Job Results:
Ligand
Structure
Job ID
195891f0cd6db7cda8666dbe4295d47d
Job name
NA
Time
2025-09-26 08:49:45
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | 3-oxo-5-beta-steroid 4-dehydrogenase | 3BUV | 7.77 | |
Target general information Gen name AKR1D1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms SRD5B1 Protein family Aldo/keto reductase family Biochemical class Oxidoreductase Function Aldo-keto reductase (NADP) activity.Delta4-3-oxosteroid 5beta-reductase activity.Steroid binding. Related diseases Congenital bile acid synthesis defect 2 (CBAS2) [MIM:235555]: A condition characterized by jaundice, intrahepatic cholestasis and hepatic failure. Patients with this liver disease show absence or low levels of chenodeoxycholic acid and cholic acid in plasma and urine. {ECO:0000269|PubMed:12970144, ECO:0000269|PubMed:15030995, ECO:0000269|PubMed:19175828, ECO:0000269|PubMed:20522910}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07557; DB07447; DB00548; DB01216; DB00741; DB06077; DB00717 Interacts with Q04828 EC number 1.3.1.3 Uniprot keywords 3D-structure; Alternative splicing; Bile acid catabolism; Cytoplasm; Disease variant; Intrahepatic cholestasis; Lipid degradation; Lipid metabolism; NADP; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37245.3 Length 325 Aromaticity 0.1 Instability index 36.43 Isoelectric point 7.26 Charge (pH=7) 0.62 2D Binding mode Binding energy (Kcal/mol) -10.6  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DLSAASHRIPLSDGNSIPIIGLGTYSEPKSTPKGACATSVKVAIDTGYRHIDGAYIYQNEHEVGEAIREKIAEGKVRREDIFYCGKLWATNHVPEMVRPTLERTLRVLQLDYVDLYIIEVPMAFKPGDEIYPRDENGKWLYHKSNLCATWEAMEACKDAGLVKSLGVSNFNRRQLELILNKPGLKHKPVSNQVECHPYFTQPKLLKFCQQHDIVITAYSPLGTSRNPIWVNVSSPPLLKDALLNSLGKRYNKTAAQIVLRFNIQRGVVVIPKSFNLERIKENFQIFDFSLTEEEMKDIEALNKNVRFVELLMWRDHPEYPFHDEY Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | LIM domain kinase-2 (LIMK-2) | 7QHG | 7.77 | |
Target general information Gen name LIMK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LIMK-2; LIM domain kinase 2 Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family Biochemical class Kinase Function Displays serine/threonine-specific phosphorylation of myelin basic protein and histone (MBP) in vitro. Related diseases Intellectual developmental disorder, autosomal recessive 80, with variant lissencephaly (MRT80) [MIM:620653]: An autosomal recessive disorder characterized by global developmental delay, mildly to moderately impaired intellectual development, attention deficit-hyperactivity disorder, hypotonia, seizure, poor social skills, and autistic traits. Brain imaging shows fronto-temporal lissencephaly and pachygyria. {ECO:0000269|PubMed:37880421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11718; DB12010 Interacts with Q16543; P08238; Q96C90; P62258 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Cytoskeleton; Kinase; LIM domain; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Serine/threonine-protein kinase; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 32109.2 Length 283 Aromaticity 0.1 Instability index 27.28 Isoelectric point 6.01 Charge (pH=7) -3.93 2D Binding mode Binding energy (Kcal/mol) -10.6 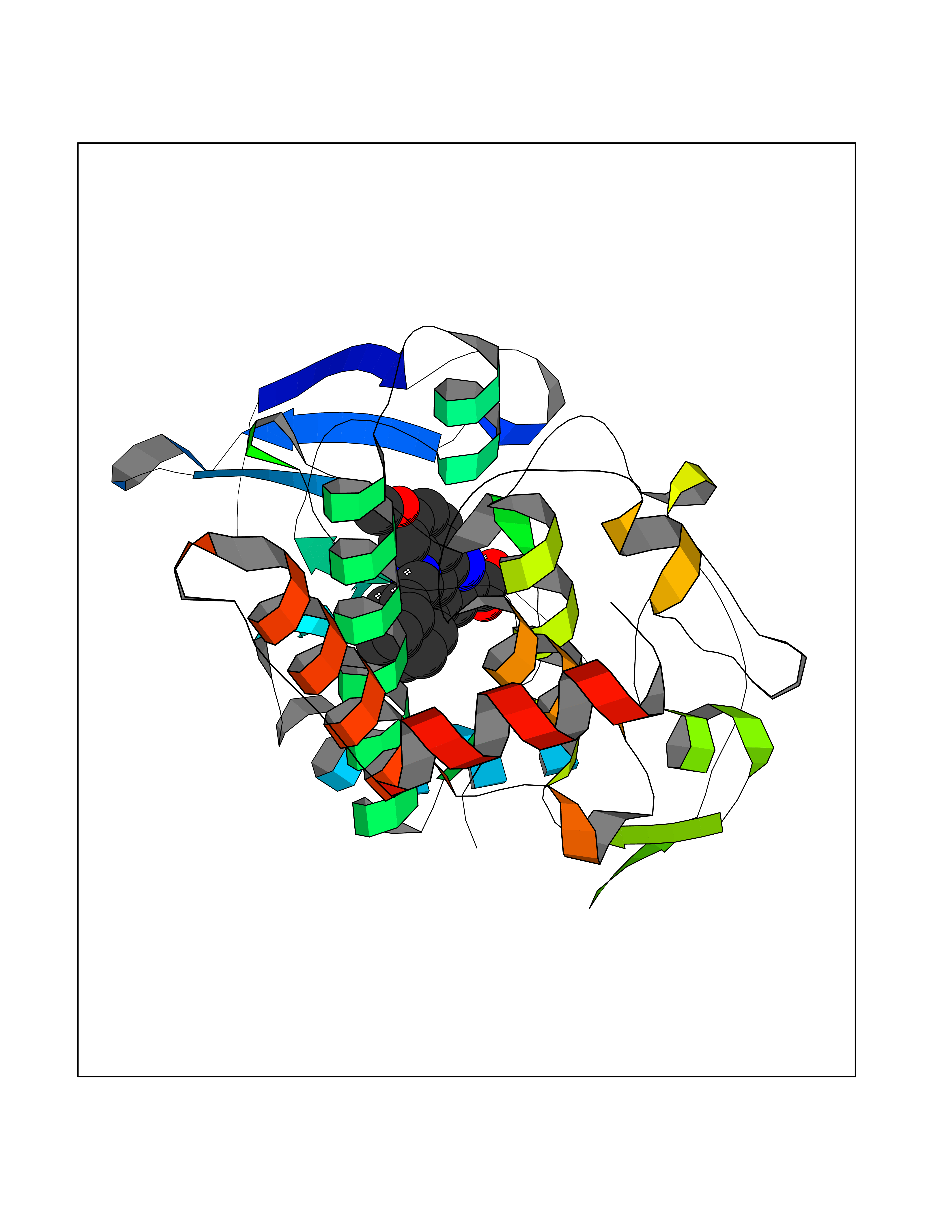 Molscript Map 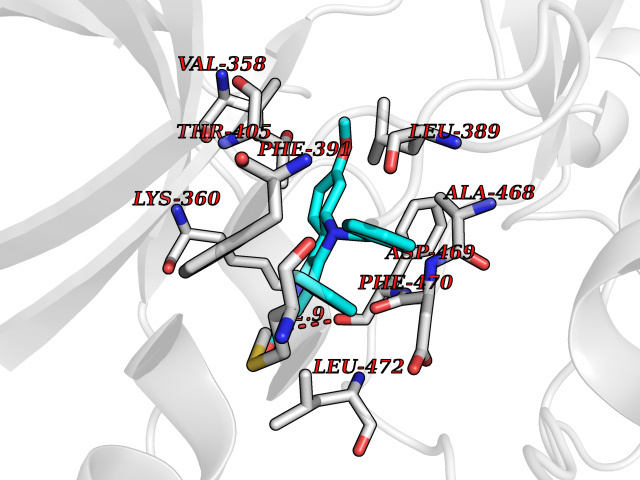 Pymol Map 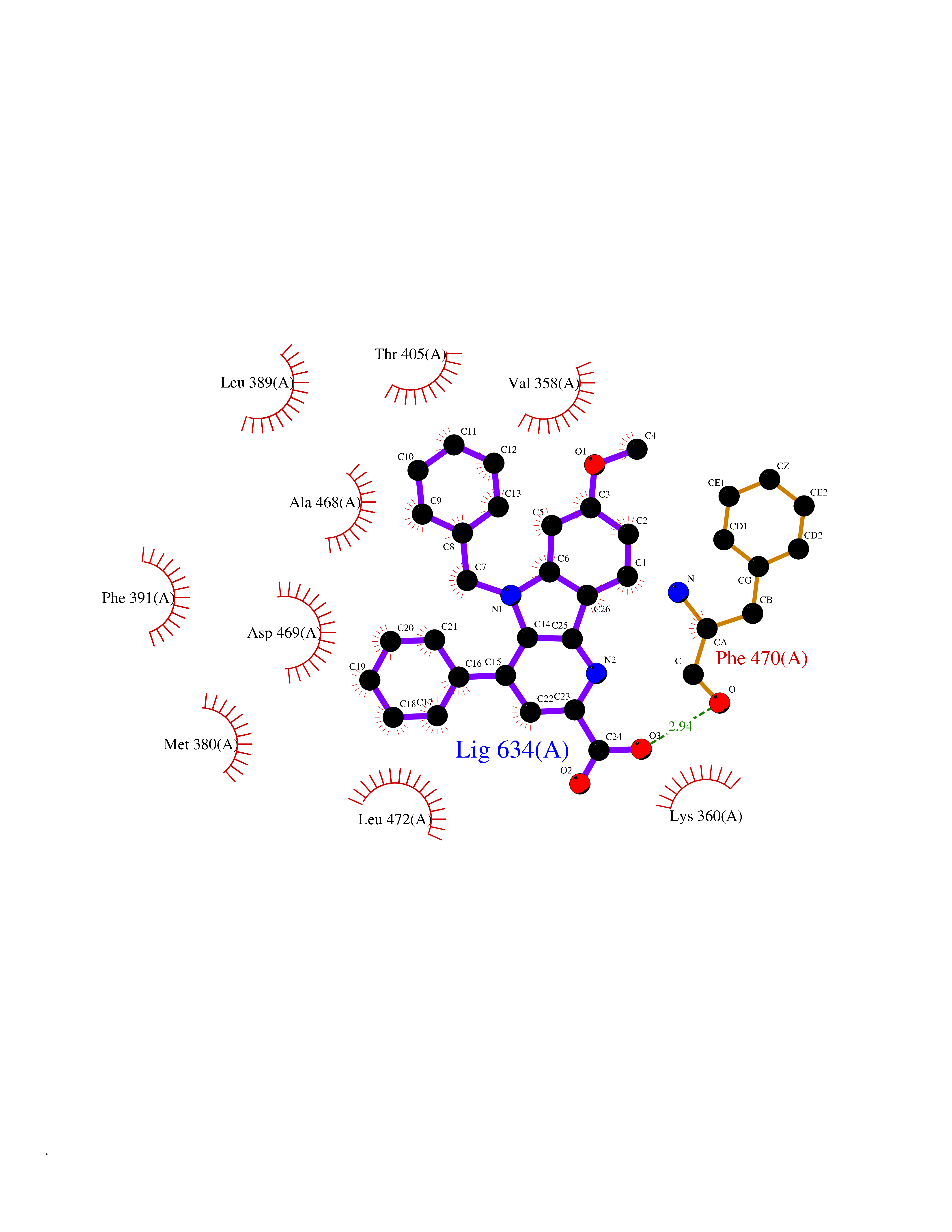 Ligplot Map 3D Binding mode Sequence MDLIHGEVLGKGFFGQAIKVTHKATGKVMVMKELIRCDEETQKTFLTEVKVMRSLDHPNVLKFIGVLYKDKKLNLLTEYIEGGTLKDFLRSMDPFPWQQKVRFAKGIASGMAYLHSMCIIHRDLNSHNCLIKLDKTVVVADFGLSRLIVDRKKRYTVVGNPYWMAPEMLNGKSYDETVDIFSFGIVLCEIIGQVYADPDCLPRTLDFGLNVKLFWEKFVPTDCPPAFFPLAAICCRLEPESRPAFSKLEDSFEALSLYLGELGIPLPAELEELDHTVSMQYGL Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Opioid receptor sigma 1 (OPRS1) | 6DJZ | 7.77 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23901 Length 212 Aromaticity 0.14 Instability index 33.12 Isoelectric point 5.61 Charge (pH=7) -5.6 2D Binding mode Binding energy (Kcal/mol) -10.59 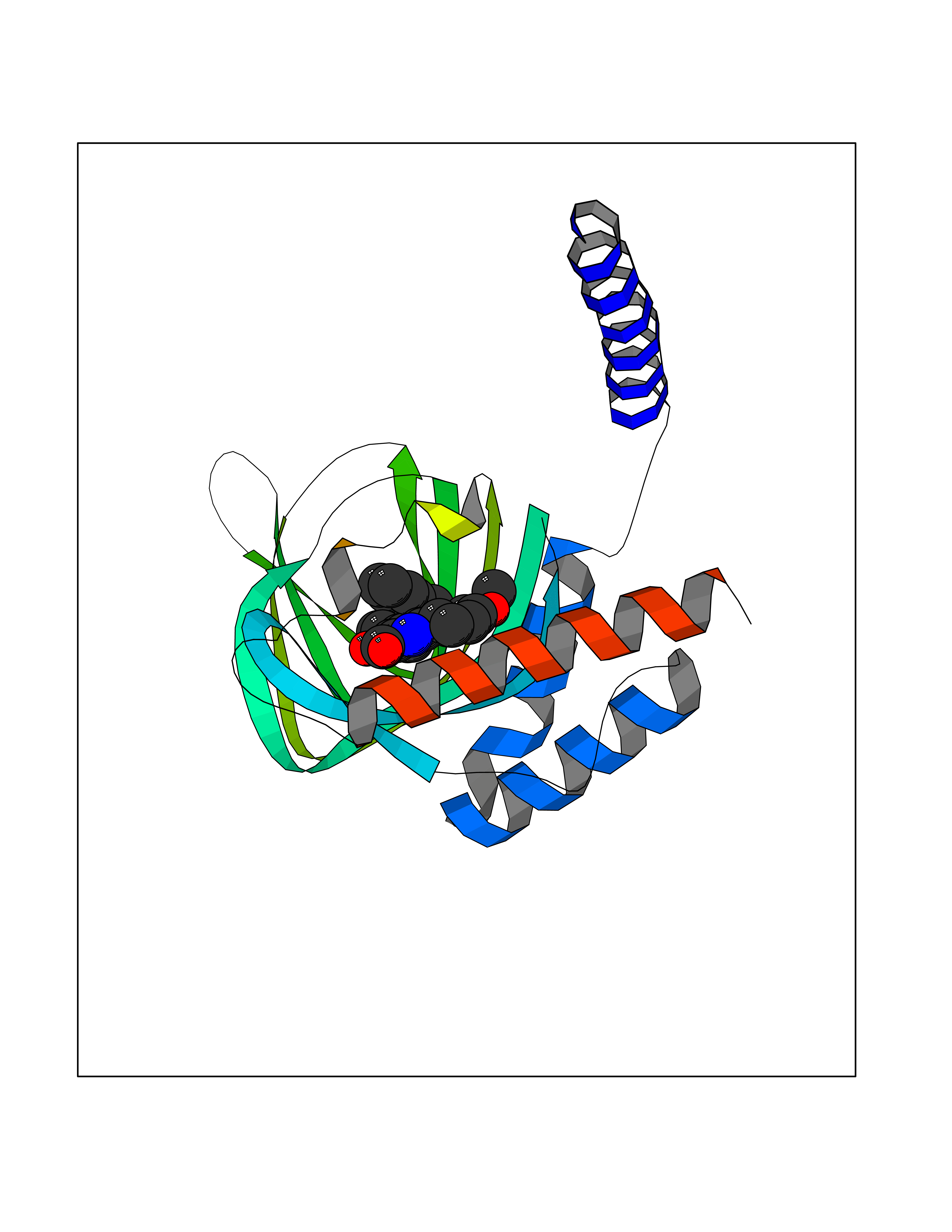 Molscript Map 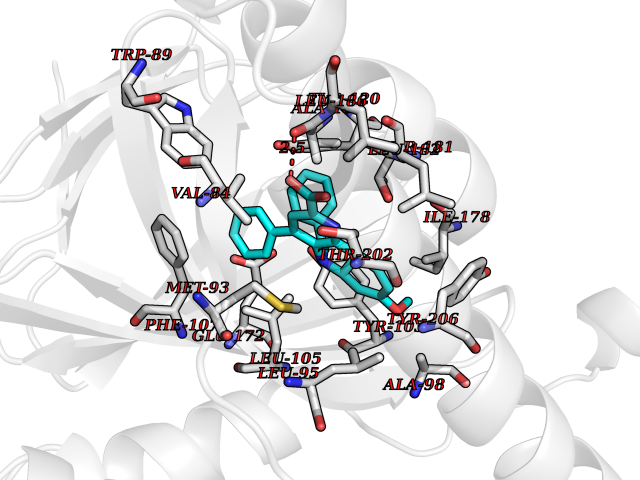 Pymol Map 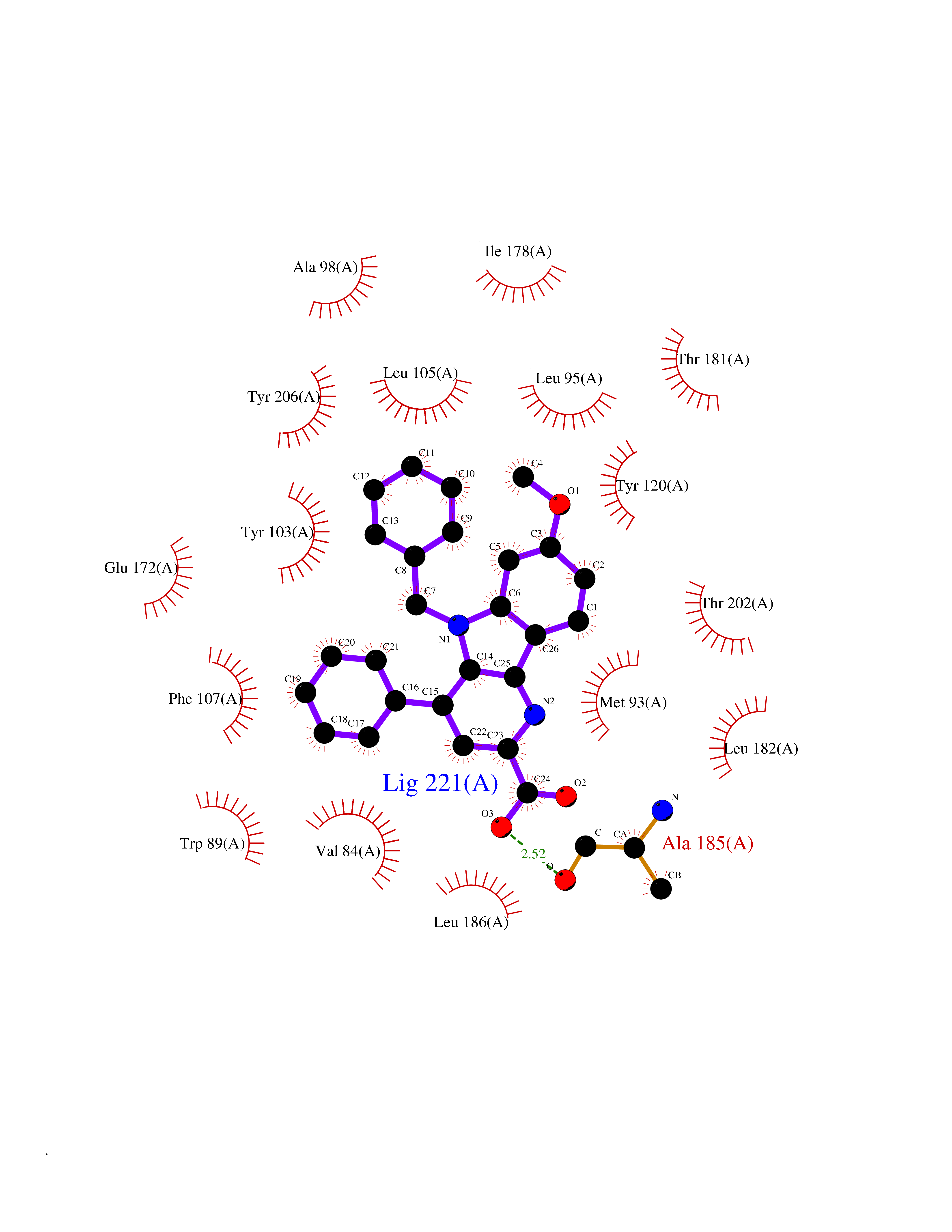 Ligplot Map 3D Binding mode Sequence RWAWAALLLAVAAVLTQVVWLWLGTQSFVFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLF Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | SEC14-like protein 2 | 4OMJ | 7.76 | |
Target general information Gen name SEC14L2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA1658;C22orf6;KIAA1186 Protein family NA Biochemical class Transport protein Function Phospholipid binding.Transporter activity.Vitamin E binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Lipid-binding; Nucleus; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 31533.3 Length 274 Aromaticity 0.1 Instability index 49.26 Isoelectric point 8.34 Charge (pH=7) 2.81 2D Binding mode Binding energy (Kcal/mol) -10.58  Molscript Map 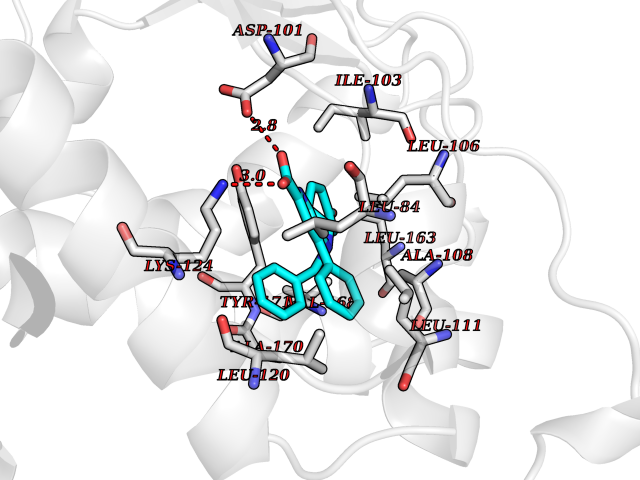 Pymol Map 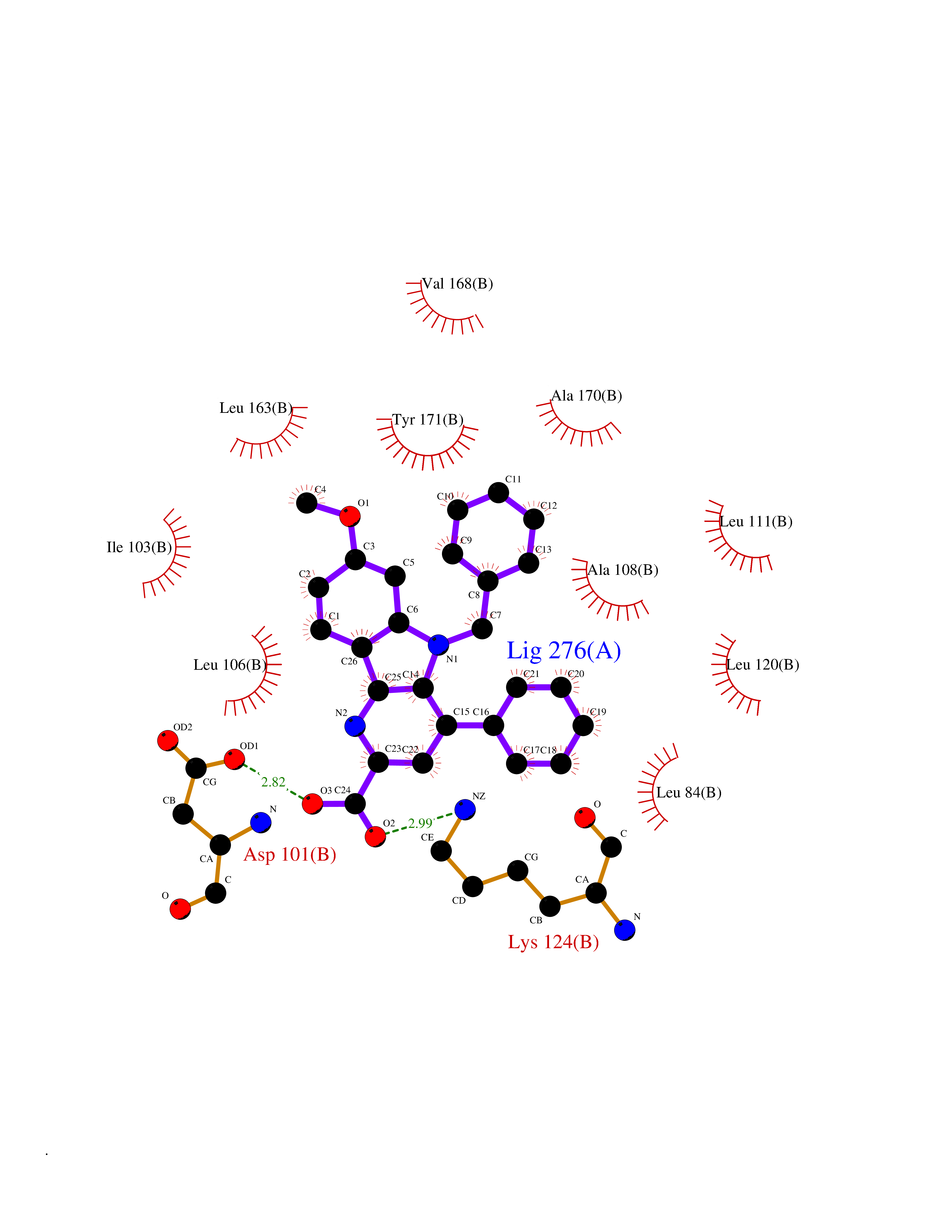 Ligplot Map 3D Binding mode Sequence MSGRVGDLSPRQKEALAKFRENVQDVLPALPNPDDYFLLRWLRARSFDLQKSEAMLRKHVEFRKQKDIDNIISWQPPEVIQQYLSGGMCGYDLDGCPVWYDIIGPLDAKGLLFSASKQDLLRTKMRECELLLQECAHQTTKLGRKVETITIIYDCEGLGLKHLWKPAVEAYGEFLCMFEENYPETLKRLFVVKAPKLFPVAYNLIKPFLSEDTRKKIMVLGANWKEVLLKHISPDQVPVEYGGTMTDPDGNPKCKSKINYGGDIPRKYYVRDQV Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Retinaldehyde-binding protein 1 | 3HX3 | 7.75 | |
Target general information Gen name RLBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CRALBP Protein family NA Biochemical class Transport protein Function 11-cis retinal binding.Retinol binding.Transporter activity. Related diseases Bothnia retinal dystrophy (BRD) [MIM:607475]: A type of retinitis punctata albescens. Affected individuals show night blindness from early childhood with features consistent with retinitis punctata albescens and macular degeneration. {ECO:0000269|PubMed:10102298}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rod-cone dystrophy Newfoundland (NFRCD) [MIM:607476]: A rod-cone dystrophy reminiscent of retinitis punctata albescens but with a substantially lower age at onset and more-rapid and distinctive progression. Rod-cone dystrophies results from initial loss of rod photoreceptors, later followed by cone photoreceptors loss. {ECO:0000269|PubMed:11868161}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Retinitis punctata albescens (RPA) [MIM:136880]: A form of fleck retina disease characterized by aggregation of white flecks posteriorly in the retina, causing night blindness and delayed dark adaptation. It differs from fundus albipunctatus in being progressive and evolving to generalized atrophy of the retina. {ECO:0000269|PubMed:10102299, ECO:0000269|PubMed:11453974, ECO:0000269|PubMed:9326942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00162 Interacts with Q9P2G9-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Disease variant; Proteomics identification; Reference proteome; Retinol-binding; Sensory transduction; Transport; Vision Protein physicochemical properties Chain ID A Molecular weight (Da) 28328.6 Length 250 Aromaticity 0.14 Instability index 52.64 Isoelectric point 4.96 Charge (pH=7) -9.87 2D Binding mode Binding energy (Kcal/mol) -10.57 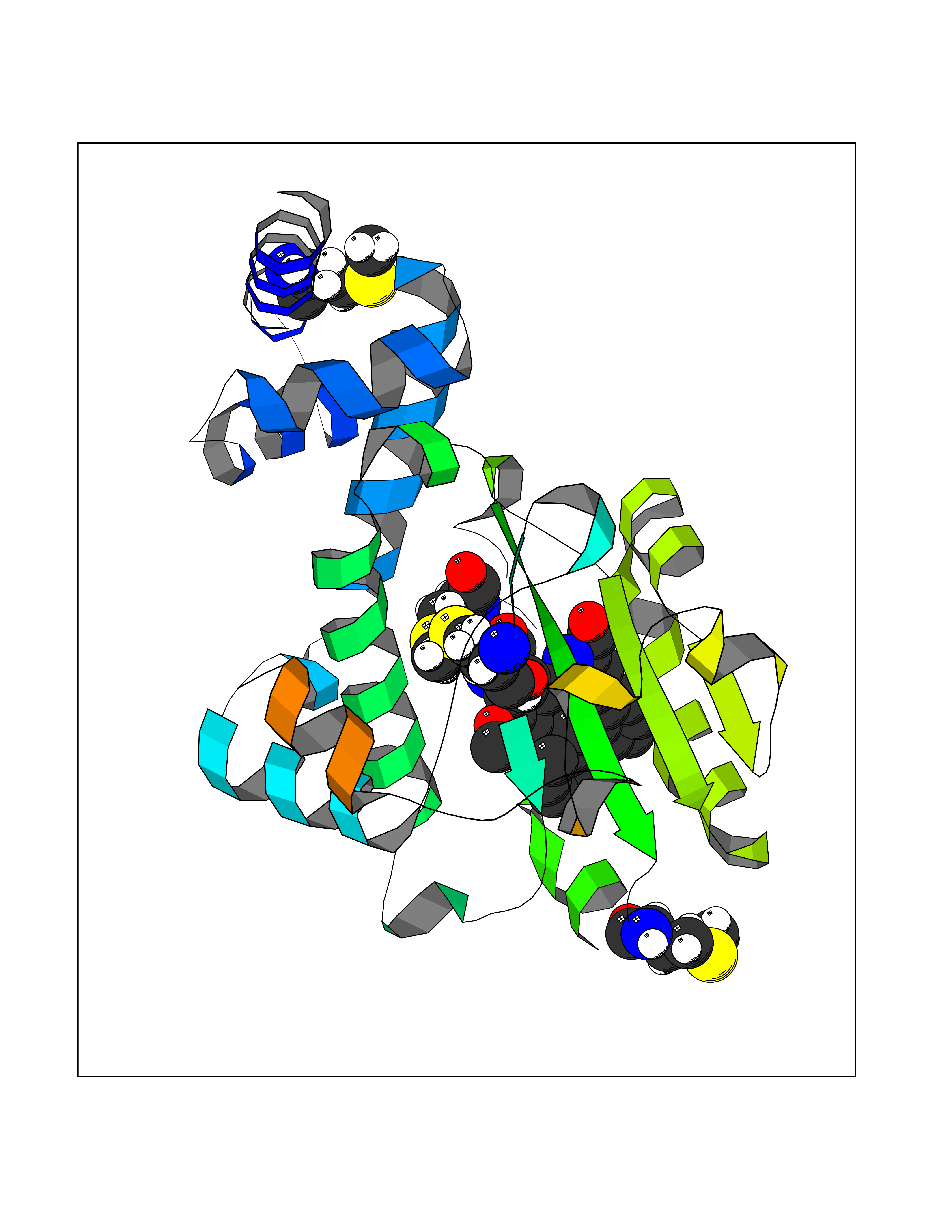 Molscript Map  Pymol Map 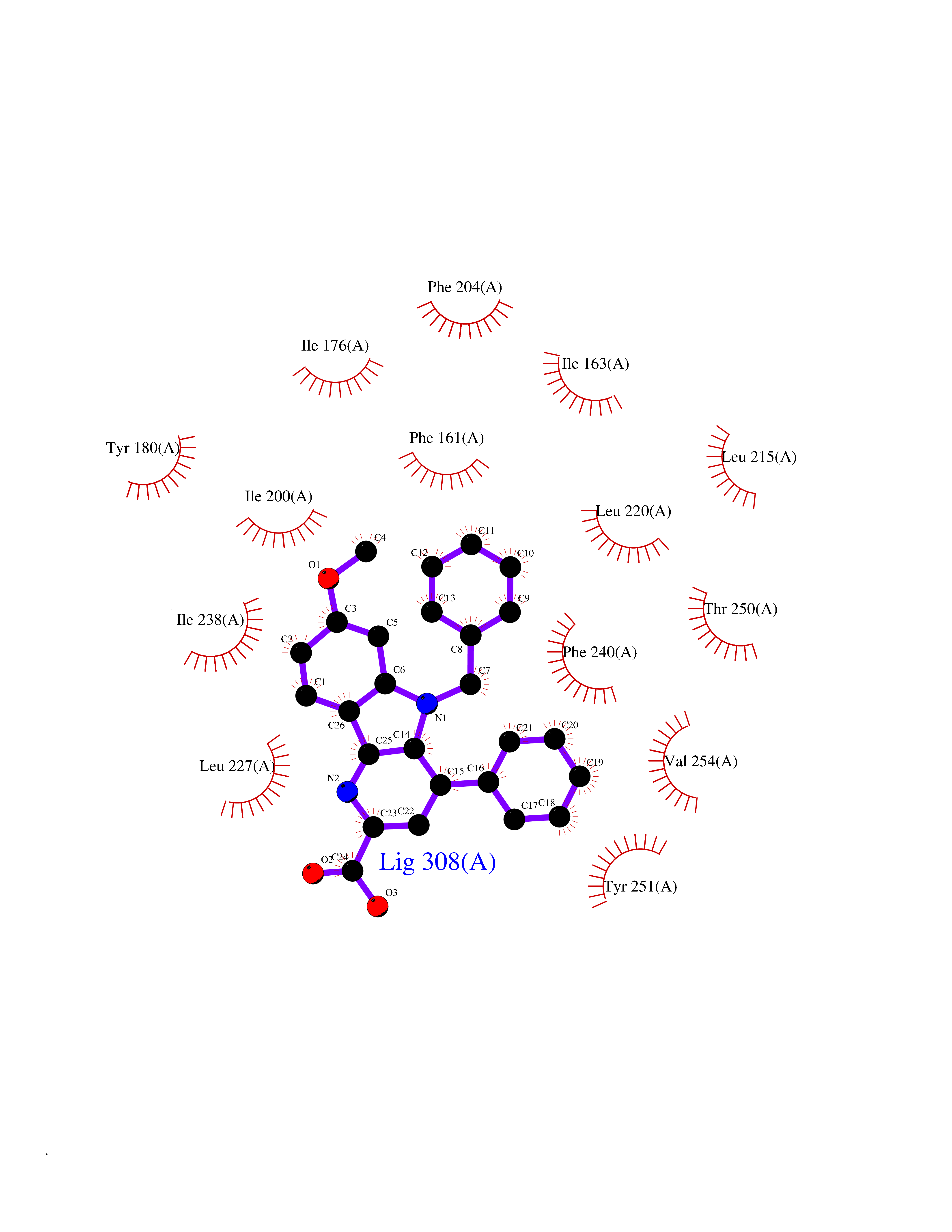 Ligplot Map 3D Binding mode Sequence ETREEAVRELQEXVQAQAASGEELAVAVAERVQEKDSGFFLRFIRARKFNVGRAYELLRGYVNFRLQYPELFDSLSPEAVRCTIEAGYPGVLSSRDKYGRVVXLFNIENWQSQEITFDEILQAYCFILEKLLENEETQINGFCIIENFKGFTXQQAASLRTSDLRKXVDXLQDSFPAWFKAIHFIHQPWYFTTTYNVVKPFLKSKLLERVFVHGDDLSGFYQEIDENILPSDFGGTLPKYDGKAVAEQLF Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Thiopurine S-methyltransferase | 2BZG | 7.75 | |
Target general information Gen name TPMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, TPMT family Biochemical class Transferase Function Thiopurine S-methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00993; DB00436; DB01327; DB01033; DB01250; DB01021 Interacts with Q8TAP4-4; Q15047-2; P61981 EC number 2.1.1.67 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 25971.5 Length 229 Aromaticity 0.12 Instability index 32.58 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -10.57 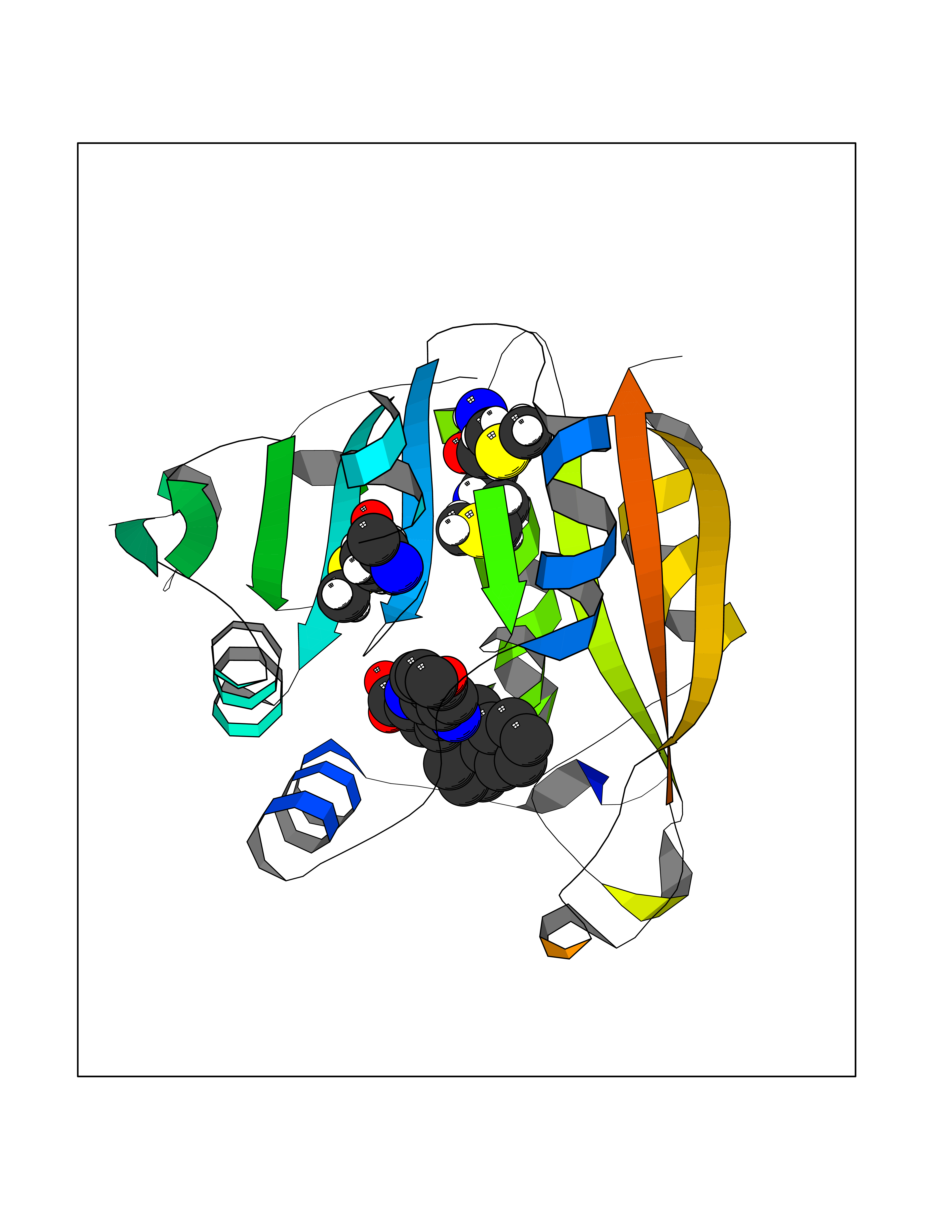 Molscript Map 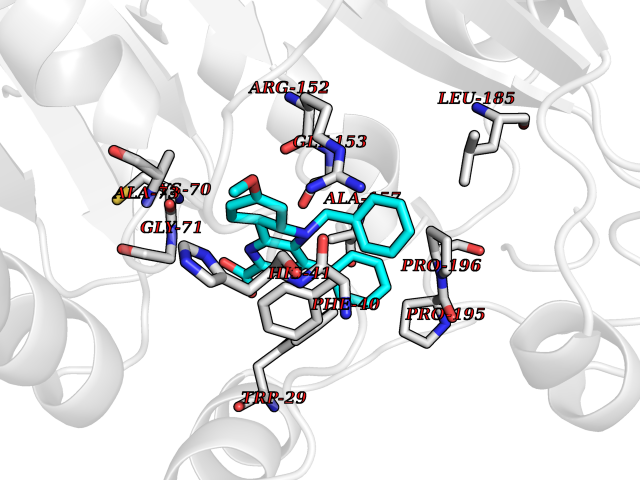 Pymol Map 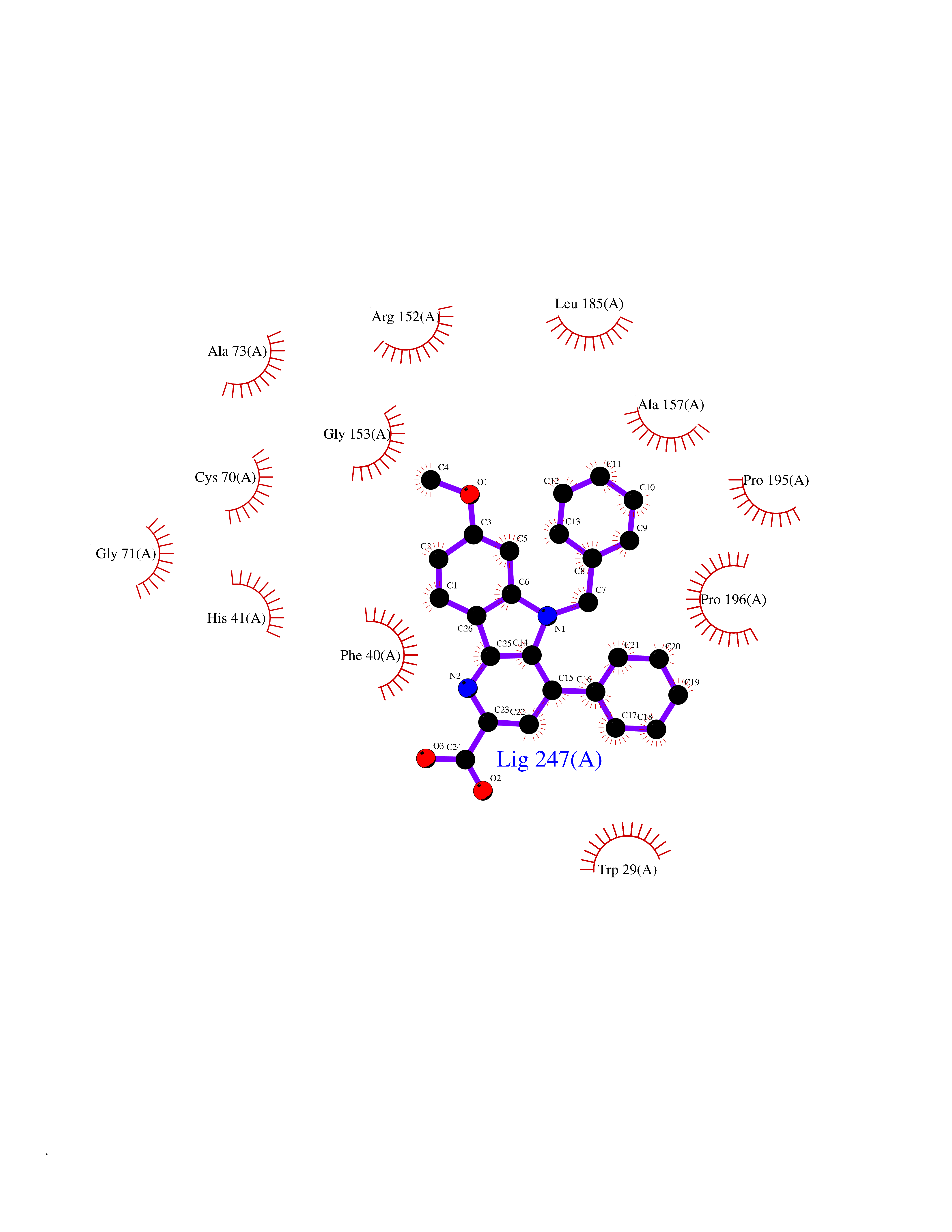 Ligplot Map 3D Binding mode Sequence EVQKNQVLTLEEWQDKWVNGKTAFHQEQGHQLLKKHLDTFLKGKSGLRVFFPLCGKAVEXKWFADRGHSVVGVEISELGIQEFFTEQNLSYSEEPITEIPGTKVFKSSSGNISLYCCSIFDLPRTNIGKFDXIWDRGALVAINPGDRKCYADTXFSLLGKKFQYLLCVLSYDPTKHPGPPFYVPHAEIERLFGKICNIRCLEKVDAFEERHKSWGIDCLFEKLYLLTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Dipeptidyl peptidase 8 (DPP-8) | 6EOP | 7.75 | |
Target general information Gen name DPP8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Prolyl dipeptidase DPP8; MSTP141; MSTP135; MSTP097; Dipeptidyl peptidase VIII; Dipeptidyl peptidase IV-related protein 1; DPRP1; DPRP-1; DPP VIII; DP8 Protein family Peptidase S9B family, DPPIV subfamily Biochemical class Peptidase Function Dipeptidyl peptidase that cleaves off N-terminal dipeptides from proteins having a Pro or Ala residue at position 2. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.14.5 Uniprot keywords 3D-structure; Alternative splicing; Aminopeptidase; Apoptosis; Cytoplasm; Hydrolase; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A,D Molecular weight (Da) 97764.9 Length 849 Aromaticity 0.12 Instability index 47.71 Isoelectric point 5.69 Charge (pH=7) -21.66 2D Binding mode Binding energy (Kcal/mol) -10.57 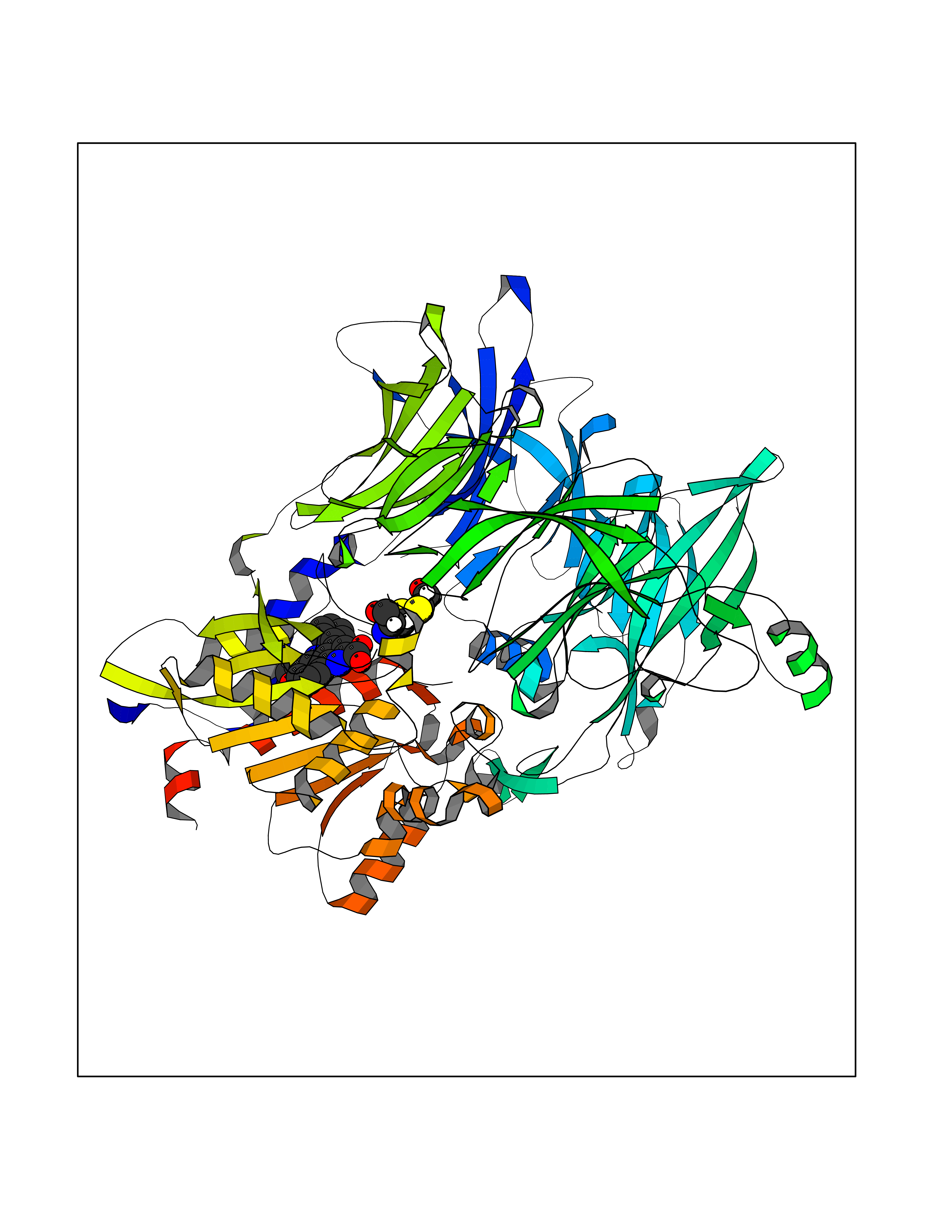 Molscript Map 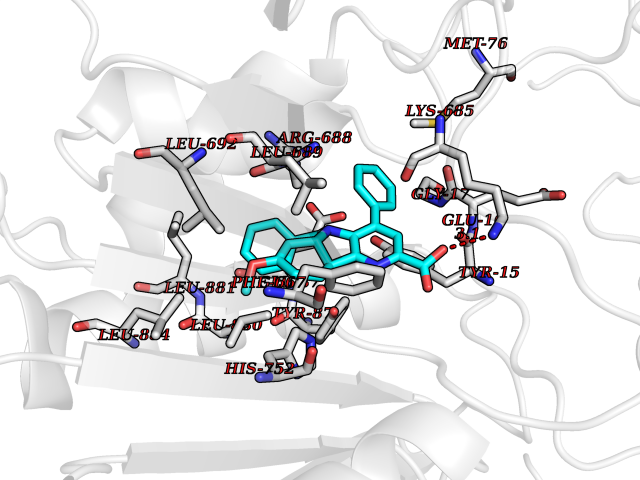 Pymol Map 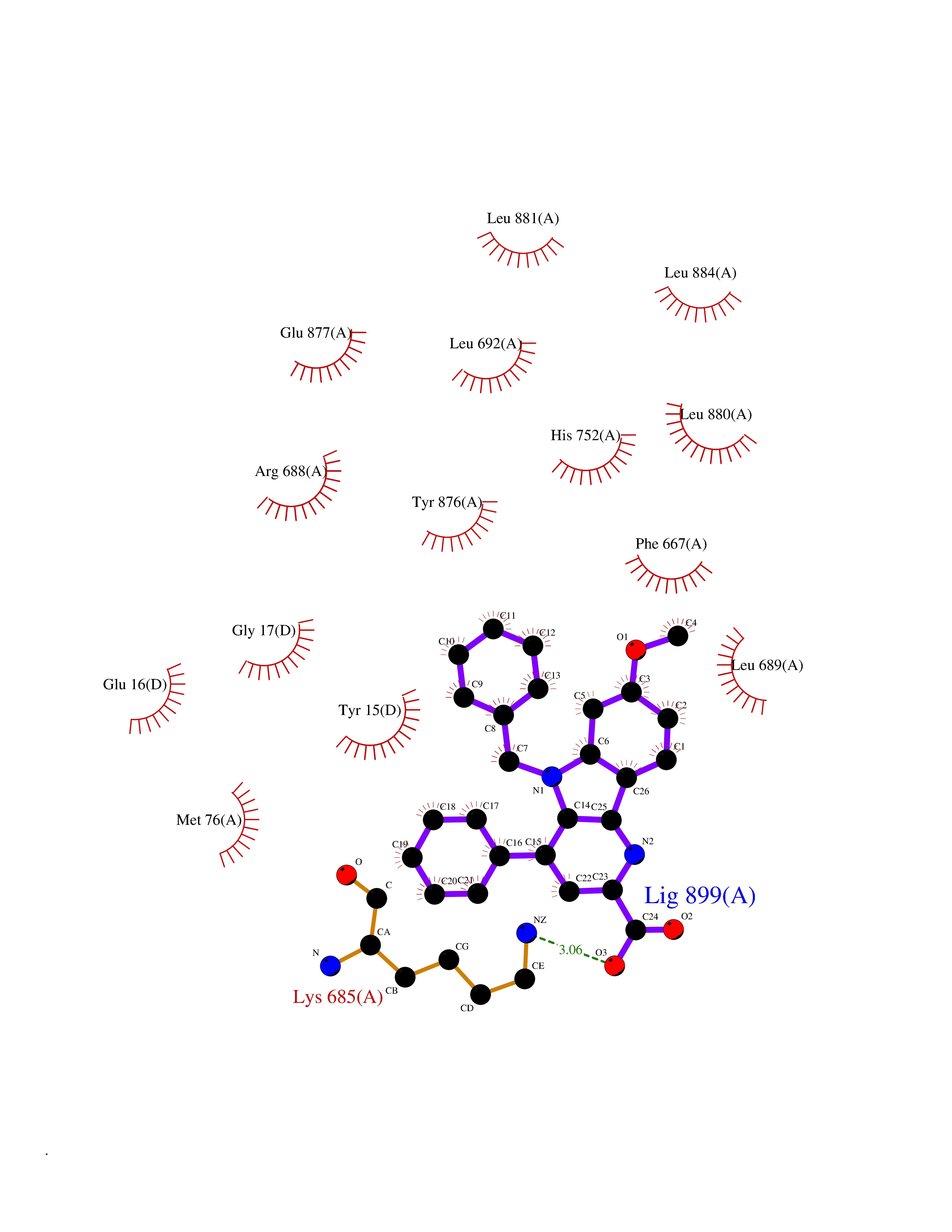 Ligplot Map 3D Binding mode Sequence LEPFYVERYSWSQLKKLLADTRKYHGYMMAKAPHDFMFVKRNDPDGPHSDRIYYLAMSNRENTLFYSEIPKTINRAAVLMLSWKPLLDLFQYSREEELLRERKRIGTVGIASYDYHQGSGTFLFQAGSGIYHVKDGGPQGFTQQPLRPNLVETSCPNIRMDPKLCPADPDWIAFIHSNDIWISNIVTREERRLTYVHNELANMEEDARSAGVATFVLQEEFDRYSGYWWCPKAETTPSGGKILRILYEENDESEVEIIHVTSPMLETRRADSFRYPKTGTANPKVTFKMSEIMIDAEGRIIDVIDKELIQPFEILFEGVEYIARAGWTPEGKYAWSILLDRSQTRLQIVLISPELFIPVEDDVMERQRLIESVPDSVTPLIIYEETTDIWINIHDIFHVFPQSHEEEIEFIFASECKTGFRHLYKITSILKESKYKRSSGGLPAPSDFKCPIKEEIAITSGEWEVLGRHGSNIQVDEVRRLVYFEGTKDSPLEHHLYVVSYVNPGEVTRLTDRGYSHSCCISQHCDFFISKYSNQKNPHCVSLYKLSSPEDDPTCKTKEFWATILDSAGPLPDYTPPEIFSFESTTGFTLYGMLYKPHDLQPGKKYPTVLFIYGGPQVQLVNNRFKGVKYFRLNTLASLGYVVVVIDNRGSXHRGLKFEGAFKYKMGQIEIDDQVEGLQYLASRYDFIDLDRVGIHGWSYGGYLSLMALMQRSDIFRVAIAGAPVTLWIFYDTGYTERYMGHPDQNEQGYYLGSVAMQAEKFPSEPNRLLLLHGFLDENVHFAHTSILLSFLVRAGKPYDLQIYPQERHSIRVPESGEHYELHLLHYLQENLGSRIAALKVSLRFLYEG Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Luteinizing hormone receptor (LHCGR) | 7FIH | 7.75 | |
Target general information Gen name LHCGR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Luteinizing hormone-releasing hormone receptor; LSH-R; LHRH receptor; LHCGR; LH/CG-R Protein family G-protein coupled receptor 1 family, FSH/LSH/TSH subfamily Biochemical class GPCR rhodopsin Function Receptor for lutropin-choriogonadotropic hormone. The activity of this receptor is mediated by G proteins which activate adenylate cyclase. Related diseases Familial male precocious puberty (FMPP) [MIM:176410]: In FMPP the receptor is constitutively activated. {ECO:0000269|PubMed:11134146, ECO:0000269|PubMed:11391350, ECO:0000269|PubMed:7629248, ECO:0000269|PubMed:7692306, ECO:0000269|PubMed:7714085, ECO:0000269|PubMed:7757065, ECO:0000269|PubMed:8281137, ECO:0000269|PubMed:8829636, ECO:0000269|PubMed:8929952, ECO:0000269|PubMed:9467560, ECO:0000269|PubMed:9661624}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Luteinizing hormone resistance (LHR) [MIM:238320]: An autosomal recessive disorder characterized by unresponsiveness to luteinizing hormone, defective sexual development in males, and defective follicular development and ovulation, amenorrhea and infertility in females. Two forms of the disorder have been defined in males. Type 1 is a severe form characterized by complete 46,XY male pseudohermaphroditism, low testosterone and high luteinizing hormone levels, total lack of responsiveness to luteinizing and chorionic gonadotropin hormones, lack of breast development, and absent development of secondary male sex characteristics. Type 2, a milder form, displays a broader range of phenotypic expression ranging from micropenis to severe hypospadias. {ECO:0000269|PubMed:12050206, ECO:0000269|PubMed:15372531, ECO:0000269|PubMed:15472221, ECO:0000269|PubMed:19551906, ECO:0000269|PubMed:7719343, ECO:0000269|PubMed:8559204, ECO:0000269|PubMed:9215288, ECO:0000269|PubMed:9514160, ECO:0000269|PubMed:9626144, ECO:0000269|PubMed:9626653}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06719; DB00050; DB00097; DB09126; DB00014; DB00044; DB00032 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; G-protein coupled receptor; Glycoprotein; Leucine-rich repeat; Lipoprotein; Membrane; Palmitate; Receptor; Reference proteome; Repeat; Signal; Sulfation; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 61996.2 Length 550 Aromaticity 0.12 Instability index 37.67 Isoelectric point 8.34 Charge (pH=7) 5.05 2D Binding mode Binding energy (Kcal/mol) -10.58  Molscript Map  Pymol Map 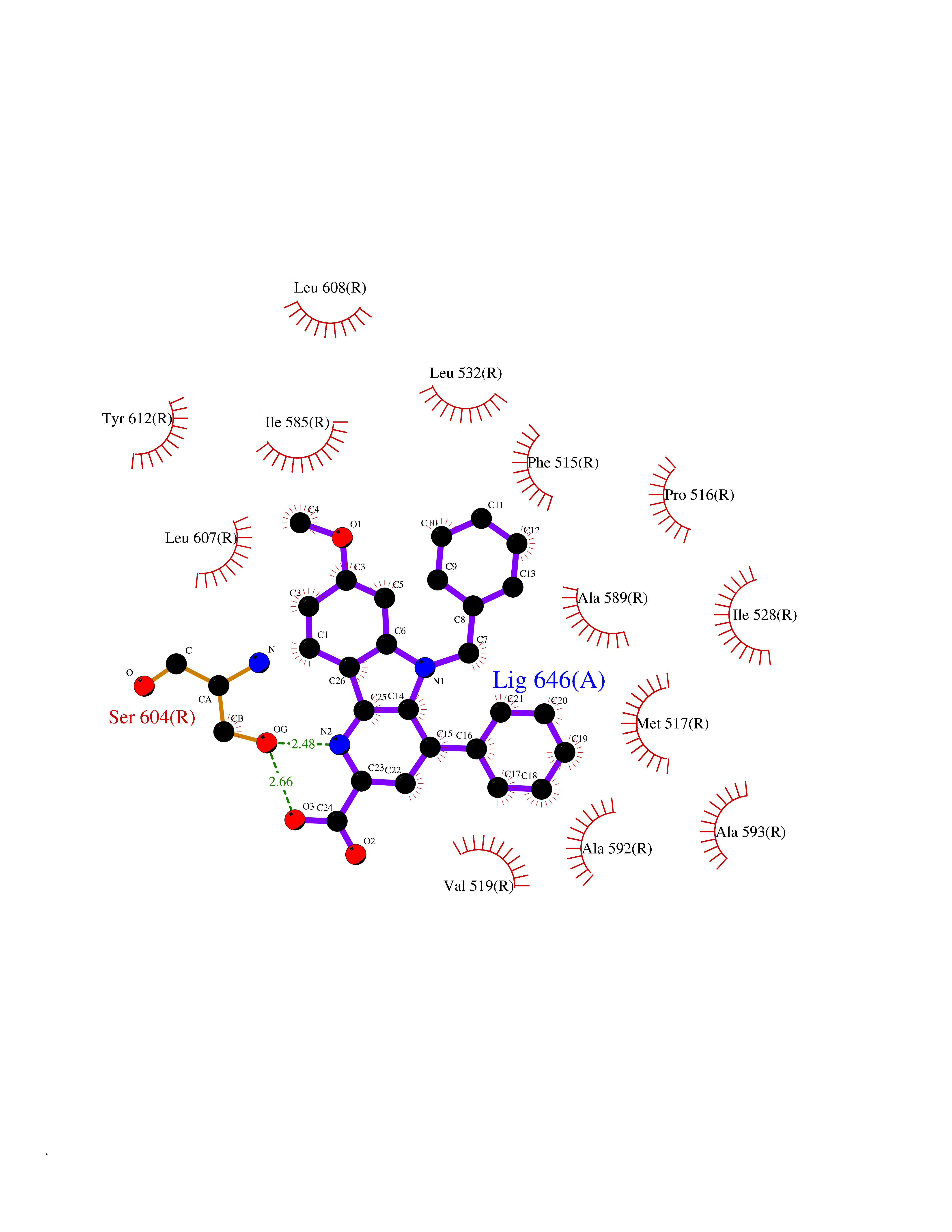 Ligplot Map 3D Binding mode Sequence TRLSLAYLPVKVIPSQAFRGLNEVIKIEISQIDSLERIEANAFDNLLNLSEILIQNTKNLRYIEPGAFINLPRLKYLSICNTGIRKFPDVTKVFSSESNFILEICDNLHITTIPGNAFQGMNNESVTLKLYGNGFEEVQSHAFNGTTLTSLELKENVHLEKMHNGAFRGATGPKTLDISSTKLQALPSYGLESIQRLIATSSYSLKKLPSRETFVNLLEATLTYPIHCCAFRNLPDYEYGFCLPKTPRCAPEPDAFNPCEDIMGYDFLRVLIWLINILAIMGNMTVLFVLLTSRYKLTVPRFLMCNLSFADFCMGLYLLLIASVDSQTKGQYYNHAIDWQTGSGCSTAGFFTVFASELSVYTLTVITLERWHTITYAIHLDQKLRLRHAILIMLGGWLFSSLIAMLPLVGVSNYMKVSICFPMDVETTLSQVYILTILILNVVAFFIICACYIKIYFAVRNPELMATNKDTKIAKKMAILIFTDFTCMAPISFFAISAAFKVPLITVTNSKVLLVLFYPINSCANPFLYAIFTKTFQRDFFLLLSKFGCC Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Urea transporter 1 (SLC14A1) | 6QD5 | 7.75 | |
Target general information Gen name SLC14A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Urea transporter, erythrocyte; UTE; UT1; Solute carrier family 14 member 1; RACH1; JK; HUT11 Protein family Urea transporter family Biochemical class Urea transporter family Function Urea channel that facilitates transmembrane urea transport down a concentration gradient. A constriction of the transmembrane channel functions as selectivity filter through which urea is expected to pass in dehydrated form. The rate of urea conduction is increased by hypotonic stress. Plays an important role in the kidney medulla collecting ducts, where it allows rapid equilibration between the lumen of the collecting ducts and the interstitium, and thereby prevents water loss driven by the high concentration of urea in the urine. Facilitates urea transport across erythrocyte membranes. May also play a role in transmembrane water transport, possibly by indirect means. Related diseases Immunodeficiency 12 (IMD12) [MIM:615468]: A primary immunodeficiency characterized by onset in infancy of recurrent bacterial and candidal infections resulting in bronchiectasis and growth delay. Manifestations include mastoiditis, aphthous ulcers, cheilitis, gingivitis, esophagitis, gastritis, duodenitis, and meningitis. Levels of absolute lymphocytes and serum immunoglobulins are normal, but specific antibody titers are low despite immunization, and T-cells show impaired proliferative responses to mitogens. {ECO:0000269|PubMed:23727036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A chromosomal aberration involving MALT1 is recurrent in low-grade mucosa-associated lymphoid tissue (MALT lymphoma). Translocation t(11;18)(q21;q21) with BIRC2. This translocation is found in approximately 50% of cytogenetically abnormal low-grade MALT lymphoma. {ECO:0000269|PubMed:10339464, ECO:0000269|PubMed:10523859, ECO:0000269|PubMed:10702396, ECO:0000269|PubMed:11090634}. Drugs (DrugBank ID) DB01005; DB03904 Interacts with Q8WVV5; Q9Y3D6; Q8WWP7; P30301; Q5QGT7; Q6UX34; P0DN84; Q9C0I4; Q5BJF2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood group antigen; Cell membrane; Glycoprotein; Membrane; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 38862.8 Length 356 Aromaticity 0.12 Instability index 40.98 Isoelectric point 7.67 Charge (pH=7) 0.95 2D Binding mode Binding energy (Kcal/mol) -10.57 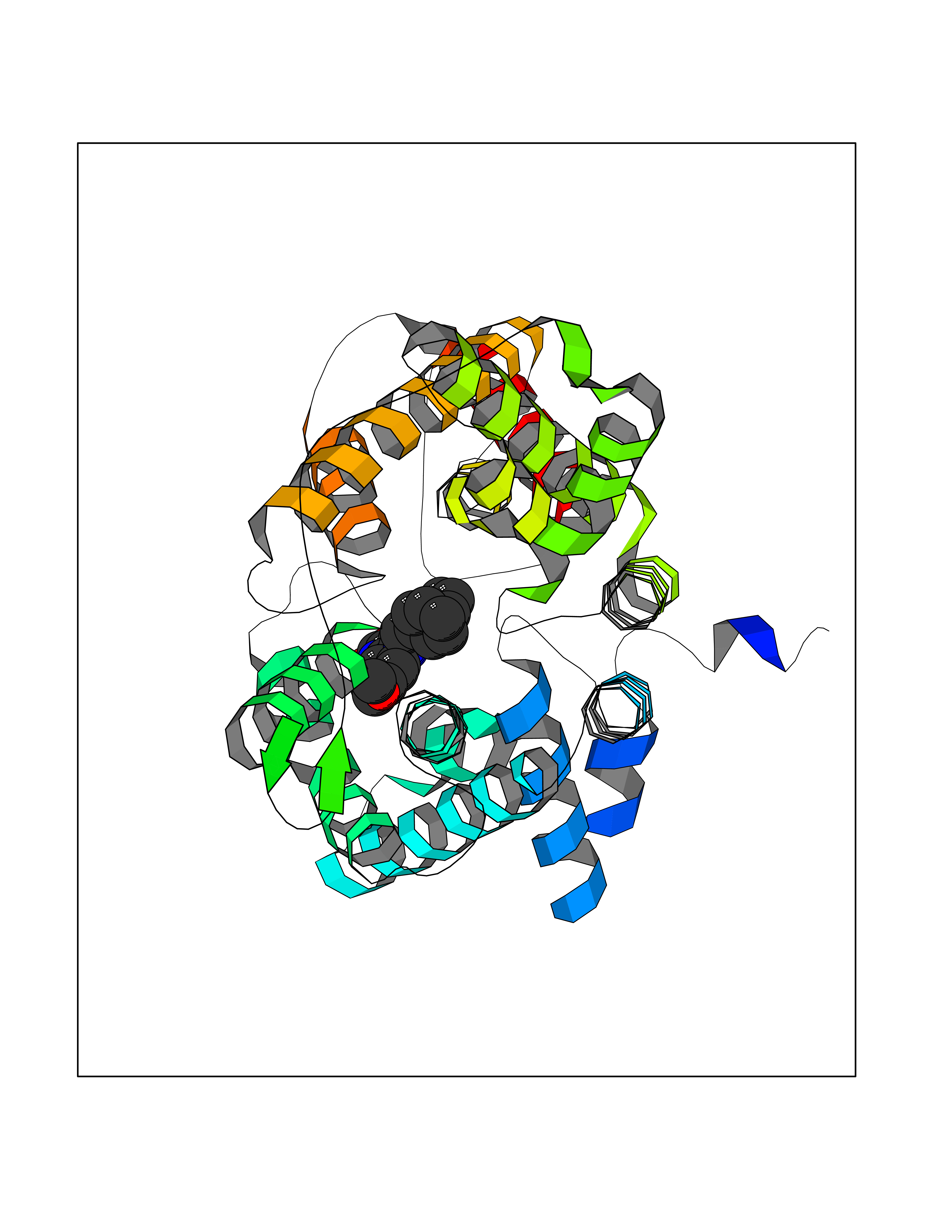 Molscript Map  Pymol Map 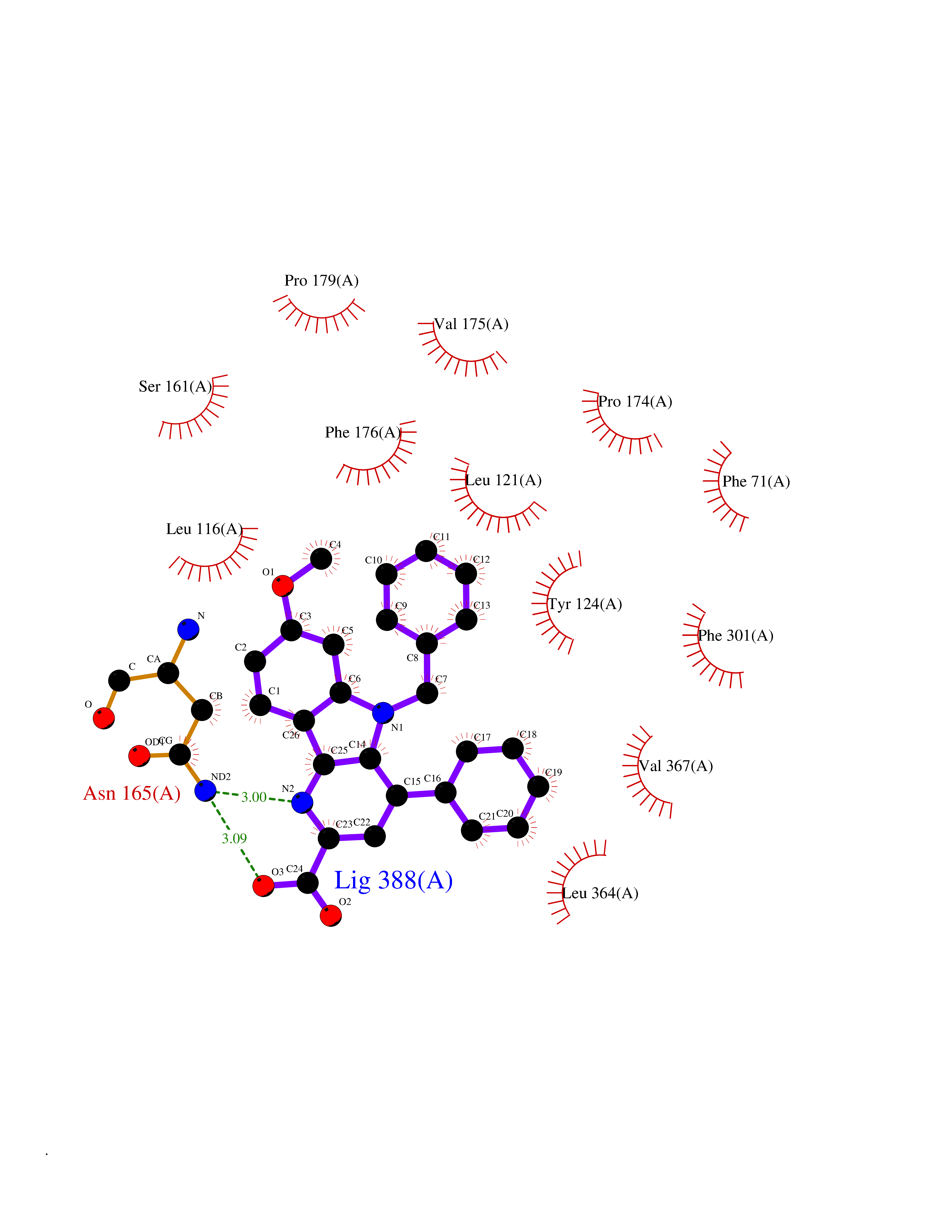 Ligplot Map 3D Binding mode Sequence FPKALGYVTGDMKELANQLKDKPVVLQFIDWILRGISQVVFVNNPVSGILILVGLLVQNPWWALTGWLGTVVSTLMALLLSQDRSLIASGLYGYNATLVGVLMAVFSDKGDYFWWLLLPVCAMSMTCPIFSSALNSVLSKWDLPVFTLPFNMALSMYLSATGHYNPFFPAKLVIPITTAPQISWSDLSALELLKSIPVGVGQIYGCDNPWTGGIFLGAILLSSPLMCLHAAIGSLLGIAAGLSLSAPFENIYFGLWGFNSSLACIAMGGMFMALTWQTHLLALGCALFTAYLGVGMANFMAEVGLPACTWPFCLATLLFLIMTTKNSNIYKMPLSKVTYPEENRIFYLQAKKRMVE Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Glutathione S-transferase theta-1 | 2C3Q | 7.73 | |
Target general information Gen name GSTT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family GST superfamily, Theta family Biochemical class Transferase Function Glutathione peroxidase activity.Glutathione transferase activity. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB00958; DB00291; DB00515; DB03619; DB00773; DB00143; DB03310; DB00526; DB09221 Interacts with P35609; Q9P2M1 EC number 2.5.1.18 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Direct protein sequencing; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27173.5 Length 239 Aromaticity 0.09 Instability index 44.08 Isoelectric point 7.87 Charge (pH=7) 1.28 2D Binding mode Binding energy (Kcal/mol) -10.55 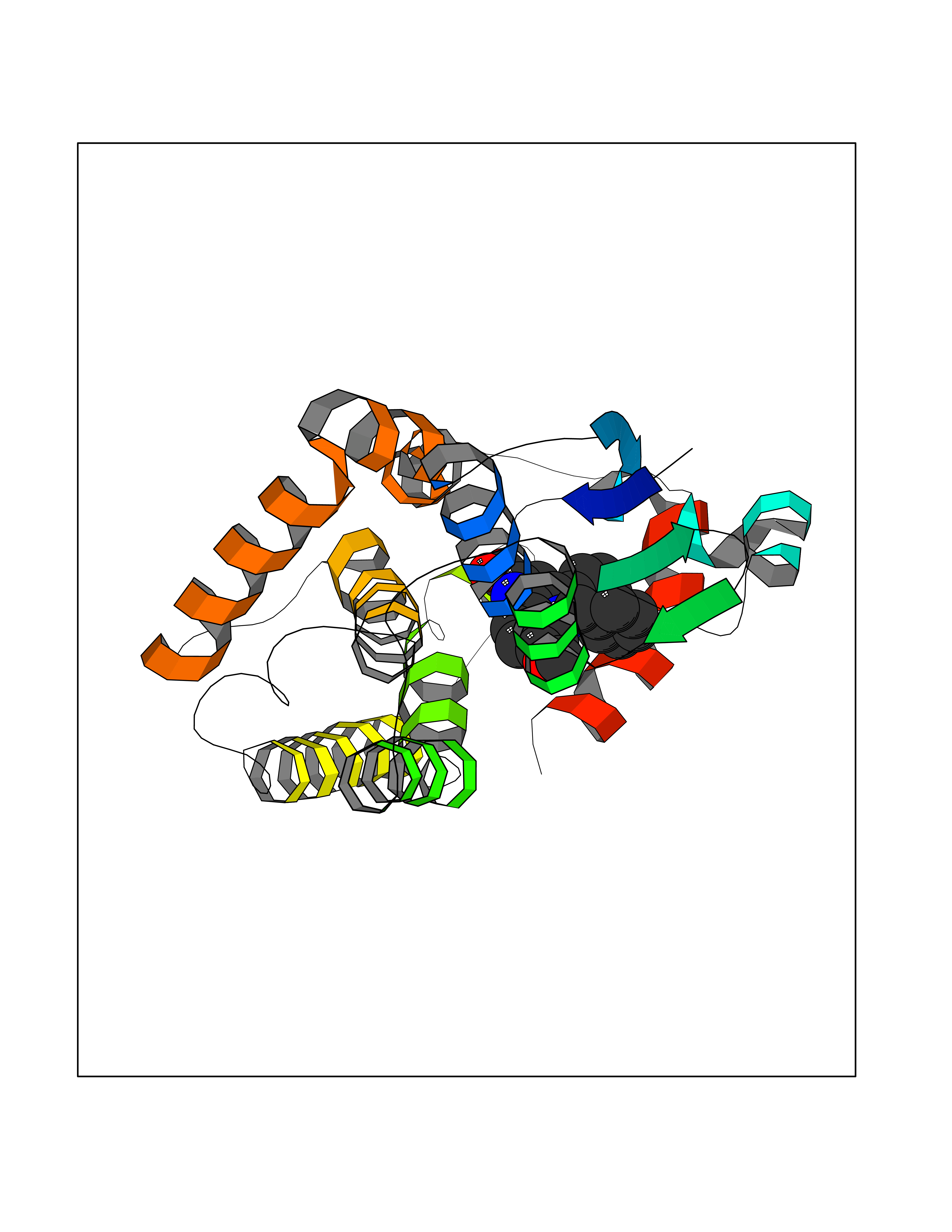 Molscript Map 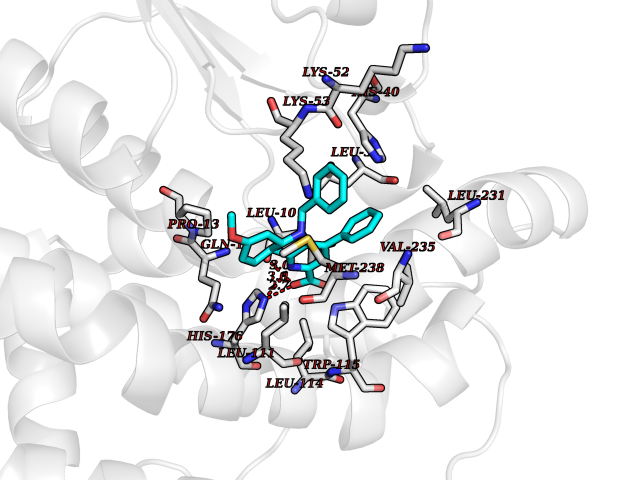 Pymol Map 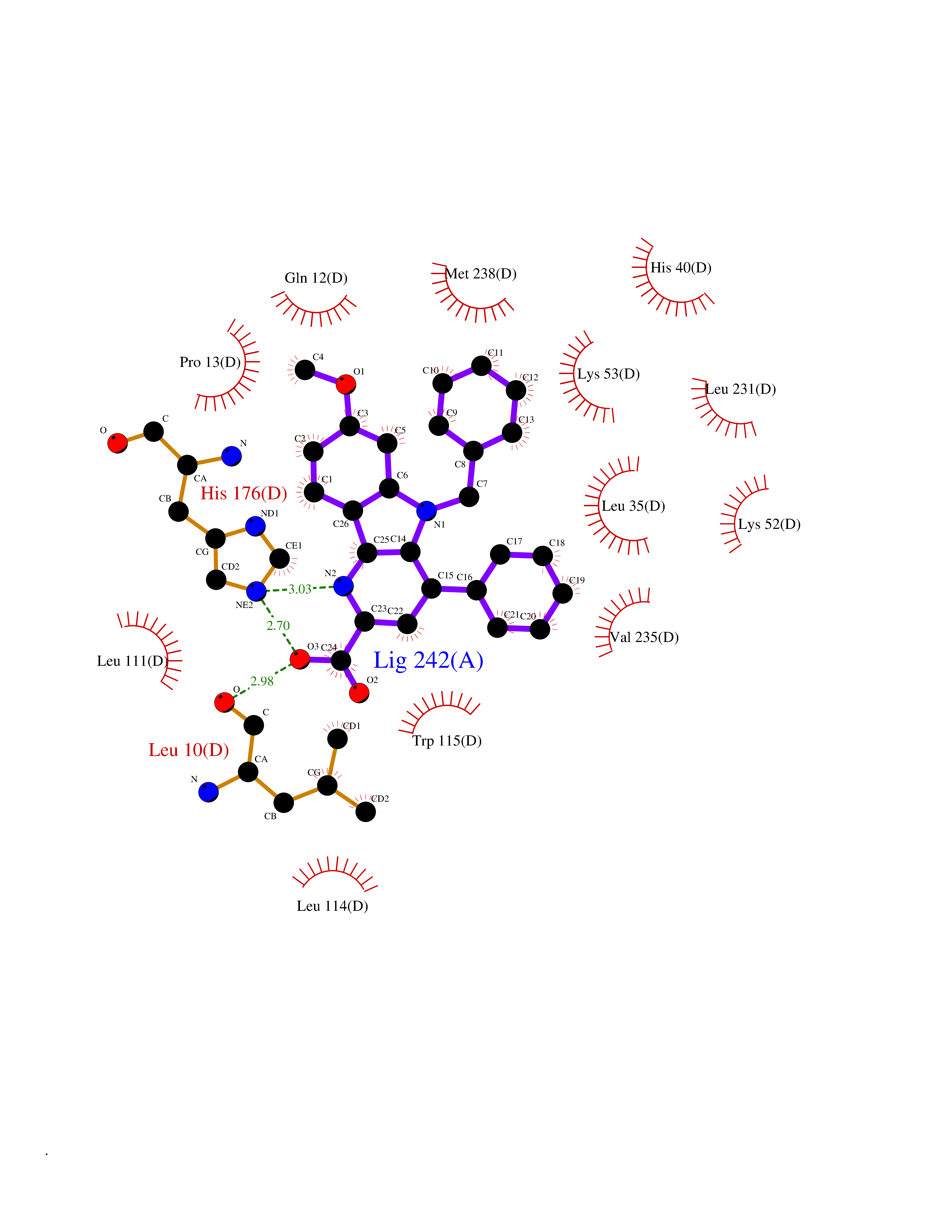 Ligplot Map 3D Binding mode Sequence GLELYLDLLSQPCRAVYIFAKKNDIPFELRIVDLIKGQHLSDAFAQVNPLKKVPALKDGDFTLTESVAILLYLTRKYKVPDYWYPQDLQARARVDEYLAWQHTTLRRSCLRALWHKVMFPVFLGEPVSPQTLAATLAELDVTLQLLEDKFLQNKAFLTGPHISLADLVAITELMHPVGAGCQVFEGRPKLATWRQRVEAAVGEDLFQEAHEVILKAKDFPPADPTIKQKLMPRVLAMIR Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Glutamate receptor ionotropic NMDA 2A (NMDAR2A) | 5KCJ | 7.73 | |
Target general information Gen name GRIN2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NR2A; NMDA receptor NR2A; N-methyl D-aspartate receptor subtype 2A; HNR2A; Glutamate receptor ionotropic, NMDA 2A; Glutamate [NMDA] receptor subunit epsilon-1; GluN2A Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, NR1/GRIN1 subfamily Biochemical class Glutamate-gated ion channel Function Channel activation requires binding of the neurotransmitter glutamate to the epsilon subunit, glycine binding to the zeta subunit, plus membrane depolarization to eliminate channel inhibition by Mg(2+). Sensitivity to glutamate and channel kinetics depend on the subunit composition; channels containing GRIN1 and GRIN2A have higher sensitivity to glutamate and faster kinetics than channels formed by GRIN1 and GRIN2B. Contributes to the slow phase of excitatory postsynaptic current, long-term synaptic potentiation, and learning. Component of NMDA receptor complexes that function as heterotetrameric, ligand-gated ion channels with high calcium permeability and voltage-dependent sensitivity to magnesium. Related diseases Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant (NDHMSD) [MIM:614254]: An autosomal dominant neurodevelopmental disorder characterized by severe intellectual disability and developmental delay, absent speech, muscular hypotonia, dyskinesia, and hyperkinetic movements. Cortical blindness, cerebral atrophy, and seizures are present in some patients. {ECO:0000269|PubMed:21376300, ECO:0000269|PubMed:25167861, ECO:0000269|PubMed:25864721, ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:28228639, ECO:0000269|PubMed:28389307, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal recessive (NDHMSR) [MIM:617820]: An autosomal recessive neurodevelopmental disorder characterized by severe intellectual disability and psychomotor developmental delay, involuntary and stereotypic movements, spasticity, and inability to walk without support. Intractable seizures manifest in some patients. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28051072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 101 (DEE101) [MIM:619814]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE101 is an autosomal recessive, severe form characterized by onset of seizures in early infancy. Death in infancy may occur. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:34611970}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01931; DB00659; DB06151; DB08838; DB01238; DB00289; DB05824; DB04620; DB03929; DB00647; DB00843; DB00228; DB11823; DB13146; DB06741; DB00142; DB00874; DB08954; DB06738; DB09409; DB09481; DB01043; DB00454; DB00333; DB04896; DB01173; DB00312; DB01174; DB01708; DB00418; DB00193 Interacts with P05067; P35637; Q12879-1; Q13224; Q62936 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Ligand-gated ion channel; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Zinc Protein physicochemical properties Chain ID B Molecular weight (Da) 53395.6 Length 469 Aromaticity 0.11 Instability index 29.84 Isoelectric point 8.72 Charge (pH=7) 5.65 2D Binding mode Binding energy (Kcal/mol) -10.54 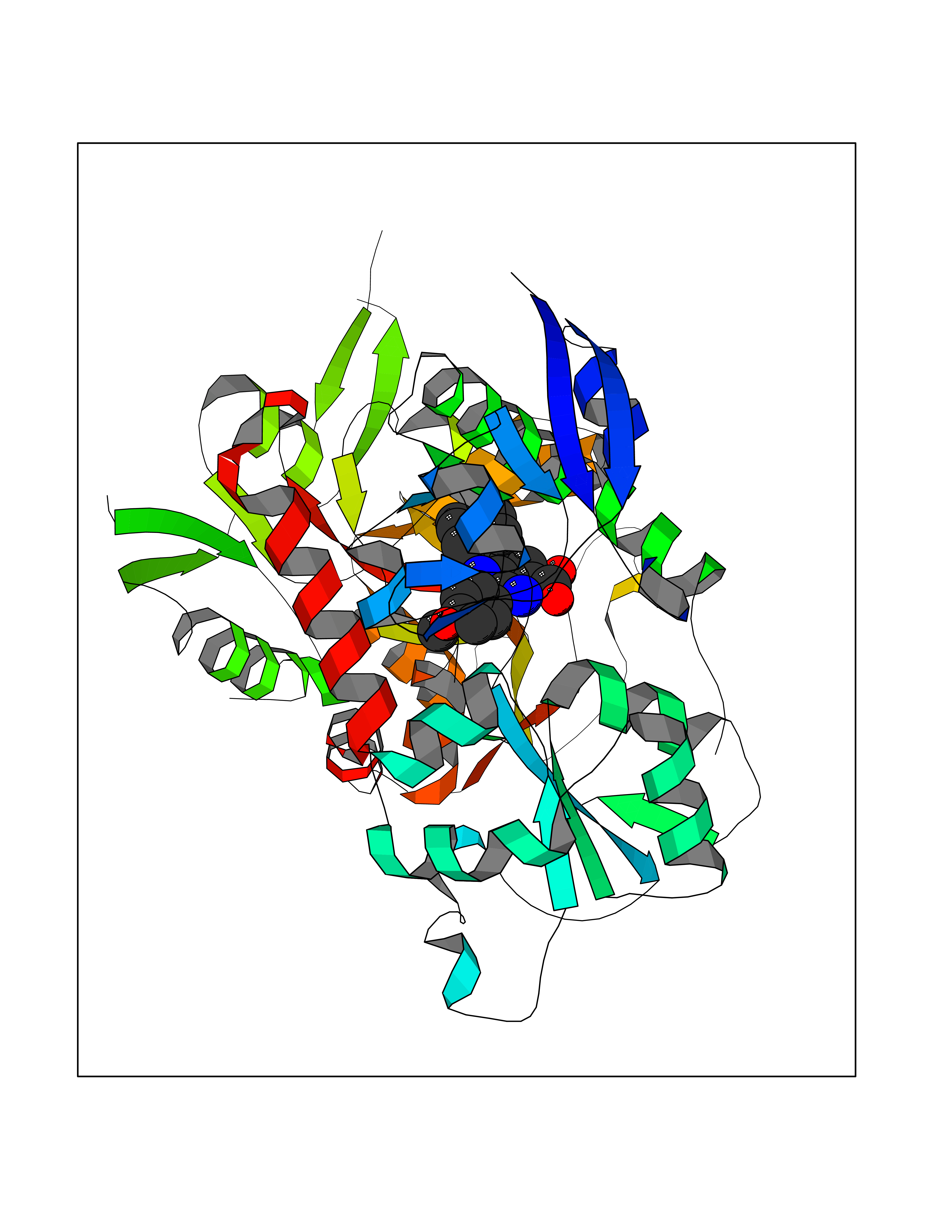 Molscript Map 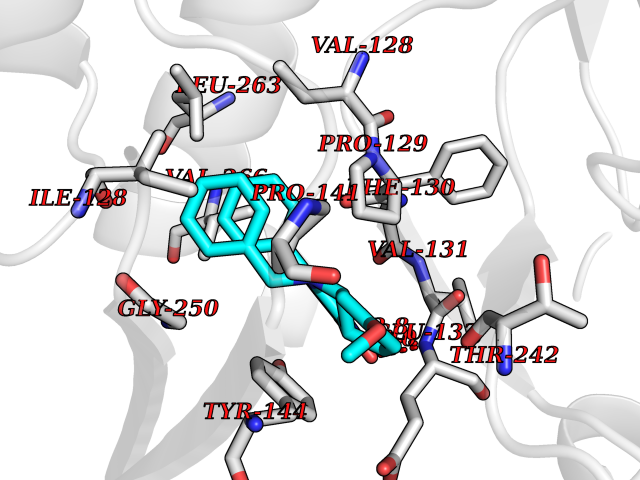 Pymol Map 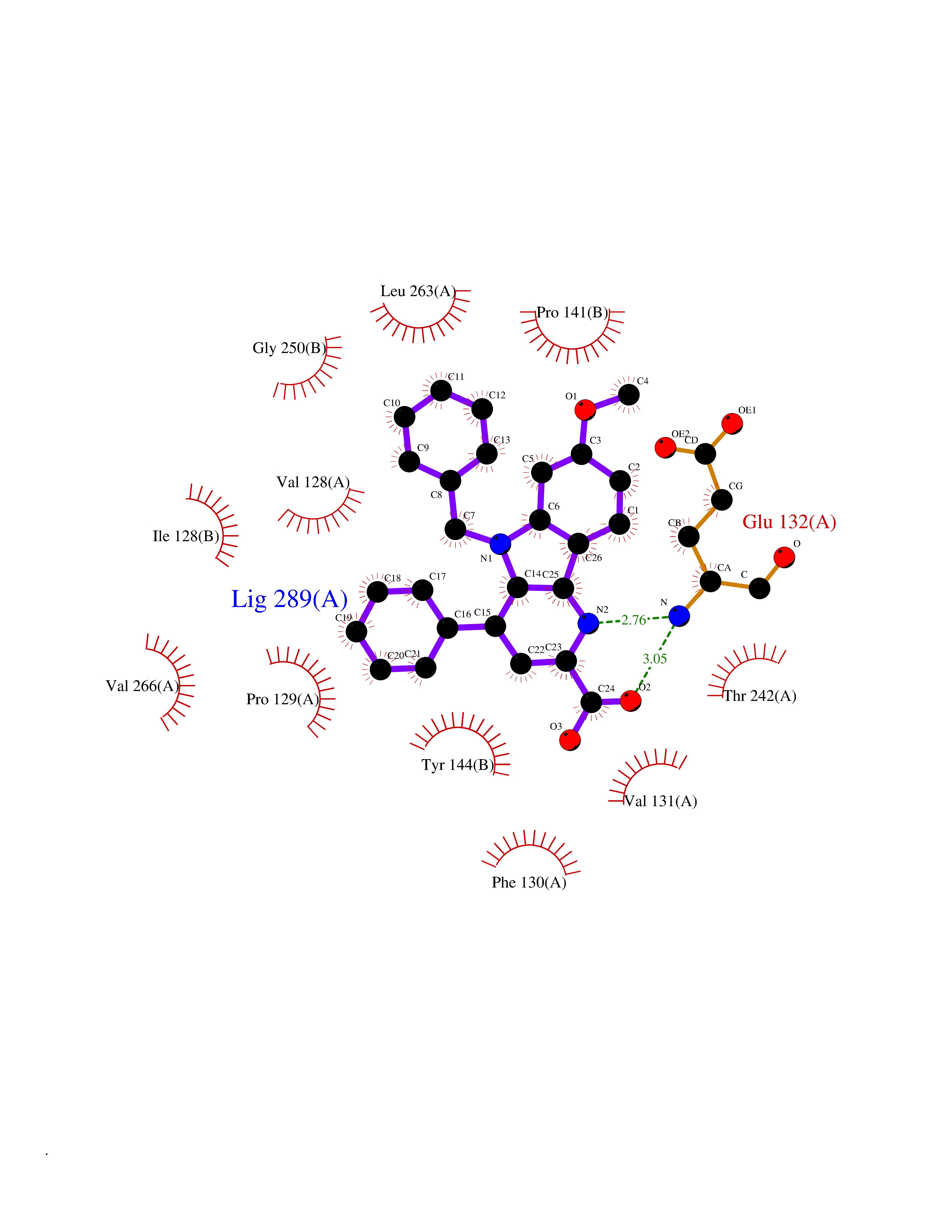 Ligplot Map 3D Binding mode Sequence DNHLSIVTLEEAPFVILKKLSRTVKFTYDLYLVTNGKHGKKVNNVWNGMIGEVVYQRAVMAVGSLTINEERSEVVDFSVPFVETGISVMVSRGTQVTGLSDKKFQRPHDYSPPFRFGTVPNGSTERNIRNNYPYMHQYMTKFNQKGVEDALVSLKTGKLDAFIYDAAVLNYKAGRDEGCKLVTIGSGYIFATTGYGIALQKGSPWKRQIDLALLQFVGDGEMEELETLWLTGICTRLKIVTIHQEPFVYYGFCIDLLIKLARTMNFTYEVHLVADGKFGTQERVNKKEWNGMMGELLSGQADMIVAPLTINNERAQYIEFSKPFKYQGLTILVKKGTRITGINDPRLRNPSDKFIYATVKQSSVDIYFRRQVELSTMYRHMEKHNYESAAEAIQAVRDNKLHAFIWDSAVLEFEASQKCDLVTTGELFFRSGFGIGMRKDSPWKQNVSLSILKSHENGFMEDLDKTWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Cholestenol delta-isomerase (EBP) | 6OHU | 7.73 | |
Target general information Gen name EBP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Emopamilbinding protein; EBP; Delta(8)Delta(7) sterol isomerase; D8D7 sterol isomerase; 3betahydroxysteroidDelta(8),Delta(7)isomerase Protein family EBP family Biochemical class Intramolecular oxidoreductases Function Catalyzes the conversion of Delta(8)-sterols to their corresponding Delta(7)-isomers. Related diseases Chondrodysplasia punctata 2, X-linked dominant (CDPX2) [MIM:302960]: A clinically and genetically heterogeneous disorder characterized by punctiform calcification of the bones. The key clinical features of CDPX2 are chondrodysplasia punctata, linear ichthyosis, cataracts and short stature. CDPX2 is a rare disorder of defective cholesterol biosynthesis, biochemically characterized by an increased amount of 8-dehydrocholesterol and cholest-8(9)-en-3-beta-ol in the plasma and tissues. {ECO:0000269|PubMed:10391218, ECO:0000269|PubMed:10391219, ECO:0000269|PubMed:10942423, ECO:0000269|PubMed:11493318, ECO:0000269|PubMed:18176751, ECO:0000269|PubMed:25814754}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: MEND syndrome (MEND) [MIM:300960]: An X-linked recessive disorder associated with a defect in sterol biosynthesis. Disease manifestations and severity are highly variable. Clinical features include intellectual disability, short stature, scoliosis, digital abnormalities, cataracts, and dermatologic abnormalities. {ECO:0000269|PubMed:12503101, ECO:0000269|PubMed:20949533, ECO:0000269|PubMed:24459067, ECO:0000269|PubMed:24700572}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00675 Interacts with O95870; Q86W74-2; Q13520; Q3SXY8; Q8N6S5; Q9H2C2; Q9HD20-3; O95393; Q12983; Q8WVV5; P06681; O14523; Q8WVX3-2; P01031; Q6UWT4; Q9P0B6; Q8NHW4; P25942; Q07108; P60033; O14735; P23141-3; Q9H9P2; Q8NHS1; O43889-2; Q96BA8; P49447; O43169; P78329; P81534; Q9H1M4; Q96LL9; Q15125; Q08426; Q9BV81; P54849; Q9UKR5; Q9Y282; Q7L5A8; Q5JX71; Q96IV6; Q9UGM5; Q9Y3D6; Q14318; Q9BWH2; Q14802-3; Q9H0Q3; Q8WWP7; Q96F15; P29033; O95452; O14653; Q8TDT2; P02724; P30519; Q7Z5P4; Q9Y5U9; Q9Y5U4; Q8N5M9; Q5T700; Q68G75; Q7L5N7; Q7Z4F1; Q96AG4; Q16873; Q6ZSS7; P50281; Q5J8X5; Q9UHE5; O95167; Q9NX14; Q99519; Q92982; Q9NZG7; Q16617; Q8IXM6; Q2M2E3; Q9P0S3; Q53FV1; Q8N138; Q7RTS5; Q9Y342; Q04941; Q8IY26; Q01453; P54315; P43378; P15151; Q8N8N0; Q5QGT7; Q96GQ5; Q9NTJ5; Q969E2; O75396; Q9Y6X1; Q8N6R1; Q9BWM7; Q8TD22; Q8IWU4; Q96G79; Q2M3R5; Q9NVC3; P08195-4; Q96JW4; Q6P1K1; Q0VAQ4; Q9NRQ5; B2RUZ4; Q9BZL3; Q6UX34; Q86Y82; P61266; Q13190; O43752; O15400; Q9UNK0; O43759-2; P57105; Q8N2H4; Q96BZ9; P07204; O14925; Q96CP7; Q96MV1; P55061; Q9NV29; P17152; Q9BXJ8; A0PK00; Q9NV12; Q9BVK8; Q9NUH8; Q9P0S9; Q9NRX6; Q8N511; Q969S6; Q9BTX3; A2RU14; Q9H0R3; Q8NBD8; Q8WW34-2; Q9NWH2; Q9BU79; Q8TBM7; Q69YG0; Q9NW97; Q9H2L4; Q6PI78; Q8N2M4; Q8N661; Q5BJF2; Q9NSU2-1; A0AVG3; Q5TGU0; A5PKU2; Q9Y385; Q9Y5Z9; Q53HI1; Q9H1C4; Q9NZ43; P23763-3; P63027; Q15836; O75379; Q9P0L0; O95292; O95070; Q9Y548; Q9BSR8; Q96EC8; Q6UX98; O95159 EC number EC 5.3.3.5 Uniprot keywords 3D-structure; Acetylation; Cataract; Cholesterol biosynthesis; Cholesterol metabolism; Cytoplasmic vesicle; Disease variant; Dwarfism; Endoplasmic reticulum; Ichthyosis; Isomerase; Lipid biosynthesis; Lipid metabolism; Membrane; Nucleus; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 24012.7 Length 206 Aromaticity 0.17 Instability index 42.52 Isoelectric point 6.19 Charge (pH=7) -3.19 2D Binding mode Binding energy (Kcal/mol) -10.55 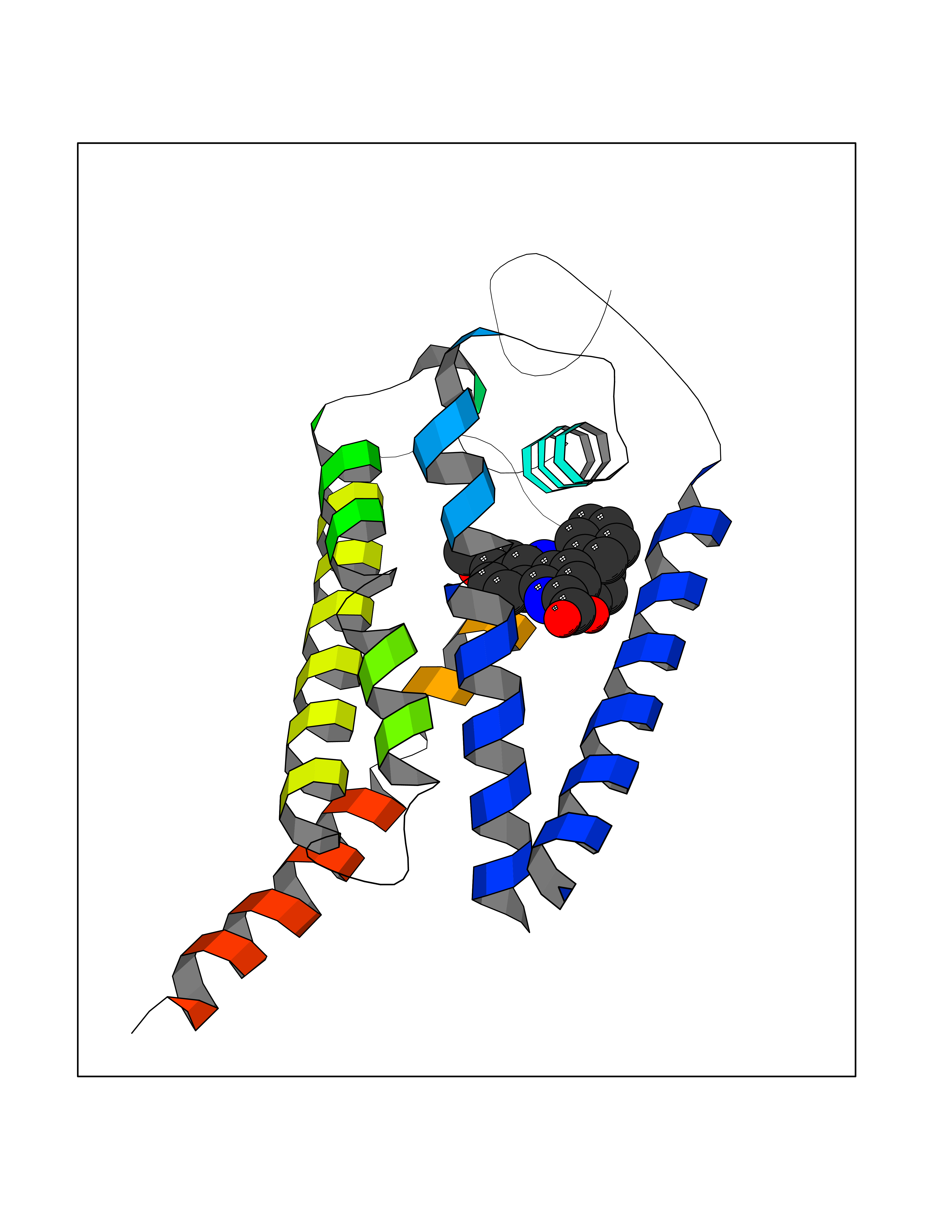 Molscript Map  Pymol Map 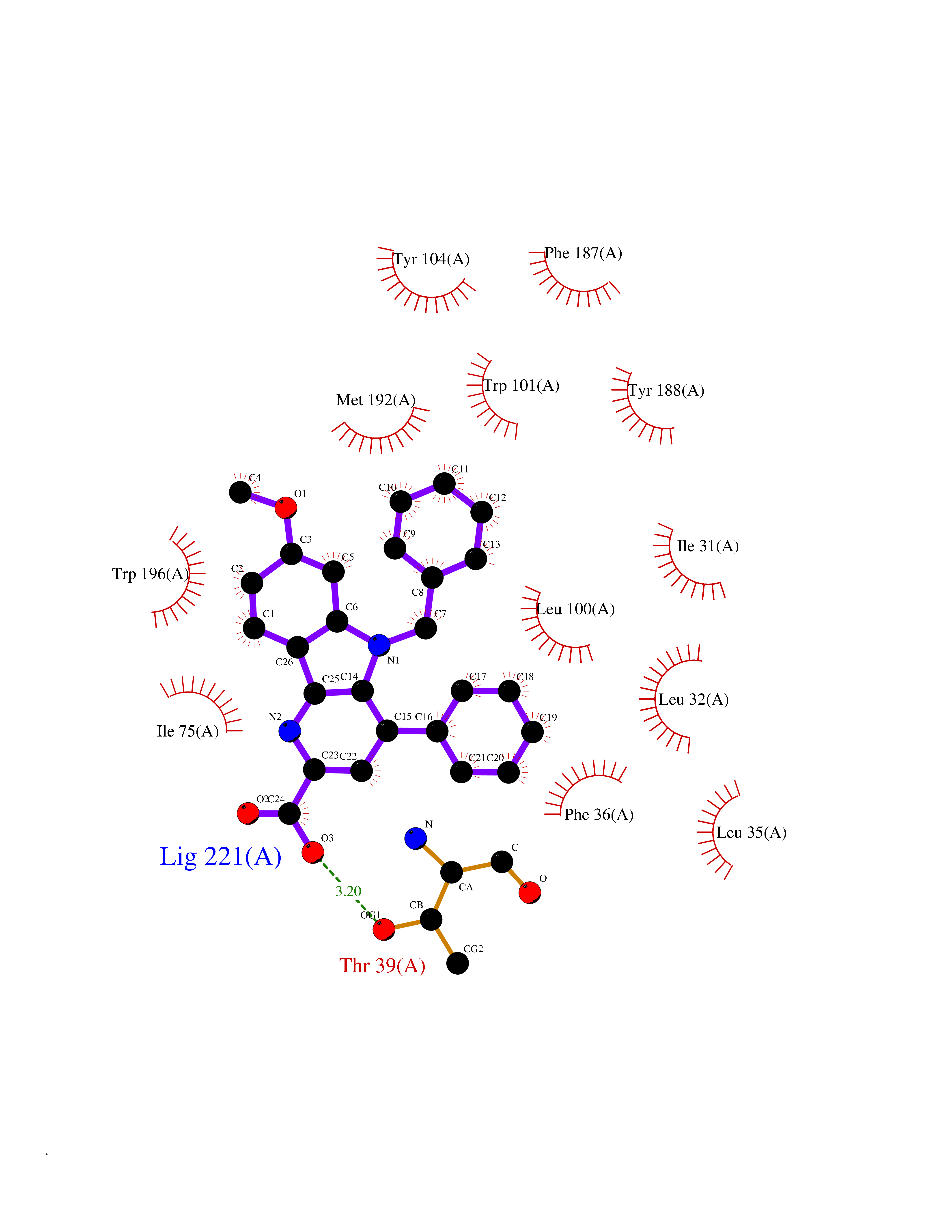 Ligplot Map 3D Binding mode Sequence PLHPYWPQHLRLDNFVPNDRPTWHILAGLFSVTGVLVVTTWLLSGRTWRRLSLCWFAVCGFIHLVIEGWFVLYYEDLLGDQAFLSQLWKEYAKGDSRYILGDNFTVCMETITACLWGPLSLWVVIAFLRQHPLRFILQLVVSVGQIYGDVLYFLTEHRDGFQHGELGHPLYFWFYFVFMNALWLVLPGVLVLDAVKHLTHAQSTLD Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Cytochrome P450 1A2 | 2HI4 | 7.72 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -10.53  Molscript Map 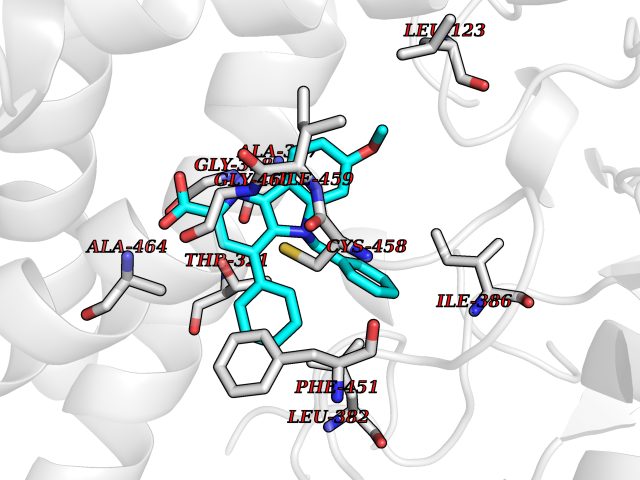 Pymol Map 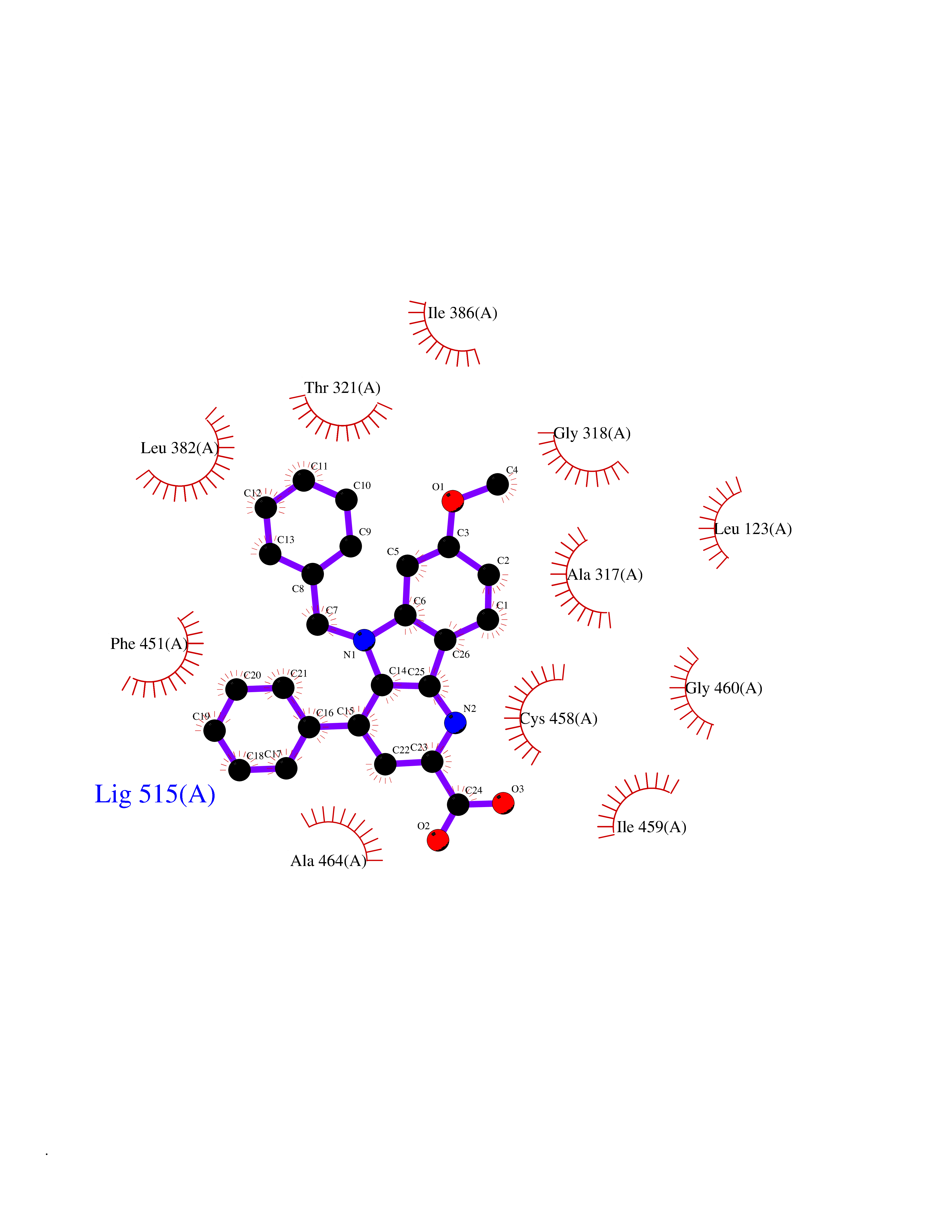 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Retinoic acid receptor RXR-beta (RXRB) | 5HJP | 7.72 | |
Target general information Gen name RXRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor beta; Nuclear receptor subfamily 2 group B member 2; NR2B2 Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE). Receptor for retinoic acid. Related diseases Noonan syndrome 13 (NS13) [MIM:619087]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. NS13 inheritance is autosomal dominant. There is considerable variability in severity. {ECO:0000269|PubMed:32721402}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08175; DB00459; DB00210; DB00523; DB00307; DB01393; DB03756; DB00926; DB01941; DB07929; DB02746; DB00412; DB00799; DB07080; DB00755 Interacts with Q00975; Q9HB07; F1D8P7; Q13133; Q13133-3; Q96RI1-1; P04150; Q9NRD5; P37231; P10276; P10276-2; P10826-2; P13631; Q6IQ16; Q13137; Q96B26; Q08379; Q6A162; Q9UJV3-2; Q13133-3; Q96RI1-1; O43586; P10276; P10826-2; Q8IUQ4-2; O75528; Q12800; Q9UBB9; Q05BL1; P14373; O94972; Q96S82 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 28845.8 Length 251 Aromaticity 0.08 Instability index 54.86 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -10.52 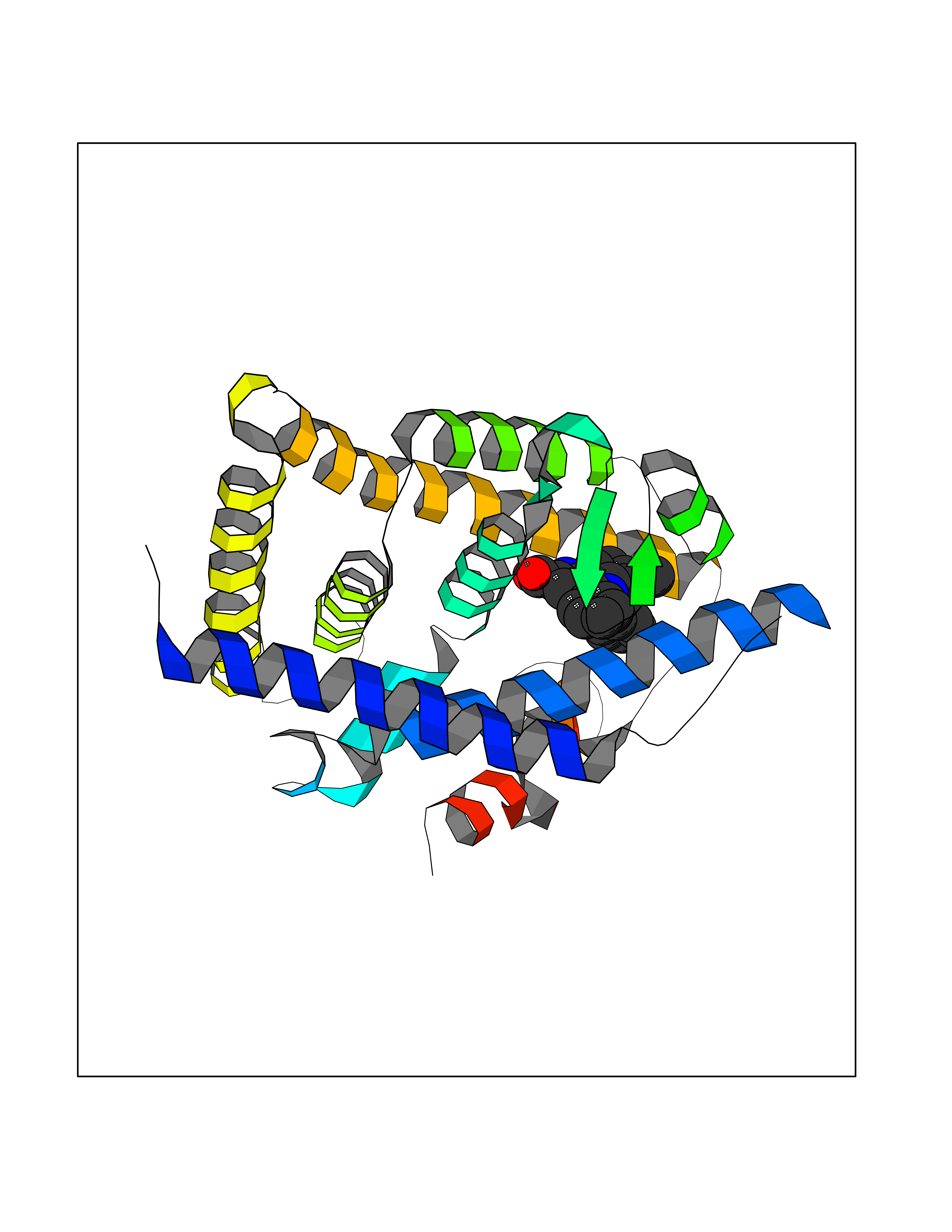 Molscript Map 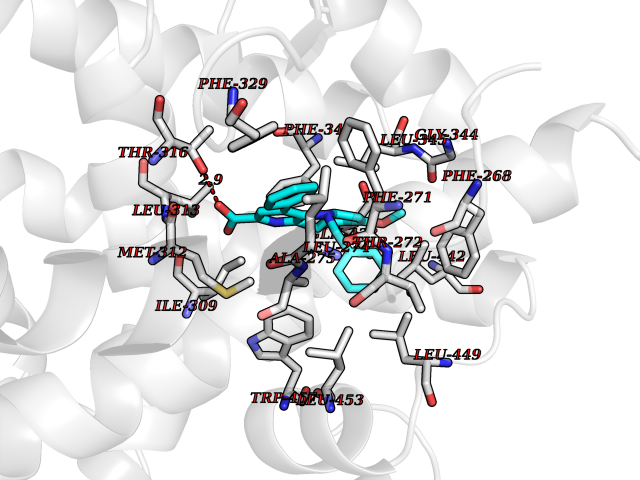 Pymol Map 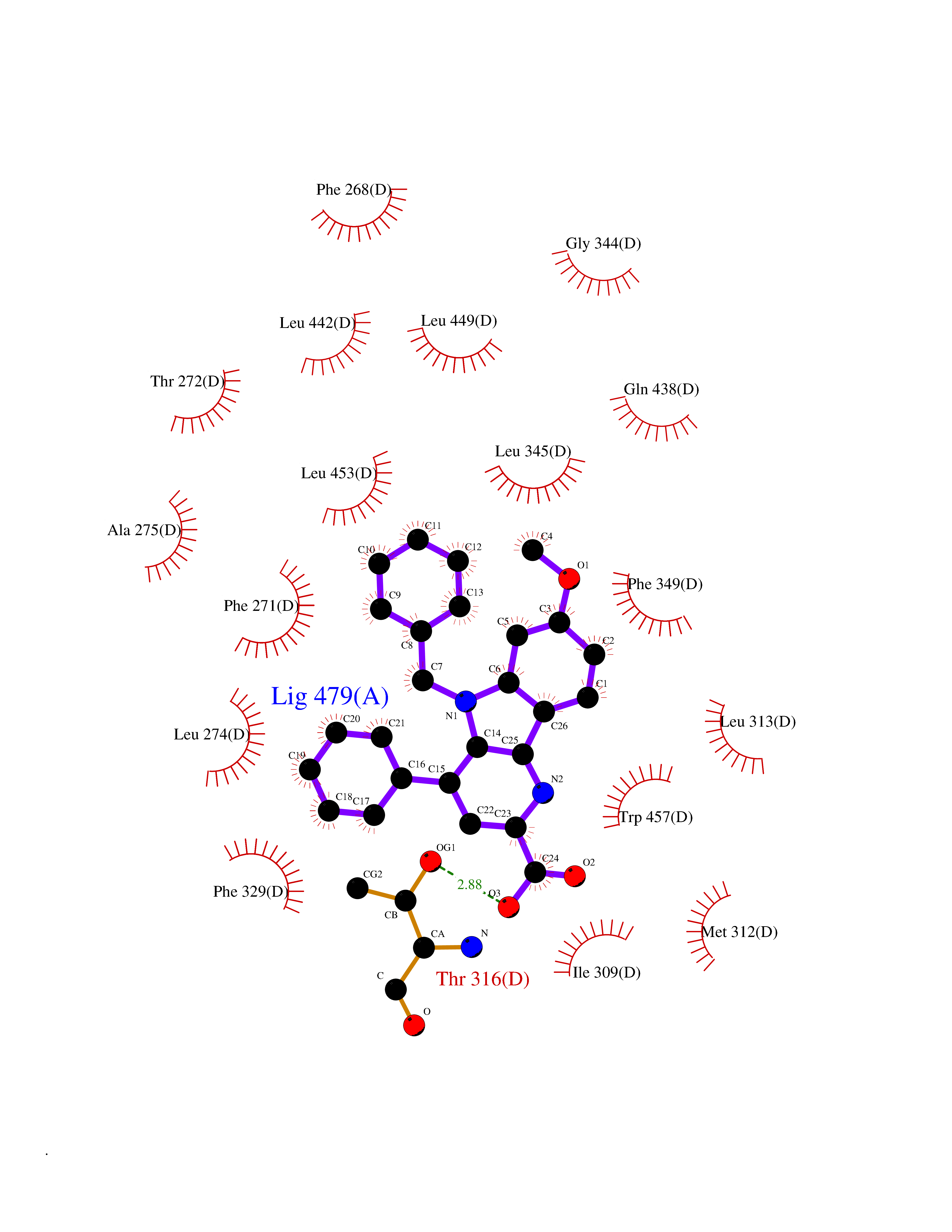 Ligplot Map 3D Binding mode Sequence QLTAAQELMIQQLVAAQLQCNKRSFSDQPKVTPWPSASQQRFAHFTELAIISVQEIVDFAKQVPGFLQLGREDQIALLKASTIEIMLLETARRYNHETECITFLKDFTYSKDDFHRAGLQVEFINPIFEFSRAMRRLGLDDAEYALLIAINIFSADRPNVQEPGRVEALQQPYVEALLSYTRIKRPQDQLRFPRMLMKLVSLRTLSSVHSEQVFALRLQDKKLPPLLSEIWDVHEGSGSGSHKILHRLLQD Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | D-amino acid oxidase (DAO) | 3ZNN | 7.71 | |
Target general information Gen name DAO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Daminoacid oxidase; DAMOX; DAAO Protein family DAMOX/DASOX family Biochemical class CH-NH(2) donor oxidoreductase Function Regulates the level of the neuromodulator D-serine in the brain. Has high activity towards D-DOPA and contributes to dopamine synthesis. Could act as a detoxifying agent which removes D-amino acids accumulated during aging. Acts on a variety of D-amino acids with a preference for those having small hydrophobic side chains followed by those bearing polar, aromatic, and basic groups. Does not act on acidic amino acids. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:12364586}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Amyotrophic lateral sclerosis (ALS) [MIM:105400]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:20368421, ECO:0000269|PubMed:20538972, ECO:0000269|PubMed:22203986, ECO:0000269|PubMed:23219954, ECO:0000269|PubMed:24138986, ECO:0000269|PubMed:25701391, ECO:0000269|PubMed:37558109, ECO:0000269|PubMed:38035964, ECO:0000269|PubMed:38134563}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07979; DB02838; DB04166; DB03793; DB03225; DB03147; DB03531; DB02988 Interacts with Q9P2K6; O43741 EC number EC 1.4.3.3 Uniprot keywords 3D-structure; Amyotrophic lateral sclerosis; Cell projection; Cytoplasm; Disease variant; FAD; Flavoprotein; Neurodegeneration; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; S-nitrosylation; Schizophrenia; Secreted; Synapse Protein physicochemical properties Chain ID A,B Molecular weight (Da) 38654.6 Length 340 Aromaticity 0.11 Instability index 29.13 Isoelectric point 6.18 Charge (pH=7) -4.45 2D Binding mode Binding energy (Kcal/mol) -10.52 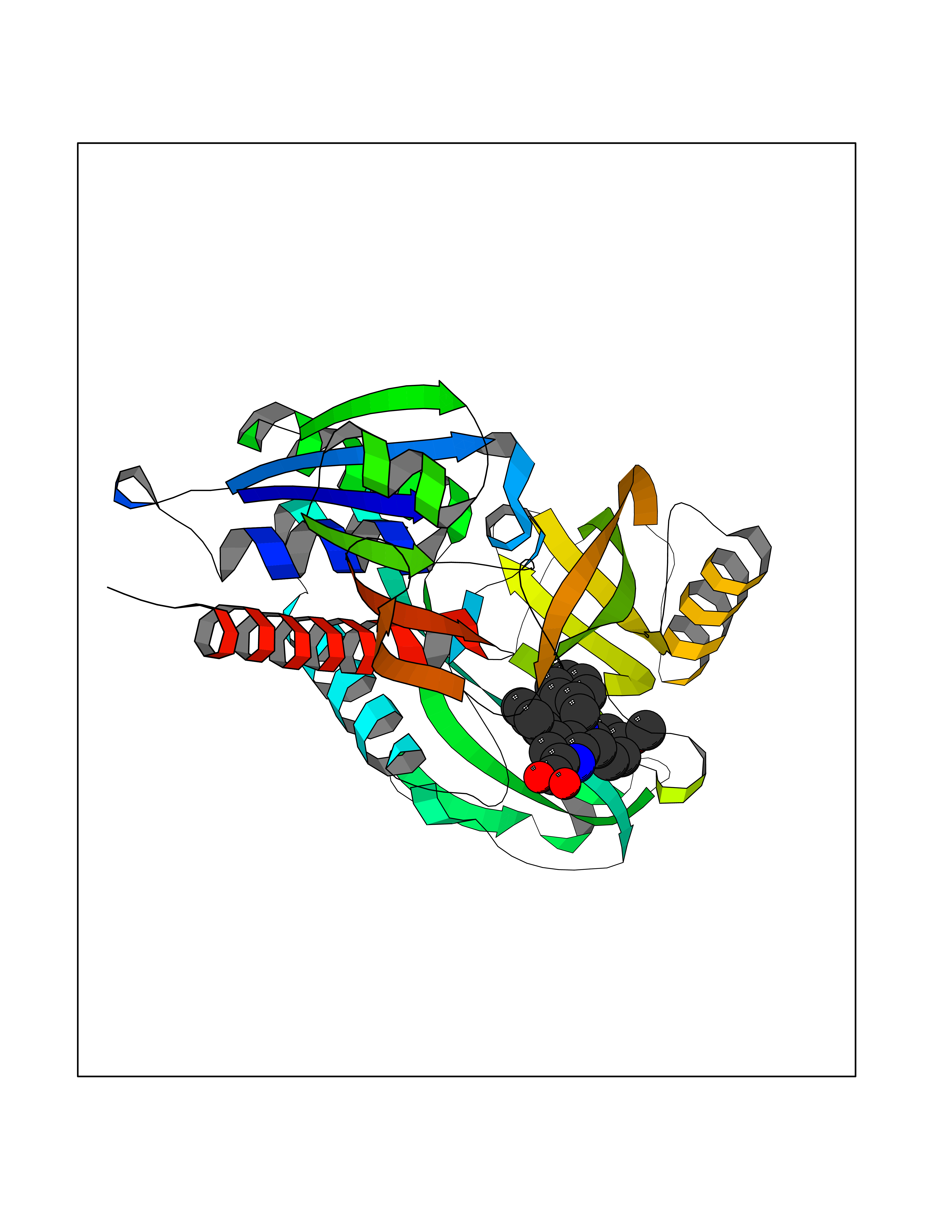 Molscript Map 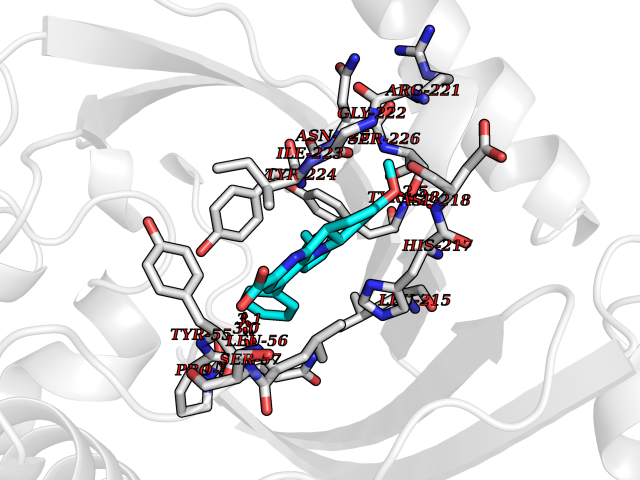 Pymol Map 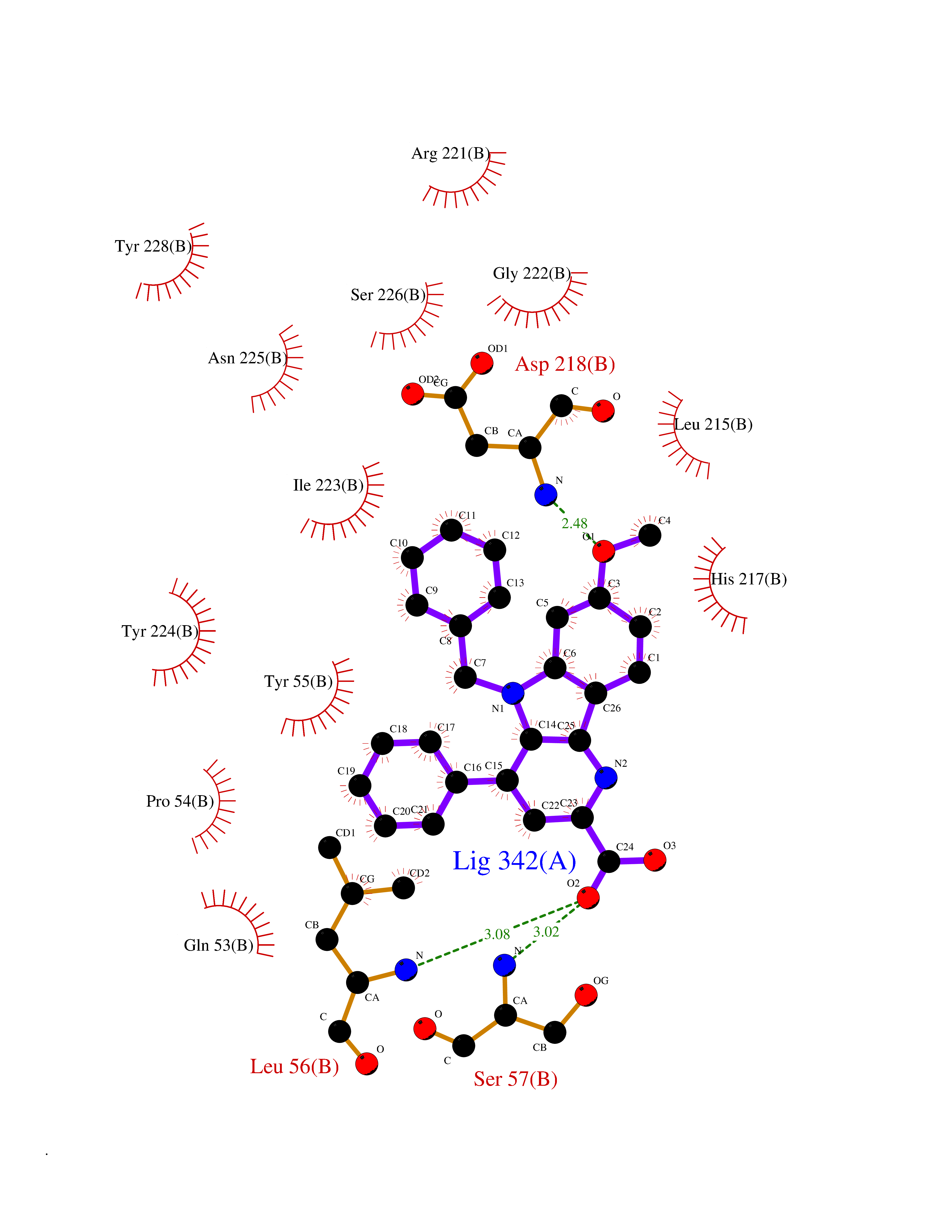 Ligplot Map 3D Binding mode Sequence MRVVVIGAGVIGLSTALCIHERYHSVLQPLDIKVYADRFTPLTTTDVAAGLWQPYLSDPNNPQEADWSQQTFDYLLSHVHSPNAENLGLFLISGYNLFHEAIPDPSWKDTVLGFRKLTPRELDMFPDYGYGWFHTSLILEGKNYLQWLTERLTERGVKFFQRKVESFEEVAREGADVIVNCTGVWAGALQRDPLLQPGRGQIMKVDAPWMKHFILTHDPERGIYNSPYIIPGTQTVTLGGIFQLGNWSELNNIQDHNTIWEGCCRLEPTLKNARIIGERTGFRPVRPQIRLEREQLRTGPSNTEVIHNYGHGGYGLTIHWGCALEAAKLFGRILEEKKLS Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Nicotinamide N-methyltransferase | 2IIP | 7.71 | |
Target general information Gen name NNMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class Transferase Function Nicotinamide N-methyltransferase activity.Pyridine N-methyltransferase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB00627 Interacts with NA EC number 2.1.1.1 Uniprot keywords 3D-structure; Acetylation; Citrullination; Cytoplasm; Direct protein sequencing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27886.8 Length 251 Aromaticity 0.1 Instability index 40.66 Isoelectric point 5.23 Charge (pH=7) -5.11 2D Binding mode Binding energy (Kcal/mol) -10.51  Molscript Map 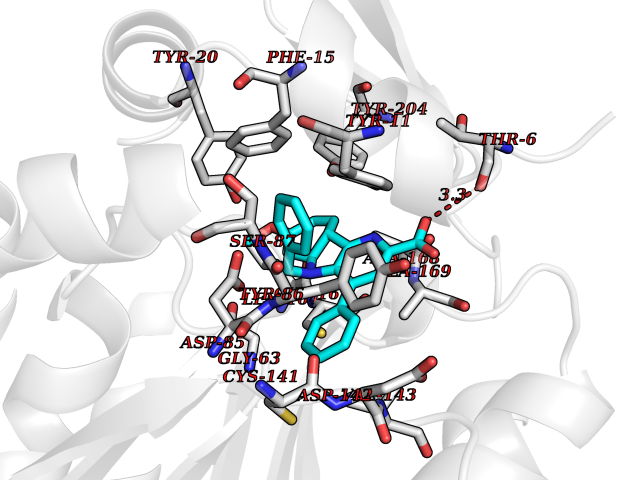 Pymol Map  Ligplot Map 3D Binding mode Sequence GFTSKDTYLSHFNPRDYLEKYYSAESQILKHLLKNLFKIFCLDGVKGDLLIDIGSGPTIYQLLSACESFKEIVVTDYSDQNLQELEKWLKAAPAAFDWSPVVTYVCDLEGNRVKGPEKEEKLRQAVKQVLKCDVTQSQPLGAVPLPPADCVLSTLCLDAACPDLPTYCRALRNLGSLLKPGGFLVIMDALKSSYYMIGEQKFSSLPLGREAVEAAVKEAGYTIEWFEVISQSYSSTMANNEGLFSLVARKL Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | NDR1 protein kinase (STK38) | 6BXI | 7.71 | |
Target general information Gen name STK38 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein kinase 38; Nuclear Dbf2-related kinase 1; NDR1 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family Biochemical class Kinase Function Converts MAP3K2 from its phosphorylated form to its non-phosphorylated form and inhibits autophosphorylation of MAP3K2. Negative regulator of MAP3K1/2 signaling. Related diseases Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:8955068}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 3 (NS3) [MIM:609942]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:16773572, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:17468812, ECO:0000269|PubMed:19396835, ECO:0000269|PubMed:20949621}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gastric cancer (GASC) [MIM:613659]: A malignant disease which starts in the stomach, can spread to the esophagus or the small intestine, and can extend through the stomach wall to nearby lymph nodes and organs. It also can metastasize to other parts of the body. The term gastric cancer or gastric carcinoma refers to adenocarcinoma of the stomach that accounts for most of all gastric malignant tumors. Two main histologic types are recognized, diffuse type and intestinal type carcinomas. Diffuse tumors are poorly differentiated infiltrating lesions, resulting in thickening of the stomach. In contrast, intestinal tumors are usually exophytic, often ulcerating, and associated with intestinal metaplasia of the stomach, most often observed in sporadic disease. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:7773929}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in KRAS are a cause of pylocytic astrocytoma (PA). Pylocytic astrocytomas are neoplasms of the brain and spinal cord derived from glial cells which vary from histologically benign forms to highly anaplastic and malignant tumors. {ECO:0000269|PubMed:16247081}.; DISEASE: Cardiofaciocutaneous syndrome 2 (CFC2) [MIM:615278]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. CFC2 patients often do not have the skin abnormalities, such as ichthyosis, hyperkeratosis, and hemangioma observed in CFC1. {ECO:0000269|PubMed:16474404, ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:20949621, ECO:0000269|PubMed:21797849}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: KRAS mutations are involved in cancer development. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:1553789, ECO:0000269|PubMed:16533793, ECO:0000269|PubMed:24623306, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:3627975, ECO:0000269|PubMed:6092920, ECO:0000269|PubMed:6695174, ECO:0000269|PubMed:7773929}.; DISEASE: Oculoectodermal syndrome (OES) [MIM:600268]: A syndrome characterized by the association of epibulbar dermoids and aplasia cutis congenita. Affected individuals show multiple, asymmetric, atrophic, non-scarring and hairless regions that may be associated with hamartomas. Ectodermal changes include linear hyperpigmentation that may follow the lines of Blaschko and rarely epidermal nevus-like lesions. Epibulbar dermoids may be uni-or bilateral. Additional ocular anomalies such as skin tags of the upper eyelid, rarely optic nerve or retinal changes, and microphthalmia can be present. The phenotypic expression is highly variable, and various other abnormalities have occasionally been reported including growth failure, lymphedema, cardiovascular defects, as well as neurodevelopmental symptoms like developmental delay, epilepsy, learning difficulties, and behavioral abnormalities. Benign tumor-like lesions such as nonossifying fibromas of the long bones and giant cell granulomas of the jaws have repeatedly been observed and appear to be age-dependent, becoming a common manifestation in individuals aged 5 years or older. {ECO:0000269|PubMed:25808193, ECO:0000269|PubMed:26970110, ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Schimmelpenning-Feuerstein-Mims syndrome (SFM) [MIM:163200]: A disease characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects. Many oral manifestations have been reported, not only including hypoplastic and malformed teeth, and mucosal papillomatosis, but also ankyloglossia, hemihyperplastic tongue, intraoral nevus, giant cell granuloma, ameloblastoma, bone cysts, follicular cysts, oligodontia, and odontodysplasia. Sebaceous nevi follow the lines of Blaschko and these can continue as linear intraoral lesions, as in mucosal papillomatosis. {ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with P49407; P32121; Q8N9N5-2; Q03135; P08238; Q9H8S9; Q70IA6; P16333; P30086; P02638 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Chromosome; Cytoplasm; Direct protein sequencing; DNA damage; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 38701.1 Length 333 Aromaticity 0.11 Instability index 38.05 Isoelectric point 5.69 Charge (pH=7) -7.8 2D Binding mode Binding energy (Kcal/mol) -10.52  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence TRLGLEDFESLKVIGRGAFGEVRLVQKKDTGHVYAMKILRKADMLEKEQVGHIRAERDILVEADSLWVVKMFYSFQDKLNLYLIMEFLPGGDMMTLLMKKDTLTEEETQFYIAETVLAIDSIHQLGFIHRDIKPDNLLLDSKGHVKLSDFGLCTGLKKAHRTEFYRNLNHSLPSDFTFQNMNSKRKAETWKRNRRQLAFSTVGTPDYIAPEVFMQTGYNKLCDWWSLGVIMYEMLIGYPPFCSETPQETYKKVMNWKETLTFPPEVPISEKAKDLILRFCCEWEHRIGAPGVEEIKSNSFFEGVDWEHIRERPAAISIEIKSIDDTSNFDEFP Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | BUB1 mitotic checkpoint serine/threonine kinase (BUB1) | 6F7B | 7.71 | |
Target general information Gen name BUB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hBUB1; Mitotic checkpoint serine/threonine-protein kinase BUB1; BUB1L; BUB1A Protein family Protein kinase superfamily, Ser/Thr protein kinase family, BUB1 subfamily Biochemical class Kinase Function Has a key role in the assembly of checkpoint proteins at the kinetochore, being required for the subsequent localization of CENPF, BUB1B, CENPE and MAD2L1. Required for the kinetochore localization of PLK1. Required for centromeric enrichment of AUKRB in prometaphase. Plays an important role in defining SGO1 localization and thereby affects sister chromatid cohesion. Acts as a substrate for anaphase-promoting complex or cyclosome (APC/C) in complex with its activator CDH1 (APC/C-Cdh1). Necessary for ensuring proper chromosome segregation and binding to BUB3 is essential for this function. Can regulate chromosome segregation in a kinetochore-independent manner. Can phosphorylate BUB3. The BUB1-BUB3 complex plays a role in the inhibition of APC/C when spindle-assembly checkpoint is activated and inhibits the ubiquitin ligase activity of APC/C by phosphorylating its activator CDC20. This complex can also phosphorylate MAD1L1. Kinase activity is essential for inhibition of APC/CCDC20 and for chromosome alignment but does not play a major role in the spindle-assembly checkpoint activity. Mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. Serine/threonine-protein kinase that performs 2 crucial functions during mitosis: it is essential for spindle-assembly checkpoint signaling and for correct chromosome alignment. Related diseases Microcephaly 30, primary, autosomal recessive (MCPH30) [MIM:620183]: A form of microcephaly, a disease defined as a head circumference more than 3 standard deviations below the age, sex and ethnically matched mean. Brain weight is markedly reduced and the cerebral cortex is disproportionately small. MCPH30 is characterized by small head, poor overall growth, and global developmental delay with variably impaired intellectual development. Affected individuals may also have variable congenital anomalies, including atrial septal defect, dysmorphic facial features, tracheal stenosis, and anomalies of the skin and teeth. {ECO:0000269|PubMed:35044816}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O95376; O60566; O43684; P46108; Q8NG31; Q8NG31-2; Q9GZQ8; P03070 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell cycle; Cell division; Centromere; Chromosome; Chromosome partition; Host-virus interaction; Intellectual disability; Kinase; Kinetochore; Mitosis; Nucleotide-binding; Nucleus; Phosphoprotein; Primary microcephaly; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39564.8 Length 343 Aromaticity 0.12 Instability index 31.17 Isoelectric point 8.49 Charge (pH=7) 4.7 2D Binding mode Binding energy (Kcal/mol) -10.51 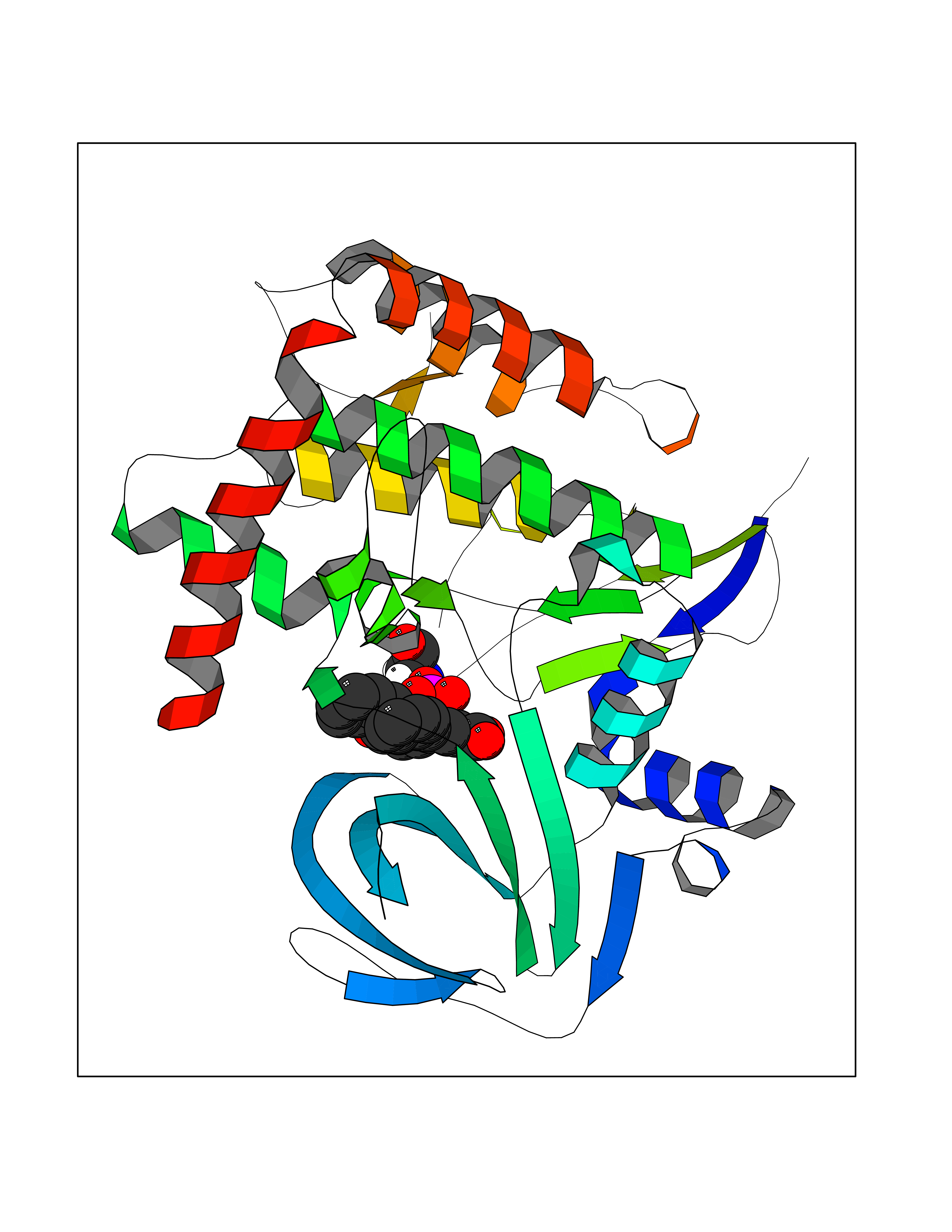 Molscript Map 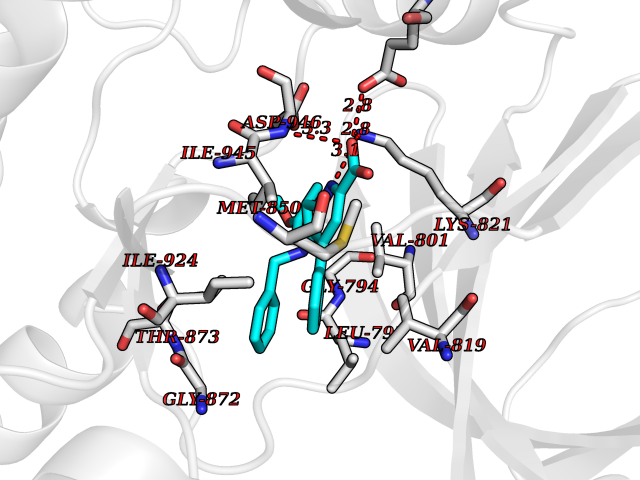 Pymol Map 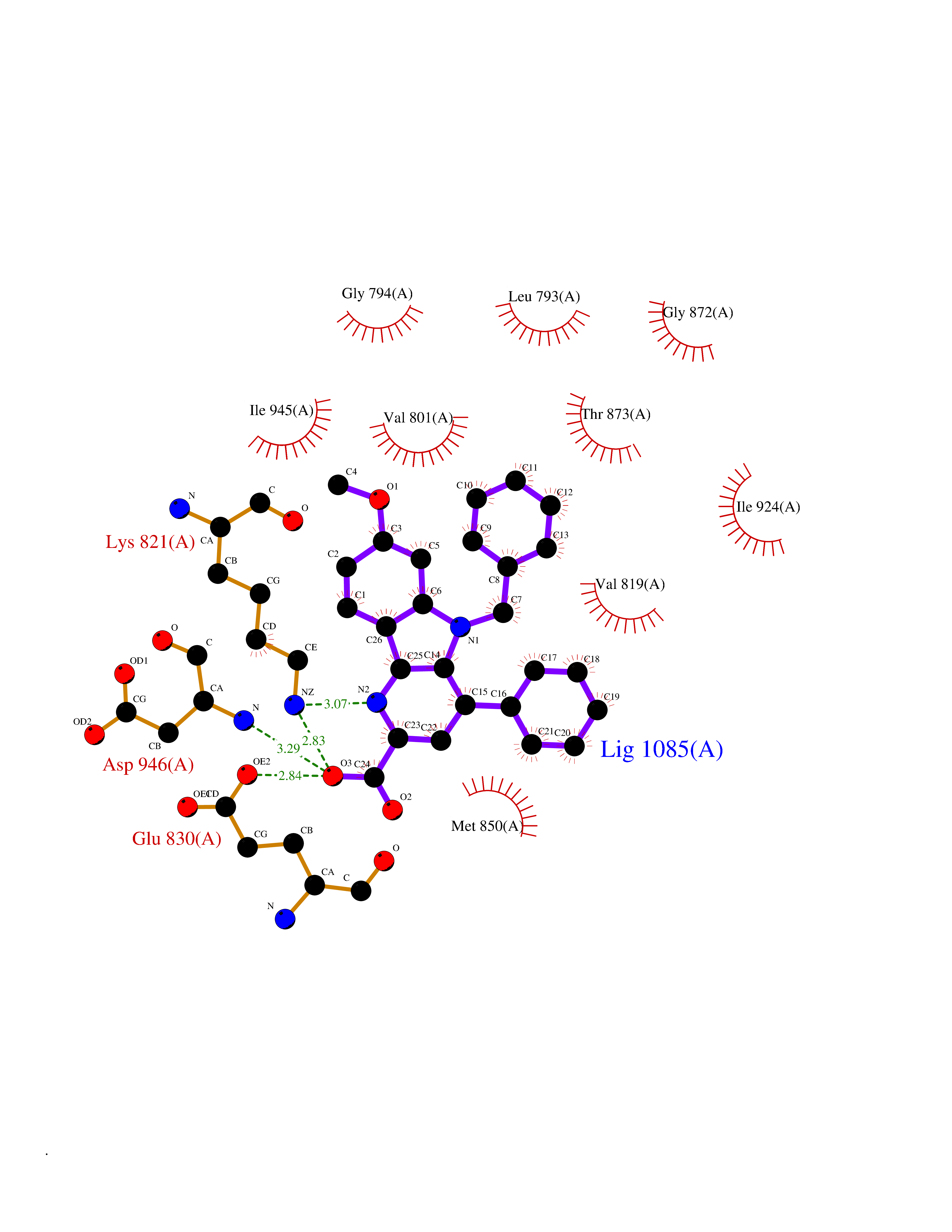 Ligplot Map 3D Binding mode Sequence APNFIVGNPWDDKLIFKLLSGLSKPVSSYPNTFEWQCKLPAIKPKTEFQLGSKLVYVHHLLGEGAFAQVYEATQKNKQKFVLKVQKPANPWEFYIGTQLMERLKPSMQHMFMKFYSAHLFQNGSVLVGELYSYGTLLNAINLYKNTPEKVMPQGLVISFAMRMLYMIEQVHDCEIIHGDIKPDNFILGNGFLEQDDEDDLSAGLALIDLGQSIDMKLFPKGTIFTAKCETXGFQCVEMLSNKPWNYQIDYFGVAATVYCMLFGTYMKVKNEECKPEGLFRRLPHLDMWNEFFHVMLNIPDCHHLPSLDLLRQKLKKVFQQHYTNKIRALRNRLIVLLLECKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Phosphodiesterase 3A (PDE3A) | 7EG0 | 7.70 | |
Target general information Gen name PDE3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-inhibited 3',5'-cyclic phosphodiesterase A; Phosphodiesterase 3A; Cyclic GMP-inhibited phosphodiesterase A; Cyclic GMP inhibited phosphodiesterase A; CGI-PDE A Protein family Cyclic nucleotide phosphodiesterase family, PDE3 subfamily Biochemical class Phosphoric diester hydrolase Function Cyclic nucleotide phosphodiesterase with a dual-specificity for the second messengers cAMP and cGMP, which are key regulators of many important physiological processes. Related diseases Hypertension and brachydactyly syndrome (HTNB) [MIM:112410]: A syndrome characterized by brachydactyly type E, severe salt-independent but age-dependent hypertension, an increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, and altered baroreflex blood pressure regulation. It results in death from stroke before age 50 years when untreated. Brachydactyly type E is characterized by shortening of the fingers mainly in the metacarpals and metatarsals. {ECO:0000269|PubMed:25961942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01223; DB01427; DB00261; DB00201; DB01166; DB04880; DB05266; DB00922; DB00235; DB01303; DB00277; DB08811; DB09283 Interacts with Q9Y6D6; Q9Y6D5 EC number EC 3.1.4.17 Uniprot keywords 3D-structure; cAMP; cGMP; Cytoplasm; Direct protein sequencing; Disease variant; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 108171 Length 939 Aromaticity 0.11 Instability index 39.75 Isoelectric point 6.53 Charge (pH=7) -4.15 2D Binding mode Binding energy (Kcal/mol) -10.5 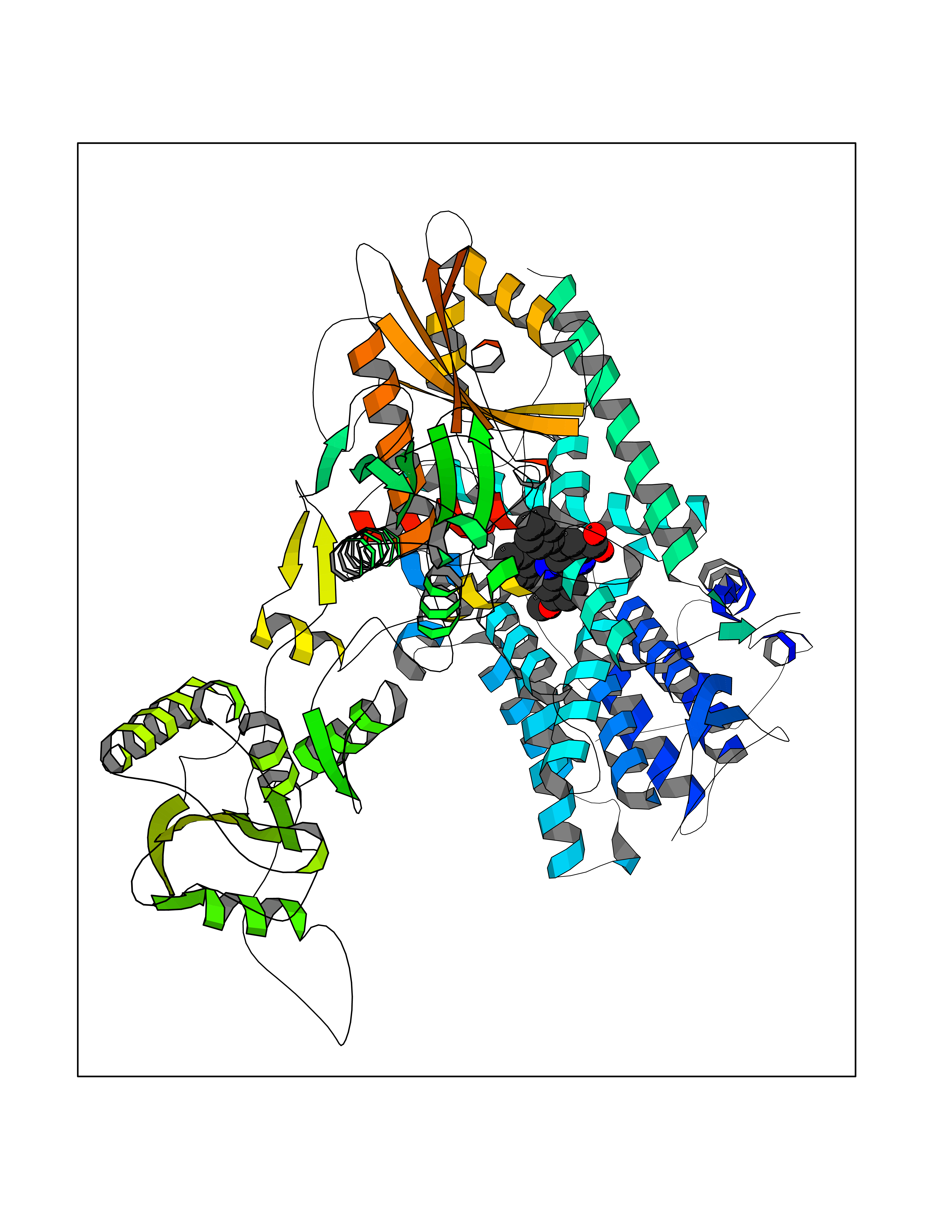 Molscript Map 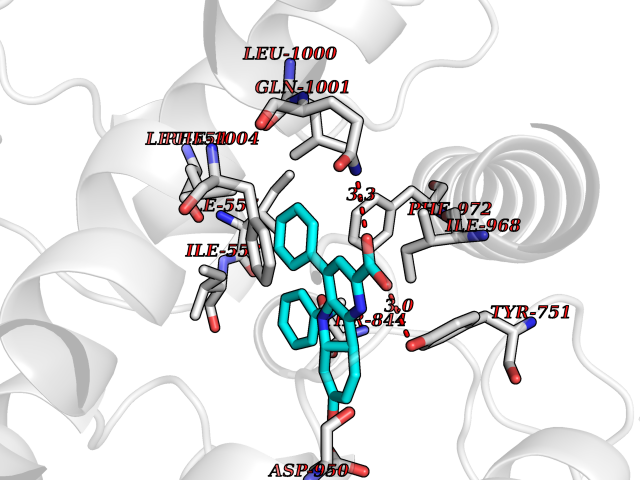 Pymol Map 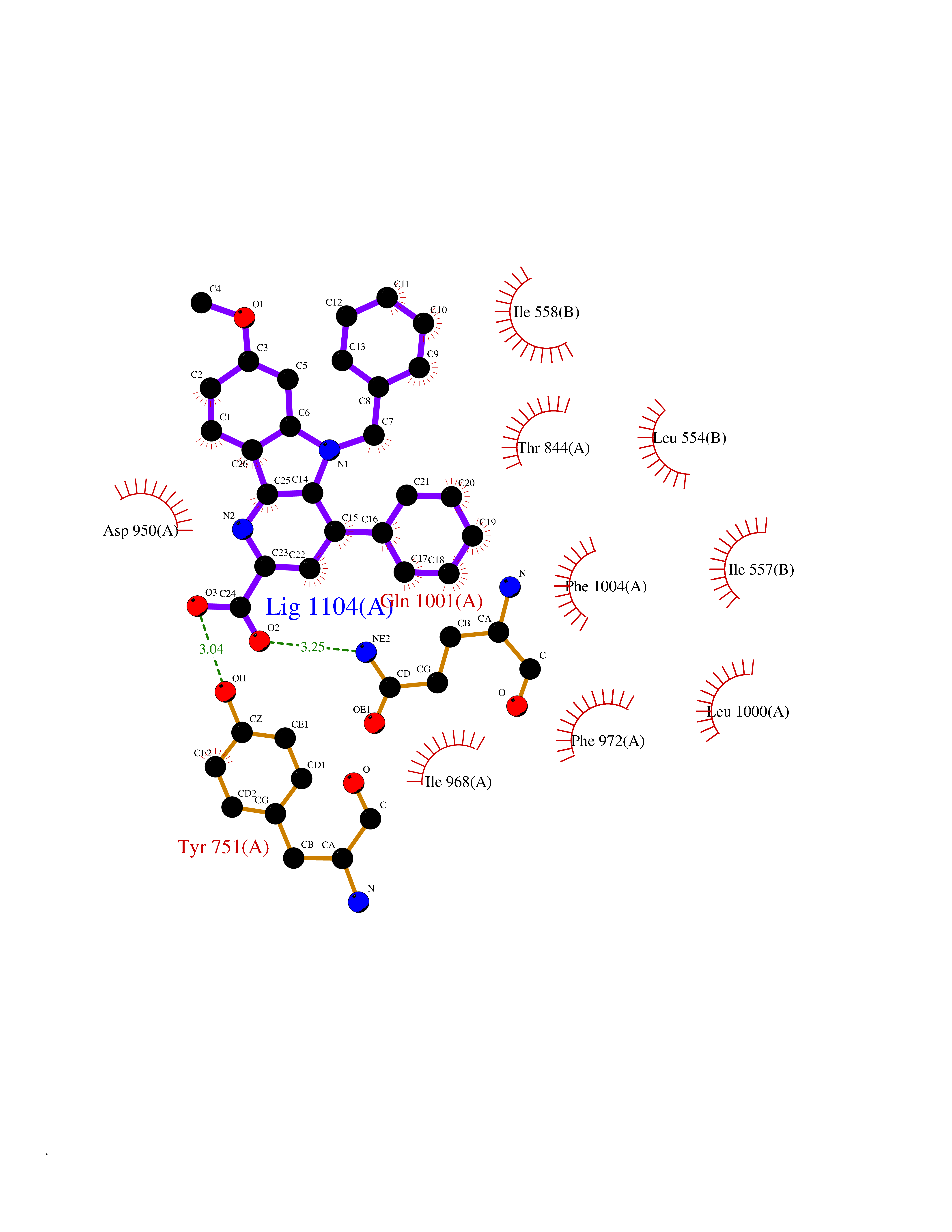 Ligplot Map 3D Binding mode Sequence KPILAPEPLVMDNLDSIMEQLNTWNFPIFDLVENIGRKCGRILSQVSYRLFEDMGLFEAFKIPIREFMNYFHALEIGYRDIPYHNRIHATDVLHAVWYLTTQPIPGLSTVGYVFSKTYNVTDDKYGCLSGNIPALELMALYVAAAMHDYDHPGRTNAFLVATSAPQAVLYNDRSVLENHHAAAAWNLFMSRPEYNFLINLDHVEFKHFRFLVIEAILATDLKKHFDFVAKFNGKVNDDVGIDWTNENDRLLVCQMCIKLADINGPAKCKELHLQWTDGIVNEFYEQGDEEASLGLPISPFMDRSAPQLANLQESFISHIVGPLCNSYDSAGLMPGKWVEKIYCQITQHLLQNHKMWKKVIEEEQRLAGIENQNISVDLETNYAELVLDVGRVTLGENSRKKMKDCKLRKKQNESVSRAMCALLNSGGGVIKAEIENEDYSYTKDGIGLDLENSFSNILLFVPEYLDFMQNGNYFLIFVKSWSLNTSGLRITTLSSNLYKRDITSAKVMNATAALEFLKDMKKTRGRLYLRPELLAKRPCVDIQEENNMKALAGVFFDRTELDRKEKLTFTESTHVEIKNFSTERLLQRIKEILPQYVSAFANTDGGYLFIGLNEDKEIIGFKAEMSDLDDLEREIEKSIRKMPVHHFCMEKKKINYSCKFLGVYDKGSLCGYVCALRVERFCCAVFAKEPDSWHVKDNRVMQLTRKEWIQFMVEAEPKFSSAYEEVISQINTSLPAPHSWPLLEWQRQRHHCPGLSGRITYTPENLCRKLFLQHEGLKQLICEEMSSVRKGSLIFSRSWSVDLGLQENHKVLCDALLISQDSPPVLYTFHMVQDEEFKGYSTQTALTLKQKLAKIGGYTKKVCVMTKIFYLSPEGMTSCQYDLRSQVIYPESYYFTRRKYLLKALFKALKRLKSLRDQFSFAENLYQIIGIDCFQKNDK Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Kynurenine 3-hydroxylase (KMO) | 5X68 | 7.69 | |
Target general information Gen name KMO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Kynurenine 3monooxygenase; Kynurenine 3-monooxygenase Protein family Aromatic-ring hydroxylase family, KMO subfamily Biochemical class Paired donor oxygen oxidoreductase Function Required for synthesis of quinolinic acid, a neurotoxic NMDA receptor antagonist and potential endogenous inhibitor of NMDA receptor signaling in axonal targeting, synaptogenesis and apoptosis during brain development. Quinolinic acid may also affect NMDA receptor signaling in pancreatic beta cells, osteoblasts, myocardial cells, and the gastrointestinal tract. Catalyzes the hydroxylation of L-kynurenine (L-Kyn) to form 3-hydroxy-L-kynurenine (L-3OHKyn). Related diseases Hirschsprung disease, cardiac defects, and autonomic dysfunction (HCAD) [MIM:613870]: A disorder characterized by skip-lesions Hirschsprung disease, craniofacial abnormalities and other dysmorphic features, cardiac defects including ductus arteriosus, small subaortic ventricular septal defect, small atrial septal defect, and autonomic dysfunction. {ECO:0000269|PubMed:9915973}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P42858 EC number EC 1.14.13.9 Uniprot keywords 3D-structure; Alternative splicing; FAD; Flavoprotein; Glycoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Monooxygenase; NADP; Oxidoreductase; Proteomics identification; Pyridine nucleotide biosynthesis; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 39424.3 Length 353 Aromaticity 0.1 Instability index 36.2 Isoelectric point 6.39 Charge (pH=7) -2.42 2D Binding mode Binding energy (Kcal/mol) -10.48 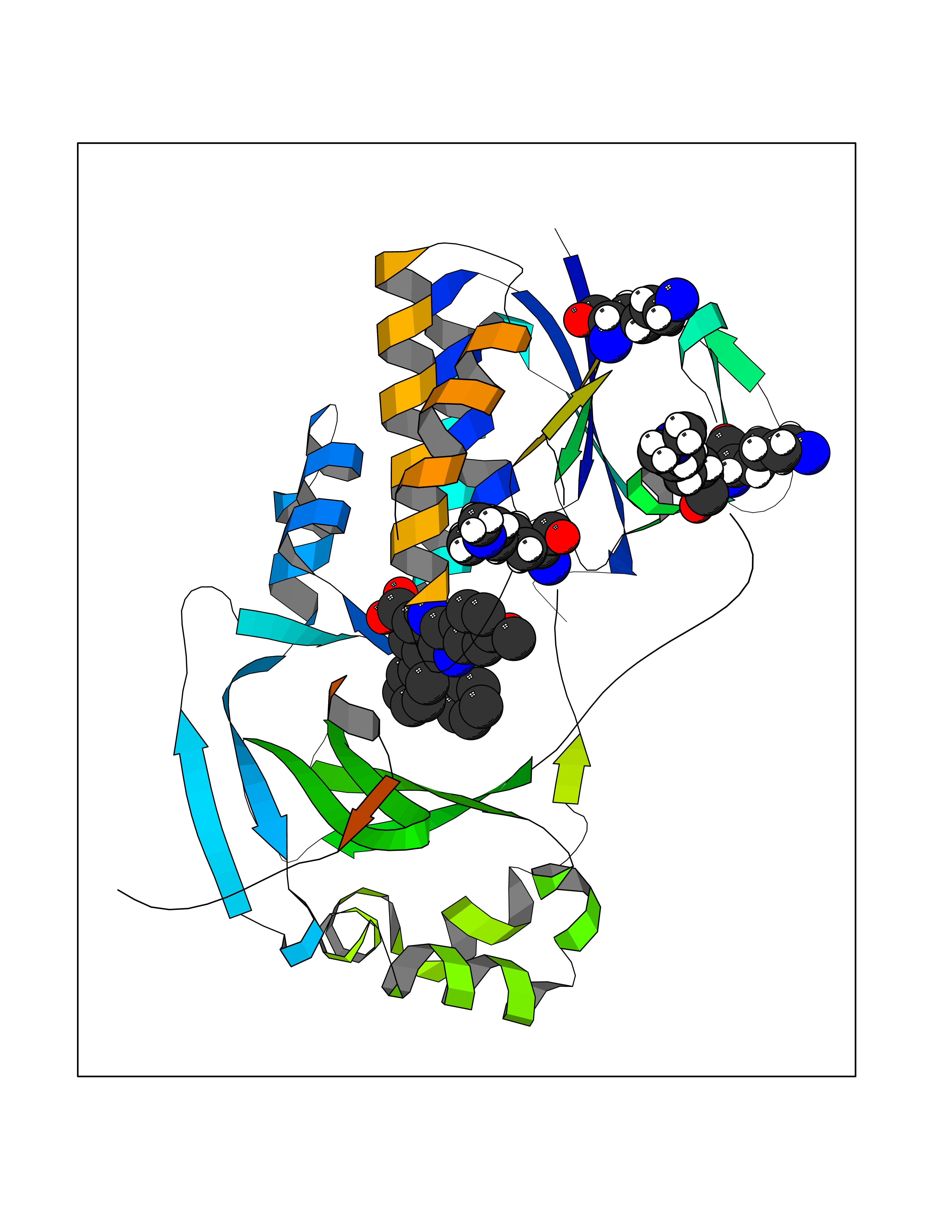 Molscript Map 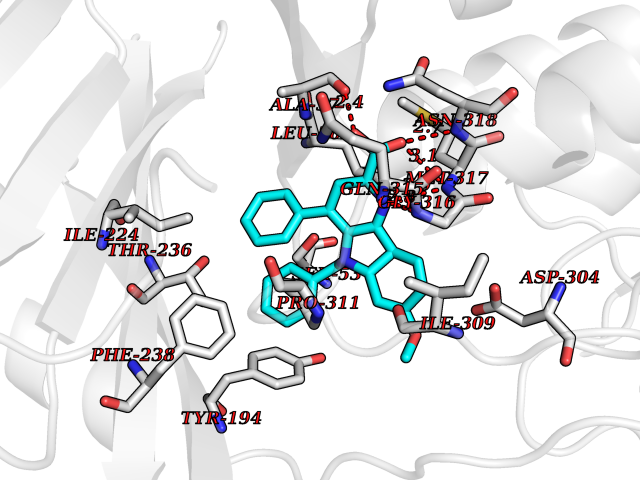 Pymol Map 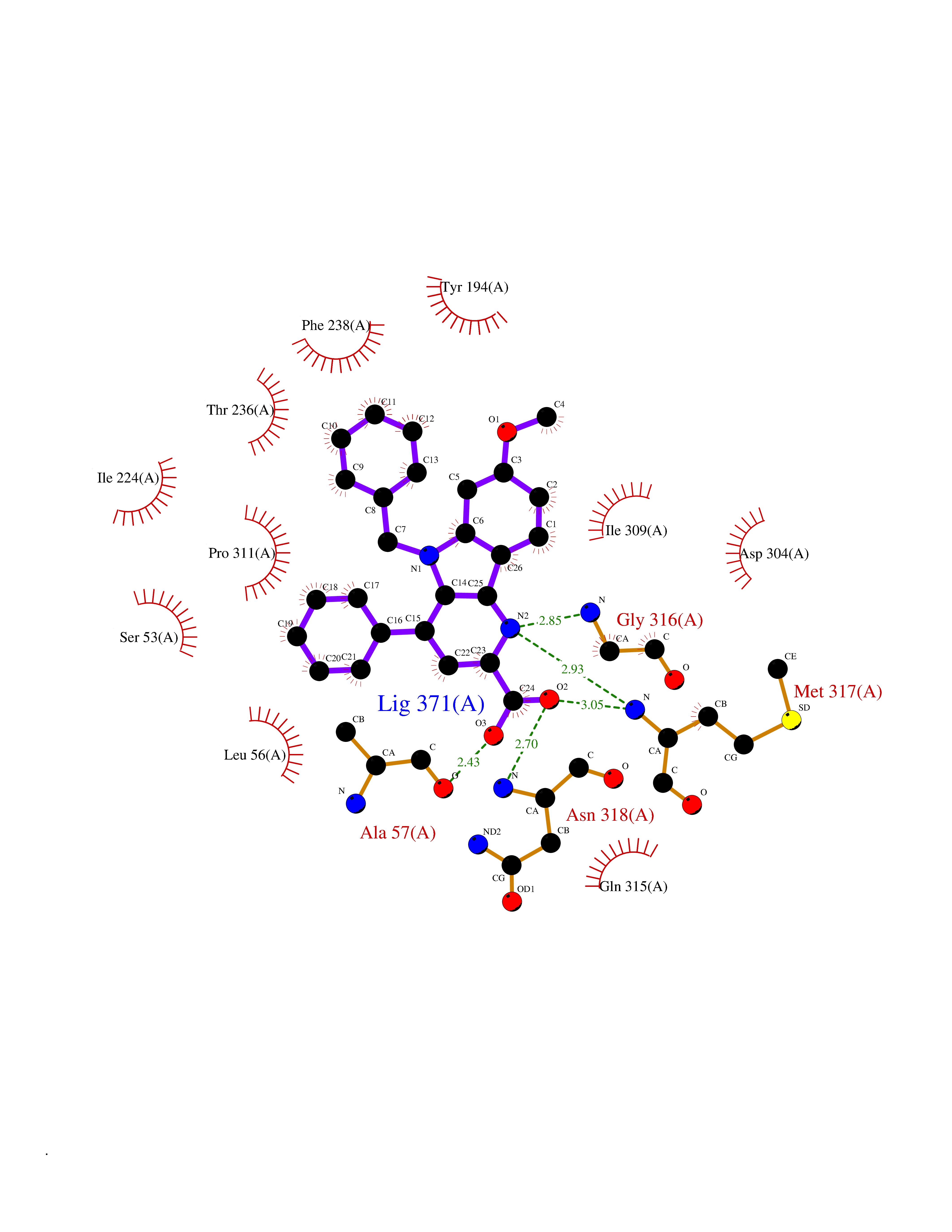 Ligplot Map 3D Binding mode Sequence RKKVAVIGGGLVGSLQACFLAKRNFQIDVYEAREDTRVASINLALSHRGRQALKAVGLEDQIVSQGIPMRARMIHSLSGKKSAIPYGTKSQYILSVSRENLNKDLLTAAEKYPNVKMHFNHRLLXCNPEEGMITVLGSDKVPKDVTCDLIVGCDGAYSTVRSHLMXKPRFDYSQQYIPHGYMELTIPPKNGDYAMEPNYLHIWPRNTFMMIALPNMNKSFTCTLFMPFEEFEKLLTSNDVVDFFQKYFPDAIPLIGEKLLVQDFFLLPAQPMISVXCSSFHFXSHCVLLGDAAHAIVPFFGQGMNAGFEDCLVFDELMDKFSNDLSLCLPVFSRLRIPDDSDLSMYNYIEMRA Hydrogen bonds contact Hydrophobic contact | ||||