Job Results:
Ligand
Structure
Job ID
5217c115ac480b5185ad5781ec63f66c
Job name
NA
Time
2026-02-27 11:51:57
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Cystathionine gamma-lyase (CTH) | 3COG | 4.57 | |
Target general information Gen name CTH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gamma-cystathionase; Cysteine-protein sulfhydrase Protein family Trans-sulfuration enzymes family Biochemical class NA Function Catalyzes the last step in the trans-sulfuration pathway from methionine to cysteine. Has broad substrate specificity. Converts cystathionine to cysteine, ammonia and 2-oxobutanoate. Converts two cysteine molecules to lanthionine and hydrogen sulfide. Can also accept homocysteine as substrate. Specificity depends on the levels of the endogenous substrates. Generates the endogenous signaling molecule hydrogen sulfide (H2S), and so contributes to the regulation of blood pressure. Acts as a cysteine-protein sulfhydrase by mediating sulfhydration of target proteins: sulfhydration consists of converting -SH groups into -SSH on specific cysteine residues of target proteins such as GAPDH, PTPN1 and NF-kappa-B subunit RELA, thereby regulating their function. Related diseases Cystathioninuria (CSTNU) [MIM:219500]: Autosomal recessive phenotype characterized by abnormal accumulation of plasma cystathionine, leading to increased urinary excretion. {ECO:0000269|PubMed:12574942, ECO:0000269|PubMed:18476726}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02328; DB03928; DB00151; DB04217; DB00114 Interacts with P32929; Q96NT3; Q96NT3-2; Q96HA8; Q6P9E2 EC number EC 4.4.1.1 Uniprot keywords 3D-structure; Alternative splicing; Amino-acid biosynthesis; Calmodulin-binding; Cysteine biosynthesis; Cytoplasm; Disease variant; Lipid metabolism; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 86026 Length 782 Aromaticity 0.08 Instability index 32.4 Isoelectric point 6.27 Charge (pH=7) -9.46 2D Binding mode Binding energy (Kcal/mol) -6.23 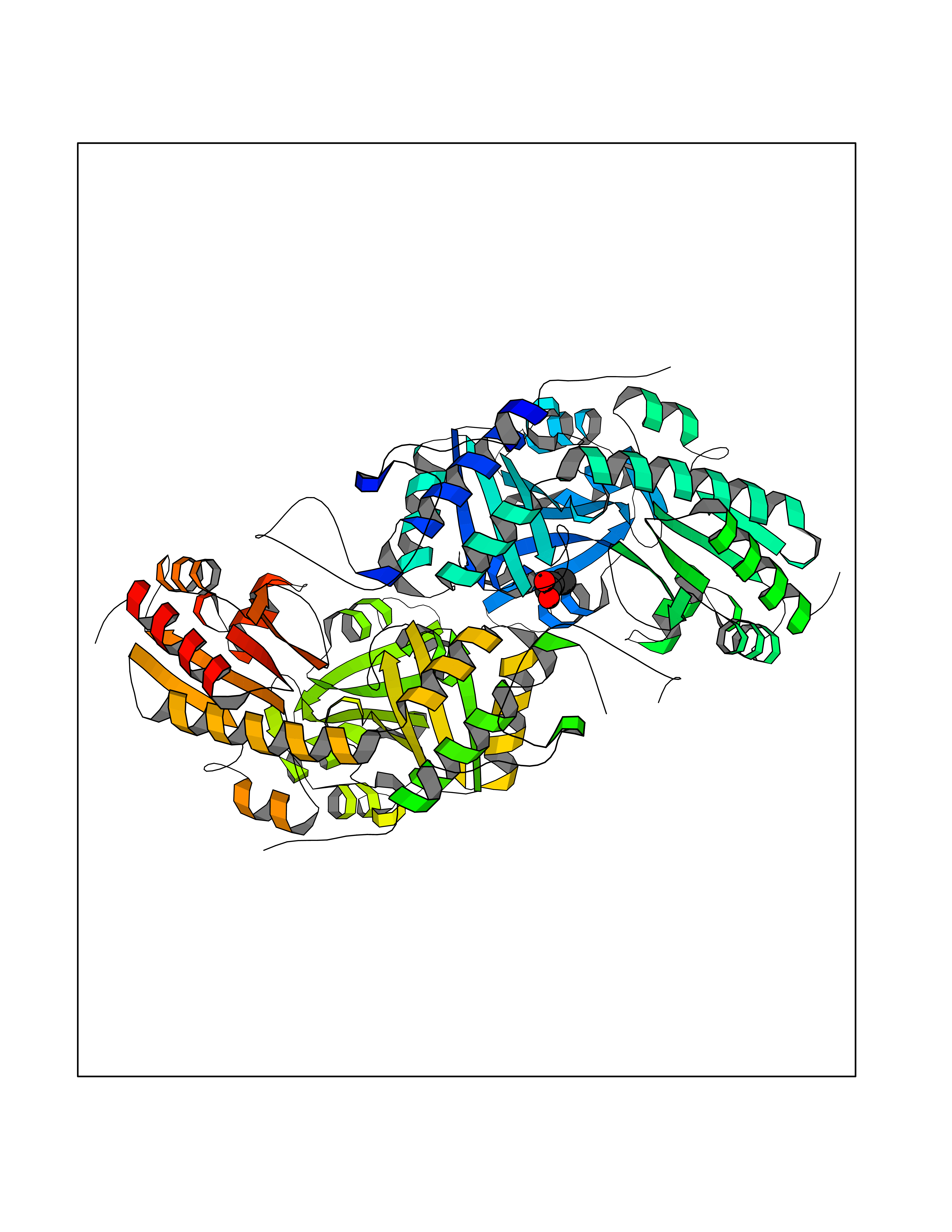 Molscript Map 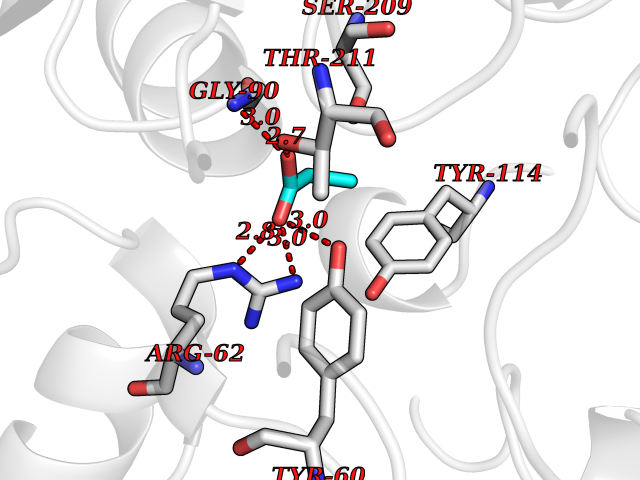 Pymol Map 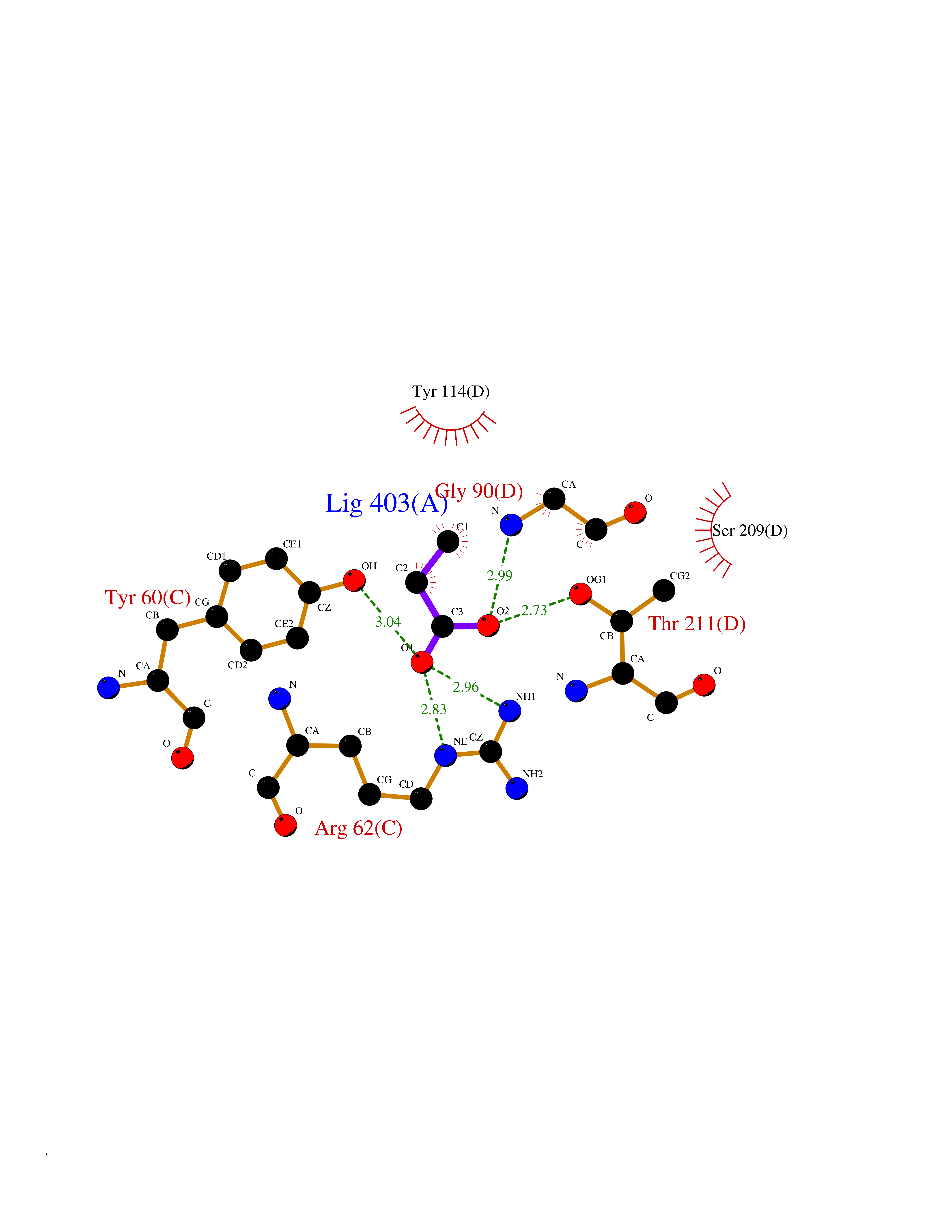 Ligplot Map 3D Binding mode Sequence GFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQHSGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPSGFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPS Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | 4-hydroxyphenylpyruvate dioxygenase | 3ISQ | 4.57 | |
Target general information Gen name HPD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PPD Protein family 4HPPD family Biochemical class Oxidoreductase Function 4-hydroxyphenylpyruvate dioxygenase activity.Metal ion binding. Related diseases Tyrosinemia 3 (TYRSN3) [MIM:276710]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, seizures and mild intellectual disability. {ECO:0000269|PubMed:10942115, ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hawkinsinuria (HWKS) [MIM:140350]: An inborn error of tyrosine metabolism characterized by failure to thrive, persistent metabolic acidosis, fine and sparse hair, and excretion of the unusual cyclic amino acid metabolite, hawkinsin, in the urine. {ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02850; DB00348 Interacts with NA EC number 1.13.11.27 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Dioxygenase; Disease variant; Endoplasmic reticulum; Golgi apparatus; Intellectual disability; Iron; Membrane; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 43164.8 Length 376 Aromaticity 0.11 Instability index 32.38 Isoelectric point 6.73 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -6.23 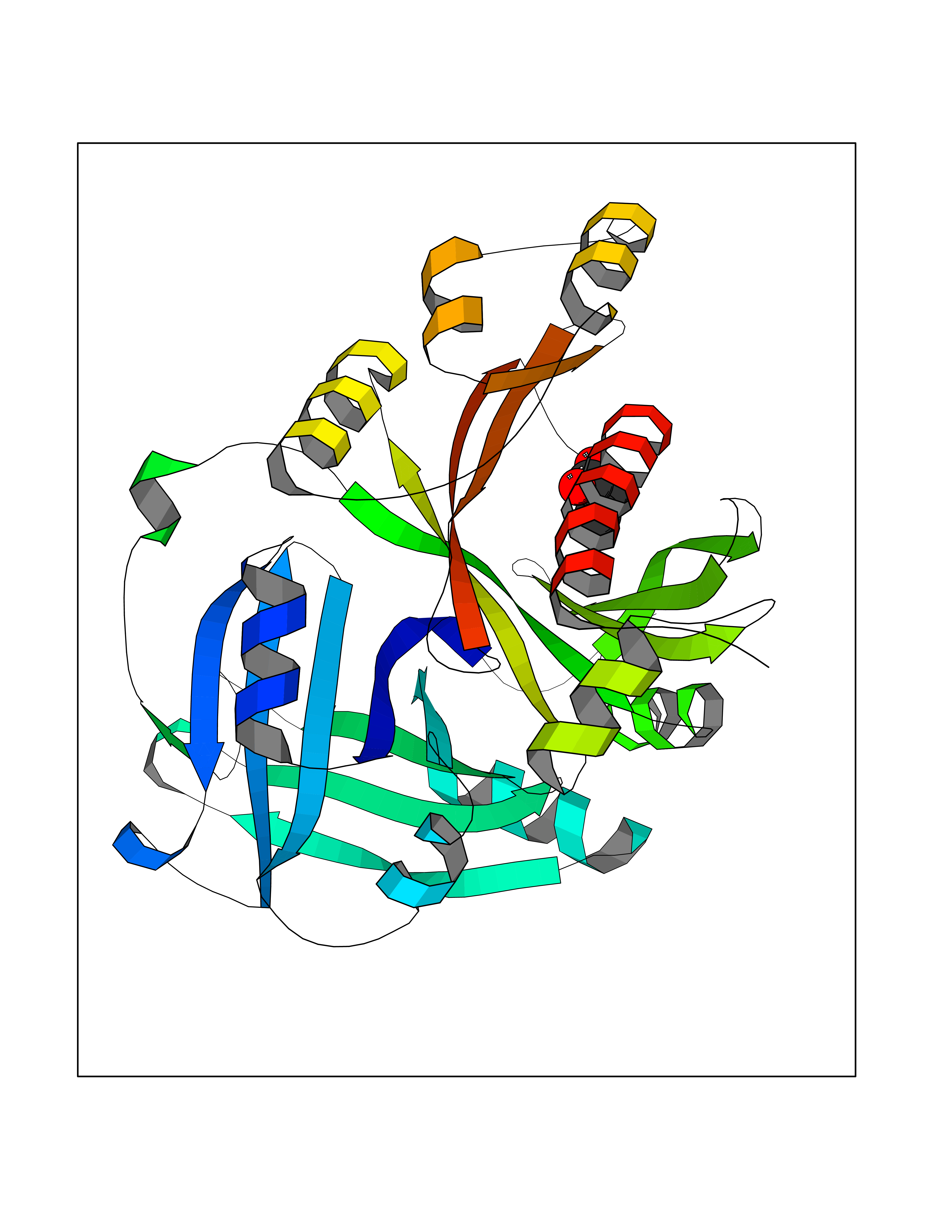 Molscript Map 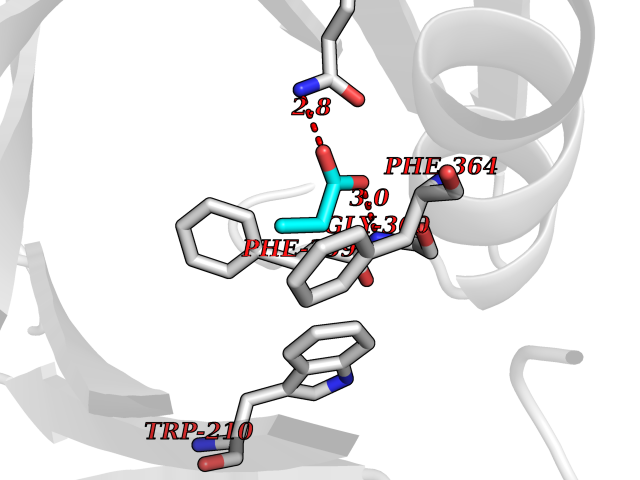 Pymol Map 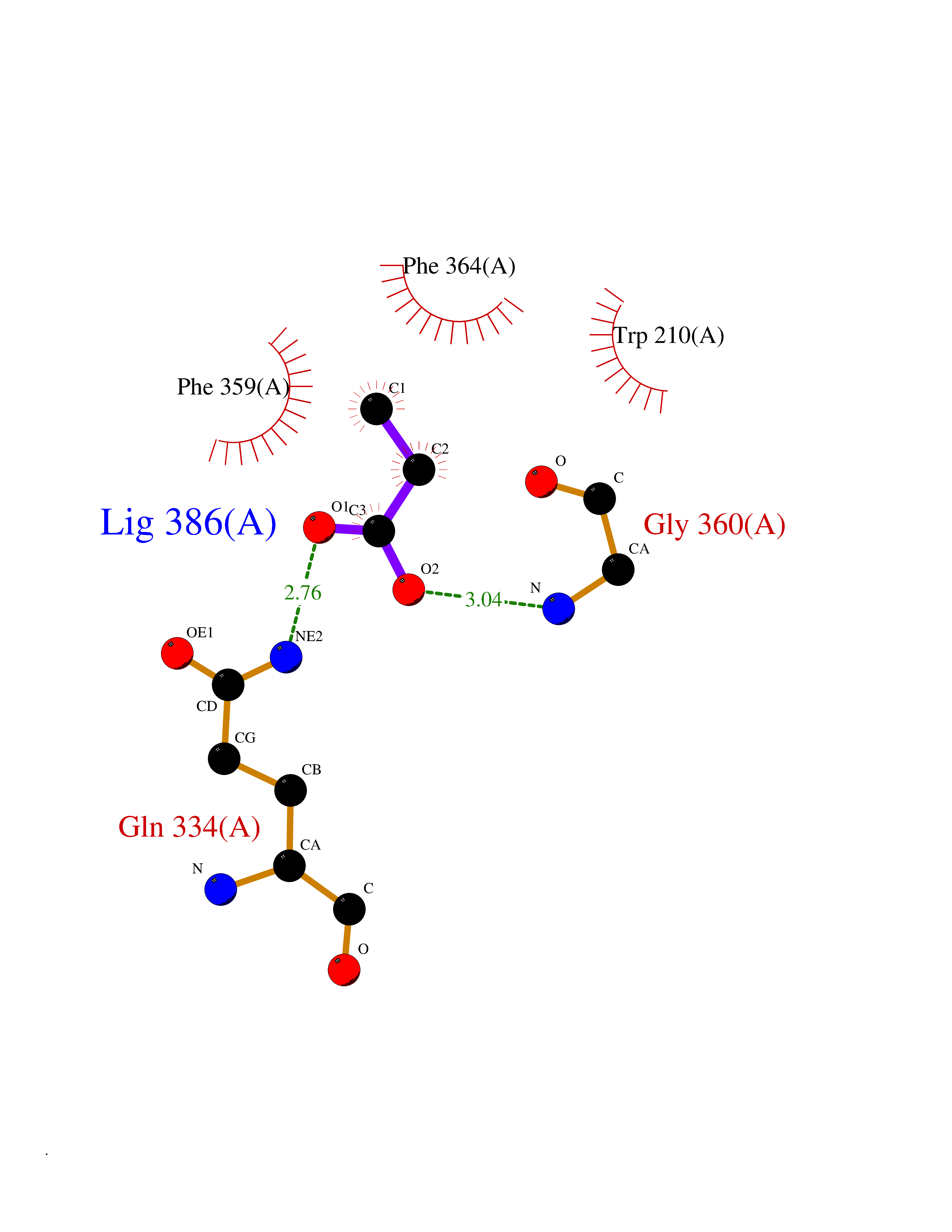 Ligplot Map 3D Binding mode Sequence AKPERGRFLHFHSVTFWVGNAKQAASFYCSKMGFEPLAYRGLETGSREVVSHVIKQGKIVFVLSSALNPWNKEMGDHLVKHGDGVKDIAFEVEDCDYIVQKARERGAKIMREPWVEQDKFGKVKFAVLQTYGDTTHTLVEKMNYIGQFLPGYEAPAFMDPLLPKLPKCSLEMIDHIVGNQPDQEMVSASEWYLKNLQFHRFWSVDDTQVHTEYSSLRSIVVANYEESIKMPINEPAPGKKKSQIQEYVDYNGGAGVQHIALKTEDIITAIRHLRERGLEFLSVPSTYYKQLREKLKTAKIKVKENIDALEELKILVDYDEKGYLLQIFTKPVQDRPTLFLEVIQRHNHQGFGAGNFNSLFKAFEEEQNLRGNLTNM Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Vascular endothelial growth factor receptor 3 | 4BSJ | 4.57 | |
Target general information Gen name FLT4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms VEGFR3 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Transferase Function ATP binding.Growth factor binding.Protein homodimerization activity.Protein phosphatase binding.Transmembrane receptor protein tyrosine kinase activity.Vascular endothelial growth factor-activated receptor activity.VEGF-C-activated receptor activity. Related diseases Lymphatic malformation 1 (LMPHM1) [MIM:153100]: A form of primary lymphedema, a disease characterized by swelling of body parts due to developmental anomalies and functional defects of the lymphatic system. Patients with lymphedema may suffer from recurrent local infections. LMPHM1 is an autosomal dominant form with variable expression and severity. Onset is usually at birth or in early childhood but can occur later. Affected individuals manifest lymphedema, predominantly in the lower limbs, and hypoplasia of lymphatic vessels. Additional features are hemangioma and nail dysplasia or papillomatosis. {ECO:0000269|PubMed:10835628, ECO:0000269|PubMed:10856194, ECO:0000269|PubMed:12881528, ECO:0000269|PubMed:15102829, ECO:0000269|PubMed:16924388, ECO:0000269|PubMed:16965327, ECO:0000269|PubMed:17458866, ECO:0000269|PubMed:19289394, ECO:0000269|PubMed:26091405, ECO:0000269|PubMed:9817924}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hemangioma, capillary infantile (HCI) [MIM:602089]: A condition characterized by dull red, firm, dome-shaped hemangiomas, sharply demarcated from surrounding skin, usually presenting at birth or occurring within the first two or three months of life. They result from highly proliferative, localized growth of capillary endothelium and generally undergo regression and involution without scarring. {ECO:0000269|PubMed:11807987}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Plays an important role in tumor lymphangiogenesis, in cancer cell survival, migration, and formation of metastases.; DISEASE: Congenital heart defects, multiple types, 7 (CHTD7) [MIM:618780]: An autosomal dominant disorder with incomplete penetrance characterized by congenital developmental abnormalities involving structures of the heart. Common defects include tetralogy of Fallot, pulmonary stenosis or atresia, absent pulmonary valve, right aortic arch, double aortic arch, and major aortopulmonary collateral arteries. {ECO:0000269|PubMed:28991257, ECO:0000269|PubMed:30232381}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06626; DB05932; DB12010; DB11679; DB06101; DB09078; DB06080; DB09079; DB06589; DB08896; DB15685; DB00398; DB01268; DB05075; DB11800; DB04879 Interacts with P08238; P35968; P49767 EC number 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 24029.2 Length 213 Aromaticity 0.1 Instability index 47.75 Isoelectric point 8.34 Charge (pH=7) 2.35 2D Binding mode Binding energy (Kcal/mol) -6.24 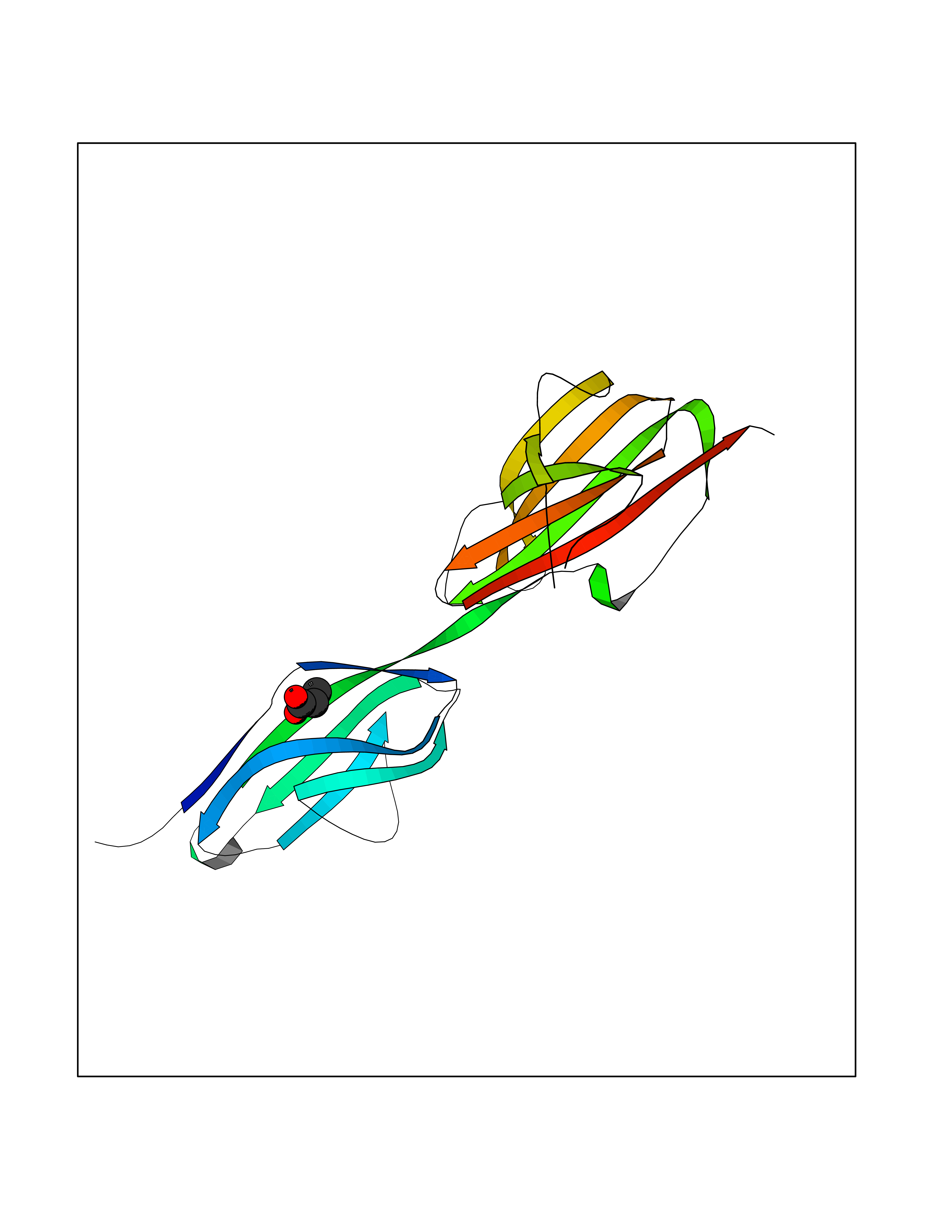 Molscript Map 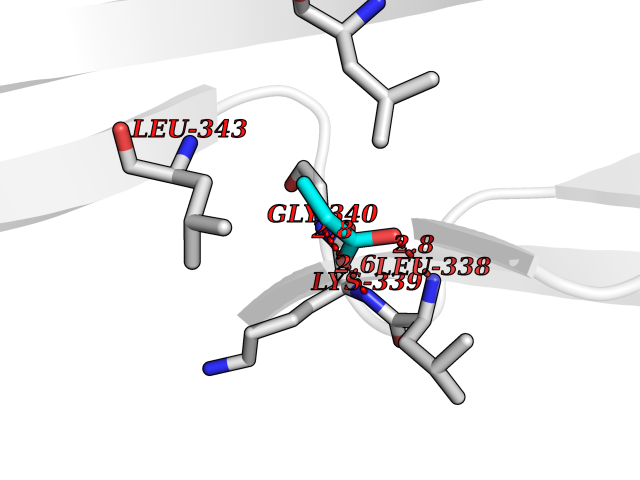 Pymol Map 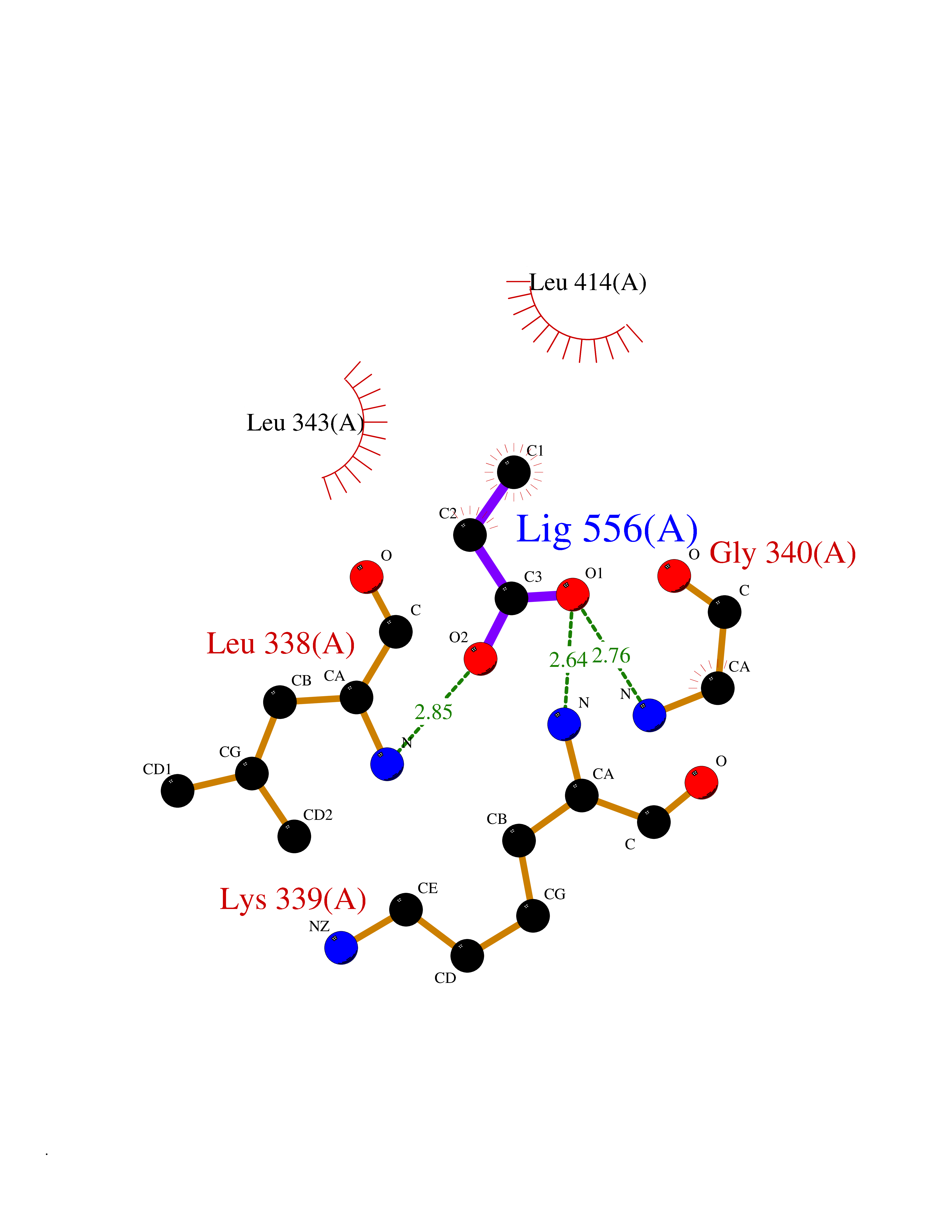 Ligplot Map 3D Binding mode Sequence DHNPFISVEWLKGPILEATAGDELVKLPVKLAAYPPPEFQWYKDGKALSGRHSPHALVLKEVTEASTGTYTLALWNSAAGLRRNISLELVVNVPPQIHEKEASSPSIYSRHSRQALTCTAYGVPLPLSIQWHWRPWTPCKMFPQCRDWRAVTTQDAVNPIESLDTWTEFVEGKNKTVSKLVIQNANVSAMYKCVVSNKVGQDERLIYFYVTTH Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Glutamate receptor ionotropic kainate 2 (GRIK2) | 5CMM | 4.57 | |
Target general information Gen name GRIK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glutamate receptor ionotropic, kainate 2; Glutamate receptor 6; GluR6; GluR-6; GluK2; Excitatory amino acid receptor 4; EAA4 Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, GRIK2 subfamily Biochemical class Glutamate-gated ion channel Function L-glutamate acts as an excitatory neurotransmitter at many synapses in the central nervous system. Binding of the excitatory neurotransmitter L-glutamate induces a conformation change, leading to the opening of the cation channel, and thereby converts the chemical signal to an electrical impulse. The receptor then desensitizes rapidly and enters a transient inactive state, characterized by the presence of bound agonist. May be involved in the transmission of light information from the retina to the hypothalamus. Modulates cell surface expression of NETO2. Ionotropic glutamate receptor. Related diseases Intellectual developmental disorder, autosomal recessive 6 (MRT6) [MIM:611092]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT6 patients display mild to severe intellectual disability and psychomotor development delay in early childhood. Patients do not have neurologic problems, congenital malformations, or facial dysmorphism. Body height, weight, and head circumference are normal. {ECO:0000269|PubMed:17847003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with impaired language and ataxia and with or without seizures (NEDLAS) [MIM:619580]: An autosomal dominant disorder characterized by axial hypotonia and global developmental delay. Affected individuals show impaired intellectual development, delayed walking, poor speech, and behavioral abnormalities. Some patients have a more severe phenotype with early-onset seizures resembling epileptic encephalopathy, inability to walk or speak, and hypomyelination on brain imaging. {ECO:0000269|PubMed:28180184, ECO:0000269|PubMed:34375587}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03425; DB01351; DB01352; DB01483; DB00237; DB00241; DB01353; DB01496; DB02852; DB00142; DB01354; DB01355; DB00463; DB00849; DB00312; DB01174; DB00794; DB02999; DB00418; DB00306; DB00599; DB00273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Isopeptide bond; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; RNA editing; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 29150.1 Length 257 Aromaticity 0.1 Instability index 35.11 Isoelectric point 5.89 Charge (pH=7) -2.05 2D Binding mode Binding energy (Kcal/mol) -6.23 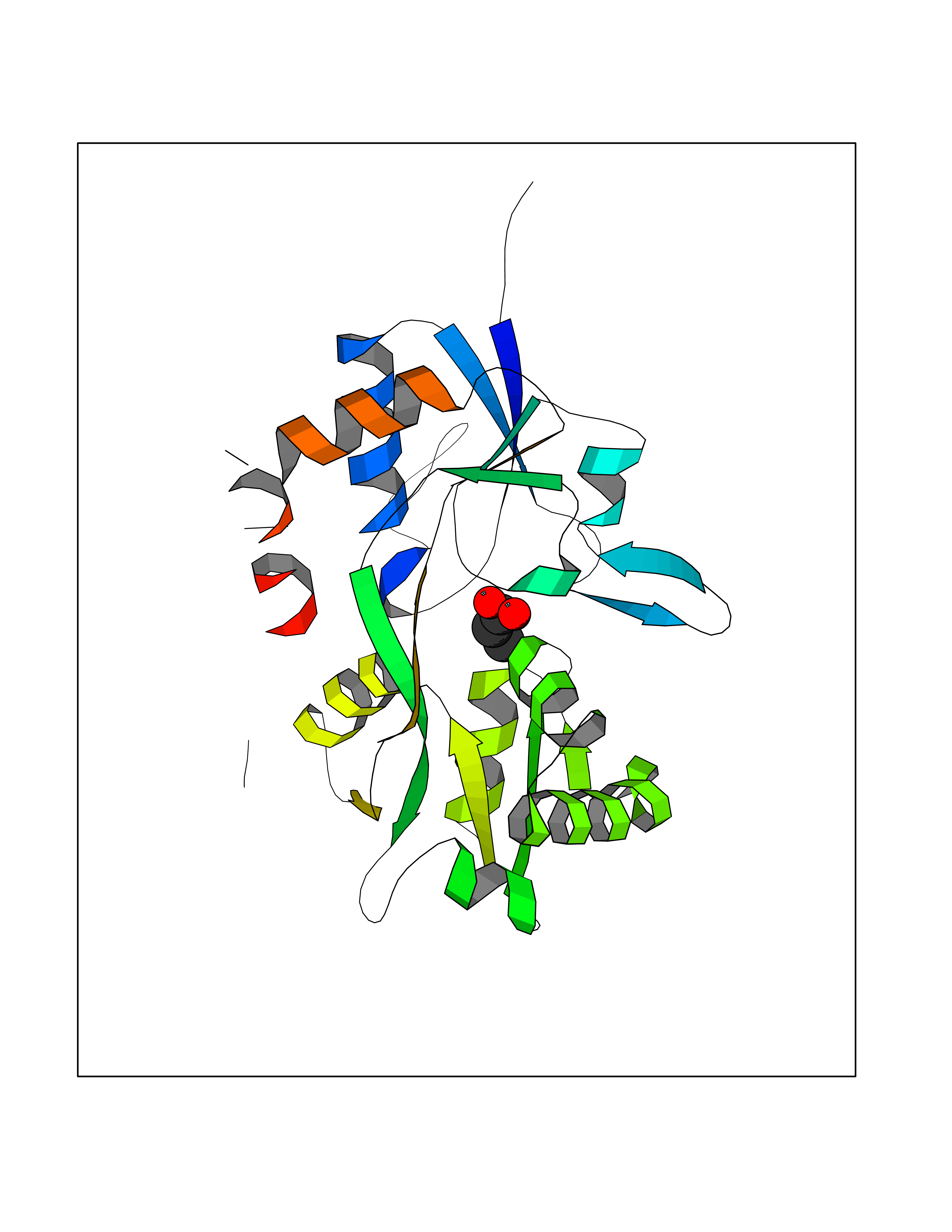 Molscript Map 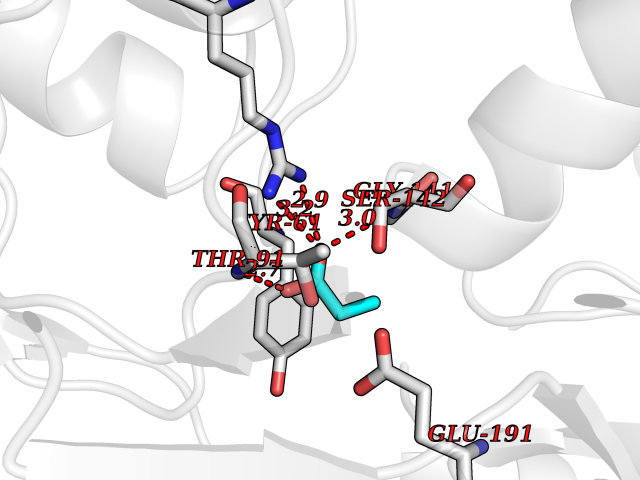 Pymol Map 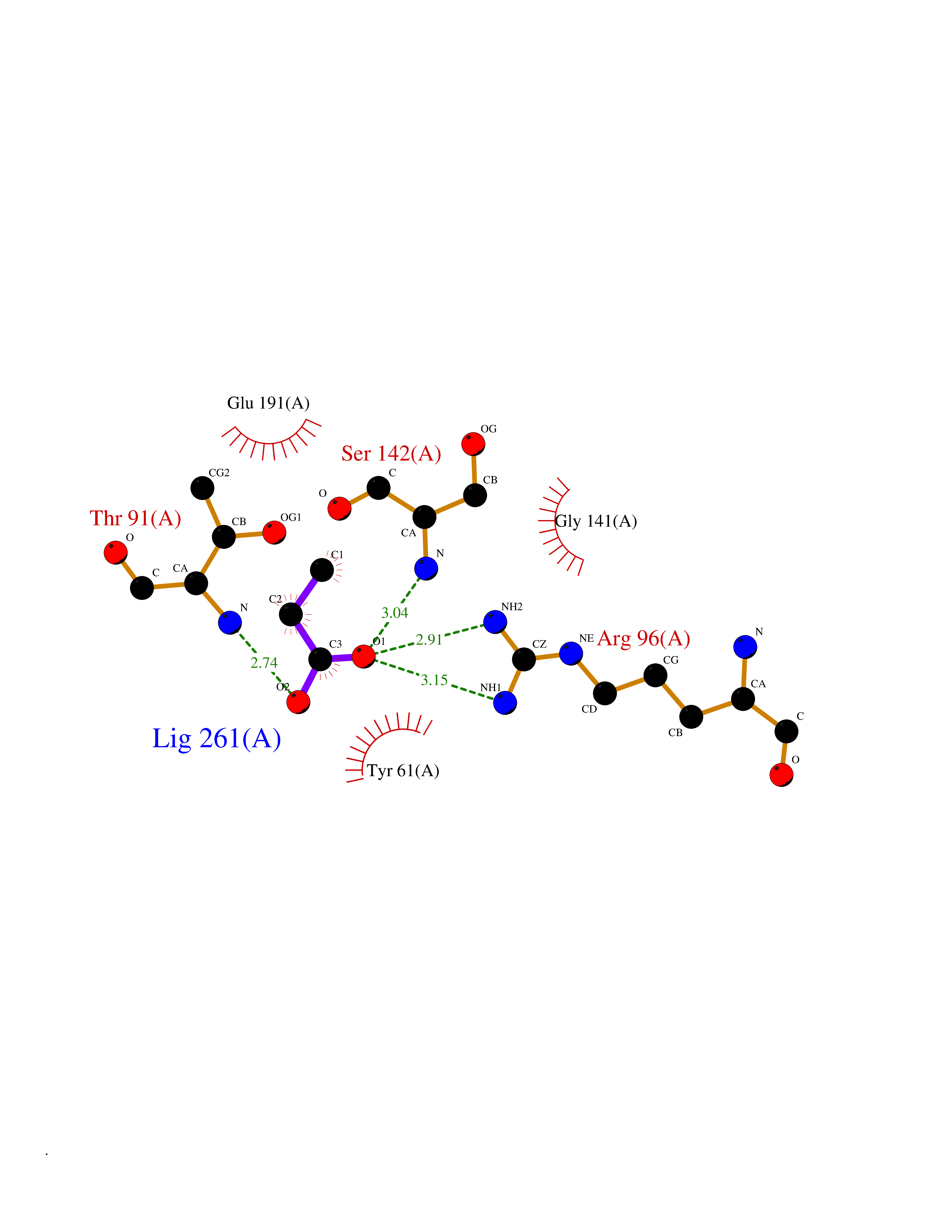 Ligplot Map 3D Binding mode Sequence GSNRSLIVTTILEEPYVLFKKSDKPLYGNDRFEGYCIDLLRELSTILGFTYEIRLVEDGKYGAQDDVNGQWNGMVRELIDHKADLAVAPLTITYVREKVIDFSKPFMTLGISILYRKGTPIDSADDLAKQTKIEYGAVEDGSTMTFFKKSKISTYDKMWAFMSSRRQSVLVKSSEEGIQRVLTSDYALLMESTTIEFVTQRNCNLTQIGGLIDSKGYGVGTPMGSPYRDKITIAILQLQEEGKLHMMKEKWWRGCPE Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Histone deacetylase 8 (HDAC8) | 5BWZ | 4.57 | |
Target general information Gen name HDAC8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histone deacetylase-8; HDACL1; HD8; CDA07 Protein family Histone deacetylase family, HD type 1 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Also involved in the deacetylation of cohesin complex protein SMC3 regulating release of cohesin complexes from chromatin. May play a role in smooth muscle cell contractility. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4). Related diseases Cornelia de Lange syndrome 5 (CDLS5) [MIM:300882]: A form of Cornelia de Lange syndrome, a clinically heterogeneous developmental disorder associated with malformations affecting multiple systems. It is characterized by facial dysmorphisms, abnormal hands and feet, growth delay, cognitive retardation, hirsutism, gastroesophageal dysfunction and cardiac, ophthalmologic and genitourinary anomalies. {ECO:0000269|PubMed:22885700, ECO:0000269|PubMed:22889856}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07350; DB02565; DB07586; DB12565; DB05015; DB08168; DB01262; DB11841; DB14490; DB14491; DB14488; DB14501; DB14489; DB12645; DB01592; DB02917; DB06603; DB06819; DB03766; DB12847; DB06176; DB04297; DB00313; DB02546; DB01593; DB14487; DB14533; DB14548 Interacts with NA EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Cytoplasm; Disease variant; Hydrolase; Intellectual disability; Metal-binding; Nucleus; Obesity; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 39018.4 Length 351 Aromaticity 0.11 Instability index 38.57 Isoelectric point 6.06 Charge (pH=7) -5.26 2D Binding mode Binding energy (Kcal/mol) -6.24 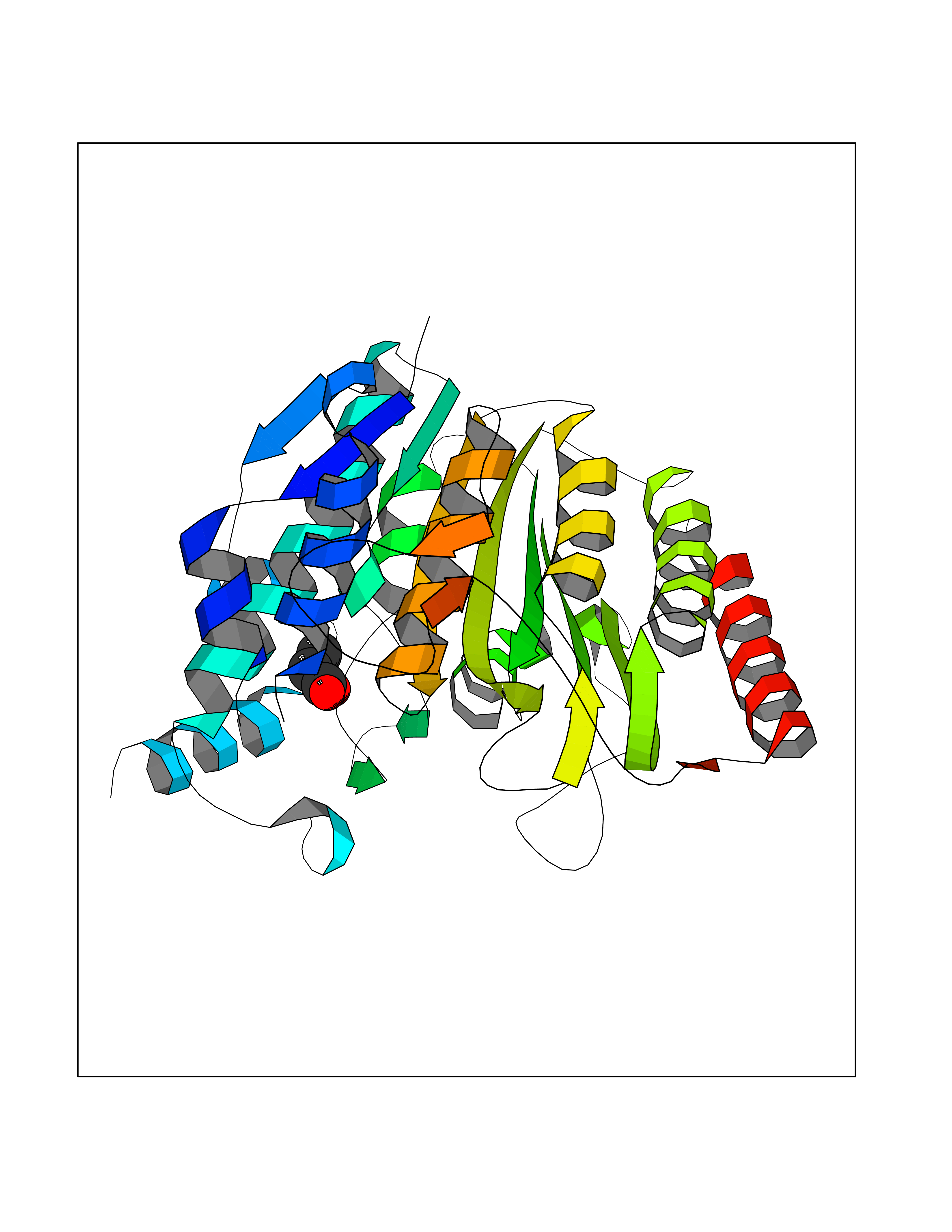 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LVPVYIYSPEYVSMCDSLPKRAEMVHSLIEAYALHKQMRIVKPKVASMEEMATFHTDAYLQHLQKVSQEYGLGYDCPATEGIFDYAAAIGGATITAAQCLIDGMCKVAINWSGGWHHAKKDEASGFCYLNDAVLGILRLRRKFERILYVDLDLHHGDGVEDAFSFTSKVMTVSLHKFSPGFFPGTGDVSDVGLGKGRYYSVNVPIQDGIQDEKYYQICESVLKEVYQAFNPKAVVLQLGADTIAGDPMCSFNMTPVGIGKCLKYILQWQLATLILGGGGYNLANTARCWTYLTGVILGKTLSSEIPDHEFFTAYGPDYVLEITPSCRPDRNEPHRIQQILNYIKGNLKHVV Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Notch-1 receptor (NOTCH1) | 5L0R | 4.57 | |
Target general information Gen name NOTCH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hN1; Translocationassociated notch protein TAN1; Translocation-associated notch protein TAN-1; TAN1; Notch 1 intracellular domain; Notch 1; Neurogenic locus notch homolog protein 1; NICD Protein family NOTCH family Biochemical class Notch protein Function Upon ligand activation through the released notch intracellular domain (NICD) it forms a transcriptional activator complex with RBPJ/RBPSUH and activates genes of the enhancer of split locus. Affects the implementation of differentiation, proliferation and apoptotic programs. Involved in angiogenesis; negatively regulates endothelial cell proliferation and migration and angiogenic sprouting. Involved in the maturation of both CD4(+) and CD8(+) cells in the thymus. Important for follicular differentiation and possibly cell fate selection within the follicle. During cerebellar development, functions as a receptor for neuronal DNER and is involved in the differentiation of Bergmann glia. Represses neuronal and myogenic differentiation. May play an essential role in postimplantation development, probably in some aspect of cell specification and/or differentiation. May be involved in mesoderm development, somite formation and neurogenesis. May enhance HIF1A function by sequestering HIF1AN away from HIF1A. Required for the THBS4 function in regulating protective astrogenesis from the subventricular zone (SVZ) niche after injury. Involved in determination of left/right symmetry by modulating the balance between motile and immotile (sensory) cilia at the left-right organiser (LRO). Functions as a receptor for membrane-bound ligands Jagged-1 (JAG1), Jagged-2 (JAG2) and Delta-1 (DLL1) to regulate cell-fate determination. Related diseases Aortic valve disease 1 (AOVD1) [MIM:109730]: A common defect in the aortic valve in which two rather than three leaflets are present. It is often associated with aortic valve calcification, stenosis and insufficiency. In extreme cases, the blood flow may be so restricted that the left ventricle fails to grow, resulting in hypoplastic left heart syndrome. {ECO:0000269|PubMed:16025100}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Adams-Oliver syndrome 5 (AOS5) [MIM:616028]: A form of Adams-Oliver syndrome, a disorder characterized by the congenital absence of skin (aplasia cutis congenita) in combination with transverse limb defects. Aplasia cutis congenita can be located anywhere on the body, but in the vast majority of the cases, it is present on the posterior parietal region where it is often associated with an underlying defect of the parietal bones. Limb abnormalities are typically limb truncation defects affecting the distal phalanges or entire digits (true ectrodactyly). Only rarely, metatarsals/metacarpals or more proximal limb structures are also affected. Apart from transverse limb defects, syndactyly, most commonly of second and third toes, can also be observed. The clinical features are highly variable and can also include cardiovascular malformations, brain abnormalities and vascular defects such as cutis marmorata and dilated scalp veins. {ECO:0000269|PubMed:25132448}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q13315; Q969H0; Q16665; P78504; O60341; Q8N423; Q92585; P19838; P46531; Q13153; Q13526; Q06330; Q06330-6; Q13573; P98170; Q8IZL2; Q96JK9 EC number NA Uniprot keywords 3D-structure; Activator; Angiogenesis; ANK repeat; Calcium; Cell membrane; Developmental protein; Differentiation; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Endosome; Glycoprotein; Hydroxylation; Isopeptide bond; Membrane; Metal-binding; Notch signaling pathway; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transcription; Transcription regulation; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 46121.2 Length 394 Aromaticity 0.13 Instability index 39.95 Isoelectric point 7.22 Charge (pH=7) 0.63 2D Binding mode Binding energy (Kcal/mol) -6.23 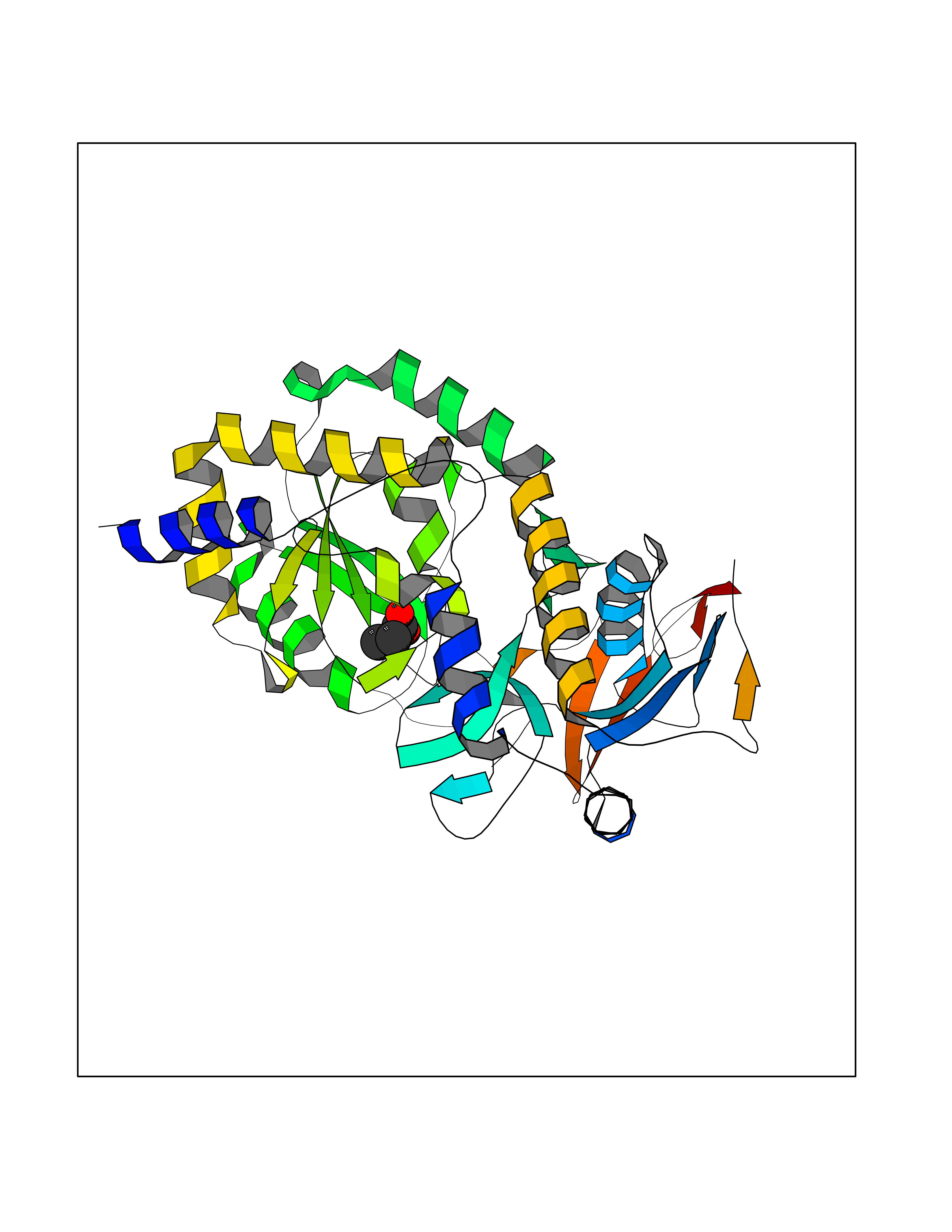 Molscript Map 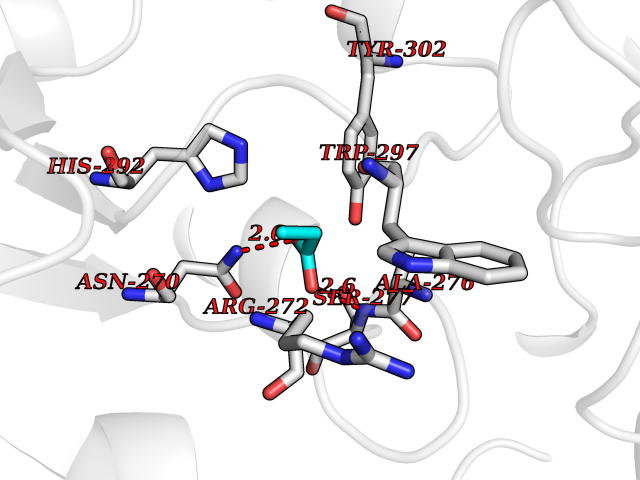 Pymol Map 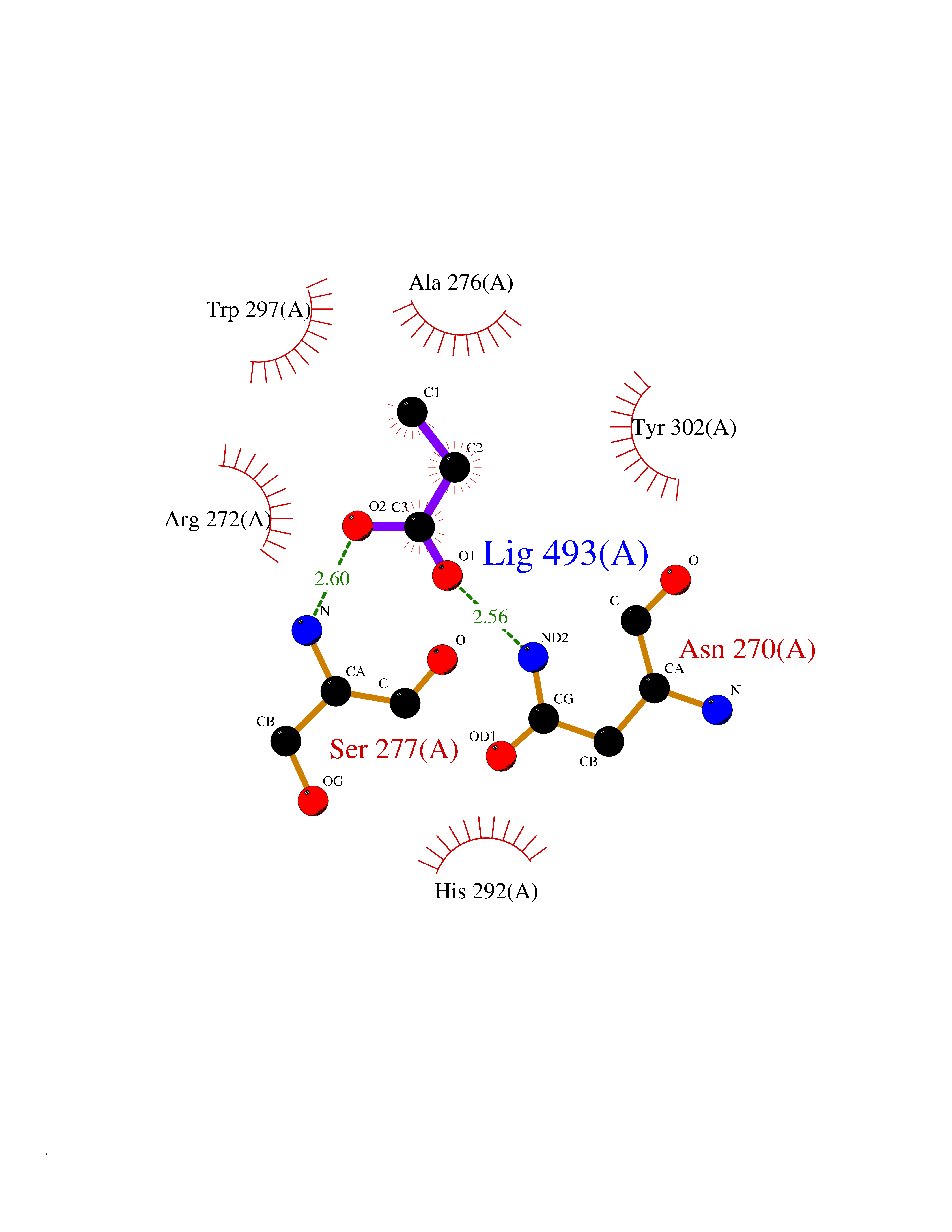 Ligplot Map 3D Binding mode Sequence KWKVFIDQINRSLENYEPCSSQNCSCYHGVIEEDLTPFRGGISRKMMAEVVRRKLGTHYQITKNRLYRENDCMFPSRCSGVEHFILEVIGRLPDMEMVINVRDYPQVPKWMEPAIPVFSFSKTSEYHDIMYPAWTFWEGGPAVWPIYPTGLGRWDLFREDLVRSAAQWPWKKKNSTAYFRGSRTSPERDPLILLSRKNPKLVDAEYTKNQAWKSMKDTLGKPAAKDVHLVDHCKYKYLFNFRGVAASFRFKHLFLCGSLVFHVGDEWLEFFYPQLKPWVHYIPVKTDLSNVQELLQFVKANDDVAQEIAERGSQFIRNHLQMDDITCYWENLLSEYSKFLSYNVTRRKGYDQIIPVNECVSNPCQNDATCLDQIGEFQCICMPGYEGVHCEVNT Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Protein kinase G1 (PRKG1) | 6BG2 | 4.57 | |
Target general information Gen name PRKG1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-dependent protein kinase I; cGMP-dependent protein kinase 1; cGKI; cGK1; cGK 1; PRKGR1B; PRKGR1A; PRKG1B Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, cGMP subfamily Biochemical class Kinase Function GMP binding activates PRKG1, which phosphorylates serines and threonines on many cellular proteins. Numerous protein targets for PRKG1 phosphorylation are implicated in modulating cellular calcium, but the contribution of each of these targets may vary substantially among cell types. Proteins that are phosphorylated by PRKG1 regulate platelet activation and adhesion, smooth muscle contraction, cardiac function, gene expression, feedback of the NO-signaling pathway, and other processes involved in several aspects of the CNS like axon guidance, hippocampal and cerebellar learning, circadian rhythm and nociception. Smooth muscle relaxation is mediated through lowering of intracellular free calcium, by desensitization of contractile proteins to calcium, and by decrease in the contractile state of smooth muscle or in platelet activation. Regulates intracellular calcium levels via several pathways: phosphorylates MRVI1/IRAG and inhibits IP3-induced Ca(2+) release from intracellular stores, phosphorylation of KCNMA1 (BKCa) channels decreases intracellular Ca(2+) levels, which leads to increased opening of this channel. PRKG1 phosphorylates the canonical transient receptor potential channel (TRPC) family which inactivates the associated inward calcium current. Another mode of action of NO/cGMP/PKGI signaling involves PKGI-mediated inactivation of the Ras homolog gene family member A (RhoA). Phosphorylation of RHOA by PRKG1 blocks the action of this protein in myriad processes: regulation of RHOA translocation; decreasing contraction; controlling vesicle trafficking, reduction of myosin light chain phosphorylation resulting in vasorelaxation. Activation of PRKG1 by NO signaling alters also gene expression in a number of tissues. In smooth muscle cells, increased cGMP and PRKG1 activity influence expression of smooth muscle-specific contractile proteins, levels of proteins in the NO/cGMP signaling pathway, down-regulation of the matrix proteins osteopontin and thrombospondin-1 to limit smooth muscle cell migration and phenotype. Regulates vasodilator-stimulated phosphoprotein (VASP) functions in platelets and smooth muscle. Serine/threonine protein kinase that acts as key mediator of the nitric oxide (NO)/cGMP signaling pathway. Related diseases Aortic aneurysm, familial thoracic 8 (AAT8) [MIM:615436]: A disease characterized by permanent dilation of the thoracic aorta usually due to degenerative changes in the aortic wall. It is primarily associated with a characteristic histologic appearance known as 'medial necrosis' or 'Erdheim cystic medial necrosis' in which there is degeneration and fragmentation of elastic fibers, loss of smooth muscle cells, and an accumulation of basophilic ground substance. {ECO:0000269|PubMed:23910461}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q13873; Q86V42; P25791; O76074; O15015; O94989; P25791-3; Q6FHY5; Q13976-2; Q9NYW8 EC number EC 2.7.11.12 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Aortic aneurysm; ATP-binding; cGMP; cGMP-binding; Coiled coil; Cytoplasm; Disease variant; Disulfide bond; Kinase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID C Molecular weight (Da) 38032 Length 334 Aromaticity 0.12 Instability index 38.26 Isoelectric point 6.23 Charge (pH=7) -2.54 2D Binding mode Binding energy (Kcal/mol) -6.24 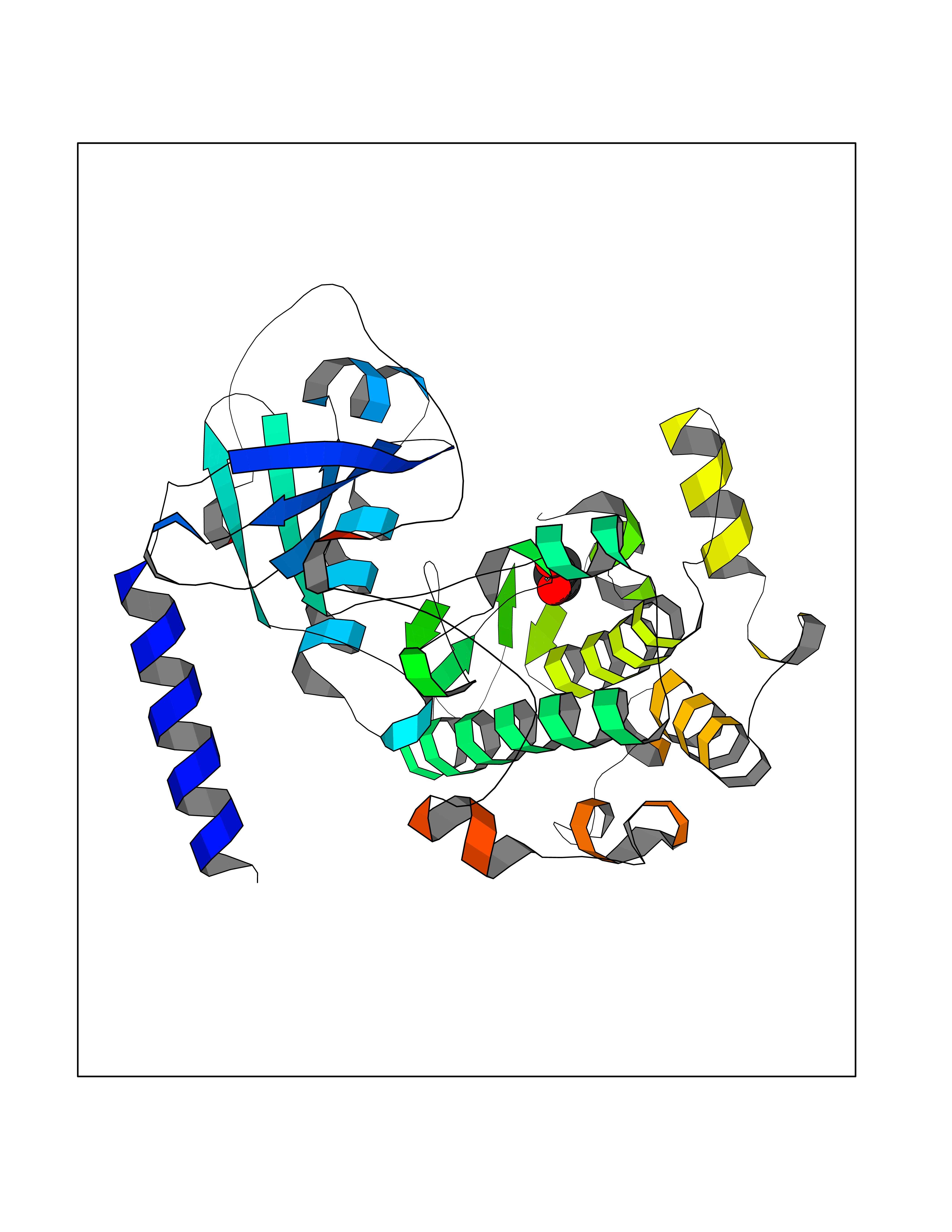 Molscript Map 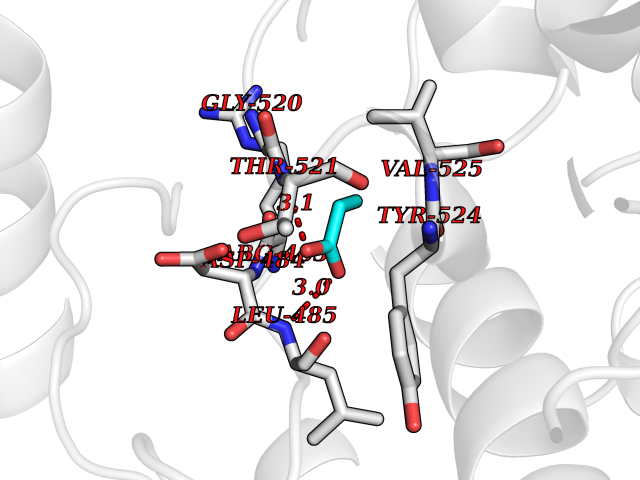 Pymol Map 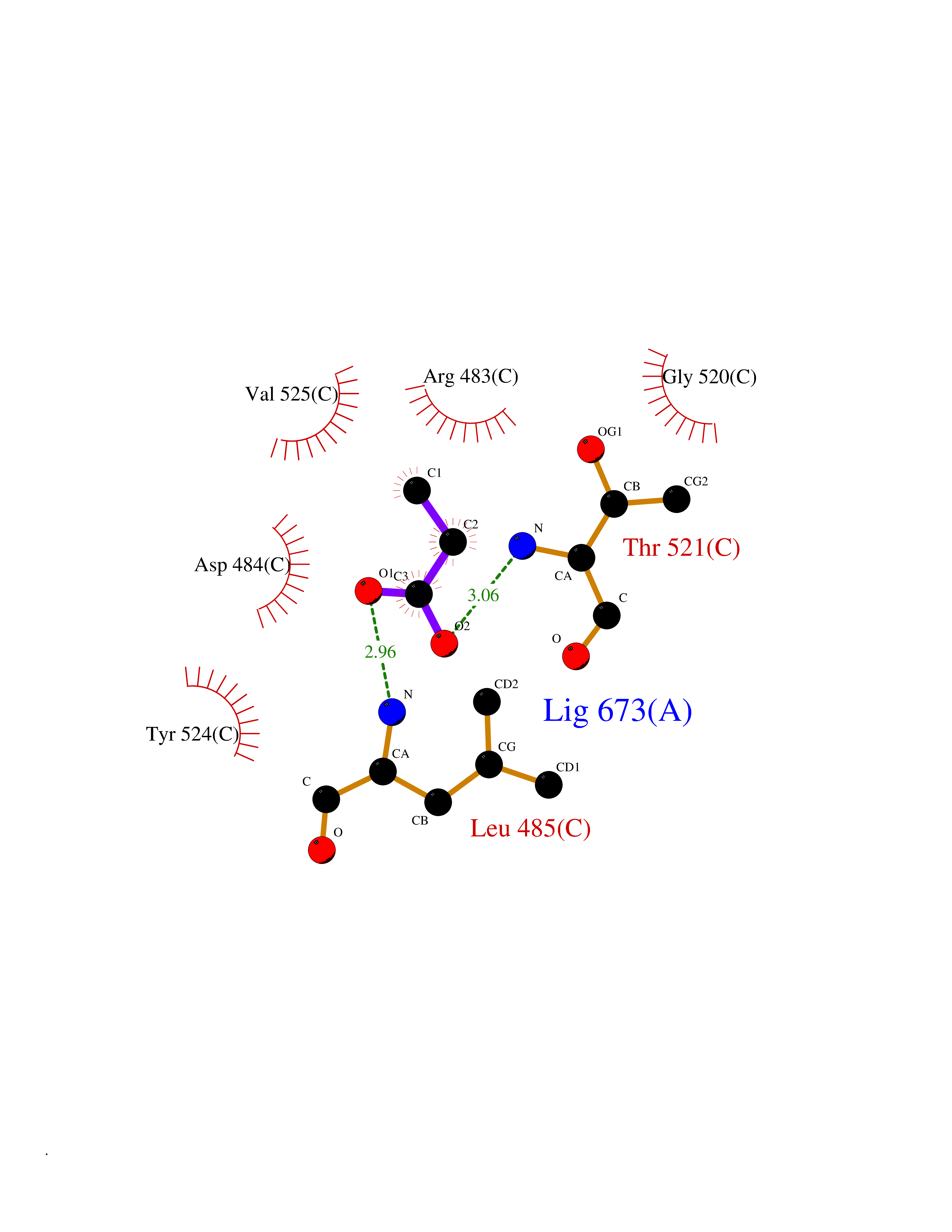 Ligplot Map 3D Binding mode Sequence EDAEAKAKYEAEAAFFANLKLSDFNIIDTLGVGGFGRVELVQLKSEESKTFAMKILKKRHIVDTRQQEHIRSEKQIMQGAHSDFIVRLYRTFKDSKYLYMLMEACLGGELWTILRDRGSFEDSTTRFYTACVVEAFAYLHSKGIIYRDLKPENLILDHRGYAKLVDFGFAKKIGFGKKTWFCGTPEYVAPEIILNKGHDISADYWSLGILMYELLTGSPPFSGPDPMKTYNIILRGIDMIEFPKKIAKNAANLIKKLCRDNPSERLGNLKNGVKDIQKHKWFEGFNWEGLRKGTLTPPIIPSVASPTDTSNFDSFPEDNDEPPPDDNSGWDIDF Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Phosphodiesterase 3A (PDE3A) | 7EG0 | 4.57 | |
Target general information Gen name PDE3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-inhibited 3',5'-cyclic phosphodiesterase A; Phosphodiesterase 3A; Cyclic GMP-inhibited phosphodiesterase A; Cyclic GMP inhibited phosphodiesterase A; CGI-PDE A Protein family Cyclic nucleotide phosphodiesterase family, PDE3 subfamily Biochemical class Phosphoric diester hydrolase Function Cyclic nucleotide phosphodiesterase with a dual-specificity for the second messengers cAMP and cGMP, which are key regulators of many important physiological processes. Related diseases Hypertension and brachydactyly syndrome (HTNB) [MIM:112410]: A syndrome characterized by brachydactyly type E, severe salt-independent but age-dependent hypertension, an increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, and altered baroreflex blood pressure regulation. It results in death from stroke before age 50 years when untreated. Brachydactyly type E is characterized by shortening of the fingers mainly in the metacarpals and metatarsals. {ECO:0000269|PubMed:25961942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01223; DB01427; DB00261; DB00201; DB01166; DB04880; DB05266; DB00922; DB00235; DB01303; DB00277; DB08811; DB09283 Interacts with Q9Y6D6; Q9Y6D5 EC number EC 3.1.4.17 Uniprot keywords 3D-structure; cAMP; cGMP; Cytoplasm; Direct protein sequencing; Disease variant; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 108171 Length 939 Aromaticity 0.11 Instability index 39.75 Isoelectric point 6.53 Charge (pH=7) -4.15 2D Binding mode Binding energy (Kcal/mol) -6.23  Molscript Map 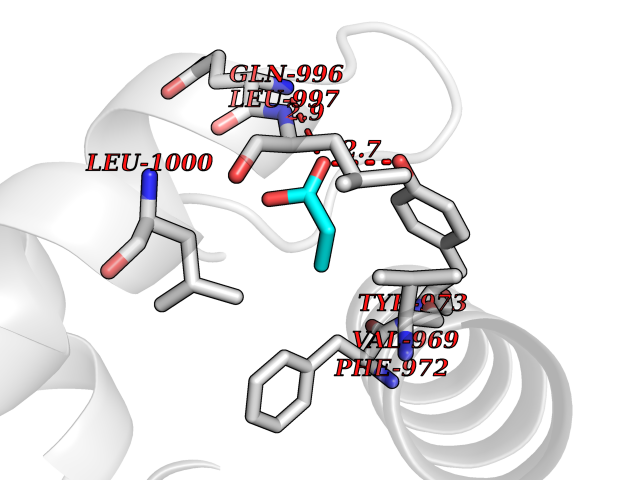 Pymol Map 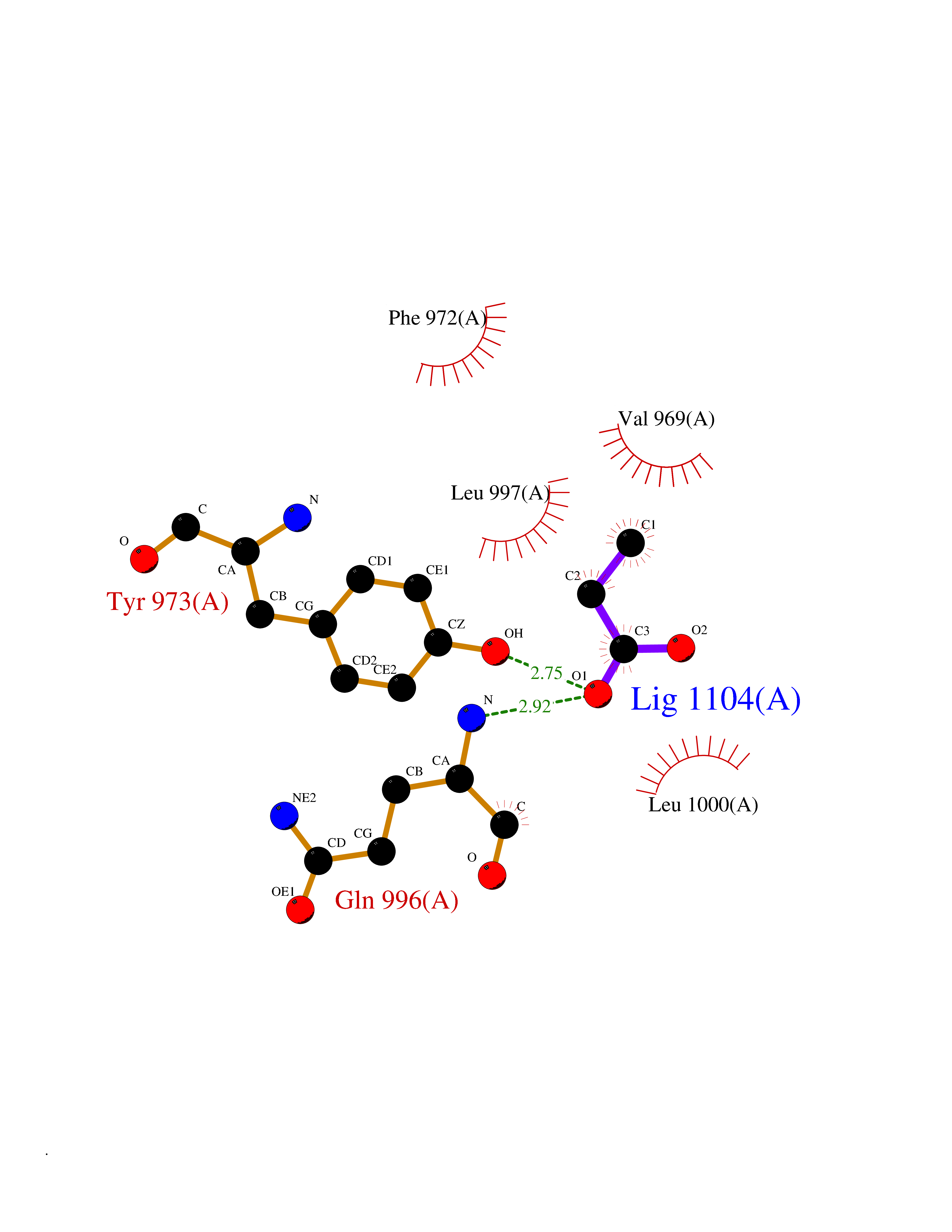 Ligplot Map 3D Binding mode Sequence KPILAPEPLVMDNLDSIMEQLNTWNFPIFDLVENIGRKCGRILSQVSYRLFEDMGLFEAFKIPIREFMNYFHALEIGYRDIPYHNRIHATDVLHAVWYLTTQPIPGLSTVGYVFSKTYNVTDDKYGCLSGNIPALELMALYVAAAMHDYDHPGRTNAFLVATSAPQAVLYNDRSVLENHHAAAAWNLFMSRPEYNFLINLDHVEFKHFRFLVIEAILATDLKKHFDFVAKFNGKVNDDVGIDWTNENDRLLVCQMCIKLADINGPAKCKELHLQWTDGIVNEFYEQGDEEASLGLPISPFMDRSAPQLANLQESFISHIVGPLCNSYDSAGLMPGKWVEKIYCQITQHLLQNHKMWKKVIEEEQRLAGIENQNISVDLETNYAELVLDVGRVTLGENSRKKMKDCKLRKKQNESVSRAMCALLNSGGGVIKAEIENEDYSYTKDGIGLDLENSFSNILLFVPEYLDFMQNGNYFLIFVKSWSLNTSGLRITTLSSNLYKRDITSAKVMNATAALEFLKDMKKTRGRLYLRPELLAKRPCVDIQEENNMKALAGVFFDRTELDRKEKLTFTESTHVEIKNFSTERLLQRIKEILPQYVSAFANTDGGYLFIGLNEDKEIIGFKAEMSDLDDLEREIEKSIRKMPVHHFCMEKKKINYSCKFLGVYDKGSLCGYVCALRVERFCCAVFAKEPDSWHVKDNRVMQLTRKEWIQFMVEAEPKFSSAYEEVISQINTSLPAPHSWPLLEWQRQRHHCPGLSGRITYTPENLCRKLFLQHEGLKQLICEEMSSVRKGSLIFSRSWSVDLGLQENHKVLCDALLISQDSPPVLYTFHMVQDEEFKGYSTQTALTLKQKLAKIGGYTKKVCVMTKIFYLSPEGMTSCQYDLRSQVIYPESYYFTRRKYLLKALFKALKRLKSLRDQFSFAENLYQIIGIDCFQKNDK Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Gamma-aminobutyric acid type B receptor subunit 1 | 4MS4 | 4.56 | |
Target general information Gen name GABBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPRC3B;GPR51 Protein family G-protein coupled receptor 3 family, GABA-B receptor subfamily Biochemical class Signaling protein / antagonist Function G-protein coupled GABA receptor activity. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08891; DB08892; DB00181; DB00363; DB02530; DB05010; DB09072 Interacts with Q9UBS5; Q9UBS5-2; P46459; Q86UR5 EC number NA Uniprot keywords 3D-structure; Cell membrane; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Intellectual disability; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 46502.1 Length 408 Aromaticity 0.12 Instability index 50.05 Isoelectric point 5.78 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -6.22 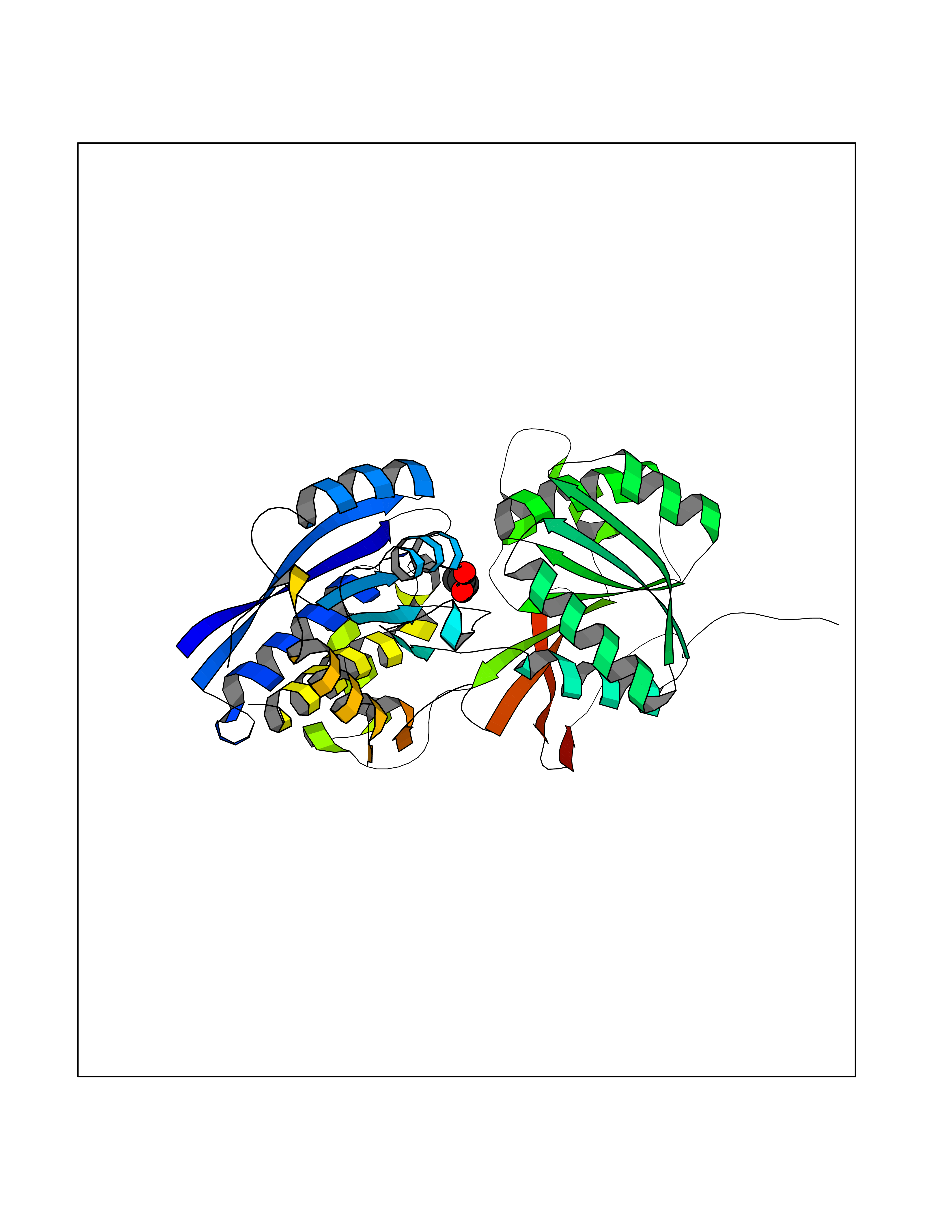 Molscript Map  Pymol Map 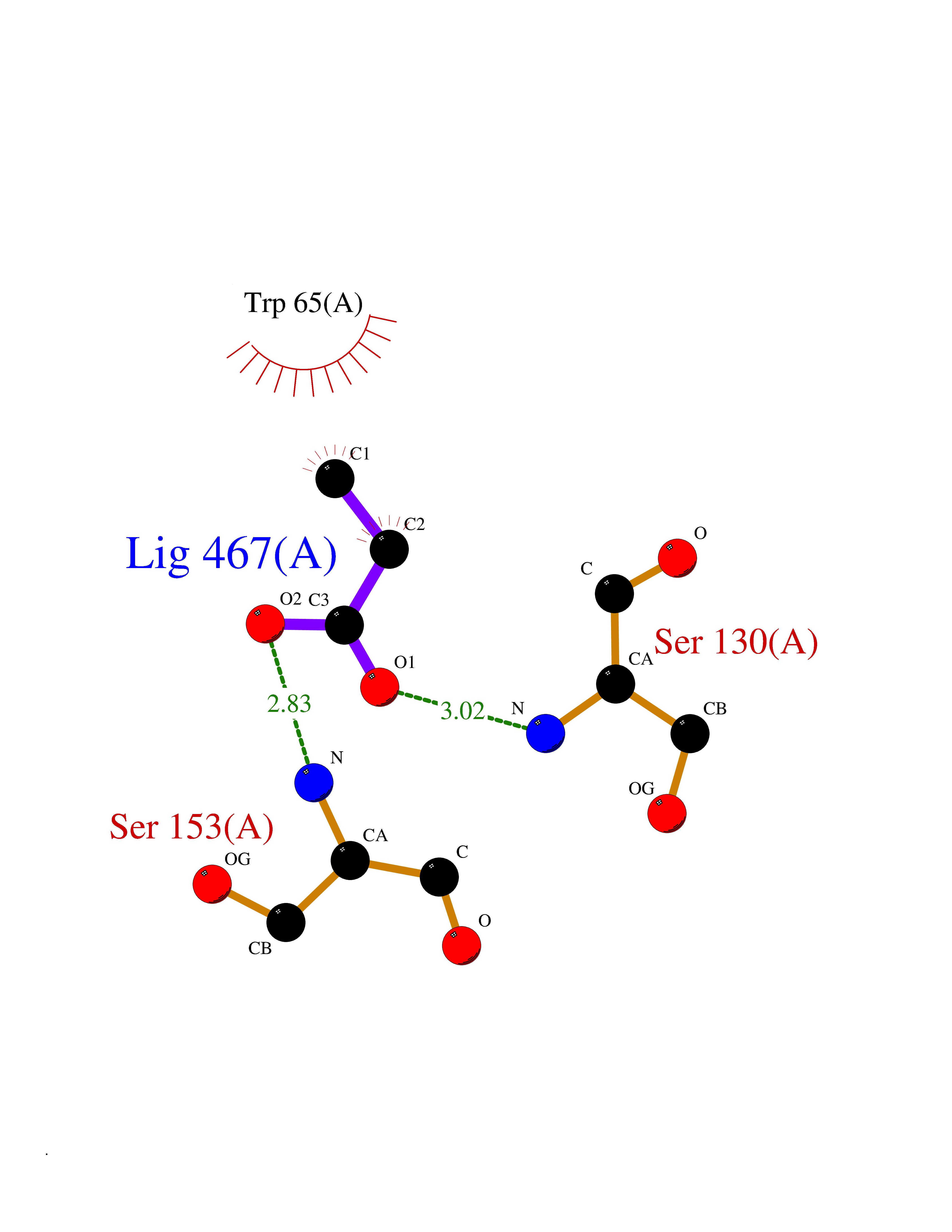 Ligplot Map 3D Binding mode Sequence RRAVYIGALFPMSGGWPGGQACQPAVEMALEDVNSRRDILPDYELKLIHHDSKCDPGQATKYLYELLYNDPIKIILMPGCSSVSTLVAEAARMWNLIVLSYGSSSPALSNRQRFPTFFRTHPSATLHNPTRVKLFEKWGWKKIATIQQTTEVFTSTLDDLEERVKEAGIEITFRQSFFSDPAVPVKNLKRQDARIIVGLFYETEARKVFCEVYKERLFGKKYVWFLIGWYADNWFKIYDPSINCTVDEMTEAVEGHITTEIVMLNPANTRSISNMTSQEFVEKLTKRLKRHPEETGGFQEAPLAYDAIWALALALNKTSRLEDFNYNNQTITDQIYRAMNSSSFEGVSGHVVFDASGSRMAWTLIEQLQGGSYKKIGYYDSTKDDLSWSKTDKWIGGSPPADDYKDDD Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | 30S ribosomal protein S3 | 4ODQ | 4.56 | |
Target general information Gen name rpsC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW3276;b3314 Protein family Universal ribosomal protein uS3 family Biochemical class Isomerase Function RRNA binding.Structural constituent of ribosome. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09093; DB12455; DB00759 Interacts with NA EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Reference proteome; Ribonucleoprotein; Ribosomal protein; RNA-binding; rRNA-binding Protein physicochemical properties Chain ID B Molecular weight (Da) 12457.7 Length 112 Aromaticity 0.05 Instability index 12.49 Isoelectric point 5.91 Charge (pH=7) -7.24 2D Binding mode Binding energy (Kcal/mol) -6.22  Molscript Map 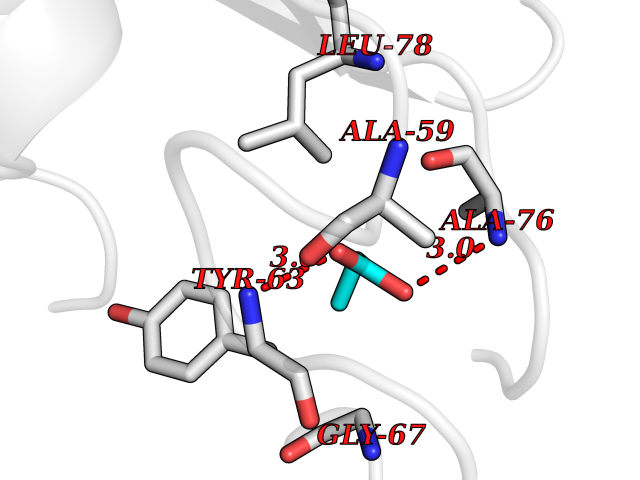 Pymol Map 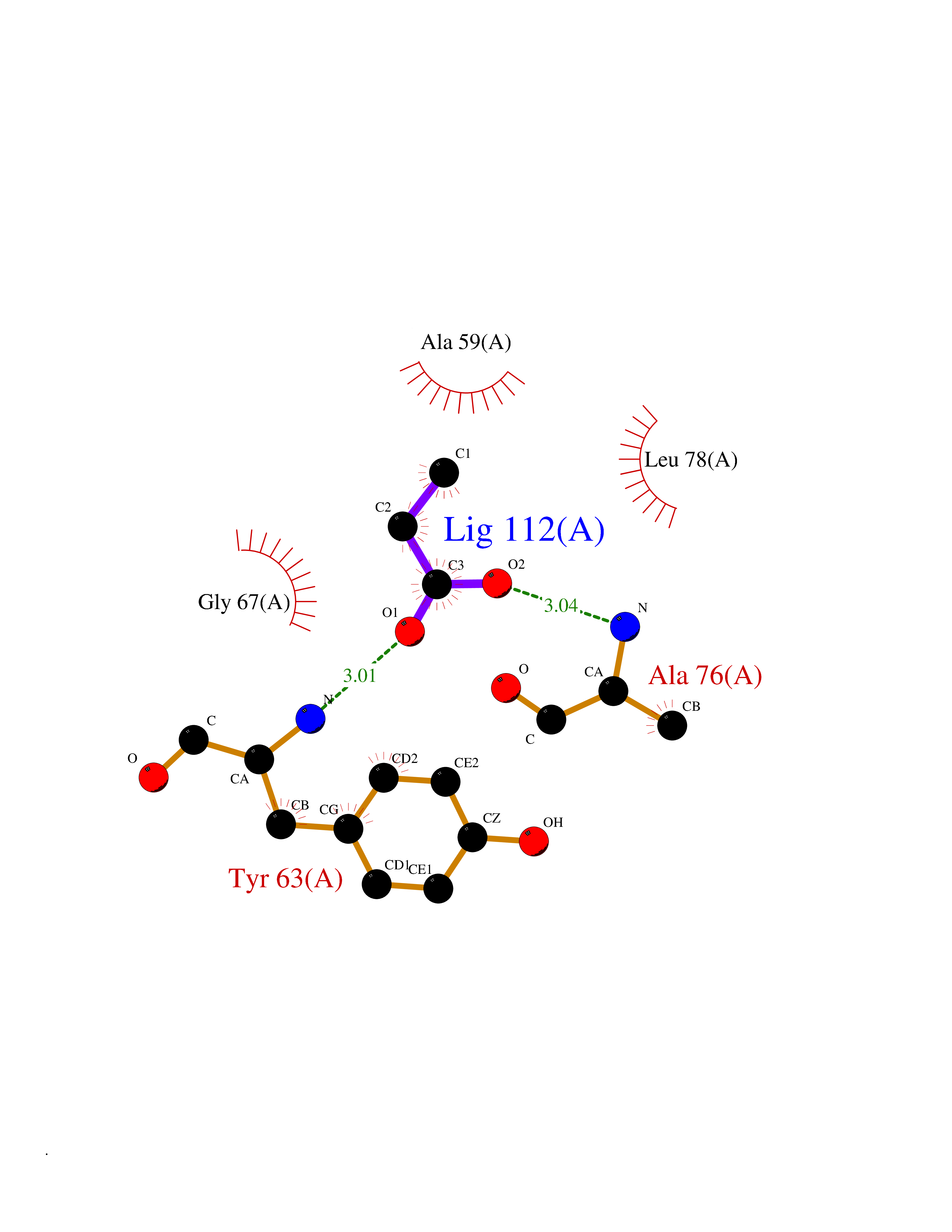 Ligplot Map 3D Binding mode Sequence MKVGQDKVVTIRYTLQVEGEVLDQGELSYLHGHRNLIPGLEEALEGREEGEAFQAHVPAEKAYGATGHPPHATLDFQVEVVKVREATPEELLHGHAHPSGHHHHHHGIVKPW Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Neprilysin | 1R1H | 4.56 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms EPN Protein family Peptidase M13 family Biochemical class Hydrolase Function Endopeptidase activity.Exopeptidase activity.Metalloendopeptidase activity.Metallopeptidase activity.Peptide binding.Zinc ion binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -6.22  Molscript Map 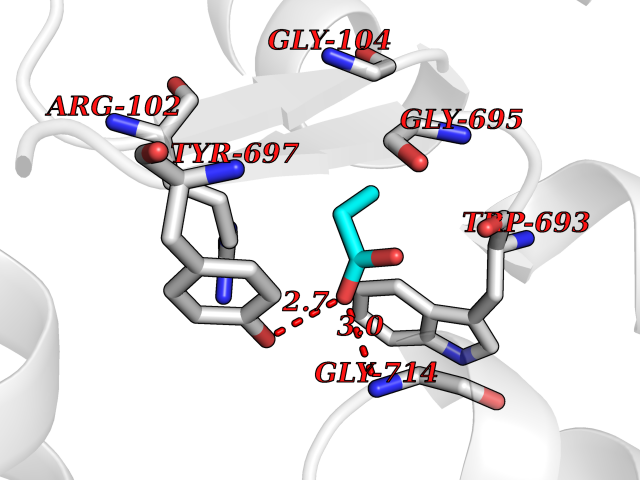 Pymol Map 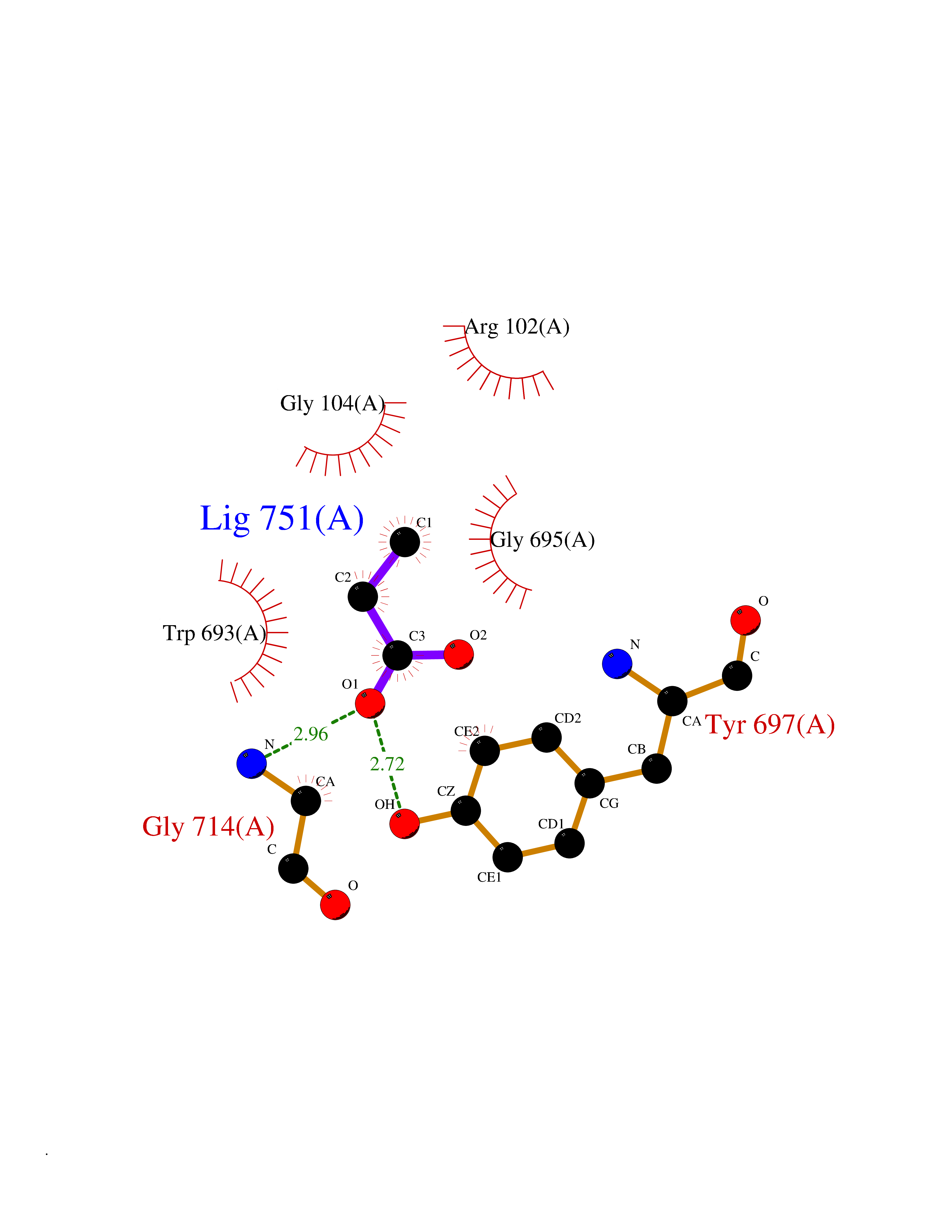 Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | mRNA-capping enzyme | 2C46 | 4.56 | |
Target general information Gen name RNGTT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CAP1A Protein family Non-receptor class of the protein-tyrosine phosphatase family; Eukaryotic GTase family Biochemical class Transferase Function GTP binding.MRNA guanylyltransferase activity.Polynucleotide 5'-phosphatase activity.Protein tyrosine/serine/threonine phosphatase activity.Protein tyrosine phosphatase activity.RNA guanylyltransferase activity.Triphosphatase activity. Related diseases Atrial fibrillation, familial, 14 (ATFB14) [MIM:615378]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:19808477}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Genetic variations in SCN2B may be involved in Brugada syndrome (PubMed:23559163). This tachyarrhythmia is characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:23559163}. Drugs (DrugBank ID) NA Interacts with Q92624; P16333-1 EC number 2.7.7.50; 3.6.1.74 Uniprot keywords 3D-structure; Alternative splicing; GTP-binding; Host-virus interaction; Hydrolase; mRNA capping; mRNA processing; Multifunctional enzyme; Nucleotide-binding; Nucleotidyltransferase; Nucleus; Protein phosphatase; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 21849.8 Length 189 Aromaticity 0.11 Instability index 53.71 Isoelectric point 5.89 Charge (pH=7) -2.91 2D Binding mode Binding energy (Kcal/mol) -6.22  Molscript Map  Pymol Map 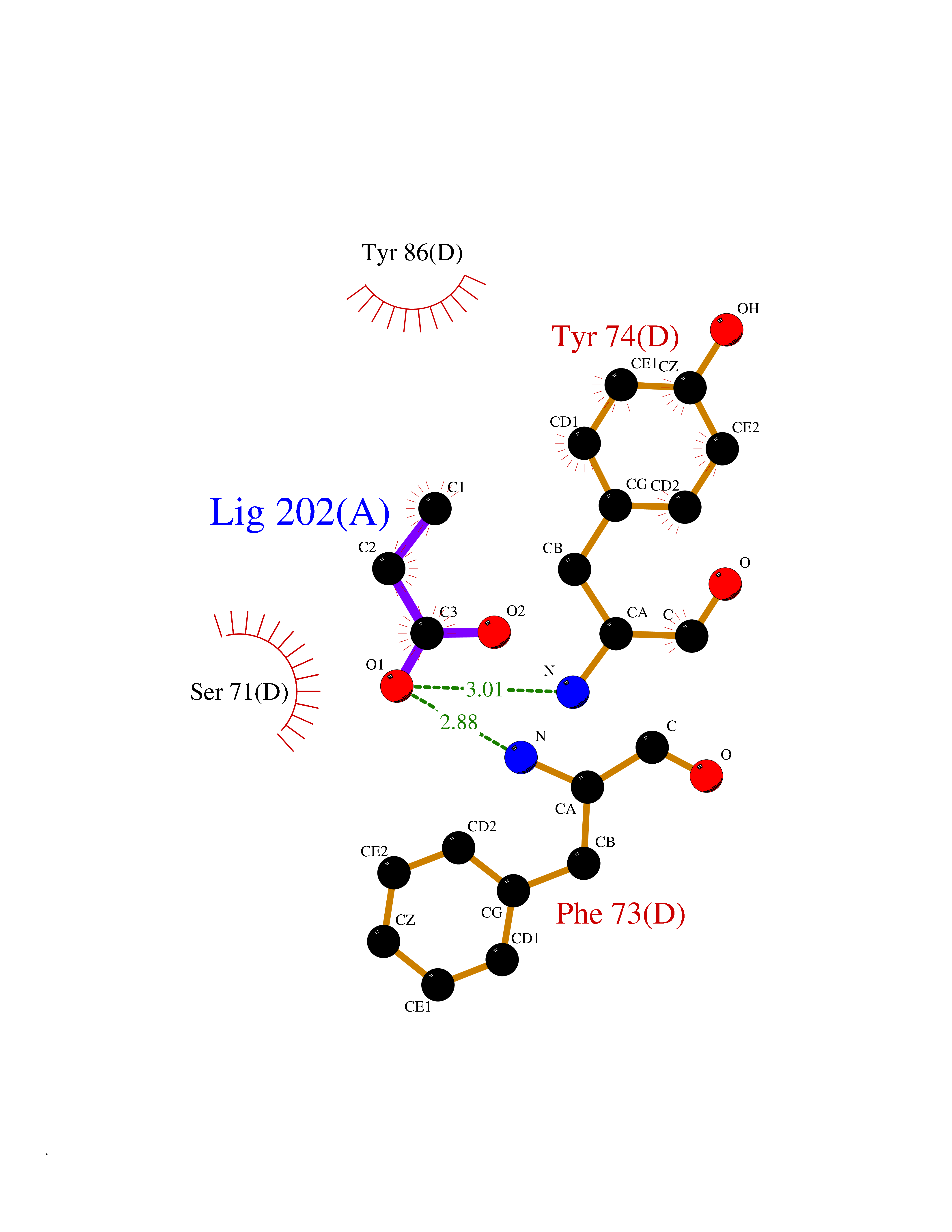 Ligplot Map 3D Binding mode Sequence NKIPPRWLNCPRRGQPVAGRFLPLKTMLGPRYDSQVAEENRFHPSMLSNYLKSVKMGLLVDLTNTSRFYDRNDIEKEGIKYIKLQCKGHGECPTTENTETFIRLCERFELIGVHCTHGFNRTGFLICAFLVEKMDWSIEAAVATFAQARPPGIYKGDYLKELFRRYGDIEEAPPPPLLPDWCFEDDEDE Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | NH(3)-dependent NAD(+) synthetase | 1KQP | 4.56 | |
Target general information Gen name nadE Organism Bacillus subtilis (strain 168) Uniprot ID TTD ID NA Synonyms outB;BSU03130 Protein family NAD synthetase family Biochemical class Ligase Function ATP binding.Metal ion binding.NAD+ synthase (glutamine-hydrolyzing) activity.NAD+ synthase activity. Related diseases Leukodystrophy, hypomyelinating, 15 (HLD15) [MIM:617951]: An autosomal recessive disorder characterized by hypomyelinating leukodystrophy with thinning of the corpus callosum. Clinical features include motor and cognitive impairment appearing in the first or second decade of life, dystonia, ataxia, spasticity, and dysphagia. Most patients develop severe optic atrophy, and some have hearing loss. {ECO:0000269|PubMed:29576217}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02596; DB04099; DB00798 Interacts with NA EC number 6.3.1.5 Uniprot keywords 3D-structure; ATP-binding; Direct protein sequencing; Ligase; Magnesium; Metal-binding; NAD; Nucleotide-binding; Phosphoprotein; Reference proteome; Sporulation; Stress response Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60509.3 Length 542 Aromaticity 0.08 Instability index 31.96 Isoelectric point 5.07 Charge (pH=7) -19.73 2D Binding mode Binding energy (Kcal/mol) -6.22 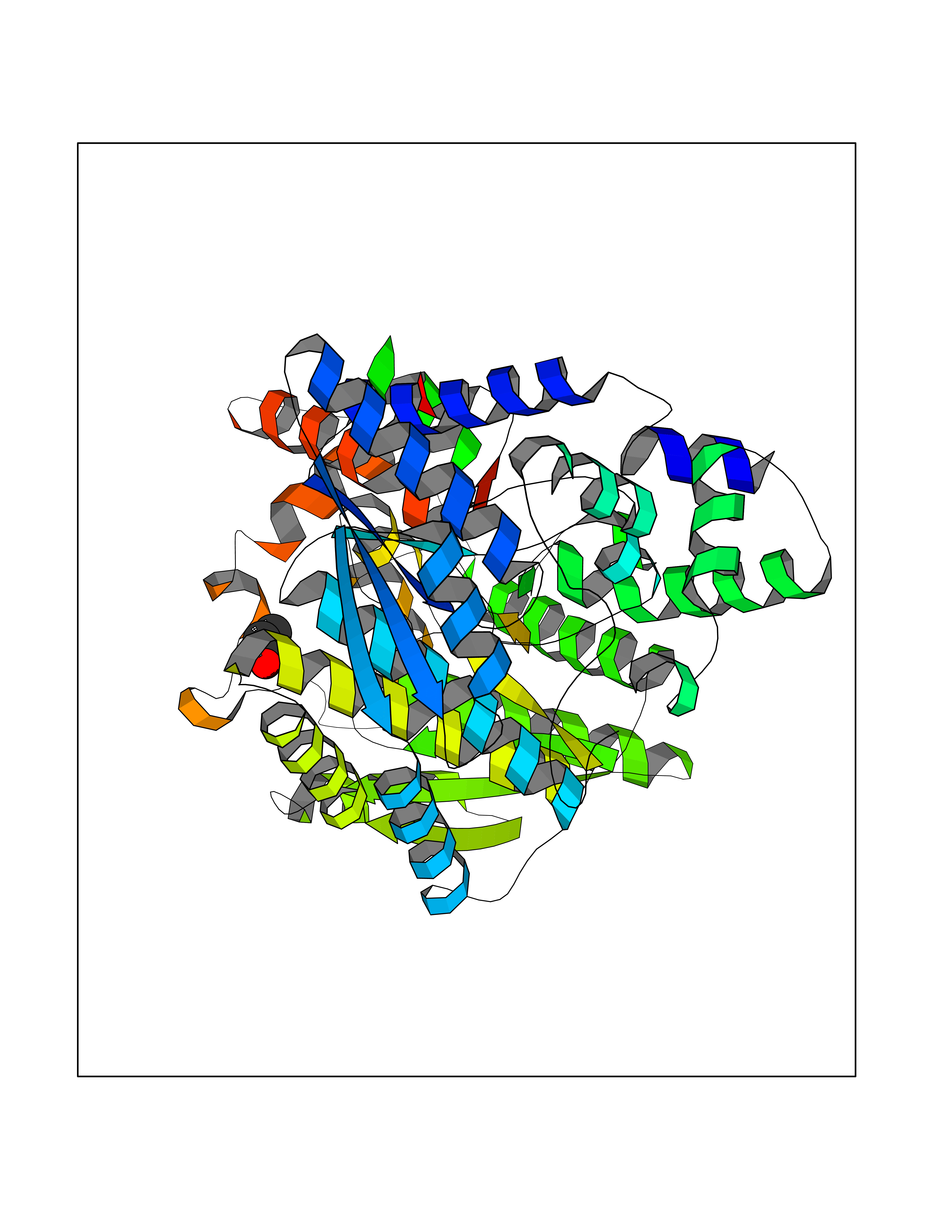 Molscript Map 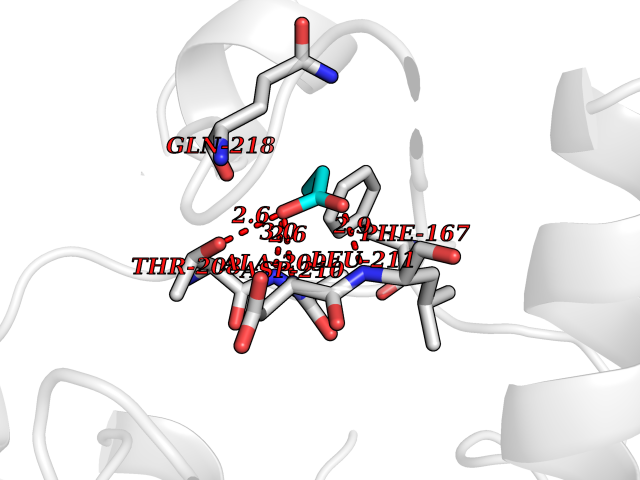 Pymol Map 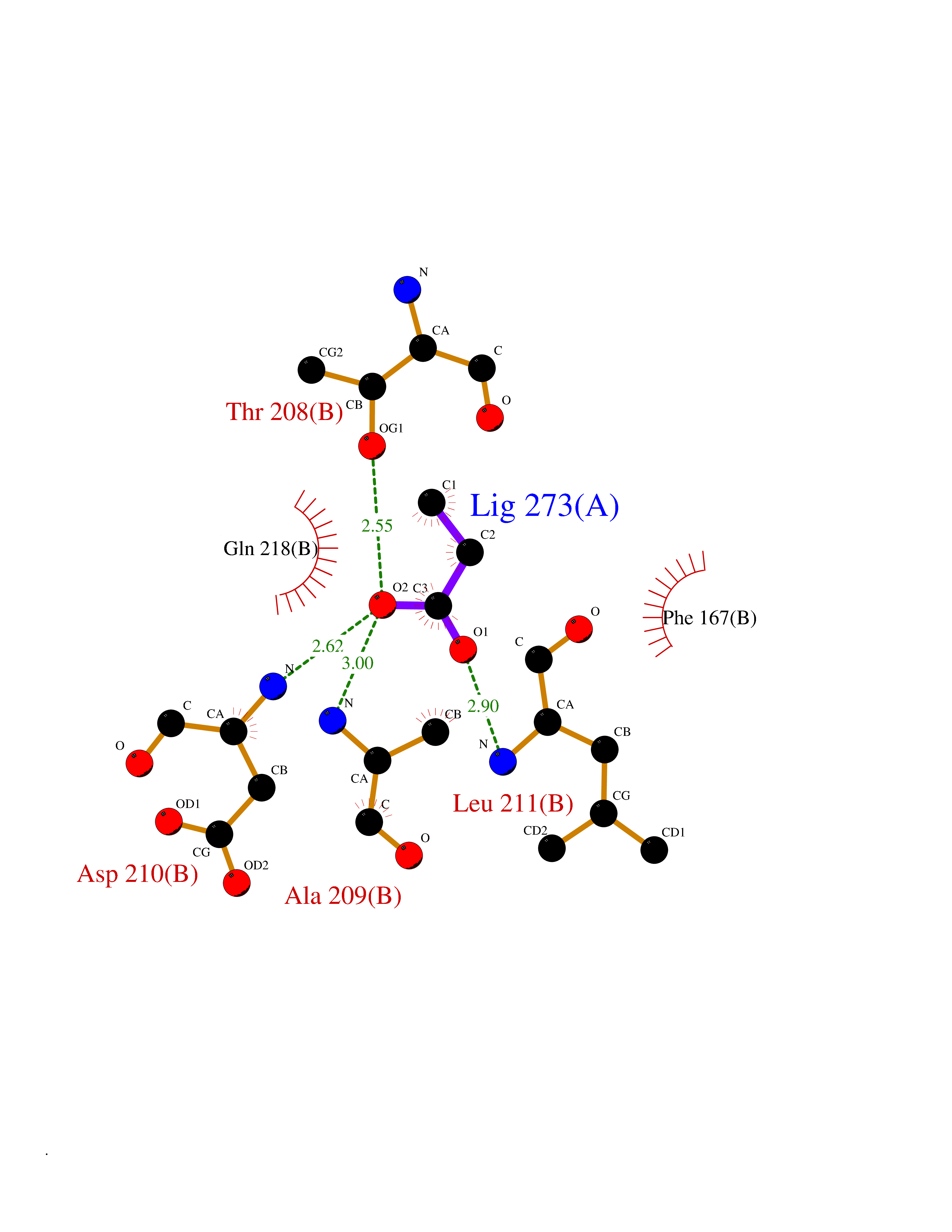 Ligplot Map 3D Binding mode Sequence SMQEKIMRELHVKPSIDPKQEIEDRVNFLKQYVKKTGAKGFVLGISGGQDSTLAGRLAQLAVESIREEGGDAQFIAVRLPHGTQQDEDDAQLALKFIKPDKSWKFDIKSTVSAFSDQYQQETGDQLTDFNKGNVKARTRMIAQYAIGGQEGLLVLGTDHAAEAVTGFFTKYGDGGADLLPLTGLTKRQGRTLLKELGAPERLYLKEPTADLLDEKPQQSDETELGISYDEIDDYLEGKEVSAKVSEALEKRYSMTEHKRQVPASMFDDWWKSMQEKIMRELHVKPSIDPKQEIEDRVNFLKQYVKKTGAKGFVLGISGGQDSTLAGRLAQLAVESIREEGGDAQFIAVRLPHGTQQDEDDAQLALKFIKPDKSWKFDIKSTVSAFSDQYQQETGDQLTDFNKGNVKARTRMIAQYAIGGQEGLLVLGTDHAAEAVTGFFTKYGDGGADLLPLTGLTKRQGRTLLKELGAPERLYLKEPTADLLDEKPQQSDETELGISYDEIDDYLEGKEVSAKVSEALEKRYSMTEHKRQVPASMFDDWWK Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Tyrosine-protein kinase ABL1 (ABL) | 5HU9 | 4.56 | |
Target general information Gen name ABL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p150; Proto-oncogene tyrosine-protein kinase ABL1; Proto-oncogene c-Abl; JTK7; C-ABL; Abl; Abelson tyrosine-protein kinase 1; Abelson murine leukemia viral oncogene homolog 1 Protein family Protein kinase superfamily, Tyr protein kinase family, ABL subfamily Biochemical class Kinase Function Coordinates actin remodeling through tyrosine phosphorylation of proteins controlling cytoskeleton dynamics like WASF3 (involved in branch formation); ANXA1 (involved in membrane anchoring); DBN1, DBNL, CTTN, RAPH1 and ENAH (involved in signaling); or MAPT and PXN (microtubule-binding proteins). Phosphorylation of WASF3 is critical for the stimulation of lamellipodia formation and cell migration. Involved in the regulation of cell adhesion and motility through phosphorylation of key regulators of these processes such as BCAR1, CRK, CRKL, DOK1, EFS or NEDD9. Phosphorylates multiple receptor tyrosine kinases and more particularly promotes endocytosis of EGFR, facilitates the formation of neuromuscular synapses through MUSK, inhibits PDGFRB-mediated chemotaxis and modulates the endocytosis of activated B-cell receptor complexes. Other substrates which are involved in endocytosis regulation are the caveolin (CAV1) and RIN1. Moreover, ABL1 regulates the CBL family of ubiquitin ligases that drive receptor down-regulation and actin remodeling. Phosphorylation of CBL leads to increased EGFR stability. Involved in late-stage autophagy by regulating positively the trafficking and function of lysosomal components. ABL1 targets to mitochondria in response to oxidative stress and thereby mediates mitochondrial dysfunction and cell death. In response to oxidative stress, phosphorylates serine/threonine kinase PRKD2 at 'Tyr-717'. ABL1 is also translocated in the nucleus where it has DNA-binding activity and is involved in DNA-damage response and apoptosis. Many substrates are known mediators of DNA repair: DDB1, DDB2, ERCC3, ERCC6, RAD9A, RAD51, RAD52 or WRN. Activates the proapoptotic pathway when the DNA damage is too severe to be repaired. Phosphorylates TP73, a primary regulator for this type of damage-induced apoptosis. Phosphorylates the caspase CASP9 on 'Tyr-153' and regulates its processing in the apoptotic response to DNA damage. Phosphorylates PSMA7 that leads to an inhibition of proteasomal activity and cell cycle transition blocks. ABL1 acts also as a regulator of multiple pathological signaling cascades during infection. Several known tyrosine-phosphorylated microbial proteins have been identified as ABL1 substrates. This is the case of A36R of Vaccinia virus, Tir (translocated intimin receptor) of pathogenic E. coli and possibly Citrobacter, CagA (cytotoxin-associated gene A) of H. pylori, or AnkA (ankyrin repeat-containing protein A) of A. phagocytophilum. Pathogens can highjack ABL1 kinase signaling to reorganize the host actin cytoskeleton for multiple purposes, like facilitating intracellular movement and host cell exit. Finally, functions as its own regulator through autocatalytic activity as well as through phosphorylation of its inhibitor, ABI1. Regulates T-cell differentiation in a TBX21-dependent manner. Phosphorylates TBX21 on tyrosine residues leading to an enhancement of its transcriptional activator activity. Non-receptor tyrosine-protein kinase that plays a role in many key processes linked to cell growth and survival such as cytoskeleton remodeling in response to extracellular stimuli, cell motility and adhesion, receptor endocytosis, autophagy, DNA damage response and apoptosis. Related diseases Leukemia, chronic myeloid (CML) [MIM:608232]: A clonal myeloproliferative disorder of a pluripotent stem cell with a specific cytogenetic abnormality, the Philadelphia chromosome (Ph), involving myeloid, erythroid, megakaryocytic, B-lymphoid, and sometimes T-lymphoid cells, but not marrow fibroblasts. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: A chromosomal aberration involving ABL1 has been found in patients with chronic myeloid leukemia. Translocation t(9;22)(q34;q11) with BCR. The translocation produces a BCR-ABL found also in acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL). {ECO:0000269|PubMed:3021337}.; DISEASE: A chromosomal aberration involving ABL1 is found in a form of acute lymphoblastic leukemia (PubMed:15361874). Translocation t(9;9)(q34;q34) with NUP214 (PubMed:15361874). {ECO:0000269|PubMed:15361874}.; DISEASE: Congenital heart defects and skeletal malformations syndrome (CHDSKM) [MIM:617602]: An autosomal dominant disorder characterized by congenital heart disease with atrial and ventricular septal defects, variable skeletal abnormalities, and failure to thrive. Skeletal defects include pectus excavatum, scoliosis, and finger contractures. Some patient exhibit joint laxity. {ECO:0000269|PubMed:28288113}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08043; DB08583; DB07831; DB08350; DB12597; DB00171; DB06616; DB12267; DB01254; DB12010; DB00619; DB13749; DB08231; DB03878; DB04868; DB08339; DB08901; DB12323; DB08896; DB14989; DB05184 Interacts with Q8IZP0; Q9NYB9; O14672; P10275; Q13315; Q4KMG0; P46108; P46109; P35222; P00533; P04626; Q03468; Q14315; P36888; P05107; P10721; Q38SD2; Q92918; Q7Z434; O43196; P15941; P15941-12; P16333; O43900; Q13905; Q86UR5; Q13671; P31947; Q15464; O75751; P37840; Q9BX66; O60504-2; Q07890; P12931; P51692; Q9Y4G6; P11387; P04637; P15498; Q9Y6W5; P62258; P61981; P63104; O35158; P37840; P48165; Q15323; P37840 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; ATP-binding; Autophagy; Cell adhesion; Chromosomal rearrangement; Cytoplasm; Cytoskeleton; Disease variant; DNA damage; DNA repair; DNA-binding; Endocytosis; Kinase; Lipoprotein; Magnesium; Manganese; Membrane; Metal-binding; Mitochondrion; Myristate; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; SH2 domain; SH3 domain; Transferase; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 31264.6 Length 270 Aromaticity 0.12 Instability index 37.99 Isoelectric point 5.42 Charge (pH=7) -7.67 2D Binding mode Binding energy (Kcal/mol) -6.22 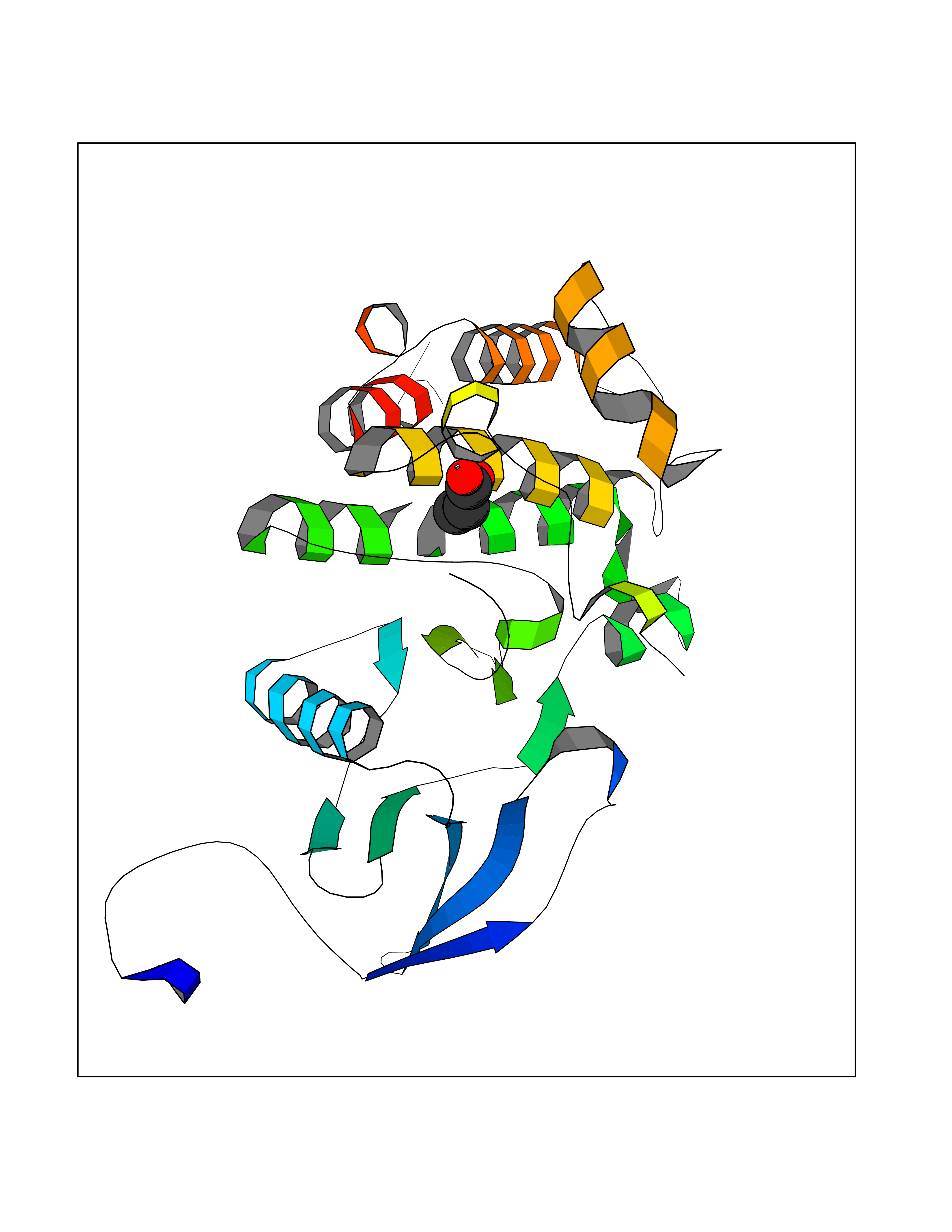 Molscript Map 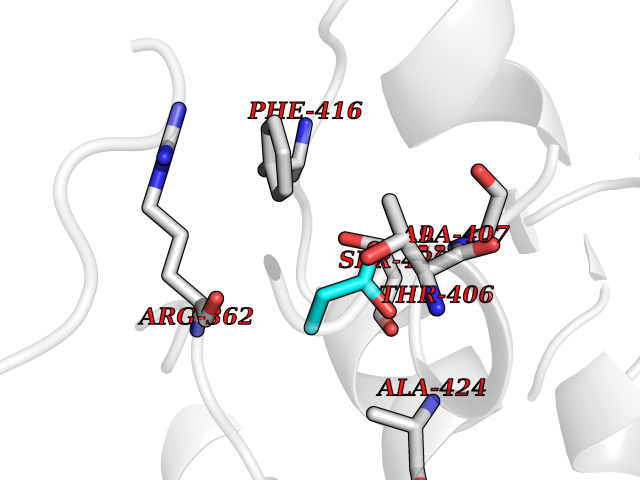 Pymol Map 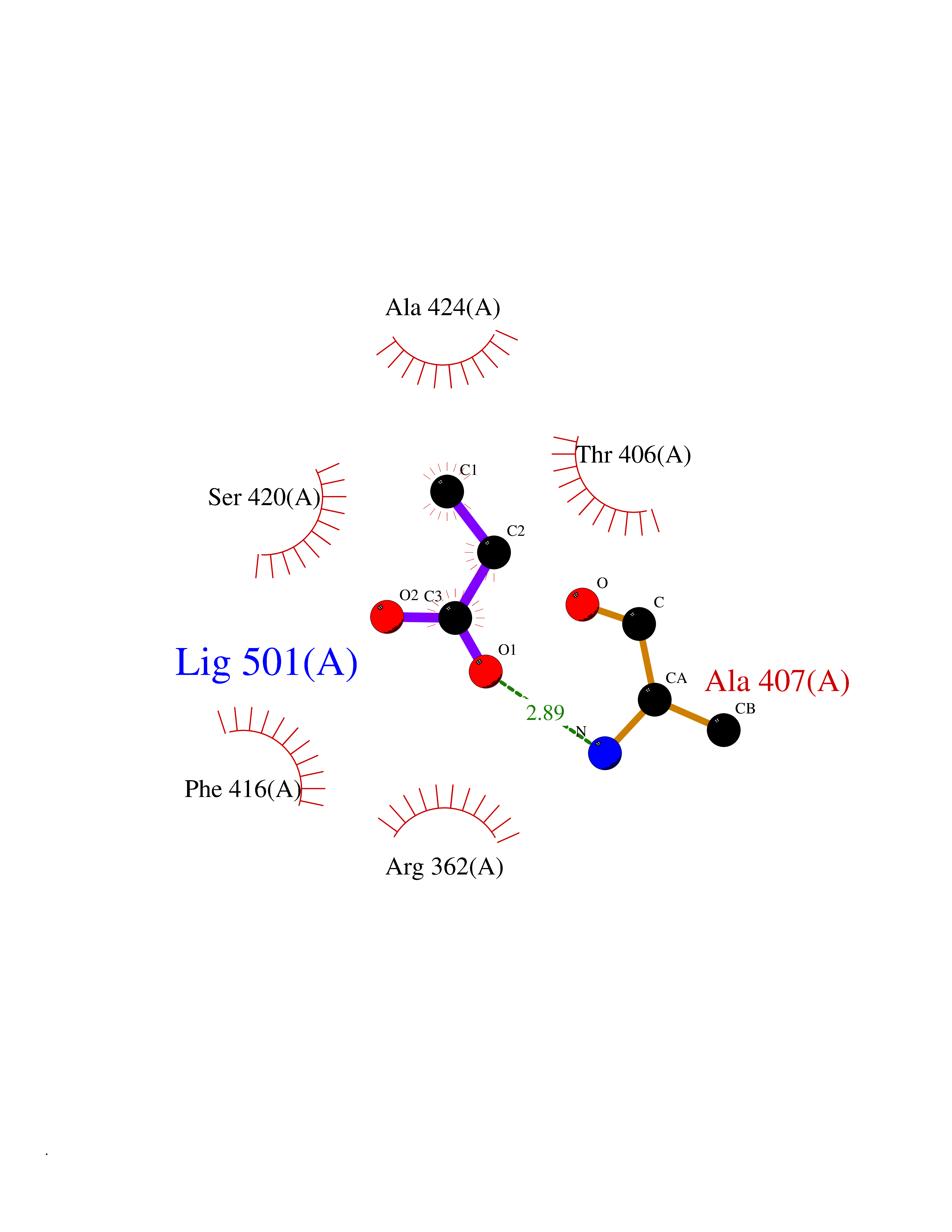 Ligplot Map 3D Binding mode Sequence AMGSSPNYDKWEMERTDITMKHKLGGGQYGEVYEGVWKKYSLTVAVKTLKEDTMEVEEFLKEAAVMKEIKHPNLVQLLGVCTREPPFYIITEFMTYGNLLDYLRECNRQEVNAVVLLYMATQISSAMEYLEKKNFIHRDLAARNCLVGENHLVKVADFGLSRLMTAHAGAKFPIKWTAPESLAYNKFSIKSDVWAFGVLLWEIATYGMSPYPGIDLSQVYELLEKDYRMERPEGCPEKVYELMRACWQWNPSDRPSFAEIHQAFETMFQE Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Catechol-O-methyl-transferase (COMT) | 3BWY | 4.56 | |
Target general information Gen name COMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms S-COMT; MB-COMT; Catechol-O-methyltransferase; COMT Protein family Class I-like SAM-binding methyltransferase superfamily, Cation-dependent O-methyltransferase family Biochemical class Methyltransferase Function Catalyzes the O-methylation, and thereby the inactivation, of catecholamine neurotransmitters and catechol hormones. Also shortens the biological half-lives of certain neuroactive drugs, like L-DOPA, alpha-methyl DOPA and isoproterenol. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07462; DB02342; DB02105; DB08049; DB00118; DB00714; DB03336; DB00286; DB00255; DB00841; DB00988; DB15488; DB00494; DB00668; DB00783; DB00977; DB01064; DB00968; DB01141; DB03907; DB04820; DB06152; DB11632; DB00252; DB01420; DB00323 Interacts with Q6P5T0; P30518; Q8NFU1; Q8NHW4; P34972; Q96BA8; P50402; Q5JX71; O14843; O00258; P08034; O75712; Q9NTQ9; O95377; Q8TDT2; Q8N6U8; O15529; P31937; Q9H2F3; O95279; Q5SR56; A6NDP7; Q0D2K0; Q7RTS5; Q9UHJ9-5; Q8IY26; Q9H6H4; Q6NTF9-3; O75783; Q99500; Q9Y6D0; Q3KNW5; O60669; P22732; Q96G79; Q5T1Q4; Q9NY26; Q9NP94; Q6P1K1; P30825; Q9UHI5; B2RUZ4; Q9UPZ6; Q96MV1; Q9NV29; A0PK00; Q9NUH8; Q9P0S9; Q14656; Q6UW68; Q9H0R3; O95807; P34981; Q15645; Q15836; O95183; O76024; P30260; Q9H816; Q92997; P29323-3; P22607; P06396; Q15323; Q6A162; P26371; O15116; P20645; O14744; Q5T160; Q9UJD0; Q2MKA7; Q8N488; O75880; Q14141; Q9UNE7; Q15645; Q9NYH9; Q8NA23-2 EC number EC 2.1.1.6 Uniprot keywords 3D-structure; Alternative initiation; Catecholamine metabolism; Cell membrane; Cytoplasm; Direct protein sequencing; Lipid metabolism; Magnesium; Membrane; Metal-binding; Methyltransferase; Neurotransmitter degradation; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Schizophrenia; Signal-anchor; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 23851.2 Length 214 Aromaticity 0.07 Instability index 25.99 Isoelectric point 5.25 Charge (pH=7) -7.75 2D Binding mode Binding energy (Kcal/mol) -6.21 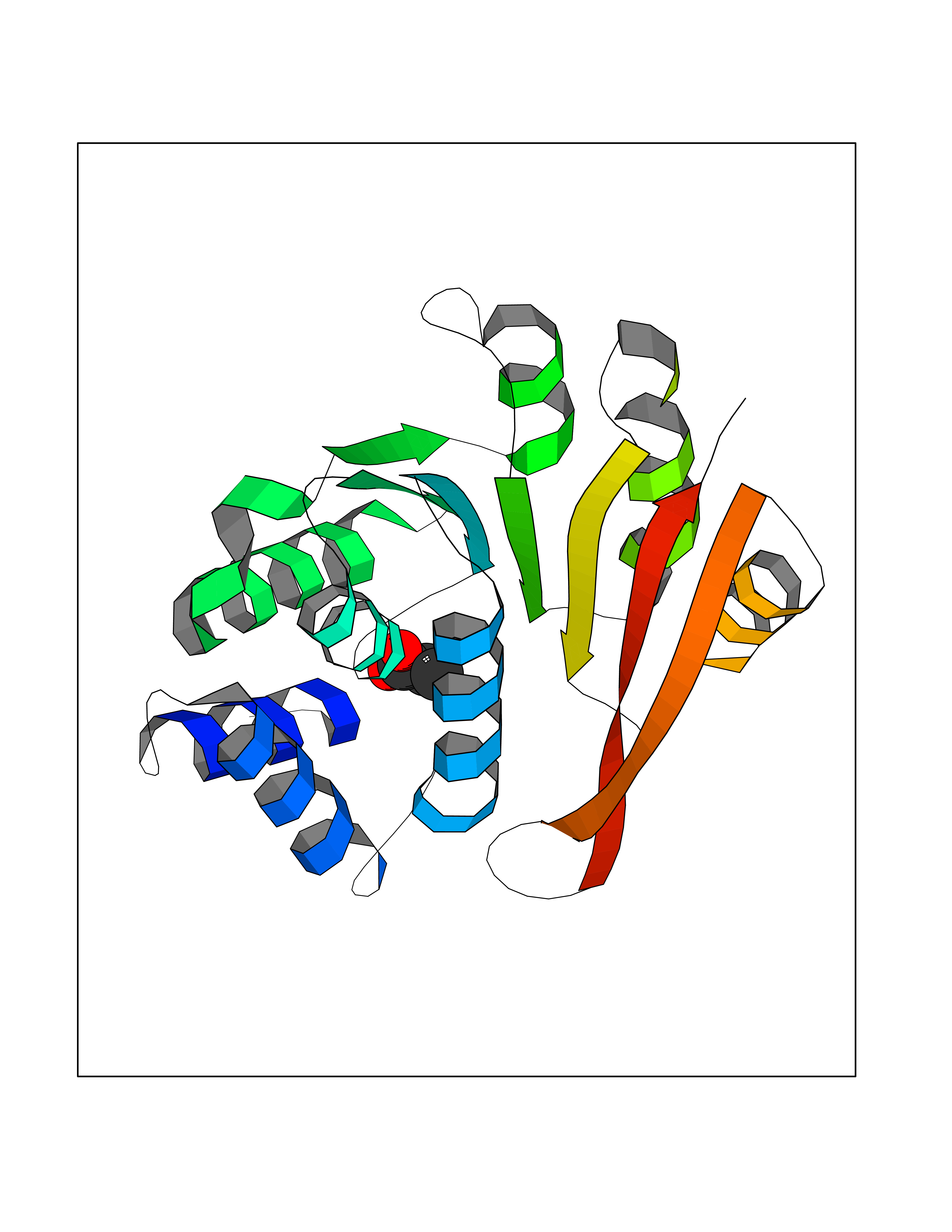 Molscript Map 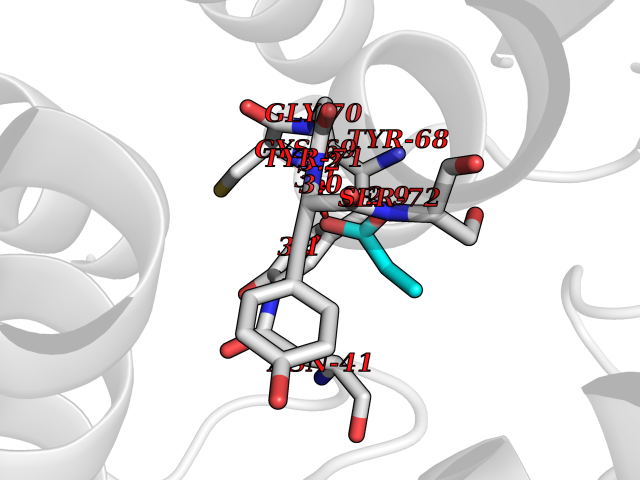 Pymol Map  Ligplot Map 3D Binding mode Sequence GDTKEQRILNHVLQHAEPGNAQSVLEAIDTYCEQKEWAMNVGDKKGKIVDAVIQEHQPSVLLELGAYCGYSAVRMARLLSPGARLITIEINPDCAAITQRMVDFAGMKDKVTLVVGASQDIIPQLKKKYDVDTLDMVFLDHWKDRYLPDTLLLEECGLLRKGTVLLADNVICPGAPDFLAHVRGSSCFECTHYQSFLEYREVVDGLEKAIYKGP Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | 2,4-dienoyl-CoA reductase | 1PS9 | 4.56 | |
Target general information Gen name fadH Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3081;ygjL;JW3052 Protein family NADH:flavin oxidoreductase/NADH oxidase family Biochemical class Oxidoreductase Function 2,4-dienoyl-CoA reductase (NADPH) activity.4 iron, 4 sulfur cluster binding.FAD binding.FMN binding.Metal ion binding. Related diseases Smith-Kingsmore syndrome (SKS) [MIM:616638]: An autosomal dominant syndrome characterized by intellectual disability, macrocephaly, seizures, umbilical hernia, and facial dysmorphic features. {ECO:0000269|PubMed:25851998, ECO:0000269|PubMed:26542245, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Focal cortical dysplasia 2 (FCORD2) [MIM:607341]: A form of focal cortical dysplasia, a malformation of cortical development that results in medically refractory epilepsy in the pediatric population and in adults. FCORD2 is a severe form, with onset usually in childhood, characterized by disrupted cortical lamination and specific cytological abnormalities. It is classified in 2 subtypes: type IIA characterized by dysmorphic neurons and lack of balloon cells; type IIB with dysmorphic neurons and balloon cells. {ECO:0000269|PubMed:25799227, ECO:0000269|PubMed:25878179, ECO:0000269|PubMed:26018084, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03698; DB03147; DB03247; DB03461 Interacts with P11349; P19318 EC number 1.3.1.34 Uniprot keywords 3D-structure; 4Fe-4S; Direct protein sequencing; FAD; Flavoprotein; FMN; Iron; Iron-sulfur; Metal-binding; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 40003.1 Length 366 Aromaticity 0.07 Instability index 33.42 Isoelectric point 5.65 Charge (pH=7) -9.48 2D Binding mode Binding energy (Kcal/mol) -6.22  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SYPSLFAPLDLGFTTLKNRVLMGSMHTGLEEYPDGAERLAAFYAERARHGVALIVSGGIAPDLTGVGMEGGAMLNDASQIPHHRTITEAVHQEGGKIALQILHTGRYSYQPHLVAPSALQAPINRFVPHELSHEEILQLIDNFARCAQLAREAGYDGVEVMGSEGYLINEFLTLRTNQRSDQWGGDYRNRMRFAVEVVRAVRERVGNDFIIIYRLSMLDLVEDGGTFAETVELAQAIEAAGATIINTGIGWHEARIPTIATPVPRGAFSWVTRKLKGHVSLPLVTTNRINDPQVADDILSRGDADMVSMARPFLADAELLSKAQSGRADEINTCIGCNQACLDQIFVGKVTSCLVNPRACHETKMP Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Fms-like tyrosine kinase 3 (FLT-3) | 1RJB | 4.56 | |
Target general information Gen name FLT3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Stem cell tyrosine kinase 1; STK1; STK-1; Receptor-type tyrosine-protein kinase FLT3; Fetal liver kinase-2; FLT-3; FLK2; FLK-2; FL cytokine receptor; CD135 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Tyrosine-protein kinase that acts as cell-surface receptor for the cytokine FLT3LG and regulates differentiation, proliferation and survival of hematopoietic progenitor cells and of dendritic cells. Promotes phosphorylation of SHC1 and AKT1, and activation of the downstream effector MTOR. Promotes activation of RAS signaling and phosphorylation of downstream kinases, including MAPK1/ERK2 and/or MAPK3/ERK1. Promotes phosphorylation of FES, FER, PTPN6/SHP, PTPN11/SHP-2, PLCG1, and STAT5A and/or STAT5B. Activation of wild-type FLT3 causes only marginal activation of STAT5A or STAT5B. Mutations that cause constitutive kinase activity promote cell proliferation and resistance to apoptosis via the activation of multiple signaling pathways. Related diseases Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:11090077, ECO:0000269|PubMed:11290608, ECO:0000269|PubMed:11442493, ECO:0000269|PubMed:14504097, ECO:0000269|PubMed:16266983, ECO:0000269|PubMed:18305215, ECO:0000269|PubMed:8946930, ECO:0000269|PubMed:9737679}. The gene represented in this entry may be involved in disease pathogenesis. Somatic mutations that lead to constitutive activation of FLT3 are frequent in AML patients. These mutations fall into two classes, the most common being in-frame internal tandem duplications of variable length in the juxtamembrane region that disrupt the normal regulation of the kinase activity. Likewise, point mutations in the activation loop of the kinase domain can result in a constitutively activated kinase. Drugs (DrugBank ID) DB12742; DB12267; DB12500; DB12010; DB12141; DB06469; DB06080; DB06595; DB11763; DB09079; DB11697; DB12978; DB08901; DB15822; DB12874; DB00398; DB01268; DB05465; DB11800; DB05014 Interacts with P00519; P42684; P46108; P46109; P06241; Q13322; Q9Y6K9; P06239; P27986; P20936; P43405; Q8R4L0 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 34209.2 Length 298 Aromaticity 0.14 Instability index 39.68 Isoelectric point 5.57 Charge (pH=7) -4.02 2D Binding mode Binding energy (Kcal/mol) -6.22 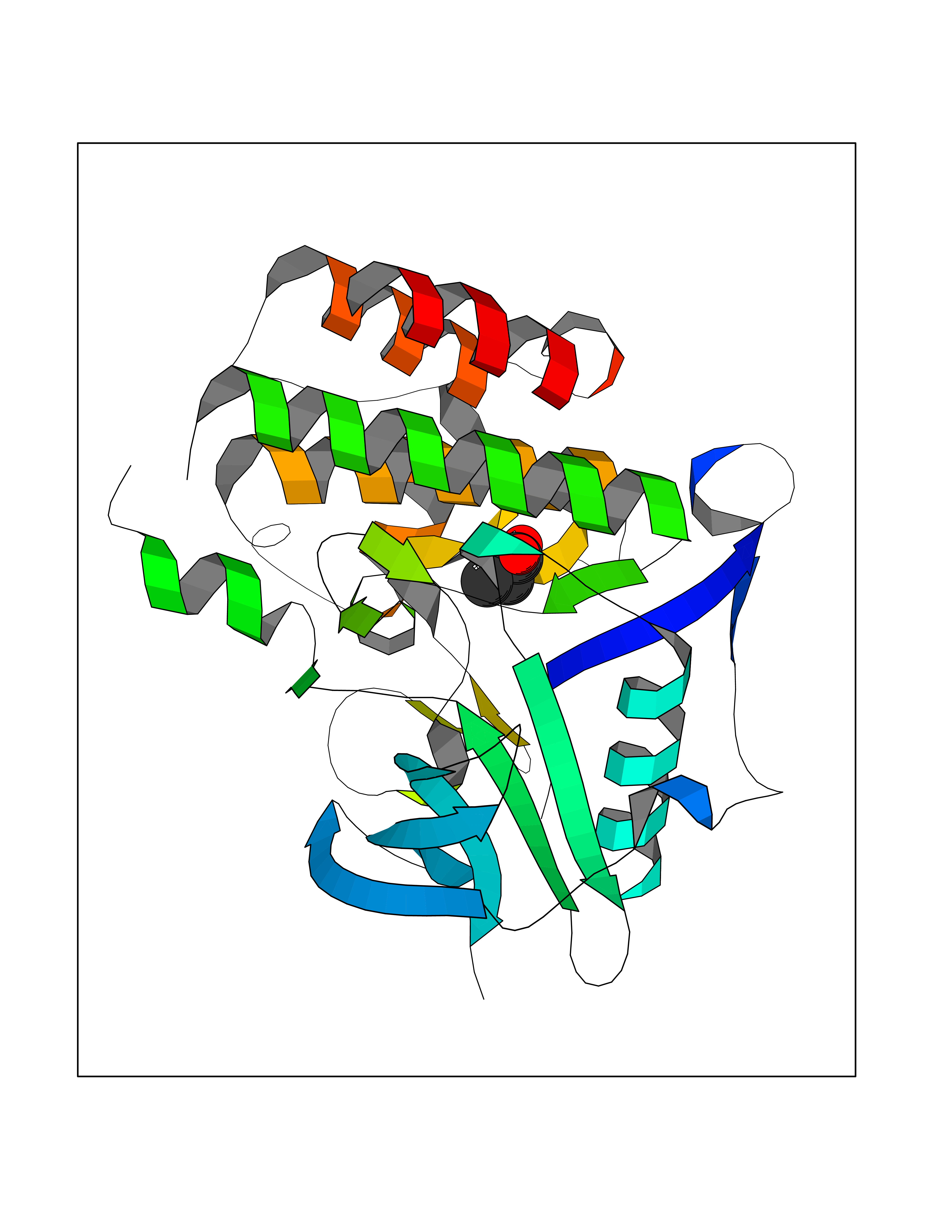 Molscript Map 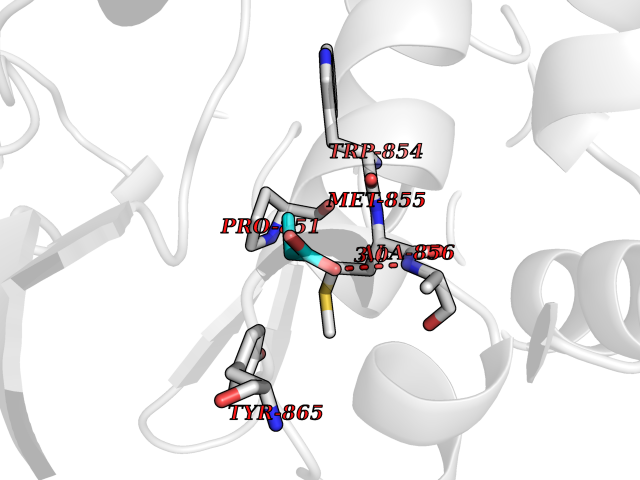 Pymol Map 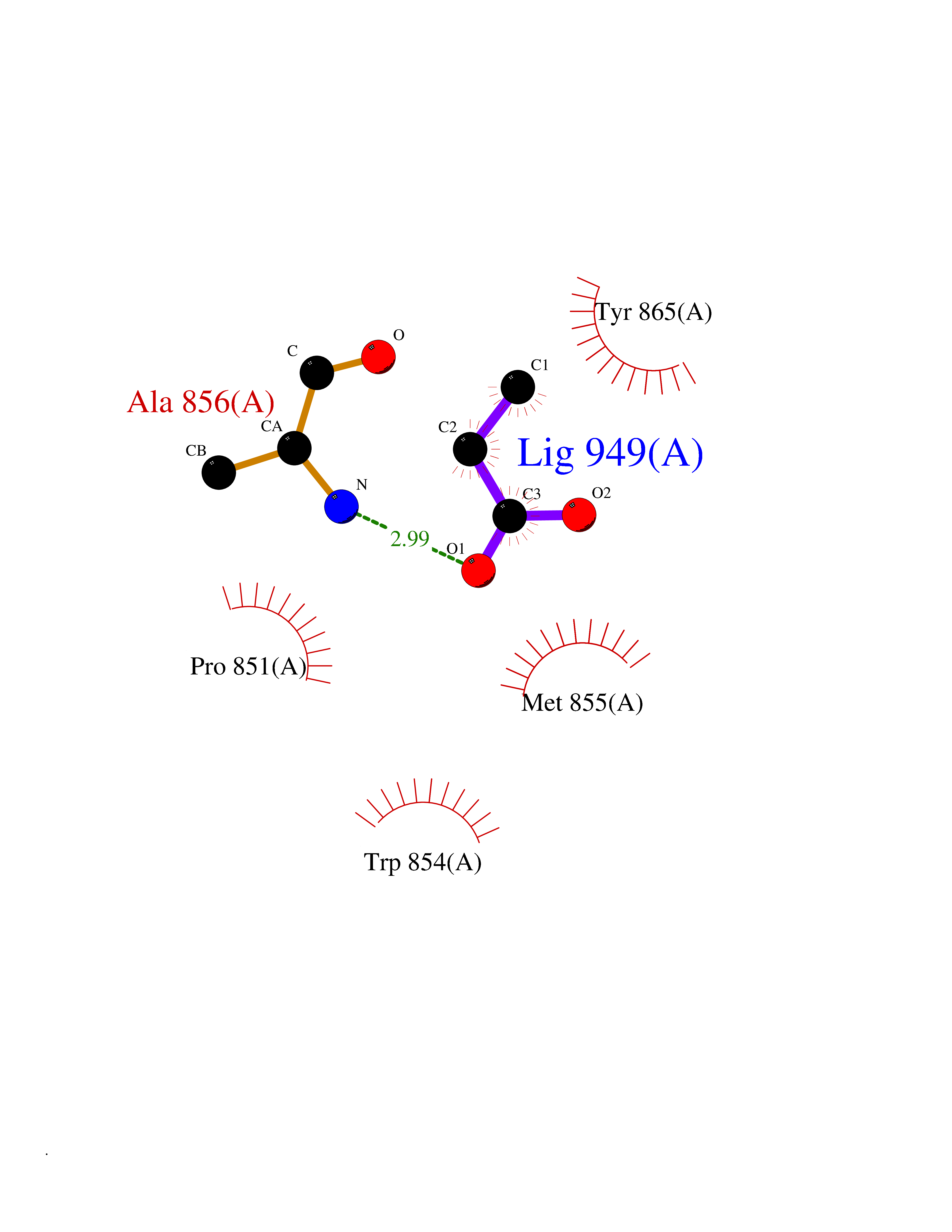 Ligplot Map 3D Binding mode Sequence YESQLQMVQVTGSSDNEYFYVDFREYEYDLKWEFPRENLEFGKVLGSGAFGKVMNATAYGISKTGVSIQVAVKMLKEREALMSELKMMTQLGSHENIVNLLGACTLSGPIYLIFEYCCYGDLLNYLRSKREKFLTFEDLLCFAYQVAKGMEFLEFKSCVHRDLAARNVLVTHGKVVKICDFGLARDIMSDSNYVVRGNARLPVKWMAPESLFEGIYTIKSDVWSYGILLWEIFSLGVNPYPGIPVDANFYKLIQNGFKMDQPFYATEEIYIIMQSCWAFDSRKRPSFPNLTSFLGCQL Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Aminoacylase-1 | 1Q7L | 4.56 | |
Target general information Gen name ACY1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase M20A family Biochemical class Hydrolase Function Aminoacylase activity.Identical protein binding.Metal ion binding.Metallopeptidase activity. Related diseases Aminoacylase-1 deficiency (ACY1D) [MIM:609924]: An enzymatic deficiency resulting in encephalopathy, unspecific psychomotor delay, psychomotor delay with atrophy of the vermis and syringomyelia, marked muscular hypotonia or normal clinical features. Epileptic seizures are a frequent feature. All affected individuals exhibit markedly increased urinary excretion of several N-acetylated amino acids. {ECO:0000269|PubMed:16274666, ECO:0000269|PubMed:16465618, ECO:0000269|PubMed:17562838, ECO:0000269|PubMed:21414403}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06151; DB00128; DB09130 Interacts with Q03154; O75934; Q96HA8; P36639; P36639-2; Q8TCT1; P0CG20; Q96A09; P54274; O43711; Q9UPN9; Q9NZC7-5 EC number 3.5.1.14 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,C Molecular weight (Da) 31172.2 Length 275 Aromaticity 0.11 Instability index 36.46 Isoelectric point 6 Charge (pH=7) -5.26 2D Binding mode Binding energy (Kcal/mol) -6.22 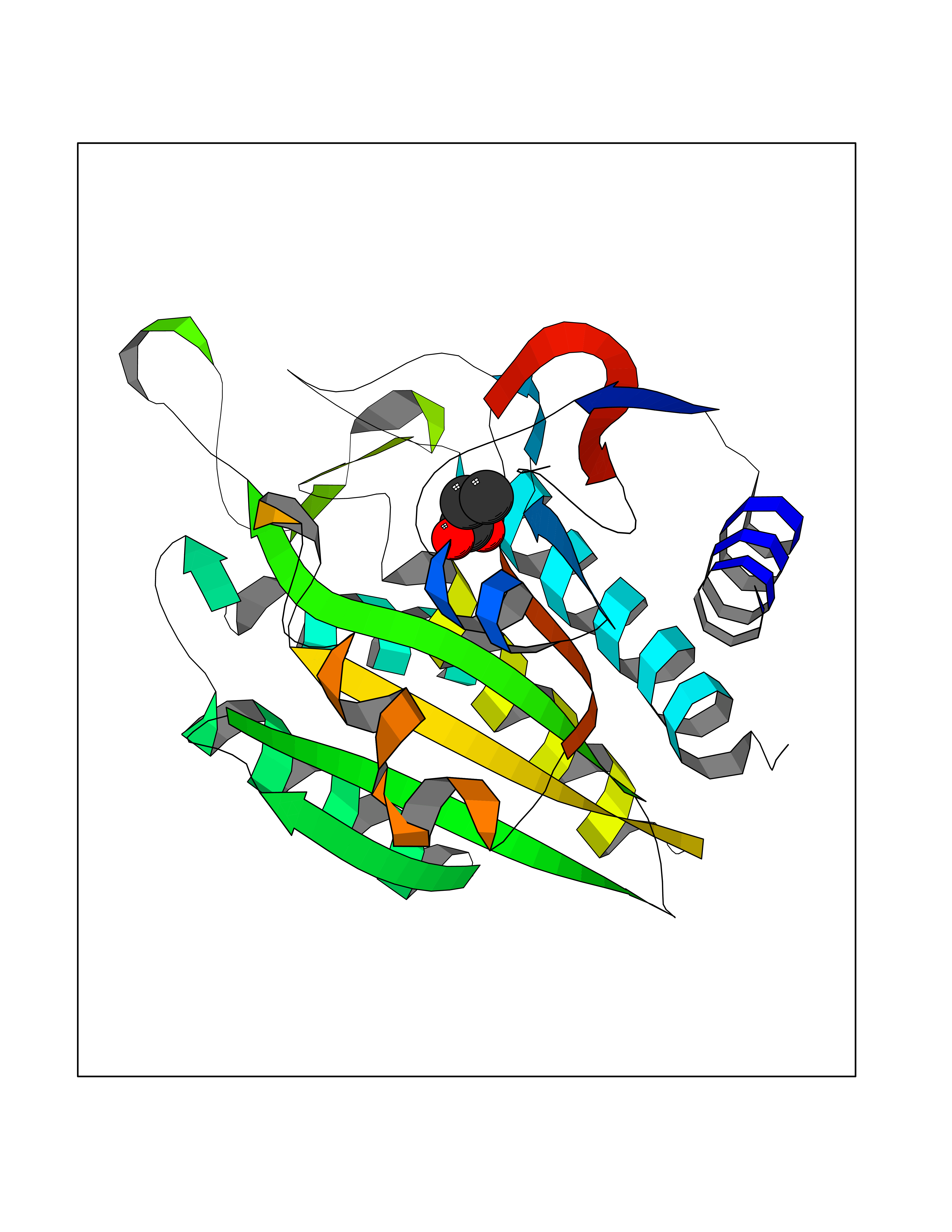 Molscript Map 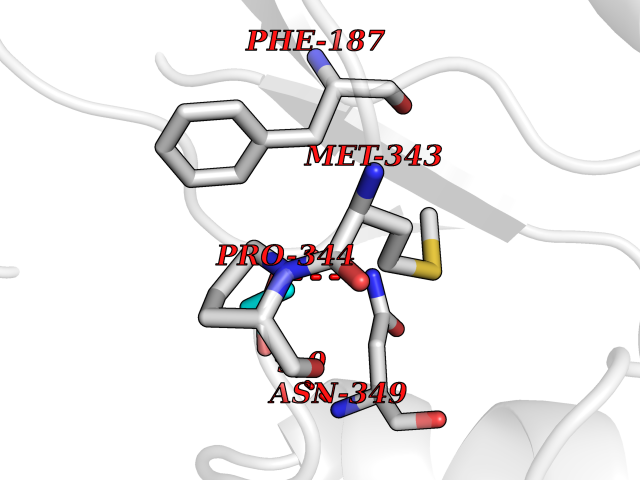 Pymol Map 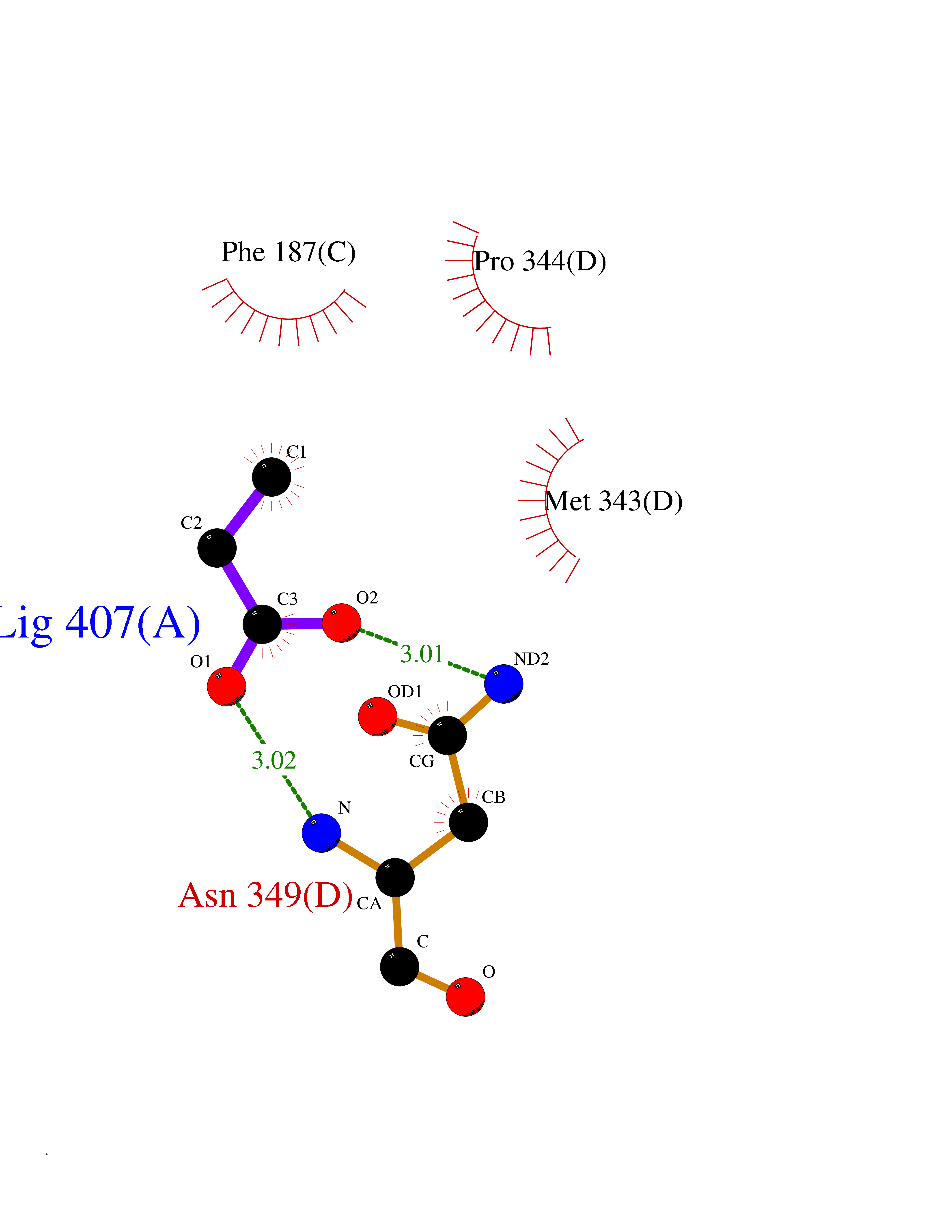 Ligplot Map 3D Binding mode Sequence NPWWAAFSRVCKDMNLTLEPEIMPAAGDNRYIRAVGVPALGFSPMNRTPVLLHDHDERLHEAVFLRGVDIYTRLLPALASVPALPEEHPSVTLFRQYLRIRTVQPKPDYGAAVAFFEETARQLGLGCQKVEVAPGYVVTVLTWPGTNPTLSSILLNSHTDVVPVFKEHWSHDPFEAFKDSEGYIYARGAQDMKCVSIQYLEAVRRLKVEGHRFPRTIHMTFVPDEEVGGHQGMELFVQRPEFHALRAGFALDEGIANPTDAFTVFYSERSPWWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 4.56 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -6.22 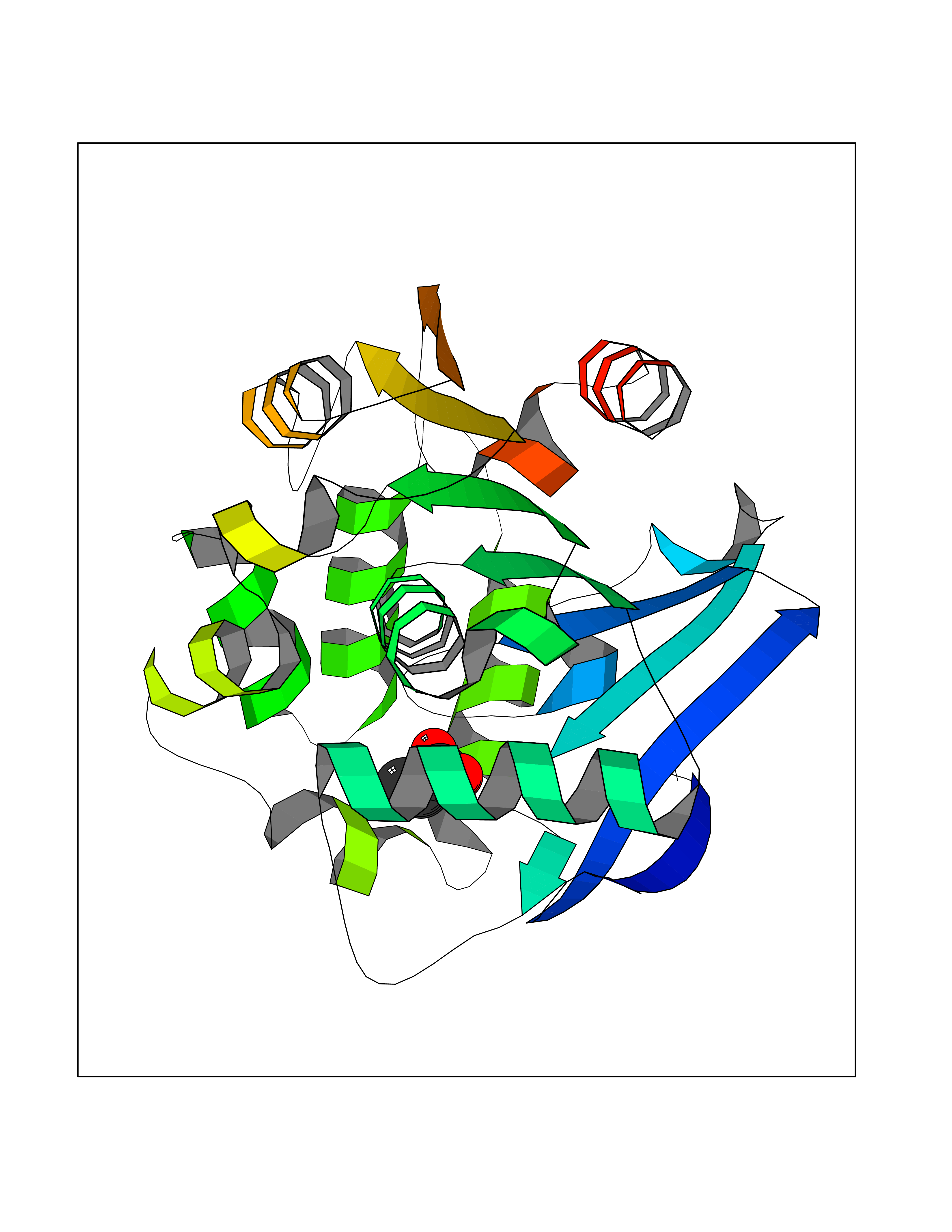 Molscript Map 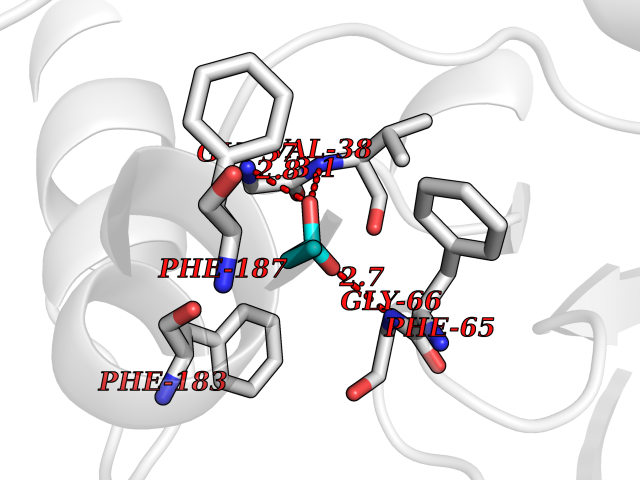 Pymol Map 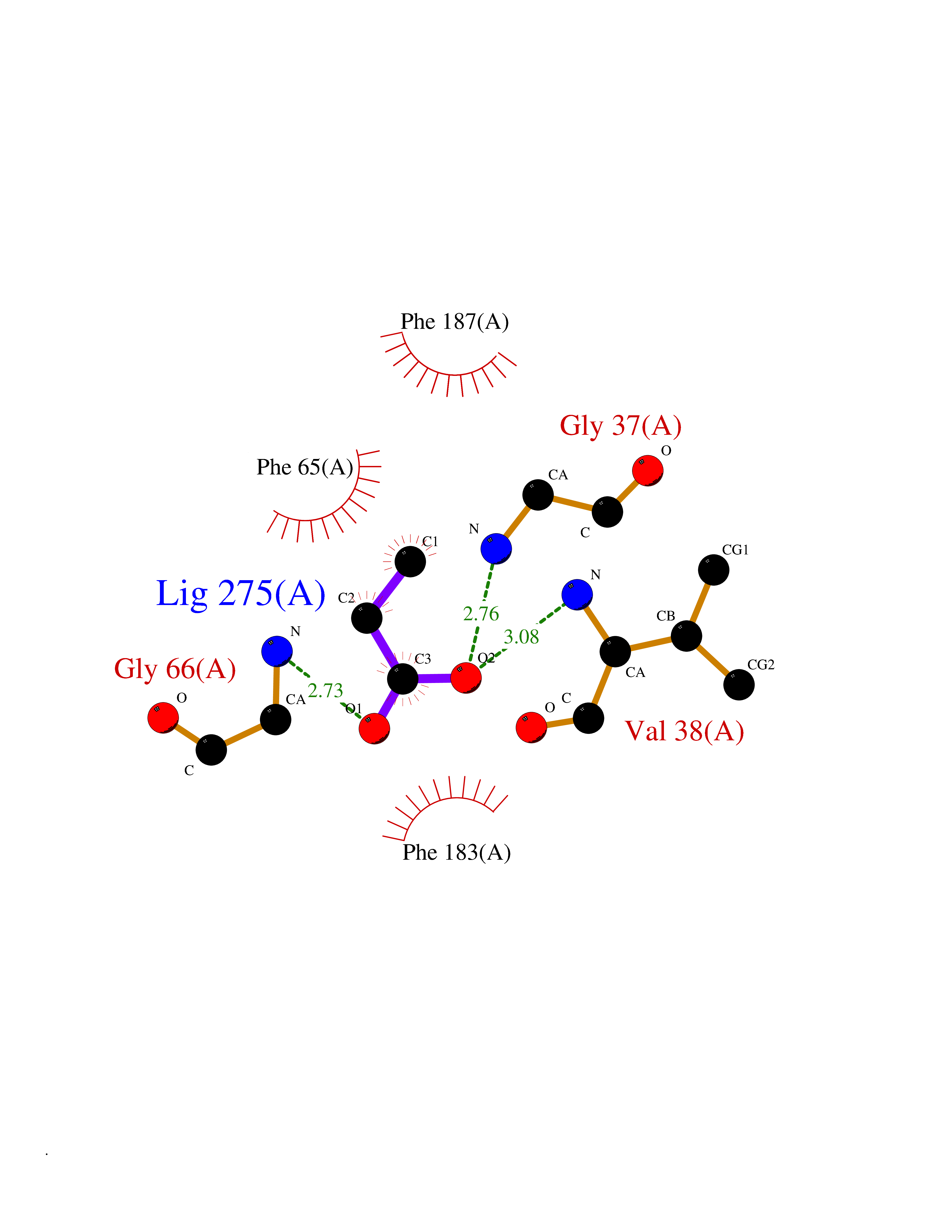 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Dihydroorotate dehydrogenase (DHODH) | 4OQV | 4.56 | |
Target general information Gen name DHODH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dihydroorotate oxidase; Dihydroorotate dehydrogenase (quinone), mitochondrial; DHOdehase; DHODH Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07559; DB07561; DB08172; DB08169; DB07443; DB07978; DB07975; DB04281; DB08249; DB07977; DB07976; DB04583; DB08008; DB01117; DB03523; DB03480; DB02613; DB04147; DB03247; DB01097; DB06481; DB08006; DB02262; DB05125; DB08880; DB07646 Interacts with Q6ZMZ0; P49638 EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Disease variant; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 38341.4 Length 353 Aromaticity 0.05 Instability index 39.27 Isoelectric point 9.28 Charge (pH=7) 5.52 2D Binding mode Binding energy (Kcal/mol) -6.22  Molscript Map 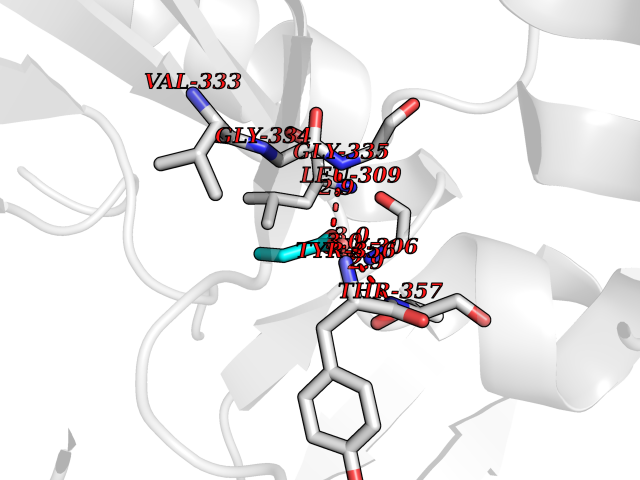 Pymol Map 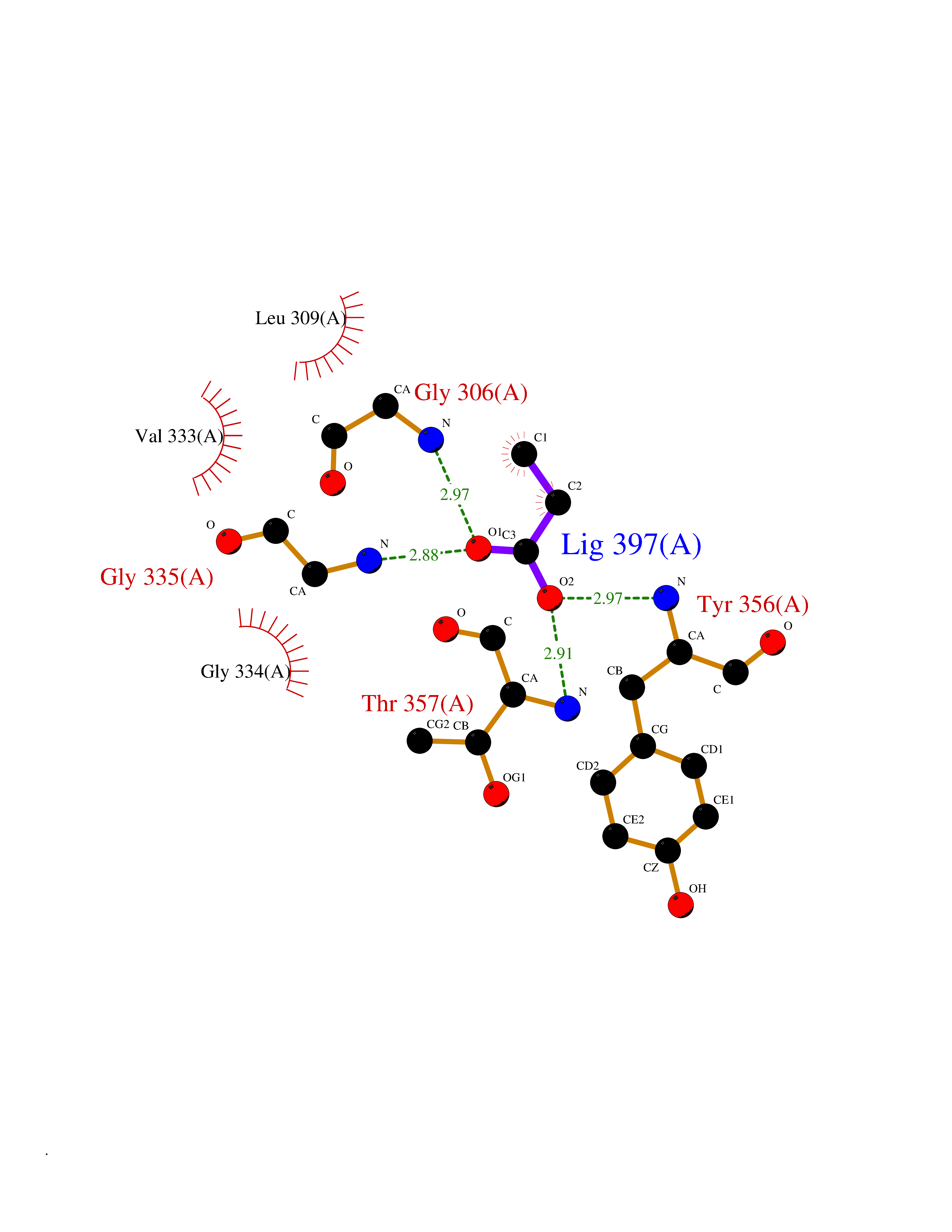 Ligplot Map 3D Binding mode Sequence DERFYAEHLMPTLQGLLDPESAHRLAVRFTSLGLLPRARFQDSDMLEVRVLGHKFRNPVGIAAGFDKHGEAVDGLYKMGFGFVEIGSVTPKPQEGNPRPRVFRLPEDQAVINRYGFNSHGLSVVEHRLRARQQKQAKLTEDGLPLGVNLGKNKTSVDAAEDYAEGVRVLGPLADYLVVNVSSPGKAELRRLLTKVLQERDGLRRVHRPAVLVKIAPDLTSQDKEDIASVVKELGIDGLIVTNTTVSRPAGLQGALRSETGGLSGKPLRDLSTQTIREMYALTQGRVPIIGVGGVSSGQDALEKIRAGASLVQLYTALTFWGPPVVGKVKRELEALLKEQGFGGVTDAIGADHR Hydrogen bonds contact Hydrophobic contact | ||||