Job Results:
Ligand
Structure
Job ID
7f371ea108ffadeed56066fb8d4cae63
Job name
NA
Time
2024-12-09 11:06:55
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Growth hormone secretagogue receptor 1 (GHSR) | 7NA8 | 4.76 | |
Target general information Gen name GHSR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Growth hormone secretagogue receptor; Ghrelin receptor; GHSR; GHS-R; GHRP; GH-releasing peptide receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for ghrelin, coupled to G-alpha-11 proteins. Stimulates growth hormone secretion. Binds also other growth hormone releasing peptides (GHRP) (e.g. Met-enkephalin and GHRP-6) as well as non-peptide, low molecular weight secretagogues (e.g. L-692,429, MK-0677, adenosine). Related diseases Growth hormone deficiency, isolated partial (GHDP) [MIM:615925]: A disorder characterized by partial growth hormone deficiency resulting in growth delay and short stature, sometimes associated with recurrent episodes of abdominal pain, vomiting, ketosis and hypoglycemia. {ECO:0000269|PubMed:16511605, ECO:0000269|PubMed:19789204}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15488; DB13074; DB12128 Interacts with Q92847-1; Q92847-2; Q99720 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Dwarfism; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32423.5 Length 285 Aromaticity 0.15 Instability index 40.42 Isoelectric point 9.47 Charge (pH=7) 13.13 2D Binding mode Binding energy (Kcal/mol) -6.5 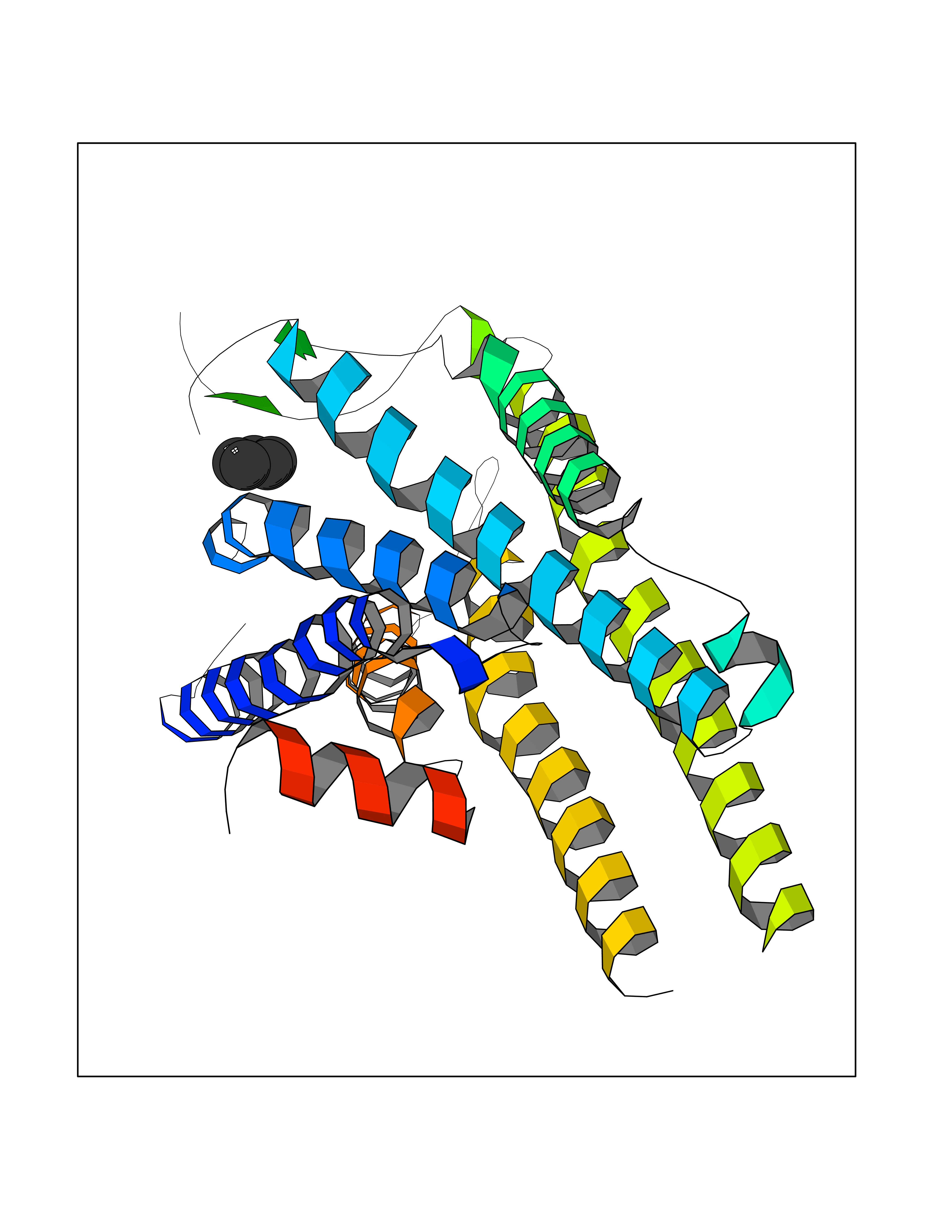 Molscript Map 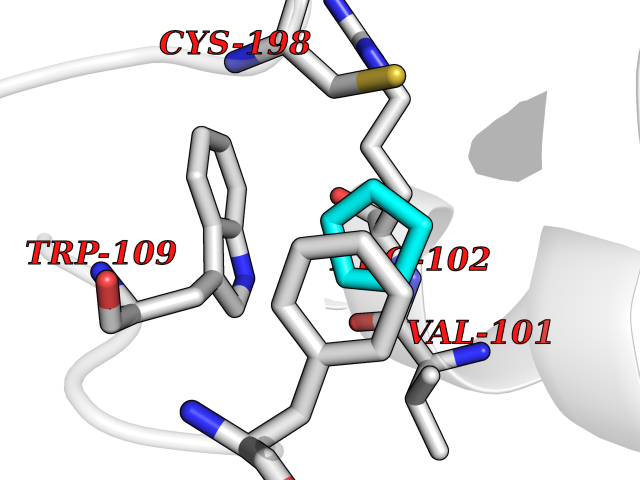 Pymol Map 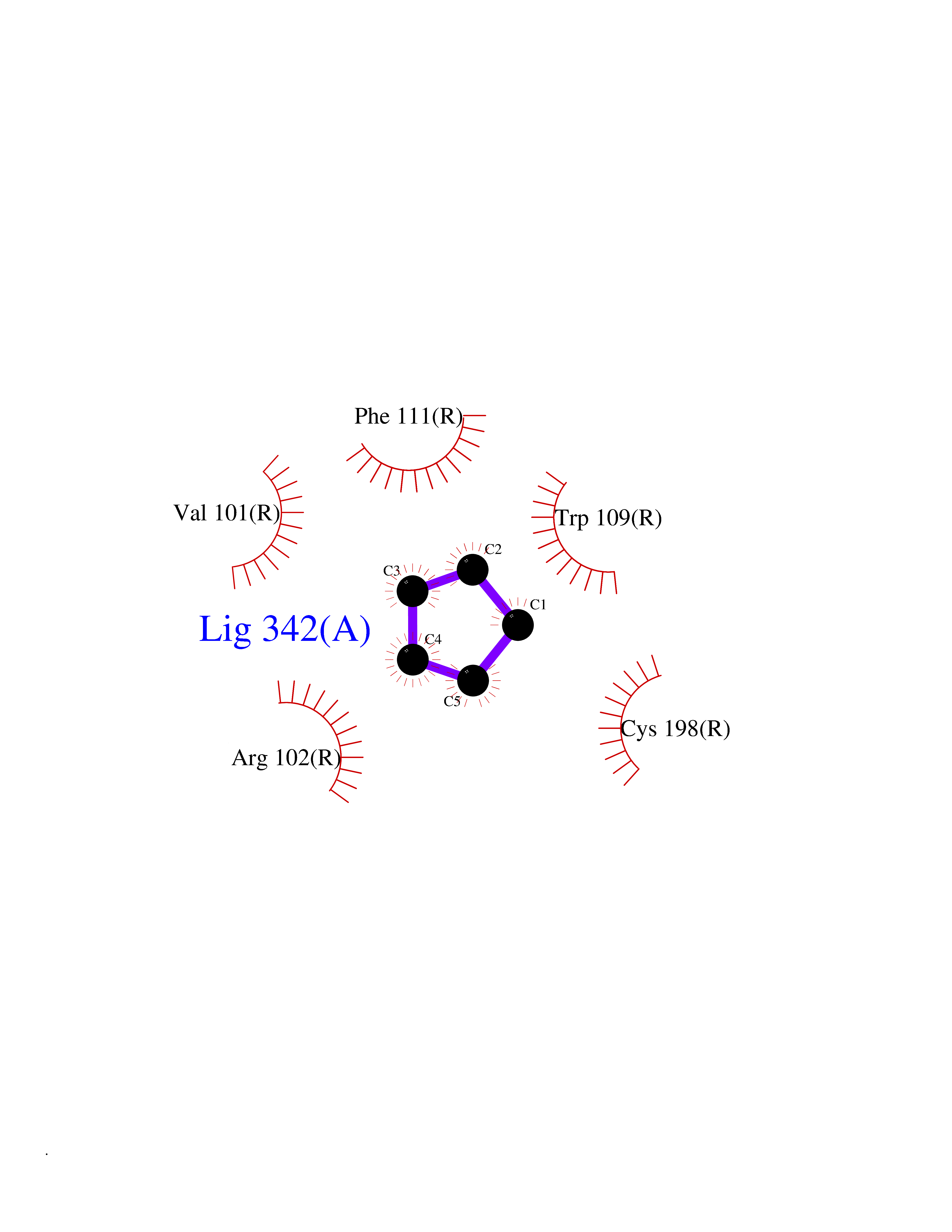 Ligplot Map 3D Binding mode Sequence PAPLLAGVTATCVALFVVGIAGNLLTMLVVSRFRELRTTTNLYLSSMAFSDLLIFLCMPLDLVRLWQPWNFGDLLCKLFQFVSESCTYAKVLTITALSVERYFAICFPLRAKVVVTKGRVKLVIFVIWAVAFCSAGPIFVLVGVEHEPWDTNECRPTEFAVRSGLLTVMVWVSSIFFFLPVFCLTVLYSLIGRKLWRRRDQNHKQTVKMLAVVVFAFILCWLPFHVGRYLFSKSFEPGSLEIAQISQYCNLVSFVLFYLSAAINPILYNIMSKKYRVAVFRLLGF Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Dipeptidyl peptidase 9 (DPP-9) | 6EOR | 4.76 | |
Target general information Gen name DPP9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dipeptidyl peptidase-like protein 9; Dipeptidyl peptidase IX; Dipeptidyl peptidase IV-related protein 2; DPRP2; DPRP-2; DPP IX; DPLP9; DP9 Protein family Peptidase S9B family, DPPIV subfamily Biochemical class Peptidase Function Dipeptidyl peptidase that cleaves off N-terminal dipeptides from proteins having a Pro or Ala residue at position 2. Related diseases Hatipoglu immunodeficiency syndrome (HATIS) [MIM:620331]: An autosomal recessive immunologic disorder manifesting in infancy or early childhood, and characterized by failure to thrive, short stature, skin pigmentation abnormalities, pancytopenia, and susceptibility to recurrent infections. {ECO:0000269|PubMed:36112693}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NXR5; Q86TI2; Q6NUP5; P46379-2; Q8WUW1; Q96A83-2; O75190-2; O14645; Q01658; P29692-2; Q06787-7; Q9Y5Q9; O14901; Q9BVL2; Q96CV9; Q06830; P14678-2; P49458; Q11203; Q13148; P14927 EC number EC 3.4.14.5 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminopeptidase; Cytoplasm; Disease variant; Hydrolase; Nucleus; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A Molecular weight (Da) 92797.4 Length 808 Aromaticity 0.12 Instability index 37.45 Isoelectric point 6.34 Charge (pH=7) -8.98 2D Binding mode Binding energy (Kcal/mol) -6.49 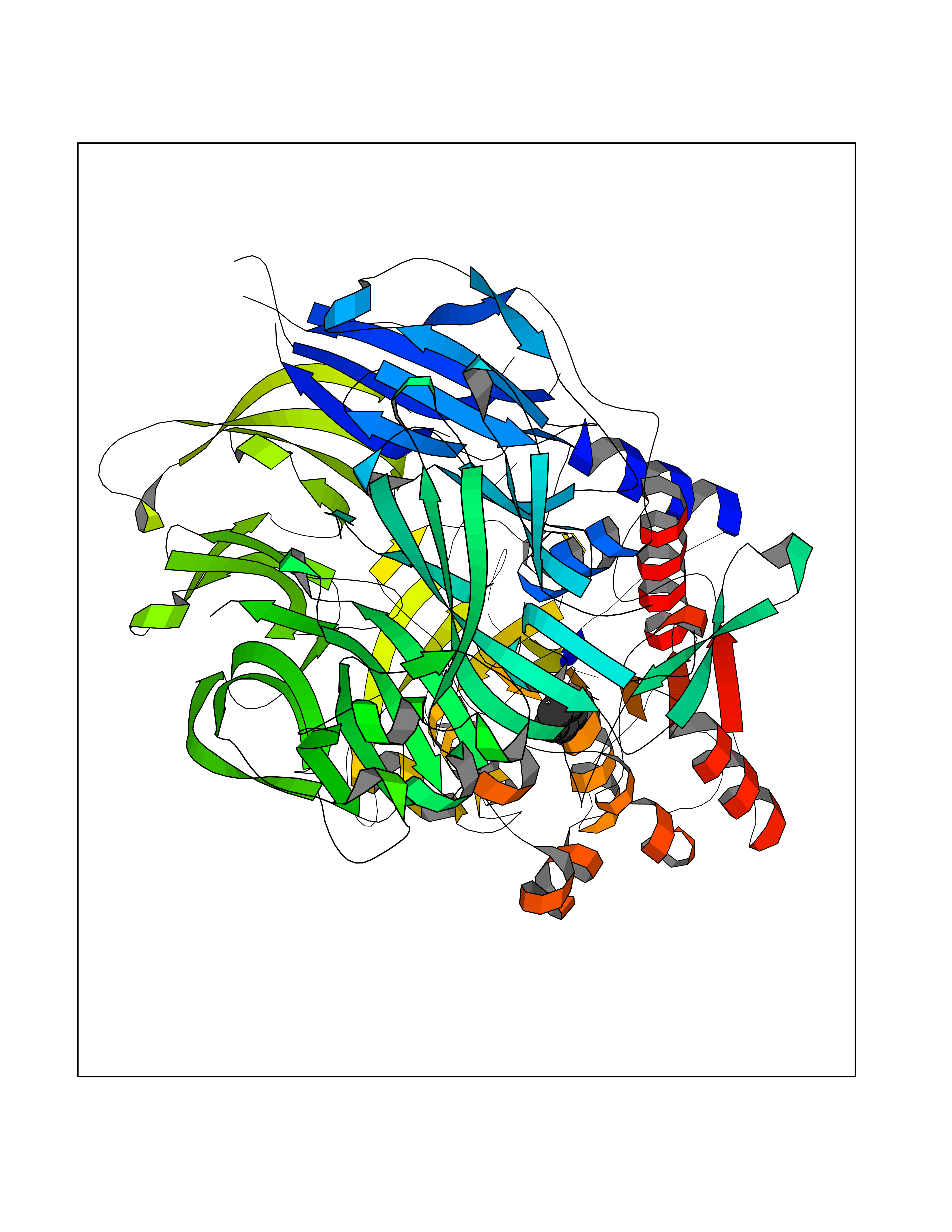 Molscript Map 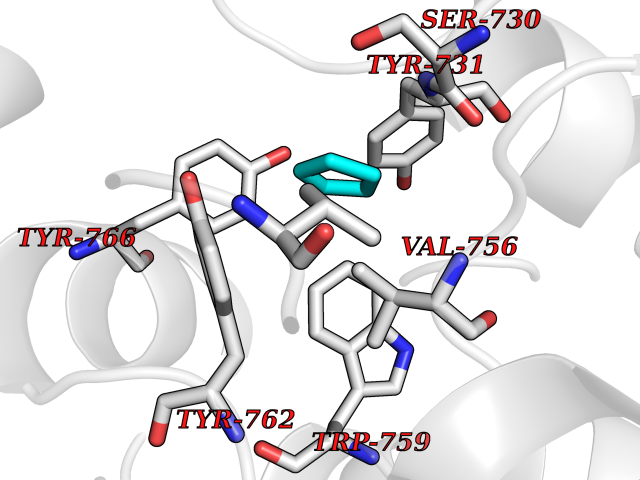 Pymol Map 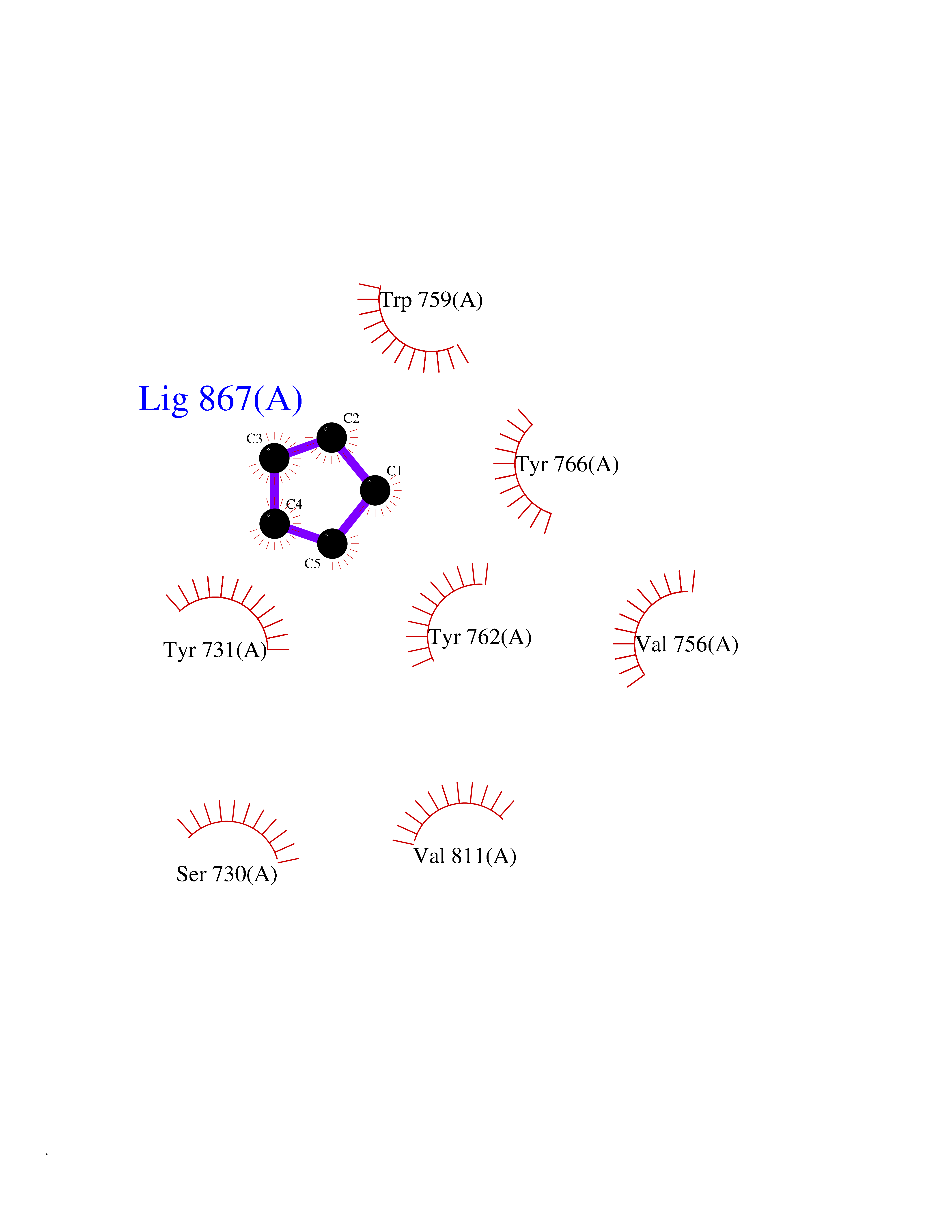 Ligplot Map 3D Binding mode Sequence AARFQVQKHSWDGLRSIIHGSRKAPHDFQFVQKSGPHSHRLYYLGMPYRENSLLYSEIPKLLLSWKQMLDHFQATPHHGVYSREEELLRERKRLGVFGITSYDFHSESGLFLFQASNSLFHCRDGGKNGFMVSPMKPLEIKTQCSGPRMDPKICPADPAFFSFINNSDLWVANIETGEERRLTFCHQNVLDDPKSAGVATFVIQEEFDRFTGYWWCPTASWEGLKTLRILYEEVDESEVEVIHVPSPALEERKTDSYRYPRTGSKNPKIALKLAEFQTDSQGKIVSTQEKELVQPFSSLFPKVEYIARAGWTRDGKYAWAMFLDRPQQWLQLVLLPPALFIPSTENEEQRLASARAVPRNVQPYVVYEEVTNVWINVHDIFYPFPQLCFLRANECKTGFCHLYKVTAVLKSQGYDWSEPFSPGEDEFKCPIKEEIALTSGEWEVLARHGSKIWVNEETKLVYFQGTKDTPLEHHLYVVSYEAAGEIVRLTTPGFSHSCSMSQNFDMFVSHYSSVSTPPCVHVYKLSGPDDDPLHKQPRFWASMMEADYVPPEIFHFHTRSDVRLYGMIYKPHALQPGKKHPTVLFVYGGPQVQLVNNSFKGIKYLRLNTLASLGYAVVVIDGRGSCQRGLRFEGALKNQMGQVEIEDQVEGLQFVAEKYGFIDLSRVAIHGWSYGGFLSLMGLIHKPQVFKVAIAGAPVTVWMAYDTGYTERYMDVPENNQHGYEAGSVALHVEKLPNEPNRLLILHGFLDENVHFFHTNFLVSQLIRAGKPYQLQIYPNERHSIRCPESGEHYEVTLLHFLQEYLHH Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Cytochrome b (Complex III subunit 3) (Complex III subunit III) (Cytochrome b-c1 complex subunit 3) (Ubiquinol-cytochrome-c reductase complex cytochrome b subunit) | 1SQB | 4.75 | |
Target general information Gen name MT-CYB Organism Bos taurus (Bovine) Uniprot ID TTD ID NA Synonyms CYTB;MTCYB;COB Protein family Cytochrome b family Biochemical class NA Function Component of the ubiquinol-cytochrome c reductase complex (complex III or cytochrome b-c1 complex) that is part of the mitochondrial respiratory chain. The b-c1 complex mediates electron transfer from ubiquinol to cytochrome c. Contributes to the generation of a proton gradient across the mitochondrial membrane that is then used for ATP synthesis. {ECO:0000269|PubMed:1327781, ECO:0000269|PubMed:20025846, ECO:0000269|PubMed:9485330, ECO:0000305|PubMed:189810}." Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Electron transport; Heme; Iron; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Reference proteome; Respiratory chain; Transmembrane; Transmembrane helix; Transport; Ubiquinone Protein physicochemical properties Chain ID C,D,F,G,H Molecular weight (Da) 99281.2 Length 866 Aromaticity 0.12 Instability index 43.81 Isoelectric point 8.32 Charge (pH=7) 7.12 2D Binding mode Binding energy (Kcal/mol) -6.48 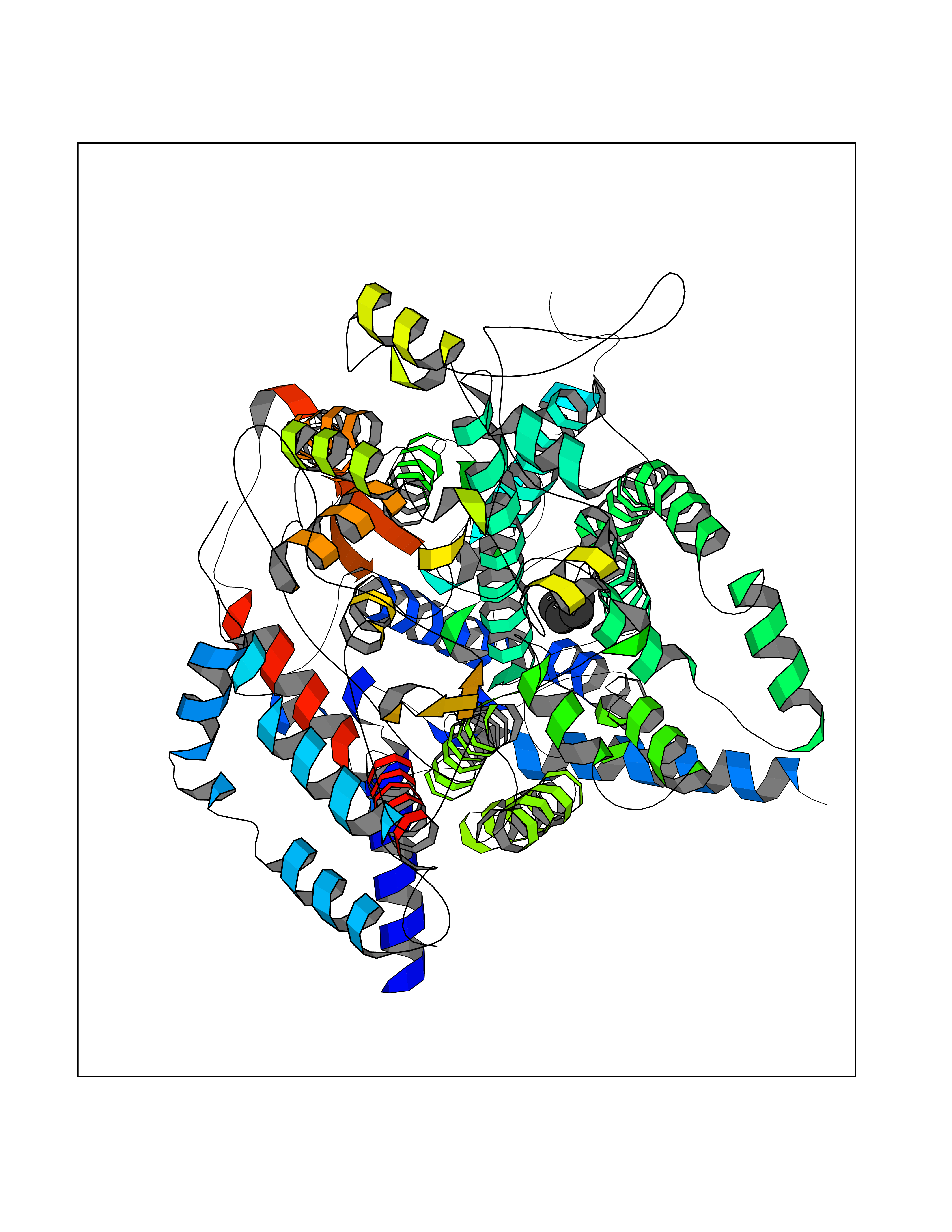 Molscript Map 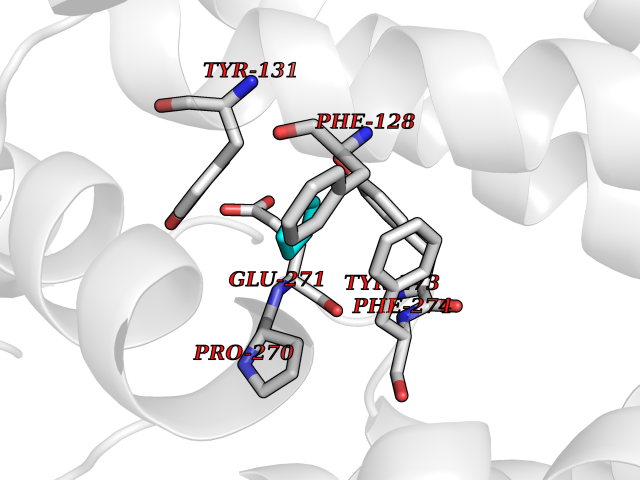 Pymol Map 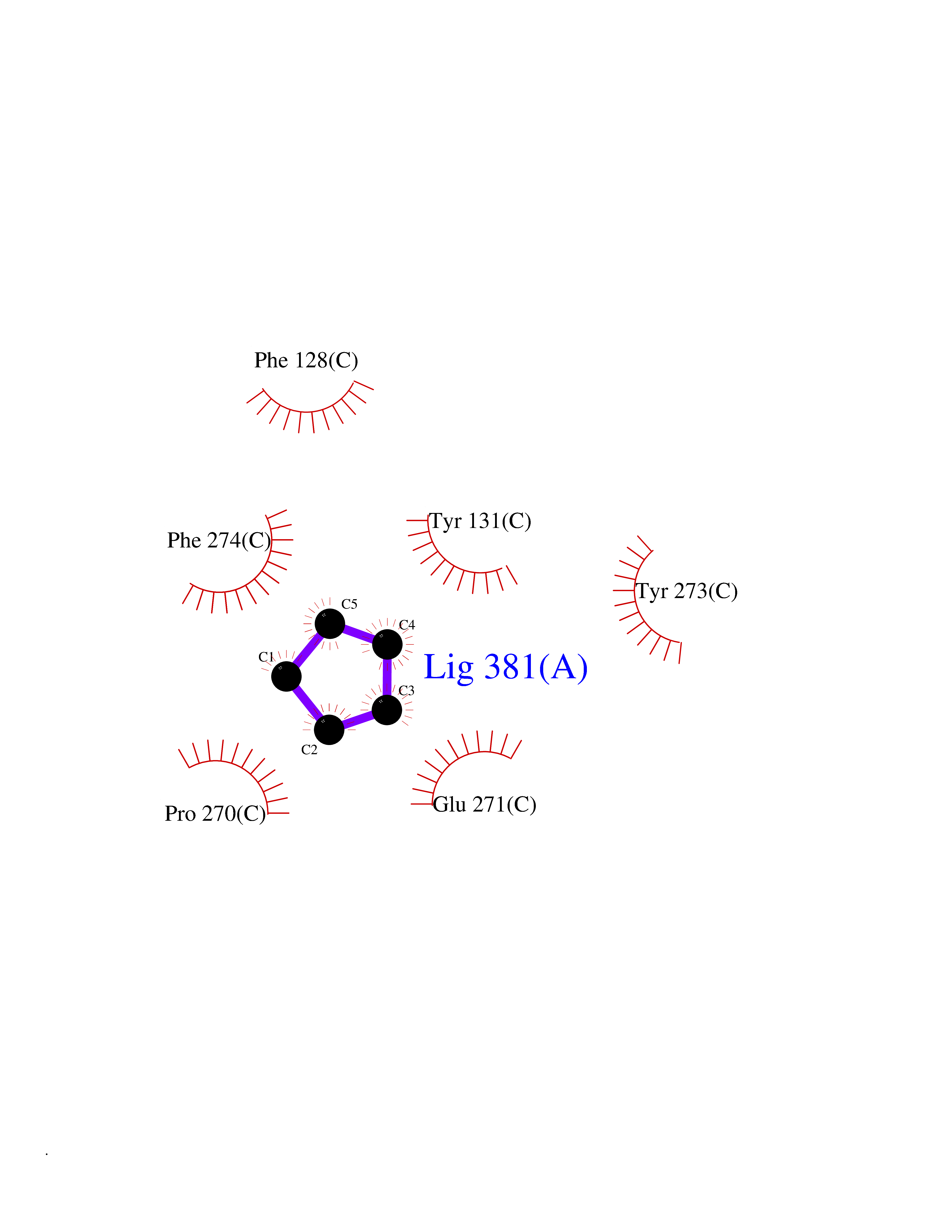 Ligplot Map 3D Binding mode Sequence VSASSRWLEGIRKWYYNAAGFNKLGLMRDDTIHENDDVKEAIRRLPENLYDDRVFRIKRALDLSMRQQILPKEQWTKYEEDKSYLEPYLKEVIRERKEREEWAKKELVDPLTTVREQCEQLEKCVKARERLELCDERVSSRSQTEEDCTEELLDFLHARDHCVAHKLFNSLKTNIRKSHPLMKIVNNAFIDLPAPSNISSWWNFGSLLGICLILQILTGLFLAMHYTSDTTTAFSSVTHICRDVNYGWIIRYMHANGASMFFICLYMHVGRGLYYGSYTFLETWNIGVILLLTVMATAFMGYVLPWGQMSFWGATVITNLLSAIPYIGTNLVEWIWGGFSVDKATLTRFFAFHFILPFIIMAIAMVHLLFLHETGSNNPTGISSDVDKIPFHPYYTIKDILGALLLILALMLLVLFAPDLLGDPDNYTPANPLNTPPHIKPEWYFLFAYAILRSIPNKLGGVLALAFSILILALIPLLHTSKQRSMMFRPLSQCLFWALVADLLTLTWIGGQPVEHPYITIGQLASVLYFLLILVLMPTAGTIENKLLKWSDLELHPPSYPWSHRGLLSSLDHTSIRRGFQVYKQVCSSCHSMDYVAYRHLVGVCYTEDEAKALAEEVEVQDGPNEDGEMFMRPGKLSDYFPKPYPNPEAARAANNGALPPDLSYIVRARHGGEDYVFSLLTGYCEPPTGVSLREGLYFNPYFPGQAIGMAPPIYNEVLEFDDGTPATMSQVAKDVCTFLRWAAEPEHDHRKRMGLKMLLMMGLLLPLVYAMKRHKWSVLKSRKLAYRPPKGRQFGHLTRVRHVITYSLSPFEQRAFPHYFSKGIPNVLRRTRACILRVAPPFVAFYLVYTWGTQEFEKSKRKNPA Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Cyclin-dependent kinase 4 | 2W96 | 4.75 | |
Target general information Gen name CDK4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Cell cycle Function ATP binding.Cyclin binding.Cyclin-dependent protein serine/threonine kinase activity.Cyclin-dependent protein serine/threonine kinase regulator activity.Protein complex binding. Related diseases Melanoma, cutaneous malignant 3 (CMM3) [MIM:609048]: A malignant neoplasm of melanocytes, arising de novo or from a pre-existing benign nevus, which occurs most often in the skin but may also involve other sites. {ECO:0000269|PubMed:7652577, ECO:0000269|PubMed:8528263, ECO:0000269|PubMed:9311594, ECO:0000269|PubMed:9425228}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12001; DB03496; DB12010; DB09073; DB02733; DB11730; DB15442 Interacts with Q9UH17; P24385; P30279; P30281; Q16543; P50613; P38936; P46527; P49918; P42771; P42772; P42773; P55273; Q9UJC3; P08238; Q9UKT9; Q0VD86; P01106; Q9ULD0; P28749; Q08999; P09936; Q8N720 EC number 2.7.11.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Cell division; Cytoplasm; Disease variant; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 30138.4 Length 267 Aromaticity 0.09 Instability index 36.2 Isoelectric point 5.78 Charge (pH=7) -5.83 2D Binding mode Binding energy (Kcal/mol) -6.48 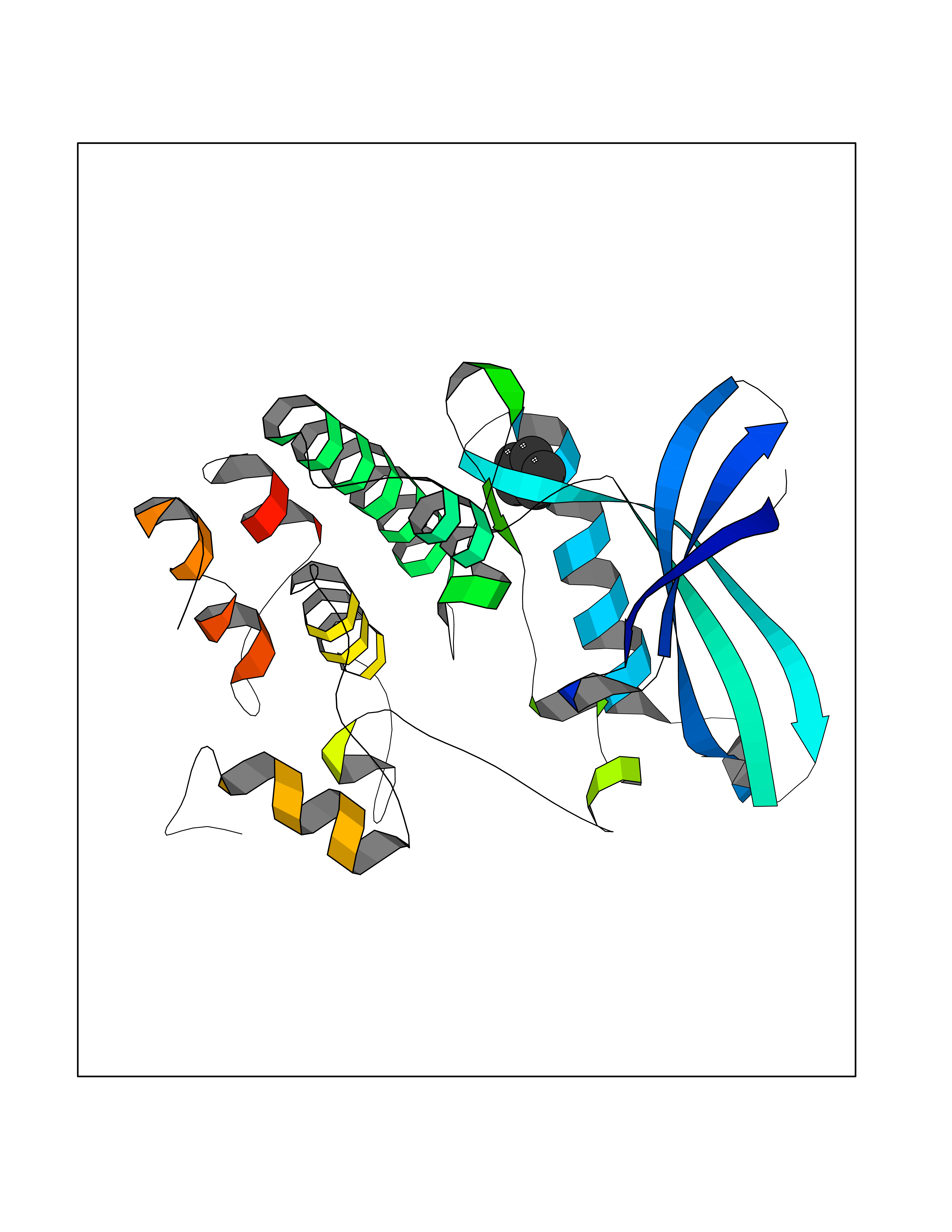 Molscript Map 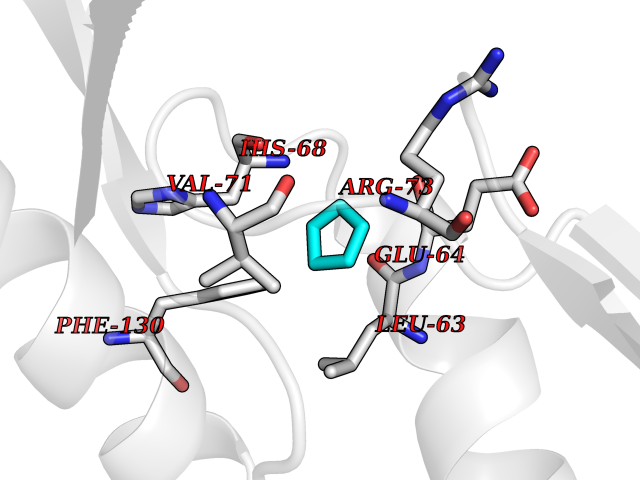 Pymol Map 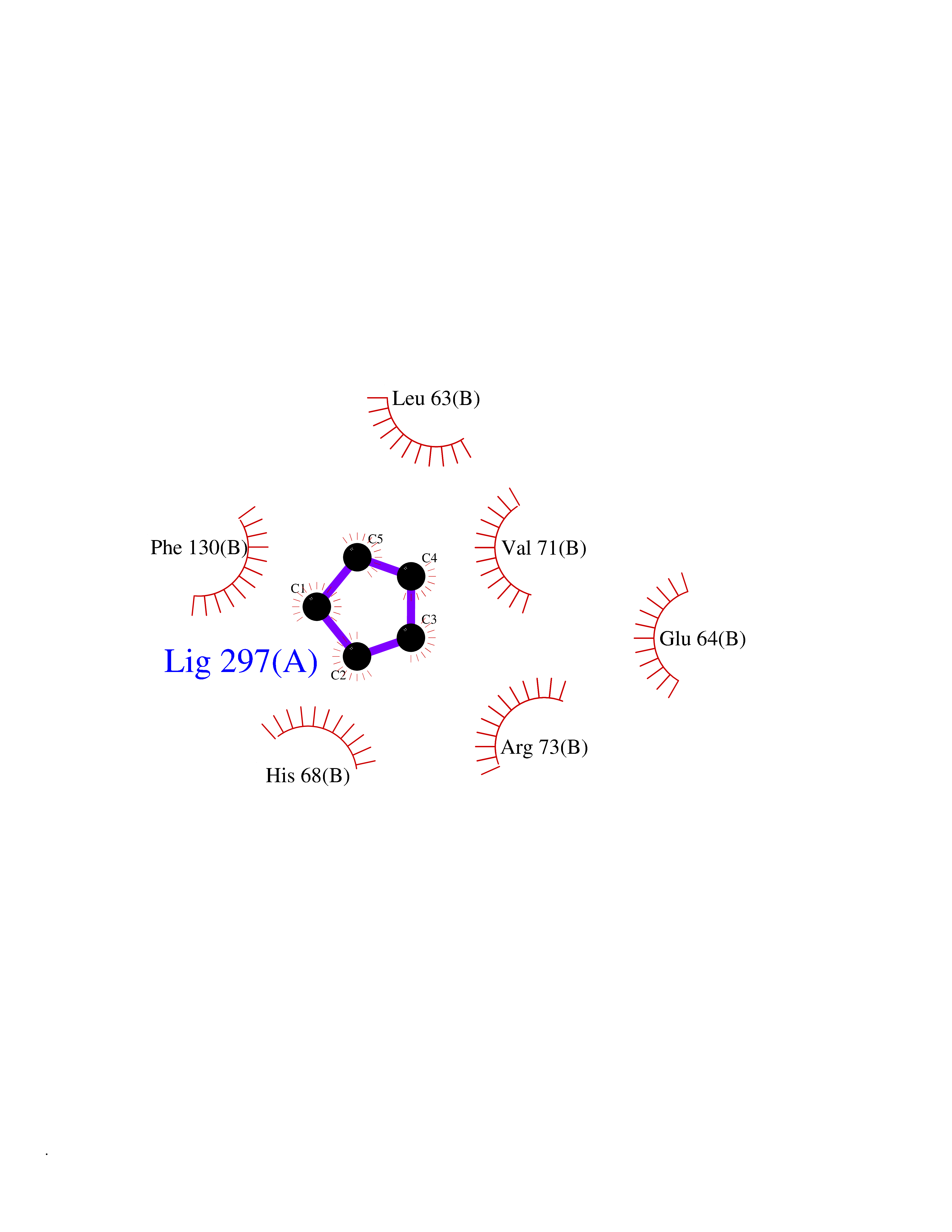 Ligplot Map 3D Binding mode Sequence SRYEPVAEIGVGAYGTVYKARDPHSGHFVALKSVRVPNGEEGLPISTVREVALLRRLEAFEHPNVVRLMDVCATSRTDREIKVTLVFEHVDQDLRTYLDKAPPPGLPAETIKDLMRQFLRGLDFLHANCIVHRDLKPENILVTSGGTVKLADFGLARIYSYQMALDPVVVTLWYRAPEVLLQSTYATPVDMWSVGCIFAEMFRRKPLFCGNSEADQLGKIFDLIGLPPEDDWVPEMEESGAQLLLEMLTFNPHKRISAFRALQHSYL Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Steroid 21-hydroxylase | 4Y8W | 4.75 | |
Target general information Gen name CYP21A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CYP21;CYP21B Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Heme binding.Iron ion binding.Steroid 21-monooxygenase activity.Steroid binding.Steroid hydroxylase activity. Related diseases Adrenal hyperplasia 3 (AH3) [MIM:201910]: A form of congenital adrenal hyperplasia, a common recessive disease due to defective synthesis of cortisol. Congenital adrenal hyperplasia is characterized by androgen excess leading to ambiguous genitalia in affected females, rapid somatic growth during childhood in both sexes with premature closure of the epiphyses and short adult stature. Four clinical types: 'salt wasting' (SW, the most severe type), 'simple virilizing' (SV, less severely affected patients), with normal aldosterone biosynthesis, 'non-classic form' or late-onset (NC or LOAH) and 'cryptic' (asymptomatic). {ECO:0000269|PubMed:10051010, ECO:0000269|PubMed:10094562, ECO:0000269|PubMed:10198222, ECO:0000269|PubMed:10364682, ECO:0000269|PubMed:10391209, ECO:0000269|PubMed:10408778, ECO:0000269|PubMed:10408786, ECO:0000269|PubMed:10443693, ECO:0000269|PubMed:10496074, ECO:0000269|PubMed:10720040, ECO:0000269|PubMed:11232002, ECO:0000269|PubMed:11598371, ECO:0000269|PubMed:11600539, ECO:0000269|PubMed:11746135, ECO:0000269|PubMed:12213891, ECO:0000269|PubMed:12222711, ECO:0000269|PubMed:12788866, ECO:0000269|PubMed:12887291, ECO:0000269|PubMed:12915679, ECO:0000269|PubMed:1406699, ECO:0000269|PubMed:1406709, ECO:0000269|PubMed:14676460, ECO:0000269|PubMed:14715874, ECO:0000269|PubMed:1496017, ECO:0000269|PubMed:15110320, ECO:0000269|PubMed:15126570, ECO:0000269|PubMed:16046588, ECO:0000269|PubMed:1644925, ECO:0000269|PubMed:16984992, ECO:0000269|PubMed:18319307, ECO:0000269|PubMed:18381579, ECO:0000269|PubMed:18445671, ECO:0000269|PubMed:1864962, ECO:0000269|PubMed:1937474, ECO:0000269|PubMed:20080860, ECO:0000269|PubMed:2072928, ECO:0000269|PubMed:21169732, ECO:0000269|PubMed:22014889, ECO:0000269|PubMed:2303461, ECO:0000269|PubMed:27721825, ECO:0000269|PubMed:29328376, ECO:0000269|PubMed:3038528, ECO:0000269|PubMed:3257825, ECO:0000269|PubMed:3260007, ECO:0000269|PubMed:3267225, ECO:0000269|PubMed:3497399, ECO:0000269|PubMed:3871526, ECO:0000269|PubMed:7749410, ECO:0000269|PubMed:8478006, ECO:0000269|PubMed:8485582, ECO:0000269|PubMed:8989258, ECO:0000269|PubMed:9067760, ECO:0000269|PubMed:9187661, ECO:0000269|PubMed:9497336}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01026; DB05667 Interacts with NA EC number 1.14.14.16 Uniprot keywords 3D-structure; Alternative splicing; Congenital adrenal hyperplasia; Disease variant; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Lipid-binding; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid-binding; Steroidogenesis Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 50326.2 Length 442 Aromaticity 0.08 Instability index 50.06 Isoelectric point 7.79 Charge (pH=7) 2.07 2D Binding mode Binding energy (Kcal/mol) -6.49  Molscript Map 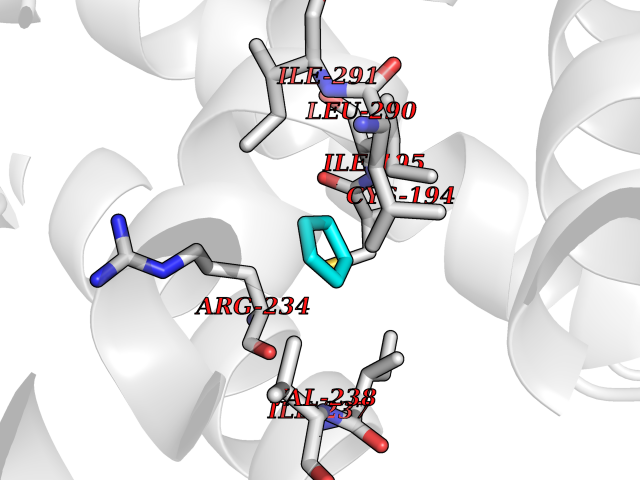 Pymol Map 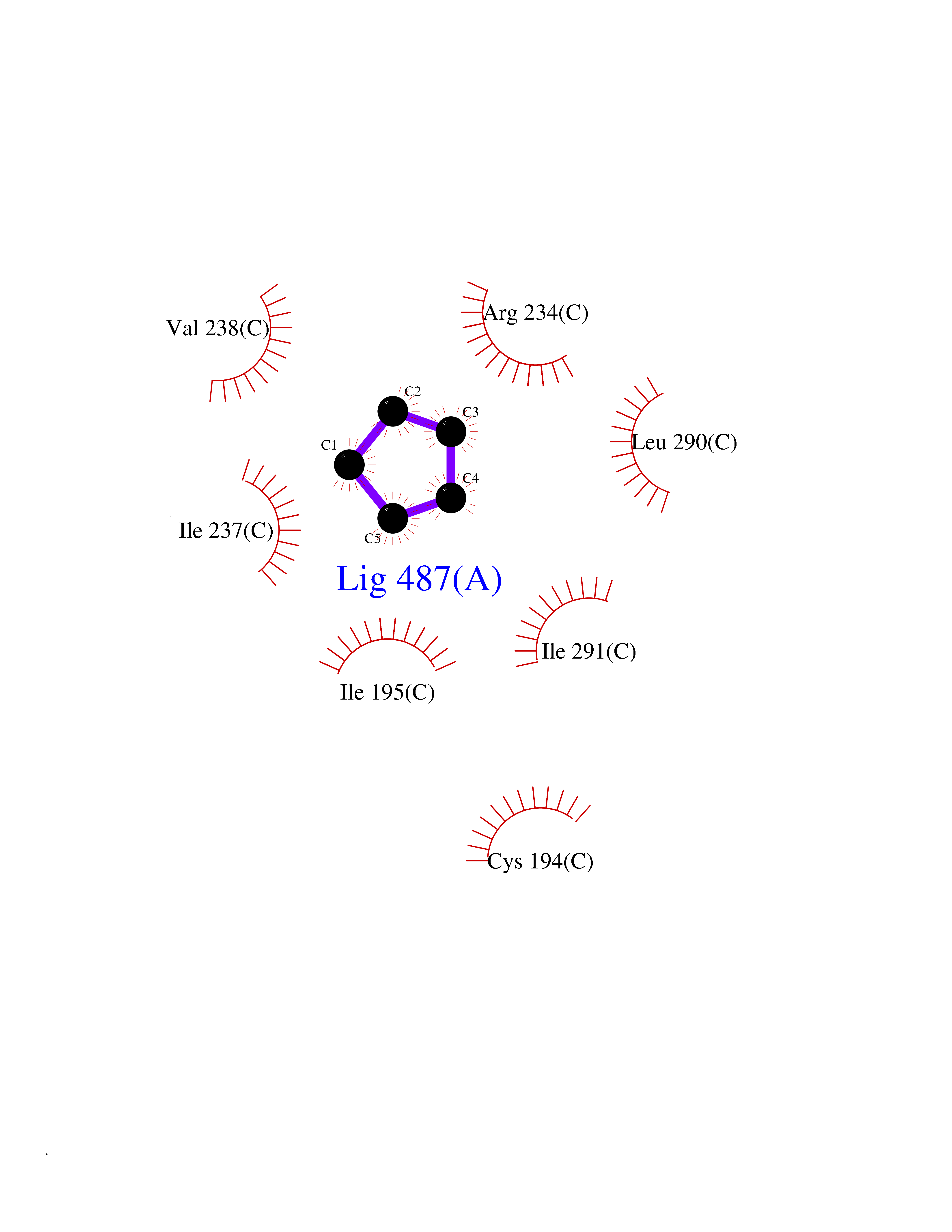 Ligplot Map 3D Binding mode Sequence KLPPLAPGFLHLLQPDLPIYLLGLTQKFGPIYRLHLGLQDVVVLNSKRTIEEAMVKKWADFAGRPEPLTYKLVSRNYPDLSLGDYSLLWKAHKKLTRSALLLGIRDSMEPVVEQLTQEFCERMRAQPGTPVAIEEEFSLLTCSIICYLTFGDKIKDDNLMPAYYKCIQEVLKTWSHWSIQIVDVIPFLRFFPNPGLRRLKQAIEKRDHIVEMQLRQHKESLVAGQWRDMMDYMLQGVAGQLLEGHVHMAAVDLLIGGTETTANTLSWAVVFLLHHPEIQQRLQEELDHESRVPYKDRARLPLLNATIAEVLRLRPVVPLALPHRTTRPSSISGYDIPEGTVIIPNLQGAHLDETVWERPHEFWPDRFLEPGKNSRALAFGCGARVCLGEPLARLELFVVLTRLLQAFTLLPSGDALPSLQPLPHCSVILKMQPFQVRLQPRG Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Angiotensin II receptor type-1 (AGTR1) | 4ZUD | 4.75 | |
Target general information Gen name AGTR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Type-1 angiotensin II receptor; Angiotensin II type-1 receptor; Angiotensin II receptor 1; Angiotensin 1 receptor; AT2R1B; AT2R1; AT1BR; AT1AR; AT1; AGTR1B; AGTR1A Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Mediates its action by association with G proteins that activate a phosphatidylinositol-calcium second messenger system. Receptor for angiotensin II. Related diseases Renal tubular dysgenesis (RTD) [MIM:267430]: Autosomal recessive severe disorder of renal tubular development characterized by persistent fetal anuria and perinatal death, probably due to pulmonary hypoplasia from early-onset oligohydramnios (the Potter phenotype). {ECO:0000269|PubMed:16116425}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11842; DB08822; DB13919; DB00796; DB05739; DB00876; DB09279; DB01342; DB01029; DB00678; DB00275; DB01347; DB01349; DB00966; DB00177 Interacts with PRO_0000032458 [P01019]; P35414; P05026; Q6ZMG9; O75937; P54368 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disease variant; Disulfide bond; G-protein coupled receptor; Glycoprotein; Host-virus interaction; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 30770.7 Length 271 Aromaticity 0.16 Instability index 28.86 Isoelectric point 8.09 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -6.48 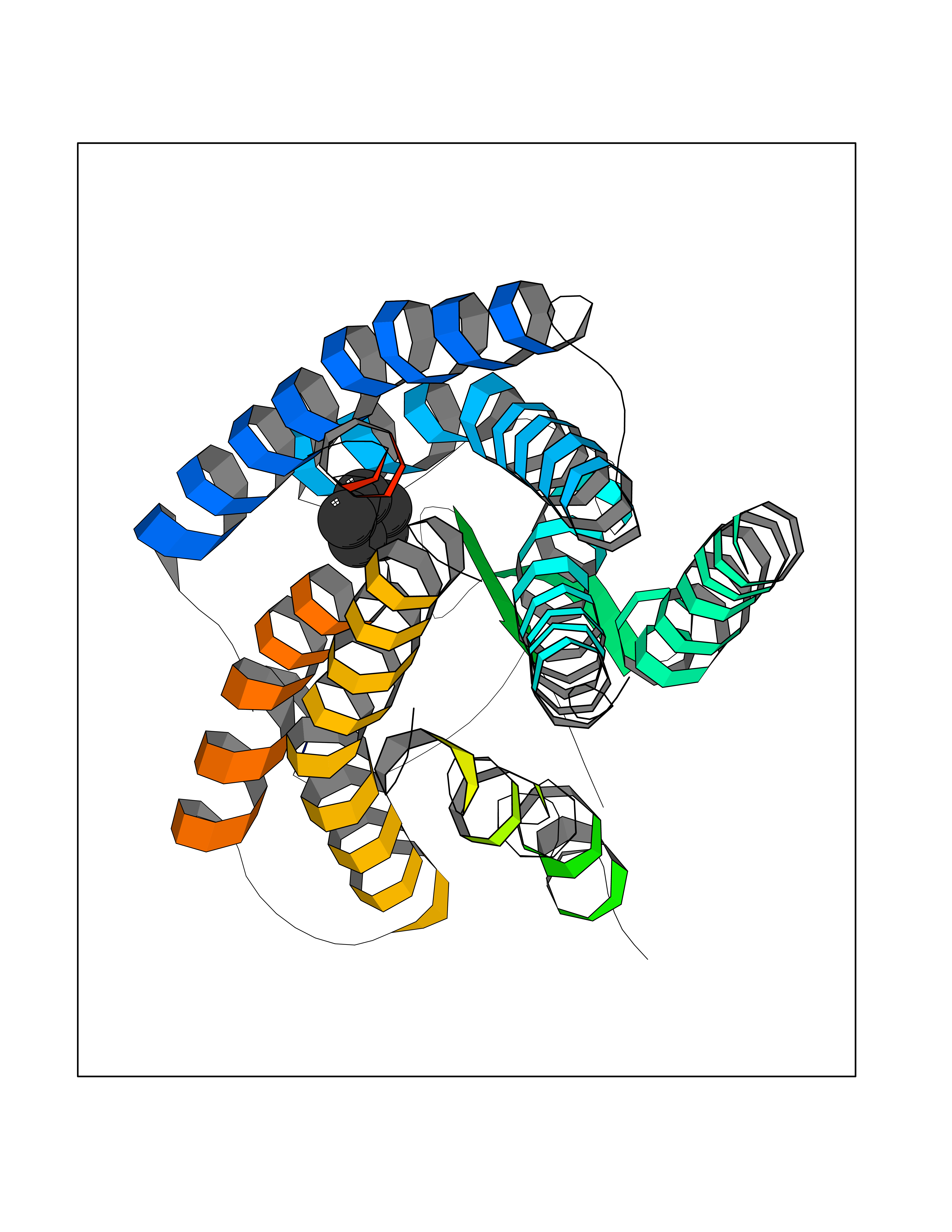 Molscript Map 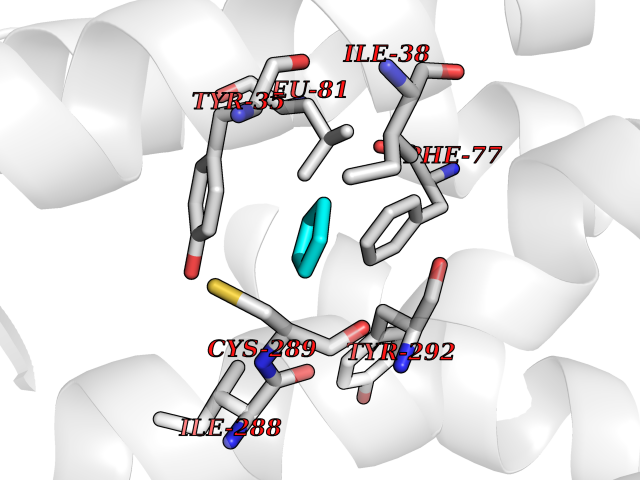 Pymol Map 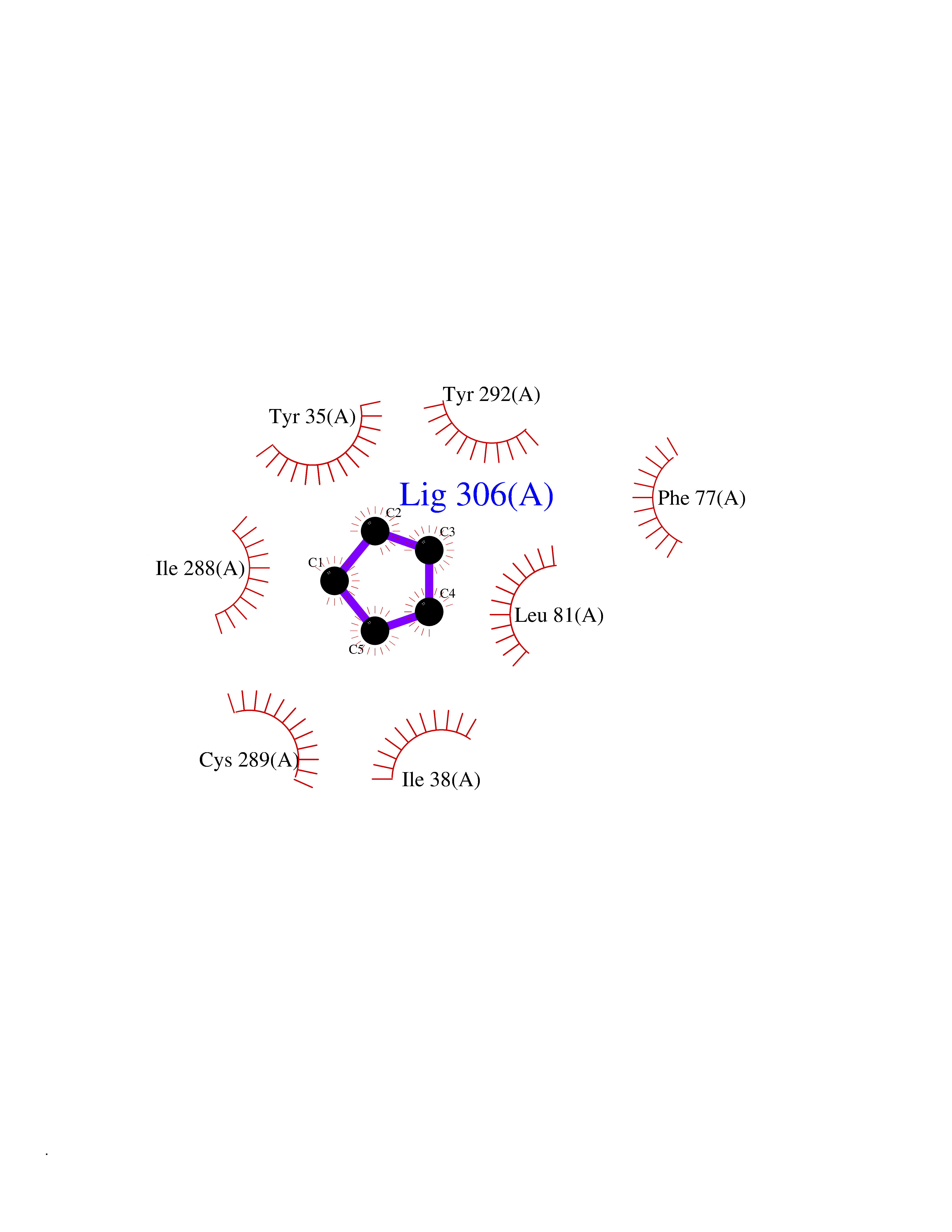 Ligplot Map 3D Binding mode Sequence ILNSSDCPKAGRHNYIFVMIPTLYSIIFVVGIFGNSLVVIVIYFYMKLKTVASVFLLNLALADLCFLLTLPLWAVYTAMEYRWPFGNYLCKIASASVSFNLYASVFLLTCLSIDRYLAIVHPTMLVAKVTCIIIWLLAGLASLPAIIHRNVFFIENTNITVCAFHYESTLPIGLGLTKNILGFLFPFLIILTSYTLIWKALNDDIFKIIMAIVLFFFFSWIPHQIFTFLDVLIQLGIIRDCRIADIVDTAMPITICIAYFNNCLNPLFYGF Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 4.75 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -6.48 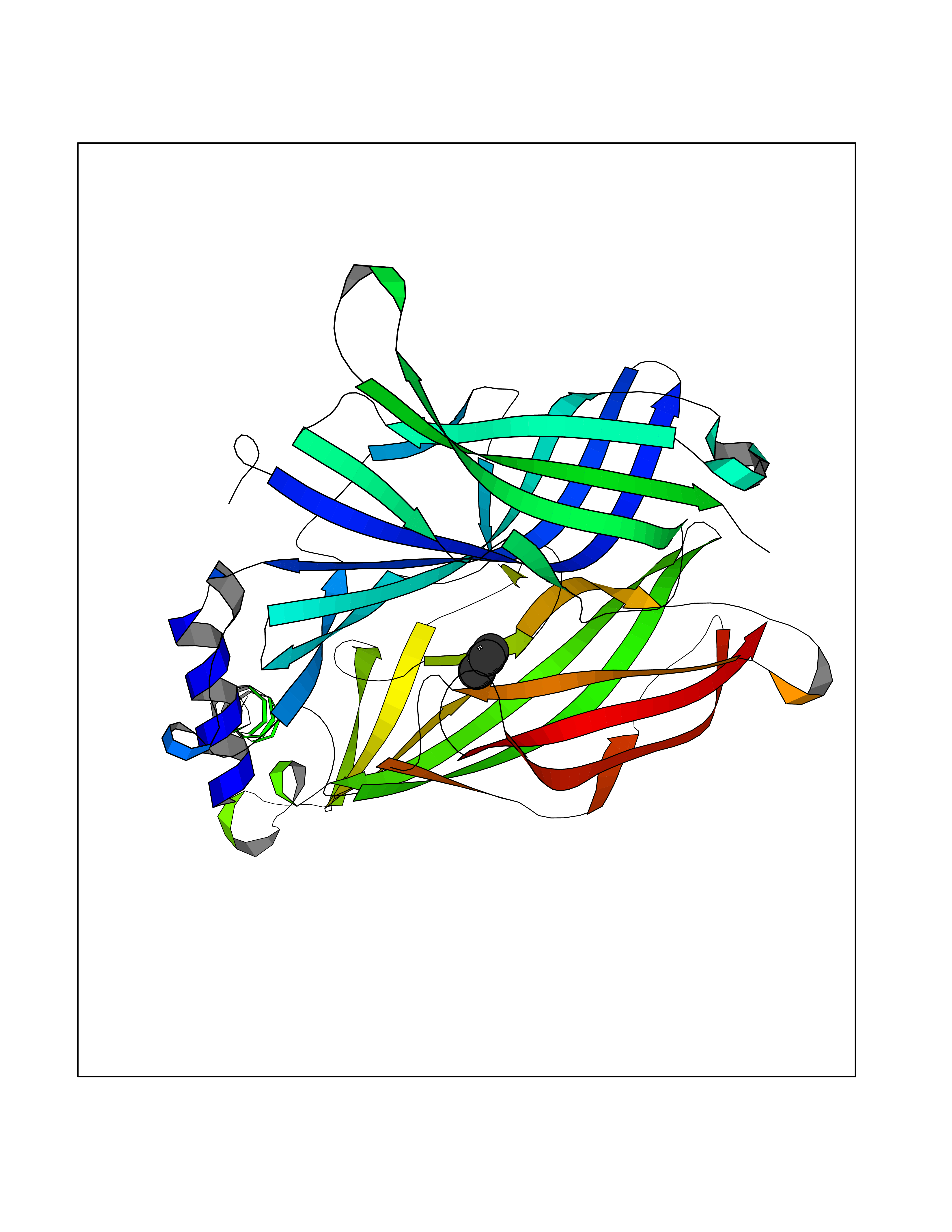 Molscript Map 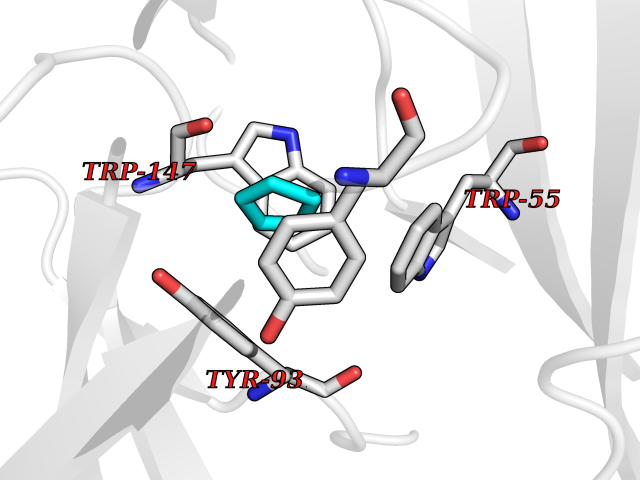 Pymol Map  Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 4.75 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -6.47 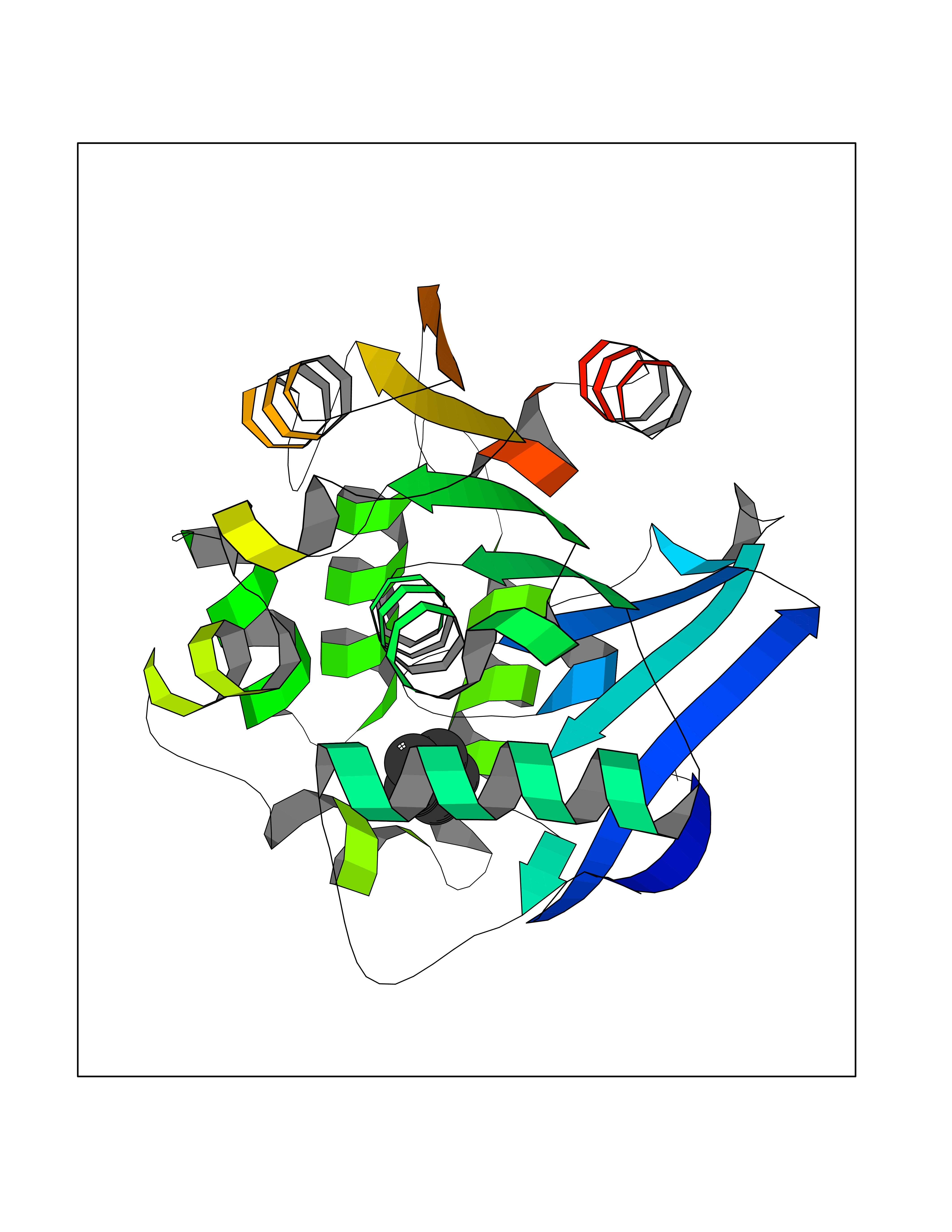 Molscript Map 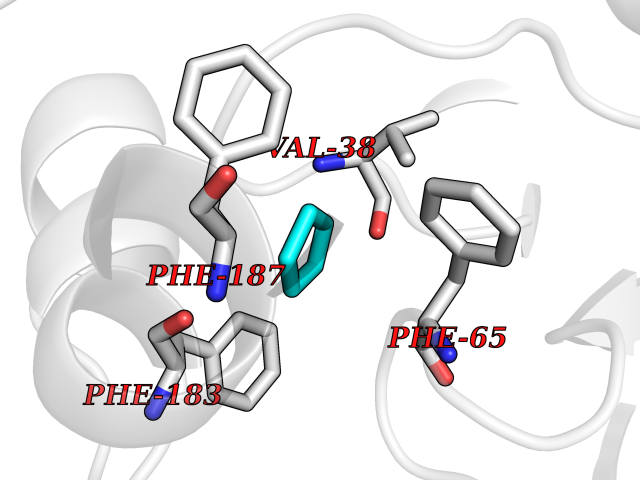 Pymol Map 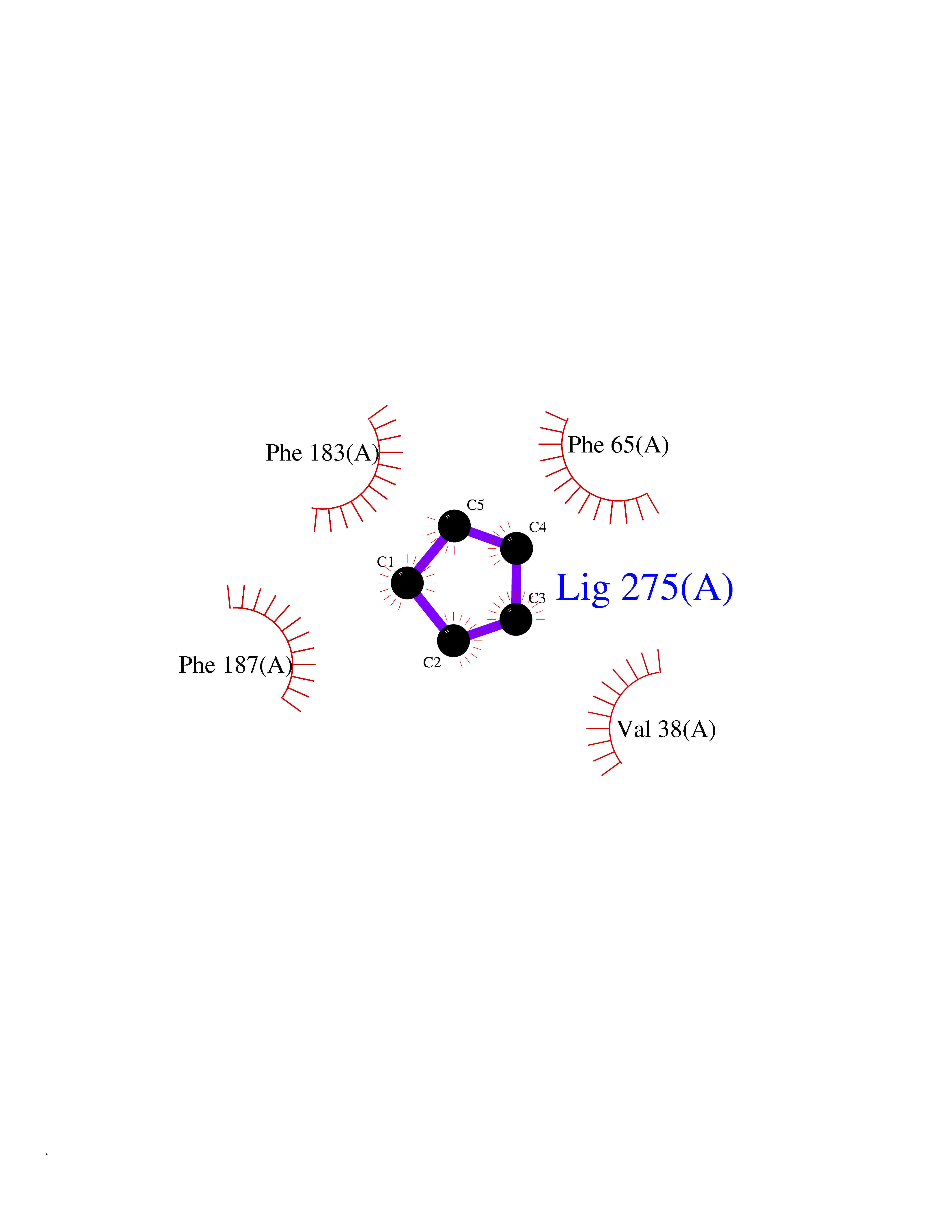 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Tyrosine-protein kinase BRK (PTK6) | 5DA3 | 4.75 | |
Target general information Gen name PTK6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein-tyrosine kinase 6; Breast tumor kinase; BRK Protein family Protein kinase superfamily, Tyr protein kinase family, BRK/PTK6/SIK subfamily Biochemical class Kinase Function Non-receptor tyrosine-protein kinase implicated in the regulation of a variety of signaling pathways that control the differentiation and maintenance of normal epithelia, as well as tumor growth. Function seems to be context dependent and differ depending on cell type, as well as its intracellular localization. A number of potential nuclear and cytoplasmic substrates have been identified. These include the RNA-binding proteins: KHDRBS1/SAM68, KHDRBS2/SLM1, KHDRBS3/SLM2 and SFPQ/PSF; transcription factors: STAT3 and STAT5A/B and a variety of signaling molecules: ARHGAP35/p190RhoGAP, PXN/paxillin, BTK/ATK, STAP2/BKS. Associates also with a variety of proteins that are likely upstream of PTK6 in various signaling pathways, or for which PTK6 may play an adapter-like role. These proteins include ADAM15, EGFR, ERBB2, ERBB3 and IRS4. In normal or non-tumorigenic tissues, PTK6 promotes cellular differentiation and apoptosis. In tumors PTK6 contributes to cancer progression by sensitizing cells to mitogenic signals and enhancing proliferation, anchorage-independent survival and migration/invasion. Association with EGFR, ERBB2, ERBB3 may contribute to mammary tumor development and growth through enhancement of EGF-induced signaling via BTK/AKT and PI3 kinase. Contributes to migration and proliferation by contributing to EGF-mediated phosphorylation of ARHGAP35/p190RhoGAP, which promotes association with RASA1/p120RasGAP, inactivating RhoA while activating RAS. EGF stimulation resulted in phosphorylation of PNX/Paxillin by PTK6 and activation of RAC1 via CRK/CrKII, thereby promoting migration and invasion. PTK6 activates STAT3 and STAT5B to promote proliferation. Nuclear PTK6 may be important for regulating growth in normal epithelia, while cytoplasmic PTK6 might activate oncogenic signaling pathways. Related diseases Periodic paralysis hypokalemic 1 (HOKPP1) [MIM:170400]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:17418573, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:7987325, ECO:0000269|PubMed:8004673}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Malignant hyperthermia 5 (MHS5) [MIM:601887]: Autosomal dominant disorder that is potentially lethal in susceptible individuals on exposure to commonly used inhalational anesthetics and depolarizing muscle relaxants. {ECO:0000269|PubMed:9199552}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Thyrotoxic periodic paralysis 1 (TTPP1) [MIM:188580]: A sporadic muscular disorder characterized by episodic weakness and hypokalemia during a thyrotoxic state. It is clinically similar to hereditary hypokalemic periodic paralysis, except for the fact that hyperthyroidism is an absolute requirement for disease manifestation. The disease presents with recurrent episodes of acute muscular weakness of the four extremities that vary in severity from paresis to complete paralysis. Attacks are triggered by ingestion of a high carbohydrate load or strenuous physical activity followed by a period of rest. Thyrotoxic periodic paralysis can occur in association with any cause of hyperthyroidism, but is most commonly associated with Graves disease. {ECO:0000269|PubMed:15001631}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 18 (CMYO18) [MIM:620246]: A congenital myopathy of variable severity, ranging from severe fetal akinesia to milder forms of muscle weakness. Most affected individuals show delayed motor development with generalized hypotonia and progressive axial and limb muscle weakness beginning soon after birth or in infancy. Additional features may include swallowing difficulties, external ophthalmoplegia, ptosis, high-arched palate, and respiratory insufficiency. Muscle biopsy shows variable morphologic abnormalities, including alveolar changes in the intermyofibrillar network, fiber size variability, focal disorganization, internal nuclei, and dilated sarcoplasmic reticulum and T-tubules. CMYO18 inheritance is autosomal dominant or recessive. {ECO:0000269|PubMed:28012042, ECO:0000269|PubMed:31227654, ECO:0000269|PubMed:33060286}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010; DB11800; DB05294; DB15035 Interacts with Q08043; Q3KP44; Q13191; Q16543; Q92841; Q8N9I9; Q5JST6; P04626; O00471; O14526; Q13480; P08238; P42858; Q9UKT9; Q5VWX1; Q5T5P2-6; P10721; O14770-4; Q13064; Q8TDC0; P78337; Q9NQX0; Q13882; Q04864; P23246; Q13239-3; O00401; Q9BYN7 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell projection; Cytoplasm; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; SH2 domain; SH3 domain; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 30240.6 Length 264 Aromaticity 0.09 Instability index 46.88 Isoelectric point 6.95 Charge (pH=7) -0.14 2D Binding mode Binding energy (Kcal/mol) -6.48 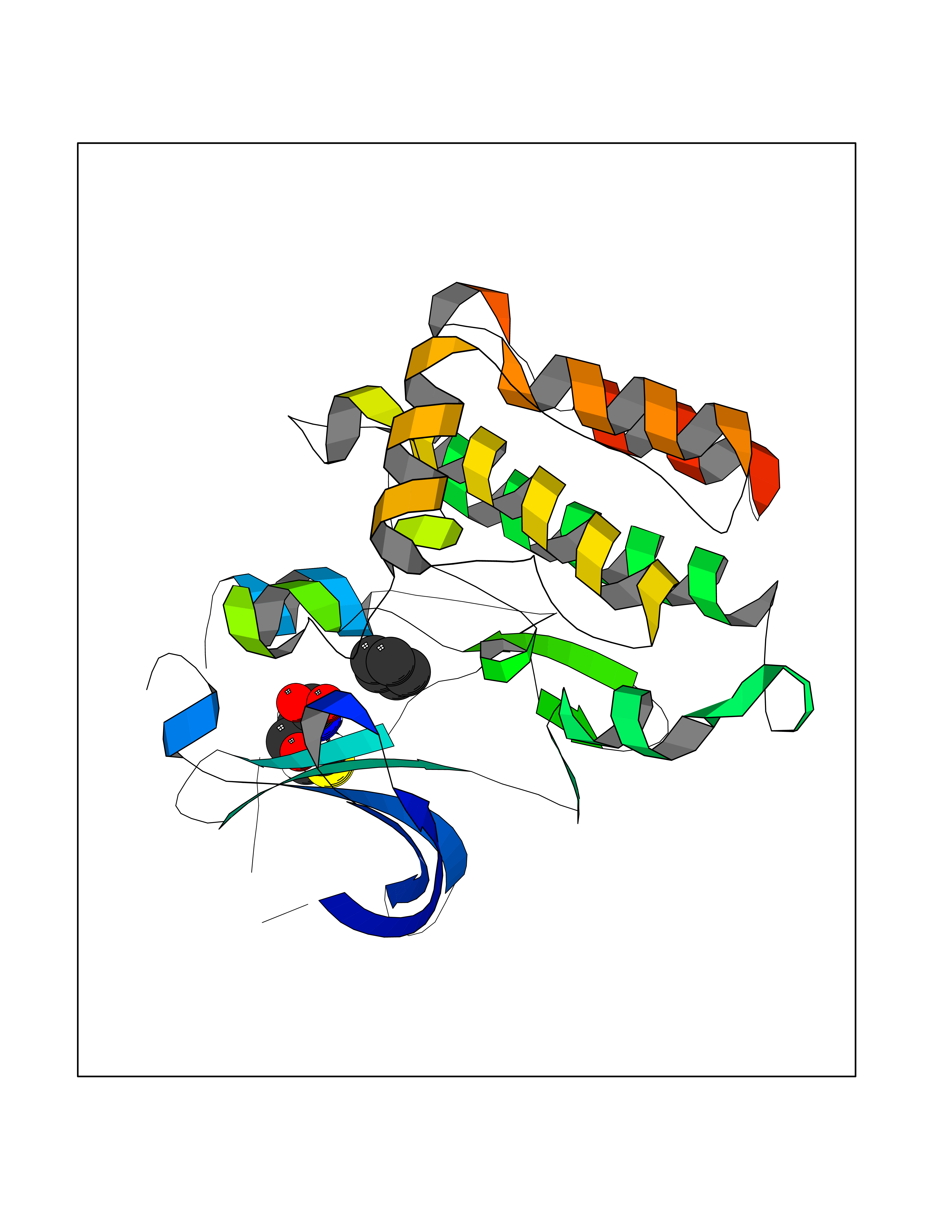 Molscript Map 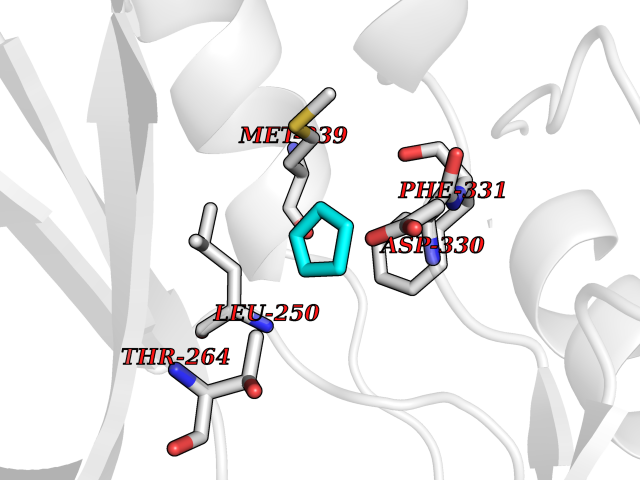 Pymol Map 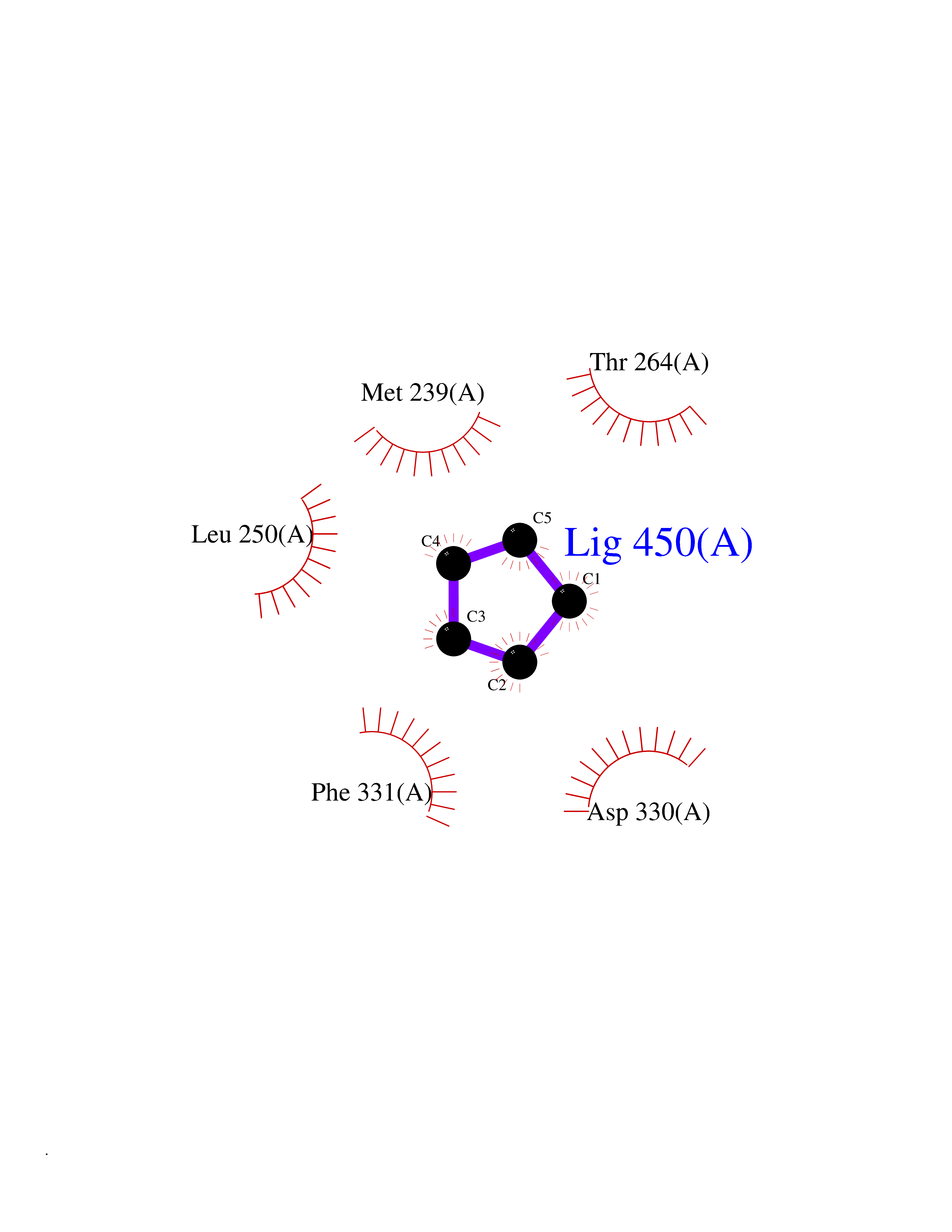 Ligplot Map 3D Binding mode Sequence XERPREEFTLCRKLGSGYFGEVFEGLWKDRVQVAIKVISRDNLLHQMLQSEIQAMKKLRHKHILALYAVVSVGDPVYIITELMAKGSLLELLRDSDEKVLPVSELLDIAWQVAEGMCYLESQNYIHRDLAARNILVGENTLCKVGDFGLARLIKEDVYLSHDHNIPYKWTAPEALSRGHYSTKSDVWSFGILLHEMFSRGQVPYPGMSNHEAFLRVDAGYRMPCPLECPPSVHKLMLTCWCRDPEQRPTFKALRERLSSFTSHH Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | NDR1 protein kinase (STK38) | 6BXI | 4.75 | |
Target general information Gen name STK38 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein kinase 38; Nuclear Dbf2-related kinase 1; NDR1 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family Biochemical class Kinase Function Converts MAP3K2 from its phosphorylated form to its non-phosphorylated form and inhibits autophosphorylation of MAP3K2. Negative regulator of MAP3K1/2 signaling. Related diseases Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:8955068}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 3 (NS3) [MIM:609942]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:16773572, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:17468812, ECO:0000269|PubMed:19396835, ECO:0000269|PubMed:20949621}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gastric cancer (GASC) [MIM:613659]: A malignant disease which starts in the stomach, can spread to the esophagus or the small intestine, and can extend through the stomach wall to nearby lymph nodes and organs. It also can metastasize to other parts of the body. The term gastric cancer or gastric carcinoma refers to adenocarcinoma of the stomach that accounts for most of all gastric malignant tumors. Two main histologic types are recognized, diffuse type and intestinal type carcinomas. Diffuse tumors are poorly differentiated infiltrating lesions, resulting in thickening of the stomach. In contrast, intestinal tumors are usually exophytic, often ulcerating, and associated with intestinal metaplasia of the stomach, most often observed in sporadic disease. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:7773929}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in KRAS are a cause of pylocytic astrocytoma (PA). Pylocytic astrocytomas are neoplasms of the brain and spinal cord derived from glial cells which vary from histologically benign forms to highly anaplastic and malignant tumors. {ECO:0000269|PubMed:16247081}.; DISEASE: Cardiofaciocutaneous syndrome 2 (CFC2) [MIM:615278]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. CFC2 patients often do not have the skin abnormalities, such as ichthyosis, hyperkeratosis, and hemangioma observed in CFC1. {ECO:0000269|PubMed:16474404, ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:20949621, ECO:0000269|PubMed:21797849}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: KRAS mutations are involved in cancer development. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:1553789, ECO:0000269|PubMed:16533793, ECO:0000269|PubMed:24623306, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:3627975, ECO:0000269|PubMed:6092920, ECO:0000269|PubMed:6695174, ECO:0000269|PubMed:7773929}.; DISEASE: Oculoectodermal syndrome (OES) [MIM:600268]: A syndrome characterized by the association of epibulbar dermoids and aplasia cutis congenita. Affected individuals show multiple, asymmetric, atrophic, non-scarring and hairless regions that may be associated with hamartomas. Ectodermal changes include linear hyperpigmentation that may follow the lines of Blaschko and rarely epidermal nevus-like lesions. Epibulbar dermoids may be uni-or bilateral. Additional ocular anomalies such as skin tags of the upper eyelid, rarely optic nerve or retinal changes, and microphthalmia can be present. The phenotypic expression is highly variable, and various other abnormalities have occasionally been reported including growth failure, lymphedema, cardiovascular defects, as well as neurodevelopmental symptoms like developmental delay, epilepsy, learning difficulties, and behavioral abnormalities. Benign tumor-like lesions such as nonossifying fibromas of the long bones and giant cell granulomas of the jaws have repeatedly been observed and appear to be age-dependent, becoming a common manifestation in individuals aged 5 years or older. {ECO:0000269|PubMed:25808193, ECO:0000269|PubMed:26970110, ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Schimmelpenning-Feuerstein-Mims syndrome (SFM) [MIM:163200]: A disease characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects. Many oral manifestations have been reported, not only including hypoplastic and malformed teeth, and mucosal papillomatosis, but also ankyloglossia, hemihyperplastic tongue, intraoral nevus, giant cell granuloma, ameloblastoma, bone cysts, follicular cysts, oligodontia, and odontodysplasia. Sebaceous nevi follow the lines of Blaschko and these can continue as linear intraoral lesions, as in mucosal papillomatosis. {ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with P49407; P32121; Q8N9N5-2; Q03135; P08238; Q9H8S9; Q70IA6; P16333; P30086; P02638 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Chromosome; Cytoplasm; Direct protein sequencing; DNA damage; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 38701.1 Length 333 Aromaticity 0.11 Instability index 38.05 Isoelectric point 5.69 Charge (pH=7) -7.8 2D Binding mode Binding energy (Kcal/mol) -6.48 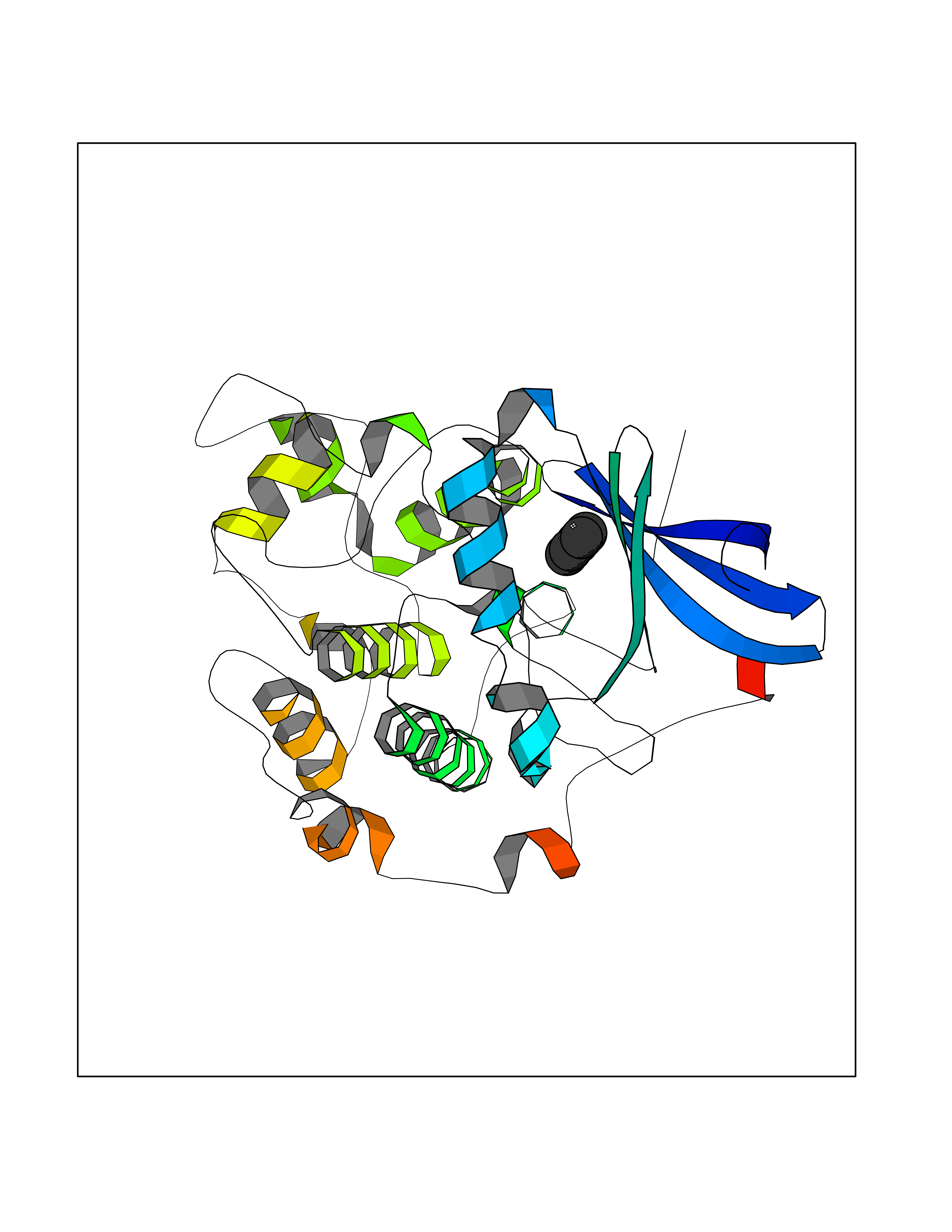 Molscript Map 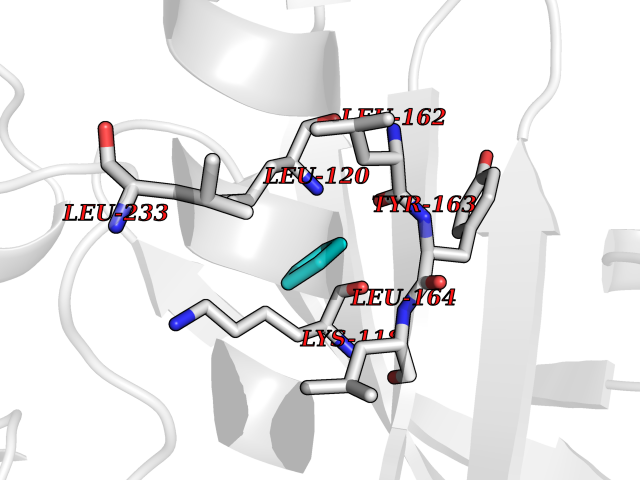 Pymol Map 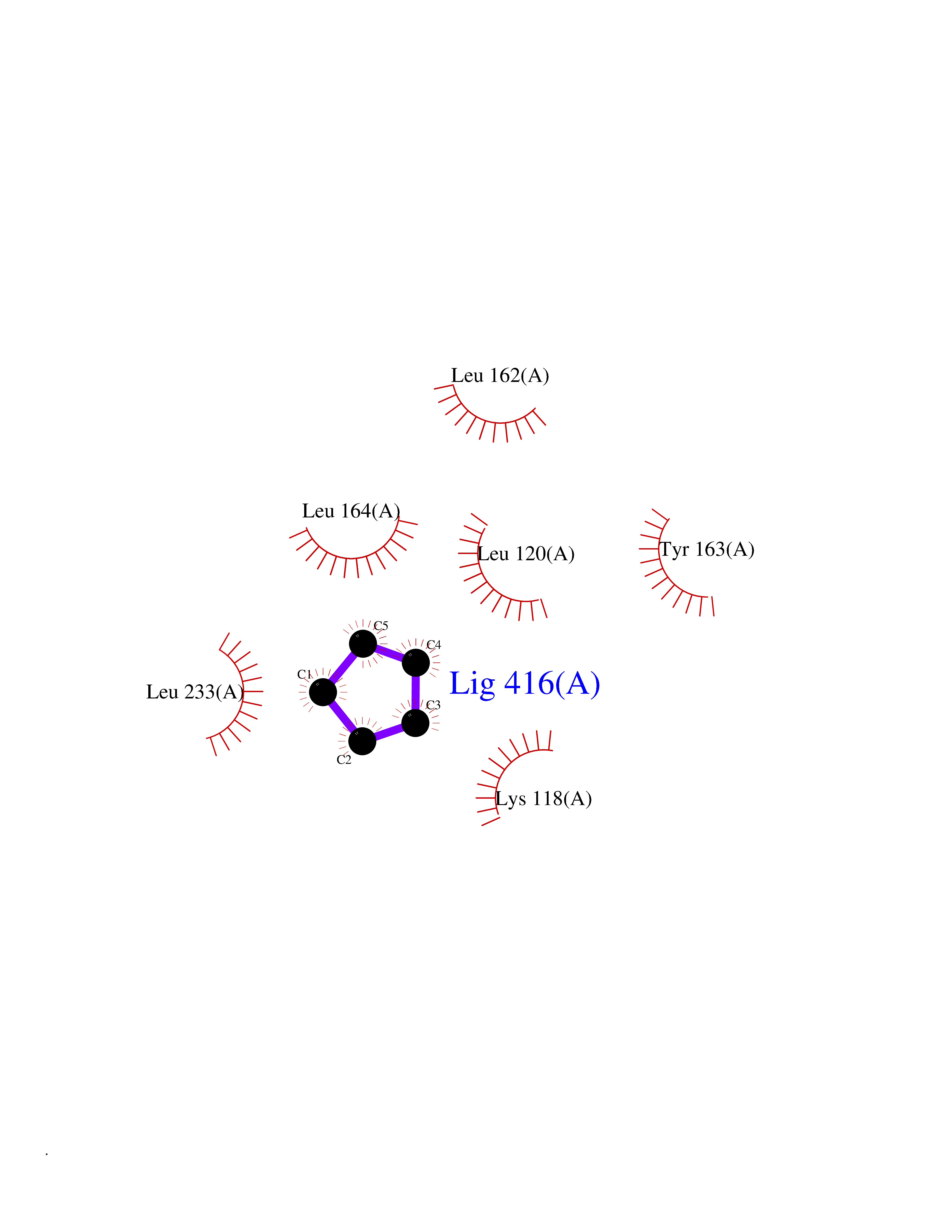 Ligplot Map 3D Binding mode Sequence TRLGLEDFESLKVIGRGAFGEVRLVQKKDTGHVYAMKILRKADMLEKEQVGHIRAERDILVEADSLWVVKMFYSFQDKLNLYLIMEFLPGGDMMTLLMKKDTLTEEETQFYIAETVLAIDSIHQLGFIHRDIKPDNLLLDSKGHVKLSDFGLCTGLKKAHRTEFYRNLNHSLPSDFTFQNMNSKRKAETWKRNRRQLAFSTVGTPDYIAPEVFMQTGYNKLCDWWSLGVIMYEMLIGYPPFCSETPQETYKKVMNWKETLTFPPEVPISEKAKDLILRFCCEWEHRIGAPGVEEIKSNSFFEGVDWEHIRERPAAISIEIKSIDDTSNFDEFP Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Bacterial Nicotinate-nucleotide adenylyltransferase (Bact nadD) | 1K4K | 4.75 | |
Target general information Gen name Bact nadD Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms nadD of Escherichia coli (strain K12); Nicotinate mononucleotide adenylyltransferase of Escherichia coli (strain K12); NaMN adenylyltransferase of Escherichia coli (strain K12); Deamido-NAD(+)Nicotina Protein family NadD family Biochemical class Kinase Function Catalyzes the reversible adenylation of nicotinate mononucleotide (namn) to nicotinic acid adenine dinucleotide (naad). Related diseases Asthma-related traits 5 (ASRT5) [MIM:611064]: Asthma-related traits include clinical symptoms of asthma, such as coughing, wheezing, dyspnea, bronchial hyperresponsiveness as assessed by methacholine challenge test, serum IgE levels, atopy and atopic dermatitis. {ECO:0000269|PubMed:17503328}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.7.7.18 Uniprot keywords 3D-structure; ATP-binding; NAD; Nucleotide-binding; Nucleotidyltransferase; Pyridine nucleotide biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 24527.6 Length 213 Aromaticity 0.09 Instability index 48.92 Isoelectric point 5.46 Charge (pH=7) -7.78 2D Binding mode Binding energy (Kcal/mol) -6.47 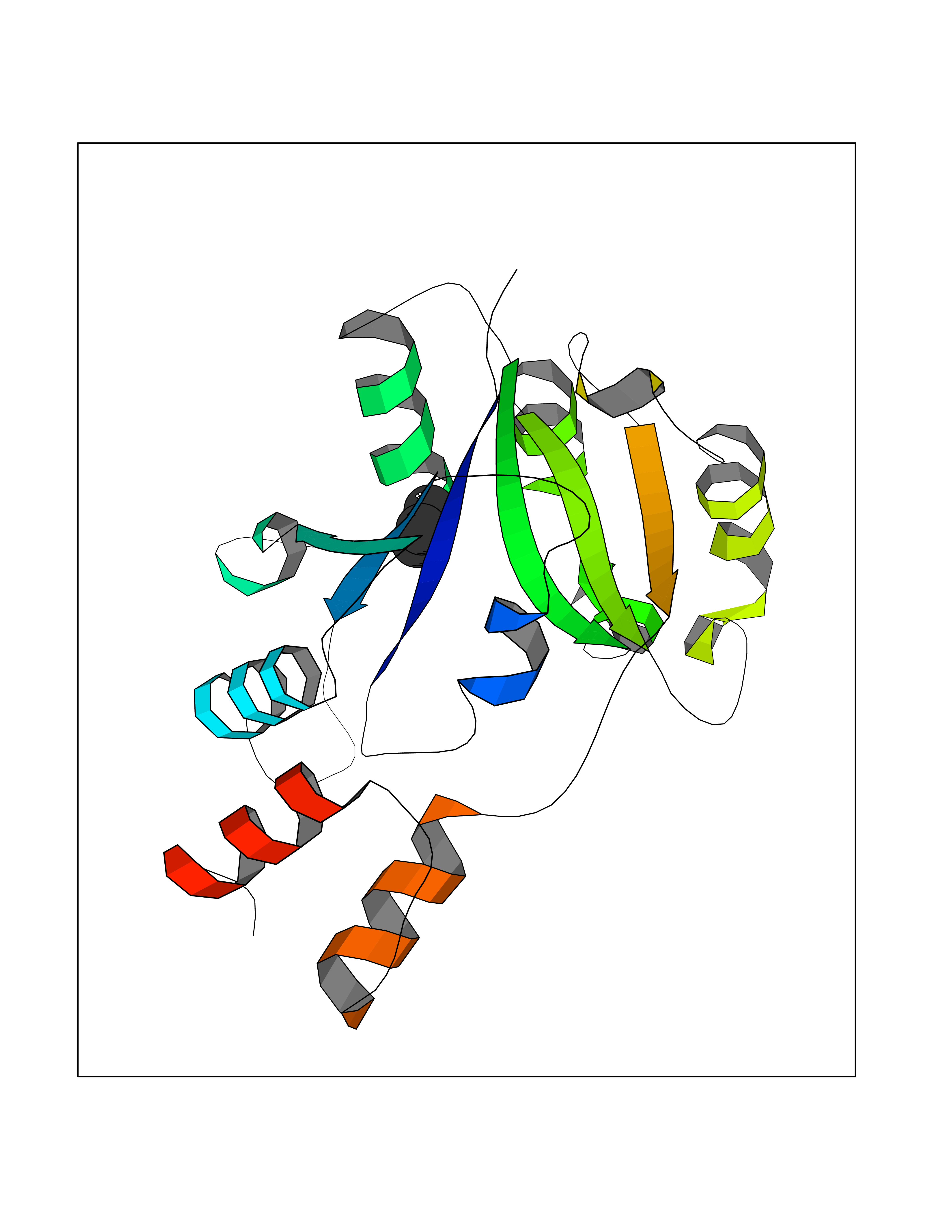 Molscript Map  Pymol Map 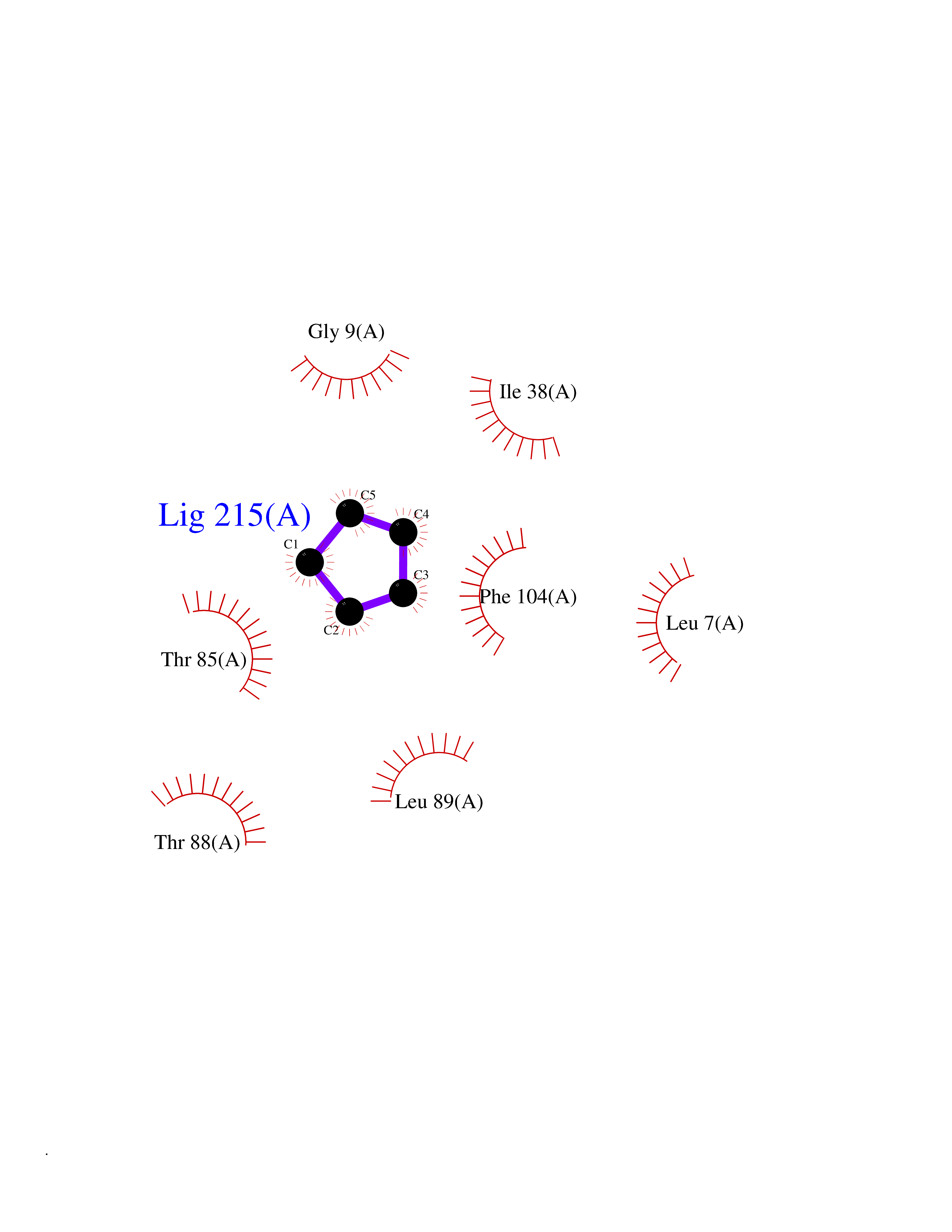 Ligplot Map 3D Binding mode Sequence MKSLQALFGGTFDPVHYGHLKPVETLANLIGLTRVTIIPNNVPPHRPQPEANSVQRKHMLELAIADKPLFTLDERELKRNAPSYTAQTLKEWRQEQGPDVPLAFIIGQDSLLTFPTWYEYETILDNAHLIVCRRPGYPLEMAQPQYQQWLEDHLTHNPEDLHLQPAGKIYLAETPWFNISATIIRERLQNGESCEDLLPEPVLTYINQQGLYR Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | MAPK signal-integrating kinase 1 (MKNK1) | 5WVD | 4.75 | |
Target general information Gen name MKNK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mnk1; MAP kinase signal-integrating kinase 1 Protein family Protein kinase superfamily, CAMK Ser/Thr protein kinase family Biochemical class Protein kinase superfamily. CAMK Ser/Thr protein kinase family Function May play a role in the response to environmental stress and cytokines. Appears to regulate translation by phosphorylating EIF4E, thus increasing the affinity of this protein for the 7-methylguanosine-containing mRNA cap. Related diseases Defects in MELK are associated with some cancers, such as brain or breast cancers. Expression is dramatically increased in aggressive undifferentiated tumors, correlating with poor patient outcome in breast and brain cancers, suggesting a role in tumor-initiating cells and proliferation via its function in cell proliferation regulation. Drugs (DrugBank ID) DB12010 Interacts with P54253; Q03060-25; P42858; P28482; Q16539; Q96CV9 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Translation regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 27536.2 Length 241 Aromaticity 0.11 Instability index 50.42 Isoelectric point 6.02 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -6.48 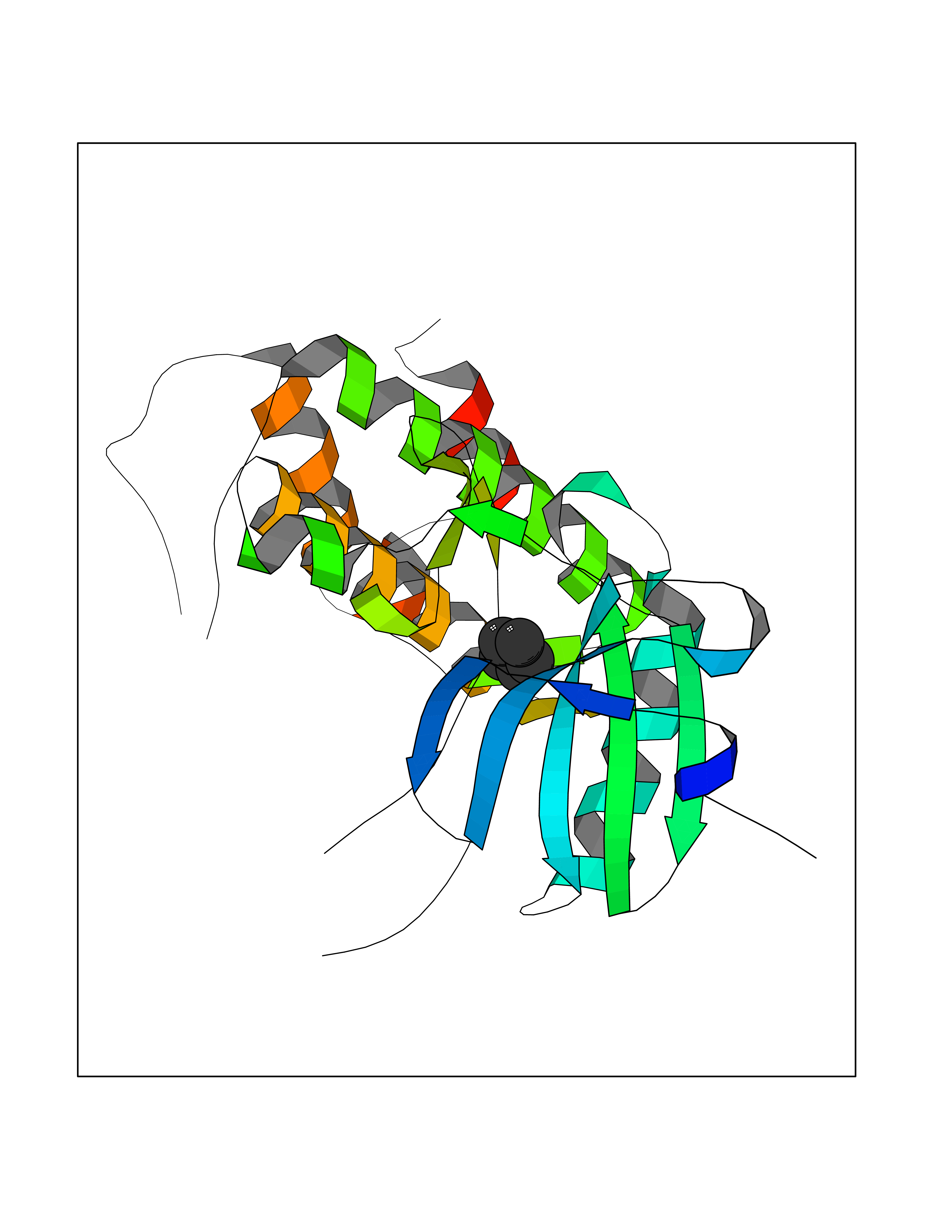 Molscript Map 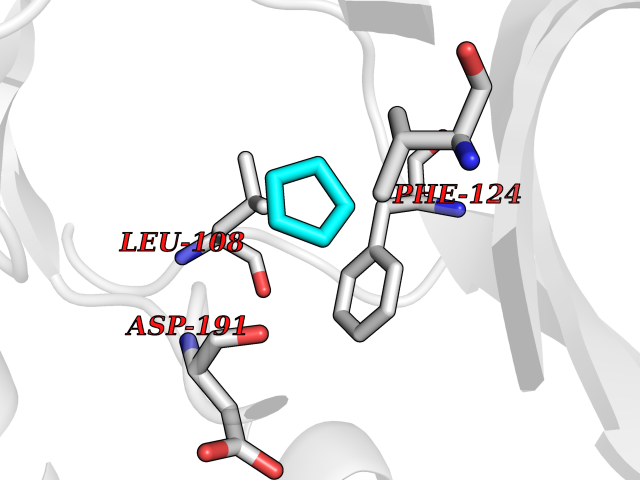 Pymol Map 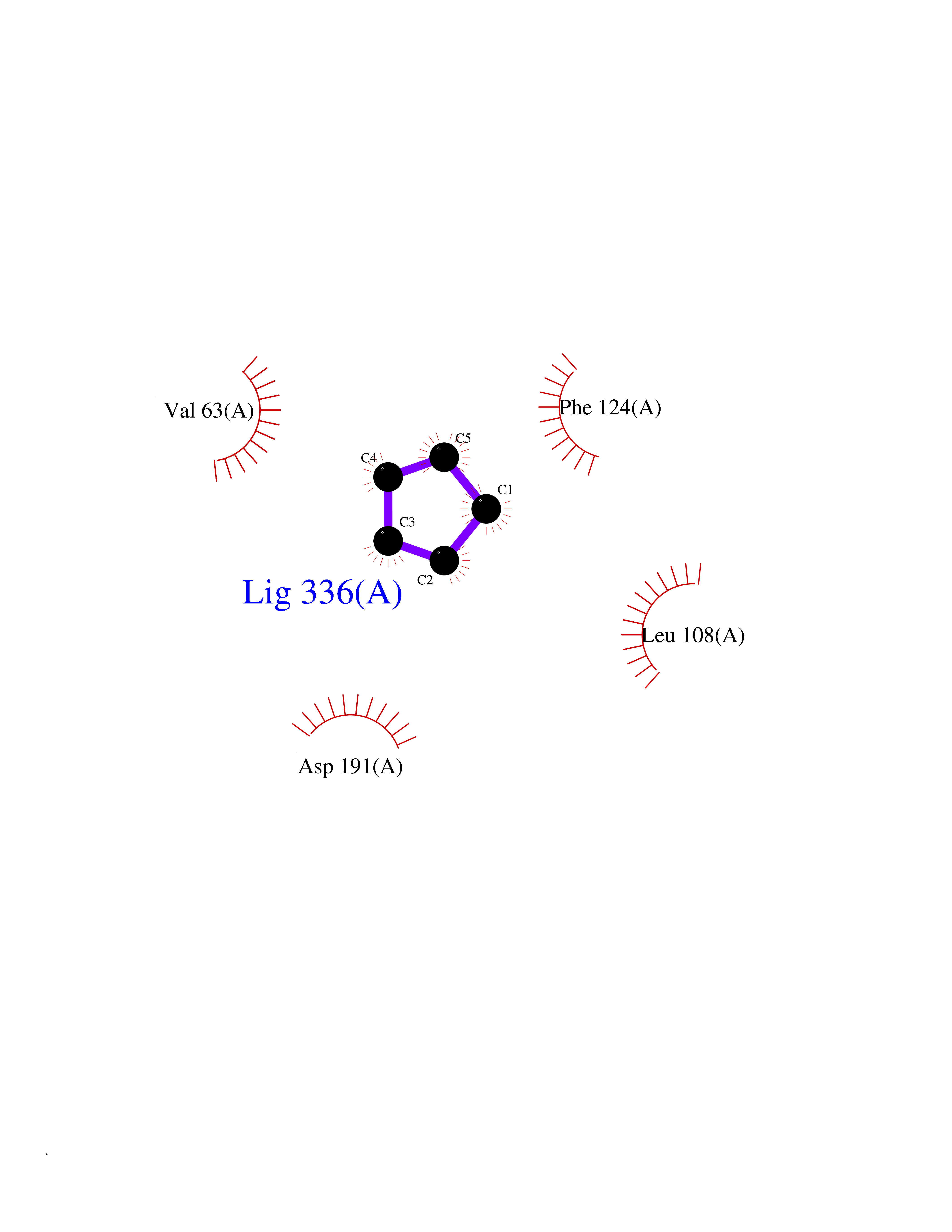 Ligplot Map 3D Binding mode Sequence PGKFEDMYKLTSELLGEGAYAKVQGAVSLQNGKEYAVKIIEKQAGHSRSRVFREVETLYQCQGNKNILELIEFFEDDTRFYLVFEKLQGGSILAHIQKQKHFNEREASRVVRDVAAALDFLHTKGIAHRDLKPENILCESPEKVSPVKICDFDLGSGYMAPEVVEVFTDQATFYDKRCDLWSLGVVLYIMLSGYPPFKYEFPDKDWAHISSEAKDLISKLLVRDAKQRLSAAQVLQHPWVQ Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Glutathione-dependent PGD synthase (HPGDS) | 2CVD | 4.74 | |
Target general information Gen name HPGDS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HPGDS; Glutathione-S-transferase; GST class-alpha Protein family GST superfamily, Sigma family Biochemical class Intramolecular oxidoreductases Function Bifunctional enzyme which catalyzes both the conversion of PGH2 to PGD2, a prostaglandin involved in smooth muscle contraction/relaxation and a potent inhibitor of platelet aggregation, and the conjugation of glutathione with a widerange of aryl halides and organic isothiocyanates. Also exhibits low glutathione-peroxidase activity towards cumene hydroperoxide. Related diseases Chromosomal aberrations involving JAK2 are found in both chronic and acute forms of eosinophilic, lymphoblastic and myeloid leukemia. Translocation t(8;9)(p22;p24) with PCM1 links the protein kinase domain of JAK2 to the major portion of PCM1. Translocation t(9;12)(p24;p13) with ETV6.; DISEASE: Budd-Chiari syndrome (BDCHS) [MIM:600880]: A syndrome caused by obstruction of hepatic venous outflow involving either the hepatic veins or the terminal segment of the inferior vena cava. Obstructions are generally caused by thrombosis and lead to hepatic congestion and ischemic necrosis. Clinical manifestations observed in the majority of patients include hepatomegaly, right upper quadrant pain and abdominal ascites. Budd-Chiari syndrome is associated with a combination of disease states including primary myeloproliferative syndromes and thrombophilia due to factor V Leiden, protein C deficiency and antithrombin III deficiency. Budd-Chiari syndrome is a rare but typical complication in patients with polycythemia vera. {ECO:0000269|PubMed:16707754}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Polycythemia vera (PV) [MIM:263300]: A myeloproliferative disorder characterized by abnormal proliferation of all hematopoietic bone marrow elements, erythroid hyperplasia, an absolute increase in total blood volume, but also by myeloid leukocytosis, thrombocytosis and splenomegaly. {ECO:0000269|PubMed:15781101, ECO:0000269|PubMed:15793561, ECO:0000269|PubMed:15858187, ECO:0000269|PubMed:16603627, ECO:0000269|PubMed:25644777}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombocythemia 3 (THCYT3) [MIM:614521]: A myeloproliferative disorder characterized by excessive platelet production, resulting in increased numbers of circulating platelets. It can be associated with spontaneous hemorrhages and thrombotic episodes. {ECO:0000269|PubMed:16325696, ECO:0000269|PubMed:22397670}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Myelofibrosis (MYELOF) [MIM:254450]: A disorder characterized by replacement of the bone marrow by fibrous tissue, occurring in association with a myeloproliferative disorder. Clinical manifestations may include anemia, pallor, splenomegaly, hypermetabolic state, petechiae, ecchymosis, bleeding, lymphadenopathy, hepatomegaly, portal hypertension. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16247455}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08790; DB01897; DB08695; DB07613; DB07917; DB07616; DB00321; DB00291; DB03619; DB00143; DB03310; DB08313; DB07614; DB07615 Interacts with Q96GS6; Q96B67; P15018; P08582-2; Q13370; Q8N1H7 EC number EC 5.3.99.2 Uniprot keywords 3D-structure; Calcium; Cytoplasm; Fatty acid biosynthesis; Fatty acid metabolism; Isomerase; Lipid biosynthesis; Lipid metabolism; Magnesium; Metal-binding; Prostaglandin biosynthesis; Prostaglandin metabolism; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 46441 Length 396 Aromaticity 0.12 Instability index 29.82 Isoelectric point 5.55 Charge (pH=7) -9.17 2D Binding mode Binding energy (Kcal/mol) -6.47 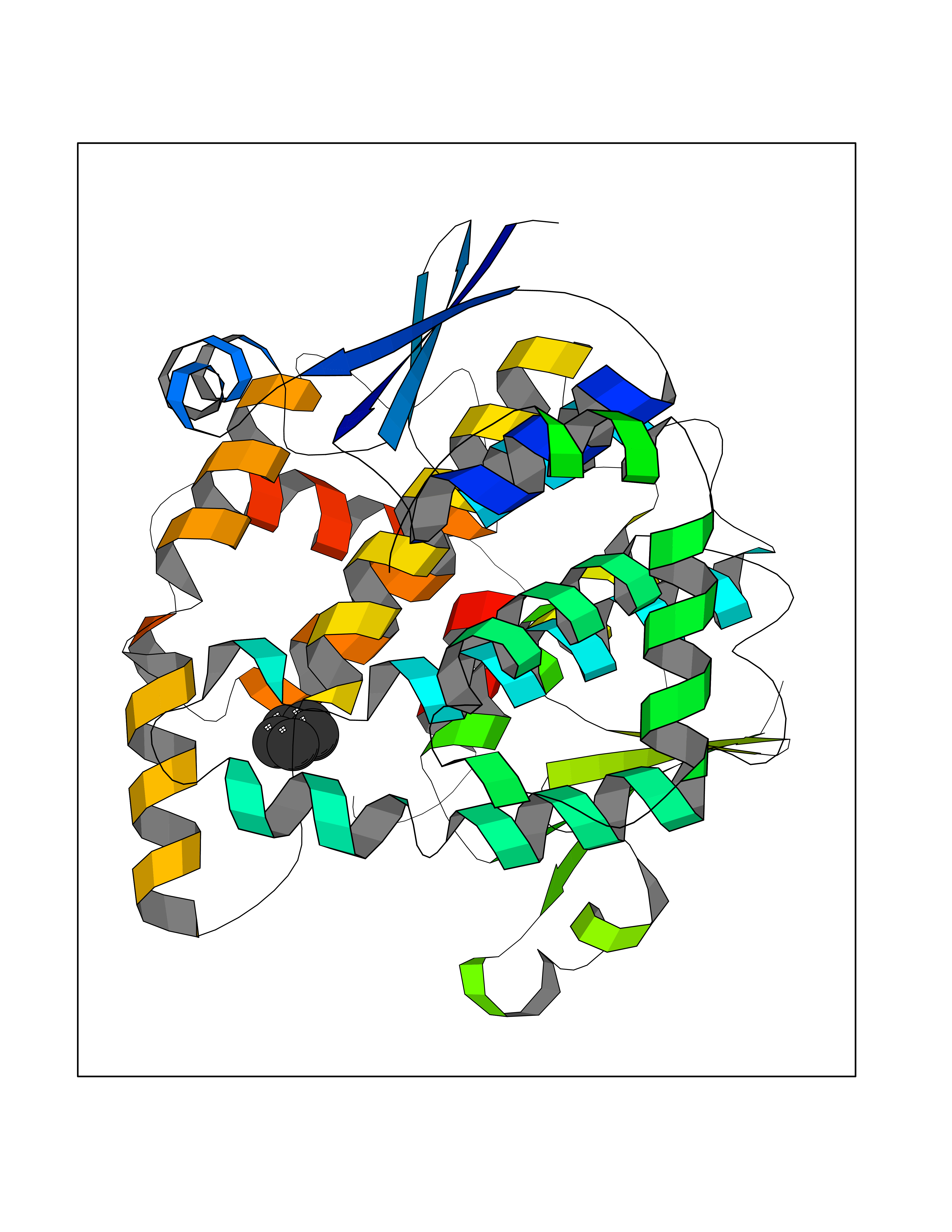 Molscript Map 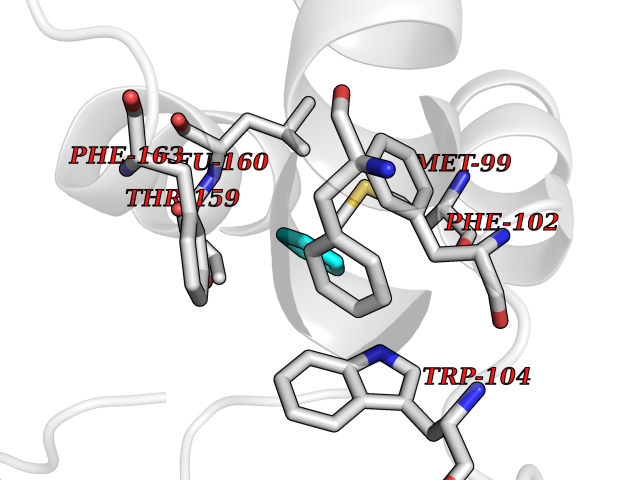 Pymol Map 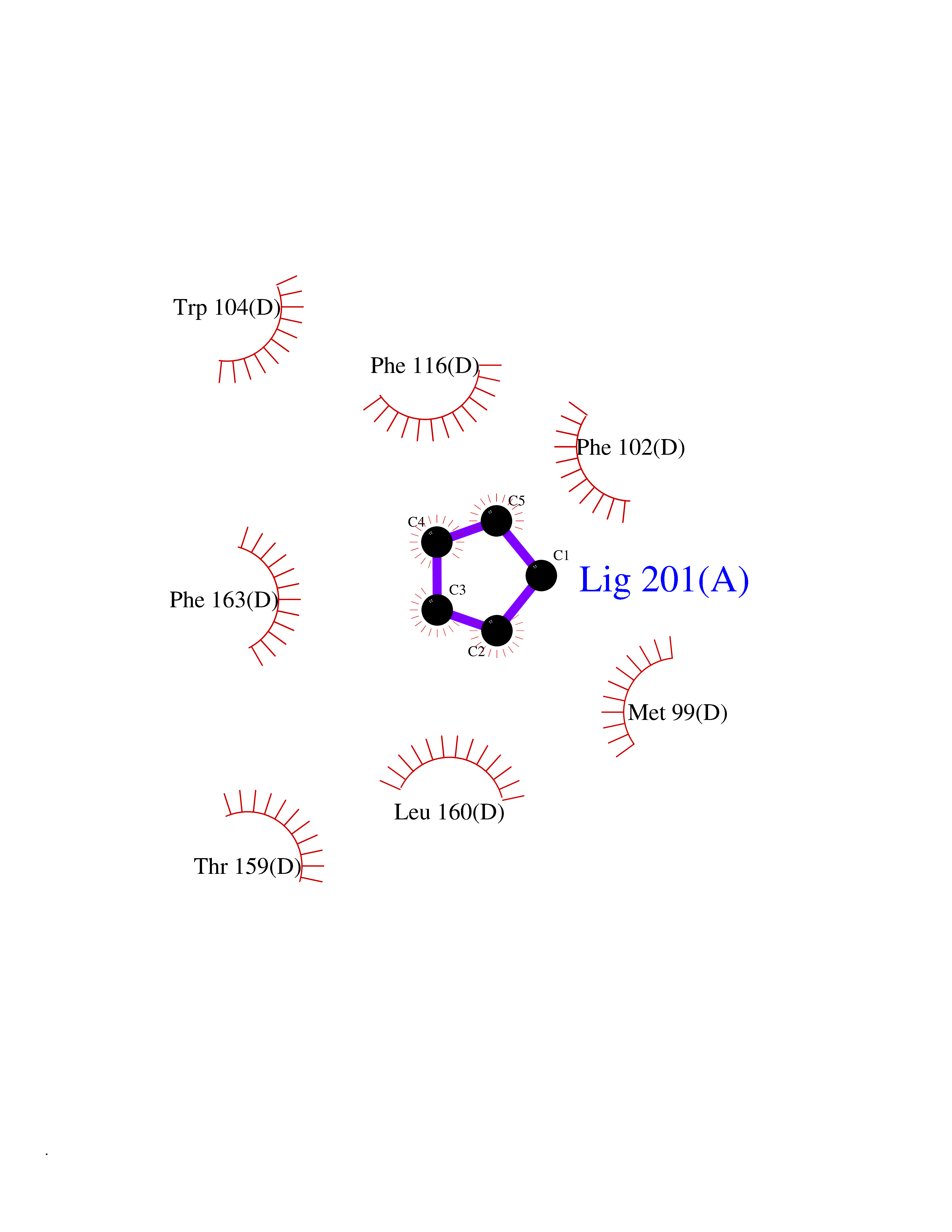 Ligplot Map 3D Binding mode Sequence PNYKLTYFNMRGRAEIIRYIFAYLDIQYEDHRIEQADWPEIKSTLPFGKIPILEVDGLTLHQSLAIARYLTKNTDLAGNTEMEQCHVDAIVDTLDDFMSCFPWAEKKQDVKEQMFNELLTYNAPHLMQDLDTYLGGREWLIGMSVTWADFYWEICSTTLLVFKPDLLDNHPRLVTLRKKVQAIPAVANWIKRRPQTKLPNYKLTYFNMRGRAEIIRYIFAYLDIQYEDHRIEQADWPEIKSTLPFGKIPILEVDGLTLHQSLAIARYLTKNTDLAGNTEMEQCHVDAIVDTLDDFMSCFPWAEKKQDVKEQMFNELLTYNAPHLMQDLDTYLGGREWLIGMSVTWADFYWEICSTTLLVFKPDLLDNHPRLVTLRKKVQAIPAVANWIKRRPQTKL Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Glutathione-dependent PGD synthase (HPGDS) | 2CVD | 4.74 | |
Target general information Gen name HPGDS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HPGDS; Glutathione-S-transferase; GST class-alpha Protein family GST superfamily, Sigma family Biochemical class Intramolecular oxidoreductases Function Bifunctional enzyme which catalyzes both the conversion of PGH2 to PGD2, a prostaglandin involved in smooth muscle contraction/relaxation and a potent inhibitor of platelet aggregation, and the conjugation of glutathione with a widerange of aryl halides and organic isothiocyanates. Also exhibits low glutathione-peroxidase activity towards cumene hydroperoxide. Related diseases Chromosomal aberrations involving JAK2 are found in both chronic and acute forms of eosinophilic, lymphoblastic and myeloid leukemia. Translocation t(8;9)(p22;p24) with PCM1 links the protein kinase domain of JAK2 to the major portion of PCM1. Translocation t(9;12)(p24;p13) with ETV6.; DISEASE: Budd-Chiari syndrome (BDCHS) [MIM:600880]: A syndrome caused by obstruction of hepatic venous outflow involving either the hepatic veins or the terminal segment of the inferior vena cava. Obstructions are generally caused by thrombosis and lead to hepatic congestion and ischemic necrosis. Clinical manifestations observed in the majority of patients include hepatomegaly, right upper quadrant pain and abdominal ascites. Budd-Chiari syndrome is associated with a combination of disease states including primary myeloproliferative syndromes and thrombophilia due to factor V Leiden, protein C deficiency and antithrombin III deficiency. Budd-Chiari syndrome is a rare but typical complication in patients with polycythemia vera. {ECO:0000269|PubMed:16707754}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Polycythemia vera (PV) [MIM:263300]: A myeloproliferative disorder characterized by abnormal proliferation of all hematopoietic bone marrow elements, erythroid hyperplasia, an absolute increase in total blood volume, but also by myeloid leukocytosis, thrombocytosis and splenomegaly. {ECO:0000269|PubMed:15781101, ECO:0000269|PubMed:15793561, ECO:0000269|PubMed:15858187, ECO:0000269|PubMed:16603627, ECO:0000269|PubMed:25644777}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombocythemia 3 (THCYT3) [MIM:614521]: A myeloproliferative disorder characterized by excessive platelet production, resulting in increased numbers of circulating platelets. It can be associated with spontaneous hemorrhages and thrombotic episodes. {ECO:0000269|PubMed:16325696, ECO:0000269|PubMed:22397670}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Myelofibrosis (MYELOF) [MIM:254450]: A disorder characterized by replacement of the bone marrow by fibrous tissue, occurring in association with a myeloproliferative disorder. Clinical manifestations may include anemia, pallor, splenomegaly, hypermetabolic state, petechiae, ecchymosis, bleeding, lymphadenopathy, hepatomegaly, portal hypertension. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16247455}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08790; DB01897; DB08695; DB07613; DB07917; DB07616; DB00321; DB00291; DB03619; DB00143; DB03310; DB08313; DB07614; DB07615 Interacts with Q96GS6; Q96B67; P15018; P08582-2; Q13370; Q8N1H7 EC number EC 5.3.99.2 Uniprot keywords 3D-structure; Calcium; Cytoplasm; Fatty acid biosynthesis; Fatty acid metabolism; Isomerase; Lipid biosynthesis; Lipid metabolism; Magnesium; Metal-binding; Prostaglandin biosynthesis; Prostaglandin metabolism; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 46441 Length 396 Aromaticity 0.12 Instability index 29.82 Isoelectric point 5.55 Charge (pH=7) -9.17 2D Binding mode Binding energy (Kcal/mol) -6.47 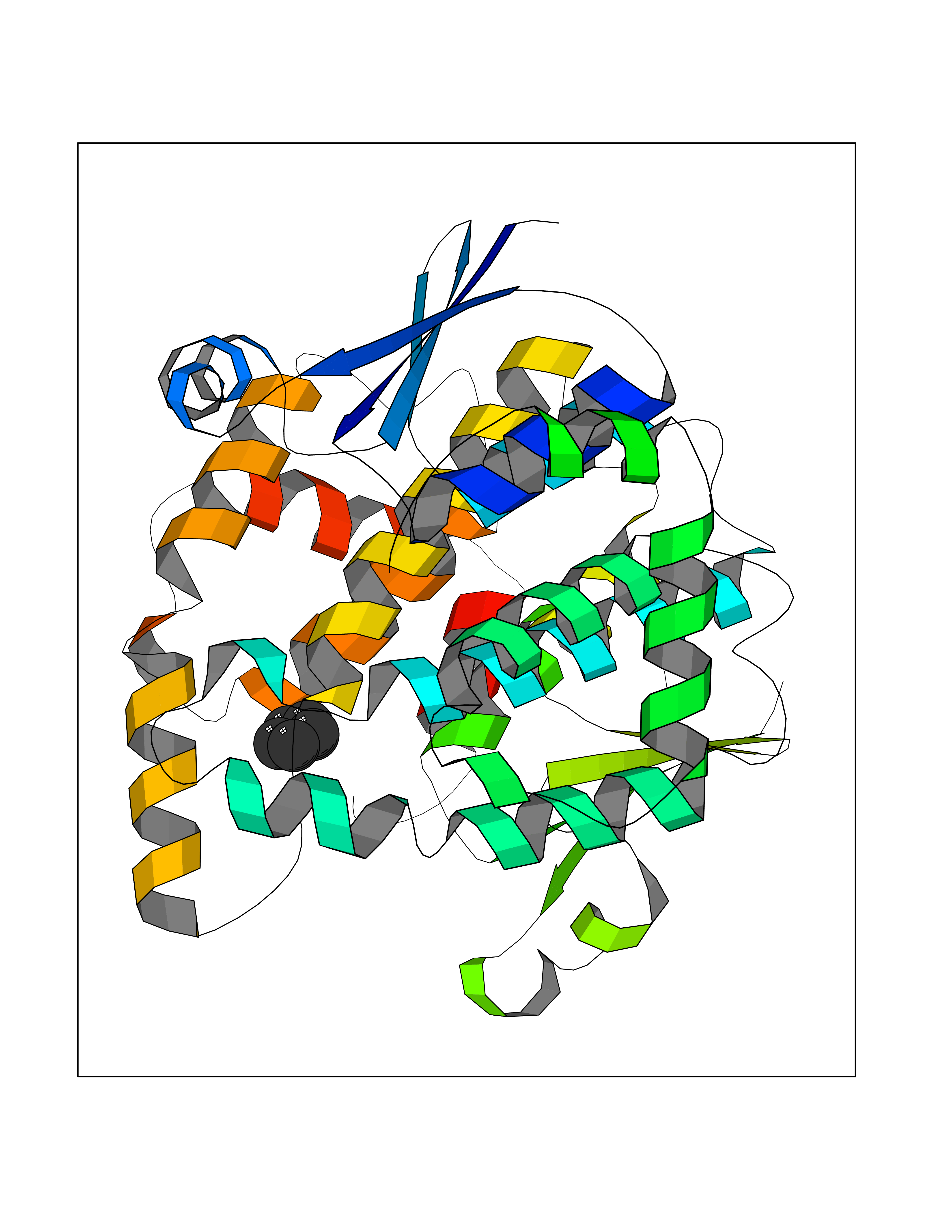 Molscript Map 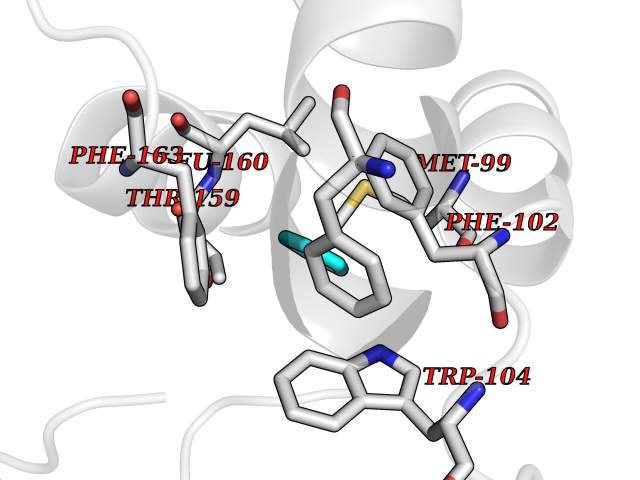 Pymol Map 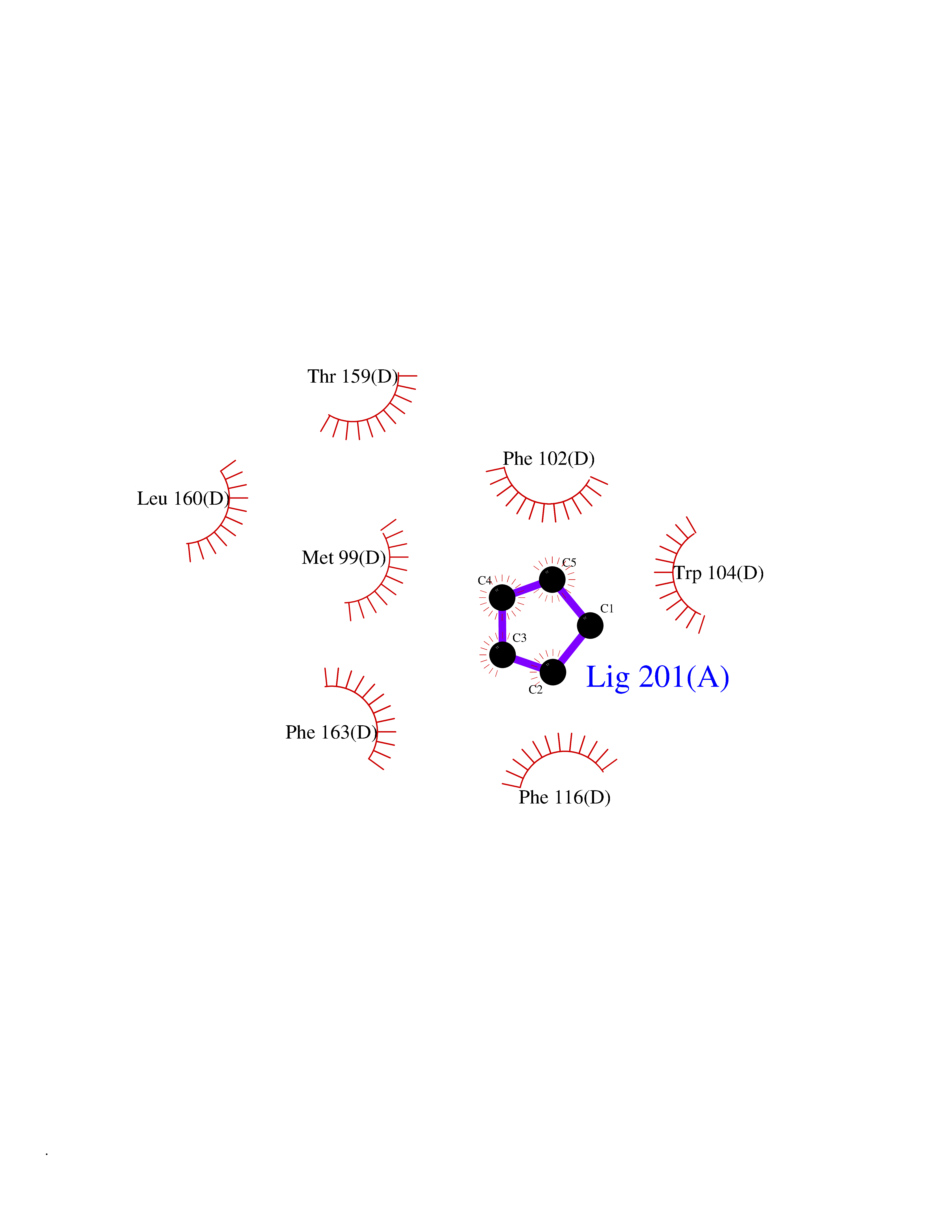 Ligplot Map 3D Binding mode Sequence PNYKLTYFNMRGRAEIIRYIFAYLDIQYEDHRIEQADWPEIKSTLPFGKIPILEVDGLTLHQSLAIARYLTKNTDLAGNTEMEQCHVDAIVDTLDDFMSCFPWAEKKQDVKEQMFNELLTYNAPHLMQDLDTYLGGREWLIGMSVTWADFYWEICSTTLLVFKPDLLDNHPRLVTLRKKVQAIPAVANWIKRRPQTKLPNYKLTYFNMRGRAEIIRYIFAYLDIQYEDHRIEQADWPEIKSTLPFGKIPILEVDGLTLHQSLAIARYLTKNTDLAGNTEMEQCHVDAIVDTLDDFMSCFPWAEKKQDVKEQMFNELLTYNAPHLMQDLDTYLGGREWLIGMSVTWADFYWEICSTTLLVFKPDLLDNHPRLVTLRKKVQAIPAVANWIKRRPQTKL Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 4.74 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -6.47 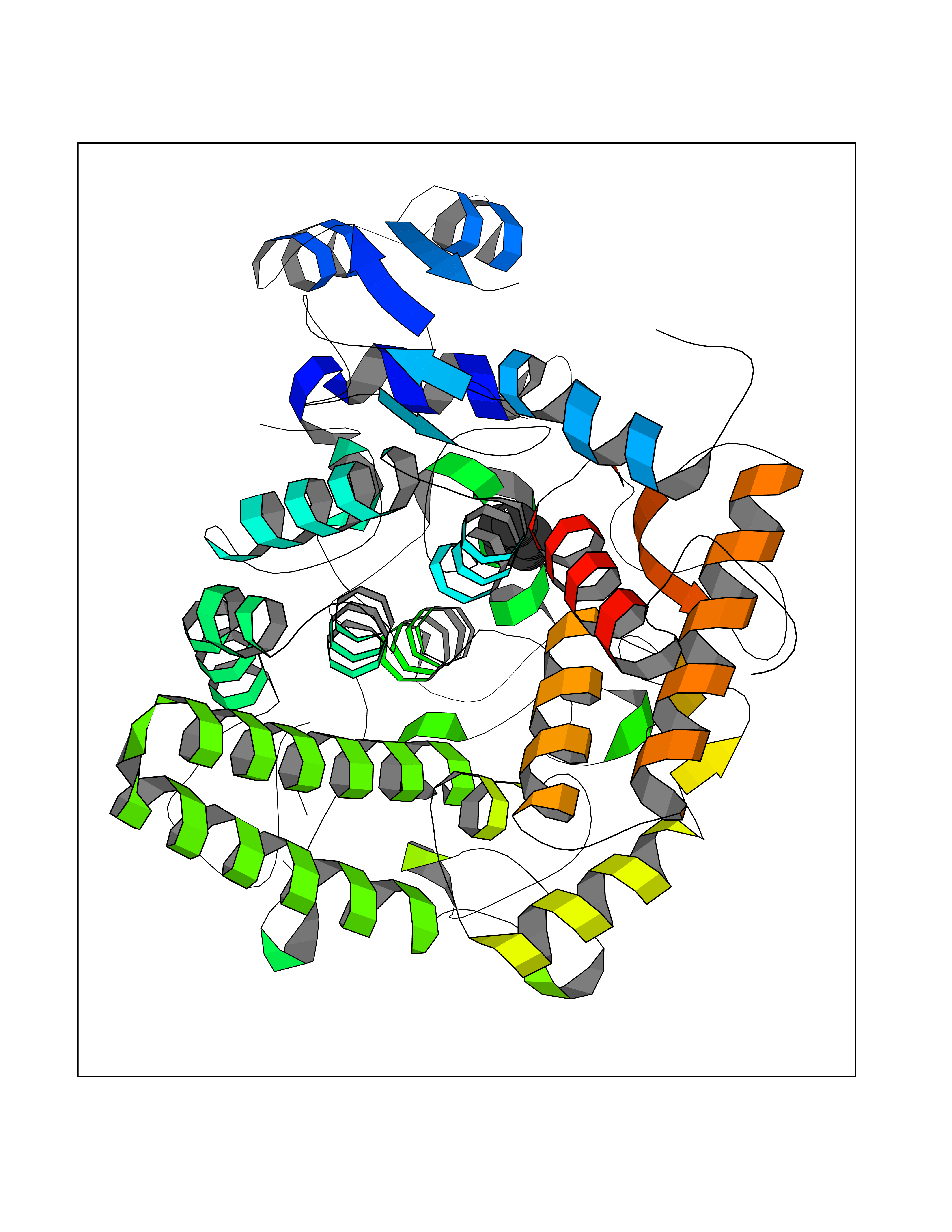 Molscript Map  Pymol Map 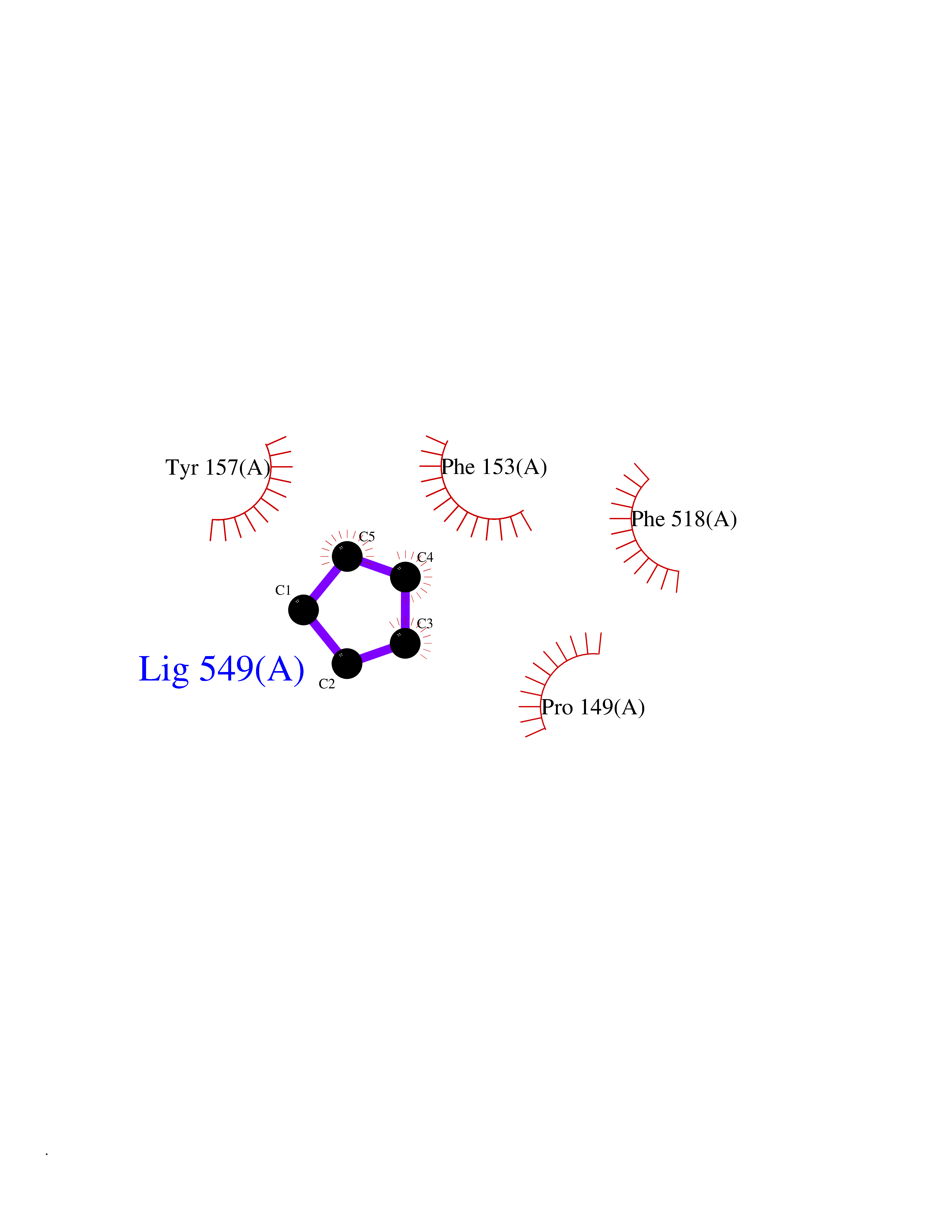 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Oxygen-insensitive NADPH nitroreductase | 1F5V | 4.74 | |
Target general information Gen name nfsA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms mda18;b0851;mdaA;ybjB;JW0835 Protein family Flavin oxidoreductase frp family Biochemical class Oxidoreductase Function Chromate reductase activity.FMN binding.Oxidoreductase activity, acting on NAD(P)H, nitrogenous group as acceptor. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03247; DB00698 Interacts with P28630 EC number 1.-.-.- Uniprot keywords 3D-structure; Flavoprotein; FMN; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 53582.7 Length 480 Aromaticity 0.07 Instability index 45.43 Isoelectric point 6.47 Charge (pH=7) -3.13 2D Binding mode Binding energy (Kcal/mol) -6.46 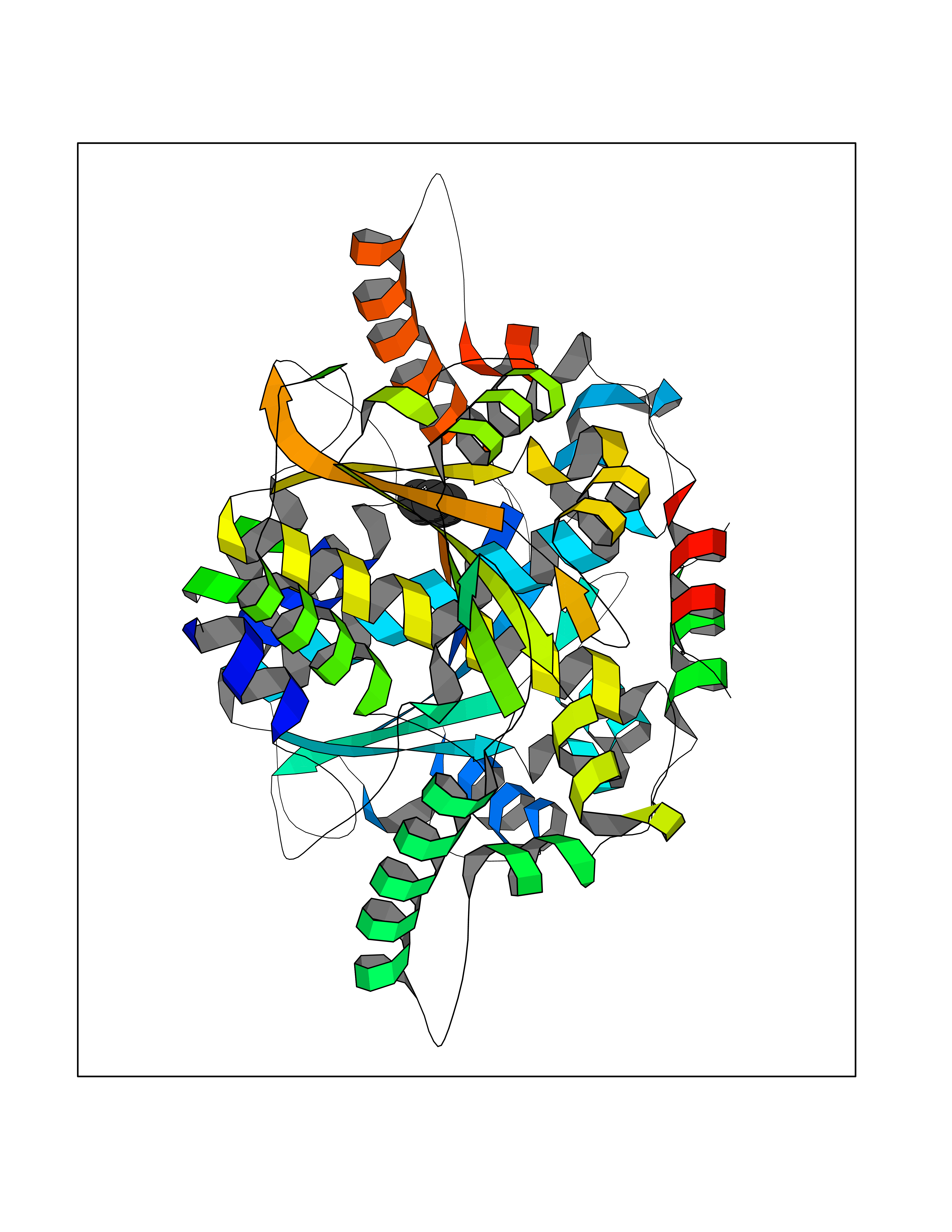 Molscript Map 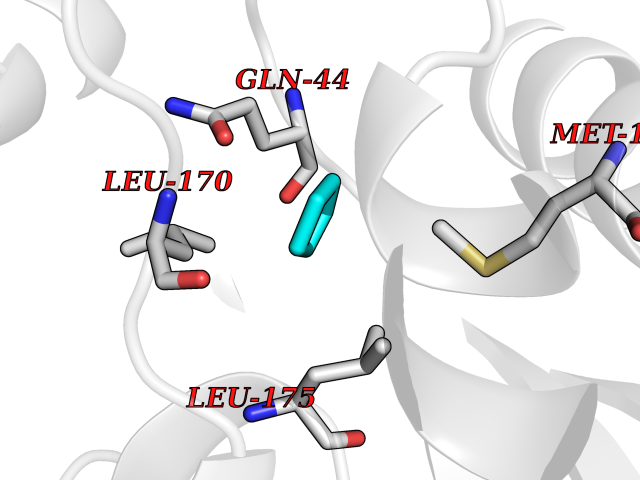 Pymol Map 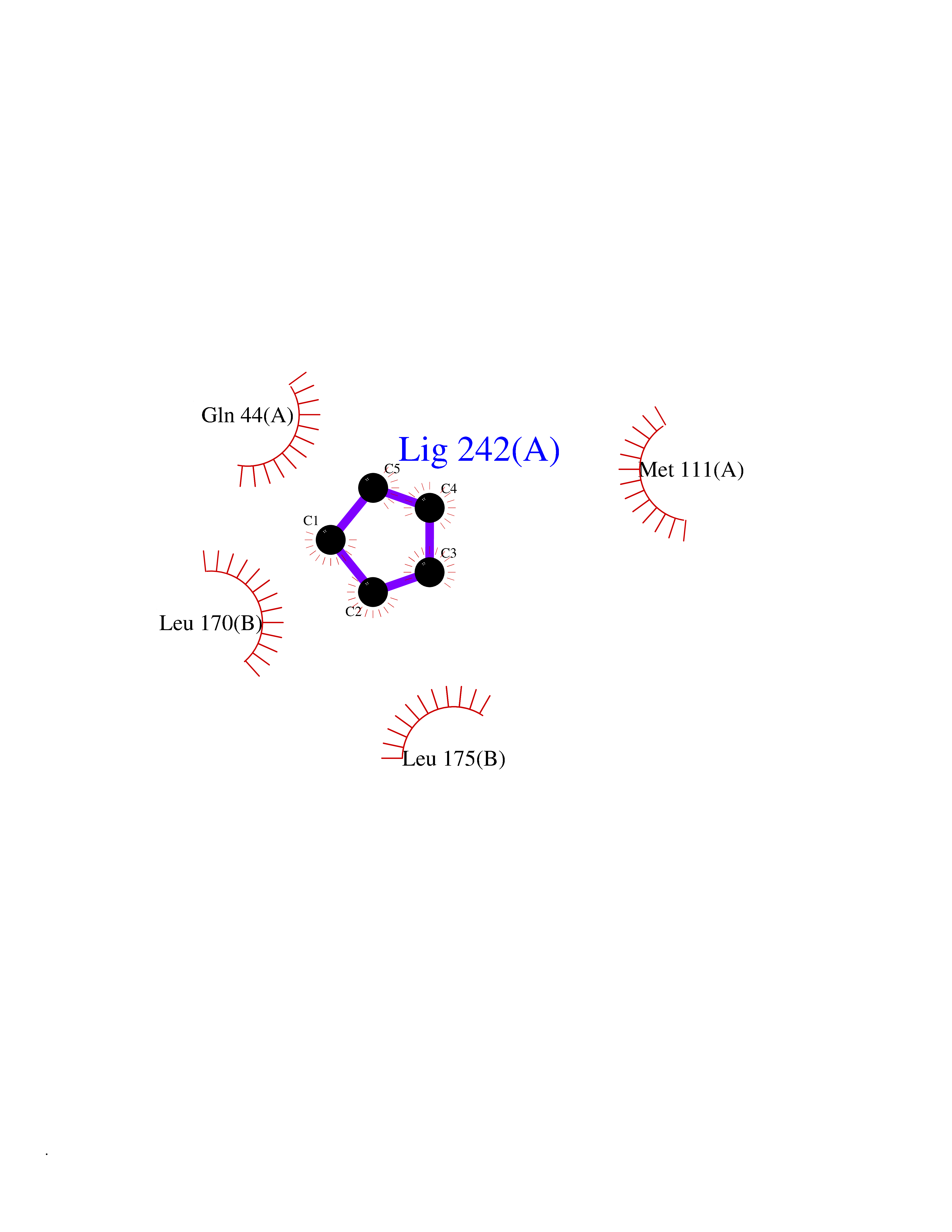 Ligplot Map 3D Binding mode Sequence MTPTIELICGHRSIRHFTDEPISEAQREAIINSARATSSSSFLQCSSIIRITDKALREELVTLTGGQKHVAQAAEFWVFCADFNRHLQICPDAQLGLAEQLLLGVVDTAMMAQNALIAAESLGLGGVYIGGLRNNIEAVTKLLKLPQHVLPLFGLCLGWPADNPDLKPRLPASILVHENSYQPLDKGALAQYDEQLAEYYLTRGSNNRRDTWSDHIRRTIIKESRPFILDYLHKQGWATRMTPTIELICGHRSIRHFTDEPISEAQREAIINSARATSSSSFLQCSSIIRITDKALREELVTLTGGQKHVAQAAEFWVFCADFNRHLQICPDAQLGLAEQLLLGVVDTAMMAQNALIAAESLGLGGVYIGGLRNNIEAVTKLLKLPQHVLPLFGLCLGWPADNPDLKPRLPASILVHENSYQPLDKGALAQYDEQLAEYYLTRGSNNRRDTWSDHIRRTIIKESRPFILDYLHKQGWATR Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Acetylcholine receptor subunit alpha | 4ZJS | 4.74 | |
Target general information Gen name CHRNA1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACHRA;CHNRA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-1/CHRNA1 sub-subfamily Biochemical class Immune system Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ion channel activity.Ligand-gated ion channel activity. Related diseases Multiple pterygium syndrome, lethal type (LMPS) [MIM:253290]: Multiple pterygia are found infrequently in children with arthrogryposis and in fetuses with fetal akinesia syndrome. In lethal multiple pterygium syndrome there is intrauterine growth retardation, multiple pterygia, and flexion contractures causing severe arthrogryposis and fetal akinesia. Subcutaneous edema can be severe, causing fetal hydrops with cystic hygroma and lung hypoplasia. Oligohydramnios and facial anomalies are frequent. {ECO:0000269|PubMed:18252226}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: The alpha subunit is the main focus for antibody binding in myasthenia gravis. Myasthenia gravis is characterized by sporadic muscular fatigability and weakness, occurring chiefly in muscles innervated by cranial nerves, and characteristically improved by cholinesterase-inhibiting drugs.; DISEASE: Myasthenic syndrome, congenital, 1A, slow-channel (CMS1A) [MIM:601462]: A common congenital myasthenic syndrome. Congenital myasthenic syndromes are characterized by muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1A is a slow-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in prolonged AChR channel opening episodes, prolonged endplate currents, and depolarization block. This is associated with calcium overload, which may contribute to subsequent degeneration of the endplate and postsynaptic membrane. {ECO:0000269|PubMed:16685696, ECO:0000269|PubMed:7619526, ECO:0000269|PubMed:8872460, ECO:0000269|PubMed:9158151, ECO:0000269|PubMed:9221765}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 1B, fast-channel (CMS1B) [MIM:608930]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1B is a fast-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in brief opening and activity of the channel, with a rapid decay in endplate current, failure to achieve threshold depolarization of the endplate and consequent failure to fire an action potential. {ECO:0000269|PubMed:10195214, ECO:0000269|PubMed:12588888, ECO:0000269|PubMed:15079006}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08838; DB00565; DB00555 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46717.8 Length 411 Aromaticity 0.11 Instability index 38.02 Isoelectric point 4.77 Charge (pH=7) -22.31 2D Binding mode Binding energy (Kcal/mol) -6.46 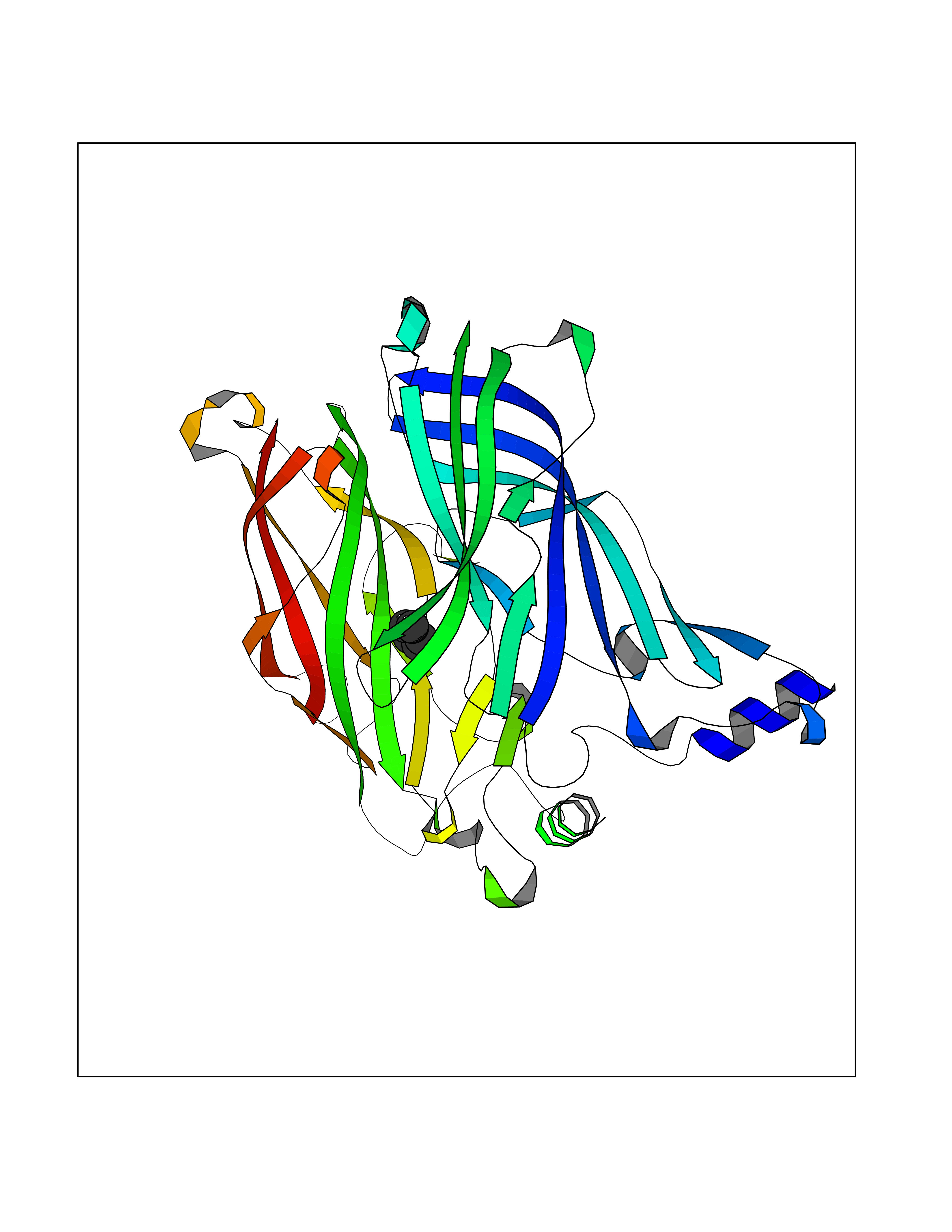 Molscript Map 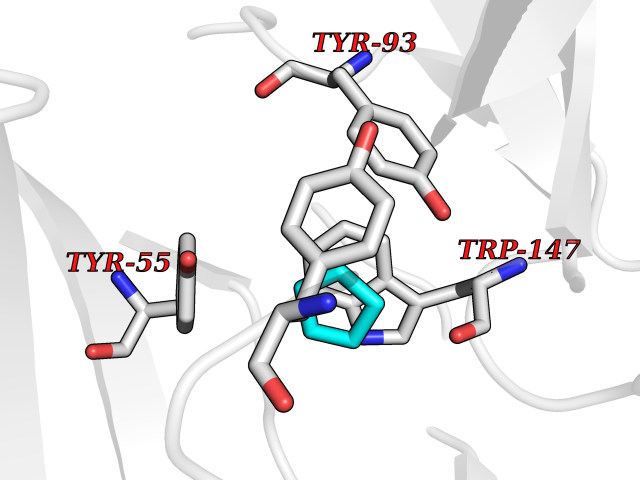 Pymol Map 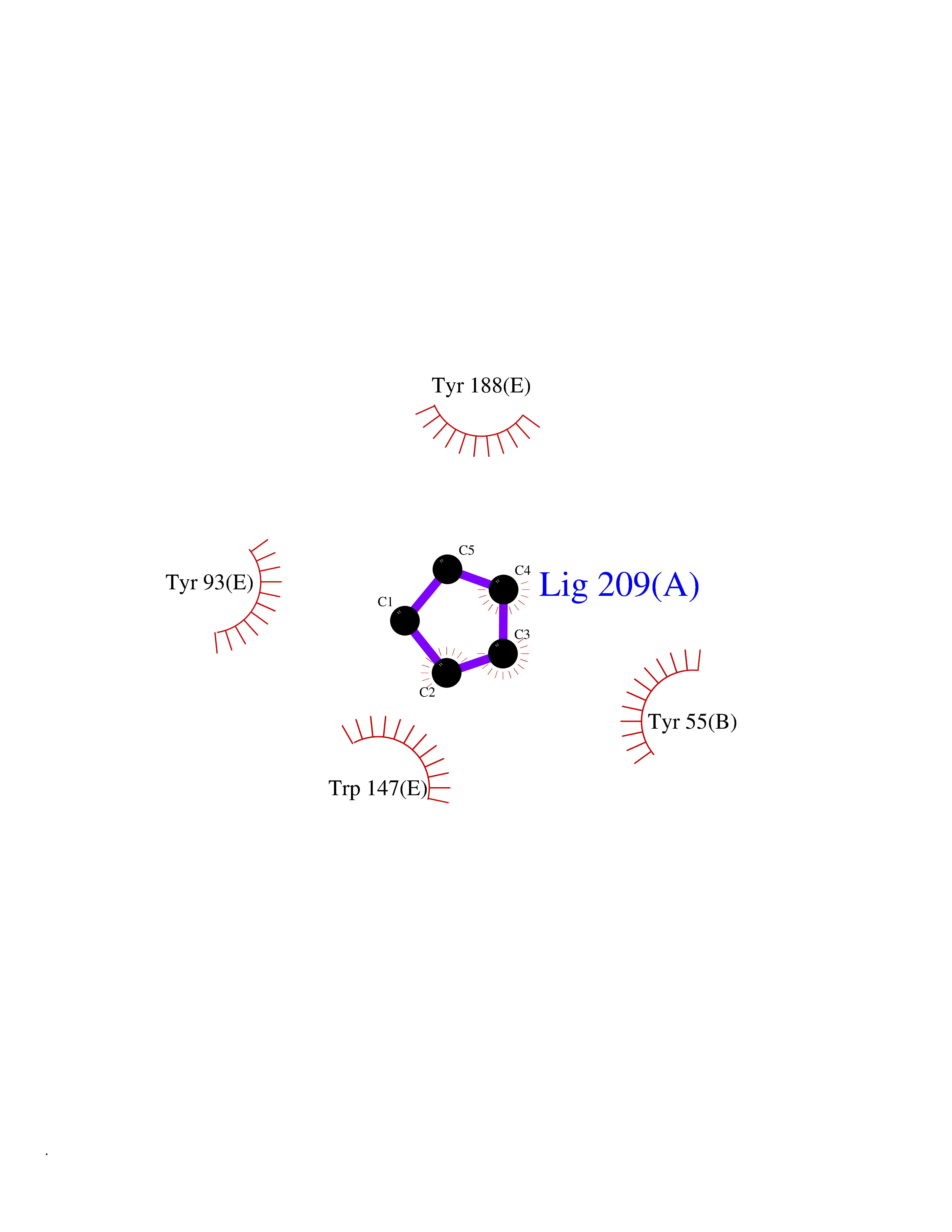 Ligplot Map 3D Binding mode Sequence EHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFREEHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFRER Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | DNA-(apurinic or apyrimidinic site) lyase | 4QHE | 4.74 | |
Target general information Gen name APEX1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms APE;APX;APE1;APEX;HAP1;REF1 Protein family DNA repair enzymes AP/ExoA family Biochemical class Lyase Function 3'-5' exonuclease activity.Chromatin DNA binding.Class I DNA-(apurinic or apyrimidinic site) lyase activity.Class III/IV DNA-(apurinic or apyrimidinic site) lyase activity.Damaged DNA binding.DNA-(apurinic or apyrimidinic site) lyase activity.DNA binding.Double-stranded DNA 3'-5' exodeoxyribonuclease activity.Double-stranded DNA exodeoxyribonuclease activity.Double-stranded telomeric DNA binding.Endodeoxyribonuclease activity.Endonuclease activity.Metal ion binding.NF-kappaB binding.Oxidoreductase activity.Phosphodiesterase I activity.Phosphoric diester hydrolase activity.Protein complex binding.RNA binding.RNA-DNA hybrid ribonuclease activity.Site-specific endodeoxyribonuclease activity, specific for altered base.Transcription coactivator activity.Transcription corepressor activity.Uracil DNA N-glycosylase activity. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB04967 Interacts with Q09472; Q8N4N3; Q16236; Q96EB6; O88846 EC number 3.1.11.2; 3.1.21.- Uniprot keywords 3D-structure; Acetylation; Activator; Cleavage on pair of basic residues; Cytoplasm; Direct protein sequencing; Disulfide bond; DNA damage; DNA recombination; DNA repair; DNA-binding; Endonuclease; Endoplasmic reticulum; Exonuclease; Hydrolase; Magnesium; Metal-binding; Mitochondrion; Nuclease; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; RNA-binding; S-nitrosylation; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 31556.6 Length 281 Aromaticity 0.1 Instability index 44.46 Isoelectric point 7.17 Charge (pH=7) 0.3 2D Binding mode Binding energy (Kcal/mol) -6.46 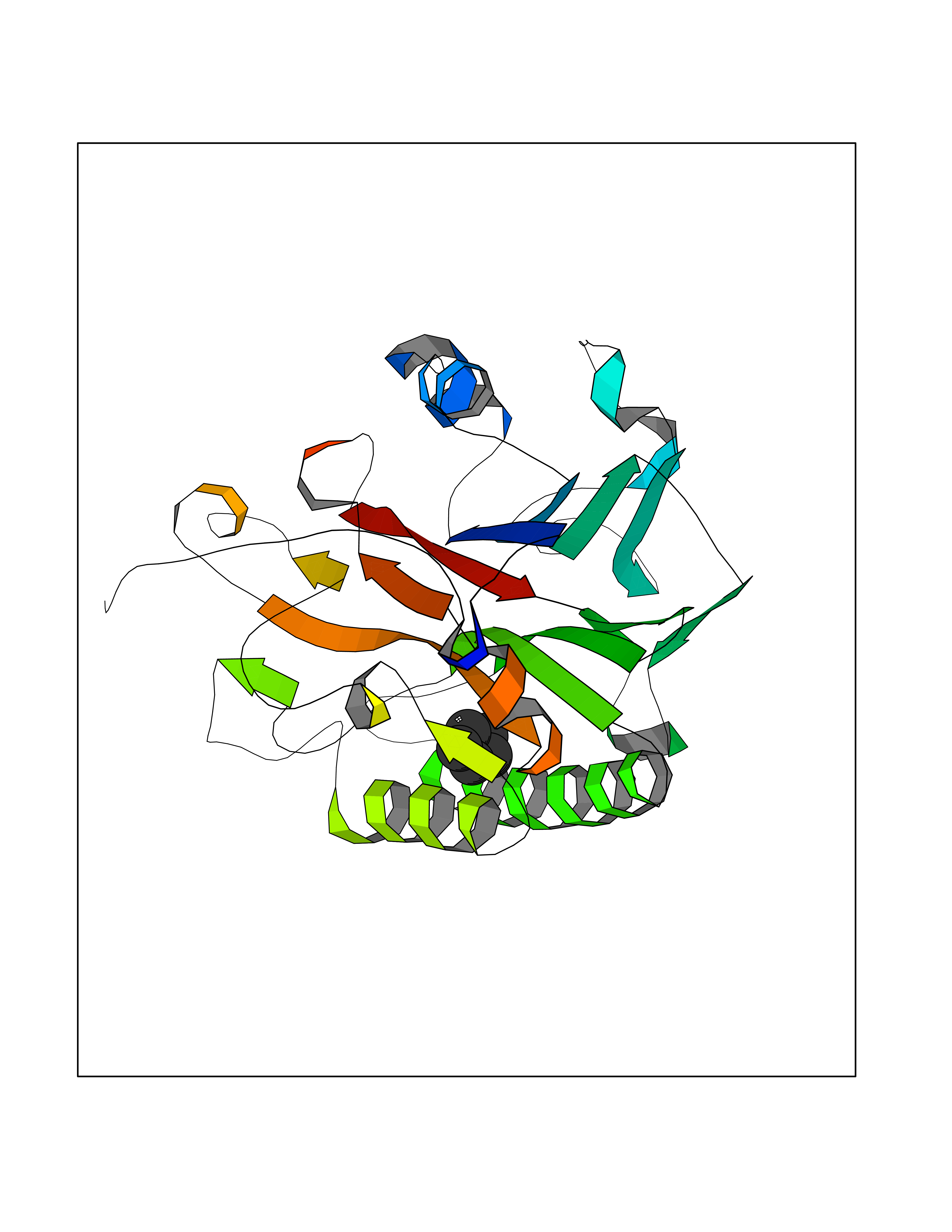 Molscript Map 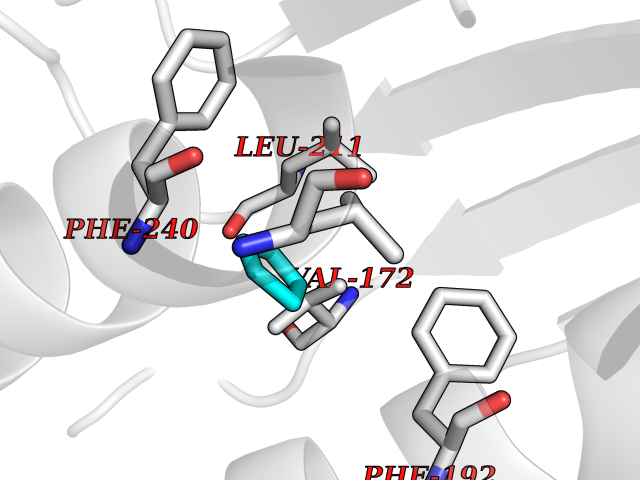 Pymol Map 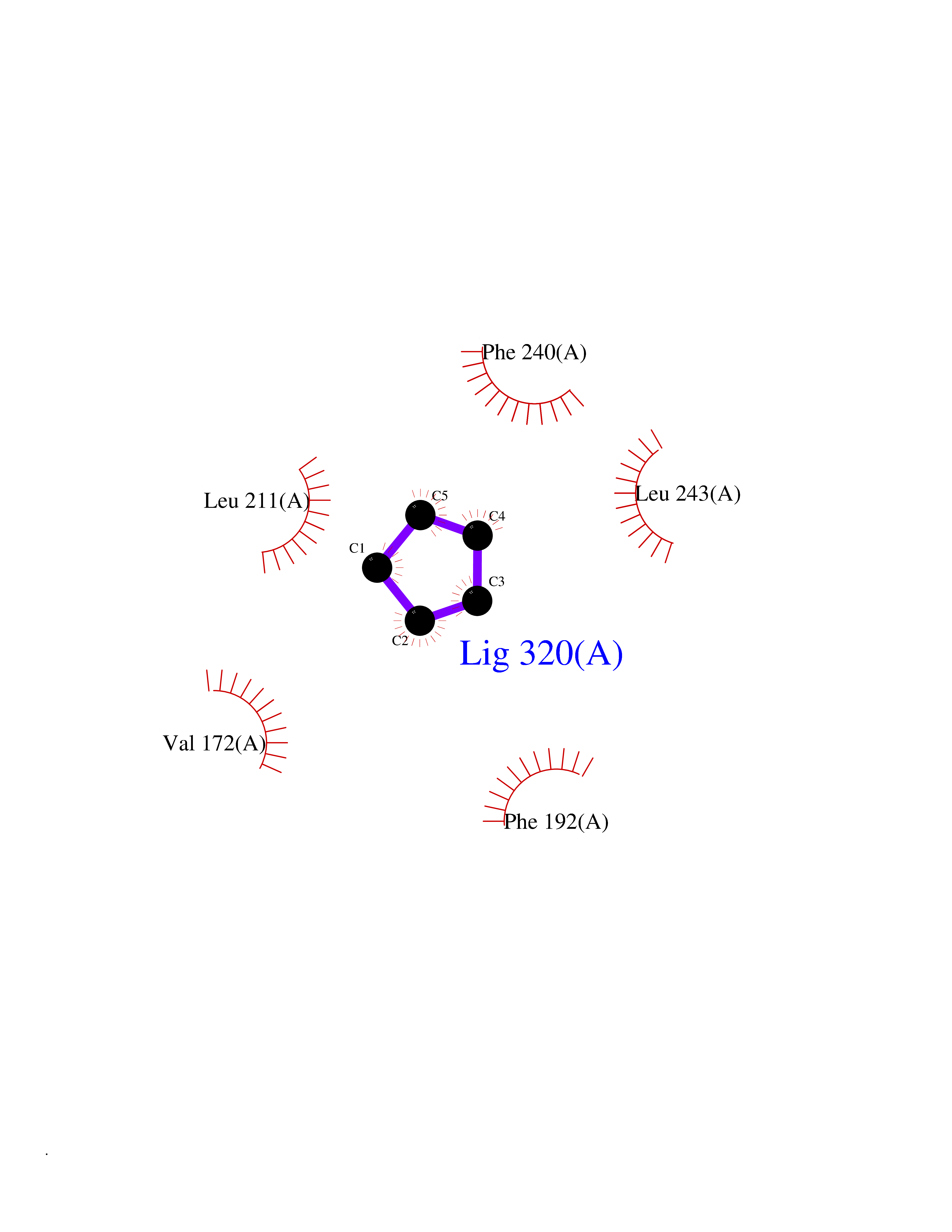 Ligplot Map 3D Binding mode Sequence ASEGPALYEDPPDQKTSPSGKPATLKICSWNVDGLRAWIKKKGLDWVKEEAPDILCLQETKCSENKLPAELQELPGLSHQYWSAPSDKEGYSGVGLLSRQAPLKVSYGIGDEEHDQEGRVIVAEFDSFVLVTAYVPNAGRGLVRLEYRQRWDEAFRKFLKGLASRKPLVLCGDLNVAHEEIDLRNPKGNKKNAGFTPQERQGFGELLQAVPLADSFRHLYPNTPYAYTFWTYMMNARSKNVGWRLDYFLLSHSLLPALCDSKIRSKALGSDHCPITLYLAL Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | C-C chemokine receptor type 5 (CCR5) | 4MBS | 4.74 | |
Target general information Gen name CCR5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HIV-1 fusion coreceptor; HIV-1 fusion co-receptor; Chemokine receptor CCR5; CMKBR5; CHEMR13; CD195 antigen; CD195; CCR-5; CC-CKR-5; C-C CKR-5 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function May play a role in the control of granulocytic lineage proliferation or differentiation. Receptor for a number of inflammatory CC-chemokines including CCL3/MIP-1-alpha, CCL4/MIP-1-beta and RANTES and subsequently transduces a signal by increasing the intracellular calcium ion level. Related diseases Type 1 diabetes mellitus 22 (T1D22) [MIM:612522]: A multifactorial disorder of glucose homeostasis that is characterized by susceptibility to ketoacidosis in the absence of insulin therapy. Clinical features are polydipsia, polyphagia and polyuria which result from hyperglycemia-induced osmotic diuresis and secondary thirst. These derangements result in long-term complications that affect the eyes, kidneys, nerves, and blood vessels. {ECO:0000269|PubMed:19073967}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06497; DB05906; DB12698; DB12960; DB05941; DB04835; DB05501; DB06652 Interacts with Q16570-2; Q92583; PRO_0000005165 [P13236]; P13501; P51681; P01730; P61073; P54849; O54081 EC number NA Uniprot keywords 3D-structure; Cell membrane; Diabetes mellitus; Disulfide bond; G-protein coupled receptor; Glycoprotein; Host cell receptor for virus entry; Host-virus interaction; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Sulfation; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 33641.8 Length 291 Aromaticity 0.16 Instability index 26.12 Isoelectric point 9.4 Charge (pH=7) 11.41 2D Binding mode Binding energy (Kcal/mol) -6.46 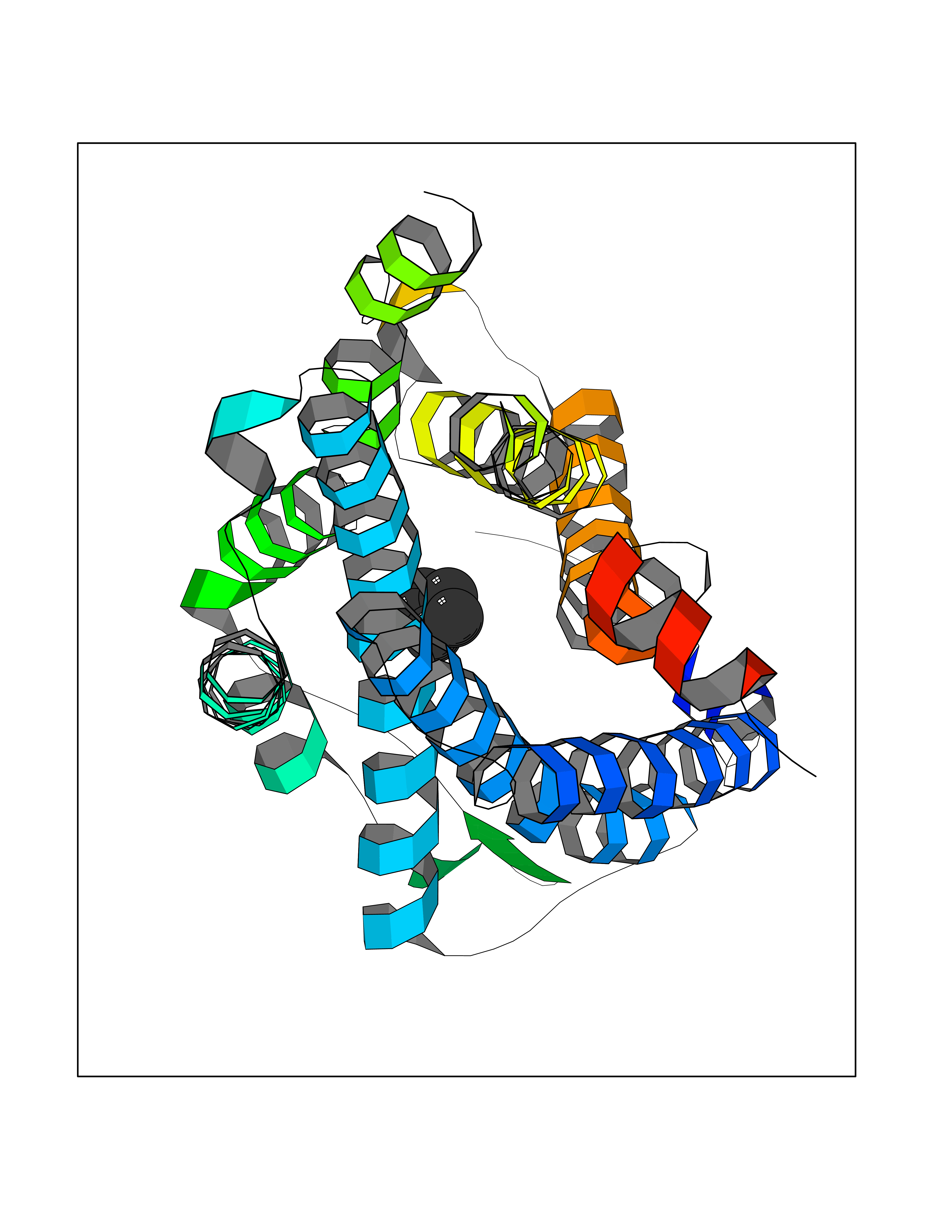 Molscript Map 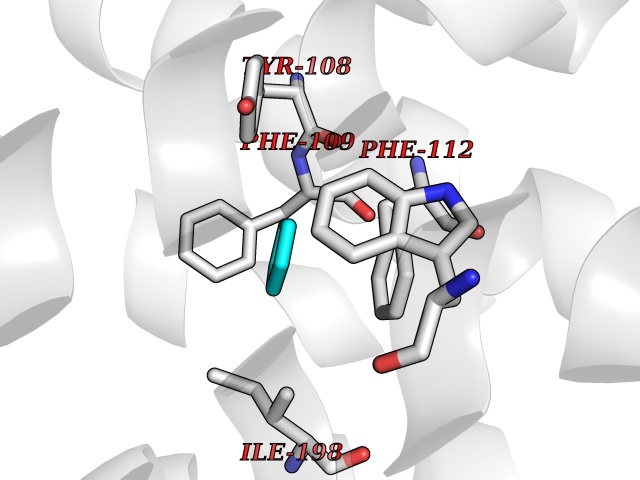 Pymol Map 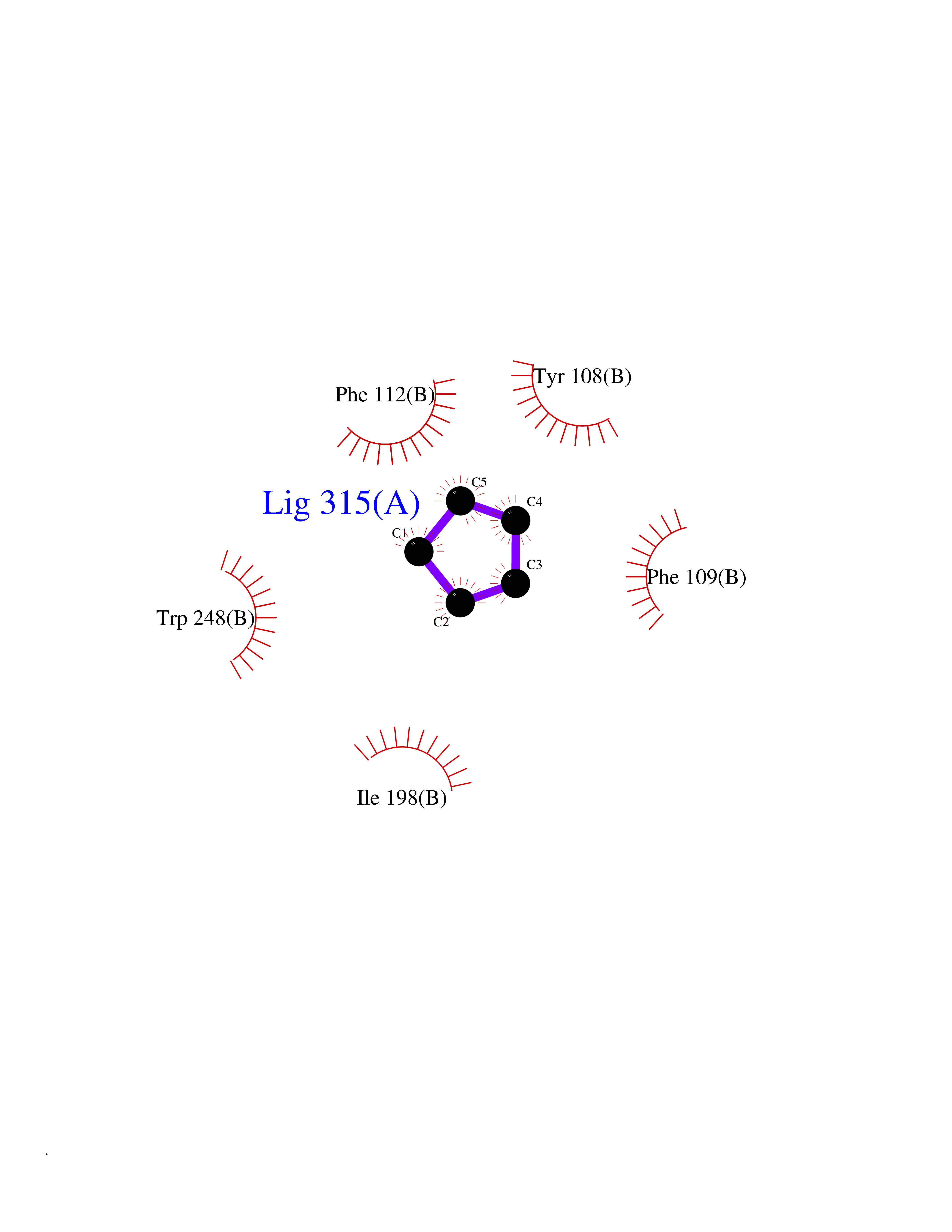 Ligplot Map 3D Binding mode Sequence PCQKINVKQIAARLLPPLYSLVFIFGFVGNMLVILILINYKRLKSMTDIYLLNLAISDLFFLLTVPFWAHYAAAQWDFGNTMCQLLTGLYFIGFFSGIFFIILLTIDRYLAVVHAVFALKARTVTFGVVTSVITWVVAVFASLPNIIFTRSQKEGLHYTCSSHFPYSQYQFWKNFQTLKIVILGLVLPLLVMVICYSGILKTLLRKKRHRDVRLIFTIMIVYFLFWAPYNIVLLLNTFQEFFGLNNCSSSNRLDQAMQVTETLGMTHCCINPIIYAFVGEEFRNYLLVFFQ Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Matrix metalloproteinase-16 (MMP-16) | 1RM8 | 4.74 | |
Target general information Gen name MMP16 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Membrane-type-3 matrix metalloproteinase; Membrane-type matrix metalloproteinase 3; MTMMP3; MT3MMP; MT3-MMP; MT-MMP 3; MMPX2; MMP-X2; C8orf57 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates progelatinase A. Involved in the matrix remodeling of blood vessels. Isoform short cleaves fibronectin and also collagen type III, but at lower rate. It has no effect on type I, II, IV and V collagen. However, upon interaction with CSPG4, it may be involved in degradation and invasion of type I collagen by melanoma cells. Endopeptidase that degrades various components of the extracellular matrix, such as collagen type III and fibronectin. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03880; DB00786 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Cleavage on pair of basic residues; Collagen degradation; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18853.6 Length 169 Aromaticity 0.12 Instability index 33.65 Isoelectric point 4.88 Charge (pH=7) -12.42 2D Binding mode Binding energy (Kcal/mol) -6.47 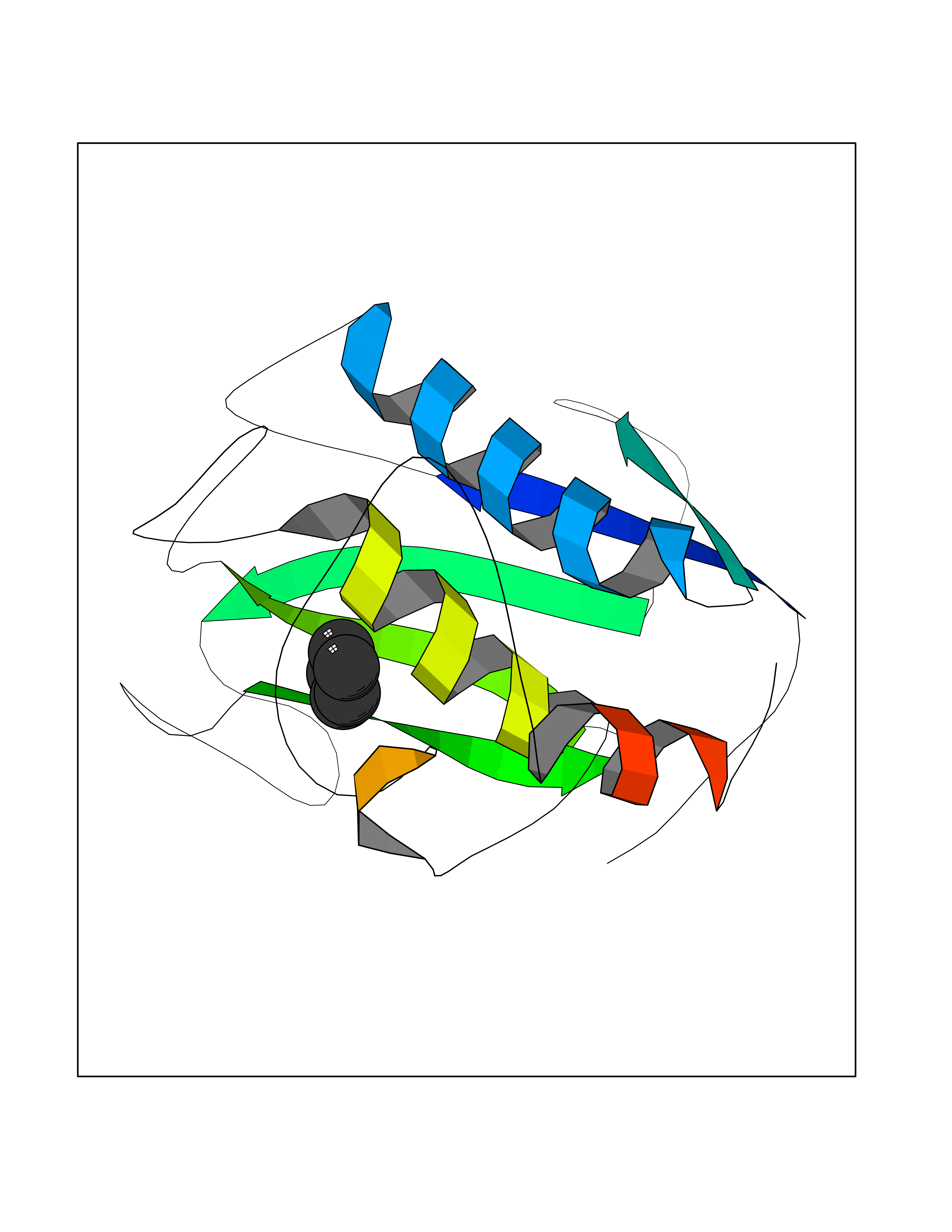 Molscript Map 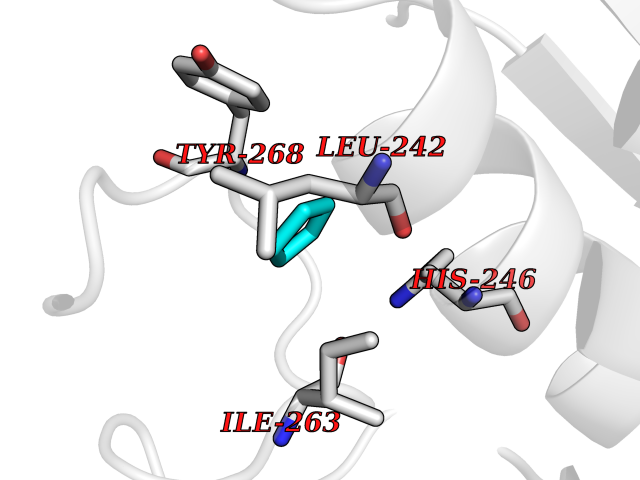 Pymol Map 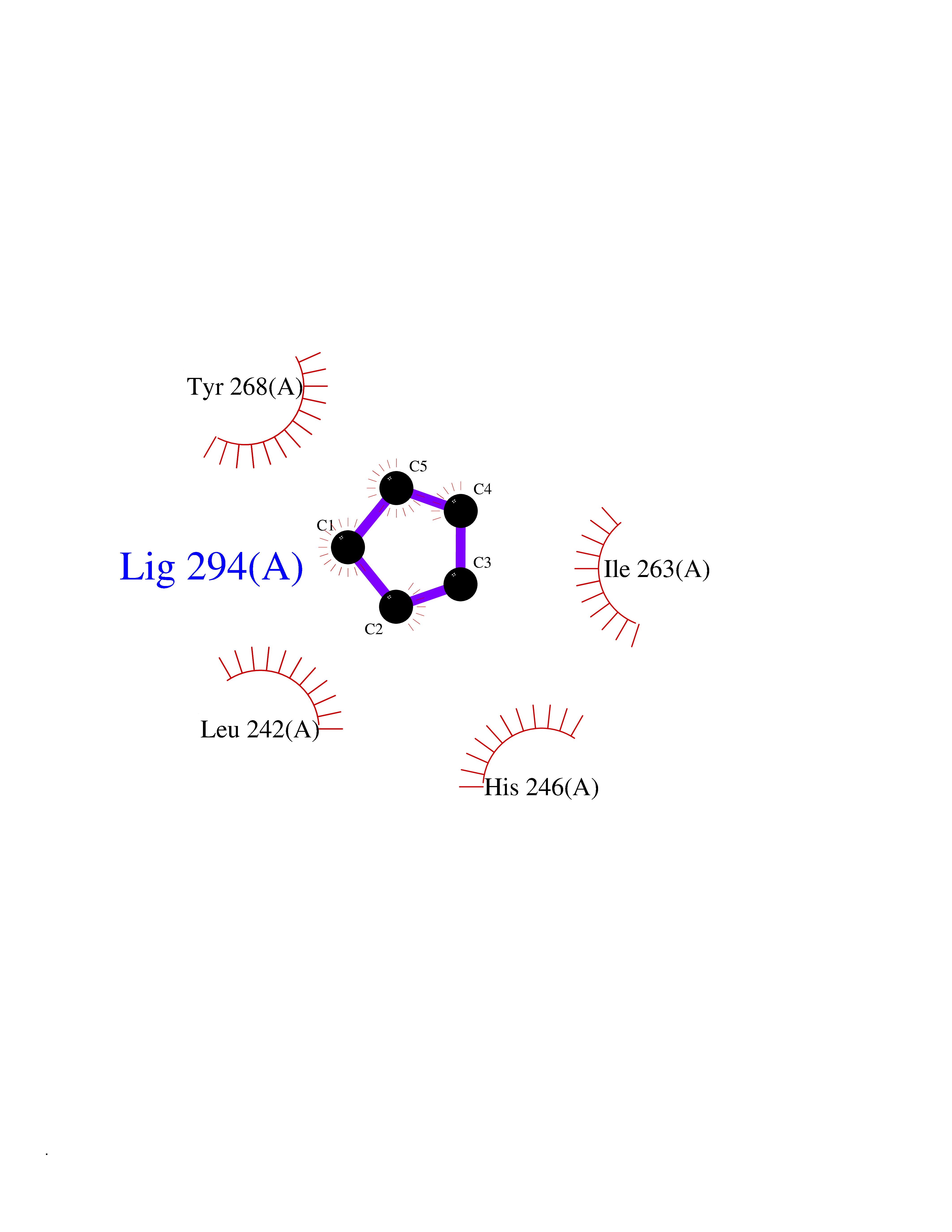 Ligplot Map 3D Binding mode Sequence GQKWQHKHITYSIKNVTPKVGDPETRKAIRRAFDVWQNVTPLTFEEVPYSELENGKRDVDITIIFASGFHGDSSPFDGEGGFLAHAYFPGPGIGGDTHFDSDEPWTLGNPNHDGNDLFLVAVHELGHALGLEHSNDPTAIMAPFYQYMETDNFKLPNDDLQGIQKIYGP Hydrogen bonds contact Hydrophobic contact | ||||