Job Results:
Ligand
Structure
Job ID
0d7fe7ad13d85a9fa0b43f4c0711e72a
Job name
NA
Time
2026-02-27 11:51:24
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Acetylcholinesterase (AChE) (EC 3.1.1.7) | 1GPK | 4.39 | |
Target general information Gen name ache Organism Tetronarce californica (Pacific electric ray) (Torpedo californica) Uniprot ID TTD ID NA Synonyms NA Protein family Type-B carboxylesterase/lipase family Biochemical class NA Function Terminates signal transduction at the neuromuscular junction by rapid hydrolysis of the acetylcholine released into the synaptic cleft. May be involved in cell-cell interactions. Related diseases Noonan syndrome 5 (NS5) [MIM:611553]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:17603482, ECO:0000269|PubMed:17603483, ECO:0000269|PubMed:20683980}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: LEOPARD syndrome 2 (LPRD2) [MIM:611554]: A disorder characterized by lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonic stenosis, abnormalities of genitalia, retardation of growth, and sensorineural deafness. {ECO:0000269|PubMed:17603483}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cardiomyopathy, dilated, 1NN (CMD1NN) [MIM:615916]: A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. {ECO:0000269|PubMed:24777450}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.1.1.7 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; Neurotransmitter degradation; Serine esterase; Signal; Synapse Protein physicochemical properties Chain ID A Molecular weight (Da) 59779.2 Length 529 Aromaticity 0.12 Instability index 48.49 Isoelectric point 5.8 Charge (pH=7) -8.48 2D Binding mode Binding energy (Kcal/mol) -5.99 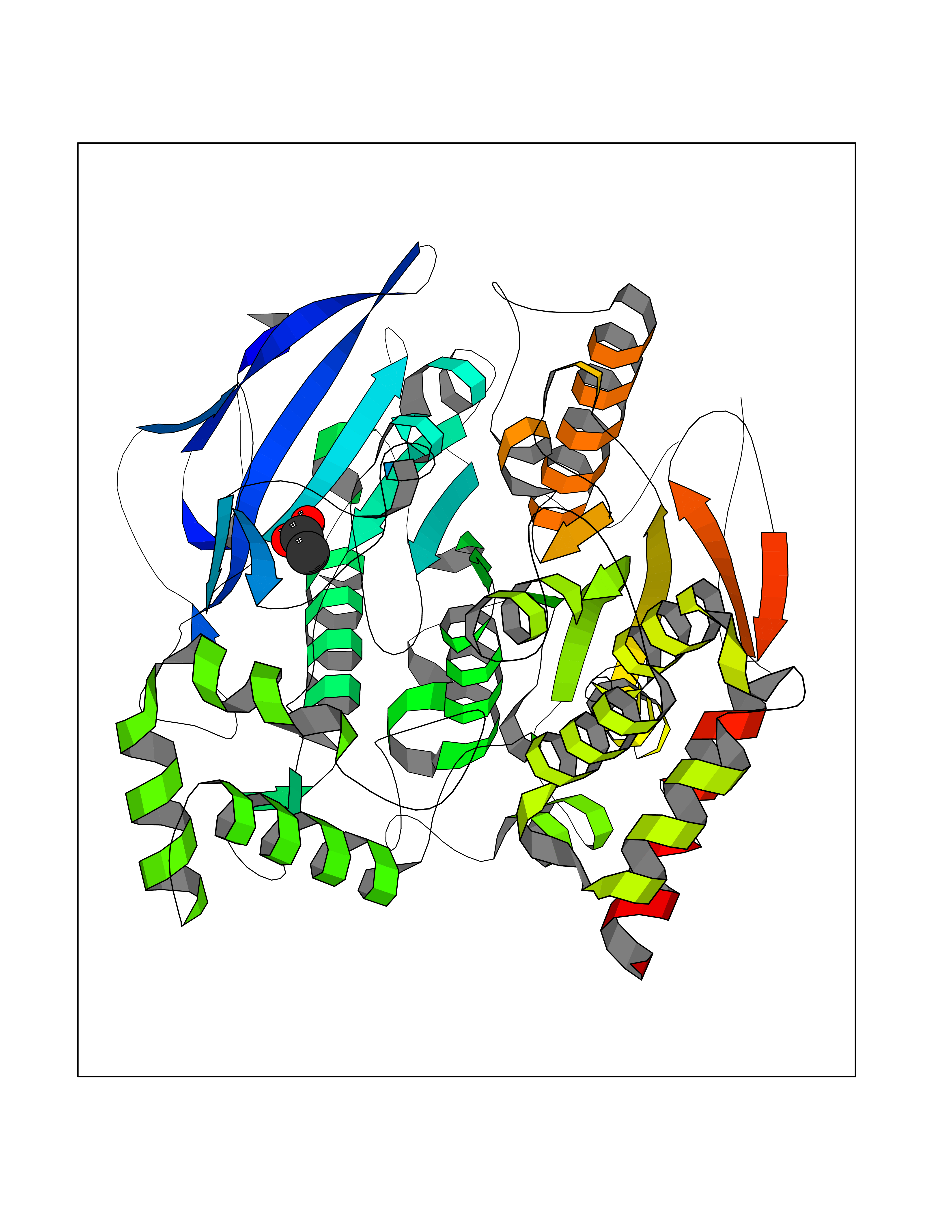 Molscript Map 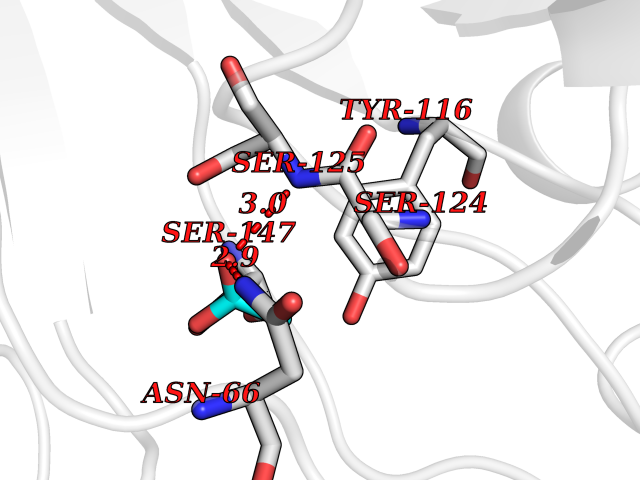 Pymol Map 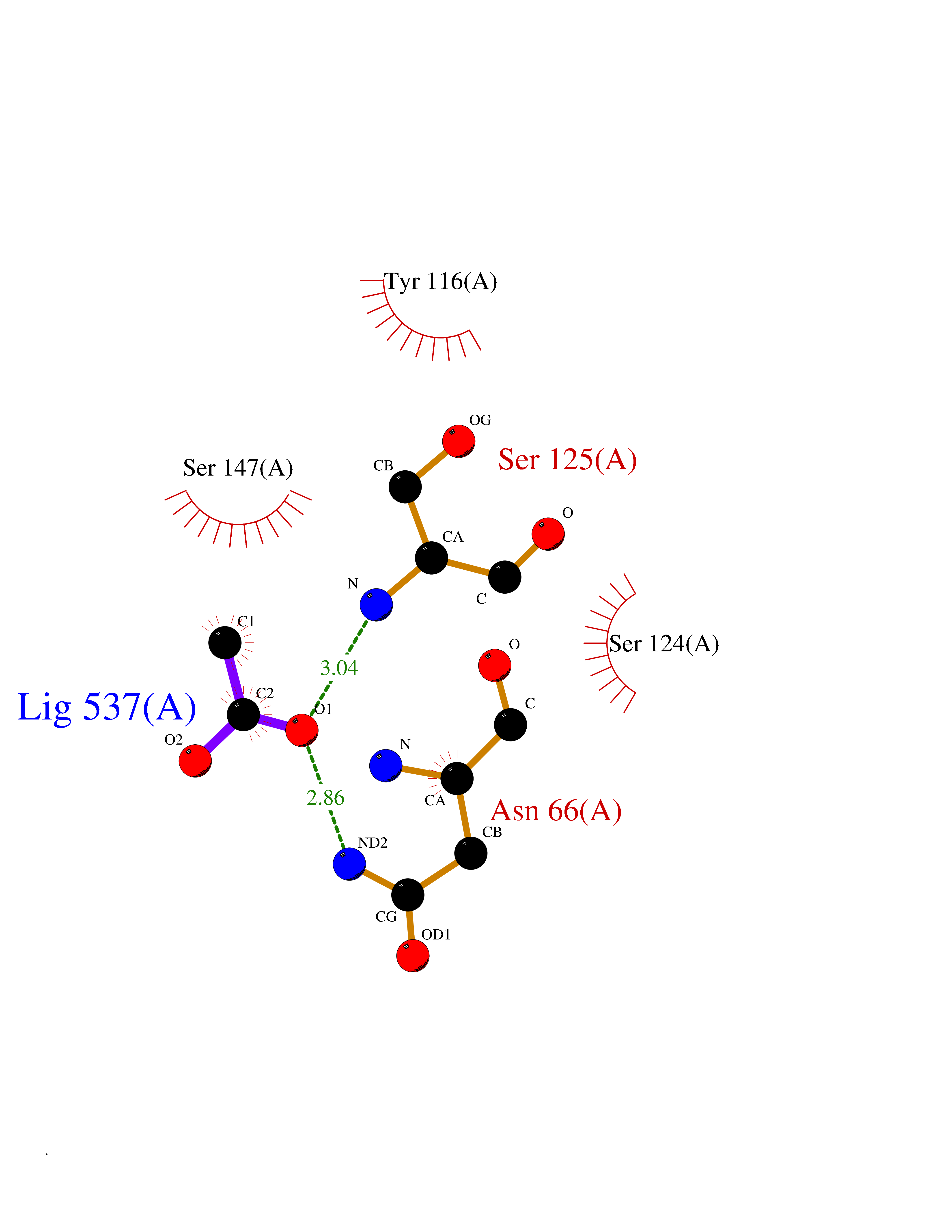 Ligplot Map 3D Binding mode Sequence SELLVNTKSGKVMGTRVPVLSSHISAFLGIPFAEPPVGNMRFRRPEPKKPWSGVWNASTYPNNCQQYVDEQFPGFSGSEMWNPNREMSEDCLYLNIWVPSPRPKSTTVMVWIYGGGFYSGSSTLDVYNGKYLAYTEEVVLVSLSYRVGAFGFLALHGSQEAPGNVGLLDQRMALQWVHDNIQFFGGDPKTVTIFGESAGGASVGMHILSPGSRDLFRRAILQSGSPNCPWASVSVAEGRRRAVELGRNLNCNLNSDEELIHCLREKKPQELIDVEWNVLPFDSIFRFSFVPVIDGEFFPTSLESMLNSGNFKKTQILLGVNKDEGSFFLLYGAPGFSKDSESKISREDFMSGVKLSVPHANDLGLDAVTLQYTDWMDDNNGIKNRDGLDDIVGDHNVICPLMHFVNKYTKFGNGTYLYFFNHRASNLVWPEWMGVIHGYEIEFVFGLPLVKELNYTAEEEALSRRIMHYWATFAKTGNPNEPESKWPLFTTKEQKFIDLNTEPMKVHQRLRVQMCVFWNQFLPKLLNAT Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Glycogen phosphorylase, liver form | 3DDS | 4.39 | |
Target general information Gen name PYGL Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Glycogen phosphorylase family Biochemical class Transferase Function AMP binding.ATP binding.Bile acid binding.Drug binding.Glucose binding.Glycogen phosphorylase activity.Linear malto-oligosaccharide phosphorylase activity.Protein homodimerization activity.Purine nucleobase binding.Pyridoxal phosphate binding.SHG alpha-glucan phosphorylase activity.Vitamin binding. Related diseases Glycogen storage disease 6 (GSD6) [MIM:232700]: A metabolic disorder characterized by mild to moderate hypoglycemia, mild ketosis, growth retardation, and prominent hepatomegaly. Heart and skeletal muscle are not affected. {ECO:0000269|PubMed:9529348}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P11216; P11217 EC number 2.4.1.1 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Carbohydrate metabolism; Cytoplasm; Disease variant; Glycogen metabolism; Glycogen storage disease; Glycosyltransferase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89481.9 Length 778 Aromaticity 0.11 Instability index 34.96 Isoelectric point 6.49 Charge (pH=7) -3.54 2D Binding mode Binding energy (Kcal/mol) -5.99  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NVAELKKSFNRHLHFTLVKDRNVATTRDYYFALAHTVRDHLVGRWIRTQQHYYDKCPKRVYYLSLEFYMGRTLQNTMINLGLQNACDEAIYQLGLDIEELEEIEEDAGLGNGGLGRLAACFLDSMATLGLAAYGYGIRYEYGIFNQKIRDGWQVEEADDWLRYGNPWEKSRPEFMLPVHFYGKVEHTNTGTKWIDTQVVLALPYDTPVPGYMNNTVNTMRLWSENISRVLYPNDNFFEGKELRLKQEYFVVAATLQDIIRRFKASKFGSTGTVFDAFPDQVAIQLNDTHPALAIPELMRIFVDIEKLPWSKAWELTQKTFAYTNHTVLPEALERWPVDLVEKLLPRHLEIIYEINQKHLDRIVALFPKDVDRLRRMSLIEEEGSKRINMAHLCIVGSHAVNGVAKIHSDIVKTKVFKDFSELEPDKFQNKTNGITPRRWLLLCNPGLAELIAEKIGEDYVKDLSQLTKLHSFLGDDVFLRELAKVKQENKLKFSQFLETEYKVKINPSSMFDVQVKRIHEYKRQLLNCLHVITMYNRIKKDPKKLFVPRTVIIGGKAAPGYHMAKMIIKLITSVADVVNNDPMVGSKLKVIFLENYRVSLAEKVIPATDLSEQISTAGTEASGTGNMKFMLNGALTIGTMDGANVEMAEEAGEENLFIFGMRIDDVAALDKKGYEAKEYYEALPELKLVIDQIDNGFFSPKQPDLFKDIINMLFYHDRFKVFADYEAYVKCQDKVSQLYMNPKAWNTMVLKNIAASGKFSSDRTIKEYAQNIWNVEPS Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Ornithine decarboxylase (ODC1) | 2OO0 | 4.39 | |
Target general information Gen name ODC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ODC Protein family Orn/Lys/Arg decarboxylase class-II family Biochemical class Carbon-carbon lyase Function Polyamines are essential for cell proliferation and are implicated in cellular processes, ranging from DNA replication to apoptosis. Catalyzes the first and rate-limiting step of polyamine biosynthesis that converts ornithine into putrescine, which is the precursor for the polyamines, spermidine and spermine. Related diseases Bachmann-Bupp syndrome (BABS) [MIM:619075]: An autosomal dominant disorder characterized by global developmental delay, alopecia, absolute or relative macrocephaly, and facial dysmorphism. Neuroimaging shows white matter abnormalities, prominent Virchow-Robin spaces, periventricular cysts, and abnormalities of the corpus callosum. {ECO:0000269|PubMed:30239107, ECO:0000269|PubMed:30475435}. The disease is caused by variants affecting the gene represented in this entry. BABS is due to truncating variants that lead to a gain of function. This phenomenon apparently results from truncation proximal to or involving the C-terminal region of ODC1 protein, distal enough to allow escape from nonsense-mediated decay. A gain of function is corroborated by elevated plasma levels of N-acetylputrescine, with otherwise normal polyamine levels, in affected individuals. {ECO:0000269|PubMed:30475435}. Drugs (DrugBank ID) DB06243; DB04263; DB03856; DB04083; DB02824; DB01917; DB00114; DB02209; DB00203; DB00127; DB00313 Interacts with Q9H8Y8; Q92993; Q9UMX2; Q9UMX2-2 EC number EC 4.1.1.17 Uniprot keywords 3D-structure; Decarboxylase; Disease variant; Hypotrichosis; Lyase; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 45682.9 Length 410 Aromaticity 0.11 Instability index 40.93 Isoelectric point 5.61 Charge (pH=7) -6.68 2D Binding mode Binding energy (Kcal/mol) -5.99  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LMNNFGNEEFDCHFLDEGFTAKDILDQKINEVSSSDDKDAFYVADLGDILKKHLRWLKALPRVTPFYAVKCNDSKAIVKTLAATGTGFDCASKTEIQLVQSLGVPPERIIYANPCKQVSQIKYAANNGVQMMTFDSEVELMKVARAHPKAKLVLRIATDDSKAVCRLSVKFGATLRTSRLLLERAKELNIDVVGVSFHVGSGCTDPETFVQAISDARCVFDMGAEVGFSMYLLDIGGGFPGSEDVKLKFEEITGVINPALDKYFPSDSGVRIIAEPGRYYVASAFTLAVNIIAKKIVLEQTFMYYVNDGVYGSFNCILYDHAHVKPLLQKRPKPDEKYYSSSIWGPTCDGLDRIVERCDLPEMHVGDWMLFENMGAYTVAAASTFNGFQRPTIYYVMSGPAWQLMQQFQN Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 4.39 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -5.98  Molscript Map  Pymol Map 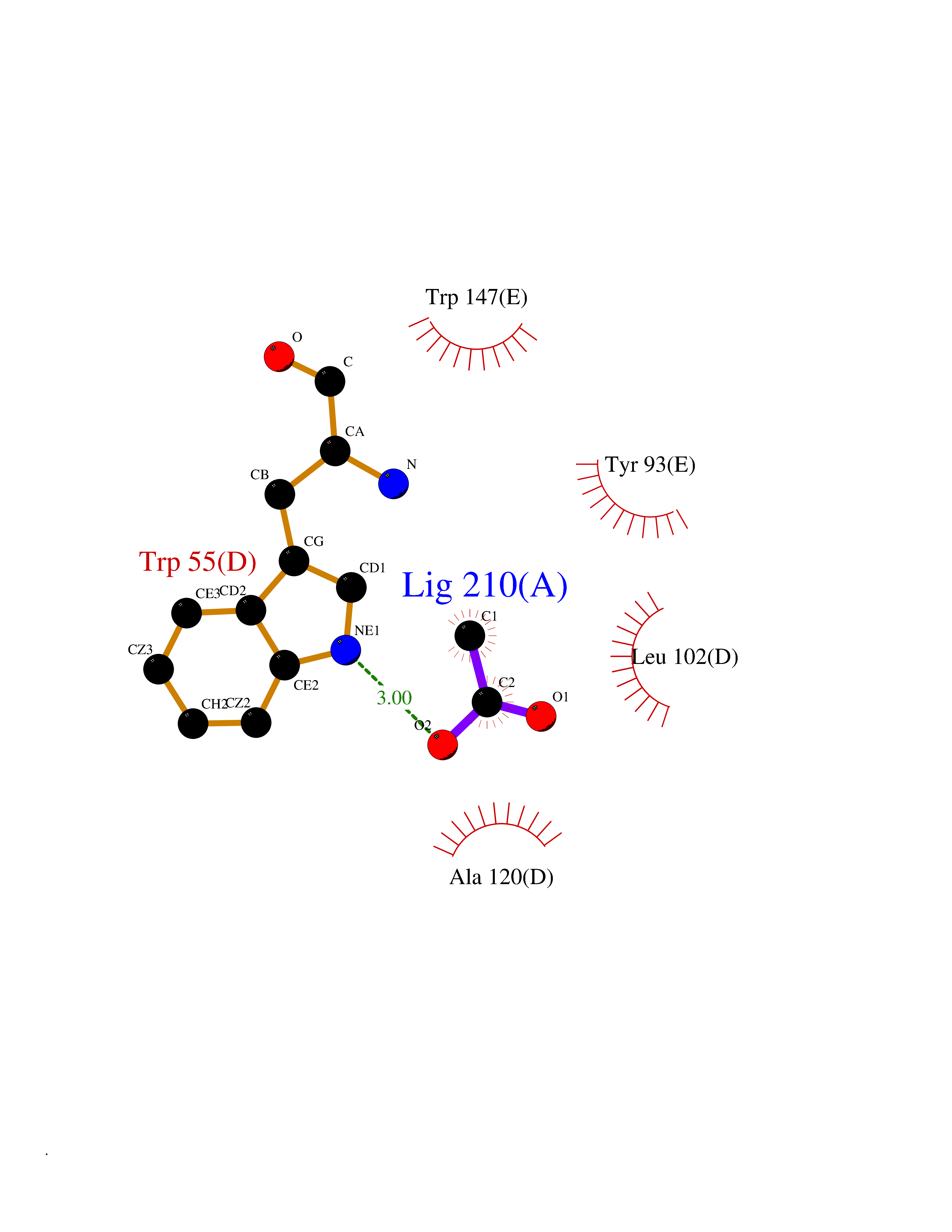 Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Vascular endothelial growth factor receptor 3 | 4BSJ | 4.39 | |
Target general information Gen name FLT4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms VEGFR3 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Transferase Function ATP binding.Growth factor binding.Protein homodimerization activity.Protein phosphatase binding.Transmembrane receptor protein tyrosine kinase activity.Vascular endothelial growth factor-activated receptor activity.VEGF-C-activated receptor activity. Related diseases Lymphatic malformation 1 (LMPHM1) [MIM:153100]: A form of primary lymphedema, a disease characterized by swelling of body parts due to developmental anomalies and functional defects of the lymphatic system. Patients with lymphedema may suffer from recurrent local infections. LMPHM1 is an autosomal dominant form with variable expression and severity. Onset is usually at birth or in early childhood but can occur later. Affected individuals manifest lymphedema, predominantly in the lower limbs, and hypoplasia of lymphatic vessels. Additional features are hemangioma and nail dysplasia or papillomatosis. {ECO:0000269|PubMed:10835628, ECO:0000269|PubMed:10856194, ECO:0000269|PubMed:12881528, ECO:0000269|PubMed:15102829, ECO:0000269|PubMed:16924388, ECO:0000269|PubMed:16965327, ECO:0000269|PubMed:17458866, ECO:0000269|PubMed:19289394, ECO:0000269|PubMed:26091405, ECO:0000269|PubMed:9817924}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hemangioma, capillary infantile (HCI) [MIM:602089]: A condition characterized by dull red, firm, dome-shaped hemangiomas, sharply demarcated from surrounding skin, usually presenting at birth or occurring within the first two or three months of life. They result from highly proliferative, localized growth of capillary endothelium and generally undergo regression and involution without scarring. {ECO:0000269|PubMed:11807987}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Plays an important role in tumor lymphangiogenesis, in cancer cell survival, migration, and formation of metastases.; DISEASE: Congenital heart defects, multiple types, 7 (CHTD7) [MIM:618780]: An autosomal dominant disorder with incomplete penetrance characterized by congenital developmental abnormalities involving structures of the heart. Common defects include tetralogy of Fallot, pulmonary stenosis or atresia, absent pulmonary valve, right aortic arch, double aortic arch, and major aortopulmonary collateral arteries. {ECO:0000269|PubMed:28991257, ECO:0000269|PubMed:30232381}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06626; DB05932; DB12010; DB11679; DB06101; DB09078; DB06080; DB09079; DB06589; DB08896; DB15685; DB00398; DB01268; DB05075; DB11800; DB04879 Interacts with P08238; P35968; P49767 EC number 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 24029.2 Length 213 Aromaticity 0.1 Instability index 47.75 Isoelectric point 8.34 Charge (pH=7) 2.35 2D Binding mode Binding energy (Kcal/mol) -5.98  Molscript Map  Pymol Map 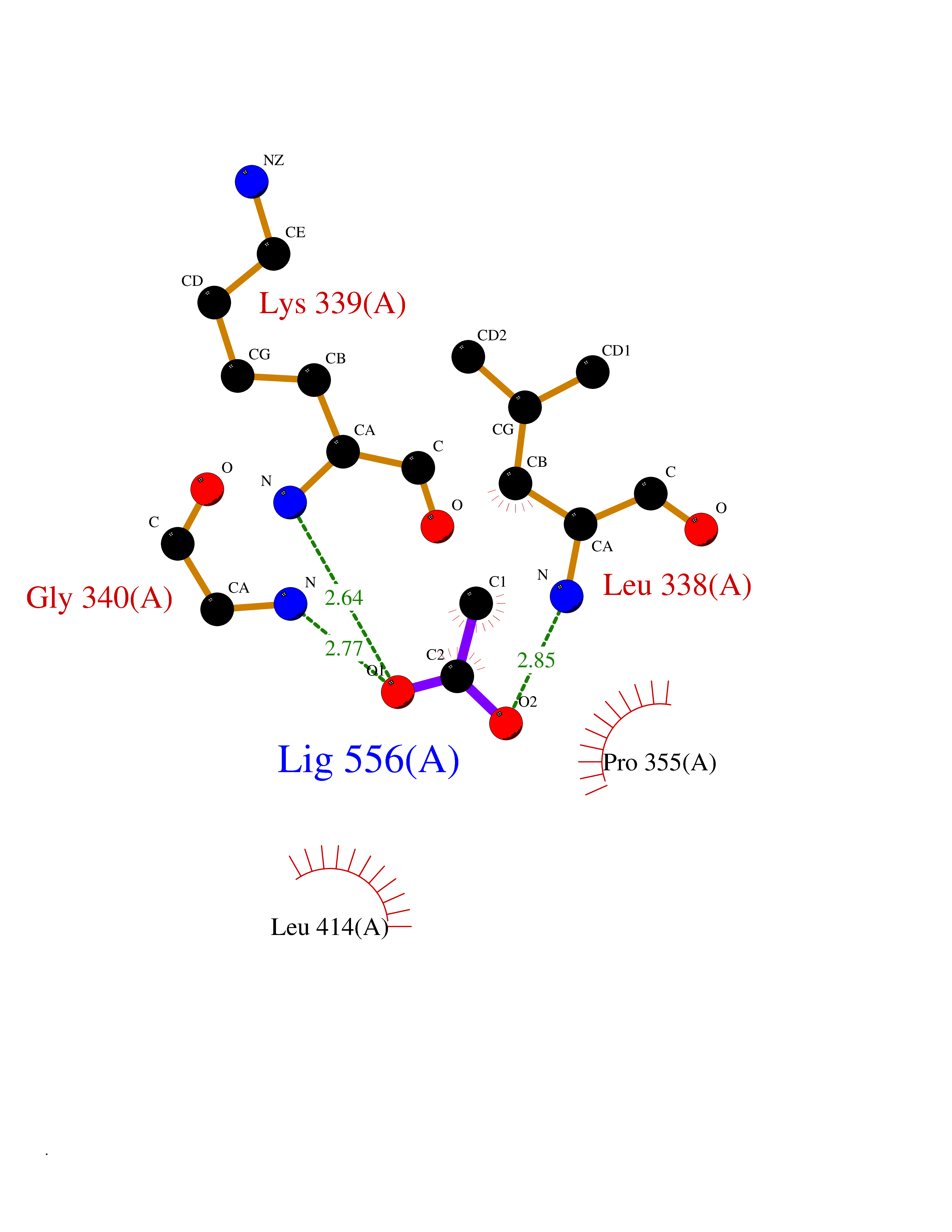 Ligplot Map 3D Binding mode Sequence DHNPFISVEWLKGPILEATAGDELVKLPVKLAAYPPPEFQWYKDGKALSGRHSPHALVLKEVTEASTGTYTLALWNSAAGLRRNISLELVVNVPPQIHEKEASSPSIYSRHSRQALTCTAYGVPLPLSIQWHWRPWTPCKMFPQCRDWRAVTTQDAVNPIESLDTWTEFVEGKNKTVSKLVIQNANVSAMYKCVVSNKVGQDERLIYFYVTTH Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Protein cereblon (CRBN) | 5FQD | 4.39 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -5.99 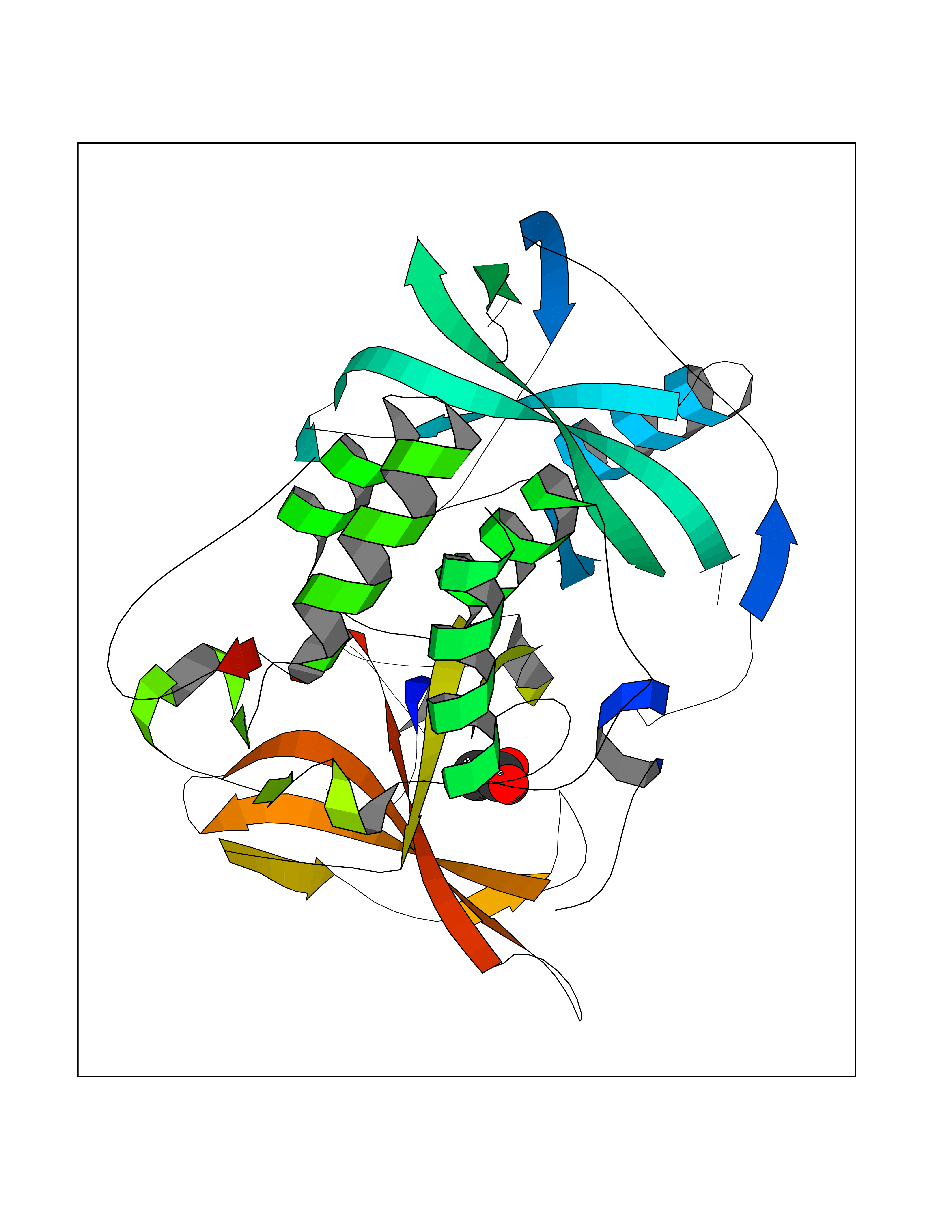 Molscript Map 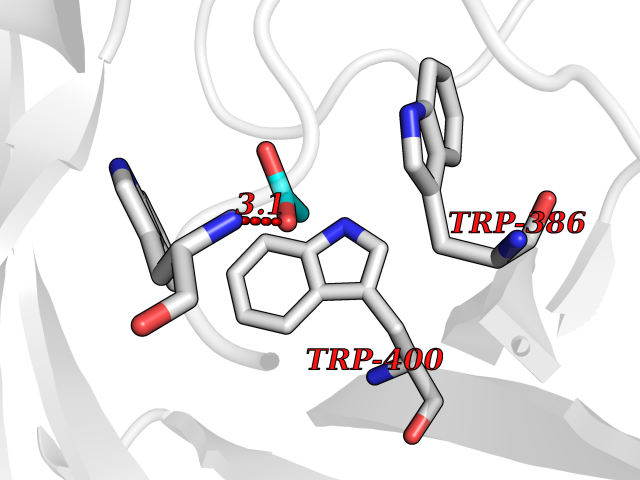 Pymol Map 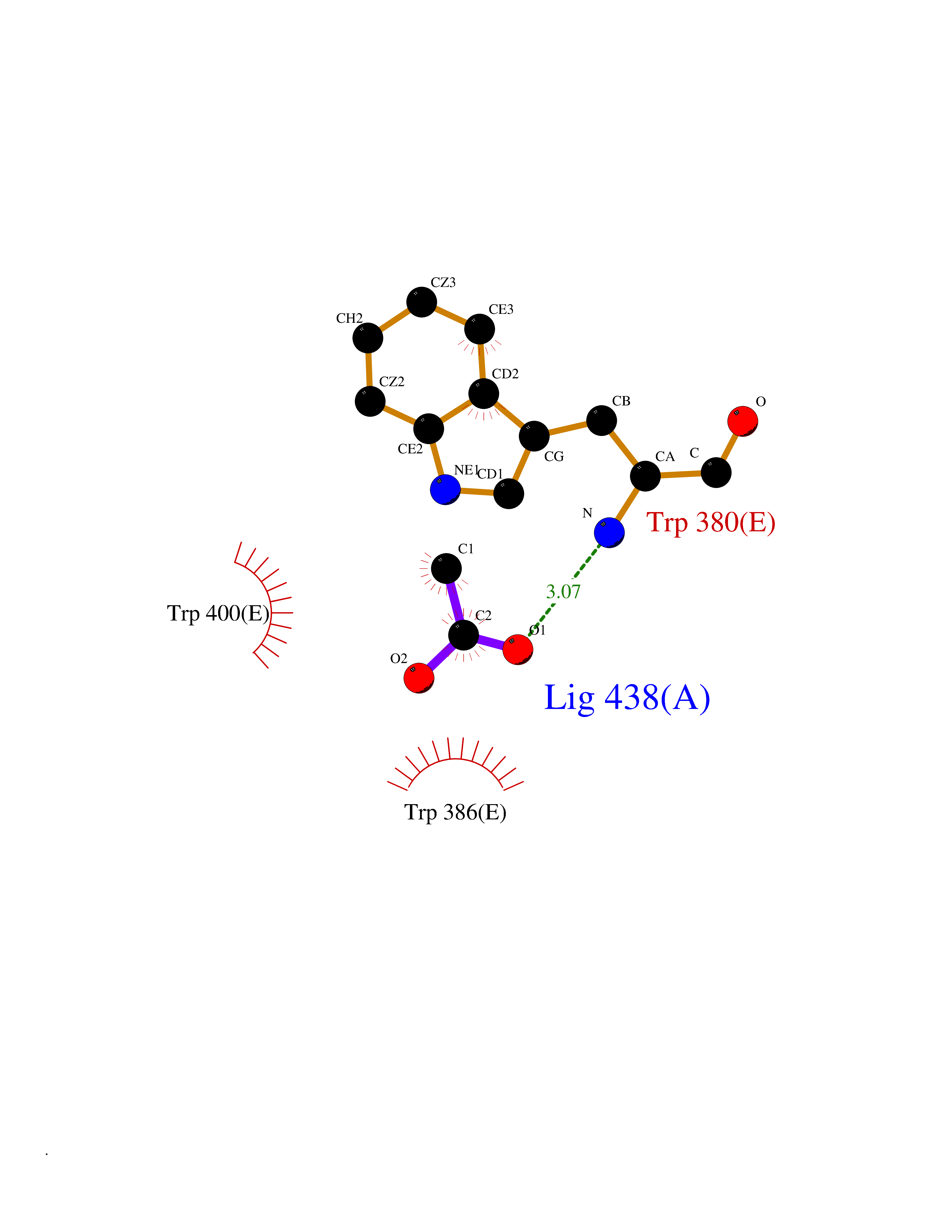 Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Pectate lyase | 1R76 | 4.39 | |
Target general information Gen name pelA Organism Niveispirillum irakense (Azospirillum irakense) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Lyase Function Lyase activity. Related diseases A chromosomal aberration involving ALK is found in a form of non-Hodgkin lymphoma. Translocation t(2;5)(p23;q35) with NPM1. The resulting chimeric NPM1-ALK protein homodimerize and the kinase becomes constitutively activated. The constitutively active fusion proteins are responsible for 5-10% of non-Hodgkin lymphomas. {ECO:0000269|PubMed:15938644}.; DISEASE: A chromosomal aberration involving ALK is associated with inflammatory myofibroblastic tumors (IMTs). Translocation t(2;11)(p23;p15) with CARS; translocation t(2;4)(p23;q21) with SEC31A. {ECO:0000269|PubMed:12112524, ECO:0000269|PubMed:16161041}.; DISEASE: A chromosomal aberration involving ALK is associated with anaplastic large-cell lymphoma (ALCL). Translocation t(2;17)(p23;q25) with ALO17. {ECO:0000269|PubMed:12112524}.; DISEASE: Neuroblastoma 3 (NBLST3) [MIM:613014]: A common neoplasm of early childhood arising from embryonic cells that form the primitive neural crest and give rise to the adrenal medulla and the sympathetic nervous system. {ECO:0000269|PubMed:18724359, ECO:0000269|PubMed:18923523, ECO:0000269|PubMed:18923525, ECO:0000269|PubMed:21242967, ECO:0000269|PubMed:22932897}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: The ALK signaling pathway plays an important role in glioblastoma, the most common malignant brain tumor of adults and one of the most lethal cancers. It regulates both glioblastoma migration and growth. {ECO:0000269|PubMed:15908427}.; DISEASE: A chromosomal aberration involving ALK is found in one subject with colorectal cancer. Translocation t(2;2)(p23.1;p23.3). A 5 million base pair tandem duplication generates an in-frame WDCP-ALK gene fusion. {ECO:0000269|PubMed:22327622}.; DISEASE: A chromosomal aberration involving ALK has been identified in a subset of patients with non-small-cell lung carcinoma. This aberration leads to the production of a fusion protein between the N-terminus of EML4 et the C-terminus of ALK. It is unclear whether the fusion protein is caused by a simple inversion within 2p (inv(2)(p21p23)) or whether the chromosome translocation involving 2p is more complex. When tested in a heterologous system, the fusion protein EML4-ALK possesses transforming activity that is dependent on ALK catalytic activity, possibly due to spontaneous dimerization mediated by the EML4 moiety, leading to ALK kinase activation. {ECO:0000269|PubMed:17625570}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Lyase; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 41907.5 Length 384 Aromaticity 0.08 Instability index 43.72 Isoelectric point 6.11 Charge (pH=7) -3.46 2D Binding mode Binding energy (Kcal/mol) -5.99 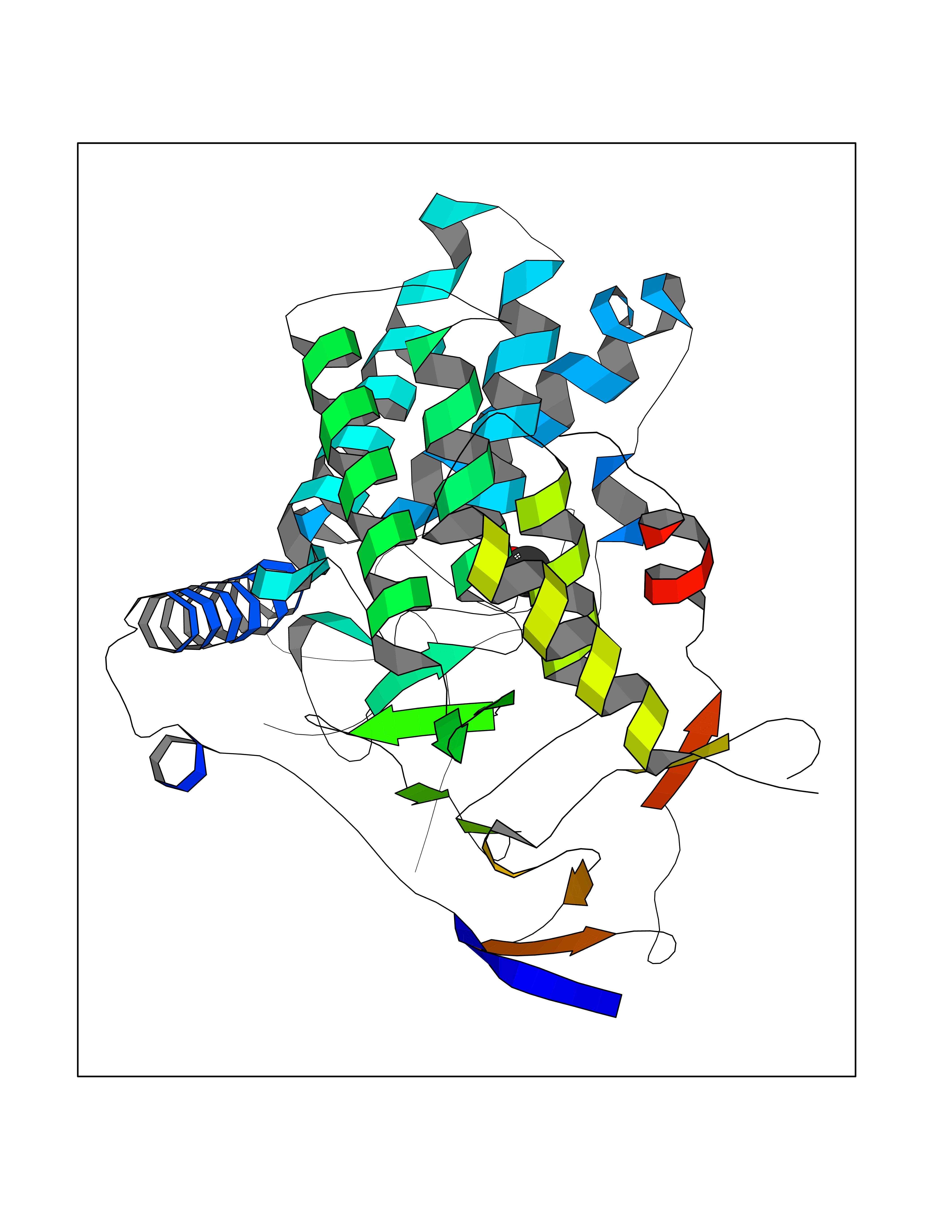 Molscript Map 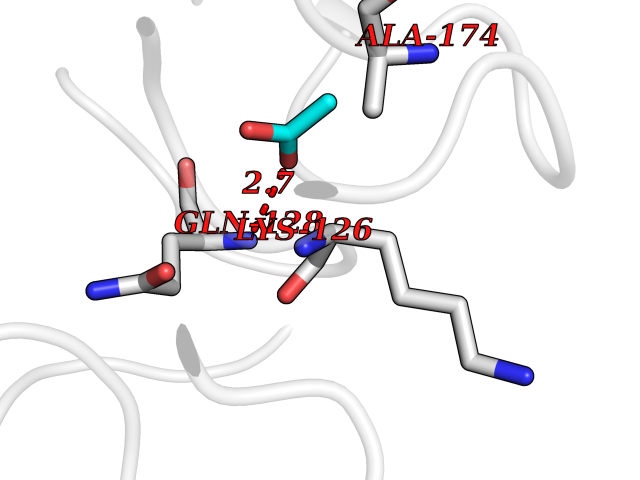 Pymol Map 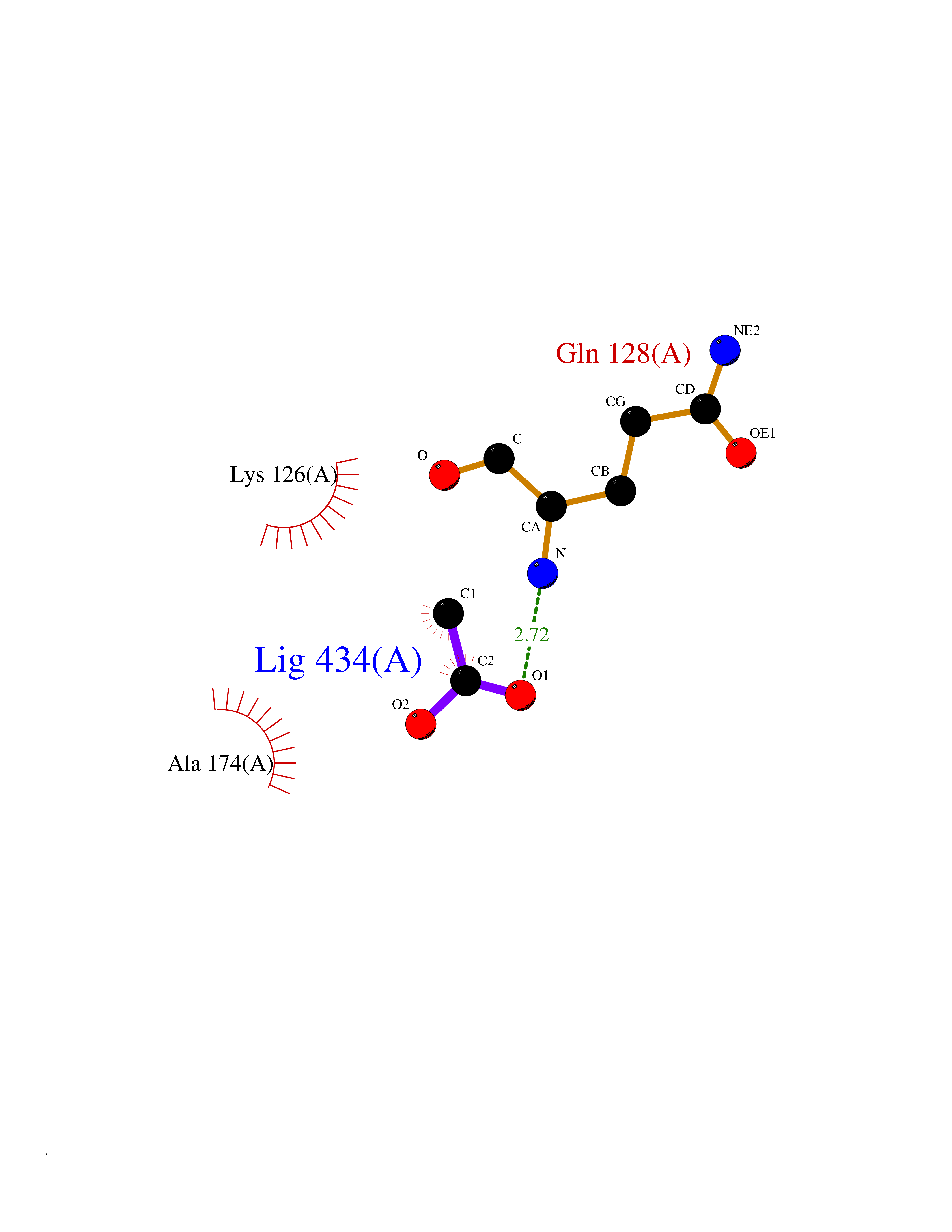 Ligplot Map 3D Binding mode Sequence AVIGMNEAASALTPSRVSSLPDTQRAAWQEYLARSEAQLSRDKASLAAELAPGQPLPPPPAEGKGADTMPLDKPAAWYTSKAARHVADVIVSFQTPAGGWGKNQPRDGALRLPGQHYTGENVAKVKRDRDWHYVGTIDNDATVTEIRFLAQVVSQLAPEEAAPYRDAALKGIEYLLASQFPNGGWPQVWPLEGGYHDAITYNDDALVHVAELLSDIAAGRDGFGFVPPAIRTRALEATNAAIHCIVETQVVQDGKRLGWGQQHDALTLRPTSARNFEPAALSSTESARILLFLMEIEAPSDAVKQAIRGGVAWLNTSVIRDQGAKPLWSRFYSLDGNKPVFGDRDKTIHDDVMGISQERRTGYAWYTTSPQKALSAFTKWEKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Oxidative stress responsive 1 (OXSR1) | 2VWI | 4.39 | |
Target general information Gen name OXSR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein kinase OSR1; Oxidative stress-responsive 1 protein; KIAA1101 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, STE20 subfamily Biochemical class NA Function Phosphorylates RELL1, RELL2 and RELT. Phosphorylates PAK1. Phosphorylates PLSCR1 in the presence of RELT. Related diseases Hyperparathyroidism, transient neonatal (HRPTTN) [MIM:618188]: An autosomal recessive disease characterized by impaired transplacental maternal-fetal transport of calcium, high serum PTH levels and signs of metabolic bone disease in the neonatal period. Skeletal anomalies include generalized osteopenia, narrow chest, short ribs with multiple healing fractures, and bowing or fractures of long bones. Affected individuals experience postnatal respiratory and feeding difficulties. The condition improves within a short time after birth once calcium is provided orally. {ECO:0000269|PubMed:29861107, ECO:0000269|PubMed:30820485}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with Q9Y376; O95747; Q8NC24; P55011; Q9H4A3; Q9Y3S1; Q96J92 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cytoplasm; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 26440.4 Length 236 Aromaticity 0.08 Instability index 44.04 Isoelectric point 6.48 Charge (pH=7) -1.55 2D Binding mode Binding energy (Kcal/mol) -5.98 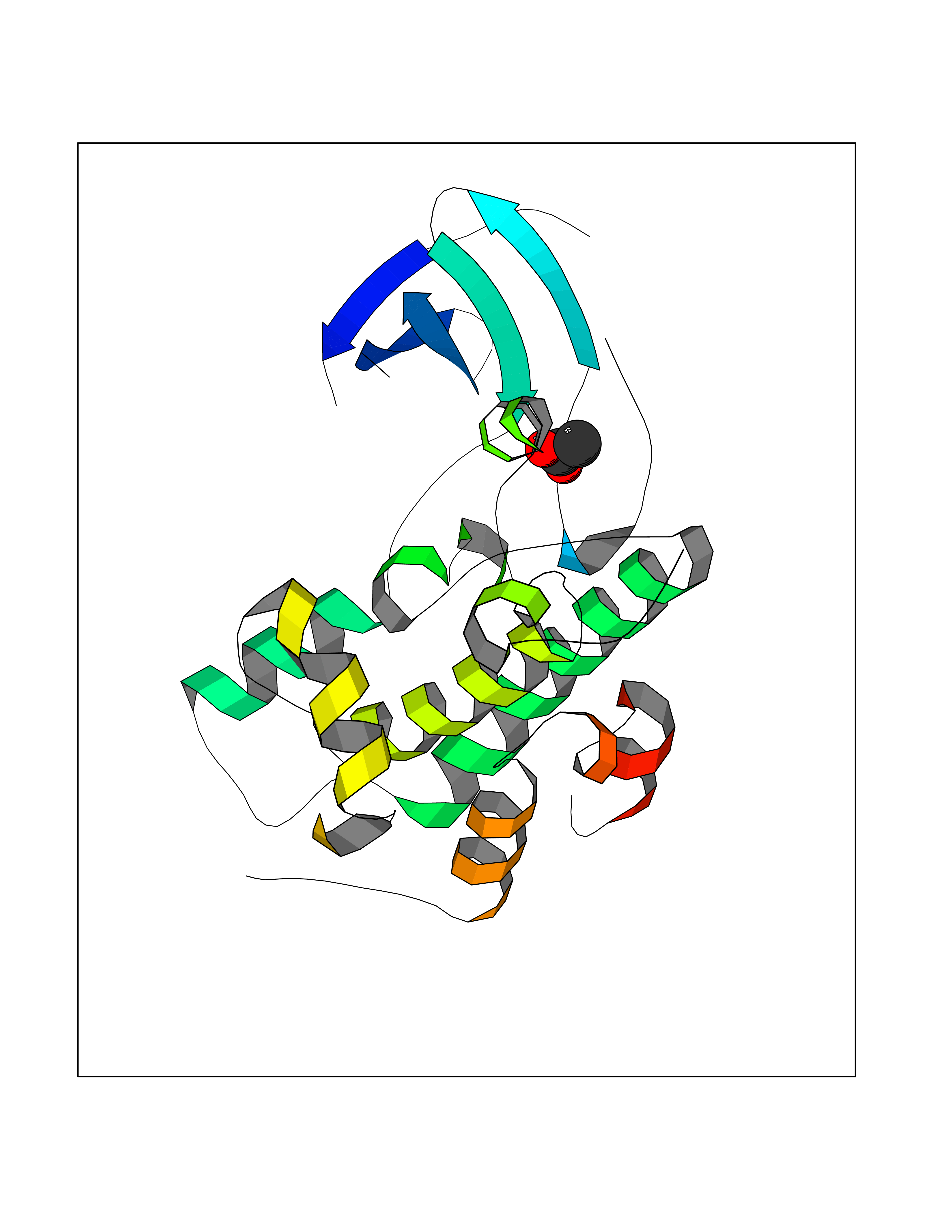 Molscript Map 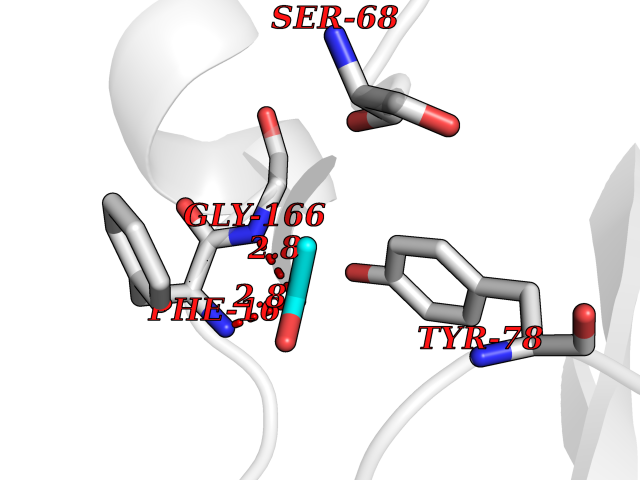 Pymol Map 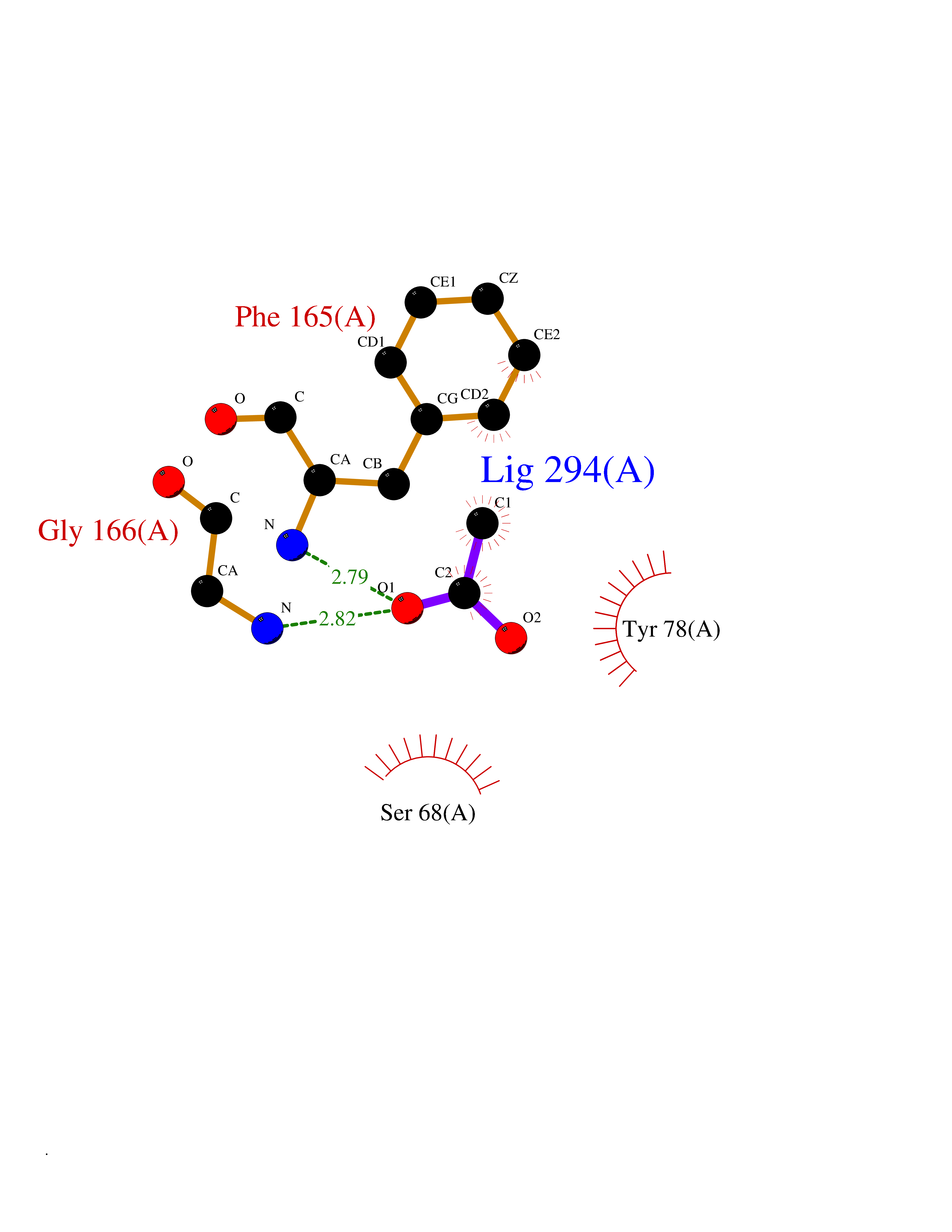 Ligplot Map 3D Binding mode Sequence NRDDYELQEVIGTAVVQAAYCEKVAIKRINAMSQCHHPNIVSYYTSFVVKDELWLVMKLLSGGSVLDIIKHIVAKGEHKSGVLDESTIATILREVLEGLEYLHKNGQIHRDVKAGNILLGEDGSVQIADFGVSAFLAGTPCWMAPEVMEQVRGYDFKADIWSFGITAIELATGAAPYHKYPPMKVLMLTLQNDPPSLETEMLKKYGKSFRKMISLCLQKDPEKRPTAAELLRHKFF Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Kallikrein-6 (KLK6) | 1LO6 | 4.39 | |
Target general information Gen name KLK6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Zyme; Serine protease 9; Serine protease 18; SP59; Protease M; PRSS9; PRSS18; Neurosin; MSP; K6 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function Shows activity against amyloid precursor protein, myelin basic protein, gelatin, casein and extracellular matrix proteins such as fibronectin, laminin, vitronectin and collagen. Degrades alpha-synuclein and prevents its polymerization, indicating that it may be involved in the pathogenesis of Parkinson disease and other synucleinopathies. May be involved in regulation of axon outgrowth following spinal cord injury. Tumor cells treated with a neutralizing KLK6 antibody migrate less than control cells, suggesting a role in invasion and metastasis. Serine protease which exhibits a preference for Arg over Lys in the substrate P1 position and for Ser or Pro in the P2 position. Related diseases Prieto syndrome (PRS) [MIM:309610]: An X-linked recessive disorder characterized by impaired intellectual development, developmental delay, autism spectrum disorder, variable epilepsy, craniofacial dysmorphism, and structural brain abnormalities including polymicrogyria and cerebral atrophy. {ECO:0000269|PubMed:35678782}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03127 Interacts with Q8NC06-3; P11117; Q53FZ2-2; Q96BT7-2; P05067; Q9NP61; Q5H9R4-2; Q8WXK3-2; Q12797-6; Q9Y6H3; P27449; O95817; Q8TBE0; Q9UQB8-3; Q9UQB8-6; P51572; O15155-2; Q9BXY8; Q7L1Q6-2; Q9H0W9-3; Q9H257-2; Q86XM0; O75309; P49336-2; Q9Y281; Q8NE62; O75508; Q9BT09; P20849; Q86WV2; P68400; P09668; P09172; P61962; Q9BTE7; Q6ZPD9-2; P63167; Q13144; P23588; P16452; Q5RHP9-3; Q6NXG1; Q6NXG1-3; Q7L5A8; Q6NZ36-4; Q14296; P62861; P31994; Q7L622; P24522; P15976-2; Q9NXC2; B2RAF7; P52790; Q9P0W2; Q4VB01; Q7LGA3-3; Q96D96-2; Q8IYA8; Q14005-2; Q0VD86; Q9BT40; Q9Y283-3; P57682; Q9Y2M5; O60259; P08727; Q14533; Q3LI72; Q3SYF9; Q8IUC2; Q92615; Q14847-2; Q6DKI2; Q8TE12-2; O95332; Q96JB6; Q99683; Q15759; P42679; Q8N6R0; Q14728; A0A0A0MR05; Q9BRA0; Q92886; P48645; Q9Y239; P06748; Q6P4D5-2; Q8WW12; Q16549; Q13371; Q9NZ53-2; Q9GZS1; P19388; Q6ZMI0-5; Q96QH2; Q86UA1; P61289; P21246; P53801; Q9NWB1-5; Q9BWF3; P52756; P47804-3; Q9H0X6; Q969K3; Q9BY12-3; Q86SQ7-2; Q9NTN9-3; Q8IUQ4-2; Q9H2B4-2; Q99717; P37840; Q5T0L3; Q496A3; Q9BUD6; Q9C004; Q99469; O75558; Q9UMX1; O43463; O60506-4; Q8TDR4; Q96A09; Q01664; Q6YHU6; Q9H808; Q8IU80-2; Q8IUR5-4; Q8WVP5; Q96KP6; O14787-2; O94900; P06753-2; Q9NX07; O60636; Q86UF1; Q5VYS8-5; Q9GZX9; Q13404; Q9H9P5-5; Q9NVA1; P61964; Q9NZC7-5; O00308; Q9HAV4; Q8N0Y2-2; Q7Z783; Q96EJ4 EC number EC 3.4.21.- Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Cleavage on pair of basic residues; Cytoplasm; Direct protein sequencing; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Hydrolase; Microsome; Mitochondrion; Nucleus; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 24300.3 Length 221 Aromaticity 0.06 Instability index 37.16 Isoelectric point 6.65 Charge (pH=7) -1.29 2D Binding mode Binding energy (Kcal/mol) -5.98 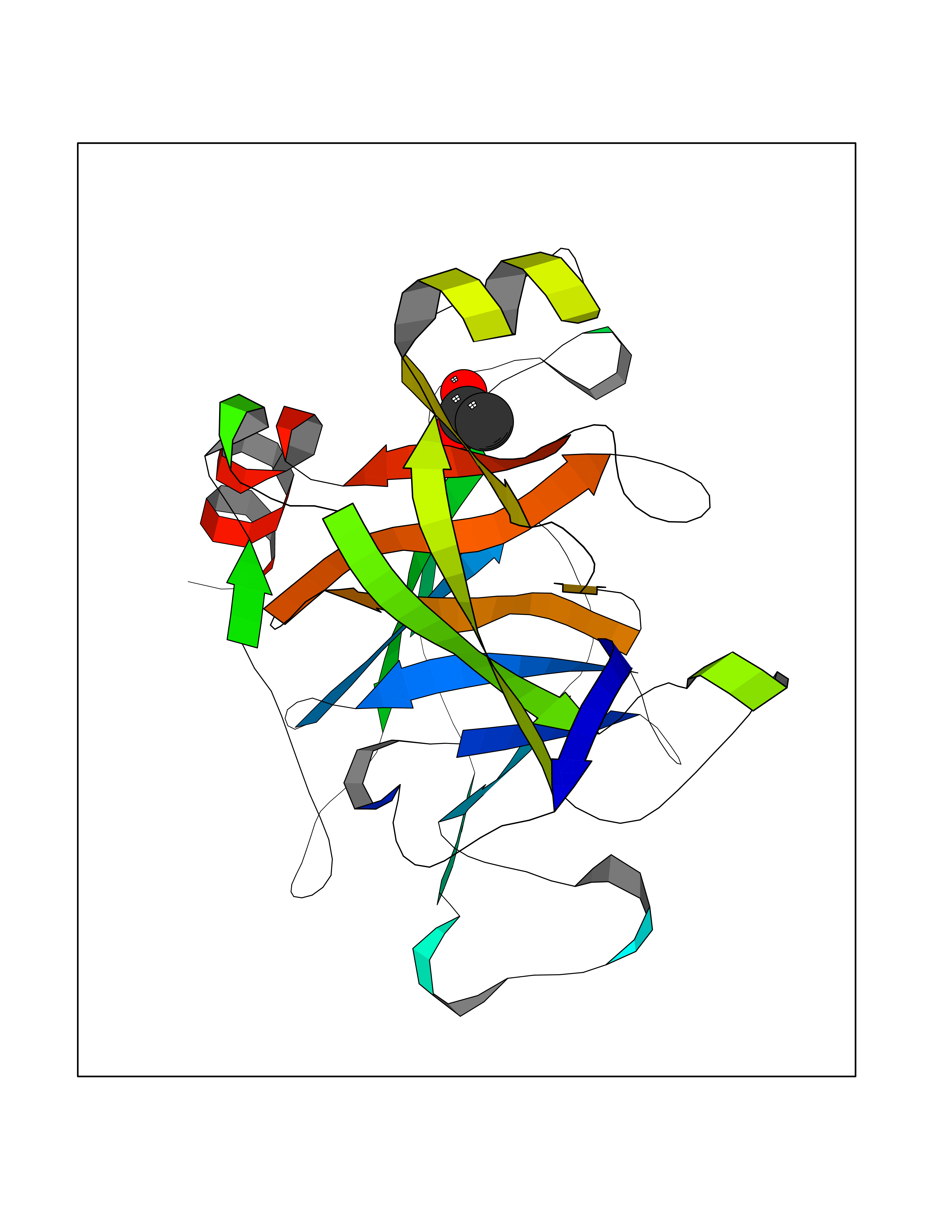 Molscript Map 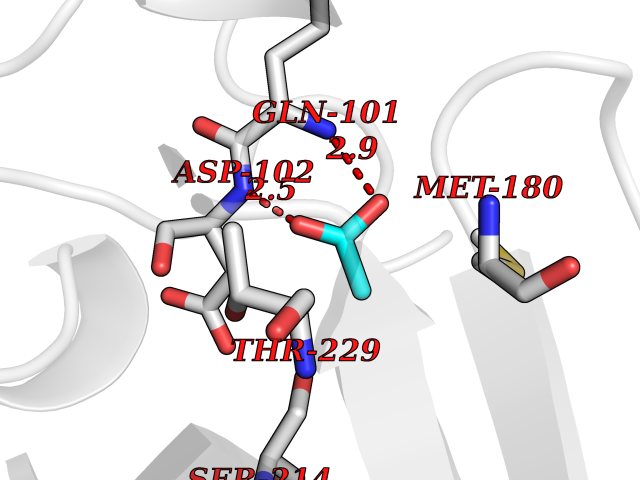 Pymol Map 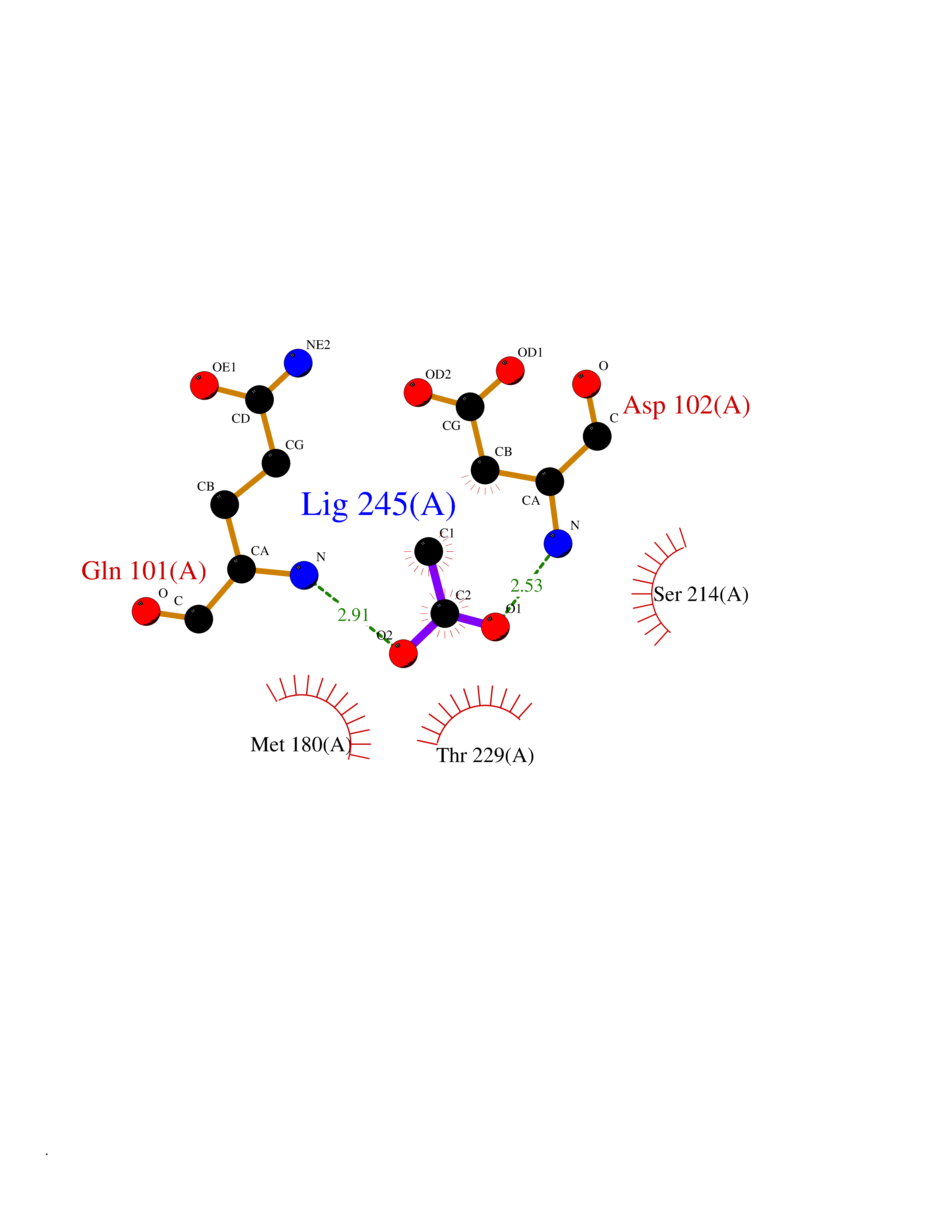 Ligplot Map 3D Binding mode Sequence LVHGGPCDKTSHPYQAALYTSGHLLCGGVLIHPLWVLTAAHCKKPNLQVFLGKHNLRQRESSQEQSSVVRAVIHPDYDAASHDQDIMLLRLARPAKLSELIQPLPLERDCSANTTSCHILGWGKTADGDFPDTIQCAYIHLVSREECEHAYPGQITQNMLCAGDEKYGKDSCQGDSGGPLVCGDHLRGLVSWGNIPCGSKEKPGVYTNVCRYTNWIQKTIQ Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Deubiquitinating enzyme 1 (USP1) | 7ZH4 | 4.39 | |
Target general information Gen name USP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hUBP; Ubiquitin-specific-processing protease 1; Ubiquitin thioesterase 1; Ubiquitin carboxyl-terminal hydrolase 1 Protein family Peptidase C19 family Biochemical class Peptidase Function Involved in PCNA-mediated translesion synthesis (TLS) by deubiquitinating monoubiquitinated PCNA. Has almost no deubiquitinating activity by itself and requires the interaction with WDR48 to have a high activity. Negative regulator of DNA damage repair which specifically deubiquitinates monoubiquitinated FANCD2. Related diseases Brachydactyly A2 (BDA2) [MIM:112600]: A form of brachydactyly. Brachydactyly defines a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. In brachydactyly type A2 shortening of the middle phalanges is confined to the index finger and the second toe, all other digits being more or less normal. Because of a rhomboid or triangular shape of the affected middle phalanx, the end of the second finger usually deviates radially. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}. The gene represented in this entry is involved in disease pathogenesis. Duplications of a cis-regulatory element located approximately 110 kb downstream of BMP2 have been found in BDA2 families. They likely cause altered BMP2 expression with pathological consequences. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}.; DISEASE: Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 1 (SSFSC1) [MIM:617877]: An autosomal dominant disorder characterized by short stature, facial dysmorphism, skeletal anomalies, and variable cardiac defects. Distinctive facial features include midface retrusion, short upturned nose, long philtrum, high-arched or cleft palate, and variable degrees of micrognathia and dental crowding. Skeletal anomalies include patterning defects of the axial skeleton, characterized by 11 pairs of ribs and brachydactyly of the fifth ray. Congenital heart defects are variably observed and appear to involve primarily the cardiac outflow tract. {ECO:0000269|PubMed:29198724}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q8TAF3; Q8TAF3-1 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Autocatalytic cleavage; DNA damage; DNA repair; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID D Molecular weight (Da) 32426 Length 285 Aromaticity 0.1 Instability index 50.73 Isoelectric point 5.85 Charge (pH=7) -4.67 2D Binding mode Binding energy (Kcal/mol) -5.99 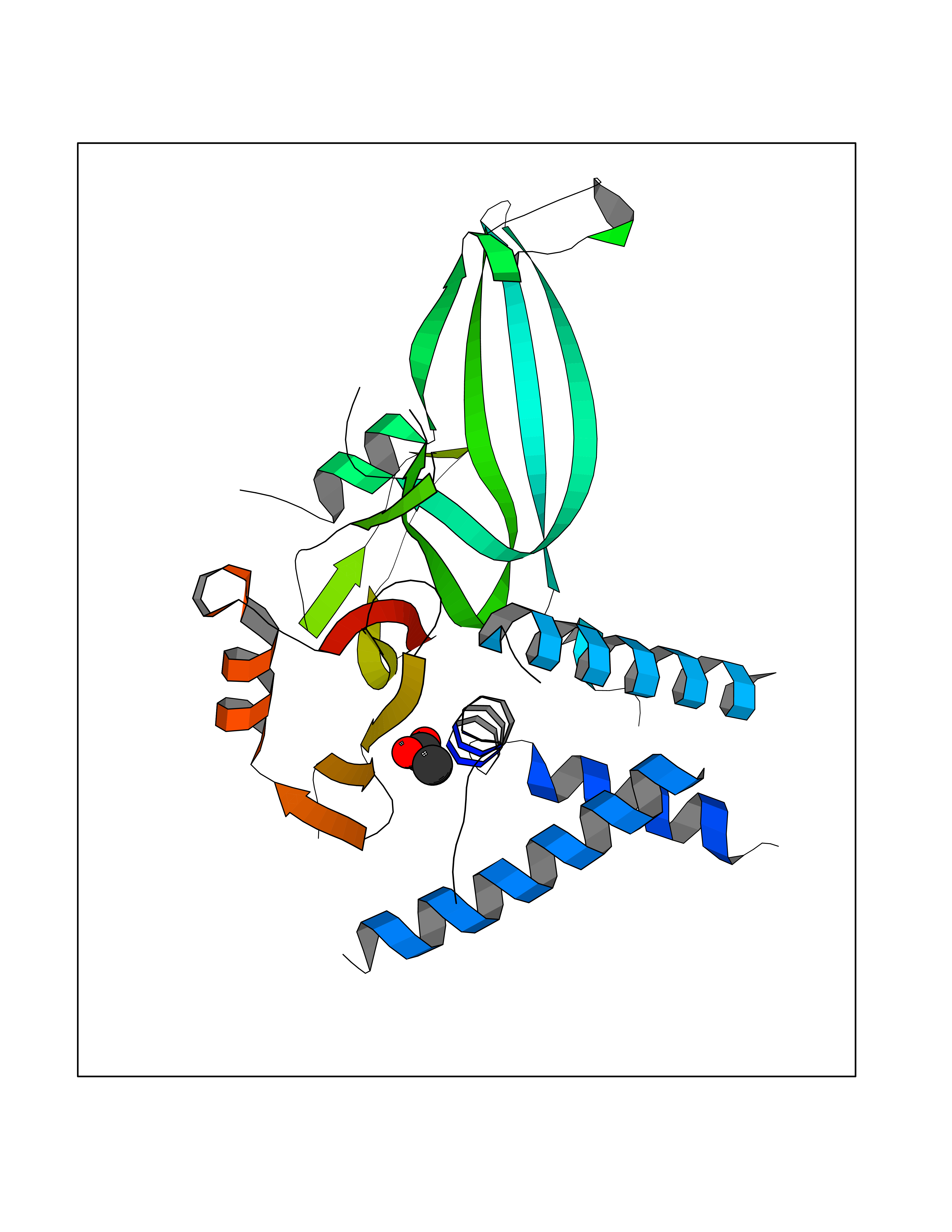 Molscript Map 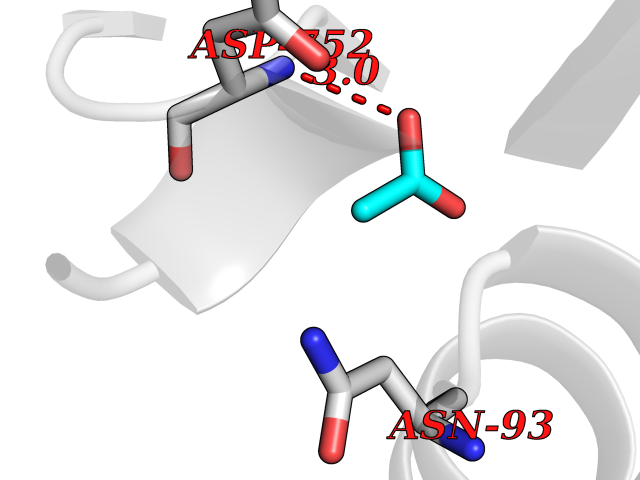 Pymol Map 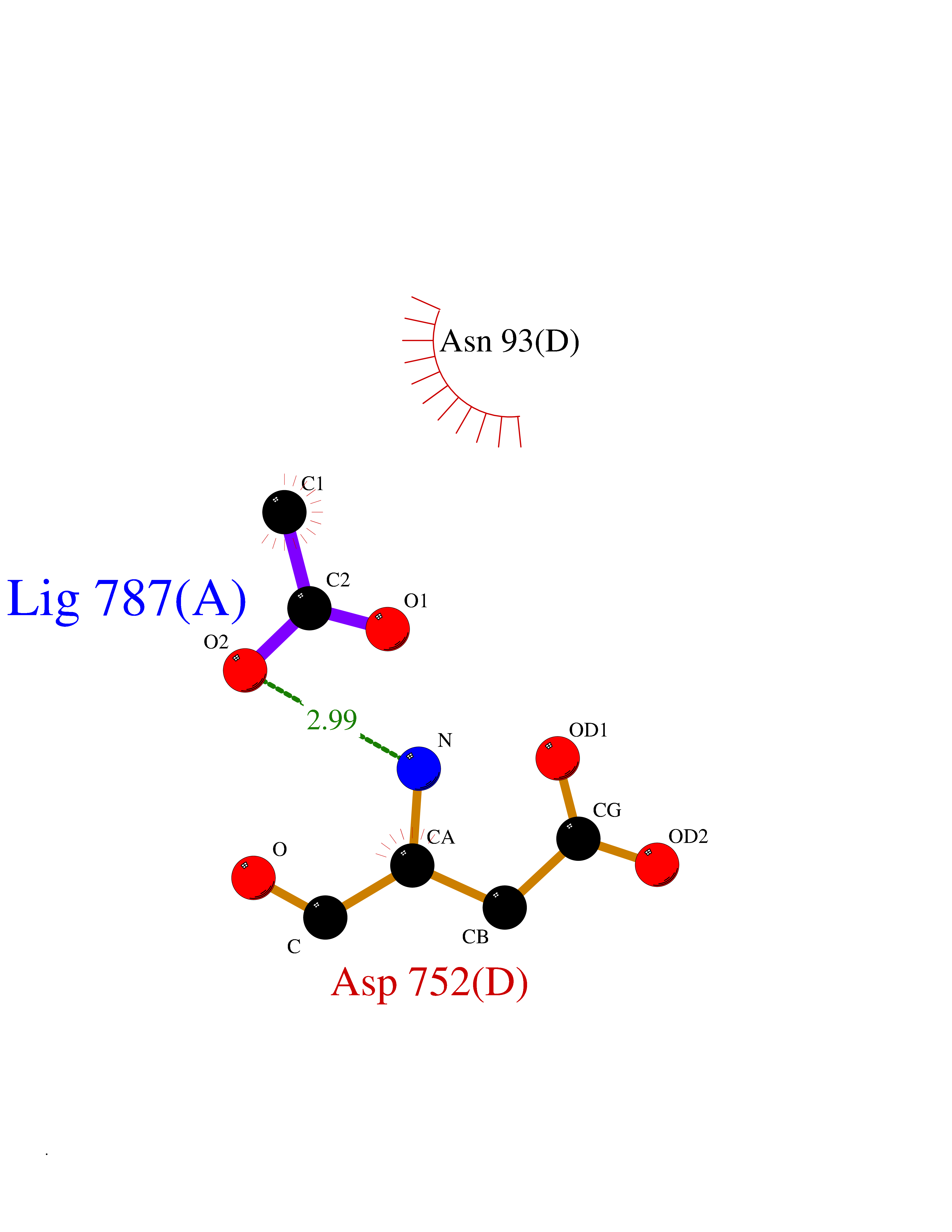 Ligplot Map 3D Binding mode Sequence GLNNLGNTSYLNSILQVLYFCPGFKSGVKHLFNIISRKKYELICSLQSLIISVEQLQASFLLNPLQHDAQEVLQCILGNIQETCQLLKKGFELVEKLFQGQLVLRTRCLECESLTERREDFQDISVPVQEDMKTLRWAISQFASVERIVGEDKYFCENCHHYTEAERSLLFDKMPEVITIHLKCFAASGLSKINTPLLTPLKLSLEEWSTKPTNDSYGLFAVVMHSGITISSGHYTASVKVTYEGKWLLFDDSEVKVTEEKDFLNSLSPSTSPTSTPYLLFYKKL Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Zinc finger protein Helios (IKZF2) | 7LPS | 4.39 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -5.98 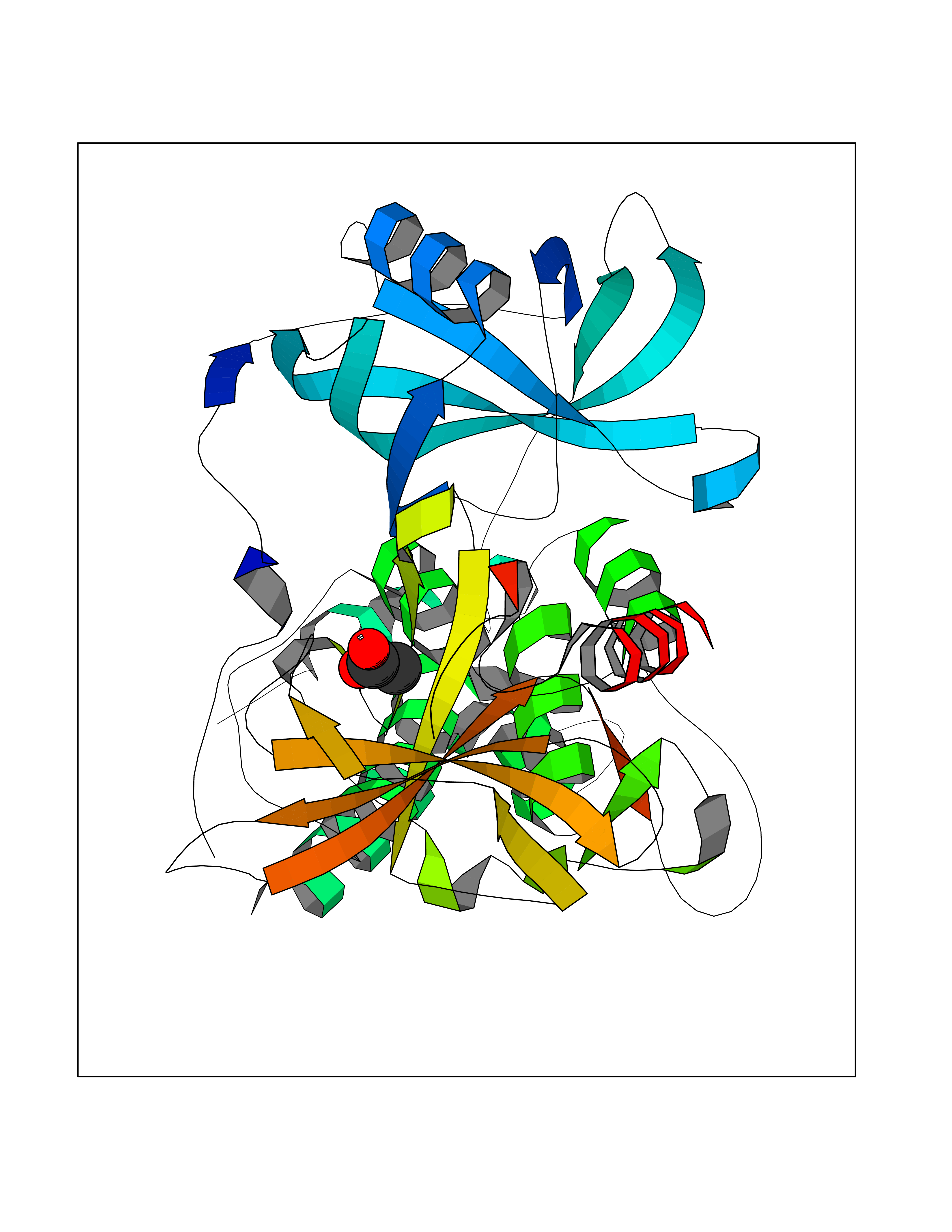 Molscript Map 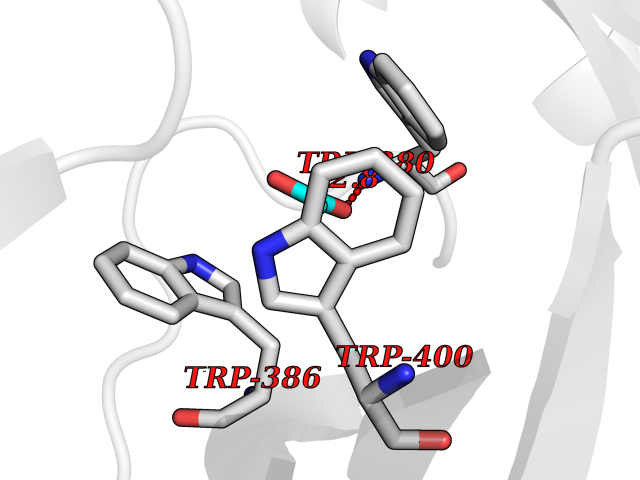 Pymol Map 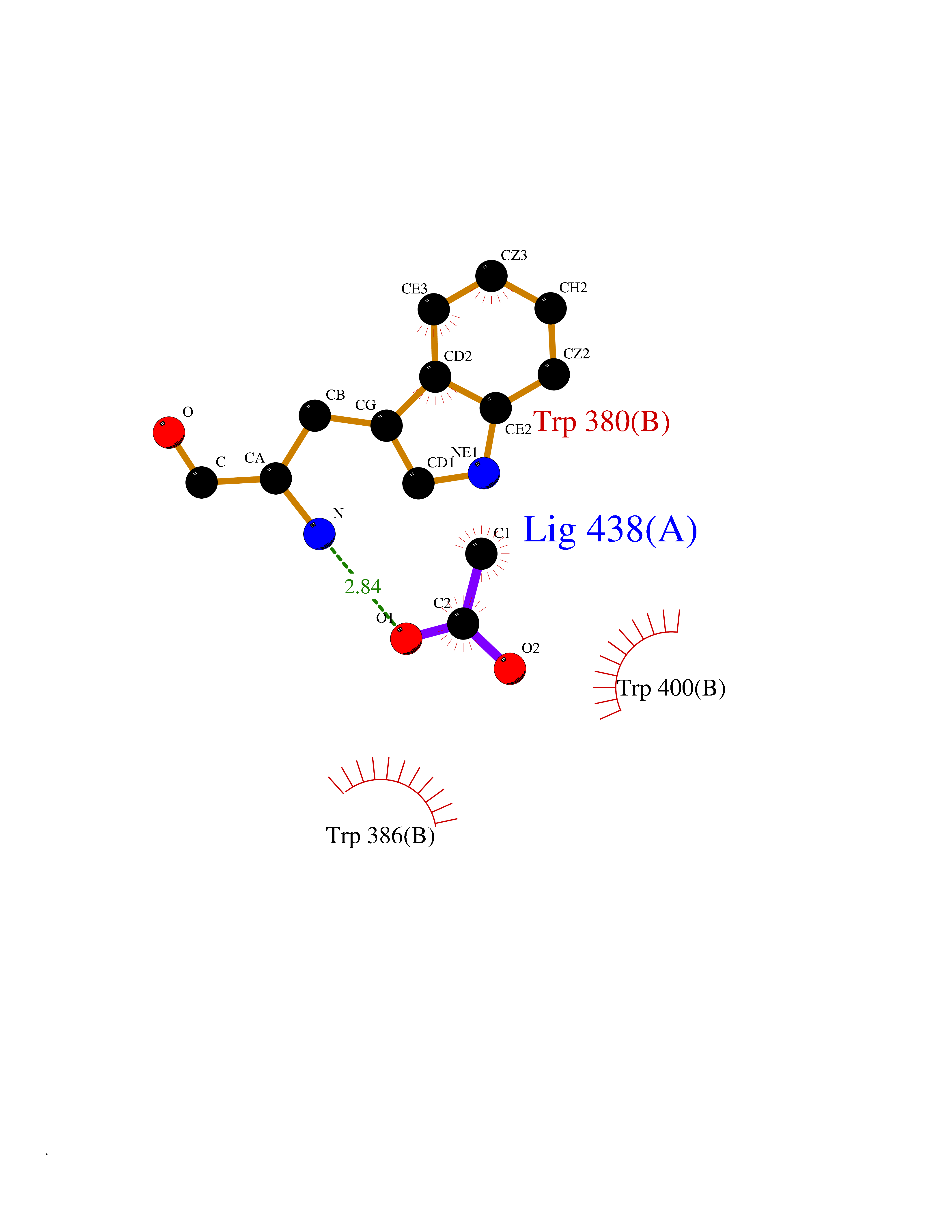 Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Glucose-dependent insulinotropic receptor (GPR119) | 7XZ6 | 4.39 | |
Target general information Gen name GPR119 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GPR119; G-protein coupled receptor 119 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the endogenous fatty-acid ethanolamide oleoylethanolamide (OEA) and lysophosphatidylcholine (LPC). Functions as a glucose-dependent insulinotropic receptor. The activity of this receptor is mediated by G proteins which activate adenylate cyclase. Seems to act through a G(s) mediated pathway. Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05166 Interacts with Q12797-6 EC number NA Uniprot keywords 3D-structure; Cell membrane; G-protein coupled receptor; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32134.1 Length 292 Aromaticity 0.12 Instability index 34.96 Isoelectric point 9.12 Charge (pH=7) 8.03 2D Binding mode Binding energy (Kcal/mol) -5.99  Molscript Map 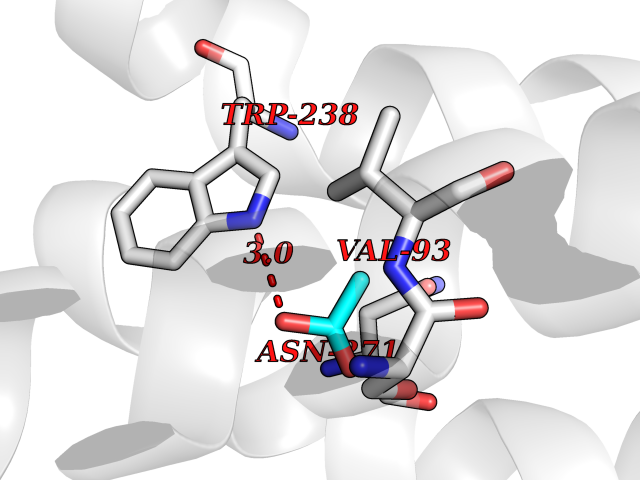 Pymol Map 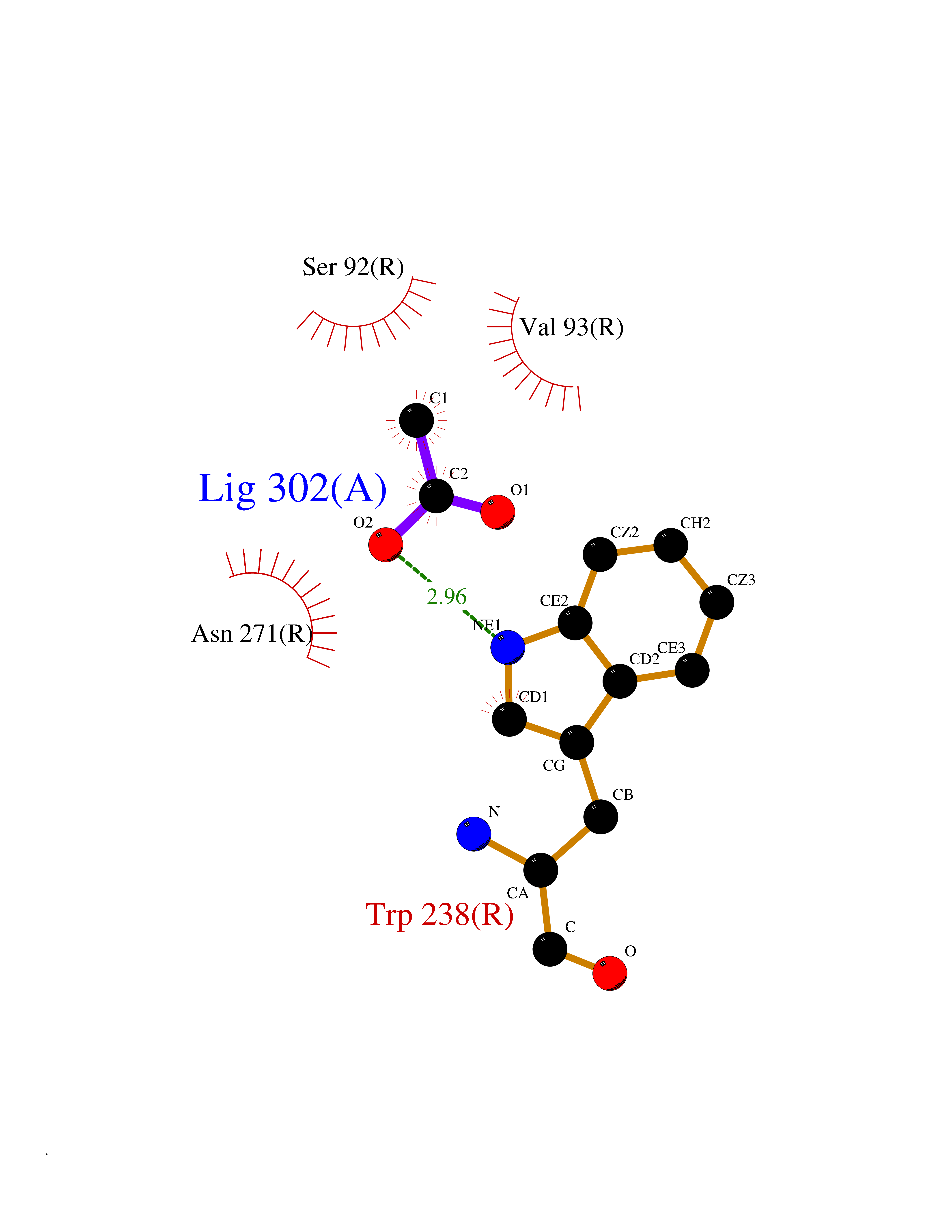 Ligplot Map 3D Binding mode Sequence MESSFSFGVILAVLASLIIATNTLVAVAVLLLIHKNDGVSLCFTLNLAVADTLIGVAISGLLTDQLSSPSRPTQKTLCSLRMAFVTSSAAASVLTVMLITFDRYLAIKQPFRYLKIMSGFVAGACIAGLWLVSYLIGFLPLGIPMFQQTAYKGQCSFFAVFHPHFVLTLSCVGFFPAMLLFVFFYCDMLKIASMHSQQIRKMEHAGAMAGSDFKALRTVSVLIGSFALSWTPFLITGIVQVACQECHLYLVLERYLWLLGVGNSLLNPLIYAYWQKEVRLQLYHMALGVKKV Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Casein kinase I epsilon (CSNK1E) | 4HNI | 4.39 | |
Target general information Gen name CSNK1E Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Casein kinase I isoform epsilon; CKIe; CKI-epsilon Protein family Protein kinase superfamily, CK1 Ser/Thr protein kinase family, Casein kinase I subfamily Biochemical class Kinase Function Can phosphorylate a large number of proteins. Participates in Wnt signaling. Phosphorylates DVL1 and DVL2. Central component of the circadian clock. In balance with PP1, determines the circadian period length, through the regulation of the speed and rhythmicity of PER1 and PER2 phosphorylation. Controls PER1 and PER2 nuclear transport and degradation. Inhibits cytokine-induced granuloytic differentiation. Casein kinases are operationally defined by their preferential utilization of acidic proteins such as caseins as substrates. Related diseases Truncation of the 3'-untranslated (3'-UTR) region of CD274 transcripts leads to elevated expression of CD274 in multiple cancers including T-cell leukemia, diffuse large B-cell lymphoma and stomach adenocarcinoma (PubMed:27281199). Disruption of 3'-UTR region is caused by structural variants that stabilize CD274 transcripts, leading to overexpression (PubMed:27281199). Increased expression in tumors promotes immune evasion and tumor cell growth by allowing malignant cells to escape destruction by the immune system (PubMed:27281199). {ECO:0000269|PubMed:27281199}. Drugs (DrugBank ID) DB06195; DB14989 Interacts with P25054; O15169; O14640; O14641; Q92997; Q9BQ89; Q1W6H9; Q86UY5; P08238; P23508; Q00987; Q16625; O15055; O75382; P62258; Q04917; Q5T7W0; O70239; Q60838 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; ATP-binding; Biological rhythms; Cytoplasm; Kinase; Methylation; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 32897.6 Length 284 Aromaticity 0.13 Instability index 40.77 Isoelectric point 9.3 Charge (pH=7) 11.34 2D Binding mode Binding energy (Kcal/mol) -5.99  Molscript Map 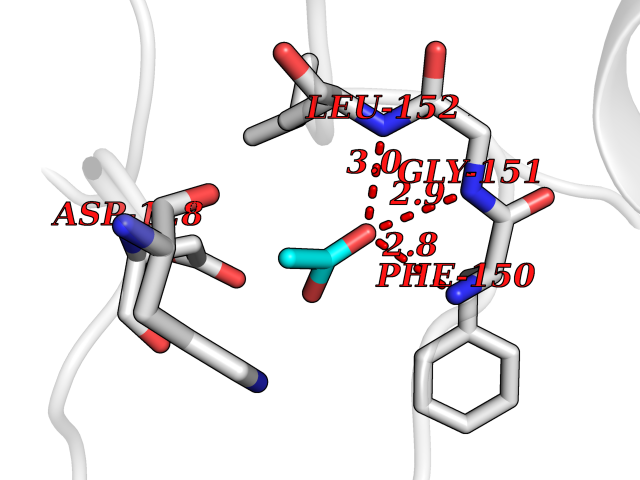 Pymol Map 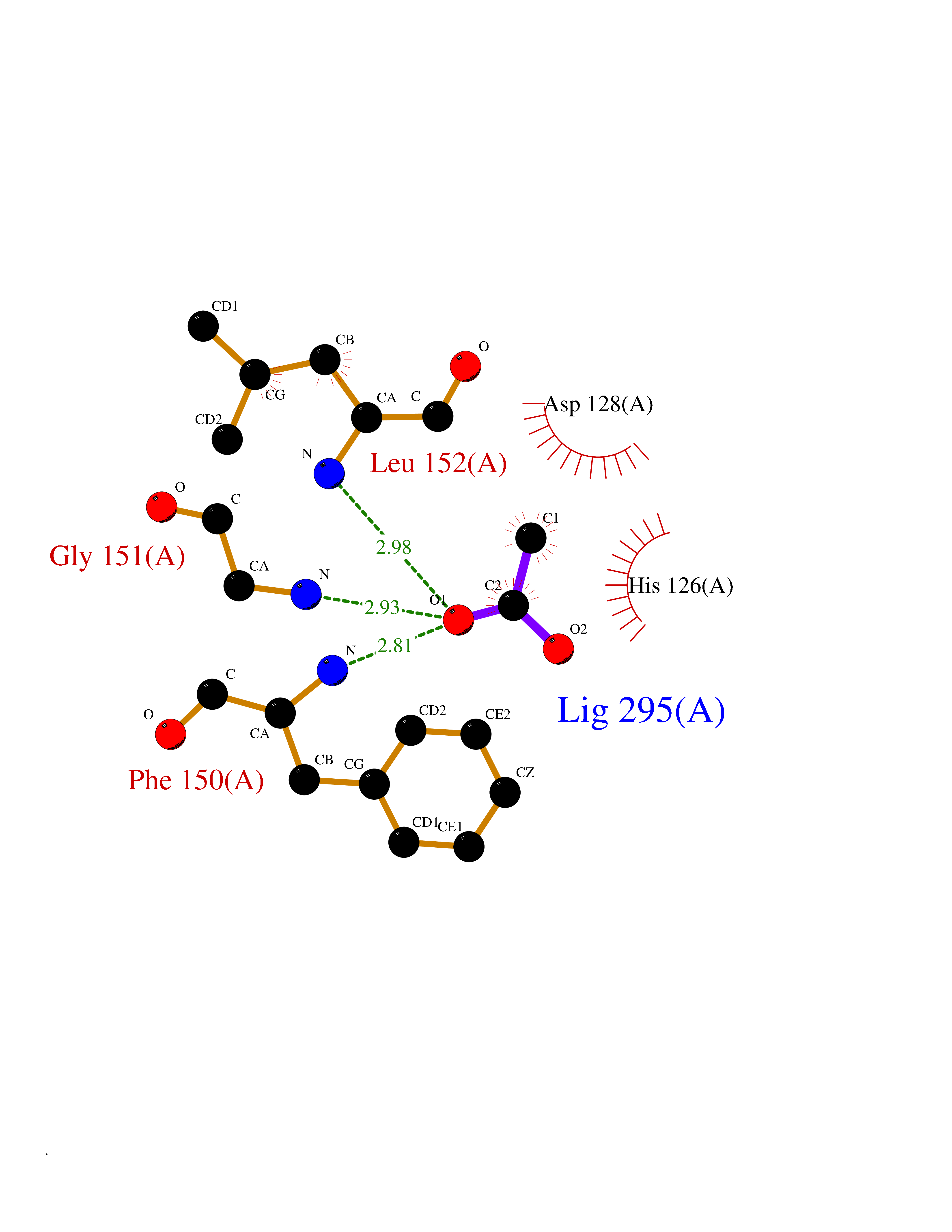 Ligplot Map 3D Binding mode Sequence LRVGNKYRLGRKIGSGSFGDIYLGANIASGEEVAIKLECVHIESKFYKMMQGGVGIPSIKWCGAEGDYNVMVMELLGPSLEDLFNFCSRKFSLKTVLLLADQMISRIEYIHSKNFIHRDVKPDNFLMGLGKKGNLVYIIDFGLAKKYRDARTHQHIPYRENKNLTGTARYASINTHLGIEQSRRDDLESLGYVLMYFNLGSLPWQGLKAATKRQKYERISEKKMSTPIEVLCKGYPSEFSTYLNFCRSLRFDDKPDYSYLRQLFRNLFHRQGFSYDYVFDWNML Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | SF3b complex (SF3b) | 3LQV | 4.38 | |
Target general information Gen name SF3B1-SF3B2-SF3B3-SF3B4-SF3B5-SF3B6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NA Protein family SF3B1 family Biochemical class NA Function NA Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14017 Interacts with Q13936; Q96I25; Q15428; Q9Y3B4; O43719; Q96I25; Q9Y3B4 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Citrullination; Isopeptide bond; mRNA processing; mRNA splicing; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; RNA-binding; Spliceosome; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 13222 Length 113 Aromaticity 0.12 Instability index 27.56 Isoelectric point 9.22 Charge (pH=7) 3.86 2D Binding mode Binding energy (Kcal/mol) -5.97 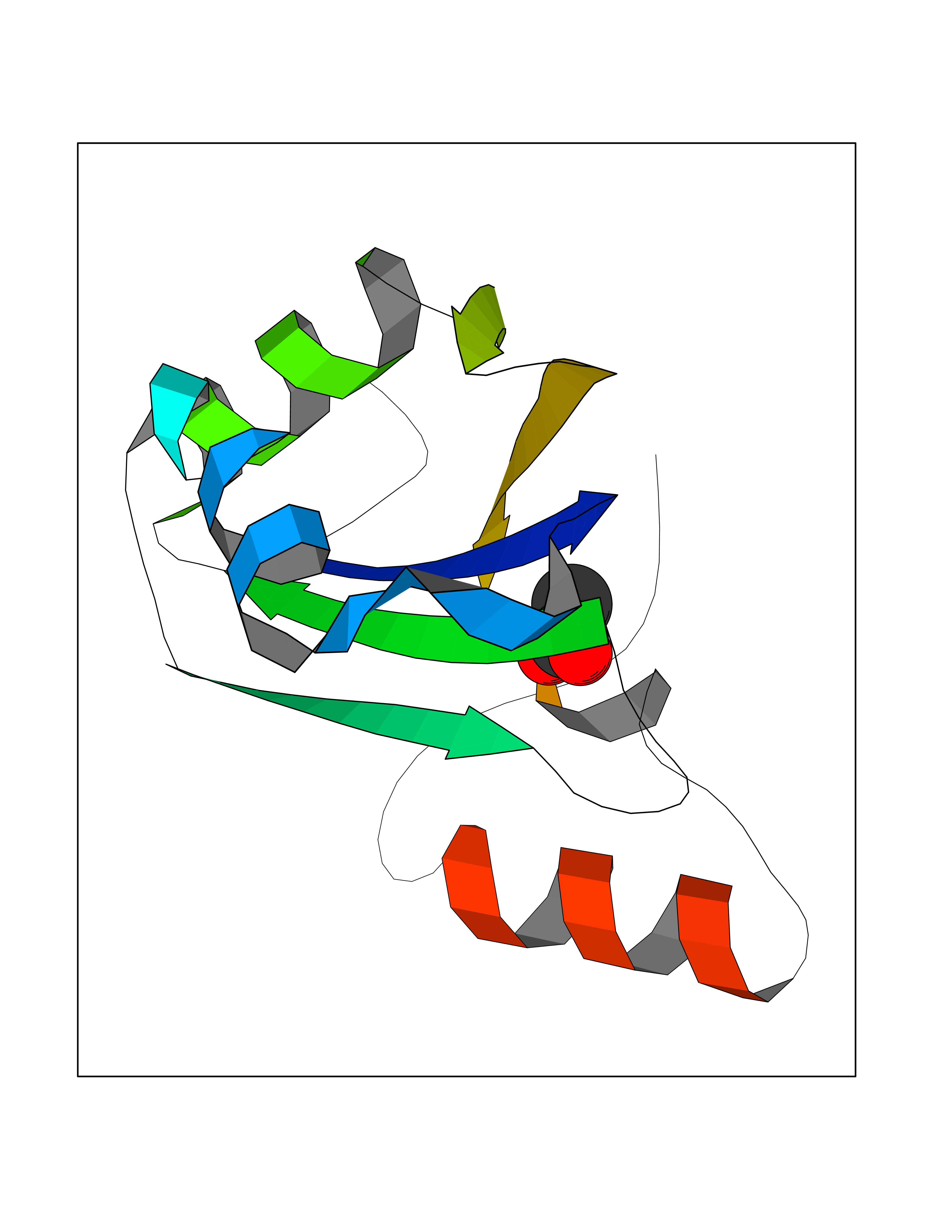 Molscript Map 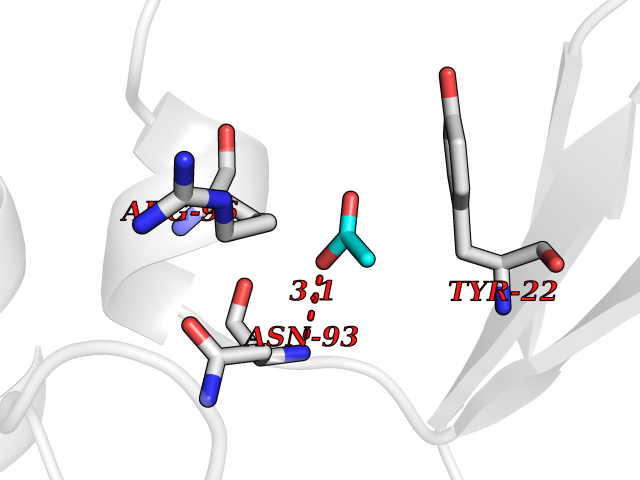 Pymol Map 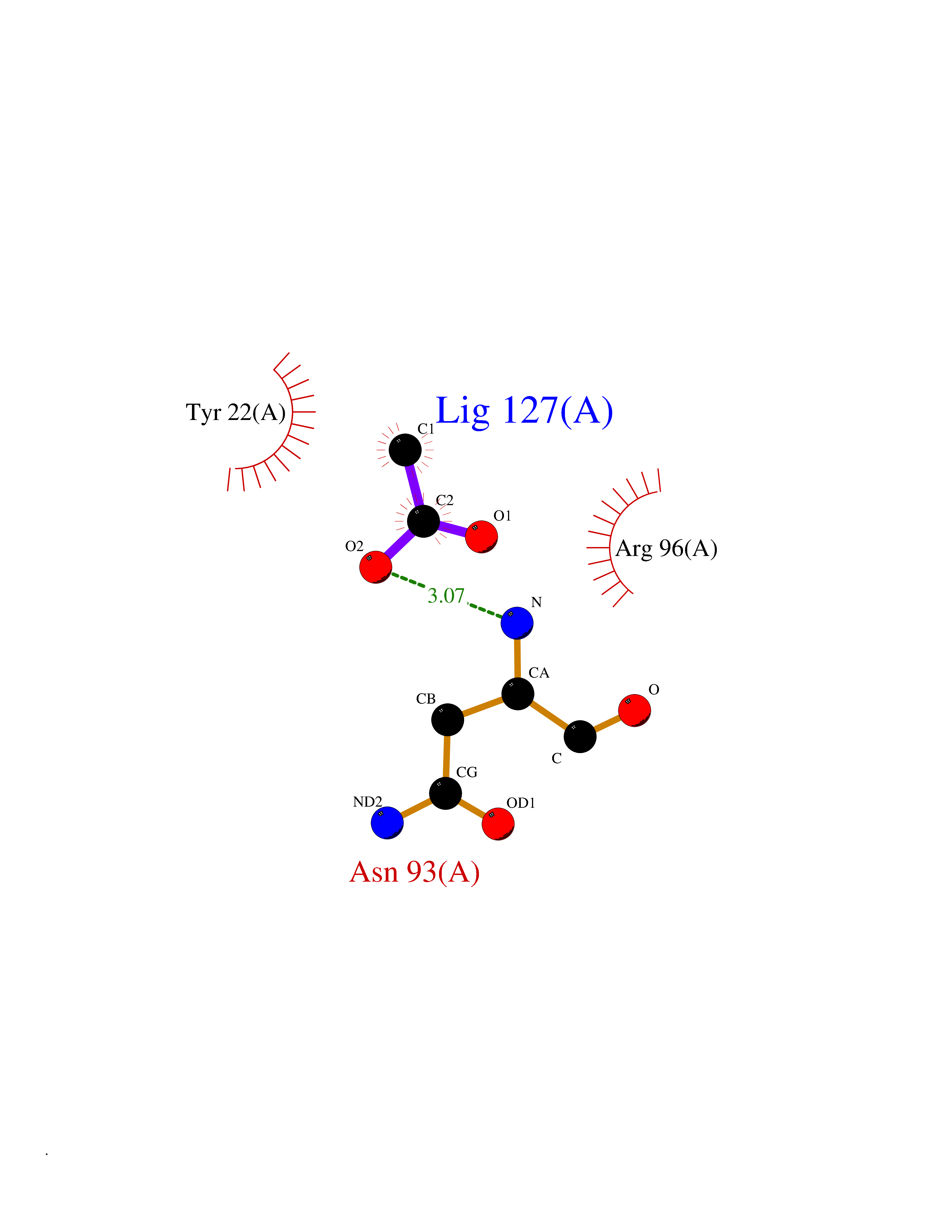 Ligplot Map 3D Binding mode Sequence LPPEVNRILYIRNLPYKITAEEMYDIFGKYGPIRQIRVGNTPETRGTAYVVYEDIFDAKNAVDHLSGFNVSNRYLVVLYYNANRAFQKMDTKKKEEQLKLLKEKYGINTDPPK Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | RAC-beta serine/threonine-protein kinase (AKT2) | 3D0E | 4.38 | |
Target general information Gen name AKT2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAC-PK-beta; Protein kinase B beta; Protein kinase Akt-2; PKB beta Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, RAC subfamily Biochemical class Kinase Function AKT2 is one of 3 closely related serine/threonine-protein kinases (AKT1, AKT2 and AKT3) called the AKT kinase, and which regulate many processes including metabolism, proliferation, cell survival, growth and angiogenesis. This is mediated through serine and/or threonine phosphorylation of a range of downstream substrates. Over 100 substrate candidates have been reported so far, but for most of them, no isoform specificity has been reported. AKT is responsible of the regulation of glucose uptake by mediating insulin-induced translocation of the SLC2A4/GLUT4 glucose transporter to the cell surface. Phosphorylation of PTPN1 at 'Ser-50' negatively modulates its phosphatase activity preventing dephosphorylation of the insulin receptor and the attenuation of insulin signaling. Phosphorylation of TBC1D4 triggers the binding of this effector to inhibitory 14-3-3 proteins, which is required for insulin-stimulated glucose transport. AKT regulates also the storage of glucose in the form of glycogen by phosphorylating GSK3A at 'Ser-21' and GSK3B at 'Ser-9', resulting in inhibition of its kinase activity. Phosphorylation of GSK3 isoforms by AKT is also thought to be one mechanism by which cell proliferation is driven. AKT regulates also cell survival via the phosphorylation of MAP3K5 (apoptosis signal-related kinase). Phosphorylation of 'Ser-83' decreases MAP3K5 kinase activity stimulated by oxidative stress and thereby prevents apoptosis. AKT mediates insulin-stimulated protein synthesis by phosphorylating TSC2 at 'Ser-939' and 'Thr-1462', thereby activating mTORC1 signaling and leading to both phosphorylation of 4E-BP1 and in activation of RPS6KB1. AKT is involved in the phosphorylation of members of the FOXO factors (Forkhead family of transcription factors), leading to binding of 14-3-3 proteins and cytoplasmic localization. In particular, FOXO1 is phosphorylated at 'Thr-24', 'Ser-256' and 'Ser-319'. FOXO3 and FOXO4 are phosphorylated on equivalent sites. AKT has an important role in the regulation of NF-kappa-B-dependent gene transcription and positively regulates the activity of CREB1 (cyclic AMP (cAMP)-response element binding protein). The phosphorylation of CREB1 induces the binding of accessory proteins that are necessary for the transcription of pro-survival genes such as BCL2 and MCL1. AKT phosphorylates 'Ser-454' on ATP citrate lyase (ACLY), thereby potentially regulating ACLY activity and fatty acid synthesis. Activates the 3B isoform of cyclic nucleotide phosphodiesterase (PDE3B) via phosphorylation of 'Ser-273', resulting in reduced cyclic AMP levels and inhibition of lipolysis. Phosphorylates PIKFYVE on 'Ser-318', which results in increased PI(3)P-5 activity. The Rho GTPase-activating protein DLC1 is another substrate and its phosphorylation is implicated in the regulation cell proliferation and cell growth. AKT plays a role as key modulator of the AKT-mTOR signaling pathway controlling the tempo of the process of newborn neurons integration during adult neurogenesis, including correct neuron positioning, dendritic development and synapse formation. Signals downstream of phosphatidylinositol 3-kinase (PI(3)K) to mediate the effects of various growth factors such as platelet-derived growth factor (PDGF), epidermal growth factor (EGF), insulin and insulin-like growth factor I (IGF-I). AKT mediates the antiapoptotic effects of IGF-I. Essential for the SPATA13-mediated regulation of cell migration and adhesion assembly and disassembly. May be involved in the regulation of the placental development. Related diseases Defects in AKT2 are a cause of susceptibility to breast cancer (BC). AKT2 promotes metastasis of tumor cells without affecting the latency of tumor development. May play a role in glioblastoma cell survival (PubMed:20167810). {ECO:0000269|PubMed:20167810}.; DISEASE: Type 2 diabetes mellitus (T2D) [MIM:125853]: A multifactorial disorder of glucose homeostasis caused by a lack of sensitivity to insulin. Affected individuals usually have an obese body habitus and manifestations of a metabolic syndrome characterized by diabetes, insulin resistance, hypertension and hypertriglyceridemia. The disease results in long-term complications that affect the eyes, kidneys, nerves, and blood vessels. {ECO:0000269|PubMed:15166380, ECO:0000269|PubMed:19164855}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Hypoinsulinemic hypoglycemia with hemihypertrophy (HIHGHH) [MIM:240900]: A disorder characterized by hypoglycemia, low insulin levels, low serum levels of ketone bodies and branched-chain amino acids, left-sided hemihypertrophy, neonatal macrosomia, reduced consciousness and hypoglycemic seizures. {ECO:0000269|PubMed:21979934}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08073; DB07859; DB12218; DB07947; DB07812 Interacts with P31749; P49841; P08238; Q6FHY5; Q9NRD5; Q04864-2; O60504; P53804; Q9C0C9; P08670; Q15118-1 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; ATP-binding; Carbohydrate metabolism; Cell membrane; Cytoplasm; Developmental protein; Diabetes mellitus; Disease variant; Disulfide bond; Endosome; Glucose metabolism; Glycogen biosynthesis; Glycogen metabolism; Glycoprotein; Kinase; Manganese; Membrane; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; Serine/threonine-protein kinase; Sugar transport; Transferase; Translation regulation; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 37380.5 Length 324 Aromaticity 0.12 Instability index 29.68 Isoelectric point 6.19 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -5.97 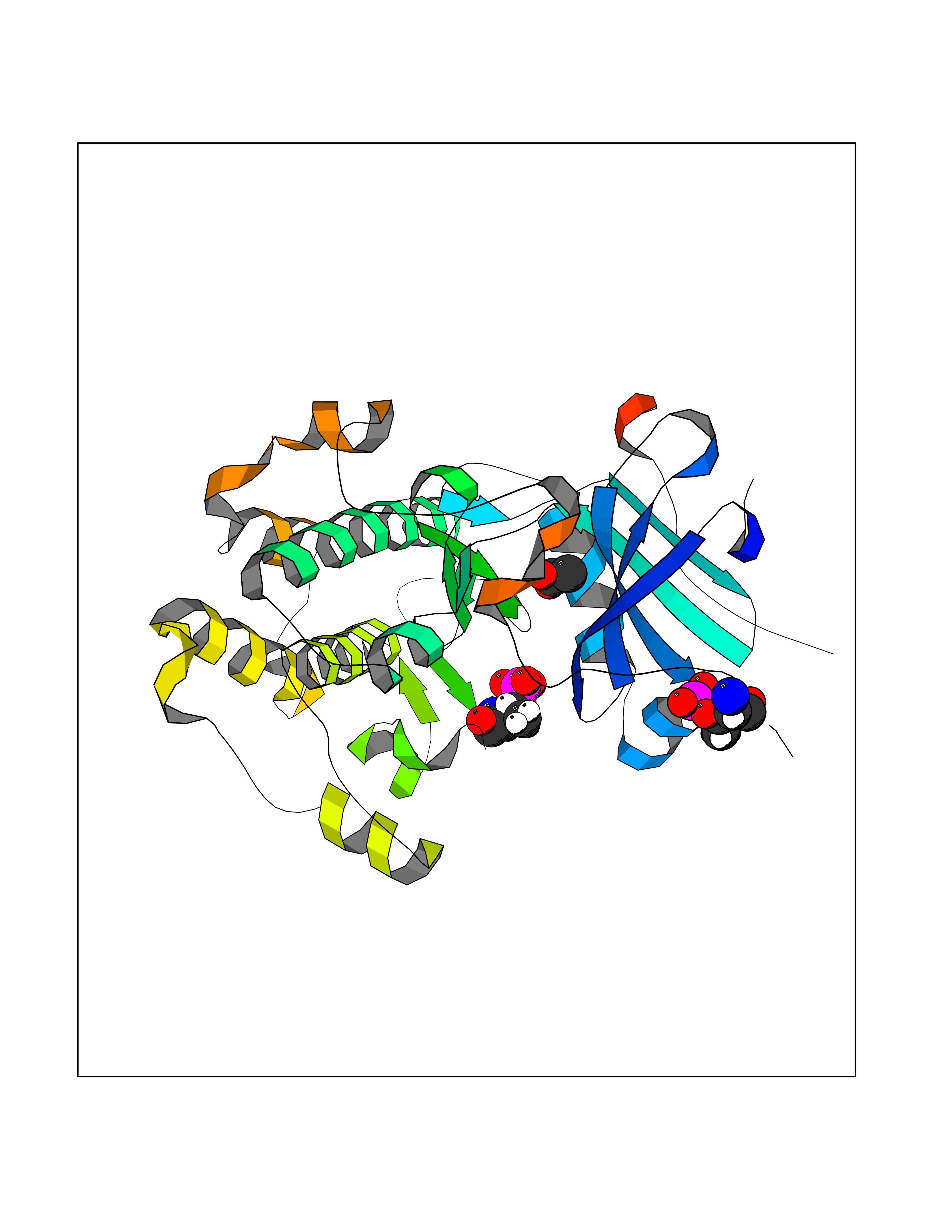 Molscript Map 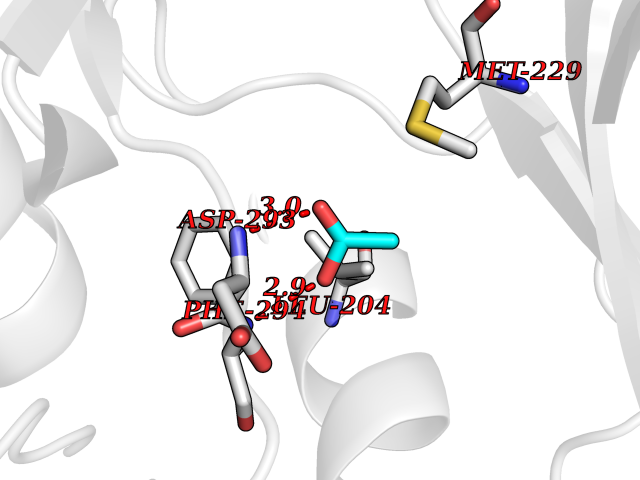 Pymol Map 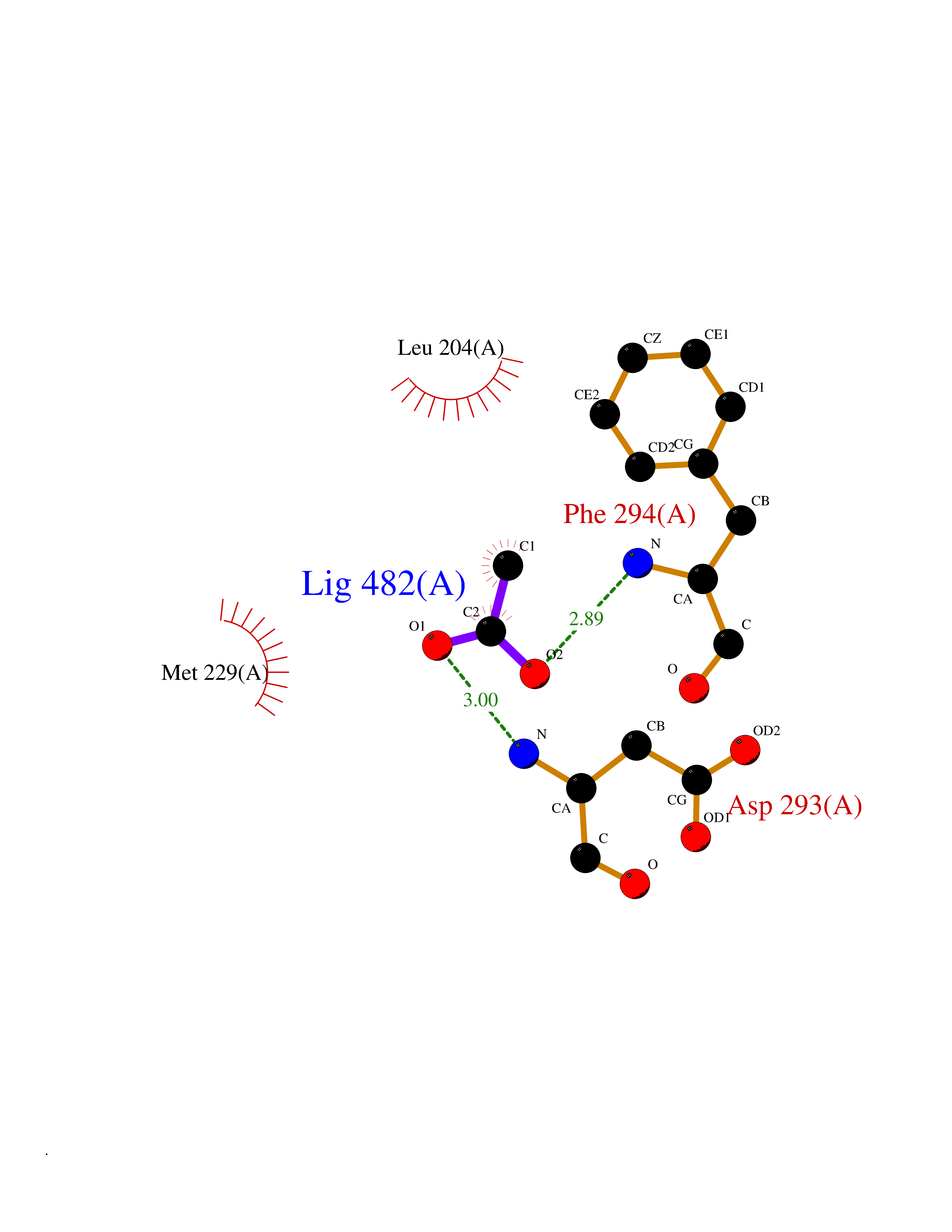 Ligplot Map 3D Binding mode Sequence KVTMNDFDYLKLLGKGTFGKVILVREKATGRYYAMKILRKEVIIAKDEVAHTVTESRVLQNTRHPFLTALKYAFQTHDRLCFVMEYANGGELFFHLSRERVFTEERARFYGAEIVSALEYLHSRDVVYRDIKLENLMLDKDGHIKITDFGLCKEGISDGATMKXFCGTPEYLAPEVLEDNDYGRAVDWWGLGVVMYEMMCGRLPFYNQDHERLFELILMEEIRFPRTLSPEAKSLLAGLLKKDPKQRLGGGPSDAKEVMEHRFFLSINWQDVVQKKLLPPFKPQVTSEVDTRYFDDEFTAQSITIXPPDQRTHFPQFDYSASIR Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Mitochondrial rRNA methyltransferase 2 (MRM2) | 2NYU | 4.38 | |
Target general information Gen name MRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms rRNA methyltransferase 2, mitochondrial; Protein ftsJ homolog 2; 16S rRNA [Um1369] 2'-O-methyltransferase; 16S rRNA (uridine(1369)-2'-O)-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, RNA methyltransferase RlmE family Biochemical class Methyltransferase Function S-adenosyl-L-methionine-dependent 2'-O-ribose methyltransferase that catalyzes the formation of 2'-O-methyluridine at position 1369 (Um1369) in the 16S mitochondrial large subunit ribosomal RNA (mtLSU rRNA), a universally conserved modification in the peptidyl transferase domain of the mtLSU rRNA. Related diseases Mitochondrial DNA depletion syndrome 17 (MTDPS17) [MIM:618567]: An autosomal recessive mitochondrial disorder characterized by childhood onset of rapidly progressive encephalopathy, stroke-like episodes, lactic acidosis, hypocitrullinemia, multiple defects of oxidative phosphorylation, mitochondrial complex I and IV deficiency, and reduced mtDNA copy number. {ECO:0000269|PubMed:28973171}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.1.1.- Uniprot keywords 3D-structure; Disease variant; Methyltransferase; Mitochondrion; Primary mitochondrial disease; Proteomics identification; Reference proteome; rRNA processing; S-adenosyl-L-methionine; Transferase; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 20008.8 Length 181 Aromaticity 0.07 Instability index 53.22 Isoelectric point 8.43 Charge (pH=7) 1.79 2D Binding mode Binding energy (Kcal/mol) -5.97 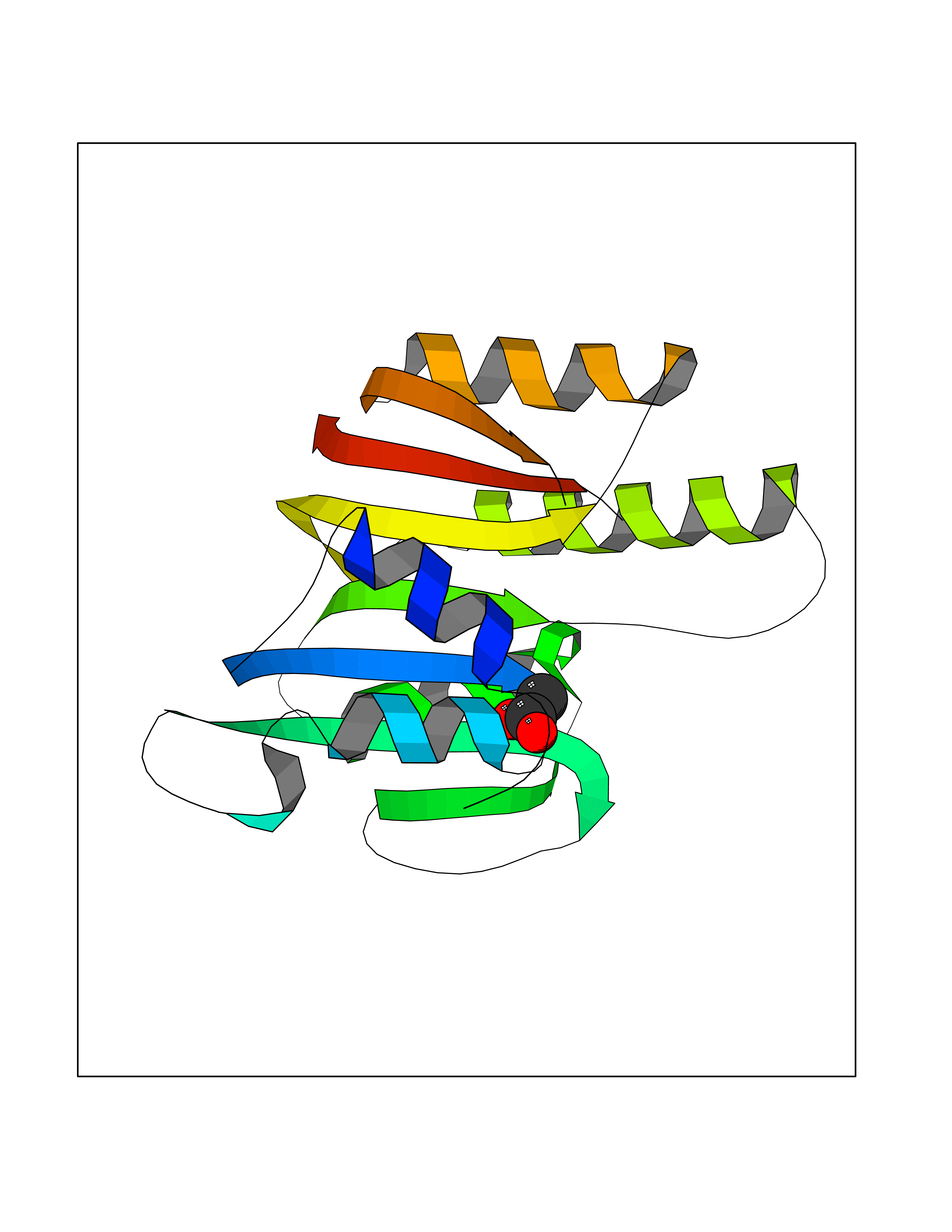 Molscript Map 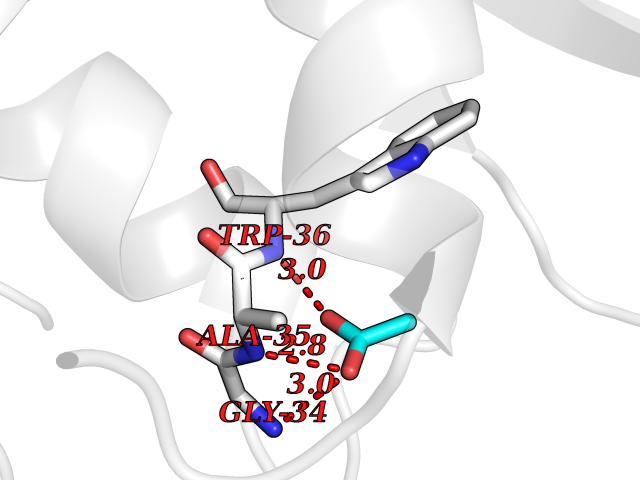 Pymol Map 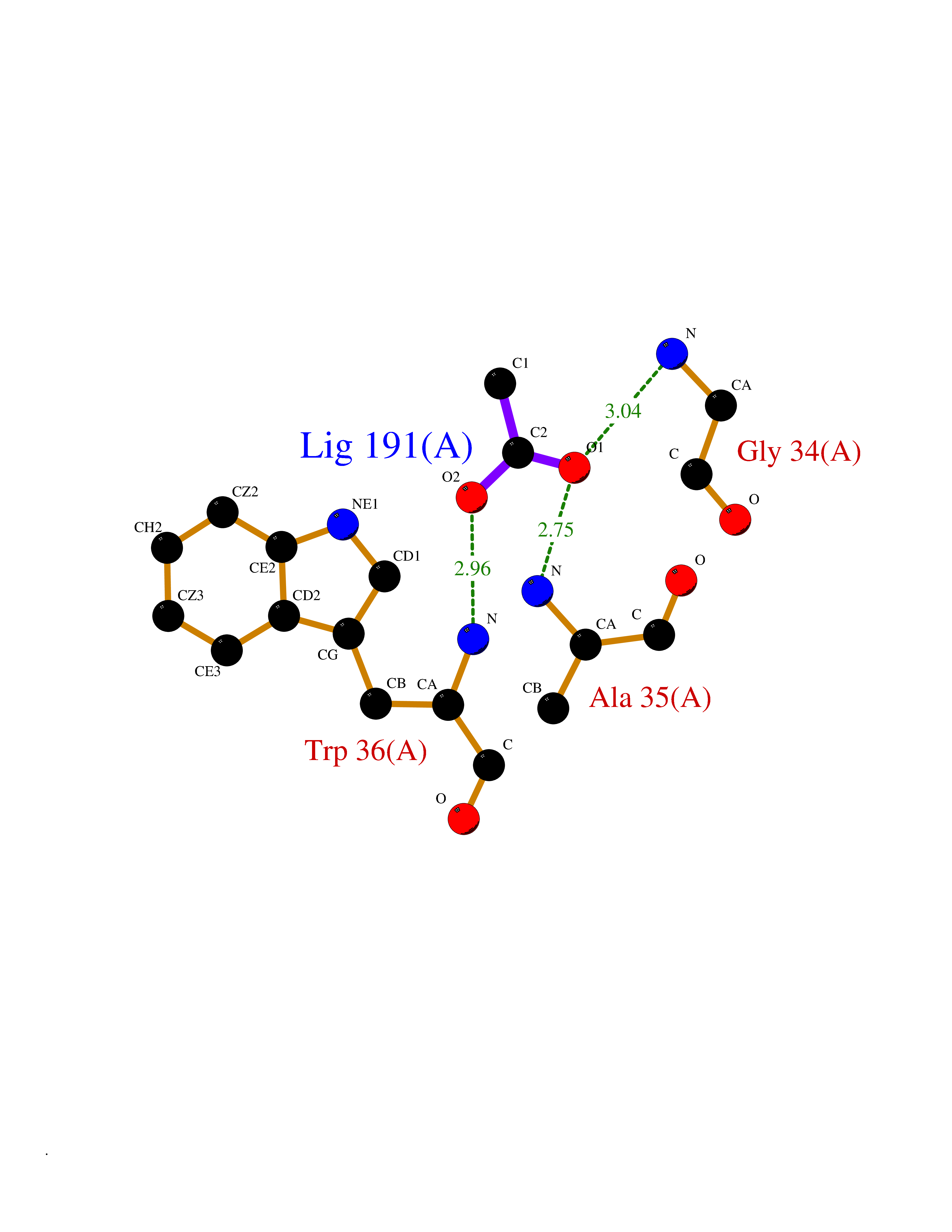 Ligplot Map 3D Binding mode Sequence SYRSRSAFKLLEVNERHQILRPGLRVLDCGAAPGAWSQVAVQKVNAAGTDPSSPVGFVLGVDLLHIFPLEGATFLCPADVTDPRTSQRILEVLPGRRADVILSDMAPNATGFRDLDHDRLISLCLTLLSVTPDILQPGGTFLCKTWAGSQSRRLQRRLTEEFQNVRIIKPEVYFLATQYHG Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Aggrecanase (ADAMTS5) | 3HY7 | 4.38 | |
Target general information Gen name ADAMTS5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aggrecanase-2; ADMP-2; ADAMTS5; ADAMTS-5; ADAM-TS5; ADAM-TS 5; ADAM-TS 11; A disintegrin and metalloproteinase with thrombospondinmotifs 5 Protein family NA Biochemical class Peptidase Function Cleaves aggrecan, a cartilage proteoglycan, and may be involved in its turnover. May play an important role in the destruction of aggrecan in arthritic diseases. May play a role in proteolytic processing mostly during the peri-implantation period. Related diseases Mucopolysaccharidosis 1H (MPS1H) [MIM:607014]: A severe form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1H usually present, within the first year of life, a combination of hepatosplenomegaly, skeletal deformities, corneal clouding and severe intellectual disability. Obstructive airways disease, respiratory infection and cardiac complications usually result in death before 10 years of age. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:1301941, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:24036510, ECO:0000269|PubMed:31194252, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:7951228, ECO:0000269|PubMed:8019563, ECO:0000269|PubMed:8328452, ECO:0000269|PubMed:8401515, ECO:0000269|Ref.20}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1H/S (MPS1H/S) [MIM:607015]: A form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. MPS1H/S represents an intermediate phenotype of the MPS1 clinical spectrum. It is characterized by relatively little neurological involvement, but most of the somatic symptoms described for severe MPS1 develop in the early to mid-teens, causing considerable loss of mobility. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8401515}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1S (MPS1S) [MIM:607016]: A mild form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1S may have little or no neurological involvement, normal stature and life span, but present development of joints stiffness, mild hepatosplenomegaly, aortic valve disease and corneal clouding. {ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:25256405, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8213840}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06837; DB03880; DB06945 Interacts with P13608 EC number EC 3.4.24.- Uniprot keywords 3D-structure; Cleavage on pair of basic residues; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 23997.8 Length 217 Aromaticity 0.06 Instability index 46.68 Isoelectric point 5.82 Charge (pH=7) -8.36 2D Binding mode Binding energy (Kcal/mol) -5.97 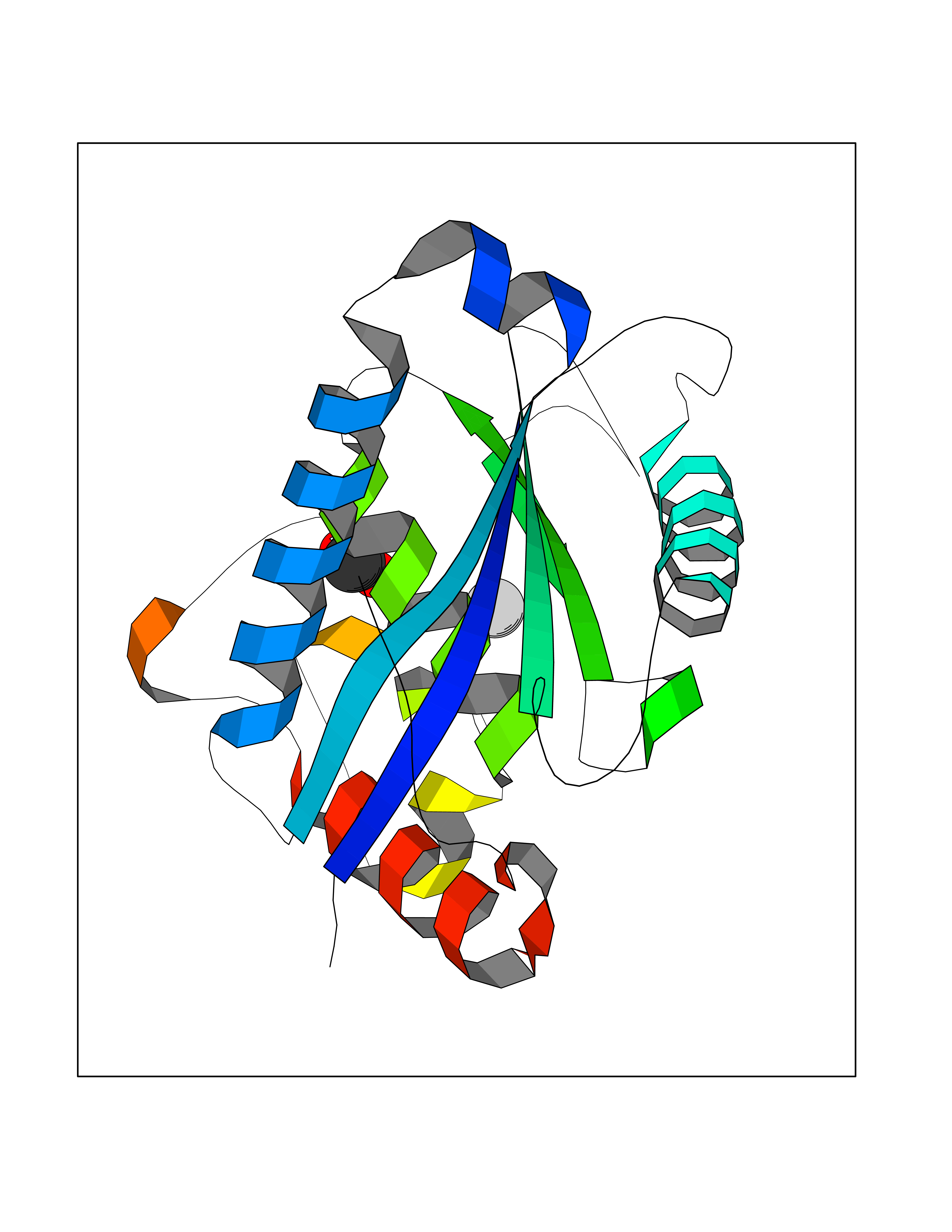 Molscript Map 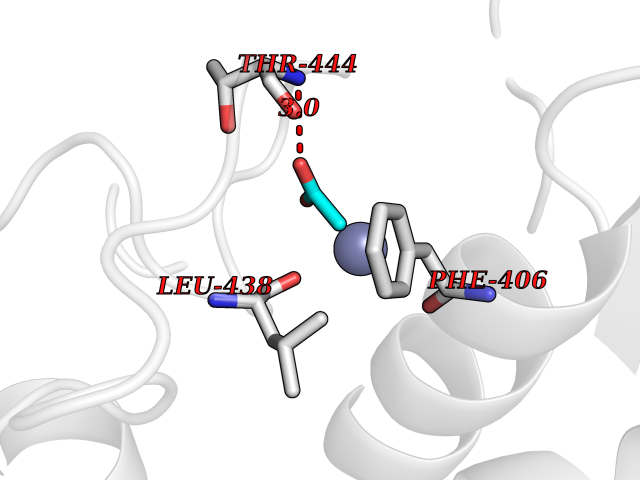 Pymol Map 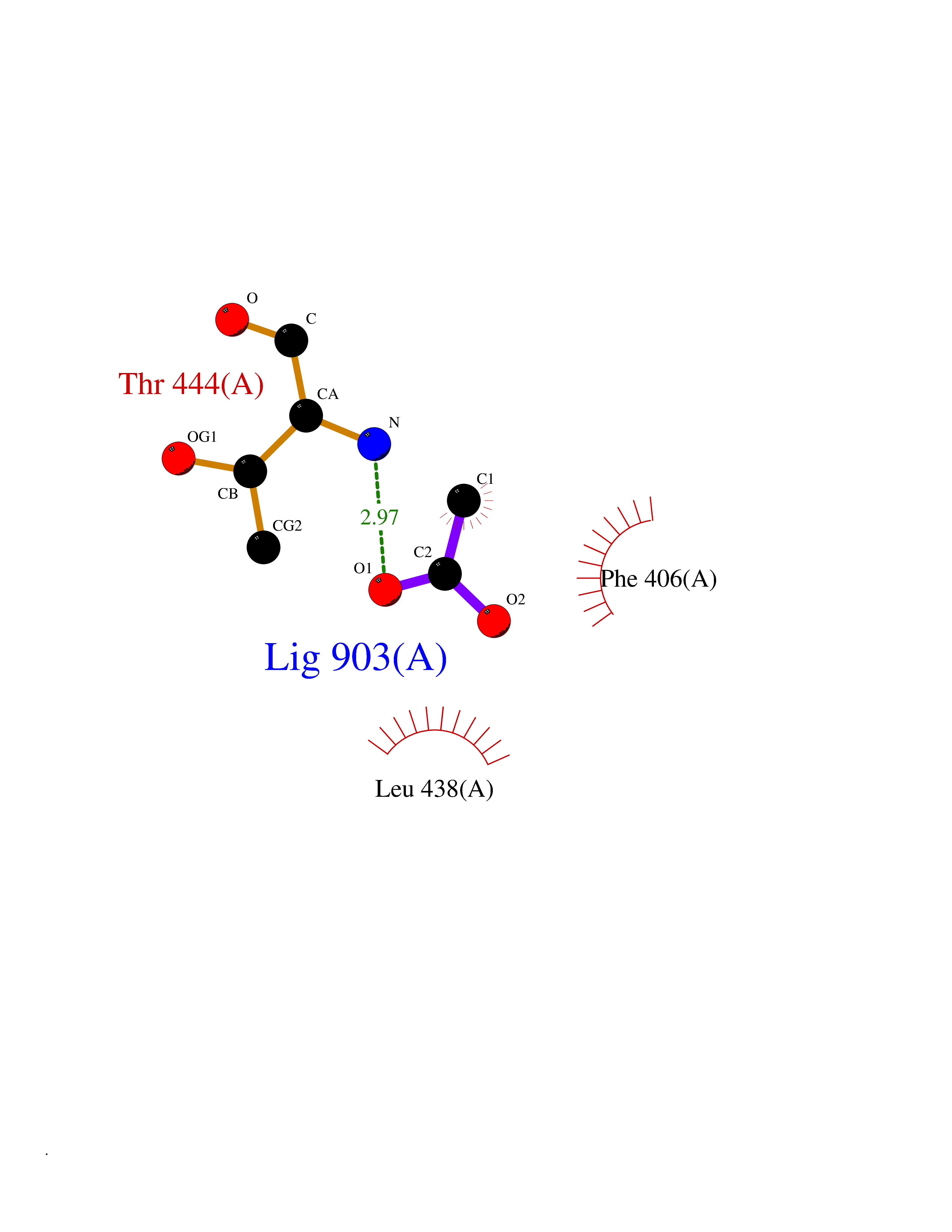 Ligplot Map 3D Binding mode Sequence SRARQVELLLVADASMARKYGRGLQHYLLTLASIANRLYSHASIENHIRLAVVKVVVLGDKDKSLEVSKNAATTLKNFCKWQHQHNQLGDDHEEHYDAAILFTREDLCGHHSCDTLGMADVGTICSPERSCAVIEDDGLHAAFTVAHEIGHLLGLSHDDSKFCEETFGSTEDKRLMSSILTSIDASKPWSKCTSATITEFLDDGHGNCLLDLPRKQI Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Histone deacetylase 3 (HDAC3) | 4A69 | 4.38 | |
Target general information Gen name HDAC3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SMAP45; RPD32; RPD3-2; HD3 Protein family Histone deacetylase family, HD type 1 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Participates in the BCL6 transcriptional repressor activity by deacetylating the H3 'Lys-27' (H3K27) on enhancer elements, antagonizing EP300 acetyltransferase activity and repressing proximal gene expression. Probably participates in the regulation of transcription through its binding to the zinc-finger transcription factor YY1; increases YY1 repression activity. Required to repress transcription of the POU1F1 transcription factor. Acts as a molecular chaperone for shuttling phosphorylated NR2C1 to PML bodies for sumoylation. Contributes, together with XBP1 isoform 1, to the activation of NFE2L2-mediated HMOX1 transcription factor gene expression in a PI(3)K/mTORC2/Akt-dependent signaling pathway leading to endothelial cell (EC) survival under disturbed flow/oxidative stress. Regulates both the transcriptional activation and repression phases of the circadian clock in a deacetylase activity-independent manner. During the activation phase, promotes the accumulation of ubiquitinated ARNTL/BMAL1 at the E-boxes and during the repression phase, blocks FBXL3-mediated CRY1/2 ubiquitination and promotes the interaction of CRY1 and ARNTL/BMAL1. The NCOR1-HDAC3 complex regulates the circadian expression of the core clock gene ARTNL/BMAL1 and the genes involved in lipid metabolism in the liver. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4), and some other non-histone substrates. Related diseases Cocoon syndrome (COCOS) [MIM:613630]: A lethal syndrome characterized by multiple fetal malformations including defective face and seemingly absent limbs, which are bound to the trunk and encased under the skin. {ECO:0000269|PubMed:20961246}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Bartsocas-Papas syndrome 2 (BPS2) [MIM:619339]: An autosomal recessive, severe form of popliteal pterygium syndrome. Popliteal pterygia syndromes have considerable variability in severity and in the associated phenotypic features but they are all characterized by cutaneous webbing across one or more major joints, cleft lip and/or palate, syndactyly, and genital malformations. {ECO:0000269|PubMed:25691407}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12565; DB05015; DB01262; DB11841; DB12645; DB11830; DB06603; DB06819; DB05223; DB03766; DB12847; DB06176; DB00313; DB02546 Interacts with O43823; Q9ULX6; Q9UKG1; P24385; P23771; Q13227; Q13547; O60341; Q969R5; P43356; Q9UIS9; P01106; O75376; Q9Y618; Q15466; P48552; P00558; P00558-1; P60510; Q15022; O09106; P08393; Q60974; Q8CBD1; P12504 EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; Chromatin regulator; Chromosome; Cytoplasm; Host-virus interaction; Hydrolase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50376.9 Length 437 Aromaticity 0.14 Instability index 29.95 Isoelectric point 6.1 Charge (pH=7) -6.84 2D Binding mode Binding energy (Kcal/mol) -5.98 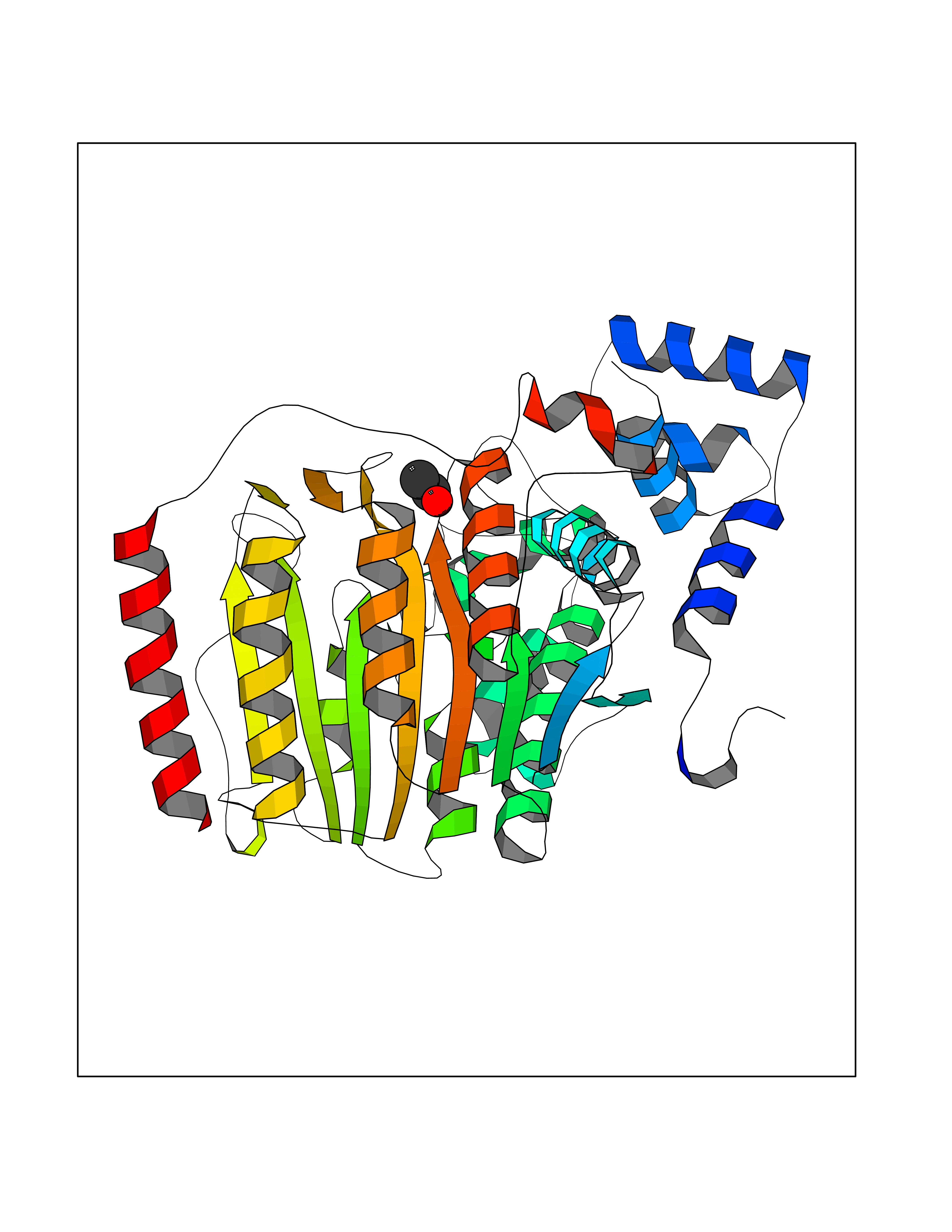 Molscript Map 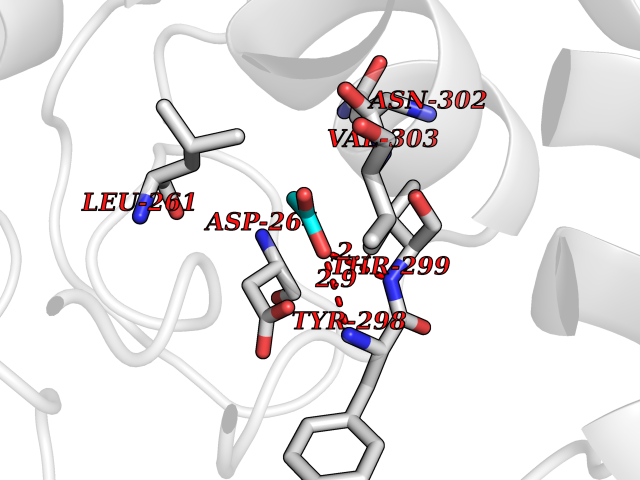 Pymol Map 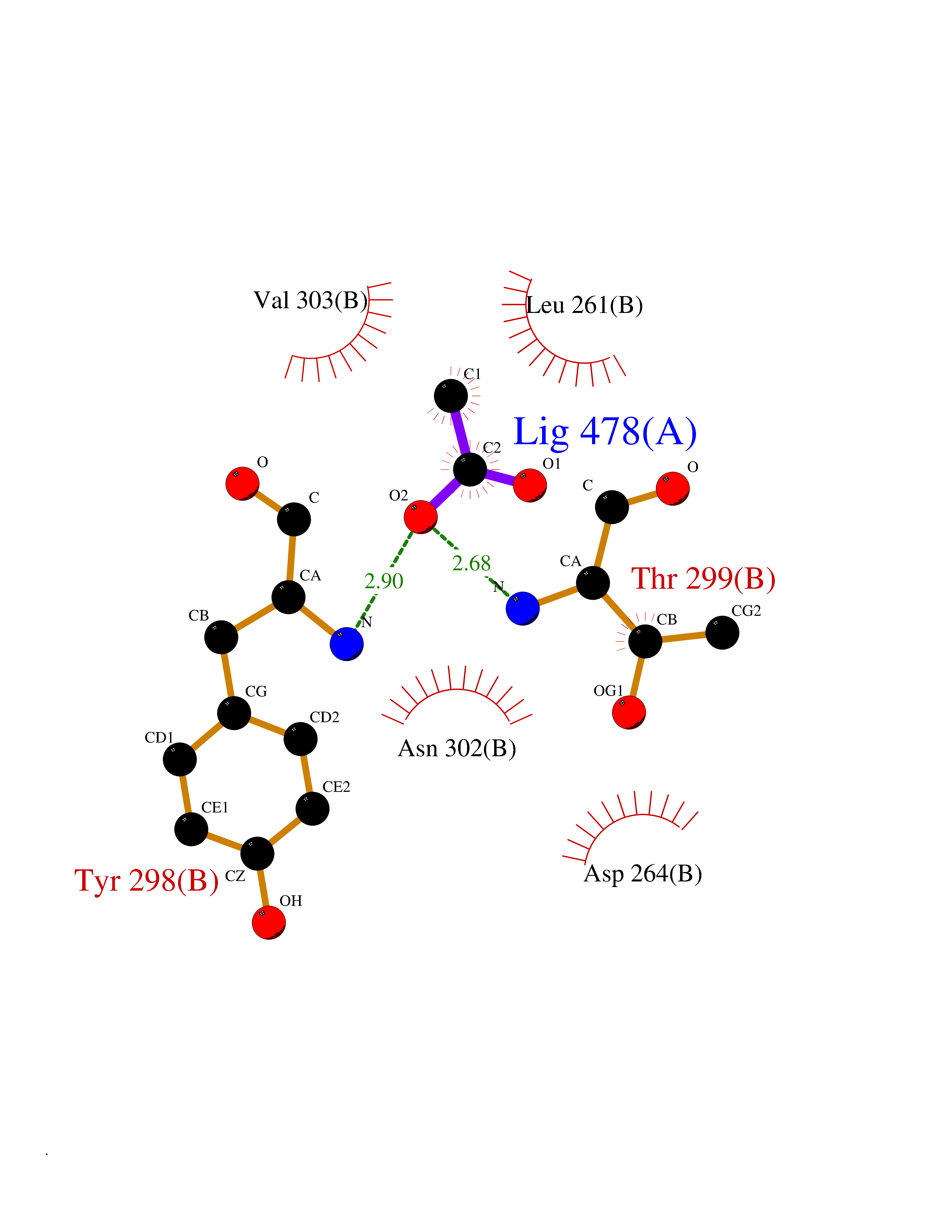 Ligplot Map 3D Binding mode Sequence KFINMNGLMADPMKVYKDRQVMNMWSEQEKETFREKFMQHPKNFGLIASFLERKTVAECVLYYYLTKKAKTVAYFYDPDVGNFHYGAGHPMKPHRLALTHSLVLHYGLYKKMIVFKPYQASQHDMCRFHSEDYIDFLQRVSPTNMQGFTKSLNAFNVGDDCPVFPGLFEFCSRYTGASLQGATQLNNKICDIAINWAGGLHHAKKFEASGFCYVNDIVIGILELLKYHPRVLYIDIDIHHGDGVQEAFYLTDRVMTVSFHKYGNYFFPGTGDMYEVGAESGRYYCLNVPLRDGIDDQSYKHLFQPVINQVVDFYQPTCIVLQCGADSLGCDRLGCFNLSIRGHGECVEYVKSFNIPLLVLGGGGYTVRNVARCWTYETSLLVEEAISEELPYSEYFEYFAPDFTLHPDVSTRIENQNSRQYLDQIRQTIFENLKMLN Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Dopamine beta-hydroxylase | 4ZEL | 4.38 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -5.98  Molscript Map 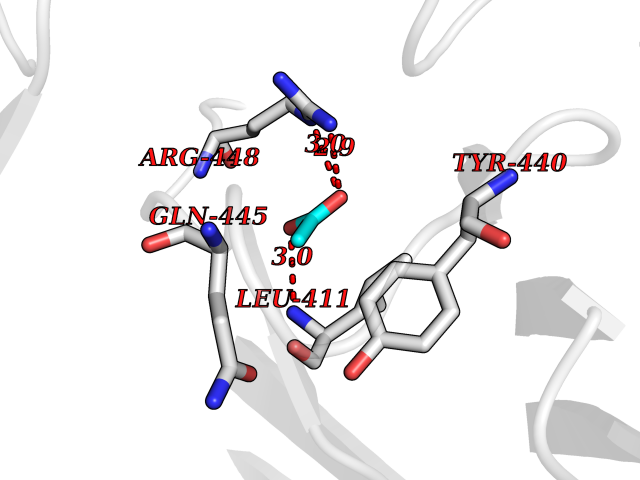 Pymol Map 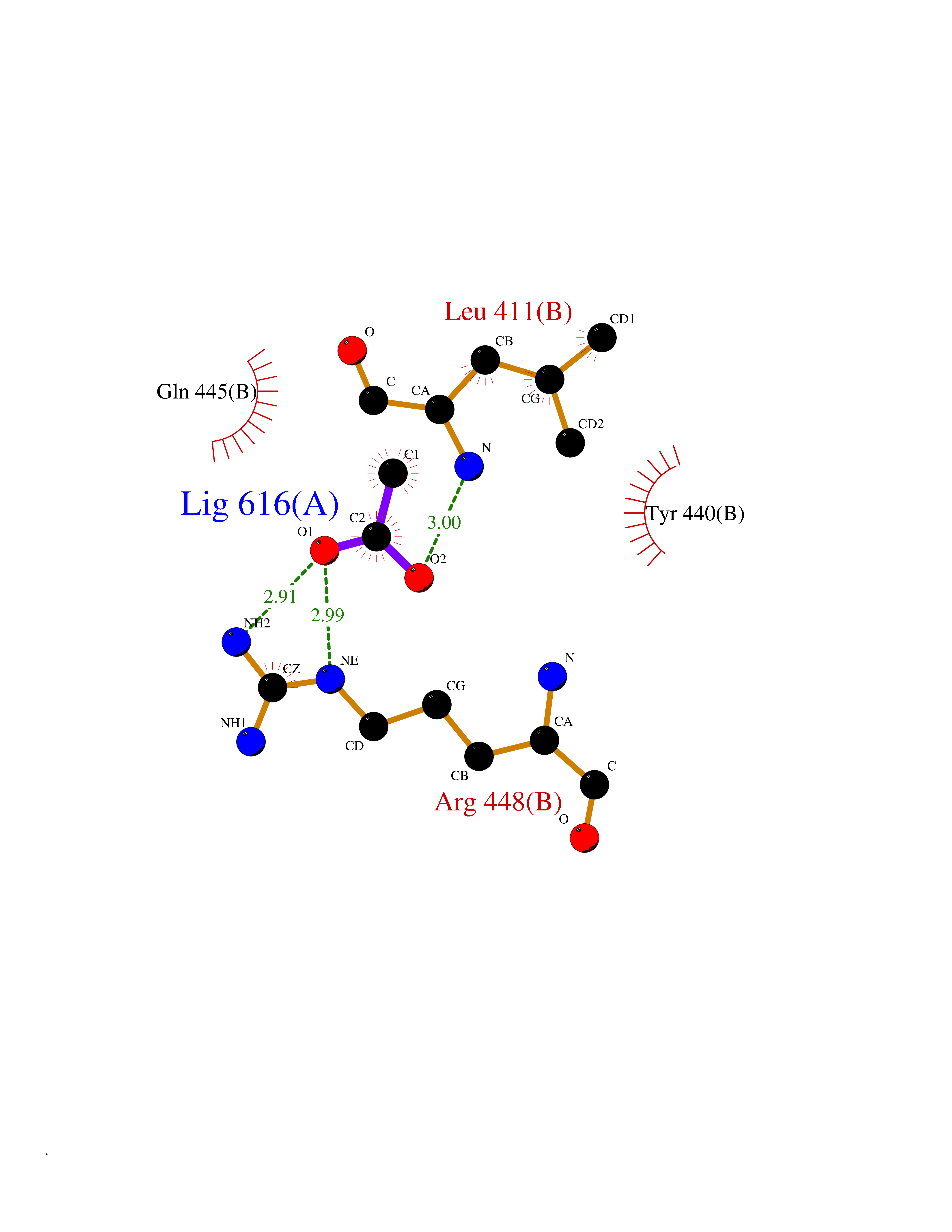 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | p-hydroxybenzoate hydroxylase | 1K0I | 4.38 | |
Target general information Gen name pobA Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID NA Synonyms PA0247 Protein family Aromatic-ring hydroxylase family Biochemical class Hydrolase Function 4-hydroxybenzoate 3-monooxygenase activity.FAD binding.Flavin adenine dinucleotide binding. Related diseases Immunodeficiency, common variable, 12, with autoimmunity (CVID12) [MIM:616576]: A primary immunodeficiency characterized by hypogammaglobulinemia and recurrent bacterial infections. About half of patients develop autoimmune features, including cytopenia, as well as generalized inflammation and lymphoproliferation manifest as lymphadenopathy or hepatosplenomegaly. {ECO:0000269|PubMed:26279205}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02839; DB04242; DB03482; DB02362; DB03147 Interacts with NA EC number 1.14.13.2 Uniprot keywords 3D-structure; Aromatic hydrocarbons catabolism; FAD; Flavoprotein; Monooxygenase; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 44295 Length 394 Aromaticity 0.09 Instability index 37.66 Isoelectric point 6.2 Charge (pH=7) -3.67 2D Binding mode Binding energy (Kcal/mol) -5.98  Molscript Map 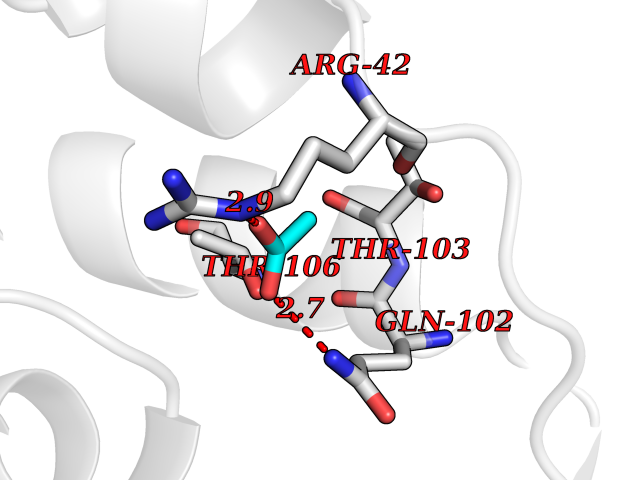 Pymol Map 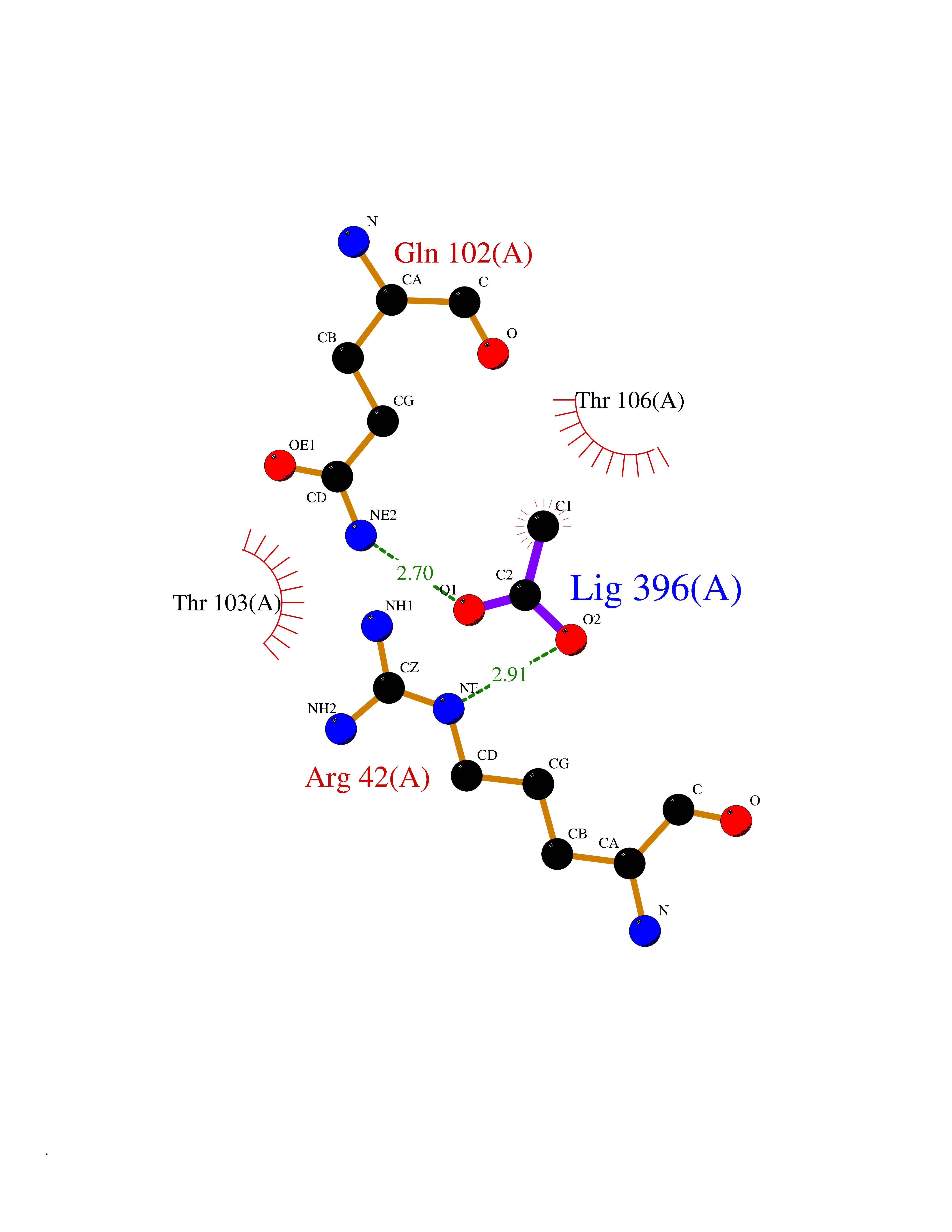 Ligplot Map 3D Binding mode Sequence MKTQVAIIGAGPSGLLLGQLLHKAGIDNVILERQTPDYVLGRIRAGVLEQGMVDLLREAGVDRRMARDGLVHEGVEIAFAGQRRRIDLKRLSGGKTVTVYGQTEVTRDLMEAREACGATTVYQAAEVRLHDLQGERPYVTFERDGERLRLDCDYIAGCDGFHGISRQSIPAERLKVFERVYPFGWLGLLADTPPVSHELIYANHPRGFALCSQRSATRSQYYVQVPLSEKVEDWSDERFWTELKARLPSEVAEKLVTGPSLEKSIAPLRSFVVEPMQHGRLFLAGDAAHIVPPTGAKGLNLAASDVSTLYRLLLKAYREGRGELLERYSAICLRRIWKAERFSWWMTSVLHRFPDTDAFSQRIQQTELEYYLGSEAGLATIAENYVGLPYEEIE Hydrogen bonds contact Hydrophobic contact | ||||