Job Results:
Ligand
Structure
Job ID
5a9d2b2db14ecd17b747d14262c01470
Job name
NA
Time
2025-04-03 16:12:50
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | S-adenosylmethionine decarboxylase proenzyme (AMD1) | 1JL0 | 5.43 | |
Target general information Gen name AMD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SamDC; S-adenosylmethioninedecarboxylase; AdoMetDC; AMD Protein family Eukaryotic AdoMetDC family Biochemical class Carbon-carbon lyase Function Promotes maintenance and self-renewal of embryonic stem cells, by maintaining spermine levels. Essential for biosynthesis of the polyamines spermidine and spermine. Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08163; DB00118; DB01917 Interacts with P17707; Q96A98; Q8WY91 EC number EC 4.1.1.50 Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Decarboxylase; Direct protein sequencing; Lyase; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Pyruvate; Reference proteome; S-adenosyl-L-methionine; Schiff base; Spermidine biosynthesis; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 35790.5 Length 311 Aromaticity 0.14 Instability index 39.47 Isoelectric point 6.03 Charge (pH=7) -2.01 2D Binding mode Binding energy (Kcal/mol) -7.41  Molscript Map 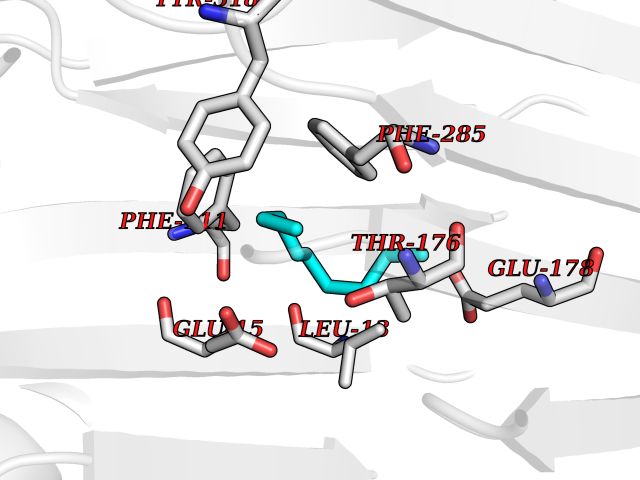 Pymol Map 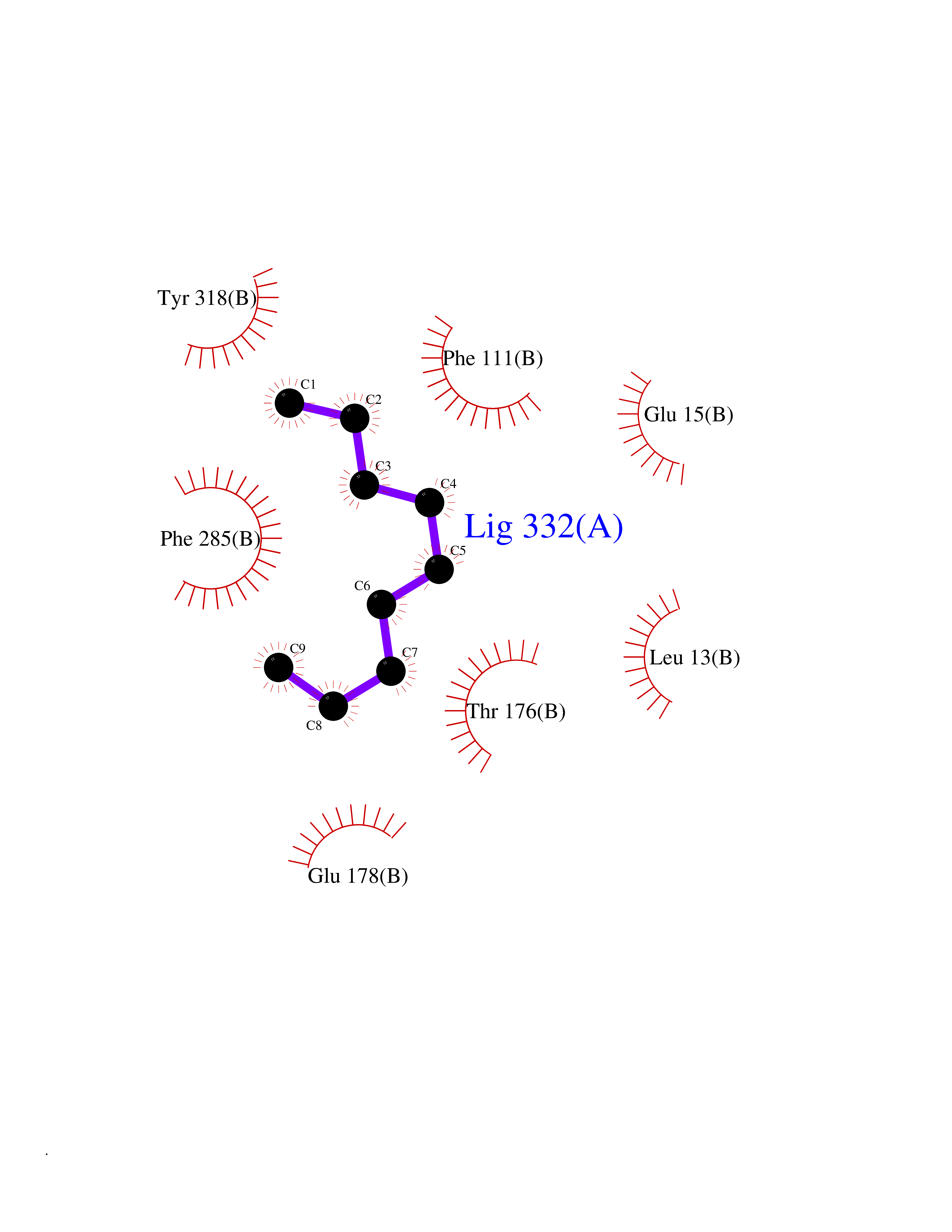 Ligplot Map 3D Binding mode Sequence HFFEGTEKLLEVWFSRQGSGDLRTIPRSEWDILLKDVQCSIISVTKTDKQEAYVLSESSMFVSKRRFILKTCGTTLLLKALVPLLKLARDYSGFDSIQSFFYSRKNFMKPSHQGYPHRNFQEEIEFLNAIFPNGAGYCMGRMNSDCWYLYTLDFRVISQPDQTLEILMSELDPAVMDQFYMKDGVTAKDVTRESGIRDLIPGSVIDATMFNPCGYSMNGMKSDGTYWTIAITPEPEFSYVSFETNLSQTSYDDLIRKVVEVFKPGKFVTTLFVNQSSKCPQKIEGFKRLDCQSAMFNDYNFVFTSFAKKQQ Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Lecithin-cholesterol acyltransferase (LCAT) | 6MVD | 5.43 | |
Target general information Gen name LCAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phospholipidcholesterolacyltransferase; Phospholipid-cholesterol acyltransferase; Phosphatidylcholinesterol acyltransferase; Phosphatidylcholine-sterol acyltransferase Protein family AB hydrolase superfamily, Lipase family Biochemical class Acyltransferase Function Synthesized mainly in the liver and secreted into plasma where it converts cholesterol and phosphatidylcholines (lecithins) to cholesteryl esters and lysophosphatidylcholines on the surface of high and low density lipoproteins (HDLs and LDLs). The cholesterol ester is then transported back to the liver. Has a preference for plasma 16:0-18:2 or 18:O-18:2 phosphatidylcholines. Also produced in the brain by primary astrocytes, and esterifies free cholesterol on nascent APOE-containing lipoproteins secreted from glia and influences cerebral spinal fluid (CSF) APOE- and APOA1 levels. Together with APOE and the cholesterol transporter ABCA1, plays a key role in the maturation of glial-derived, nascent lipoproteins. Required for remodeling high-density lipoprotein particles into their spherical forms. Central enzyme in the extracellular metabolism of plasma lipoproteins. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P02647; O76024 EC number EC 2.3.1.43 Uniprot keywords 3D-structure; Acyltransferase; Cholesterol metabolism; Corneal dystrophy; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipid metabolism; Proteomics identification; Reference proteome; Secreted; Signal; Steroid metabolism; Sterol metabolism; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42715.4 Length 376 Aromaticity 0.12 Instability index 42.05 Isoelectric point 5.69 Charge (pH=7) -9.12 2D Binding mode Binding energy (Kcal/mol) -7.41  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence HTRPVILVPGCLGNQLEAKLDKPDVVNWMCYRKTEDFFTIWLDLNMFLPLGVDCWIDNTRVVYNRSSGLVSNAPGVQIRVPGFGKTYSVEYLDSSKLAGYLHTLVQNLVNNGYVRDETVRAAPYDWRLEPGQQEEYYRKLAGLVEEMHAAYGKPVFLIGHSLGCLHLLYFLLRQPQAWKDRFIDGFISLGAPWGGSIKPMLVLASGDNQGIPIMSSIKEEQRITTTSPWMFPSRMAWPEDHVFISTPSFNYTGRDFQRFFADLHFEEGWYMWLQSRDLLAGLPAPGVEVYCLYGVGLPTPRTYIYDHGFPYTDPVGVLYEDGDDTVATRSTELCGLWQGRQPQPVHLLPLHGIQHLNMVFSNLTLEHINAILLGAH Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Protein-tyrosine phosphatase SHP-2 (PTPN11) | 2SHP | 5.43 | |
Target general information Gen name PTPN11 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein phosphatase non-receptor type 11; SHPTP2; SHP2; SHP-2; SH-PTP3; SH-PTP2; Protein-tyrosine phosphatase SHP2; Protein-tyrosine phosphatase 2C; Protein-tyrosine phosphatase 1D; PTP2C; PT Protein family Protein-tyrosine phosphatase family, Non-receptor class 2 subfamily Biochemical class Phosphoric monoester hydrolase Function Positively regulates MAPK signal transduction pathway. Dephosphorylates GAB1, ARHGAP35 and EGFR. Dephosphorylates ROCK2 at 'Tyr-722' resulting in stimulatation of its RhoA binding activity. Dephosphorylates CDC73. Acts downstream of various receptor and cytoplasmic protein tyrosine kinases to participate in the signal transduction from the cell surface to the nucleus. Related diseases LEOPARD syndrome 1 (LPRD1) [MIM:151100]: A disorder characterized by lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonic stenosis, abnormalities of genitalia, retardation of growth, and sensorineural deafness. {ECO:0000269|PubMed:12058348, ECO:0000269|PubMed:14961557, ECO:0000269|PubMed:15121796, ECO:0000269|PubMed:15389709, ECO:0000269|PubMed:15520399, ECO:0000269|PubMed:15690106, ECO:0000269|PubMed:16679933, ECO:0000269|PubMed:16733669, ECO:0000269|PubMed:24891296, ECO:0000269|PubMed:26742426}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 1 (NS1) [MIM:163950]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. Some patients with NS1 develop multiple giant cell lesions of the jaw or other bony or soft tissues, which are classified as pigmented villonodular synovitis (PVNS) when occurring in the jaw or joints. {ECO:0000269|PubMed:11704759, ECO:0000269|PubMed:11992261, ECO:0000269|PubMed:12161469, ECO:0000269|PubMed:12325025, ECO:0000269|PubMed:12529711, ECO:0000269|PubMed:12634870, ECO:0000269|PubMed:12717436, ECO:0000269|PubMed:12739139, ECO:0000269|PubMed:12960218, ECO:0000269|PubMed:15384080, ECO:0000269|PubMed:15889278, ECO:0000269|PubMed:15948193, ECO:0000269|PubMed:19020799, ECO:0000269|PubMed:24891296, ECO:0000269|PubMed:28074573}. The disease is caused by variants affecting the gene represented in this entry. Mutations in PTPN11 account for more than 50% of the cases.; DISEASE: Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:12717436, ECO:0000269|PubMed:26742426}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metachondromatosis (MC) [MIM:156250]: A skeletal disorder with radiologic features of both multiple exostoses and Ollier disease, characterized by the presence of exostoses, commonly of the bones of the hands and feet, and enchondromas of the metaphyses of long bones and iliac crest. {ECO:0000269|PubMed:20577567}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02779 Interacts with P10275; P32239; Q9BZW8; P20138; Q08345; P00533; P29317; P04626; Q8WU20; Q13480; Q9UQC2; P62993; P08069; P06213; P35568; P43628; P10721; P08581; O95297; Q15116; P09619; P16284; P49023; P49247; Q13049; P68105; Q71V39; P35570; P97710; Q6P1J9; Q13480; O75496; Q9UKI8 EC number EC 3.1.3.48 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Deafness; Disease variant; Hydrolase; Nucleus; Phosphoprotein; Protein phosphatase; Proteomics identification; Reference proteome; Repeat; SH2 domain Protein physicochemical properties Chain ID A Molecular weight (Da) 56341.2 Length 491 Aromaticity 0.09 Instability index 41.37 Isoelectric point 7.76 Charge (pH=7) 2.43 2D Binding mode Binding energy (Kcal/mol) -7.41  Molscript Map 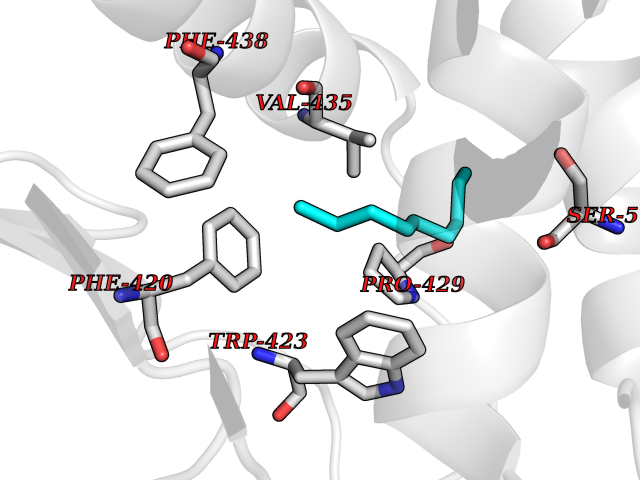 Pymol Map 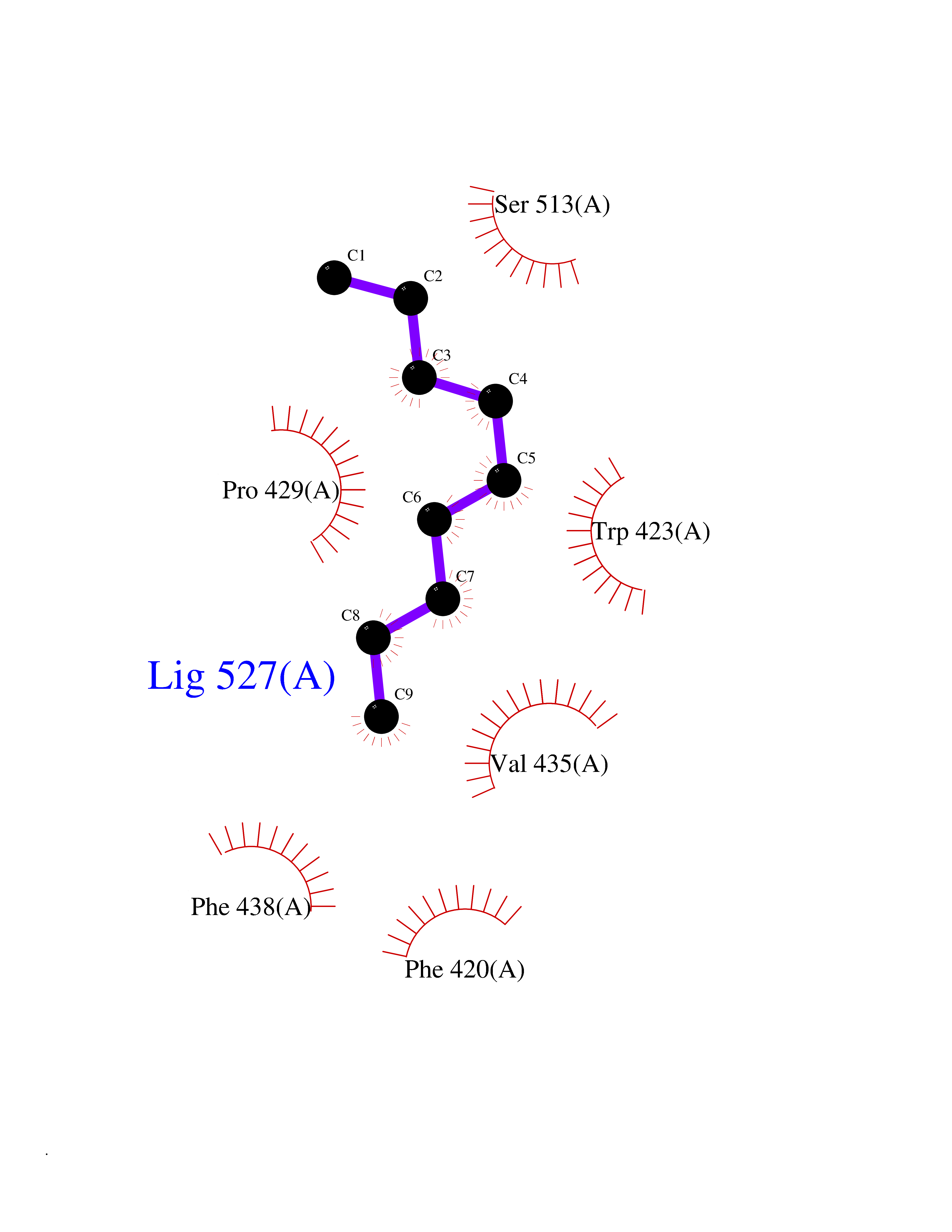 Ligplot Map 3D Binding mode Sequence KSRRWFHPNITGVEAENLLLTRGVDGSFLARPSKSNPGDLTLSVRRNGAVTHIKIQNTGDYYDLYGGEKFATLAELVQYYMEHHGQLKEKNGDVIELKYPLNCADPTSERWFHGHLSGKEAEKLLTEKGKHGSFLVRESQSHPGDFVLSVRTGDNDGKSKVTHVMIRCQELKYDVGGGERFDSLTDLVEHYKKNPMVETLGTVLQLKQPLNTTRINAAEIESRVRELSKGFWEEFETLQQQECKLLYSRKEGQRQENKNKNRYKNILPFDHTRVVLHDSDYINANIIMPKKSYIATQGCLQNTVNDFWRMVFQENSRVIVMTTKEVERGKSKCVKYWPDEYALKEYGVMRVRNVKESAAHDYTLRELKLSKVGQGNTERTVWQYHFRTWPDHGVPSDPGGVLDFLEEVHHKQESIMDAGPVVVHCSAGIGRTGTFIVIDILIDIIREKGVDCDIDVPKTIQMVRSQRSGMVQTEAQYRSIYMAVQHYIETL Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Tumor-associated calcium signal transducer 1 (EPCAM) | 4MZV | 5.43 | |
Target general information Gen name EPCAM Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hEGP314; TROP1; TACSTD1; Major gastrointestinal tumor-associated protein GA733-2; MIC18; M4S1; M1S2; KSA; KS 1/4 antigen; Gastrointestinal carcinoma antigen GA733; GA733-2; Epithelial glycoprotein 314 Protein family EPCAM family Biochemical class NA Function Plays a role in embryonic stem cells proliferation and differentiation. Up-regulates the expression of FABP5, MYC and cyclins A and E. May act as a physical homophilic interaction molecule between intestinal epithelial cells (IECs) and intraepithelial lymphocytes (IELs) at the mucosal epithelium for providing immunological barrier as a first line of defense against mucosal infection. Related diseases Diarrhea 5, with tufting enteropathy, congenital (DIAR5) [MIM:613217]: An intractable diarrhea of infancy characterized by villous atrophy and absence of inflammation, with intestinal epithelial cell dysplasia manifesting as focal epithelial tufts in the duodenum and jejunum. {ECO:0000269|PubMed:18572020, ECO:0000269|PubMed:24142340}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Lynch syndrome 8 (LYNCH8) [MIM:613244]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:19098912}. The disease is caused by variants affecting the gene represented in this entry. LYNCH8 results from heterozygous deletion of 3-prime exons of EPCAM and intergenic regions directly upstream of MSH2, resulting in transcriptional read-through and epigenetic silencing of MSH2 in tissues expressing EPCAM. Drugs (DrugBank ID) DB06607; DB11075; DB05831; DB05319; DB09336 Interacts with P27797; P12830; Q15078; P36957; Q8TDX7 EC number NA Uniprot keywords 3D-structure; Cell junction; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hereditary nonpolyposis colorectal cancer; Membrane; Proteomics identification; Reference proteome; Repeat; Signal; Tight junction; Transmembrane; Transmembrane helix; Tumor antigen Protein physicochemical properties Chain ID A Molecular weight (Da) 27439.8 Length 243 Aromaticity 0.07 Instability index 33.77 Isoelectric point 6.03 Charge (pH=7) -1.99 2D Binding mode Binding energy (Kcal/mol) -7.41 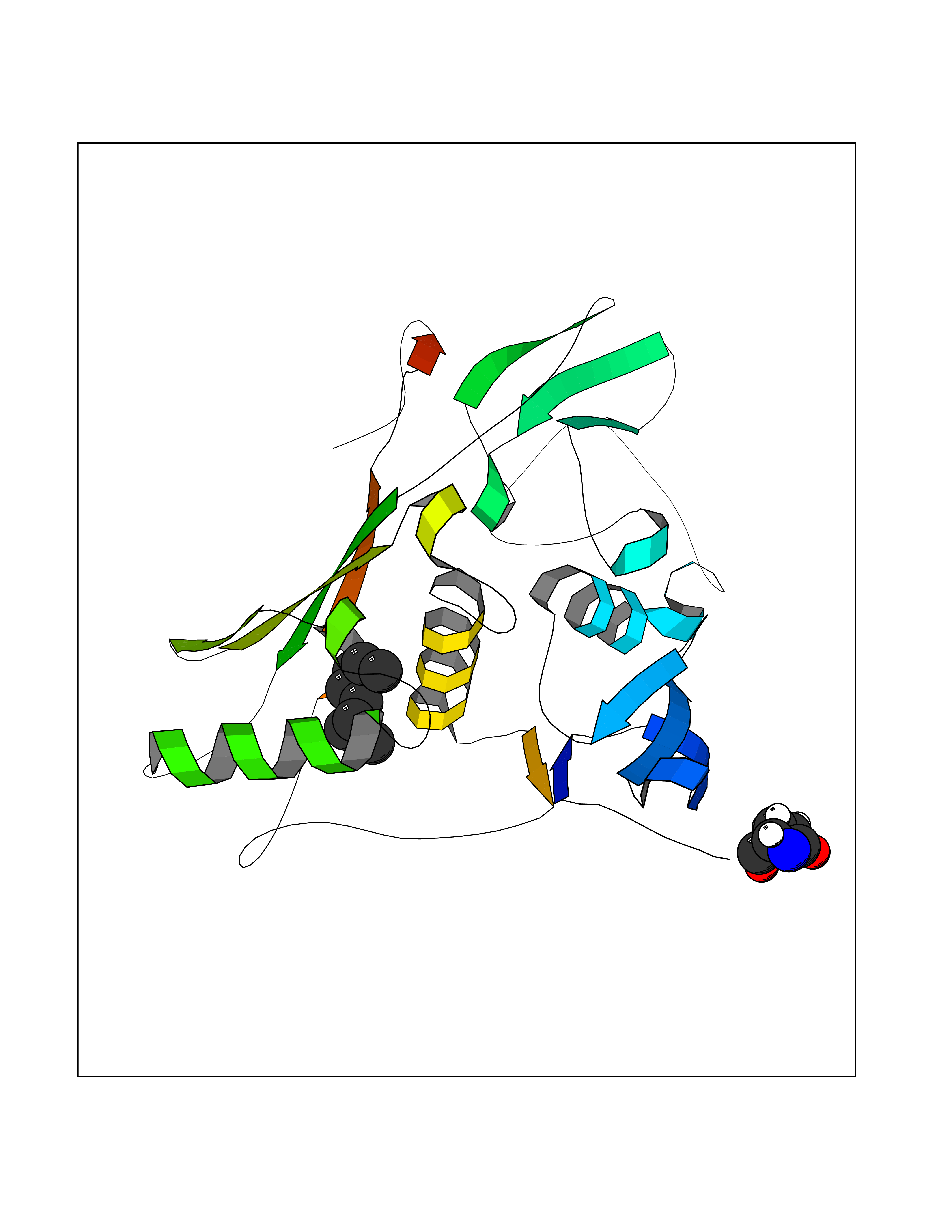 Molscript Map 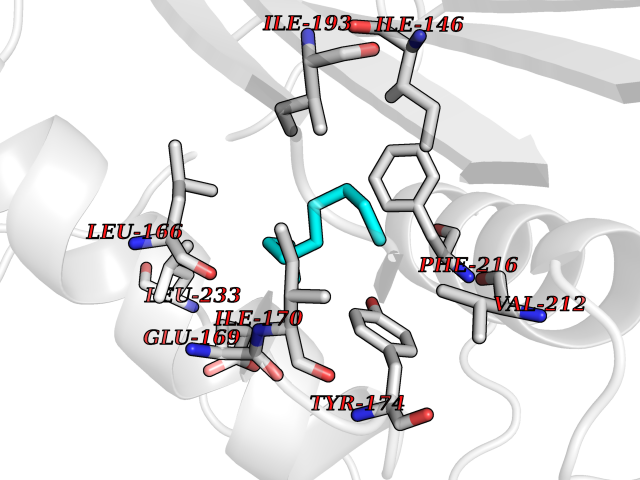 Pymol Map 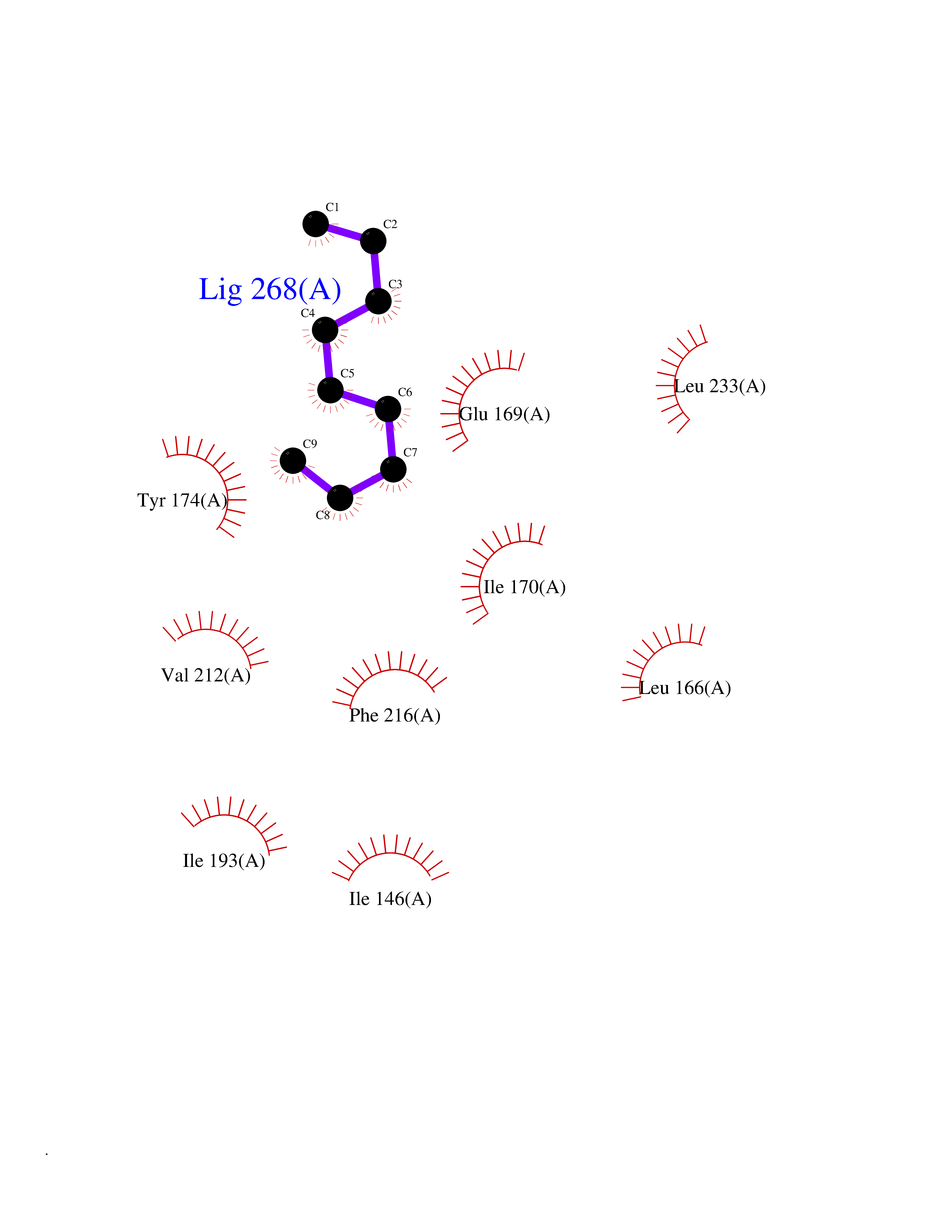 Ligplot Map 3D Binding mode Sequence XEECVCENYKLAVNCFVNNNRQCQCTSVGAQNTVICSKLAAKCLVMKAEMQGSKLGRRAKPEGALQNNDGLYDPDCDESGLFKAKQCQGTSTCWCVNTAGVRRTDKDTEITCSERVRTYWIIIELKHKAREKPYDSKSLRTALQKEITTRYQLDPKFITSILYENNVITIDLVQQSSQKTQNDVDIADVAYYFEKDVKGESLFHSKKMDLTVNGEQLDLDPGQTLIYYVDEKAPEFSMQGLKH Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Caspase-7 (CASP7) | 1SHJ | 5.43 | |
Target general information Gen name CASP7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH3; ICE-like apoptotic protease 3; ICE-LAP3; CMH-1; CASP-7; Apoptotic protease Mch-3 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves and activates sterol regulatory element binding proteins (SREBPs). Proteolytically cleaves poly(ADP-ribose) polymerase (PARP) at a '216-Asp-|-Gly-217' bond. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Pregnancy loss, recurrent, 3 (RPRGL3) [MIM:614391]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:17339269}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05408; DB03384; DB06255 Interacts with Q13490; P83105; P42858; Q8N4N3-2; P43364; Q16236; Q9GZT8; Q13177; P27986-2; P21673; Q86WV1-2; P17405; P98170 EC number EC 3.4.22.60 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Apoptosis; Cytoplasm; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; RNA-binding; Secreted; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 47441.5 Length 417 Aromaticity 0.11 Instability index 20.98 Isoelectric point 8.38 Charge (pH=7) 6.12 2D Binding mode Binding energy (Kcal/mol) -7.4 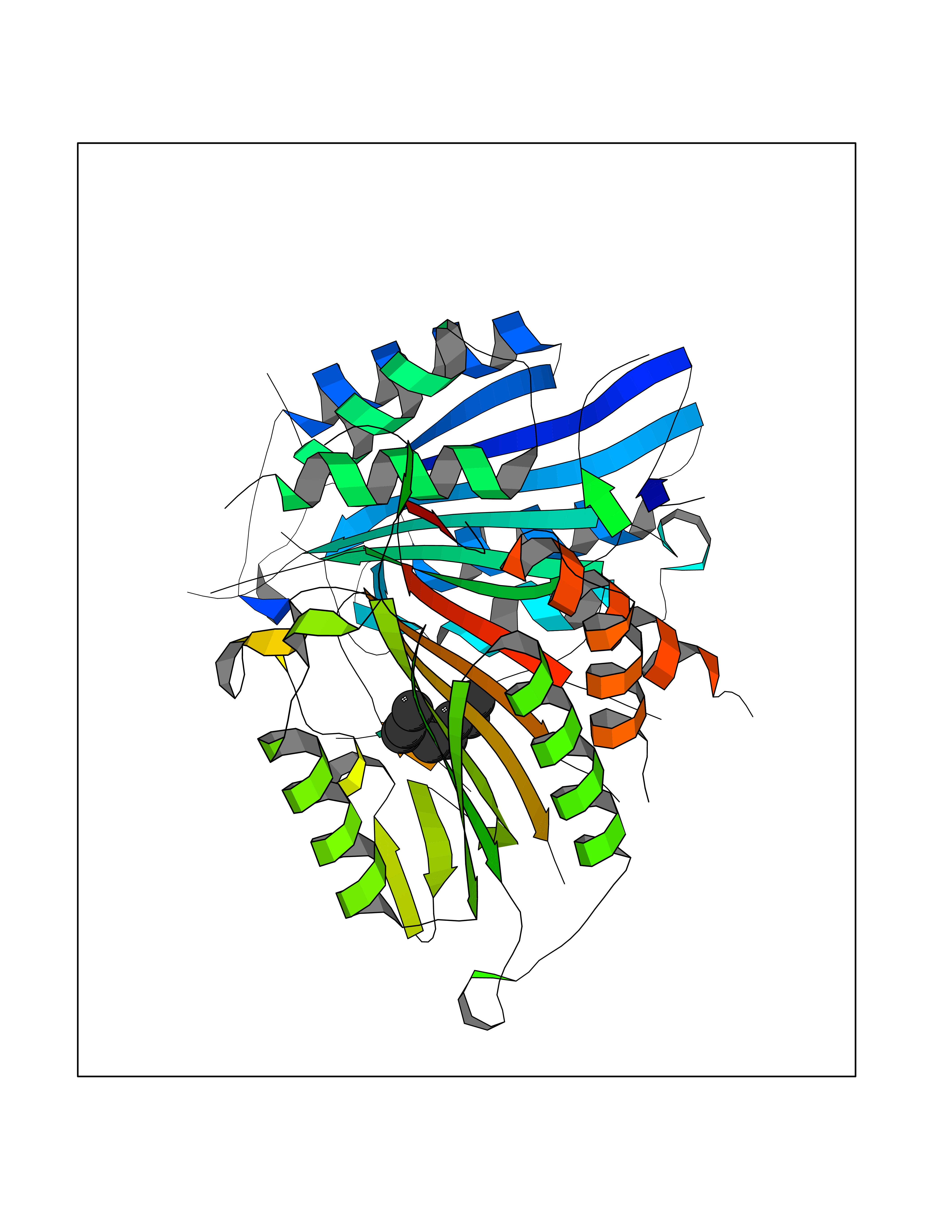 Molscript Map 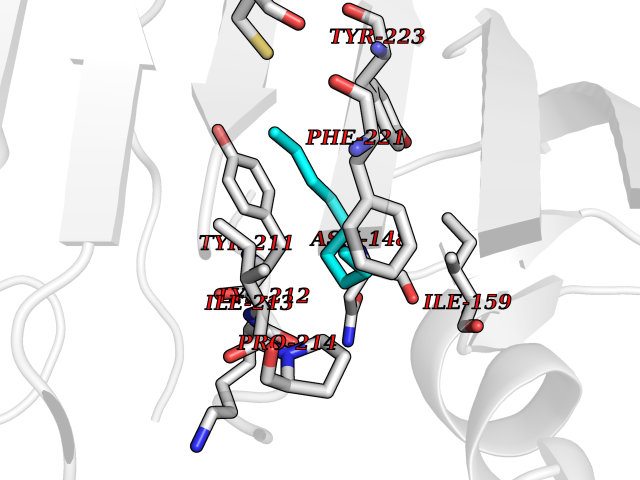 Pymol Map 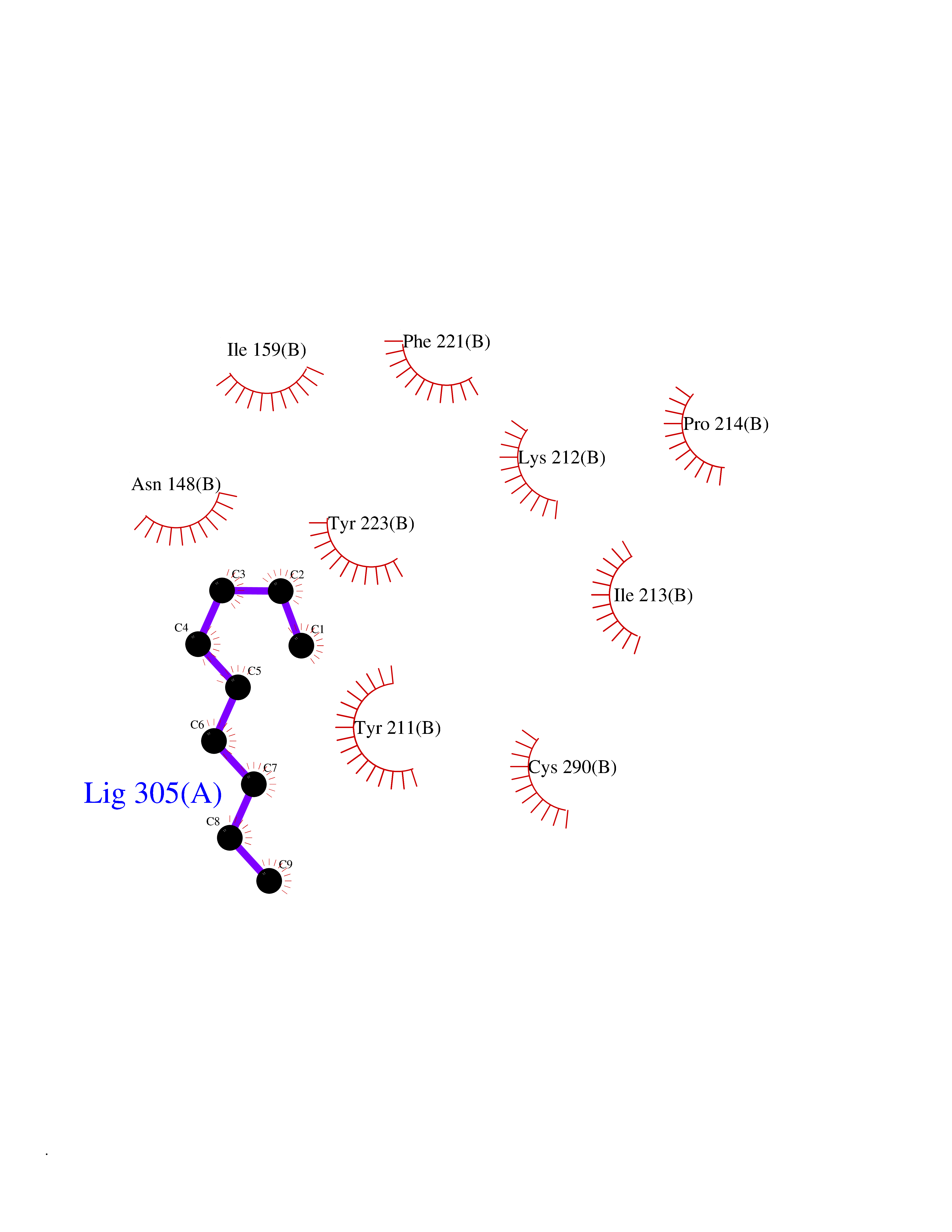 Ligplot Map 3D Binding mode Sequence TYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGTEPRYKIPVEADFLFAYSTVRGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFKKQIPCVVSMLTKELYFSQVPTYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGPRYKIPVEADFLFAYSTVPGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFESKQIPCVVSMLTKELYFSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 5.42 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -7.39 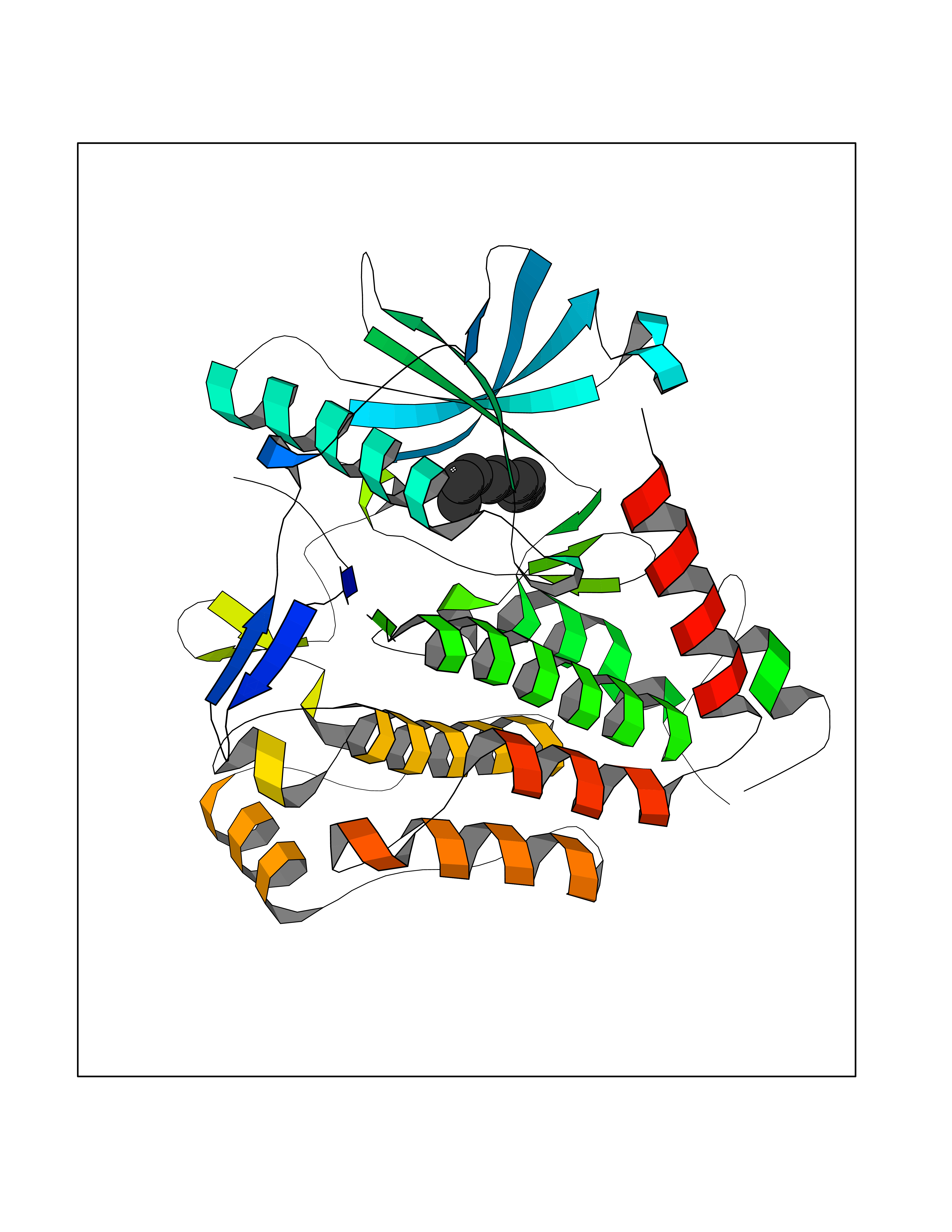 Molscript Map 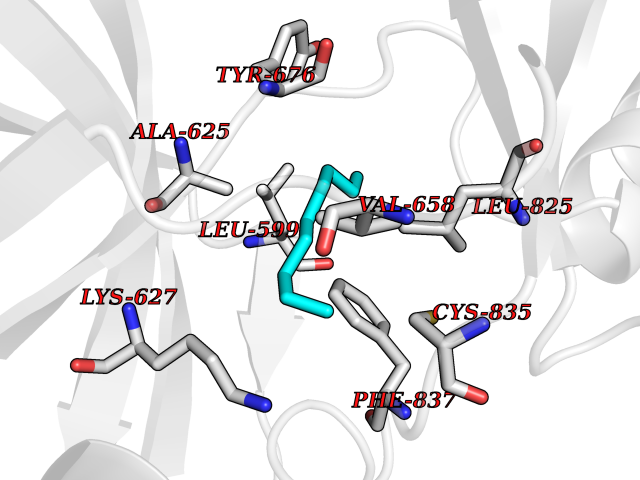 Pymol Map 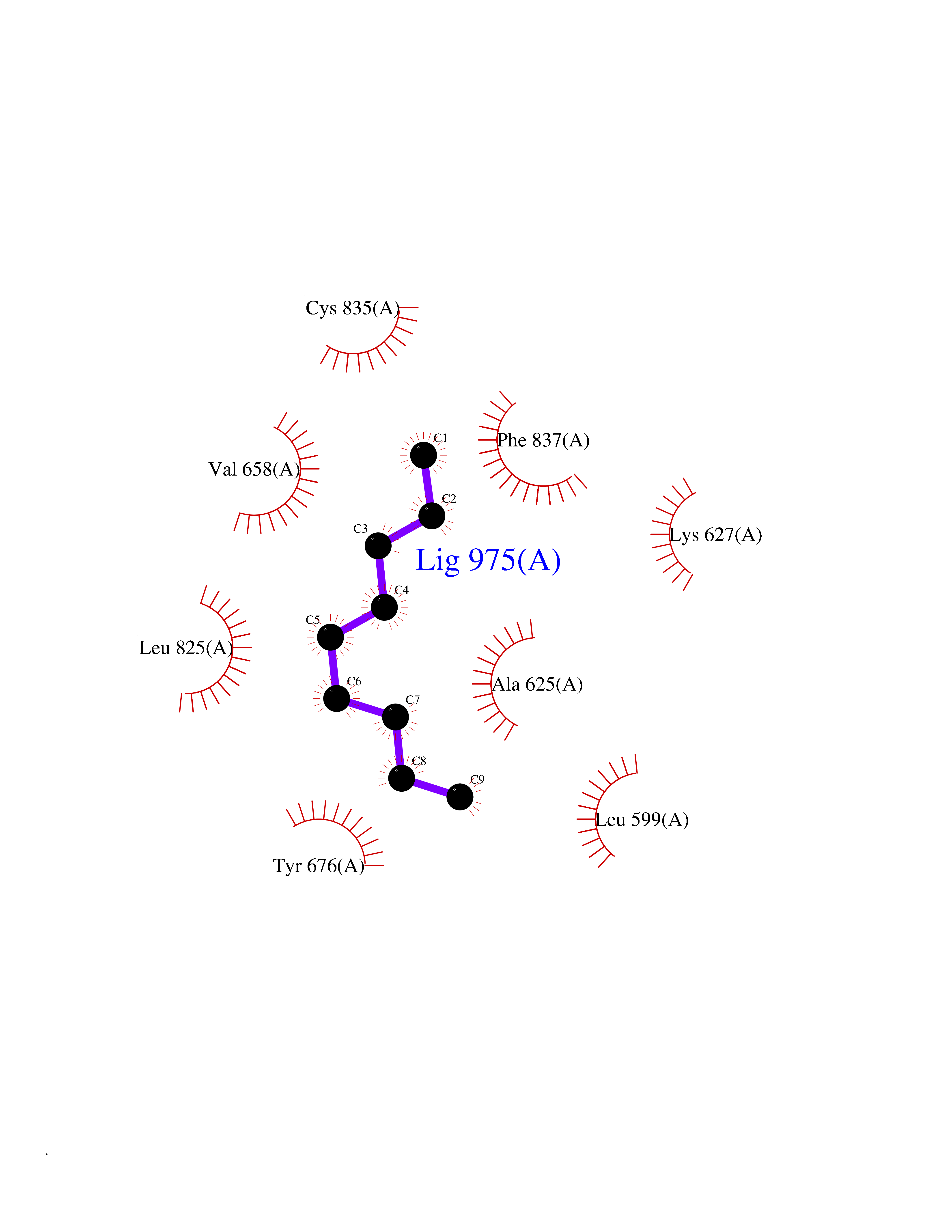 Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Cytochrome P450 1A2 | 2HI4 | 5.42 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -7.39 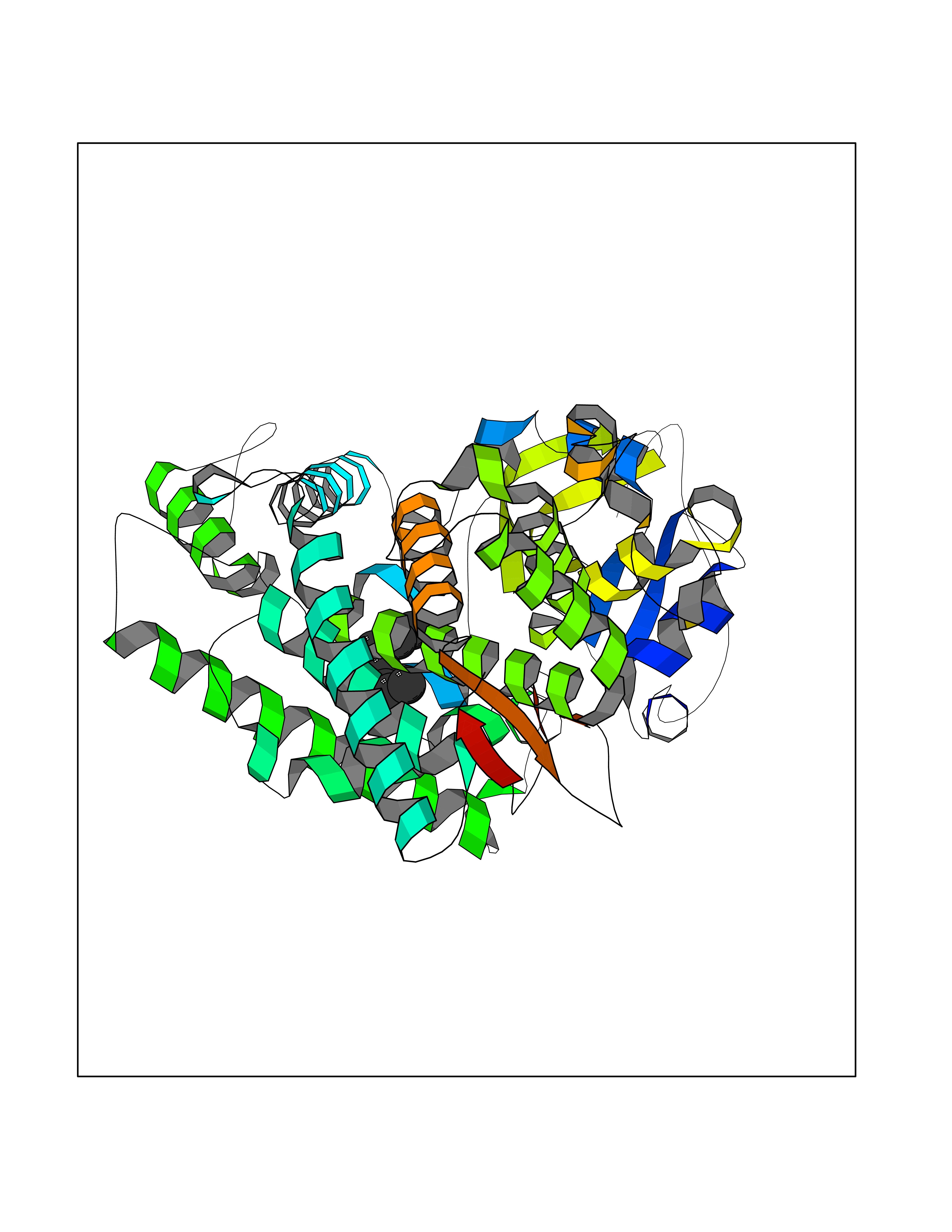 Molscript Map 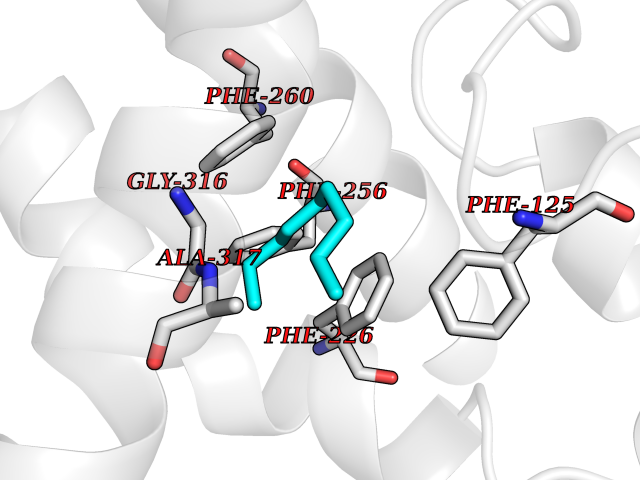 Pymol Map 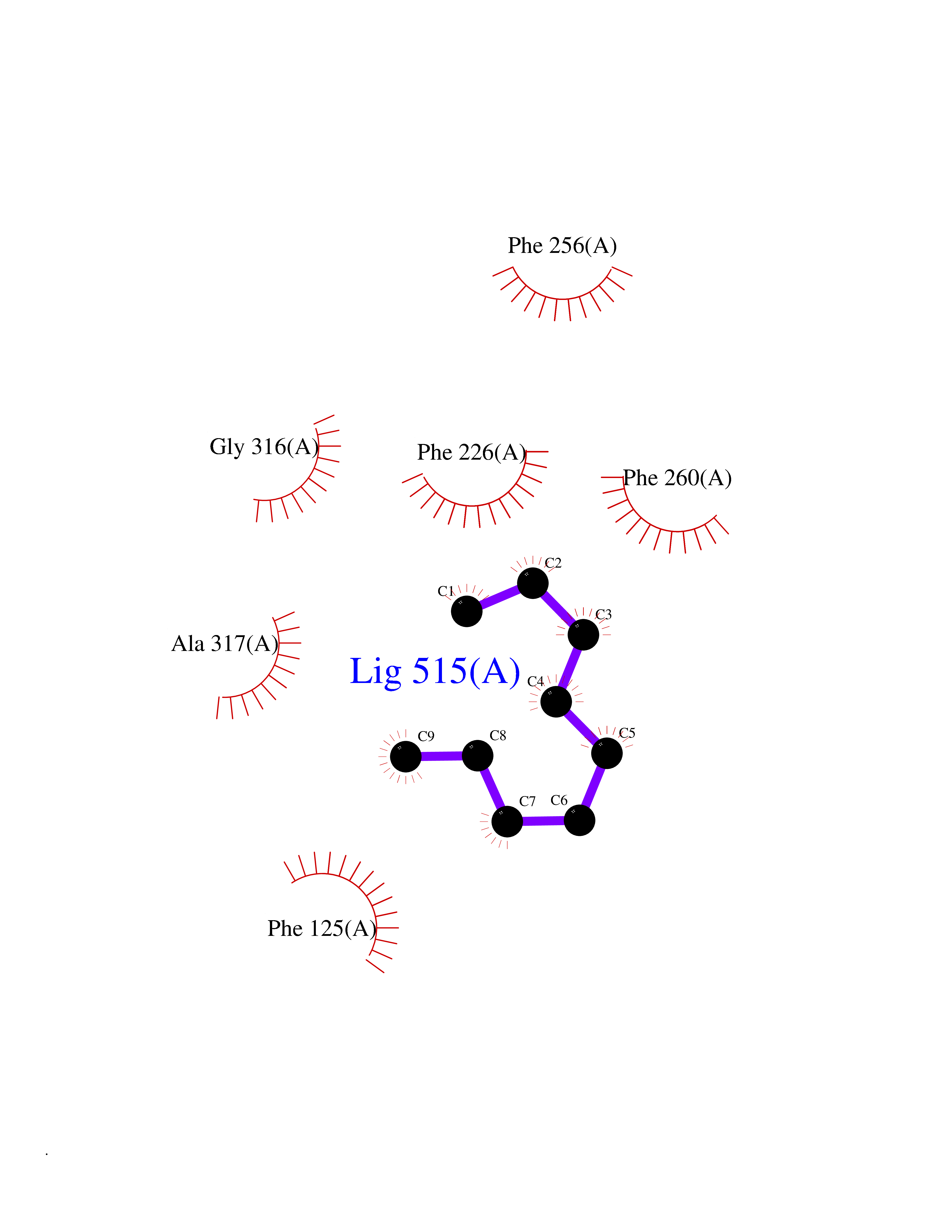 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 5.42 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -7.4 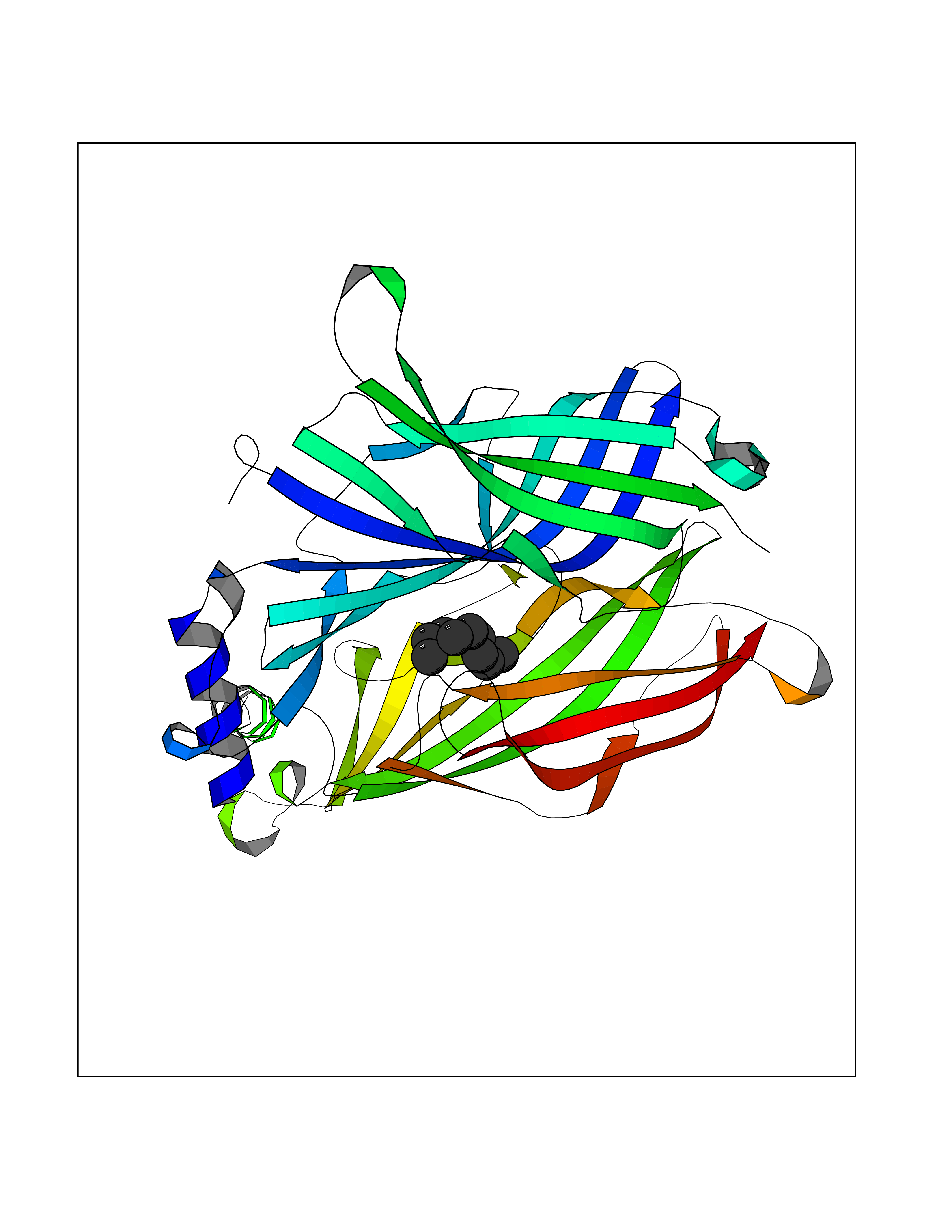 Molscript Map 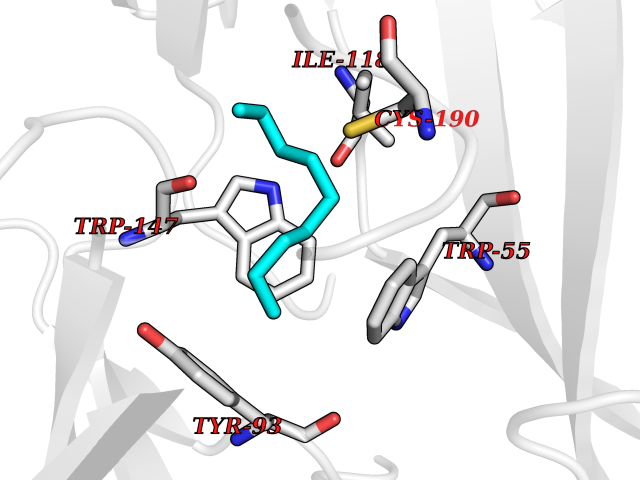 Pymol Map 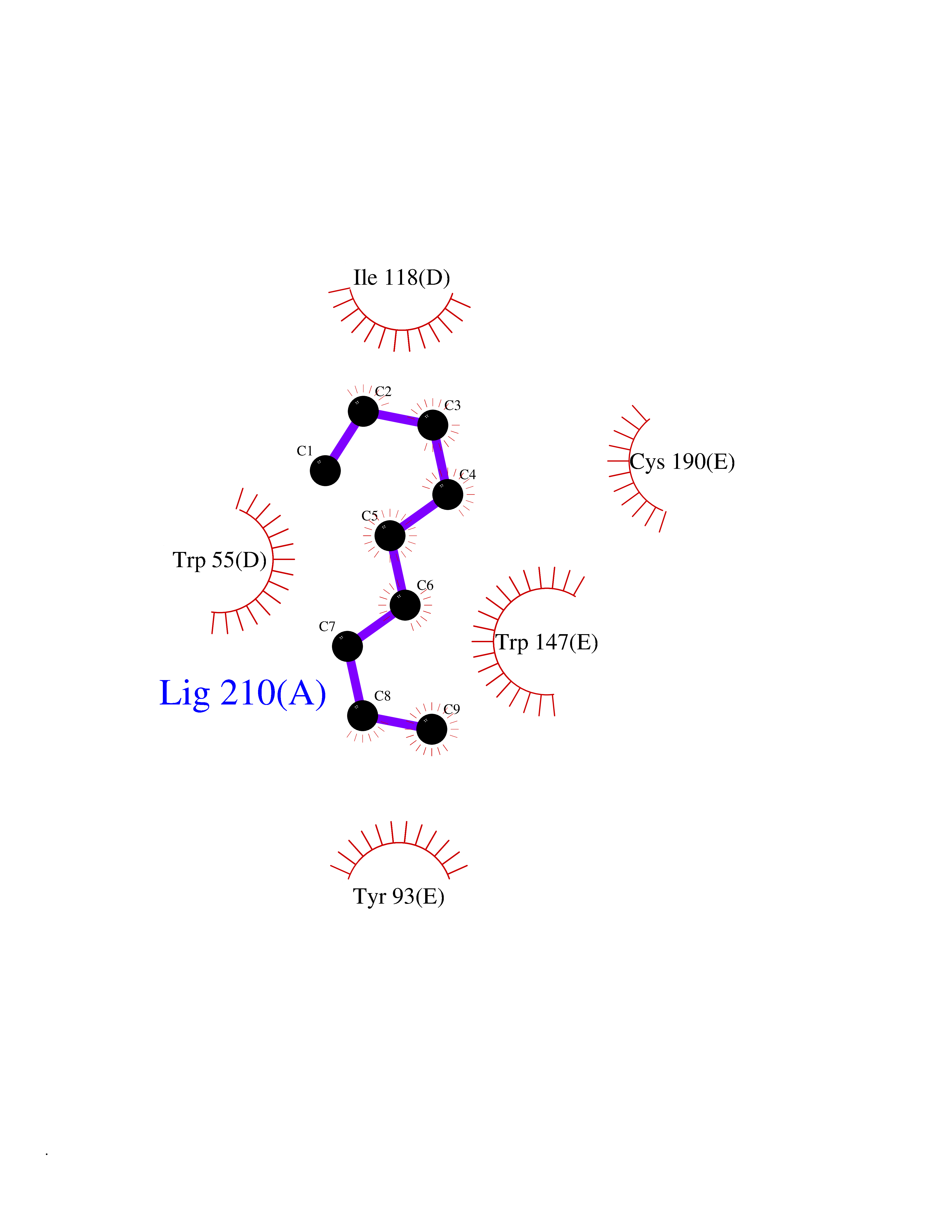 Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Serine hydroxymethyltransferase, mitochondrial | 3OU5 | 5.42 | |
Target general information Gen name SHMT2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SHMT family Biochemical class Transferase Function Amino acid binding.Chromatin binding.Glycine hydroxymethyltransferase activity.Identical protein binding.L-allo-threonine aldolase activity.Pyridoxal phosphate binding. Related diseases Neurodevelopmental disorder with cardiomyopathy, spasticity, and brain abnormalities (NEDCASB) [MIM:619121]: An autosomal recessive neurodevelopmental disorder characterized by global developmental delay, moderate to severe intellectual disability, spastic paraparesis, ataxia, and/or peripheral neuropathy. Patients also exhibit dysmorphic features and congenital microcephaly. Most affected individuals develop progressive hypertrophic cardiomyopathy in childhood or have cardiac developmental anomalies. Brain imaging shows corpus callosum abnormalities in all patients, and perisylvian polymicrogyria-like pattern in some individuals. {ECO:0000269|PubMed:33015733}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11638; DB00145; DB00114; DB00116 Interacts with Q15041; P46736; Q9Y376; Q96DZ9; Q96DZ9-2; Q8IZU9; Q969L2; P34897 EC number 2.1.2.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cardiomyopathy; Cytoplasm; Disease variant; Intellectual disability; Membrane; Mitochondrion; Mitochondrion inner membrane; Mitochondrion nucleoid; Nucleus; One-carbon metabolism; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 46797.9 Length 422 Aromaticity 0.07 Instability index 38.89 Isoelectric point 7.24 Charge (pH=7) 0.55 2D Binding mode Binding energy (Kcal/mol) -7.4 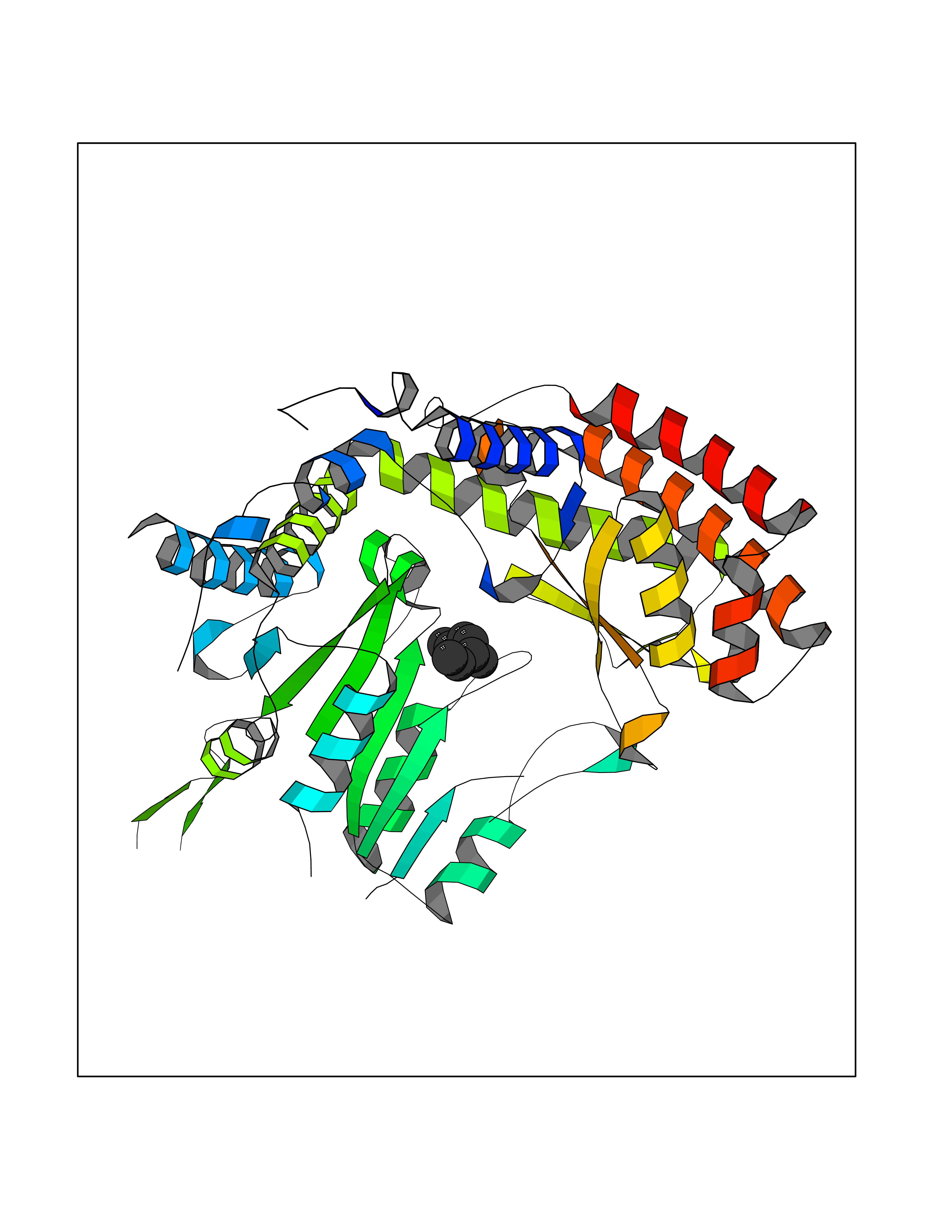 Molscript Map 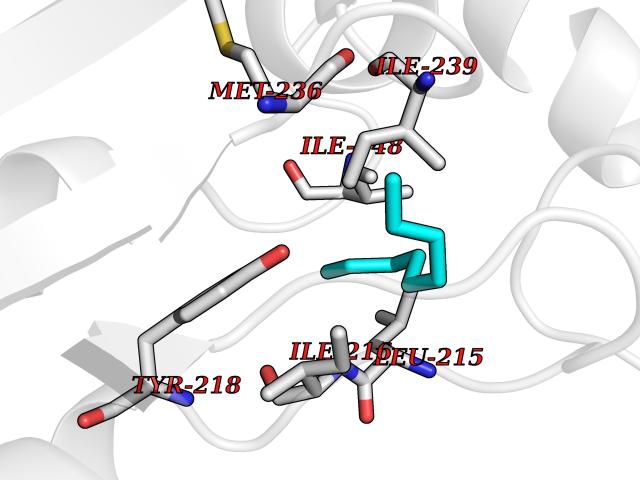 Pymol Map 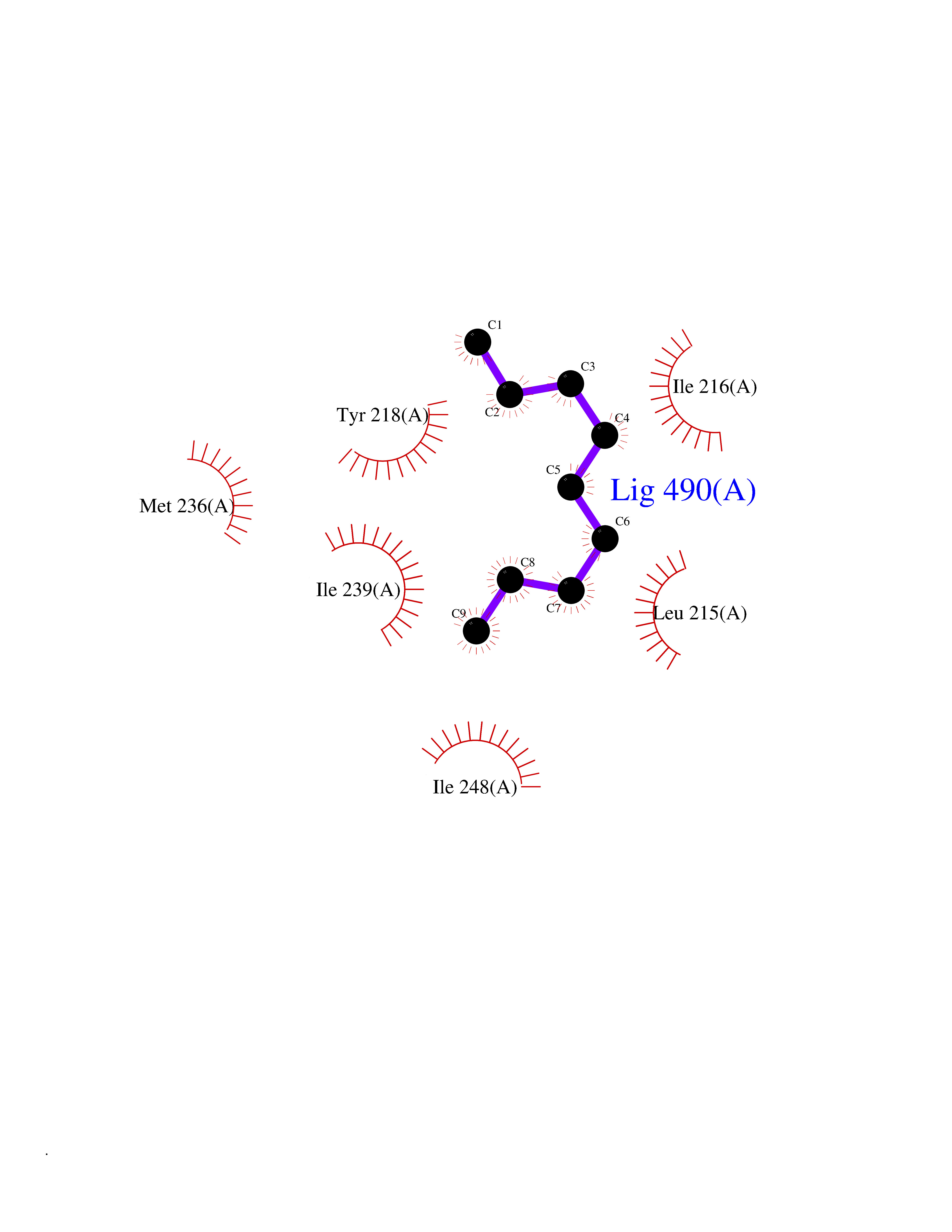 Ligplot Map 3D Binding mode Sequence GWTGQESLSDSDPEMWELLQREKDRQCRGLELIASENFCSRAALEALGSCLNNKYSEGEVVDEIELLCQRRALEAFDLDPAQWGVNVQPYSGSPANLAVYTALLQHDRIMGLDPYKLNPKTGLIDYNQLALTARLFRPRLIIAGTSAYARLIDYARMREVCDEVKAHLLADMAHISGLVAAKVIPSPFKHADIVTTTTHKTLRGARSGLIFYRKGVKAVDPGREIPYTFEDRINFAVFPSLQGGPHNHAIAAVAVALKQACTPMFREYSLQVLKNARAMADALLERGYSLVSGGTDNHLVLVDLRPKGLDGARAERVLELVSITANKNTCPGDRSAITPGGLRLGAPALTSRQFREDDFRRVVDFIDEGVNIGLEVKSKTAKLQDFKSFLLKDSETSQRLANLRQRVEQFARAFPMPGFDEH Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 5.42 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -7.4 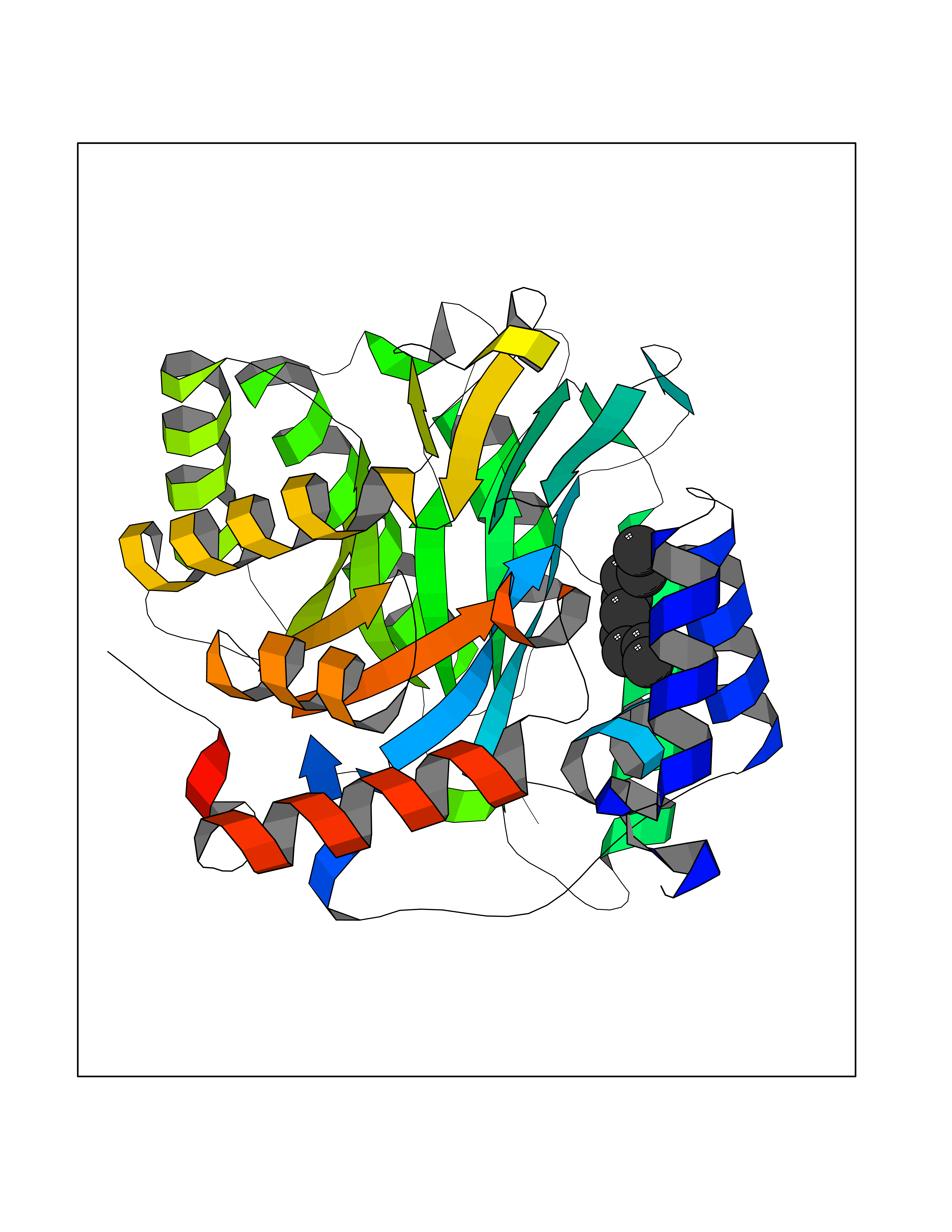 Molscript Map 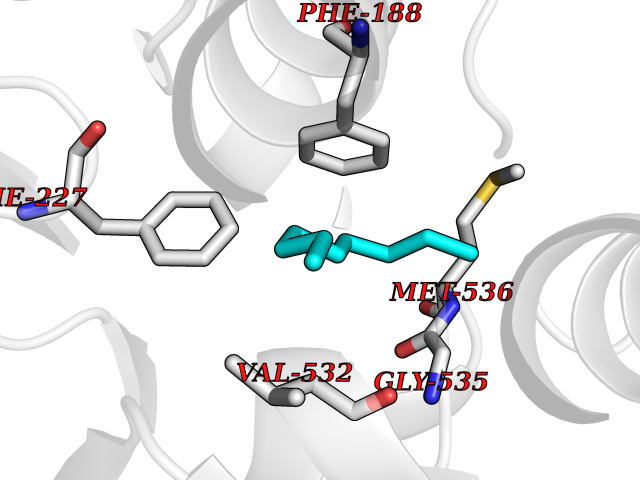 Pymol Map 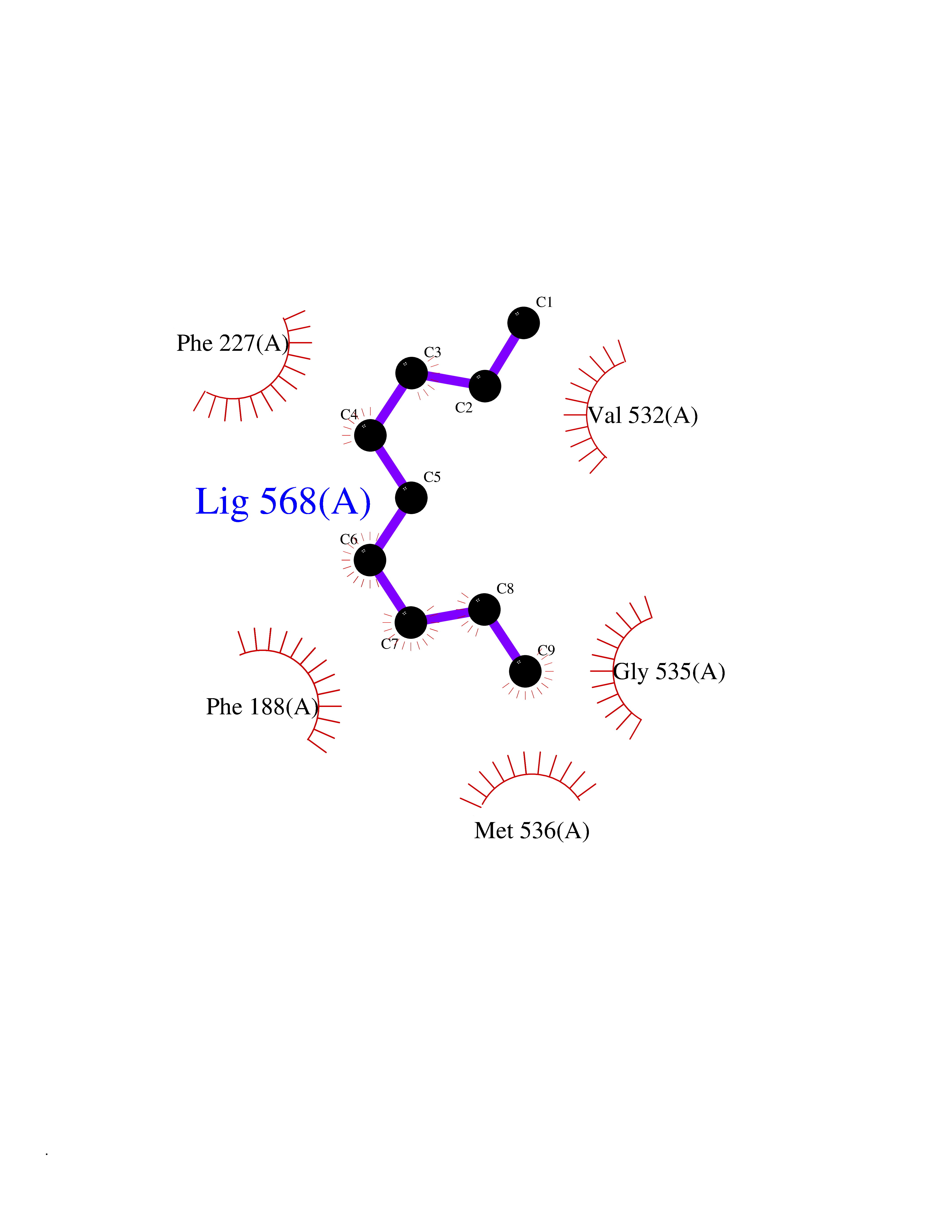 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Euchromatic histone-lysine N-methyltransferase 1 (EHMT1) | 5TTG | 5.42 | |
Target general information Gen name EHMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms EHMT1 Protein family Class V-like SAM-binding methyltransferase superfamily Biochemical class NA Function Histone methyltransferase that specifically mono- and dimethylates 'Lys-9' of histone H3 (H3K9me1 and H3K9me2, respectively) in euchromatin. H3K9me represents a specific tag for epigenetic transcriptional repression by recruiting HP1 proteins to methylated histones. Also weakly methylates 'Lys-27' of histone H3 (H3K27me). Also required for DNA methylation, the histone methyltransferase activity is not required for DNA methylation, suggesting that these 2 activities function independently. Probably targeted to histone H3 by different DNA-binding proteins like E2F6, MGA, MAX and/or DP1. During G0 phase, it probably contributes to silencing of MYC- and E2F-responsive genes, suggesting a role in G0/G1 transition in cell cycle. In addition to the histone methyltransferase activity, also methylates non-histone proteins: mediates dimethylation of 'Lys-373' of p53/TP53. Related diseases Kleefstra syndrome 1 (KLEFS1) [MIM:610253]: A form of Kleefstra syndrome, an autosomal dominant disease characterized by variable intellectual disability, psychomotor developmental delay, seizures, behavioral abnormalities, and facial dysmorphisms. KLEFS1 patients additionally manifest brachy(micro)cephaly, congenital heart defects, and urogenital defects. {ECO:0000269|PubMed:16826528, ECO:0000269|PubMed:19264732}. The disease is caused by variants affecting the gene represented in this entry. The syndrome can be either caused by intragenic EHMT1 mutations leading to haploinsufficiency of the EHMT1 gene or by a submicroscopic 9q34.3 deletion. Although it is not known if and to what extent other genes in the 9q34.3 region contribute to the syndrome observed in deletion cases, EHMT1 seems to be the major determinant of the core disease phenotype (PubMed:19264732). {ECO:0000269|PubMed:16826528, ECO:0000269|PubMed:19264732}. Drugs (DrugBank ID) NA Interacts with Q99549; Q04206; Q04207 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ANK repeat; Chromatin regulator; Chromosome; Disease variant; Intellectual disability; Isopeptide bond; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-adenosyl-L-methionine; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 30066.9 Length 260 Aromaticity 0.11 Instability index 49.19 Isoelectric point 5.73 Charge (pH=7) -4.88 2D Binding mode Binding energy (Kcal/mol) -7.4 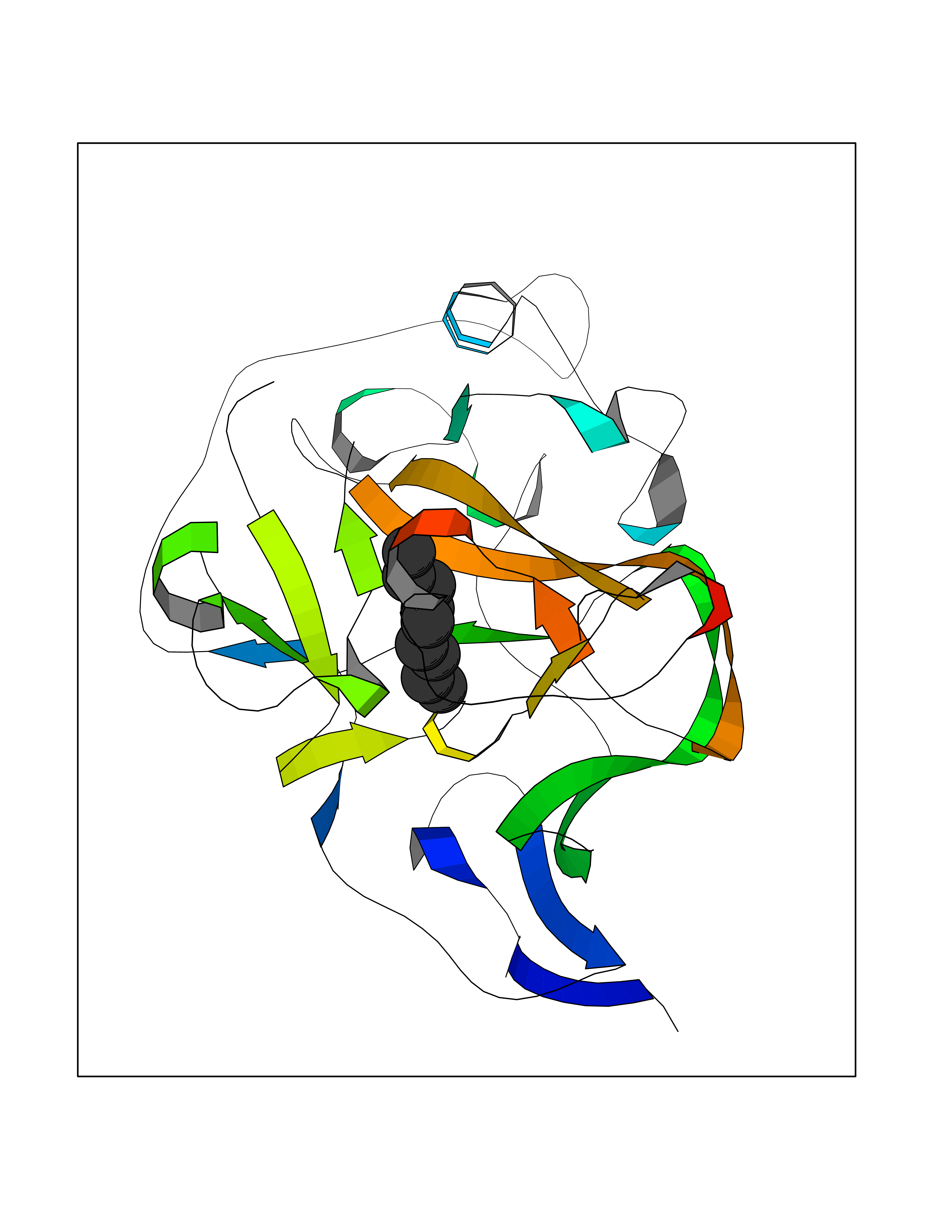 Molscript Map 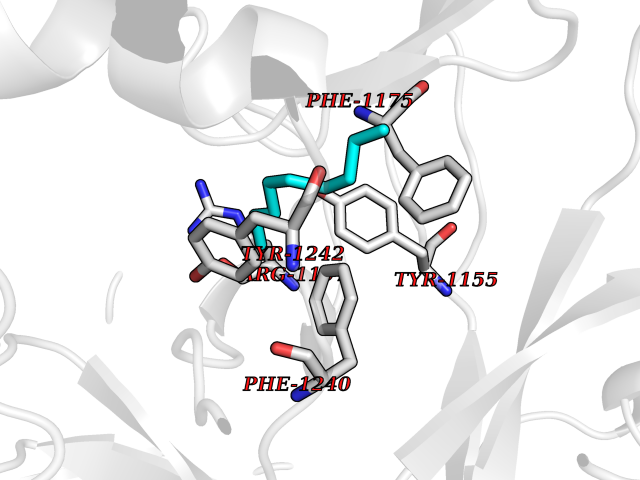 Pymol Map 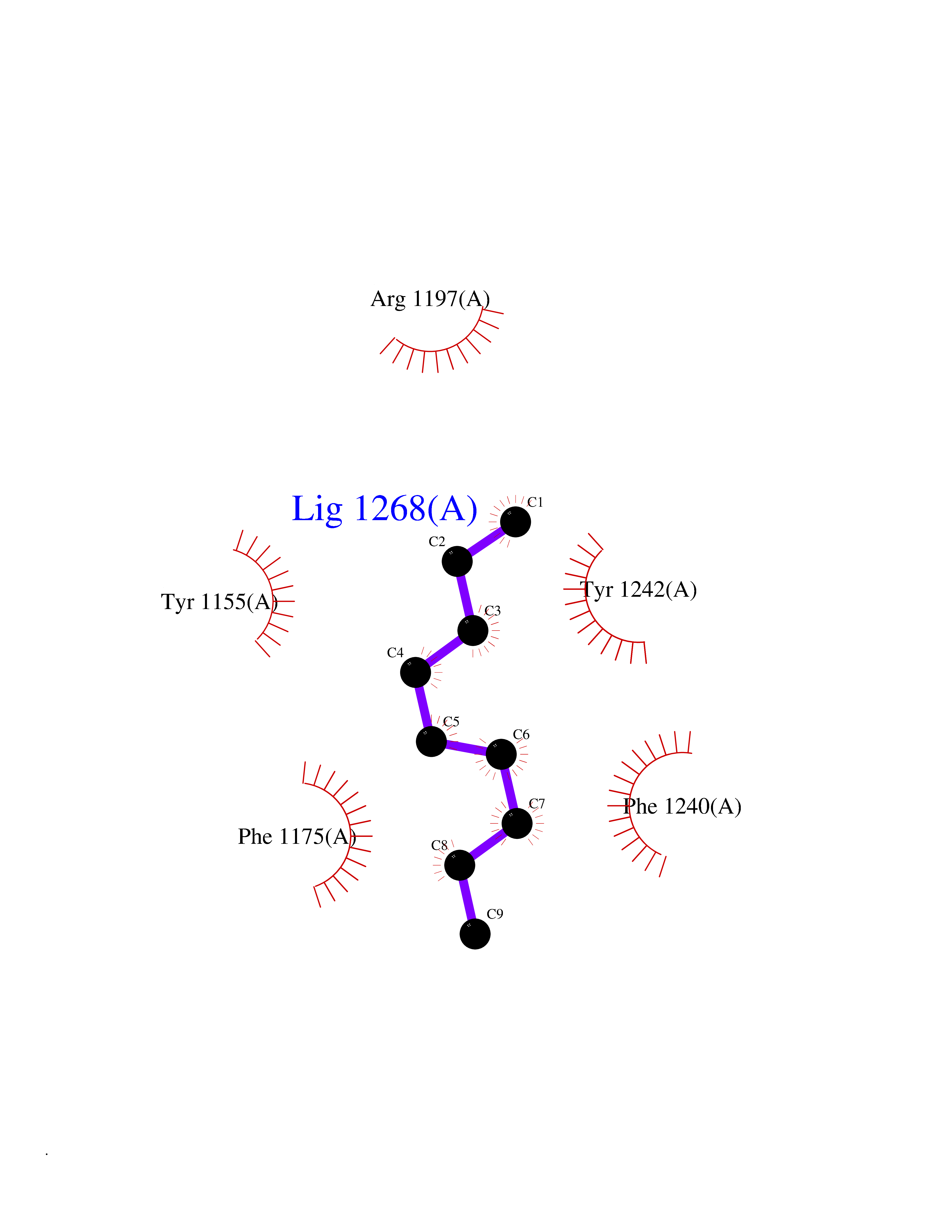 Ligplot Map 3D Binding mode Sequence VERIVSRDIARGYERIPIPCVNAVDSEPCPSNYKYVSQNCVTSPMNIDRNITHLQYCVCIDDCSSSNCMCGQLSMRCWYDKDGRLLPEFNMAEPPLIFECNHACSCWRNCRNRVVQNGLRARLQLYRTRDMGWGVRSLQDIPPGTFVCEYVGELISDSEADVREEDSYLFDLDNDGEVYCIDARFYGNVSRFINHHCEPNLVPVRVFMAHQDLRFPRIAFFSTRLIEAGEQLGFDYGERFWDIKGKLFSCRCGSPKCRHS Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | MAPK signal-integrating kinase 1 (MKNK1) | 5WVD | 5.42 | |
Target general information Gen name MKNK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mnk1; MAP kinase signal-integrating kinase 1 Protein family Protein kinase superfamily, CAMK Ser/Thr protein kinase family Biochemical class Protein kinase superfamily. CAMK Ser/Thr protein kinase family Function May play a role in the response to environmental stress and cytokines. Appears to regulate translation by phosphorylating EIF4E, thus increasing the affinity of this protein for the 7-methylguanosine-containing mRNA cap. Related diseases Defects in MELK are associated with some cancers, such as brain or breast cancers. Expression is dramatically increased in aggressive undifferentiated tumors, correlating with poor patient outcome in breast and brain cancers, suggesting a role in tumor-initiating cells and proliferation via its function in cell proliferation regulation. Drugs (DrugBank ID) DB12010 Interacts with P54253; Q03060-25; P42858; P28482; Q16539; Q96CV9 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Translation regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 27536.2 Length 241 Aromaticity 0.11 Instability index 50.42 Isoelectric point 6.02 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -7.39 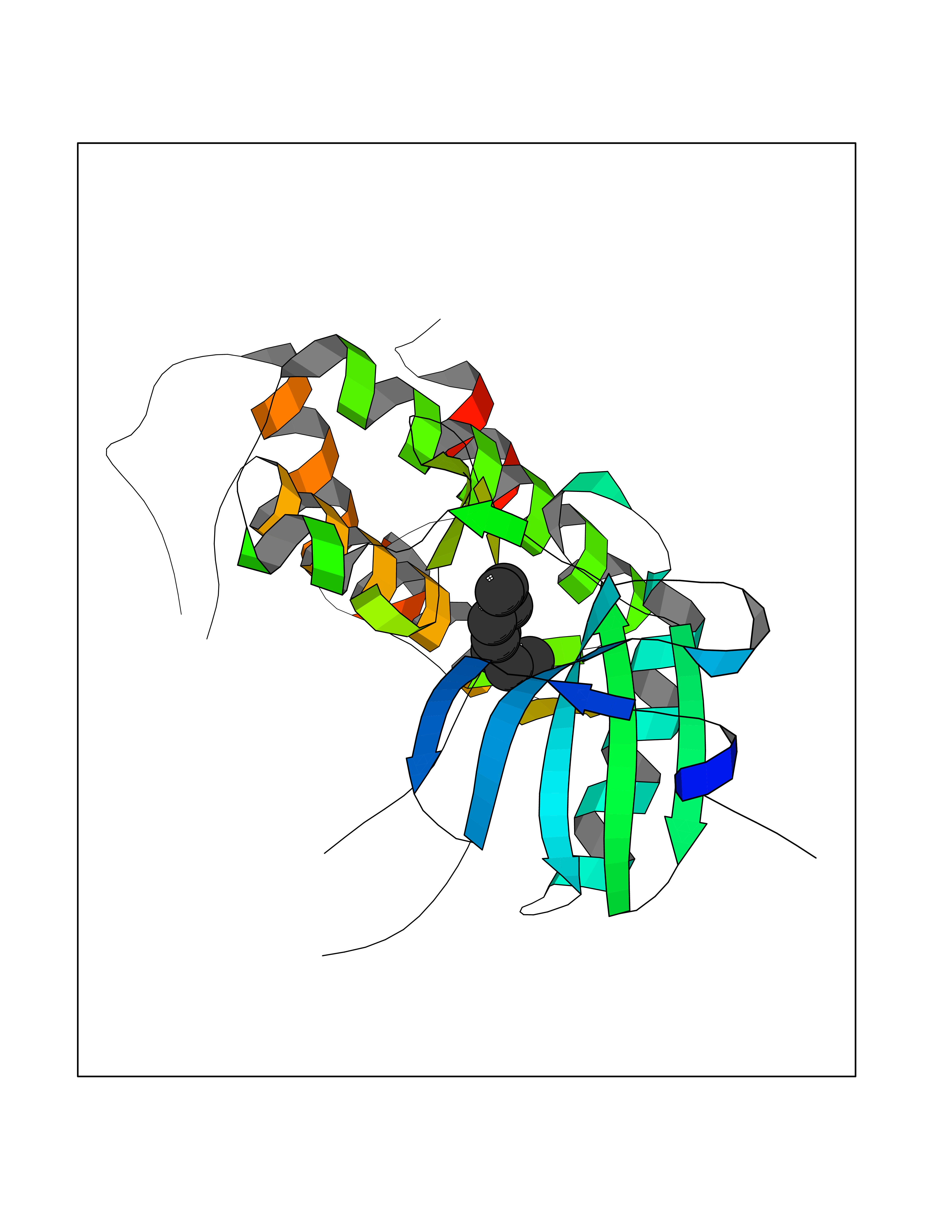 Molscript Map 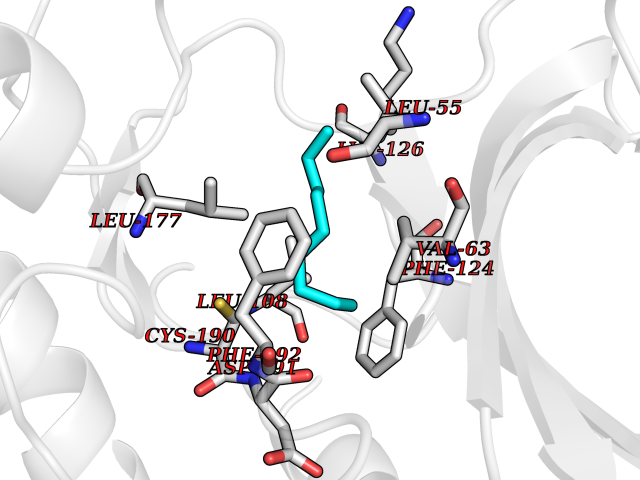 Pymol Map 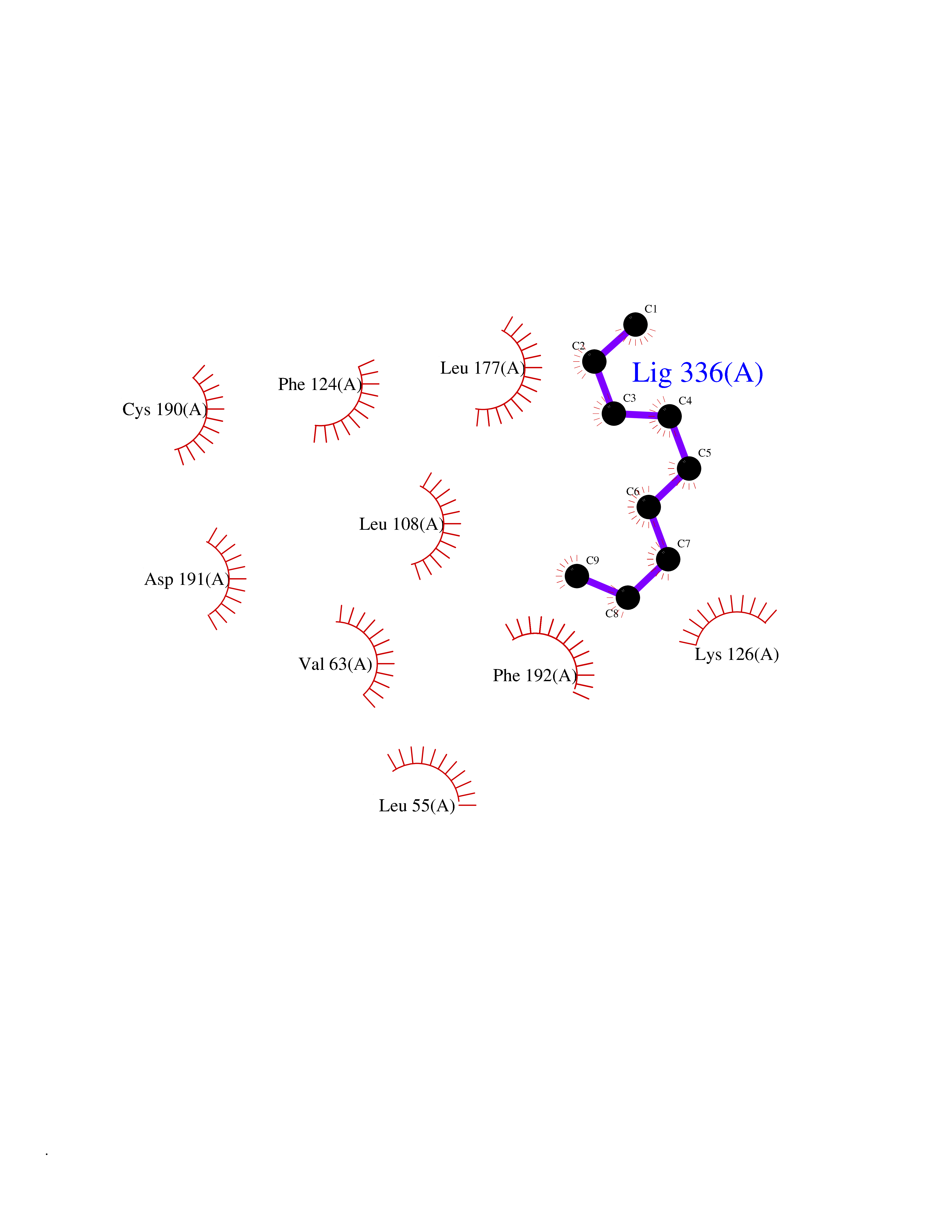 Ligplot Map 3D Binding mode Sequence PGKFEDMYKLTSELLGEGAYAKVQGAVSLQNGKEYAVKIIEKQAGHSRSRVFREVETLYQCQGNKNILELIEFFEDDTRFYLVFEKLQGGSILAHIQKQKHFNEREASRVVRDVAAALDFLHTKGIAHRDLKPENILCESPEKVSPVKICDFDLGSGYMAPEVVEVFTDQATFYDKRCDLWSLGVVLYIMLSGYPPFKYEFPDKDWAHISSEAKDLISKLLVRDAKQRLSAAQVLQHPWVQ Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Programmed cell death 1 ligand 1 (PD-L1) | 5J89 | 5.42 | |
Target general information Gen name CD274 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hPD-L1; Programmed death ligand 1; PDL1; PDCD1LG1; PDCD1L1; PDCD1 ligand 1; B7H1; B7-H1; B7 homolog 1 Protein family Immunoglobulin superfamily, BTN/MOG family Biochemical class Immunoglobulin Function As a ligand for the inhibitory receptor PDCD1/PD-1, modulates the activation threshold of T-cells and limits T-cell effector response. Through a yet unknown activating receptor, may costimulate T-cell subsets that predominantly produce interleukin-10 (IL10). Plays a critical role in induction and maintenance of immune tolerance to self. Related diseases Truncation of the 3'-untranslated (3'-UTR) region of CD274 transcripts leads to elevated expression of CD274 in multiple cancers including T-cell leukemia, diffuse large B-cell lymphoma and stomach adenocarcinoma (PubMed:27281199). Disruption of 3'-UTR region is caused by structural variants that stabilize CD274 transcripts, leading to overexpression (PubMed:27281199). Increased expression in tumors promotes immune evasion and tumor cell growth by allowing malignant cells to escape destruction by the immune system (PubMed:27281199). {ECO:0000269|PubMed:27281199}. Drugs (DrugBank ID) DB15773; DB11595; DB15771; DB11945; DB15772; DB14776; DB15770; DB11714; DB15769; DB09035; DB09037; DB00203; DB00313 Interacts with P33681; Q8IZR5; Q9NX76; Q15116; Q15116 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Endosome; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Nucleus; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID C,D Molecular weight (Da) 28335.2 Length 249 Aromaticity 0.1 Instability index 35.39 Isoelectric point 6.15 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -7.4 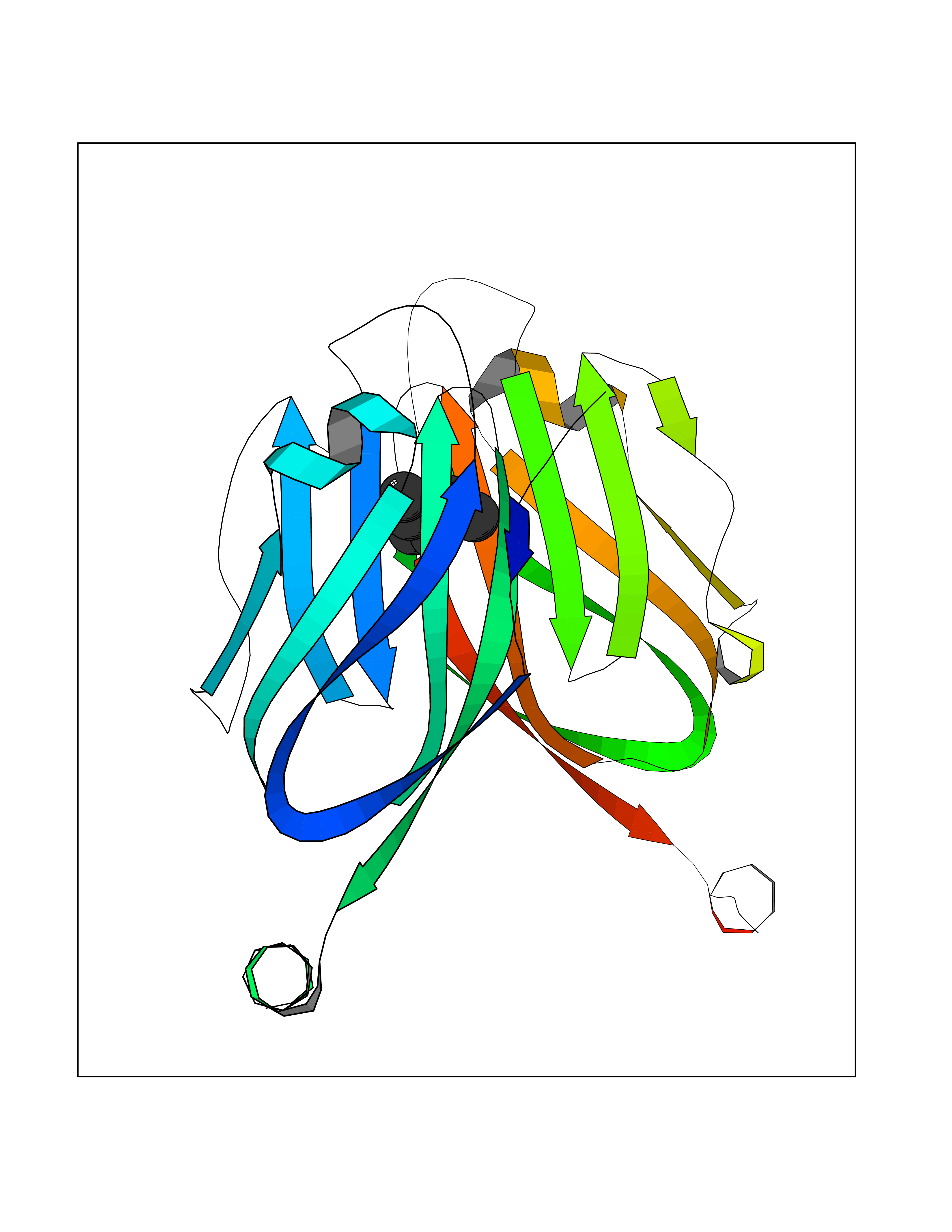 Molscript Map 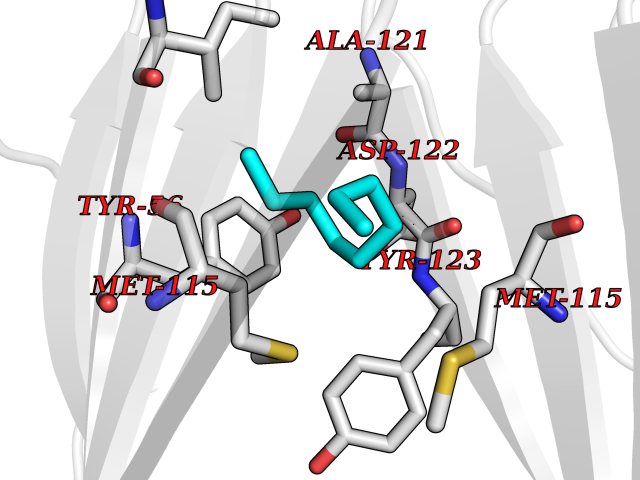 Pymol Map 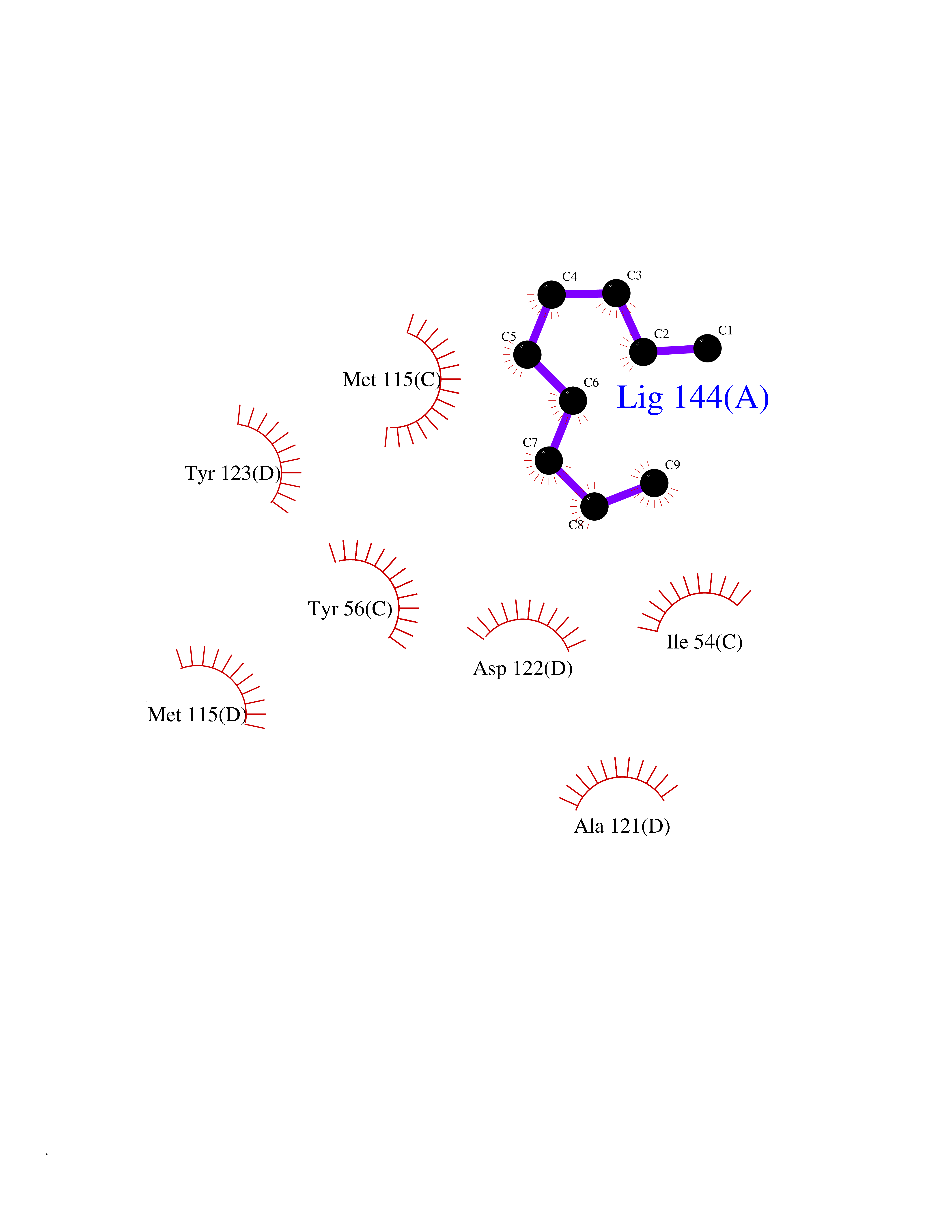 Ligplot Map 3D Binding mode Sequence AFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHHHAFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHH Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Xylose isomerase | 4XIA | 5.41 | |
Target general information Gen name xylA Organism Arthrobacter sp. (strain NRRL B3728) Uniprot ID TTD ID NA Synonyms NA Protein family Xylose isomerase family Biochemical class Isomerase(intramolecular oxidoreductase) Function Metal ion binding.Xylose isomerase activity. Related diseases Bachmann-Bupp syndrome (BABS) [MIM:619075]: An autosomal dominant disorder characterized by global developmental delay, alopecia, absolute or relative macrocephaly, and facial dysmorphism. Neuroimaging shows white matter abnormalities, prominent Virchow-Robin spaces, periventricular cysts, and abnormalities of the corpus callosum. {ECO:0000269|PubMed:30239107, ECO:0000269|PubMed:30475435}. The disease is caused by variants affecting the gene represented in this entry. BABS is due to truncating variants that lead to a gain of function. This phenomenon apparently results from truncation proximal to or involving the C-terminal region of ODC1 protein, distal enough to allow escape from nonsense-mediated decay. A gain of function is corroborated by elevated plasma levels of N-acetylputrescine, with otherwise normal polyamine levels, in affected individuals. {ECO:0000269|PubMed:30475435}. Drugs (DrugBank ID) DB02172; DB03206; DB11195 Interacts with NA EC number 5.3.1.5 Uniprot keywords 3D-structure; Carbohydrate metabolism; Cytoplasm; Direct protein sequencing; Isomerase; Magnesium; Metal-binding; Xylose metabolism Protein physicochemical properties Chain ID A,B Molecular weight (Da) 35700.7 Length 325 Aromaticity 0.11 Instability index 20.63 Isoelectric point 5.19 Charge (pH=7) -14.16 2D Binding mode Binding energy (Kcal/mol) -7.38  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence VQPTPADHFTFGLWTVGWTGADPFGVATRANLDPVEAVHKLAELGAYGITFHDNDLIPFDATAAEREKILGDFNQALADTGLKVPMVTTNLFSHPVFKDGGFTSNDRSIRRFALAKVLHNIDLAAEMGAETFVMWGGREGSEYDGSKDLAAALDRMREGVDTAAGYIKDKGYNLRIALEPKPNEPRGDIFLPTVGHGLAFIEQLEHGDIVGLNPETGHEQMAGLNFTHGIAQALWAEKLFHIDLNGQRGIKYDQDLVFGHGDLTSAFFTVDLLENGFPNGGPKYTGPRHFDYKPSRTDGYDGVWDSAKANMSMYLLLKERALAFR Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | SEC14-like protein 2 | 4OMJ | 5.41 | |
Target general information Gen name SEC14L2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA1658;C22orf6;KIAA1186 Protein family NA Biochemical class Transport protein Function Phospholipid binding.Transporter activity.Vitamin E binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Lipid-binding; Nucleus; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 31533.3 Length 274 Aromaticity 0.1 Instability index 49.26 Isoelectric point 8.34 Charge (pH=7) 2.81 2D Binding mode Binding energy (Kcal/mol) -7.38  Molscript Map 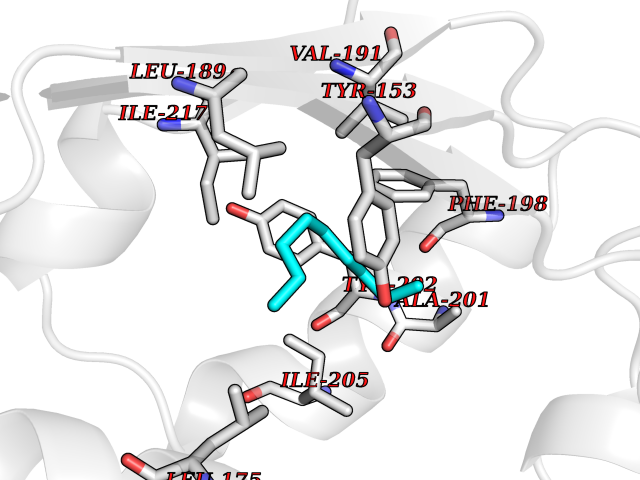 Pymol Map 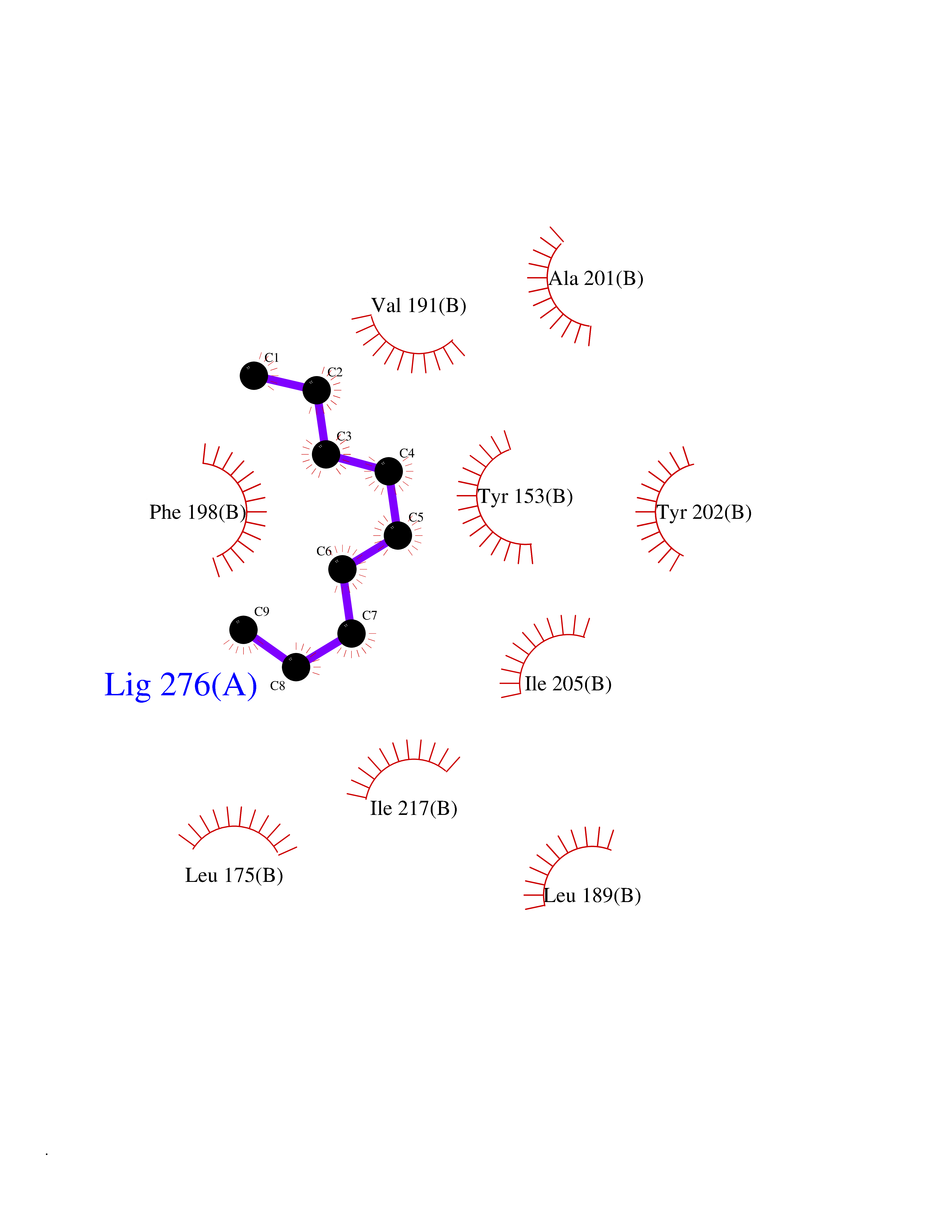 Ligplot Map 3D Binding mode Sequence MSGRVGDLSPRQKEALAKFRENVQDVLPALPNPDDYFLLRWLRARSFDLQKSEAMLRKHVEFRKQKDIDNIISWQPPEVIQQYLSGGMCGYDLDGCPVWYDIIGPLDAKGLLFSASKQDLLRTKMRECELLLQECAHQTTKLGRKVETITIIYDCEGLGLKHLWKPAVEAYGEFLCMFEENYPETLKRLFVVKAPKLFPVAYNLIKPFLSEDTRKKIMVLGANWKEVLLKHISPDQVPVEYGGTMTDPDGNPKCKSKINYGGDIPRKYYVRDQV Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Dual specificity mitogen-activated protein kinase kinase 1 | 3EQC | 5.41 | |
Target general information Gen name MAP2K1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PRKMK1;MEK1 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase subfamily Biochemical class Transferase Function ATP binding.MAP kinase kinase activity.Protein C-terminus binding.Protein kinase activity.Protein N-terminus binding.Protein serine/threonine/tyrosine kinase activity.Protein serine/threonine kinase activator activity.Protein serine/threonine kinase activity.Protein tyrosine kinase activity.Signal transducer, downstream of receptor, with protein tyrosine phosphatase activity. Related diseases Cardiofaciocutaneous syndrome 3 (CFC3) [MIM:615279]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. Distinctive features of CFC3 include macrostomia and horizontal shape of palpebral fissures. {ECO:0000269|PubMed:16439621, ECO:0000269|PubMed:18042262}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melorheostosis, isolated (MEL) [MIM:155950]: A sclerosing bone disorder characterized by hyperostosis of the cortex of tubular bones, frequently involving one limb. The lesions may be accompanied by abnormalities of adjacent soft tissue, joint contractures, sclerodermatous skin lesions, muscle atrophy, or hemangioma. {ECO:0000269|PubMed:29643386}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06892; DB07046; DB08208; DB03115; DB11967; DB06616; DB05239; DB02152; DB07101; DB08130; DB14904; DB11689; DB08911 Interacts with Q8N9N5; Q8N9N5-2; Q9NR09; P15056; Q9Y297; O15519-1; P28482; P27361; Q13526; Q9H8W4; P04049; Q8WWU5-7; Q86Y07; Q86Y07-1; P46937 EC number 2.7.12.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cardiomyopathy; Cytoplasm; Cytoskeleton; Direct protein sequencing; Disease variant; Ectodermal dysplasia; Intellectual disability; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 34949.2 Length 312 Aromaticity 0.07 Instability index 45.3 Isoelectric point 5.96 Charge (pH=7) -4.47 2D Binding mode Binding energy (Kcal/mol) -7.38  Molscript Map 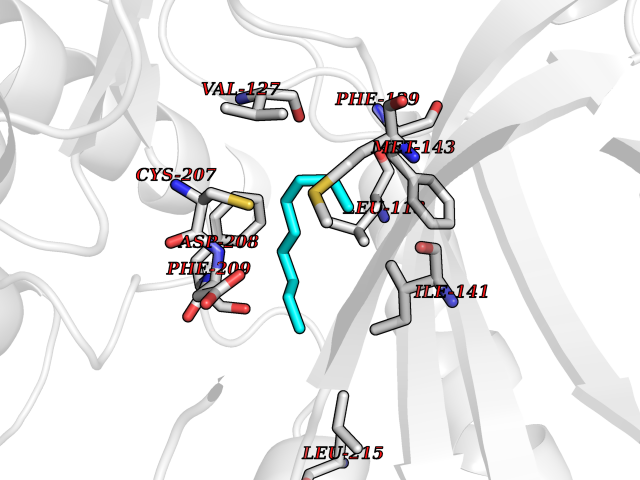 Pymol Map 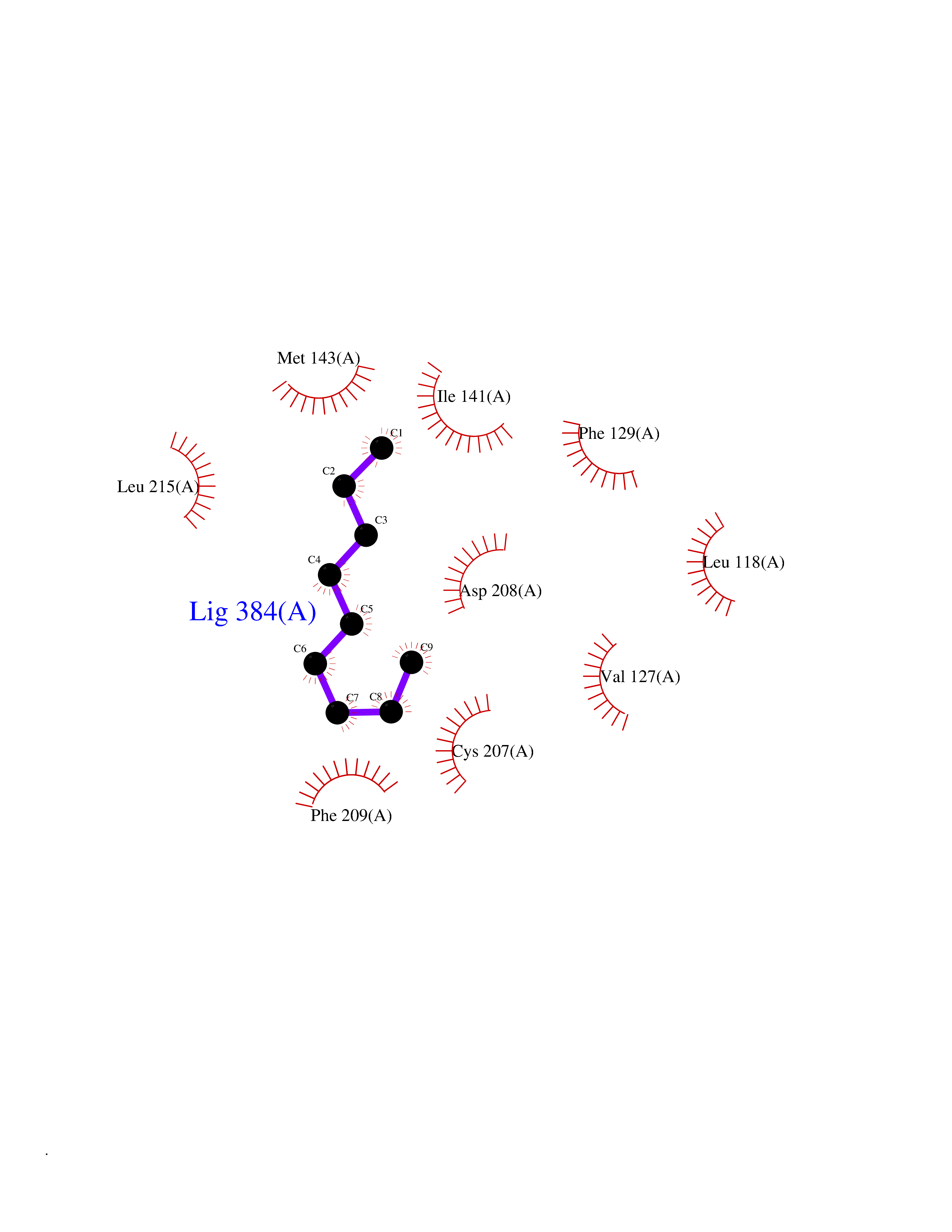 Ligplot Map 3D Binding mode Sequence ELELDEQQRKRLEAFLTQKQKVGELKDDDFEKISELGAGNGGVVFKVSHKPSGLVMARKLIHLEIKPAIRNQIIRELQVLHECNSPYIVGFYGAFYSDGEISICMEHMDGGSLDQVLKKAGRIPEQILGKVSIAVIKGLTYLREKHKIMHRDVKPSNILVNSRGEIKLCDFGVSGQLIDSMAVGTRSYMSPERLQGTHYSVQSDIWSMGLSLVEMAVGRYPIPPPDAKELELMFGCPMAIFELLDYIVNEPPPKLPSGVFSLEFQDFVNKCLIKNPAERADLKQLMVHAFIKRSDAEEVDFAGWLCSTIGLN Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | SEC14-like protein 4 | 4TLG | 5.41 | |
Target general information Gen name SEC14L4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TAP3 Protein family NA Biochemical class Transport protein Function Lipid binding.Transporter activity. Related diseases Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia (CDP-PBHM) [MIM:300863]: A disease characterized by chondrodysplasia, severe platyspondyly, hydrocephaly, and facial features with microphthalmia. Bone abnormalities include a distinctive metaphyseal cupping of the metacarpals, metatarsals, and phalanges. Affected females show a milder phenotype with small stature, sometimes associated with body asymmetry and mild intellectual disability. {ECO:0000269|PubMed:20181727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB11635; DB11251; DB00163 Interacts with Q96LC9; O43186; P78358; Q9NYQ3; Q0VD86; Q15323; O76011; P50221; Q6FHY5; Q02548; P26367; Q9H8W4; Q04864; Q04864-2; Q9UHV2; P15884; P15884-3; Q96N21; Q9BYV2; Q8N6Y0; Q9H0C1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 23947.6 Length 210 Aromaticity 0.1 Instability index 50.84 Isoelectric point 5.55 Charge (pH=7) -3.11 2D Binding mode Binding energy (Kcal/mol) -7.38 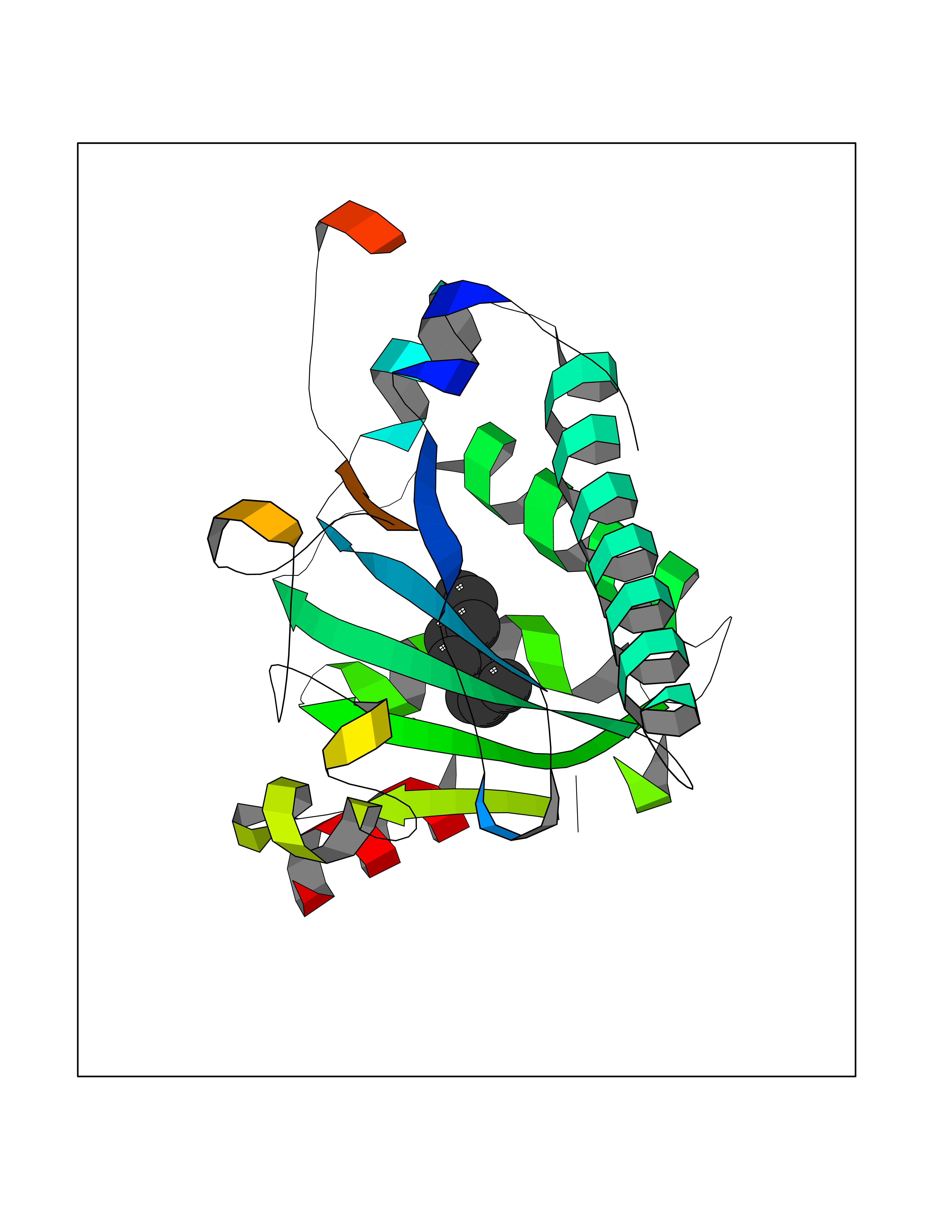 Molscript Map 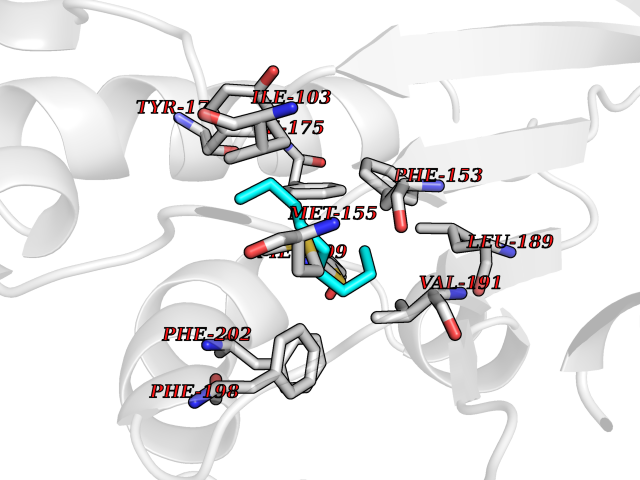 Pymol Map 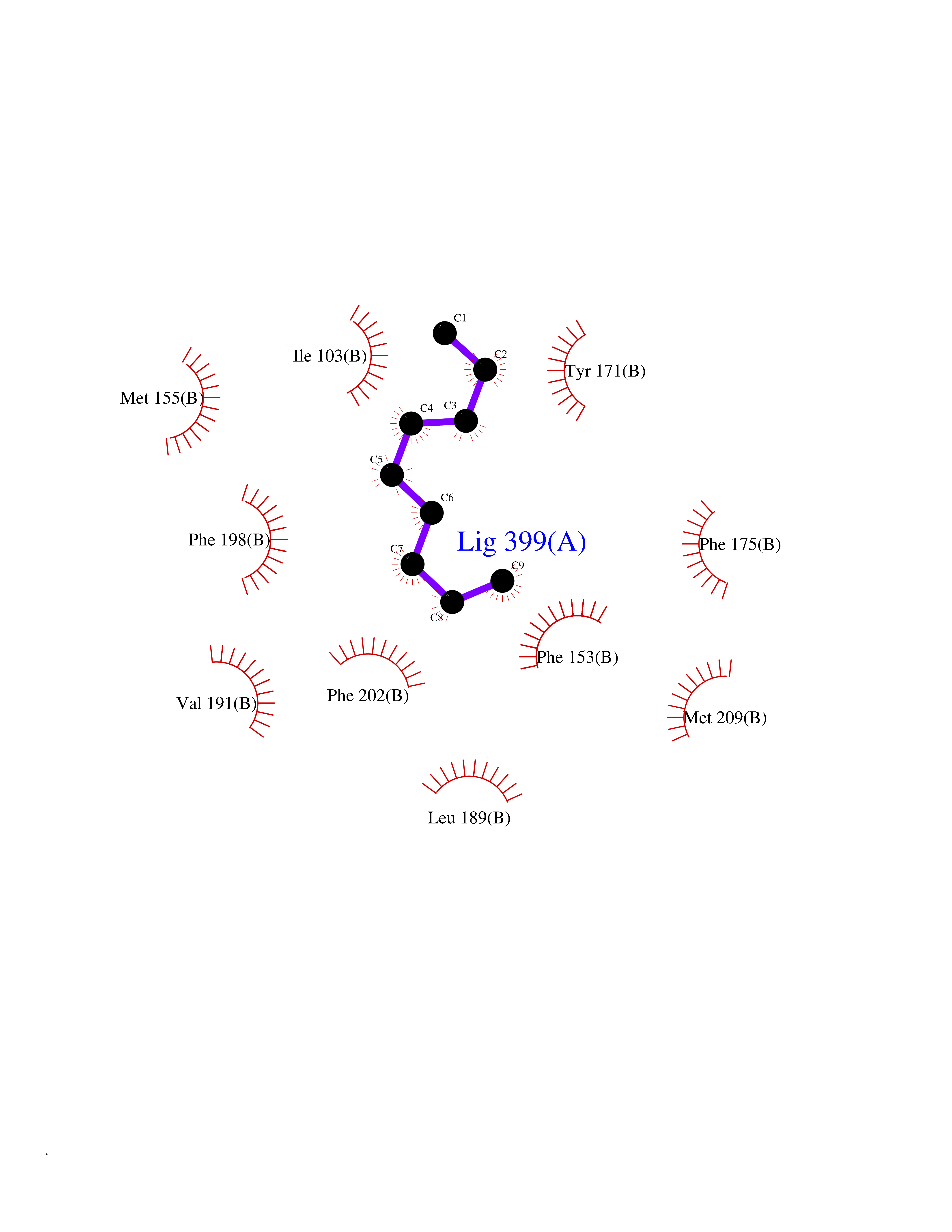 Ligplot Map 3D Binding mode Sequence VTWQPPEVIQLYDSGGLCGYDYEGCPVYFNIIGSLDPKGLLLSASKQDMIRKRIKVCELLLHECELQTQKLGRKIEMALMVFDMEGLSLKHLWKPAVEVYQQFFSILEANYPETLKNLIVIRAPKLFPVAFNLVKSFMSEETRRKIVILGDNWKQELTKFISPDQLPVEFGGTMTDPDGNPKCLTKINYGGEVPKSYYPDKASEETLQSM Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Mycobacterium Isocitrate lyase (MycB icl) | 1F8M | 5.41 | |
Target general information Gen name MycB icl Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID Synonyms Isocitratase; Isocitrase; ICL Protein family Isocitrate lyase/PEP mutase superfamily, Isocitrate lyase family Biochemical class Carbon-carbon lyase Function Catalyzes the formation of succinate and glyoxylate from isocitrate, a key step of the glyoxylate cycle. May be involved in the assimilation of one-carbon compounds via the isocitrate lyase- positive serine pathway. Related diseases Intellectual developmental disorder, autosomal recessive 80, with variant lissencephaly (MRT80) [MIM:620653]: An autosomal recessive disorder characterized by global developmental delay, mildly to moderately impaired intellectual development, attention deficit-hyperactivity disorder, hypotonia, seizure, poor social skills, and autistic traits. Brain imaging shows fronto-temporal lissencephaly and pachygyria. {ECO:0000269|PubMed:37880421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04343 Interacts with NA EC number EC 4.1.3.1 Uniprot keywords 3D-structure; Glyoxylate bypass; Isopeptide bond; Lyase; Magnesium; Manganese; Metal-binding; Reference proteome; Tricarboxylic acid cycle; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 93759.5 Length 854 Aromaticity 0.09 Instability index 28.02 Isoelectric point 4.98 Charge (pH=7) -34.53 2D Binding mode Binding energy (Kcal/mol) -7.38  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ASVVGTPKSAEQIQQEWDTNPRWKDVTRTYSAEDVVALQGSVVEEHTLARRGAEVLWEQLHDLEWVNALGALTGNMAVQQVRAGLKAIYLSGWQVAGDANLSGHTYPDQSLYPANSVPQVVRRINNALQRADQIAKIEGDTSVENWLAPIVADGEAGFGGALNVYELQKALIAAGVAGSHWEDQLASEKKCGHLGGKVLIPTQQHIRTLTSARLAADVADVPTVVIARTDAEAATLITSDVDERDQPFITGERTREGFYRTKNGIEPCIARAKAYAPFADLIWMETGTPDLEAARQFSEAVKAEYPDQMLAYNCSPSFNWKKHLDDATIAKFQKELAAMGFKFQFITLAGFHALNYSMFDLAYGYAQNQMSAYVELQEREFAAEERGYTATKHQREVGAGYFDRIATTVDPNSSTTALTGSTEEGQFASVVGTPKSAEQIQQEWDTNPRWKDVTRTYSAEDVVALQGSVVEEHTLARRGAEVLWEQLHDLEWVNALGALTGNMAVQQVRAGLKAIYLSGWQVAGDANLSGHTYPDQSLYPANSVPQVVRRINNALQRADQIAKIEGDTSVENWLAPIVADGEAGFGGALNVYELQKALIAAGVAGSHWEDQLASEKKCGHLGGKVLIPTQQHIRTLTSARLAADVADVPTVVIARTDAEAATLITSDVDERDQPFITGERTREGFYRTKNGIEPCIARAKAYAPFADLIWMETGTPDLEAARQFSEAVKAEYPDQMLAYNCSPSFNWKKHLDDATIAKFQKELAAMGFKFQFITLAGFHALNYSMFDLAYGYAQNQMSAYVELQEREFAAEERGYTATKHQREVGAGYFDRIATTVDPNSSTTALTGSTEEGQF Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Extracellular lysophospholipase D (E-NPP2) | 4ZGA | 5.41 | |
Target general information Gen name ENPP2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LysoPLD; Ectonucleotide pyrophosphatase/phosphodiesterase family member 2; E-NPP 2; Autotaxin; ATX Protein family Nucleotide pyrophosphatase/phosphodiesterase family Biochemical class Phosphoric diester hydrolase Function Hydrolyzes lysophospholipids to produce the signaling molecule lysophosphatidic acid (LPA) in extracellular fluids. Major substrate is lysophosphatidylcholine. Also can act on sphingosylphosphorylcholine producing sphingosine-1-phosphate, a modulator of cell motility. Can hydrolyze, in vitro, bis-pNPP, to some extent pNP-TMP, and barely ATP. Involved in several motility-related processes such as angiogenesis and neurite outgrowth. Acts as an angiogenic factor by stimulating migration of smooth muscle cells and microtubule formation. Stimulates migration of melanoma cells, probably via a pertussis toxin-sensitive G protein. May have a role in induction of parturition. Possible involvement in cell proliferation and adipose tissue development (Probable). Tumor cell motility-stimulating factor. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.1.4.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Chemotaxis; Cleavage on pair of basic residues; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lipid degradation; Lipid metabolism; Metal-binding; Obesity; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 85397.4 Length 740 Aromaticity 0.12 Instability index 58.63 Isoelectric point 7.16 Charge (pH=7) 1.02 2D Binding mode Binding energy (Kcal/mol) -7.38  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GSCKGRCFELCRCDNLCKSYTSCCHDFDELCLKTARGWECTKDRCGNEENACHCEDCLARGDCCTNYQVVCKGESHWVDDDCEEIKAAECPAGFVRPPLIIFSVDGFRASYMKKGSKVMPNIEKLRSCGTHSPYMRPVYPTKTFPNLYTLATGLYPESHGIVGNSMYDPVFDATFHLRGREKFNHRWWGGQPLWITATKQGVKAGTFFWSVVIPHERRILTILQWLTLPDHERPSVYAFYSEQPDFSGHKYGPFGPEMTNPLREIDKIVGQLMDGLKQLKLHRCVNVIFVGDHGMEDVTCDRTEFLSNYLTNVDDITLVPGTLGRIRSKFDPKAIIANLTCKKPDQHFKPYLKQHLPKRLHYANNRRIEDIHLLVERRWHVARKPFFQGDHGFDNKVNSMQTVFVGYGSTFKYKTKVPPFENIELYNVMCDLLGLKPAPNNGTHGSLNHLLRTNTFRPTMPEEVTRPNYPGIMYLQSDFDLGTEERHLLYGRPAVLYRTRYDILYHTDFESGYSEIFLMPLWTSYTVSKQACVRPDVRVSPSFSQNCLAYKNDKQMSYGFLFPPYLSSSPEAKYDAFLVTNMVPMYPAFKRVWNYFQRVLVKKYASERNGVNVISGPIFDYDYDGLHDTEDKIKQYVEGSSIPVPTHYYSIITSCLDFTQPADKCDGPLSVSSFILPHRPDNEESCNSSEDESKWVEELMKMHTARVRDIEHLTSLDFFRKTSRSYPEILTLKTYLHTYE Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Neuronal acetylcholine receptor alpha-4 (CHRNA4) | 6CNJ | 5.41 | |
Target general information Gen name CHRNA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor alpha4; CHRNA4; Alpha-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-4/CHRNA4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasmamembrane permeable to sodium ions. Related diseases Epilepsy, nocturnal frontal lobe, 1 (ENFL1) [MIM:600513]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:10563623, ECO:0000269|PubMed:14623738, ECO:0000269|PubMed:7550350}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01351; DB01352; DB00572; DB01483; DB00237; DB00241; DB01353; DB00564; DB00565; DB09028; DB01245; DB00514; DB01496; DB07720; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00898; DB01354; DB01355; DB00753; DB00657; DB00333; DB00463; DB00849; DB00184; DB00312; DB01174; DB00981; DB05458; DB00794; DB05740; DB00747; DB00418; DB00202; DB00306; DB00599; DB01273 Interacts with Q6UY14-3; P05067; P83916; Q6UXH1-1; Q6UXH1-3; P20042; Q9NZR2; Q92673; P17787 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -7.38  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||