Job Results:
Ligand
Structure
Job ID
369cc27f15dbe7b3a0e0dfe22b2eb6d9
Job name
NA
Time
2025-03-05 09:37:27
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Quinone-dependent D-lactate dehydrogenase | 1F0X | 4.01 | |
Target general information Gen name dld Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW2121;b2133 Protein family Quinone-dependent D-lactate dehydrogenase family Biochemical class Oxidoreductase Function (R)-2-hydroxyglutarate dehydrogenase activity.D-lactate dehydrogenase (cytochrome) activity.Electron carrier activity.FAD binding.Flavin adenine dinucleotide binding.NAD binding.Oxidoreductase activity, acting on the CH-OH group of donors, quinone or similar compound as acceptor.Quinone binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB00756 Interacts with NA EC number 1.1.5.12 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Membrane; Oxidoreductase; Quinone; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56475.2 Length 502 Aromaticity 0.09 Instability index 32.5 Isoelectric point 5.97 Charge (pH=7) -10.38 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 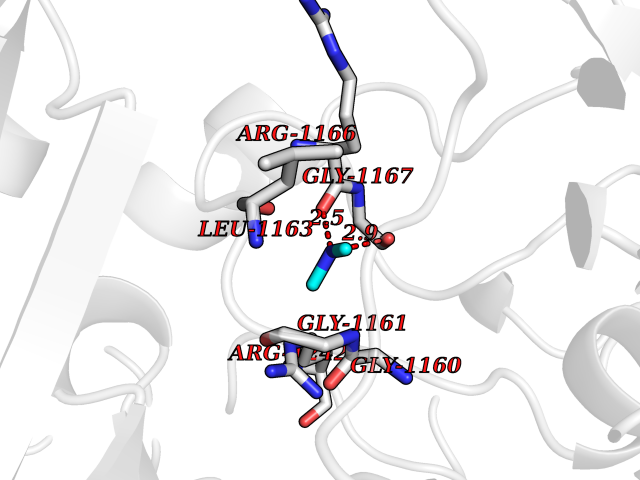 Pymol Map 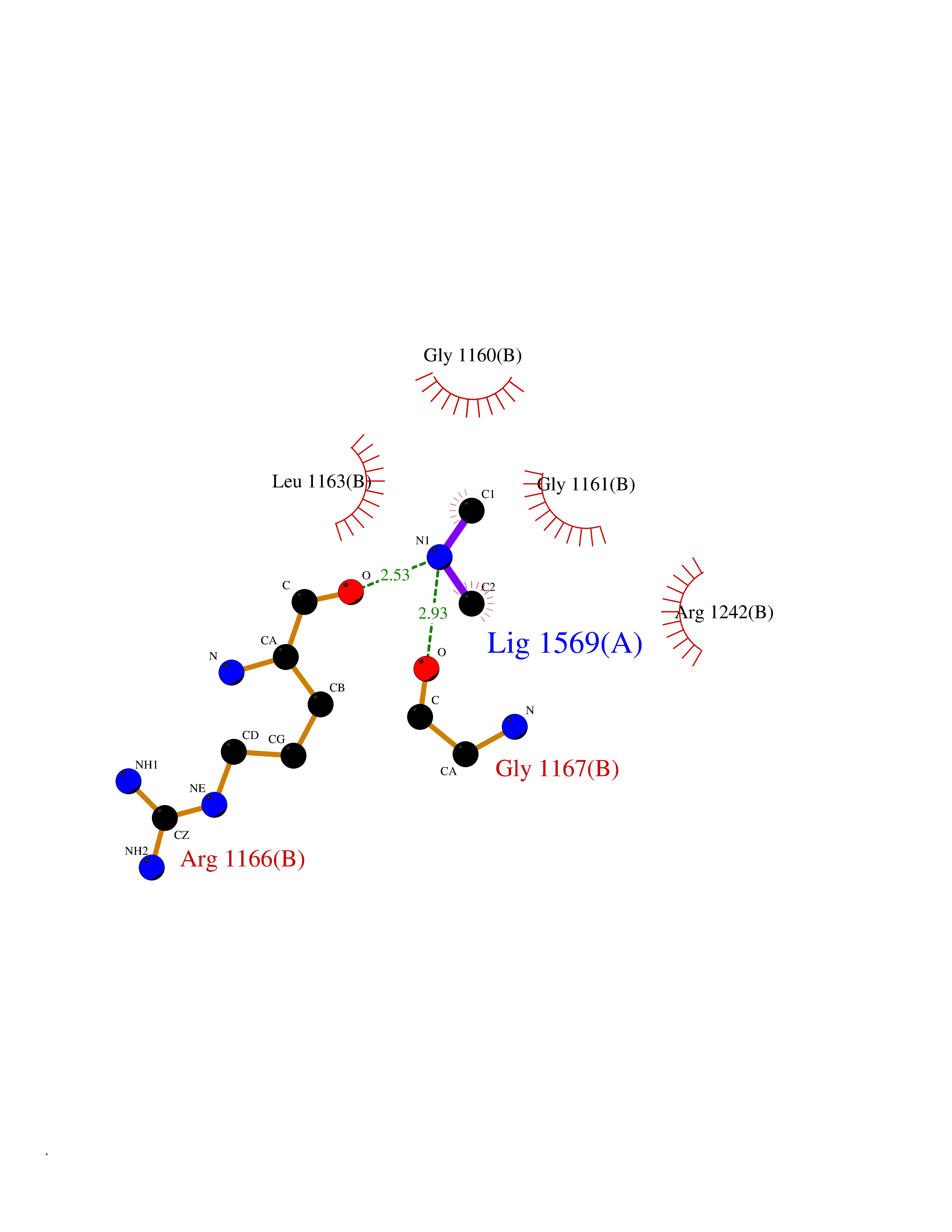 Ligplot Map 3D Binding mode Sequence NKAFLNELARLVGSSHLLTDPAKTARYRKGFRSGQGDALAVVFPGSLLELWRVLKACVTADKIILMQAANTGLTEGSTPNGNDYDRDVVIISTLRLDKLHVLGKGEQVLAYPGTTLYSLEKALKPLGREPHSVIGSSCIGASVIGGICNNSGGSLVQRGPAYTEMSLFARINEDGKLTLVNHLGIDLGETPEQILSKLDDDRIKDDDVRHDGRHAHDYDYVHRVRDIEADTPARYNADPDRLFESSGCAGKLAVFAVRLDTFEAEKNQQVFYIGTNQPEVLTEIRRHILANFENLPVAGEYMHRDIYDIAELPPRMKNWRDKYEHHLLLKMAGDGVGEAKSWLVDYFKQAEGDFFVCTPEEGSKAFLHRFAAAGAAIRYQAVHSDEVEDILALDIALRRNDTEWYEHLPPEIDSQLVHKLYYGHFMCYVFHQDYIVKKGVDVHALKEQMLELLQQRGAQYPAEHNVGHLYKAPETLQKFYRENDPTNSMNPGIGKTSKRKNW Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | DNA topoisomerase II alpha (TOP2A) | 1ZXM | 4.01 | |
Target general information Gen name TOP2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms DNA topoisomerase II, alpha isozyme; DNA topoisomerase 2alpha; DNA topoisomerase 2-alpha Protein family Type II topoisomerase family Biochemical class Topoisomerase Function Topoisomerase II makes double-strand breaks. Essential during mitosis and meiosis for proper segregation of daughter chromosomes. May play a role in regulating the period length of ARNTL/BMAL1 transcriptional oscillation. Control of topological states of DNA by transient breakage and subsequent rejoining of DNA strands. Related diseases A chromosomal aberration involving TOP1 is found in a form of therapy-related myelodysplastic syndrome. Translocation t(11;20)(p15;q11) with NUP98. {ECO:0000269|PubMed:10556215}. Drugs (DrugBank ID) DB05706; DB06013; DB05022; DB06263; DB00276; DB06420; DB04975; DB06362; DB00537; DB00970; DB00694; DB06421; DB00380; DB00997; DB05129; DB00467; DB00445; DB00773; DB09047; DB04576; DB01645; DB01177; DB00978; DB04967; DB01204; DB00218; DB01059; DB01165; DB00487; DB01179; DB05920; DB04978; DB01208; DB00444; DB00685; DB00385; DB06042 Interacts with O14497-1; P38398; P35222; Q05655 EC number EC 5.6.2.3 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Biological rhythms; Cytoplasm; Direct protein sequencing; DNA-binding; Isomerase; Isopeptide bond; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Topoisomerase; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42640.6 Length 373 Aromaticity 0.1 Instability index 33.34 Isoelectric point 8.64 Charge (pH=7) 5.05 2D Binding mode Binding energy (Kcal/mol) -5.46 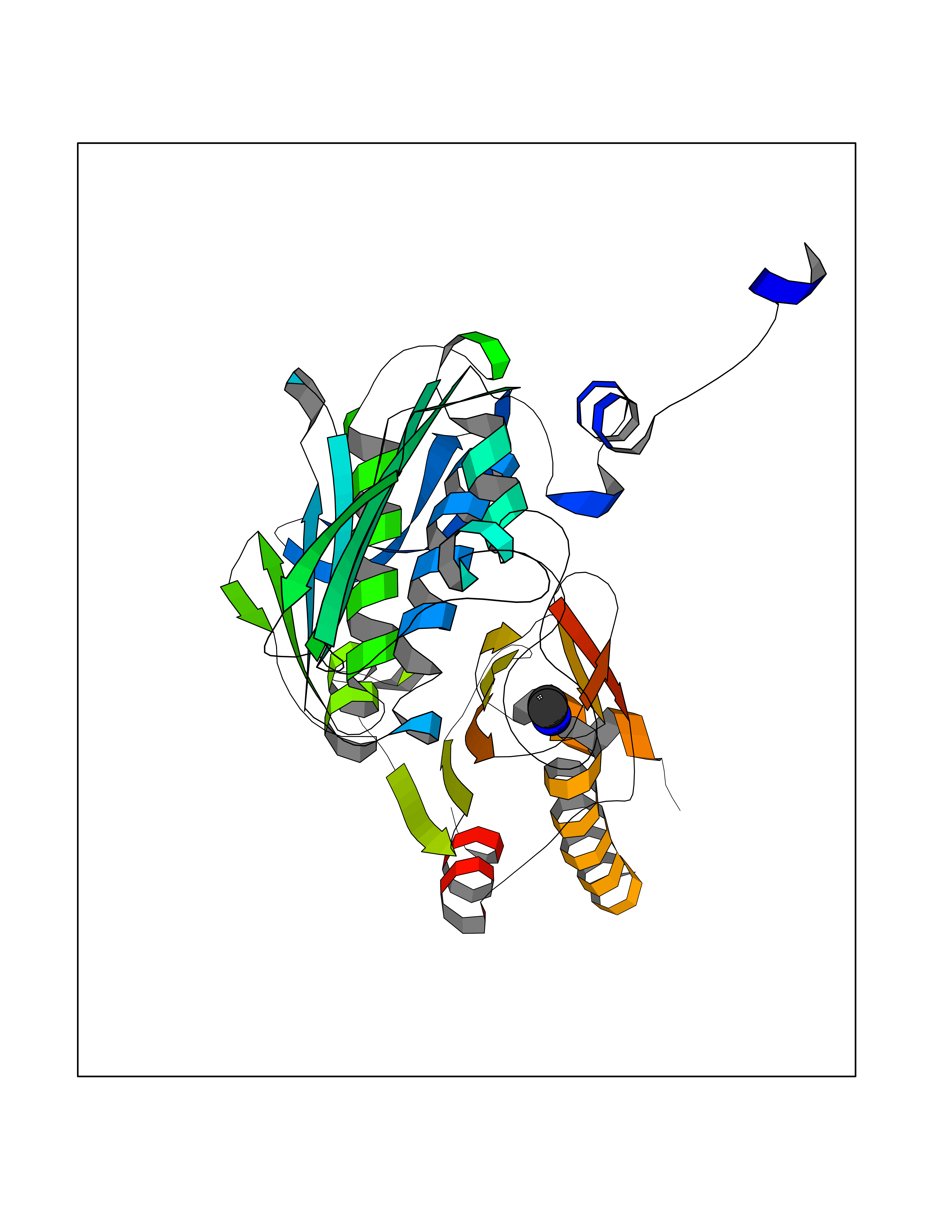 Molscript Map 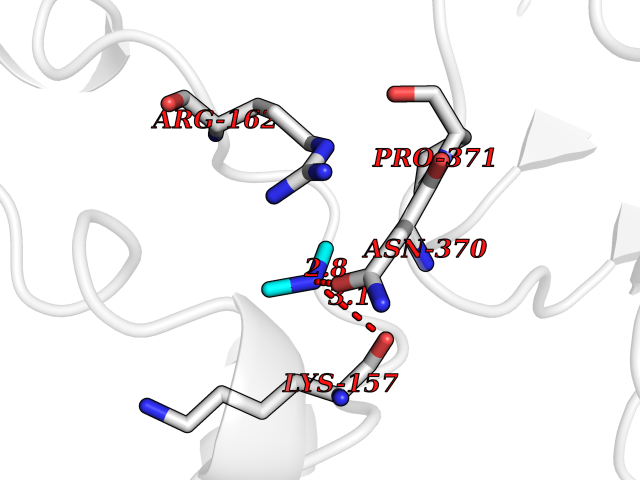 Pymol Map 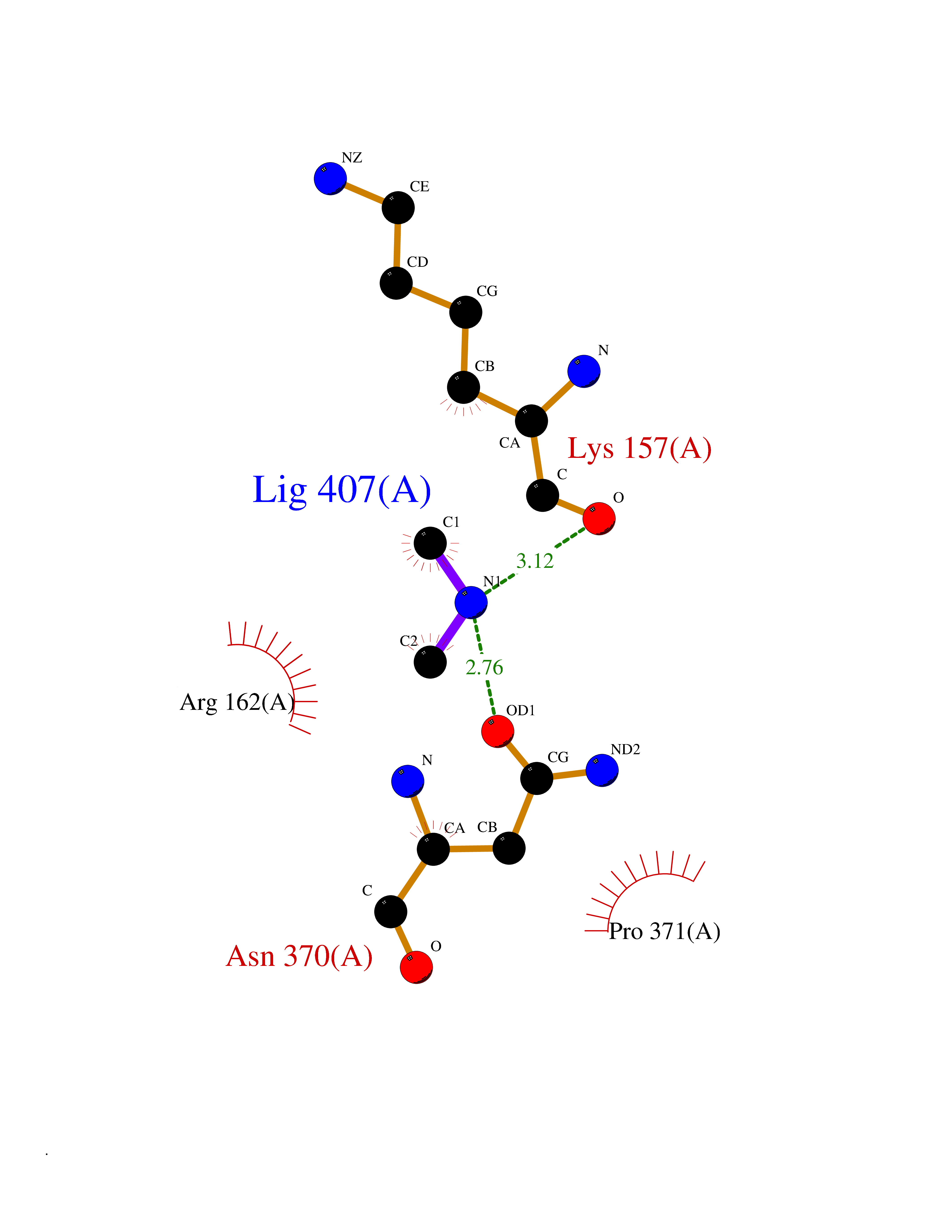 Ligplot Map 3D Binding mode Sequence SVERIYQKKTQLEHILLRPDTYIGSVELVTQQMWVYDEDVGINYREVTFVPGLYKIFDEILVNAADNKQRDPKMSCIRVTIDPENNLISIWNNGKGIPVVEHKVEKMYVPALIFGQLLTSSNYDDDEKKVTGGRNGYGAKLCNIFSTKFTVETASREYKKMFKQTWMDNMGRAGEMELKPFNGEDYTCITFQPDLSKFKMQSLDKDIVALMVRRAYDIAGSTKDVKVFLNGNKLPVKGFRSYVDMYLKDKLDETGNSLKVIHEQVNHRWEVCLTMSEKGFQQISFVNSIATSKGGRHVDYVADQIVTKLVDVVKKKNAVKAHQVKNHMWIFVNALIENPTFDSQTKENMTLQPKSFGSTCQLSEKFIKAAIGC Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | mRNA-capping enzyme | 2C46 | 4.01 | |
Target general information Gen name RNGTT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CAP1A Protein family Non-receptor class of the protein-tyrosine phosphatase family; Eukaryotic GTase family Biochemical class Transferase Function GTP binding.MRNA guanylyltransferase activity.Polynucleotide 5'-phosphatase activity.Protein tyrosine/serine/threonine phosphatase activity.Protein tyrosine phosphatase activity.RNA guanylyltransferase activity.Triphosphatase activity. Related diseases Atrial fibrillation, familial, 14 (ATFB14) [MIM:615378]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:19808477}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Genetic variations in SCN2B may be involved in Brugada syndrome (PubMed:23559163). This tachyarrhythmia is characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:23559163}. Drugs (DrugBank ID) NA Interacts with Q92624; P16333-1 EC number 2.7.7.50; 3.6.1.74 Uniprot keywords 3D-structure; Alternative splicing; GTP-binding; Host-virus interaction; Hydrolase; mRNA capping; mRNA processing; Multifunctional enzyme; Nucleotide-binding; Nucleotidyltransferase; Nucleus; Protein phosphatase; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 21849.8 Length 189 Aromaticity 0.11 Instability index 53.71 Isoelectric point 5.89 Charge (pH=7) -2.91 2D Binding mode Binding energy (Kcal/mol) -5.47 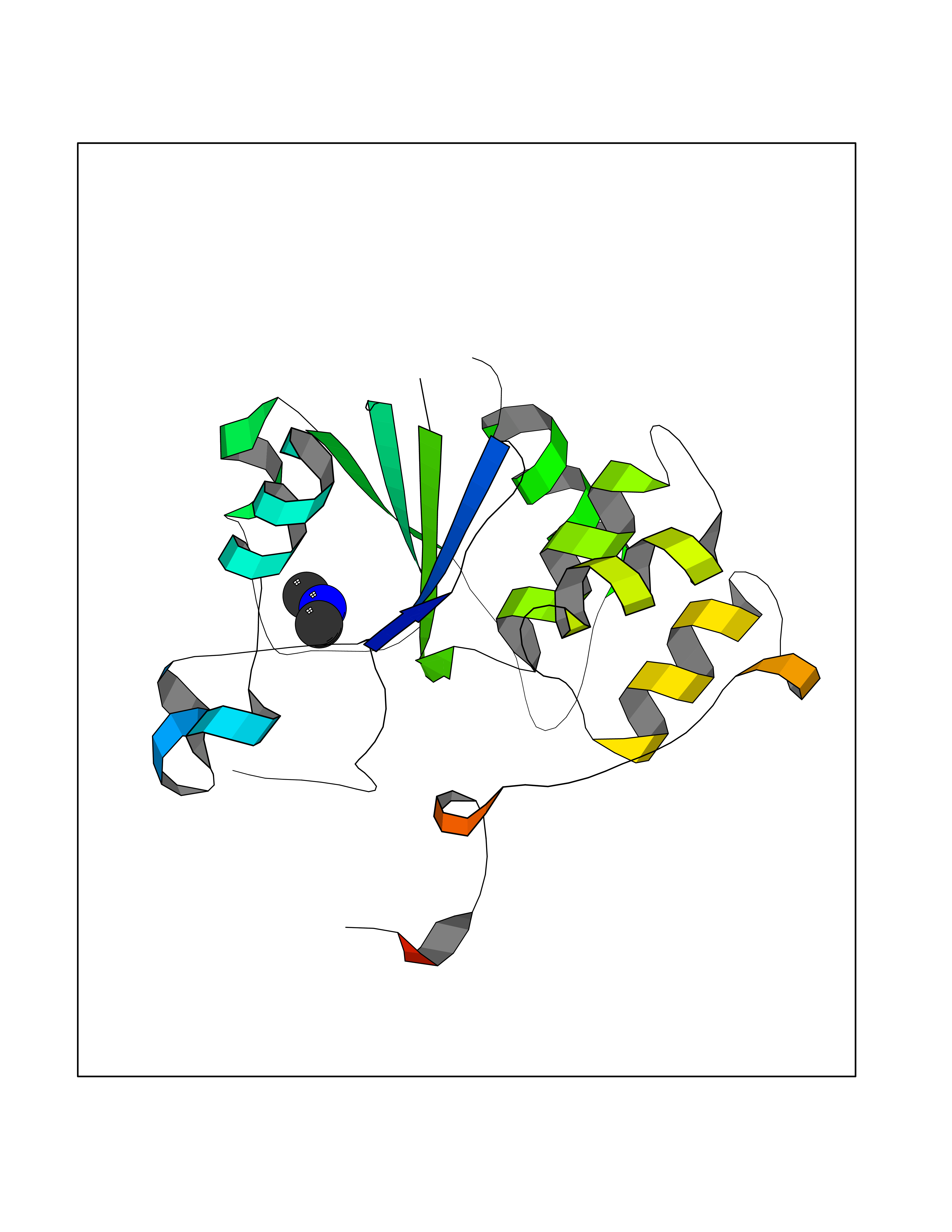 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NKIPPRWLNCPRRGQPVAGRFLPLKTMLGPRYDSQVAEENRFHPSMLSNYLKSVKMGLLVDLTNTSRFYDRNDIEKEGIKYIKLQCKGHGECPTTENTETFIRLCERFELIGVHCTHGFNRTGFLICAFLVEKMDWSIEAAVATFAQARPPGIYKGDYLKELFRRYGDIEEAPPPPLLPDWCFEDDEDE Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Isovaleryl-CoA dehydrogenase, mitochondrial | 1IVH | 4.01 | |
Target general information Gen name IVD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Isovaleryl-CoA dehydrogenase activity. Related diseases Isovaleric acidemia (IVA) [MIM:243500]: A metabolic disorder characterized by retarded psychomotor development, a peculiar odor resembling sweaty feet, an aversion to dietary protein, and pernicious vomiting, leading to acidosis and coma. The acute neonatal form leads to massive metabolic acidosis from the first days of life and rapid death. {ECO:0000269|PubMed:2063866, ECO:0000269|PubMed:22004070, ECO:0000269|PubMed:22350545, ECO:0000269|PubMed:23587913, ECO:0000269|PubMed:28535199, ECO:0000269|PubMed:9665741}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04036; DB03147 Interacts with Q08043; Q9Y4H4 EC number 1.3.8.1; 1.3.8.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; FAD; Fatty acid metabolism; Flavoprotein; Lipid metabolism; Mitochondrion; Oxidoreductase; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 84454.2 Length 774 Aromaticity 0.08 Instability index 30.01 Isoelectric point 6.85 Charge (pH=7) -0.77 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map  Pymol Map 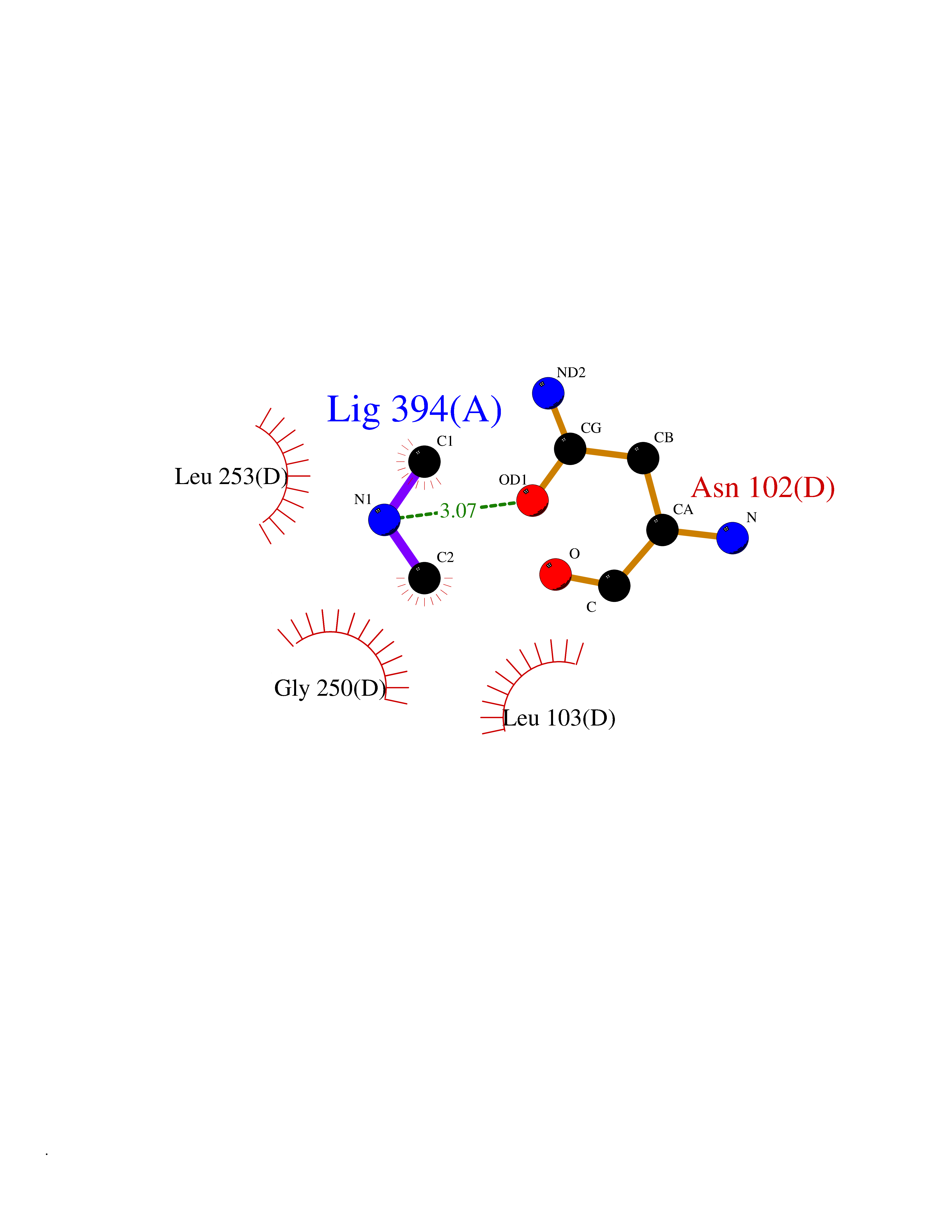 Ligplot Map 3D Binding mode Sequence VDDAINGLSEEQRQLRQTMAKFLQEHLAPKAQEIDRSNEFKNLREFWKQLGNLGVLGITAPVQYGGSGLGYLEHVLVMEEISRASGAVGLSYGAHSNLCINQLVRNGNEAQKEKYLPKLISGEYIGALAMSEPNAGSDVVSMKLKAEKKGNHYILNGNKFWITNGPDADVLIVYAKTDLAAVPASRGITAFIVEKGMPGFSTSKKLDKLGMRGSNTCELIFEDCKIPAANILGHENKGVYVLMSGLDLERLVLAGGPLGLMQAVLDHTIPYLHVREAFGQKIGHFQLMQGKMADMYTRLMACRQYVYNVAKACDEGHCTAKDCAGVILYSAECATQVALDGIQCFGGNGYINDFPMGRFLRDAKLYEIGAGTSEVRRLVIGRAFNADVDDAINGLSEEQRQLRQTMAKFLQEHLAPKAQEIDRSNEFKNLREFWKQLGNLGVLGITAPVQYGGSGLGYLEHVLVMEEISRASGAVGLSYGAHSNLCINQLVRNGNEAQKEKYLPKLISGEYIGALAMSEPNAGSDVVSMKLKAEKKGNHYILNGNKFWITNGPDADVLIVYAKTDLAAVPASRGITAFIVEKGMPGFSTSKKLDKLGMRGSNTCELIFEDCKIPAANILGHENKGVYVLMSGLDLERLVLAGGPLGLMQAVLDHTIPYLHVREAFGQKIGHFQLMQGKMADMYTRLMACRQYVYNVAKACDEGHCTAKDCAGVILYSAECATQVALDGIQCFGGNGYINDFPMGRFLRDAKLYEIGAGTSEVRRLVIGRAFNAD Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Phosphoribosylaminoimidazolecarboxamide formyltransferase (ATIC) | 1PKX | 4.01 | |
Target general information Gen name ATIC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PURH; OK/SW-cl.86; Bifunctional purine biosynthesis protein PURH Protein family PurH family Biochemical class Methyltransferase Function Bifunctional enzyme that catalyzes 2 steps in purine biosynthesis. Related diseases AICA-ribosuria due to ATIC deficiency (AICAR) [MIM:608688]: A neurologically devastating inborn error of purine biosynthesis. Patients excrete massive amounts of AICA-riboside in the urine and accumulate AICA-ribotide and its derivatives in erythrocytes and fibroblasts. Clinical features include profound intellectual disability, epilepsy, dysmorphic features and congenital blindness. AICAR inheritance is autosomal recessive. {ECO:0000269|PubMed:15114530}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02309; DB03442; DB01700; DB01972; DB00563; DB04057; DB00642; DB00116 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Epilepsy; Hydrolase; Intellectual disability; Multifunctional enzyme; Proteomics identification; Purine biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 41584.9 Length 386 Aromaticity 0.06 Instability index 42 Isoelectric point 5.6 Charge (pH=7) -7.51 2D Binding mode Binding energy (Kcal/mol) -5.47 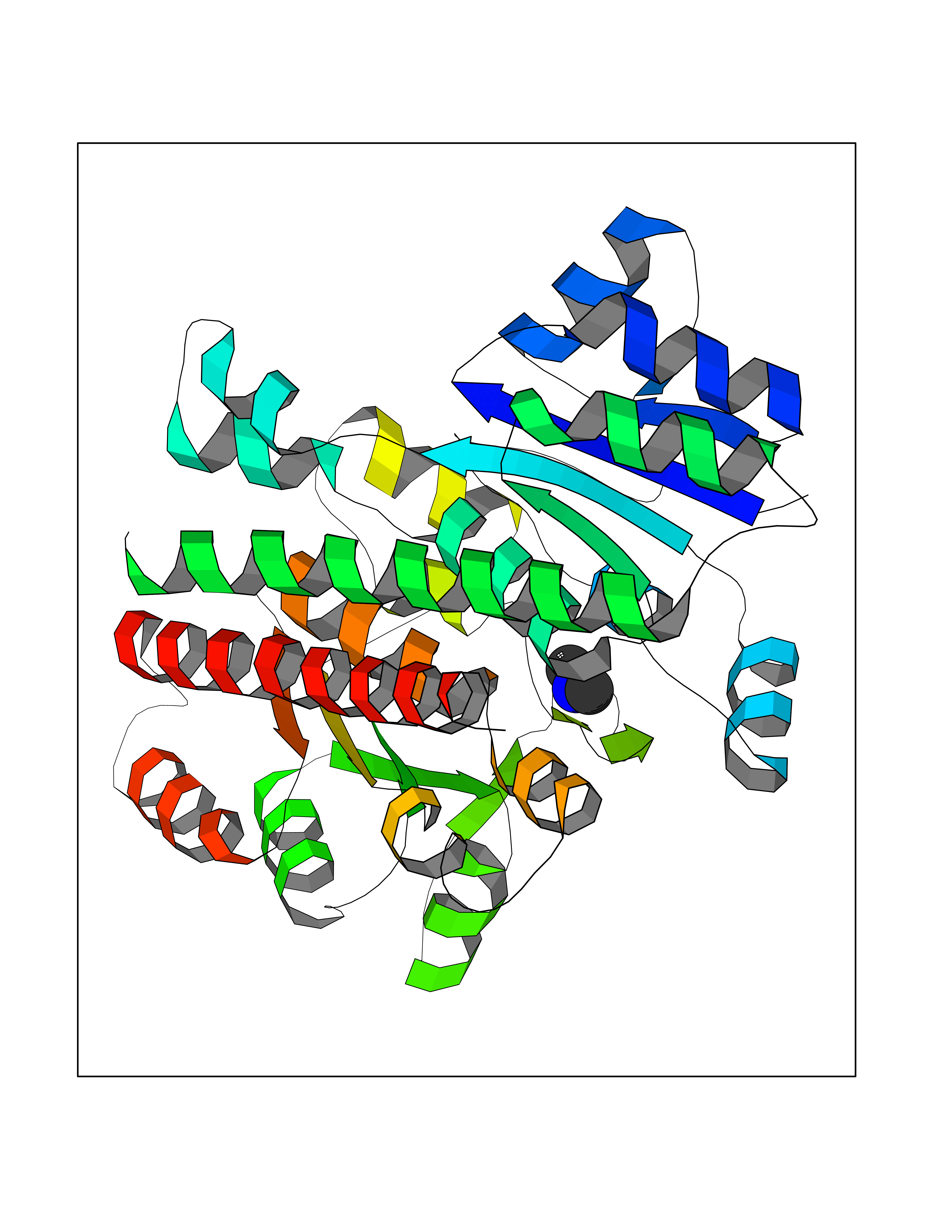 Molscript Map 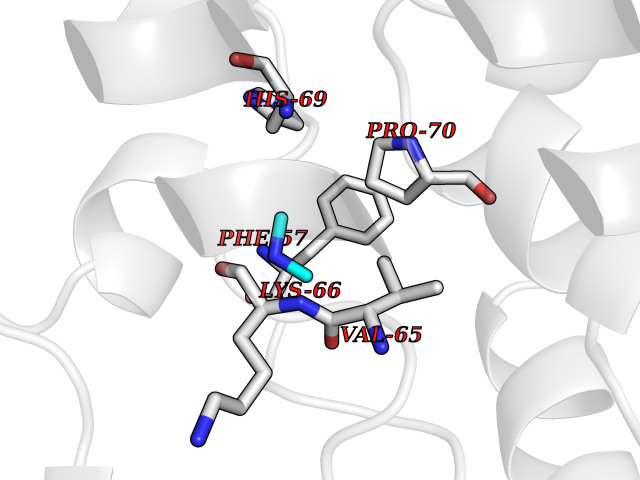 Pymol Map 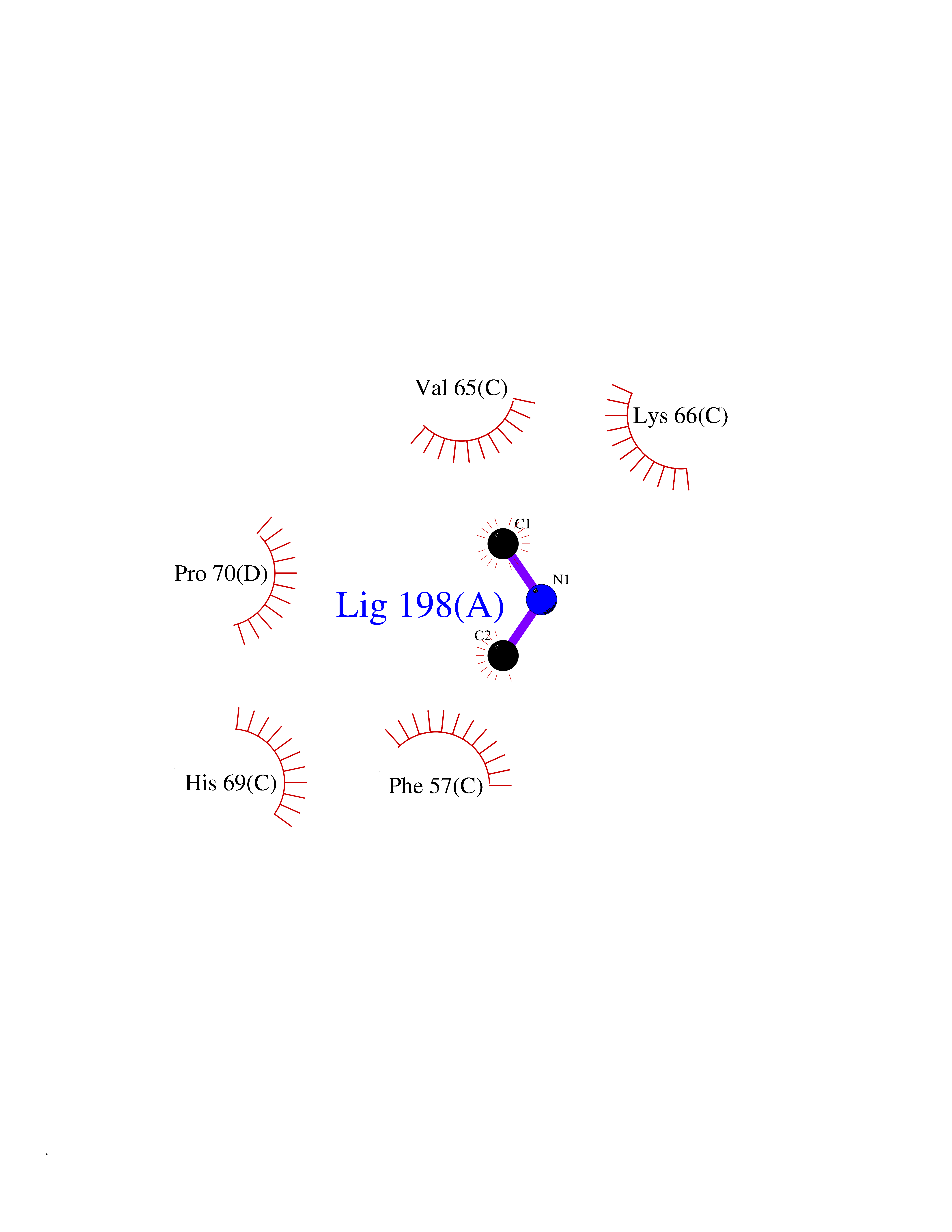 Ligplot Map 3D Binding mode Sequence GQLALFSVSDKTGLVEFARNLTALGLNLVASGGTAKALRDAGLAVRDVSELTGFPEMLGGRVKTLHPAVHAGILARNIPEDNADMARLDFNLIRVVACNLYPFVKTVASPGVTVEEAVEQIDIGGVTLLRAAAKNHARVTVVCEPEDYVVVSTEMQSSESKDTSLETRRQLALKAFTHTAQYDEAISDYFRKQGQLALFSVSDKTGLVEFARNLTALGLNLVASGGTAKALRDAGLAVRDVSELTGFPEMLGGRVKTLHPAVHAGILARNIPEDNADMARLDFNLIRVVACNLYPFVKTVASPGVTVEEAVEQIDIGGVTLLRAAAKNHARVTVVCEPEDYVVVSTEMQSSESKDTSLETRRQLALKAFTHTAQYDEAISDYFRKQ Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Aminoacylase-1 | 1Q7L | 4.01 | |
Target general information Gen name ACY1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase M20A family Biochemical class Hydrolase Function Aminoacylase activity.Identical protein binding.Metal ion binding.Metallopeptidase activity. Related diseases Aminoacylase-1 deficiency (ACY1D) [MIM:609924]: An enzymatic deficiency resulting in encephalopathy, unspecific psychomotor delay, psychomotor delay with atrophy of the vermis and syringomyelia, marked muscular hypotonia or normal clinical features. Epileptic seizures are a frequent feature. All affected individuals exhibit markedly increased urinary excretion of several N-acetylated amino acids. {ECO:0000269|PubMed:16274666, ECO:0000269|PubMed:16465618, ECO:0000269|PubMed:17562838, ECO:0000269|PubMed:21414403}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06151; DB00128; DB09130 Interacts with Q03154; O75934; Q96HA8; P36639; P36639-2; Q8TCT1; P0CG20; Q96A09; P54274; O43711; Q9UPN9; Q9NZC7-5 EC number 3.5.1.14 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,C Molecular weight (Da) 31172.2 Length 275 Aromaticity 0.11 Instability index 36.46 Isoelectric point 6 Charge (pH=7) -5.26 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NPWWAAFSRVCKDMNLTLEPEIMPAAGDNRYIRAVGVPALGFSPMNRTPVLLHDHDERLHEAVFLRGVDIYTRLLPALASVPALPEEHPSVTLFRQYLRIRTVQPKPDYGAAVAFFEETARQLGLGCQKVEVAPGYVVTVLTWPGTNPTLSSILLNSHTDVVPVFKEHWSHDPFEAFKDSEGYIYARGAQDMKCVSIQYLEAVRRLKVEGHRFPRTIHMTFVPDEEVGGHQGMELFVQRPEFHALRAGFALDEGIANPTDAFTVFYSERSPWWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Kynureninase (KYNU) | 3E9K | 4.01 | |
Target general information Gen name KYNU Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms L-kynurenine hydrolase; KYNU Protein family Kynureninase family Biochemical class Carbon-carbon bonds hydrolase Function Catalyzes the cleavage of L-kynurenine (L-Kyn) and L-3- hydroxykynurenine (L-3OHKyn) into anthranilic acid (AA) and 3- hydroxyanthranilic acid (3-OHAA), respectively. Has a preference for the L-3-hydroxy form. Also has cysteine-conjugate-beta-lyase activity. Related diseases Hydroxykynureninuria (HYXKY) [MIM:236800]: An inborn error of amino acid metabolism characterized by massive urinary excretion of large amounts of kynurenine, 3-hydroxykynurenine and xanthurenic acid. Affected individuals manifest renal tubular dysfunction, metabolic acidosis, psychomotor retardation, non-progressive encephalopathy, and muscular hypertonia. {ECO:0000269|PubMed:17334708, ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Vertebral, cardiac, renal, and limb defects syndrome 2 (VCRL2) [MIM:617661]: An autosomal recessive congenital malformation syndrome characterized by vertebral segmentation abnormalities, congenital cardiac defects, renal defects, and distal mild limb defects. {ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00160; DB07069; DB00114 Interacts with Q8WUE5; P56545-3; Q9NVL1-2; P61968; P59942; Q8TDC0; P78356-2; Q86WH2 EC number EC 3.7.1.3 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Disease variant; Hydrolase; Proteomics identification; Pyridine nucleotide biosynthesis; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 50204.5 Length 446 Aromaticity 0.09 Instability index 39.91 Isoelectric point 6.36 Charge (pH=7) -4.59 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 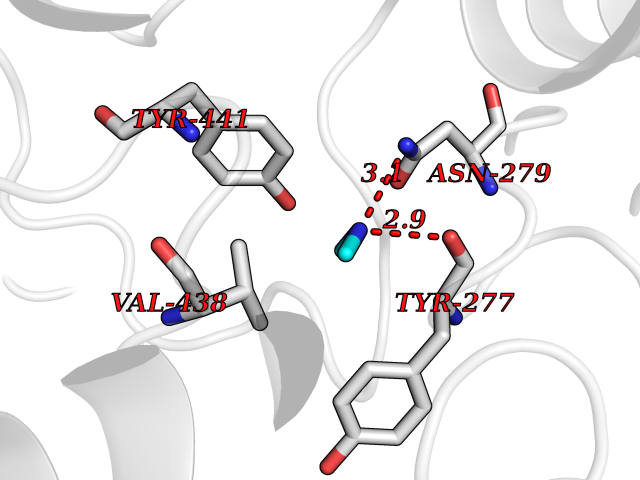 Pymol Map 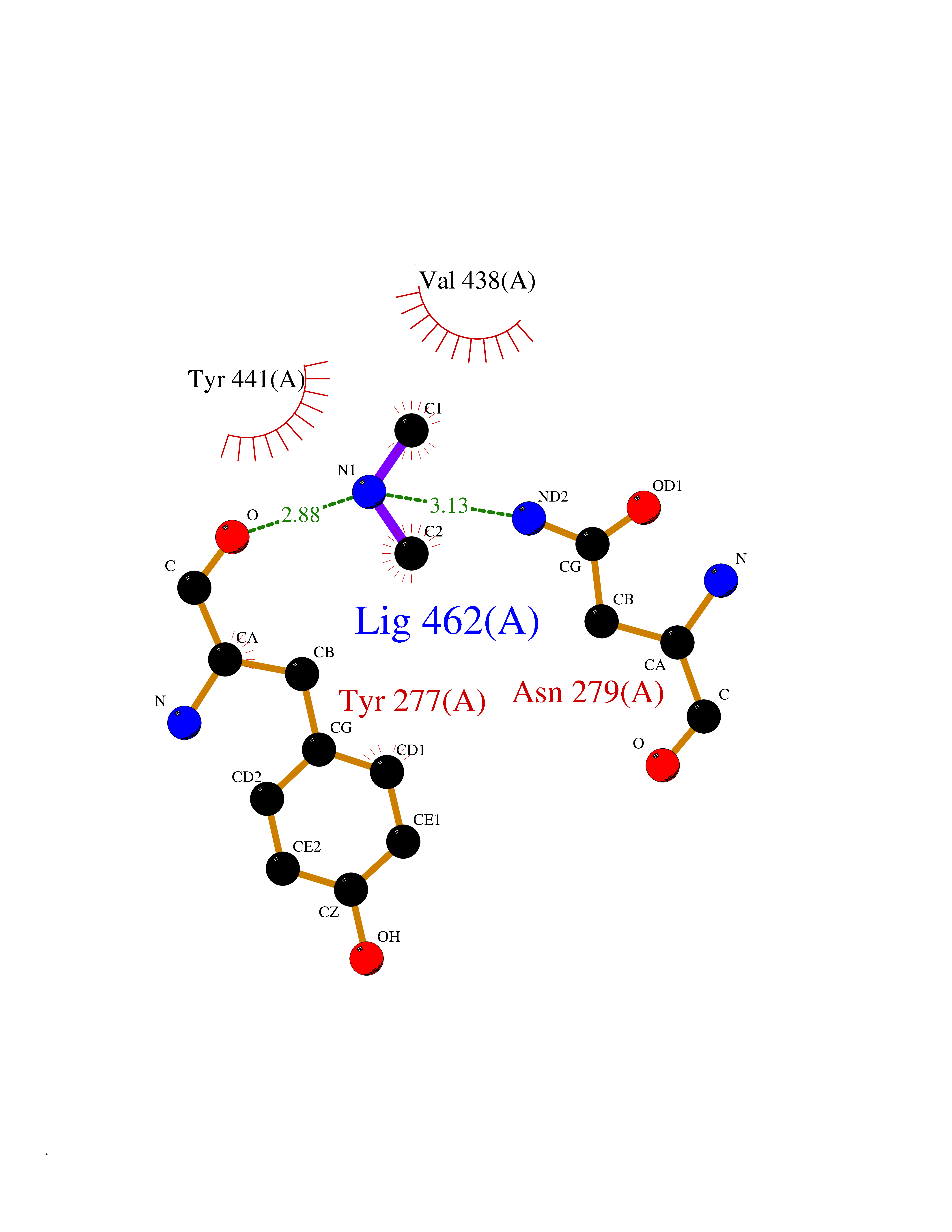 Ligplot Map 3D Binding mode Sequence LELPADTVQRIAAELKCHPTDERVALHLDEEDKLRHFREXFYIPKIQDLPPVDLSLVNKDENAIYFLGNSLGLQPKMVKTYLEEELDKWAKIAAYGHEVGKRPWITGDESIVGLMKDIVGANEKEIALMNALTVNLHLLMLSFFKPTPKRYKILLEAKAFPSDHYAIESQLQLHGLNIEESMRMIKPREGEETLRIEDILEVIEKEGDSIAVILFSGVHFYTGQHFNIPAITKAGQAKGCYVGFDLAHAVGNVELYLHDWGVDFACWCSYKYLNAGAGGIAGAFIHEKHAHTIKPALVGWFGHELSTRFKMDNKLQLIPGVCGFRISNPPILLVCSLHASLEIFKQATMKALRKKSVLLTGYLEYLIKHNYGVVNIITPSHVEERGCQLTITFSVPNKDVFQELEKRGVVCDKRNPNGIRVAPVPLYNSFHDVYKFTNLLTSILDS Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Lanosterol 14-alpha-demethylase (EC 1.14.13.70) | 4G3J | 4.01 | |
Target general information Gen name Tb11.02.4080 Organism Trypanosoma brucei brucei (strain 927/4 GUTat10.1) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class NA Function NA Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) NA Interacts with NA EC number 1.14.13.70 Uniprot keywords 3D-structure; Heme; Iron; Metal-binding; Monooxygenase; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID D Molecular weight (Da) 50691.4 Length 448 Aromaticity 0.08 Instability index 54.09 Isoelectric point 6.99 Charge (pH=7) -0.03 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 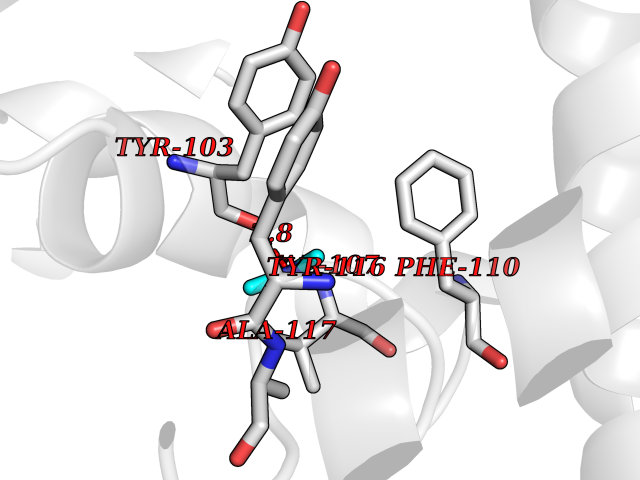 Pymol Map 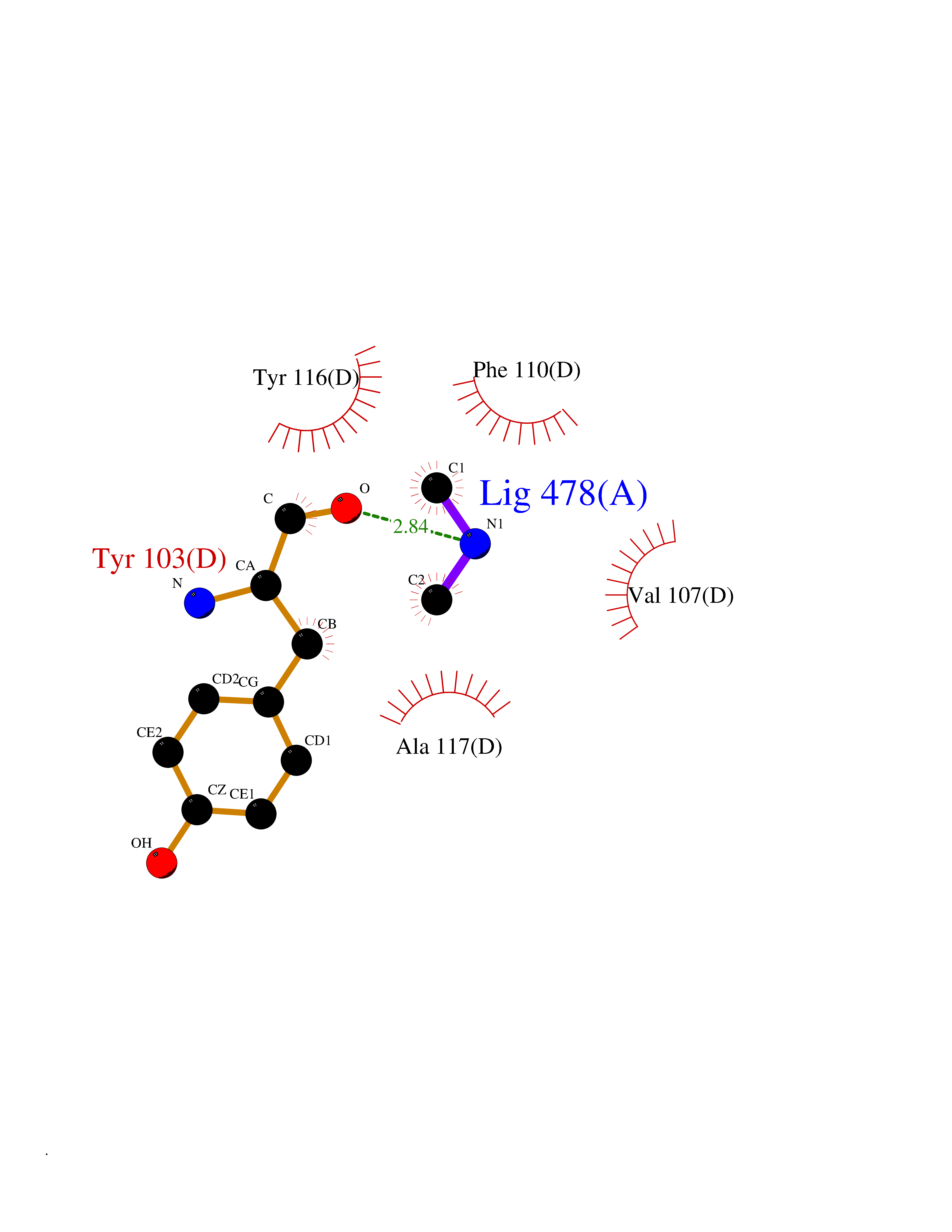 Ligplot Map 3D Binding mode Sequence GKLPPVYPVTVPILGHIIQFGKSPLGFMQECKRQLKSGIFTINIVGKRVTIVGDPHEHSRFFLPRNEVLSPREVYSFMVPVFGEGVAYAAPYPRMREQLNFLAEELTIAKFQNFVPAIQHEVRKFMAANWDKDEGEINLLEDCSTMIINTACQCLFGEDLRKRLDARRFAQLLAKMESSLIPAAVFLPILLKLPLPQSARCHEARTELQKILSEIIIARKEEEVNKDSSTSDLLSGLLSAVYRDGTPMSLHEVCGMIVAAMFAGQHTSSITTTWSMLHLMHPANVKHLEALRKEIEEFPAQLNYNNVMDEMPFAERCARESIRRDPPLLMLMRKVMADVKVGSYVVPKGDIIACSPLLSHHDEEAFPEPRRWDPERDEKVEGAFIGFGAGVHKCIGQKFGLLQVKTILATAFRSYDFQLLRDEVPDPDYHTMVVGPTASQCRVKYIRR Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Alr1529 protein | 1Z8H | 4.01 | |
Target general information Gen name alr1529 Organism Nostoc sp. (strain PCC 7120 / SAG 25.82 / UTEX 2576) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function NA Related diseases Desbuquois dysplasia 1 (DBQD1) [MIM:251450]: A chondrodysplasia characterized by severe prenatal and postnatal growth retardation (less than -5 SD), joint laxity, short extremities, progressive scoliosis, round face, midface hypoplasia, prominent bulging eyes. The main radiologic features are short long bones with metaphyseal splay, a 'Swedish key' appearance of the proximal femur (exaggerated trochanter), and advance carpal and tarsal bone age. Two forms of Desbuquois dysplasia are distinguished on the basis of the presence or absence of characteristic hand anomalies: an extra ossification center distal to the second metacarpal, delta phalanx, bifid distal thumb phalanx, and phalangeal dislocations. {ECO:0000269|PubMed:19853239, ECO:0000269|PubMed:20425819, ECO:0000269|PubMed:21037275, ECO:0000269|PubMed:21412251, ECO:0000269|PubMed:21654728, ECO:0000269|PubMed:22539336}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epiphyseal dysplasia, multiple, 7 (EDM7) [MIM:617719]: A form of multiple epiphyseal dysplasia, a generalized skeletal dysplasia associated with significant morbidity. Joint pain, joint deformity, waddling gait, and short stature are the main clinical signs and symptoms. Radiological examination of the skeleton shows delayed, irregular mineralization of the epiphyseal ossification centers and of the centers of the carpal and tarsal bones. Multiple epiphyseal dysplasia is broadly categorized into the more severe Fairbank and the milder Ribbing types. The Fairbank type is characterized by shortness of stature, short and stubby fingers, small epiphyses in several joints, including the knee, ankle, hand, and hip. The Ribbing type is confined predominantly to the hip joints and is characterized by hands that are normal and stature that is normal or near-normal. EDM7 inheritance is autosomal recessive. {ECO:0000269|PubMed:28742282}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 45603.2 Length 401 Aromaticity 0.11 Instability index 49.26 Isoelectric point 7.67 Charge (pH=7) 1.26 2D Binding mode Binding energy (Kcal/mol) -5.46 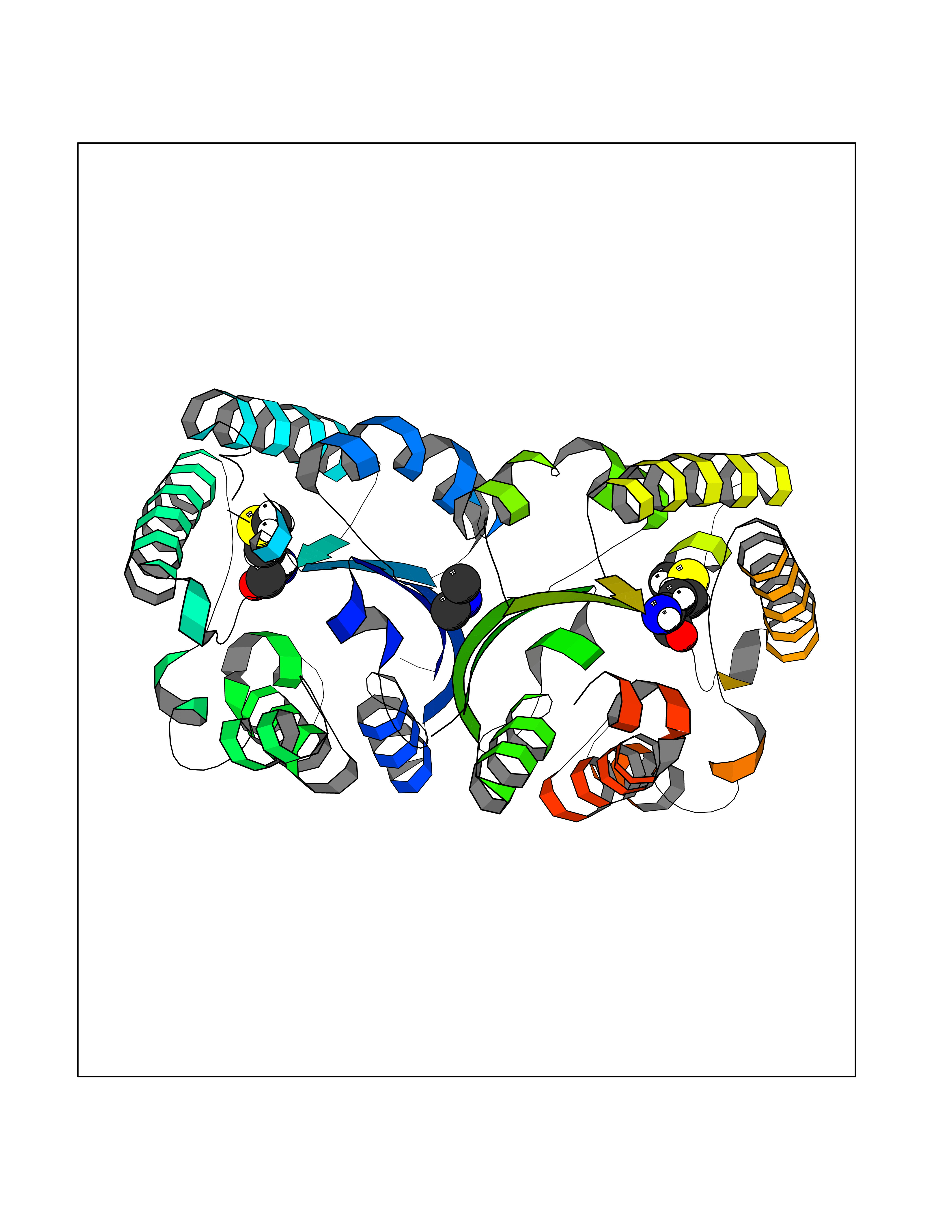 Molscript Map  Pymol Map 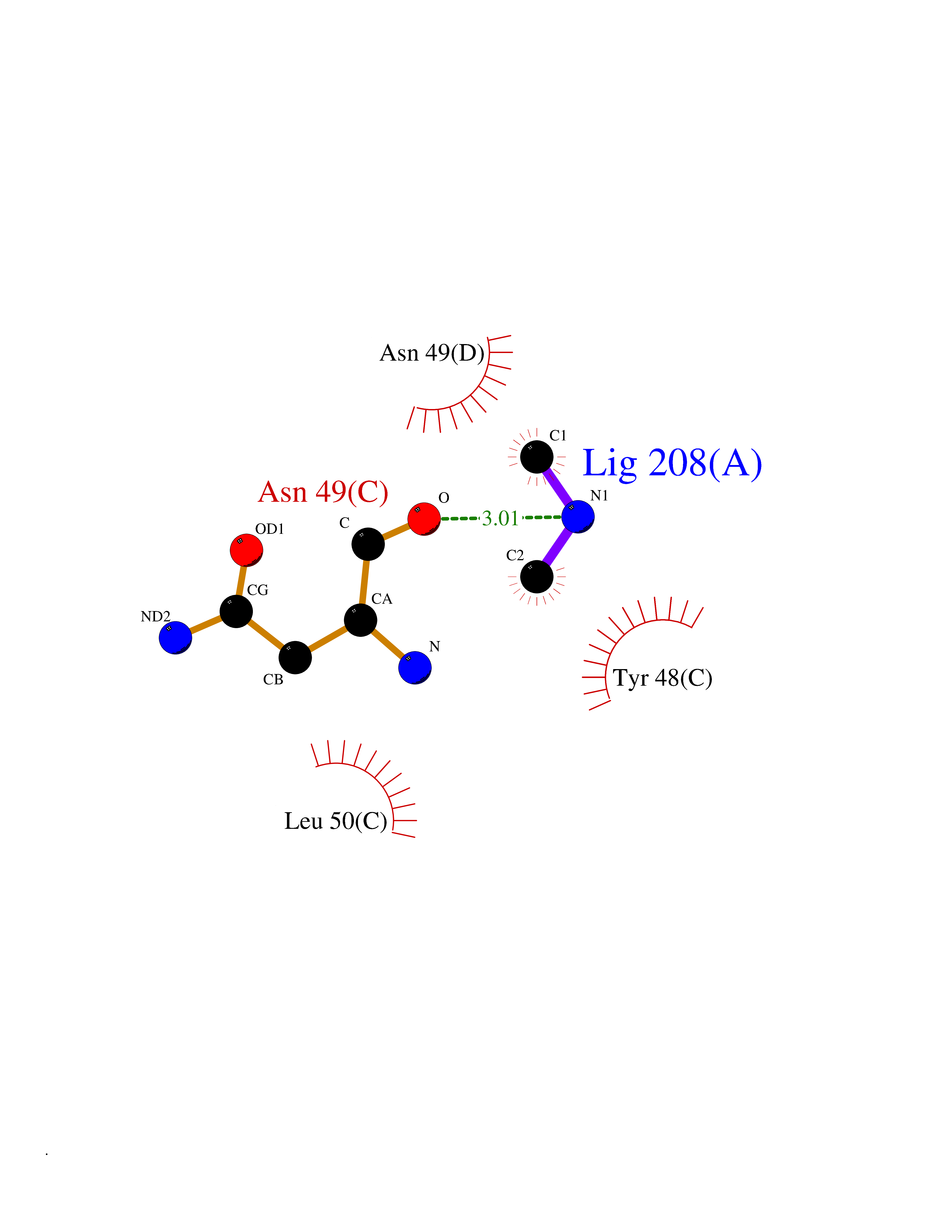 Ligplot Map 3D Binding mode Sequence KTQIRICFVGDSFVNGTGDPECLGWTGRVCVNANKKGYDVTYYNLGIRRDTSSDIAKRWLQEVSLRLHKEYNSLVVFSFGLNDTTLENGKPRVSIAETIKNTREILTQAKKLYPVLXISPAPYIEQQDPGRRRRTIDLSQQLALVCQDLDVPYLDVFPLLEKPSVWLHEAKANDGVHPQAGGYTEFARIVENWDAWLNWFRKTQIRICFVGDSFVNGTGDPECLGWTGRVCVNANKKGYDVTYYNLGIRRDTSSDIAKRWLQEVSLRLHKEYNSLVVFSFGLNDTTLENGKPRVSIAETIKNTREILTQAKKLYPVLXISPAPYIEQQDPGRRRRTIDLSQQLALVCQDLDVPYLDVFPLLEKPSVWLHEAKANDGVHPQAGGYTEFARIVENWDAWLNWF Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Protein cereblon (CRBN) | 5FQD | 4.01 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Peroxisomal trans-2-enoyl-CoA reductase | 1YXM | 4.01 | |
Target general information Gen name PECR Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PRO1004;SDR29C1 Protein family Short-chain dehydrogenases/reductases (SDR) family Biochemical class Oxidoreductase Function Receptor binding.Trans-2-enoyl-CoA reductase (NADPH) activity. Related diseases Cornelia de Lange syndrome 5 (CDLS5) [MIM:300882]: A form of Cornelia de Lange syndrome, a clinically heterogeneous developmental disorder associated with malformations affecting multiple systems. It is characterized by facial dysmorphisms, abnormal hands and feet, growth delay, cognitive retardation, hirsutism, gastroesophageal dysfunction and cardiac, ophthalmologic and genitourinary anomalies. {ECO:0000269|PubMed:22885700, ECO:0000269|PubMed:22889856}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00173 Interacts with NA EC number 1.3.1.38 Uniprot keywords 3D-structure; Alternative splicing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 30349.5 Length 283 Aromaticity 0.08 Instability index 38.34 Isoelectric point 8.89 Charge (pH=7) 5.26 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 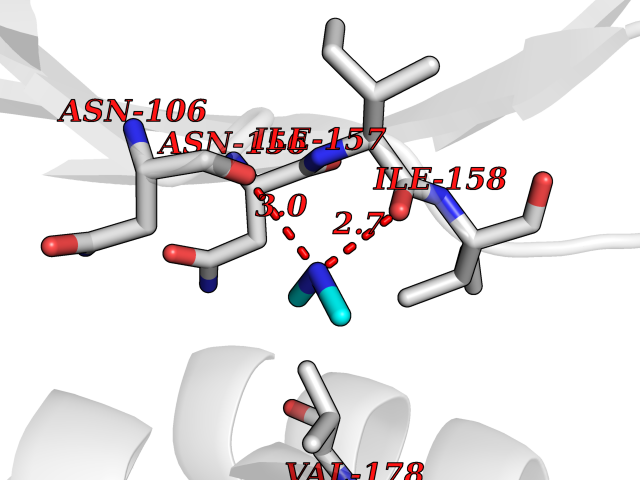 Pymol Map 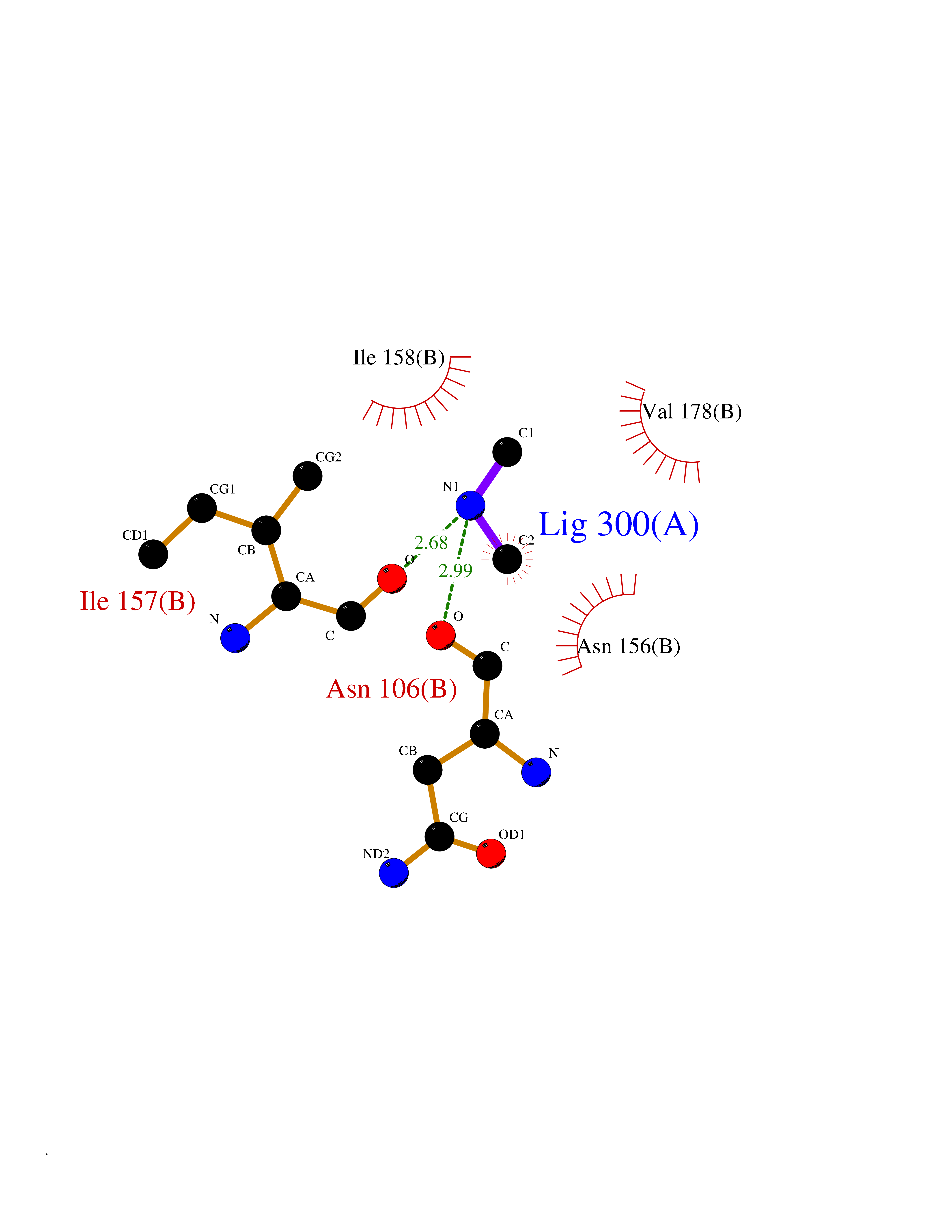 Ligplot Map 3D Binding mode Sequence RSYLAPGLLQGQVAIVTGGATGIGKAIVKELLELGSNVVIASRKLERLKSAADELQANLPPTKQARVIPIQCNIRNEEEVNNLVKSTLDTFGKINFLVNNGGGQFLSPAEHISSKGWHAVLETNLTGTFYMCKAVYSSWMKEHGGSIVNIIVPTKAGFPLAVHSGAARAGVYNLTKSLALEWACSGIRINCVAPGVIYSQTAQSFFEGSFQKIPAKRIGVPEEVSSVVCFLLSPAASFITGQSVDVDGGRSLYTHSYEVPDHDNWPKGAGDLSVVKKMKETFK Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Neutral endopeptidase (MME) | 6XVP | 4.01 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Skin fibroblast elastase; SFE; Neutral endopeptidase 24.11; Neprilysin; NEP protein; Enkephalinase; EPN; Common acute lymphocytic leukemia antigen; CD10; CALLA; Atriopeptidase Protein family Peptidase M13 family Biochemical class Peptidase Function Biologically important in the destruction of opioid peptides such as Met- and Leu-enkephalins by cleavage of a Gly-Phe bond. Able to cleave angiotensin-1, angiotensin-2 and angiotensin 1-9. Involved in the degradation of atrial natriuretic factor (ANF). Displays UV-inducible elastase activity toward skin preelastic and elastic fibers. Thermolysin-like specificity, but is almost confined on acting on polypeptides of up to 30 amino acids. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number EC 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -5.47 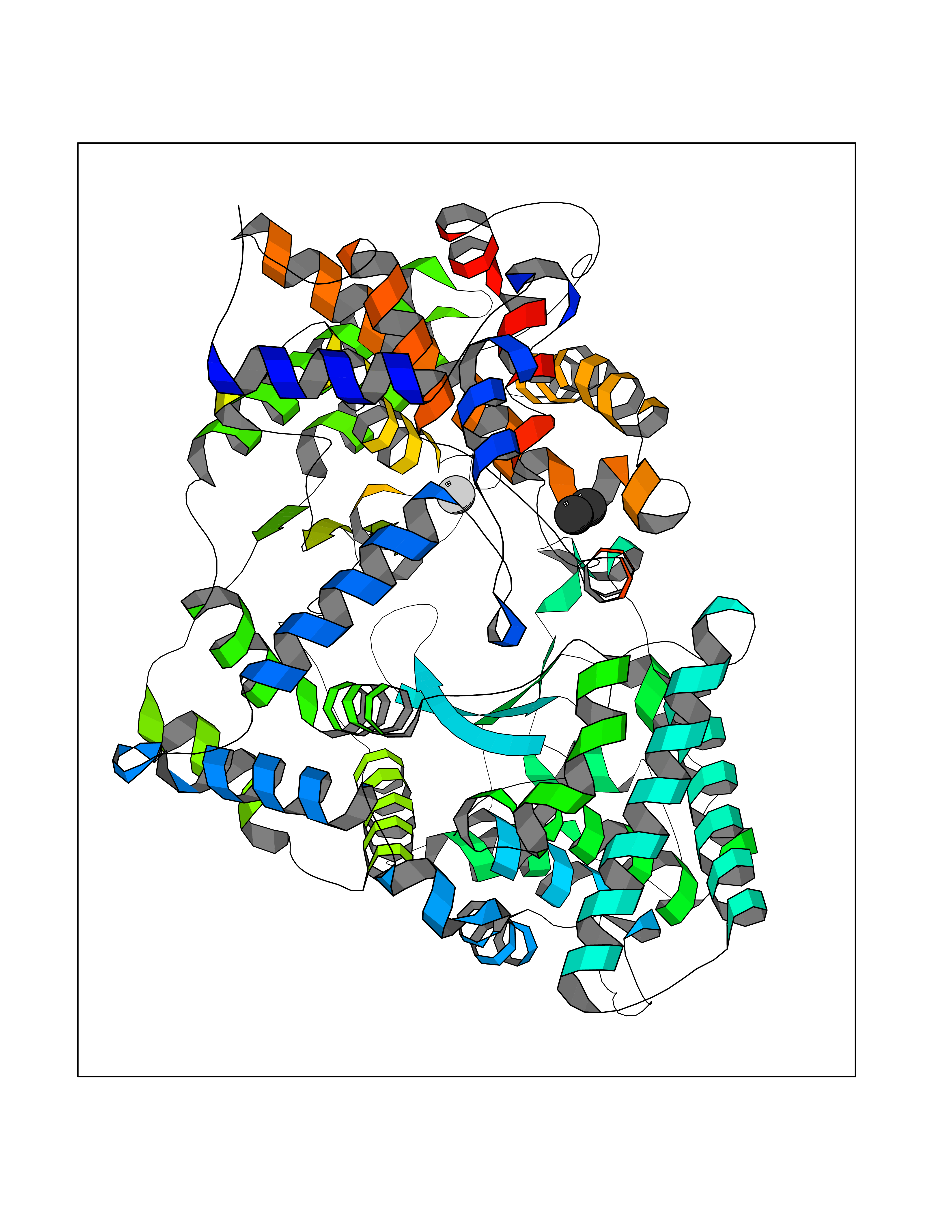 Molscript Map 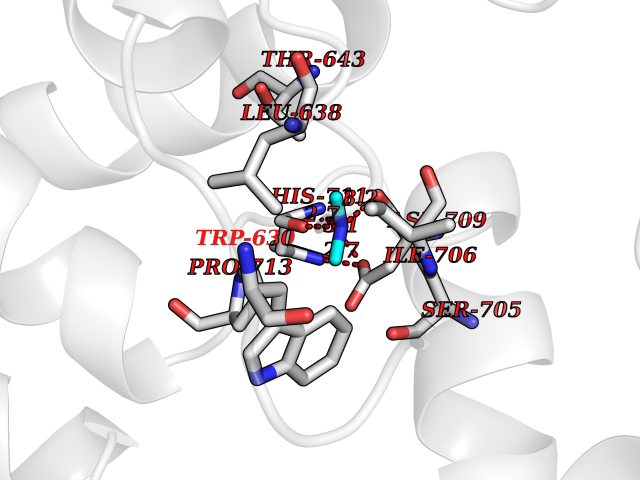 Pymol Map 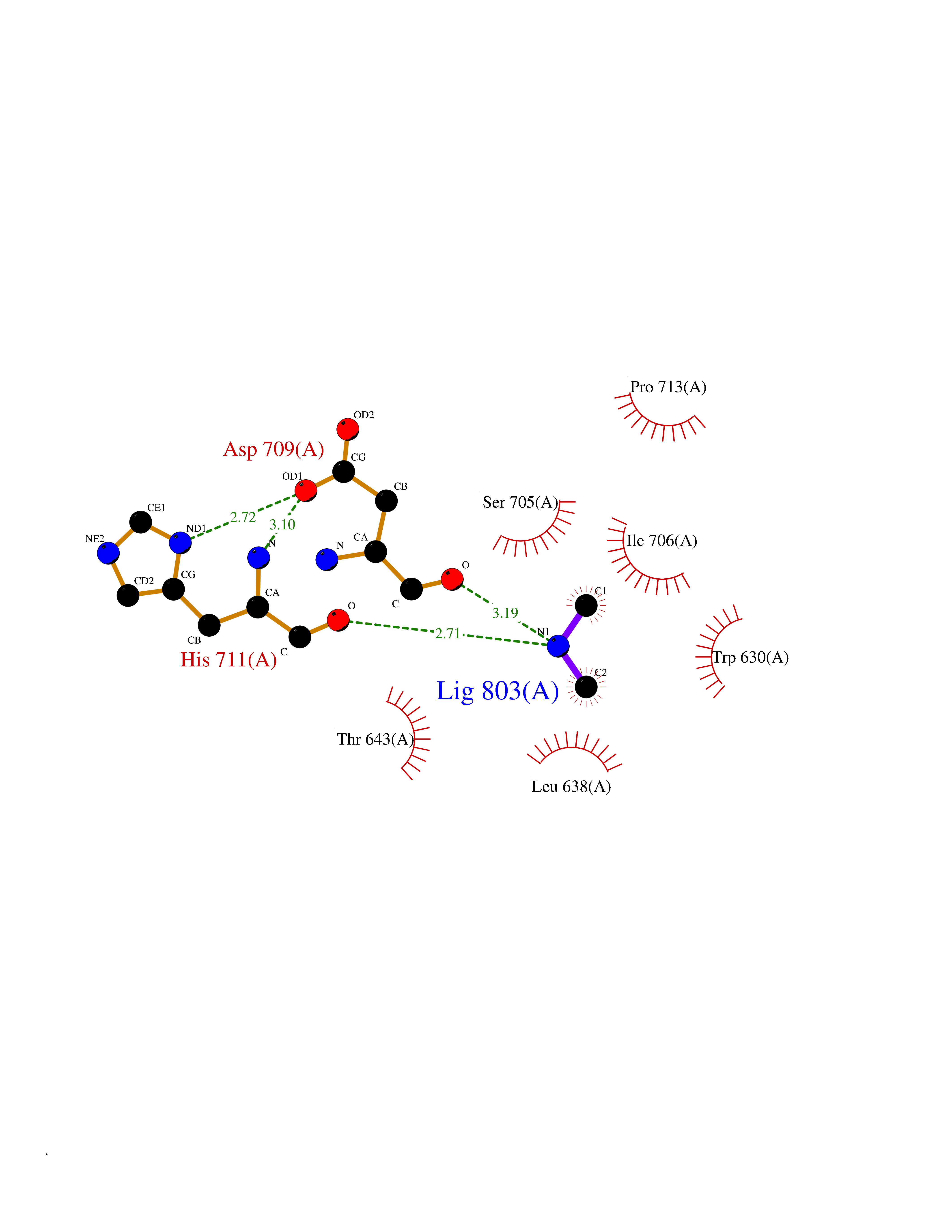 Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Pseudomonas Methionine gamma-lyase (Pseudo mdeA) | 1PG8 | 4.01 | |
Target general information Gen name Pseudo mdeA Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID Synonyms Pseudo MGL; L-methionine gamma-lyase; L-methioninase; Homocysteine desulfhydrase Protein family Trans-sulfuration enzymes family, L-methionine gamma-lyase subfamily Biochemical class Carbon-sulfur lyases Function Catalyzes the alpha,gamma-elimination of L-methionine to produce methanethiol, 2-oxobutanoate and ammonia. Is involved in L-methionine catabolism. In fact, shows a multicatalytic function since it also catalyzes gamma-replacement of L-methionine with thiol compounds, alpha,gamma-elimination and gamma-replacement reactions of L-homocysteine and its S-substituted derivatives, O-substituted-L-homoserines and DL-selenomethionine, and, to a lesser extent, alpha,beta-elimination and beta-replacement reactions of L-cysteine, S-methyl-L-cysteine, and O-acetyl-L-serine. Also catalyzes deamination and gamma-addition reactions of L-vinylglycine. Thus, the enzyme is able to cleave C-S, C-Se, and C-O bonds of sulfur, selenium, and oxygen amino acids, respectively. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04083 Interacts with NA EC number EC 4.4.1.11 Uniprot keywords 3D-structure; Direct protein sequencing; Lyase; Pyridoxal phosphate Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85234.4 Length 796 Aromaticity 0.07 Instability index 37.1 Isoelectric point 6.21 Charge (pH=7) -11.34 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map  Pymol Map 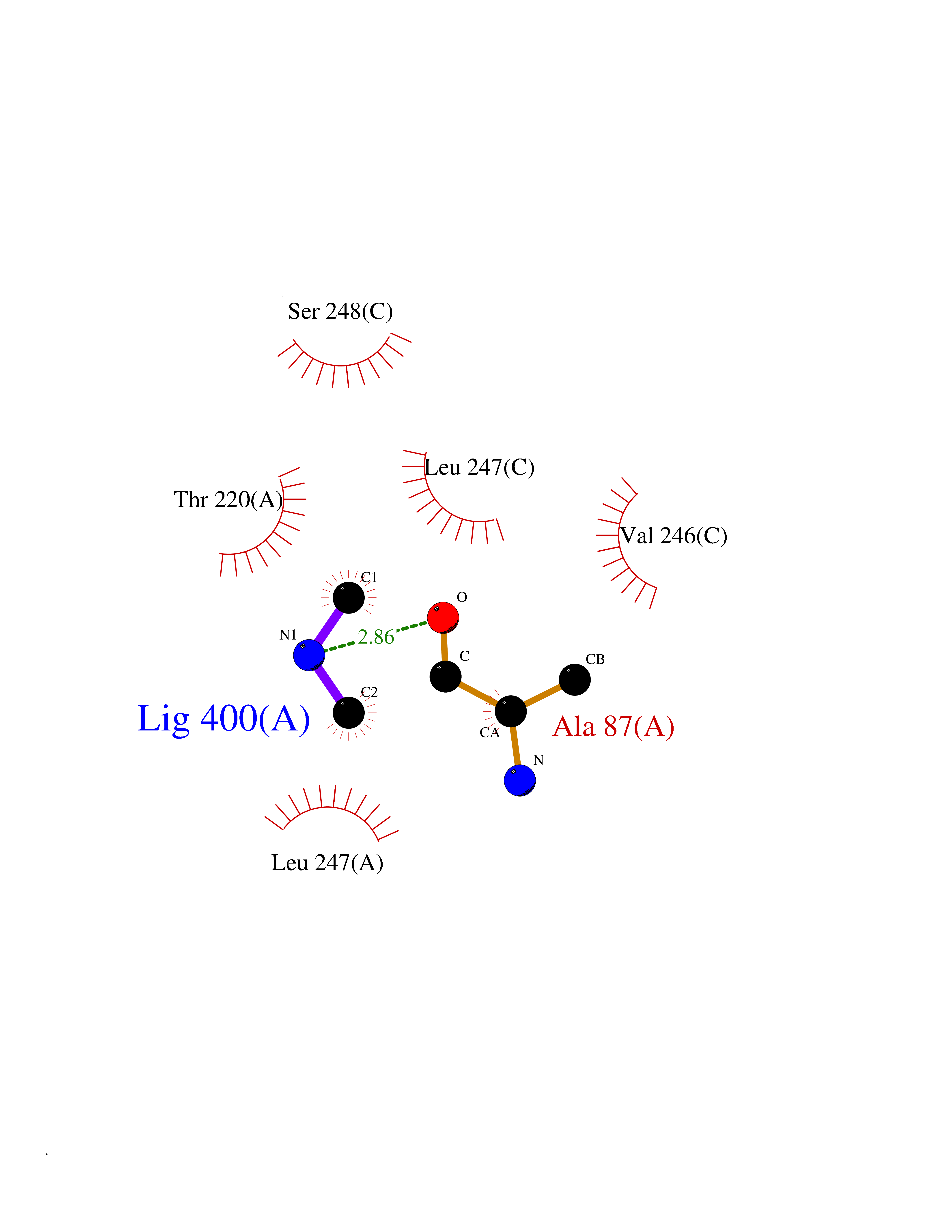 Ligplot Map 3D Binding mode Sequence MHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASAMHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASA Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Orotidine 5'-monophosphate decarboxylase (UMPS) | 3MI2 | 4.01 | |
Target general information Gen name UMPS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Uridine 5'-monophosphate synthase; UMP synthase Protein family Purine/pyrimidine phosphoribosyltransferase family; OMP decarboxylase family Biochemical class Pentosyltransferase Function Catalyses the formation of uridine monophosphate (UMP), an energy-carrying molecule in many important biosynthetic pathways. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02890; DB00544 Interacts with P54764; P11172-1 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Decarboxylase; Disease variant; Glycosyltransferase; Lyase; Multifunctional enzyme; Phosphoprotein; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 55844 Length 514 Aromaticity 0.06 Instability index 22.7 Isoelectric point 6.44 Charge (pH=7) -2.99 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 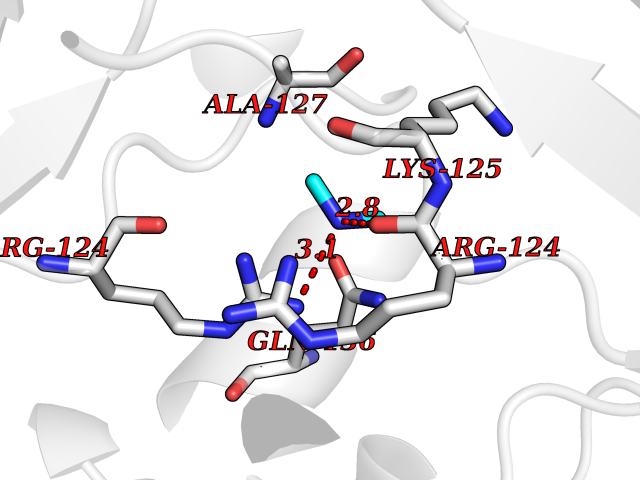 Pymol Map 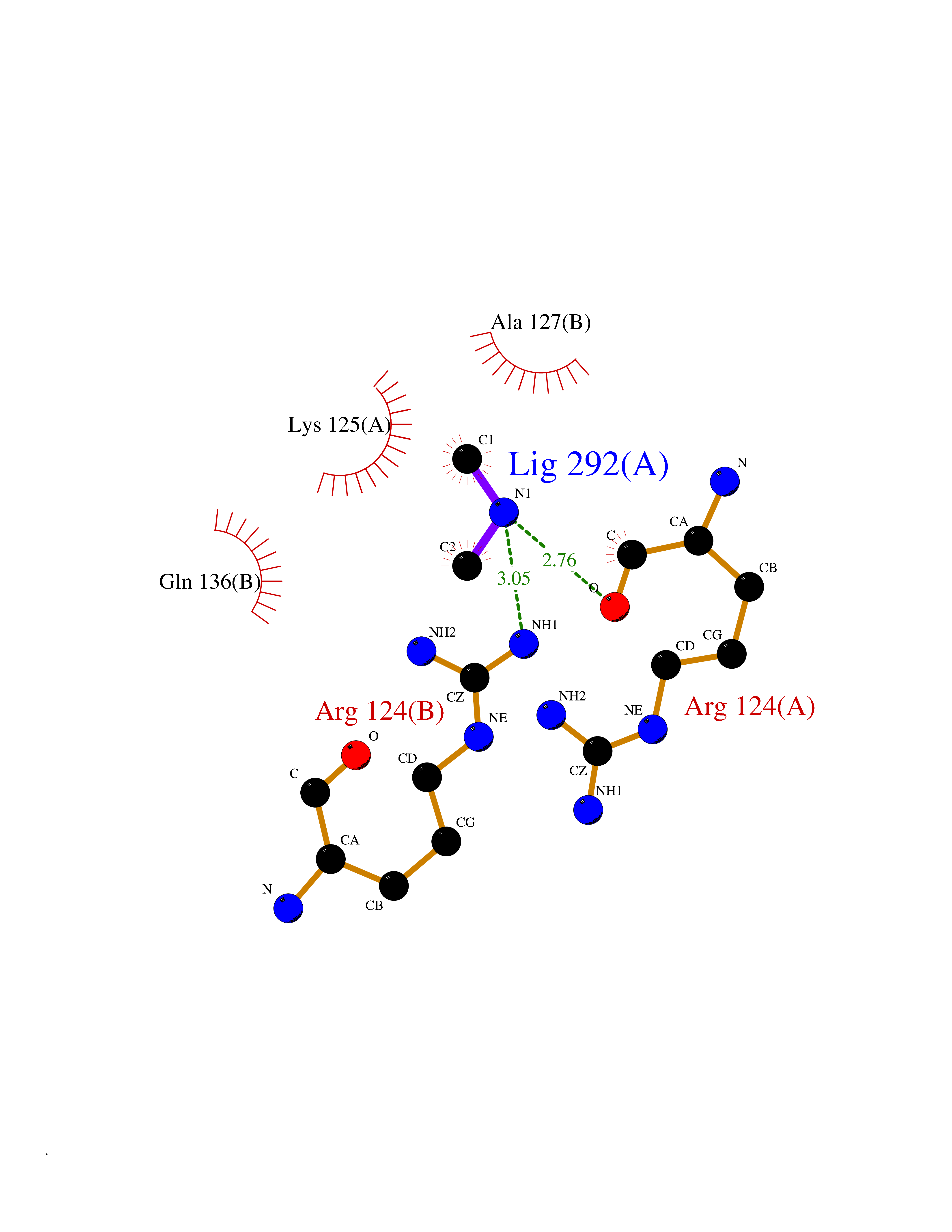 Ligplot Map 3D Binding mode Sequence KELSFGARAELPRIHPVASKLLRLMQKKETNLCLSADVSLARELLQLADALGPSICMLKTHVDILNDFTLDVMKELITLAKXHEFLIFEDRKFADIGNTVKKQYEGGIFKIASWADLVNAHVVPGSGVVKGLQEVGLPLHRGCLLIAEMSSTGSLATGDYTRAAVRMAEEHSEFVVGFISGSRVSMKPEFLHLTPGVQLEAGGDNLGQQYNSPQEVIGKRGSDIIIVGRGIISAADRLEAAEMYRKAAWEAYLSRLGKELSFGARAELPRIHPVASKLLRLMQKKETNLCLSADVSLARELLQLADALGPSICMLKTHVDILNDFTLDVMKELITLAKXHEFLIFEDRKFADIGNTVKKQYEGGIFKIASWADLVNAHVVPGSGVVKGLQEVGLPLHRGCLLIAEMSSTGSLATGDYTRAAVRMAEEHSEFVVGFISGSRVSMKPEFLHLTPGVQLEAGGDNLGQQYNSPQEVIGKRGSDIIIVGRGIISAADRLEAAEMYRKAAWEAYLSRLG Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Haemophilus influenzae NadR protein (Hae-influ nadR) | 1LW7 | 4.01 | |
Target general information Gen name Hae-influ nadR Organism Haemophilus influenzae (strain ATCC 51907 / DSM 11121 / KW20 / Rd) Uniprot ID TTD ID Synonyms nadR; Transcriptional regulator nadR Protein family Bacterial NMN adenylyltransferase family; Bacterial RNK family Biochemical class Nicotinamide ribonucleoside uptake permease Function This enzyme has twoactivities: nicotinamide mononucleotide (NMN) adenylyltransferase and ribosylnicotinamide (RN) kinase. The RN kinase activity catalyzes the phosphorylation of RN to form nicotinamide ribonucleotide. The NMN adenylyltransferase activity catalyzes the transfer of the AMP moiety of ATP to nicotinamide ribonucleotide to form NAD(+). Related diseases Involved in the epigenetic regulation of ESR1 expression in breast cancer in a TFAP2C, IFI16 and HDAC4/5/6-dependent manner. {ECO:0000269|PubMed:24413532}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative initiation; ATP-binding; Cell membrane; Cytoplasm; Kinase; Membrane; Multifunctional enzyme; NAD; Nucleotide-binding; Pyridine nucleotide biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 39581.5 Length 344 Aromaticity 0.14 Instability index 41.39 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 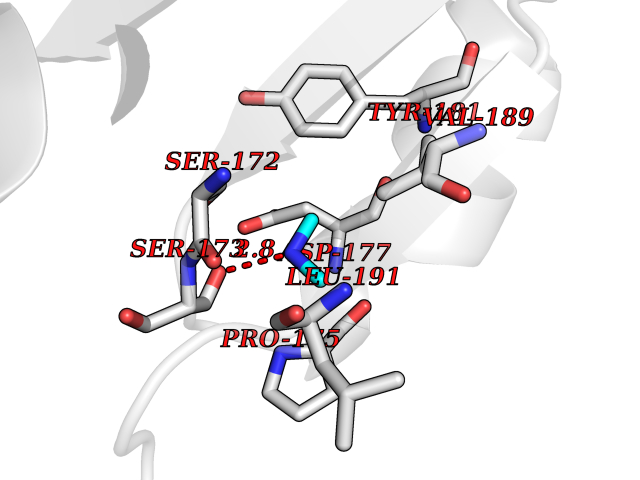 Pymol Map 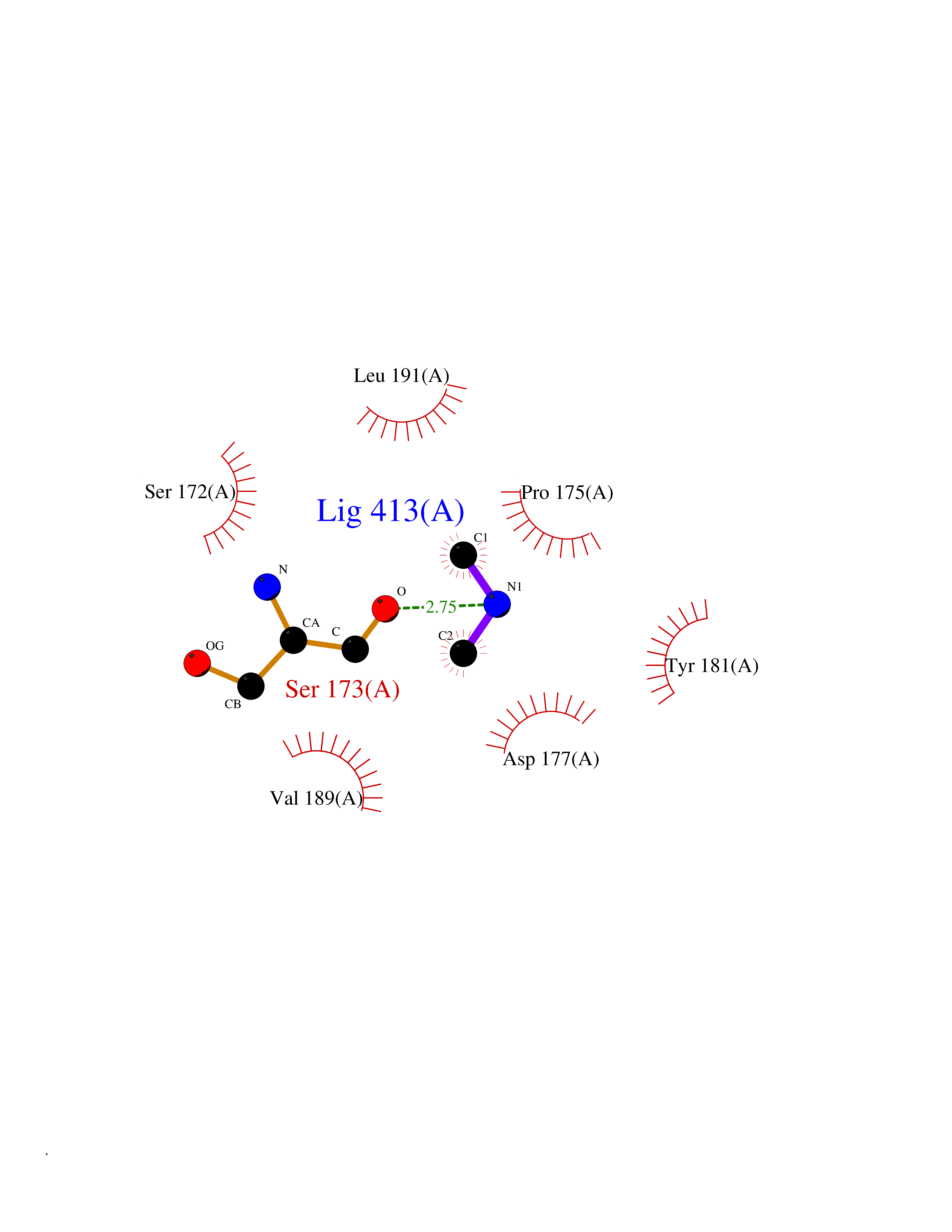 Ligplot Map 3D Binding mode Sequence EKKVGVIFGKFYPVHTGHINXIYEAFSKVDELHVIVCSDTVRDLKLFYDSKXKRXPTVQDRLRWXQQIFKYQKNQIFIHHLVEDGIPSYPNGWQSWSEAVKTLFHEKHFEPSIVFSSEPQDKAPYEKYLGLEVSLVDPDRTFFNVSATKIRTTPFQYWKFIPKEARPFFAKTVAILGGESSGKSVLVNKLAAVFNTTSAWEYGREFVFEKLGGDEQAMQYSDYPQXALGHQRYIDYAVRHSHKIAFIDTDFITTQAFCIQYEGKAHPFLDSXIKEYPFDVTILLKNNTEQKQRQQFQQLLKKLLDKYKVPYIEIESPSYLDRYNQVKAVIEKVLNEEEISELQN Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Soluble epoxide hydrolase (EPHX2) | 1ZD3 | 4.01 | |
Target general information Gen name EPHX2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Bifunctional epoxide hydrolase 2 Protein family AB hydrolase superfamily, Epoxide hydrolase family Biochemical class Ether bond hydrolase Function Bifunctional enzyme. The C-terminal domain has epoxide hydrolase activity and acts on epoxides (alkene oxides, oxiranes) and arene oxides. Plays a role in xenobiotic metabolism by degrading potentially toxic epoxides (By similarity). Also determines steady-state levels of physiological mediators. The N-terminal domain has lipid phosphatase activity, with the highest activity towards threo-9,10-phosphonooxy-hydroxy-octadecanoic acid, followed by erythro-9,10-phosphonooxy-hydroxy-octadecanoic acid, 12-phosphonooxy-octadec-9Z-enoic acid and 12-phosphonooxy-octadec-9E-enoic acid. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08257; DB08258; DB08259; DB06345; DB12610; DB08256; DB02029; DB04213; DB03677 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aromatic hydrocarbons catabolism; Cytoplasm; Detoxification; Direct protein sequencing; Hydrolase; Lipid metabolism; Lipoprotein; Magnesium; Metal-binding; Multifunctional enzyme; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 61744.9 Length 547 Aromaticity 0.09 Instability index 43.97 Isoelectric point 5.81 Charge (pH=7) -7.76 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 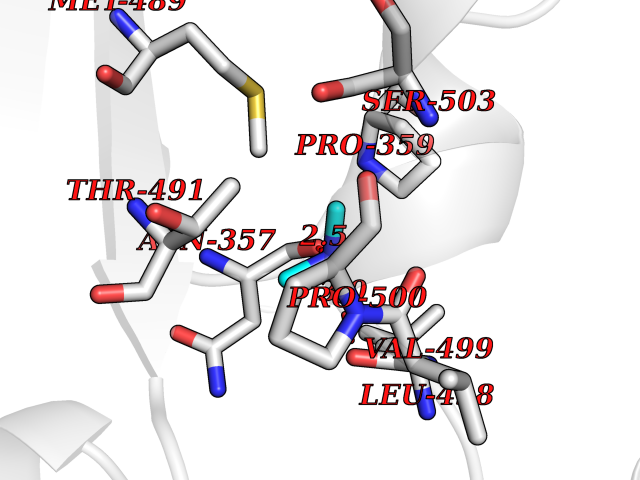 Pymol Map 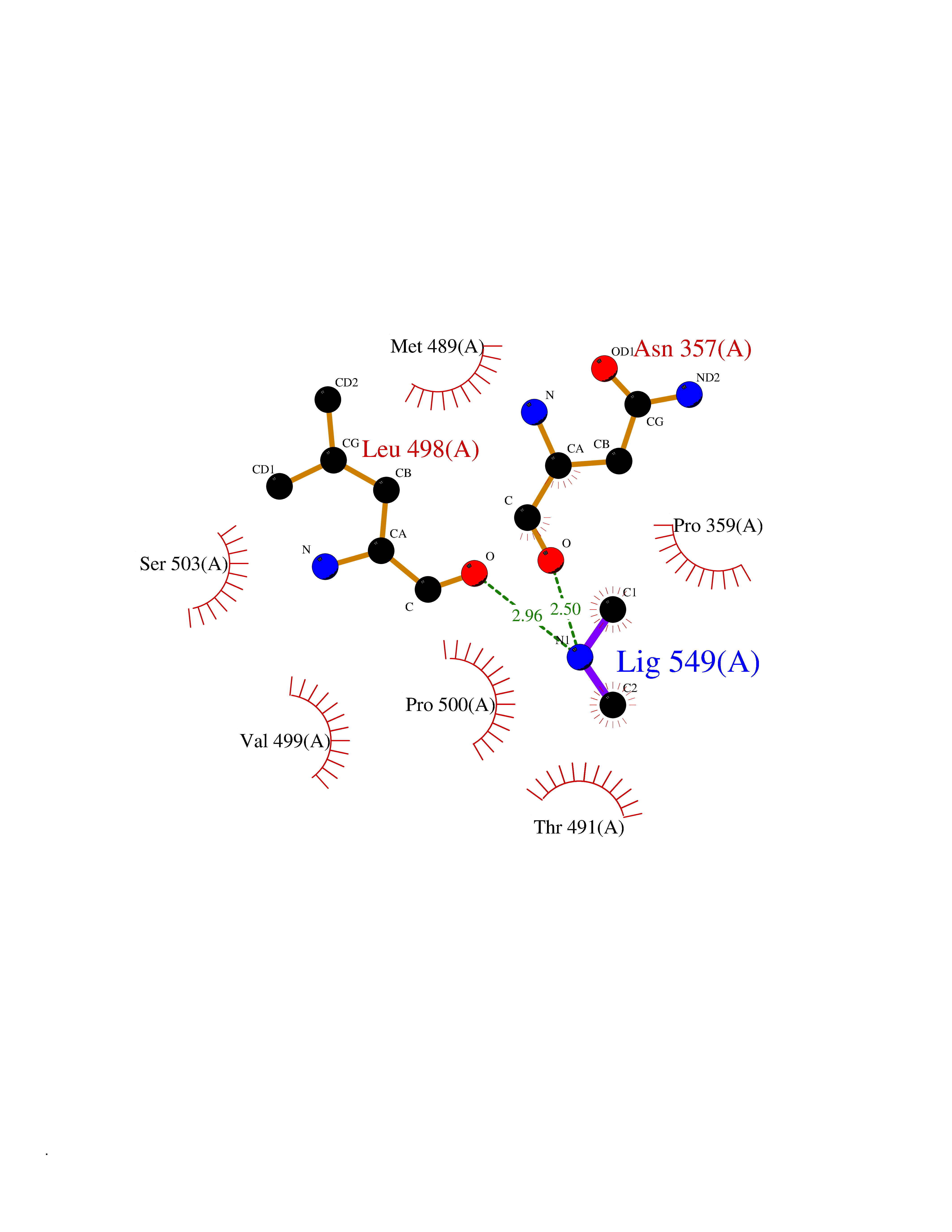 Ligplot Map 3D Binding mode Sequence TLRAAVFDLDGVLALPAVFGVLGRTEEALALPRGLLNDAFQKGGPEGATTRLMKGEITLSQWIPLMEENCRKCSETAKVCLPKNFSIKEIFDKAISARKINRPMLQAALMLRKKGFTTAILTNTWLDDRAERDGLAQLMCELKMHFDFLIESCQVGMVKPEPQIYKFLLDTLKASPSEVVFLDDIGANLKPARDLGMVTILVQDTDTALKELEKVTGIQLLNTPAPLPTSCNPSDMSHGYVTVKPRVRLHFVELGSGPAVCLCHGFPESWYSWRYQIPALAQAGYRVLAMDMKGYGESSAPPEIEEYCMEVLCKEMVTFLDKLGLSQAVFIGHDWGGMLVWYMALFYPERVRAVASLNTPFIPANPNMSPLESIKANPVFDYQLYFQEPGVAEAELEQNLSRTFKSLFRASDESVLSMHKVCEAGGLFVNSPEEPSLSRMVTEEEIQFYVQQFKKSGFRGPLNWYRNMERNWKWACKSLGRKILIPALMVTAEKDFVLVPQMSQHMEDWIPHLKRGHIEDCGHWTQMDKPTEVNQILIKWLDSDARN Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | 6YND | 4.01 | |
Target general information Gen name GAPDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peptidyl-cysteine S-nitrosylase GAPDH; OK/SW-cl.12; GAPD; D-glyceraldehyde-3-phosphate dehydrogenase; CDABP0047 Protein family Glyceraldehyde-3-phosphate dehydrogenase family Biochemical class Aldehyde/oxo donor oxidoreductase Function Participates in nuclear events including transcription, RNA transport, DNA replication and apoptosis. Nuclear functions are probably due to the nitrosylase activity that mediates cysteine S-nitrosylation of nuclear target proteins such as SIRT1, HDAC2 and PRKDC. Modulates the organization and assembly of the cytoskeleton. Facilitates the CHP1-dependent microtubule and membrane associations through its ability to stimulate the binding of CHP1 to microtubules. Glyceraldehyde-3-phosphate dehydrogenase is a key enzyme in glycolysis that catalyzes the first step of the pathway by converting D-glyceraldehyde 3-phosphate (G3P) into 3-phospho-D-glyceroyl phosphate. Component of the GAIT (gamma interferon-activated inhibitor of translation) complex which mediates interferon-gamma-induced transcript-selective translation inhibition in inflammation processes. Upon interferon-gamma treatment assembles into the GAIT complex which binds to stem loop-containing GAIT elements in the 3'-UTR of diverse inflammatory mRNAs (such as ceruplasmin) and suppresses their translation. Has both glyceraldehyde-3-phosphate dehydrogenase and nitrosylase activities, thereby playing a role in glycolysis and nuclear functions, respectively. Related diseases Bone marrow failure and diabetes mellitus syndrome (BMFDMS) [MIM:620044]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFDMS is an autosomal recessive form characterized by various degrees of bone marrow failure, ranging from dyserythropoiesis to bone marrow aplasia, with onset in infancy or early childhood, and non-autoimmune insulin-dependent diabetes mellitus appearing in the first or second decades. Many patients show pigmentary skin abnormalities and short stature. {ECO:0000269|PubMed:28073829, ECO:0000269|PubMed:35611808, ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07347; DB02059; DB11638; DB09130; DB00157; DB03893; DB09092 Interacts with Q6UY14-3; Q9UIJ7; P05067; Q9UQM7; Q14194; P35222; Q9BPW9-4; P00533; O00471; O75344; P06241; P04406; O14556; Q8NEA9; P42858; Q92993-2; P42695; P35228; P12004; P00558; P48147; P17612; Q8WUY3; Q9UHX1-2; P15927; P05109; Q96GZ6; P00441; Q9BSI4; P10599 EC number EC 1.2.1.12 Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Alternative splicing; Apoptosis; Cytoplasm; Cytoskeleton; Direct protein sequencing; Glycolysis; Glycoprotein; Immunity; Innate immunity; Isopeptide bond; Membrane; Methylation; NAD; Nucleus; Oxidation; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; S-nitrosylation; Transferase; Translation regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35818.4 Length 333 Aromaticity 0.08 Instability index 13.69 Isoelectric point 8.64 Charge (pH=7) 3.64 2D Binding mode Binding energy (Kcal/mol) -5.46 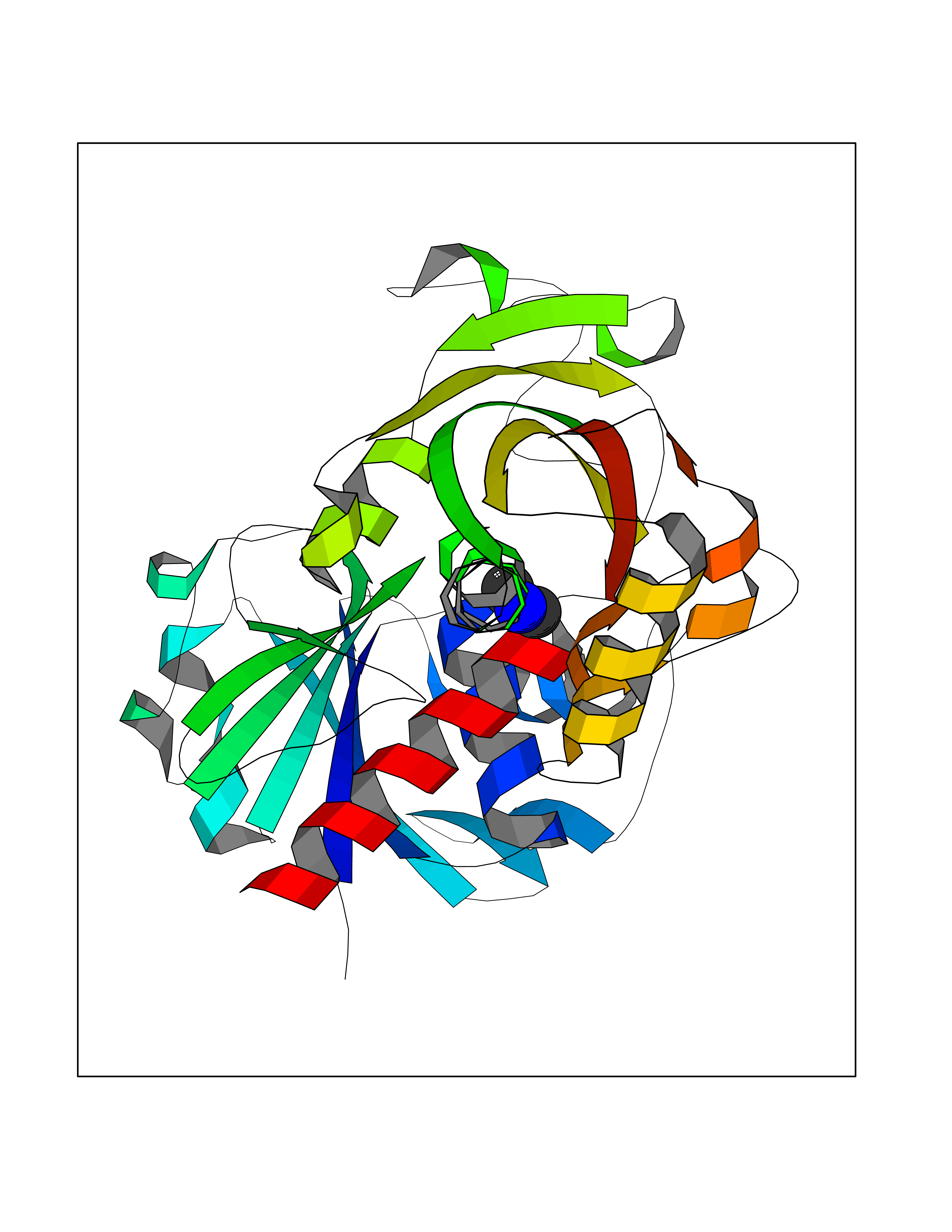 Molscript Map 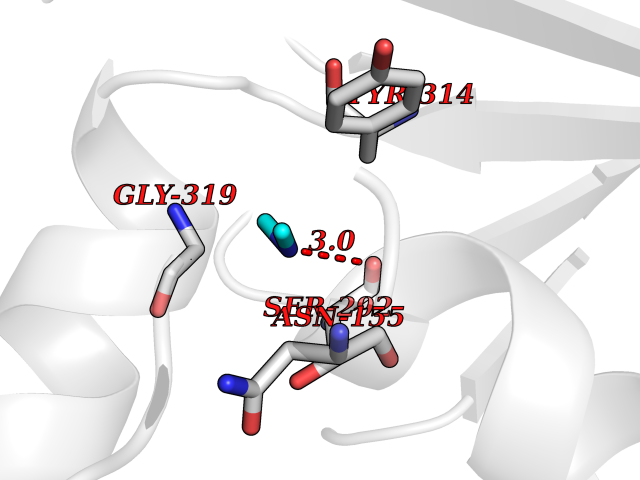 Pymol Map 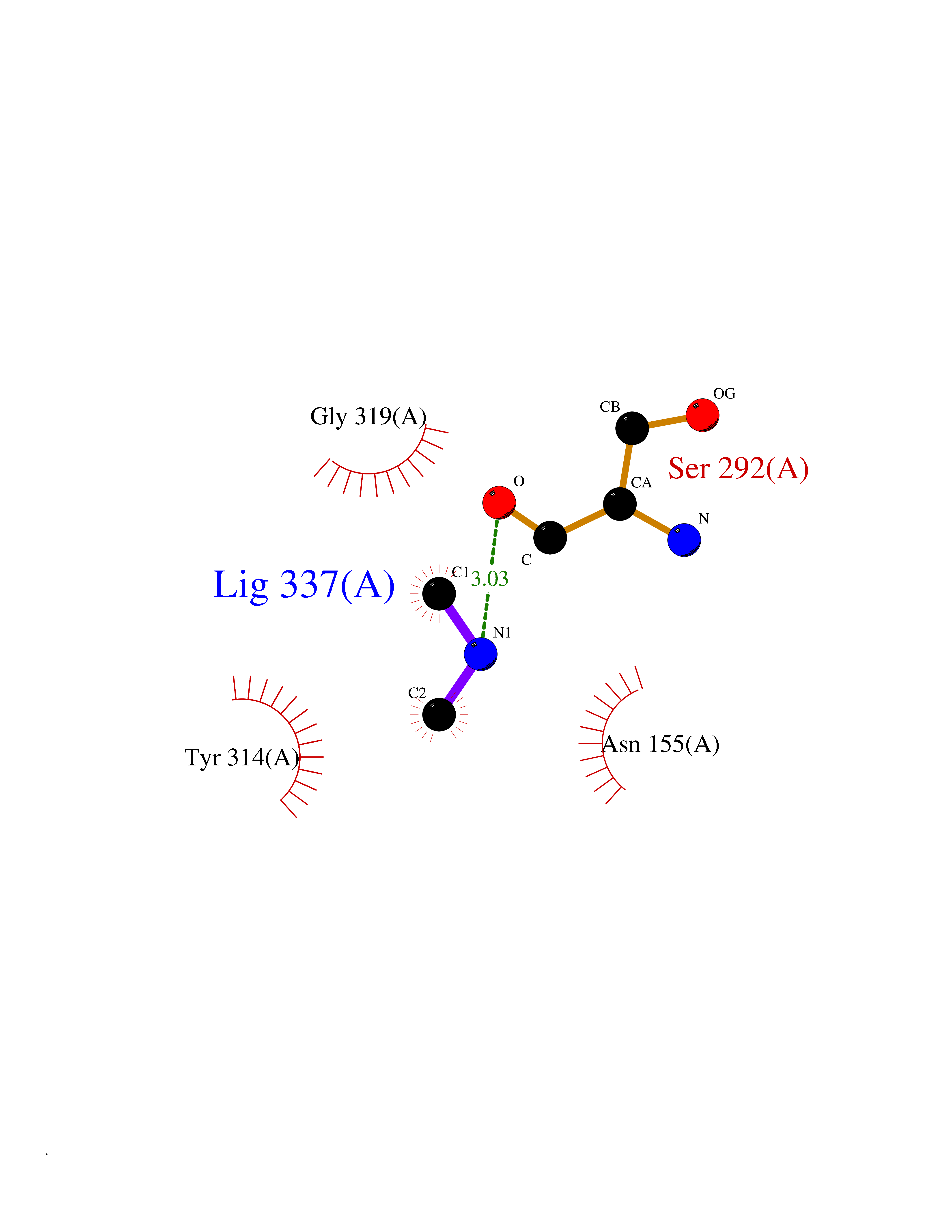 Ligplot Map 3D Binding mode Sequence GKVKVGVNGFGRIGRLVTRAAFNSGKVDIVAINDPFIDLNYMVYMFQYDSTHGKFHGTVKAENGKLVINGNPITIFQERDPSKIKWGDAGAEYVVESTGVFTTMEKAGAHLQGGAKRVIISAPSADAPMFVMGVNHEKYDNSLKIISNASTTNCLAPLAKVIHDNFGIVEGLMTTVHAITATQKTVDGPSGKLWRDGRGALQNIIPASTGAAKAVGKVIPELNGKLTGMAFRVPTANVSVVDLTCRLEKPAKYDDIKKVVKQASEGPLKGILGYTEHQVVSSDFNSDTHSSTFDAGAGIALNDHFVKLISWYDNEFGYSNRVVDLMAHMASKE Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Triggering receptor expressed on monocytes 1 (TREM1) | 1Q8M | 4.01 | |
Target general information Gen name Trem1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Trem1; TREM-1 Protein family NA Biochemical class NA Function Stimulates neutrophil and monocyte-mediated inflammatory responses. Triggers release of pro-inflammatory chemokines and cytokines, as well as increased surface expression of cell activation markers. Amplifier of inflammatory responses that are triggered by bacterial and fungal infections and is a crucial mediator of septic shock. Related diseases GLUT1 deficiency syndrome 1 (GLUT1DS1) [MIM:606777]: A neurologic disorder showing wide phenotypic variability. The most severe 'classic' phenotype comprises infantile-onset epileptic encephalopathy associated with delayed development, acquired microcephaly, motor incoordination, and spasticity. Onset of seizures, usually characterized by apneic episodes, staring spells, and episodic eye movements, occurs within the first 4 months of life. Other paroxysmal findings include intermittent ataxia, confusion, lethargy, sleep disturbance, and headache. Varying degrees of cognitive impairment can occur, ranging from learning disabilities to severe intellectual disability. {ECO:0000269|PubMed:10227690, ECO:0000269|PubMed:10980529, ECO:0000269|PubMed:11136715, ECO:0000269|PubMed:11603379, ECO:0000269|PubMed:12325075, ECO:0000269|PubMed:15622525, ECO:0000269|PubMed:19901175, ECO:0000269|PubMed:20129935, ECO:0000269|PubMed:20221955, ECO:0000269|PubMed:20574033, ECO:0000269|PubMed:24847886, ECO:0000269|PubMed:25982116, ECO:0000269|PubMed:30197081}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GLUT1 deficiency syndrome 2 (GLUT1DS2) [MIM:612126]: A clinically variable disorder characterized primarily by onset in childhood of paroxysmal exercise-induced dyskinesia. The dyskinesia involves transient abnormal involuntary movements, such as dystonia and choreoathetosis, induced by exercise or exertion, and affecting the exercised limbs. Some patients may also have epilepsy, most commonly childhood absence epilepsy. Mild intellectual disability may also occur. In some patients involuntary exertion-induced dystonic, choreoathetotic, and ballistic movements may be associated with macrocytic hemolytic anemia. {ECO:0000269|PubMed:14605501, ECO:0000269|PubMed:18451999, ECO:0000269|PubMed:19630075, ECO:0000269|PubMed:19798636, ECO:0000269|PubMed:20129935, ECO:0000269|PubMed:20574033, ECO:0000269|PubMed:20621801, ECO:0000269|PubMed:20830593, ECO:0000269|PubMed:21204808}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epilepsy, idiopathic generalized 12 (EIG12) [MIM:614847]: A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. In some EIG12 patients seizures may remit with age. {ECO:0000269|PubMed:19798636, ECO:0000269|PubMed:22282645, ECO:0000269|PubMed:23280796, ECO:0000269|PubMed:25982116}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Dystonia 9 (DYT9) [MIM:601042]: An autosomal dominant neurologic disorder characterized by childhood onset of paroxysmal choreoathetosis and progressive spastic paraplegia. Most patients show some degree of cognitive impairment. Other variable features may include seizures, migraine headaches, and ataxia. {ECO:0000269|PubMed:21832227}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Stomatin-deficient cryohydrocytosis with neurologic defects (SDCHCN) [MIM:608885]: A rare form of stomatocytosis characterized by episodic hemolytic anemia, cold-induced red cells cation leak, erratic hyperkalemia, neonatal hyperbilirubinemia, hepatosplenomegaly, cataracts, seizures, intellectual disability, and movement disorder. {ECO:0000269|PubMed:21791420, ECO:0000269|PubMed:22492876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01694 Interacts with NA EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Innate immunity; Membrane; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 28186.2 Length 242 Aromaticity 0.07 Instability index 45.2 Isoelectric point 6.1 Charge (pH=7) -3.8 2D Binding mode Binding energy (Kcal/mol) -5.47 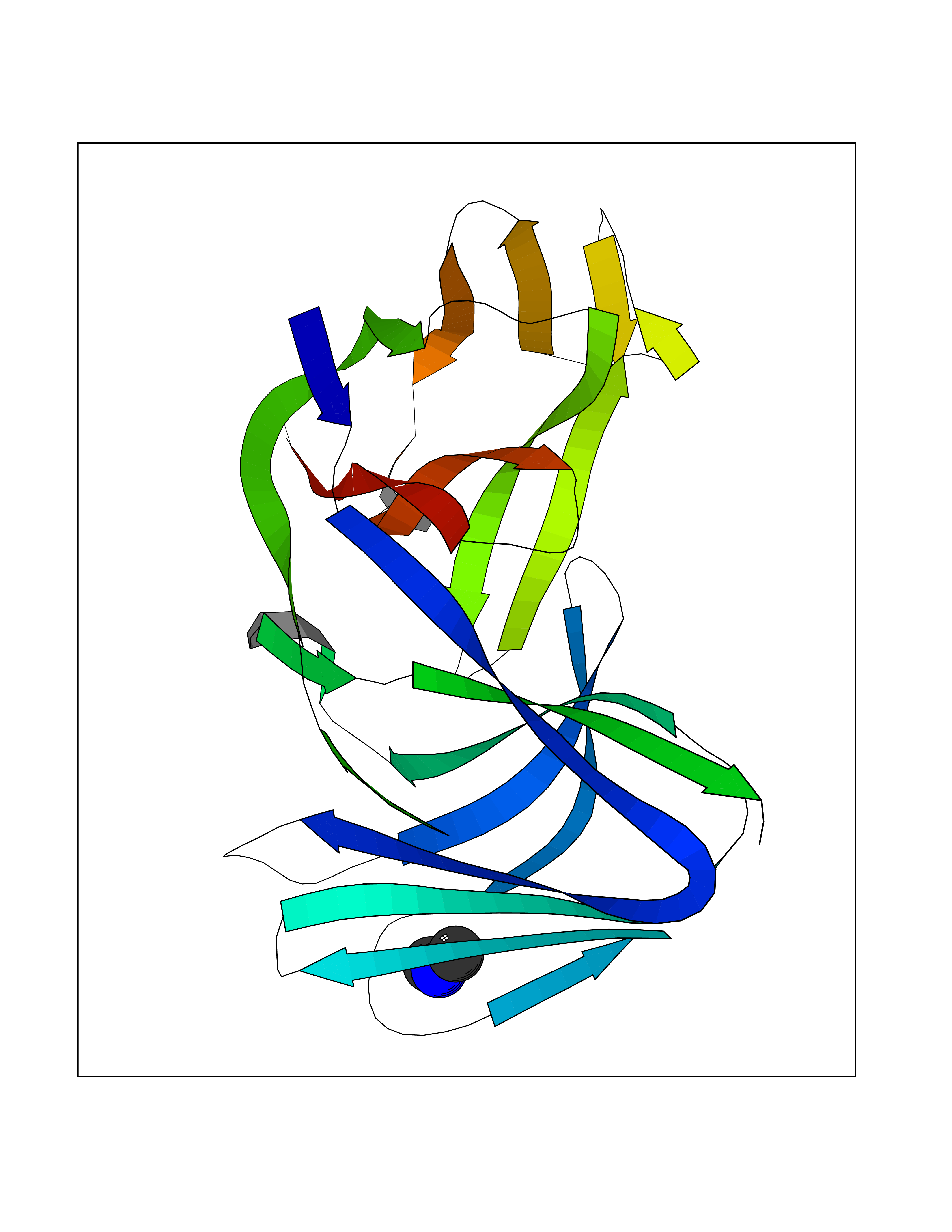 Molscript Map 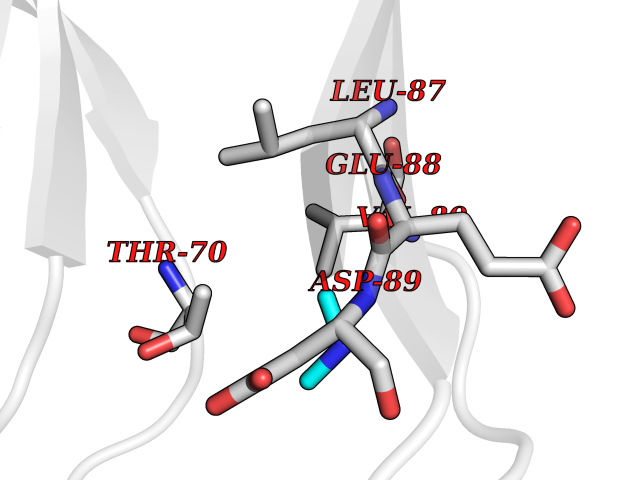 Pymol Map 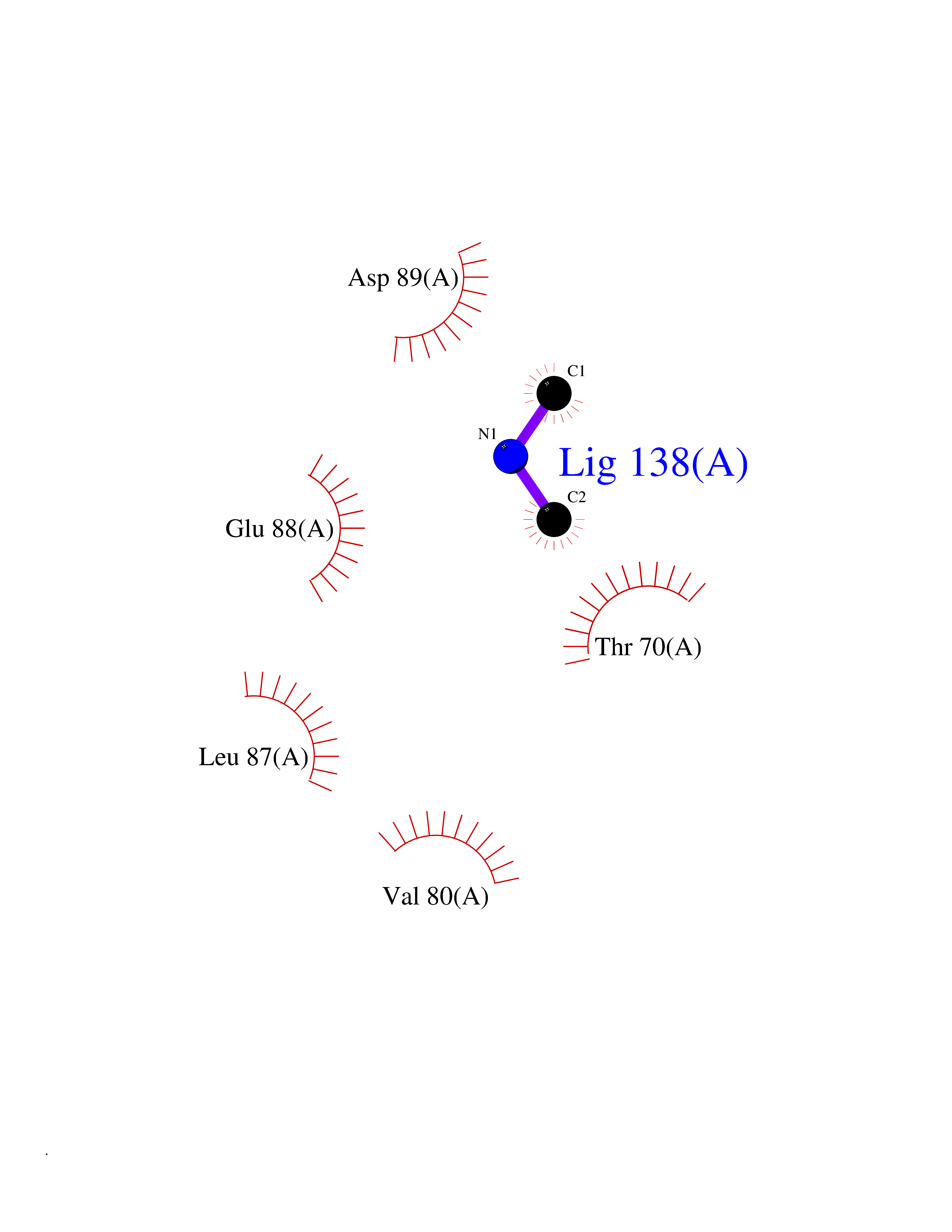 Ligplot Map 3D Binding mode Sequence MELRAATKLTEEKYELKEGQTLDVKCDYTLEKFASSQKAWQIIRDGEMPKTLACTERPSKNSHPVQVGRIILEDYHDHGLLRVRMVNLQVEDSGLYQCVIYQPPKEPHMLFDRIRLVVTLEMELRAATKLTEEKYELKEGQTLDVKCDYTLEKFASSQKAWQIIRDGEMPKTLACTERPSKNSHPVQVGRIILEDYHDHGLLRVRMVNLQVEDSGLYQCVIYQPPKEPHMLFDRIRLVVTLE Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Hydrogen peroxide-inducible genes activator | 1I69 | 4.00 | |
Target general information Gen name oxyR Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3961;mor;momR;JW3933 Protein family LysR transcriptional regulatory family Biochemical class Transcription Function Bacterial-type RNA polymerase core promoter proximal region sequence-specific DNA binding.Transcription factor activity, bacterial-type RNA polymerase core promoter proximal region sequence-specific binding. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; Direct protein sequencing; Disulfide bond; DNA-binding; Glutathionylation; Oxidation; Reference proteome; Repressor; S-nitrosylation; Stress response; Transcription; Transcription regulation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 46013 Length 412 Aromaticity 0.07 Instability index 44.76 Isoelectric point 5.57 Charge (pH=7) -14.52 2D Binding mode Binding energy (Kcal/mol) -5.46 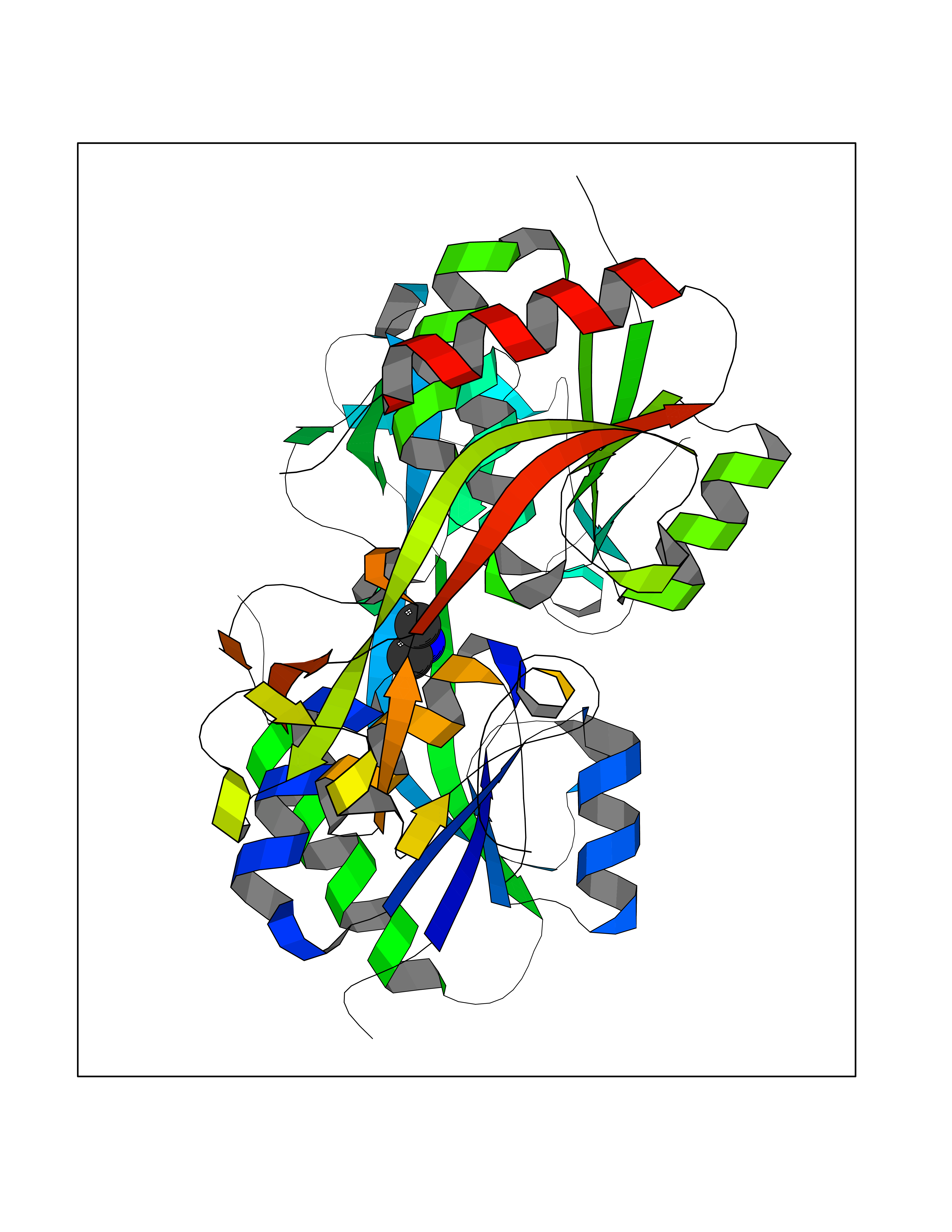 Molscript Map 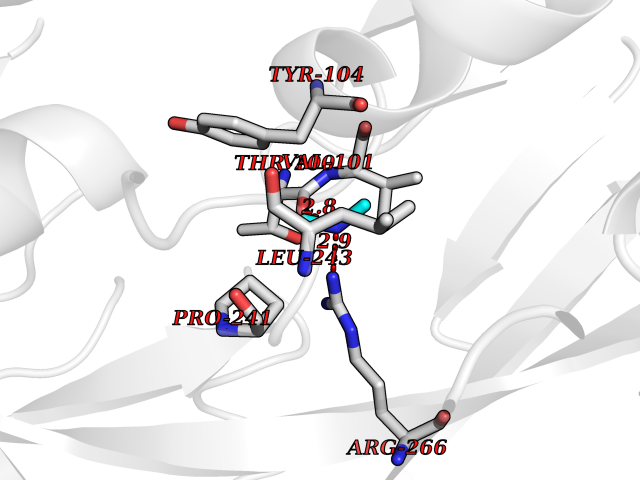 Pymol Map 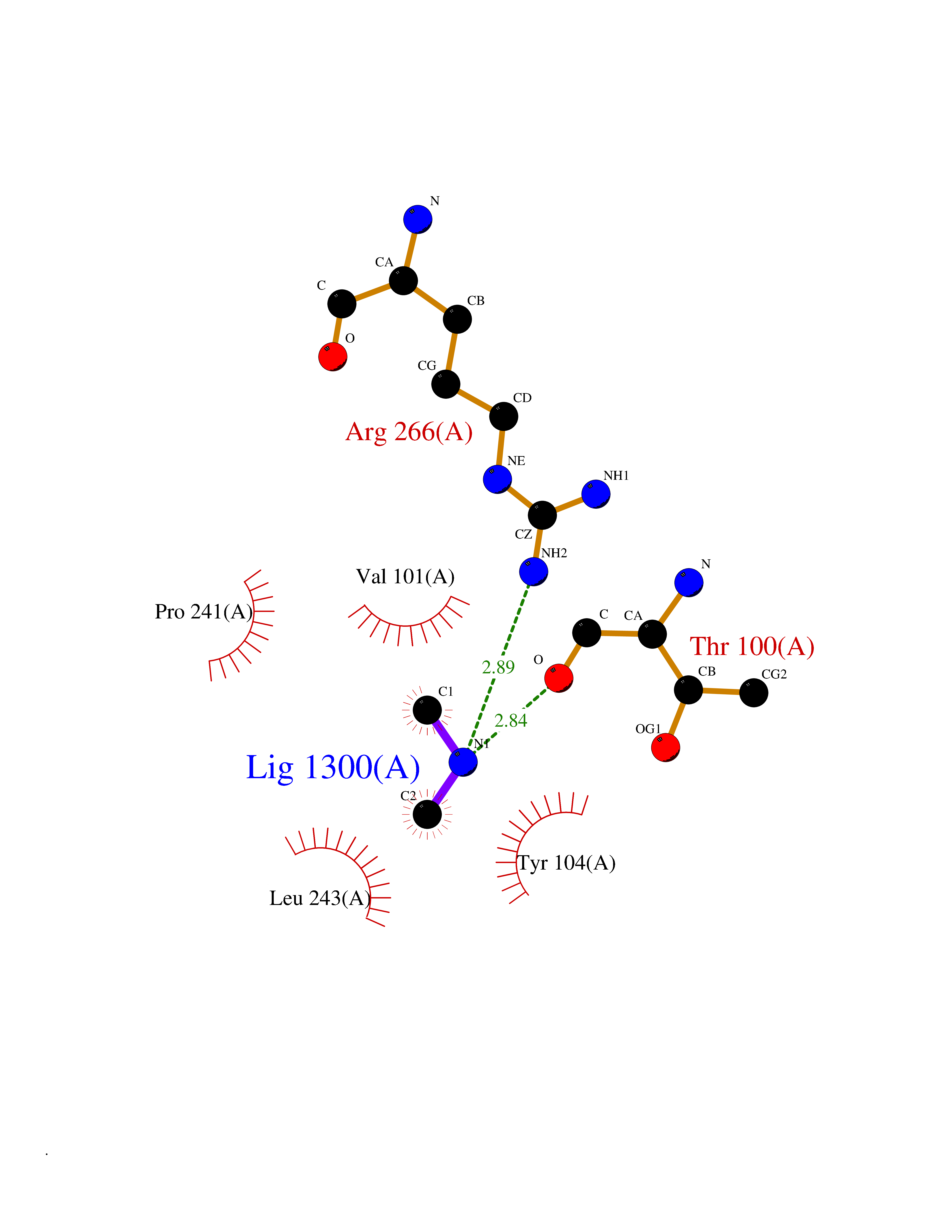 Ligplot Map 3D Binding mode Sequence ETMSGPLHIGLIPTVGPYLLPHIIPMLHQTFPKLEMYLHEAQTHQLLAQLDSGKLDAVILALVKESEAFIEVPLFDEPMLLAIYEDHPWANREAVPMADLAGEKLLMLEDGHSLRDQAMGFCFDTHFRATSLETLRNMVAAGSGITLLPALAVPPERKRDGVVYLPAIKPEPRRTIGLVYRPGSPLRSRYEQLAEAIRARMDGHFDETMSGPLHIGLIPTVGPYLLPHIIPMLHQTFPKLEMYLHEAQTHQLLAQLDSGKLDAVILALVKESEAFIEVPLFDEPMLLAIYEDHPWANREAVPMADLAGEKLLMLEDGHSLRDQAMGFCFETHFRATSLETLRNMVAAGSGITLLPALAVPPERKRDGVVYLPAIKPEPRRTIGLVYRPGSPLRSRYEQLAEAIRARMDGHFD Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Alcohol dehydrogenase 1B | 1U3U | 4.00 | |
Target general information Gen name ADH1B Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ADH2 Protein family Zinc-containing alcohol dehydrogenase family Biochemical class Oxidoreductase Function Alcohol dehydrogenase activity, zinc-dependent.Ethanol binding.Zinc ion binding. Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02721; DB03703; DB00898; DB01213; DB09462; DB02481; DB04105; DB00157; DB03461 Interacts with P00326 EC number 1.1.1.105 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Lipid metabolism; Metal-binding; NAD; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 39722.9 Length 374 Aromaticity 0.06 Instability index 18.9 Isoelectric point 8.63 Charge (pH=7) 6.96 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map 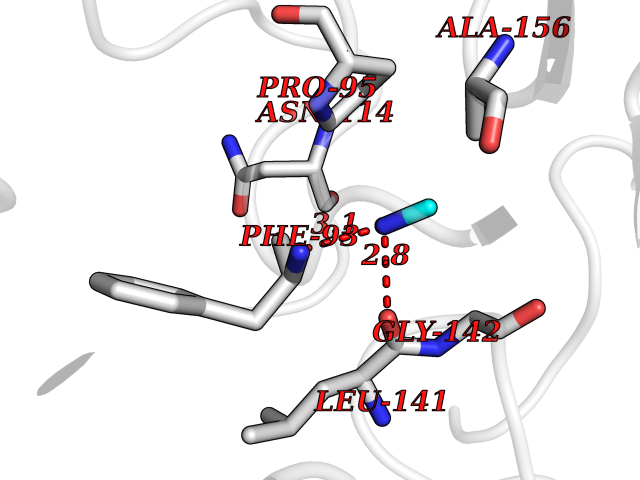 Pymol Map 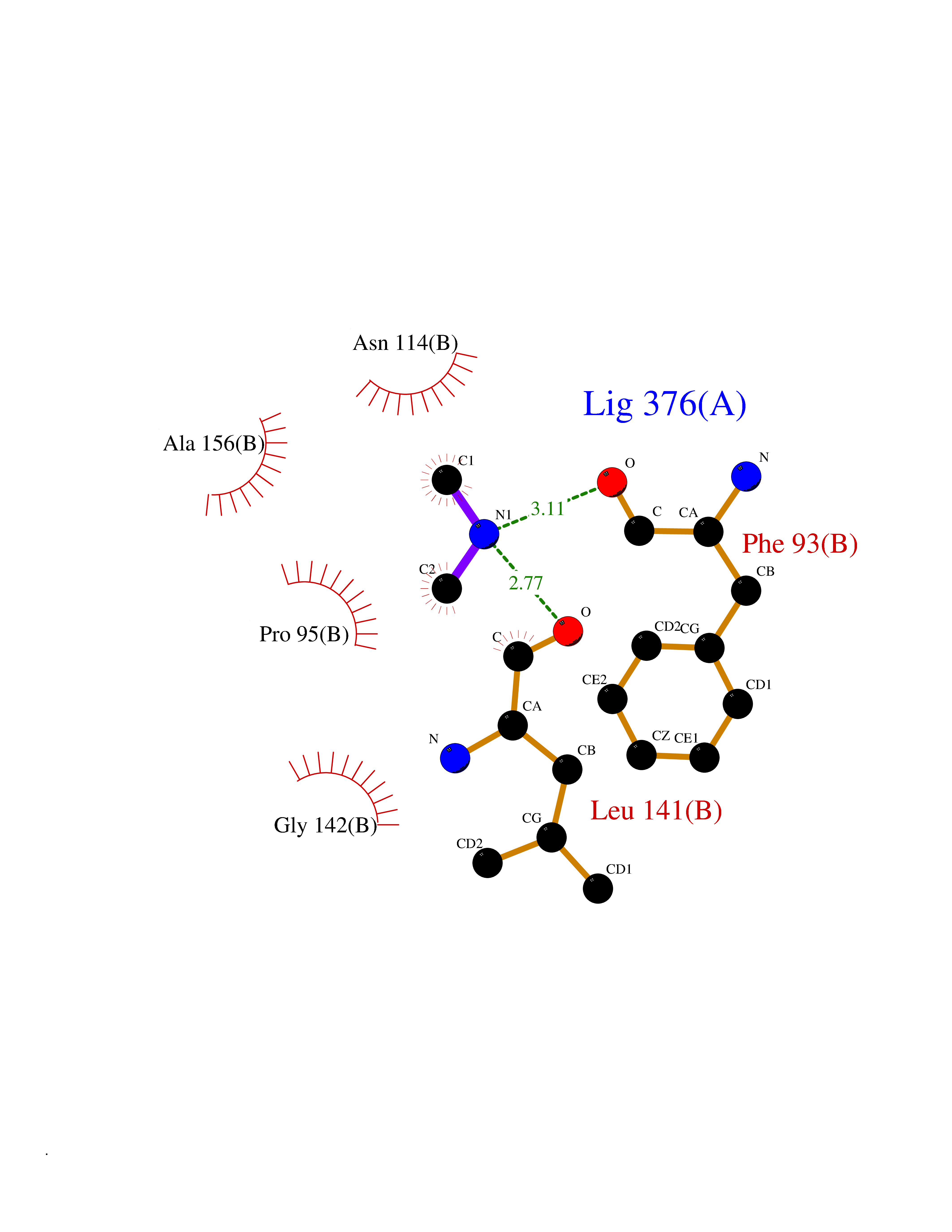 Ligplot Map 3D Binding mode Sequence STAGKVIKCKAAVLWEVKKPFSIEDVEVAPPKAYEVRIKMVAVGICRTDDHVVSGNLVTPLPVILGHEAAGIVESVGEGVTTVKPGDKVIPLFTPQCGKCRVCKNPESNYCLKNDLGNPRGTLQDGTRRFTCRGKPIHHFLGTSTFSQYTVVDENAVAKIDAASPLEKVCLIGCGFSTGYGSAVNVAKVTPGSTCAVFGLGGVGLSAVMGCKAAGAARIIAVDINKDKFAKAKELGATECINPQDYKKPIQEVLKEMTDGGVDFSFEVIGRLDTMMASLLCCHEACGTSVIVGVPPASQNLSINPMLLLTGRTWKGAVYGGFKSKEGIPKLVADFMAKKFSLDALITHVLPFEKINEGFDLLHSGKSIRTVLTF Hydrogen bonds contact Hydrophobic contact | ||||