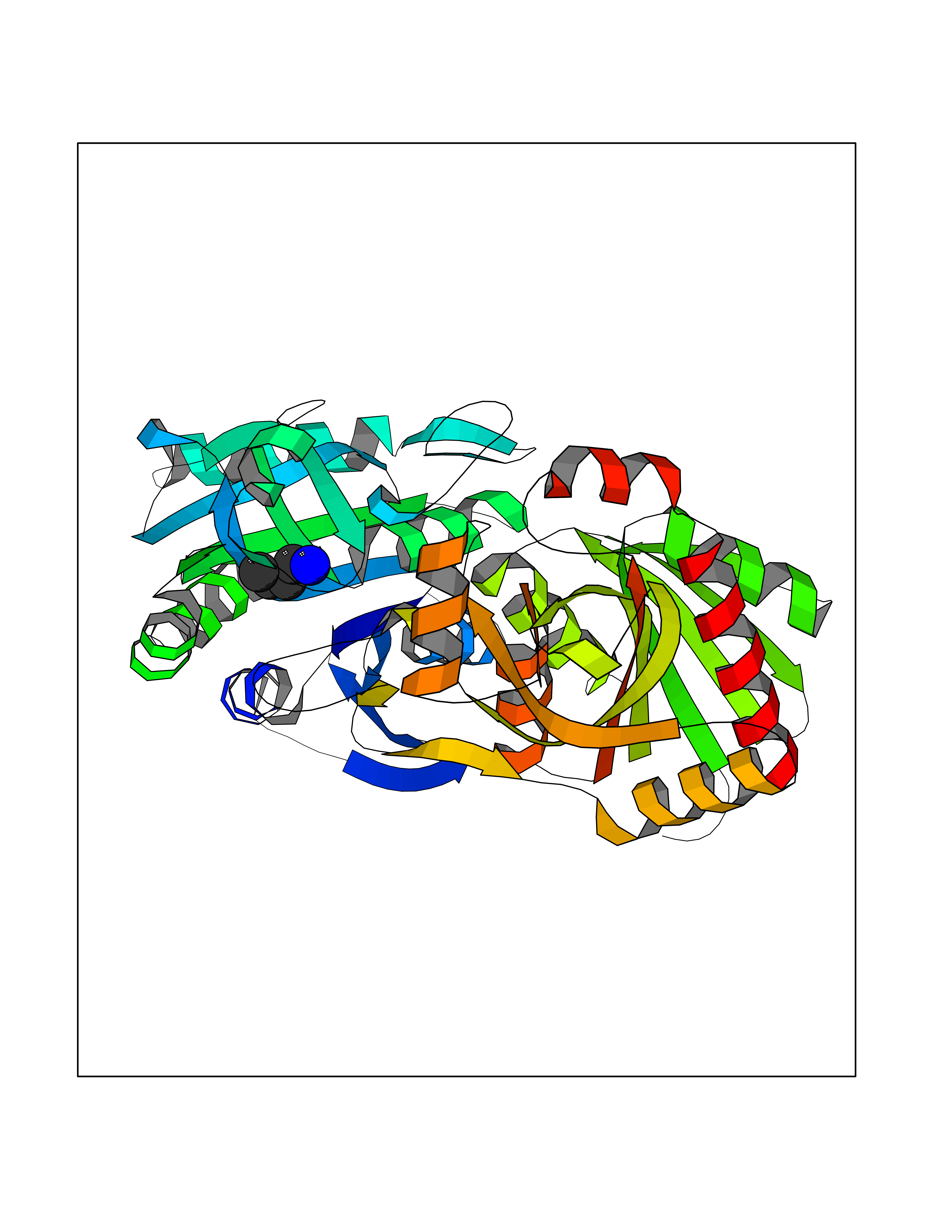 Molscript Map 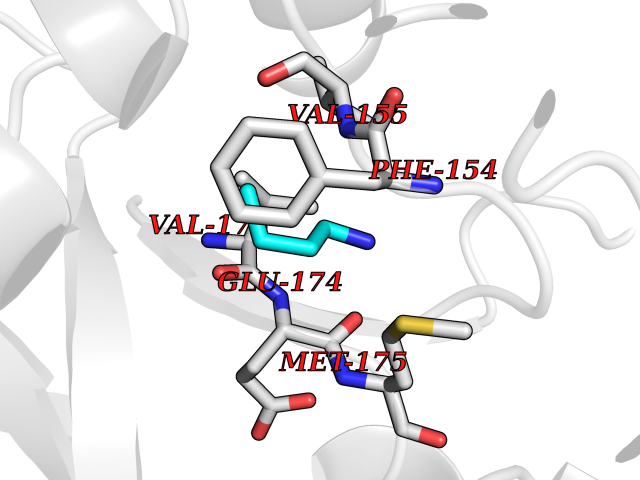 Pymol Map 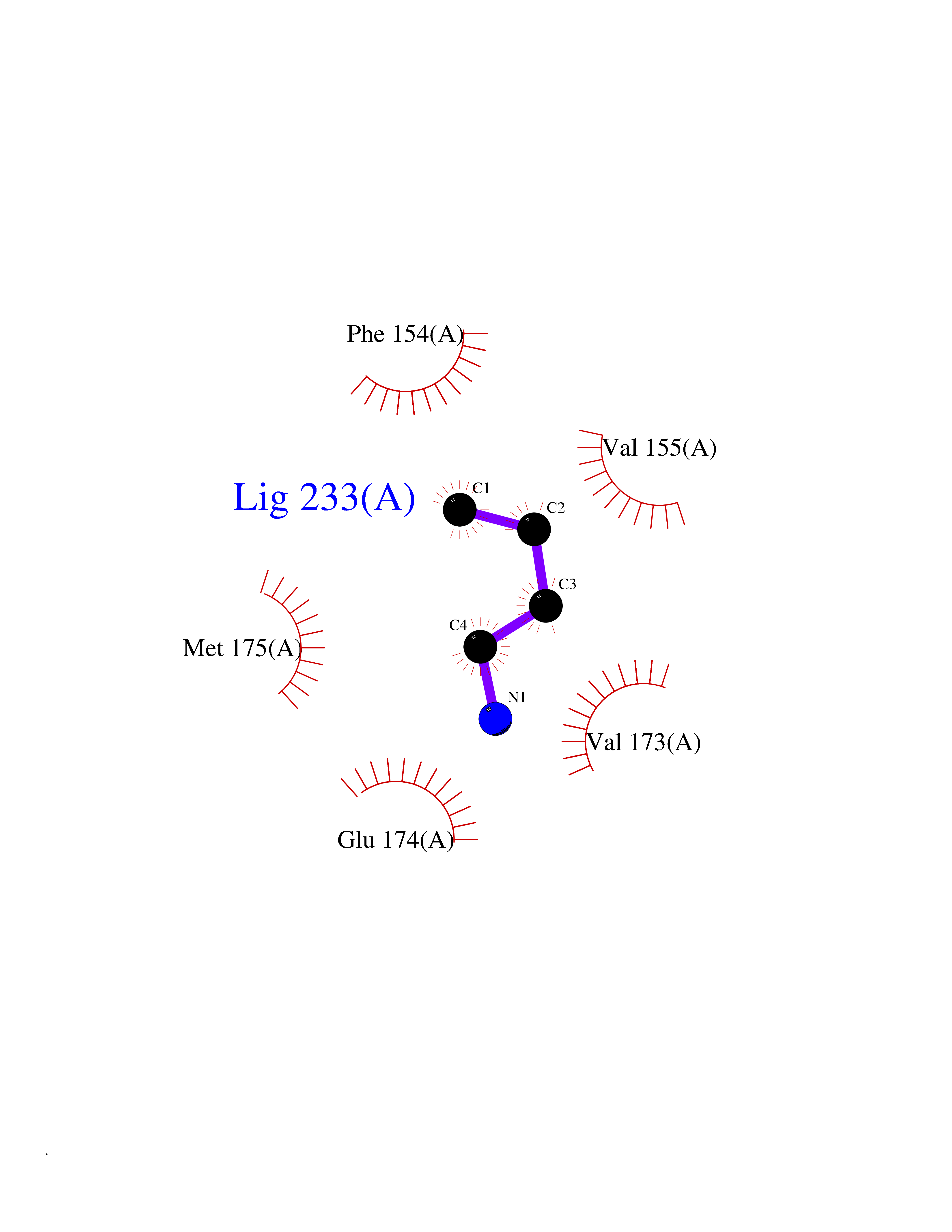 Ligplot Map 3D Binding mode Sequence VQKIGILGAMREEITPILELFGVDFEEIPLGGNVFHKGVYHNKEIIVAYSKIGKVHSTLTTTSMILAFGVQKVLFSGVAGSLVKDLKINDLLVAIQLVQHDVDLSAFDHPLGFIPESAIFIETSESLNALAKEVANEQHIVLKEGVIASGDQFVHSKERKEFLVSEFKASAVEMEGASVAFVCQKFGVPCCVLRSISNNADEEANMSFDAFLEKSAQTSAKFLKSMVDELGSHMVQKIGILGAMREEITPILELFGVDFEEIPLGGNVFHKGVYHNKEIIVAYSKIGKVHSTLTTTSMILAFGVQKVLFSGVAGSLVKDLKINDLLVAIQLVQHDVDLSAFDHPLGFIPESAIFIETSESLNALAKEVANEQHIVLKEGVIASGDQFVHSKERKEFLVSEFKASAVEMEGASVAFVCQKFGVPCCVLRSISNNADEEANMSFDAFLEKSAQTSAKFLKSMVDEL Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Endolysin | 1AM7 | 4.60 | |
Target general information Gen name R Organism Escherichia phage lambda (Bacteriophage lambda) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 24 family Biochemical class Glycosidase Function Lyase activity.Lysozyme activity.Lytic transglycosylase activity. Related diseases Estrogen resistance (ESTRR) [MIM:615363]: A disorder characterized by partial or complete resistance to estrogens, in the presence of elevated estrogen serum levels. Clinical features include absence of the pubertal growth spurt, delayed bone maturation, unfused epiphyses, reduced bone mineral density, osteoporosis, continued growth into adulthood and very tall adult stature. Glucose intolerance, hyperinsulinemia and lipid abnormalities may also be present. {ECO:0000269|PubMed:23841731, ECO:0000269|PubMed:27754803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04206 Interacts with NA EC number 4.2.2.n2 Uniprot keywords 3D-structure; Antimicrobial; Bacteriolytic enzyme; Cytolysis; Direct protein sequencing; Host cell lysis by virus; Host cytoplasm; Lyase; Reference proteome; Viral release from host cell Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 49834.9 Length 462 Aromaticity 0.07 Instability index 18.78 Isoelectric point 9.6 Charge (pH=7) 18.29 2D Binding mode Binding energy (Kcal/mol) -6.28 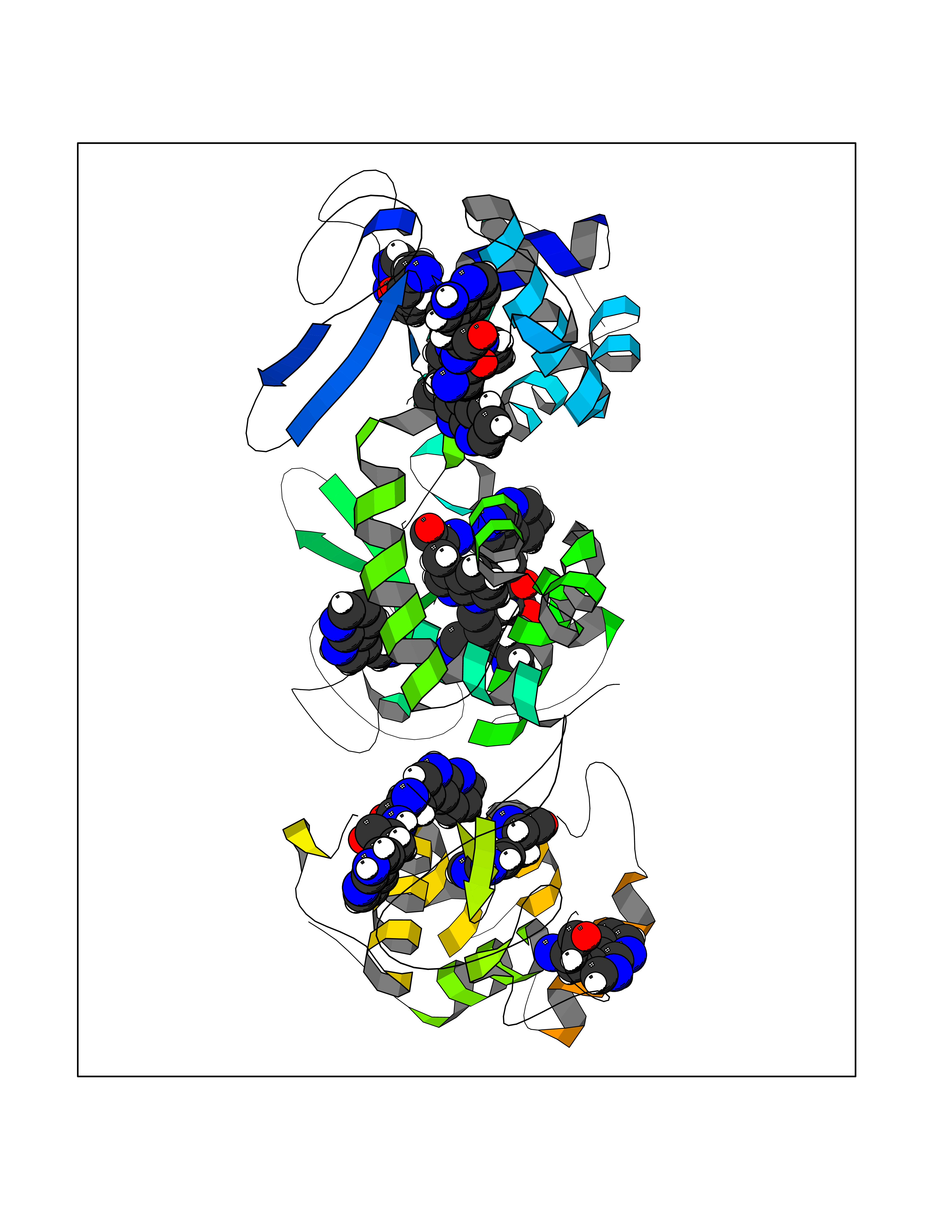 Molscript Map 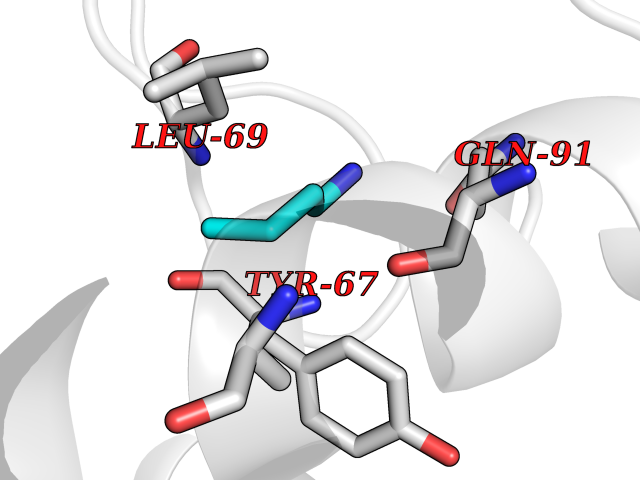 Pymol Map 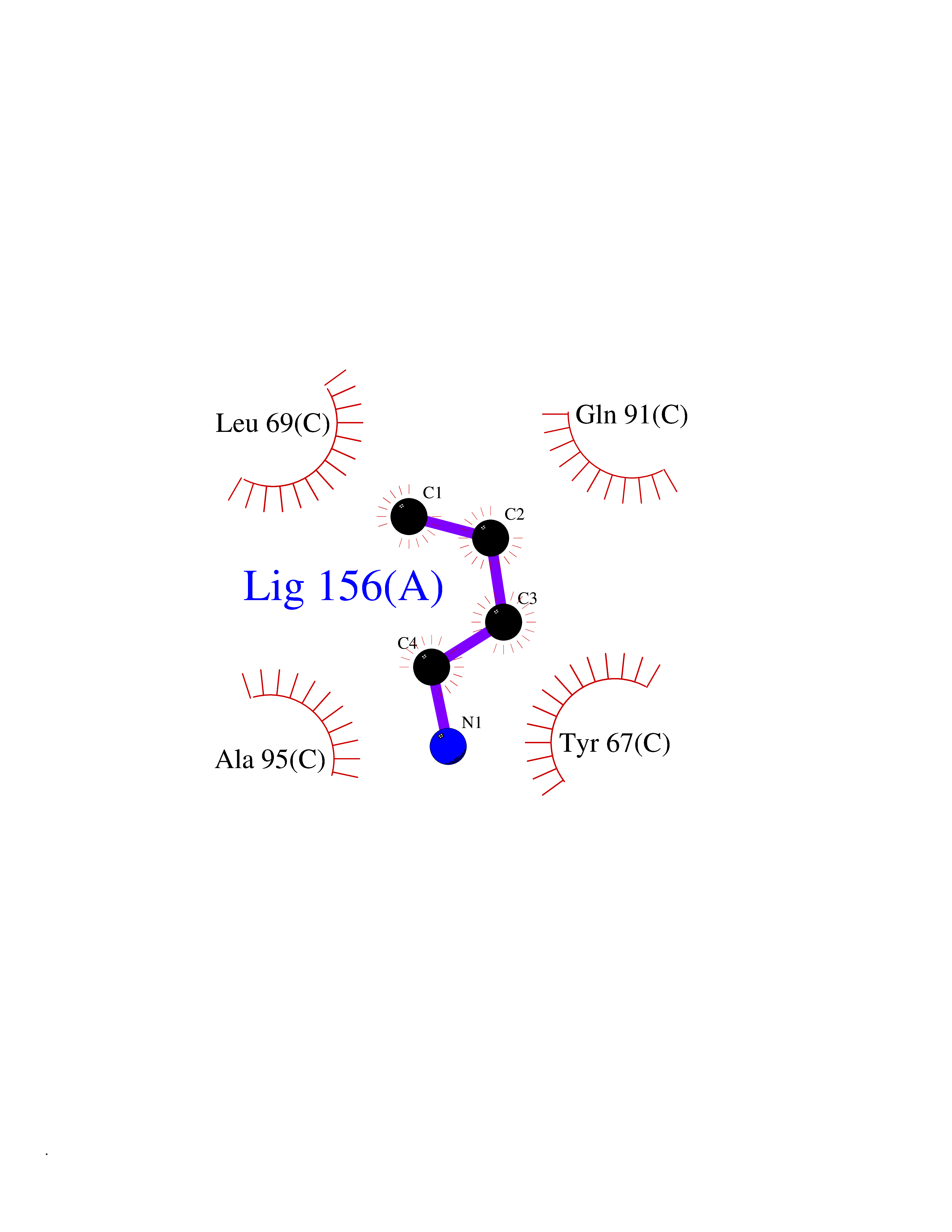 Ligplot Map 3D Binding mode Sequence MVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVR Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Chromodomain-helicase-DNA-binding protein 1 | 4O42 | 4.60 | |
Target general information Gen name CHD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SNF2/RAD54 helicase family Biochemical class Dna binding protein / viral protein Function ATP binding.ATP-dependent DNA helicase activity.DNA binding.Methylated histone binding. Related diseases Pilarowski-Bjornsson syndrome (PILBOS) [MIM:617682]: An autosomal dominant disorder characterized by developmental delay, speech apraxia, intellectual disability, autism, and facial dysmorphic features. Some patients may have seizures. {ECO:0000269|PubMed:28866611}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60341-1; B2BUF1; P28799; O76024 EC number 3.6.4.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromatin regulator; Cytoplasm; Disease variant; DNA-binding; Helicase; Hydrolase; Intellectual disability; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 20969.1 Length 180 Aromaticity 0.12 Instability index 46.35 Isoelectric point 5.88 Charge (pH=7) -2.83 2D Binding mode Binding energy (Kcal/mol) -6.27 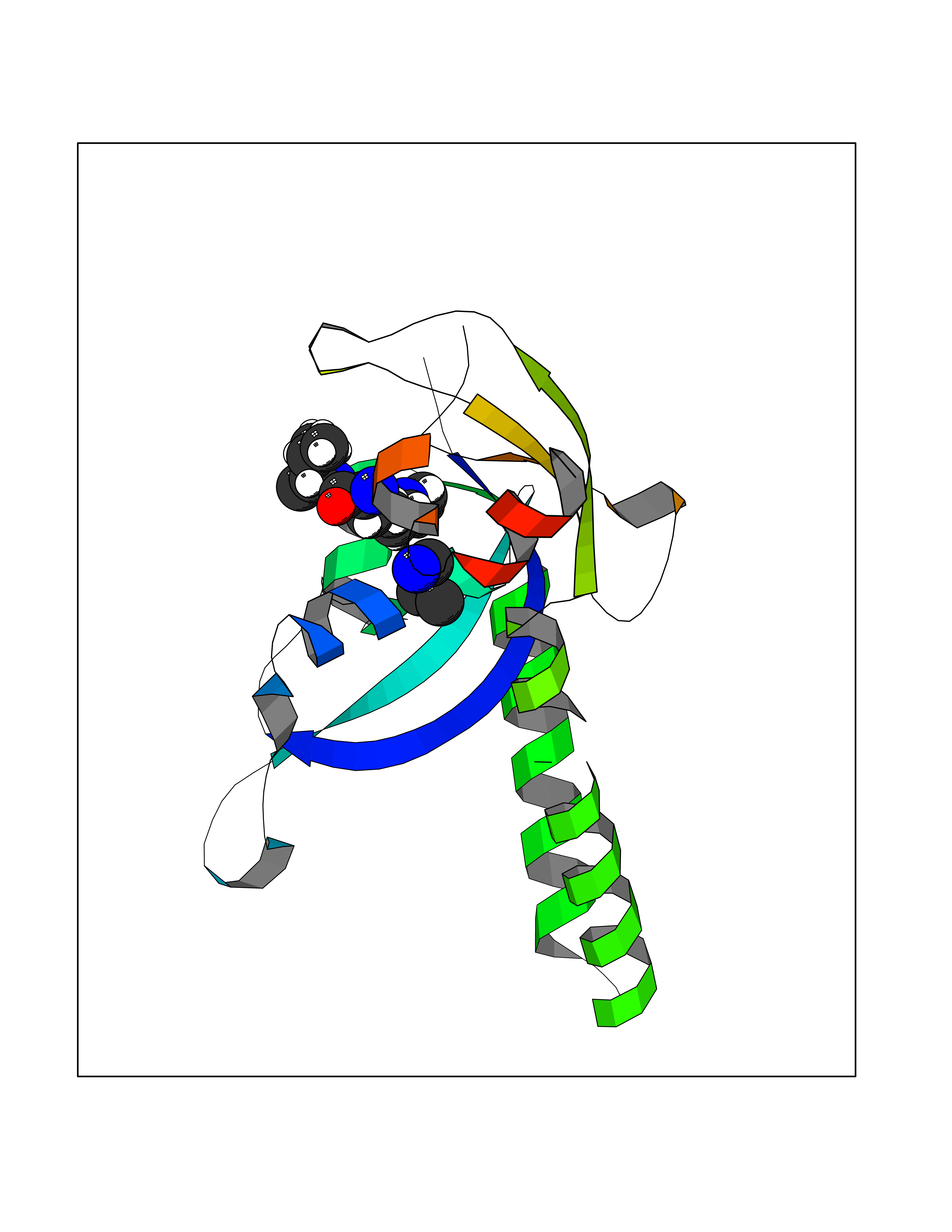 Molscript Map 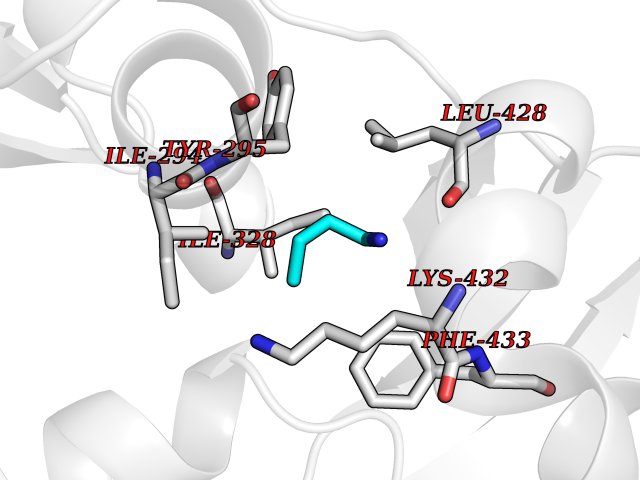 Pymol Map 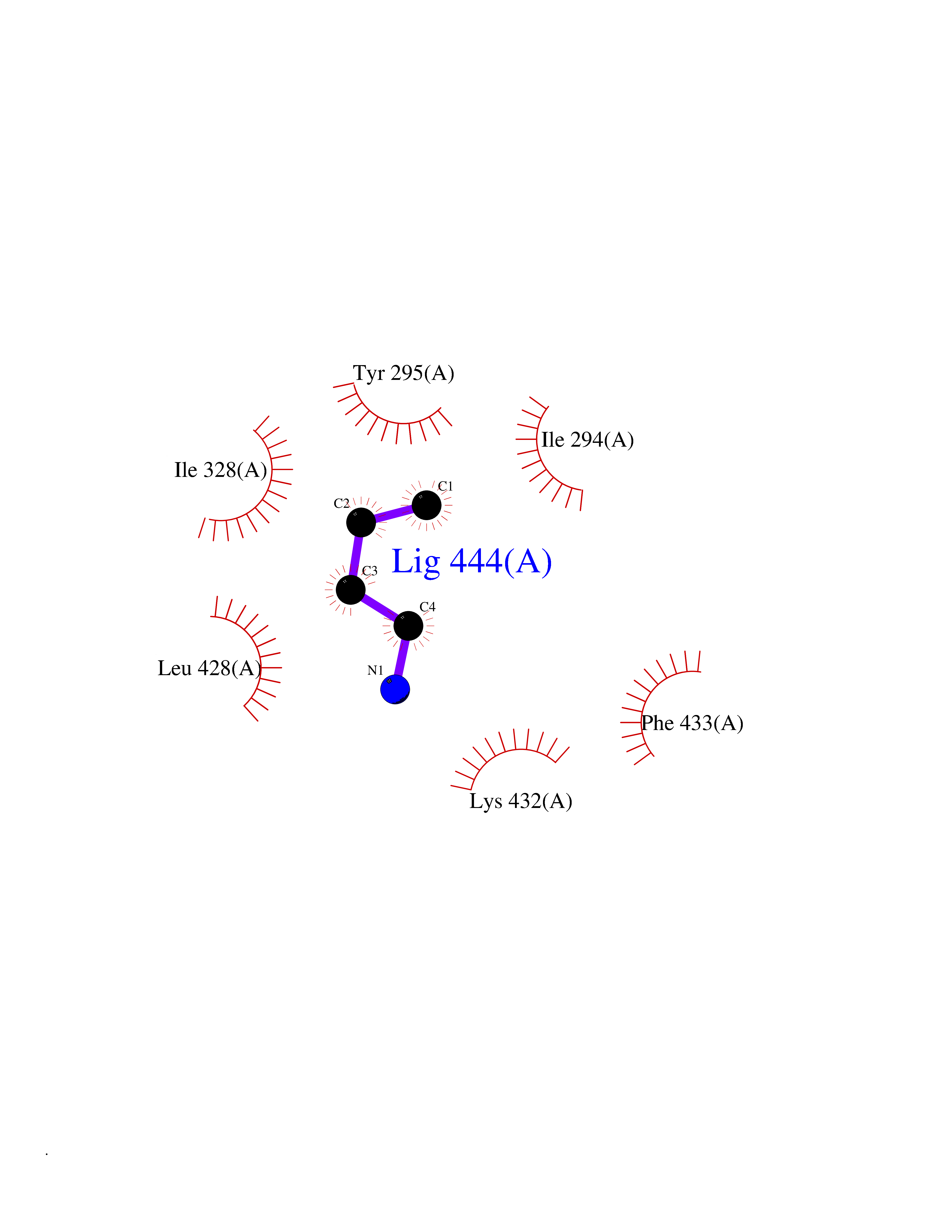 Ligplot Map 3D Binding mode Sequence EFETIERFMDCRIGRKGATGATTTIYAVEADGDPNAGFEKNKEPGEIQYLIKWKGWSHIHNTWETEETLKQQNVRGMKKLDNYKKKDQETKRWLKNASPEDVEYYNCQQELTDDLHKQYQIVERIIAHSNQKSAAGYPDYYCKWQGLPYSECSWEDGALISKKFQACIDEYFSRTARSXV Hydrogen bonds contact Hydrophobic contact | ||||