Job Results:
Ligand
Structure
Job ID
7eae9f0de32fdedcd28c1404448b5ea8
Job name
NA
Time
2025-04-07 15:33:07
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Acetyl-CoA carboxylase 1 | 2YL2 | 4.28 | |
Target general information Gen name ACACA Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACAC;ACCA;ACC1 Protein family NA Biochemical class Ligase Function Acetyl-CoA carboxylase activity.ATP binding.Biotin carboxylase activity.Identical protein binding.Metal ion binding. Related diseases Acetyl-CoA carboxylase-alpha deficiency (ACACAD) [MIM:613933]: An autosomal recessive inborn error of de novo fatty acid synthesis associated with severe brain damage, persistent myopathy and poor growth. {ECO:0000269|PubMed:6114432}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00121 Interacts with Q13085; O60218; P38398; Q96EB6; Q9CQ20; P02654; Q92915-2; Q6NTF9-3 EC number 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative promoter usage; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Magnesium; Manganese; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 54237.7 Length 486 Aromaticity 0.09 Instability index 39.18 Isoelectric point 6.37 Charge (pH=7) -2.46 2D Binding mode Binding energy (Kcal/mol) -5.84 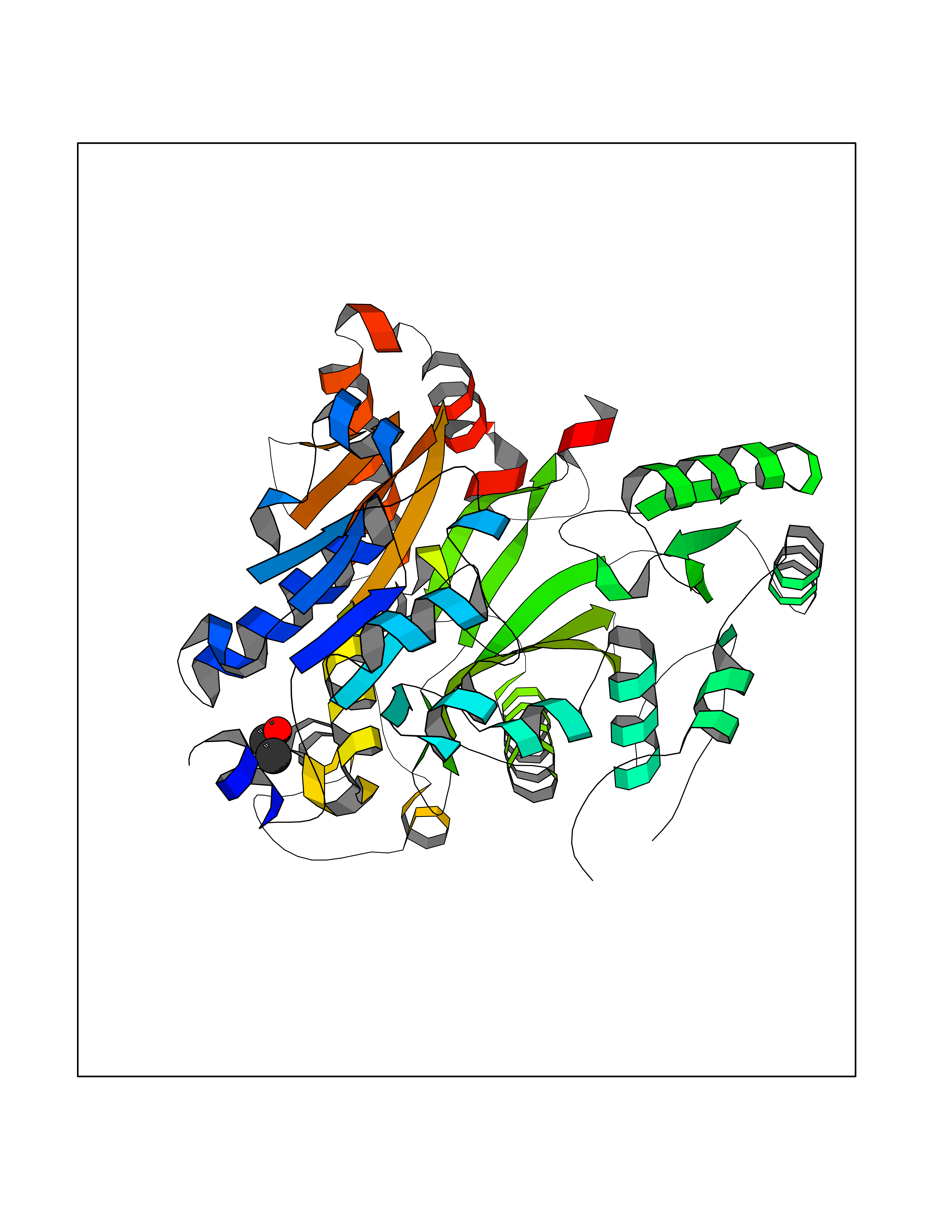 Molscript Map 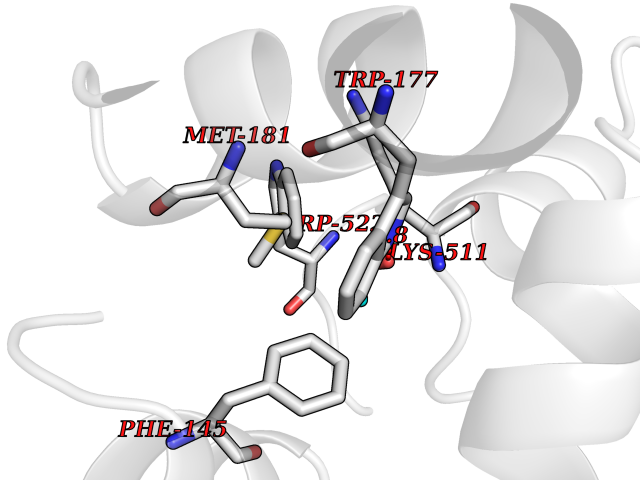 Pymol Map 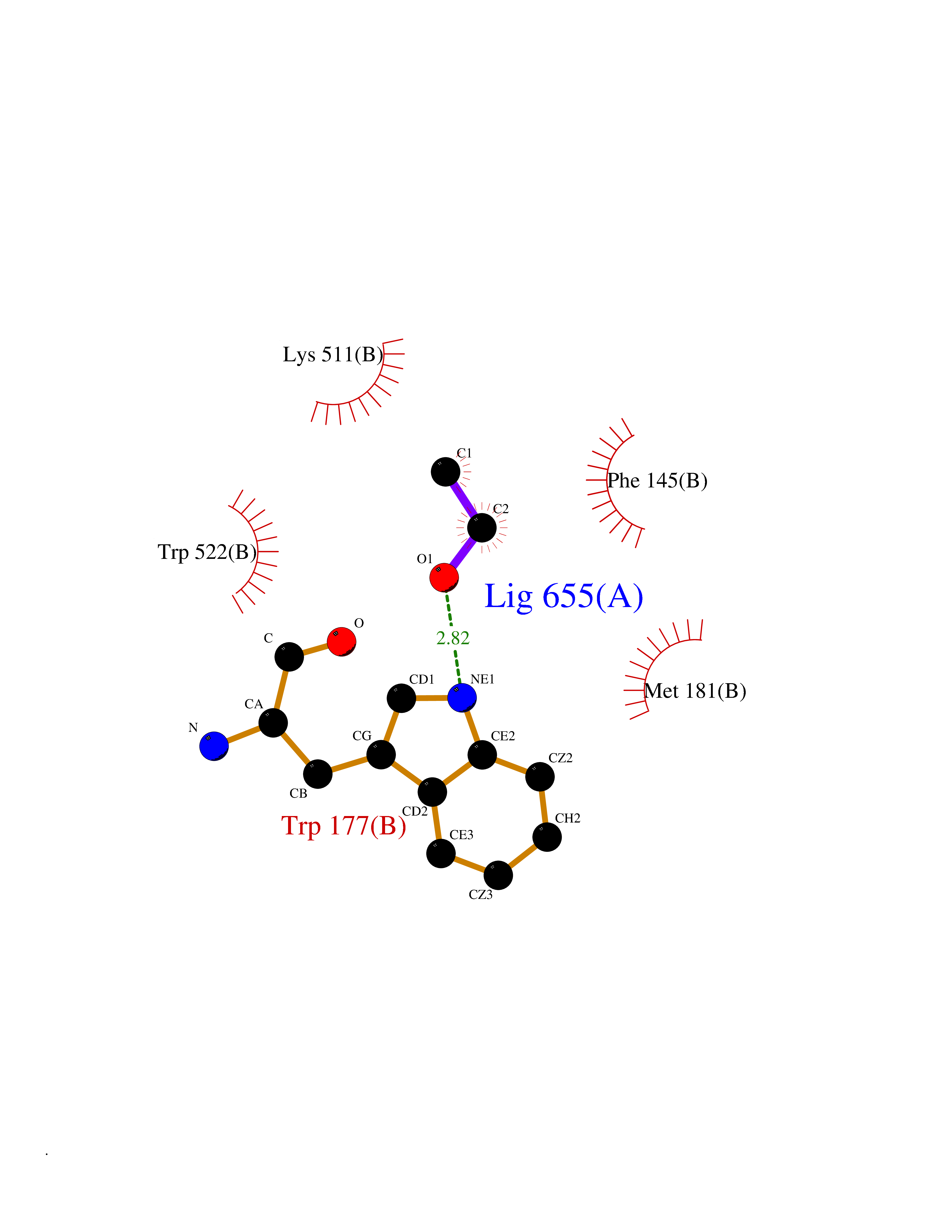 Ligplot Map 3D Binding mode Sequence VASPAEFVTRFGGNKVIEKVLIANNGIAAVKCMRSIRRWSYEMFRNERAIRFVVMVTPEDLKANAEYIKMADHYVPVPGGPNNNNYANVELILDIAKRIPVQAVWAGWGHASENPKLPELLLKNGIAFMGPPSQAMWALGDKIASSIVAQTAGIPTLPWSGSGLRVDWSKRILNVPQELYEKGYVKDVDDGLQAAEEVGYPVMIKASEGGGGKGIRKVNNADDFPNLFRQVQAEVPGSPIFVMRLAKQSRHLEVQILADQYGNAISLFGRDCSVQRRHQKIIEEAPATIATPAVFEHMEQCAVKLAKMVGYVSAGTVEYLYSQDGSFYFLELNPRLQVEHPCTEMVADVNLPAAQLQIAMGIPLYRIKDIRMMYGVSPWGDSPIDFEDSAHVPCPRGHVIAARITGTVQELNFRSNKNVWGYFSVQFGHCFSWGENREEAISNMVVALKELSIRGDFRTTVEYLIKLLETESFQMNRIDTGWLDRL Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Soluble calcium-activated nucleotidase 1 | 1S1D | 4.28 | |
Target general information Gen name CANT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms SHAPY Protein family Apyrase family Biochemical class Hydrolase Function Adenosine-diphosphatase activity.Calcium ion binding.Guanosine-diphosphatase activity.Protein homodimerization activity.Signal transducer activity.Uridine-diphosphatase activity. Related diseases Desbuquois dysplasia 1 (DBQD1) [MIM:251450]: A chondrodysplasia characterized by severe prenatal and postnatal growth retardation (less than -5 SD), joint laxity, short extremities, progressive scoliosis, round face, midface hypoplasia, prominent bulging eyes. The main radiologic features are short long bones with metaphyseal splay, a 'Swedish key' appearance of the proximal femur (exaggerated trochanter), and advance carpal and tarsal bone age. Two forms of Desbuquois dysplasia are distinguished on the basis of the presence or absence of characteristic hand anomalies: an extra ossification center distal to the second metacarpal, delta phalanx, bifid distal thumb phalanx, and phalangeal dislocations. {ECO:0000269|PubMed:19853239, ECO:0000269|PubMed:20425819, ECO:0000269|PubMed:21037275, ECO:0000269|PubMed:21412251, ECO:0000269|PubMed:21654728, ECO:0000269|PubMed:22539336}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epiphyseal dysplasia, multiple, 7 (EDM7) [MIM:617719]: A form of multiple epiphyseal dysplasia, a generalized skeletal dysplasia associated with significant morbidity. Joint pain, joint deformity, waddling gait, and short stature are the main clinical signs and symptoms. Radiological examination of the skeleton shows delayed, irregular mineralization of the epiphyseal ossification centers and of the centers of the carpal and tarsal bones. Multiple epiphyseal dysplasia is broadly categorized into the more severe Fairbank and the milder Ribbing types. The Fairbank type is characterized by shortness of stature, short and stubby fingers, small epiphyses in several joints, including the knee, ankle, hand, and hip. The Ribbing type is confined predominantly to the hip joints and is characterized by hands that are normal and stature that is normal or near-normal. EDM7 inheritance is autosomal recessive. {ECO:0000269|PubMed:28742282}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03486 Interacts with P58418; Q5JX71 EC number 3.6.1.6 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Dwarfism; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Hydrolase; Membrane; Metal-binding; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 35143.9 Length 315 Aromaticity 0.11 Instability index 21.87 Isoelectric point 4.95 Charge (pH=7) -13.67 2D Binding mode Binding energy (Kcal/mol) -5.83 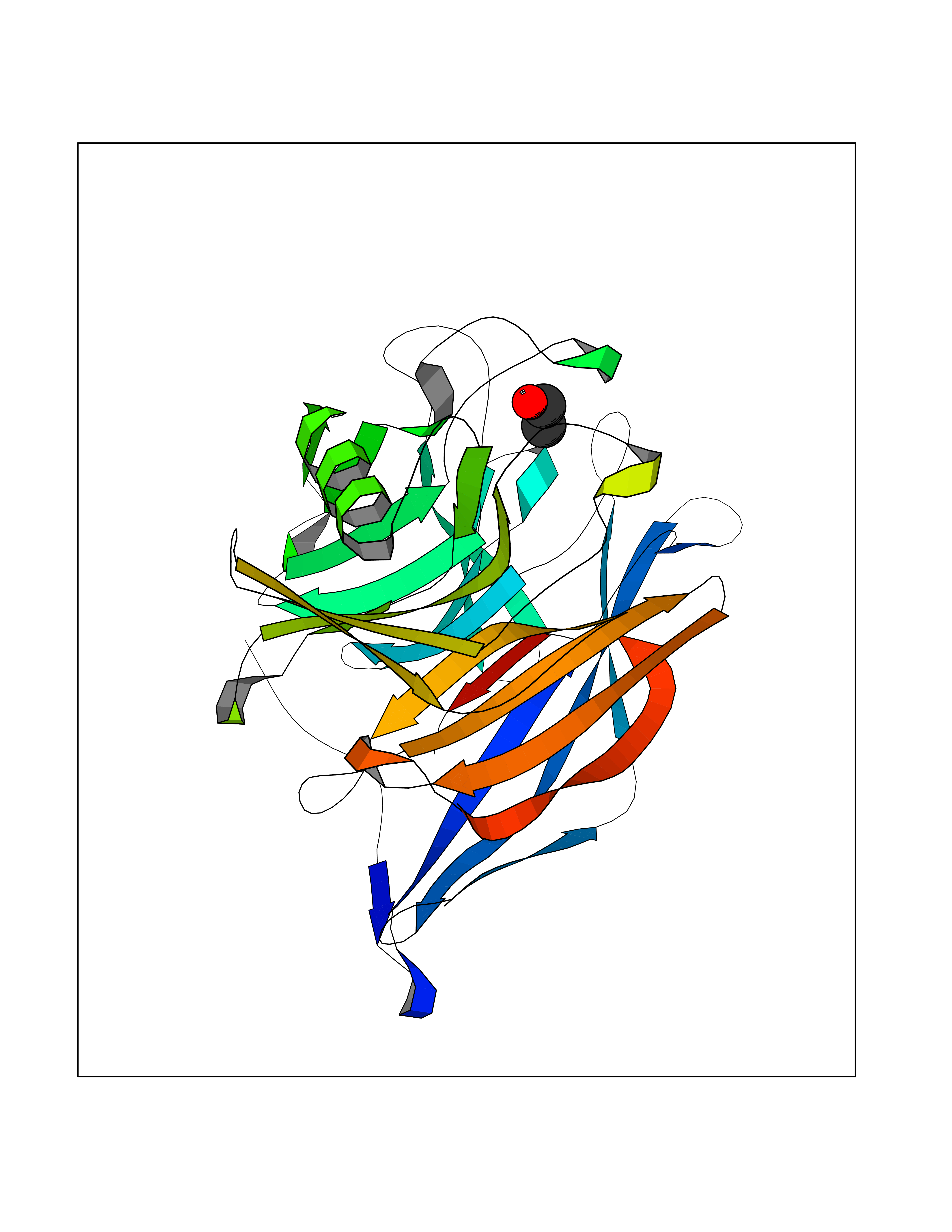 Molscript Map 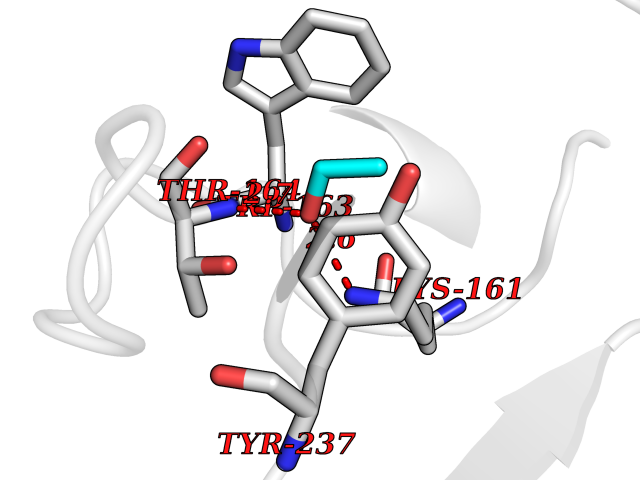 Pymol Map 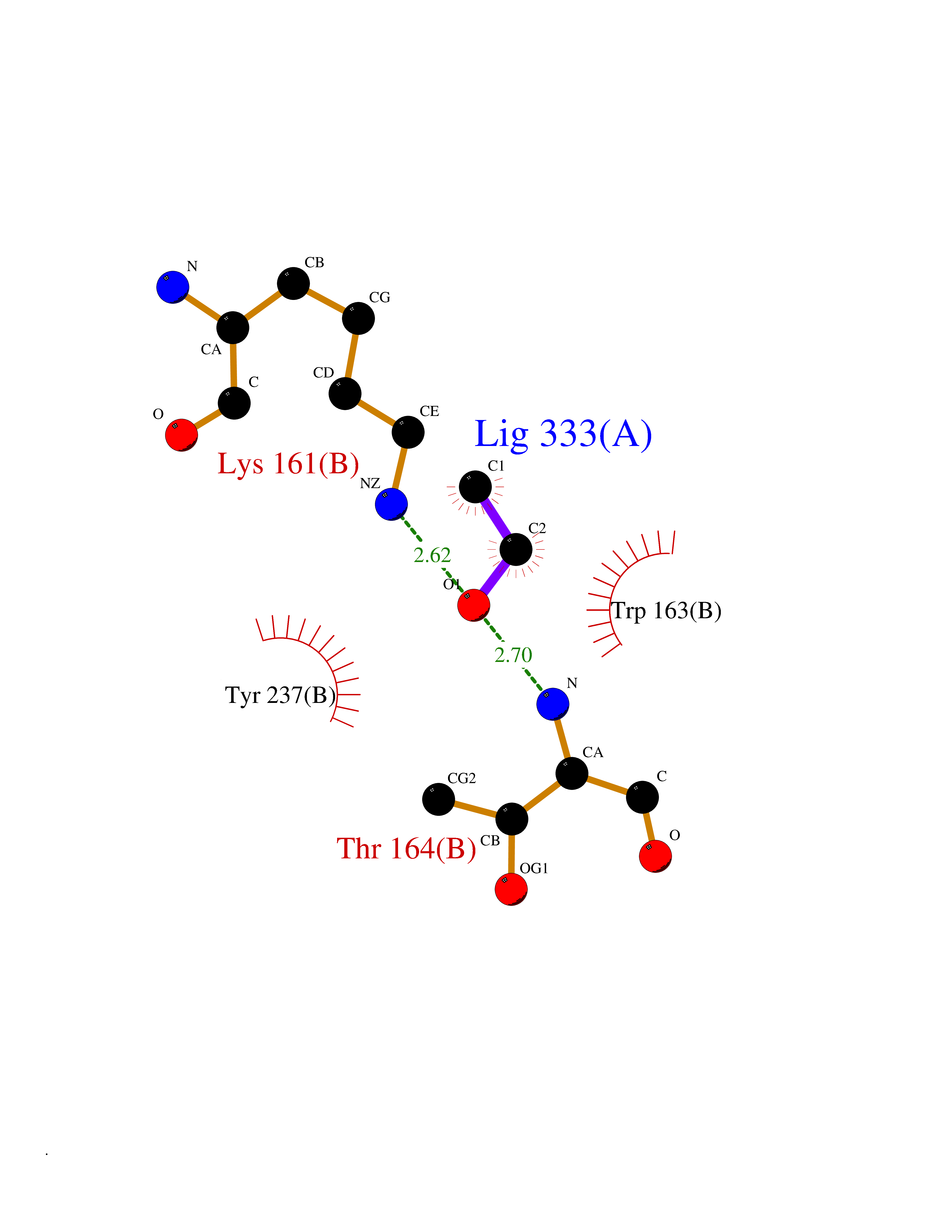 Ligplot Map 3D Binding mode Sequence YNDTYPLSPPQRTPAGIRYRIAVIADLDTESRAQEENTWFSYLKKGYLTLSDSGDKVAVEWDKDHGVLESHLAEKGRGMELSDLIVFNGKLYSVDDRTGVVYQIEGSKAVPWVILSDGDGTVEKGFKAEWLAVKDERLYVGGLGKEWTTTTGDVVNENPEWVKVVGYKGSVDHENWVSNYNALRAAAGIQPPGYLIHESACWSDTLQRWFFLPRRASQERYSEKDDERKGANLLLSASPDFGDIAVSHVGAVVPTHGFSSFKFIPNTDDQIIVALKSEEDSGRVASYIMAFTLDGRFLLPETKIGSVKYEGIEFI Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Protein-arginine deiminase type-6 | 4DAT | 4.28 | |
Target general information Gen name PADI6 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PAD6 Protein family Protein arginine deiminase family Biochemical class Signaling protein / protein binding Function Calcium ion binding.Protein-arginine deiminase activity. Related diseases Oocyte/zygote/embryo maturation arrest 16 (OZEMA16) [MIM:617234]: A rare cause of female primary infertility. In affected women, ovulation and fertilization proceed normally but embryos are arrested at early stages of development. Inheritance is autosomal recessive. {ECO:0000269|PubMed:27545678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00155 Interacts with Q96D03; O95198; O43463; Q8TF50 EC number 3.5.3.15 Uniprot keywords 3D-structure; Calcium; Cytoplasm; Cytoplasmic vesicle; Disease variant; Hydrolase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID B Molecular weight (Da) 25395.5 Length 225 Aromaticity 0.08 Instability index 46.3 Isoelectric point 5 Charge (pH=7) -10.23 2D Binding mode Binding energy (Kcal/mol) -5.84 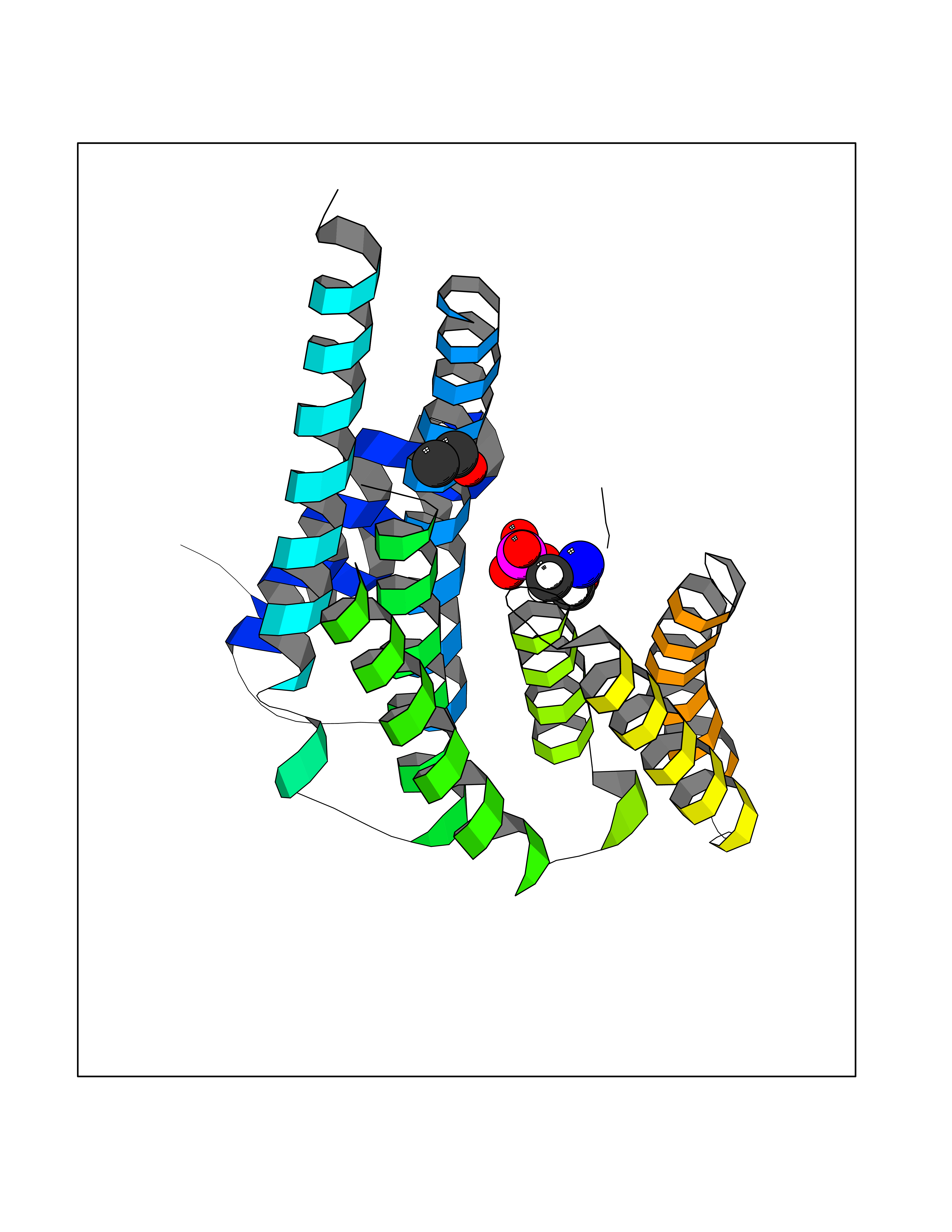 Molscript Map 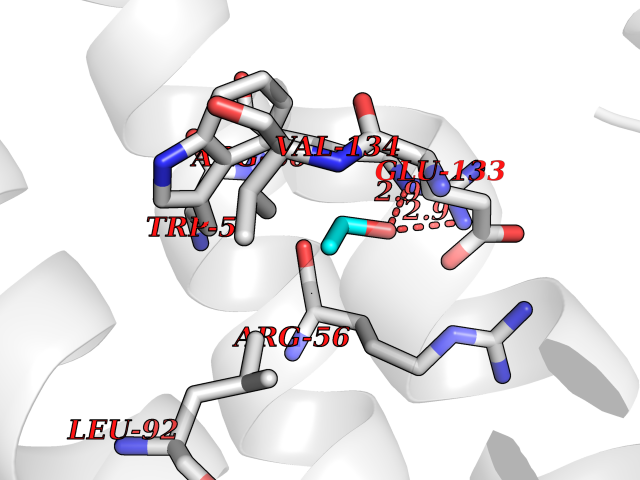 Pymol Map 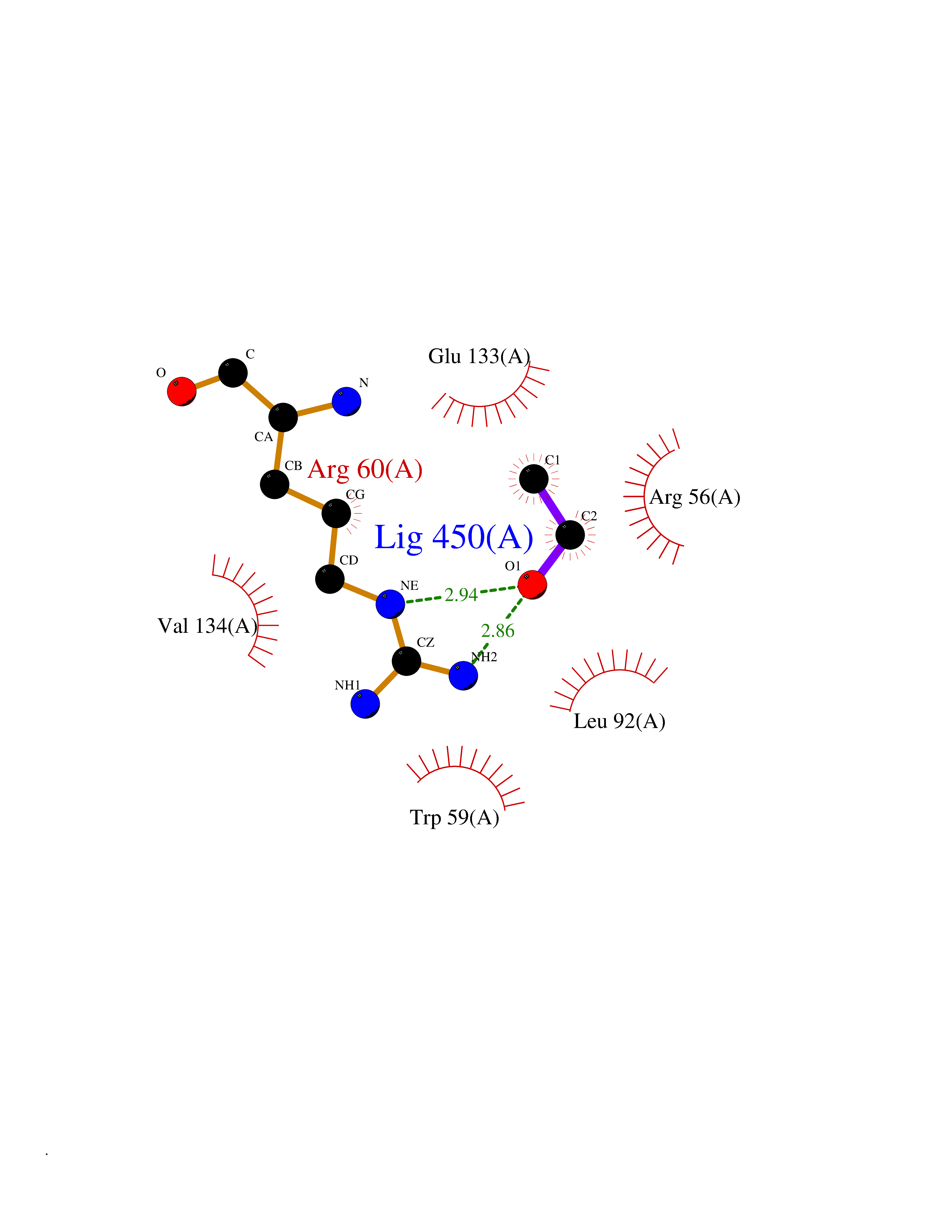 Ligplot Map 3D Binding mode Sequence SMERASLIQKAKLAEQAERYEDMAAFMKGAVEKGEELSCEERNLLSVAYKNVVGGQRAAWRVLSSIEQGPEVREYREKVETELQGVCDTVLGLLDSHLIKEAGDAESRVFYLKMKGDYYRYLAEVATKKRIIDSARSAYQEAMDISKKEMPPTNPIRLGLALNFSVFHYEIANSPEEAISLAKTTFDEAMADLHTLSEDSYKDSTLIMQLLRDNLTLWTFYPXAE Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Cocaine esterase | 3I2K | 4.28 | |
Target general information Gen name cocE Organism Rhodococcus sp. (strain MB1 Bresler) Uniprot ID TTD ID NA Synonyms NA Protein family CocE/NonD hydrolase family Biochemical class Hydrolase Function Carboxylic ester hydrolase activity.Dipeptidyl-peptidase activity. Related diseases Thiamine metabolism dysfunction syndrome 5, episodic encephalopathy type (THMD5) [MIM:614458]: An autosomal recessive metabolic disorder due to an inborn error of thiamine metabolism. The phenotype is highly variable, but in general, affected individuals have onset in early childhood of acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive neurologic dysfunction manifest as gait disturbances, ataxia, dystonia, and spasticity, which in some cases may result in loss of ability to walk. Cognitive function is usually preserved, although mildly delayed development has been reported. These episodes are usually associated with infection and metabolic decompensation. Some patients may have recovery of some neurologic deficits. {ECO:0000269|PubMed:22152682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03793; DB01795 Interacts with NA EC number 3.1.1.84 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Hydrolase; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 62127.9 Length 574 Aromaticity 0.09 Instability index 26.62 Isoelectric point 4.56 Charge (pH=7) -33.24 2D Binding mode Binding energy (Kcal/mol) -5.84 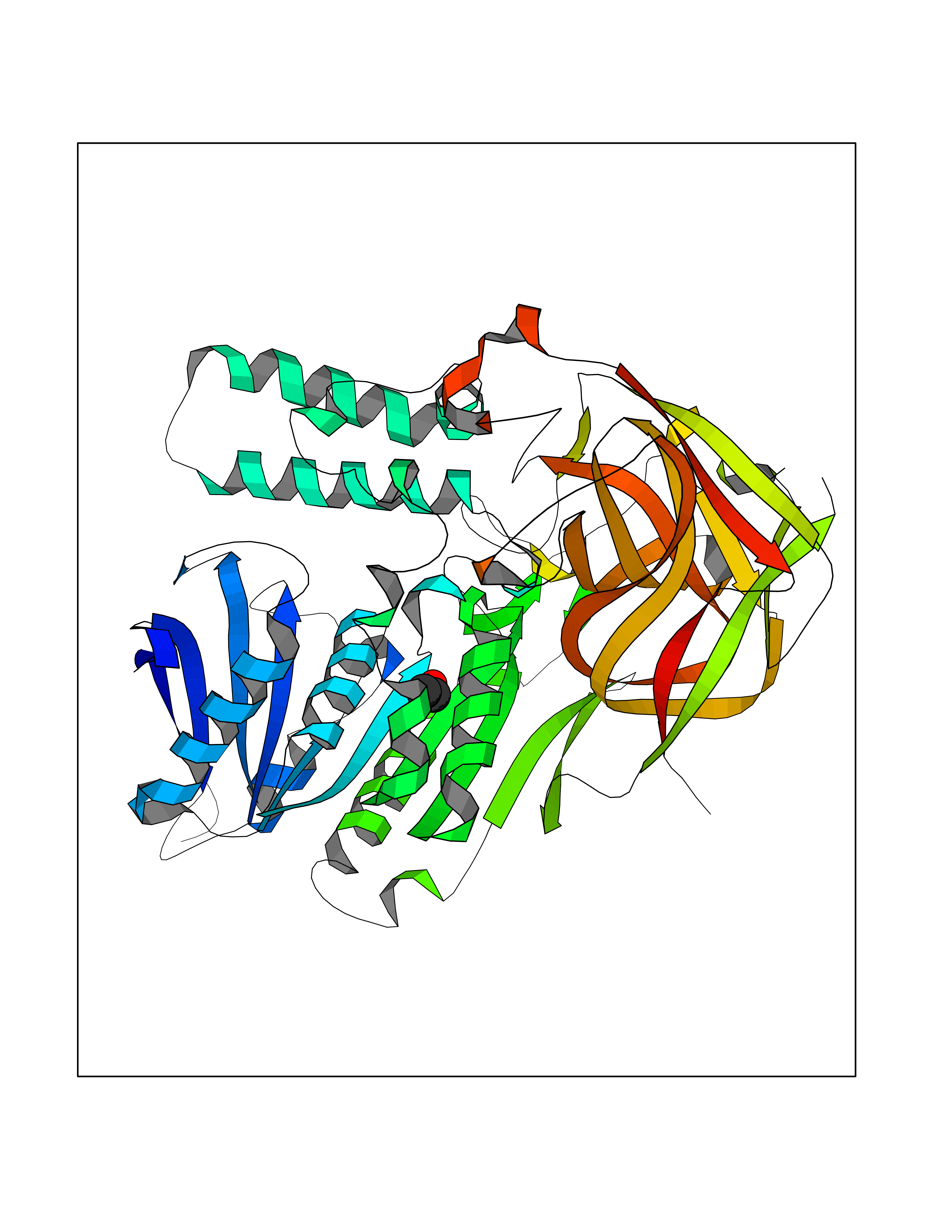 Molscript Map 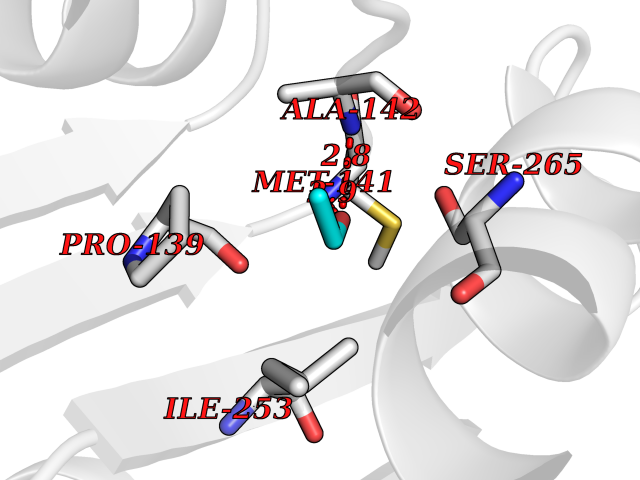 Pymol Map 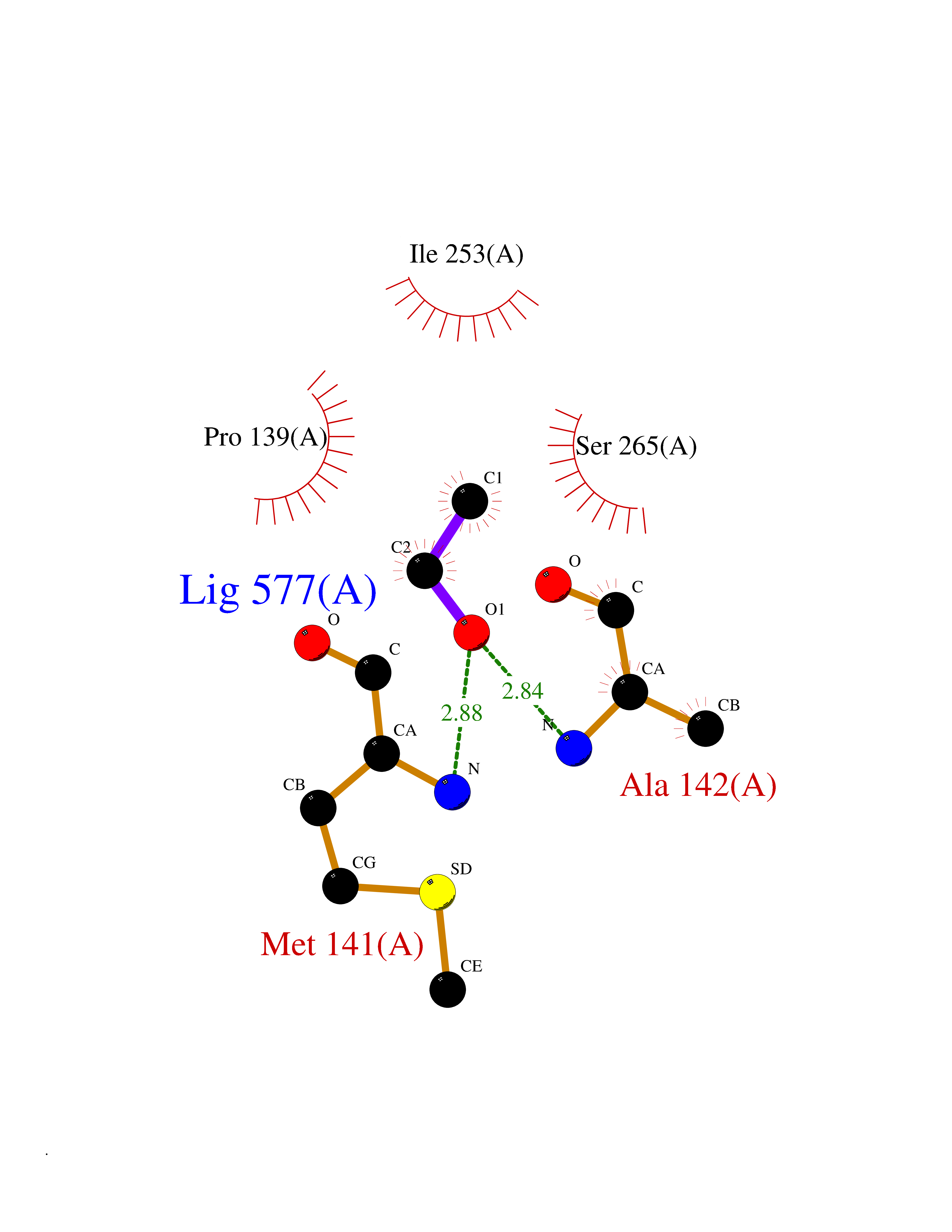 Ligplot Map 3D Binding mode Sequence VDGNYSVASNVMVPMRDGVRLAVDLYRPDADGPVPVLLVRNPYDKFDVFAWSTQSTNWLEFVRDGYAVVIQDTRGLFASEGEFVPHVDDEADAEDTLSWILEQAWCDGNVGMFGVSYLGVTQWQAAVSGVGGLKAIAPSMASADLYRAPWYGPGGALSVEALLGWSALIGTGLITSRSDARPEDAADFVQLAAILNDVAGAASVTPLAEQPLLGRLIPWVIDQVVDHPDNDESWQSISLFERLGGLATPALITAGWYDGFVGESLRTFVAVKDNADARLVVGPWSHSNLTGRNADRKFGIAATYPIQEATTMHKAFFDRHLRGETDALAGVPKVRLFVMGIDEWRDETDWPLPDTAYTPFYLGGSGAANTSTGGGTLSTSISGTESADTYLYDPADPVPSLGGTLLFHNGDNGPADQRPIHDRDDVLCYSTEVLTDPVEVTGTVSARLFVSSSAVDTDFTAKLVDVFPDGRAIALCDGIVRMRYRETLVNPTLIEAGEIYEVAIDMLATSNVFLPGHRIMVQVSSSNFPKYDRNSNTGGVIAREQLEEMCTAVNRIHRGPEHPSHIVLPIIKRK Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Bromodomain and PHD finger containing 1 (BRPF1) | 4UYE | 4.28 | |
Target general information Gen name BRPF1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Br140; Peregrin; Bromodomain and PHD finger-containing protein 1; BR140 Protein family NA Biochemical class NA Function Component of the MOZ/MORF complex which has a histone H3 acetyltransferase activity. Preferentially mediates histone H3-K23 acetylation. Positively regulates the transcription of RUNX1 and RUNX2. Related diseases Intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) [MIM:617333]: An autosomal dominant neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, delayed language, and facial dysmorphisms, most notably ptosis. Additional features may include poor growth, hypotonia, and seizures. {ECO:0000269|PubMed:27939639, ECO:0000269|PubMed:27939640}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P0C0S8; P62805; Q96GN5; Q96MT8-3; Q8NHQ1; Q8WYH8; Q2KHM9; Q8N987; Q8N6Y0; Q9Y2K6 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; Bromodomain; Chromatin regulator; Chromosome; Cytoplasm; Disease variant; DNA-binding; Intellectual disability; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 12891.6 Length 109 Aromaticity 0.12 Instability index 51.42 Isoelectric point 5.47 Charge (pH=7) -2.14 2D Binding mode Binding energy (Kcal/mol) -5.84 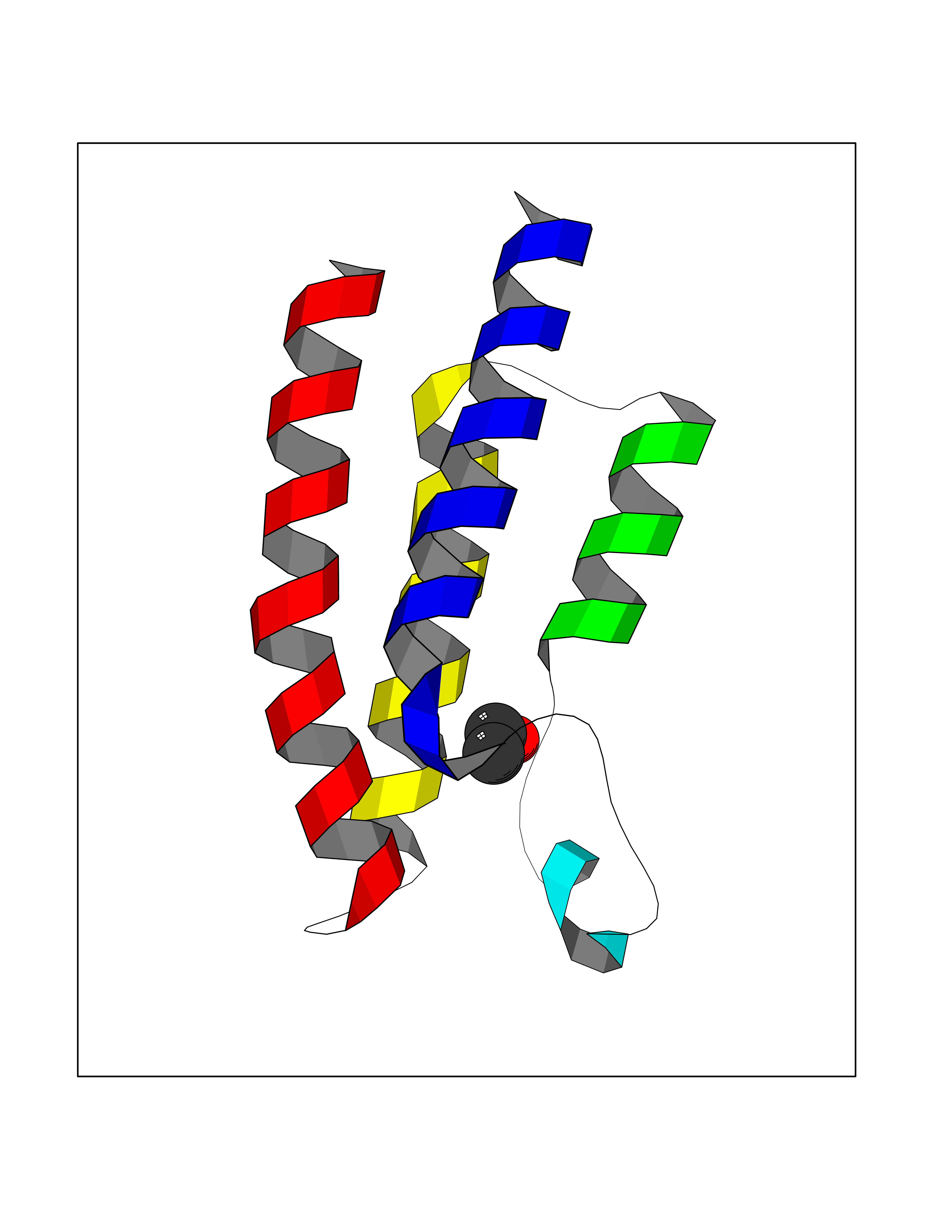 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence QLTPFLILLRKTLEQLQEKDTGNIFSEPVPLSEVPDYLDHIKKPMDFFTMKQNLEAYRYLNFDDFEEDFNLIVSNCLKYNAKDTIFYRAAVRLREQGGAVLRQARRQAE Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Mycobacterium Nicotinate-nucleotide pyrophosphorylase (MycB nadC) | 1QPR | 4.28 | |
Target general information Gen name MycB nadC Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID Synonyms nadC; Quinolinic acid phosphoribosyltransferase; Quinolinate phosphoribosyltransferase [decarboxylating]; QAPRTase Protein family NadC/ModD family Biochemical class Pentosyltransferase Function Involved in the catabolism of quinolinic acid (QA). Related diseases Oculodentodigital dysplasia (ODDD) [MIM:164200]: A disease characterized by a typical facial appearance and variable involvement of the eyes, dentition, and fingers. Characteristic facial features include a narrow, pinched nose with hypoplastic alae nasi, prominent columella and thin anteverted nares together with a narrow nasal bridge, and prominent epicanthic folds giving the impression of hypertelorism. The teeth are usually small and carious. Typical eye findings include microphthalmia and microcornea. The characteristic digital malformation is complete syndactyly of the fourth and fifth fingers (syndactyly type III) but the third finger may be involved and associated camptodactyly is a common finding. Cardiac abnormalities are observed in rare instances. {ECO:0000269|PubMed:16816024}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Oculodentodigital dysplasia, autosomal recessive (ODDD-AR) [MIM:257850]: A disease characterized by a typical facial appearance and variable involvement of the eyes, dentition, and fingers. Characteristic facial features include a narrow, pinched nose with hypoplastic alae nasi, prominent columella and thin anteverted nares together with a narrow nasal bridge, and prominent epicanthic folds giving the impression of hypertelorism. The teeth are usually small and carious. Typical eye findings include microphthalmia and microcornea. The characteristic digital malformation is complete syndactyly of the fourth and fifth fingers (syndactyly type III) but the third finger may be involved and associated camptodactyly is a common finding. Cardiac abnormalities are observed in rare instances. {ECO:0000269|PubMed:16816024}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Syndactyly 3 (SDTY3) [MIM:186100]: A form of syndactyly, a congenital anomaly of the hand or foot marked by persistence of the webbing between adjacent digits that are more or less completely attached. In SDTY3, there is usually complete and bilateral syndactyly between the fourth and fifth fingers. Usually it is soft tissue syndactyly but occasionally the distal phalanges are fused. The fifth finger is short with absent or rudimentary middle phalanx. The feet are not affected. {ECO:0000269|PubMed:14729836}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Hypoplastic left heart syndrome 1 (HLHS1) [MIM:241550]: A syndrome due to defective development of the aorta proximal to the entrance of the ductus arteriosus, and hypoplasia of the left ventricle and mitral valve. As a result of the abnormal circulation, the ductus arteriosus and foramen ovale are patent and the right atrium, right ventricle, and pulmonary artery are enlarged. {ECO:0000269|PubMed:11470490}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Hallermann-Streiff syndrome (HSS) [MIM:234100]: A disorder characterized by a typical skull shape (brachycephaly with frontal bossing), hypotrichosis, microphthalmia, cataracts, beaked nose, micrognathia, skin atrophy, dental anomalies and proportionate short stature. Intellectual disability is present in a minority of cases. {ECO:0000269|PubMed:14974090}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Craniometaphyseal dysplasia, autosomal recessive (CMDR) [MIM:218400]: An osteochondrodysplasia characterized by hyperostosis and sclerosis of the craniofacial bones associated with abnormal modeling of the metaphyses. Sclerosis of the skull may lead to asymmetry of the mandible, as well as to cranial nerve compression, that may finally result in hearing loss and facial palsy. {ECO:0000269|PubMed:23951358}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythrokeratodermia variabilis et progressiva 3 (EKVP3) [MIM:617525]: A form of erythrokeratodermia variabilis et progressiva, a genodermatosis characterized by the coexistence of two independent skin lesions: transient erythema and hyperkeratosis that is usually localized but occasionally occurs in its generalized form. Clinical presentation varies significantly within a family and from one family to another. Palmoplantar keratoderma is present in around 50% of cases. {ECO:0000269|PubMed:25398053}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Palmoplantar keratoderma and congenital alopecia 1 (PPKCA1) [MIM:104100]: A rare autosomal dominant disorder characterized by severe hyperkeratosis of the palms and soles, and congenital hypotrichosis or alopecia. Dystrophic nail changes occur in some patients. {ECO:0000269|PubMed:25168385}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04294; DB02382; DB02746; DB01796 Interacts with NA EC number EC 2.4.2.19 Uniprot keywords 3D-structure; Glycosyltransferase; Pyridine nucleotide biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 59621.1 Length 568 Aromaticity 0.03 Instability index 20.43 Isoelectric point 5.09 Charge (pH=7) -15.46 2D Binding mode Binding energy (Kcal/mol) -5.83 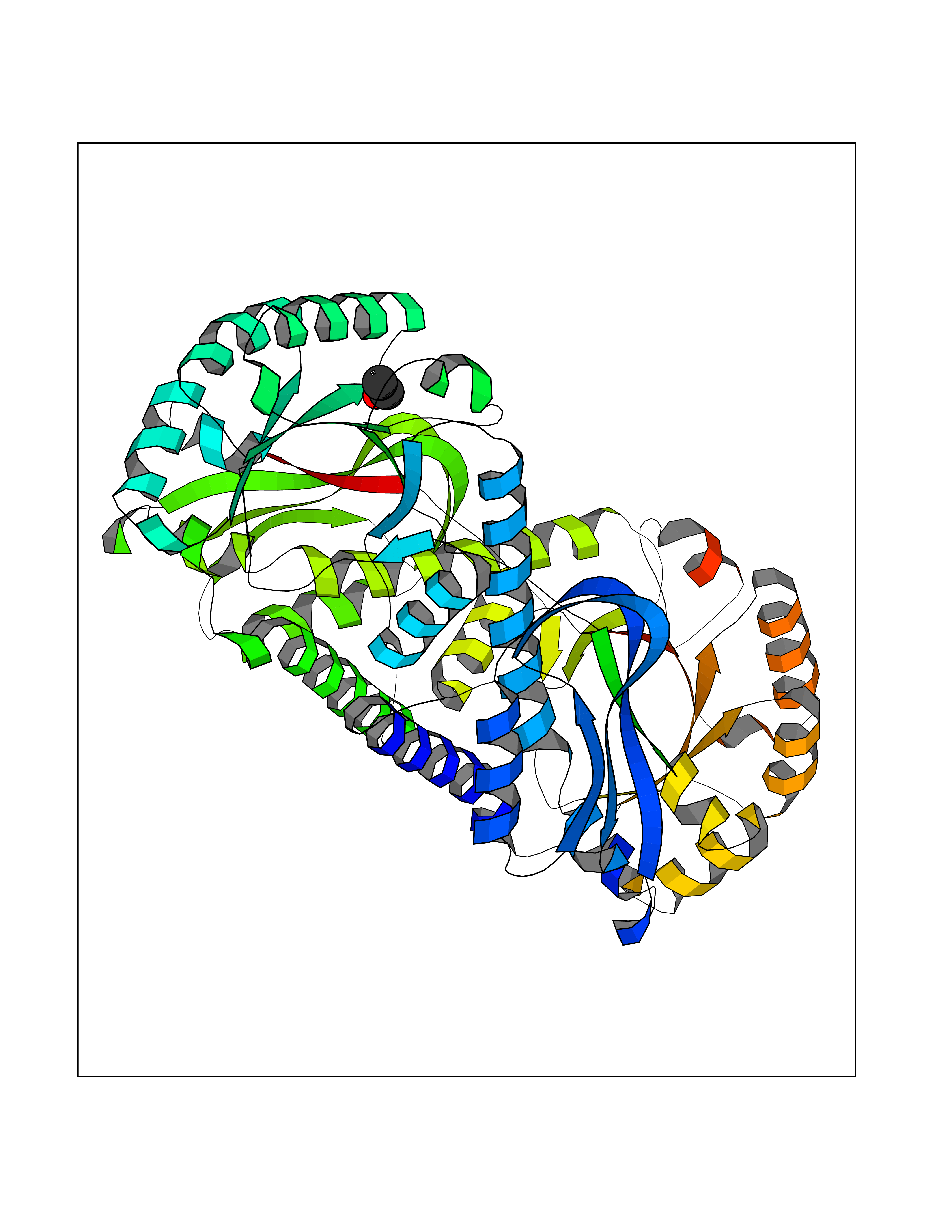 Molscript Map 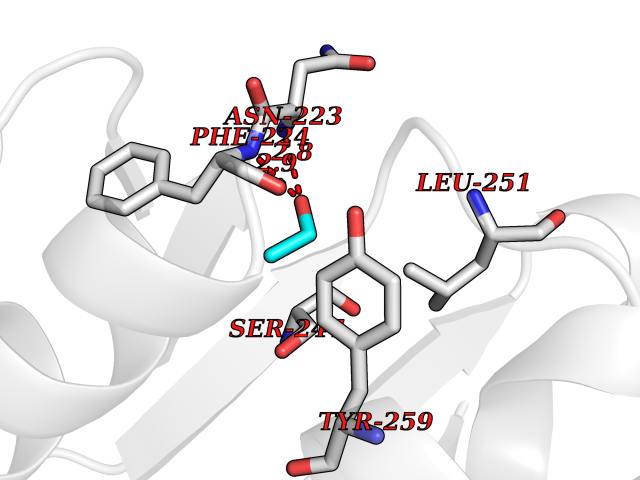 Pymol Map 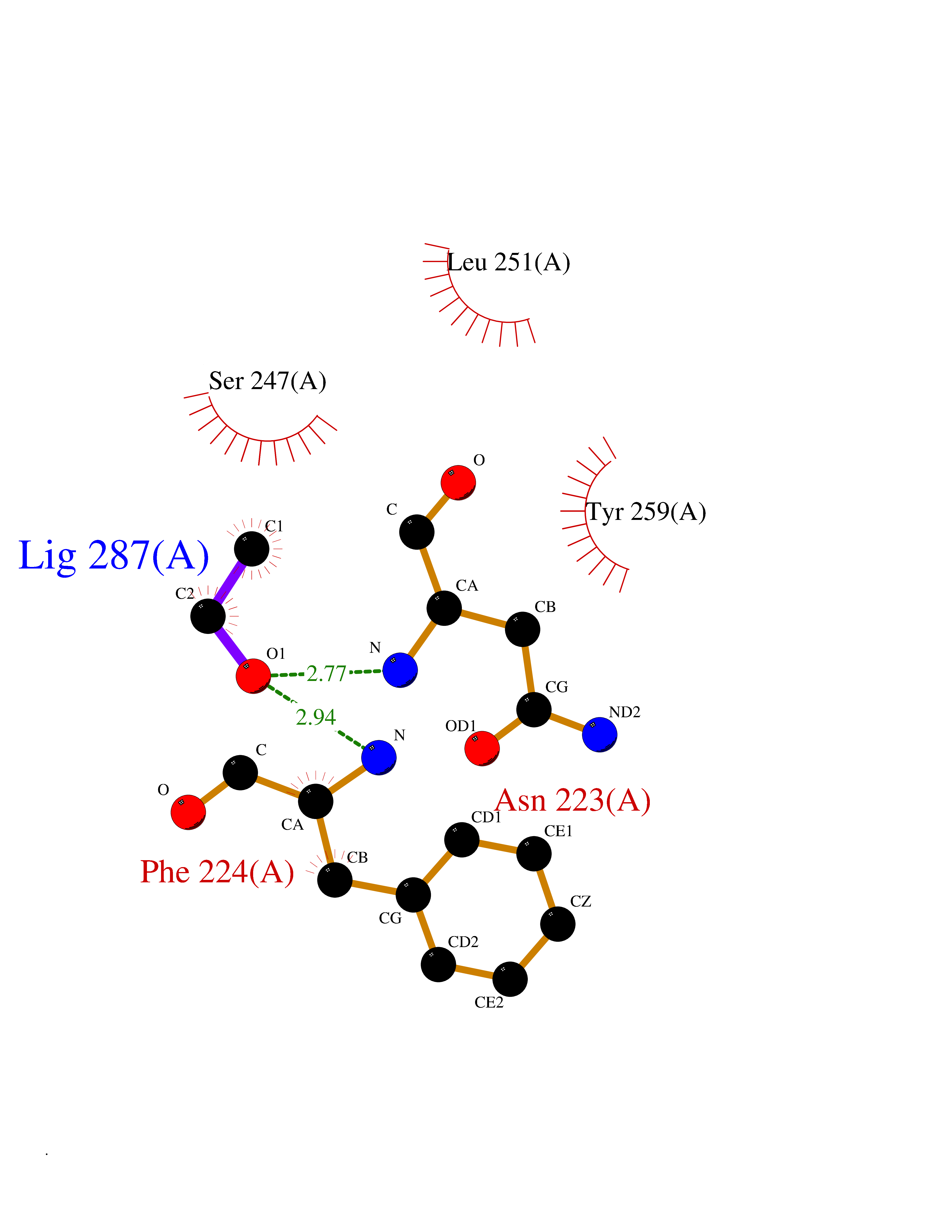 Ligplot Map 3D Binding mode Sequence GLSDWELAAARAAIARGLDEDLRYGPDVTTLATVPASATTTASLVTREAGVVAGLDVALLTLNEVLGTNGYRVLDRVEDGARVPPGEALMTLEAQTRGLLTAERTMLNLVGHLSGIATATAAWVDAVRGTKAKIRDTRKTLPGLRALQKYAVRTGGGVNHRLGLGDAALIKDNHVAAAGSVVDALRAVRNAAPDLPCEVEVDSLEQLDAVLPEKPELILLDNFAVWQTQTAVQRRDSRAPTVMLESSGGLSLQTAATYAETGVDYLAVGALTHSVRVLDIGLDMGLSDWELAAARAAIARGLDEDLRYGPDVTTLATVPASATTTASLVTREAGVVAGLDVALLTLNEVLGTNGYRVLDRVEDGARVPPGEALMTLEAQTRGLLTAERTMLNLVGHLSGIATATAAWVDAVRGTKAKIRDTRKTLPGLRALQKYAVRTGGGVNHRLGLGDAALIKDNHVAAAGSVVDALRAVRNAAPDLPCEVEVDSLEQLDAVLPEKPELILLDNFAVWQTQTAVQRRDSRAPTVMLESSGGLSLQTAATYAETGVDYLAVGALTHSVRVLDIGLDM Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Schistosoma Purine nucleoside phosphorylase (sch PNP) | 3F8W | 4.28 | |
Target general information Gen name sch PNP Organism Schistosoma mansoni (Blood fluke) Uniprot ID TTD ID Synonyms sch Purine nucleoside phosphorylase; sch Inosine-guanosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class NA Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta-(deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Hemolytic anemia, non-spherocytic, due to glucose phosphate isomerase deficiency (HA-GPID) [MIM:613470]: A form of anemia in which there is no abnormal hemoglobin or spherocytosis. It is caused by glucose phosphate isomerase deficiency. {ECO:0000269|PubMed:28803808, ECO:0000269|PubMed:7989588, ECO:0000269|PubMed:8499925, ECO:0000269|PubMed:8822952, ECO:0000269|PubMed:8822954, ECO:0000269|PubMed:9446754, ECO:0000269|PubMed:9856489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Glycosyltransferase; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 61140.6 Length 564 Aromaticity 0.06 Instability index 29.8 Isoelectric point 7.6 Charge (pH=7) 1.82 2D Binding mode Binding energy (Kcal/mol) -5.84 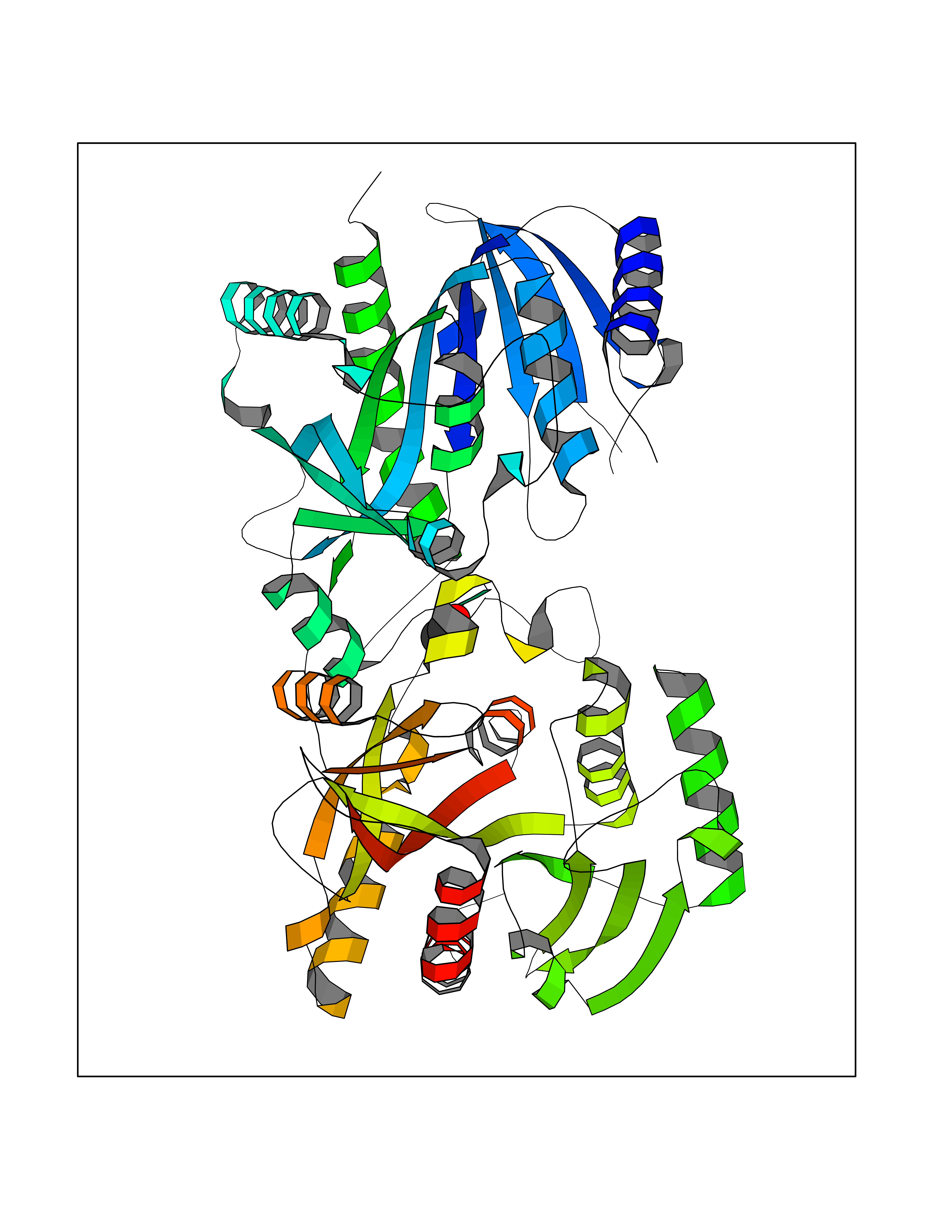 Molscript Map 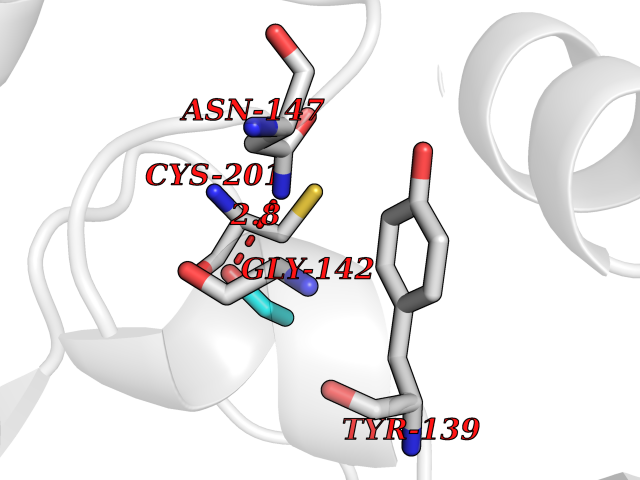 Pymol Map 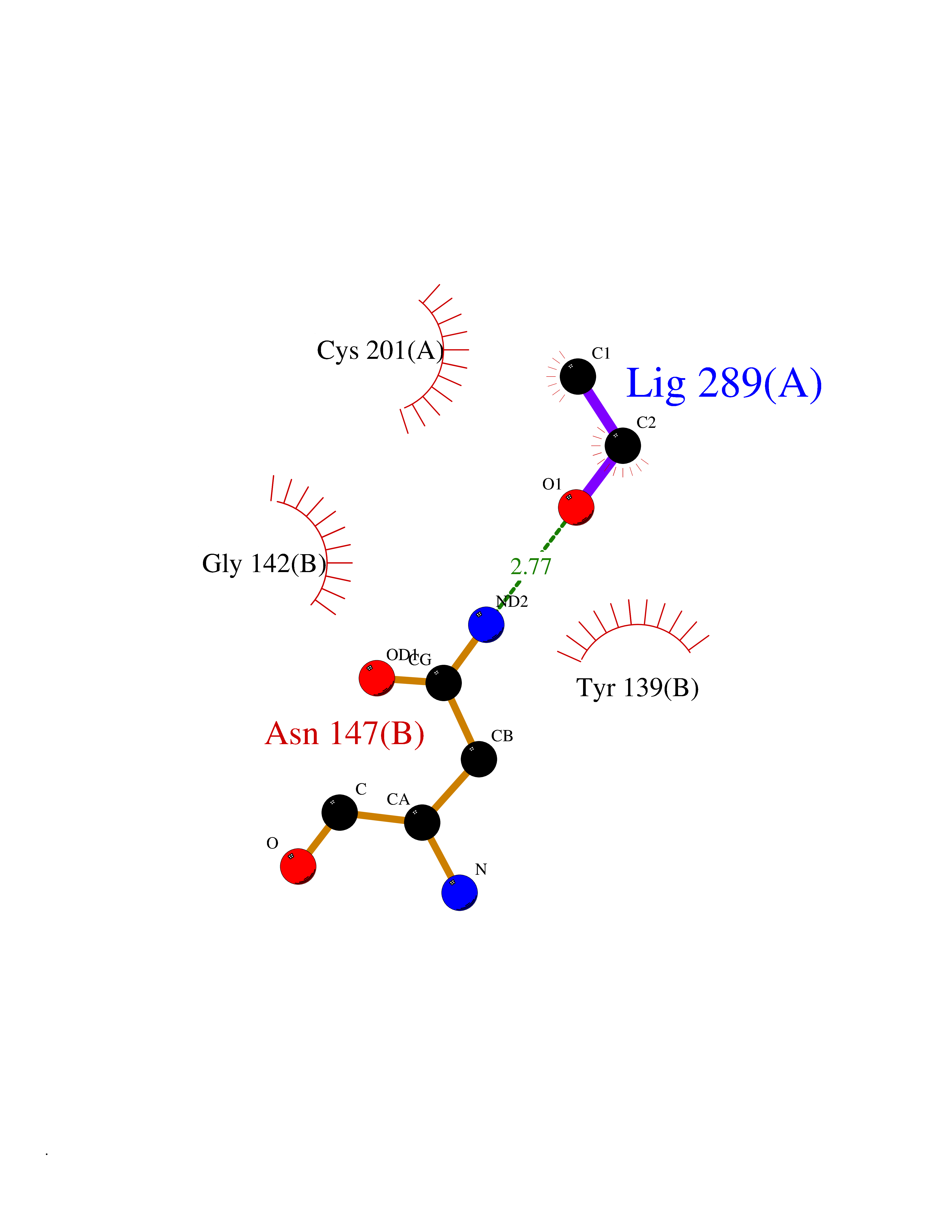 Ligplot Map 3D Binding mode Sequence SVTANIENVKKVAHHIQKLTSIVPEIGIICGSGLGKLADGVKDKITIPYTKIPNFPQTSHSGNLIFGTLSGRKVVVMQGRFHMYEGYSNDTVALPIRVMKLLGVKILMVSNAAGGLNRSLKLGDFVILKDHIYLPGLGLNNILVGPNQEAFGTRFPALSNAYDRDLRKLAVQVAEENGFGNLVHQGVYVMNGGPCYETPAECTMLLNMGCDVVGMSTIPEVVIARHCGIQVFAVSLVTNISVLDVESDLKPNHEEVLATGAQRAELMQSWFEKIIEKLPKSVTANIENVKKVAHHIQKLTSIVPEIGIICGSGLGKLADGVKDKITIPYTKIPNFPQTSVVGHSGNLIFGTLSGRKVVVMQGRFHMYEGYSNDTVALPIRVMKLLGVKILMVSNAAGGLNRSLKLGDFVILKDHIYLPGLGLNNILVGPNQEAFGTRFPALSNAYDRDLRKLAVQVAEENGFGNLVHQGVYVMNGGPCYETPAECTMLLNMGCDVVGMSTIPEVVIARHCGIQVFAVSLVTNISVLDVESDLKPNHEEVLATGAQRAELMQSWFEKIIEKLPKD Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Protein tyrosine phosphatase beta (PTPRB) | 2I4G | 4.28 | |
Target general information Gen name PTPRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vascular endothelial protein tyrosine phosphatase; VE-PTP; Receptor-type tyrosine-protein phosphatase beta; R-PTP-beta; PTPRB Protein family Protein-tyrosine phosphatase family, Receptor class 3 subfamily Biochemical class Phosphoric monoester hydrolase Function Plays an important role in blood vessel remodeling and angiogenesis. Not necessary for the initial formation of blood vessels, but is essential for their maintenance and remodeling. Can induce dephosphorylation of TEK/TIE2, CDH5/VE-cadherin and KDR/VEGFR-2. Regulates angiopoietin-TIE2 signaling in endothelial cells. Acts as a negative regulator of TIE2, and controls TIE2 driven endothelial cell proliferation, which in turn affects blood vessel remodeling during embryonic development and determines blood vessel size during perinatal growth. Essential for the maintenance of endothelial cell contact integrity and for the adhesive function of VE-cadherin in endothelial cells and this requires the presence of plakoglobin. Related diseases WHIM syndrome 2 (WHIMS2) [MIM:619407]: An autosomal recessive form of WHIM syndrome, a primary immunodeficiency disorder characterized by warts, hypogammaglobulinemia, infections, and myelokathexis. Myelokathexis is a unique form of non-cyclic severe congenital neutropenia caused by accumulation of mature and degenerating neutrophils in the bone marrow. Monocytopenia and lymphopenia, especially B lymphopenia, also commonly occur. There is significant phenotypic variation among patients, such that some individuals may have an incomplete form of the disorder in which one or more of the classic tetrad features are not present. {ECO:0000269|PubMed:24777453}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08678; DB07068; DB16353; DB07127; DB06989 Interacts with P00533; P04626; P10912; P27361; P08581; Q02763 EC number EC 3.1.3.48 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; Glycoprotein; Hydrolase; Membrane; Phosphoprotein; Protein phosphatase; Proteomics identification; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 33576.8 Length 289 Aromaticity 0.1 Instability index 39.59 Isoelectric point 7.28 Charge (pH=7) 0.75 2D Binding mode Binding energy (Kcal/mol) -5.84 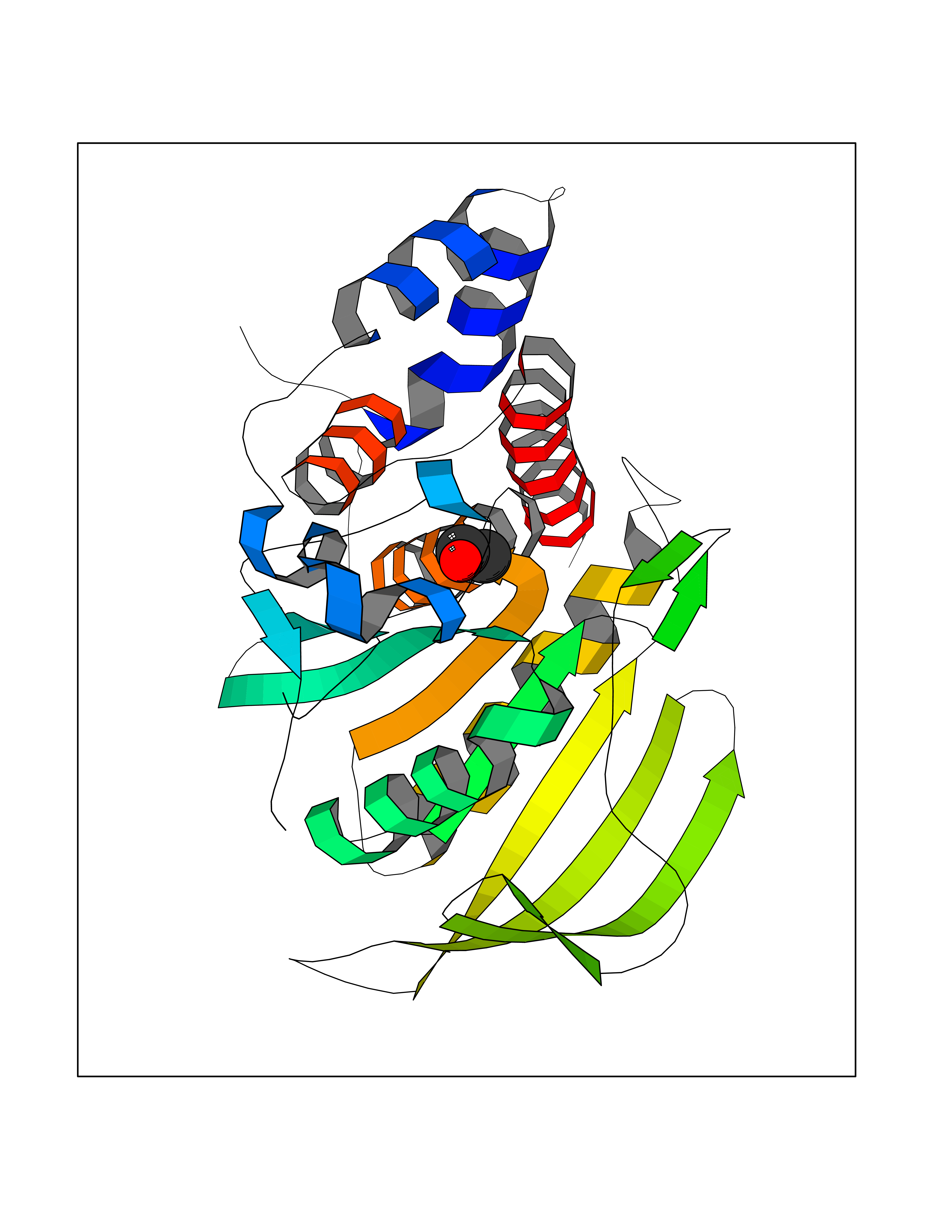 Molscript Map 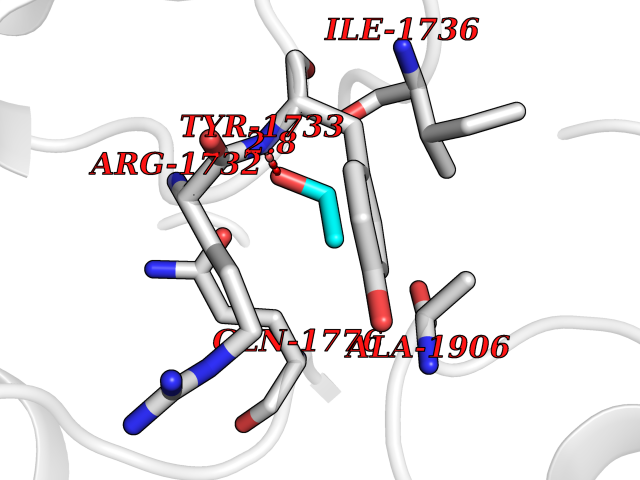 Pymol Map 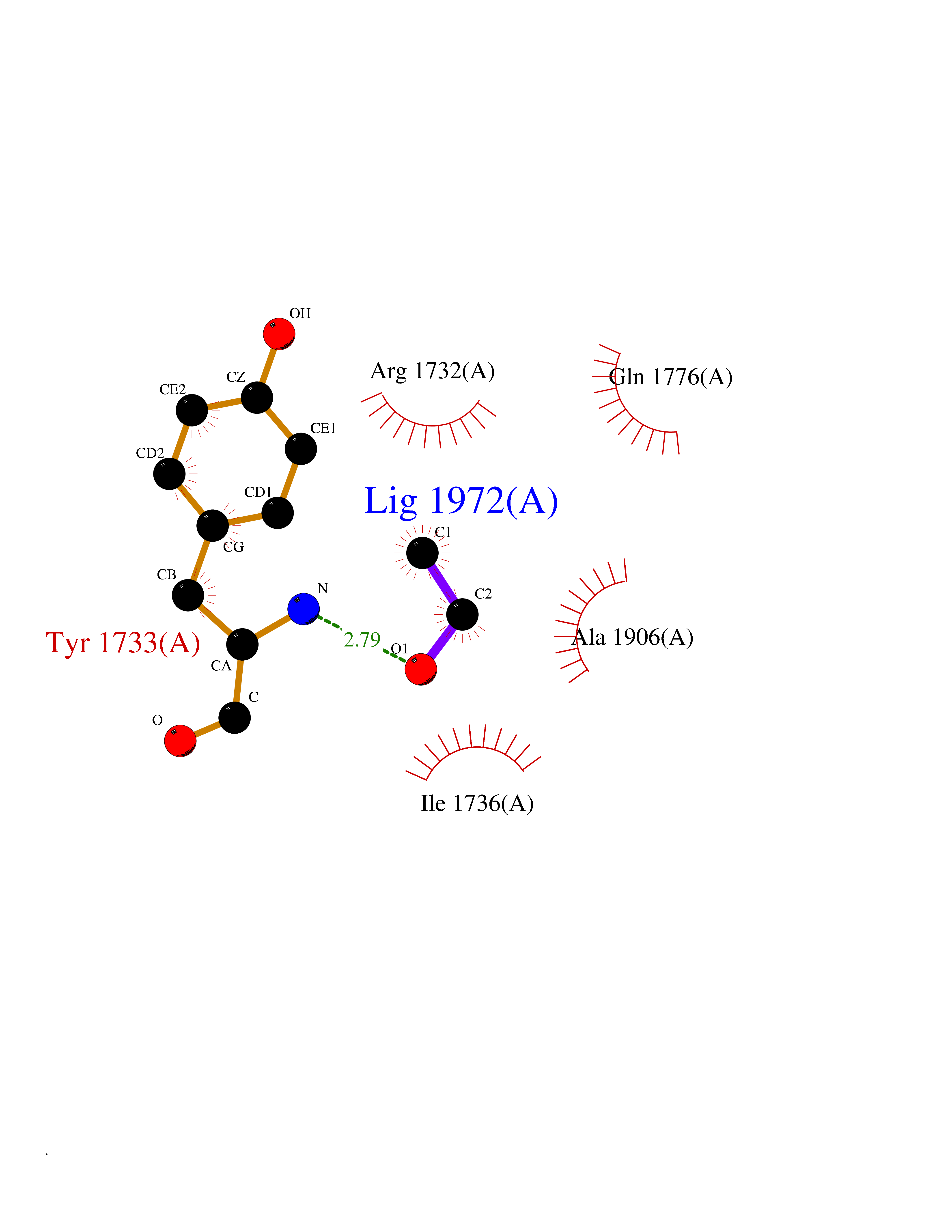 Ligplot Map 3D Binding mode Sequence KTSCPIKINQFEGHFMKLQADSNYLLSKEYEELKDVGRNQSCDIALLPENRGKNRYNNILPYDATRVKLSNVCSDYINASYIPGNNFRREYIVTQGPLPGTKDDFWKMVWEQNVHNIVMVTQCVEKGRVKCDHYWPADQDSLYYGDLILQMLSESVLPEWTIREFKICGEEQLDAHRLIRHFHYTVWPDHGVPETTQSLIQFVRTVRDYINRSPGAGPTVVHCSAGVGRTGTFIALDRILQQLDSKDSVDIYGAVHDLRLHRVHMVQTECQYVYLHQCVRDVLRARKLR Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Positive transcription elongation factor b (P-TEFb) | 4OGR | 4.28 | |
Target general information Gen name CDK9-CCNT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms P-TEFb Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class S-phase kinase-associated protein Function The elongin BC complex seems to be involved as an adapter protein in the proteasomal degradation of target proteins via different E3 ubiquitin ligase complexes, including the von Hippel-Lindau ubiquitination complex CBC(VHL). By binding to BC- box motifs it seems to link target recruitment subunits, like VHL and members of the SOCS box family, to Cullin/RBX1 modules that activate E2 ubiquitination enzymes. Related diseases Chronic activation of CDK9 causes cardiac myocyte enlargement leading to cardiac hypertrophy and confers predisposition to heart failure. Drugs (DrugBank ID) DB03496; DB06195; DB15442; DB16656 Interacts with Q13535; Q8WXE1; O60885-1; O60563; O60583; O60583-1; O60583-2; Q16543; Q9HAW4; Q9NR30; Q13451; O94992; P07900; P08238; Q6NYC1; Q4G0J3; P53041; P40763; P04608; P28799; P28799-2; Q6XYB7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cytoplasm; DNA damage; DNA repair; Host-virus interaction; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 65613.1 Length 569 Aromaticity 0.09 Instability index 38.98 Isoelectric point 7.94 Charge (pH=7) 3.26 2D Binding mode Binding energy (Kcal/mol) -5.84 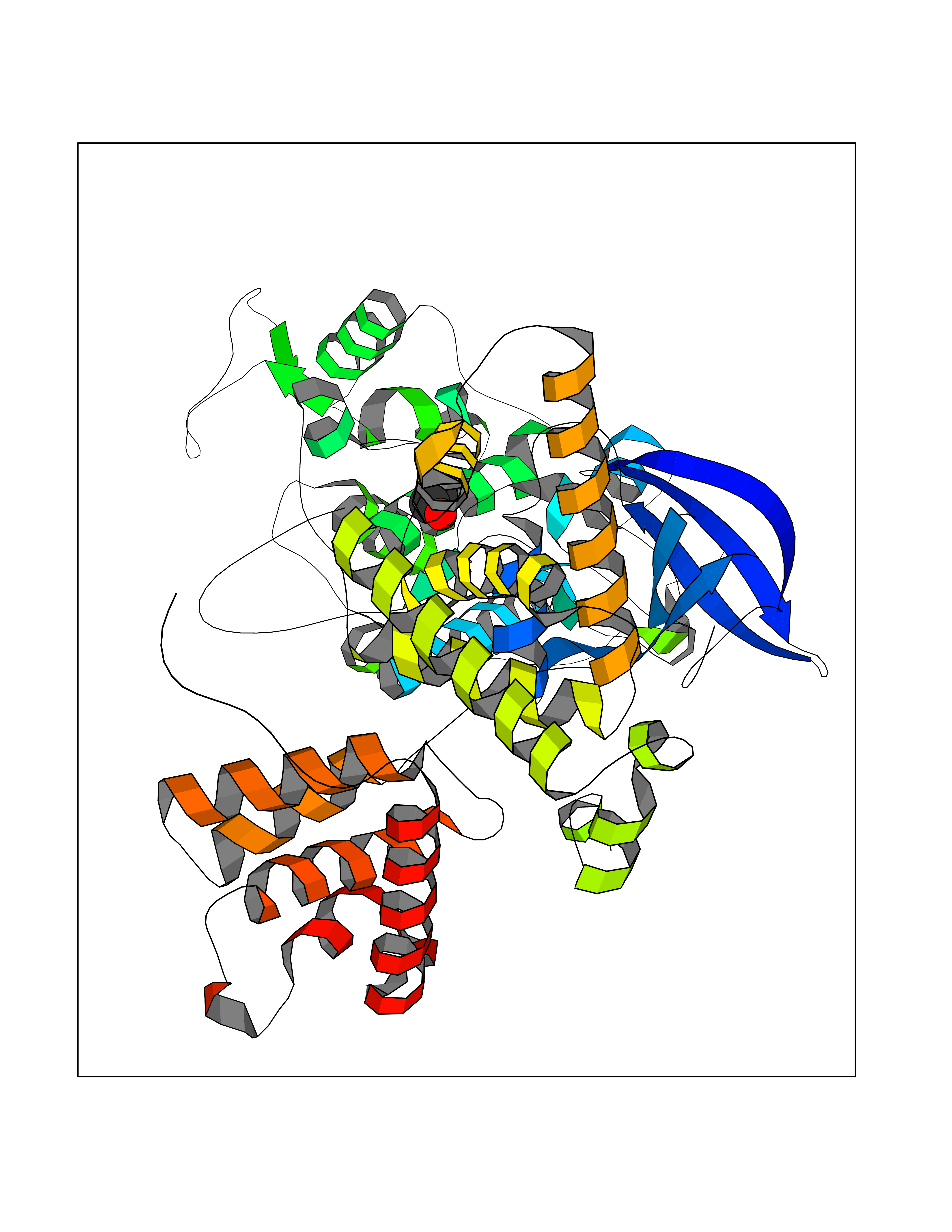 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence VECPFCDEVSKYEKLAKIGQGTFGEVFKARHRKTGQKVALKKVLMENEKEGFPITALREIKILQLLKHENVVNLIEICRTKGSIYLVFDFCEHDLAGLLSNVLVKFTLSEIKRVMQMLLNGLYYIHRNKILHRDMKAANVLITRDGVLKLADFGLARAFSLAKNSQPNRYNRVVTLWYRPPELLLGERDYGPPIDLWGAGCIMAEMWTRSPIMQGNTEQHQLALISQLCGSITPEVWPNVDNYELYEKLELVKGQKRKVKDRLKAYVRDPYALDLIDKLLVLDPAQRIDSDDALNHDFFWSDPMPSDLKGMLSTNNNKRWYFTREQLENSPSRRFGVDPDKELSYRQQAANLLQDMGQRLNVSQLTINTAIVYMHRFYMIQSFTQFPGNSVAPAALFLAAKVEEQPKKLEHVIKVAHTCLHPQESLPDTRSEAYLQQVQDLVILESIILQTLGFELTIDHPHTHVVKCTQLVRASKDLAQTSYFMATNSLHLTTFSLQYTPPVVACVCIHLACKWSNWEIPVSTDGKHWWEYVDATVTLELLDELTHEFLQILEKTPNRLKRIWNWRAC Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Zinc finger protein Helios (IKZF2) | 7LPS | 4.28 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -5.83 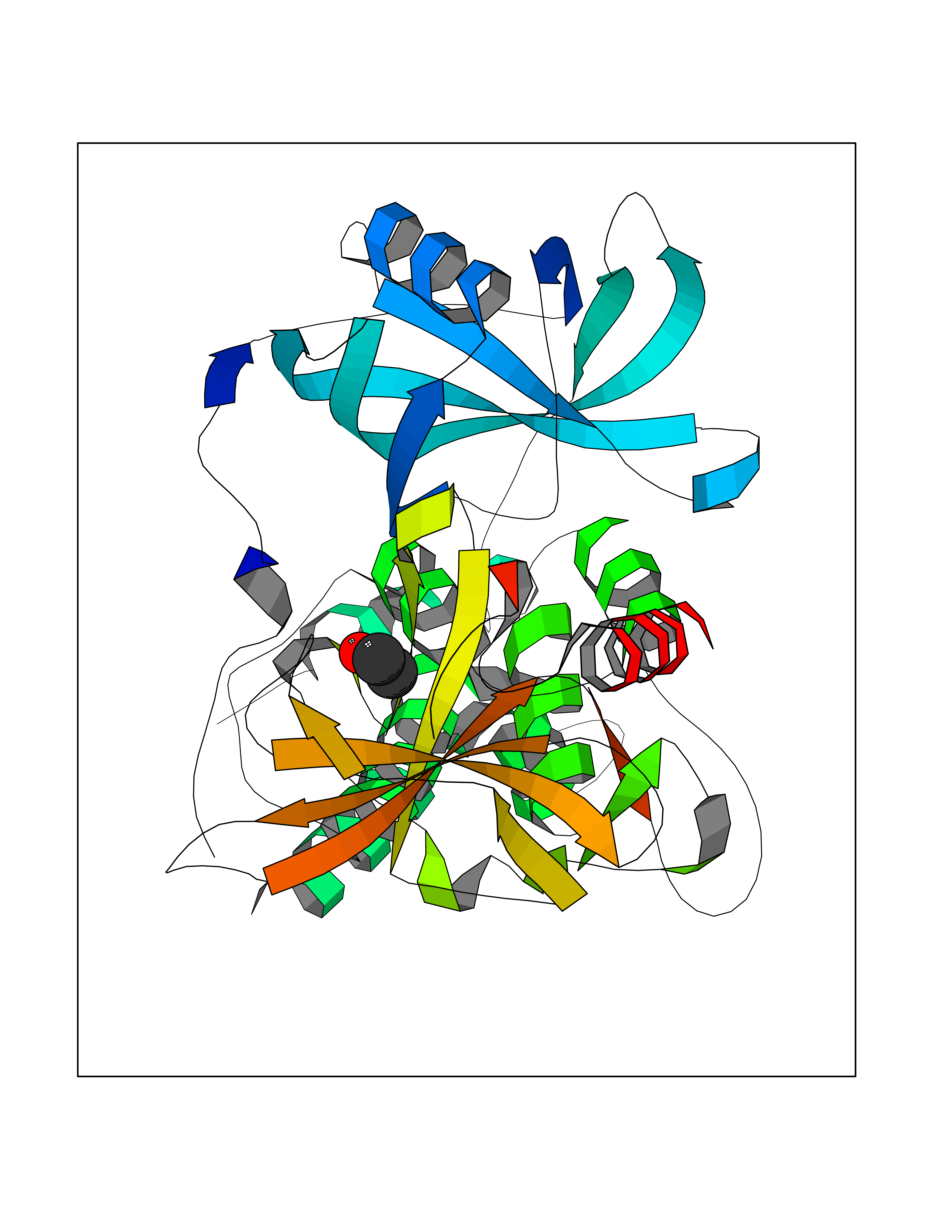 Molscript Map 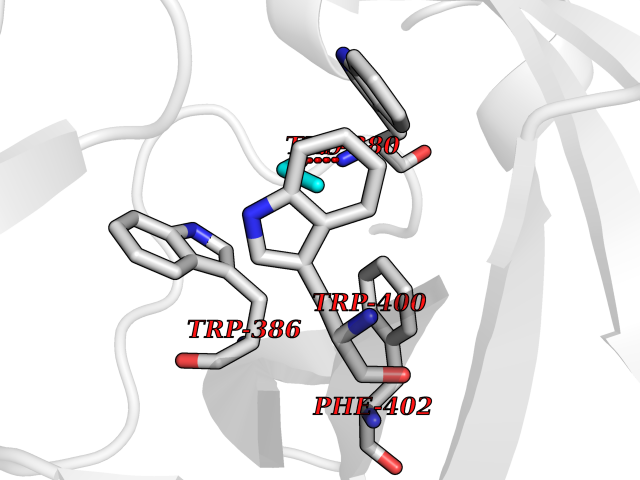 Pymol Map 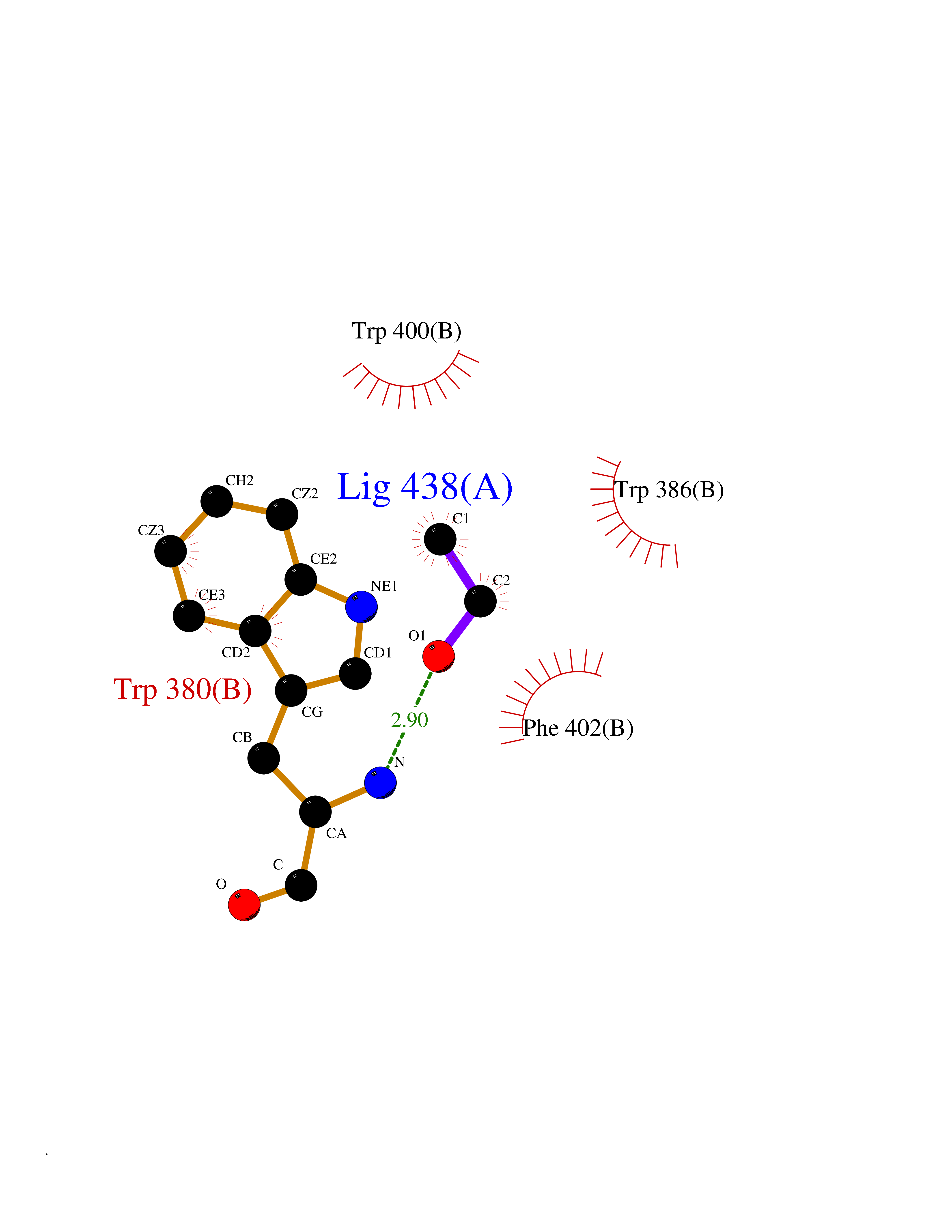 Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Trypanosoma Trypanothione reductase (Trypano TPR) | 2WBA | 4.28 | |
Target general information Gen name Trypano TPR Organism Trypanosoma brucei brucei Uniprot ID TTD ID Synonyms TRYR; TPR; Parasite-specific trypanothione reductase; N(1),N(8)-bis(glutathionyl)spermidine reductase Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Sulfur donor oxidoreductase Function Trypanothione is the parasite analog of glutathione; this enzyme is the equivalent of glutathione reductase. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.8.1.12 Uniprot keywords 3D-structure; Cytoplasm; Disulfide bond; FAD; Flavoprotein; NADP; Oxidoreductase; Redox-active center Protein physicochemical properties Chain ID A Molecular weight (Da) 52797.9 Length 489 Aromaticity 0.08 Instability index 33.55 Isoelectric point 6.24 Charge (pH=7) -3.67 2D Binding mode Binding energy (Kcal/mol) -5.84 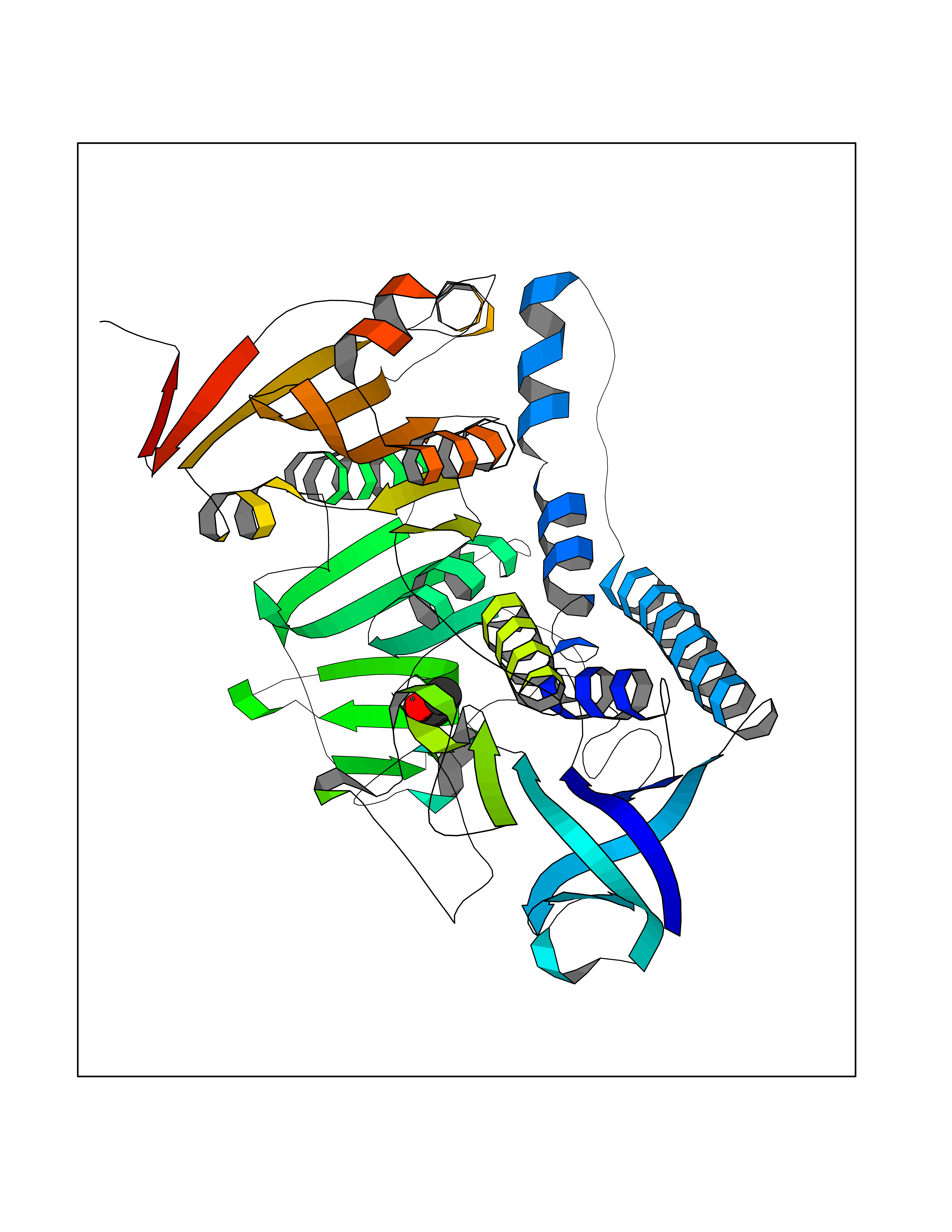 Molscript Map 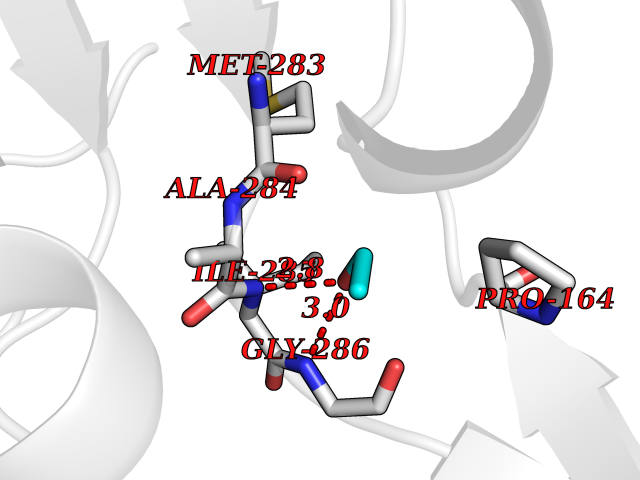 Pymol Map 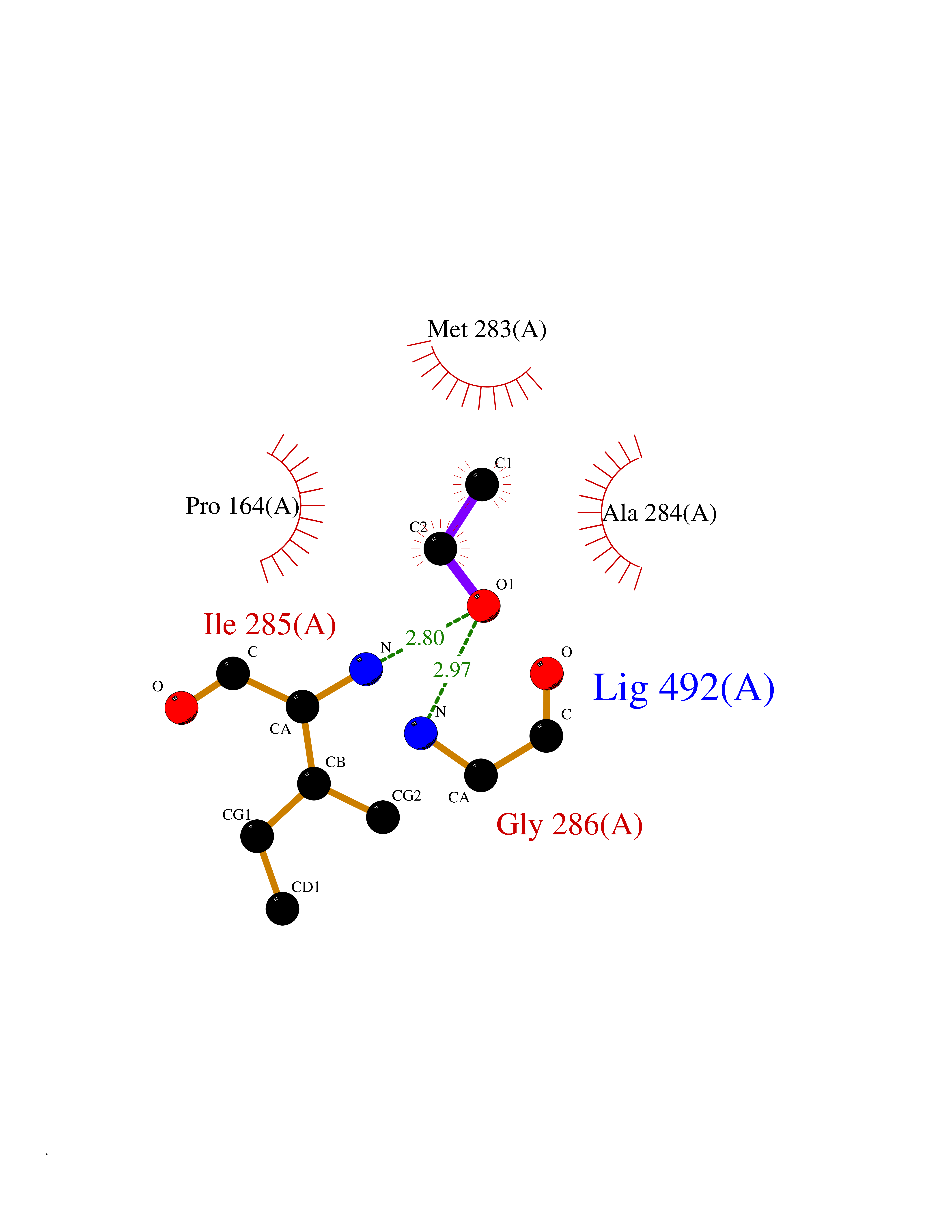 Ligplot Map 3D Binding mode Sequence SKAFDLVVIGAGSGGLEAGWNAATLYGKRVAVVDVQTSHGPPFYAALGGTCVNVGCVPKKLMVTGAQYMDHLRESAGFGWEFDGSSVKANWKKLIAAKNEAVLDINKSYEGMFNDTEGLDFFLGWGSLESKNVVVVRETADPKSAVKERLQADHILLATGSWPQMPAIPGIEHCISSNEAFYLPEPPRRVLTVGGGFISVEFAGIFNAYKPPGGKVTLCYRNNLILRGFDETIREEVTKQLTANGIEIMTNENPAKVSLNTDGSKHVTFESGKTLDVDVVMMAIGRIPRTNDLQLGNVGVKLTPKGGVQVDEFSRTNVPNIYAIGDITDRLMLTPVAINEGAALVDTVFGNKPRKTDHTRVASAVFSIPPIGTCGLIEEVAAKEFEKVAVYMSSFTPLMHNISGSKYKKFVAKIVTNHSDGTVLGVHLLGDGAPEIIQAVGVCLRLNAKISDFYNTIGVHPTSAEELCSMRTPSYYYVKGEKMEKLPDS Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Macrophage colony-stimulating factor 1 receptor (CSF1R) | 2I1M | 4.27 | |
Target general information Gen name CSF1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Proto-oncogene c-Fms; M-CSF-R; FMS; CSF-1R; CSF-1-R; CSF-1 receptor; CD115 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Promotes the release of proinflammatory chemokines in response to IL34 and CSF1, and thereby plays an important role in innate immunity and in inflammatory processes. Plays an important role in the regulation of osteoclast proliferation and differentiation, the regulation of bone resorption, and is required for normal bone and tooth development. Required for normal male and female fertility, and for normal development of milk ducts and acinar structures in the mammary gland during pregnancy. Promotes reorganization of the actin cytoskeleton, regulates formation of membrane ruffles, cell adhesion and cell migration, and promotes cancer cell invasion. Activates several signaling pathways in response to ligand binding. Phosphorylates PIK3R1, PLCG2, GRB2, SLA2 and CBL. Activation of PLCG2 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, that then lead to the activation of protein kinase C family members, especially PRKCD. Phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, leads to activation of the AKT1 signaling pathway. Activated CSF1R also mediates activation of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1, and of the SRC family kinases SRC, FYN and YES1. Activated CSF1R transmits signals both via proteins that directly interact with phosphorylated tyrosine residues in its intracellular domain, or via adapter proteins, such as GRB2. Promotes activation of STAT family members STAT3, STAT5A and/or STAT5B. Promotes tyrosine phosphorylation of SHC1 and INPP5D/SHIP-1. Receptor signaling is down-regulated by protein phosphatases, such as INPP5D/SHIP-1, that dephosphorylate the receptor and its downstream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as cell-surface receptor for CSF1 and IL34 and plays an essential role in the regulation of survival, proliferation and differentiation of hematopoietic precursor cells, especially mononuclear phagocytes, such as macrophages and monocytes. Related diseases Aberrant expression of CSF1 or CSF1R can promote cancer cell proliferation, invasion and formation of metastases. Overexpression of CSF1 or CSF1R is observed in a significant percentage of breast, ovarian, prostate, and endometrial cancers.; DISEASE: Aberrant expression of CSF1 or CSF1R may play a role in inflammatory diseases, such as rheumatoid arthritis, glomerulonephritis, atherosclerosis, and allograft rejection.; DISEASE: Leukoencephalopathy, hereditary diffuse, with spheroids 1 (HDLS1) [MIM:221820]: An autosomal dominant adult-onset rapidly progressive neurodegenerative disorder characterized by variable behavioral, cognitive, and motor changes. Patients often die of dementia within 6 years of onset. Brain imaging shows patchy abnormalities in the cerebral white matter, predominantly affecting the frontal and parietal lobes. {ECO:0000269|PubMed:22197934, ECO:0000269|PubMed:23408870, ECO:0000269|PubMed:24336230, ECO:0000269|PubMed:24532199}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brain abnormalities, neurodegeneration, and dysosteosclerosis (BANDDOS) [MIM:618476]: An autosomal recessive disease with variable manifestations. Main features are brain malformations with calcifying leukoencephalopathy, progressive neurodegeneration, and bone sclerotic features. The age at onset ranges from infancy to early adulthood. Neurologic features include loss of previous motor and language skills, cognitive impairment, spasticity, and focal seizures. Brain imaging shows periventricular white matter abnormalities and calcifications, large cisterna magna or Dandy-Walker malformation, and sometimes agenesis of the corpus callosum. {ECO:0000269|PubMed:30982608, ECO:0000269|PubMed:30982609}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07167; DB07202; DB12147; DB12010; DB00619; DB06080; DB12978; DB01268 Interacts with P09603; Q15375; P29323; Q6ZMJ4-1 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Inflammatory response; Innate immunity; Kinase; Membrane; Neurodegeneration; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35082.9 Length 311 Aromaticity 0.11 Instability index 44.6 Isoelectric point 8.13 Charge (pH=7) 2.42 2D Binding mode Binding energy (Kcal/mol) -5.83 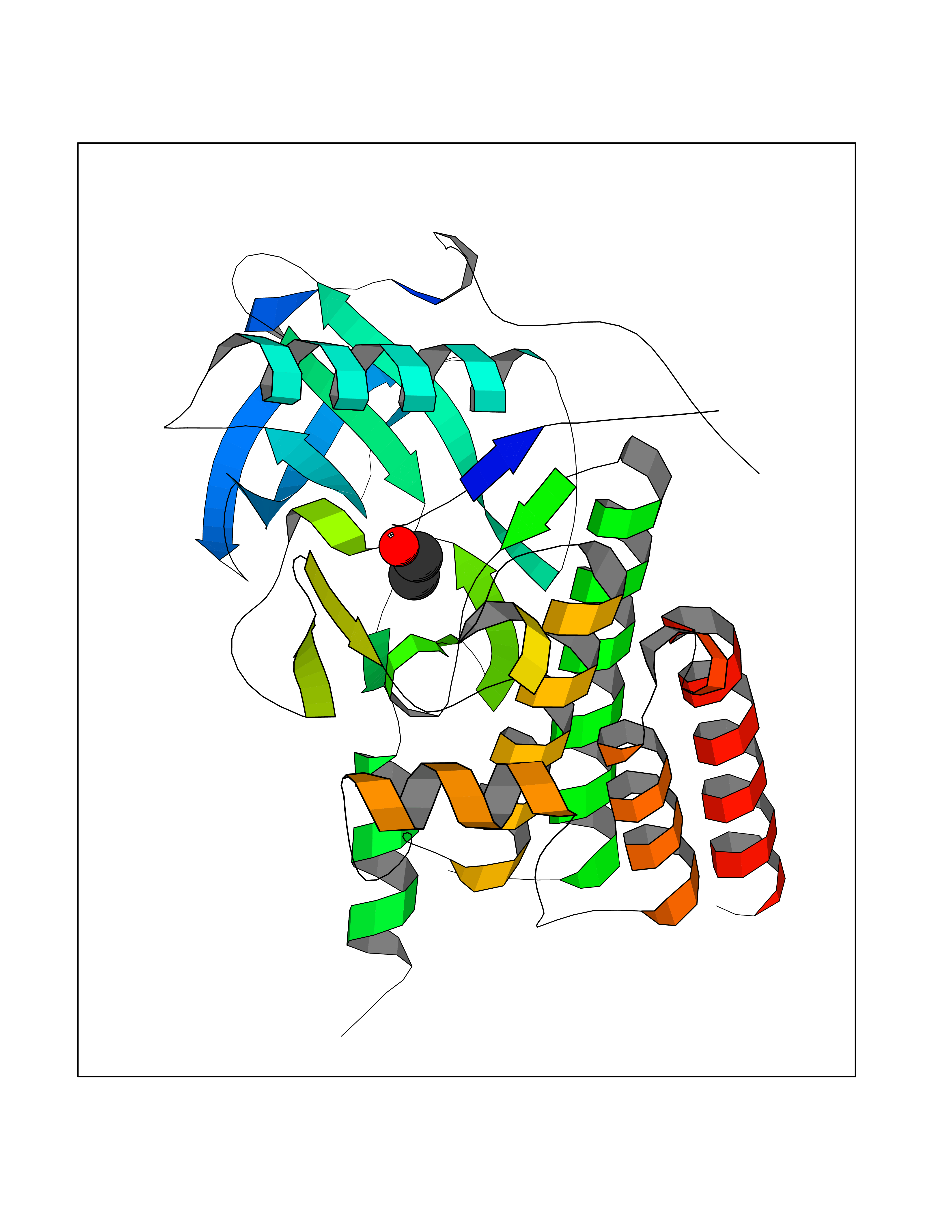 Molscript Map 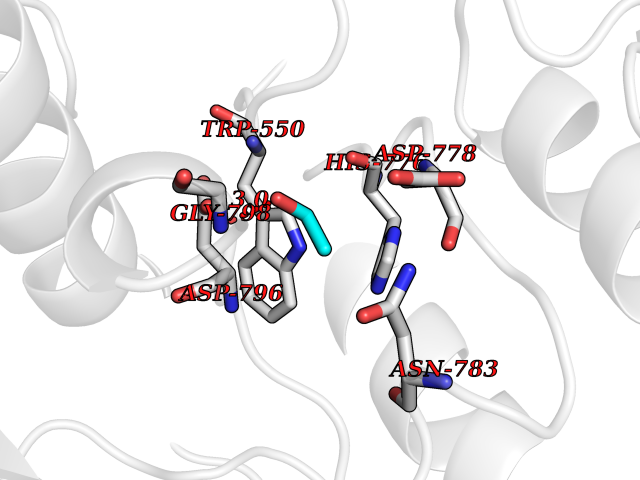 Pymol Map 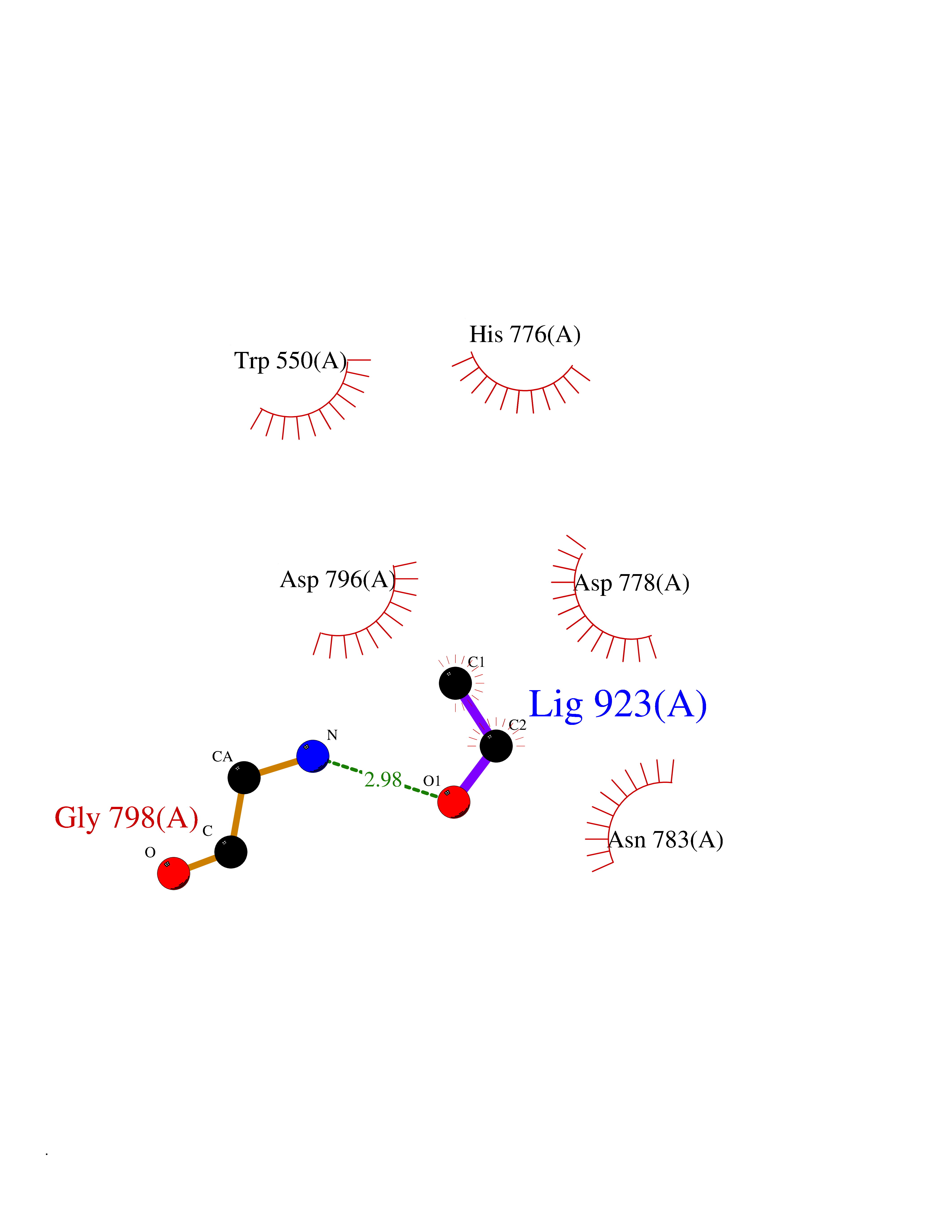 Ligplot Map 3D Binding mode Sequence QVRWKIIESYNSYTFIDPTQLPYNEKWEFPRNNLQFGKTLGAGAFGKVVEATAFGLGKEDAVLKVAVKMLKSTAHADEKEALMSELKIMSHLGQHENIVNLLGACTHGGPVLVITEYCCYGDLLNFLRRKSRVLETDSTASTRDLLHFSSQVAQGMAFLASKNCIHRDVAARNVLLTNGHVAKIGDFGLARDIMNDSNYIVKGNARLPVKWMAPESIFDCVYTVQSDVWSYGILLWEIFSLGLNPYPGILVNSKFYKLVKDGYQMAQPAFAPKNIYSIMQACWALEPTHRPTFQQICSFLQEQAQEDRRER Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Tyrosine 3-monooxygenase (TH) | 2XSN | 4.27 | |
Target general information Gen name TH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine 3-hydroxylase; TH Protein family Biopterin-dependent aromatic amino acid hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Plays an important role in the physiology of adrenergic neurones. Related diseases Segawa syndrome autosomal recessive (ARSEGS) [MIM:605407]: A form of DOPA-responsive dystonia presenting in infancy or early childhood. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Some cases present with parkinsonian symptoms in infancy. Unlike all other forms of dystonia, it is an eminently treatable condition, due to a favorable response to L-DOPA. {ECO:0000269|PubMed:10585338, ECO:0000269|PubMed:11196107, ECO:0000269|PubMed:11246459, ECO:0000269|PubMed:15505183, ECO:0000269|PubMed:15747353, ECO:0000269|PubMed:16049992, ECO:0000269|PubMed:17696123, ECO:0000269|PubMed:18058633, ECO:0000269|PubMed:18554280, ECO:0000269|PubMed:19491146, ECO:0000269|PubMed:20056467, ECO:0000269|PubMed:20430833, ECO:0000269|PubMed:21940685, ECO:0000269|PubMed:22264700, ECO:0000269|PubMed:22815559, ECO:0000269|PubMed:23762320, ECO:0000269|PubMed:23939262, ECO:0000269|PubMed:24753243, ECO:0000269|PubMed:7814018, ECO:0000269|PubMed:8528210, ECO:0000269|PubMed:8817341, ECO:0000269|PubMed:9613851, ECO:0000269|PubMed:9703425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: May play a role in the pathogenesis of Parkinson disease (PD). A genome-wide copy number variation analysis has identified a 34 kilobase deletion over the TH gene in a PD patient but not in any controls. {ECO:0000269|PubMed:20809526}. Drugs (DrugBank ID) DB03552; DB04400; DB00765; DB00120; DB00360; DB00135 Interacts with P29762; P61978-2; Q99750; P08651-5; O75928-2; Q9UHX1-2; P0DJD3-2; P07101-3; Q9UJ04; C9J7I0; Q5MCW4 EC number EC 1.14.16.2 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Cell projection; Cytoplasm; Cytoplasmic vesicle; Disease variant; Dystonia; Iron; Metal-binding; Monooxygenase; Neurotransmitter biosynthesis; Nucleus; Oxidoreductase; Parkinson disease; Parkinsonism; Phosphoprotein; Proteomics identification; Reference proteome; Synapse Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 34997 Length 306 Aromaticity 0.12 Instability index 42.59 Isoelectric point 5.32 Charge (pH=7) -12.31 2D Binding mode Binding energy (Kcal/mol) -5.83 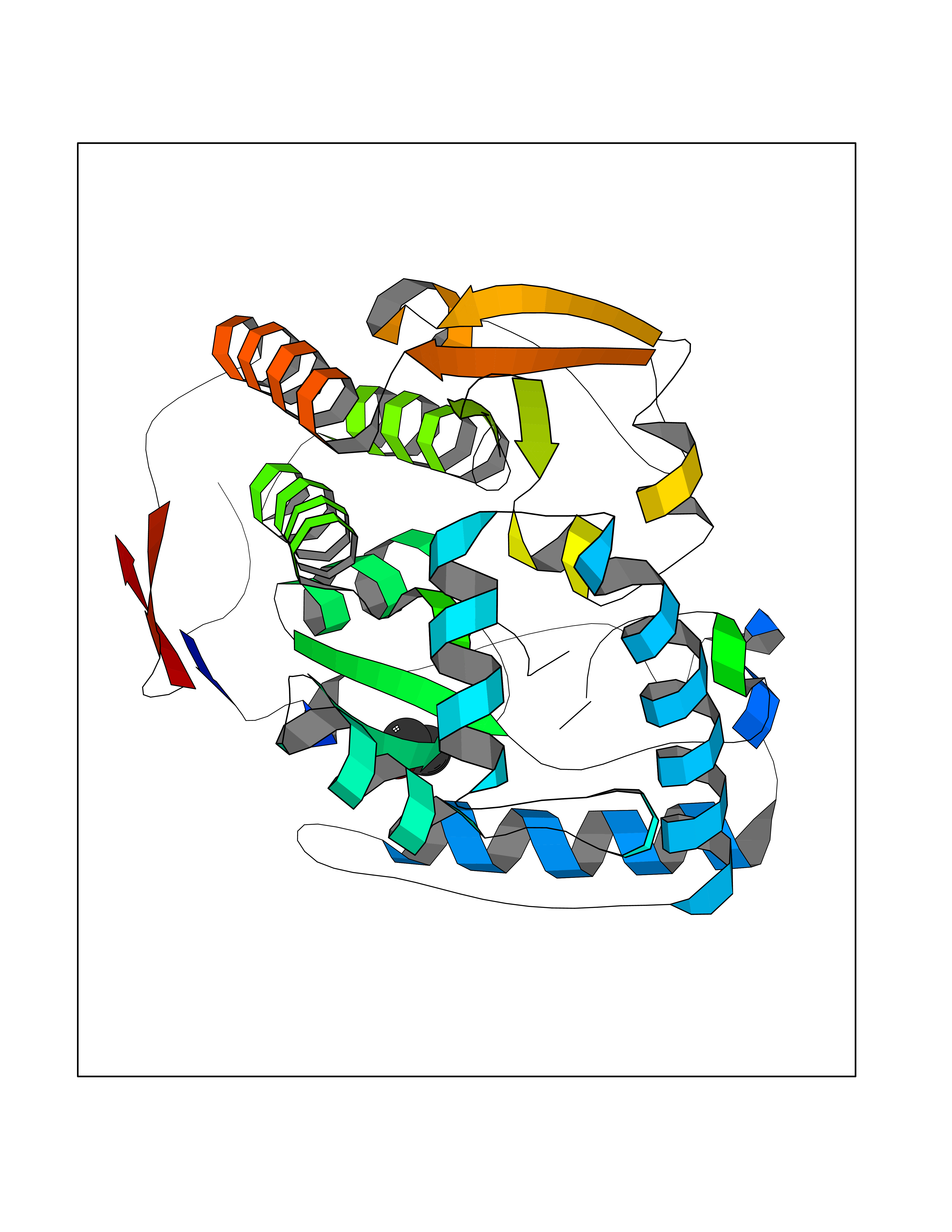 Molscript Map 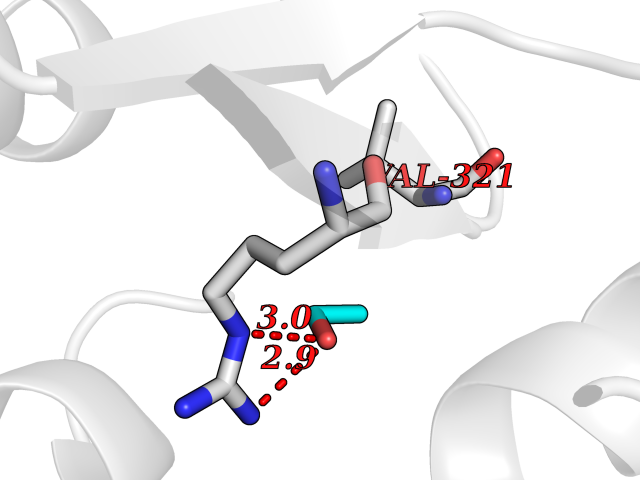 Pymol Map 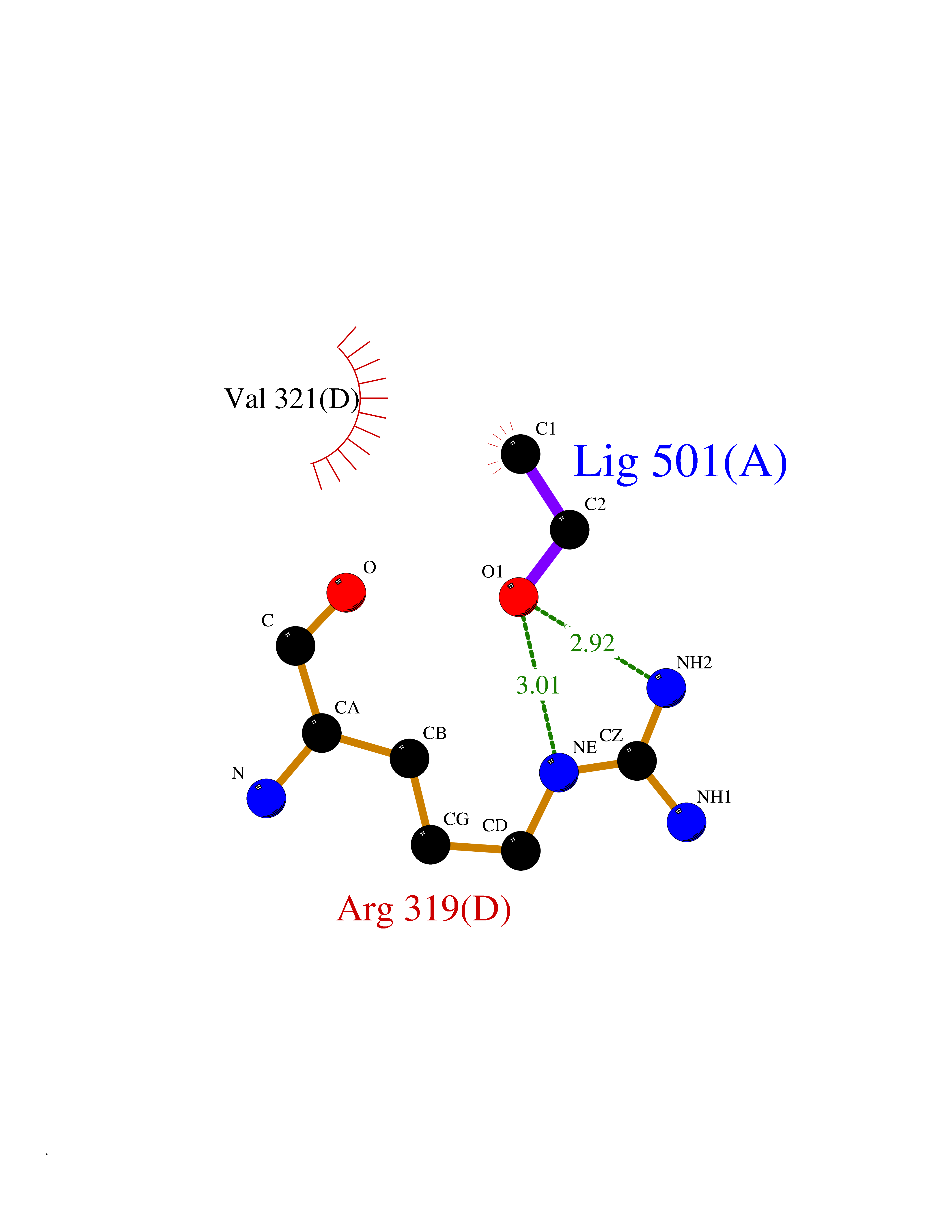 Ligplot Map 3D Binding mode Sequence VPWFPRKVSELDKCHHLVTKFDPDLDLDHPGFSDQVYRQRRKLIAEIAFQYRHGDPIPRVEYTAEEIATWKEVYTTLKGLYATHACGEHLEAFALLERFSGYREDNIPQLEDVSRFLKERTGFQLRPVAGLLSARDFLASLAFRVFQCTQYIRHASSPMHSPEPDCCHELLGHVPMLADRTFAQFSQDIGLASLGASDEEIEKLSTLYWFTVEFGLCKQNGEVKAYGAGLLSSYGELLHCLSEEPEIRAFDPEAAAVQPYQDQTYQSVYFVSESFSDAKDKLRSYASRIQRPFSVKFDPYTLAIDV Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Cytochrome P450 1A2 | 2HI4 | 4.27 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -5.83  Molscript Map 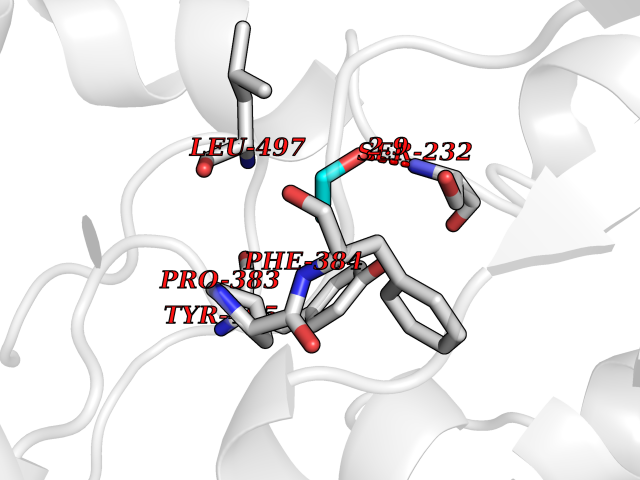 Pymol Map 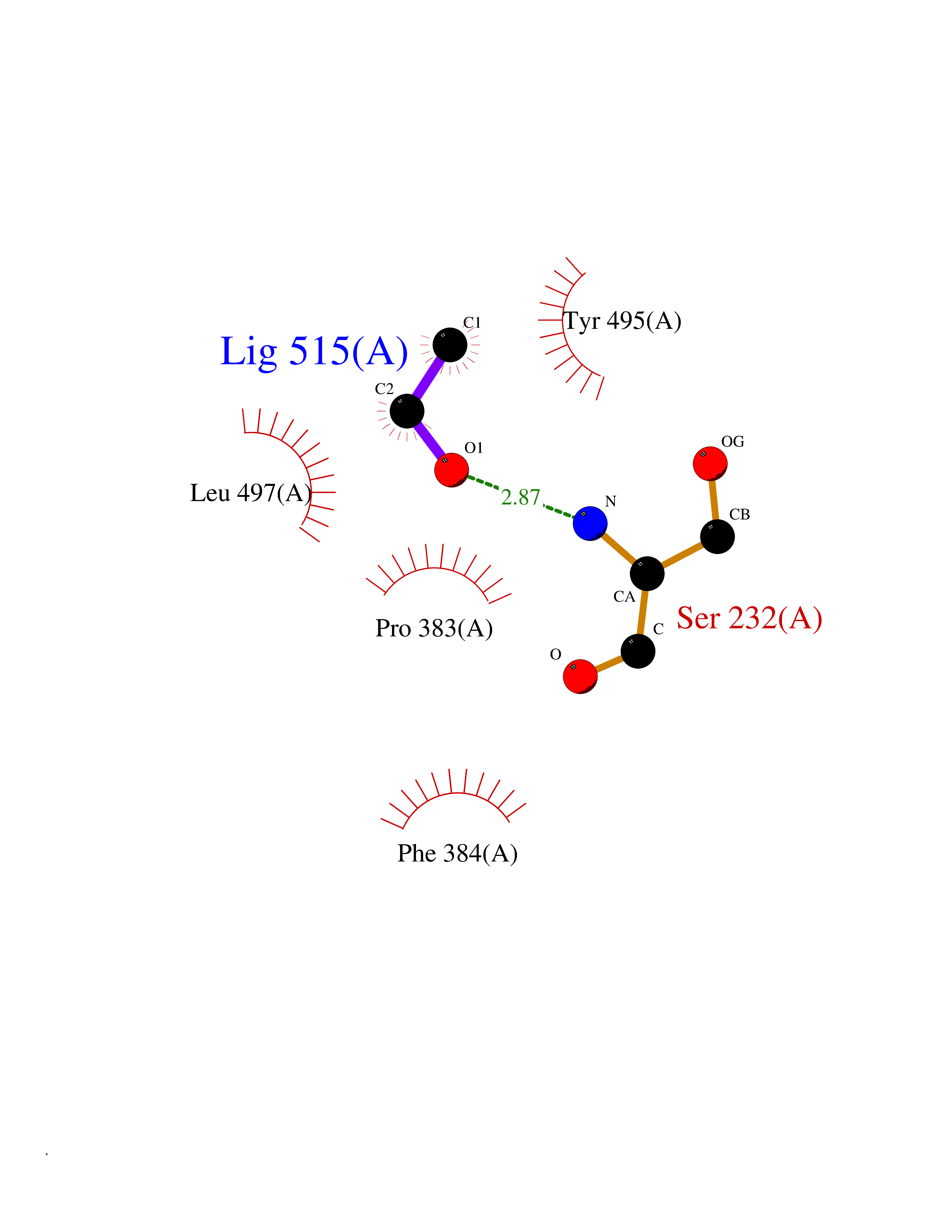 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Cytochrome P450 2D6 | 3TBG | 4.27 | |
Target general information Gen name CYP2D6 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CYP2DL1 Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Drug binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding.Steroid hydroxylase activity. Related diseases A chromosomal aberration involving BCL2 has been found in chronic lymphatic leukemia. Translocation t(14;18)(q32;q21) with immunoglobulin gene regions. BCL2 mutations found in non-Hodgkin lymphomas carrying the chromosomal translocation could be attributed to the Ig somatic hypermutation mechanism resulting in nucleotide transitions. {ECO:0000269|PubMed:2875799, ECO:0000269|PubMed:3285301}. Drugs (DrugBank ID) DB01562; DB01472; DB14010; DB12001; DB05812; DB01193; DB00316; DB15568; DB00918; DB06203; DB00866; DB01424; DB01118; DB00321; DB00381; DB00613; DB00543; DB00182; DB00701; DB11785; DB01435; DB01429; DB01274; DB01238; DB14185; DB09204; DB11638; DB06216; DB00637; DB11586; DB00335; DB00289; DB01076; DB00972; DB04957; DB09013; DB16703; DB01086; DB06770; DB01244; DB15982; DB00195; DB01295; DB12236; DB01128; DB04889; DB00810; DB13975; DB08807; DB00188; DB09128; DB12151; DB12752; DB06726; DB00297; DB08808; DB00921; DB01156; DB00490; DB09173; DB00201; DB09061; DB14737; DB06016; DB00521; DB01136; DB00482; DB04846; DB00439; DB00185; DB00608; DB01114; DB00477; DB00356; DB01410; DB01166; DB00501; DB01012; DB00568; DB00604; DB00215; DB12499; DB00283; DB04920; DB14025; DB00349; DB00845; DB01242; DB00575; DB13508; DB00257; DB00363; DB09065; DB05239; DB00907; DB00318; DB11672; DB14635; DB00924; DB00091; DB11963; DB06292; DB04884; DB00496; DB01264; DB09183; DB04840; DB00705; DB06512; DB01151; DB06700; DB16650; DB12161; DB13679; DB09555; DB01191; DB00633; DB01576; DB00514; DB00647; DB11994; DB01551; DB00343; DB01093; DB01075; DB00757; DB01184; DB00843; DB09167; DB00590; DB01142; DB00997; DB00470; DB04855; DB00476; DB00625; DB11979; DB00216; DB15444; DB09039; DB13874; DB01228; DB06735; DB11718; DB00494; DB13757; DB00751; DB00530; DB13443; DB01175; DB06678; DB00187; DB00330; DB01466; DB01628; DB01590; DB12500; DB01023; DB00574; DB06702; DB12265; DB01195; DB04841; DB00472; DB00623; DB01095; DB00176; DB00983; DB02703; DB15149; DB00674; DB05087; DB00317; DB08909; DB00986; DB01218; DB00502; DB00956; DB01611; DB00557; DB09053; DB01177; DB04946; DB00619; DB00458; DB08952; DB00224; DB06370; DB13293; DB04818; DB16200; DB11633; DB06636; DB00951; DB11757; DB00602; DB09570; DB01026; DB00598; DB12212; DB00448; DB11732; DB16217; DB09078; DB00528; DB12070; DB09351; DB01210; DB08918; DB00281; DB04948; DB01206; DB00836; DB01601; DB00455; DB04871; DB09195; DB06708; DB04829; DB09238; DB00934; DB14921; DB00737; DB14009; DB09224; DB00170; DB00454; DB00532; DB13530; DB06691; DB01071; DB00933; DB01577; DB00333; DB00763; DB01403; DB01028; DB09241; DB01214; DB01233; DB00264; DB00379; DB06148; DB01388; DB01110; DB00211; DB01454; DB06595; DB00834; DB00805; DB08893; DB00370; DB12523; DB01171; DB00745; DB14011; DB09049; DB00731; DB04861; DB01149; DB00220; DB09048; DB00238; DB00627; DB00622; DB00699; DB02701; DB00184; DB01115; DB04868; DB12005; DB00540; DB00334; DB14881; DB00338; DB00904; DB11130; DB04911; DB01173; DB11837; DB04938; DB01096; DB01580; DB01062; DB00497; DB06412; DB01192; DB01267; DB00377; DB06603; DB00715; DB06589; DB00022; DB01359; DB00738; DB01074; DB08922; DB00850; DB03783; DB00780; DB00914; DB00252; DB05316; DB01100; DB00960; DB00592; DB01621; DB04951; DB17472; DB11642; DB08901; DB01297; DB15822; DB01087; DB01035; DB00433; DB00396; DB01131; DB00420; DB01069; DB09288; DB01182; DB00571; DB04216; DB01224; DB00908; DB00468; DB01129; DB00863; DB00243; DB00234; DB14761; DB00409; DB06506; DB02709; DB11855; DB13174; DB11753; DB08864; DB14840; DB00734; DB12693; DB00503; DB00953; DB09291; DB15119; DB00412; DB05271; DB12332; DB11614; DB06654; DB01232; DB01037; DB06144; DB01104; DB00203; DB00641; DB01591; DB00398; DB12713; DB00489; DB06727; DB01323; DB09118; DB06820; DB06729; DB06608; DB11770; DB00675; DB00706; DB06204; DB06083; DB01079; DB12095; DB06287; DB00857; DB00342; DB13775; DB04905; DB04844; DB11712; DB00277; DB00679; DB01623; DB00208; DB00373; DB01409; DB00932; DB06137; DB01036; DB05109; DB00193; DB00752; DB00656; DB12245; DB00726; DB00792; DB00209; DB15328; DB09076; DB13609; DB15091; DB11915; DB00862; DB08881; DB00285; DB00661; DB06217; DB06684; DB09185; DB00570; DB00361; DB11739; DB09068; DB01392; DB00549; DB15688; DB00425; DB01624 Interacts with NA EC number 1.14.14.- Uniprot keywords 3D-structure; Alternative splicing; Cholesterol metabolism; Endoplasmic reticulum; Fatty acid metabolism; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 51178.2 Length 456 Aromaticity 0.09 Instability index 44.04 Isoelectric point 6.57 Charge (pH=7) -1.99 2D Binding mode Binding energy (Kcal/mol) -5.82  Molscript Map 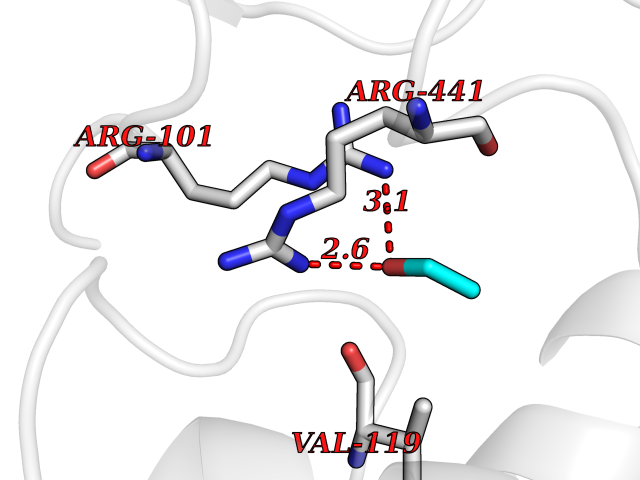 Pymol Map 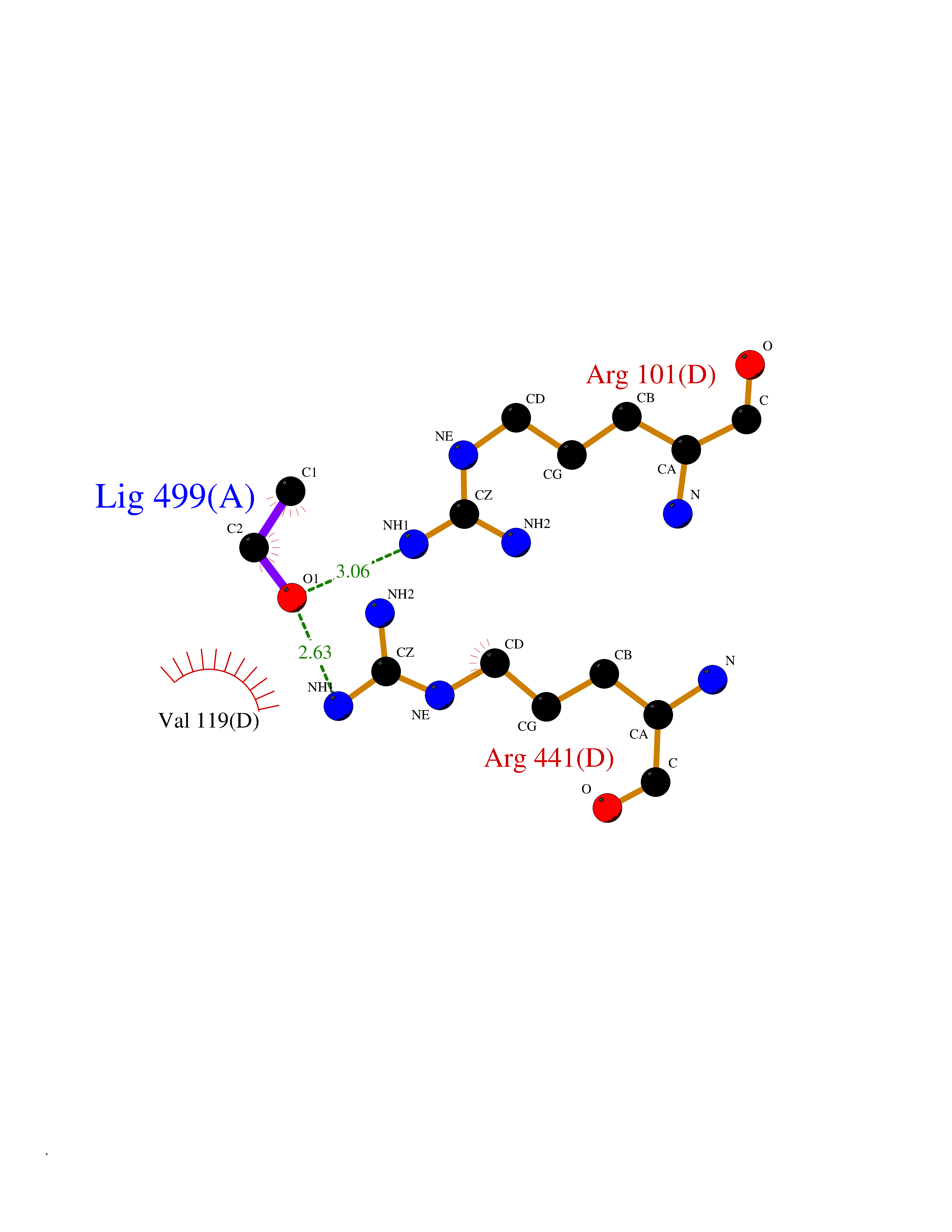 Ligplot Map 3D Binding mode Sequence KLPPGPLPDFQNTPYCFDQLRRRFGDVFSLQLAWTPVVVLNGLAAVREALVTHGEDTADRPPVPITQILGFGPRSQGVFLARYGPAWREQRRFSVSTLRNLGLGKKSLEQWVTEEAACLCAAFANHSGRPFRPNGLLDKAVSNVIASLTCGRRFEYDDPRFLRLLDLAQEGLKEESGFLREVLNAVPVLLHIPALAGKVLRFQKAFLTQLDELLTEHRMTWDPAQPPRDLTEAFLAEMEKAKGNPESSFNDENLRIVVADLFSAGMVTTSTTLAWGLLLMILHPDVQRRVQQEIDDVIGQVRRPEMGDQAHMPYTTAVIHEVQRFGDIVPLGVTHMTSRDIEVQGFRIPKGTTLITNLSSVLKDEAVWEKPFRFHPEHFLDAQGHFVKPEAFLPFSAGRRACLGEPLARMELFLFFTSLLQHFSFSVPTGQPRPSHHGVFAFLVSPSPYELCAVPR Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Chromodomain-helicase-DNA-binding protein 1 | 4O42 | 4.27 | |
Target general information Gen name CHD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SNF2/RAD54 helicase family Biochemical class Dna binding protein / viral protein Function ATP binding.ATP-dependent DNA helicase activity.DNA binding.Methylated histone binding. Related diseases Pilarowski-Bjornsson syndrome (PILBOS) [MIM:617682]: An autosomal dominant disorder characterized by developmental delay, speech apraxia, intellectual disability, autism, and facial dysmorphic features. Some patients may have seizures. {ECO:0000269|PubMed:28866611}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60341-1; B2BUF1; P28799; O76024 EC number 3.6.4.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromatin regulator; Cytoplasm; Disease variant; DNA-binding; Helicase; Hydrolase; Intellectual disability; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 20969.1 Length 180 Aromaticity 0.12 Instability index 46.35 Isoelectric point 5.88 Charge (pH=7) -2.83 2D Binding mode Binding energy (Kcal/mol) -5.82 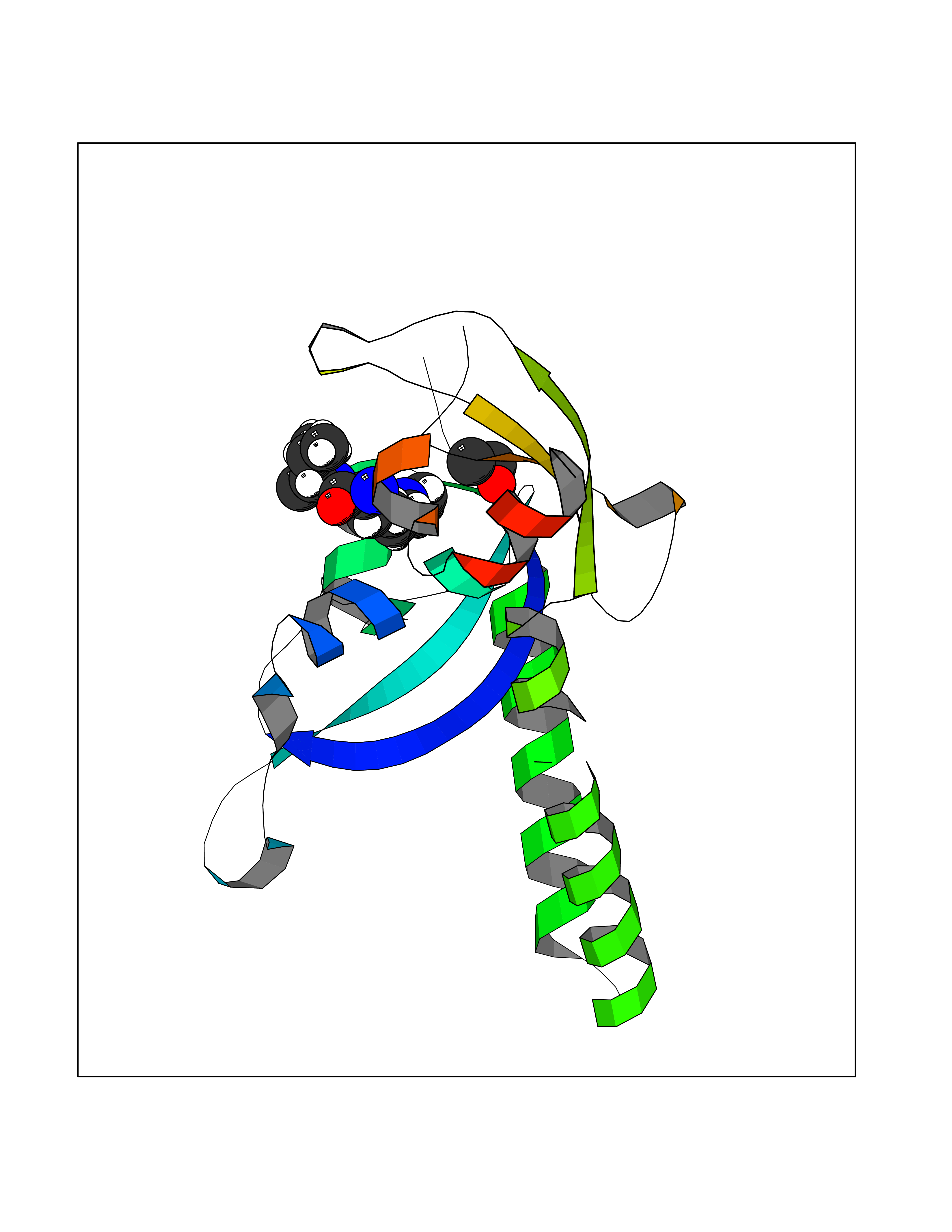 Molscript Map  Pymol Map 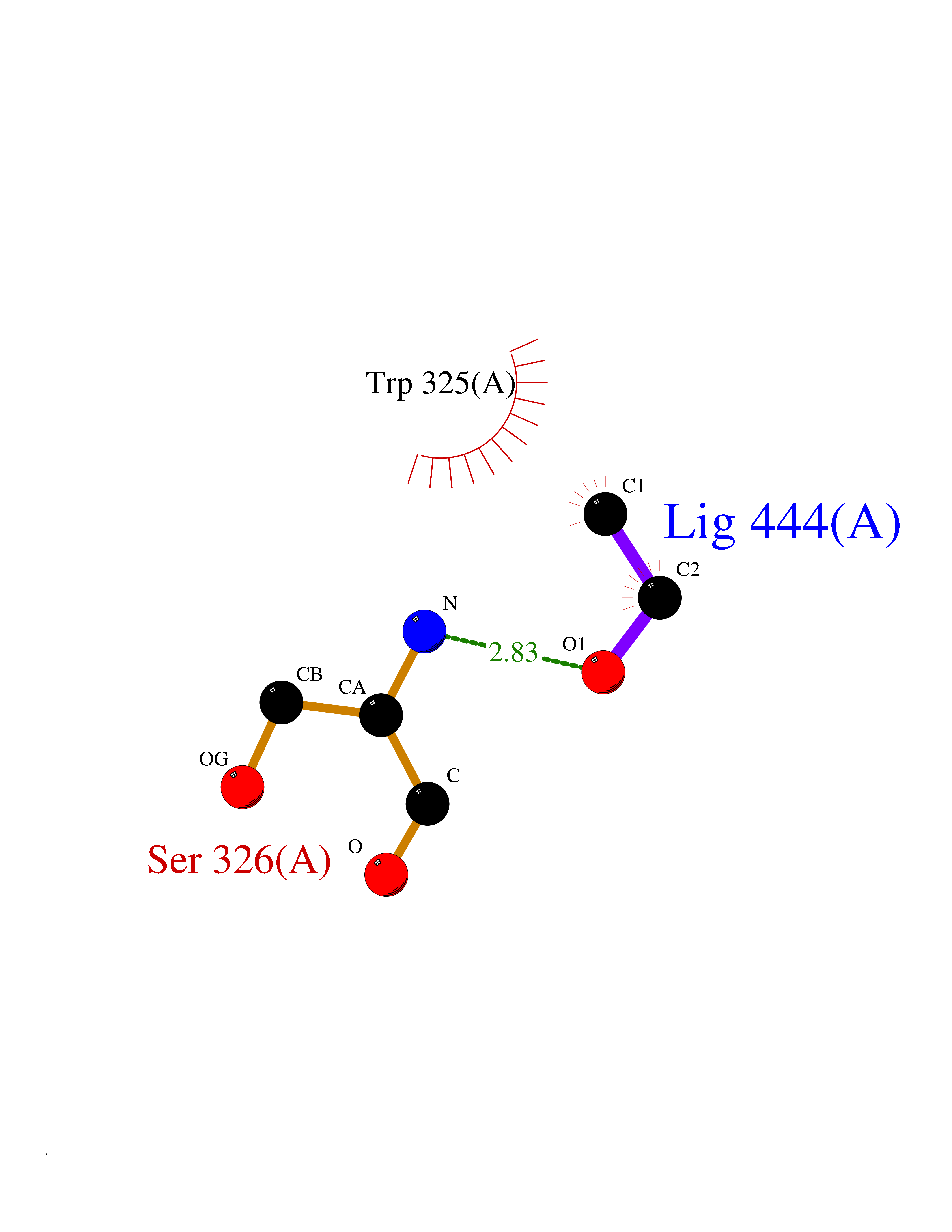 Ligplot Map 3D Binding mode Sequence EFETIERFMDCRIGRKGATGATTTIYAVEADGDPNAGFEKNKEPGEIQYLIKWKGWSHIHNTWETEETLKQQNVRGMKKLDNYKKKDQETKRWLKNASPEDVEYYNCQQELTDDLHKQYQIVERIIAHSNQKSAAGYPDYYCKWQGLPYSECSWEDGALISKKFQACIDEYFSRTARSXV Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Alkyl hydroperoxide reductase subunit F | 1HYU | 4.27 | |
Target general information Gen name ahpF Organism Salmonella typhimurium (strain LT2 / SGSC1412 / ATCC 700720) Uniprot ID TTD ID NA Synonyms STM0609 Protein family Class-II pyridine nucleotide-disulfide oxidoreductase family Biochemical class Oxidoreductase Function Alkyl hydroperoxide reductase activity.Electron carrier activity.Flavin adenine dinucleotide binding.NAD binding.Protein disulfide oxidoreductase activity. Related diseases Glutathionuria (GLUTH) [MIM:231950]: A very rare, autosomal recessive metabolic disorder characterized by the presence of glutathione in the urine, due to generalized gamma-glutamyl transpeptidase deficiency. Most patients manifest mild to moderate intellectual disability, and behavioral disturbance. Seizures, tremor, marfanoid features and strabismus are observed in some patients. {ECO:0000269|PubMed:29483667}. The disease is caused by variants affecting the gene represented in this entry. A large homozygous deletion that removes several exons of all isoforms of GGT1 has been found in one family affected by glutathionuria. {ECO:0000269|PubMed:29483667}. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.8.1.- Uniprot keywords 3D-structure; Direct protein sequencing; Disulfide bond; FAD; Flavoprotein; NAD; NADP; Oxidoreductase; Redox-active center; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 55949.2 Length 521 Aromaticity 0.06 Instability index 25.77 Isoelectric point 5.62 Charge (pH=7) -9.6 2D Binding mode Binding energy (Kcal/mol) -5.83 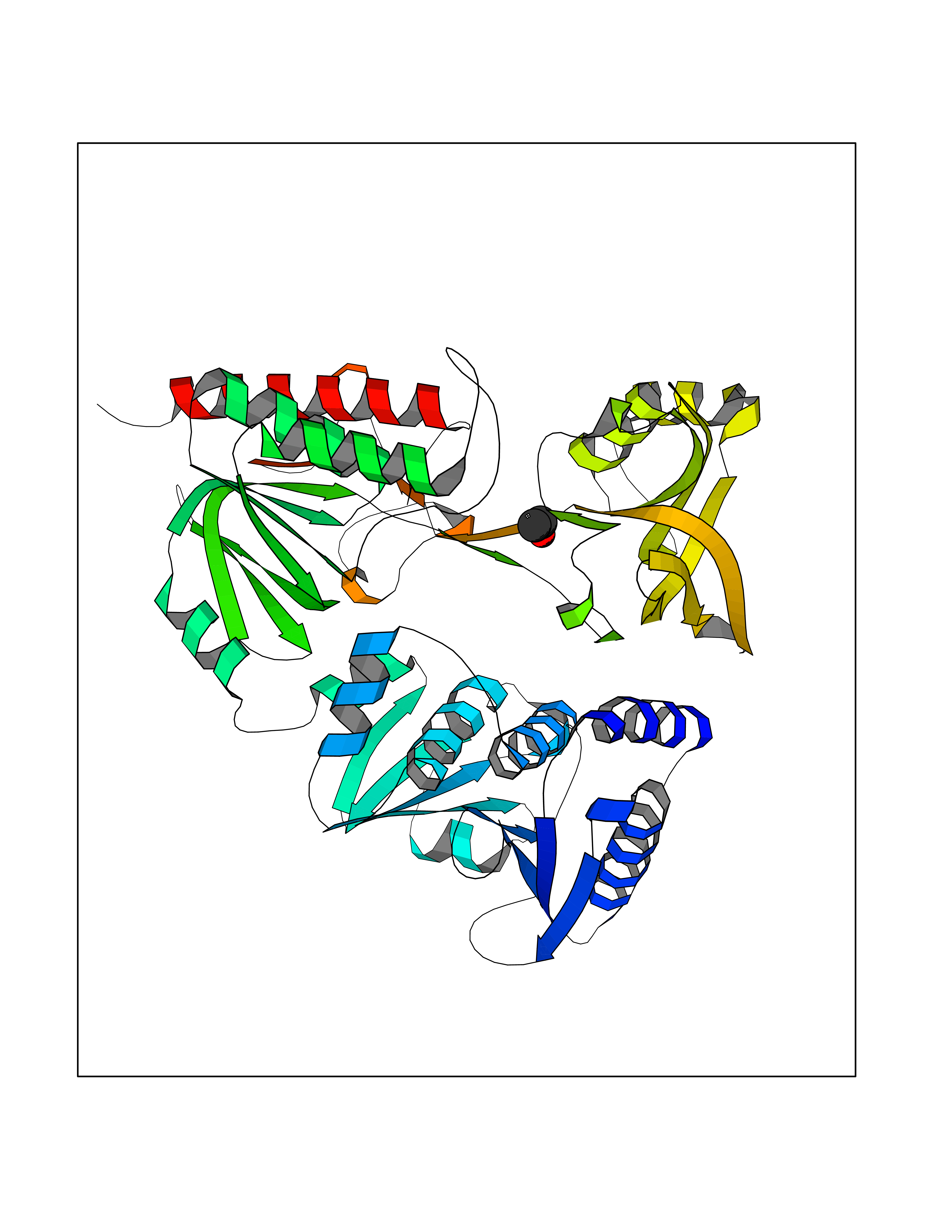 Molscript Map 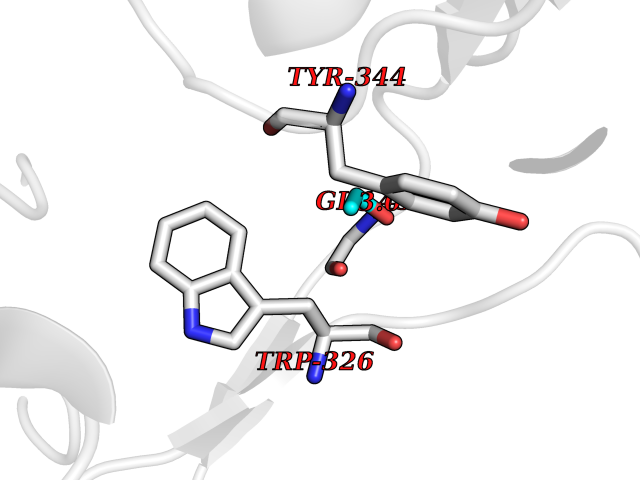 Pymol Map 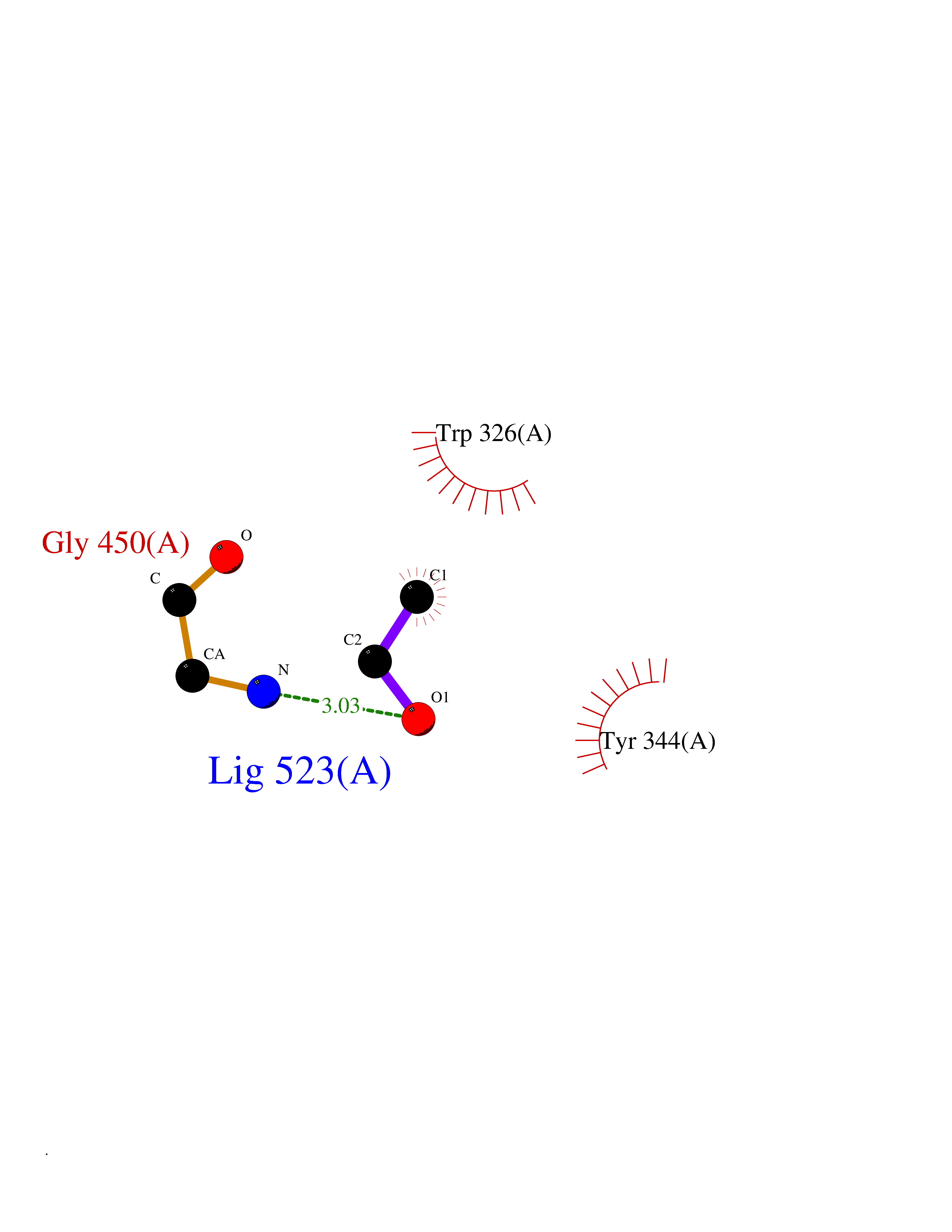 Ligplot Map 3D Binding mode Sequence MLDTNMKTQLRAYLEKLTKPVELIATLDDSAKSAEIKELLAEIAELSDKVTFKEDNTLPVRKPSFLITNPGSQQGPRFAGSPLGHEFTSLVLALLWTGGHPSKEAQSLLEQIRDIDGDFEFETYYSLSCHNCPDVVQALNLMAVLNPRIKHTAIDGGTFQNEITERNVMGVPAVFVNGKEFGQGRMTLTEIVAKVDTGAEKRAAEALNKRDAYDVLIVGSGPAGAAAAVYSARKGIRTGLMGERFGGQVLDTVDIENYISVPKTEGQKLAGALKAHVSDYDVDVIDSQSASKLVPAATEGGLHQIETASGAVLKARSIIIATGAKWRNMNVPGEDQYRTKGVTYCPHCDGPLFKGKRVAVIGGGNSGVEAAIDLAGIVEHVTLLEFAPEMKADQVLQDKVRSLKNVDIILNAQTTEVKGDGSKVVGLEYRDRVSGDIHSVALAGIFVQIGLLPNTHWLEGALERNRMGEIIIDAKCETSVKGVFAAGDCTTVPYKQIIIATGEGAKASLSAFDYLIRTKIA Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Thioredoxin reductase (PRDX5) | 3MNG | 4.27 | |
Target general information Gen name PRDX5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Thioredoxin peroxidase PMP20; TPx type VI; SBBI10; PrxV; Prx-V; Peroxisomal antioxidant enzyme; Peroxiredoxin5, mitochondrial; Peroxiredoxin-5, mitochondrial; Peroxiredoxin V; PLP; Liver tissue 2Dpage Protein family Peroxiredoxin family, Prx5 subfamily Biochemical class Peroxide acceptor oxidoreductase Function Plays a role in cell protection against oxidative stress by detoxifying peroxides and as sensor of hydrogen peroxide-mediated signaling events. Thiol-specific peroxidase that catalyzes the reduction of hydrogen peroxide and organic hydroperoxides to water and alcohols, respectively. Related diseases Variation Asp-298 in NOS3 may be associated with susceptibility to coronary spasm. {ECO:0000269|PubMed:11740345, ECO:0000269|PubMed:9737779}. Drugs (DrugBank ID) DB00995; DB03608; DB09221 Interacts with P55273; Q7Z417; P49901; P00441; P30044-2 EC number EC 1.11.1.15 Uniprot keywords 3D-structure; Acetylation; Alternative initiation; Alternative splicing; Antioxidant; Cytoplasm; Direct protein sequencing; Disulfide bond; Lipoprotein; Mitochondrion; Oxidoreductase; Palmitate; Peroxidase; Peroxisome; Phosphoprotein; Proteomics identification; Redox-active center; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 16899.3 Length 161 Aromaticity 0.06 Instability index 19.82 Isoelectric point 6.96 Charge (pH=7) -0.04 2D Binding mode Binding energy (Kcal/mol) -5.82 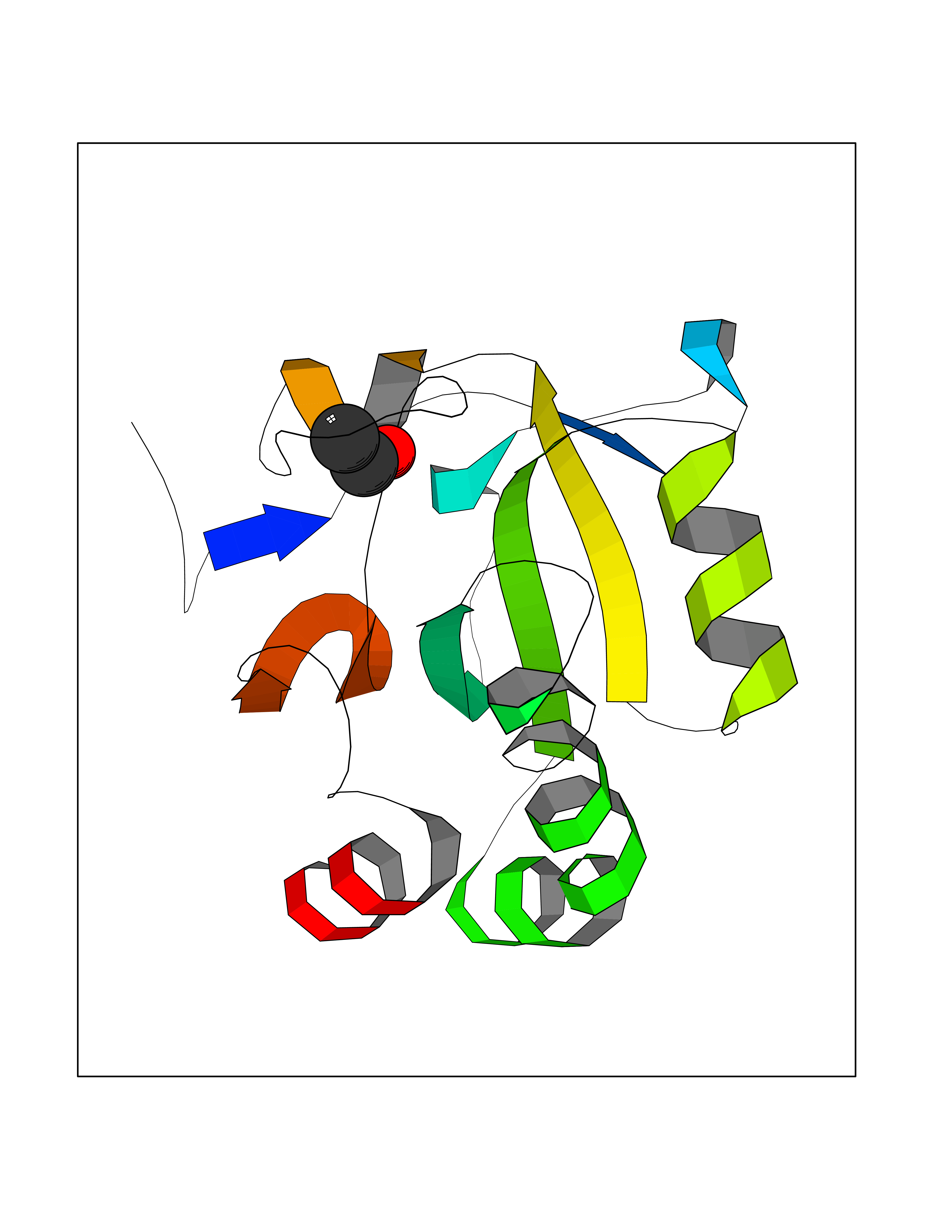 Molscript Map 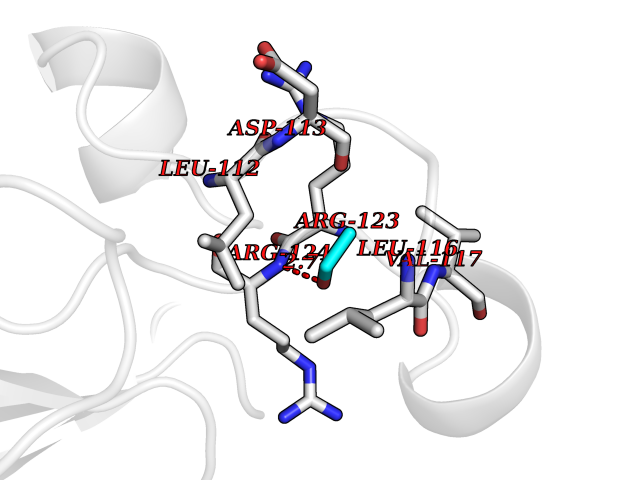 Pymol Map 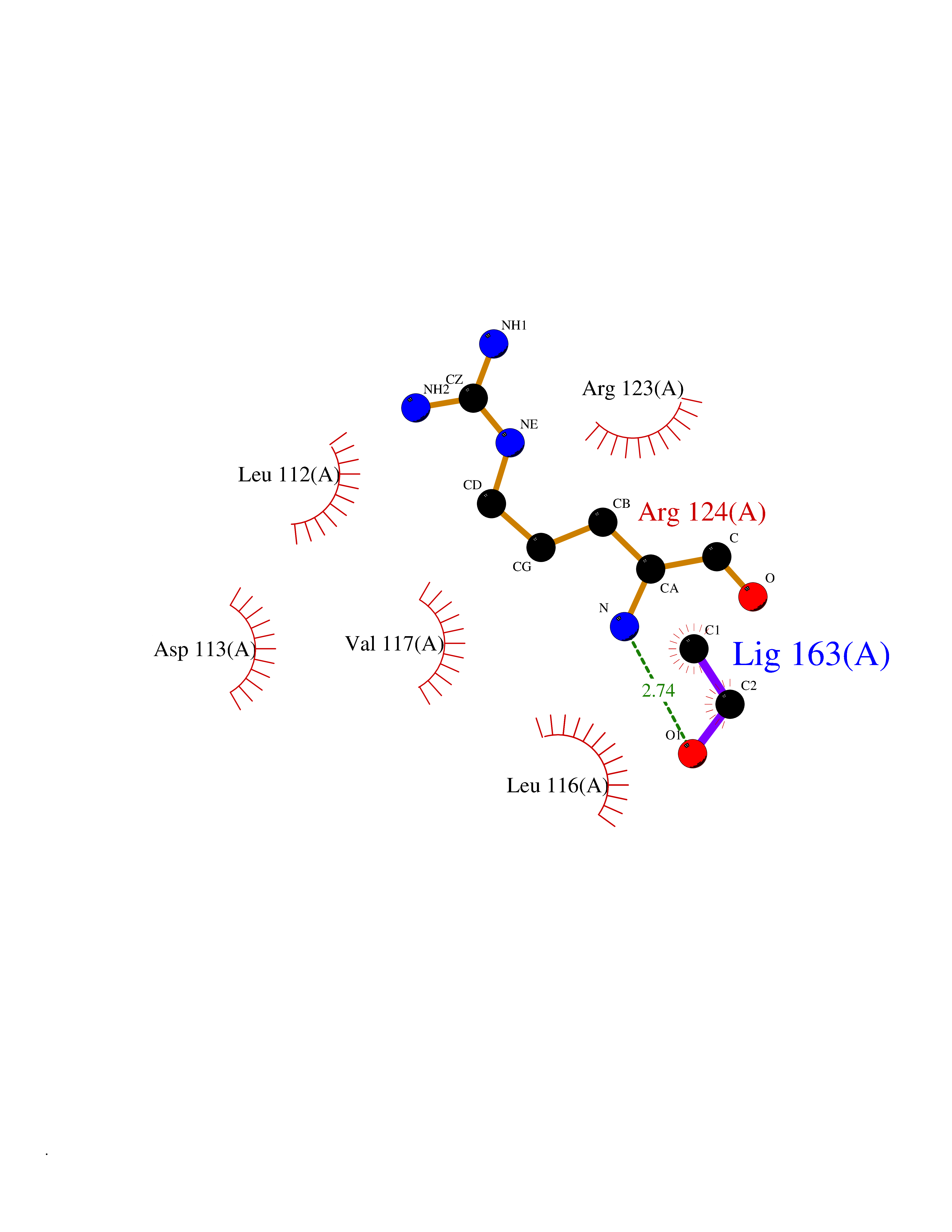 Ligplot Map 3D Binding mode Sequence APIKVGDAIPAVEVFEGEPGNKVNLAELFKGKKGVLFGVPGAFTPGCSKTHLPGFVEQAEALKAKGVQVVACLSVNDAFVTGEWGRAHKAEGKVRLLADPTGAFGKETDLLLDDSLVSIFGNRRLKRFSMVVQDGIVKALNVEPDGTGLTCSLAPNIISQL Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Thiopurine S-methyltransferase | 2BZG | 4.27 | |
Target general information Gen name TPMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, TPMT family Biochemical class Transferase Function Thiopurine S-methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00993; DB00436; DB01327; DB01033; DB01250; DB01021 Interacts with Q8TAP4-4; Q15047-2; P61981 EC number 2.1.1.67 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 25971.5 Length 229 Aromaticity 0.12 Instability index 32.58 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -5.82 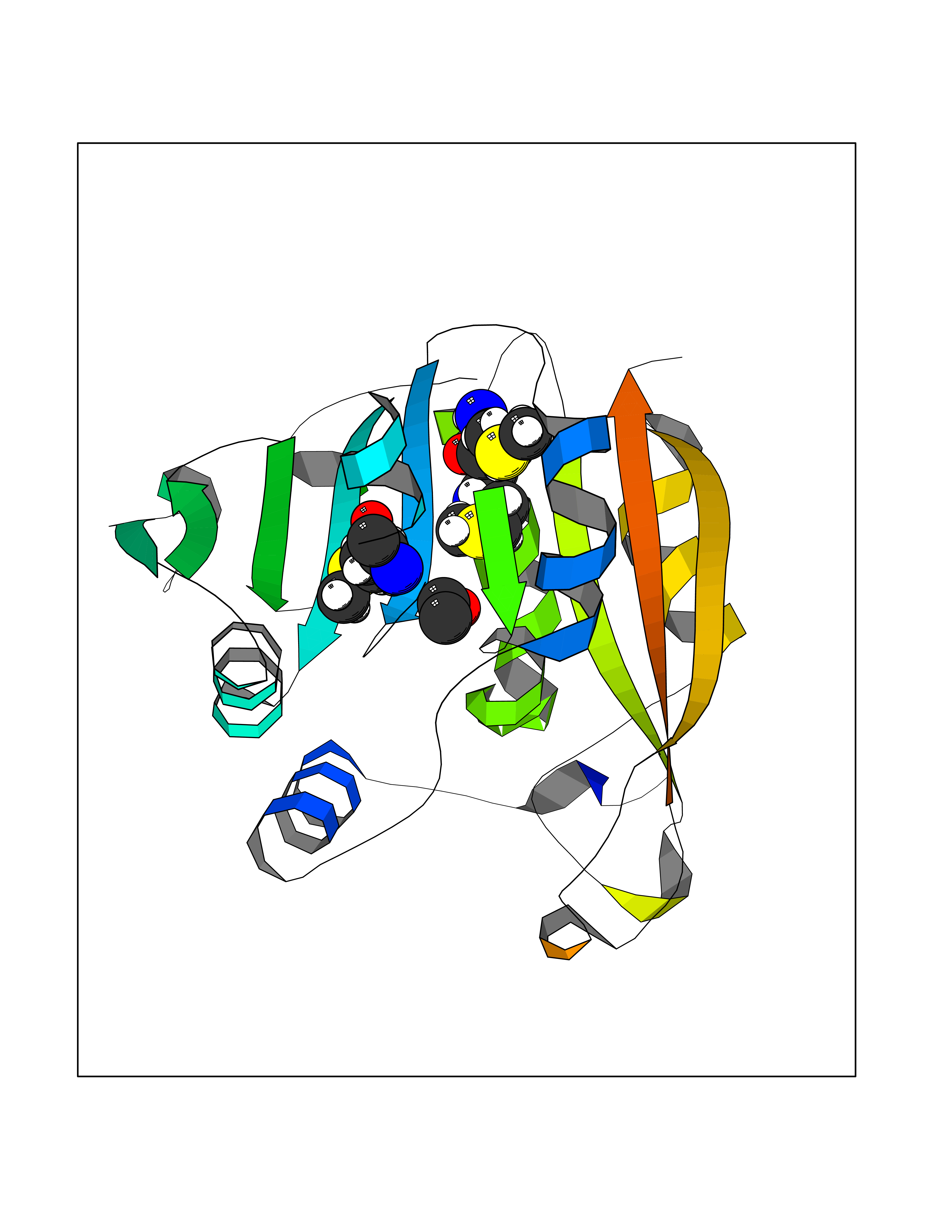 Molscript Map 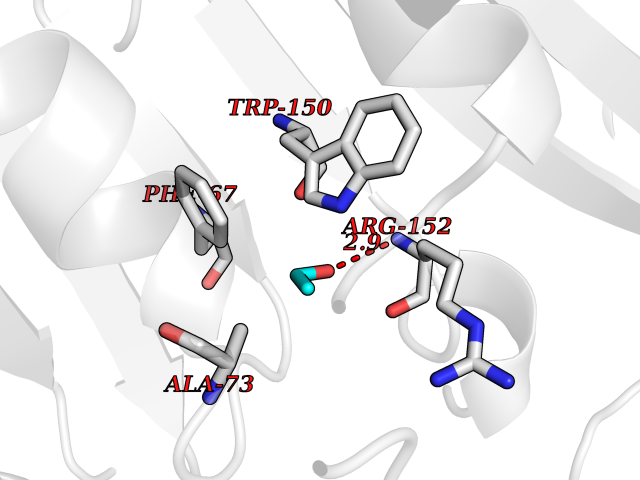 Pymol Map 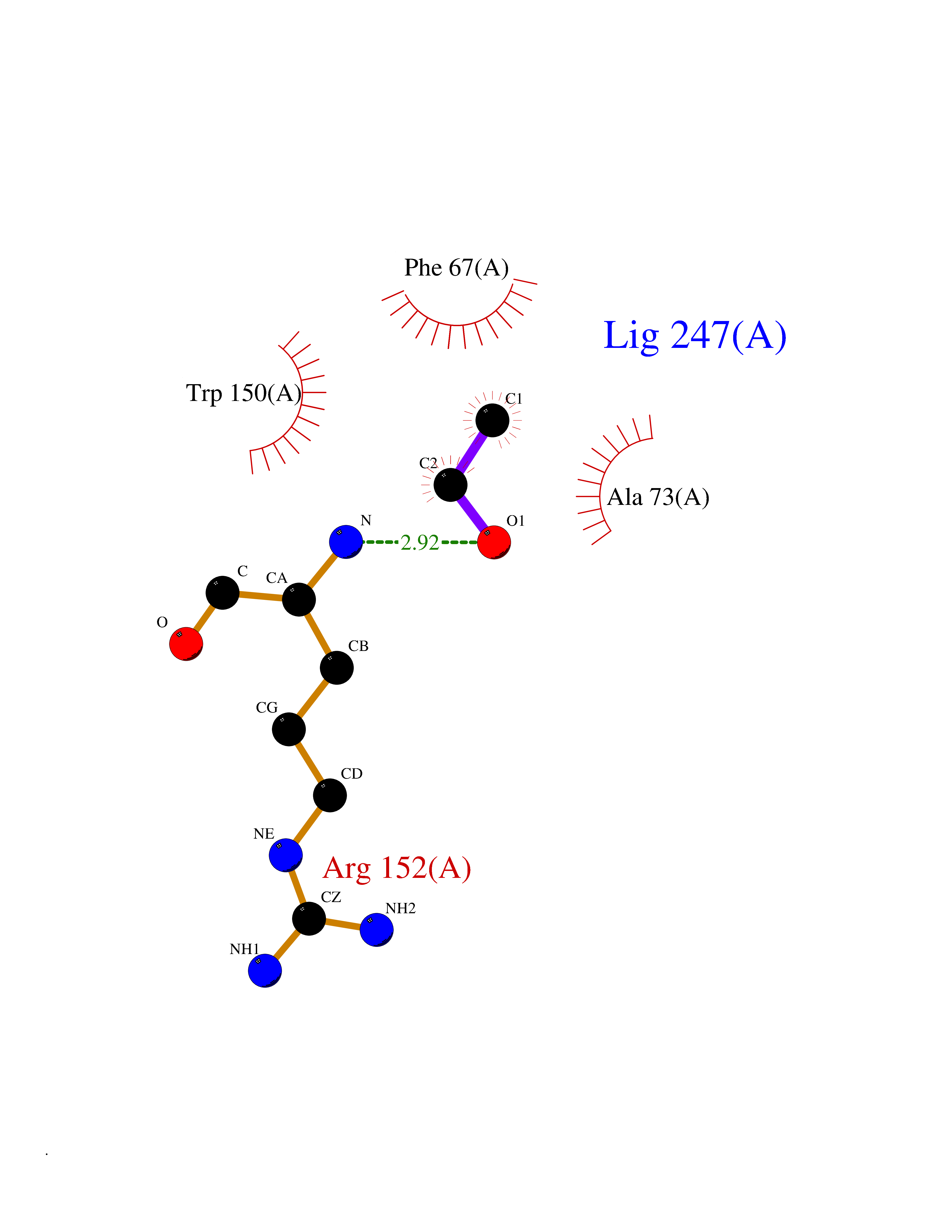 Ligplot Map 3D Binding mode Sequence EVQKNQVLTLEEWQDKWVNGKTAFHQEQGHQLLKKHLDTFLKGKSGLRVFFPLCGKAVEXKWFADRGHSVVGVEISELGIQEFFTEQNLSYSEEPITEIPGTKVFKSSSGNISLYCCSIFDLPRTNIGKFDXIWDRGALVAINPGDRKCYADTXFSLLGKKFQYLLCVLSYDPTKHPGPPFYVPHAEIERLFGKICNIRCLEKVDAFEERHKSWGIDCLFEKLYLLTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 4.27 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -5.82  Molscript Map 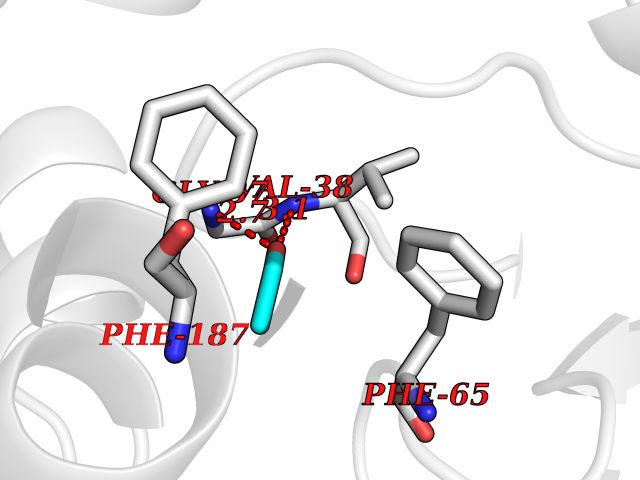 Pymol Map 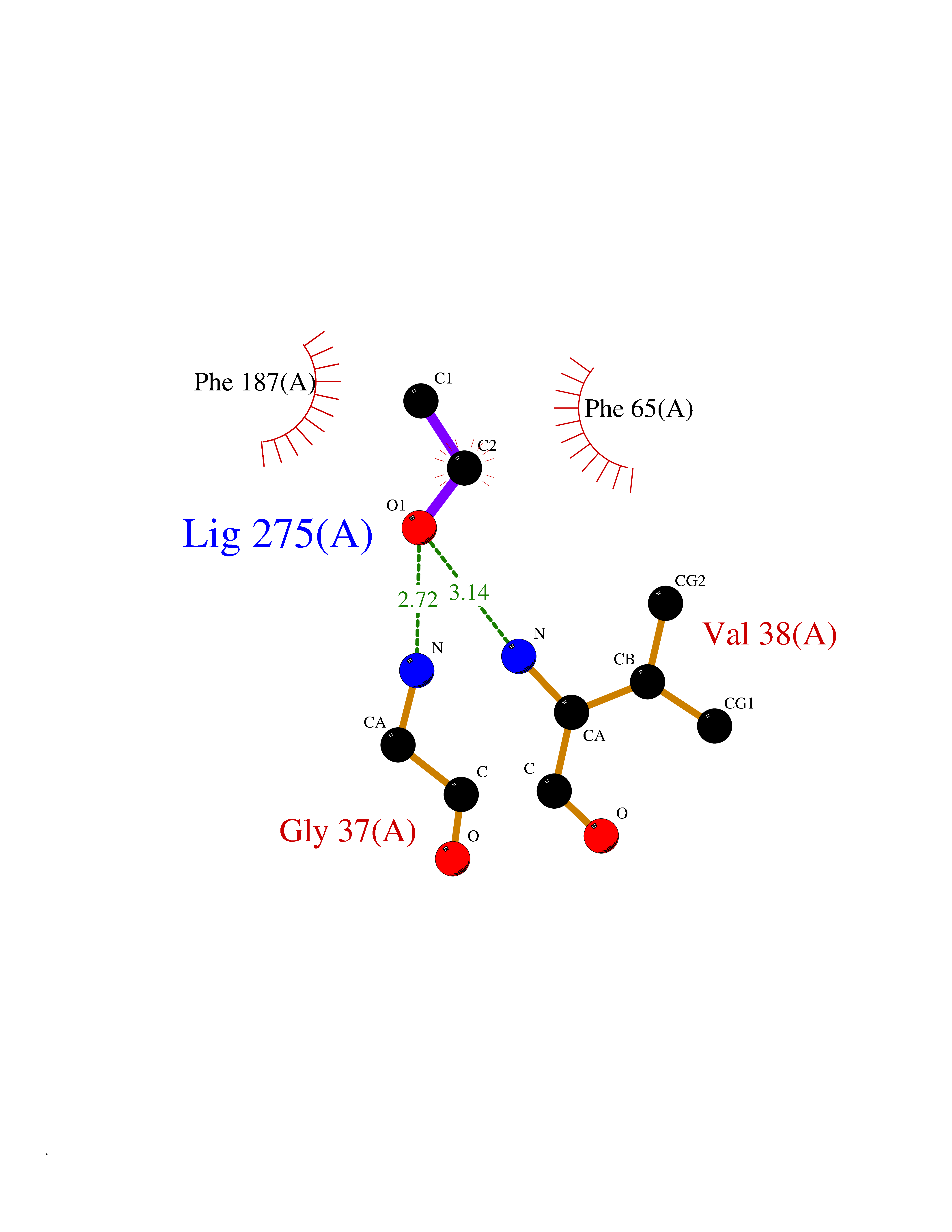 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||