Job Results:
Ligand
Structure
Job ID
c3579b2488a0044c00296f575f551b42
Job name
NA
Time
2025-02-13 15:23:33
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Phosphoinositide dependent protein kinase-1 (PDPK1) | 5LVO | 4.01 | |
Target general information Gen name PDPK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PDK1; HPDK1; 3-phosphoinositide-dependent protein kinase 1; 3-Phosphoinositide-dependent kinase-1; 3'-phosphoinositide dependent kinase 1 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, PDPK1 subfamily Biochemical class Kinase Function Its targets include: protein kinase B (PKB/AKT1, PKB/AKT2, PKB/AKT3), p70 ribosomal protein S6 kinase (RPS6KB1), p90 ribosomal protein S6 kinase (RPS6KA1, RPS6KA2 and RPS6KA3), cyclic AMP-dependent protein kinase (PRKACA), protein kinase C (PRKCD and PRKCZ), serum and glucocorticoid-inducible kinase (SGK1, SGK2 and SGK3), p21-activated kinase-1 (PAK1), protein kinase PKN (PKN1 and PKN2). Plays a central role in the transduction of signals from insulin by providing the activating phosphorylation to PKB/AKT1, thus propagating the signal to downstream targets controlling cell proliferation and survival, as well as glucose and amino acid uptake and storage. Negatively regulates the TGF-beta-induced signaling by: modulating the association of SMAD3 and SMAD7 with TGF-beta receptor, phosphorylating SMAD2, SMAD3, SMAD4 and SMAD7, preventing the nuclear translocation of SMAD3 and SMAD4 and the translocation of SMAD7 from the nucleus to the cytoplasm in response to TGF-beta. Activates PPARG transcriptional activity and promotes adipocyte differentiation. Activates the NF-kappa-B pathway via phosphorylation of IKKB. The tyrosine phosphorylated form is crucial for the regulation of focal adhesions by angiotensin II. Controls proliferation, survival, and growth of developing pancreatic cells. Participates in the regulation of Ca(2+) entry and Ca(2+)-activated K(+) channels of mast cells. Essential for the motility of vascular endothelial cells (ECs) and is involved in the regulation of their chemotaxis. Plays a critical role in cardiac homeostasis by serving as a dual effector for cell survival and beta-adrenergic response. Plays an important role during thymocyte development by regulating the expression of key nutrient receptors on the surface of pre-T cells and mediating Notch-induced cell growth and proliferative responses. Provides negative feedback inhibition to toll-like receptor-mediated NF-kappa-B activation in macrophages. Isoform 3 is catalytically inactive. Serine/threonine kinase which acts as a master kinase, phosphorylating and activating a subgroup of the AGC family of protein kinases. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB07132; DB06932; DB07300; DB07456; DB07457; DB07033; DB01933; DB03777; DB01946; DB00482; DB04522; DB12010; DB01863; DB02010 Interacts with P31749; Q00005; Q9Y4P3; O75385; P54252; P42858; Q8WXH2; Q8IUH5 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; ATP-binding; Cell junction; Cell membrane; Cytoplasm; Direct protein sequencing; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32750.5 Length 286 Aromaticity 0.12 Instability index 40.9 Isoelectric point 8.55 Charge (pH=7) 3.58 2D Binding mode Binding energy (Kcal/mol) -5.48 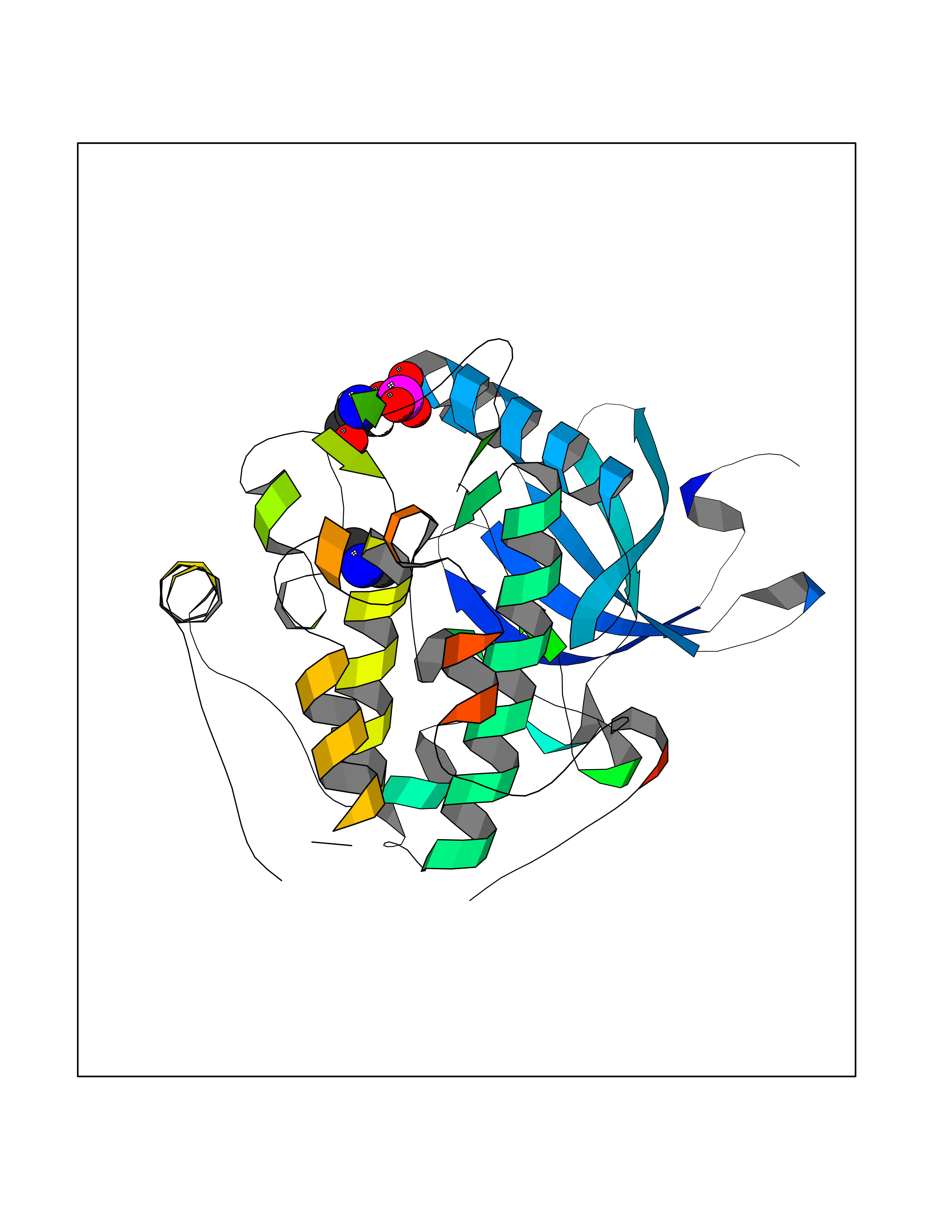 Molscript Map 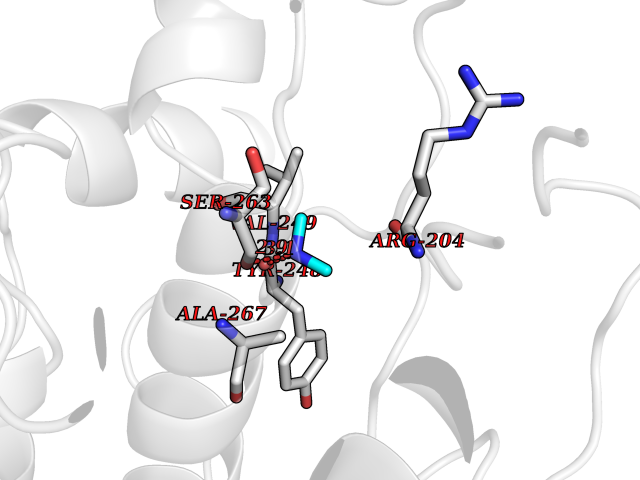 Pymol Map 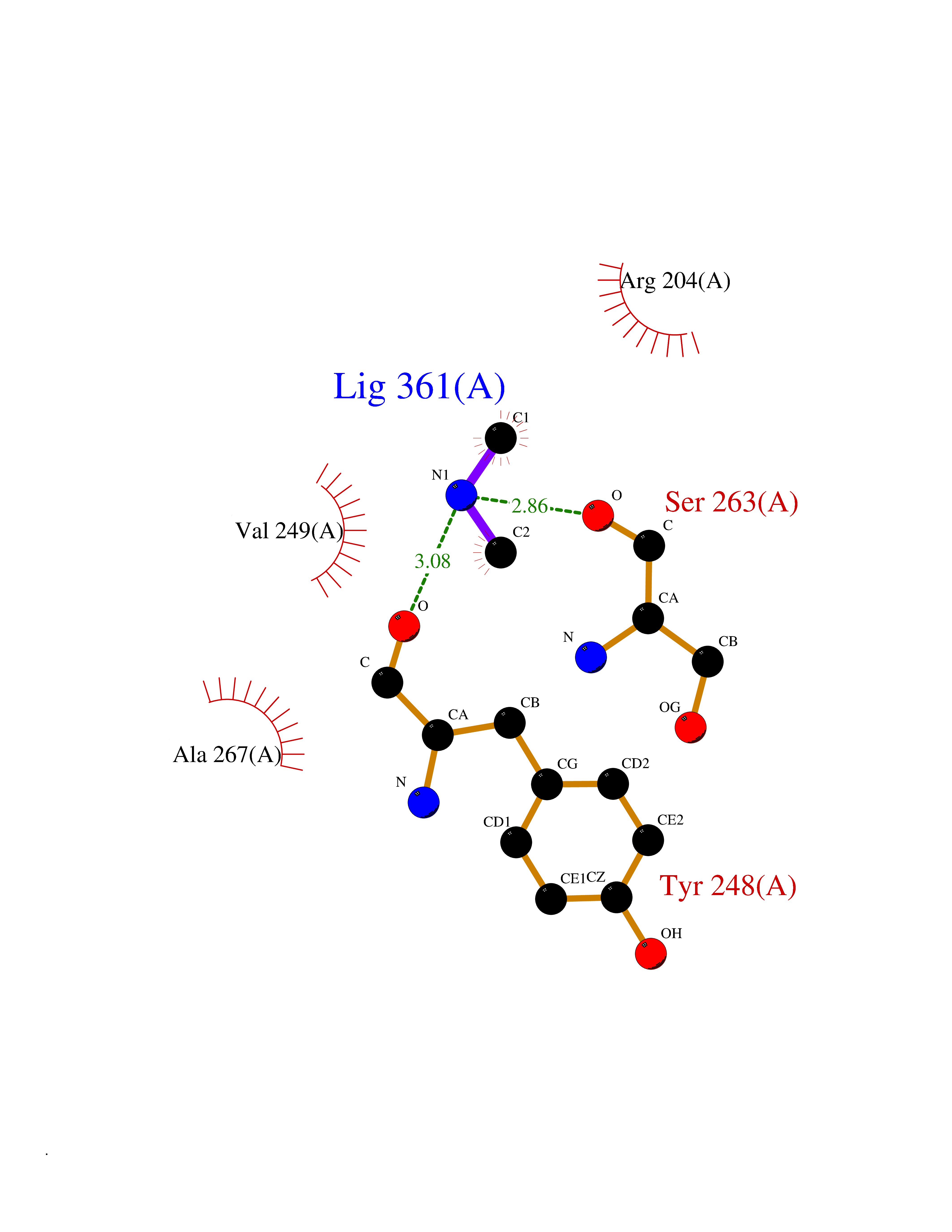 Ligplot Map 3D Binding mode Sequence PRKKRPEDFKFGKILGEGSFSTVVLARELATSREYAIKILEKRHIIKENKVPYVTRERDVMSRLDHPFFVKLYFTFQDDEKLYFGLSYAKNGELLKYIRKIGSFDETCTRFYTAEIVSALEYLHGKGIIHRDLKPENILLNEDMHIQITDFGTAKVLSPESKQARANXFVGTAQYVSPELLTEKSACKSSDLWALGCIIYQLVAGLPPFRAGNEGLIFAKIIKLEYDFPEKFFPKARDLVEKLLVLDATKRLGCEEMEGYGPLKAHPFFESVTWENLHQQTPPKLT Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Beta-glucosidase A | 1E4I | 4.01 | |
Target general information Gen name bglA Organism Paenibacillus polymyxa (Bacillus polymyxa) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 1 family Biochemical class Hydrolase Function Beta-glucosidase activity.Scopolin beta-glucosidase activity. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02658; DB04282; DB04304 Interacts with NA EC number 3.2.1.21 Uniprot keywords 3D-structure; Carbohydrate metabolism; Cellulose degradation; Glycosidase; Hydrolase; Polysaccharide degradation Protein physicochemical properties Chain ID A Molecular weight (Da) 51515.2 Length 447 Aromaticity 0.14 Instability index 38.44 Isoelectric point 5.28 Charge (pH=7) -18.1 2D Binding mode Binding energy (Kcal/mol) -5.47 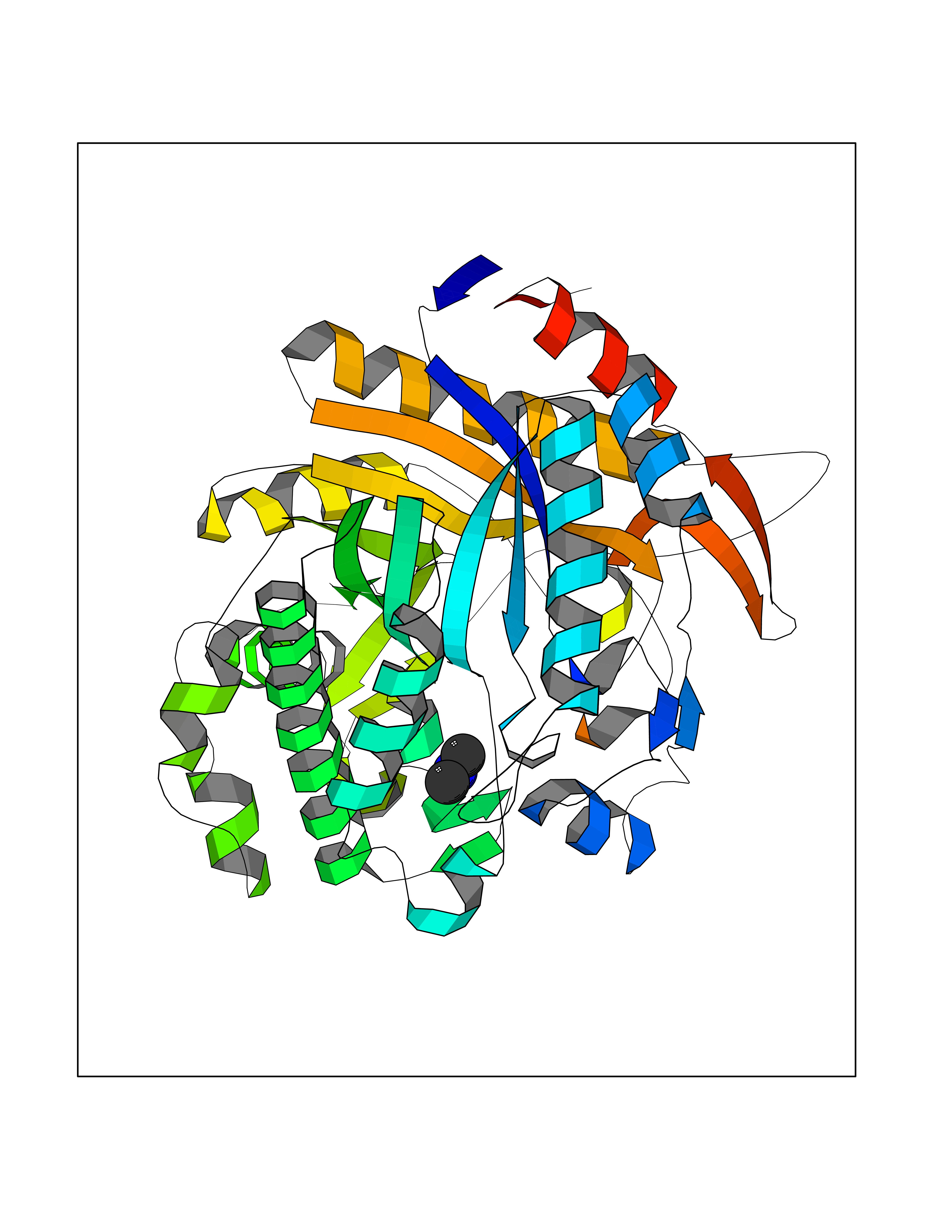 Molscript Map 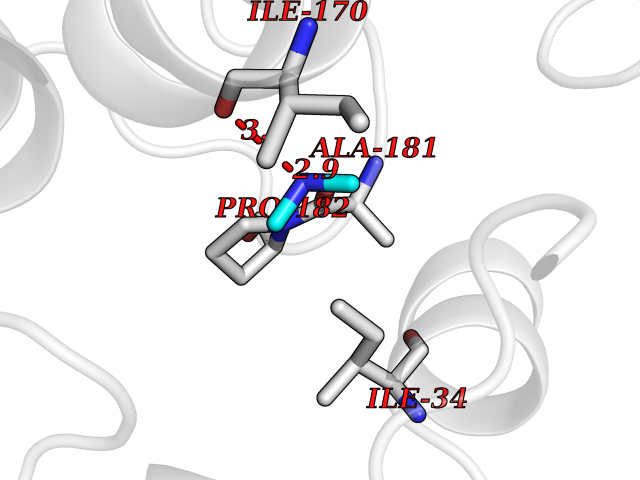 Pymol Map 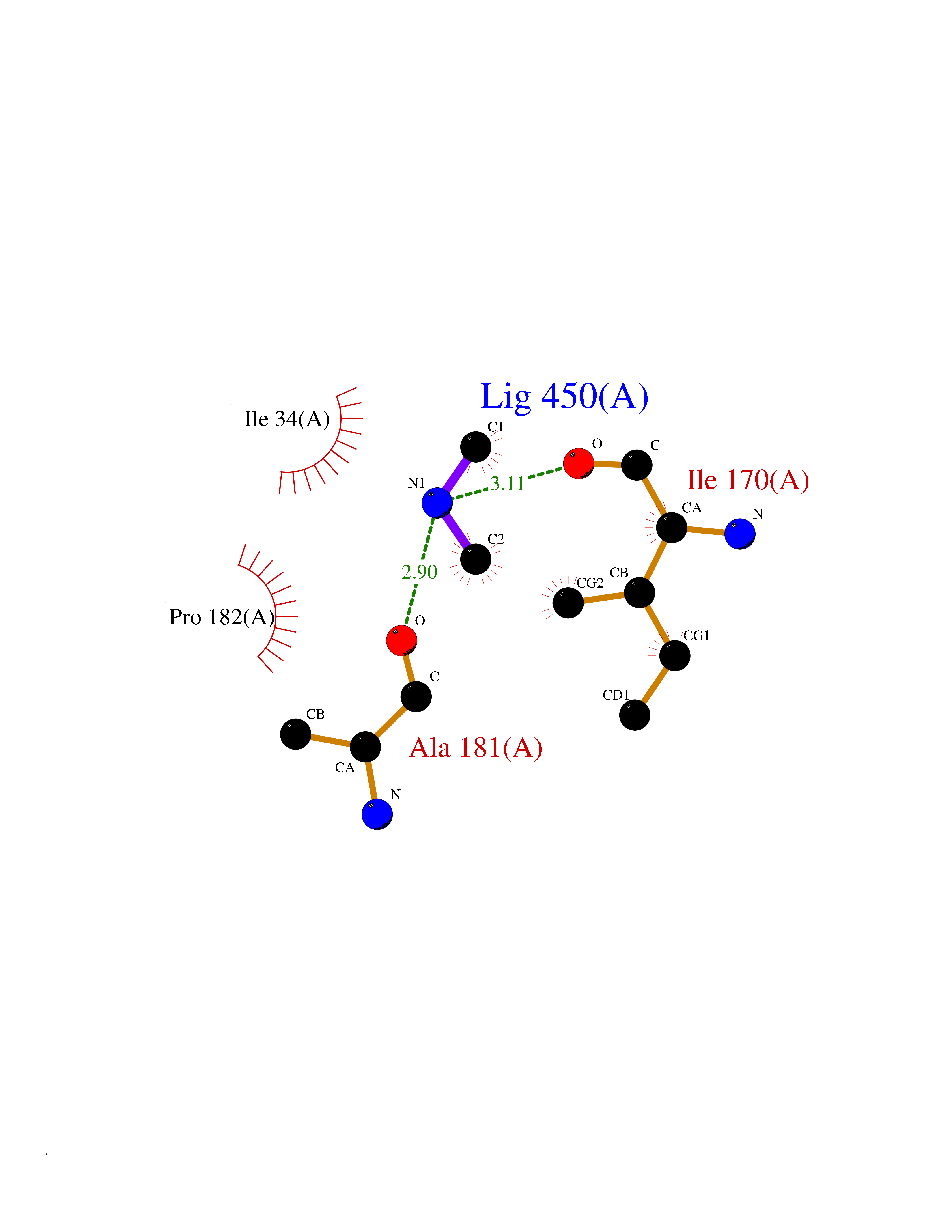 Ligplot Map 3D Binding mode Sequence TIFQFPQDFMWGTATAAYQIEGAYQEDGRGLSIWDTFAHTPGKVFNGDNGNVACDSYHRYEEDIRLMKELGIRTYRFSVSWPRIFPNGDGEVNQKGLDYYHRVVDLLNDNGIEPFCTLYHWDLPQALQDAGGWGNRRTIQAFVQFAETMFREFHGKIQHWLTFNEPWCIAFLSNMLGVHAPGLTNLQTAIDVGHHLLVAHGLSVRRFRELGTSGQIGIAPNVSWAVPYSTSEEDKAACARTISLHSDWFLQPIYQGSYPQFLVDWFAEQGATVPIQDGDMDIIGEPIDMIGINYYSMSVNRFNPEAGFLQSEEINMGLPVTDIGWPVESRGLYEVLHYLQKYGNIDIYITENGACINDEVVNGKVQDDRRISYMQQHLVQVHRTIHDGLHVKGYMAWSLLDNFEWAEGYNMRFGMIHVDFRTQVRTPKQSYYWYRNVVSNNWLETRR Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Phosphoribosylaminoimidazolecarboxamide formyltransferase (ATIC) | 1PKX | 4.01 | |
Target general information Gen name ATIC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PURH; OK/SW-cl.86; Bifunctional purine biosynthesis protein PURH Protein family PurH family Biochemical class Methyltransferase Function Bifunctional enzyme that catalyzes 2 steps in purine biosynthesis. Related diseases AICA-ribosuria due to ATIC deficiency (AICAR) [MIM:608688]: A neurologically devastating inborn error of purine biosynthesis. Patients excrete massive amounts of AICA-riboside in the urine and accumulate AICA-ribotide and its derivatives in erythrocytes and fibroblasts. Clinical features include profound intellectual disability, epilepsy, dysmorphic features and congenital blindness. AICAR inheritance is autosomal recessive. {ECO:0000269|PubMed:15114530}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02309; DB03442; DB01700; DB01972; DB00563; DB04057; DB00642; DB00116 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Epilepsy; Hydrolase; Intellectual disability; Multifunctional enzyme; Proteomics identification; Purine biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 41584.9 Length 386 Aromaticity 0.06 Instability index 42 Isoelectric point 5.6 Charge (pH=7) -7.51 2D Binding mode Binding energy (Kcal/mol) -5.47 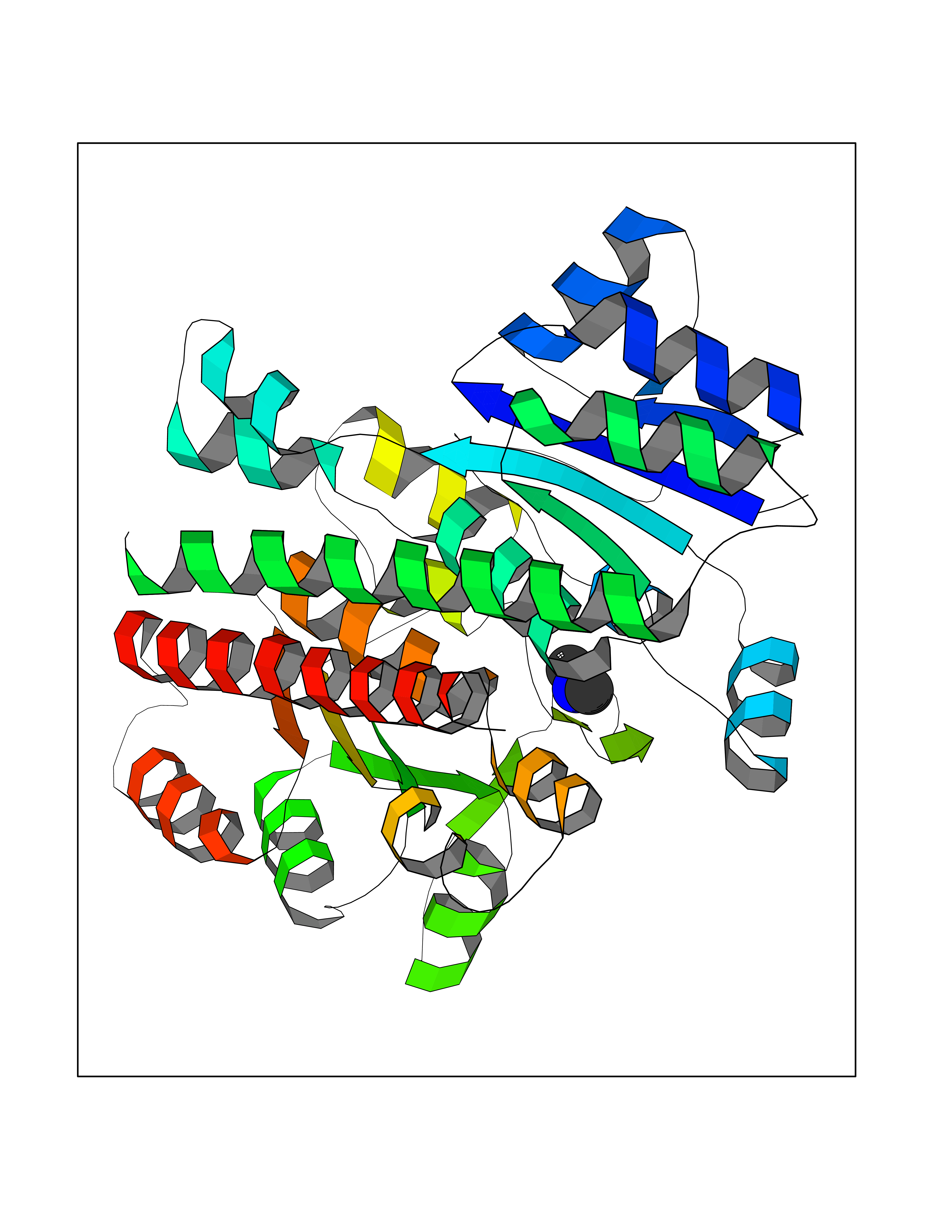 Molscript Map 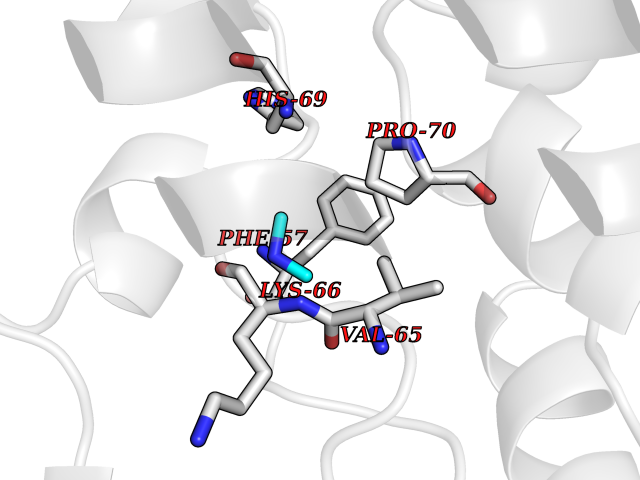 Pymol Map 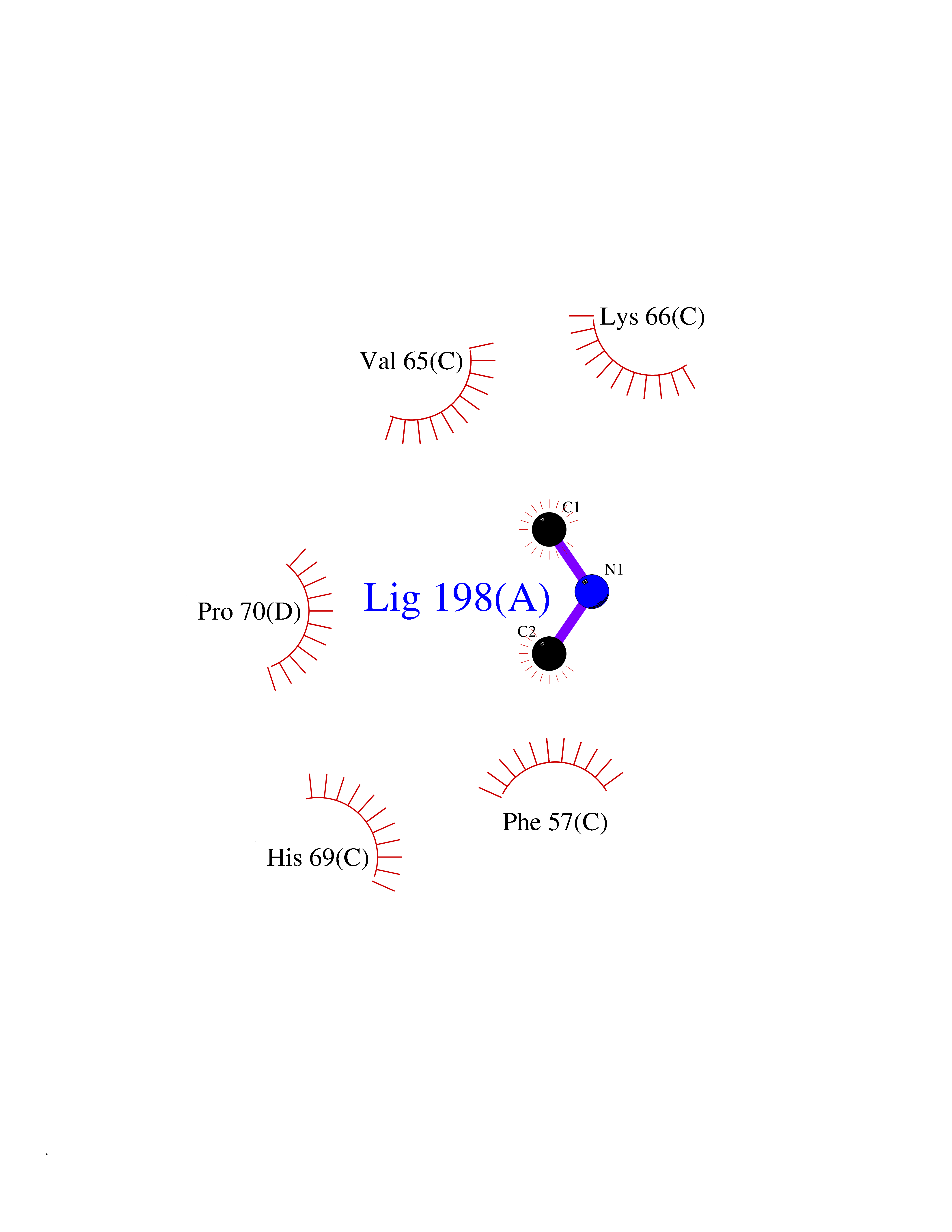 Ligplot Map 3D Binding mode Sequence GQLALFSVSDKTGLVEFARNLTALGLNLVASGGTAKALRDAGLAVRDVSELTGFPEMLGGRVKTLHPAVHAGILARNIPEDNADMARLDFNLIRVVACNLYPFVKTVASPGVTVEEAVEQIDIGGVTLLRAAAKNHARVTVVCEPEDYVVVSTEMQSSESKDTSLETRRQLALKAFTHTAQYDEAISDYFRKQGQLALFSVSDKTGLVEFARNLTALGLNLVASGGTAKALRDAGLAVRDVSELTGFPEMLGGRVKTLHPAVHAGILARNIPEDNADMARLDFNLIRVVACNLYPFVKTVASPGVTVEEAVEQIDIGGVTLLRAAAKNHARVTVVCEPEDYVVVSTEMQSSESKDTSLETRRQLALKAFTHTAQYDEAISDYFRKQ Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 4.01 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map  Pymol Map 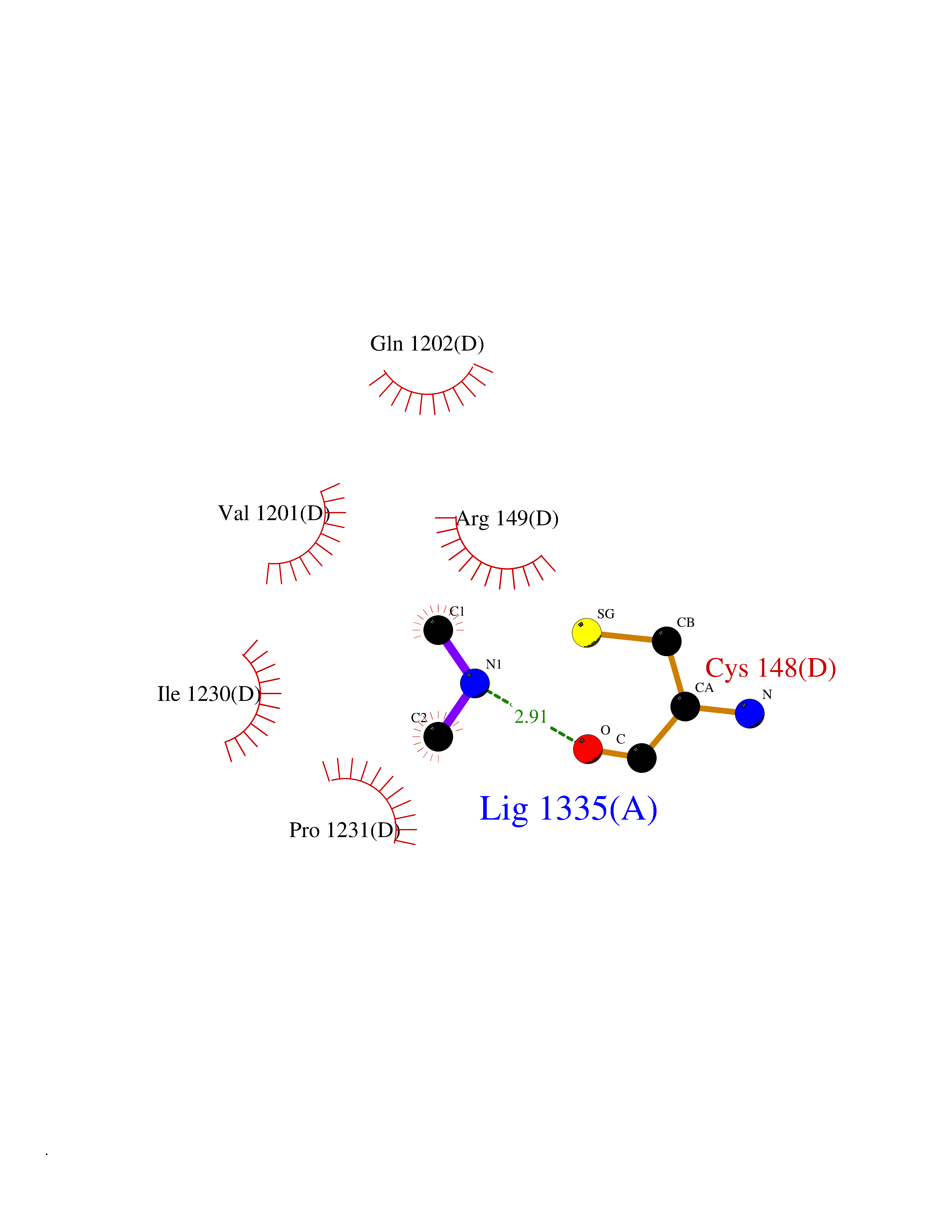 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Beta-lactamase TEM | 1M40 | 4.01 | |
Target general information Gen name bla Organism Escherichia coli Uniprot ID TTD ID NA Synonyms blaT-4;blaT-6;blaT-5;blaT-3 Protein family Class-A beta-lactamase family Biochemical class Hydrolase Function Beta-lactamase activity. Related diseases WHIM syndrome 1 (WHIMS1) [MIM:193670]: An autosomal dominant immunologic disease characterized by neutropenia, hypogammaglobulinemia and extensive human papillomavirus (HPV) infection. Despite the peripheral neutropenia, bone marrow aspirates from affected individuals contain abundant mature myeloid cells, a condition termed myelokathexis. {ECO:0000269|PubMed:12692554, ECO:0000269|PubMed:15536153}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: CXCR4 mutations play a role in the pathogenesis of Waldenstroem macroglobulinemia (WM) and influence disease presentation and outcome, as well as response to therapy. WM is a B-cell lymphoma characterized by accumulation of malignant lymphoplasmacytic cells in the bone marrow, lymph nodes and spleen, and hypersecretion of monoclonal immunoglobulin M (IgM). Excess IgM production results in serum hyperviscosity, tissue infiltration, and autoimmune-related pathology. {ECO:0000269|PubMed:24366360, ECO:0000269|PubMed:24553177}. Drugs (DrugBank ID) DB07466; DB07464; DB02614; DB04430; DB08551; DB07599; DB02841; DB02642; DB09060; DB01053; DB04035; DB01598; DB04037; DB12377; DB12107 Interacts with P35804 EC number 3.5.2.6 Uniprot keywords 3D-structure; Antibiotic resistance; Direct protein sequencing; Disulfide bond; Hydrolase; Plasmid; Signal; Transposable element Protein physicochemical properties Chain ID A Molecular weight (Da) 28876.6 Length 263 Aromaticity 0.05 Instability index 37.98 Isoelectric point 5.46 Charge (pH=7) -6.68 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 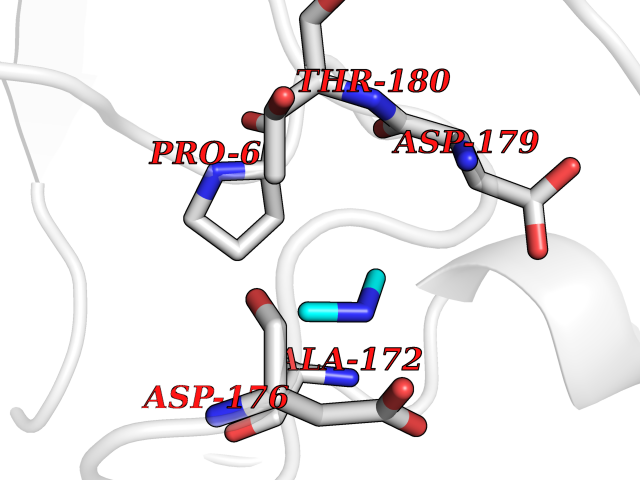 Pymol Map 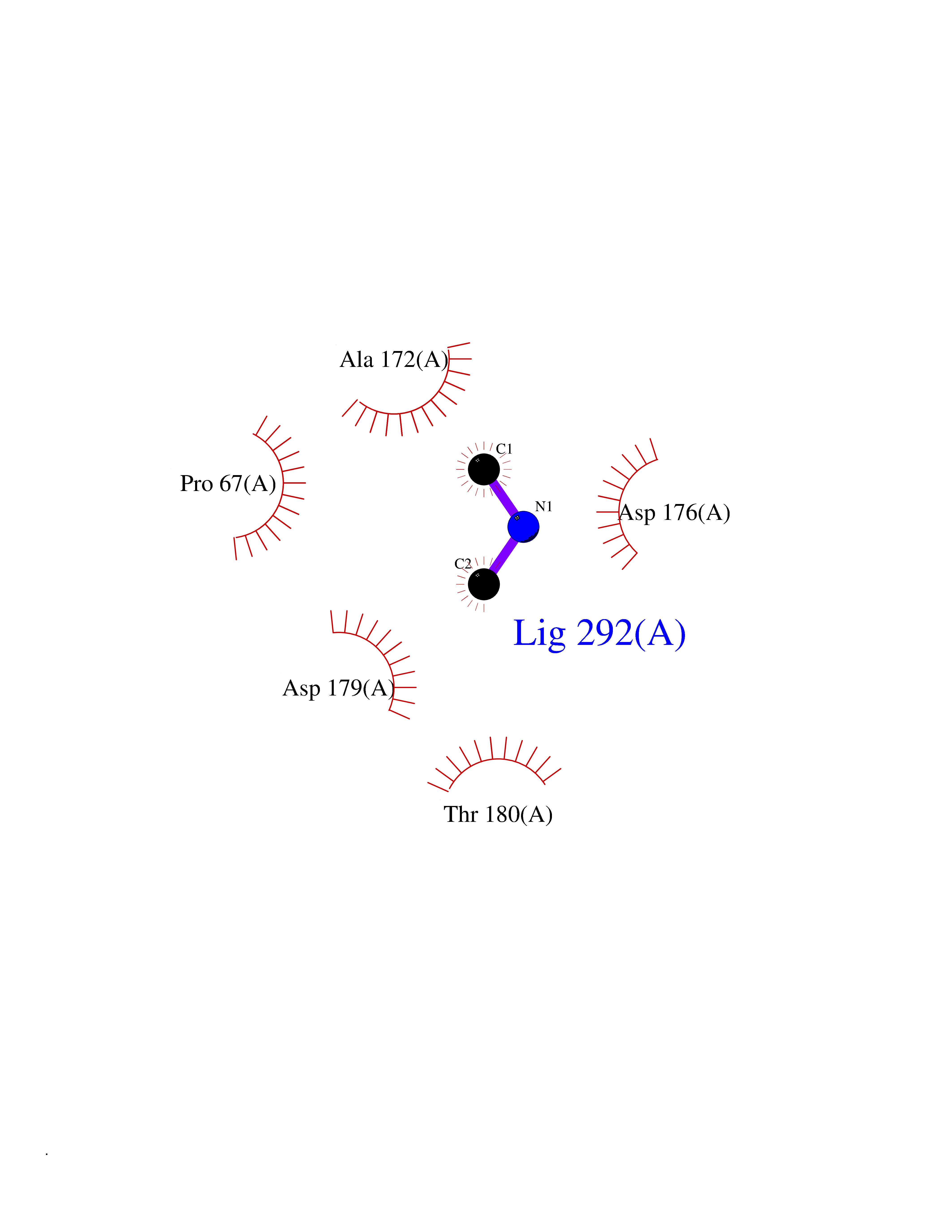 Ligplot Map 3D Binding mode Sequence HPETLVKVKDAEDQLGARVGYIELDLNSGKILESFRPEERFPMMSTFKVLLCGAVLSRVDAGQEQLGRRIHYSQNDLVEYSPVTEKHLTDGMTVRELCSAAITMSDNTAANLLLTTIGGPKELTAFLHNMGDHVTRLDRWEPELNEAIPNDERDTTTPAAMATTLRKLLTGELLTLASRQQLIDWMEADKVAGPLLRSALPAGWFIADKSGAGERGSRGIIAALGPDGKPSRIVVIYTTGSQATMDERNRQIAEIGASLIKHW Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Lanosterol 14-alpha-demethylase (EC 1.14.13.70) | 4G3J | 4.01 | |
Target general information Gen name Tb11.02.4080 Organism Trypanosoma brucei brucei (strain 927/4 GUTat10.1) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class NA Function NA Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) NA Interacts with NA EC number 1.14.13.70 Uniprot keywords 3D-structure; Heme; Iron; Metal-binding; Monooxygenase; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID D Molecular weight (Da) 50691.4 Length 448 Aromaticity 0.08 Instability index 54.09 Isoelectric point 6.99 Charge (pH=7) -0.03 2D Binding mode Binding energy (Kcal/mol) -5.46 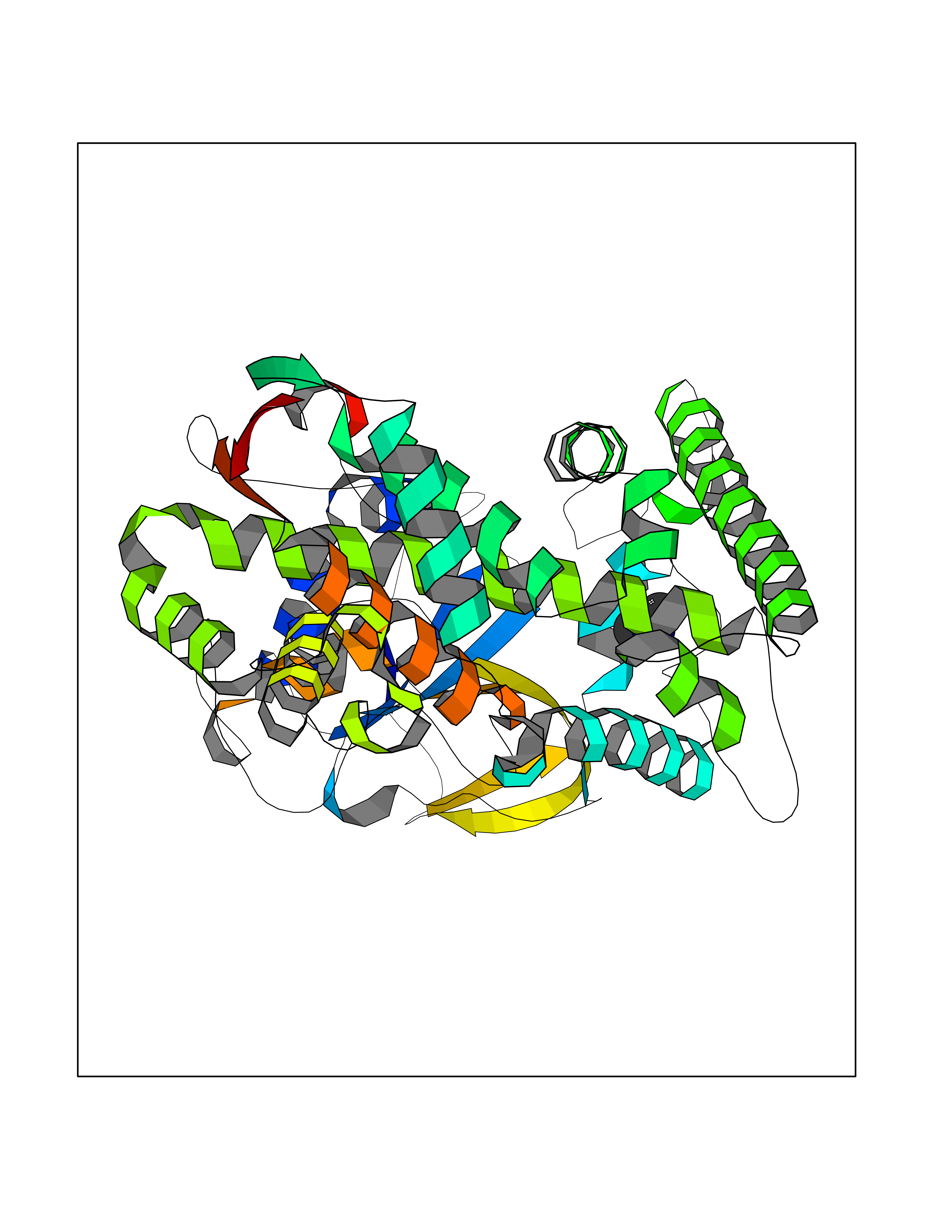 Molscript Map 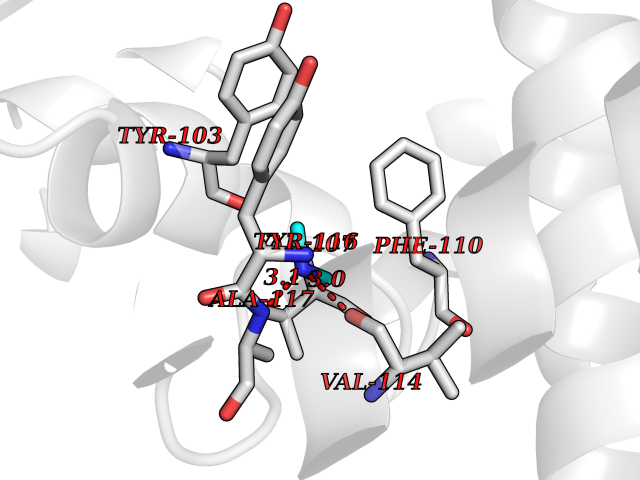 Pymol Map 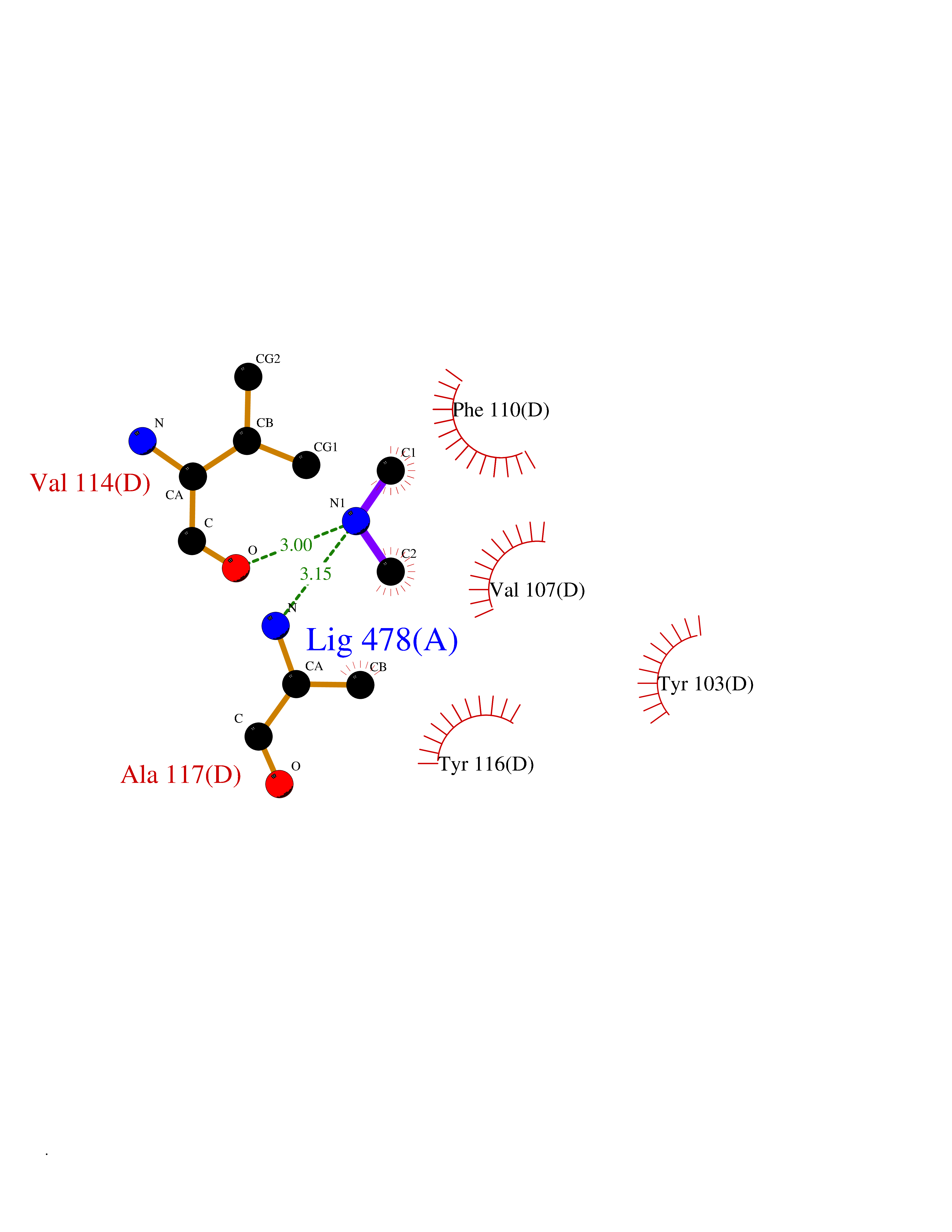 Ligplot Map 3D Binding mode Sequence GKLPPVYPVTVPILGHIIQFGKSPLGFMQECKRQLKSGIFTINIVGKRVTIVGDPHEHSRFFLPRNEVLSPREVYSFMVPVFGEGVAYAAPYPRMREQLNFLAEELTIAKFQNFVPAIQHEVRKFMAANWDKDEGEINLLEDCSTMIINTACQCLFGEDLRKRLDARRFAQLLAKMESSLIPAAVFLPILLKLPLPQSARCHEARTELQKILSEIIIARKEEEVNKDSSTSDLLSGLLSAVYRDGTPMSLHEVCGMIVAAMFAGQHTSSITTTWSMLHLMHPANVKHLEALRKEIEEFPAQLNYNNVMDEMPFAERCARESIRRDPPLLMLMRKVMADVKVGSYVVPKGDIIACSPLLSHHDEEAFPEPRRWDPERDEKVEGAFIGFGAGVHKCIGQKFGLLQVKTILATAFRSYDFQLLRDEVPDPDYHTMVVGPTASQCRVKYIRR Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Leukotriene C4 synthase (LTC4S) | 3PCV | 4.01 | |
Target general information Gen name LTC4S Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Leukotriene-C(4)Leukotriene C4 synthase synthase; Leukotriene-C(4) synthase; LTC4 synthase Protein family MAPEG family Biochemical class Carbon-sulfur lyase Function Catalyzes the conjugation of leukotriene A4 with reduced glutathione to form leukotriene C4. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB00972; DB00143 Interacts with P11912; Q96BA8; Q15125; O14843; Q14802-3; Q8TDT2; O15529; Q16873; Q8TBB6; Q4KMG9 EC number EC 4.4.1.20 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Leukotriene biosynthesis; Lipid biosynthesis; Lipid metabolism; Lyase; Membrane; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 16099 Length 146 Aromaticity 0.13 Instability index 38.66 Isoelectric point 10.2 Charge (pH=7) 7.66 2D Binding mode Binding energy (Kcal/mol) -5.47 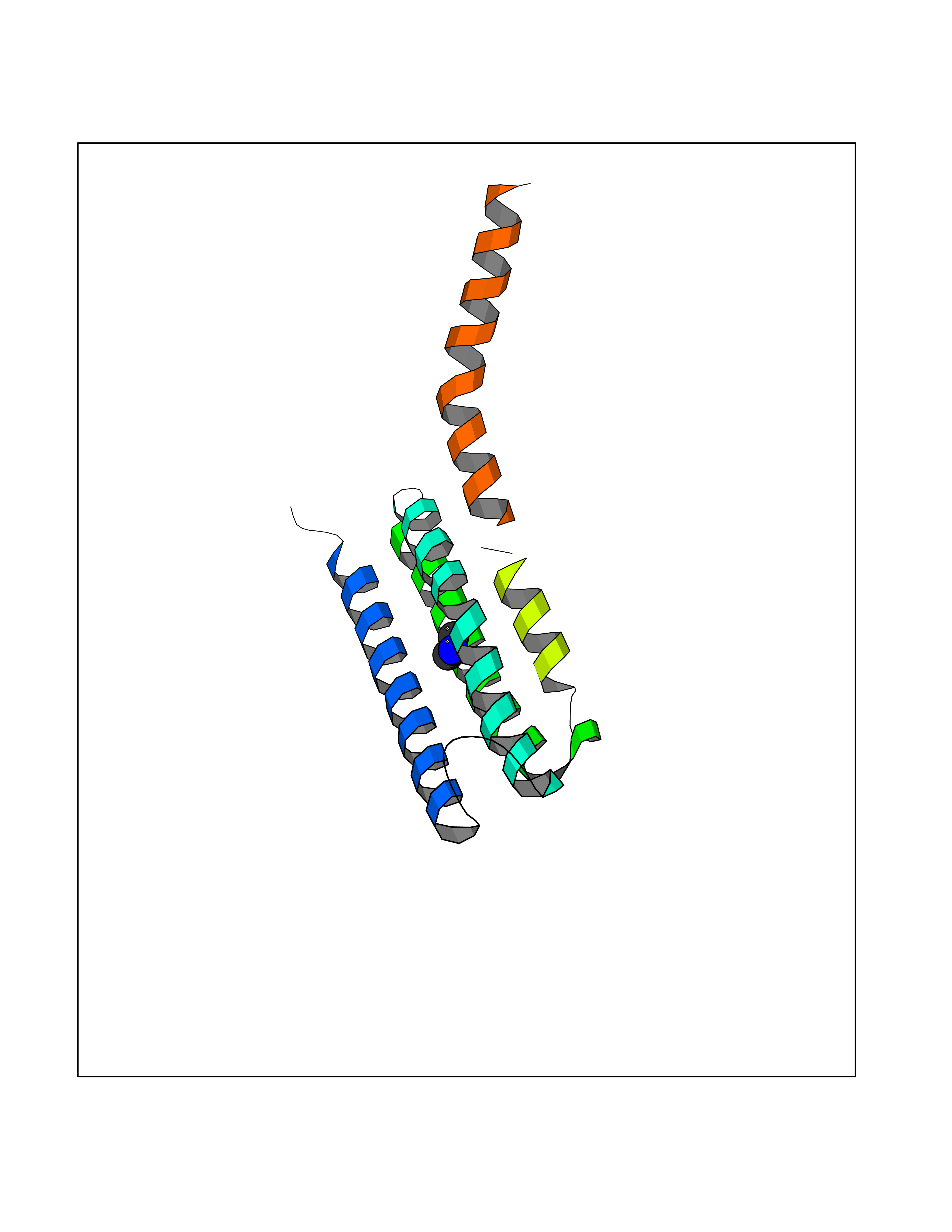 Molscript Map 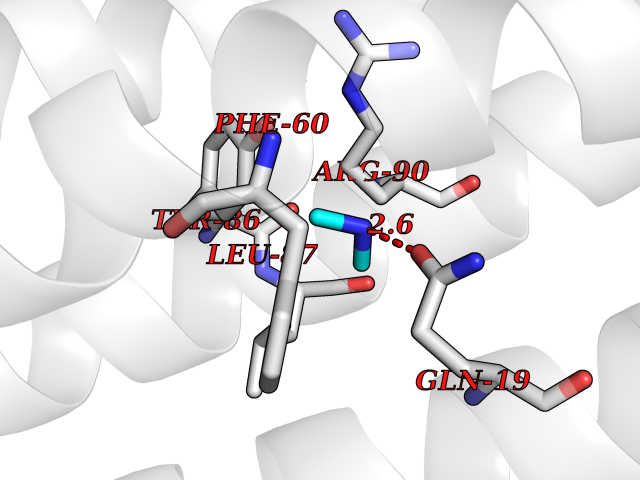 Pymol Map 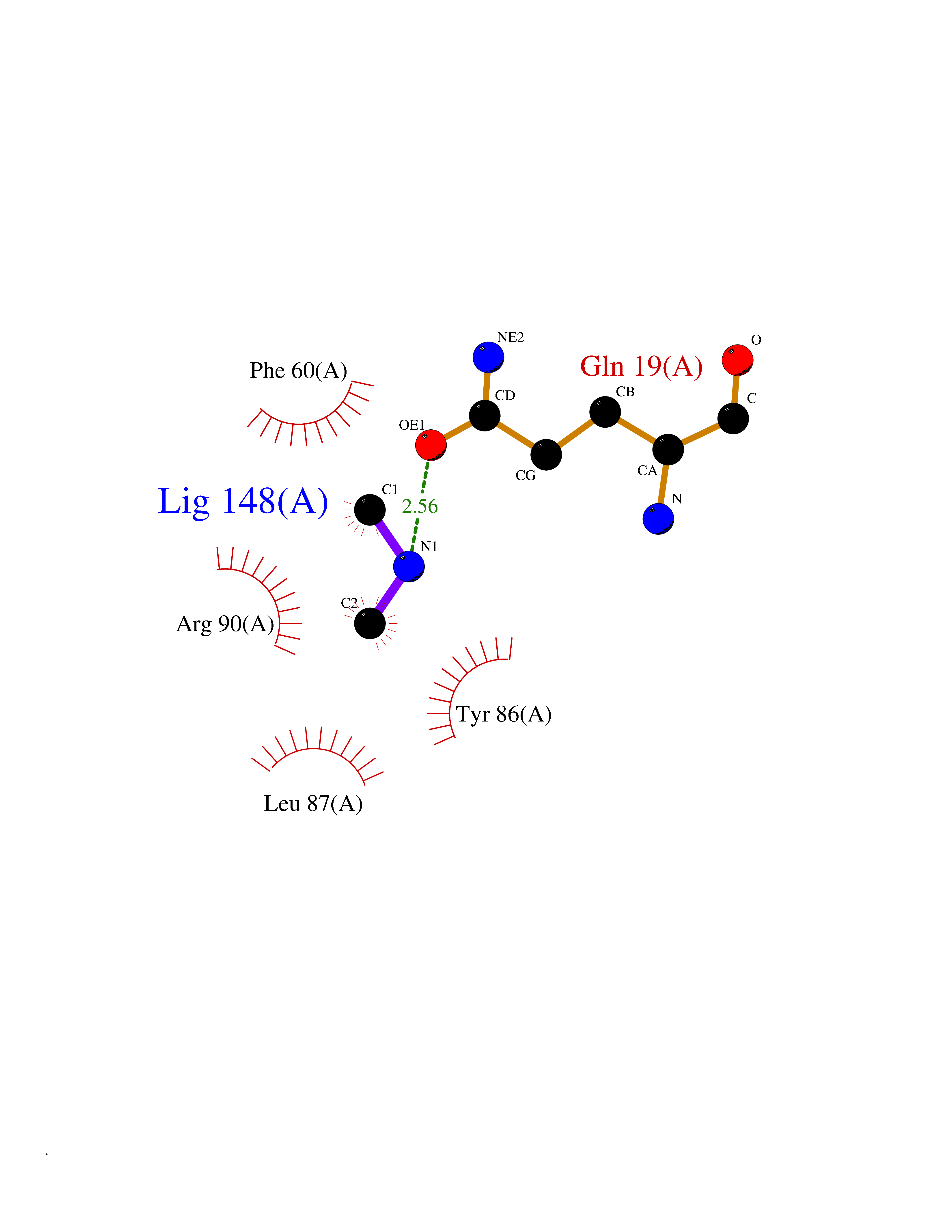 Ligplot Map 3D Binding mode Sequence MKDEVALLAAVTLLGVLLQAYFSLQVISARRAFRVSPPLTTGPPEFERVYRAQVNCSEYFPLFLATLWVAGIFFHEGAAALCGLVYLFARLRYFQGYARSAQLRLAPLYASARALWLLVALAALGLLAHFLPAALRAALLGRLRTL Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Neutral endopeptidase (MME) | 6XVP | 4.01 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Skin fibroblast elastase; SFE; Neutral endopeptidase 24.11; Neprilysin; NEP protein; Enkephalinase; EPN; Common acute lymphocytic leukemia antigen; CD10; CALLA; Atriopeptidase Protein family Peptidase M13 family Biochemical class Peptidase Function Biologically important in the destruction of opioid peptides such as Met- and Leu-enkephalins by cleavage of a Gly-Phe bond. Able to cleave angiotensin-1, angiotensin-2 and angiotensin 1-9. Involved in the degradation of atrial natriuretic factor (ANF). Displays UV-inducible elastase activity toward skin preelastic and elastic fibers. Thermolysin-like specificity, but is almost confined on acting on polypeptides of up to 30 amino acids. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number EC 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 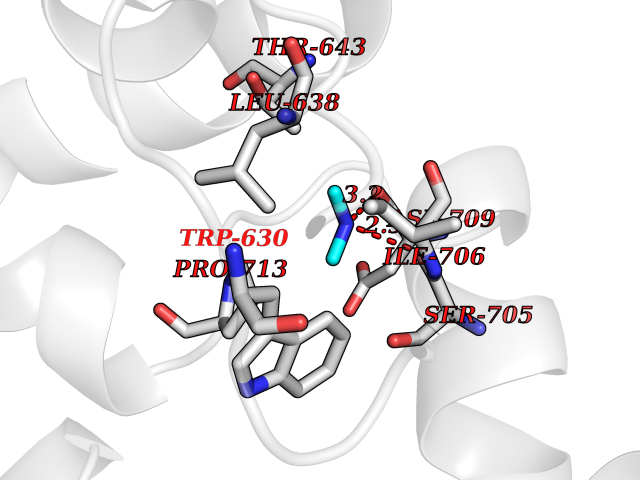 Pymol Map 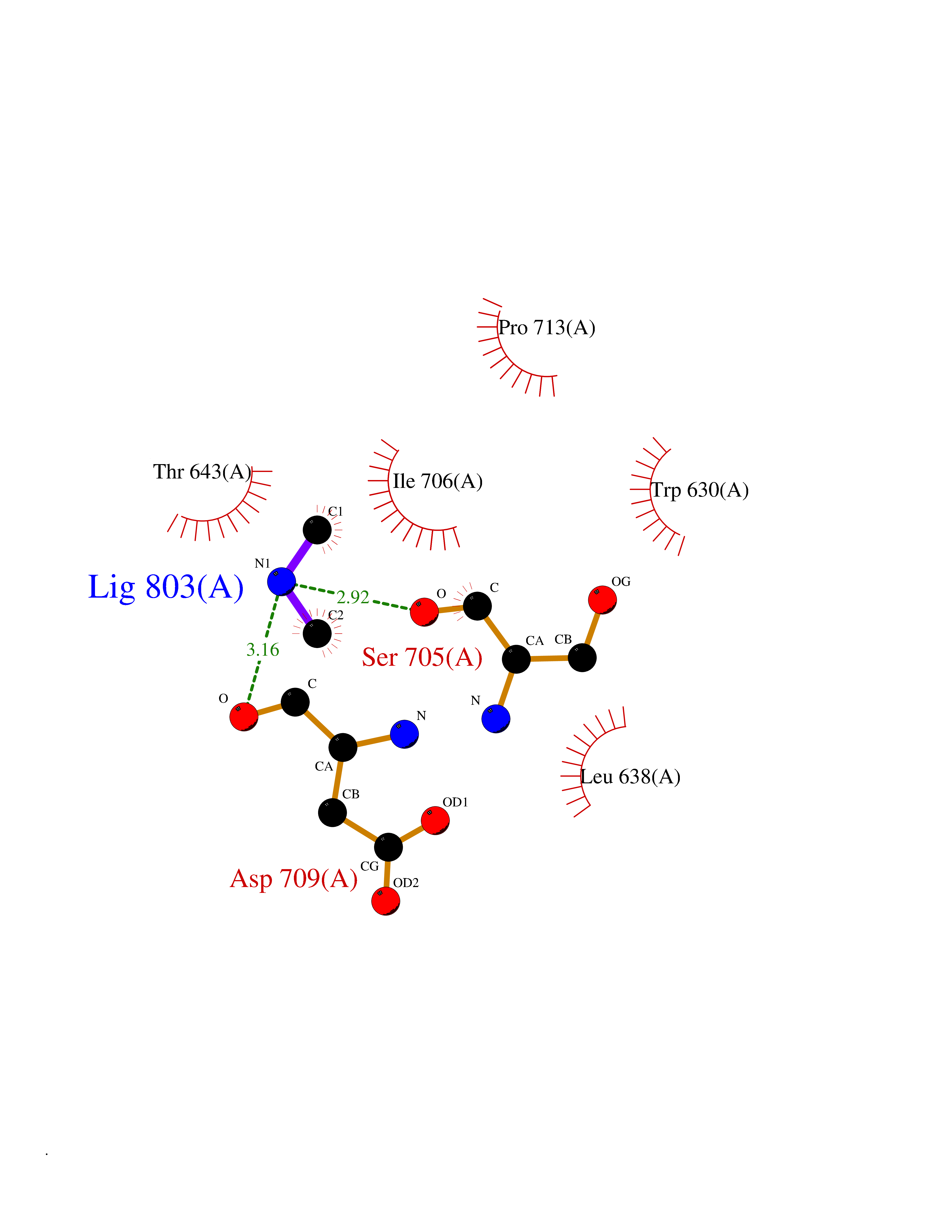 Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | N-acetylgalactosamine 6 sulfatase (GALNS) | 4FDJ | 4.01 | |
Target general information Gen name GALNS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms N-acetylgalactosamine-6-sulfate sulfatase; N-acetylgalactosamine-6-sulfatase; Galactose-6-sulfate sulfatase; GalNAc6S sulfatase; GalN6S; Chondroitinsulfatase; Chondroitinase Protein family Sulfatase family Biochemical class Sulfuric ester hydrolase Function Catalyzes the chemical reaction of cleaving off the 6-sulfate groups of the N-acetyl-D-galactosamine 6-sulfate units of the macromolecule chondroitin sulfate and, similarly, of the D-galactose 6-sulfate units of the macromolecule keratan sulfate. Related diseases Mucopolysaccharidosis 4A (MPS4A) [MIM:253000]: A form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life. {ECO:0000269|PubMed:1522213, ECO:0000269|PubMed:16287098, ECO:0000269|PubMed:24726177, ECO:0000269|PubMed:7581409, ECO:0000269|PubMed:7633425, ECO:0000269|PubMed:7668283, ECO:0000269|PubMed:7795586, ECO:0000269|PubMed:8651279, ECO:0000269|PubMed:8826435, ECO:0000269|PubMed:9298823, ECO:0000269|PubMed:9375852, ECO:0000269|PubMed:9521421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09301 Interacts with NA EC number EC 3.1.6.4 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Dwarfism; Glycoprotein; Hydrolase; Lysosome; Metal-binding; Mucopolysaccharidosis; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 55013.8 Length 493 Aromaticity 0.11 Instability index 35.46 Isoelectric point 6.14 Charge (pH=7) -6.48 2D Binding mode Binding energy (Kcal/mol) -5.48 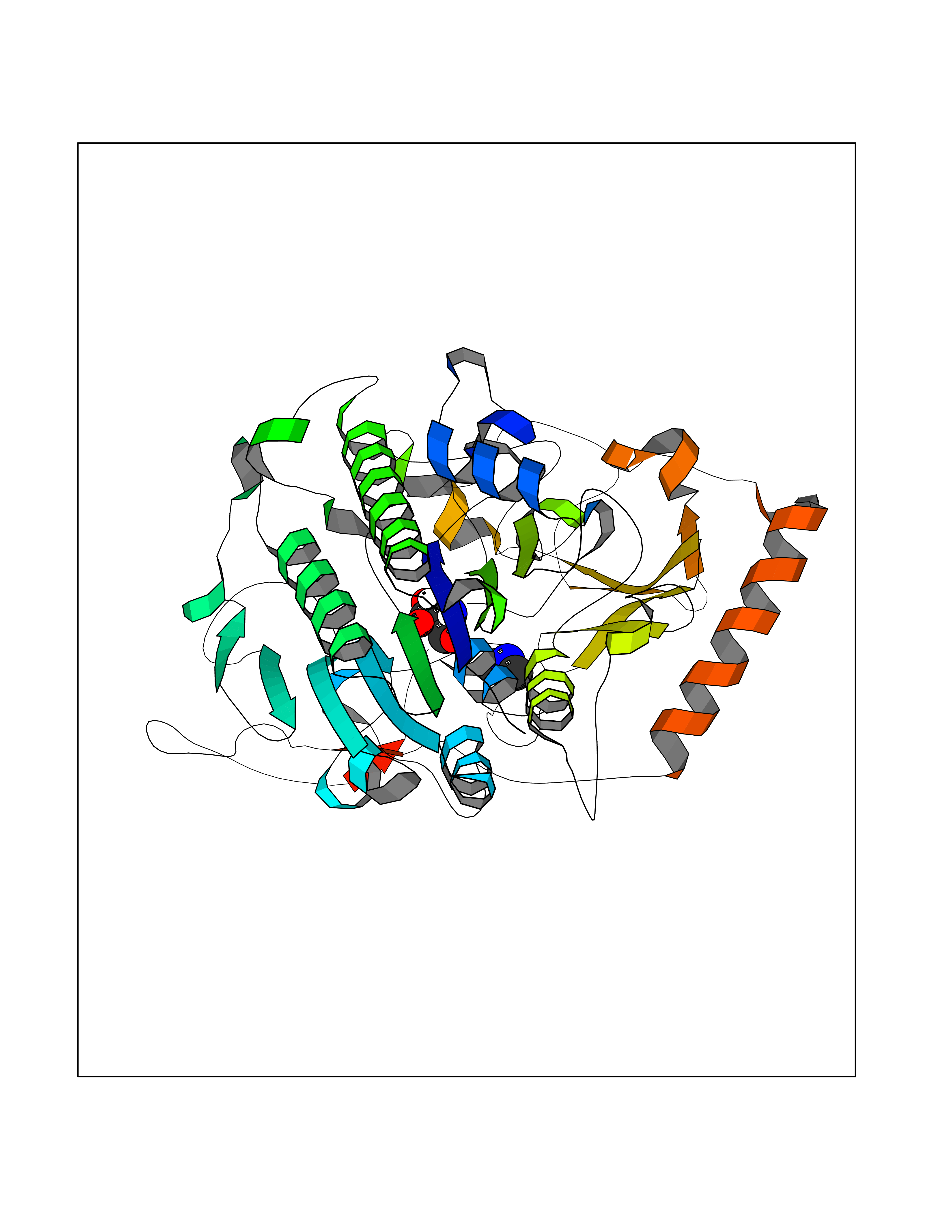 Molscript Map  Pymol Map 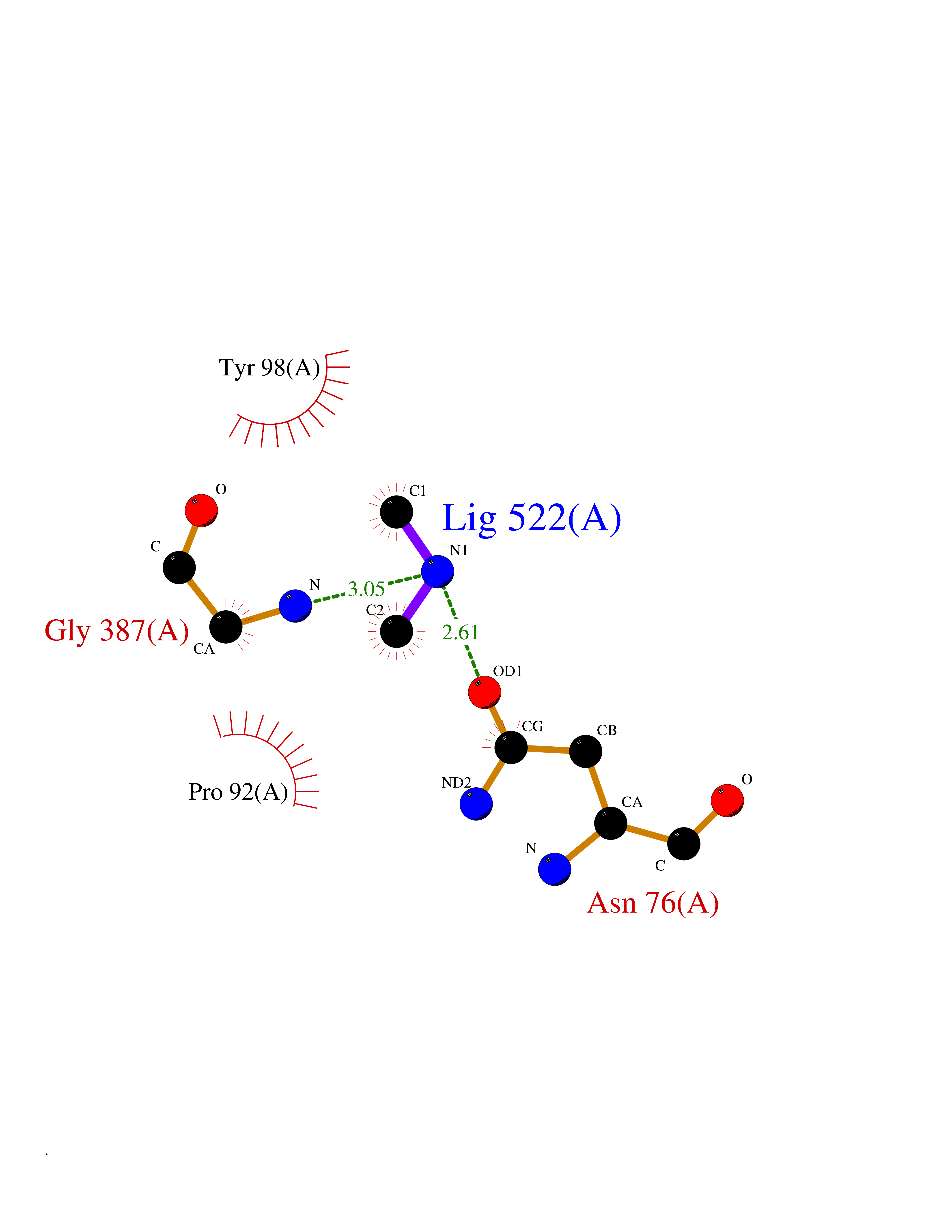 Ligplot Map 3D Binding mode Sequence PQPPNILLLLMDDMGWGDLGVYGEPSRETPNLDRMAAEGLLFPNFYSANPLXSPSRAALLTGRLPIRNGFYTTNAHARNAYTPQEIVGGIPDSEQLLPELLKKAGYVSKIVGKWHLGHRPQFHPLKHGFDEWFGSPNCHFGPYDNKARPNIPVYRDWEMVGRYYEEFPINLKTGEANLTQIYLQEALDFIKRQARHHPFFLYWAVDATHAPVYASKPFLGTSQRGRYGDAVREIDDSIGKILELLQDLHVADNTFVFFTSDNGAALISAPEQGGSNGPFLCGKQTTFEGGMREPALAWWPGHVTAGQVSHQLGSIMDLFTTSLALAGLTPPSDRAIDGLNLLPTLLQGRLMDRPIFYYRGDTLMAATLGQHKAHFWTWTNSWENFRQGIDFCPGQNVSGVTTHNLEDHTKLPLIFHLGRDPGERFPLSFASAEYQEALSRITSVVQQHQEALVPAQPQLNVCNWAVMNWAPPGCEKLGKCLTPPESIPKKCLW Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Haemophilus influenzae NadR protein (Hae-influ nadR) | 1LW7 | 4.01 | |
Target general information Gen name Hae-influ nadR Organism Haemophilus influenzae (strain ATCC 51907 / DSM 11121 / KW20 / Rd) Uniprot ID TTD ID Synonyms nadR; Transcriptional regulator nadR Protein family Bacterial NMN adenylyltransferase family; Bacterial RNK family Biochemical class Nicotinamide ribonucleoside uptake permease Function This enzyme has twoactivities: nicotinamide mononucleotide (NMN) adenylyltransferase and ribosylnicotinamide (RN) kinase. The RN kinase activity catalyzes the phosphorylation of RN to form nicotinamide ribonucleotide. The NMN adenylyltransferase activity catalyzes the transfer of the AMP moiety of ATP to nicotinamide ribonucleotide to form NAD(+). Related diseases Involved in the epigenetic regulation of ESR1 expression in breast cancer in a TFAP2C, IFI16 and HDAC4/5/6-dependent manner. {ECO:0000269|PubMed:24413532}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative initiation; ATP-binding; Cell membrane; Cytoplasm; Kinase; Membrane; Multifunctional enzyme; NAD; Nucleotide-binding; Pyridine nucleotide biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 39581.5 Length 344 Aromaticity 0.14 Instability index 41.39 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -5.47 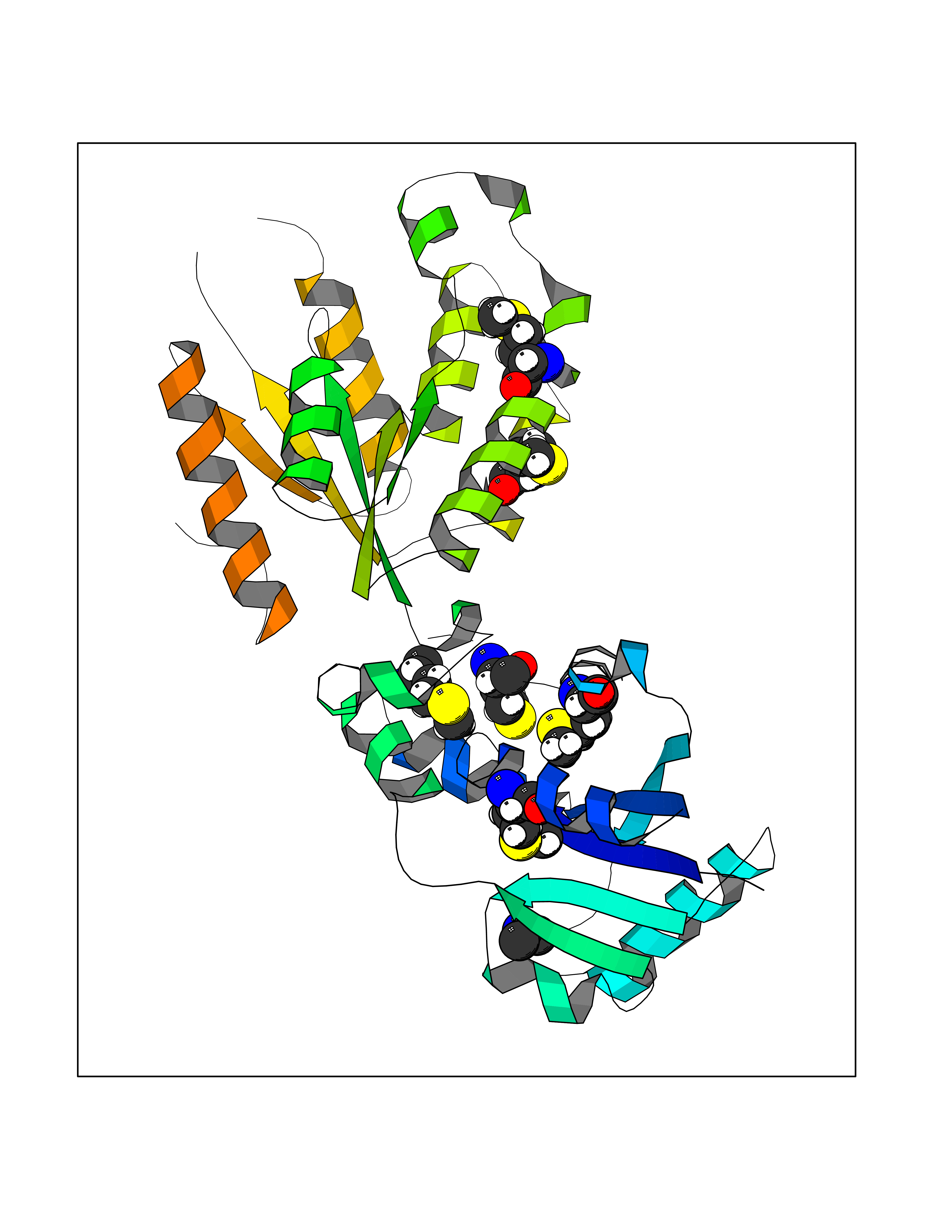 Molscript Map 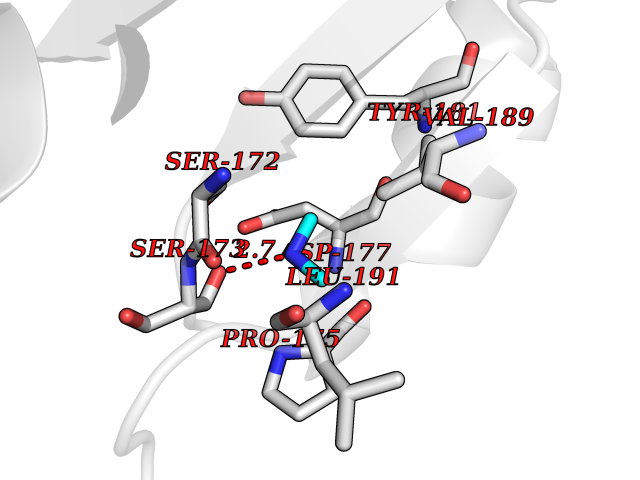 Pymol Map 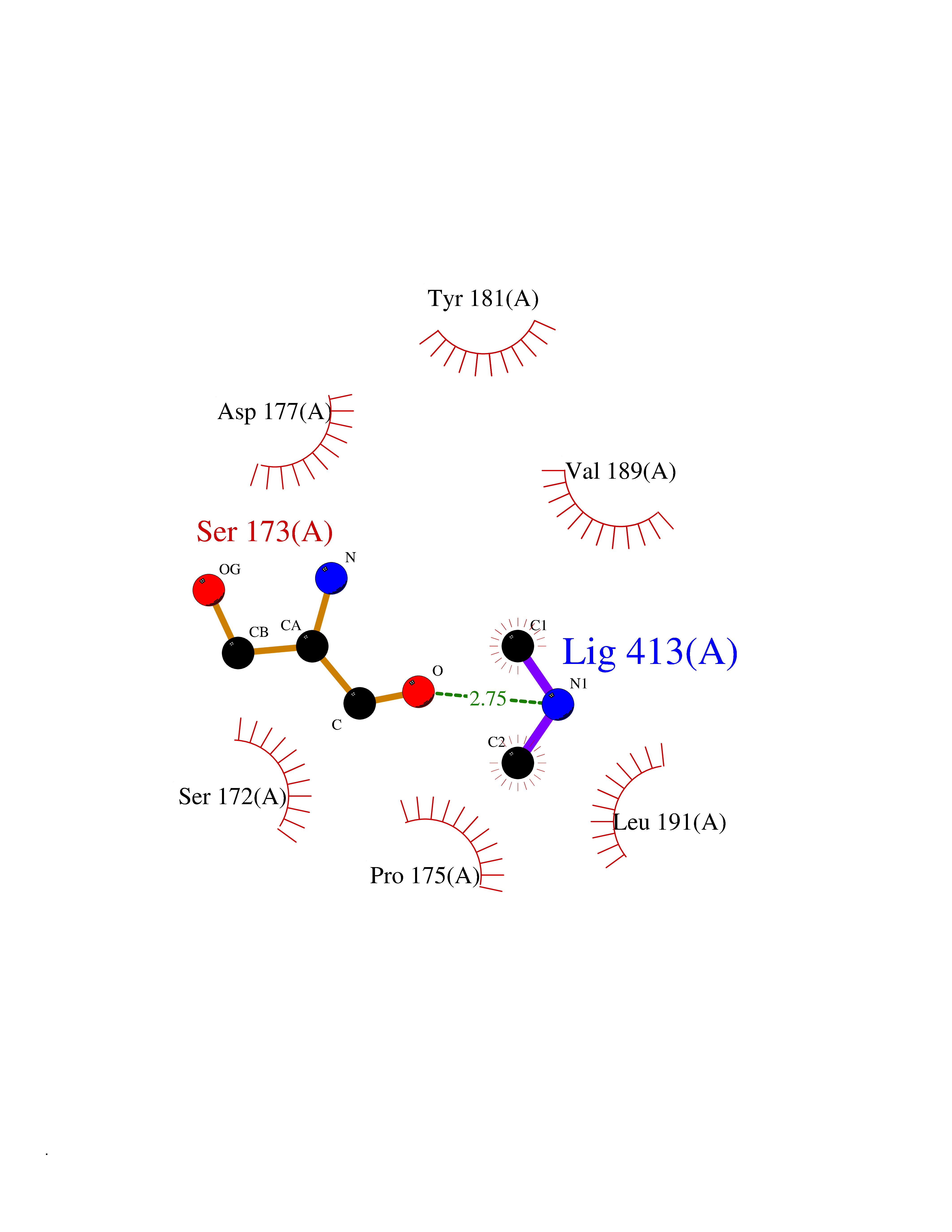 Ligplot Map 3D Binding mode Sequence EKKVGVIFGKFYPVHTGHINXIYEAFSKVDELHVIVCSDTVRDLKLFYDSKXKRXPTVQDRLRWXQQIFKYQKNQIFIHHLVEDGIPSYPNGWQSWSEAVKTLFHEKHFEPSIVFSSEPQDKAPYEKYLGLEVSLVDPDRTFFNVSATKIRTTPFQYWKFIPKEARPFFAKTVAILGGESSGKSVLVNKLAAVFNTTSAWEYGREFVFEKLGGDEQAMQYSDYPQXALGHQRYIDYAVRHSHKIAFIDTDFITTQAFCIQYEGKAHPFLDSXIKEYPFDVTILLKNNTEQKQRQQFQQLLKKLLDKYKVPYIEIESPSYLDRYNQVKAVIEKVLNEEEISELQN Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | 6YND | 4.01 | |
Target general information Gen name GAPDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peptidyl-cysteine S-nitrosylase GAPDH; OK/SW-cl.12; GAPD; D-glyceraldehyde-3-phosphate dehydrogenase; CDABP0047 Protein family Glyceraldehyde-3-phosphate dehydrogenase family Biochemical class Aldehyde/oxo donor oxidoreductase Function Participates in nuclear events including transcription, RNA transport, DNA replication and apoptosis. Nuclear functions are probably due to the nitrosylase activity that mediates cysteine S-nitrosylation of nuclear target proteins such as SIRT1, HDAC2 and PRKDC. Modulates the organization and assembly of the cytoskeleton. Facilitates the CHP1-dependent microtubule and membrane associations through its ability to stimulate the binding of CHP1 to microtubules. Glyceraldehyde-3-phosphate dehydrogenase is a key enzyme in glycolysis that catalyzes the first step of the pathway by converting D-glyceraldehyde 3-phosphate (G3P) into 3-phospho-D-glyceroyl phosphate. Component of the GAIT (gamma interferon-activated inhibitor of translation) complex which mediates interferon-gamma-induced transcript-selective translation inhibition in inflammation processes. Upon interferon-gamma treatment assembles into the GAIT complex which binds to stem loop-containing GAIT elements in the 3'-UTR of diverse inflammatory mRNAs (such as ceruplasmin) and suppresses their translation. Has both glyceraldehyde-3-phosphate dehydrogenase and nitrosylase activities, thereby playing a role in glycolysis and nuclear functions, respectively. Related diseases Bone marrow failure and diabetes mellitus syndrome (BMFDMS) [MIM:620044]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFDMS is an autosomal recessive form characterized by various degrees of bone marrow failure, ranging from dyserythropoiesis to bone marrow aplasia, with onset in infancy or early childhood, and non-autoimmune insulin-dependent diabetes mellitus appearing in the first or second decades. Many patients show pigmentary skin abnormalities and short stature. {ECO:0000269|PubMed:28073829, ECO:0000269|PubMed:35611808, ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07347; DB02059; DB11638; DB09130; DB00157; DB03893; DB09092 Interacts with Q6UY14-3; Q9UIJ7; P05067; Q9UQM7; Q14194; P35222; Q9BPW9-4; P00533; O00471; O75344; P06241; P04406; O14556; Q8NEA9; P42858; Q92993-2; P42695; P35228; P12004; P00558; P48147; P17612; Q8WUY3; Q9UHX1-2; P15927; P05109; Q96GZ6; P00441; Q9BSI4; P10599 EC number EC 1.2.1.12 Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Alternative splicing; Apoptosis; Cytoplasm; Cytoskeleton; Direct protein sequencing; Glycolysis; Glycoprotein; Immunity; Innate immunity; Isopeptide bond; Membrane; Methylation; NAD; Nucleus; Oxidation; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; S-nitrosylation; Transferase; Translation regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35818.4 Length 333 Aromaticity 0.08 Instability index 13.69 Isoelectric point 8.64 Charge (pH=7) 3.64 2D Binding mode Binding energy (Kcal/mol) -5.46 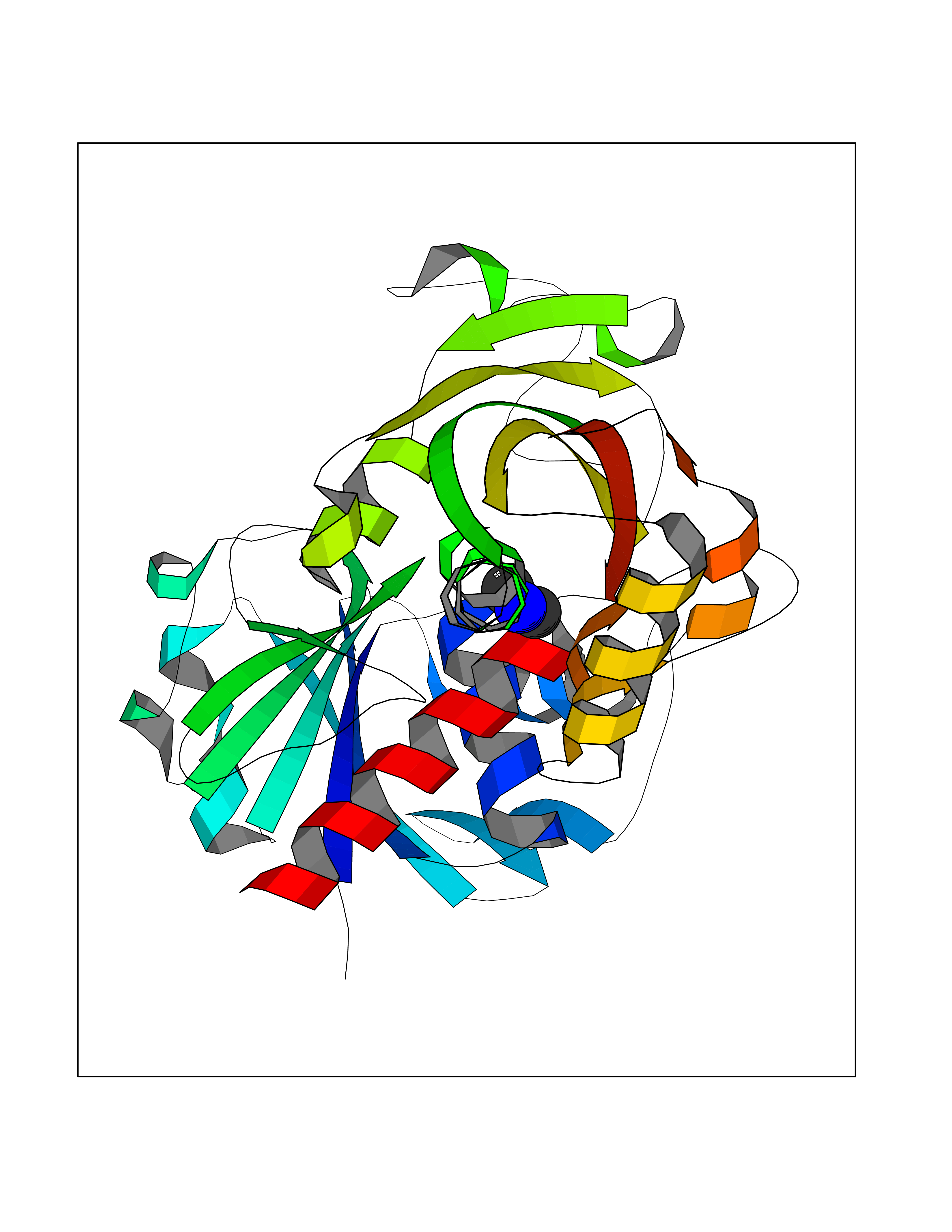 Molscript Map 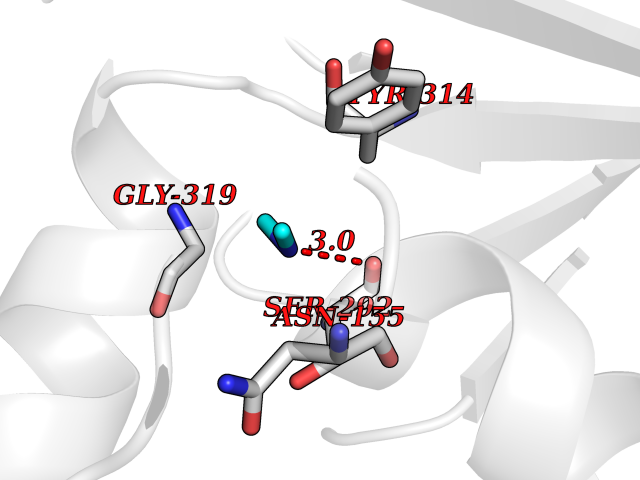 Pymol Map 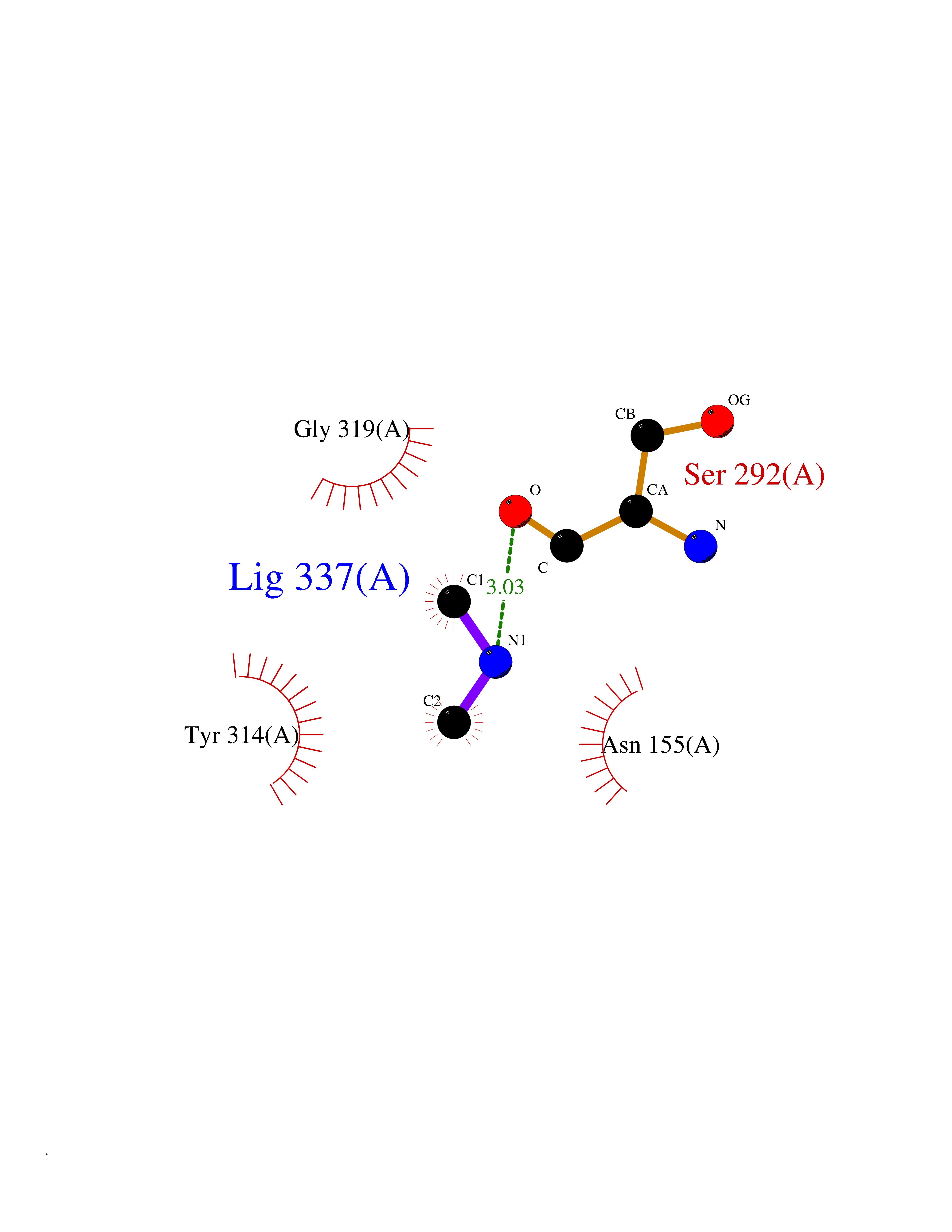 Ligplot Map 3D Binding mode Sequence GKVKVGVNGFGRIGRLVTRAAFNSGKVDIVAINDPFIDLNYMVYMFQYDSTHGKFHGTVKAENGKLVINGNPITIFQERDPSKIKWGDAGAEYVVESTGVFTTMEKAGAHLQGGAKRVIISAPSADAPMFVMGVNHEKYDNSLKIISNASTTNCLAPLAKVIHDNFGIVEGLMTTVHAITATQKTVDGPSGKLWRDGRGALQNIIPASTGAAKAVGKVIPELNGKLTGMAFRVPTANVSVVDLTCRLEKPAKYDDIKKVVKQASEGPLKGILGYTEHQVVSSDFNSDTHSSTFDAGAGIALNDHFVKLISWYDNEFGYSNRVVDLMAHMASKE Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Aspartyl aminopeptidase (DNPEP) | 4DYO | 4.01 | |
Target general information Gen name DNPEP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms DAP; ASPEP Protein family Peptidase M18 family Biochemical class Peptidase Function Likely to play an important role in intracellular protein and peptide metabolism. Aminopeptidase with specificity towards an acidic amino acid at the N-terminus. Related diseases Cohen-Gibson syndrome (COGIS) [MIM:617561]: An autosomal dominant overgrowth disorder characterized by accelerated osseous maturation, advanced bone age, skeletal abnormalities including flaring of the metaphyses of the long bones, large hands with long fingers and camptodactyly, scoliosis, cervical spine anomalies, dysmorphic facial features, and variable intellectual disability. {ECO:0000269|PubMed:25787343, ECO:0000269|PubMed:27193220, ECO:0000269|PubMed:27868325, ECO:0000269|PubMed:28229514, ECO:0000269|PubMed:28475857}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00142 Interacts with Q9ULA0; Q8TBB1; Q00013; Q9UPN6 EC number EC 3.4.11.21 Uniprot keywords 3D-structure; Acetylation; Alternative initiation; Aminopeptidase; Cytoplasm; Hydrolase; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 50574.6 Length 457 Aromaticity 0.07 Instability index 55.73 Isoelectric point 7.73 Charge (pH=7) 2.3 2D Binding mode Binding energy (Kcal/mol) -5.47 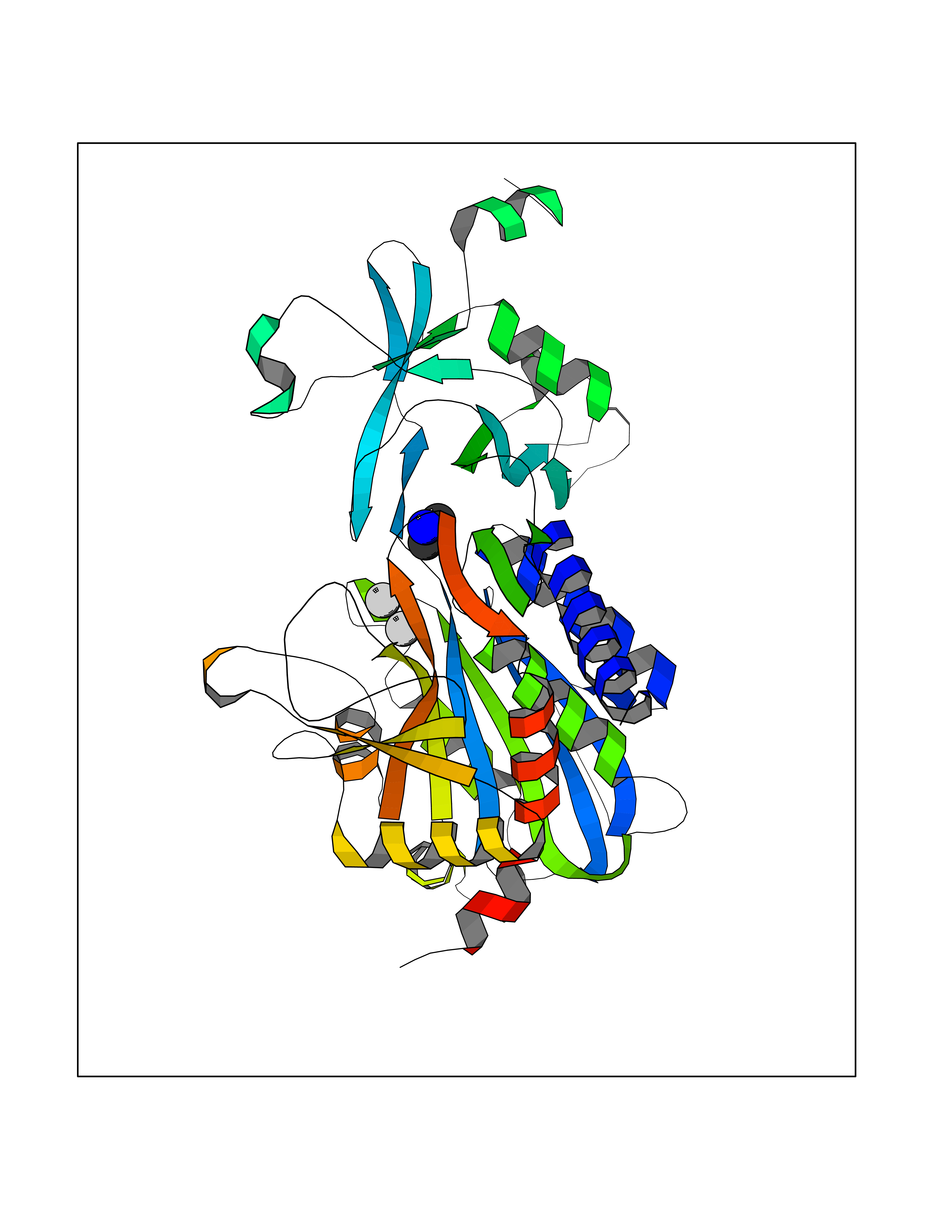 Molscript Map 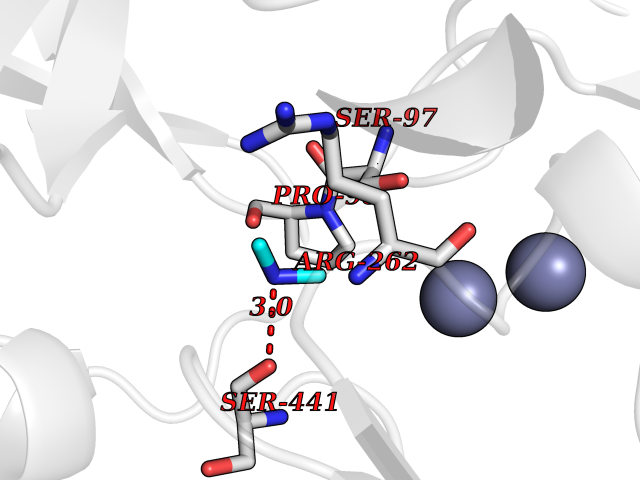 Pymol Map 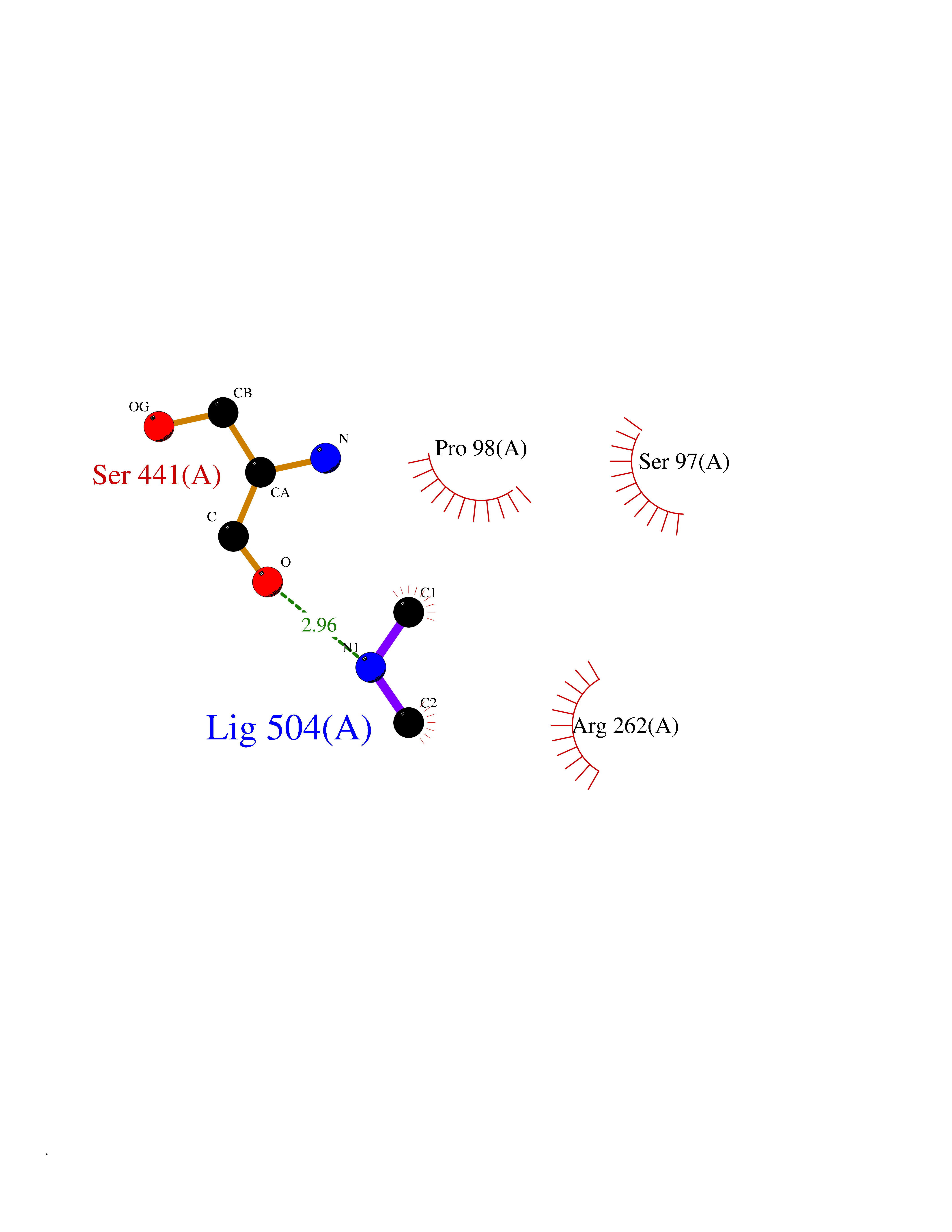 Ligplot Map 3D Binding mode Sequence GKARKEAVQTAAKELLKFVNRSPSPFHAVAECRNRLLQAGFSELKETEKWNIKPESKYFMTRNSSTIIAFAVGGQYVPGNGFSLIGAHTDSPCLRVKRRSRRSQVGFQQVGVETYGGGIWSTWFDRDLTLAGRVIVKCPTSGRLEQQLVHVERPILRIPHLAIHLQRNINENFGPNTEMHLVPILATAIQEELEKGTERHHSVLMSLLCAHLGLSPKDIVEMELCLADTQPAVLGGAYDEFIFAPRLDNLHSCFCALQALIDSCAGPGSLATEPHVRMVTLYDNEEVGSESAQGAQSLLTELVLRRISASCQHPTAFEEAIPKSFMISADMAHAVHPNYLDKHEENHRPLFHKGPVIKVNSKQRYASNAVSEALIREVANKVKVPLQDLMVRNDTPCGTTIGPILASRLGLRVLDLGSPQLAMHSIREMACTTGVLQTLTLFKGFFELFPSLAENLY Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Lysine-specific demethylase 6B (KDM6B) | 6F6D | 4.01 | |
Target general information Gen name KDM6B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine demethylase 6B; KIAA0346; Jumonji domain-containing protein 3; JmjC domain-containing protein 3; JMJD3 Protein family UTX family Biochemical class NA Function Histone demethylase that specifically demethylates 'Lys-27' of histone H3, thereby playing a central role in histone code. Demethylates trimethylated and dimethylated H3 'Lys-27'. Plays a central role in regulation of posterior development, by regulating HOX gene expression. Involved in inflammatory response by participating in macrophage differentiation in case of inflammation by regulating gene expression and macrophage differentiation. Plays a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression by acting as a link between T-box factors and the SMARCA4-containing SWI/SNF remodeling complex (By similarity). Related diseases Stolerman neurodevelopmental syndrome (NEDSST) [MIM:618505]: An autosomal dominant disorder characterized by global developmental delay, variable intellectual disability, poor language acquisition, and dysmorphic facial features including a prominent nasal bridge and coarse features. Some patients manifest autism spectrum disorder. Musculoskeletal features may be present and include widened and thickened hands and fingers, joint hypermobility, clinodactyly of the fifth fingers, and toe syndactyly. {ECO:0000269|PubMed:31124279}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P03372 EC number EC 1.14.11.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Dioxygenase; Disease variant; Inflammatory response; Intellectual disability; Iron; Isopeptide bond; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 53008.8 Length 467 Aromaticity 0.1 Instability index 44.23 Isoelectric point 8.37 Charge (pH=7) 4.9 2D Binding mode Binding energy (Kcal/mol) -5.47 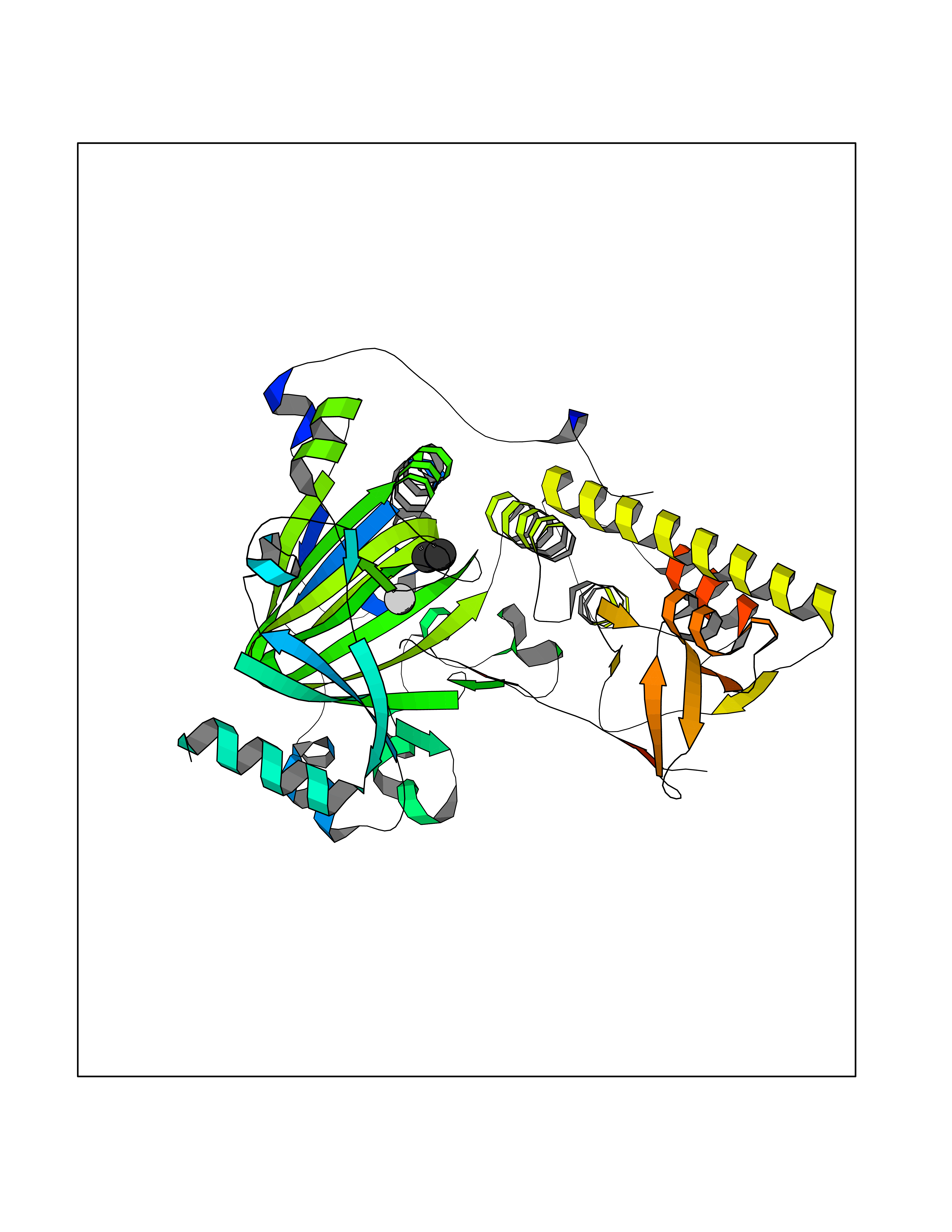 Molscript Map 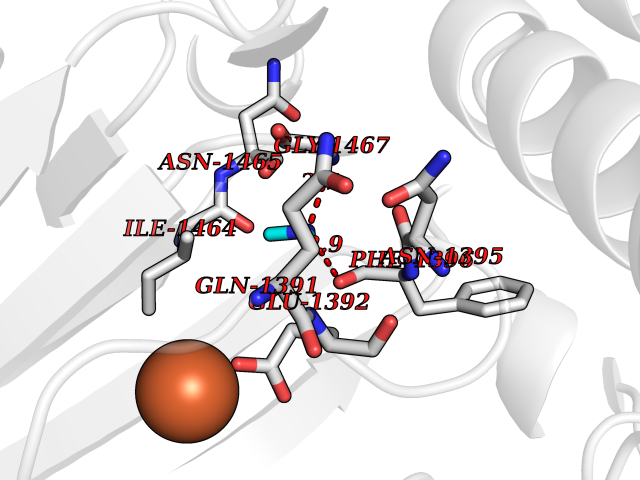 Pymol Map 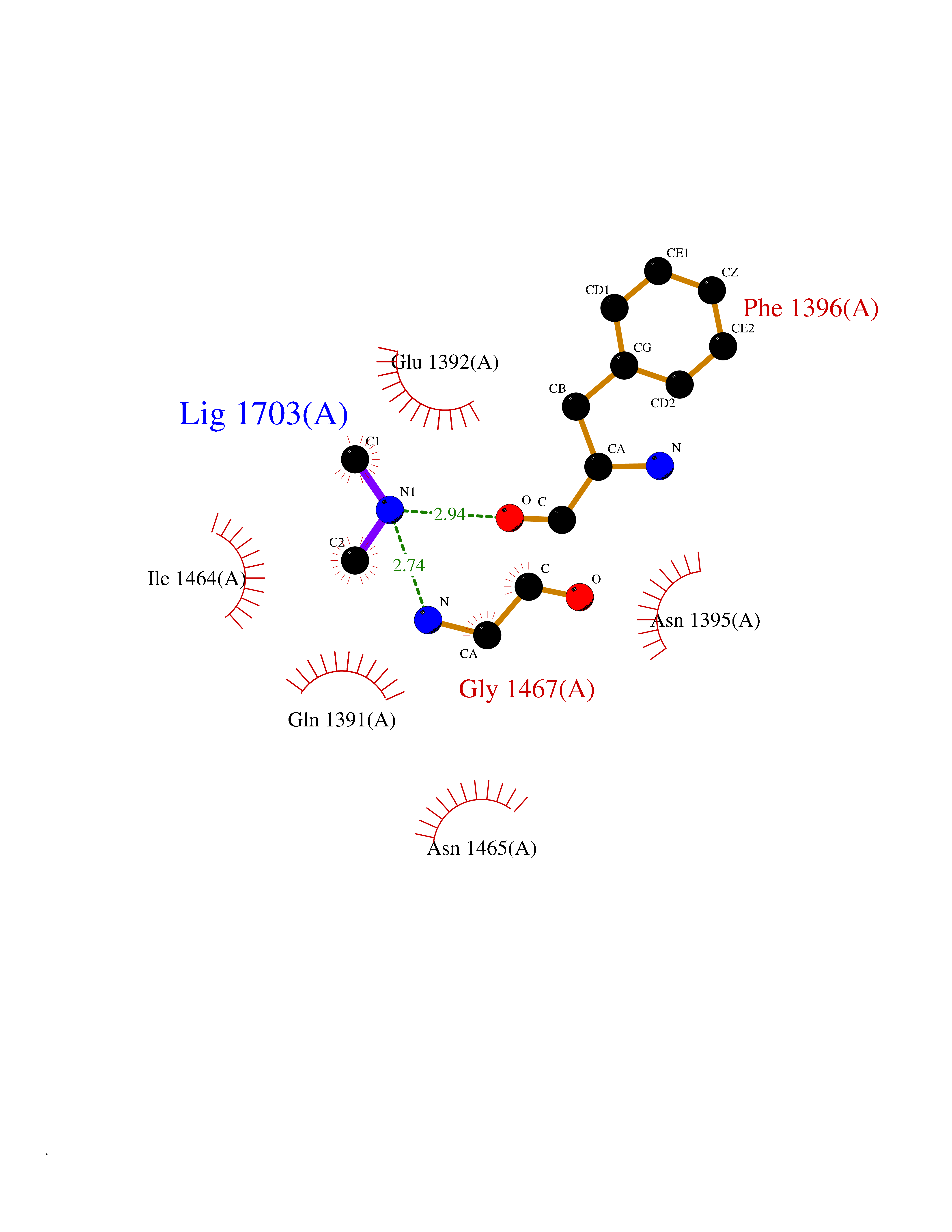 Ligplot Map 3D Binding mode Sequence ESYLSPAQSVKPKINTEEKLPREKLNPPTPSIYLESKRDAFSPVLLQFCTDPRNPITVIRGLAGSLRLNLGLFSTKTLVEASGEHTVEVRTQVQQPSDENWDLTGTRQIWPCESSRSHTTIAKYAQYQASSFQESHIIKFGTNIDLSDAKRWKPQLQELLKLPAFMRVTSTGNMLSHVGHTILGMNTVQLYMKVPGSRTPGHQENNNFCSVNINIGPGDCEWFAVHEHYWETISAFCDRHGVDYLTGSWWPILDDLYASNIPVYRFVQRPGDLVWINAGTVHWVQATGWCNNIAWNVGPLTAYQYQLALERYEWNEVKNVKSIVPMIHVSWNVARTVKISDPDLFKMIKFCLLQSMKHCQVQRESLVRAGKKIAYQGRVKDEPAYYCNECDVEVFNILFVTSENGSRNTYLVHCEGCARRRSAGLQGVVVLEQYRTEELAQAYDAFTLAPRIQLMTKAARKSAPATG Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Staphylococcus Peptide deformylase (Stap-coc def) | 1Q1Y | 4.01 | |
Target general information Gen name Stap-coc def Organism Staphylococcus aureus Uniprot ID TTD ID Synonyms def; Stap-coc Polypeptide deformylase Protein family Polypeptide deformylase family Biochemical class Carbon-nitrogen hydrolase Function Removes the formyl group from the N-terminal Met of newly synthesized proteins. Requires at least a dipeptide for an efficient rate of reaction. N-terminal L-methionine is a prerequisite for activity but the enzyme has broad specificity at other positions. Related diseases Immunodeficiency 43 (IMD43) [MIM:241600]: A disorder characterized by marked reduction in serum concentrations of immunoglobulins and albumin, and hypoproteinemia due to hypercatabolism. Patients may suffer from recurrent respiratory tract infections and severe skin disease. {ECO:0000269|PubMed:16549777}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Amyloidosis, hereditary systemic 6 (AMYLD6) [MIM:620659]: A form of hereditary systemic amyloidosis, a disorder characterized by amyloid deposition in multiple tissues resulting in a wide clinical spectrum. AMYLD6 is mainly characterized by gastrointestinal and cardiac symptoms. Neurologic involvement, sicca syndrome, and carpal tunnel syndrome may also be present. Inheritance is autosomal dominant. {ECO:0000269|PubMed:22693999}. The disease is caused by variants affecting the gene represented in this entry. Apart from the presence of causative mutations, beta-2-microglobulin may adopt the fibrillar configuration of amyloid, resulting in amyloidosis, when its serum levels are persistently high, as seen in patients on long-term hemodialysis (PubMed:7918443). In contrast to patients with dialysis-related amyloidosis, patients with hereditary amyloidosis have normal circulating concentrations of beta2-microglobulin (PubMed:22693999). {ECO:0000269|PubMed:22693999, ECO:0000269|PubMed:7918443}. Drugs (DrugBank ID) DB04310 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase; Iron; Metal-binding; Protein biosynthesis Protein physicochemical properties Chain ID A Molecular weight (Da) 20835.6 Length 186 Aromaticity 0.05 Instability index 42.9 Isoelectric point 5.65 Charge (pH=7) -7.59 2D Binding mode Binding energy (Kcal/mol) -5.47 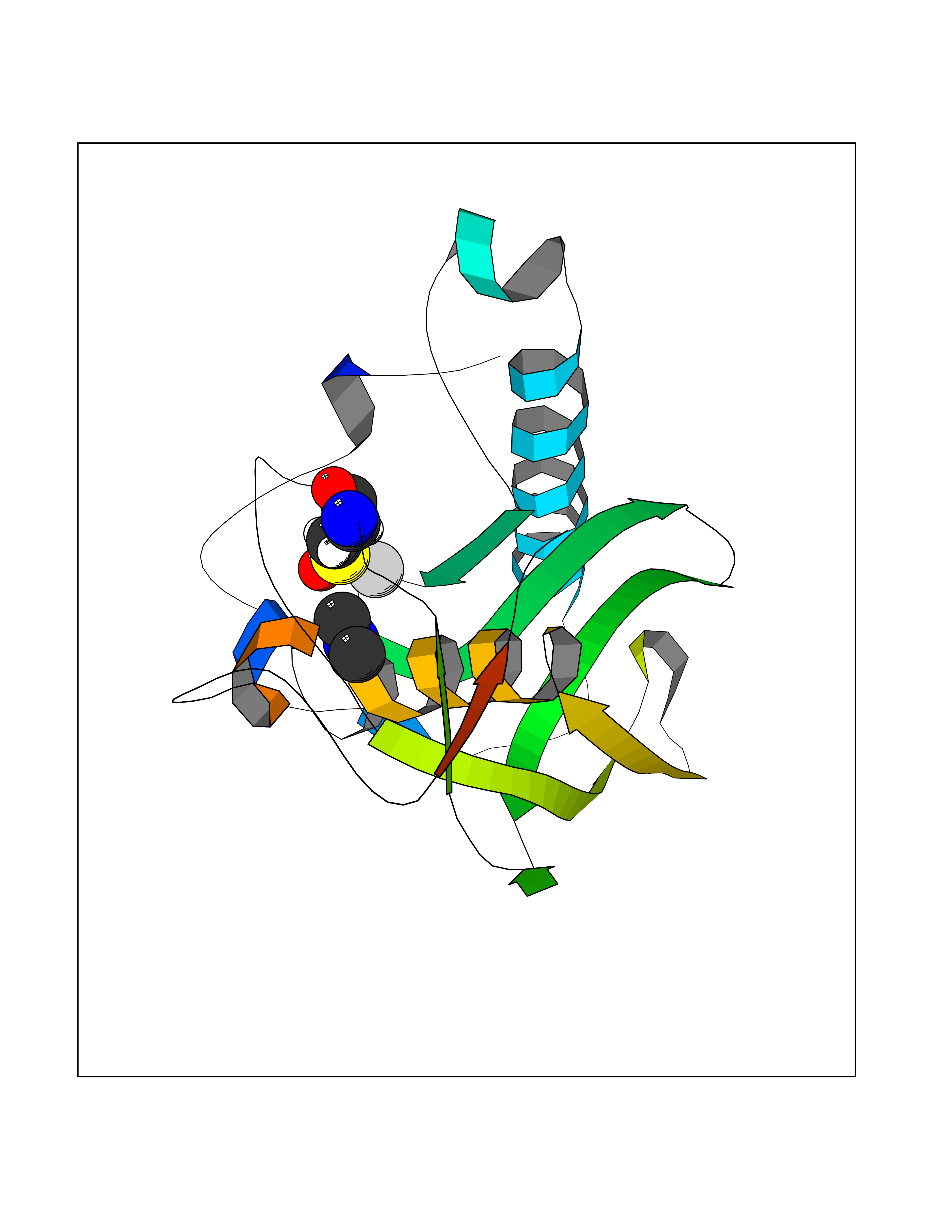 Molscript Map 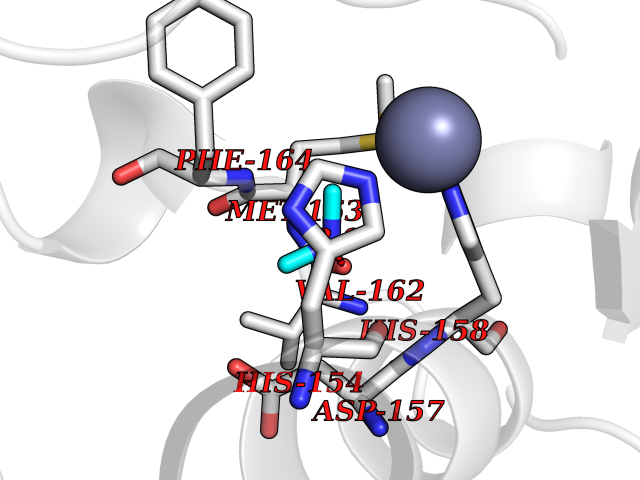 Pymol Map 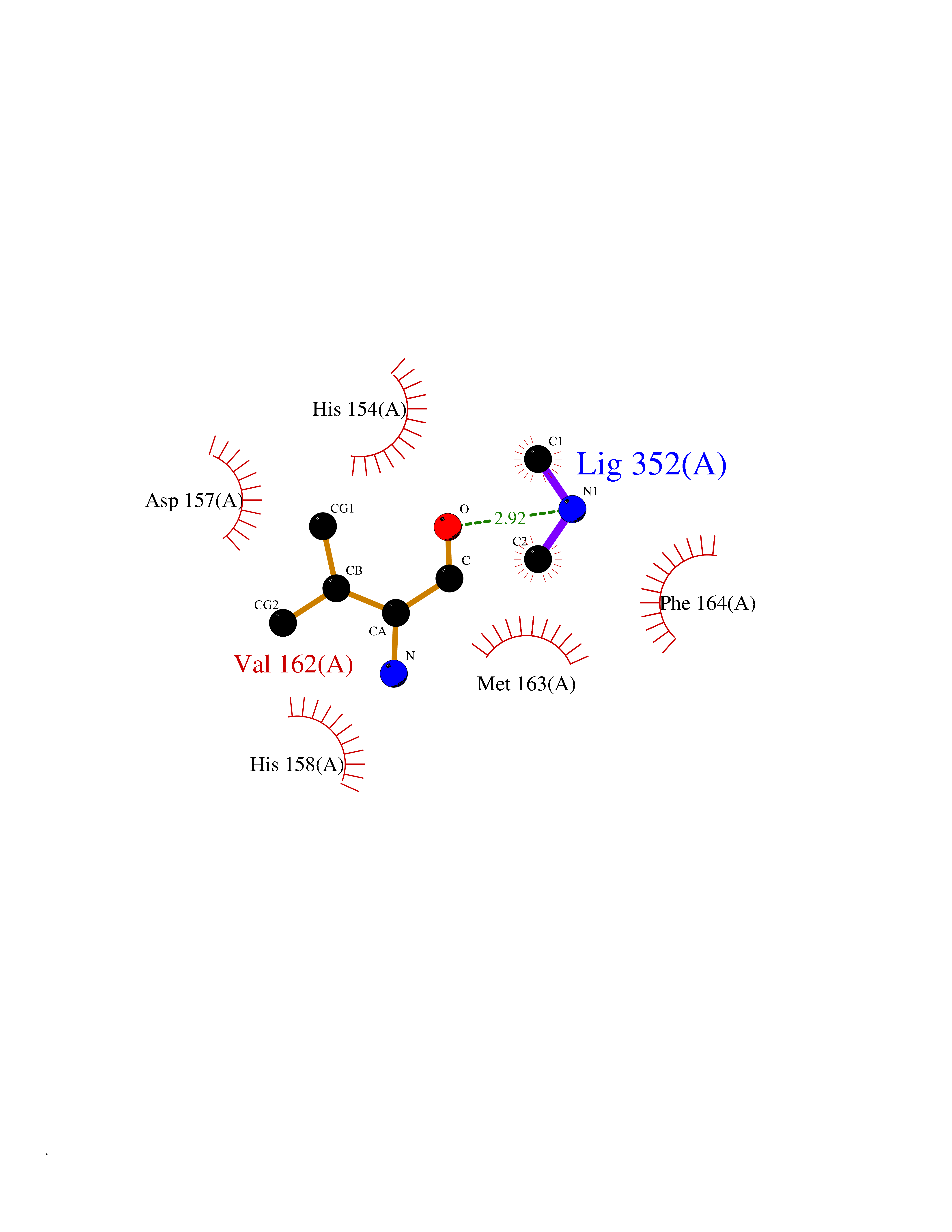 Ligplot Map 3D Binding mode Sequence MLTMKDIIRDGHPTLRQKAAELELPLTKEEKETLIAMREFLVNSQDEEIAKRYGLRSGVGLAAPQINISKRMIAVLIPDDGSGKSYDYMLVNPKIVSHSVQEAYLPTGEGXLSVDDNVAGLVHRHNRITIKAKDIEGNDIQLRLKGYPAIVFQHEIDHLNGVMFYDHIDKDHPLQPHTDAVEVLEH Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Plasma kallikrein (KLKB1) | 6T7P | 4.01 | |
Target general information Gen name KLKB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Plasma prekallikrein; Plasma kallikrein light chain; Plasma kallikrein heavy chain; PKK; Kininogenin; KLK3; Fletcher factor Protein family Peptidase S1 family, Plasma kallikrein subfamily Biochemical class Peptidase Function It activates, in a reciprocal reaction, factor XII after its binding to a negatively charged surface. It also releases bradykinin from HMW kininogen and may also play a role in the renin-angiotensin system by converting prorenin into renin. The enzyme cleaves Lys-Arg and Arg-Ser bonds. Related diseases Prekallikrein deficiency (PKKD) [MIM:612423]: An autosomal recessive condition characterized by a clotting defect due to prolongation of activated partial thromboplastin time. Affected individuals are clinically asymptomatic. {ECO:0000269|PubMed:14652634, ECO:0000269|PubMed:17598838, ECO:0000269|PubMed:34847617}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15982; DB09228; DB05311; DB12831; DB06404; DB14597; DB01593; DB14487; DB14533; DB14548 Interacts with Q9UI10; O00746; C9J082; O14744; Q8TAS3; O00233; Q8IYM2; Q9UMY4; O43493-5; Q8NFB2; Q8N0U8 EC number EC 3.4.21.34 Uniprot keywords 3D-structure; Blood coagulation; Direct protein sequencing; Disease variant; Disulfide bond; Fibrinolysis; Glycoprotein; Hemostasis; Hydrolase; Inflammatory response; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 26696.2 Length 237 Aromaticity 0.1 Instability index 34.33 Isoelectric point 8.07 Charge (pH=7) 2.21 2D Binding mode Binding energy (Kcal/mol) -5.47 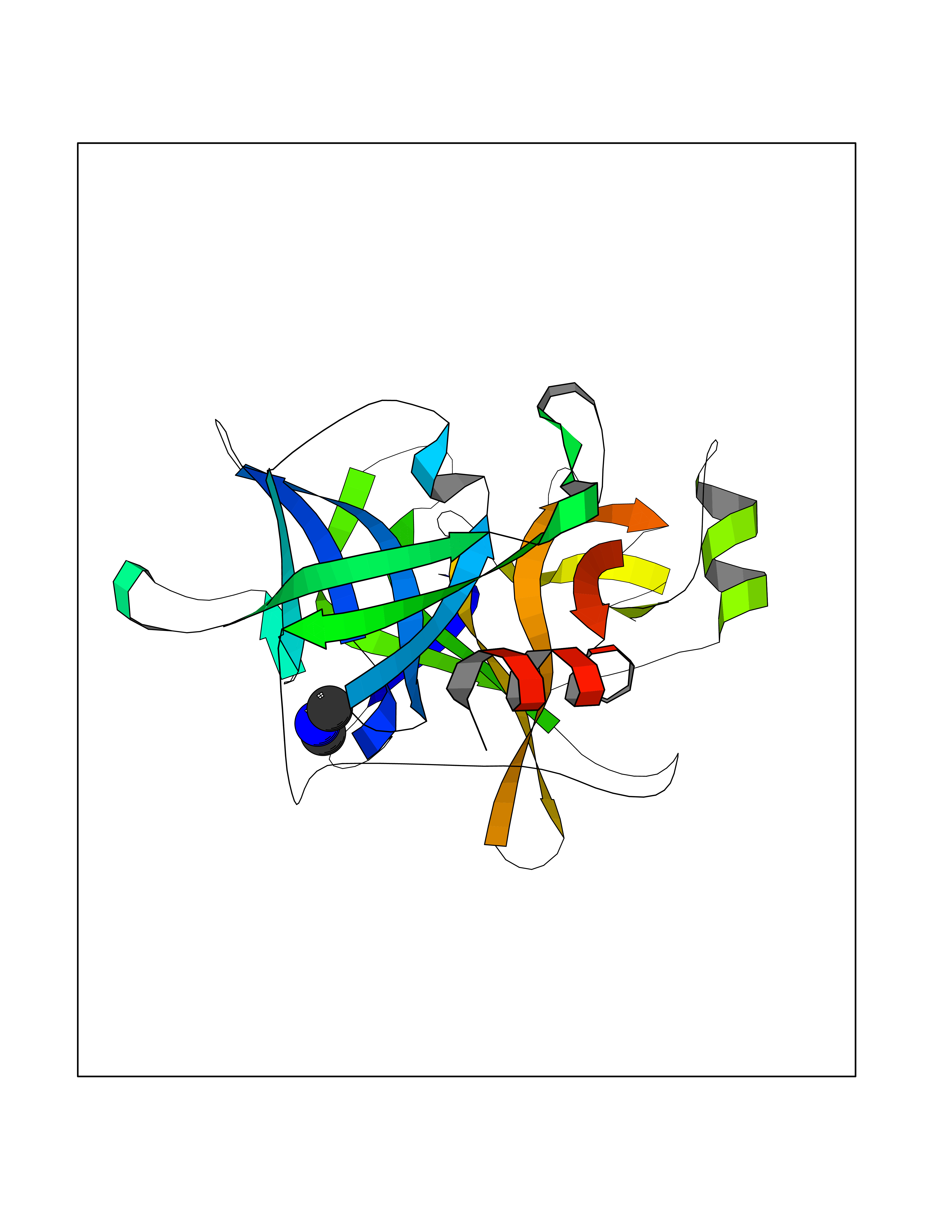 Molscript Map 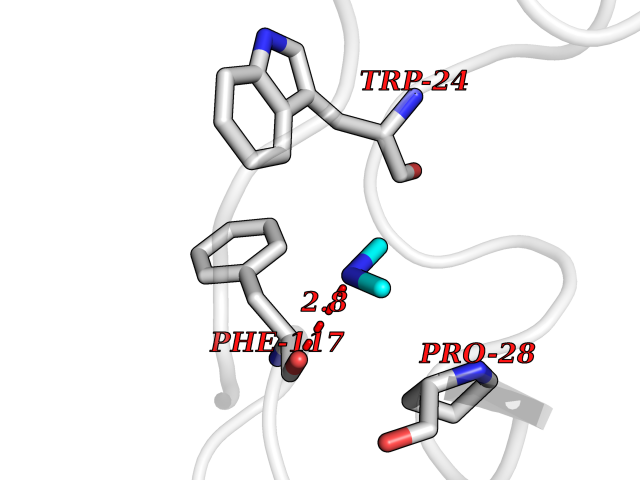 Pymol Map 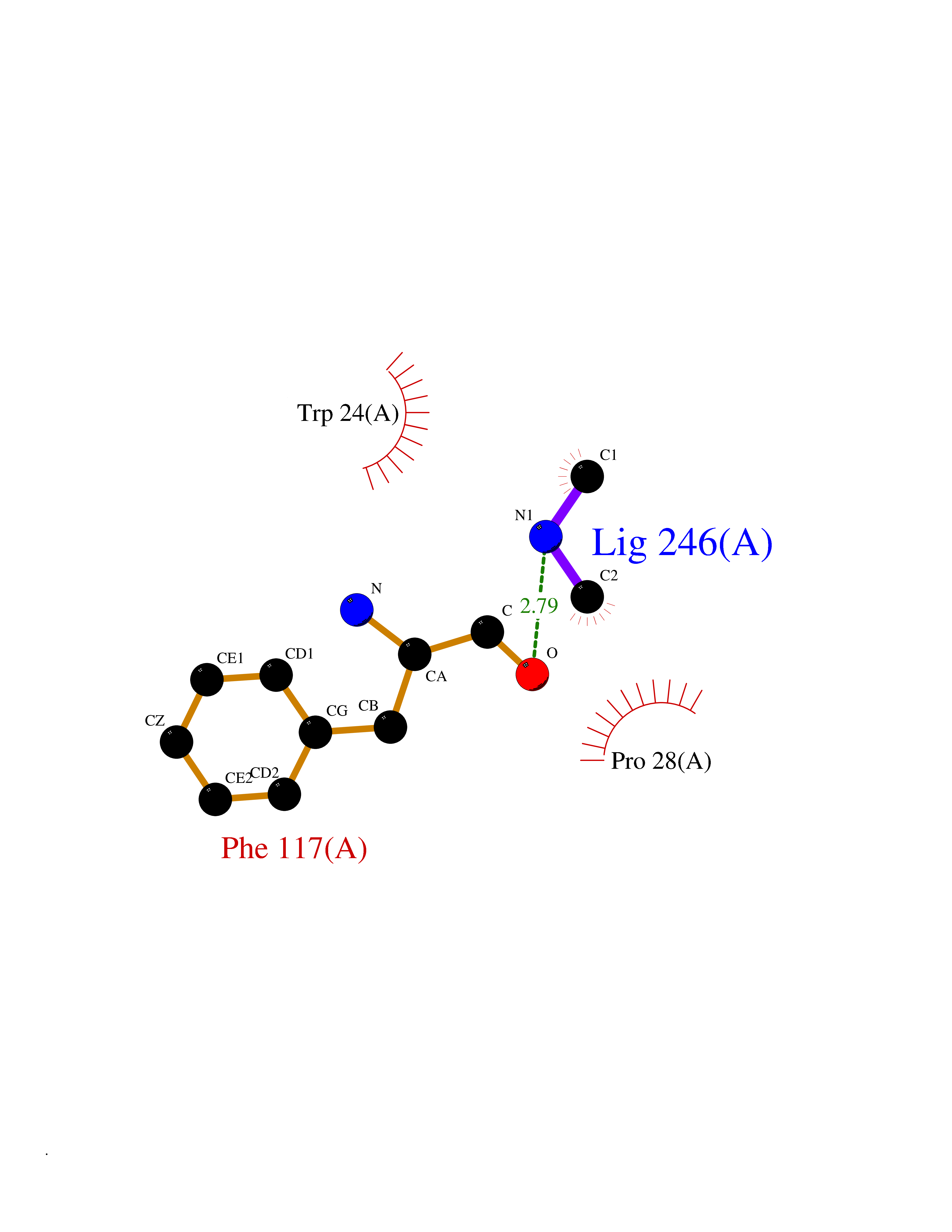 Ligplot Map 3D Binding mode Sequence IVGGTNSSWGEWPWQVSLQVKLTAQRHLCGGSLIGHQWVLTAAHCFDGLPLQDVWRIYSGILNLSDITKDTPFSQIKEIIIHQNYKVSEGNHDIALIKLQAPLNYTEFQKPICLPSKGDTSTIYTNCWVTGWGFSKEKGEIQNILQKVNIPLVTNEECQKRYQDYKITQRMVCAGYKEGGKDACKGDSGGPLVCKHNGMWRLVGITSWGEGCARREQPGVYTKVAEYMDWILEKTQS Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Triggering receptor expressed on monocytes 1 (TREM1) | 1Q8M | 4.01 | |
Target general information Gen name Trem1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Trem1; TREM-1 Protein family NA Biochemical class NA Function Stimulates neutrophil and monocyte-mediated inflammatory responses. Triggers release of pro-inflammatory chemokines and cytokines, as well as increased surface expression of cell activation markers. Amplifier of inflammatory responses that are triggered by bacterial and fungal infections and is a crucial mediator of septic shock. Related diseases GLUT1 deficiency syndrome 1 (GLUT1DS1) [MIM:606777]: A neurologic disorder showing wide phenotypic variability. The most severe 'classic' phenotype comprises infantile-onset epileptic encephalopathy associated with delayed development, acquired microcephaly, motor incoordination, and spasticity. Onset of seizures, usually characterized by apneic episodes, staring spells, and episodic eye movements, occurs within the first 4 months of life. Other paroxysmal findings include intermittent ataxia, confusion, lethargy, sleep disturbance, and headache. Varying degrees of cognitive impairment can occur, ranging from learning disabilities to severe intellectual disability. {ECO:0000269|PubMed:10227690, ECO:0000269|PubMed:10980529, ECO:0000269|PubMed:11136715, ECO:0000269|PubMed:11603379, ECO:0000269|PubMed:12325075, ECO:0000269|PubMed:15622525, ECO:0000269|PubMed:19901175, ECO:0000269|PubMed:20129935, ECO:0000269|PubMed:20221955, ECO:0000269|PubMed:20574033, ECO:0000269|PubMed:24847886, ECO:0000269|PubMed:25982116, ECO:0000269|PubMed:30197081}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GLUT1 deficiency syndrome 2 (GLUT1DS2) [MIM:612126]: A clinically variable disorder characterized primarily by onset in childhood of paroxysmal exercise-induced dyskinesia. The dyskinesia involves transient abnormal involuntary movements, such as dystonia and choreoathetosis, induced by exercise or exertion, and affecting the exercised limbs. Some patients may also have epilepsy, most commonly childhood absence epilepsy. Mild intellectual disability may also occur. In some patients involuntary exertion-induced dystonic, choreoathetotic, and ballistic movements may be associated with macrocytic hemolytic anemia. {ECO:0000269|PubMed:14605501, ECO:0000269|PubMed:18451999, ECO:0000269|PubMed:19630075, ECO:0000269|PubMed:19798636, ECO:0000269|PubMed:20129935, ECO:0000269|PubMed:20574033, ECO:0000269|PubMed:20621801, ECO:0000269|PubMed:20830593, ECO:0000269|PubMed:21204808}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epilepsy, idiopathic generalized 12 (EIG12) [MIM:614847]: A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. In some EIG12 patients seizures may remit with age. {ECO:0000269|PubMed:19798636, ECO:0000269|PubMed:22282645, ECO:0000269|PubMed:23280796, ECO:0000269|PubMed:25982116}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Dystonia 9 (DYT9) [MIM:601042]: An autosomal dominant neurologic disorder characterized by childhood onset of paroxysmal choreoathetosis and progressive spastic paraplegia. Most patients show some degree of cognitive impairment. Other variable features may include seizures, migraine headaches, and ataxia. {ECO:0000269|PubMed:21832227}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Stomatin-deficient cryohydrocytosis with neurologic defects (SDCHCN) [MIM:608885]: A rare form of stomatocytosis characterized by episodic hemolytic anemia, cold-induced red cells cation leak, erratic hyperkalemia, neonatal hyperbilirubinemia, hepatosplenomegaly, cataracts, seizures, intellectual disability, and movement disorder. {ECO:0000269|PubMed:21791420, ECO:0000269|PubMed:22492876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01694 Interacts with NA EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Innate immunity; Membrane; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 28186.2 Length 242 Aromaticity 0.07 Instability index 45.2 Isoelectric point 6.1 Charge (pH=7) -3.8 2D Binding mode Binding energy (Kcal/mol) -5.47 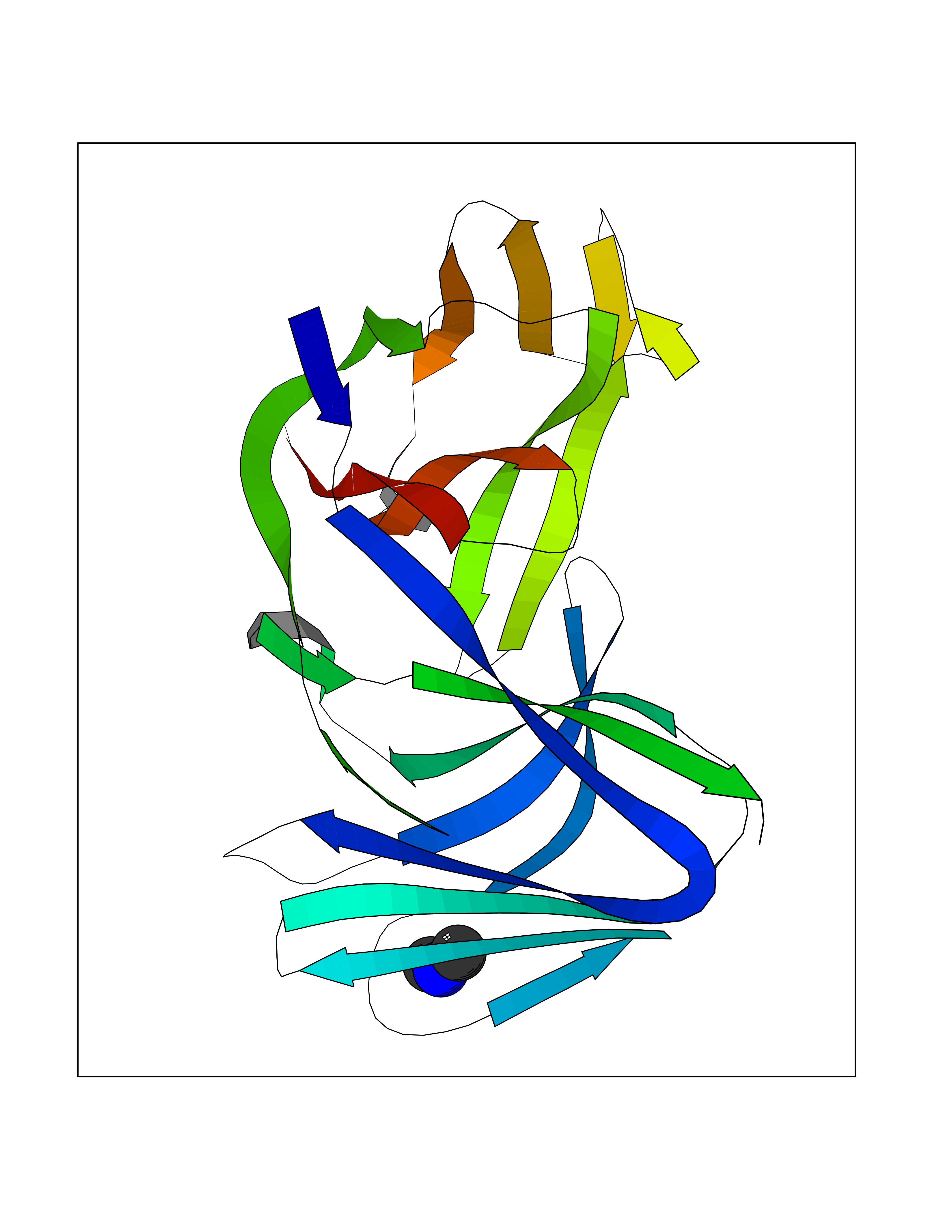 Molscript Map 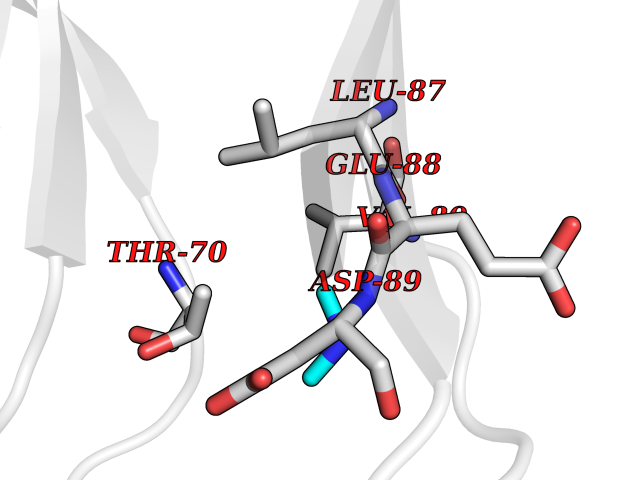 Pymol Map 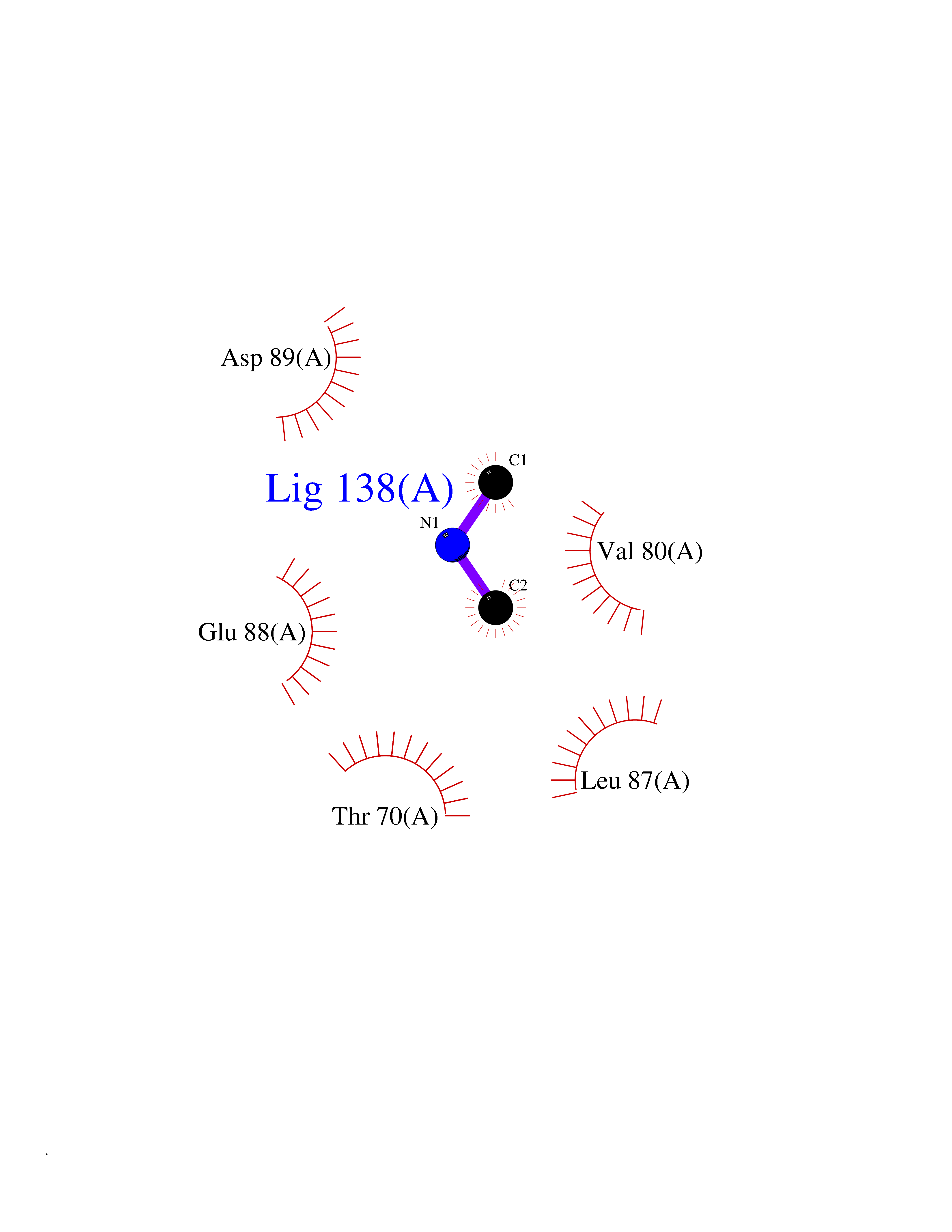 Ligplot Map 3D Binding mode Sequence MELRAATKLTEEKYELKEGQTLDVKCDYTLEKFASSQKAWQIIRDGEMPKTLACTERPSKNSHPVQVGRIILEDYHDHGLLRVRMVNLQVEDSGLYQCVIYQPPKEPHMLFDRIRLVVTLEMELRAATKLTEEKYELKEGQTLDVKCDYTLEKFASSQKAWQIIRDGEMPKTLACTERPSKNSHPVQVGRIILEDYHDHGLLRVRMVNLQVEDSGLYQCVIYQPPKEPHMLFDRIRLVVTLE Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Cerebroside-sulfatase (ARSA) | 1E2S | 4.01 | |
Target general information Gen name ARSA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cerebrosidesulfatase; Arylsulfatase A component C; ASA; ARSA Protein family Sulfatase family Biochemical class Sulfuric ester hydrolase Function Hydrolyzes cerebroside sulfate. Related diseases Metachromatic leukodystrophy (MLD) [MIM:250100]: An autosomal recessive disease caused by abnormal intralysosomal accumulation of cerebroside-3-sulfate in central and peripheral nervous systems, as well as other organs. MLD is clinically characterized by leukodystrophy, progressive demyelination and a variety of neurological symptoms, including gait disturbances, ataxias, optical atrophy, dementia, seizures, and spastic tetraparesis. Decreased arylsulfatase A activity is detected in urine, leukocytes, and fibroblasts of affected individuals. Several forms of the disease can be distinguished according to the age at onset and disease severity: late infantile, juvenile and adult forms, partial cerebroside sulfate deficiency, and pseudoarylsulfatase A deficiency. Individuals with pseudoarylsulfatase A deficiency have low arylsulfatase A activity but lack neurological manifestations and are apparently healthy. {ECO:0000269|PubMed:10220151, ECO:0000269|PubMed:10381328, ECO:0000269|PubMed:10477432, ECO:0000269|PubMed:10533072, ECO:0000269|PubMed:10751093, ECO:0000269|PubMed:11020646, ECO:0000269|PubMed:11061266, ECO:0000269|PubMed:11456299, ECO:0000269|PubMed:11941485, ECO:0000269|PubMed:12503099, ECO:0000269|PubMed:12788103, ECO:0000269|PubMed:1353340, ECO:0000269|PubMed:14517960, ECO:0000269|PubMed:14680985, ECO:0000269|PubMed:15026521, ECO:0000269|PubMed:15326627, ECO:0000269|PubMed:15710861, ECO:0000269|PubMed:1670590, ECO:0000269|PubMed:1673291, ECO:0000269|PubMed:1678251, ECO:0000269|PubMed:18693274, ECO:0000269|PubMed:19606494, ECO:0000269|PubMed:20339381, ECO:0000269|PubMed:21265945, ECO:0000269|PubMed:2574462, ECO:0000269|PubMed:7581401, ECO:0000269|PubMed:7825603, ECO:0000269|PubMed:7860068, ECO:0000269|PubMed:7902317, ECO:0000269|PubMed:7906588, ECO:0000269|PubMed:7909527, ECO:0000269|PubMed:8095918, ECO:0000269|PubMed:8101038, ECO:0000269|PubMed:8101083, ECO:0000269|PubMed:8104633, ECO:0000269|PubMed:8891236, ECO:0000269|PubMed:9090526, ECO:0000269|PubMed:9272717, ECO:0000269|PubMed:9452102, ECO:0000269|PubMed:9490297, ECO:0000269|PubMed:9600244, ECO:0000269|PubMed:9819708}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Multiple sulfatase deficiency (MSD) [MIM:272200]: A clinically and biochemically heterogeneous disorder caused by the simultaneous impairment of all sulfatases, due to defective post-translational modification and activation. It combines features of individual sulfatase deficiencies such as metachromatic leukodystrophy, mucopolysaccharidosis, chondrodysplasia punctata, hydrocephalus, ichthyosis, neurologic deterioration and developmental delay. {ECO:0000269|PubMed:15146462}. The protein represented in this entry is involved in disease pathogenesis. Arylsulfatase A activity is impaired in multiple sulfatase deficiency due to mutations in SUMF1 (PubMed:15146462). SUMF1 mutations result in defective post-translational modification of ARSA at residue Cys-69 that is not converted to 3-oxoalanine (PubMed:7628016). {ECO:0000269|PubMed:15146462, ECO:0000269|PubMed:7628016}. Drugs (DrugBank ID) DB03821; DB01800; DB01141; DB04786 Interacts with P50995; Q6P5X5; Q13554-3; O60826; Q96D98; Q9H0I2; Q12951-2; Q16512; P28069; O75360; Q9BQY4; Q15645; O95231 EC number EC 3.1.6.8 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Hydrolase; Ichthyosis; Leukodystrophy; Lipid metabolism; Lysosome; Metachromatic leukodystrophy; Metal-binding; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID P Molecular weight (Da) 51173.7 Length 481 Aromaticity 0.09 Instability index 47.42 Isoelectric point 5.59 Charge (pH=7) -12.84 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 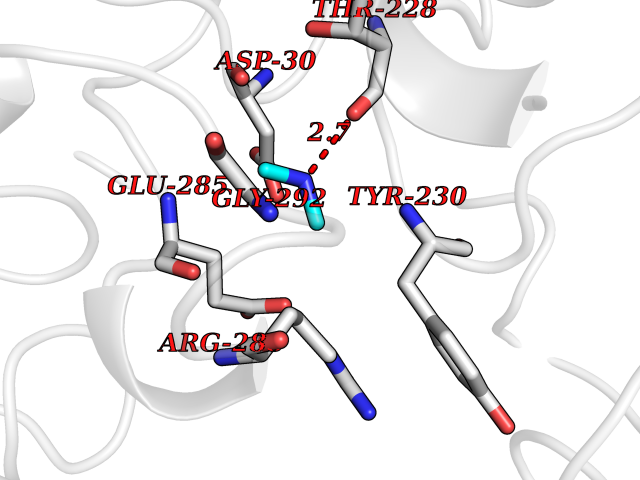 Pymol Map 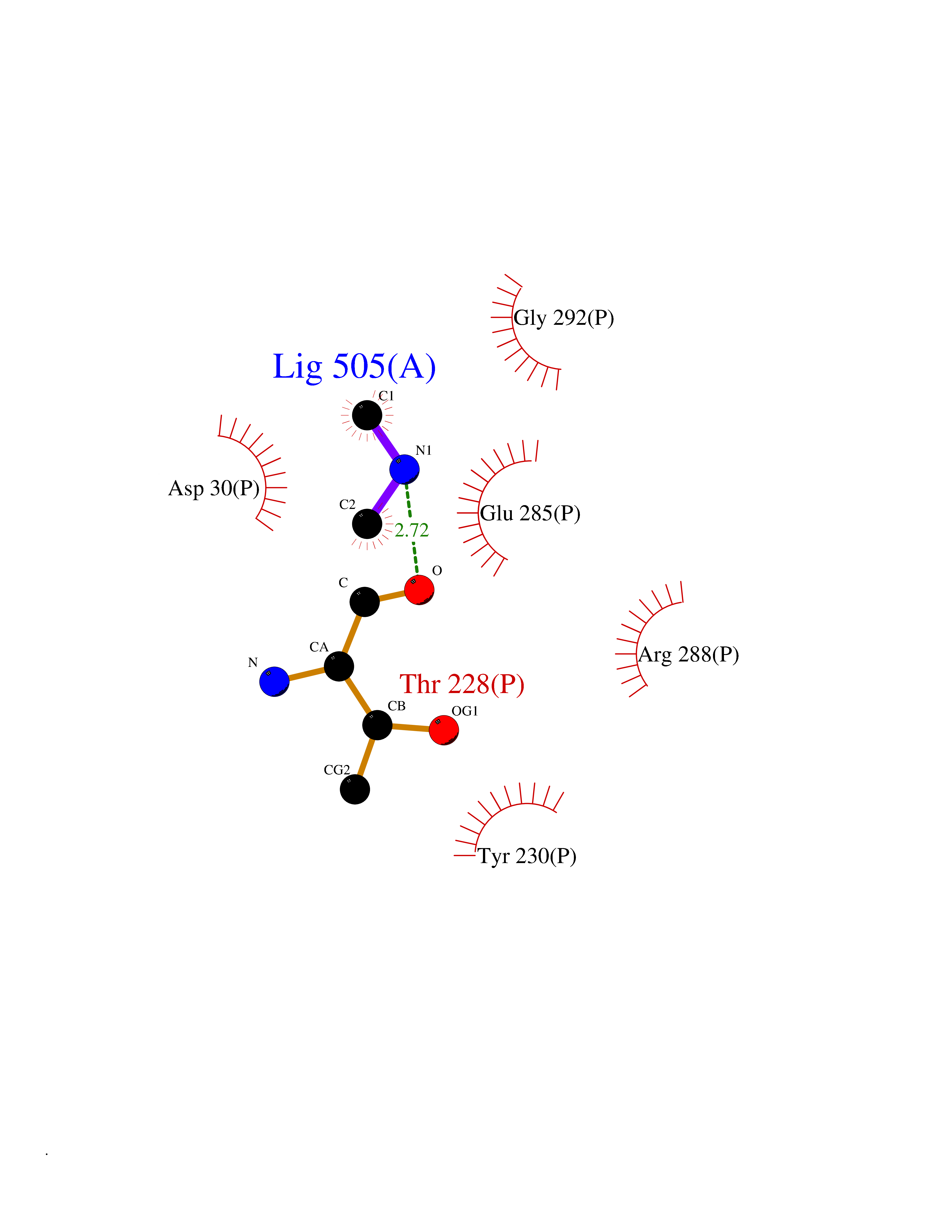 Ligplot Map 3D Binding mode Sequence RPPNIVLIFADDLGYGDLGCYGHPSSTTPNLDQLAAGGLRFTDFYVPVSLATPSRAALLTGRLPVRMGMYPGVLVPSSRGGLPLEEVTVAEVLAARGYLTGMAGKWHLGVGPEGAFLPPHQGFHRFLGIPYSHDQGPCQNLTCFPPATPCDGGCDQGLVPIPLLANLSVEAQPPWLPGLEARYMAFAHDLMADAQRQDRPFFLYYASHHTHYPQFSGQSFAERSGRGPFGDSLMELDAAVGTLMTAIGDLGLLEETLVIFTADNGPETMRMSRGGCSGLLRCGKGTTYEGGVREPALAFWPGHIAPGVTHELASSLDLLPTLAALAGAPLPNVTLDGFDLSPLLLGTGKSPRQSLFFYPSYPDEVRGVFAVRTGKYKAHFFTQGSAHSDTTADPACHASSSLTAHEPPLLYDLSKDPGENYNLLGATPEVLQALKQLQLLKAQLDAAVTFGPSQVARGEDPALQICCHPGCTPRPACCHCP Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | 5,10-methylenetetrahydrofolate reductase | 3FST | 4.00 | |
Target general information Gen name metF Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3941;JW3913 Protein family Methylenetetrahydrofolate reductase family Biochemical class Oxidoreductase Function FAD binding.Methylenetetrahydrofolate reductase (NAD(P)H) activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.5.1.54 Uniprot keywords 3D-structure; Amino-acid biosynthesis; FAD; Flavoprotein; Methionine biosynthesis; NAD; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,C,E Molecular weight (Da) 30855.9 Length 274 Aromaticity 0.09 Instability index 27.54 Isoelectric point 5.84 Charge (pH=7) -4.61 2D Binding mode Binding energy (Kcal/mol) -5.46 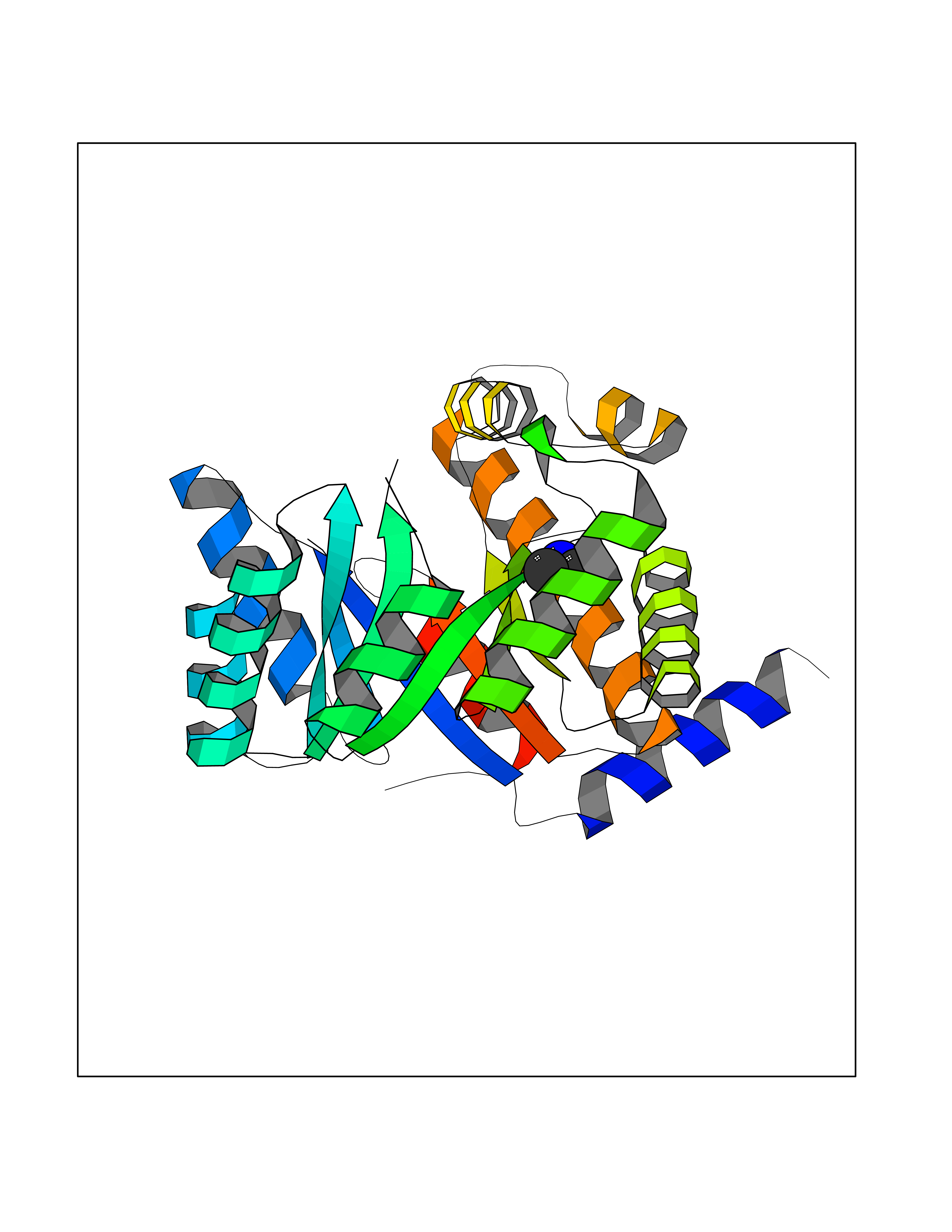 Molscript Map 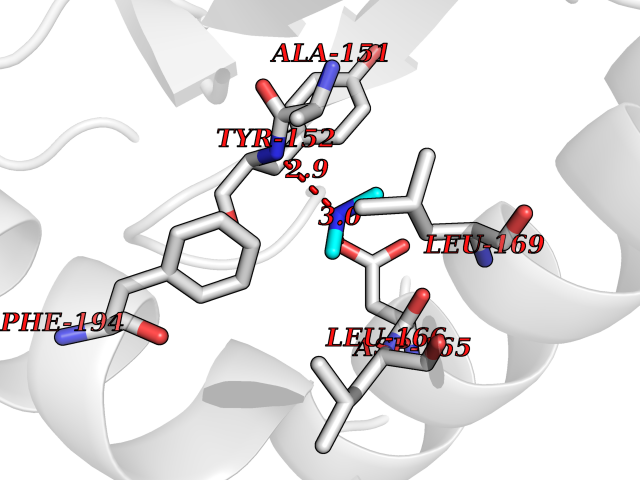 Pymol Map 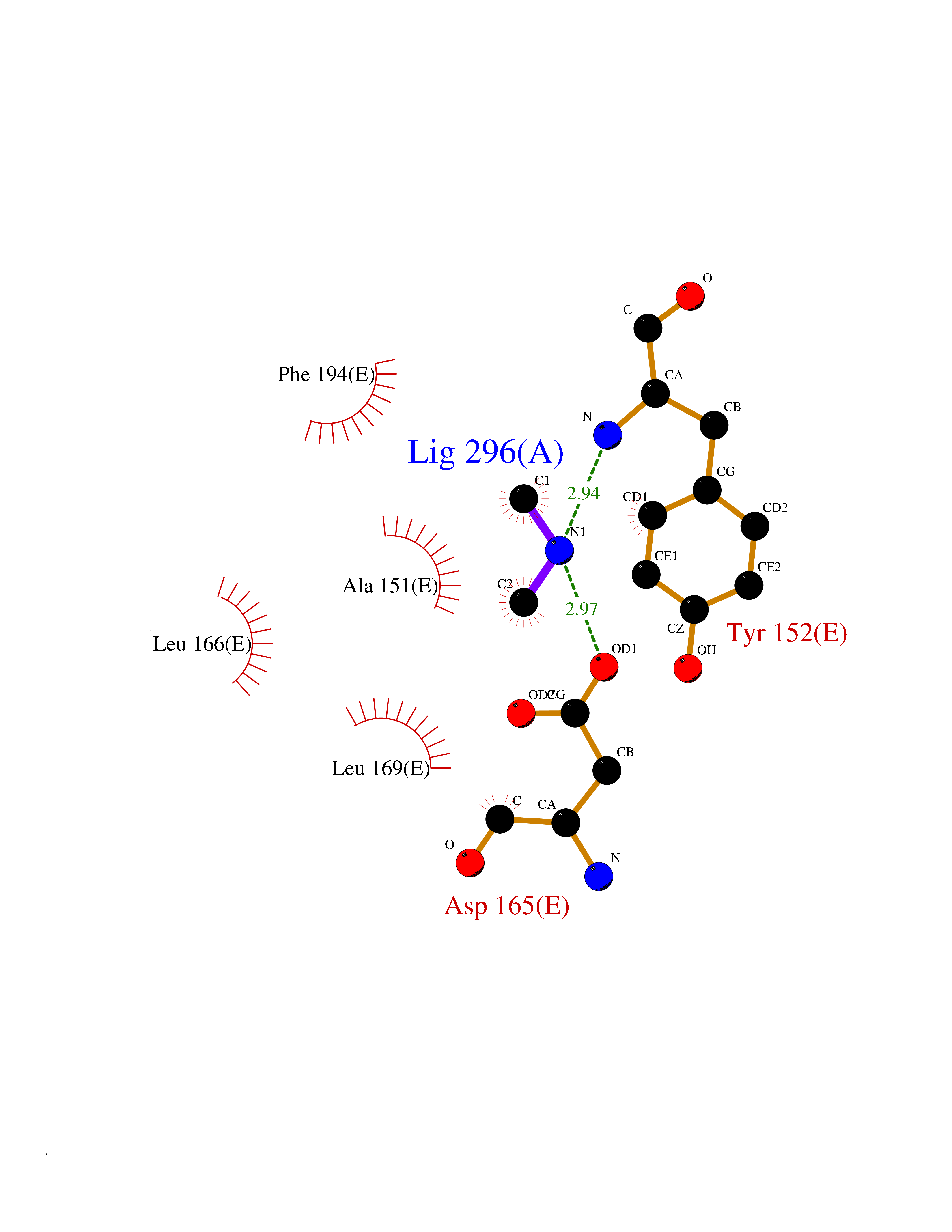 Ligplot Map 3D Binding mode Sequence FHASQRDALNQSLAEVQGQINVSFEFFPPRTSEMEQTLWNSIDRLSSLKPKFVSVTYTHSIIKGIKDRTGLEAAPHLTCIDATPDELRTIARDYWNNGIRHIVALRGDEMYASDLVTLLKEVADFDISVAAYPEVHPEAKSAQADLLNLKRKVDAGANRAITQFFFDVESYLRFRDRCVSAGIDVEIIPGILPVSNFKQAKKLADMTNVRIPAWMAQMFDGLDDDAETRKLVGANIAMDMVKILSREGVKDFHFYTLNRAEMSYAICHTLGVRP Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Endolysin | 1AM7 | 4.00 | |
Target general information Gen name R Organism Escherichia phage lambda (Bacteriophage lambda) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 24 family Biochemical class Glycosidase Function Lyase activity.Lysozyme activity.Lytic transglycosylase activity. Related diseases Estrogen resistance (ESTRR) [MIM:615363]: A disorder characterized by partial or complete resistance to estrogens, in the presence of elevated estrogen serum levels. Clinical features include absence of the pubertal growth spurt, delayed bone maturation, unfused epiphyses, reduced bone mineral density, osteoporosis, continued growth into adulthood and very tall adult stature. Glucose intolerance, hyperinsulinemia and lipid abnormalities may also be present. {ECO:0000269|PubMed:23841731, ECO:0000269|PubMed:27754803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04206 Interacts with NA EC number 4.2.2.n2 Uniprot keywords 3D-structure; Antimicrobial; Bacteriolytic enzyme; Cytolysis; Direct protein sequencing; Host cell lysis by virus; Host cytoplasm; Lyase; Reference proteome; Viral release from host cell Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 49834.9 Length 462 Aromaticity 0.07 Instability index 18.78 Isoelectric point 9.6 Charge (pH=7) 18.29 2D Binding mode Binding energy (Kcal/mol) -5.45 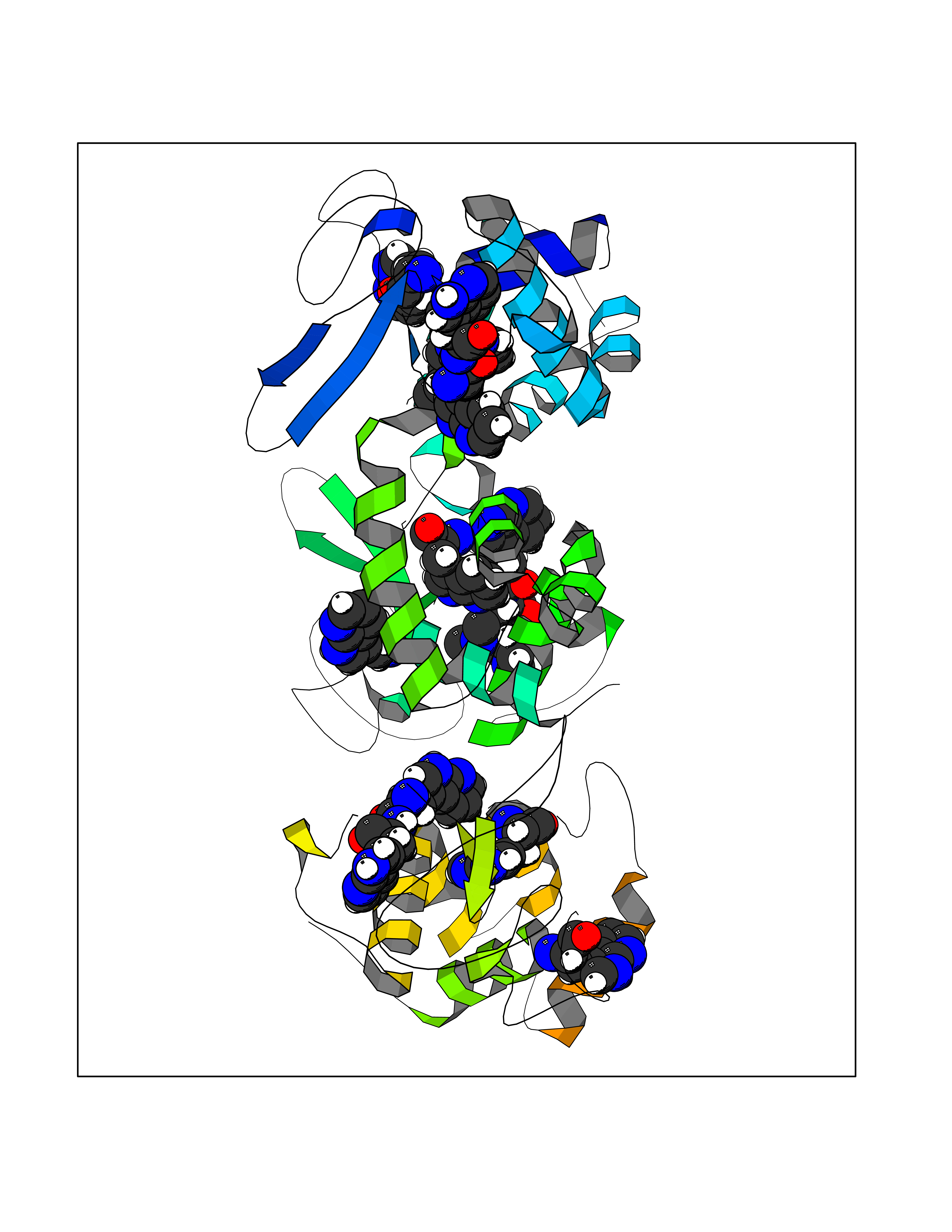 Molscript Map 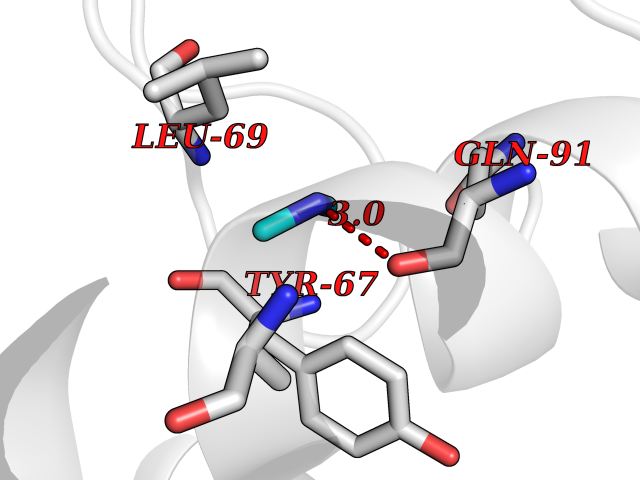 Pymol Map 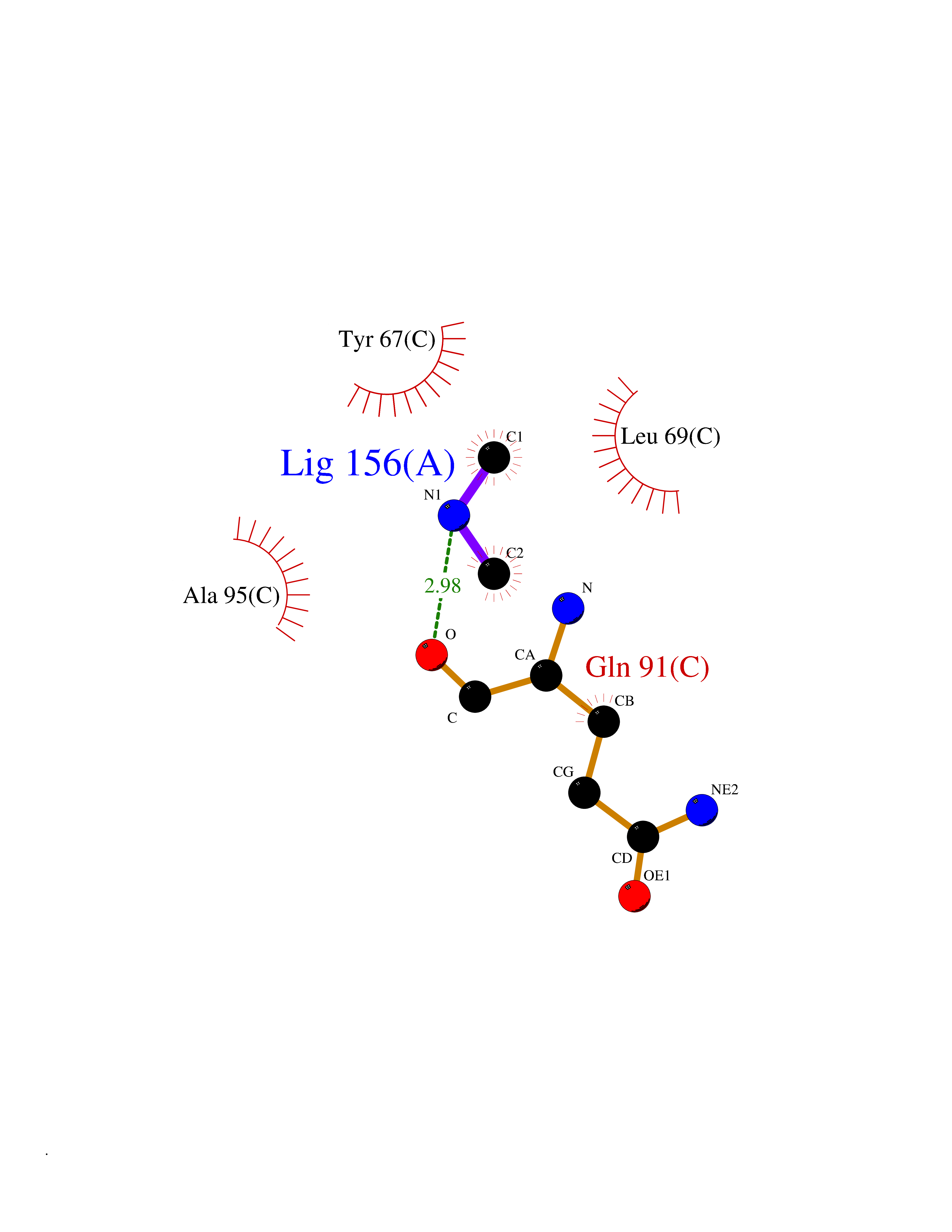 Ligplot Map 3D Binding mode Sequence MVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVR Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Cytochrome b (Complex III subunit 3) (Complex III subunit III) (Cytochrome b-c1 complex subunit 3) (Ubiquinol-cytochrome-c reductase complex cytochrome b subunit) | 4G6H | 4.00 | |
Target general information Gen name MT-CYB Organism Bos taurus (Bovine) Uniprot ID TTD ID NA Synonyms CYTB;MTCYB;COB Protein family Cytochrome b family Biochemical class NA Function Component of the ubiquinol-cytochrome c reductase complex (complex III or cytochrome b-c1 complex) that is part of the mitochondrial respiratory chain. The b-c1 complex mediates electron transfer from ubiquinol to cytochrome c. Contributes to the generation of a proton gradient across the mitochondrial membrane that is then used for ATP synthesis. {ECO:0000269|PubMed:1327781, ECO:0000269|PubMed:20025846, ECO:0000269|PubMed:9485330, ECO:0000305|PubMed:189810}." Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Electron transport; Heme; Iron; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Reference proteome; Respiratory chain; Transmembrane; Transmembrane helix; Transport; Ubiquinone Protein physicochemical properties Chain ID A,B Molecular weight (Da) 105644 Length 944 Aromaticity 0.1 Instability index 37 Isoelectric point 9.24 Charge (pH=7) 19.42 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map 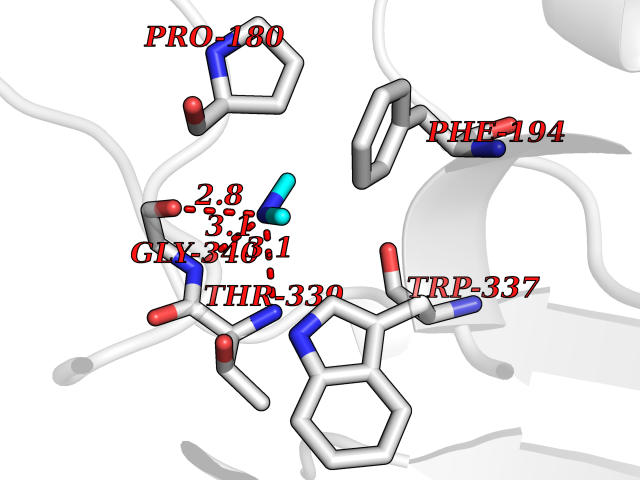 Pymol Map 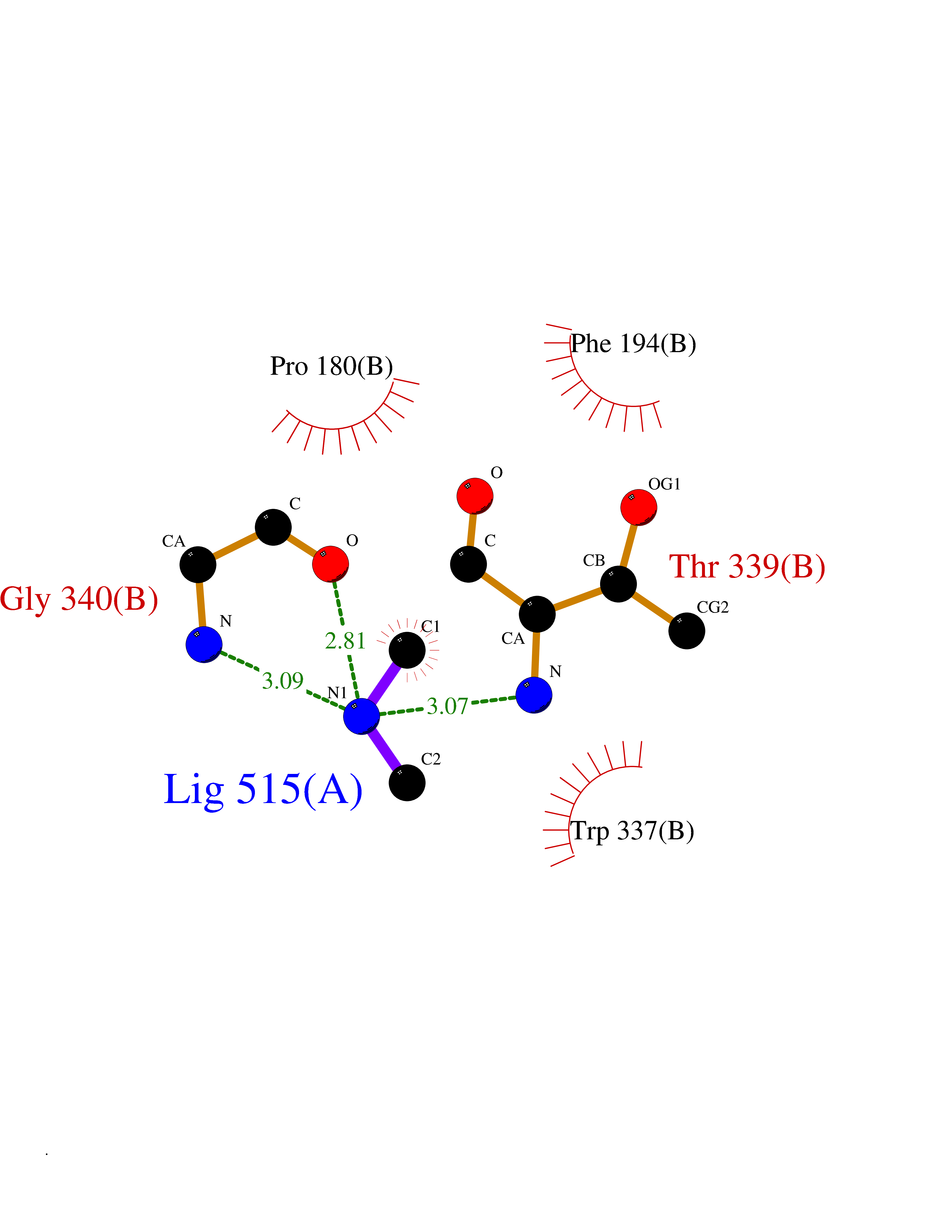 Ligplot Map 3D Binding mode Sequence TMKVIDPQHSDKPNVLILGSGWGAISFLKHIDTKKYNVSIISPRSYFLFTPLLPSAPVGTVDEKSIIEPIVNFALKKKGNVTYYEAEATSINPDRNTVTIKSLSAVSQLYQPENHLGLHQAEPAEIKYDYLISAVGAEPNTFGIPGVTDYGHFLKEIPNSLEIRRTFAANLEKANLLPKGDPERRRLLSIVVVGGGPTGVEAAGELQDYVHQDLRKFLPALAEEVQIHLVEALPIVLNMFEKKLSSYAQSHLENTSIKVHLRTAVAKVEEKQLLAKTKHEDGKITEETIPYGTLIWATGNKARPVITDLFKKIPEQNSSKRGLAVNDFLQVKGSNNIFAIGDNAFAGLPPTAQVAHQEAEYLAKNFDKMAQIPNFQKNLSSRKDKIDLLFEENNFKPFKYNDLGALAYLGSERAIATIRSGKRTFYTGGGLMTFYLWRILYLSMILSARSRLKVFFDWIKLAFFKRDFFKGLTMKVIDPQHSDKPNVLILGSGWGAISFLKHIDTKKYNVSIISPRSYFLFTPLLPSAPVGTVDEKSIIEPIVNFALKKKGNVTYYEAEATSINPDRNTVTIKSLSAVSQLYQPENHLGLHQAEPAEIKYDYLISAVGAEPNTFGIPGVTDYGHFLKEIPNSLEIRRTFAANLEKANLLPKGDPERRRLLSIVVVGGGPTGVEAAGELQDYVHQDLRKFLPALAEEVQIHLVEALPIVLNMFEKKLSSYAQSHLENTSIKVHLRTAVAKVEEKQLLAKTKHEDGKITEETIPYGTLIWATGNKARPVITDLFKKIPEQNSSKRGLAVNDFLQVKGSNNIFAIGDNAFAGLPPTAQVAHQEAEYLAKNFDKMAQIPNFQKNLSSRKDKIDLLFEENNFKPFKYNDLGALAYLGSERAIATIRSGKRTFYTGGGLMTFYLWRILYLSMILSARSRLKVFFDWIKLAFFKRDFFKGL Hydrogen bonds contact Hydrophobic contact | ||||