Job Results:
Ligand
Structure
Job ID
f5eac8ba4ac90e4335358bfb735bd782
Job name
NA
Time
2025-10-13 17:37:26
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Glutathione S-transferase A2 (GSTA2) | 2VCT | 5.71 | |
Target general information Gen name GSTA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GTH2; GSTA2-2; GST2; GST-gamma; GST class-alpha member 2; GST HA subunit 2 Protein family GST superfamily, Alpha family Biochemical class NA Function Conjugation of reduced glutathione to a wide number of exogenous and endogenous hydrophobic electrophiles. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14001; DB00321; DB00993; DB01008; DB00291; DB00608; DB00636; DB14002; DB03619; DB00903; DB00143; DB03310; DB00163 Interacts with O15217; Q7RTV2 EC number EC 2.5.1.18 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 51074.9 Length 442 Aromaticity 0.1 Instability index 44.47 Isoelectric point 8.69 Charge (pH=7) 4.39 2D Binding mode Binding energy (Kcal/mol) -7.79  Molscript Map 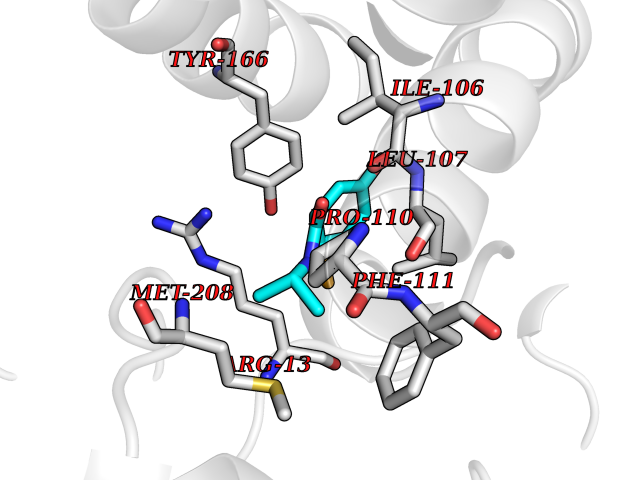 Pymol Map 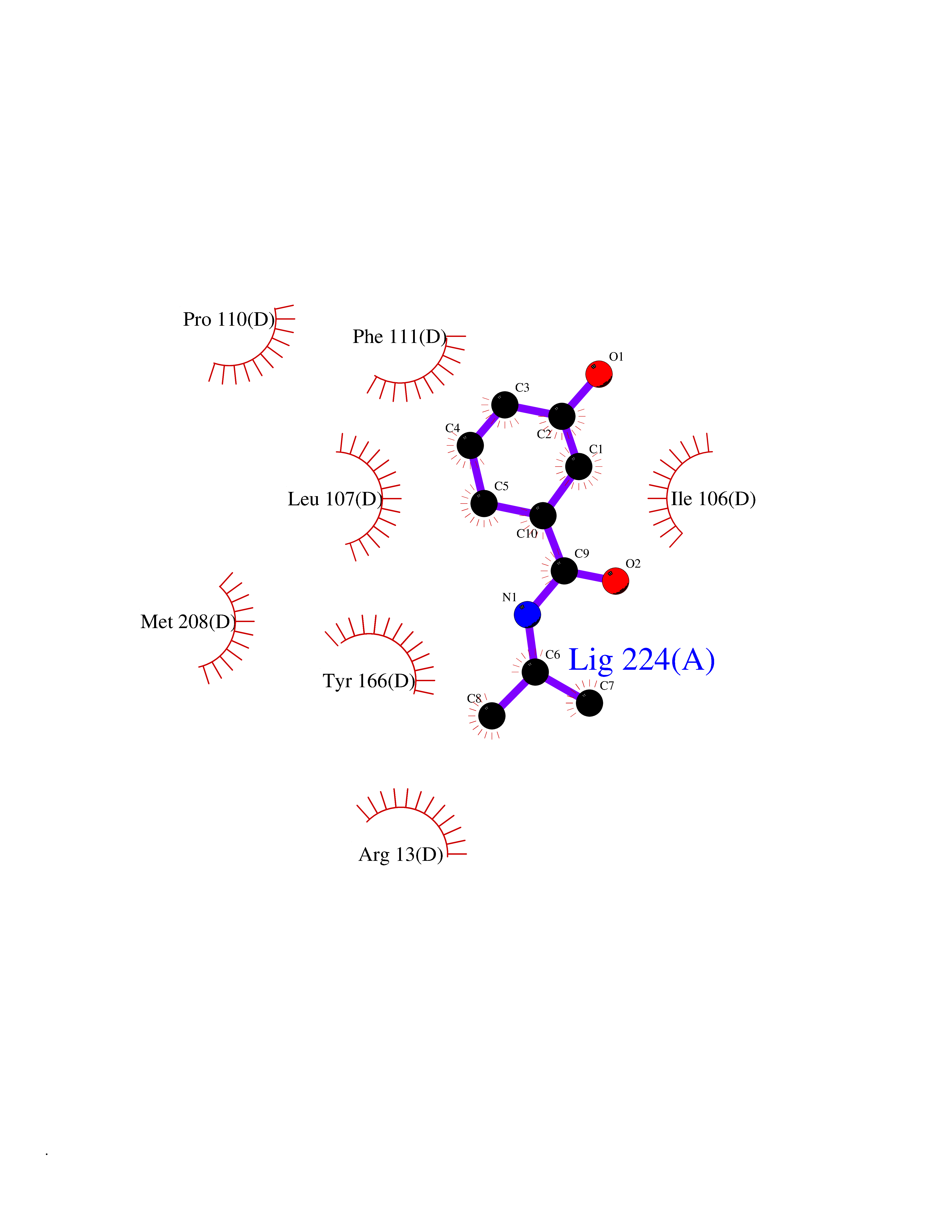 Ligplot Map 3D Binding mode Sequence AEKPKLHYSNIRGRMESIRWLLAAAGVEFEEKFIKSAEDLDKLRNDGYLMFQQVPMVEIDGMKLVQTRAILNYIASKYNLYGKDIKEKALIDMYIEGIADLGEMILLLPFTQPEEQDAKLALIQEKTKNRYFPAFEKVLKSHGQDYLVGNKLSRADIHLVELLYYVEELDSSLISSFPLLKALKTRISNLPTVKKFLQPGSPRKPPMDEKSLEESRKIFRFAEKPKLHYSNIRGRMESIRWLLAAAGVEFEEKFIKSAEDLDKLRNDGYLMFQQVPMVEIDGMKLVQTRAILNYIASKYNLYGKDIKEKALIDMYIEGIADLGEMILLLPFTQPEEQDAKLALIQEKTKNRYFPAFEKVLKSHGQDYLVGNKLSRADIHLVELLYYVEELDSSLISSFPLLKALKTRISNLPTVKKFLQPGSPRKPPMDEKSLEESRKIFRF Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | L-aspartate oxidase | 1KNR | 5.70 | |
Target general information Gen name nadB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW2558;nicB;b2574 Protein family FAD-dependent oxidoreductase 2 family, NadB subfamily Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.L-aspartate:fumarate oxidoreductase activity.L-aspartate oxidase activity. Related diseases Thyroid hormone resistance, generalized, autosomal dominant (GRTHD) [MIM:188570]: An autosomal dominant disease characterized by high levels of circulating thyroid hormones (T3-T4), goiter, abnormal mental functions, increased susceptibility to infections, abnormal growth and bone maturation, tachycardia and deafness. Affected individuals may also have attention deficit-hyperactivity disorders (ADHD) and language difficulties. Patients have normal or slightly elevated thyroid stimulating hormone (TSH). {ECO:0000269|PubMed:10660344, ECO:0000269|PubMed:12511610, ECO:0000269|PubMed:12554782, ECO:0000269|PubMed:1314846, ECO:0000269|PubMed:1324420, ECO:0000269|PubMed:1563081, ECO:0000269|PubMed:1587388, ECO:0000269|PubMed:1619012, ECO:0000269|PubMed:1661299, ECO:0000269|PubMed:16804041, ECO:0000269|PubMed:1846005, ECO:0000269|PubMed:19268523, ECO:0000269|PubMed:2153155, ECO:0000269|PubMed:2510172, ECO:0000269|PubMed:7833659, ECO:0000269|PubMed:8175986, ECO:0000269|PubMed:8514853, ECO:0000269|PubMed:8664910, ECO:0000269|PubMed:8889584}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid hormone resistance, generalized, autosomal recessive (GRTHR) [MIM:274300]: An autosomal recessive disorder characterized by goiter, clinical euthyroidism, end-organ unresponsiveness to thyroid hormone, abnormal growth and bone maturation, and deafness. Patients also have high levels of circulating thyroid hormones, with elevated thyroid stimulating hormone. {ECO:0000269|PubMed:1653889}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Selective pituitary thyroid hormone resistance (PRTH) [MIM:145650]: Variant form of thyroid hormone resistance and is characterized by clinical hyperthyroidism, with elevated free thyroid hormones, but inappropriately normal serum TSH. Unlike GRTH, where the syndrome usually segregates with a dominant allele, the mode of inheritance in PRTH has not been established. {ECO:0000269|PubMed:7528740, ECO:0000269|PubMed:8381821}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.4.3.16; 1.5.99.- Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase; Pyridine nucleotide biosynthesis; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 58993.2 Length 529 Aromaticity 0.07 Instability index 36.96 Isoelectric point 5.76 Charge (pH=7) -15.84 2D Binding mode Binding energy (Kcal/mol) -7.77 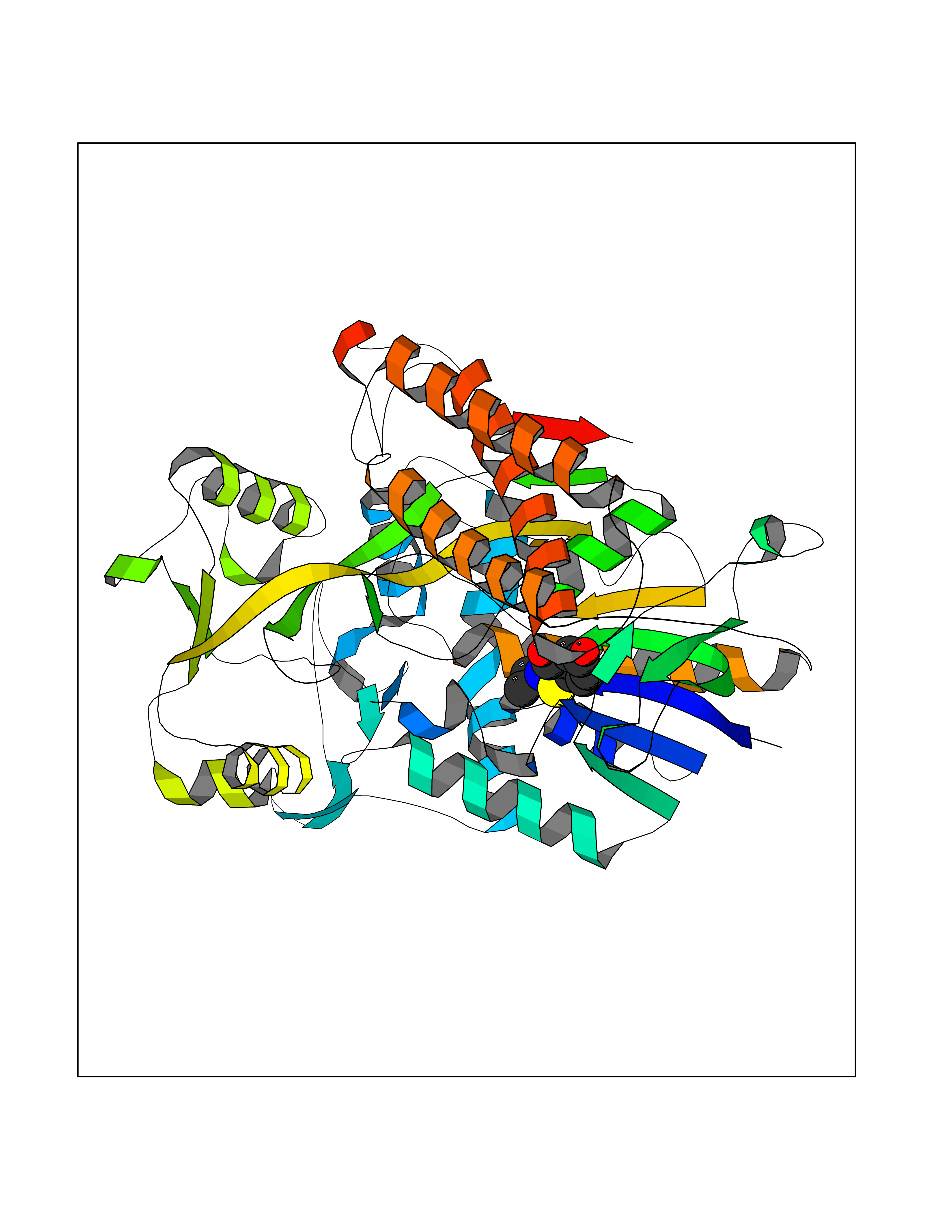 Molscript Map 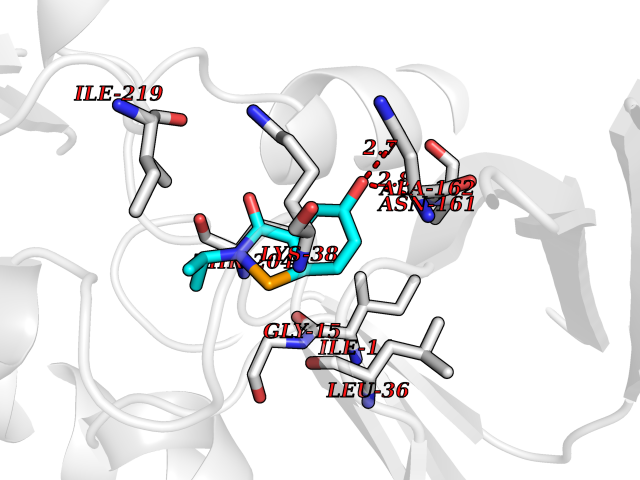 Pymol Map 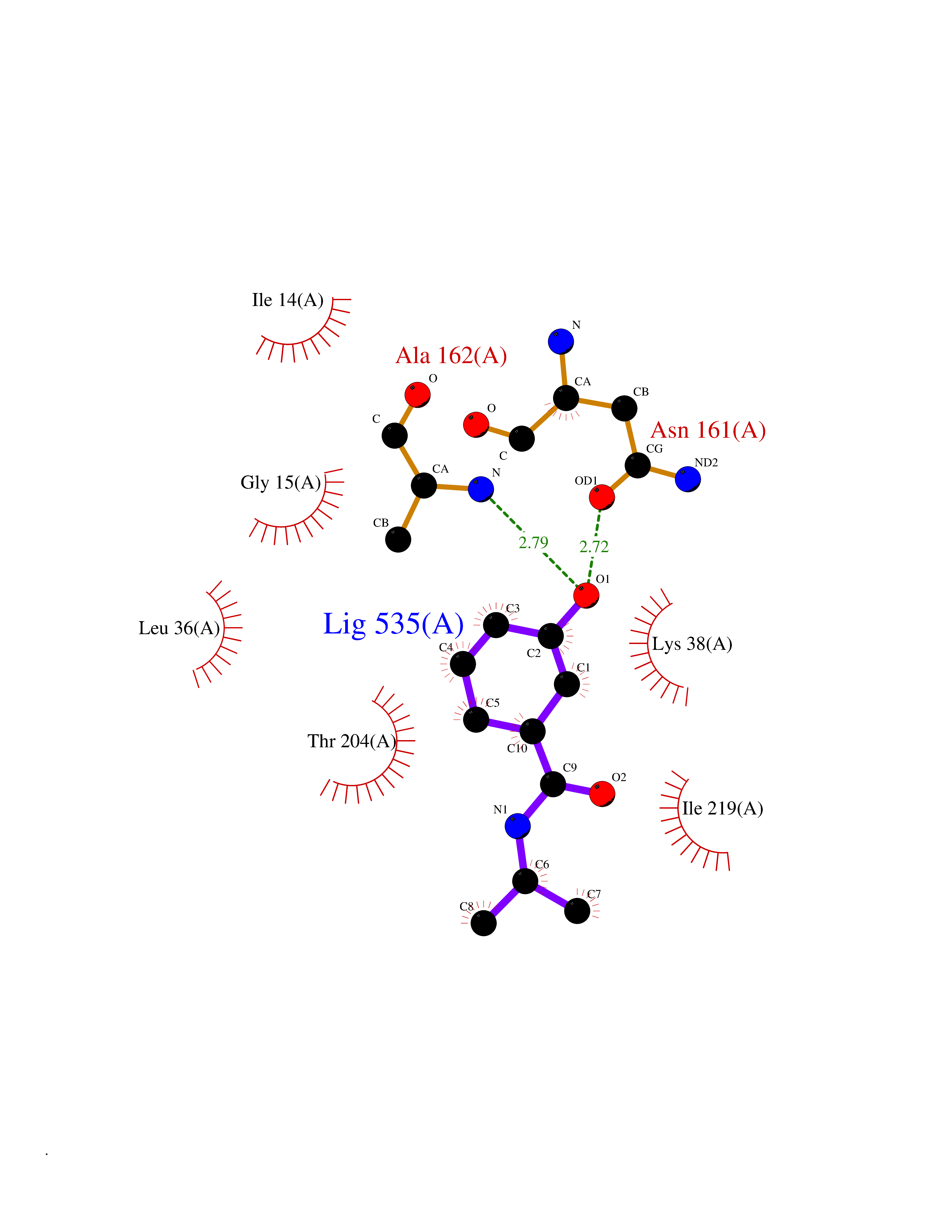 Ligplot Map 3D Binding mode Sequence PEHSCDVLIIGSGAAGLSLALRLADQHQVIVLSKGPVTEGSTFYAQGGIAAVFDETDSIDSHVEDTLIAGAGICDRHAVEFVASNARSCVQWLIDQGVLFDTHIQPNGEESYHLTREGGHSHRRILHAADATGREVETTLVSKALNHPNIRVLERTNAVDLIVSDKIGLPGTRRVVGAWVWNRNKETVETCHAKAVVLATGGASKVYQYTTNPDISSGDGIAMAWRAGCRVANLEFNQFHPTALYHPQARNFLLTEALRGEGAYLKRPDGTRFMPDFDERGELAPRDIVARAIDHEMKRLGADCMFLDISHKPADFIRQHFPMIYEKLLGLGIDLTQEPVPIVPAAHYTCGGVMVDDHGRTDVEGLYAIGEVSYTGLHGANLMASNSLLECLVYGWSAAEDITRRMPYAHDISTLPPWDESRVENPDERVVIQHNWHELRLFMWDYVGIVRTTKRLERALRRITMLQQEIDEYYAHFRVSNNLLELRNLVQVAELIVRCAMMRKESRGLHFTLDYPELLTHSGPSILSP Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Retinoic acid receptor RXR-alpha (RXRA) | 2P1T | 5.70 | |
Target general information Gen name RXRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor alpha; RXRalpha; Nuclear receptor subfamily 2 group B member 1; NR2B1 Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. The high affinity ligand for RXRs is 9-cis retinoic acid. RXRA serves as a common heterodimeric partner for a number of nuclear receptors. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. The RXRA/PPARA heterodimer is required for PPARA transcriptional activity on fatty acid oxidation genes such as ACOX1 and the P450 system genes. Receptor for retinoic acid. Related diseases Lichtenstein-Knorr syndrome (LIKNS) [MIM:616291]: An autosomal recessive neurologic disorder characterized by progressive cerebellar ataxia and severe progressive sensorineural hearing loss. {ECO:0000269|PubMed:25205112, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08063; DB08402; DB07863; DB07557; DB00459; DB00210; DB01436; DB00523; DB00132; DB04557; DB00307; DB01393; DB03756; DB00749; DB00926; DB05956; DB04224; DB02746; DB00412; DB00755; DB08601 Interacts with O14503; P35637; Q15648; Q71SY5; Q15788; Q15596; P55055; P55055-1; Q13133; P27986; P37231; P37231-1; P10276; P42224; P11473; P97792-1; Q9JLI4; P04625; PRO_0000278730 [Q03463] EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; DNA-binding; Host-virus interaction; Isopeptide bond; Metal-binding; Mitochondrion; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 24958.9 Length 221 Aromaticity 0.07 Instability index 39.47 Isoelectric point 6.16 Charge (pH=7) -3.45 2D Binding mode Binding energy (Kcal/mol) -7.77 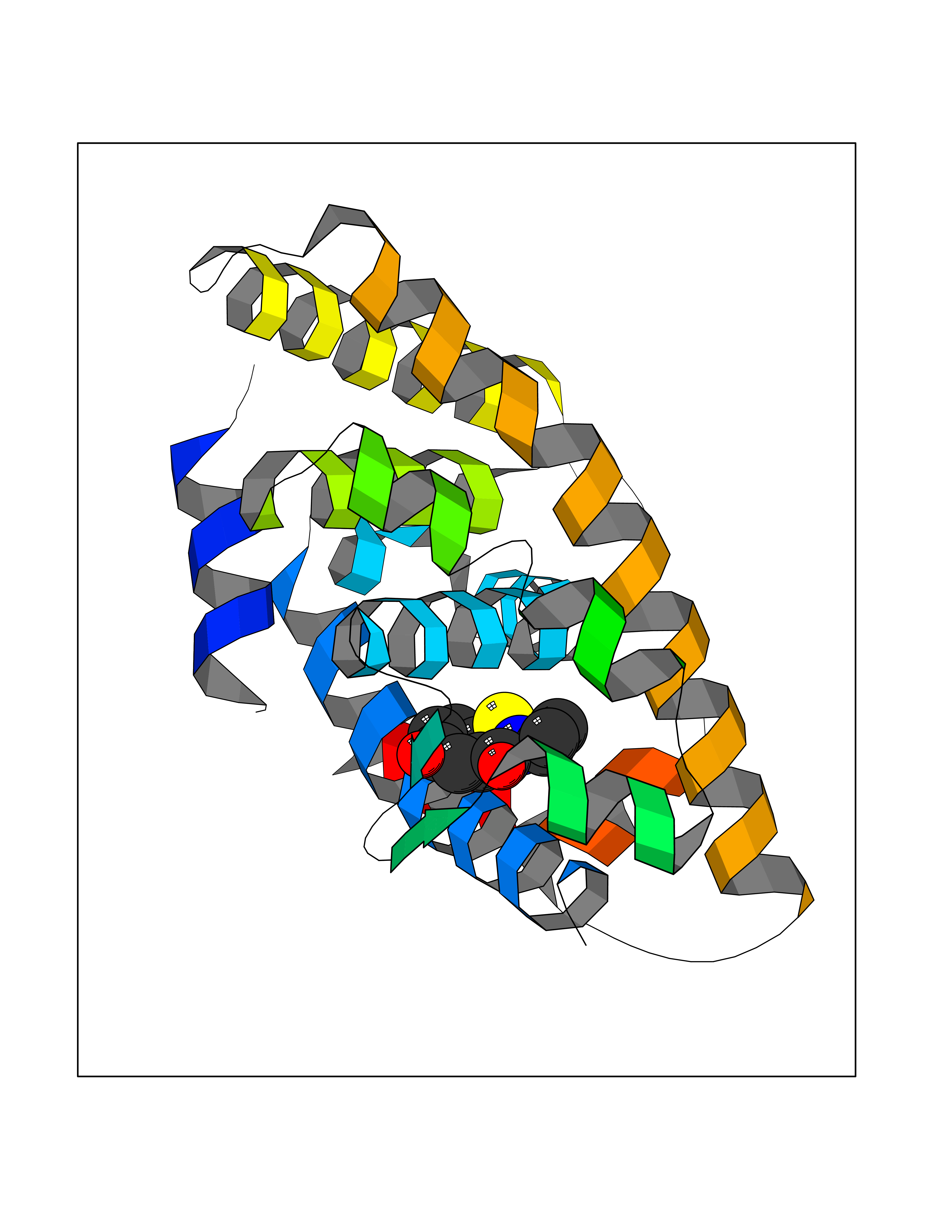 Molscript Map 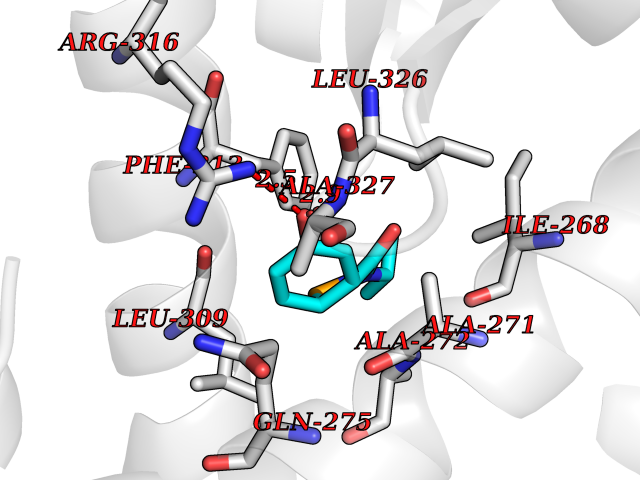 Pymol Map 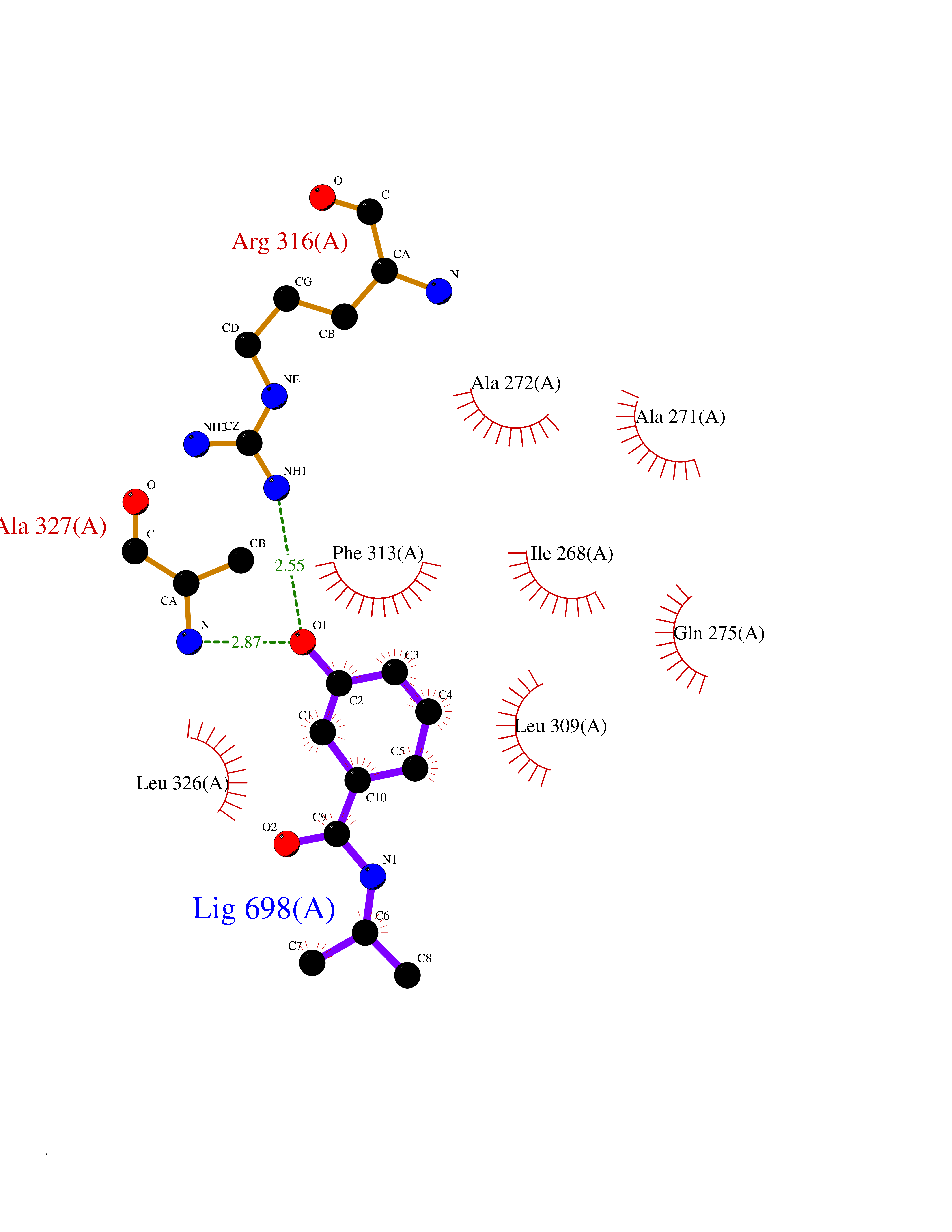 Ligplot Map 3D Binding mode Sequence DMPVERILEAELAVEDPVTNICQAADKQLFTLVEWAKRIPHFSELPLDDQVILLRAGWNELLIASFSHRSIAVKDGILLATGLHVHRNSAHSAGVGAIFDRVLTELVSKMRDMQMDKTELGCLRAIVLFNPDSKGLSNPAEVEALREKVYASLEAYCKHKYPEQPGRFAKLLLRLPALRSIGLKCLEHLFFFKLIGDTPIDTFLMEMLEAPHKILHRLLQD Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Pyruvate kinase PKLR | 4IP7 | 5.70 | |
Target general information Gen name PKLR Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PKL;PK1 Protein family Pyruvate kinase family Biochemical class Transferase Function ATP binding.Kinase activity.Magnesium ion binding.Potassium ion binding.Pyruvate kinase activity. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02726; DB00787; DB04551; DB16236; DB00119 Interacts with Q9UBL6-2 EC number 2.7.1.40 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Disease variant; Glycolysis; Hereditary hemolytic anemia; Kinase; Magnesium; Manganese; Metal-binding; Nucleotide-binding; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 45695.1 Length 421 Aromaticity 0.06 Instability index 34.44 Isoelectric point 6.88 Charge (pH=7) -0.35 2D Binding mode Binding energy (Kcal/mol) -7.77 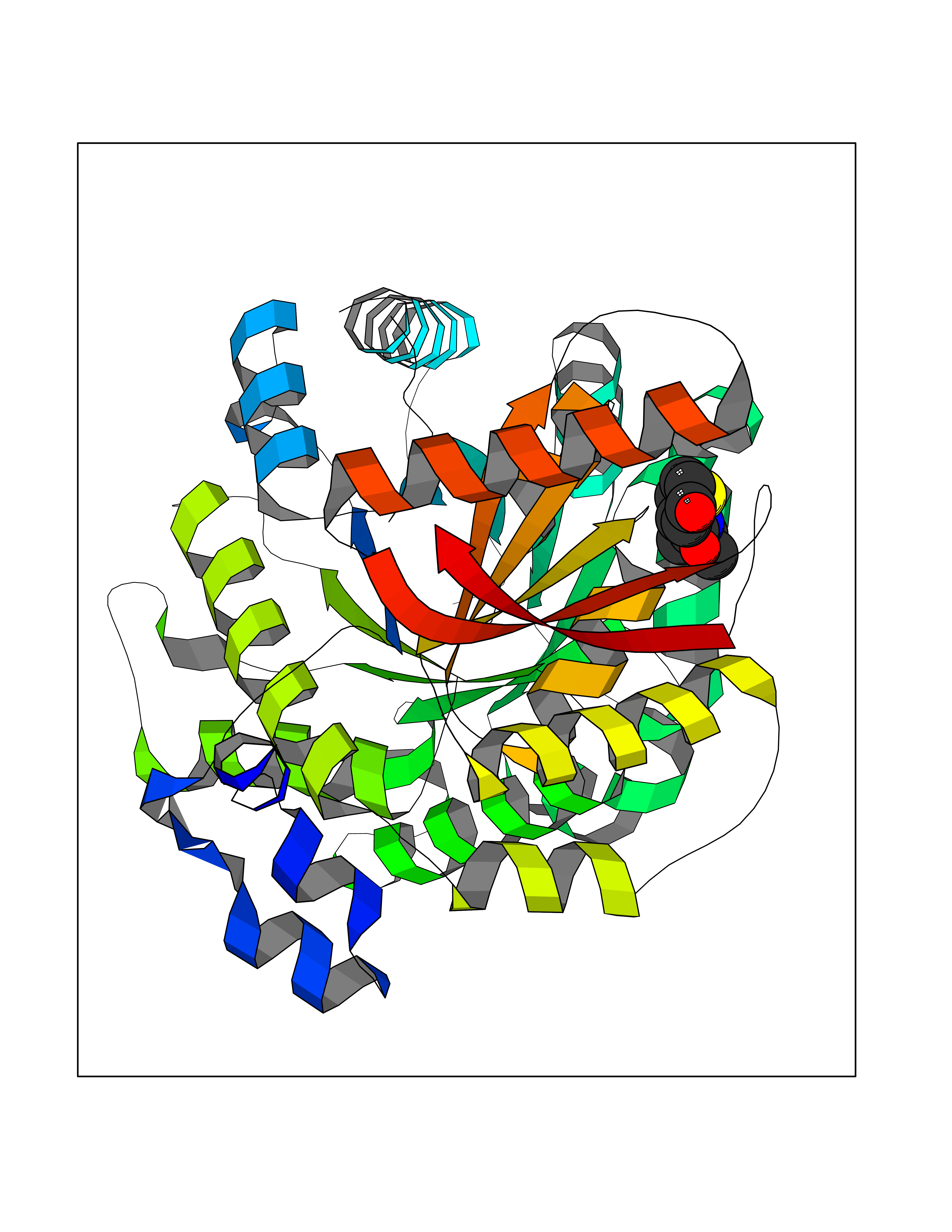 Molscript Map 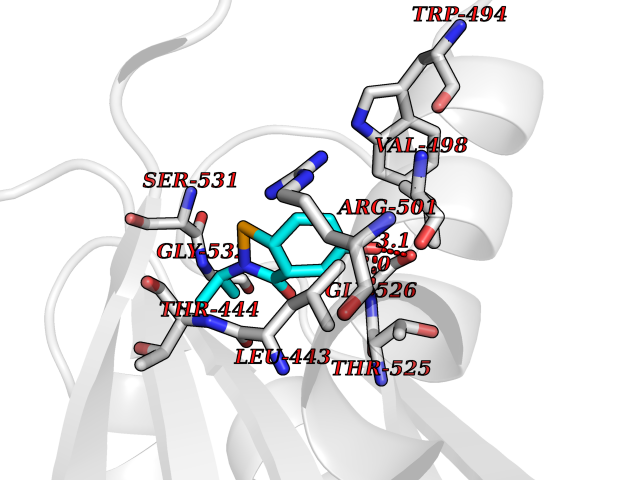 Pymol Map 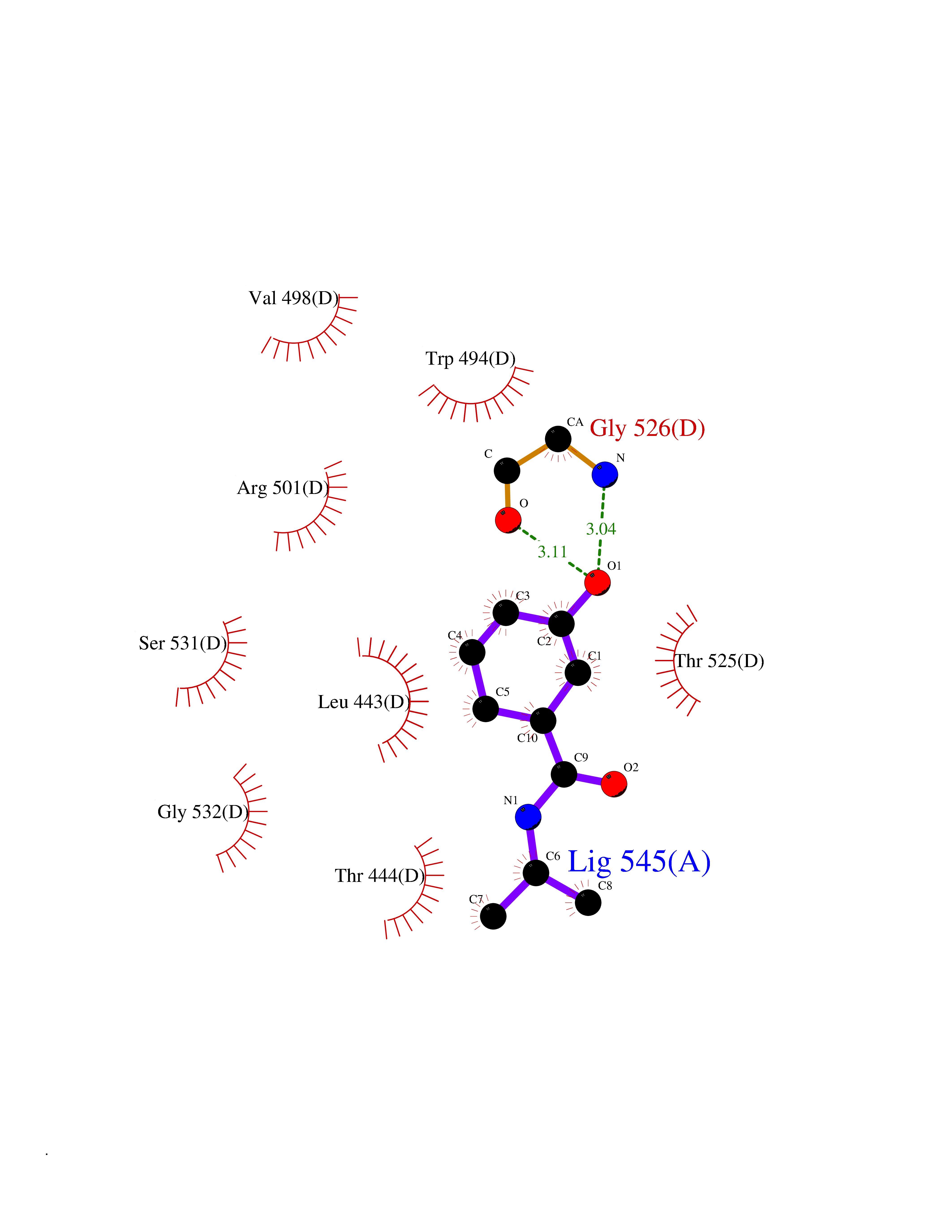 Ligplot Map 3D Binding mode Sequence GTAFFQQQQLPAAMADTFLEHLCLLDIDSEPVAARSTSIIATIGPASRSVERLKEMIKAGMNIARLNFSHGSHEYHAESIANVREAVESFSPLSYRPVAIALDTKGPEIGLSEQDVRDLRFGVEHGVDIVFASFVRKASDVAAVRAALGPEGHGIKIISKIENHEGVKRFDEILEVSDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNLAGKPVVCATQMLESMITKPRPTRAETSDVANAVLDGADCIMLSGETAKGNFPVEAVKMQHAIAREAEAAVYHRQLFEELRRAAPLSRDPTEVTAIGAVEAAFKCCAAAIIVLTTTGRSAQLLSRYRPRAAVIAVTRSAQAARQVHLCRGVFPLLYREPPEAIWADDVDRRVQFGIESGKLRGFLRVGDLVIVVTGWRPGSGYTNIMRVLSIS Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 5.70 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -7.77 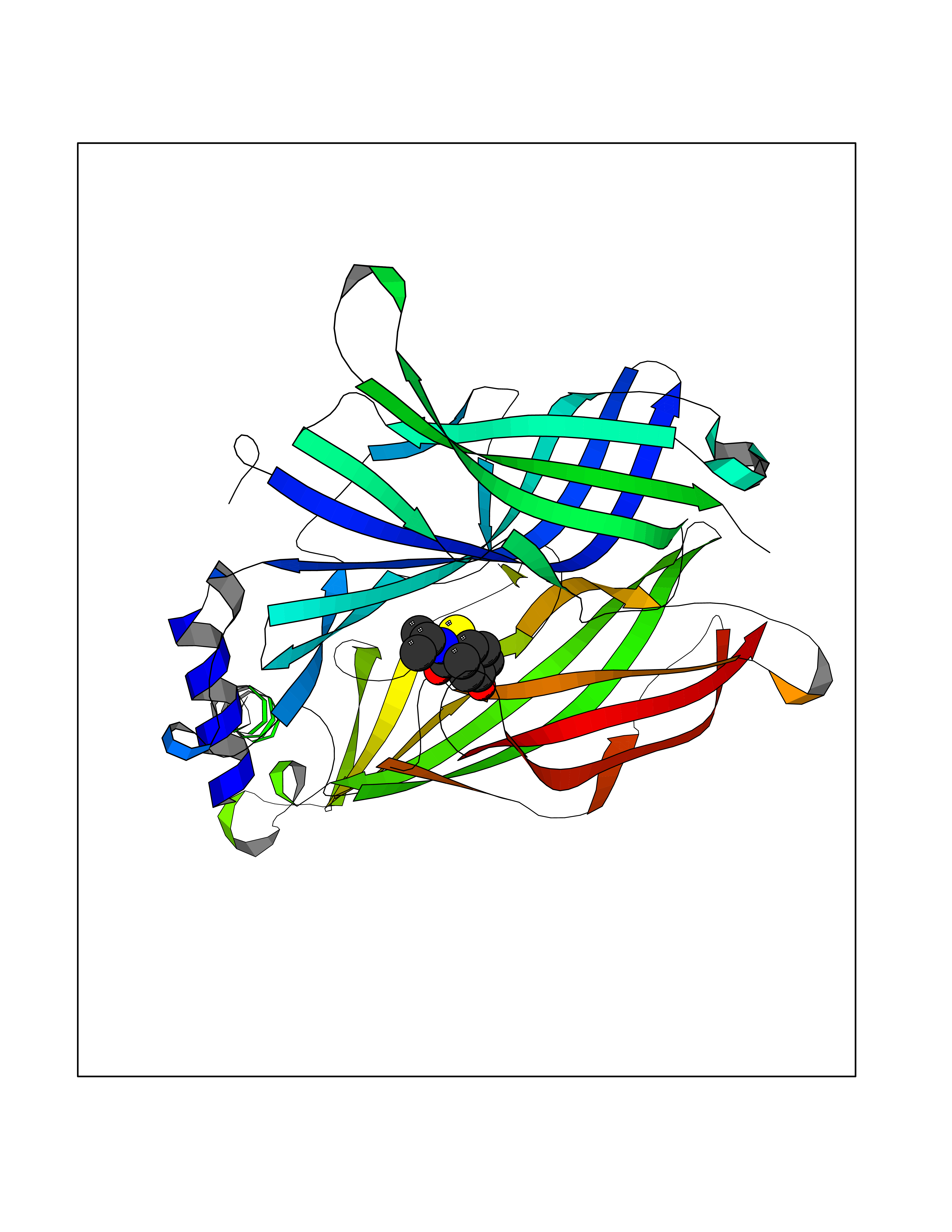 Molscript Map 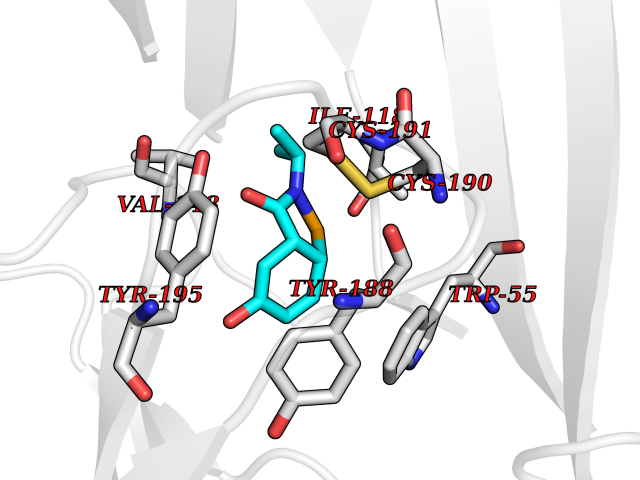 Pymol Map 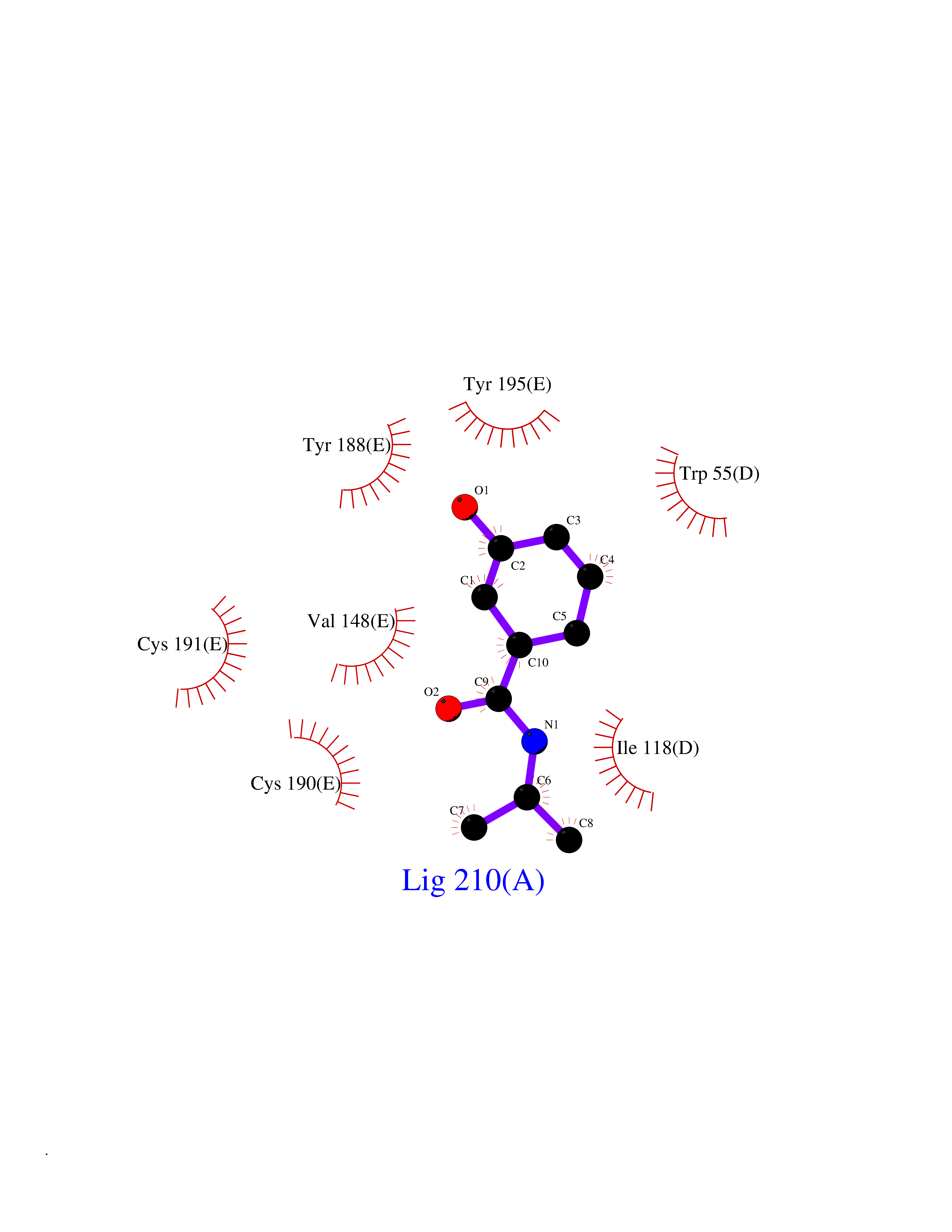 Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Spermine synthase | 3C6K | 5.70 | |
Target general information Gen name SMS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Spermidine/spermine synthase family Biochemical class Transferase Function Spermine synthase activity. Related diseases Intellectual developmental disorder, X-linked, syndromic, Snyder-Robinson type (MRXSSR) [MIM:309583]: An X-linked intellectual disability syndrome characterized by a collection of clinical features including facial asymmetry, marfanoid habitus, hypertonia, osteoporosis and unsteady gait. {ECO:0000269|PubMed:14508504, ECO:0000269|PubMed:18550699, ECO:0000269|PubMed:19206178, ECO:0000269|PubMed:22612257, ECO:0000269|PubMed:23696453, ECO:0000269|PubMed:23897707}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00127 Interacts with NA EC number 2.5.1.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Intellectual disability; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27177.1 Length 238 Aromaticity 0.11 Instability index 41.31 Isoelectric point 4.82 Charge (pH=7) -9.19 2D Binding mode Binding energy (Kcal/mol) -7.78 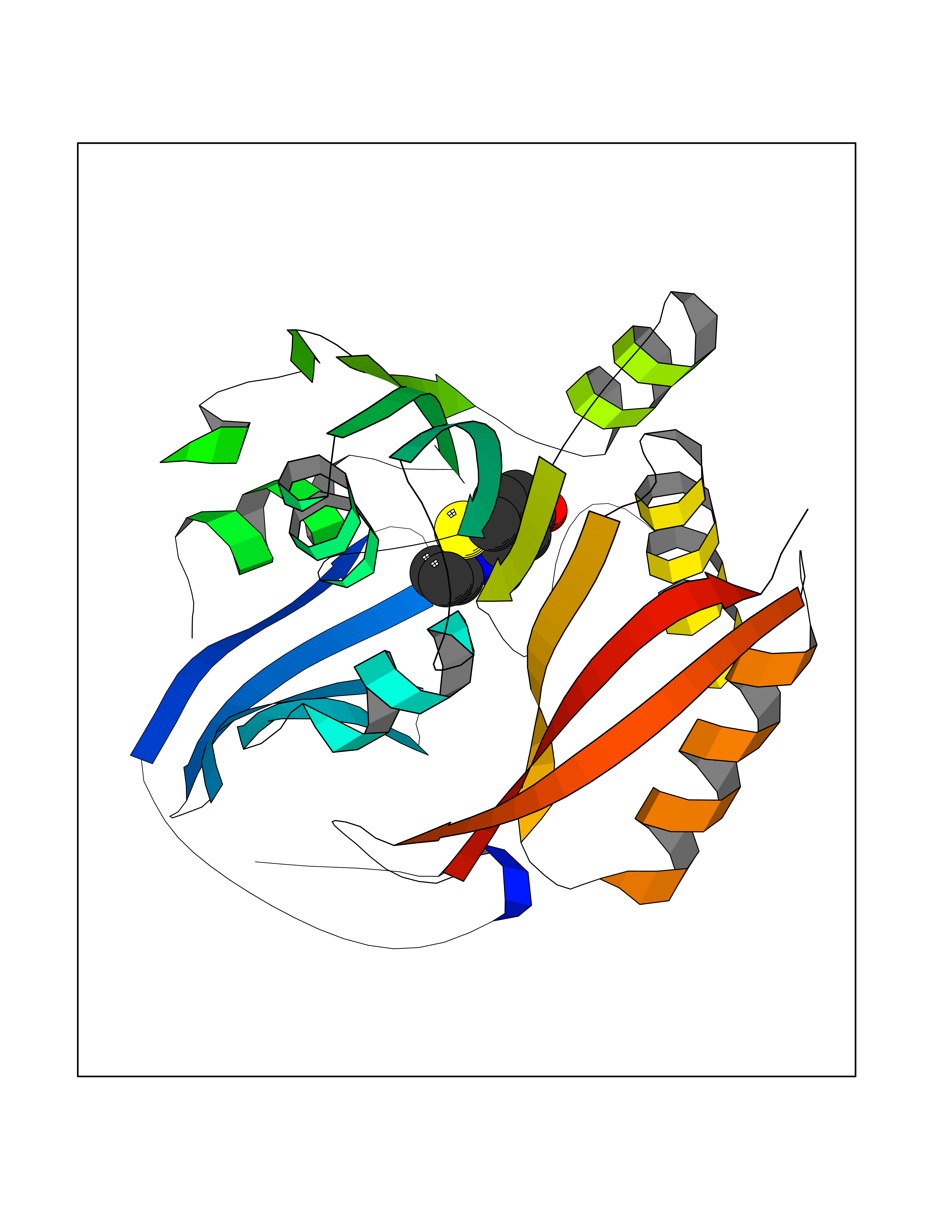 Molscript Map 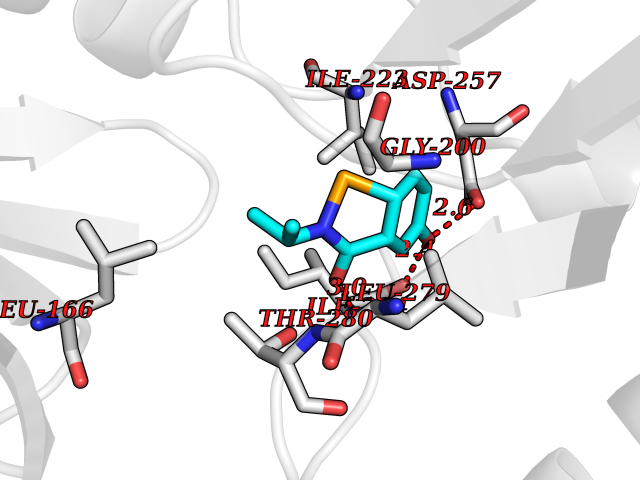 Pymol Map 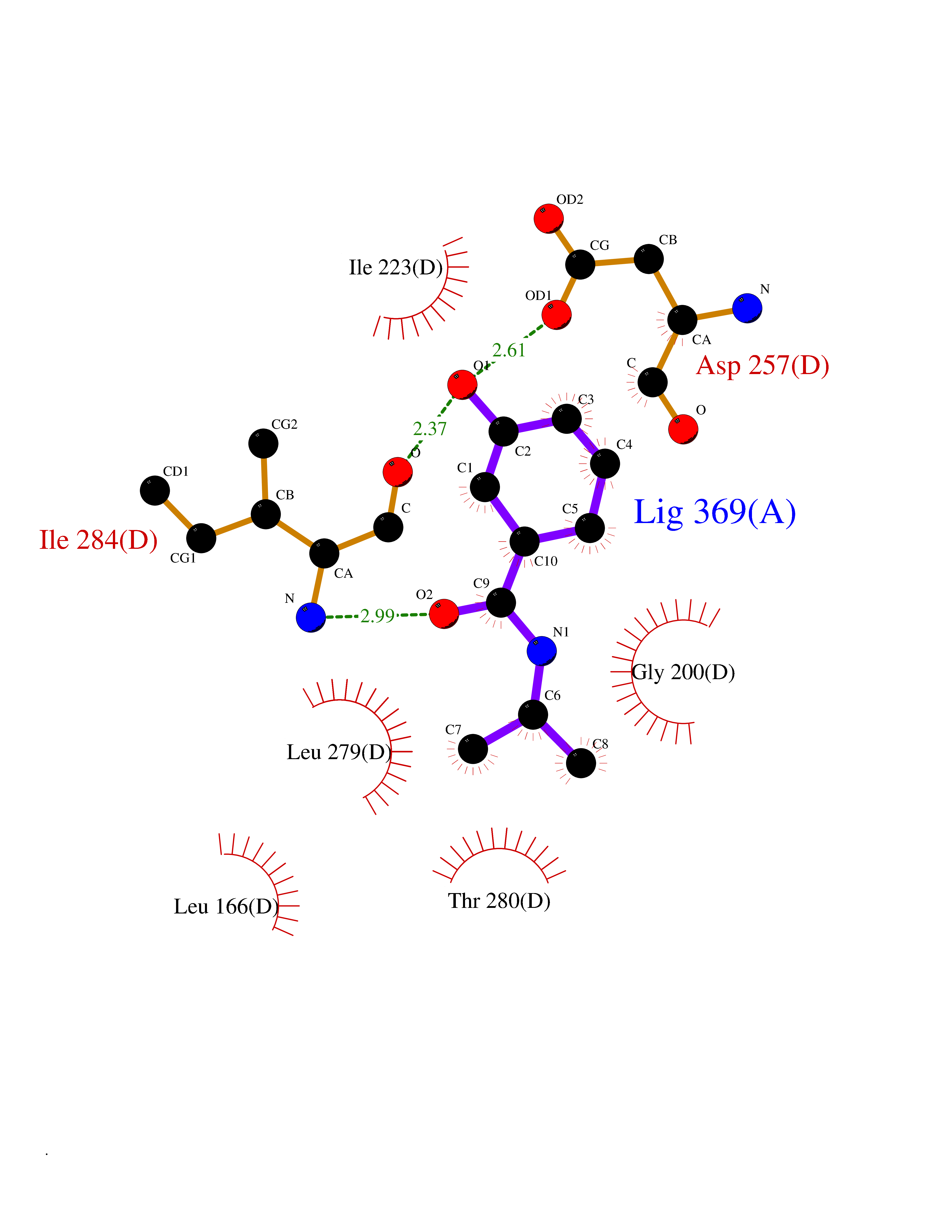 Ligplot Map 3D Binding mode Sequence RYWPTADGRLVEYDIDEVVYDEDSPYQNIKILHSKQFGNILILSGDVNLAESDLAYTRAIMGSGKEDYTGKDVLILGGGDGGILCEIVKLKPKMVTMVEIDQMVIDGCKKYMRKDVLDNLKGDCYQVLIEDCIPVLKRYAKEGREFDYVINDLTAVPISTSPSTWEFLRLILDLSMKVLKQDGKYFTQGNCVNLTEALSLYEEQLGRLYCPVEFSKEIVCVPSYLELWVFYTVWKKAK Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | cAMP-dependent protein kinase A type I (PRKAR1A) | 5KJZ | 5.70 | |
Target general information Gen name PRKAR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-dependent protein kinase type I-alpha regulatory subunit; Tissue-specific extinguisher 1; TSE1; PRKAR1; PKR1 Protein family CAMP-dependent kinase regulatory chain family Biochemical class Kinase Function Regulatory subunit of the cAMP-dependent protein kinases involved in cAMP signaling in cells. Related diseases Carney complex 1 (CNC1) [MIM:160980]: CNC is a multiple neoplasia syndrome characterized by spotty skin pigmentation, cardiac and other myxomas, endocrine tumors, and psammomatous melanotic schwannomas. {ECO:0000269|PubMed:15371594, ECO:0000269|PubMed:18241045, ECO:0000269|PubMed:22785148, ECO:0000269|PubMed:23323113, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Intracardiac myxoma (INTMYX) [MIM:255960]: Inheritance is autosomal recessive. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Primary pigmented nodular adrenocortical disease 1 (PPNAD1) [MIM:610489]: A rare bilateral adrenal defect causing ACTH-independent Cushing syndrome. Macroscopic appearance of the adrenals is characteristic with small pigmented micronodules observed in the cortex. Clinical manifestations of Cushing syndrome include facial and truncal obesity, abdominal striae, muscular weakness, osteoporosis, arterial hypertension, diabetes. PPNAD1 is most often diagnosed in patients with Carney complex, a multiple neoplasia syndrome. However it can also be observed in patients without other manifestations. {ECO:0000269|PubMed:12213893}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Acrodysostosis 1, with or without hormone resistance (ACRDYS1) [MIM:101800]: A form of skeletal dysplasia characterized by short stature, severe brachydactyly, facial dysostosis, and nasal hypoplasia. Affected individuals often have advanced bone age and obesity. Laboratory studies show resistance to multiple hormones, including parathyroid, thyrotropin, calcitonin, growth hormone-releasing hormone, and gonadotropin. However, not all patients show endocrine abnormalities. {ECO:0000269|PubMed:21651393, ECO:0000269|PubMed:22464250, ECO:0000269|PubMed:22464252, ECO:0000269|PubMed:22723333, ECO:0000269|PubMed:23043190, ECO:0000269|PubMed:23425300, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01790; DB02527; DB02315; DB05798 Interacts with Q9GZX7; P24588; O43687-2; Q9BSF0; Q9H6J7-2; Q86Y01; P0C7A2-2; Q9H0R8; Q9H8W4; P17612; P31321; P51817; P35250; Q86UC2; Q01105; Q8N0X7; O96006; P03259-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; cAMP; cAMP-binding; Cell membrane; Cushing syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat Protein physicochemical properties Chain ID A Molecular weight (Da) 17007.4 Length 149 Aromaticity 0.08 Instability index 49.36 Isoelectric point 6.36 Charge (pH=7) -0.52 2D Binding mode Binding energy (Kcal/mol) -7.77 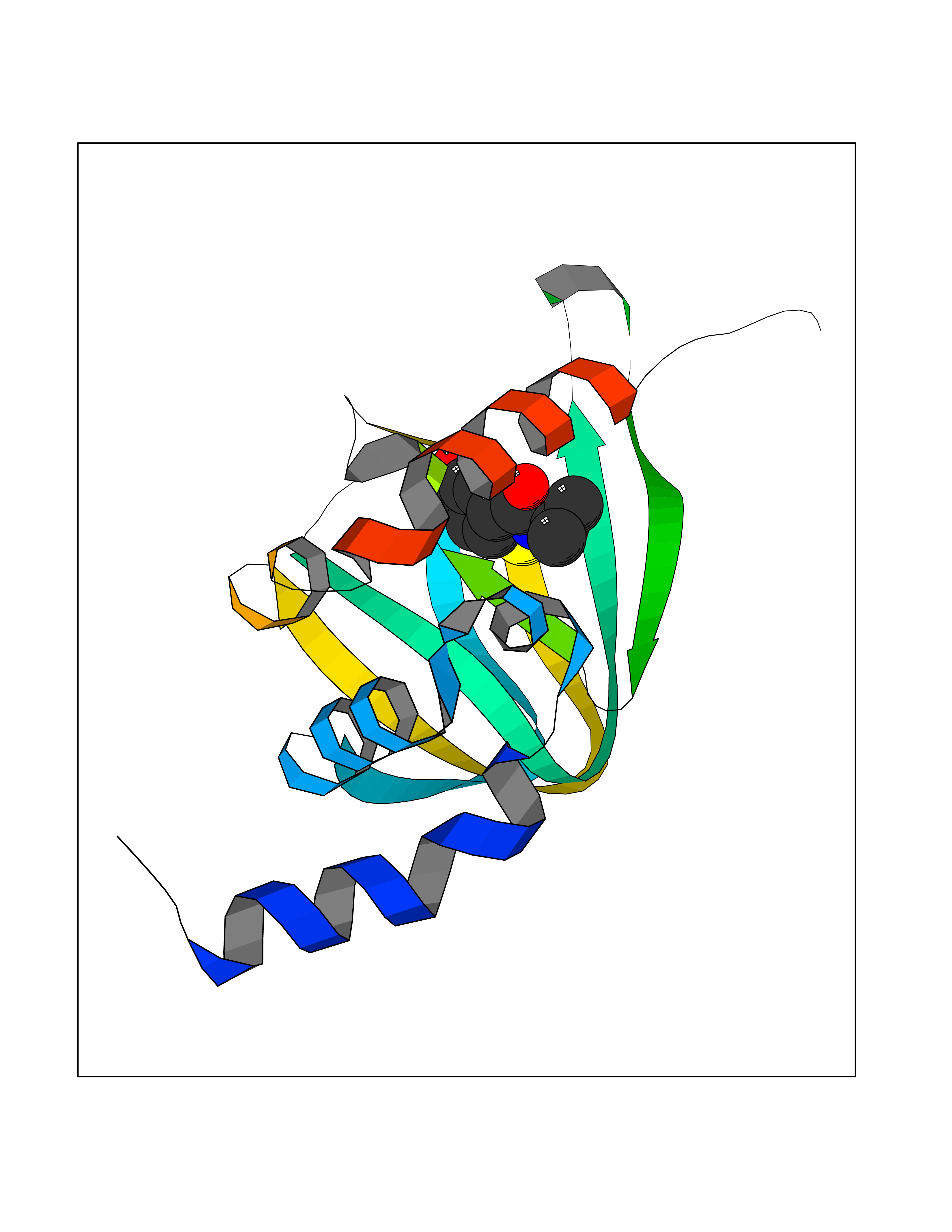 Molscript Map 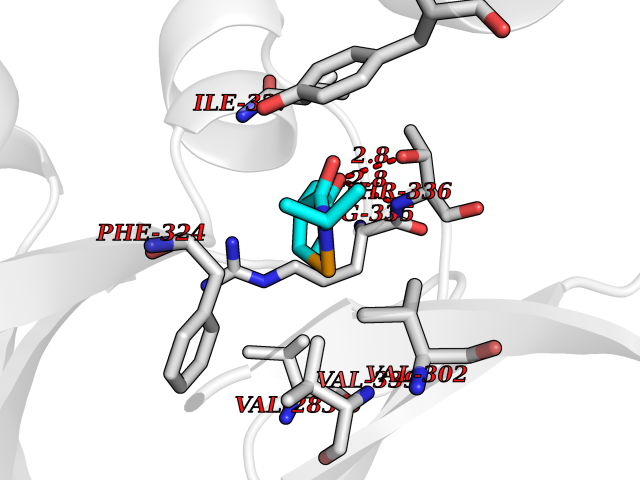 Pymol Map 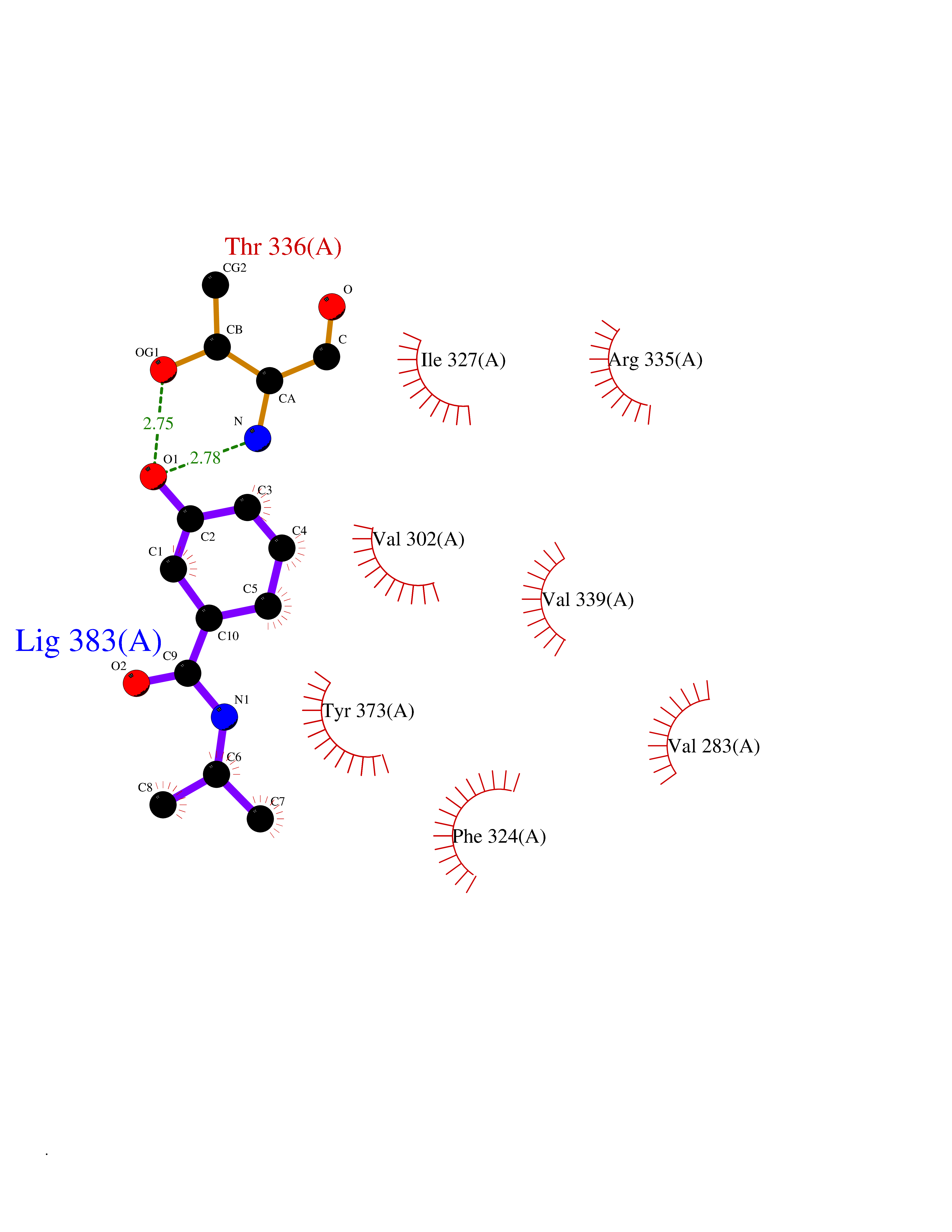 Ligplot Map 3D Binding mode Sequence SILMGSTLRKRKMYEEFLSKVSILESLDKWERLTVADALEPVQFEDGQKIVVQGEPGDEFFIILEGSAAVLQRRSENEEFVEVRRLGPSDYFGEIALLMNRPRTATVVARGPLKCVKLDRPRFERVLGPCSDILKRNIQQYNSFVSLSV Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Bromodomain and PHD finger containing 1 (BRPF1) | 4UYE | 5.70 | |
Target general information Gen name BRPF1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Br140; Peregrin; Bromodomain and PHD finger-containing protein 1; BR140 Protein family NA Biochemical class NA Function Component of the MOZ/MORF complex which has a histone H3 acetyltransferase activity. Preferentially mediates histone H3-K23 acetylation. Positively regulates the transcription of RUNX1 and RUNX2. Related diseases Intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) [MIM:617333]: An autosomal dominant neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, delayed language, and facial dysmorphisms, most notably ptosis. Additional features may include poor growth, hypotonia, and seizures. {ECO:0000269|PubMed:27939639, ECO:0000269|PubMed:27939640}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P0C0S8; P62805; Q96GN5; Q96MT8-3; Q8NHQ1; Q8WYH8; Q2KHM9; Q8N987; Q8N6Y0; Q9Y2K6 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; Bromodomain; Chromatin regulator; Chromosome; Cytoplasm; Disease variant; DNA-binding; Intellectual disability; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 12891.6 Length 109 Aromaticity 0.12 Instability index 51.42 Isoelectric point 5.47 Charge (pH=7) -2.14 2D Binding mode Binding energy (Kcal/mol) -7.78 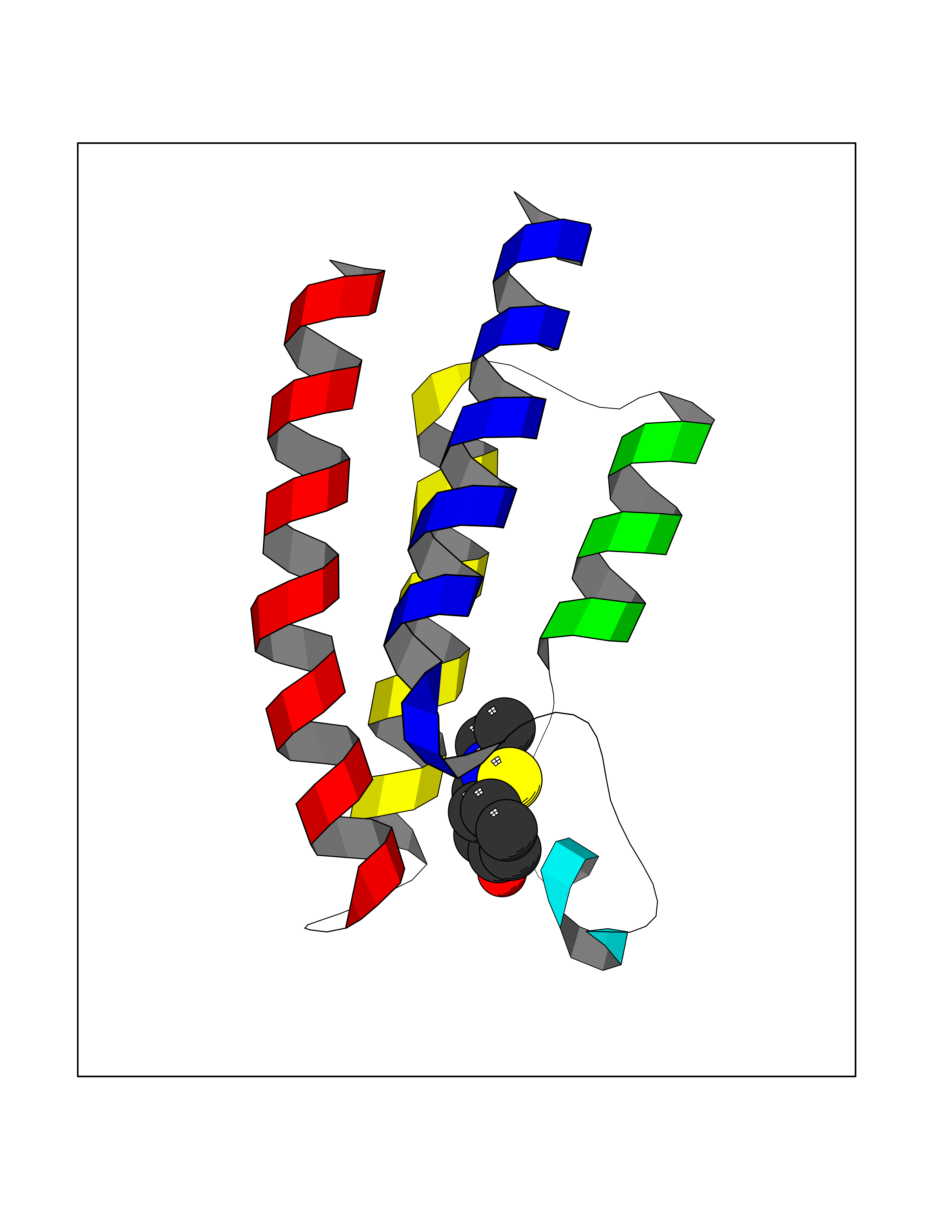 Molscript Map  Pymol Map 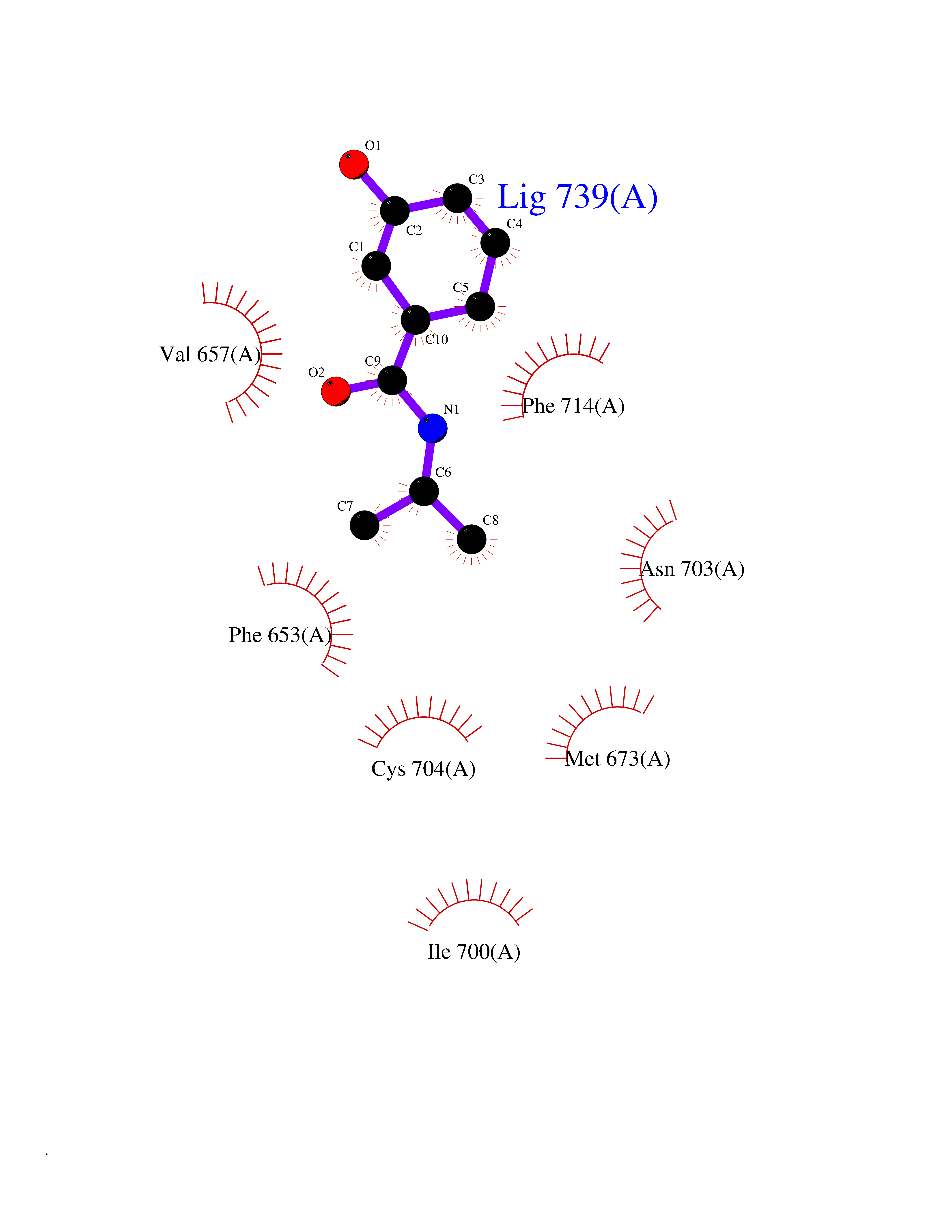 Ligplot Map 3D Binding mode Sequence QLTPFLILLRKTLEQLQEKDTGNIFSEPVPLSEVPDYLDHIKKPMDFFTMKQNLEAYRYLNFDDFEEDFNLIVSNCLKYNAKDTIFYRAAVRLREQGGAVLRQARRQAE Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Schistosoma Purine nucleoside phosphorylase (sch PNP) | 3F8W | 5.70 | |
Target general information Gen name sch PNP Organism Schistosoma mansoni (Blood fluke) Uniprot ID TTD ID Synonyms sch Purine nucleoside phosphorylase; sch Inosine-guanosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class NA Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta-(deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Hemolytic anemia, non-spherocytic, due to glucose phosphate isomerase deficiency (HA-GPID) [MIM:613470]: A form of anemia in which there is no abnormal hemoglobin or spherocytosis. It is caused by glucose phosphate isomerase deficiency. {ECO:0000269|PubMed:28803808, ECO:0000269|PubMed:7989588, ECO:0000269|PubMed:8499925, ECO:0000269|PubMed:8822952, ECO:0000269|PubMed:8822954, ECO:0000269|PubMed:9446754, ECO:0000269|PubMed:9856489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Glycosyltransferase; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 61140.6 Length 564 Aromaticity 0.06 Instability index 29.8 Isoelectric point 7.6 Charge (pH=7) 1.82 2D Binding mode Binding energy (Kcal/mol) -7.78 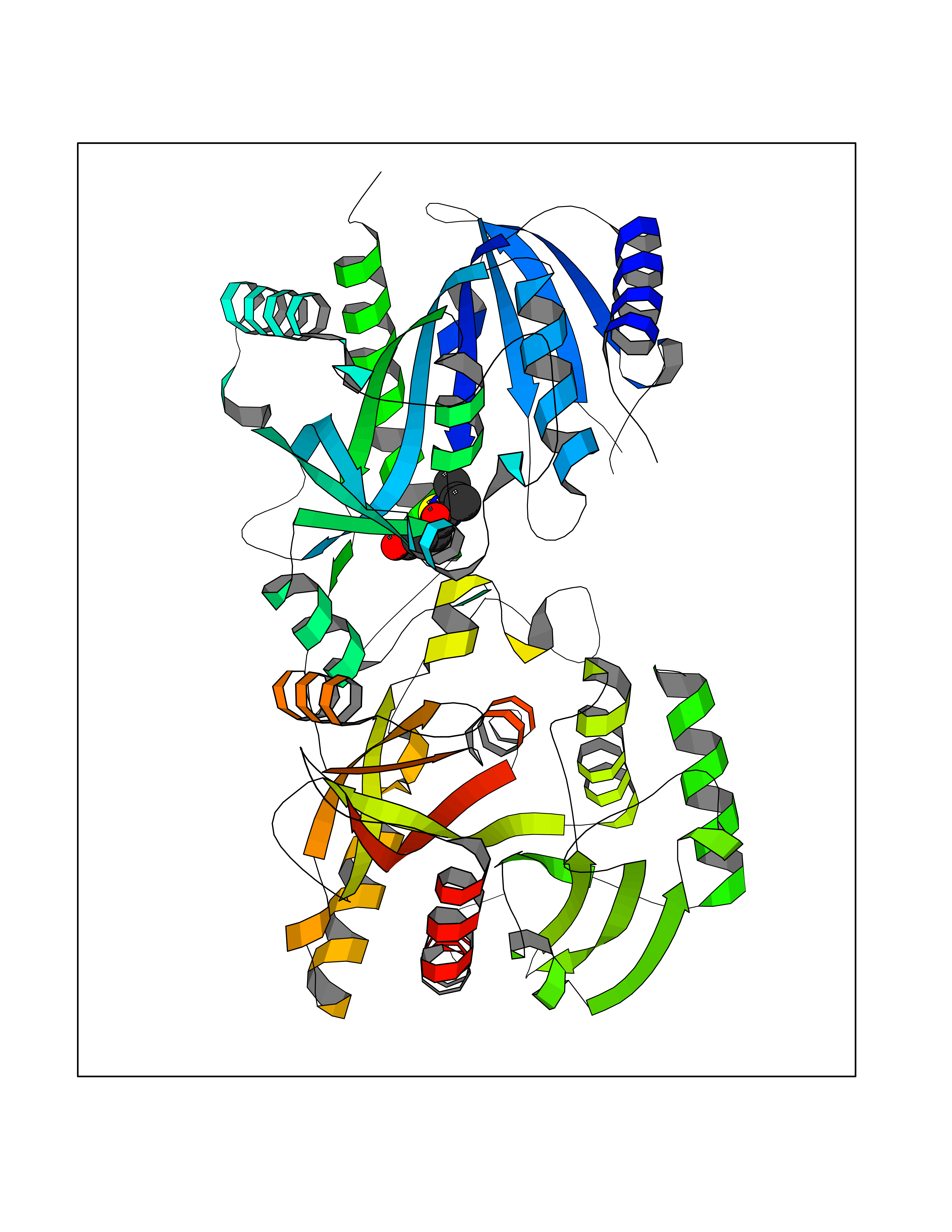 Molscript Map 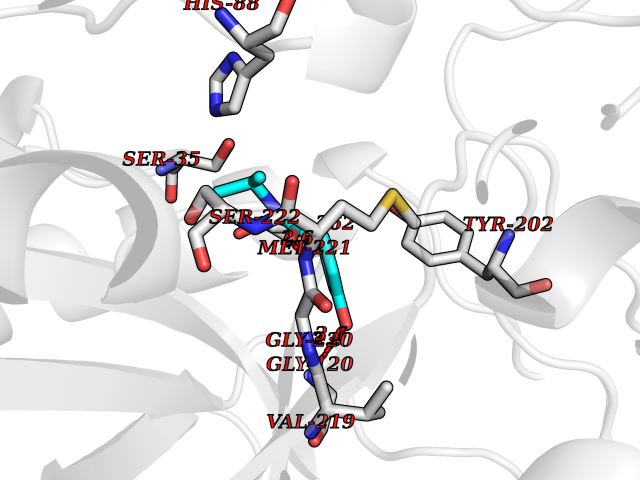 Pymol Map 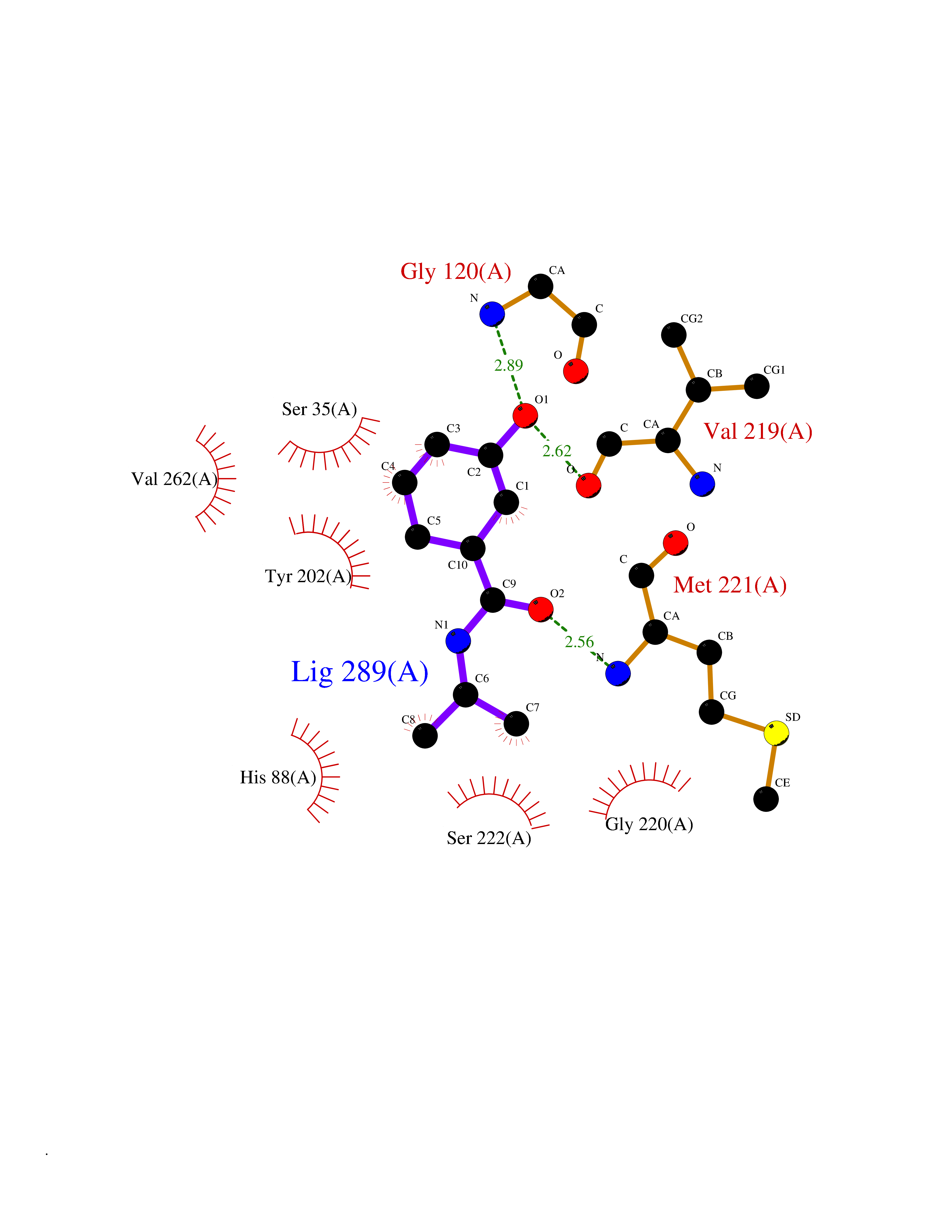 Ligplot Map 3D Binding mode Sequence SVTANIENVKKVAHHIQKLTSIVPEIGIICGSGLGKLADGVKDKITIPYTKIPNFPQTSHSGNLIFGTLSGRKVVVMQGRFHMYEGYSNDTVALPIRVMKLLGVKILMVSNAAGGLNRSLKLGDFVILKDHIYLPGLGLNNILVGPNQEAFGTRFPALSNAYDRDLRKLAVQVAEENGFGNLVHQGVYVMNGGPCYETPAECTMLLNMGCDVVGMSTIPEVVIARHCGIQVFAVSLVTNISVLDVESDLKPNHEEVLATGAQRAELMQSWFEKIIEKLPKSVTANIENVKKVAHHIQKLTSIVPEIGIICGSGLGKLADGVKDKITIPYTKIPNFPQTSVVGHSGNLIFGTLSGRKVVVMQGRFHMYEGYSNDTVALPIRVMKLLGVKILMVSNAAGGLNRSLKLGDFVILKDHIYLPGLGLNNILVGPNQEAFGTRFPALSNAYDRDLRKLAVQVAEENGFGNLVHQGVYVMNGGPCYETPAECTMLLNMGCDVVGMSTIPEVVIARHCGIQVFAVSLVTNISVLDVESDLKPNHEEVLATGAQRAELMQSWFEKIIEKLPKD Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Neuronal acetylcholine receptor beta-2 (CHRNB2) | 6CNJ | 5.70 | |
Target general information Gen name CHRNB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor beta2; Nicotinic acetylcholine receptor beta 2-subunit protein; CHRNB2; Beta-2 nAChR; Alpha-4/beta-2 nicotinic receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-2/CHRNB2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane permeable to sodiun ions. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00572; DB00237; DB00565; DB09028; DB01245; DB00514; DB07720; DB00898; DB00753; DB00657; DB00333; DB00184; DB00981; DB05458; DB05855; DB05740; DB00747; DB00202; DB01273 Interacts with P43681-1; P30532 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -7.77 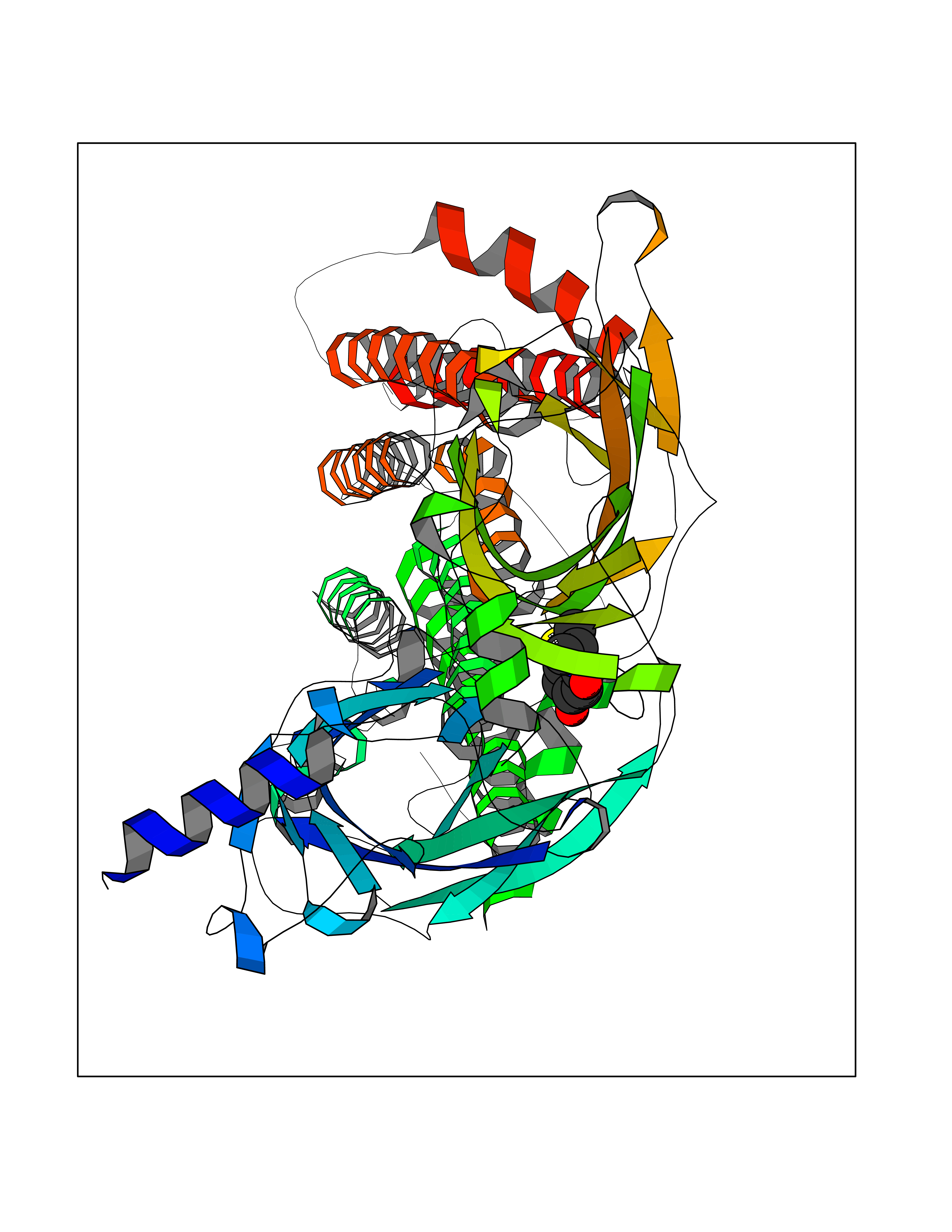 Molscript Map 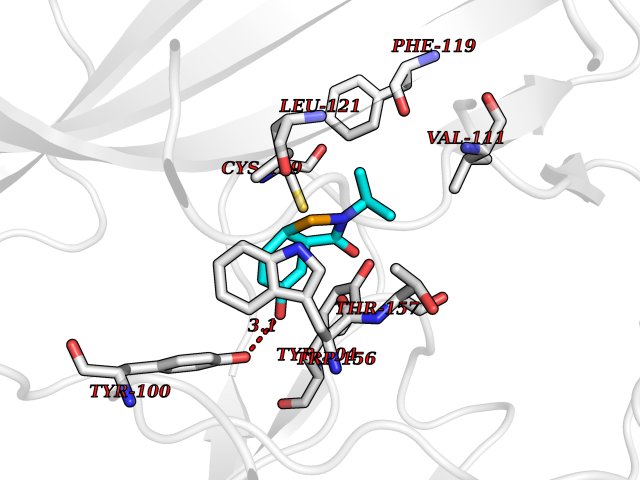 Pymol Map 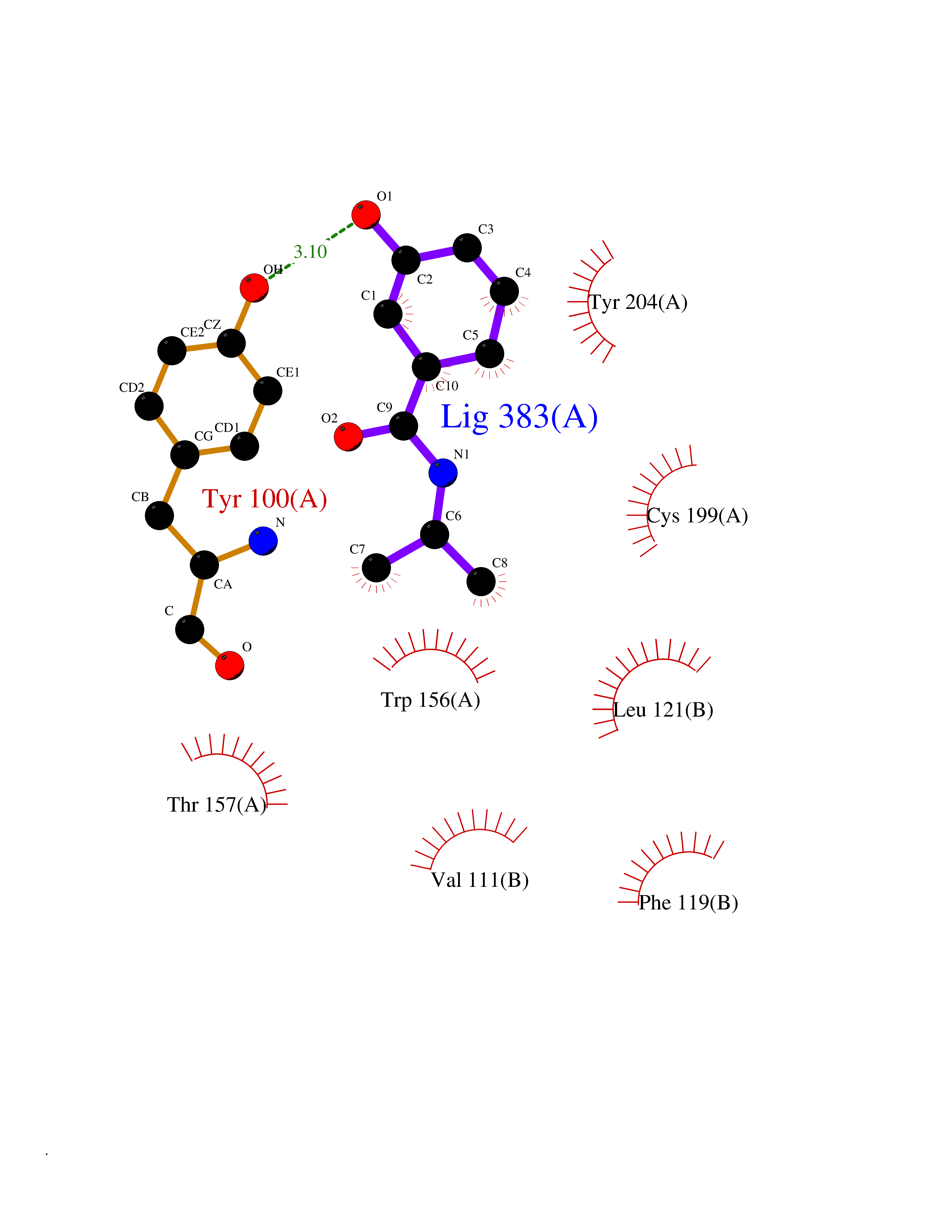 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Cytochrome P450 1B1 (CYP1B1) | 3PM0 | 5.70 | |
Target general information Gen name CYP1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CYPIB1 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function In liver microsomes, this enzyme is involved in an NADPH-dependent electron transport pathway. It oxidizes a variety of structurally unrelated compounds, including steroids, fatty acids, retinoid and xenobiotics. Preferentially oxidizes 17beta-estradiol to the carcinogenic 4-hydroxy derivative, and a variety of procarcinogenic compounds to their activated forms, including polycyclic aromatic hydrocarbons. Promotes angiogenesis by removing cellular oxygenation products, thereby decreasing oxidative stress, release of antiangiogenic factor THBS2, then allowing endothelial cells migration, cell adhesion and capillary morphogenesis. These changes are concommitant with the endothelial nitric oxide synthase activity and nitric oxide synthesis. Plays an important role in the regulation of perivascular cell proliferation, migration, and survival through modulation of the intracellular oxidative state and NF-kappa-B expression and/or activity, during angiogenesis. Contributes to oxidative homeostasis and ultrastructural organization and function of trabecular meshwork tissue through modulation of POSTN expression. Cytochromes P450 are a group of heme-thiolate monooxygenases. Related diseases Anterior segment dysgenesis 6 (ASGD6) [MIM:617315]: A form of anterior segment dysgenesis, a group of defects affecting anterior structures of the eye including cornea, iris, lens, trabecular meshwork, and Schlemm canal. Anterior segment dysgeneses result from abnormal migration or differentiation of the neural crest derived mesenchymal cells that give rise to components of the anterior chamber during eye development. Different anterior segment anomalies may exist alone or in combination, including iris hypoplasia, enlarged or reduced corneal diameter, corneal vascularization and opacity, posterior embryotoxon, corectopia, polycoria, abnormal iridocorneal angle, ectopia lentis, and anterior synechiae between the iris and posterior corneal surface. Clinical conditions falling within the phenotypic spectrum of anterior segment dysgeneses include aniridia, Axenfeld anomaly, Reiger anomaly/syndrome, Peters anomaly, and iridogoniodysgenesis. ASGD6 patients predominantly manifest Peters anomaly. Peters anomaly consists of corneal leukoma, defects in the posterior structures of the cornea such as absence of the posterior corneal stroma and Descemet membrane, and a variable degree of iridocorneal and/or keratolenticular adhesions. Over 50% of patients develop glaucoma in childhood. {ECO:0000269|PubMed:11403040}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glaucoma 3, primary congenital, A (GLC3A) [MIM:231300]: An autosomal recessive form of primary congenital glaucoma (PCG). PCG is characterized by marked increase of intraocular pressure at birth or early childhood, large ocular globes (buphthalmos) and corneal edema. It results from developmental defects of the trabecular meshwork and anterior chamber angle of the eye that prevent adequate drainage of aqueous humor. {ECO:0000269|PubMed:10227395, ECO:0000269|PubMed:10655546, ECO:0000269|PubMed:11184479, ECO:0000269|PubMed:11527932, ECO:0000269|PubMed:11774072, ECO:0000269|PubMed:11980847, ECO:0000269|PubMed:12036985, ECO:0000269|PubMed:12525557, ECO:0000269|PubMed:14635112, ECO:0000269|PubMed:14640114, ECO:0000269|PubMed:15255109, ECO:0000269|PubMed:15342693, ECO:0000269|PubMed:15475877, ECO:0000269|PubMed:16490498, ECO:0000269|PubMed:16688110, ECO:0000269|PubMed:16735994, ECO:0000269|PubMed:16862072, ECO:0000269|PubMed:18470941, ECO:0000269|PubMed:9463332, ECO:0000269|PubMed:9497261}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Glaucoma 1, open angle, A (GLC1A) [MIM:137750]: A form of primary open angle glaucoma (POAG). POAG is characterized by a specific pattern of optic nerve and visual field defects. The angle of the anterior chamber of the eye is open, and usually the intraocular pressure is increased. However, glaucoma can occur at any intraocular pressure. The disease is generally asymptomatic until the late stages, by which time significant and irreversible optic nerve damage has already taken place. {ECO:0000269|PubMed:11774072}. The gene represented in this entry acts as a disease modifier. Digenic mutations in CYP1B1 and MYOC have been found in a family segregating both primary adult-onset and juvenile forms of open angle glaucoma (PubMed:11774072). All affected family members with mutations in both MYOC and CYP1B1 had juvenile glaucoma, whereas those with only the MYOC mutation had the adult-onset form (PubMed:11774072). {ECO:0000269|PubMed:11774072}. Drugs (DrugBank ID) DB02342; DB00613; DB06732; DB00443; DB00121; DB01222; DB00201; DB09061; DB14737; DB01254; DB00694; DB01248; DB00997; DB00470; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB07776; DB00499; DB01645; DB01381; DB00741; DB01064; DB01026; DB00448; DB14009; DB01065; DB00170; DB00959; DB01204; DB14011; DB03467; DB00338; DB01229; DB14631; DB00635; DB01087; DB00396; DB00818; DB04216; DB02709; DB00675; DB00624; DB13946; DB00277; DB12245; DB11155 Interacts with Q02763 EC number EC 1.14.14.- Uniprot keywords 3D-structure; Disease variant; Endoplasmic reticulum; Fatty acid metabolism; Glaucoma; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Mitochondrion; Monooxygenase; Oxidoreductase; Peters anomaly; Proteomics identification; Reference proteome; Steroid metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 51875.9 Length 459 Aromaticity 0.1 Instability index 34.16 Isoelectric point 8.64 Charge (pH=7) 4.89 2D Binding mode Binding energy (Kcal/mol) -7.77 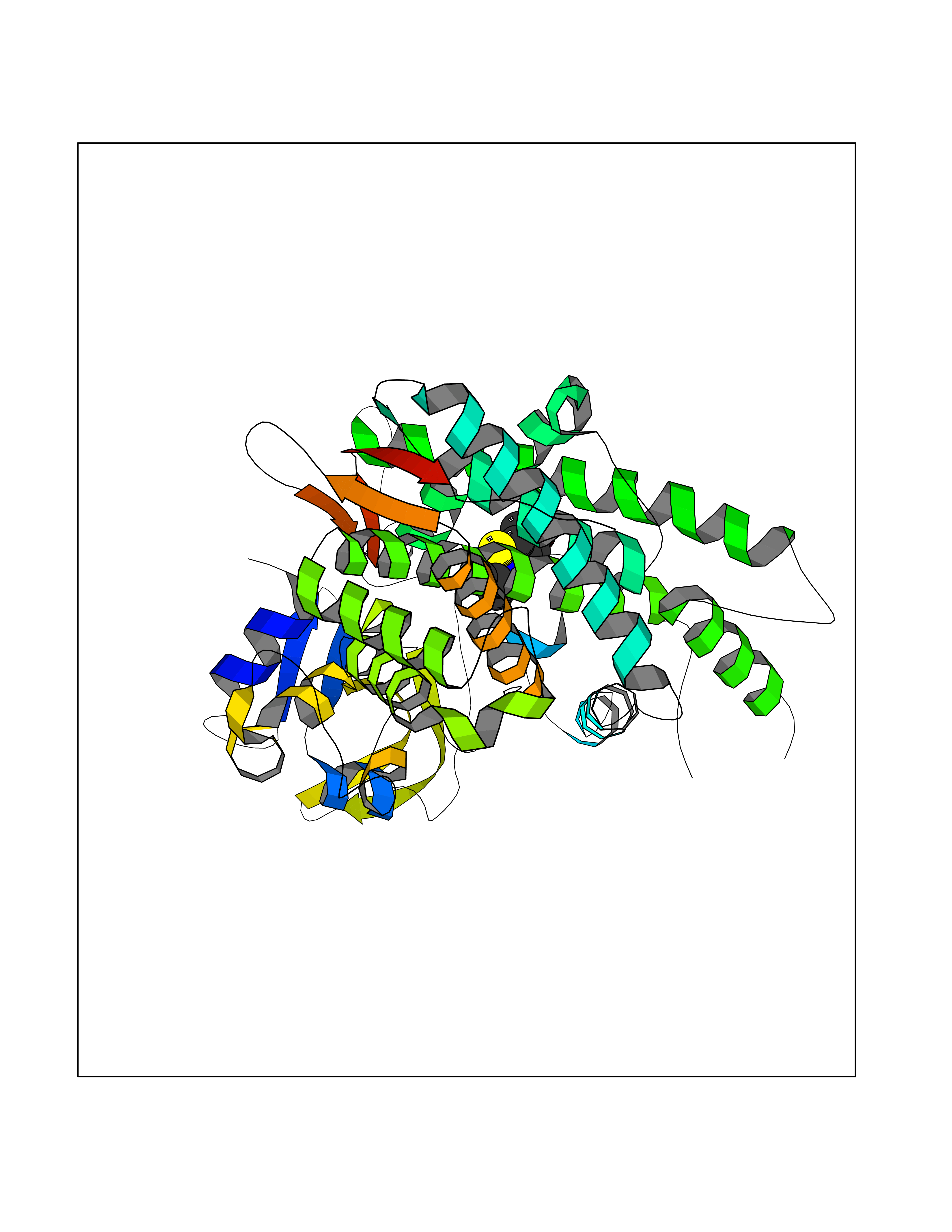 Molscript Map 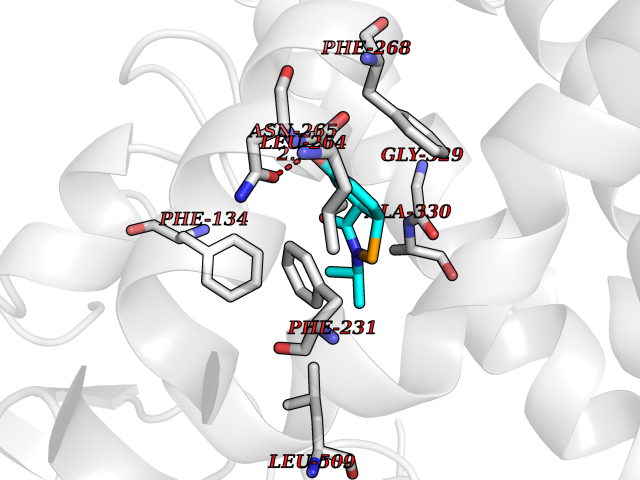 Pymol Map 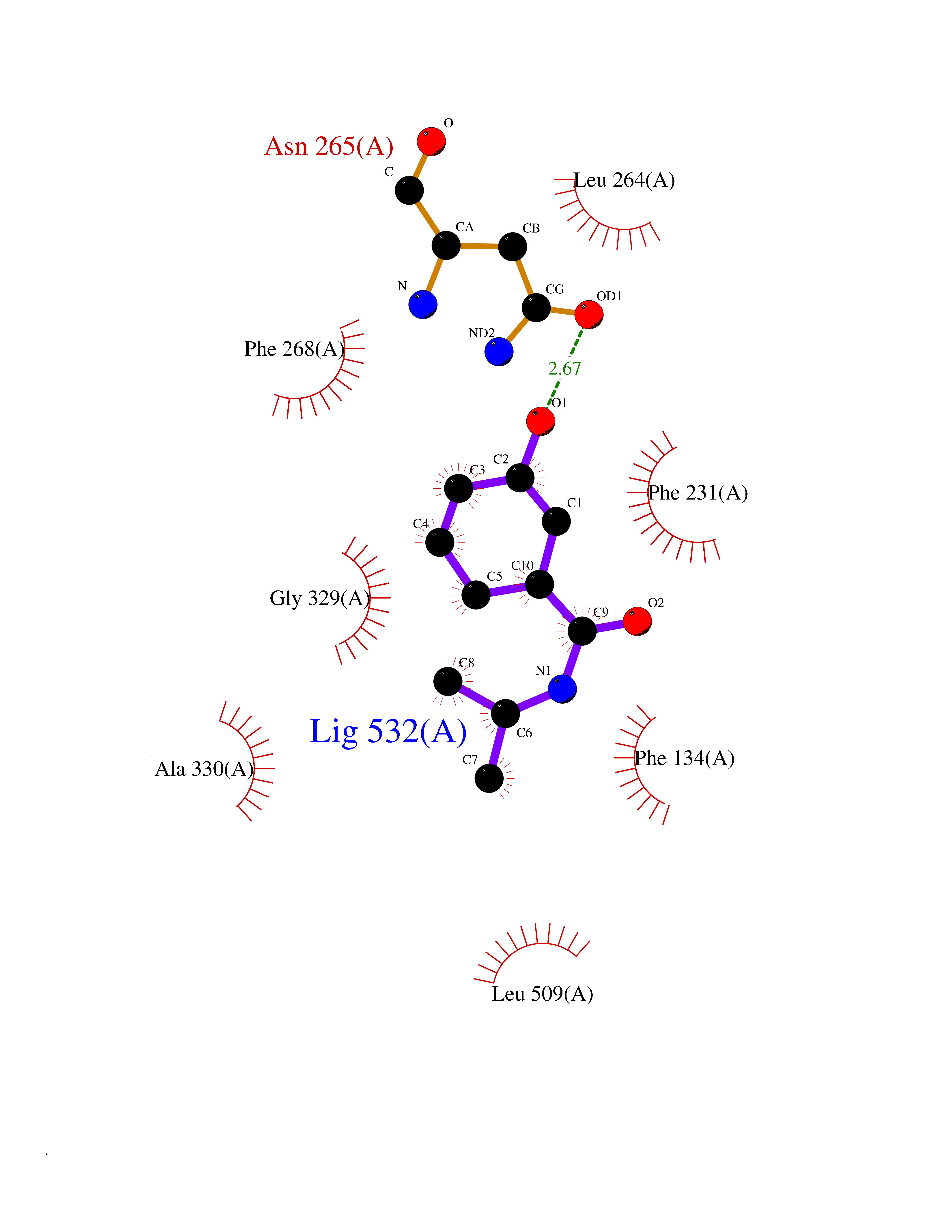 Ligplot Map 3D Binding mode Sequence QAAHLSFARLARRYGDVFQIRLGSCPIVVLNGERAIHQALVQQGSAFADRPSFASFRVVSGGRSMAFGHYSEHWKVQRRAAHSMMRNFFTRQPRSRQVLEGHVLSEARELVALLVRGSADGAFLDPRPLTVVAVANVMSAVCFGCRYSHDDPEFRELLSHNEEFGRTVGAGSLVDVMPWLQYFPNPVRTVFREFEQLNRNFSNFILDKFLRHCESLRPGAAPRDMMDAFILSAEKKAAGDGARLDLENVPATITDIFGASQDTLSTALQWLLLLFTRYPDVQTRVQAELDQVVGRDRLPCMGDQPNLPYVLAFLYEAMRFSSFVPVTIPHATTANTSVLGYHIPKDTVVFVNQWSVNHDPLKWPNPENFDPARFLDKDGLINKDLTSRVMIFSVGKRRCIGEELSKMQLFLFISILAHQCDFRANPNEPAKMNFSYGLTIKPKSFKVNVTLRESMELLD Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Dopamine D3 receptor (D3R) | 3PBL | 5.69 | |
Target general information Gen name DRD3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms D(3) dopamine receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Dopamine receptor whose activity is mediated by G proteins which inhibit adenylyl cyclase. Promotes cell proliferation. Related diseases Tremor, hereditary essential 1 (ETM1) [MIM:190300]: A common movement disorder mainly characterized by postural tremor of the arms. Head, legs, trunk, voice, jaw, and facial muscles may also be involved. The condition can be aggravated by emotions, hunger, fatigue and temperature extremes, and may cause a functional disability or even incapacitation. Inheritance is autosomal dominant. {ECO:0000269|PubMed:16650084, ECO:0000269|PubMed:16809426}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Glycine at position 9 results in gain of function and is associated with susceptibility to essential tremor. {ECO:0000269|PubMed:16650084, ECO:0000269|PubMed:16809426}.; DISEASE: Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:1362221, ECO:0000269|PubMed:9514583}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Glycine at position 9 results in gain of function and may be a risk factor for schizophrenia. {ECO:0000269|PubMed:1362221, ECO:0000269|PubMed:9514583}. Drugs (DrugBank ID) DB06288; DB00543; DB00714; DB01238; DB14185; DB09207; DB06216; DB09223; DB09128; DB01200; DB00490; DB00248; DB09014; DB06016; DB00477; DB01239; DB00363; DB11274; DB13345; DB00320; DB01184; DB00988; DB11275; DB01049; DB00875; DB00502; DB04946; DB01235; DB00589; DB00408; DB01403; DB06148; DB08804; DB05766; DB00334; DB01267; DB12061; DB01186; DB01100; DB09286; DB12478; DB00413; DB01224; DB00409; DB00268; DB05271; DB06454; DB00391; DB06477; DB09289; DB13025; DB01392; DB00246 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Receptor; Reference proteome; Schizophrenia; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 29257.6 Length 263 Aromaticity 0.13 Instability index 40.85 Isoelectric point 8.79 Charge (pH=7) 6.89 2D Binding mode Binding energy (Kcal/mol) -7.76 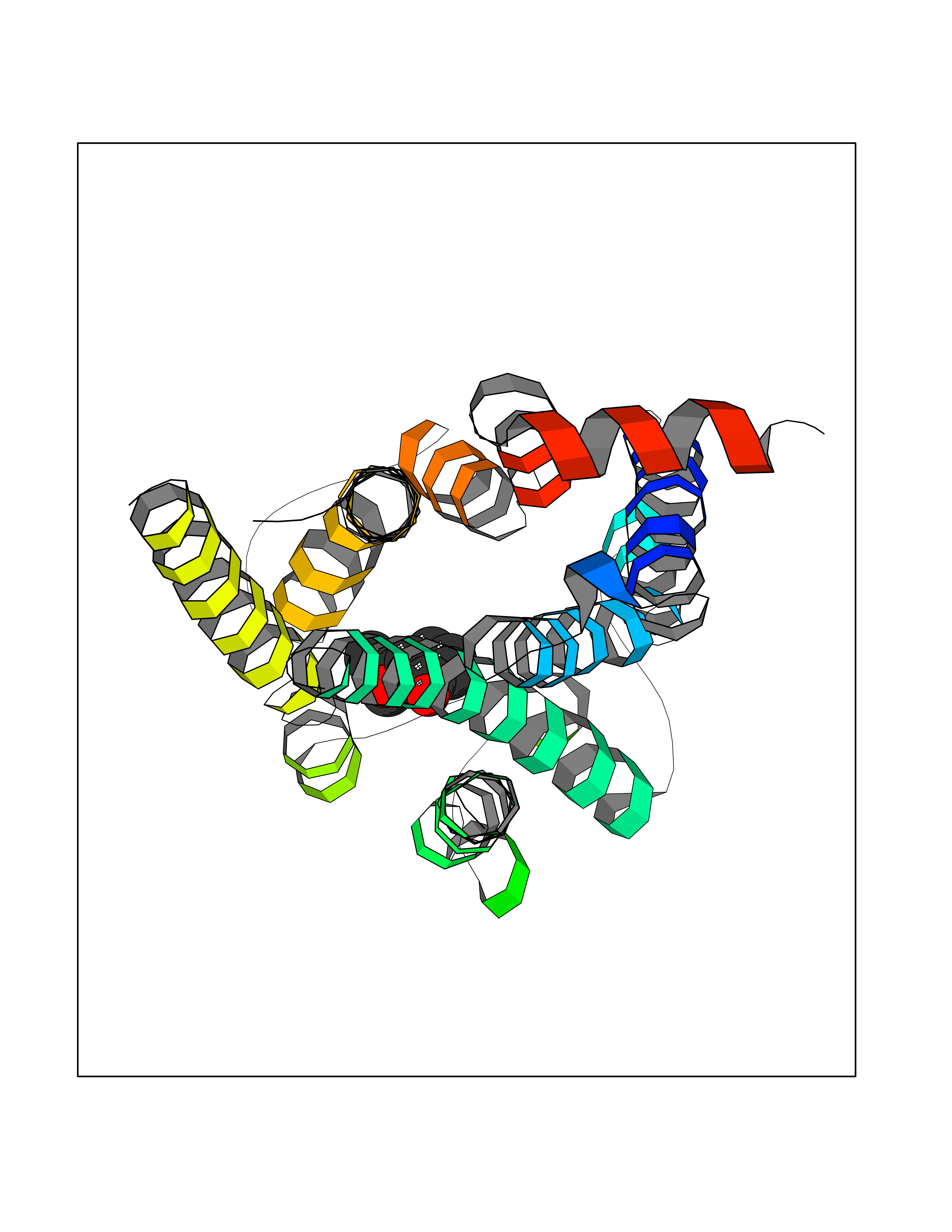 Molscript Map  Pymol Map 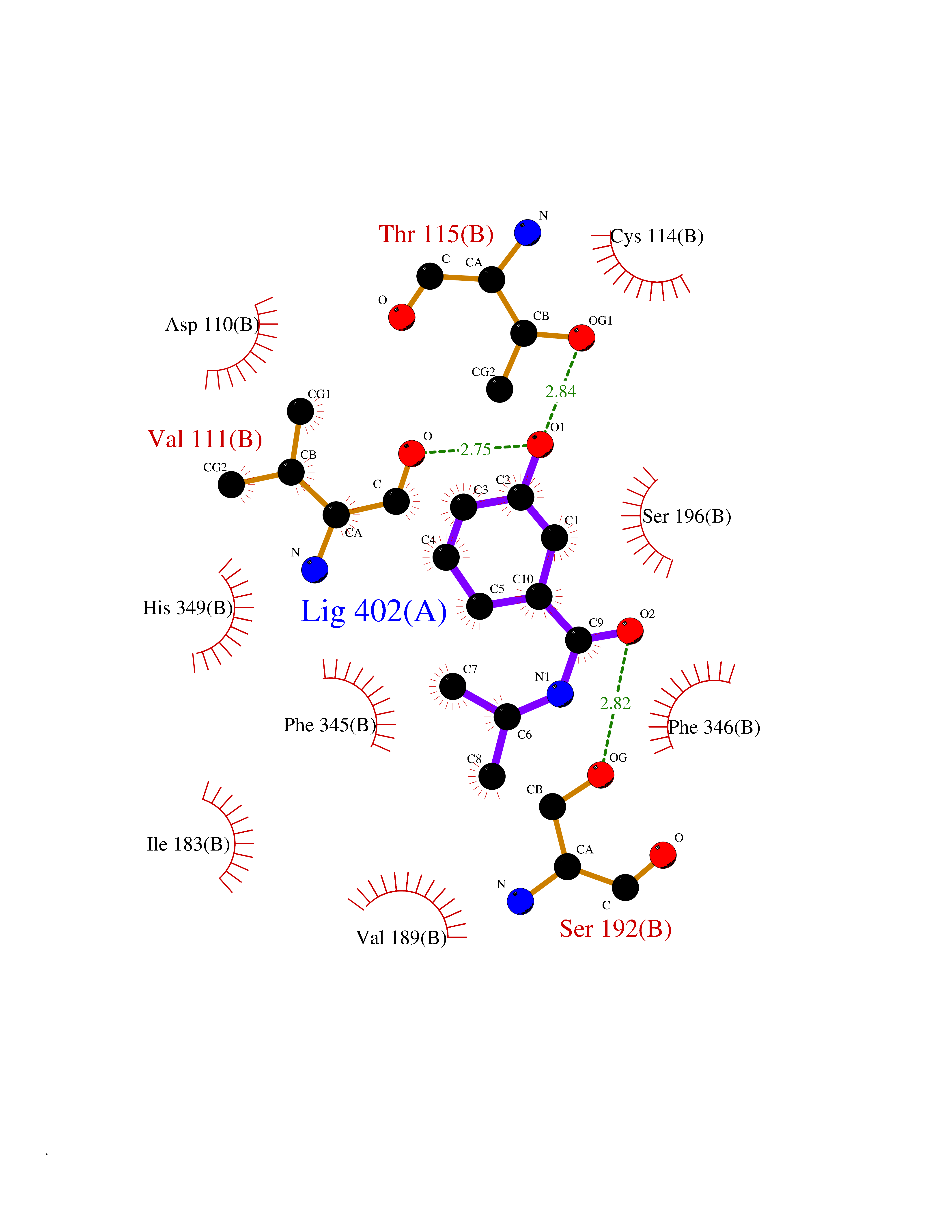 Ligplot Map 3D Binding mode Sequence YALSYCALILAIVFGNGLVCMAVLKERALQTTTNYLVVSLAVADLLVATLVMPWVVYLEVTGGVWNFSRICCDVFVTLDVMMCTASIWNLCAISIDRYTAVVMPSSCRRVALMITAVWVLAFAVSCPLLFGFNTTGDPTVCSISNPDFVIYSSVVSFYLPFGVTVLVYARIYVVLKQRRRKGVPLREKKATQMVAIVLGAFIVCWLPFFLTHVLNTHCQTCHVSPELYSATTWLGYVNSALNPVIYTTFNIEFRKAFLKILSC Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Monoglyceride lipase (MAGL) | 3PE6 | 5.69 | |
Target general information Gen name MGLL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoacylglycerol lipase; MGL; Lysophospholipaselike; Lysophospholipase-like; Lysophospholipase homolog; HUK5; HU-K5 Protein family AB hydrolase superfamily, Monoacylglycerol lipase family Biochemical class Carboxylic ester hydrolase Function Hydrolyzes the endocannabinoid 2-arachidonoylglycerol, and thereby contributes to the regulation of endocannabinoid signaling, nociperception and perception of pain. Regulates the levels of fatty acids that serve as signaling molecules and promote cancer cell migration, invasion and tumor growth. Converts monoacylglycerides to free fatty acids and glycerol. Related diseases Systemic lupus erythematosus 9 (SLEB9) [MIM:610927]: A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. {ECO:0000269|PubMed:17360460}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency, common variable, 7 (CVID7) [MIM:614699]: A primary immunodeficiency characterized by antibody deficiency, hypogammaglobulinemia, recurrent bacterial infections and an inability to mount an antibody response to antigen. The defect results from a failure of B-cell differentiation and impaired secretion of immunoglobulins; the numbers of circulating B-cells is usually in the normal range, but can be low. {ECO:0000269|PubMed:22035880}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07550; P37235 EC number EC 3.1.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Lipid biosynthesis; Lipid degradation; Lipid metabolism; Membrane; Nitration; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 31808.4 Length 289 Aromaticity 0.08 Instability index 29.7 Isoelectric point 6.73 Charge (pH=7) -0.91 2D Binding mode Binding energy (Kcal/mol) -7.76 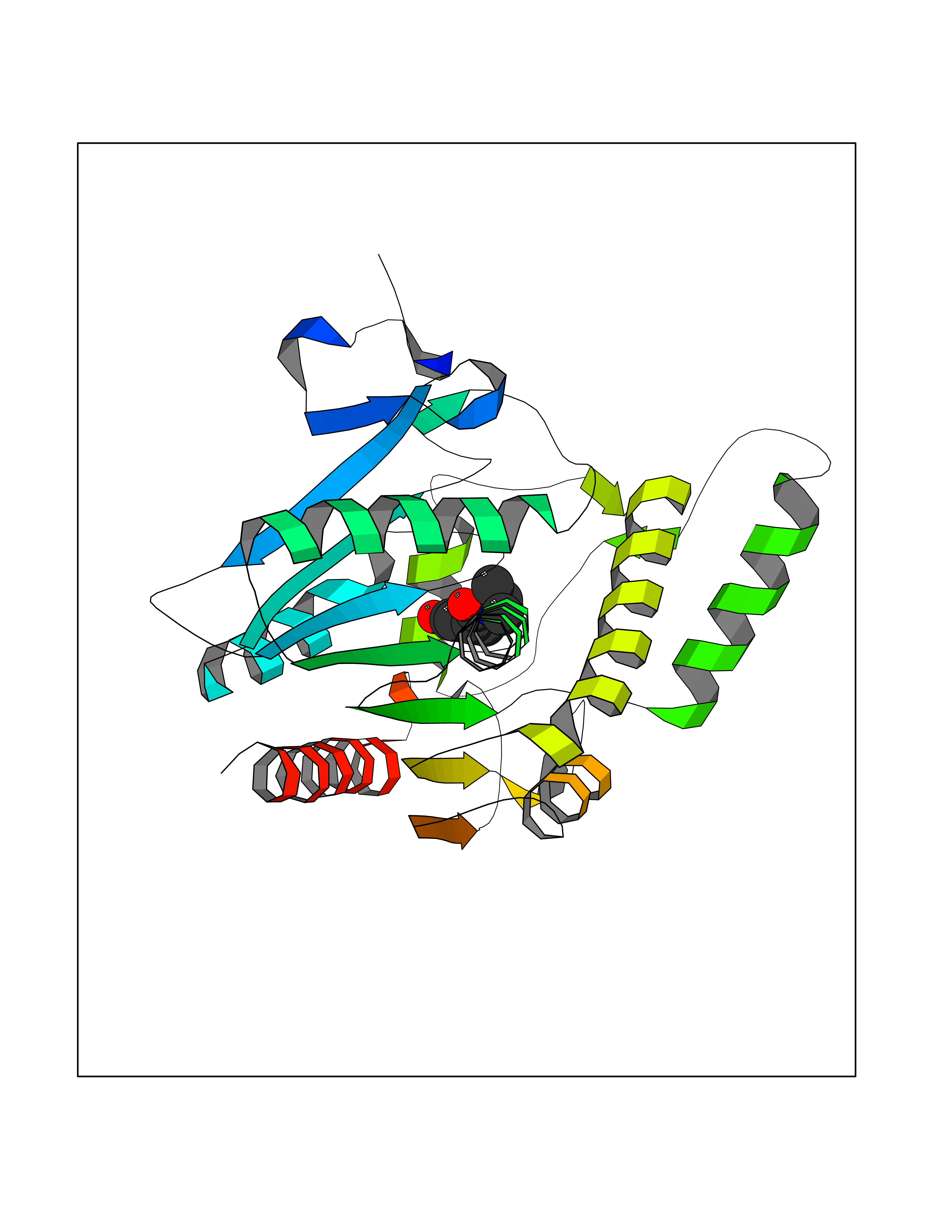 Molscript Map 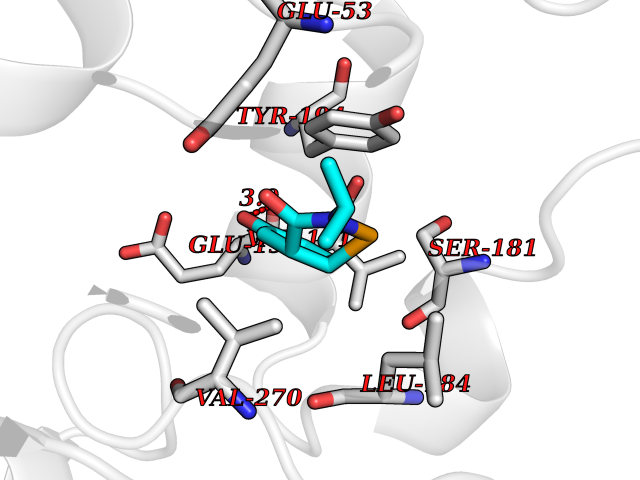 Pymol Map 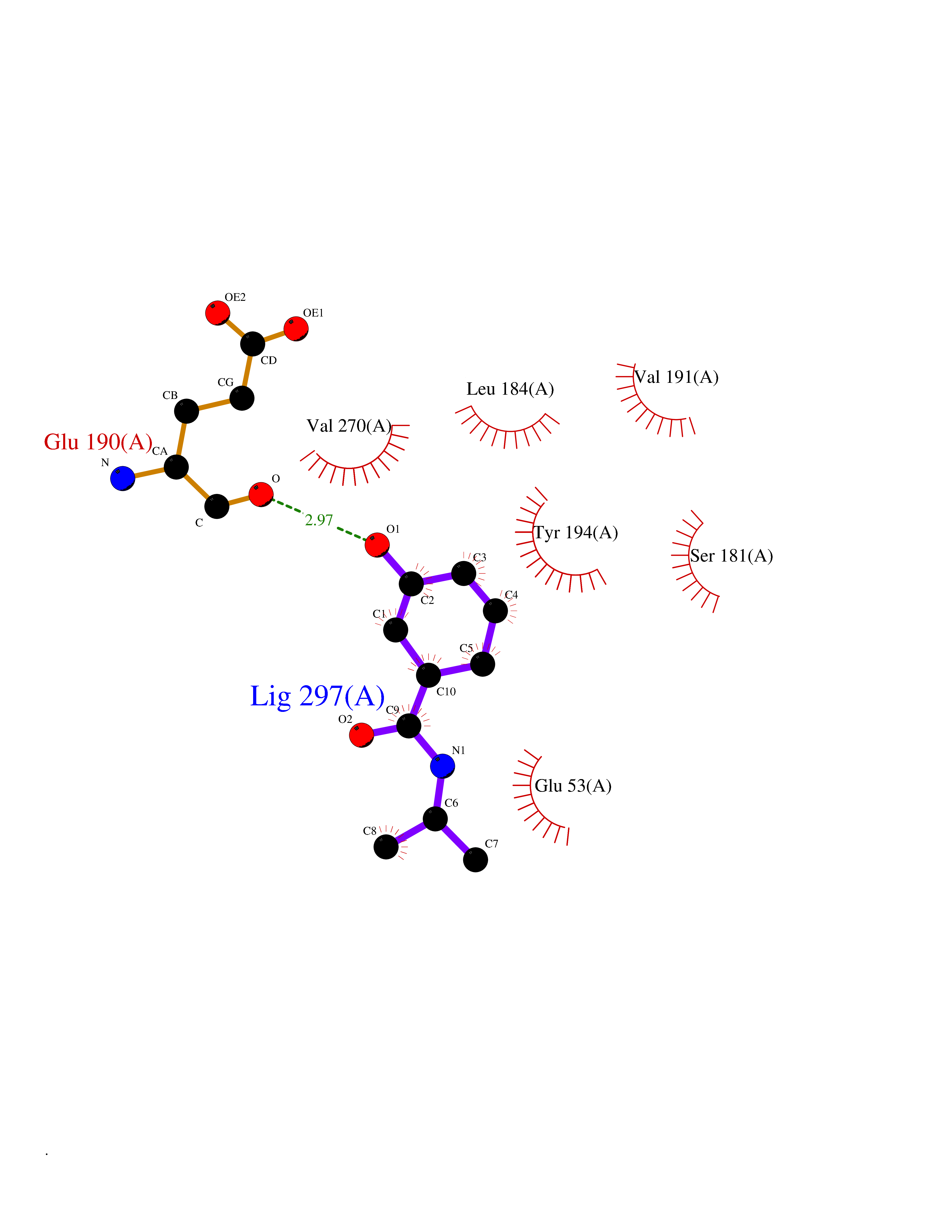 Ligplot Map 3D Binding mode Sequence PRRTPQSIPYQDLPHLVNADGQYLFCRYWAPTGTPKALIFVSHGAGEHSGRYEELARMLMGLDLLVFAHDHVGHGQSEGERMVVSDFHVFVRDVLQHVDSMQKDYPGLPVFLLGHSMGGAIAILTAAERPGHFAGMVLISPLVLANPESATTFKVLAAKVLNSVLPNLSSGPIDSSVLSRNKTEVDIYNSDPLICRAGLKVCFGIQLLNAVSRVERALPKLTVPFLLLQGSADRLCDSKGAYLLMELAKSQDKTLKIYEGAYHVLHKELPEVTNSVFHEINMWVSQRTA Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | p53-binding protein Mdm4 (MDM4) | 6Q9Y | 5.69 | |
Target general information Gen name MDM4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Mdmx; Mdm2-like p53-binding protein; Double minute 4 protein Protein family MDM2/MDM4 family Biochemical class MDM2/MDM4 family Function Inhibits p53/TP53- and TP73/p73-mediated cell cycle arrest and apoptosis by binding its transcriptional activation domain. Inhibits degradation of MDM2. Can reverse MDM2-targeted degradation of TP53 while maintaining suppression of TP53 transactivation and apoptotic functions. Related diseases Bone marrow failure syndrome 6 (BMFS6) [MIM:618849]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFS6 is an autosomal dominant form characterized by intermittent neutropenia, lymphopenia, or anemia associated with hypocellular bone marrow, and increased susceptibility to cancer. {ECO:0000269|PubMed:32300648}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NX04; P10415; Q7Z479; O95971; P48729; Q00987; Q13064; P41227; P06400; Q9Y4L5; P23297; P29034; P33763; P04271; P31947; P04637; P62837; Q93009; O14972; P61964; P62258; P61981; P63104; Q9BRR0; A0A0S2Z6X0; Q3YBA8; P03255-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 19722 Length 173 Aromaticity 0.08 Instability index 50.78 Isoelectric point 8.48 Charge (pH=7) 2.27 2D Binding mode Binding energy (Kcal/mol) -7.76 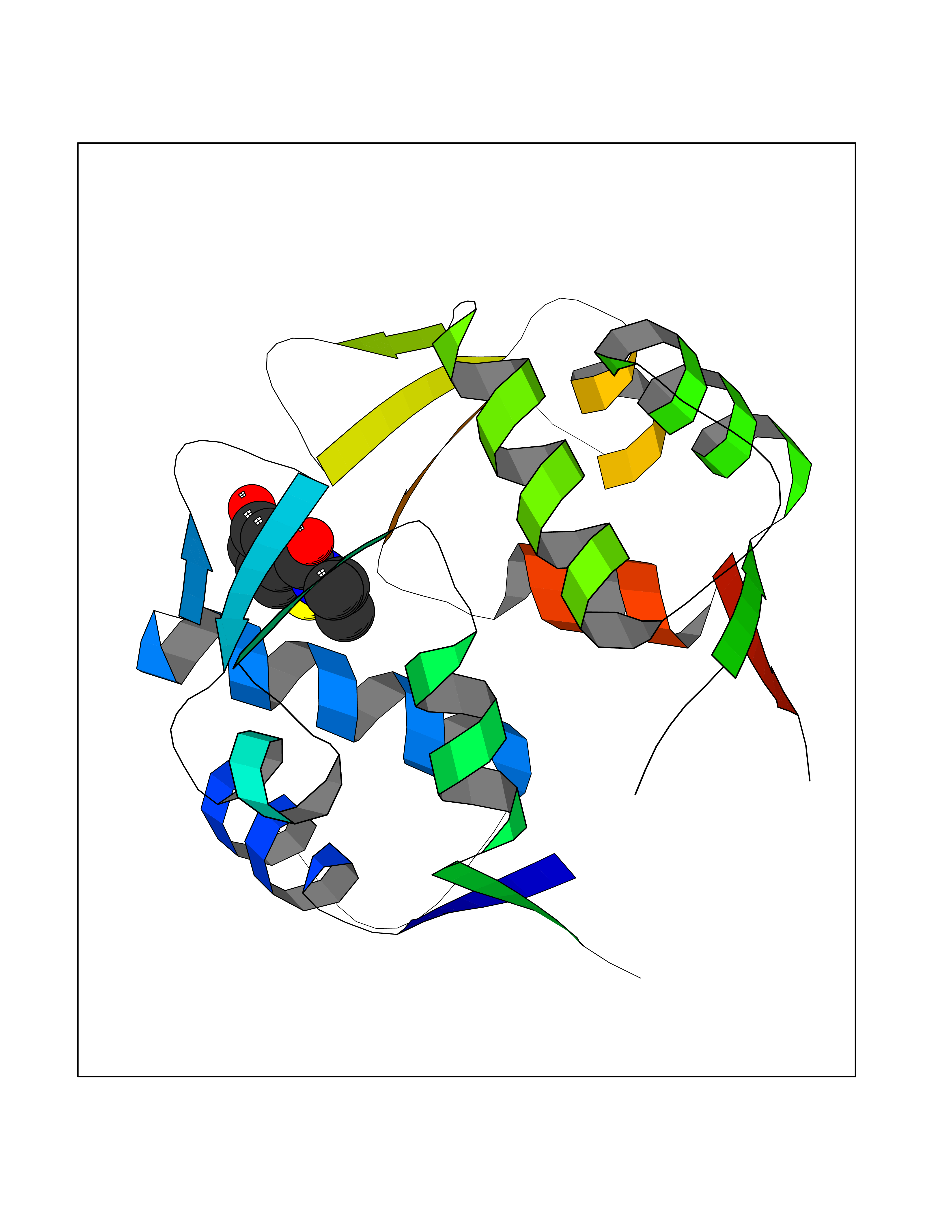 Molscript Map 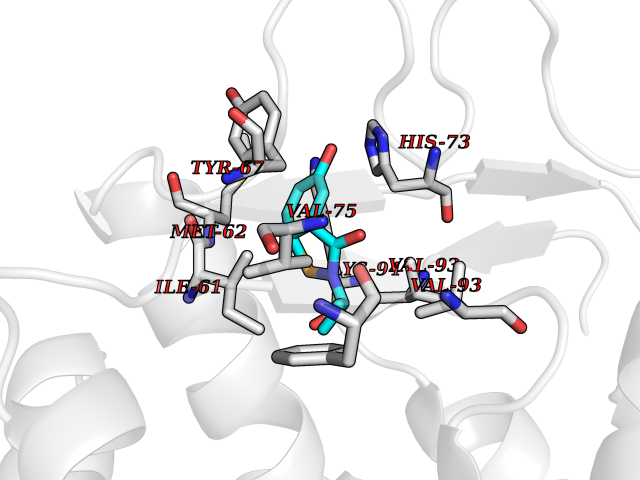 Pymol Map 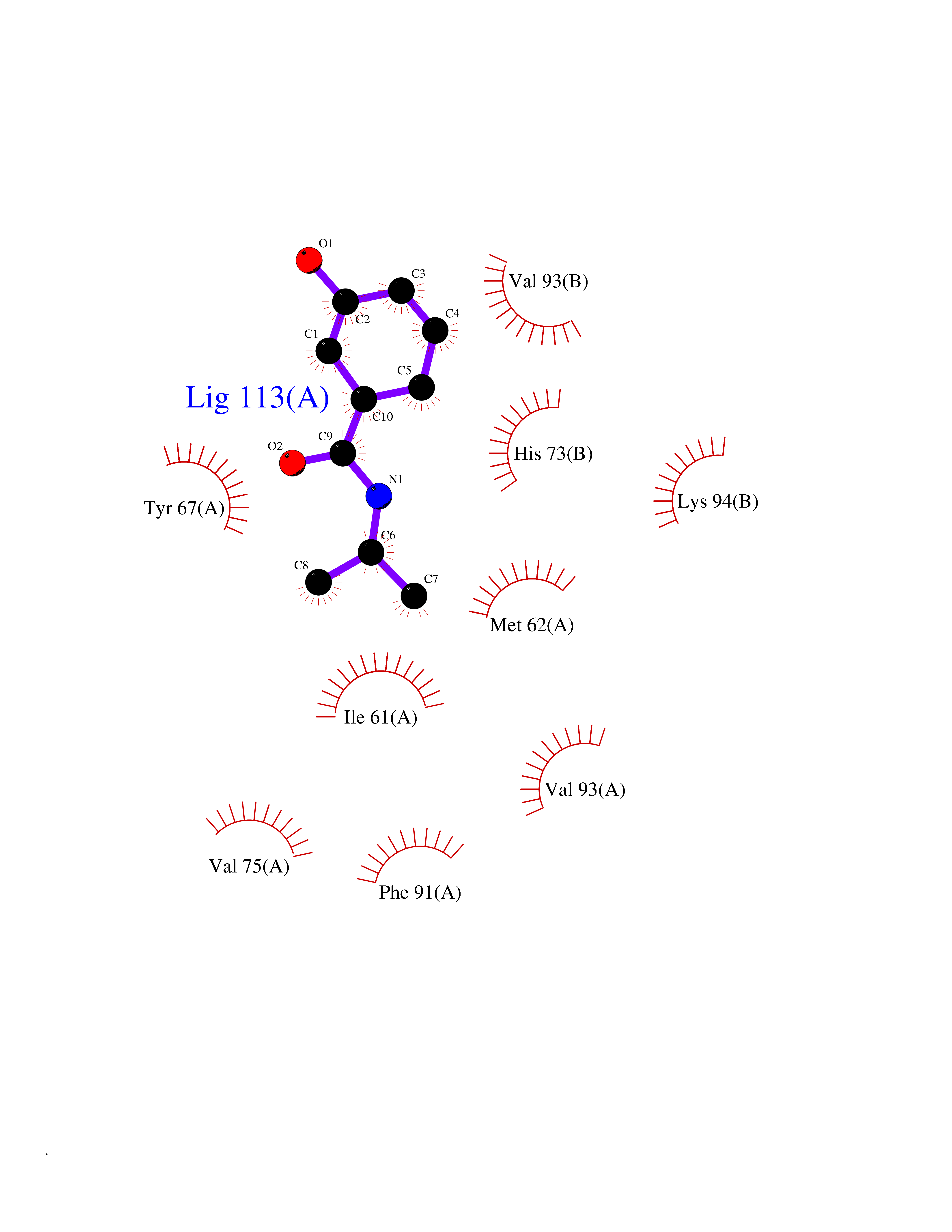 Ligplot Map 3D Binding mode Sequence QVRPKLPLLKILHAAGAQGEMFTVKEVMHYLGQYIMVKQLYDQQEQHMVYCGGDLLGELLGRQSFSVKDPSPLYDMLRKNLVTLAQINQVRPKLPLLKILHAAGAQGEMFTVKEVMHYLGQYIMVKQLYDQQEQHMVYCGGDLLGELLGRQSFSVKDPSPLYDMLRKNLVTLA Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Neuronal acetylcholine receptor alpha-4 (CHRNA4) | 6CNJ | 5.69 | |
Target general information Gen name CHRNA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor alpha4; CHRNA4; Alpha-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-4/CHRNA4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasmamembrane permeable to sodium ions. Related diseases Epilepsy, nocturnal frontal lobe, 1 (ENFL1) [MIM:600513]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:10563623, ECO:0000269|PubMed:14623738, ECO:0000269|PubMed:7550350}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01351; DB01352; DB00572; DB01483; DB00237; DB00241; DB01353; DB00564; DB00565; DB09028; DB01245; DB00514; DB01496; DB07720; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00898; DB01354; DB01355; DB00753; DB00657; DB00333; DB00463; DB00849; DB00184; DB00312; DB01174; DB00981; DB05458; DB00794; DB05740; DB00747; DB00418; DB00202; DB00306; DB00599; DB01273 Interacts with Q6UY14-3; P05067; P83916; Q6UXH1-1; Q6UXH1-3; P20042; Q9NZR2; Q92673; P17787 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -7.77 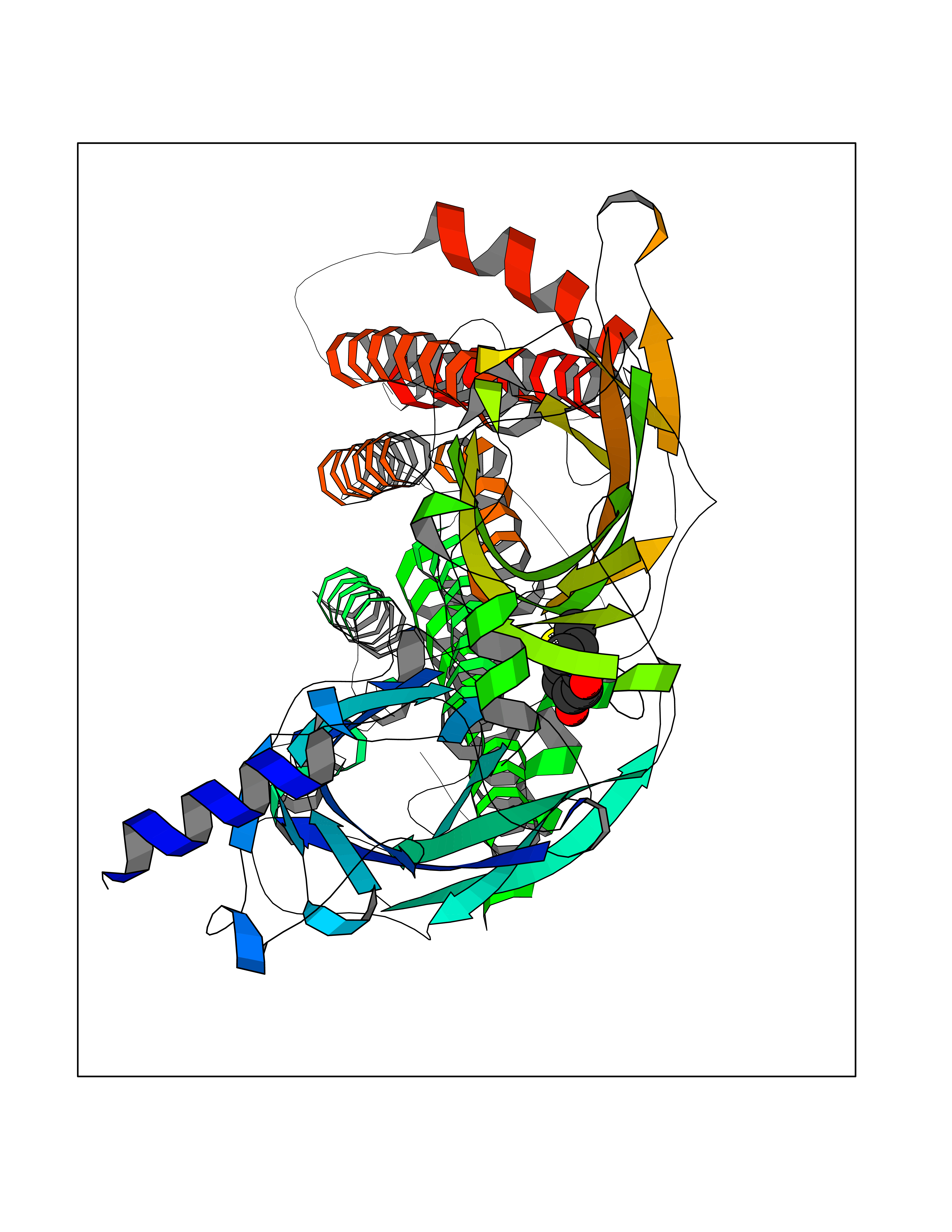 Molscript Map 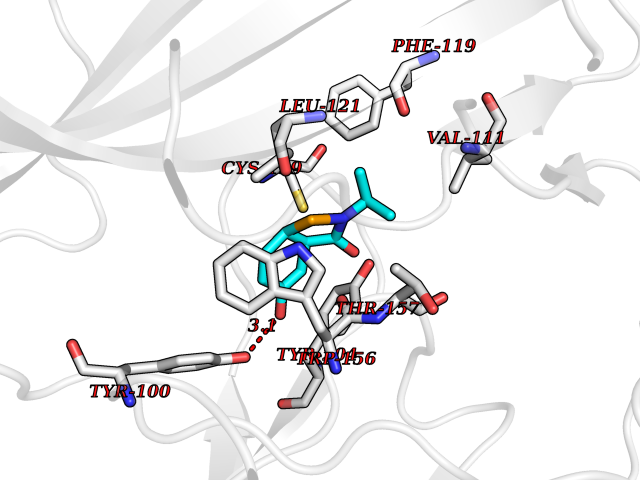 Pymol Map 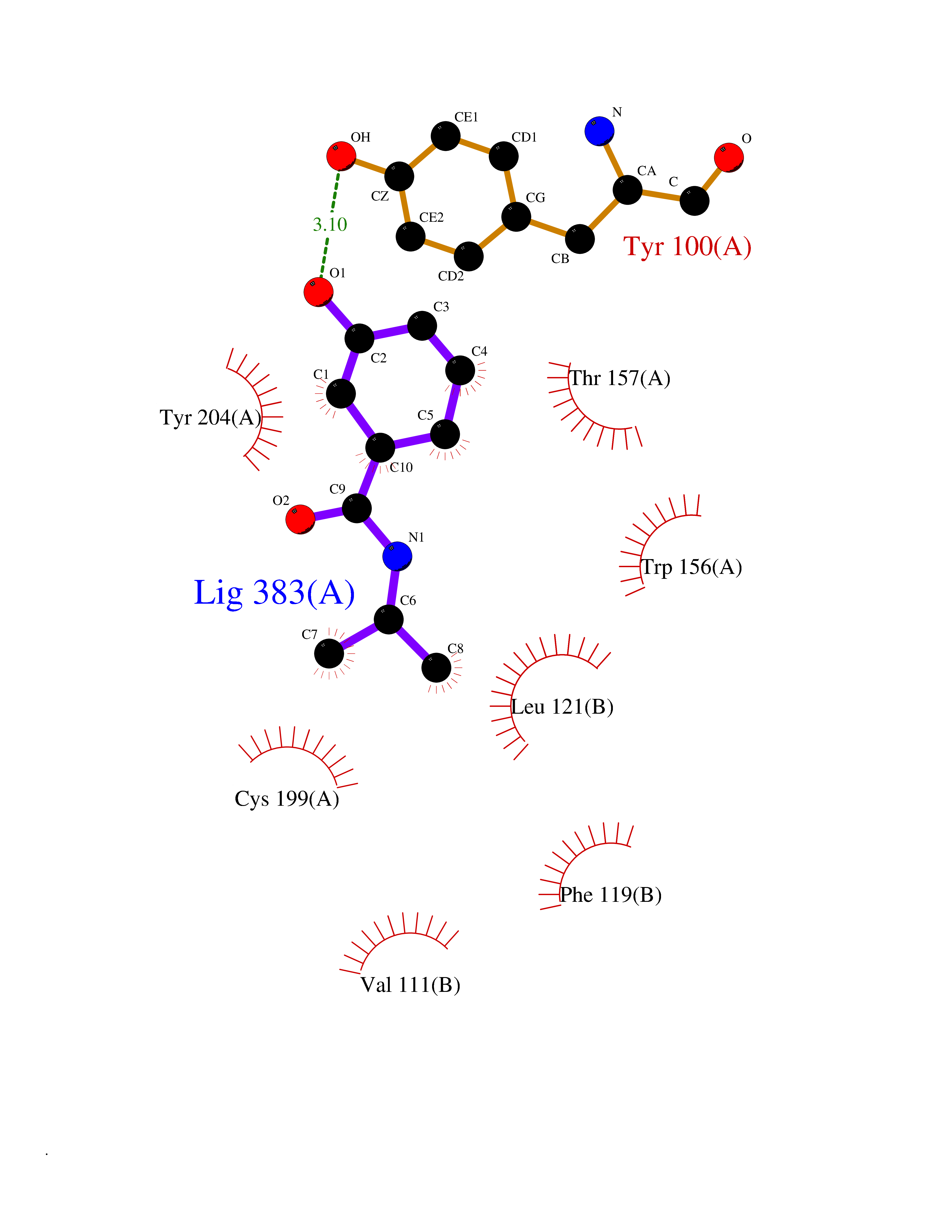 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Aggrecanase (ADAMTS5) | 3HY7 | 5.69 | |
Target general information Gen name ADAMTS5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aggrecanase-2; ADMP-2; ADAMTS5; ADAMTS-5; ADAM-TS5; ADAM-TS 5; ADAM-TS 11; A disintegrin and metalloproteinase with thrombospondinmotifs 5 Protein family NA Biochemical class Peptidase Function Cleaves aggrecan, a cartilage proteoglycan, and may be involved in its turnover. May play an important role in the destruction of aggrecan in arthritic diseases. May play a role in proteolytic processing mostly during the peri-implantation period. Related diseases Mucopolysaccharidosis 1H (MPS1H) [MIM:607014]: A severe form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1H usually present, within the first year of life, a combination of hepatosplenomegaly, skeletal deformities, corneal clouding and severe intellectual disability. Obstructive airways disease, respiratory infection and cardiac complications usually result in death before 10 years of age. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:1301941, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:24036510, ECO:0000269|PubMed:31194252, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:7951228, ECO:0000269|PubMed:8019563, ECO:0000269|PubMed:8328452, ECO:0000269|PubMed:8401515, ECO:0000269|Ref.20}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1H/S (MPS1H/S) [MIM:607015]: A form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. MPS1H/S represents an intermediate phenotype of the MPS1 clinical spectrum. It is characterized by relatively little neurological involvement, but most of the somatic symptoms described for severe MPS1 develop in the early to mid-teens, causing considerable loss of mobility. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8401515}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1S (MPS1S) [MIM:607016]: A mild form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1S may have little or no neurological involvement, normal stature and life span, but present development of joints stiffness, mild hepatosplenomegaly, aortic valve disease and corneal clouding. {ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:25256405, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8213840}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06837; DB03880; DB06945 Interacts with P13608 EC number EC 3.4.24.- Uniprot keywords 3D-structure; Cleavage on pair of basic residues; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 23997.8 Length 217 Aromaticity 0.06 Instability index 46.68 Isoelectric point 5.82 Charge (pH=7) -8.36 2D Binding mode Binding energy (Kcal/mol) -7.76  Molscript Map 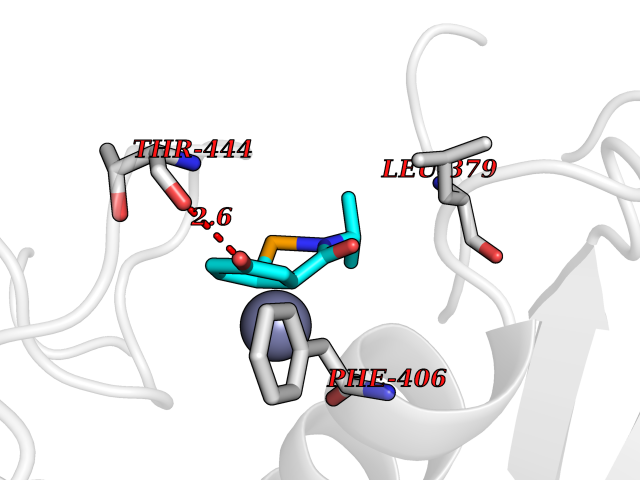 Pymol Map  Ligplot Map 3D Binding mode Sequence SRARQVELLLVADASMARKYGRGLQHYLLTLASIANRLYSHASIENHIRLAVVKVVVLGDKDKSLEVSKNAATTLKNFCKWQHQHNQLGDDHEEHYDAAILFTREDLCGHHSCDTLGMADVGTICSPERSCAVIEDDGLHAAFTVAHEIGHLLGLSHDDSKFCEETFGSTEDKRLMSSILTSIDASKPWSKCTSATITEFLDDGHGNCLLDLPRKQI Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | T-cell receptor beta constant 1 (TRBC1) | 4LCC | 5.69 | |
Target general information Gen name TRBC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TCRBC1; BV05S1J2.2 Protein family NA Biochemical class NA Function Alpha-beta T cell receptors are antigen specific receptors which are essential to the immune response and are present on the cell surface of T lymphocytes. Recognize peptide-major histocompatibility (MH) (pMH) complexes that are displayed by antigen presenting cells (APC), a prerequisite for efficient T cell adaptive immunity against pathogens. Binding of alpha-beta TR to pMH complex initiates TR-CD3 clustering on the cell surface and intracellular activation of LCK that phosphorylates the ITAM motifs of CD3G, CD3D, CD3E and CD247 enabling the recruitment of ZAP70. In turn, ZAP70 phosphorylates LAT, which recruits numerous signaling molecules to form the LAT signalosome. The LAT signalosome propagates signal branching to three major signaling pathways, the calcium, the mitogen-activated protein kinase (MAPK) kinase and the nuclear factor NF-kappa-B (NF-kB) pathways, leading to the mobilization of transcription factors that are critical for gene expression and essential for T cell growth and differentiation. The T cell repertoire is generated in the thymus, by V-(D)-J rearrangement. This repertoire is then shaped by intrathymic selection events to generate a peripheral T cell pool of self-MH restricted, non-autoaggressive T cells. Post-thymic interaction of alpha-beta TR with the pMH complexes shapes TR structural and functional avidity. Constant region of T cell receptor (TR) alpha chain. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02740 Interacts with NA EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Cell membrane; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Proteomics identification; Receptor; Reference proteome; T cell receptor; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID C,B,A Molecular weight (Da) 84668.2 Length 736 Aromaticity 0.13 Instability index 39.96 Isoelectric point 5.8 Charge (pH=7) -14.36 2D Binding mode Binding energy (Kcal/mol) -7.76  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence IQRPPKIQVYSRHPPNYLNCYVYGFHPPQIEIDLLKIKSEQSDLSFSKDWSFYLLSHATPNSKDQYSCRVKHVTLEQPRIVKWDRTHSLRYFRLGISEIPEFISAGYVDSHPITMYNSVSQLKEPRALWMEENLAPDHWERYTQLLRGWQQMFKVELKQLQHHYNHSGFHTYQRMIGCELLEDGSITGFLQYAYDGQDFLIFNKDTLSWMAMDNVADIIRRVWEANQHELLYQKNWLEEECIAWLKRFLEYGKDALQRTEPPKVRVNHKTTLYCRAYGFYPPEISINWMKNGEEIFQDTDYGGILPSGDGTYQTWVSVELGDIYSCHVEHGGVHMVLQGFQQNIDQPTEMTATEGAIVQINCTYQTSGFNGLFWYQQHAGEAPTFLSYNVLDGLEEKGRFSSFLSRSKGYSYLLLKELQMKDSASYLCAVKDSNYQLIWGAGTKLIIKPNIQNPDPAVYQLRDSKSSDKSVCLFTDFDKDSDVYITDKKSNSAVAWSNAGVTQTPKFQVLKTGQSMTLQCAQDMNHNSMYWYRQDPGMGLRLIYYSASEGTTDKGEVPNGYNVSRLNKREFSLRLESAAPSQTSVYFCASSVWTGEGSGELFFGEGSRLTVLEDLKNVFPPEVAVFEPSEAEISHTQKATLVCLATGFYPDHVELSWWVNGKEVHSGVCTDPQPLKEQPALNDSRYALSSRLRVSATFWQNPRNHFRCQVQFYGLSENDEWKPVTQIVSAEAWGRA Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Cytosolic 10-formyltetrahydrofolate dehydrogenase | 2CFI | 5.68 | |
Target general information Gen name ALDH1L1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FTHFD Protein family GART family; Aldehyde dehydrogenase family, ALDH1L subfamily Biochemical class Oxidoreductase Function Aldehyde dehydrogenase (NAD) activity.Catalytic activity.Formyltetrahydrofolate dehydrogenase activity.Hydroxymethyl-, formyl- and related transferase activity. Related diseases Developmental and epileptic encephalopathy 39 with leukodystrophy (DEE39) [MIM:612949]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE39 is characterized by global hypomyelination of the central nervous system, with the gray matter appearing relatively unaffected. Inheritance is autosomal recessive. {ECO:0000269|PubMed:19641205, ECO:0000269|PubMed:24515575}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00116 Interacts with Q3SY69; Q92624 EC number 1.5.1.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; NADP; One-carbon metabolism; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33869.5 Length 308 Aromaticity 0.08 Instability index 30.42 Isoelectric point 6.09 Charge (pH=7) -3.83 2D Binding mode Binding energy (Kcal/mol) -7.75 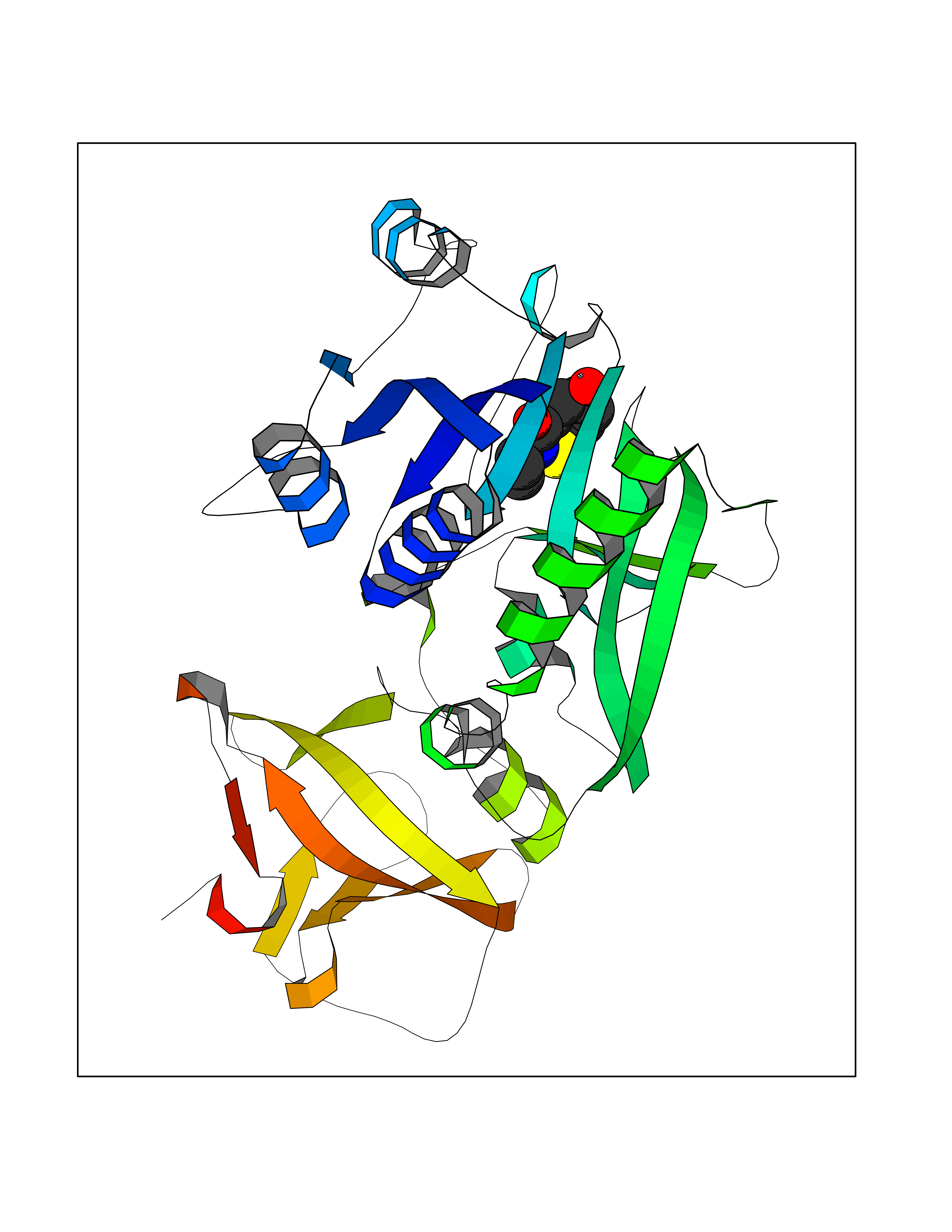 Molscript Map 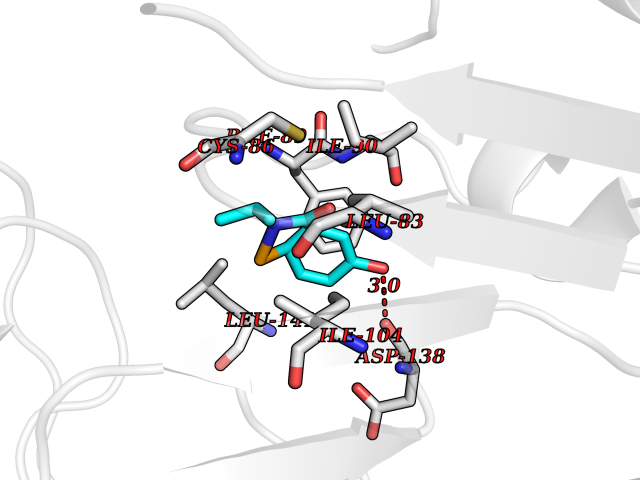 Pymol Map 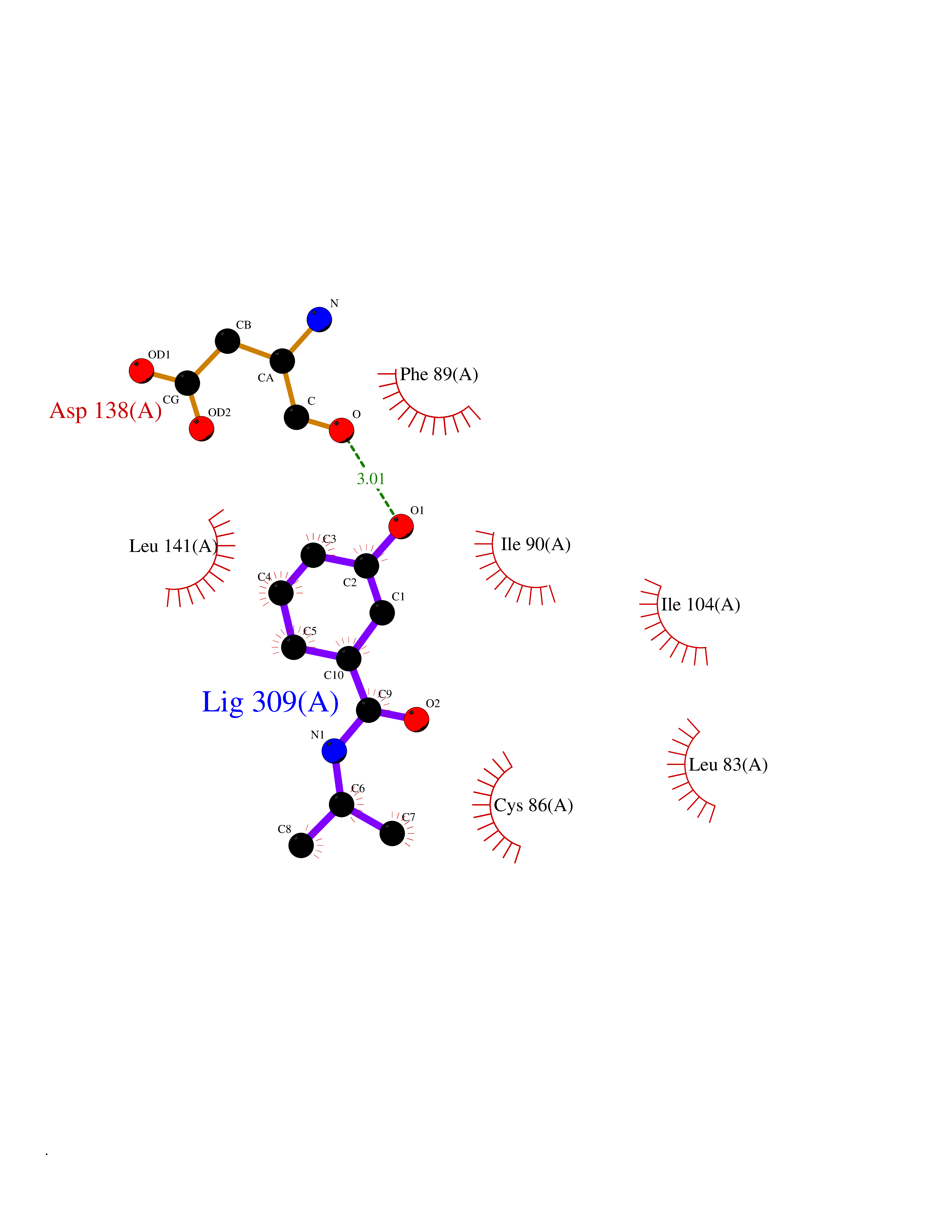 Ligplot Map 3D Binding mode Sequence SMKIAVIGQSLFGQEVYCHLRKEGHEVVGVFTVPDKDGKADPLGLEAEKDGVPVFKYSRWRAKGQALPDVVAKYQALGAELNVLPFCSQFIPMEIISAPRHGSIIYHPSLLPRHRGASAINWTLIHGDKKGGFSIFWADDGLDTGDLLLQKECEVLPDDTVSTLYNRFLFPEGIKGMVQAVRLIAEGKAPRLPQPEEGATYEGIQKKETAKINWDQPAEAIHNWIRGNDKVPGAWTEACEQKLTFFNSTLNTSGLVPEGDALPIPGAHRPGVVTKAGLILFGNDDKMLLVKNIQLEDGKMILASNFFK Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Cytochrome b (Complex III subunit 3) (Complex III subunit III) (Cytochrome b-c1 complex subunit 3) (Ubiquinol-cytochrome-c reductase complex cytochrome b subunit) | 1SQB | 5.68 | |
Target general information Gen name MT-CYB Organism Bos taurus (Bovine) Uniprot ID TTD ID NA Synonyms CYTB;MTCYB;COB Protein family Cytochrome b family Biochemical class NA Function Component of the ubiquinol-cytochrome c reductase complex (complex III or cytochrome b-c1 complex) that is part of the mitochondrial respiratory chain. The b-c1 complex mediates electron transfer from ubiquinol to cytochrome c. Contributes to the generation of a proton gradient across the mitochondrial membrane that is then used for ATP synthesis. {ECO:0000269|PubMed:1327781, ECO:0000269|PubMed:20025846, ECO:0000269|PubMed:9485330, ECO:0000305|PubMed:189810}." Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Electron transport; Heme; Iron; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Reference proteome; Respiratory chain; Transmembrane; Transmembrane helix; Transport; Ubiquinone Protein physicochemical properties Chain ID C,D,F,G,H Molecular weight (Da) 99281.2 Length 866 Aromaticity 0.12 Instability index 43.81 Isoelectric point 8.32 Charge (pH=7) 7.12 2D Binding mode Binding energy (Kcal/mol) -7.75 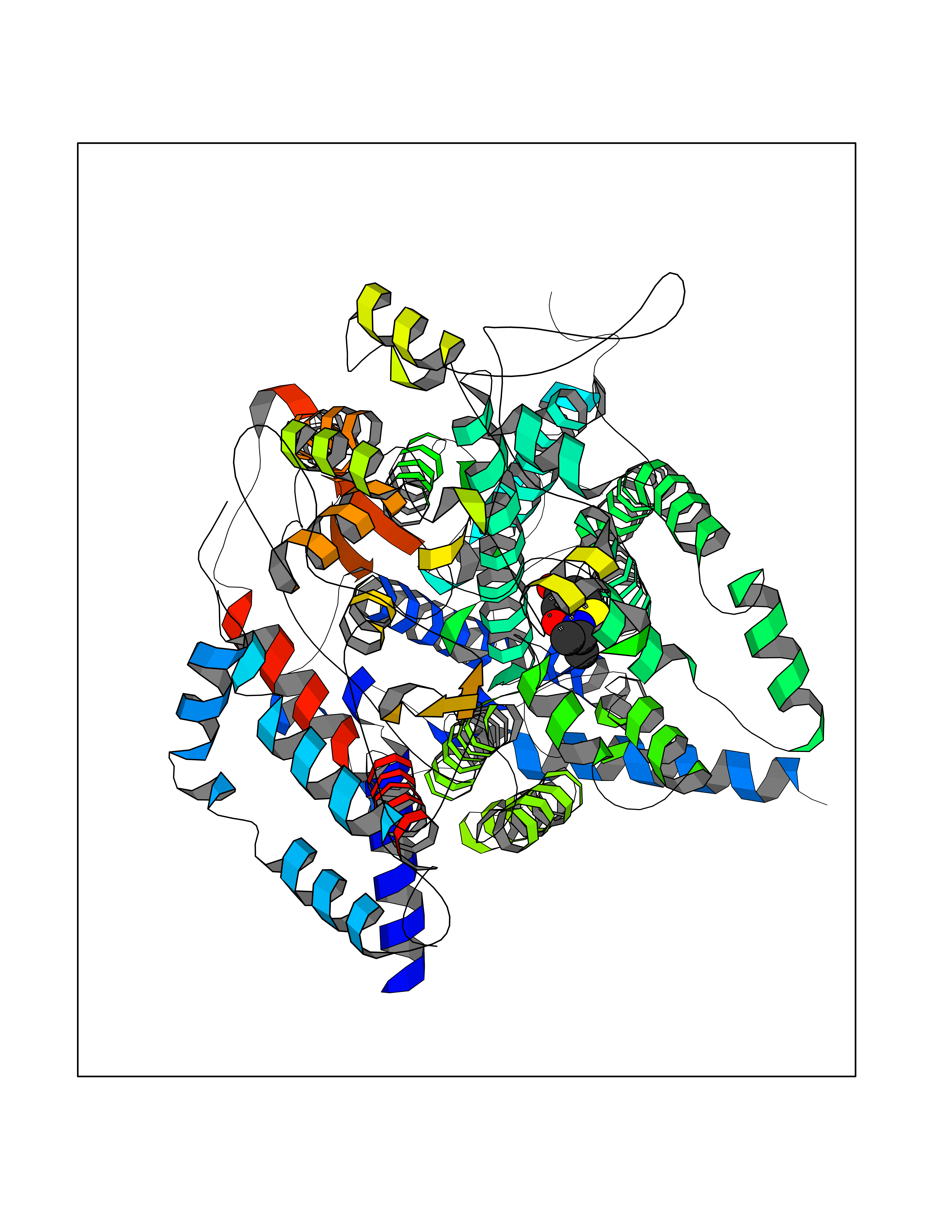 Molscript Map 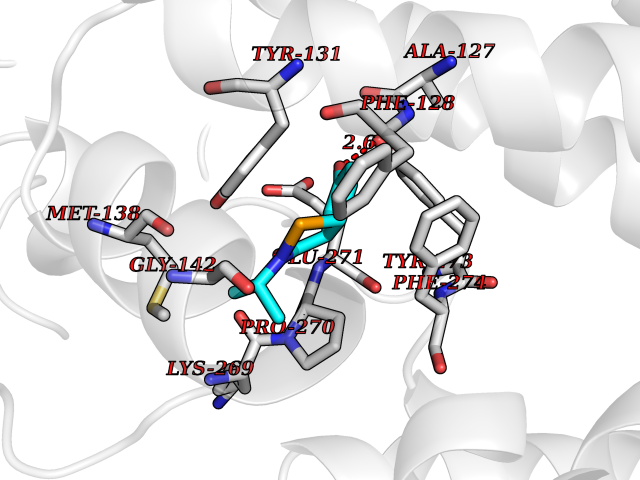 Pymol Map 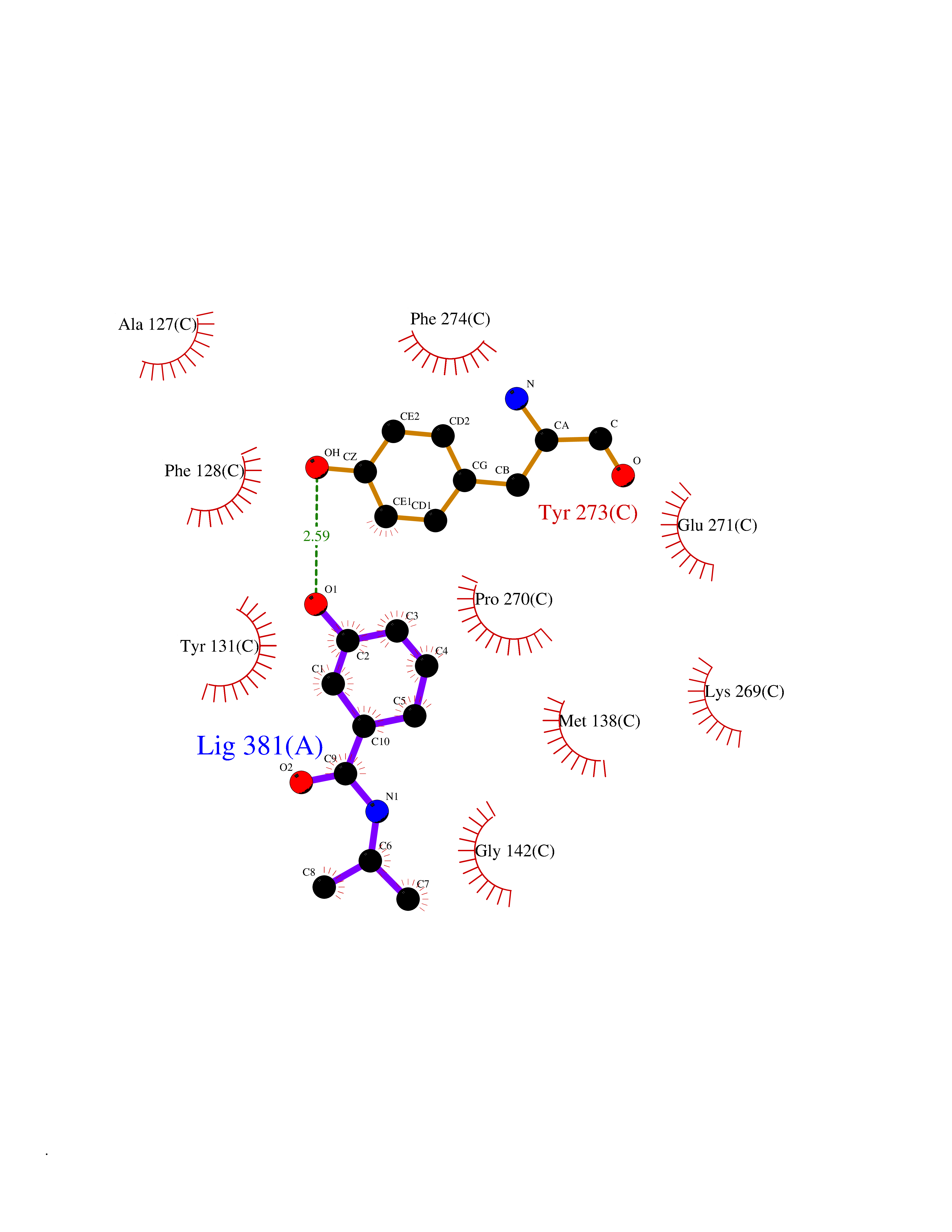 Ligplot Map 3D Binding mode Sequence VSASSRWLEGIRKWYYNAAGFNKLGLMRDDTIHENDDVKEAIRRLPENLYDDRVFRIKRALDLSMRQQILPKEQWTKYEEDKSYLEPYLKEVIRERKEREEWAKKELVDPLTTVREQCEQLEKCVKARERLELCDERVSSRSQTEEDCTEELLDFLHARDHCVAHKLFNSLKTNIRKSHPLMKIVNNAFIDLPAPSNISSWWNFGSLLGICLILQILTGLFLAMHYTSDTTTAFSSVTHICRDVNYGWIIRYMHANGASMFFICLYMHVGRGLYYGSYTFLETWNIGVILLLTVMATAFMGYVLPWGQMSFWGATVITNLLSAIPYIGTNLVEWIWGGFSVDKATLTRFFAFHFILPFIIMAIAMVHLLFLHETGSNNPTGISSDVDKIPFHPYYTIKDILGALLLILALMLLVLFAPDLLGDPDNYTPANPLNTPPHIKPEWYFLFAYAILRSIPNKLGGVLALAFSILILALIPLLHTSKQRSMMFRPLSQCLFWALVADLLTLTWIGGQPVEHPYITIGQLASVLYFLLILVLMPTAGTIENKLLKWSDLELHPPSYPWSHRGLLSSLDHTSIRRGFQVYKQVCSSCHSMDYVAYRHLVGVCYTEDEAKALAEEVEVQDGPNEDGEMFMRPGKLSDYFPKPYPNPEAARAANNGALPPDLSYIVRARHGGEDYVFSLLTGYCEPPTGVSLREGLYFNPYFPGQAIGMAPPIYNEVLEFDDGTPATMSQVAKDVCTFLRWAAEPEHDHRKRMGLKMLLMMGLLLPLVYAMKRHKWSVLKSRKLAYRPPKGRQFGHLTRVRHVITYSLSPFEQRAFPHYFSKGIPNVLRRTRACILRVAPPFVAFYLVYTWGTQEFEKSKRKNPA Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | L-serine dehydratase/L-threonine deaminase | 1P5J | 5.68 | |
Target general information Gen name SDS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms SDH Protein family Serine/threonine dehydratase family Biochemical class Lyase Function L-serine ammonia-lyase activity.L-threonine ammonia-lyase activity.Protein homodimerization activity.Pyridoxal phosphate binding. Related diseases Immunodeficiency, common variable, 12, with autoimmunity (CVID12) [MIM:616576]: A primary immunodeficiency characterized by hypogammaglobulinemia and recurrent bacterial infections. About half of patients develop autoimmune features, including cytopenia, as well as generalized inflammation and lymphoproliferation manifest as lymphadenopathy or hepatosplenomegaly. {ECO:0000269|PubMed:26279205}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00114; DB00133 Interacts with Q8WTU0; O14964; Q96PF1 EC number 4.3.1.17; 4.3.1.19 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Gluconeogenesis; Lipid metabolism; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33559.7 Length 319 Aromaticity 0.06 Instability index 34.65 Isoelectric point 7.2 Charge (pH=7) 0.42 2D Binding mode Binding energy (Kcal/mol) -7.75 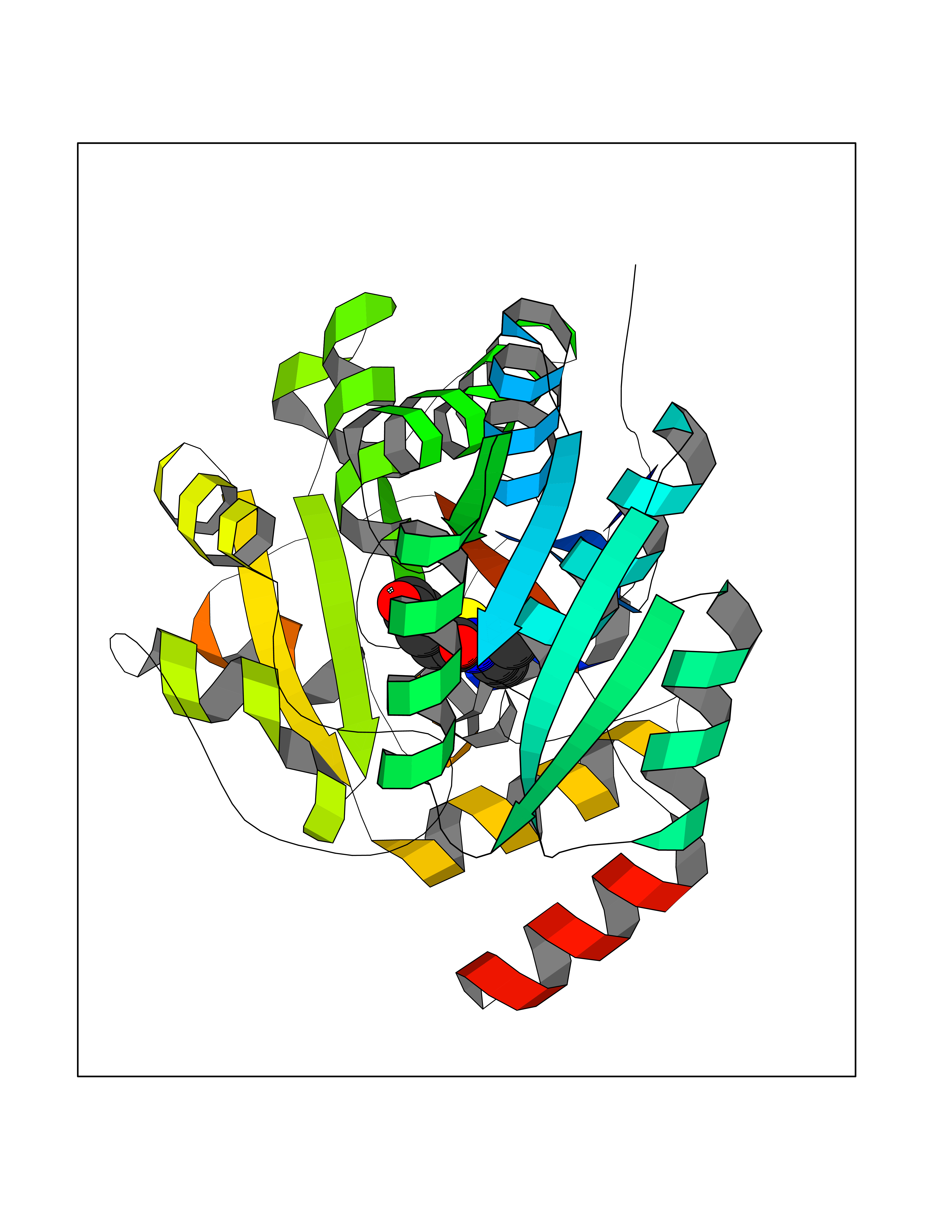 Molscript Map 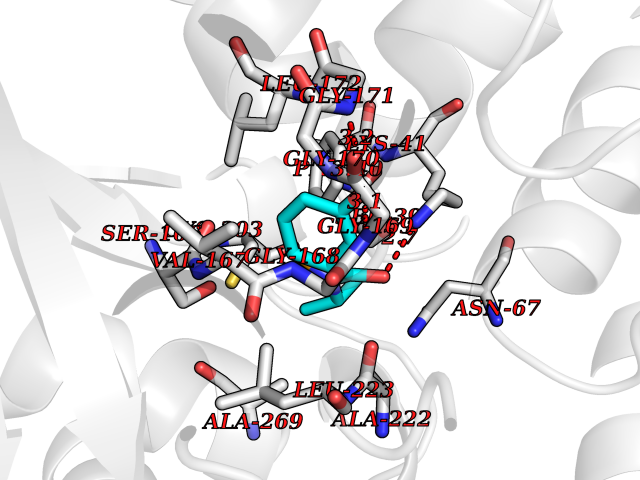 Pymol Map 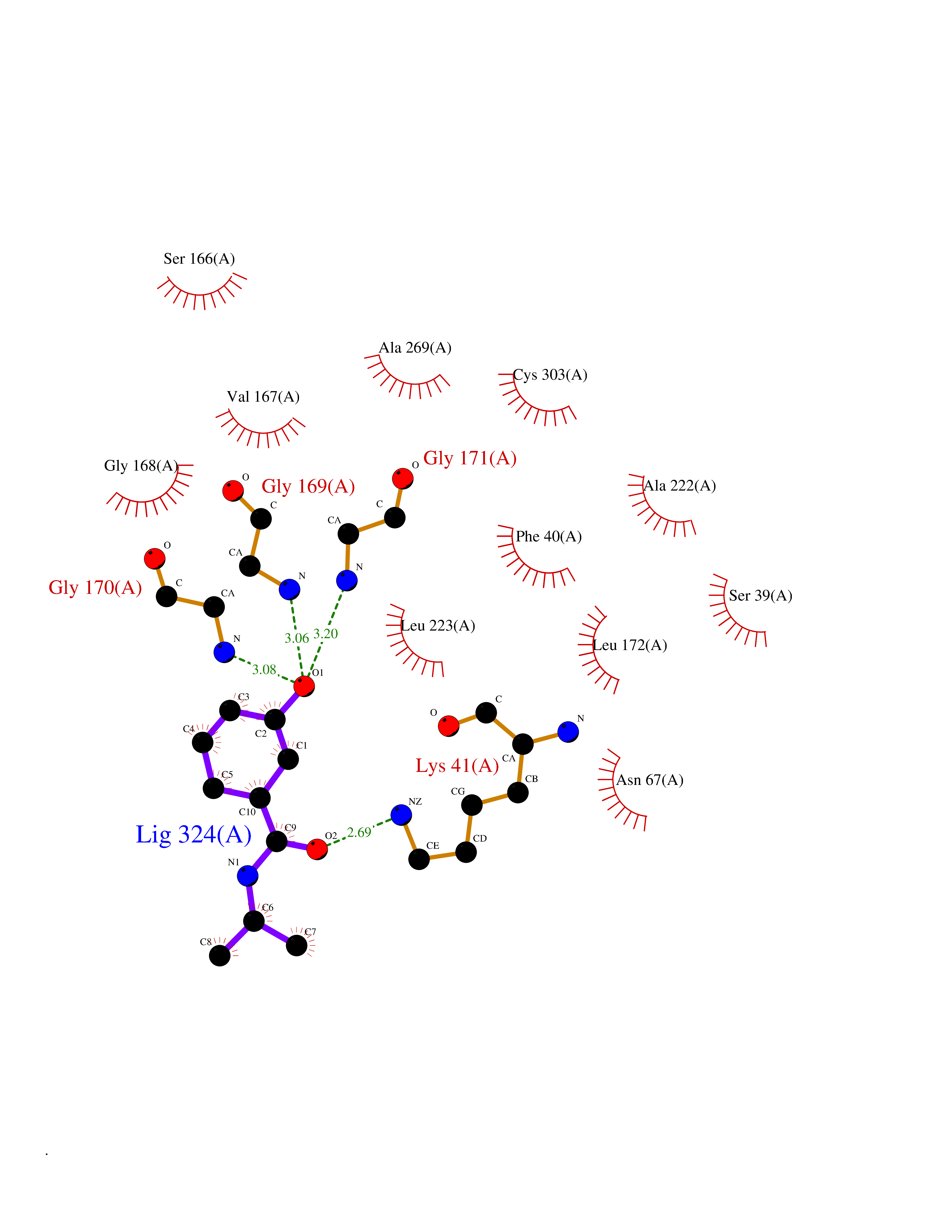 Ligplot Map 3D Binding mode Sequence GEPLHVKTPIRDSMALSKMAGTSVYLKMDSAQPSGSFKIRGIGHFCKRWAKQGCAHFVCSSAGNAGMAAAYAARQLGVPATIVVPGTTPALTIERLKNEGATCKVVGELLDEAFELAKALAKNNPGWVYIPPFDDPLIWEGHASIVKELKETLWEKPGAIALSVGGGGLLCGVVQGLQECGWGDVPVIAMETFGAHSFHAATTAGKLVSLPKITSVAKALGVKTVGSQALKLFQEHPIFSEVISDQEAVAAIEKFVDDEKILVEPACGAALAAVYSHVIQKLQLEGNLRTPLPSLVVIVCGGSNISLAQLRALKEQLGM Hydrogen bonds contact Hydrophobic contact | ||||