Job Results:
Ligand
Structure
Job ID
3c17296c67deee94355bca3744afb8a3
Job name
NA
Time
2025-02-13 15:25:56
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 4.01 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 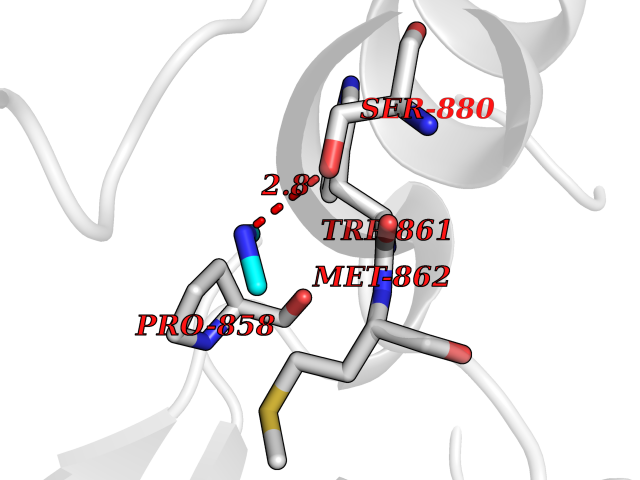 Pymol Map 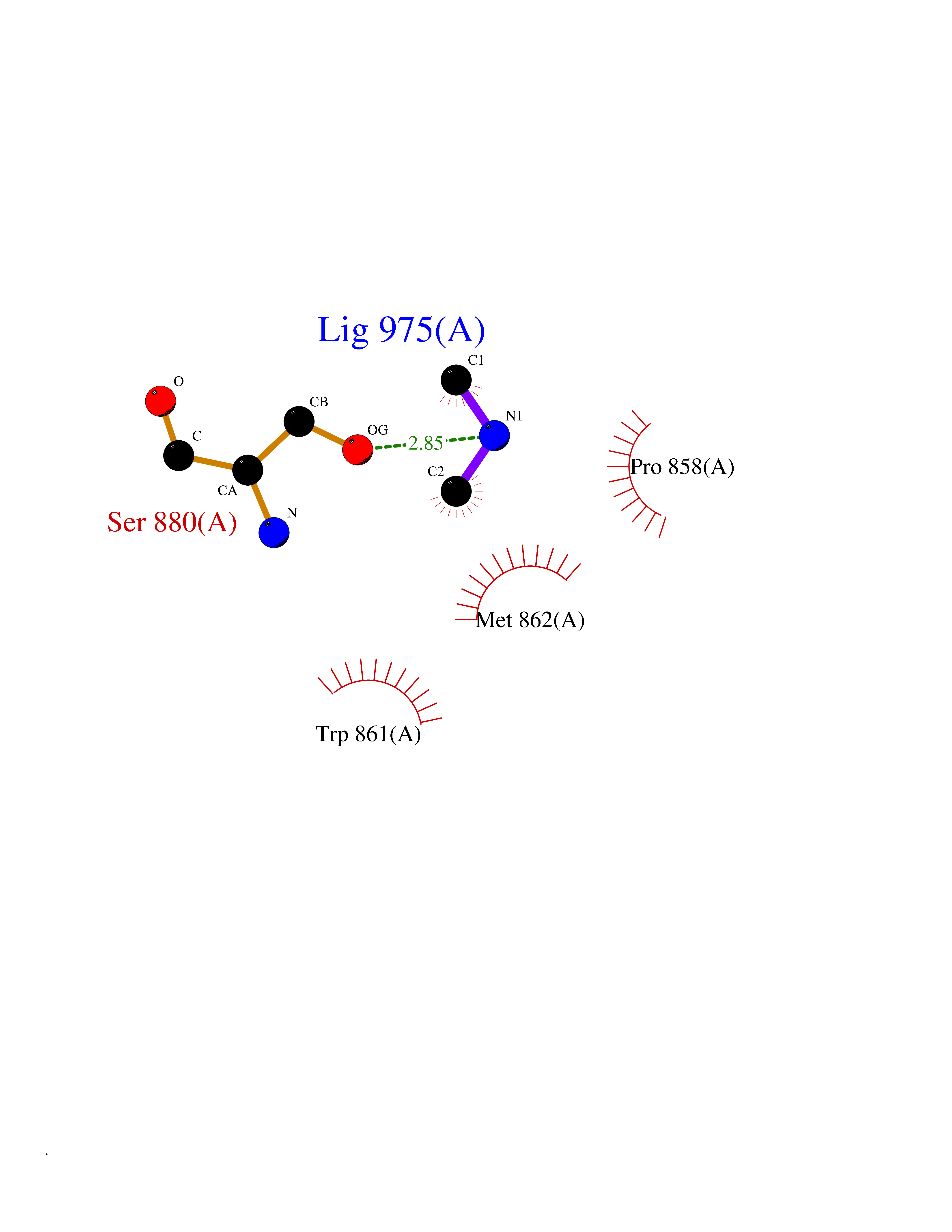 Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Quinone-dependent D-lactate dehydrogenase | 1F0X | 4.01 | |
Target general information Gen name dld Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW2121;b2133 Protein family Quinone-dependent D-lactate dehydrogenase family Biochemical class Oxidoreductase Function (R)-2-hydroxyglutarate dehydrogenase activity.D-lactate dehydrogenase (cytochrome) activity.Electron carrier activity.FAD binding.Flavin adenine dinucleotide binding.NAD binding.Oxidoreductase activity, acting on the CH-OH group of donors, quinone or similar compound as acceptor.Quinone binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB00756 Interacts with NA EC number 1.1.5.12 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Membrane; Oxidoreductase; Quinone; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56475.2 Length 502 Aromaticity 0.09 Instability index 32.5 Isoelectric point 5.97 Charge (pH=7) -10.38 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 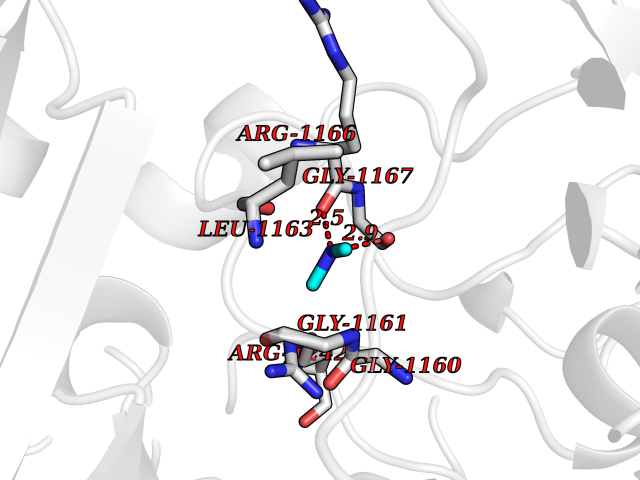 Pymol Map 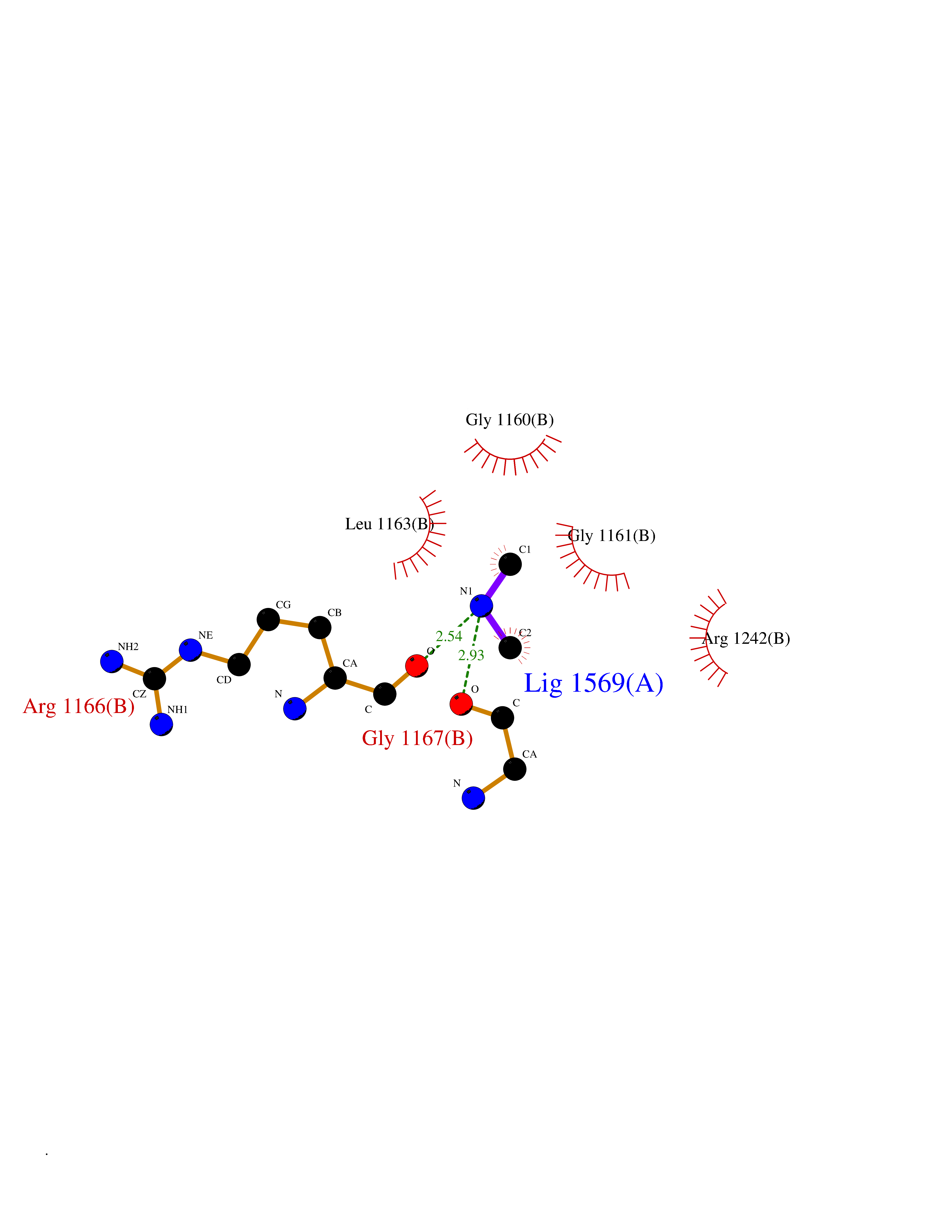 Ligplot Map 3D Binding mode Sequence NKAFLNELARLVGSSHLLTDPAKTARYRKGFRSGQGDALAVVFPGSLLELWRVLKACVTADKIILMQAANTGLTEGSTPNGNDYDRDVVIISTLRLDKLHVLGKGEQVLAYPGTTLYSLEKALKPLGREPHSVIGSSCIGASVIGGICNNSGGSLVQRGPAYTEMSLFARINEDGKLTLVNHLGIDLGETPEQILSKLDDDRIKDDDVRHDGRHAHDYDYVHRVRDIEADTPARYNADPDRLFESSGCAGKLAVFAVRLDTFEAEKNQQVFYIGTNQPEVLTEIRRHILANFENLPVAGEYMHRDIYDIAELPPRMKNWRDKYEHHLLLKMAGDGVGEAKSWLVDYFKQAEGDFFVCTPEEGSKAFLHRFAAAGAAIRYQAVHSDEVEDILALDIALRRNDTEWYEHLPPEIDSQLVHKLYYGHFMCYVFHQDYIVKKGVDVHALKEQMLELLQQRGAQYPAEHNVGHLYKAPETLQKFYRENDPTNSMNPGIGKTSKRKNW Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Beta-glucosidase A | 1E4I | 4.01 | |
Target general information Gen name bglA Organism Paenibacillus polymyxa (Bacillus polymyxa) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 1 family Biochemical class Hydrolase Function Beta-glucosidase activity.Scopolin beta-glucosidase activity. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02658; DB04282; DB04304 Interacts with NA EC number 3.2.1.21 Uniprot keywords 3D-structure; Carbohydrate metabolism; Cellulose degradation; Glycosidase; Hydrolase; Polysaccharide degradation Protein physicochemical properties Chain ID A Molecular weight (Da) 51515.2 Length 447 Aromaticity 0.14 Instability index 38.44 Isoelectric point 5.28 Charge (pH=7) -18.1 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 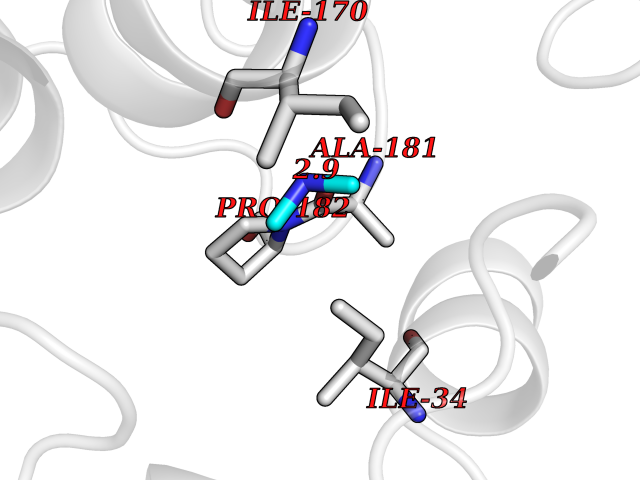 Pymol Map 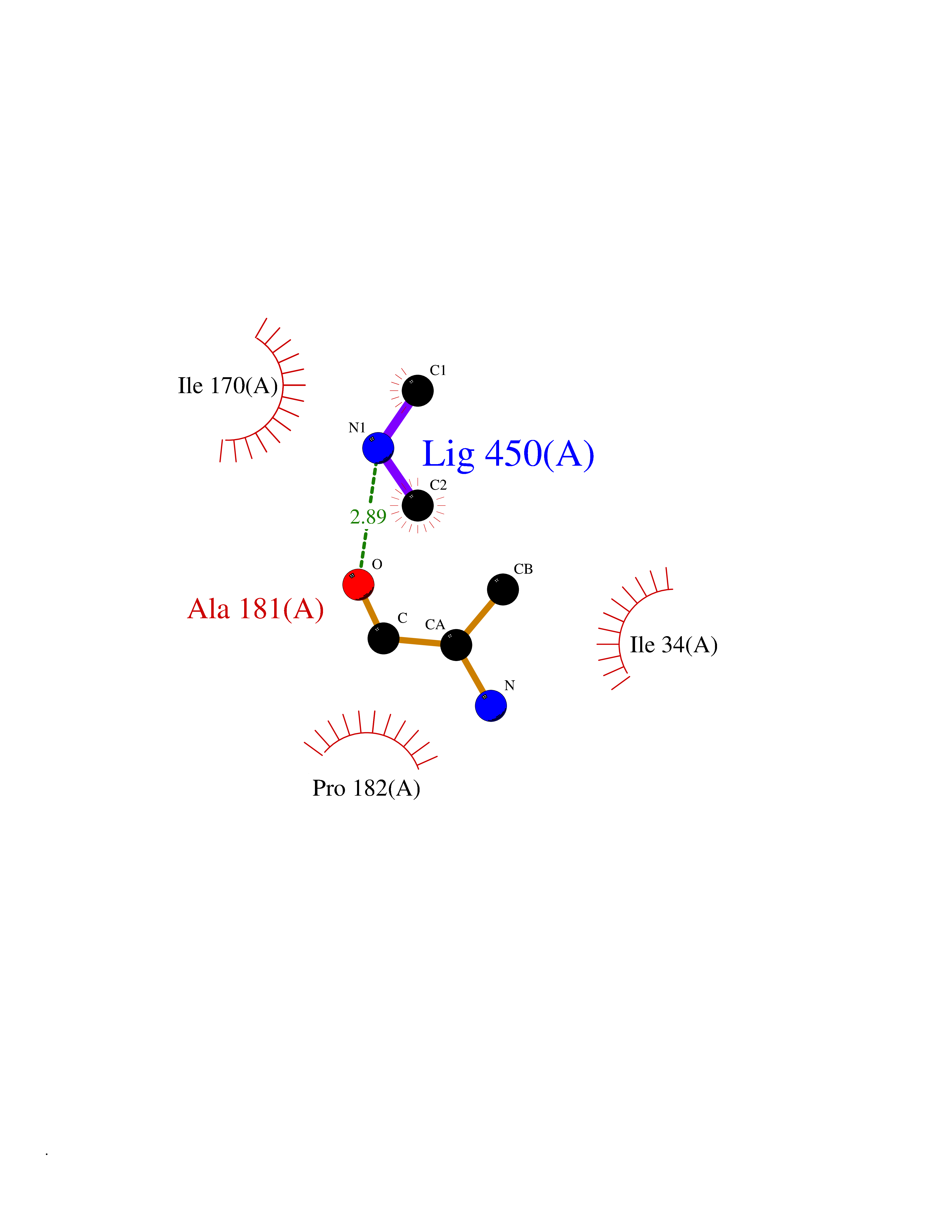 Ligplot Map 3D Binding mode Sequence TIFQFPQDFMWGTATAAYQIEGAYQEDGRGLSIWDTFAHTPGKVFNGDNGNVACDSYHRYEEDIRLMKELGIRTYRFSVSWPRIFPNGDGEVNQKGLDYYHRVVDLLNDNGIEPFCTLYHWDLPQALQDAGGWGNRRTIQAFVQFAETMFREFHGKIQHWLTFNEPWCIAFLSNMLGVHAPGLTNLQTAIDVGHHLLVAHGLSVRRFRELGTSGQIGIAPNVSWAVPYSTSEEDKAACARTISLHSDWFLQPIYQGSYPQFLVDWFAEQGATVPIQDGDMDIIGEPIDMIGINYYSMSVNRFNPEAGFLQSEEINMGLPVTDIGWPVESRGLYEVLHYLQKYGNIDIYITENGACINDEVVNGKVQDDRRISYMQQHLVQVHRTIHDGLHVKGYMAWSLLDNFEWAEGYNMRFGMIHVDFRTQVRTPKQSYYWYRNVVSNNWLETRR Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Phosphoribosylaminoimidazolecarboxamide formyltransferase (ATIC) | 1PKX | 4.01 | |
Target general information Gen name ATIC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PURH; OK/SW-cl.86; Bifunctional purine biosynthesis protein PURH Protein family PurH family Biochemical class Methyltransferase Function Bifunctional enzyme that catalyzes 2 steps in purine biosynthesis. Related diseases AICA-ribosuria due to ATIC deficiency (AICAR) [MIM:608688]: A neurologically devastating inborn error of purine biosynthesis. Patients excrete massive amounts of AICA-riboside in the urine and accumulate AICA-ribotide and its derivatives in erythrocytes and fibroblasts. Clinical features include profound intellectual disability, epilepsy, dysmorphic features and congenital blindness. AICAR inheritance is autosomal recessive. {ECO:0000269|PubMed:15114530}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02309; DB03442; DB01700; DB01972; DB00563; DB04057; DB00642; DB00116 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Epilepsy; Hydrolase; Intellectual disability; Multifunctional enzyme; Proteomics identification; Purine biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 41584.9 Length 386 Aromaticity 0.06 Instability index 42 Isoelectric point 5.6 Charge (pH=7) -7.51 2D Binding mode Binding energy (Kcal/mol) -5.47 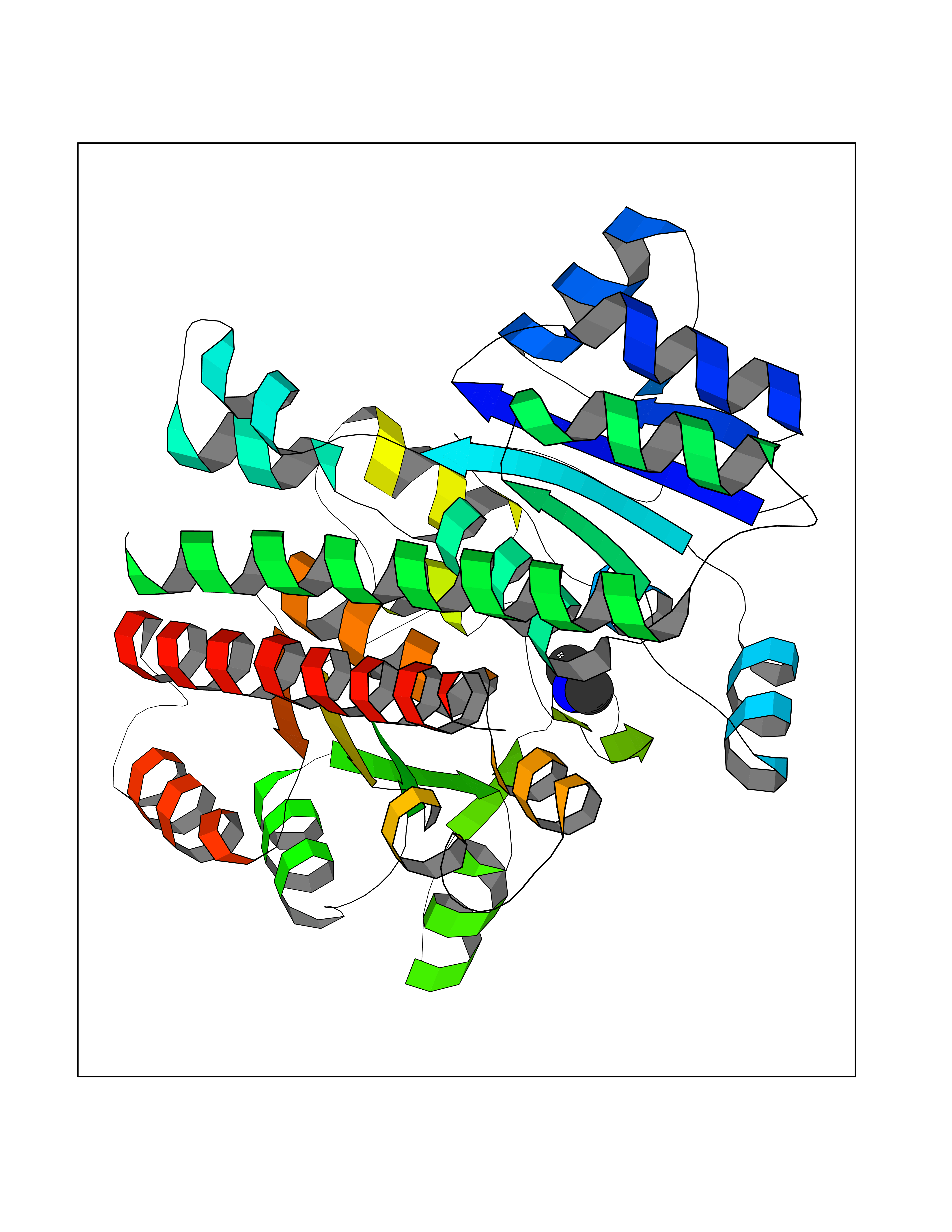 Molscript Map 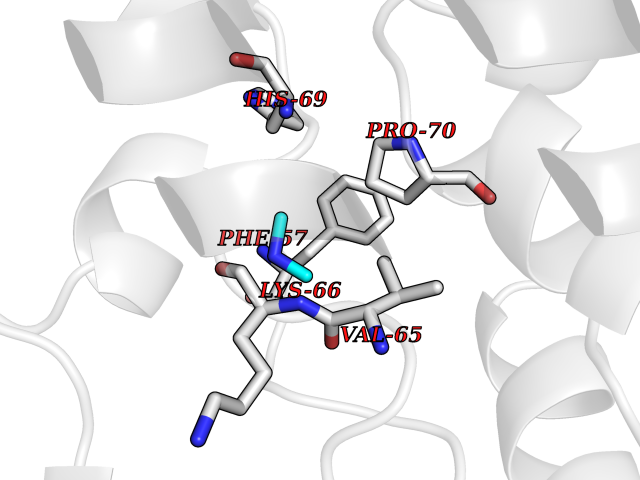 Pymol Map  Ligplot Map 3D Binding mode Sequence GQLALFSVSDKTGLVEFARNLTALGLNLVASGGTAKALRDAGLAVRDVSELTGFPEMLGGRVKTLHPAVHAGILARNIPEDNADMARLDFNLIRVVACNLYPFVKTVASPGVTVEEAVEQIDIGGVTLLRAAAKNHARVTVVCEPEDYVVVSTEMQSSESKDTSLETRRQLALKAFTHTAQYDEAISDYFRKQGQLALFSVSDKTGLVEFARNLTALGLNLVASGGTAKALRDAGLAVRDVSELTGFPEMLGGRVKTLHPAVHAGILARNIPEDNADMARLDFNLIRVVACNLYPFVKTVASPGVTVEEAVEQIDIGGVTLLRAAAKNHARVTVVCEPEDYVVVSTEMQSSESKDTSLETRRQLALKAFTHTAQYDEAISDYFRKQ Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Monomeric sarcosine oxidase | 2GF3 | 4.01 | |
Target general information Gen name soxA Organism Bacillus sp. (strain B-0618) Uniprot ID TTD ID NA Synonyms sox Protein family MSOX/MTOX family, MSOX subfamily Biochemical class Oxidoreductase Function Sarcosine oxidase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB03098; DB01918; DB03517; DB03147; DB03366; DB02083; DB02543 Interacts with NA EC number 1.5.3.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42606.4 Length 385 Aromaticity 0.1 Instability index 26.97 Isoelectric point 5.27 Charge (pH=7) -17.18 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence STHFDVIVVGAGSMGMAAGYQLAKQGVKTLLVDAFDPPHTNGSHHGDTRIIRHAYGEGREYVPLALRSQELWYELEKETHHKIFTKTGVLVFGPKGESAFVAETMEAAKEHSLTVDLLEGDEINKRWPGITVPENYNAIFEPNSGVLFSENCIRAYRELAEARGAKVLTHTRVEDFDISPDSVKIETANGSYTADKLIVSMGAWNSKLLSKLNLDIPLQPYRQVVGFFESDESKYSNDIDFPGFMVEVPNGIYYGFPSFGGCGLKLGYHTFGQKIDPDTINREFGVYPEDESNLRAFLEEYMPGANGELKRGAVCMYTKTLDEHFIIDLHPEHSNVVIAAGFSGHGFKFSSGVGEVLSQLALTGKTEHDISIFSINRPALKESLQ Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Cytosolic purine 5'-nucleotidase | 2JC9 | 4.01 | |
Target general information Gen name NT5C2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PNT5;NT5B;NT5CP Protein family 5'(3')-deoxyribonucleotidase family Biochemical class Hydrolase Function 5'-nucleotidase activity.Metal ion binding.Nucleoside phosphotransferase activity.Nucleotide binding. Related diseases Spastic paraplegia 45, autosomal recessive (SPG45) [MIM:613162]: A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. Some SPG45 patients manifest intellectual disability, contractures and learning disability. {ECO:0000269|PubMed:24482476, ECO:0000269|PubMed:28884889}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00171; DB00811; DB06408 Interacts with P48047; P51116; Q8IVS8; Q7L9L4; Q86TA1; Q70IA8; Q9Y5B8; Q6ZVK8; O00560; Q9NRS6 EC number 2.7.1.77; 3.1.3.5; 3.1.3.99 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Cytoplasm; Disease variant; Hereditary spastic paraplegia; Hydrolase; Magnesium; Metal-binding; Neurodegeneration; Nucleotide metabolism; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 53978.4 Length 467 Aromaticity 0.14 Instability index 30.87 Isoelectric point 8.25 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 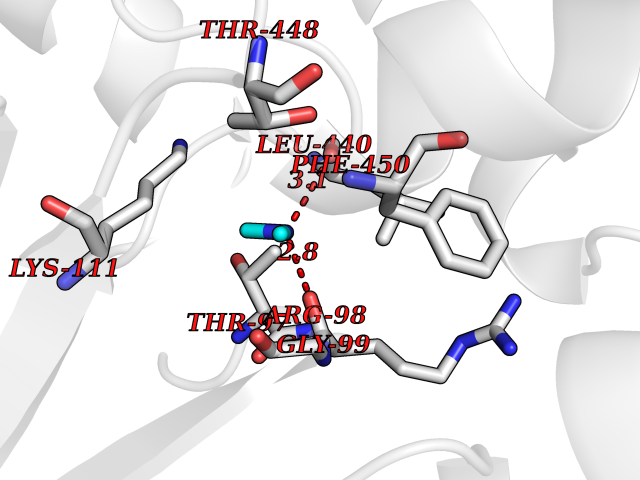 Pymol Map 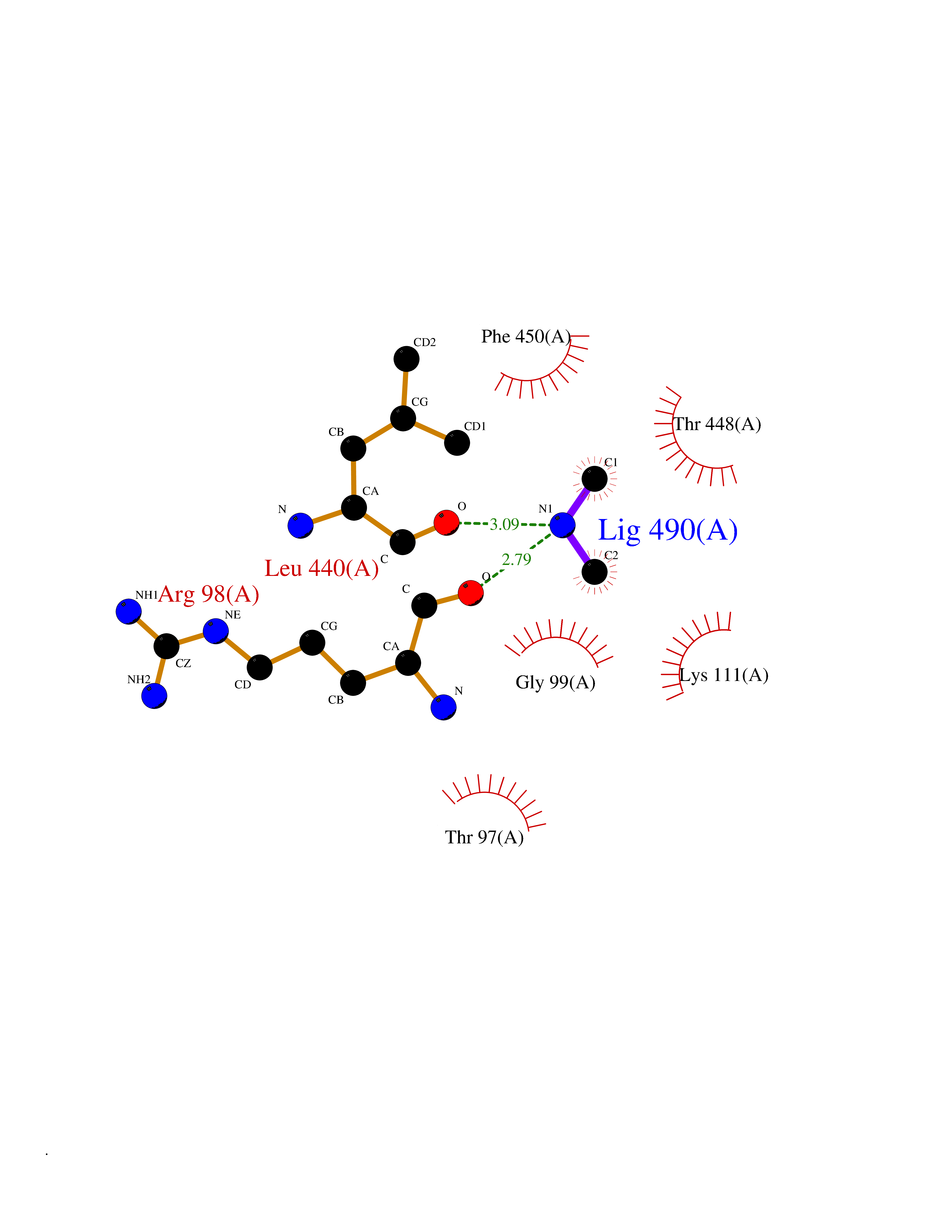 Ligplot Map 3D Binding mode Sequence TSWSDRLQNAADMPANMDKHALKKYRREAYHRVFVNRSLAMEKIKCFGFDMDYTLAVYKSPEYESLGFELTVERLVSIGYPQELLSFAYDSTFPTRGLVFDTLYGNLLKVDAYGNLLVCAHGFNFIRGPETREQYPNKFIQRDDTERFYILNTLFNLPETYLLACLVDFFTNCPRYTSCETGFKDGDLFMSYRSMFQDVRDAVDWVHYKGSLKEKTVENLEKYVVKDGKLPLLLSRMKEVGKVFLATNSDYKYTDKIMTYLFDFPHGPKPGSSHRPWQSYFDLILVDARKPLFFGEGTVLRQVDTKTGKLKIGTYTGPLQHGIVYSGGSSDTICDLLGAKGKDILYIGDHIFGDILKSKKRQGWRTFLVIPELAQELHVWTDKSSLFEELQSLDIFLAQRRIKKVTHDMDMCYGMMGSLFRSGSRQTLFASQVMRYADLYAASFINLLYYPFSYLFRAAHVLMPHES Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Lanosterol 14-alpha-demethylase (EC 1.14.13.70) | 4G3J | 4.01 | |
Target general information Gen name Tb11.02.4080 Organism Trypanosoma brucei brucei (strain 927/4 GUTat10.1) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class NA Function NA Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) NA Interacts with NA EC number 1.14.13.70 Uniprot keywords 3D-structure; Heme; Iron; Metal-binding; Monooxygenase; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID D Molecular weight (Da) 50691.4 Length 448 Aromaticity 0.08 Instability index 54.09 Isoelectric point 6.99 Charge (pH=7) -0.03 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 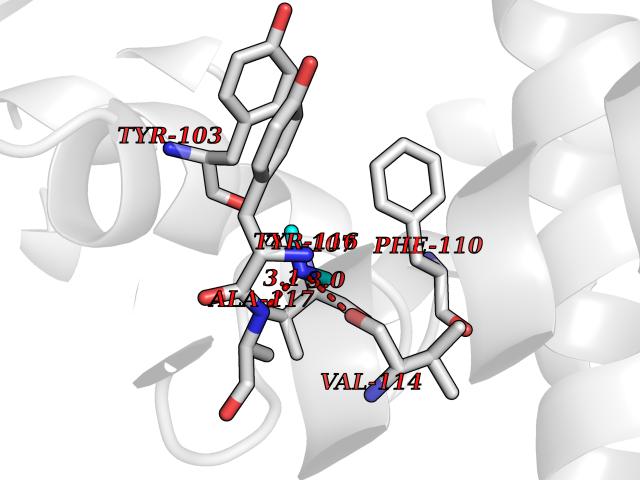 Pymol Map 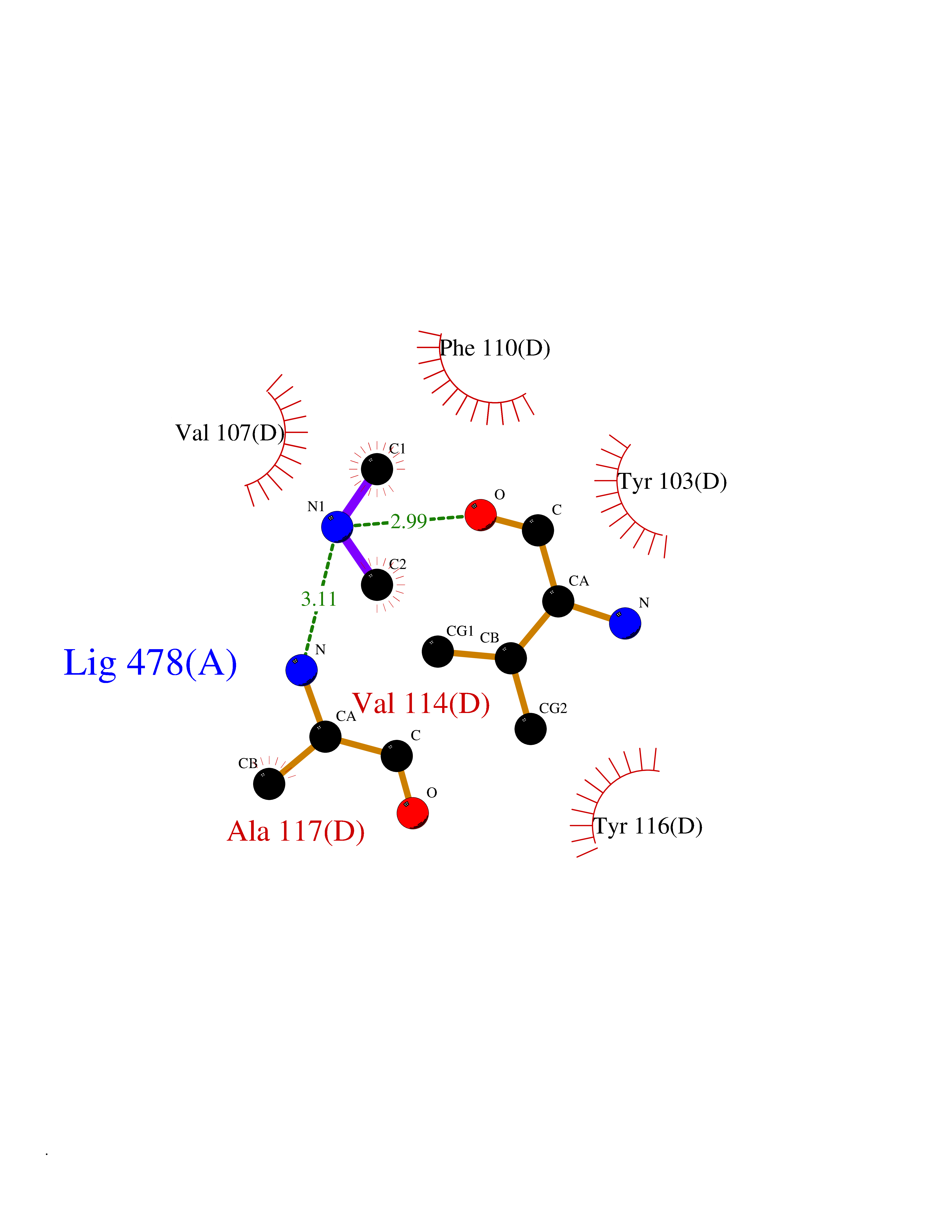 Ligplot Map 3D Binding mode Sequence GKLPPVYPVTVPILGHIIQFGKSPLGFMQECKRQLKSGIFTINIVGKRVTIVGDPHEHSRFFLPRNEVLSPREVYSFMVPVFGEGVAYAAPYPRMREQLNFLAEELTIAKFQNFVPAIQHEVRKFMAANWDKDEGEINLLEDCSTMIINTACQCLFGEDLRKRLDARRFAQLLAKMESSLIPAAVFLPILLKLPLPQSARCHEARTELQKILSEIIIARKEEEVNKDSSTSDLLSGLLSAVYRDGTPMSLHEVCGMIVAAMFAGQHTSSITTTWSMLHLMHPANVKHLEALRKEIEEFPAQLNYNNVMDEMPFAERCARESIRRDPPLLMLMRKVMADVKVGSYVVPKGDIIACSPLLSHHDEEAFPEPRRWDPERDEKVEGAFIGFGAGVHKCIGQKFGLLQVKTILATAFRSYDFQLLRDEVPDPDYHTMVVGPTASQCRVKYIRR Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Protein cereblon (CRBN) | 5FQD | 4.01 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 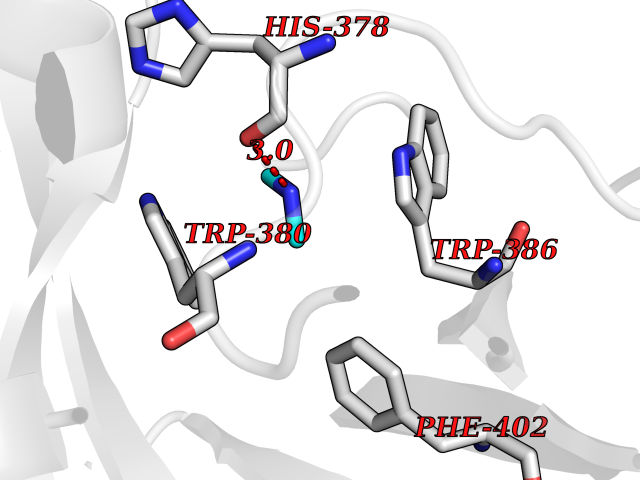 Pymol Map 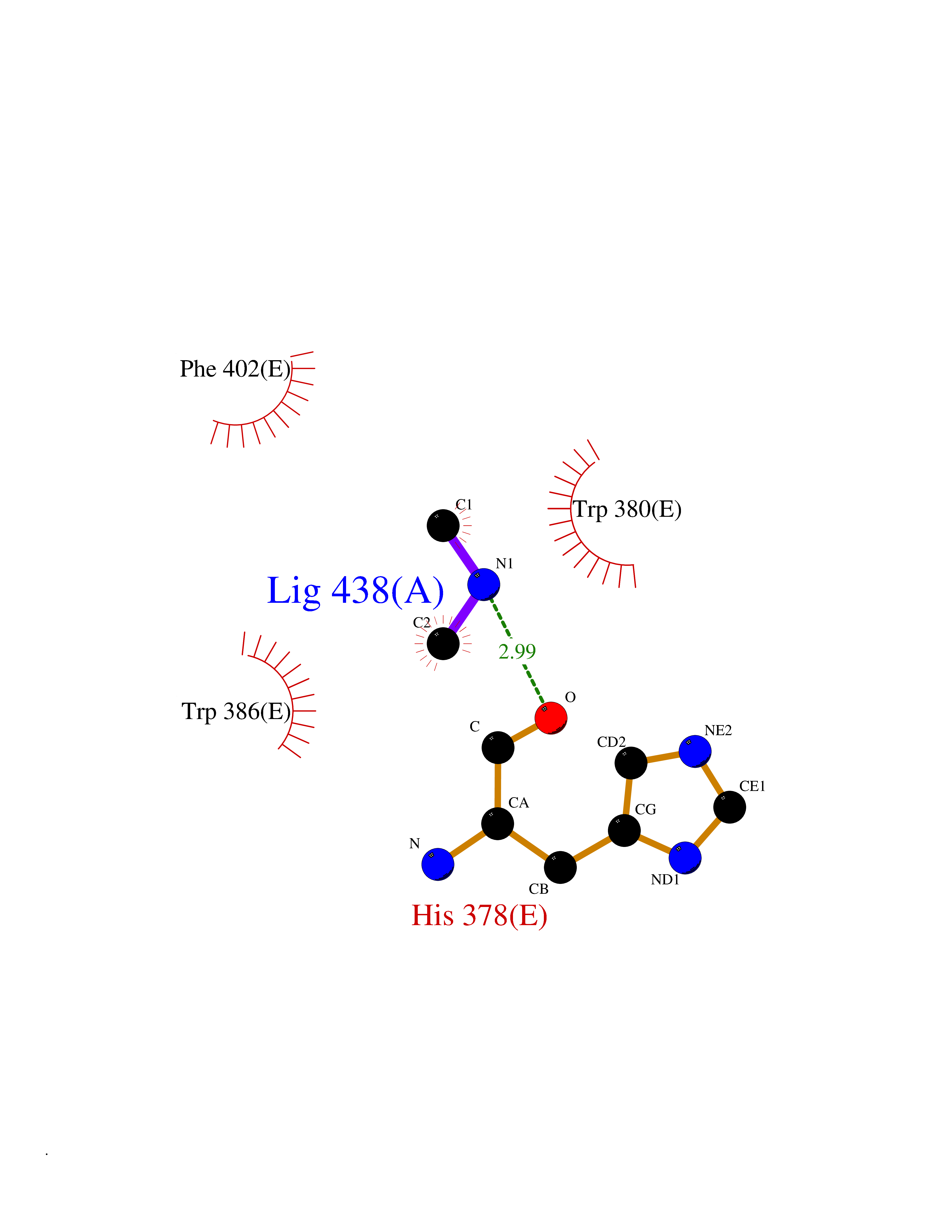 Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | N-acetylgalactosamine 6 sulfatase (GALNS) | 4FDJ | 4.01 | |
Target general information Gen name GALNS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms N-acetylgalactosamine-6-sulfate sulfatase; N-acetylgalactosamine-6-sulfatase; Galactose-6-sulfate sulfatase; GalNAc6S sulfatase; GalN6S; Chondroitinsulfatase; Chondroitinase Protein family Sulfatase family Biochemical class Sulfuric ester hydrolase Function Catalyzes the chemical reaction of cleaving off the 6-sulfate groups of the N-acetyl-D-galactosamine 6-sulfate units of the macromolecule chondroitin sulfate and, similarly, of the D-galactose 6-sulfate units of the macromolecule keratan sulfate. Related diseases Mucopolysaccharidosis 4A (MPS4A) [MIM:253000]: A form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life. {ECO:0000269|PubMed:1522213, ECO:0000269|PubMed:16287098, ECO:0000269|PubMed:24726177, ECO:0000269|PubMed:7581409, ECO:0000269|PubMed:7633425, ECO:0000269|PubMed:7668283, ECO:0000269|PubMed:7795586, ECO:0000269|PubMed:8651279, ECO:0000269|PubMed:8826435, ECO:0000269|PubMed:9298823, ECO:0000269|PubMed:9375852, ECO:0000269|PubMed:9521421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09301 Interacts with NA EC number EC 3.1.6.4 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Dwarfism; Glycoprotein; Hydrolase; Lysosome; Metal-binding; Mucopolysaccharidosis; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 55013.8 Length 493 Aromaticity 0.11 Instability index 35.46 Isoelectric point 6.14 Charge (pH=7) -6.48 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 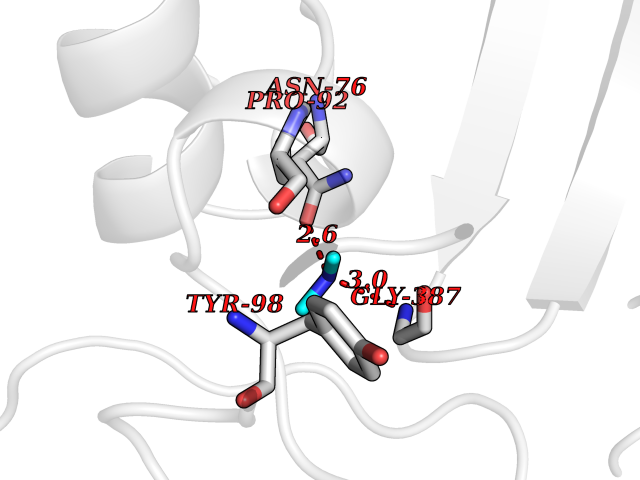 Pymol Map 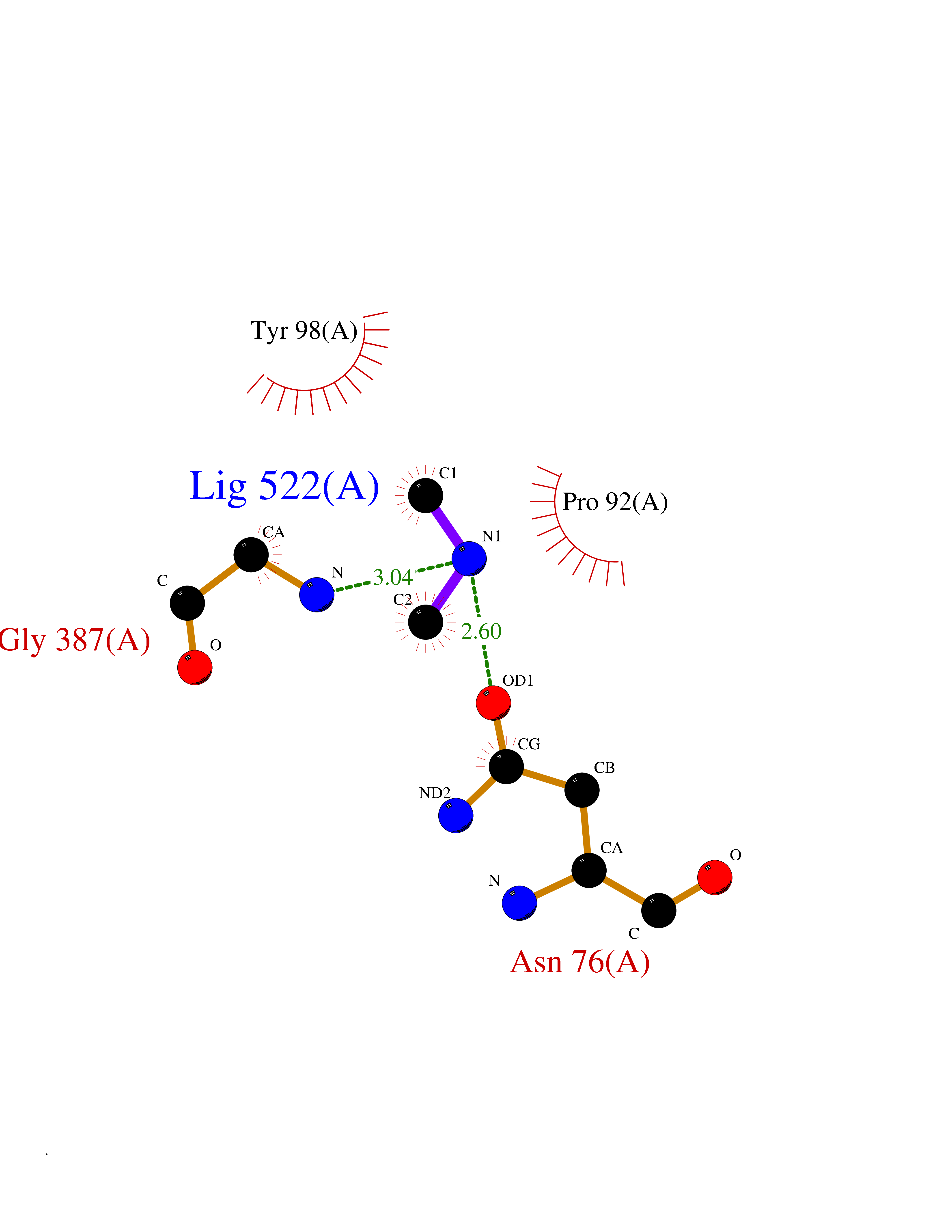 Ligplot Map 3D Binding mode Sequence PQPPNILLLLMDDMGWGDLGVYGEPSRETPNLDRMAAEGLLFPNFYSANPLXSPSRAALLTGRLPIRNGFYTTNAHARNAYTPQEIVGGIPDSEQLLPELLKKAGYVSKIVGKWHLGHRPQFHPLKHGFDEWFGSPNCHFGPYDNKARPNIPVYRDWEMVGRYYEEFPINLKTGEANLTQIYLQEALDFIKRQARHHPFFLYWAVDATHAPVYASKPFLGTSQRGRYGDAVREIDDSIGKILELLQDLHVADNTFVFFTSDNGAALISAPEQGGSNGPFLCGKQTTFEGGMREPALAWWPGHVTAGQVSHQLGSIMDLFTTSLALAGLTPPSDRAIDGLNLLPTLLQGRLMDRPIFYYRGDTLMAATLGQHKAHFWTWTNSWENFRQGIDFCPGQNVSGVTTHNLEDHTKLPLIFHLGRDPGERFPLSFASAEYQEALSRITSVVQQHQEALVPAQPQLNVCNWAVMNWAPPGCEKLGKCLTPPESIPKKCLW Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Haemophilus influenzae NadR protein (Hae-influ nadR) | 1LW7 | 4.01 | |
Target general information Gen name Hae-influ nadR Organism Haemophilus influenzae (strain ATCC 51907 / DSM 11121 / KW20 / Rd) Uniprot ID TTD ID Synonyms nadR; Transcriptional regulator nadR Protein family Bacterial NMN adenylyltransferase family; Bacterial RNK family Biochemical class Nicotinamide ribonucleoside uptake permease Function This enzyme has twoactivities: nicotinamide mononucleotide (NMN) adenylyltransferase and ribosylnicotinamide (RN) kinase. The RN kinase activity catalyzes the phosphorylation of RN to form nicotinamide ribonucleotide. The NMN adenylyltransferase activity catalyzes the transfer of the AMP moiety of ATP to nicotinamide ribonucleotide to form NAD(+). Related diseases Involved in the epigenetic regulation of ESR1 expression in breast cancer in a TFAP2C, IFI16 and HDAC4/5/6-dependent manner. {ECO:0000269|PubMed:24413532}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative initiation; ATP-binding; Cell membrane; Cytoplasm; Kinase; Membrane; Multifunctional enzyme; NAD; Nucleotide-binding; Pyridine nucleotide biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 39581.5 Length 344 Aromaticity 0.14 Instability index 41.39 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 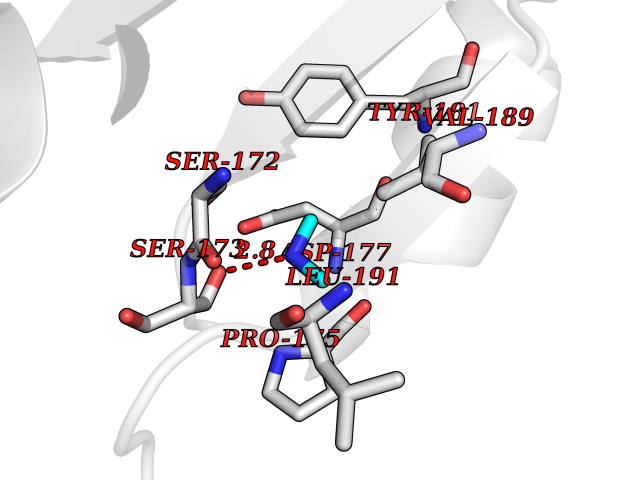 Pymol Map 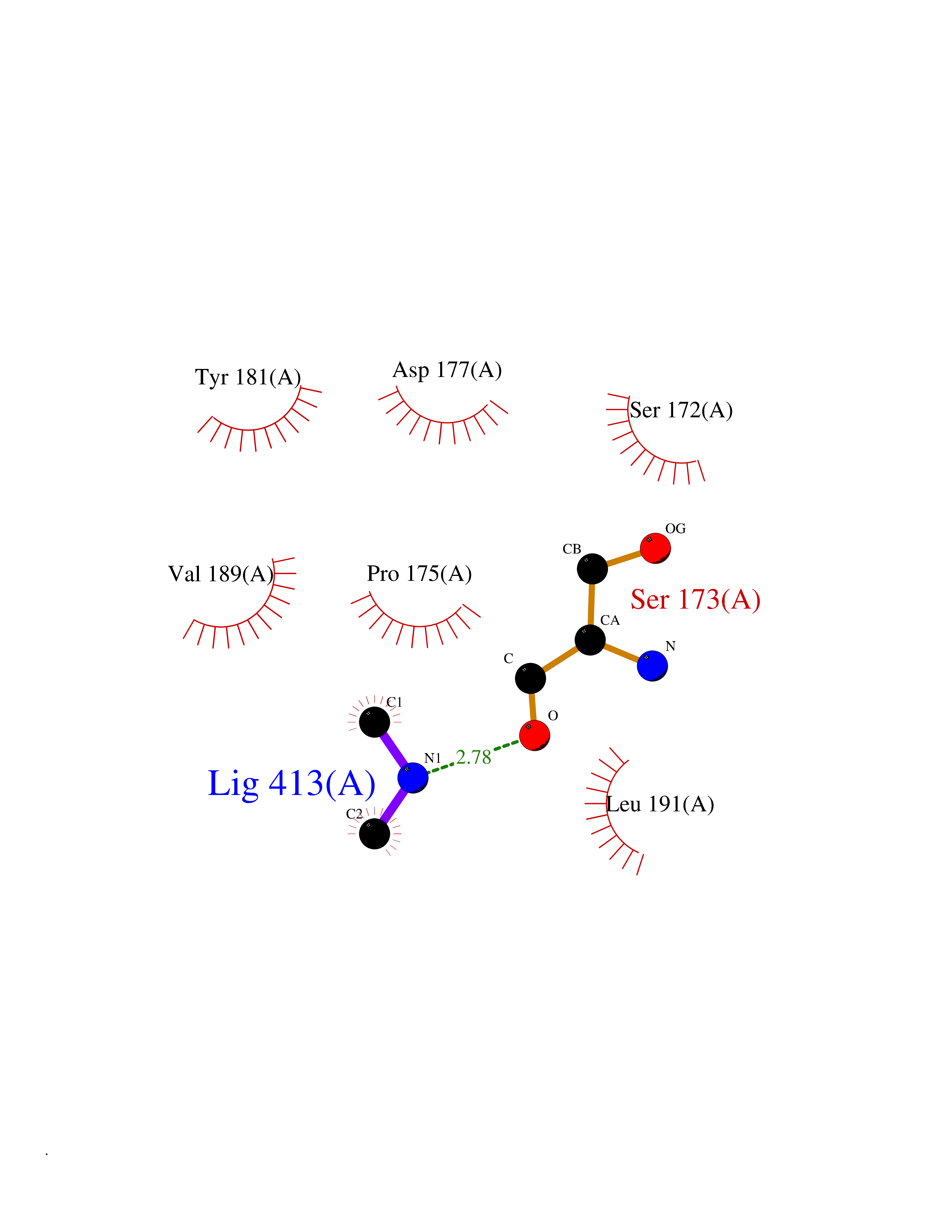 Ligplot Map 3D Binding mode Sequence EKKVGVIFGKFYPVHTGHINXIYEAFSKVDELHVIVCSDTVRDLKLFYDSKXKRXPTVQDRLRWXQQIFKYQKNQIFIHHLVEDGIPSYPNGWQSWSEAVKTLFHEKHFEPSIVFSSEPQDKAPYEKYLGLEVSLVDPDRTFFNVSATKIRTTPFQYWKFIPKEARPFFAKTVAILGGESSGKSVLVNKLAAVFNTTSAWEYGREFVFEKLGGDEQAMQYSDYPQXALGHQRYIDYAVRHSHKIAFIDTDFITTQAFCIQYEGKAHPFLDSXIKEYPFDVTILLKNNTEQKQRQQFQQLLKKLLDKYKVPYIEIESPSYLDRYNQVKAVIEKVLNEEEISELQN Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Suppressor of tumorigenicity 14 protein (ST14) | 3P8G | 4.01 | |
Target general information Gen name ST14 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tumor-associated differentially-expressed gene 15 protein; Tumor associated differentially-expressed gene-15 protein; TADG15; Serine protease TADG-15; Serine protease 14; SNC19; Prostamin; PRSS14; Mem Protein family Peptidase S1 family Biochemical class Peptidase Function Proposed to play a role in breast cancer invasion and metastasis. Exhibits trypsin-like activity as defined by cleavage of synthetic substrates with Arg or Lys as the P1 site. Involved in the terminal differentiation of keratinocytes through prostasin (PRSS8) activation and filaggrin (FLG) processing. Degrades extracellular matrix. Related diseases Ichthyosis, congenital, autosomal recessive 11 (ARCI11) [MIM:602400]: A form of autosomal recessive congenital ichthyosis, a disorder of keratinization with abnormal differentiation and desquamation of the epidermis, resulting in abnormal skin scaling over the whole body. The main skin phenotypes are lamellar ichthyosis (LI) and non-bullous congenital ichthyosiform erythroderma (NCIE), although phenotypic overlap within the same patient or among patients from the same family can occur. Lamellar ichthyosis is a condition often associated with an embedment in a collodion-like membrane at birth; skin scales later develop, covering the entire body surface. Non-bullous congenital ichthyosiform erythroderma characterized by fine whitish scaling on an erythrodermal background; larger brownish scales are present on the buttocks, neck and legs. {ECO:0000269|PubMed:17273967, ECO:0000269|PubMed:18843291}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03127; DB13729; DB00013 Interacts with NA EC number EC 3.4.21.109 Uniprot keywords 3D-structure; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Hypotrichosis; Ichthyosis; Membrane; Protease; Proteomics identification; Reference proteome; Repeat; Serine protease; Signal-anchor; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 26451.5 Length 241 Aromaticity 0.1 Instability index 30.45 Isoelectric point 5.6 Charge (pH=7) -5.69 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 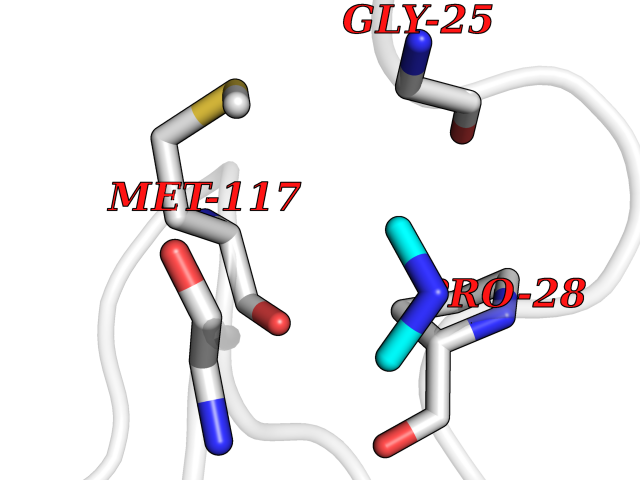 Pymol Map 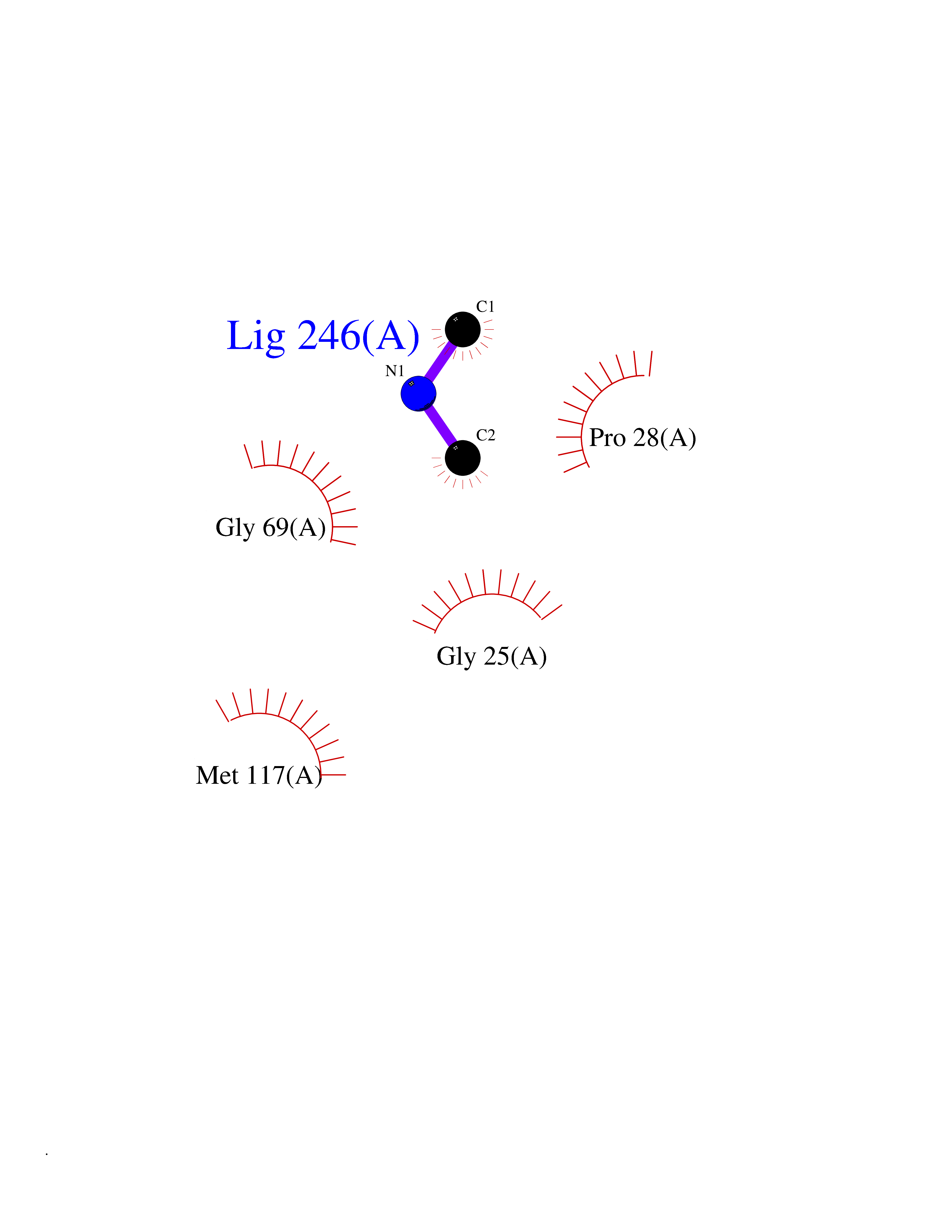 Ligplot Map 3D Binding mode Sequence VVGGTDADEGEWPWQVSLHALGQGHICGASLISPNWLVSAAHCYIDDRGFRYSDPTQWTAFLGLHDQSQRSAPGVQERRLKRIISHPFFNDFTFDYDIALLELEKPAEYSSMVRPICLPDASHVFPAGKAIWVTGWGHTQYGGTGALILQKGEIRVIQQTTCENLLPQQITPRMMCVGFLSGGVDSCQGDSGGPLSSVEADGRIFQAGVVSWGDGCAQRNKPGVYTRLPLFRDWIKENTGV Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Soluble epoxide hydrolase (EPHX2) | 1ZD3 | 4.01 | |
Target general information Gen name EPHX2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Bifunctional epoxide hydrolase 2 Protein family AB hydrolase superfamily, Epoxide hydrolase family Biochemical class Ether bond hydrolase Function Bifunctional enzyme. The C-terminal domain has epoxide hydrolase activity and acts on epoxides (alkene oxides, oxiranes) and arene oxides. Plays a role in xenobiotic metabolism by degrading potentially toxic epoxides (By similarity). Also determines steady-state levels of physiological mediators. The N-terminal domain has lipid phosphatase activity, with the highest activity towards threo-9,10-phosphonooxy-hydroxy-octadecanoic acid, followed by erythro-9,10-phosphonooxy-hydroxy-octadecanoic acid, 12-phosphonooxy-octadec-9Z-enoic acid and 12-phosphonooxy-octadec-9E-enoic acid. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08257; DB08258; DB08259; DB06345; DB12610; DB08256; DB02029; DB04213; DB03677 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aromatic hydrocarbons catabolism; Cytoplasm; Detoxification; Direct protein sequencing; Hydrolase; Lipid metabolism; Lipoprotein; Magnesium; Metal-binding; Multifunctional enzyme; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 61744.9 Length 547 Aromaticity 0.09 Instability index 43.97 Isoelectric point 5.81 Charge (pH=7) -7.76 2D Binding mode Binding energy (Kcal/mol) -5.48 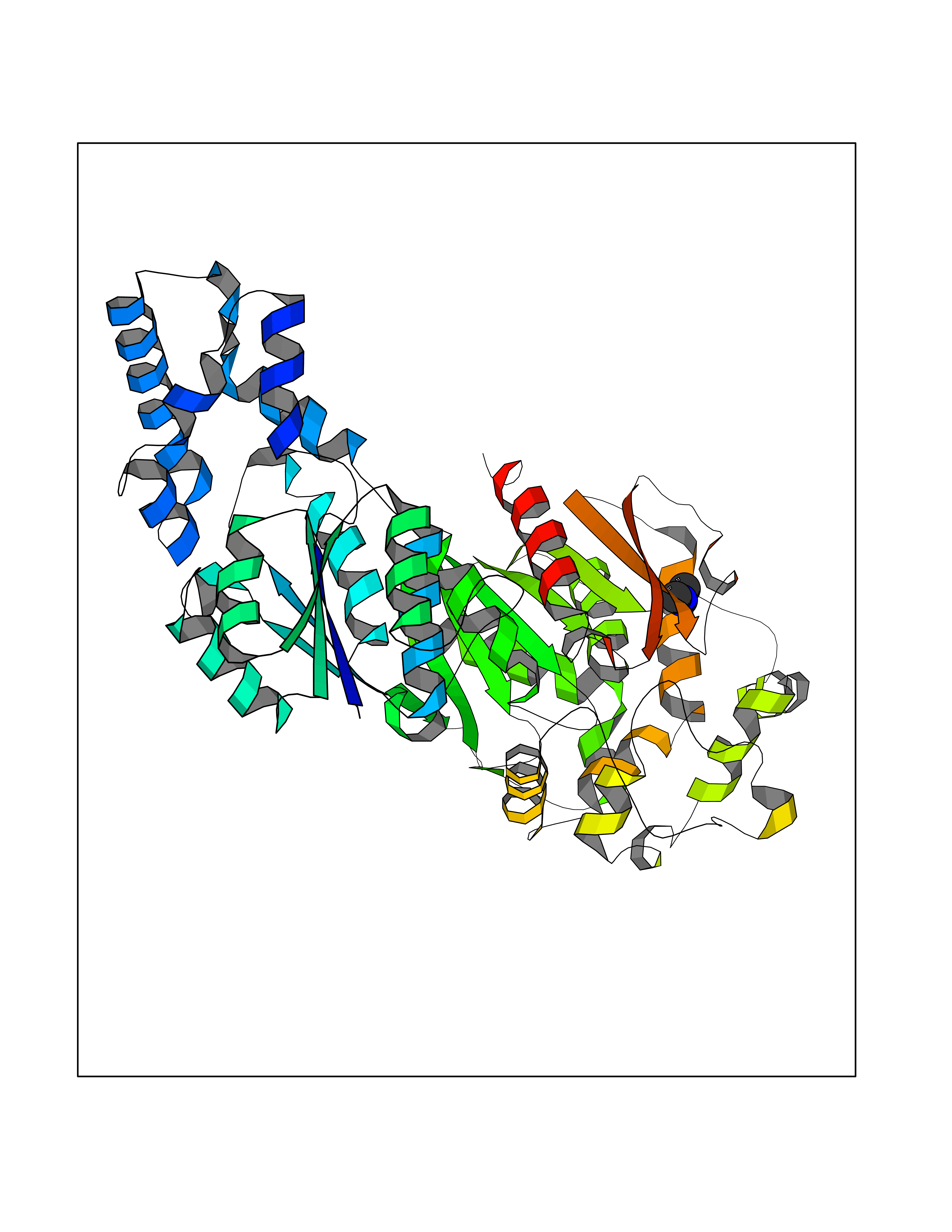 Molscript Map  Pymol Map 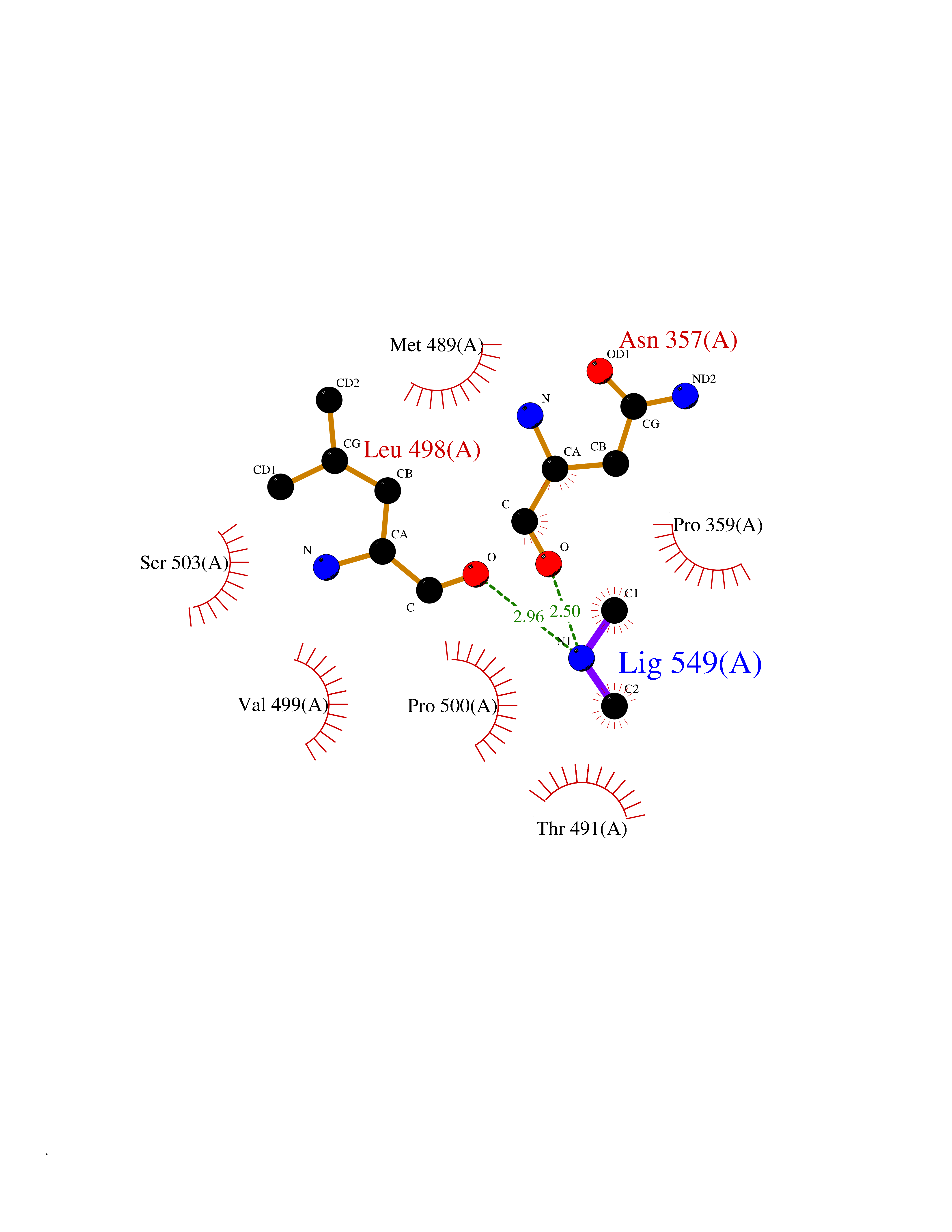 Ligplot Map 3D Binding mode Sequence TLRAAVFDLDGVLALPAVFGVLGRTEEALALPRGLLNDAFQKGGPEGATTRLMKGEITLSQWIPLMEENCRKCSETAKVCLPKNFSIKEIFDKAISARKINRPMLQAALMLRKKGFTTAILTNTWLDDRAERDGLAQLMCELKMHFDFLIESCQVGMVKPEPQIYKFLLDTLKASPSEVVFLDDIGANLKPARDLGMVTILVQDTDTALKELEKVTGIQLLNTPAPLPTSCNPSDMSHGYVTVKPRVRLHFVELGSGPAVCLCHGFPESWYSWRYQIPALAQAGYRVLAMDMKGYGESSAPPEIEEYCMEVLCKEMVTFLDKLGLSQAVFIGHDWGGMLVWYMALFYPERVRAVASLNTPFIPANPNMSPLESIKANPVFDYQLYFQEPGVAEAELEQNLSRTFKSLFRASDESVLSMHKVCEAGGLFVNSPEEPSLSRMVTEEEIQFYVQQFKKSGFRGPLNWYRNMERNWKWACKSLGRKILIPALMVTAEKDFVLVPQMSQHMEDWIPHLKRGHIEDCGHWTQMDKPTEVNQILIKWLDSDARN Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | 6YND | 4.01 | |
Target general information Gen name GAPDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peptidyl-cysteine S-nitrosylase GAPDH; OK/SW-cl.12; GAPD; D-glyceraldehyde-3-phosphate dehydrogenase; CDABP0047 Protein family Glyceraldehyde-3-phosphate dehydrogenase family Biochemical class Aldehyde/oxo donor oxidoreductase Function Participates in nuclear events including transcription, RNA transport, DNA replication and apoptosis. Nuclear functions are probably due to the nitrosylase activity that mediates cysteine S-nitrosylation of nuclear target proteins such as SIRT1, HDAC2 and PRKDC. Modulates the organization and assembly of the cytoskeleton. Facilitates the CHP1-dependent microtubule and membrane associations through its ability to stimulate the binding of CHP1 to microtubules. Glyceraldehyde-3-phosphate dehydrogenase is a key enzyme in glycolysis that catalyzes the first step of the pathway by converting D-glyceraldehyde 3-phosphate (G3P) into 3-phospho-D-glyceroyl phosphate. Component of the GAIT (gamma interferon-activated inhibitor of translation) complex which mediates interferon-gamma-induced transcript-selective translation inhibition in inflammation processes. Upon interferon-gamma treatment assembles into the GAIT complex which binds to stem loop-containing GAIT elements in the 3'-UTR of diverse inflammatory mRNAs (such as ceruplasmin) and suppresses their translation. Has both glyceraldehyde-3-phosphate dehydrogenase and nitrosylase activities, thereby playing a role in glycolysis and nuclear functions, respectively. Related diseases Bone marrow failure and diabetes mellitus syndrome (BMFDMS) [MIM:620044]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFDMS is an autosomal recessive form characterized by various degrees of bone marrow failure, ranging from dyserythropoiesis to bone marrow aplasia, with onset in infancy or early childhood, and non-autoimmune insulin-dependent diabetes mellitus appearing in the first or second decades. Many patients show pigmentary skin abnormalities and short stature. {ECO:0000269|PubMed:28073829, ECO:0000269|PubMed:35611808, ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07347; DB02059; DB11638; DB09130; DB00157; DB03893; DB09092 Interacts with Q6UY14-3; Q9UIJ7; P05067; Q9UQM7; Q14194; P35222; Q9BPW9-4; P00533; O00471; O75344; P06241; P04406; O14556; Q8NEA9; P42858; Q92993-2; P42695; P35228; P12004; P00558; P48147; P17612; Q8WUY3; Q9UHX1-2; P15927; P05109; Q96GZ6; P00441; Q9BSI4; P10599 EC number EC 1.2.1.12 Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Alternative splicing; Apoptosis; Cytoplasm; Cytoskeleton; Direct protein sequencing; Glycolysis; Glycoprotein; Immunity; Innate immunity; Isopeptide bond; Membrane; Methylation; NAD; Nucleus; Oxidation; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; S-nitrosylation; Transferase; Translation regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35818.4 Length 333 Aromaticity 0.08 Instability index 13.69 Isoelectric point 8.64 Charge (pH=7) 3.64 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 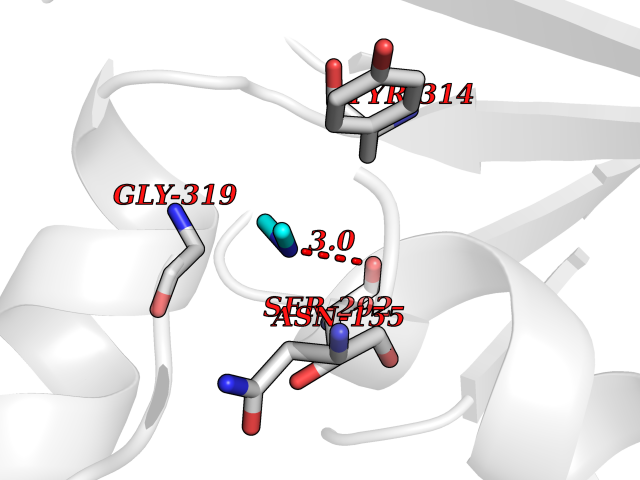 Pymol Map 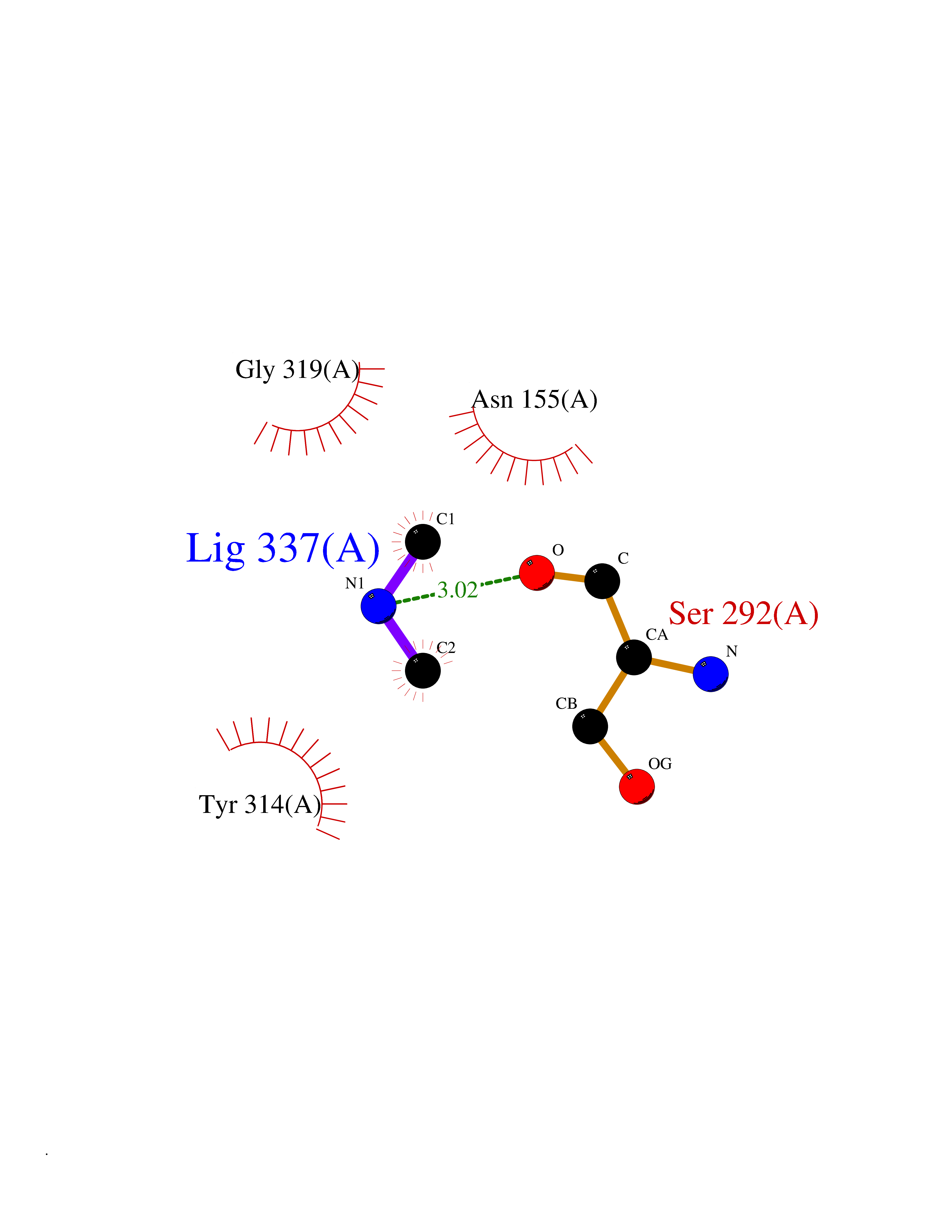 Ligplot Map 3D Binding mode Sequence GKVKVGVNGFGRIGRLVTRAAFNSGKVDIVAINDPFIDLNYMVYMFQYDSTHGKFHGTVKAENGKLVINGNPITIFQERDPSKIKWGDAGAEYVVESTGVFTTMEKAGAHLQGGAKRVIISAPSADAPMFVMGVNHEKYDNSLKIISNASTTNCLAPLAKVIHDNFGIVEGLMTTVHAITATQKTVDGPSGKLWRDGRGALQNIIPASTGAAKAVGKVIPELNGKLTGMAFRVPTANVSVVDLTCRLEKPAKYDDIKKVVKQASEGPLKGILGYTEHQVVSSDFNSDTHSSTFDAGAGIALNDHFVKLISWYDNEFGYSNRVVDLMAHMASKE Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Cathepsin G (CTSG) | 1KYN | 4.01 | |
Target general information Gen name CTSG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CG Protein family Peptidase S1 family Biochemical class Peptidase Function Cleaves complement C3. Has antibacterial activity against the Gram-negative bacterium P. aeruginosa, antibacterial activity is inhibited by LPS from P. aeruginosa, Z-Gly-Leu-Phe-CH2Cl and phenylmethylsulfonyl fluoride. Serine protease with trypsin- and chymotrypsin-like specificity. Related diseases Lethal congenital contracture syndrome 2 (LCCS2) [MIM:607598]: A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS2 patients manifest craniofacial/ocular findings, lack of hydrops, multiple pterygia, and fractures, as well as a normal duration of pregnancy and a unique feature of a markedly distended urinary bladder (neurogenic bladder defect). The phenotype suggests a spinal cord neuropathic etiology. {ECO:0000269|PubMed:17701904}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythroleukemia, familial (FERLK) [MIM:133180]: An autosomal dominant myeloproliferative disorder characterized by neoplastic proliferation of erythroblastic and myeloblastic elements with atypical erythroblasts and myeloblasts in the peripheral blood. Disease penetrance is incomplete. {ECO:0000269|PubMed:27416908}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Visceral neuropathy, familial, 1, autosomal recessive (VSCN1) [MIM:243180]: An autosomal recessive disorder characterized by intestinal dysmotility due to aganglionosis (Hirschsprung disease), hypoganglionosis, and/or chronic intestinal pseudoobstruction. Additional variable features are progressive peripheral neuropathy, arthrogryposis, hypoplasia or aplasia of the olfactory bulb and of the external auditory canals, microtia or anotia, and facial dysmorphism. Some patients present structural cardiac anomalies and arthrogryposis with multiple pterygia. {ECO:0000269|PubMed:33497358}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04016; DB02360 Interacts with A8MQ03; Q15323; Q7Z3S9; Q9NRD5 EC number EC 3.4.21.20 Uniprot keywords 3D-structure; Antibiotic; Antimicrobial; Cell membrane; Chemotaxis; Cytoplasm; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lysosome; Membrane; Nucleus; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 25353.8 Length 223 Aromaticity 0.06 Instability index 66.1 Isoelectric point 11.51 Charge (pH=7) 23.24 2D Binding mode Binding energy (Kcal/mol) -5.48 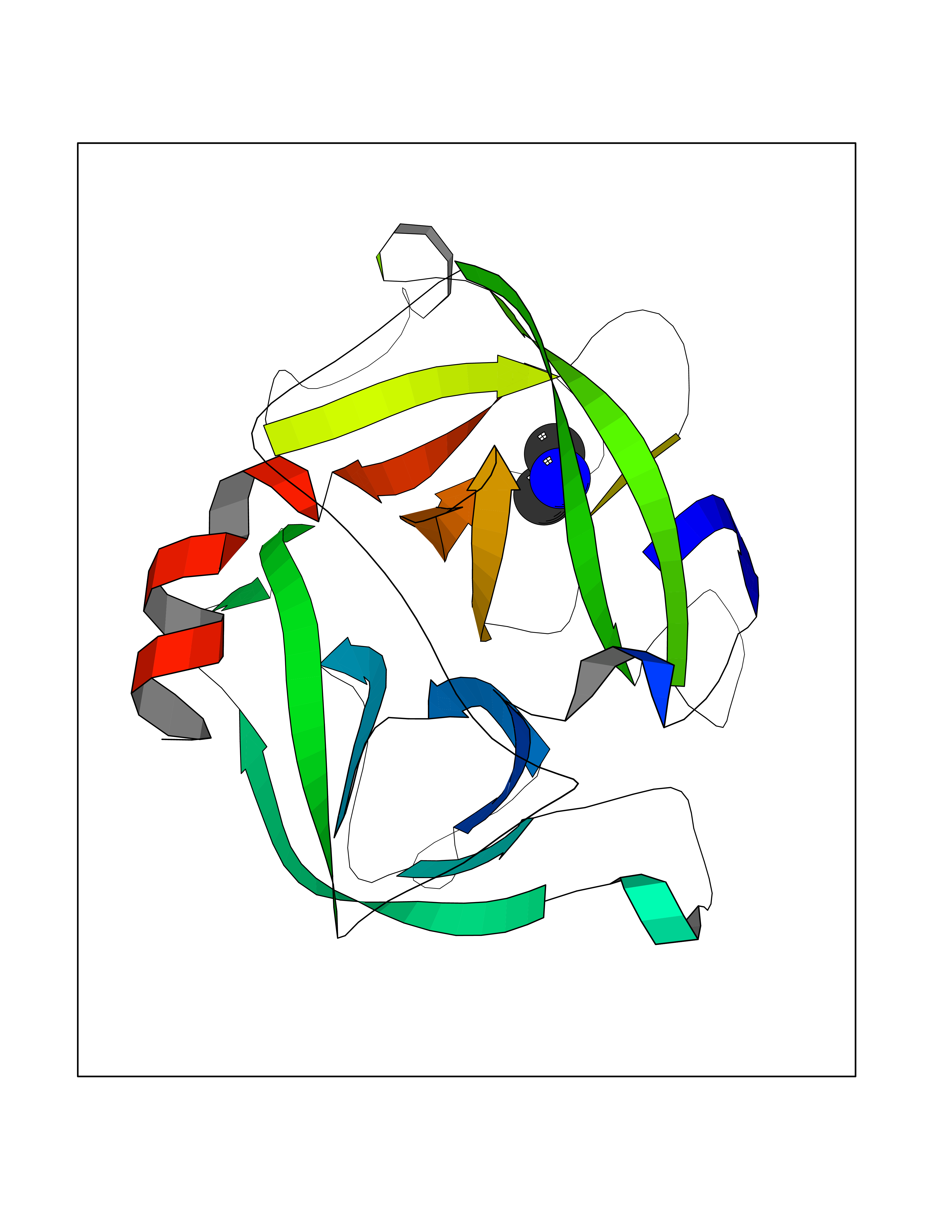 Molscript Map 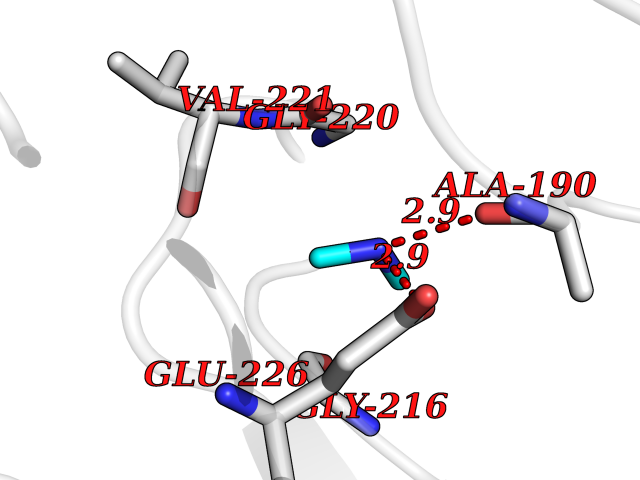 Pymol Map 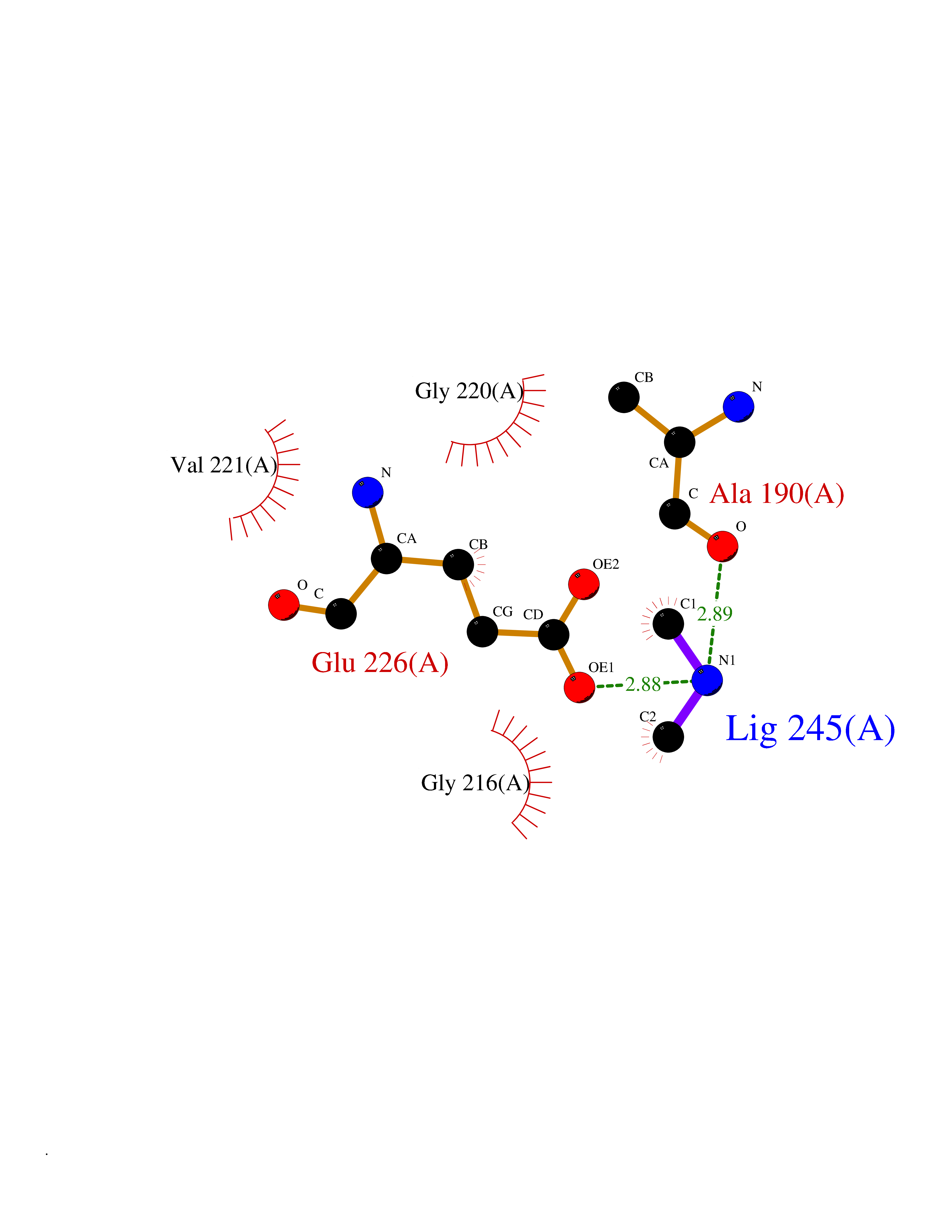 Ligplot Map 3D Binding mode Sequence IIGGRESRPHSRPYMAYLQIQSPAGQSRCGGFLVREDFVLTAAHCWGSNINVTLGAHNIQRRENTQQHITARRAIRHPQYNQRTIQNDIMLLQLSRRVRRNRNVNPVALPRAQEGLRPGTLCTVAGWGRVSMRRGTDTLREVQLRVQRDRQCLRIFGSYDPRRQICVGDRRERKAAFKGDSGGPLLCNNVAHGIVSYGKSSGVPPEVFTRVSSFLPWIRTTMR Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Triggering receptor expressed on monocytes 1 (TREM1) | 1Q8M | 4.01 | |
Target general information Gen name Trem1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Trem1; TREM-1 Protein family NA Biochemical class NA Function Stimulates neutrophil and monocyte-mediated inflammatory responses. Triggers release of pro-inflammatory chemokines and cytokines, as well as increased surface expression of cell activation markers. Amplifier of inflammatory responses that are triggered by bacterial and fungal infections and is a crucial mediator of septic shock. Related diseases GLUT1 deficiency syndrome 1 (GLUT1DS1) [MIM:606777]: A neurologic disorder showing wide phenotypic variability. The most severe 'classic' phenotype comprises infantile-onset epileptic encephalopathy associated with delayed development, acquired microcephaly, motor incoordination, and spasticity. Onset of seizures, usually characterized by apneic episodes, staring spells, and episodic eye movements, occurs within the first 4 months of life. Other paroxysmal findings include intermittent ataxia, confusion, lethargy, sleep disturbance, and headache. Varying degrees of cognitive impairment can occur, ranging from learning disabilities to severe intellectual disability. {ECO:0000269|PubMed:10227690, ECO:0000269|PubMed:10980529, ECO:0000269|PubMed:11136715, ECO:0000269|PubMed:11603379, ECO:0000269|PubMed:12325075, ECO:0000269|PubMed:15622525, ECO:0000269|PubMed:19901175, ECO:0000269|PubMed:20129935, ECO:0000269|PubMed:20221955, ECO:0000269|PubMed:20574033, ECO:0000269|PubMed:24847886, ECO:0000269|PubMed:25982116, ECO:0000269|PubMed:30197081}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GLUT1 deficiency syndrome 2 (GLUT1DS2) [MIM:612126]: A clinically variable disorder characterized primarily by onset in childhood of paroxysmal exercise-induced dyskinesia. The dyskinesia involves transient abnormal involuntary movements, such as dystonia and choreoathetosis, induced by exercise or exertion, and affecting the exercised limbs. Some patients may also have epilepsy, most commonly childhood absence epilepsy. Mild intellectual disability may also occur. In some patients involuntary exertion-induced dystonic, choreoathetotic, and ballistic movements may be associated with macrocytic hemolytic anemia. {ECO:0000269|PubMed:14605501, ECO:0000269|PubMed:18451999, ECO:0000269|PubMed:19630075, ECO:0000269|PubMed:19798636, ECO:0000269|PubMed:20129935, ECO:0000269|PubMed:20574033, ECO:0000269|PubMed:20621801, ECO:0000269|PubMed:20830593, ECO:0000269|PubMed:21204808}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epilepsy, idiopathic generalized 12 (EIG12) [MIM:614847]: A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. In some EIG12 patients seizures may remit with age. {ECO:0000269|PubMed:19798636, ECO:0000269|PubMed:22282645, ECO:0000269|PubMed:23280796, ECO:0000269|PubMed:25982116}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Dystonia 9 (DYT9) [MIM:601042]: An autosomal dominant neurologic disorder characterized by childhood onset of paroxysmal choreoathetosis and progressive spastic paraplegia. Most patients show some degree of cognitive impairment. Other variable features may include seizures, migraine headaches, and ataxia. {ECO:0000269|PubMed:21832227}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Stomatin-deficient cryohydrocytosis with neurologic defects (SDCHCN) [MIM:608885]: A rare form of stomatocytosis characterized by episodic hemolytic anemia, cold-induced red cells cation leak, erratic hyperkalemia, neonatal hyperbilirubinemia, hepatosplenomegaly, cataracts, seizures, intellectual disability, and movement disorder. {ECO:0000269|PubMed:21791420, ECO:0000269|PubMed:22492876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01694 Interacts with NA EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Innate immunity; Membrane; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 28186.2 Length 242 Aromaticity 0.07 Instability index 45.2 Isoelectric point 6.1 Charge (pH=7) -3.8 2D Binding mode Binding energy (Kcal/mol) -5.47 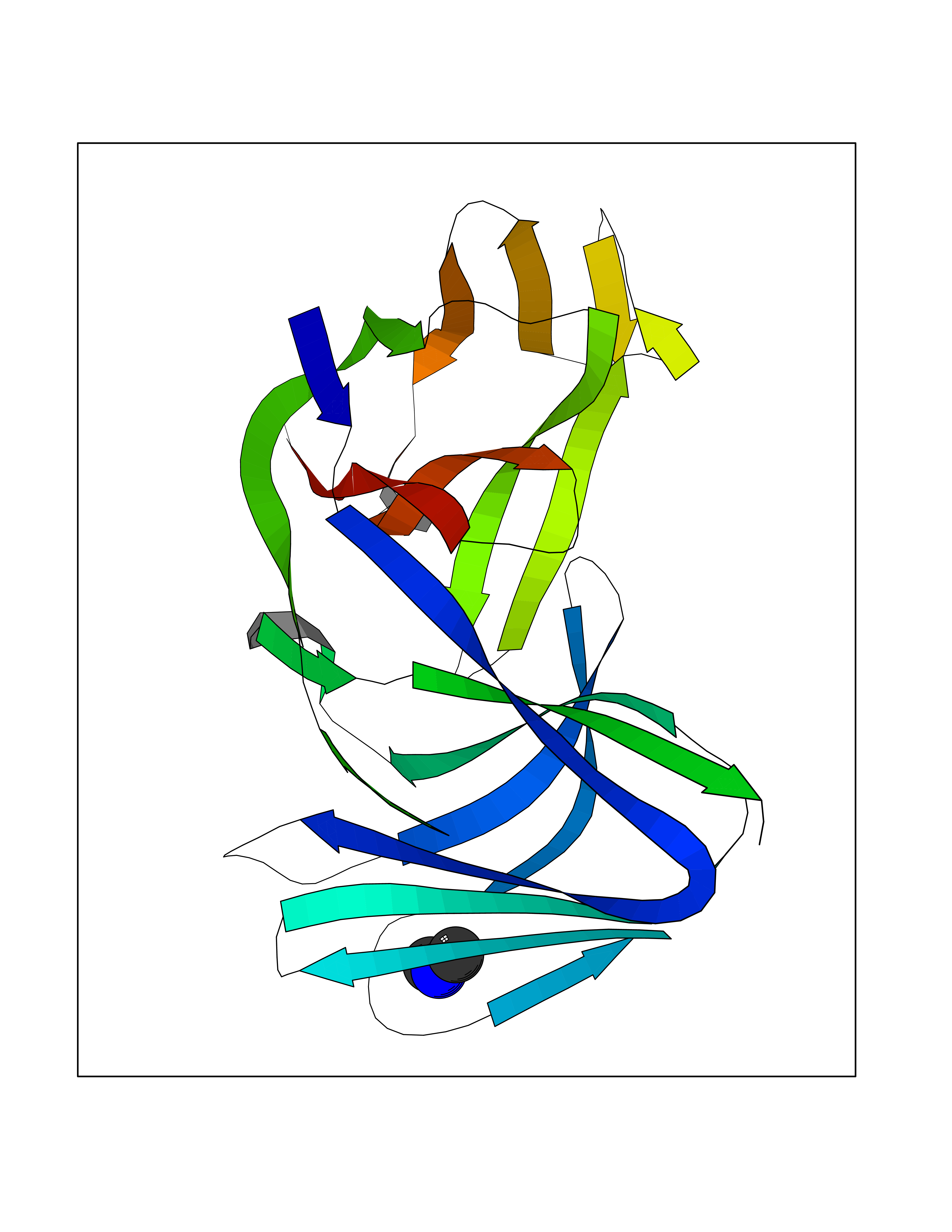 Molscript Map 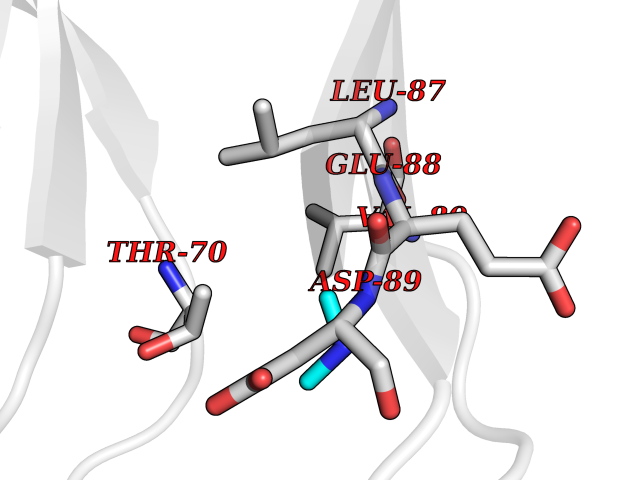 Pymol Map 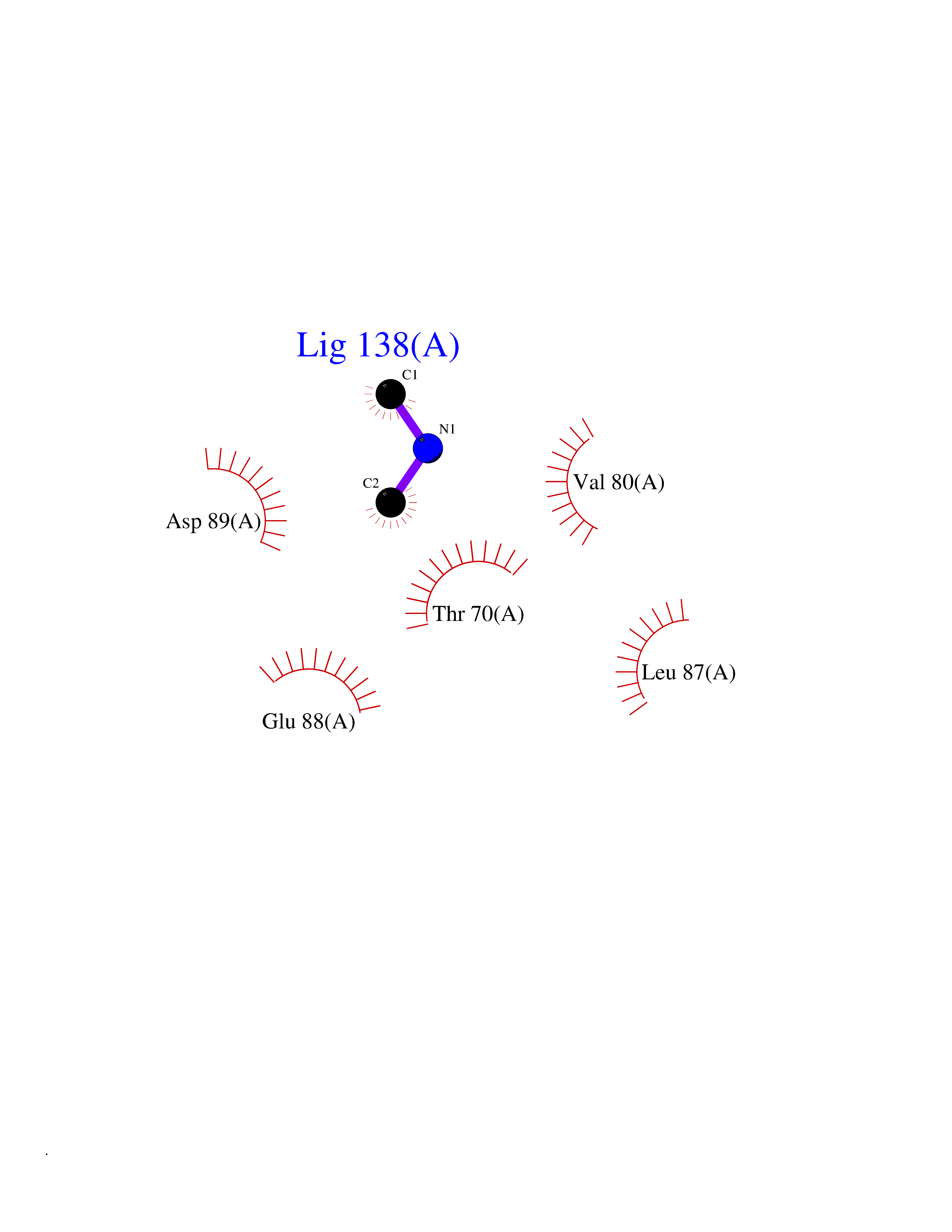 Ligplot Map 3D Binding mode Sequence MELRAATKLTEEKYELKEGQTLDVKCDYTLEKFASSQKAWQIIRDGEMPKTLACTERPSKNSHPVQVGRIILEDYHDHGLLRVRMVNLQVEDSGLYQCVIYQPPKEPHMLFDRIRLVVTLEMELRAATKLTEEKYELKEGQTLDVKCDYTLEKFASSQKAWQIIRDGEMPKTLACTERPSKNSHPVQVGRIILEDYHDHGLLRVRMVNLQVEDSGLYQCVIYQPPKEPHMLFDRIRLVVTLE Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Endolysin | 1AM7 | 4.00 | |
Target general information Gen name R Organism Escherichia phage lambda (Bacteriophage lambda) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 24 family Biochemical class Glycosidase Function Lyase activity.Lysozyme activity.Lytic transglycosylase activity. Related diseases Estrogen resistance (ESTRR) [MIM:615363]: A disorder characterized by partial or complete resistance to estrogens, in the presence of elevated estrogen serum levels. Clinical features include absence of the pubertal growth spurt, delayed bone maturation, unfused epiphyses, reduced bone mineral density, osteoporosis, continued growth into adulthood and very tall adult stature. Glucose intolerance, hyperinsulinemia and lipid abnormalities may also be present. {ECO:0000269|PubMed:23841731, ECO:0000269|PubMed:27754803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04206 Interacts with NA EC number 4.2.2.n2 Uniprot keywords 3D-structure; Antimicrobial; Bacteriolytic enzyme; Cytolysis; Direct protein sequencing; Host cell lysis by virus; Host cytoplasm; Lyase; Reference proteome; Viral release from host cell Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 49834.9 Length 462 Aromaticity 0.07 Instability index 18.78 Isoelectric point 9.6 Charge (pH=7) 18.29 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map  Pymol Map 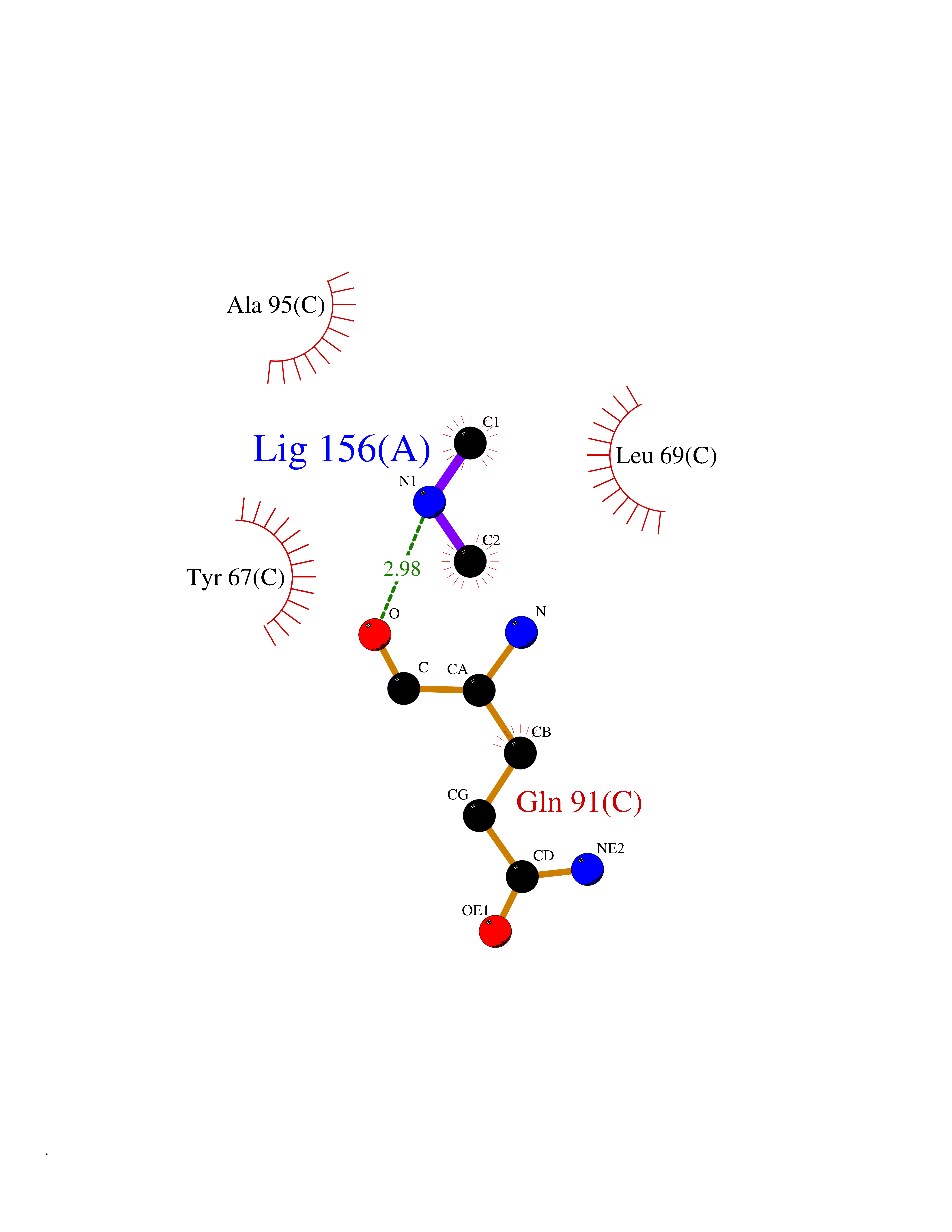 Ligplot Map 3D Binding mode Sequence MVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVR Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Fructose-1,6-bisphosphatase 1 | 2JJK | 4.00 | |
Target general information Gen name FBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FBP Protein family FBPase class 1 family Biochemical class Hydrolase Function AMP binding.Fructose 1,6-bisphosphate 1-phosphatase activity.Identical protein binding.Metal ion binding.Monosaccharide binding. Related diseases Fructose-1,6-bisphosphatase deficiency (FBP1D) [MIM:229700]: An autosomal recessive metabolic disorder characterized by impaired gluconeogenesis, and episodes of hypoglycemia and metabolic acidosis that can be lethal in newborn infants or young children. {ECO:0000269|PubMed:12126934, ECO:0000269|PubMed:25601412, ECO:0000269|PubMed:9382095}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02778; DB07312; DB07321; DB08484; DB00131; DB04493; DB05518; DB05053; DB04175; DB07270; DB02848 Interacts with O00499; Q99814; P09467; O00757; P51116; Q16665; P42858 EC number 3.1.3.11 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Carbohydrate metabolism; Direct protein sequencing; Disease variant; Gluconeogenesis; Hydrolase; Magnesium; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 69197.4 Length 634 Aromaticity 0.08 Instability index 25.2 Isoelectric point 7.84 Charge (pH=7) 2.55 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 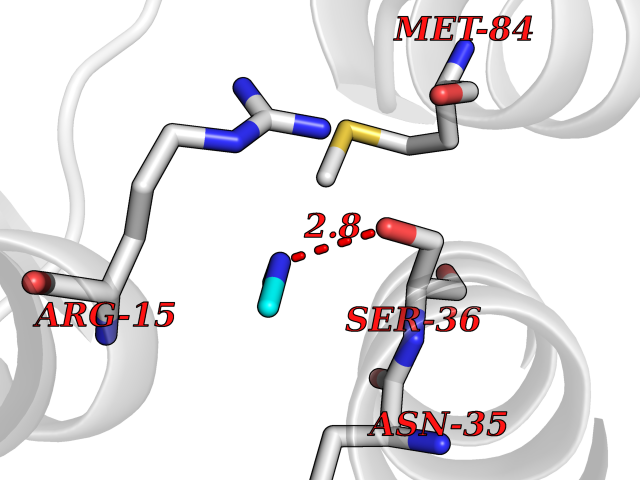 Pymol Map 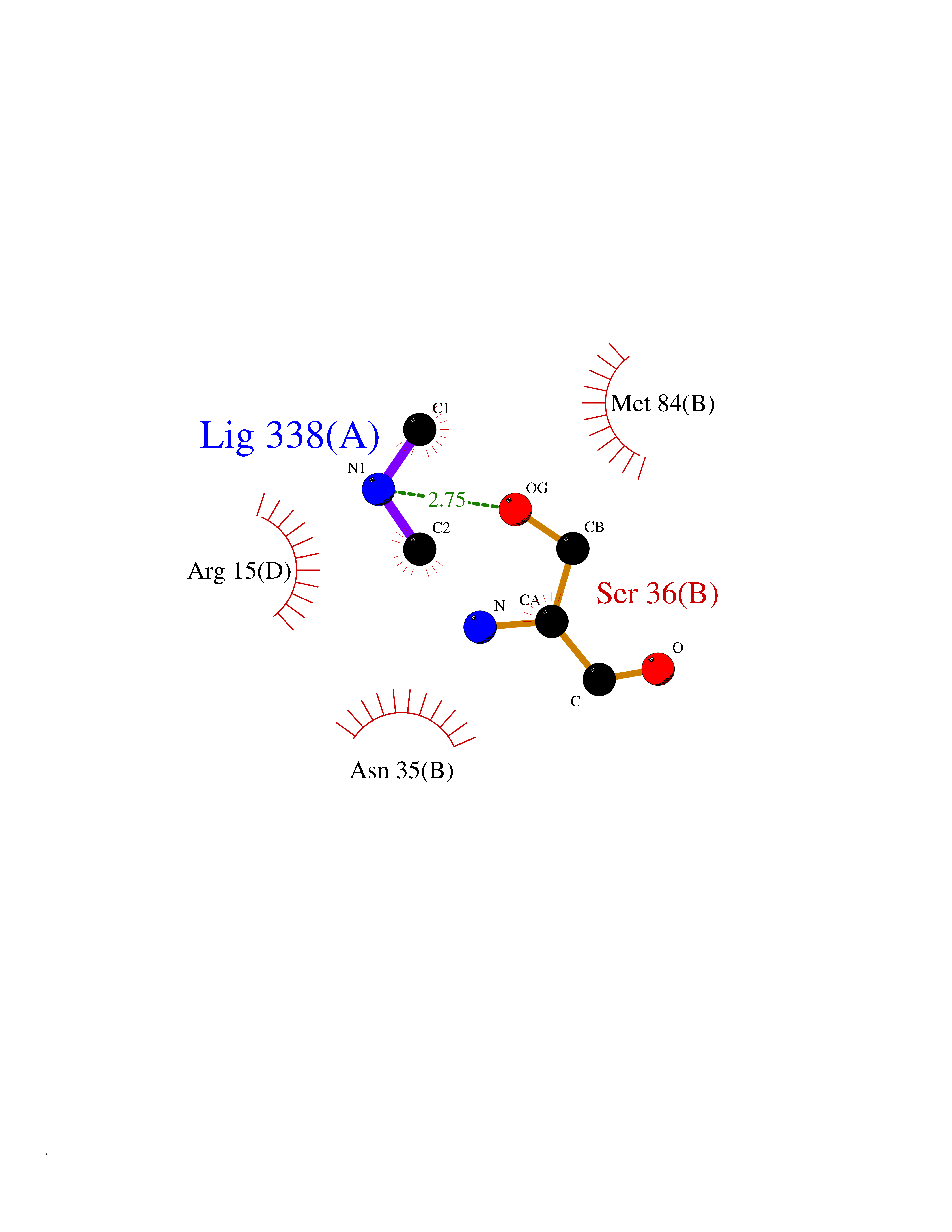 Ligplot Map 3D Binding mode Sequence DVNTLTRFVMEEGRKARGTGELTQLLNSLCTAVKAISSAVRKAGIAHLYGIAGKLDVLSNDLVMNMLKSSFATCVLVSEEDKHAIIVEPEKRGKYVVCFDPLDGSSNIDCLVSVGTIFGIYRKKSTDEPSEKDALQPGRNLVAAGYALYGSATMLVLAMDCGVNCFMLDPAIGEFILVDKDVKIKKKGKIYSLNEGYAKDFDPAVTEYIQRKKFPPDNSAPYGARYVGSMVADVHRTLVYGGIFLYPANKKSPNGKLRLLYECNPMAYVMEKAGGMATTGKEAVLDVIPTDIHQRAPVILGSPDDVLEFLKVYEKHSDVNTLTRFVMEEGRKARGTGELTQLLNSLCTAVKAISSAVRKAGIAHLYGIAGKLDVLSNDLVMNMLKSSFATCVLVSEEDKHAIIVEPEKRGKYVVCFDPLDGSSNIDCLVSVGTIFGIYRKKSTDEPSEKDALQPGRNLVAAGYALYGSATMLVLAMDCGVNCFMLDPAIGEFILVDKDVKIKKKGKIYSLNEGYAKDFDPAVTEYIQRKKFPPDNSAPYGARYVGSMVADVHRTLVYGGIFLYPANKKSPNGKLRLLYECNPMAYVMEKAGGMATTGKEAVLDVIPTDIHQRAPVILGSPDDVLEFLKVYEKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | mRNA-capping enzyme | 2C46 | 4.00 | |
Target general information Gen name RNGTT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CAP1A Protein family Non-receptor class of the protein-tyrosine phosphatase family; Eukaryotic GTase family Biochemical class Transferase Function GTP binding.MRNA guanylyltransferase activity.Polynucleotide 5'-phosphatase activity.Protein tyrosine/serine/threonine phosphatase activity.Protein tyrosine phosphatase activity.RNA guanylyltransferase activity.Triphosphatase activity. Related diseases Atrial fibrillation, familial, 14 (ATFB14) [MIM:615378]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:19808477}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Genetic variations in SCN2B may be involved in Brugada syndrome (PubMed:23559163). This tachyarrhythmia is characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:23559163}. Drugs (DrugBank ID) NA Interacts with Q92624; P16333-1 EC number 2.7.7.50; 3.6.1.74 Uniprot keywords 3D-structure; Alternative splicing; GTP-binding; Host-virus interaction; Hydrolase; mRNA capping; mRNA processing; Multifunctional enzyme; Nucleotide-binding; Nucleotidyltransferase; Nucleus; Protein phosphatase; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 21849.8 Length 189 Aromaticity 0.11 Instability index 53.71 Isoelectric point 5.89 Charge (pH=7) -2.91 2D Binding mode Binding energy (Kcal/mol) -5.46 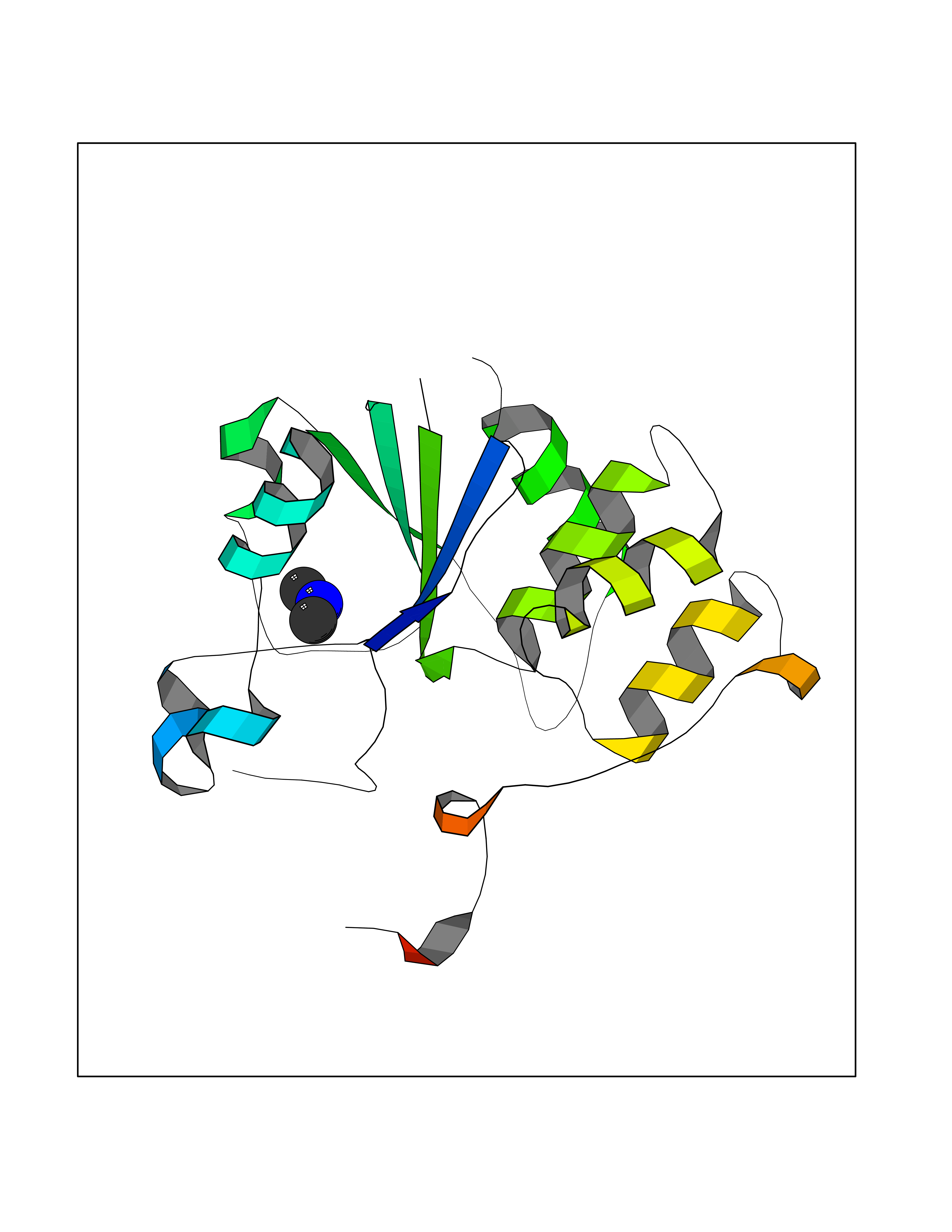 Molscript Map 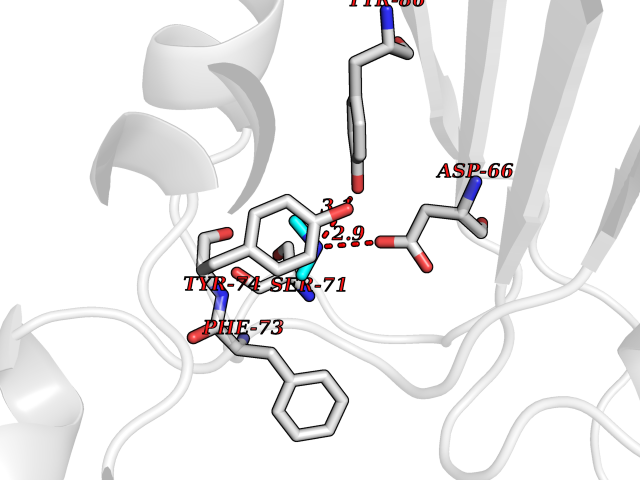 Pymol Map 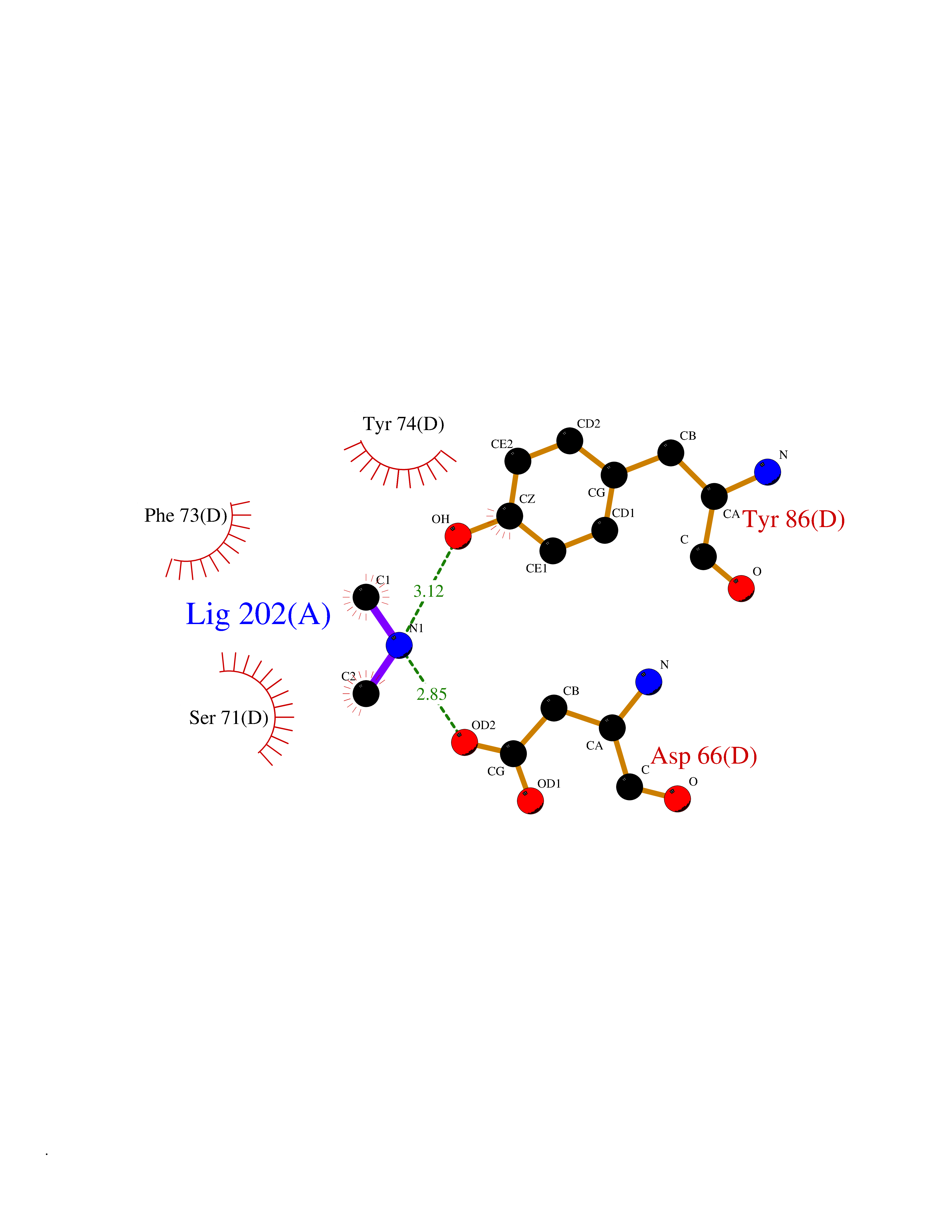 Ligplot Map 3D Binding mode Sequence NKIPPRWLNCPRRGQPVAGRFLPLKTMLGPRYDSQVAEENRFHPSMLSNYLKSVKMGLLVDLTNTSRFYDRNDIEKEGIKYIKLQCKGHGECPTTENTETFIRLCERFELIGVHCTHGFNRTGFLICAFLVEKMDWSIEAAVATFAQARPPGIYKGDYLKELFRRYGDIEEAPPPPLLPDWCFEDDEDE Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Leukotriene A-4 hydrolase (LTA4H) | 3U9W | 4.00 | |
Target general information Gen name LTA4H Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Leukotriene A4 hydrolase; Leukotriene A(4)Leukotriene A-4 hydrolase hydrolase; Leukotriene A(4) hydrolase; LTA4; LTA-H; LTA-4hydrolase; LTA-4 hydrolase Protein family Peptidase M1 family Biochemical class Ether bond hydrolase Function Has also aminopeptidase activity. Epoxide hydrolase that catalyzes the final step in the biosynthesis of the proinflammatory mediator leukotriene B4. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07102; DB06917; DB07258; DB07094; DB07259; DB02352; DB07292; DB07104; DB06828; DB08466; DB01197; DB05177; DB03366; DB08040; DB06851; DB02062; DB07099; DB07260; DB07196; DB11781; DB03424; DB07237 Interacts with Q9BSI4 EC number EC 3.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Leukotriene biosynthesis; Lipid metabolism; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 68927 Length 608 Aromaticity 0.1 Instability index 38.84 Isoelectric point 5.87 Charge (pH=7) -9.86 2D Binding mode Binding energy (Kcal/mol) -5.45 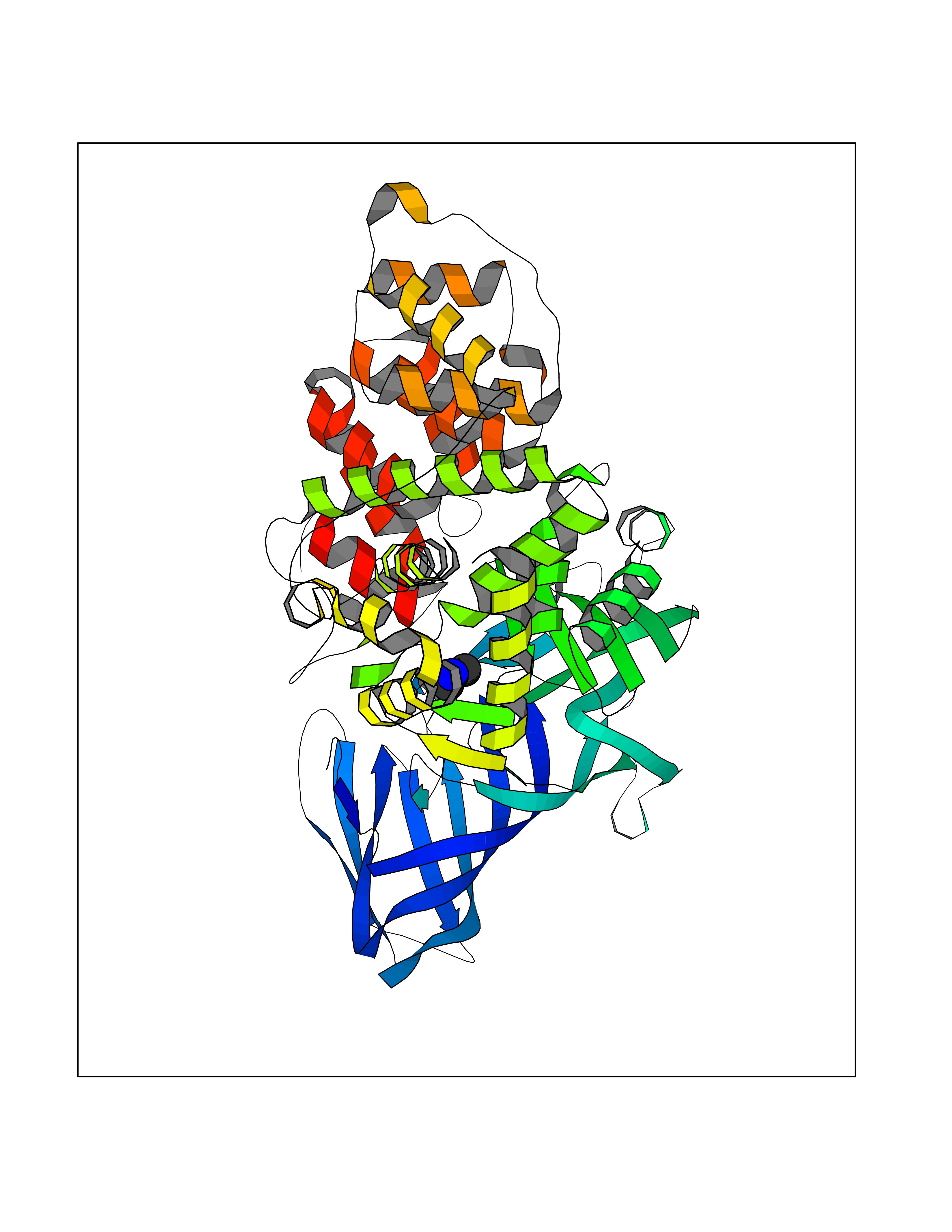 Molscript Map 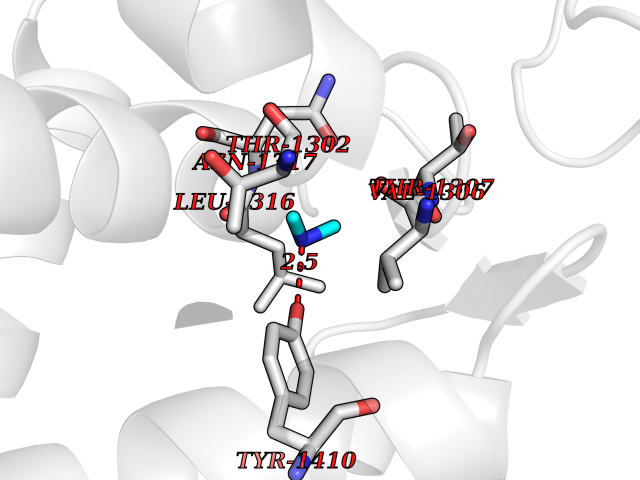 Pymol Map 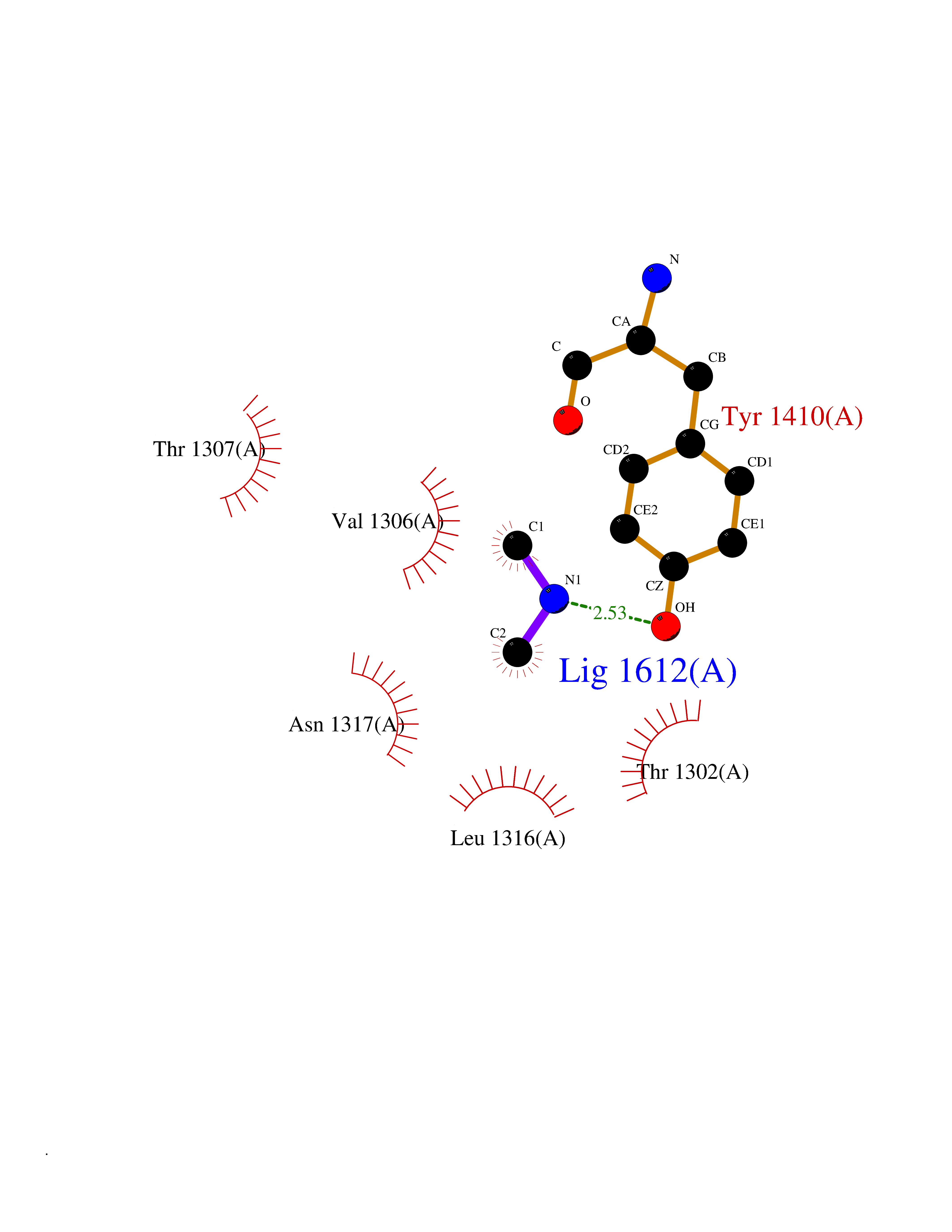 Ligplot Map 3D Binding mode Sequence IVDTCSLASPASVCRTKHLHLRCSVDFTRRTLTGTAALTVQSQEDNLRSLVLDTKDLTIEKVVINGQEVKYALGERQSYKGSPMEISLPIALSKNQEIVIEISFETSPKSSALQWLTPEQTSGKEHPYLFSQCQAIHCRAILPCQDTPSVKLTYTAEVSVPKELVALMSAIRDGETPDPEDPSRKIYKFIQKVPIPCYLIALVVGALESRQIGPRTLVWSEKEQVEKSAYEFSETESMLKIAEDLGGPYVWGQYDLLVLPPSFPYGGMENPCLTFVTPTLLAGDKSLSNVIAHEISHSWTGNLVTNKTWDHFWLNEGHTVYLERHICGRLFGEKFRHFNALGGWGELQNSVKTFGETHPFTKLVVDLTDIDPDVAYSSVPYEKGFALLFYLEQLLGGPEIFLGFLKAYVEKFSYKSITTDDWKDFLYSYFKDKVDVLNQVDWNAWLYSPGLPPIKPNYDMTLTNACIALSQRWITAKEDDLNSFNATDLKDLSSHQLNEFLAQTLQRAPLPLGHIKRMQEVYNFNAINNSEIRFRWLRLCIQSKWEDAIPLALKMATEQGRMKFTRPLFKDLAAFDKSHDQAVRTYQEHKASMHPVTAMLVGKDLKVD Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | DNA topoisomerase 4 subunit A | 1ZVT | 4.00 | |
Target general information Gen name parC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3019;JW2987 Protein family Type II topoisomerase GyrA/ParC subunit family, ParC type 1 subfamily Biochemical class Isomerase Function ATP binding.DNA binding.DNA topoisomerase type II (ATP-hydrolyzing) activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11943; DB12924; DB00817 Interacts with P22523; P0A7K2 EC number 5.6.2.2 Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; DNA-binding; Isomerase; Membrane; Reference proteome; Topoisomerase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 26490.3 Length 246 Aromaticity 0.04 Instability index 46.03 Isoelectric point 8.94 Charge (pH=7) 2.83 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 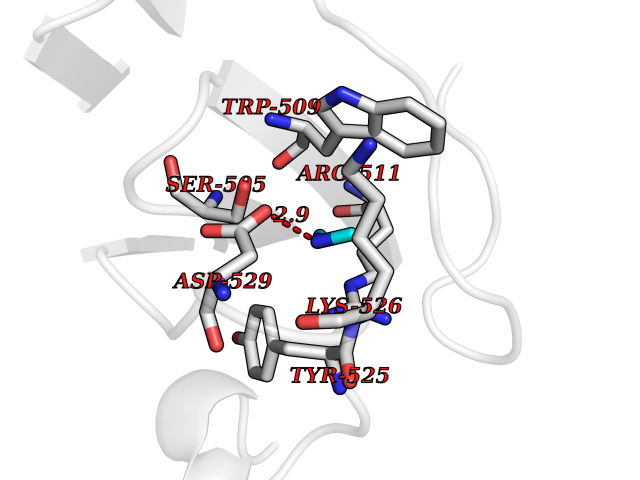 Pymol Map 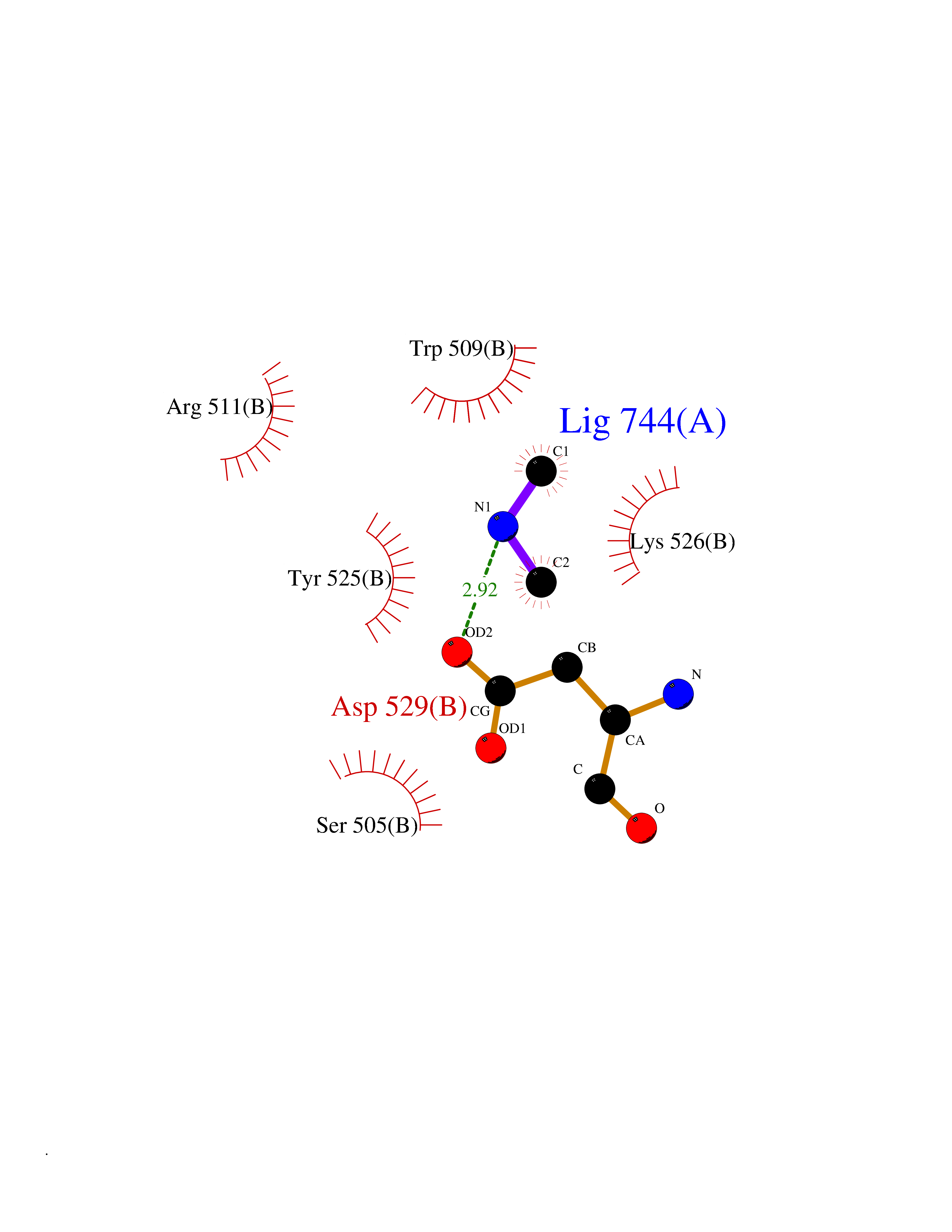 Ligplot Map 3D Binding mode Sequence SEPVTIVLSQMGWVRSAKGHDIDAPGLNYKAGDSFKAAVKGKSNQPVVFVDSTGRSYAIDPITLPSARGQGEPLTGKLTLPPGATVDHMLMESDDQKLLMASDAGYGFVCTFNDLVARNRAGKALITLPENAHVMPPVVIEDASDMLLAITQAGRMLMFPVSDLPQLSKGKGNKIINIPSAEAARGEDGLAQLYVLPPQSTLTIHVGKRKIKLRPEELQKVTGERGRRGTLMRGLQRIDRVEIDSP Hydrogen bonds contact Hydrophobic contact | ||||