Job Results:
Ligand
Structure
Job ID
a99a6085d597d3b02a857114a6fe159a
Job name
NA
Time
2026-01-11 00:01:47
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Cyclin-dependent kinase 8 (CDK8) | 3RGF | 7.51 | |
Target general information Gen name CDK8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein kinase K35; Mediator of RNA polymerase II transcription subunit CDK8; Mediator complex subunit CDK8; Cell division protein kinase 8 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Kinase Function Mediator functions as a bridge to convey information from gene-specific regulatory proteins to the basal RNA polymerase II transcription machinery. Mediator is recruited to promoters by direct interactions with regulatory proteins and serves as a scaffold for the assembly of a functional preinitiation complex with RNA polymerase II and the general transcription factors. Phosphorylates the CTD (C-terminal domain) of the large subunit of RNA polymerase II (RNAp II), which may inhibit the formation of a transcription initiation complex. Phosphorylates CCNH leading to down-regulation of the TFIIH complex and transcriptional repression. Recruited through interaction with MAML1 to hyperphosphorylate the intracellular domain of NOTCH, leading to its degradation. Component of the Mediator complex, a coactivator involved in regulated gene transcription of nearly all RNA polymerase II-dependent genes. Related diseases Intellectual developmental disorder with hypotonia and behavioral abnormalities (IDDHBA) [MIM:618748]: An autosomal dominant neurodevelopmental disorder with onset in infancy. IDDHBA is characterized by hypotonia, global developmental delay, learning disability, and behavioral abnormalities, such as autistic features and attention deficit-hyperactivity disorder. Additional variable features may include non-specific facial dysmorphism, congenital heart defects, ocular anomalies, and poor feeding. {ECO:0000269|PubMed:30905399}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03496 Interacts with P24863; Q01094; P02489; Q14204; Q92876 EC number EC 2.7.11.22 Uniprot keywords 3D-structure; Activator; Alternative splicing; ATP-binding; Autism spectrum disorder; Disease variant; Intellectual disability; Kinase; Nucleotide-binding; Nucleus; Proteomics identification; Reference proteome; Repressor; Serine/threonine-protein kinase; Transcription; Transcription regulation; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 37655.2 Length 321 Aromaticity 0.11 Instability index 37.04 Isoelectric point 8.56 Charge (pH=7) 5.13 2D Binding mode Binding energy (Kcal/mol) -10.25  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DKMDYDFKVKLSSERERVEDLFEYEGCKVGHVYKAKRKDGKDDKDYALKQIEGTGISMSACREIALLRELKHPNVISLQKVFLSHADRKVWLLFDYAEHDLWHIIKFHRASKLPRGMVKSLLYQILDGIHYLHANWVLHRDLKPANILVMGEGPERGRVKIADMGFARVTFWYRAPELLLGARHYTKAIDIWAIGCIFAELLTSEPIFHCRQNPYHHDQLDRIFNVMGFPADKDWEDIKKMPEHSTLMKDFRRNTYTNCSLIKYMEKHKVKPDSKAFHLLQKLLTMDPIKRITSEQAMQDPYFLEDPLPTSDVFAGCQIPY Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Glutamate receptor ionotropic NMDA 2A (NMDAR2A) | 5H8Q | 7.50 | |
Target general information Gen name GRIN2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NR2A; NMDA receptor NR2A; N-methyl D-aspartate receptor subtype 2A; HNR2A; Glutamate receptor ionotropic, NMDA 2A; Glutamate [NMDA] receptor subunit epsilon-1; GluN2A Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, NR1/GRIN1 subfamily Biochemical class Glutamate-gated ion channel Function Channel activation requires binding of the neurotransmitter glutamate to the epsilon subunit, glycine binding to the zeta subunit, plus membrane depolarization to eliminate channel inhibition by Mg(2+). Sensitivity to glutamate and channel kinetics depend on the subunit composition; channels containing GRIN1 and GRIN2A have higher sensitivity to glutamate and faster kinetics than channels formed by GRIN1 and GRIN2B. Contributes to the slow phase of excitatory postsynaptic current, long-term synaptic potentiation, and learning. Component of NMDA receptor complexes that function as heterotetrameric, ligand-gated ion channels with high calcium permeability and voltage-dependent sensitivity to magnesium. Related diseases Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant (NDHMSD) [MIM:614254]: An autosomal dominant neurodevelopmental disorder characterized by severe intellectual disability and developmental delay, absent speech, muscular hypotonia, dyskinesia, and hyperkinetic movements. Cortical blindness, cerebral atrophy, and seizures are present in some patients. {ECO:0000269|PubMed:21376300, ECO:0000269|PubMed:25167861, ECO:0000269|PubMed:25864721, ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:28228639, ECO:0000269|PubMed:28389307, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal recessive (NDHMSR) [MIM:617820]: An autosomal recessive neurodevelopmental disorder characterized by severe intellectual disability and psychomotor developmental delay, involuntary and stereotypic movements, spasticity, and inability to walk without support. Intractable seizures manifest in some patients. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28051072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 101 (DEE101) [MIM:619814]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE101 is an autosomal recessive, severe form characterized by onset of seizures in early infancy. Death in infancy may occur. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:34611970}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01931; DB00659; DB06151; DB08838; DB01238; DB00289; DB05824; DB04620; DB03929; DB00647; DB00843; DB00228; DB11823; DB13146; DB06741; DB00142; DB00874; DB08954; DB06738; DB09409; DB09481; DB01043; DB00454; DB00333; DB04896; DB01173; DB00312; DB01174; DB01708; DB00418; DB00193 Interacts with P05067; P35637; Q12879-1; Q13224; Q62936 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Ligand-gated ion channel; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Zinc Protein physicochemical properties Chain ID B Molecular weight (Da) 63014.6 Length 557 Aromaticity 0.1 Instability index 28.81 Isoelectric point 8.59 Charge (pH=7) 7.66 2D Binding mode Binding energy (Kcal/mol) -10.22  Molscript Map 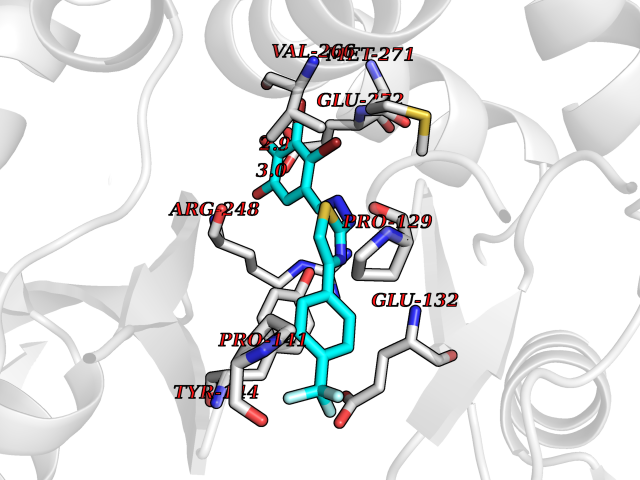 Pymol Map 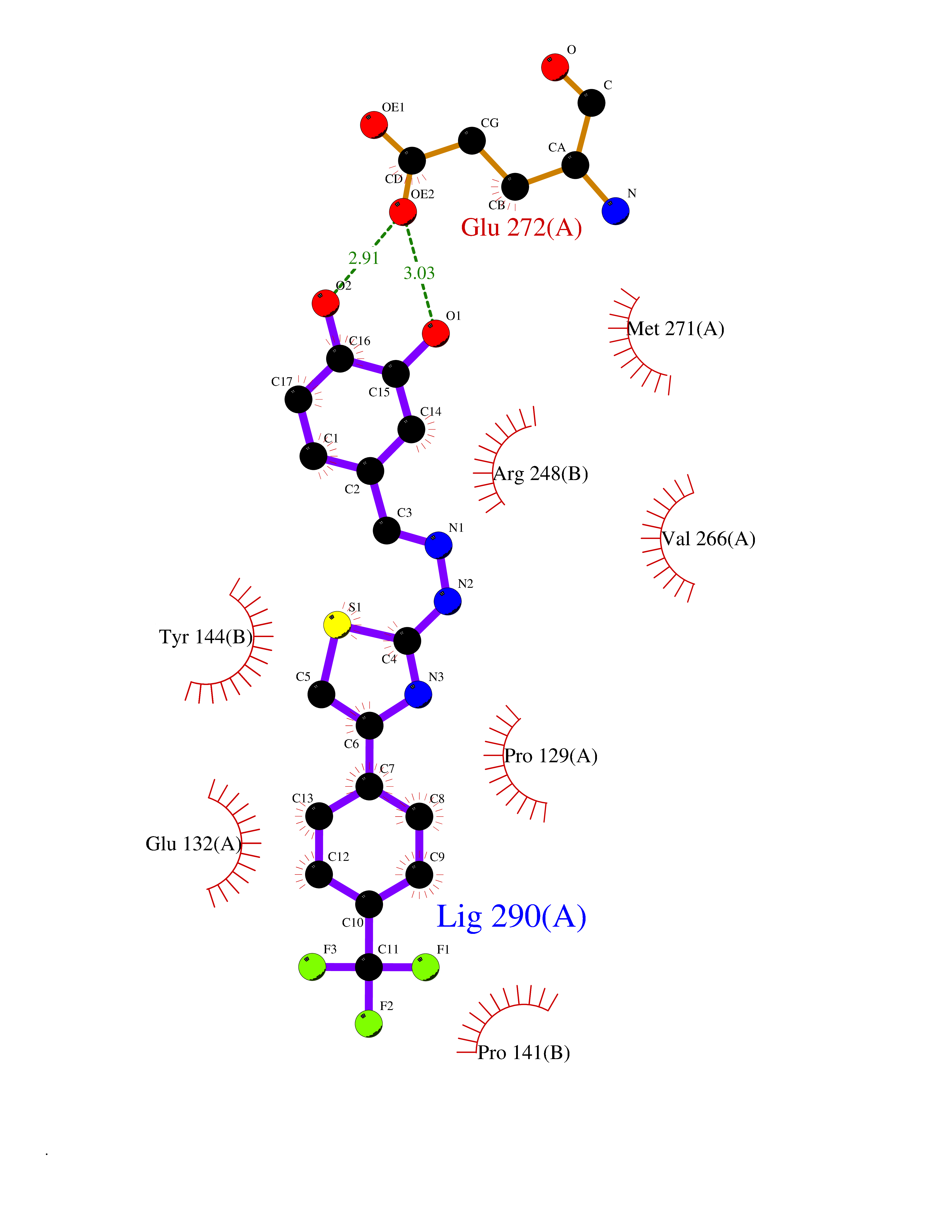 Ligplot Map 3D Binding mode Sequence NHLSIVTLEEAPFVIVEDIDPETCVRNTVPCRKFVKINNSTNEGMNVKKCCKGFCIDILKKLSRTVKFTYDLYLVTNGKHGKKVNNVWNGMIGEVVYQRAVMAVGSLTINEERSEVVDFSVPFVETGISVMVSRGTQVTGLSDKKFQRPHDYSPPFRFGTVPNGSTERNIRNNYPYMHQYMTKFNQKGVEDALVSLKTGKLDAFIYDAAVLNYKAGRDEGCKLVTIGSGYIFATTGYGIALQKGSPWKRQIDLALLQFVGDGEMEELETLWLTGICTRLKIVTIHQEPFVYVKPTLSDGTCKEEFTVNGDPVKKVICTGPNDTSPGSPRHTVPQCCYGFCIDLLIKLARTMNFTYEVHLVADGKFGTQERVNKKEWNGMMGELLSGQADMIVAPLTINNERAQYIEFSKPFKYQGLTILVKKGTRITGINDPRLRNPSDKFIYATVKQSSVDIYFRRQVELSTMYRHMEKHNYESAAEAIQAVRDNKLHAFIWDSAVLEFEASQKCDLVTTGELFFRSGFGIGMRKDSPWKQNVSLSILKSHENGFMEDLDKTWVRY Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | P2Y purinoceptor 12 | 4PXZ | 7.50 | |
Target general information Gen name P2RY12 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HORK3 Protein family G-protein coupled receptor 1 family Biochemical class Membrane protein Function ADP receptor activity.G-protein coupled adenosine receptor activity.Guanyl-nucleotide exchange factor activity. Related diseases Bleeding disorder, platelet-type, 8 (BDPLT8) [MIM:609821]: A condition characterized by mild to moderate mucocutaneous bleeding, and excessive bleeding after surgery or trauma. The defect is due to severe impairment of platelet response to ADP resulting in defective platelet aggregation. {ECO:0000269|PubMed:11196645, ECO:0000269|PubMed:12578987, ECO:0000269|PubMed:25428217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06441; DB00758; DB06350; DB01240; DB06209; DB01069; DB05553; DB15163; DB08816; DB00208; DB00374; DB16349 Interacts with NA EC number NA Uniprot keywords 3D-structure; Blood coagulation; Cell membrane; Disease variant; Disulfide bond; G-protein coupled receptor; Glycoprotein; Hemostasis; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 28830.4 Length 248 Aromaticity 0.17 Instability index 24.19 Isoelectric point 9.39 Charge (pH=7) 10.55 2D Binding mode Binding energy (Kcal/mol) -10.23  Molscript Map 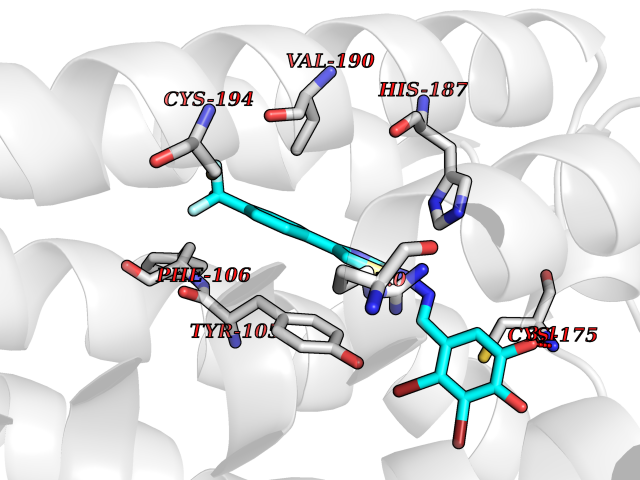 Pymol Map 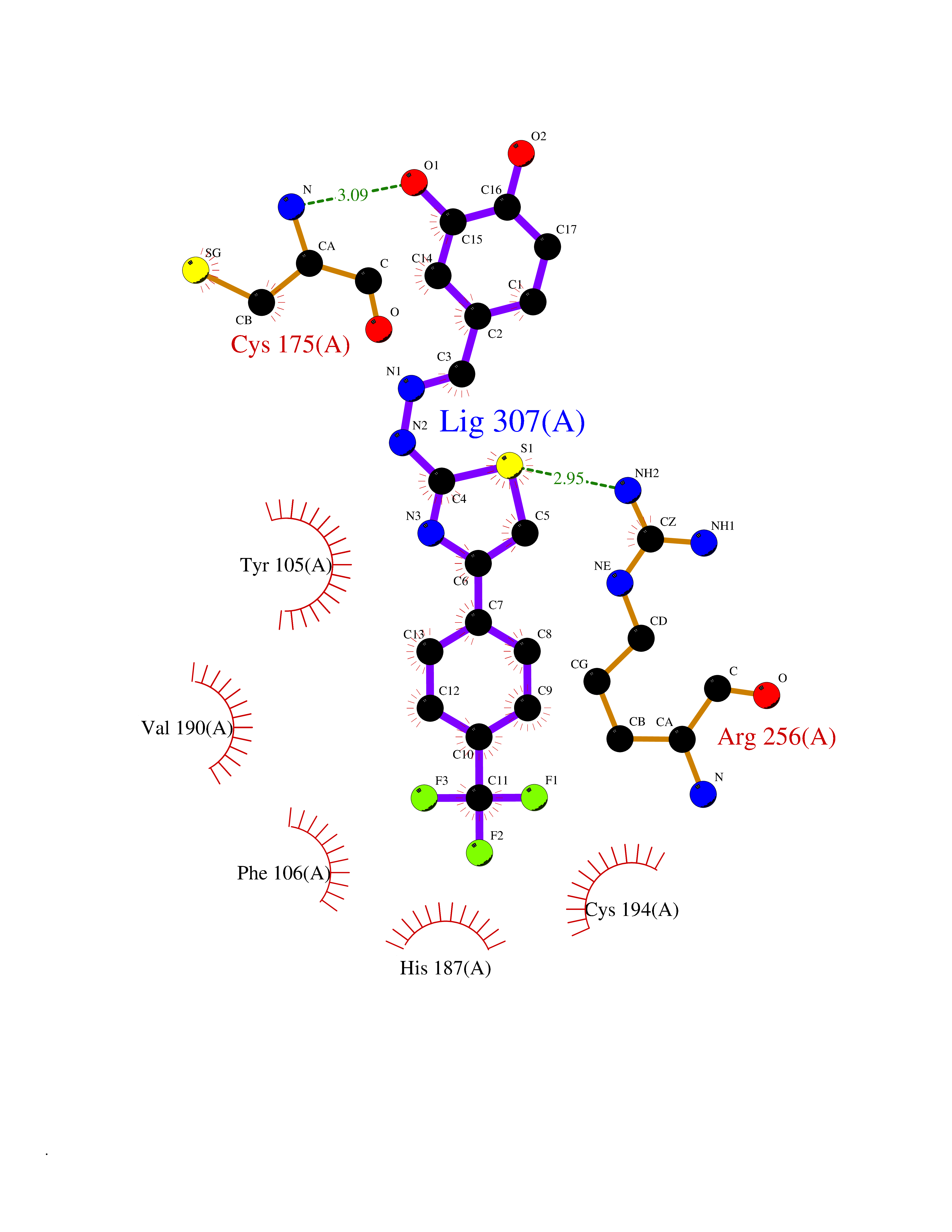 Ligplot Map 3D Binding mode Sequence SLCTRDYKITQVLFPLLYTVLFFVGLITNGLAMRIFFQIRSKSNFIIFLKNTVISDLLMILTFPFKILSDAKLGTGPLRTFVCQVTSVIFYFTMYISISFLGLITIDPKNLLGAKILSVVIWAFMFLLSLPNMILTNRQPRDKNVKKCSFLKSEFGLVWHEIVNYICQVIFWINFLIVVKVFIIIAVFFICFVPFHFARIPYTLSQTRDVFDCTAENTLFYVKESTLWLTSLNACLNPFIYFFLCKSF Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 7.50 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -10.23 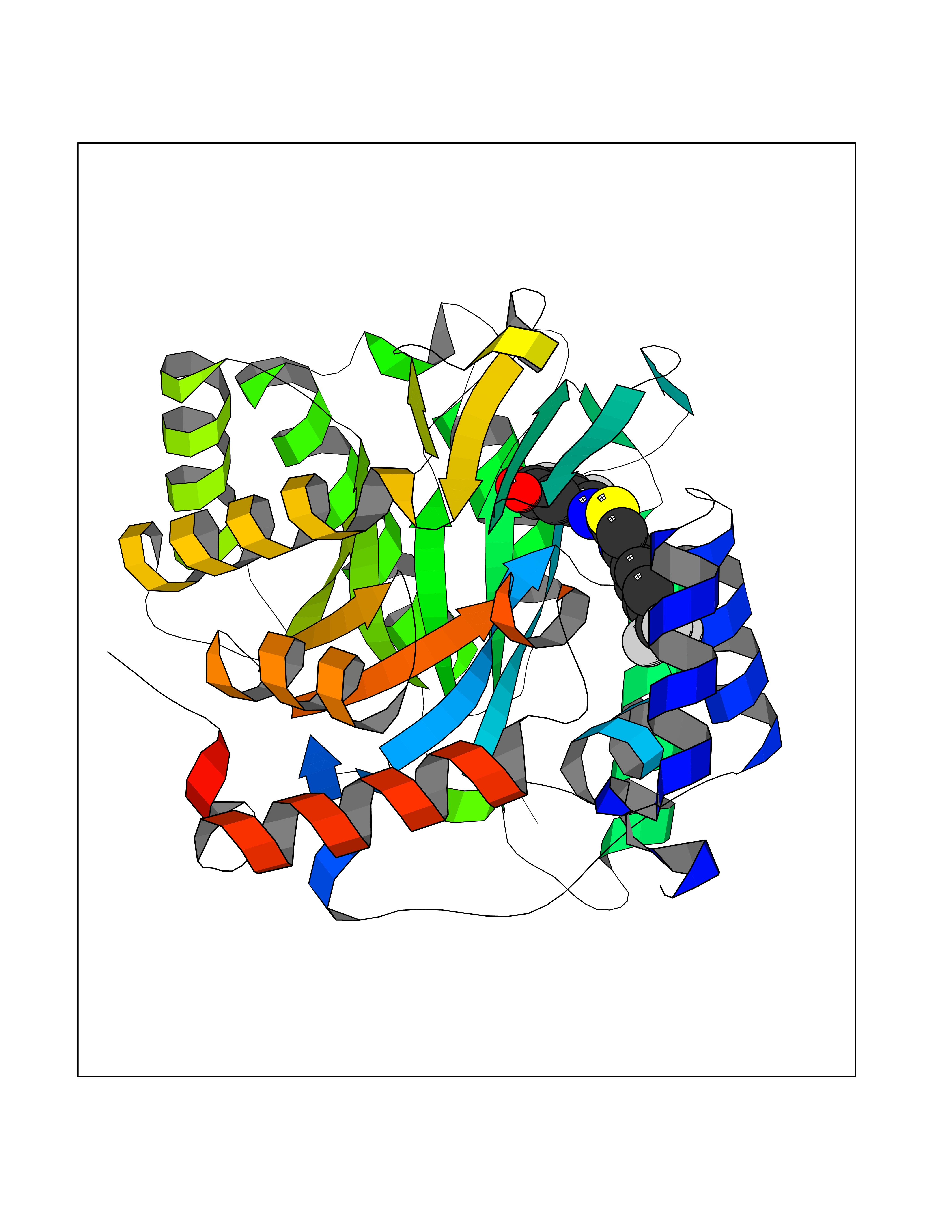 Molscript Map 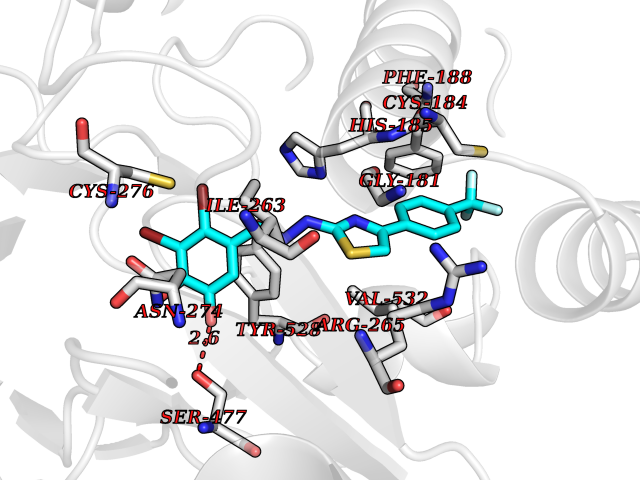 Pymol Map 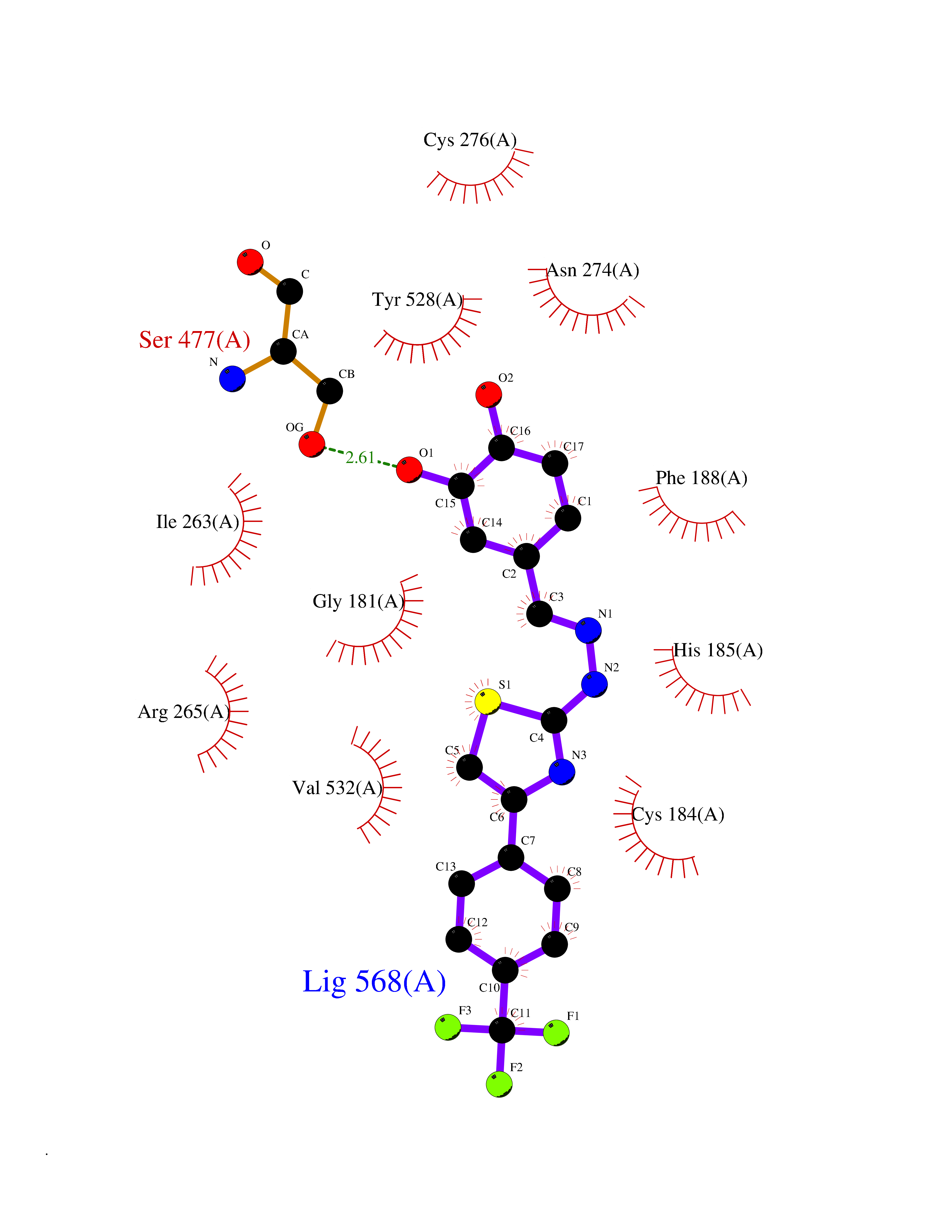 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Pyruvate kinase M2 (PKM) | 3GR4 | 7.49 | |
Target general information Gen name PKM Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p58; Tumor M2-PK; Thyroid hormone-binding protein 1; THBP1; Pyruvate kinase muscle isozyme; Pyruvate kinase isozymes M1/M2; Pyruvate kinase PKM; Pyruvate kinase 2/3; PKM2; PK3; PK2; Opa-interacting pr Protein family Pyruvate kinase family Biochemical class Kinase Function Stimulates POU5F1-mediated transcriptional activation. Plays a general role in caspase independent cell death of tumor cells. The ratio between the highly active tetrameric form and nearly inactive dimeric form determines whether glucose carbons are channeled to biosynthetic processes or used for glycolytic ATP production. The transition between the 2 forms contributes to the control of glycolysis and is important for tumor cell proliferation and survival. Promotes in a STAT1-dependent manner, the expression of the immune checkpoint protein CD274 in ARNTL/BMAL1-deficient macrophages. Glycolytic enzyme that catalyzes the transfer of a phosphoryl group from phosphoenolpyruvate (PEP) to ADP, generating ATP. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07697; DB07692; DB02726; DB07628; DB00787; DB11638; DB09130; DB08951; DB01733; DB11263; DB00119 Interacts with P49407; P32121; Q96IK1-2; P35222; P53355; P22607; P42858; P04049; Q8N488; Q7Z699; Q9BSI4; Q9UMX0; Q9Y649; Q9WMX2; P35222; P53355; Q9H6Z9; P68431; Q16665; P27361 EC number EC 2.7.1.40 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; ATP-binding; Cytoplasm; Direct protein sequencing; Glycolysis; Hydroxylation; Isopeptide bond; Kinase; Magnesium; Metal-binding; Methylation; Nucleotide-binding; Nucleus; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; S-nitrosylation; Transferase; Translation regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 112053 Length 1024 Aromaticity 0.05 Instability index 27.06 Isoelectric point 7.34 Charge (pH=7) 1.66 2D Binding mode Binding energy (Kcal/mol) -10.22 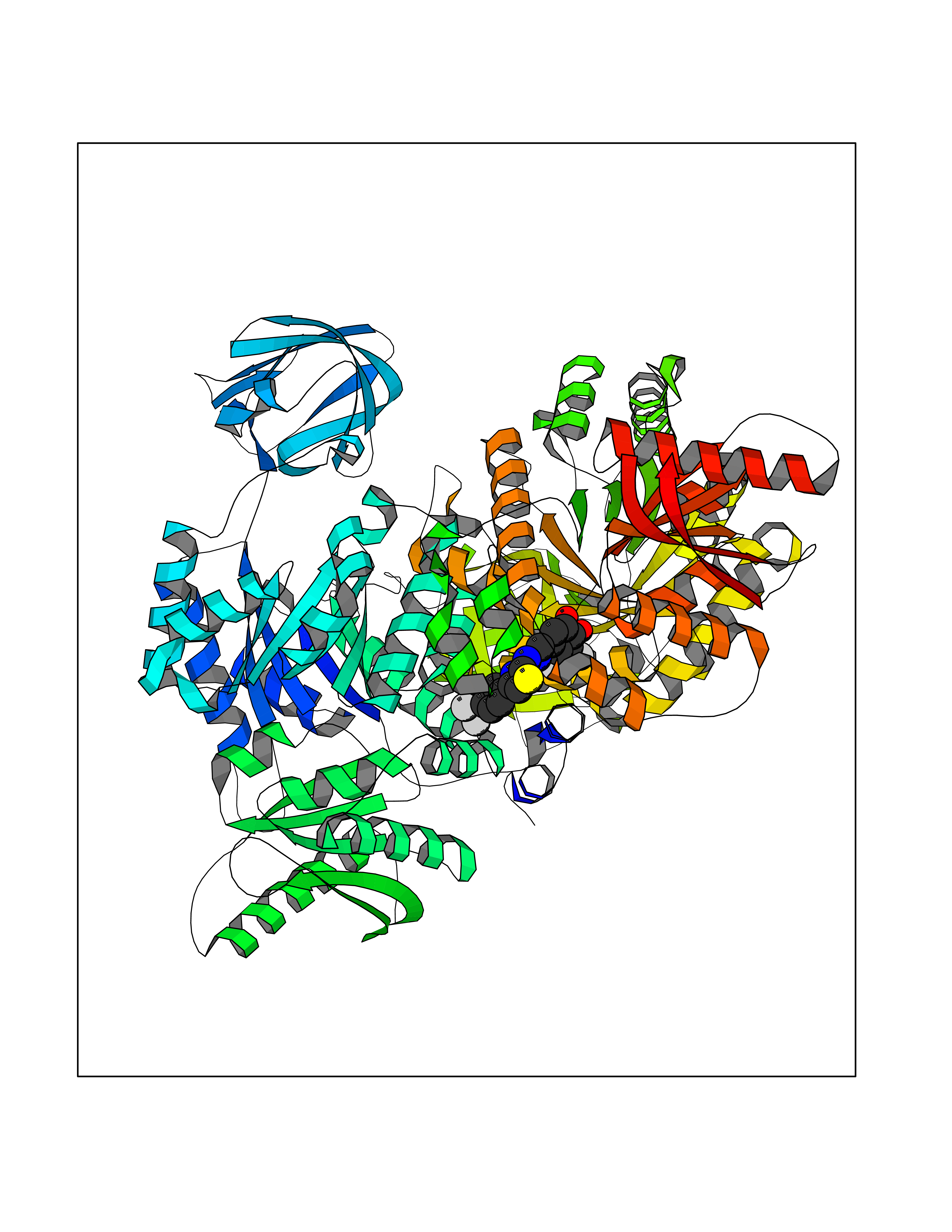 Molscript Map 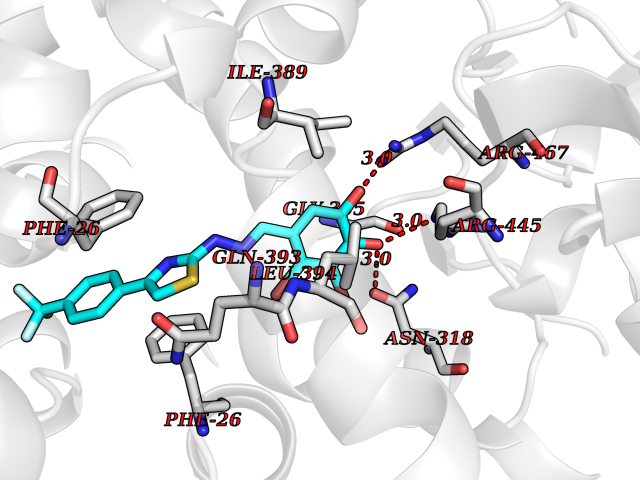 Pymol Map 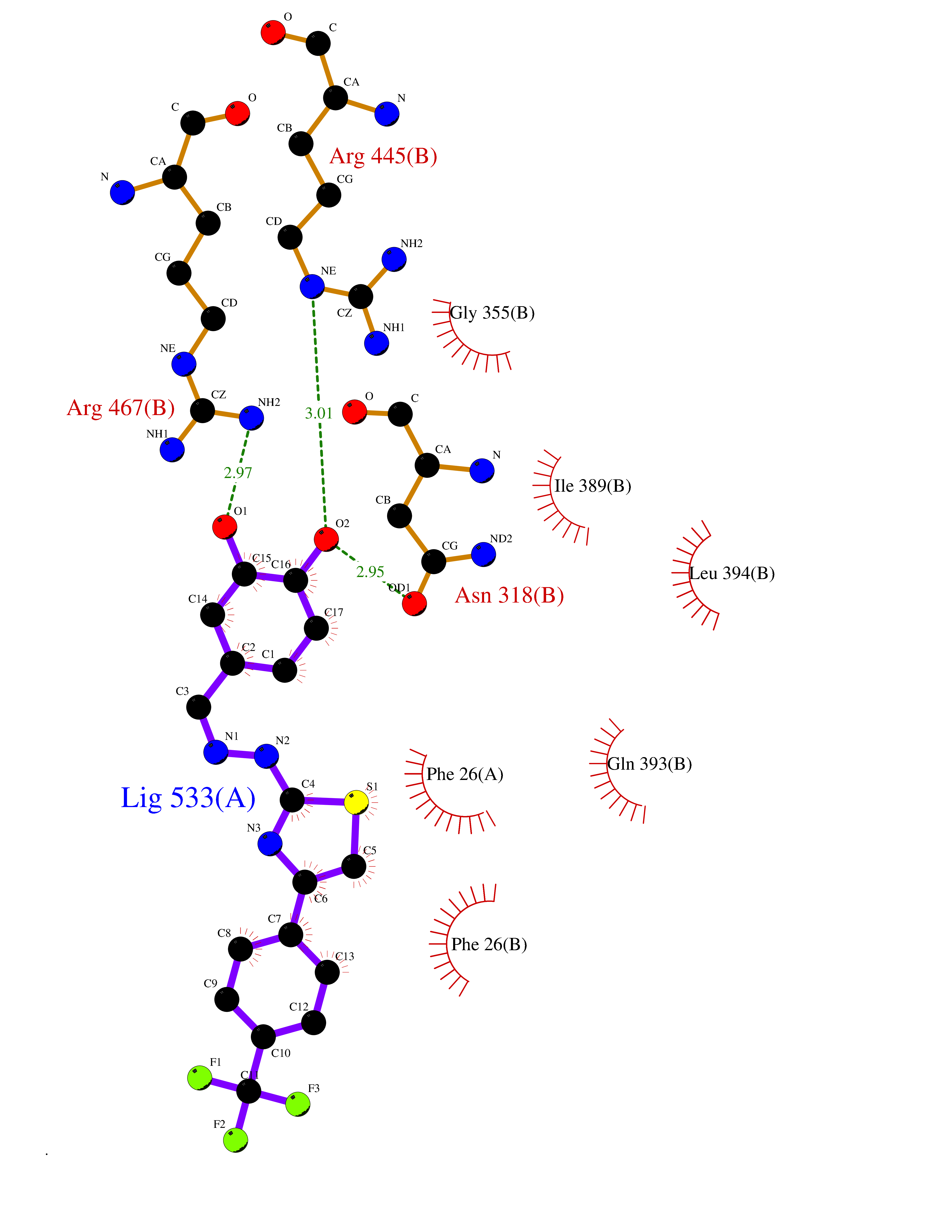 Ligplot Map 3D Binding mode Sequence IQTQQLHAAMADTFLEHMCRLDIDSPPITARNTGIICTIGPASRSVETLKEMIKSGMNVARLNFSHGTHEYHAETIKNVRTATESFASDPILYRPVAVALDTKGPEIRTGLIKGSGTAEVELKKGATLKITLDNAYMEKCDENILWLDYKNICKVVEVGSKIYVDDGLISLQVKQKGADFLVTEVENGGSLGSKKGVNLPGAAVDLPAVSEKDIQDLKFGVEQDVDMVFASFIRKASDVHEVRKVLGEKGKNIKIISKIENHEGVRRFDEILEASDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNRAGKPVICATQMLESMIKKPRPTRAEGSDVANAVLDGADCIMLSGETAKGDYPLEAVRMQHLIAREAEAAIYHLQLFEELRRLAPITSDPTEATAVGAVEASFKCCSGAIIVLTKSGRSAHQVARYRPRAPIIAVTRNPQTARQAHLYRGIFPVLCKDPVQEAWAEDVDLRVNFAMNVGKARGFFKKGDVVIVLTGWRPGSGFTNTMRVVPVPIQTQQLHAAMADTFLEHMCRLDIDSPPITARNTGIICTIGPASRSVETLKEMIKSGMNVARLNFSHGTHEYHAETIKNVRTATESFASDPILYRPVAVALDTKGPEIRTGLIKEVEATLKITLDNAYMEKCDENILWLDYKNICKVVEVGSKIYVDDGLISLQVDFLVTEVENGGSLGSKKGVNLPGAAVDLPAVSEKDIQDLKFGVEQDVDMVFASFIRKASDVHEVRKVLGEKGKNIKIISKIENHEGVRRFDEILEASDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNRAGKPVICATQMLESMIKKPRPTRAEGSDVANAVLDGADCIMLSGETAKGDYPLEAVRMQHLIAREAEAAIYHLQLFEELRRLAPITSDPTEATAVGAVEASFKCCSGAIIVLTKSGRSAHQVARYRPRAPIIAVTRNPQTARQAHLYRGIFPVLCKDPVQEAWAEDVDLRVNFAMNVGKARGFFKKGDVVIVLTGWRPGSGFTNTMRVVPVP Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Camphor 5-monooxygenase | 4L4E | 7.48 | |
Target general information Gen name camC Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID NA Synonyms cyp101 Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Camphor 5-monooxygenase activity.Heme binding.Iron ion binding. Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03836; DB02617; DB02817; DB03627; DB04032; DB03031; DB02125; DB04501; DB01744; DB01663; DB01011; DB01703; DB01826; DB03540; DB02851 Interacts with P00259 EC number 1.14.15.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Heme; Iron; Metal-binding; Monooxygenase; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 45446.3 Length 405 Aromaticity 0.08 Instability index 45.33 Isoelectric point 5.23 Charge (pH=7) -16.08 2D Binding mode Binding energy (Kcal/mol) -10.21 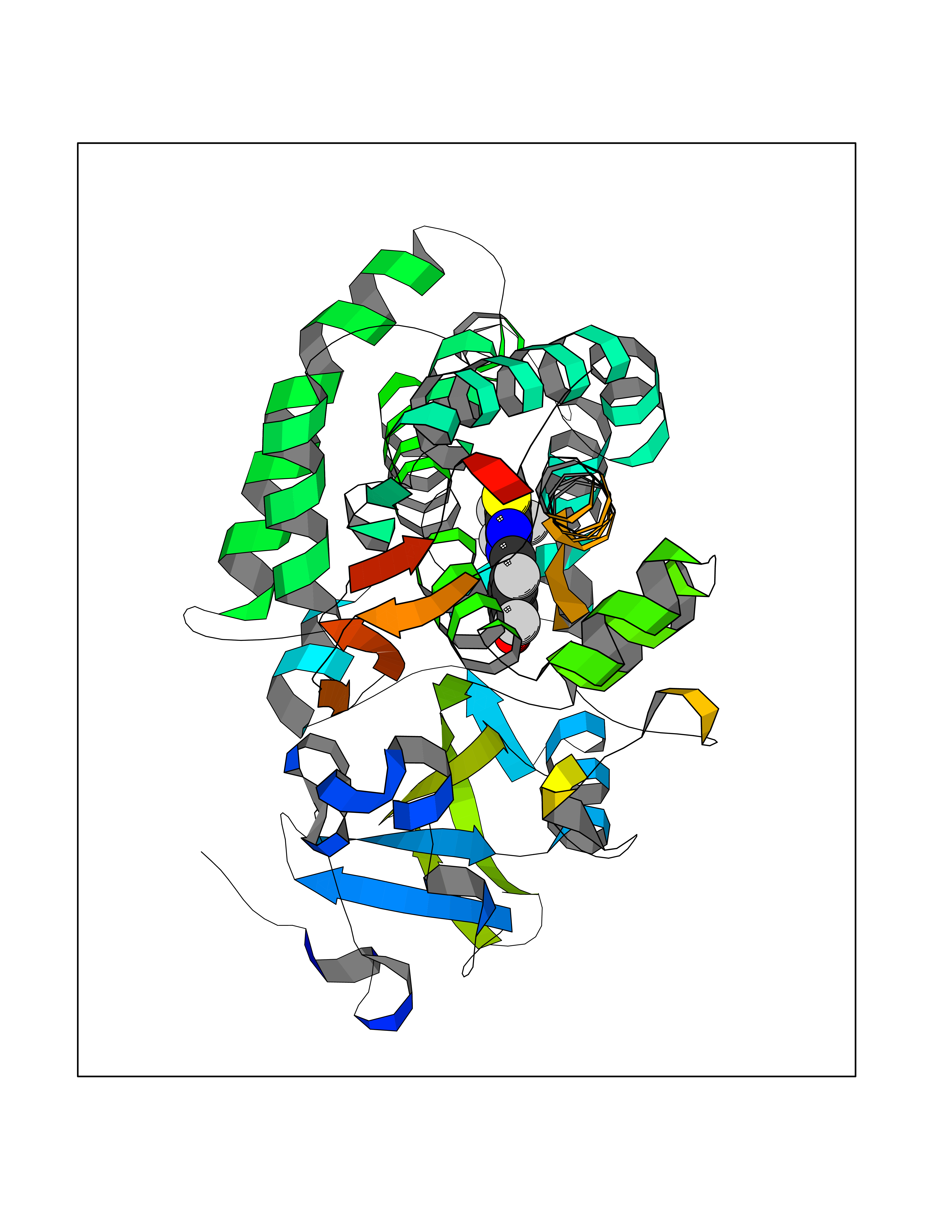 Molscript Map 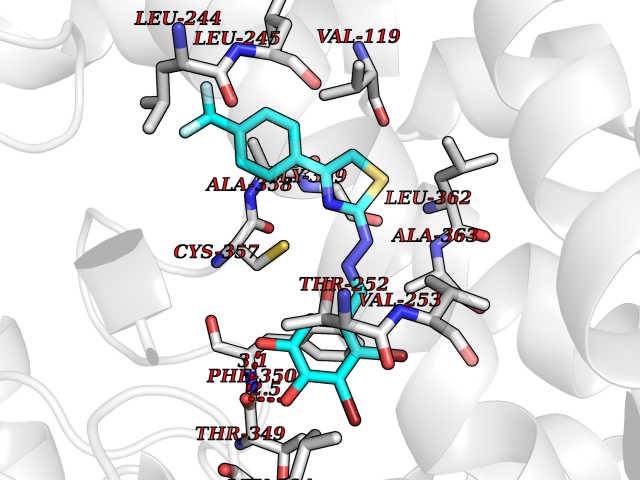 Pymol Map 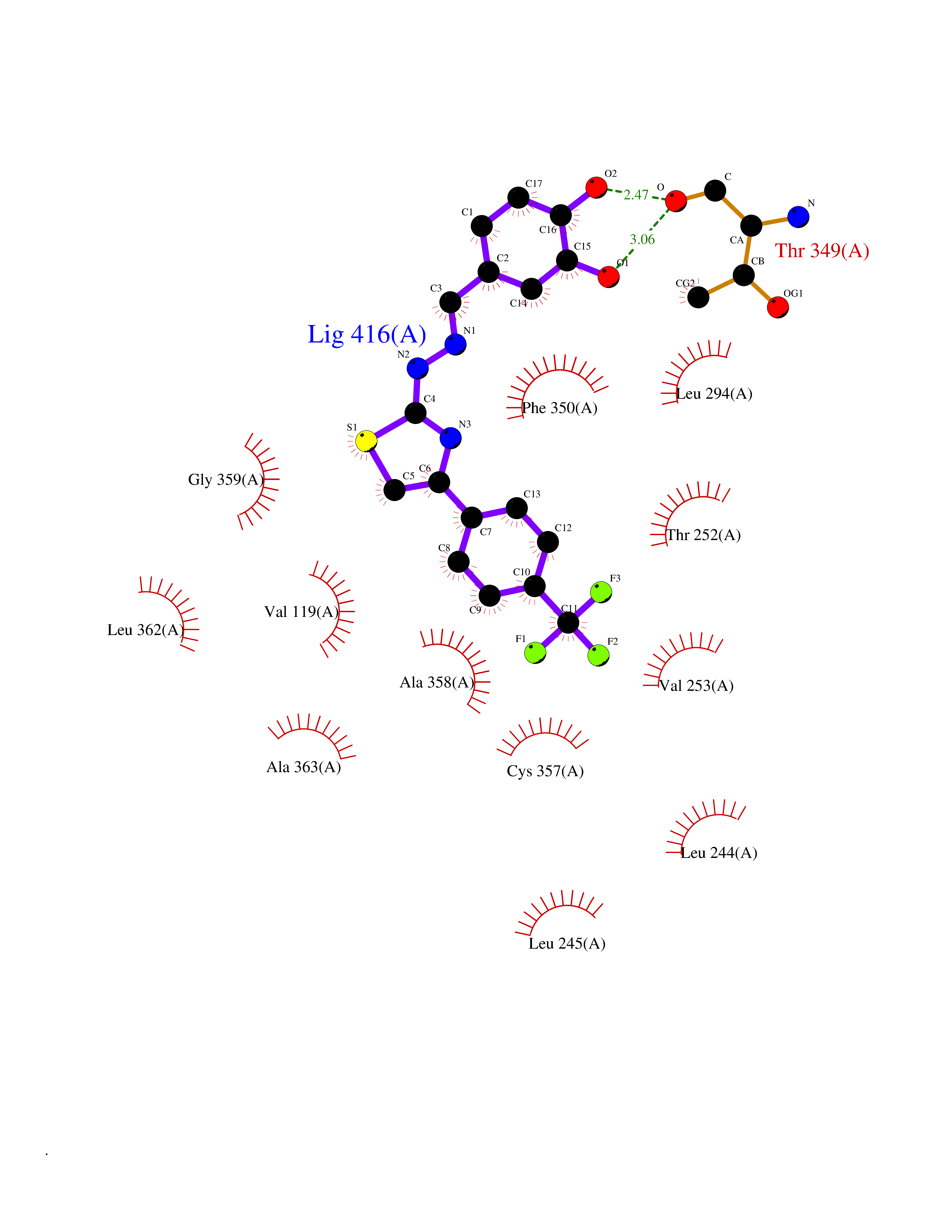 Ligplot Map 3D Binding mode Sequence NLAPLPPHVPEHLVFDFDMYNPSNLSAGVQEAWAVLQESNVPDLVWTRCNGGHWIATRGQLIREAYEDYRHFSSECPFIPREAGEAYDFIPTSMDPPEQRQFRALANQVVGMPVVDKLENRIQELACSLIESLRPQGQCNFTEDYAEPFPIRIFMLLAGLPEEDIPHLGYLTDQMTRPDGSMTFAEAKEALYDYLIPIIEQRRQKPGTDAISIVANGQVNGRPITSDEAKRMCGLLLVGGLDTVVNFLSFSMEFLAKSPEHRQELIERPERIPAACEELLRRFSLVADGRILTSDYEFHGVQLKKGDQILLPQMLSGLDERENAAPMHVDFSRQKVSHTTFGHGSHLCAGQHLARREIIVTLKEWLTRIPDFSIAPGAQIQHKSGIVSGVQALPLVWDPATTKAV Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | LIM domain kinase-2 (LIMK-2) | 7QHG | 7.48 | |
Target general information Gen name LIMK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LIMK-2; LIM domain kinase 2 Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family Biochemical class Kinase Function Displays serine/threonine-specific phosphorylation of myelin basic protein and histone (MBP) in vitro. Related diseases Intellectual developmental disorder, autosomal recessive 80, with variant lissencephaly (MRT80) [MIM:620653]: An autosomal recessive disorder characterized by global developmental delay, mildly to moderately impaired intellectual development, attention deficit-hyperactivity disorder, hypotonia, seizure, poor social skills, and autistic traits. Brain imaging shows fronto-temporal lissencephaly and pachygyria. {ECO:0000269|PubMed:37880421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11718; DB12010 Interacts with Q16543; P08238; Q96C90; P62258 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Cytoskeleton; Kinase; LIM domain; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Serine/threonine-protein kinase; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 32109.2 Length 283 Aromaticity 0.1 Instability index 27.28 Isoelectric point 6.01 Charge (pH=7) -3.93 2D Binding mode Binding energy (Kcal/mol) -10.2 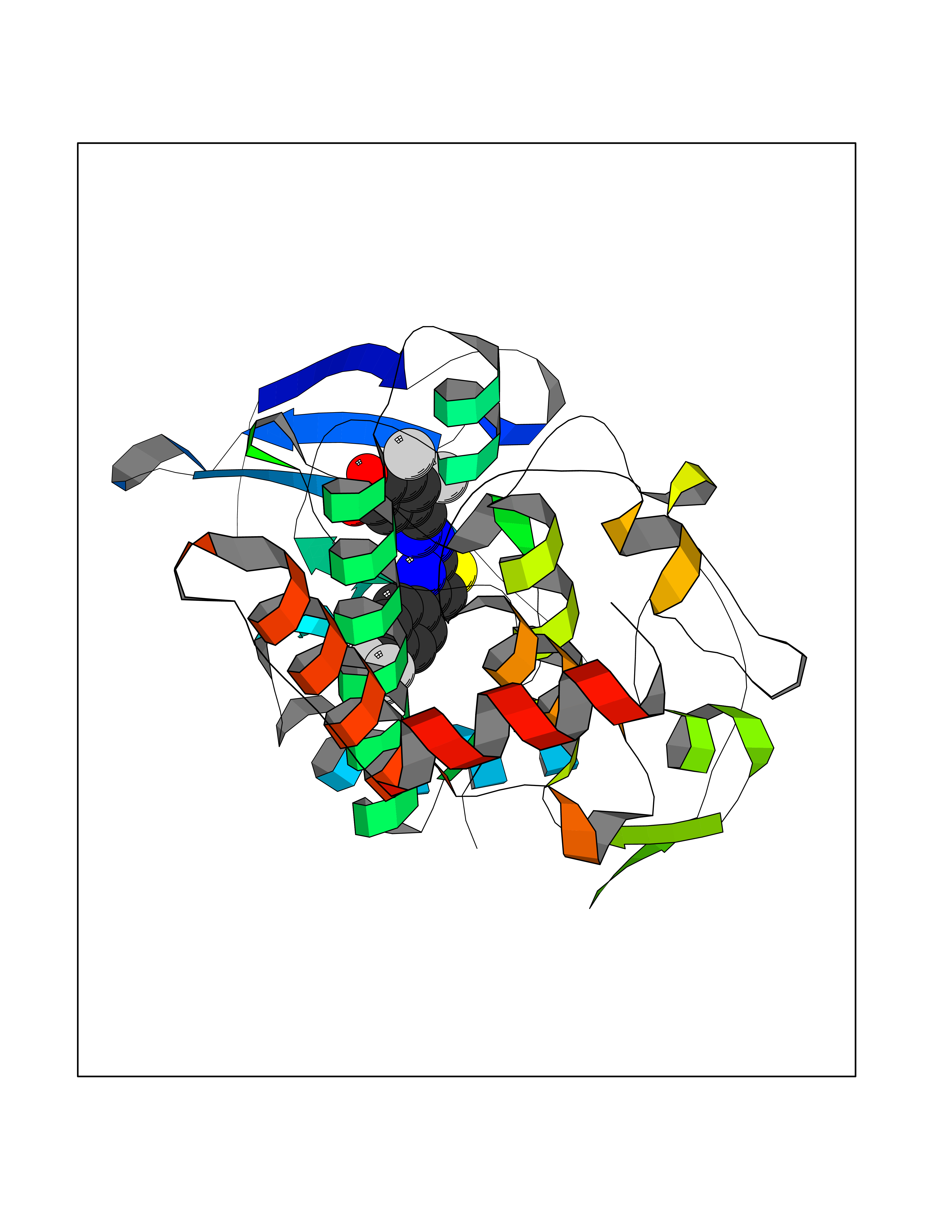 Molscript Map 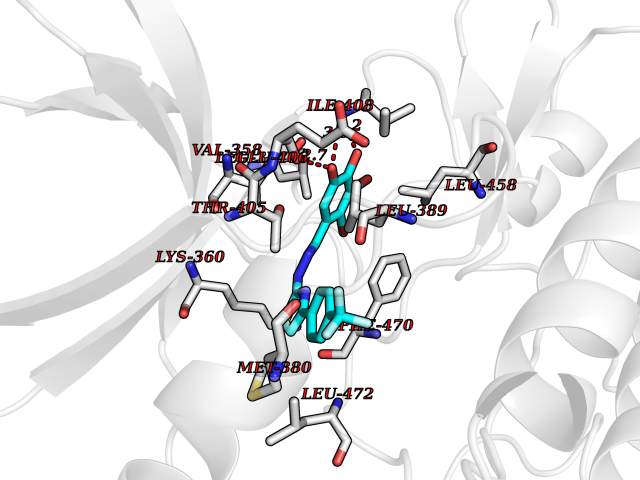 Pymol Map 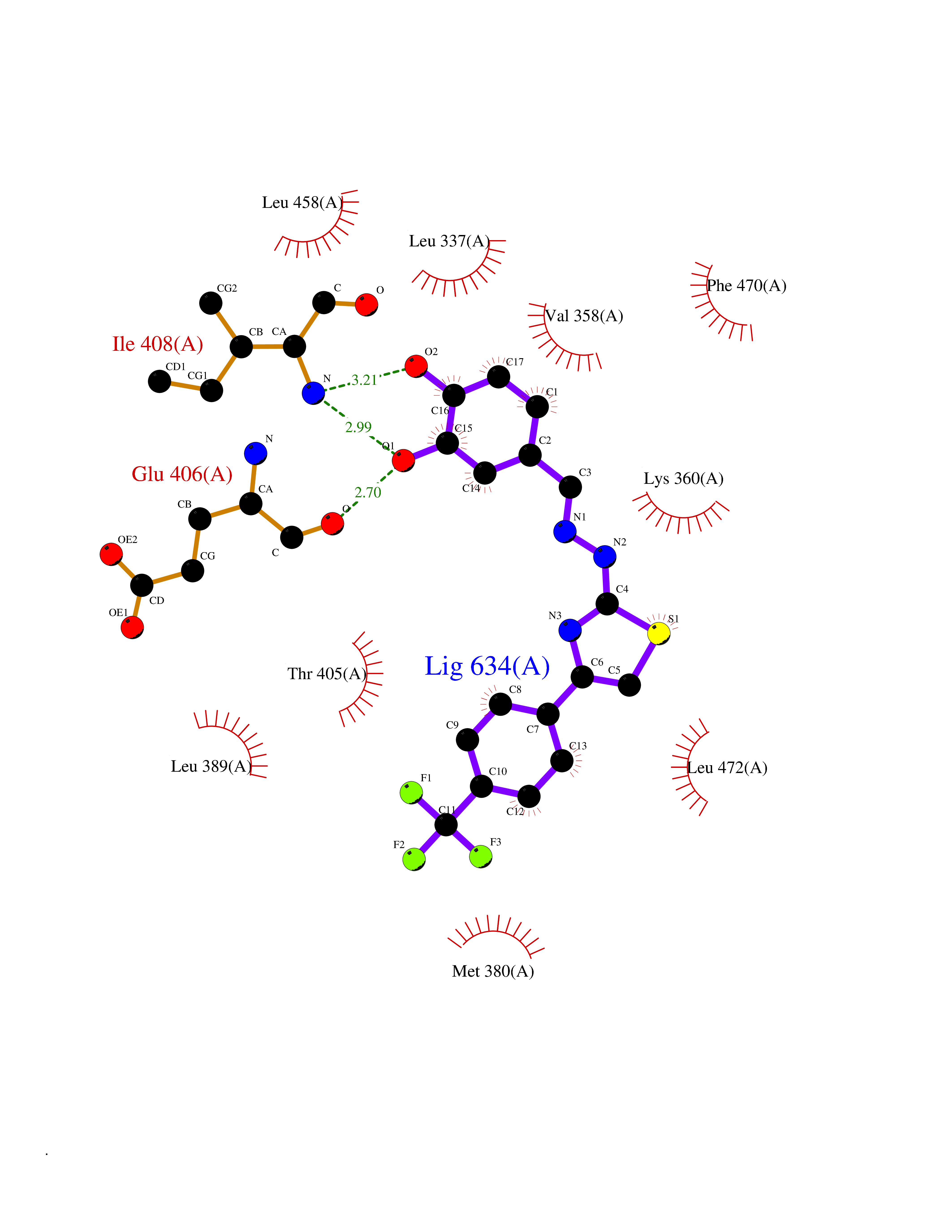 Ligplot Map 3D Binding mode Sequence MDLIHGEVLGKGFFGQAIKVTHKATGKVMVMKELIRCDEETQKTFLTEVKVMRSLDHPNVLKFIGVLYKDKKLNLLTEYIEGGTLKDFLRSMDPFPWQQKVRFAKGIASGMAYLHSMCIIHRDLNSHNCLIKLDKTVVVADFGLSRLIVDRKKRYTVVGNPYWMAPEMLNGKSYDETVDIFSFGIVLCEIIGQVYADPDCLPRTLDFGLNVKLFWEKFVPTDCPPAFFPLAAICCRLEPESRPAFSKLEDSFEALSLYLGELGIPLPAELEELDHTVSMQYGL Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Vitamin K epoxide reductase complex 1 (VKORC1) | 6WV3 | 7.48 | |
Target general information Gen name VKORC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin K1 2,3-epoxide reductase subunit 1; VKORC1; VKOR; UNQ308/PRO351; MSTP576; MSTP134 Protein family VKOR family Biochemical class Short-chain dehydrogenases reductase Function Involved invitamin K metabolism. Catalytic subunit of the vitamin K epoxide reductase (VKOR) complex which reduces inactive vitamin K 2,3-epoxide to active vitamin K. Vitamin K is required for the gamma-carboxylation of various proteins, including clotting factors, and is required for normal blood coagulation, but also for normal bone development. Related diseases Combined deficiency of vitamin K-dependent clotting factors 2 (VKCFD2) [MIM:607473]: VKCFD leads to a bleeding tendency that is usually reversed by oral administration of vitamin K. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:16270630}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Coumarin resistance (CMRES) [MIM:122700]: A condition characterized by partial or complete resistance to warfarin or other 4-hydroxycoumarin derivatives. These drugs are used as anti-coagulants for the prevention of thromboembolic diseases in subjects with deep vein thrombosis, atrial fibrillation, or mechanical heart valve replacement. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:20946155}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01418; DB00266; DB09332; DB00170; DB00498; DB00946; DB01022; DB00682 Interacts with Q13323; Q7Z7G2; Q96BA8; Q9Y282; Q5JX71; Q96KR6; Q5T7V8; Q8TDT2; Q9NQG1; P15941-11; Q96TC7; Q9NR31; A0A0S2Z4U3; Q8TBB6; O15393-2; Q19QW4 EC number EC 1.17.4.4 Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Disulfide bond; Endoplasmic reticulum; Membrane; Oxidoreductase; Proteomics identification; Quinone; Redox-active center; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 42656.4 Length 381 Aromaticity 0.1 Instability index 32.12 Isoelectric point 7.73 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -10.21  Molscript Map 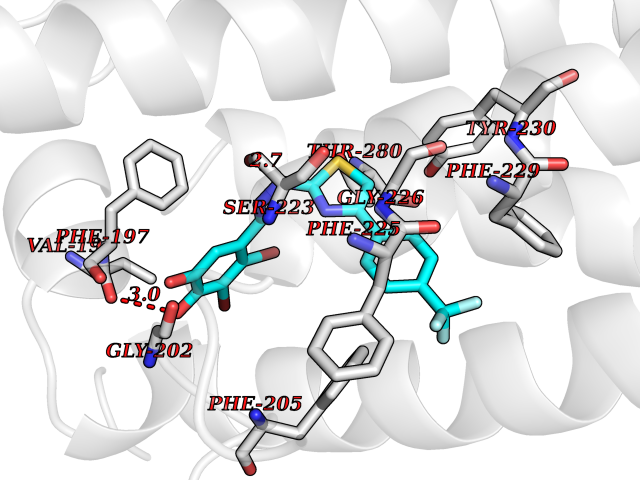 Pymol Map 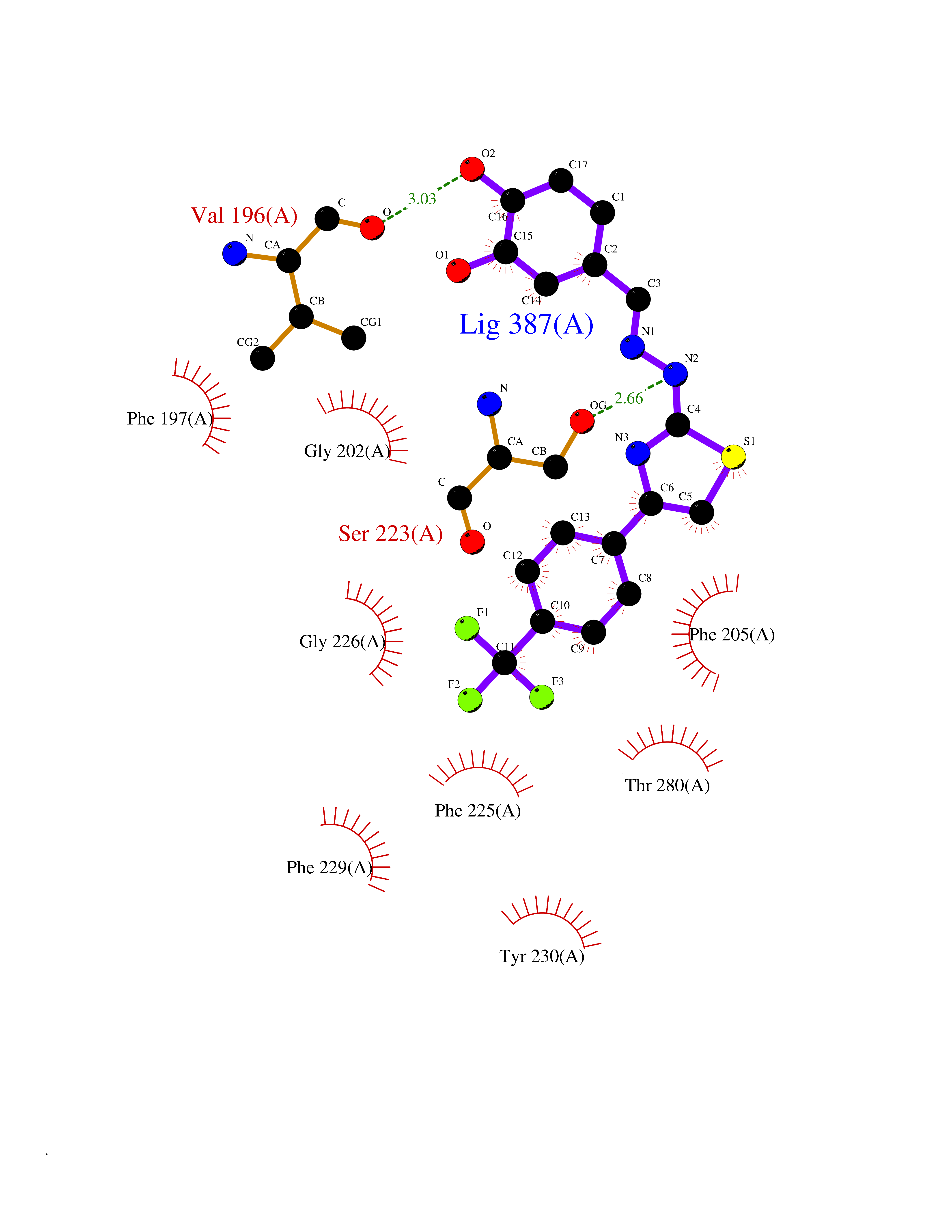 Ligplot Map 3D Binding mode Sequence KGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLXVQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNSTWGSPGWVRLALCLTGLVLSLYALHVKAARARDRDYRALCDVGTAISCSRVFSSRWGRGFGLVEHVLGQDSILNQSNSIFGCIFYTLQLLLGCLRTRWASVLMLLSSLVSLAGSVYLAWILFFVLYDFCIVCITTYAINVSLMWLSFRKVQENSHNVYITADKQKNGIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHHH Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Sphingosine kinase 1 (SPHK1) | 3VZB | 7.48 | |
Target general information Gen name SPHK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SPK 1; SPK; SPHK1; SK 1; Acetyltransferase SPHK1 Protein family NA Biochemical class Kinase Function Acts on D-erythro-sphingosine and to a lesser extent sphinganine, but not other lipids, such as D,L-threo-dihydrosphingosine, N,N-dimethylsphingosine, diacylglycerol, ceramide, or phosphatidylinositol. In contrast to proapoptotic SPHK2, has a negative effect on intracellular ceramide levels, enhances cell growth and inhibits apoptosis. Involved in the regulation of inflammatory response and neuroinflammation. Via the product sphingosine 1-phosphate, stimulates TRAF2 E3 ubiquitin ligase activity, and promotes activation of NF-kappa-B in response to TNF signaling leading to IL17 secretion. In response to TNF and in parallel to NF-kappa-B activation, negatively regulates RANTES inducion through p38 MAPK signaling pathway. Involved in endocytic membrane trafficking induced by sphingosine, recruited to dilate endosomes, also plays a role on later stages of endosomal maturation and membrane fusion independently of its kinase activity. In Purkinje cells, seems to be also involved in the regulation of autophagosome-lysosome fusion upon VEGFA. Catalyzes the phosphorylation of sphingosine to form sphingosine 1-phosphate (SPP), a lipid mediator with both intra- and extracellular functions. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08868 Interacts with P07858; P68104; Q14192; Q2M3C7; Q9Y4K3; P13473-2; Q9Y371 EC number EC 2.7.1.91 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Calmodulin-binding; Cell membrane; Coated pit; Cytoplasm; Endosome; Kinase; Lipid metabolism; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Synapse; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 39813 Length 360 Aromaticity 0.08 Instability index 43.79 Isoelectric point 7.34 Charge (pH=7) 0.84 2D Binding mode Binding energy (Kcal/mol) -10.21 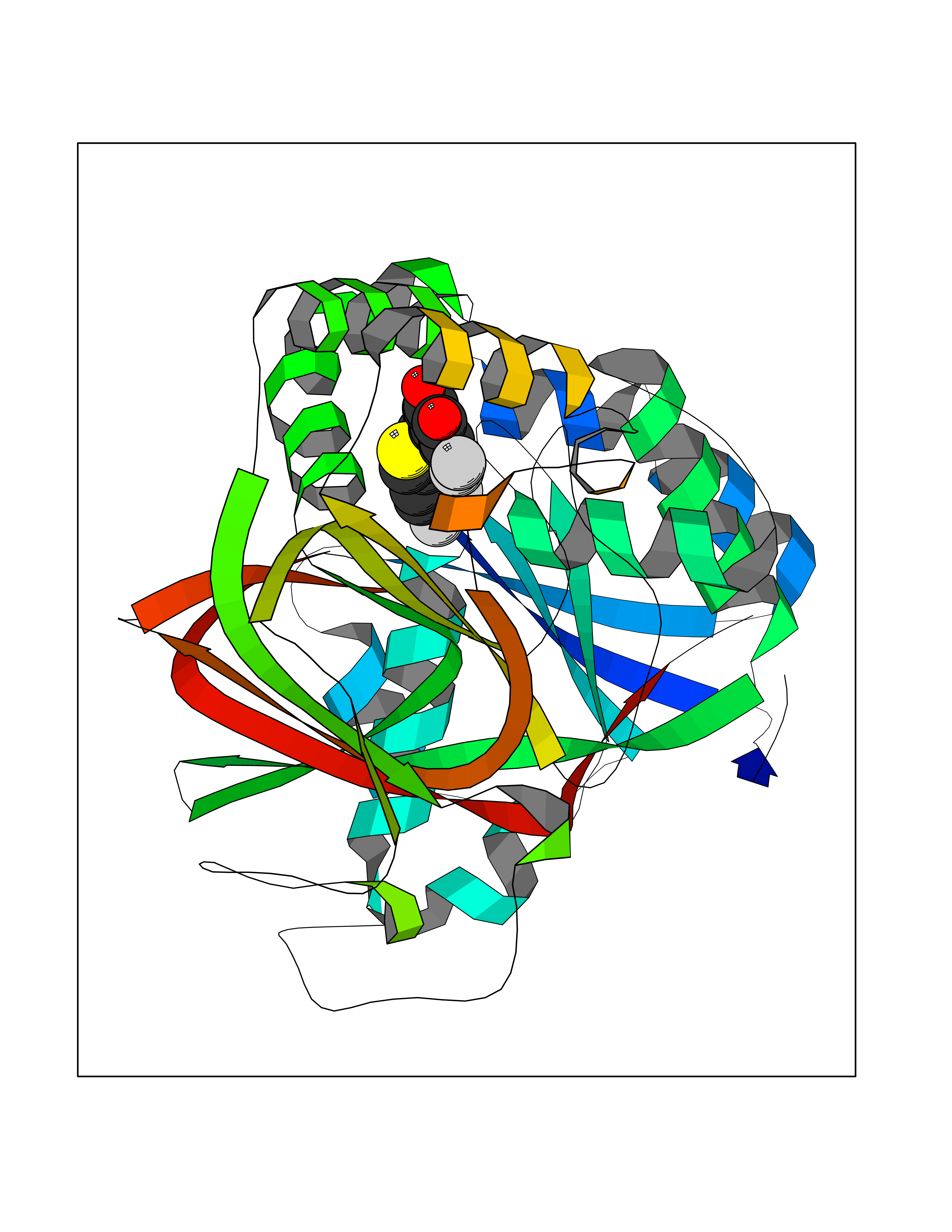 Molscript Map 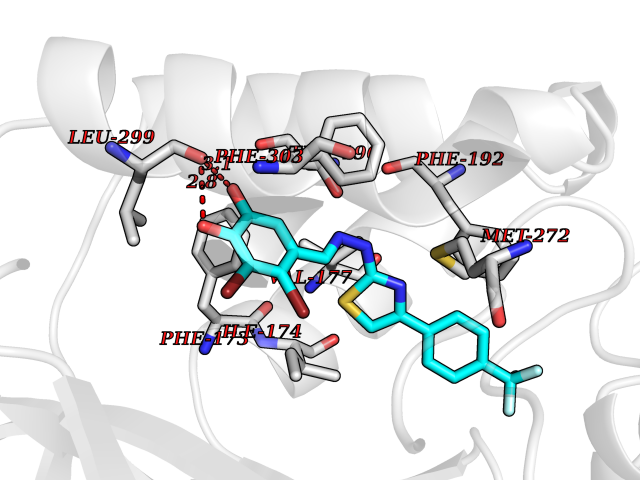 Pymol Map 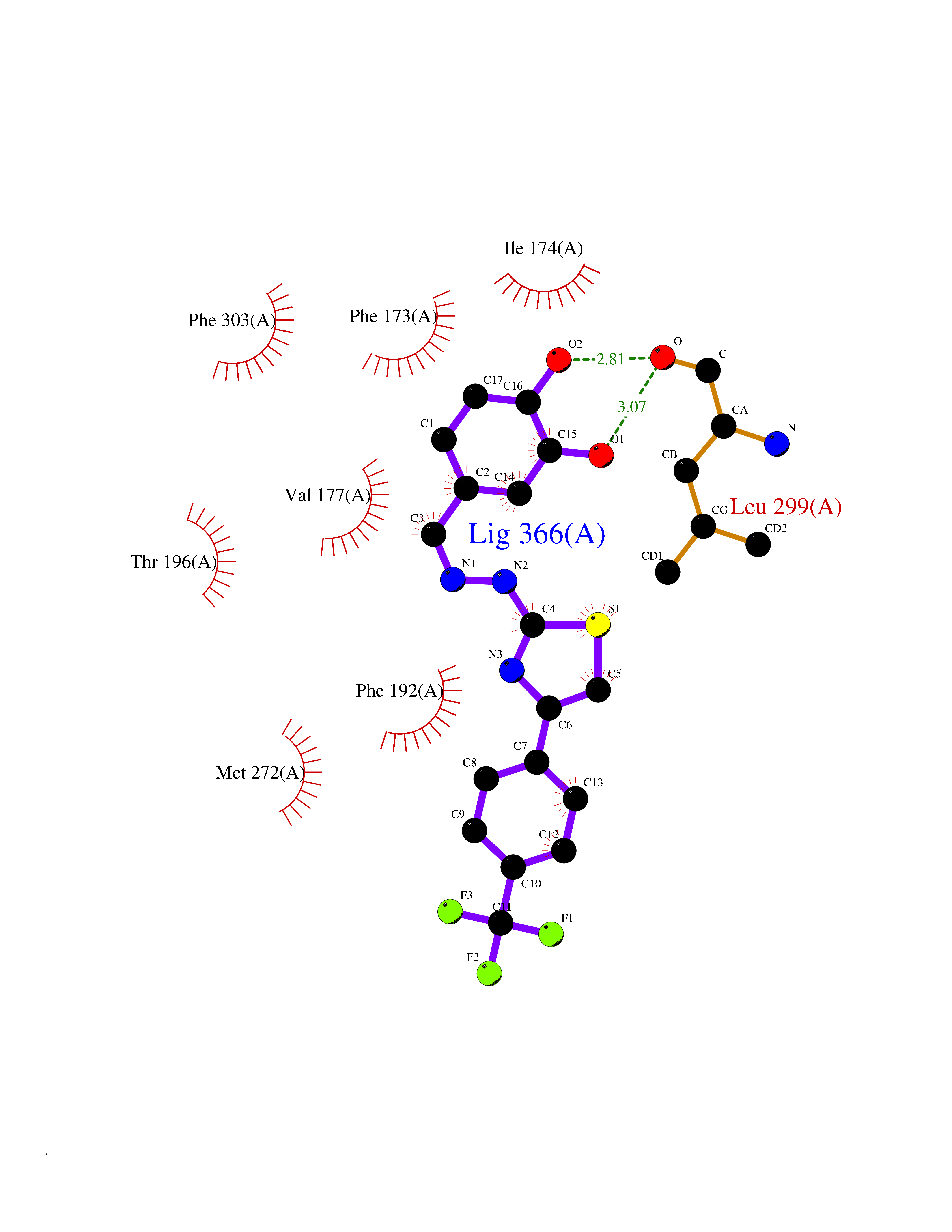 Ligplot Map 3D Binding mode Sequence AMGSGVLPRPCRVLVLLNPRGGKGKALQLFRSHVQPLLAEAEISFTLMLTERRNHARELVRSEELGRWDALVVMSGDGLMHEVVNGLMERPDWETAIQKPLCSLPAGSGNALAASLNHYAGYEQVTNEDLLTNCTLLLCRRLLSPMNLLSLHTASGLRLFSVLSLAWGFIADVDLESEKYRRLGEMRFTLGTFLRLAALRTYRGRLAYLPVGRVGSKTPASPVVVQQGPVDAHLVPLEEPVPSHWTVVPDEDFVLVLALLHSHLGSEMFAAPMGRCAAGVMHLFYVRAGVSRAMLLRLFLAMEKGRHMEYECPYLVYVPVVAFRLEPKDGKGVFAVDGELMVSEAVQGQVHPNYFWMVSG Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Glycine oxidase | 1RYI | 7.47 | |
Target general information Gen name thiO Organism Bacillus subtilis (strain 168) Uniprot ID TTD ID NA Synonyms goxB;yjbR;BSU11670 Protein family DAO family, ThiO subfamily Biochemical class Oxidoreductase Function FAD binding.Glycine oxidase activity.Oxidoreductase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB02713; DB03147 Interacts with NA EC number 1.4.3.19 Uniprot keywords 3D-structure; Cytoplasm; FAD; Flavoprotein; Herbicide resistance; Oxidoreductase; Reference proteome; Thiamine biosynthesis Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 40395.8 Length 364 Aromaticity 0.1 Instability index 36.07 Isoelectric point 6.02 Charge (pH=7) -6.35 2D Binding mode Binding energy (Kcal/mol) -10.19  Molscript Map 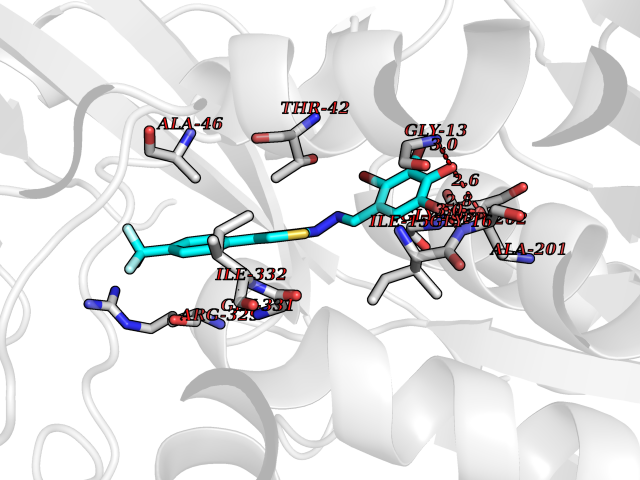 Pymol Map 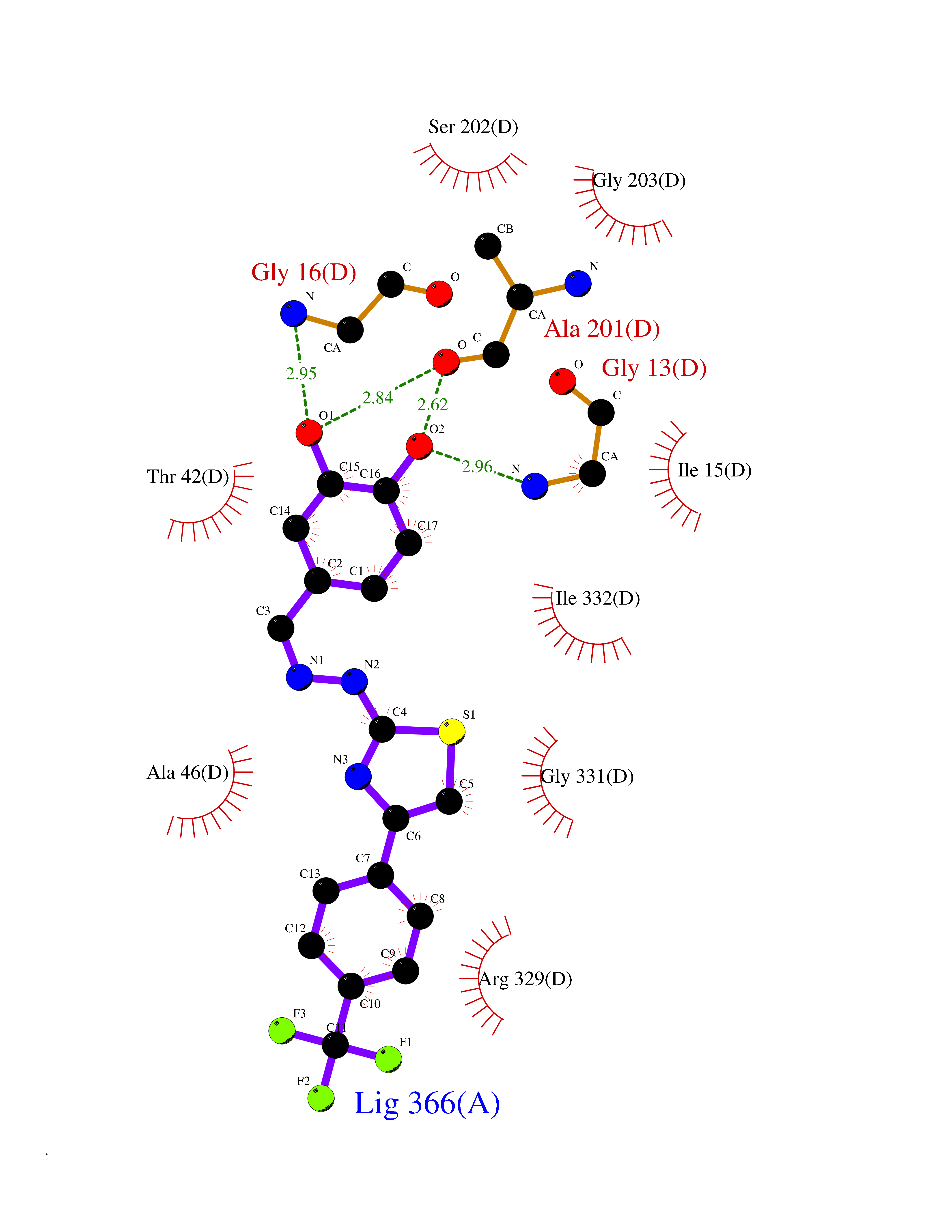 Ligplot Map 3D Binding mode Sequence MKRHYEAVVIGGGIIGSAIAYYLAKENKNTALFESGTMGGRTTSAAAGMLGAHAECEERDAFFDFAMHSQRLYKGLGEELYALSGVDIRQHNGGMFKLAFSEEDVLQLRQMDDLDSVSWYSKEEVLEKEPYASGDIFGASFIQDDVHVEPYFVCKAYVKAAKMLGAEIFEHTPVLHVERDGEALFIKTPSGDVWANHVVVASGVWSGMFFKQLGLNNAFLPVKGECLSVWNDDIPLTKTLYHDHCYIVPRKSGRLVVGATMKPGDWSETPDLGGLESVMKKAKTMLPAIQNMKVDRFWAGLRPGTKDGKPYIGRHPEDSRILFAAGHFRNGILLAPATGALISDLIMNKEVNQDWLHAFRIDRK Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Matrix metalloproteinase-10 (MMP-10) | 1Q3A | 7.47 | |
Target general information Gen name MMP10 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Transin-2; Stromelysin-2; STMY2; SL-2 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates procollagenase. Can degrade fibronectin, gelatins of type I, III, IV, and V; weakly collagens III, IV, and V. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00786; DB08271 Interacts with NA EC number EC 3.4.24.22 Uniprot keywords 3D-structure; Calcium; Collagen degradation; Disulfide bond; Extracellular matrix; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 52822 Length 471 Aromaticity 0.12 Instability index 21.13 Isoelectric point 4.83 Charge (pH=7) -35.32 2D Binding mode Binding energy (Kcal/mol) -10.19  Molscript Map 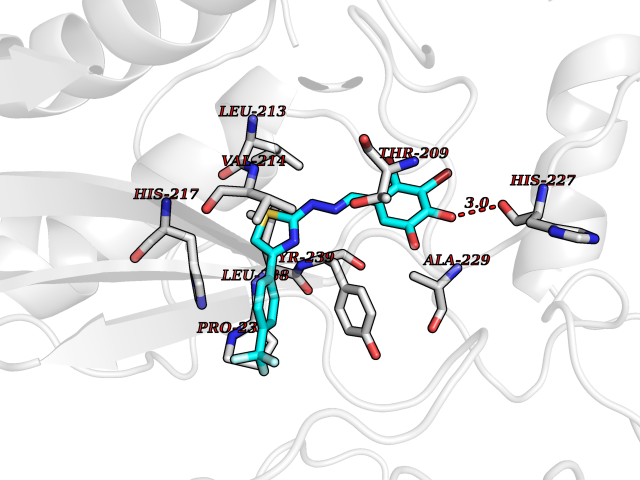 Pymol Map 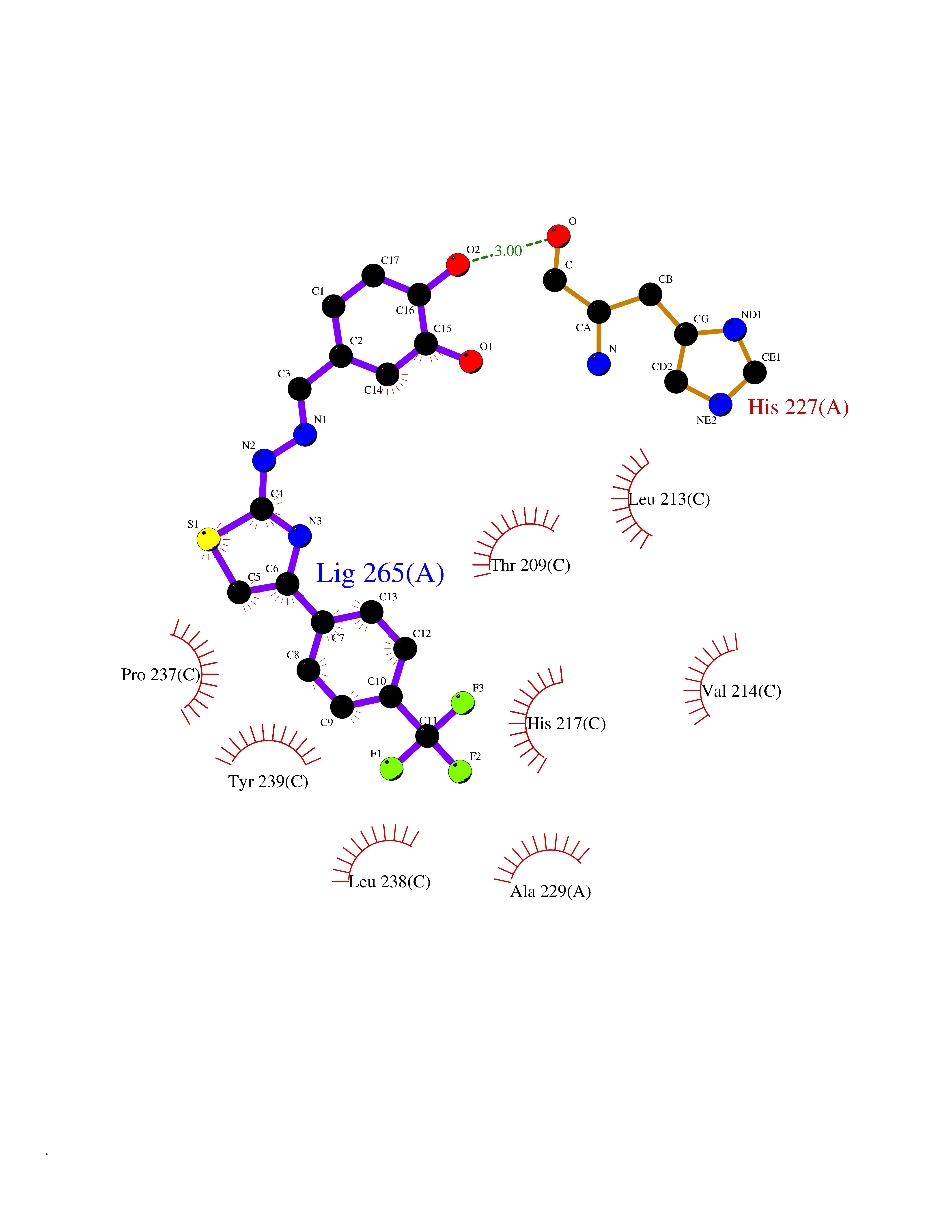 Ligplot Map 3D Binding mode Sequence MPKWRKTHLTYRIVNYTPDLPRDAVDSAIEKALKVWEEVTPLTFSRLYEGEADIMISFAVKEHGDNYSFDGPGHSLAHAYPPGPGLYGDIHFDDDEKWTEDASGTNLFLVAAHELGHSLGLFHSANTEALMYPLYNSLAQFRLSQDDVNGIQSLYGPKWRKTHLTYRIVNYTPDLPRDAVDSAIEKALKVWEEVTPLTFSRLYEGEADIMISFAVKEHGDNYSFDGPGHSLAHAYPPGPGLYGDIHFDDDEKWTEDASGTNLFLVAAHELGHSLGLFHSANTEALMYPLYNSLAQFRLSQDDVNGIQSLYGGMPKWRKTHLTYRIVNYTPDLPRDAVDSAIEKALKVWEEVTPLTFSRLYEGEADIMISFAVKEHGDNYSFDGPGHSLAHAYPPGPGLYGDIHFDDDEKWTEDASGTNLFLVAAHELGHSLGLFHSANTEALMYPLYNSFTELAQFRLSQDDVNGIQSLYG Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Oxalosuccinate decarboxylase (IDH1) | 6ADG | 7.47 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD; NADP(+)-specific ICDH; Isocitrate dehydrogenase [NADP] cytoplasmic; IDP; IDH; Cytosolic NADP-isocitrate dehydrogenase Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -10.19 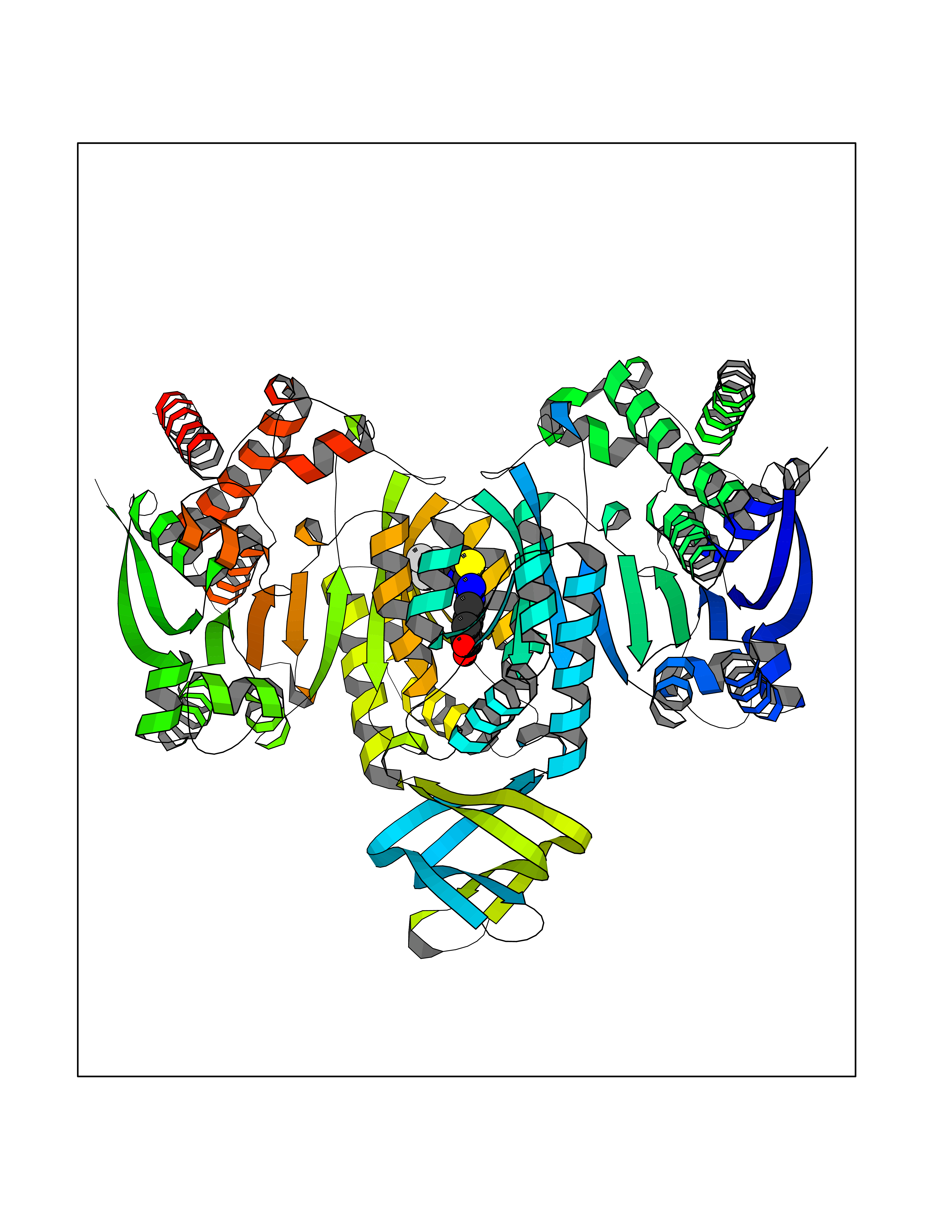 Molscript Map 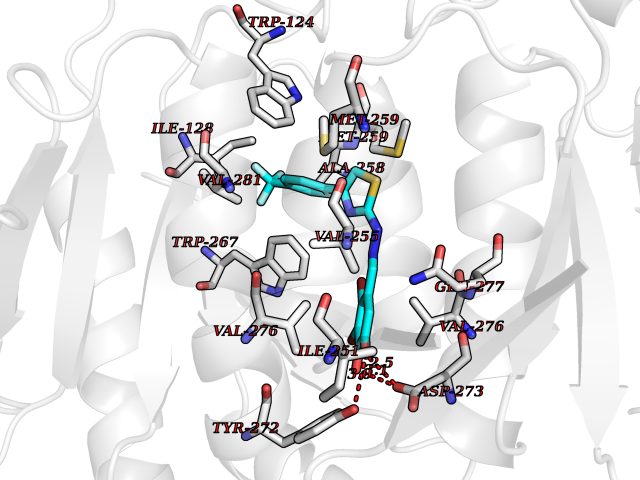 Pymol Map 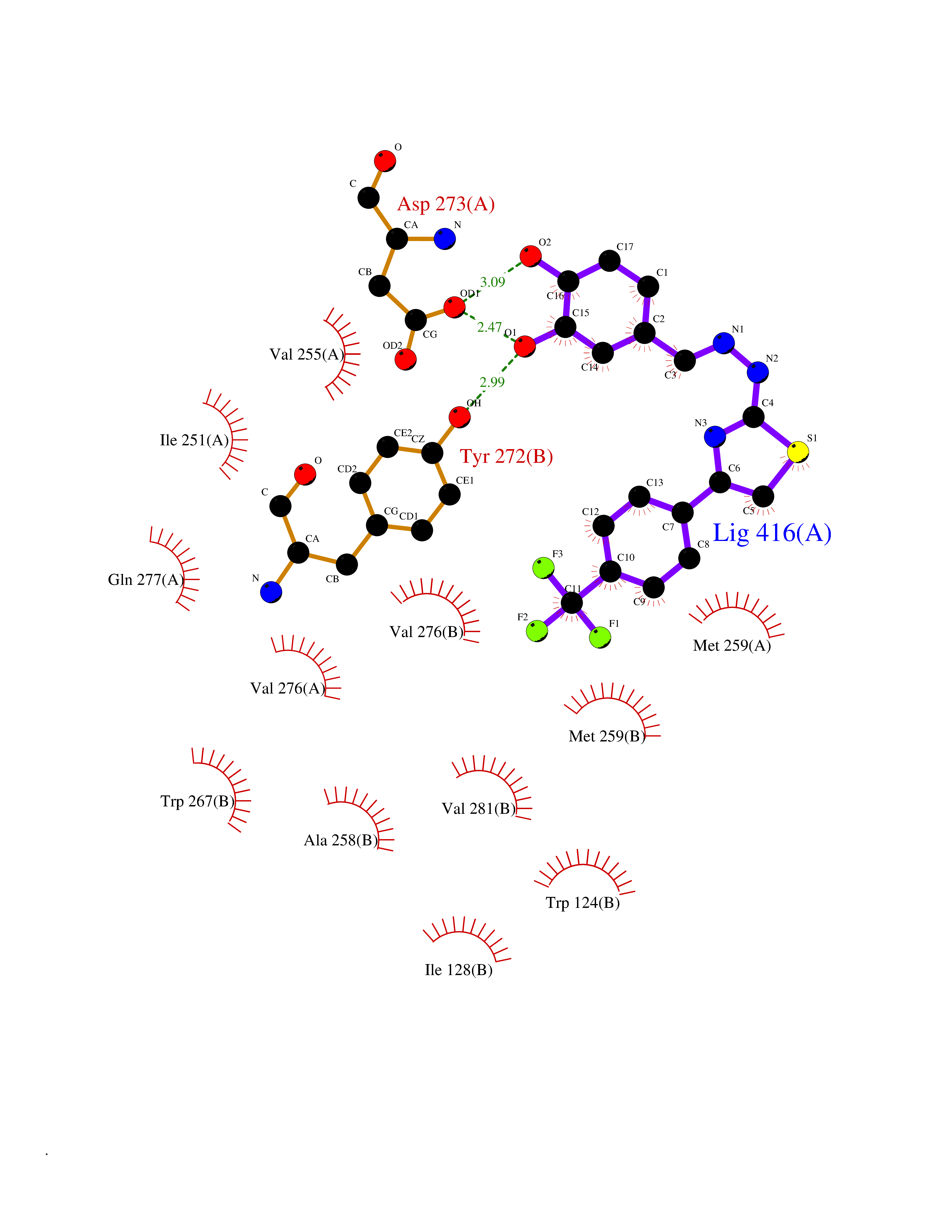 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Metabotropic glutamate receptor 5 (mGluR5) | 4OO9 | 7.46 | |
Target general information Gen name GRM5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MGLUR5; GPRC1E Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function G-protein coupled receptor for glutamate. Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling activates a phosphatidylinositol-calcium second messenger system and generates a calcium-activated chloride current. Plays an important role in the regulation of synaptic plasticity and the modulation of the neural network activity. Related diseases Charcot-Marie-Tooth disease, axonal, 2D (CMT2D) [MIM:601472]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:17101916, ECO:0000269|PubMed:17663003, ECO:0000269|PubMed:20169446, ECO:0000269|PubMed:24604904, ECO:0000269|PubMed:25168514, ECO:0000269|PubMed:26244500, ECO:0000269|PubMed:26503042, ECO:0000269|PubMed:31173493}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 5 (HMND5) [MIM:600794]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:23279345, ECO:0000269|PubMed:24627108, ECO:0000269|PubMed:26503042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinal muscular atrophy, infantile, James type (SMAJI) [MIM:619042]: An autosomal dominant form of spinal muscular atrophy, a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. SMAJI is a severe disease characterized by hypotonia manifesting in the first weeks or months of life, delayed motor development, motor regression, and muscle weakness and atrophy primarily affecting distal muscles. Additional variable features include feeding difficulties, poor overall growth, foot deformities, kyphosis, hyperlordosis, scoliosis, vocal cord dysfunction, and respiratory insufficiency. {ECO:0000269|PubMed:32181591}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00659; DB05070; DB12733; DB06201 Interacts with P41594; Q7Z6G3 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Methylation; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 27065.4 Length 247 Aromaticity 0.13 Instability index 42.92 Isoelectric point 9.24 Charge (pH=7) 11.34 2D Binding mode Binding energy (Kcal/mol) -10.18  Molscript Map 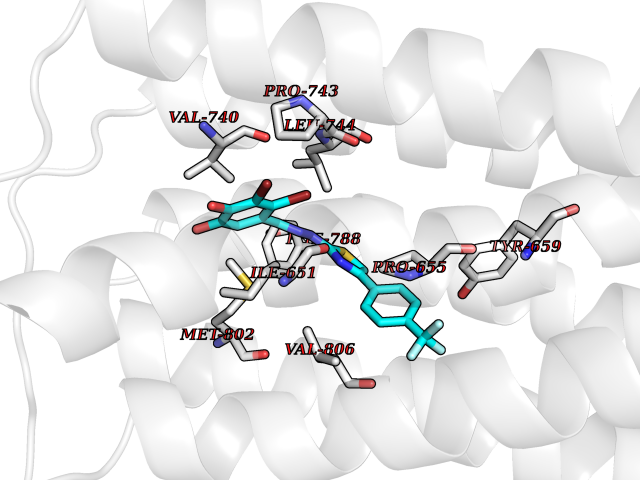 Pymol Map 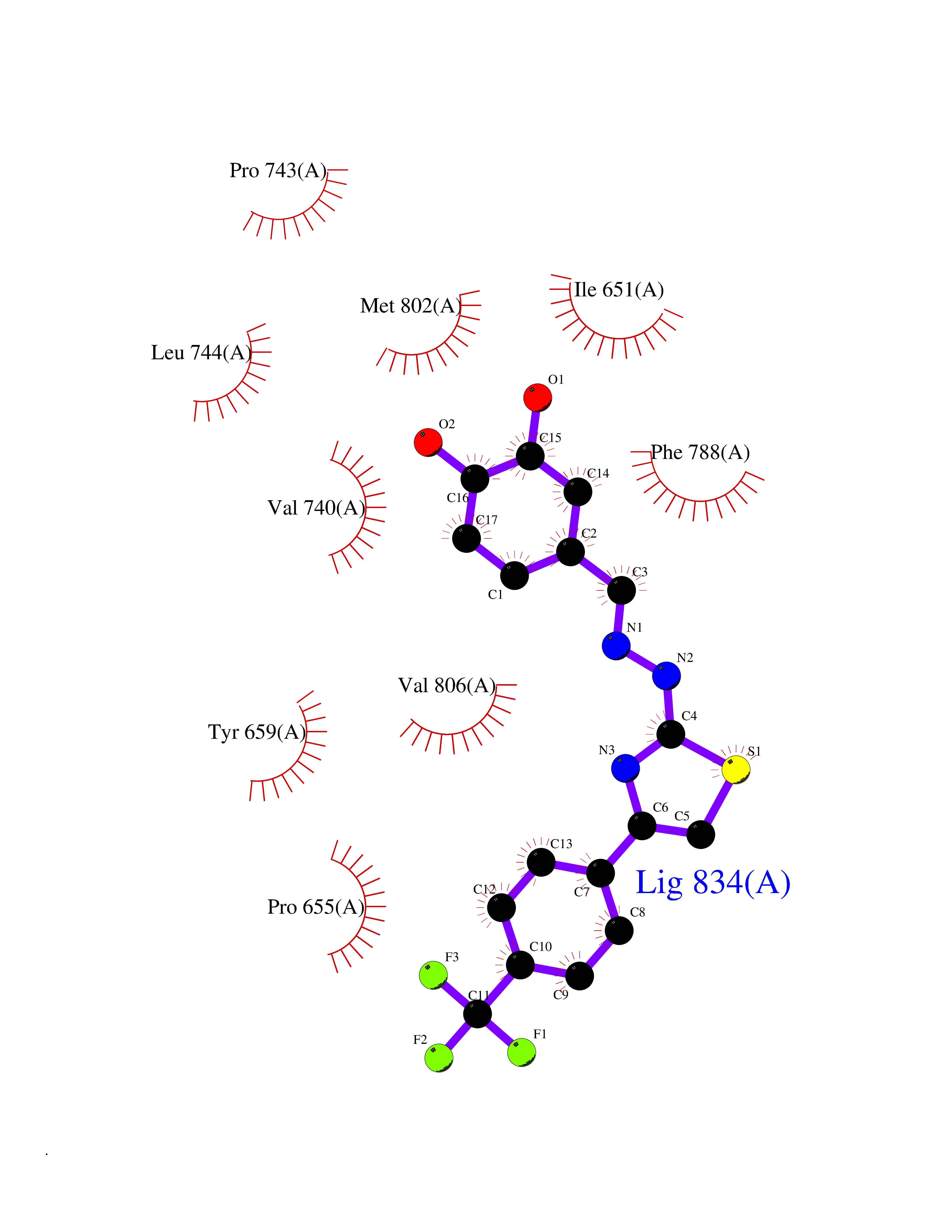 Ligplot Map 3D Binding mode Sequence SPVQYLRWGDPAPIAAVVFACLGLLATLFVTVVFIIYRDTPVVKSSSRELCYIILAGICLGYLCTFXLIAKPKQIYCYLQRIGIGLSPAMSYSALVTKTYRAARILAMSKKSAXAQLVIAFILICIQLGIIVALFIMEPPDIMVYLICNTTNLGVVAPLGYNGLLILACTFYAFKTRNVPANFNEAKYIAFTMYTTCIIWLAFVPIYFGSNYKIITMCFSVSLSATVALGCMFVPKVYIILAKPERN Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Mutated oxalosuccinate decarboxylase (mIDH1) | 6ADG | 7.46 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD (mutated); Oxalosuccinate decarboxylase (mutated); NADP(+)-specific ICDH (mutated); Isocitrate dehydrogenase [NADP] cytoplasmic (mutated); IDP (mutated); IDH (mutated); Cytosolic NADP-isocitrate Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -10.17 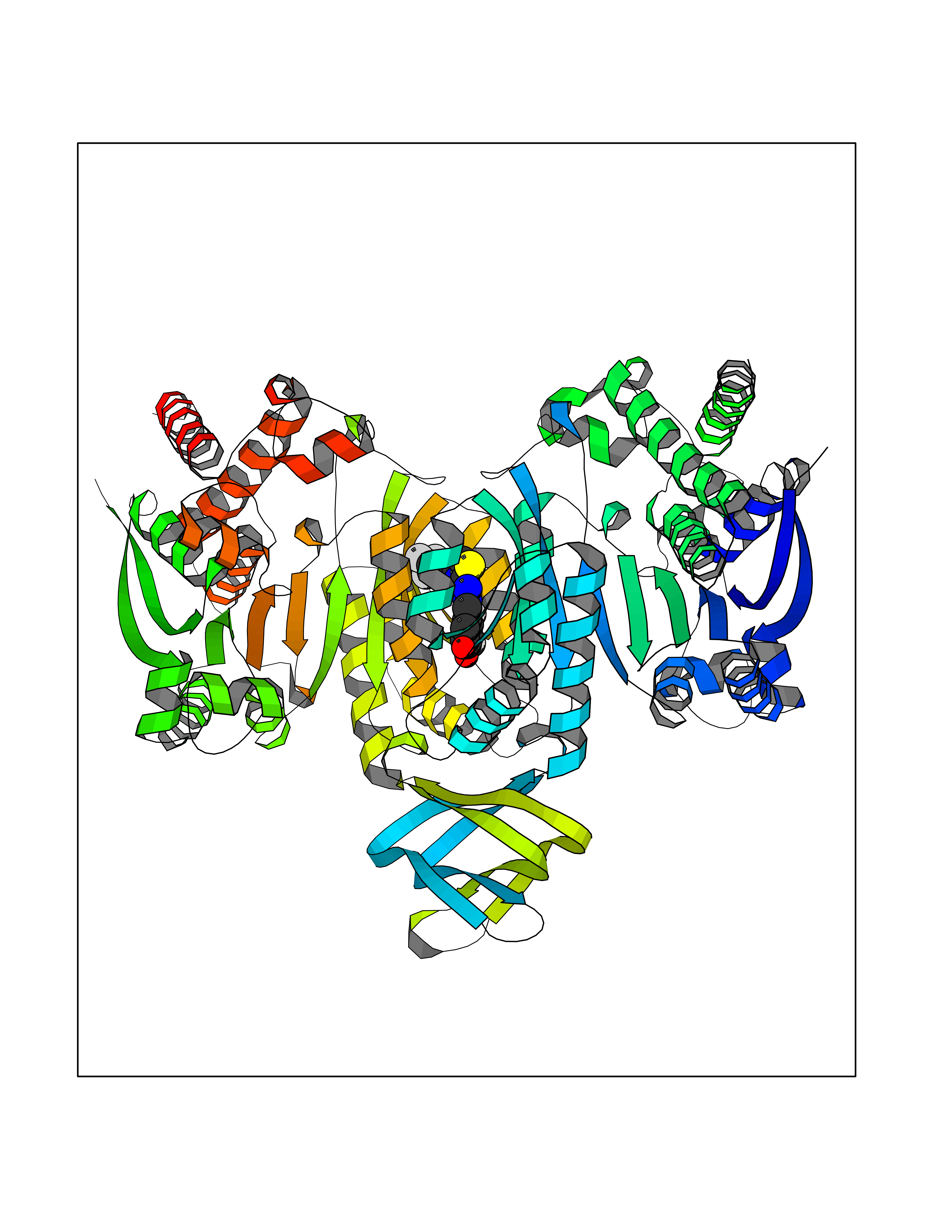 Molscript Map 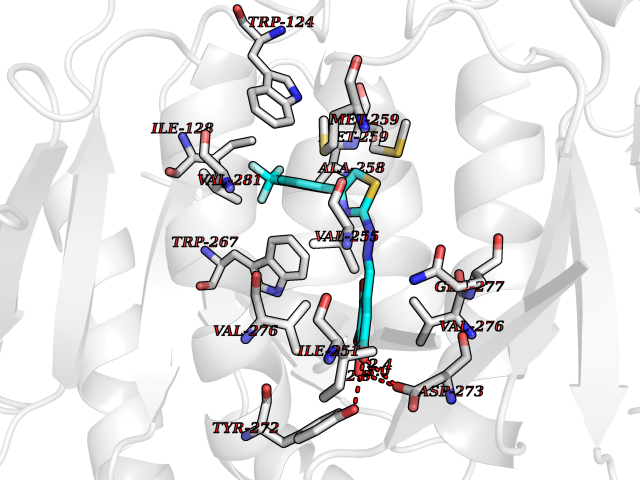 Pymol Map 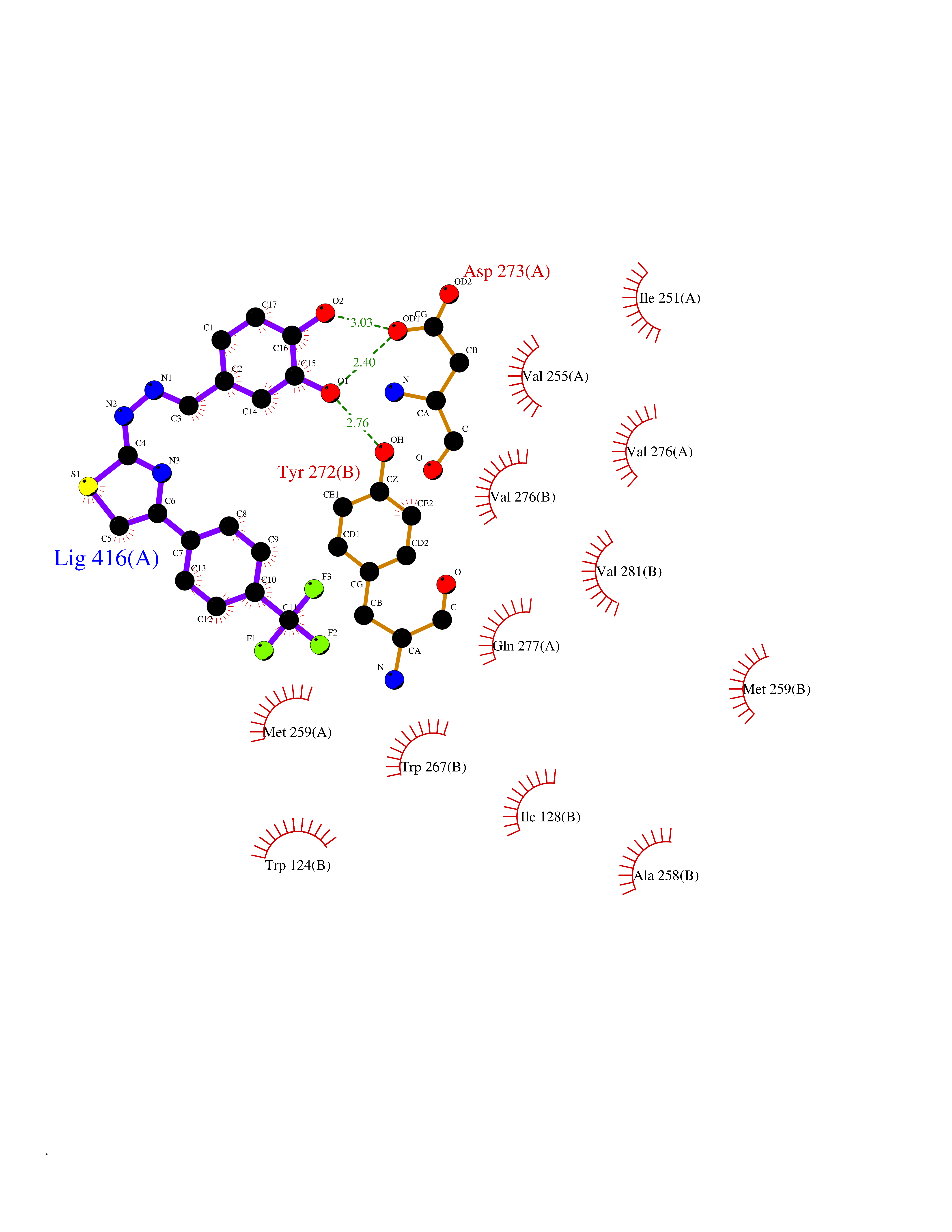 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Retinoic acid receptor RXR-alpha (RXRA) | 2P1T | 7.45 | |
Target general information Gen name RXRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor alpha; RXRalpha; Nuclear receptor subfamily 2 group B member 1; NR2B1 Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. The high affinity ligand for RXRs is 9-cis retinoic acid. RXRA serves as a common heterodimeric partner for a number of nuclear receptors. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. The RXRA/PPARA heterodimer is required for PPARA transcriptional activity on fatty acid oxidation genes such as ACOX1 and the P450 system genes. Receptor for retinoic acid. Related diseases Lichtenstein-Knorr syndrome (LIKNS) [MIM:616291]: An autosomal recessive neurologic disorder characterized by progressive cerebellar ataxia and severe progressive sensorineural hearing loss. {ECO:0000269|PubMed:25205112, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08063; DB08402; DB07863; DB07557; DB00459; DB00210; DB01436; DB00523; DB00132; DB04557; DB00307; DB01393; DB03756; DB00749; DB00926; DB05956; DB04224; DB02746; DB00412; DB00755; DB08601 Interacts with O14503; P35637; Q15648; Q71SY5; Q15788; Q15596; P55055; P55055-1; Q13133; P27986; P37231; P37231-1; P10276; P42224; P11473; P97792-1; Q9JLI4; P04625; PRO_0000278730 [Q03463] EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; DNA-binding; Host-virus interaction; Isopeptide bond; Metal-binding; Mitochondrion; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 24958.9 Length 221 Aromaticity 0.07 Instability index 39.47 Isoelectric point 6.16 Charge (pH=7) -3.45 2D Binding mode Binding energy (Kcal/mol) -10.17 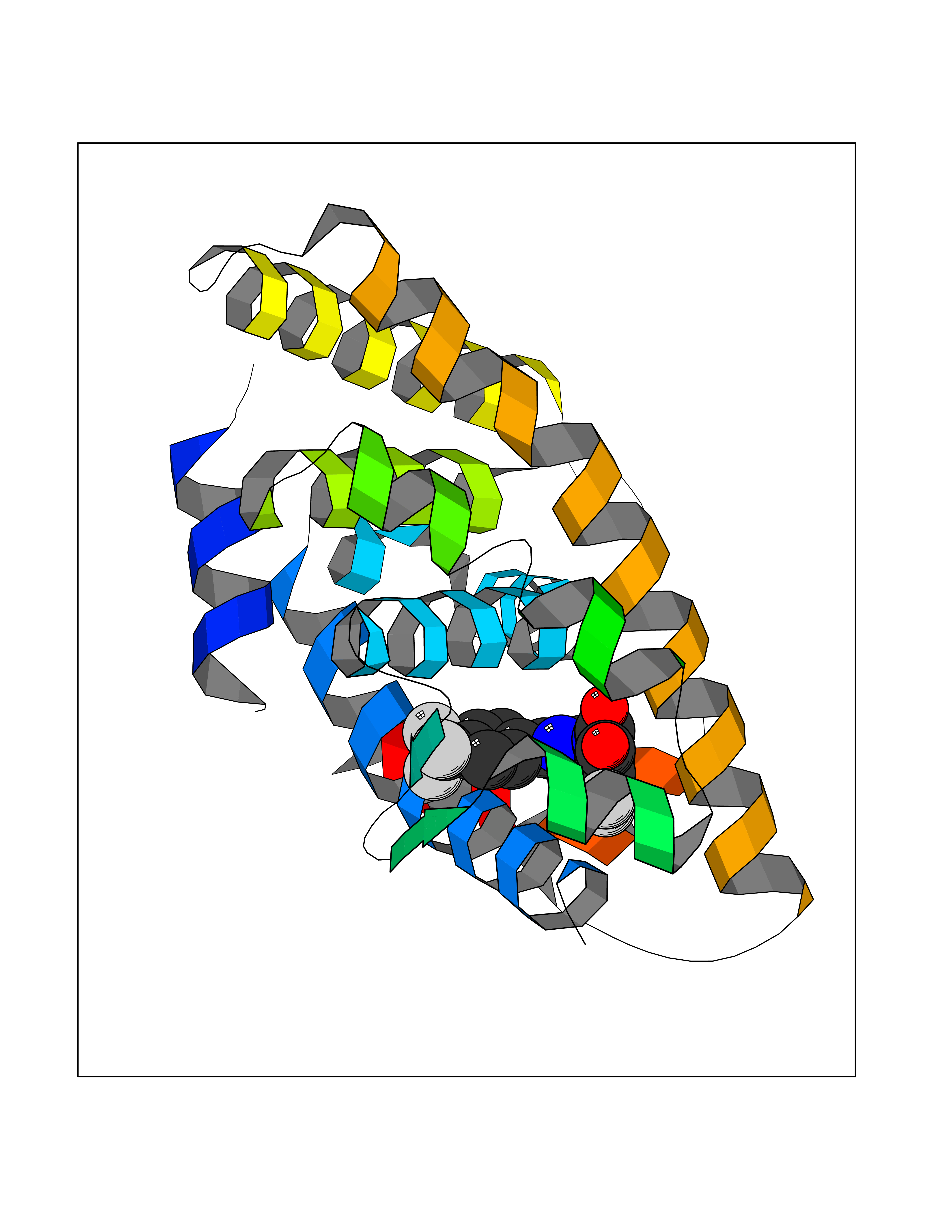 Molscript Map 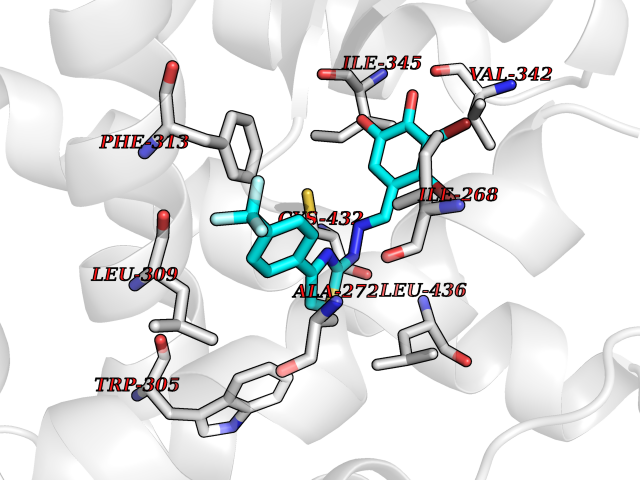 Pymol Map 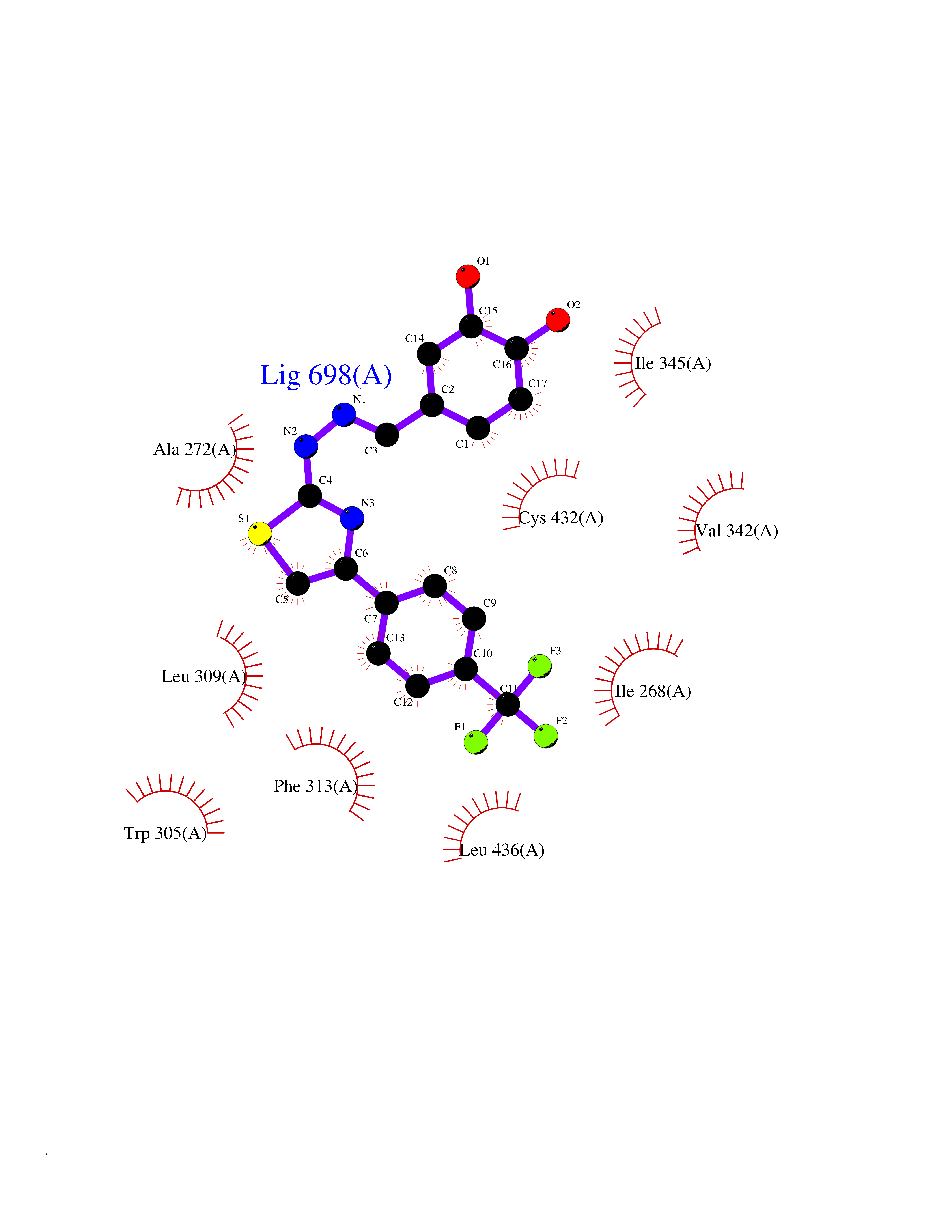 Ligplot Map 3D Binding mode Sequence DMPVERILEAELAVEDPVTNICQAADKQLFTLVEWAKRIPHFSELPLDDQVILLRAGWNELLIASFSHRSIAVKDGILLATGLHVHRNSAHSAGVGAIFDRVLTELVSKMRDMQMDKTELGCLRAIVLFNPDSKGLSNPAEVEALREKVYASLEAYCKHKYPEQPGRFAKLLLRLPALRSIGLKCLEHLFFFKLIGDTPIDTFLMEMLEAPHKILHRLLQD Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Acetyl-CoA carboxylase (ACC) (EC 6.4.1.2) (Fatty acid synthetase 3) (mRNA transport-defective protein 7) [Includes: Biotin carboxylase (EC 6.3.4.14)] | 1UYS | 7.45 | |
Target general information Gen name ACC1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms MTR7;YNR016C;N3175;ABP2;FAS3 Protein family NA Biochemical class NA Function Carries out three functions: biotin carboxyl carrier protein, biotin carboxylase and carboxyltransferase. Involved in the synthesis of very-long-chain fatty acid synthesis which is required to maintain a functional nuclear envelope. Required for acylation and vacuolar membrane association of VAC8 which is necessary to maintain a normal morphology of the vacuole. {ECO:0000269|PubMed:10757783, ECO:0000269|PubMed:12730220, ECO:0000269|PubMed:6103540, ECO:0000269|PubMed:6108218, ECO:0000269|PubMed:8943372}." Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q00955 EC number 6.3.4.14; 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Endoplasmic reticulum; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Manganese; Membrane; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome Protein physicochemical properties Chain ID B,C Molecular weight (Da) 145619 Length 1328 Aromaticity 0.1 Instability index 30.31 Isoelectric point 5.32 Charge (pH=7) -26.79 2D Binding mode Binding energy (Kcal/mol) -10.16  Molscript Map  Pymol Map 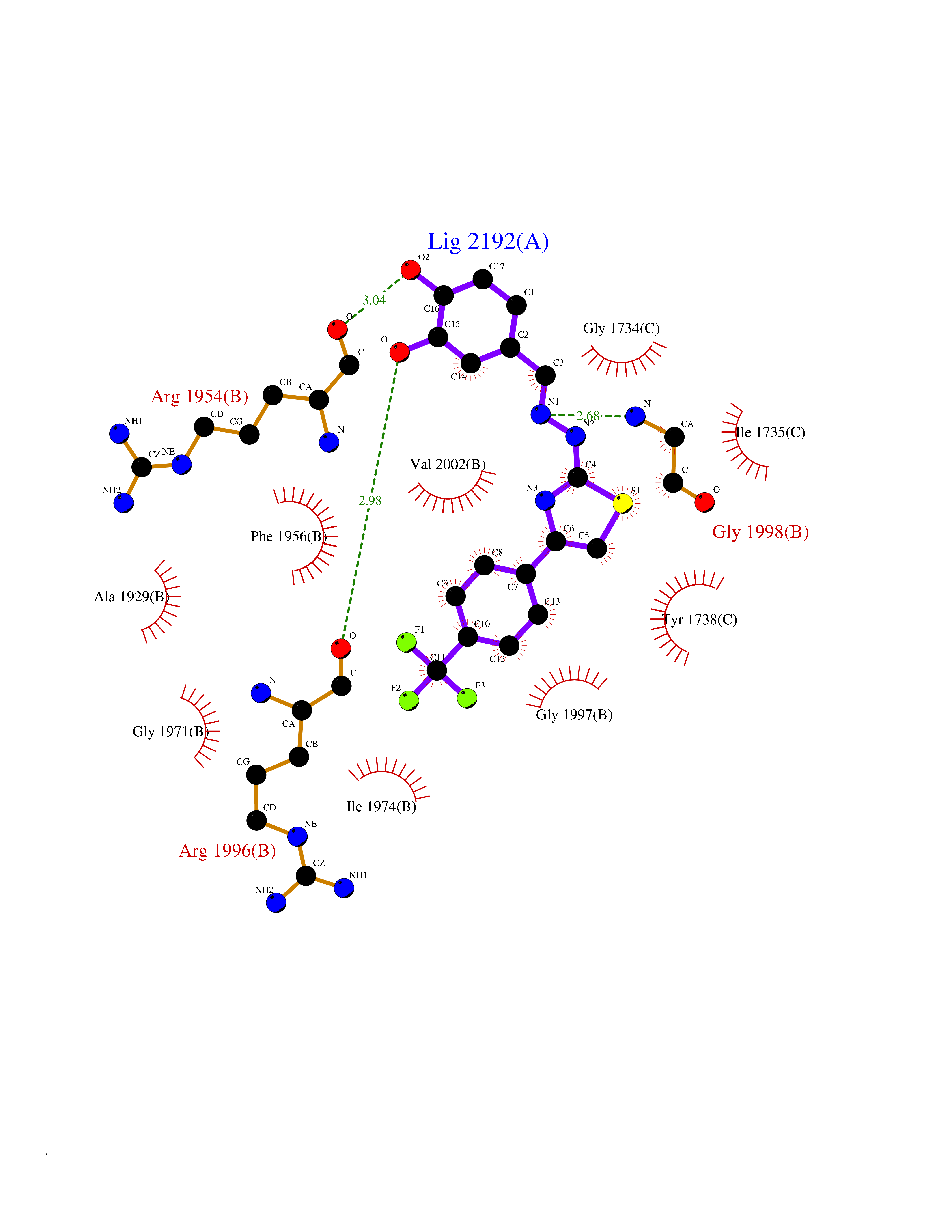 Ligplot Map 3D Binding mode Sequence WLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGLPIATPYPVKEWLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGL Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Smoothened homolog (SMO) | 4JKV | 7.45 | |
Target general information Gen name SMO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Smo-D473H; SMOH; Protein Gx Protein family G-protein coupled receptor Fz/Smo family Biochemical class GPCR frizzled Function Binding of sonic hedgehog (SHH) to its receptor patched is thought to prevent normal inhibition by patched of smoothened (SMO). Required for the accumulation of KIF7, GLI2 and GLI3 in the cilia. Interacts with DLG5 at the ciliary base to induce the accumulation of KIF7 and GLI2 at the ciliary tip for GLI2 activation. G protein-coupled receptor that probably associates with the patched protein (PTCH) to transduce the hedgehog's proteins signal. Related diseases Curry-Jones syndrome (CRJS) [MIM:601707]: A multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. The disease is caused by variants affecting the gene represented in this entry. 8 individuals have been identified with the disease-causing mutation Phe-412 and all were mosaic. The mutation could not be reliably detected in blood, greatest success rates were obtained with affected tissues obtained by invasive procedures. It is thought that the mutation has arisen postzygotically early during embryonic development (PubMed:27236920). This mutation has also been identified in ameloblastoma, medulloblastoma, meningioma, and basal cell carcinoma, and has been reported as the oncogenic driver in some of these tumors (PubMed:24859340). {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. Drugs (DrugBank ID) DB01047; DB11978; DB06786; DB09143; DB08828 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; Developmental protein; Disease variant; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37420.1 Length 333 Aromaticity 0.16 Instability index 25.17 Isoelectric point 6.65 Charge (pH=7) -0.76 2D Binding mode Binding energy (Kcal/mol) -10.17  Molscript Map 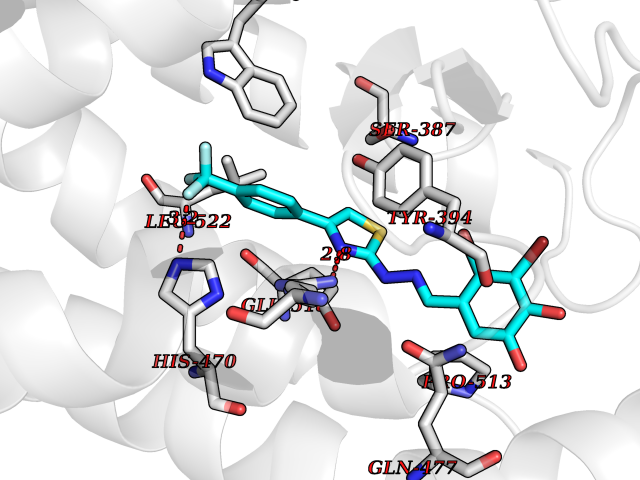 Pymol Map 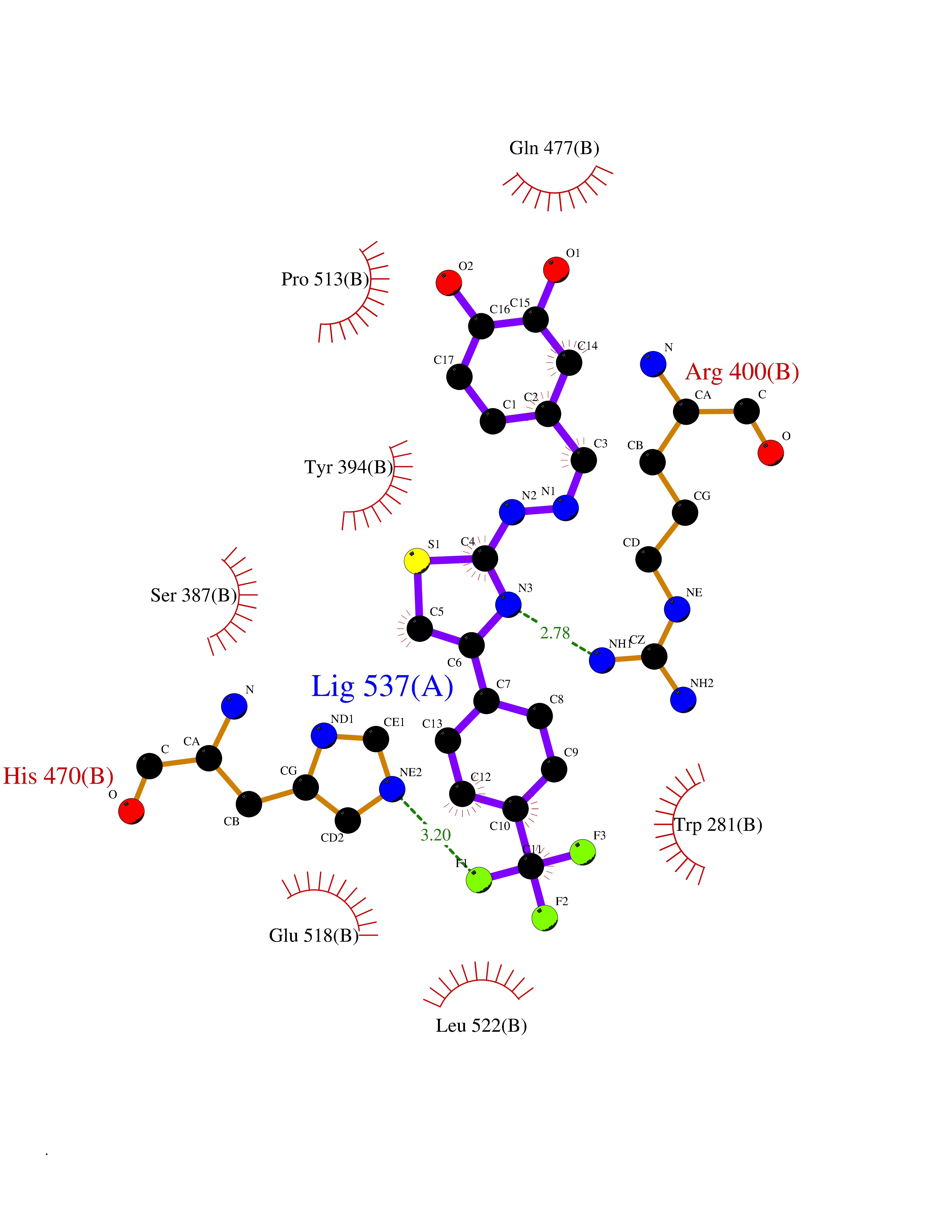 Ligplot Map 3D Binding mode Sequence AYIQKYLSGQCEVPLVRTDNPKSWYEDVEGCGIQCQNPLFTEAEHQDMHSYIAAFGAVTGLCTLFTLATFVADWRNSNRYPAVILFYVNACFFVGSIGWLAQFMDGARREIVCRADGTMRLGEPTSNETLSCVIIFVIVYYALMAGVVWFVVLTYAWHTSFKALGKTSYFHLLTWSLPFVLTVAILAVAQVDGDSVSGICFVGYKNYRYRAGFVLAPIGLVLIVGGYFLIRGVMTLFSIKSNHPGLLSEKAASKINETMLRLGIFGFLAFGFVLITFSCHFYDFFNQAEWERSFRDYVLCQANDCEIKNRPSLLVEKINLFAMFGTGIAMSTW Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Opioid receptor sigma 1 (OPRS1) | 5HK1 | 7.44 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 20805.3 Length 185 Aromaticity 0.14 Instability index 31.72 Isoelectric point 5.44 Charge (pH=7) -6.63 2D Binding mode Binding energy (Kcal/mol) -10.15  Molscript Map 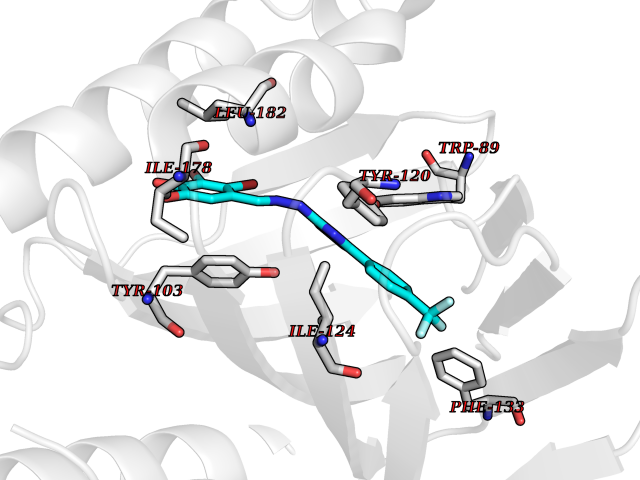 Pymol Map 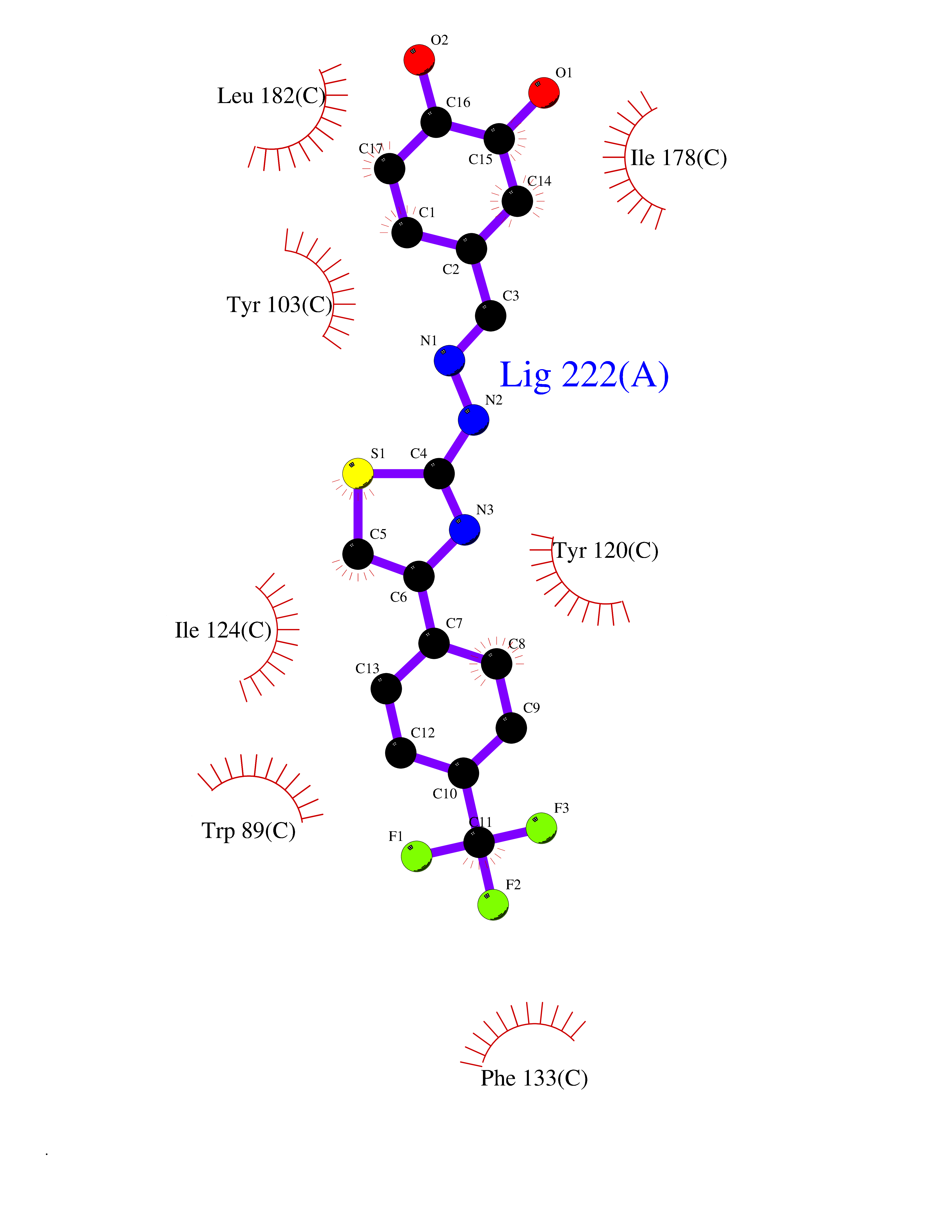 Ligplot Map 3D Binding mode Sequence VFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLFG Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Gastric alcohol dehydrogenase (ADH7) | 1D1T | 7.44 | |
Target general information Gen name ADH7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Alcohol dehydrogenase class IV mu/sigma chain; Alcohol dehydrogenase class 4 mu/sigma chain; ADH7 Protein family Zinc-containing alcohol dehydrogenase family, Class-IV subfamily Biochemical class Short-chain dehydrogenases reductase Function Could function in retinol oxidation for the synthesis of retinoic acid, a hormone important for cellular differentiation. Medium-chain (octanol) and aromatic (m-nitrobenzaldehyde) compounds are the best substrates. Ethanol is not a good substrate but at the high ethanol concentrations reached in the digestive tract, it plays a role in the ethanol oxidation and contributes to the first pass ethanol metabolism. Related diseases Intellectual developmental disorder, autosomal recessive 80, with variant lissencephaly (MRT80) [MIM:620653]: An autosomal recessive disorder characterized by global developmental delay, mildly to moderately impaired intellectual development, attention deficit-hyperactivity disorder, hypotonia, seizure, poor social skills, and autistic traits. Brain imaging shows fronto-temporal lissencephaly and pachygyria. {ECO:0000269|PubMed:37880421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00898; DB00157 Interacts with NA EC number EC 1.1.1.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Direct protein sequencing; Lipid metabolism; Metal-binding; NAD; Oxidoreductase; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 79693.9 Length 746 Aromaticity 0.06 Instability index 28.65 Isoelectric point 8.38 Charge (pH=7) 8.79 2D Binding mode Binding energy (Kcal/mol) -10.15  Molscript Map 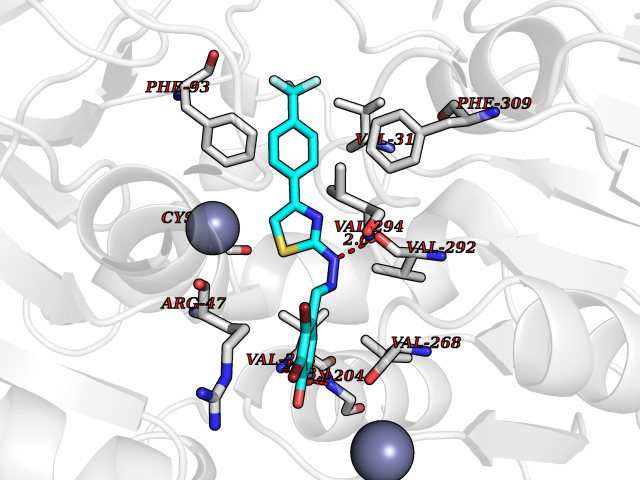 Pymol Map 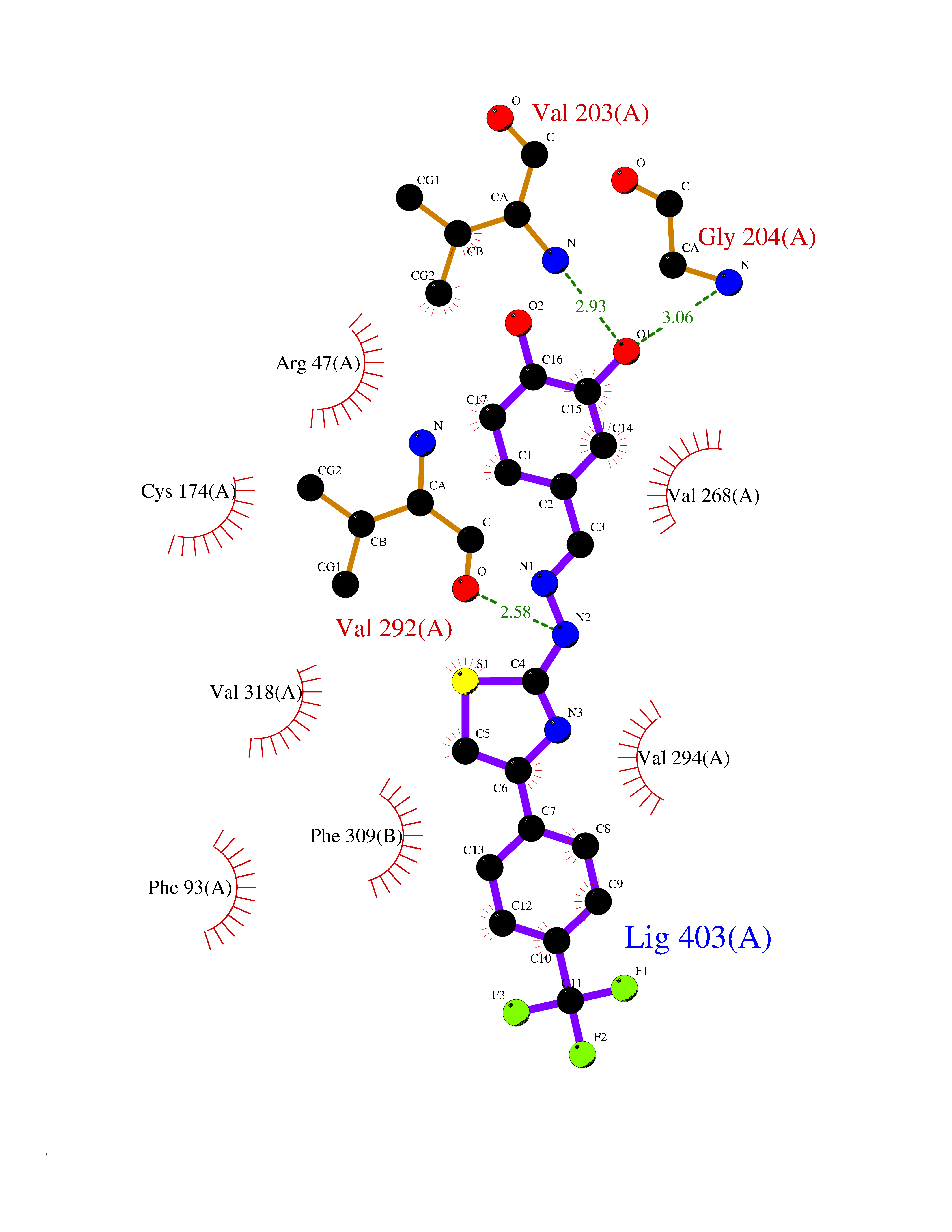 Ligplot Map 3D Binding mode Sequence GTAGKVIKCKAAVLWEQKQPFSIEEIEVAPPKTKEVRIKILATGICRTDDHVIKGTMVSKFPVIVGHEATGIVESIGEGVTTVKPGDKVIPLFLPQCRECNACRNPDGNLCIRSDITGRGVLADGTTRFTCKGKPVHHFLNTSTFTEYTVVDESSVAKIDDAAPPEKVCLIGCGFSTGYGAAVKTGKVKPGSTCVVFGLGGVGLSVIMGCKSAGASRIIGIDLNKDKFEKAMAVGATECISPKDSTKPISEVLSEMTGNNVGYTFEVIGHLETMIDALASCHMNYGTSVVVGVPPSAKMLTYDPMLLFTGRTWKGCVFGGLKSRDDVPKLVTEFLAKKFDLDQLITHVLPFKKISEGFELLNSGQSIRTVLTFGTAGKVIKCKAAVLWEQKQPFSIEEIEVAPPKTKEVRIKILATGICRTDDHVIKGTMVSKFPVIVGHEATGIVESIGEGVTTVKPGDKVIPLFLPQCRECNACRNPDGNLCIRSDITGRGVLADGTTRFTCKGKPVHHFLNTSTFTEYTVVDESSVAKIDDAAPPEKVCLIGCGFSTGYGAAVKTGKVKPGSTCVVFGLGGVGLSVIMGCKSAGASRIIGIDLNKDKFEKAMAVGATECISPKDSTKPISEVLSEMTGNNVGYTFEVIGHLETMIDALASCHMNYGTSVVVGVPPSAKMLTYDPMLLFTGRTWKGCVFGGLKSRDDVPKLVTEFLAKKFDLDQLITHVLPFKKISEGFELLNSGQSIRTVLTF Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Branched-chain-amino-acid transaminase 2 (BCAT2) | 5CR5 | 7.43 | |
Target general information Gen name BCAT2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Placental protein 18; PP18; ECA39 protein; Branched-chain amino acid aminotransferase; BCAT2; BCAT(m); BCAT Protein family Class-IV pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Catalyzes the first reaction in the catabolism of the essential branched chain amino acids leucine, isoleucine, and valine. May also function as a transporter of branched chain alpha-keto acids. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB04074; DB00142; DB00167; DB00149; DB02635; DB00114; DB02142 Interacts with P10809 EC number EC 2.6.1.42 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Amino-acid biosynthesis; Aminotransferase; Branched-chain amino acid biosynthesis; Disease variant; Lipid metabolism; Mitochondrion; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40725 Length 359 Aromaticity 0.09 Instability index 39.14 Isoelectric point 8.46 Charge (pH=7) 3.24 2D Binding mode Binding energy (Kcal/mol) -10.13 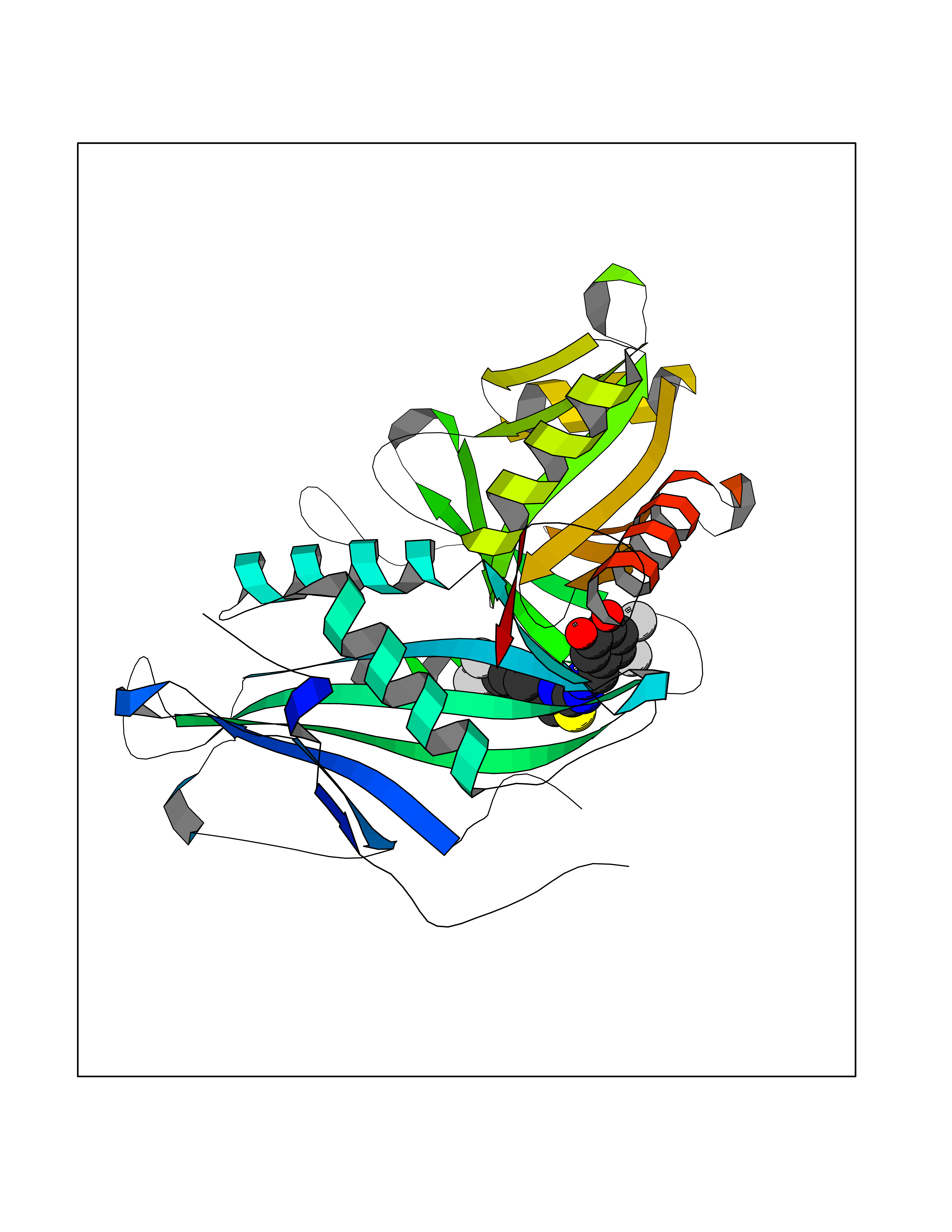 Molscript Map 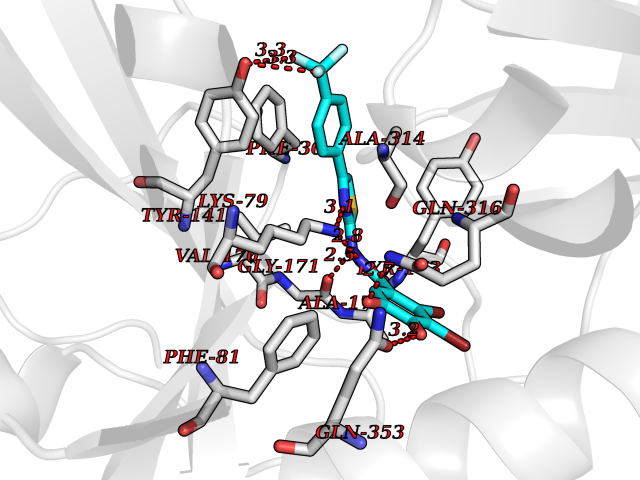 Pymol Map 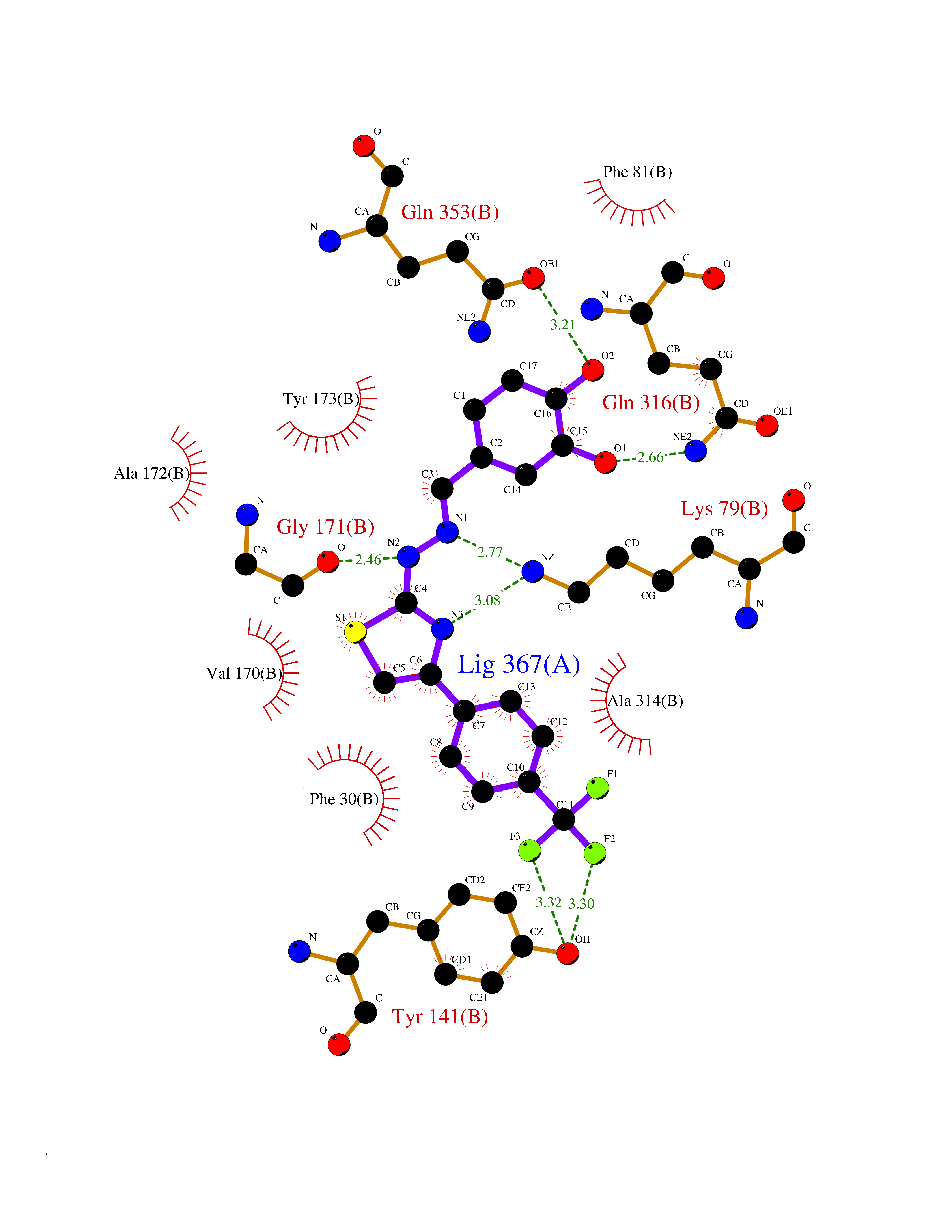 Ligplot Map 3D Binding mode Sequence SSFKAADLQLEMTQKPHKKPGLVFGKTFTDHMLMVEWNDKGWGQPRIQPFQNLTLHPASSSLHYSLQLFEGMKAFKGKDQQVRLFRPWLNMDRMLRSAMRLCLPSFDKLELLECIRRLIEVDKDWVPDAAGTSLYVRPVLIGNEPSLGVSQPTRALLFVILCPVGAYFPGGSVTPVSLLADPAFIRAWVGGVGNYKLGGNYGPTVLVQQEALKRGCEQVLWLYGPDHQLTEVGTMNIFVYWTHEDGVLELVTPPLNGVILPGVVRQSLLDMAQTWGEFRVVERTITMKQLLRALEEGRVREVFGSGTACQVCPVHRILYKDRNLHIPTMENGPELILRFQKELKEIQYGIRAHEWMFPV Hydrogen bonds contact Hydrophobic contact | ||||