Job Results:
Ligand
Structure
Job ID
a9ea1a8efdae070d2ad25867c2a2c1f3
Job name
NA
Time
2026-02-24 16:12:06
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Cerebron E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase) | 4CI1 | 5.63 | |
Target general information Gen name CUL4A/CUL4B-DDB1-CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NA Protein family Cullin family Biochemical class NA Function NA Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; Q86VP6; Q16531; Q92466; P08238; O94888; P55072 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; DNA damage; DNA repair; Host-virus interaction; Isopeptide bond; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID B Molecular weight (Da) 42669.7 Length 368 Aromaticity 0.1 Instability index 44.94 Isoelectric point 8.72 Charge (pH=7) 6.58 2D Binding mode Binding energy (Kcal/mol) -7.68  Molscript Map 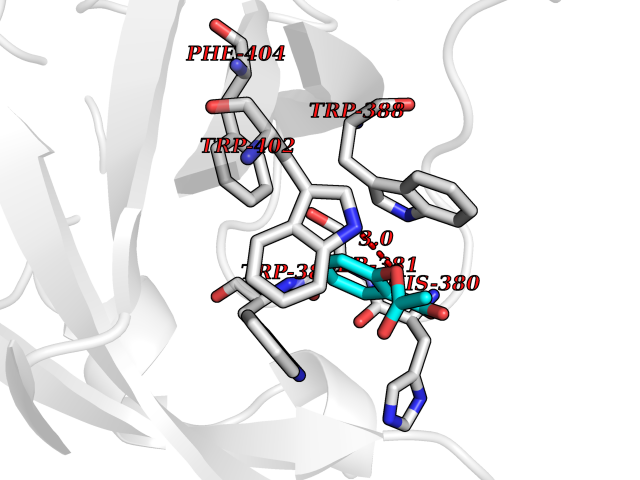 Pymol Map 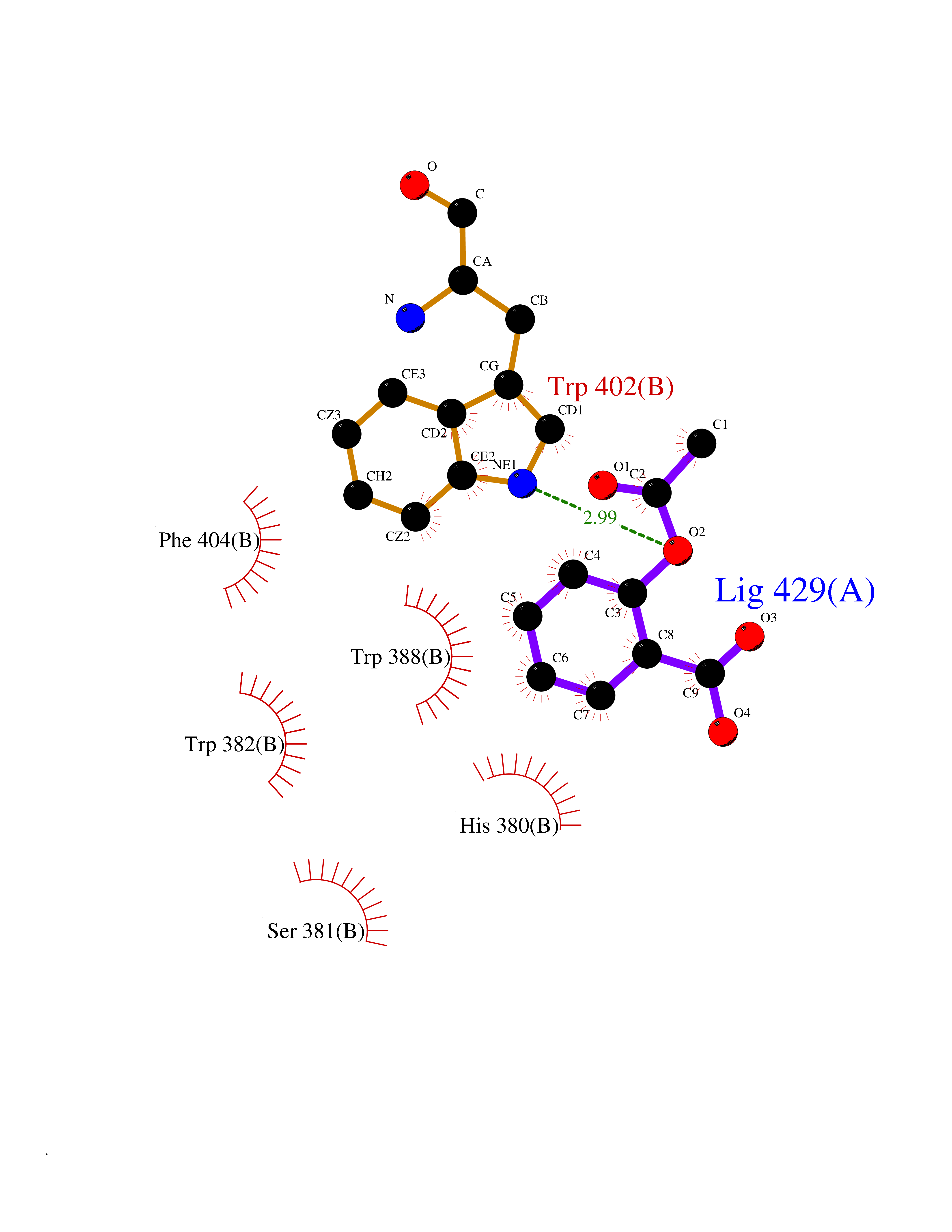 Ligplot Map 3D Binding mode Sequence MINFDTSLPTSHMYLGSDMEEFHGRTLHDDDSCQVIPVLPHVMVMLIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVREREAHFGTTAEIYAYREEQEYGIETVKVKAIGRQRFKVLEIRTQSDGIQQAKVQILPERVLPSTMSAVQLQSLSRRHIRAFRQWWQKYQKRKFHCASLTSWPPWLYSLYDAETLMERVKRQLHEWDENLKDESLPTNPIDFSYRVAACLPIDDALRIQLLKIGSAIQRLRELDIMNKTSLCCKQCQDTEITTKNEIFSLSLCGPMAAYVNPHGYIHETLTVYKACNLNLSGRPSTEHSWFPGYAWTIAQCRICGNHMGWKFTATKKDMSPQKFWGLTRSALLPR Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Folate receptor alpha (FOLR1) | 4LRH | 5.63 | |
Target general information Gen name FOLR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ovarian tumorassociated antigen MOv18; KB cells FBP; Folate receptor, adult; Folate receptor 1; FRalpha; FOLR1; Adult folatebinding protein Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pHafter receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Required for normal embryonic development and normal cell proliferation. Related diseases Neurodegeneration due to cerebral folate transport deficiency (NCFTD) [MIM:613068]: An autosomal recessive neurodegenerative disorder resulting from brain-specific folate deficiency early in life. Onset is apparent in late infancy with severe developmental regression, movement disturbances, epilepsy and leukodystrophy. {ECO:0000269|PubMed:19732866}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05595; DB00158; DB00563; DB12489; DB15413; DB05168 Interacts with Q8N357 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Endosome; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Neurodegeneration; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 24216 Length 207 Aromaticity 0.13 Instability index 49.36 Isoelectric point 8.14 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -7.67 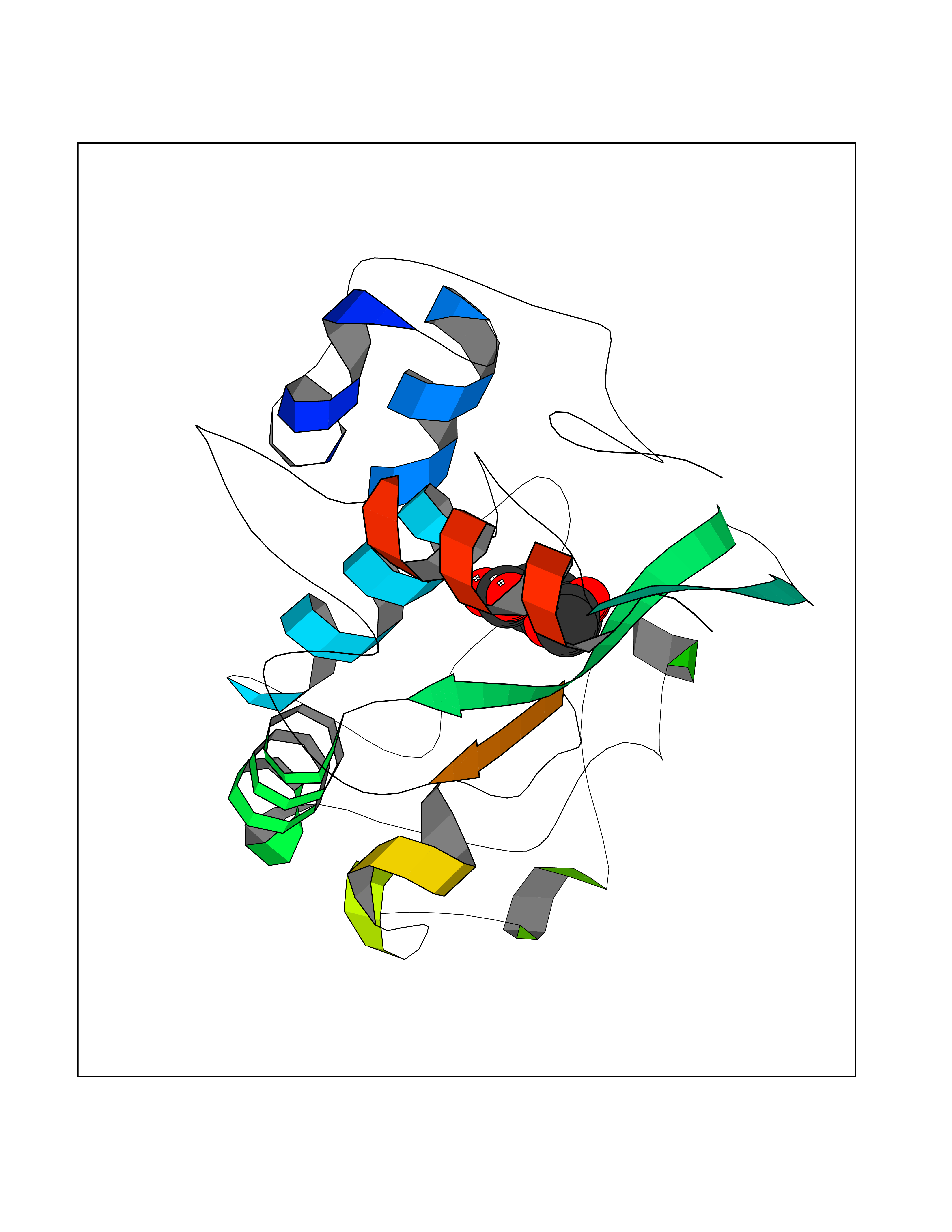 Molscript Map 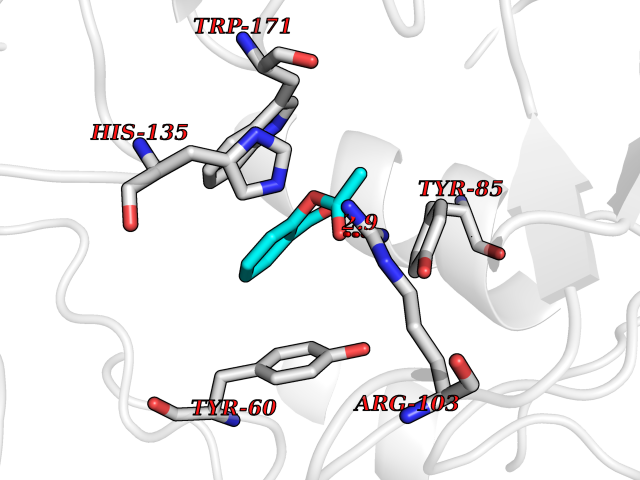 Pymol Map 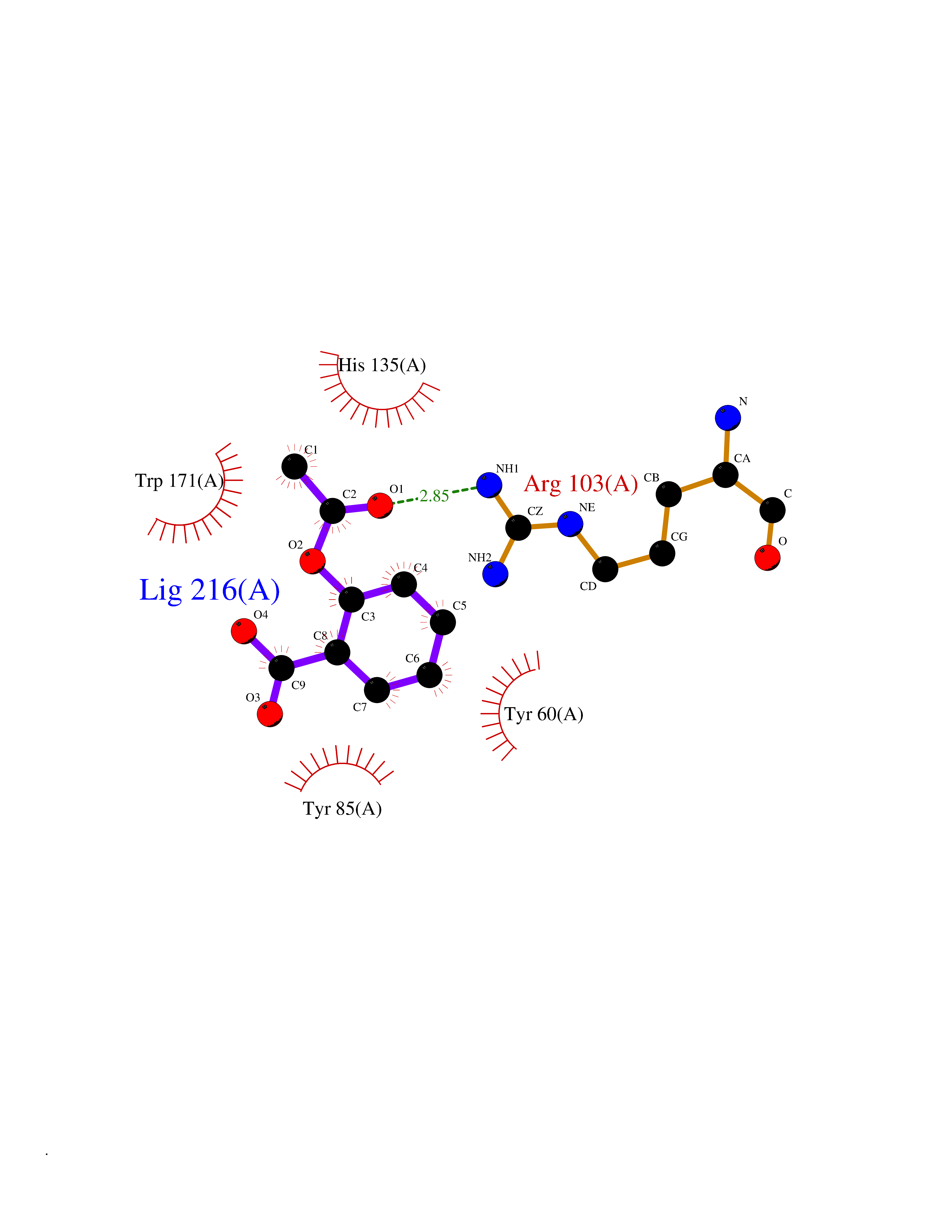 Ligplot Map 3D Binding mode Sequence RTELLNVCMNAKHHKEKPGPEDKLHEQCRPWRKNACCSTNTSQEAHKDVSYLYRFNWNHCGEMAPACKRHFIQDTCLYECSPNLGPWIQQVDQSWRKERVLNVPLCKEDCEQWWEDCRTSYTCKSNWHKGWNWTSGFNKCAVGAACQPFHFYFPTPTVLCNEIWTHSYKVSNYSRGSGRCIQMWFDPAQGNPNEEVARFYAAAMSGT Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | p-hydroxybenzoate hydroxylase | 1PBE | 5.62 | |
Target general information Gen name pobA Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family Aromatic-ring hydroxylase family Biochemical class Oxidoreductase Function 4-hydroxybenzoate 3-monooxygenase activity.FAD binding.Flavin adenine dinucleotide binding. Related diseases Mitochondrial complex IV deficiency (MT-C4D) [MIM:220110]: A disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations, ranging from isolated myopathy to severe multisystem disease affecting several tissues and organs. Features include hypertrophic cardiomyopathy, hepatomegaly and liver dysfunction, hypotonia, muscle weakness, exercise intolerance, developmental delay, delayed motor development and intellectual disability. Some affected individuals manifest a fatal hypertrophic cardiomyopathy resulting in neonatal death. A subset of patients manifest Leigh syndrome. {ECO:0000269|PubMed:10486321}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02839; DB04242; DB02059; DB02362; DB03147 Interacts with NA EC number 1.14.13.2 Uniprot keywords 3D-structure; Aromatic hydrocarbons catabolism; Direct protein sequencing; FAD; Flavoprotein; Monooxygenase; NADP; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 43949.7 Length 391 Aromaticity 0.09 Instability index 34.92 Isoelectric point 6.78 Charge (pH=7) -0.68 2D Binding mode Binding energy (Kcal/mol) -7.67 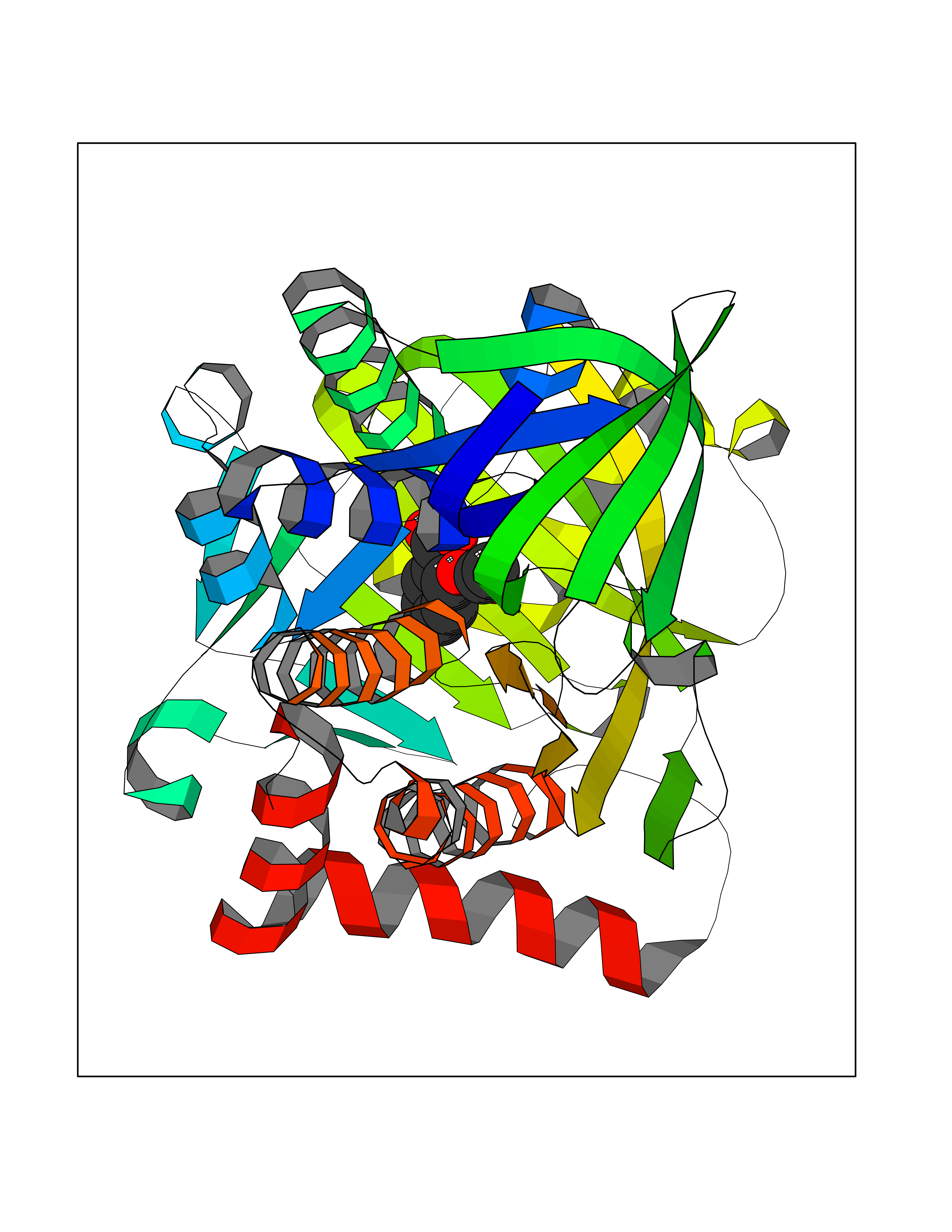 Molscript Map 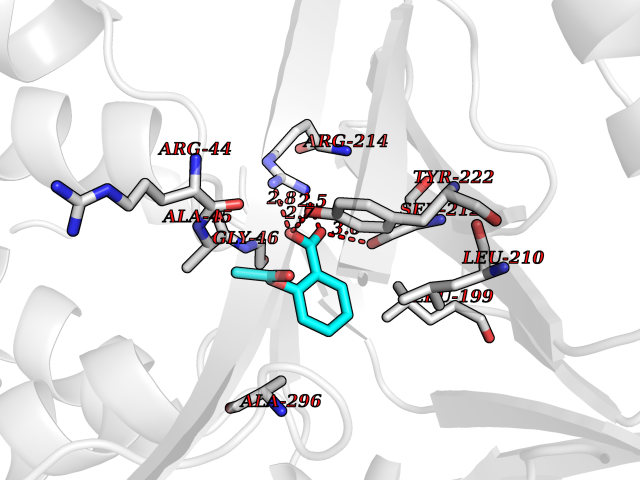 Pymol Map 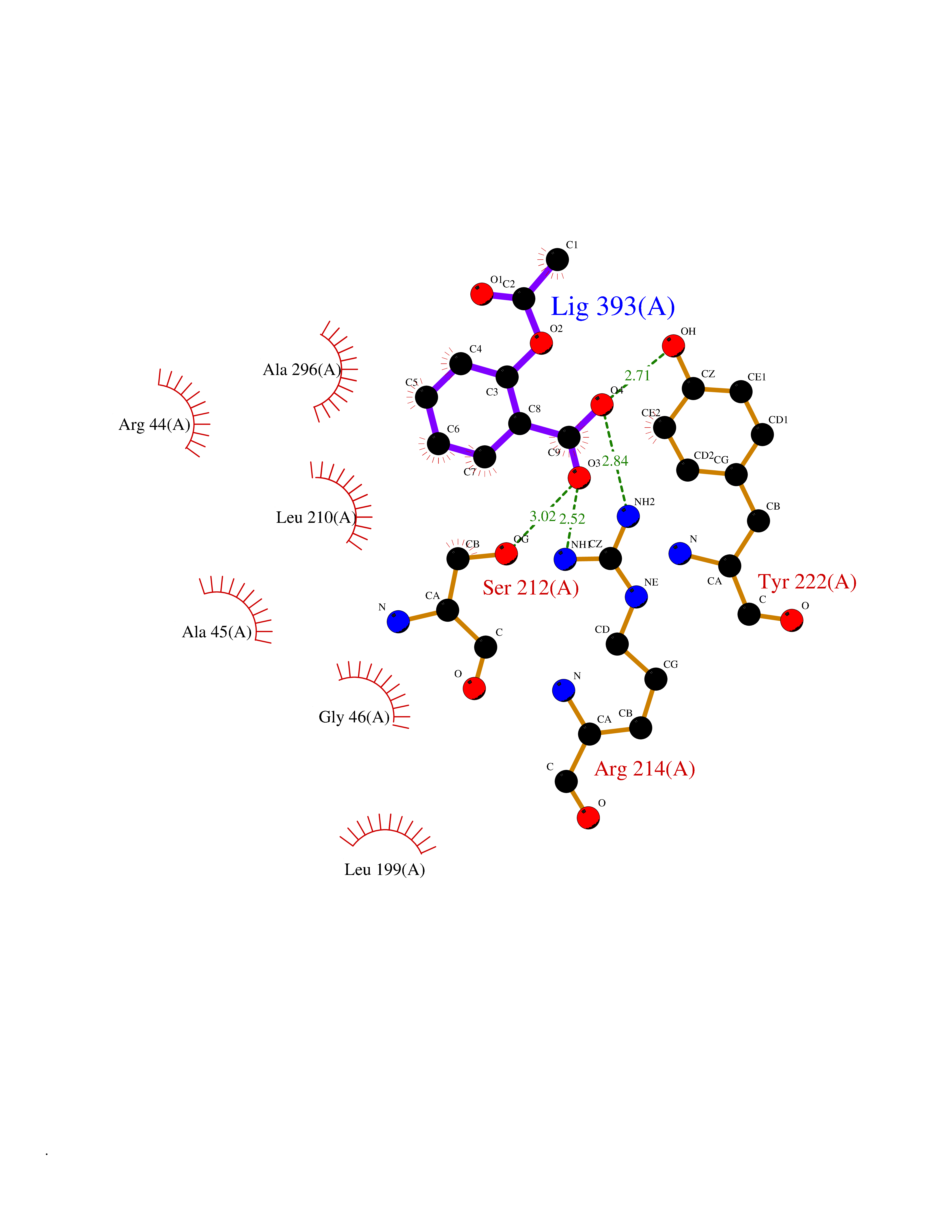 Ligplot Map 3D Binding mode Sequence MKTQVAIIGAGPSGLLLGQLLHKAGIDNVILERQTPDYVLGRIRAGVLEQGMVDLLREAGVDRRMARDGLVHEGVEIAFAGQRRRIDLKRLSGGKTVTVYGQTEVTRDLMEAREACGATTVYQAAEVRLHDLQGERPYVTFERDGERLRLDCDYIAGCDGFHGISRQSIPAERLKVFERVYPFGWLGLLADTPPVSHELIYANHPRGFALCSQRSATRSRYYVQVPLTEKVEDWSDERFWTELKARLPAEVAEKLVTGPSLEKSIAPLRSFVVEPMQHGRLFLAGDAAHIVPPTGAKGLNLAASDVSTLYRLLLKAYREGRGELLERYSAICLRRIWKAERFSWWMTSVLHRFPDTDAFSQRIQQTELEYYLGSEAGLATIAENYVGLPYE Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | NADH peroxidase | 1NHP | 5.62 | |
Target general information Gen name npr Organism Enterococcus faecalis (strain ATCC 700802 / V583) Uniprot ID TTD ID NA Synonyms EF_1211 Protein family Class-III pyridine nucleotide-disulfide oxidoreductase family Biochemical class Oxidoreductase (h2o2(a)) Function Flavin adenine dinucleotide binding.NADH peroxidase activity. Related diseases Telangiectasia, hereditary hemorrhagic, 2 (HHT2) [MIM:600376]: A multisystemic vascular dysplasia leading to dilation of permanent blood vessels and arteriovenous malformations of skin, mucosa, and viscera. The disease is characterized by recurrent epistaxis and gastro-intestinal hemorrhage. Visceral involvement includes arteriovenous malformations of the lung, liver, and brain. {ECO:0000269|PubMed:10694922, ECO:0000269|PubMed:10767348, ECO:0000269|PubMed:11170071, ECO:0000269|PubMed:11484689, ECO:0000269|PubMed:14684682, ECO:0000269|PubMed:15024723, ECO:0000269|PubMed:15712270, ECO:0000269|PubMed:16525724, ECO:0000269|PubMed:16752392, ECO:0000269|PubMed:20414677, ECO:0000269|PubMed:26176610, ECO:0000269|PubMed:8640225, ECO:0000269|PubMed:9245985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB03382 Interacts with NA EC number 1.11.1.1 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; NAD; Oxidation; Oxidoreductase; Peroxidase; Redox-active center; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 49518.8 Length 447 Aromaticity 0.09 Instability index 27.53 Isoelectric point 4.83 Charge (pH=7) -19.06 2D Binding mode Binding energy (Kcal/mol) -7.67  Molscript Map 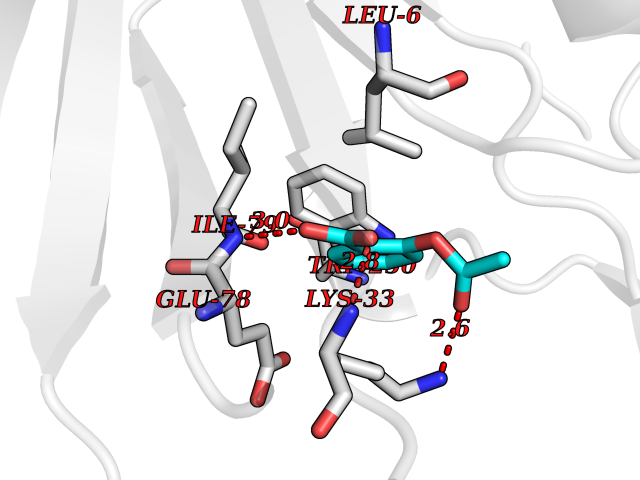 Pymol Map 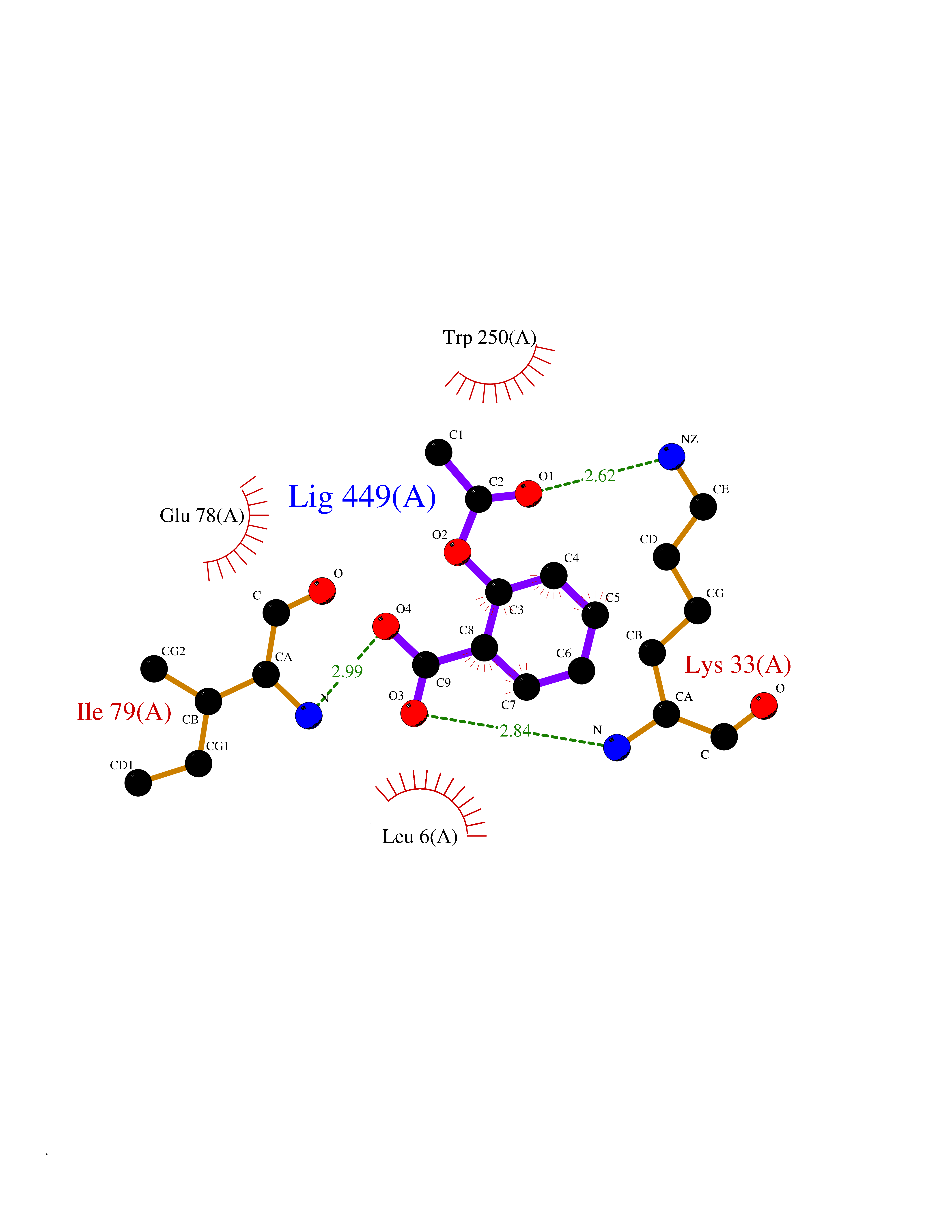 Ligplot Map 3D Binding mode Sequence MKVIVLGSSHGGYEAVEELLNLHPDAEIQWYEKGDFISFLSAGMQLYLEGKVKDVNSVRYMTGEKMESRGVNVFSNTEITAIQPKEHQVTVKDLVSGEERVENYDKLIISPGAVPFELDIPGKDLDNIYLMRGRQWAIKLKQKTVDPEVNNVVVIGSGYIGIEAAEAFAKAGKKVTVIDILDRPLGVYLDKEFTDVLTEEMEANNITIATGETVERYEGDGRVQKVVTDKNAYDADLVVVAVGVRPNTAWLKGTLELHPNGLIKTDEYMRTSEPDVFAVGDATLIKYNPADTEVNIALATNARKQGRFAVKNLEEPVKPFPGVQGSSGLAVFDYKFASTGINEVMAQKLGKETKAVTVVEDYLMDFNPDKQKAWFKLVYDPETTQILGAQLMSKADLTANINAISLAIQAKMTIEDLAYADFFFQPAFDKPWNIINTAALEAVKQER Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 5.62 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -7.67  Molscript Map 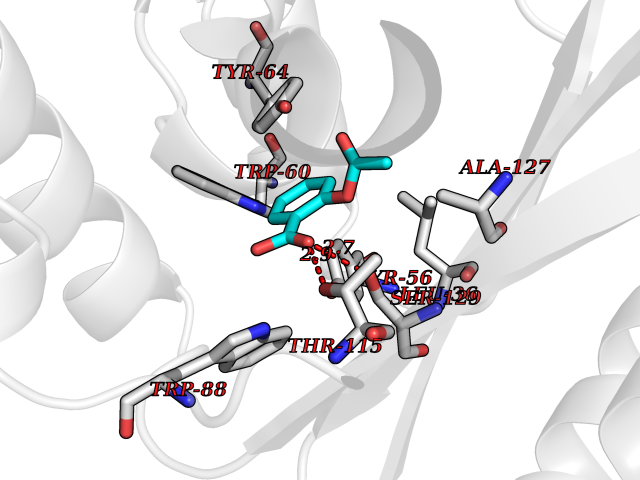 Pymol Map 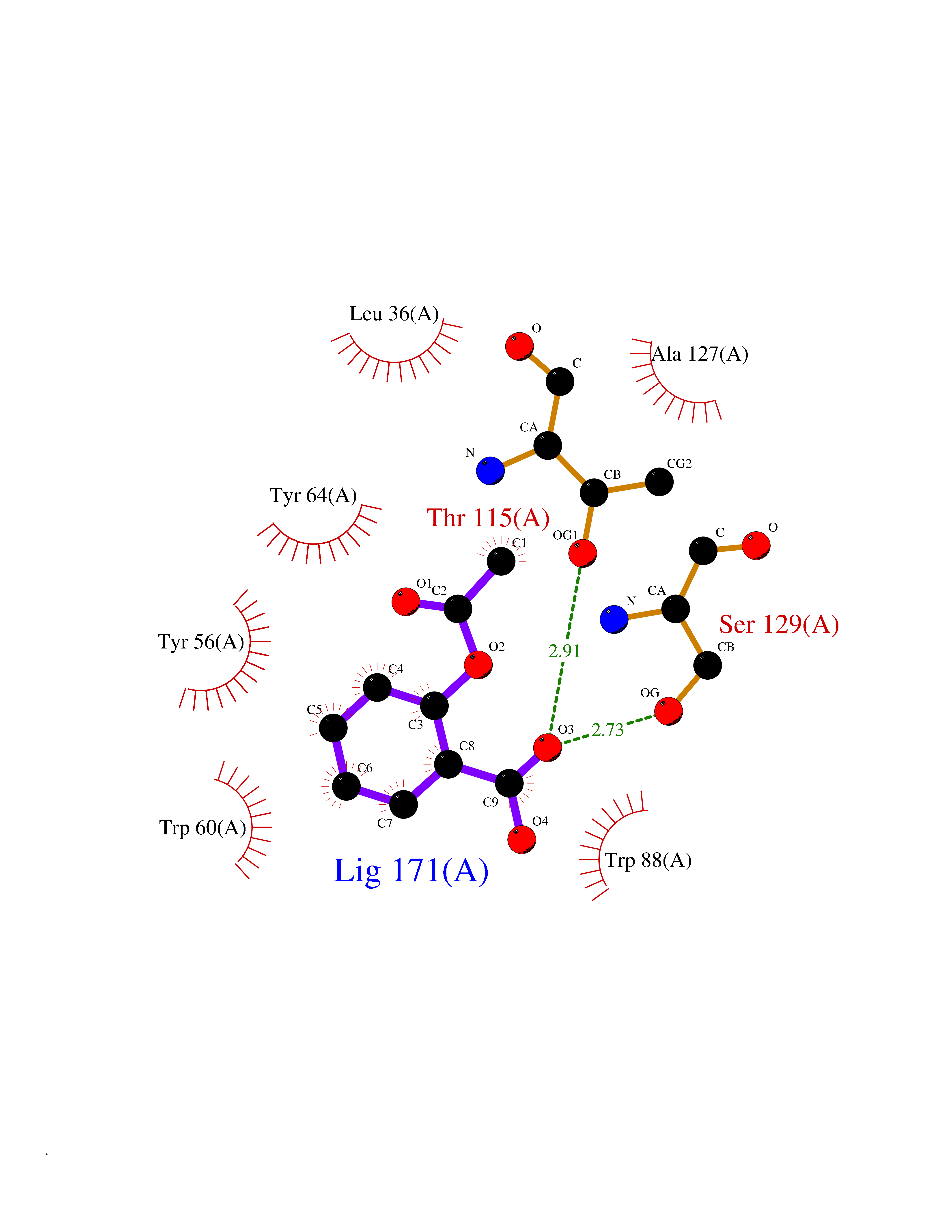 Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Bacterial DD-carboxypeptidase (Bact vanYB) | 5ZHW | 5.62 | |
Target general information Gen name Bact vanYB Organism Enterococcus faecalis (strain ATCC 700802 / V583) Uniprot ID TTD ID Synonyms vanYB; DD-peptidase; DD-carboxypeptidase; D-alanyl-D-alanine carboxypeptidase-transpeptidase Protein family Peptidase M15B family Biochemical class Peptidase Function Vancomycin-inducible, penicillin-resistant, DD- carboxypeptidase that hydrolyzes depsipeptide- and D-alanyl-D- alanine-containing peptidoglycan precursors. Insensitive to beta- lactams. Related diseases Brachyolmia 3 (BCYM3) [MIM:113500]: A form of brachyolmia, a clinically and genetically heterogeneous skeletal dysplasia primarily affecting the spine and characterized by a short trunk, short stature, and platyspondyly. BCYM3 is an autosomal dominant form with severe scoliosis with or without kyphosis, and flattened irregular cervical vertebrae. {ECO:0000269|PubMed:18587396}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondylometaphyseal dysplasia Kozlowski type (SMDK) [MIM:184252]: A form of spondylometaphyseal dysplasia, a group of short stature disorders distinguished by abnormalities in the vertebrae and the metaphyses of the tubular bones. It is characterized by postnatal dwarfism, significant scoliosis and mild metaphyseal abnormalities in the pelvis. The vertebrae exhibit platyspondyly and overfaced pedicles. {ECO:0000269|PubMed:19232556, ECO:0000269|PubMed:20577006, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metatropic dysplasia (MTD) [MIM:156530]: A severe spondyloepimetaphyseal dysplasia characterized by short limbs with limitation and enlargement of joints and usually severe kyphoscoliosis. Radiologic features include severe platyspondyly, severe metaphyseal enlargement and shortening of long bones. {ECO:0000269|PubMed:19232556, ECO:0000269|PubMed:20425821, ECO:0000269|PubMed:20577006, ECO:0000269|PubMed:22702953, ECO:0000269|PubMed:26249260, ECO:0000269|Ref.6}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 8 (HMND8) [MIM:600175]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:20037588, ECO:0000269|PubMed:22526352, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2C (CMT2C) [MIM:606071]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:20037586, ECO:0000269|PubMed:20037587, ECO:0000269|PubMed:20037588, ECO:0000269|PubMed:21115951, ECO:0000269|PubMed:21288981, ECO:0000269|PubMed:22702953, ECO:0000269|PubMed:25256292}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Scapuloperoneal spinal muscular atrophy (SPSMA) [MIM:181405]: A clinically variable neuromuscular disorder characterized by neurogenic scapuloperoneal amyotrophy, laryngeal palsy, congenital absence of muscles, progressive scapuloperoneal atrophy and progressive distal weakness and amyotrophy. {ECO:0000269|PubMed:20037587, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepiphyseal dysplasia Maroteaux type (SEDM) [MIM:184095]: A clinically variable spondyloepiphyseal dysplasia with manifestations limited to the musculoskeletal system. Clinical features include short stature, brachydactyly, platyspondyly, short and stubby hands and feet, epiphyseal hypoplasia of the large joints, and iliac hypoplasia. Intelligence is normal. {ECO:0000269|PubMed:20503319, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parastremmatic dwarfism (PSTD) [MIM:168400]: A bone dysplasia characterized by severe dwarfism, kyphoscoliosis, distortion and bowing of the extremities, and contractures of the large joints. Radiographically, the disease is characterized by a combination of decreased bone density, bowing of the long bones, platyspondyly and striking irregularities of endochondral ossification with areas of calcific stippling and streaking in radiolucent epiphyses, metaphyses and apophyses. {ECO:0000269|PubMed:20503319}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Digital arthropathy-brachydactyly, familial (FDAB) [MIM:606835]: A disorder characterized by irregularities in the proximal articular surfaces of the distal interphalangeal joints of the hand. Individuals appear normal at birth, with no clinical or radiographic evidence of a developmental skeletal dysplasia. The earliest changes appear during the first decade of life. By adulthood, all interphalangeal, metacarpophalangeal, and metatarsophalangeal joints are affected by a deforming, painful osteoarthritis. The remainder of the skeleton is clinically and radiographically unaffected. {ECO:0000269|PubMed:21964574}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Avascular necrosis of the femoral head, primary 2 (ANFH2) [MIM:617383]: A disease characterized by mechanical failure of the subchondral bone, and degeneration of the hip joint. It usually leads to destruction of the hip joint in the third to fifth decade of life. The clinical manifestations, such as pain on exertion, a limping gait, and a discrepancy in leg length, cause considerable disability. {ECO:0000269|PubMed:27330106}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.16.4 Uniprot keywords 3D-structure; Antibiotic resistance; Carboxypeptidase; Cell membrane; Cell shape; Cell wall biogenesis/degradation; Hydrolase; Membrane; Metal-binding; Peptidoglycan synthesis; Protease; Reference proteome; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 20547.5 Length 181 Aromaticity 0.11 Instability index 33.61 Isoelectric point 4.81 Charge (pH=7) -10.78 2D Binding mode Binding energy (Kcal/mol) -7.66  Molscript Map 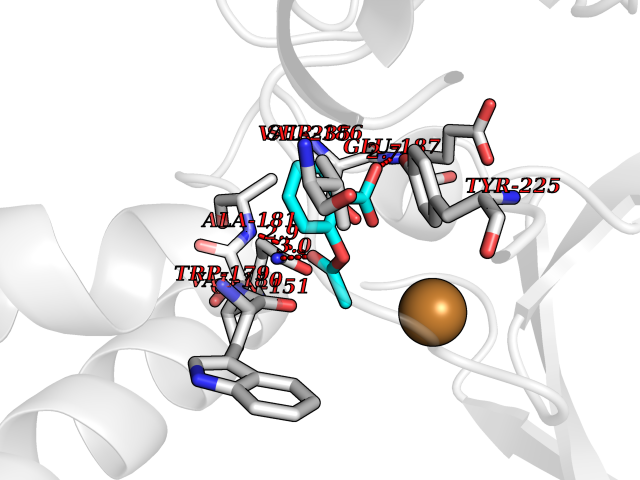 Pymol Map 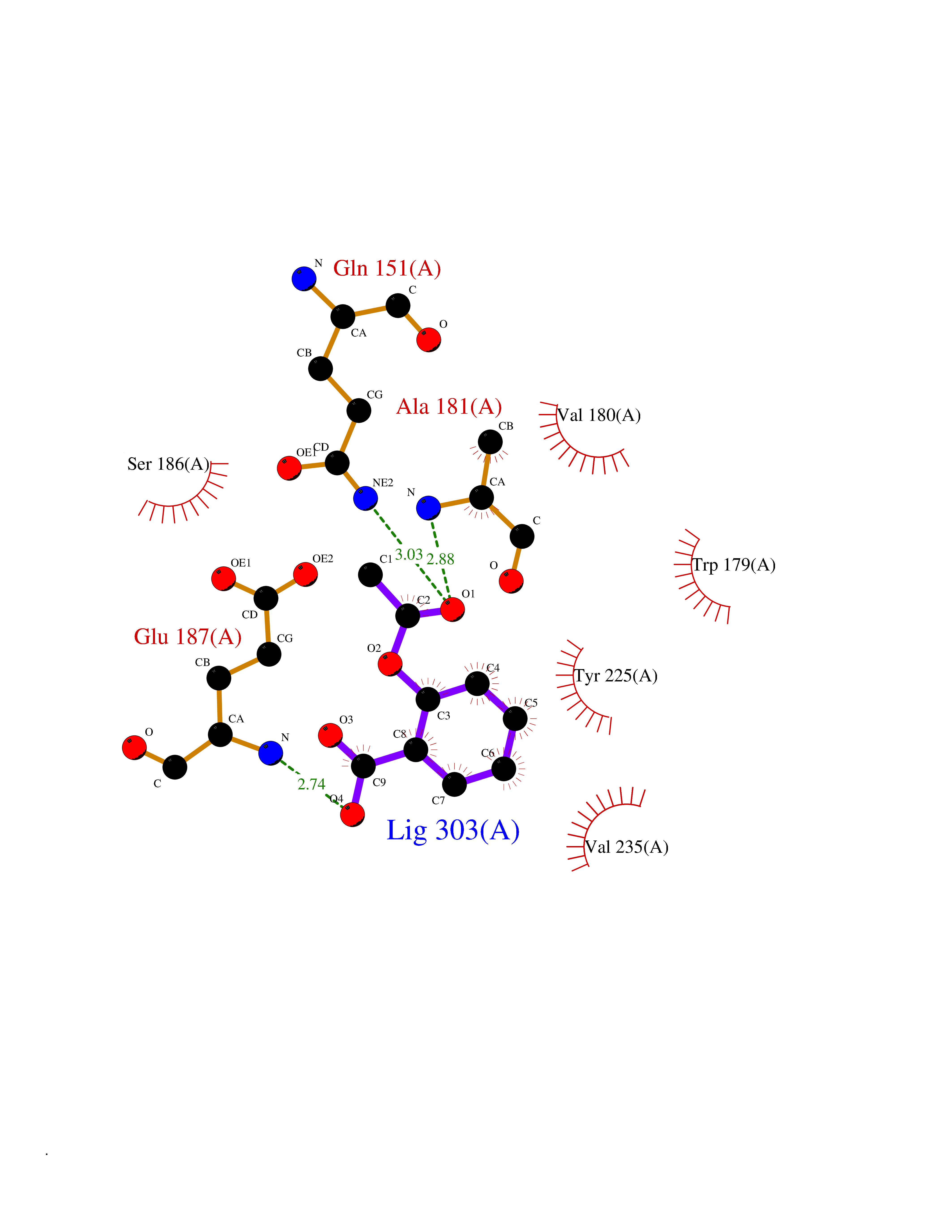 Ligplot Map 3D Binding mode Sequence EWSLILVNRQNPIPAQYDVELEQLSNGERIDIRISPYLQDLFDAARADGVYPIVASGYRTTEKQQEIXDEKVAEYKAKGYTSAQAKAEAETWVAVPGTSEHQLGLAVDINADGIHSTGNEVYRWLDENSYRFGFIRRYPPDKTEITGVSNEPWHYRYVGIEAATKIYHQGLCLEEYLNTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Zinc finger protein Helios (IKZF2) | 7LPS | 5.62 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -7.66  Molscript Map 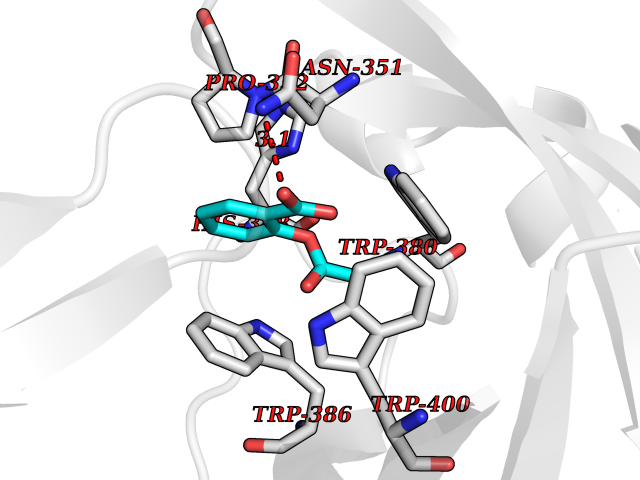 Pymol Map 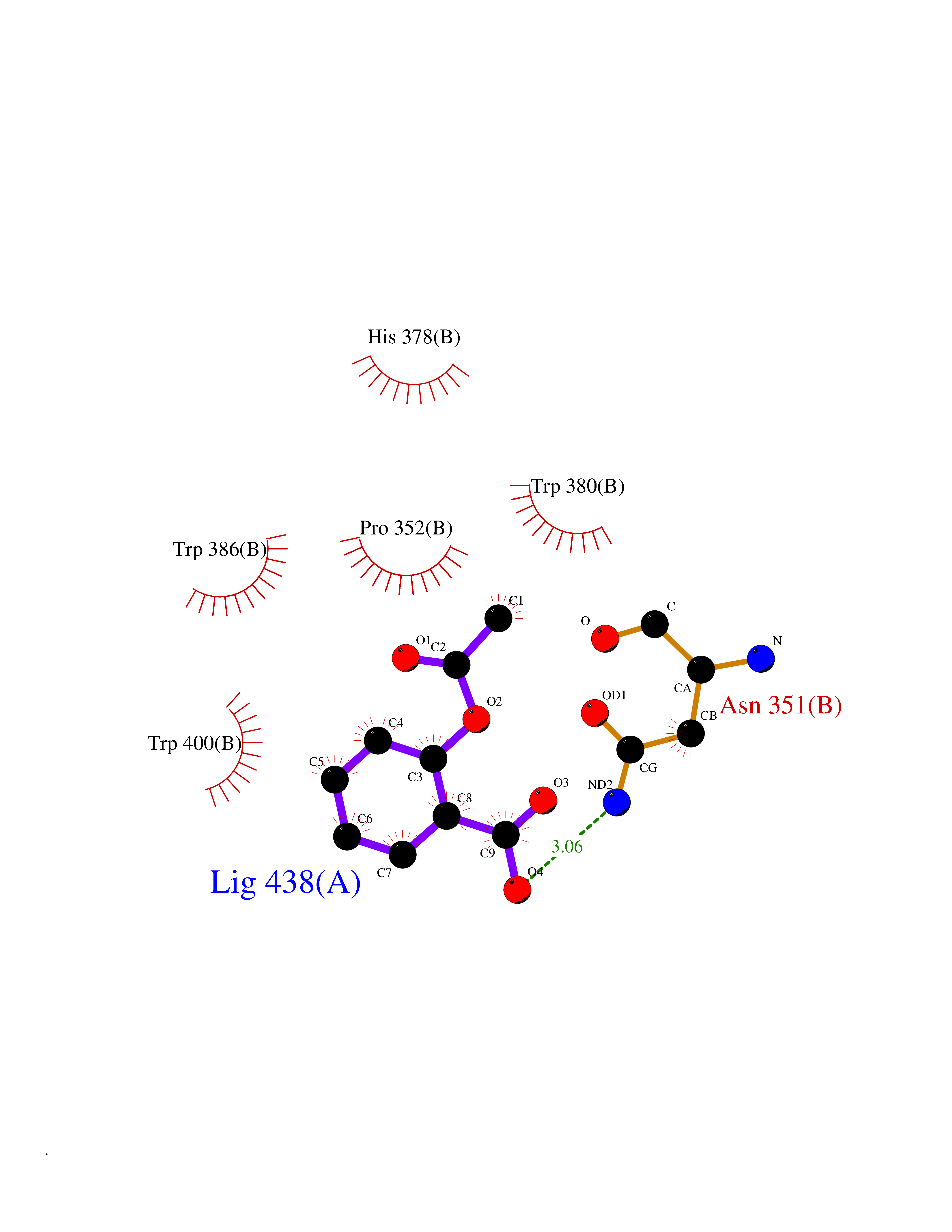 Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Glucose-dependent insulinotropic receptor (GPR119) | 7XZ6 | 5.62 | |
Target general information Gen name GPR119 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GPR119; G-protein coupled receptor 119 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the endogenous fatty-acid ethanolamide oleoylethanolamide (OEA) and lysophosphatidylcholine (LPC). Functions as a glucose-dependent insulinotropic receptor. The activity of this receptor is mediated by G proteins which activate adenylate cyclase. Seems to act through a G(s) mediated pathway. Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05166 Interacts with Q12797-6 EC number NA Uniprot keywords 3D-structure; Cell membrane; G-protein coupled receptor; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32134.1 Length 292 Aromaticity 0.12 Instability index 34.96 Isoelectric point 9.12 Charge (pH=7) 8.03 2D Binding mode Binding energy (Kcal/mol) -7.67 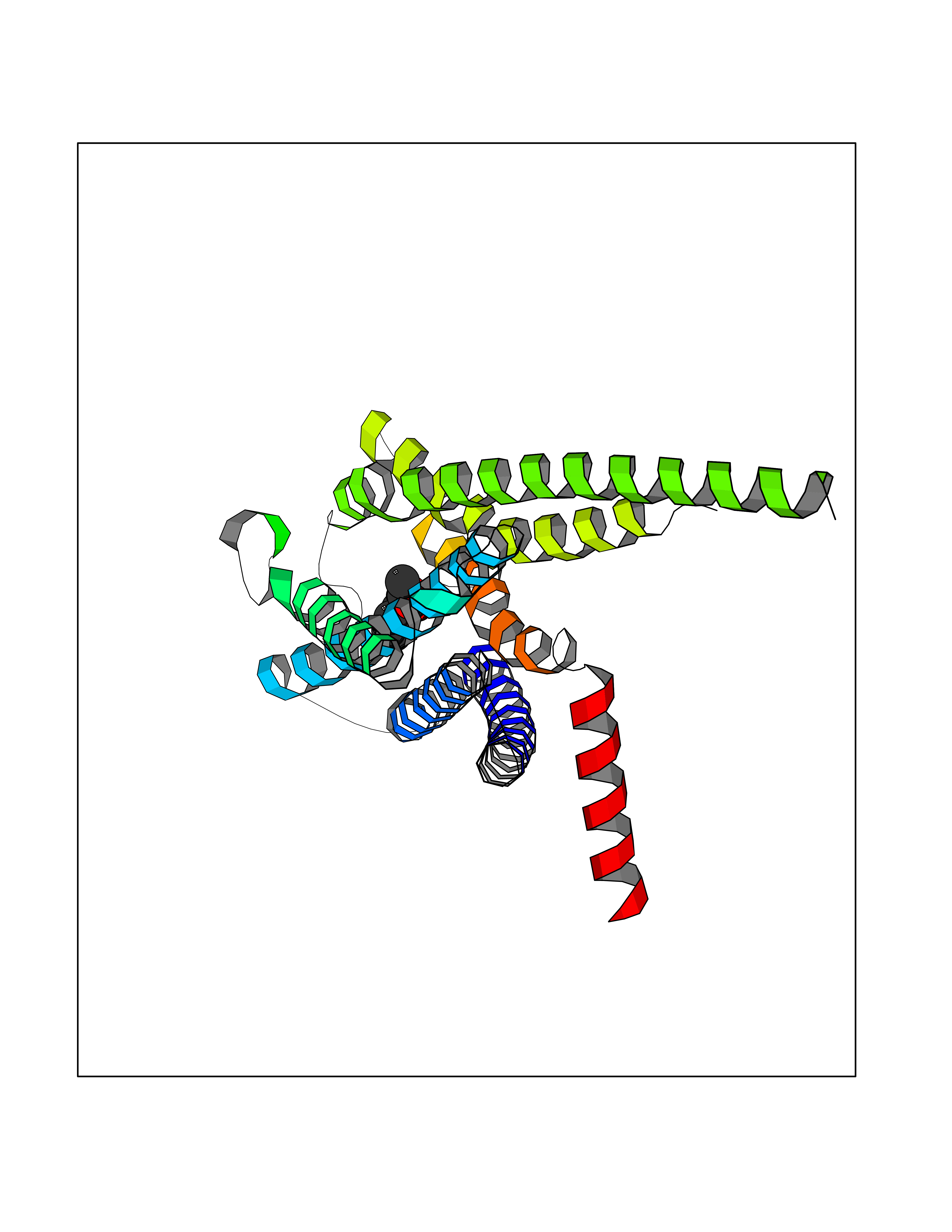 Molscript Map 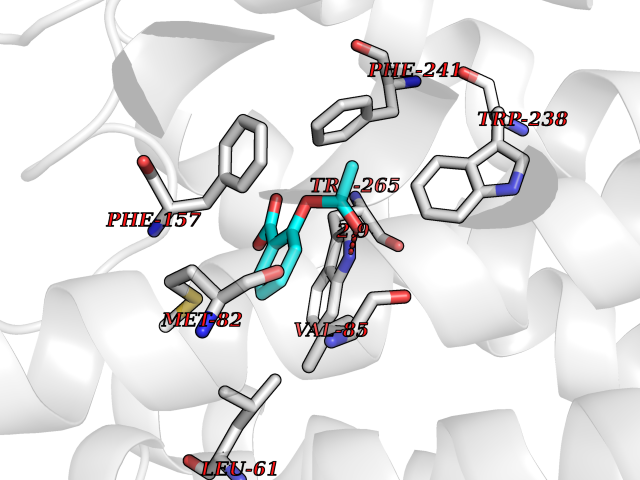 Pymol Map 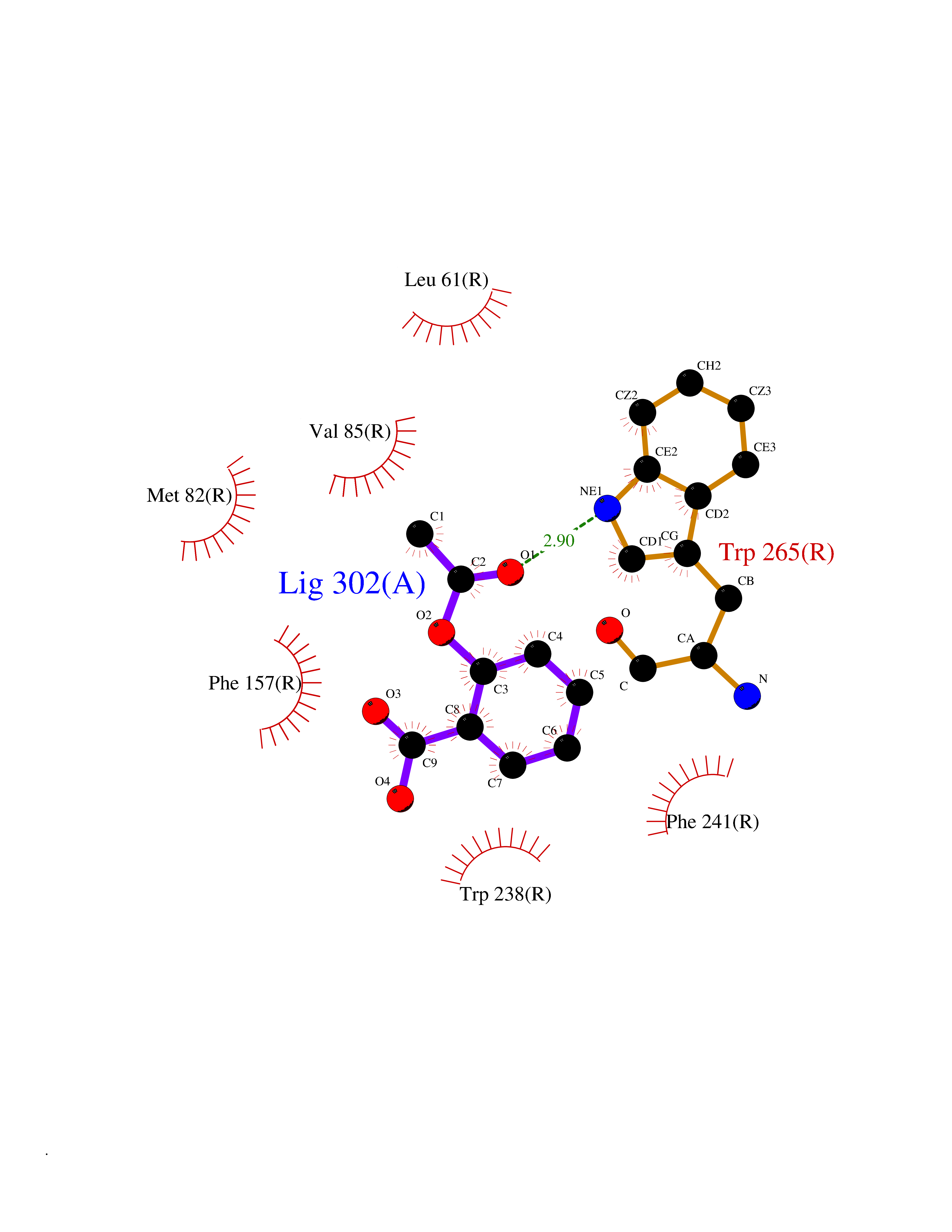 Ligplot Map 3D Binding mode Sequence MESSFSFGVILAVLASLIIATNTLVAVAVLLLIHKNDGVSLCFTLNLAVADTLIGVAISGLLTDQLSSPSRPTQKTLCSLRMAFVTSSAAASVLTVMLITFDRYLAIKQPFRYLKIMSGFVAGACIAGLWLVSYLIGFLPLGIPMFQQTAYKGQCSFFAVFHPHFVLTLSCVGFFPAMLLFVFFYCDMLKIASMHSQQIRKMEHAGAMAGSDFKALRTVSVLIGSFALSWTPFLITGIVQVACQECHLYLVLERYLWLLGVGNSLLNPLIYAYWQKEVRLQLYHMALGVKKV Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Trypanosoma Trypanothione reductase (Trypano TPR) | 2WBA | 5.62 | |
Target general information Gen name Trypano TPR Organism Trypanosoma brucei brucei Uniprot ID TTD ID Synonyms TRYR; TPR; Parasite-specific trypanothione reductase; N(1),N(8)-bis(glutathionyl)spermidine reductase Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Sulfur donor oxidoreductase Function Trypanothione is the parasite analog of glutathione; this enzyme is the equivalent of glutathione reductase. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.8.1.12 Uniprot keywords 3D-structure; Cytoplasm; Disulfide bond; FAD; Flavoprotein; NADP; Oxidoreductase; Redox-active center Protein physicochemical properties Chain ID A,B Molecular weight (Da) 105578 Length 978 Aromaticity 0.08 Instability index 33.76 Isoelectric point 6.25 Charge (pH=7) -6.81 2D Binding mode Binding energy (Kcal/mol) -7.67  Molscript Map 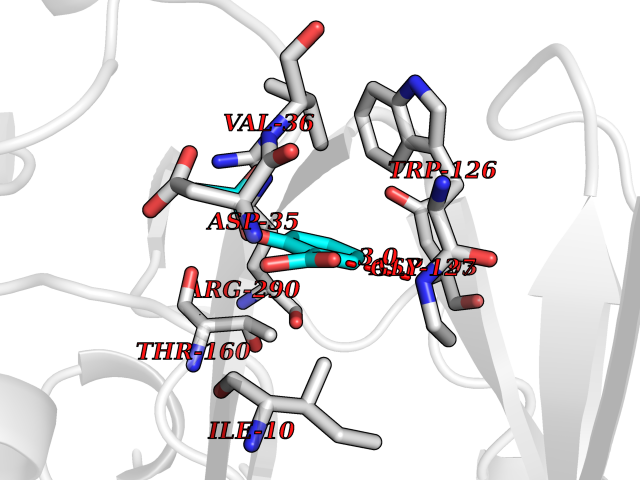 Pymol Map 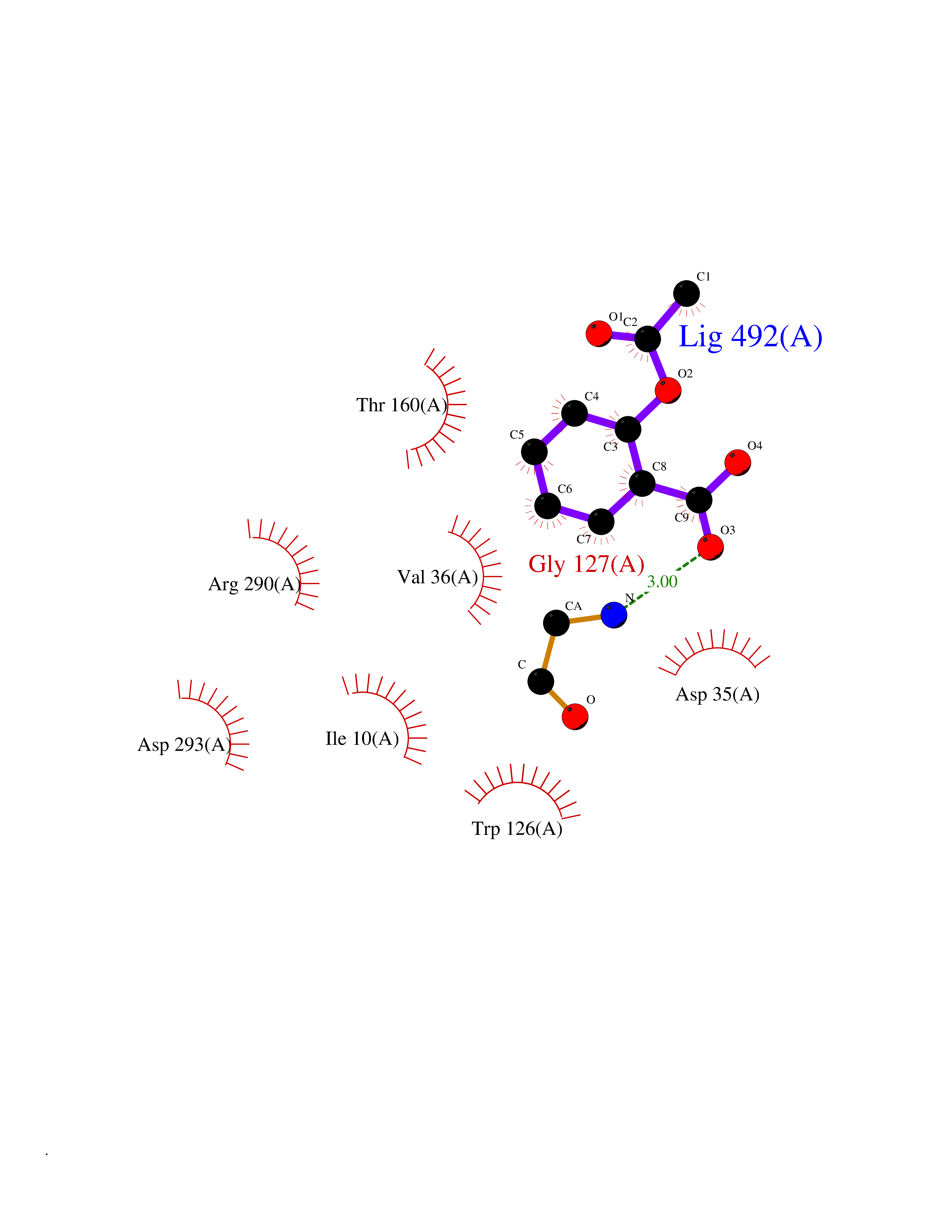 Ligplot Map 3D Binding mode Sequence SKAFDLVVIGAGSGGLEAGWNAATLYGKRVAVVDVQTSHGPPFYAALGGTCVNVGCVPKKLMVTGAQYMDHLRESAGFGWEFDGSSVKANWKKLIAAKNEAVLDINKSYEGMFNDTEGLDFFLGWGSLESKNVVVVRETADPKSAVKERLQADHILLATGSWPQMPAIPGIEHCISSNEAFYLPEPPRRVLTVGGGFISVEFAGIFNAYKPPGGKVTLCYRNNLILRGFDETIREEVTKQLTANGIEIMTNENPAKVSLNTDGSKHVTFESGKTLDVDVVMMAIGRIPRTNDLQLGNVGVKLTPKGGVQVDEFSRTNVPNIYAIGDITDRLMLTPVAINEGAALVDTVFGNKPRKTDHTRVASAVFSIPPIGTCGLIEEVAAKEFEKVAVYMSSFTPLMHNISGSKYKKFVAKIVTNHSDGTVLGVHLLGDGAPEIIQAVGVCLRLNAKISDFYNTIGVHPTSAEELCSMRTPSYYYVKGEKMEKLPDSSKAFDLVVIGAGSGGLEAGWNAATLYGKRVAVVDVQTSHGPPFYAALGGTCVNVGCVPKKLMVTGAQYMDHLRESAGFGWEFDGSSVKANWKKLIAAKNEAVLDINKSYEGMFNDTEGLDFFLGWGSLESKNVVVVRETADPKSAVKERLQADHILLATGSWPQMPAIPGIEHCISSNEAFYLPEPPRRVLTVGGGFISVEFAGIFNAYKPPGGKVTLCYRNNLILRGFDETIREEVTKQLTANGIEIMTNENPAKVSLNTDGSKHVTFESGKTLDVDVVMMAIGRIPRTNDLQLGNVGVKLTPKGGVQVDEFSRTNVPNIYAIGDITDRLMLTPVAINEGAALVDTVFGNKPRKTDHTRVASAVFSIPPIGTCGLIEEVAAKEFEKVAVYMSSFTPLMHNISGSKYKKFVAKIVTNHSDGTVLGVHLLGDGAPEIIQAVGVCLRLNAKISDFYNTIGVHPTSAEELCSMRTPSYYYVKGEKMEKLPDS Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Orexin receptor type 1 (HCRTR1) | 4ZJ8 | 5.61 | |
Target general information Gen name HCRTR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ox1r; Ox1-R; Ox-1-R; Orexin-1 receptor; Hypocretin receptor type 1; HFGAN72 receptor; 7-transmembrane G-protein coupledneuropeptide receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Triggers an increase in cytoplasmic Ca(2+) levels in response to orexin-A binding. Moderately selective excitatory receptor for orexin-A and, with a lower affinity, for orexin-B neuropeptide. Related diseases Hyperchlorhidrosis, isolated (HYCHL) [MIM:143860]: An autosomal recessive disorder characterized by excessive sweating and increased sweat chloride levels. Affected individuals suffer from episodes of hyponatremic dehydration and report increased amounts of visible salt precipitates in sweat. {ECO:0000269|PubMed:21035102, ECO:0000269|PubMed:21184099, ECO:0000269|PubMed:26911677}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15031; DB11951; DB09034 Interacts with P35414 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 35359.5 Length 307 Aromaticity 0.15 Instability index 36.85 Isoelectric point 9.32 Charge (pH=7) 12.2 2D Binding mode Binding energy (Kcal/mol) -7.65  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DYEDEFLRYLWRDYLYPKQYEWVLIAAYVAVFVVALVGNTLVCLAVWRNHHMRTVTNYFIVNLSLADVLVTAICLPASLLVDITESWLFGHALCKVIPYLQAVSVSVAVLTLSFIALDRWYAICHPLLFKSTARRARGSILGIWAVSLAIMVPQAAVMECSSVLPELANRTRLFSVCDERWADDLYPKIYHSCFFIVTYLAPLGLMAMAYFQIFRKLWGRQKQMRARRKTAKMLMVVLLVFALCYLPISVLNILKRVFGMFRQASDREAVYACFTFSHWLVYANSAANPIIYNFLSGKFREQFKAAF Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Aldose reductase (AKR1B1) | 1US0 | 5.61 | |
Target general information Gen name AKR1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde reductase; AKR1B1 Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies. Related diseases Glutamine deficiency, congenital (GLND) [MIM:610015]: An autosomal recessive disorder characterized by variable brain malformations, encephalopathy, severe developmental delay, seizures, and decreased glutamine levels in bodily fluids. Death in early infancy may occur. {ECO:0000269|PubMed:16267323, ECO:0000269|PubMed:26711351, ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 116 (DEE116) [MIM:620806]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE116 is autosomal dominant form characterized by severe developmental delay, seizures, and white matter abnormalities. {ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry. DEE116 is caused by variants that disrupt the canonical translation start codon in GLUL resulting in initiation of translation at Met-18 (PubMed:38579670). The resulting protein is enzymatically competent but insensitive to negative feedback regulation via glutamine-induced degradation. {ECO:0000269|PubMed:38579670}. Drugs (DrugBank ID) DB07028; DB07030; DB07450; DB02101; DB08449; DB08000; DB07139; DB07498; DB02007; DB02020; DB11859; DB02994; DB04272; DB07187; DB00694; DB00997; DB06246; DB01039; DB02021; DB16707; DB00143; DB02834; DB08084; DB01689; DB07063; DB06077; DB02518; DB00157; DB03461; DB05383; DB05533; DB05327; DB02712; DB00605; DB02383; DB02132; DB08772; DB07093; DB07999; DB08098 Interacts with Q9BUY7 EC number EC 1.1.1.300 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35447.6 Length 313 Aromaticity 0.09 Instability index 36.41 Isoelectric point 7.1 Charge (pH=7) 0.26 2D Binding mode Binding energy (Kcal/mol) -7.66  Molscript Map 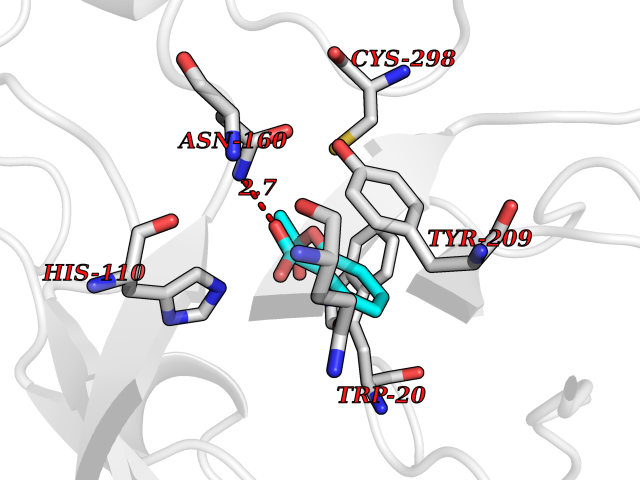 Pymol Map 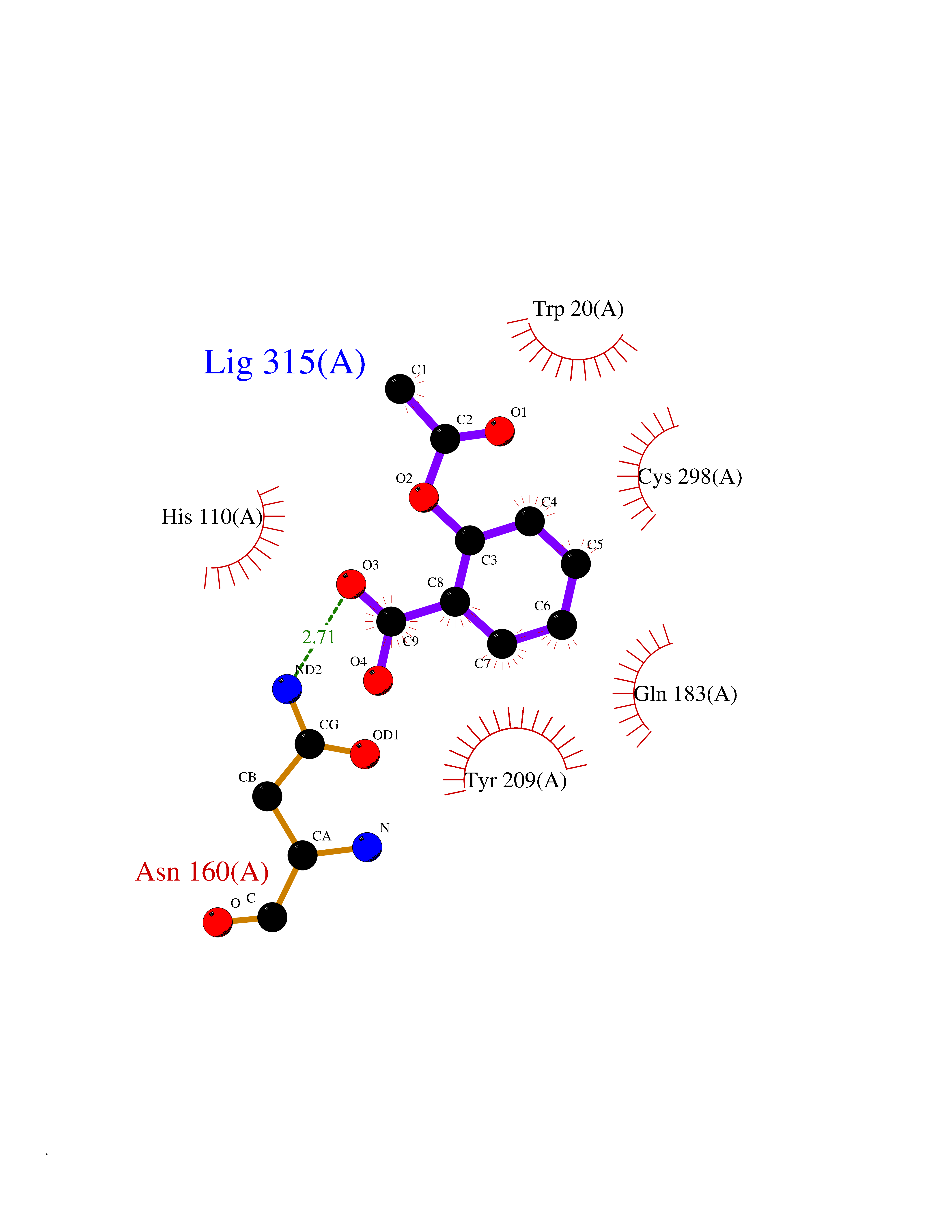 Ligplot Map 3D Binding mode Sequence MASRILLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEKLREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEFFPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAVNQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHNKTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRNWRVCALLSCTSHKDYPFH Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Ornithine transcarbamylase (OTC) | 1OTH | 5.61 | |
Target general information Gen name OTC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms OTCase; Ornithine carbamoyltransferase, mitochondrial Protein family Aspartate/ornithine carbamoyltransferase superfamily, OTCase family Biochemical class NA Function Catalyzes the second step of the urea cycle, the condensation of carbamoyl phosphate with L-ornithine to form L-citrulline. The urea cycle ensures the detoxification of ammonia by converting it to urea for excretion. Related diseases Ornithine carbamoyltransferase deficiency (OTCD) [MIM:311250]: An X-linked disorder of the urea cycle which causes a form of hyperammonemia. Mutations with no residual enzyme activity are always expressed in hemizygote males by a very severe neonatal hyperammonemic coma that generally proves to be fatal. Heterozygous females are either asymptomatic or express orotic aciduria spontaneously or after protein intake. The disorder is treatable with supplemental dietary arginine and low protein diet. The arbitrary classification of patients into the 'neonatal' group (clinical hyperammonemia in the first few days of life) and 'late' onset (clinical presentation after the neonatal period) has been used to differentiate severe from mild forms. {ECO:0000269|PubMed:10070627, ECO:0000269|PubMed:10502831, ECO:0000269|PubMed:10737985, ECO:0000269|PubMed:11793483, ECO:0000269|PubMed:1480464, ECO:0000269|PubMed:1671317, ECO:0000269|PubMed:1721894, ECO:0000269|PubMed:2347583, ECO:0000269|PubMed:2474822, ECO:0000269|PubMed:2556444, ECO:0000269|PubMed:3170748, ECO:0000269|PubMed:7474905, ECO:0000269|PubMed:7951259, ECO:0000269|PubMed:8019569, ECO:0000269|PubMed:8081373, ECO:0000269|PubMed:8081398, ECO:0000269|PubMed:8099056, ECO:0000269|PubMed:8112735, ECO:0000269|PubMed:8530002, ECO:0000269|PubMed:8807340, ECO:0000269|PubMed:8830175, ECO:0000269|PubMed:8956038, ECO:0000269|PubMed:8956045, ECO:0000269|PubMed:9065786, ECO:0000269|PubMed:9143919, ECO:0000269|PubMed:9266388, ECO:0000269|PubMed:9286441, ECO:0000269|PubMed:9452024, ECO:0000269|PubMed:9452049, ECO:0000269|PubMed:9452065, ECO:0000269|Ref.32, ECO:0000269|Ref.43}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00155; DB02011; DB04185; DB00129 Interacts with NA EC number EC 2.1.3.3 Uniprot keywords 3D-structure; Acetylation; Amino-acid biosynthesis; Arginine biosynthesis; Disease variant; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Transit peptide; Urea cycle Protein physicochemical properties Chain ID A Molecular weight (Da) 36060.2 Length 321 Aromaticity 0.08 Instability index 36.44 Isoelectric point 7.87 Charge (pH=7) 1.48 2D Binding mode Binding energy (Kcal/mol) -7.66 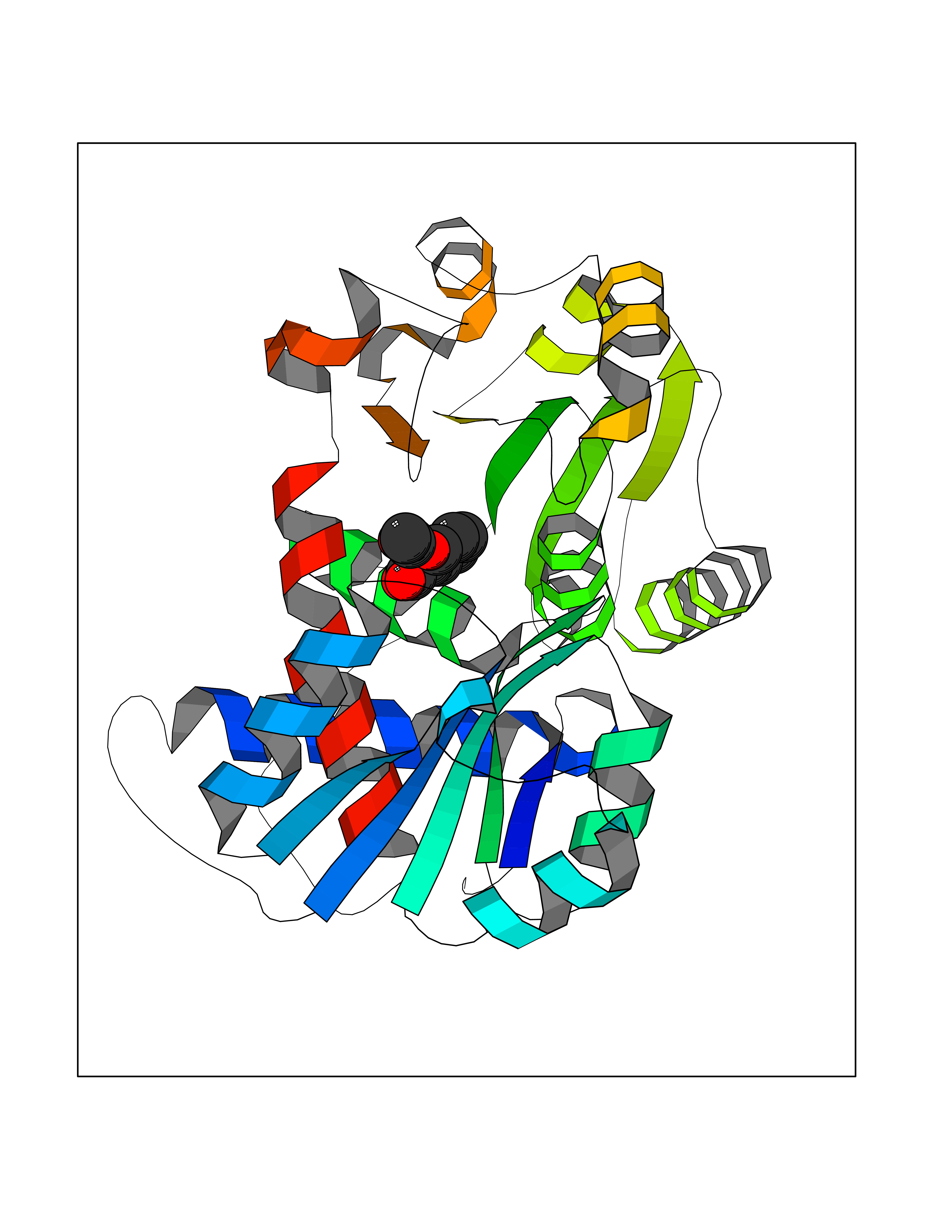 Molscript Map 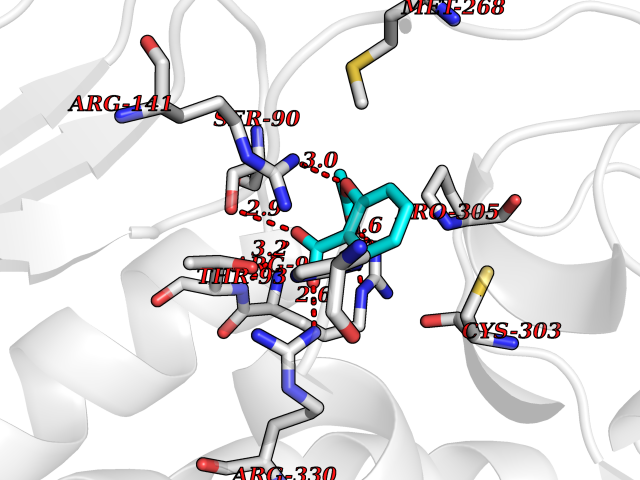 Pymol Map 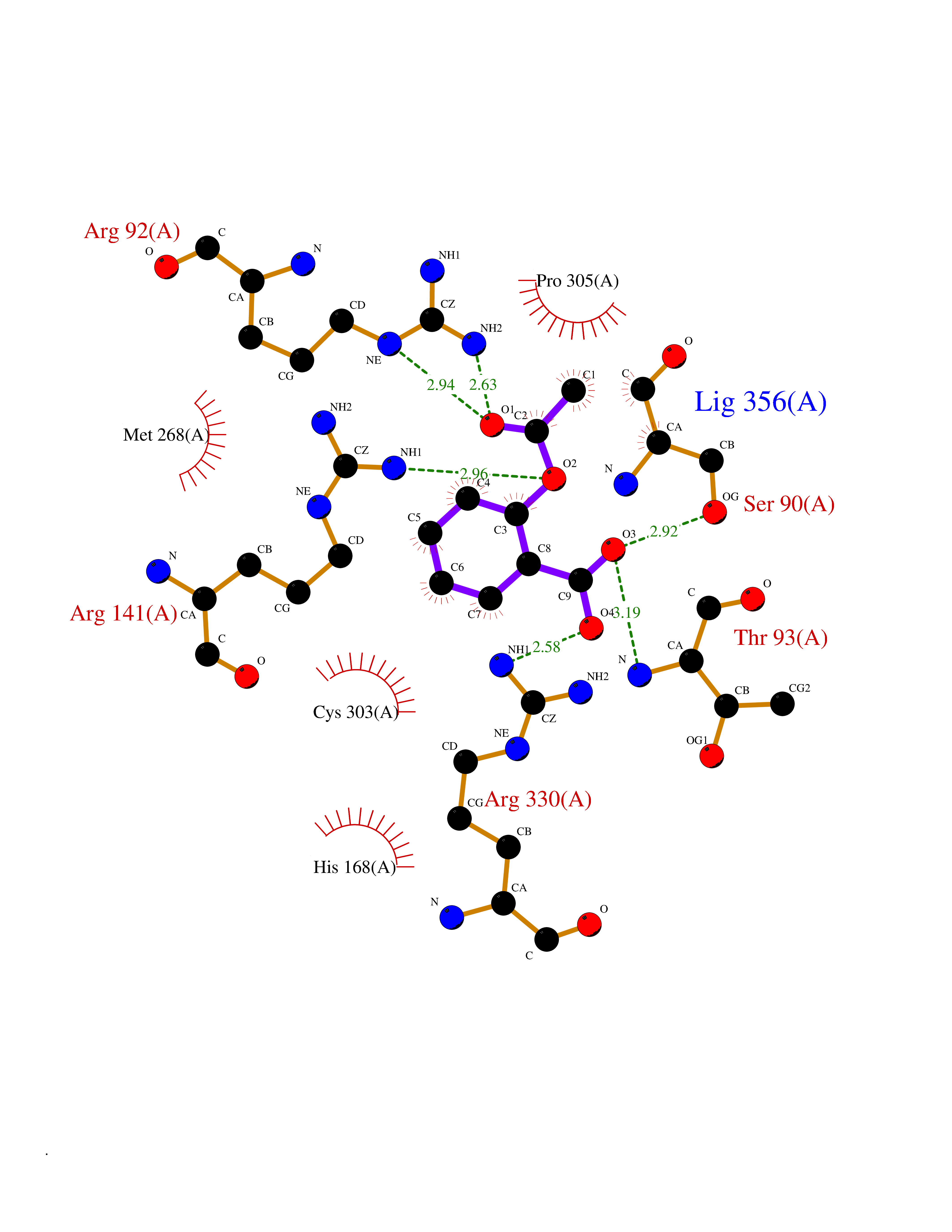 Ligplot Map 3D Binding mode Sequence KVQLKGRDLLTLKNFTGEEIKYMLWLSADLKFRIKQKGEYLPLLQGKSLGMIFEKRSTRTRLSTETGFALLGGHPCFLTTQDIHLGVNESLTDTARVLSSMADAVLARVYKQSDLDTLAKEASIPIINGLSDLYHPIQILADYLTLQEHYSSLKGLTLSWIGDGNNILHSIMMSAAKFGMHLQAATPKGYEPDASVTKLAEQYAKENGTKLLLTNDPLEAAHGGNVLITDTWISMGREEEKKKRLQAFQGYQVTMKTAKVAASDWTFLHCLPRKPEEVDDEVFYSPRSLVFPEAENRKWTIMAVMVSLLTDYSPQLQKPKF Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Dopamine beta-hydroxylase | 4ZEL | 5.61 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -7.66  Molscript Map 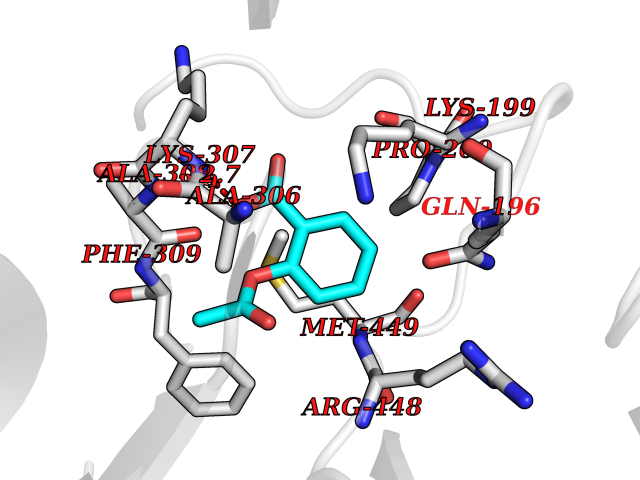 Pymol Map 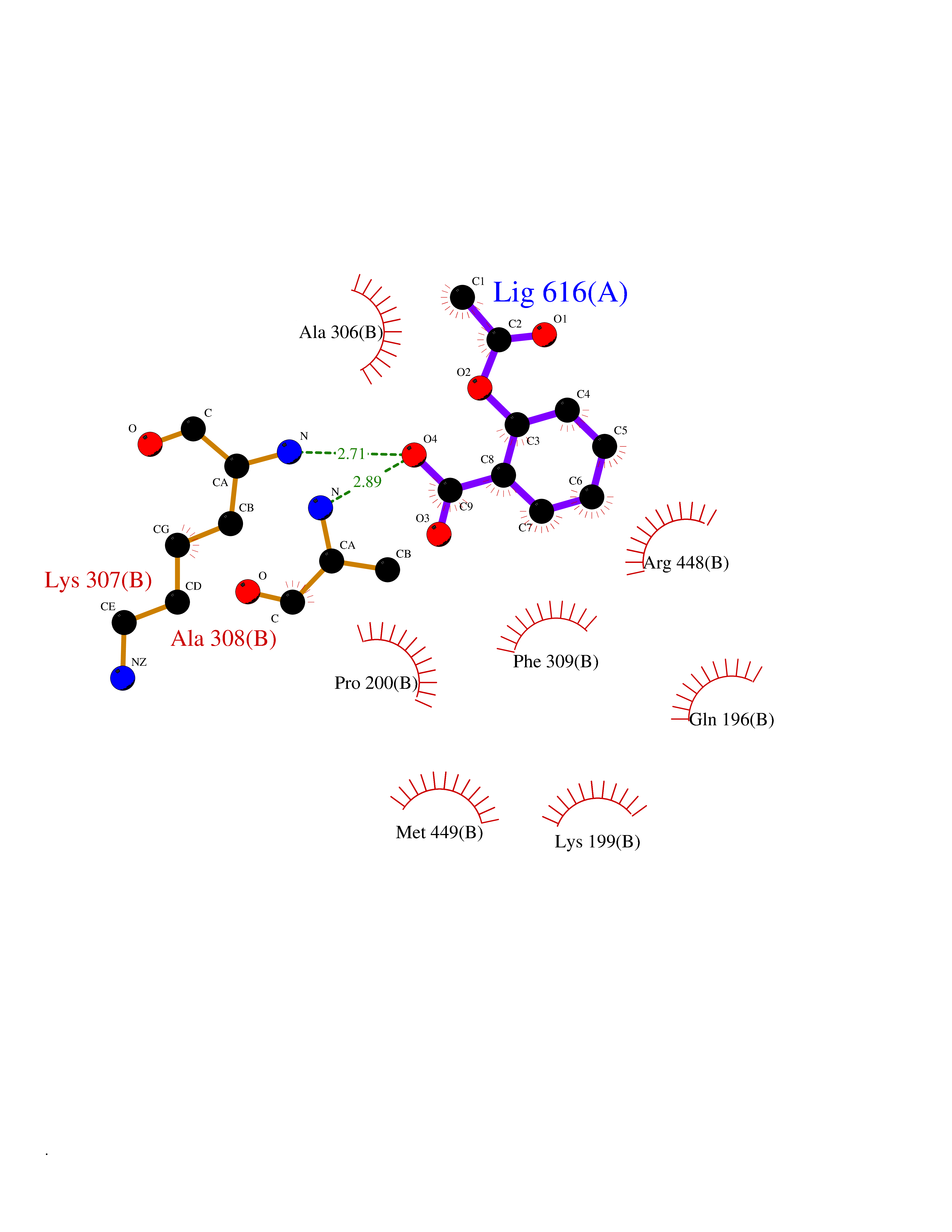 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Insulin-degrading enzyme (IDE) | 4PES | 5.61 | |
Target general information Gen name IDE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Insulysin; Insulinase; Insulin protease; Abeta-degrading protease Protein family Peptidase M16 family Biochemical class Peptidase Function Substrate binding induces important conformation changes, making it possible to bind and degrade larger substrates, such as insulin. Contributes to the regulation of peptide hormone signaling cascades and regulation of blood glucose homeostasis via its role in the degradation of insulin, glucagon and IAPP. Plays a role in the degradation and clearance of APP-derived amyloidogenic peptides that are secreted by neurons and microglia. Involved in antigen processing. Produces both the N terminus and the C terminus of MAGEA3-derived antigenic peptide (EVDPIGHLY) that is presented to cytotoxic T lymphocytes by MHC class I. Plays a role in the cellular breakdown of insulin, APP peptides, IAPP peptides, glucagon, bradykinin, kallidin and other peptides, and thereby plays a role in intercellular peptide signaling. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00626; DB09456; DB09564; DB01307; DB00030; DB00046; DB00071; DB14487; DB14533; DB14548 Interacts with P05067; P55773; P10147; P02686; PRO_0000000093 [P05067]; P01275; P10997; P14735-1; P01308; Q9J3M8 EC number EC 3.4.24.56 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Host cell receptor for virus entry; Host-virus interaction; Hydrolase; Membrane; Metal-binding; Metalloprotease; Nucleotide-binding; Protease; Proteomics identification; Receptor; Reference proteome; Secreted; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 55426 Length 483 Aromaticity 0.11 Instability index 30.18 Isoelectric point 5.79 Charge (pH=7) -12.48 2D Binding mode Binding energy (Kcal/mol) -7.65 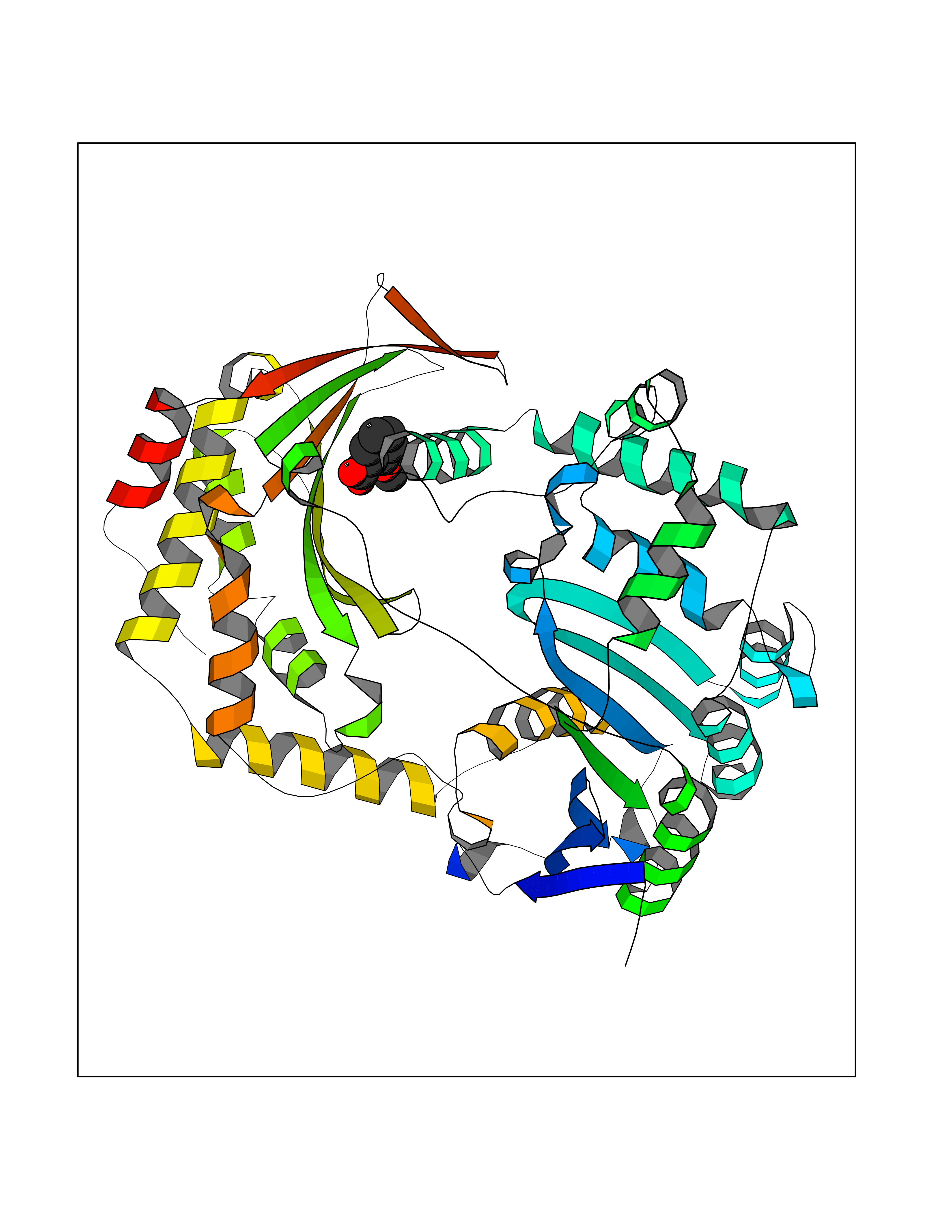 Molscript Map 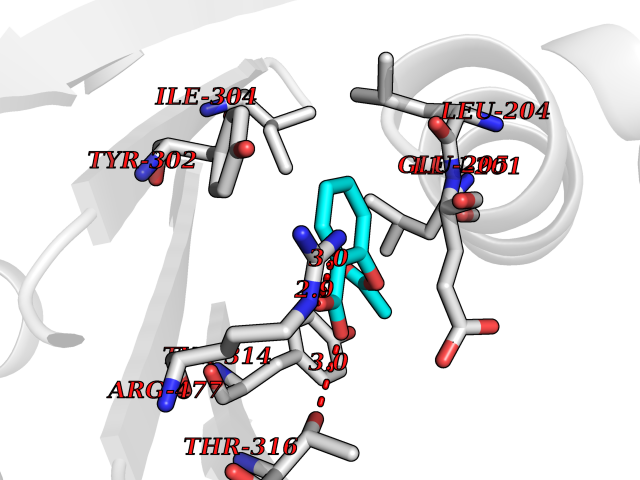 Pymol Map 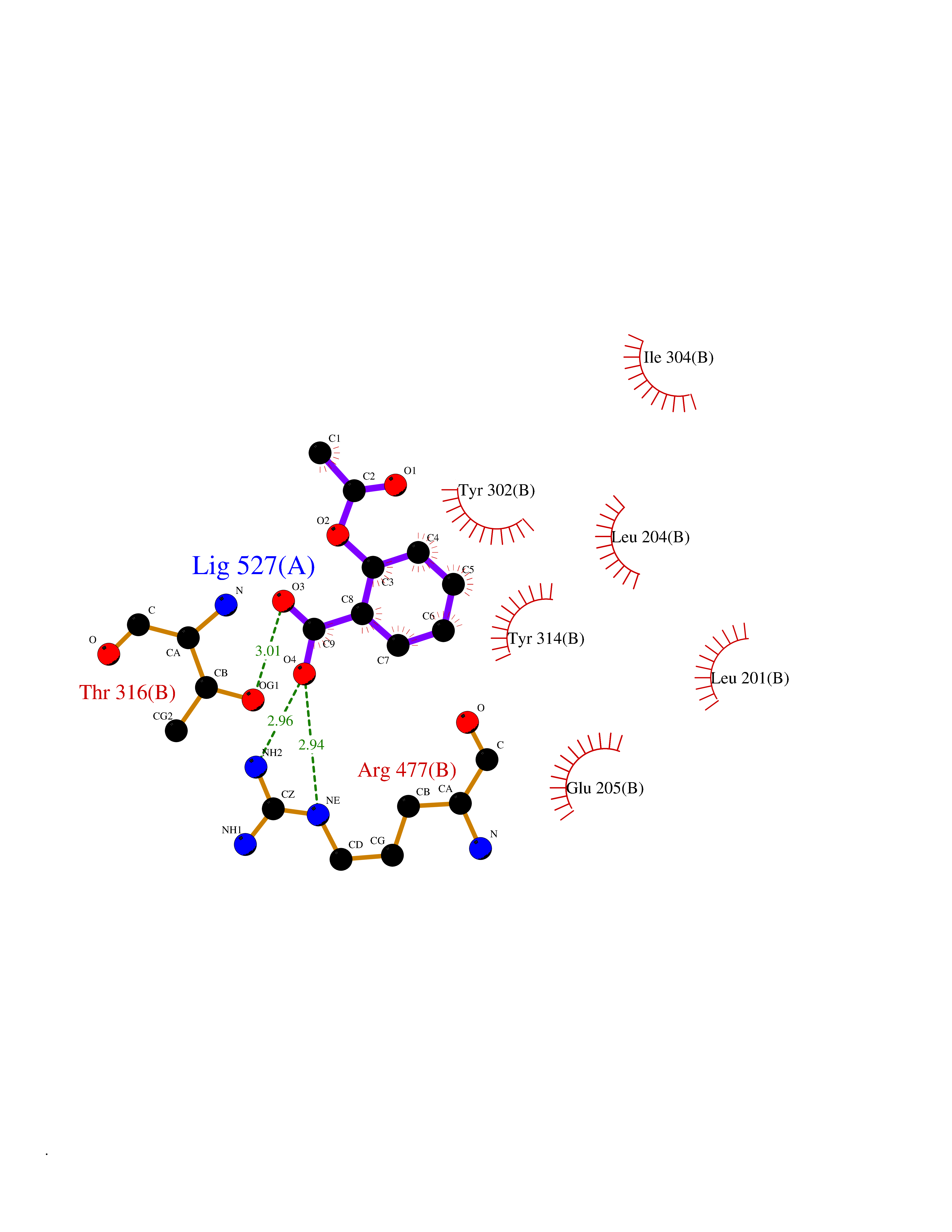 Ligplot Map 3D Binding mode Sequence NNPAIKRIGNHITKSPEDKREYRGLELANGIKVLLISDPTTDKSSAALDVHIGSLSDPPNIAGLSHFLQHMLFLGTKKYPKENEYSQFLSEHAGSSNAFTSGEHTNYYFDVSHEHLEGALDRFAQFFLSPLFDESAKDREVNAVDSEHEKNVMNDAWRLFQLEKATGNPKHPFSKFGTGNKYTLETRPNQEGIDVRQELLKFHSAYYSSNLMAVVVLGRESLDDLTNLVVKLFSEVENKNVPLPEFPEHPFQEEHLKQLYKIVPIKDIRNLYVTFPIPDLQKYYKSNPGHYLGHLIGHEGPGSLLSELKSKGWVNTLVGGQKEGARGFMFFIINVDLTEEGLLHVEDIILHMFQYIQKLRAEGPQEWVFQELKDLNAVAFRFKDKERPRGYTSKIAGILHYYPLEEVLTAEYLLEEFRPDLIEMVLDKLRPENVRVAIVSKSFEGKTDRTEEWYGTQYKQEAIPDEVIKKWQNADLNGKFKLP Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Spermine synthase | 3C6K | 5.61 | |
Target general information Gen name SMS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Spermidine/spermine synthase family Biochemical class Transferase Function Spermine synthase activity. Related diseases Intellectual developmental disorder, X-linked, syndromic, Snyder-Robinson type (MRXSSR) [MIM:309583]: An X-linked intellectual disability syndrome characterized by a collection of clinical features including facial asymmetry, marfanoid habitus, hypertonia, osteoporosis and unsteady gait. {ECO:0000269|PubMed:14508504, ECO:0000269|PubMed:18550699, ECO:0000269|PubMed:19206178, ECO:0000269|PubMed:22612257, ECO:0000269|PubMed:23696453, ECO:0000269|PubMed:23897707}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00127 Interacts with NA EC number 2.5.1.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Intellectual disability; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27177.1 Length 238 Aromaticity 0.11 Instability index 41.31 Isoelectric point 4.82 Charge (pH=7) -9.19 2D Binding mode Binding energy (Kcal/mol) -7.65 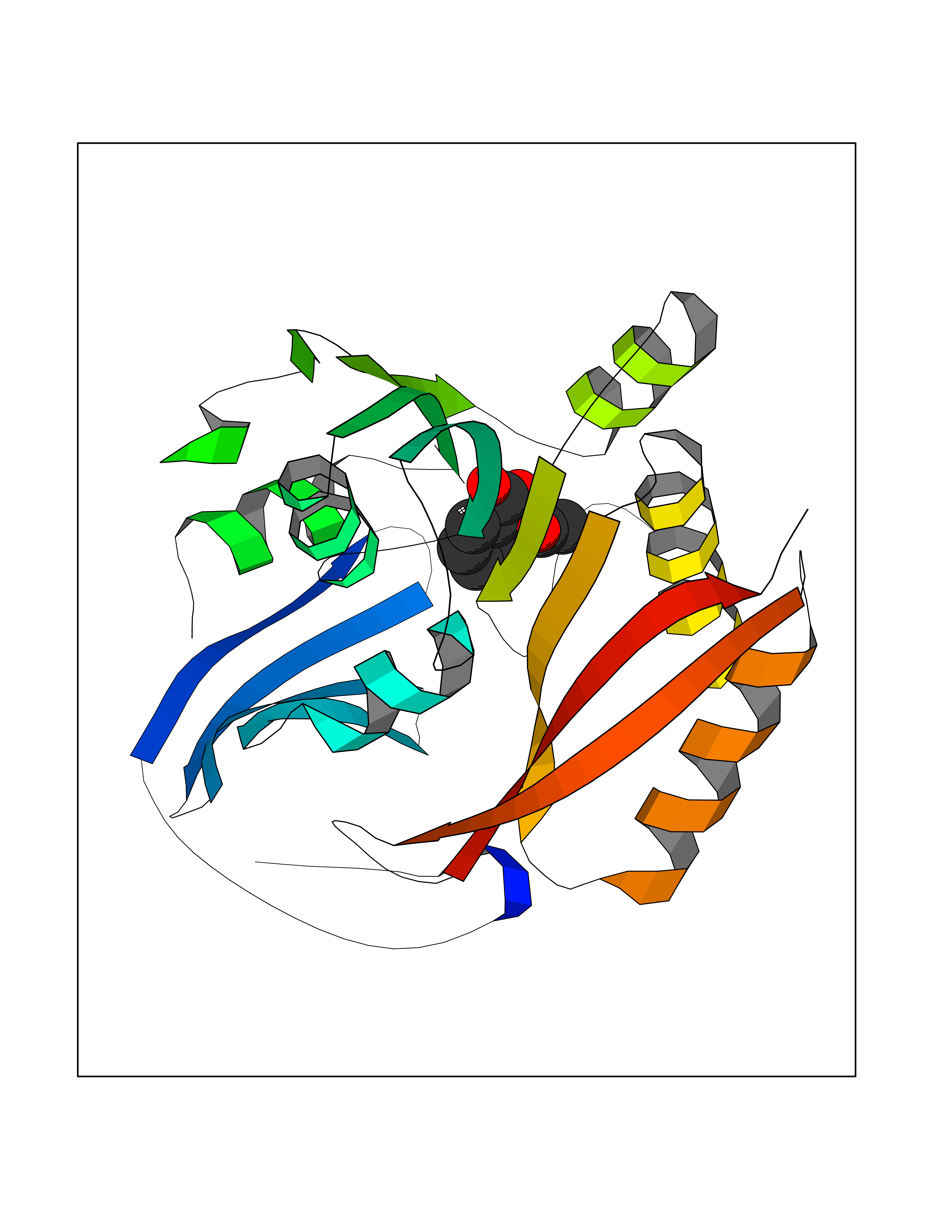 Molscript Map 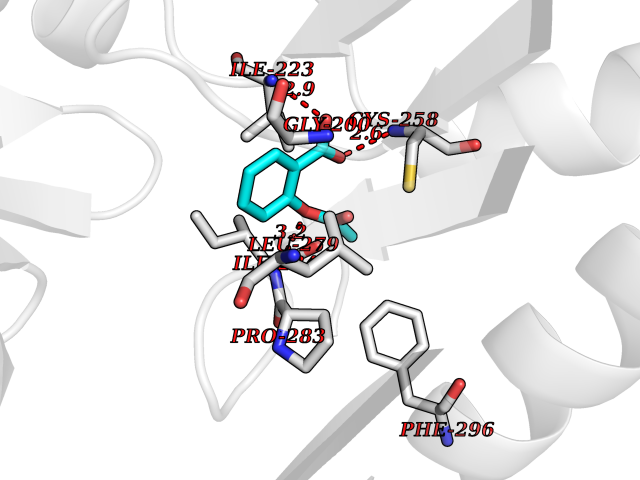 Pymol Map 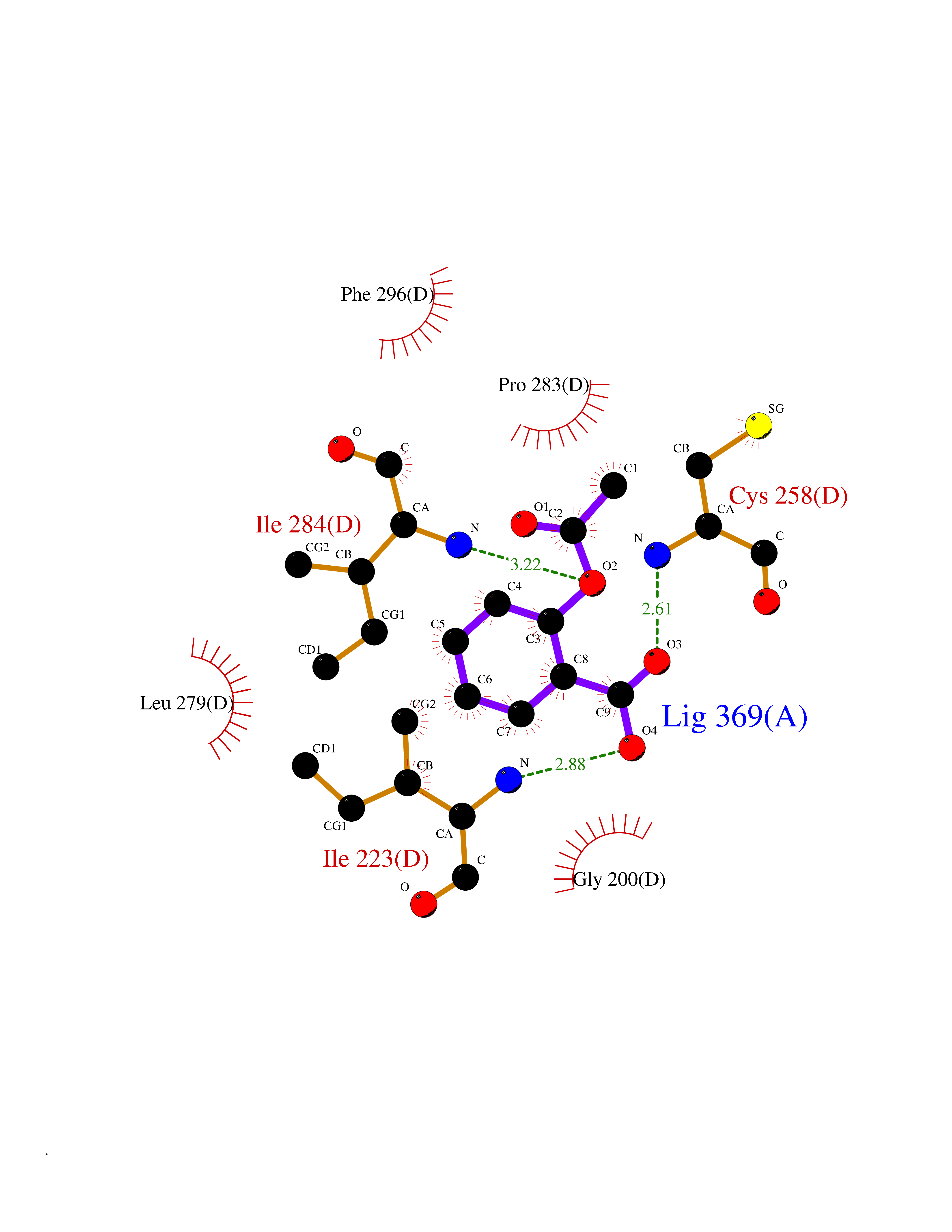 Ligplot Map 3D Binding mode Sequence RYWPTADGRLVEYDIDEVVYDEDSPYQNIKILHSKQFGNILILSGDVNLAESDLAYTRAIMGSGKEDYTGKDVLILGGGDGGILCEIVKLKPKMVTMVEIDQMVIDGCKKYMRKDVLDNLKGDCYQVLIEDCIPVLKRYAKEGREFDYVINDLTAVPISTSPSTWEFLRLILDLSMKVLKQDGKYFTQGNCVNLTEALSLYEEQLGRLYCPVEFSKEIVCVPSYLELWVFYTVWKKAK Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 5.61 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -7.65  Molscript Map 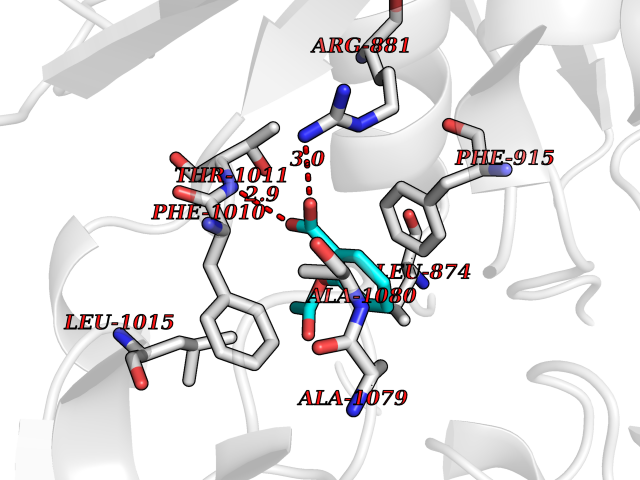 Pymol Map  Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Influenza Polymerase acidic endonuclease (Influ PA) | 4ZI0 | 5.61 | |
Target general information Gen name Influ PA Organism Influenza A virus (strain A/Puerto Rico/8/1934 H1N1) Uniprot ID TTD ID Synonyms RNA-directed RNA polymerase subunit P2; Polymerase acidic protein Protein family Influenza viruses PA family Biochemical class NA Function Plays an essential role in viral RNA transcription and replication by forming the heterotrimeric polymerase complex together with PB1 and PB2 subunits. The complex transcribes viral mRNAs by using a unique mechanism called cap-snatching. It consists in the hijacking and cleavage of host capped pre-mRNAs. These short capped RNAs are then used as primers for viral mRNAs. The PB2 subunit is responsible for the binding of the 5' cap of cellular pre-mRNAs which are subsequently cleaved after 10-13 nucleotides by the PA subunit that carries the endonuclease activity. Related diseases Lymphatic malformation 8 (LMPHM8) [MIM:618773]: A form of primary lymphedema, a disease characterized by swelling of body parts due to developmental anomalies and functional defects of the lymphatic system. Adult patients with lymphedema may suffer from recurrent local infections. Impaired lymphatic drainage in the fetus can develop into hydrops fetalis, a severe condition characterized by excessive fluid accumulation in more than two fetal extra-vascular compartments and body cavities, placental enlargement and edema, pericardial or pleural effusion, or ascites. LMPHM8 is an autosomal recessive form characterized by onset in utero and fetal death due to non-immune hydrops fetalis. {ECO:0000269|PubMed:30115739}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB13997 Interacts with P03485; P03466; P03431 EC number EC 3.1.-.- Uniprot keywords 3D-structure; Cap snatching; Endonuclease; Eukaryotic host gene expression shutoff by virus; Eukaryotic host transcription shutoff by virus; Host cytoplasm; Host gene expression shutoff by virus; Host nucleus; Host-virus interaction; Hydrolase; Inhibition of host RNA polymerase II by virus; Manganese; Metal-binding; Nuclease; Phosphoprotein; Reference proteome; Ribosomal frameshifting Protein physicochemical properties Chain ID A Molecular weight (Da) 21288.1 Length 181 Aromaticity 0.11 Instability index 49.23 Isoelectric point 6.27 Charge (pH=7) -1.8 2D Binding mode Binding energy (Kcal/mol) -7.65  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GPLGSMEDFVRQCFNPMIVELAEKTMKEYGEDLKIETNKFAAICTHLEVCFMYSDASKHRFEIIEGRDRTMAWTVVNSICNTTGAEKPKFLPDLYDYKENRFIEIGVTRREVHIYYLEKANKIKSEKTHIHIFSFTGEEMATKADYTLDEESRARIKTRLFTIRQEMASRGLWDSFRQSER Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Dual-specificity tyrosine-phosphorylation regulated kinase 2 (DYRK2) | 6HDR | 5.61 | |
Target general information Gen name DYRK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dual specificity tyrosine-phosphorylation-regulated kinase 2 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, MNB/DYRK subfamily Biochemical class Kinase Function Functions in part via its role in ubiquitin-dependent proteasomal protein degradation. Functions downstream of ATM and phosphorylates p53/TP53 at 'Ser-46', and thereby contributes to the induction of apoptosis in response to DNA damage. Phosphorylates NFATC1, and thereby inhibits its accumulation in the nucleus and its transcription factor activity. Phosphorylates EIF2B5 at 'Ser-544', enabling its subsequent phosphorylation and inhibition by GSK3B. Likewise, phosphorylation of NFATC1, CRMP2/DPYSL2 and CRMP4/DPYSL3 promotes their subsequent phosphorylation by GSK3B. May play a general role in the priming of GSK3 substrates. Inactivates GYS1 by phosphorylation at 'Ser-641', and potentially also a second phosphorylation site, thus regulating glycogen synthesis. Mediates EDVP E3 ligase complex formation and is required for the phosphorylation and subsequent degradation of KATNA1. Phosphorylates TERT at 'Ser-457', promoting TERT ubiquitination by the EDVP complex. Phosphorylates SIAH2, and thereby increases its ubiquitin ligase activity. Promotes the proteasomal degradation of MYC and JUN, and thereby regulates progress through the mitotic cell cycle and cell proliferation. Promotes proteasomal degradation of GLI2 and GLI3, and thereby plays a role in smoothened and sonic hedgehog signaling. Plays a role in cytoskeleton organization and neurite outgrowth via its phosphorylation of DCX and DPYSL2. Phosphorylates CRMP2/DPYSL2, CRMP4/DPYSL3, DCX, EIF2B5, EIF4EBP1, GLI2, GLI3, GYS1, JUN, MDM2, MYC, NFATC1, p53/TP53, TAU/MAPT and KATNA1. Can phosphorylate histone H1, histone H3 and histone H2B (in vitro). Can phosphorylate CARHSP1 (in vitro). Serine/threonine-protein kinase involved in the regulation of the mitotic cell cycle, cell proliferation, apoptosis, organization of the cytoskeleton and neurite outgrowth. Related diseases Bone marrow failure and diabetes mellitus syndrome (BMFDMS) [MIM:620044]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFDMS is an autosomal recessive form characterized by various degrees of bone marrow failure, ranging from dyserythropoiesis to bone marrow aplasia, with onset in infancy or early childhood, and non-autoimmune insulin-dependent diabetes mellitus appearing in the first or second decades. Many patients show pigmentary skin abnormalities and short stature. {ECO:0000269|PubMed:28073829, ECO:0000269|PubMed:35611808, ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NR20; Q13422; Q9BQD3; Q9BRK4; P23497; O43379; P62258; Q96C00 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cytoplasm; Kinase; Magnesium; Manganese; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 46422.1 Length 407 Aromaticity 0.09 Instability index 44.91 Isoelectric point 9.09 Charge (pH=7) 12.37 2D Binding mode Binding energy (Kcal/mol) -7.65 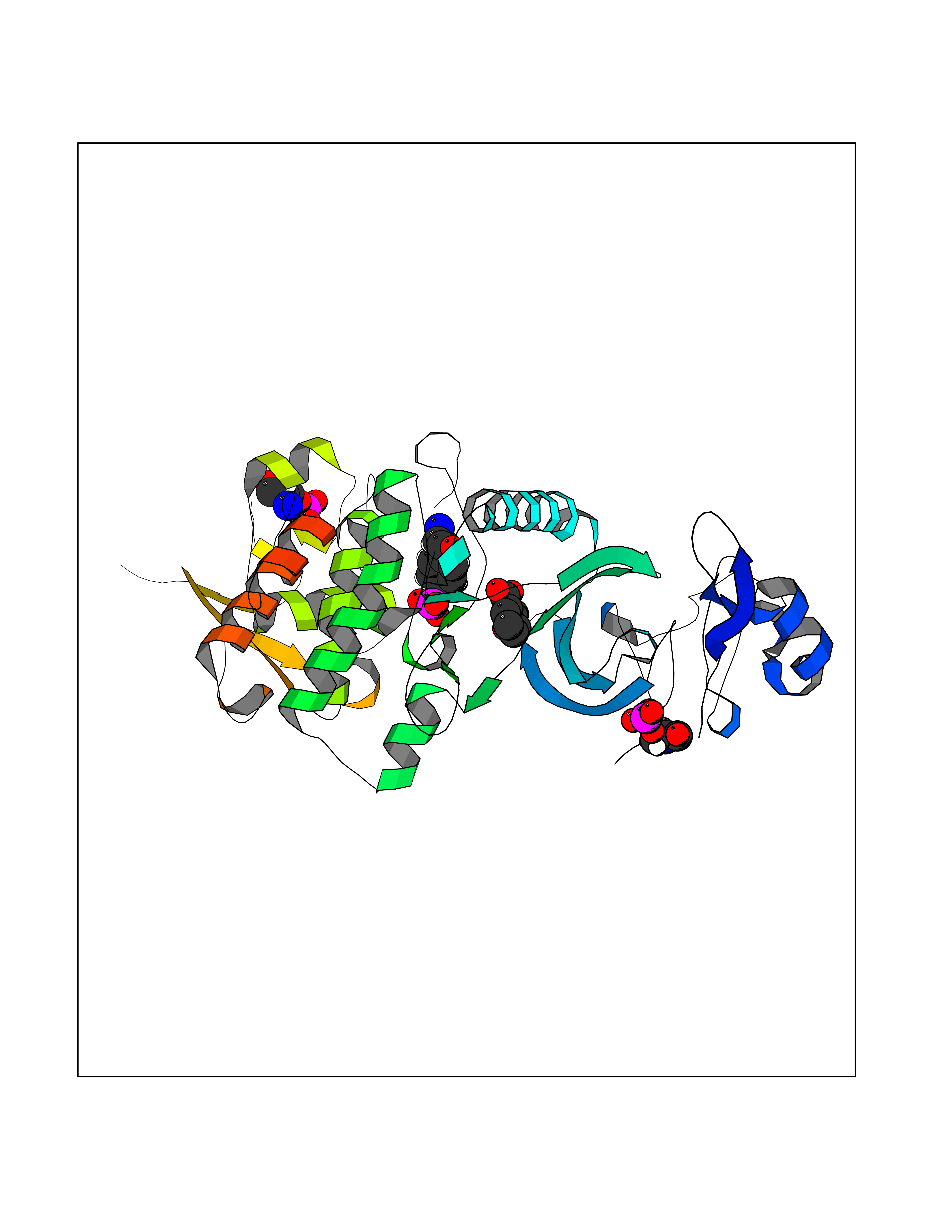 Molscript Map 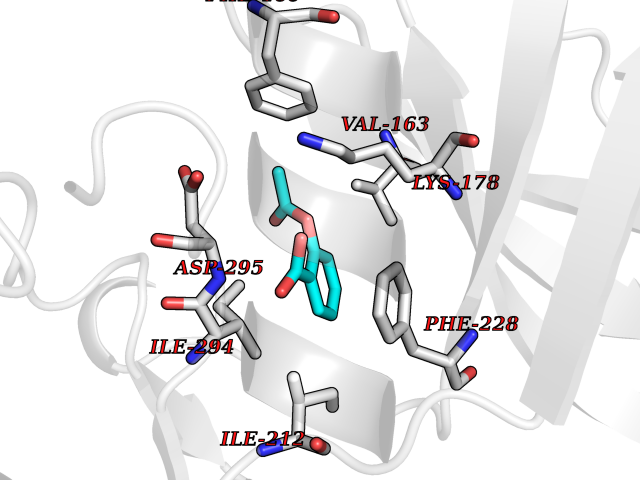 Pymol Map 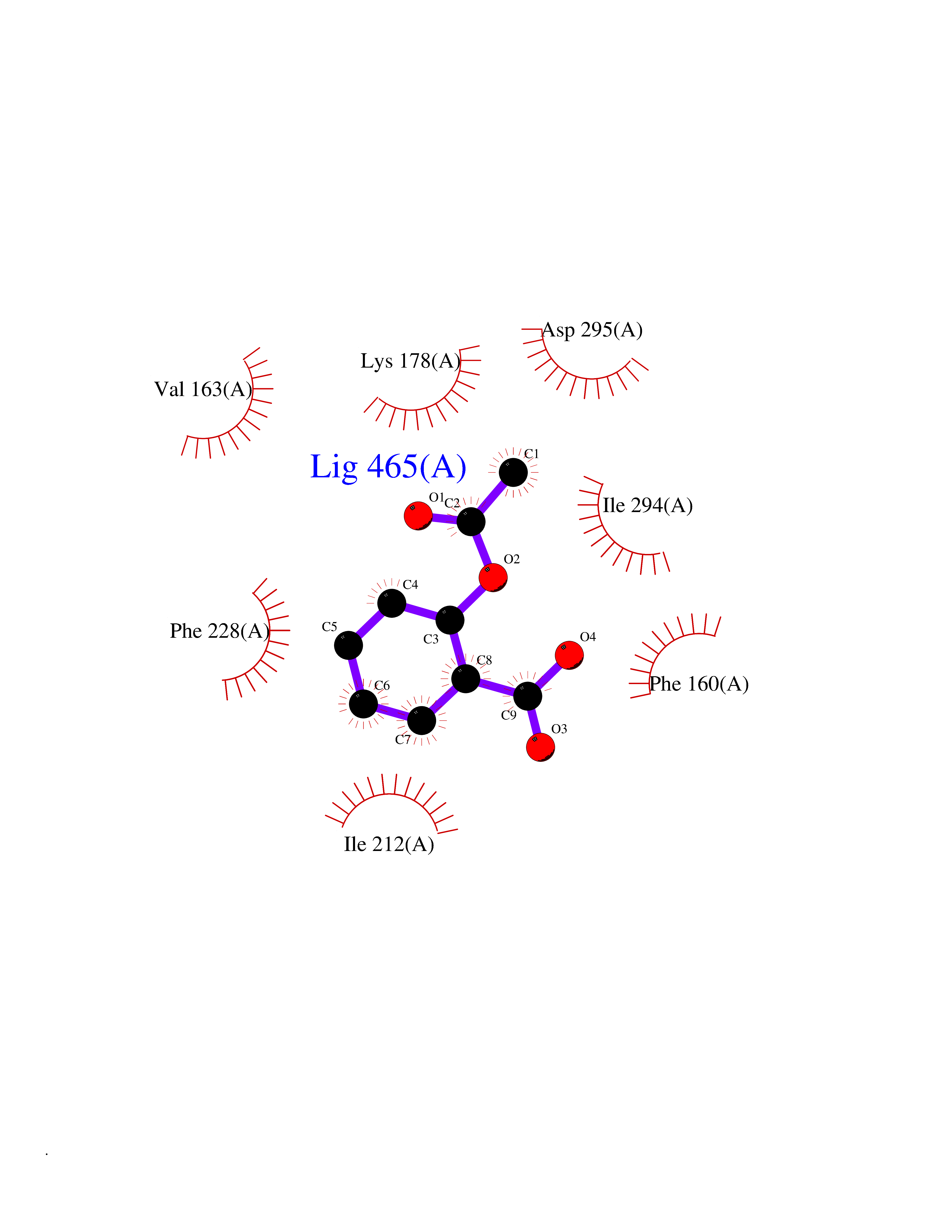 Ligplot Map 3D Binding mode Sequence HHHSXGVDLGTENLYFQSMGKVKATPMTPEQAMKQYMQKLTAFEHHEIFSYPEIYFLGLNAKKRQGMTGGPNNGGYDDDQGSYVQVPHDHVAYRYEVLKVIGKGSFGQVVKAYDHKVHQHVALKMVRNEKRFHRQAAEEIRILEHLRKQDKDNTMNVIHMLENFTFRNHICMTFELLSMNLYELIKKNKFQGFSLPLVRKFAHSILQCLDALHKNRIIHCDLKPENILLKQQGRSGIKVIDFGSSCYEHQRVYTXIQSRFYRAPEVILGARYGMPIDMWSLGCILAELLTGYPLLPGEDEGDQLACMIELLGMPSQKLLDASKRAKNFVSXKGYPRYCTVTTLSDVVLNGGRSRRGKLRGPPESREWGNALKGCDDPLFLDFLKQCLEWDPAVRMTPGQALRHPWLR Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Histone-lysine N-methyltransferase (HLNM) | 3QOW | 5.61 | |
Target general information Gen name DOT1L Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 4; KMT4; KIAA1814; Histone-lysine N-methyltransferase, H3 lysine-79 specific; Histone H3-K79 methyltransferase; H3-K79-HMTase; DOT1-like protein Protein family Class I-like SAM-binding methyltransferase superfamily, DOT1 family Biochemical class Methyltransferase Function Histone methyltransferase. Methylates 'Lys-79' of histone H3. Nucleosomes are preferred as substrate compared to free histones. Binds to DNA. Related diseases Defects in DOTL1 are associated with an autosomal dominant form of global developmental delay and intellectual disability, with or without one or more major congenital anomalies (PubMed:37827158). The patient phenotypes are characterized by central nervous system (CNS) dysfunction, such as mild motor delay and significant speech and language delay, and a range of congenital anomalies, including brain structural anomalies, cardiac defects, varied urogenital features and growth restriction (PubMed:37827158). Variants may cause a gain-of-function effect leading to an increase in cellular H3K79 methylation levels (PubMed:37827158). {ECO:0000269|PubMed:37827158}. Drugs (DrugBank ID) NA Interacts with Q03111; P42568 EC number EC 2.1.1.43 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Disease variant; DNA-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 37367.1 Length 322 Aromaticity 0.11 Instability index 32.71 Isoelectric point 6.03 Charge (pH=7) -5.25 2D Binding mode Binding energy (Kcal/mol) -7.65 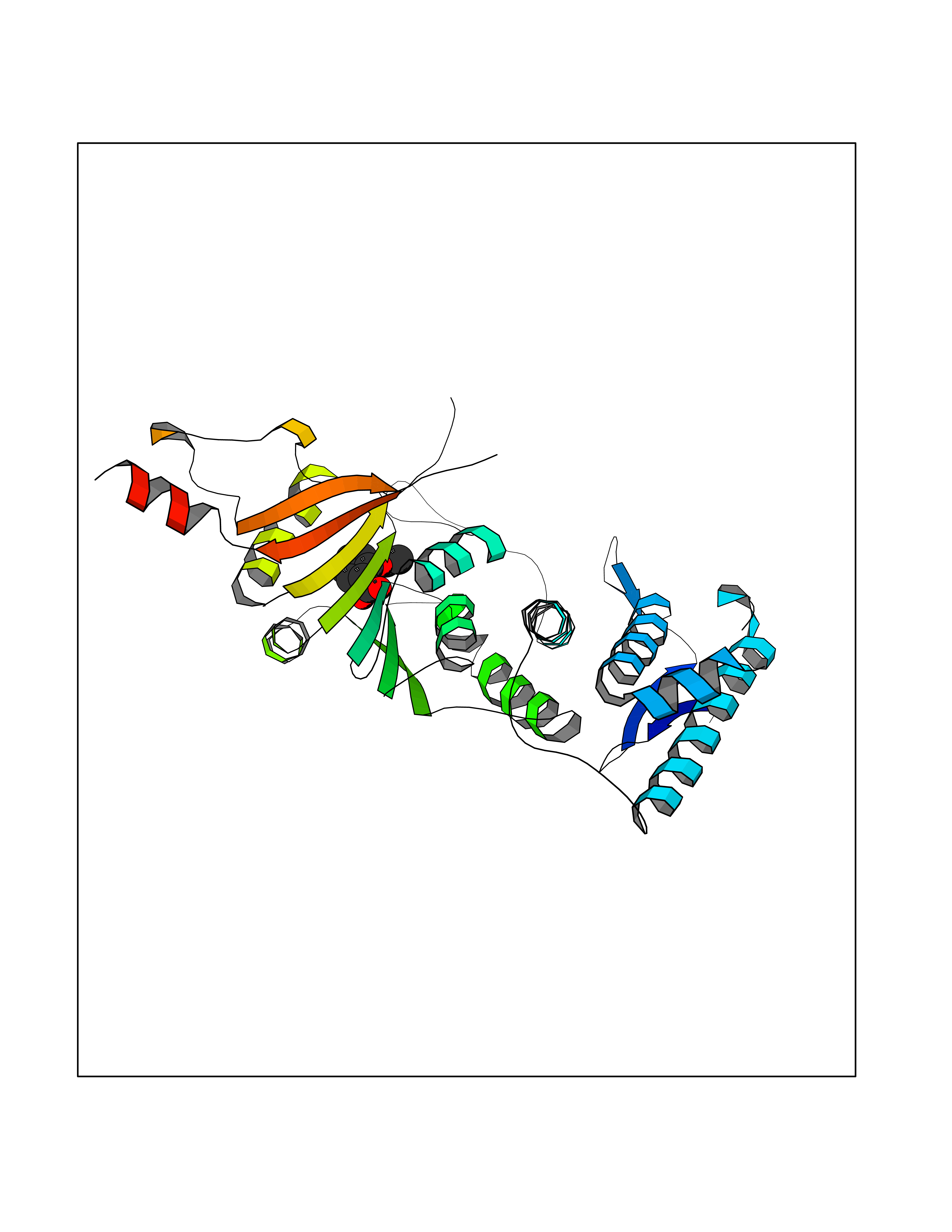 Molscript Map 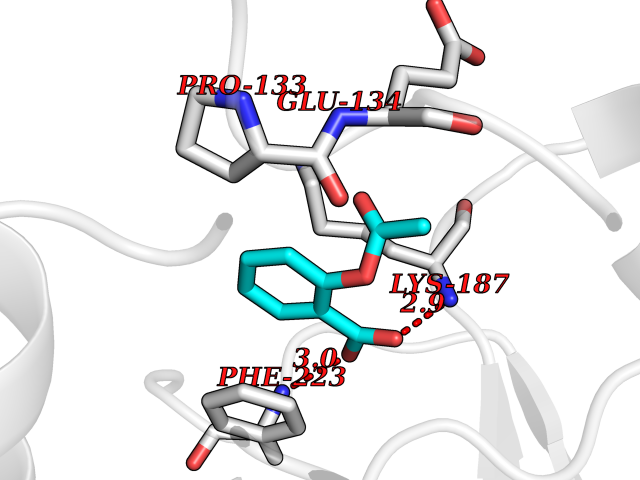 Pymol Map 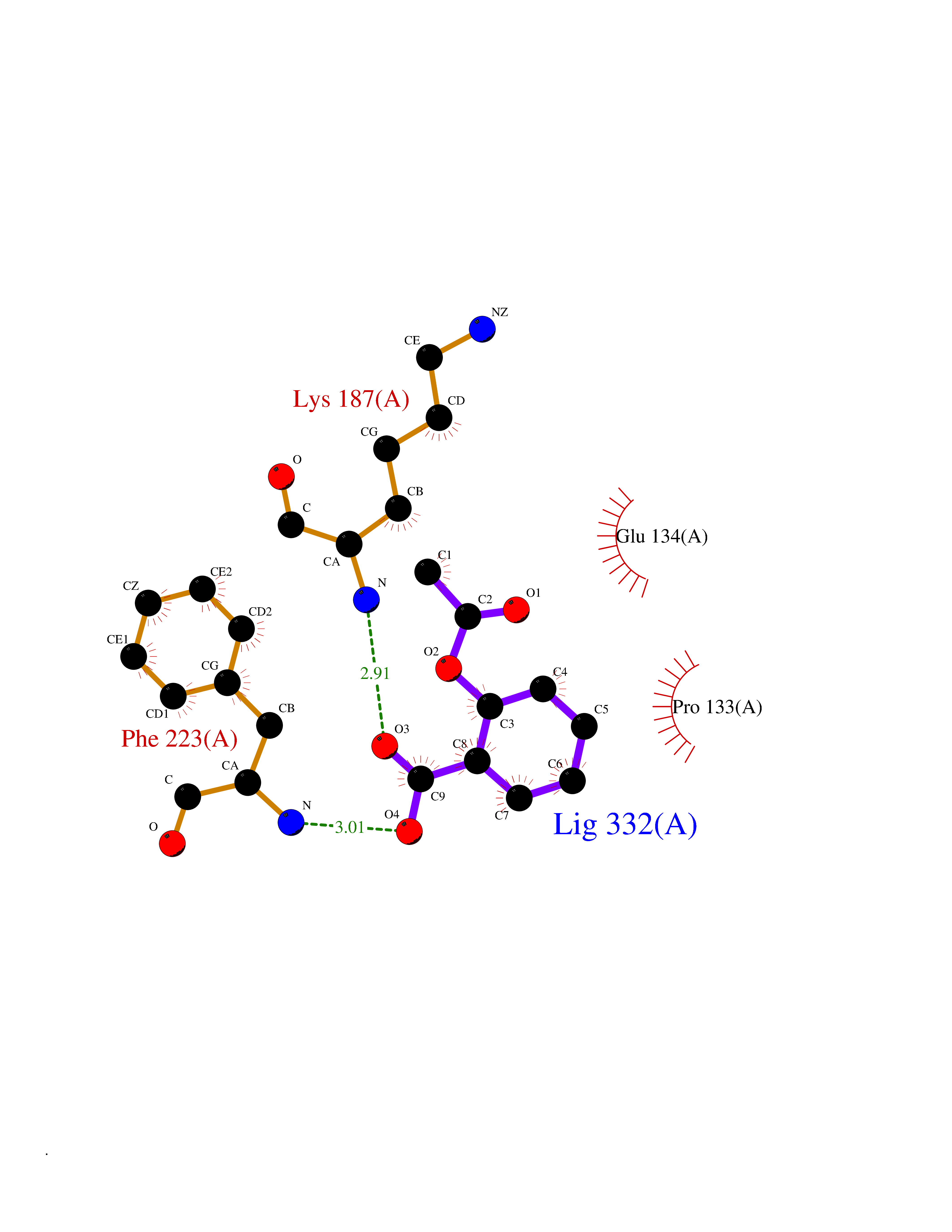 Ligplot Map 3D Binding mode Sequence KLELRLKSPVGAEPAVYPWPLPVYDKHHDAAHEIIETIRWVCEEIPDLKLAMENYLIDYDTKSFESMQRLCDKYNRAIDSIHQLWKGTTQPMKLNTRPSTGLLRHILQQVYNHSVTDPEKLNNYEPFSPEVYGETSFDLVAQMIDEIKMTDDDLFVDLGSGVGQVVLQVAAATNCKHHYGVEKADIPAKYAETMDREFRKWMKWYGKKHAEYTLERGDFLSEEWRERIANTSVIFVNNFAFGPEVDHQLKERFANMKEGGRIVSSKPFAPLNFRINSRNLSDIGTIMRVVELSPLKWTGKPVSYYLHTIDRTILENYFSSLK Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Fumarate reductase flavoprotein subunit | 1KF6 | 5.60 | |
Target general information Gen name frdA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW4115;b4154 Protein family FAD-dependent oxidoreductase 2 family, FRD/SDH subfamily Biochemical class Oxidoreductase Function Electron carrier activity.FAD binding.Fumarate reductase (menaquinone).Succinate dehydrogenase activity. Related diseases Glycogen storage disease 11 (GSD11) [MIM:612933]: A metabolic disorder that results in exertional myoglobinuria, pain, cramps and easy fatigue. {ECO:0000269|PubMed:2334430}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07490; DB07918; DB00730 Interacts with P0AC47; P0ACB4; P76111 EC number 1.3.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; Electron transport; FAD; Flavoprotein; Membrane; Nucleotide-binding; Oxidoreductase; Reference proteome; Transport Protein physicochemical properties Chain ID A,M Molecular weight (Da) 90370.7 Length 820 Aromaticity 0.08 Instability index 28.88 Isoelectric point 5.86 Charge (pH=7) -16.21 2D Binding mode Binding energy (Kcal/mol) -7.63  Molscript Map 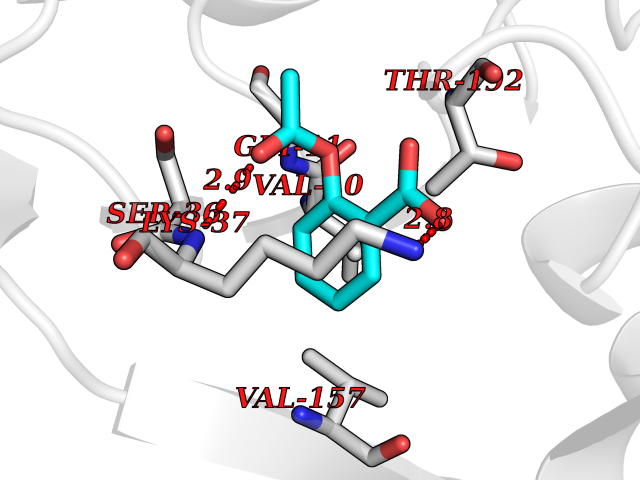 Pymol Map 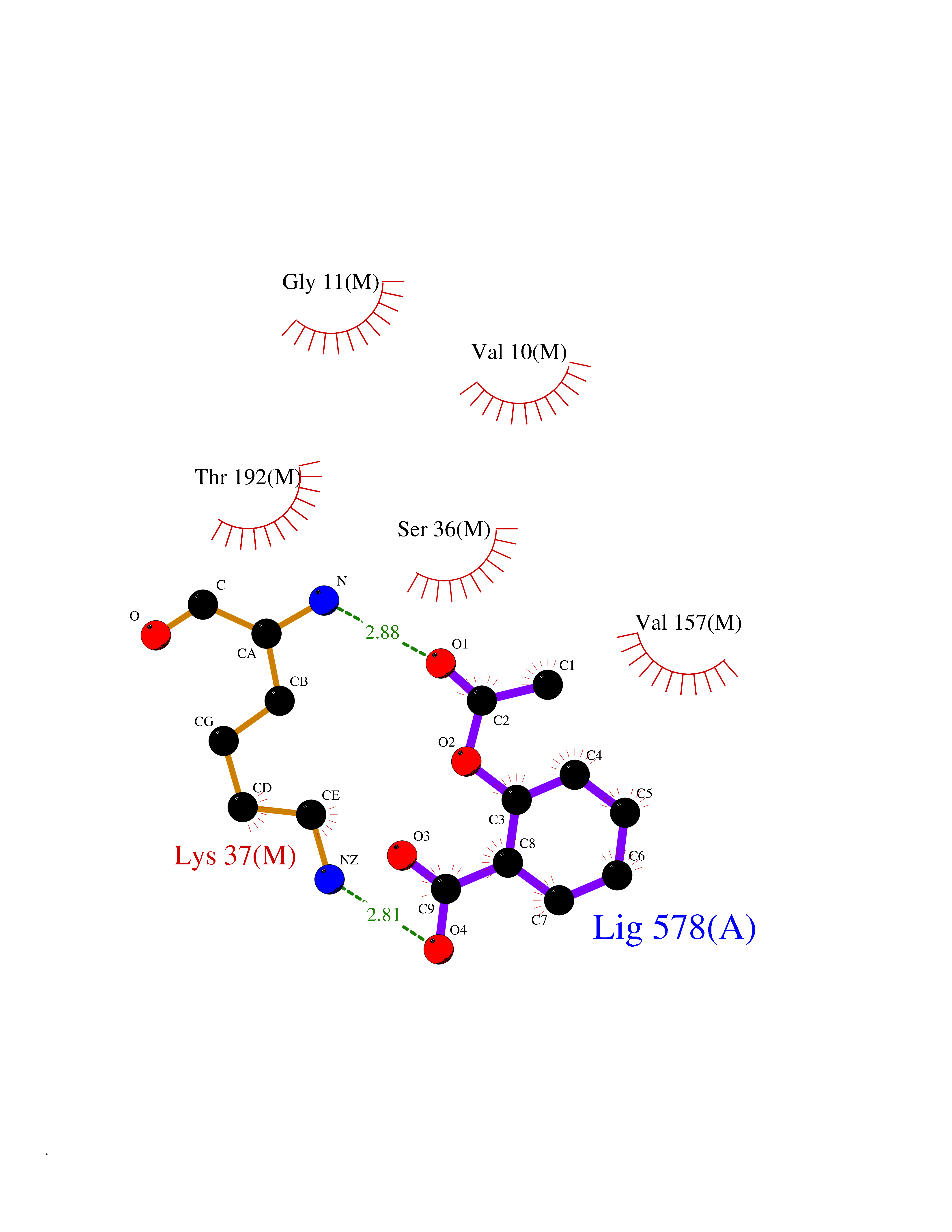 Ligplot Map 3D Binding mode Sequence MQTFQADLAIVGAGGAGLRAAIAAAQANPNAKIALISKVYPMRSHTVAAEGGSAAVAQDHDSFEYHFHDTVAGGDWLCEQDVVDYFVHHCPTEMTQLELWGCPWSRRPDGSVNVRRFGGMKIERTWFAADKTGFHMLHTLFQTSLQFPQIQRFDEHFVLDILVDDGHVRGLVAMNMMEGTLVQIRANAVVMATGGAGRVYRYNTNGGIVTGDGMGMALSHGVPLRDMEFVQYHPTGLPGSGILMTEGCRGEGGILVNKNGYRYLQDYGMGPETPLGEPKNKYMELGPRDKVSQAFWHEWRKGNTISTPRGDVVYLDLRHLGEKKLHERLPFICELAKAYVGVDPVKEPIPVRPTAHYTMGGIETDQNCETRIKGLFAVGECSSVGLHGANRLGSNSLAELVVFGRLAGEQATERAATAGNGNEAAIEAQAAGVEQRLKDLVNQDGGENWAKIRDEMGLAMEEGCGIYRTPELMQKTIDKLAELQERFKRVRITDTSSVFNTDLLYTIELGHGLNVAECMAHSAMARKESRGAHQRLDEGCTERDDVNFLKHTLAFRDADGTTRLEYSDVKITTLPPAAEMKNLKIEVVRYNPEVDTAPHSAFYEVPYDATTSLLDALGYIKDNLAPDLSYRWSCRMAICGSCGMMVNNVPKLACKTFLRDYTDGMKVEALANFPIERDLVVDMTHFIESLEAIKPYIIGNSRTADQGTNIQTPAQMAKYHQFSGCINCGLCYAACPQFGLNPEFIGPAAITLAHRYNEDSRDHGKKERMAQLNSQNGVWSCTFVGYCSEVCPKHVDPAAAIQQGKVESSKDFLIATLKPR Hydrogen bonds contact Hydrophobic contact | ||||