Job Results:
Ligand
Structure
Job ID
9069676bc0da30cae69182dac941f7de
Job name
NA
Time
2026-02-27 11:56:18
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Bacterial Pyruvate decarboxylase (Bact aceE) | 1L8A | 3.94 | |
Target general information Gen name Bact aceE Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms aceE; Pyruvate decarboxylase E1 component Protein family NA Biochemical class Aldehyde/oxo donor oxidoreductase Function The pyruvate dehydrogenase complex catalyzes the overall conversion of pyruvate to acetyl-coa and co(2). It contains multiple copies of three enzymatic components: pyruvate dehydrogenase (e1), dihydrolipoamide acetyltransferase (e2) and lipoamide dehydrogenase. Related diseases Pseudovaginal perineoscrotal hypospadias (PPSH) [MIM:264600]: A form of male pseudohermaphroditism in which 46,XY males show ambiguous genitalia at birth, including perineal hypospadias and a blind perineal pouch, and develop masculinization at puberty. The name of the disorder stems from the finding of a blind-ending perineal opening resembling a vagina and a severely hypospadiac penis with the urethra opening onto the perineum. {ECO:0000269|PubMed:10718838, ECO:0000269|PubMed:10898110, ECO:0000269|PubMed:10999800, ECO:0000269|PubMed:12843198, ECO:0000269|PubMed:15064320, ECO:0000269|PubMed:1522235, ECO:0000269|PubMed:15528927, ECO:0000269|PubMed:15770495, ECO:0000269|PubMed:16098368, ECO:0000269|PubMed:16181229, ECO:0000269|PubMed:7554313, ECO:0000269|PubMed:8626825, ECO:0000269|PubMed:8768837, ECO:0000269|PubMed:9208814, ECO:0000269|PubMed:9745434, ECO:0000269|PubMed:9843052}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01987 Interacts with P0AFG8; P0A8N7; P77439; P46889; P0A6F5; P0AEV9; P0AB83; P0AB89; P0AG40; P05100; P26602; P0A9U1; P0A8L7; P76049; P76170 EC number NA Uniprot keywords 3D-structure; Acetylation; Direct protein sequencing; Magnesium; Metal-binding; Oxidoreductase; Pyruvate; Reference proteome; Thiamine pyrophosphate Protein physicochemical properties Chain ID A,B Molecular weight (Da) 179825 Length 1602 Aromaticity 0.1 Instability index 37.46 Isoelectric point 5.51 Charge (pH=7) -42.23 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ISNYINTIPVEEQPEYPGNLELERRIRSAIRWNAIMTVLRASKKDLELGGHMASFQSSATIYDVCFNHFFRARNEQDGGDLVYFQGHISPGVYARAFLEGRLTQEQLDNFRQEVHGNGLSSYPHPKLMPEFWQFPTVSMGLGPIGAIYQAKFLKYLEHRGLKDTSKQTVYAFLGDGEMDEPESKGAITIATREKLDNLVFVINCNLQRLDGPVTGNGKIINELEGIFEGAGWNVIKVMWGSRWDELLRKDTSGKLIQLMNETVDGDYQTFKSKDGAYVREHFFGKYPETAALVADWTDEQIWALNRGGHDPKKIYAAFKKAQETKGKATVILAHTIKGYGMGDAAMDGVRHIRDRFNVPVSDADIEKLPYITFPEGSEEHTYLHAQRQKLHGYLPSRQPNFTEKLELPSLQDFGALLEEQSKEISTTIAFVRALNVMLKNKSIKDRLVPIIADEARTFGMEGLFRQIGIYSPEDEKGQILQEGINELGAGCSWLAAATSYSTNNLPMIPFYIYYSMFGFQRIGDLCWAAGDQQARGFLIGGTSGRTTLNGEGLQHEDGHSHIQSLTIPNCISYDPAYAYEVAVIMHDGLERMYGEKQENVYYYITTLNENYHMPAMPEGAEEGIRKGIYKLETIEGSKGKVQLLGSGSILRHVREAAEILAKDYGVGSDVYSVTSFTELARDGQDCERWNMLHPLETPRVPYIAQVMNDAPAVASTDYMKLFAEQVRTYVPADDYRVLGTDGFGRSDSRENLRHHFEVDASYVVVAALGELAKRGEIDKKVVADAIAKFNIDADKVNPRLAISNYINTIPVEEQPEYPGNLELERRIRSAIRWNAIMTVLRASKKDLELGGHMASFQSSATIYDVCFNHFFRARNEQDGGDLVYFQGHISPGVYARAFLEGRLTQEQLDNFRQEVHGNGLSSYPHPKLMPEFWQFPTVSMGLGPIGAIYQAKFLKYLEHRGLKDTSKQTVYAFLGDGEMDEPESKGAITIATREKLDNLVFVINCNLQRLDGPVTGNGKIINELEGIFEGAGWNVIKVMWGSRWDELLRKDTSGKLIQLMNETVDGDYQTFKSKDGAYVREHFFGKYPETAALVADWTDEQIWALNRGGHDPKKIYAAFKKAQETKGKATVILAHTIKGYGMGDAAMDGVRHIRDRFNVPVSDADIEKLPYITFPEGSEEHTYLHAQRQKLHGYLPSRQPNFTEKLELPSLQDFGALLEEQSKEISTTIAFVRALNVMLKNKSIKDRLVPIIADEARTFGMEGLFRQIGIYSPEDEKGQILQEGINELGAGCSWLAAATSYSTNNLPMIPFYIYYSMFGFQRIGDLCWAAGDQQARGFLIGGTSGRTTLNGEGLQHEDGHSHIQSLTIPNCISYDPAYAYEVAVIMHDGLERMYGEKQENVYYYITTLNENYHMPAMPEGAEEGIRKGIYKLETIEGSKGKVQLLGSGSILRHVREAAEILAKDYGVGSDVYSVTSFTELARDGQDCERWNMLHPLETPRVPYIAQVMNDAPAVASTDYMKLFAEQVRTYVPADDYRVLGTDGFGRSDSRENLRHHFEVDASYVVVAALGELAKRGEIDKKVVADAIAKFNIDADKVNPRLA Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Beta-galactosidase (GLB1) | 3THD | 3.94 | |
Target general information Gen name GLB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lactase; Elastin receptor 1; ELNR1; Acid beta-galactosidase Protein family Glycosyl hydrolase 35 family Biochemical class NA Function Isoform 1: Cleaves beta-linked terminal galactosyl residues from gangliosides, glycoproteins, and glycosaminoglycans. Related diseases GM1-gangliosidosis 1 (GM1G1) [MIM:230500]: An autosomal recessive lysosomal storage disease marked by the accumulation of GM1 gangliosides, glycoproteins and keratan sulfate primarily in neurons of the central nervous system. GM1-gangliosidosis type 1 is characterized by onset within the first three months of life, central nervous system degeneration, coarse facial features, hepatosplenomegaly, skeletal dysmorphology reminiscent of Hurler syndrome, and rapidly progressive psychomotor deterioration. Urinary oligosaccharide levels are high. It leads to death usually between the first and second year of life. {ECO:0000269|PubMed:10338095, ECO:0000269|PubMed:10737981, ECO:0000269|PubMed:10839995, ECO:0000269|PubMed:1487238, ECO:0000269|PubMed:15365997, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:15791924, ECO:0000269|PubMed:16538002, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:1928092, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8213816, ECO:0000269|Ref.28, ECO:0000269|Ref.31}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GM1-gangliosidosis 2 (GM1G2) [MIM:230600]: A gangliosidosis characterized by onset between ages 1 and 5. The main symptom is locomotor ataxia, ultimately leading to a state of decerebration with epileptic seizures. Patients do not display the skeletal changes associated with the infantile form, but they nonetheless excrete elevated amounts of beta-linked galactose-terminal oligosaccharides. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10737981, ECO:0000269|PubMed:12644936, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8213816}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GM1-gangliosidosis 3 (GM1G3) [MIM:230650]: A gangliosidosis with a variable phenotype. Patients show mild skeletal abnormalities, dysarthria, gait disturbance, dystonia and visual impairment. Visceromegaly is absent. Intellectual deficit can initially be mild or absent but progresses over time. Inheritance is autosomal recessive. {ECO:0000269|PubMed:11511921, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:15986423, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8198123, ECO:0000269|Ref.28, ECO:0000269|Ref.30}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 4B (MPS4B) [MIM:253010]: A form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life. {ECO:0000269|PubMed:11511921, ECO:0000269|PubMed:12393180, ECO:0000269|PubMed:16538002, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1928092, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:7586649}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04465 Interacts with Q8NBJ4; Q3KNW5; Q9BRI3; P30825 EC number EC 3.2.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Gangliosidosis; Glycoprotein; Glycosidase; Hydrolase; Lysosome; Mucopolysaccharidosis; Proteomics identification; Reference proteome; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 67980.6 Length 605 Aromaticity 0.13 Instability index 40.91 Isoelectric point 5.81 Charge (pH=7) -9.05 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map 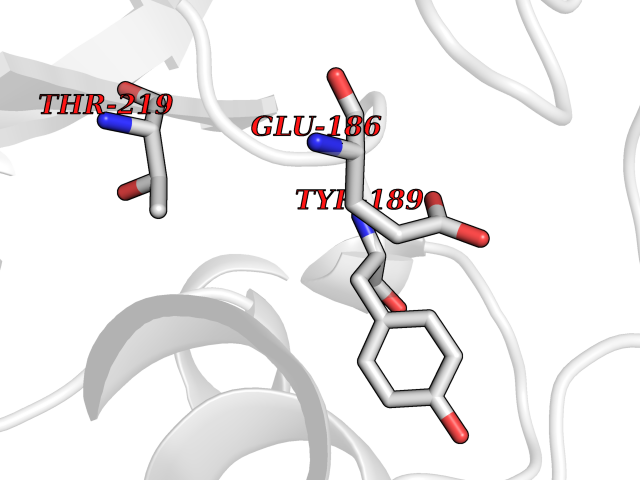 Pymol Map 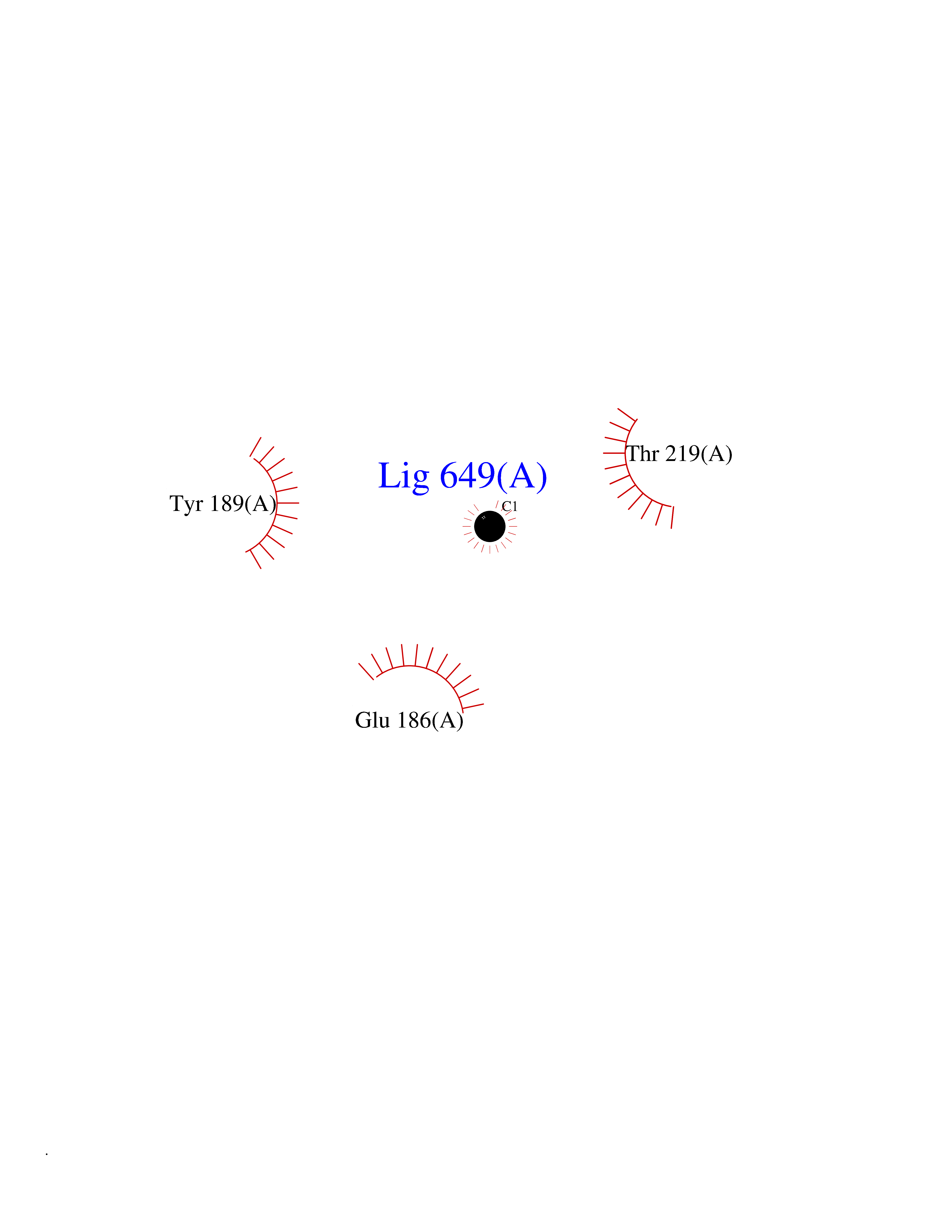 Ligplot Map 3D Binding mode Sequence QRMFEIDYSRDSFLKDGQPFRYISGSIHYSRVPRFYWKDRLLKMKMAGLNAIQTYVPWNFHEPWPGQYQFSEDHDVEYFLRLAHELGLLVILRPGPYICAEWEMGGLPAWLLEKESILLRSSDPDYLAAVDKWLGVLLPKMKPLLYQNGGPVITVQVENEYGSYFACDFDYLRFLQKRFRHHLGDDVVLFTTDGAHKTFLKCGALQGLYTTVDFGTGSNITDAFLSQRKCEPKGPLINSEFYTGWLDHWGQPHSTIKTEAVASSLYDILARGASVNLYMFIGGTNFAYWNGANSPYAAQPTSYDYDAPLSEAGDLTEKYFALRNIIQKFEKVPEGPIPPSTPKFAYGKVTLEKLKTVGAALDILCPSGPIKSLYPLTFIQVKQHYGFVLYRTTLPQDCSNPAPLSSPLNGVHDRAYVAVDGIPQGVLERNNVITLNITGKAGATLDLLVENMGRVNYGAYINDFKGLVSNLTLSSNILTDWTIFPLDTEDAVRSHLGGWGHRNYTLPAFYMGNFSIPSGIPDLPQDTFIQFPGWTKGQVWINGFNLGRYWPARGPQLTLFVPQHILMTSAPNTITVLELEWAPCSSDDPELCAVTFVDRPVIGSS Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Thioredoxin reductase (PRDX5) | 4K7I | 3.94 | |
Target general information Gen name PRDX5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Thioredoxin peroxidase PMP20; TPx type VI; SBBI10; PrxV; Prx-V; Peroxisomal antioxidant enzyme; Peroxiredoxin5, mitochondrial; Peroxiredoxin-5, mitochondrial; Peroxiredoxin V; PLP; Liver tissue 2Dpage Protein family Peroxiredoxin family, Prx5 subfamily Biochemical class Peroxide acceptor oxidoreductase Function Plays a role in cell protection against oxidative stress by detoxifying peroxides and as sensor of hydrogen peroxide-mediated signaling events. Thiol-specific peroxidase that catalyzes the reduction of hydrogen peroxide and organic hydroperoxides to water and alcohols, respectively. Related diseases Variation Asp-298 in NOS3 may be associated with susceptibility to coronary spasm. {ECO:0000269|PubMed:11740345, ECO:0000269|PubMed:9737779}. Drugs (DrugBank ID) DB00995; DB03608; DB09221 Interacts with P55273; Q7Z417; P49901; P00441; P30044-2 EC number EC 1.11.1.15 Uniprot keywords 3D-structure; Acetylation; Alternative initiation; Alternative splicing; Antioxidant; Cytoplasm; Direct protein sequencing; Disulfide bond; Lipoprotein; Mitochondrion; Oxidoreductase; Palmitate; Peroxidase; Peroxisome; Phosphoprotein; Proteomics identification; Redox-active center; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,C Molecular weight (Da) 33752.7 Length 322 Aromaticity 0.06 Instability index 20.09 Isoelectric point 7.57 Charge (pH=7) 0.8 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map 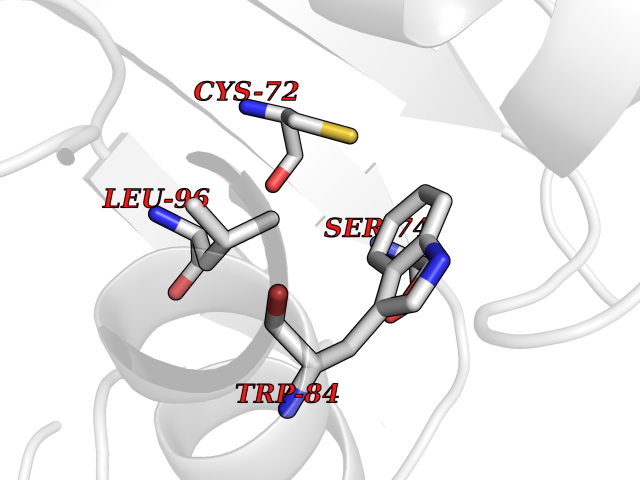 Pymol Map 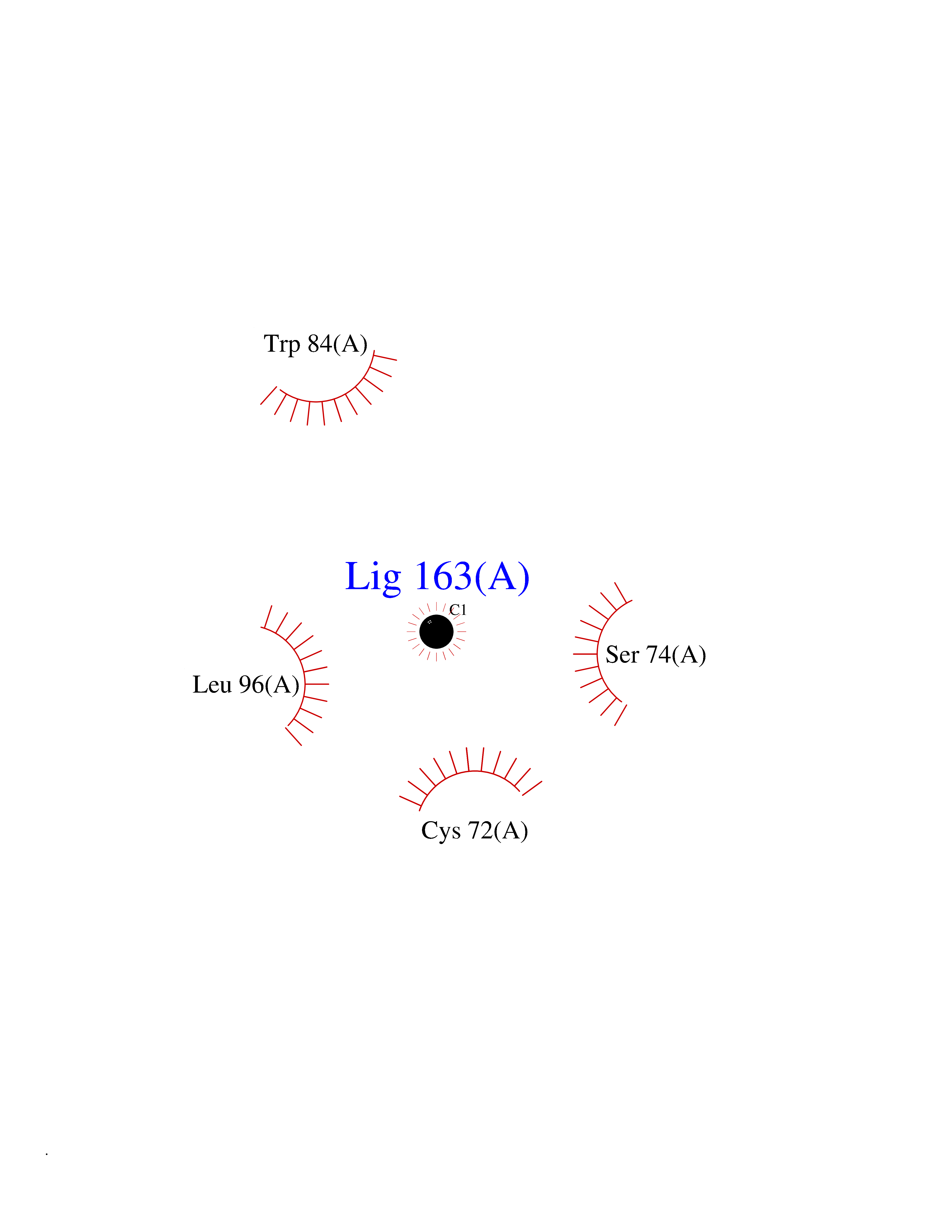 Ligplot Map 3D Binding mode Sequence SAPIKVGDAIPAVEVFEGEPGNKVNLAELFKGKKGVLFGVPGAFTPGCSKTHLPGFVEQAEALKAKGVQVVACLSVNDAFVTGEWGRAHKAEGKVRLLADPTGAFGKETDLLLDDSLVSIFGNRRLKRFSMVVQDGIVKALNVEPDGTGLTCSLAPNIISQLAPIKVGDAIPAVEVFEGEPGNKVNLAELFKGKKGVLFGVPGAFTPGCSKTHLPGFVEQAEALKAKGVQVVACLSVNDAFVTGEWGRAHKAEGKVRLLADPTGAFGKETDLLLDDSLVSIFGNRRLKRFSMVVQDGIVKALNVEPGTGLTCSLAPNIISQL Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | GABA(A) receptor beta-2 (GABRB2) | 6X3T | 3.94 | |
Target general information Gen name GABRB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GABRB2; GABA(A) receptor subunit beta-2 Protein family Ligand-gated ion channel (TC 1.A.9) family, Gamma-aminobutyric acid receptor (TC 1.A.9.5) subfamily, GABRB2 sub-subfamily Biochemical class Neurotransmitter receptor Function Component of the heteropentameric receptor for GABA, the major inhibitory neurotransmitter in the vertebrate brain. Functions also as histamine receptor and mediates cellular responses to histamine. Functions as receptor for diazepines and various anesthetics, such as pentobarbital; these are bound at a separate allosteric effector binding site. Functions as ligand- gated chloride channel. Related diseases Epileptic encephalopathy, infantile or early childhood, 2 (IECEE2) [MIM:617829]: A form of epileptic encephalopathy, a heterogeneous group of severe childhood onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. IECEE2 is an autosomal dominant condition with variable age at seizure onset, ranging from early infancy to 6 years. {ECO:0000269|PubMed:25124326, ECO:0000269|PubMed:27789573, ECO:0000269|PubMed:29100083}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12537; DB00546; DB00404; DB00543; DB11901; DB14719; DB11859; DB01558; DB09017; DB00237; DB00241; DB01489; DB00395; DB00475; DB14715; DB01594; DB00349; DB01068; DB00628; DB01559; DB01553; DB01511; DB01189; DB00829; DB13837; DB00228; DB01215; DB00402; DB00898; DB00189; DB01545; DB09166; DB00292; DB01567; DB01205; DB01544; DB00690; DB06716; DB05087; DB01381; DB01437; DB00801; DB01159; DB00753; DB01587; DB00555; DB13643; DB00186; DB13872; DB13437; DB00603; DB01043; DB00371; DB00463; DB01028; DB01107; DB15489; DB00683; DB01595; DB14028; DB00842; DB14672; DB00312; DB00252; DB13335; DB01708; DB01588; DB00794; DB00818; DB01589; DB12404; DB01236; DB09118; DB00306; DB01956; DB00231; DB11582; DB00897; DB15490 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Chloride; Chloride channel; Cytoplasmic vesicle; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 77460 Length 672 Aromaticity 0.14 Instability index 35.95 Isoelectric point 8.75 Charge (pH=7) 7.31 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map 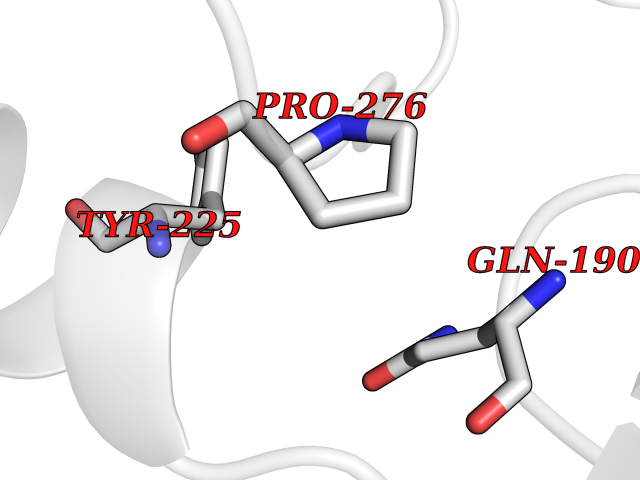 Pymol Map 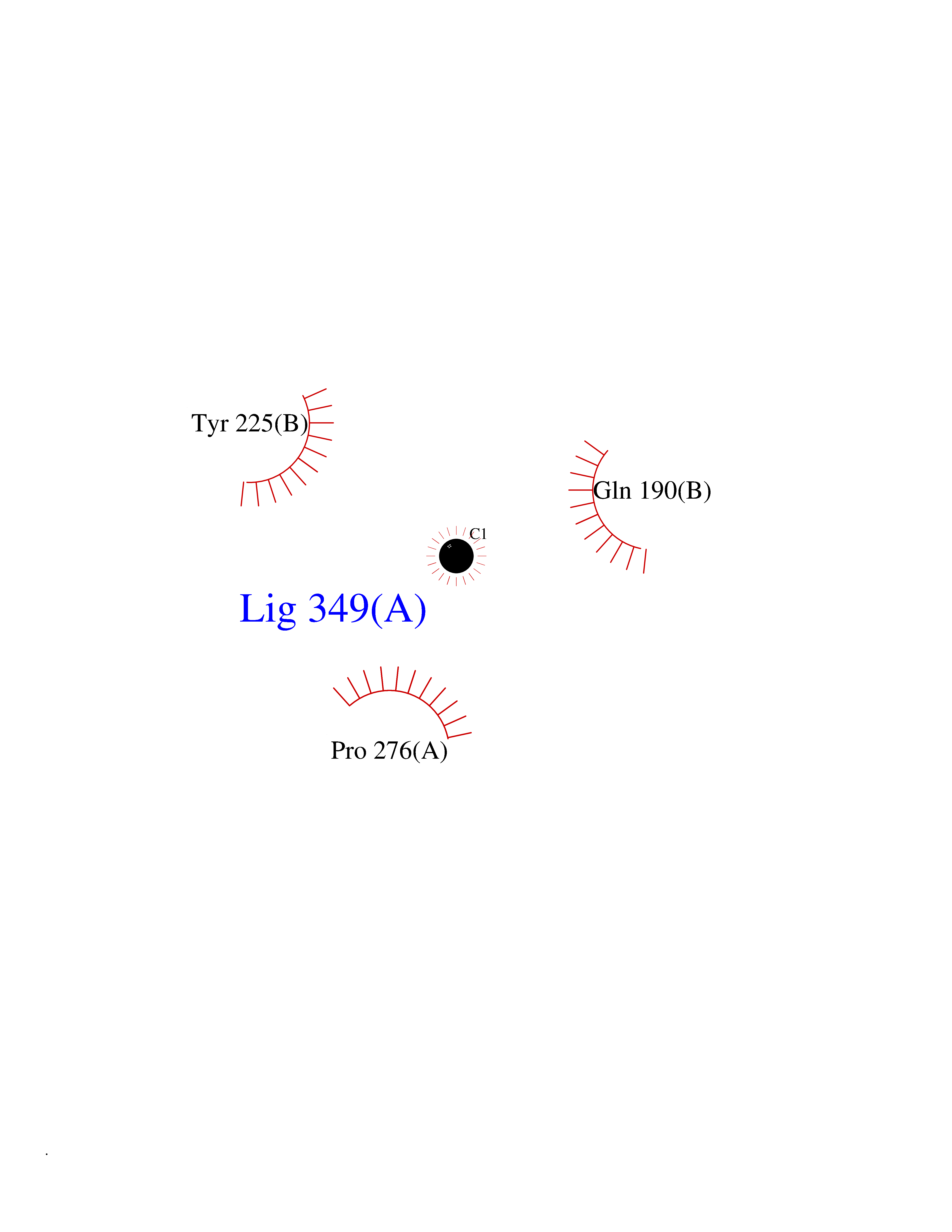 Ligplot Map 3D Binding mode Sequence SNMSLVKETVDRLLKGYDIRLRPDFGGPPVAVGMNIDIASIDMVSEVNMDYTLTMYFQQAWRDKRLSYNVIPLNLTLDNRVADQLWVPDTYFLNDKKSFVHGVTVKNRMIRLHPDGTVLYGLRITTTAACMMDLRRYPLDEQNCTLEIESYGYTTDDIEFYWRGDDNAVTGVTKIELPQFSIVDYKLITKKVVFSTGSYPRLSLSFKLKRNIGYFILQTYMPSILITILSWVSFWINYDASAARVALGITTVLTMTTINTHLRETLPKIPYVKAIDMYLMGCFVFVFMALLEYALVNYIFFSQPARAAAIDRWSRIFFPVVFSFFNIVYWLYYVDNTTVFTRILDRLLDGYDNRLRPGLGERVTEVKTDIFVTSFGPVSDHDMEYTIDVFFRQSWKDERLKFKGPMTVLRLNNLMASKIWTPDTFFHNGKKSVAHNMTMPNKLLRITEDGTLLYTMRLTVRAECPMHLEDFPMDAHACPLKFGSYAYTRAEVVYEWTREPARSVVVAEDGSRLNQYDLLGQTVDSGIVQSSTGEYVVMTTHFHLKRKIGYFVIQTYLPCIMTVILSQVSFWLNRESVPARTVFGVTTVLTMTTLSISARNSLPKVAYATAMDWFIAVCYAFVFSALIEFATVNYFTKSQPARAAKIDRLSRIAFPLLFGIFNLVYWATYLNR Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Activin receptor type IIB (ACVR2B) | 2QLU | 3.94 | |
Target general information Gen name ACVR2B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Activin receptor type2B; Activin receptor type-2B; ACTRIIB; ACTR-IIB Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family, TGFB receptor subfamily Biochemical class Kinase Function Transduces the activin signal from the cell surface to the cytoplasm and is thus regulating many physiological and pathological processes including neuronal differentiation and neuronal survival, hair follicle development and cycling, FSH production by the pituitary gland, wound healing, extracellular matrix production, immunosuppression and carcinogenesis. Activin is also thought to have a paracrine or autocrine role in follicular development in the ovary. Within the receptor complex, the type-2 receptors act as a primary activin receptors (binds activin-A/INHBA, activin-B/INHBB as well as inhibin-A/INHA-INHBA). The type-1 receptors like ACVR1B act as downstream transducers of activin signals. Activin binds to type-2 receptor at the plasma membrane and activates its serine-threonine kinase. The activated receptor type-2 then phosphorylates and activates the type-1 receptor. Once activated, the type-1 receptor binds and phosphorylates the SMAD proteins SMAD2 and SMAD3, on serine residues of the C-terminal tail. Soon after their association with the activin receptor and subsequent phosphorylation, SMAD2 and SMAD3 are released into the cytoplasm where they interact with the common partner SMAD4. This SMAD complex translocates into the nucleus where it mediates activin-induced transcription. Inhibitory SMAD7, which is recruited to ACVR1B through FKBP1A, can prevent the association of SMAD2 and SMAD3 with the activin receptor complex, thereby blocking the activin signal. Activin signal transduction is also antagonized by the binding to the receptor of inhibin-B via the IGSF1 inhibin coreceptor. Transmembrane serine/threonine kinase activin type-2 receptor forming an activin receptor complex with activin type-1 serine/threonine kinase receptors (ACVR1, ACVR1B or ACVR1c). Related diseases Heterotaxy, visceral, 4, autosomal (HTX4) [MIM:613751]: A form of visceral heterotaxy, a complex disorder due to disruption of the normal left-right asymmetry of the thoracoabdominal organs. Visceral heterotaxy or situs ambiguus results in randomization of the placement of visceral organs, including the heart, lungs, liver, spleen, and stomach. The organs are oriented randomly with respect to the left-right axis and with respect to one another. It can be associated with a variety of congenital defects including cardiac malformations. HTX4 clinical features include dextrocardia, right aortic arch and a right-sided spleen, anomalies of the inferior and the superior vena cava, atrial ventricular canal defect with dextro-transposed great arteries, pulmonary stenosis, polysplenia and midline liver. {ECO:0000269|PubMed:9916847}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O95390; O14793 EC number EC 2.7.11.30 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Heterotaxy; Kinase; Magnesium; Manganese; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Serine/threonine-protein kinase; Signal; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 33819.5 Length 297 Aromaticity 0.08 Instability index 49.31 Isoelectric point 5.97 Charge (pH=7) -6.19 2D Binding mode Binding energy (Kcal/mol) -5.37 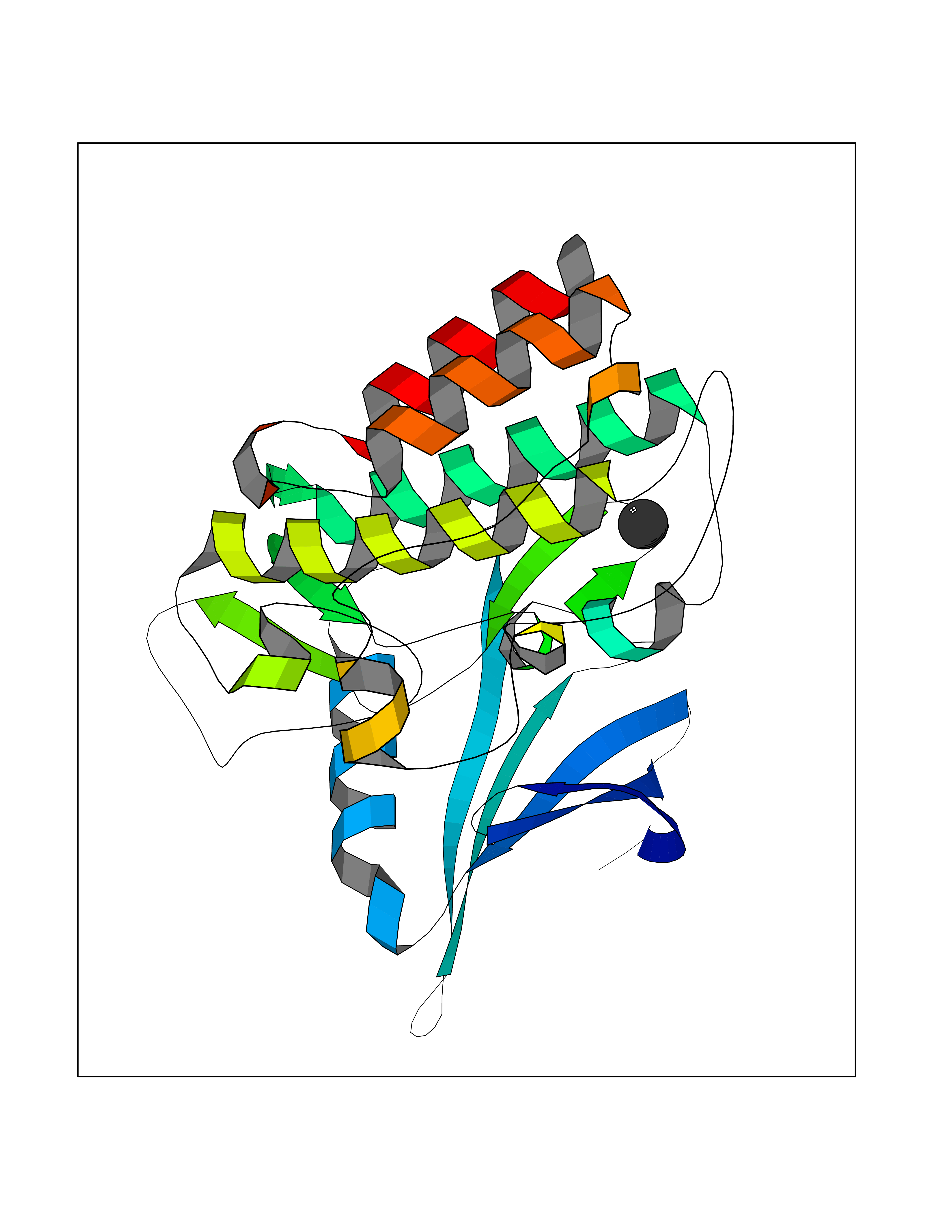 Molscript Map 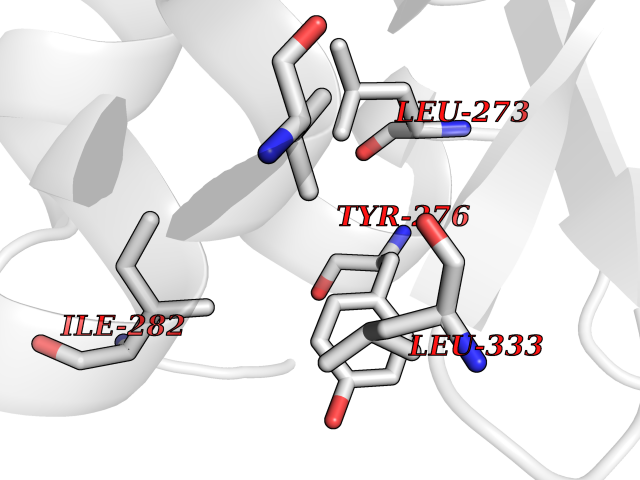 Pymol Map 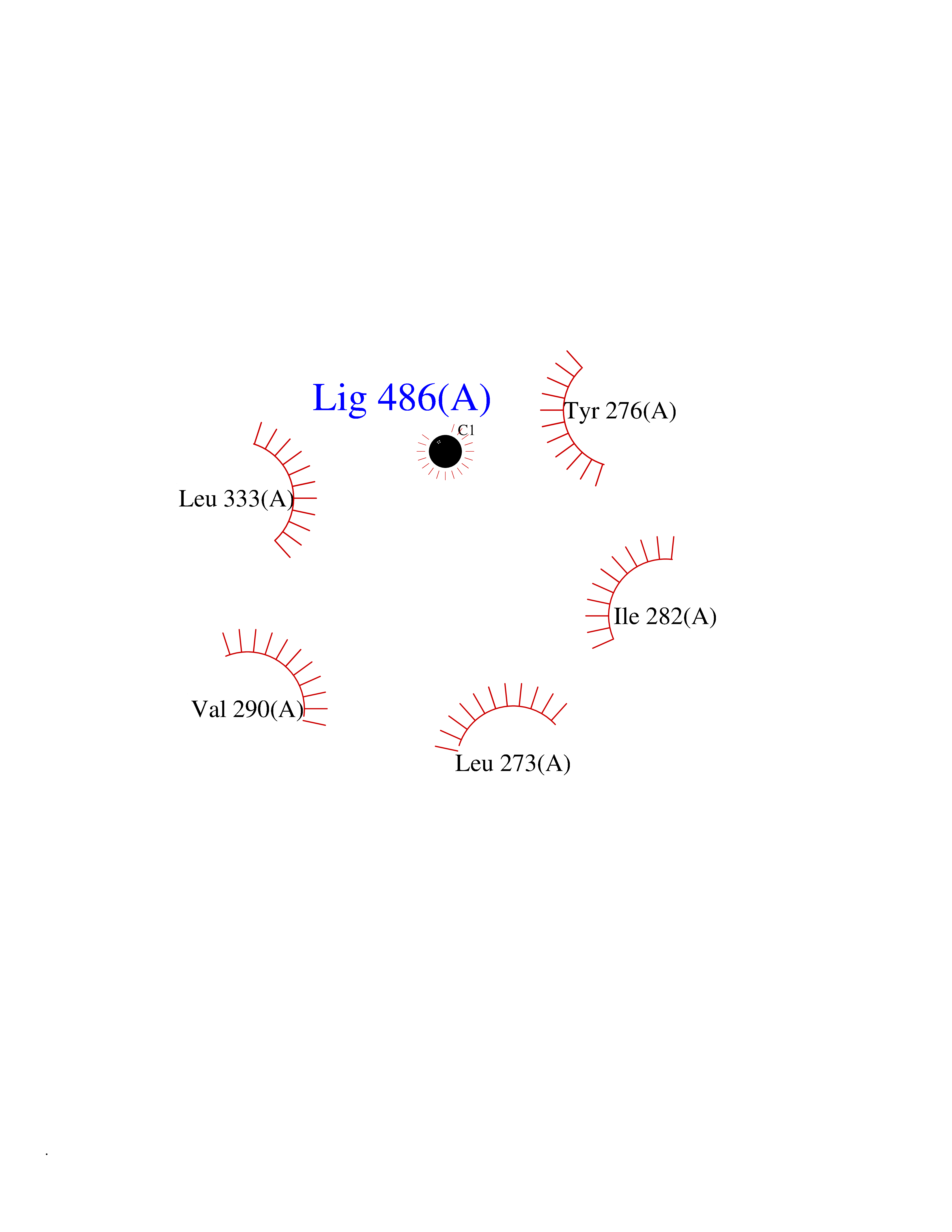 Ligplot Map 3D Binding mode Sequence GSLQLLEIKARGRFGCVWKAQLMNDFVAVKIFPLQDKQSWQSEREIFSTPGMKHENLLQFIAAEKRGSNLEVELWLITAFHDKGSLTDYLKGNIITWNELCHVAETMSRGLSYLHEDVPWCRGEGHKPSIAHRDFKSKNVLLKSDLTAVLADFGLAVRFEPGKPPGDTHGQVGTRRYMAPEVLEGAINFQRDAFLRIDMYAMGLVLWELVSRCKAADGPVDEYMLPFEEEIGQHPSLEELQEVVVHKKMRPTIKDHWLKHPGLAQLCVTIEECWDHDAEARLSAGCVEERVSLIRRS Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Neuronal acetylcholine receptor beta-2 (CHRNB2) | 6CNJ | 3.94 | |
Target general information Gen name CHRNB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor beta2; Nicotinic acetylcholine receptor beta 2-subunit protein; CHRNB2; Beta-2 nAChR; Alpha-4/beta-2 nicotinic receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-2/CHRNB2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane permeable to sodiun ions. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00572; DB00237; DB00565; DB09028; DB01245; DB00514; DB07720; DB00898; DB00753; DB00657; DB00333; DB00184; DB00981; DB05458; DB05855; DB05740; DB00747; DB00202; DB01273 Interacts with P43681-1; P30532 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Kynurenine oxoglutarate transaminase II (AADAT) | 2XH1 | 3.94 | |
Target general information Gen name AADAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Kynurenineoxoglutarate transaminase II; Kynurenineoxoglutarate transaminase 2; Kynurenineoxoglutarate aminotransferase II; Kynurenine/alphaaminoadipate aminotransferase, mitochondrial; Kynurenine amin Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Transaminase with broad substrate specificity. Has transaminase activity towards aminoadipate, kynurenine, methionine and glutamate. Shows activity also towards tryptophan, aspartate and hydroxykynurenine. Accepts a variety of oxo-acids as amino- group acceptors, with a preference for 2-oxoglutarate, 2- oxocaproic acid, phenylpyruvate and alpha-oxo-gamma-methiol butyric acid. Can also use glyoxylate as amino-group acceptor (in vitro). Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00142; DB00114 Interacts with NA EC number EC 2.6.1.39 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminotransferase; Mitochondrion; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A,B Molecular weight (Da) 91792.6 Length 824 Aromaticity 0.09 Instability index 47.71 Isoelectric point 5.96 Charge (pH=7) -8.73 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map 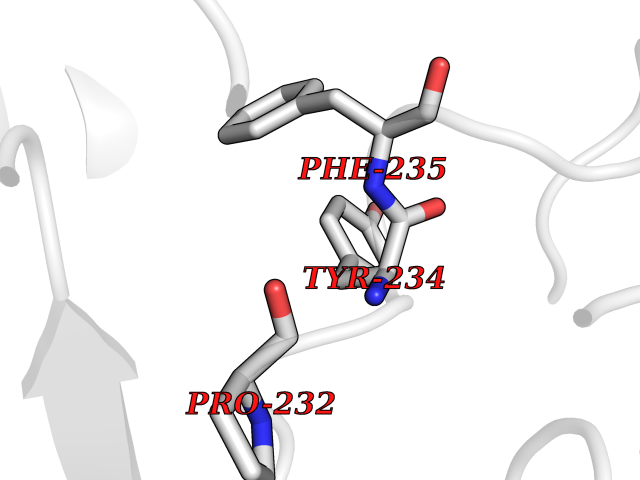 Pymol Map 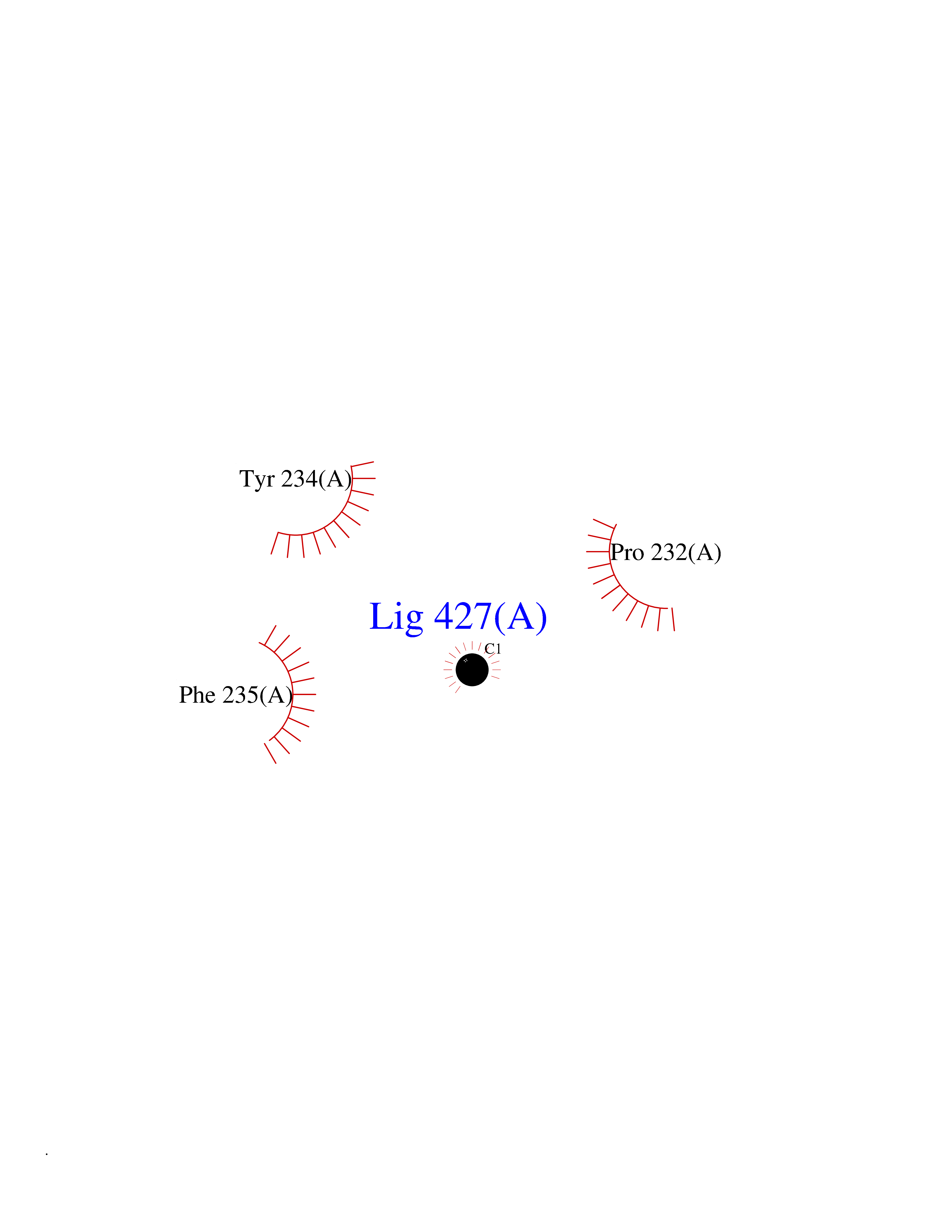 Ligplot Map 3D Binding mode Sequence ENYARFITAASAARNPSPIMISLAGGLPNPNMFPFKTAVITVENGKTIQFGEEMMKRALQYSPSAGIPELLSWLKQLQIKLHNPPTIHYPPSQGQMDLCVTSGSQQGLCKVFEMIINPGDNVLLDEPAYSGTLQSLHPLGCNIINVASDESGIVPDSLRDILSRWKPEDAKNPQKNTPKFLYTVPNGNNPTGNSLTSERKKEIYELARKYDFLIIEDDPYYFLQFNKFRVPTFLSMDVDGRVIRADSFSKIISSGLRIGFLTGPKPLIERVILHIQVSTLHPSTFNQLMISQLLHEWGEEGFMAHVDRVIDFYSNQKDAILAAADKWLTGLAEWHVPAAGMFLWIKVKGINDVKELIEEKAVKMGVLMLPGNAFYVDSSAPSPYLRASFSSASPEQMDVAFQVLAQLIKESLENYARFITAASAARNPSPIMISLAGGLPNPNMFPFKTAVITVENGKTIQFGEEMMKRALQYSPSAGIPELLSWLKQLQIKLHNPPTIHYPPSQGQMDLCVTSGSQQGLCKVFEMIINPGDNVLLDEPAYSGTLQSLHPLGCNIINVASDESGIVPDSLRDILSRWKPEDAKNPQKNTPKFLYTVPNGNNPTGNSLTSERKKEIYELARKYDFLIIEDDPYYFLQFNKFRVPTFLSMDVDGRVIRADSFSKIISSGLRIGFLTGPKPLIERVILHIQVSTLHPSTFNQLMISQLLHEWGEEGFMAHVDRVIDFYSNQKDAILAAADKWLTGLAEWHVPAAGMFLWIKVKGINDVKELIEEKAVKMGVLMLPGNAFYVDSSAPSPYLRASFSSASPEQMDVAFQVLAQLIKESL Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Urea transporter 1 (SLC14A1) | 6QD5 | 3.94 | |
Target general information Gen name SLC14A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Urea transporter, erythrocyte; UTE; UT1; Solute carrier family 14 member 1; RACH1; JK; HUT11 Protein family Urea transporter family Biochemical class Urea transporter family Function Urea channel that facilitates transmembrane urea transport down a concentration gradient. A constriction of the transmembrane channel functions as selectivity filter through which urea is expected to pass in dehydrated form. The rate of urea conduction is increased by hypotonic stress. Plays an important role in the kidney medulla collecting ducts, where it allows rapid equilibration between the lumen of the collecting ducts and the interstitium, and thereby prevents water loss driven by the high concentration of urea in the urine. Facilitates urea transport across erythrocyte membranes. May also play a role in transmembrane water transport, possibly by indirect means. Related diseases Immunodeficiency 12 (IMD12) [MIM:615468]: A primary immunodeficiency characterized by onset in infancy of recurrent bacterial and candidal infections resulting in bronchiectasis and growth delay. Manifestations include mastoiditis, aphthous ulcers, cheilitis, gingivitis, esophagitis, gastritis, duodenitis, and meningitis. Levels of absolute lymphocytes and serum immunoglobulins are normal, but specific antibody titers are low despite immunization, and T-cells show impaired proliferative responses to mitogens. {ECO:0000269|PubMed:23727036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A chromosomal aberration involving MALT1 is recurrent in low-grade mucosa-associated lymphoid tissue (MALT lymphoma). Translocation t(11;18)(q21;q21) with BIRC2. This translocation is found in approximately 50% of cytogenetically abnormal low-grade MALT lymphoma. {ECO:0000269|PubMed:10339464, ECO:0000269|PubMed:10523859, ECO:0000269|PubMed:10702396, ECO:0000269|PubMed:11090634}. Drugs (DrugBank ID) DB01005; DB03904 Interacts with Q8WVV5; Q9Y3D6; Q8WWP7; P30301; Q5QGT7; Q6UX34; P0DN84; Q9C0I4; Q5BJF2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood group antigen; Cell membrane; Glycoprotein; Membrane; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 38862.8 Length 356 Aromaticity 0.12 Instability index 40.98 Isoelectric point 7.67 Charge (pH=7) 0.95 2D Binding mode Binding energy (Kcal/mol) -5.38 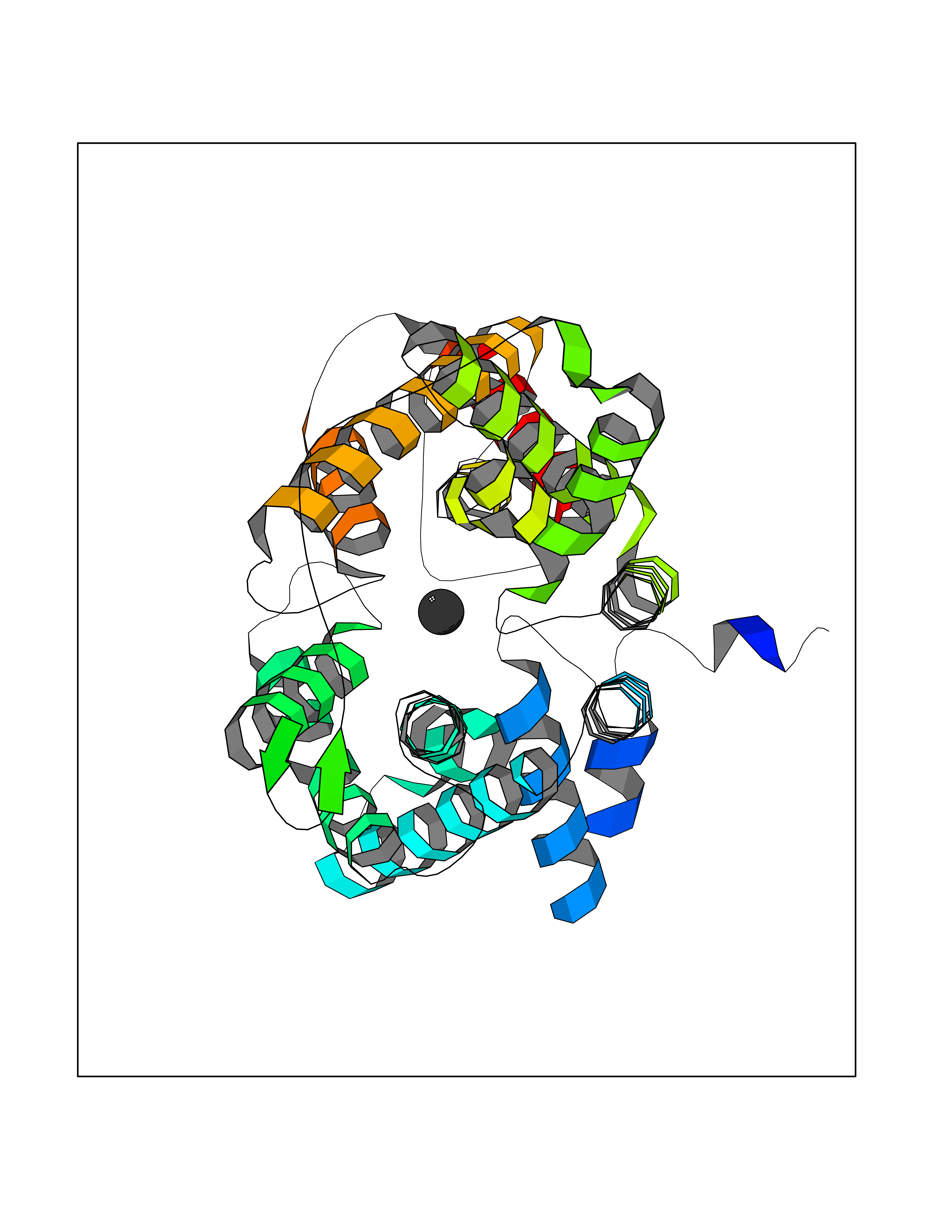 Molscript Map 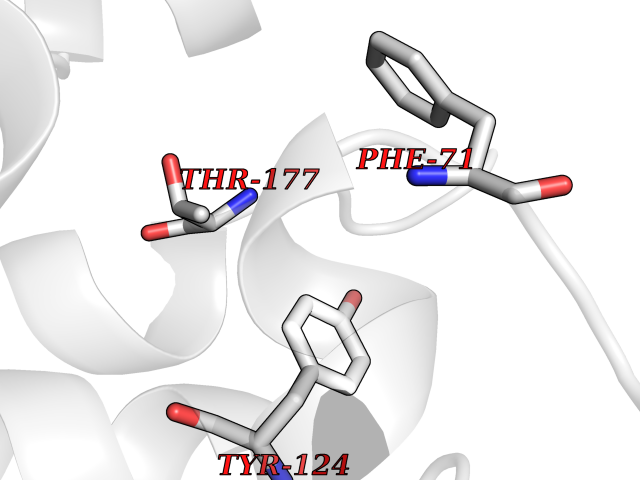 Pymol Map  Ligplot Map 3D Binding mode Sequence FPKALGYVTGDMKELANQLKDKPVVLQFIDWILRGISQVVFVNNPVSGILILVGLLVQNPWWALTGWLGTVVSTLMALLLSQDRSLIASGLYGYNATLVGVLMAVFSDKGDYFWWLLLPVCAMSMTCPIFSSALNSVLSKWDLPVFTLPFNMALSMYLSATGHYNPFFPAKLVIPITTAPQISWSDLSALELLKSIPVGVGQIYGCDNPWTGGIFLGAILLSSPLMCLHAAIGSLLGIAAGLSLSAPFENIYFGLWGFNSSLACIAMGGMFMALTWQTHLLALGCALFTAYLGVGMANFMAEVGLPACTWPFCLATLLFLIMTTKNSNIYKMPLSKVTYPEENRIFYLQAKKRMVE Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Melatonin receptor type 1A (MTNR1A) | 7DB6 | 3.94 | |
Target general information Gen name MTNR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mel1a receptor; Mel1AR; Mel-1A-R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Likely to mediate the reproductive and circadian actions of melatonin. The activity of this receptor is mediated by pertussis toxin sensitive G proteins that inhibit adenylate cyclase activity. High affinity receptor for melatonin. Related diseases Spermatogenic failure 5 (SPGF5) [MIM:243060]: An infertility disorder caused by spermatogenesis defects. Semen from affected men show close to 100% morphologically abnormal multiflagellar spermatozoa with low motility, oversized irregular heads, and abnormal midpiece and acrosome. {ECO:0000269|PubMed:17435757, ECO:0000269|PubMed:21733974}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06594; DB01065; DB00980; DB02709; DB09071 Interacts with P27797; A8MQ03; Q8IUG1; P49286; O76081; P57088 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID D Molecular weight (Da) 31301 Length 276 Aromaticity 0.15 Instability index 37.33 Isoelectric point 9.22 Charge (pH=7) 9.92 2D Binding mode Binding energy (Kcal/mol) -5.38  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RPSWLASALACVLIFTIVVDILGNLLVILSVYRNKKLRNAGNIFVVSLAVADLVVAIYPYPLVLMSIFNNGWNLGYLHCQVSGFLMGLSVIGSIFNITGIAINRYCYICHSLKYDKLYSSKNSLCYVLLIWLLTLAAVLPNLRAGTLQYDPRIYSCTFAQSVSSAYTIAVVVFHFLVPMIIVIFCYLRIWILVLQVRQRVPQDFRNFVTMFVVFVLFAICWAPLNFIGLAVASDPASMVPRIPEWLFVASYYMAYFNSCLNAIIYGLLNQNFRKEY Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Cerebroside-sulfatase (ARSA) | 1E2S | 3.94 | |
Target general information Gen name ARSA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cerebrosidesulfatase; Arylsulfatase A component C; ASA; ARSA Protein family Sulfatase family Biochemical class Sulfuric ester hydrolase Function Hydrolyzes cerebroside sulfate. Related diseases Metachromatic leukodystrophy (MLD) [MIM:250100]: An autosomal recessive disease caused by abnormal intralysosomal accumulation of cerebroside-3-sulfate in central and peripheral nervous systems, as well as other organs. MLD is clinically characterized by leukodystrophy, progressive demyelination and a variety of neurological symptoms, including gait disturbances, ataxias, optical atrophy, dementia, seizures, and spastic tetraparesis. Decreased arylsulfatase A activity is detected in urine, leukocytes, and fibroblasts of affected individuals. Several forms of the disease can be distinguished according to the age at onset and disease severity: late infantile, juvenile and adult forms, partial cerebroside sulfate deficiency, and pseudoarylsulfatase A deficiency. Individuals with pseudoarylsulfatase A deficiency have low arylsulfatase A activity but lack neurological manifestations and are apparently healthy. {ECO:0000269|PubMed:10220151, ECO:0000269|PubMed:10381328, ECO:0000269|PubMed:10477432, ECO:0000269|PubMed:10533072, ECO:0000269|PubMed:10751093, ECO:0000269|PubMed:11020646, ECO:0000269|PubMed:11061266, ECO:0000269|PubMed:11456299, ECO:0000269|PubMed:11941485, ECO:0000269|PubMed:12503099, ECO:0000269|PubMed:12788103, ECO:0000269|PubMed:1353340, ECO:0000269|PubMed:14517960, ECO:0000269|PubMed:14680985, ECO:0000269|PubMed:15026521, ECO:0000269|PubMed:15326627, ECO:0000269|PubMed:15710861, ECO:0000269|PubMed:1670590, ECO:0000269|PubMed:1673291, ECO:0000269|PubMed:1678251, ECO:0000269|PubMed:18693274, ECO:0000269|PubMed:19606494, ECO:0000269|PubMed:20339381, ECO:0000269|PubMed:21265945, ECO:0000269|PubMed:2574462, ECO:0000269|PubMed:7581401, ECO:0000269|PubMed:7825603, ECO:0000269|PubMed:7860068, ECO:0000269|PubMed:7902317, ECO:0000269|PubMed:7906588, ECO:0000269|PubMed:7909527, ECO:0000269|PubMed:8095918, ECO:0000269|PubMed:8101038, ECO:0000269|PubMed:8101083, ECO:0000269|PubMed:8104633, ECO:0000269|PubMed:8891236, ECO:0000269|PubMed:9090526, ECO:0000269|PubMed:9272717, ECO:0000269|PubMed:9452102, ECO:0000269|PubMed:9490297, ECO:0000269|PubMed:9600244, ECO:0000269|PubMed:9819708}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Multiple sulfatase deficiency (MSD) [MIM:272200]: A clinically and biochemically heterogeneous disorder caused by the simultaneous impairment of all sulfatases, due to defective post-translational modification and activation. It combines features of individual sulfatase deficiencies such as metachromatic leukodystrophy, mucopolysaccharidosis, chondrodysplasia punctata, hydrocephalus, ichthyosis, neurologic deterioration and developmental delay. {ECO:0000269|PubMed:15146462}. The protein represented in this entry is involved in disease pathogenesis. Arylsulfatase A activity is impaired in multiple sulfatase deficiency due to mutations in SUMF1 (PubMed:15146462). SUMF1 mutations result in defective post-translational modification of ARSA at residue Cys-69 that is not converted to 3-oxoalanine (PubMed:7628016). {ECO:0000269|PubMed:15146462, ECO:0000269|PubMed:7628016}. Drugs (DrugBank ID) DB03821; DB01800; DB01141; DB04786 Interacts with P50995; Q6P5X5; Q13554-3; O60826; Q96D98; Q9H0I2; Q12951-2; Q16512; P28069; O75360; Q9BQY4; Q15645; O95231 EC number EC 3.1.6.8 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Hydrolase; Ichthyosis; Leukodystrophy; Lipid metabolism; Lysosome; Metachromatic leukodystrophy; Metal-binding; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID P Molecular weight (Da) 51173.7 Length 481 Aromaticity 0.09 Instability index 47.42 Isoelectric point 5.59 Charge (pH=7) -12.84 2D Binding mode Binding energy (Kcal/mol) -5.38 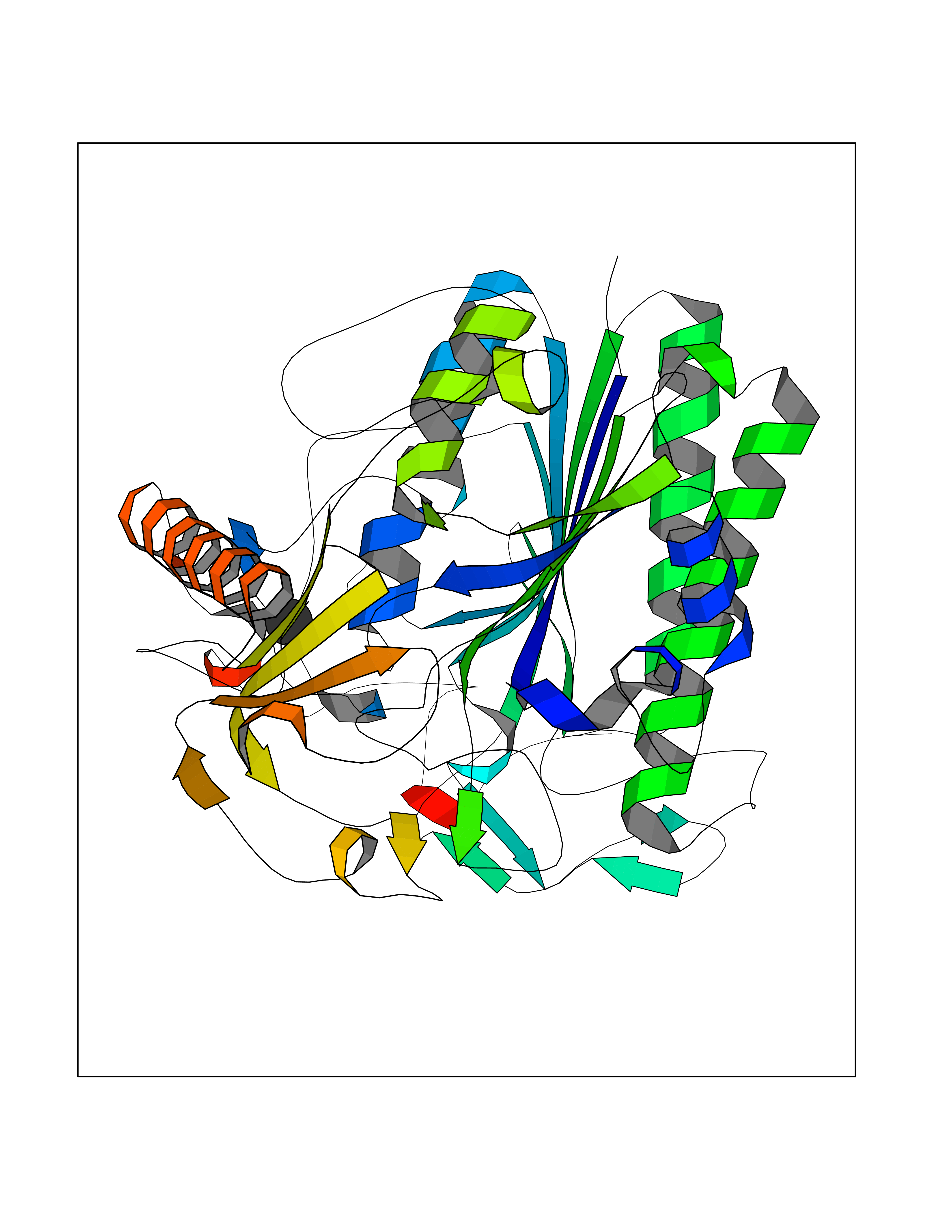 Molscript Map 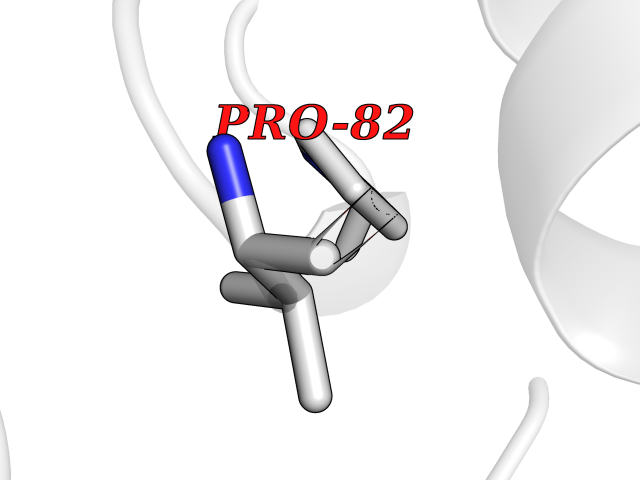 Pymol Map 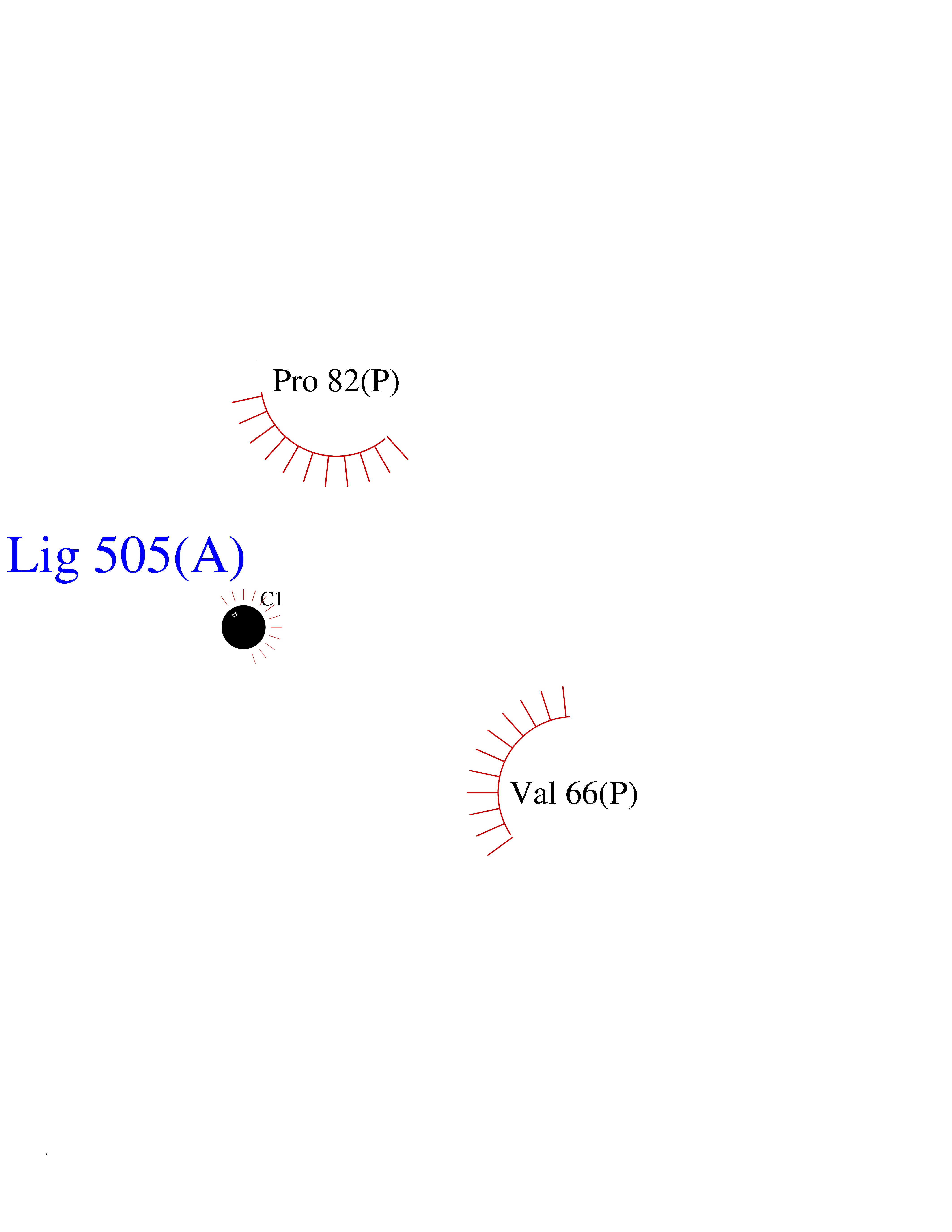 Ligplot Map 3D Binding mode Sequence RPPNIVLIFADDLGYGDLGCYGHPSSTTPNLDQLAAGGLRFTDFYVPVSLATPSRAALLTGRLPVRMGMYPGVLVPSSRGGLPLEEVTVAEVLAARGYLTGMAGKWHLGVGPEGAFLPPHQGFHRFLGIPYSHDQGPCQNLTCFPPATPCDGGCDQGLVPIPLLANLSVEAQPPWLPGLEARYMAFAHDLMADAQRQDRPFFLYYASHHTHYPQFSGQSFAERSGRGPFGDSLMELDAAVGTLMTAIGDLGLLEETLVIFTADNGPETMRMSRGGCSGLLRCGKGTTYEGGVREPALAFWPGHIAPGVTHELASSLDLLPTLAALAGAPLPNVTLDGFDLSPLLLGTGKSPRQSLFFYPSYPDEVRGVFAVRTGKYKAHFFTQGSAHSDTTADPACHASSSLTAHEPPLLYDLSKDPGENYNLLGATPEVLQALKQLQLLKAQLDAAVTFGPSQVARGEDPALQICCHPGCTPRPACCHCP Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Two pore calcium channel protein 2 (TPC2) | 6NQ2 | 3.94 | |
Target general information Gen name TPCN2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Voltage-dependent calcium channel protein TPC2; TPC2 Protein family Calcium channel alpha-1 subunit (TC 1.A.1.11) family, Two pore calcium channel subfamily Biochemical class NA Function Nicotinic acid adenine dinucleotide phosphate (NAADP) receptor that may function as one of the major voltage-gated Ca(2+) channels (VDCC) across the lysosomal membrane. May be involved in smooth muscle contraction. Related diseases Spermatogenic failure 5 (SPGF5) [MIM:243060]: An infertility disorder caused by spermatogenesis defects. Semen from affected men show close to 100% morphologically abnormal multiflagellar spermatozoa with low motility, oversized irregular heads, and abnormal midpiece and acrosome. {ECO:0000269|PubMed:17435757, ECO:0000269|PubMed:21733974}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O00165; P42345; Q9ULQ1; Q8NHX9 EC number NA Uniprot keywords 3D-structure; Calcium; Calcium channel; Calcium transport; Endosome; Glycoprotein; Ion channel; Ion transport; Lysosome; Membrane; Proteomics identification; Reference proteome; Repeat; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 71128.5 Length 621 Aromaticity 0.14 Instability index 35.3 Isoelectric point 8.84 Charge (pH=7) 8.43 2D Binding mode Binding energy (Kcal/mol) -5.37 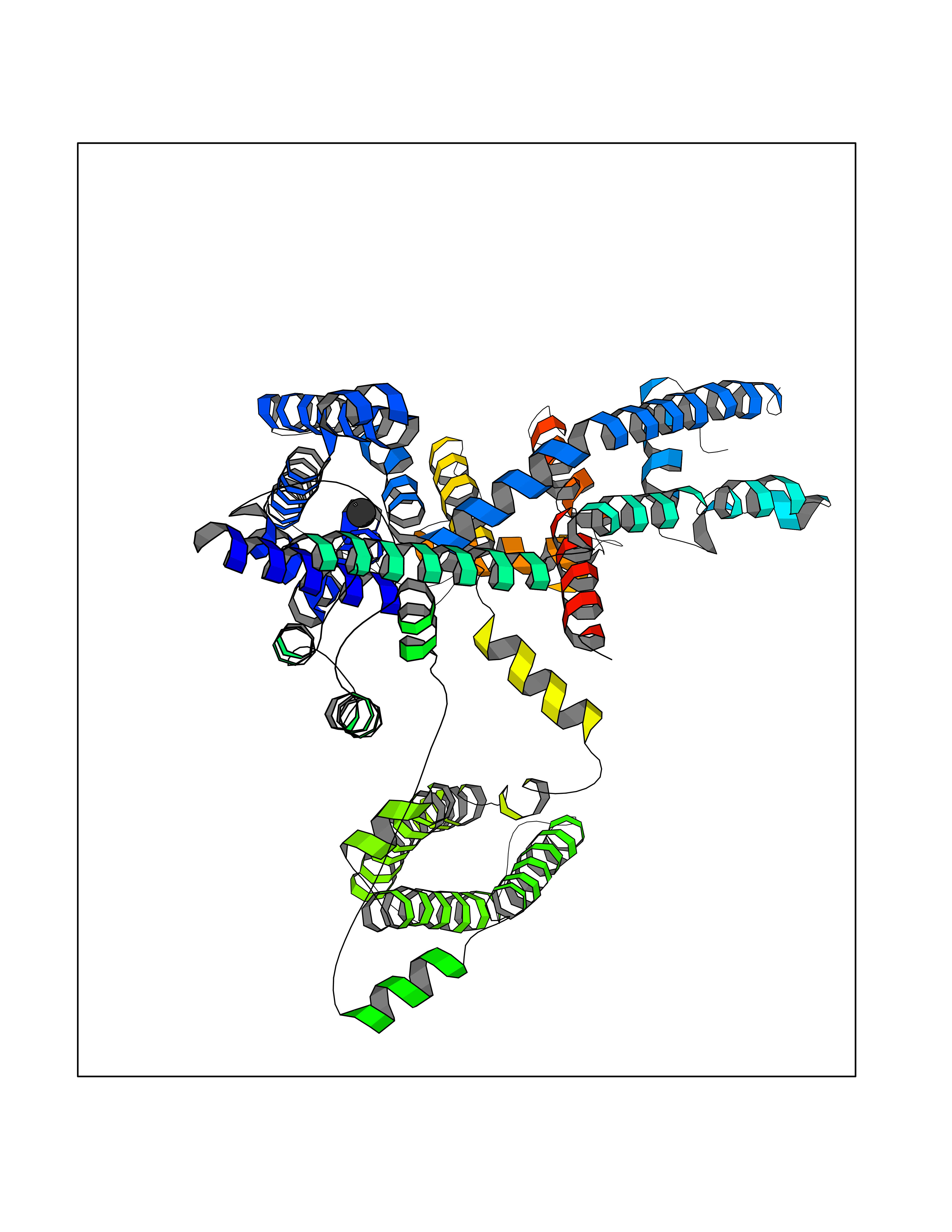 Molscript Map 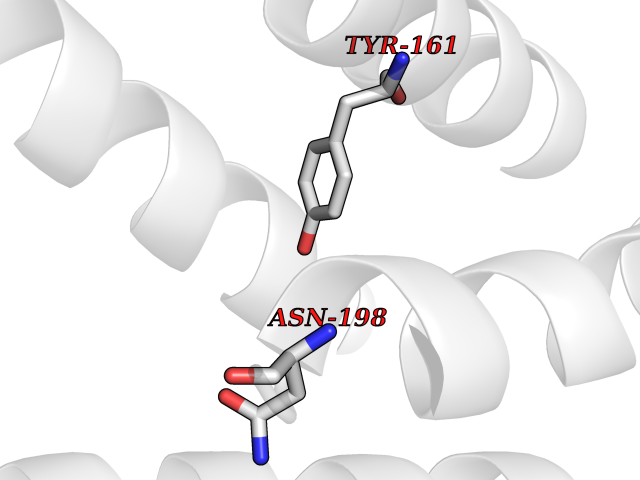 Pymol Map  Ligplot Map 3D Binding mode Sequence AARWDLCIDQAVVFIEDAIQYRSINHRVDASSMWLYRRYYSNVCQRTLSFTIFLILFLAFIETPSSLTSTADVRYRAAPWEPPCGLTESVEVLCLLVFAADLSVKGYLFGWAHFQKNLWLLGYLVVLVVSLVDWTVSLSLVCHEPLRIRRLLRPFFLLQNSSMMKKTLKCIRWSLPEMASVGLLLAIHLCLFTMFGMLLFAGLTYFQNLPESLTSLLVLLTTANNPDVMIPAYSKNRAYAIFFIVFTVIGSLFLMNLLTAIIYSQFRGYLMKSLQTSLFRRRLGTRAAFEVLSSMVGAVGVKPQNLLQVLQKVQLDSSHKQAMMEKVRSYGSVLLSAEEFQKLFNELDRSVVKEHPPRPEYQSPFLQSAQFLFGHYYFDYLGNLIALANLVSICVFLVLDADVLPAERDDFILGILNCVFIVYYLLEMLLKVFALGLRGYLSYPSNVFDGLLTVVLLVLEISTLAVYRLSLWDMTRMLNMLIVFRFLRIIPSMKPMAVVASTVLGLVQNMRAFGGILVVVYYVFAIIGINLFRGVIVALSAPCGSFEQLEYWANNFDDFAAALVTLWNLMVVNNWQVFLDAYRRYSGPWSKIYFVLWWLVSSVIWVNLFLALILENFLHKW Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Dopamine beta-hydroxylase | 4ZEL | 3.94 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -5.38  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | N(G),N(G)-dimethylarginine dimethylaminohydrolase 1 | 3I4A | 3.94 | |
Target general information Gen name DDAH1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms DDAH Protein family DDAH family Biochemical class Hydrolase Function Amino acid binding.Catalytic activity.Dimethylargininase activity.Metal ion binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00155; DB05351; DB00736; DB00213 Interacts with NA EC number 3.5.3.18 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; S-nitrosylation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 30156.3 Length 275 Aromaticity 0.05 Instability index 38.25 Isoelectric point 5.56 Charge (pH=7) -7.51 2D Binding mode Binding energy (Kcal/mol) -5.38  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence AAFGRATHAVVRALPESLGQHALRSAKGEEVDVARAERQHQLYVGVLGSKLGLQVVELPADESLPDCVFVEDVAVVCEETALITRPGAPSRRKEVDMMKEALEKLQLNIVEMKDENATLDGGDVLFTGREFFVGLSKRTNQRGAEILADTFKDYAVSTVPVADGLHLKSFCSMAGPNLIAIGSSESAQKALKIMQQMSDHRYDKLTVPDDIAANCIYLNIPNKGHVLLHRTPEEYPESAKVYEKLKDHMLIPVSMSELEKVDGLLTCCSVLINKK Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Hypoxanthine-guanine phosphoribosyltransferase | 5HIA | 3.94 | |
Target general information Gen name HPRT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HPRT Protein family Purine/pyrimidine phosphoribosyltransferase family Biochemical class Transferase Function Guanine phosphoribosyltransferase activity.Hypoxanthine phosphoribosyltransferase activity.Identical protein binding.Magnesium ion binding.Nucleotide binding.Protein homodimerization activity. Related diseases Lesch-Nyhan syndrome (LNS) [MIM:300322]: Characterized by complete lack of enzymatic activity that results in hyperuricemia, choreoathetosis, intellectual disability, and compulsive self-mutilation. {ECO:0000269|PubMed:15571223, ECO:0000269|PubMed:17027311, ECO:0000269|PubMed:20544509, ECO:0000269|PubMed:2071157, ECO:0000269|PubMed:2246854, ECO:0000269|PubMed:2347587, ECO:0000269|PubMed:2358296, ECO:0000269|PubMed:24940672, ECO:0000269|PubMed:2572141, ECO:0000269|PubMed:2910902, ECO:0000269|PubMed:3265398, ECO:0000269|PubMed:3384338, ECO:0000269|PubMed:6853716, ECO:0000269|PubMed:7627191, ECO:0000269|PubMed:9452051}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperuricemia, HPRT-related (HRH) [MIM:300323]: An X-linked metabolic disorder characterized by uric acid excess in the blood, renal stones, uric acid nephropathy, and renal obstruction. After puberty, the hyperuricemia may cause gout. {ECO:0000269|PubMed:15571223, ECO:0000269|PubMed:17027311, ECO:0000269|PubMed:20544509, ECO:0000269|PubMed:24940672, ECO:0000269|PubMed:2896620, ECO:0000269|PubMed:2909537, ECO:0000269|PubMed:3198771, ECO:0000269|PubMed:3358423, ECO:0000269|PubMed:6572373, ECO:0000269|PubMed:6706936, ECO:0000269|PubMed:6853490, ECO:0000269|PubMed:7987318}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03153; DB02309; DB01632; DB04356; DB00993; DB01033; DB00352 Interacts with Q96G04; Q8N7B9-2; Q14749; P00492; P42858; Q9H1K1; Q96HA8; Q9NRG1; O00560; Q9NZD8; Q7Z699; Q8N9Q2 EC number 2.4.2.8 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Gout; Isopeptide bond; Magnesium; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 23099.5 Length 205 Aromaticity 0.09 Instability index 18.85 Isoelectric point 6.2 Charge (pH=7) -2.09 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SPGVVISDDEPGYDLDLFAIPNHYAEDLERVFIPHGLIMDRTERLARDVMKEMGGHHIVALCVLKGGYKFFADLLDYIKALNRNSDRSIPMTVDFIRLKDIKVIGGDDLSTLTGKNVLIVEDIIDTGKTMQTLLSLVRQYNPKMVKVASLLVKRTPRSVGYKPDFVGFEIPDKFVVGYALDYNEYFRDLNHVAVISETGKAKYKA Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Tyrosine 3-monooxygenase (TH) | 2XSN | 3.94 | |
Target general information Gen name TH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine 3-hydroxylase; TH Protein family Biopterin-dependent aromatic amino acid hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Plays an important role in the physiology of adrenergic neurones. Related diseases Segawa syndrome autosomal recessive (ARSEGS) [MIM:605407]: A form of DOPA-responsive dystonia presenting in infancy or early childhood. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Some cases present with parkinsonian symptoms in infancy. Unlike all other forms of dystonia, it is an eminently treatable condition, due to a favorable response to L-DOPA. {ECO:0000269|PubMed:10585338, ECO:0000269|PubMed:11196107, ECO:0000269|PubMed:11246459, ECO:0000269|PubMed:15505183, ECO:0000269|PubMed:15747353, ECO:0000269|PubMed:16049992, ECO:0000269|PubMed:17696123, ECO:0000269|PubMed:18058633, ECO:0000269|PubMed:18554280, ECO:0000269|PubMed:19491146, ECO:0000269|PubMed:20056467, ECO:0000269|PubMed:20430833, ECO:0000269|PubMed:21940685, ECO:0000269|PubMed:22264700, ECO:0000269|PubMed:22815559, ECO:0000269|PubMed:23762320, ECO:0000269|PubMed:23939262, ECO:0000269|PubMed:24753243, ECO:0000269|PubMed:7814018, ECO:0000269|PubMed:8528210, ECO:0000269|PubMed:8817341, ECO:0000269|PubMed:9613851, ECO:0000269|PubMed:9703425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: May play a role in the pathogenesis of Parkinson disease (PD). A genome-wide copy number variation analysis has identified a 34 kilobase deletion over the TH gene in a PD patient but not in any controls. {ECO:0000269|PubMed:20809526}. Drugs (DrugBank ID) DB03552; DB04400; DB00765; DB00120; DB00360; DB00135 Interacts with P29762; P61978-2; Q99750; P08651-5; O75928-2; Q9UHX1-2; P0DJD3-2; P07101-3; Q9UJ04; C9J7I0; Q5MCW4 EC number EC 1.14.16.2 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Cell projection; Cytoplasm; Cytoplasmic vesicle; Disease variant; Dystonia; Iron; Metal-binding; Monooxygenase; Neurotransmitter biosynthesis; Nucleus; Oxidoreductase; Parkinson disease; Parkinsonism; Phosphoprotein; Proteomics identification; Reference proteome; Synapse Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 34997 Length 306 Aromaticity 0.12 Instability index 42.59 Isoelectric point 5.32 Charge (pH=7) -12.31 2D Binding mode Binding energy (Kcal/mol) -5.38  Molscript Map 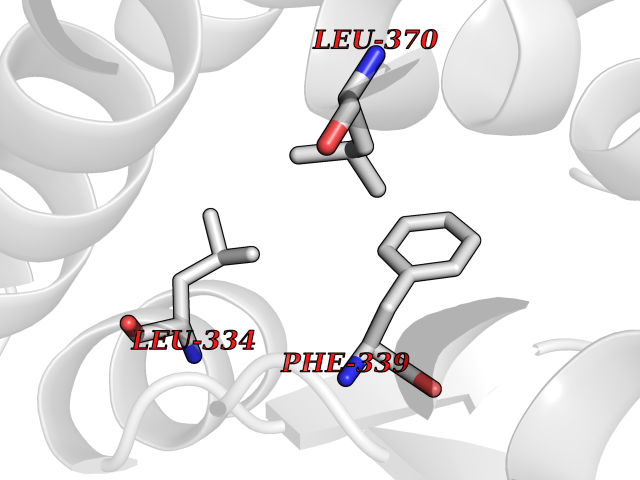 Pymol Map 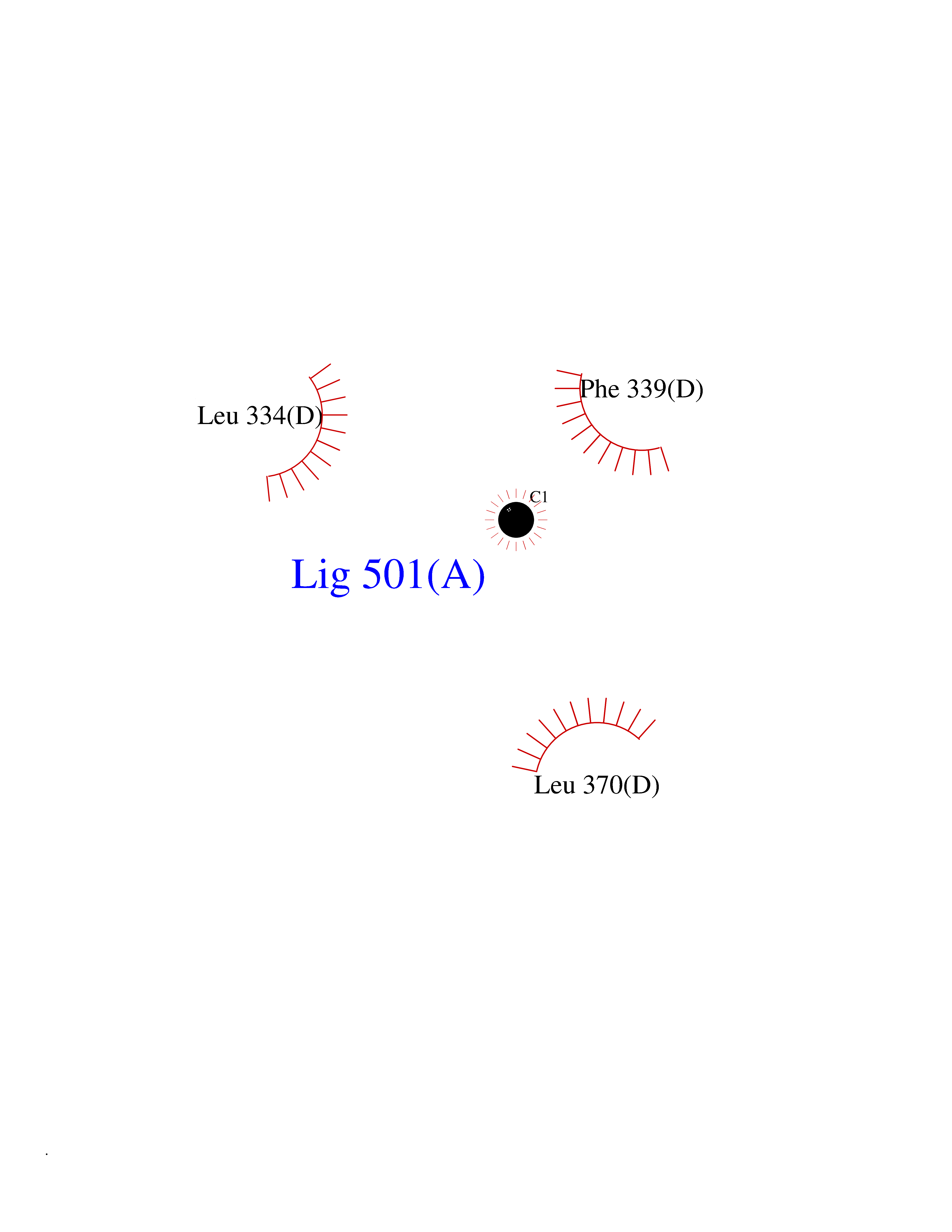 Ligplot Map 3D Binding mode Sequence VPWFPRKVSELDKCHHLVTKFDPDLDLDHPGFSDQVYRQRRKLIAEIAFQYRHGDPIPRVEYTAEEIATWKEVYTTLKGLYATHACGEHLEAFALLERFSGYREDNIPQLEDVSRFLKERTGFQLRPVAGLLSARDFLASLAFRVFQCTQYIRHASSPMHSPEPDCCHELLGHVPMLADRTFAQFSQDIGLASLGASDEEIEKLSTLYWFTVEFGLCKQNGEVKAYGAGLLSSYGELLHCLSEEPEIRAFDPEAAAVQPYQDQTYQSVYFVSESFSDAKDKLRSYASRIQRPFSVKFDPYTLAIDV Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Pyruvate dehydrogenase [ubiquinone] | 3EYA | 3.94 | |
Target general information Gen name poxB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b0871;JW0855 Protein family TPP enzyme family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Identical protein binding.Lipid binding.Magnesium ion binding.Pyruvate dehydrogenase (quinone) activity.Thiamine pyrophosphate binding. Related diseases Glycogen storage disease 6 (GSD6) [MIM:232700]: A metabolic disorder characterized by mild to moderate hypoglycemia, mild ketosis, growth retardation, and prominent hepatomegaly. Heart and skeletal muscle are not affected. {ECO:0000269|PubMed:9529348}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07003 EC number 1.2.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Lipid-binding; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Oxidoreductase; Pyruvate; Reference proteome; Thiamine pyrophosphate; Ubiquinone Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I,J,K,L Molecular weight (Da) 113027 Length 1046 Aromaticity 0.07 Instability index 35.99 Isoelectric point 5.75 Charge (pH=7) -24.38 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map 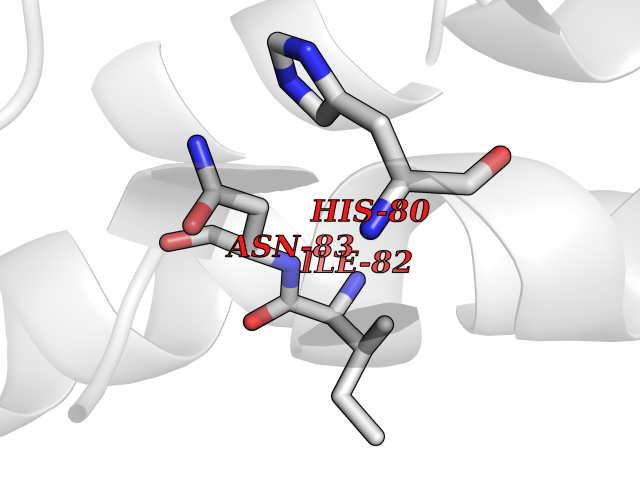 Pymol Map 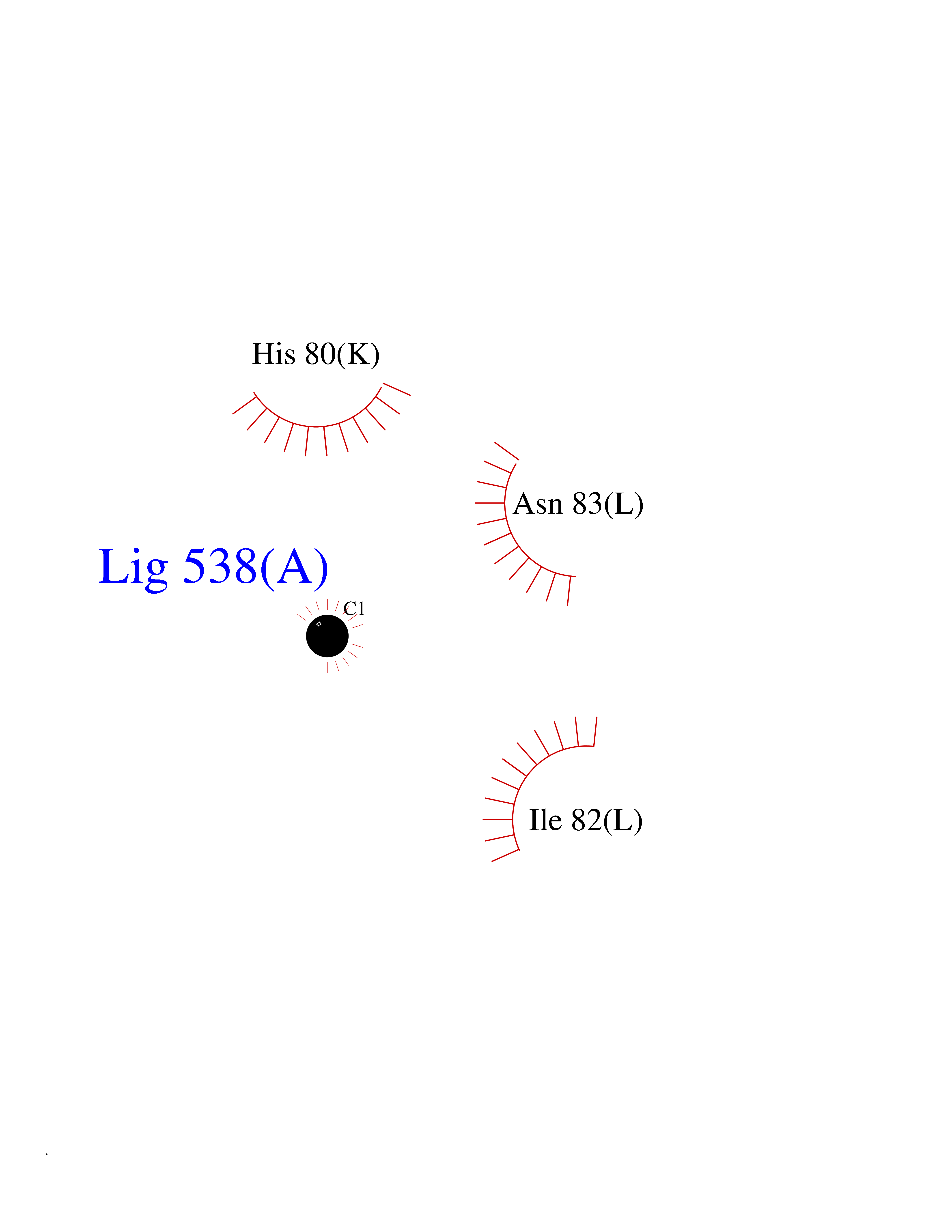 Ligplot Map 3D Binding mode Sequence MKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFDGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIPMKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFVGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIP Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Tyrosine 3-monooxygenase (TH) | 2XSN | 3.94 | |
Target general information Gen name TH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine 3-hydroxylase; TH Protein family Biopterin-dependent aromatic amino acid hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Plays an important role in the physiology of adrenergic neurones. Related diseases Segawa syndrome autosomal recessive (ARSEGS) [MIM:605407]: A form of DOPA-responsive dystonia presenting in infancy or early childhood. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Some cases present with parkinsonian symptoms in infancy. Unlike all other forms of dystonia, it is an eminently treatable condition, due to a favorable response to L-DOPA. {ECO:0000269|PubMed:10585338, ECO:0000269|PubMed:11196107, ECO:0000269|PubMed:11246459, ECO:0000269|PubMed:15505183, ECO:0000269|PubMed:15747353, ECO:0000269|PubMed:16049992, ECO:0000269|PubMed:17696123, ECO:0000269|PubMed:18058633, ECO:0000269|PubMed:18554280, ECO:0000269|PubMed:19491146, ECO:0000269|PubMed:20056467, ECO:0000269|PubMed:20430833, ECO:0000269|PubMed:21940685, ECO:0000269|PubMed:22264700, ECO:0000269|PubMed:22815559, ECO:0000269|PubMed:23762320, ECO:0000269|PubMed:23939262, ECO:0000269|PubMed:24753243, ECO:0000269|PubMed:7814018, ECO:0000269|PubMed:8528210, ECO:0000269|PubMed:8817341, ECO:0000269|PubMed:9613851, ECO:0000269|PubMed:9703425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: May play a role in the pathogenesis of Parkinson disease (PD). A genome-wide copy number variation analysis has identified a 34 kilobase deletion over the TH gene in a PD patient but not in any controls. {ECO:0000269|PubMed:20809526}. Drugs (DrugBank ID) DB03552; DB04400; DB00765; DB00120; DB00360; DB00135 Interacts with P29762; P61978-2; Q99750; P08651-5; O75928-2; Q9UHX1-2; P0DJD3-2; P07101-3; Q9UJ04; C9J7I0; Q5MCW4 EC number EC 1.14.16.2 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Cell projection; Cytoplasm; Cytoplasmic vesicle; Disease variant; Dystonia; Iron; Metal-binding; Monooxygenase; Neurotransmitter biosynthesis; Nucleus; Oxidoreductase; Parkinson disease; Parkinsonism; Phosphoprotein; Proteomics identification; Reference proteome; Synapse Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 34997 Length 306 Aromaticity 0.12 Instability index 42.59 Isoelectric point 5.32 Charge (pH=7) -12.31 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map 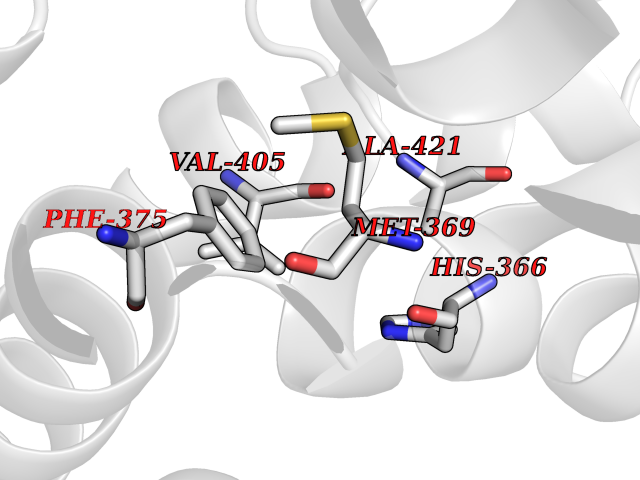 Pymol Map 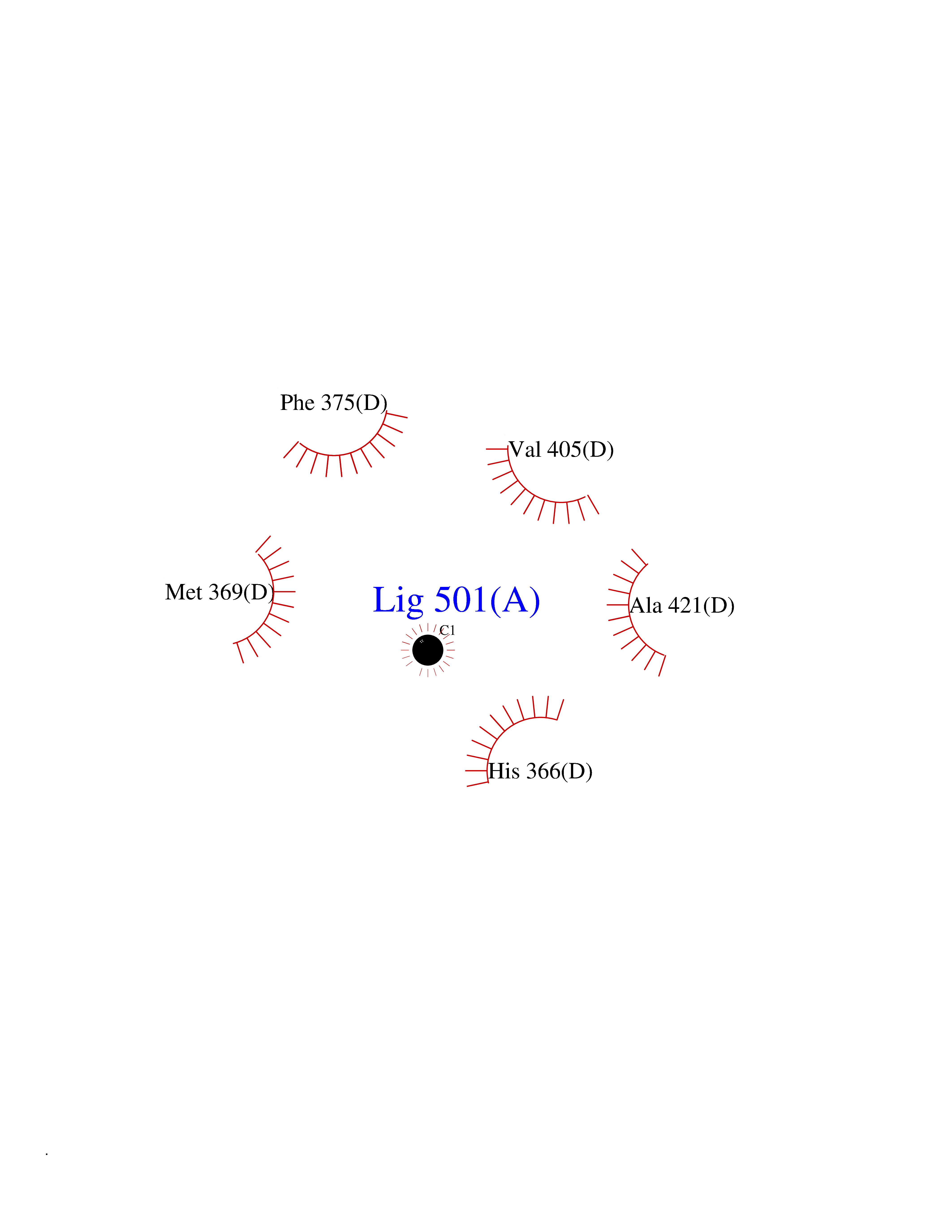 Ligplot Map 3D Binding mode Sequence VPWFPRKVSELDKCHHLVTKFDPDLDLDHPGFSDQVYRQRRKLIAEIAFQYRHGDPIPRVEYTAEEIATWKEVYTTLKGLYATHACGEHLEAFALLERFSGYREDNIPQLEDVSRFLKERTGFQLRPVAGLLSARDFLASLAFRVFQCTQYIRHASSPMHSPEPDCCHELLGHVPMLADRTFAQFSQDIGLASLGASDEEIEKLSTLYWFTVEFGLCKQNGEVKAYGAGLLSSYGELLHCLSEEPEIRAFDPEAAAVQPYQDQTYQSVYFVSESFSDAKDKLRSYASRIQRPFSVKFDPYTLAIDV Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Protein kinase C beta (PRKCB) | 2I0E | 3.94 | |
Target general information Gen name PRKCB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein kinase C beta type; PRKCB1; PKCB; PKC-beta; PKC-B Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, PKC subfamily Biochemical class Kinase Function Plays a key role in B-cell activation by regulating BCR-induced NF-kappa-B activation. Mediates the activation of the canonical NF-kappa-B pathway (NFKB1) by direct phosphorylation of CARD11/CARMA1 at 'Ser-559', 'Ser-644' and 'Ser-652'. Phosphorylation induces CARD11/CARMA1 association with lipid rafts and recruitment of the BCL10-MALT1 complex as well as MAP3K7/TAK1, which then activates IKK complex, resulting in nuclear translocation and activation of NFKB1. Plays a direct role in the negative feedback regulation of the BCR signaling, by down-modulating BTK function via direct phosphorylation of BTK at 'Ser-180', which results in the alteration of BTK plasma membrane localization and in turn inhibition of BTK activity. Involved in apoptosis following oxidative damage: in case of oxidative conditions, specifically phosphorylates 'Ser-36' of isoform p66Shc of SHC1, leading to mitochondrial accumulation of p66Shc, where p66Shc acts as a reactive oxygen species producer. Acts as a coactivator of androgen receptor (ANDR)-dependent transcription, by being recruited to ANDR target genes and specifically mediating phosphorylation of 'Thr-6' of histone H3 (H3T6ph), a specific tag for epigenetic transcriptional activation that prevents demethylation of histone H3 'Lys-4' (H3K4me) by LSD1/KDM1A. In insulin signaling, may function downstream of IRS1 in muscle cells and mediate insulin-dependent DNA synthesis through the RAF1-MAPK/ERK signaling cascade. May participate in the regulation of glucose transport in adipocytes by negatively modulating the insulin-stimulated translocation of the glucose transporter SLC2A4/GLUT4. Under high glucose in pancreatic beta-cells, is probably involved in the inhibition of the insulin gene transcription, via regulation of MYC expression. In endothelial cells, activation of PRKCB induces increased phosphorylation of RB1, increased VEGFA-induced cell proliferation, and inhibits PI3K/AKT-dependent nitric oxide synthase (NOS3/eNOS) regulation by insulin, which causes endothelial dysfunction. Also involved in triglyceride homeostasis. Phosphorylates ATF2 which promotes cooperation between ATF2 and JUN, activating transcription. Calcium-activated, phospholipid- and diacylglycerol (DAG)-dependent serine/threonine-protein kinase involved in various cellular processes such as regulation of the B-cell receptor (BCR) signalosome, oxidative stress-induced apoptosis, androgen receptor-dependent transcription regulation, insulin signaling and endothelial cells proliferation. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14001; DB09096; DB08862; DB14002; DB04209; DB08846; DB06486; DB01738; DB02010; DB00675; DB00163 Interacts with Q8NEM2; Q9BSI4; O60341-1; Q6DHV7-2; Q9UQM7; Q6ZQX7-4; O60346; Q6ZVD8 EC number EC 2.7.11.13 Uniprot keywords 3D-structure; Acetylation; Adaptive immunity; Alternative splicing; Apoptosis; ATP-binding; Calcium; Chromatin regulator; Cytoplasm; Direct protein sequencing; Immunity; Kinase; Membrane; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Serine/threonine-protein kinase; Transcription; Transcription regulation; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34383.3 Length 302 Aromaticity 0.13 Instability index 34.04 Isoelectric point 5.22 Charge (pH=7) -10.63 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map 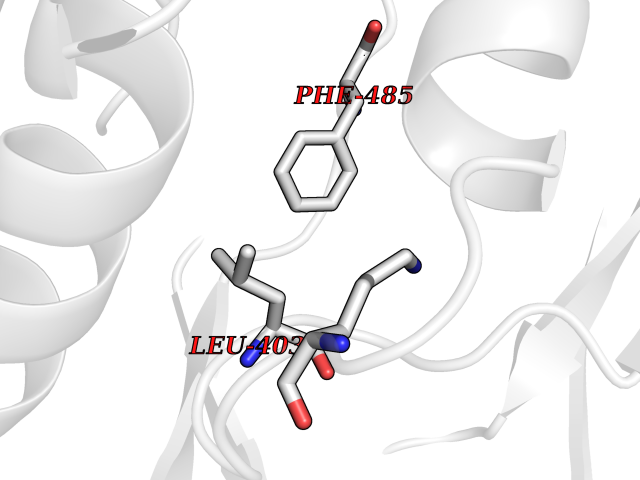 Pymol Map 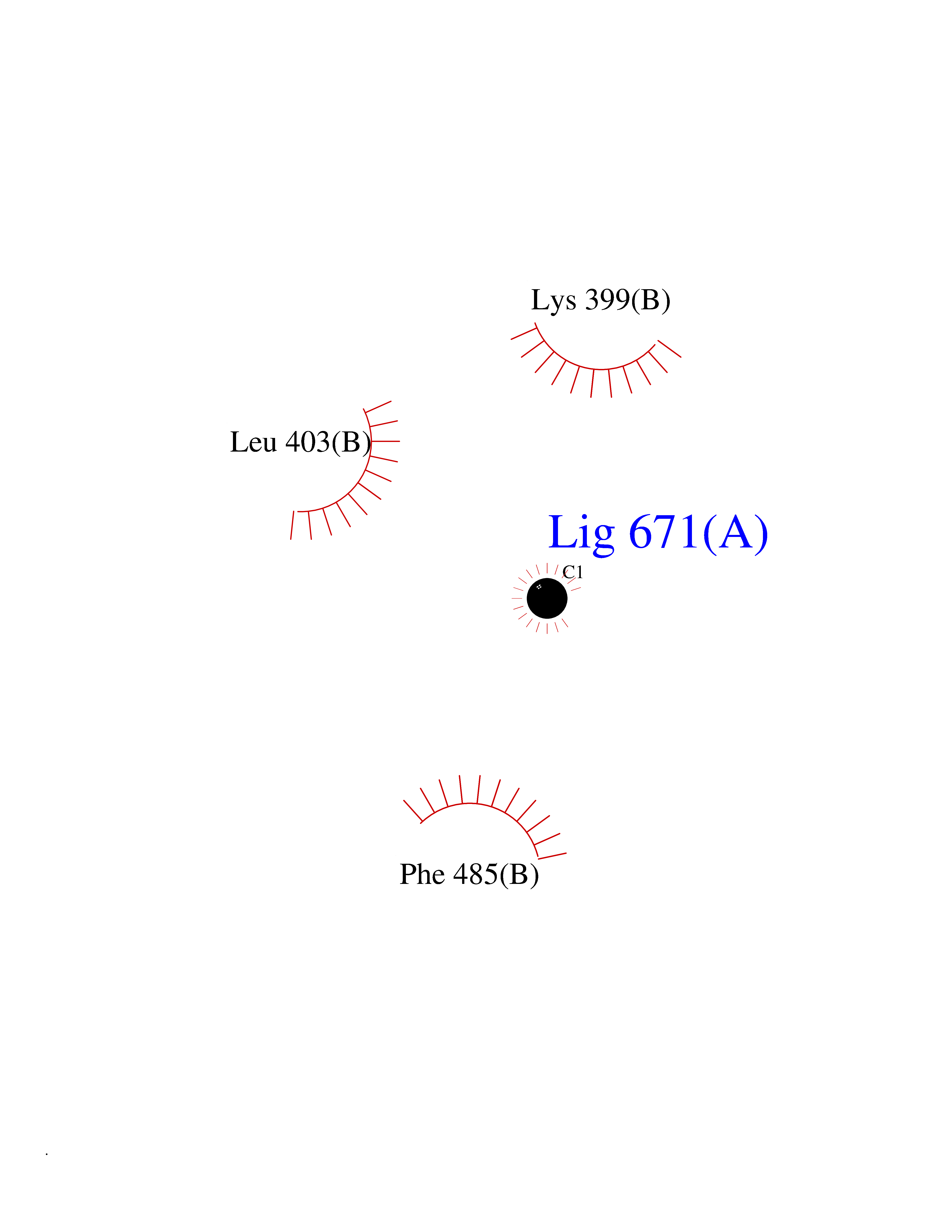 Ligplot Map 3D Binding mode Sequence LTDFNFLMVLGKGSFGKVMLSERKGTDELYAVKILKKDVVIQDDDVECTMVEKRVLALPGKPPFLTQLHSCFQTMDRLYFVMEYVNGGDLMYHIQQVGRFKEPHAVFYAAEIAIGLFFLQSKGIIYRDLKLDNVMLDSEGHIKIADFGMCKENIWDGVTTKXFCGTPDYIAPEIIAYQPYGKSVDWWAFGVLLYEMLAGQAPFEGEDEDELFQSIMEHNVAYPKSMSKEAVAICKGLMTKHPGKRLGCGPEGERDIKEHAFFRYIDWEKLERKEIQPPYKPKAIDQSEFEGFXFVNSEFLKP Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Chromodomain-helicase-DNA-binding protein 1 | 4O42 | 3.94 | |
Target general information Gen name CHD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SNF2/RAD54 helicase family Biochemical class Dna binding protein / viral protein Function ATP binding.ATP-dependent DNA helicase activity.DNA binding.Methylated histone binding. Related diseases Pilarowski-Bjornsson syndrome (PILBOS) [MIM:617682]: An autosomal dominant disorder characterized by developmental delay, speech apraxia, intellectual disability, autism, and facial dysmorphic features. Some patients may have seizures. {ECO:0000269|PubMed:28866611}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60341-1; B2BUF1; P28799; O76024 EC number 3.6.4.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromatin regulator; Cytoplasm; Disease variant; DNA-binding; Helicase; Hydrolase; Intellectual disability; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 20969.1 Length 180 Aromaticity 0.12 Instability index 46.35 Isoelectric point 5.88 Charge (pH=7) -2.83 2D Binding mode Binding energy (Kcal/mol) -5.38  Molscript Map 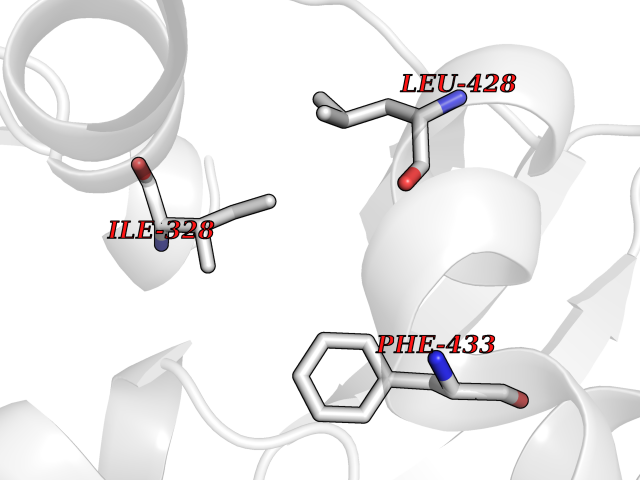 Pymol Map 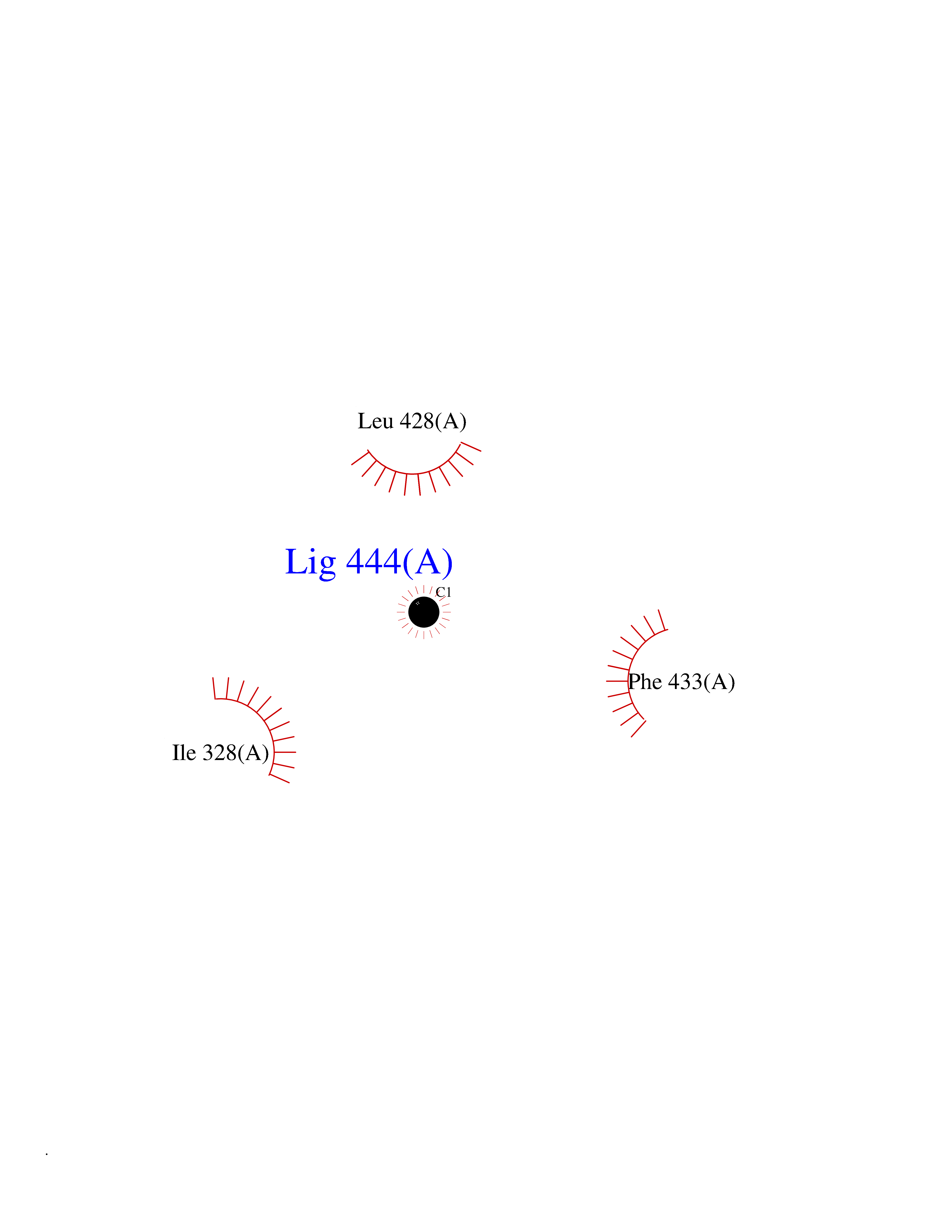 Ligplot Map 3D Binding mode Sequence EFETIERFMDCRIGRKGATGATTTIYAVEADGDPNAGFEKNKEPGEIQYLIKWKGWSHIHNTWETEETLKQQNVRGMKKLDNYKKKDQETKRWLKNASPEDVEYYNCQQELTDDLHKQYQIVERIIAHSNQKSAAGYPDYYCKWQGLPYSECSWEDGALISKKFQACIDEYFSRTARSXV Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Natriuretic peptides B | 1YK1 | 3.94 | |
Target general information Gen name NPPB Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Natriuretic peptide family Biochemical class Hormone / growth factor receptor Function Diuretic hormone activity.Hormone activity.Peptide hormone receptor binding.Receptor binding. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01136; DB06412 Interacts with A8MQ03; P57678; Q6A162; P60411; Q7Z3S9; P25788; Q9UJW9 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disulfide bond; Glycoprotein; Hormone; Pharmaceutical; Proteoglycan; Proteomics identification; Reference proteome; Secreted; Signal; Vasoactive; Vasodilator Protein physicochemical properties Chain ID E Molecular weight (Da) 46353.1 Length 415 Aromaticity 0.1 Instability index 37.91 Isoelectric point 5.51 Charge (pH=7) -12.09 2D Binding mode Binding energy (Kcal/mol) -5.38 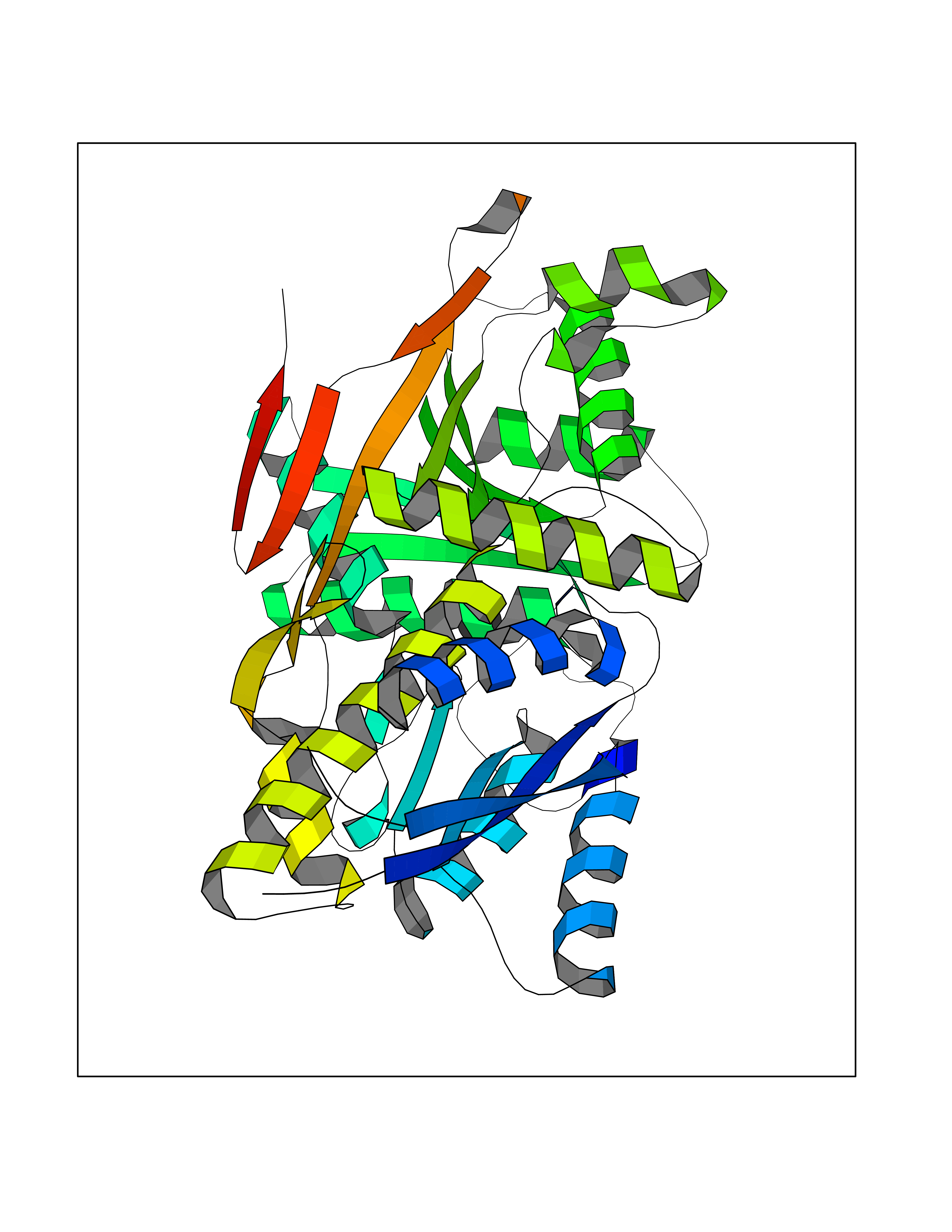 Molscript Map 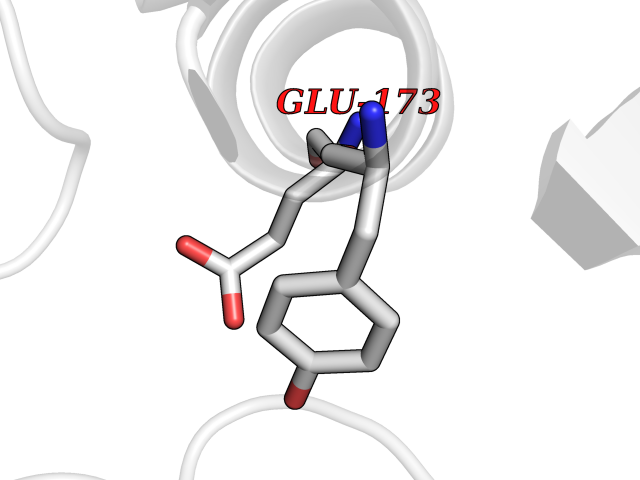 Pymol Map 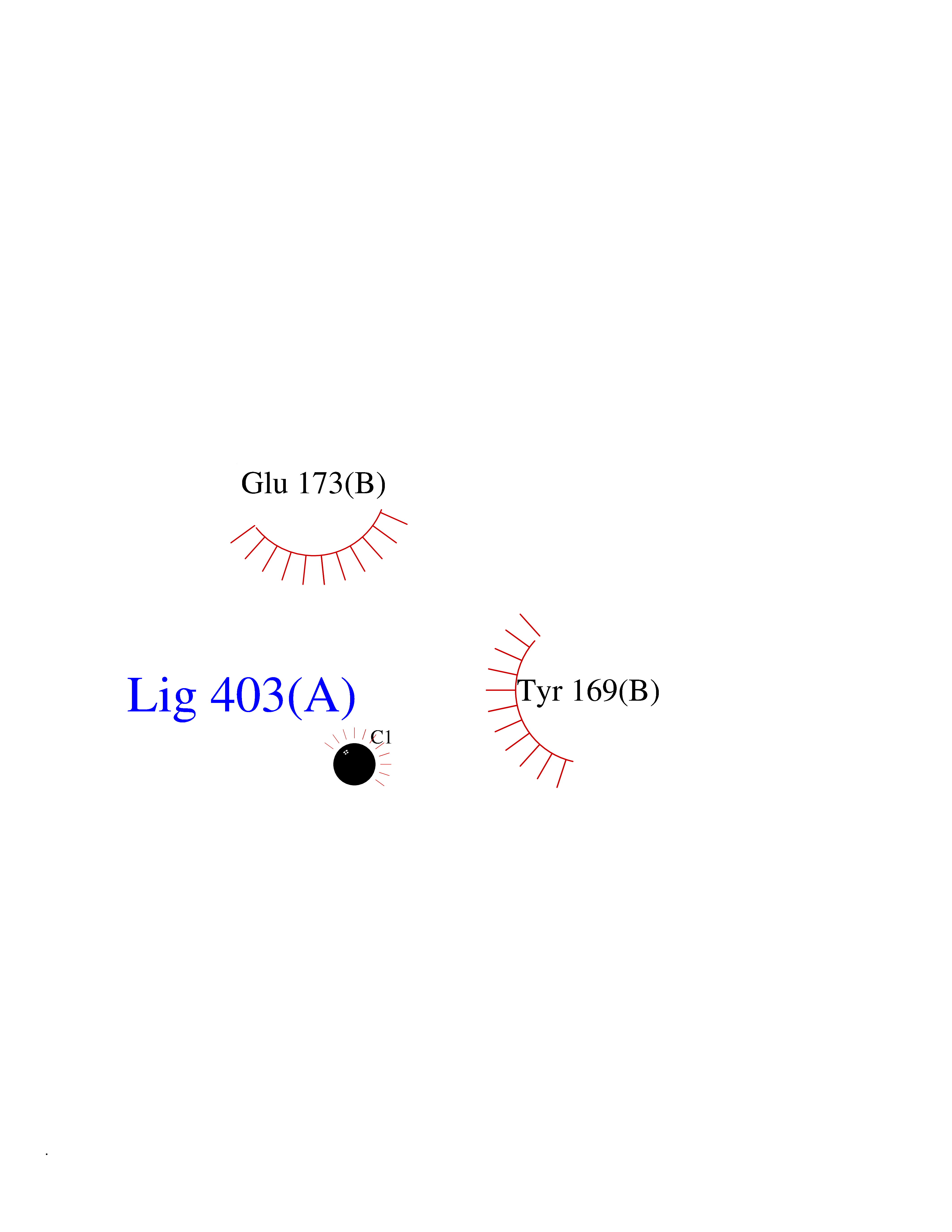 Ligplot Map 3D Binding mode Sequence GCFGRKMDRISSSSGLGCKVLALPPQKIEVLVLLPQDDSYLFSLTRVRPAIEYALRSVEGLLPPGTRFQVAYEDSDCGNRALFSLVDRVAAARGAKPDLILGPVCEYAAAPVARLASHWDLPMLSAGALAAGFQHKDSEYSHLTRVAPAYAKMGEMMLALFRHHHWSRAALVYSDDKLERNCYFTLEGVHEVFQEEGLHTSIYSFDETKDLDLEDIVRNIQASERVVIMCASSDTIRSIMLVAHRHGMTSGDYAFFNIELFNSSSYGDGSWKRGDKHDFEAKQAYSSLQTVTLLRTVKPEFEKFSMEVKSSVEKQGLNMEDYVNMFVEGFHDAILLYVLALHEVLRAGYSKKDGGKIIQQTWNRTFEGIAGQVSIDANGDRYGDFSVIAMTDVEAGTQEVIGDYFGKEGRFEMRP Hydrogen bonds contact Hydrophobic contact | ||||