Job Results:
Ligand
Structure
Job ID
43a62498dd267475554cbec83e92e777
Job name
NA
Time
2025-02-13 15:08:06
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Cathepsin G (CTSG) | 1KYN | 4.02 | |
Target general information Gen name CTSG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CG Protein family Peptidase S1 family Biochemical class Peptidase Function Cleaves complement C3. Has antibacterial activity against the Gram-negative bacterium P. aeruginosa, antibacterial activity is inhibited by LPS from P. aeruginosa, Z-Gly-Leu-Phe-CH2Cl and phenylmethylsulfonyl fluoride. Serine protease with trypsin- and chymotrypsin-like specificity. Related diseases Lethal congenital contracture syndrome 2 (LCCS2) [MIM:607598]: A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS2 patients manifest craniofacial/ocular findings, lack of hydrops, multiple pterygia, and fractures, as well as a normal duration of pregnancy and a unique feature of a markedly distended urinary bladder (neurogenic bladder defect). The phenotype suggests a spinal cord neuropathic etiology. {ECO:0000269|PubMed:17701904}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythroleukemia, familial (FERLK) [MIM:133180]: An autosomal dominant myeloproliferative disorder characterized by neoplastic proliferation of erythroblastic and myeloblastic elements with atypical erythroblasts and myeloblasts in the peripheral blood. Disease penetrance is incomplete. {ECO:0000269|PubMed:27416908}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Visceral neuropathy, familial, 1, autosomal recessive (VSCN1) [MIM:243180]: An autosomal recessive disorder characterized by intestinal dysmotility due to aganglionosis (Hirschsprung disease), hypoganglionosis, and/or chronic intestinal pseudoobstruction. Additional variable features are progressive peripheral neuropathy, arthrogryposis, hypoplasia or aplasia of the olfactory bulb and of the external auditory canals, microtia or anotia, and facial dysmorphism. Some patients present structural cardiac anomalies and arthrogryposis with multiple pterygia. {ECO:0000269|PubMed:33497358}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04016; DB02360 Interacts with A8MQ03; Q15323; Q7Z3S9; Q9NRD5 EC number EC 3.4.21.20 Uniprot keywords 3D-structure; Antibiotic; Antimicrobial; Cell membrane; Chemotaxis; Cytoplasm; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lysosome; Membrane; Nucleus; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 25353.8 Length 223 Aromaticity 0.06 Instability index 66.1 Isoelectric point 11.51 Charge (pH=7) 23.24 2D Binding mode Binding energy (Kcal/mol) -5.48 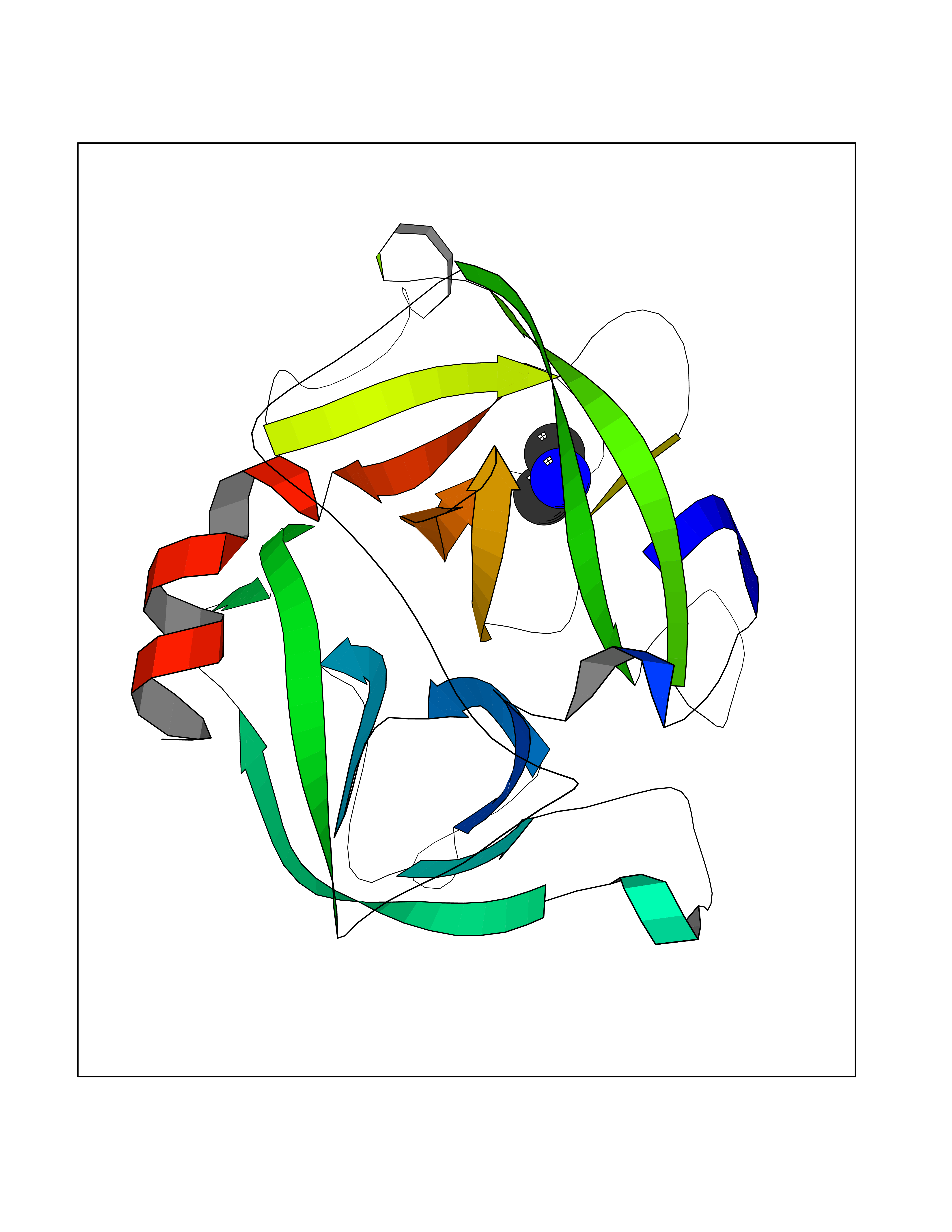 Molscript Map 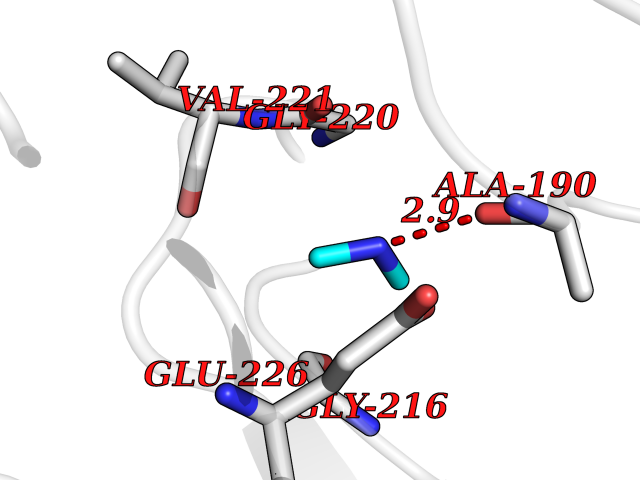 Pymol Map 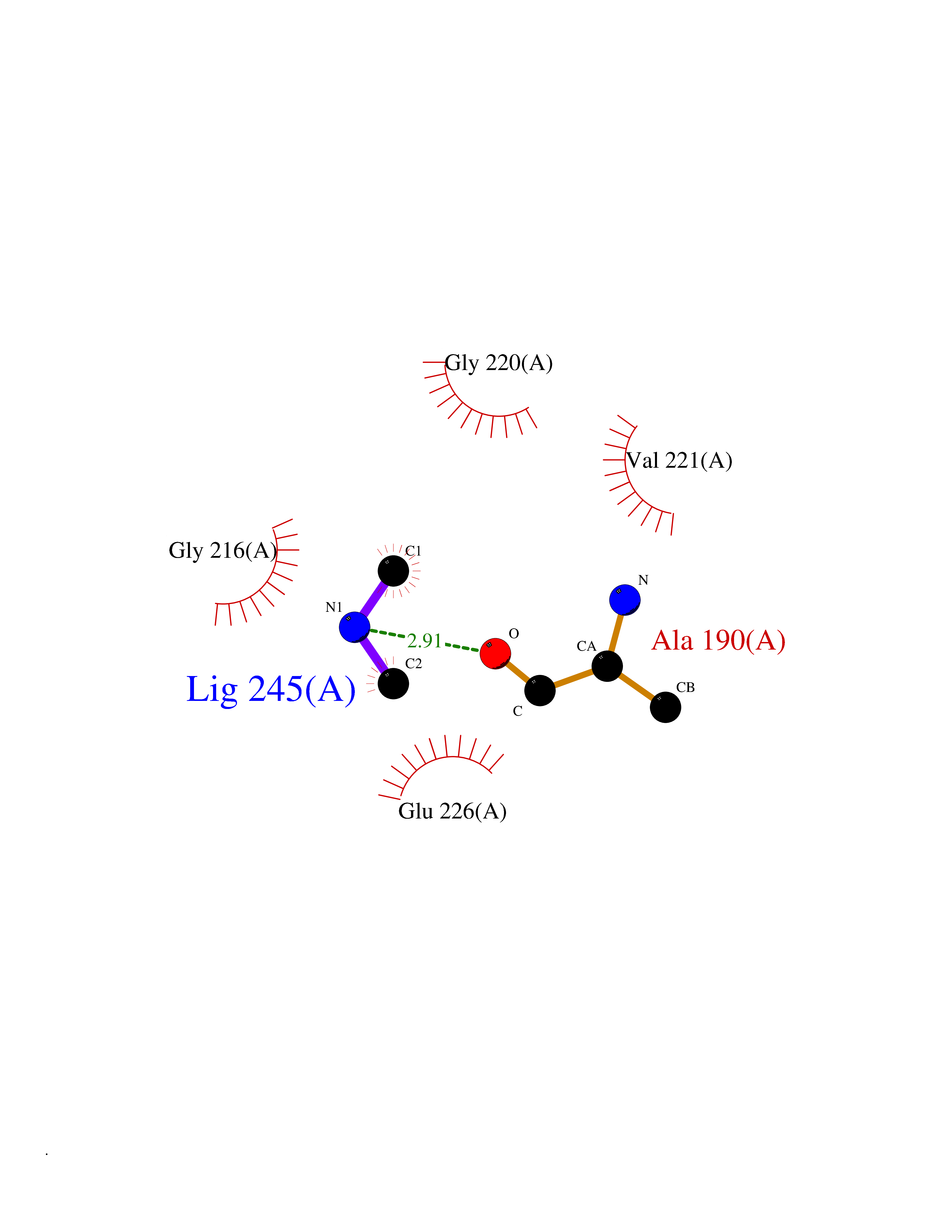 Ligplot Map 3D Binding mode Sequence IIGGRESRPHSRPYMAYLQIQSPAGQSRCGGFLVREDFVLTAAHCWGSNINVTLGAHNIQRRENTQQHITARRAIRHPQYNQRTIQNDIMLLQLSRRVRRNRNVNPVALPRAQEGLRPGTLCTVAGWGRVSMRRGTDTLREVQLRVQRDRQCLRIFGSYDPRRQICVGDRRERKAAFKGDSGGPLLCNNVAHGIVSYGKSSGVPPEVFTRVSSFLPWIRTTMR Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Enoyl-[acyl-carrier-protein] reductase [NADH] FabI | 2PD4 | 4.01 | |
Target general information Gen name fabI Organism Helicobacter pylori (strain ATCC 700392 / 26695) (Campylobacter pylori) Uniprot ID TTD ID NA Synonyms HP_0195 Protein family Short-chain dehydrogenases/reductases (SDR) family, FabI subfamily Biochemical class Oxidoreductase Function Enoyl-[acyl-carrier-protein] reductase (NADH) activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB04393; DB08604 Interacts with NA EC number 1.3.1.9 Uniprot keywords 3D-structure; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; NAD; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27671.5 Length 256 Aromaticity 0.09 Instability index 24 Isoelectric point 8.49 Charge (pH=7) 2.38 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GFLKGKKGLIVGVANNKSIAYGIAQSCFNQGATLAFTYLNESLEKRVRPIAQELNSPYVYELDVSKEEHFKSLYNSVKKDLGSLDFIVHSVAFAPKEALEGSLLETSKSAFNTAMEISVYSLIELTNTLKPLLNNGASVLTLSYLGSTKYMAHYNVMGLAKAALESAVRYLAVDLGKHHIRVNALSAGPIRTLASSGIADFRMILKWNEINAPLRKNVSLEEVGNAGMYLLSSLSSGVSGEVHFVDAGYHVMGMGA Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Neprilysin | 1R1H | 4.01 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms EPN Protein family Peptidase M13 family Biochemical class Hydrolase Function Endopeptidase activity.Exopeptidase activity.Metalloendopeptidase activity.Metallopeptidase activity.Peptide binding.Zinc ion binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map  Pymol Map 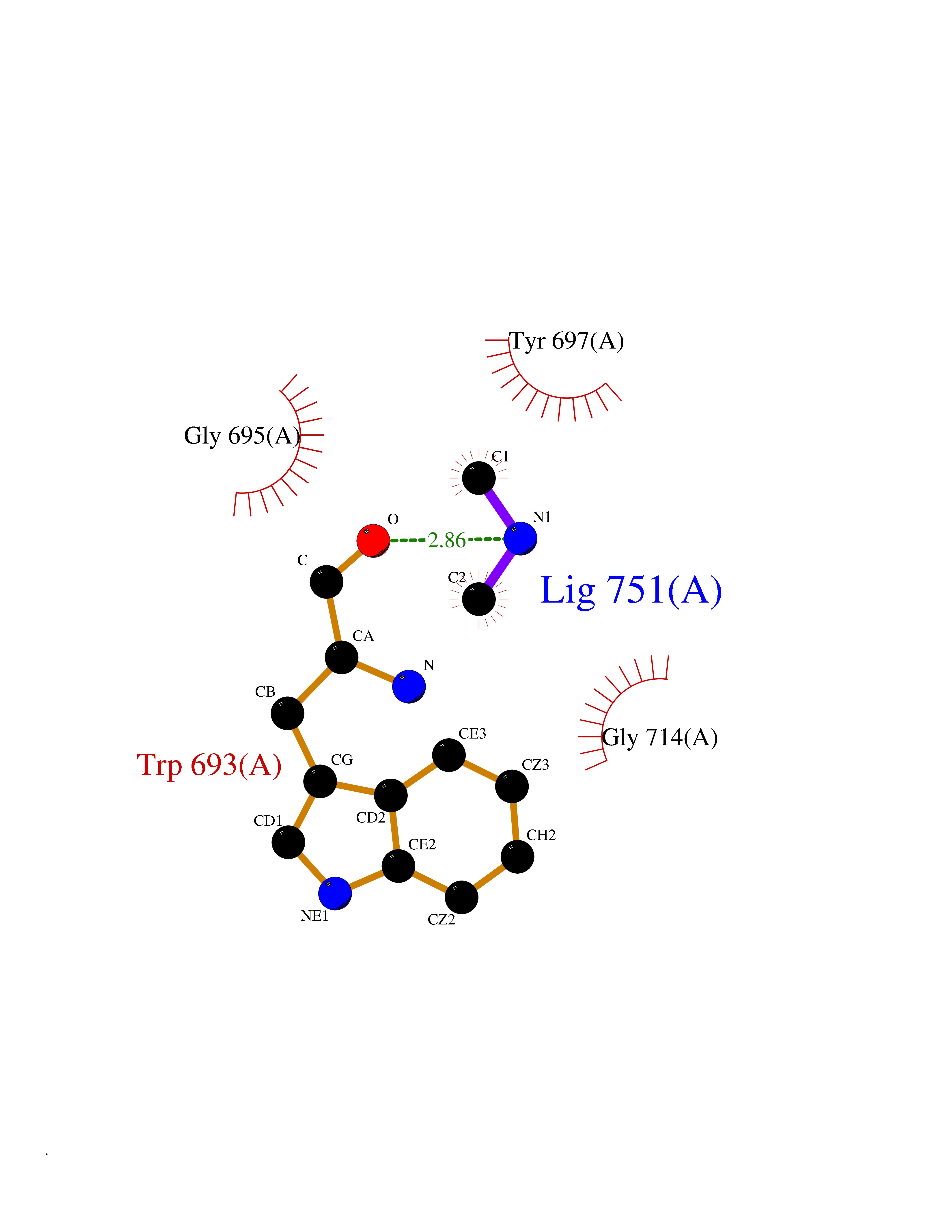 Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Leukotriene A-4 hydrolase (LTA4H) | 3U9W | 4.01 | |
Target general information Gen name LTA4H Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Leukotriene A4 hydrolase; Leukotriene A(4)Leukotriene A-4 hydrolase hydrolase; Leukotriene A(4) hydrolase; LTA4; LTA-H; LTA-4hydrolase; LTA-4 hydrolase Protein family Peptidase M1 family Biochemical class Ether bond hydrolase Function Has also aminopeptidase activity. Epoxide hydrolase that catalyzes the final step in the biosynthesis of the proinflammatory mediator leukotriene B4. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07102; DB06917; DB07258; DB07094; DB07259; DB02352; DB07292; DB07104; DB06828; DB08466; DB01197; DB05177; DB03366; DB08040; DB06851; DB02062; DB07099; DB07260; DB07196; DB11781; DB03424; DB07237 Interacts with Q9BSI4 EC number EC 3.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Leukotriene biosynthesis; Lipid metabolism; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 68927 Length 608 Aromaticity 0.1 Instability index 38.84 Isoelectric point 5.87 Charge (pH=7) -9.86 2D Binding mode Binding energy (Kcal/mol) -5.46 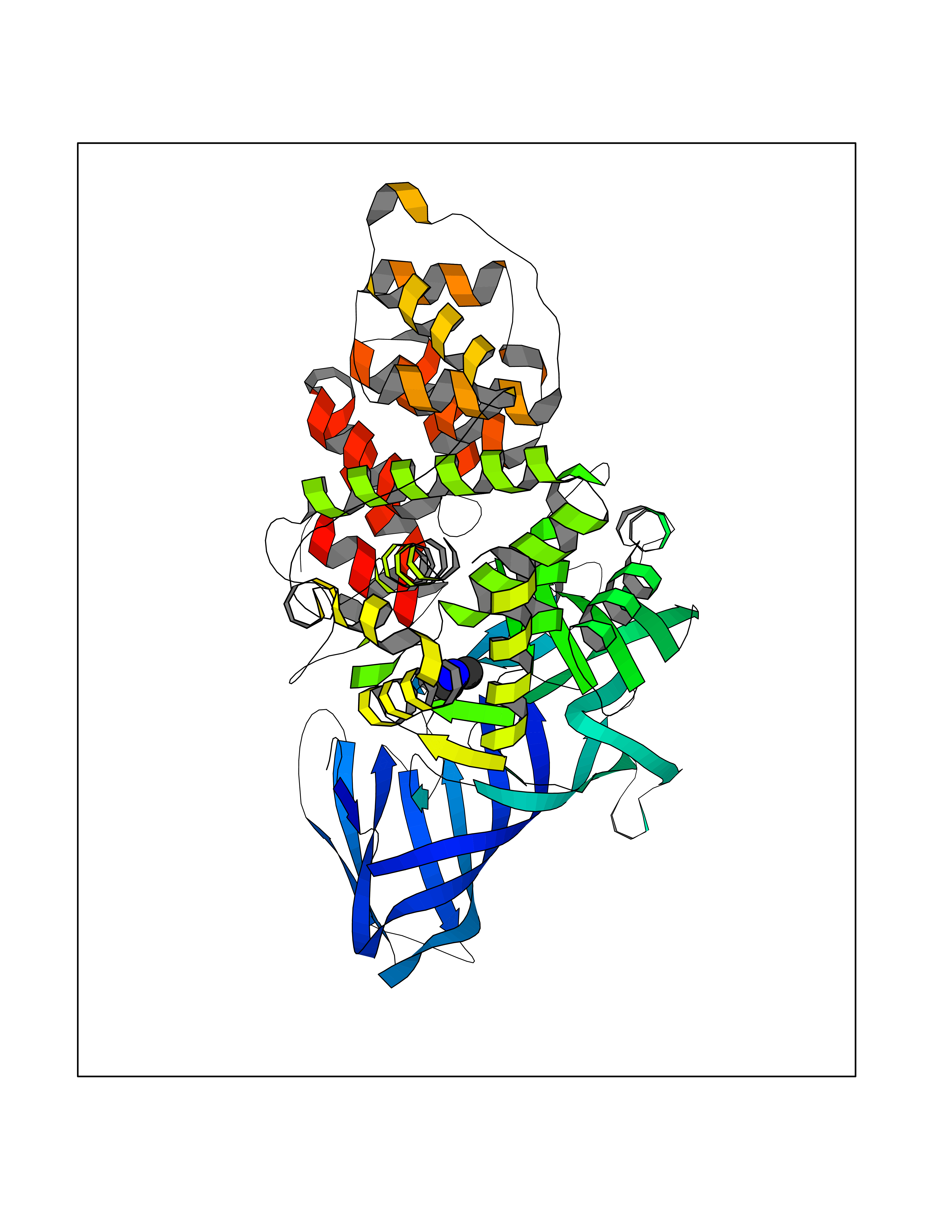 Molscript Map 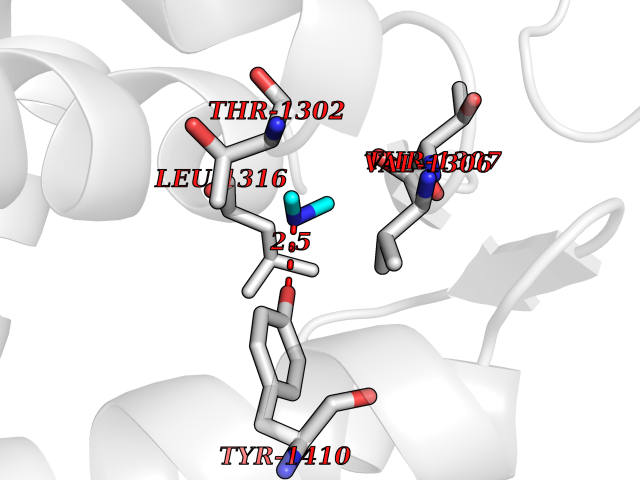 Pymol Map 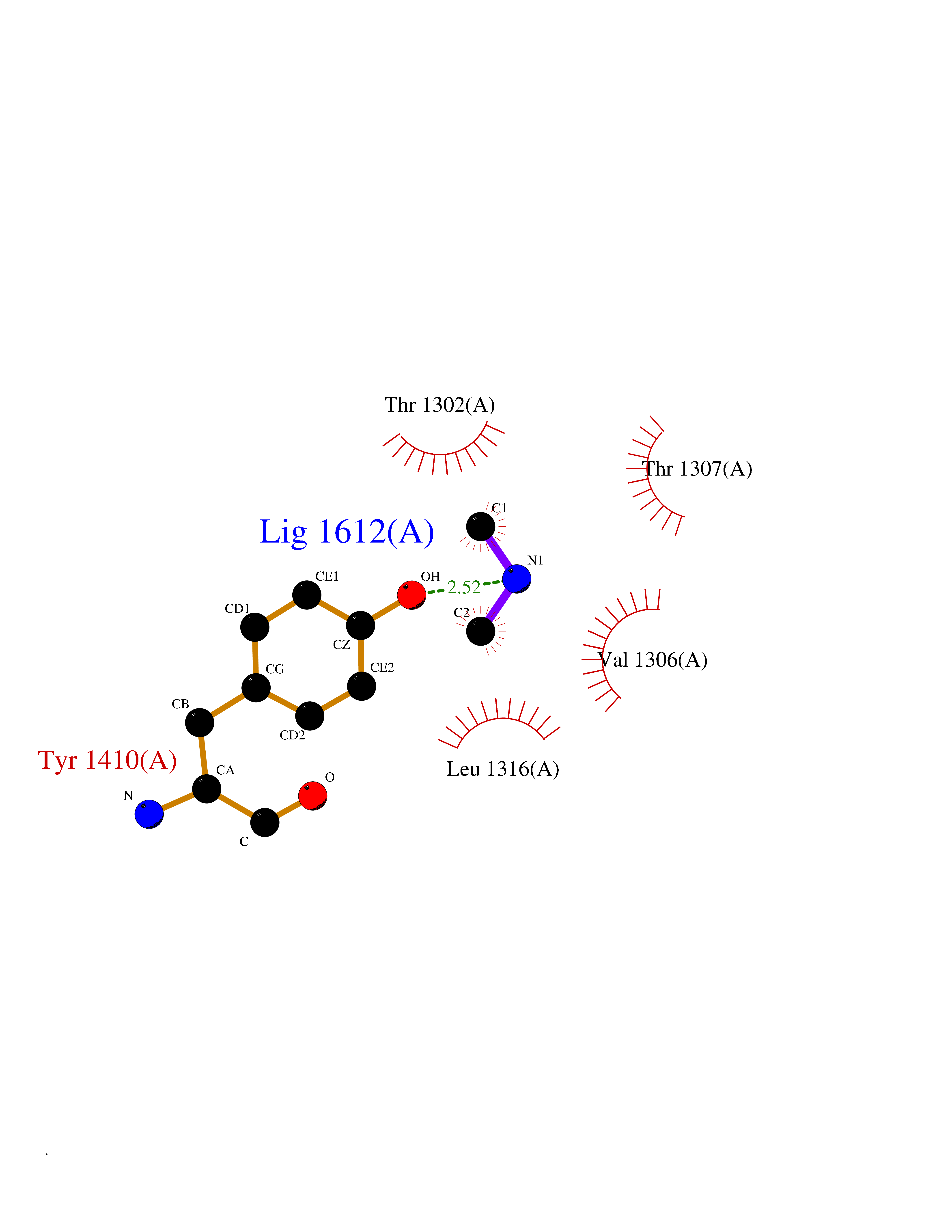 Ligplot Map 3D Binding mode Sequence IVDTCSLASPASVCRTKHLHLRCSVDFTRRTLTGTAALTVQSQEDNLRSLVLDTKDLTIEKVVINGQEVKYALGERQSYKGSPMEISLPIALSKNQEIVIEISFETSPKSSALQWLTPEQTSGKEHPYLFSQCQAIHCRAILPCQDTPSVKLTYTAEVSVPKELVALMSAIRDGETPDPEDPSRKIYKFIQKVPIPCYLIALVVGALESRQIGPRTLVWSEKEQVEKSAYEFSETESMLKIAEDLGGPYVWGQYDLLVLPPSFPYGGMENPCLTFVTPTLLAGDKSLSNVIAHEISHSWTGNLVTNKTWDHFWLNEGHTVYLERHICGRLFGEKFRHFNALGGWGELQNSVKTFGETHPFTKLVVDLTDIDPDVAYSSVPYEKGFALLFYLEQLLGGPEIFLGFLKAYVEKFSYKSITTDDWKDFLYSYFKDKVDVLNQVDWNAWLYSPGLPPIKPNYDMTLTNACIALSQRWITAKEDDLNSFNATDLKDLSSHQLNEFLAQTLQRAPLPLGHIKRMQEVYNFNAINNSEIRFRWLRLCIQSKWEDAIPLALKMATEQGRMKFTRPLFKDLAAFDKSHDQAVRTYQEHKASMHPVTAMLVGKDLKVD Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | DNA topoisomerase II alpha (TOP2A) | 1ZXM | 4.01 | |
Target general information Gen name TOP2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms DNA topoisomerase II, alpha isozyme; DNA topoisomerase 2alpha; DNA topoisomerase 2-alpha Protein family Type II topoisomerase family Biochemical class Topoisomerase Function Topoisomerase II makes double-strand breaks. Essential during mitosis and meiosis for proper segregation of daughter chromosomes. May play a role in regulating the period length of ARNTL/BMAL1 transcriptional oscillation. Control of topological states of DNA by transient breakage and subsequent rejoining of DNA strands. Related diseases A chromosomal aberration involving TOP1 is found in a form of therapy-related myelodysplastic syndrome. Translocation t(11;20)(p15;q11) with NUP98. {ECO:0000269|PubMed:10556215}. Drugs (DrugBank ID) DB05706; DB06013; DB05022; DB06263; DB00276; DB06420; DB04975; DB06362; DB00537; DB00970; DB00694; DB06421; DB00380; DB00997; DB05129; DB00467; DB00445; DB00773; DB09047; DB04576; DB01645; DB01177; DB00978; DB04967; DB01204; DB00218; DB01059; DB01165; DB00487; DB01179; DB05920; DB04978; DB01208; DB00444; DB00685; DB00385; DB06042 Interacts with O14497-1; P38398; P35222; Q05655 EC number EC 5.6.2.3 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Biological rhythms; Cytoplasm; Direct protein sequencing; DNA-binding; Isomerase; Isopeptide bond; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Topoisomerase; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42640.6 Length 373 Aromaticity 0.1 Instability index 33.34 Isoelectric point 8.64 Charge (pH=7) 5.05 2D Binding mode Binding energy (Kcal/mol) -5.47 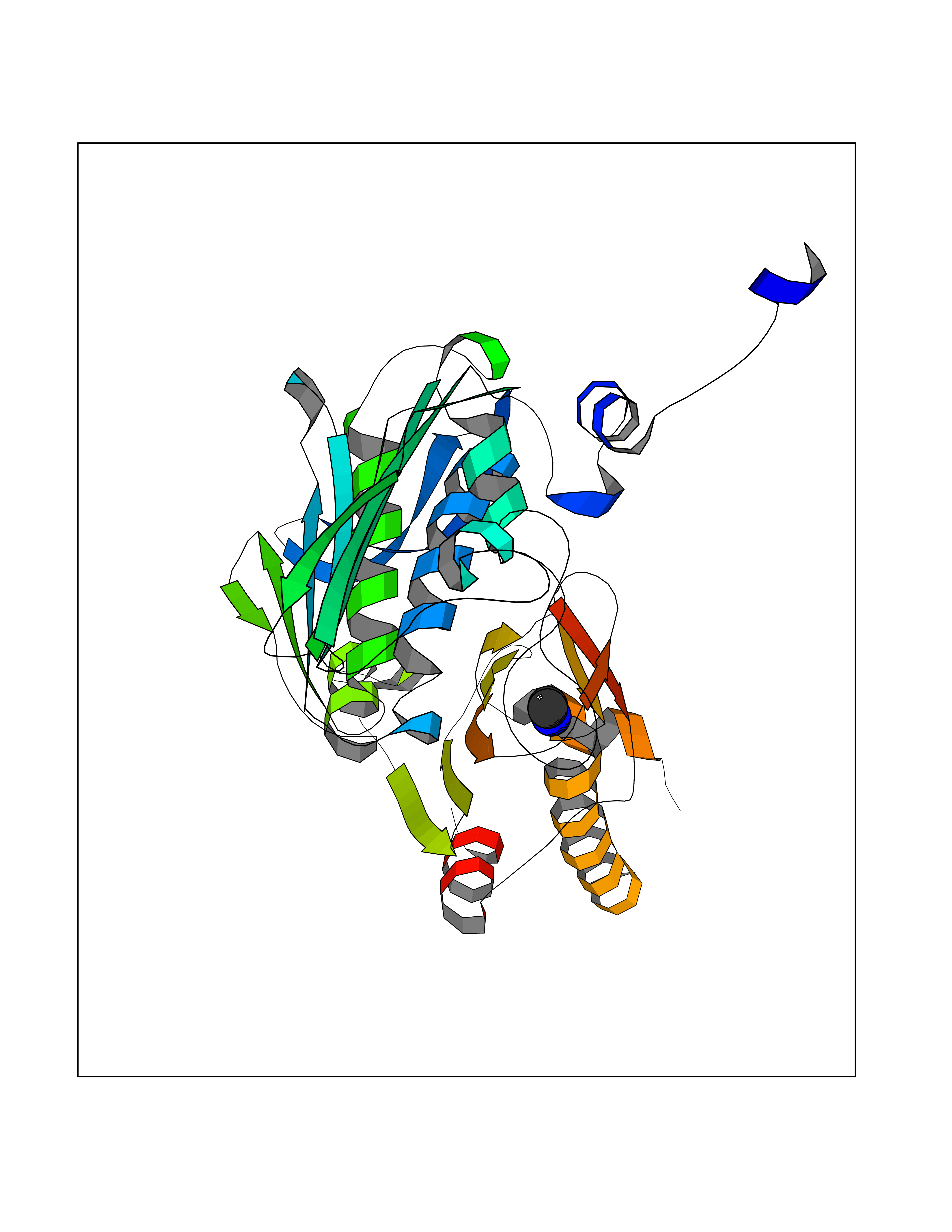 Molscript Map 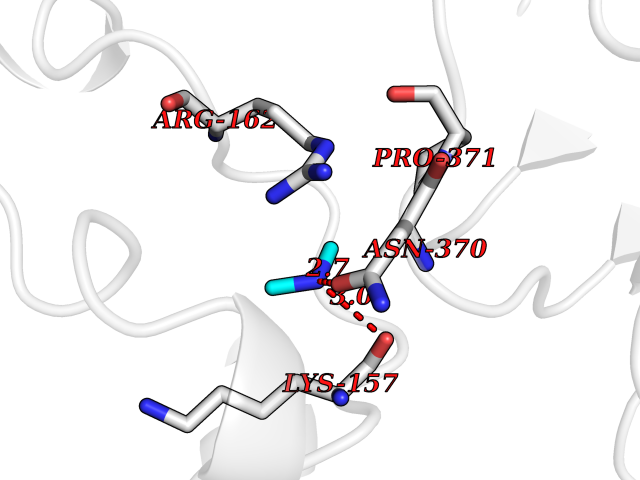 Pymol Map 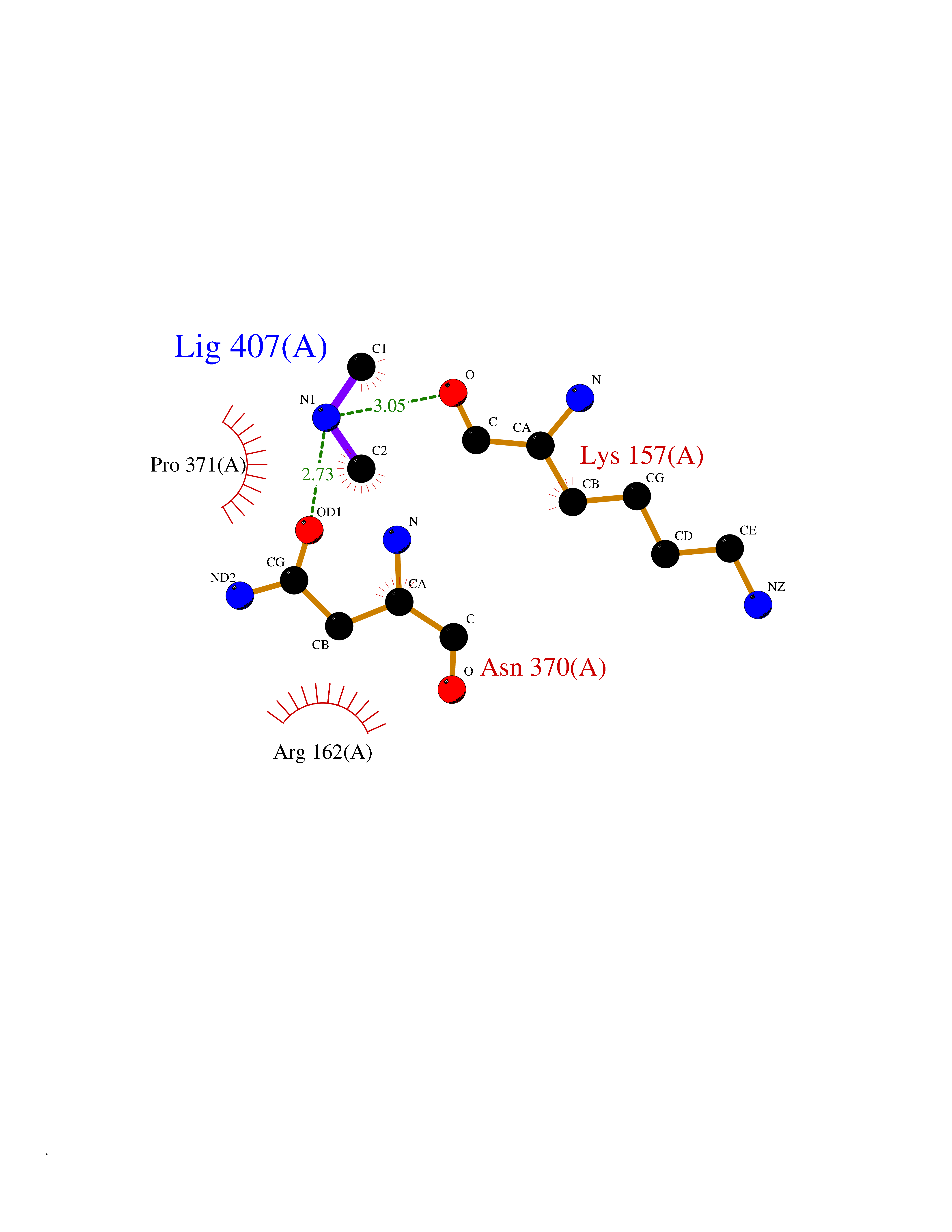 Ligplot Map 3D Binding mode Sequence SVERIYQKKTQLEHILLRPDTYIGSVELVTQQMWVYDEDVGINYREVTFVPGLYKIFDEILVNAADNKQRDPKMSCIRVTIDPENNLISIWNNGKGIPVVEHKVEKMYVPALIFGQLLTSSNYDDDEKKVTGGRNGYGAKLCNIFSTKFTVETASREYKKMFKQTWMDNMGRAGEMELKPFNGEDYTCITFQPDLSKFKMQSLDKDIVALMVRRAYDIAGSTKDVKVFLNGNKLPVKGFRSYVDMYLKDKLDETGNSLKVIHEQVNHRWEVCLTMSEKGFQQISFVNSIATSKGGRHVDYVADQIVTKLVDVVKKKNAVKAHQVKNHMWIFVNALIENPTFDSQTKENMTLQPKSFGSTCQLSEKFIKAAIGC Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Urease subunit alpha | 1FWE | 4.01 | |
Target general information Gen name ureC Organism Klebsiella aerogenes (Enterobacter aerogenes) Uniprot ID TTD ID NA Synonyms NA Protein family Metallo-dependent hydrolases superfamily, Urease alpha subunit family Biochemical class Hydrolase Function Nickel cation binding.Urease activity. Related diseases Can contribute to cancer cell survival, proliferation, migration, and invasion, and tumor angiogenesis and metastasis. May contribute to cancer pathogenesis by promoting inflammatory responses and recruitment of tumor-infiltrating macrophages.; DISEASE: Abnormally high expression of soluble isoforms (isoform 2, isoform 3 or isoform 4) may be a cause of preeclampsia. Drugs (DrugBank ID) DB00551; DB05265 Interacts with P18316 EC number 3.5.1.5 Uniprot keywords 3D-structure; Cytoplasm; Hydrolase; Metal-binding; Nickel Protein physicochemical properties Chain ID C Molecular weight (Da) 80688.3 Length 753 Aromaticity 0.06 Instability index 28.4 Isoelectric point 5.58 Charge (pH=7) -21.17 2D Binding mode Binding energy (Kcal/mol) -5.47 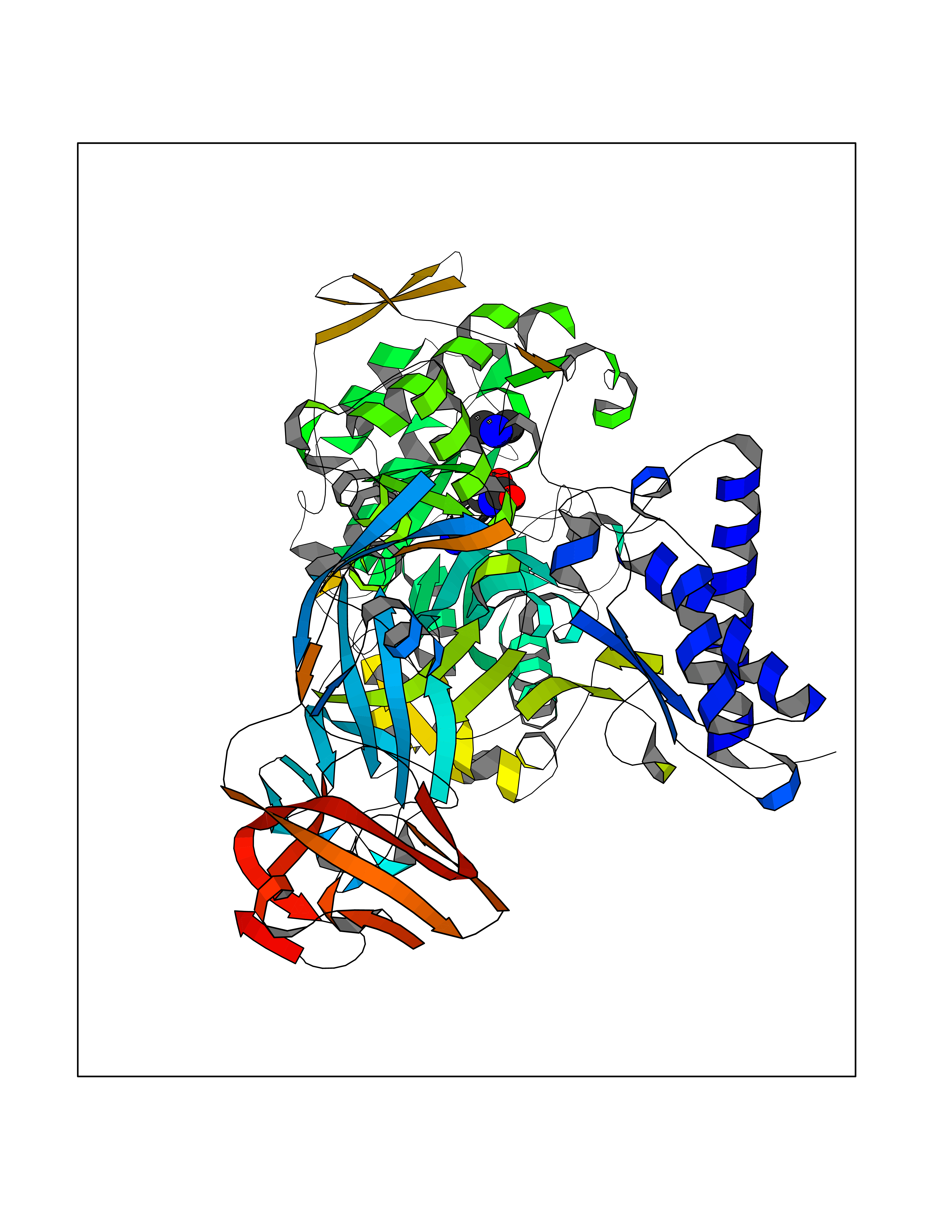 Molscript Map 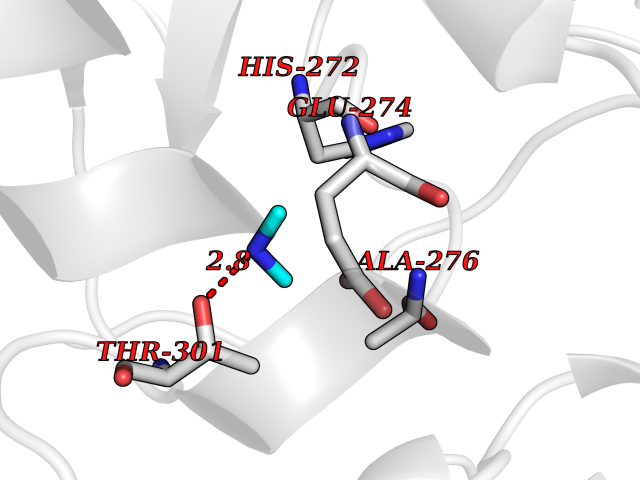 Pymol Map 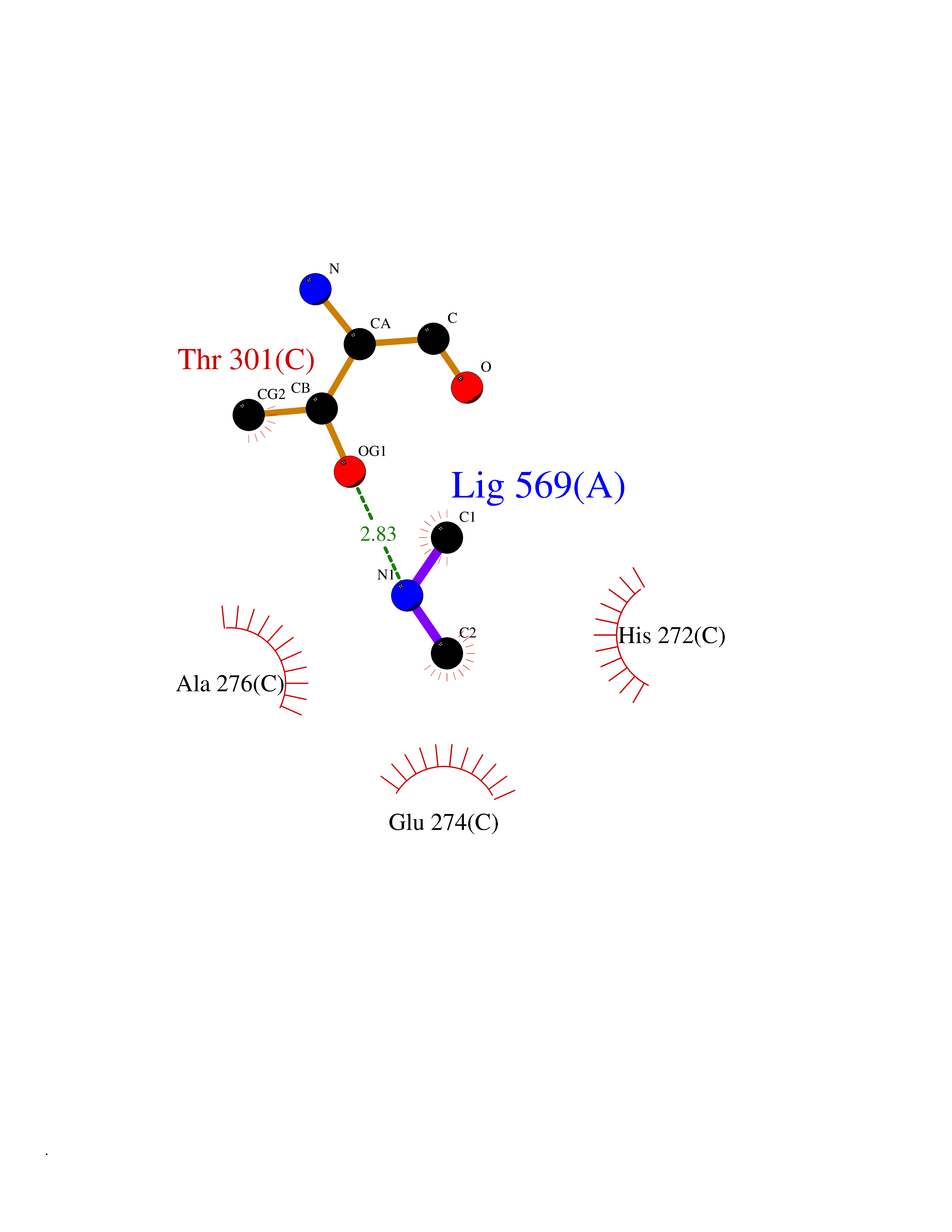 Ligplot Map 3D Binding mode Sequence MELTPREKDKLLLFTAALVAERRLARGLKLNYPESVALISAFIMEGARDGKSVASLMEEGRHVLTREQVMEGVPEMIPDIQVEATFPDGSKLVTVHNPIISNISRQAYADMFGPTVGDKVRLADTELWIEVEDDLTTYGEEVKFGGGKVIRDGMGQGQMLAADCVDLVLTNALIVDHWGIVKADIGVKDGRIFAIGKAGNPDIQPNVTIPIGAATEVIAAEGKIVTAGGIDTHIHWICPQQAEEALVSGVTTMVGGGTGPAAGTHATTCTPGPWYISRMLQAADSLPVNIGLLGKGNVSQPDALREQVAAGVIGLXIHEDWGATPAAIDCALTVADEMDIQVALHSDTLNESGFVEDTLAAIGGRTIHTFHTEGAGGGHAPDIITACAHPNILPSSTNPTLPYTLNTIDEHLDMLMFAESRIRRETIAAEDVLHDLGAFSLTSSDSQAMGRVGEVILRTWQVAHRMKVQRGALAEETGDNDNFRVKRYIAKYTINPALTHGIAHEVGSIEVGKLADLVVWSPAFFGVKPATVIKGGMIAIAPMGDINASIPTPQPVHYRPMFGALGSARHHCRLTFLSQAAAANGVAERLNLRSAIAVVKGCRTVQKADMVHNSLQPNITVDAQTYEVRVDGELITSEPADVLPMAQRYFLFMIPGEYHVKPGQIALNTGRATCRVVVENHGDRPIQVGSHYHFAEVNPALKFDRQQAAGYRLNIPAGTAVRFEPGQKREVELVAFAGHRAVFGFRGEVMGPL Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Isovaleryl-CoA dehydrogenase, mitochondrial | 1IVH | 4.01 | |
Target general information Gen name IVD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Isovaleryl-CoA dehydrogenase activity. Related diseases Isovaleric acidemia (IVA) [MIM:243500]: A metabolic disorder characterized by retarded psychomotor development, a peculiar odor resembling sweaty feet, an aversion to dietary protein, and pernicious vomiting, leading to acidosis and coma. The acute neonatal form leads to massive metabolic acidosis from the first days of life and rapid death. {ECO:0000269|PubMed:2063866, ECO:0000269|PubMed:22004070, ECO:0000269|PubMed:22350545, ECO:0000269|PubMed:23587913, ECO:0000269|PubMed:28535199, ECO:0000269|PubMed:9665741}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04036; DB03147 Interacts with Q08043; Q9Y4H4 EC number 1.3.8.1; 1.3.8.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; FAD; Fatty acid metabolism; Flavoprotein; Lipid metabolism; Mitochondrion; Oxidoreductase; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 84454.2 Length 774 Aromaticity 0.08 Instability index 30.01 Isoelectric point 6.85 Charge (pH=7) -0.77 2D Binding mode Binding energy (Kcal/mol) -5.47 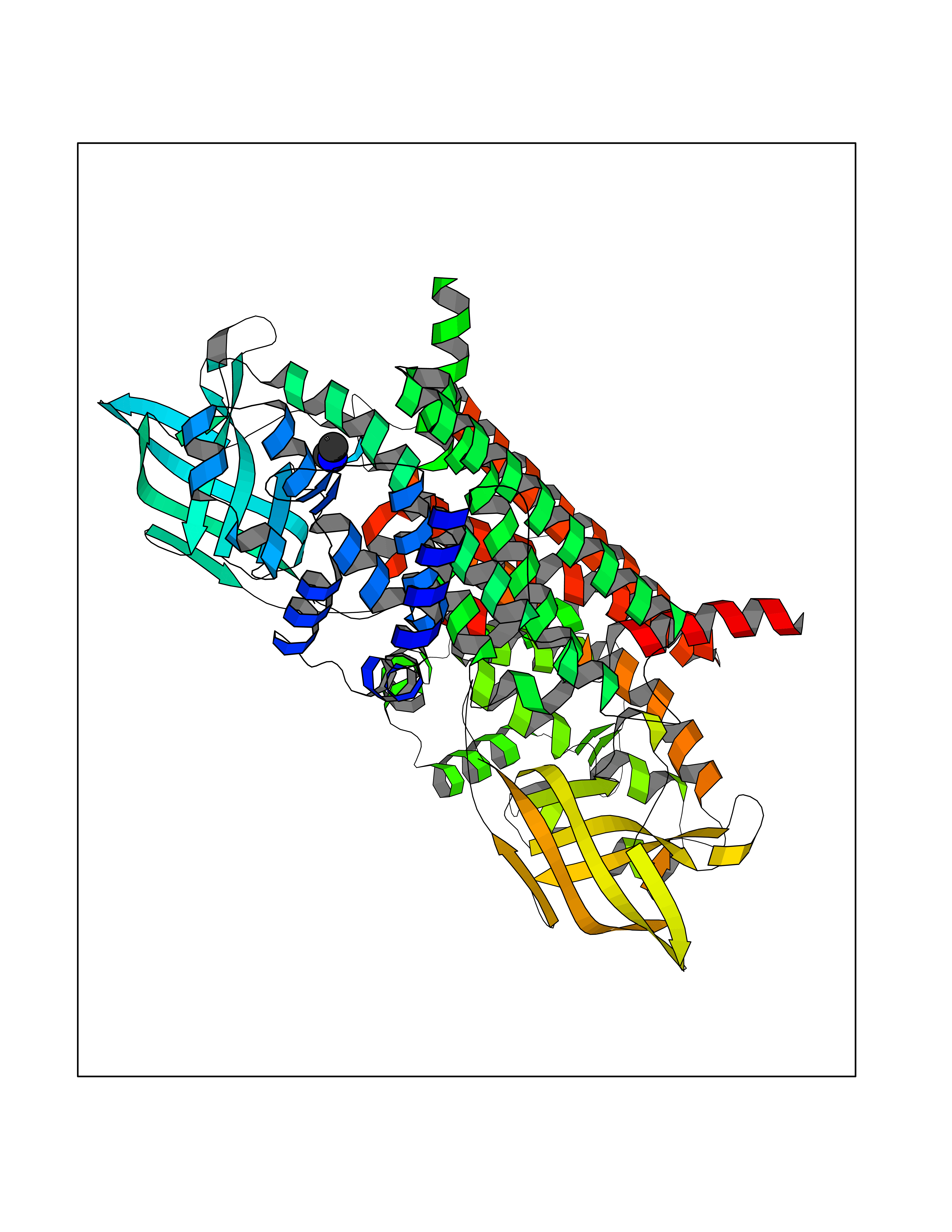 Molscript Map 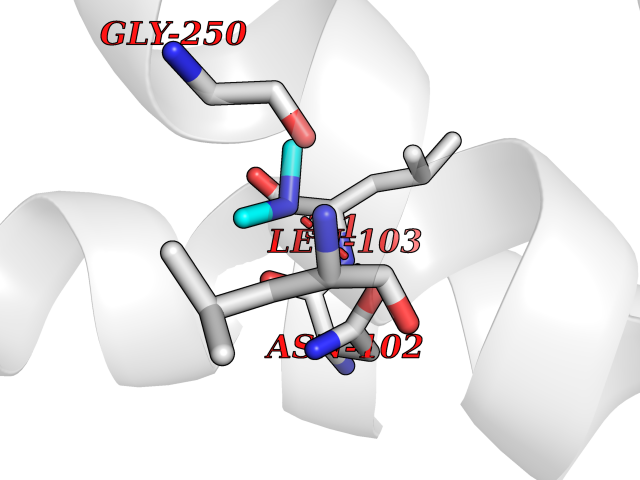 Pymol Map 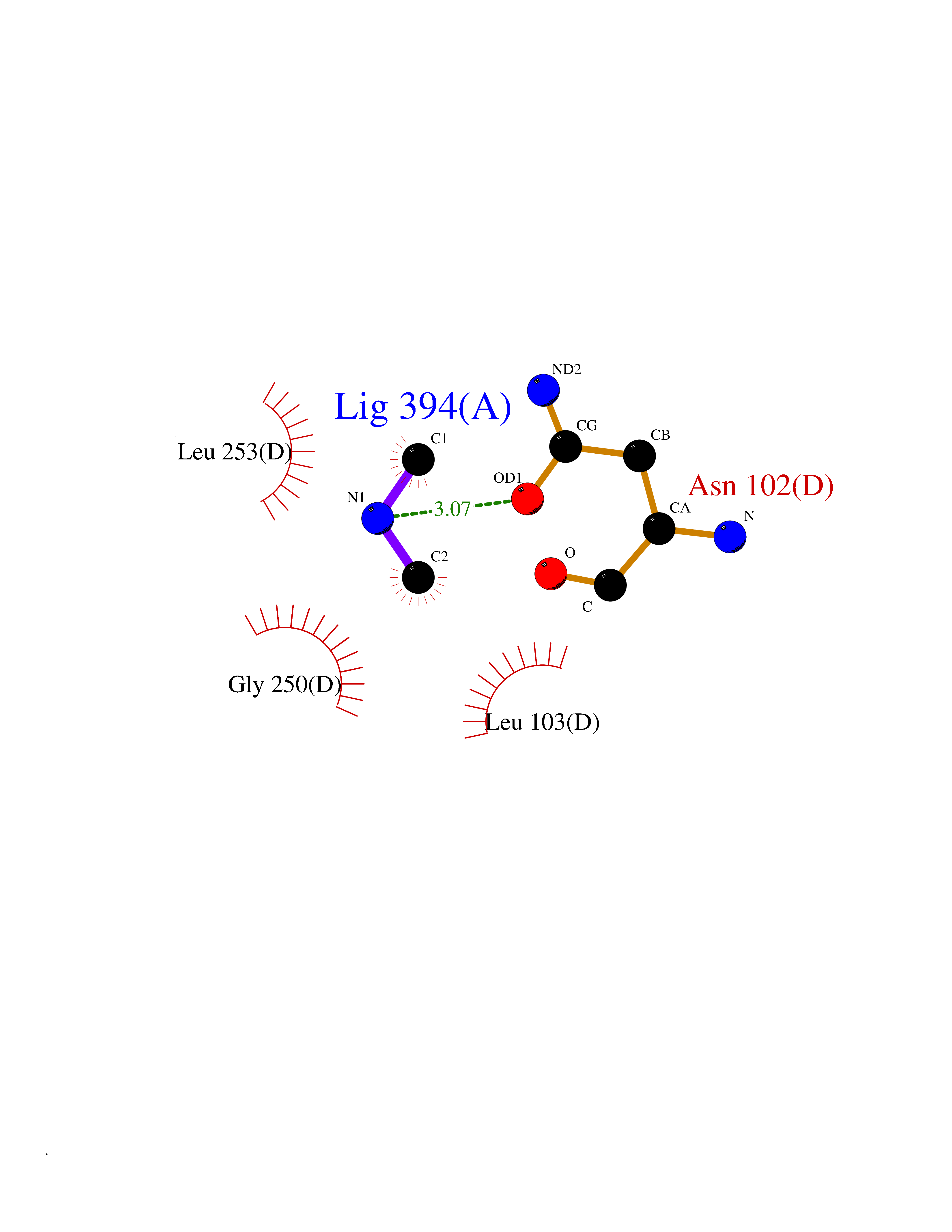 Ligplot Map 3D Binding mode Sequence VDDAINGLSEEQRQLRQTMAKFLQEHLAPKAQEIDRSNEFKNLREFWKQLGNLGVLGITAPVQYGGSGLGYLEHVLVMEEISRASGAVGLSYGAHSNLCINQLVRNGNEAQKEKYLPKLISGEYIGALAMSEPNAGSDVVSMKLKAEKKGNHYILNGNKFWITNGPDADVLIVYAKTDLAAVPASRGITAFIVEKGMPGFSTSKKLDKLGMRGSNTCELIFEDCKIPAANILGHENKGVYVLMSGLDLERLVLAGGPLGLMQAVLDHTIPYLHVREAFGQKIGHFQLMQGKMADMYTRLMACRQYVYNVAKACDEGHCTAKDCAGVILYSAECATQVALDGIQCFGGNGYINDFPMGRFLRDAKLYEIGAGTSEVRRLVIGRAFNADVDDAINGLSEEQRQLRQTMAKFLQEHLAPKAQEIDRSNEFKNLREFWKQLGNLGVLGITAPVQYGGSGLGYLEHVLVMEEISRASGAVGLSYGAHSNLCINQLVRNGNEAQKEKYLPKLISGEYIGALAMSEPNAGSDVVSMKLKAEKKGNHYILNGNKFWITNGPDADVLIVYAKTDLAAVPASRGITAFIVEKGMPGFSTSKKLDKLGMRGSNTCELIFEDCKIPAANILGHENKGVYVLMSGLDLERLVLAGGPLGLMQAVLDHTIPYLHVREAFGQKIGHFQLMQGKMADMYTRLMACRQYVYNVAKACDEGHCTAKDCAGVILYSAECATQVALDGIQCFGGNGYINDFPMGRFLRDAKLYEIGAGTSEVRRLVIGRAFNAD Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Kynureninase (KYNU) | 3E9K | 4.01 | |
Target general information Gen name KYNU Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms L-kynurenine hydrolase; KYNU Protein family Kynureninase family Biochemical class Carbon-carbon bonds hydrolase Function Catalyzes the cleavage of L-kynurenine (L-Kyn) and L-3- hydroxykynurenine (L-3OHKyn) into anthranilic acid (AA) and 3- hydroxyanthranilic acid (3-OHAA), respectively. Has a preference for the L-3-hydroxy form. Also has cysteine-conjugate-beta-lyase activity. Related diseases Hydroxykynureninuria (HYXKY) [MIM:236800]: An inborn error of amino acid metabolism characterized by massive urinary excretion of large amounts of kynurenine, 3-hydroxykynurenine and xanthurenic acid. Affected individuals manifest renal tubular dysfunction, metabolic acidosis, psychomotor retardation, non-progressive encephalopathy, and muscular hypertonia. {ECO:0000269|PubMed:17334708, ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Vertebral, cardiac, renal, and limb defects syndrome 2 (VCRL2) [MIM:617661]: An autosomal recessive congenital malformation syndrome characterized by vertebral segmentation abnormalities, congenital cardiac defects, renal defects, and distal mild limb defects. {ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00160; DB07069; DB00114 Interacts with Q8WUE5; P56545-3; Q9NVL1-2; P61968; P59942; Q8TDC0; P78356-2; Q86WH2 EC number EC 3.7.1.3 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Disease variant; Hydrolase; Proteomics identification; Pyridine nucleotide biosynthesis; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 50204.5 Length 446 Aromaticity 0.09 Instability index 39.91 Isoelectric point 6.36 Charge (pH=7) -4.59 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map  Pymol Map 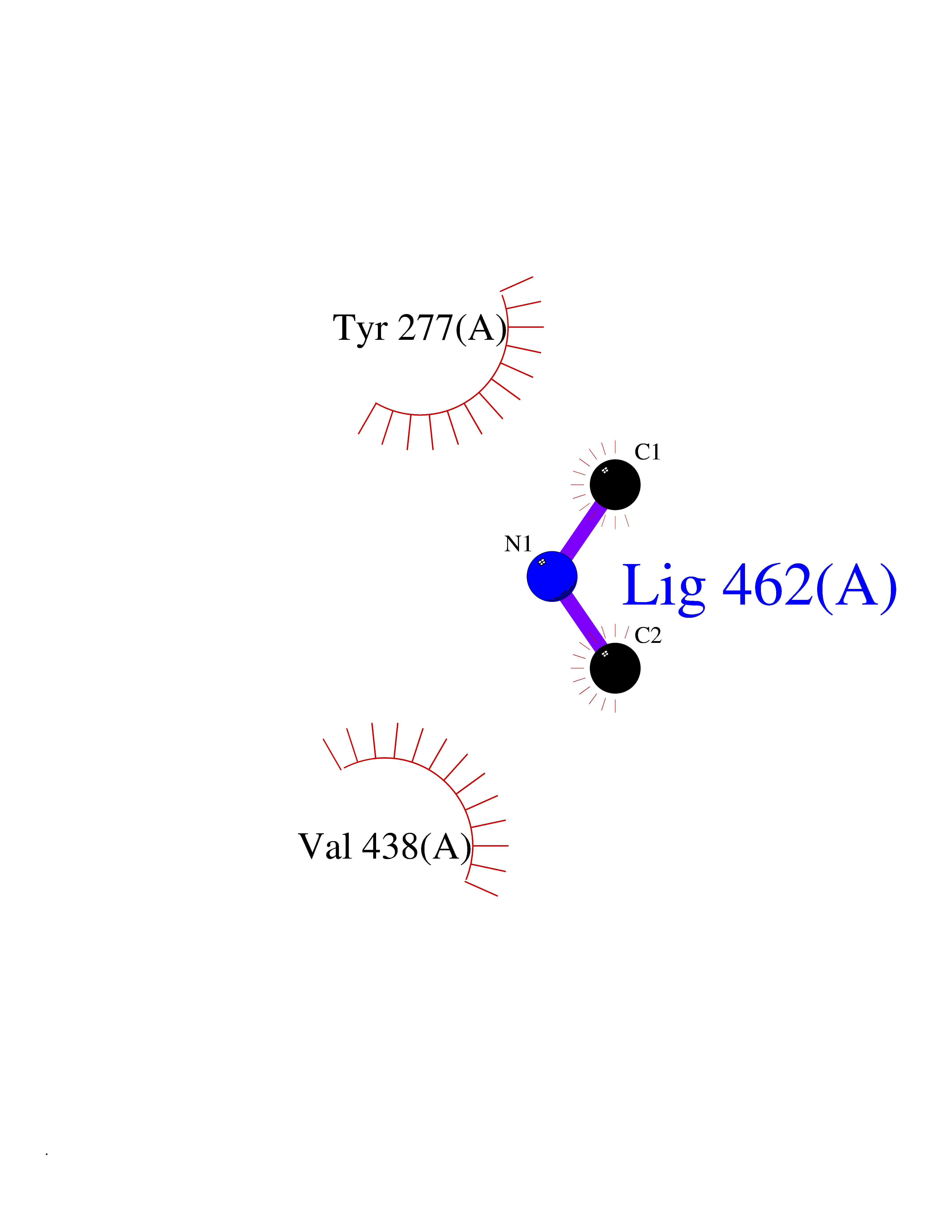 Ligplot Map 3D Binding mode Sequence LELPADTVQRIAAELKCHPTDERVALHLDEEDKLRHFREXFYIPKIQDLPPVDLSLVNKDENAIYFLGNSLGLQPKMVKTYLEEELDKWAKIAAYGHEVGKRPWITGDESIVGLMKDIVGANEKEIALMNALTVNLHLLMLSFFKPTPKRYKILLEAKAFPSDHYAIESQLQLHGLNIEESMRMIKPREGEETLRIEDILEVIEKEGDSIAVILFSGVHFYTGQHFNIPAITKAGQAKGCYVGFDLAHAVGNVELYLHDWGVDFACWCSYKYLNAGAGGIAGAFIHEKHAHTIKPALVGWFGHELSTRFKMDNKLQLIPGVCGFRISNPPILLVCSLHASLEIFKQATMKALRKKSVLLTGYLEYLIKHNYGVVNIITPSHVEERGCQLTITFSVPNKDVFQELEKRGVVCDKRNPNGIRVAPVPLYNSFHDVYKFTNLLTSILDS Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Lanosterol 14-alpha-demethylase (EC 1.14.13.70) | 4G3J | 4.01 | |
Target general information Gen name Tb11.02.4080 Organism Trypanosoma brucei brucei (strain 927/4 GUTat10.1) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class NA Function NA Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) NA Interacts with NA EC number 1.14.13.70 Uniprot keywords 3D-structure; Heme; Iron; Metal-binding; Monooxygenase; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID D Molecular weight (Da) 50691.4 Length 448 Aromaticity 0.08 Instability index 54.09 Isoelectric point 6.99 Charge (pH=7) -0.03 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GKLPPVYPVTVPILGHIIQFGKSPLGFMQECKRQLKSGIFTINIVGKRVTIVGDPHEHSRFFLPRNEVLSPREVYSFMVPVFGEGVAYAAPYPRMREQLNFLAEELTIAKFQNFVPAIQHEVRKFMAANWDKDEGEINLLEDCSTMIINTACQCLFGEDLRKRLDARRFAQLLAKMESSLIPAAVFLPILLKLPLPQSARCHEARTELQKILSEIIIARKEEEVNKDSSTSDLLSGLLSAVYRDGTPMSLHEVCGMIVAAMFAGQHTSSITTTWSMLHLMHPANVKHLEALRKEIEEFPAQLNYNNVMDEMPFAERCARESIRRDPPLLMLMRKVMADVKVGSYVVPKGDIIACSPLLSHHDEEAFPEPRRWDPERDEKVEGAFIGFGAGVHKCIGQKFGLLQVKTILATAFRSYDFQLLRDEVPDPDYHTMVVGPTASQCRVKYIRR Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Alr1529 protein | 1Z8H | 4.01 | |
Target general information Gen name alr1529 Organism Nostoc sp. (strain PCC 7120 / SAG 25.82 / UTEX 2576) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function NA Related diseases Desbuquois dysplasia 1 (DBQD1) [MIM:251450]: A chondrodysplasia characterized by severe prenatal and postnatal growth retardation (less than -5 SD), joint laxity, short extremities, progressive scoliosis, round face, midface hypoplasia, prominent bulging eyes. The main radiologic features are short long bones with metaphyseal splay, a 'Swedish key' appearance of the proximal femur (exaggerated trochanter), and advance carpal and tarsal bone age. Two forms of Desbuquois dysplasia are distinguished on the basis of the presence or absence of characteristic hand anomalies: an extra ossification center distal to the second metacarpal, delta phalanx, bifid distal thumb phalanx, and phalangeal dislocations. {ECO:0000269|PubMed:19853239, ECO:0000269|PubMed:20425819, ECO:0000269|PubMed:21037275, ECO:0000269|PubMed:21412251, ECO:0000269|PubMed:21654728, ECO:0000269|PubMed:22539336}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epiphyseal dysplasia, multiple, 7 (EDM7) [MIM:617719]: A form of multiple epiphyseal dysplasia, a generalized skeletal dysplasia associated with significant morbidity. Joint pain, joint deformity, waddling gait, and short stature are the main clinical signs and symptoms. Radiological examination of the skeleton shows delayed, irregular mineralization of the epiphyseal ossification centers and of the centers of the carpal and tarsal bones. Multiple epiphyseal dysplasia is broadly categorized into the more severe Fairbank and the milder Ribbing types. The Fairbank type is characterized by shortness of stature, short and stubby fingers, small epiphyses in several joints, including the knee, ankle, hand, and hip. The Ribbing type is confined predominantly to the hip joints and is characterized by hands that are normal and stature that is normal or near-normal. EDM7 inheritance is autosomal recessive. {ECO:0000269|PubMed:28742282}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 45603.2 Length 401 Aromaticity 0.11 Instability index 49.26 Isoelectric point 7.67 Charge (pH=7) 1.26 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 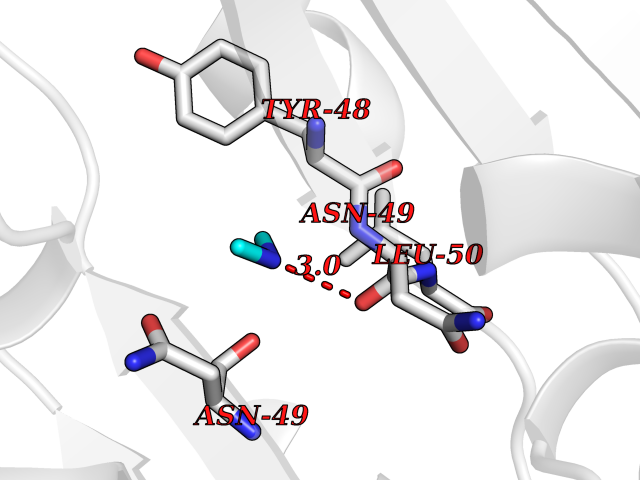 Pymol Map 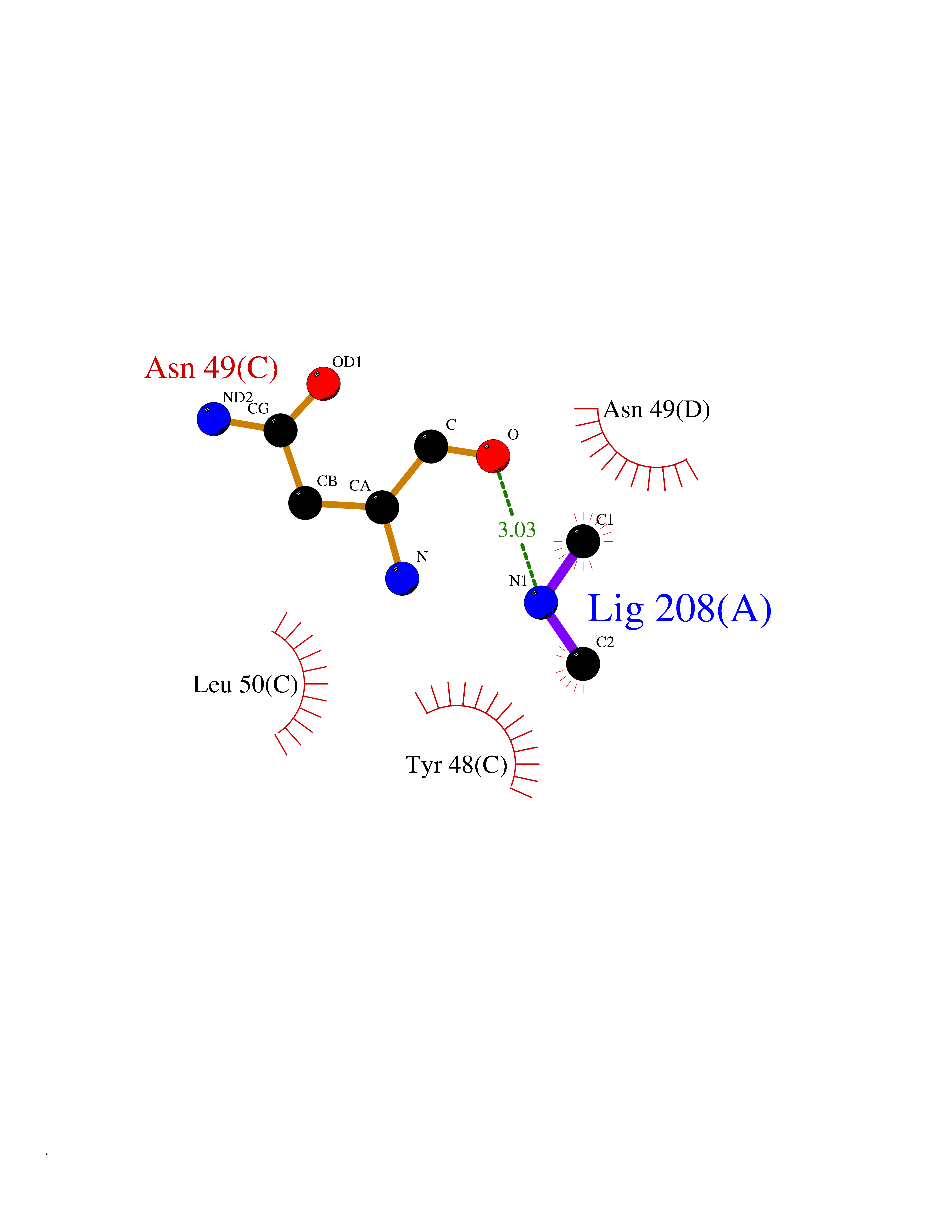 Ligplot Map 3D Binding mode Sequence KTQIRICFVGDSFVNGTGDPECLGWTGRVCVNANKKGYDVTYYNLGIRRDTSSDIAKRWLQEVSLRLHKEYNSLVVFSFGLNDTTLENGKPRVSIAETIKNTREILTQAKKLYPVLXISPAPYIEQQDPGRRRRTIDLSQQLALVCQDLDVPYLDVFPLLEKPSVWLHEAKANDGVHPQAGGYTEFARIVENWDAWLNWFRKTQIRICFVGDSFVNGTGDPECLGWTGRVCVNANKKGYDVTYYNLGIRRDTSSDIAKRWLQEVSLRLHKEYNSLVVFSFGLNDTTLENGKPRVSIAETIKNTREILTQAKKLYPVLXISPAPYIEQQDPGRRRRTIDLSQQLALVCQDLDVPYLDVFPLLEKPSVWLHEAKANDGVHPQAGGYTEFARIVENWDAWLNWF Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Protein cereblon (CRBN) | 5FQD | 4.01 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -5.47 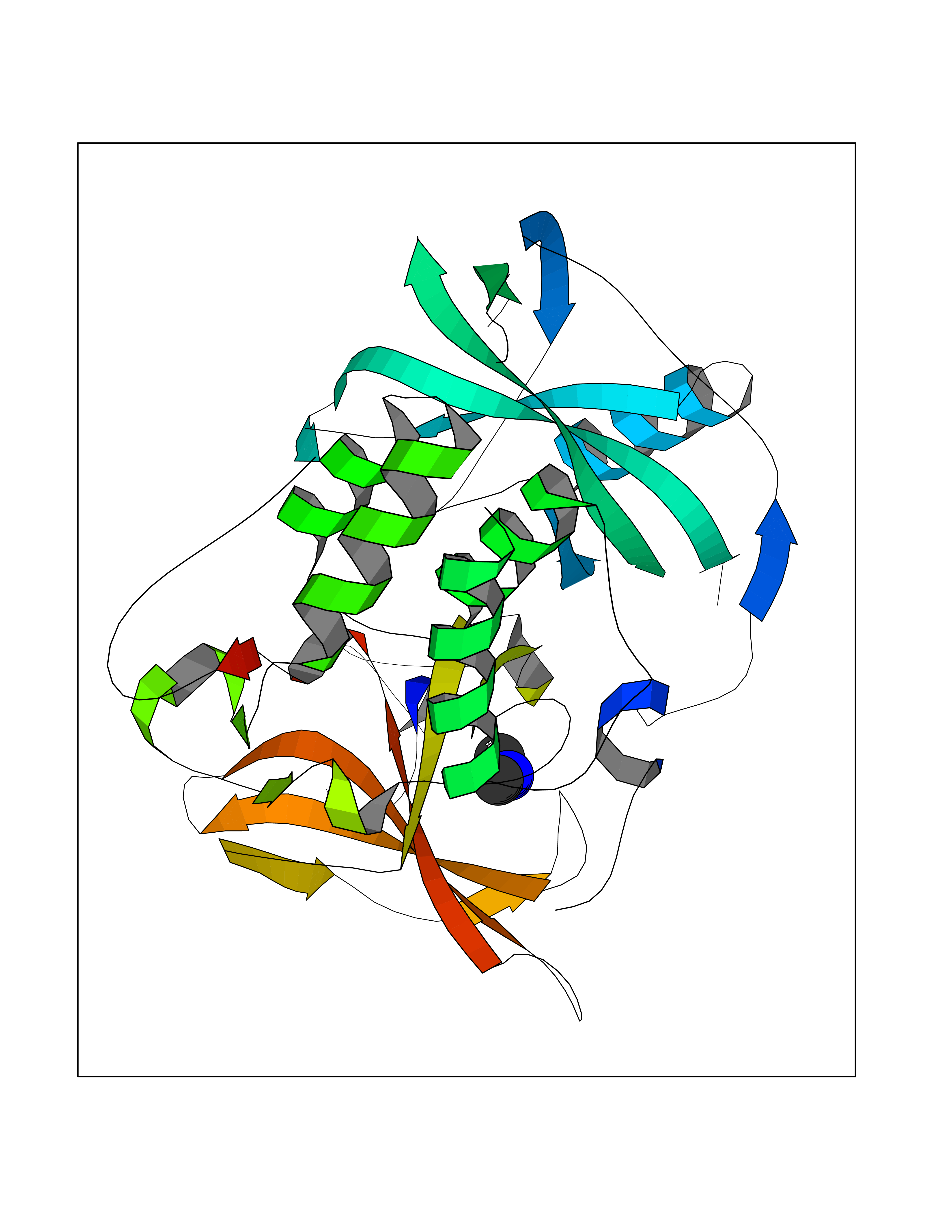 Molscript Map  Pymol Map 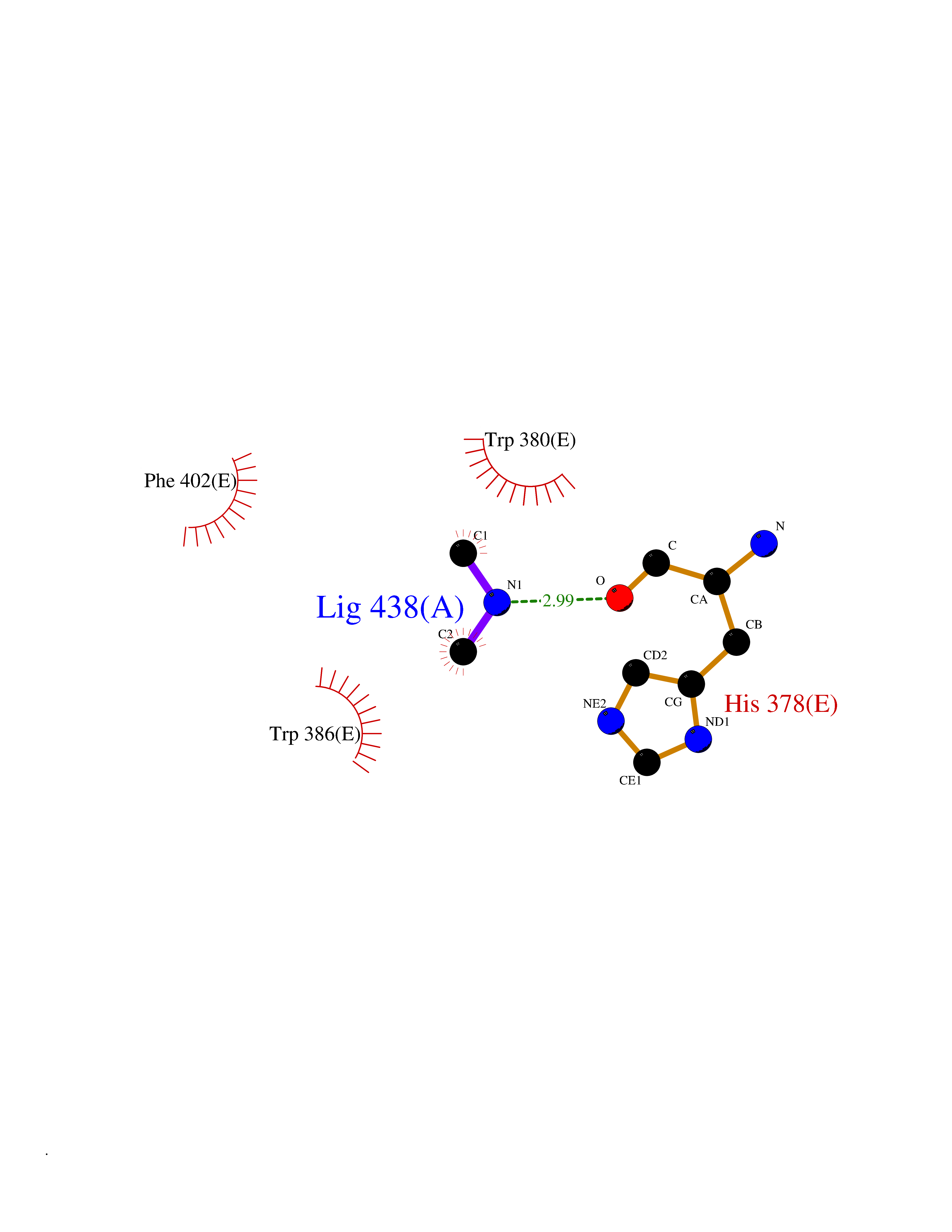 Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Kallikrein-5 (KLK5) | 6QFE | 4.01 | |
Target general information Gen name KLK5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ570/PRO1132; Stratum corneum tryptic enzyme; SCTE; Kallikrein-like protein 2; KLK-L2 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function May be involved in desquamation. Related diseases Lipodystrophy, familial partial, 8 (FPLD8) [MIM:620679]: An autosomal dominant form of partial lipodystrophy, a disorder characterized by abnormal subcutaneous fat distribution. FPLD8 patients show selective loss of subcutaneous adipose tissue from the limbs, beginning around 13 to 15 years of age, and abnormal accumulation of subcutaneous adipose tissue in the dorsal neck and face, as well as in the posterior thoracic and abdominal regions. The disorder is associated with metabolic abnormalities, including diabetes mellitus and hyperlipidemia. {ECO:0000269|PubMed:27376152}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P20930; Q9NQG1 EC number EC 3.4.21.- Uniprot keywords 3D-structure; Disulfide bond; Glycoprotein; Hydrolase; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50299.2 Length 454 Aromaticity 0.07 Instability index 40.74 Isoelectric point 9.25 Charge (pH=7) 23.09 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 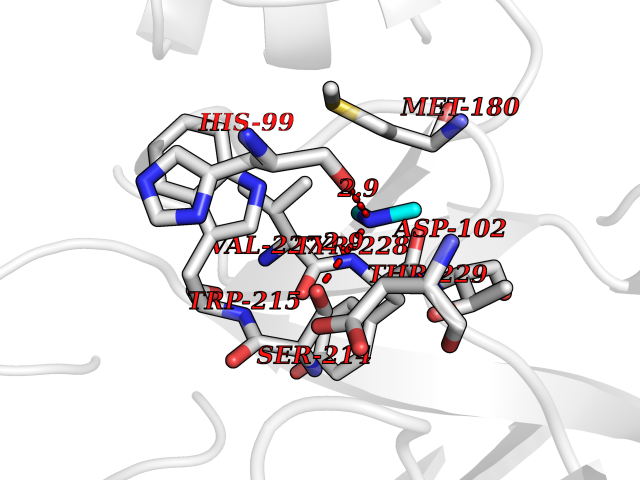 Pymol Map 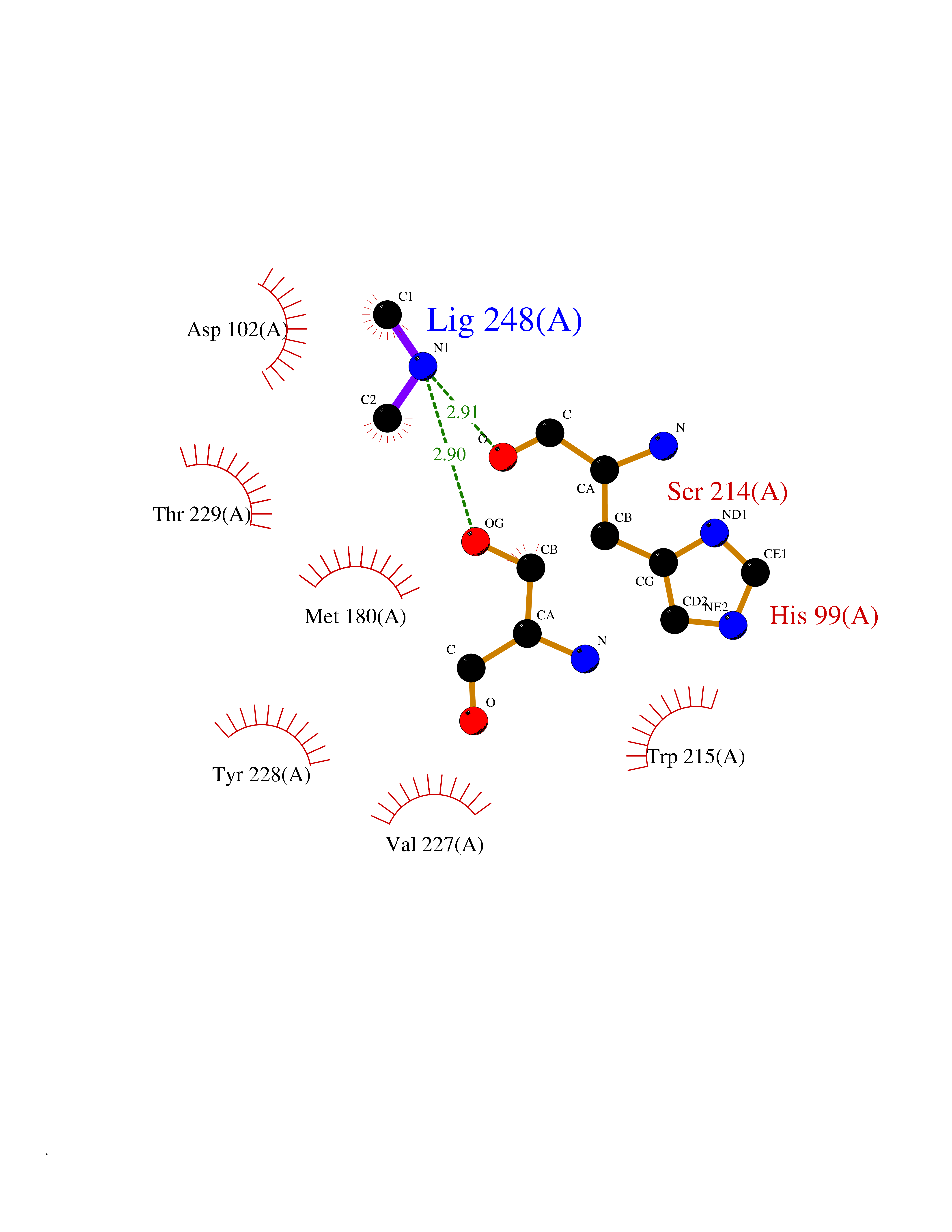 Ligplot Map 3D Binding mode Sequence IINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANSIINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANS Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Orotidine 5'-monophosphate decarboxylase (UMPS) | 3MI2 | 4.01 | |
Target general information Gen name UMPS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Uridine 5'-monophosphate synthase; UMP synthase Protein family Purine/pyrimidine phosphoribosyltransferase family; OMP decarboxylase family Biochemical class Pentosyltransferase Function Catalyses the formation of uridine monophosphate (UMP), an energy-carrying molecule in many important biosynthetic pathways. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02890; DB00544 Interacts with P54764; P11172-1 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Decarboxylase; Disease variant; Glycosyltransferase; Lyase; Multifunctional enzyme; Phosphoprotein; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 55844 Length 514 Aromaticity 0.06 Instability index 22.7 Isoelectric point 6.44 Charge (pH=7) -2.99 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 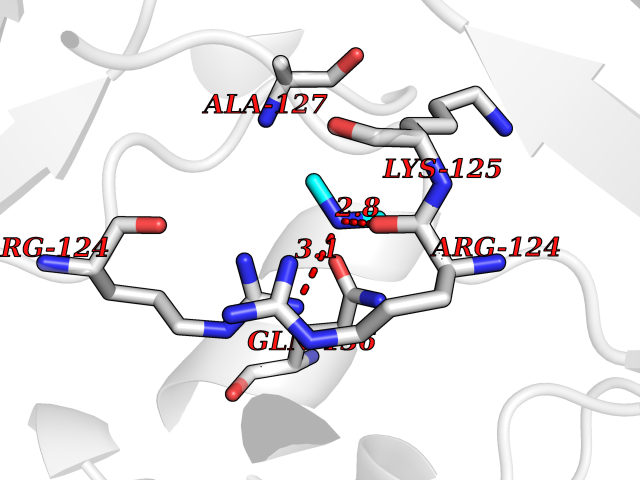 Pymol Map 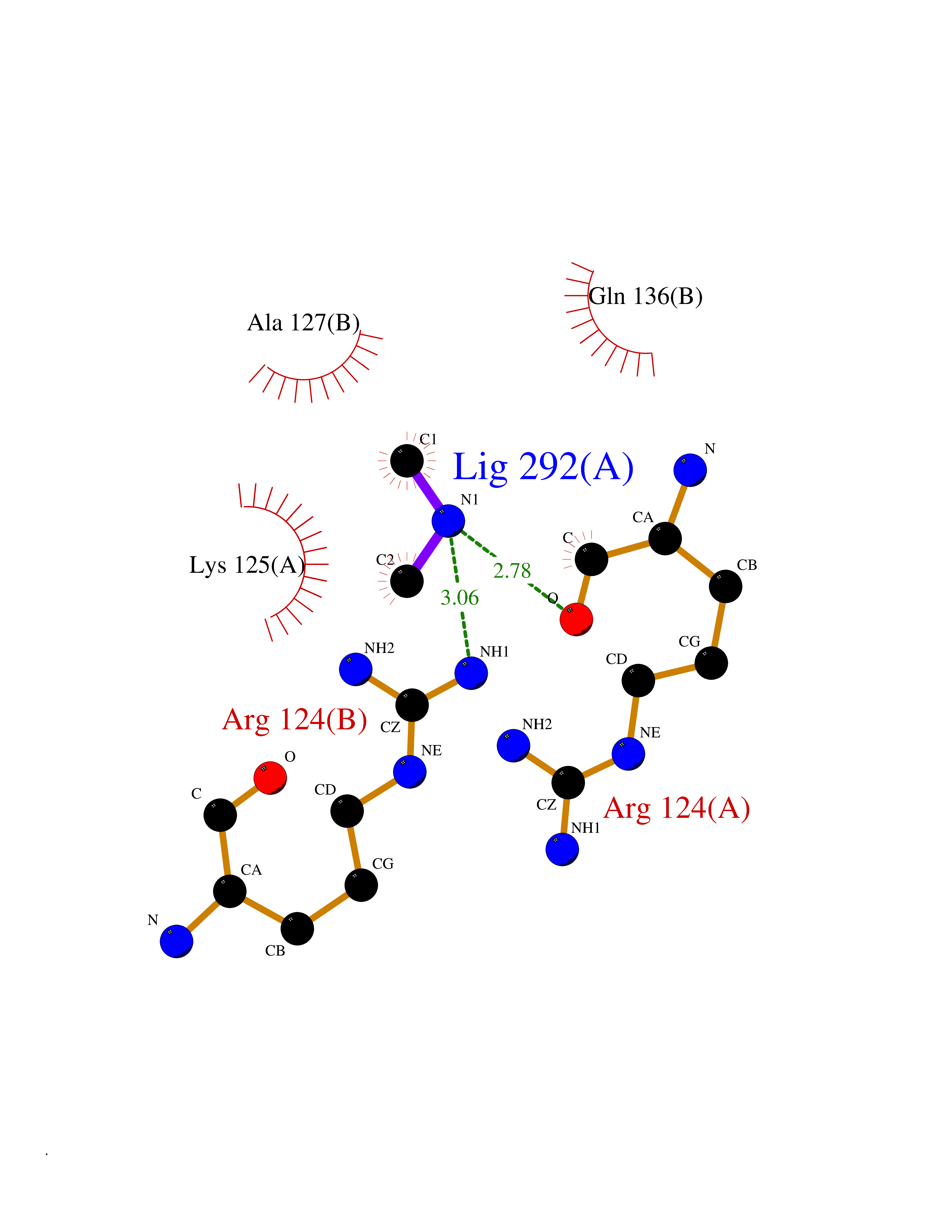 Ligplot Map 3D Binding mode Sequence KELSFGARAELPRIHPVASKLLRLMQKKETNLCLSADVSLARELLQLADALGPSICMLKTHVDILNDFTLDVMKELITLAKXHEFLIFEDRKFADIGNTVKKQYEGGIFKIASWADLVNAHVVPGSGVVKGLQEVGLPLHRGCLLIAEMSSTGSLATGDYTRAAVRMAEEHSEFVVGFISGSRVSMKPEFLHLTPGVQLEAGGDNLGQQYNSPQEVIGKRGSDIIIVGRGIISAADRLEAAEMYRKAAWEAYLSRLGKELSFGARAELPRIHPVASKLLRLMQKKETNLCLSADVSLARELLQLADALGPSICMLKTHVDILNDFTLDVMKELITLAKXHEFLIFEDRKFADIGNTVKKQYEGGIFKIASWADLVNAHVVPGSGVVKGLQEVGLPLHRGCLLIAEMSSTGSLATGDYTRAAVRMAEEHSEFVVGFISGSRVSMKPEFLHLTPGVQLEAGGDNLGQQYNSPQEVIGKRGSDIIIVGRGIISAADRLEAAEMYRKAAWEAYLSRLG Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | N-acetylgalactosamine 6 sulfatase (GALNS) | 4FDJ | 4.01 | |
Target general information Gen name GALNS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms N-acetylgalactosamine-6-sulfate sulfatase; N-acetylgalactosamine-6-sulfatase; Galactose-6-sulfate sulfatase; GalNAc6S sulfatase; GalN6S; Chondroitinsulfatase; Chondroitinase Protein family Sulfatase family Biochemical class Sulfuric ester hydrolase Function Catalyzes the chemical reaction of cleaving off the 6-sulfate groups of the N-acetyl-D-galactosamine 6-sulfate units of the macromolecule chondroitin sulfate and, similarly, of the D-galactose 6-sulfate units of the macromolecule keratan sulfate. Related diseases Mucopolysaccharidosis 4A (MPS4A) [MIM:253000]: A form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life. {ECO:0000269|PubMed:1522213, ECO:0000269|PubMed:16287098, ECO:0000269|PubMed:24726177, ECO:0000269|PubMed:7581409, ECO:0000269|PubMed:7633425, ECO:0000269|PubMed:7668283, ECO:0000269|PubMed:7795586, ECO:0000269|PubMed:8651279, ECO:0000269|PubMed:8826435, ECO:0000269|PubMed:9298823, ECO:0000269|PubMed:9375852, ECO:0000269|PubMed:9521421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09301 Interacts with NA EC number EC 3.1.6.4 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Dwarfism; Glycoprotein; Hydrolase; Lysosome; Metal-binding; Mucopolysaccharidosis; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 55013.8 Length 493 Aromaticity 0.11 Instability index 35.46 Isoelectric point 6.14 Charge (pH=7) -6.48 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 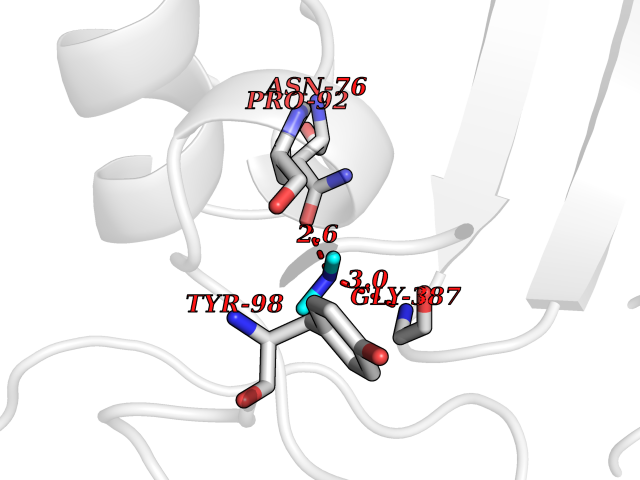 Pymol Map 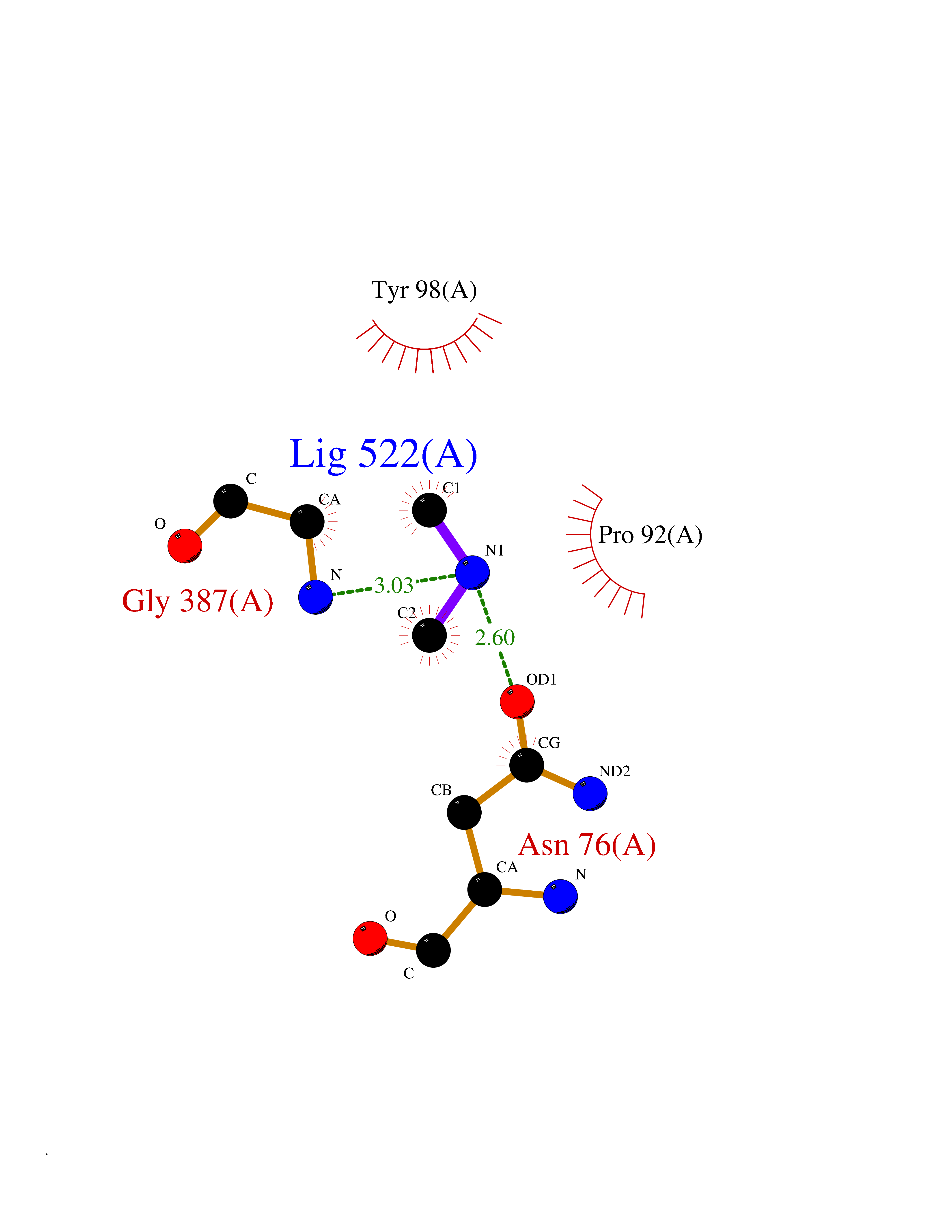 Ligplot Map 3D Binding mode Sequence PQPPNILLLLMDDMGWGDLGVYGEPSRETPNLDRMAAEGLLFPNFYSANPLXSPSRAALLTGRLPIRNGFYTTNAHARNAYTPQEIVGGIPDSEQLLPELLKKAGYVSKIVGKWHLGHRPQFHPLKHGFDEWFGSPNCHFGPYDNKARPNIPVYRDWEMVGRYYEEFPINLKTGEANLTQIYLQEALDFIKRQARHHPFFLYWAVDATHAPVYASKPFLGTSQRGRYGDAVREIDDSIGKILELLQDLHVADNTFVFFTSDNGAALISAPEQGGSNGPFLCGKQTTFEGGMREPALAWWPGHVTAGQVSHQLGSIMDLFTTSLALAGLTPPSDRAIDGLNLLPTLLQGRLMDRPIFYYRGDTLMAATLGQHKAHFWTWTNSWENFRQGIDFCPGQNVSGVTTHNLEDHTKLPLIFHLGRDPGERFPLSFASAEYQEALSRITSVVQQHQEALVPAQPQLNVCNWAVMNWAPPGCEKLGKCLTPPESIPKKCLW Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Haemophilus influenzae NadR protein (Hae-influ nadR) | 1LW7 | 4.01 | |
Target general information Gen name Hae-influ nadR Organism Haemophilus influenzae (strain ATCC 51907 / DSM 11121 / KW20 / Rd) Uniprot ID TTD ID Synonyms nadR; Transcriptional regulator nadR Protein family Bacterial NMN adenylyltransferase family; Bacterial RNK family Biochemical class Nicotinamide ribonucleoside uptake permease Function This enzyme has twoactivities: nicotinamide mononucleotide (NMN) adenylyltransferase and ribosylnicotinamide (RN) kinase. The RN kinase activity catalyzes the phosphorylation of RN to form nicotinamide ribonucleotide. The NMN adenylyltransferase activity catalyzes the transfer of the AMP moiety of ATP to nicotinamide ribonucleotide to form NAD(+). Related diseases Involved in the epigenetic regulation of ESR1 expression in breast cancer in a TFAP2C, IFI16 and HDAC4/5/6-dependent manner. {ECO:0000269|PubMed:24413532}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative initiation; ATP-binding; Cell membrane; Cytoplasm; Kinase; Membrane; Multifunctional enzyme; NAD; Nucleotide-binding; Pyridine nucleotide biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 39581.5 Length 344 Aromaticity 0.14 Instability index 41.39 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -5.47 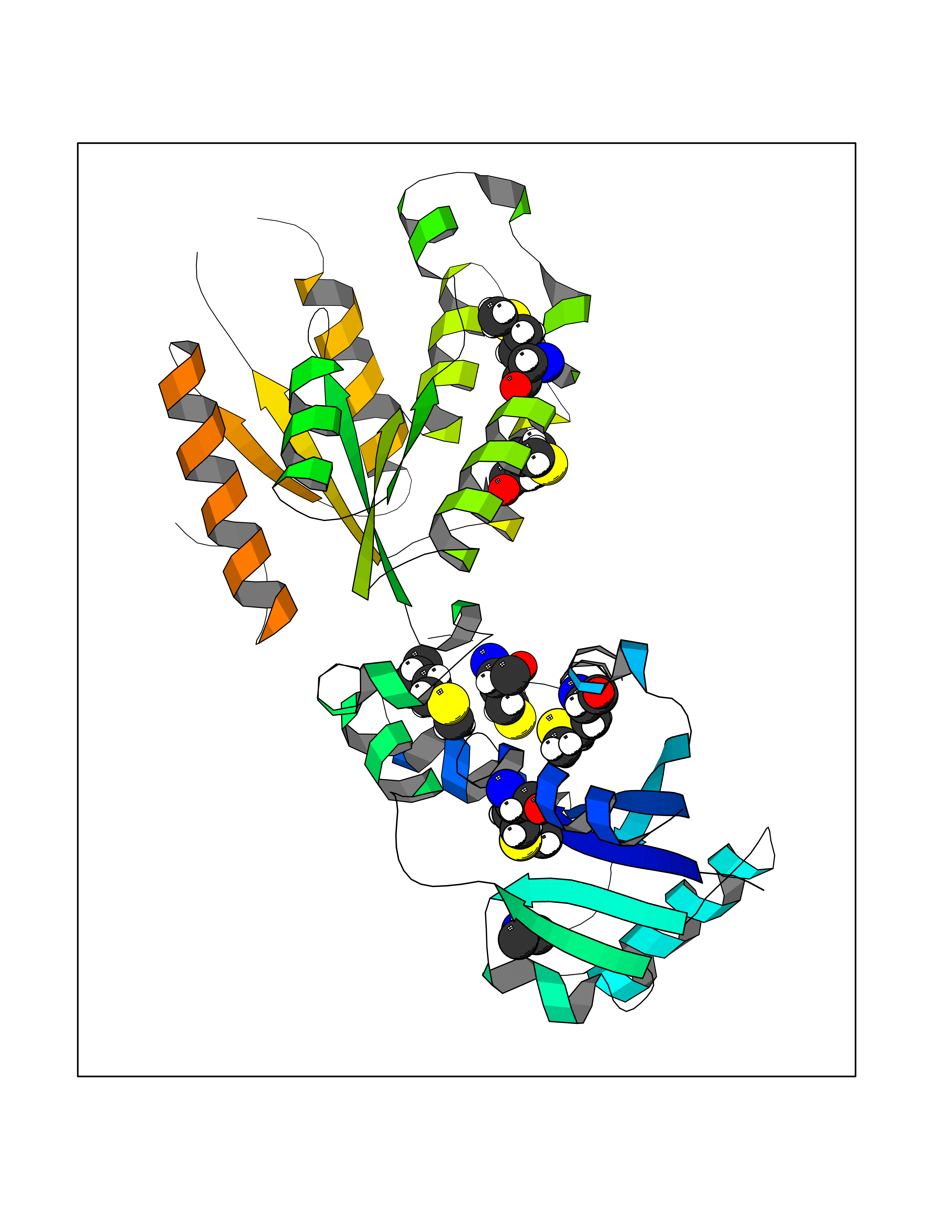 Molscript Map 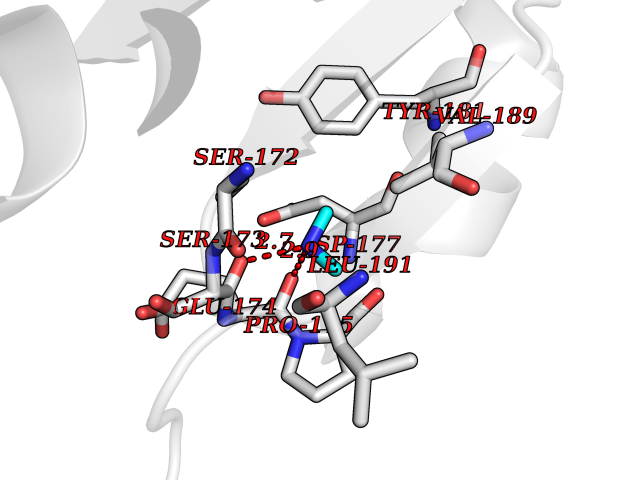 Pymol Map 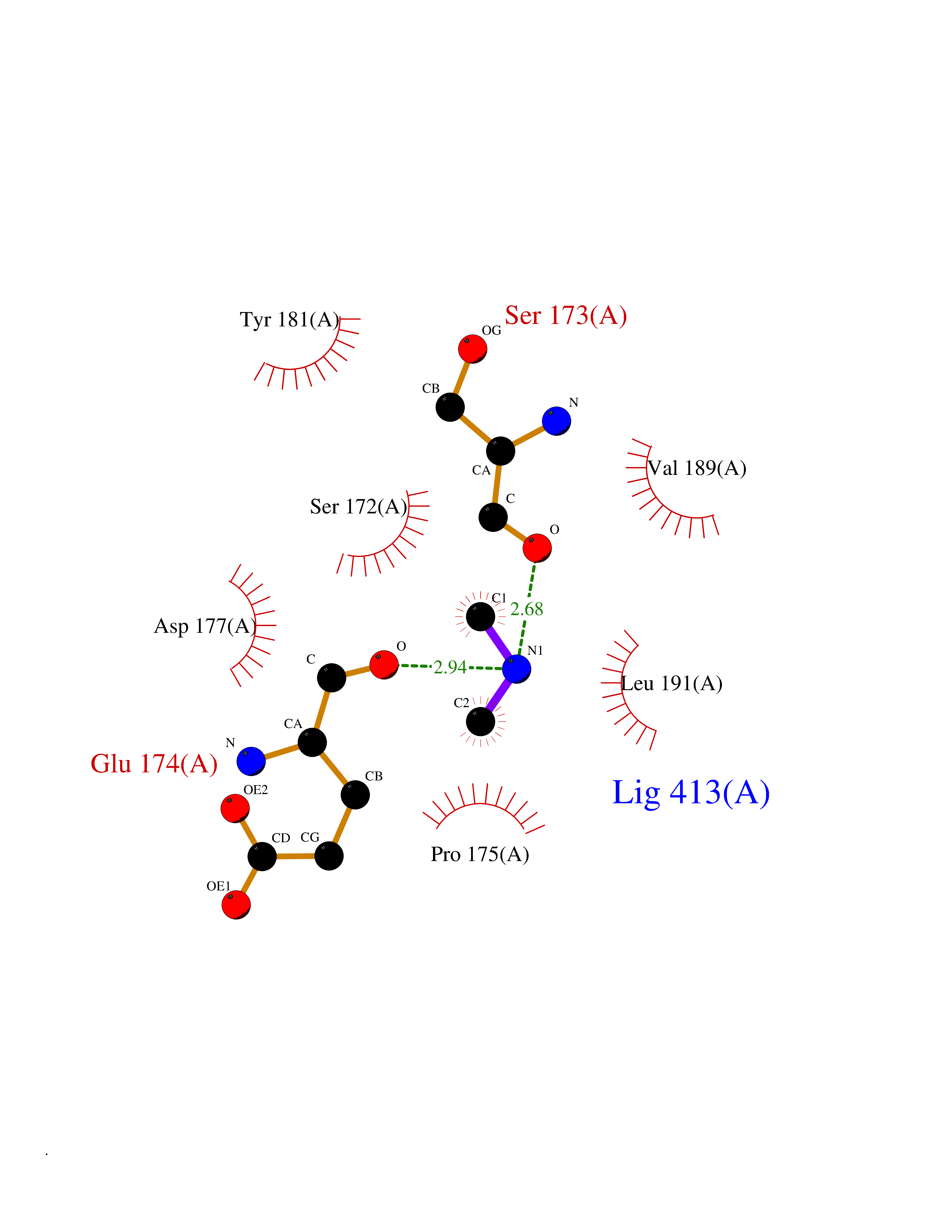 Ligplot Map 3D Binding mode Sequence EKKVGVIFGKFYPVHTGHINXIYEAFSKVDELHVIVCSDTVRDLKLFYDSKXKRXPTVQDRLRWXQQIFKYQKNQIFIHHLVEDGIPSYPNGWQSWSEAVKTLFHEKHFEPSIVFSSEPQDKAPYEKYLGLEVSLVDPDRTFFNVSATKIRTTPFQYWKFIPKEARPFFAKTVAILGGESSGKSVLVNKLAAVFNTTSAWEYGREFVFEKLGGDEQAMQYSDYPQXALGHQRYIDYAVRHSHKIAFIDTDFITTQAFCIQYEGKAHPFLDSXIKEYPFDVTILLKNNTEQKQRQQFQQLLKKLLDKYKVPYIEIESPSYLDRYNQVKAVIEKVLNEEEISELQN Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Proto-oncogene c-Fes (FES) | 3CBL | 4.01 | |
Target general information Gen name FES Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p93c-fes; Tyrosine-protein kinase Fes/Fps; Proto-oncogene c-Fps; Feline sarcoma/Fujinami avian sarcoma oncogene homolog; FPS Protein family Protein kinase superfamily, Tyr protein kinase family, Fes/fps subfamily Biochemical class Kinase Function Plays a role in FCER1 (high affinity immunoglobulin epsilon receptor)-mediated signaling in mast cells. Acts down-stream of the activated FCER1 receptor and the mast/stem cell growth factor receptor KIT. Plays a role in the regulation of mast cell degranulation. Plays a role in the regulation of cell differentiation and promotes neurite outgrowth in response to NGF signaling. Plays a role in cell scattering and cell migration in response to HGF-induced activation of EZR. Phosphorylates BCR and down-regulates BCR kinase activity. Phosphorylates HCLS1/HS1, PECAM1, STAT3 and TRIM28. Tyrosine-protein kinase that acts downstream of cell surface receptors and plays a role in the regulation of the actin cytoskeleton, microtubule assembly, cell attachment and cell spreading. Related diseases Has been shown to act as proto-oncogene in some types of cancer, possibly due to abnormal activation of the kinase. Has been shown to act as tumor suppressor in other types of cancer. Expressed and present as activated kinase in a subset of acute myeloid leukemia patients; promotes survival of leukemia cells (PubMed:20111072). Expression is absent in K562 leukemia cells; ectopic expression of FSP/FES restores myeloid differentiation (PubMed:2656706). May function as tumor suppressor in colorectal cancer; expression is reduced or absent in samples from some colon cancer patients (PubMed:16455651). May function as tumor suppressor in melanoma by preventing melanoma cell proliferation; expression is reduced or absent in samples from some melanoma patients (PubMed:28463229). Ectopic expression of FSP/FES suppresses anchorage-independent growth in colon cancer cell lines (PubMed:16455651). Up-regulated in prostate cancer, and might be a predictor of recurrence after radical surgery (PubMed:16455651). May promote growth of renal carcinoma cells (PubMed:19082481). {ECO:0000269|PubMed:16455651, ECO:0000269|PubMed:19082481, ECO:0000269|PubMed:20111072, ECO:0000269|PubMed:2656706, ECO:0000269|PubMed:28463229}. Drugs (DrugBank ID) DB12010 Interacts with P10275; P15924; P15311; Q13480; P10721; P54274 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell junction; Cell membrane; Coiled coil; Cytoplasm; Cytoplasmic vesicle; Cytoskeleton; Golgi apparatus; Kinase; Lipid-binding; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; SH2 domain; Transferase; Tumor suppressor; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 40460.2 Length 356 Aromaticity 0.09 Instability index 42.56 Isoelectric point 7.12 Charge (pH=7) 0.32 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 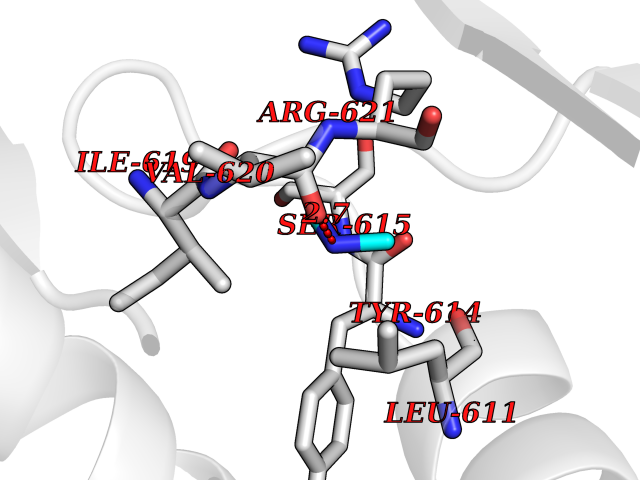 Pymol Map 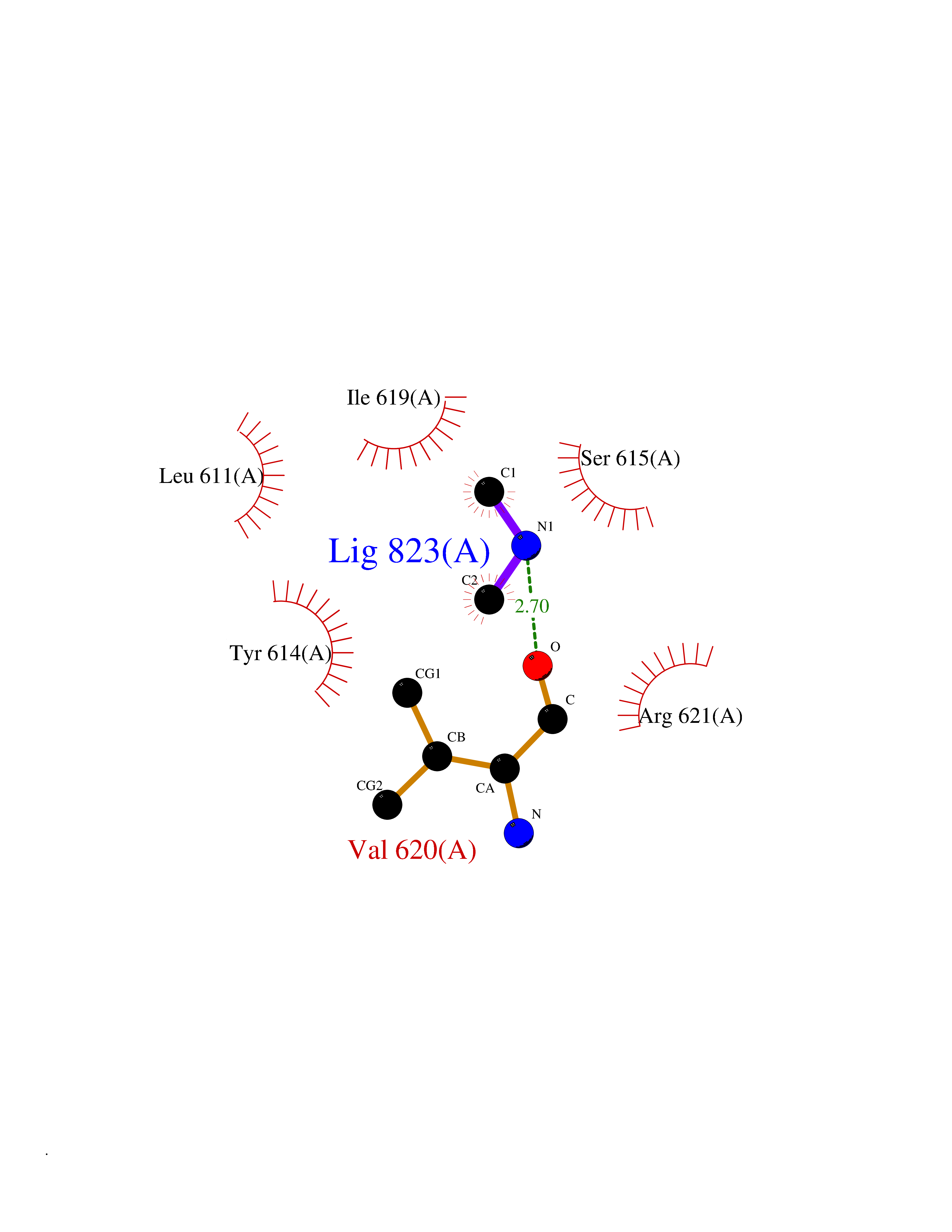 Ligplot Map 3D Binding mode Sequence SMIPEVQKPLHEQLWYHGAIPRAEVAELLVHSGDFLVRESQQEYVLSVPRHFIINLYRLFPSIPLLIDHLLSTQQPLVVLHRAVPKDKWVLNHEDLVLGEQIGRGNFGEVFSGRLRADNTLVAVKSCRETLPPDLKAKFLQEARILKQYSHPNIVRLIGVCTQKQPIYIVMELVQGGDFLTFLRTEGARLRVKTLLQMVGDAAAGMEYLESKCCIHRDLAARNCLVTEKNVLKISDFGMSREEADGVYAASGGLRQVPVKWTAPEALNYGRYSSESDVWSFGILLWETFSLGASPYPNLSNQQTREFVEKGGRLPCPELCPDAVFRLMEQCWAYEPGQRPSFSTIYQELQSIRKRH Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Mycobacterium Nicotinate-nucleotide pyrophosphorylase (MycB nadC) | 1QPR | 4.01 | |
Target general information Gen name MycB nadC Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID Synonyms nadC; Quinolinic acid phosphoribosyltransferase; Quinolinate phosphoribosyltransferase [decarboxylating]; QAPRTase Protein family NadC/ModD family Biochemical class Pentosyltransferase Function Involved in the catabolism of quinolinic acid (QA). Related diseases Oculodentodigital dysplasia (ODDD) [MIM:164200]: A disease characterized by a typical facial appearance and variable involvement of the eyes, dentition, and fingers. Characteristic facial features include a narrow, pinched nose with hypoplastic alae nasi, prominent columella and thin anteverted nares together with a narrow nasal bridge, and prominent epicanthic folds giving the impression of hypertelorism. The teeth are usually small and carious. Typical eye findings include microphthalmia and microcornea. The characteristic digital malformation is complete syndactyly of the fourth and fifth fingers (syndactyly type III) but the third finger may be involved and associated camptodactyly is a common finding. Cardiac abnormalities are observed in rare instances. {ECO:0000269|PubMed:16816024}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Oculodentodigital dysplasia, autosomal recessive (ODDD-AR) [MIM:257850]: A disease characterized by a typical facial appearance and variable involvement of the eyes, dentition, and fingers. Characteristic facial features include a narrow, pinched nose with hypoplastic alae nasi, prominent columella and thin anteverted nares together with a narrow nasal bridge, and prominent epicanthic folds giving the impression of hypertelorism. The teeth are usually small and carious. Typical eye findings include microphthalmia and microcornea. The characteristic digital malformation is complete syndactyly of the fourth and fifth fingers (syndactyly type III) but the third finger may be involved and associated camptodactyly is a common finding. Cardiac abnormalities are observed in rare instances. {ECO:0000269|PubMed:16816024}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Syndactyly 3 (SDTY3) [MIM:186100]: A form of syndactyly, a congenital anomaly of the hand or foot marked by persistence of the webbing between adjacent digits that are more or less completely attached. In SDTY3, there is usually complete and bilateral syndactyly between the fourth and fifth fingers. Usually it is soft tissue syndactyly but occasionally the distal phalanges are fused. The fifth finger is short with absent or rudimentary middle phalanx. The feet are not affected. {ECO:0000269|PubMed:14729836}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Hypoplastic left heart syndrome 1 (HLHS1) [MIM:241550]: A syndrome due to defective development of the aorta proximal to the entrance of the ductus arteriosus, and hypoplasia of the left ventricle and mitral valve. As a result of the abnormal circulation, the ductus arteriosus and foramen ovale are patent and the right atrium, right ventricle, and pulmonary artery are enlarged. {ECO:0000269|PubMed:11470490}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Hallermann-Streiff syndrome (HSS) [MIM:234100]: A disorder characterized by a typical skull shape (brachycephaly with frontal bossing), hypotrichosis, microphthalmia, cataracts, beaked nose, micrognathia, skin atrophy, dental anomalies and proportionate short stature. Intellectual disability is present in a minority of cases. {ECO:0000269|PubMed:14974090}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Craniometaphyseal dysplasia, autosomal recessive (CMDR) [MIM:218400]: An osteochondrodysplasia characterized by hyperostosis and sclerosis of the craniofacial bones associated with abnormal modeling of the metaphyses. Sclerosis of the skull may lead to asymmetry of the mandible, as well as to cranial nerve compression, that may finally result in hearing loss and facial palsy. {ECO:0000269|PubMed:23951358}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythrokeratodermia variabilis et progressiva 3 (EKVP3) [MIM:617525]: A form of erythrokeratodermia variabilis et progressiva, a genodermatosis characterized by the coexistence of two independent skin lesions: transient erythema and hyperkeratosis that is usually localized but occasionally occurs in its generalized form. Clinical presentation varies significantly within a family and from one family to another. Palmoplantar keratoderma is present in around 50% of cases. {ECO:0000269|PubMed:25398053}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Palmoplantar keratoderma and congenital alopecia 1 (PPKCA1) [MIM:104100]: A rare autosomal dominant disorder characterized by severe hyperkeratosis of the palms and soles, and congenital hypotrichosis or alopecia. Dystrophic nail changes occur in some patients. {ECO:0000269|PubMed:25168385}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04294; DB02382; DB02746; DB01796 Interacts with NA EC number EC 2.4.2.19 Uniprot keywords 3D-structure; Glycosyltransferase; Pyridine nucleotide biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 59621.1 Length 568 Aromaticity 0.03 Instability index 20.43 Isoelectric point 5.09 Charge (pH=7) -15.46 2D Binding mode Binding energy (Kcal/mol) -5.47 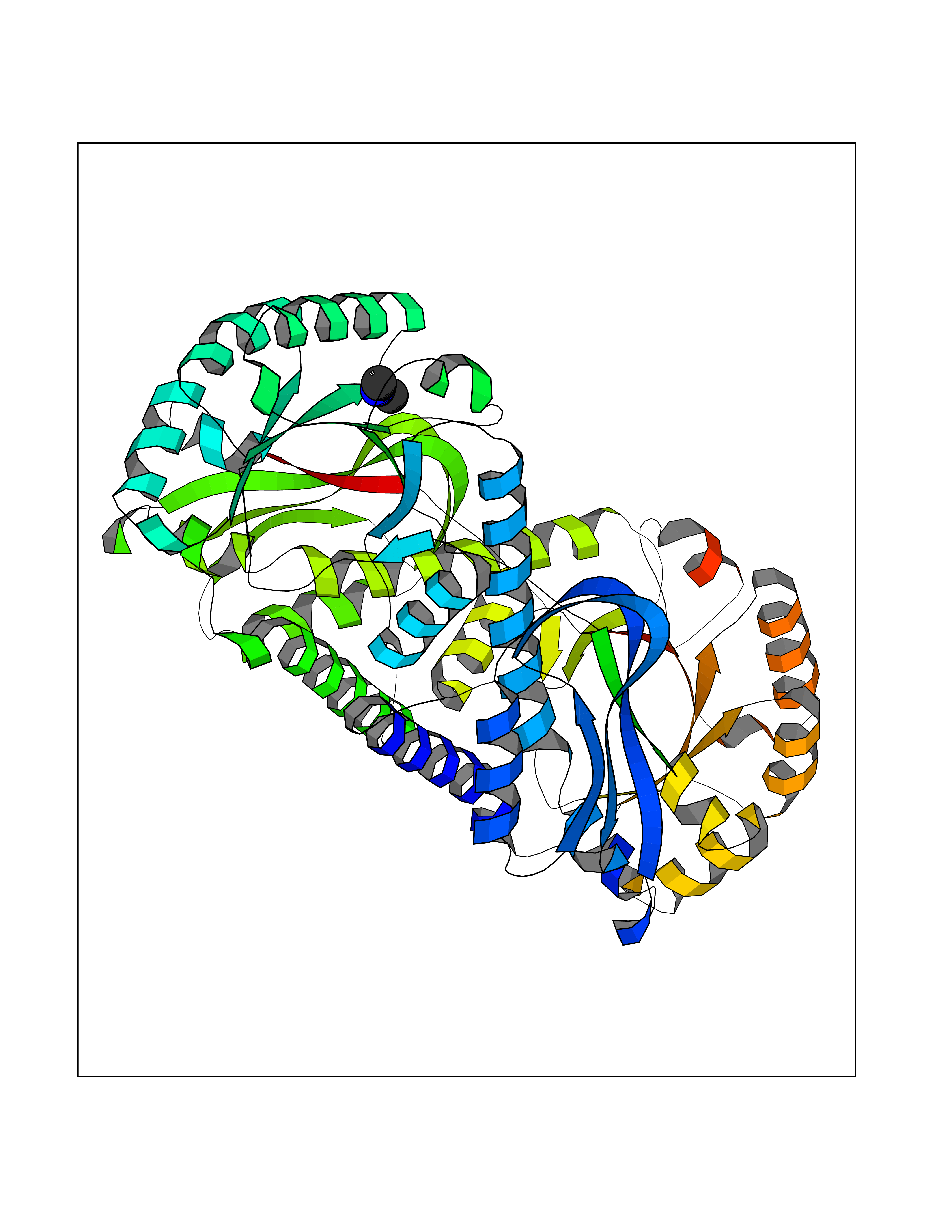 Molscript Map 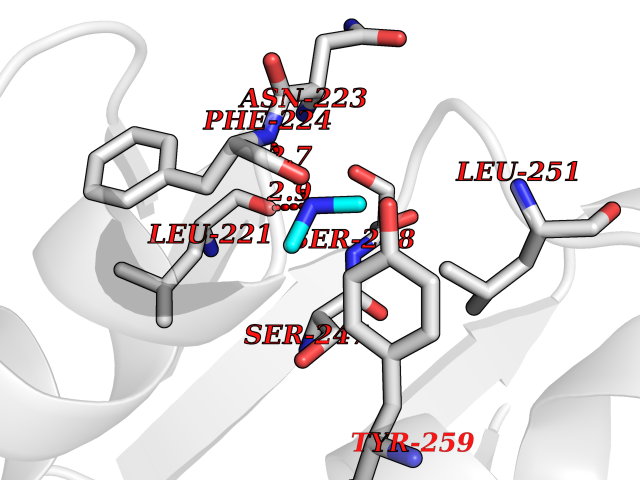 Pymol Map 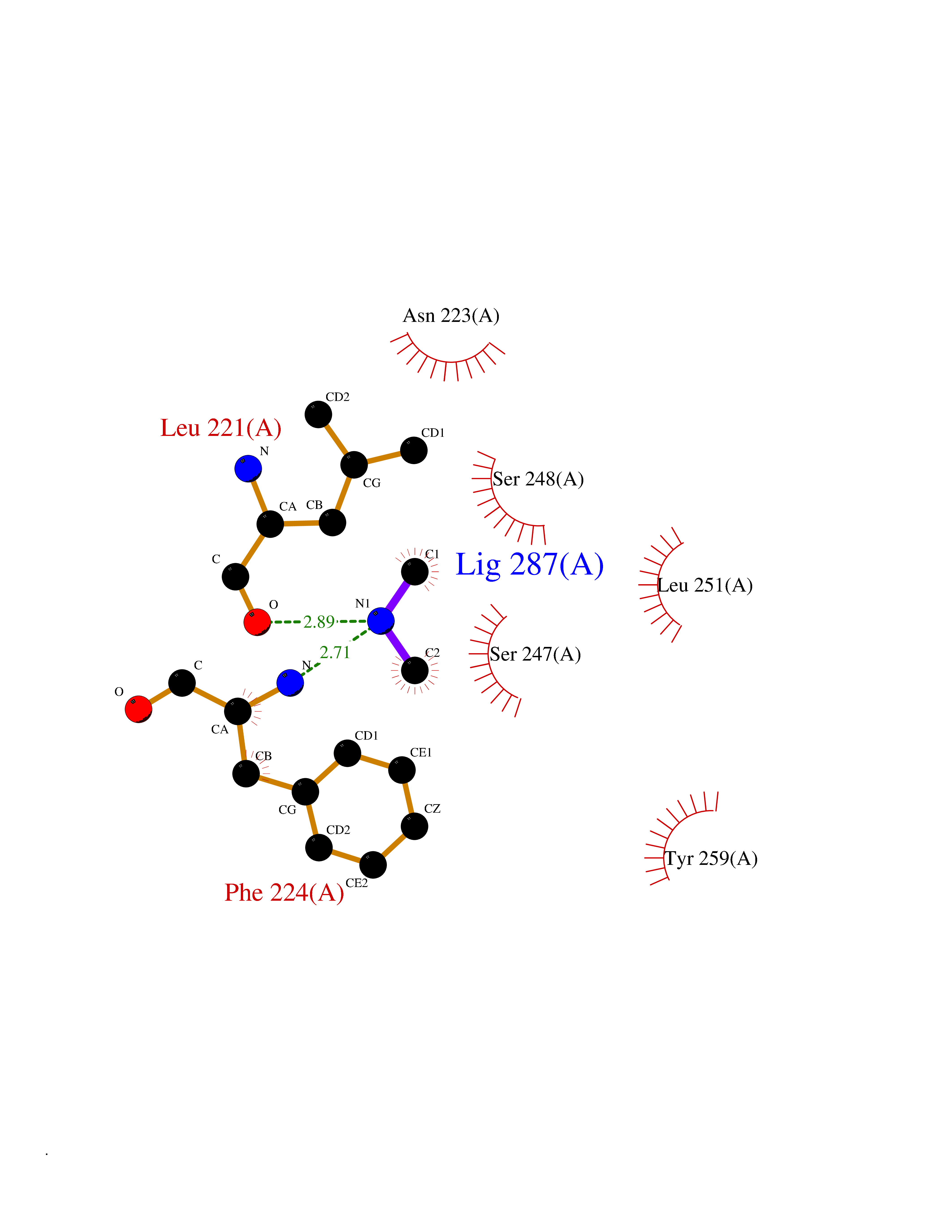 Ligplot Map 3D Binding mode Sequence GLSDWELAAARAAIARGLDEDLRYGPDVTTLATVPASATTTASLVTREAGVVAGLDVALLTLNEVLGTNGYRVLDRVEDGARVPPGEALMTLEAQTRGLLTAERTMLNLVGHLSGIATATAAWVDAVRGTKAKIRDTRKTLPGLRALQKYAVRTGGGVNHRLGLGDAALIKDNHVAAAGSVVDALRAVRNAAPDLPCEVEVDSLEQLDAVLPEKPELILLDNFAVWQTQTAVQRRDSRAPTVMLESSGGLSLQTAATYAETGVDYLAVGALTHSVRVLDIGLDMGLSDWELAAARAAIARGLDEDLRYGPDVTTLATVPASATTTASLVTREAGVVAGLDVALLTLNEVLGTNGYRVLDRVEDGARVPPGEALMTLEAQTRGLLTAERTMLNLVGHLSGIATATAAWVDAVRGTKAKIRDTRKTLPGLRALQKYAVRTGGGVNHRLGLGDAALIKDNHVAAAGSVVDALRAVRNAAPDLPCEVEVDSLEQLDAVLPEKPELILLDNFAVWQTQTAVQRRDSRAPTVMLESSGGLSLQTAATYAETGVDYLAVGALTHSVRVLDIGLDM Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Triggering receptor expressed on monocytes 1 (TREM1) | 1Q8M | 4.01 | |
Target general information Gen name Trem1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Trem1; TREM-1 Protein family NA Biochemical class NA Function Stimulates neutrophil and monocyte-mediated inflammatory responses. Triggers release of pro-inflammatory chemokines and cytokines, as well as increased surface expression of cell activation markers. Amplifier of inflammatory responses that are triggered by bacterial and fungal infections and is a crucial mediator of septic shock. Related diseases GLUT1 deficiency syndrome 1 (GLUT1DS1) [MIM:606777]: A neurologic disorder showing wide phenotypic variability. The most severe 'classic' phenotype comprises infantile-onset epileptic encephalopathy associated with delayed development, acquired microcephaly, motor incoordination, and spasticity. Onset of seizures, usually characterized by apneic episodes, staring spells, and episodic eye movements, occurs within the first 4 months of life. Other paroxysmal findings include intermittent ataxia, confusion, lethargy, sleep disturbance, and headache. Varying degrees of cognitive impairment can occur, ranging from learning disabilities to severe intellectual disability. {ECO:0000269|PubMed:10227690, ECO:0000269|PubMed:10980529, ECO:0000269|PubMed:11136715, ECO:0000269|PubMed:11603379, ECO:0000269|PubMed:12325075, ECO:0000269|PubMed:15622525, ECO:0000269|PubMed:19901175, ECO:0000269|PubMed:20129935, ECO:0000269|PubMed:20221955, ECO:0000269|PubMed:20574033, ECO:0000269|PubMed:24847886, ECO:0000269|PubMed:25982116, ECO:0000269|PubMed:30197081}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GLUT1 deficiency syndrome 2 (GLUT1DS2) [MIM:612126]: A clinically variable disorder characterized primarily by onset in childhood of paroxysmal exercise-induced dyskinesia. The dyskinesia involves transient abnormal involuntary movements, such as dystonia and choreoathetosis, induced by exercise or exertion, and affecting the exercised limbs. Some patients may also have epilepsy, most commonly childhood absence epilepsy. Mild intellectual disability may also occur. In some patients involuntary exertion-induced dystonic, choreoathetotic, and ballistic movements may be associated with macrocytic hemolytic anemia. {ECO:0000269|PubMed:14605501, ECO:0000269|PubMed:18451999, ECO:0000269|PubMed:19630075, ECO:0000269|PubMed:19798636, ECO:0000269|PubMed:20129935, ECO:0000269|PubMed:20574033, ECO:0000269|PubMed:20621801, ECO:0000269|PubMed:20830593, ECO:0000269|PubMed:21204808}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epilepsy, idiopathic generalized 12 (EIG12) [MIM:614847]: A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. In some EIG12 patients seizures may remit with age. {ECO:0000269|PubMed:19798636, ECO:0000269|PubMed:22282645, ECO:0000269|PubMed:23280796, ECO:0000269|PubMed:25982116}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Dystonia 9 (DYT9) [MIM:601042]: An autosomal dominant neurologic disorder characterized by childhood onset of paroxysmal choreoathetosis and progressive spastic paraplegia. Most patients show some degree of cognitive impairment. Other variable features may include seizures, migraine headaches, and ataxia. {ECO:0000269|PubMed:21832227}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Stomatin-deficient cryohydrocytosis with neurologic defects (SDCHCN) [MIM:608885]: A rare form of stomatocytosis characterized by episodic hemolytic anemia, cold-induced red cells cation leak, erratic hyperkalemia, neonatal hyperbilirubinemia, hepatosplenomegaly, cataracts, seizures, intellectual disability, and movement disorder. {ECO:0000269|PubMed:21791420, ECO:0000269|PubMed:22492876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01694 Interacts with NA EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Innate immunity; Membrane; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 28186.2 Length 242 Aromaticity 0.07 Instability index 45.2 Isoelectric point 6.1 Charge (pH=7) -3.8 2D Binding mode Binding energy (Kcal/mol) -5.46 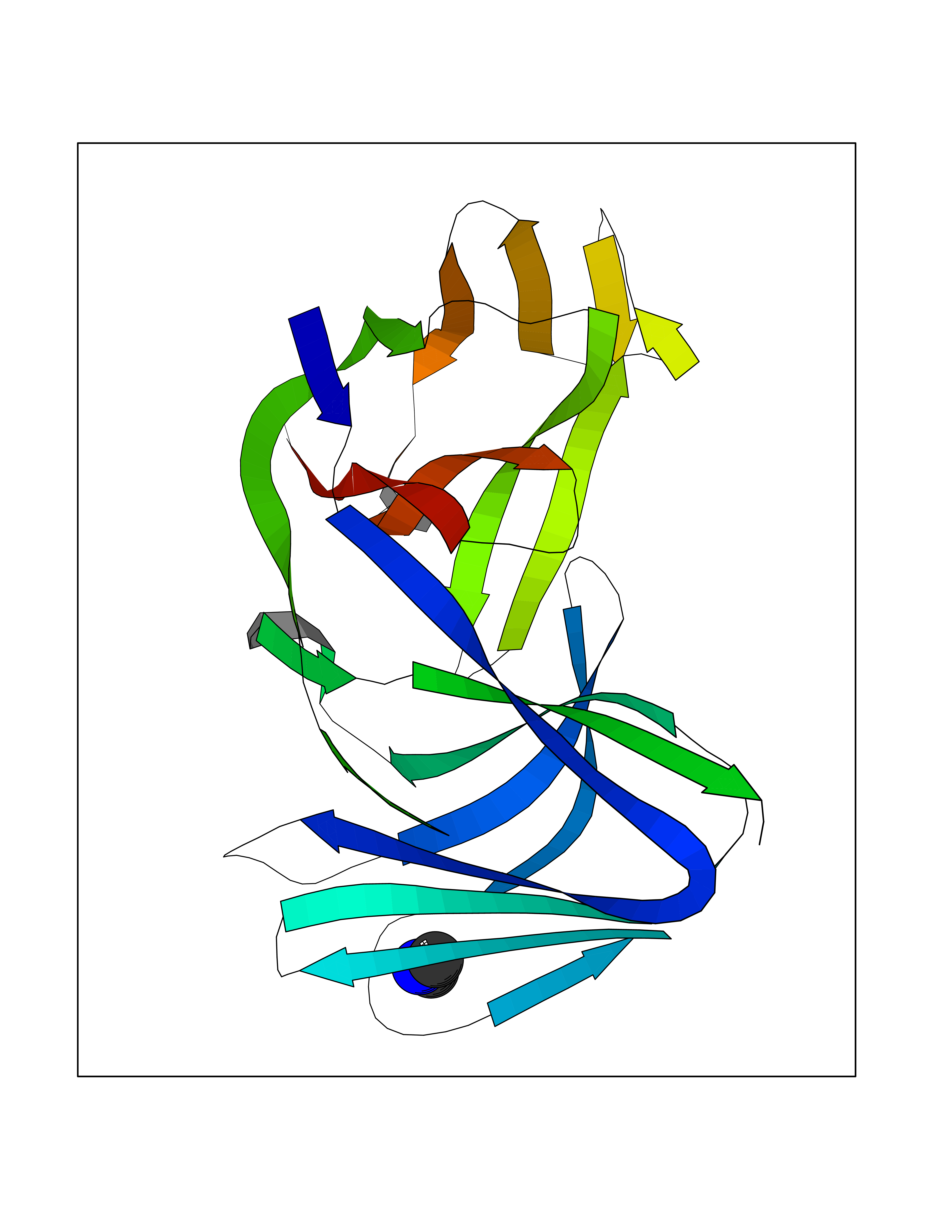 Molscript Map 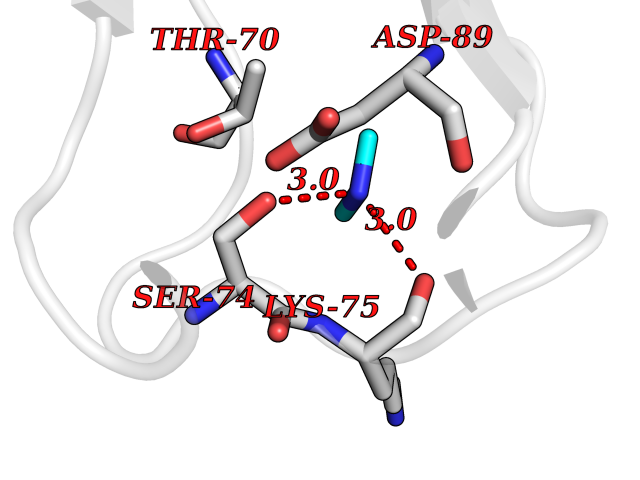 Pymol Map 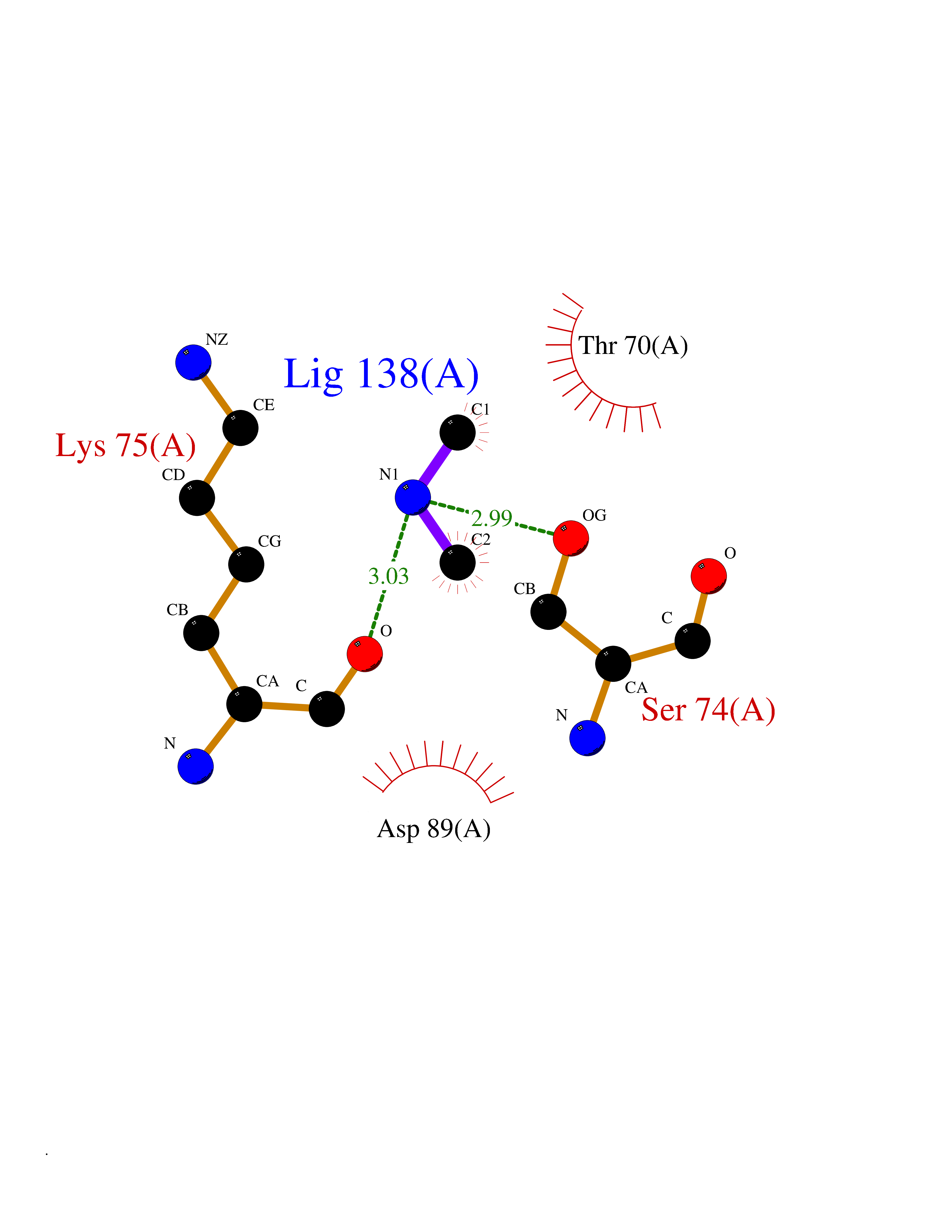 Ligplot Map 3D Binding mode Sequence MELRAATKLTEEKYELKEGQTLDVKCDYTLEKFASSQKAWQIIRDGEMPKTLACTERPSKNSHPVQVGRIILEDYHDHGLLRVRMVNLQVEDSGLYQCVIYQPPKEPHMLFDRIRLVVTLEMELRAATKLTEEKYELKEGQTLDVKCDYTLEKFASSQKAWQIIRDGEMPKTLACTERPSKNSHPVQVGRIILEDYHDHGLLRVRMVNLQVEDSGLYQCVIYQPPKEPHMLFDRIRLVVTLE Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | NAPE-hydrolyzing phospholipase D (NAPE-PLD) | 4QN9 | 4.01 | |
Target general information Gen name NAPEPLD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NAPE-PLD; N-acyl-phosphatidylethanolamine-hydrolyzing phospholipase D; N-acyl phosphatidylethanolamine phospholipase D; C7orf18 Protein family NAPE-PLD family Biochemical class NA Function Hydrolyzes N-acyl-phosphatidylethanolamines (NAPEs) to produce N-acylethanolamines (NAEs) and phosphatidic acid. Responsible for the generation of these bioactive fatty acid ethanolamides (FAEs), including anandamide (N-arachidonoylethanolamine), the ligand of cannabinoid and vanilloid receptors. As a regulator of lipid metabolism in the adipose tissue, mediates the crosstalk between adipocytes, gut microbiota and immune cells to control body temperature and weight. In particular, regulates energy homeostasis by promoting cold-induced brown or beige adipocyte differentiation program to generate heat from fatty acids and glucose (By similarity). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) DB14009 Interacts with Q6IQ20 EC number EC 3.1.4.54 Uniprot keywords 3D-structure; Acetylation; Endosome; Golgi apparatus; Hydrolase; Lipid degradation; Lipid metabolism; Membrane; Metal-binding; Nucleus; Phospholipid degradation; Phospholipid metabolism; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 74256.5 Length 643 Aromaticity 0.13 Instability index 48.34 Isoelectric point 5.65 Charge (pH=7) -17.61 2D Binding mode Binding energy (Kcal/mol) -5.47 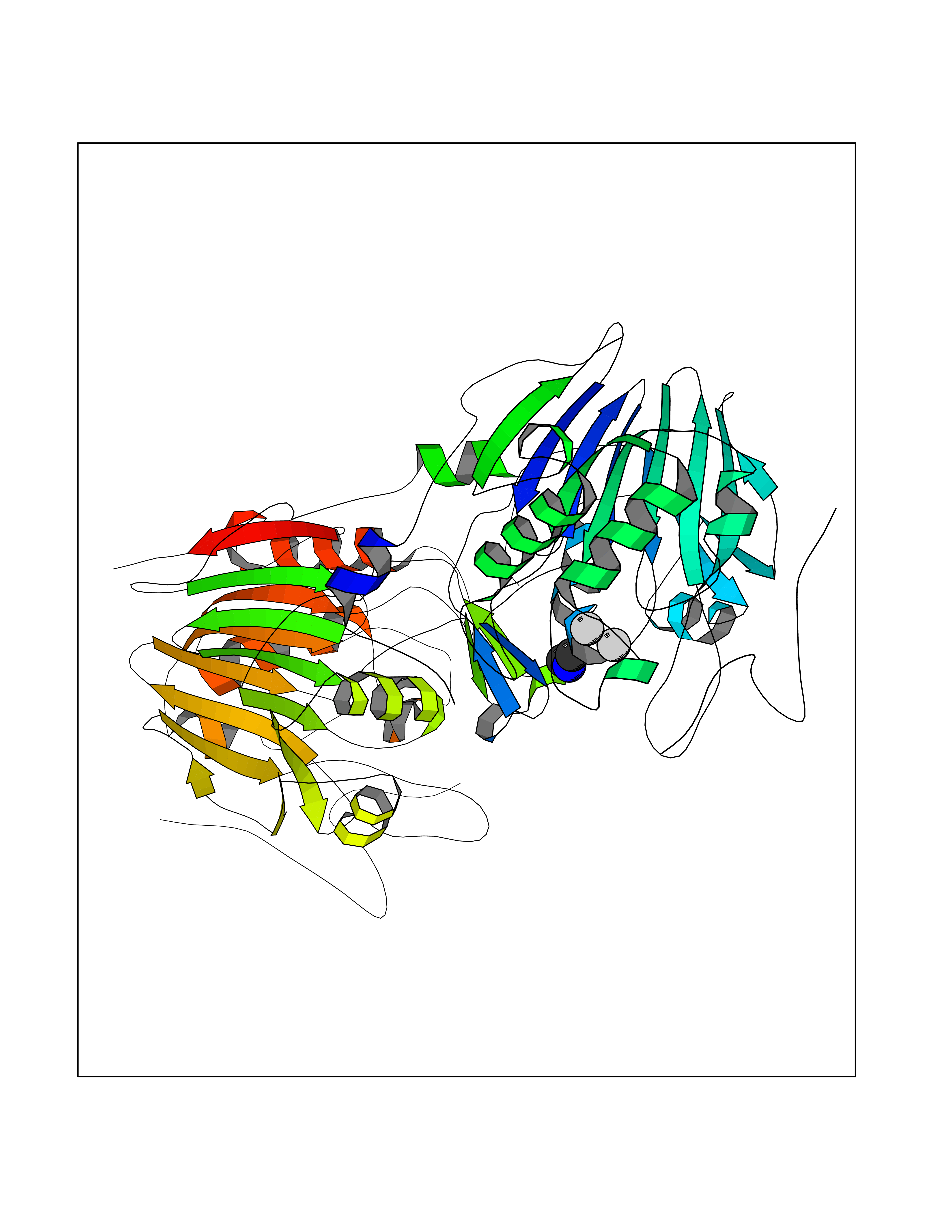 Molscript Map 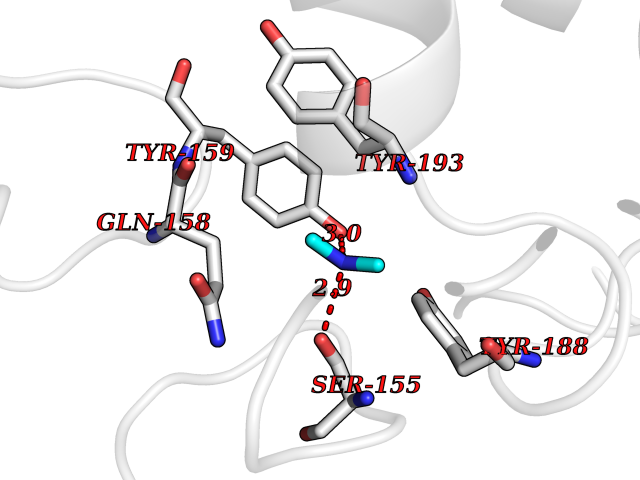 Pymol Map 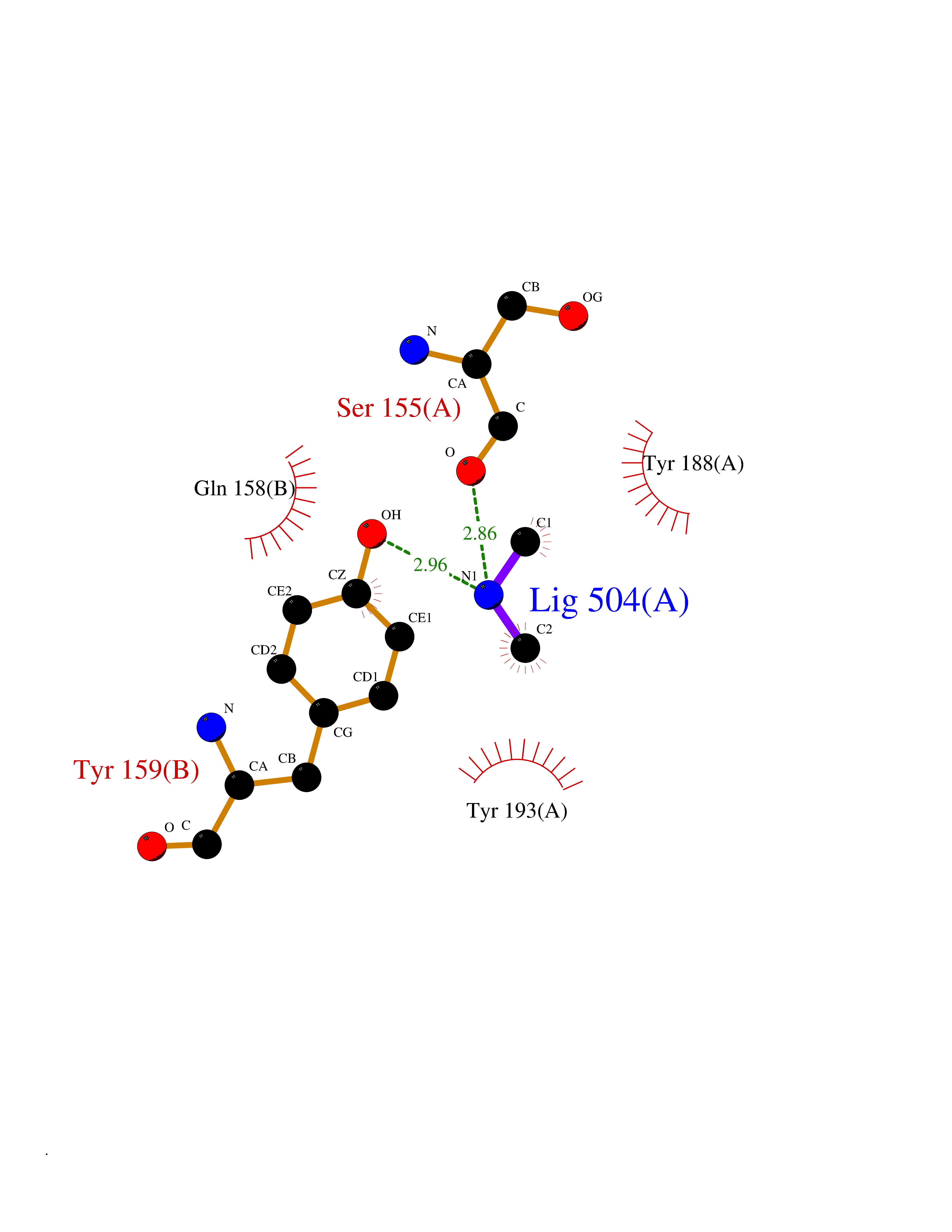 Ligplot Map 3D Binding mode Sequence SKKGKDGRFVNPWPTWKNPSIPNSSVPSSKEELDKELPVLKPYFITNPEEAGVREAGLRVTWLGHATVMVEMDELIFLTDPIFSSRASPSQYMGPKRFRRSPCTISELPPIDAVLISHNHYDHLDYNSVIALNERFGNELRWFVPLGLLDWMQKCGCENVIELDWWEENCVPGHDKVTFVFTPSQHWCKRTLMDDNKVLWGSWSVLGPWNRFFFAGDTGYCPAFEEIGKRFGPFDLAAIPIGAYEPRWFMKYQHVDPEEAVRIHTDVQTKKSMAIHWGTFALANEHYLEPPVKLNEALERYGLNAEDFFVLKHGESRYLNNSKKGKDGRFVNPWPTWKNPSIPNSSVPSSKEELDKELPVLKPYFITNPEEAGVREAGLRVTWLGHATVMVEMDELIFLTDPIFSSRASPSQYMGPKRFRRSPCTISELPPIDAVLISHNHYDHLDYNSVIALNERFGNELRWFVPLGLLDWMQKCGCENVIELDWWEENCVPGHDKVTFVFTPSQHWCKRTLMDDNKVLWGSWSVLGPWNRFFFAGDTGYCPAFEEIGKRFGPFDLAAIPIGAYEPRWFMKYQHVDPEEAVRIHTDVQTKKSMAIHWGTFALANEHYLEPPVKLNEALERYGLNAEDFFVLKHGESRYLNND Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Cerebroside-sulfatase (ARSA) | 1E2S | 4.01 | |
Target general information Gen name ARSA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cerebrosidesulfatase; Arylsulfatase A component C; ASA; ARSA Protein family Sulfatase family Biochemical class Sulfuric ester hydrolase Function Hydrolyzes cerebroside sulfate. Related diseases Metachromatic leukodystrophy (MLD) [MIM:250100]: An autosomal recessive disease caused by abnormal intralysosomal accumulation of cerebroside-3-sulfate in central and peripheral nervous systems, as well as other organs. MLD is clinically characterized by leukodystrophy, progressive demyelination and a variety of neurological symptoms, including gait disturbances, ataxias, optical atrophy, dementia, seizures, and spastic tetraparesis. Decreased arylsulfatase A activity is detected in urine, leukocytes, and fibroblasts of affected individuals. Several forms of the disease can be distinguished according to the age at onset and disease severity: late infantile, juvenile and adult forms, partial cerebroside sulfate deficiency, and pseudoarylsulfatase A deficiency. Individuals with pseudoarylsulfatase A deficiency have low arylsulfatase A activity but lack neurological manifestations and are apparently healthy. {ECO:0000269|PubMed:10220151, ECO:0000269|PubMed:10381328, ECO:0000269|PubMed:10477432, ECO:0000269|PubMed:10533072, ECO:0000269|PubMed:10751093, ECO:0000269|PubMed:11020646, ECO:0000269|PubMed:11061266, ECO:0000269|PubMed:11456299, ECO:0000269|PubMed:11941485, ECO:0000269|PubMed:12503099, ECO:0000269|PubMed:12788103, ECO:0000269|PubMed:1353340, ECO:0000269|PubMed:14517960, ECO:0000269|PubMed:14680985, ECO:0000269|PubMed:15026521, ECO:0000269|PubMed:15326627, ECO:0000269|PubMed:15710861, ECO:0000269|PubMed:1670590, ECO:0000269|PubMed:1673291, ECO:0000269|PubMed:1678251, ECO:0000269|PubMed:18693274, ECO:0000269|PubMed:19606494, ECO:0000269|PubMed:20339381, ECO:0000269|PubMed:21265945, ECO:0000269|PubMed:2574462, ECO:0000269|PubMed:7581401, ECO:0000269|PubMed:7825603, ECO:0000269|PubMed:7860068, ECO:0000269|PubMed:7902317, ECO:0000269|PubMed:7906588, ECO:0000269|PubMed:7909527, ECO:0000269|PubMed:8095918, ECO:0000269|PubMed:8101038, ECO:0000269|PubMed:8101083, ECO:0000269|PubMed:8104633, ECO:0000269|PubMed:8891236, ECO:0000269|PubMed:9090526, ECO:0000269|PubMed:9272717, ECO:0000269|PubMed:9452102, ECO:0000269|PubMed:9490297, ECO:0000269|PubMed:9600244, ECO:0000269|PubMed:9819708}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Multiple sulfatase deficiency (MSD) [MIM:272200]: A clinically and biochemically heterogeneous disorder caused by the simultaneous impairment of all sulfatases, due to defective post-translational modification and activation. It combines features of individual sulfatase deficiencies such as metachromatic leukodystrophy, mucopolysaccharidosis, chondrodysplasia punctata, hydrocephalus, ichthyosis, neurologic deterioration and developmental delay. {ECO:0000269|PubMed:15146462}. The protein represented in this entry is involved in disease pathogenesis. Arylsulfatase A activity is impaired in multiple sulfatase deficiency due to mutations in SUMF1 (PubMed:15146462). SUMF1 mutations result in defective post-translational modification of ARSA at residue Cys-69 that is not converted to 3-oxoalanine (PubMed:7628016). {ECO:0000269|PubMed:15146462, ECO:0000269|PubMed:7628016}. Drugs (DrugBank ID) DB03821; DB01800; DB01141; DB04786 Interacts with P50995; Q6P5X5; Q13554-3; O60826; Q96D98; Q9H0I2; Q12951-2; Q16512; P28069; O75360; Q9BQY4; Q15645; O95231 EC number EC 3.1.6.8 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Hydrolase; Ichthyosis; Leukodystrophy; Lipid metabolism; Lysosome; Metachromatic leukodystrophy; Metal-binding; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID P Molecular weight (Da) 51173.7 Length 481 Aromaticity 0.09 Instability index 47.42 Isoelectric point 5.59 Charge (pH=7) -12.84 2D Binding mode Binding energy (Kcal/mol) -5.47 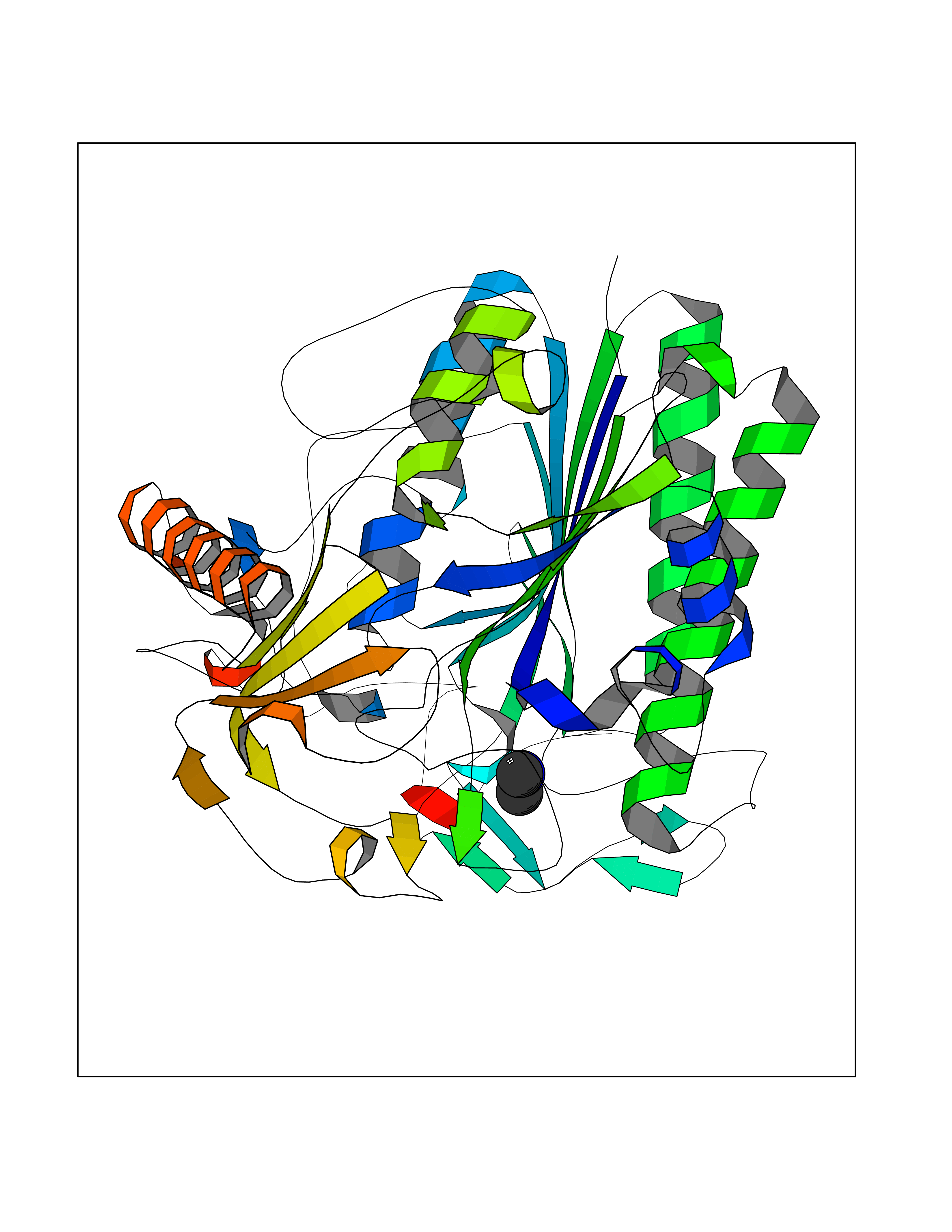 Molscript Map 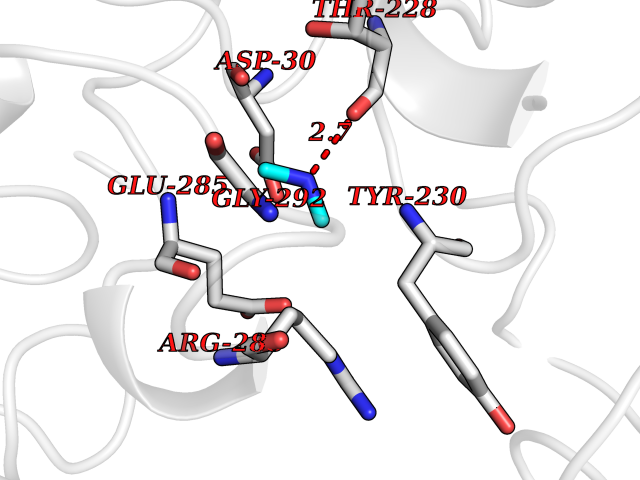 Pymol Map 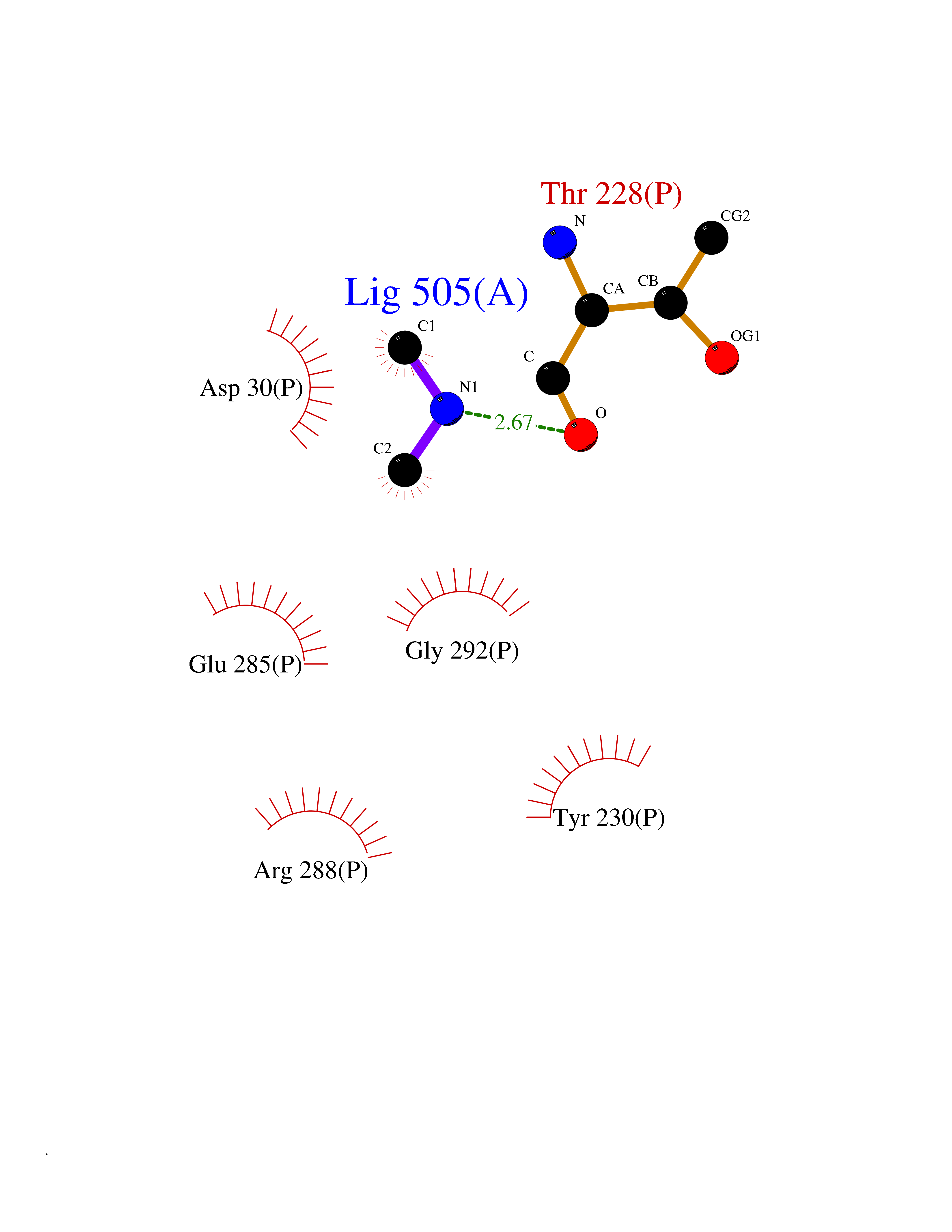 Ligplot Map 3D Binding mode Sequence RPPNIVLIFADDLGYGDLGCYGHPSSTTPNLDQLAAGGLRFTDFYVPVSLATPSRAALLTGRLPVRMGMYPGVLVPSSRGGLPLEEVTVAEVLAARGYLTGMAGKWHLGVGPEGAFLPPHQGFHRFLGIPYSHDQGPCQNLTCFPPATPCDGGCDQGLVPIPLLANLSVEAQPPWLPGLEARYMAFAHDLMADAQRQDRPFFLYYASHHTHYPQFSGQSFAERSGRGPFGDSLMELDAAVGTLMTAIGDLGLLEETLVIFTADNGPETMRMSRGGCSGLLRCGKGTTYEGGVREPALAFWPGHIAPGVTHELASSLDLLPTLAALAGAPLPNVTLDGFDLSPLLLGTGKSPRQSLFFYPSYPDEVRGVFAVRTGKYKAHFFTQGSAHSDTTADPACHASSSLTAHEPPLLYDLSKDPGENYNLLGATPEVLQALKQLQLLKAQLDAAVTFGPSQVARGEDPALQICCHPGCTPRPACCHCP Hydrogen bonds contact Hydrophobic contact | ||||