Job Results:
Ligand
Structure
Job ID
0fdb6be2dae96620ab05e05b4d1adc2b
Job name
NA
Time
2025-04-03 17:50:26
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Vascular endothelial growth factor receptor 2 (KDR) | 2XIR | 7.03 | |
Target general information Gen name KDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms VEGFR2; VEGFR-2; VEGF-2 receptor; Protein-tyrosine kinase receptor flk-1; Kinase insert domain receptor; Fetal liver kinase 1; FLK1; FLK-1; CD309 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Plays an essential role in the regulation of angiogenesis, vascular development, vascular permeability, and embryonic hematopoiesis. Promotes proliferation, survival, migration and differentiation of endothelial cells. Promotes reorganization of the actin cytoskeleton. Isoforms lacking a transmembrane domain, such as isoform 2 and isoform 3, may function as decoy receptors for VEGFA, VEGFC and/or VEGFD. Isoform 2 plays an important role as negative regulator of VEGFA- and VEGFC-mediated lymphangiogenesis by limiting the amount of free VEGFA and/or VEGFC and preventing their binding to FLT4. Modulates FLT1 and FLT4 signaling by forming heterodimers. Binding of vascular growth factors to isoform 1 leads to the activation of several signaling cascades. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate and the activation of protein kinase C. Mediates activation of MAPK1/ERK2, MAPK3/ERK1 and the MAP kinase signaling pathway, as well as of the AKT1 signaling pathway. Mediates phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, reorganization of the actin cytoskeleton and activation of PTK2/FAK1. Required for VEGFA-mediated induction of NOS2 and NOS3, leading to the production of the signaling molecule nitric oxide (NO) by endothelial cells. Phosphorylates PLCG1. Promotes phosphorylation of FYN, NCK1, NOS3, PIK3R1, PTK2/FAK1 and SRC. Tyrosine-protein kinase that acts as a cell-surface receptor for VEGFA, VEGFC and VEGFD. Related diseases Hemangioma, capillary infantile (HCI) [MIM:602089]: A condition characterized by dull red, firm, dome-shaped hemangiomas, sharply demarcated from surrounding skin, usually presenting at birth or occurring within the first two or three months of life. They result from highly proliferative, localized growth of capillary endothelium and generally undergo regression and involution without scarring. {ECO:0000269|PubMed:11807987, ECO:0000269|PubMed:18931684}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Plays a major role in tumor angiogenesis. In case of HIV-1 infection, the interaction with extracellular viral Tat protein seems to enhance angiogenesis in Kaposi's sarcoma lesions. Drugs (DrugBank ID) DB04727; DB07514; DB07528; DB06938; DB07326; DB06626; DB08875; DB04849; DB05198; DB12147; DB12307; DB12010; DB11679; DB06101; DB09078; DB06080; DB06595; DB07537; DB07183; DB07333; DB07334; DB07274; DB09079; DB08519; DB08042; DB16265; DB06589; DB05931; DB08901; DB15822; DB05984; DB05578; DB08896; DB14840; DB06436; DB00398; DB01268; DB05075; DB11800; DB04879; DB05146; DB05014 Interacts with P35916; O60565; P98160; PRO_0000391621 [P98160]; PRO_0000391622 [P98160]; P17301; P35968; P09382; P08581; P16333; O14786; O75340; P09619; P29350; Q12913; P12931; P15692; P15692-4; P49767; Q9MYV3-3 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; ATP-binding; Cell junction; Cell membrane; Cytoplasm; Cytoplasmic vesicle; Developmental protein; Differentiation; Disulfide bond; Endoplasmic reticulum; Endosome; Glycoprotein; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33863.9 Length 296 Aromaticity 0.1 Instability index 40.01 Isoelectric point 8.5 Charge (pH=7) 4.59 2D Binding mode Binding energy (Kcal/mol) -9.59  Molscript Map 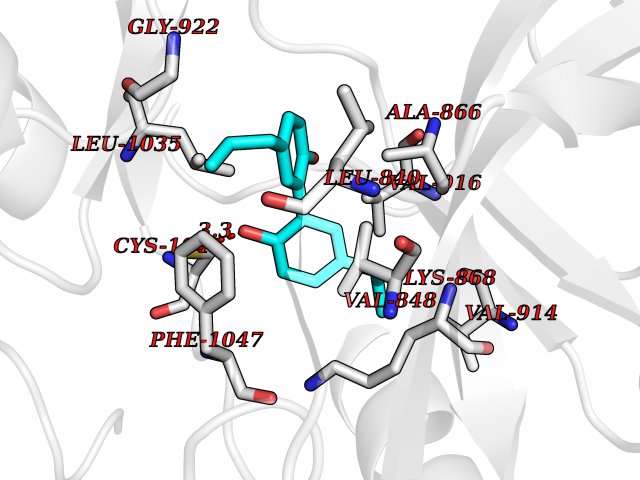 Pymol Map 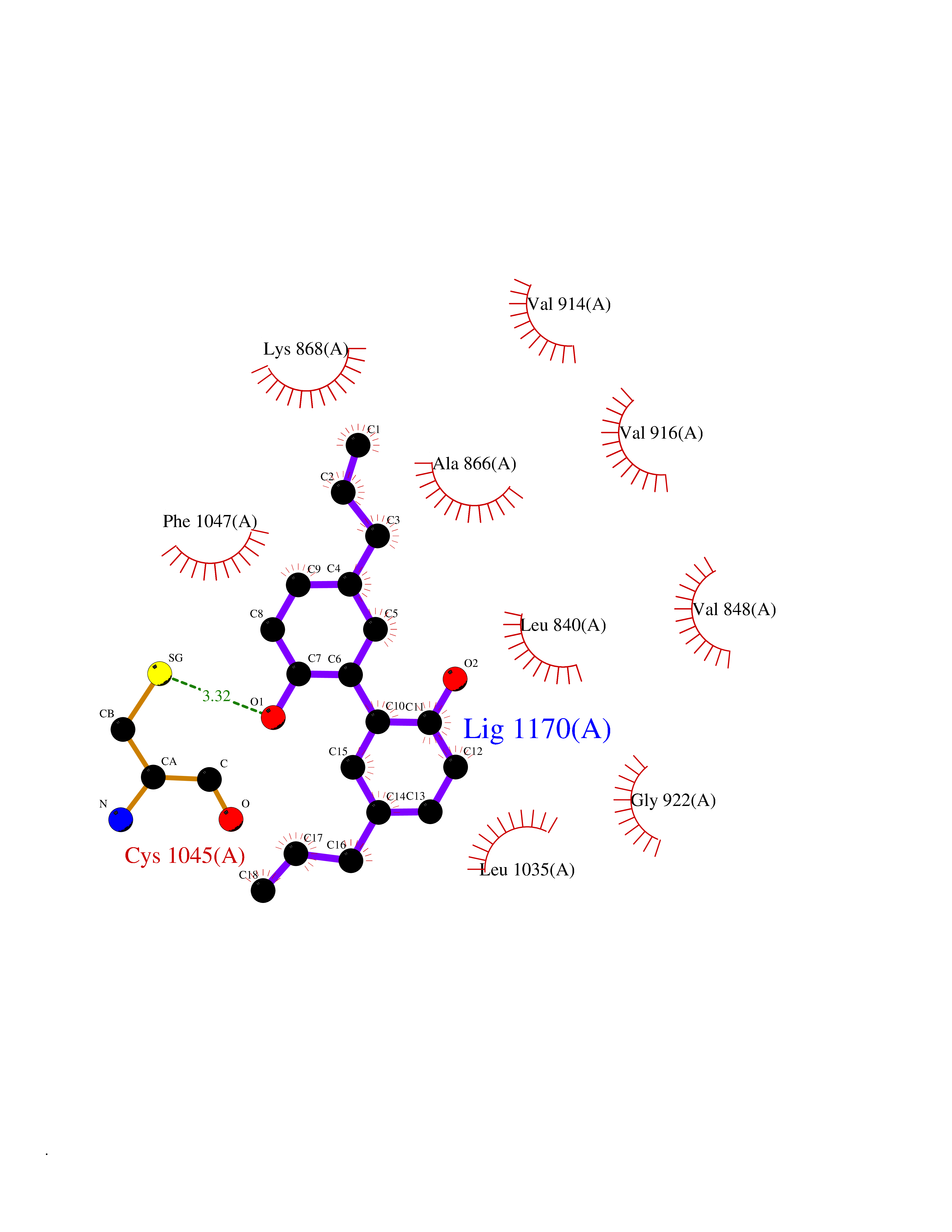 Ligplot Map 3D Binding mode Sequence HCERLPYDASKWEFPRDRLKLGKPLGRGAFGQVIEADAFGIDKTATCRTVAVKMLKEGATHSEHRALMSELKILIHIGHHLNVVNLLGACTKPGGPLMVIVEFCKFGNLSTYLRSKRNEFVPYYKDFLTLEHLICYSFQVAKGMEFLASRKCIHRDLAARNILLSEKNVVKICDFGLARDIYKDPDYVRKGDARLPLKWMAPETIFDRVYTIQSDVWSFGVLLWEIFSLGASPYPGVKIDEEFCRRLKEGTRMRAPDYTTPEMYQTMLDCWHGEPSQRPTFSELVEHLGNLLQANA Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 7.03 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -9.59 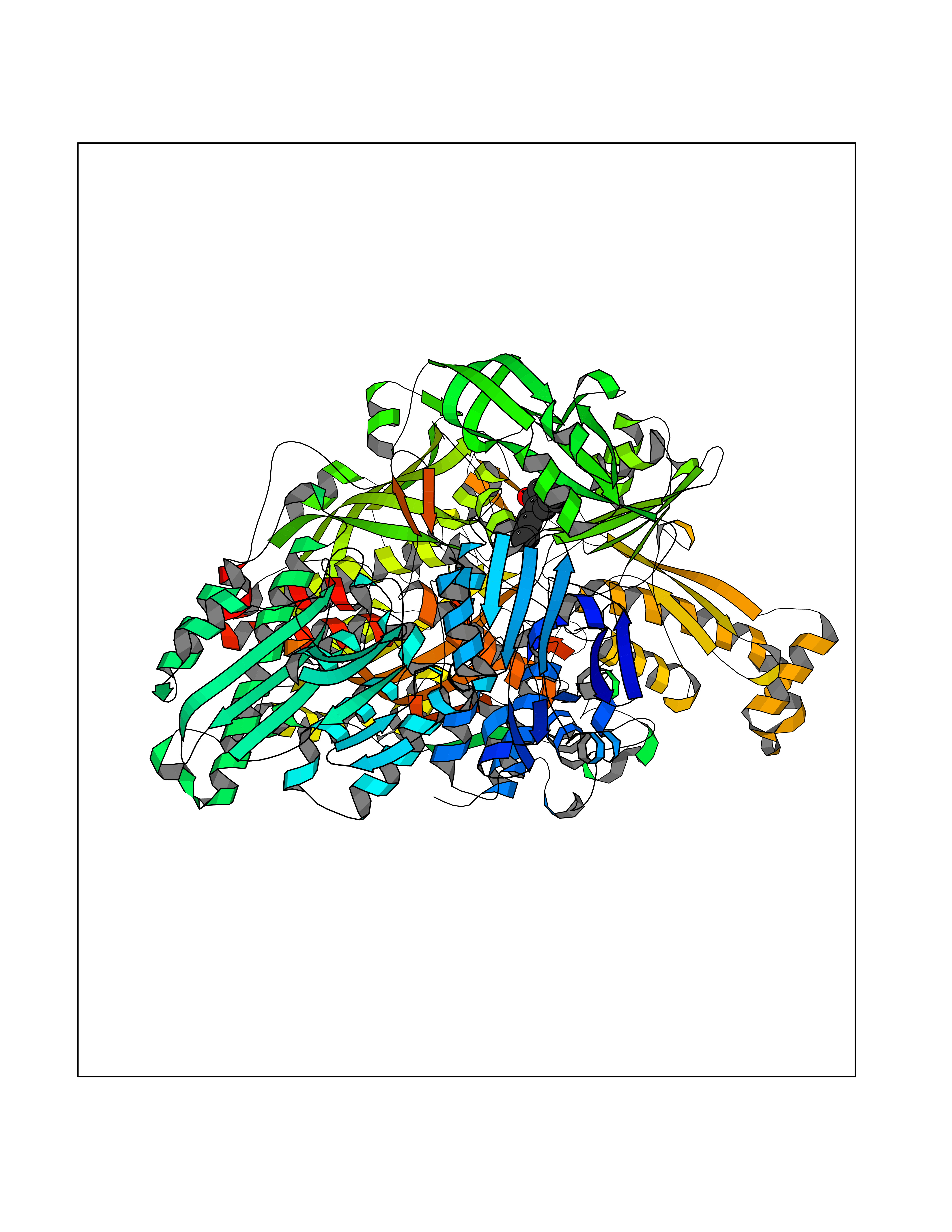 Molscript Map 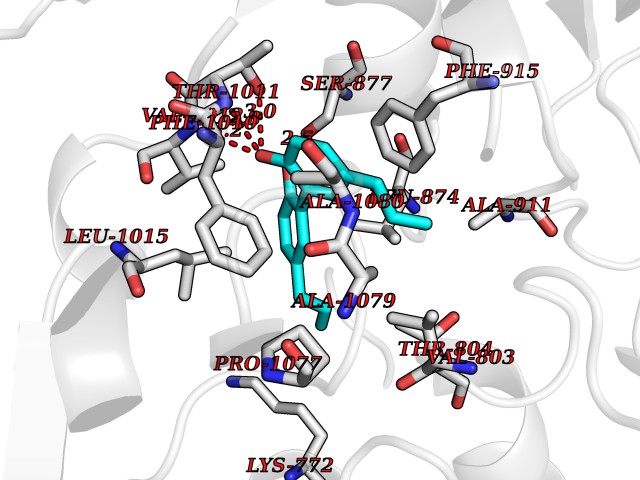 Pymol Map 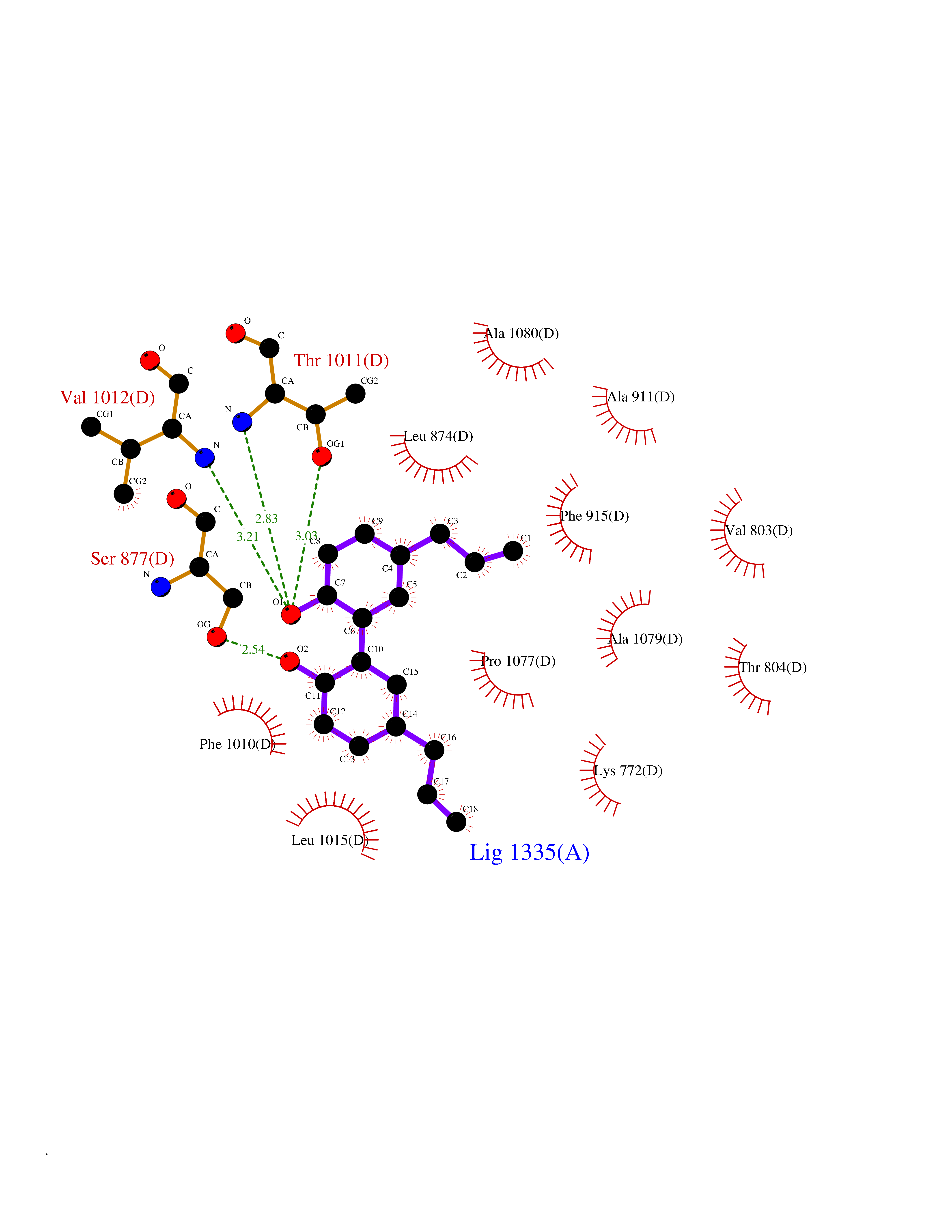 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Glycolipid transfer protein | 3RZN | 7.03 | |
Target general information Gen name GLTP Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family GLTP family Biochemical class Lipid transport Function Glycolipid binding.Glycolipid transporter activity.Identical protein binding.Intermembrane lipid transfer activity.Lipid binding. Related diseases Brugada syndrome 7 (BRGDA7) [MIM:613120]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:20031595}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Atrial fibrillation, familial, 16 (ATFB16) [MIM:613120]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:20558140, ECO:0000269|PubMed:21051419}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03600; DB04465; DB03017; DB03203 Interacts with Q96DZ9; Q9NZD2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Lipid transport; Proteomics identification; Reference proteome; Repeat; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23534.1 Length 206 Aromaticity 0.11 Instability index 36.45 Isoelectric point 7.08 Charge (pH=7) 0.1 2D Binding mode Binding energy (Kcal/mol) -9.59 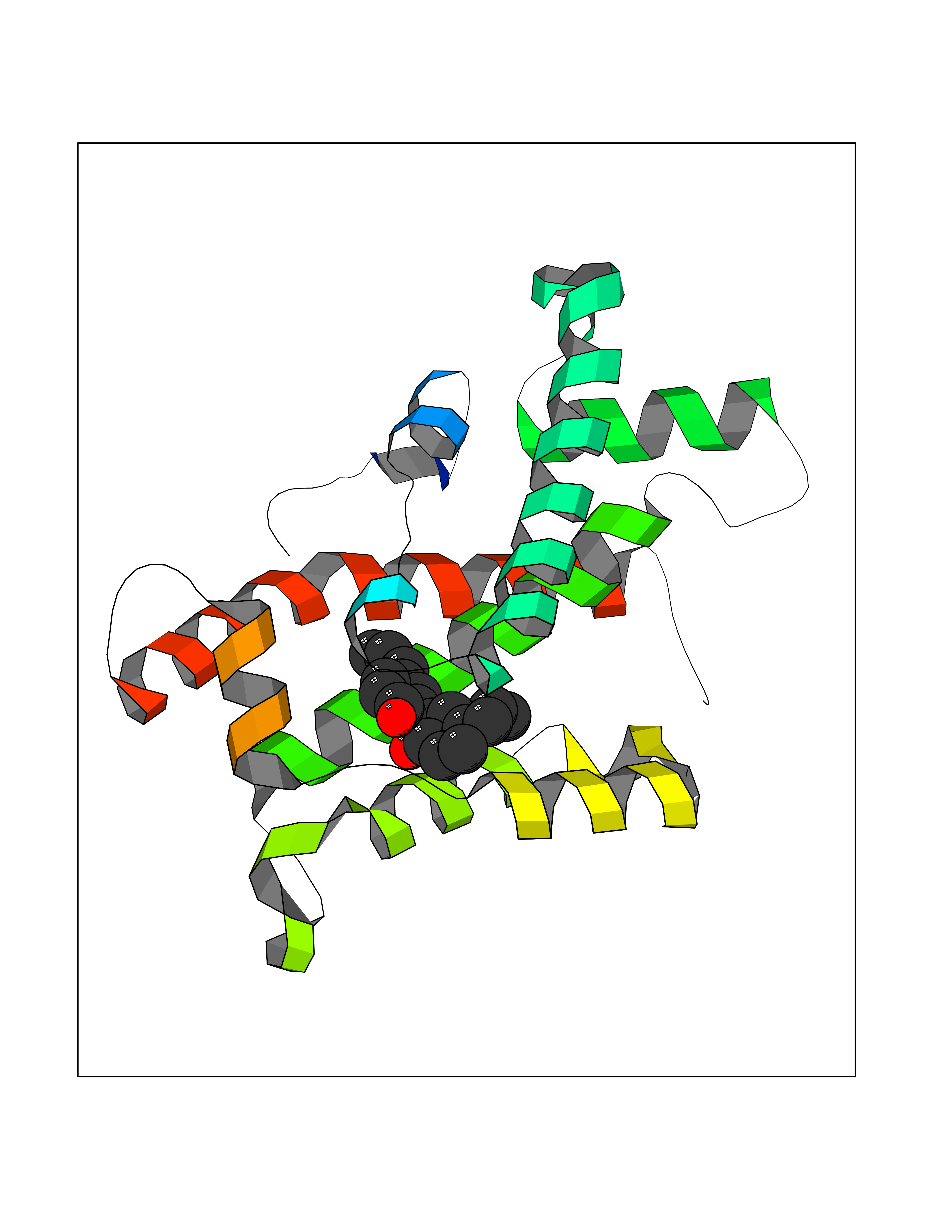 Molscript Map 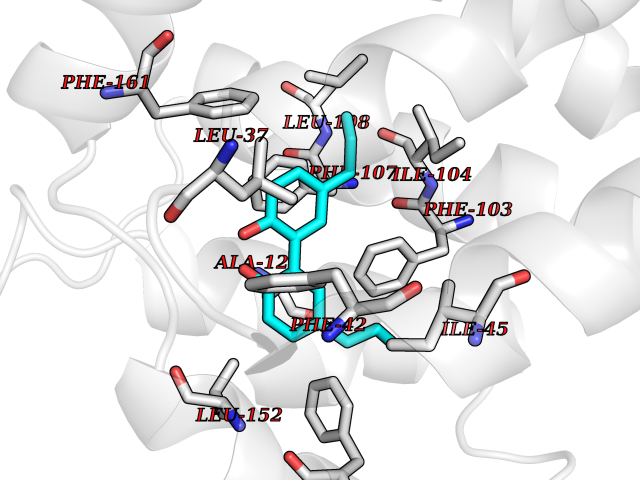 Pymol Map 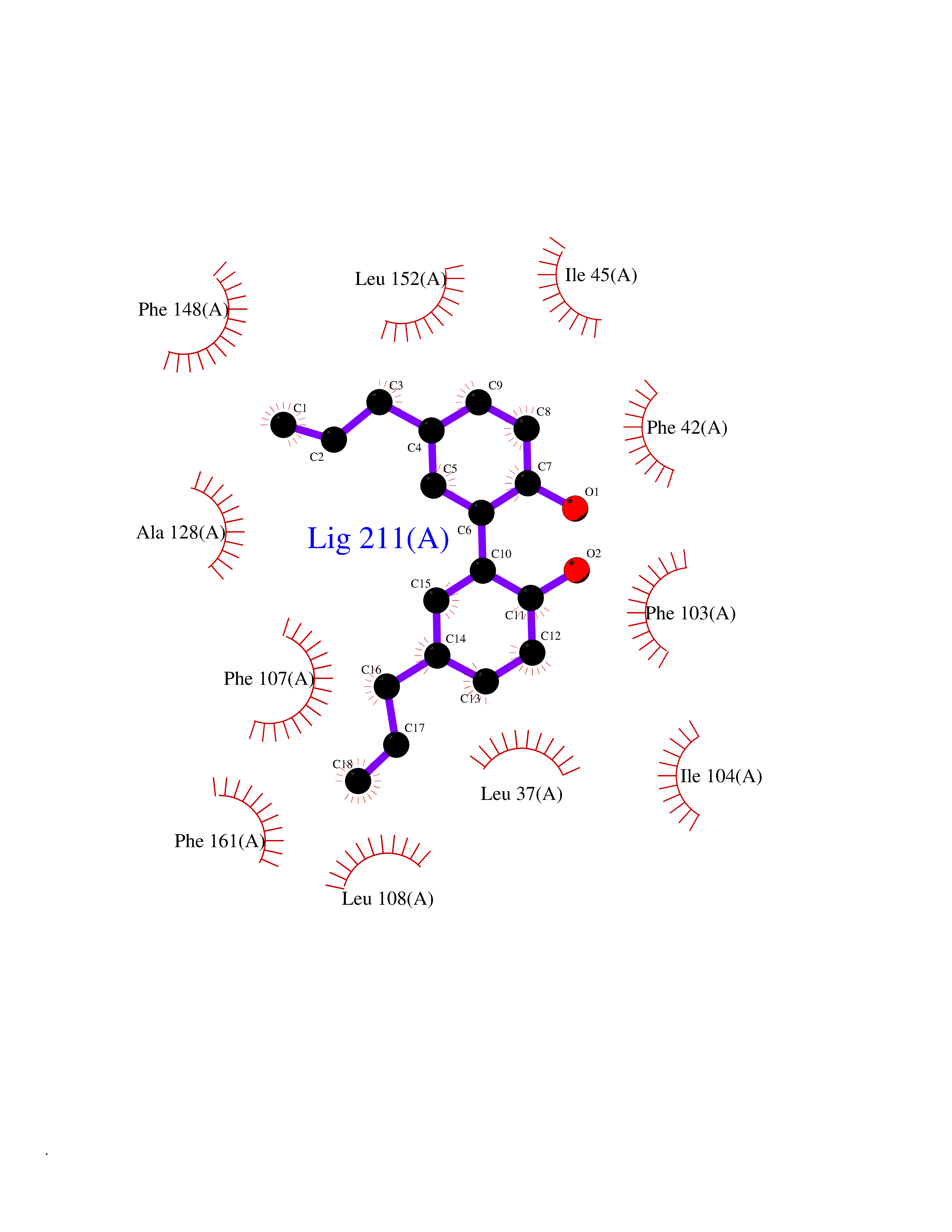 Ligplot Map 3D Binding mode Sequence LAEHLLKPLPADKQIETGPFLEAVSHLPPFFDCLGSPVFTPIKADISGNITKIKAVYDTNPAKFRTLQNILEVEKEMYGAEWPKVGATLALMWLKRGLRFIQVFLQSICDGERDENHPNLIRVNATKAYEMALKKYHGWIVQKIFQAALYAAPYKSDFLKALSKGQNVTEEECLEKIRLFLVNYTATIDVIYEMYTQMNAELNYKV Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Zinc finger protein Helios (IKZF2) | 7LPS | 7.03 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -9.59  Molscript Map 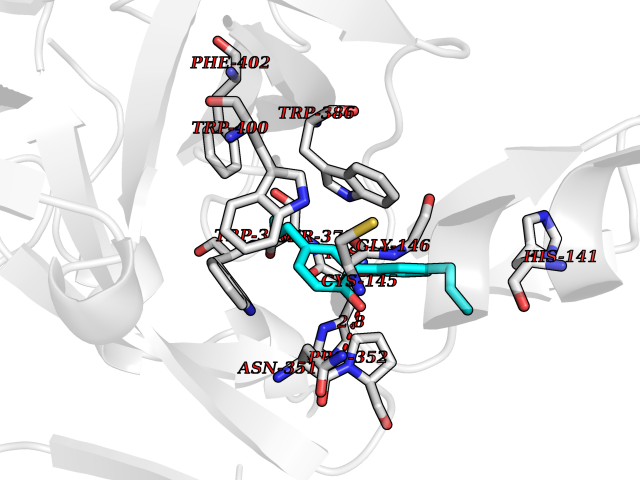 Pymol Map 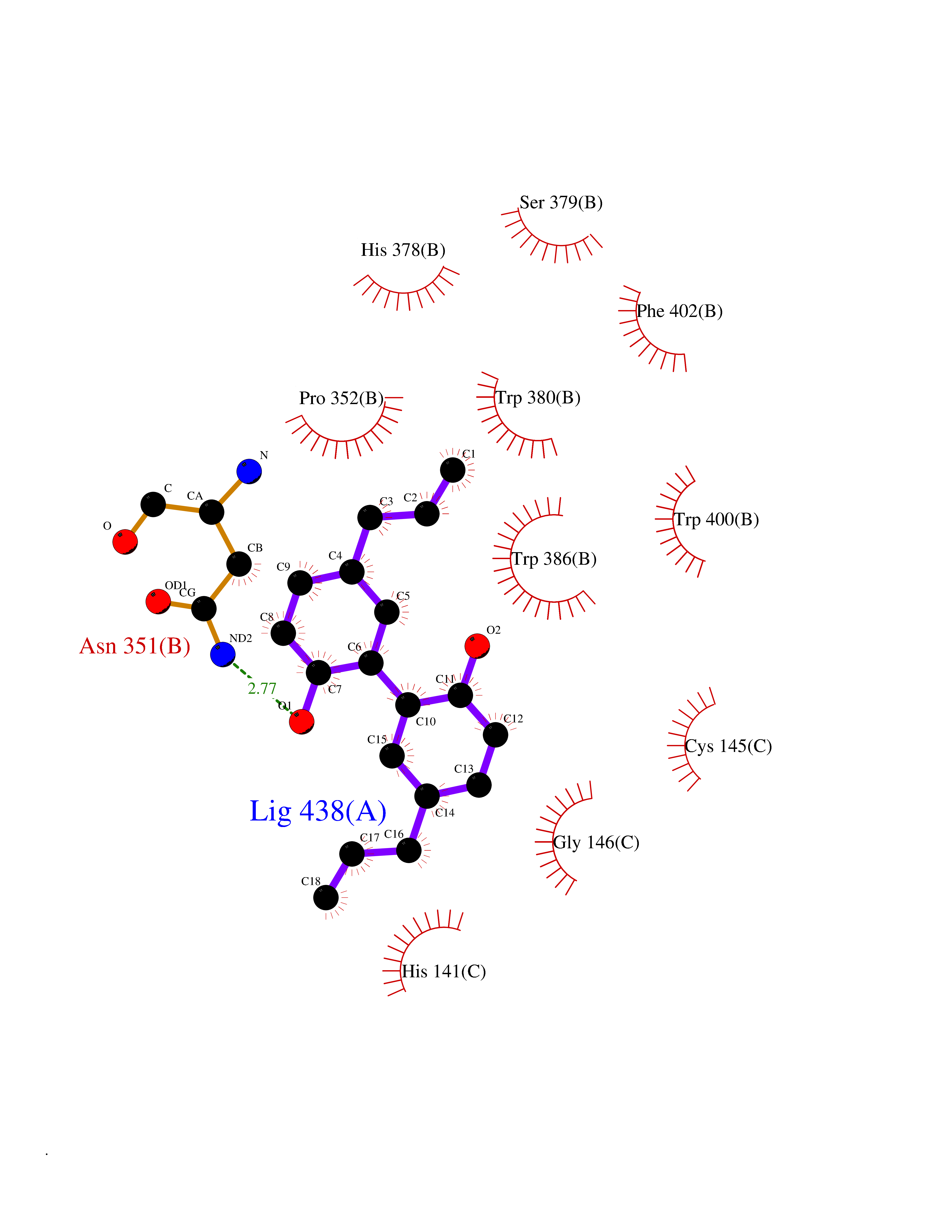 Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | SEC14-like protein 4 | 4TLG | 7.02 | |
Target general information Gen name SEC14L4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TAP3 Protein family NA Biochemical class Transport protein Function Lipid binding.Transporter activity. Related diseases Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia (CDP-PBHM) [MIM:300863]: A disease characterized by chondrodysplasia, severe platyspondyly, hydrocephaly, and facial features with microphthalmia. Bone abnormalities include a distinctive metaphyseal cupping of the metacarpals, metatarsals, and phalanges. Affected females show a milder phenotype with small stature, sometimes associated with body asymmetry and mild intellectual disability. {ECO:0000269|PubMed:20181727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB11635; DB11251; DB00163 Interacts with Q96LC9; O43186; P78358; Q9NYQ3; Q0VD86; Q15323; O76011; P50221; Q6FHY5; Q02548; P26367; Q9H8W4; Q04864; Q04864-2; Q9UHV2; P15884; P15884-3; Q96N21; Q9BYV2; Q8N6Y0; Q9H0C1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 23947.6 Length 210 Aromaticity 0.1 Instability index 50.84 Isoelectric point 5.55 Charge (pH=7) -3.11 2D Binding mode Binding energy (Kcal/mol) -9.57  Molscript Map 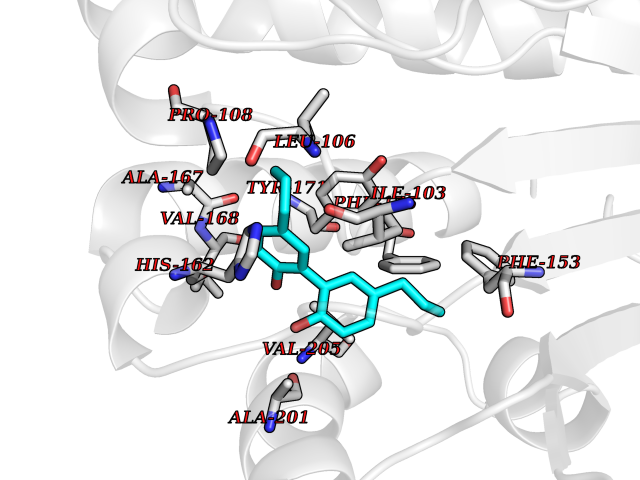 Pymol Map 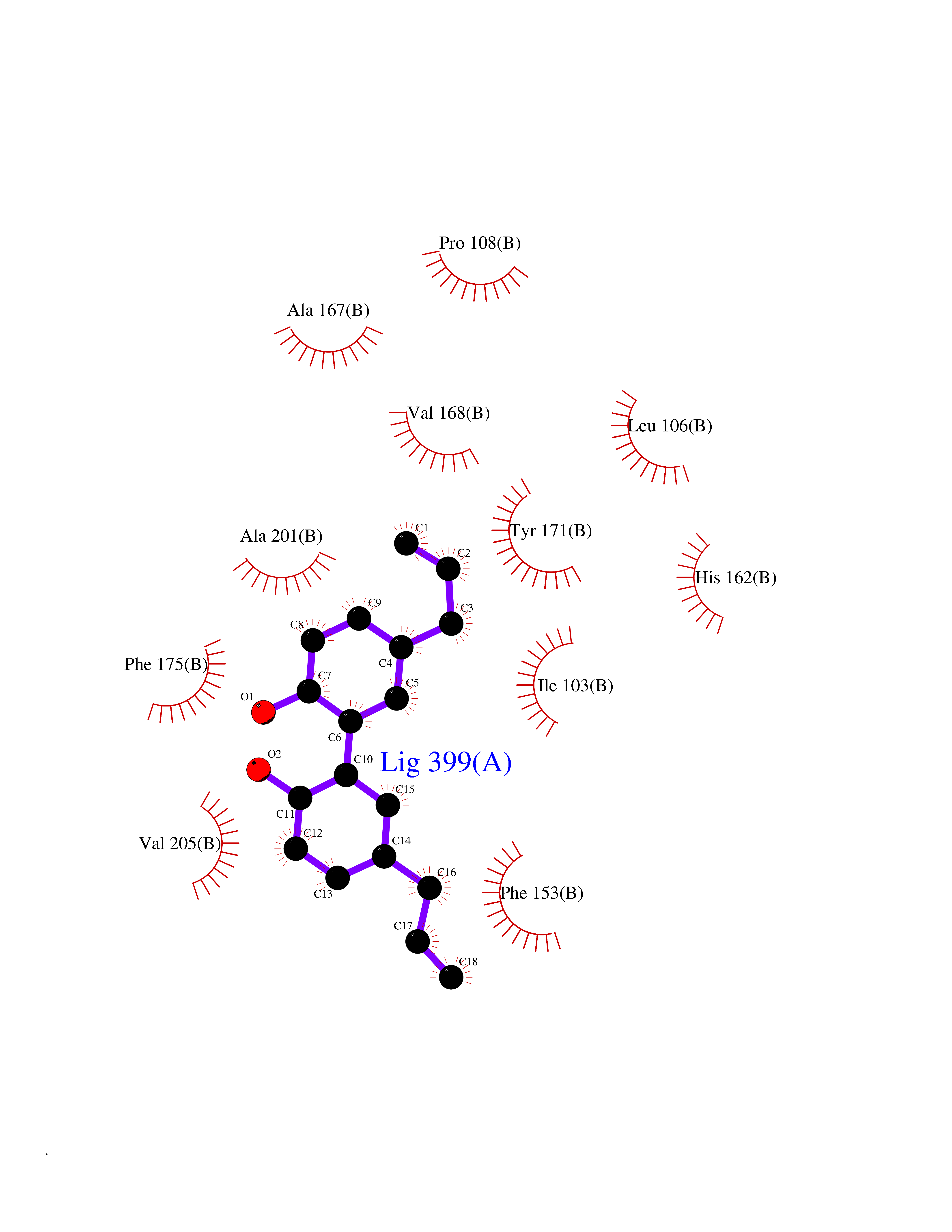 Ligplot Map 3D Binding mode Sequence VTWQPPEVIQLYDSGGLCGYDYEGCPVYFNIIGSLDPKGLLLSASKQDMIRKRIKVCELLLHECELQTQKLGRKIEMALMVFDMEGLSLKHLWKPAVEVYQQFFSILEANYPETLKNLIVIRAPKLFPVAFNLVKSFMSEETRRKIVILGDNWKQELTKFISPDQLPVEFGGTMTDPDGNPKCLTKINYGGEVPKSYYPDKASEETLQSM Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | PRKR-like endoplasmic reticulum kinase (PERK) | 4G31 | 7.02 | |
Target general information Gen name EIF2AK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PEK Protein family Protein kinase superfamily, Ser/Thr protein kinase family, GCN2 subfamily Biochemical class Kinase Function Converts phosphorylated eIF-2-alpha/EIF2S1 either in a global protein synthesis inhibitor, leading to a reduced overall utilization of amino acids, or to a translation initiation activator of specific mRNAs, such as the transcriptional activator ATF4, and hence allowing ATF4-mediated reprogramming of amino acid biosynthetic gene expression to alleviate nutrient depletion. Serves as a critical effector of unfolded protein response (UPR)-induced G1 growth arrest due to the loss of cyclin-D1 (CCND1). Involved in control of mitochondrial morphology and function. Metabolic-stress sensing protein kinase that phosphorylates the alpha subunit of eukaryotic translation initiation factor 2 (eIF-2-alpha/EIF2S1) on 'Ser-52' during the unfolded protein response (UPR) and in response to low amino acid availability. Related diseases Wolcott-Rallison syndrome (WRS) [MIM:226980]: A rare autosomal recessive disorder, characterized by permanent neonatal or early infancy insulin-dependent diabetes and, at a later age, epiphyseal dysplasia, osteoporosis, growth retardation and other multisystem manifestations, such as hepatic and renal dysfunctions, intellectual disability and cardiovascular abnormalities. {ECO:0000269|PubMed:10932183, ECO:0000269|PubMed:12086964, ECO:0000269|PubMed:12960215, ECO:0000269|PubMed:16813601, ECO:0000269|PubMed:24168455, ECO:0000269|PubMed:24194294, ECO:0000269|PubMed:27145240, ECO:0000269|PubMed:28220546, ECO:0000269|PubMed:30906465, ECO:0000269|PubMed:30922274, ECO:0000269|PubMed:32216767, ECO:0000269|PubMed:34123975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NZJ5; P11021 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; ADP-ribosylation; ATP-binding; Diabetes mellitus; Disease variant; Endoplasmic reticulum; Glycoprotein; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Signal; Stress response; Transferase; Translation regulation; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Unfolded protein response Protein physicochemical properties Chain ID A Molecular weight (Da) 29033.5 Length 248 Aromaticity 0.1 Instability index 43.71 Isoelectric point 7.75 Charge (pH=7) 1.27 2D Binding mode Binding energy (Kcal/mol) -9.58 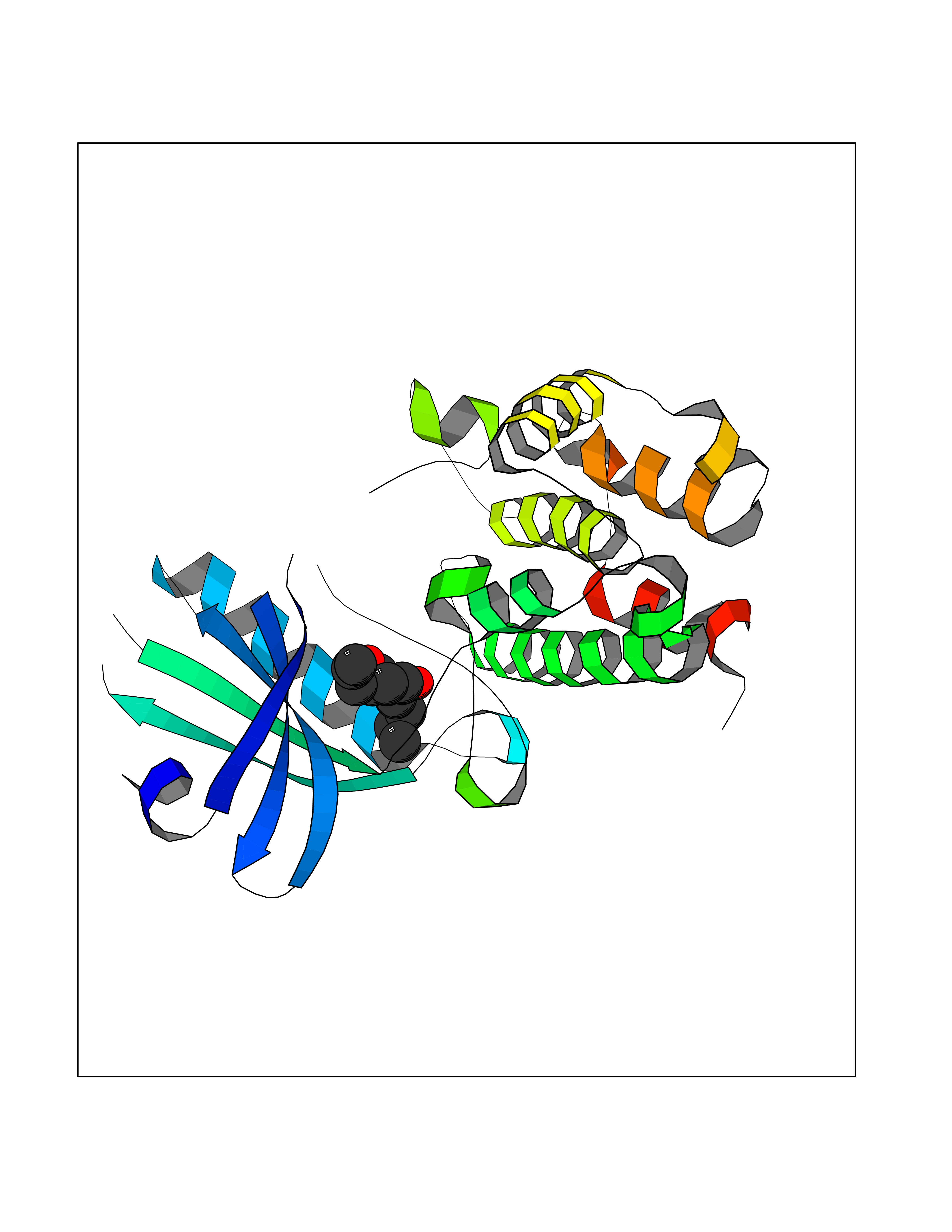 Molscript Map 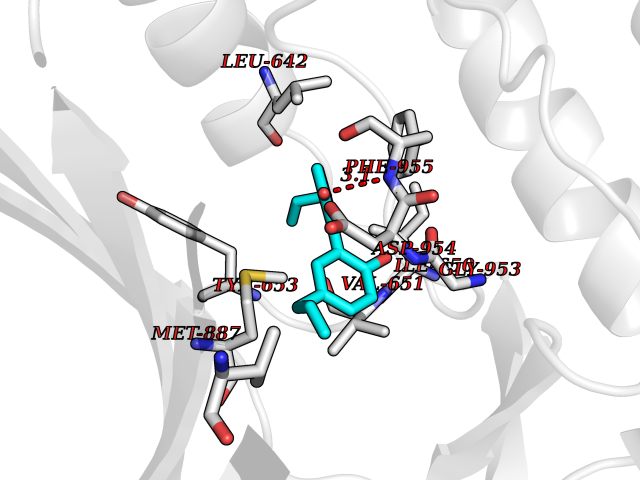 Pymol Map 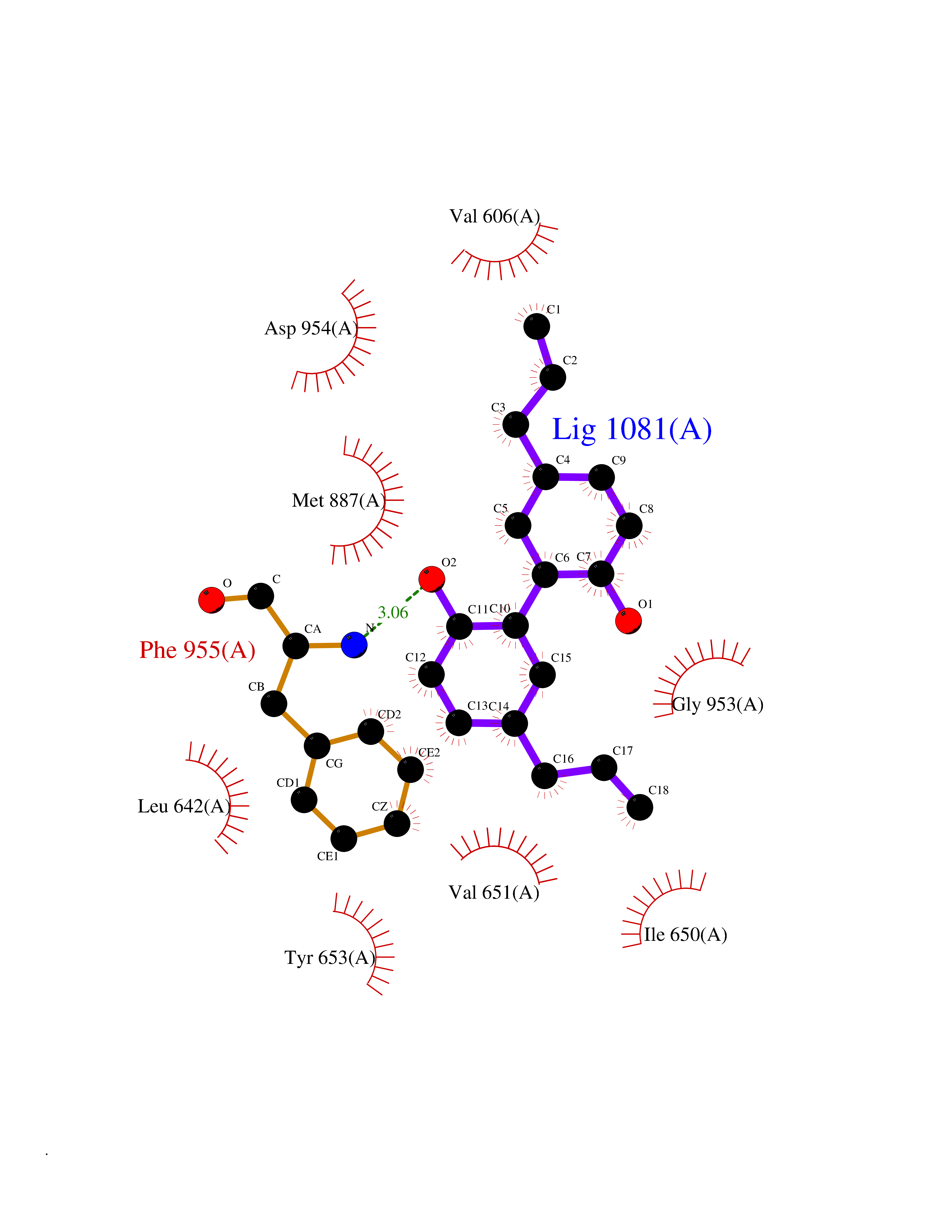 Ligplot Map 3D Binding mode Sequence GRYLTDFEPIQCLGRGGVVFEAKNKVDDCNYAIKRIRLPNRELAREKVMREVKALAKLEHPGIVRYFNAWLEKNKVYLYIQMQLCRKENLKDWMNGRCTIEERERSVCLHIFLQIAEAVEFLHSKGLMHRDLKPSNIFFTMDDVVKVGDFGLVGTKLYMSPEQIHGNSYSHKVDIFSLGLILFELLYPFSTQMERVRTLTDVRNLKFPPLFTQKYPCEYVMVQDMLSPSPMERPEAINIIENAVFEDL Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Neuronal acetylcholine receptor alpha-3/beta-4 (CHRNA3/B4) | 6PV7 | 7.02 | |
Target general information Gen name CHRNA3-CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Neuronal acetylcholine receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Neurotransmitter receptor Function A type of nicotinic acetylcholine receptor, consisting of 3 and 4 subunits. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -9.57 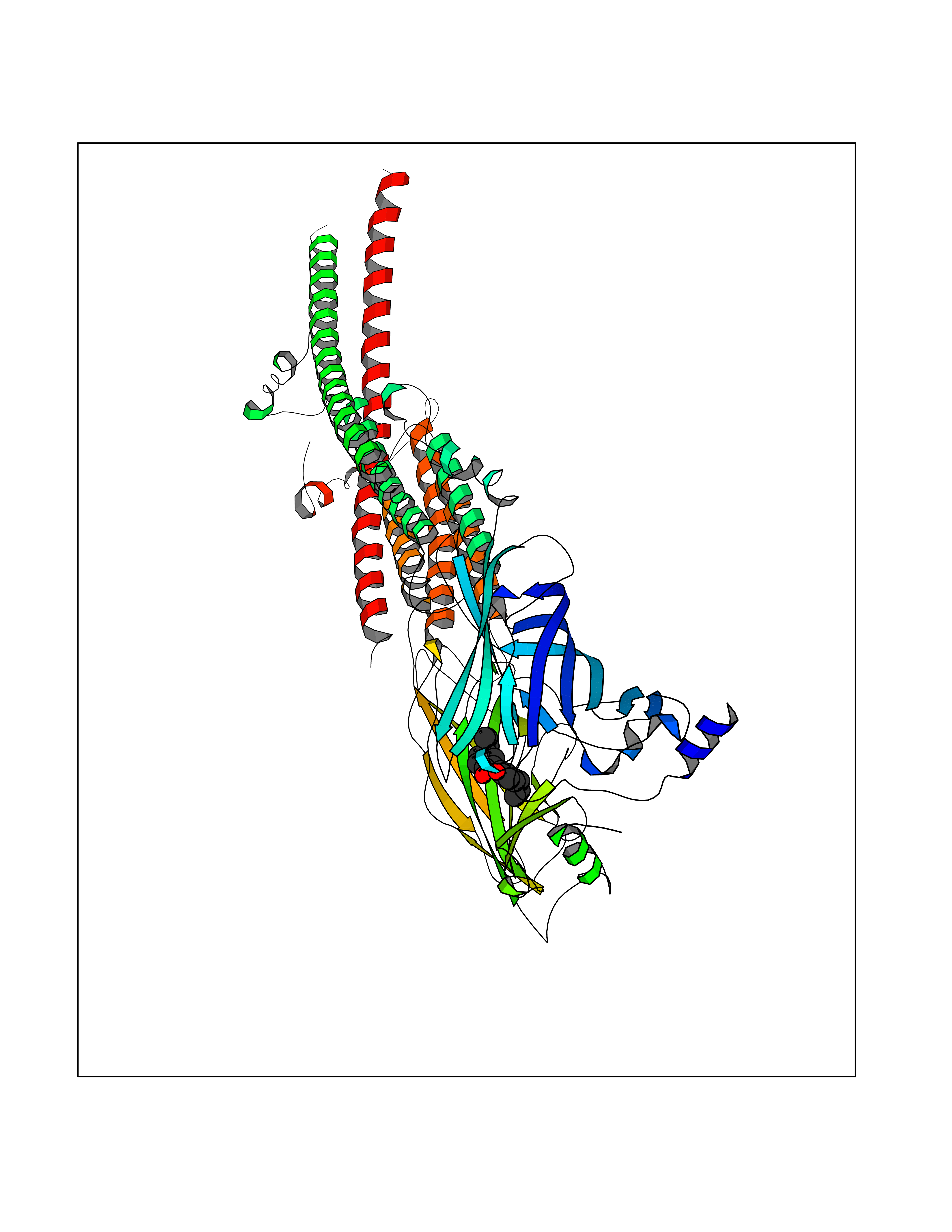 Molscript Map 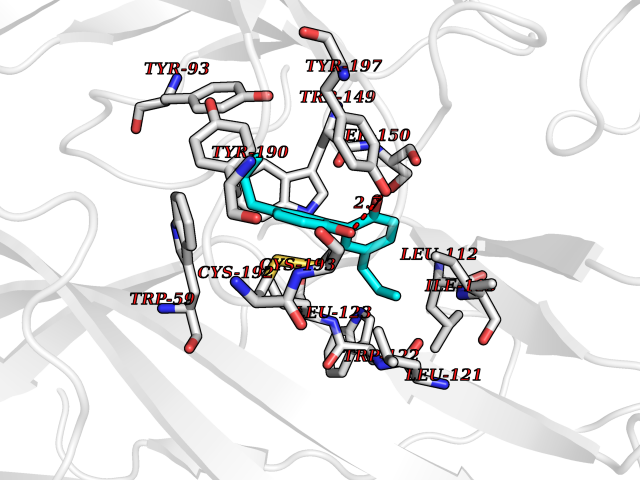 Pymol Map 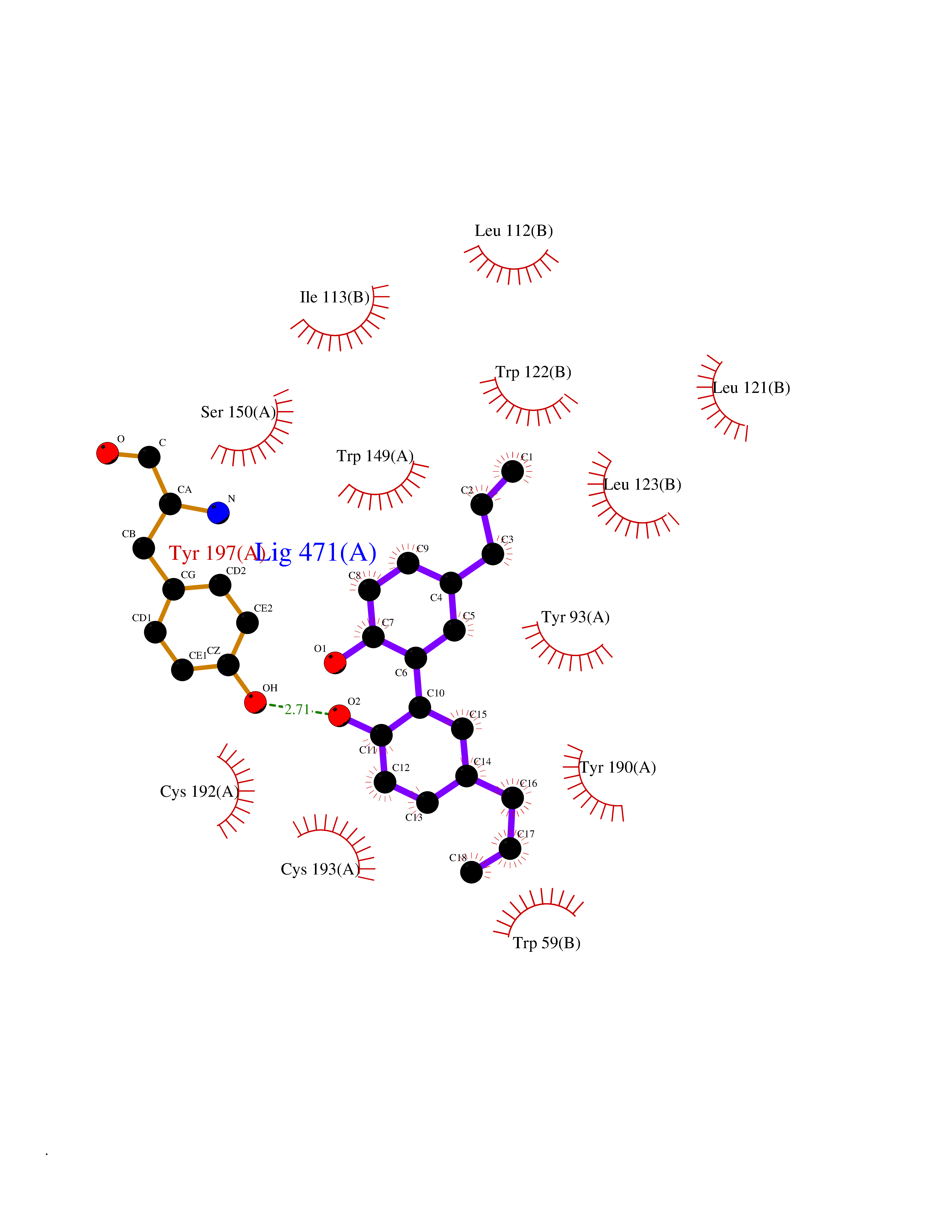 Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Cytochrome b (Complex III subunit 3) (Complex III subunit III) (Cytochrome b-c1 complex subunit 3) (Ubiquinol-cytochrome-c reductase complex cytochrome b subunit) | 1SQB | 7.01 | |
Target general information Gen name MT-CYB Organism Bos taurus (Bovine) Uniprot ID TTD ID NA Synonyms CYTB;MTCYB;COB Protein family Cytochrome b family Biochemical class NA Function Component of the ubiquinol-cytochrome c reductase complex (complex III or cytochrome b-c1 complex) that is part of the mitochondrial respiratory chain. The b-c1 complex mediates electron transfer from ubiquinol to cytochrome c. Contributes to the generation of a proton gradient across the mitochondrial membrane that is then used for ATP synthesis. {ECO:0000269|PubMed:1327781, ECO:0000269|PubMed:20025846, ECO:0000269|PubMed:9485330, ECO:0000305|PubMed:189810}." Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Electron transport; Heme; Iron; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Reference proteome; Respiratory chain; Transmembrane; Transmembrane helix; Transport; Ubiquinone Protein physicochemical properties Chain ID C,D,F,G,H Molecular weight (Da) 99281.2 Length 866 Aromaticity 0.12 Instability index 43.81 Isoelectric point 8.32 Charge (pH=7) 7.12 2D Binding mode Binding energy (Kcal/mol) -9.56  Molscript Map 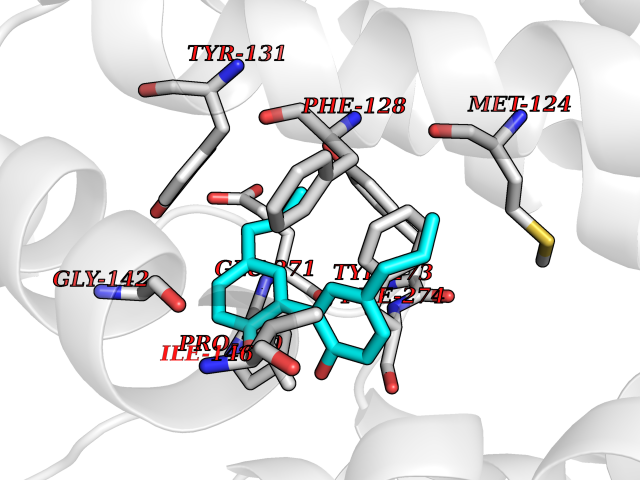 Pymol Map 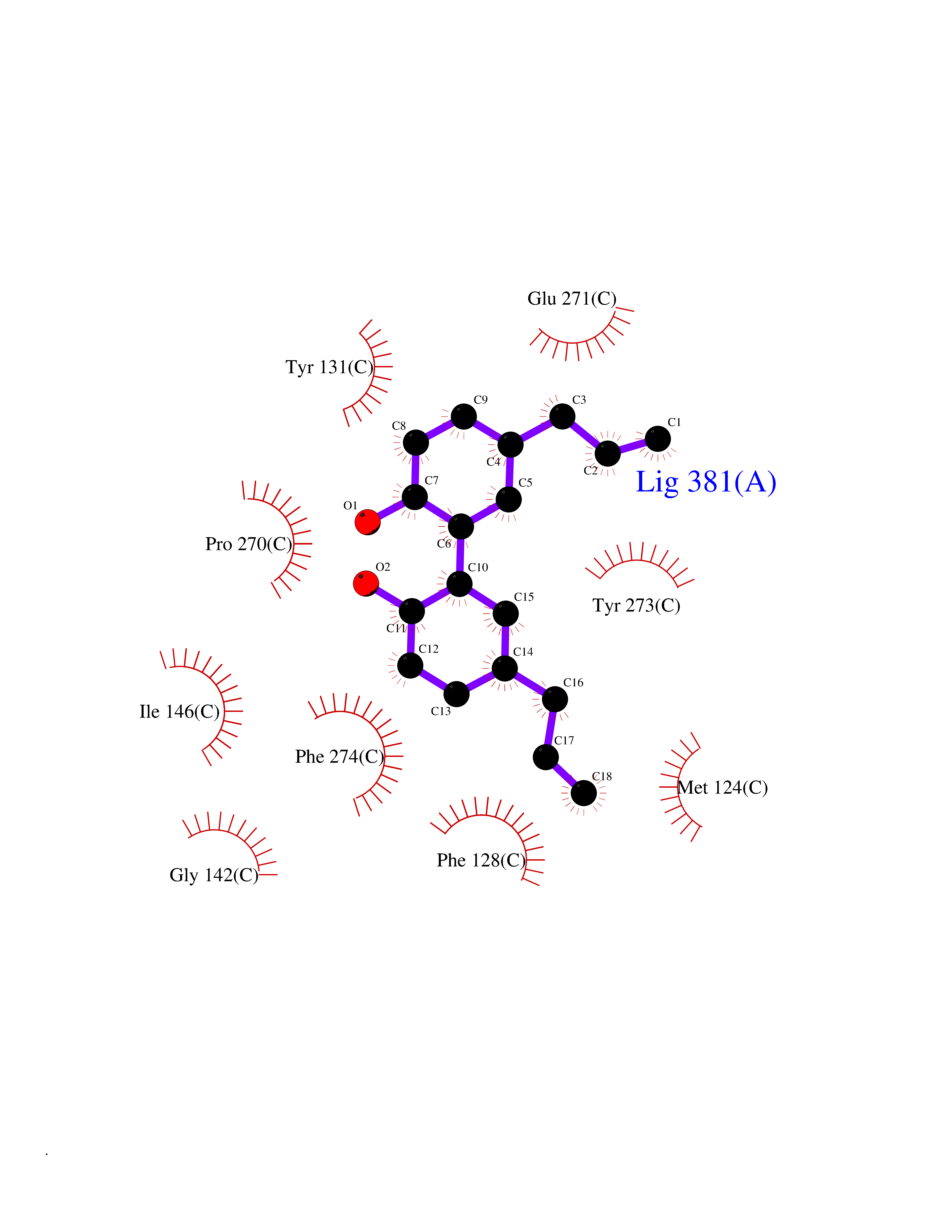 Ligplot Map 3D Binding mode Sequence VSASSRWLEGIRKWYYNAAGFNKLGLMRDDTIHENDDVKEAIRRLPENLYDDRVFRIKRALDLSMRQQILPKEQWTKYEEDKSYLEPYLKEVIRERKEREEWAKKELVDPLTTVREQCEQLEKCVKARERLELCDERVSSRSQTEEDCTEELLDFLHARDHCVAHKLFNSLKTNIRKSHPLMKIVNNAFIDLPAPSNISSWWNFGSLLGICLILQILTGLFLAMHYTSDTTTAFSSVTHICRDVNYGWIIRYMHANGASMFFICLYMHVGRGLYYGSYTFLETWNIGVILLLTVMATAFMGYVLPWGQMSFWGATVITNLLSAIPYIGTNLVEWIWGGFSVDKATLTRFFAFHFILPFIIMAIAMVHLLFLHETGSNNPTGISSDVDKIPFHPYYTIKDILGALLLILALMLLVLFAPDLLGDPDNYTPANPLNTPPHIKPEWYFLFAYAILRSIPNKLGGVLALAFSILILALIPLLHTSKQRSMMFRPLSQCLFWALVADLLTLTWIGGQPVEHPYITIGQLASVLYFLLILVLMPTAGTIENKLLKWSDLELHPPSYPWSHRGLLSSLDHTSIRRGFQVYKQVCSSCHSMDYVAYRHLVGVCYTEDEAKALAEEVEVQDGPNEDGEMFMRPGKLSDYFPKPYPNPEAARAANNGALPPDLSYIVRARHGGEDYVFSLLTGYCEPPTGVSLREGLYFNPYFPGQAIGMAPPIYNEVLEFDDGTPATMSQVAKDVCTFLRWAAEPEHDHRKRMGLKMLLMMGLLLPLVYAMKRHKWSVLKSRKLAYRPPKGRQFGHLTRVRHVITYSLSPFEQRAFPHYFSKGIPNVLRRTRACILRVAPPFVAFYLVYTWGTQEFEKSKRKNPA Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Multidrug efflux pump subunit AcrB | 5ENQ | 7.01 | |
Target general information Gen name acrB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b0462;acrE;JW0451 Protein family Resistance-nodulation-cell division (RND) (TC 2.A.6) family Biochemical class Transport protein Function Drug:proton antiporter activity.Drug transmembrane transporter activity.Identical protein binding. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03619; DB04209; DB03825 Interacts with P31224; P0AAW9; P0ADZ7 EC number NA Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Membrane; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 60242.4 Length 553 Aromaticity 0.08 Instability index 29.1 Isoelectric point 4.76 Charge (pH=7) -18.86 2D Binding mode Binding energy (Kcal/mol) -9.55 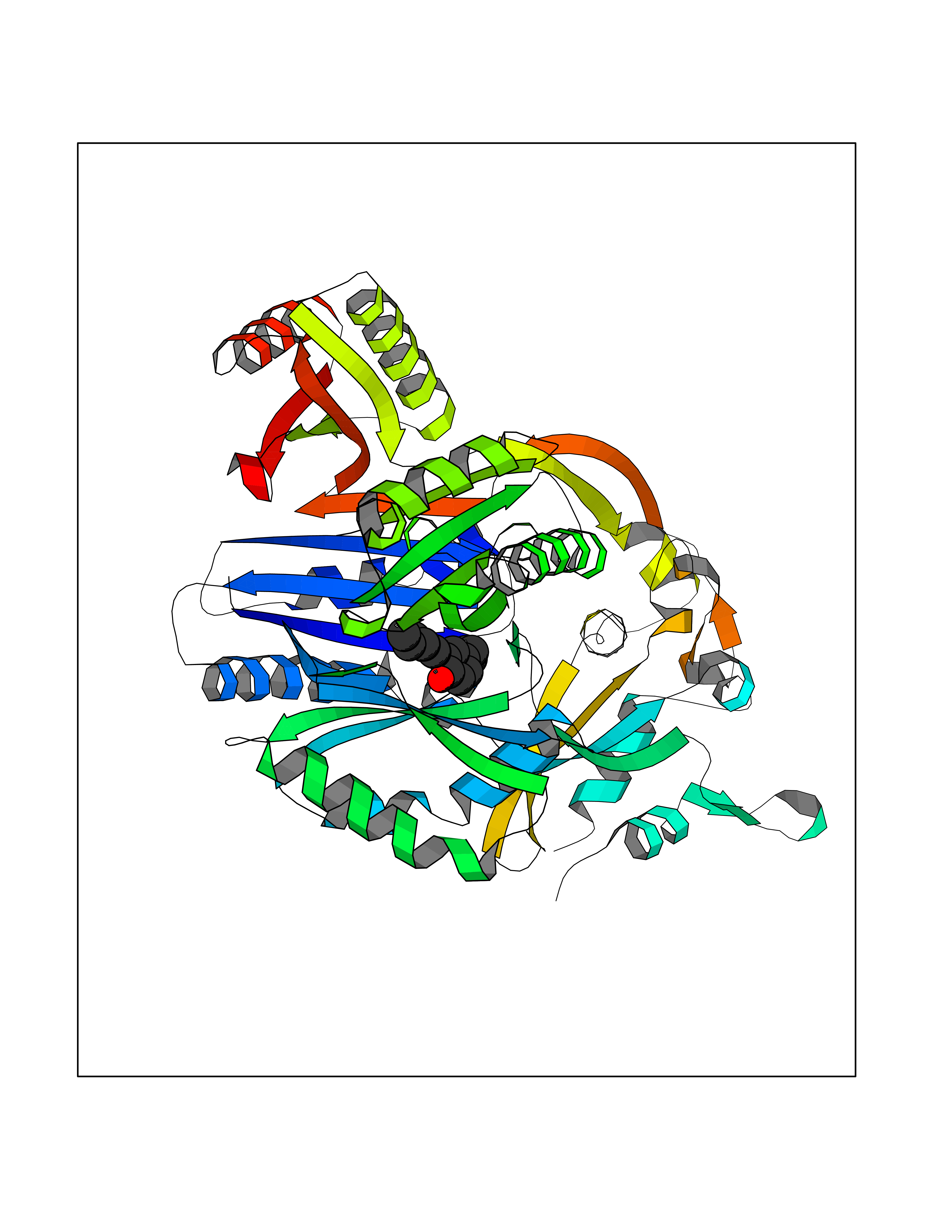 Molscript Map 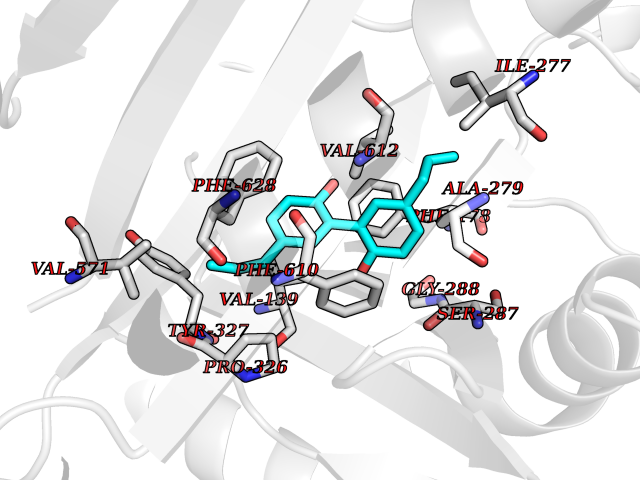 Pymol Map 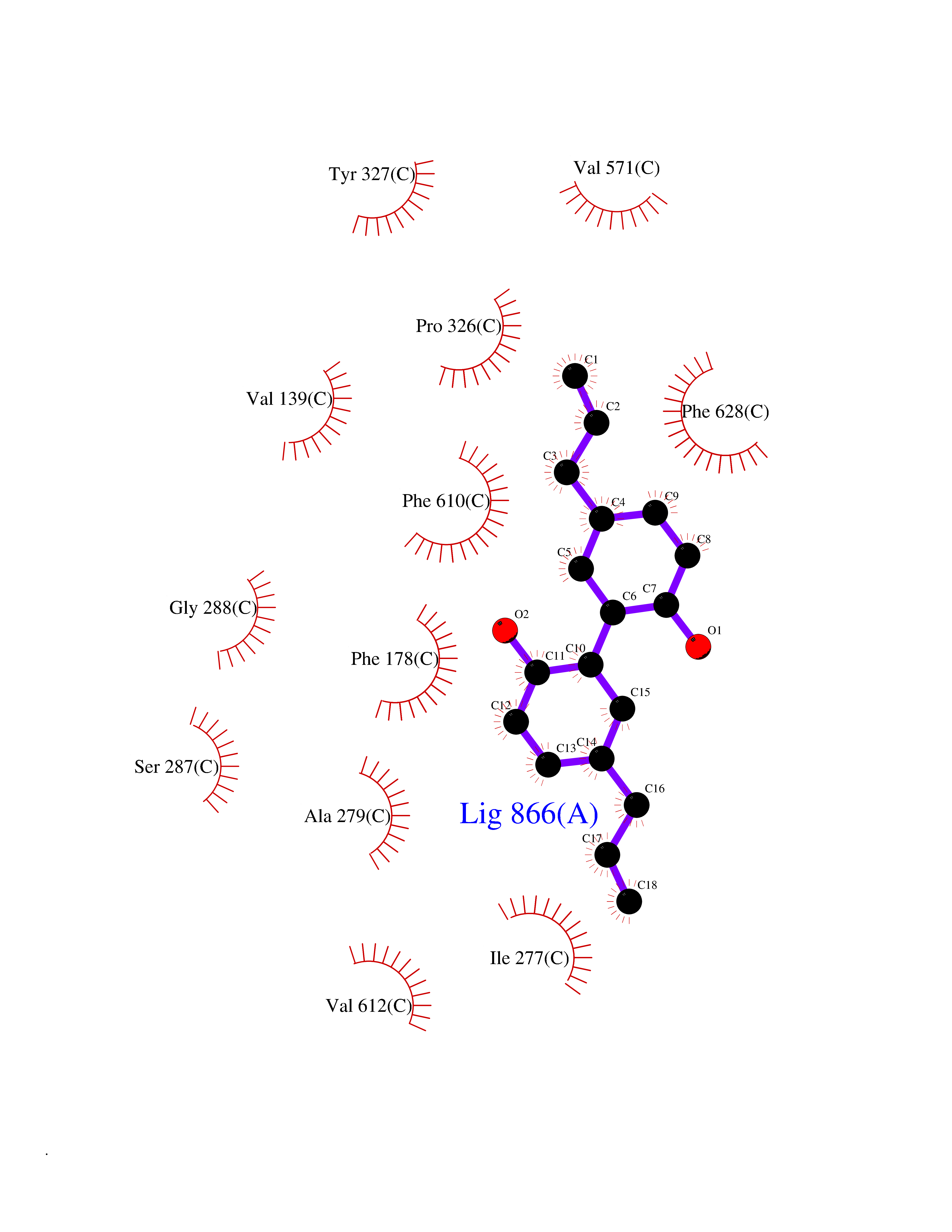 Ligplot Map 3D Binding mode Sequence APPAVTISASYPGADAKTVQDTVTQVIEQNMNGIDNLMYMSSNSDSTGTVQITLTFESGTDADIAQVQVQNKLQLAMPLLPQEVQQQGVSVEKSSSSFLMVVGVINTDGTMTQEDISDYVAANMKDAISRTSGVGDVQLFGSQYAMRIWMNPNELNKFQLTPVDVITAIKAQNATRLTSTEEFGKILLKVNQDGSRVLLRDVAKIELGGENYDIIAEFNGQPASGLGIKLATGANALDTAAAIRAELAKMEPFFPSGLKIVYPYDTGVFMTMVQLPAGATQERTQKVLNEVTHYYLTKEKNNVESVFAVNGFGFAGRGQNTGIAFVSLKDWADRPGEENKVEAITMRATRAFSQIKDAMVFAFNLATGFDFELIDQAGLGHEKLTQARNQLLAEAAKHPDMLTSVRPNGLEDTPQFKIDIDQEKAQALGVSINDINTTLGAAWGGSYVNDFIDRGRVKKVYVMSEAKYRMLPDDIGDWYVRAADGQMVPFSAFSSSRWEYGSPRLERYNGLPSMEILGQAAPGKSTGEAMELMEQLASKLPTGVGYDWTGMSY Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Histamine N-methyltransferase (HNMT) | 2AOT | 7.01 | |
Target general information Gen name HNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine-N-methyltransferase; HNMT; HMT Protein family Class I-like SAM-binding methyltransferase superfamily, HNMT family Biochemical class Methyltransferase Function Inactivates histamine by N-methylation. Plays an important role in degrading histamine and in regulating the airway response to histamine. Related diseases Intellectual developmental disorder, autosomal recessive 51 (MRT51) [MIM:616739]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:26206890}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. Drugs (DrugBank ID) DB00613; DB13875; DB05381; DB04655; DB01103; DB01752; DB07106 Interacts with NA EC number EC 2.1.1.8 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32712 Length 288 Aromaticity 0.1 Instability index 36.38 Isoelectric point 5.18 Charge (pH=7) -9.97 2D Binding mode Binding energy (Kcal/mol) -9.56 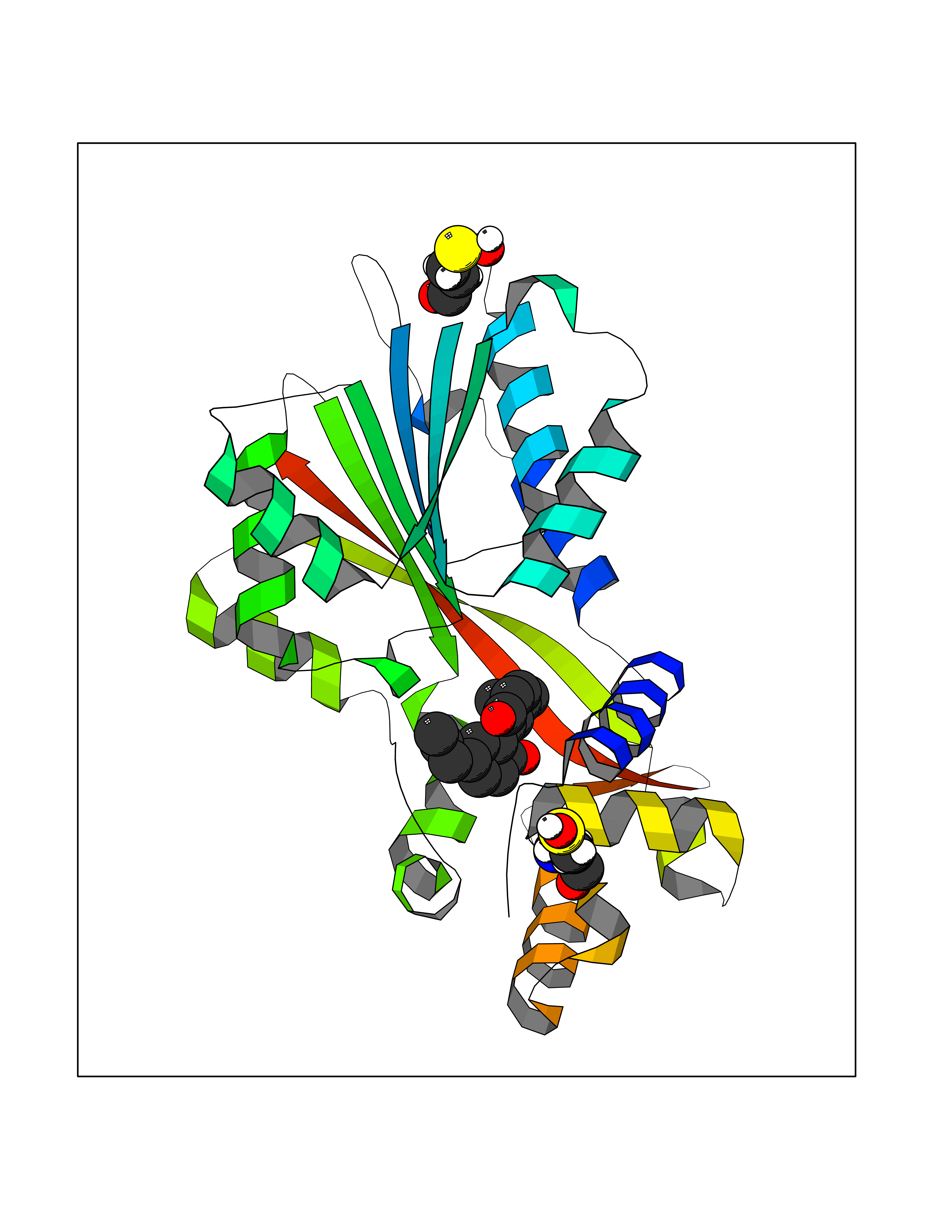 Molscript Map 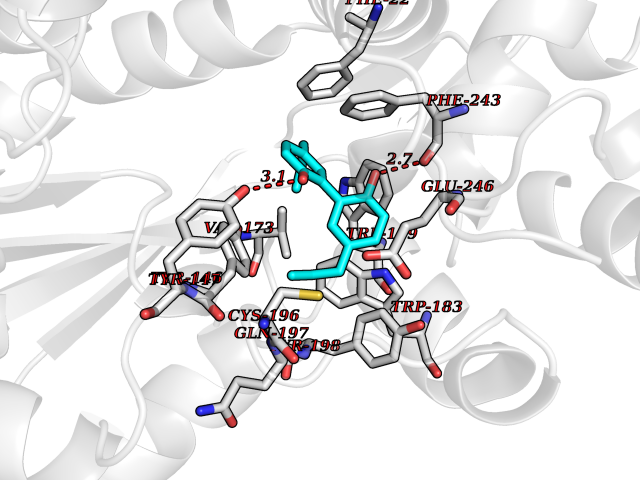 Pymol Map 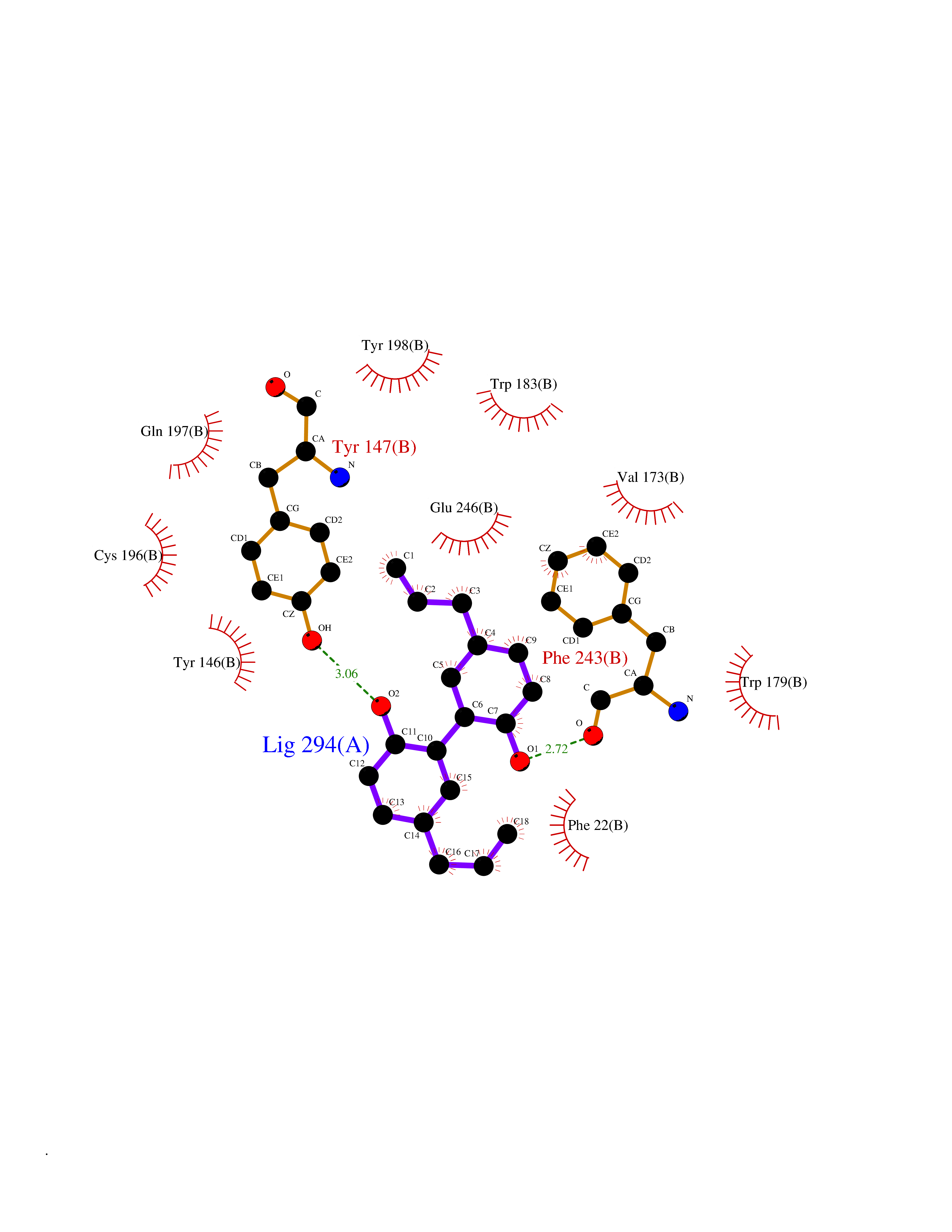 Ligplot Map 3D Binding mode Sequence MRSLFSDHGKYVESFRRFLNHSTEHQCMQEFMDKKLPGIIGRIGDTKSEIKILSIGGGAGEIDLQILSKVQAQYPGVXINNEVVEPSAEQIAKYKELVAKTSNLENVKFAWHKETSSEYQSRMLEKKELQKWDFIHMIQMLYYVKDIPATLKFFHSLLGTNAKMLIIVVSGSSGWDKLWKKYGSRFPQDDLCQYITSDDLTQMLDNLGLKYECYDLLSTMDISDCFIDGNENGDLLWDFLTETXNFNATAPPDLRAELGKDLQEPEFSAKKEGKVLFNNTLSFIVIEA Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Dihydroorotate dehydrogenase (quinone), mitochondrial | 4CQ8 | 7.01 | |
Target general information Gen name PFF0160c Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class Oxidoreductase Function Dihydroorotate dehydrogenase activity. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42573.5 Length 378 Aromaticity 0.1 Instability index 36.63 Isoelectric point 8.21 Charge (pH=7) 3.17 2D Binding mode Binding energy (Kcal/mol) -9.56 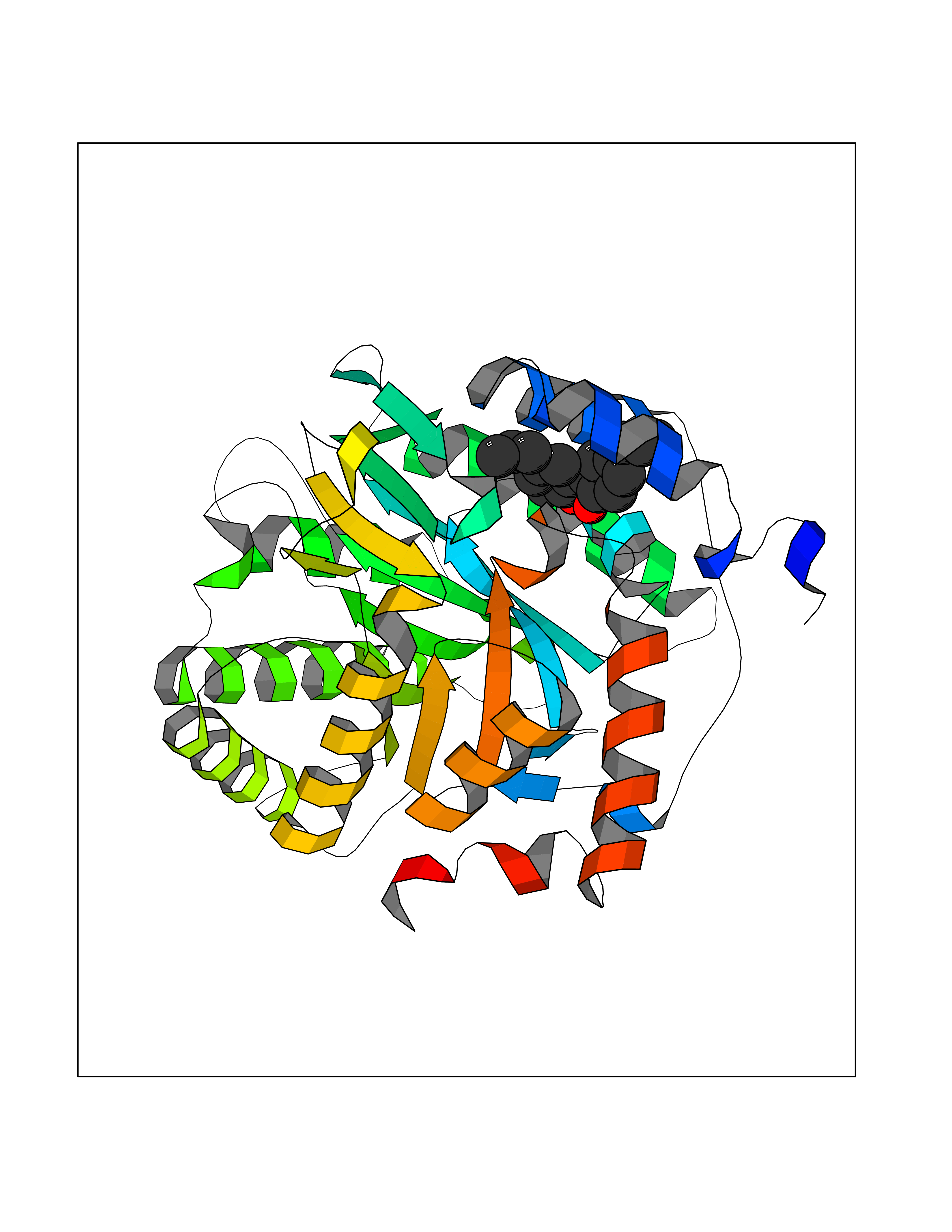 Molscript Map 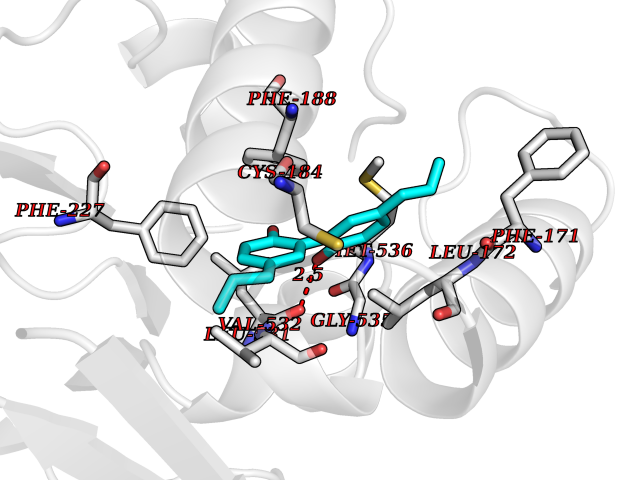 Pymol Map 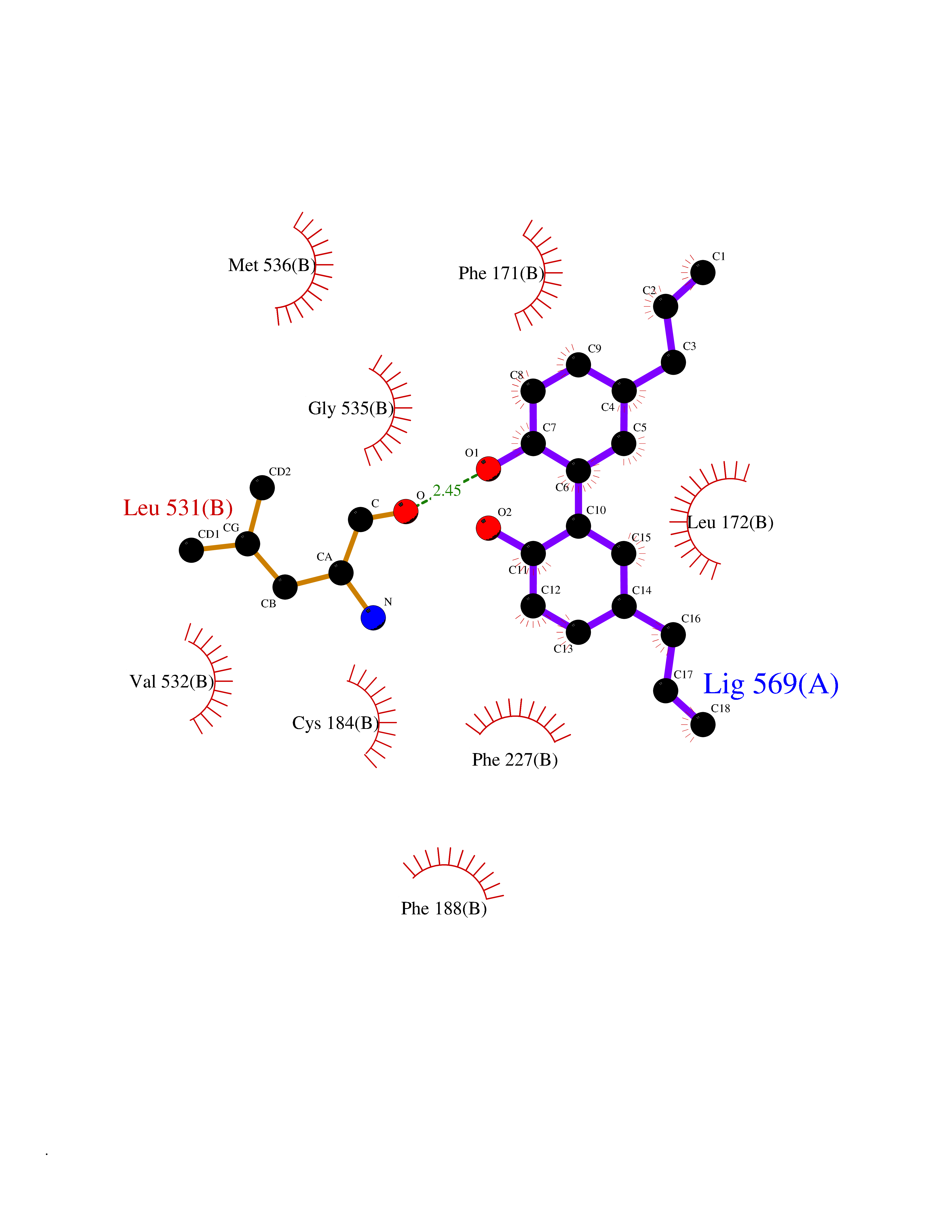 Ligplot Map 3D Binding mode Sequence ADPFESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEKNNFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Histone deacetylase 2 (HDAC2) | 4LY1 | 7.01 | |
Target general information Gen name HDAC2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HD2 Protein family Histone deacetylase family, HD type 1 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Forms transcriptional repressor complexes by associating with MAD, SIN3, YY1 and N-COR. Interacts in the late S-phase of DNA-replication with DNMT1 in the other transcriptional repressor complex composed of DNMT1, DMAP1, PCNA, CAF1. Deacetylates TSHZ3 and regulates its transcriptional repressor activity. Component of a RCOR/GFI/KDM1A/HDAC complex that suppresses, via histone deacetylase (HDAC) recruitment, a number of genes implicated in multilineage blood cell development. May be involved in the transcriptional repression of circadian target genes, such as PER1, mediated by CRY1 through histone deacetylation. Involved in MTA1-mediated transcriptional corepression of TFF1 and CDKN1A. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4). Related diseases Ventricular tachycardia, catecholaminergic polymorphic, 1, with or without atrial dysfunction and/or dilated cardiomyopathy (CPVT1) [MIM:604772]: An arrhythmogenic disorder characterized by stress-induced, bidirectional ventricular tachycardia that may degenerate into cardiac arrest and cause sudden death. Patients present with recurrent syncope, seizures, or sudden death after physical activity or emotional stress. CPVT1 inheritance is autosomal dominant. {ECO:0000269|PubMed:11157710, ECO:0000269|PubMed:11159936, ECO:0000269|PubMed:11208676, ECO:0000269|PubMed:12093772, ECO:0000269|PubMed:12106942, ECO:0000269|PubMed:14571276, ECO:0000269|PubMed:15046072, ECO:0000269|PubMed:15046073, ECO:0000269|PubMed:15466642, ECO:0000269|PubMed:15544015, ECO:0000269|PubMed:16188589, ECO:0000269|PubMed:24793461, ECO:0000269|PubMed:25372681, ECO:0000269|PubMed:27733687}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ventricular arrhythmias due to cardiac ryanodine receptor calcium release deficiency syndrome (VACRDS) [MIM:115000]: An autosomal dominant arrhythmogenic disorder characterized by syncope, cardiac arrest and/or sudden unexpected death, often in association with physical exertion or acute emotional stress. Patients who survive manifest polymorphic ventricular tachycardia and ventricular fibrillation. Unlike typical catecholaminergic ventricular tachycardia, arrhythmias are not reproducible on exercise stress testing or adrenaline challenge. {ECO:0000269|PubMed:12093772, ECO:0000269|PubMed:17984046, ECO:0000269|PubMed:33536282}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12565; DB01223; DB01076; DB05015; DB01262; DB11841; DB01095; DB12645; DB00227; DB11830; DB01303; DB06603; DB06819; DB05223; DB00175; DB03766; DB12847; DB06176; DB00641; DB00277; DB09091; DB00313; DB02546 Interacts with Q9C0K0; Q9HCU9; P68400; Q9UER7; P51610; Q13547; Q9UIS9; Q13330; P01106; P06748; P48382; Q96ST3; O95863; Q9HD15; O43463; Q9H3M7; Q92618; Q17R98; Q2HR82; PRO_0000449623 [P0DTD1] EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Cytoplasm; Hydrolase; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-nitrosylation; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 42020.5 Length 366 Aromaticity 0.13 Instability index 29.52 Isoelectric point 6.52 Charge (pH=7) -2.16 2D Binding mode Binding energy (Kcal/mol) -9.57 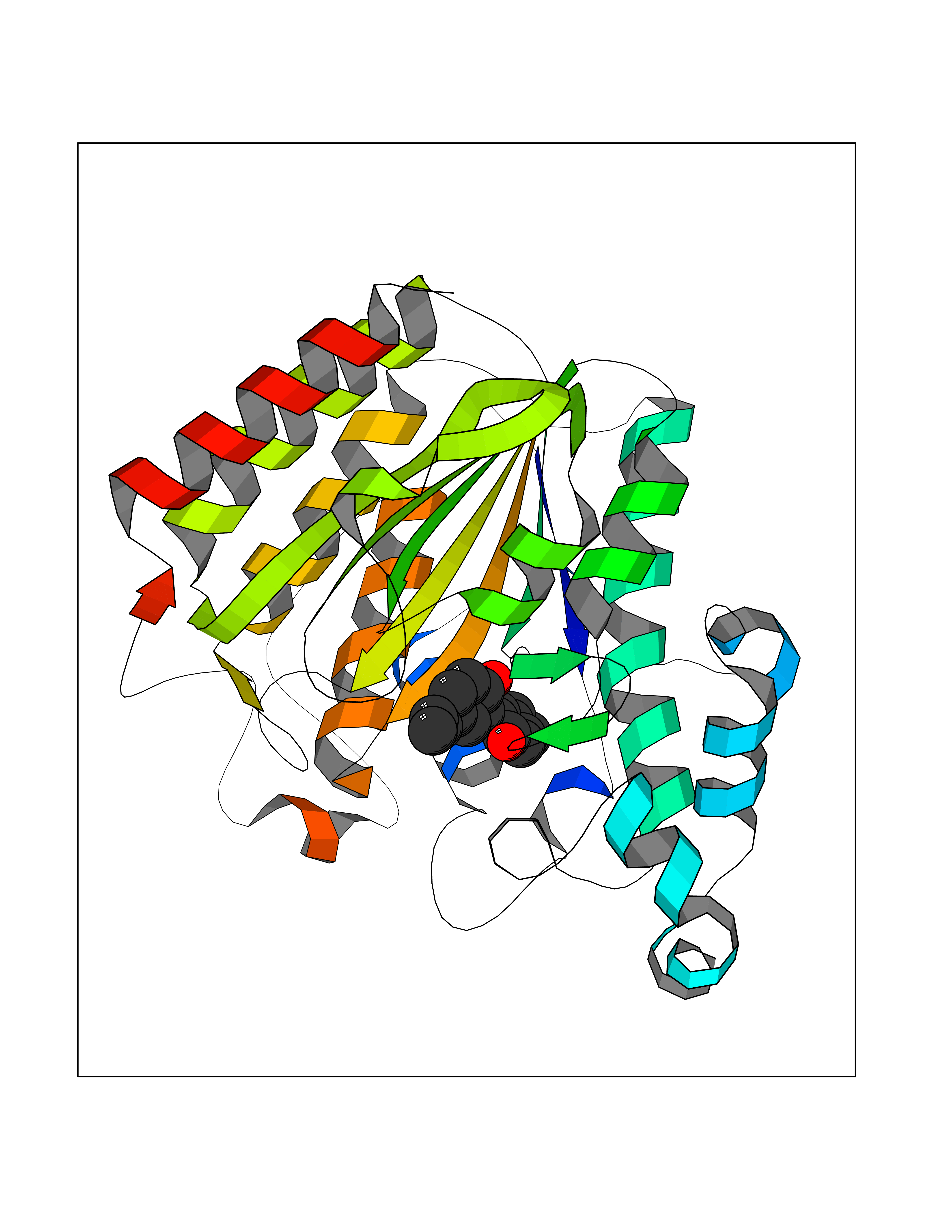 Molscript Map 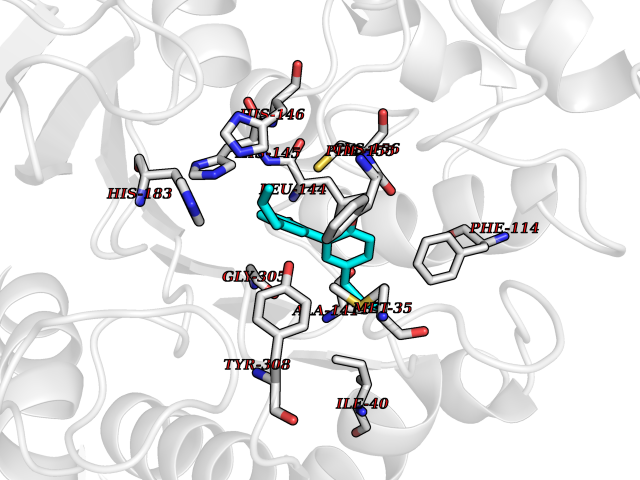 Pymol Map 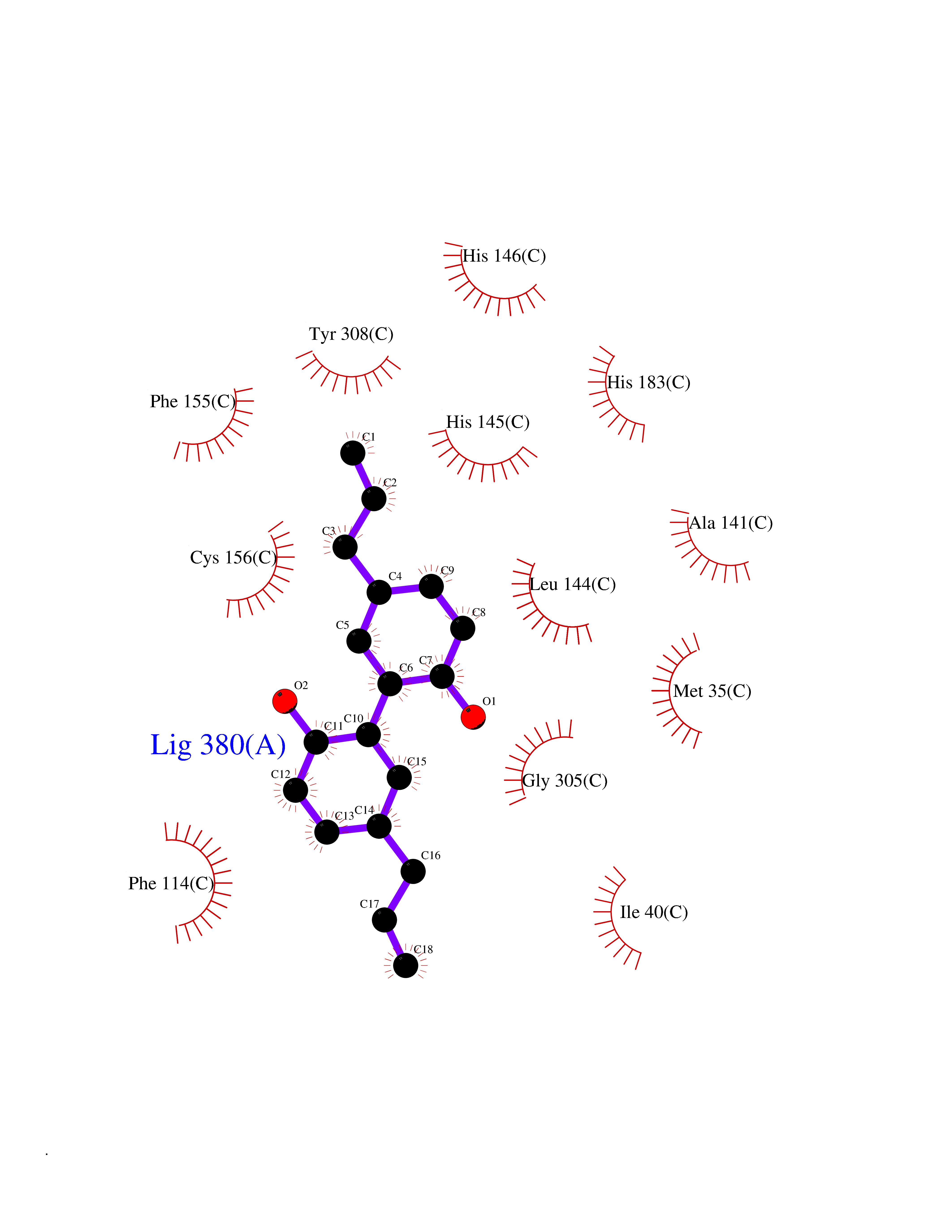 Ligplot Map 3D Binding mode Sequence KKKVCYYYDGDIGNYYYGQGHPMKPHRIRMTHNLLLNYGLYRKMEIYRPHKATAEEMTKYHSDEYIKFLRSIRPDNMSEYSKQMQRFNVGEDCPVFDGLFEFCQLSTGGSVAGAVKLNRQQTDMAVNWAGGLHHAKKSEASGFCYVNDIVLAILELLKYHQRVLYIDIDIHHGDGVEEAFYTTDRVMTVSFHKYGEYFPGTGDLRDIGAGKGKYYAVNFPMRDGIDDESYGQIFKPIISKVMEMYQPSAVVLQCGADSLSGDRLGCFNLTVKGHAKCVEVVKTFNLPLLMLGGGGYTIRNVARCWTYETAVALDCEIPNELPYNDYFEYFGPDFKLHISPSNMTNQNTPEYMEKIKQRLFENLRML Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | BUB1 mitotic checkpoint serine/threonine kinase (BUB1) | 6F7B | 7.01 | |
Target general information Gen name BUB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hBUB1; Mitotic checkpoint serine/threonine-protein kinase BUB1; BUB1L; BUB1A Protein family Protein kinase superfamily, Ser/Thr protein kinase family, BUB1 subfamily Biochemical class Kinase Function Has a key role in the assembly of checkpoint proteins at the kinetochore, being required for the subsequent localization of CENPF, BUB1B, CENPE and MAD2L1. Required for the kinetochore localization of PLK1. Required for centromeric enrichment of AUKRB in prometaphase. Plays an important role in defining SGO1 localization and thereby affects sister chromatid cohesion. Acts as a substrate for anaphase-promoting complex or cyclosome (APC/C) in complex with its activator CDH1 (APC/C-Cdh1). Necessary for ensuring proper chromosome segregation and binding to BUB3 is essential for this function. Can regulate chromosome segregation in a kinetochore-independent manner. Can phosphorylate BUB3. The BUB1-BUB3 complex plays a role in the inhibition of APC/C when spindle-assembly checkpoint is activated and inhibits the ubiquitin ligase activity of APC/C by phosphorylating its activator CDC20. This complex can also phosphorylate MAD1L1. Kinase activity is essential for inhibition of APC/CCDC20 and for chromosome alignment but does not play a major role in the spindle-assembly checkpoint activity. Mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. Serine/threonine-protein kinase that performs 2 crucial functions during mitosis: it is essential for spindle-assembly checkpoint signaling and for correct chromosome alignment. Related diseases Microcephaly 30, primary, autosomal recessive (MCPH30) [MIM:620183]: A form of microcephaly, a disease defined as a head circumference more than 3 standard deviations below the age, sex and ethnically matched mean. Brain weight is markedly reduced and the cerebral cortex is disproportionately small. MCPH30 is characterized by small head, poor overall growth, and global developmental delay with variably impaired intellectual development. Affected individuals may also have variable congenital anomalies, including atrial septal defect, dysmorphic facial features, tracheal stenosis, and anomalies of the skin and teeth. {ECO:0000269|PubMed:35044816}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O95376; O60566; O43684; P46108; Q8NG31; Q8NG31-2; Q9GZQ8; P03070 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell cycle; Cell division; Centromere; Chromosome; Chromosome partition; Host-virus interaction; Intellectual disability; Kinase; Kinetochore; Mitosis; Nucleotide-binding; Nucleus; Phosphoprotein; Primary microcephaly; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39564.8 Length 343 Aromaticity 0.12 Instability index 31.17 Isoelectric point 8.49 Charge (pH=7) 4.7 2D Binding mode Binding energy (Kcal/mol) -9.56 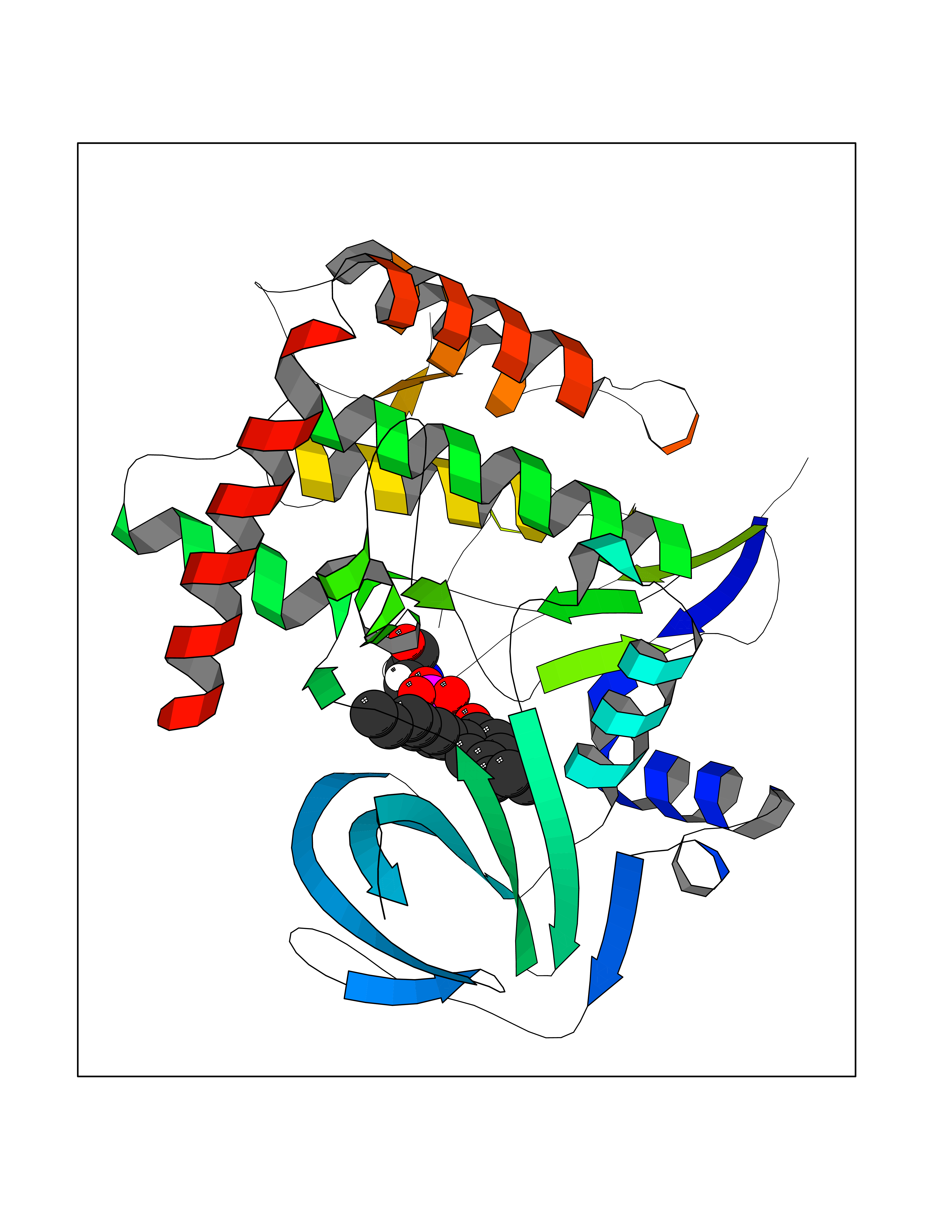 Molscript Map 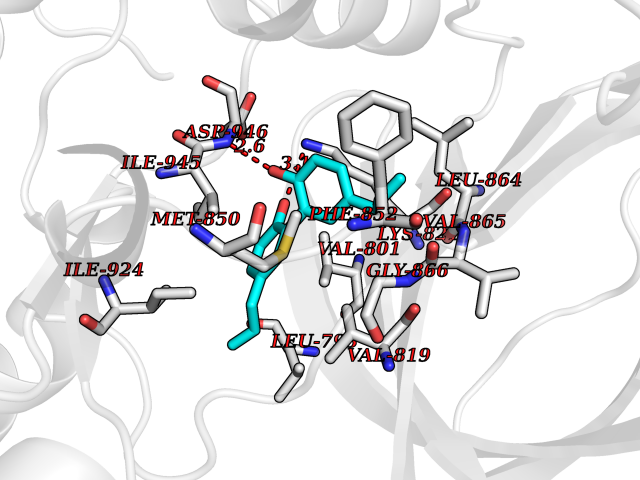 Pymol Map 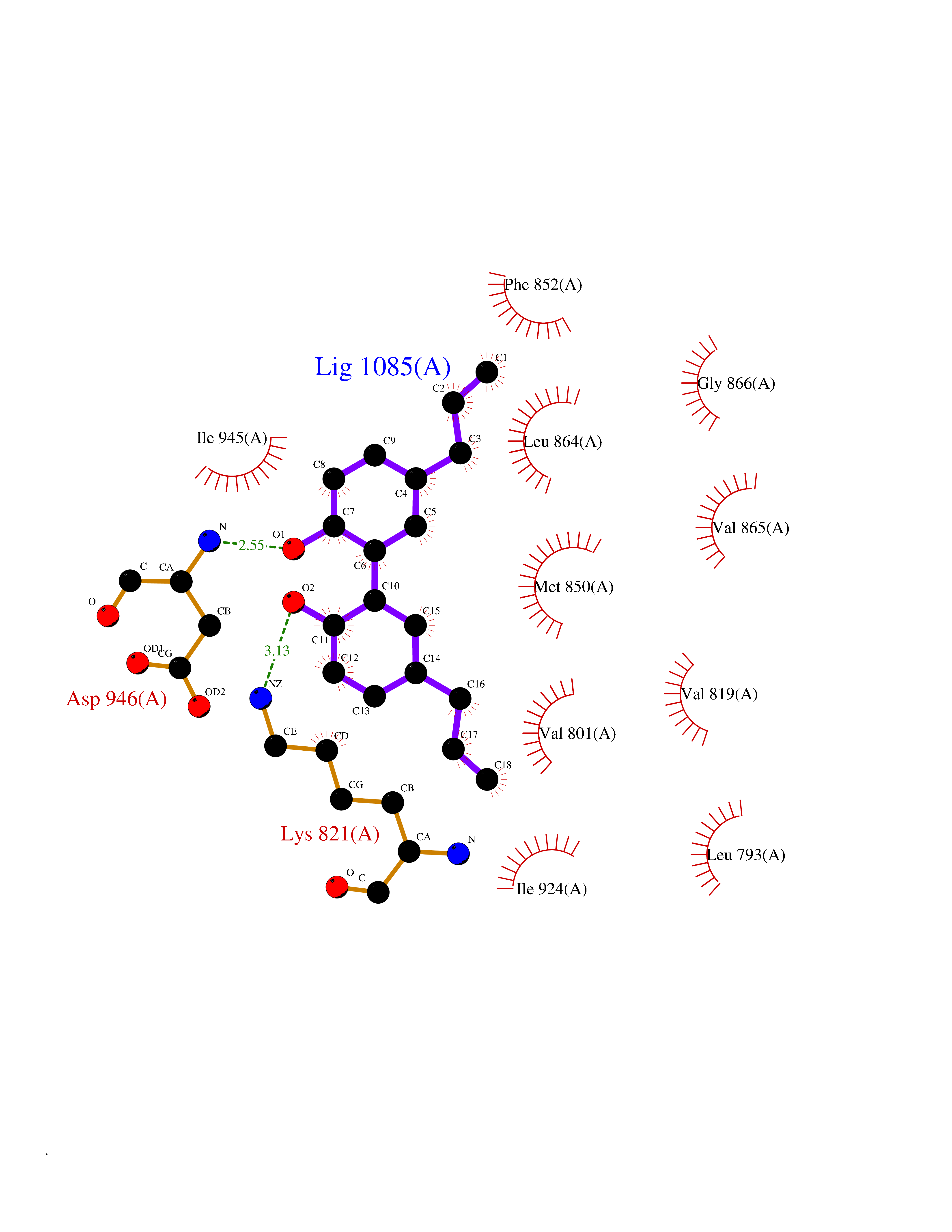 Ligplot Map 3D Binding mode Sequence APNFIVGNPWDDKLIFKLLSGLSKPVSSYPNTFEWQCKLPAIKPKTEFQLGSKLVYVHHLLGEGAFAQVYEATQKNKQKFVLKVQKPANPWEFYIGTQLMERLKPSMQHMFMKFYSAHLFQNGSVLVGELYSYGTLLNAINLYKNTPEKVMPQGLVISFAMRMLYMIEQVHDCEIIHGDIKPDNFILGNGFLEQDDEDDLSAGLALIDLGQSIDMKLFPKGTIFTAKCETXGFQCVEMLSNKPWNYQIDYFGVAATVYCMLFGTYMKVKNEECKPEGLFRRLPHLDMWNEFFHVMLNIPDCHHLPSLDLLRQKLKKVFQQHYTNKIRALRNRLIVLLLECKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Sphingosine-1-phosphate receptor 2 (S1PR2) | 7T6B | 7.01 | |
Target general information Gen name S1PR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Sphingosine 1-phosphate receptor Edg-5; S1PR2; S1P2; S1P receptor Edg-5; S1P receptor 2; Endothelial differentiation G-protein coupled receptor 5 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the lysosphingolipid sphingosine 1- phosphate (S1P). S1P is a bioactive lysophospholipid that elicits diverse physiological effect on most types of cells and tissues. When expressed in rat HTC4 hepatoma cells, is capable of mediating S1P-induced cell proliferation and suppression of apoptosis. Related diseases Deafness, autosomal recessive, 68 (DFNB68) [MIM:610419]: A form of non-syndromic sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:26805784}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P16144; Q9JK11-1 EC number NA Uniprot keywords 3D-structure; Cell membrane; Deafness; Disease variant; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Non-syndromic deafness; Palmitate; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 28917.3 Length 264 Aromaticity 0.11 Instability index 38.95 Isoelectric point 9.11 Charge (pH=7) 9.27 2D Binding mode Binding energy (Kcal/mol) -9.57 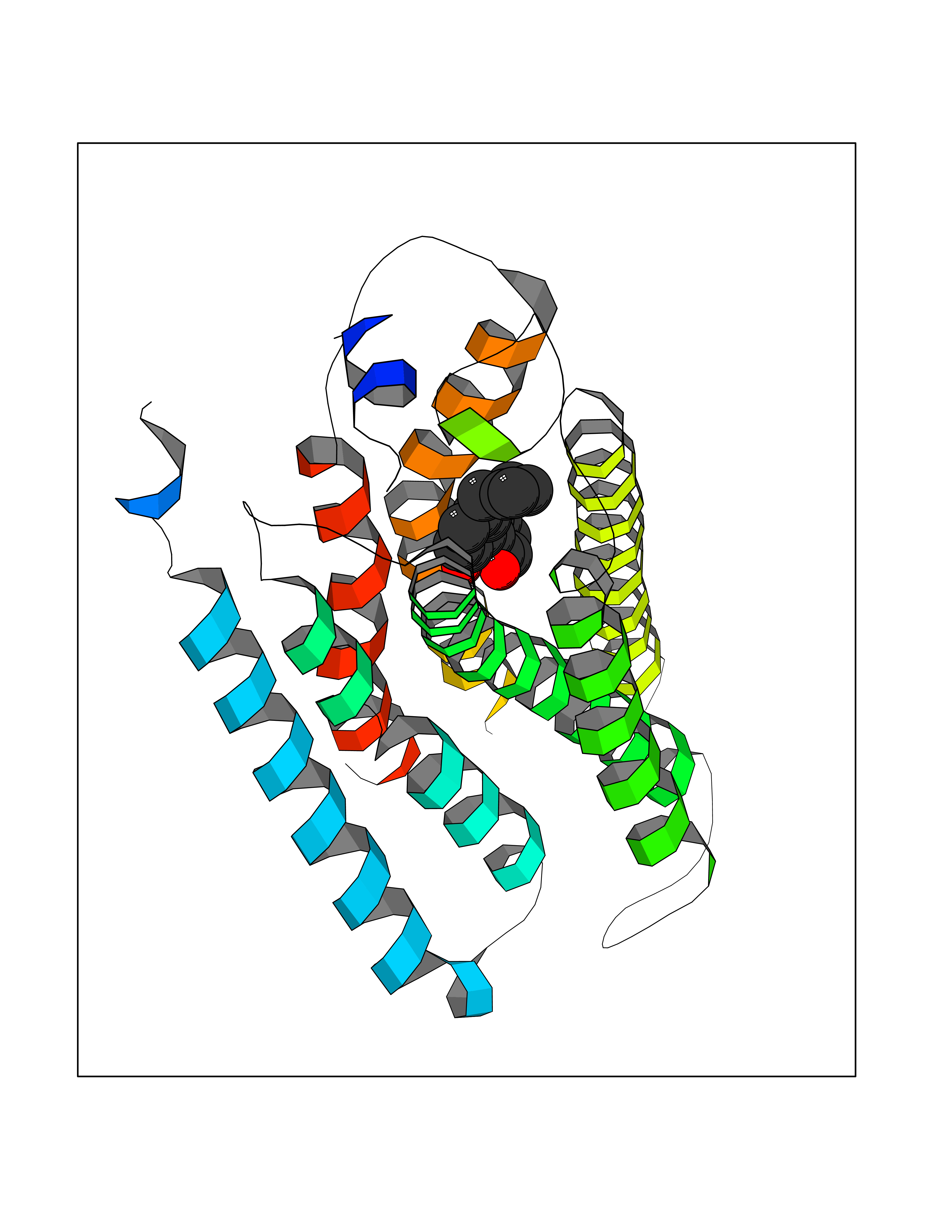 Molscript Map 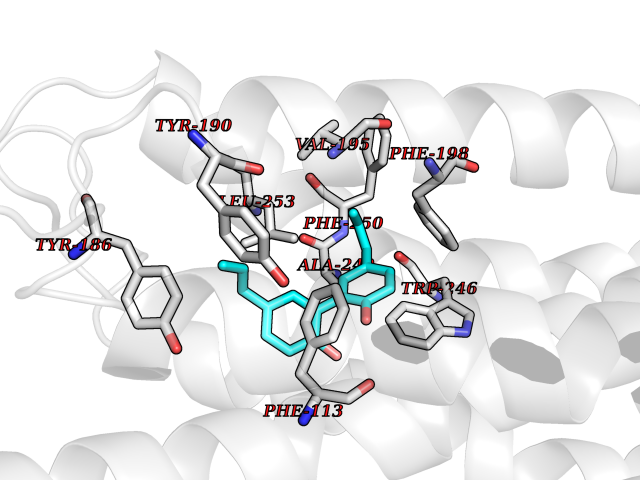 Pymol Map 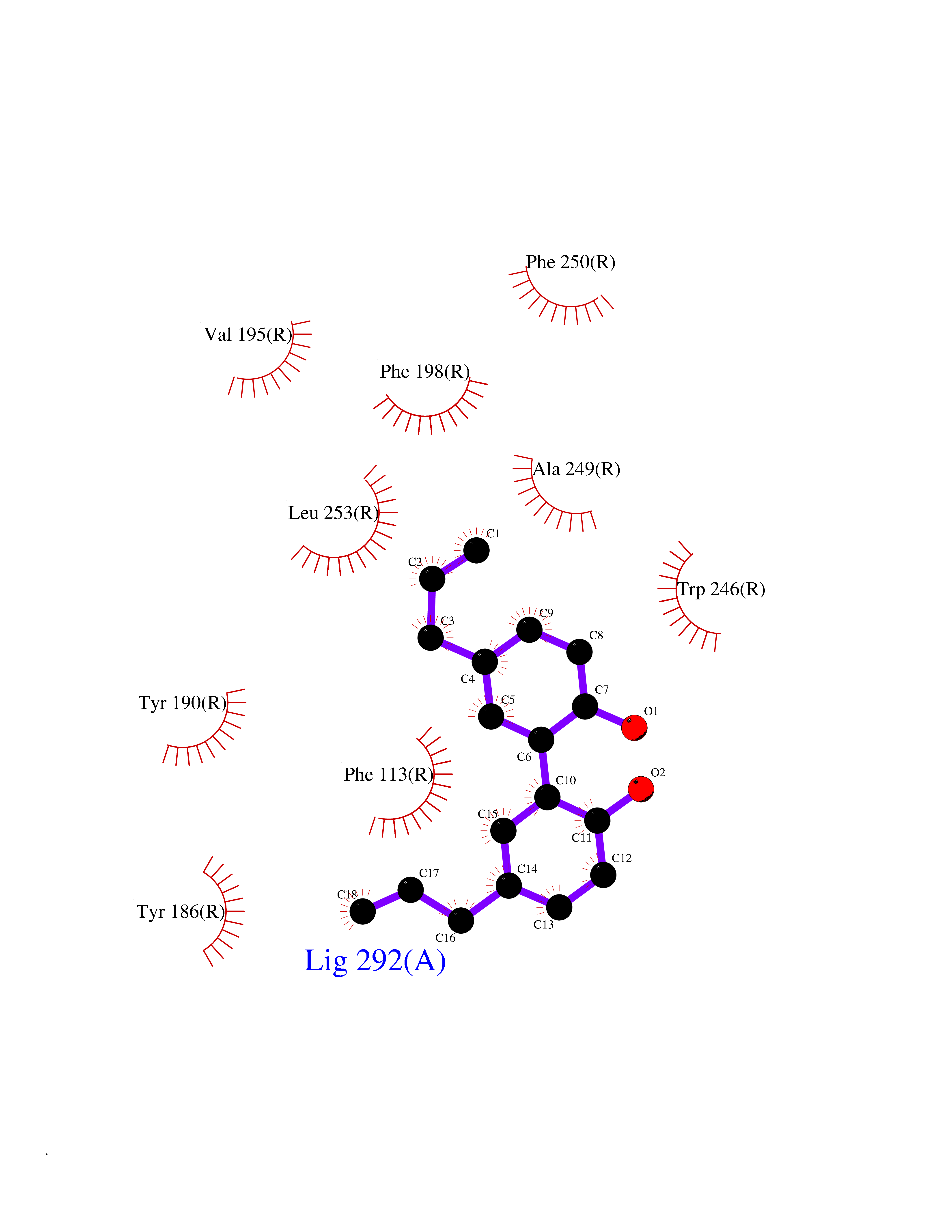 Ligplot Map 3D Binding mode Sequence NKVQEHYNYTKTSRQVASAFIVILCCAIVVENLLVLIAVARNSKFHSAMYLFLGNLAASDLLAGVAFVANTLLSGSVTLRLTPVQWFAREGSAFITLSASVFSLLAIAIERHVAIAKVKLYGSDKSCRMLLLIGASWLISLVLGGLPILGWNCLGHLEACSTVLPLYAKHYVLCVVTIFSIILLAIVALYVRIYCVVRSSQTLALLKTVTIVLGVFIVCWLPAFSILLLDYACPVHSCPILYKAHYFFAVSTLNSLLNPVIYTW Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Tyrosine-protein kinase Kit (KIT) | 1T46 | 7.00 | |
Target general information Gen name KIT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog; p145 c-kit; Proto-oncogene tyrosine-protein kinase Kit; Proto-oncogene c-Kit; Piebald trait protein; PBT; Mast/stem cell growth factor re Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function In response to KITLG/SCF binding, KIT can activate several signaling pathways. Phosphorylates PIK3R1, PLCG1, SH2B2/APS and CBL. Activates the AKT1 signaling pathway by phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase. Activated KIT also transmits signals via GRB2 and activation of RAS, RAF1 and the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3, STAT5A and STAT5B. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate. KIT signaling is modulated by protein phosphatases, and by rapid internalization and degradation of the receptor. Activated KIT promotes phosphorylation of the protein phosphatases PTPN6/SHP-1 and PTPRU, and of the transcription factors STAT1, STAT3, STAT5A and STAT5B. Promotes phosphorylation of PIK3R1, CBL, CRK (isoform Crk-II), LYN, MAPK1/ERK2 and/or MAPK3/ERK1, PLCG1, SRC and SHC1. Tyrosine-protein kinase that acts as cell-surface receptor for the cytokine KITLG/SCF and plays an essential role in the regulation of cell survival and proliferation, hematopoiesis, stem cell maintenance, gametogenesis, mast cell development, migration and function, and in melanogenesis. Related diseases Piebald trait (PBT) [MIM:172800]: Autosomal dominant genetic developmental abnormality of pigmentation characterized by congenital patches of white skin and hair that lack melanocytes. {ECO:0000269|PubMed:11074500, ECO:0000269|PubMed:1370874, ECO:0000269|PubMed:1376329, ECO:0000269|PubMed:1717985, ECO:0000269|PubMed:7687267, ECO:0000269|PubMed:8680409, ECO:0000269|PubMed:9450866, ECO:0000269|PubMed:9699740}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:11505412, ECO:0000269|PubMed:15824741, ECO:0000269|PubMed:9438854, ECO:0000269|PubMed:9697690}. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Testicular germ cell tumor (TGCT) [MIM:273300]: A common malignancy in males representing 95% of all testicular neoplasms. TGCTs have various pathologic subtypes including: unclassified intratubular germ cell neoplasia, seminoma (including cases with syncytiotrophoblastic cells), spermatocytic seminoma, embryonal carcinoma, yolk sac tumor, choriocarcinoma, and teratoma. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. The gene represented in this entry is involved in disease pathogenesis. Somatic mutations that lead to constitutive activation of KIT are detected in AML patients. These mutations fall into two classes, the most common being in-frame internal tandem duplications of variable length in the juxtamembrane region that disrupt the normal regulation of the kinase activity. Likewise, point mutations in the kinase domain can result in a constitutively activated kinase.; DISEASE: Mastocytosis, cutaneous (MASTC) [MIM:154800]: A form of mastocytosis, a heterogeneous group of disorders associated with abnormal proliferation and accumulation of mast cells in various tissues, especially in the skin and hematopoietic organs. MASTC is an autosomal dominant form characterized by macules, papules, nodules, or diffuse infiltration of the skin, often associated with localized hyperpigmentation. Gentle rubbing of the lesions induces histamine release from mechanically activated mast cells, causing local wheals, erythema, and often pruritus, a phenomenon termed Darier sign. {ECO:0000269|PubMed:15173254, ECO:0000269|PubMed:19865100, ECO:0000269|PubMed:21689725, ECO:0000269|PubMed:24289326, ECO:0000269|PubMed:9990072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mastocytosis, systemic (MASTSYS) [MIM:154800]: A severe form of mastocytosis characterized by abnormal proliferation and accumulation of mast cells in several organs, resulting in a systemic disease that may affect bone, gastrointestinal tract, lymphatics, spleen, and liver. In some cases, it is associated with a clonal hematologic non-mast-cell lineage disease, such as a myelodysplastic or myeloproliferative disorder. It can also lead to mast cell leukemia, which carries a high risk of mortality. {ECO:0000269|PubMed:9990072}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB09103; DB15233; DB01254; DB12147; DB12010; DB00619; DB09078; DB06080; DB06595; DB04868; DB05913; DB06589; DB12978; DB01962; DB08901; DB08896; DB14840; DB00398; DB01268; DB11800; DB05146 Interacts with P00519; P42684; O75815; P51451; Q8WV28; P46108; P07332; P09769; O75791; P62993; Q14451; P08631; Q96JZ2; P21583; P06239; P07948; P16333; O43639; P27986; O00459; Q92569; P19174; P16885; Q13882; Q06124; Q92729; P20936; Q9UQQ2; O14796; Q9NP31; Q8N5H7; P78314; Q15464; P29353; P98077; Q92529; Q9H6Q3; O14508; O14543; O14544; P12931; Q9ULZ2; Q9HBL0; Q63HR2; Q68CZ2; P42681; P07947; P43403; Q8VBX6; P35235 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Immunoglobulin domain; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33575.6 Length 297 Aromaticity 0.11 Instability index 45.37 Isoelectric point 8.37 Charge (pH=7) 3.24 2D Binding mode Binding energy (Kcal/mol) -9.55 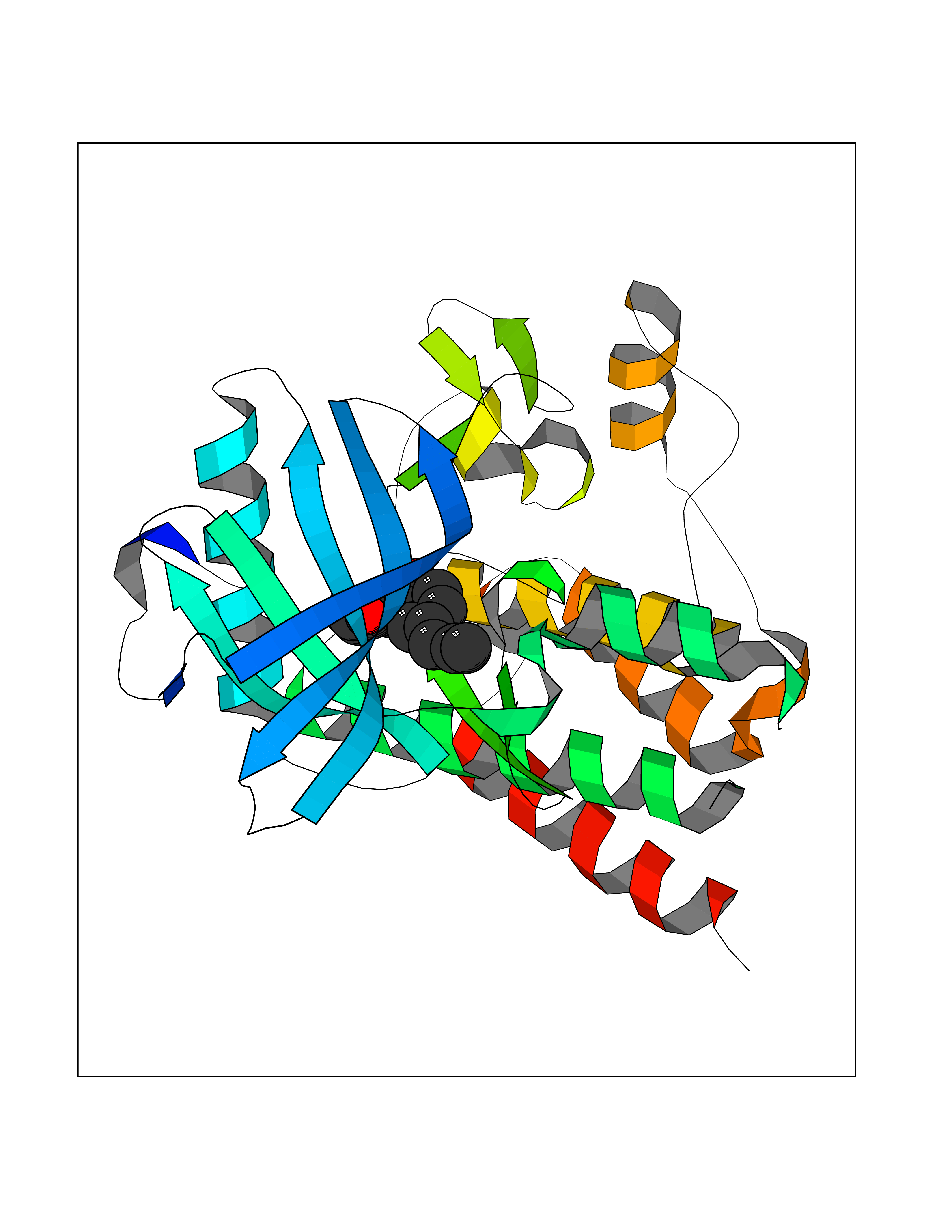 Molscript Map 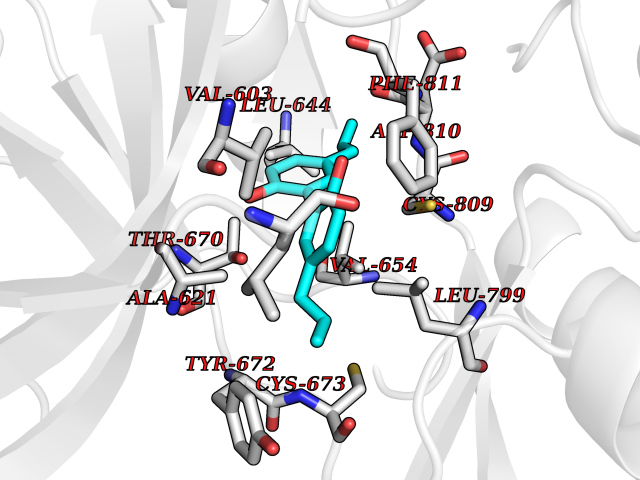 Pymol Map 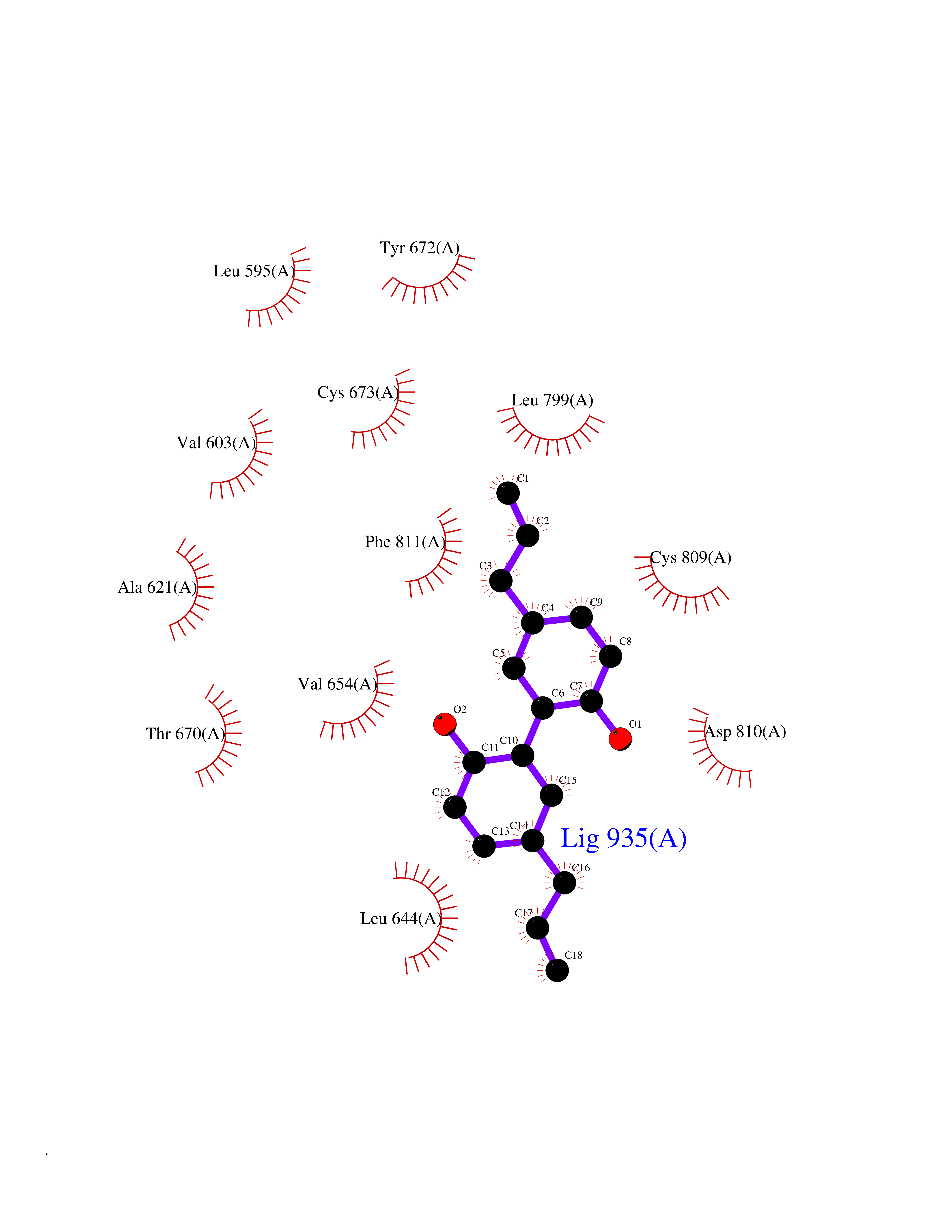 Ligplot Map 3D Binding mode Sequence GNNYVYIDPTQLPYDHKWEFPRNRLSFGKTLGAGAFGKVVEATAYGLIKSDAAMTVAVKMLKPSAHLTEREALMSELKVLSYLGNHMNIVNLLGACTIGGPTLVITEYCCYGDLLNFLRRKRDSFLALDLEDLLSFSYQVAKGMAFLASKNCIHRDLAARNILLTHGRITKICDFGLARDIKNDSNYVVKGNARLPVKWMAPESIFNCVYTFESDVWSYGIFLWELFSLGSSPYPGMPVDSKFYKMIKEGFRMLSPEHAPAEMYDIMKTCWDADPLKRPTFKQIVQLIEKQISESTN Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Tyrosine 3-monooxygenase (TH) | 2XSN | 7.00 | |
Target general information Gen name TH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine 3-hydroxylase; TH Protein family Biopterin-dependent aromatic amino acid hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Plays an important role in the physiology of adrenergic neurones. Related diseases Segawa syndrome autosomal recessive (ARSEGS) [MIM:605407]: A form of DOPA-responsive dystonia presenting in infancy or early childhood. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Some cases present with parkinsonian symptoms in infancy. Unlike all other forms of dystonia, it is an eminently treatable condition, due to a favorable response to L-DOPA. {ECO:0000269|PubMed:10585338, ECO:0000269|PubMed:11196107, ECO:0000269|PubMed:11246459, ECO:0000269|PubMed:15505183, ECO:0000269|PubMed:15747353, ECO:0000269|PubMed:16049992, ECO:0000269|PubMed:17696123, ECO:0000269|PubMed:18058633, ECO:0000269|PubMed:18554280, ECO:0000269|PubMed:19491146, ECO:0000269|PubMed:20056467, ECO:0000269|PubMed:20430833, ECO:0000269|PubMed:21940685, ECO:0000269|PubMed:22264700, ECO:0000269|PubMed:22815559, ECO:0000269|PubMed:23762320, ECO:0000269|PubMed:23939262, ECO:0000269|PubMed:24753243, ECO:0000269|PubMed:7814018, ECO:0000269|PubMed:8528210, ECO:0000269|PubMed:8817341, ECO:0000269|PubMed:9613851, ECO:0000269|PubMed:9703425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: May play a role in the pathogenesis of Parkinson disease (PD). A genome-wide copy number variation analysis has identified a 34 kilobase deletion over the TH gene in a PD patient but not in any controls. {ECO:0000269|PubMed:20809526}. Drugs (DrugBank ID) DB03552; DB04400; DB00765; DB00120; DB00360; DB00135 Interacts with P29762; P61978-2; Q99750; P08651-5; O75928-2; Q9UHX1-2; P0DJD3-2; P07101-3; Q9UJ04; C9J7I0; Q5MCW4 EC number EC 1.14.16.2 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Cell projection; Cytoplasm; Cytoplasmic vesicle; Disease variant; Dystonia; Iron; Metal-binding; Monooxygenase; Neurotransmitter biosynthesis; Nucleus; Oxidoreductase; Parkinson disease; Parkinsonism; Phosphoprotein; Proteomics identification; Reference proteome; Synapse Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 34997 Length 306 Aromaticity 0.12 Instability index 42.59 Isoelectric point 5.32 Charge (pH=7) -12.31 2D Binding mode Binding energy (Kcal/mol) -9.55  Molscript Map 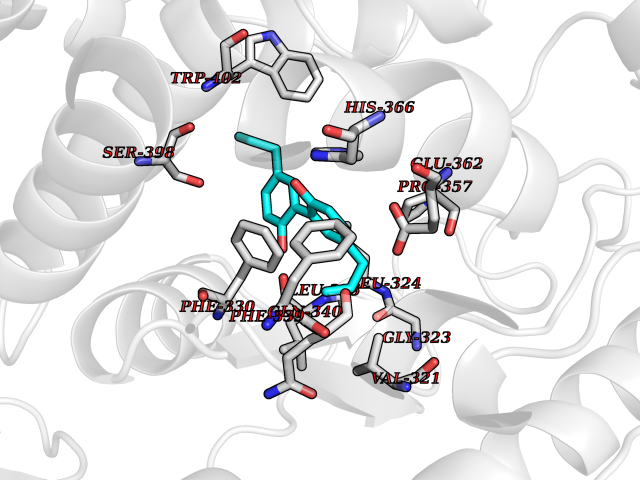 Pymol Map 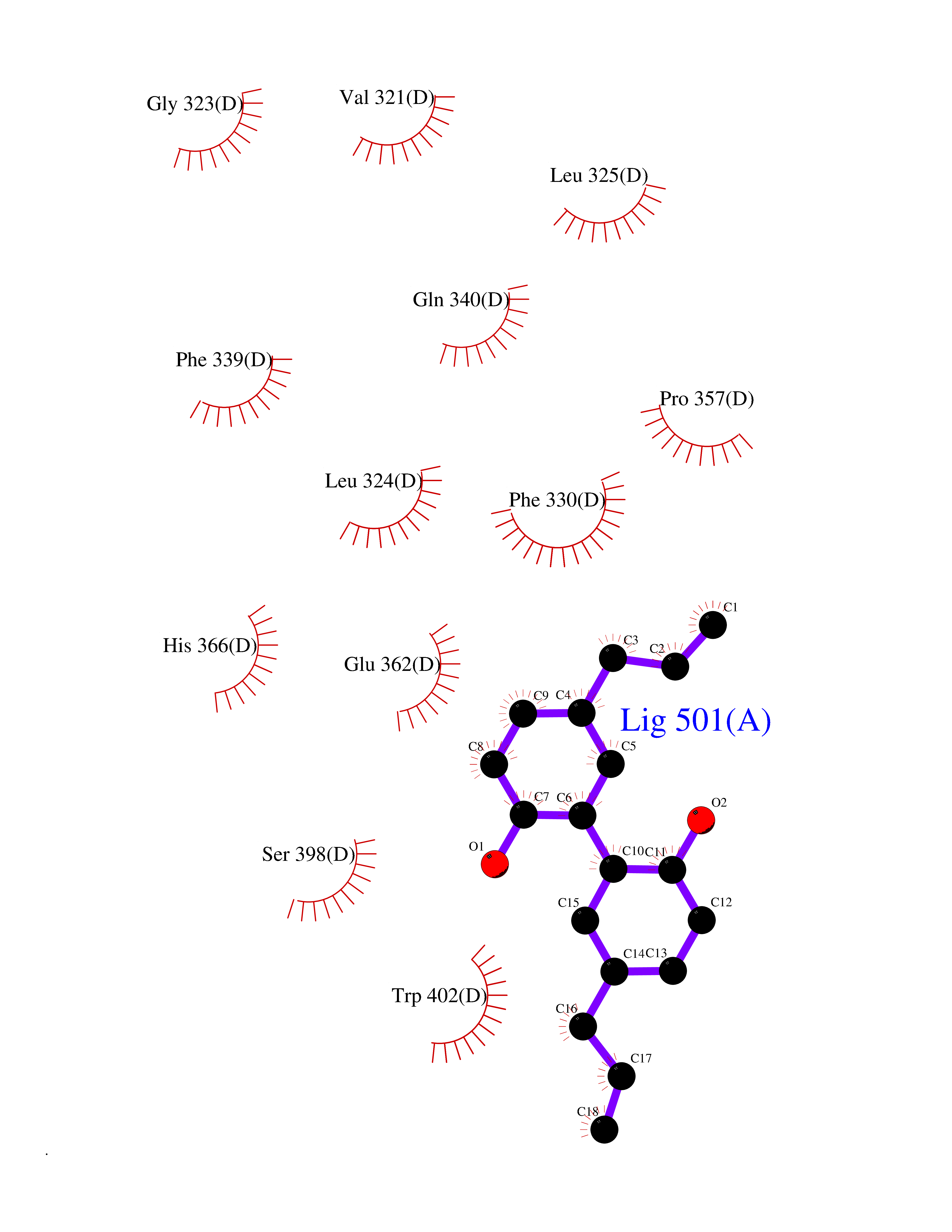 Ligplot Map 3D Binding mode Sequence VPWFPRKVSELDKCHHLVTKFDPDLDLDHPGFSDQVYRQRRKLIAEIAFQYRHGDPIPRVEYTAEEIATWKEVYTTLKGLYATHACGEHLEAFALLERFSGYREDNIPQLEDVSRFLKERTGFQLRPVAGLLSARDFLASLAFRVFQCTQYIRHASSPMHSPEPDCCHELLGHVPMLADRTFAQFSQDIGLASLGASDEEIEKLSTLYWFTVEFGLCKQNGEVKAYGAGLLSSYGELLHCLSEEPEIRAFDPEAAAVQPYQDQTYQSVYFVSESFSDAKDKLRSYASRIQRPFSVKFDPYTLAIDV Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Matrix metalloproteinase-10 (MMP-10) | 1Q3A | 7.00 | |
Target general information Gen name MMP10 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Transin-2; Stromelysin-2; STMY2; SL-2 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates procollagenase. Can degrade fibronectin, gelatins of type I, III, IV, and V; weakly collagens III, IV, and V. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00786; DB08271 Interacts with NA EC number EC 3.4.24.22 Uniprot keywords 3D-structure; Calcium; Collagen degradation; Disulfide bond; Extracellular matrix; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 52822 Length 471 Aromaticity 0.12 Instability index 21.13 Isoelectric point 4.83 Charge (pH=7) -35.32 2D Binding mode Binding energy (Kcal/mol) -9.55 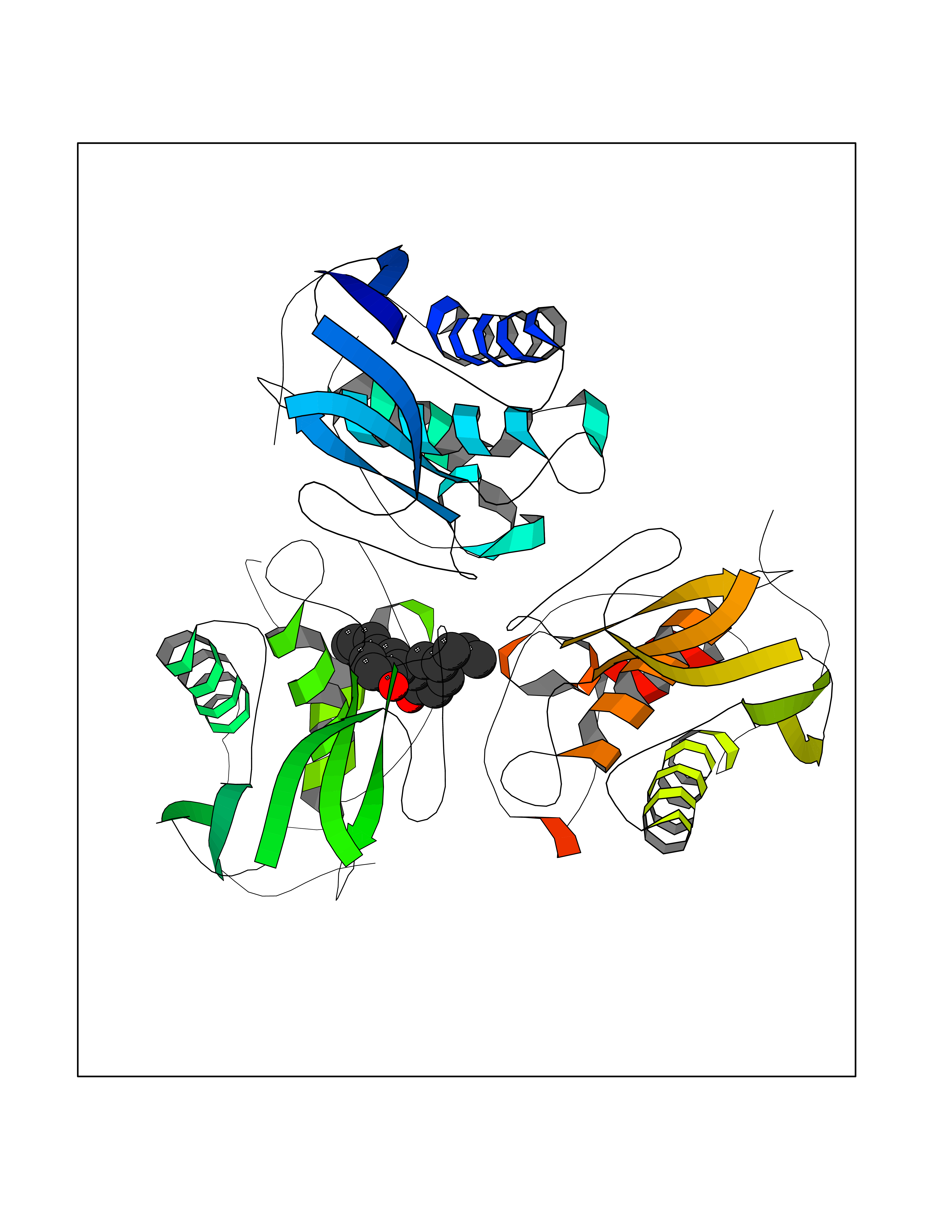 Molscript Map 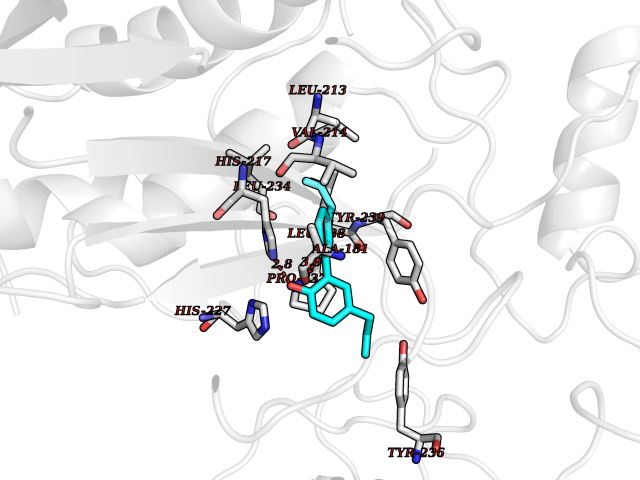 Pymol Map 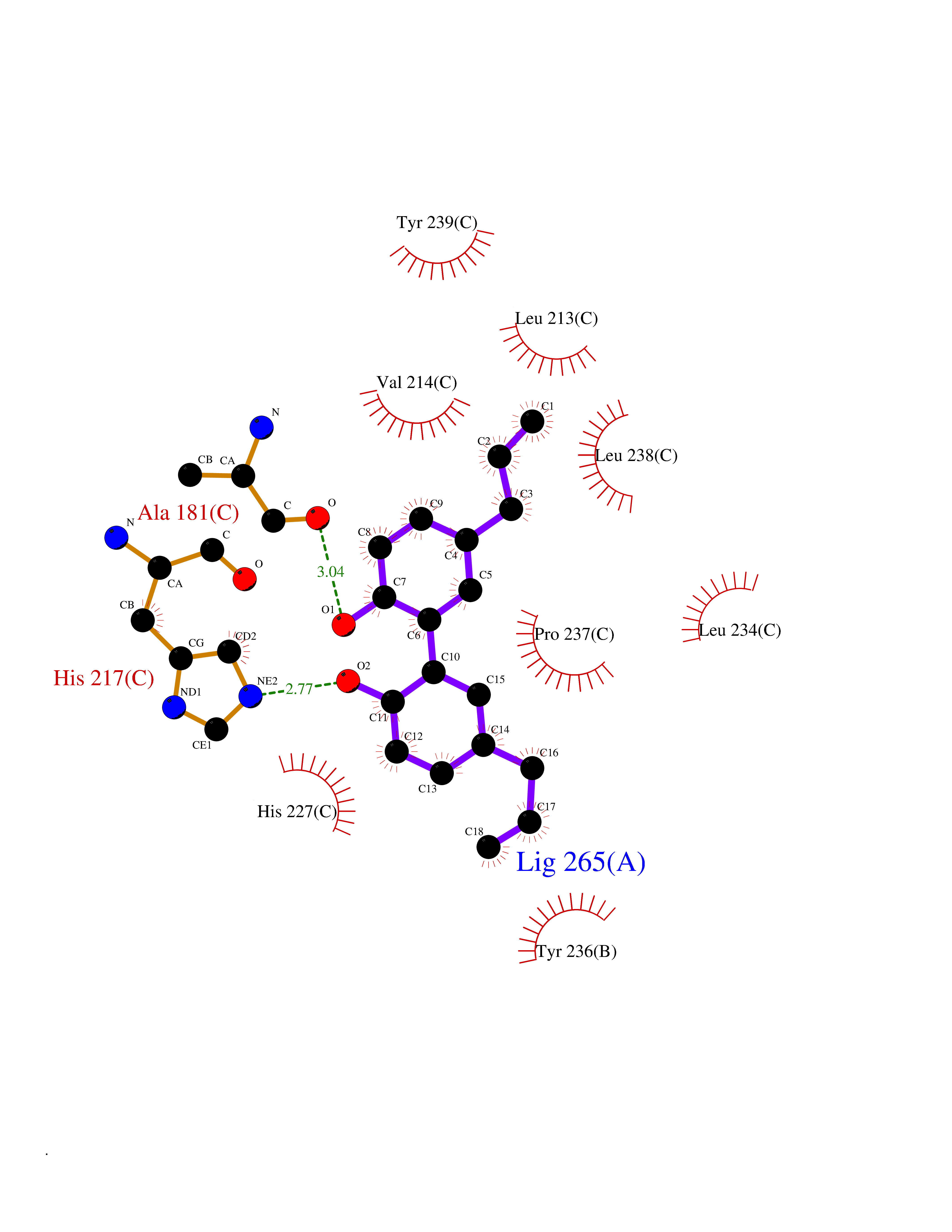 Ligplot Map 3D Binding mode Sequence MPKWRKTHLTYRIVNYTPDLPRDAVDSAIEKALKVWEEVTPLTFSRLYEGEADIMISFAVKEHGDNYSFDGPGHSLAHAYPPGPGLYGDIHFDDDEKWTEDASGTNLFLVAAHELGHSLGLFHSANTEALMYPLYNSLAQFRLSQDDVNGIQSLYGPKWRKTHLTYRIVNYTPDLPRDAVDSAIEKALKVWEEVTPLTFSRLYEGEADIMISFAVKEHGDNYSFDGPGHSLAHAYPPGPGLYGDIHFDDDEKWTEDASGTNLFLVAAHELGHSLGLFHSANTEALMYPLYNSLAQFRLSQDDVNGIQSLYGGMPKWRKTHLTYRIVNYTPDLPRDAVDSAIEKALKVWEEVTPLTFSRLYEGEADIMISFAVKEHGDNYSFDGPGHSLAHAYPPGPGLYGDIHFDDDEKWTEDASGTNLFLVAAHELGHSLGLFHSANTEALMYPLYNSFTELAQFRLSQDDVNGIQSLYG Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Tryptophan 5-hydroxylase 1 (TPH1) | 5TPG | 7.00 | |
Target general information Gen name TPH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tryptophan 5-monooxygenase 1; TRPH; TPRH Protein family Biopterin-dependent aromatic amino acid hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Responsible for addition of the -HO group (hydroxylation) to the 5 position to form the amino acid 5-hydroxytryptophan (5-HTP), which is the initial and rate-limiting step in the synthesis of the neurotransmitter serotonin. Related diseases Tyrosinemia 2 (TYRSN2) [MIM:276600]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, and oculocutaneous manifestations. Typical features include palmoplantar keratosis, painful corneal ulcers, and intellectual disability. {ECO:0000269|PubMed:1357662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05199; DB00360; DB12095; DB00150 Interacts with Q14457; Q96IK1-2; Q9UKB3; Q9H8Y8; O43586; O95789-4 EC number EC 1.14.16.4 Uniprot keywords 3D-structure; Alternative splicing; Iron; Metal-binding; Monooxygenase; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Serotonin biosynthesis; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 31138.2 Length 271 Aromaticity 0.13 Instability index 43.43 Isoelectric point 6.73 Charge (pH=7) -0.86 2D Binding mode Binding energy (Kcal/mol) -9.55 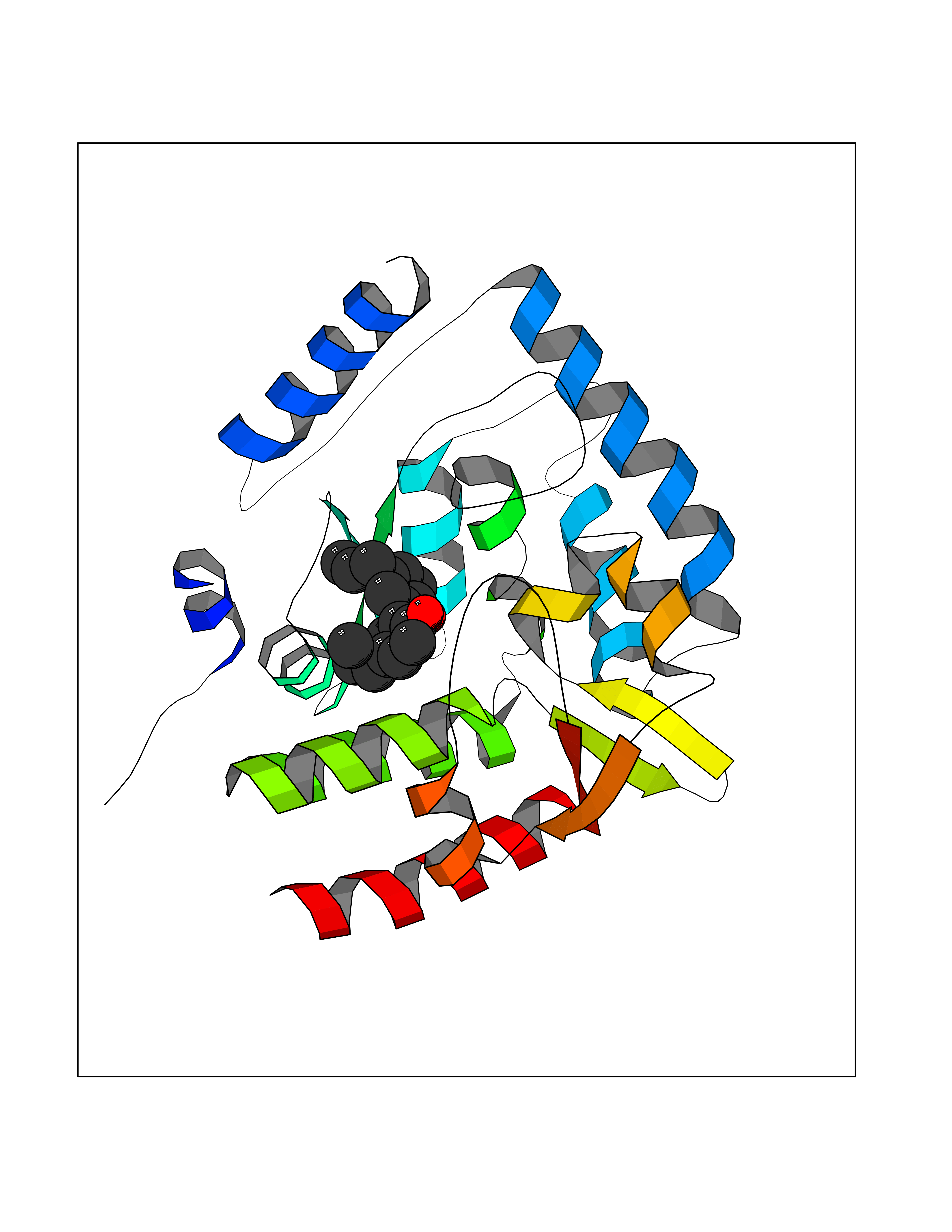 Molscript Map 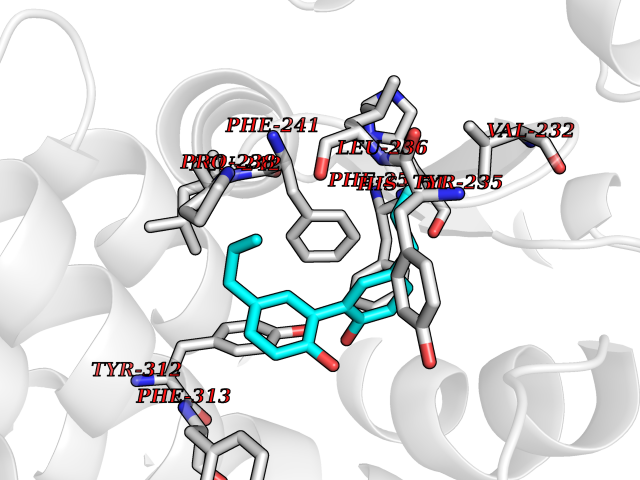 Pymol Map 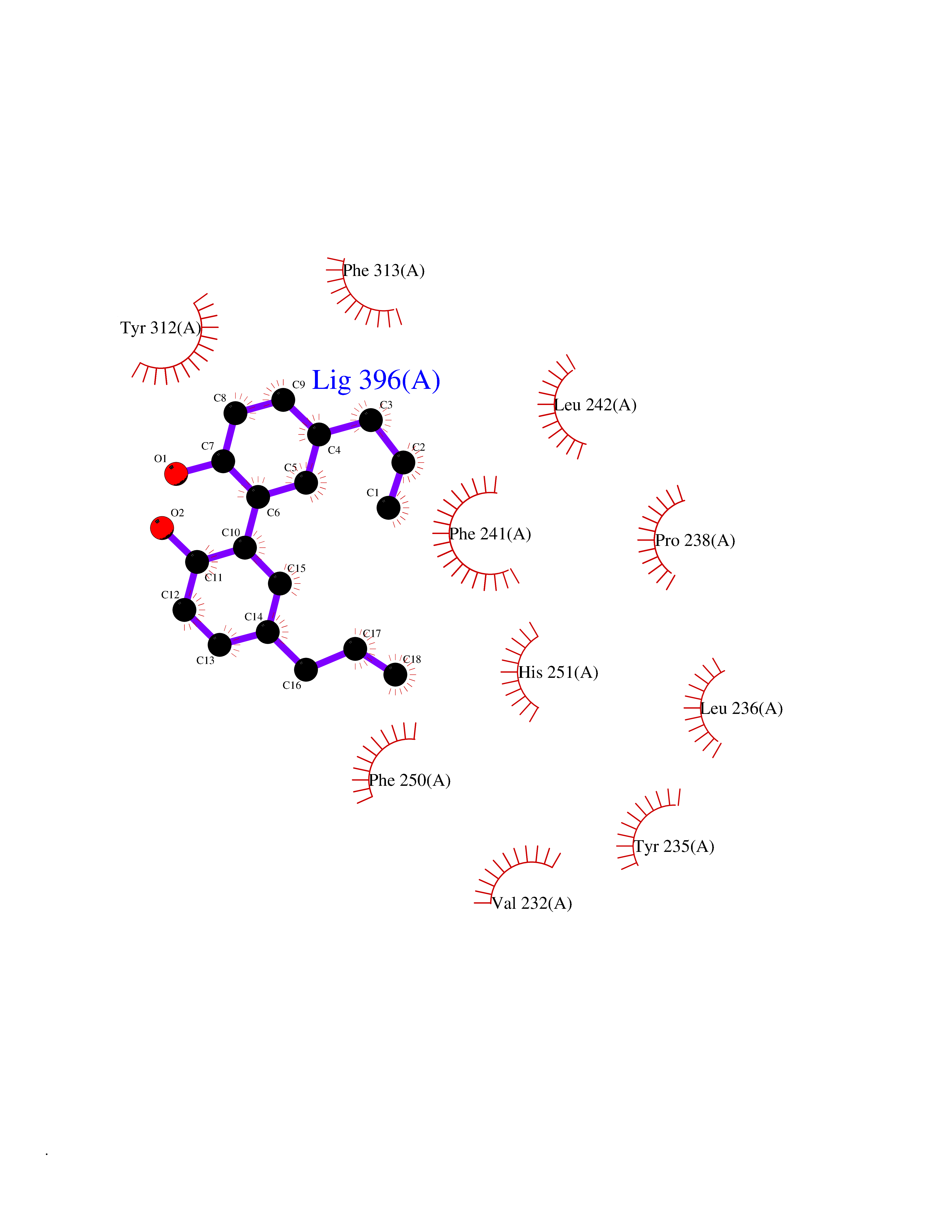 Ligplot Map 3D Binding mode Sequence TVPWFPKKISDLDHCNVYRKRRKYFADLAMNYKHGDPIPKVEFTEEEIKTWGTVFQELNKLYPTHACREYLKNLPLLSKYCGYREDNIPQLEDVSNFLKERTGFSIRPVAGYLSPRDFLSGLAFRVFHCTQYVRHSSDPFYTPEPDTCHELLGHVPLLAEPSFAQFSQEIGLASLGASEEAVQKLATCYFFTVEFGLCKQDGQLRVFGAGLLSSISELKHALSGHAKVKPFDPKITCKQECLITTFQDVYFVSESFEDAKEKMREFTKTIK Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Urea transporter 1 (SLC14A1) | 6QD5 | 7.00 | |
Target general information Gen name SLC14A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Urea transporter, erythrocyte; UTE; UT1; Solute carrier family 14 member 1; RACH1; JK; HUT11 Protein family Urea transporter family Biochemical class Urea transporter family Function Urea channel that facilitates transmembrane urea transport down a concentration gradient. A constriction of the transmembrane channel functions as selectivity filter through which urea is expected to pass in dehydrated form. The rate of urea conduction is increased by hypotonic stress. Plays an important role in the kidney medulla collecting ducts, where it allows rapid equilibration between the lumen of the collecting ducts and the interstitium, and thereby prevents water loss driven by the high concentration of urea in the urine. Facilitates urea transport across erythrocyte membranes. May also play a role in transmembrane water transport, possibly by indirect means. Related diseases Immunodeficiency 12 (IMD12) [MIM:615468]: A primary immunodeficiency characterized by onset in infancy of recurrent bacterial and candidal infections resulting in bronchiectasis and growth delay. Manifestations include mastoiditis, aphthous ulcers, cheilitis, gingivitis, esophagitis, gastritis, duodenitis, and meningitis. Levels of absolute lymphocytes and serum immunoglobulins are normal, but specific antibody titers are low despite immunization, and T-cells show impaired proliferative responses to mitogens. {ECO:0000269|PubMed:23727036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A chromosomal aberration involving MALT1 is recurrent in low-grade mucosa-associated lymphoid tissue (MALT lymphoma). Translocation t(11;18)(q21;q21) with BIRC2. This translocation is found in approximately 50% of cytogenetically abnormal low-grade MALT lymphoma. {ECO:0000269|PubMed:10339464, ECO:0000269|PubMed:10523859, ECO:0000269|PubMed:10702396, ECO:0000269|PubMed:11090634}. Drugs (DrugBank ID) DB01005; DB03904 Interacts with Q8WVV5; Q9Y3D6; Q8WWP7; P30301; Q5QGT7; Q6UX34; P0DN84; Q9C0I4; Q5BJF2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood group antigen; Cell membrane; Glycoprotein; Membrane; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 38862.8 Length 356 Aromaticity 0.12 Instability index 40.98 Isoelectric point 7.67 Charge (pH=7) 0.95 2D Binding mode Binding energy (Kcal/mol) -9.54 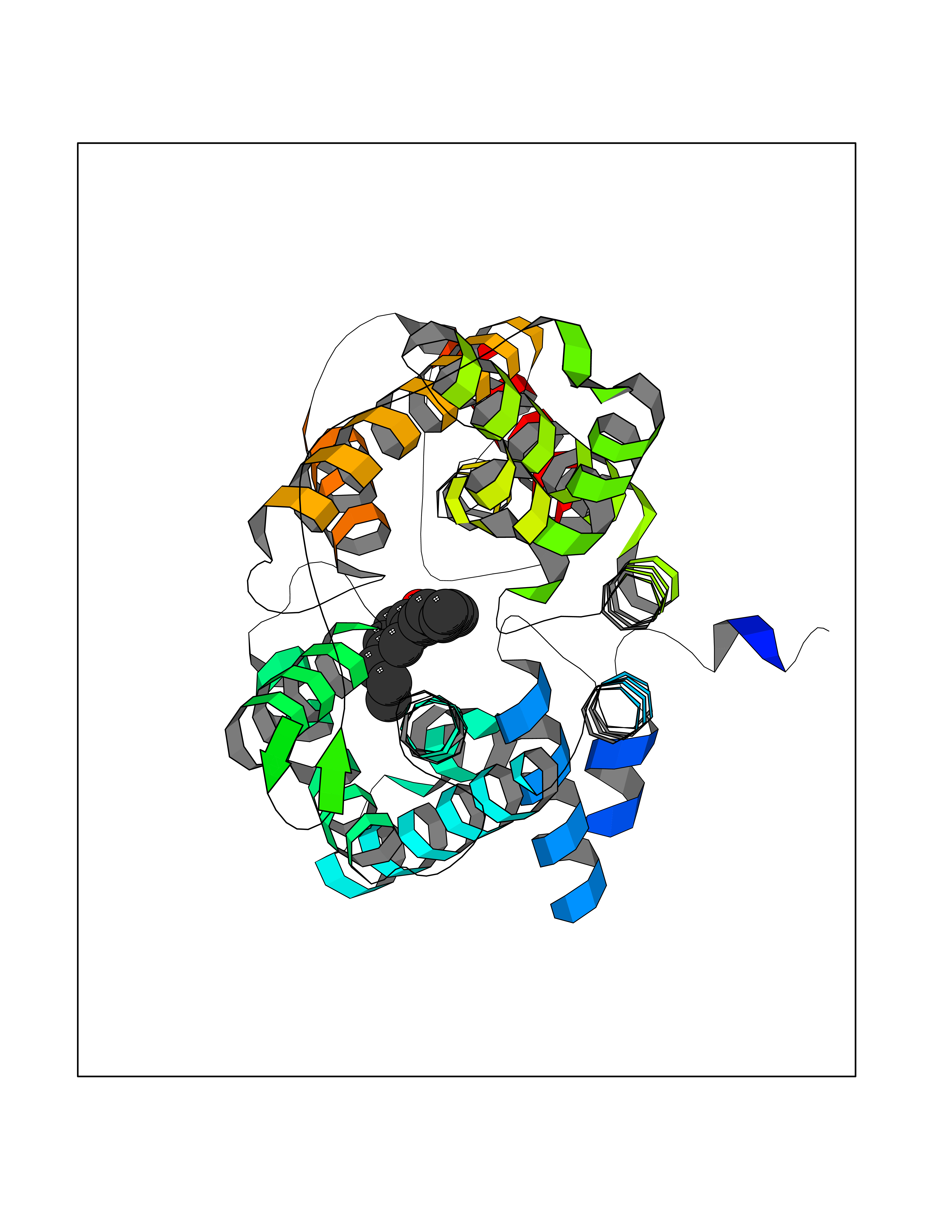 Molscript Map 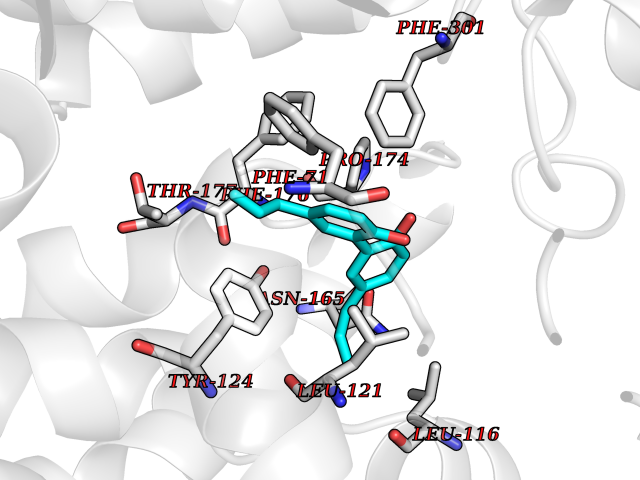 Pymol Map 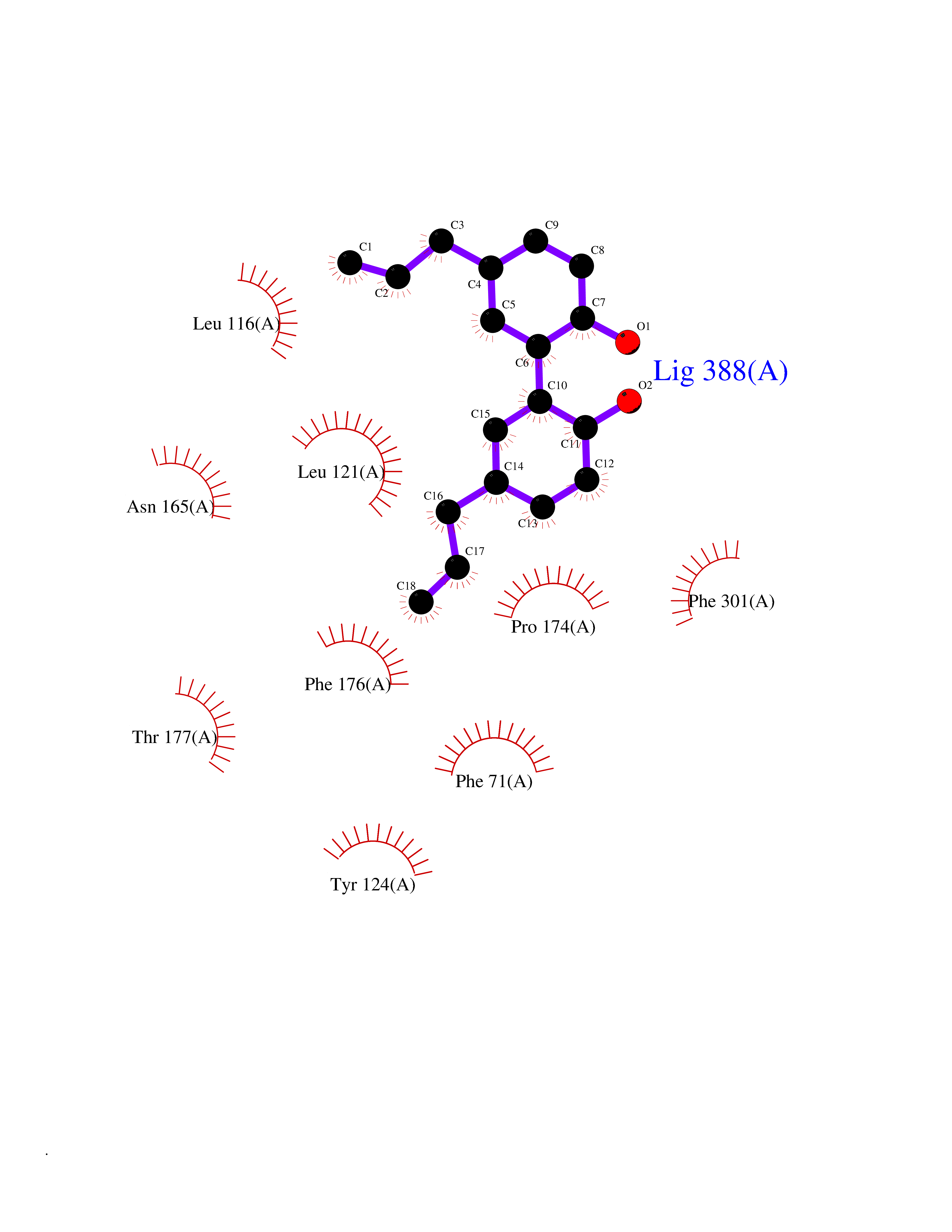 Ligplot Map 3D Binding mode Sequence FPKALGYVTGDMKELANQLKDKPVVLQFIDWILRGISQVVFVNNPVSGILILVGLLVQNPWWALTGWLGTVVSTLMALLLSQDRSLIASGLYGYNATLVGVLMAVFSDKGDYFWWLLLPVCAMSMTCPIFSSALNSVLSKWDLPVFTLPFNMALSMYLSATGHYNPFFPAKLVIPITTAPQISWSDLSALELLKSIPVGVGQIYGCDNPWTGGIFLGAILLSSPLMCLHAAIGSLLGIAAGLSLSAPFENIYFGLWGFNSSLACIAMGGMFMALTWQTHLLALGCALFTAYLGVGMANFMAEVGLPACTWPFCLATLLFLIMTTKNSNIYKMPLSKVTYPEENRIFYLQAKKRMVE Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Cytochrome c | 3ZOO | 6.99 | |
Target general information Gen name CYCS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CYC Protein family Cytochrome c family Biochemical class Oxidoreductase Function Electron transporter, transferring electrons from CoQH2-cytochrome c reductase complex and cytochrome c oxidase complex activity.Heme binding.Metal ion binding. Related diseases Thrombocytopenia 4 (THC4) [MIM:612004]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. {ECO:0000269|PubMed:18345000}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11638; DB03317; DB03366; DB01017; DB02110; DB03977; DB03934; DB04249 Interacts with O14727; P05067; Q6XD76; Q9NSI6-4; Q3SXR2; Q96BR5; Q9UKG9-2; O00303; Q8IZU1; Q3SYB3; P06241; Q8N5Z5; Q6A162; Q1L5Z9; P02750; Q8IYG6; Q6FHY5; A0A0A0MR05; Q9BUL5; Q6ZMI0-5; Q66K80; Q9NTN9-3; P37840; Q13573; Q92797-2; O43829; Q9FKS5 EC number NA Uniprot keywords 3D-structure; Acetylation; Apoptosis; Direct protein sequencing; Disease variant; Electron transport; Heme; Iron; Metal-binding; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Respiratory chain; Transport Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 11601.4 Length 104 Aromaticity 0.09 Instability index 12.21 Isoelectric point 9.61 Charge (pH=7) 9.01 2D Binding mode Binding energy (Kcal/mol) -9.53  Molscript Map 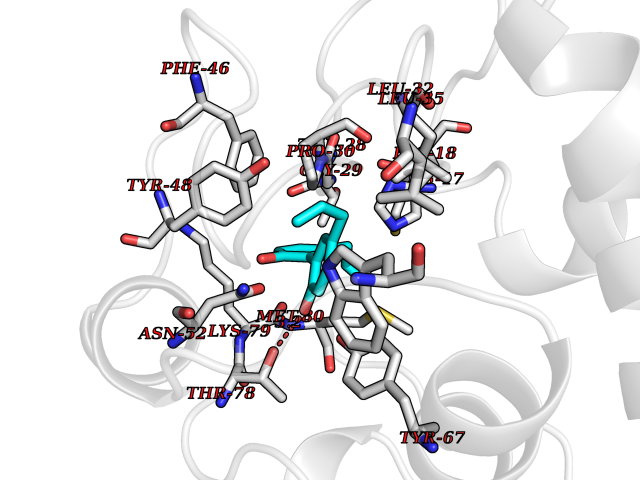 Pymol Map 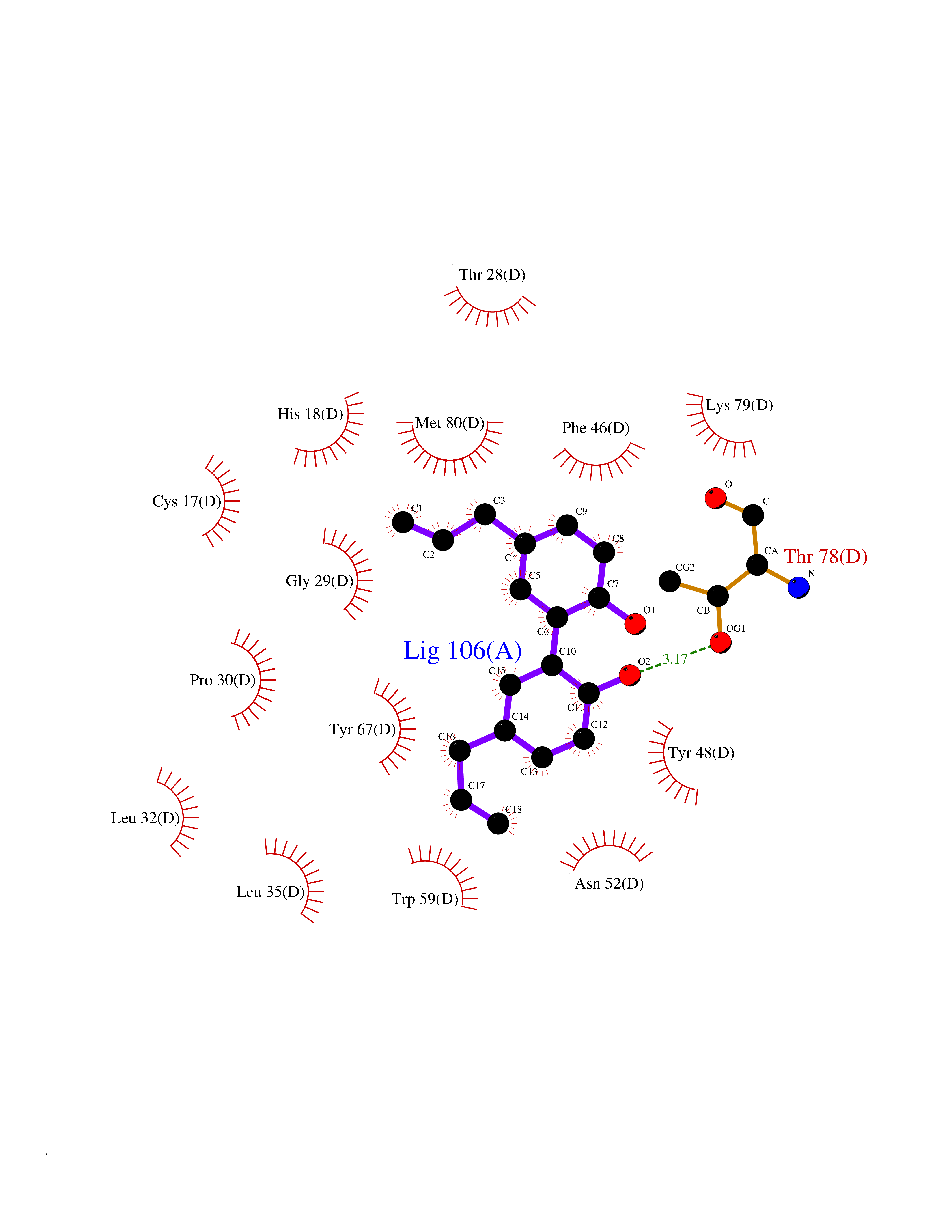 Ligplot Map 3D Binding mode Sequence GDVEKGKKIFIMKCSQCHTVEKGGKHKTGPNLHGLFGRKTGQAPGFSYTAANKNKGIIWGEDTLMEYLENPKKYIPGTKMIFVGIKKKEERADLIAYLKKATNE Hydrogen bonds contact Hydrophobic contact | ||||