Job Results:
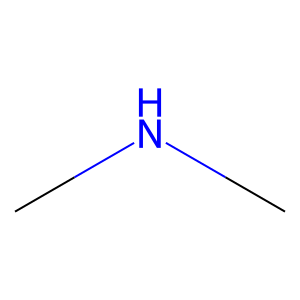
Ligand
Structure
Job ID
7689e25b6c3c7fc348a8f90b8451d3c4
Job name
NA
Time
2025-02-18 14:12:25
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Alcohol dehydrogenase 1B | 1U3U | 4.01 | |
Target general information Gen name ADH1B Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ADH2 Protein family Zinc-containing alcohol dehydrogenase family Biochemical class Oxidoreductase Function Alcohol dehydrogenase activity, zinc-dependent.Ethanol binding.Zinc ion binding. Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02721; DB03703; DB00898; DB01213; DB09462; DB02481; DB04105; DB00157; DB03461 Interacts with P00326 EC number 1.1.1.105 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Lipid metabolism; Metal-binding; NAD; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 39722.9 Length 374 Aromaticity 0.06 Instability index 18.9 Isoelectric point 8.63 Charge (pH=7) 6.96 2D Binding mode Binding energy (Kcal/mol) -5.47 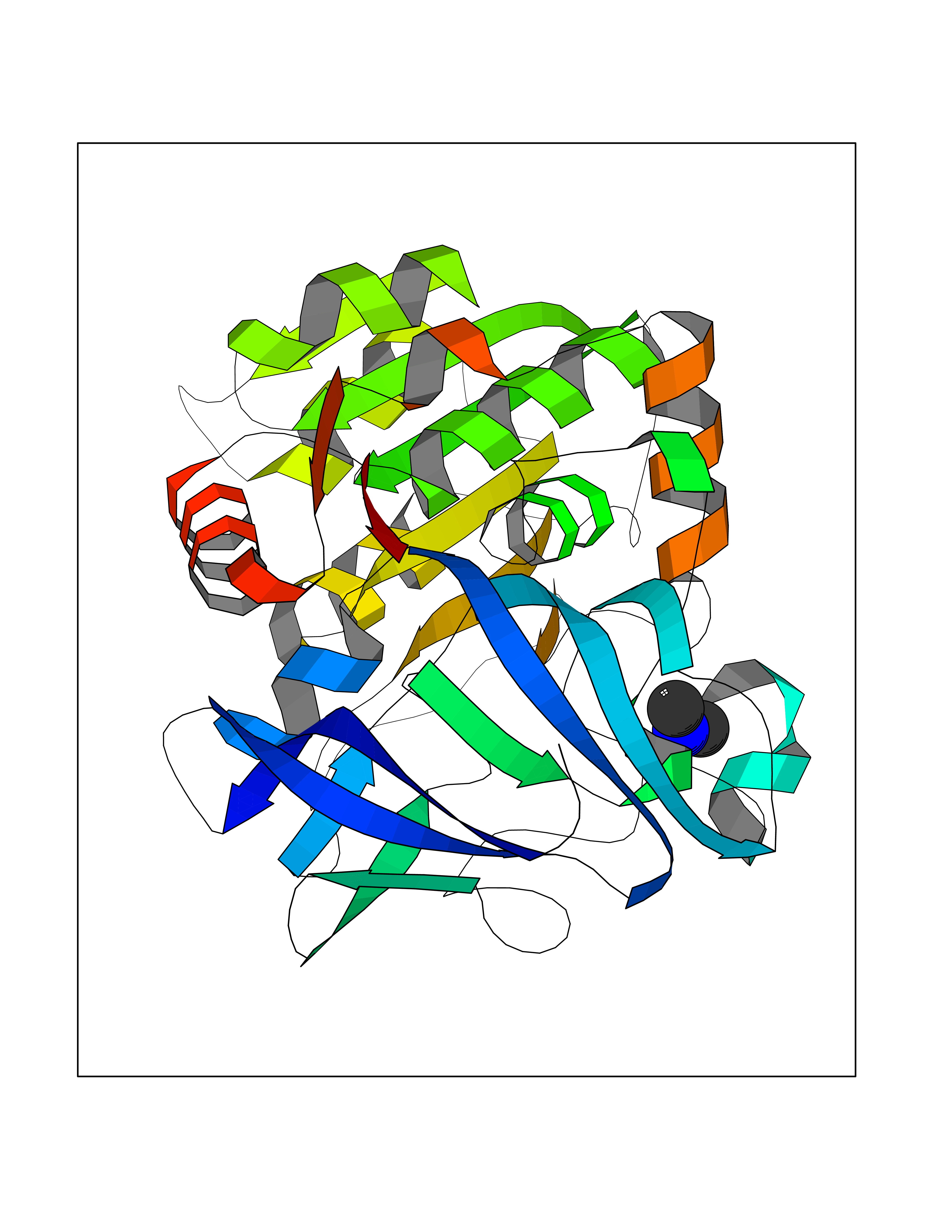 Molscript Map 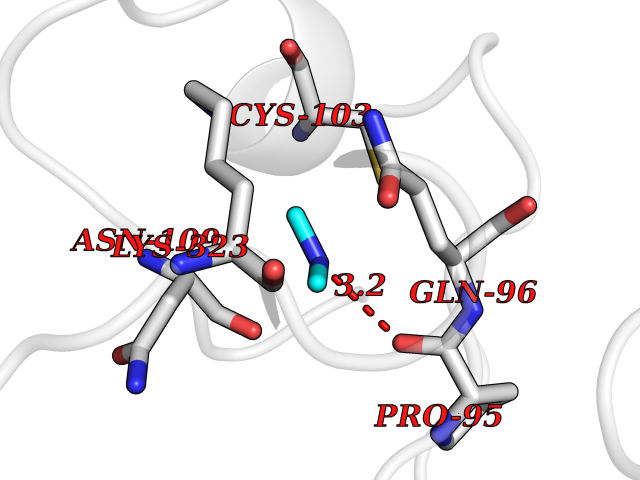 Pymol Map 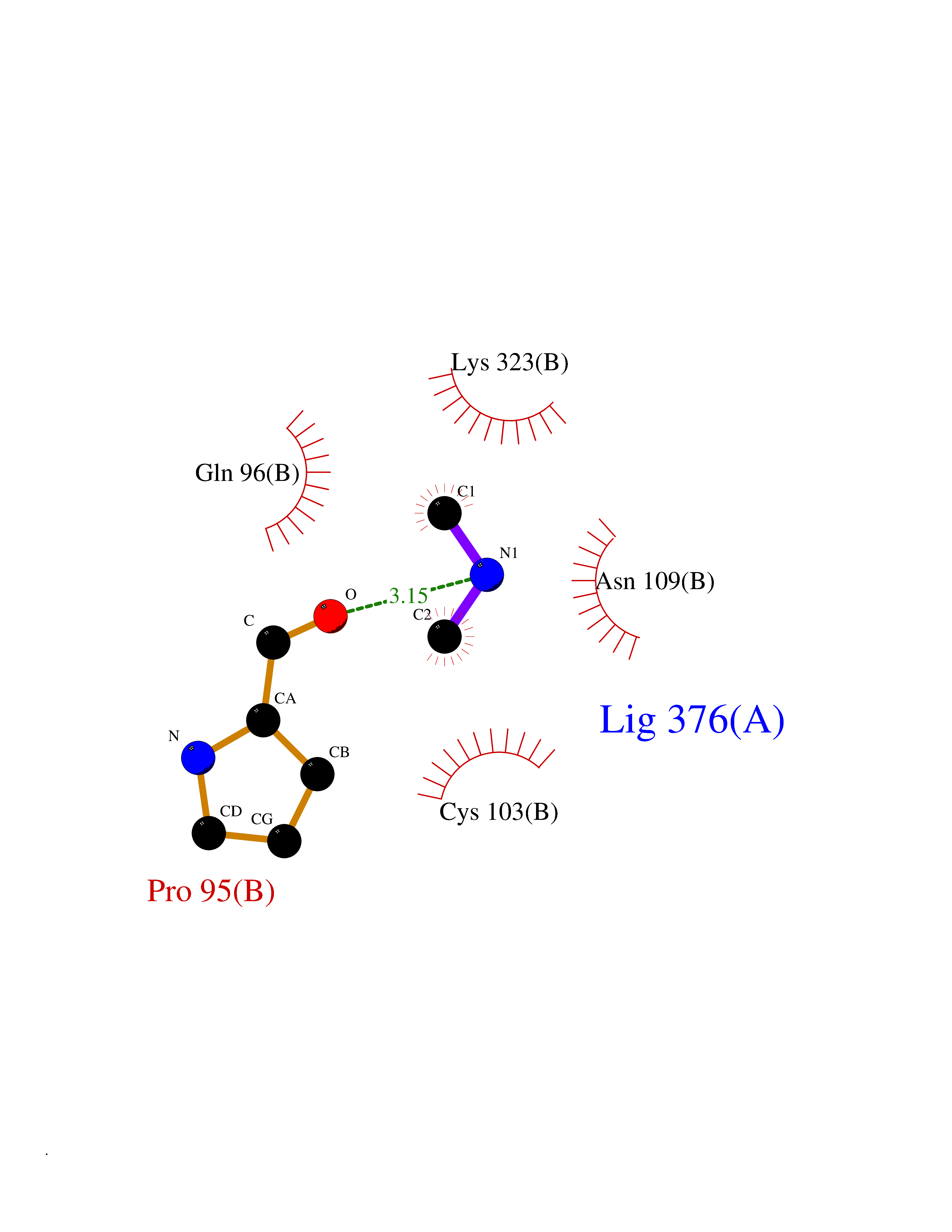 Ligplot Map 3D Binding mode Sequence STAGKVIKCKAAVLWEVKKPFSIEDVEVAPPKAYEVRIKMVAVGICRTDDHVVSGNLVTPLPVILGHEAAGIVESVGEGVTTVKPGDKVIPLFTPQCGKCRVCKNPESNYCLKNDLGNPRGTLQDGTRRFTCRGKPIHHFLGTSTFSQYTVVDENAVAKIDAASPLEKVCLIGCGFSTGYGSAVNVAKVTPGSTCAVFGLGGVGLSAVMGCKAAGAARIIAVDINKDKFAKAKELGATECINPQDYKKPIQEVLKEMTDGGVDFSFEVIGRLDTMMASLLCCHEACGTSVIVGVPPASQNLSINPMLLLTGRTWKGAVYGGFKSKEGIPKLVADFMAKKFSLDALITHVLPFEKINEGFDLLHSGKSIRTVLTF Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Fumarate reductase flavoprotein subunit | 1KF6 | 4.01 | |
Target general information Gen name frdA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW4115;b4154 Protein family FAD-dependent oxidoreductase 2 family, FRD/SDH subfamily Biochemical class Oxidoreductase Function Electron carrier activity.FAD binding.Fumarate reductase (menaquinone).Succinate dehydrogenase activity. Related diseases Glycogen storage disease 11 (GSD11) [MIM:612933]: A metabolic disorder that results in exertional myoglobinuria, pain, cramps and easy fatigue. {ECO:0000269|PubMed:2334430}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07490; DB07918; DB00730 Interacts with P0AC47; P0ACB4; P76111 EC number 1.3.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; Electron transport; FAD; Flavoprotein; Membrane; Nucleotide-binding; Oxidoreductase; Reference proteome; Transport Protein physicochemical properties Chain ID A,M Molecular weight (Da) 90370.7 Length 820 Aromaticity 0.08 Instability index 28.88 Isoelectric point 5.86 Charge (pH=7) -16.21 2D Binding mode Binding energy (Kcal/mol) -5.48 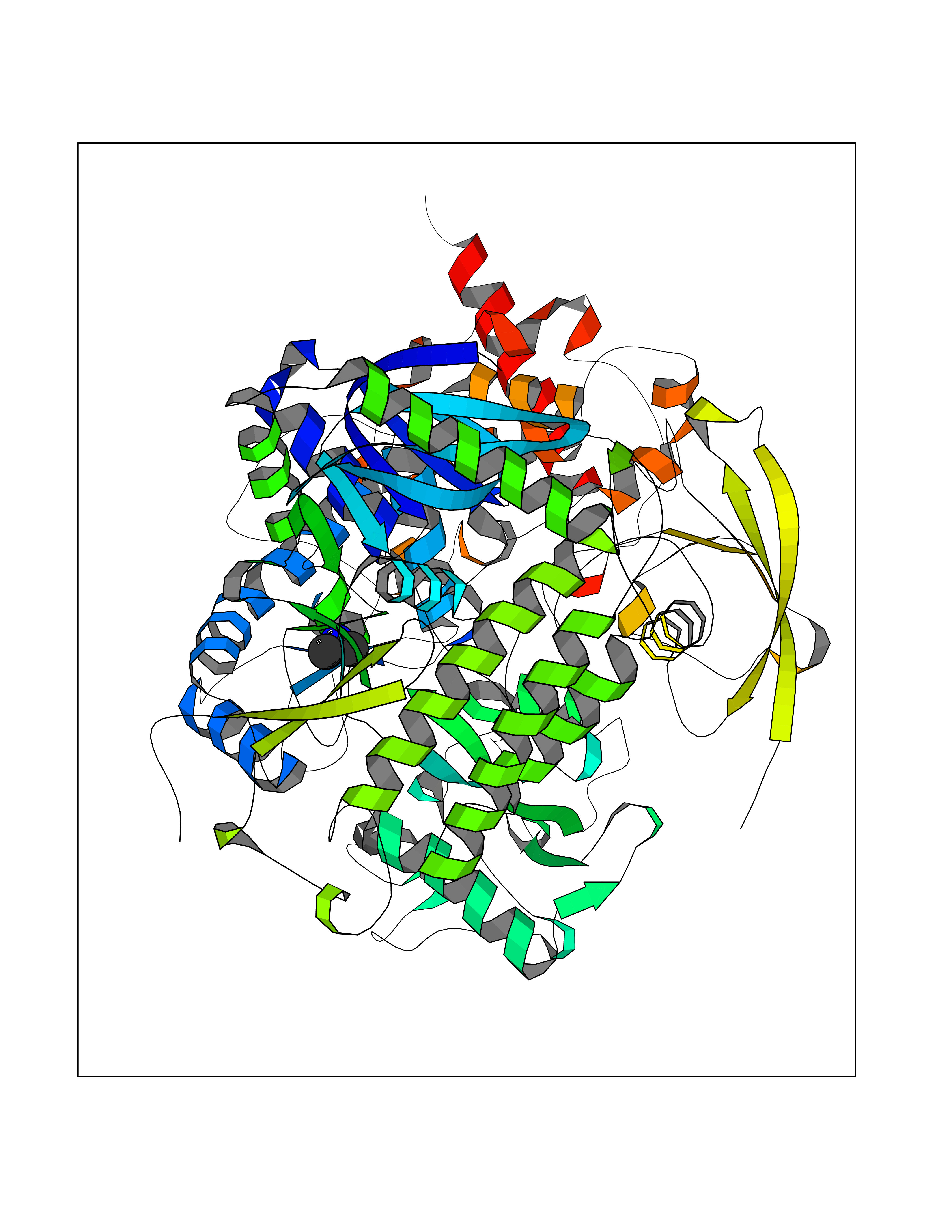 Molscript Map 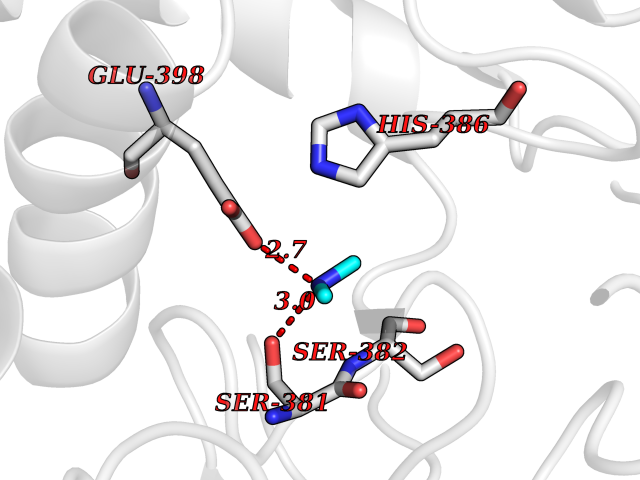 Pymol Map 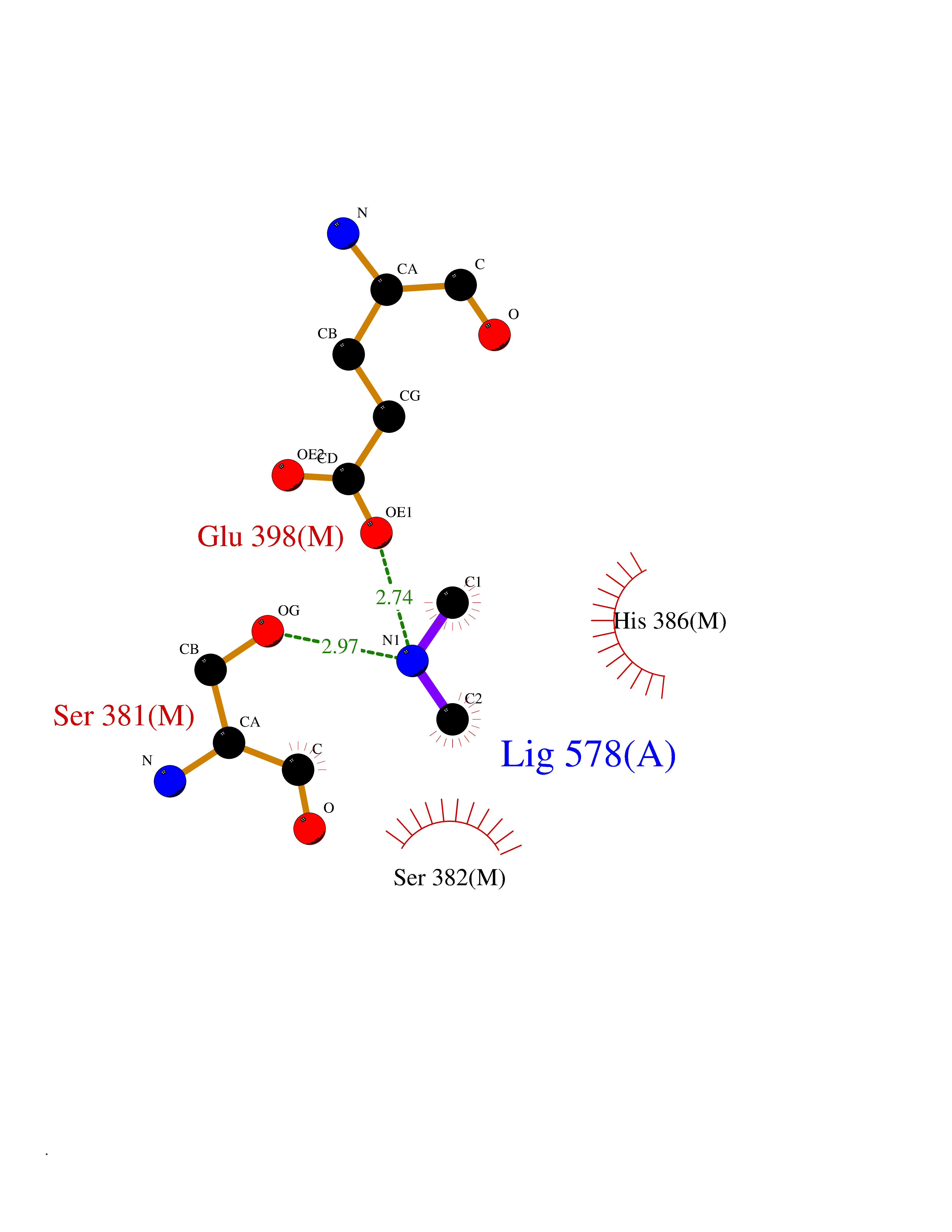 Ligplot Map 3D Binding mode Sequence MQTFQADLAIVGAGGAGLRAAIAAAQANPNAKIALISKVYPMRSHTVAAEGGSAAVAQDHDSFEYHFHDTVAGGDWLCEQDVVDYFVHHCPTEMTQLELWGCPWSRRPDGSVNVRRFGGMKIERTWFAADKTGFHMLHTLFQTSLQFPQIQRFDEHFVLDILVDDGHVRGLVAMNMMEGTLVQIRANAVVMATGGAGRVYRYNTNGGIVTGDGMGMALSHGVPLRDMEFVQYHPTGLPGSGILMTEGCRGEGGILVNKNGYRYLQDYGMGPETPLGEPKNKYMELGPRDKVSQAFWHEWRKGNTISTPRGDVVYLDLRHLGEKKLHERLPFICELAKAYVGVDPVKEPIPVRPTAHYTMGGIETDQNCETRIKGLFAVGECSSVGLHGANRLGSNSLAELVVFGRLAGEQATERAATAGNGNEAAIEAQAAGVEQRLKDLVNQDGGENWAKIRDEMGLAMEEGCGIYRTPELMQKTIDKLAELQERFKRVRITDTSSVFNTDLLYTIELGHGLNVAECMAHSAMARKESRGAHQRLDEGCTERDDVNFLKHTLAFRDADGTTRLEYSDVKITTLPPAAEMKNLKIEVVRYNPEVDTAPHSAFYEVPYDATTSLLDALGYIKDNLAPDLSYRWSCRMAICGSCGMMVNNVPKLACKTFLRDYTDGMKVEALANFPIERDLVVDMTHFIESLEAIKPYIIGNSRTADQGTNIQTPAQMAKYHQFSGCINCGLCYAACPQFGLNPEFIGPAAITLAHRYNEDSRDHGKKERMAQLNSQNGVWSCTFVGYCSEVCPKHVDPAAAIQQGKVESSKDFLIATLKPR Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Prothrombin | 4UD9 | 4.01 | |
Target general information Gen name F2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase S1 family Biochemical class Hydrolase Function Calcium ion binding.Growth factor activity.Heparin binding.Lipopolysaccharide binding.Receptor binding.Serine-type endopeptidase activity.Thrombospondin receptor activity. Related diseases Factor II deficiency (FA2D) [MIM:613679]: A very rare blood coagulation disorder characterized by mucocutaneous bleeding symptoms. The severity of the bleeding manifestations correlates with blood factor II levels. {ECO:0000269|PubMed:1349838, ECO:0000269|PubMed:1354985, ECO:0000269|PubMed:1421398, ECO:0000269|PubMed:14962227, ECO:0000269|PubMed:2719946, ECO:0000269|PubMed:3242619, ECO:0000269|PubMed:3567158, ECO:0000269|PubMed:3771562, ECO:0000269|PubMed:3801671, ECO:0000269|PubMed:6405779, ECO:0000269|PubMed:7792730, ECO:0000269|PubMed:7865694}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Thrombophilia due to thrombin defect (THPH1) [MIM:188050]: A multifactorial disorder of hemostasis characterized by abnormal platelet aggregation in response to various agents and recurrent thrombi formation. {ECO:0000269|PubMed:2825773}. The disease is caused by variants affecting the gene represented in this entry. A common genetic variation in the 3-prime untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increased risk of venous thrombosis.; DISEASE: Pregnancy loss, recurrent, 2 (RPRGL2) [MIM:614390]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:11506076}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07211; DB07796; DB07016; DB07521; DB06850; DB07091; DB06845; DB07088; DB07131; DB07095; DB07515; DB07897; DB06878; DB06947; DB08624; DB06869; DB06929; DB07400; DB04771; DB04772; DB02287; DB07277; DB07550; DB07549; DB07548; DB07105; DB04722; DB07366; DB08254; DB01725; DB08062; DB07639; DB07461; DB07120; DB07190; DB07741; DB07353; DB07508; DB07809; DB08546; DB08061; DB07718; DB03136; DB02723; DB07440; DB07376; DB06861; DB06866; DB06865; DB03865; DB06841; DB07934; DB08422; DB07659; DB07660; DB07658; DB13151; DB00025; DB11166; DB00278; DB01766; DB07083; DB00006; DB00100; DB13152; DB09228; DB09130; DB03159; DB06911; DB06996; DB06919; DB07027; DB07133; DB07143; DB07005; DB06695; DB00055; DB01225; DB05714; DB12831; DB03847; DB07278; DB01767; DB06404; DB09332; DB00001; DB13998; DB04136; DB00170; DB06838; DB13999; DB06868; DB06942; DB06936; DB07165; DB07527; DB07522; DB07665; DB07946; DB06859; DB06853; DB06858; DB07279; DB08187; DB04591; DB07944; DB07128; DB12598; DB01123; DB04786; DB05777; DB04697; DB09109; DB14738; DB04898; DB01593; DB14487; DB08152 Interacts with P05067; P07204; Q846V4; PRO_0000032489 [P01008] EC number 3.4.21.5 Uniprot keywords 3D-structure; Acute phase; Blood coagulation; Calcium; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; Gamma-carboxyglutamic acid; Glycoprotein; Hemostasis; Hydrolase; Kringle; Pharmaceutical; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Thrombophilia; Zymogen Protein physicochemical properties Chain ID H Molecular weight (Da) 29321.6 Length 254 Aromaticity 0.1 Instability index 39.57 Isoelectric point 8.56 Charge (pH=7) 4.16 2D Binding mode Binding energy (Kcal/mol) -5.47 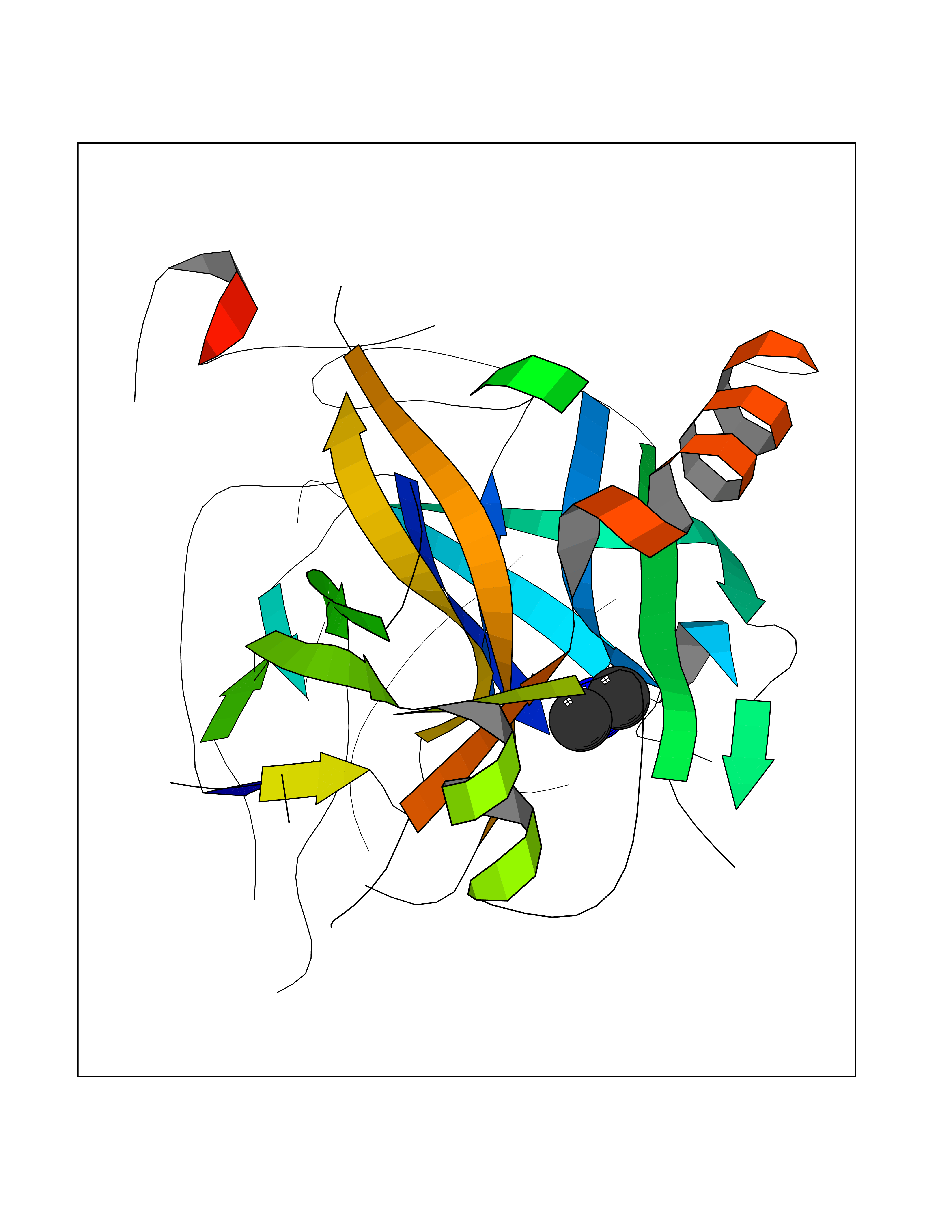 Molscript Map 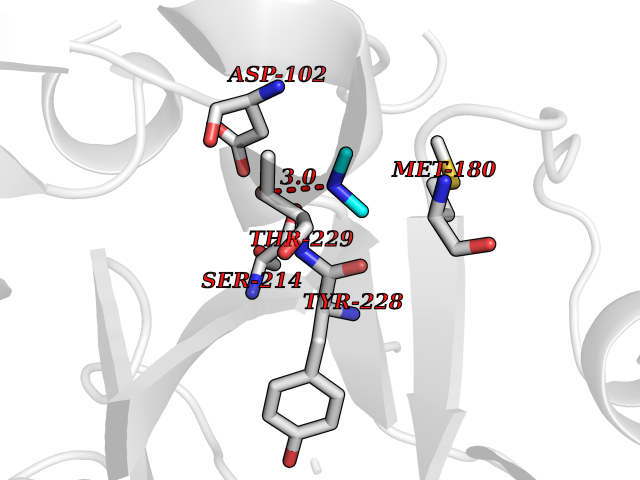 Pymol Map 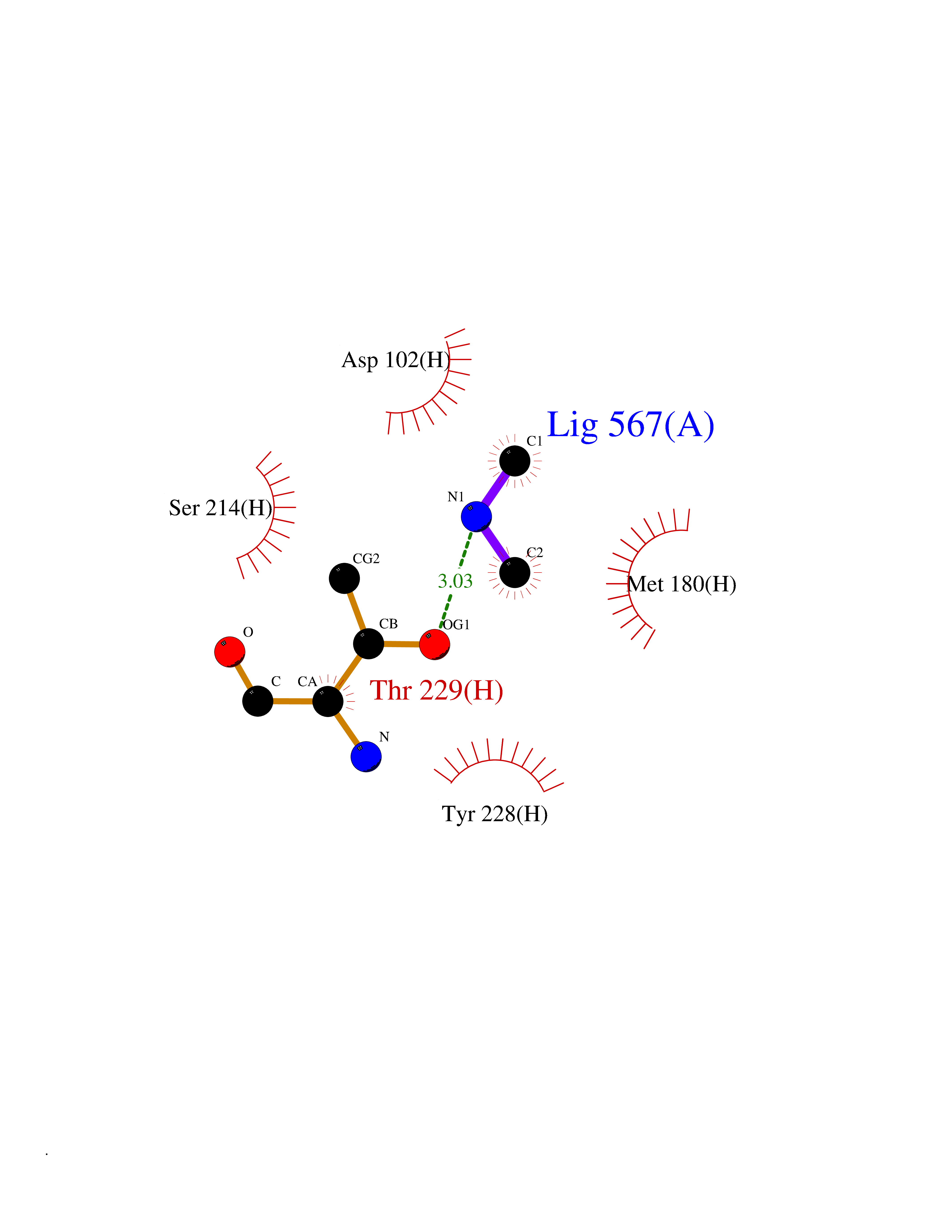 Ligplot Map 3D Binding mode Sequence IVEGSDAEIGMSPWQVMLFRSPQELLCGASLISDRWVLTAAHCLLTENDLLVRIGKHSRTRYRNIEKISMLEKIYIHPRYNWENLDRDIALMKLKKPVAFSDYIHPVCLPDRETLLQAGYKGRVTGWGNLKETWGQPSVLQVVNLPIVERPVCKDSTRIRITDNMFCAYKKRGDACEGDSGGPFVMKSNNRWYQMGIVSWGEGCRDGKYGFYTHVFRLKKWIQKVIDQFGGDFEEIPEELQCGLRPLFEKKSLE Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | mRNA-capping enzyme | 2C46 | 4.01 | |
Target general information Gen name RNGTT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CAP1A Protein family Non-receptor class of the protein-tyrosine phosphatase family; Eukaryotic GTase family Biochemical class Transferase Function GTP binding.MRNA guanylyltransferase activity.Polynucleotide 5'-phosphatase activity.Protein tyrosine/serine/threonine phosphatase activity.Protein tyrosine phosphatase activity.RNA guanylyltransferase activity.Triphosphatase activity. Related diseases Atrial fibrillation, familial, 14 (ATFB14) [MIM:615378]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:19808477}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Genetic variations in SCN2B may be involved in Brugada syndrome (PubMed:23559163). This tachyarrhythmia is characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:23559163}. Drugs (DrugBank ID) NA Interacts with Q92624; P16333-1 EC number 2.7.7.50; 3.6.1.74 Uniprot keywords 3D-structure; Alternative splicing; GTP-binding; Host-virus interaction; Hydrolase; mRNA capping; mRNA processing; Multifunctional enzyme; Nucleotide-binding; Nucleotidyltransferase; Nucleus; Protein phosphatase; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 21849.8 Length 189 Aromaticity 0.11 Instability index 53.71 Isoelectric point 5.89 Charge (pH=7) -2.91 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map  Pymol Map 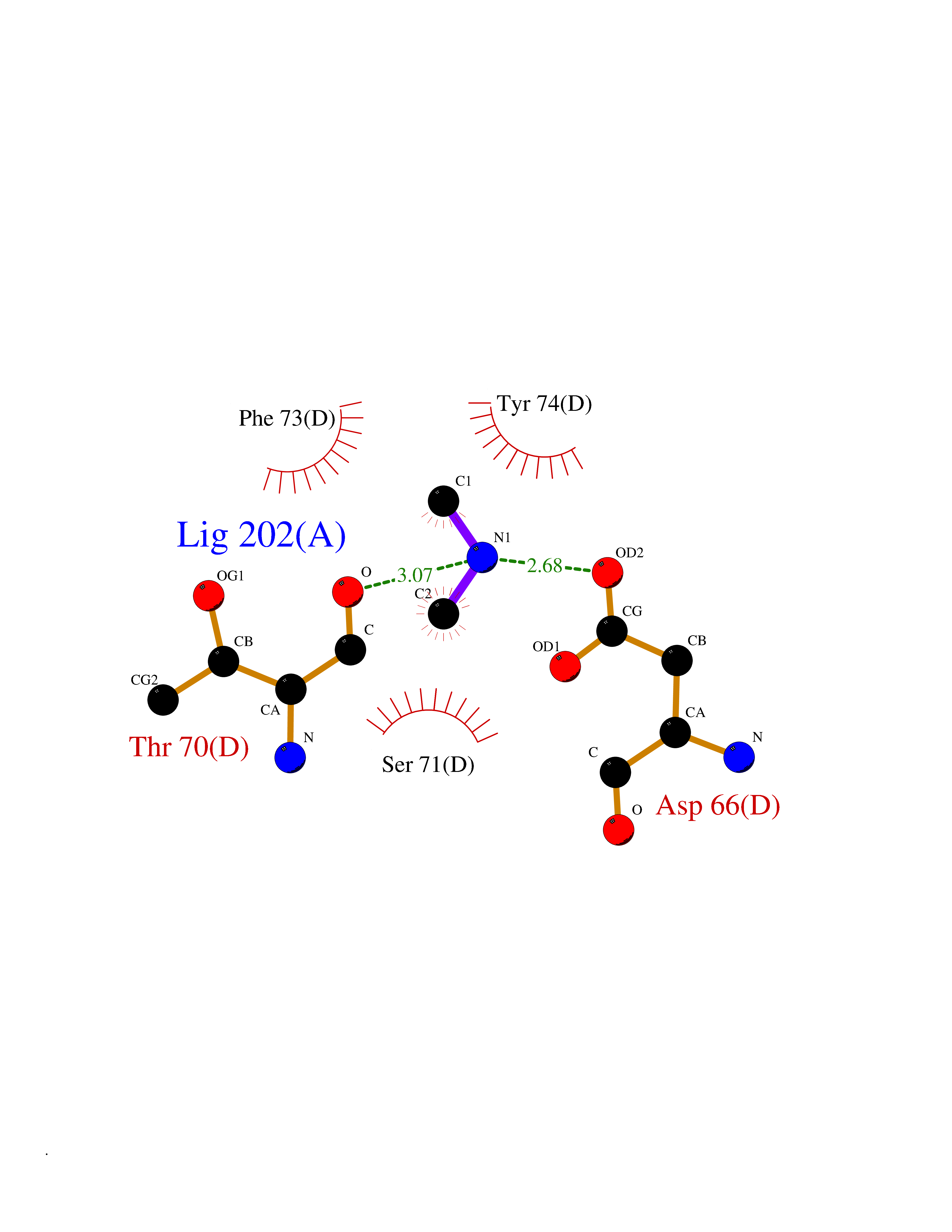 Ligplot Map 3D Binding mode Sequence NKIPPRWLNCPRRGQPVAGRFLPLKTMLGPRYDSQVAEENRFHPSMLSNYLKSVKMGLLVDLTNTSRFYDRNDIEKEGIKYIKLQCKGHGECPTTENTETFIRLCERFELIGVHCTHGFNRTGFLICAFLVEKMDWSIEAAVATFAQARPPGIYKGDYLKELFRRYGDIEEAPPPPLLPDWCFEDDEDE Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Beta-glucosidase A | 1E4I | 4.01 | |
Target general information Gen name bglA Organism Paenibacillus polymyxa (Bacillus polymyxa) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 1 family Biochemical class Hydrolase Function Beta-glucosidase activity.Scopolin beta-glucosidase activity. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02658; DB04282; DB04304 Interacts with NA EC number 3.2.1.21 Uniprot keywords 3D-structure; Carbohydrate metabolism; Cellulose degradation; Glycosidase; Hydrolase; Polysaccharide degradation Protein physicochemical properties Chain ID A Molecular weight (Da) 51515.2 Length 447 Aromaticity 0.14 Instability index 38.44 Isoelectric point 5.28 Charge (pH=7) -18.1 2D Binding mode Binding energy (Kcal/mol) -5.48 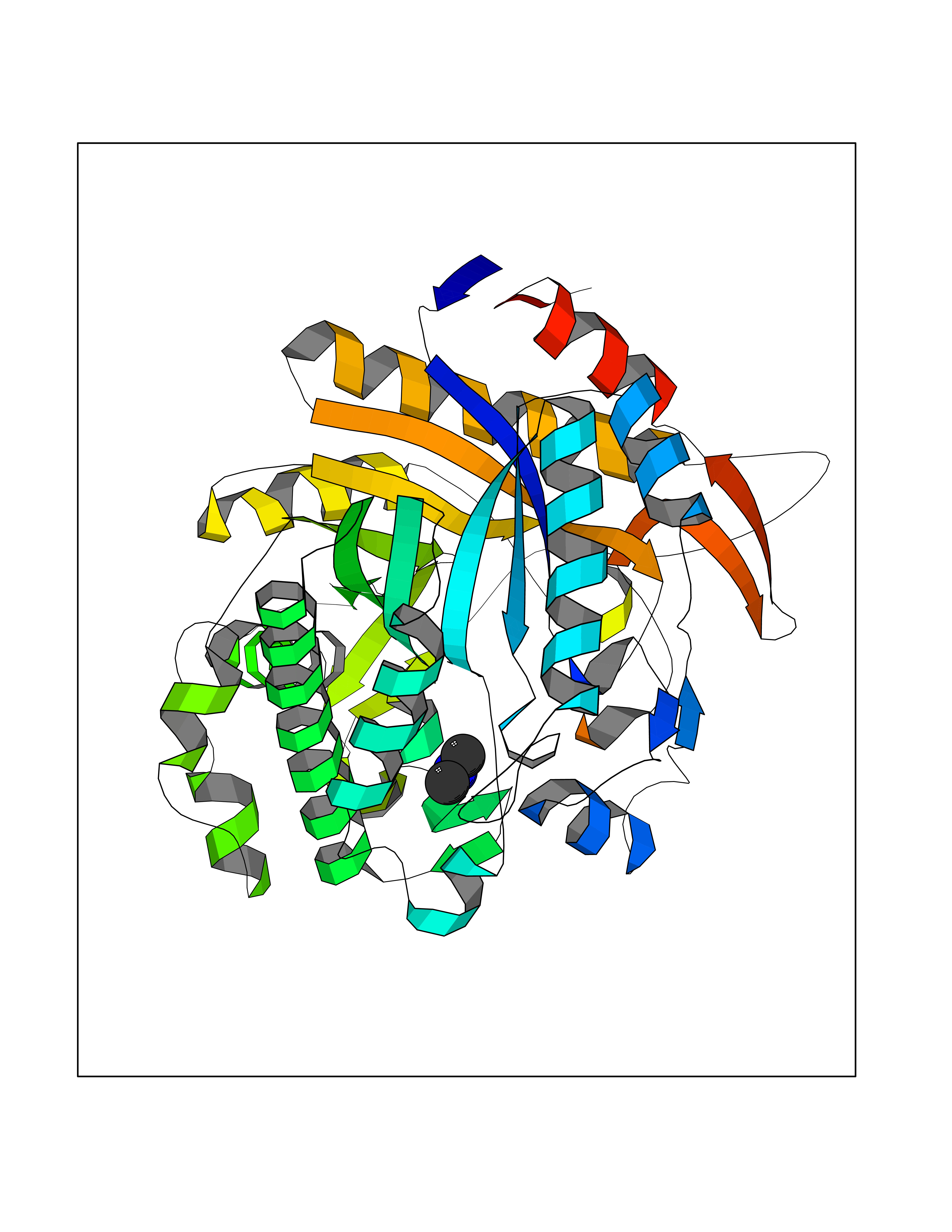 Molscript Map  Pymol Map 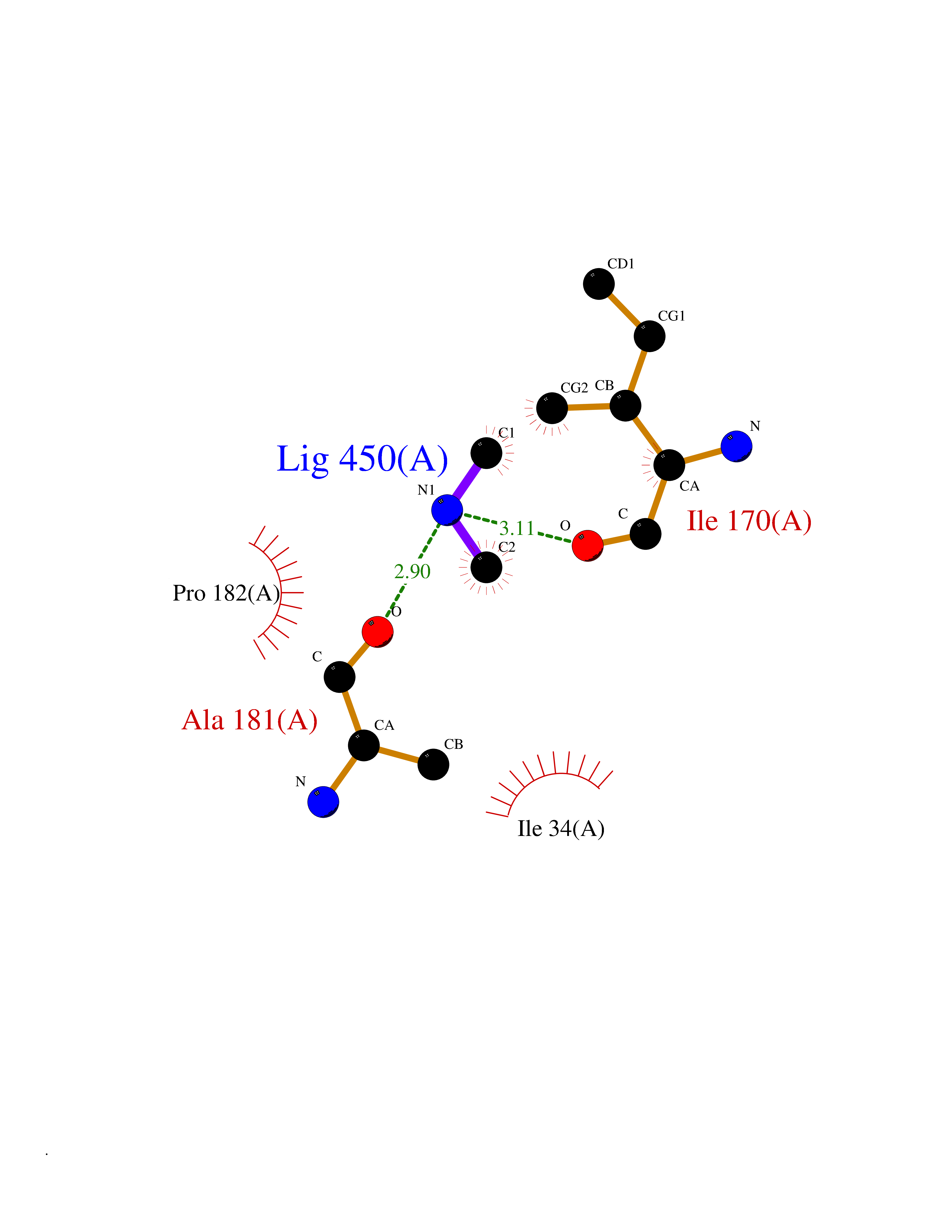 Ligplot Map 3D Binding mode Sequence TIFQFPQDFMWGTATAAYQIEGAYQEDGRGLSIWDTFAHTPGKVFNGDNGNVACDSYHRYEEDIRLMKELGIRTYRFSVSWPRIFPNGDGEVNQKGLDYYHRVVDLLNDNGIEPFCTLYHWDLPQALQDAGGWGNRRTIQAFVQFAETMFREFHGKIQHWLTFNEPWCIAFLSNMLGVHAPGLTNLQTAIDVGHHLLVAHGLSVRRFRELGTSGQIGIAPNVSWAVPYSTSEEDKAACARTISLHSDWFLQPIYQGSYPQFLVDWFAEQGATVPIQDGDMDIIGEPIDMIGINYYSMSVNRFNPEAGFLQSEEINMGLPVTDIGWPVESRGLYEVLHYLQKYGNIDIYITENGACINDEVVNGKVQDDRRISYMQQHLVQVHRTIHDGLHVKGYMAWSLLDNFEWAEGYNMRFGMIHVDFRTQVRTPKQSYYWYRNVVSNNWLETRR Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | S-adenosylmethionine decarboxylase proenzyme (AMD1) | 1JL0 | 4.01 | |
Target general information Gen name AMD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SamDC; S-adenosylmethioninedecarboxylase; AdoMetDC; AMD Protein family Eukaryotic AdoMetDC family Biochemical class Carbon-carbon lyase Function Promotes maintenance and self-renewal of embryonic stem cells, by maintaining spermine levels. Essential for biosynthesis of the polyamines spermidine and spermine. Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08163; DB00118; DB01917 Interacts with P17707; Q96A98; Q8WY91 EC number EC 4.1.1.50 Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Decarboxylase; Direct protein sequencing; Lyase; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Pyruvate; Reference proteome; S-adenosyl-L-methionine; Schiff base; Spermidine biosynthesis; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 35790.5 Length 311 Aromaticity 0.14 Instability index 39.47 Isoelectric point 6.03 Charge (pH=7) -2.01 2D Binding mode Binding energy (Kcal/mol) -5.46 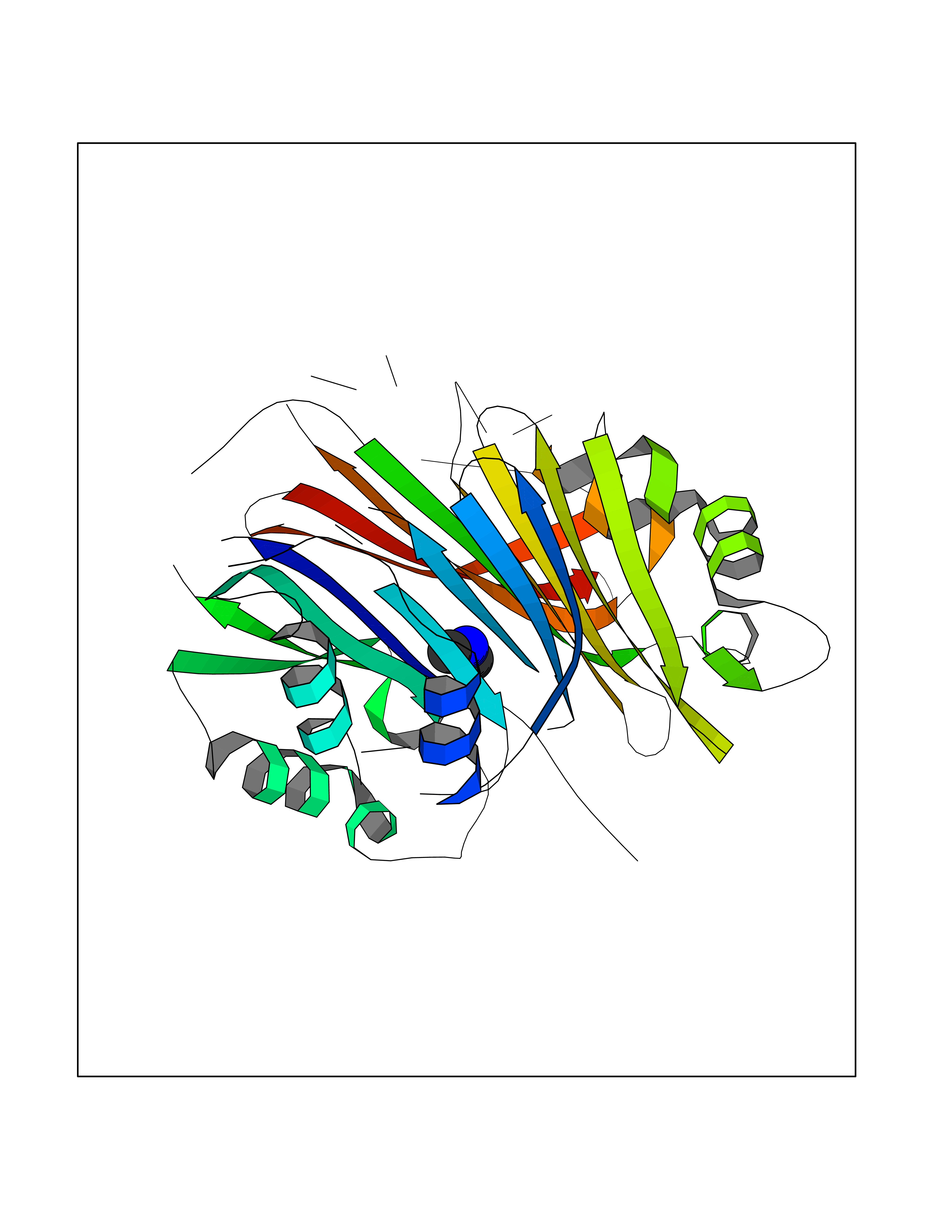 Molscript Map 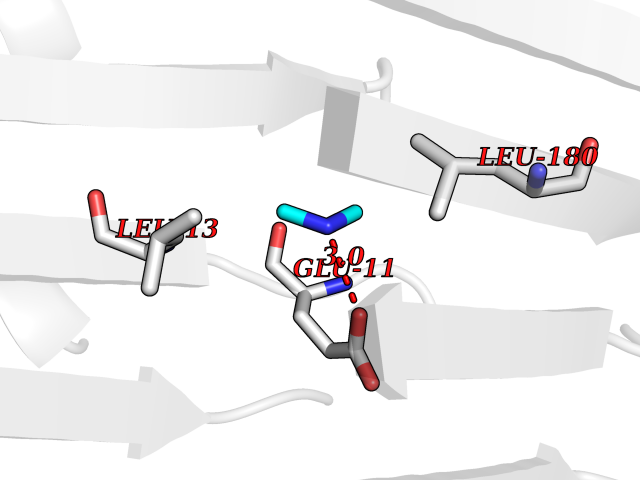 Pymol Map 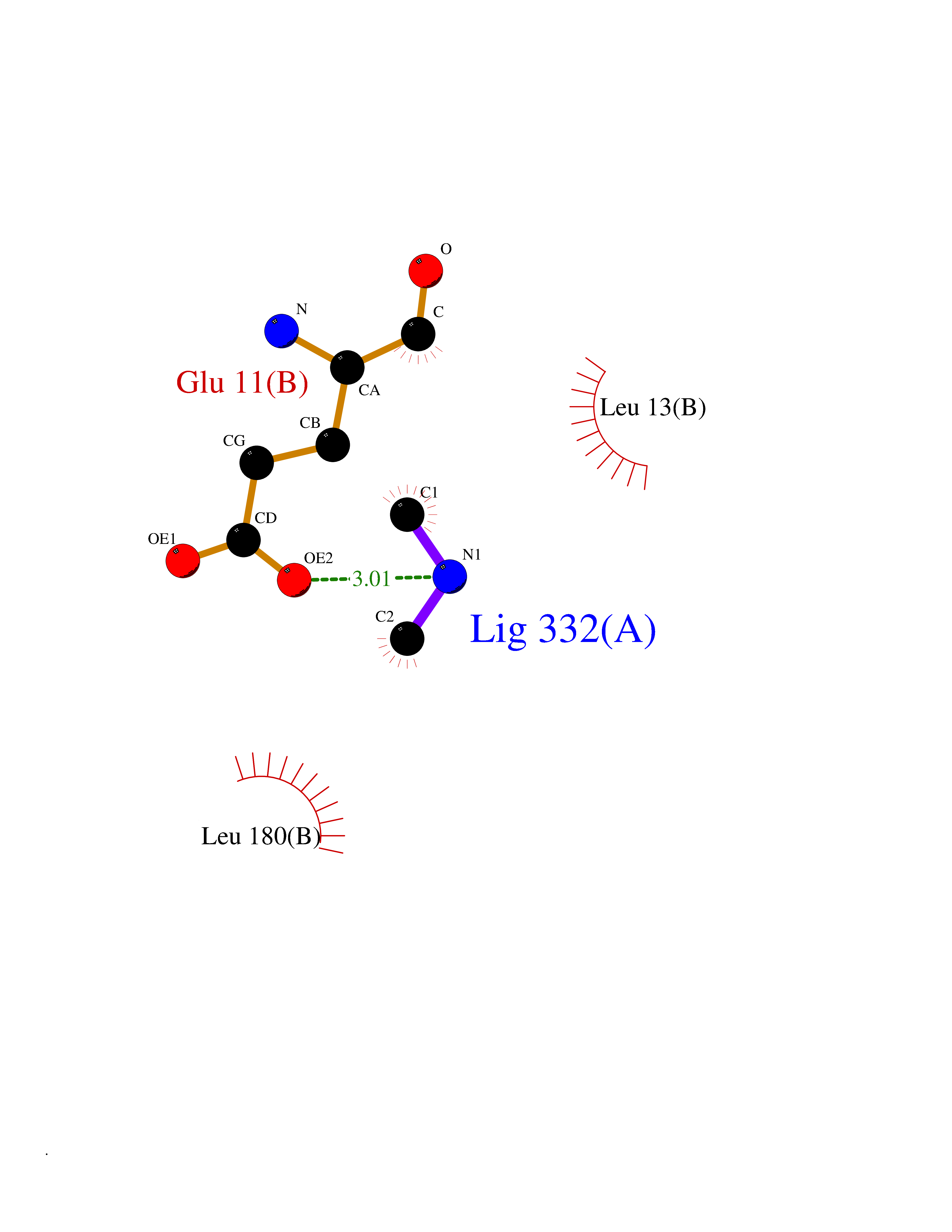 Ligplot Map 3D Binding mode Sequence HFFEGTEKLLEVWFSRQGSGDLRTIPRSEWDILLKDVQCSIISVTKTDKQEAYVLSESSMFVSKRRFILKTCGTTLLLKALVPLLKLARDYSGFDSIQSFFYSRKNFMKPSHQGYPHRNFQEEIEFLNAIFPNGAGYCMGRMNSDCWYLYTLDFRVISQPDQTLEILMSELDPAVMDQFYMKDGVTAKDVTRESGIRDLIPGSVIDATMFNPCGYSMNGMKSDGTYWTIAITPEPEFSYVSFETNLSQTSYDDLIRKVVEVFKPGKFVTTLFVNQSSKCPQKIEGFKRLDCQSAMFNDYNFVFTSFAKKQQ Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Gamma-aminobutyric acid receptor subunit beta-3 | 4COF | 4.01 | |
Target general information Gen name GABRB3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Ligand-gated ion channel (TC 1.A.9) family, Gamma-aminobutyric acid receptor (TC 1.A.9.5) subfamily, GABRB3 sub-subfamily Biochemical class Transport protein Function GABA-A receptor activity.GABA-gated chloride ion channel activity.Identical protein binding. Related diseases Epilepsy, childhood absence 5 (ECA5) [MIM:612269]: A subtype of idiopathic generalized epilepsy characterized by an onset at age 6-7 years, frequent absence seizures (several per day) and bilateral, synchronous, symmetric 3-Hz spike waves on EEG. Tonic-clonic seizures often develop in adolescence. Absence seizures may either remit or persist into adulthood. {ECO:0000269|PubMed:18514161, ECO:0000269|PubMed:22303015}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 43 (DEE43) [MIM:617113]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE43 inheritance is autosomal dominant. {ECO:0000269|PubMed:23934111, ECO:0000269|PubMed:25356899, ECO:0000269|PubMed:26950270, ECO:0000269|PubMed:26993267, ECO:0000269|PubMed:27476654, ECO:0000269|PubMed:27864847}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12537; DB00546; DB00404; DB00543; DB11901; DB14719; DB11859; DB01558; DB09017; DB00237; DB00241; DB01489; DB00475; DB14715; DB01594; DB00349; DB01068; DB00628; DB01559; DB01553; DB01511; DB01189; DB00829; DB13837; DB00228; DB01215; DB00402; DB00898; DB00189; DB01545; DB09166; DB00292; DB01567; DB01205; DB01544; DB00690; DB06716; DB05087; DB01437; DB00801; DB01159; DB00753; DB01587; DB00555; DB00431; DB13643; DB00186; DB13872; DB13437; DB00603; DB01043; DB00371; DB00463; DB01028; DB01107; DB15489; DB00683; DB12458; DB01595; DB14028; DB00842; DB14672; DB00312; DB00252; DB13335; DB00592; DB01708; DB01588; DB00794; DB00818; DB01589; DB12404; DB01236; DB09118; DB00306; DB01956; DB00231; DB11582; DB00897; DB15490 Interacts with P28472 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Chloride; Chloride channel; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 76970.6 Length 663 Aromaticity 0.15 Instability index 39.4 Isoelectric point 8.37 Charge (pH=7) 4.12 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 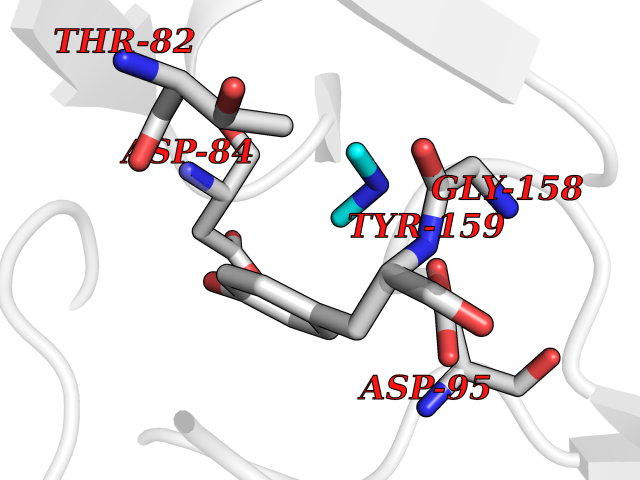 Pymol Map  Ligplot Map 3D Binding mode Sequence SFVKETVDKLLKGYDIRLRPDFGGPPVCVGMNIDIASIDMVSEVNMDYTLTMYFQQYWRDKRLAYSGIPLNLTLDNRVADQLWVPDTYFLNDKKSFVHGVTVKNRMIRLHPDGTVLYGLRITTTAACMMDLRRYPLDEQNCTLEIESYGYTTDDIEFYWRGGDKAVTGVERIELPQFSIVEHRLVSRNVVFATGAYPRLSLSFRLKRNIGYFILQTYMPSILITILSWVSFWINYDASAARVALGITTVLTMTTINTHLRETLPKIPYVKAIDMYLMGCFVFVFLALLEYAFVNYIFFSQPARAAAIDRWSRIVFPFTFSLFNLVYWLYYVNSFVKETVDKLLKGYDIRLRPDFGGPPVCVGMNIDIASIDMVSEVNMDYTLTMYFQQYWRDKRLAYSGIPLNLTLDNRVADQLWVPDTYFLNDKKSFVHGVTVKNRMIRLHPDGTVLYGLRITTTAACMMDLRRYPLDEQNCTLEIESYGYTTDDIEFYWRGGDKAVTGVERIELPQFSIVEHRLVSRNVVFATGAYPRLSLSFRLKRNIGYFILQTYMPSILITILSWVSFWINYDASAARVALGITTVLTMTTINTHLRETLPKIPYVKAIDMYLMGCFVFVFLALLEYAFVNYIFFSQPARAAAIDRWSRIVFPFTFSLFNLVYWLYYV Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Isovaleryl-CoA dehydrogenase, mitochondrial | 1IVH | 4.01 | |
Target general information Gen name IVD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Isovaleryl-CoA dehydrogenase activity. Related diseases Isovaleric acidemia (IVA) [MIM:243500]: A metabolic disorder characterized by retarded psychomotor development, a peculiar odor resembling sweaty feet, an aversion to dietary protein, and pernicious vomiting, leading to acidosis and coma. The acute neonatal form leads to massive metabolic acidosis from the first days of life and rapid death. {ECO:0000269|PubMed:2063866, ECO:0000269|PubMed:22004070, ECO:0000269|PubMed:22350545, ECO:0000269|PubMed:23587913, ECO:0000269|PubMed:28535199, ECO:0000269|PubMed:9665741}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04036; DB03147 Interacts with Q08043; Q9Y4H4 EC number 1.3.8.1; 1.3.8.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; FAD; Fatty acid metabolism; Flavoprotein; Lipid metabolism; Mitochondrion; Oxidoreductase; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 84454.2 Length 774 Aromaticity 0.08 Instability index 30.01 Isoelectric point 6.85 Charge (pH=7) -0.77 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence VDDAINGLSEEQRQLRQTMAKFLQEHLAPKAQEIDRSNEFKNLREFWKQLGNLGVLGITAPVQYGGSGLGYLEHVLVMEEISRASGAVGLSYGAHSNLCINQLVRNGNEAQKEKYLPKLISGEYIGALAMSEPNAGSDVVSMKLKAEKKGNHYILNGNKFWITNGPDADVLIVYAKTDLAAVPASRGITAFIVEKGMPGFSTSKKLDKLGMRGSNTCELIFEDCKIPAANILGHENKGVYVLMSGLDLERLVLAGGPLGLMQAVLDHTIPYLHVREAFGQKIGHFQLMQGKMADMYTRLMACRQYVYNVAKACDEGHCTAKDCAGVILYSAECATQVALDGIQCFGGNGYINDFPMGRFLRDAKLYEIGAGTSEVRRLVIGRAFNADVDDAINGLSEEQRQLRQTMAKFLQEHLAPKAQEIDRSNEFKNLREFWKQLGNLGVLGITAPVQYGGSGLGYLEHVLVMEEISRASGAVGLSYGAHSNLCINQLVRNGNEAQKEKYLPKLISGEYIGALAMSEPNAGSDVVSMKLKAEKKGNHYILNGNKFWITNGPDADVLIVYAKTDLAAVPASRGITAFIVEKGMPGFSTSKKLDKLGMRGSNTCELIFEDCKIPAANILGHENKGVYVLMSGLDLERLVLAGGPLGLMQAVLDHTIPYLHVREAFGQKIGHFQLMQGKMADMYTRLMACRQYVYNVAKACDEGHCTAKDCAGVILYSAECATQVALDGIQCFGGNGYINDFPMGRFLRDAKLYEIGAGTSEVRRLVIGRAFNAD Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Lanosterol 14-alpha-demethylase (EC 1.14.13.70) | 4G3J | 4.01 | |
Target general information Gen name Tb11.02.4080 Organism Trypanosoma brucei brucei (strain 927/4 GUTat10.1) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class NA Function NA Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) NA Interacts with NA EC number 1.14.13.70 Uniprot keywords 3D-structure; Heme; Iron; Metal-binding; Monooxygenase; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID D Molecular weight (Da) 50691.4 Length 448 Aromaticity 0.08 Instability index 54.09 Isoelectric point 6.99 Charge (pH=7) -0.03 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GKLPPVYPVTVPILGHIIQFGKSPLGFMQECKRQLKSGIFTINIVGKRVTIVGDPHEHSRFFLPRNEVLSPREVYSFMVPVFGEGVAYAAPYPRMREQLNFLAEELTIAKFQNFVPAIQHEVRKFMAANWDKDEGEINLLEDCSTMIINTACQCLFGEDLRKRLDARRFAQLLAKMESSLIPAAVFLPILLKLPLPQSARCHEARTELQKILSEIIIARKEEEVNKDSSTSDLLSGLLSAVYRDGTPMSLHEVCGMIVAAMFAGQHTSSITTTWSMLHLMHPANVKHLEALRKEIEEFPAQLNYNNVMDEMPFAERCARESIRRDPPLLMLMRKVMADVKVGSYVVPKGDIIACSPLLSHHDEEAFPEPRRWDPERDEKVEGAFIGFGAGVHKCIGQKFGLLQVKTILATAFRSYDFQLLRDEVPDPDYHTMVVGPTASQCRVKYIRR Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Leukotriene C4 synthase (LTC4S) | 3PCV | 4.01 | |
Target general information Gen name LTC4S Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Leukotriene-C(4)Leukotriene C4 synthase synthase; Leukotriene-C(4) synthase; LTC4 synthase Protein family MAPEG family Biochemical class Carbon-sulfur lyase Function Catalyzes the conjugation of leukotriene A4 with reduced glutathione to form leukotriene C4. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB00972; DB00143 Interacts with P11912; Q96BA8; Q15125; O14843; Q14802-3; Q8TDT2; O15529; Q16873; Q8TBB6; Q4KMG9 EC number EC 4.4.1.20 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Leukotriene biosynthesis; Lipid biosynthesis; Lipid metabolism; Lyase; Membrane; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 16099 Length 146 Aromaticity 0.13 Instability index 38.66 Isoelectric point 10.2 Charge (pH=7) 7.66 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MKDEVALLAAVTLLGVLLQAYFSLQVISARRAFRVSPPLTTGPPEFERVYRAQVNCSEYFPLFLATLWVAGIFFHEGAAALCGLVYLFARLRYFQGYARSAQLRLAPLYASARALWLLVALAALGLLAHFLPAALRAALLGRLRTL Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Neutral endopeptidase (MME) | 6XVP | 4.01 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Skin fibroblast elastase; SFE; Neutral endopeptidase 24.11; Neprilysin; NEP protein; Enkephalinase; EPN; Common acute lymphocytic leukemia antigen; CD10; CALLA; Atriopeptidase Protein family Peptidase M13 family Biochemical class Peptidase Function Biologically important in the destruction of opioid peptides such as Met- and Leu-enkephalins by cleavage of a Gly-Phe bond. Able to cleave angiotensin-1, angiotensin-2 and angiotensin 1-9. Involved in the degradation of atrial natriuretic factor (ANF). Displays UV-inducible elastase activity toward skin preelastic and elastic fibers. Thermolysin-like specificity, but is almost confined on acting on polypeptides of up to 30 amino acids. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number EC 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map  Pymol Map 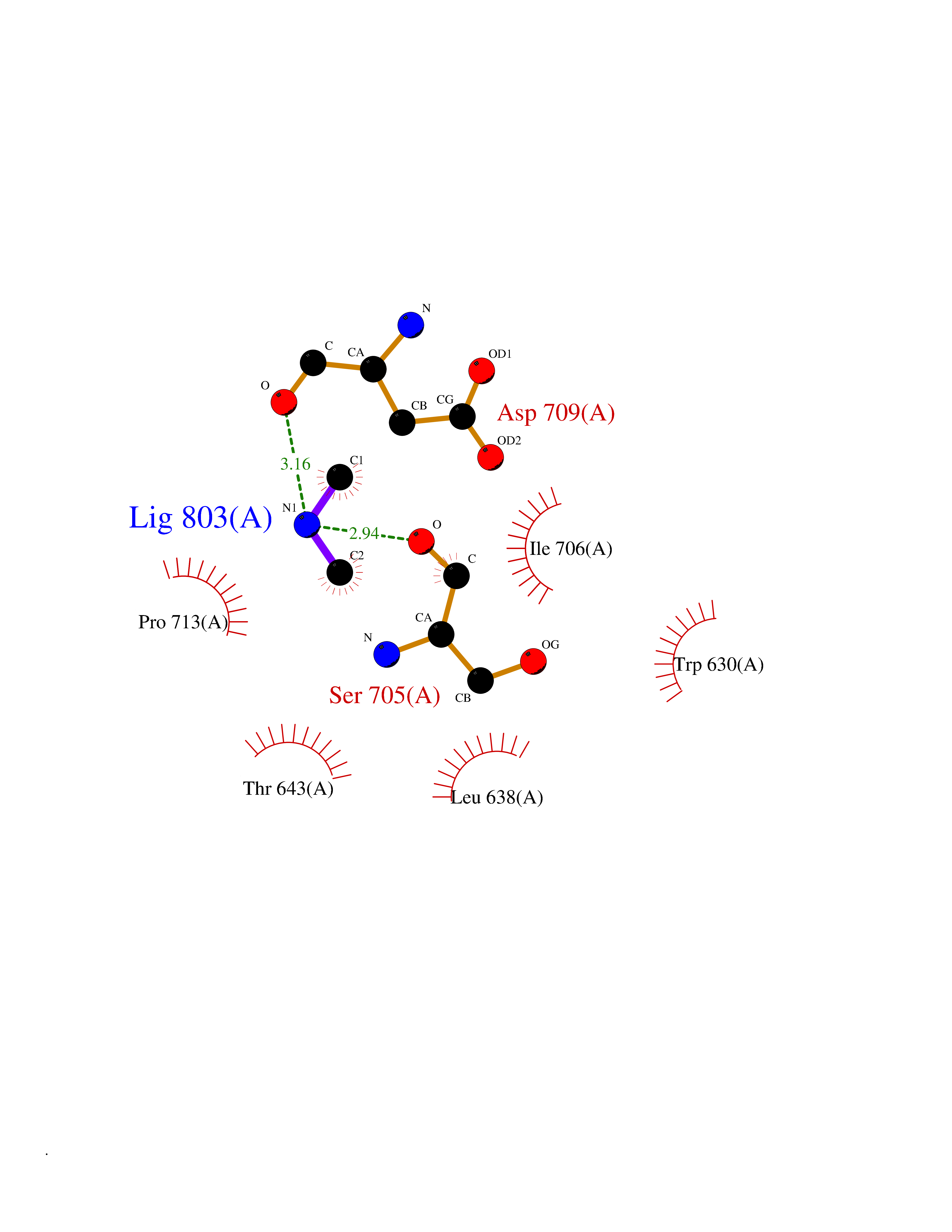 Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Kallikrein-5 (KLK5) | 6QFE | 4.01 | |
Target general information Gen name KLK5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ570/PRO1132; Stratum corneum tryptic enzyme; SCTE; Kallikrein-like protein 2; KLK-L2 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function May be involved in desquamation. Related diseases Lipodystrophy, familial partial, 8 (FPLD8) [MIM:620679]: An autosomal dominant form of partial lipodystrophy, a disorder characterized by abnormal subcutaneous fat distribution. FPLD8 patients show selective loss of subcutaneous adipose tissue from the limbs, beginning around 13 to 15 years of age, and abnormal accumulation of subcutaneous adipose tissue in the dorsal neck and face, as well as in the posterior thoracic and abdominal regions. The disorder is associated with metabolic abnormalities, including diabetes mellitus and hyperlipidemia. {ECO:0000269|PubMed:27376152}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P20930; Q9NQG1 EC number EC 3.4.21.- Uniprot keywords 3D-structure; Disulfide bond; Glycoprotein; Hydrolase; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50299.2 Length 454 Aromaticity 0.07 Instability index 40.74 Isoelectric point 9.25 Charge (pH=7) 23.09 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 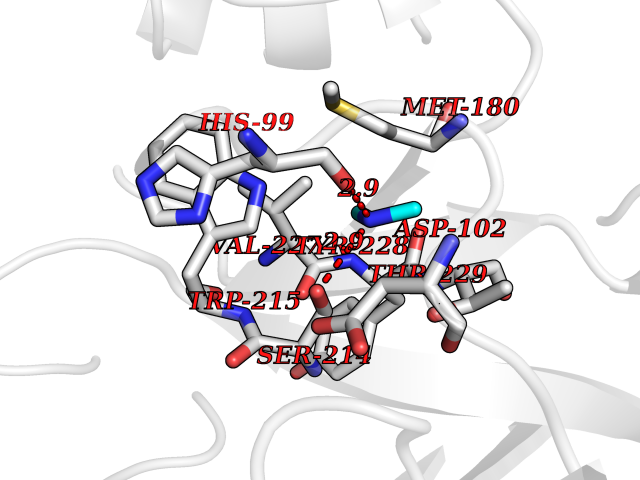 Pymol Map 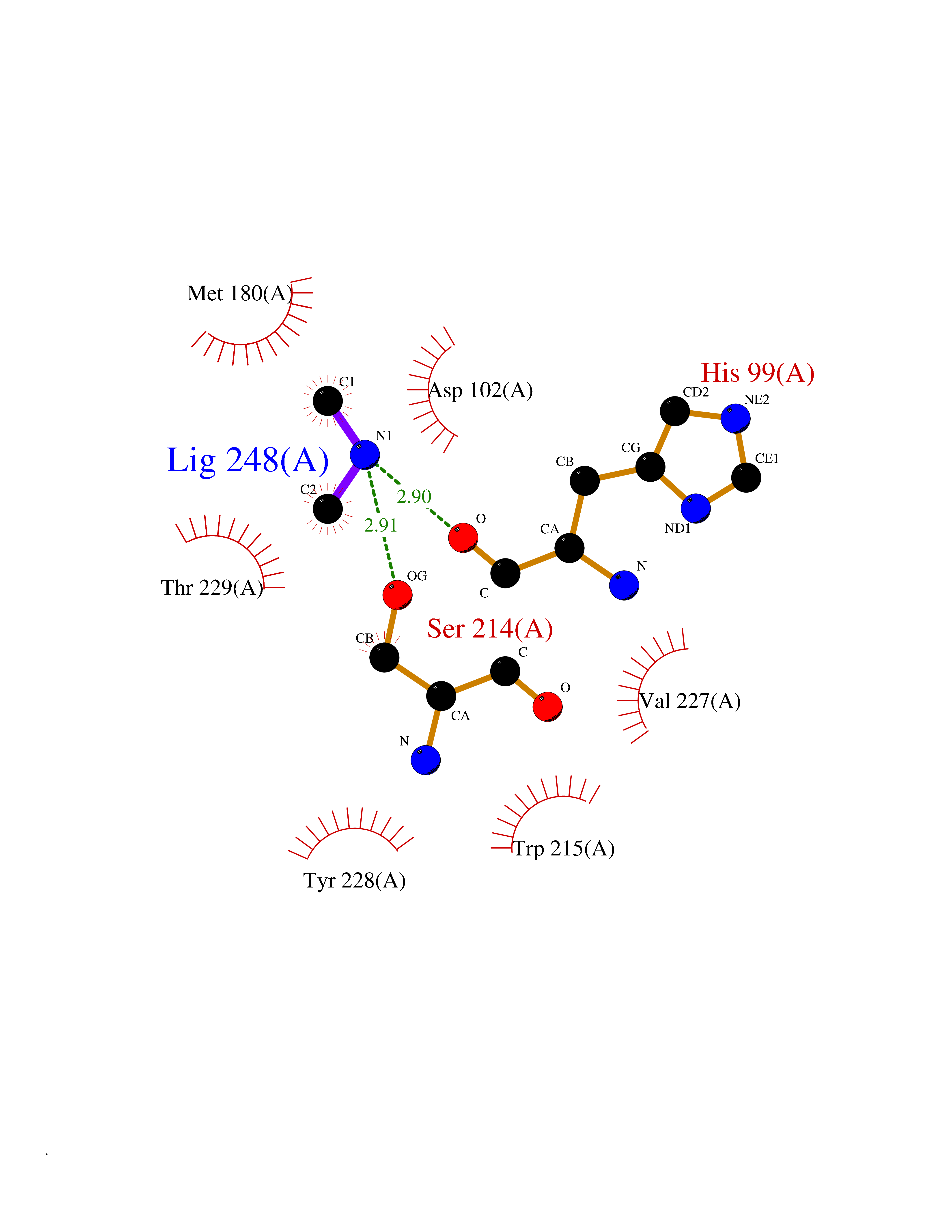 Ligplot Map 3D Binding mode Sequence IINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANSIINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANS Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Orotidine 5'-monophosphate decarboxylase (UMPS) | 3MI2 | 4.01 | |
Target general information Gen name UMPS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Uridine 5'-monophosphate synthase; UMP synthase Protein family Purine/pyrimidine phosphoribosyltransferase family; OMP decarboxylase family Biochemical class Pentosyltransferase Function Catalyses the formation of uridine monophosphate (UMP), an energy-carrying molecule in many important biosynthetic pathways. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02890; DB00544 Interacts with P54764; P11172-1 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Decarboxylase; Disease variant; Glycosyltransferase; Lyase; Multifunctional enzyme; Phosphoprotein; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 55844 Length 514 Aromaticity 0.06 Instability index 22.7 Isoelectric point 6.44 Charge (pH=7) -2.99 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KELSFGARAELPRIHPVASKLLRLMQKKETNLCLSADVSLARELLQLADALGPSICMLKTHVDILNDFTLDVMKELITLAKXHEFLIFEDRKFADIGNTVKKQYEGGIFKIASWADLVNAHVVPGSGVVKGLQEVGLPLHRGCLLIAEMSSTGSLATGDYTRAAVRMAEEHSEFVVGFISGSRVSMKPEFLHLTPGVQLEAGGDNLGQQYNSPQEVIGKRGSDIIIVGRGIISAADRLEAAEMYRKAAWEAYLSRLGKELSFGARAELPRIHPVASKLLRLMQKKETNLCLSADVSLARELLQLADALGPSICMLKTHVDILNDFTLDVMKELITLAKXHEFLIFEDRKFADIGNTVKKQYEGGIFKIASWADLVNAHVVPGSGVVKGLQEVGLPLHRGCLLIAEMSSTGSLATGDYTRAAVRMAEEHSEFVVGFISGSRVSMKPEFLHLTPGVQLEAGGDNLGQQYNSPQEVIGKRGSDIIIVGRGIISAADRLEAAEMYRKAAWEAYLSRLG Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | N-acetylgalactosamine 6 sulfatase (GALNS) | 4FDJ | 4.01 | |
Target general information Gen name GALNS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms N-acetylgalactosamine-6-sulfate sulfatase; N-acetylgalactosamine-6-sulfatase; Galactose-6-sulfate sulfatase; GalNAc6S sulfatase; GalN6S; Chondroitinsulfatase; Chondroitinase Protein family Sulfatase family Biochemical class Sulfuric ester hydrolase Function Catalyzes the chemical reaction of cleaving off the 6-sulfate groups of the N-acetyl-D-galactosamine 6-sulfate units of the macromolecule chondroitin sulfate and, similarly, of the D-galactose 6-sulfate units of the macromolecule keratan sulfate. Related diseases Mucopolysaccharidosis 4A (MPS4A) [MIM:253000]: A form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life. {ECO:0000269|PubMed:1522213, ECO:0000269|PubMed:16287098, ECO:0000269|PubMed:24726177, ECO:0000269|PubMed:7581409, ECO:0000269|PubMed:7633425, ECO:0000269|PubMed:7668283, ECO:0000269|PubMed:7795586, ECO:0000269|PubMed:8651279, ECO:0000269|PubMed:8826435, ECO:0000269|PubMed:9298823, ECO:0000269|PubMed:9375852, ECO:0000269|PubMed:9521421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09301 Interacts with NA EC number EC 3.1.6.4 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Dwarfism; Glycoprotein; Hydrolase; Lysosome; Metal-binding; Mucopolysaccharidosis; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 55013.8 Length 493 Aromaticity 0.11 Instability index 35.46 Isoelectric point 6.14 Charge (pH=7) -6.48 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PQPPNILLLLMDDMGWGDLGVYGEPSRETPNLDRMAAEGLLFPNFYSANPLXSPSRAALLTGRLPIRNGFYTTNAHARNAYTPQEIVGGIPDSEQLLPELLKKAGYVSKIVGKWHLGHRPQFHPLKHGFDEWFGSPNCHFGPYDNKARPNIPVYRDWEMVGRYYEEFPINLKTGEANLTQIYLQEALDFIKRQARHHPFFLYWAVDATHAPVYASKPFLGTSQRGRYGDAVREIDDSIGKILELLQDLHVADNTFVFFTSDNGAALISAPEQGGSNGPFLCGKQTTFEGGMREPALAWWPGHVTAGQVSHQLGSIMDLFTTSLALAGLTPPSDRAIDGLNLLPTLLQGRLMDRPIFYYRGDTLMAATLGQHKAHFWTWTNSWENFRQGIDFCPGQNVSGVTTHNLEDHTKLPLIFHLGRDPGERFPLSFASAEYQEALSRITSVVQQHQEALVPAQPQLNVCNWAVMNWAPPGCEKLGKCLTPPESIPKKCLW Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Soluble epoxide hydrolase (EPHX2) | 1ZD3 | 4.01 | |
Target general information Gen name EPHX2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Bifunctional epoxide hydrolase 2 Protein family AB hydrolase superfamily, Epoxide hydrolase family Biochemical class Ether bond hydrolase Function Bifunctional enzyme. The C-terminal domain has epoxide hydrolase activity and acts on epoxides (alkene oxides, oxiranes) and arene oxides. Plays a role in xenobiotic metabolism by degrading potentially toxic epoxides (By similarity). Also determines steady-state levels of physiological mediators. The N-terminal domain has lipid phosphatase activity, with the highest activity towards threo-9,10-phosphonooxy-hydroxy-octadecanoic acid, followed by erythro-9,10-phosphonooxy-hydroxy-octadecanoic acid, 12-phosphonooxy-octadec-9Z-enoic acid and 12-phosphonooxy-octadec-9E-enoic acid. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08257; DB08258; DB08259; DB06345; DB12610; DB08256; DB02029; DB04213; DB03677 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aromatic hydrocarbons catabolism; Cytoplasm; Detoxification; Direct protein sequencing; Hydrolase; Lipid metabolism; Lipoprotein; Magnesium; Metal-binding; Multifunctional enzyme; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 61744.9 Length 547 Aromaticity 0.09 Instability index 43.97 Isoelectric point 5.81 Charge (pH=7) -7.76 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map  Pymol Map 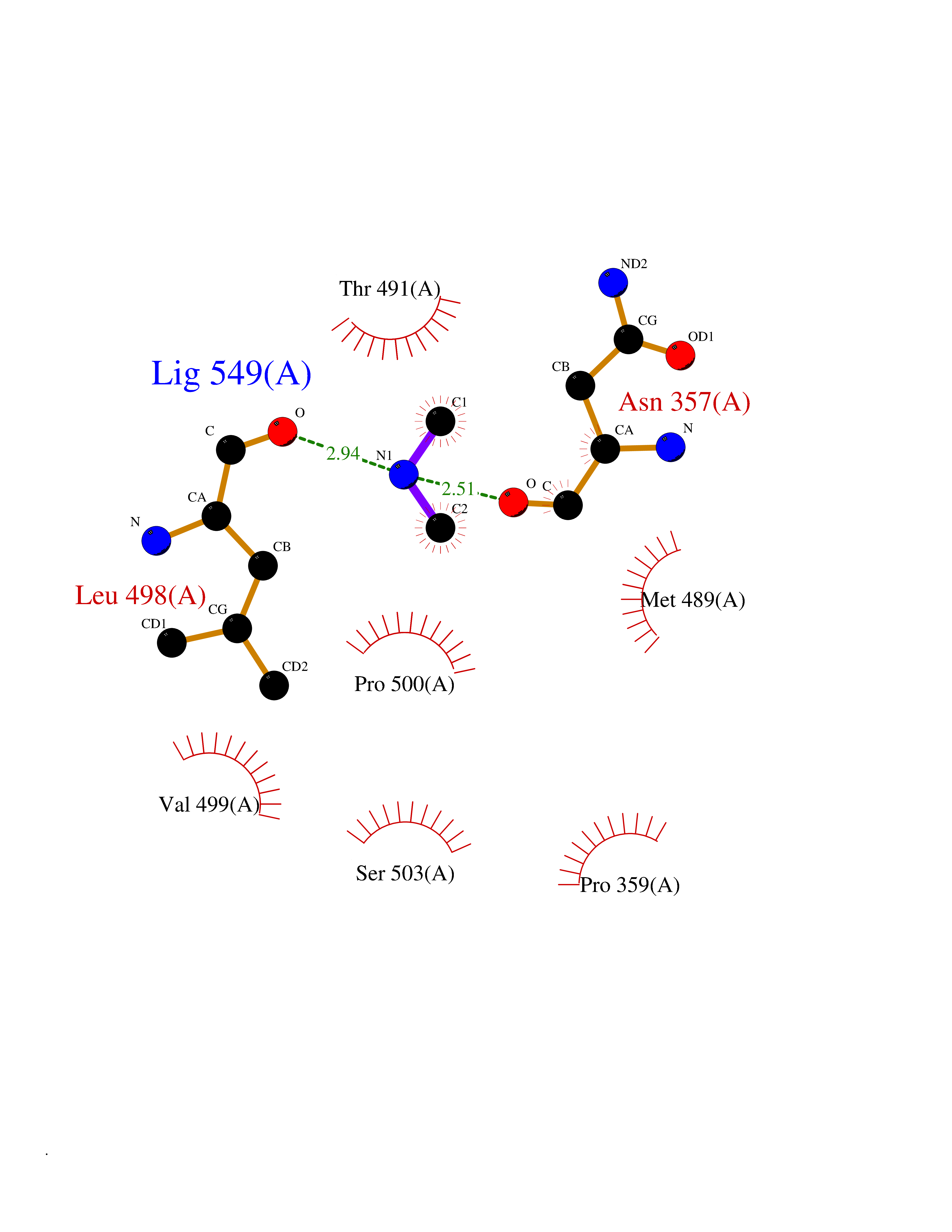 Ligplot Map 3D Binding mode Sequence TLRAAVFDLDGVLALPAVFGVLGRTEEALALPRGLLNDAFQKGGPEGATTRLMKGEITLSQWIPLMEENCRKCSETAKVCLPKNFSIKEIFDKAISARKINRPMLQAALMLRKKGFTTAILTNTWLDDRAERDGLAQLMCELKMHFDFLIESCQVGMVKPEPQIYKFLLDTLKASPSEVVFLDDIGANLKPARDLGMVTILVQDTDTALKELEKVTGIQLLNTPAPLPTSCNPSDMSHGYVTVKPRVRLHFVELGSGPAVCLCHGFPESWYSWRYQIPALAQAGYRVLAMDMKGYGESSAPPEIEEYCMEVLCKEMVTFLDKLGLSQAVFIGHDWGGMLVWYMALFYPERVRAVASLNTPFIPANPNMSPLESIKANPVFDYQLYFQEPGVAEAELEQNLSRTFKSLFRASDESVLSMHKVCEAGGLFVNSPEEPSLSRMVTEEEIQFYVQQFKKSGFRGPLNWYRNMERNWKWACKSLGRKILIPALMVTAEKDFVLVPQMSQHMEDWIPHLKRGHIEDCGHWTQMDKPTEVNQILIKWLDSDARN Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | 6YND | 4.01 | |
Target general information Gen name GAPDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peptidyl-cysteine S-nitrosylase GAPDH; OK/SW-cl.12; GAPD; D-glyceraldehyde-3-phosphate dehydrogenase; CDABP0047 Protein family Glyceraldehyde-3-phosphate dehydrogenase family Biochemical class Aldehyde/oxo donor oxidoreductase Function Participates in nuclear events including transcription, RNA transport, DNA replication and apoptosis. Nuclear functions are probably due to the nitrosylase activity that mediates cysteine S-nitrosylation of nuclear target proteins such as SIRT1, HDAC2 and PRKDC. Modulates the organization and assembly of the cytoskeleton. Facilitates the CHP1-dependent microtubule and membrane associations through its ability to stimulate the binding of CHP1 to microtubules. Glyceraldehyde-3-phosphate dehydrogenase is a key enzyme in glycolysis that catalyzes the first step of the pathway by converting D-glyceraldehyde 3-phosphate (G3P) into 3-phospho-D-glyceroyl phosphate. Component of the GAIT (gamma interferon-activated inhibitor of translation) complex which mediates interferon-gamma-induced transcript-selective translation inhibition in inflammation processes. Upon interferon-gamma treatment assembles into the GAIT complex which binds to stem loop-containing GAIT elements in the 3'-UTR of diverse inflammatory mRNAs (such as ceruplasmin) and suppresses their translation. Has both glyceraldehyde-3-phosphate dehydrogenase and nitrosylase activities, thereby playing a role in glycolysis and nuclear functions, respectively. Related diseases Bone marrow failure and diabetes mellitus syndrome (BMFDMS) [MIM:620044]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFDMS is an autosomal recessive form characterized by various degrees of bone marrow failure, ranging from dyserythropoiesis to bone marrow aplasia, with onset in infancy or early childhood, and non-autoimmune insulin-dependent diabetes mellitus appearing in the first or second decades. Many patients show pigmentary skin abnormalities and short stature. {ECO:0000269|PubMed:28073829, ECO:0000269|PubMed:35611808, ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07347; DB02059; DB11638; DB09130; DB00157; DB03893; DB09092 Interacts with Q6UY14-3; Q9UIJ7; P05067; Q9UQM7; Q14194; P35222; Q9BPW9-4; P00533; O00471; O75344; P06241; P04406; O14556; Q8NEA9; P42858; Q92993-2; P42695; P35228; P12004; P00558; P48147; P17612; Q8WUY3; Q9UHX1-2; P15927; P05109; Q96GZ6; P00441; Q9BSI4; P10599 EC number EC 1.2.1.12 Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Alternative splicing; Apoptosis; Cytoplasm; Cytoskeleton; Direct protein sequencing; Glycolysis; Glycoprotein; Immunity; Innate immunity; Isopeptide bond; Membrane; Methylation; NAD; Nucleus; Oxidation; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; S-nitrosylation; Transferase; Translation regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35818.4 Length 333 Aromaticity 0.08 Instability index 13.69 Isoelectric point 8.64 Charge (pH=7) 3.64 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map  Pymol Map 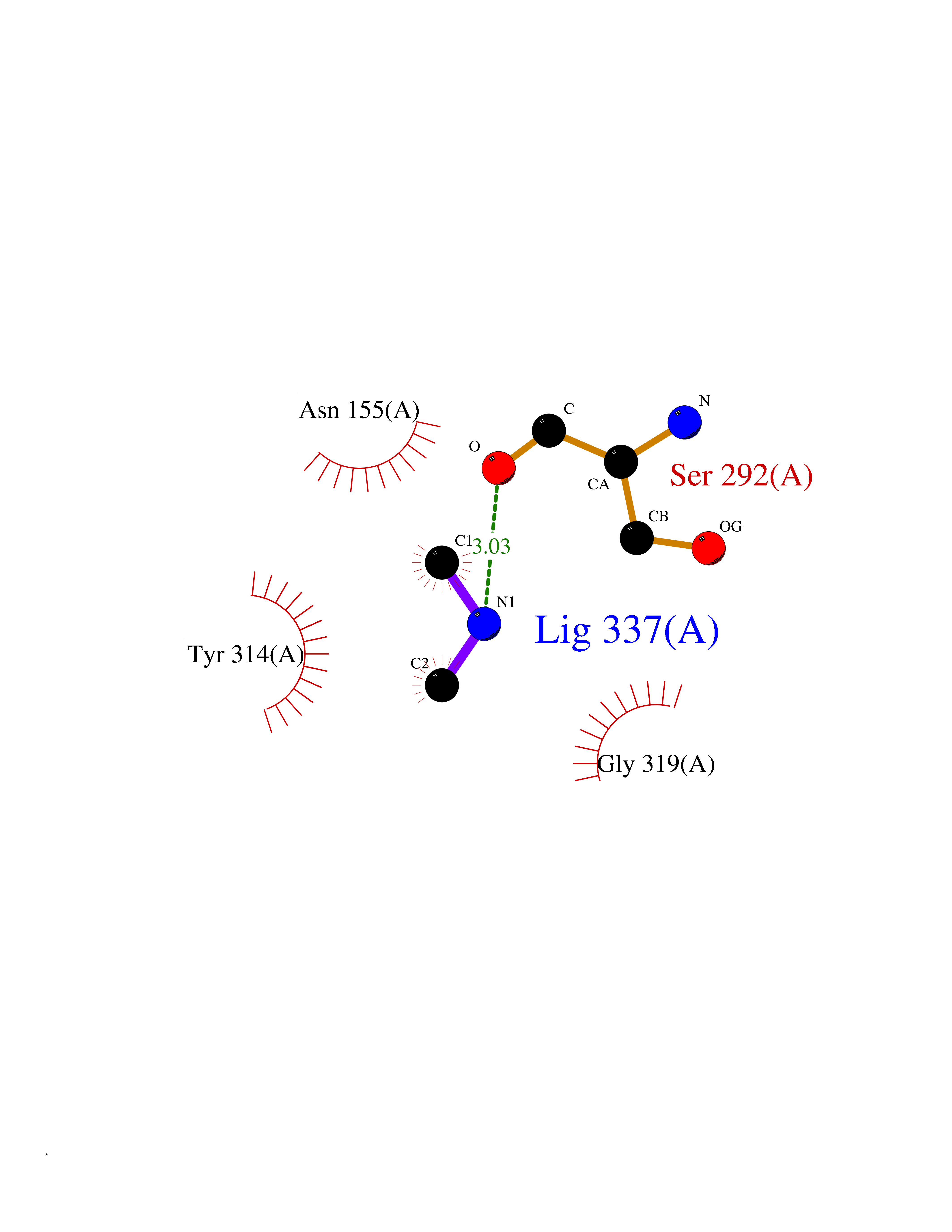 Ligplot Map 3D Binding mode Sequence GKVKVGVNGFGRIGRLVTRAAFNSGKVDIVAINDPFIDLNYMVYMFQYDSTHGKFHGTVKAENGKLVINGNPITIFQERDPSKIKWGDAGAEYVVESTGVFTTMEKAGAHLQGGAKRVIISAPSADAPMFVMGVNHEKYDNSLKIISNASTTNCLAPLAKVIHDNFGIVEGLMTTVHAITATQKTVDGPSGKLWRDGRGALQNIIPASTGAAKAVGKVIPELNGKLTGMAFRVPTANVSVVDLTCRLEKPAKYDDIKKVVKQASEGPLKGILGYTEHQVVSSDFNSDTHSSTFDAGAGIALNDHFVKLISWYDNEFGYSNRVVDLMAHMASKE Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Triggering receptor expressed on monocytes 1 (TREM1) | 1Q8M | 4.01 | |
Target general information Gen name Trem1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Trem1; TREM-1 Protein family NA Biochemical class NA Function Stimulates neutrophil and monocyte-mediated inflammatory responses. Triggers release of pro-inflammatory chemokines and cytokines, as well as increased surface expression of cell activation markers. Amplifier of inflammatory responses that are triggered by bacterial and fungal infections and is a crucial mediator of septic shock. Related diseases GLUT1 deficiency syndrome 1 (GLUT1DS1) [MIM:606777]: A neurologic disorder showing wide phenotypic variability. The most severe 'classic' phenotype comprises infantile-onset epileptic encephalopathy associated with delayed development, acquired microcephaly, motor incoordination, and spasticity. Onset of seizures, usually characterized by apneic episodes, staring spells, and episodic eye movements, occurs within the first 4 months of life. Other paroxysmal findings include intermittent ataxia, confusion, lethargy, sleep disturbance, and headache. Varying degrees of cognitive impairment can occur, ranging from learning disabilities to severe intellectual disability. {ECO:0000269|PubMed:10227690, ECO:0000269|PubMed:10980529, ECO:0000269|PubMed:11136715, ECO:0000269|PubMed:11603379, ECO:0000269|PubMed:12325075, ECO:0000269|PubMed:15622525, ECO:0000269|PubMed:19901175, ECO:0000269|PubMed:20129935, ECO:0000269|PubMed:20221955, ECO:0000269|PubMed:20574033, ECO:0000269|PubMed:24847886, ECO:0000269|PubMed:25982116, ECO:0000269|PubMed:30197081}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GLUT1 deficiency syndrome 2 (GLUT1DS2) [MIM:612126]: A clinically variable disorder characterized primarily by onset in childhood of paroxysmal exercise-induced dyskinesia. The dyskinesia involves transient abnormal involuntary movements, such as dystonia and choreoathetosis, induced by exercise or exertion, and affecting the exercised limbs. Some patients may also have epilepsy, most commonly childhood absence epilepsy. Mild intellectual disability may also occur. In some patients involuntary exertion-induced dystonic, choreoathetotic, and ballistic movements may be associated with macrocytic hemolytic anemia. {ECO:0000269|PubMed:14605501, ECO:0000269|PubMed:18451999, ECO:0000269|PubMed:19630075, ECO:0000269|PubMed:19798636, ECO:0000269|PubMed:20129935, ECO:0000269|PubMed:20574033, ECO:0000269|PubMed:20621801, ECO:0000269|PubMed:20830593, ECO:0000269|PubMed:21204808}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epilepsy, idiopathic generalized 12 (EIG12) [MIM:614847]: A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. In some EIG12 patients seizures may remit with age. {ECO:0000269|PubMed:19798636, ECO:0000269|PubMed:22282645, ECO:0000269|PubMed:23280796, ECO:0000269|PubMed:25982116}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Dystonia 9 (DYT9) [MIM:601042]: An autosomal dominant neurologic disorder characterized by childhood onset of paroxysmal choreoathetosis and progressive spastic paraplegia. Most patients show some degree of cognitive impairment. Other variable features may include seizures, migraine headaches, and ataxia. {ECO:0000269|PubMed:21832227}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Stomatin-deficient cryohydrocytosis with neurologic defects (SDCHCN) [MIM:608885]: A rare form of stomatocytosis characterized by episodic hemolytic anemia, cold-induced red cells cation leak, erratic hyperkalemia, neonatal hyperbilirubinemia, hepatosplenomegaly, cataracts, seizures, intellectual disability, and movement disorder. {ECO:0000269|PubMed:21791420, ECO:0000269|PubMed:22492876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01694 Interacts with NA EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Innate immunity; Membrane; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 28186.2 Length 242 Aromaticity 0.07 Instability index 45.2 Isoelectric point 6.1 Charge (pH=7) -3.8 2D Binding mode Binding energy (Kcal/mol) -5.47 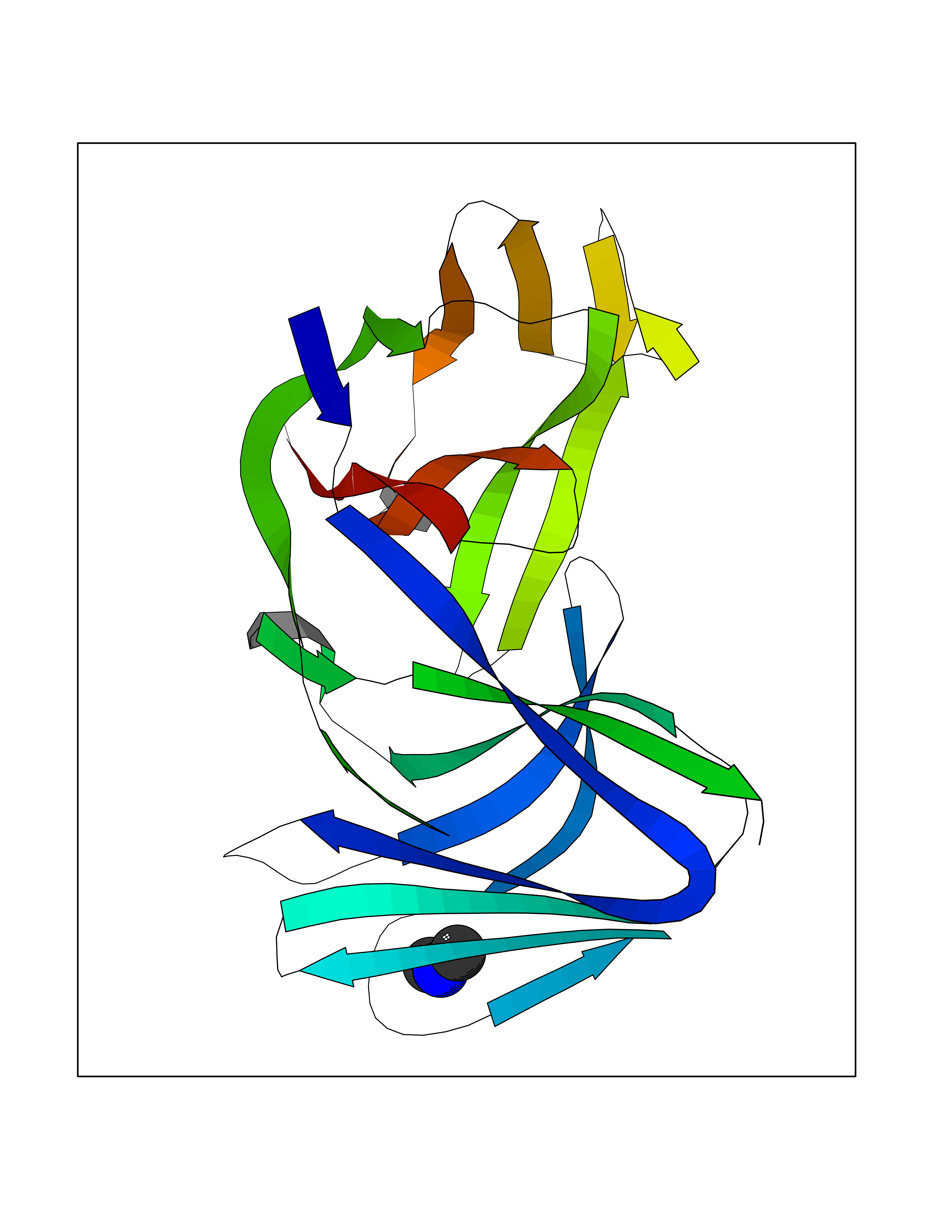 Molscript Map 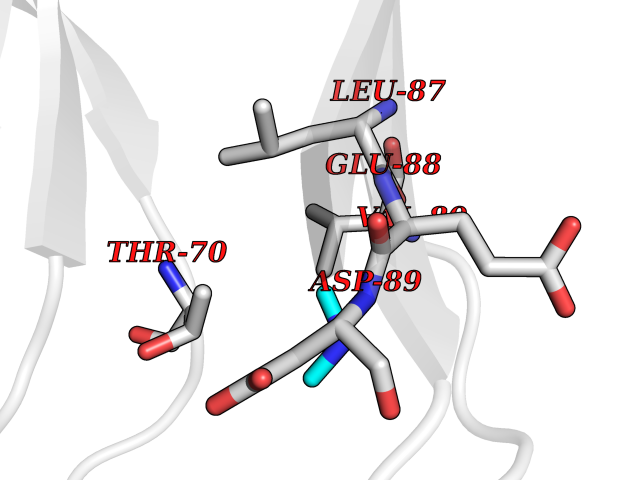 Pymol Map 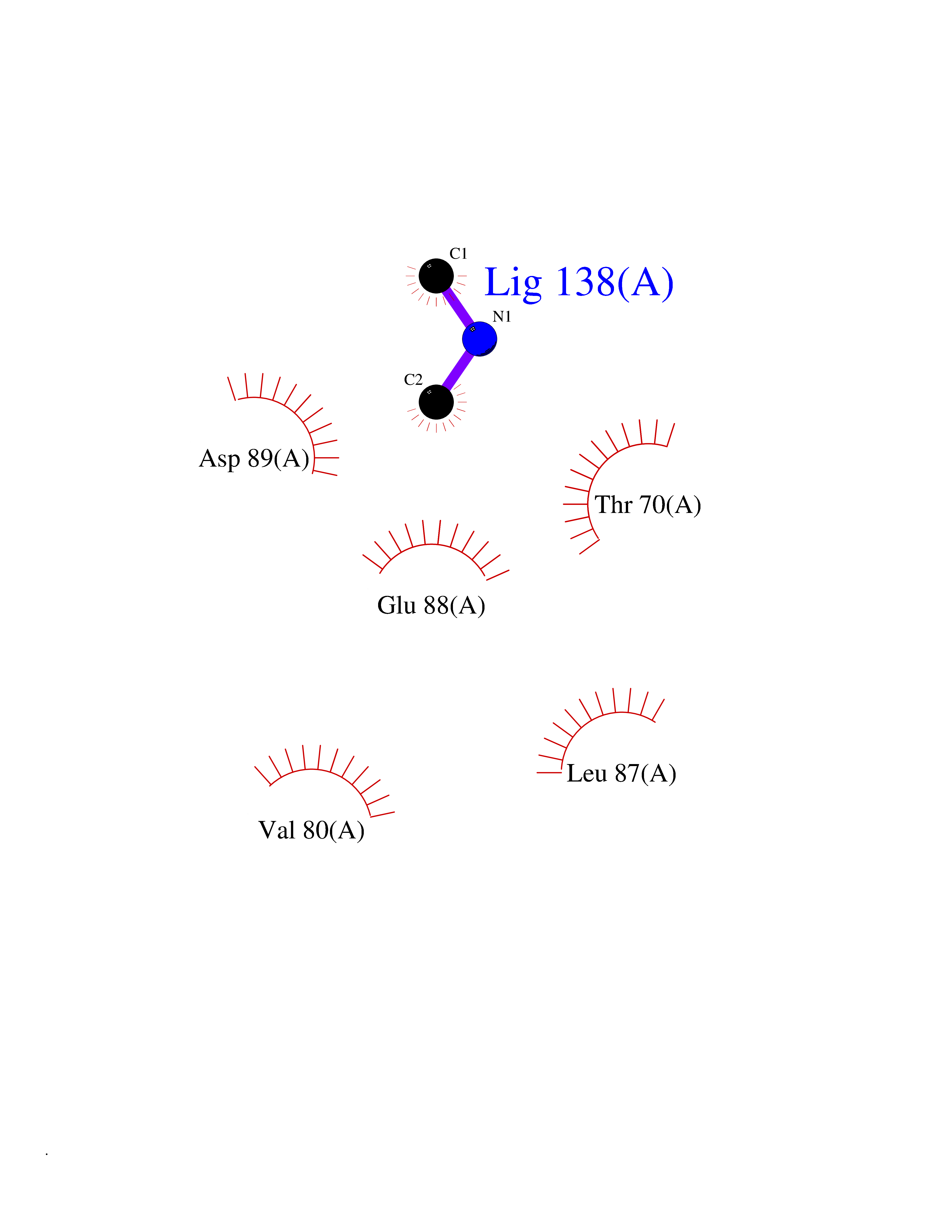 Ligplot Map 3D Binding mode Sequence MELRAATKLTEEKYELKEGQTLDVKCDYTLEKFASSQKAWQIIRDGEMPKTLACTERPSKNSHPVQVGRIILEDYHDHGLLRVRMVNLQVEDSGLYQCVIYQPPKEPHMLFDRIRLVVTLEMELRAATKLTEEKYELKEGQTLDVKCDYTLEKFASSQKAWQIIRDGEMPKTLACTERPSKNSHPVQVGRIILEDYHDHGLLRVRMVNLQVEDSGLYQCVIYQPPKEPHMLFDRIRLVVTLE Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | NAPE-hydrolyzing phospholipase D (NAPE-PLD) | 4QN9 | 4.01 | |
Target general information Gen name NAPEPLD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NAPE-PLD; N-acyl-phosphatidylethanolamine-hydrolyzing phospholipase D; N-acyl phosphatidylethanolamine phospholipase D; C7orf18 Protein family NAPE-PLD family Biochemical class NA Function Hydrolyzes N-acyl-phosphatidylethanolamines (NAPEs) to produce N-acylethanolamines (NAEs) and phosphatidic acid. Responsible for the generation of these bioactive fatty acid ethanolamides (FAEs), including anandamide (N-arachidonoylethanolamine), the ligand of cannabinoid and vanilloid receptors. As a regulator of lipid metabolism in the adipose tissue, mediates the crosstalk between adipocytes, gut microbiota and immune cells to control body temperature and weight. In particular, regulates energy homeostasis by promoting cold-induced brown or beige adipocyte differentiation program to generate heat from fatty acids and glucose (By similarity). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) DB14009 Interacts with Q6IQ20 EC number EC 3.1.4.54 Uniprot keywords 3D-structure; Acetylation; Endosome; Golgi apparatus; Hydrolase; Lipid degradation; Lipid metabolism; Membrane; Metal-binding; Nucleus; Phospholipid degradation; Phospholipid metabolism; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 74256.5 Length 643 Aromaticity 0.13 Instability index 48.34 Isoelectric point 5.65 Charge (pH=7) -17.61 2D Binding mode Binding energy (Kcal/mol) -5.47 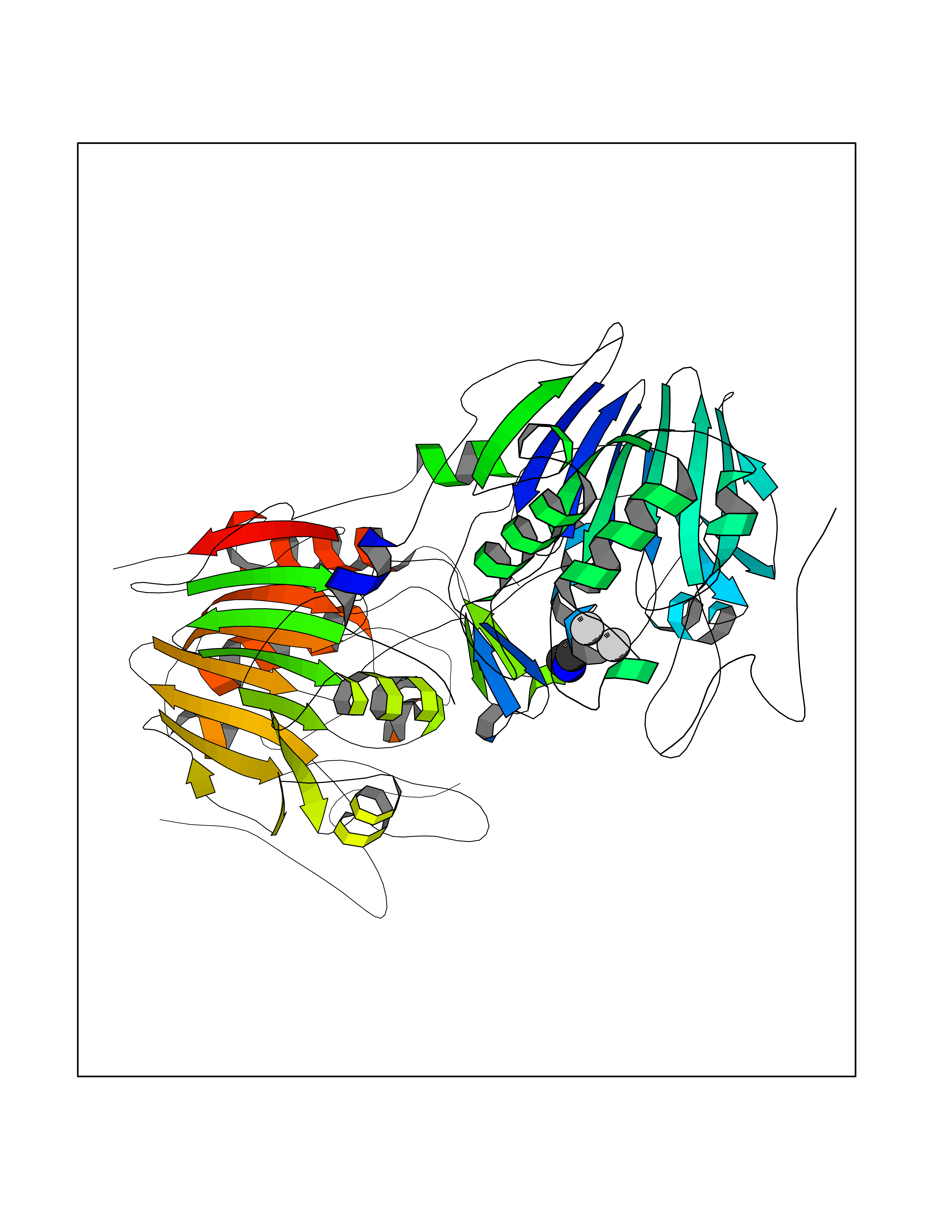 Molscript Map 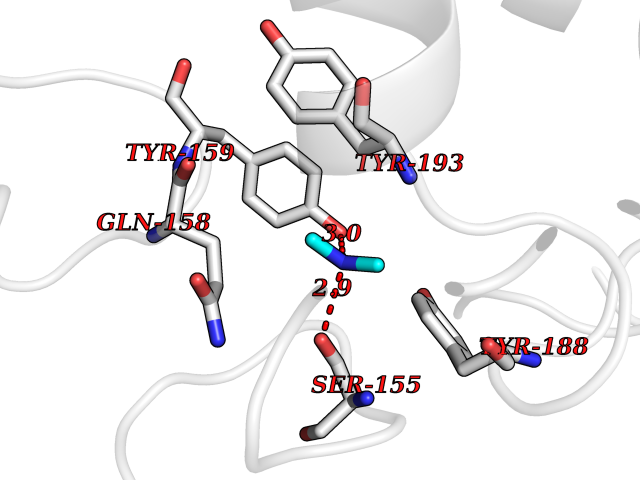 Pymol Map 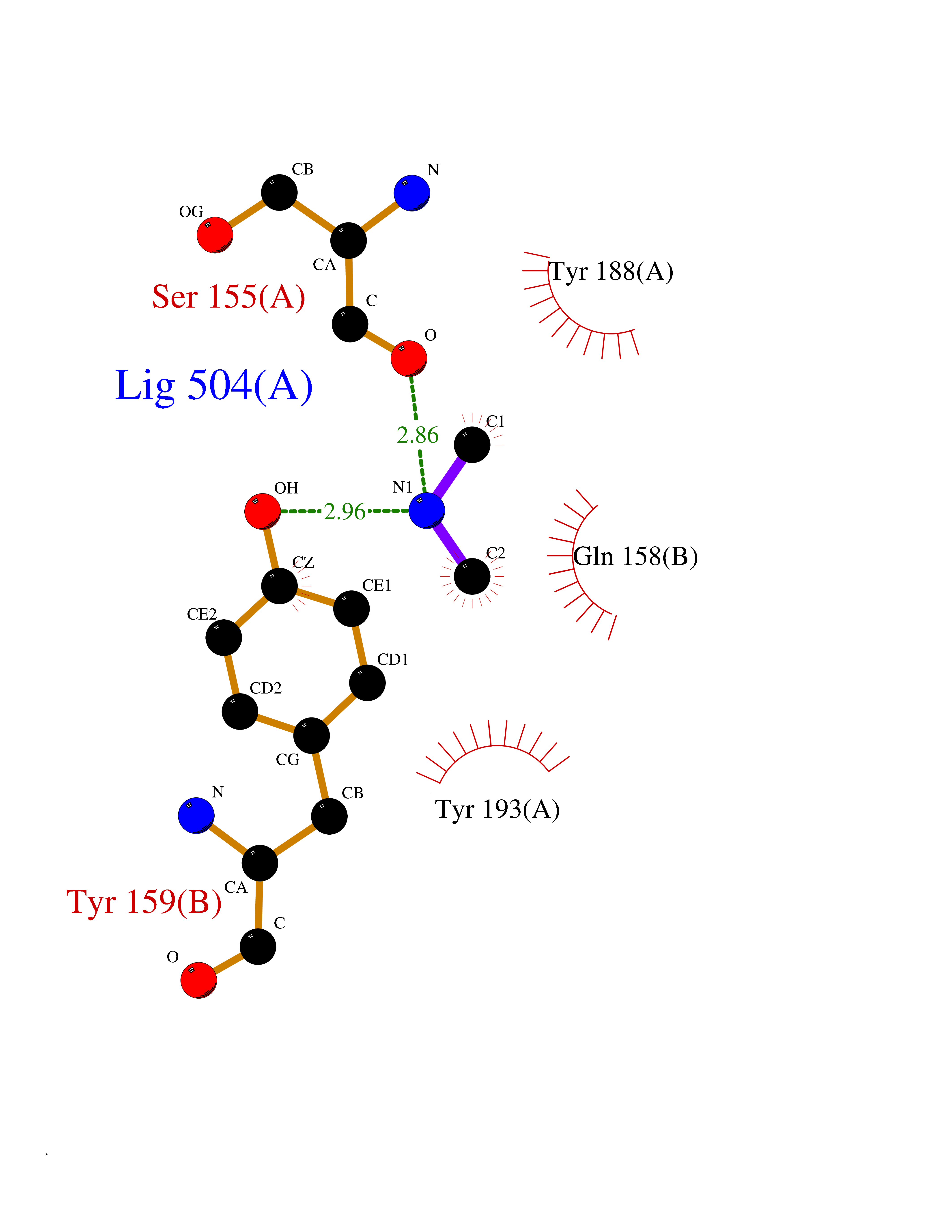 Ligplot Map 3D Binding mode Sequence SKKGKDGRFVNPWPTWKNPSIPNSSVPSSKEELDKELPVLKPYFITNPEEAGVREAGLRVTWLGHATVMVEMDELIFLTDPIFSSRASPSQYMGPKRFRRSPCTISELPPIDAVLISHNHYDHLDYNSVIALNERFGNELRWFVPLGLLDWMQKCGCENVIELDWWEENCVPGHDKVTFVFTPSQHWCKRTLMDDNKVLWGSWSVLGPWNRFFFAGDTGYCPAFEEIGKRFGPFDLAAIPIGAYEPRWFMKYQHVDPEEAVRIHTDVQTKKSMAIHWGTFALANEHYLEPPVKLNEALERYGLNAEDFFVLKHGESRYLNNSKKGKDGRFVNPWPTWKNPSIPNSSVPSSKEELDKELPVLKPYFITNPEEAGVREAGLRVTWLGHATVMVEMDELIFLTDPIFSSRASPSQYMGPKRFRRSPCTISELPPIDAVLISHNHYDHLDYNSVIALNERFGNELRWFVPLGLLDWMQKCGCENVIELDWWEENCVPGHDKVTFVFTPSQHWCKRTLMDDNKVLWGSWSVLGPWNRFFFAGDTGYCPAFEEIGKRFGPFDLAAIPIGAYEPRWFMKYQHVDPEEAVRIHTDVQTKKSMAIHWGTFALANEHYLEPPVKLNEALERYGLNAEDFFVLKHGESRYLNND Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Tyrosine-protein kinase ABL1 (ABL) | 5HU9 | 4.00 | |
Target general information Gen name ABL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p150; Proto-oncogene tyrosine-protein kinase ABL1; Proto-oncogene c-Abl; JTK7; C-ABL; Abl; Abelson tyrosine-protein kinase 1; Abelson murine leukemia viral oncogene homolog 1 Protein family Protein kinase superfamily, Tyr protein kinase family, ABL subfamily Biochemical class Kinase Function Coordinates actin remodeling through tyrosine phosphorylation of proteins controlling cytoskeleton dynamics like WASF3 (involved in branch formation); ANXA1 (involved in membrane anchoring); DBN1, DBNL, CTTN, RAPH1 and ENAH (involved in signaling); or MAPT and PXN (microtubule-binding proteins). Phosphorylation of WASF3 is critical for the stimulation of lamellipodia formation and cell migration. Involved in the regulation of cell adhesion and motility through phosphorylation of key regulators of these processes such as BCAR1, CRK, CRKL, DOK1, EFS or NEDD9. Phosphorylates multiple receptor tyrosine kinases and more particularly promotes endocytosis of EGFR, facilitates the formation of neuromuscular synapses through MUSK, inhibits PDGFRB-mediated chemotaxis and modulates the endocytosis of activated B-cell receptor complexes. Other substrates which are involved in endocytosis regulation are the caveolin (CAV1) and RIN1. Moreover, ABL1 regulates the CBL family of ubiquitin ligases that drive receptor down-regulation and actin remodeling. Phosphorylation of CBL leads to increased EGFR stability. Involved in late-stage autophagy by regulating positively the trafficking and function of lysosomal components. ABL1 targets to mitochondria in response to oxidative stress and thereby mediates mitochondrial dysfunction and cell death. In response to oxidative stress, phosphorylates serine/threonine kinase PRKD2 at 'Tyr-717'. ABL1 is also translocated in the nucleus where it has DNA-binding activity and is involved in DNA-damage response and apoptosis. Many substrates are known mediators of DNA repair: DDB1, DDB2, ERCC3, ERCC6, RAD9A, RAD51, RAD52 or WRN. Activates the proapoptotic pathway when the DNA damage is too severe to be repaired. Phosphorylates TP73, a primary regulator for this type of damage-induced apoptosis. Phosphorylates the caspase CASP9 on 'Tyr-153' and regulates its processing in the apoptotic response to DNA damage. Phosphorylates PSMA7 that leads to an inhibition of proteasomal activity and cell cycle transition blocks. ABL1 acts also as a regulator of multiple pathological signaling cascades during infection. Several known tyrosine-phosphorylated microbial proteins have been identified as ABL1 substrates. This is the case of A36R of Vaccinia virus, Tir (translocated intimin receptor) of pathogenic E. coli and possibly Citrobacter, CagA (cytotoxin-associated gene A) of H. pylori, or AnkA (ankyrin repeat-containing protein A) of A. phagocytophilum. Pathogens can highjack ABL1 kinase signaling to reorganize the host actin cytoskeleton for multiple purposes, like facilitating intracellular movement and host cell exit. Finally, functions as its own regulator through autocatalytic activity as well as through phosphorylation of its inhibitor, ABI1. Regulates T-cell differentiation in a TBX21-dependent manner. Phosphorylates TBX21 on tyrosine residues leading to an enhancement of its transcriptional activator activity. Non-receptor tyrosine-protein kinase that plays a role in many key processes linked to cell growth and survival such as cytoskeleton remodeling in response to extracellular stimuli, cell motility and adhesion, receptor endocytosis, autophagy, DNA damage response and apoptosis. Related diseases Leukemia, chronic myeloid (CML) [MIM:608232]: A clonal myeloproliferative disorder of a pluripotent stem cell with a specific cytogenetic abnormality, the Philadelphia chromosome (Ph), involving myeloid, erythroid, megakaryocytic, B-lymphoid, and sometimes T-lymphoid cells, but not marrow fibroblasts. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: A chromosomal aberration involving ABL1 has been found in patients with chronic myeloid leukemia. Translocation t(9;22)(q34;q11) with BCR. The translocation produces a BCR-ABL found also in acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL). {ECO:0000269|PubMed:3021337}.; DISEASE: A chromosomal aberration involving ABL1 is found in a form of acute lymphoblastic leukemia (PubMed:15361874). Translocation t(9;9)(q34;q34) with NUP214 (PubMed:15361874). {ECO:0000269|PubMed:15361874}.; DISEASE: Congenital heart defects and skeletal malformations syndrome (CHDSKM) [MIM:617602]: An autosomal dominant disorder characterized by congenital heart disease with atrial and ventricular septal defects, variable skeletal abnormalities, and failure to thrive. Skeletal defects include pectus excavatum, scoliosis, and finger contractures. Some patient exhibit joint laxity. {ECO:0000269|PubMed:28288113}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08043; DB08583; DB07831; DB08350; DB12597; DB00171; DB06616; DB12267; DB01254; DB12010; DB00619; DB13749; DB08231; DB03878; DB04868; DB08339; DB08901; DB12323; DB08896; DB14989; DB05184 Interacts with Q8IZP0; Q9NYB9; O14672; P10275; Q13315; Q4KMG0; P46108; P46109; P35222; P00533; P04626; Q03468; Q14315; P36888; P05107; P10721; Q38SD2; Q92918; Q7Z434; O43196; P15941; P15941-12; P16333; O43900; Q13905; Q86UR5; Q13671; P31947; Q15464; O75751; P37840; Q9BX66; O60504-2; Q07890; P12931; P51692; Q9Y4G6; P11387; P04637; P15498; Q9Y6W5; P62258; P61981; P63104; O35158; P37840; P48165; Q15323; P37840 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; ATP-binding; Autophagy; Cell adhesion; Chromosomal rearrangement; Cytoplasm; Cytoskeleton; Disease variant; DNA damage; DNA repair; DNA-binding; Endocytosis; Kinase; Lipoprotein; Magnesium; Manganese; Membrane; Metal-binding; Mitochondrion; Myristate; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; SH2 domain; SH3 domain; Transferase; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 31264.6 Length 270 Aromaticity 0.12 Instability index 37.99 Isoelectric point 5.42 Charge (pH=7) -7.67 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map  Pymol Map 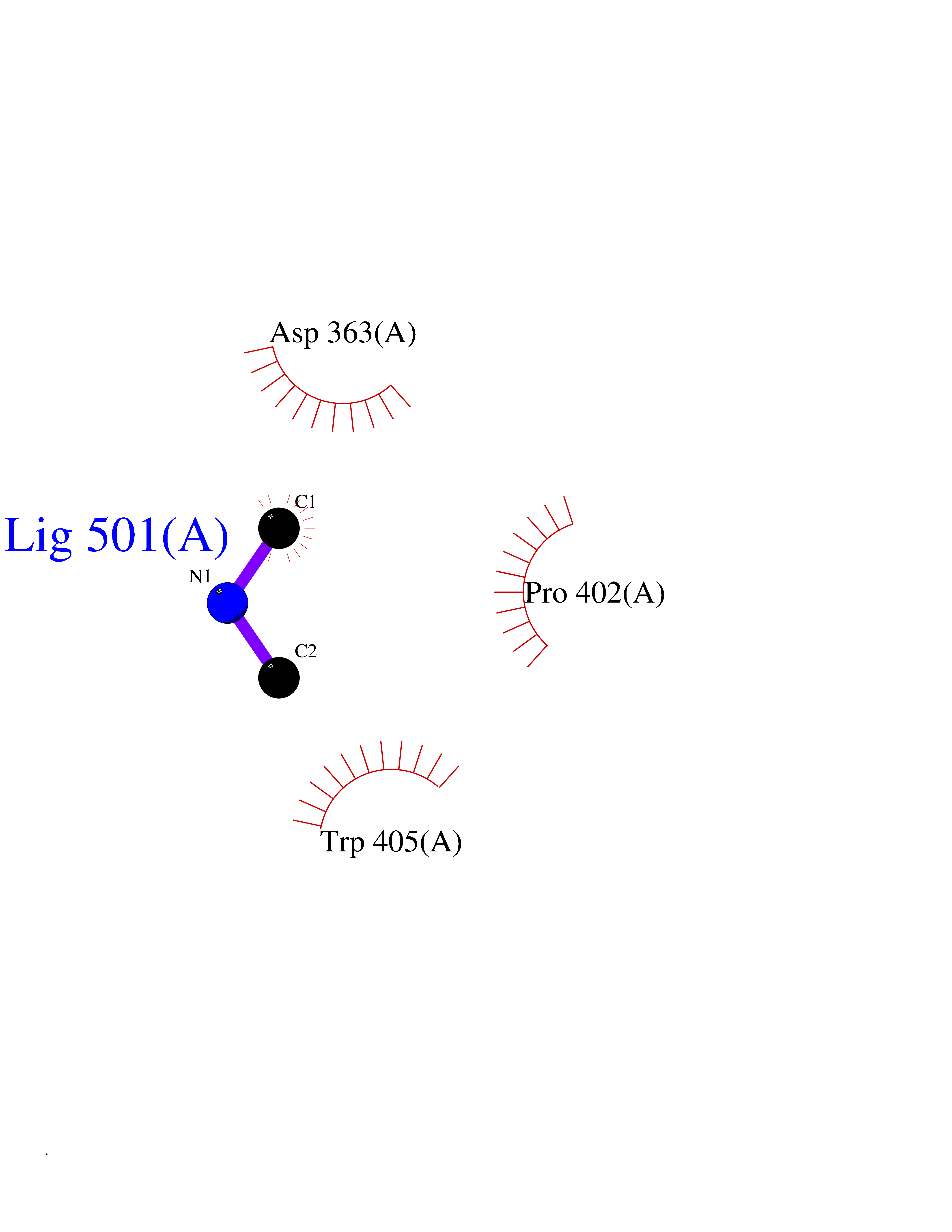 Ligplot Map 3D Binding mode Sequence AMGSSPNYDKWEMERTDITMKHKLGGGQYGEVYEGVWKKYSLTVAVKTLKEDTMEVEEFLKEAAVMKEIKHPNLVQLLGVCTREPPFYIITEFMTYGNLLDYLRECNRQEVNAVVLLYMATQISSAMEYLEKKNFIHRDLAARNCLVGENHLVKVADFGLSRLMTAHAGAKFPIKWTAPESLAYNKFSIKSDVWAFGVLLWEIATYGMSPYPGIDLSQVYELLEKDYRMERPEGCPEKVYELMRACWQWNPSDRPSFAEIHQAFETMFQE Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Quinone-dependent D-lactate dehydrogenase | 1F0X | 4.00 | |
Target general information Gen name dld Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW2121;b2133 Protein family Quinone-dependent D-lactate dehydrogenase family Biochemical class Oxidoreductase Function (R)-2-hydroxyglutarate dehydrogenase activity.D-lactate dehydrogenase (cytochrome) activity.Electron carrier activity.FAD binding.Flavin adenine dinucleotide binding.NAD binding.Oxidoreductase activity, acting on the CH-OH group of donors, quinone or similar compound as acceptor.Quinone binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB00756 Interacts with NA EC number 1.1.5.12 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Membrane; Oxidoreductase; Quinone; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56475.2 Length 502 Aromaticity 0.09 Instability index 32.5 Isoelectric point 5.97 Charge (pH=7) -10.38 2D Binding mode Binding energy (Kcal/mol) -5.46 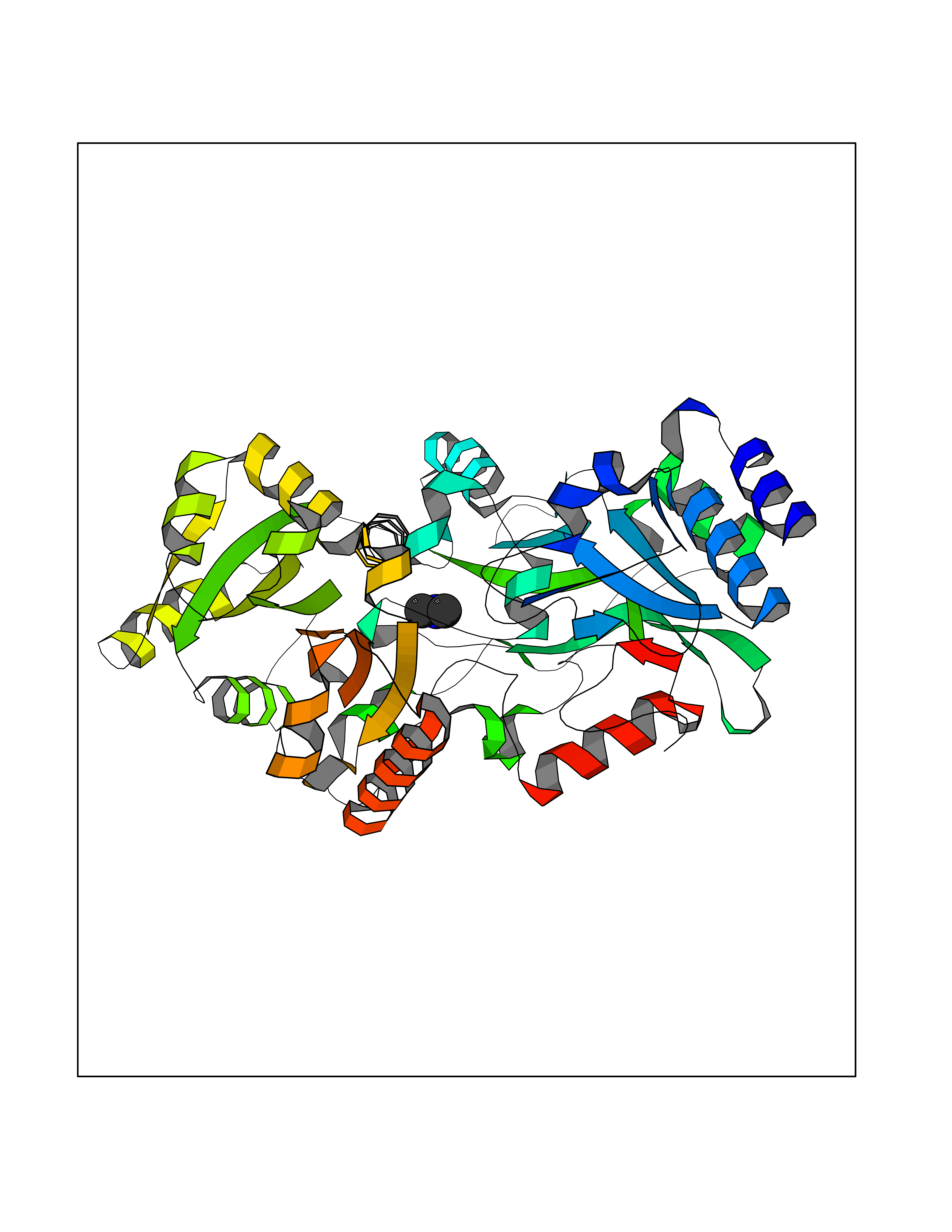 Molscript Map 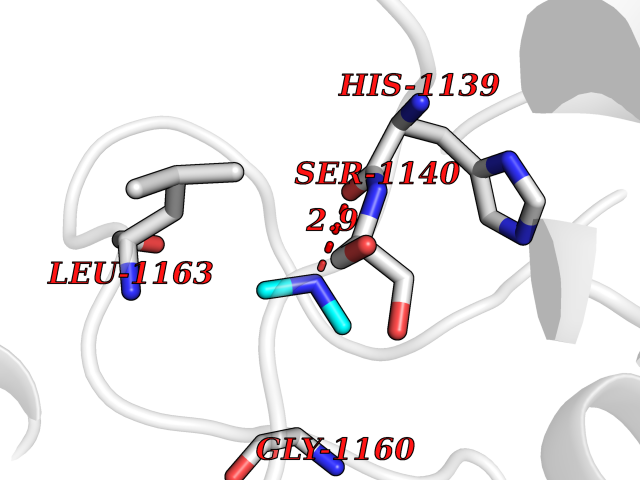 Pymol Map 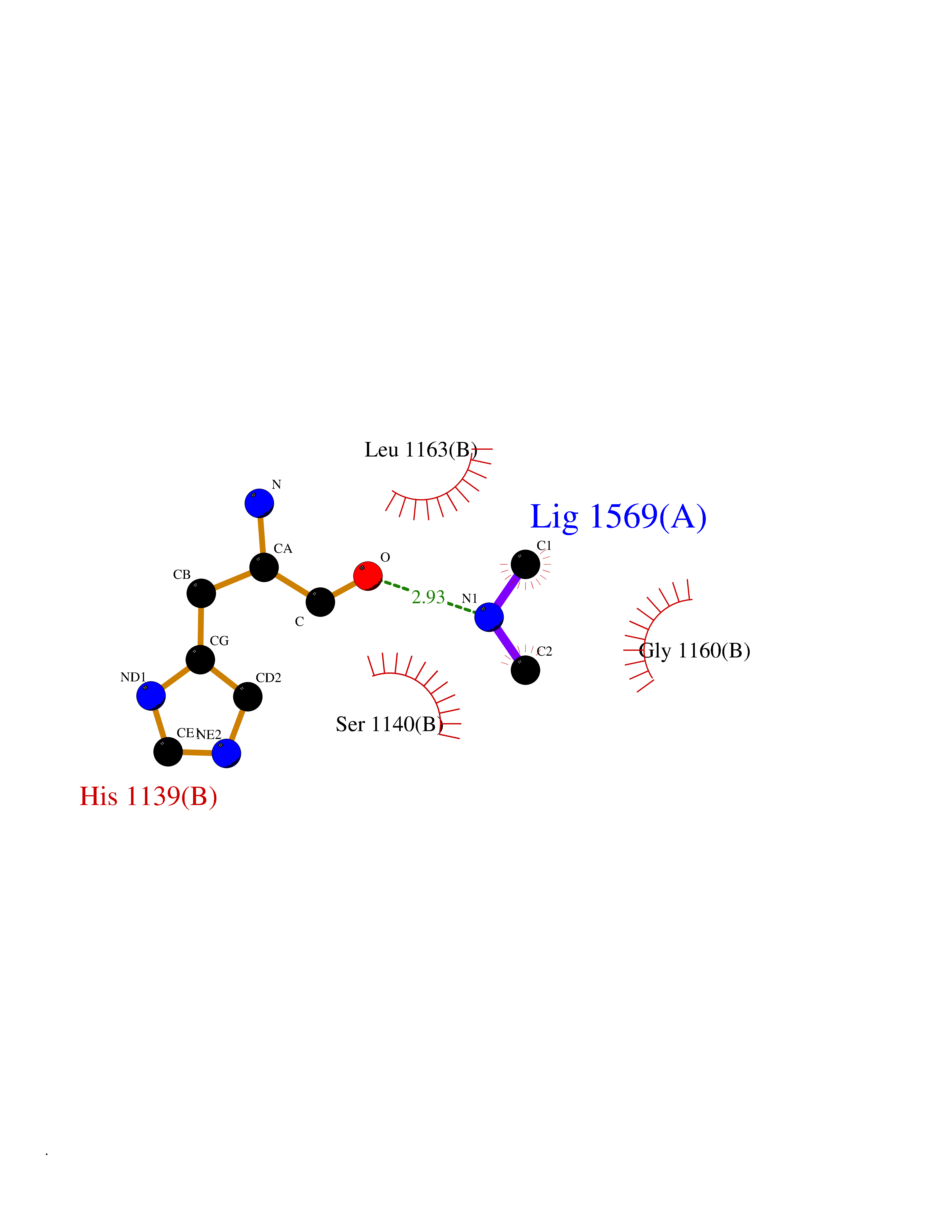 Ligplot Map 3D Binding mode Sequence NKAFLNELARLVGSSHLLTDPAKTARYRKGFRSGQGDALAVVFPGSLLELWRVLKACVTADKIILMQAANTGLTEGSTPNGNDYDRDVVIISTLRLDKLHVLGKGEQVLAYPGTTLYSLEKALKPLGREPHSVIGSSCIGASVIGGICNNSGGSLVQRGPAYTEMSLFARINEDGKLTLVNHLGIDLGETPEQILSKLDDDRIKDDDVRHDGRHAHDYDYVHRVRDIEADTPARYNADPDRLFESSGCAGKLAVFAVRLDTFEAEKNQQVFYIGTNQPEVLTEIRRHILANFENLPVAGEYMHRDIYDIAELPPRMKNWRDKYEHHLLLKMAGDGVGEAKSWLVDYFKQAEGDFFVCTPEEGSKAFLHRFAAAGAAIRYQAVHSDEVEDILALDIALRRNDTEWYEHLPPEIDSQLVHKLYYGHFMCYVFHQDYIVKKGVDVHALKEQMLELLQQRGAQYPAEHNVGHLYKAPETLQKFYRENDPTNSMNPGIGKTSKRKNW Hydrogen bonds contact Hydrophobic contact | ||||