Job Results:
Ligand
Structure
Job ID
215921f481371e928c7811c49ba9b3ae
Job name
NA
Time
2025-01-23 16:39:47
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Cytochrome P450 1A2 | 2HI4 | 7.11 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -9.7  Molscript Map  Pymol Map 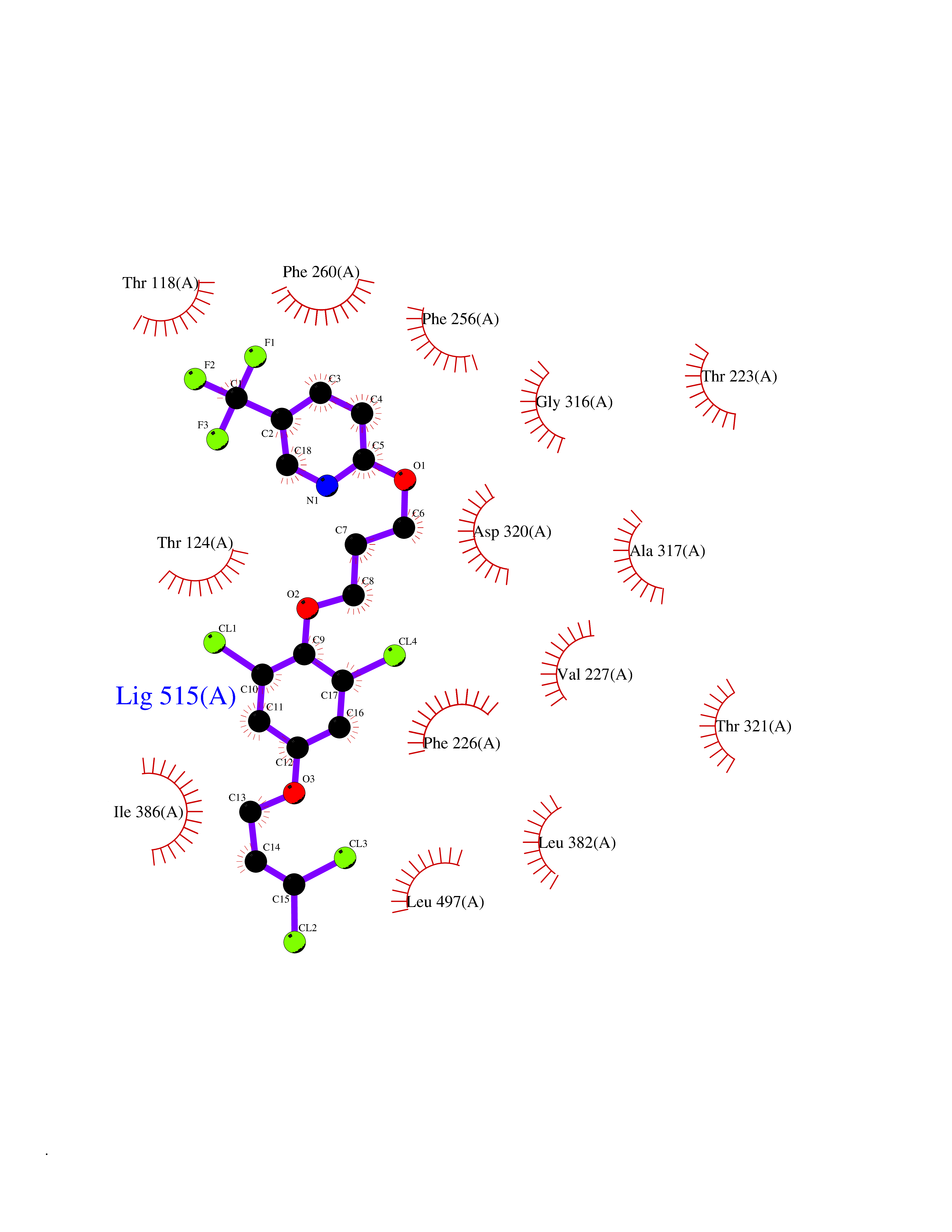 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Monoamine oxidase type B (MAO-B) | 2V5Z | 7.11 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -9.7 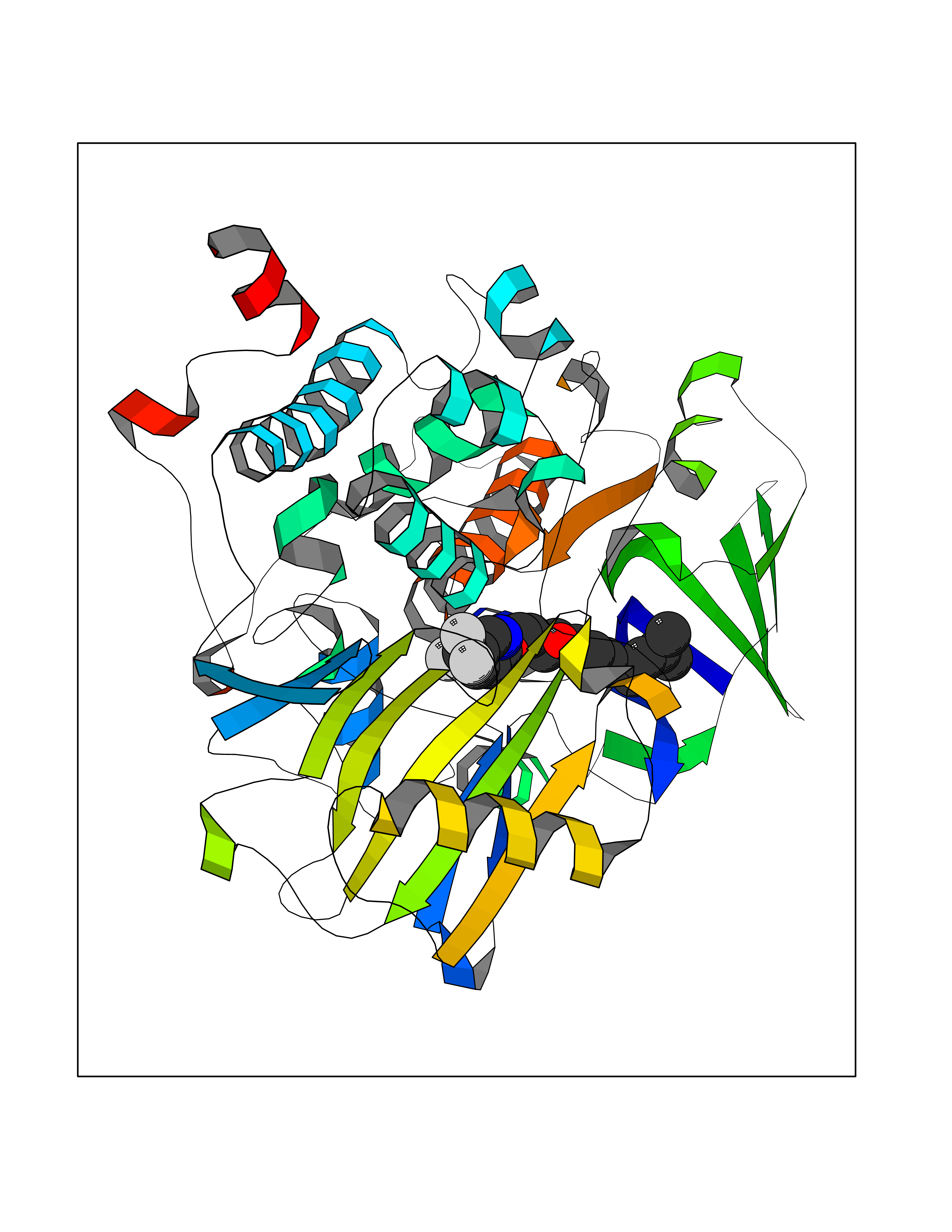 Molscript Map 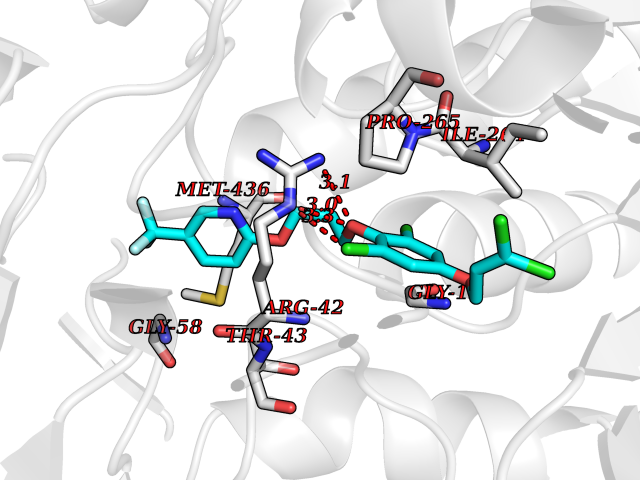 Pymol Map 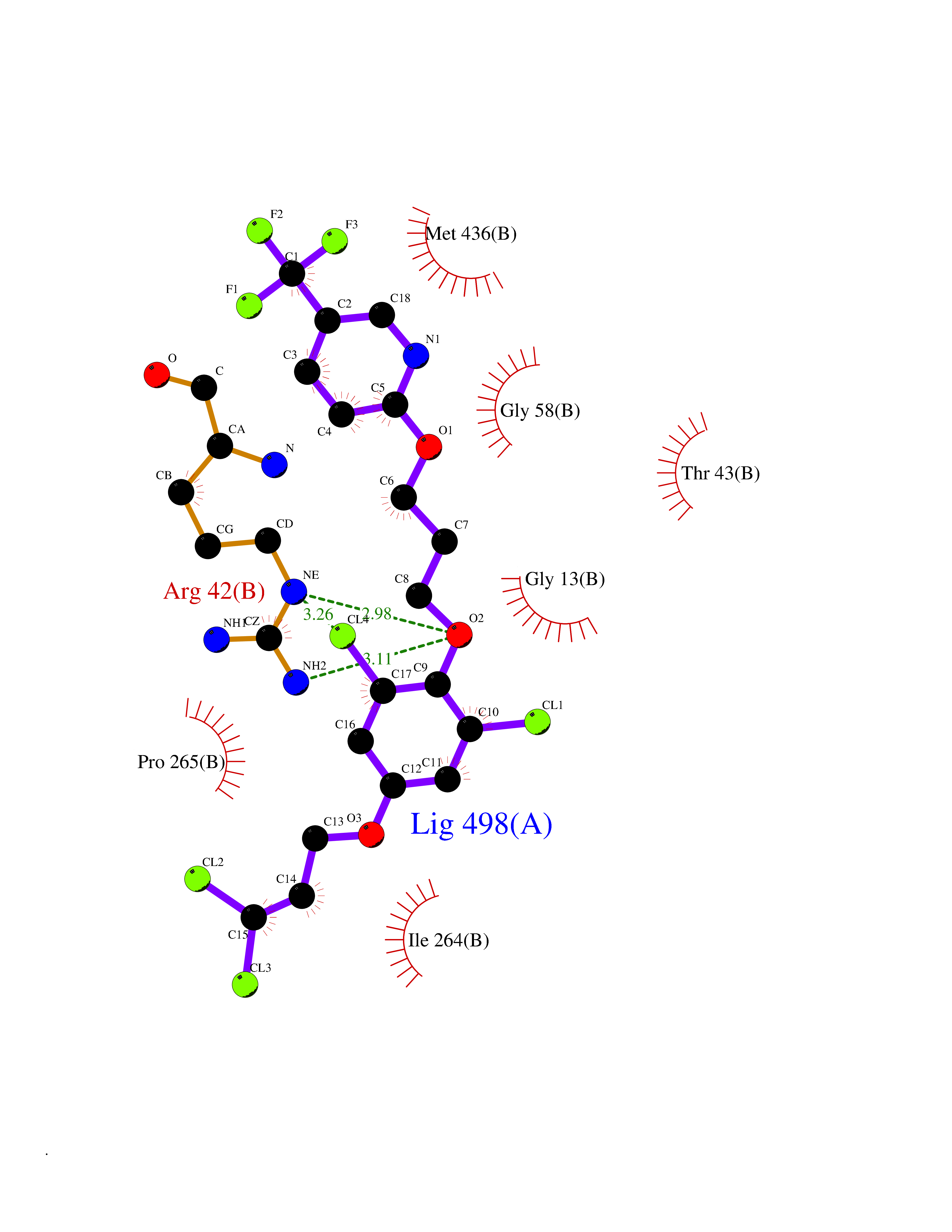 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Thyroid hormone receptor beta (THRB) | 1N46 | 7.10 | |
Target general information Gen name THRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms c-erbA-beta; c-erbA-2; THR1; Nuclear receptor subfamily 1 group A member 2; NR1A2; ERBA2 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function High affinity receptor for thyroid hormones, including triiodothyronine and thyroxine. Nuclear hormone receptor that can act as a repressor or activator of transcription. Related diseases Thyroid hormone resistance, generalized, autosomal dominant (GRTHD) [MIM:188570]: An autosomal dominant disease characterized by high levels of circulating thyroid hormones (T3-T4), goiter, abnormal mental functions, increased susceptibility to infections, abnormal growth and bone maturation, tachycardia and deafness. Affected individuals may also have attention deficit-hyperactivity disorders (ADHD) and language difficulties. Patients have normal or slightly elevated thyroid stimulating hormone (TSH). {ECO:0000269|PubMed:10660344, ECO:0000269|PubMed:12511610, ECO:0000269|PubMed:12554782, ECO:0000269|PubMed:1314846, ECO:0000269|PubMed:1324420, ECO:0000269|PubMed:1563081, ECO:0000269|PubMed:1587388, ECO:0000269|PubMed:1619012, ECO:0000269|PubMed:1661299, ECO:0000269|PubMed:16804041, ECO:0000269|PubMed:1846005, ECO:0000269|PubMed:19268523, ECO:0000269|PubMed:2153155, ECO:0000269|PubMed:2510172, ECO:0000269|PubMed:7833659, ECO:0000269|PubMed:8175986, ECO:0000269|PubMed:8514853, ECO:0000269|PubMed:8664910, ECO:0000269|PubMed:8889584}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid hormone resistance, generalized, autosomal recessive (GRTHR) [MIM:274300]: An autosomal recessive disorder characterized by goiter, clinical euthyroidism, end-organ unresponsiveness to thyroid hormone, abnormal growth and bone maturation, and deafness. Patients also have high levels of circulating thyroid hormones, with elevated thyroid stimulating hormone. {ECO:0000269|PubMed:1653889}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Selective pituitary thyroid hormone resistance (PRTH) [MIM:145650]: Variant form of thyroid hormone resistance and is characterized by clinical hyperthyroidism, with elevated free thyroid hormones, but inappropriately normal serum TSH. Unlike GRTH, where the syndrome usually segregates with a dominant allele, the mode of inheritance in PRTH has not been established. {ECO:0000269|PubMed:7528740, ECO:0000269|PubMed:8381821}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08085; DB03181; DB02106; DB01118; DB00509; DB05035; DB03788; DB03176; DB00451; DB00279; DB01583; DB05192; DB07425; DB09100; DB03604 Interacts with Q60974; Q9Y618 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Deafness; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 27235.4 Length 239 Aromaticity 0.09 Instability index 43.29 Isoelectric point 5.42 Charge (pH=7) -8.55 2D Binding mode Binding energy (Kcal/mol) -9.68 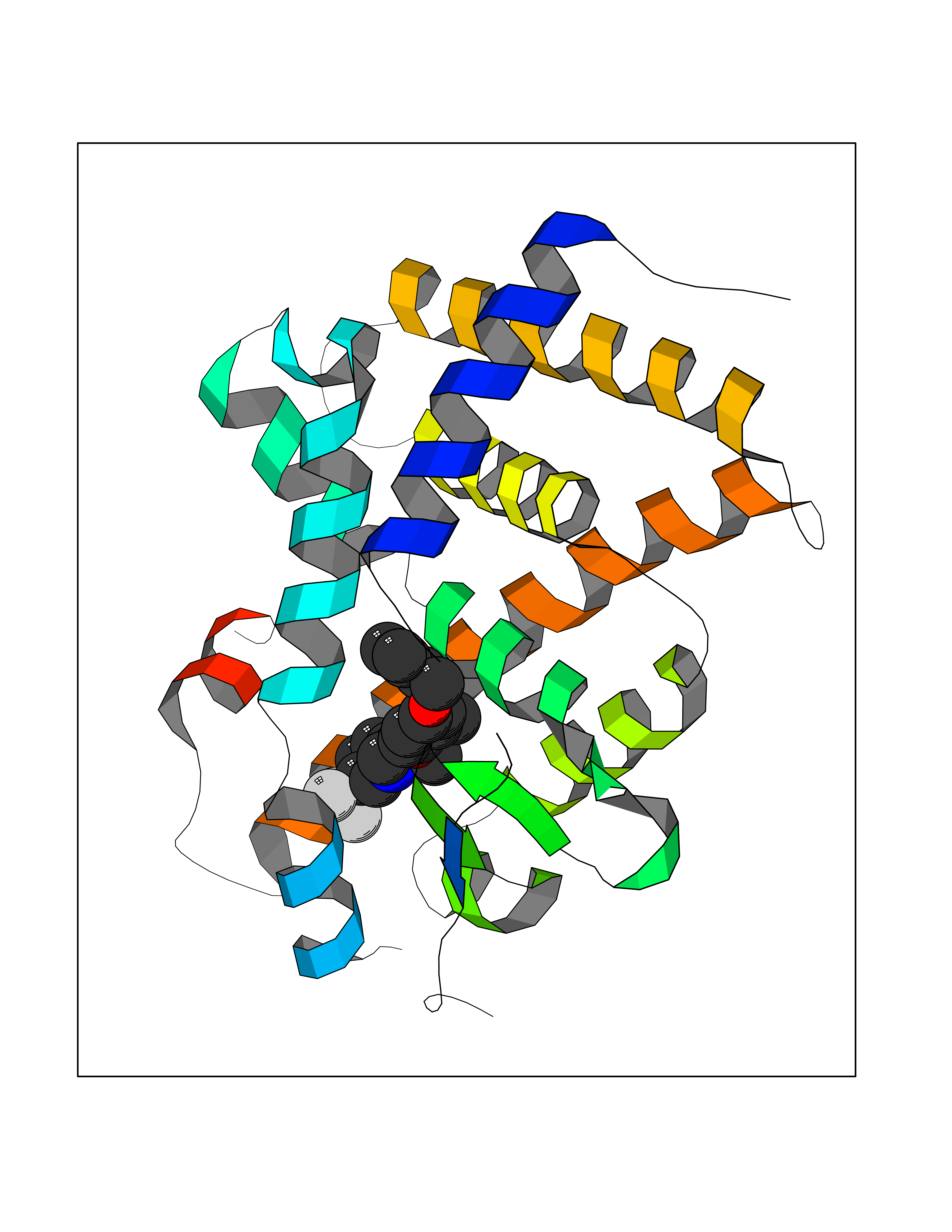 Molscript Map 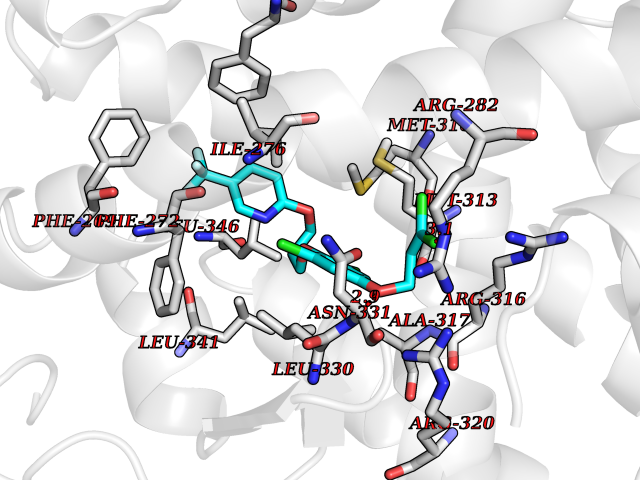 Pymol Map 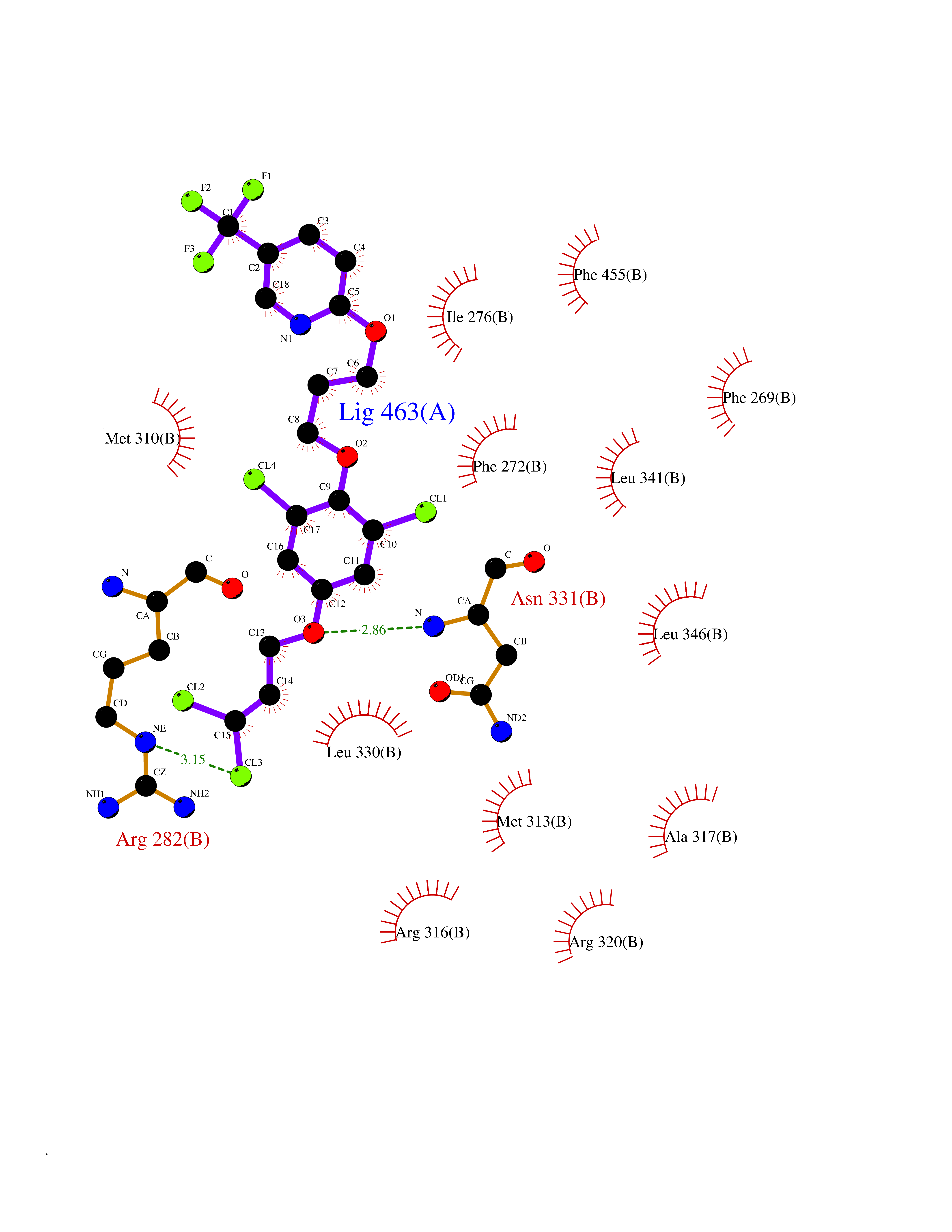 Ligplot Map 3D Binding mode Sequence KPEPTDEEWELIKTVTEAHVATNAQWKQKRKFLPEDIGQAKVDLEAFSHFTKIITPAITRVVDFAKKLPMFCELPCEDQIILLKGCCMEIMSLRAAVRYDPESETLTLNGEMAVTRGQLKNGGLGVVSDAIFDLGMSLSSFNLDDTEVALLQAVLLMSSDRPGLACVERIEKYQDSFLLAFEHYINYRKHHVTHFWPKLLMKVTDLRMIGACHASRFLHMKVECPTELFPPLFLEVFED Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Glutathione reductase (GR) | 3DK9 | 7.09 | |
Target general information Gen name GSR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glutathione reductase, mitochondrial; GRase; GRD1; GLUR Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Sulfur donor oxidoreductase Function Maintains high levels of reduced glutathione in the cytosol. Related diseases Hemolytic anemia due to glutathione reductase deficiency (HAGRD) [MIM:618660]: An autosomal recessive disease characterized by hemolytic anemia and impaired activity of glutathione reductase. Patients experience hemolytic anemia in response to oxidative stress or ingestion of fava beans. {ECO:0000269|PubMed:17185460}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07393; DB01644; DB02895; DB03867; DB02153; DB07714; DB09061; DB00262; DB09130; DB03147; DB00143; DB03310; DB02553; DB14009; DB14011; DB00157; DB11135; DB11590 Interacts with NA EC number EC 1.8.1.7 Uniprot keywords 3D-structure; Acetylation; Alternative initiation; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; FAD; Flavoprotein; Hereditary hemolytic anemia; Mitochondrion; NADP; Oxidoreductase; Proteomics identification; Redox-active center; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 49983.1 Length 462 Aromaticity 0.06 Instability index 38.04 Isoelectric point 7.73 Charge (pH=7) 2.16 2D Binding mode Binding energy (Kcal/mol) -9.67  Molscript Map 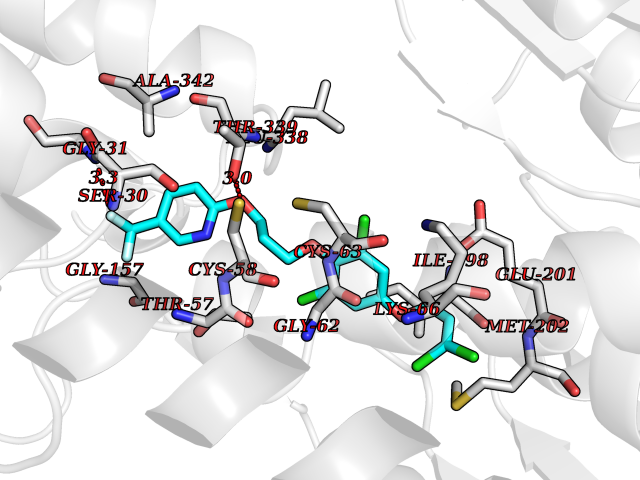 Pymol Map 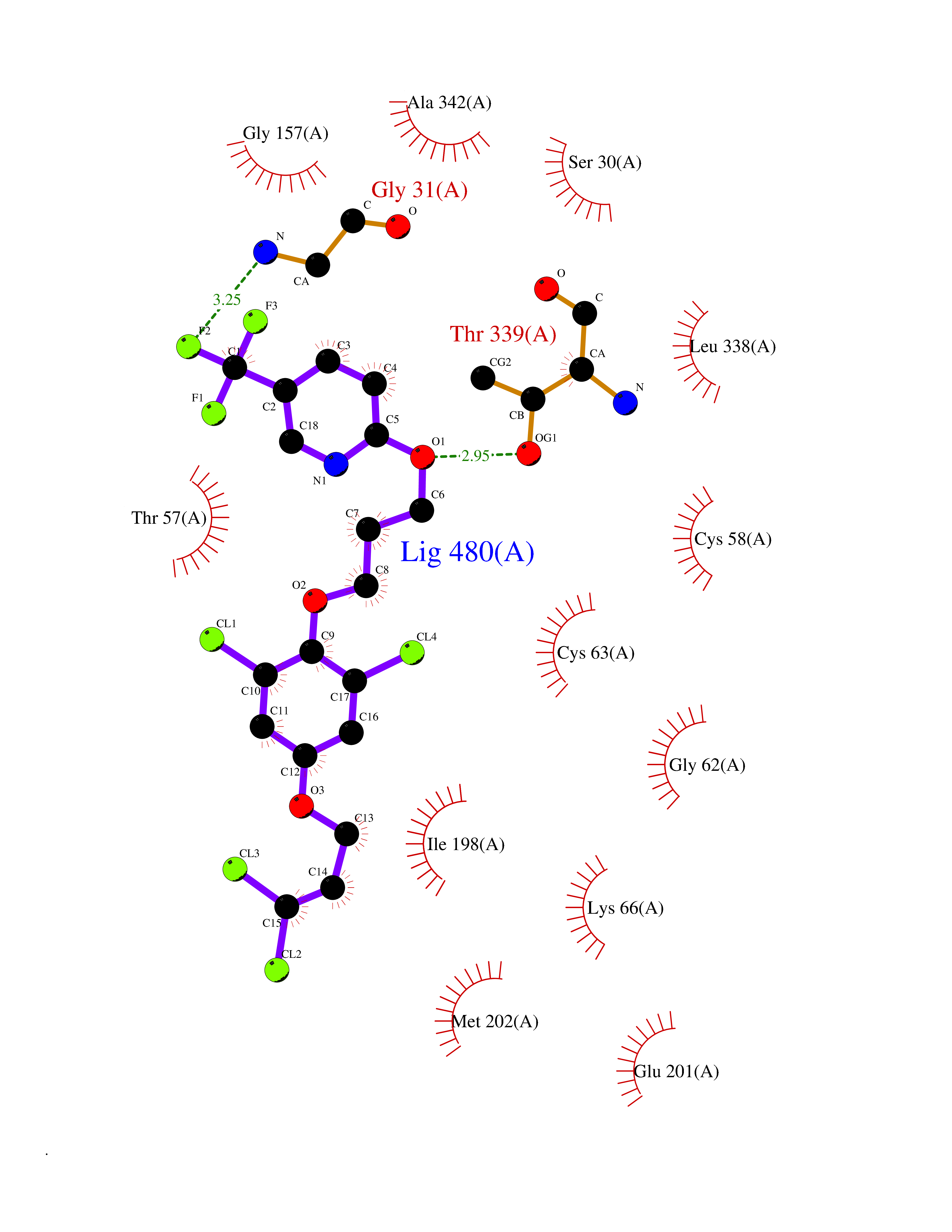 Ligplot Map 3D Binding mode Sequence AVASYDYLVIGGGSGGLASARRAAELGARAAVVESHKLGGTCVNVGCVPKKVMWNTAVHSEFMHDHADYGFPSCEGKFNWRVIKEKRDAYVSRLNAIYQNNLTKSHIEIIRGHAAFTSDPKPTIEVSGKKYTAPHILIATGGMPSTPHESQIPGASLGITSDGFFQLEELPGRSVIVGAGYIAVEMAGILSALGSKTSLMIRHDKVLRSFDSMISTNCTEELENAGVEVLKFSQVKEVKKTLSGLEVSMVTAVPGRLPVMTMIPDVDCLLWAIGRVPNTKDLSLNKLGIQTDDKGHIIVDEFQNTNVKGIYAVGDVCGKALLTPVAIAAGRKLAHRLFEYKEDSKLDYNNIPTVVFSHPPIGTVGLTEDEAIHKYGIENVKTYSTSFTPMYHAVTKRKTKCVMKMVCANKEEKVVGIHMQGLGCDEMLQGFAVAVKMGATKADFDNTVAIHPTSSEELVTLR Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Retinoic acid receptor RXR-alpha (RXRA) | 2P1T | 7.09 | |
Target general information Gen name RXRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor alpha; RXRalpha; Nuclear receptor subfamily 2 group B member 1; NR2B1 Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. The high affinity ligand for RXRs is 9-cis retinoic acid. RXRA serves as a common heterodimeric partner for a number of nuclear receptors. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. The RXRA/PPARA heterodimer is required for PPARA transcriptional activity on fatty acid oxidation genes such as ACOX1 and the P450 system genes. Receptor for retinoic acid. Related diseases Lichtenstein-Knorr syndrome (LIKNS) [MIM:616291]: An autosomal recessive neurologic disorder characterized by progressive cerebellar ataxia and severe progressive sensorineural hearing loss. {ECO:0000269|PubMed:25205112, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08063; DB08402; DB07863; DB07557; DB00459; DB00210; DB01436; DB00523; DB00132; DB04557; DB00307; DB01393; DB03756; DB00749; DB00926; DB05956; DB04224; DB02746; DB00412; DB00755; DB08601 Interacts with O14503; P35637; Q15648; Q71SY5; Q15788; Q15596; P55055; P55055-1; Q13133; P27986; P37231; P37231-1; P10276; P42224; P11473; P97792-1; Q9JLI4; P04625; PRO_0000278730 [Q03463] EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; DNA-binding; Host-virus interaction; Isopeptide bond; Metal-binding; Mitochondrion; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 24958.9 Length 221 Aromaticity 0.07 Instability index 39.47 Isoelectric point 6.16 Charge (pH=7) -3.45 2D Binding mode Binding energy (Kcal/mol) -9.68  Molscript Map 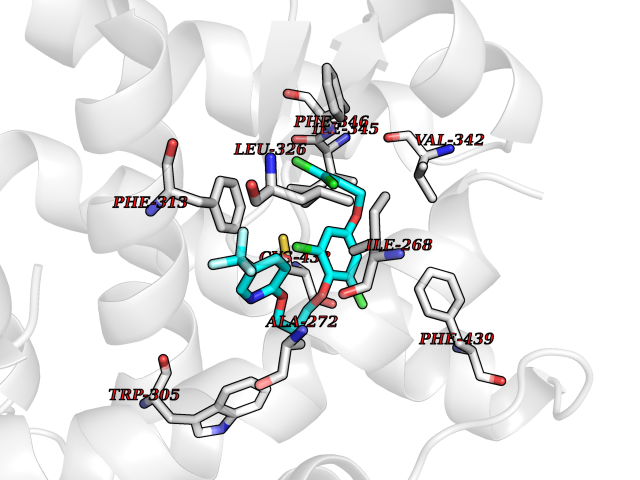 Pymol Map 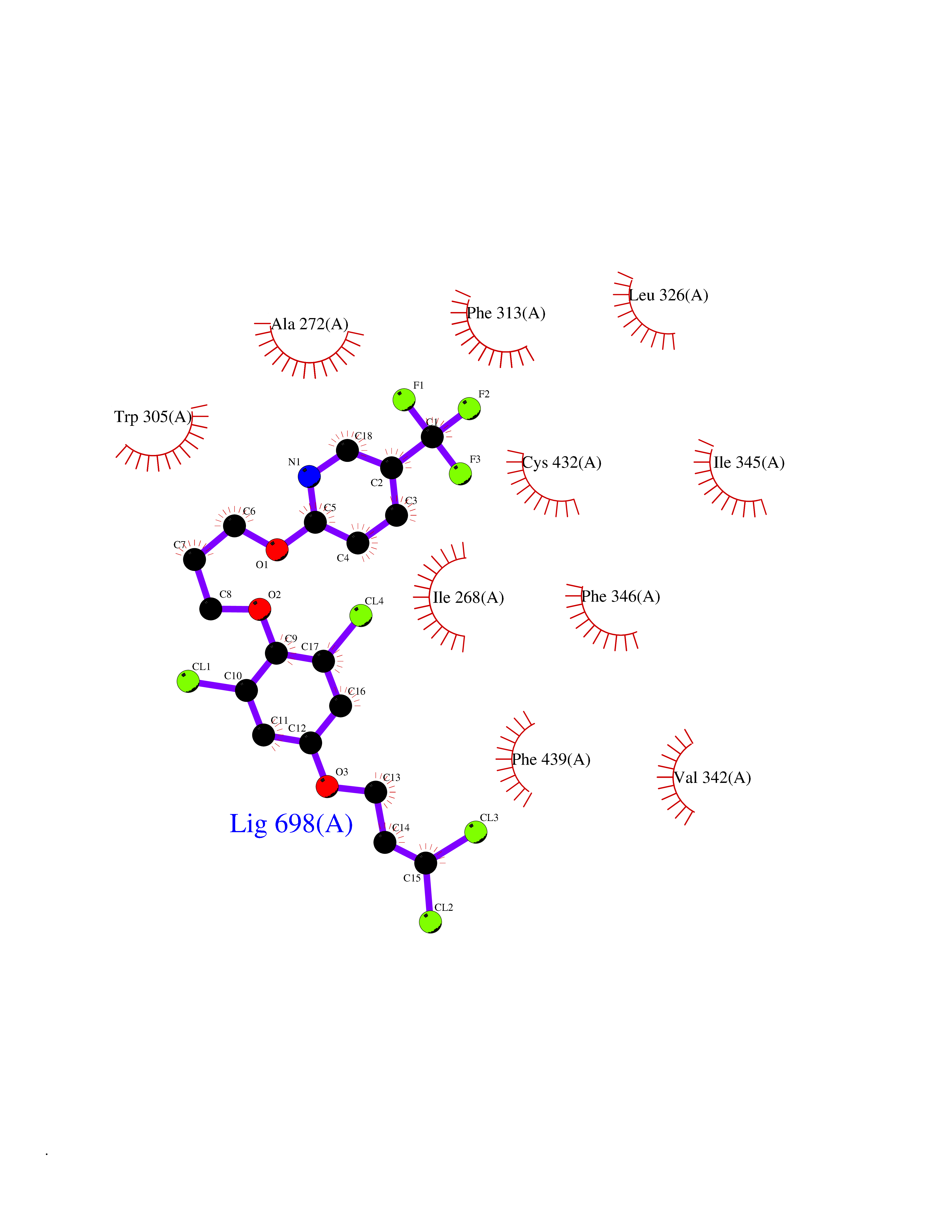 Ligplot Map 3D Binding mode Sequence DMPVERILEAELAVEDPVTNICQAADKQLFTLVEWAKRIPHFSELPLDDQVILLRAGWNELLIASFSHRSIAVKDGILLATGLHVHRNSAHSAGVGAIFDRVLTELVSKMRDMQMDKTELGCLRAIVLFNPDSKGLSNPAEVEALREKVYASLEAYCKHKYPEQPGRFAKLLLRLPALRSIGLKCLEHLFFFKLIGDTPIDTFLMEMLEAPHKILHRLLQD Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Bacterial Flavohemoglobin (Bact hmp) | 1GVH | 7.09 | |
Target general information Gen name Bact hmp Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms Nitric oxide dioxygenase; NOD; NO oxygenase; Hemoglobin-like protein; HMP; Ferrisiderophore reductase B; Dihydropteridine reductase Protein family Globin family, Two-domain flavohemoproteins subfamily; Flavoprotein pyridine nucleotide cytochrome reductase family Biochemical class Paired donor oxygen oxidoreductase Function Various electron acceptors arealso reduced by HMP in vitro, including dihydropterine, ferrisiderophores, ferric citrate, cytochrome c, nitrite, S-nitrosoglutathione, and alkylhydroperoxides. However, it is unknown if these reactions are of any biological significance in vivo. Related diseases Ovarian dysgenesis 1 (ODG1) [MIM:233300]: An autosomal recessive disease characterized by primary amenorrhea, variable development of secondary sex characteristics, poorly developed streak ovaries, and high serum levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH). {ECO:0000269|PubMed:10551778, ECO:0000269|PubMed:11889179, ECO:0000269|PubMed:12571157, ECO:0000269|PubMed:12915623, ECO:0000269|PubMed:7553856, ECO:0000269|PubMed:9769327, ECO:0000269|PubMed:9851774}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ovarian hyperstimulation syndrome (OHSS) [MIM:608115]: Disorder which occurs either spontaneously or most often as an iatrogenic complication of ovarian stimulation treatments for in vitro fertilization. The clinical manifestations vary from abdominal distention and discomfort to potentially life-threatening, massive ovarian enlargement and capillary leak with fluid sequestration. Pathologic features of this syndrome include the presence of multiple serous and hemorrhagic follicular cysts lined by luteinized cells, a condition called hyperreactio luteinalis. {ECO:0000269|PubMed:12930927, ECO:0000269|PubMed:12930928, ECO:0000269|PubMed:15080154, ECO:0000269|PubMed:16278261, ECO:0000269|PubMed:17721928, ECO:0000269|PubMed:24058690, ECO:0000269|PubMed:25581598}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number EC 1.14.12.17 Uniprot keywords 3D-structure; Cytoplasm; Detoxification; Direct protein sequencing; FAD; Flavoprotein; Heme; Iron; Metal-binding; NAD; NADP; Oxidoreductase; Oxygen transport; Reference proteome; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 43867.1 Length 396 Aromaticity 0.1 Instability index 28.85 Isoelectric point 5.48 Charge (pH=7) -12.32 2D Binding mode Binding energy (Kcal/mol) -9.67 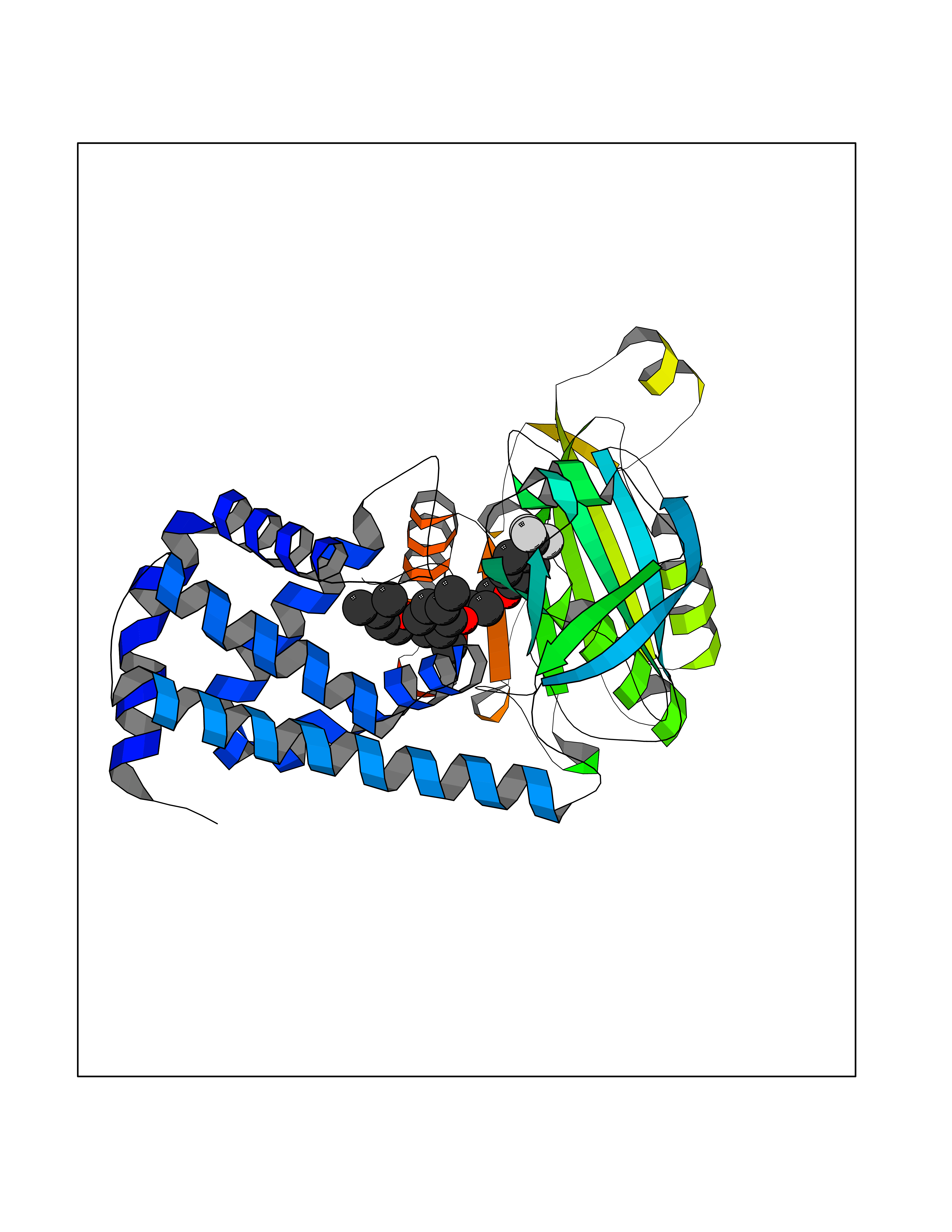 Molscript Map 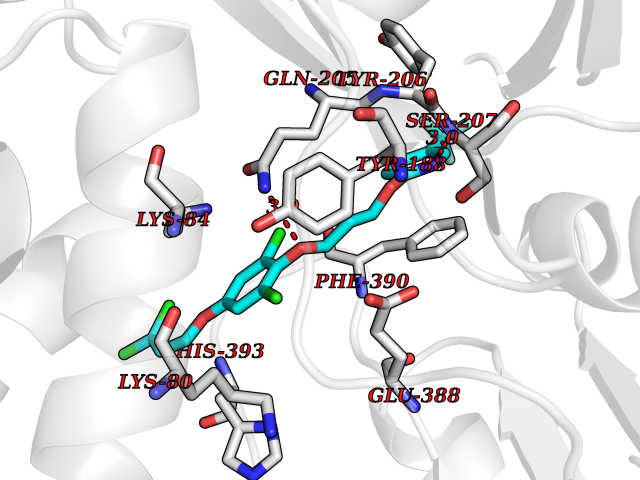 Pymol Map 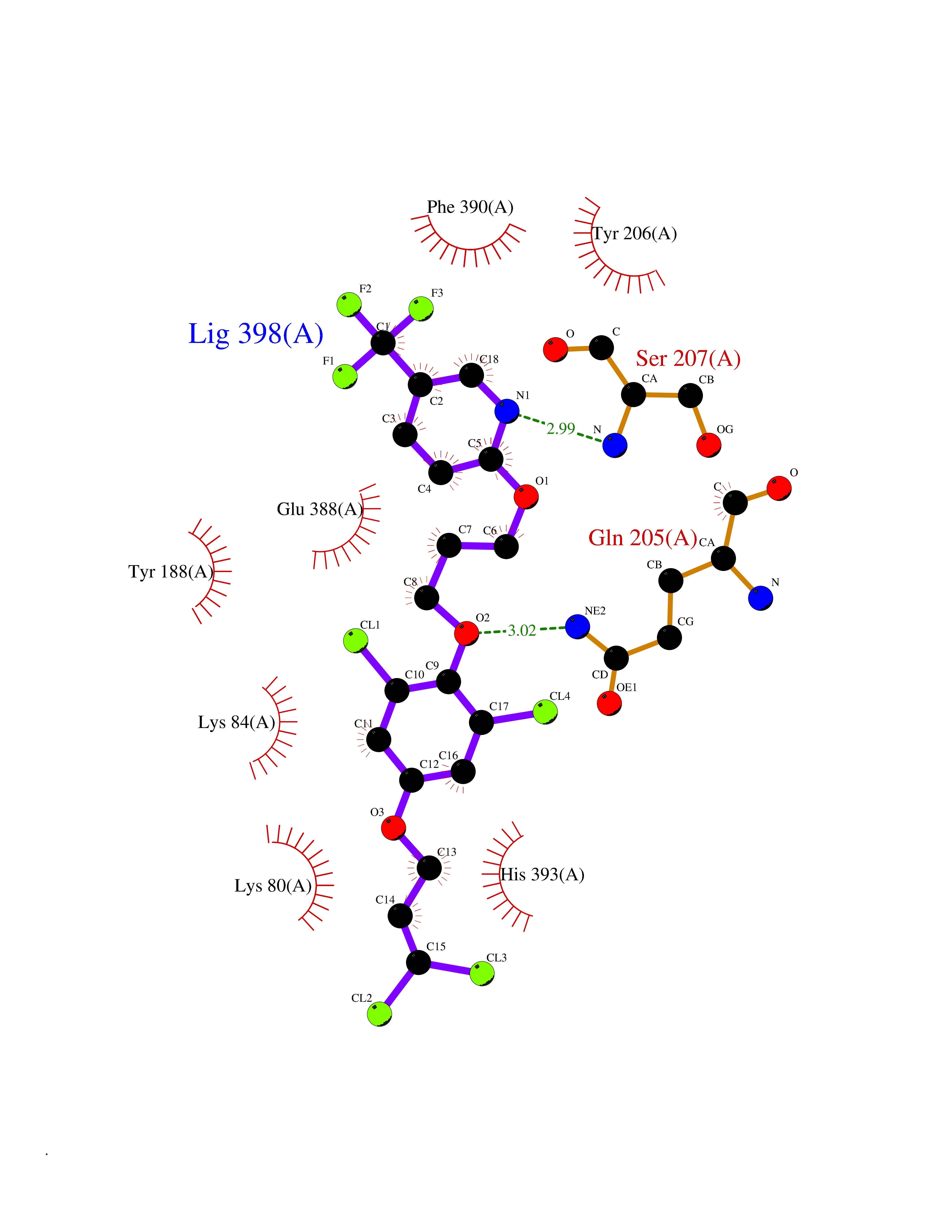 Ligplot Map 3D Binding mode Sequence MLDAQTIATVKATIPLLVETGPKLTAHFYDRMFTHNPELKEIFNMSNQRNGDQREALFNAIAAYASNIENLPALLPAVEKIAQKHTSFQIKPEQYNIVGEHLLATLDEMFSPGQEVLDAWGKAYGVLANVFINREAEIYNENASKAGGWEGTRDFRIVAKTPRSALITSFELEPVDGGAVAEYRPGQYLGVWLKPEGFPHQEIRQYSLTRKPDGKGYRIAVKREEGGQVSNWLHNHANVGDVVKLVAPAGDFFMAVADDTPVTLISAGVGQTPMLAMLDTLAKAGHTAQVNWFHAAENGDVHAFADEVKELGQSLPRFTAHTWYRQPSEADRAKGQFDSEGLMDLSKLEGAFSDPTMQFYLCGPVGFMQFTAKQLVDLGVKQENIHYECFGPHKVL Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Dimethylglycine oxidase | 1PJ5 | 7.09 | |
Target general information Gen name dmg Organism Arthrobacter globiformis Uniprot ID TTD ID NA Synonyms NA Protein family GcvT family Biochemical class Oxidoreductase Function Dimethylglycine oxidase activity.Nucleotide binding. Related diseases Curry-Jones syndrome (CRJS) [MIM:601707]: A multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. The disease is caused by variants affecting the gene represented in this entry. 8 individuals have been identified with the disease-causing mutation Phe-412 and all were mosaic. The mutation could not be reliably detected in blood, greatest success rates were obtained with affected tissues obtained by invasive procedures. It is thought that the mutation has arisen postzygotically early during embryonic development (PubMed:27236920). This mutation has also been identified in ameloblastoma, medulloblastoma, meningioma, and basal cell carcinoma, and has been reported as the oncogenic driver in some of these tumors (PubMed:24859340). {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. Drugs (DrugBank ID) DB03256; DB03147 Interacts with NA EC number 1.5.3.10 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 45912.2 Length 427 Aromaticity 0.07 Instability index 43.46 Isoelectric point 4.83 Charge (pH=7) -20.69 2D Binding mode Binding energy (Kcal/mol) -9.68 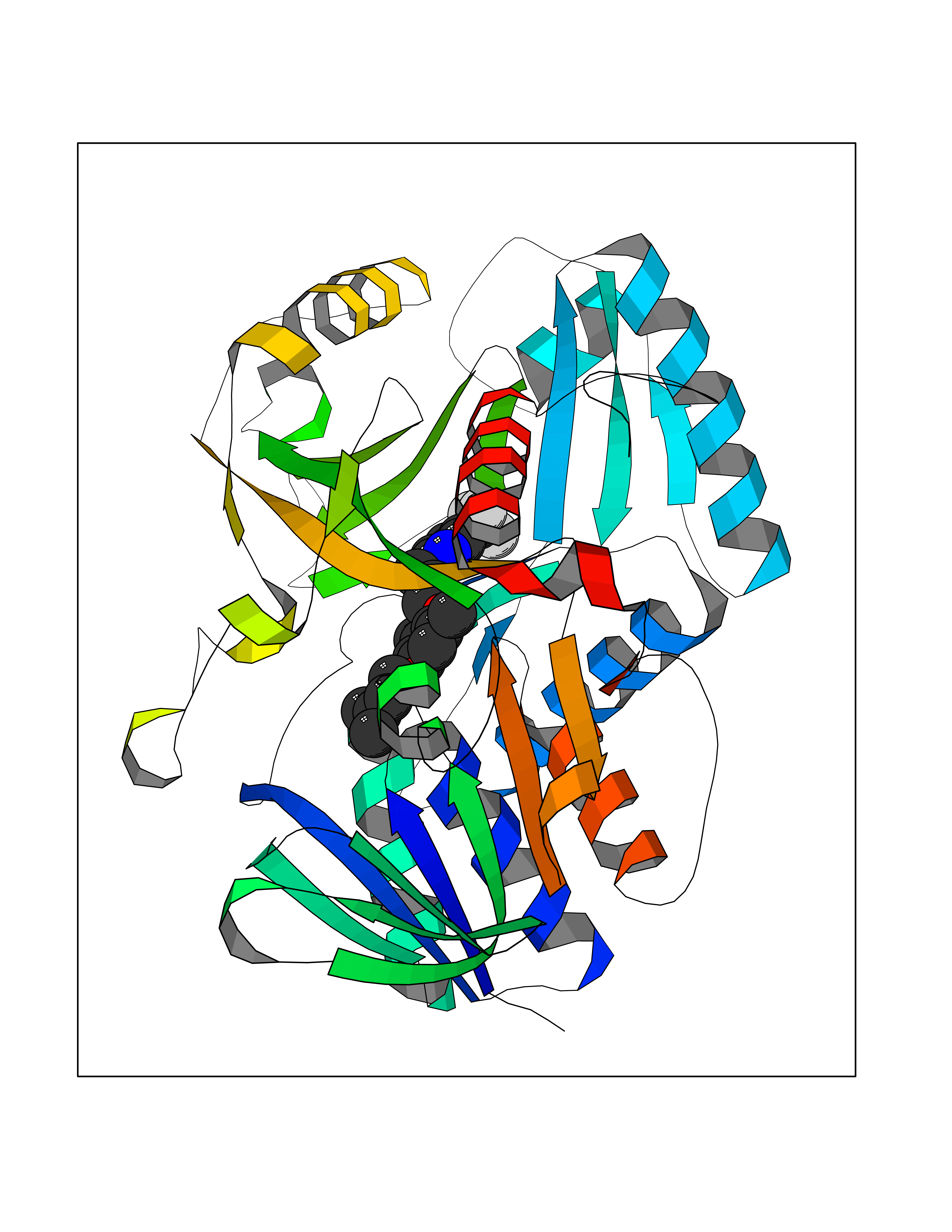 Molscript Map 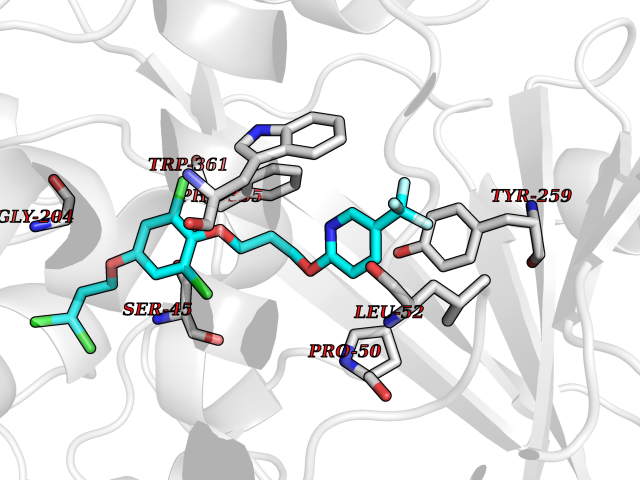 Pymol Map 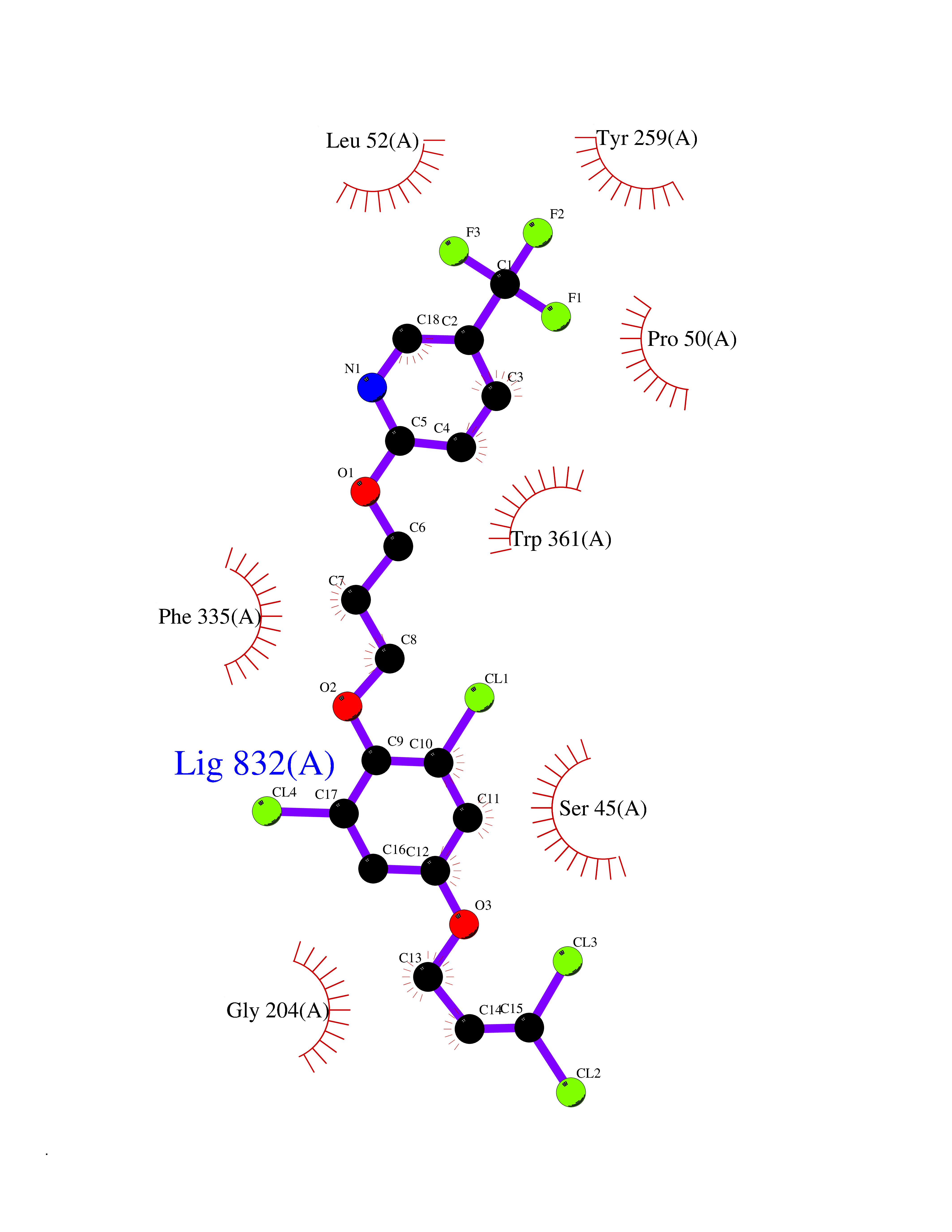 Ligplot Map 3D Binding mode Sequence TPRIVIIGAGIVGTNLADELVTRGWNNITVLDQGPLNMPGGSTSHAPGLVFQTNPSKTMASFAKYTVEKLLSLTEDGVSCFNQVGGLEVATTETRLADLKRKLGYAAAWGIEGRLLSPAECQELYPLLDGENILGGLHVPSDGLASAARAVQLLIKRTESAGVTYRGSTTVTGIEQSGGRVTGVQTADGVIPADIVVSCAGFWGAKIGAMIGMAVPLLPLAHQYVKTTPVPAQQGRNDQPNGARLPILRHQDQDLYYREHGDRYGIGSYAHRPMPVDVDTLGAYAPETVSEHHMPSRLDFTLEDFLPAWEATKQLLPALADSEIEDGFNGIFSFTPDGGPLLGESKELDGFYVAEAVWVTHSAGVAKAMAELLTTGRSETDLGECDITRFEDVQLTPEYVSETSQQNFVEIYDVLHPLQPRLSPRNL Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | BUB1 mitotic checkpoint serine/threonine kinase (BUB1) | 6F7B | 7.09 | |
Target general information Gen name BUB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hBUB1; Mitotic checkpoint serine/threonine-protein kinase BUB1; BUB1L; BUB1A Protein family Protein kinase superfamily, Ser/Thr protein kinase family, BUB1 subfamily Biochemical class Kinase Function Has a key role in the assembly of checkpoint proteins at the kinetochore, being required for the subsequent localization of CENPF, BUB1B, CENPE and MAD2L1. Required for the kinetochore localization of PLK1. Required for centromeric enrichment of AUKRB in prometaphase. Plays an important role in defining SGO1 localization and thereby affects sister chromatid cohesion. Acts as a substrate for anaphase-promoting complex or cyclosome (APC/C) in complex with its activator CDH1 (APC/C-Cdh1). Necessary for ensuring proper chromosome segregation and binding to BUB3 is essential for this function. Can regulate chromosome segregation in a kinetochore-independent manner. Can phosphorylate BUB3. The BUB1-BUB3 complex plays a role in the inhibition of APC/C when spindle-assembly checkpoint is activated and inhibits the ubiquitin ligase activity of APC/C by phosphorylating its activator CDC20. This complex can also phosphorylate MAD1L1. Kinase activity is essential for inhibition of APC/CCDC20 and for chromosome alignment but does not play a major role in the spindle-assembly checkpoint activity. Mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. Serine/threonine-protein kinase that performs 2 crucial functions during mitosis: it is essential for spindle-assembly checkpoint signaling and for correct chromosome alignment. Related diseases Microcephaly 30, primary, autosomal recessive (MCPH30) [MIM:620183]: A form of microcephaly, a disease defined as a head circumference more than 3 standard deviations below the age, sex and ethnically matched mean. Brain weight is markedly reduced and the cerebral cortex is disproportionately small. MCPH30 is characterized by small head, poor overall growth, and global developmental delay with variably impaired intellectual development. Affected individuals may also have variable congenital anomalies, including atrial septal defect, dysmorphic facial features, tracheal stenosis, and anomalies of the skin and teeth. {ECO:0000269|PubMed:35044816}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O95376; O60566; O43684; P46108; Q8NG31; Q8NG31-2; Q9GZQ8; P03070 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell cycle; Cell division; Centromere; Chromosome; Chromosome partition; Host-virus interaction; Intellectual disability; Kinase; Kinetochore; Mitosis; Nucleotide-binding; Nucleus; Phosphoprotein; Primary microcephaly; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39564.8 Length 343 Aromaticity 0.12 Instability index 31.17 Isoelectric point 8.49 Charge (pH=7) 4.7 2D Binding mode Binding energy (Kcal/mol) -9.67 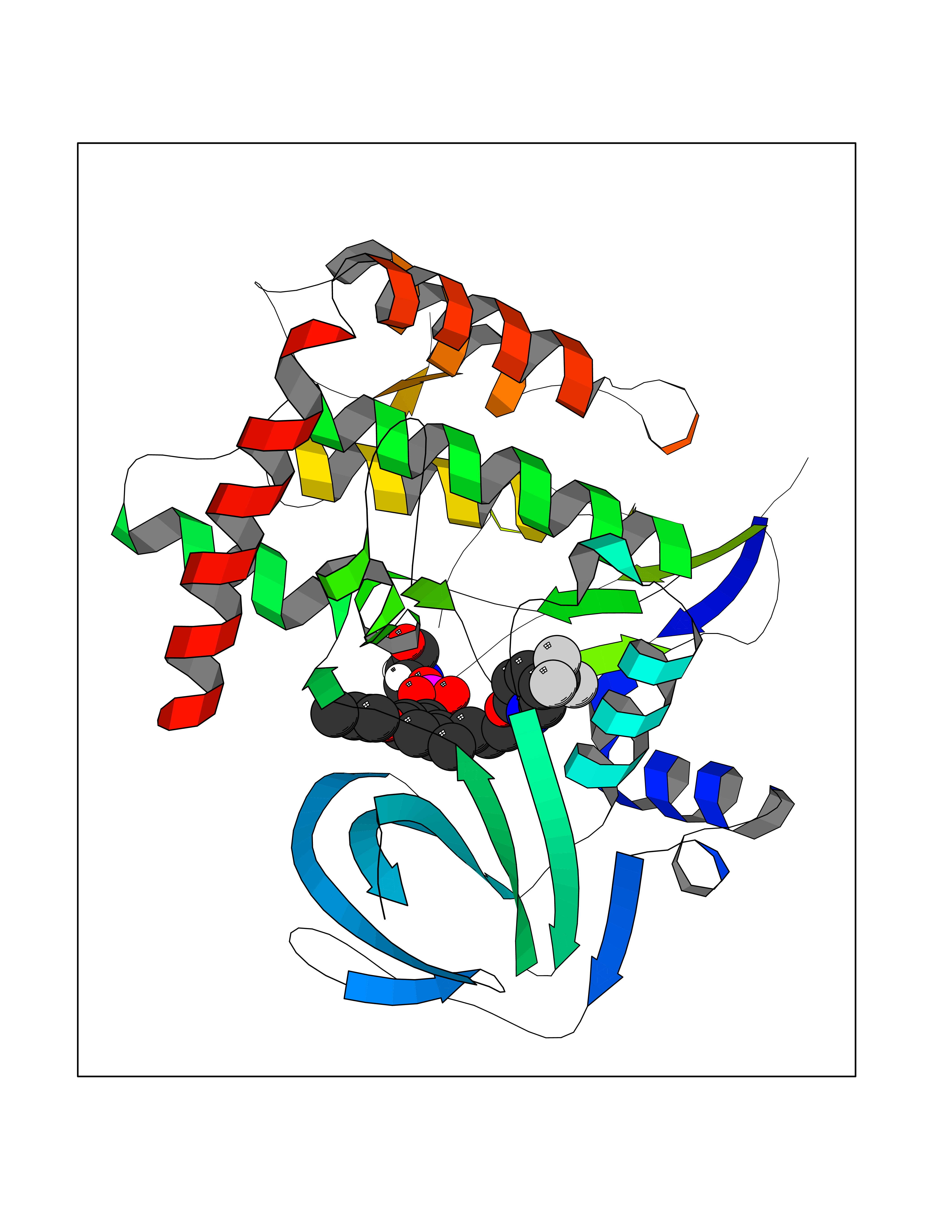 Molscript Map 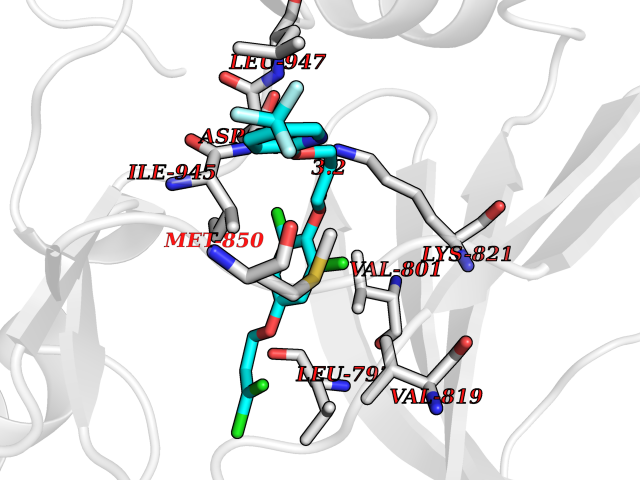 Pymol Map 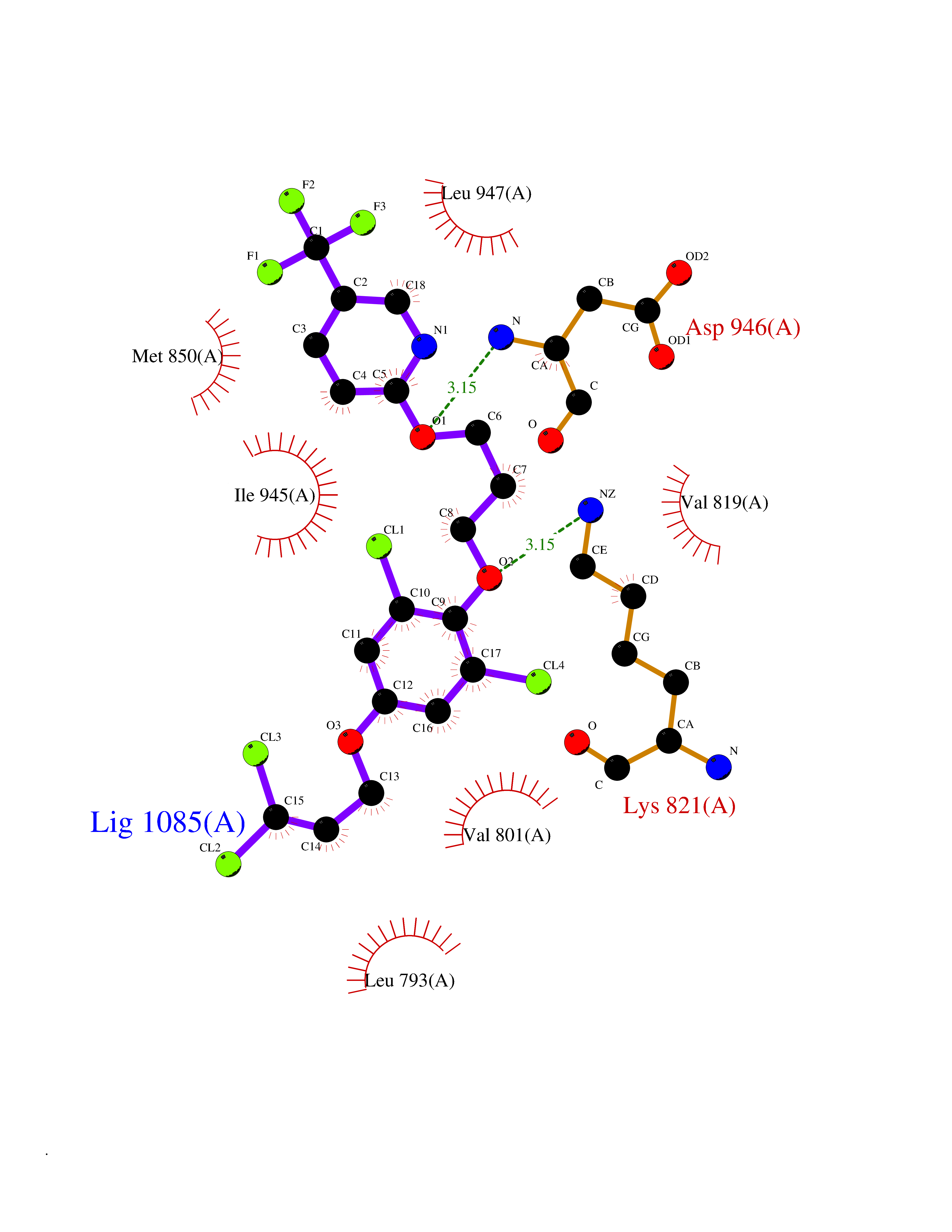 Ligplot Map 3D Binding mode Sequence APNFIVGNPWDDKLIFKLLSGLSKPVSSYPNTFEWQCKLPAIKPKTEFQLGSKLVYVHHLLGEGAFAQVYEATQKNKQKFVLKVQKPANPWEFYIGTQLMERLKPSMQHMFMKFYSAHLFQNGSVLVGELYSYGTLLNAINLYKNTPEKVMPQGLVISFAMRMLYMIEQVHDCEIIHGDIKPDNFILGNGFLEQDDEDDLSAGLALIDLGQSIDMKLFPKGTIFTAKCETXGFQCVEMLSNKPWNYQIDYFGVAATVYCMLFGTYMKVKNEECKPEGLFRRLPHLDMWNEFFHVMLNIPDCHHLPSLDLLRQKLKKVFQQHYTNKIRALRNRLIVLLLECKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Sphingosine kinase 1 (SPHK1) | 3VZB | 7.09 | |
Target general information Gen name SPHK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SPK 1; SPK; SPHK1; SK 1; Acetyltransferase SPHK1 Protein family NA Biochemical class Kinase Function Acts on D-erythro-sphingosine and to a lesser extent sphinganine, but not other lipids, such as D,L-threo-dihydrosphingosine, N,N-dimethylsphingosine, diacylglycerol, ceramide, or phosphatidylinositol. In contrast to proapoptotic SPHK2, has a negative effect on intracellular ceramide levels, enhances cell growth and inhibits apoptosis. Involved in the regulation of inflammatory response and neuroinflammation. Via the product sphingosine 1-phosphate, stimulates TRAF2 E3 ubiquitin ligase activity, and promotes activation of NF-kappa-B in response to TNF signaling leading to IL17 secretion. In response to TNF and in parallel to NF-kappa-B activation, negatively regulates RANTES inducion through p38 MAPK signaling pathway. Involved in endocytic membrane trafficking induced by sphingosine, recruited to dilate endosomes, also plays a role on later stages of endosomal maturation and membrane fusion independently of its kinase activity. In Purkinje cells, seems to be also involved in the regulation of autophagosome-lysosome fusion upon VEGFA. Catalyzes the phosphorylation of sphingosine to form sphingosine 1-phosphate (SPP), a lipid mediator with both intra- and extracellular functions. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08868 Interacts with P07858; P68104; Q14192; Q2M3C7; Q9Y4K3; P13473-2; Q9Y371 EC number EC 2.7.1.91 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Calmodulin-binding; Cell membrane; Coated pit; Cytoplasm; Endosome; Kinase; Lipid metabolism; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Synapse; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 39813 Length 360 Aromaticity 0.08 Instability index 43.79 Isoelectric point 7.34 Charge (pH=7) 0.84 2D Binding mode Binding energy (Kcal/mol) -9.67 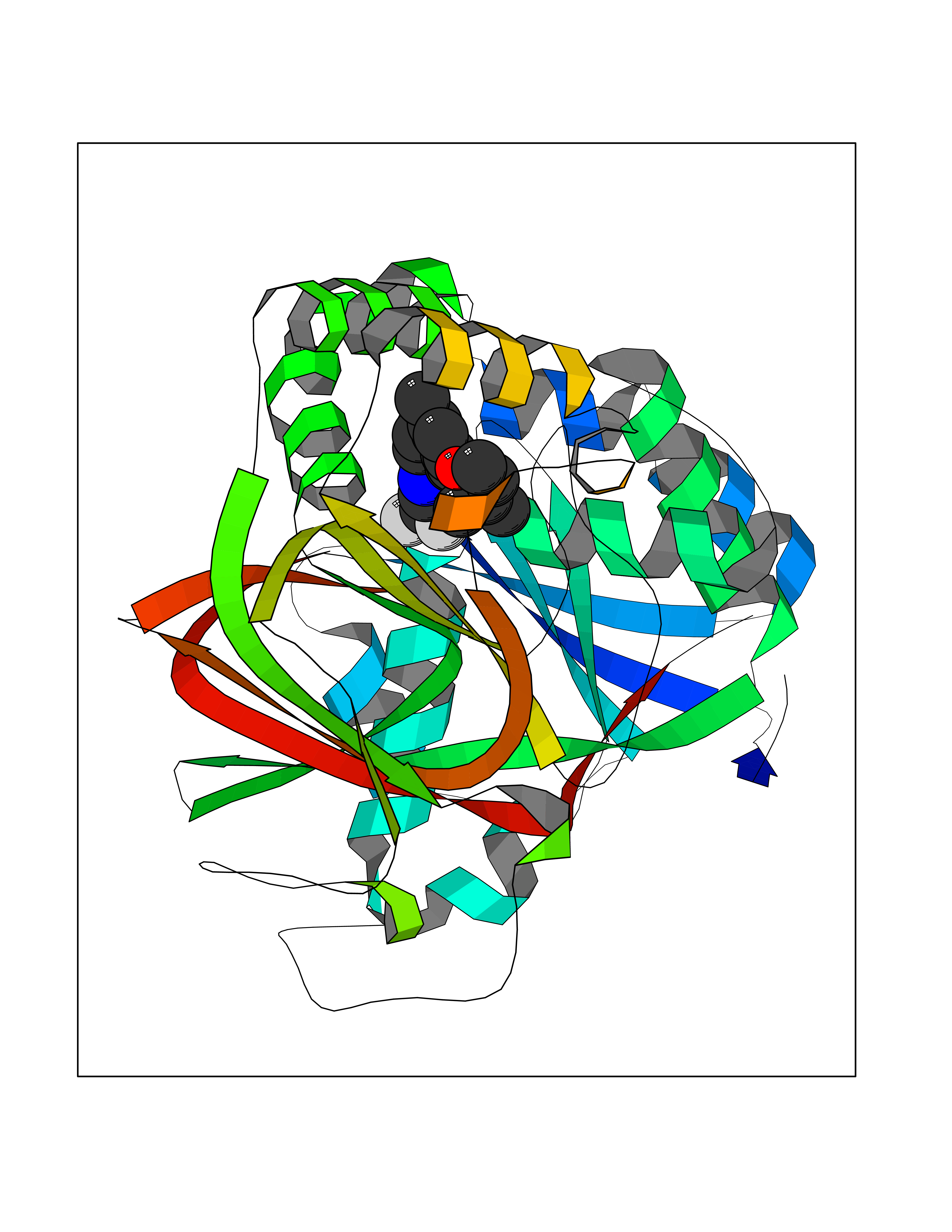 Molscript Map 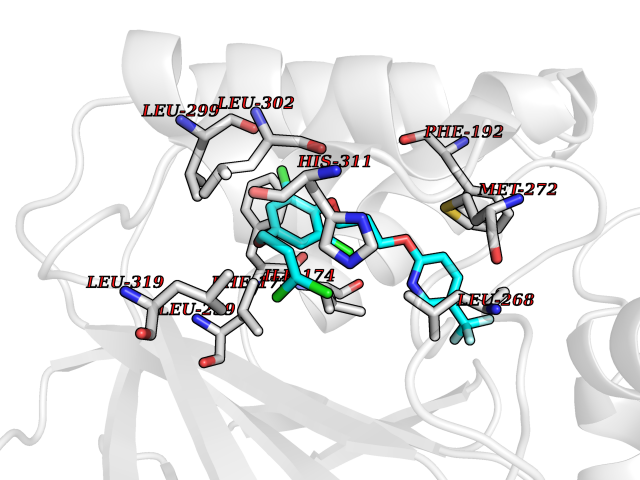 Pymol Map 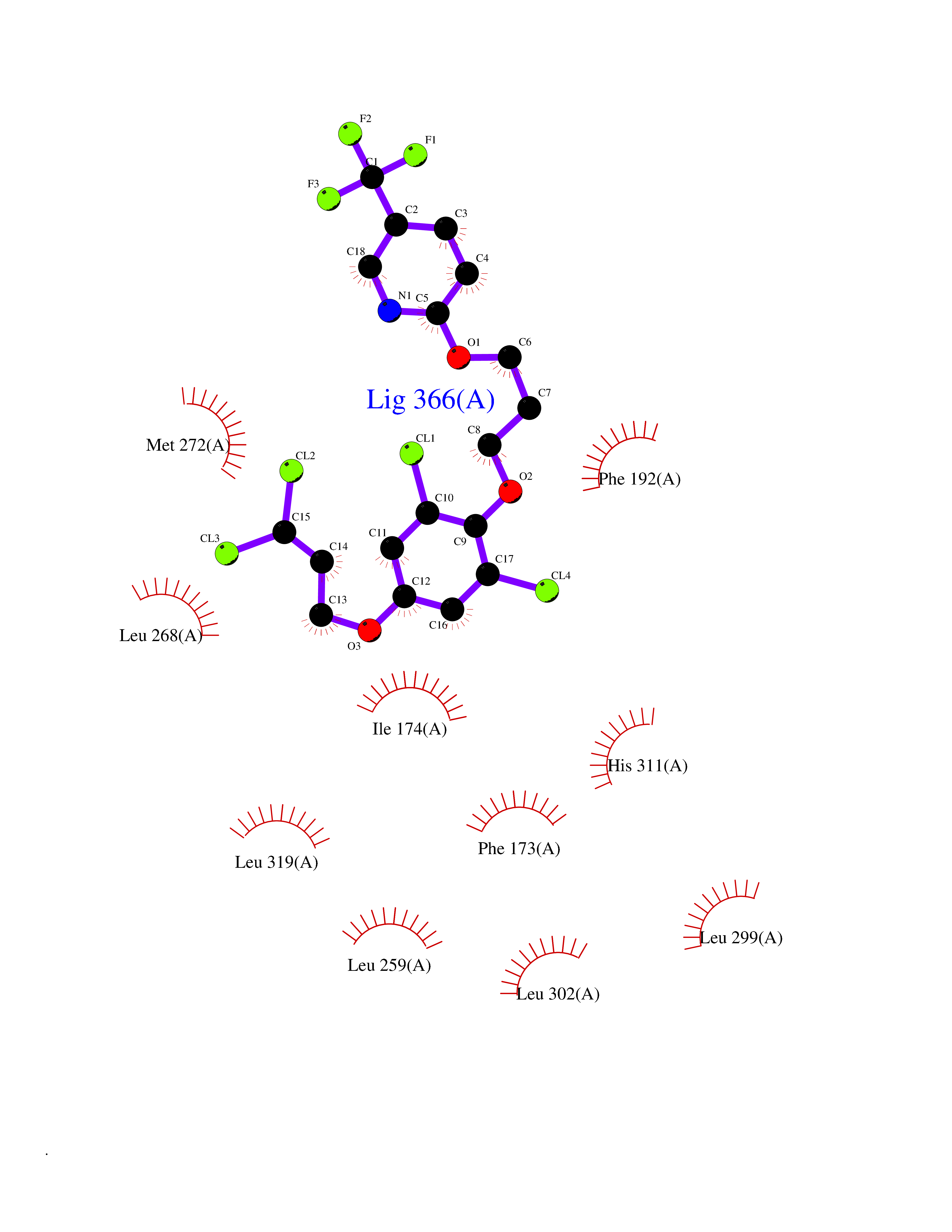 Ligplot Map 3D Binding mode Sequence AMGSGVLPRPCRVLVLLNPRGGKGKALQLFRSHVQPLLAEAEISFTLMLTERRNHARELVRSEELGRWDALVVMSGDGLMHEVVNGLMERPDWETAIQKPLCSLPAGSGNALAASLNHYAGYEQVTNEDLLTNCTLLLCRRLLSPMNLLSLHTASGLRLFSVLSLAWGFIADVDLESEKYRRLGEMRFTLGTFLRLAALRTYRGRLAYLPVGRVGSKTPASPVVVQQGPVDAHLVPLEEPVPSHWTVVPDEDFVLVLALLHSHLGSEMFAAPMGRCAAGVMHLFYVRAGVSRAMLLRLFLAMEKGRHMEYECPYLVYVPVVAFRLEPKDGKGVFAVDGELMVSEAVQGQVHPNYFWMVSG Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Mineralocorticoid receptor (MR) | 4PF3 | 7.08 | |
Target general information Gen name NR3C2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor subfamily 3 group C member 2; Mineralocorticoid receptor; MLR; MCR; Inner ear mineralocorticoid receptor; Delta Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Binds to mineralocorticoid response elements (MRE) and transactivates target genes. The effect of MC is to increase ion and water transport and thus raise extracellular fluid volume and blood pressure and lower potassium levels. Receptor for both mineralocorticoids (MC) such as aldosterone and glucocorticoids (GC) such as corticosterone or cortisol. Related diseases Pseudohypoaldosteronism 1, autosomal dominant (PHA1A) [MIM:177735]: A salt wasting disease resulting from target organ unresponsiveness to mineralocorticoids. PHA1A is a mild form characterized by target organ defects confined to kidney. Patients may present with neonatal renal salt wasting with hyperkalaemic acidosis despite high aldosterone levels. These patients improve with age and usually become asymptomatic without treatment. {ECO:0000269|PubMed:11134129, ECO:0000269|PubMed:12788847, ECO:0000269|PubMed:16954160, ECO:0000269|PubMed:16972228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Early-onset hypertension with severe exacerbation in pregnancy (EOHSEP) [MIM:605115]: Inheritance is autosomal dominant. The disease is characterized by the onset of severe hypertension before the age of 20, and by suppression of aldosterone secretion. {ECO:0000269|PubMed:10884226, ECO:0000269|PubMed:15908963, ECO:0000269|PubMed:15967794}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04630; DB01013; DB04652; DB06780; DB01134; DB01395; DB00700; DB01023; DB16165; DB00687; DB13867; DB08906; DB00588; DB02998; DB00393; DB00396; DB00421; DB02901; DB13951; DB00624; DB13943; DB13944; DB15114 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Endoplasmic reticulum; Lipid-binding; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Steroid-binding; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29012.4 Length 249 Aromaticity 0.12 Instability index 51.27 Isoelectric point 6.3 Charge (pH=7) -2.08 2D Binding mode Binding energy (Kcal/mol) -9.66  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence TPSPVMVLENIEPEIVYAGYDSSKPDTAENLLSTLNRLAGKQMIQVVKWAKVLPGFKNLPLEDQITLIQYSWMSLLSFALSWRSYKHTNSQFLYFAPDLVFNEEKMHQSAMYELCQGMHQISLQFVRLQLTFEEYTIMKVLLLLSTIPKDGLKSQAAFEEMRTNYIKELRKMVTKCPNNSGQSWQRFYQLTKLLDSMHDLVSDLLEFCFYTFRESHALKVEFPAMLVEIISDQLPKVESGNVKPLYFHR Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Retinoic acid receptor RXR-beta (RXRB) | 5HJP | 7.08 | |
Target general information Gen name RXRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor beta; Nuclear receptor subfamily 2 group B member 2; NR2B2 Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE). Receptor for retinoic acid. Related diseases Noonan syndrome 13 (NS13) [MIM:619087]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. NS13 inheritance is autosomal dominant. There is considerable variability in severity. {ECO:0000269|PubMed:32721402}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08175; DB00459; DB00210; DB00523; DB00307; DB01393; DB03756; DB00926; DB01941; DB07929; DB02746; DB00412; DB00799; DB07080; DB00755 Interacts with Q00975; Q9HB07; F1D8P7; Q13133; Q13133-3; Q96RI1-1; P04150; Q9NRD5; P37231; P10276; P10276-2; P10826-2; P13631; Q6IQ16; Q13137; Q96B26; Q08379; Q6A162; Q9UJV3-2; Q13133-3; Q96RI1-1; O43586; P10276; P10826-2; Q8IUQ4-2; O75528; Q12800; Q9UBB9; Q05BL1; P14373; O94972; Q96S82 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 28845.8 Length 251 Aromaticity 0.08 Instability index 54.86 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -9.66  Molscript Map 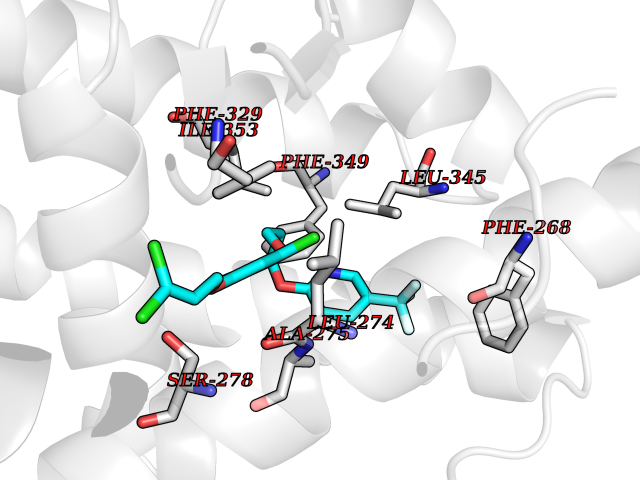 Pymol Map 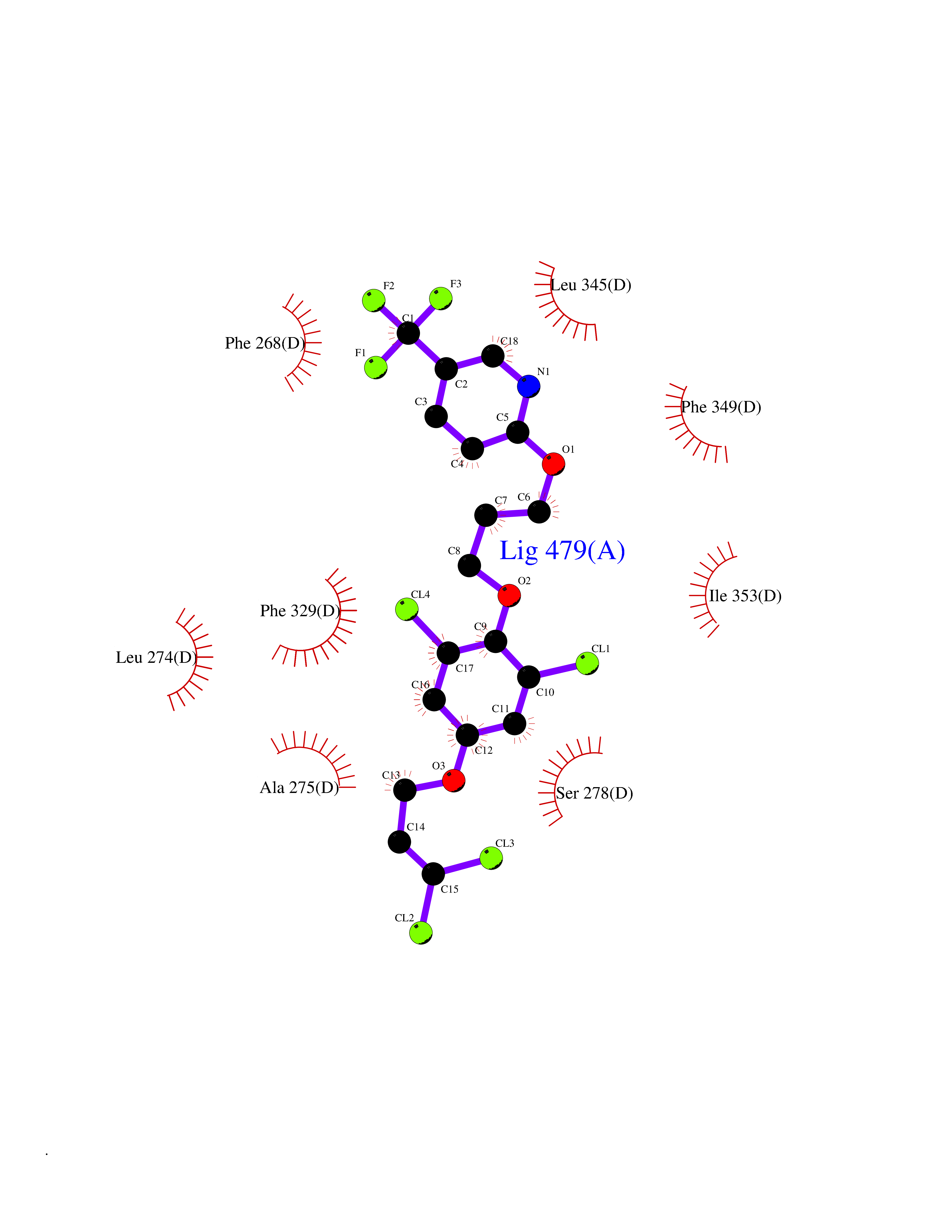 Ligplot Map 3D Binding mode Sequence QLTAAQELMIQQLVAAQLQCNKRSFSDQPKVTPWPSASQQRFAHFTELAIISVQEIVDFAKQVPGFLQLGREDQIALLKASTIEIMLLETARRYNHETECITFLKDFTYSKDDFHRAGLQVEFINPIFEFSRAMRRLGLDDAEYALLIAINIFSADRPNVQEPGRVEALQQPYVEALLSYTRIKRPQDQLRFPRMLMKLVSLRTLSSVHSEQVFALRLQDKKLPPLLSEIWDVHEGSGSGSHKILHRLLQD Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Glycolipid transfer protein | 3RZN | 7.08 | |
Target general information Gen name GLTP Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family GLTP family Biochemical class Lipid transport Function Glycolipid binding.Glycolipid transporter activity.Identical protein binding.Intermembrane lipid transfer activity.Lipid binding. Related diseases Brugada syndrome 7 (BRGDA7) [MIM:613120]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:20031595}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Atrial fibrillation, familial, 16 (ATFB16) [MIM:613120]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:20558140, ECO:0000269|PubMed:21051419}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03600; DB04465; DB03017; DB03203 Interacts with Q96DZ9; Q9NZD2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Lipid transport; Proteomics identification; Reference proteome; Repeat; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23534.1 Length 206 Aromaticity 0.11 Instability index 36.45 Isoelectric point 7.08 Charge (pH=7) 0.1 2D Binding mode Binding energy (Kcal/mol) -9.65 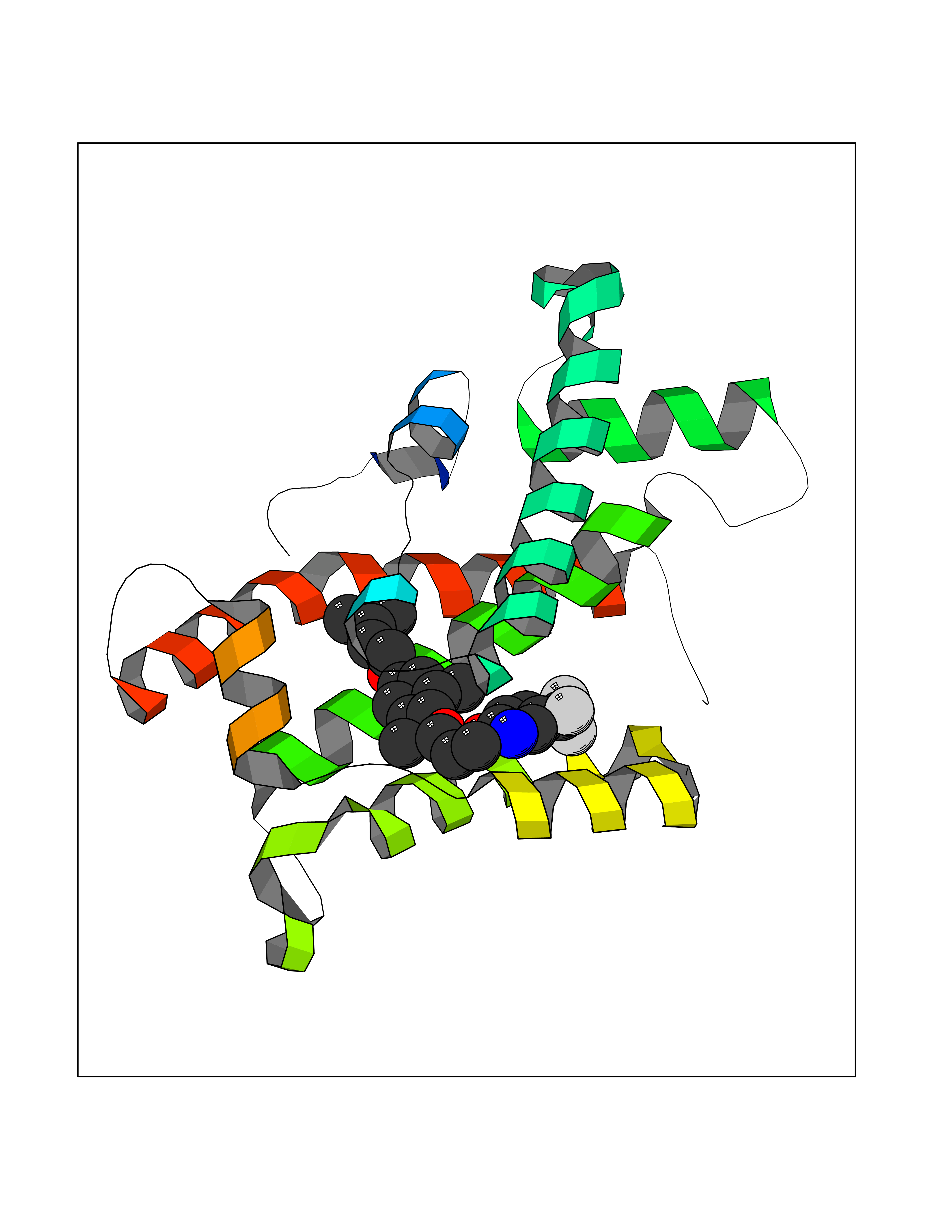 Molscript Map 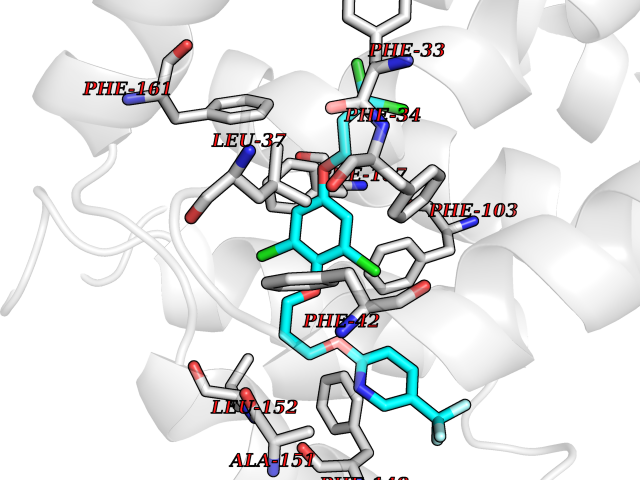 Pymol Map 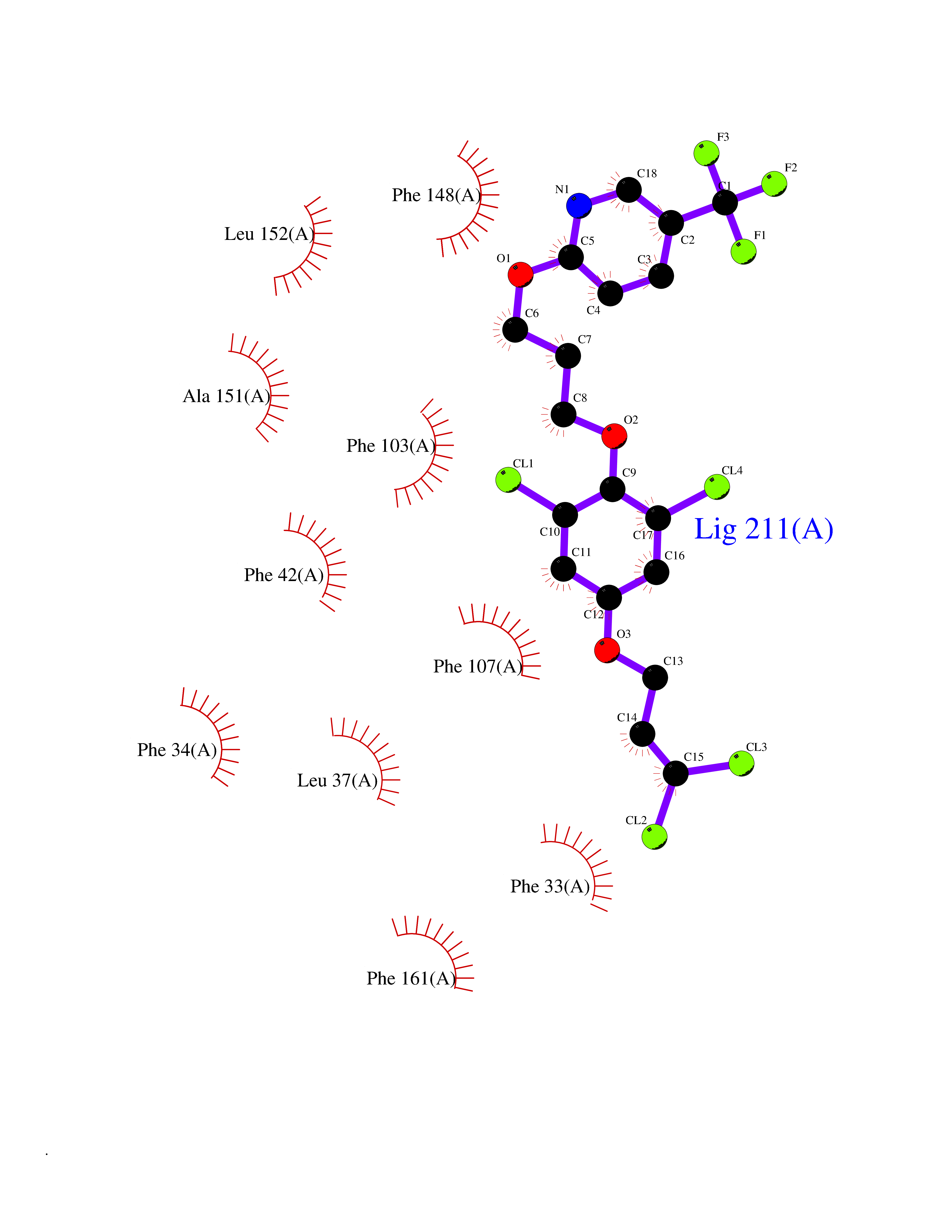 Ligplot Map 3D Binding mode Sequence LAEHLLKPLPADKQIETGPFLEAVSHLPPFFDCLGSPVFTPIKADISGNITKIKAVYDTNPAKFRTLQNILEVEKEMYGAEWPKVGATLALMWLKRGLRFIQVFLQSICDGERDENHPNLIRVNATKAYEMALKKYHGWIVQKIFQAALYAAPYKSDFLKALSKGQNVTEEECLEKIRLFLVNYTATIDVIYEMYTQMNAELNYKV Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | 4-cresol dehydrogenase [hydroxylating] flavoprotein subunit | 1WVF | 7.07 | |
Target general information Gen name pchF Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Oxidoreductase Function 4-cresol dehydrogenase (hydroxylating) activity.Flavin adenine dinucleotide binding.Oxidoreductase activity, acting on CH-OH group of donors. Related diseases Dihydrolipoamide dehydrogenase deficiency (DLDD) [MIM:246900]: An autosomal recessive metabolic disorder characterized biochemically by a combined deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC), pyruvate dehydrogenase complex (PDC), and alpha-ketoglutarate dehydrogenase complex (KGDC). Clinically, affected individuals have lactic acidosis and neurologic deterioration due to sensitivity of the central nervous system to defects in oxidative metabolism. {ECO:0000269|PubMed:10448086, ECO:0000269|PubMed:11687750, ECO:0000269|PubMed:12925875, ECO:0000269|PubMed:15712224, ECO:0000269|PubMed:16442803, ECO:0000269|PubMed:16770810, ECO:0000269|PubMed:17404228, ECO:0000269|PubMed:20160912, ECO:0000269|PubMed:8506365, ECO:0000269|PubMed:8968745, ECO:0000269|PubMed:9540846, ECO:0000269|PubMed:9934985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.17.9.1 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase; Plasmid Protein physicochemical properties Chain ID A Molecular weight (Da) 57240.8 Length 515 Aromaticity 0.1 Instability index 30.94 Isoelectric point 6.06 Charge (pH=7) -4.42 2D Binding mode Binding energy (Kcal/mol) -9.64 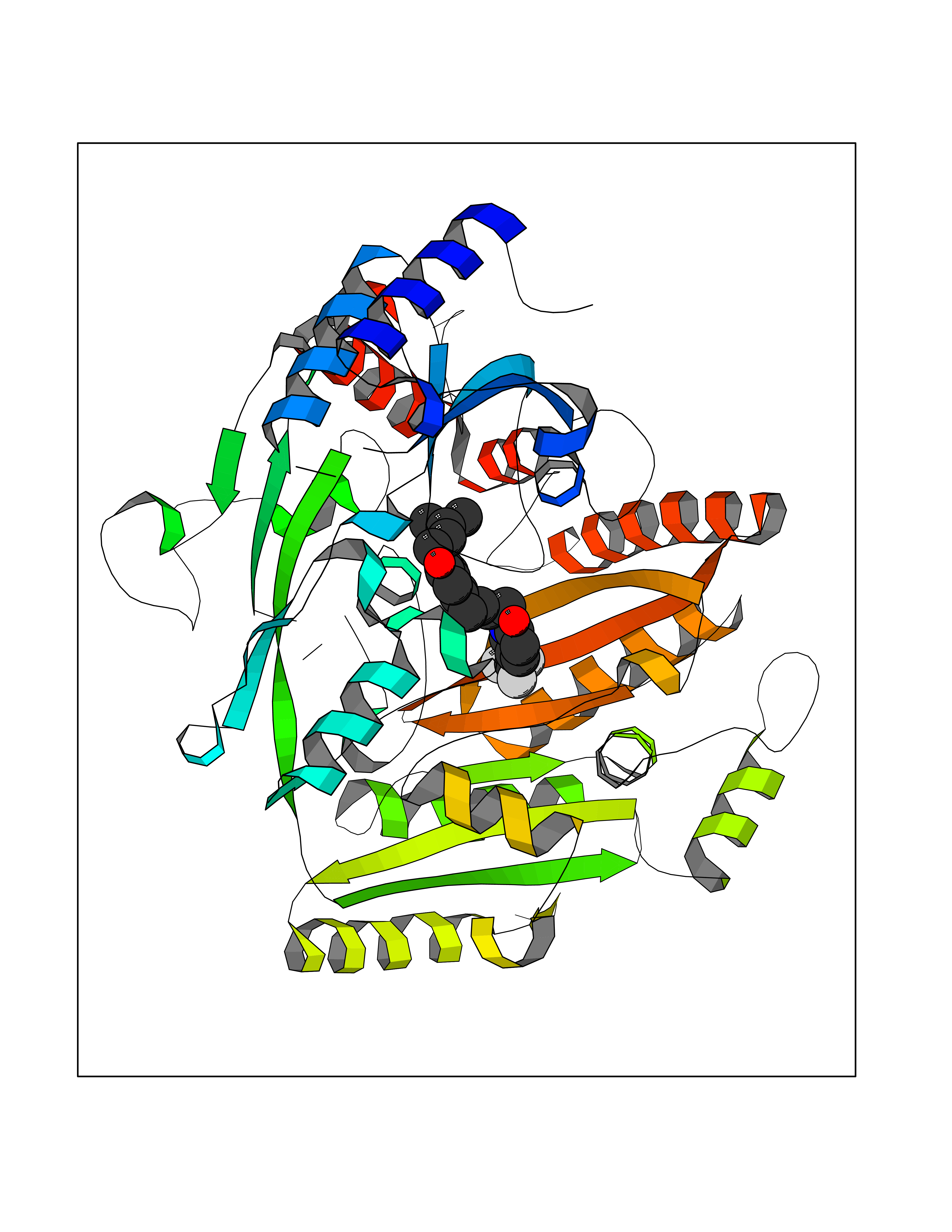 Molscript Map 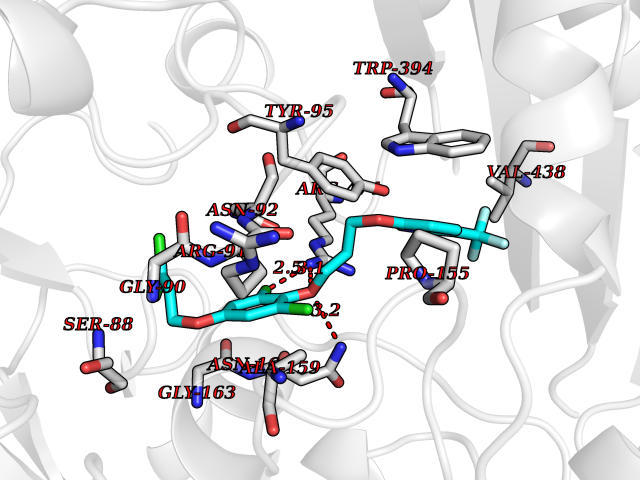 Pymol Map 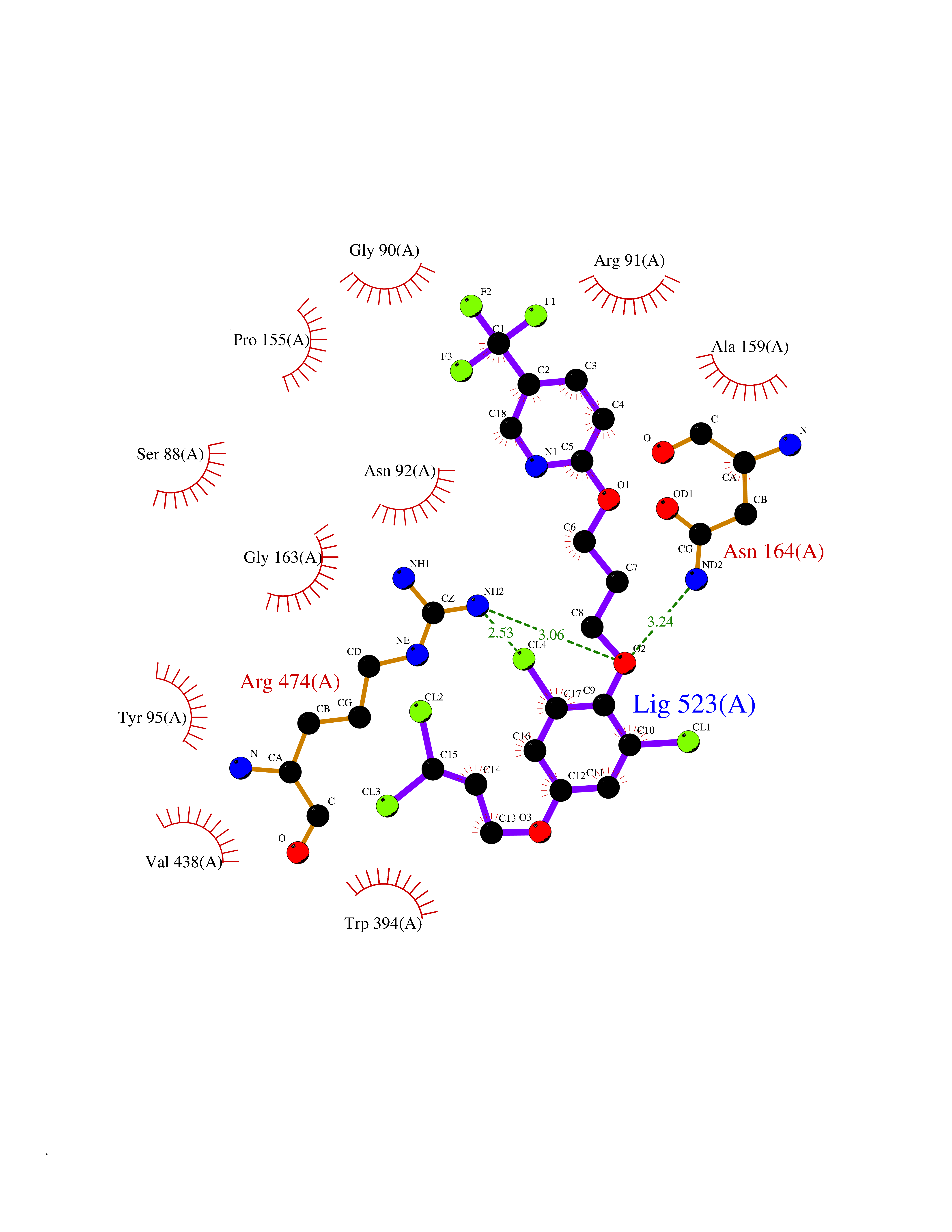 Ligplot Map 3D Binding mode Sequence AVLPKGVTQGEFNKAVQKFRALLGDDNVLVESDQLVPYNKIMMPVENAAHAPSAAVTATTVEQVQGVVKICNEHKIPIWTISTGRNFGYGSAAPVQRGQVILDLKKMNKIIKIDPEMCYALVEPGVTFGQMYDYIQENNLPVMLSFSAPSAIAGPVGNTMDRGVGYTPYGEHFMMQCGMEVVLANGDVYRTGMGGVPGSNTWQIFKWGYGPTLDGMFTQANYGICTKMGFWLMPKPPVFKPFEVIFEDEADIVEIVDALRPLRMSNTIPNSVVIASTLWEAGSAHLTRAQYTTEPGHTPDSVIKQMQKDTGMGAWNLYAALYGTQEQVDVNWKIVTDVFKKLGKGRIVTQEEAGDTQPFKYRAQLMSGVPNLQEFGLYNWRGGGGSMWFAPVSEARGSECKKQAAMAKRVLHKYGLDYVAEFIVAPRDMHHVIDVLYDRTNPEETKRADACFNELLDEFEKEGYAVYRVNTRFQDRVAQSYGPVKRKLEHAIKRAVDPNNILAPGRSGIDLNNDF Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Tyrosine-protein kinase Kit (KIT) | 1T46 | 7.07 | |
Target general information Gen name KIT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog; p145 c-kit; Proto-oncogene tyrosine-protein kinase Kit; Proto-oncogene c-Kit; Piebald trait protein; PBT; Mast/stem cell growth factor re Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function In response to KITLG/SCF binding, KIT can activate several signaling pathways. Phosphorylates PIK3R1, PLCG1, SH2B2/APS and CBL. Activates the AKT1 signaling pathway by phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase. Activated KIT also transmits signals via GRB2 and activation of RAS, RAF1 and the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3, STAT5A and STAT5B. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate. KIT signaling is modulated by protein phosphatases, and by rapid internalization and degradation of the receptor. Activated KIT promotes phosphorylation of the protein phosphatases PTPN6/SHP-1 and PTPRU, and of the transcription factors STAT1, STAT3, STAT5A and STAT5B. Promotes phosphorylation of PIK3R1, CBL, CRK (isoform Crk-II), LYN, MAPK1/ERK2 and/or MAPK3/ERK1, PLCG1, SRC and SHC1. Tyrosine-protein kinase that acts as cell-surface receptor for the cytokine KITLG/SCF and plays an essential role in the regulation of cell survival and proliferation, hematopoiesis, stem cell maintenance, gametogenesis, mast cell development, migration and function, and in melanogenesis. Related diseases Piebald trait (PBT) [MIM:172800]: Autosomal dominant genetic developmental abnormality of pigmentation characterized by congenital patches of white skin and hair that lack melanocytes. {ECO:0000269|PubMed:11074500, ECO:0000269|PubMed:1370874, ECO:0000269|PubMed:1376329, ECO:0000269|PubMed:1717985, ECO:0000269|PubMed:7687267, ECO:0000269|PubMed:8680409, ECO:0000269|PubMed:9450866, ECO:0000269|PubMed:9699740}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:11505412, ECO:0000269|PubMed:15824741, ECO:0000269|PubMed:9438854, ECO:0000269|PubMed:9697690}. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Testicular germ cell tumor (TGCT) [MIM:273300]: A common malignancy in males representing 95% of all testicular neoplasms. TGCTs have various pathologic subtypes including: unclassified intratubular germ cell neoplasia, seminoma (including cases with syncytiotrophoblastic cells), spermatocytic seminoma, embryonal carcinoma, yolk sac tumor, choriocarcinoma, and teratoma. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. The gene represented in this entry is involved in disease pathogenesis. Somatic mutations that lead to constitutive activation of KIT are detected in AML patients. These mutations fall into two classes, the most common being in-frame internal tandem duplications of variable length in the juxtamembrane region that disrupt the normal regulation of the kinase activity. Likewise, point mutations in the kinase domain can result in a constitutively activated kinase.; DISEASE: Mastocytosis, cutaneous (MASTC) [MIM:154800]: A form of mastocytosis, a heterogeneous group of disorders associated with abnormal proliferation and accumulation of mast cells in various tissues, especially in the skin and hematopoietic organs. MASTC is an autosomal dominant form characterized by macules, papules, nodules, or diffuse infiltration of the skin, often associated with localized hyperpigmentation. Gentle rubbing of the lesions induces histamine release from mechanically activated mast cells, causing local wheals, erythema, and often pruritus, a phenomenon termed Darier sign. {ECO:0000269|PubMed:15173254, ECO:0000269|PubMed:19865100, ECO:0000269|PubMed:21689725, ECO:0000269|PubMed:24289326, ECO:0000269|PubMed:9990072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mastocytosis, systemic (MASTSYS) [MIM:154800]: A severe form of mastocytosis characterized by abnormal proliferation and accumulation of mast cells in several organs, resulting in a systemic disease that may affect bone, gastrointestinal tract, lymphatics, spleen, and liver. In some cases, it is associated with a clonal hematologic non-mast-cell lineage disease, such as a myelodysplastic or myeloproliferative disorder. It can also lead to mast cell leukemia, which carries a high risk of mortality. {ECO:0000269|PubMed:9990072}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB09103; DB15233; DB01254; DB12147; DB12010; DB00619; DB09078; DB06080; DB06595; DB04868; DB05913; DB06589; DB12978; DB01962; DB08901; DB08896; DB14840; DB00398; DB01268; DB11800; DB05146 Interacts with P00519; P42684; O75815; P51451; Q8WV28; P46108; P07332; P09769; O75791; P62993; Q14451; P08631; Q96JZ2; P21583; P06239; P07948; P16333; O43639; P27986; O00459; Q92569; P19174; P16885; Q13882; Q06124; Q92729; P20936; Q9UQQ2; O14796; Q9NP31; Q8N5H7; P78314; Q15464; P29353; P98077; Q92529; Q9H6Q3; O14508; O14543; O14544; P12931; Q9ULZ2; Q9HBL0; Q63HR2; Q68CZ2; P42681; P07947; P43403; Q8VBX6; P35235 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Immunoglobulin domain; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33575.6 Length 297 Aromaticity 0.11 Instability index 45.37 Isoelectric point 8.37 Charge (pH=7) 3.24 2D Binding mode Binding energy (Kcal/mol) -9.65 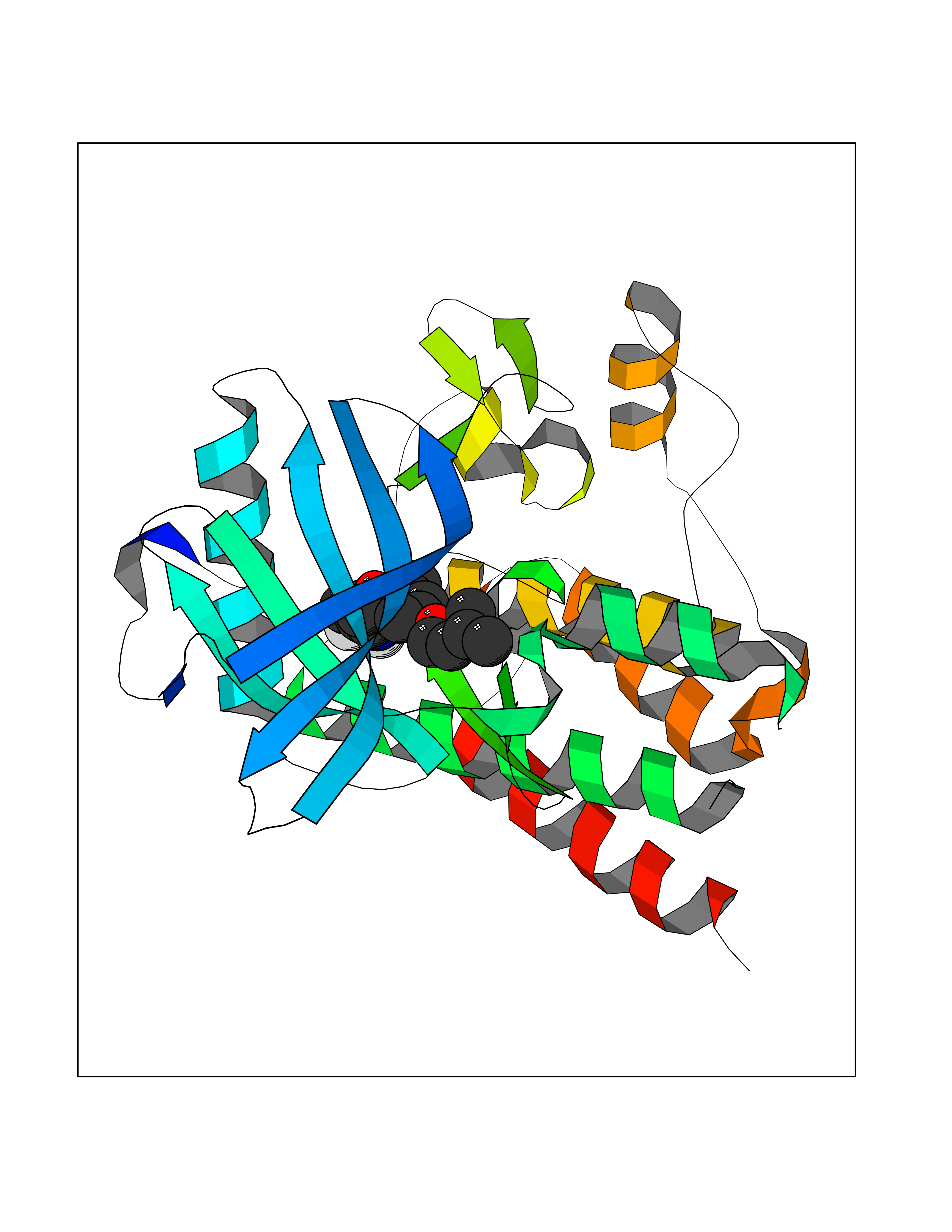 Molscript Map 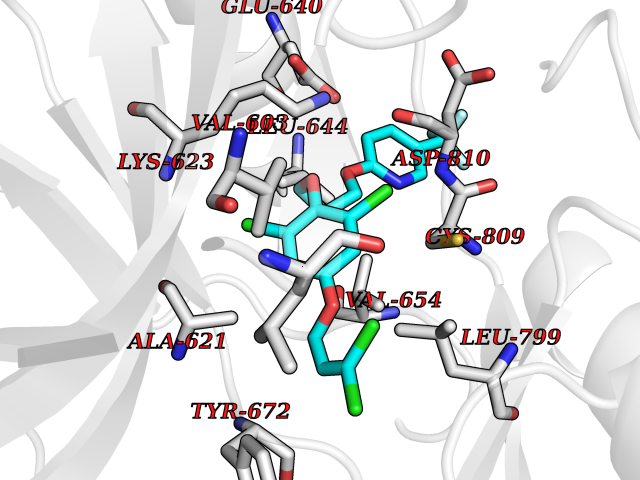 Pymol Map 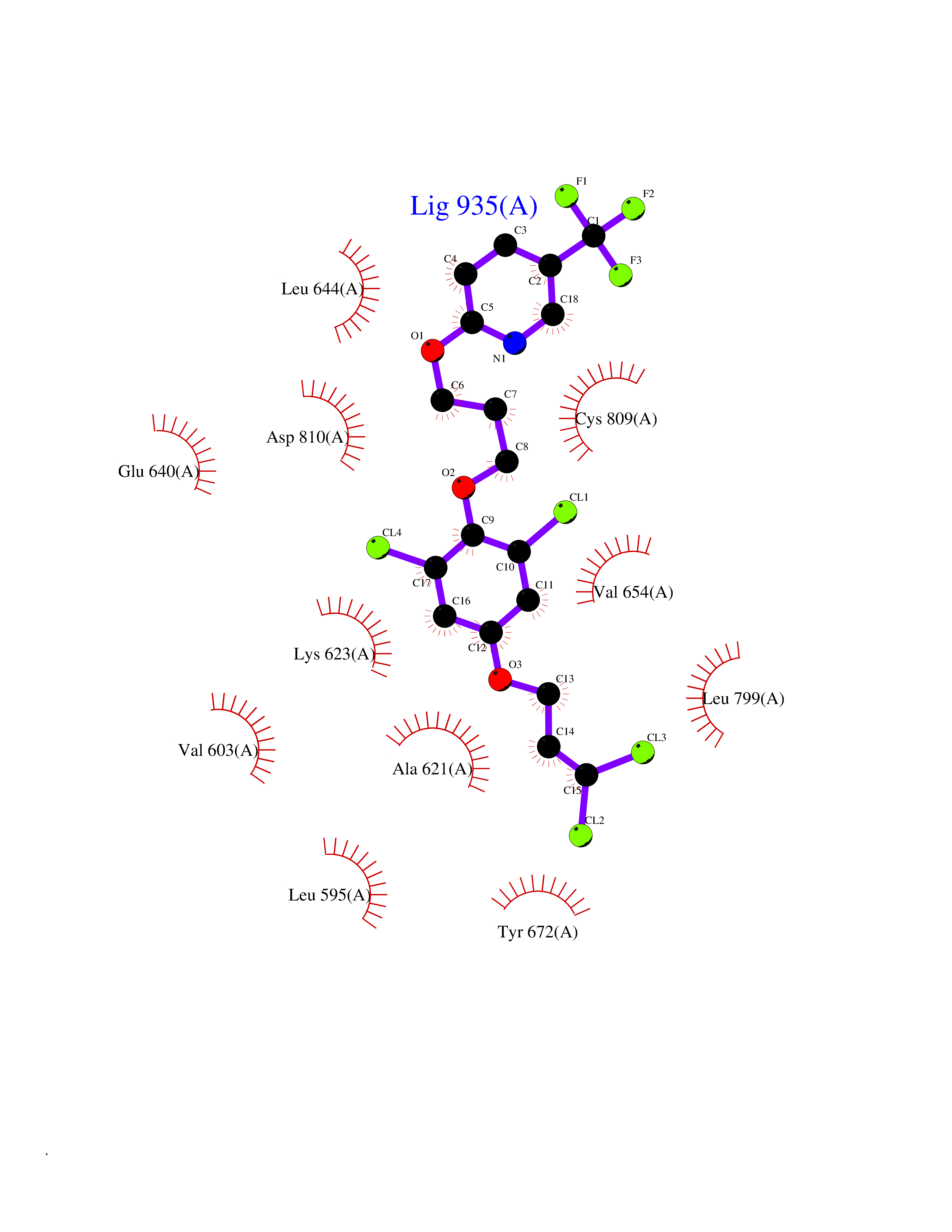 Ligplot Map 3D Binding mode Sequence GNNYVYIDPTQLPYDHKWEFPRNRLSFGKTLGAGAFGKVVEATAYGLIKSDAAMTVAVKMLKPSAHLTEREALMSELKVLSYLGNHMNIVNLLGACTIGGPTLVITEYCCYGDLLNFLRRKRDSFLALDLEDLLSFSYQVAKGMAFLASKNCIHRDLAARNILLTHGRITKICDFGLARDIKNDSNYVVKGNARLPVKWMAPESIFNCVYTFESDVWSYGIFLWELFSLGSSPYPGMPVDSKFYKMIKEGFRMLSPEHAPAEMYDIMKTCWDADPLKRPTFKQIVQLIEKQISESTN Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Rhinovirus Protease 3C (HRV P3C) | 1FPN | 7.07 | |
Target general information Gen name HRV P3C Organism Human rhinovirus 2 (HRV-2) Uniprot ID TTD ID Synonyms Rhinovirus P3C Protein family Picornaviruses polyprotein family Biochemical class NA Function Capsid protein VP1: Forms an icosahedral capsid of pseudo T=3 symmetry with capsid proteins VP2 and VP3. The capsid is 300 Angstroms in diameter, composed of 60 copies of each capsid protein and enclosing the viral positive strand RNA genome. Capsid protein VP1 mainly forms the vertices of the capsid. Capsid protein VP1 interacts with host VLDLR to provide virion attachment to target host cells. This attachment induces virion internalization. Tyrosine kinases are probably involved in the entry process. After binding to its receptor, the capsid undergoes conformational changes. Capsid protein VP1 N-terminus (that contains an amphipathic alpha-helix) and capsid protein VP4 are externalized. Together, they shape a pore in the host membrane through which viral genome is translocated to host cell cytoplasm. After genome has been released, the channel shrinks (By similarity). Related diseases Charcot-Marie-Tooth disease, axonal, 2DD (CMT2DD) [MIM:618036]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:29499166}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypomagnesemia, seizures, and impaired intellectual development 2 (HOMGSMR2) [MIM:618314]: An autosomal dominant disease characterized by generalized seizures in infancy, severe hypomagnesemia, and renal magnesium wasting. Seizures persist despite magnesium supplementation and are associated with significant intellectual disability. {ECO:0000269|PubMed:30388404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02313; DB03017 Interacts with NA EC number EC 3.4.22.28 Uniprot keywords 3D-structure; Activation of host autophagy by virus; ATP-binding; Autocatalytic cleavage; Capsid protein; Covalent protein-RNA linkage; DNA replication; Eukaryotic host gene expression shutoff by virus; Eukaryotic host translation shutoff by virus; Helicase; Host cytoplasm; Host cytoplasmic vesicle; Host gene expression shutoff by virus; Host membrane; Host mRNA suppression by virus; Host nucleus; Host-virus interaction; Hydrolase; Inhibition of host innate immune response by virus; Inhibition of host mRNA nuclear export by virus; Inhibition of host RIG-I by virus; Inhibition of host RLR pathway by virus; Ion channel; Ion transport; Lipoprotein; Magnesium; Membrane; Metal-binding; Myristate; Nucleotide-binding; Nucleotidyltransferase; Phosphoprotein; Pore-mediated penetration of viral genome into host cell; Protease; Repeat; RNA-binding; RNA-directed RNA polymerase; T=pseudo3 icosahedral capsid protein; Thiol protease; Transferase; Transport; Viral attachment to host cell; Viral immunoevasion; Viral ion channel; Viral penetration into host cytoplasm; Viral RNA replication; Virion; Virus endocytosis by host; Virus entry into host cell; Zinc; Zinc-finger Protein physicochemical properties Chain ID 1 Molecular weight (Da) 30610.1 Length 269 Aromaticity 0.1 Instability index 55.66 Isoelectric point 6.86 Charge (pH=7) -0.38 2D Binding mode Binding energy (Kcal/mol) -9.65 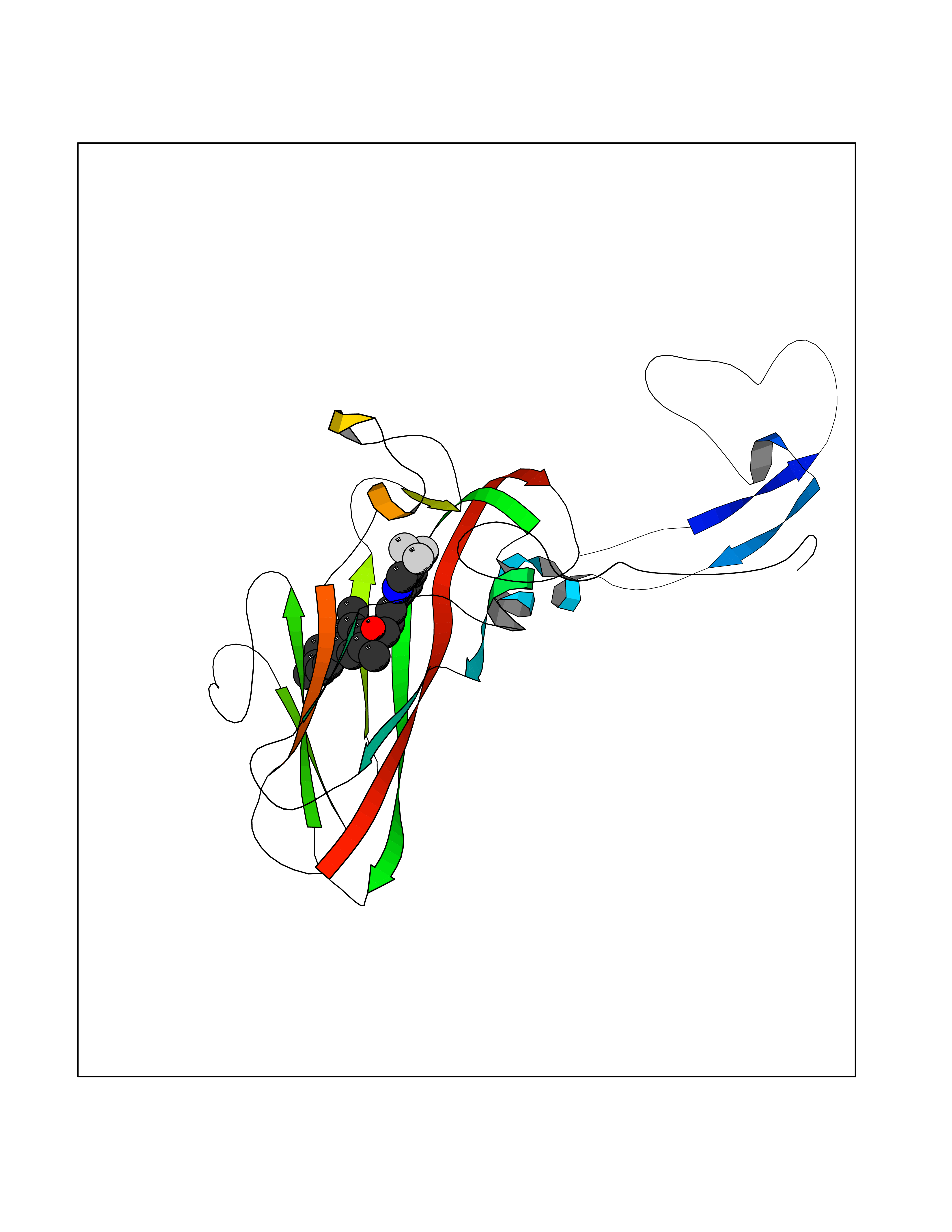 Molscript Map 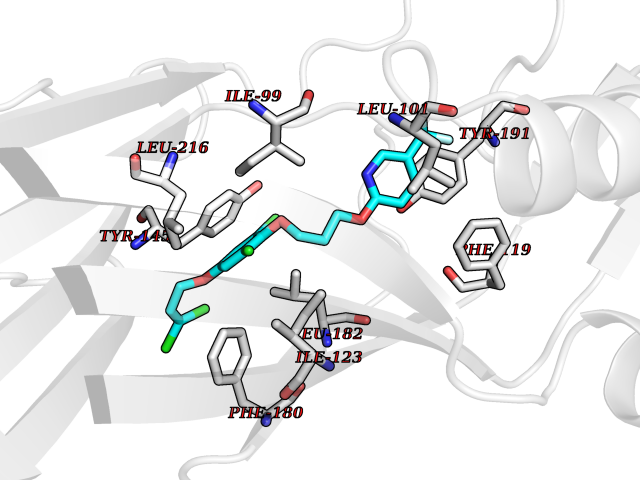 Pymol Map 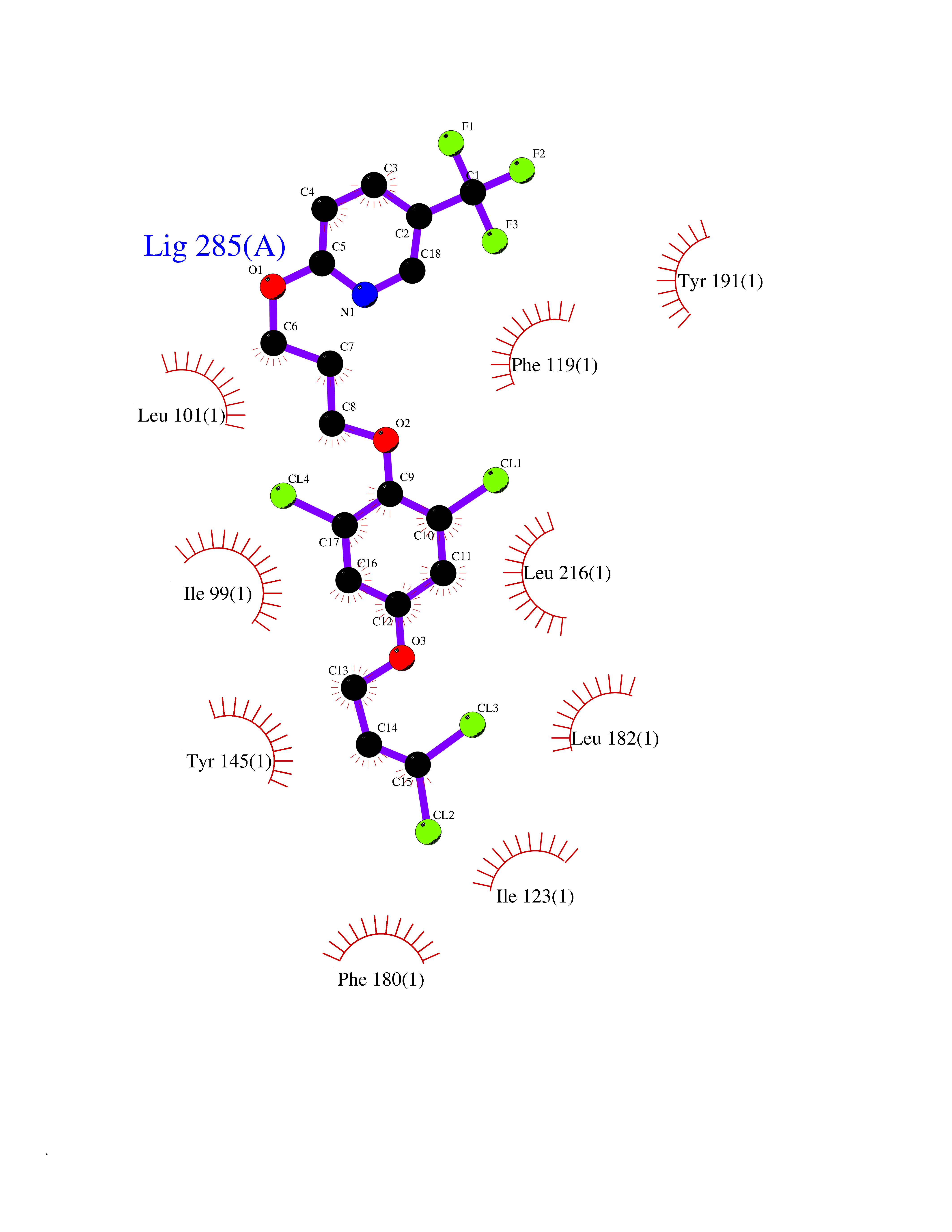 Ligplot Map 3D Binding mode Sequence LVVPNINSSNPTTSNSAPALDAAETGHTSSVQPEDVIETRYVQTSQTRDEMSLESFLGRSGCIHESKLEVTLANYNKENFTVWAINLQEMAQIRRKFELFTYTRFDSEITLVPCISALSQDIGHITMQYMYVPPGAPVPNSRDDYAWQSGTNASVFWQHGQAYPRFSLPFLSVASAYYMFYDGYDEQDQNYGTANTNNMGSLCSRIVTEKHIHKVHIMTRIYHKAKHVKAWCPRPPRALEYTRAHRTNFKIEDRSIQTAIVTRPIITTA Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Fumarate reductase flavoprotein subunit | 1KF6 | 7.06 | |
Target general information Gen name frdA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW4115;b4154 Protein family FAD-dependent oxidoreductase 2 family, FRD/SDH subfamily Biochemical class Oxidoreductase Function Electron carrier activity.FAD binding.Fumarate reductase (menaquinone).Succinate dehydrogenase activity. Related diseases Glycogen storage disease 11 (GSD11) [MIM:612933]: A metabolic disorder that results in exertional myoglobinuria, pain, cramps and easy fatigue. {ECO:0000269|PubMed:2334430}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07490; DB07918; DB00730 Interacts with P0AC47; P0ACB4; P76111 EC number 1.3.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; Electron transport; FAD; Flavoprotein; Membrane; Nucleotide-binding; Oxidoreductase; Reference proteome; Transport Protein physicochemical properties Chain ID A,M Molecular weight (Da) 90370.7 Length 820 Aromaticity 0.08 Instability index 28.88 Isoelectric point 5.86 Charge (pH=7) -16.21 2D Binding mode Binding energy (Kcal/mol) -9.62 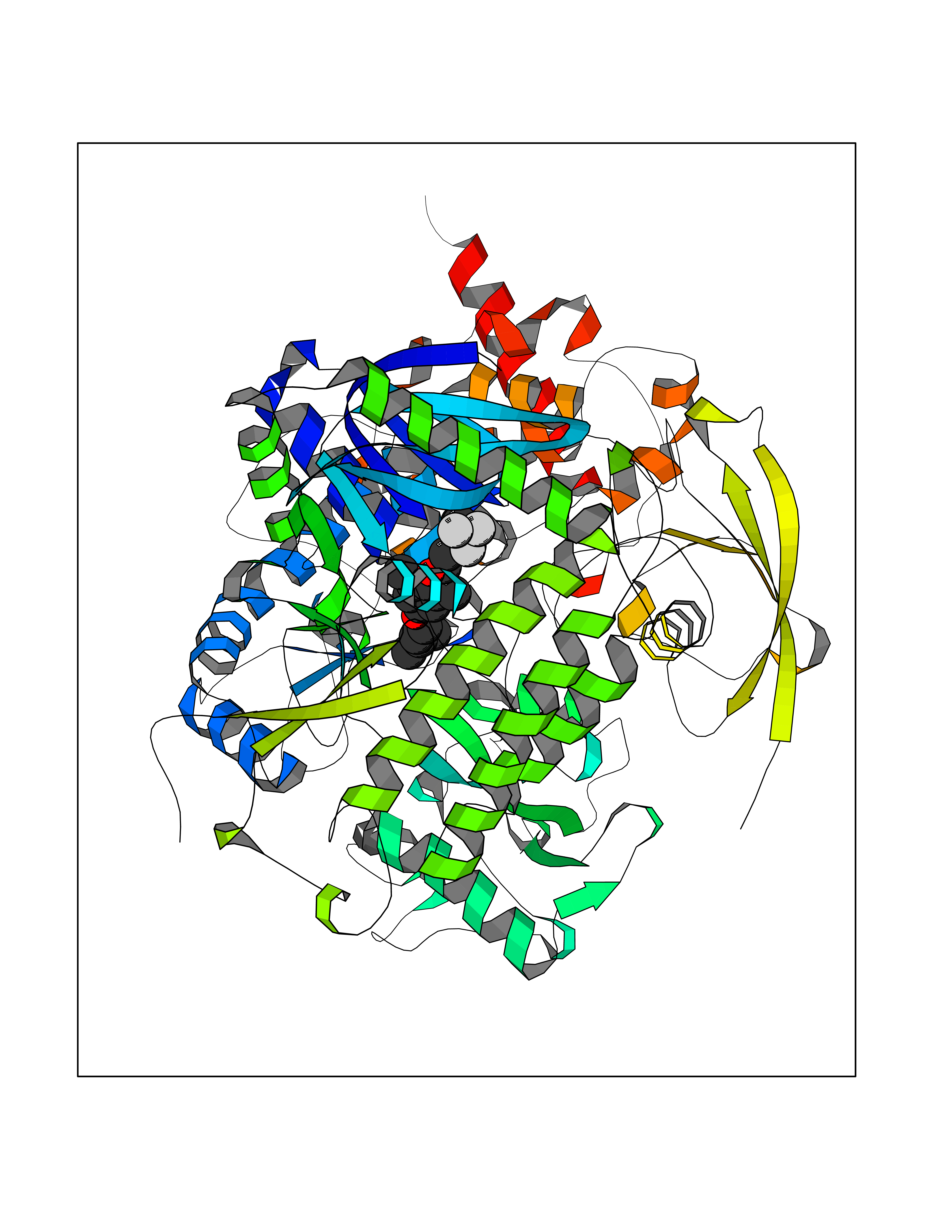 Molscript Map 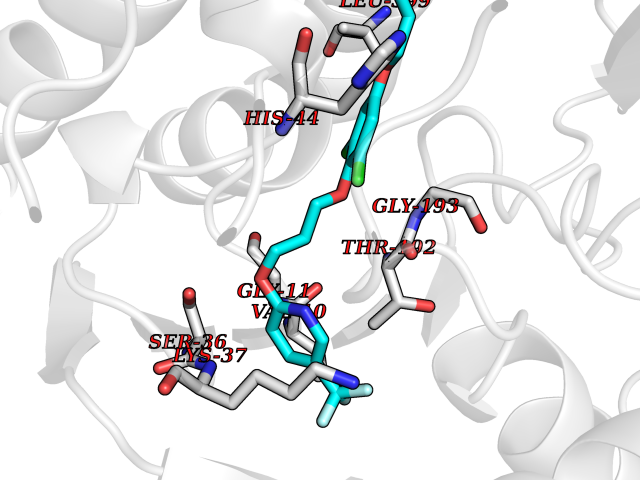 Pymol Map 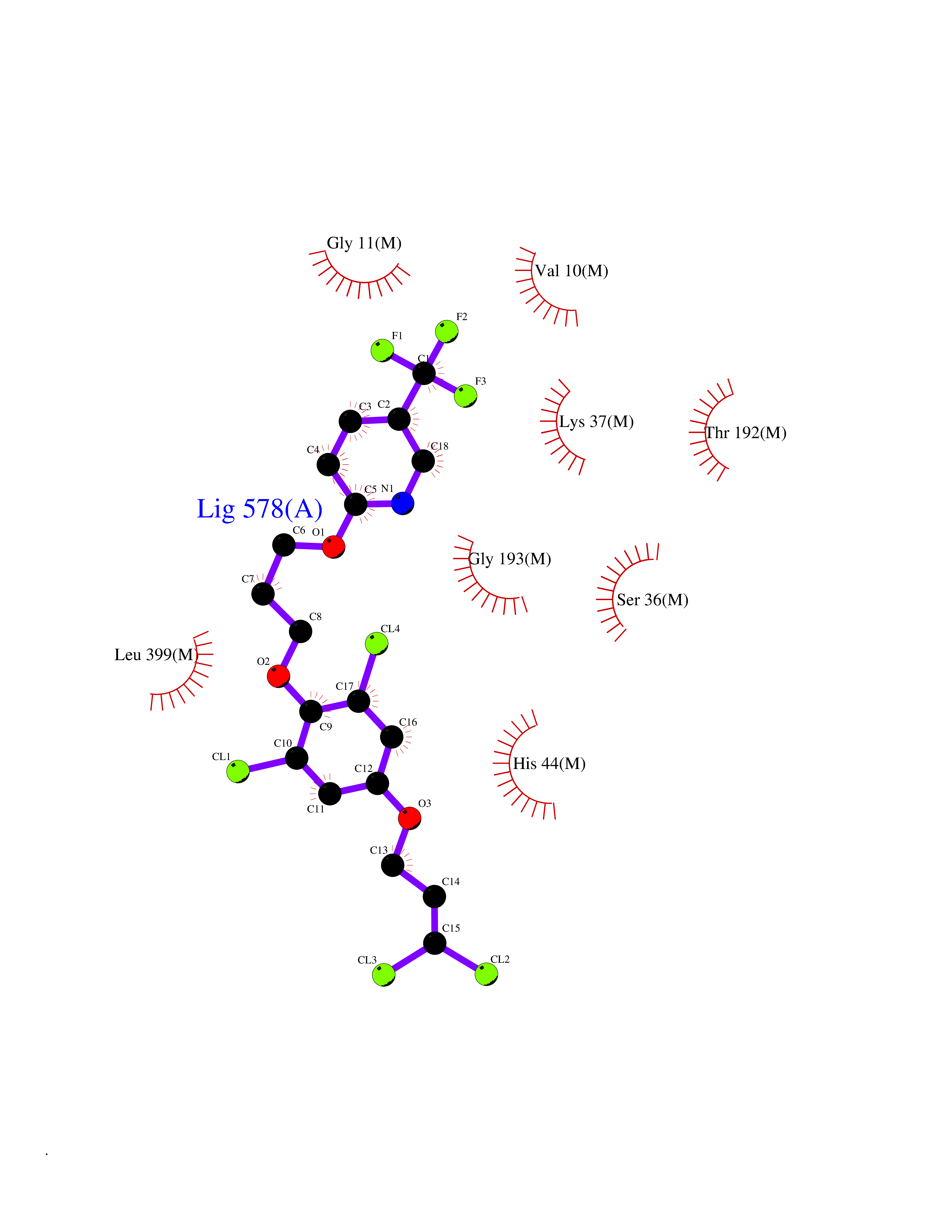 Ligplot Map 3D Binding mode Sequence MQTFQADLAIVGAGGAGLRAAIAAAQANPNAKIALISKVYPMRSHTVAAEGGSAAVAQDHDSFEYHFHDTVAGGDWLCEQDVVDYFVHHCPTEMTQLELWGCPWSRRPDGSVNVRRFGGMKIERTWFAADKTGFHMLHTLFQTSLQFPQIQRFDEHFVLDILVDDGHVRGLVAMNMMEGTLVQIRANAVVMATGGAGRVYRYNTNGGIVTGDGMGMALSHGVPLRDMEFVQYHPTGLPGSGILMTEGCRGEGGILVNKNGYRYLQDYGMGPETPLGEPKNKYMELGPRDKVSQAFWHEWRKGNTISTPRGDVVYLDLRHLGEKKLHERLPFICELAKAYVGVDPVKEPIPVRPTAHYTMGGIETDQNCETRIKGLFAVGECSSVGLHGANRLGSNSLAELVVFGRLAGEQATERAATAGNGNEAAIEAQAAGVEQRLKDLVNQDGGENWAKIRDEMGLAMEEGCGIYRTPELMQKTIDKLAELQERFKRVRITDTSSVFNTDLLYTIELGHGLNVAECMAHSAMARKESRGAHQRLDEGCTERDDVNFLKHTLAFRDADGTTRLEYSDVKITTLPPAAEMKNLKIEVVRYNPEVDTAPHSAFYEVPYDATTSLLDALGYIKDNLAPDLSYRWSCRMAICGSCGMMVNNVPKLACKTFLRDYTDGMKVEALANFPIERDLVVDMTHFIESLEAIKPYIIGNSRTADQGTNIQTPAQMAKYHQFSGCINCGLCYAACPQFGLNPEFIGPAAITLAHRYNEDSRDHGKKERMAQLNSQNGVWSCTFVGYCSEVCPKHVDPAAAIQQGKVESSKDFLIATLKPR Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Smoothened homolog (SMO) | 4JKV | 7.06 | |
Target general information Gen name SMO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Smo-D473H; SMOH; Protein Gx Protein family G-protein coupled receptor Fz/Smo family Biochemical class GPCR frizzled Function Binding of sonic hedgehog (SHH) to its receptor patched is thought to prevent normal inhibition by patched of smoothened (SMO). Required for the accumulation of KIF7, GLI2 and GLI3 in the cilia. Interacts with DLG5 at the ciliary base to induce the accumulation of KIF7 and GLI2 at the ciliary tip for GLI2 activation. G protein-coupled receptor that probably associates with the patched protein (PTCH) to transduce the hedgehog's proteins signal. Related diseases Curry-Jones syndrome (CRJS) [MIM:601707]: A multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. The disease is caused by variants affecting the gene represented in this entry. 8 individuals have been identified with the disease-causing mutation Phe-412 and all were mosaic. The mutation could not be reliably detected in blood, greatest success rates were obtained with affected tissues obtained by invasive procedures. It is thought that the mutation has arisen postzygotically early during embryonic development (PubMed:27236920). This mutation has also been identified in ameloblastoma, medulloblastoma, meningioma, and basal cell carcinoma, and has been reported as the oncogenic driver in some of these tumors (PubMed:24859340). {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. Drugs (DrugBank ID) DB01047; DB11978; DB06786; DB09143; DB08828 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; Developmental protein; Disease variant; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37420.1 Length 333 Aromaticity 0.16 Instability index 25.17 Isoelectric point 6.65 Charge (pH=7) -0.76 2D Binding mode Binding energy (Kcal/mol) -9.63 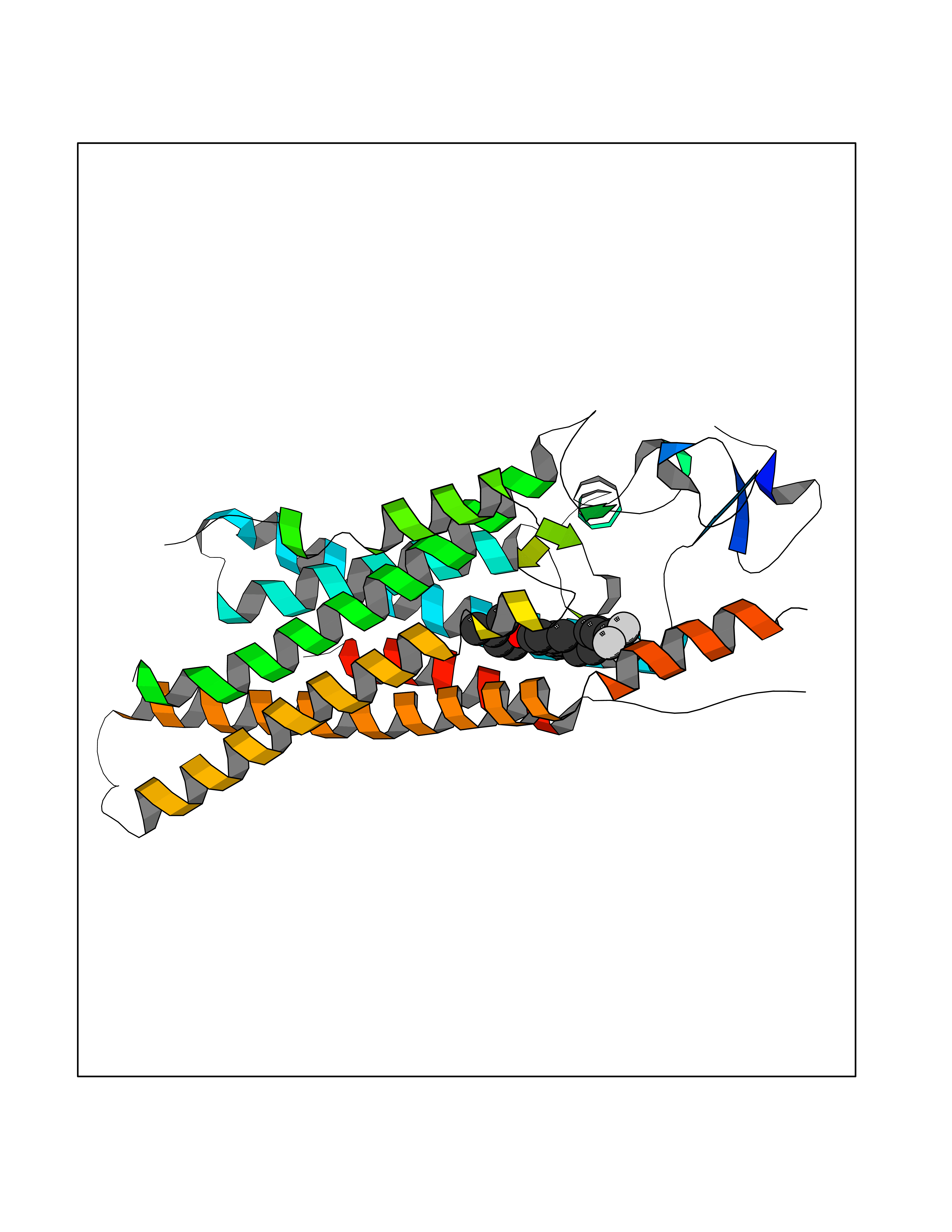 Molscript Map 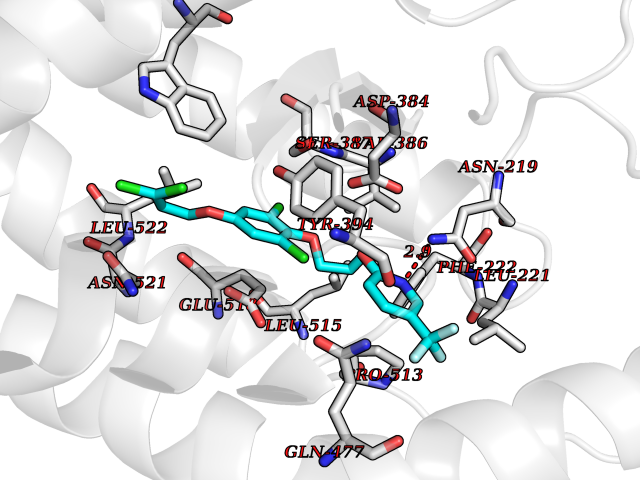 Pymol Map 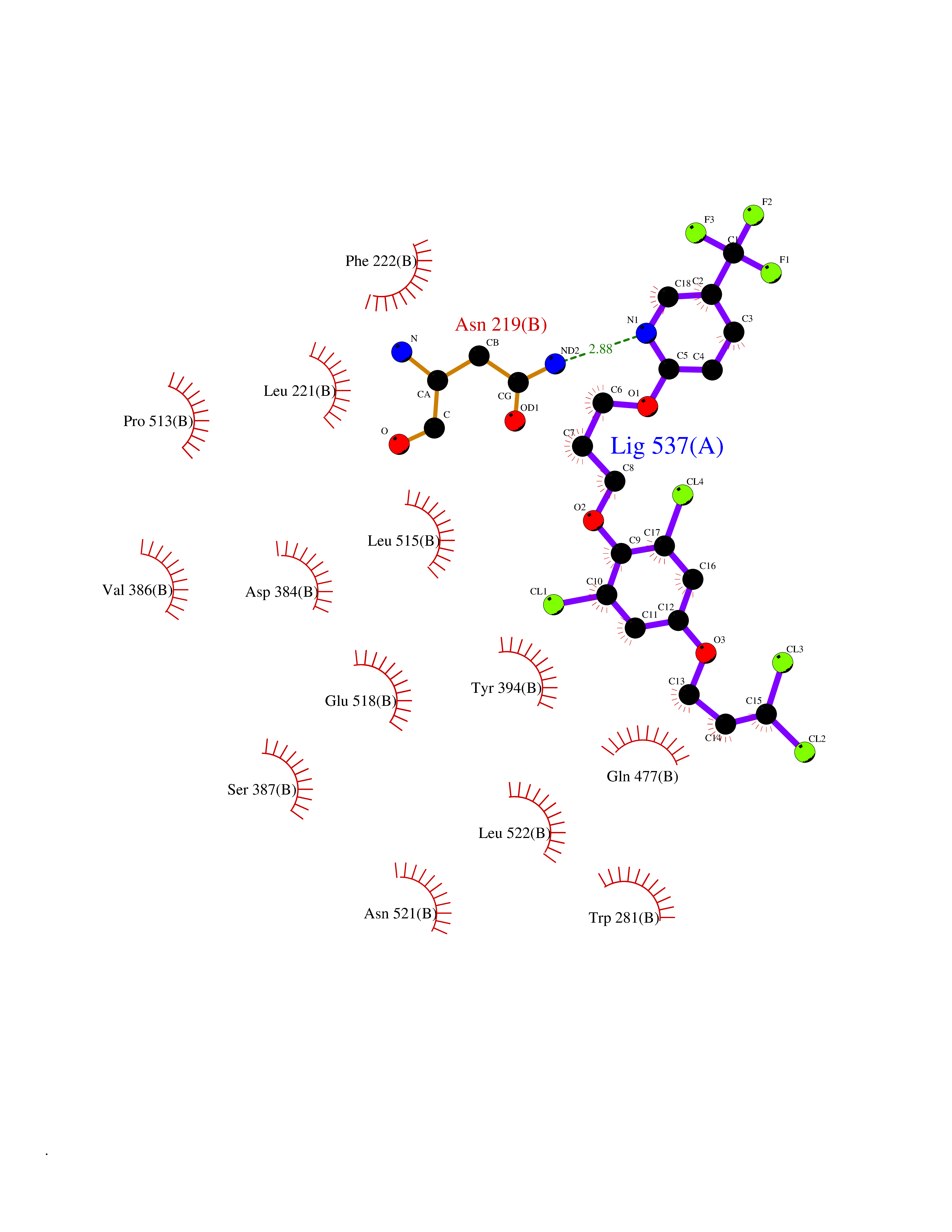 Ligplot Map 3D Binding mode Sequence AYIQKYLSGQCEVPLVRTDNPKSWYEDVEGCGIQCQNPLFTEAEHQDMHSYIAAFGAVTGLCTLFTLATFVADWRNSNRYPAVILFYVNACFFVGSIGWLAQFMDGARREIVCRADGTMRLGEPTSNETLSCVIIFVIVYYALMAGVVWFVVLTYAWHTSFKALGKTSYFHLLTWSLPFVLTVAILAVAQVDGDSVSGICFVGYKNYRYRAGFVLAPIGLVLIVGGYFLIRGVMTLFSIKSNHPGLLSEKAASKINETMLRLGIFGFLAFGFVLITFSCHFYDFFNQAEWERSFRDYVLCQANDCEIKNRPSLLVEKINLFAMFGTGIAMSTW Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Dipeptidyl peptidase 8 (DPP-8) | 6EOP | 7.06 | |
Target general information Gen name DPP8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Prolyl dipeptidase DPP8; MSTP141; MSTP135; MSTP097; Dipeptidyl peptidase VIII; Dipeptidyl peptidase IV-related protein 1; DPRP1; DPRP-1; DPP VIII; DP8 Protein family Peptidase S9B family, DPPIV subfamily Biochemical class Peptidase Function Dipeptidyl peptidase that cleaves off N-terminal dipeptides from proteins having a Pro or Ala residue at position 2. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.14.5 Uniprot keywords 3D-structure; Alternative splicing; Aminopeptidase; Apoptosis; Cytoplasm; Hydrolase; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A,D Molecular weight (Da) 97764.9 Length 849 Aromaticity 0.12 Instability index 47.71 Isoelectric point 5.69 Charge (pH=7) -21.66 2D Binding mode Binding energy (Kcal/mol) -9.63 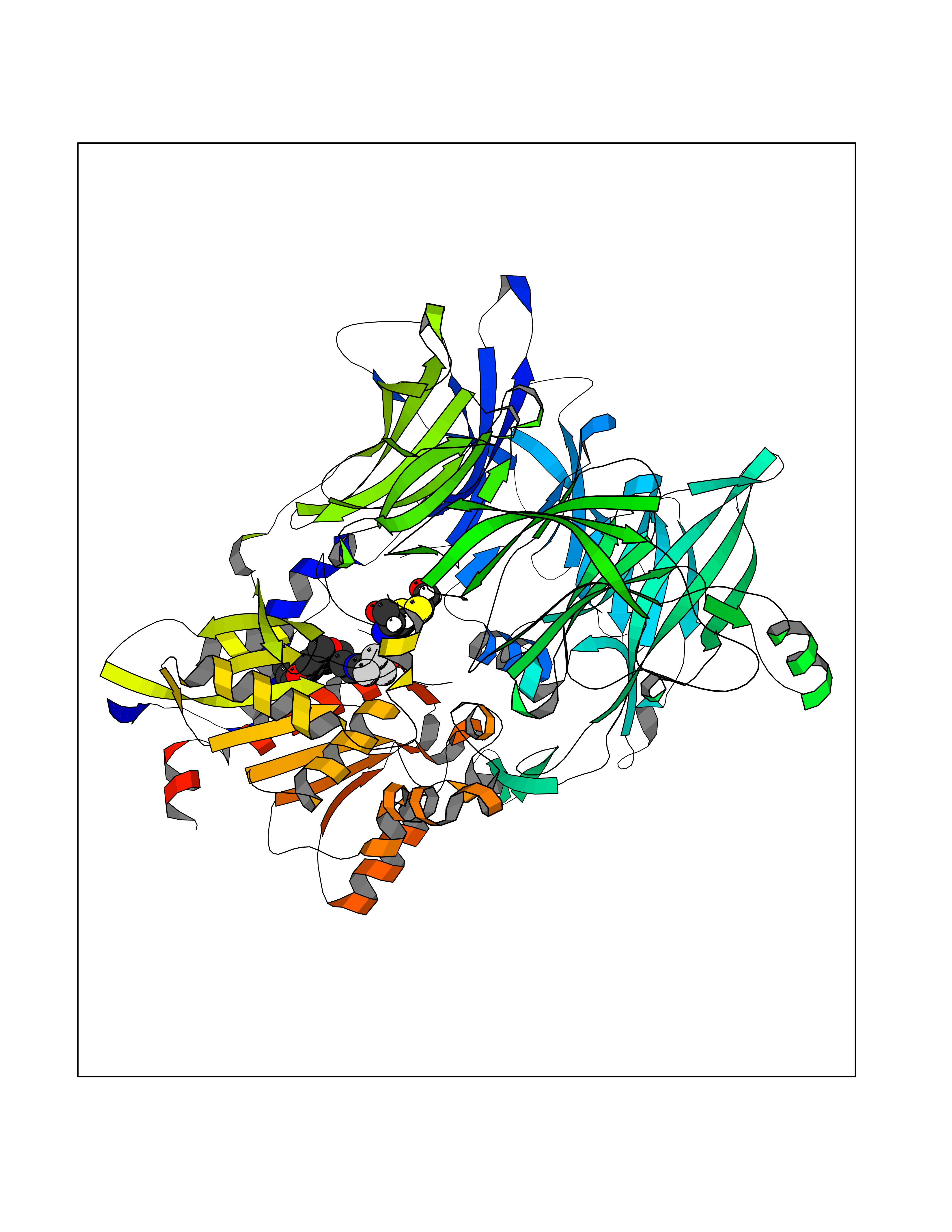 Molscript Map 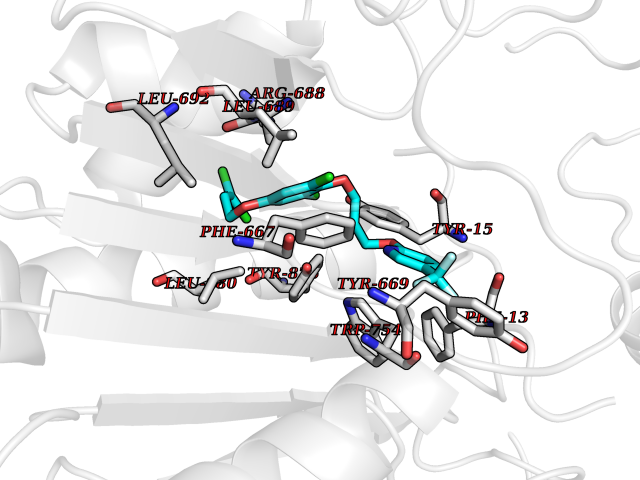 Pymol Map 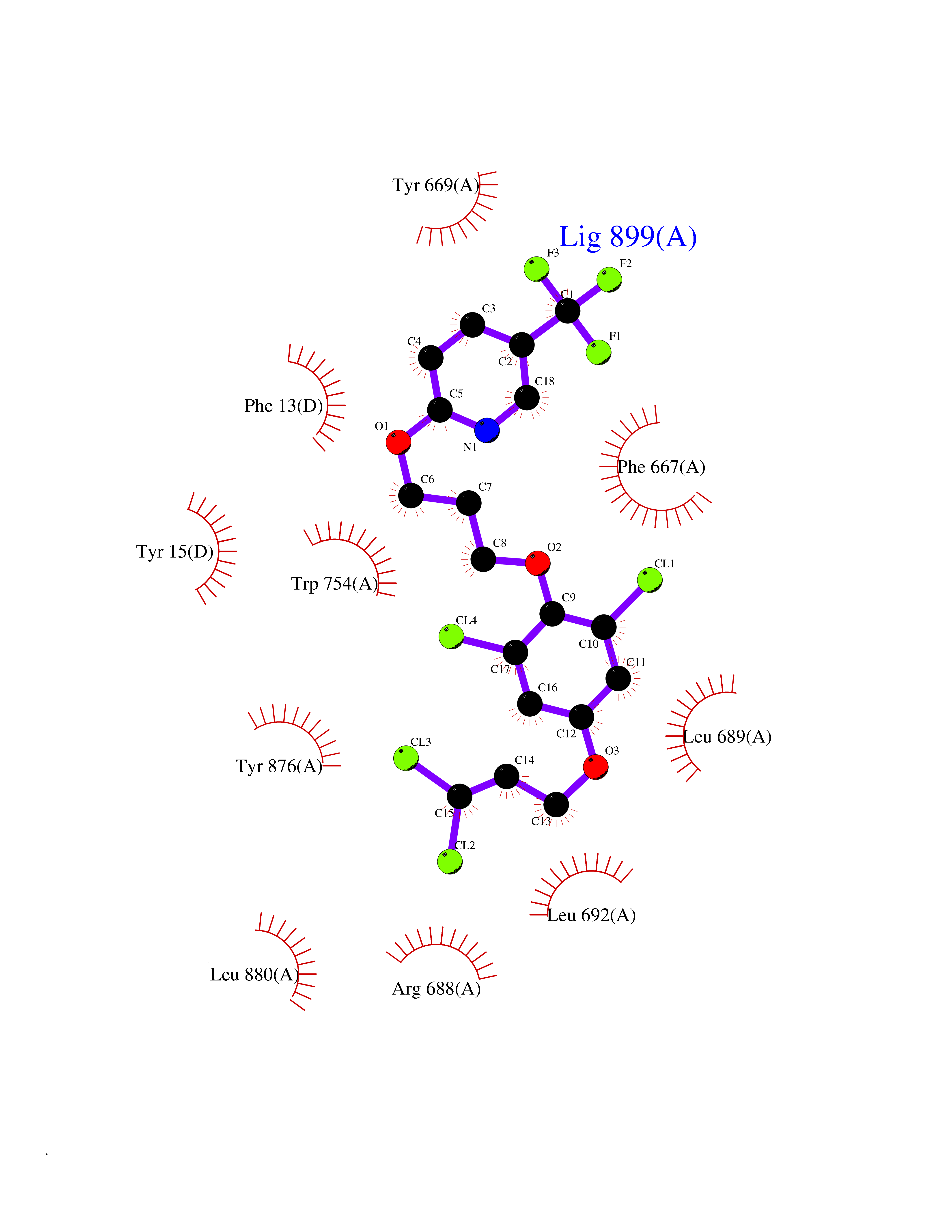 Ligplot Map 3D Binding mode Sequence LEPFYVERYSWSQLKKLLADTRKYHGYMMAKAPHDFMFVKRNDPDGPHSDRIYYLAMSNRENTLFYSEIPKTINRAAVLMLSWKPLLDLFQYSREEELLRERKRIGTVGIASYDYHQGSGTFLFQAGSGIYHVKDGGPQGFTQQPLRPNLVETSCPNIRMDPKLCPADPDWIAFIHSNDIWISNIVTREERRLTYVHNELANMEEDARSAGVATFVLQEEFDRYSGYWWCPKAETTPSGGKILRILYEENDESEVEIIHVTSPMLETRRADSFRYPKTGTANPKVTFKMSEIMIDAEGRIIDVIDKELIQPFEILFEGVEYIARAGWTPEGKYAWSILLDRSQTRLQIVLISPELFIPVEDDVMERQRLIESVPDSVTPLIIYEETTDIWINIHDIFHVFPQSHEEEIEFIFASECKTGFRHLYKITSILKESKYKRSSGGLPAPSDFKCPIKEEIAITSGEWEVLGRHGSNIQVDEVRRLVYFEGTKDSPLEHHLYVVSYVNPGEVTRLTDRGYSHSCCISQHCDFFISKYSNQKNPHCVSLYKLSSPEDDPTCKTKEFWATILDSAGPLPDYTPPEIFSFESTTGFTLYGMLYKPHDLQPGKKYPTVLFIYGGPQVQLVNNRFKGVKYFRLNTLASLGYVVVVIDNRGSXHRGLKFEGAFKYKMGQIEIDDQVEGLQYLASRYDFIDLDRVGIHGWSYGGYLSLMALMQRSDIFRVAIAGAPVTLWIFYDTGYTERYMGHPDQNEQGYYLGSVAMQAEKFPSEPNRLLLLHGFLDENVHFAHTSILLSFLVRAGKPYDLQIYPQERHSIRVPESGEHYELHLLHYLQENLGSRIAALKVSLRFLYEG Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Mutated Histone H3.3 (H3F3A) | 4GUS | 7.06 | |
Target general information Gen name H3F3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PP781; Histone H3.3; H3F3; H3.3B; H3.3A Protein family Histone H3 family Biochemical class NA Function Variant histone H3 which replaces conventional H3 in a wide range of nucleosomes in active genes. Constitutes the predominant form of histone H3 in non-dividing cells and is incorporated into chromatin independently of DNA synthesis. Deposited at sites of nucleosomal displacement throughout transcribed genes, suggesting that it represents an epigenetic imprint of transcriptionally active chromatin. Nucleosomes wrap and compact DNA into chromatin, limiting DNA accessibility to the cellular machineries which require DNA as a template. Histones thereby play a central role in transcription regulation, DNA repair, DNA replication and chromosomal stability. DNA accessibility is regulated via a complex set of post-translational modifications of histones, also called histone code, and nucleosome remodeling. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) NA Interacts with Q9NVP2; P45973; Q13111; Q9UER7; Q9UER7-1; Q9Y6K1; P62805; P49321-2; Q8IZL8; Q5VWG9; Q9VK33; Q8R5C8 EC number NA Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Chromosome; Citrullination; Direct protein sequencing; Disease variant; DNA-binding; Hydroxylation; Intellectual disability; Lipoprotein; Methylation; Nucleosome core; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,C Molecular weight (Da) 86148.9 Length 766 Aromaticity 0.1 Instability index 37.57 Isoelectric point 8.25 Charge (pH=7) 7.16 2D Binding mode Binding energy (Kcal/mol) -9.62 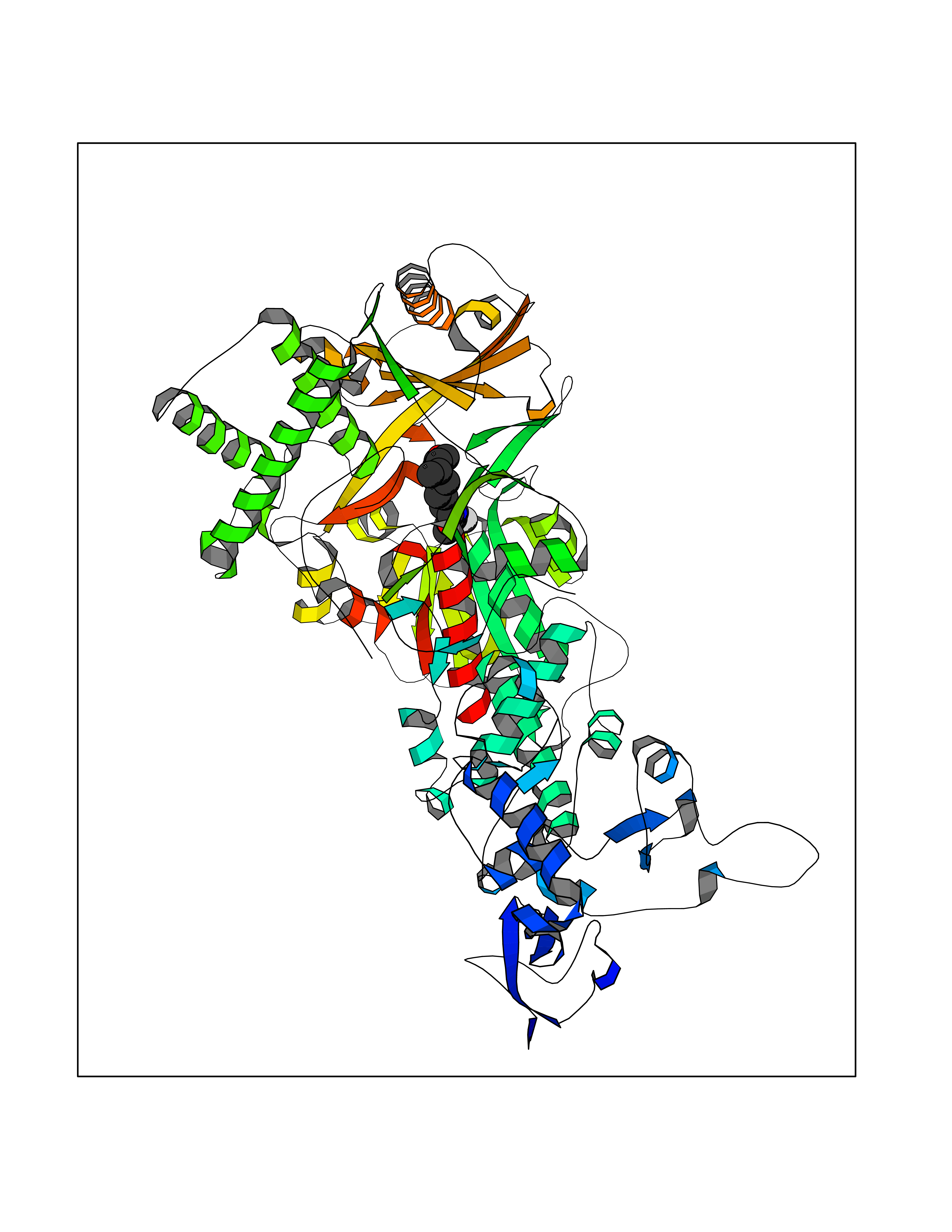 Molscript Map 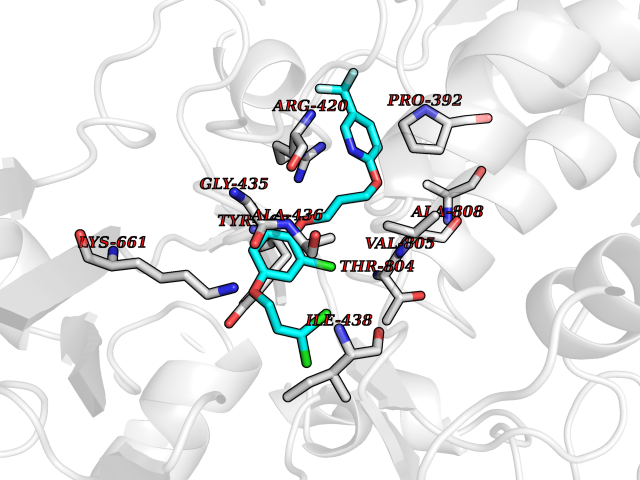 Pymol Map 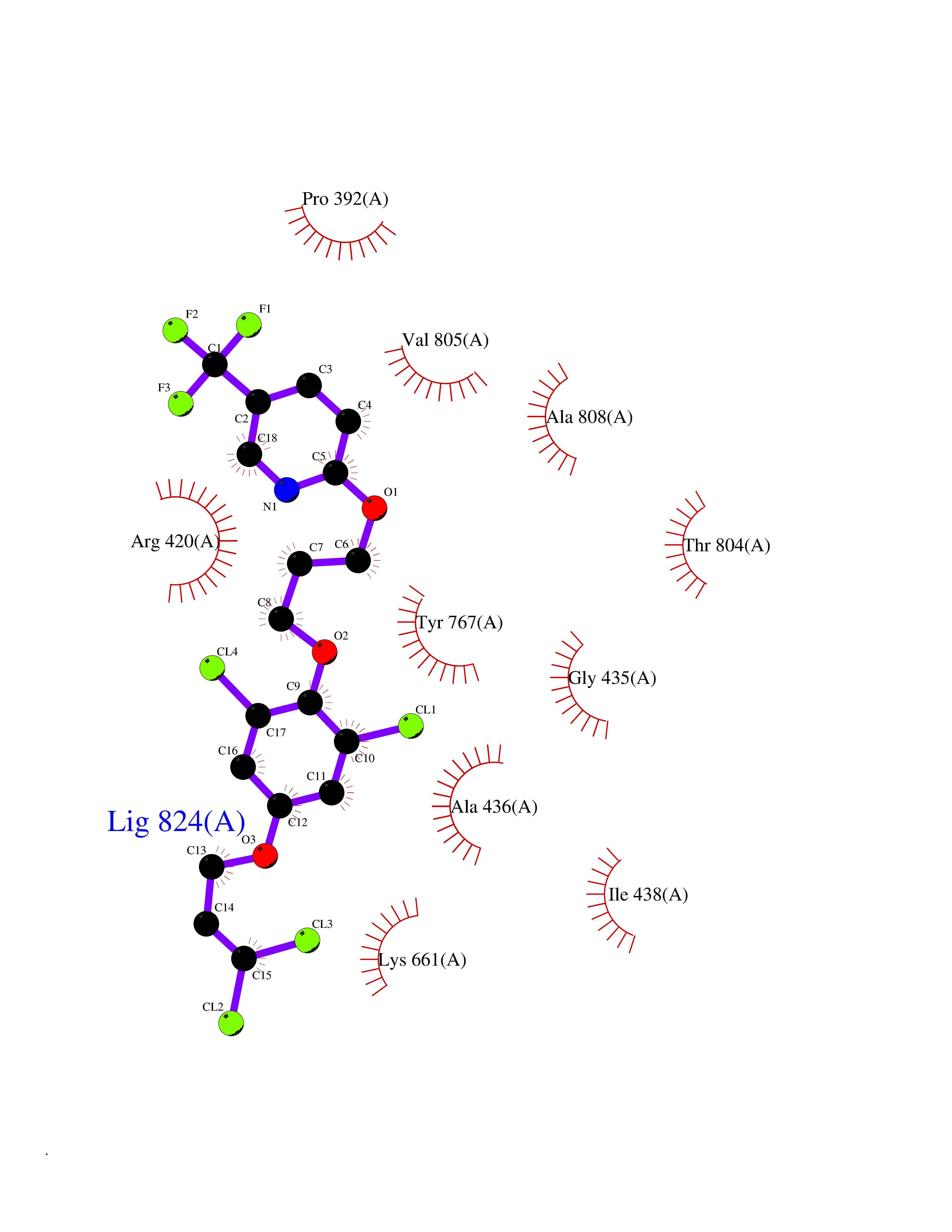 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKPNTAIKPETSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQL Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Gamma-butyrobetaine dioxygenase | 4C5W | 7.05 | |
Target general information Gen name BBOX1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms BBH;BBOX Protein family Gamma-BBH/TMLD family Biochemical class Oxidoreductase Function Gamma-butyrobetaine dioxygenase activity.Identical protein binding.Iron ion binding.Zinc ion binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126 Interacts with O75936; A0MZ66-7 EC number 1.14.11.1 Uniprot keywords 3D-structure; Carnitine biosynthesis; Cytoplasm; Dioxygenase; Iron; Metal-binding; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 31642.5 Length 275 Aromaticity 0.12 Instability index 35.68 Isoelectric point 6.33 Charge (pH=7) -2.46 2D Binding mode Binding energy (Kcal/mol) -9.61 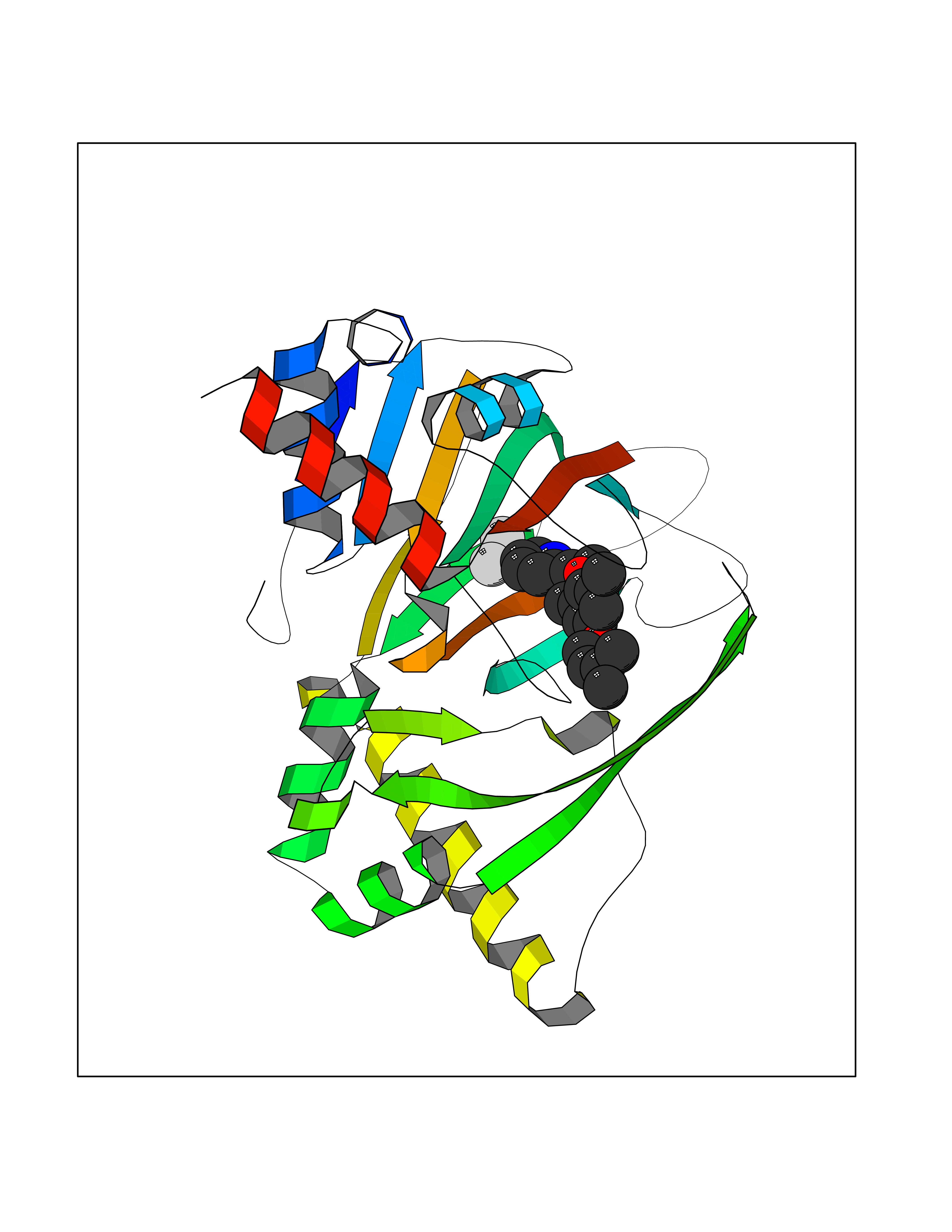 Molscript Map 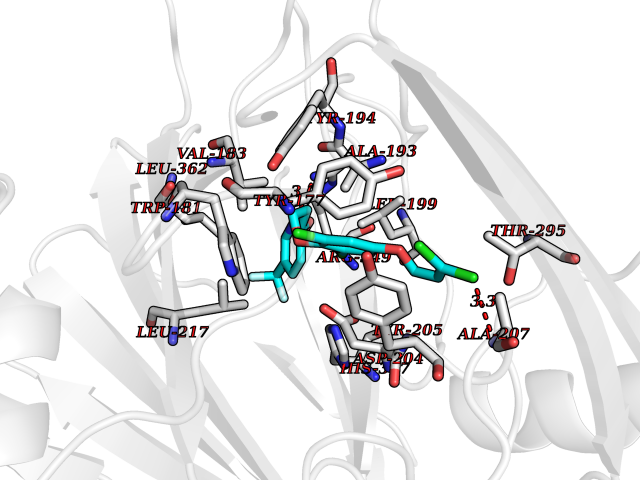 Pymol Map 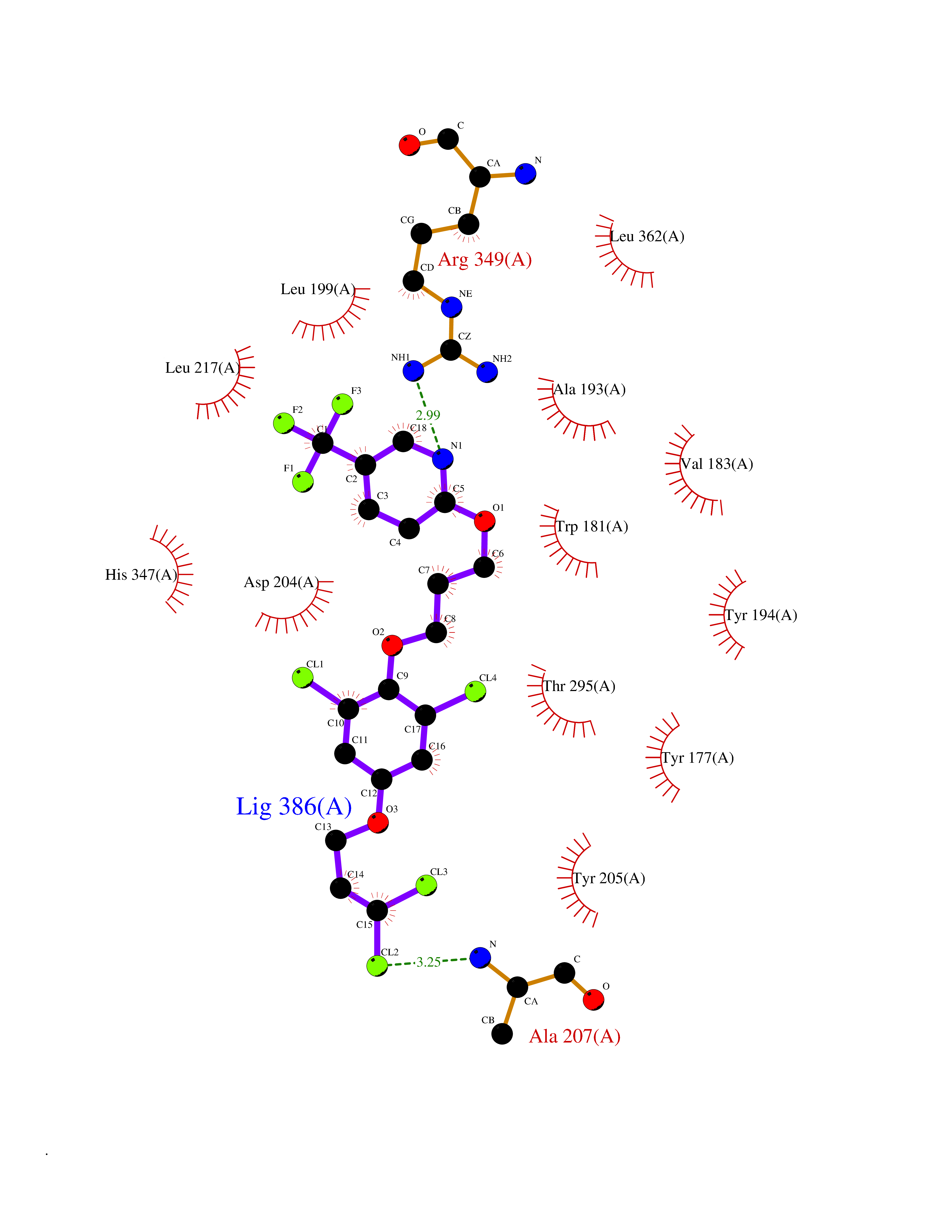 Ligplot Map 3D Binding mode Sequence FPECQYWGSELQLPTLDFEDVLRYDEHAYKWLSTLKKVGIVRLTGASDKPGEVSKLGKRMGFLYLTFYGHTWQVQDKIDANNVAYTTGKLSFHTDYPALHHPPGVQLLHCIKQTVTGGDSEIVDGFNVCQKLKKNNPQAFQILSSTFVDFTDIGVDYCDFSVQSKHKIIELDDKGQVVRINFNNATRDTIFDVPVERVQPFYAALKEFVDLMNSKESKFTFKMNPGDVITFDNWRLLHGRRSYEAGTEISRHLEGAYADWDVVMSRLRILRQRVE Hydrogen bonds contact Hydrophobic contact | ||||