Job Results:
Ligand
Structure
Job ID
1aab99f222a82120b630ad6d44e2ad5f
Job name
NA
Time
2026-02-27 11:58:02
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 4.57 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -6.24 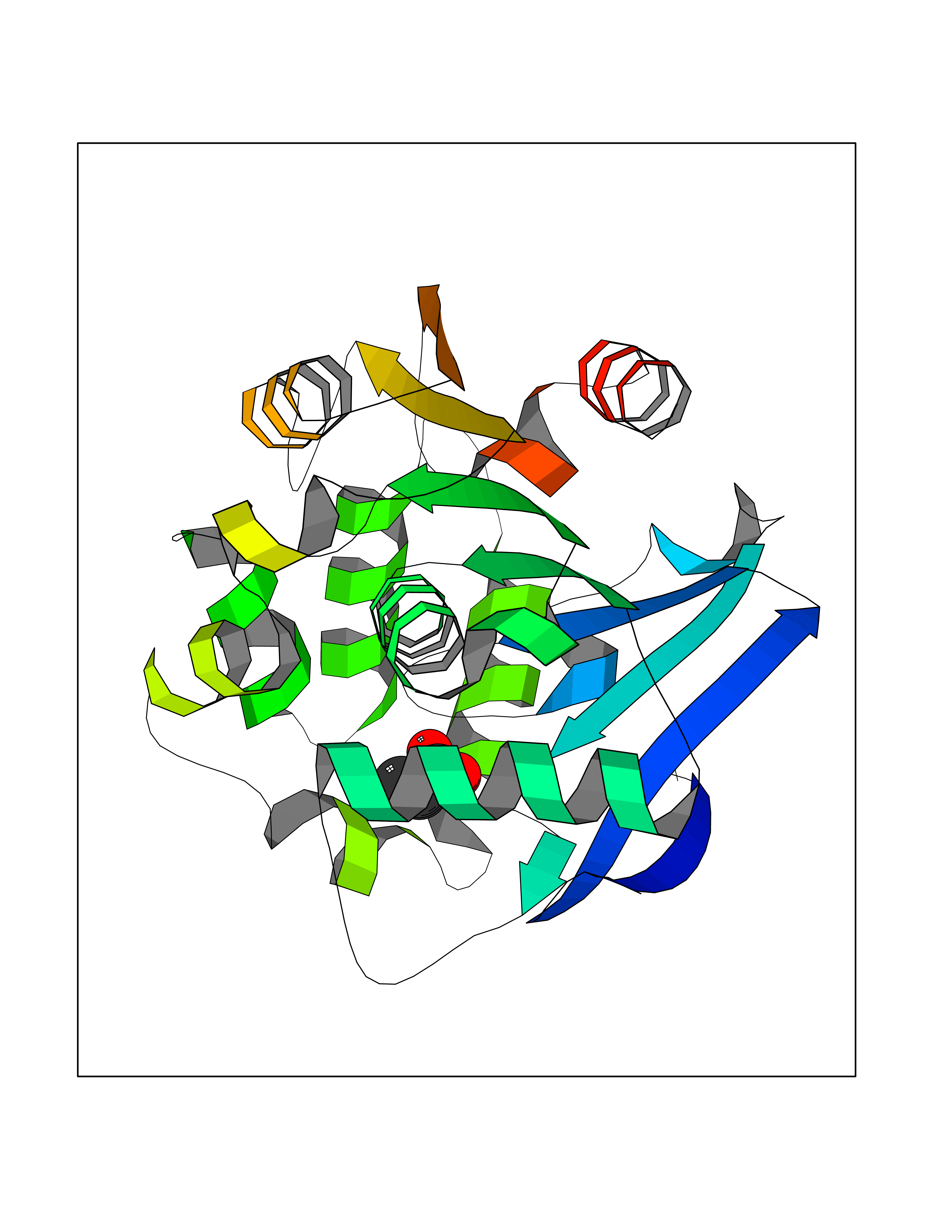 Molscript Map 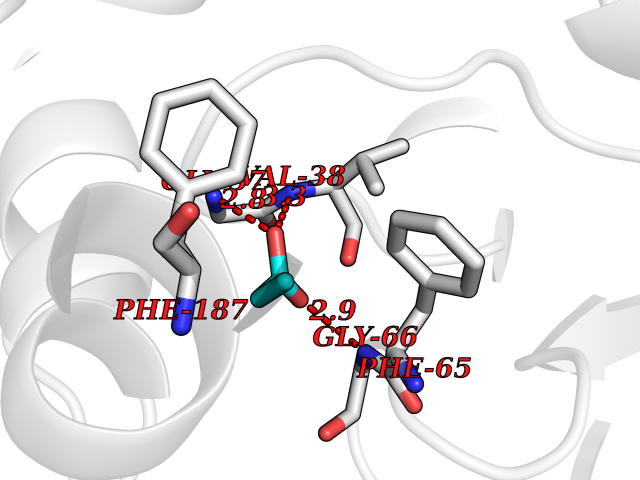 Pymol Map 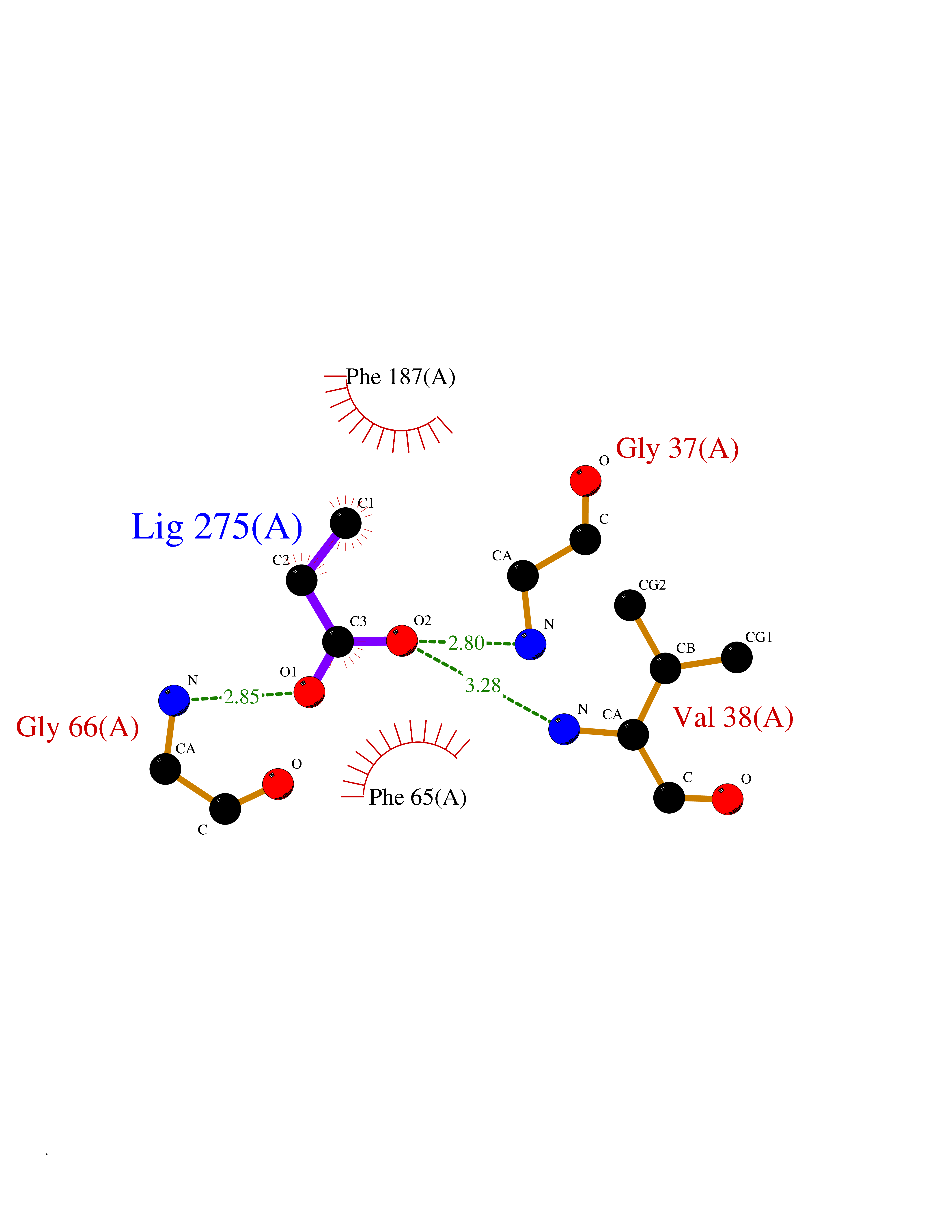 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 4.57 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -6.24 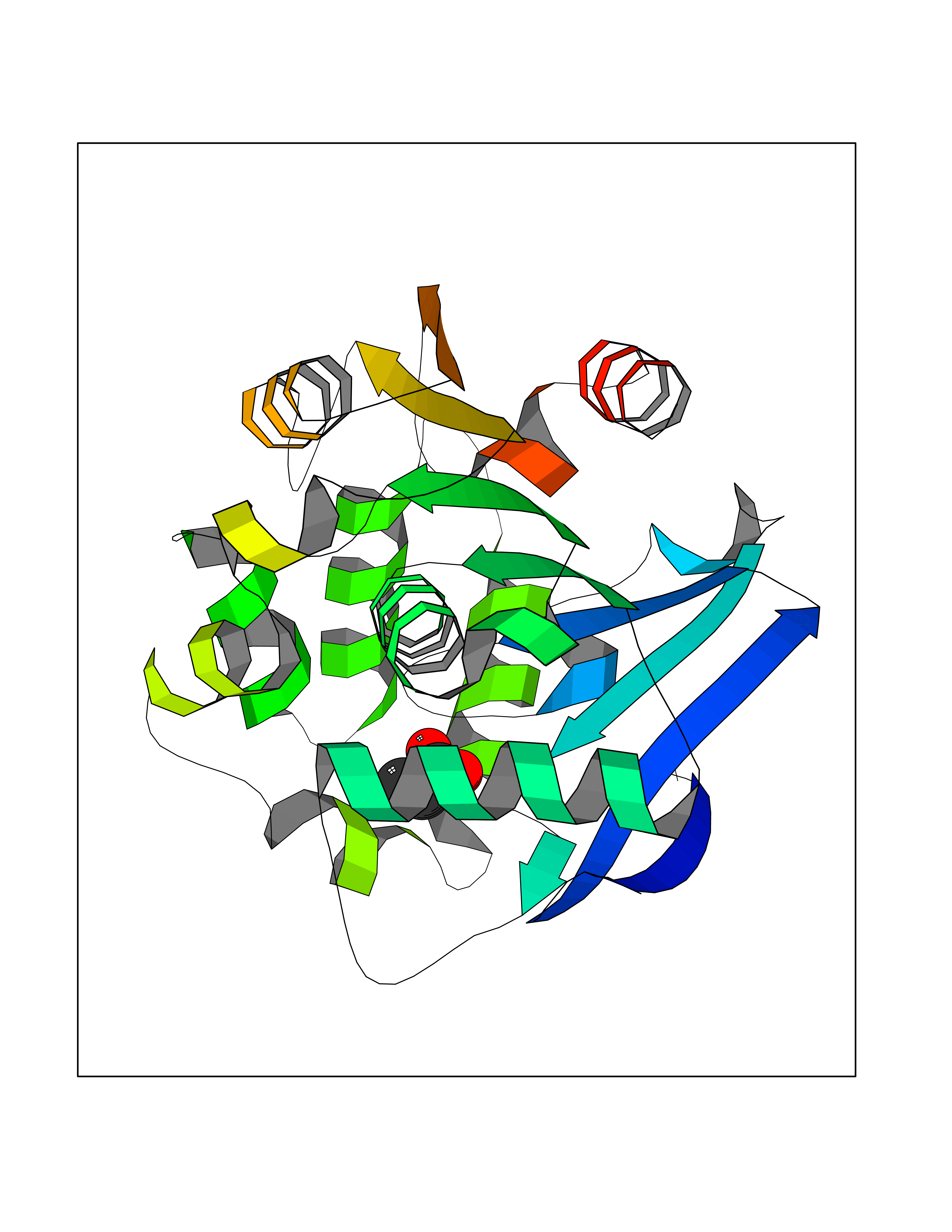 Molscript Map 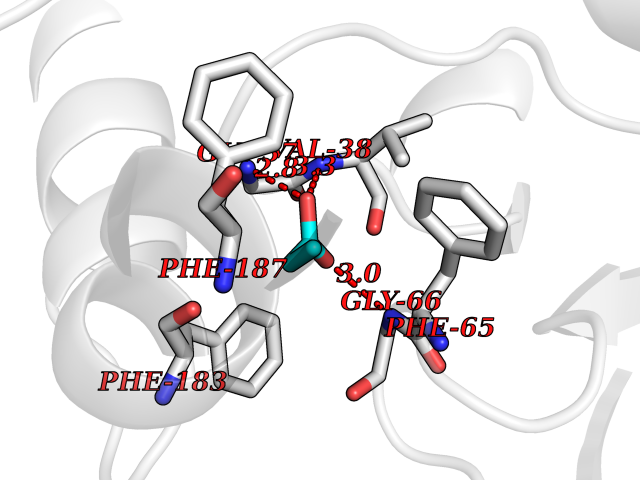 Pymol Map 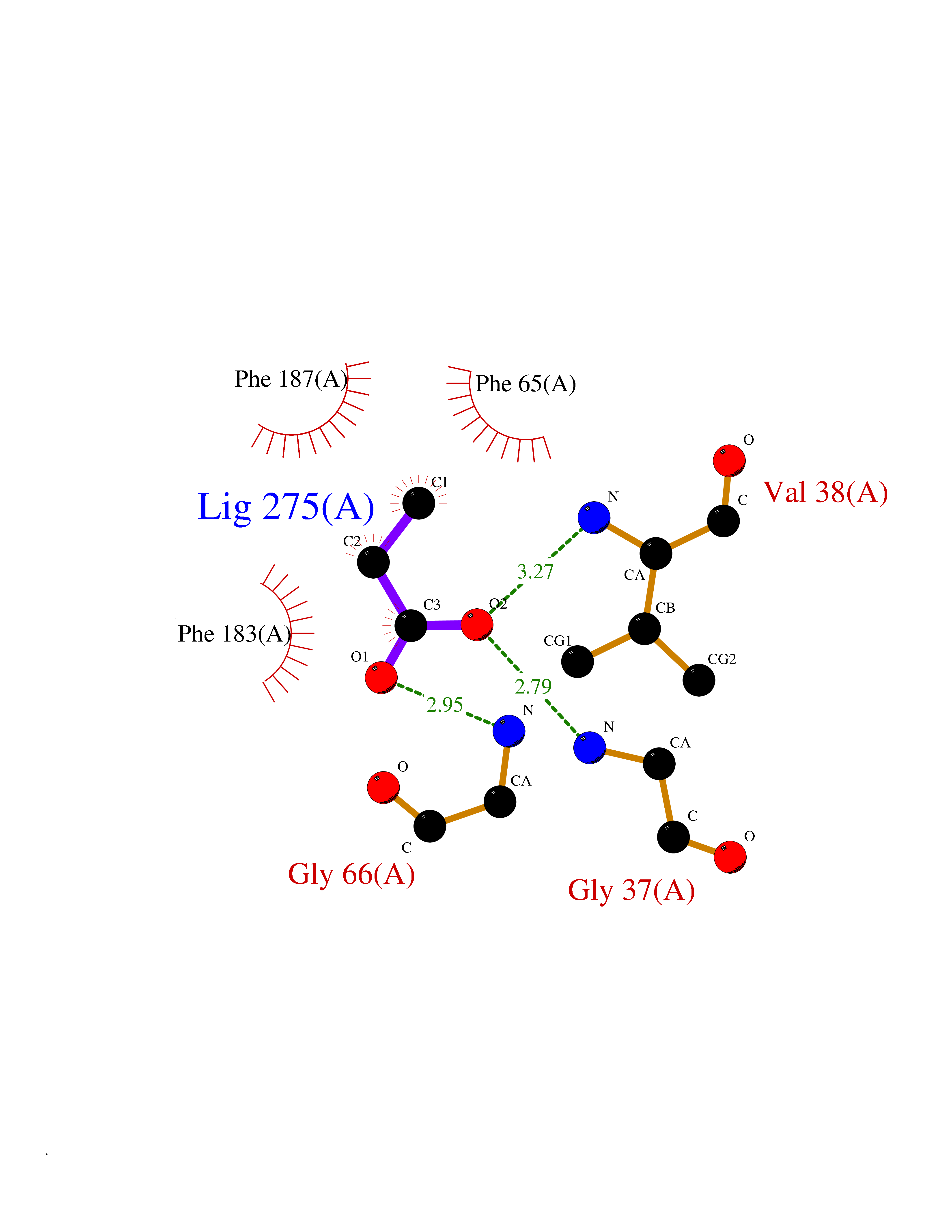 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | cAMP-specific 3',5'-cyclic phosphodiesterase 4C | 2QYM | 4.57 | |
Target general information Gen name PDE4C Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms DPDE1 Protein family Cyclic nucleotide phosphodiesterase family, PDE4 subfamily Biochemical class Hydrolase Function 3',5'-cyclic-AMP phosphodiesterase activity.Metal ion binding. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01427; DB00201; DB05219; DB00651; DB06246; DB05266; DB01088; DB01791; DB01656; DB01954; DB09283 Interacts with Q96D03; O15499; P26718; P50221; Q6FHY5; Q9UJX0; P26367; P30626; P59817; P30626 EC number 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cell projection; Cilium; Hydrolase; Manganese; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 33042.2 Length 291 Aromaticity 0.08 Instability index 23.45 Isoelectric point 4.91 Charge (pH=7) -18.1 2D Binding mode Binding energy (Kcal/mol) -6.24 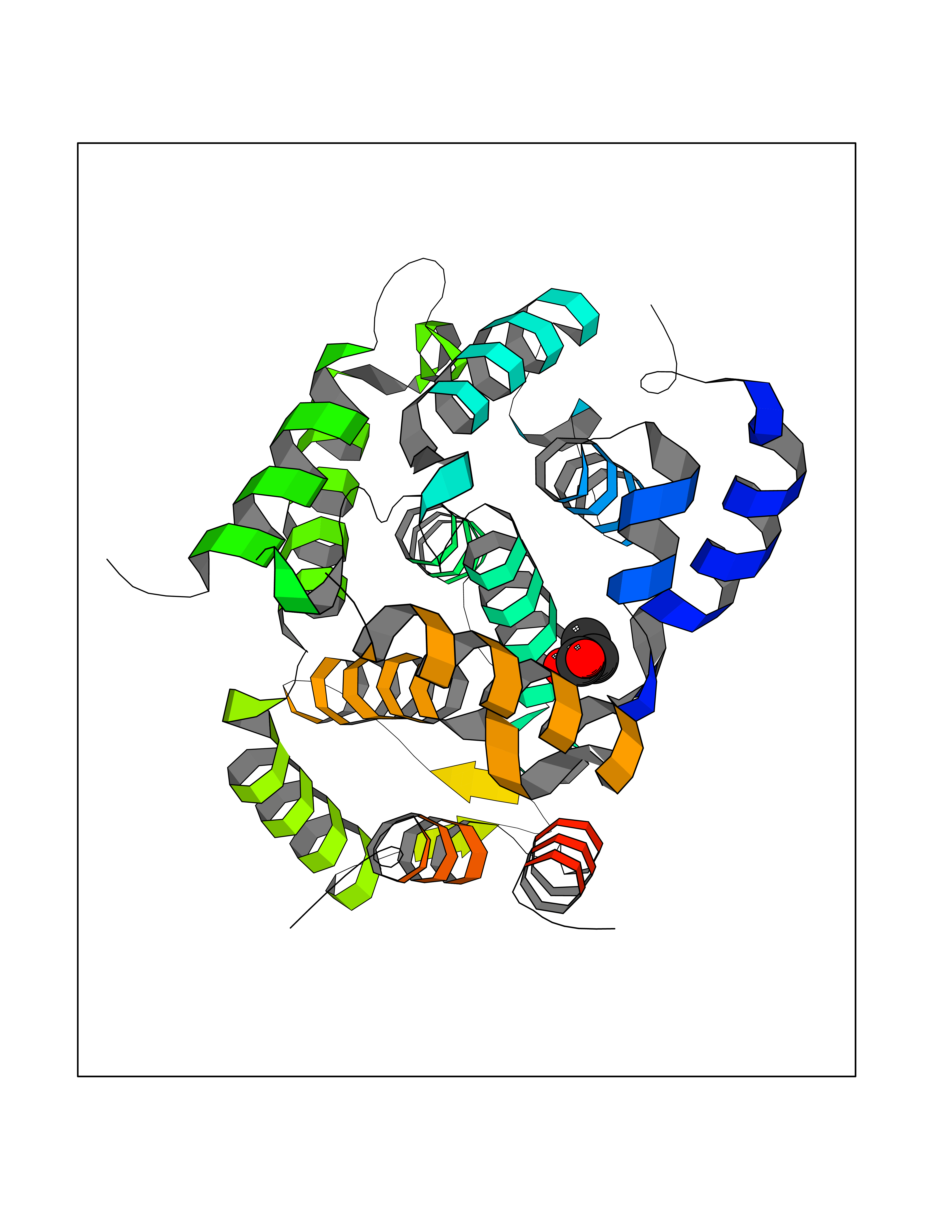 Molscript Map 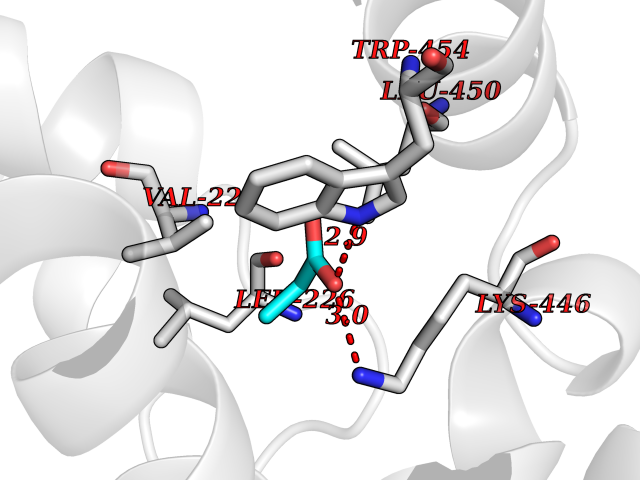 Pymol Map 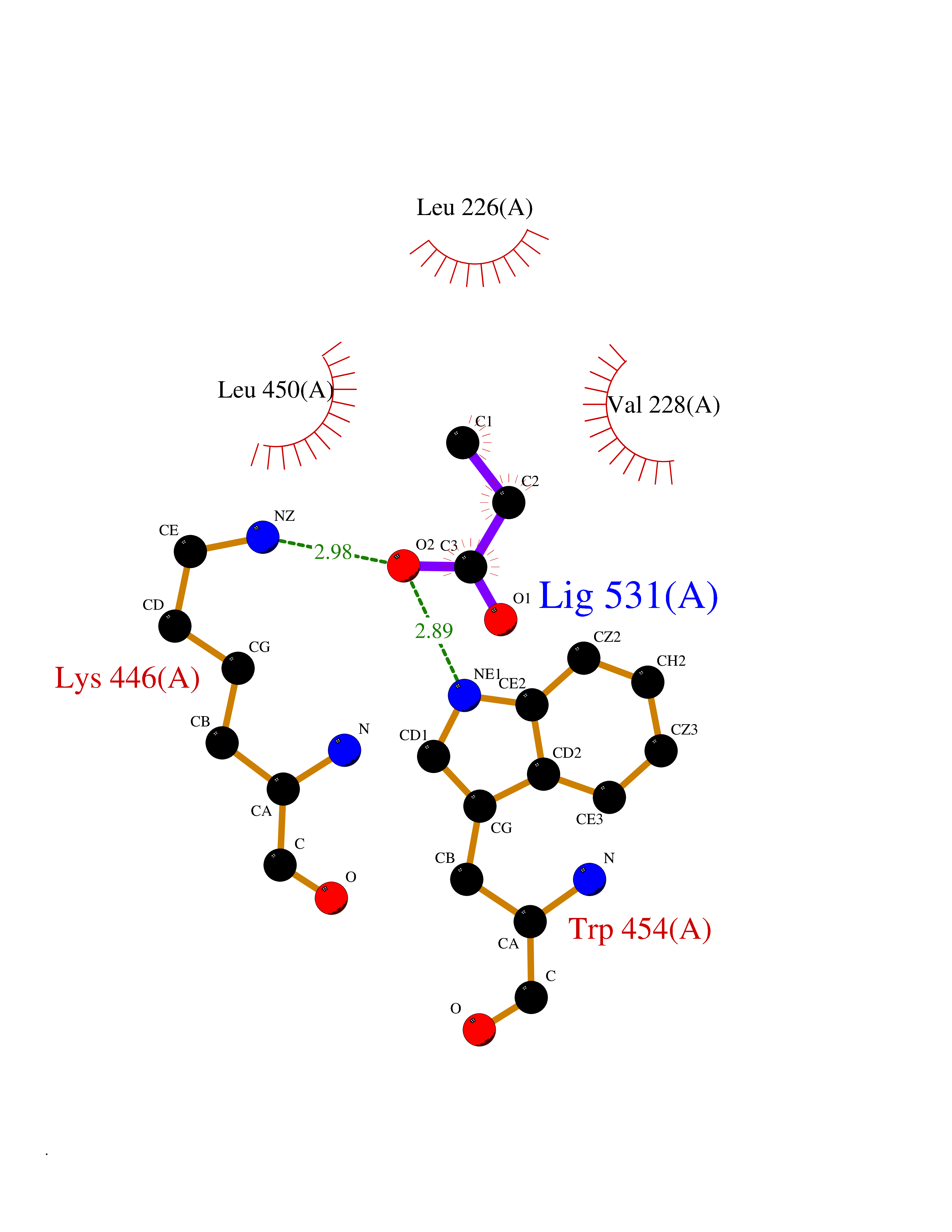 Ligplot Map 3D Binding mode Sequence VPRFGVQTDQEEQLAKELEDTNKWGLDVFKVAELSGNRPLTAIIFSIFQERDLLKTFQIPADTLATYLLMLEGHYHANVAYHNSLHAADVAQSTHVLLATPALEAVFTDLEILAALFASAIHDVDHPGVSNQNDASVLENHHLAVGFKLLQAENCDIFQNLSAKQRLSLRRMVIDMVLATDMSKHMNLLADLKTMVETKKVTSLGVLLLDNYSDRIQVLQNLVHCADLSNPTKPLPLYRQWTDRIMAEFFQQQVGFIDYIAHPLWETWADLVHPDAQDLLDTLEDNREWYQ Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Glutamate receptor ionotropic NMDA 2A (NMDAR2A) | 5KCJ | 4.57 | |
Target general information Gen name GRIN2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NR2A; NMDA receptor NR2A; N-methyl D-aspartate receptor subtype 2A; HNR2A; Glutamate receptor ionotropic, NMDA 2A; Glutamate [NMDA] receptor subunit epsilon-1; GluN2A Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, NR1/GRIN1 subfamily Biochemical class Glutamate-gated ion channel Function Channel activation requires binding of the neurotransmitter glutamate to the epsilon subunit, glycine binding to the zeta subunit, plus membrane depolarization to eliminate channel inhibition by Mg(2+). Sensitivity to glutamate and channel kinetics depend on the subunit composition; channels containing GRIN1 and GRIN2A have higher sensitivity to glutamate and faster kinetics than channels formed by GRIN1 and GRIN2B. Contributes to the slow phase of excitatory postsynaptic current, long-term synaptic potentiation, and learning. Component of NMDA receptor complexes that function as heterotetrameric, ligand-gated ion channels with high calcium permeability and voltage-dependent sensitivity to magnesium. Related diseases Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant (NDHMSD) [MIM:614254]: An autosomal dominant neurodevelopmental disorder characterized by severe intellectual disability and developmental delay, absent speech, muscular hypotonia, dyskinesia, and hyperkinetic movements. Cortical blindness, cerebral atrophy, and seizures are present in some patients. {ECO:0000269|PubMed:21376300, ECO:0000269|PubMed:25167861, ECO:0000269|PubMed:25864721, ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:28228639, ECO:0000269|PubMed:28389307, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal recessive (NDHMSR) [MIM:617820]: An autosomal recessive neurodevelopmental disorder characterized by severe intellectual disability and psychomotor developmental delay, involuntary and stereotypic movements, spasticity, and inability to walk without support. Intractable seizures manifest in some patients. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28051072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 101 (DEE101) [MIM:619814]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE101 is an autosomal recessive, severe form characterized by onset of seizures in early infancy. Death in infancy may occur. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:34611970}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01931; DB00659; DB06151; DB08838; DB01238; DB00289; DB05824; DB04620; DB03929; DB00647; DB00843; DB00228; DB11823; DB13146; DB06741; DB00142; DB00874; DB08954; DB06738; DB09409; DB09481; DB01043; DB00454; DB00333; DB04896; DB01173; DB00312; DB01174; DB01708; DB00418; DB00193 Interacts with P05067; P35637; Q12879-1; Q13224; Q62936 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Ligand-gated ion channel; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Zinc Protein physicochemical properties Chain ID B Molecular weight (Da) 53395.6 Length 469 Aromaticity 0.11 Instability index 29.84 Isoelectric point 8.72 Charge (pH=7) 5.65 2D Binding mode Binding energy (Kcal/mol) -6.23  Molscript Map 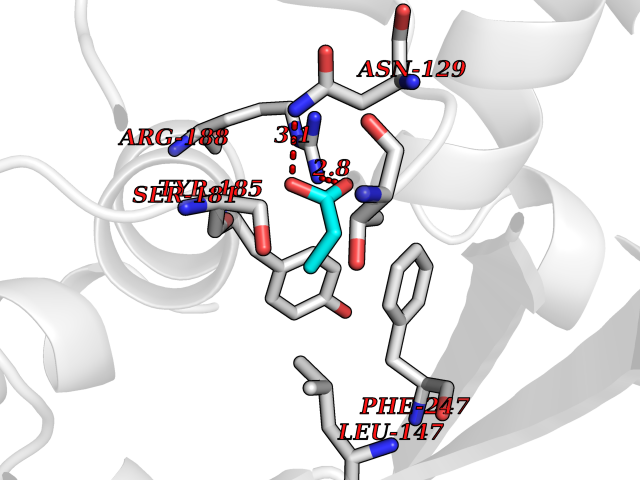 Pymol Map 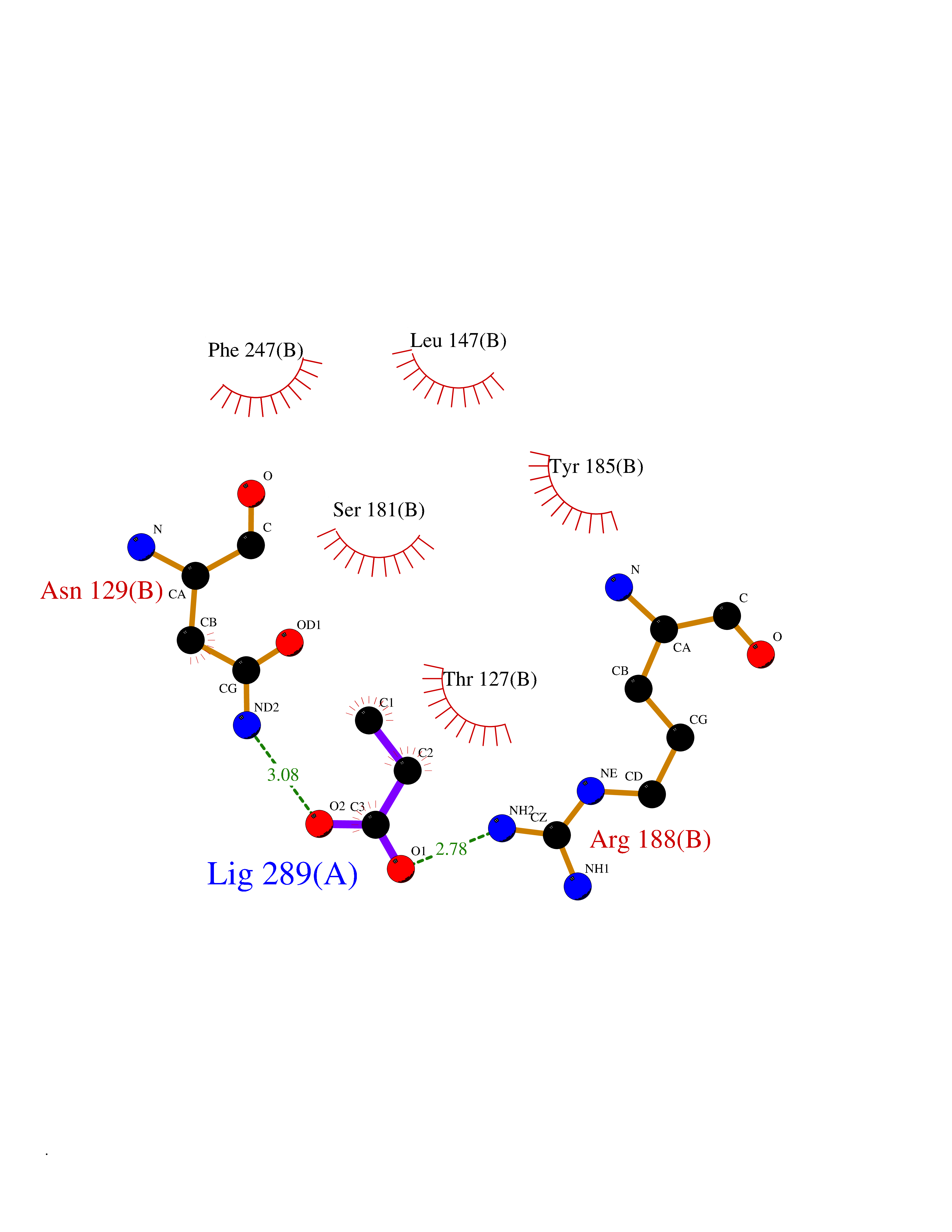 Ligplot Map 3D Binding mode Sequence DNHLSIVTLEEAPFVILKKLSRTVKFTYDLYLVTNGKHGKKVNNVWNGMIGEVVYQRAVMAVGSLTINEERSEVVDFSVPFVETGISVMVSRGTQVTGLSDKKFQRPHDYSPPFRFGTVPNGSTERNIRNNYPYMHQYMTKFNQKGVEDALVSLKTGKLDAFIYDAAVLNYKAGRDEGCKLVTIGSGYIFATTGYGIALQKGSPWKRQIDLALLQFVGDGEMEELETLWLTGICTRLKIVTIHQEPFVYYGFCIDLLIKLARTMNFTYEVHLVADGKFGTQERVNKKEWNGMMGELLSGQADMIVAPLTINNERAQYIEFSKPFKYQGLTILVKKGTRITGINDPRLRNPSDKFIYATVKQSSVDIYFRRQVELSTMYRHMEKHNYESAAEAIQAVRDNKLHAFIWDSAVLEFEASQKCDLVTTGELFFRSGFGIGMRKDSPWKQNVSLSILKSHENGFMEDLDKTWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Histone deacetylase 8 (HDAC8) | 5BWZ | 4.57 | |
Target general information Gen name HDAC8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histone deacetylase-8; HDACL1; HD8; CDA07 Protein family Histone deacetylase family, HD type 1 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Also involved in the deacetylation of cohesin complex protein SMC3 regulating release of cohesin complexes from chromatin. May play a role in smooth muscle cell contractility. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4). Related diseases Cornelia de Lange syndrome 5 (CDLS5) [MIM:300882]: A form of Cornelia de Lange syndrome, a clinically heterogeneous developmental disorder associated with malformations affecting multiple systems. It is characterized by facial dysmorphisms, abnormal hands and feet, growth delay, cognitive retardation, hirsutism, gastroesophageal dysfunction and cardiac, ophthalmologic and genitourinary anomalies. {ECO:0000269|PubMed:22885700, ECO:0000269|PubMed:22889856}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07350; DB02565; DB07586; DB12565; DB05015; DB08168; DB01262; DB11841; DB14490; DB14491; DB14488; DB14501; DB14489; DB12645; DB01592; DB02917; DB06603; DB06819; DB03766; DB12847; DB06176; DB04297; DB00313; DB02546; DB01593; DB14487; DB14533; DB14548 Interacts with NA EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Cytoplasm; Disease variant; Hydrolase; Intellectual disability; Metal-binding; Nucleus; Obesity; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 39018.4 Length 351 Aromaticity 0.11 Instability index 38.57 Isoelectric point 6.06 Charge (pH=7) -5.26 2D Binding mode Binding energy (Kcal/mol) -6.23 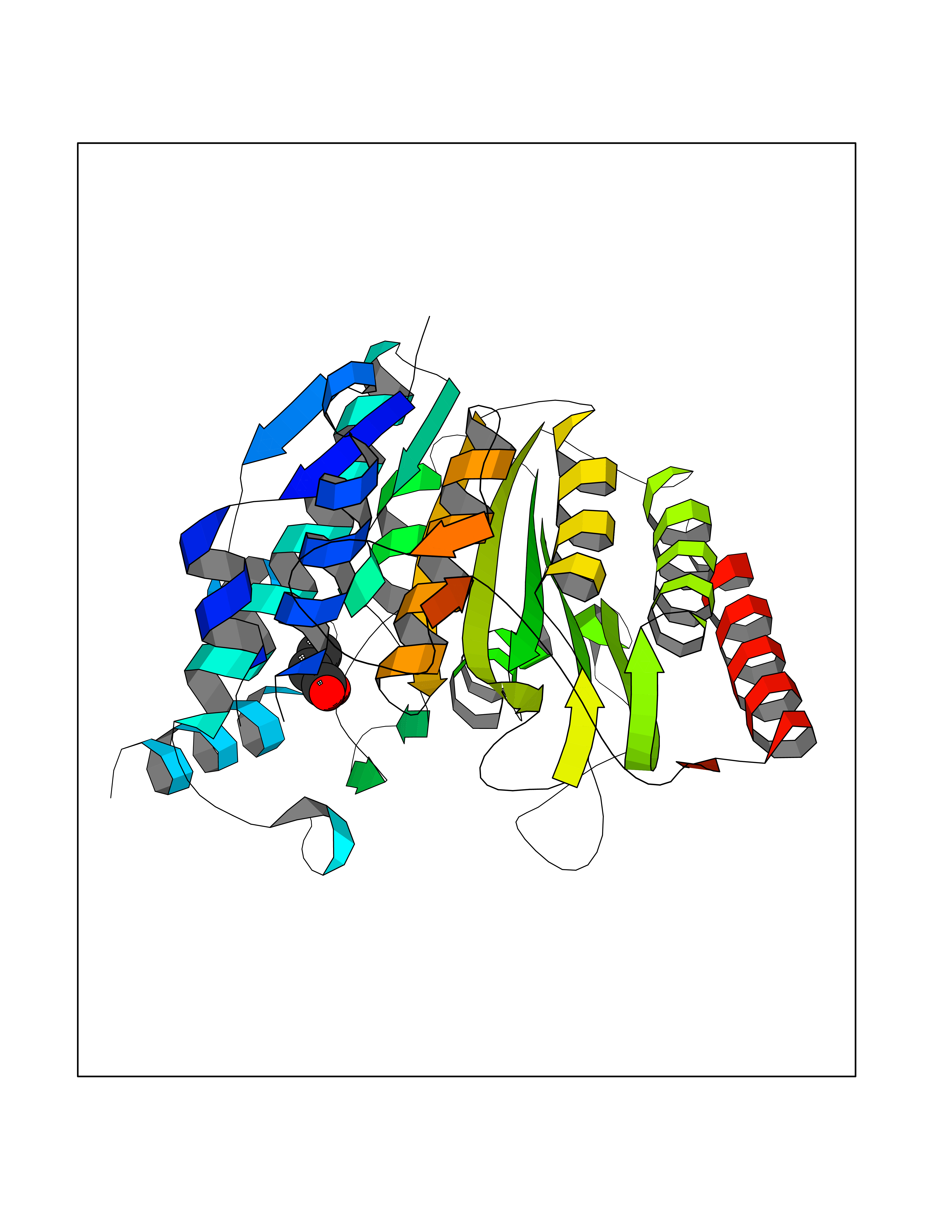 Molscript Map 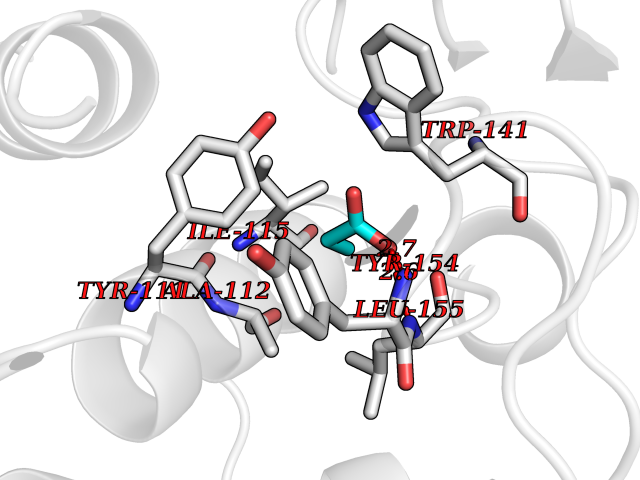 Pymol Map 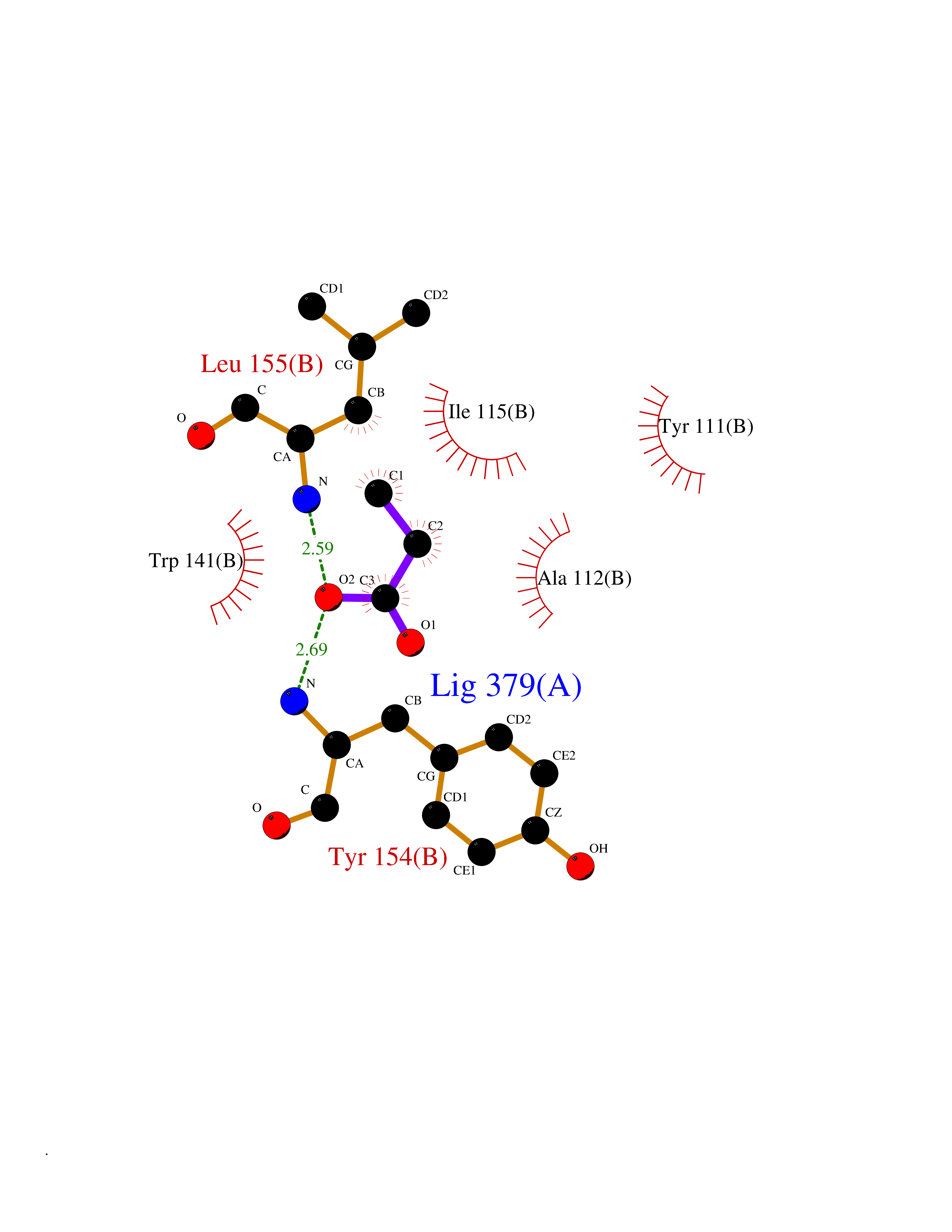 Ligplot Map 3D Binding mode Sequence LVPVYIYSPEYVSMCDSLPKRAEMVHSLIEAYALHKQMRIVKPKVASMEEMATFHTDAYLQHLQKVSQEYGLGYDCPATEGIFDYAAAIGGATITAAQCLIDGMCKVAINWSGGWHHAKKDEASGFCYLNDAVLGILRLRRKFERILYVDLDLHHGDGVEDAFSFTSKVMTVSLHKFSPGFFPGTGDVSDVGLGKGRYYSVNVPIQDGIQDEKYYQICESVLKEVYQAFNPKAVVLQLGADTIAGDPMCSFNMTPVGIGKCLKYILQWQLATLILGGGGYNLANTARCWTYLTGVILGKTLSSEIPDHEFFTAYGPDYVLEITPSCRPDRNEPHRIQQILNYIKGNLKHVV Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Protein kinase G2 (PRKG2) | 5BV6 | 4.57 | |
Target general information Gen name PRKG2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-dependent protein kinase II; cGMP-dependent protein kinase 2; cGKII; cGK2; cGK 2; PRKGR2 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, cGMP subfamily Biochemical class NA Function Crucial regulator of intestinal secretion and bone growth (By similarity). Phosphorylates and activates CFTR on the plasma membrane. Plays a key role in intestinal secretion by regulating cGMP-dependent translocation of CFTR in jejunum (By similarity). Acts downstream of NMDAR to activate the plasma membrane accumulation of GRIA1/GLUR1 in synapse and increase synaptic plasticity. Phosphorylates GRIA1/GLUR1 at Ser-863 (By similarity). Acts as regulator of gene expression and activator of the extracellular signal-regulated kinases MAPK3/ERK1 and MAPK1/ERK2 in mechanically stimulated osteoblasts. Under fluid shear stress, mediates ERK activation and subsequent induction of FOS, FOSL1/FRA1, FOSL2/FRA2 and FOSB that play a key role in the osteoblast anabolic response to mechanical stimulation (By similarity). Related diseases Spondylometaphyseal dysplasia, Pagnamenta type (SMDP) [MIM:619638]: A form of spondylometaphyseal dysplasia, a group of short stature disorders distinguished by abnormalities in the vertebrae and the metaphyses of the tubular bones. SMDP is an autosomal recessive form characterized by short stature and mild platyspondyly with no disproportion between the limbs. Mild metaphyseal changes are present. {ECO:0000269|PubMed:34782440}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Acromesomelic dysplasia 4 (AMD4) [MIM:619636]: A form of acromesomelic dysplasia, a skeletal disorder characterized by short stature, very short limbs and hand/foot malformations. The severity of limb abnormalities increases from proximal to distal with profoundly affected hands and feet showing brachydactyly and/or rudimentary fingers (knob-like fingers). AMD4 radiographic hallmarks include mild to moderate platyspondyly, moderate brachydactyly, iliac flaring, and metaphyseal alterations of the long bones that progressively increase with age. AMD4 inheritance is autosomal recessive. {ECO:0000269|PubMed:33106379, ECO:0000269|PubMed:34782440}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with NA EC number EC 2.7.11.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; cGMP; cGMP-binding; Coiled coil; Disease variant; Dwarfism; Kinase; Lipoprotein; Membrane; Myristate; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 17227.1 Length 150 Aromaticity 0.09 Instability index 43.7 Isoelectric point 4.88 Charge (pH=7) -8.41 2D Binding mode Binding energy (Kcal/mol) -6.24 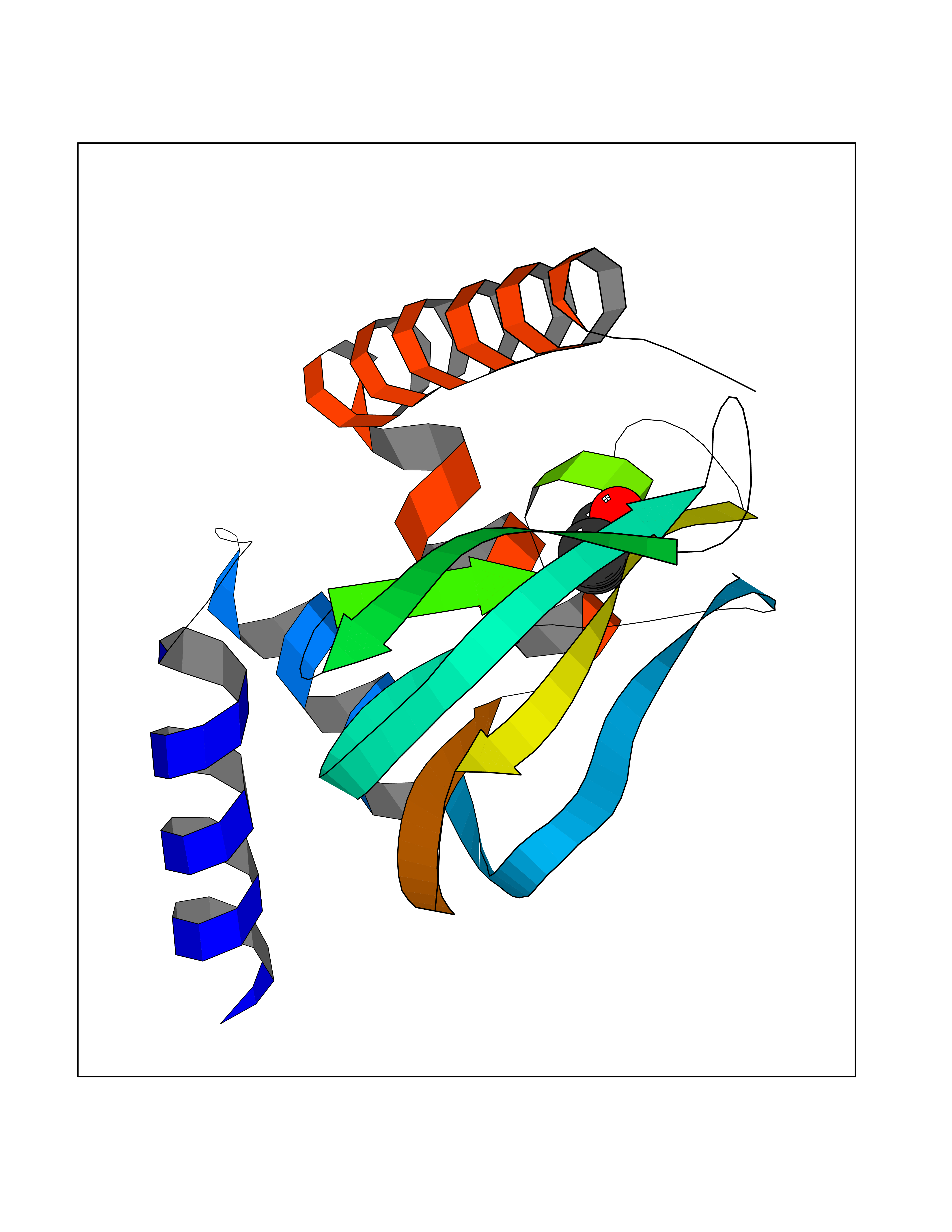 Molscript Map 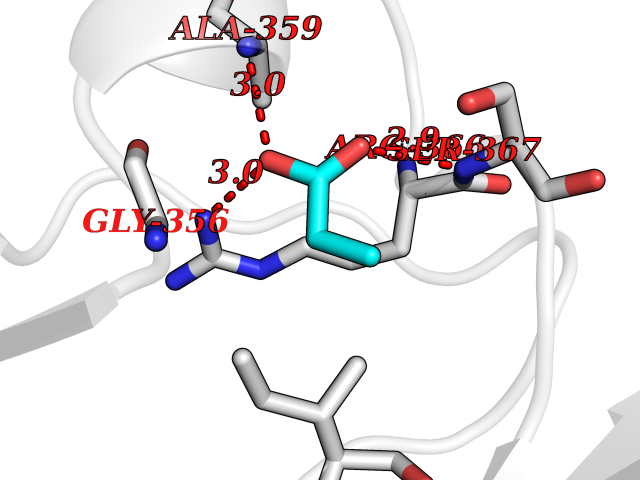 Pymol Map 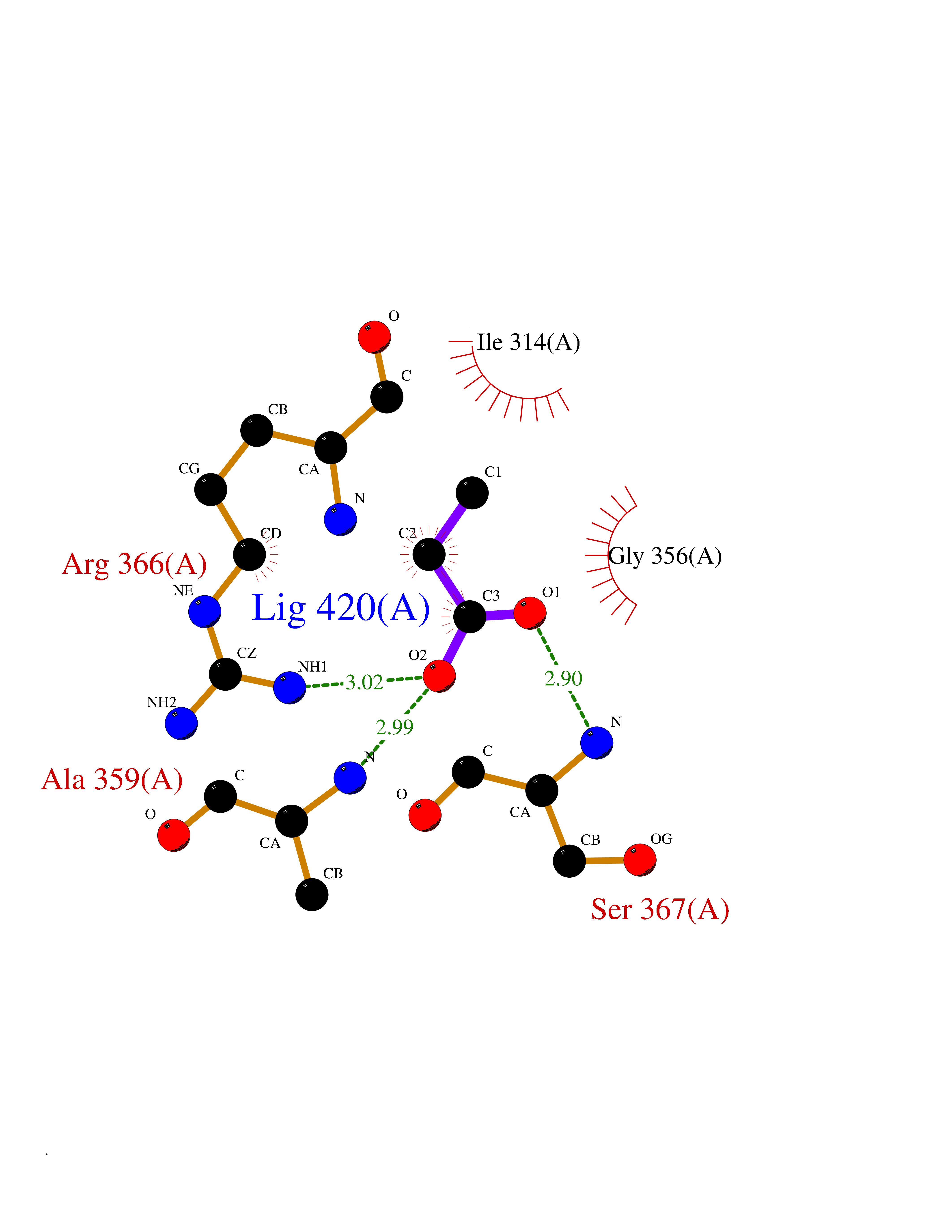 Ligplot Map 3D Binding mode Sequence TAQARDEQYRNFLRSVSLLKNLPEDKLTKIIDCLEVEYYDKGDYIIREGEEGSTFFILAKGKVKVTQSTEGHDQPQLIKTLQKGEYFGEKALISDDVRSANIIAEENDVACLVIDRETFNQTVGTFEELQKYLEGYVANLNRDDEKRHAK Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Plasmodium Adenylosuccinate synthetase (Malaria Adss) | 1P9B | 4.57 | |
Target general information Gen name Malaria Adss Organism Plasmodium falciparum Uniprot ID TTD ID Synonyms IMP--aspartate ligase; Adenylosuccinate synthase; AdSS; AMPSase Protein family Adenylosuccinate synthetase family Biochemical class Carbon-nitrogen ligase Function Plays an important role in the salvage pathway for purine nucleotide biosynthesis. Catalyzes the first committed step in the biosynthesis of AMP from IMP. Related diseases Hypertension and brachydactyly syndrome (HTNB) [MIM:112410]: A syndrome characterized by brachydactyly type E, severe salt-independent but age-dependent hypertension, an increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, and altered baroreflex blood pressure regulation. It results in death from stroke before age 50 years when untreated. Brachydactyly type E is characterized by shortening of the fingers mainly in the metacarpals and metatarsals. {ECO:0000269|PubMed:25961942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03510; DB04315; DB02109 Interacts with NA EC number EC 6.3.4.4 Uniprot keywords 3D-structure; Cytoplasm; GTP-binding; Ligase; Magnesium; Metal-binding; Nucleotide-binding; Purine biosynthesis Protein physicochemical properties Chain ID A Molecular weight (Da) 47877.9 Length 424 Aromaticity 0.09 Instability index 31.72 Isoelectric point 7.63 Charge (pH=7) 1.58 2D Binding mode Binding energy (Kcal/mol) -6.23 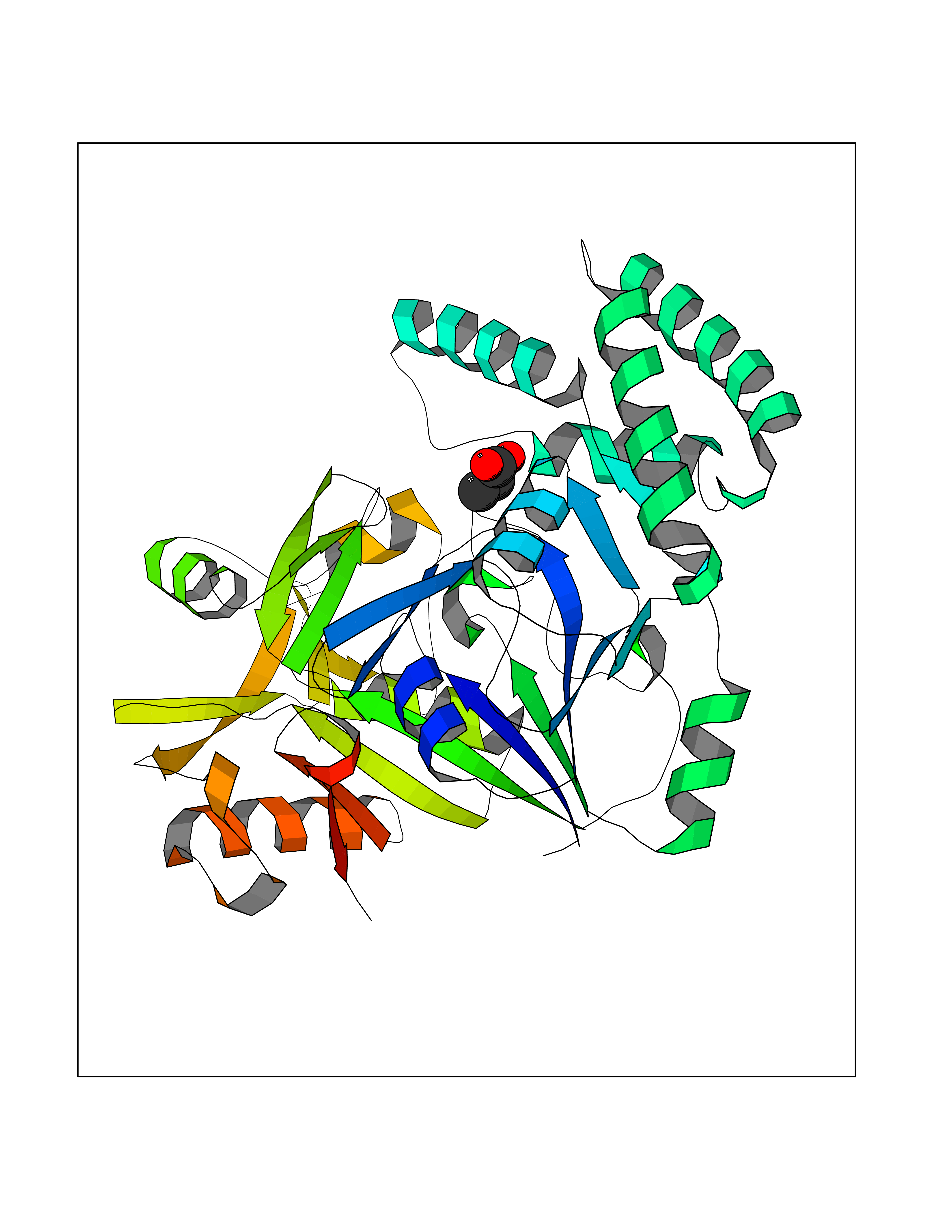 Molscript Map 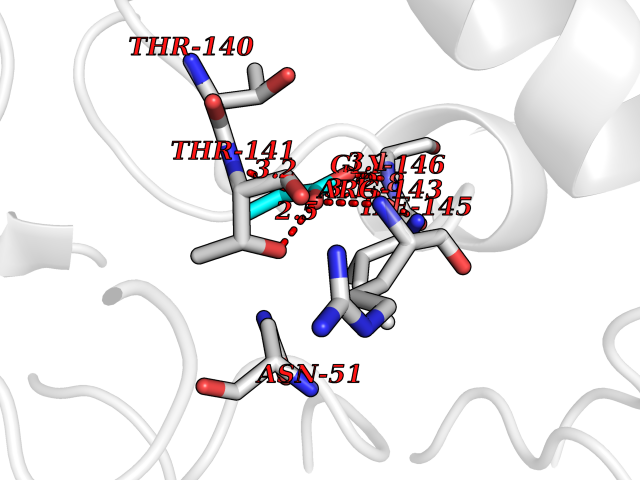 Pymol Map 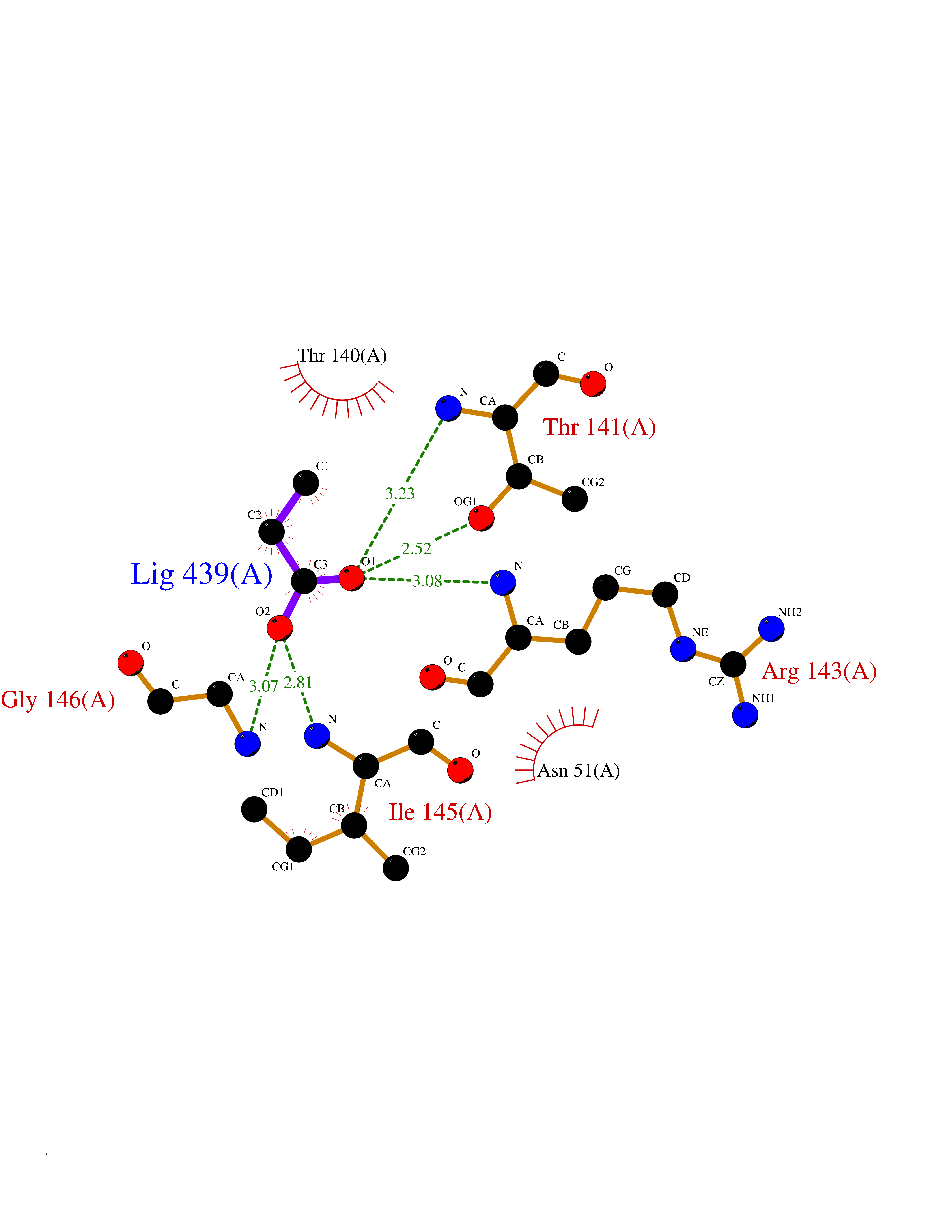 Ligplot Map 3D Binding mode Sequence GNVVAILGAQWGDEGKGKIIDMLSEYSDITCRFNGGANAGHTISVNDKKYALHLLPCGVLYDNNISVLGNGMVIHVKSLMEEIESVGGKLLDRLYLSNKAHILFDIHQIIDSIQETKKLKEGKQIGTTKRGIGPCYSTKASRIGIRLGTLKNFENFKNMYSKLIDHLMDLYNITEYDKEKELNLFYNYHIKLRDRIVDVISFMNTNLENNKKVLIEGANAAMLDIDFGTYPYVTSSCTTVGGVFSGLGIHHKKLNLVVGVVKSYLTRVGCGPFLTELNNDVGQYLREKGHEYGTTTKRPRRCGWLDIPMLLYVKCINSIDMINLTKLDVLSGLEEILLCVNFKNKKTGELLEKGCYPVEEEISEEYEPVYEKFSGWKEDISTCNEFDELPENAKKYILAIEKYLKTPIVWIGVGPNRKNMIVKK Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Phosphodiesterase 3A (PDE3A) | 7EG0 | 4.57 | |
Target general information Gen name PDE3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-inhibited 3',5'-cyclic phosphodiesterase A; Phosphodiesterase 3A; Cyclic GMP-inhibited phosphodiesterase A; Cyclic GMP inhibited phosphodiesterase A; CGI-PDE A Protein family Cyclic nucleotide phosphodiesterase family, PDE3 subfamily Biochemical class Phosphoric diester hydrolase Function Cyclic nucleotide phosphodiesterase with a dual-specificity for the second messengers cAMP and cGMP, which are key regulators of many important physiological processes. Related diseases Hypertension and brachydactyly syndrome (HTNB) [MIM:112410]: A syndrome characterized by brachydactyly type E, severe salt-independent but age-dependent hypertension, an increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, and altered baroreflex blood pressure regulation. It results in death from stroke before age 50 years when untreated. Brachydactyly type E is characterized by shortening of the fingers mainly in the metacarpals and metatarsals. {ECO:0000269|PubMed:25961942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01223; DB01427; DB00261; DB00201; DB01166; DB04880; DB05266; DB00922; DB00235; DB01303; DB00277; DB08811; DB09283 Interacts with Q9Y6D6; Q9Y6D5 EC number EC 3.1.4.17 Uniprot keywords 3D-structure; cAMP; cGMP; Cytoplasm; Direct protein sequencing; Disease variant; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 108171 Length 939 Aromaticity 0.11 Instability index 39.75 Isoelectric point 6.53 Charge (pH=7) -4.15 2D Binding mode Binding energy (Kcal/mol) -6.24  Molscript Map 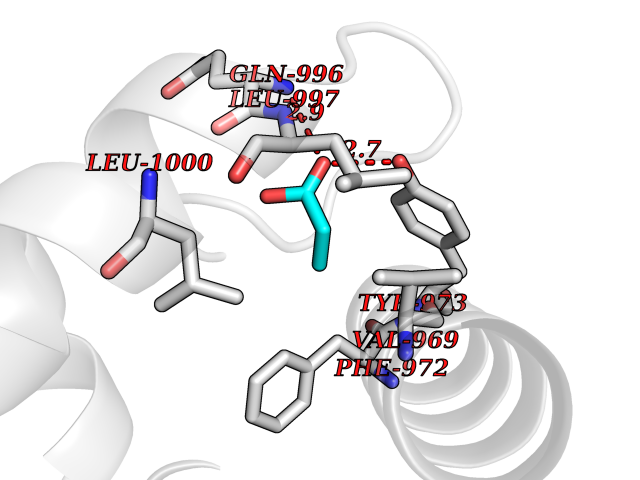 Pymol Map 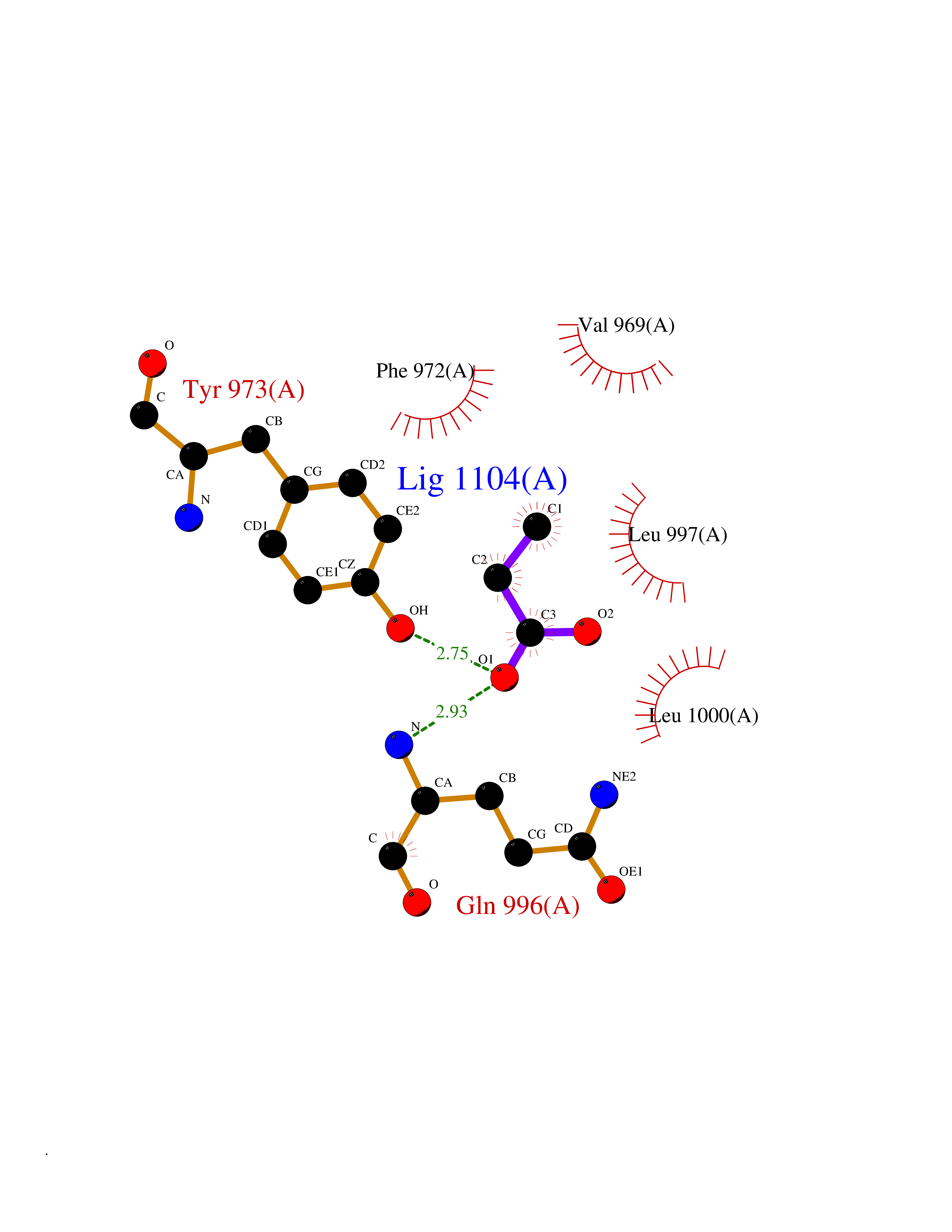 Ligplot Map 3D Binding mode Sequence KPILAPEPLVMDNLDSIMEQLNTWNFPIFDLVENIGRKCGRILSQVSYRLFEDMGLFEAFKIPIREFMNYFHALEIGYRDIPYHNRIHATDVLHAVWYLTTQPIPGLSTVGYVFSKTYNVTDDKYGCLSGNIPALELMALYVAAAMHDYDHPGRTNAFLVATSAPQAVLYNDRSVLENHHAAAAWNLFMSRPEYNFLINLDHVEFKHFRFLVIEAILATDLKKHFDFVAKFNGKVNDDVGIDWTNENDRLLVCQMCIKLADINGPAKCKELHLQWTDGIVNEFYEQGDEEASLGLPISPFMDRSAPQLANLQESFISHIVGPLCNSYDSAGLMPGKWVEKIYCQITQHLLQNHKMWKKVIEEEQRLAGIENQNISVDLETNYAELVLDVGRVTLGENSRKKMKDCKLRKKQNESVSRAMCALLNSGGGVIKAEIENEDYSYTKDGIGLDLENSFSNILLFVPEYLDFMQNGNYFLIFVKSWSLNTSGLRITTLSSNLYKRDITSAKVMNATAALEFLKDMKKTRGRLYLRPELLAKRPCVDIQEENNMKALAGVFFDRTELDRKEKLTFTESTHVEIKNFSTERLLQRIKEILPQYVSAFANTDGGYLFIGLNEDKEIIGFKAEMSDLDDLEREIEKSIRKMPVHHFCMEKKKINYSCKFLGVYDKGSLCGYVCALRVERFCCAVFAKEPDSWHVKDNRVMQLTRKEWIQFMVEAEPKFSSAYEEVISQINTSLPAPHSWPLLEWQRQRHHCPGLSGRITYTPENLCRKLFLQHEGLKQLICEEMSSVRKGSLIFSRSWSVDLGLQENHKVLCDALLISQDSPPVLYTFHMVQDEEFKGYSTQTALTLKQKLAKIGGYTKKVCVMTKIFYLSPEGMTSCQYDLRSQVIYPESYYFTRRKYLLKALFKALKRLKSLRDQFSFAENLYQIIGIDCFQKNDK Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | NAPE-hydrolyzing phospholipase D (NAPE-PLD) | 4QN9 | 4.57 | |
Target general information Gen name NAPEPLD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NAPE-PLD; N-acyl-phosphatidylethanolamine-hydrolyzing phospholipase D; N-acyl phosphatidylethanolamine phospholipase D; C7orf18 Protein family NAPE-PLD family Biochemical class NA Function Hydrolyzes N-acyl-phosphatidylethanolamines (NAPEs) to produce N-acylethanolamines (NAEs) and phosphatidic acid. Responsible for the generation of these bioactive fatty acid ethanolamides (FAEs), including anandamide (N-arachidonoylethanolamine), the ligand of cannabinoid and vanilloid receptors. As a regulator of lipid metabolism in the adipose tissue, mediates the crosstalk between adipocytes, gut microbiota and immune cells to control body temperature and weight. In particular, regulates energy homeostasis by promoting cold-induced brown or beige adipocyte differentiation program to generate heat from fatty acids and glucose (By similarity). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) DB14009 Interacts with Q6IQ20 EC number EC 3.1.4.54 Uniprot keywords 3D-structure; Acetylation; Endosome; Golgi apparatus; Hydrolase; Lipid degradation; Lipid metabolism; Membrane; Metal-binding; Nucleus; Phospholipid degradation; Phospholipid metabolism; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 74256.5 Length 643 Aromaticity 0.13 Instability index 48.34 Isoelectric point 5.65 Charge (pH=7) -17.61 2D Binding mode Binding energy (Kcal/mol) -6.24 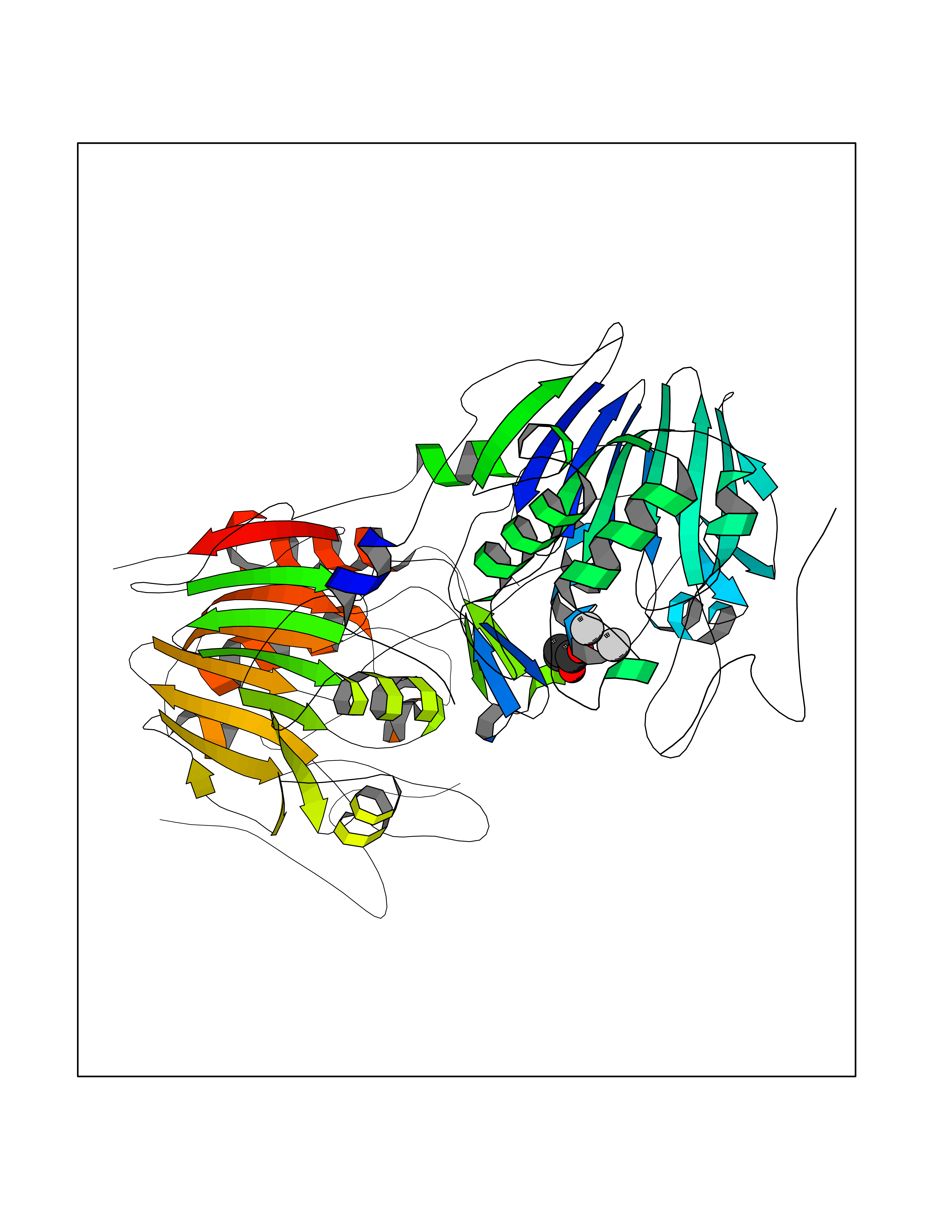 Molscript Map 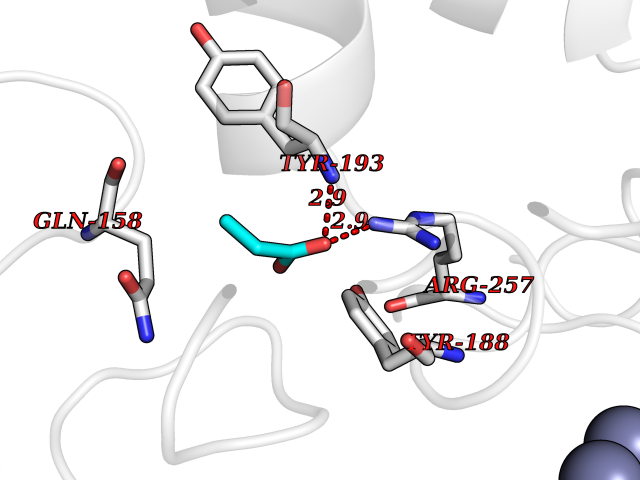 Pymol Map 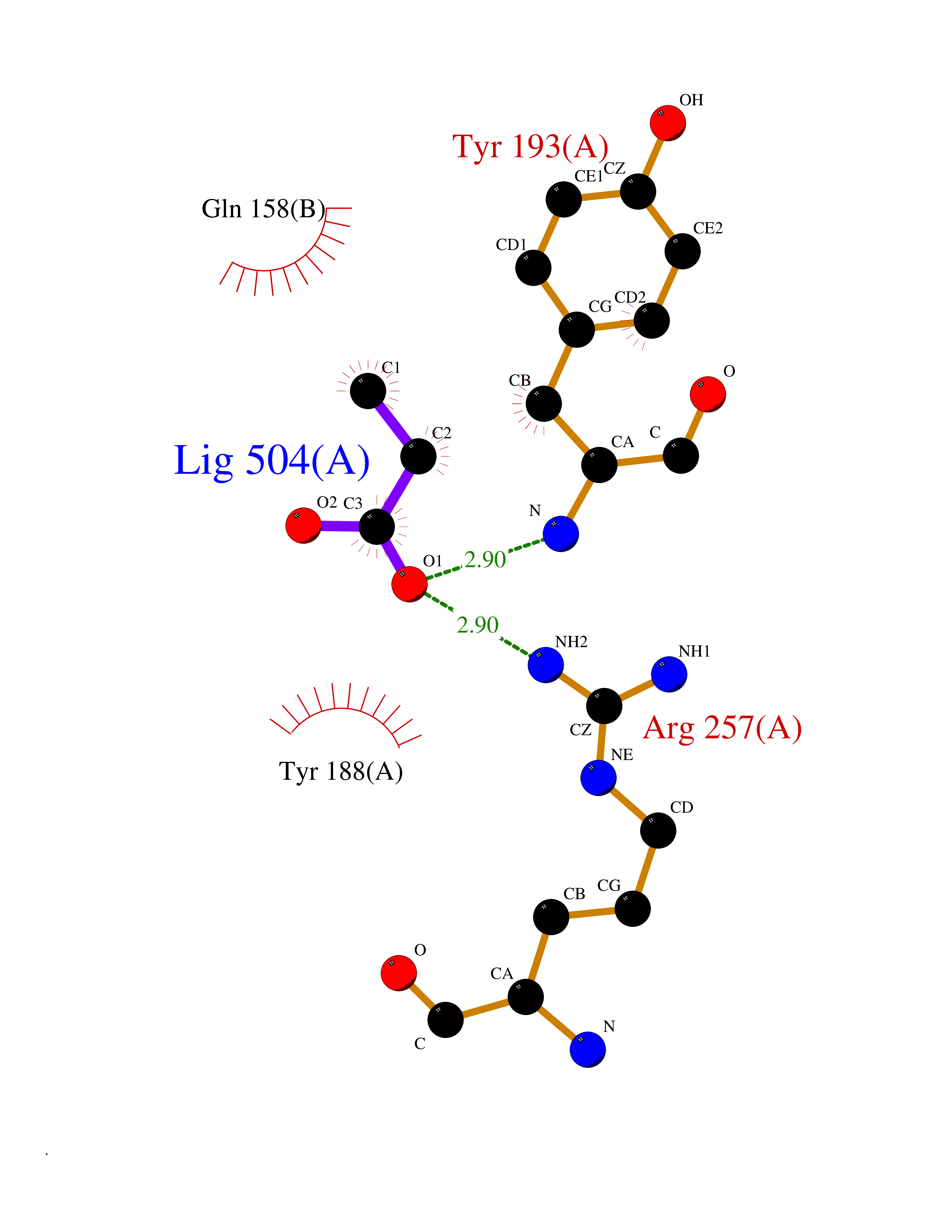 Ligplot Map 3D Binding mode Sequence SKKGKDGRFVNPWPTWKNPSIPNSSVPSSKEELDKELPVLKPYFITNPEEAGVREAGLRVTWLGHATVMVEMDELIFLTDPIFSSRASPSQYMGPKRFRRSPCTISELPPIDAVLISHNHYDHLDYNSVIALNERFGNELRWFVPLGLLDWMQKCGCENVIELDWWEENCVPGHDKVTFVFTPSQHWCKRTLMDDNKVLWGSWSVLGPWNRFFFAGDTGYCPAFEEIGKRFGPFDLAAIPIGAYEPRWFMKYQHVDPEEAVRIHTDVQTKKSMAIHWGTFALANEHYLEPPVKLNEALERYGLNAEDFFVLKHGESRYLNNSKKGKDGRFVNPWPTWKNPSIPNSSVPSSKEELDKELPVLKPYFITNPEEAGVREAGLRVTWLGHATVMVEMDELIFLTDPIFSSRASPSQYMGPKRFRRSPCTISELPPIDAVLISHNHYDHLDYNSVIALNERFGNELRWFVPLGLLDWMQKCGCENVIELDWWEENCVPGHDKVTFVFTPSQHWCKRTLMDDNKVLWGSWSVLGPWNRFFFAGDTGYCPAFEEIGKRFGPFDLAAIPIGAYEPRWFMKYQHVDPEEAVRIHTDVQTKKSMAIHWGTFALANEHYLEPPVKLNEALERYGLNAEDFFVLKHGESRYLNND Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Phosphatidylethanolamine-binding protein 1 (PEBP1) | 2QYQ | 4.57 | |
Target general information Gen name PEBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Raf kinase inhibitor protein; RKIP; Prostatic-binding protein; PEBP-1; PEBP; PBP; Neuropolypeptide h3; Hippocampal cholinergic neurostimulating peptide; HCNPpp; HCNP Protein family Phosphatidylethanolamine-binding protein family Biochemical class Phosphatidylethanolamine-binding protein family Function Binds ATP, opioids and phosphatidylethanolamine. Has lower affinity for phosphatidylinositol and phosphatidylcholine. Serine protease inhibitor which inhibits thrombin, neuropsin and chymotrypsin but not trypsin, tissue type plasminogen activator and elastase. Inhibits the kinase activity of RAF1 by inhibiting its activation and by dissociating the RAF1/MEK complex and acting as a competitive inhibitor of MEK phosphorylation. Related diseases Retinitis pigmentosa 49 (RP49) [MIM:613756]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:15570217, ECO:0000269|PubMed:7479749}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09130; DB09568 Interacts with P16050; Q9NRD5; P04049; Q15208; Q9NS68; Q9JLL3 EC number NA Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Direct protein sequencing; Disulfide bond; Lipid-binding; Nucleotide-binding; Phosphoprotein; Protease inhibitor; Proteomics identification; Reference proteome; Serine protease inhibitor Protein physicochemical properties Chain ID A Molecular weight (Da) 20928.3 Length 186 Aromaticity 0.1 Instability index 24.05 Isoelectric point 6.59 Charge (pH=7) -0.98 2D Binding mode Binding energy (Kcal/mol) -6.24 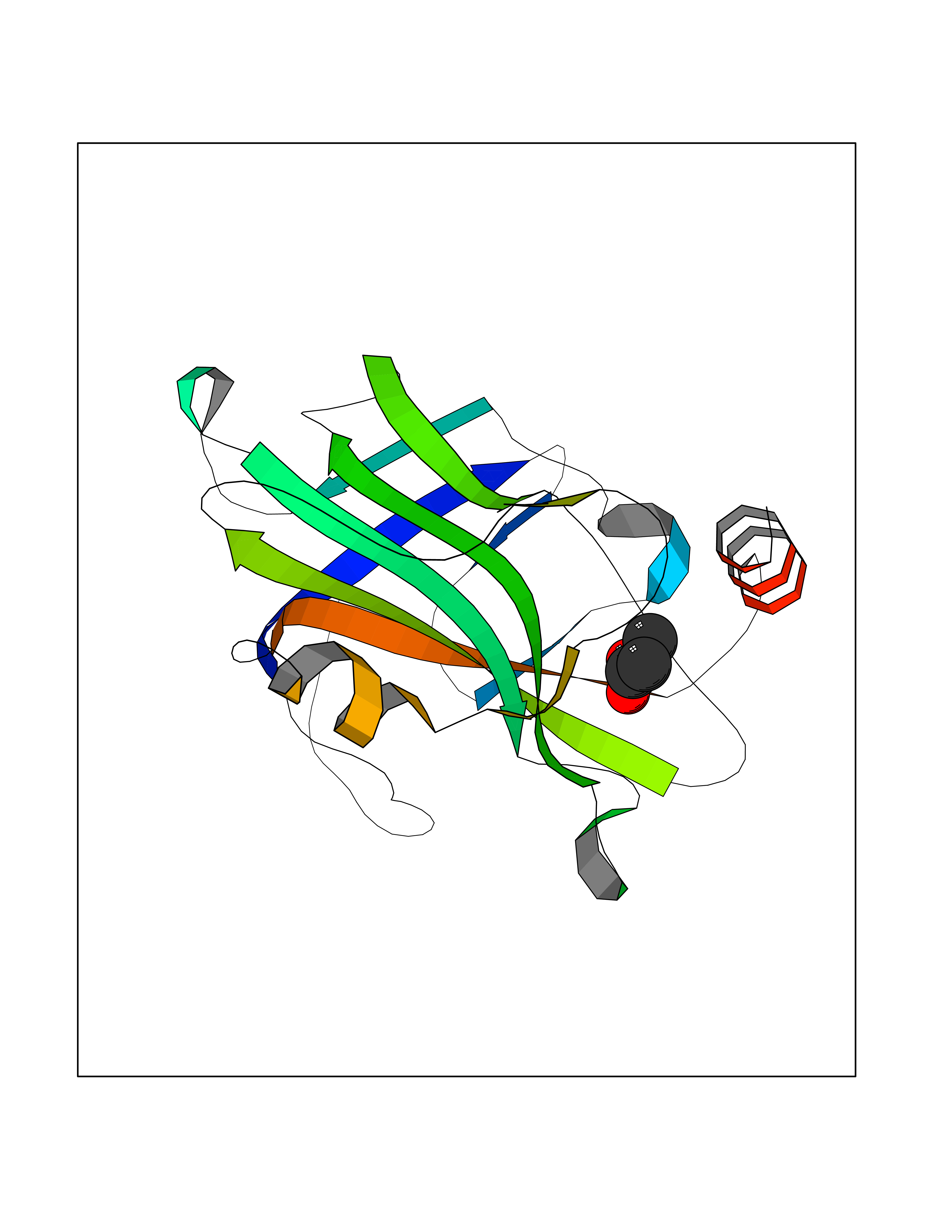 Molscript Map 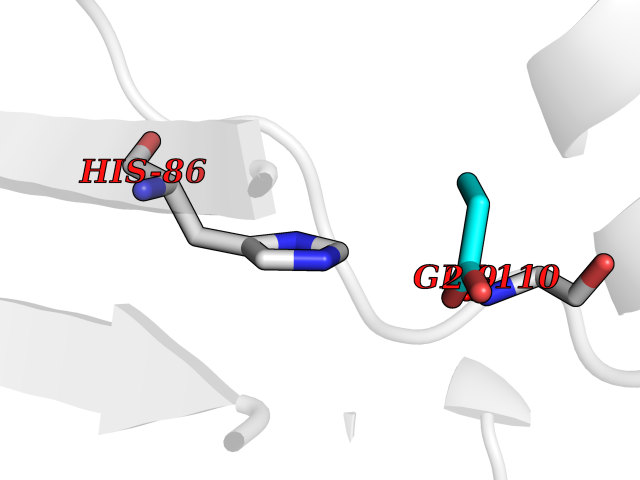 Pymol Map 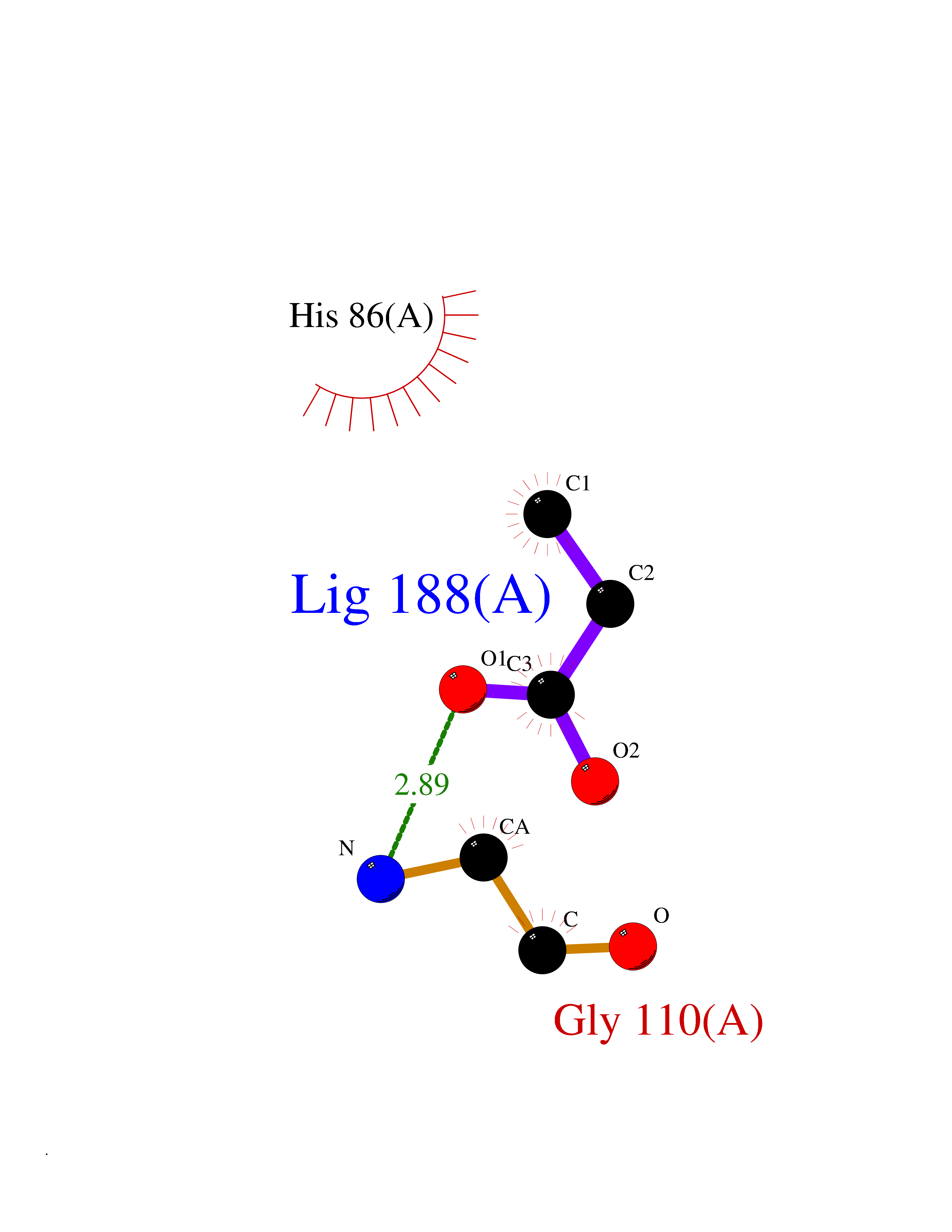 Ligplot Map 3D Binding mode Sequence MPVDLSKWSGPLSLQEVDEQPQHPLHVTYAGAAVDELGKVLTPTQVKNRPTSISWDGLDSGKLYTLVLTDPDAPSRKDPKYREWHHFLVVNMKGNDISSGTVLSDYVGSGPPKGTGLHRYVWLVYEQDRPLKCDEPILSNRSGDHRGKFKVASFRKKYELRAPVAGTCYQAEWDDYVPKLYEQLSG Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Neprilysin | 1R1H | 4.56 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms EPN Protein family Peptidase M13 family Biochemical class Hydrolase Function Endopeptidase activity.Exopeptidase activity.Metalloendopeptidase activity.Metallopeptidase activity.Peptide binding.Zinc ion binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -6.22  Molscript Map 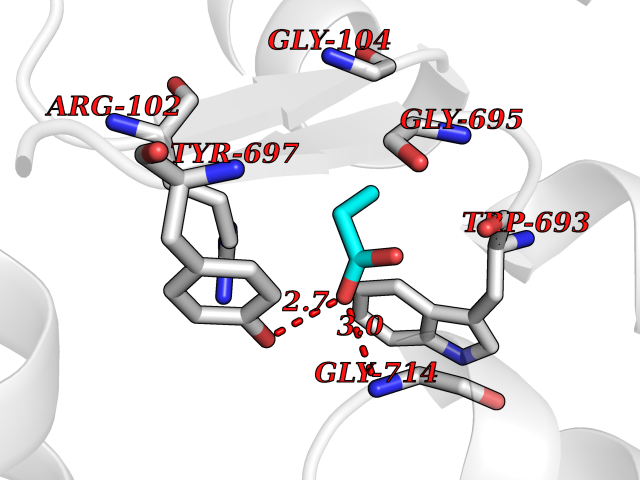 Pymol Map 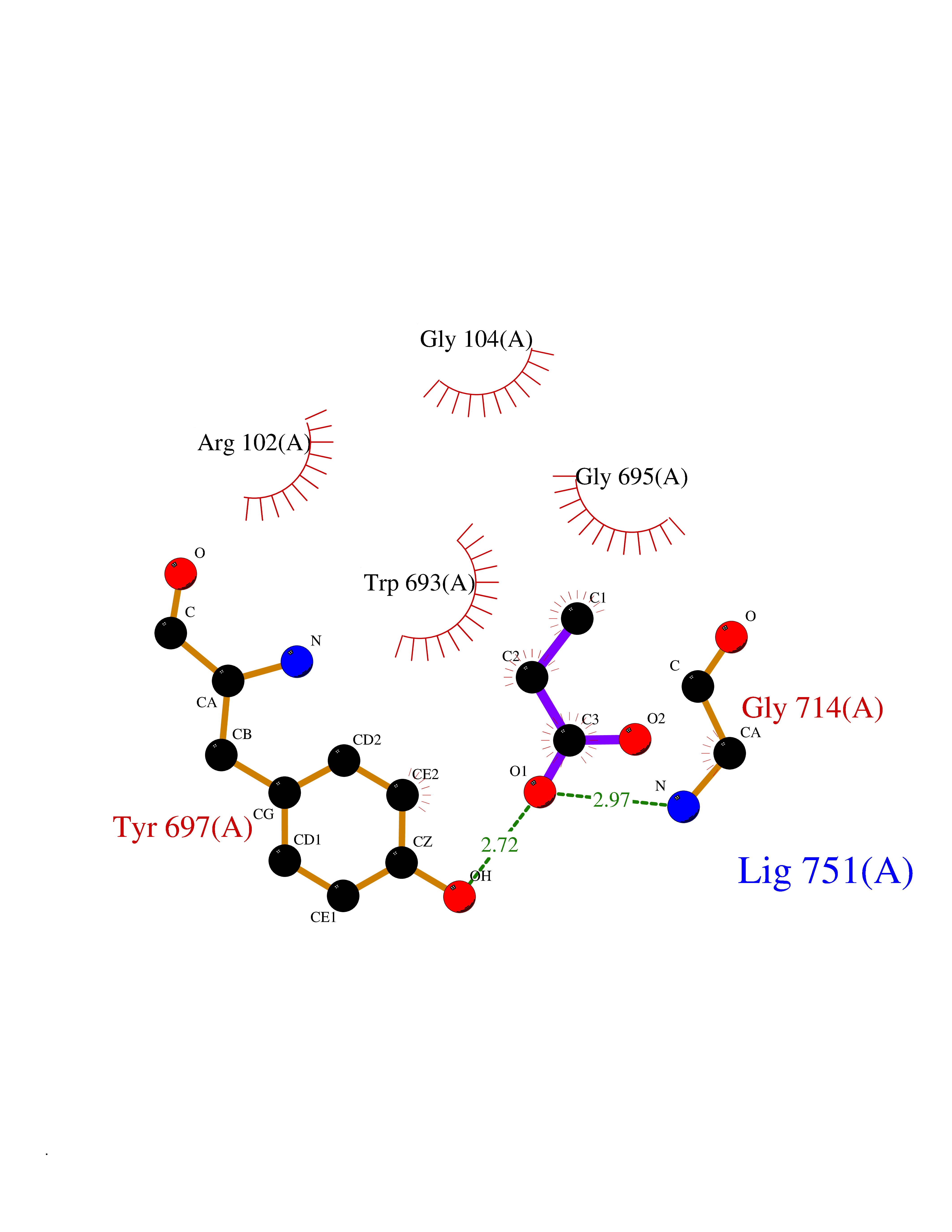 Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Xylose isomerase | 1XIM | 4.56 | |
Target general information Gen name xylA Organism Actinoplanes missouriensis (strain ATCC 14538 / DSM 43046 / CBS 188.64 / JCM 3121 / NBRC 102363 / NCIMB 12654 / NRRL B-3342 / UNCC 431) Uniprot ID TTD ID NA Synonyms AMIS_10350;XI Protein family Xylose isomerase family Biochemical class Isomerase(intramolecular oxidoreductase) Function Metal ion binding.Xylose isomerase activity. Related diseases Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Renal tubular dysgenesis (RTD) [MIM:267430]: Autosomal recessive severe disorder of renal tubular development characterized by persistent fetal anuria and perinatal death, probably due to pulmonary hypoplasia from early-onset oligohydramnios (the Potter phenotype). {ECO:0000269|PubMed:16116425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Microvascular complications of diabetes 3 (MVCD3) [MIM:612624]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. {ECO:0000269|PubMed:10099885}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Intracerebral hemorrhage (ICH) [MIM:614519]: A pathological condition characterized by bleeding into one or both cerebral hemispheres including the basal ganglia and the cerebral cortex. It is often associated with hypertension and craniocerebral trauma. Intracerebral bleeding is a common cause of stroke. {ECO:0000269|PubMed:15277638}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11195 Interacts with NA EC number 5.3.1.5 Uniprot keywords 3D-structure; Carbohydrate metabolism; Cytoplasm; Isomerase; Magnesium; Metal-binding; Reference proteome; Xylose metabolism Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 35820.8 Length 322 Aromaticity 0.1 Instability index 32.29 Isoelectric point 5.32 Charge (pH=7) -11.33 2D Binding mode Binding energy (Kcal/mol) -6.22 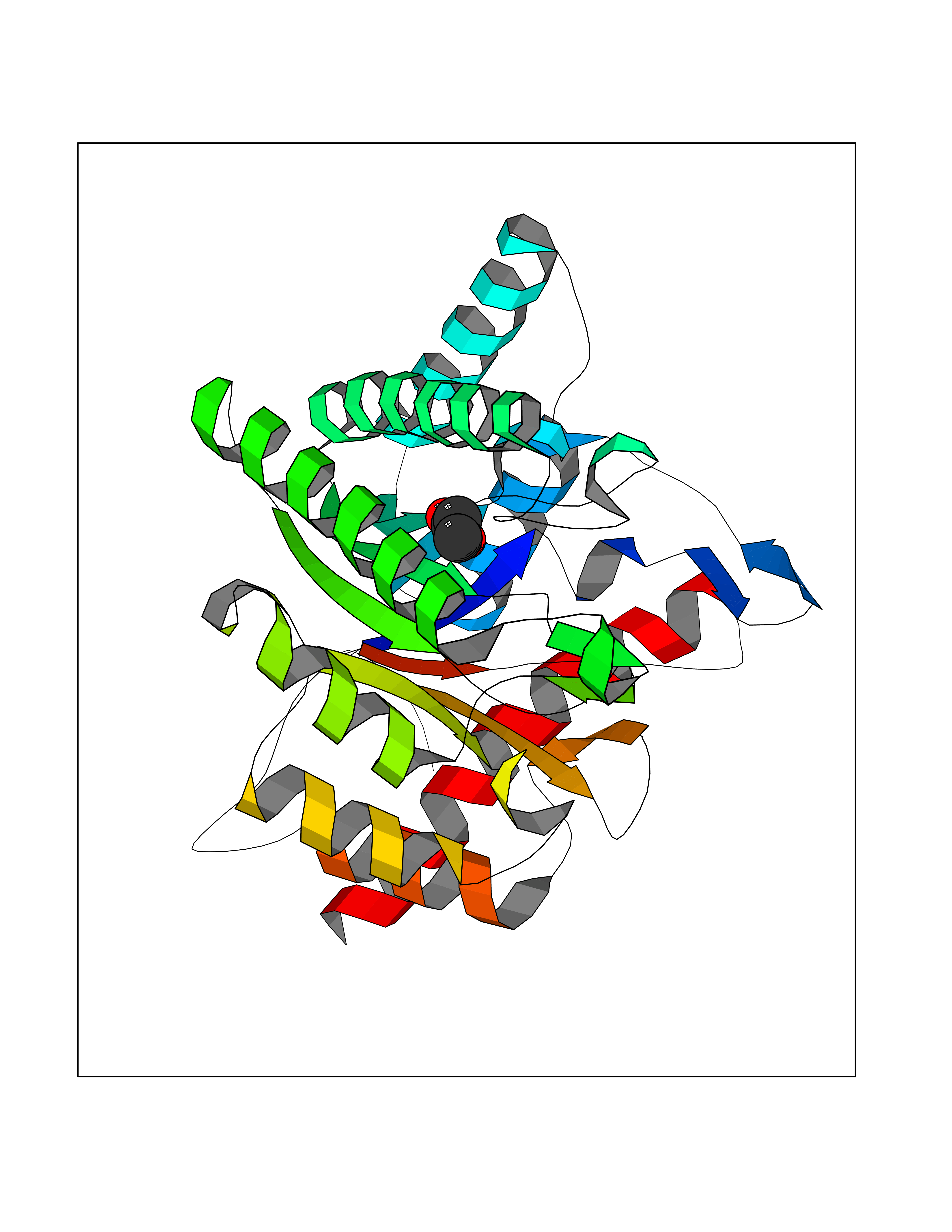 Molscript Map 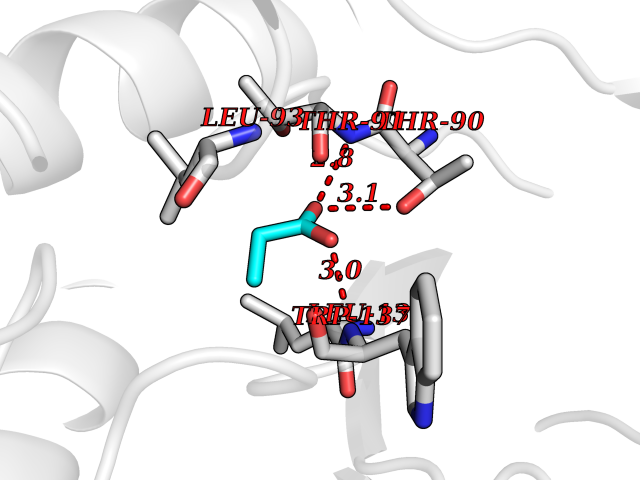 Pymol Map 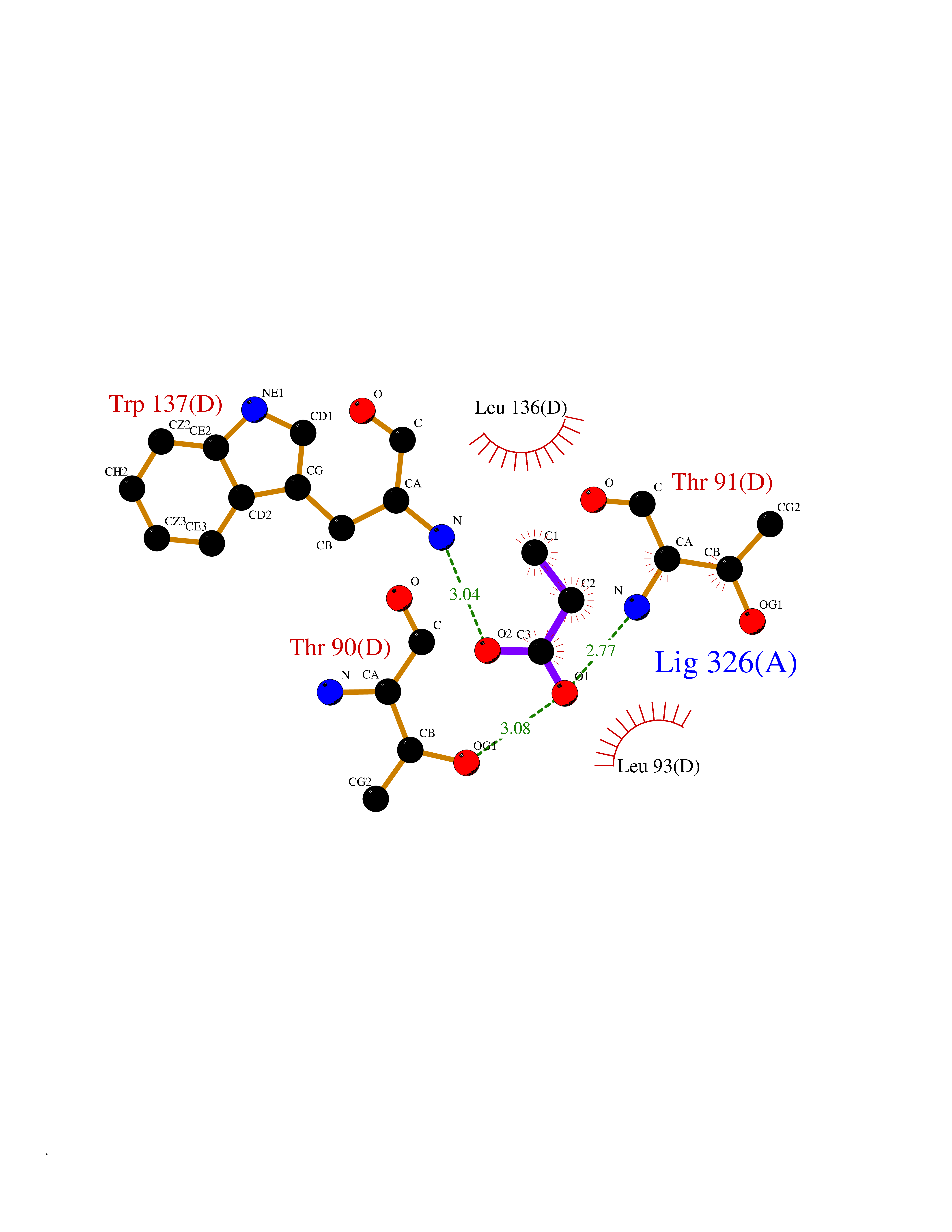 Ligplot Map 3D Binding mode Sequence VQATREDKFSFGLWTVGWQARDAFGDATRTALDPVEAVHKLAEIGAYGITFHDDDLVPFGSDAQTRDGIIAGFKKALDETGLIVPMVTTNLFTHPVFKDGGFTSNDRSVRRYAIRKVLRQMDLGAELGAKTLVLWGGREGAEYDSAKDVSAALDRYREALNLLAQYSEDRGYGLRFAIEPKPNEPRGDILLPTAGHAIAFVQELERPELFGINPETGHEQMSNLNFTQGIAQALWHKKLFHIDLNGQHGPKFDQDLVFGHGDLLNAFSLVDLLENGPDGAPAYDGPRHFDYKPSRTEDYDGVWESAKANIRMYLLLKERAKA Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | NH(3)-dependent NAD(+) synthetase | 1KQP | 4.56 | |
Target general information Gen name nadE Organism Bacillus subtilis (strain 168) Uniprot ID TTD ID NA Synonyms outB;BSU03130 Protein family NAD synthetase family Biochemical class Ligase Function ATP binding.Metal ion binding.NAD+ synthase (glutamine-hydrolyzing) activity.NAD+ synthase activity. Related diseases Leukodystrophy, hypomyelinating, 15 (HLD15) [MIM:617951]: An autosomal recessive disorder characterized by hypomyelinating leukodystrophy with thinning of the corpus callosum. Clinical features include motor and cognitive impairment appearing in the first or second decade of life, dystonia, ataxia, spasticity, and dysphagia. Most patients develop severe optic atrophy, and some have hearing loss. {ECO:0000269|PubMed:29576217}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02596; DB04099; DB00798 Interacts with NA EC number 6.3.1.5 Uniprot keywords 3D-structure; ATP-binding; Direct protein sequencing; Ligase; Magnesium; Metal-binding; NAD; Nucleotide-binding; Phosphoprotein; Reference proteome; Sporulation; Stress response Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60509.3 Length 542 Aromaticity 0.08 Instability index 31.96 Isoelectric point 5.07 Charge (pH=7) -19.73 2D Binding mode Binding energy (Kcal/mol) -6.22  Molscript Map 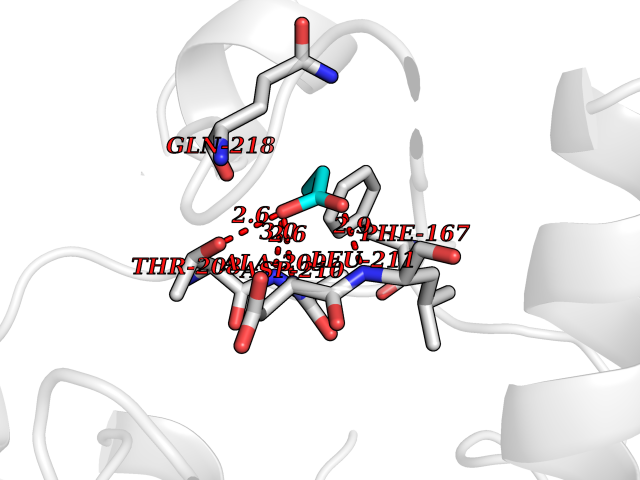 Pymol Map 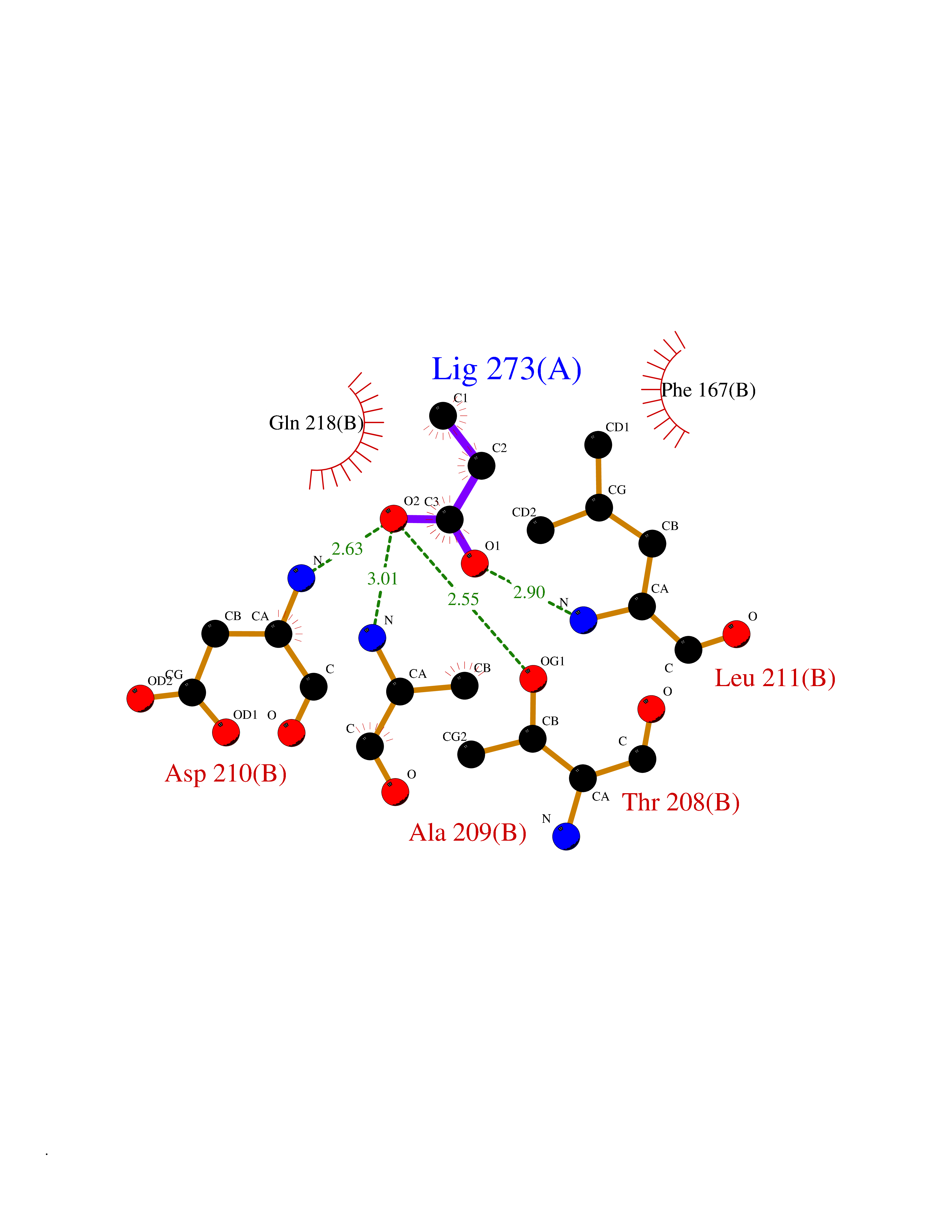 Ligplot Map 3D Binding mode Sequence SMQEKIMRELHVKPSIDPKQEIEDRVNFLKQYVKKTGAKGFVLGISGGQDSTLAGRLAQLAVESIREEGGDAQFIAVRLPHGTQQDEDDAQLALKFIKPDKSWKFDIKSTVSAFSDQYQQETGDQLTDFNKGNVKARTRMIAQYAIGGQEGLLVLGTDHAAEAVTGFFTKYGDGGADLLPLTGLTKRQGRTLLKELGAPERLYLKEPTADLLDEKPQQSDETELGISYDEIDDYLEGKEVSAKVSEALEKRYSMTEHKRQVPASMFDDWWKSMQEKIMRELHVKPSIDPKQEIEDRVNFLKQYVKKTGAKGFVLGISGGQDSTLAGRLAQLAVESIREEGGDAQFIAVRLPHGTQQDEDDAQLALKFIKPDKSWKFDIKSTVSAFSDQYQQETGDQLTDFNKGNVKARTRMIAQYAIGGQEGLLVLGTDHAAEAVTGFFTKYGDGGADLLPLTGLTKRQGRTLLKELGAPERLYLKEPTADLLDEKPQQSDETELGISYDEIDDYLEGKEVSAKVSEALEKRYSMTEHKRQVPASMFDDWWK Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Non-heme chloroperoxidase | 1A8U | 4.56 | |
Target general information Gen name cpo Organism Kitasatospora aureofaciens (Streptomyces aureofaciens) Uniprot ID TTD ID NA Synonyms cpoT Protein family AB hydrolase superfamily, Bacterial non-heme haloperoxidase / perhydrolase family Biochemical class Haloperoxidase Function Chloride peroxidase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB03793 Interacts with NA EC number 1.11.1.- Uniprot keywords 3D-structure; Chloride; Oxidoreductase; Peroxidase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60428.4 Length 554 Aromaticity 0.13 Instability index 31.26 Isoelectric point 4.65 Charge (pH=7) -32.72 2D Binding mode Binding energy (Kcal/mol) -6.21  Molscript Map 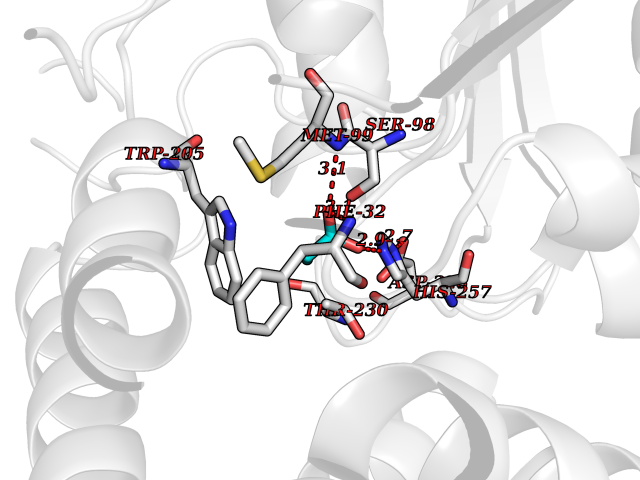 Pymol Map 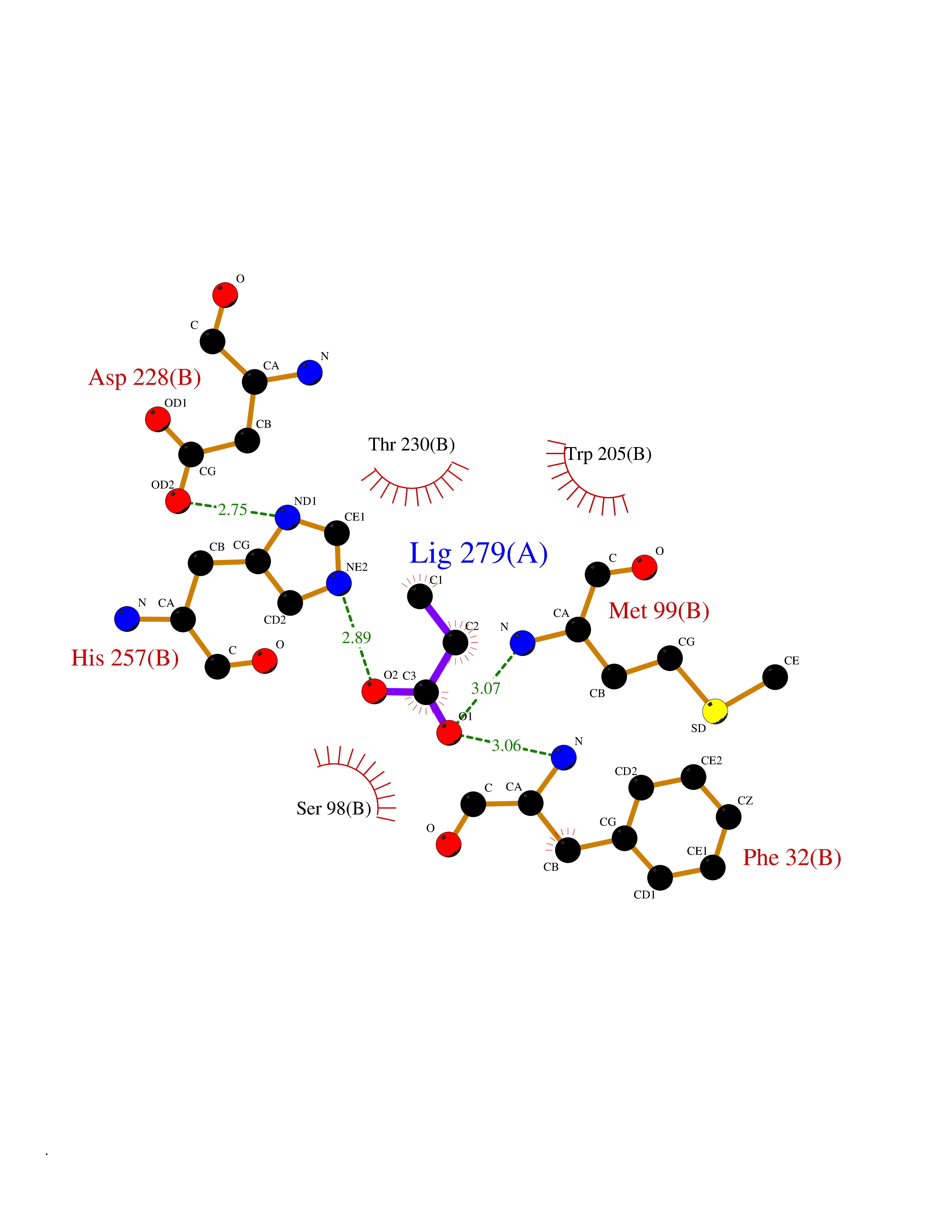 Ligplot Map 3D Binding mode Sequence PFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAKPFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | 2,4-dienoyl-CoA reductase | 1PS9 | 4.56 | |
Target general information Gen name fadH Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3081;ygjL;JW3052 Protein family NADH:flavin oxidoreductase/NADH oxidase family Biochemical class Oxidoreductase Function 2,4-dienoyl-CoA reductase (NADPH) activity.4 iron, 4 sulfur cluster binding.FAD binding.FMN binding.Metal ion binding. Related diseases Smith-Kingsmore syndrome (SKS) [MIM:616638]: An autosomal dominant syndrome characterized by intellectual disability, macrocephaly, seizures, umbilical hernia, and facial dysmorphic features. {ECO:0000269|PubMed:25851998, ECO:0000269|PubMed:26542245, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Focal cortical dysplasia 2 (FCORD2) [MIM:607341]: A form of focal cortical dysplasia, a malformation of cortical development that results in medically refractory epilepsy in the pediatric population and in adults. FCORD2 is a severe form, with onset usually in childhood, characterized by disrupted cortical lamination and specific cytological abnormalities. It is classified in 2 subtypes: type IIA characterized by dysmorphic neurons and lack of balloon cells; type IIB with dysmorphic neurons and balloon cells. {ECO:0000269|PubMed:25799227, ECO:0000269|PubMed:25878179, ECO:0000269|PubMed:26018084, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03698; DB03147; DB03247; DB03461 Interacts with P11349; P19318 EC number 1.3.1.34 Uniprot keywords 3D-structure; 4Fe-4S; Direct protein sequencing; FAD; Flavoprotein; FMN; Iron; Iron-sulfur; Metal-binding; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 40003.1 Length 366 Aromaticity 0.07 Instability index 33.42 Isoelectric point 5.65 Charge (pH=7) -9.48 2D Binding mode Binding energy (Kcal/mol) -6.22  Molscript Map 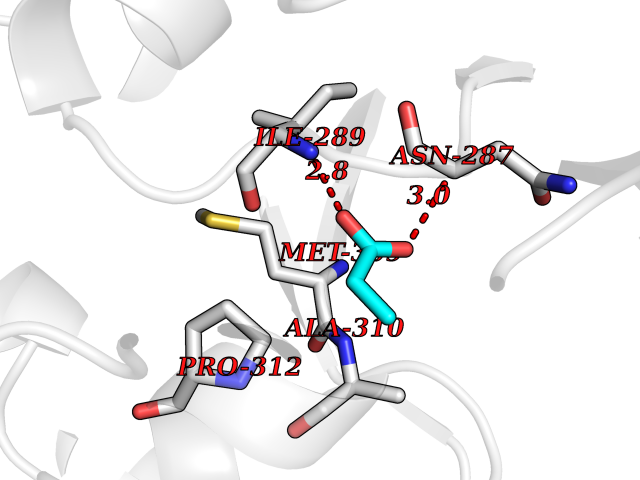 Pymol Map 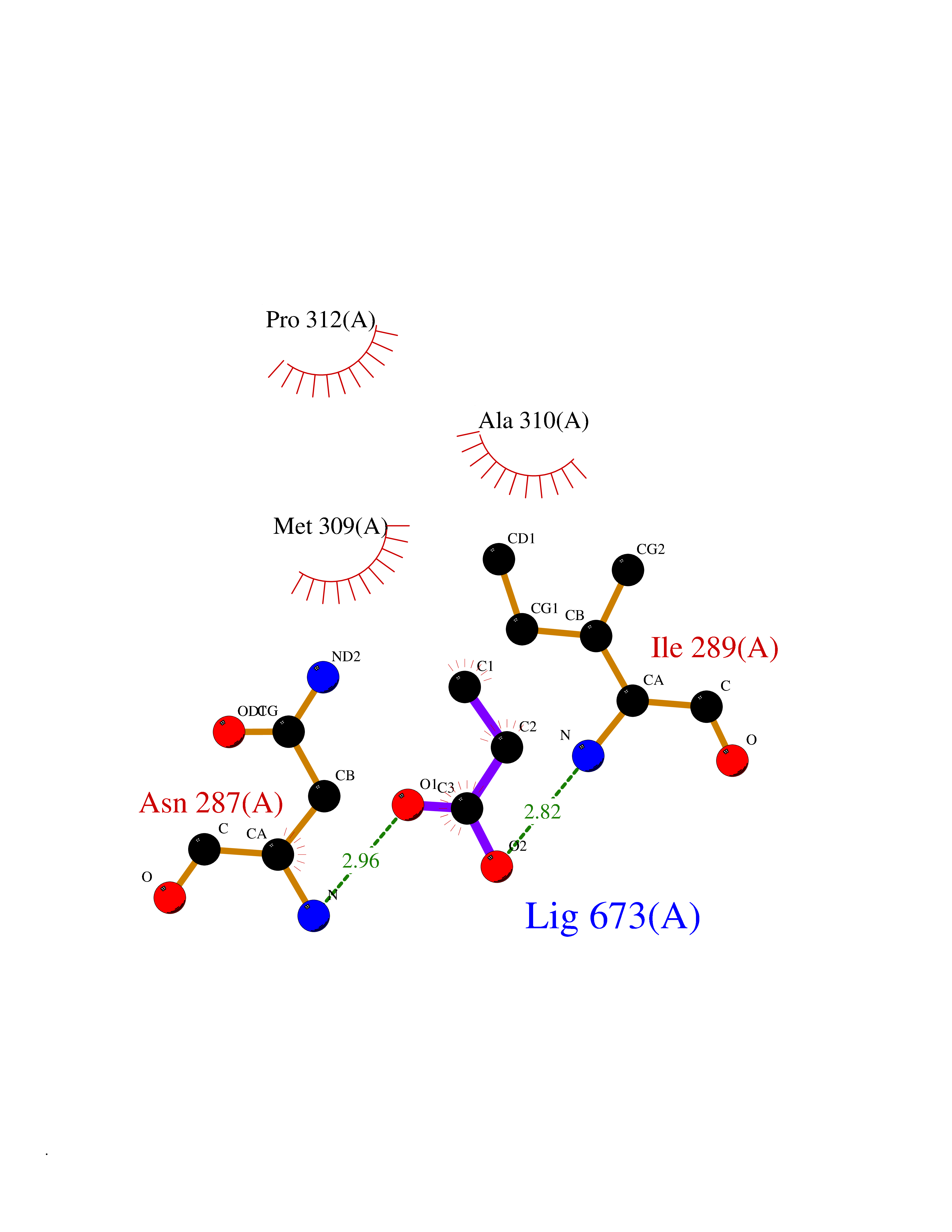 Ligplot Map 3D Binding mode Sequence SYPSLFAPLDLGFTTLKNRVLMGSMHTGLEEYPDGAERLAAFYAERARHGVALIVSGGIAPDLTGVGMEGGAMLNDASQIPHHRTITEAVHQEGGKIALQILHTGRYSYQPHLVAPSALQAPINRFVPHELSHEEILQLIDNFARCAQLAREAGYDGVEVMGSEGYLINEFLTLRTNQRSDQWGGDYRNRMRFAVEVVRAVRERVGNDFIIIYRLSMLDLVEDGGTFAETVELAQAIEAAGATIINTGIGWHEARIPTIATPVPRGAFSWVTRKLKGHVSLPLVTTNRINDPQVADDILSRGDADMVSMARPFLADAELLSKAQSGRADEINTCIGCNQACLDQIFVGKVTSCLVNPRACHETKMP Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Aminoacylase-1 | 1Q7L | 4.56 | |
Target general information Gen name ACY1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase M20A family Biochemical class Hydrolase Function Aminoacylase activity.Identical protein binding.Metal ion binding.Metallopeptidase activity. Related diseases Aminoacylase-1 deficiency (ACY1D) [MIM:609924]: An enzymatic deficiency resulting in encephalopathy, unspecific psychomotor delay, psychomotor delay with atrophy of the vermis and syringomyelia, marked muscular hypotonia or normal clinical features. Epileptic seizures are a frequent feature. All affected individuals exhibit markedly increased urinary excretion of several N-acetylated amino acids. {ECO:0000269|PubMed:16274666, ECO:0000269|PubMed:16465618, ECO:0000269|PubMed:17562838, ECO:0000269|PubMed:21414403}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06151; DB00128; DB09130 Interacts with Q03154; O75934; Q96HA8; P36639; P36639-2; Q8TCT1; P0CG20; Q96A09; P54274; O43711; Q9UPN9; Q9NZC7-5 EC number 3.5.1.14 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,C Molecular weight (Da) 31172.2 Length 275 Aromaticity 0.11 Instability index 36.46 Isoelectric point 6 Charge (pH=7) -5.26 2D Binding mode Binding energy (Kcal/mol) -6.22 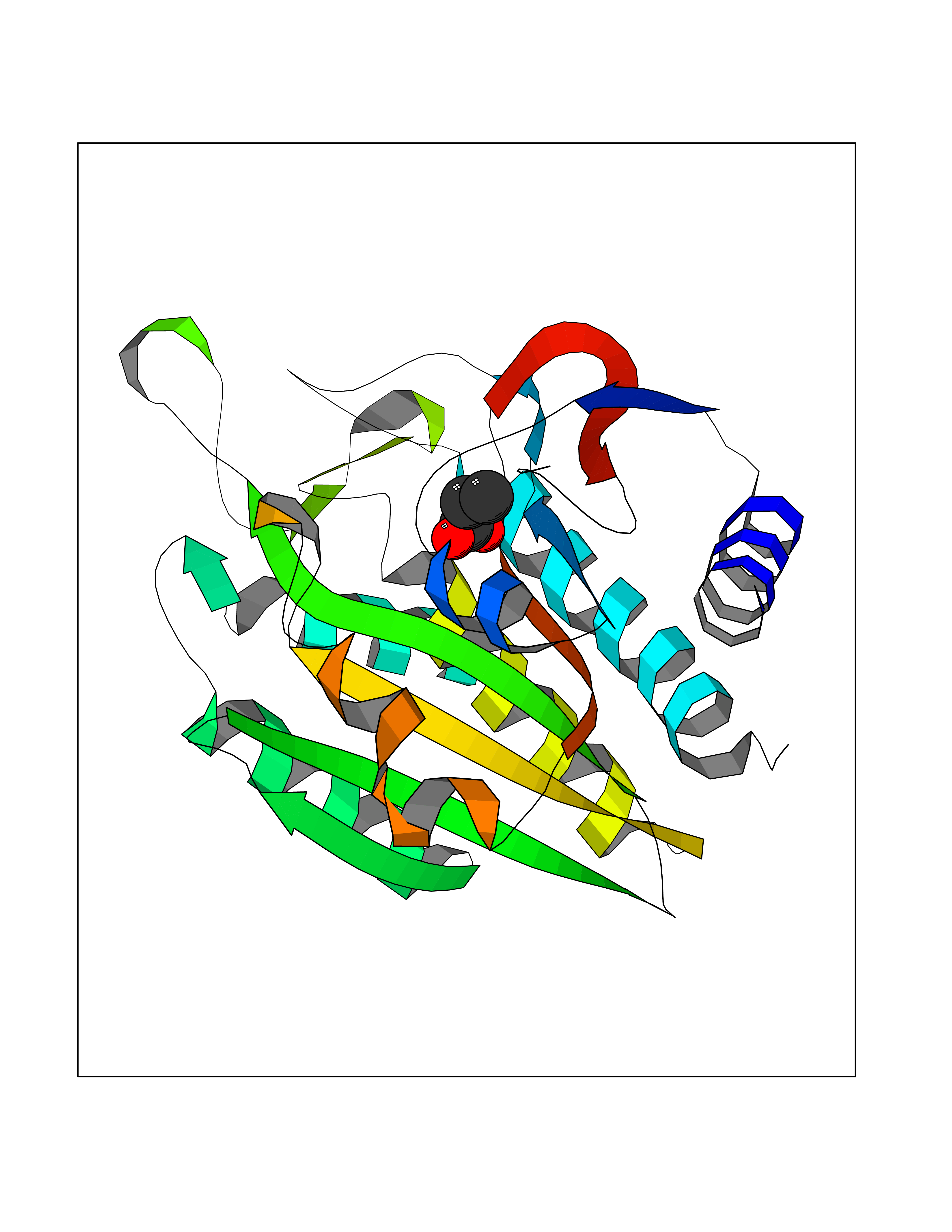 Molscript Map 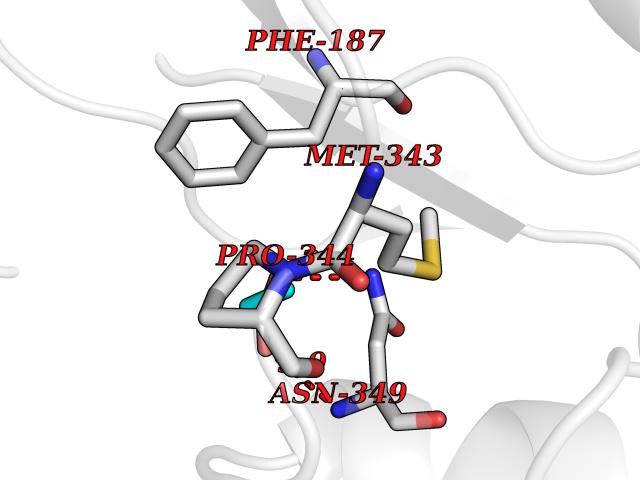 Pymol Map 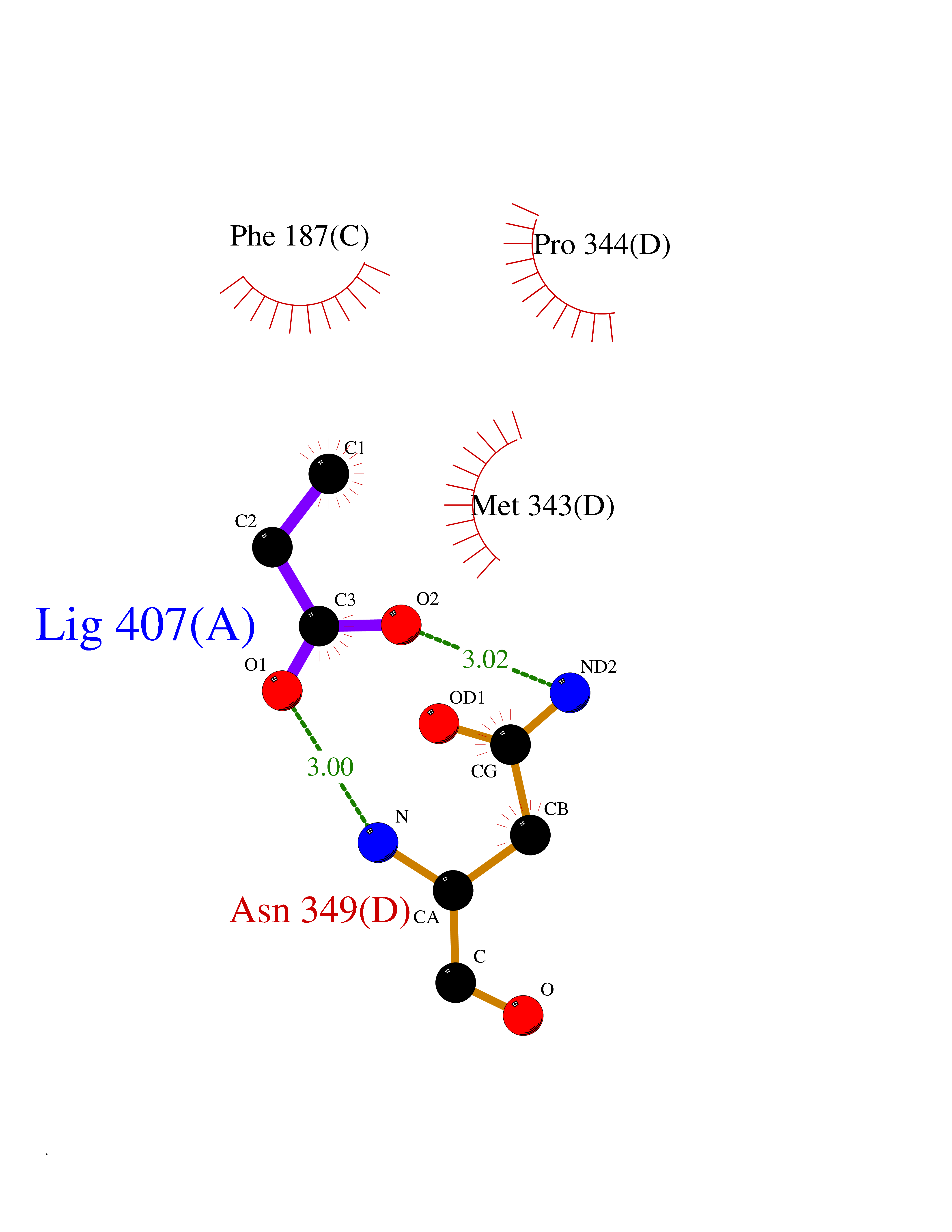 Ligplot Map 3D Binding mode Sequence NPWWAAFSRVCKDMNLTLEPEIMPAAGDNRYIRAVGVPALGFSPMNRTPVLLHDHDERLHEAVFLRGVDIYTRLLPALASVPALPEEHPSVTLFRQYLRIRTVQPKPDYGAAVAFFEETARQLGLGCQKVEVAPGYVVTVLTWPGTNPTLSSILLNSHTDVVPVFKEHWSHDPFEAFKDSEGYIYARGAQDMKCVSIQYLEAVRRLKVEGHRFPRTIHMTFVPDEEVGGHQGMELFVQRPEFHALRAGFALDEGIANPTDAFTVFYSERSPWWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Dihydroorotate dehydrogenase (DHODH) | 4OQV | 4.56 | |
Target general information Gen name DHODH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dihydroorotate oxidase; Dihydroorotate dehydrogenase (quinone), mitochondrial; DHOdehase; DHODH Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07559; DB07561; DB08172; DB08169; DB07443; DB07978; DB07975; DB04281; DB08249; DB07977; DB07976; DB04583; DB08008; DB01117; DB03523; DB03480; DB02613; DB04147; DB03247; DB01097; DB06481; DB08006; DB02262; DB05125; DB08880; DB07646 Interacts with Q6ZMZ0; P49638 EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Disease variant; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 38341.4 Length 353 Aromaticity 0.05 Instability index 39.27 Isoelectric point 9.28 Charge (pH=7) 5.52 2D Binding mode Binding energy (Kcal/mol) -6.21 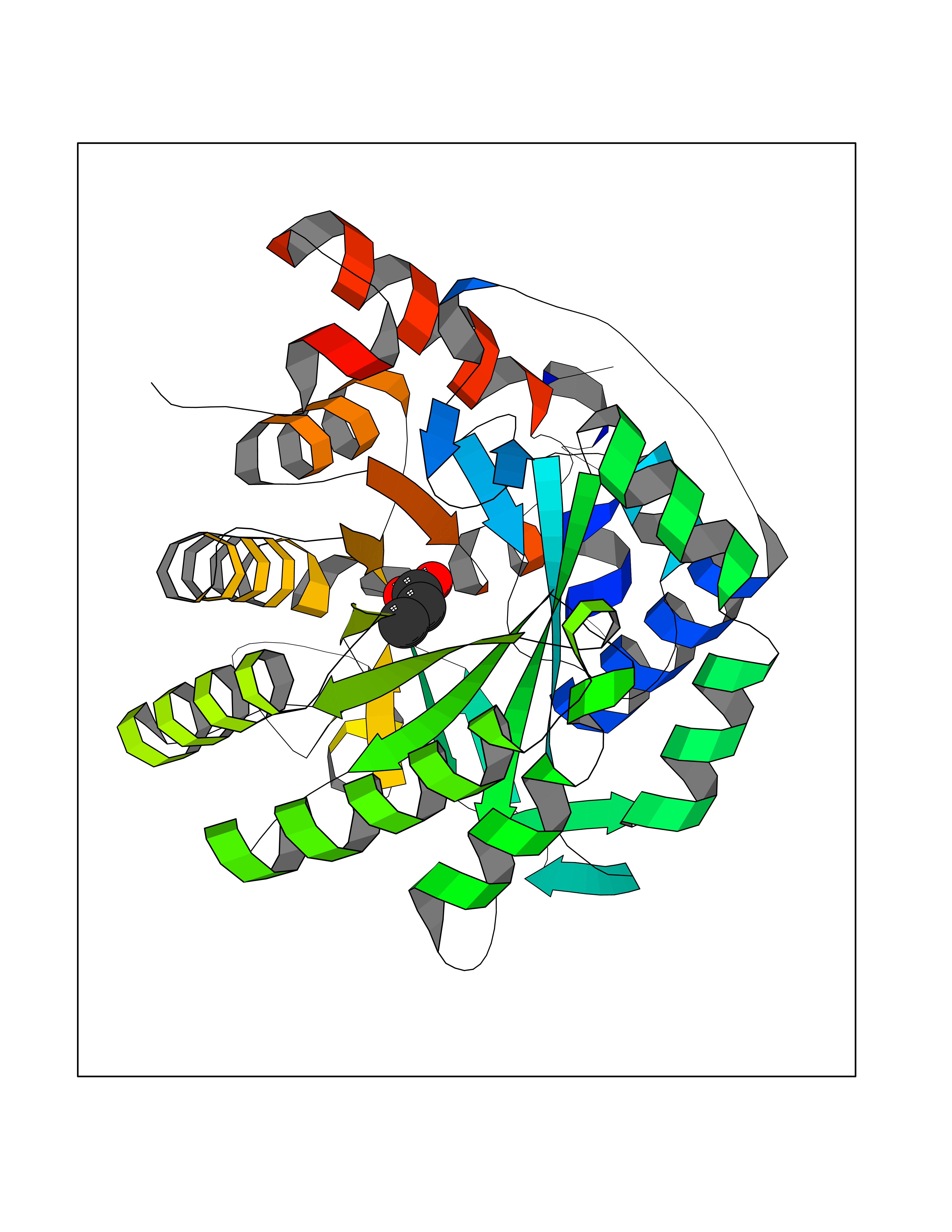 Molscript Map 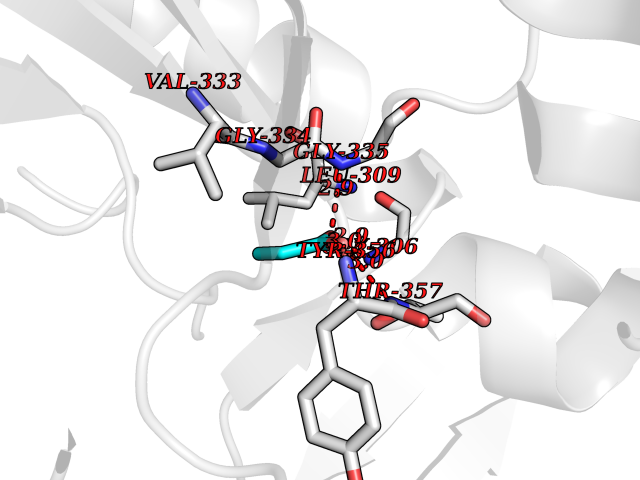 Pymol Map 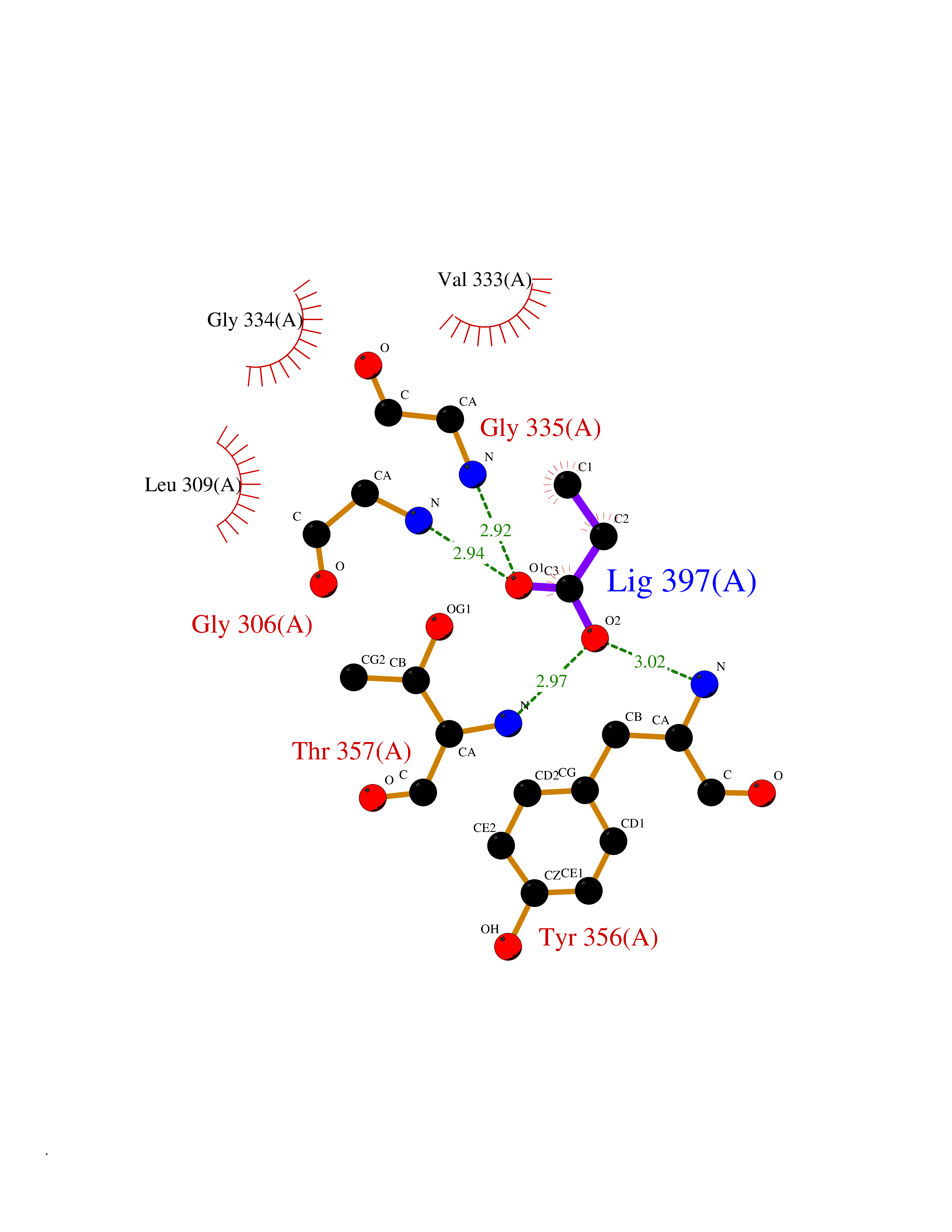 Ligplot Map 3D Binding mode Sequence DERFYAEHLMPTLQGLLDPESAHRLAVRFTSLGLLPRARFQDSDMLEVRVLGHKFRNPVGIAAGFDKHGEAVDGLYKMGFGFVEIGSVTPKPQEGNPRPRVFRLPEDQAVINRYGFNSHGLSVVEHRLRARQQKQAKLTEDGLPLGVNLGKNKTSVDAAEDYAEGVRVLGPLADYLVVNVSSPGKAELRRLLTKVLQERDGLRRVHRPAVLVKIAPDLTSQDKEDIASVVKELGIDGLIVTNTTVSRPAGLQGALRSETGGLSGKPLRDLSTQTIREMYALTQGRVPIIGVGGVSSGQDALEKIRAGASLVQLYTALTFWGPPVVGKVKRELEALLKEQGFGGVTDAIGADHR Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Dihydroorotate dehydrogenase (quinone), mitochondrial | 4CQ8 | 4.56 | |
Target general information Gen name PFF0160c Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class Oxidoreductase Function Dihydroorotate dehydrogenase activity. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42573.5 Length 378 Aromaticity 0.1 Instability index 36.63 Isoelectric point 8.21 Charge (pH=7) 3.17 2D Binding mode Binding energy (Kcal/mol) -6.22 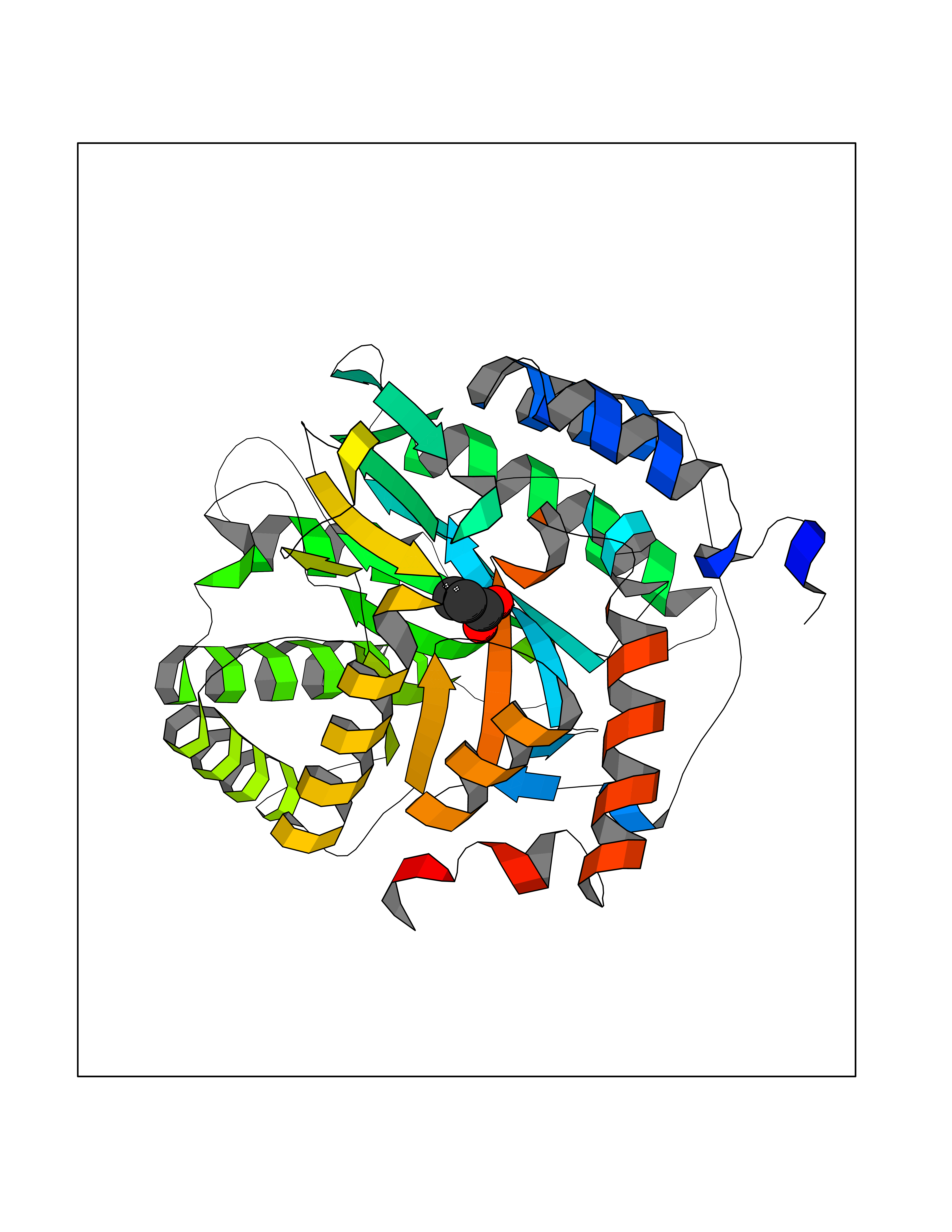 Molscript Map 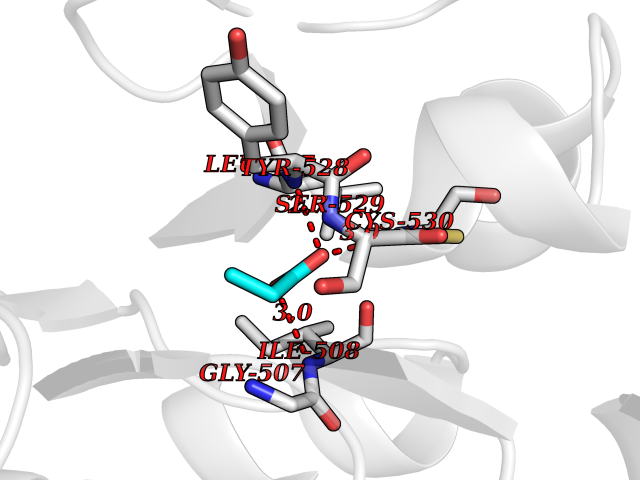 Pymol Map 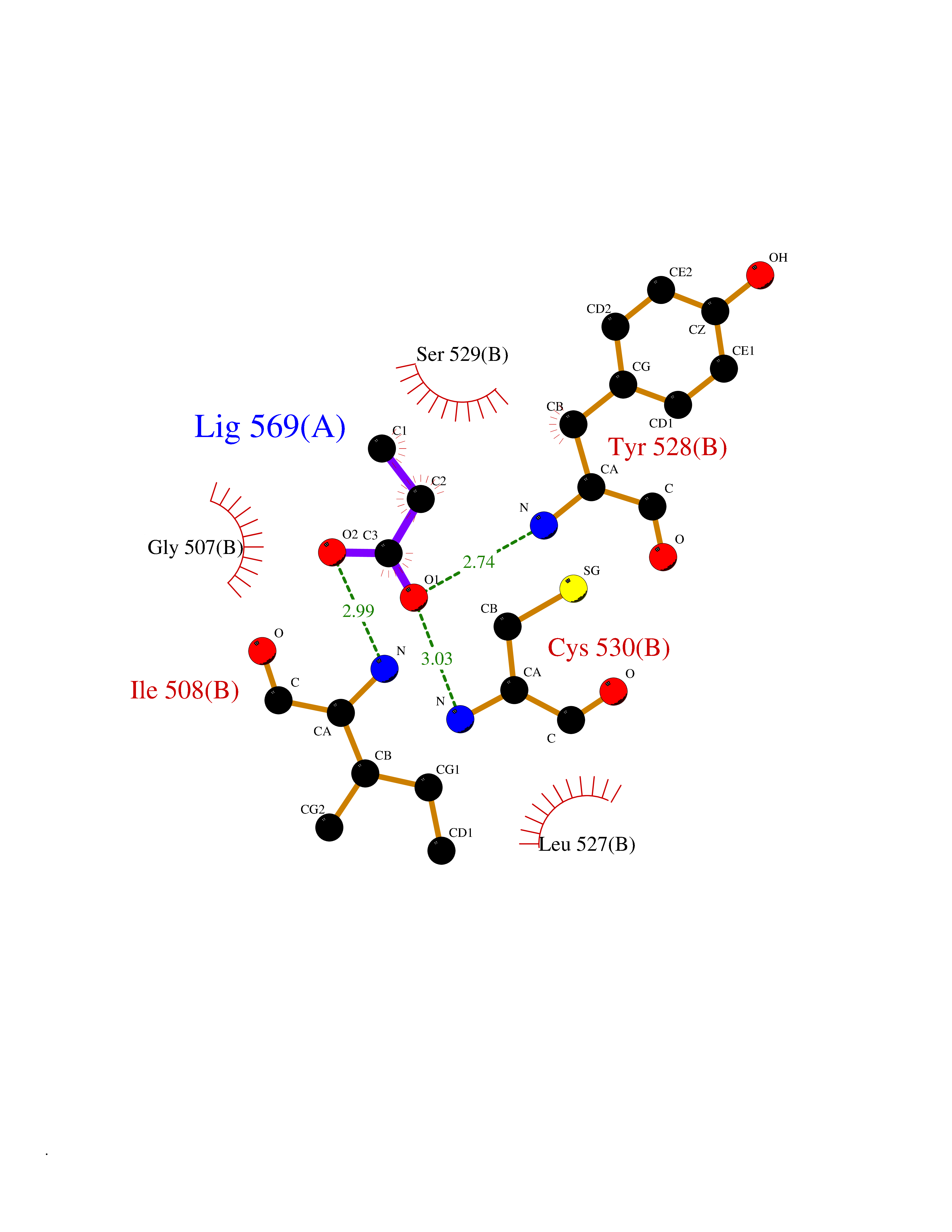 Ligplot Map 3D Binding mode Sequence ADPFESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEKNNFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | cAMP-dependent protein kinase A type I (PRKAR1A) | 5KJZ | 4.56 | |
Target general information Gen name PRKAR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-dependent protein kinase type I-alpha regulatory subunit; Tissue-specific extinguisher 1; TSE1; PRKAR1; PKR1 Protein family CAMP-dependent kinase regulatory chain family Biochemical class Kinase Function Regulatory subunit of the cAMP-dependent protein kinases involved in cAMP signaling in cells. Related diseases Carney complex 1 (CNC1) [MIM:160980]: CNC is a multiple neoplasia syndrome characterized by spotty skin pigmentation, cardiac and other myxomas, endocrine tumors, and psammomatous melanotic schwannomas. {ECO:0000269|PubMed:15371594, ECO:0000269|PubMed:18241045, ECO:0000269|PubMed:22785148, ECO:0000269|PubMed:23323113, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Intracardiac myxoma (INTMYX) [MIM:255960]: Inheritance is autosomal recessive. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Primary pigmented nodular adrenocortical disease 1 (PPNAD1) [MIM:610489]: A rare bilateral adrenal defect causing ACTH-independent Cushing syndrome. Macroscopic appearance of the adrenals is characteristic with small pigmented micronodules observed in the cortex. Clinical manifestations of Cushing syndrome include facial and truncal obesity, abdominal striae, muscular weakness, osteoporosis, arterial hypertension, diabetes. PPNAD1 is most often diagnosed in patients with Carney complex, a multiple neoplasia syndrome. However it can also be observed in patients without other manifestations. {ECO:0000269|PubMed:12213893}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Acrodysostosis 1, with or without hormone resistance (ACRDYS1) [MIM:101800]: A form of skeletal dysplasia characterized by short stature, severe brachydactyly, facial dysostosis, and nasal hypoplasia. Affected individuals often have advanced bone age and obesity. Laboratory studies show resistance to multiple hormones, including parathyroid, thyrotropin, calcitonin, growth hormone-releasing hormone, and gonadotropin. However, not all patients show endocrine abnormalities. {ECO:0000269|PubMed:21651393, ECO:0000269|PubMed:22464250, ECO:0000269|PubMed:22464252, ECO:0000269|PubMed:22723333, ECO:0000269|PubMed:23043190, ECO:0000269|PubMed:23425300, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01790; DB02527; DB02315; DB05798 Interacts with Q9GZX7; P24588; O43687-2; Q9BSF0; Q9H6J7-2; Q86Y01; P0C7A2-2; Q9H0R8; Q9H8W4; P17612; P31321; P51817; P35250; Q86UC2; Q01105; Q8N0X7; O96006; P03259-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; cAMP; cAMP-binding; Cell membrane; Cushing syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat Protein physicochemical properties Chain ID A Molecular weight (Da) 17007.4 Length 149 Aromaticity 0.08 Instability index 49.36 Isoelectric point 6.36 Charge (pH=7) -0.52 2D Binding mode Binding energy (Kcal/mol) -6.22 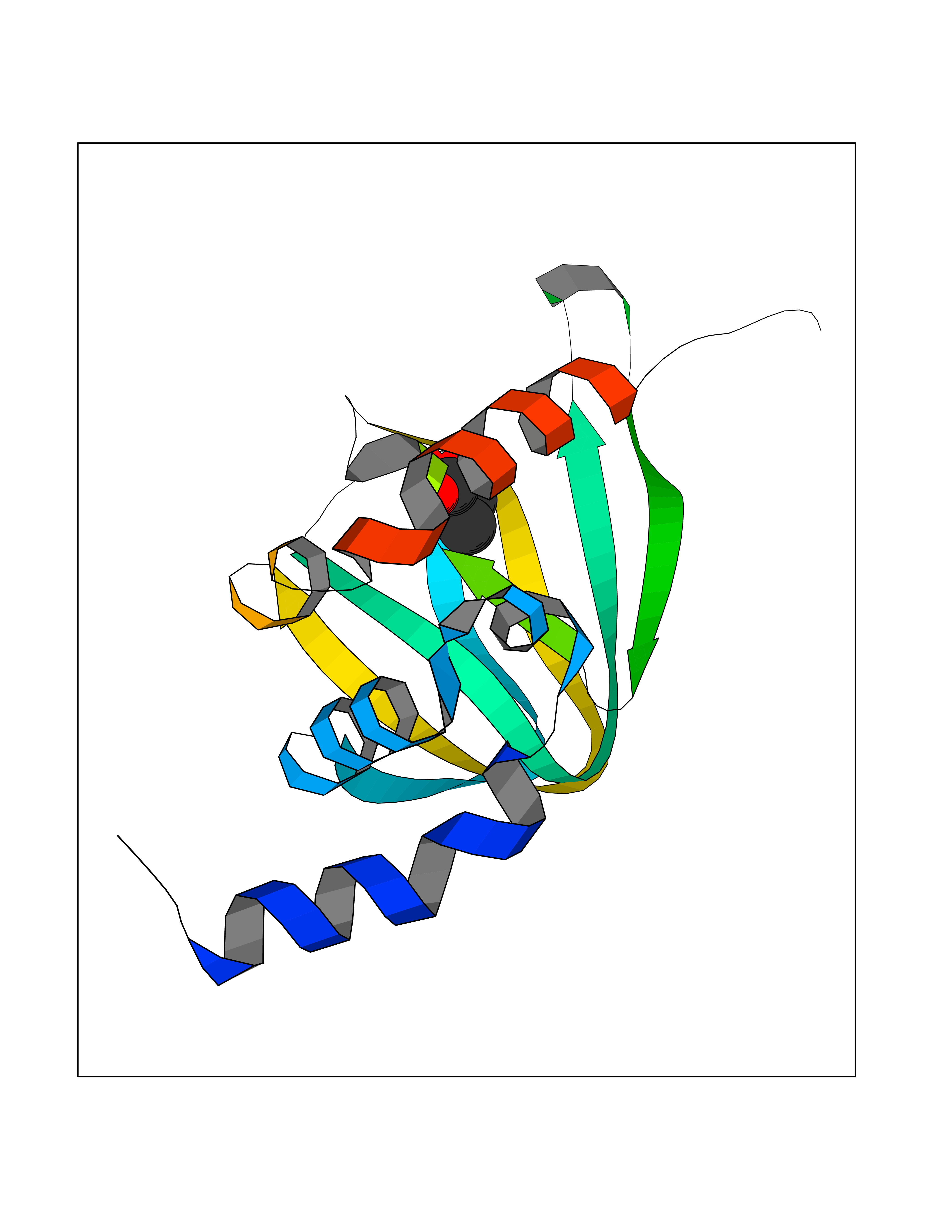 Molscript Map 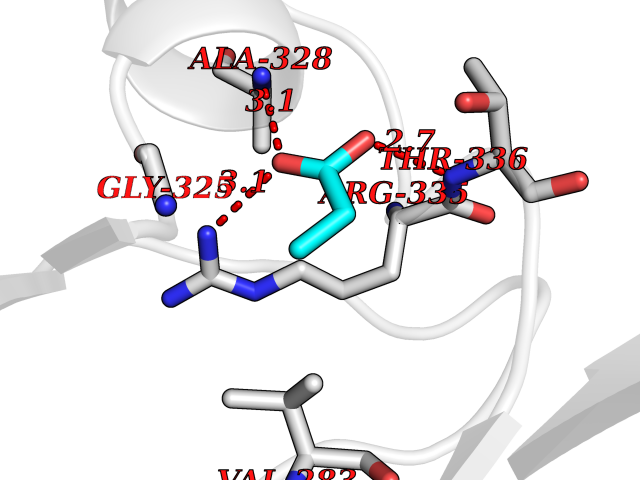 Pymol Map 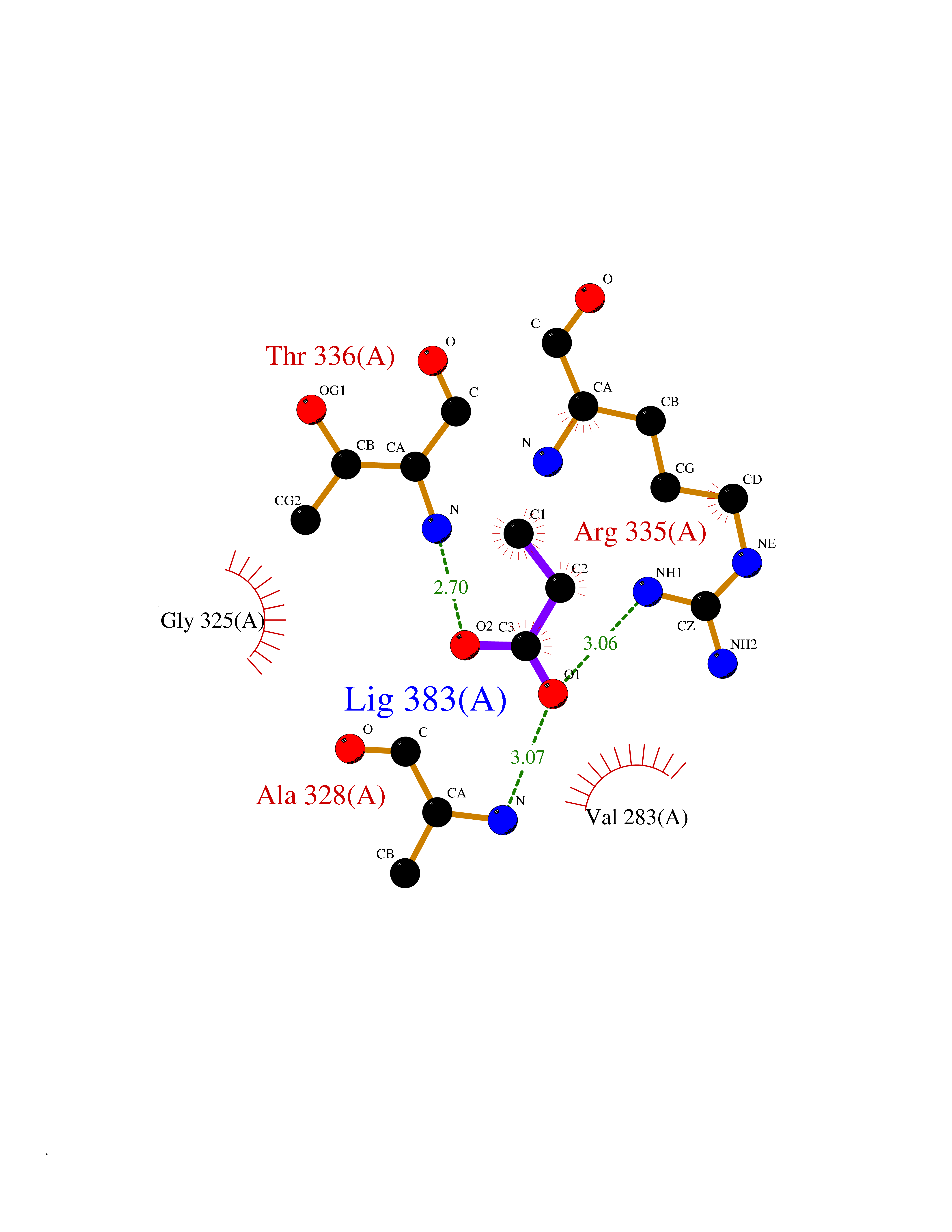 Ligplot Map 3D Binding mode Sequence SILMGSTLRKRKMYEEFLSKVSILESLDKWERLTVADALEPVQFEDGQKIVVQGEPGDEFFIILEGSAAVLQRRSENEEFVEVRRLGPSDYFGEIALLMNRPRTATVVARGPLKCVKLDRPRFERVLGPCSDILKRNIQQYNSFVSLSV Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Hyperpolarization cyclic nucleotide-gated channel 4 (HCN4) | 3OTF | 4.56 | |
Target general information Gen name HCN4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 4 Protein family Potassium channel HCN family Biochemical class Voltage-gated ion channel Function Contributes to the native pacemaker currents in heart (If) that regulate the rhythm of heart beat. May contribute to the native pacemaker currents in neurons (Ih). May mediate responses to sour stimuli. Hyperpolarization-activated ion channel with very slow activation and inactivation exhibiting weak selectivity for potassium over sodium ions. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60741; Q9Y3Q4 EC number NA Uniprot keywords 3D-structure; Brugada syndrome; cAMP; cAMP-binding; Cell membrane; Disease variant; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Nucleotide-binding; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 23211.2 Length 197 Aromaticity 0.12 Instability index 42.69 Isoelectric point 8.67 Charge (pH=7) 3.11 2D Binding mode Binding energy (Kcal/mol) -6.22 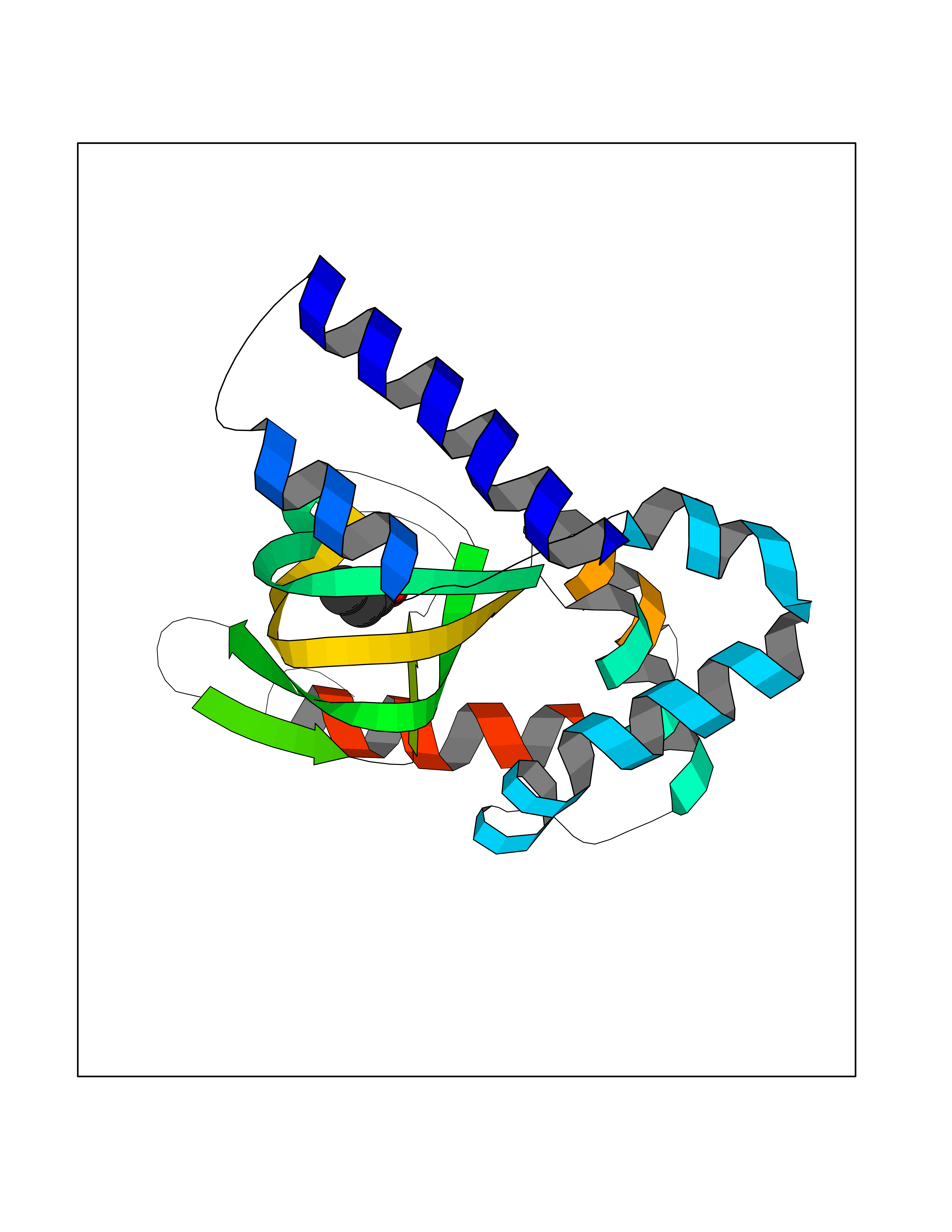 Molscript Map 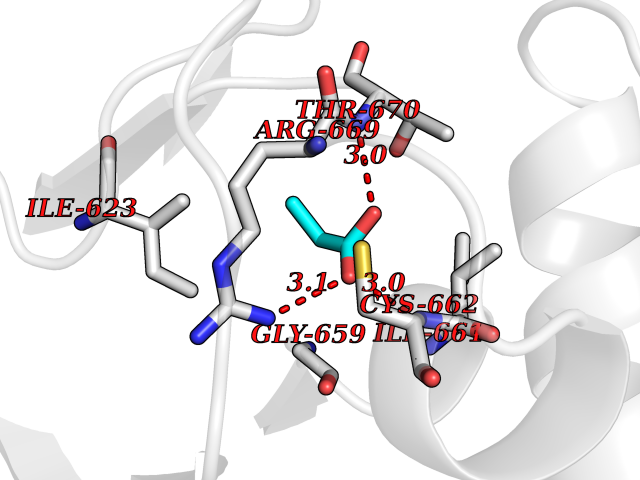 Pymol Map 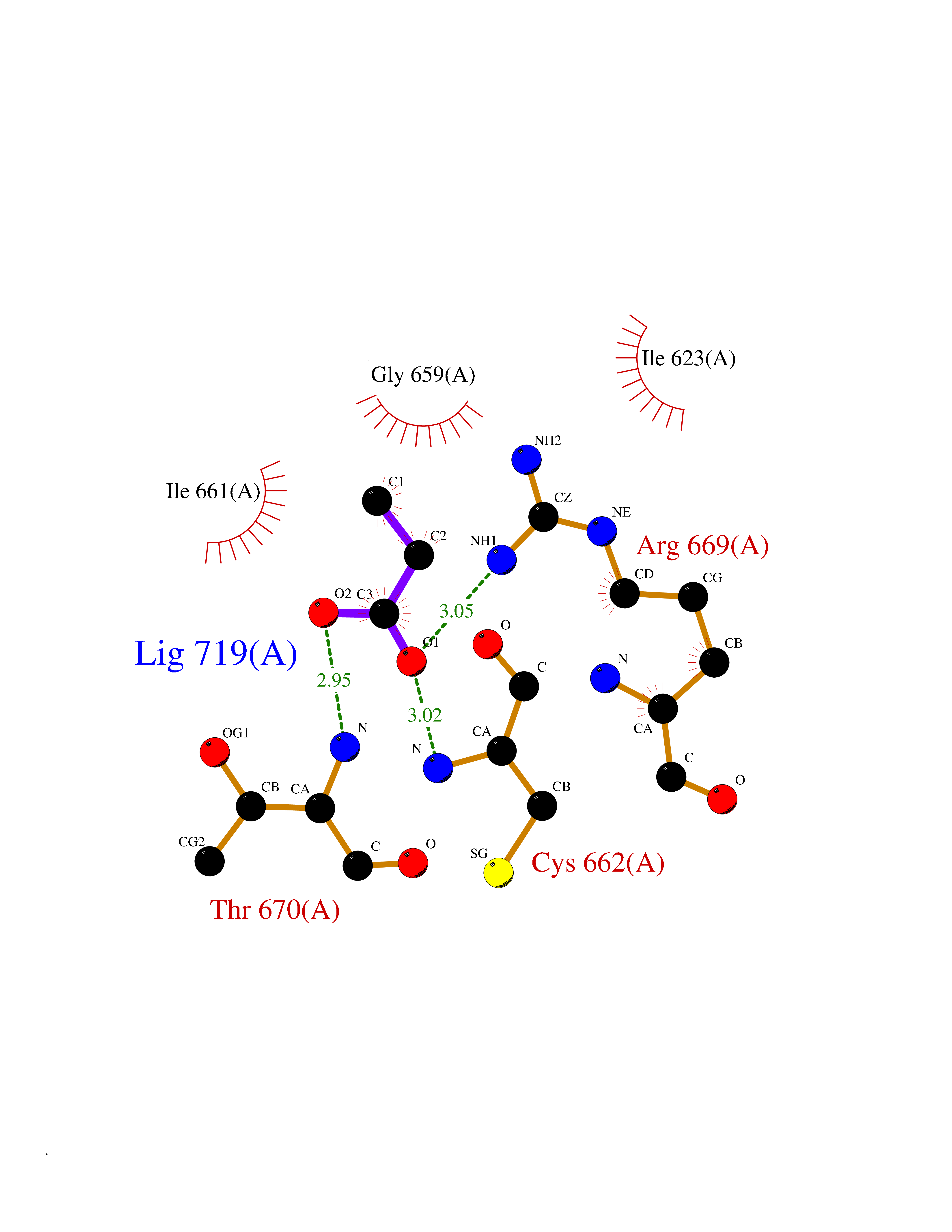 Ligplot Map 3D Binding mode Sequence DSSRRQYQEKYKQVEQYMSFHKLPPDTRQRIHDYYEHRYQGKMFDEESILGELSEPLREEIINFNCRKLVASMPLFANADPNFVTSMLTKLRFEVFQPGDYIIREGTIGKKMYFIQHGVVSVLTKGNKETKLADGSYFGEICLLTRGRRTASVRADTYCRLYSLSVDNFNEVLEEYPMMRRAFETVALDRLDRIGKK Hydrogen bonds contact Hydrophobic contact | ||||