Job Results:
Ligand
Structure
Job ID
f5a9e34d4d6e0fad4e04e6dd0d3ae3aa
Job name
NA
Time
2026-01-10 22:50:50
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Dipeptidyl peptidase 8 (DPP-8) | 6EOP | 7.07 | |
Target general information Gen name DPP8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Prolyl dipeptidase DPP8; MSTP141; MSTP135; MSTP097; Dipeptidyl peptidase VIII; Dipeptidyl peptidase IV-related protein 1; DPRP1; DPRP-1; DPP VIII; DP8 Protein family Peptidase S9B family, DPPIV subfamily Biochemical class Peptidase Function Dipeptidyl peptidase that cleaves off N-terminal dipeptides from proteins having a Pro or Ala residue at position 2. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.14.5 Uniprot keywords 3D-structure; Alternative splicing; Aminopeptidase; Apoptosis; Cytoplasm; Hydrolase; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A,D Molecular weight (Da) 97764.9 Length 849 Aromaticity 0.12 Instability index 47.71 Isoelectric point 5.69 Charge (pH=7) -21.66 2D Binding mode Binding energy (Kcal/mol) -9.65  Molscript Map 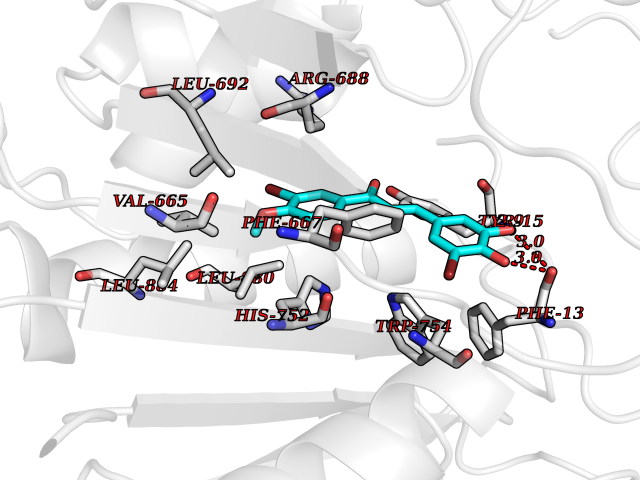 Pymol Map 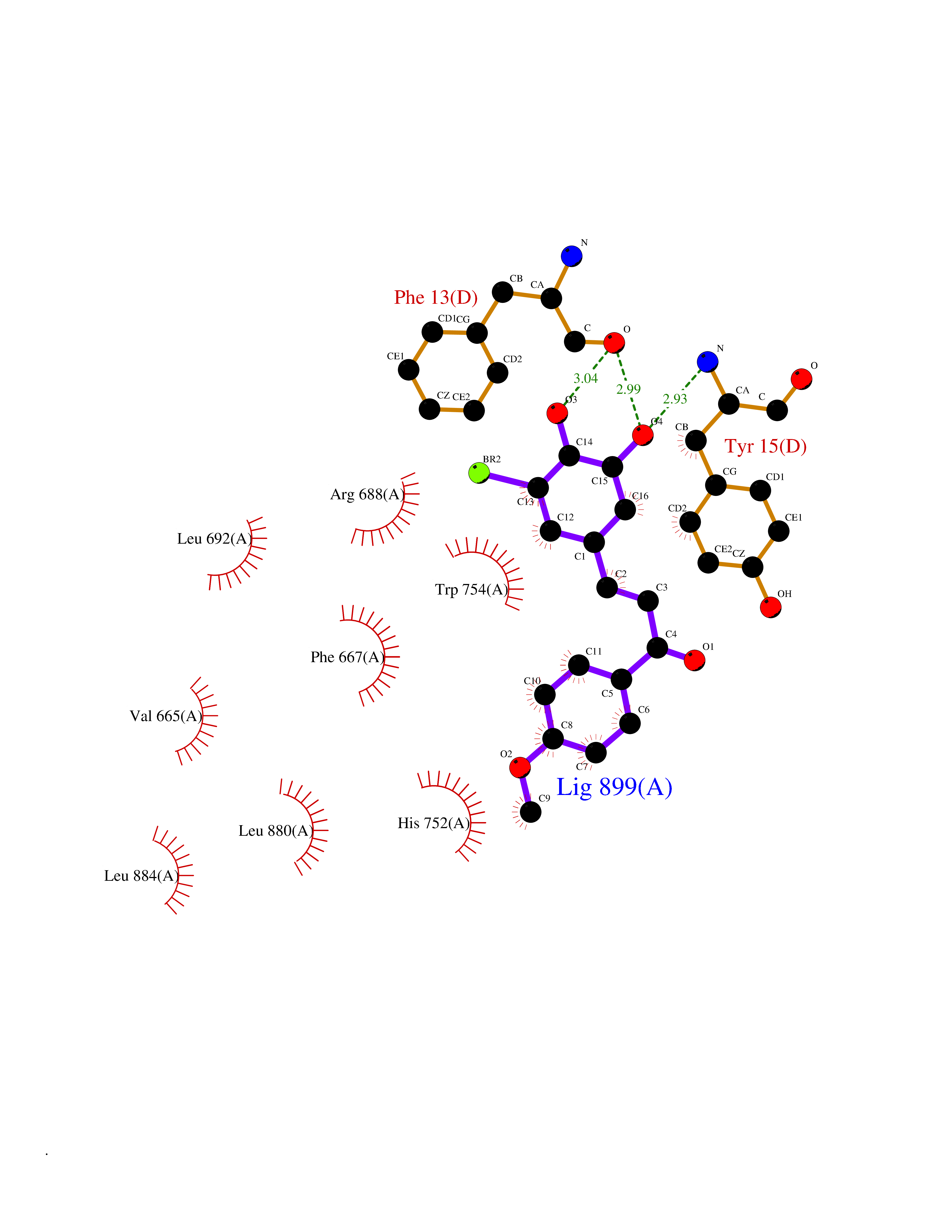 Ligplot Map 3D Binding mode Sequence LEPFYVERYSWSQLKKLLADTRKYHGYMMAKAPHDFMFVKRNDPDGPHSDRIYYLAMSNRENTLFYSEIPKTINRAAVLMLSWKPLLDLFQYSREEELLRERKRIGTVGIASYDYHQGSGTFLFQAGSGIYHVKDGGPQGFTQQPLRPNLVETSCPNIRMDPKLCPADPDWIAFIHSNDIWISNIVTREERRLTYVHNELANMEEDARSAGVATFVLQEEFDRYSGYWWCPKAETTPSGGKILRILYEENDESEVEIIHVTSPMLETRRADSFRYPKTGTANPKVTFKMSEIMIDAEGRIIDVIDKELIQPFEILFEGVEYIARAGWTPEGKYAWSILLDRSQTRLQIVLISPELFIPVEDDVMERQRLIESVPDSVTPLIIYEETTDIWINIHDIFHVFPQSHEEEIEFIFASECKTGFRHLYKITSILKESKYKRSSGGLPAPSDFKCPIKEEIAITSGEWEVLGRHGSNIQVDEVRRLVYFEGTKDSPLEHHLYVVSYVNPGEVTRLTDRGYSHSCCISQHCDFFISKYSNQKNPHCVSLYKLSSPEDDPTCKTKEFWATILDSAGPLPDYTPPEIFSFESTTGFTLYGMLYKPHDLQPGKKYPTVLFIYGGPQVQLVNNRFKGVKYFRLNTLASLGYVVVVIDNRGSXHRGLKFEGAFKYKMGQIEIDDQVEGLQYLASRYDFIDLDRVGIHGWSYGGYLSLMALMQRSDIFRVAIAGAPVTLWIFYDTGYTERYMGHPDQNEQGYYLGSVAMQAEKFPSEPNRLLLLHGFLDENVHFAHTSILLSFLVRAGKPYDLQIYPQERHSIRVPESGEHYELHLLHYLQENLGSRIAALKVSLRFLYEG Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Benzoate 1,2-dioxygenase electron transfer component | 1KRH | 7.06 | |
Target general information Gen name benC Organism Acinetobacter baylyi (strain ATCC 33305 / BD413 / ADP1) Uniprot ID TTD ID NA Synonyms ACIAD1438 Protein family Bacterial ring-hydroxylating dioxygenase ferredoxin reductase family Biochemical class Oxidoreductase Function 2 iron, 2 sulfur cluster binding.Electron carrier activity.Ferredoxin-NAD+ reductase activity.Metal ion binding. Related diseases Adenine phosphoribosyltransferase deficiency (APRTD) [MIM:614723]: An enzymatic deficiency that can lead to urolithiasis and renal failure. Patients have 2,8-dihydroxyadenine (DHA) urinary stones. {ECO:0000269|PubMed:11243733, ECO:0000269|PubMed:1353080, ECO:0000269|PubMed:15571218, ECO:0000269|PubMed:1746557, ECO:0000269|PubMed:21635362, ECO:0000269|PubMed:3343350, ECO:0000269|PubMed:3680503, ECO:0000269|PubMed:7915931}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.18.1.3 Uniprot keywords 2Fe-2S; 3D-structure; Aromatic hydrocarbons catabolism; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; NAD; Oxidoreductase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37496.7 Length 337 Aromaticity 0.1 Instability index 47.02 Isoelectric point 4.75 Charge (pH=7) -18.87 2D Binding mode Binding energy (Kcal/mol) -9.63  Molscript Map 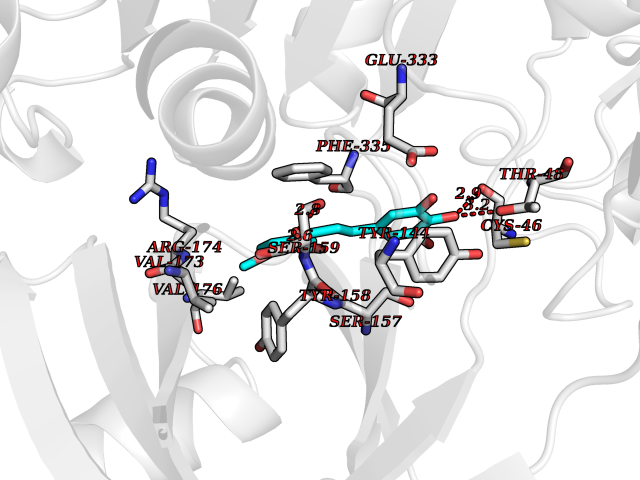 Pymol Map 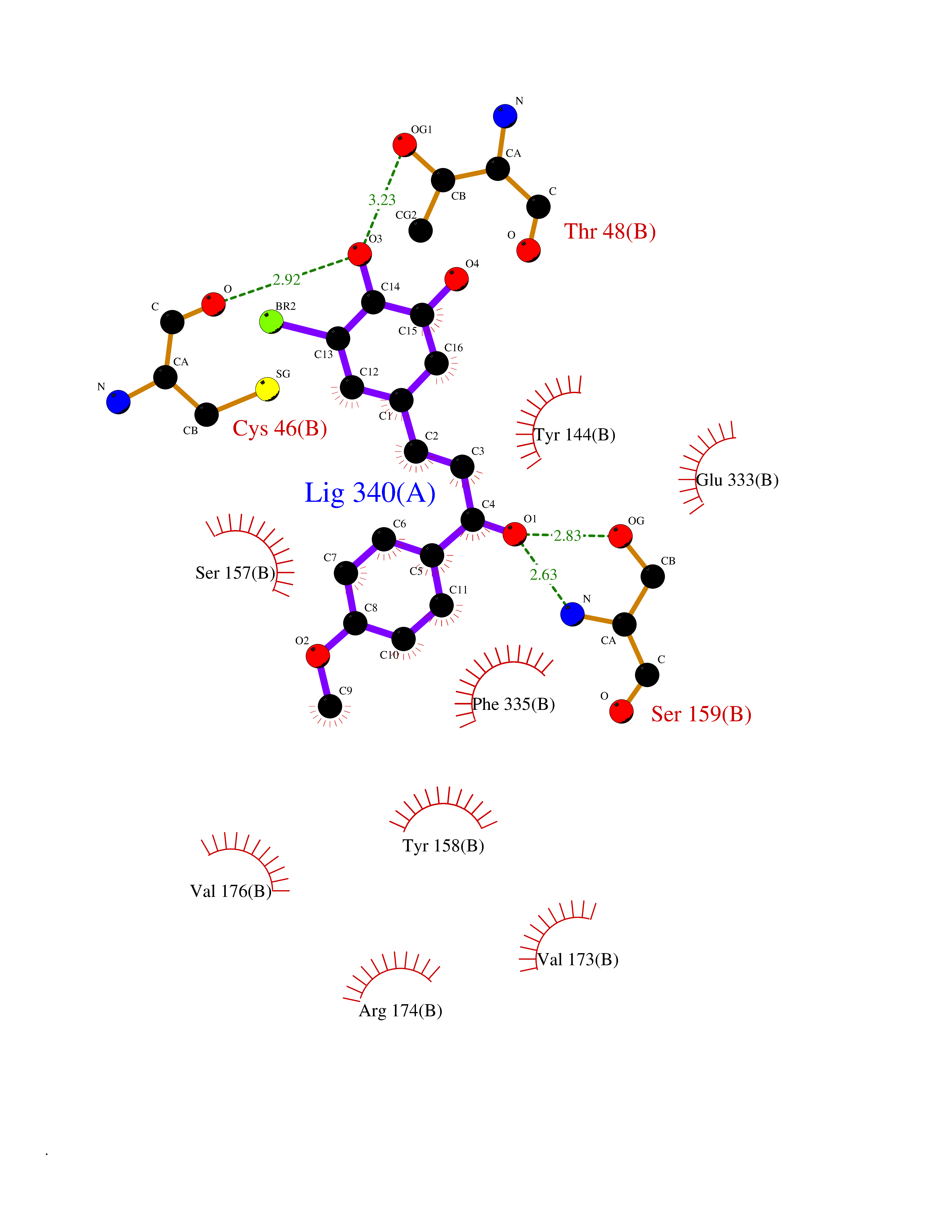 Ligplot Map 3D Binding mode Sequence SNHQVALQFEDGVTRFICIAQGETLSDAAYRQQINIPMDCREGECGTCRAFCESGNYDMPEDNYIEDALTPEEAQQGYVLACQCRPTSDAVFQIQASSEVCKTKIHHFEGTLARVENLSDSTITFDIQLDDGQPDIHFLAGQYVNVTLPGTTETRSYSFSSQPGNRLTGFVVRNVPQGKMSEYLSVQAKAGDKMSFTGPFGSFYLRDVKRPVLMLAGGTGIAPFLSMLQVLEQKGSEHPVRLVFGVTQDCDLVALEQLDALQQKLPWFEYRTVVAHAESQHERKGYVTGHIEYDWLNGGEVDVYLCGPVPMVEAVRSWLDTQGIQPANFLFEKFSAN Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Cytochrome P450 2B6 (CYP2B6) | 3IBD | 7.06 | |
Target general information Gen name CYP2B6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450 IIB1; CYPIIB6; 1,4-cineole 2-exo-monooxygenase Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function In liver microsomes, this enzyme is involved in an NADPH-dependent electron transport pathway. It oxidizes a variety of structurally unrelated compounds, including steroids, fatty acids, and xenobiotics. Acts as a 1,4-cineole 2-exo-monooxygenase. Cytochromes P450 are a group of heme-thiolate monooxygenases. Related diseases Aromatic L-amino-acid decarboxylase deficiency (AADCD) [MIM:608643]: An inborn error in neurotransmitter metabolism that leads to combined serotonin and catecholamine deficiency. It causes developmental and psychomotor delay, poor feeding, lethargy, ptosis, intermittent hypothermia, gastrointestinal disturbances. The onset is early in infancy and inheritance is autosomal recessive. {ECO:0000269|PubMed:14991824, ECO:0000269|PubMed:15079002, ECO:0000269|Ref.12}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08369; DB02974; DB11932; DB12001; DB14973; DB15568; DB00321; DB00381; DB00701; DB01435; DB00714; DB06413; DB06697; DB13132; DB11586; DB01076; DB15011; DB00972; DB08822; DB13997; DB04975; DB01086; DB00865; DB00443; DB04794; DB12151; DB01194; DB05541; DB01222; DB01156; DB09061; DB14737; DB00564; DB06119; DB00439; DB00568; DB00604; DB00515; DB12499; DB00349; DB06470; DB00758; DB01559; DB00257; DB01394; DB05219; DB08865; DB11672; DB14635; DB04664; DB00531; DB08912; DB01151; DB16650; DB01234; DB14649; DB04856; DB00514; DB00829; DB00586; DB01184; DB00997; DB00470; DB00476; DB00625; DB15444; DB13874; DB11718; DB08899; DB00751; DB11823; DB00655; DB00898; DB01466; DB00574; DB12265; DB01544; DB00472; DB01095; DB00176; DB01320; DB05087; DB00986; DB01159; DB00956; DB00741; DB09054; DB01181; DB00458; DB00762; DB11633; DB06636; DB00753; DB11757; DB01167; DB14568; DB09570; DB01221; DB06738; DB01026; DB11951; DB09078; DB12070; DB05667; DB00281; DB00836; DB01601; DB00455; DB04871; DB12130; DB09280; DB00772; DB09238; DB14921; DB14009; DB01043; DB00170; DB00454; DB00532; DB04817; DB00333; DB00763; DB01028; DB09241; DB00849; DB00959; DB06710; DB00379; DB06148; DB01110; DB06595; DB16236; DB00745; DB11763; DB00220; DB00238; DB00622; DB00184; DB01115; DB04868; DB12005; DB00435; DB00957; DB09074; DB16267; DB11632; DB01173; DB11837; DB04938; DB05467; DB00715; DB08883; DB01074; DB04930; DB12978; DB03575; DB01174; DB00252; DB13941; DB17472; DB11642; DB06209; DB14631; DB00635; DB01069; DB00818; DB00908; DB00481; DB08896; DB11853; DB16826; DB02709; DB00615; DB01045; DB11753; DB01201; DB08864; DB00503; DB06176; DB00296; DB00412; DB00778; DB01037; DB06739; DB01104; DB01236; DB00641; DB00398; DB15569; DB12548; DB09118; DB06729; DB01138; DB00675; DB12020; DB12095; DB00231; DB00624; DB13943; DB13944; DB13946; DB11712; DB01041; DB09499; DB04572; DB08816; DB00208; DB06137; DB00193; DB00755; DB12245; DB12808; DB00197; DB12255; DB00313; DB11613; DB08881; DB00661; DB09185; DB11739; DB00582; DB09068; DB14975; DB15035 Interacts with NA EC number EC 1.14.13.- Uniprot keywords 3D-structure; Alternative splicing; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 53293 Length 465 Aromaticity 0.12 Instability index 38.79 Isoelectric point 8.63 Charge (pH=7) 5.35 2D Binding mode Binding energy (Kcal/mol) -9.63  Molscript Map 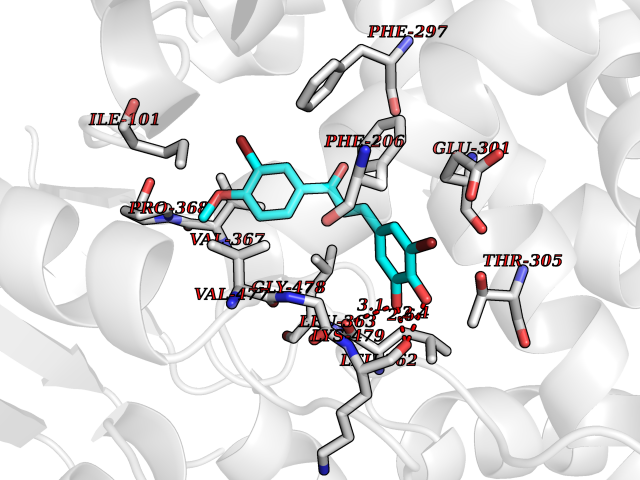 Pymol Map 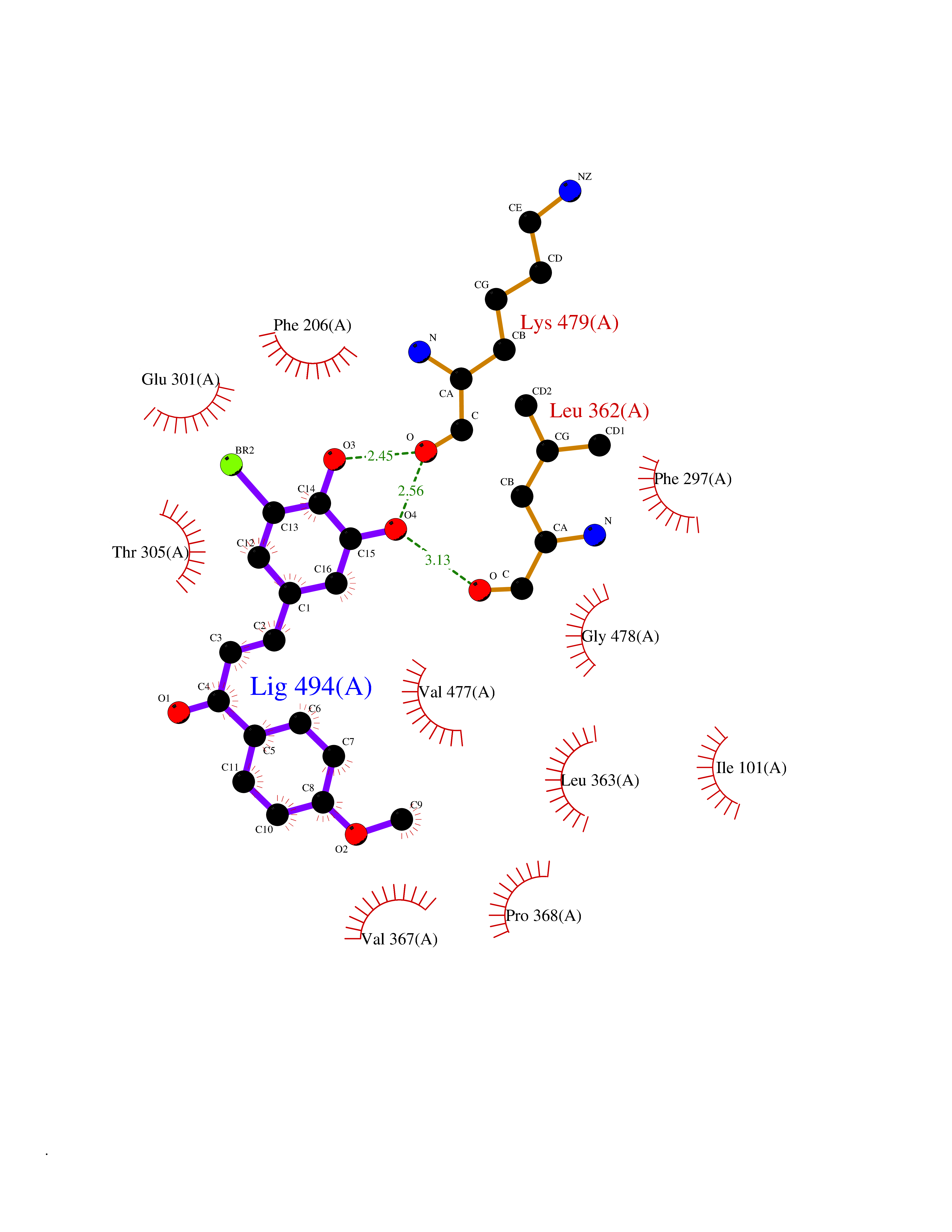 Ligplot Map 3D Binding mode Sequence GKLPPGPRPLPLLGNLLQMDRRGLLKSFLRFREKYGDVFTVHLGPRPVVMLCGVEAIREALVDKAEAFSGRGKIAMVDPFFRGYGVIFANGNRWKVLRRFSVTTMRDFGMGKRSVEERIQEEAQCLIEELRKSKGALMDPTFLFQSITANIICSIVFGKRFHYQDQEFLKMLNLFYQTFSLISSVFGQLFELFSGFLKHFPGAHRQVYKNLQEINAYIGHSVEKHRETLDPSAPRDLIDTYLLHMEKEKSNAHSEFSHQNLNLNTLSLFFAGTETTSTTLRYGFLLMLKYPHVAERVYREIEQVIGPHRPPELHDRAKMPYTEAVIYEIQRFSDLLPMGVPHIVTQHTSFRGYIIPKDTEVFLILSTALHDPHYFEKPDAFNPDHFLDANGALKKTEAFIPFSLGKRICLGEGIARAELFLFFTTILQNFSMASPVAPEDIDLTPQECGVGKIPPTYQIRFLPRH Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Guanidinoacetate N-methyltransferase | 3ORH | 7.06 | |
Target general information Gen name GAMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, RMT2 methyltransferase family Biochemical class Transferase Function Guanidinoacetate N-methyltransferase activity.Methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 2 (CCDS2) [MIM:612736]: An autosomal recessive disorder characterized by developmental delay and regression, intellectual disability, severe disturbance of expressive and cognitive speech, intractable seizures, movement disturbances, severe depletion of creatine and phosphocreatine in the brain, and accumulation of guanidinoacetic acid in brain and body fluids. {ECO:0000269|PubMed:12468279, ECO:0000269|PubMed:15108290, ECO:0000269|PubMed:15651030, ECO:0000269|PubMed:16293431, ECO:0000269|PubMed:16855203, ECO:0000269|PubMed:17101918, ECO:0000269|PubMed:17466557, ECO:0000269|PubMed:19388150, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:24415674, ECO:0000269|PubMed:8651275}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00148; DB02751; DB00536; DB13191; DB01752 Interacts with O95363; Q969Q5; Q9HCM9-2 EC number 2.1.1.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Disease variant; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 24656 Length 219 Aromaticity 0.11 Instability index 46.5 Isoelectric point 5.91 Charge (pH=7) -4.34 2D Binding mode Binding energy (Kcal/mol) -9.63  Molscript Map 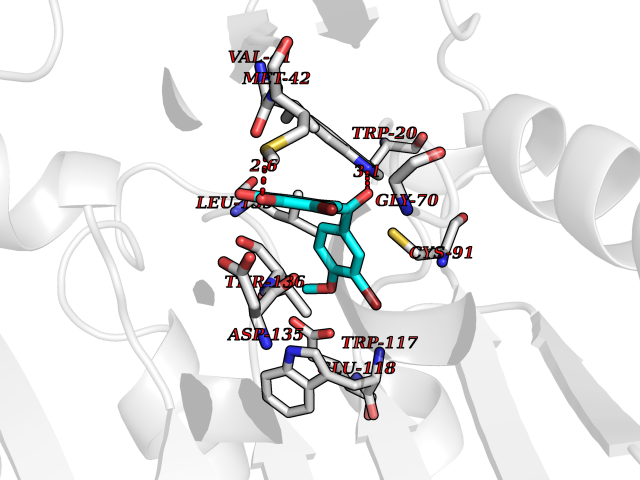 Pymol Map 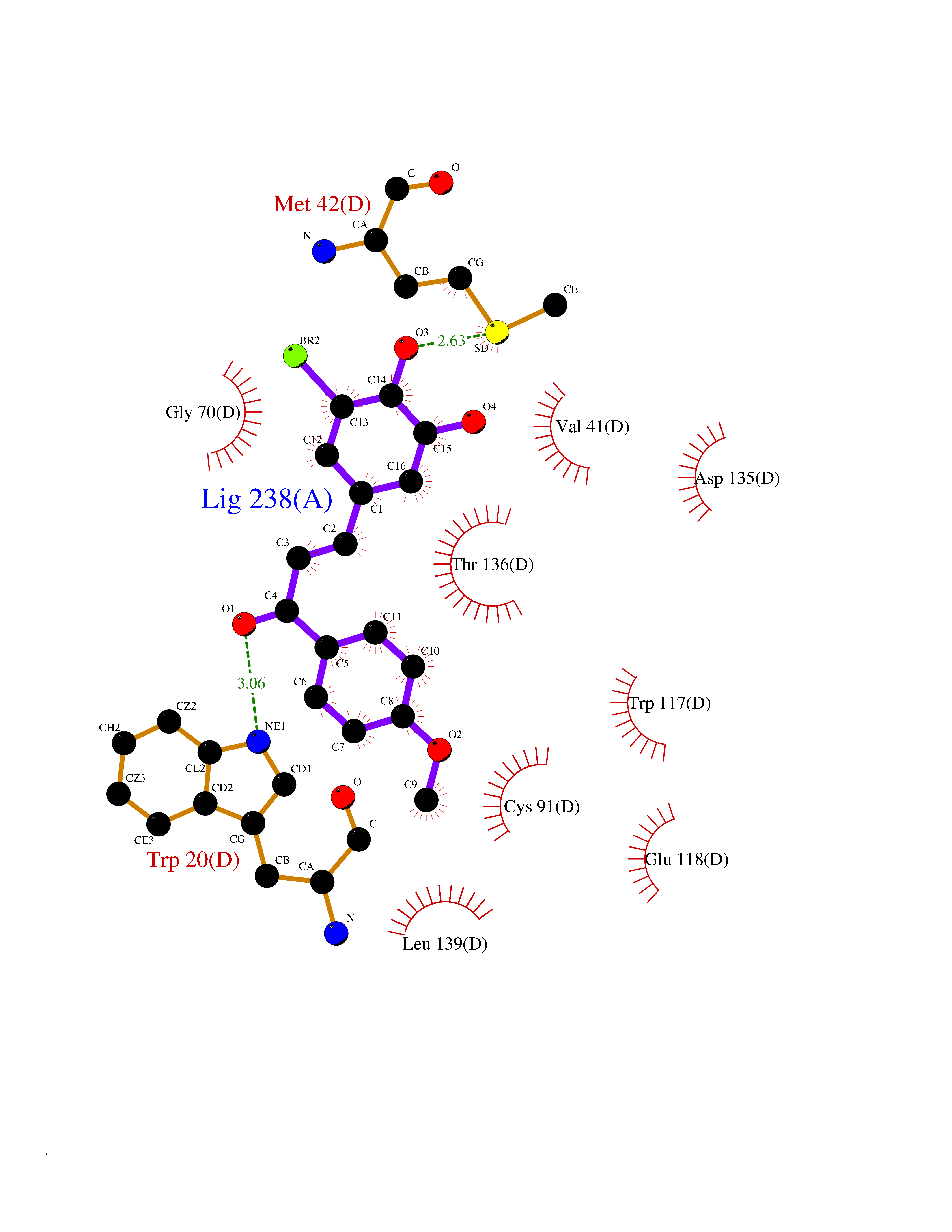 Ligplot Map 3D Binding mode Sequence PAWGAAPAAYDAADTHLRILGKPVMERWETPYMHALAAAASSKGGRVLEVGFGMAIAASKVQEAPIDEHWIIECNDGVFQRLRDWAPRQTHKVIPLKGLWEDVAPTLPDGHFDGILYDTYPLSEETWHTHQFNFIKNHAFRLLKPGGVLTYCNLTSWGELMKSKYSDITIMFEETQVPALLEAGFRRENIRTEVMALVPPADCRYYAFPQMITPLVTKG Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | WNK lysine-deficient protein kinase 3 (WNK3) | 5O2B | 7.06 | |
Target general information Gen name WNK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein kinase WNK3; Protein kinase with no lysine 3; Protein kinase lysine-deficient 3; KIAA1566 Protein family Protein kinase superfamily, Ser/Thr protein kinase family, WNK subfamily Biochemical class NA Function Serine/threonine kinase which plays an important role in the regulation of electrolyte homeostasis, cell signaling, survival and proliferation. Acts as an activator and inhibitor of sodium-coupled chloride cotransporters and potassium-coupled chloride cotransporters respectively. Phosphorylates WNK4. Regulates the phosphorylation of SLC12A1 and SLC12A2. Increases Ca(2+) influx mediated by TRPV5 and TRPV6 by enhancing their membrane expression level via a kinase-dependent pathway. Inhibits the activity of KCNJ1 by decreasing its expression at the cell membrane in a non-catalytic manner. Related diseases Prieto syndrome (PRS) [MIM:309610]: An X-linked recessive disorder characterized by impaired intellectual development, developmental delay, autism spectrum disorder, variable epilepsy, craniofacial dysmorphism, and structural brain abnormalities including polymicrogyria and cerebral atrophy. {ECO:0000269|PubMed:35678782}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P42574; P52954; Q04864-2 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Disease variant; Intellectual disability; Kinase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30818.4 Length 269 Aromaticity 0.1 Instability index 41.03 Isoelectric point 6.27 Charge (pH=7) -2.07 2D Binding mode Binding energy (Kcal/mol) -9.64 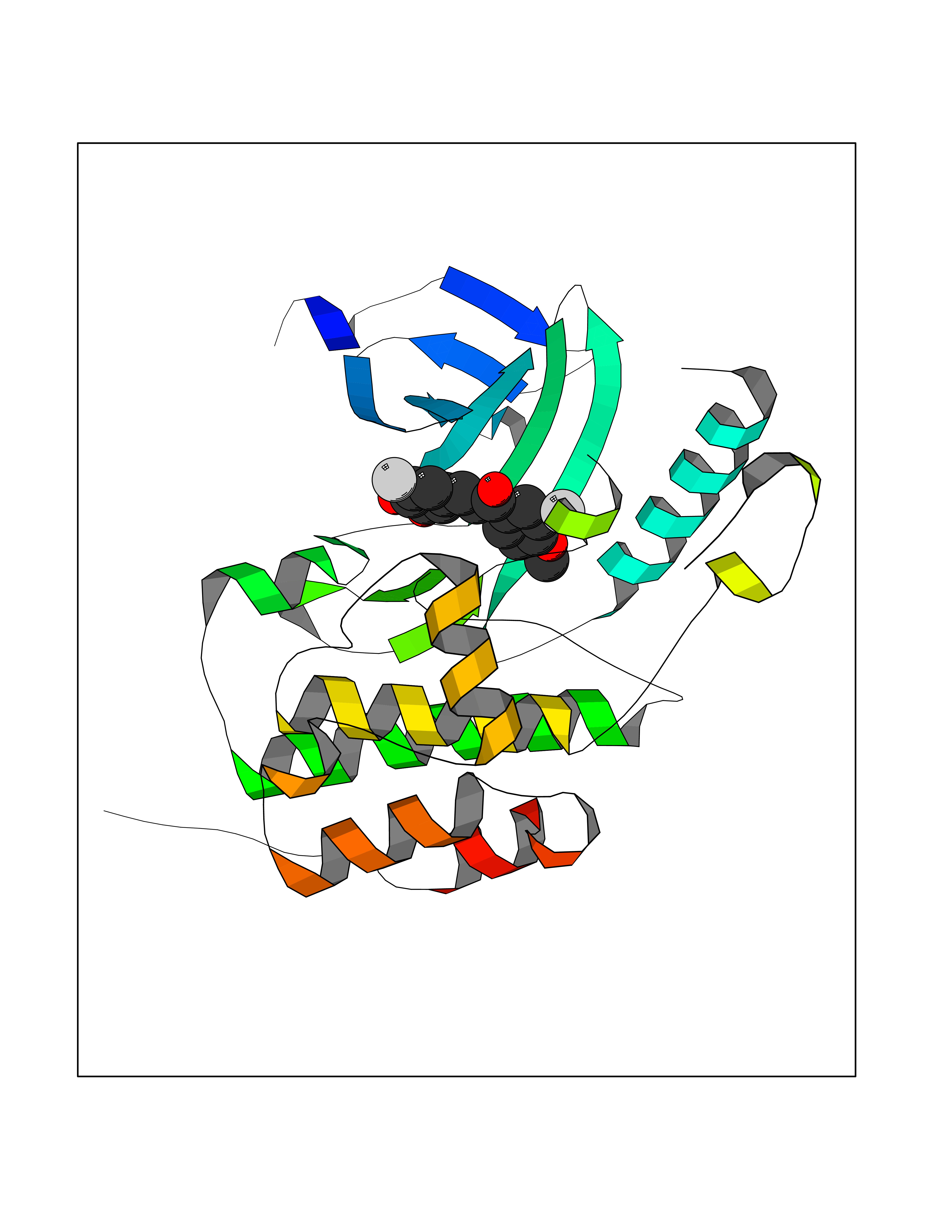 Molscript Map 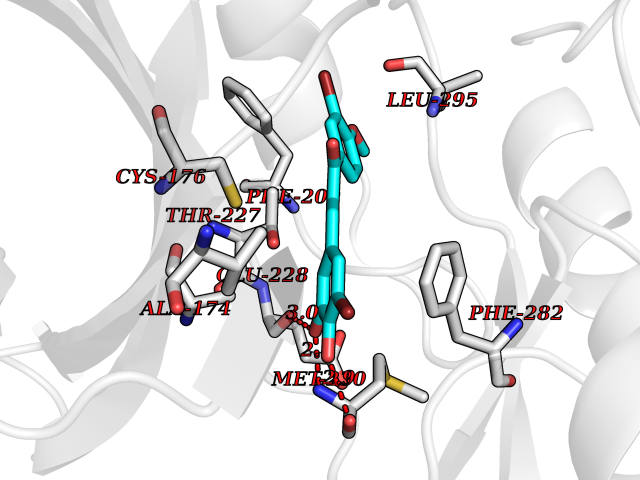 Pymol Map 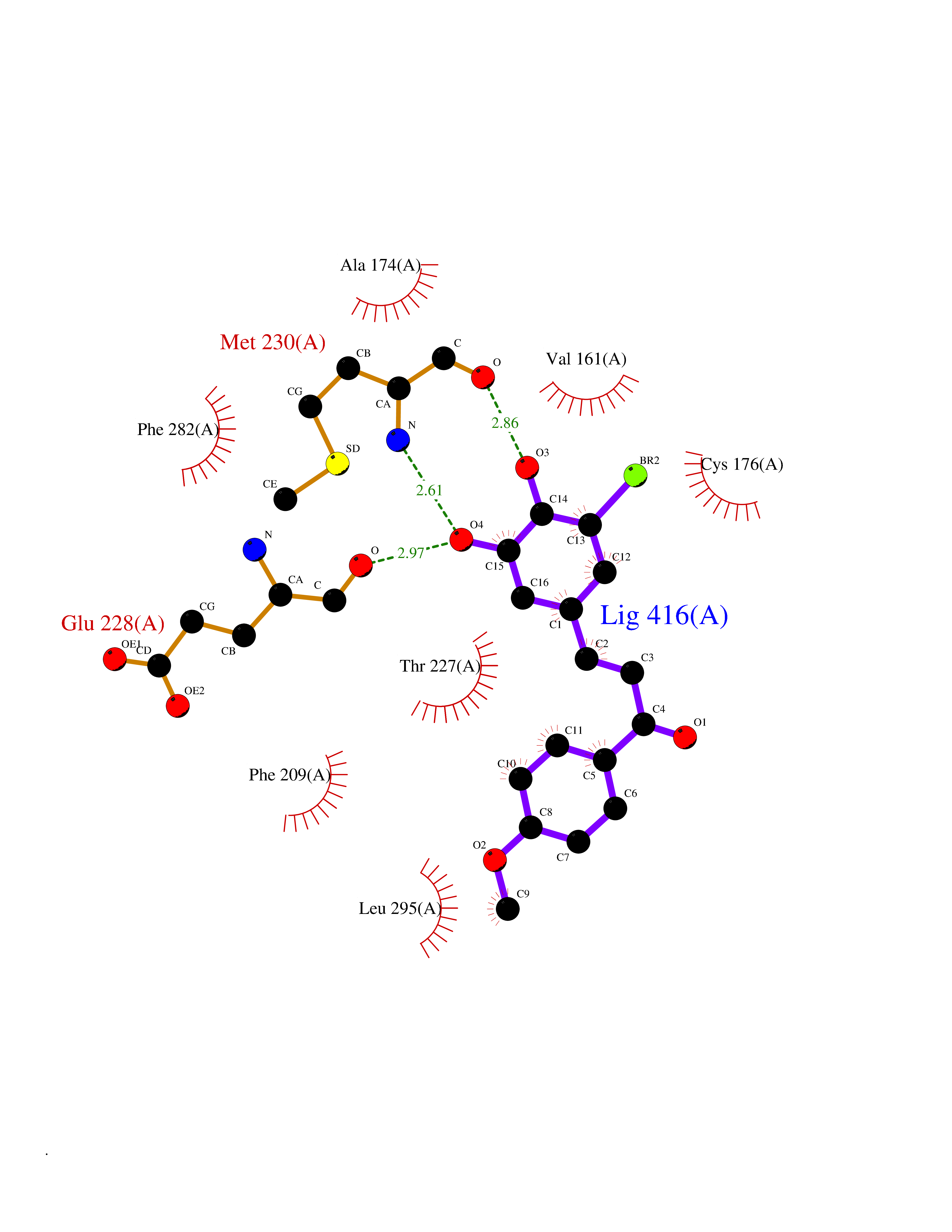 Ligplot Map 3D Binding mode Sequence MEAEMKAVATSPSGRFLKFDIELGRGAFKTVYKGLDTETWVEVAWCELQLTKAEQQRFKEEAEMLKGLQHPNIVRFYDSWESIKCIVLVTELMTSGTLKTYLKRFKVMKPKVLRSWCRQILKGLQFLHTRTPPIIHRDLKCDNIFITGPTGSVKIGDLGLATLMIGTPEFMAPEMYEEHYDESVDVYAFGMCMLEMATSEYPYSECQNAAQIYRKVTSGIKPASFNKVTDPEVKEIIEGCIRQNKSERLSIRDLLNHAFFAEDTGLRVE Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Aggrecanase-1 (ADAMTS4) | 2RJP | 7.06 | |
Target general information Gen name ADAMTS4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aggrecanase 1; ADMP-1; ADAMTS4; ADAM-TS4; ADAM-TS 4; A disintegrin and metalloproteinase with thrombospondin motifs 4 Protein family NA Biochemical class Peptidase Function Cleaves aggrecan, a cartilage proteoglycan, and may be involved in its turnover. May play an important role in the destruction of aggrecan in arthritic diseases. Could also be a critical factor in the exacerbation of neurodegeneration in Alzheimer disease. Cleaves aggrecan at the '392-Glu-|-Ala-393' site. Related diseases Familial male precocious puberty (FMPP) [MIM:176410]: In FMPP the receptor is constitutively activated. {ECO:0000269|PubMed:11134146, ECO:0000269|PubMed:11391350, ECO:0000269|PubMed:7629248, ECO:0000269|PubMed:7692306, ECO:0000269|PubMed:7714085, ECO:0000269|PubMed:7757065, ECO:0000269|PubMed:8281137, ECO:0000269|PubMed:8829636, ECO:0000269|PubMed:8929952, ECO:0000269|PubMed:9467560, ECO:0000269|PubMed:9661624}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Luteinizing hormone resistance (LHR) [MIM:238320]: An autosomal recessive disorder characterized by unresponsiveness to luteinizing hormone, defective sexual development in males, and defective follicular development and ovulation, amenorrhea and infertility in females. Two forms of the disorder have been defined in males. Type 1 is a severe form characterized by complete 46,XY male pseudohermaphroditism, low testosterone and high luteinizing hormone levels, total lack of responsiveness to luteinizing and chorionic gonadotropin hormones, lack of breast development, and absent development of secondary male sex characteristics. Type 2, a milder form, displays a broader range of phenotypic expression ranging from micropenis to severe hypospadias. {ECO:0000269|PubMed:12050206, ECO:0000269|PubMed:15372531, ECO:0000269|PubMed:15472221, ECO:0000269|PubMed:19551906, ECO:0000269|PubMed:7719343, ECO:0000269|PubMed:8559204, ECO:0000269|PubMed:9215288, ECO:0000269|PubMed:9514160, ECO:0000269|PubMed:9626144, ECO:0000269|PubMed:9626653}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06822 Interacts with NA EC number EC 3.4.24.82 Uniprot keywords 3D-structure; Alternative splicing; Cleavage on pair of basic residues; Direct protein sequencing; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 31309.4 Length 291 Aromaticity 0.07 Instability index 42.35 Isoelectric point 5.97 Charge (pH=7) -7.02 2D Binding mode Binding energy (Kcal/mol) -9.62 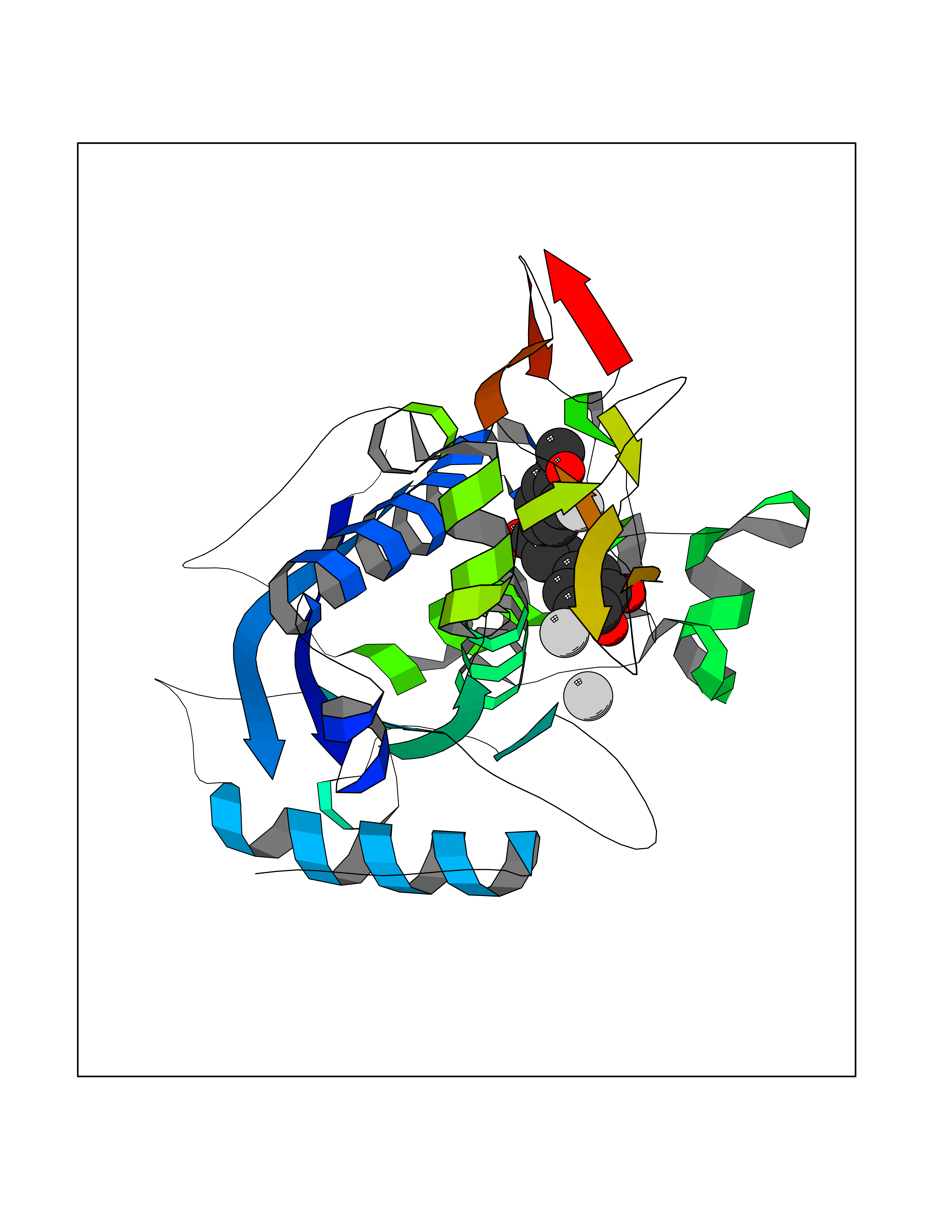 Molscript Map 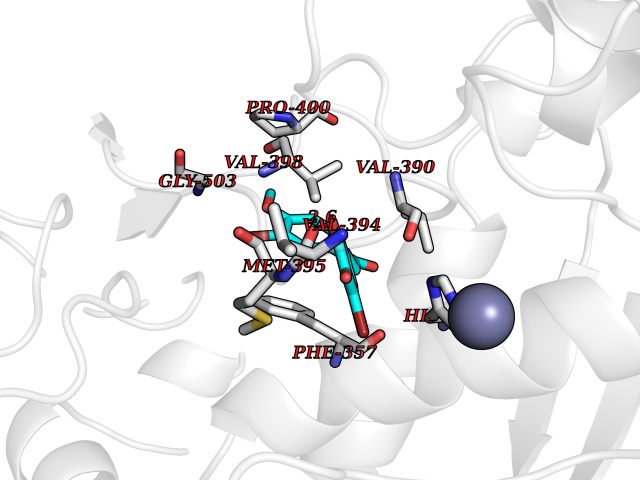 Pymol Map 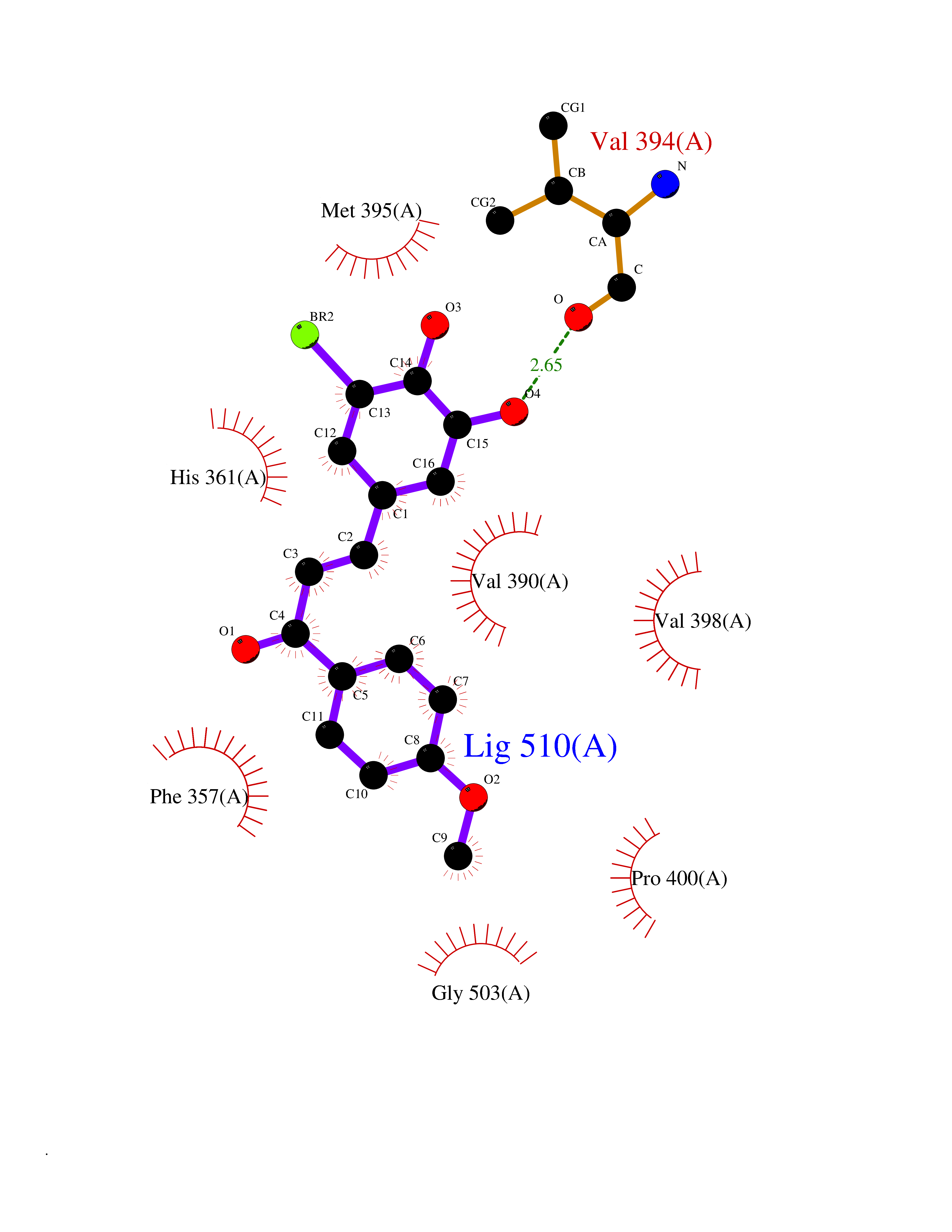 Ligplot Map 3D Binding mode Sequence ASLSRFVETLVVADDKMAAFHGAGLKRYLLTVMAAAAKAFKHPSIRNPVSLVVTRLVILEGPQVGPSAAQTLRSFCAWQRGLNTPEDSDPDHFDTAILFTRQDLCGVSTCDTLGMADVGTVCDPARSCAIVEDDGLQSAFTAAHQLGHVFNMLHDNSKPCISLNGPLSTSRHVMAPVMAHVDPEEPWSPCSARFITDFLDNGYGHCLLDKPEAPLHLPVTFPGKDYDADRQCQLTFGPDSRHCPQLPPPCAALWCSGHLNGHAMCQTKHSPWADGTPCGPAQACMGGRCLH Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | T-cell receptor beta constant 1 (TRBC1) | 4LCC | 7.06 | |
Target general information Gen name TRBC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TCRBC1; BV05S1J2.2 Protein family NA Biochemical class NA Function Alpha-beta T cell receptors are antigen specific receptors which are essential to the immune response and are present on the cell surface of T lymphocytes. Recognize peptide-major histocompatibility (MH) (pMH) complexes that are displayed by antigen presenting cells (APC), a prerequisite for efficient T cell adaptive immunity against pathogens. Binding of alpha-beta TR to pMH complex initiates TR-CD3 clustering on the cell surface and intracellular activation of LCK that phosphorylates the ITAM motifs of CD3G, CD3D, CD3E and CD247 enabling the recruitment of ZAP70. In turn, ZAP70 phosphorylates LAT, which recruits numerous signaling molecules to form the LAT signalosome. The LAT signalosome propagates signal branching to three major signaling pathways, the calcium, the mitogen-activated protein kinase (MAPK) kinase and the nuclear factor NF-kappa-B (NF-kB) pathways, leading to the mobilization of transcription factors that are critical for gene expression and essential for T cell growth and differentiation. The T cell repertoire is generated in the thymus, by V-(D)-J rearrangement. This repertoire is then shaped by intrathymic selection events to generate a peripheral T cell pool of self-MH restricted, non-autoaggressive T cells. Post-thymic interaction of alpha-beta TR with the pMH complexes shapes TR structural and functional avidity. Constant region of T cell receptor (TR) alpha chain. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02740 Interacts with NA EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Cell membrane; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Proteomics identification; Receptor; Reference proteome; T cell receptor; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID C,B,A Molecular weight (Da) 84668.2 Length 736 Aromaticity 0.13 Instability index 39.96 Isoelectric point 5.8 Charge (pH=7) -14.36 2D Binding mode Binding energy (Kcal/mol) -9.62 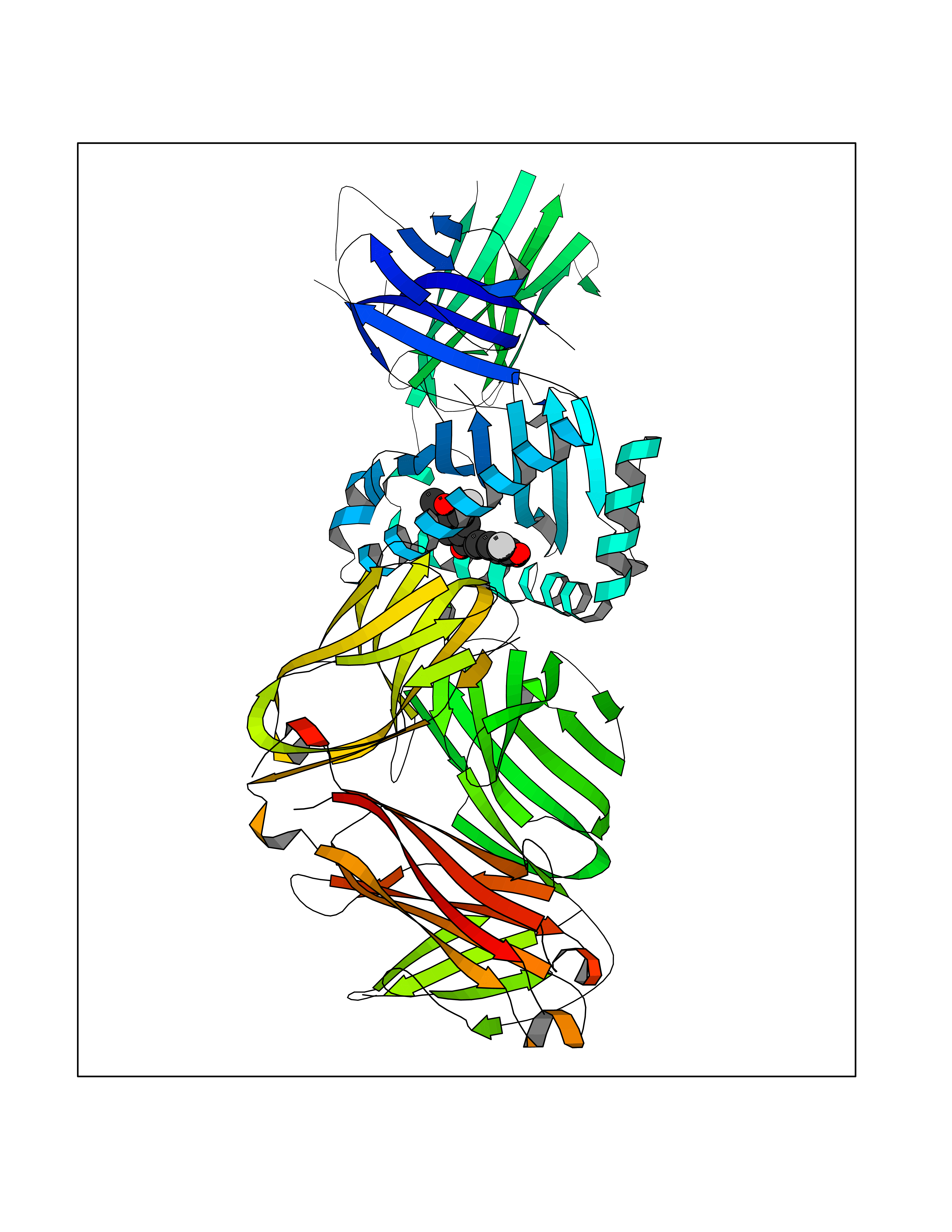 Molscript Map  Pymol Map 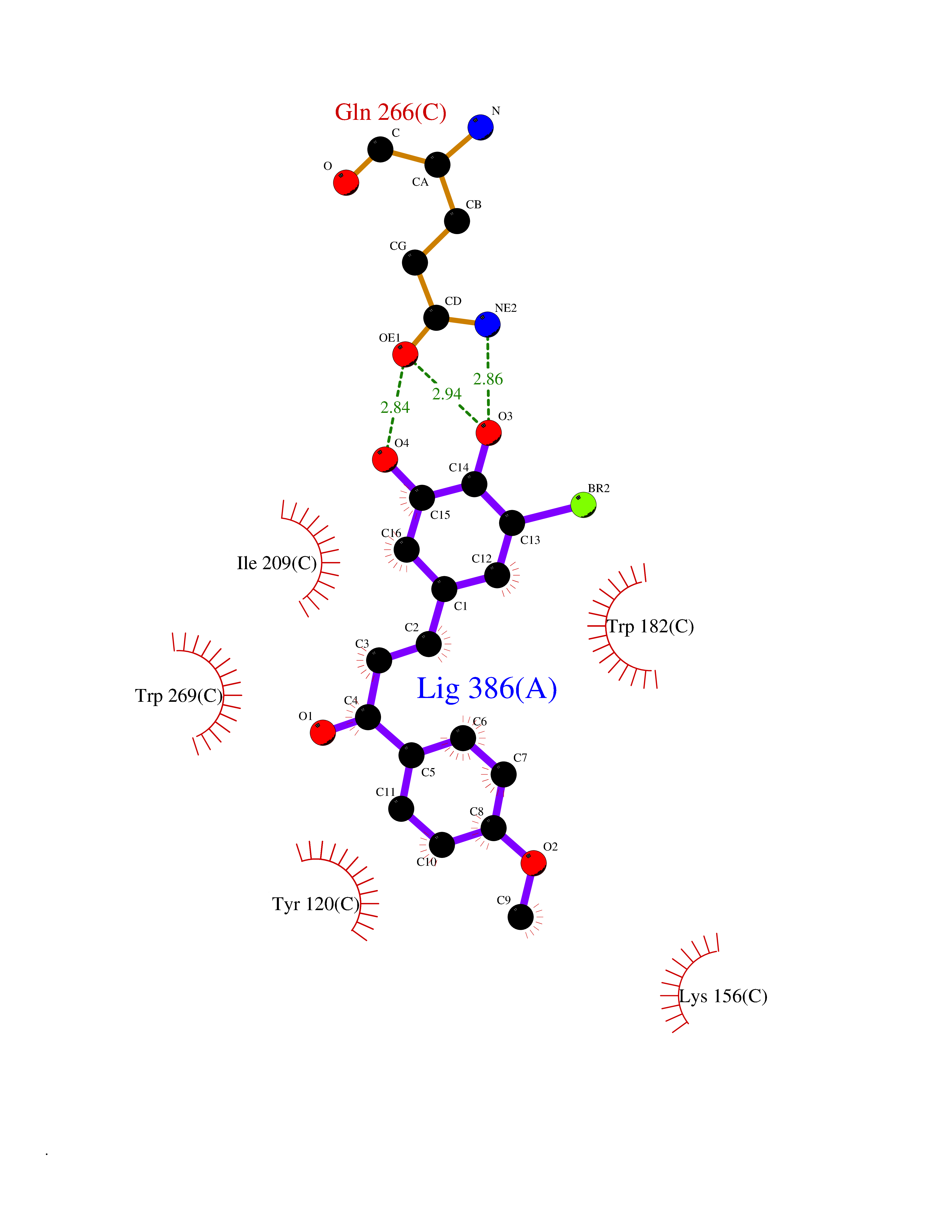 Ligplot Map 3D Binding mode Sequence IQRPPKIQVYSRHPPNYLNCYVYGFHPPQIEIDLLKIKSEQSDLSFSKDWSFYLLSHATPNSKDQYSCRVKHVTLEQPRIVKWDRTHSLRYFRLGISEIPEFISAGYVDSHPITMYNSVSQLKEPRALWMEENLAPDHWERYTQLLRGWQQMFKVELKQLQHHYNHSGFHTYQRMIGCELLEDGSITGFLQYAYDGQDFLIFNKDTLSWMAMDNVADIIRRVWEANQHELLYQKNWLEEECIAWLKRFLEYGKDALQRTEPPKVRVNHKTTLYCRAYGFYPPEISINWMKNGEEIFQDTDYGGILPSGDGTYQTWVSVELGDIYSCHVEHGGVHMVLQGFQQNIDQPTEMTATEGAIVQINCTYQTSGFNGLFWYQQHAGEAPTFLSYNVLDGLEEKGRFSSFLSRSKGYSYLLLKELQMKDSASYLCAVKDSNYQLIWGAGTKLIIKPNIQNPDPAVYQLRDSKSSDKSVCLFTDFDKDSDVYITDKKSNSAVAWSNAGVTQTPKFQVLKTGQSMTLQCAQDMNHNSMYWYRQDPGMGLRLIYYSASEGTTDKGEVPNGYNVSRLNKREFSLRLESAAPSQTSVYFCASSVWTGEGSGELFFGEGSRLTVLEDLKNVFPPEVAVFEPSEAEISHTQKATLVCLATGFYPDHVELSWWVNGKEVHSGVCTDPQPLKEQPALNDSRYALSSRLRVSATFWQNPRNHFRCQVQFYGLSENDEWKPVTQIVSAEAWGRA Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Proteinase-activated receptor 1 | 3VW7 | 7.05 | |
Target general information Gen name F2R Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TR;PAR1;CF2R Protein family G-protein coupled receptor 1 family Biochemical class Signaling protein / antagonist Function G-protein alpha-subunit binding.G-protein beta-subunit binding.G-protein coupled receptor activity.Receptor binding.Thrombin-activated receptor activity. Related diseases 3-ketothiolase deficiency (3KTD) [MIM:203750]: An autosomal recessive inborn error of isoleucine catabolism characterized by intermittent ketoacidotic attacks associated with unconsciousness. Some patients die during an attack or are mentally retarded. Urinary excretion of 2-methyl-3-hydroxybutyric acid, 2-methylacetoacetic acid, triglylglycine, butanone is increased. It seems likely that the severity of this disease correlates better with the environmental or acquired factors than with the ACAT1 genotype. {ECO:0000269|PubMed:1346617, ECO:0000269|PubMed:1715688, ECO:0000269|PubMed:7728148, ECO:0000269|PubMed:9744475}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05361; DB00086; DB11300; DB09030 Interacts with Q03135; Q9UNN8 EC number NA Uniprot keywords 3D-structure; Blood coagulation; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Hemostasis; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 31193.7 Length 282 Aromaticity 0.16 Instability index 36.7 Isoelectric point 8.2 Charge (pH=7) 2.98 2D Binding mode Binding energy (Kcal/mol) -9.61  Molscript Map  Pymol Map 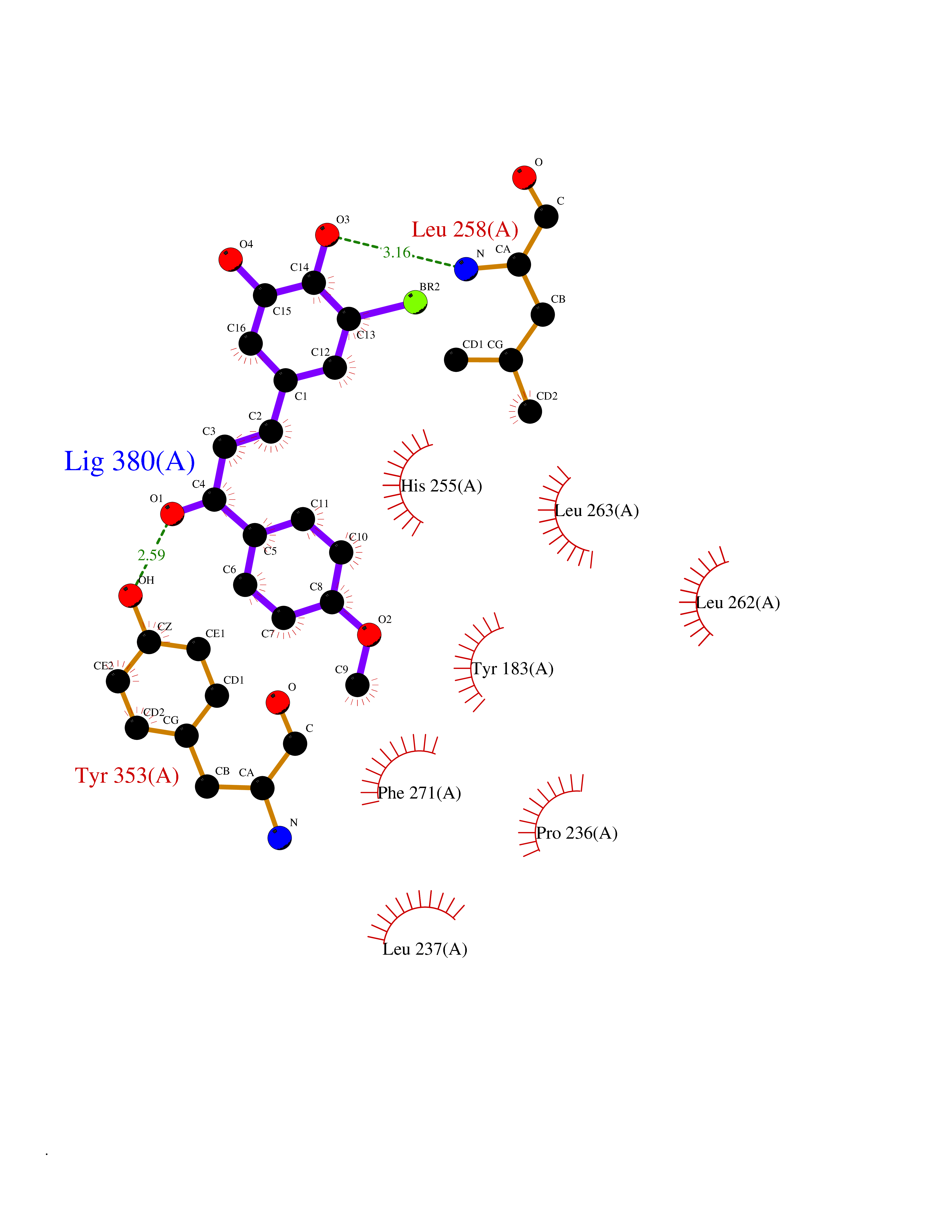 Ligplot Map 3D Binding mode Sequence DASGYLTSSWLTLFVPSVYTGVFVVSLPLNIMAIVVFILKMKVKKPAVVYMLHLATADVLFVSVLPFKISYYFSGSDWQFGSELCRFVTAAFYCNMYASILLMTVISIDRFLAVVYPMRTLGRASFTCLAIWALAIAGVVPLLLKEQTIQVPGLGITTCHDVLSETLLEGYYAYYFSAFSAVFFFVPLIISTVCYVSIIRCLSSSAANRSKKSRALFLSAAVFCIFIICFGPTNVLLIAHYSFLSHTSTTEAAYFAYLLCVCVSSISCCIDPLIYYYASSEC Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Acetyl-CoA carboxylase (ACC) (EC 6.4.1.2) (Fatty acid synthetase 3) (mRNA transport-defective protein 7) [Includes: Biotin carboxylase (EC 6.3.4.14)] | 1UYS | 7.05 | |
Target general information Gen name ACC1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms MTR7;YNR016C;N3175;ABP2;FAS3 Protein family NA Biochemical class NA Function Carries out three functions: biotin carboxyl carrier protein, biotin carboxylase and carboxyltransferase. Involved in the synthesis of very-long-chain fatty acid synthesis which is required to maintain a functional nuclear envelope. Required for acylation and vacuolar membrane association of VAC8 which is necessary to maintain a normal morphology of the vacuole. {ECO:0000269|PubMed:10757783, ECO:0000269|PubMed:12730220, ECO:0000269|PubMed:6103540, ECO:0000269|PubMed:6108218, ECO:0000269|PubMed:8943372}." Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q00955 EC number 6.3.4.14; 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Endoplasmic reticulum; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Manganese; Membrane; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome Protein physicochemical properties Chain ID B,C Molecular weight (Da) 145619 Length 1328 Aromaticity 0.1 Instability index 30.31 Isoelectric point 5.32 Charge (pH=7) -26.79 2D Binding mode Binding energy (Kcal/mol) -9.62 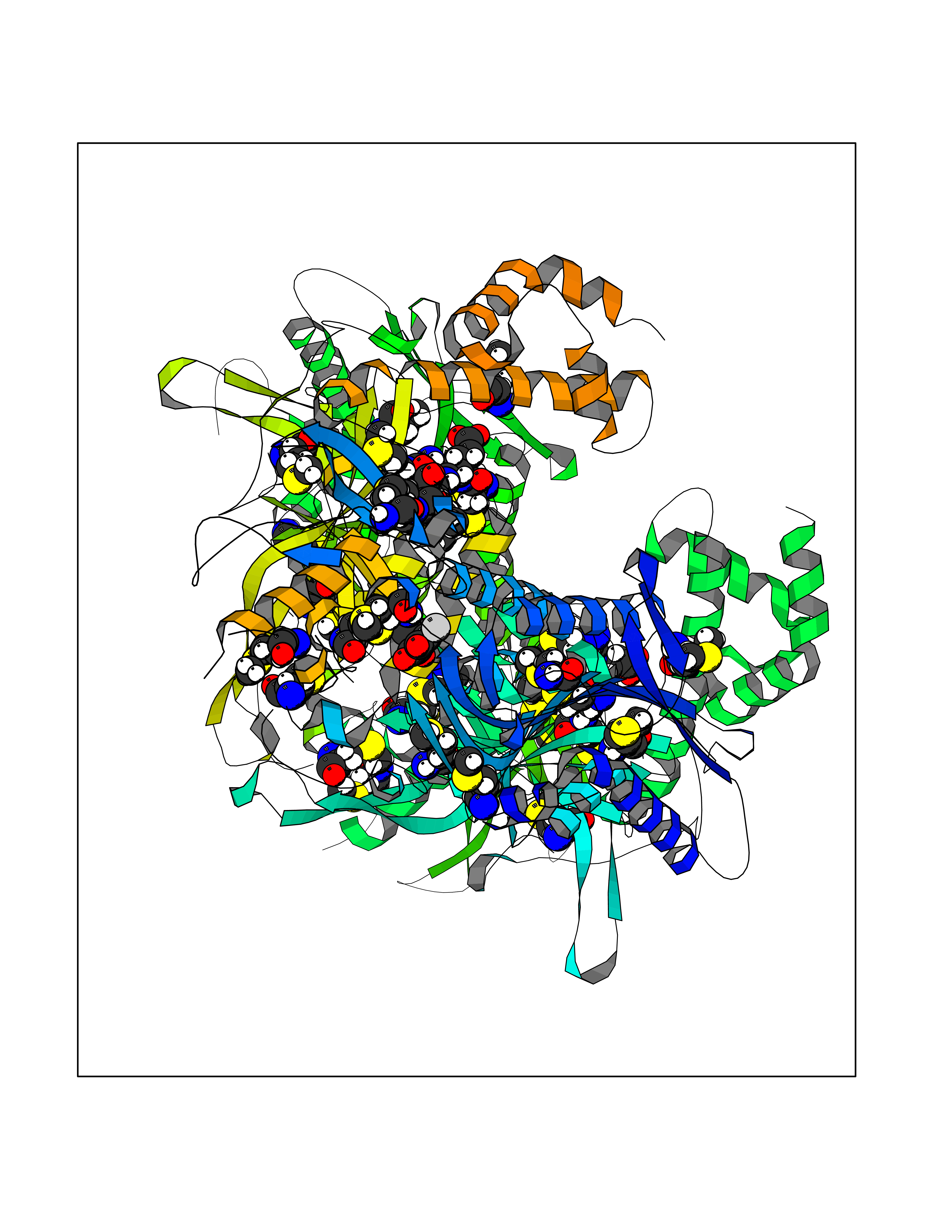 Molscript Map  Pymol Map 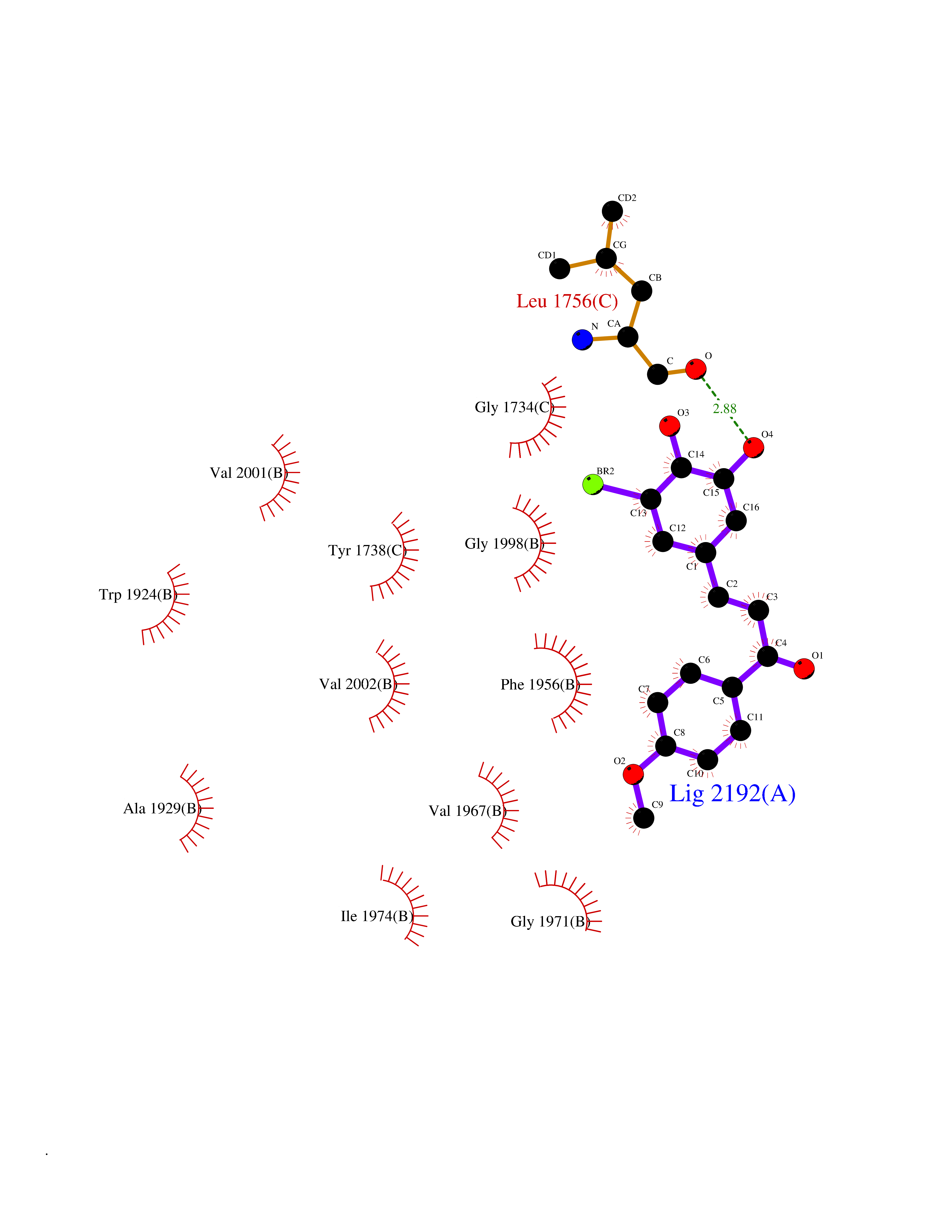 Ligplot Map 3D Binding mode Sequence WLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGLPIATPYPVKEWLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGL Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Glutamate receptor ionotropic NMDA 2A (NMDAR2A) | 5I2N | 7.05 | |
Target general information Gen name GRIN2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NR2A; NMDA receptor NR2A; N-methyl D-aspartate receptor subtype 2A; HNR2A; Glutamate receptor ionotropic, NMDA 2A; Glutamate [NMDA] receptor subunit epsilon-1; GluN2A Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, NR1/GRIN1 subfamily Biochemical class Glutamate-gated ion channel Function Channel activation requires binding of the neurotransmitter glutamate to the epsilon subunit, glycine binding to the zeta subunit, plus membrane depolarization to eliminate channel inhibition by Mg(2+). Sensitivity to glutamate and channel kinetics depend on the subunit composition; channels containing GRIN1 and GRIN2A have higher sensitivity to glutamate and faster kinetics than channels formed by GRIN1 and GRIN2B. Contributes to the slow phase of excitatory postsynaptic current, long-term synaptic potentiation, and learning. Component of NMDA receptor complexes that function as heterotetrameric, ligand-gated ion channels with high calcium permeability and voltage-dependent sensitivity to magnesium. Related diseases Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant (NDHMSD) [MIM:614254]: An autosomal dominant neurodevelopmental disorder characterized by severe intellectual disability and developmental delay, absent speech, muscular hypotonia, dyskinesia, and hyperkinetic movements. Cortical blindness, cerebral atrophy, and seizures are present in some patients. {ECO:0000269|PubMed:21376300, ECO:0000269|PubMed:25167861, ECO:0000269|PubMed:25864721, ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:28228639, ECO:0000269|PubMed:28389307, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal recessive (NDHMSR) [MIM:617820]: An autosomal recessive neurodevelopmental disorder characterized by severe intellectual disability and psychomotor developmental delay, involuntary and stereotypic movements, spasticity, and inability to walk without support. Intractable seizures manifest in some patients. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28051072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 101 (DEE101) [MIM:619814]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE101 is an autosomal recessive, severe form characterized by onset of seizures in early infancy. Death in infancy may occur. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:34611970}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01931; DB00659; DB06151; DB08838; DB01238; DB00289; DB05824; DB04620; DB03929; DB00647; DB00843; DB00228; DB11823; DB13146; DB06741; DB00142; DB00874; DB08954; DB06738; DB09409; DB09481; DB01043; DB00454; DB00333; DB04896; DB01173; DB00312; DB01174; DB01708; DB00418; DB00193 Interacts with P05067; P35637; Q12879-1; Q13224; Q62936 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Ligand-gated ion channel; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Zinc Protein physicochemical properties Chain ID B Molecular weight (Da) 63184.8 Length 559 Aromaticity 0.1 Instability index 29.67 Isoelectric point 8.51 Charge (pH=7) 6.66 2D Binding mode Binding energy (Kcal/mol) -9.62 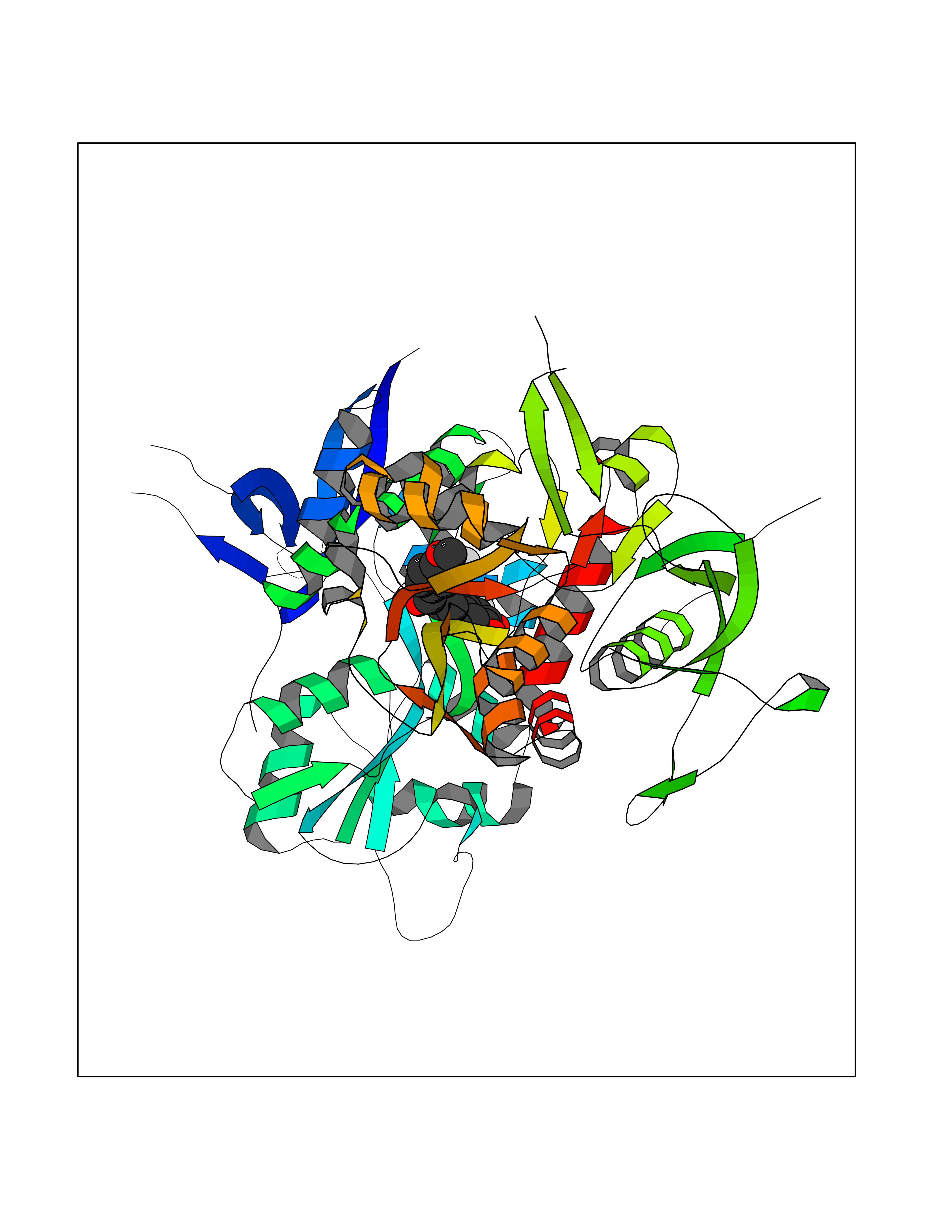 Molscript Map 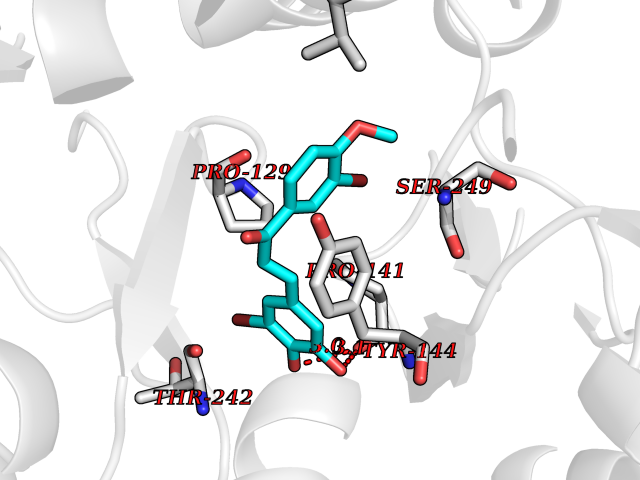 Pymol Map 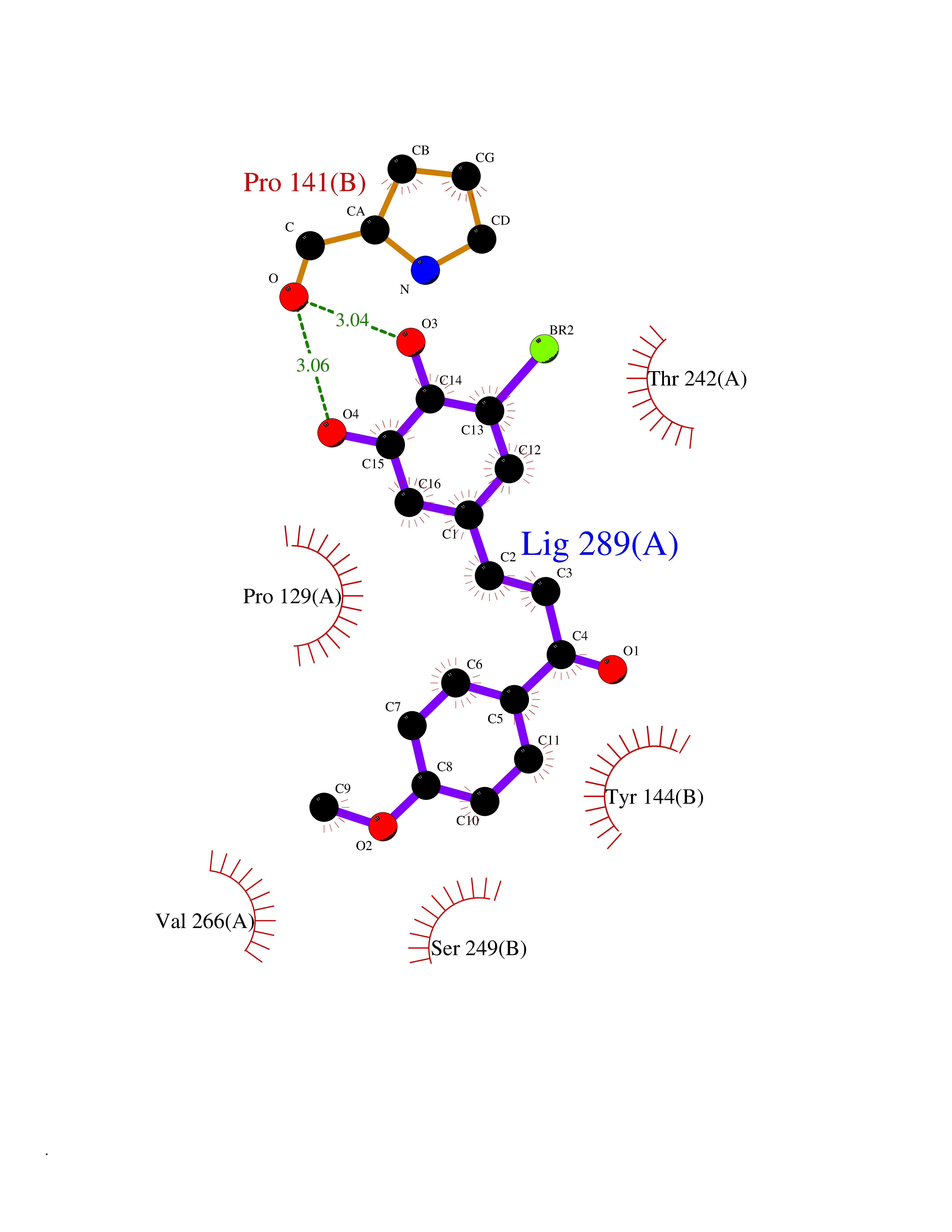 Ligplot Map 3D Binding mode Sequence DNHLSIVTLEEAPFVIVEDIDPETCVRNTVPCRKFVKINNSTNEGMNVKKCCKGFCIDILKKLSRTVKFTYDLYLVTNGKHGKKVNNVWNGMIGEVVYQRAVMAVGSLTINEERSEVVDFSVPFVETGISVMVSRGTQVTGLSDKKFQRPHDYSPPFRFGTVPNGSTERNIRNNYPYMHQYMTKFNQKGVEDALVSLKTGKLDAFIYDAAVLNYKAGRDEGCKLVTIGSGYIFATTGYGIALQKGSPWKRQIDLALLQFVGDGEMEELETLWLTGICMSTRLKIVTIHQEPFVYVKPTLSDGTCKEEFTVNGDPVKKVICTGPNDTSPGSPRHTVPQCCYGFCIDLLIKLARTMNFTYEVHLVADGKFGTQERVNKKEWNGMMGELLSGQADMIVAPLTINNERAQYIEFSKPFKYQGLTILVKKGTRITGINDPRLRNPSDKFIYATVKQSSVDIYFRRQVELSTMYRHMEKHNYESAAEAIQAVRDNKLHAFIWDSAVLEFEASQKCDLVTTGELFFRSGFGIGMRKDSPWKQNVSLSILKSHENGFMEDLDKTWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Dihydroorotate dehydrogenase (DHODH) | 4OQV | 7.05 | |
Target general information Gen name DHODH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dihydroorotate oxidase; Dihydroorotate dehydrogenase (quinone), mitochondrial; DHOdehase; DHODH Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07559; DB07561; DB08172; DB08169; DB07443; DB07978; DB07975; DB04281; DB08249; DB07977; DB07976; DB04583; DB08008; DB01117; DB03523; DB03480; DB02613; DB04147; DB03247; DB01097; DB06481; DB08006; DB02262; DB05125; DB08880; DB07646 Interacts with Q6ZMZ0; P49638 EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Disease variant; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 38341.4 Length 353 Aromaticity 0.05 Instability index 39.27 Isoelectric point 9.28 Charge (pH=7) 5.52 2D Binding mode Binding energy (Kcal/mol) -9.62  Molscript Map 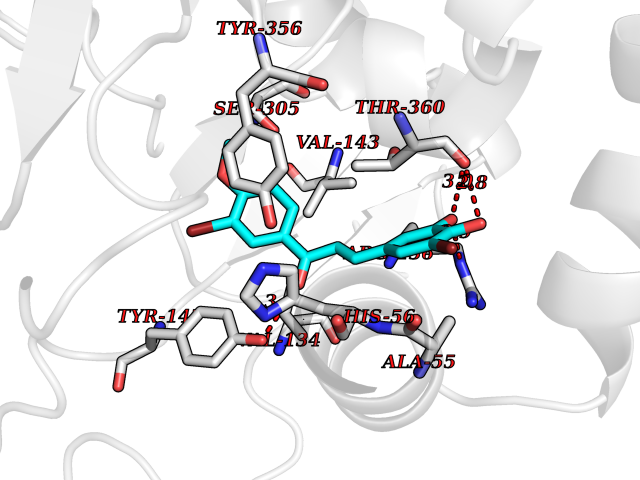 Pymol Map 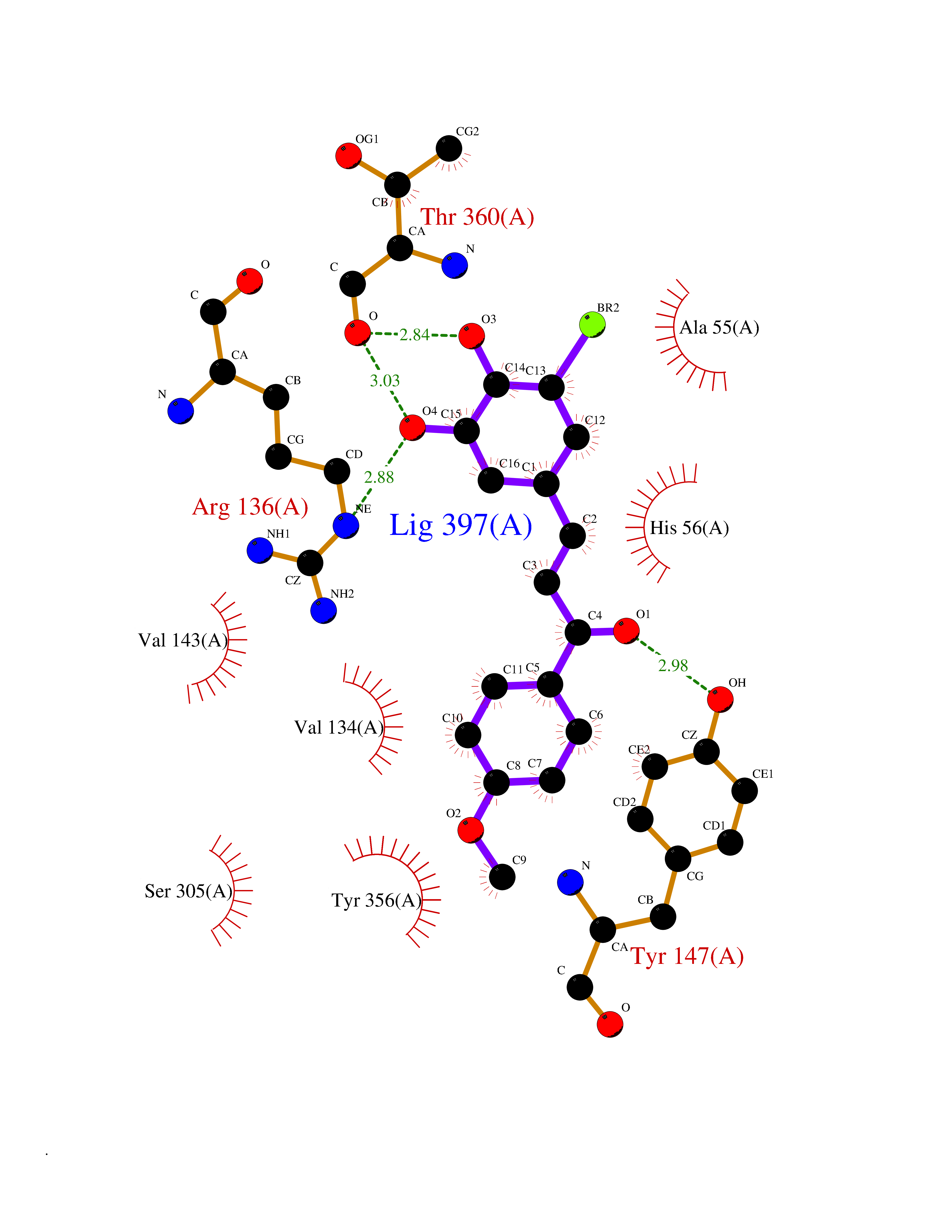 Ligplot Map 3D Binding mode Sequence DERFYAEHLMPTLQGLLDPESAHRLAVRFTSLGLLPRARFQDSDMLEVRVLGHKFRNPVGIAAGFDKHGEAVDGLYKMGFGFVEIGSVTPKPQEGNPRPRVFRLPEDQAVINRYGFNSHGLSVVEHRLRARQQKQAKLTEDGLPLGVNLGKNKTSVDAAEDYAEGVRVLGPLADYLVVNVSSPGKAELRRLLTKVLQERDGLRRVHRPAVLVKIAPDLTSQDKEDIASVVKELGIDGLIVTNTTVSRPAGLQGALRSETGGLSGKPLRDLSTQTIREMYALTQGRVPIIGVGGVSSGQDALEKIRAGASLVQLYTALTFWGPPVVGKVKRELEALLKEQGFGGVTDAIGADHR Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Rhinovirus Protease 3C (HRV P3C) | 1FPN | 7.05 | |
Target general information Gen name HRV P3C Organism Human rhinovirus 2 (HRV-2) Uniprot ID TTD ID Synonyms Rhinovirus P3C Protein family Picornaviruses polyprotein family Biochemical class NA Function Capsid protein VP1: Forms an icosahedral capsid of pseudo T=3 symmetry with capsid proteins VP2 and VP3. The capsid is 300 Angstroms in diameter, composed of 60 copies of each capsid protein and enclosing the viral positive strand RNA genome. Capsid protein VP1 mainly forms the vertices of the capsid. Capsid protein VP1 interacts with host VLDLR to provide virion attachment to target host cells. This attachment induces virion internalization. Tyrosine kinases are probably involved in the entry process. After binding to its receptor, the capsid undergoes conformational changes. Capsid protein VP1 N-terminus (that contains an amphipathic alpha-helix) and capsid protein VP4 are externalized. Together, they shape a pore in the host membrane through which viral genome is translocated to host cell cytoplasm. After genome has been released, the channel shrinks (By similarity). Related diseases Charcot-Marie-Tooth disease, axonal, 2DD (CMT2DD) [MIM:618036]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:29499166}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypomagnesemia, seizures, and impaired intellectual development 2 (HOMGSMR2) [MIM:618314]: An autosomal dominant disease characterized by generalized seizures in infancy, severe hypomagnesemia, and renal magnesium wasting. Seizures persist despite magnesium supplementation and are associated with significant intellectual disability. {ECO:0000269|PubMed:30388404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02313; DB03017 Interacts with NA EC number EC 3.4.22.28 Uniprot keywords 3D-structure; Activation of host autophagy by virus; ATP-binding; Autocatalytic cleavage; Capsid protein; Covalent protein-RNA linkage; DNA replication; Eukaryotic host gene expression shutoff by virus; Eukaryotic host translation shutoff by virus; Helicase; Host cytoplasm; Host cytoplasmic vesicle; Host gene expression shutoff by virus; Host membrane; Host mRNA suppression by virus; Host nucleus; Host-virus interaction; Hydrolase; Inhibition of host innate immune response by virus; Inhibition of host mRNA nuclear export by virus; Inhibition of host RIG-I by virus; Inhibition of host RLR pathway by virus; Ion channel; Ion transport; Lipoprotein; Magnesium; Membrane; Metal-binding; Myristate; Nucleotide-binding; Nucleotidyltransferase; Phosphoprotein; Pore-mediated penetration of viral genome into host cell; Protease; Repeat; RNA-binding; RNA-directed RNA polymerase; T=pseudo3 icosahedral capsid protein; Thiol protease; Transferase; Transport; Viral attachment to host cell; Viral immunoevasion; Viral ion channel; Viral penetration into host cytoplasm; Viral RNA replication; Virion; Virus endocytosis by host; Virus entry into host cell; Zinc; Zinc-finger Protein physicochemical properties Chain ID 1 Molecular weight (Da) 30610.1 Length 269 Aromaticity 0.1 Instability index 55.66 Isoelectric point 6.86 Charge (pH=7) -0.38 2D Binding mode Binding energy (Kcal/mol) -9.62 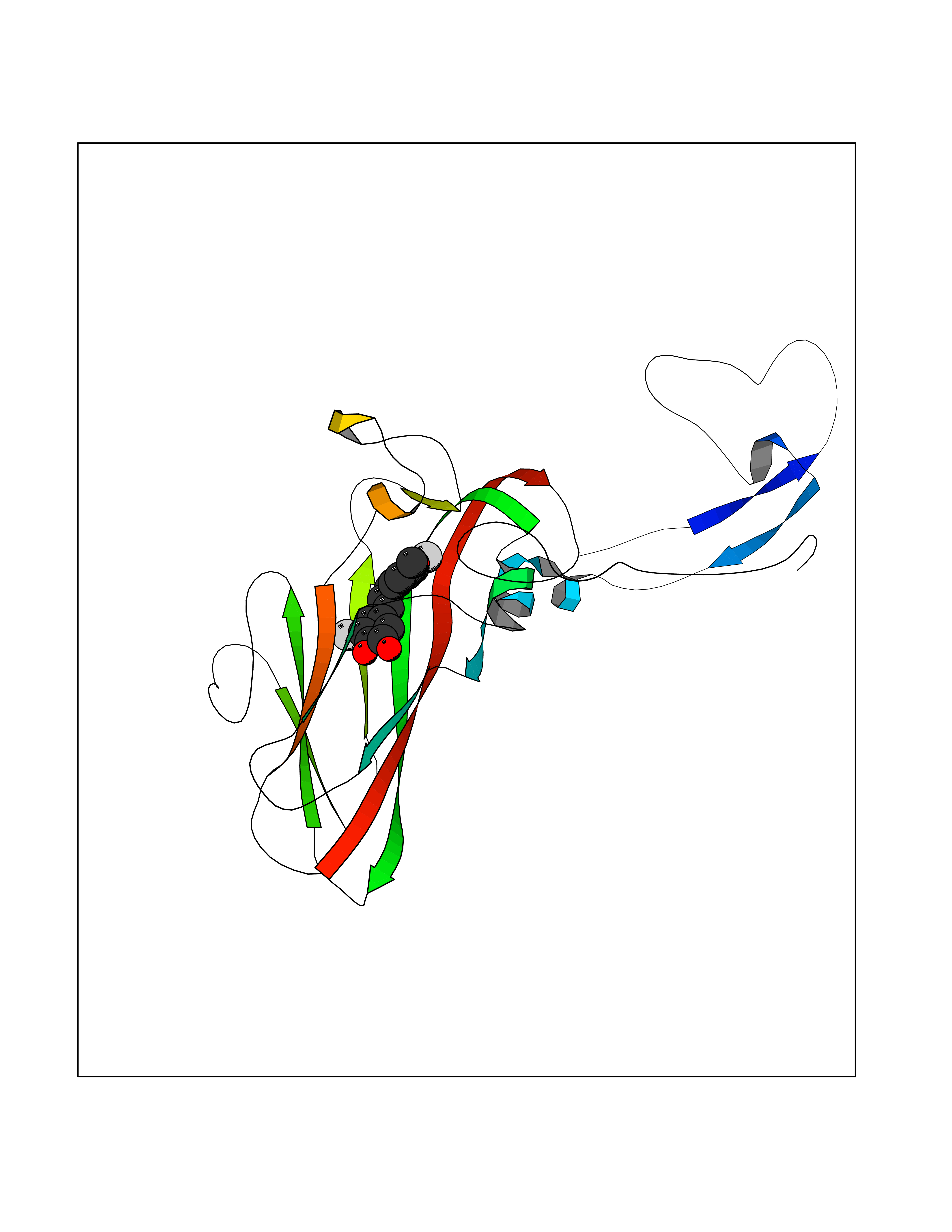 Molscript Map 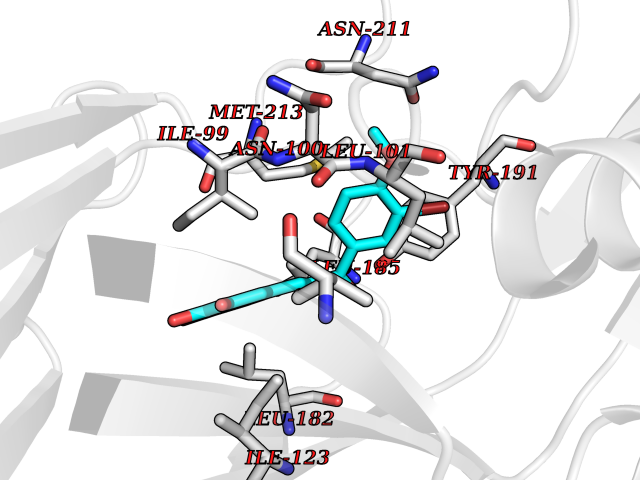 Pymol Map 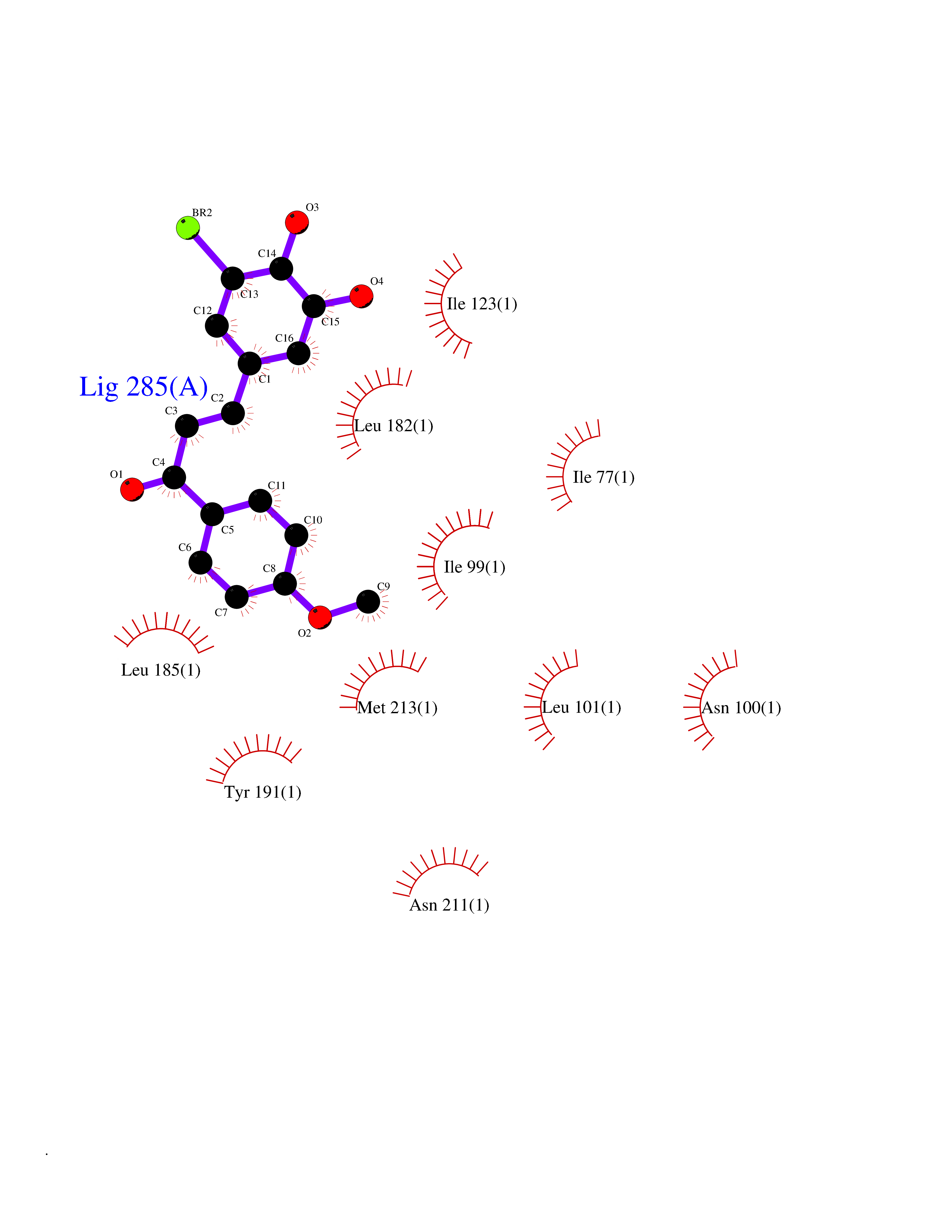 Ligplot Map 3D Binding mode Sequence LVVPNINSSNPTTSNSAPALDAAETGHTSSVQPEDVIETRYVQTSQTRDEMSLESFLGRSGCIHESKLEVTLANYNKENFTVWAINLQEMAQIRRKFELFTYTRFDSEITLVPCISALSQDIGHITMQYMYVPPGAPVPNSRDDYAWQSGTNASVFWQHGQAYPRFSLPFLSVASAYYMFYDGYDEQDQNYGTANTNNMGSLCSRIVTEKHIHKVHIMTRIYHKAKHVKAWCPRPPRALEYTRAHRTNFKIEDRSIQTAIVTRPIITTA Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Thyroid hormone receptor beta (THRB) | 1N46 | 7.04 | |
Target general information Gen name THRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms c-erbA-beta; c-erbA-2; THR1; Nuclear receptor subfamily 1 group A member 2; NR1A2; ERBA2 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function High affinity receptor for thyroid hormones, including triiodothyronine and thyroxine. Nuclear hormone receptor that can act as a repressor or activator of transcription. Related diseases Thyroid hormone resistance, generalized, autosomal dominant (GRTHD) [MIM:188570]: An autosomal dominant disease characterized by high levels of circulating thyroid hormones (T3-T4), goiter, abnormal mental functions, increased susceptibility to infections, abnormal growth and bone maturation, tachycardia and deafness. Affected individuals may also have attention deficit-hyperactivity disorders (ADHD) and language difficulties. Patients have normal or slightly elevated thyroid stimulating hormone (TSH). {ECO:0000269|PubMed:10660344, ECO:0000269|PubMed:12511610, ECO:0000269|PubMed:12554782, ECO:0000269|PubMed:1314846, ECO:0000269|PubMed:1324420, ECO:0000269|PubMed:1563081, ECO:0000269|PubMed:1587388, ECO:0000269|PubMed:1619012, ECO:0000269|PubMed:1661299, ECO:0000269|PubMed:16804041, ECO:0000269|PubMed:1846005, ECO:0000269|PubMed:19268523, ECO:0000269|PubMed:2153155, ECO:0000269|PubMed:2510172, ECO:0000269|PubMed:7833659, ECO:0000269|PubMed:8175986, ECO:0000269|PubMed:8514853, ECO:0000269|PubMed:8664910, ECO:0000269|PubMed:8889584}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid hormone resistance, generalized, autosomal recessive (GRTHR) [MIM:274300]: An autosomal recessive disorder characterized by goiter, clinical euthyroidism, end-organ unresponsiveness to thyroid hormone, abnormal growth and bone maturation, and deafness. Patients also have high levels of circulating thyroid hormones, with elevated thyroid stimulating hormone. {ECO:0000269|PubMed:1653889}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Selective pituitary thyroid hormone resistance (PRTH) [MIM:145650]: Variant form of thyroid hormone resistance and is characterized by clinical hyperthyroidism, with elevated free thyroid hormones, but inappropriately normal serum TSH. Unlike GRTH, where the syndrome usually segregates with a dominant allele, the mode of inheritance in PRTH has not been established. {ECO:0000269|PubMed:7528740, ECO:0000269|PubMed:8381821}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08085; DB03181; DB02106; DB01118; DB00509; DB05035; DB03788; DB03176; DB00451; DB00279; DB01583; DB05192; DB07425; DB09100; DB03604 Interacts with Q60974; Q9Y618 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Deafness; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 27235.4 Length 239 Aromaticity 0.09 Instability index 43.29 Isoelectric point 5.42 Charge (pH=7) -8.55 2D Binding mode Binding energy (Kcal/mol) -9.6 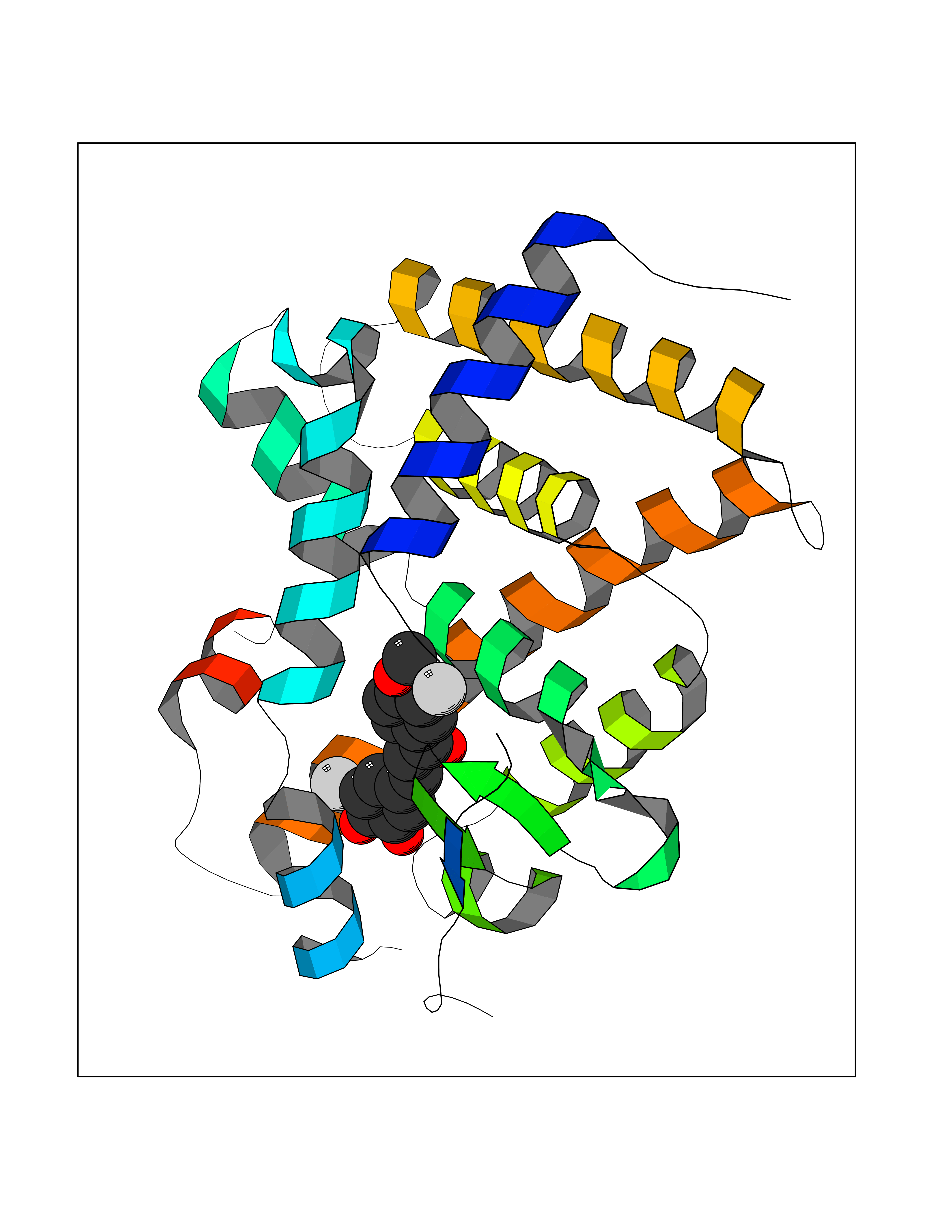 Molscript Map 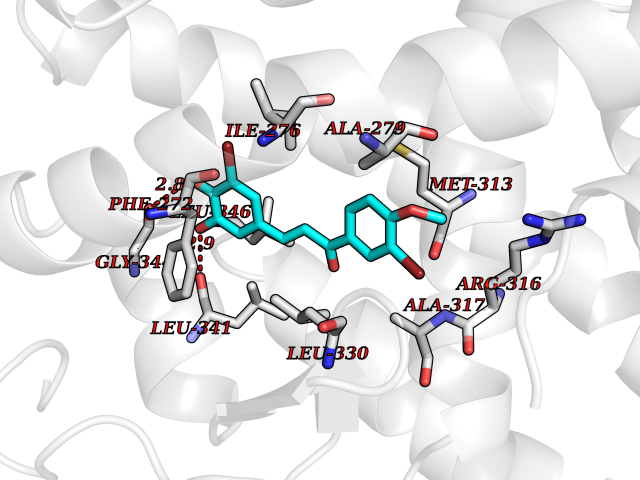 Pymol Map 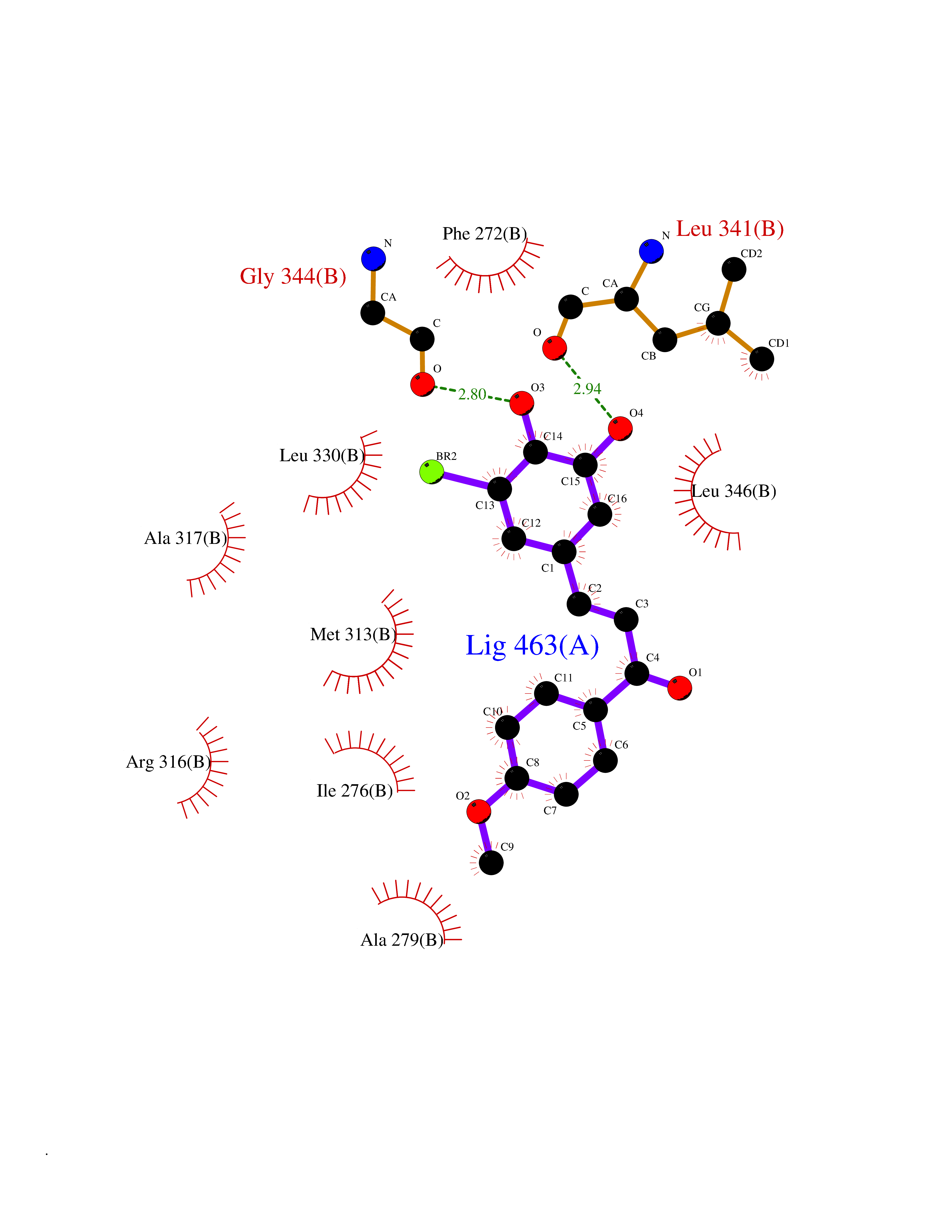 Ligplot Map 3D Binding mode Sequence KPEPTDEEWELIKTVTEAHVATNAQWKQKRKFLPEDIGQAKVDLEAFSHFTKIITPAITRVVDFAKKLPMFCELPCEDQIILLKGCCMEIMSLRAAVRYDPESETLTLNGEMAVTRGQLKNGGLGVVSDAIFDLGMSLSSFNLDDTEVALLQAVLLMSSDRPGLACVERIEKYQDSFLLAFEHYINYRKHHVTHFWPKLLMKVTDLRMIGACHASRFLHMKVECPTELFPPLFLEVFED Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 7.04 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -9.6 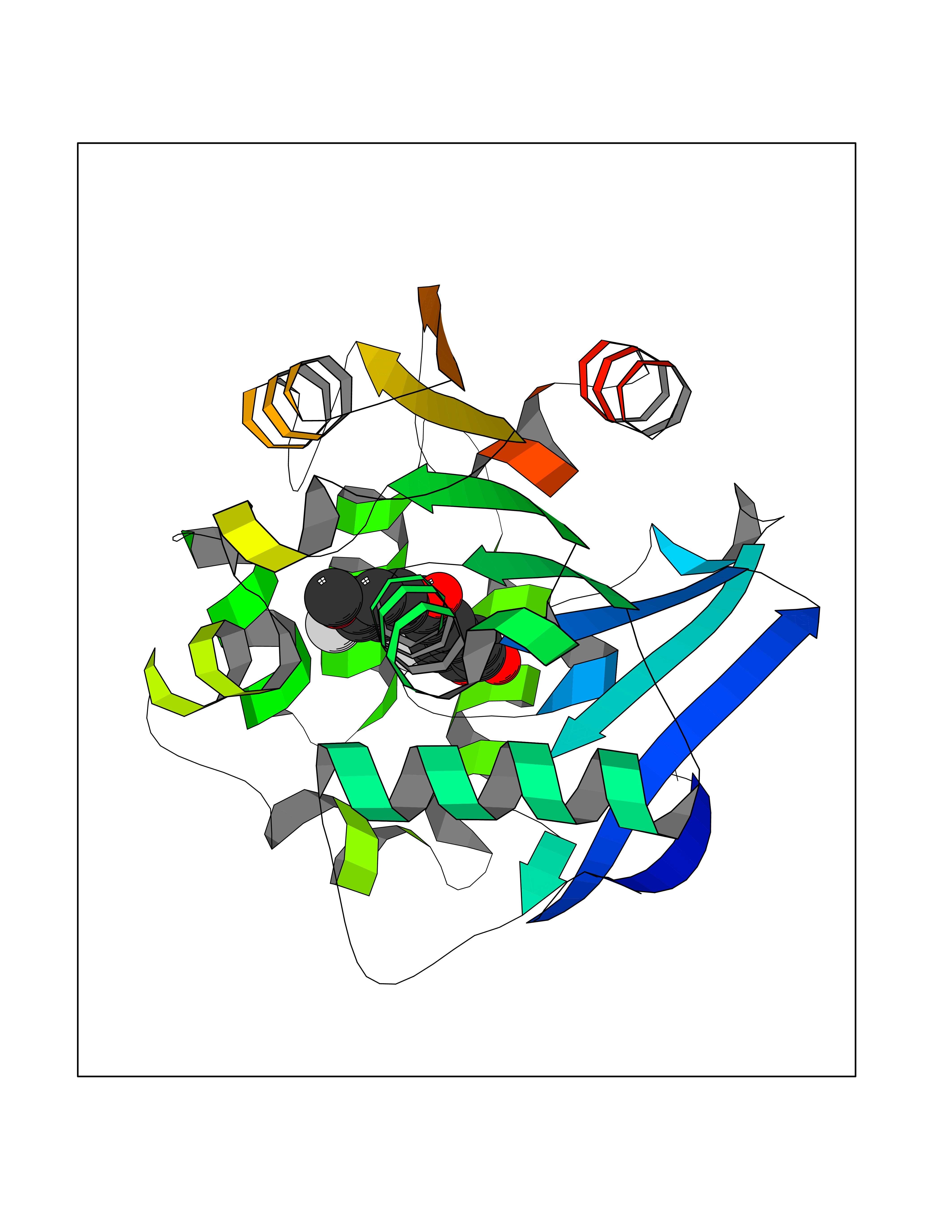 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Opioid receptor sigma 1 (OPRS1) | 5HK1 | 7.04 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 20805.3 Length 185 Aromaticity 0.14 Instability index 31.72 Isoelectric point 5.44 Charge (pH=7) -6.63 2D Binding mode Binding energy (Kcal/mol) -9.61  Molscript Map  Pymol Map 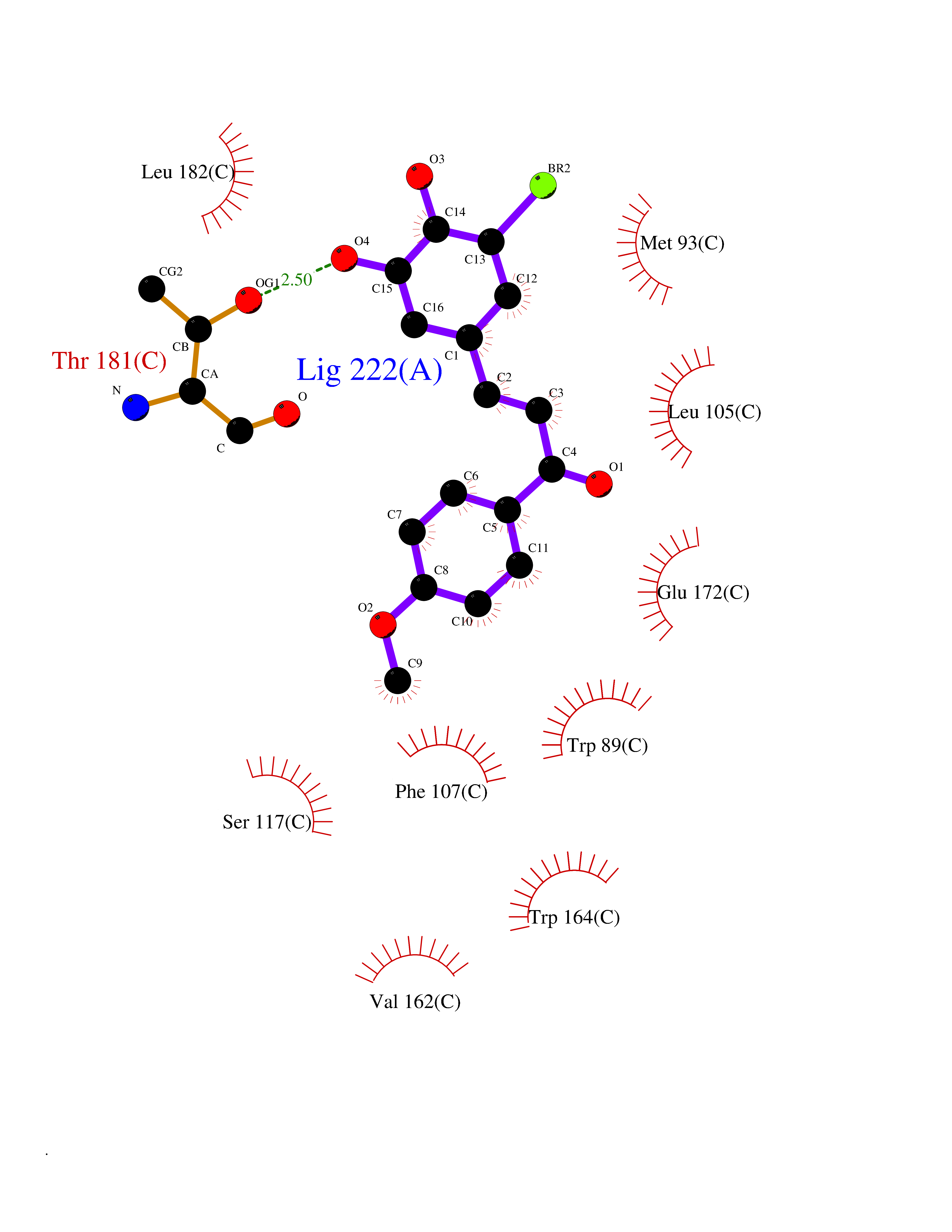 Ligplot Map 3D Binding mode Sequence VFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLFG Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | SEC14-like protein 3 | 4UYB | 7.04 | |
Target general information Gen name SEC14L3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TAP2 Protein family NA Biochemical class Transport protein Function Lipid binding.Transporter activity. Related diseases Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia (CDP-PBHM) [MIM:300863]: A disease characterized by chondrodysplasia, severe platyspondyly, hydrocephaly, and facial features with microphthalmia. Bone abnormalities include a distinctive metaphyseal cupping of the metacarpals, metatarsals, and phalanges. Affected females show a milder phenotype with small stature, sometimes associated with body asymmetry and mild intellectual disability. {ECO:0000269|PubMed:20181727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 46148.7 Length 401 Aromaticity 0.1 Instability index 45.19 Isoelectric point 5.79 Charge (pH=7) -5.94 2D Binding mode Binding energy (Kcal/mol) -9.61 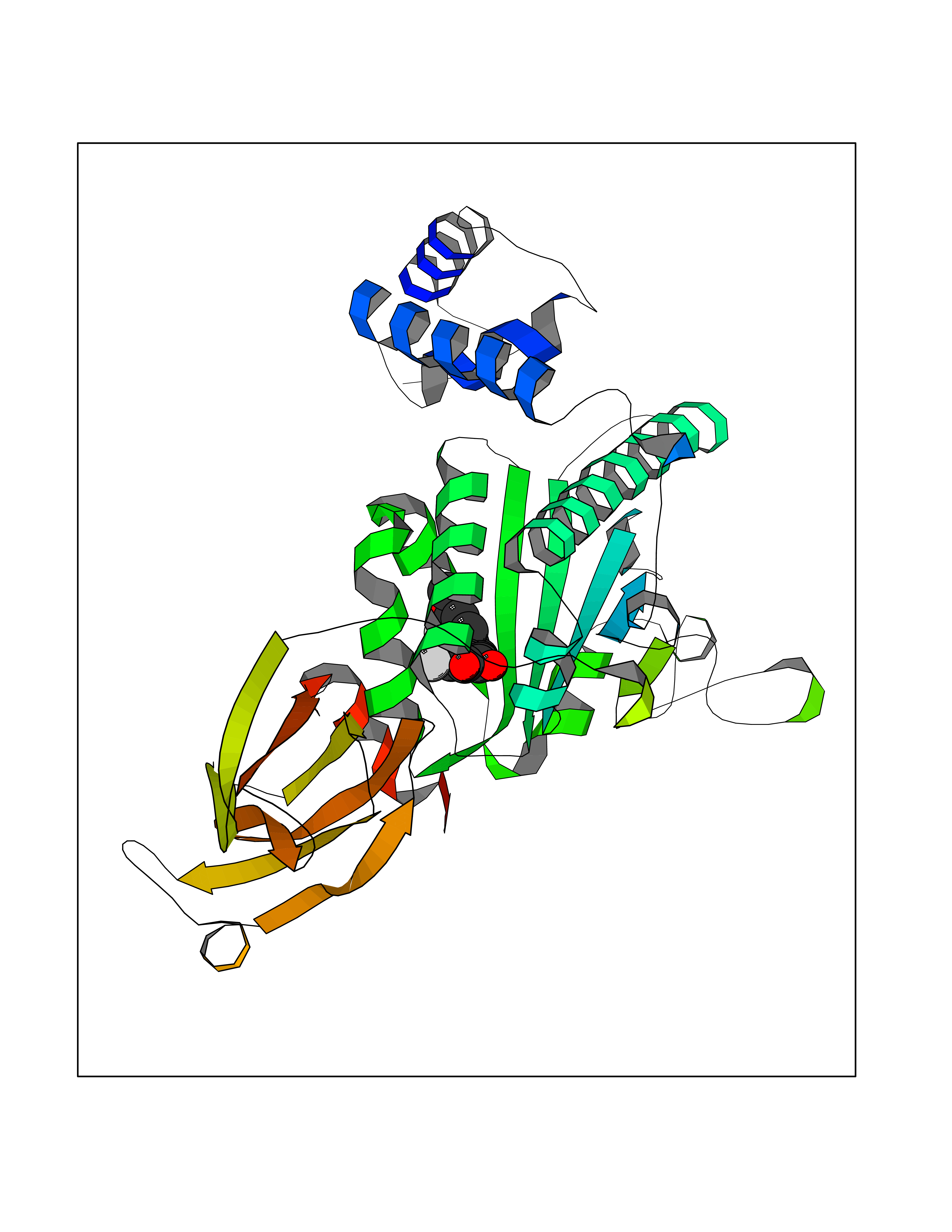 Molscript Map 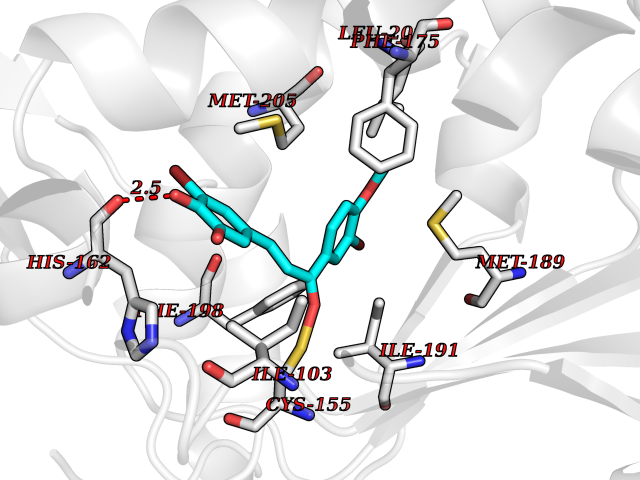 Pymol Map 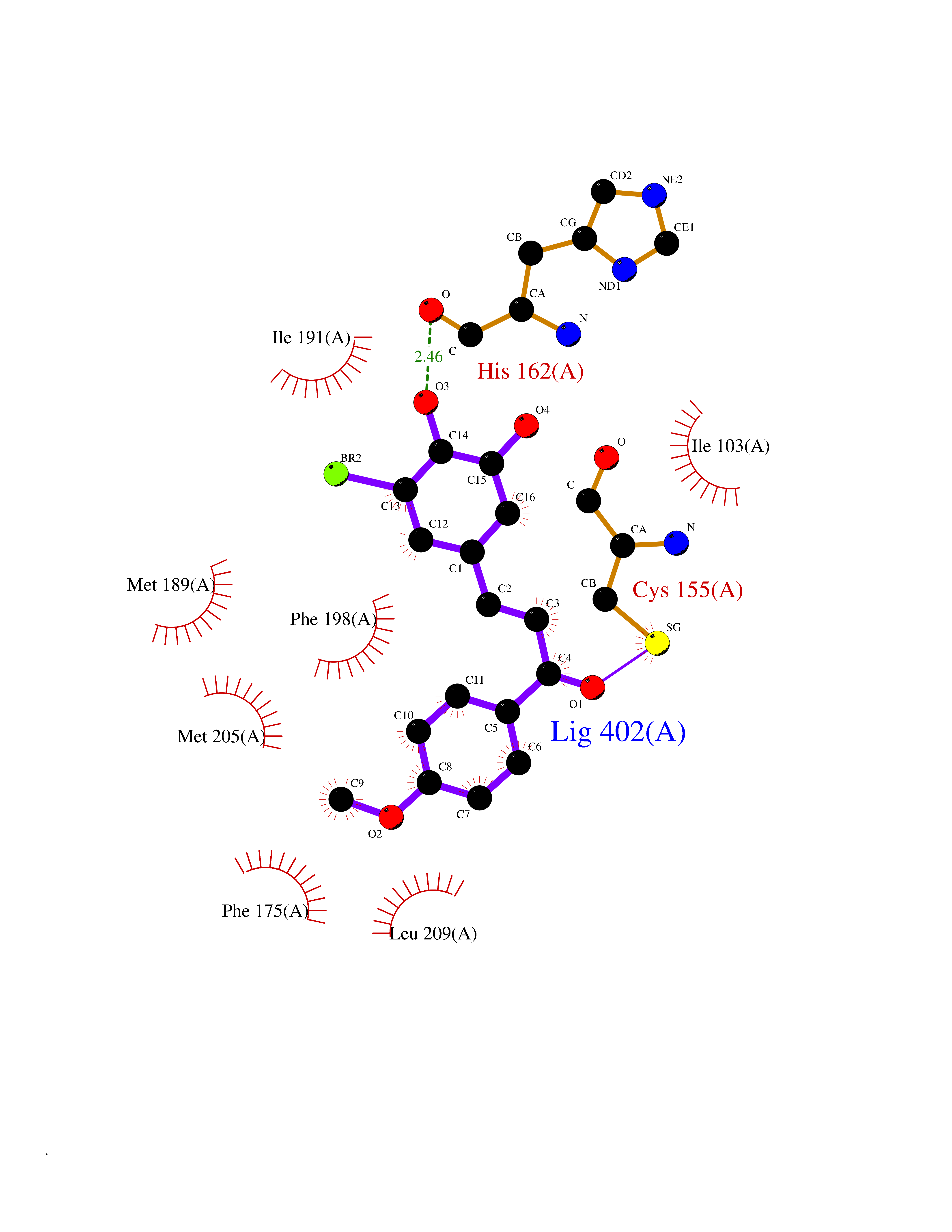 Ligplot Map 3D Binding mode Sequence SMSGRVGDLSPKQAETLAKFRENVQDVLPALPNPDDYFLLRWLRARNFDLQKSEALLRKYMEFRKTMDIDHILDWQPPEVIQKYMPGGLCGYDRDGCPVWYDIIGPLDPKGLLFSVTKQDLLKTKMRDCERILHECDLQTERLGKKIETIVMIFDCEGLGLKHFWKPLVEVYQEFFGLLEENYPETLKFMLIVKATKLFPVGYNLMKPFLSEDTRRKIIVLGNNWKEGLLKLISPEELPAQFGGTLTDPDGNPKCLTKINYGGEIPKSMYVRDQVKTQYEHSVQINRGSSHQVEYEILFPGCVLRWQFSSDGADIGFGVFLKTKMGERQRAGEMTEVLPSQRYNAHMVPEDGNLTCSEAGVYVLRFDNTYSFVHAKKVSFTVEVLLPDEGMQKYDKELTPV Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Sodium/glucose cotransporter 1 (SGLT1) | 7WMV | 7.04 | |
Target general information Gen name SLC5A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 5 member 1; Na(+)/glucose cotransporter 1; NAGT; High affinity sodium-glucose cotransporter Protein family Sodium:solute symporter (SSF) (TC 2.A.21) family Biochemical class Solute:sodium symporter Function Efficient substrate transport in mammalian kidney is provided by the concerted action of a low affinity high capacity and a high affinity low capacity Na(+)/glucose cotransporter arranged in series along kidney proximal tubules. Actively transports glucose into cells by Na(+) cotransport with a Na(+) to glucose coupling ratio of 2:1. Related diseases Congenital glucose/galactose malabsorption (GGM) [MIM:606824]: Intestinal monosaccharide transporter deficiency. It is an autosomal recessive disorder manifesting itself within the first weeks of life. It is characterized by severe diarrhea and dehydration which are usually fatal unless glucose and galactose are eliminated from the diet. {ECO:0000269|PubMed:10036327, ECO:0000269|PubMed:11406349, ECO:0000269|PubMed:2008213, ECO:0000269|PubMed:8195156, ECO:0000269|PubMed:8563765}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00766; DB01914; DB09341; DB05018; DB12713 Interacts with P00533 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Sodium; Sodium transport; Sugar transport; Symport; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 66451.3 Length 602 Aromaticity 0.12 Instability index 35.84 Isoelectric point 8.3 Charge (pH=7) 4.43 2D Binding mode Binding energy (Kcal/mol) -9.6  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ETHELIRNAADISIIVIYFVVVMAVGLWAMFSTNRGTVGGFFLAGRSMVWWPIGASLFASNIGSGHFVGLAGTGAASGIAIGGFEWNALVLVVVLGWLFVPIYIKAGVVTMPEYLRKRFGGQRIQVYLSLLSLLLYIFTKISADIFSGAIFINLALGLNLYLAIFLLLAITALYTITGGLAAVIYTDTLQTVIMLVGSLILTGFAFHEVGGYDAFMEKYMKAIPTIVSDGNTTFQEKCYTPRADSFHIFRDPLTGDLPWPGFIFGMSILTLWYWCTDQVIVQRCLSAKNMSHVKGGCILCGYLKLMPMFIMVMPGMISRILYTEKIACVVPSECEKYCGTKVGCTNIAYPTLVVELMPNGLRGLMLSVMLASLMSSLTSIFNSASTLFTMDIYAKVRKRASEKELMIAGRLFILVLIGISIAWVPIVQSAQSGQLFDYIQSITSYLGPPIAAVFLLAIFWKRVNEPGAFWGLILGLLIGISRMITEFAYGTGSCMEPSNCPTIICGVHYLYFAIILFAISFITIVVISLLTKPIPDVHLYRLCWSLRNSKEERIDLDATEEEEKAMKMKMTDTSEKPLWRTVLNVNGIILVTVAVFCHAYFA Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | 3-oxoacyl-[acyl-carrier-protein] synthase 2 | 3I8P | 7.03 | |
Target general information Gen name fabF Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1081;b1095;fabJ Protein family Thiolase-like superfamily, Beta-ketoacyl-ACP synthases family Biochemical class Transferase Function 3-oxoacyl-[acyl-carrier-protein] synthase activity.Beta-ketoacyl-acyl-carrier-protein synthase II activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08366; DB01034; DB03017; DB08407 Interacts with P0A6Y8 EC number 2.3.1.179 Uniprot keywords 3D-structure; Acyltransferase; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42794.9 Length 411 Aromaticity 0.06 Instability index 31.41 Isoelectric point 5.72 Charge (pH=7) -7.34 2D Binding mode Binding energy (Kcal/mol) -9.59  Molscript Map 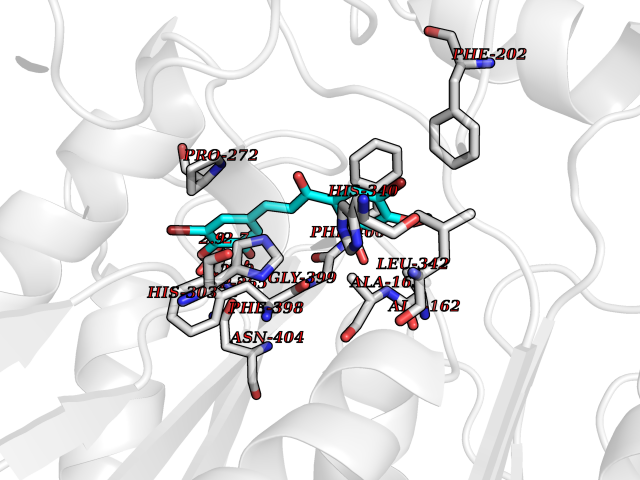 Pymol Map 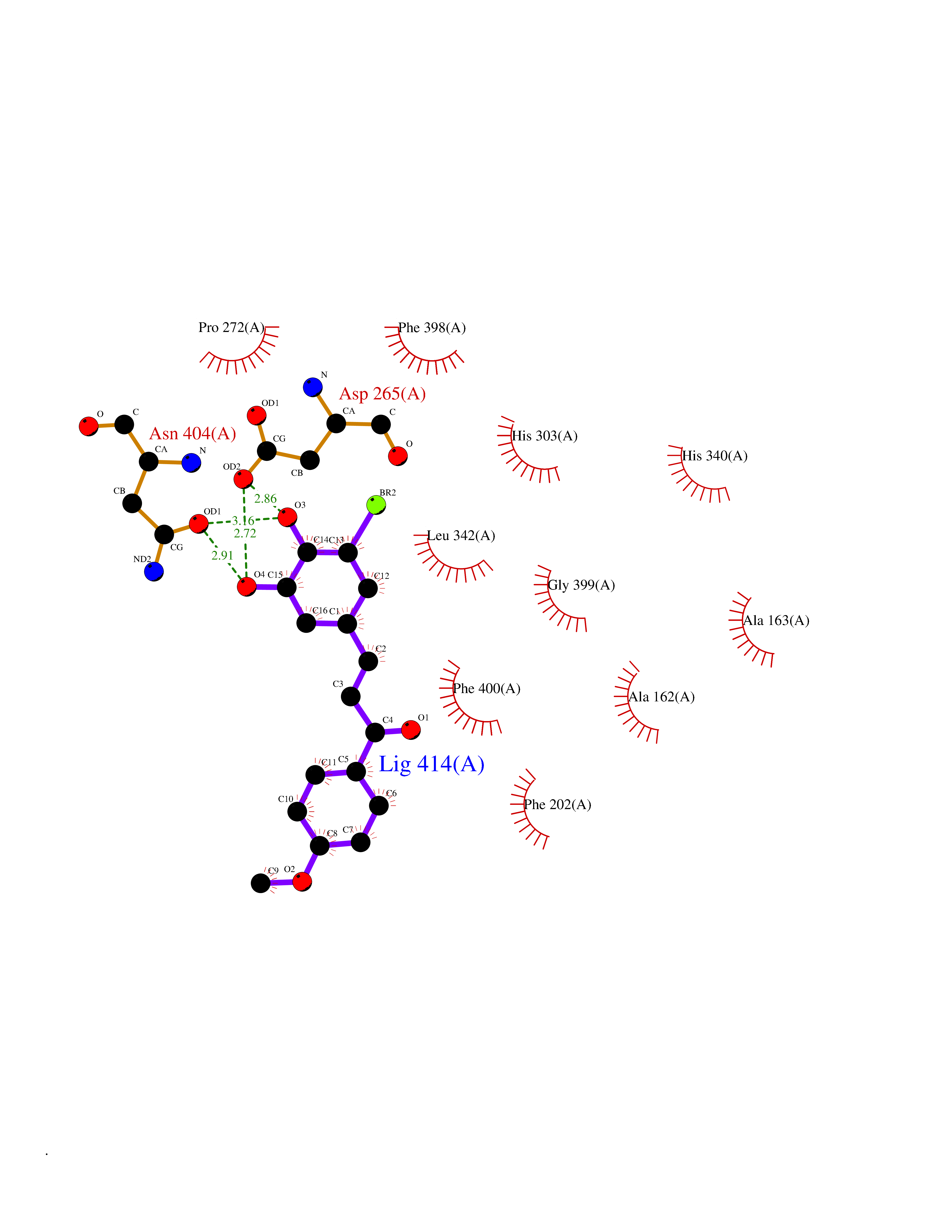 Ligplot Map 3D Binding mode Sequence KRRVVVTGLGMLSPVGNTVESTWKALLAGQSGISLIDHFDTSAYATKFAGLVKDFNCEDIISRKEQRKMDAFIQYGIVAGVQAMQDSGLEITEENATRIGAAIGSGIGGLGLIEENHTSLMNGGPRKISPFFVPSTIVNMVAGHLTIMYGLRGPSISIATAATSGVHNIGHAARIIAYGDADVMVAGGAEKASTPLGVGGFGAARALSTRNDNPQAASRPWDKERDGFVLGDGAGMLVLEEYEHAKKRGAKIYAELVGFGMSSDAYHMTSPPENGAGAALAMANALRDAGIEASQIGYVNAHGTSTPAGDKAEAQAVKTIFGEAASRVLVSSTKSMTGHLLGAAGAVESIYSILALRDQAVPPTINLDNPDEGCDLDFVPHEARQVSGMEYTLCNSFGFGGTNGSLIFKKI Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | 1C3R | 7.03 | |
Target general information Gen name murA Organism Aquifex aeolicus (strain VF5) Uniprot ID TTD ID NA Synonyms aq_1023 Protein family Histone deacetylase family Biochemical class Lyase Function UDP-N-acetylglucosamine 1-carboxyvinyltransferase activity. Related diseases Atrial fibrillation, familial, 14 (ATFB14) [MIM:615378]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:19808477}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Genetic variations in SCN2B may be involved in Brugada syndrome (PubMed:23559163). This tachyarrhythmia is characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:23559163}. Drugs (DrugBank ID) DB04297; DB02546 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetoin catabolism; Metal-binding; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42338.9 Length 372 Aromaticity 0.12 Instability index 35.74 Isoelectric point 5.77 Charge (pH=7) -6.5 2D Binding mode Binding energy (Kcal/mol) -9.58 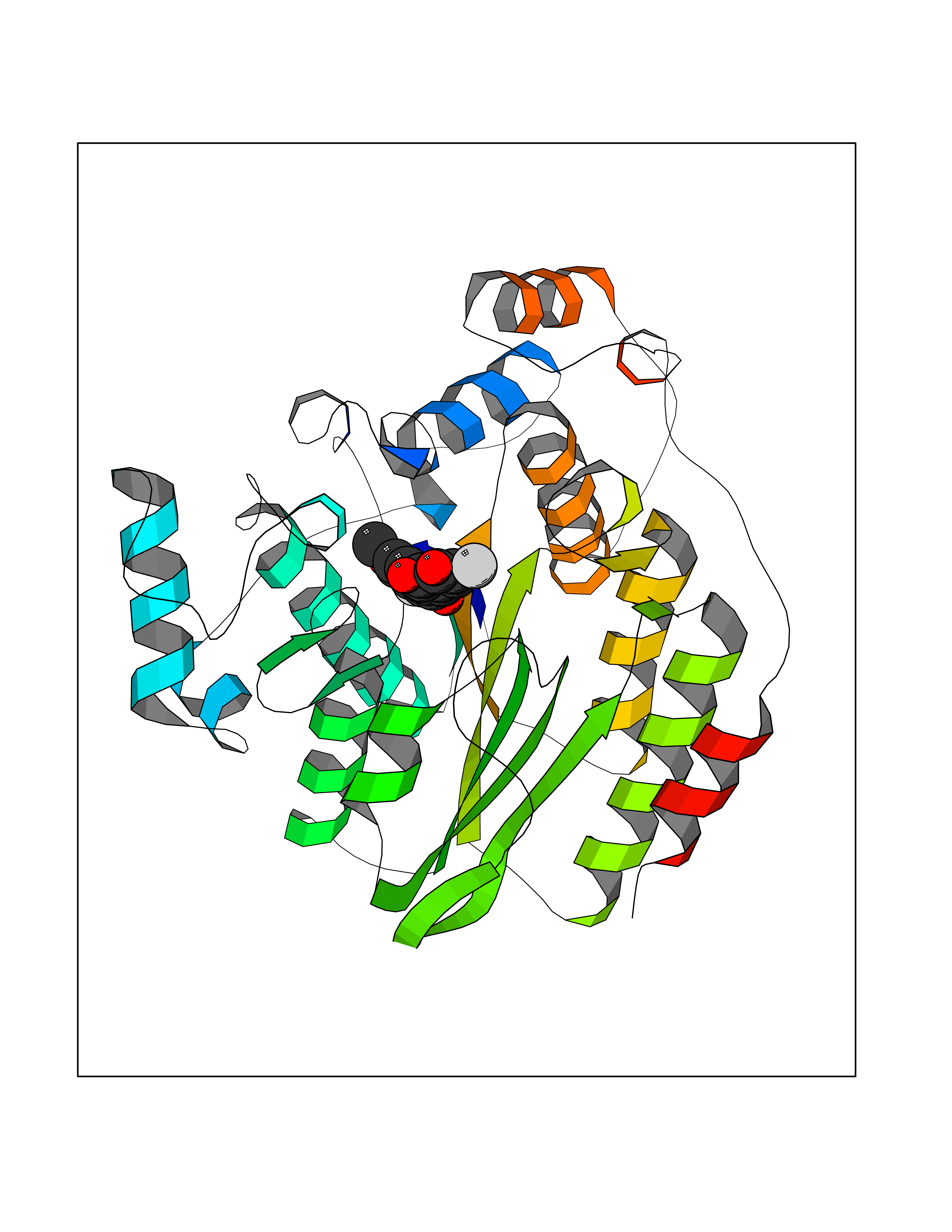 Molscript Map 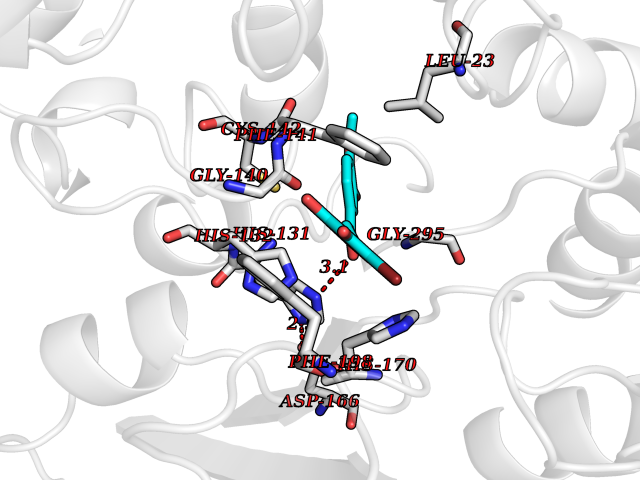 Pymol Map 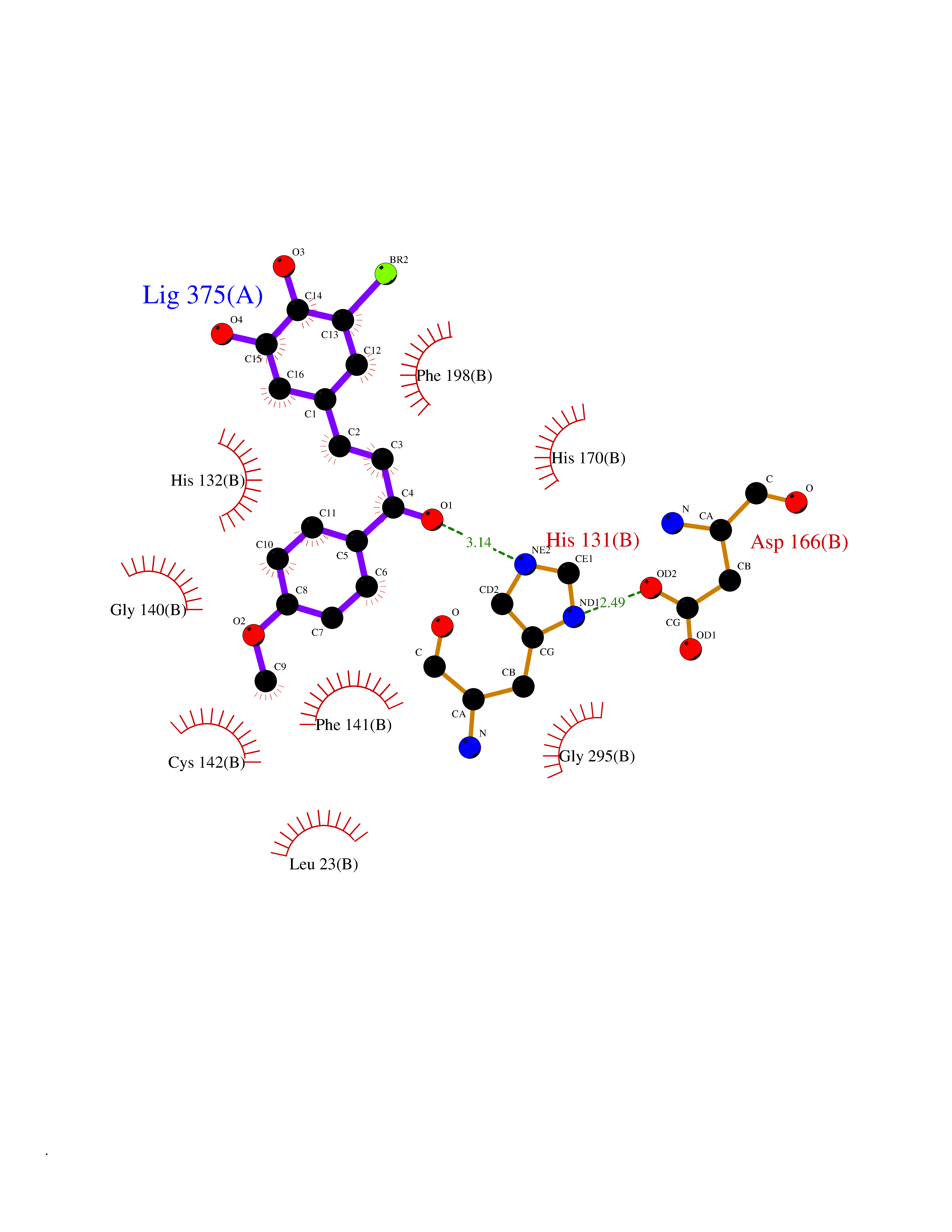 Ligplot Map 3D Binding mode Sequence KKVKLIGTLDYGKYRYPKNHPLKIPRVSLLLRFKDAMNLIDEKELIKSRPATKEELLLFHTEDYINTLMEAERSQSVPKGAREKYNIGGYENPVSYAMFTGSSLATGSTVQAIEEFLKGNVAFNPAGGMHHAFKSRANGFCYINNPAVGIEYLRKKGFKRILYIDLDAHHCDGVQEAFYDTDQVFVLSLHQSPEYAFPFEKGFLEEIGEGKGKGYNLNIPLPKGLNDNEFLFALEKSLEIVKEVFEPEVYLLQLGTDPLLEDYLSKFNLSNVAFLKAFNIVREVFGEGVYLGGGGYHPYALARAWTLIWCELSGREVPEKLNNKAKELLKSIDFEEFDDEVDRSYMLETLKDPWRGGEVRKEVKDTLEKAKA Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Nitric-oxide synthase inducible (NOS2) | 3E7G | 7.03 | |
Target general information Gen name NOS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms iNOS; Peptidyl-cysteine S-nitrosylase NOS2; Nitric oxide synthase, inducible; NOS2A; NOS type II; Inducible NOS; Inducible NO synthase; Hepatocyte NOS; HEP-NOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function Produces nitric oxide (NO) which is a messenger molecule with diverse functions throughout the body. In macrophages, NO mediates tumoricidal and bactericidal actions. Also has nitrosylase activity and mediates cysteine S-nitrosylation of cytoplasmic target proteins such PTGS2/COX2 (By similarity). As component of the iNOS-S100A8/9 transnitrosylase complex involved in the selective inflammatory stimulus-dependent S-nitrosylation of GAPDH on 'Cys-247' implicated in regulation of the GAIT complex activity and probably multiple targets including ANXA5, EZR, MSN and VIM. Involved in inflammation, enhances the synthesis of proinflammatory mediators such as IL6 and IL8. Related diseases Cerebellar ataxia, impaired intellectual development, and dysequilibrium syndrome 3 (CAMRQ3) [MIM:613227]: An autosomal recessive, congenital cerebellar ataxia associated with dysarthia, quadrupedal gait and intellectual disability. {ECO:0000269|PubMed:19461874}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07003; DB07007; DB07011; DB07405; DB08750; DB01997; DB07029; DB07008; DB08214; DB07002; DB01835; DB06879; DB04534; DB03100; DB02207; DB00125; DB00155; DB01234; DB14649; DB11327; DB00997; DB07306; DB07388; DB05252; DB01381; DB03366; DB05214; DB04400; DB09237; DB00244; DB01110; DB01017; DB03144; DB01686; DB03449; DB06916; DB07318; DB07389; DB02044; DB02644; DB05383; DB02234; DB03953; DB02462; DB08814 Interacts with P04406 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calmodulin-binding; Cytoplasm; FAD; Flavoprotein; FMN; Heme; Iron; Metal-binding; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 48633 Length 421 Aromaticity 0.12 Instability index 46.5 Isoelectric point 6.75 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -9.58  Molscript Map 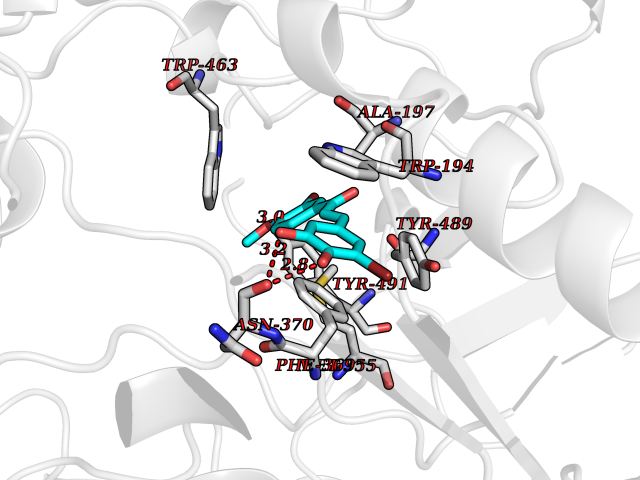 Pymol Map 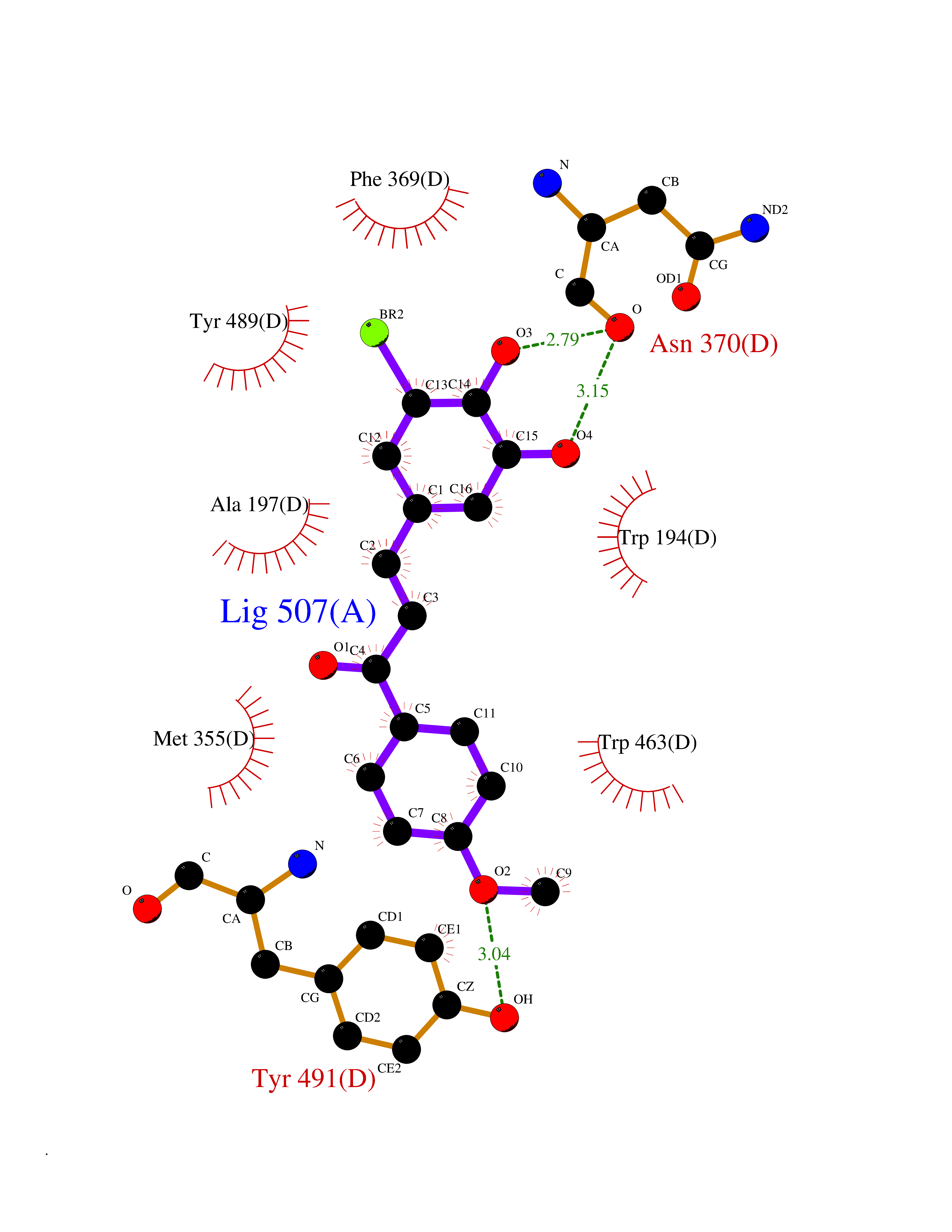 Ligplot Map 3D Binding mode Sequence RHVRIKNWGSGMTFQDTLHHKAKGILTCRSKSCLGSIMTPKSLTRGPRDKPTPPDELLPQAIEFVNQYYGSFKEAKIEEHLARVEAVTKEIETTGTYQLTGDELIFATKQAWRNAPRCIGRIQWSNLQVFDARSCSTAREMFEHICRHVRYSTNNGNIRSAITVFPQRSDGKHDFRVWNAQLIRYAGYQMPDGSIRGDPANVEFTQLCIDLGWKPKYGRFDVVPLVLQANGRDPELFEIPPDLVLEVAMEHPKYEWFRELELKWYALPAVANMLLEVGGLEFPGCPFNGWYMGTEIGVRDFCDVQRYNILEEVGRRMGLETHKLASLWKDQAVVEINIAVLHSFQKQNVTIMDHHSAAESFMKYMQNEYRSRGGCPADWIWLVPPMSGSITPVFHQEMLNYVLSPFYYYQVEAWKTHVWQD Hydrogen bonds contact Hydrophobic contact | ||||