Job Results:
Ligand
Structure
Job ID
e848ab91d8ae28235d6e66c6c8c71011
Job name
NA
Time
2024-09-30 12:19:50
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Dibasic-processing enzyme (Furin) | 7LCU | 6.60 | |
Target general information Gen name FURIN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Paired basic amino acid residuecleaving enzyme; Paired basic amino acid residue-cleaving enzyme; PCSK3; PACE; FUR; Dibasicprocessing enzyme Protein family Peptidase S8 family, Furin subfamily Biochemical class Peptidase Function Mediates processing of TGFB1, an essential step in TGF-beta-1 activation. Ubiquitous endoprotease within constitutive secretory pathways capable of cleavage at the RX(K/R)R consensus motif. Related diseases Brachydactyly A2 (BDA2) [MIM:112600]: A form of brachydactyly. Brachydactyly defines a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. In brachydactyly type A2 shortening of the middle phalanges is confined to the index finger and the second toe, all other digits being more or less normal. Because of a rhomboid or triangular shape of the affected middle phalanx, the end of the second finger usually deviates radially. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}. The gene represented in this entry is involved in disease pathogenesis. Duplications of a cis-regulatory element located approximately 110 kb downstream of BMP2 have been found in BDA2 families. They likely cause altered BMP2 expression with pathological consequences. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}.; DISEASE: Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 1 (SSFSC1) [MIM:617877]: An autosomal dominant disorder characterized by short stature, facial dysmorphism, skeletal anomalies, and variable cardiac defects. Distinctive facial features include midface retrusion, short upturned nose, long philtrum, high-arched or cleft palate, and variable degrees of micrognathia and dental crowding. Skeletal anomalies include patterning defects of the axial skeleton, characterized by 11 pairs of ribs and brachydactyly of the fifth ray. Congenital heart defects are variably observed and appear to involve primarily the cardiac outflow tract. {ECO:0000269|PubMed:29198724}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03600 Interacts with P05067; P50281; Q9H239; O14793; K9N5Q8; P0DTC2; Q91QT1 EC number EC 3.4.21.75 Uniprot keywords 3D-structure; Autocatalytic cleavage; Calcium; Cell membrane; Cleavage on pair of basic residues; Disulfide bond; Endosome; Glycoprotein; Golgi apparatus; Heparin-binding; Host-virus interaction; Hydrolase; Membrane; Metal-binding; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Transmembrane; Transmembrane helix; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 51029.8 Length 470 Aromaticity 0.08 Instability index 26.23 Isoelectric point 5.23 Charge (pH=7) -16.94 2D Binding mode Binding energy (Kcal/mol) -6.99  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence YQEPTDPKFPQQWYLSGVTQRDLNVKAAWAQGYTGHGIVVSILDDGIEKNHPDLAGNYDPGASFDVNDQDPDPQPRYTQMNDNRHGTRCAGEVAAVANNGVCGVGVAYNARIGGVRMLDGEVTDAVEARSLGLNPNHIHIYSASWGPEDDGKTVDGPARLAEEAFFRGVSQGRGGLGSIFVWASGNGGREHDSCNCDGYTNSIYTLSISSATQFGNVPWYSEACSSTLATTYSSGNQNEKQIVTTDLRQKCTESHTGTSASAPLAAGIIALTLEANKDLTWRDMQHLVVQTSKPAHLNANDWATNGVGRKVSHSYGYGLLDAGAMVALAQDWTTVAPQRKCIIDILTEPKDIGKRLEVRKTVTACLGEPNHITRLEHAQARLTLSYNRRGDLAIHLVSPMGTRSTLLAARPHDYSADGFNDWAFMTTHSWDEDPSGEWVLEIENTSEANNYGTLTKFTLVLYGTAGENLY Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | SET domain containing 8 (KMT5A) | 5TEG | 6.60 | |
Target general information Gen name KMT5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SETD8; SET8; SET07; SET domain-containing protein 8; PRSET7; PR/SET07; PR/SET domain-containing protein 07; PR-Set7; N-lysine methyltransferase KMT5A; Lysine-specific methylase 5A; Lysine N-methyltran Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, PR/SET subfamily Biochemical class Methyltransferase Function Specifically monomethylates 'Lys-20' of histone H4 (H4K20me1). H4K20me1 is enriched during mitosis and represents a specific tag for epigenetic transcriptional repression. Mainly functions in euchromatin regions, thereby playing a central role in the silencing of euchromatic genes. Required for cell proliferation, probably by contributing to the maintenance of proper higher-order structure of DNA during mitosis. Involved in chromosome condensation and proper cytokinesis. Nucleosomes are preferred as substrate compared to free histones. Mediates monomethylation of p53/TP53 at 'Lys-382', leading to repress p53/TP53-target genes. Plays a negative role in TGF-beta response regulation and a positive role in cell migration. Protein-lysine N-methyltransferase that monomethylates both histones and non-histone proteins. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P62805; P07910; Q15672 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell cycle; Cell division; Chromatin regulator; Chromosome; Coiled coil; Direct protein sequencing; Methyltransferase; Mitosis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 19129.4 Length 168 Aromaticity 0.08 Instability index 49.18 Isoelectric point 7.88 Charge (pH=7) 1.37 2D Binding mode Binding energy (Kcal/mol) -7.3  Molscript Map 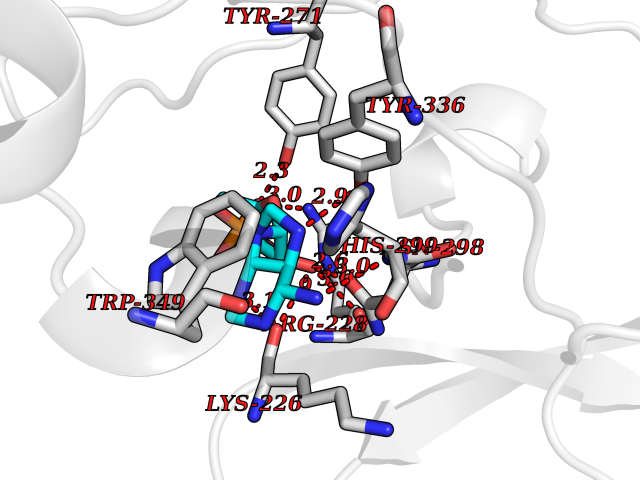 Pymol Map 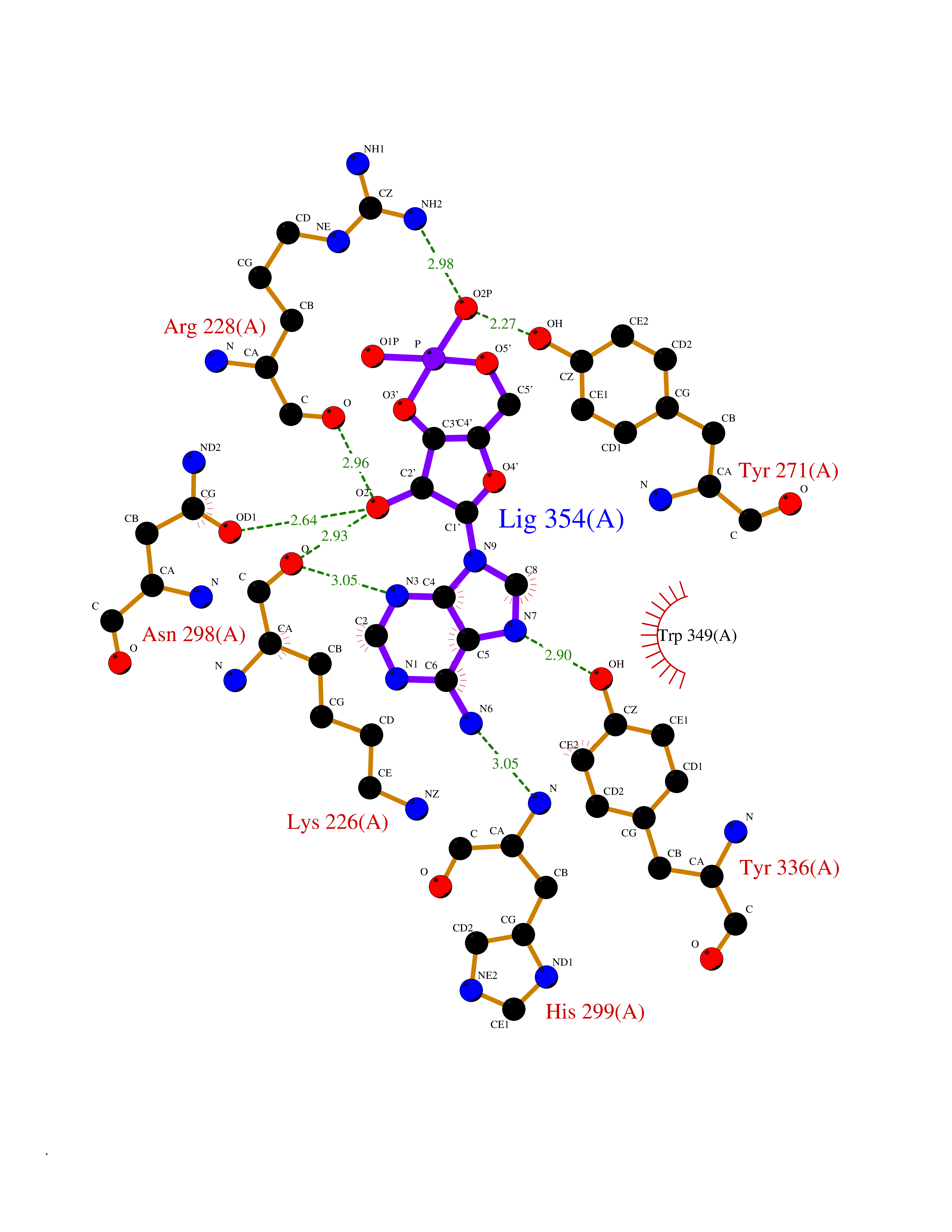 Ligplot Map 3D Binding mode Sequence KSKAELQSEERKRIDELIESGKEEGMKIDLIDGKGRGVIATKQFSRGDFVVEYHGDLIEITDAKKREALYAQDPSTGCYMYYFQYLSKTYCVDATRETNRLGRLINHSKSGNCQTKLHDIDGVPHLILIASRDIAAGEELLYDYGDRSKASIEAHPWLKHKRHRXVLR Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Metabotropic glutamate receptor 2 (mGluR2) | 7E9G | 6.60 | |
Target general information Gen name GRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR2; Group II metabotropic glutamate receptor; Glutamate receptor mGLU2; GPRC1B Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors, such as adenylate cyclase. Signaling inhibits adenylate cyclase activity. May mediate suppression of neurotransmission or may be involved in synaptogenesis or synaptic stabilization. G-protein coupled receptor for glutamate. Related diseases Oocyte/zygote/embryo maturation arrest 21 (OZEMA21) [MIM:620610]: An autosomal dominant, female infertility disorder characterized by zygote development arrest due to failure of pronuclei fusion. {ECO:0000269|PubMed:33948904, ECO:0000269|PubMed:33953335}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with Q5T8D3-2; Q9BYF1; Q13520; Q13323; Q8WV48; P57739; O95484; Q7Z7G2; P00387; P27487; P28223-1; Q5SR56; O14880; Q8N4V1; Q58DX5; Q13113; Q9NR31; Q8IWU4; Q9H2H9; P27105; Q8N3G9; Q96Q45-2; Q9NWD8; Q8WUV1; Q9UMX0-2; P0DTC2 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 85146.2 Length 769 Aromaticity 0.11 Instability index 36.84 Isoelectric point 8.48 Charge (pH=7) 9.7 2D Binding mode Binding energy (Kcal/mol) -7.6  Molscript Map 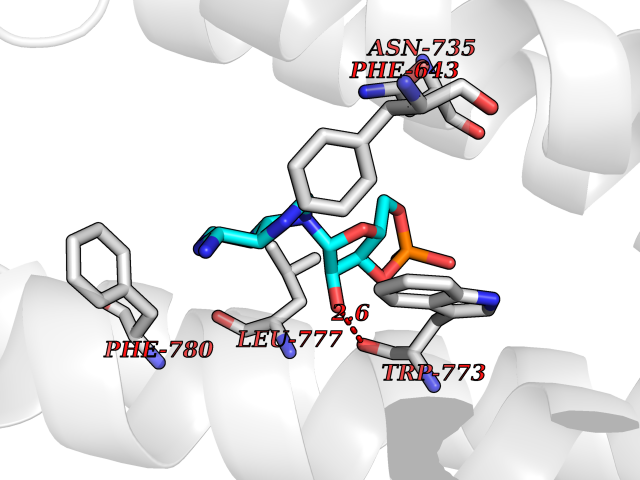 Pymol Map 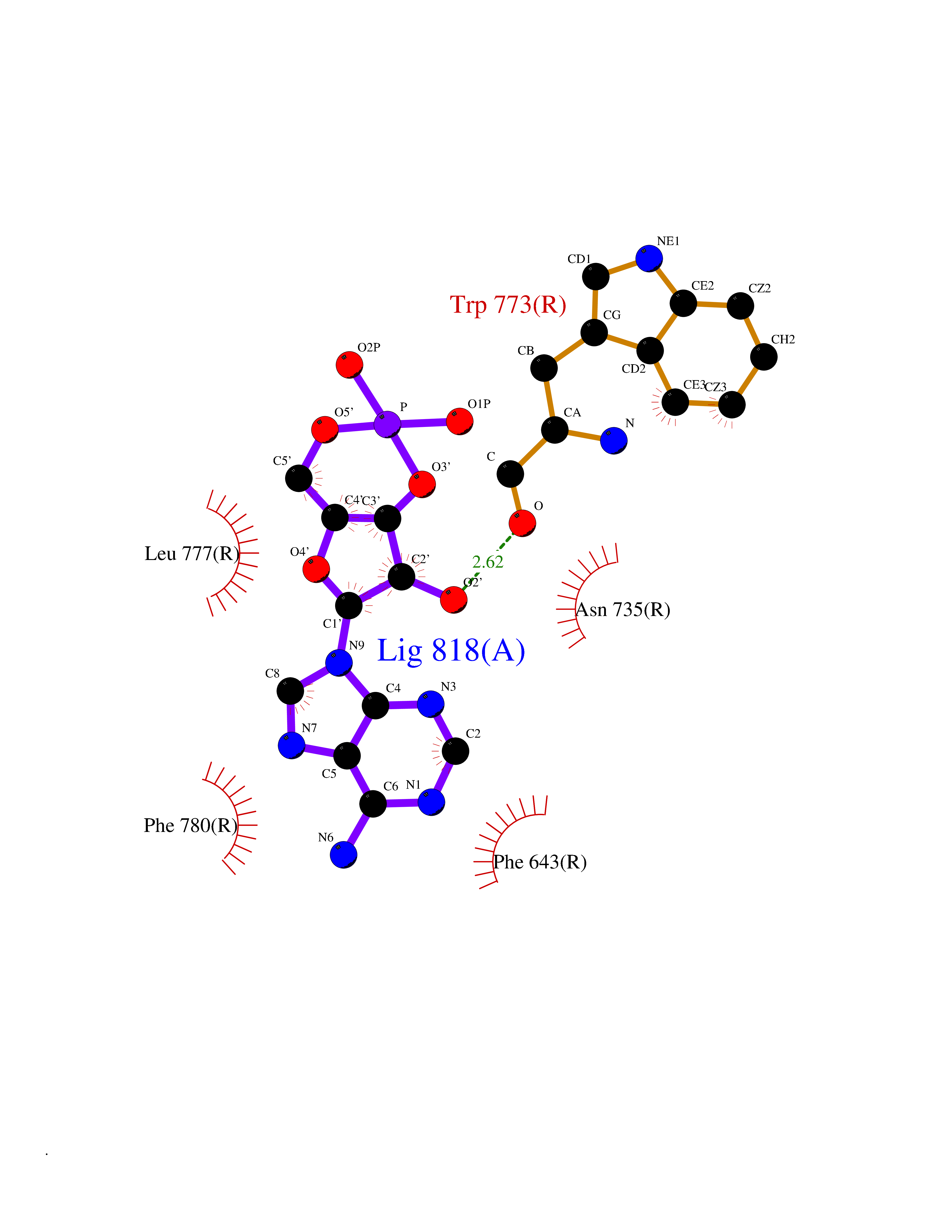 Ligplot Map 3D Binding mode Sequence KKVLTLEGDLVLGGLFPVHQKGGPAEDCGPVNEHRGIQRLEAMLFALDRINRDPHLLPGVRLGAHILDSCSKDTHALEQALDFVRASLITGVIGGSYSDVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFFQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFELEARARNICVATSEKVGRAMSRAAFEGVVRALLQKPSARVAVLFTRSEDARELLAASQRLNASFTWVASDGWGALESVVAGSEGAAEGAITIELASYPISDFASYFQSLDPWNNSRNPWFREFWEQRFRCSFRQRDCAAHSLRAVPFEQESKIMFVVNAVYAMAHALHNMHRALCPNTTRLCDAMRPVNGRRLYKDFVLNVKFDAPFRPADTHNEVRFDRFGDGIGRYNIFTYLRAGSGRYRYQKVGYWAEGLTLDTSLIPWASPSAGPLPASRCSEPCLQNEVKSVQPGEVCCWLCIPCQPYEYRLDEFTCADCGLGYWPNASLTGCFELPQEYIRWGDAWAVGPVTIACLGALATLFVLGVFVRHNATPVVKAAGRELCYILLGGVFLCYCMTFIFIAKPSTAVCTLRRLGLGTAFSVCYSALLTKTNRIARIFGGAREGAQRPRFISPASQVAICLALISGQLLIVVAWLVVEAPGTGKETAPERREVVTLRCNHRDASMLGSLAYNVLLIALCTLYAFKTRKCPENFNEAKFIGFTMYTTCIIWLAFLPIFYVTSSDYRVQTTTMCVSVSLSGSVVLGCLFAPKLHI Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Voltage-gated sodium channel alpha Nav1.7 (SCN9A) | 7W9M | 6.60 | |
Target general information Gen name SCN9A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hNE-Na; Voltage-gated sodium channel subunit alpha Nav1.7; Sodium channel proteintype IX subunit alpha; Sodium channel proteintype 9 subunit alpha; Sodium channel protein type IX subunit alpha; Sodium Protein family Sodium channel (TC 1.A.1.10) family, Nav1.7/SCN9A subfamily Biochemical class Voltage-gated ion channel Function Assuming opened or closed conformations in response to the voltage difference across the membrane, the protein forms a sodium-selective channel through which Na(+) ions may pass in accordance with their electrochemical gradient. It is a tetrodotoxin-sensitive Na(+) channel isoform. Plays a role in pain mechanisms, especially in the development of inflammatory pain. Mediates the voltage-dependent sodium ion permeability of excitable membranes. Related diseases Primary erythermalgia (PERYTHM) [MIM:133020]: Autosomal dominant disease characterized by recurrent episodes of severe pain associated with redness and warmth in the feet or hands. {ECO:0000269|PubMed:14985375, ECO:0000269|PubMed:15385606, ECO:0000269|PubMed:15955112, ECO:0000269|PubMed:15958509, ECO:0000269|PubMed:16216943, ECO:0000269|PubMed:16392115, ECO:0000269|PubMed:16702558, ECO:0000269|PubMed:16988069, ECO:0000269|PubMed:18945915, ECO:0000269|PubMed:19369487, ECO:0000269|PubMed:24311784}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Indifference to pain, congenital, autosomal recessive (CIP) [MIM:243000]: A disorder characterized by congenital inability to perceive any form of pain, in any part of the body. All other sensory modalities are preserved and the peripheral and central nervous systems are apparently intact. Patients perceive the sensations of touch, warm and cold temperature, proprioception, tickle and pressure, but not painful stimuli. There is no evidence of a motor or sensory neuropathy, either axonal or demyelinating. {ECO:0000269|PubMed:20635406}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Paroxysmal extreme pain disorder (PEXPD) [MIM:167400]: An autosomal dominant paroxysmal disorder of pain and autonomic dysfunction. The distinctive features are paroxysmal episodes of burning pain in the rectal, ocular, and mandibular areas accompanied by autonomic manifestations such as skin flushing. {ECO:0000269|PubMed:17145499, ECO:0000269|PubMed:18945915, ECO:0000269|PubMed:25285947}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09088; DB13746; DB05541; DB00564; DB01161; DB00907; DB13269; DB13961; DB06218; DB00555; DB00281; DB00776; DB11186; DB09345; DB01069; DB09342; DB00243; DB06201; DB09085; DB00273; DB00313; DB00909 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Ubl conjugation; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 162402 Length 1418 Aromaticity 0.14 Instability index 35.66 Isoelectric point 6.72 Charge (pH=7) -1.33 2D Binding mode Binding energy (Kcal/mol) -8.03  Molscript Map  Pymol Map 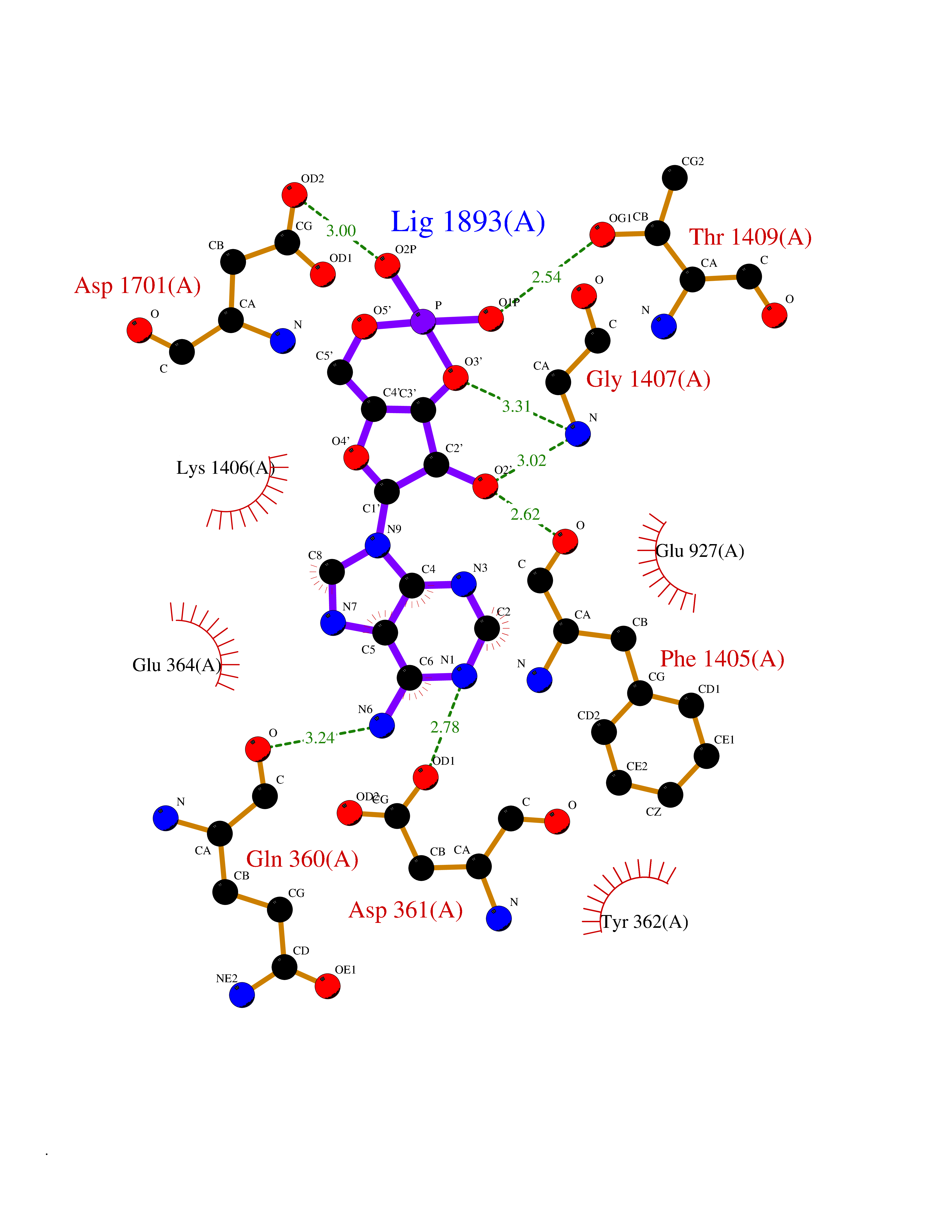 Ligplot Map 3D Binding mode Sequence GPQSFVHFTKQSLALIEQRIAERKSKEPKPSSDLEAGKQLPFIYGDIPPGMVSEPLEDLDPYYADKKTFIVLNKGKTIFRFNATPALYMLSPFSPLRRISIKILVHSLFSMLIMCTILTNCIFMTMNNPPDWTKNVEYTFTGIYTFESLVKILARGFCVGEFTFLRDPWNWLDFVVIVFAYLTEFVNNVSALRTFRVLRALKTISVIPGLKTIVGALIQSVKKLSDVMILTVFCLSVFALIGLQLFMGNLKHKCFRNSLENNETLESIMNTLESEEDFRKYFYYLEGSKDALLCGFSTDSGQCPEGYTCVKIGRNPDYGYTSFDTFSWAFLALFRLMTQDYWENLYQQTLRAAGKTYMIFFVVVIFLGSFYLINLILAVVAMAYKEQNQANIEEAKQKELEFQQMLDRLKKEQEPYWIKFKKCIYFIVMDPFVDLAITICIVLNTLFMAMEHHPMTEEFKNVLAIGNLVFTGIFAAEMVLKLIAMDPYEYFQVGWNIFDSLIVTLSLVELFLADVEGLSVLRSFRLLRVFKLAKSWPTLNMLIKIIGNSVGALGNLTLVLAIIVFIFAVVGMQLFGKSYKECVCKINDDCTLPRWHMNDFFHSFLIVFRVLCGEWIETMWDCMEVAGQAMCLIVYMMVMVIGNLVVLNLFLALLLSSFSSDNLTAIEEDPDANNLQIAVTRIKKGINYVKQTLREFILKAFGKIWWNIRKTCYKIVEHSWFESFIVLMILLSSGALAFEDIYIERKKTIKIILEYADKIFTYIFILEMLLKWIAYGYKTYFTNAWCWLDFLIVDVSLVTLVANTLGYSDLGPIKSLRTLRALRPLRALSRFEGMRVVVNALIGAIPSIMNVLLVCLIFWLIFSIMGVNLFAGKFYECINTTDGSRFPASQVPNRSECFALMNVSQNVRWKNLKVNFDNVGLGYLSLLQVATFKGWTIIMYAAVDSVNVDKQPKYEYSLYMYIYFVVFIIFGSFFTLNLFIGVIIDNFNQQKKKLGGQDIFMTEEQKKYYNAMKKLGSKKPQKPIPRPGNKIQGCIFDLVTNQAFDISIMVLICLNMVTMMVEKEGQSQHMTEVLYWINVVFIILFTGECVLKLISLRHYYFTVGWNIFDFVVVIISIVGMFLADLIETYFVSPTLFRVIRLARIGRILRLVKGAKGIRTLLFALMMSLPALFNIGLLLFLVMFIYAIFGMSNFAYVKKEDGINDMFNFETFGNSMICLFQITTSAGWDGLLAPILNSKPPDCDPKKVHPGSSVEGDCGNPSVGIFYFVSYIIISFLVVVNMYIAVILENFSVATEESTEPLSEDDFEMFYEVWEKFDPDATQFIEFSKLSDFAAALDPPLLIAKPNKVQLIAMDLPMVSGDRIHCLDILFAFTKRVLGESGEMDSLRSQMEERFMSANPSKVSYEPITTTLKRKQEDV Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Proto-oncogene c-Fes (FES) | 3CBL | 6.60 | |
Target general information Gen name FES Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p93c-fes; Tyrosine-protein kinase Fes/Fps; Proto-oncogene c-Fps; Feline sarcoma/Fujinami avian sarcoma oncogene homolog; FPS Protein family Protein kinase superfamily, Tyr protein kinase family, Fes/fps subfamily Biochemical class Kinase Function Plays a role in FCER1 (high affinity immunoglobulin epsilon receptor)-mediated signaling in mast cells. Acts down-stream of the activated FCER1 receptor and the mast/stem cell growth factor receptor KIT. Plays a role in the regulation of mast cell degranulation. Plays a role in the regulation of cell differentiation and promotes neurite outgrowth in response to NGF signaling. Plays a role in cell scattering and cell migration in response to HGF-induced activation of EZR. Phosphorylates BCR and down-regulates BCR kinase activity. Phosphorylates HCLS1/HS1, PECAM1, STAT3 and TRIM28. Tyrosine-protein kinase that acts downstream of cell surface receptors and plays a role in the regulation of the actin cytoskeleton, microtubule assembly, cell attachment and cell spreading. Related diseases Has been shown to act as proto-oncogene in some types of cancer, possibly due to abnormal activation of the kinase. Has been shown to act as tumor suppressor in other types of cancer. Expressed and present as activated kinase in a subset of acute myeloid leukemia patients; promotes survival of leukemia cells (PubMed:20111072). Expression is absent in K562 leukemia cells; ectopic expression of FSP/FES restores myeloid differentiation (PubMed:2656706). May function as tumor suppressor in colorectal cancer; expression is reduced or absent in samples from some colon cancer patients (PubMed:16455651). May function as tumor suppressor in melanoma by preventing melanoma cell proliferation; expression is reduced or absent in samples from some melanoma patients (PubMed:28463229). Ectopic expression of FSP/FES suppresses anchorage-independent growth in colon cancer cell lines (PubMed:16455651). Up-regulated in prostate cancer, and might be a predictor of recurrence after radical surgery (PubMed:16455651). May promote growth of renal carcinoma cells (PubMed:19082481). {ECO:0000269|PubMed:16455651, ECO:0000269|PubMed:19082481, ECO:0000269|PubMed:20111072, ECO:0000269|PubMed:2656706, ECO:0000269|PubMed:28463229}. Drugs (DrugBank ID) DB12010 Interacts with P10275; P15924; P15311; Q13480; P10721; P54274 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell junction; Cell membrane; Coiled coil; Cytoplasm; Cytoplasmic vesicle; Cytoskeleton; Golgi apparatus; Kinase; Lipid-binding; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; SH2 domain; Transferase; Tumor suppressor; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 40460.2 Length 356 Aromaticity 0.09 Instability index 42.56 Isoelectric point 7.12 Charge (pH=7) 0.32 2D Binding mode Binding energy (Kcal/mol) -7.11 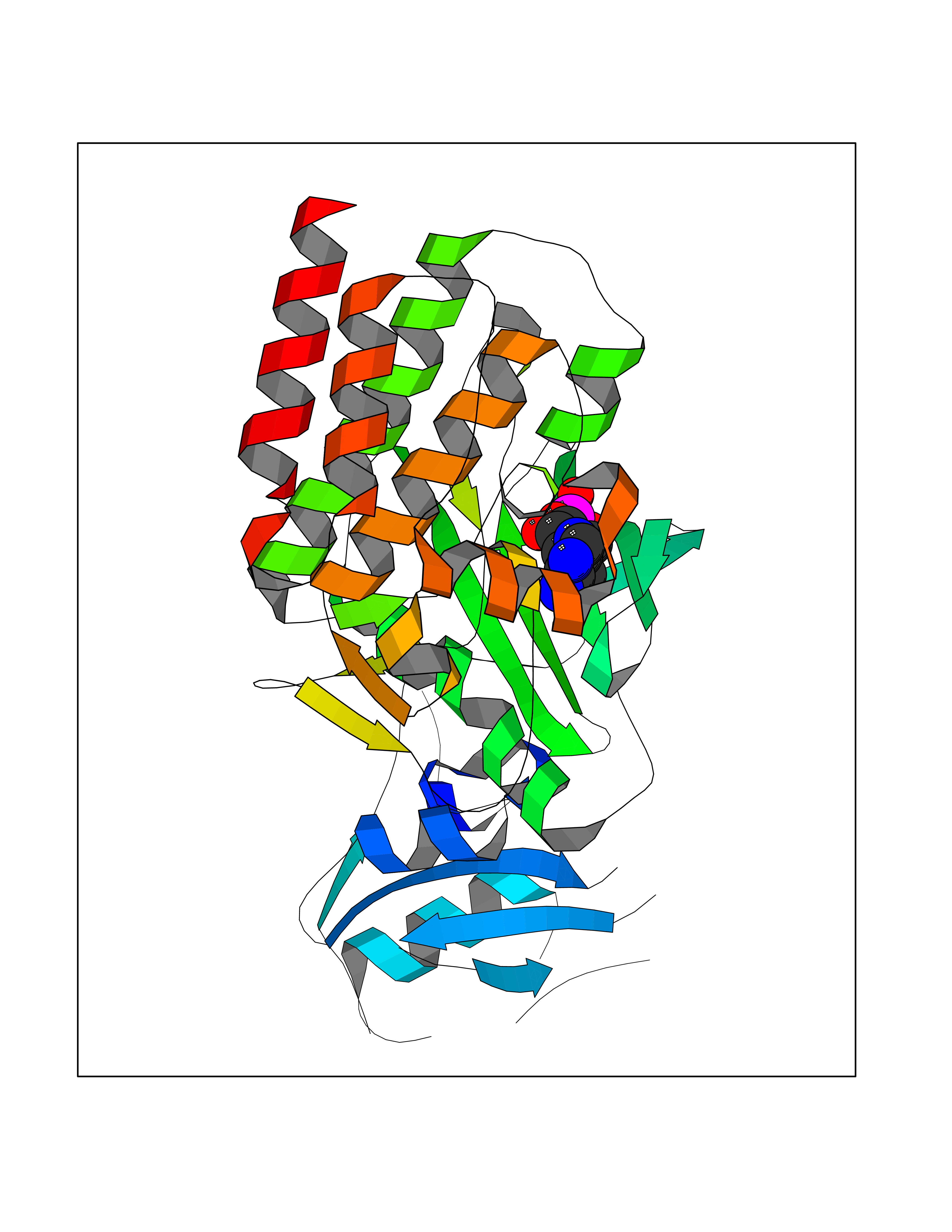 Molscript Map 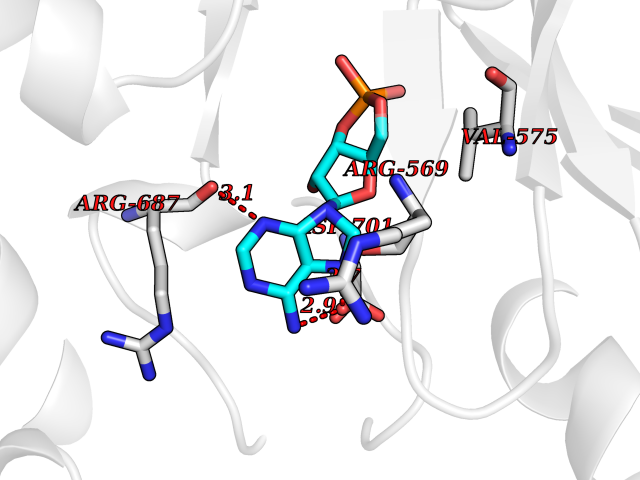 Pymol Map 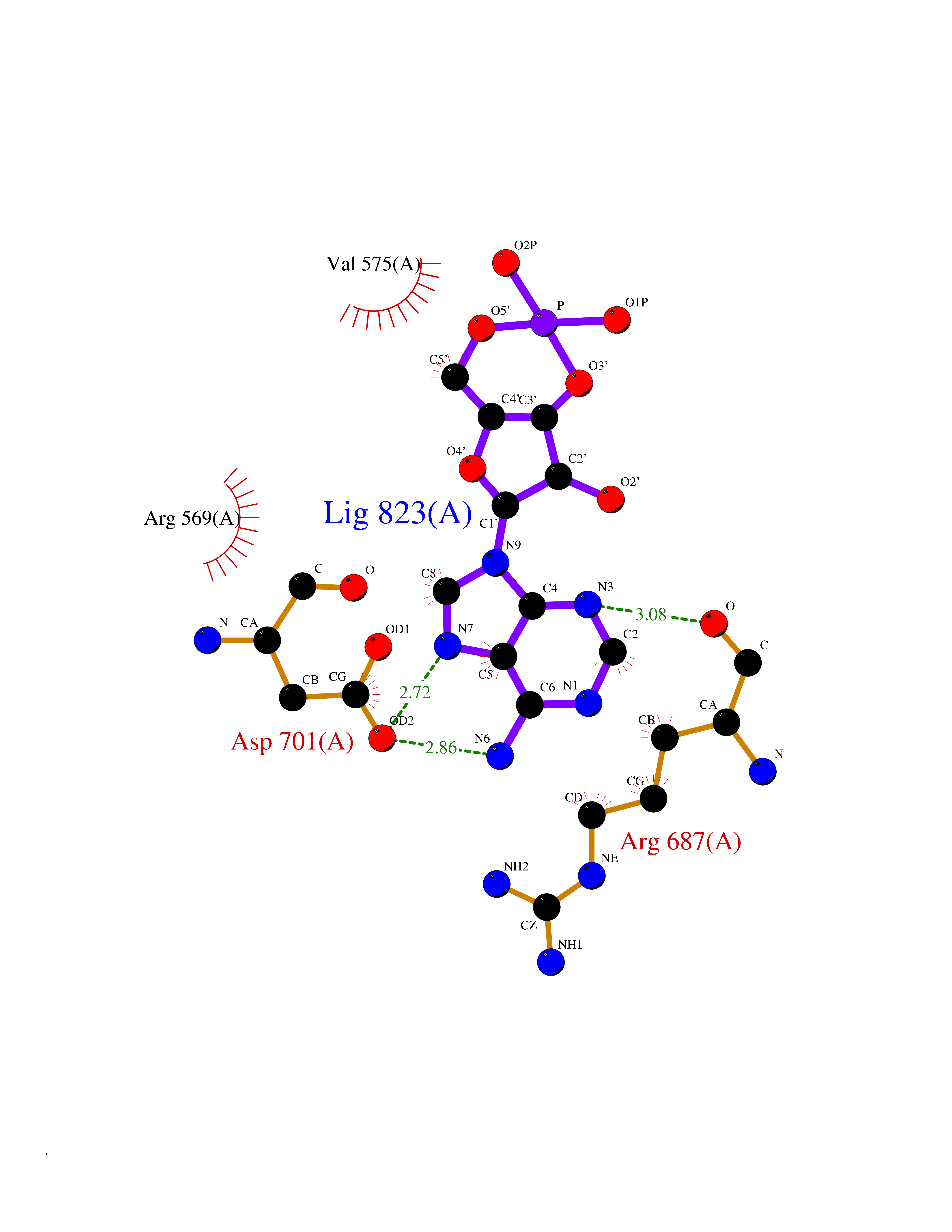 Ligplot Map 3D Binding mode Sequence SMIPEVQKPLHEQLWYHGAIPRAEVAELLVHSGDFLVRESQQEYVLSVPRHFIINLYRLFPSIPLLIDHLLSTQQPLVVLHRAVPKDKWVLNHEDLVLGEQIGRGNFGEVFSGRLRADNTLVAVKSCRETLPPDLKAKFLQEARILKQYSHPNIVRLIGVCTQKQPIYIVMELVQGGDFLTFLRTEGARLRVKTLLQMVGDAAAGMEYLESKCCIHRDLAARNCLVTEKNVLKISDFGMSREEADGVYAASGGLRQVPVKWTAPEALNYGRYSSESDVWSFGILLWETFSLGASPYPNLSNQQTREFVEKGGRLPCPELCPDAVFRLMEQCWAYEPGQRPSFSTIYQELQSIRKRH Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Histone acetyltransferase p300 (EP300) | 5LKX | 6.59 | |
Target general information Gen name EP300 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p300 HAT; Protein propionyltransferase p300; P300; Histone crotonyltransferase p300; Histone butyryltransferase p300; E1Aassociated protein p300; E1A-associated protein p300 Protein family NA Biochemical class Acyltransferase Function Acetylates all four core histones in nucleosomes. Histone acetylation gives an epigenetic tag for transcriptional activation. Mediates cAMP-gene regulation by binding specifically to phosphorylated CREB protein. Mediates acetylation of histone H3 at 'Lys-122' (H3K122ac), a modification that localizes at the surface of the histone octamer and stimulates transcription, possibly by promoting nucleosome instability. Mediates acetylation of histone H3 at 'Lys-27' (H3K27ac). Also functions as acetyltransferase for non-histone targets, such as ALX1, HDAC1, PRMT1 or SIRT2. Acetylates 'Lys-131' of ALX1 and acts as its coactivator. Acetylates SIRT2 and is proposed to indirectly increase the transcriptional activity of TP53 through acetylation and subsequent attenuation of SIRT2 deacetylase function. Acetylates HDAC1 leading to its inactivation and modulation of transcription. Acts as a TFAP2A-mediated transcriptional coactivator in presence of CITED2. Plays a role as a coactivator of NEUROD1-dependent transcription of the secretin and p21 genes and controls terminal differentiation of cells in the intestinal epithelium. Promotes cardiac myocyte enlargement. Can also mediate transcriptional repression. Acetylates FOXO1 and enhances its transcriptional activity. Acetylates BCL6 wich disrupts its ability to recruit histone deacetylases and hinders its transcriptional repressor activity. Participates in CLOCK or NPAS2-regulated rhythmic gene transcription; exhibits a circadian association with CLOCK or NPAS2, correlating with increase in PER1/2 mRNA and histone H3 acetylation on the PER1/2 promoter. Acetylates MTA1 at 'Lys-626' which is essential for its transcriptional coactivator activity. Acetylates XBP1 isoform 2; acetylation increases protein stability of XBP1 isoform 2 and enhances its transcriptional activity. Acetylates PCNA; acetylation promotes removal of chromatin-bound PCNA and its degradation during nucleotide excision repair (NER). Acetylates MEF2D. Acetylates and stabilizes ZBTB7B protein by antagonizing ubiquitin conjugation and degragation, this mechanism may be involved in CD4/CD8 lineage differentiation. In addition to protein acetyltransferase, can use different acyl-CoA substrates, such as (2E)-butenoyl-CoA (crotonyl-CoA), butanoyl-CoA (butyryl-CoA) or propanoyl-CoA (propionyl-CoA), and is able to mediate protein crotonylation, butyrylation or propionylation, respectively. Acts as a histone crotonyltransferase; crotonylation marks active promoters and enhancers and confers resistance to transcriptional repressors. Histone crotonyltransferase activity is dependent on the concentration of (2E)-butenoyl-CoA (crotonyl-CoA) substrate and such activity is weak when (E)-but-2-enoyl-CoA (crotonyl-CoA) concentration is low. Also acts as a histone butyryltransferase; butyrylation marks active promoters. Functions as a transcriptional coactivator for SMAD4 in the TGF-beta signaling pathway. Acetylates PCK1 and promotes PCK1 anaplerotic activity. Functions as histone acetyltransferase and regulates transcription via chromatin remodeling. Related diseases Defects in EP300 may play a role in epithelial cancer.; DISEASE: Chromosomal aberrations involving EP300 may be a cause of acute myeloid leukemias. Translocation t(8;22)(p11;q13) with KAT6A.; DISEASE: Rubinstein-Taybi syndrome 2 (RSTS2) [MIM:613684]: A disorder characterized by craniofacial abnormalities, postnatal growth deficiency, broad thumbs, broad big toes, intellectual disability and a propensity for development of malignancies. Some individuals with RSTS2 have less severe mental impairment, more severe microcephaly, and a greater degree of changes in facial bone structure than RSTS1 patients. {ECO:0000269|PubMed:15706485}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Menke-Hennekam syndrome 2 (MKHK2) [MIM:618333]: A form of Menke-Hennekam syndrome, a congenital autosomal dominant disease characterized by developmental delay, growth retardation, and craniofacial dysmorphism. Patients have intellectual disability of variable severity, speech delay, autistic behavior, short stature and microcephaly. Main facial characteristics include short palpebral fissures, telecanthi, depressed nasal ridge, short nose, anteverted nares, short columella and long philtrum. {ECO:0000269|PubMed:29460469}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NXW9; P27695; Q9UBL3; Q8WXX7; Q9NPI1; P24941; Q99967; P61201; P16220-1; P17844; Q01844; P35637; Q00403; Q16665; Q9H2X6; Q92831; P55209; O60934; P20265; Q96KQ4; Q8WUF5; Q13761; Q96EB6; Q13309; O95863; P42226; Q9UL17; P56279; P05549; P04637; Q13625; O15350; P11473; P67809; K4P3M7; P03122; P06422; P06790; Q61221; Q9QXM1; P04608; P03070; P03255; P03255-2; P03259 EC number EC 2.3.1.48 Uniprot keywords 3D-structure; Acetylation; Acyltransferase; Biological rhythms; Bromodomain; Cell cycle; Chromosomal rearrangement; Chromosome; Citrullination; Cytoplasm; Direct protein sequencing; Disease variant; Host-virus interaction; Intellectual disability; Isopeptide bond; Metal-binding; Methylation; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 64477.2 Length 554 Aromaticity 0.12 Instability index 45.78 Isoelectric point 7.01 Charge (pH=7) 0.05 2D Binding mode Binding energy (Kcal/mol) -7.74 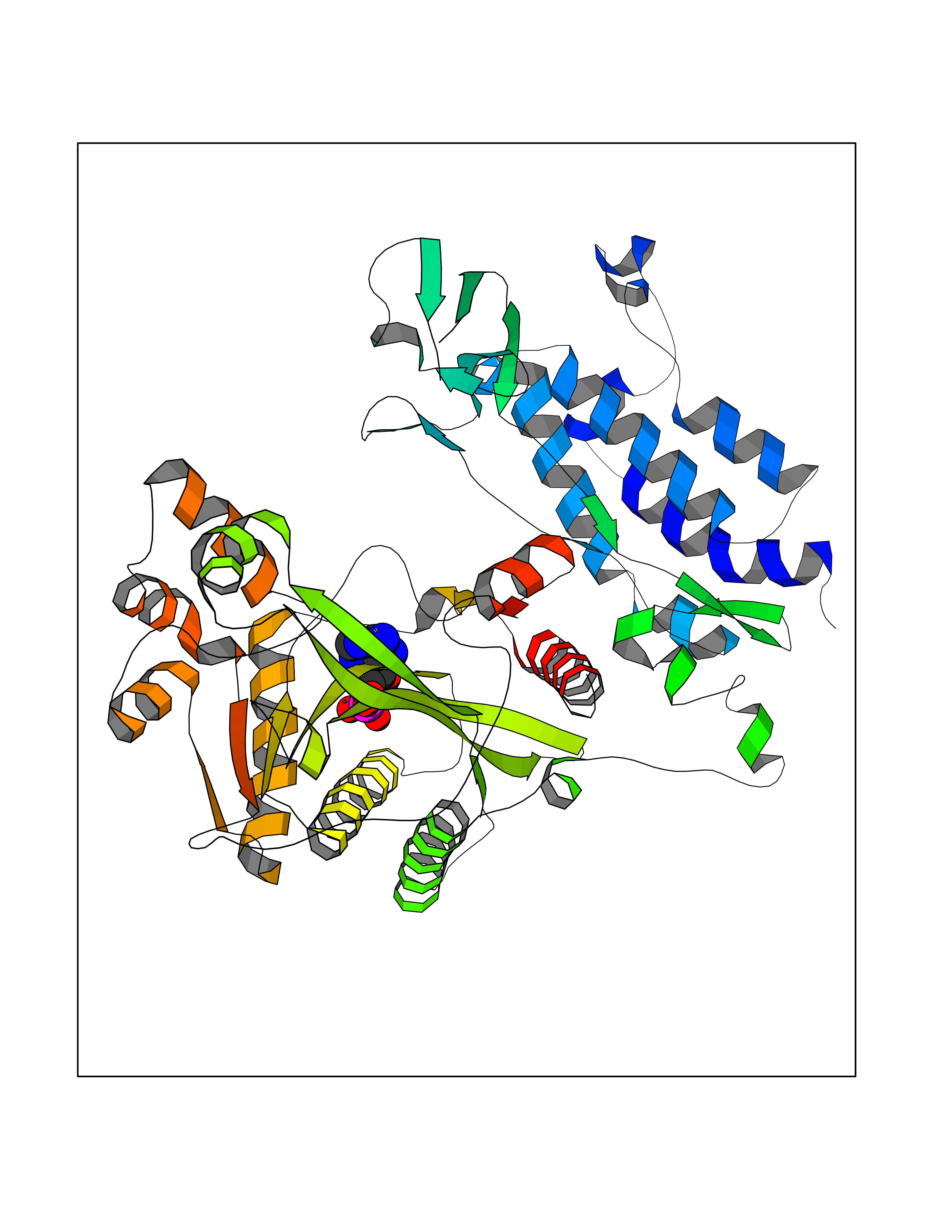 Molscript Map 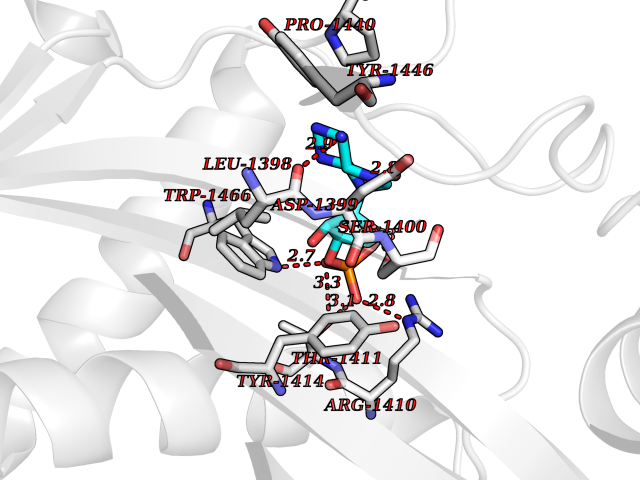 Pymol Map 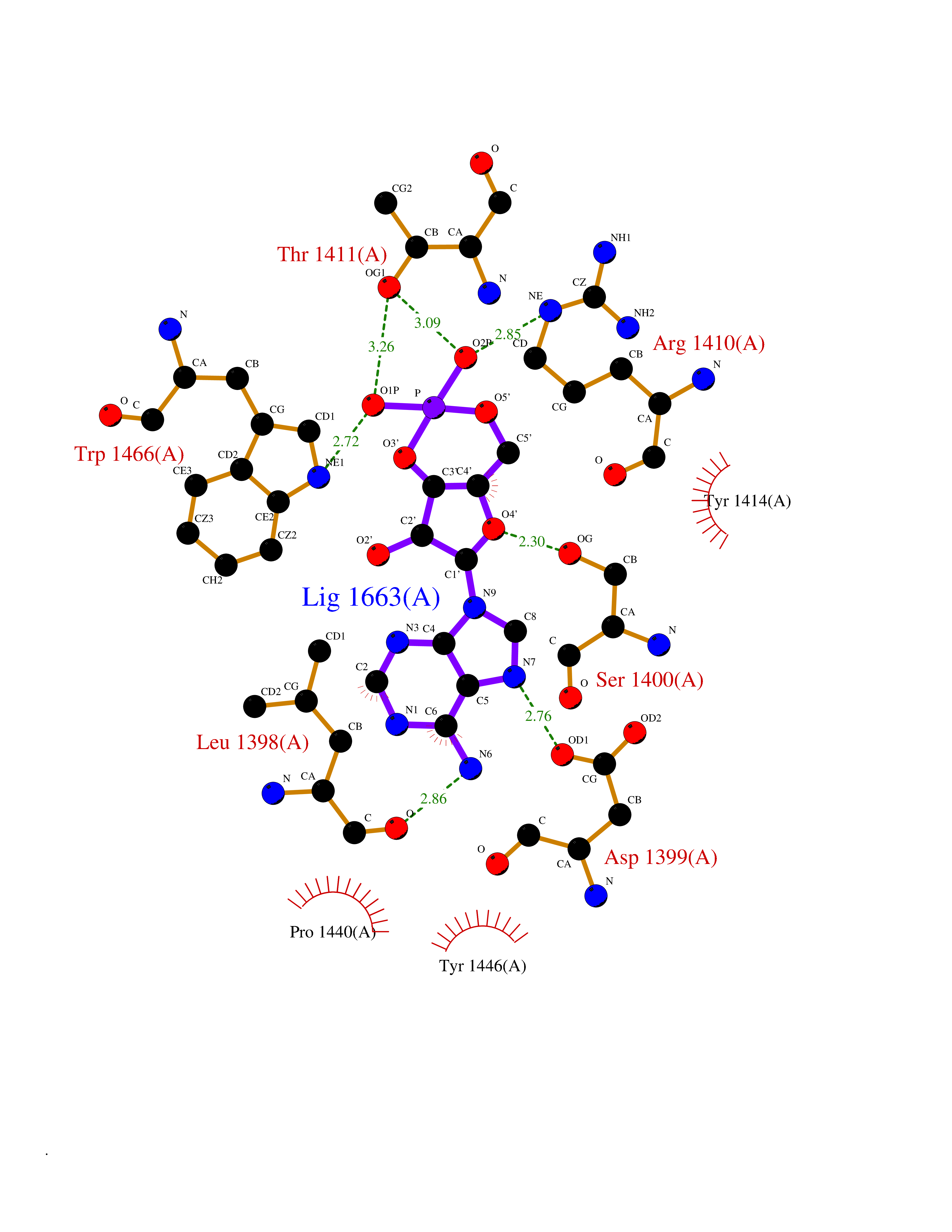 Ligplot Map 3D Binding mode Sequence KKIFKPEELRQALMPTLEALYRQDPESLPFRQPVDPQLLGIPDYFDIVKSPMDLSTIKRKLDTGQYQEPWQYVDDIWLMFNNAWLYNRKTSRVYKYCSKLSEVFEQEIDPVMQSLGYCCGRKLEFSPQTLCCYGKQLCTIPRDATYYSYQNRYHFCEKCFNEIQGESVSLGQTTINKEQFSKRKNDTLDPELFVECTECGRKMHQICVLHHEIIWPAGFVCDGCLKKSARTRKENKFSAKRLPSTRLGTFLENRVNDFLRRQNHPESGEVTVRVVHASDKTVEVKPGMKARFVDSGEMAESFPYRTKALFAFEEIDGVDLCFFGMHVQEYGSDCPPPNQRRVYISYLDSVHFFRPKCLRTAVYHEILIGYLEYVKKLGYTTGHIWACPPSEGDDYIFHCHPPDQKIPKPKRLQEWFKKMLDKAVSERIVHDYKDIFKQATEDRLTSAKELPYFEGDFWPNVLEESIKESGGSGSQKLYATMEKHKEVFFVIRLIAGPAANSLPPIVDPDPLIPCDLMDGRDAFLTLARDKHLEFSSLRRAQWSTMCMLVELHTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Vascular endothelial growth factor receptor 2 (KDR) | 2XIR | 6.57 | |
Target general information Gen name KDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms VEGFR2; VEGFR-2; VEGF-2 receptor; Protein-tyrosine kinase receptor flk-1; Kinase insert domain receptor; Fetal liver kinase 1; FLK1; FLK-1; CD309 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Plays an essential role in the regulation of angiogenesis, vascular development, vascular permeability, and embryonic hematopoiesis. Promotes proliferation, survival, migration and differentiation of endothelial cells. Promotes reorganization of the actin cytoskeleton. Isoforms lacking a transmembrane domain, such as isoform 2 and isoform 3, may function as decoy receptors for VEGFA, VEGFC and/or VEGFD. Isoform 2 plays an important role as negative regulator of VEGFA- and VEGFC-mediated lymphangiogenesis by limiting the amount of free VEGFA and/or VEGFC and preventing their binding to FLT4. Modulates FLT1 and FLT4 signaling by forming heterodimers. Binding of vascular growth factors to isoform 1 leads to the activation of several signaling cascades. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate and the activation of protein kinase C. Mediates activation of MAPK1/ERK2, MAPK3/ERK1 and the MAP kinase signaling pathway, as well as of the AKT1 signaling pathway. Mediates phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, reorganization of the actin cytoskeleton and activation of PTK2/FAK1. Required for VEGFA-mediated induction of NOS2 and NOS3, leading to the production of the signaling molecule nitric oxide (NO) by endothelial cells. Phosphorylates PLCG1. Promotes phosphorylation of FYN, NCK1, NOS3, PIK3R1, PTK2/FAK1 and SRC. Tyrosine-protein kinase that acts as a cell-surface receptor for VEGFA, VEGFC and VEGFD. Related diseases Hemangioma, capillary infantile (HCI) [MIM:602089]: A condition characterized by dull red, firm, dome-shaped hemangiomas, sharply demarcated from surrounding skin, usually presenting at birth or occurring within the first two or three months of life. They result from highly proliferative, localized growth of capillary endothelium and generally undergo regression and involution without scarring. {ECO:0000269|PubMed:11807987, ECO:0000269|PubMed:18931684}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Plays a major role in tumor angiogenesis. In case of HIV-1 infection, the interaction with extracellular viral Tat protein seems to enhance angiogenesis in Kaposi's sarcoma lesions. Drugs (DrugBank ID) DB04727; DB07514; DB07528; DB06938; DB07326; DB06626; DB08875; DB04849; DB05198; DB12147; DB12307; DB12010; DB11679; DB06101; DB09078; DB06080; DB06595; DB07537; DB07183; DB07333; DB07334; DB07274; DB09079; DB08519; DB08042; DB16265; DB06589; DB05931; DB08901; DB15822; DB05984; DB05578; DB08896; DB14840; DB06436; DB00398; DB01268; DB05075; DB11800; DB04879; DB05146; DB05014 Interacts with P35916; O60565; P98160; PRO_0000391621 [P98160]; PRO_0000391622 [P98160]; P17301; P35968; P09382; P08581; P16333; O14786; O75340; P09619; P29350; Q12913; P12931; P15692; P15692-4; P49767; Q9MYV3-3 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; ATP-binding; Cell junction; Cell membrane; Cytoplasm; Cytoplasmic vesicle; Developmental protein; Differentiation; Disulfide bond; Endoplasmic reticulum; Endosome; Glycoprotein; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33863.9 Length 296 Aromaticity 0.1 Instability index 40.01 Isoelectric point 8.5 Charge (pH=7) 4.59 2D Binding mode Binding energy (Kcal/mol) -7.64  Molscript Map 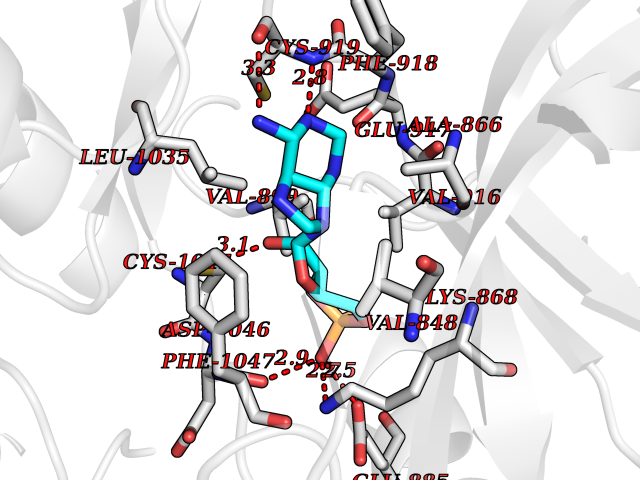 Pymol Map 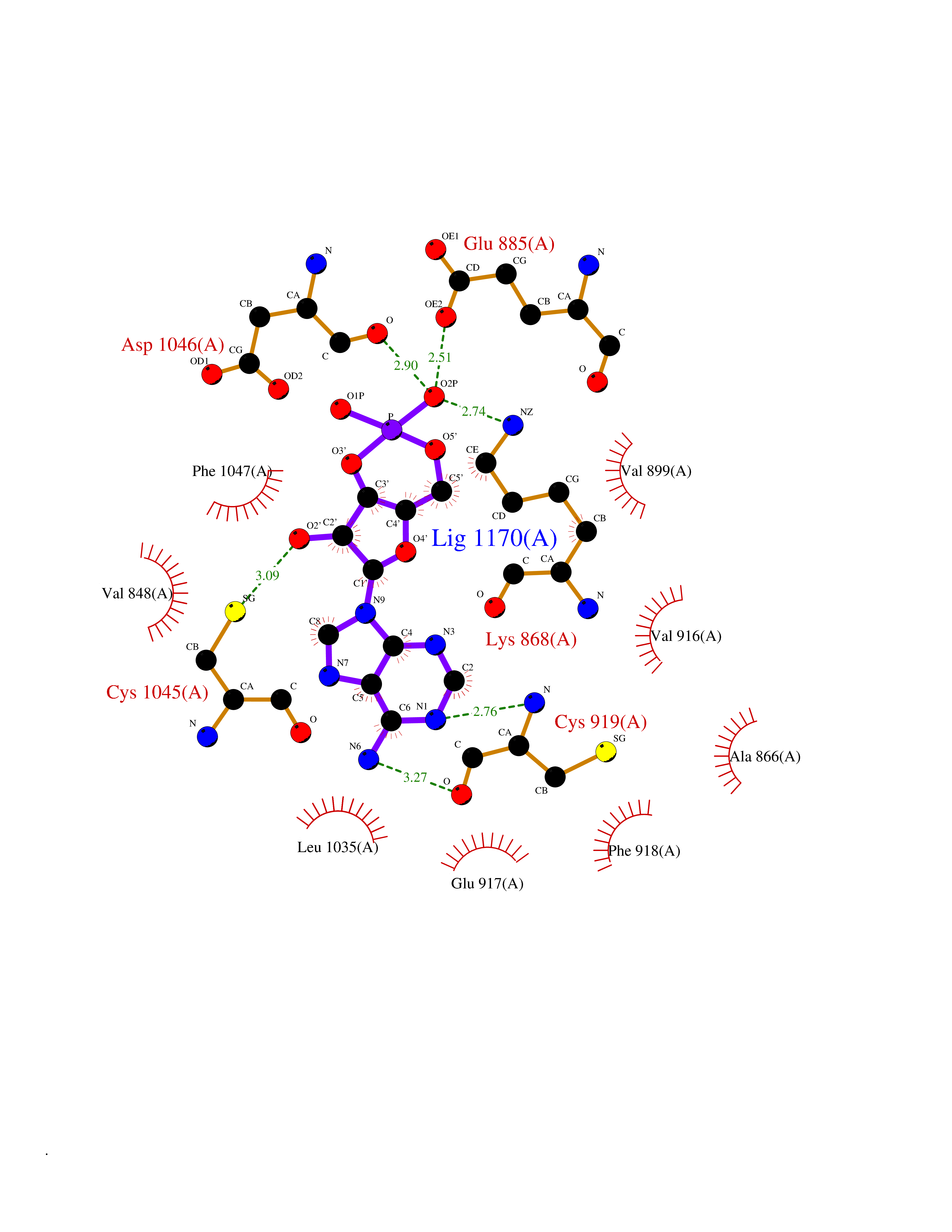 Ligplot Map 3D Binding mode Sequence HCERLPYDASKWEFPRDRLKLGKPLGRGAFGQVIEADAFGIDKTATCRTVAVKMLKEGATHSEHRALMSELKILIHIGHHLNVVNLLGACTKPGGPLMVIVEFCKFGNLSTYLRSKRNEFVPYYKDFLTLEHLICYSFQVAKGMEFLASRKCIHRDLAARNILLSEKNVVKICDFGLARDIYKDPDYVRKGDARLPLKWMAPETIFDRVYTIQSDVWSFGVLLWEIFSLGASPYPGVKIDEEFCRRLKEGTRMRAPDYTTPEMYQTMLDCWHGEPSQRPTFSELVEHLGNLLQANA Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | NIMA-related kinase 7 (NEK7) | 2WQN | 6.57 | |
Target general information Gen name NEK7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein kinase Nek7; NimA-related protein kinase 7; Never in mitosis A-related kinase 7 Protein family Protein kinase superfamily, NEK Ser/Thr protein kinase family, NIMA subfamily Biochemical class NA Function Protein kinase which plays an important role in mitotic cell cycle progression. Required for microtubule nucleation activity of the centrosome, robust mitotic spindle formation and cytokinesis. Phosphorylates RPS6KB1. Related diseases Bone marrow failure and diabetes mellitus syndrome (BMFDMS) [MIM:620044]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFDMS is an autosomal recessive form characterized by various degrees of bone marrow failure, ranging from dyserythropoiesis to bone marrow aplasia, with onset in infancy or early childhood, and non-autoimmune insulin-dependent diabetes mellitus appearing in the first or second decades. Many patients show pigmentary skin abnormalities and short stature. {ECO:0000269|PubMed:28073829, ECO:0000269|PubMed:35611808, ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q8N1W1-4; Q8TAB5; O94983-5; O75175; Q6UXH1-2; Q96D03; P49184; Q8IY82; Q99944; P16422; Q9NVM1; Q96GK7; Q10981; P36382; P09471; P79483; P14060; Q7Z6Z7-2; O75874; Q9ULR0; A0JP07; P26715; Q6P5S2; P09382; O00214-2; P43356; Q7Z434; Q8NCR3; Q8TB02; Q96EY8; A2RUH7; Q9NPC7; Q9HC98-4; Q8NCF5-2; Q96P20; Q8N323; Q6P4D5-2; P00558; Q8N2W9; Q8NBT0; O14829; Q96QH2; Q9Y617; P43686; Q13200; Q9Y3Y4; Q13636; Q96E17; P61224; P50749; Q96I25; P52756; Q02978; Q3SY56; Q96L03; Q7Z698; F6Y2X3; Q13428-5; P48775; Q9BT49; P49746; P42166; Q96JJ7-2; Q9H2S6-2; Q68CL5-3; Q9ULW0; Q9NX07; P07437; Q7Z780; O75317; Q96EF9; Q9BQ29; Q96P20-1 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cytoplasm; Cytoskeleton; Kinase; Magnesium; Metal-binding; Microtubule; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 30018.6 Length 259 Aromaticity 0.1 Instability index 61.02 Isoelectric point 8.34 Charge (pH=7) 3.44 2D Binding mode Binding energy (Kcal/mol) -7.11 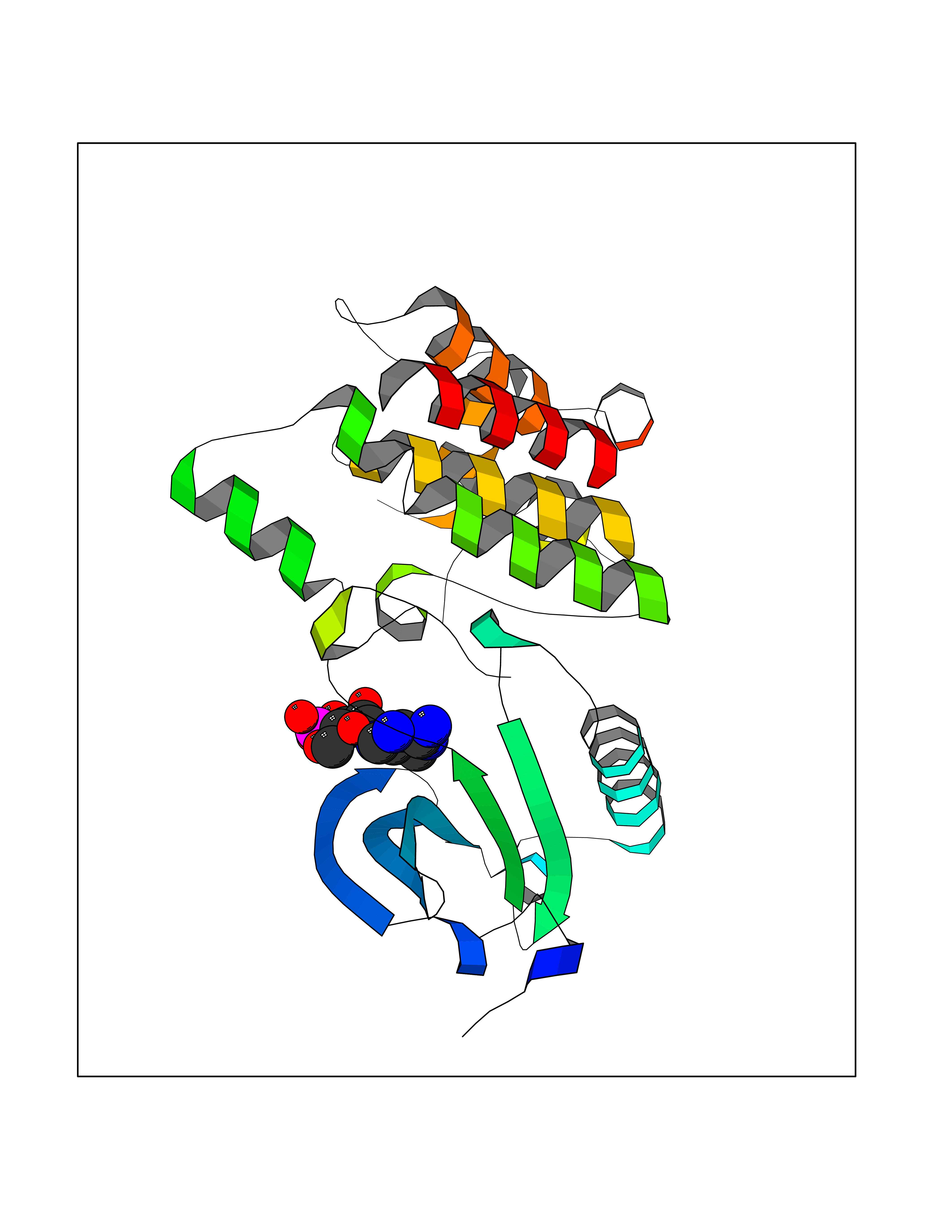 Molscript Map 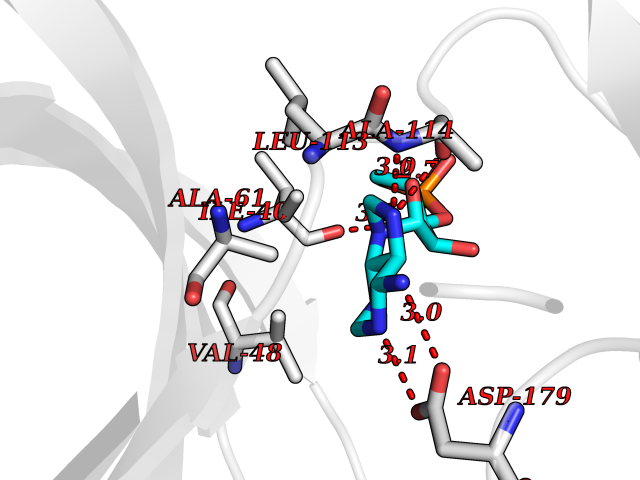 Pymol Map 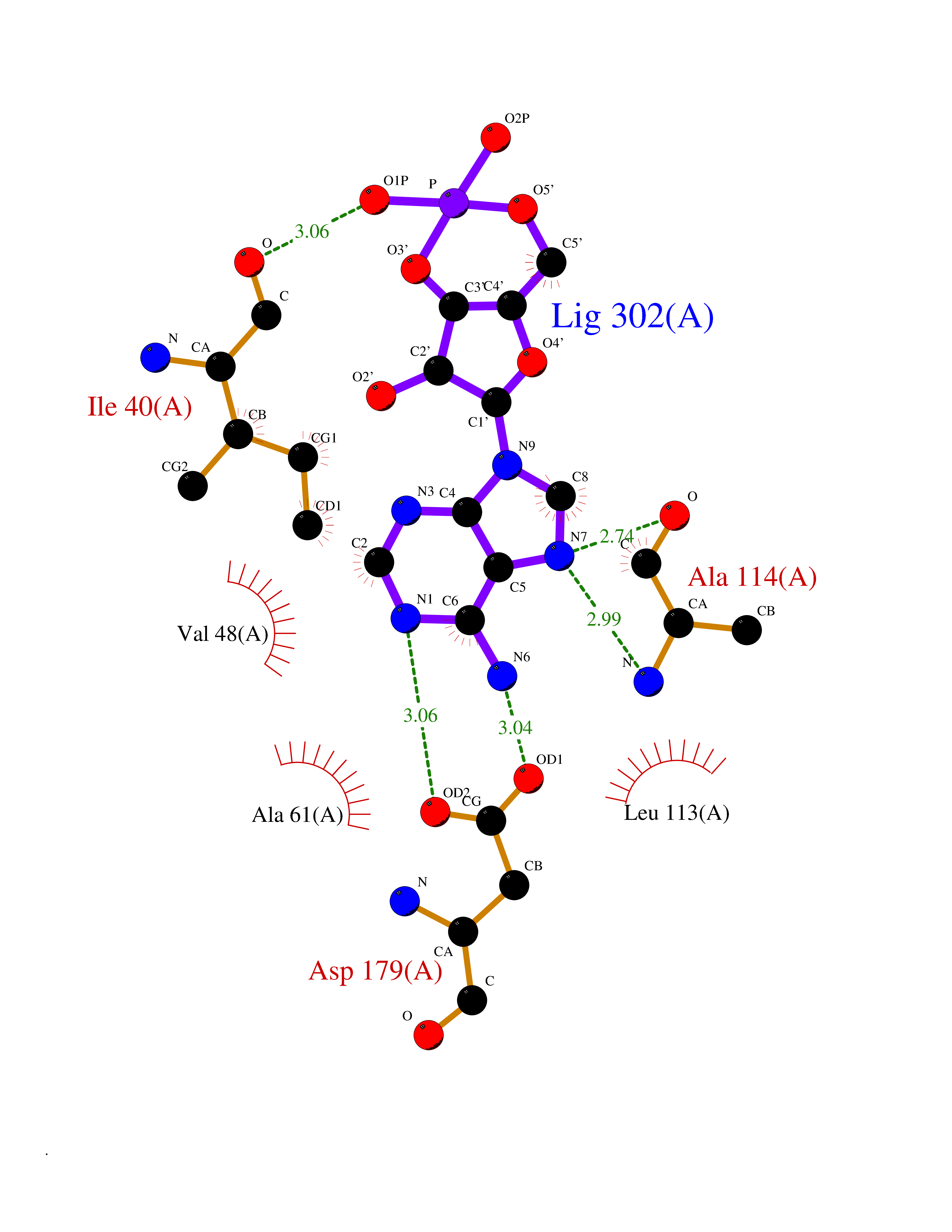 Ligplot Map 3D Binding mode Sequence ALRPDMGYNTLANFRIEKKIGRGQFSEVYRAACLLDGVPVALKKVQIFDLMDAKARADCIKEIDLLKQLNHPNVIKYYASFIEDNELNIVLELADAGDLSRMIKHFKKQKRLIPERTVWKYFVQLCSALEHMHSRRVMHRDIKPANVFITATGVVKLGDLTPYYMSPERIHENGYNFKSDIWSLGCLLYEMAALQSPFYMNLYSLCKKIEQCDYPPLPSDHYSEELRQLVNMCINPDPEKRPDVTYVYDVAKRMHACTA Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Beta-klotho (KLB) | 5VAN | 6.56 | |
Target general information Gen name KLB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Klotho beta-like protein; BetaKlotho; BKL Protein family Glycosyl hydrolase 1 family, Klotho subfamily Biochemical class Glycosylase Function Probably inactive as a glycosidase. Increases the ability of FGFR1 and FGFR4 to bind FGF21. Contributes to the transcriptional repression of cholesterol 7-alpha-hydroxylase (CYP7A1), the rate-limiting enzyme in bile acid synthesis. Related diseases Charcot-Marie-Tooth disease, axonal, 2B (CMT2B) [MIM:600882]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:12545426, ECO:0000269|PubMed:15455439, ECO:0000269|PubMed:17060578, ECO:0000269|PubMed:20028791, ECO:0000269|PubMed:21151572, ECO:0000269|PubMed:23179371}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NSA1 EC number NA Uniprot keywords 3D-structure; Cell membrane; Glycoprotein; Membrane; Proteomics identification; Reference proteome; Repeat; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 99301.9 Length 861 Aromaticity 0.14 Instability index 32.93 Isoelectric point 8.85 Charge (pH=7) 12.12 2D Binding mode Binding energy (Kcal/mol) -5.92  Molscript Map 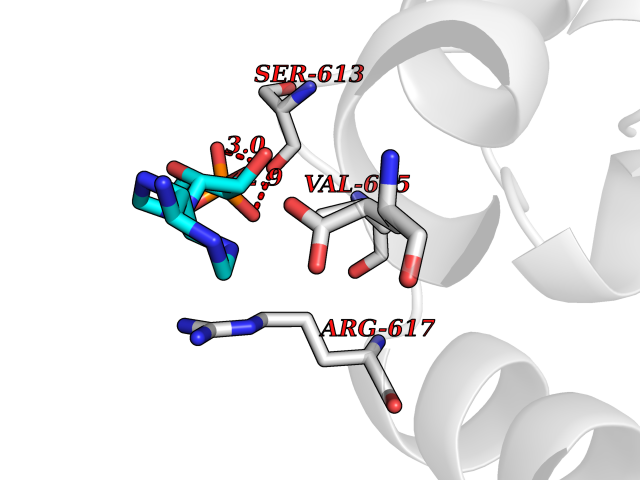 Pymol Map 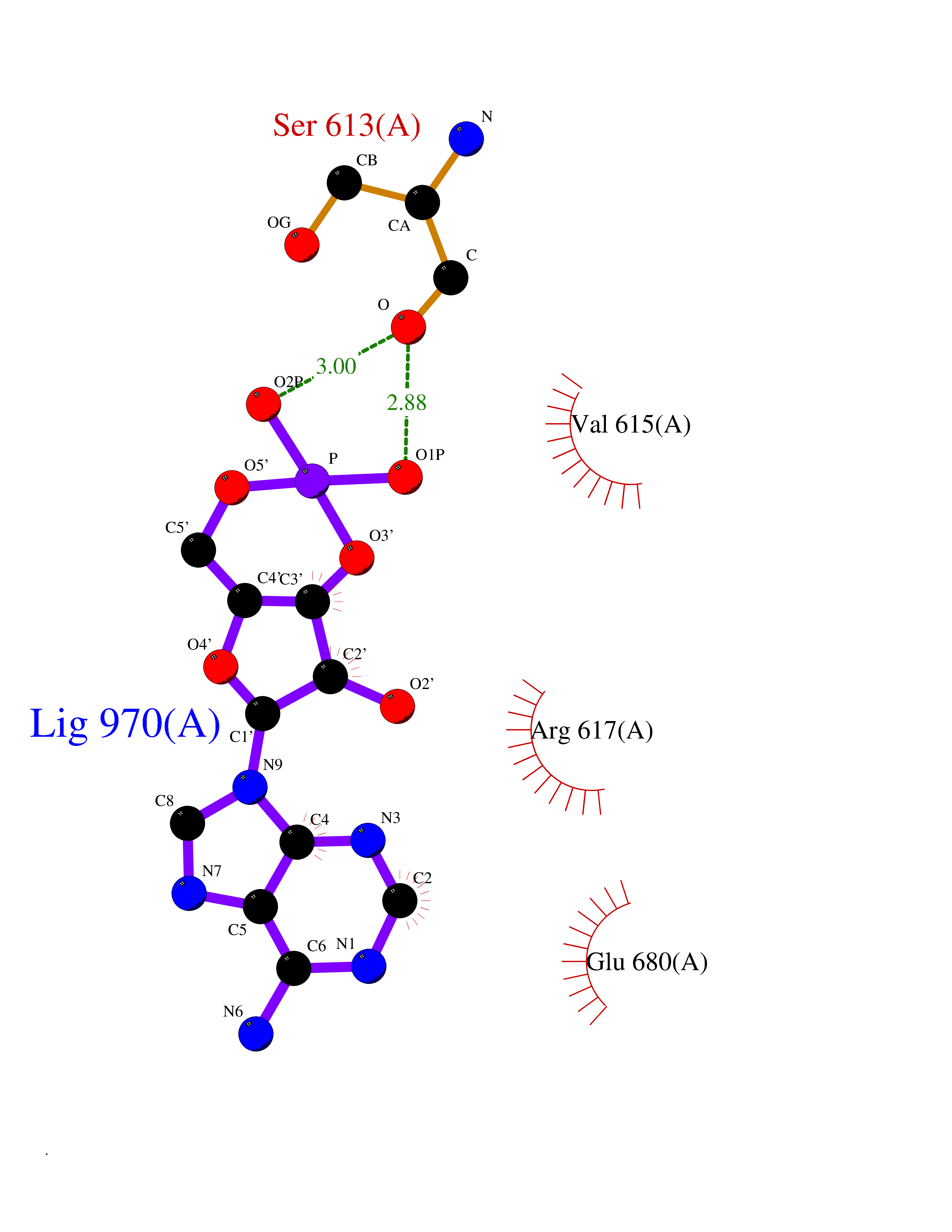 Ligplot Map 3D Binding mode Sequence FSGDGRAIWSQLFLYDTFPKNFFWGIGTGALQVEGSWKKDGKGPSIWDHFIHTHLGSSDSYIFLEKDLSALDFIGVSFYQFSISWPRLFPDGIVTVANAKGLQYYSTLLDALVLRNIEPIVTLYHWDLPLALQEKYGGWKNDTIIDIFNDYATYCFQMFGDRVKYWITIHNPYLVAWHGYGTGMHAPGEKGNLAAVYTVGHNLIKAHSKVWHNYNTHFRPHQKGWLSITLGSHWIEPQRSENTMDIFKCQQSMVSVLGWFANPIHGDGDYPEGMRKKLFSVLPIFSEAEKHEMRGTADFFAFSFGPNNFKPLNTMAKMGQNVSLNLREALNWIKLEYNNPRILIAENGWFTDSRVKTEDTTAIYMMKNFLSQVLQAIRLDEIRVFGYTAWSLLDGFEWQDAYTIRRGLFYVDFNSKQKERKPKSSAHYYKQIIRENGFSLKESTPDVQGQFPCDFSWGVTESVLKPEQCTDFVNIKKQLEMLARMKVTHYRFALDWASVLPTGQLSAVNRQALRYYRCVVSEGLKLGISAMVTLYYPTHAHLGLPEPLLHADGWLNPSTAEAFQAYAGLCFQELGDLVKLWITINEPNRLSDIYNRSGNDTYGAAHNLLVAHALAWRLYDRQFRPSQRGAVSLSLHADWAEPANPYADSHWRAAERFLQFEIAWFAEPLFKTGDYPAAMREYIASKHRRGLSSSALPRLTEAERRLLKGTVDFCALNHFTTRFVMHEQLAGSRYDSDRDIQFLQDITRLSSPTRLAVIPWGVRKLLRWVRRNYGDMDIYITASGIDDQALEDDRLRKYYLGKYLQEVLKAYLIDKVRIKGYYAFKLAEEKSKPRFGFFTSDFKAKSSIQFYNKVISSRGFP Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Probable glutathione peroxidase 8 | 3KIJ | 6.56 | |
Target general information Gen name GPX8 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms UNQ847/PRO1785 Protein family Glutathione peroxidase family Biochemical class Oxidoreductase Function Glutathione peroxidase activity.Peroxidase activity. Related diseases Neurodevelopmental disorder with spastic paraplegia and microcephaly (NEDSPM) [MIM:616281]: An autosomal recessive syndrome characterized by severe psychomotor developmental delay, dysarthria, walking difficulties, moderately to severely impaired intellectual development, poor or absent speech, and progressive microcephaly. {ECO:0000269|PubMed:25758935}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09096; DB00143; DB03310 Interacts with Q6RW13-2; Q9NVV5-2; Q9BVK2; P02656; P05090; P29972; P41181; Q92482; P07306; Q12797-6; Q92843; O15155; Q13323; O95393; Q12982; Q8WVV5; Q06432; Q8IX05; P19397; P60033; O14735; O95674; Q9BXR6; O43916; Q8N6F1-2; Q8NC01; Q6UVW9; Q96DZ9-2; O95406; Q8TBE1; P29400-2; Q4VAQ0; Q9Y5Q5; P49447; Q8NBI2; O14569; Q96LL9; Q9UPQ8; P56851; Q9UKR5; Q7L5A8; Q92520; Q96IV6; O14556; O43681; O14653; Q8TDV0; O60883; Q7Z429; P02724; Q9HCP6; P30519; P24593; Q9Y5U4; P43628; Q96E93; Q86VI4; O95214; Q8TAF8; Q9UIQ6-2; Q9UBY5; Q9Y2E5; Q9P0N8; Q9NX47; Q6N075; Q6ZSS7; Q99735; O14880; P30301; Q15546; A6NDP7; Q99519; Q92982; Q6P499; Q16617; Q8N912; Q8IXM6; Q16625; P09466; Q9NXK6; Q6TCH4; Q9UHJ9-5; Q9Y342; P26678; Q04941; Q5VZY2; Q8IY26; P54315; A5D903; Q8N0V3; Q92730; Q5QGT7; Q14108; Q14162; O00767; O75396; Q9Y6D0; Q9NRX5; Q9Y6X1; A2A2V5; P11686; Q8NHU3; Q8WWT9; Q99726; Q8N130; P78382; Q969S0; Q96JW4; Q0VAQ4; Q6UX34; Q96JF0-2; Q13277; Q9UNK0; Q9BQS2-2; P02787; P07204; Q9NPL8; P48230; P55061; Q6UX40; Q9BVC6; A0PK00; Q5BJH2-2; Q9NUH8; Q96HH6; A2RU14; Q8NBD8; Q9BU79; Q8TBM7; Q69YG0; P56557; Q9H2L4; Q8N661; Q5BJF2; Q9NRS4; Q71RG4; Q8N609; Q86UF1; Q53HI1; O75841; Q15836; O75379; O95183; Q8N0U8; Q6UX27-3; Q9UEU0; O95070; Q96EC8; Q8N966 EC number 1.11.1.9 Uniprot keywords 3D-structure; Acetylation; Membrane; Oxidoreductase; Peroxidase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 18580.2 Length 161 Aromaticity 0.13 Instability index 38.6 Isoelectric point 9.39 Charge (pH=7) 6.73 2D Binding mode Binding energy (Kcal/mol) -6.31  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SFYAFEVKDAKGRTVSLEKYKGKVSLVVNVASDCQLTDRNYLGLKELHKEFGPSHFSVLAFPCNQFGESEPRPSKEVESFARKNYGVTFPIFHKIKILGSEGEPAFRFLVDSSKKEPRWNFWKYLVNPEGQVVKFWRPEEPIEVIRPDIAALVRQVIIKKK Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Multidrug resistance protein 3 (ABCB4) | 6S7P | 6.55 | |
Target general information Gen name ABCB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PGY3; MDR3; ATP-binding cassette sub-family B member 4; ABCB4 Protein family ABC transporter superfamily, ABCB family, Multidrug resistance exporter (TC 3.A.1.201) subfamily Biochemical class Acid anhydrides hydrolase Function Mediates ATP-dependent export of organic anions and drugs from the cytoplasm. Hydrolyzes ATP with low efficiency. Not capable of conferring drug resistance. Mediates the translocation of phosphatidylcholine across the canalicular membrane of the hepatocyte. Related diseases Cholestasis, progressive familial intrahepatic, 3 (PFIC3) [MIM:602347]: A disorder characterized by early onset of cholestasis that progresses to hepatic fibrosis, cirrhosis, and end-stage liver disease before adulthood. PFIC3 inheritance is autosomal recessive. {ECO:0000269|PubMed:11313315, ECO:0000269|PubMed:12671900, ECO:0000269|PubMed:17726488, ECO:0000269|PubMed:21119540, ECO:0000269|PubMed:24045840, ECO:0000269|PubMed:24594635, ECO:0000269|PubMed:24806754, ECO:0000269|PubMed:28012258, ECO:0000269|PubMed:9419367}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cholestasis of pregnancy, intrahepatic 3 (ICP3) [MIM:614972]: A liver disorder of pregnancy. It presents during the second or, more commonly, the third trimester of pregnancy with intense pruritus which becomes more severe with advancing gestation and cholestasis. It causes fetal distress, spontaneous premature delivery and intrauterine death. Patients have spontaneous and progressive disappearance of cholestasis after delivery. Cholestasis results from abnormal biliary transport from the liver into the small intestine. {ECO:0000269|PubMed:10767346, ECO:0000269|PubMed:12746424, ECO:0000269|PubMed:15077010}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gallbladder disease 1 (GBD1) [MIM:600803]: One of the major digestive diseases. Gallstones composed of cholesterol (cholelithiasis) are the common manifestations in western countries. Most people with gallstones, however, remain asymptomatic through their lifetimes. {ECO:0000269|PubMed:11313316, ECO:0000269|PubMed:12891548, ECO:0000269|PubMed:22331132, ECO:0000269|PubMed:23533021, ECO:0000269|PubMed:24723470, ECO:0000269|PubMed:28012258, ECO:0000269|PubMed:28587926, ECO:0000269|Ref.2}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06414; DB06207 Interacts with NA EC number EC 7.6.2.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cytoplasm; Cytoplasmic vesicle; Disease variant; Glycoprotein; Intrahepatic cholestasis; Lipid transport; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 123919 Length 1128 Aromaticity 0.1 Instability index 29.44 Isoelectric point 8.78 Charge (pH=7) 11.08 2D Binding mode Binding energy (Kcal/mol) -7.54  Molscript Map  Pymol Map 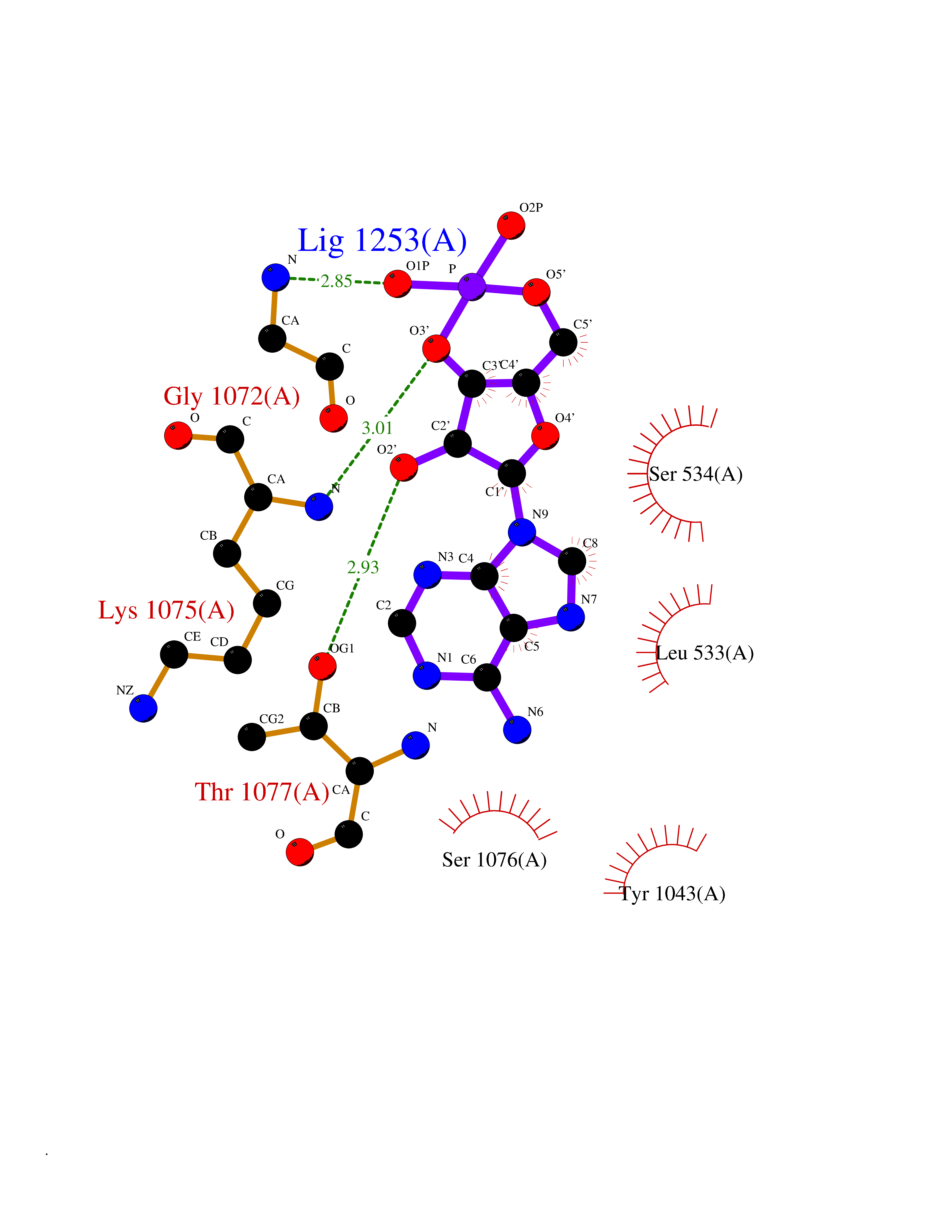 Ligplot Map 3D Binding mode Sequence VLTLFRYSDWQDKLFMSLGTIMAIAHGSGLPLMMIVFGEMTDKPGKILEEEMTRYAYYYSGLGAGVLVAAYIQVSFWTLAAGRQIRKIRQKFFHAILRQEIGWFDINDTTELNTRLTDDISKISEGIGDKVGMFFQAVATFFAGFIVGFIRGWKLTLVIMAISPILGLSAAVWAKILSAFSDKELAAYAKAGAVAEEALGAIRTVIAFGGQNKELERYQKHLENAKEIGIKKAISANISMGIAFLLIYASYALAFWYGSTLVISKEYTIGNAMTVFFSILIGAFSVGQAAPCIDAFANARGAAYVIFDIIDNNPKIDSFSERGHKPDSIKGNLEFNDVHFSYPSRANVKILKGLNLKVQSGQTVALVGSSGCGKSTTVQLIQRLYDPDEGTINIDGQDIRNFNVNYLREIIGVVSQEPVLFSTTIAENICYGRGNVTMDEIKKAVKEANAYEFIMKLPQKFDTLVGERGAQLSGGQKQRIAIARALVRNPKILLLDQATSALDTESEAEVQAALDKAREGRTTIVIAHRLSTVRNADVIAGFEDGVIVEQGSHSELMKKEGVYFKLVNVPPVSFLKVLKLNKTEWPYFVVGTVCAIANGGLQPAFSVIFSEIIAIFGPGDDAVKQQKCNIFSLIFLFLGIISFFTFFLQGFTFGKAGEILTRRLRSMAFKAMLRQDMSWFDDHKNSTGALSTRLATDAAQVQGATGTRLALIAQNIANLGTGIIISFIYGWQLTLLLLAVVPIIAVSGIVEMKLLAGNAKRDKKELEAAGKIATEAIENIRTVVSLTQERKFESMYVEKLYGPYRNSVQKAHIYGITFSISQAFMYFSYAGCFRFGAYLIVNGHMRFRDVILVFSAIVFGAVALGHASSFAPDYAKAKLSAAHLFMLFERQPLIDSYSEEGLKPDKFEGNITFNEVVFNYPTRANVPVLQGLSLEVKKGQTLALVGSSGCGKSTVVQLLERFYDPLAGTVLLDGQEAKKLNVQWLRAQLGIVSQEPILFDCSIAENIAYGDNSRVVSQDEIVSAAKAANIHPFIETLPHKYETRVGDKGTQLSGGQKQRIAIARALIRQPQILLLDQATSALDTESEKVVQEALDKAREGRTCIVIAHRLSTIQNADLIVVFQNGRVK Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Bifunctional aminoacyl-tRNA synthetase (EPRS) | 4HVC | 6.55 | |
Target general information Gen name EPRS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms QPRS; QARS; ProlinetRNA ligase; PIG32; GlutamatylprolyltRNA synthetase; Glutamatyl-prolyl-tRNA synthetase; GluRS; EPRS; Cell proliferationinducing gene 32 protein; Cell proliferation-inducing gene 32 Protein family Class-I aminoacyl-tRNA synthetase family, Glutamate--tRNA ligase type 2 subfamily; Class-II aminoacyl-tRNA synthetase family Biochemical class Carbon-oxygen ligase Function The phosphorylation of EPRS, induced by interferon-gamma, dissociates the protein from the aminoacyl-tRNA synthetase multienzyme complex and recruits it to the GAIT complex that binds to stem loop-containing GAIT elements in the 3'-UTR of diverse inflammatory mRNAs (such as ceruplasmin), suppressing their translation. Interferon-gamma can therefore redirect, in specific cells, the EPRS function from protein synthesis to translation inhibition. Also functions as an effector of the mTORC1 signaling pathway by promoting, through SLC27A1, the uptake of long-chain fatty acid by adipocytes. Thereby, it also plays a role in fat metabolism and more indirectly influences lifespan. Multifunctional protein which is primarily part of the aminoacyl-tRNA synthetase multienzyme complex, also know as multisynthetase complex, that catalyzes the attachment of the cognate amino acid to the corresponding tRNA in a two-step reaction: the amino acid is first activated by ATP to form a covalent intermediate with AMP and is then transferred to the acceptor end of the cognate tRNA. Related diseases Leukodystrophy, hypomyelinating, 15 (HLD15) [MIM:617951]: An autosomal recessive disorder characterized by hypomyelinating leukodystrophy with thinning of the corpus callosum. Clinical features include motor and cognitive impairment appearing in the first or second decade of life, dystonia, ataxia, spasticity, and dysphagia. Most patients develop severe optic atrophy, and some have hearing loss. {ECO:0000269|PubMed:29576217}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02684; DB02510; DB03376; DB00142; DB00172 Interacts with P07814; Q8IWL3; P41252; Q15046; P42695; P54136; O60506 EC number NA Uniprot keywords 3D-structure; Acetylation; Aminoacyl-tRNA synthetase; ATP-binding; Cytoplasm; Disease variant; Leukodystrophy; Ligase; Membrane; Metal-binding; Methylation; Multifunctional enzyme; Neurodegeneration; Nucleotide-binding; Phosphoprotein; Protein biosynthesis; Proteomics identification; Reference proteome; Repeat; RNA-binding; Translation regulation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 55039.7 Length 483 Aromaticity 0.1 Instability index 39.15 Isoelectric point 5.92 Charge (pH=7) -6.4 2D Binding mode Binding energy (Kcal/mol) -7.71  Molscript Map 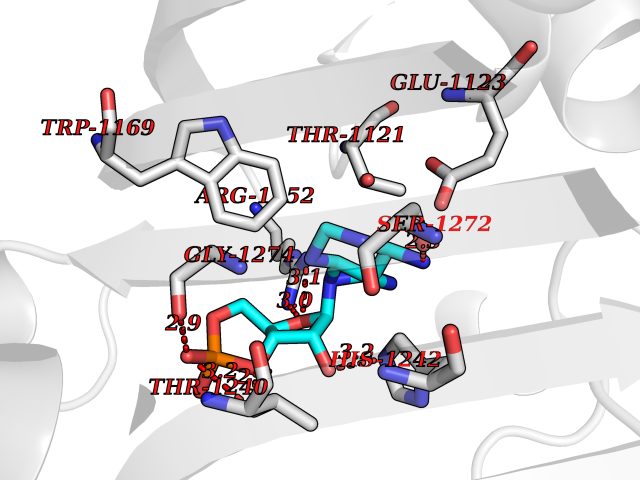 Pymol Map 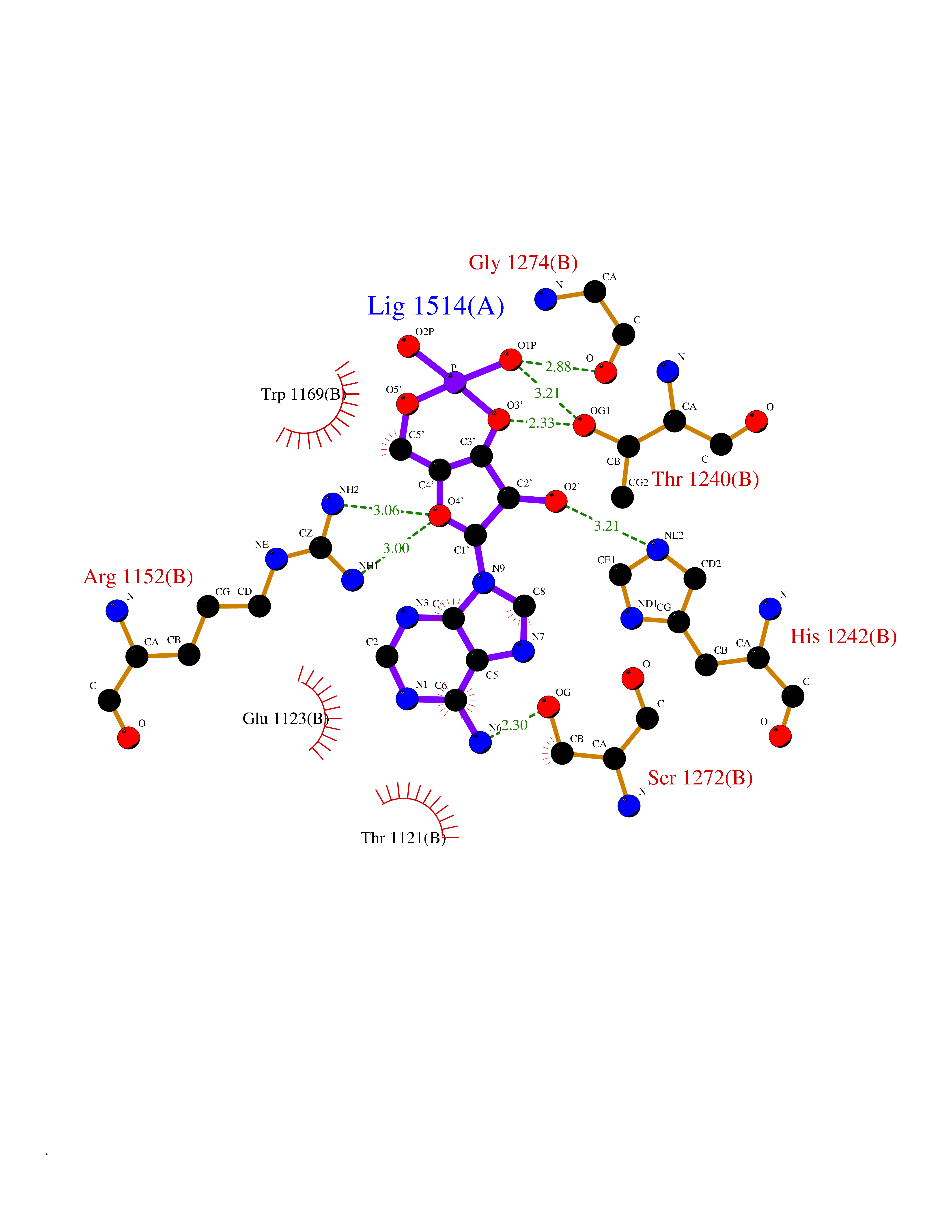 Ligplot Map 3D Binding mode Sequence GLEAKKEENLADWYSQVITKSEMIEYHDISGCYILRPWAYAIWEAIKDFFDAEIKKLGVENCYFPMFVSQSALEKEKTHVADFAPEVAWVTRSGKTELAEPIAIRPTSETVMYPAYAKWVQSHRDLPIKLNQWCNVVRWEFKHPQPFLRTREFLWQEGHSAFATMEEAAEEVLQILDLYAQVYEELLAIPVVKGRKTEKEKFAGGDYTTTIEAFISASGRAIQGGTSHHLGQNFSKMFEIVFEDPKIPGEKQFAYQNSWGLTTRTIGVMTMVHGDNMGLVLPPRVACVQVVIIPCGISEEDKEALIAKCNDYRRRLLSVNIRVRADLRDNYSPGWKFNHWELKGVPIRLEVGPRDMKSCQFVAVRRDTGEKLTVAENEAETKLQAILEDIQVTLFTRASEDLKTHMVVANTMEDFQKILDSGKIVQIPFCGEIDCEDWIKKTTAMGAKSLCIPFKPLCELQPGAKCVCGKNPAKYYTLFGRSY Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Soluble epoxide hydrolase (EPHX2) | 1ZD3 | 6.54 | |
Target general information Gen name EPHX2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Bifunctional epoxide hydrolase 2 Protein family AB hydrolase superfamily, Epoxide hydrolase family Biochemical class Ether bond hydrolase Function Bifunctional enzyme. The C-terminal domain has epoxide hydrolase activity and acts on epoxides (alkene oxides, oxiranes) and arene oxides. Plays a role in xenobiotic metabolism by degrading potentially toxic epoxides (By similarity). Also determines steady-state levels of physiological mediators. The N-terminal domain has lipid phosphatase activity, with the highest activity towards threo-9,10-phosphonooxy-hydroxy-octadecanoic acid, followed by erythro-9,10-phosphonooxy-hydroxy-octadecanoic acid, 12-phosphonooxy-octadec-9Z-enoic acid and 12-phosphonooxy-octadec-9E-enoic acid. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08257; DB08258; DB08259; DB06345; DB12610; DB08256; DB02029; DB04213; DB03677 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aromatic hydrocarbons catabolism; Cytoplasm; Detoxification; Direct protein sequencing; Hydrolase; Lipid metabolism; Lipoprotein; Magnesium; Metal-binding; Multifunctional enzyme; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 61744.9 Length 547 Aromaticity 0.09 Instability index 43.97 Isoelectric point 5.81 Charge (pH=7) -7.76 2D Binding mode Binding energy (Kcal/mol) -7.86  Molscript Map 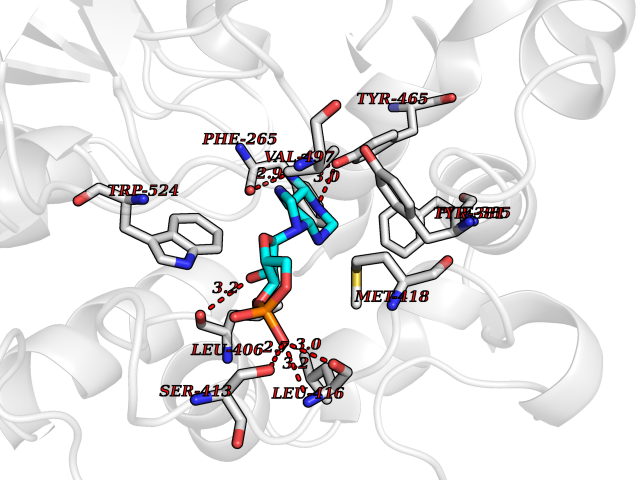 Pymol Map 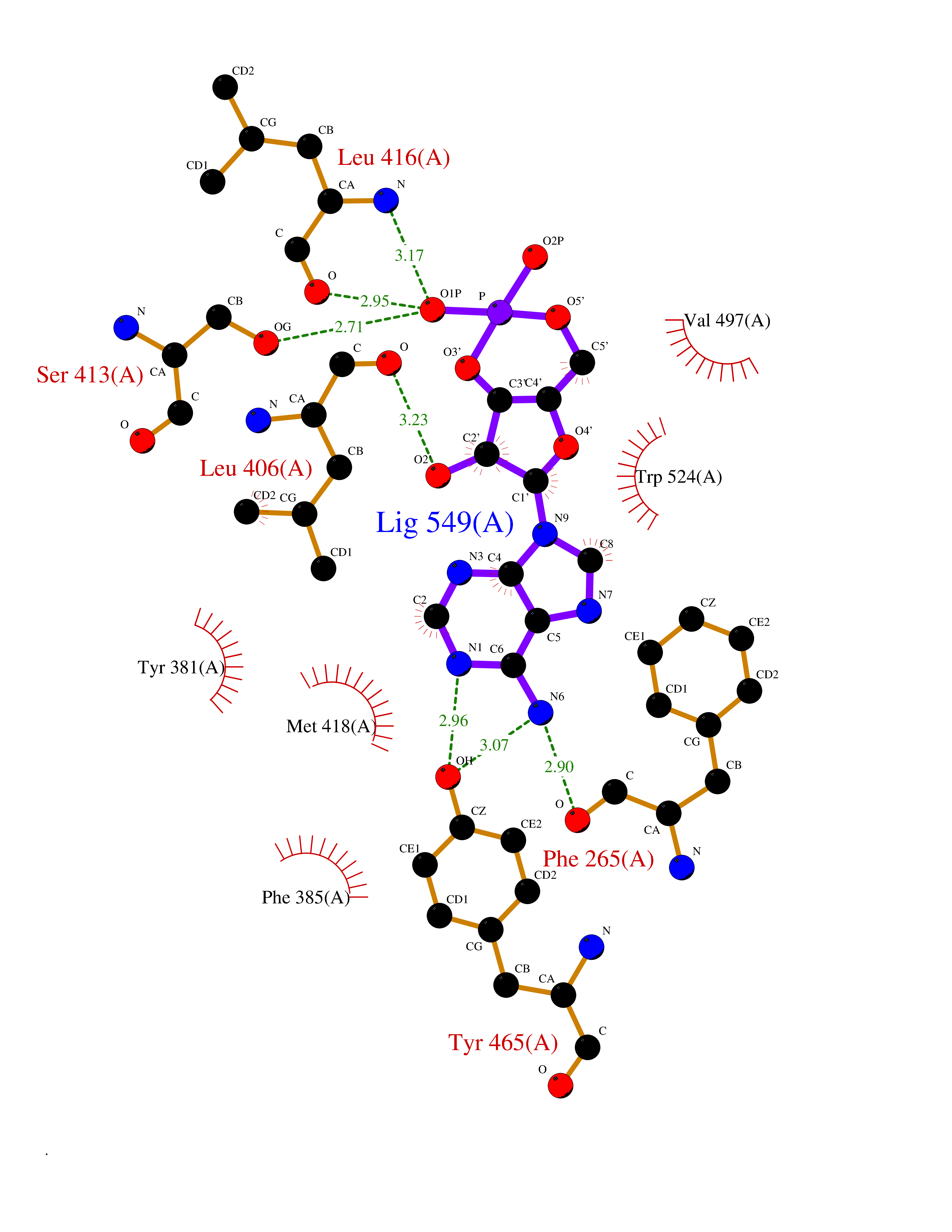 Ligplot Map 3D Binding mode Sequence TLRAAVFDLDGVLALPAVFGVLGRTEEALALPRGLLNDAFQKGGPEGATTRLMKGEITLSQWIPLMEENCRKCSETAKVCLPKNFSIKEIFDKAISARKINRPMLQAALMLRKKGFTTAILTNTWLDDRAERDGLAQLMCELKMHFDFLIESCQVGMVKPEPQIYKFLLDTLKASPSEVVFLDDIGANLKPARDLGMVTILVQDTDTALKELEKVTGIQLLNTPAPLPTSCNPSDMSHGYVTVKPRVRLHFVELGSGPAVCLCHGFPESWYSWRYQIPALAQAGYRVLAMDMKGYGESSAPPEIEEYCMEVLCKEMVTFLDKLGLSQAVFIGHDWGGMLVWYMALFYPERVRAVASLNTPFIPANPNMSPLESIKANPVFDYQLYFQEPGVAEAELEQNLSRTFKSLFRASDESVLSMHKVCEAGGLFVNSPEEPSLSRMVTEEEIQFYVQQFKKSGFRGPLNWYRNMERNWKWACKSLGRKILIPALMVTAEKDFVLVPQMSQHMEDWIPHLKRGHIEDCGHWTQMDKPTEVNQILIKWLDSDARN Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Alanine--tRNA ligase, cytoplasmic | 4XEM | 6.54 | |
Target general information Gen name AARS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AARS Protein family Class-II aminoacyl-tRNA synthetase family Biochemical class Ligase Function Alanine-tRNA ligase activity.Amino acid binding.Aminoacyl-tRNA editing activity.ATP binding.Metal ion binding.TRNA binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2N (CMT2N) [MIM:613287]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:20045102, ECO:0000269|PubMed:22009580, ECO:0000269|PubMed:22206013, ECO:0000269|PubMed:35911843, ECO:0000269|PubMed:35971119}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 29 (DEE29) [MIM:616339]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE29 patients manifest severe infantile epileptic encephalopathy, clubfoot, absent deep tendon reflexes, extrapyramidal symptoms, and persistently deficient myelination. {ECO:0000269|PubMed:25817015, ECO:0000269|PubMed:28493438}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukoencephalopathy, hereditary diffuse, with spheroids 2 (HDLS2) [MIM:619661]: An autosomal dominant neurodegenerative disorder characterized by progressive cognitive and executive dysfunction, psychiatric disturbances, and neurologic symptoms, such as gait abnormalities, paresis, seizures, and rigidity. Symptom onset is usually in adulthood, although earlier onset has been reported. Some patients have an acute encephalopathic course with severe neurologic decline resulting in early death, whereas other patients have a more protracted and chronic disease course. Neuropathologic examination shows a leukoencephalopathy with axonal spheroids and myelination defects. {ECO:0000269|PubMed:31775912, ECO:0000269|PubMed:37106376}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Trichothiodystrophy 8, non-photosensitive (TTD8) [MIM:619691]: A form of trichothiodystrophy, a disease characterized by sulfur-deficient brittle hair and multisystem variable abnormalities. The spectrum of clinical features varies from mild disease with only hair involvement to severe disease with cutaneous, neurologic and profound developmental defects. Ichthyosis, intellectual and developmental disabilities, decreased fertility, abnormal characteristics at birth, ocular abnormalities, short stature, and infections are common manifestations. There are both photosensitive and non-photosensitive forms of the disorder. TTD8 is an autosomal recessive, non-photosensitive form characterized by brittle hair and nails, scaly skin, accompanied by failure to thrive, microcephaly, and neuromotor developmental delay. {ECO:0000269|PubMed:33909043}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00160 Interacts with NA EC number 6.-.-.-; 6.1.1.7 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminoacyl-tRNA synthetase; ATP-binding; Charcot-Marie-Tooth disease; Cytoplasm; Direct protein sequencing; Disease variant; Epilepsy; Ligase; Metal-binding; Methylation; Neurodegeneration; Neuropathy; Nucleotide-binding; Nucleus; Phosphoprotein; Protein biosynthesis; Proteomics identification; Reference proteome; RNA-binding; tRNA-binding; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 42385.6 Length 377 Aromaticity 0.1 Instability index 27.33 Isoelectric point 5.15 Charge (pH=7) -13.8 2D Binding mode Binding energy (Kcal/mol) -7.75  Molscript Map 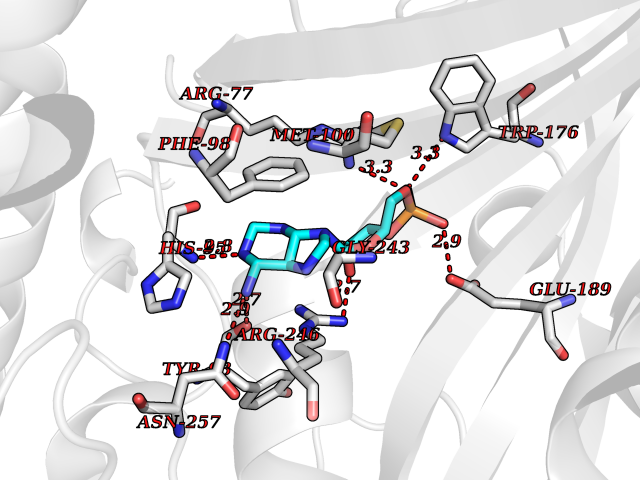 Pymol Map 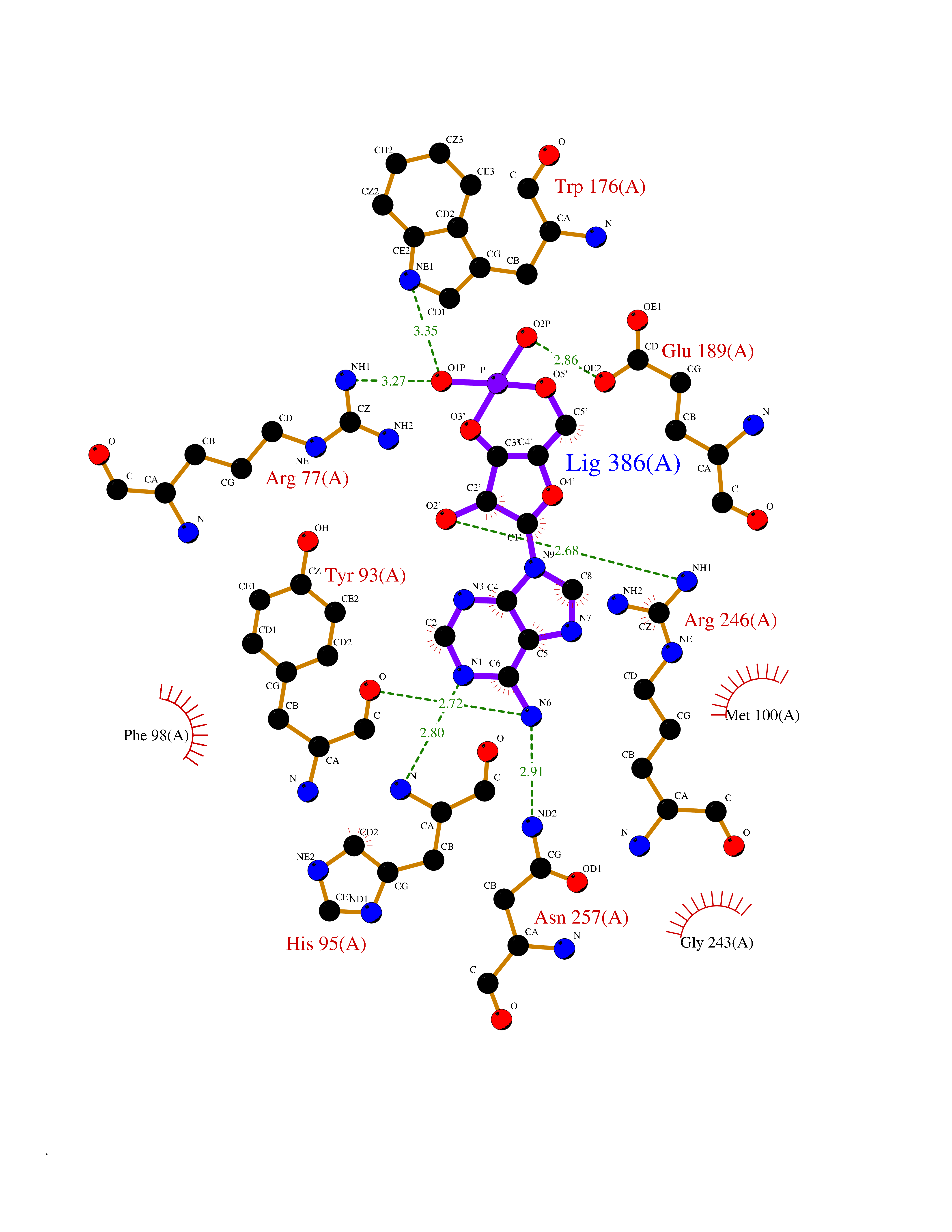 Ligplot Map 3D Binding mode Sequence TLTASEIRQRFIDFFKRNEHTYVHSSATIPLDDPTLLFANAGMNQFKPIFLNTIDPSHPMAKLSRAANTQKCIRAGDLDDVGKDVYHHTFFEMLGSWSFGDYFKELACKMALELLTQEFGIPIERLYVTYFGGDEAAGLEADLECKQIWQNLGLDDTKILPGNMKDNFWEMGDTGPCGPCSEIHYDRIGGRDAAHLVNQDDPNVLEIWNLVFIQYNREADGILKPLPKKSIDTGMGLERLVSVLQNKMSNYDTDLFVPYFEAIQKGTGARPYTGKVGAEDADGIDMAYRVLADHARTITVALADGGRPDNTGRGYVLRRILRRAVRYAHEKLNASRGFFATLVDVVVQSLGDAFPELKKDPDMVKDIINEEEVQFLK Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Prothrombin | 4UD9 | 6.54 | |
Target general information Gen name F2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase S1 family Biochemical class Hydrolase Function Calcium ion binding.Growth factor activity.Heparin binding.Lipopolysaccharide binding.Receptor binding.Serine-type endopeptidase activity.Thrombospondin receptor activity. Related diseases Factor II deficiency (FA2D) [MIM:613679]: A very rare blood coagulation disorder characterized by mucocutaneous bleeding symptoms. The severity of the bleeding manifestations correlates with blood factor II levels. {ECO:0000269|PubMed:1349838, ECO:0000269|PubMed:1354985, ECO:0000269|PubMed:1421398, ECO:0000269|PubMed:14962227, ECO:0000269|PubMed:2719946, ECO:0000269|PubMed:3242619, ECO:0000269|PubMed:3567158, ECO:0000269|PubMed:3771562, ECO:0000269|PubMed:3801671, ECO:0000269|PubMed:6405779, ECO:0000269|PubMed:7792730, ECO:0000269|PubMed:7865694}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Thrombophilia due to thrombin defect (THPH1) [MIM:188050]: A multifactorial disorder of hemostasis characterized by abnormal platelet aggregation in response to various agents and recurrent thrombi formation. {ECO:0000269|PubMed:2825773}. The disease is caused by variants affecting the gene represented in this entry. A common genetic variation in the 3-prime untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increased risk of venous thrombosis.; DISEASE: Pregnancy loss, recurrent, 2 (RPRGL2) [MIM:614390]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:11506076}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07211; DB07796; DB07016; DB07521; DB06850; DB07091; DB06845; DB07088; DB07131; DB07095; DB07515; DB07897; DB06878; DB06947; DB08624; DB06869; DB06929; DB07400; DB04771; DB04772; DB02287; DB07277; DB07550; DB07549; DB07548; DB07105; DB04722; DB07366; DB08254; DB01725; DB08062; DB07639; DB07461; DB07120; DB07190; DB07741; DB07353; DB07508; DB07809; DB08546; DB08061; DB07718; DB03136; DB02723; DB07440; DB07376; DB06861; DB06866; DB06865; DB03865; DB06841; DB07934; DB08422; DB07659; DB07660; DB07658; DB13151; DB00025; DB11166; DB00278; DB01766; DB07083; DB00006; DB00100; DB13152; DB09228; DB09130; DB03159; DB06911; DB06996; DB06919; DB07027; DB07133; DB07143; DB07005; DB06695; DB00055; DB01225; DB05714; DB12831; DB03847; DB07278; DB01767; DB06404; DB09332; DB00001; DB13998; DB04136; DB00170; DB06838; DB13999; DB06868; DB06942; DB06936; DB07165; DB07527; DB07522; DB07665; DB07946; DB06859; DB06853; DB06858; DB07279; DB08187; DB04591; DB07944; DB07128; DB12598; DB01123; DB04786; DB05777; DB04697; DB09109; DB14738; DB04898; DB01593; DB14487; DB08152 Interacts with P05067; P07204; Q846V4; PRO_0000032489 [P01008] EC number 3.4.21.5 Uniprot keywords 3D-structure; Acute phase; Blood coagulation; Calcium; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; Gamma-carboxyglutamic acid; Glycoprotein; Hemostasis; Hydrolase; Kringle; Pharmaceutical; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Thrombophilia; Zymogen Protein physicochemical properties Chain ID H Molecular weight (Da) 29321.6 Length 254 Aromaticity 0.1 Instability index 39.57 Isoelectric point 8.56 Charge (pH=7) 4.16 2D Binding mode Binding energy (Kcal/mol) -7.66  Molscript Map 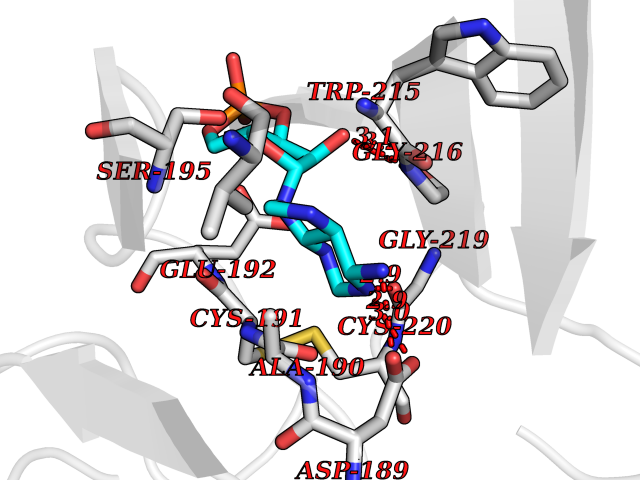 Pymol Map 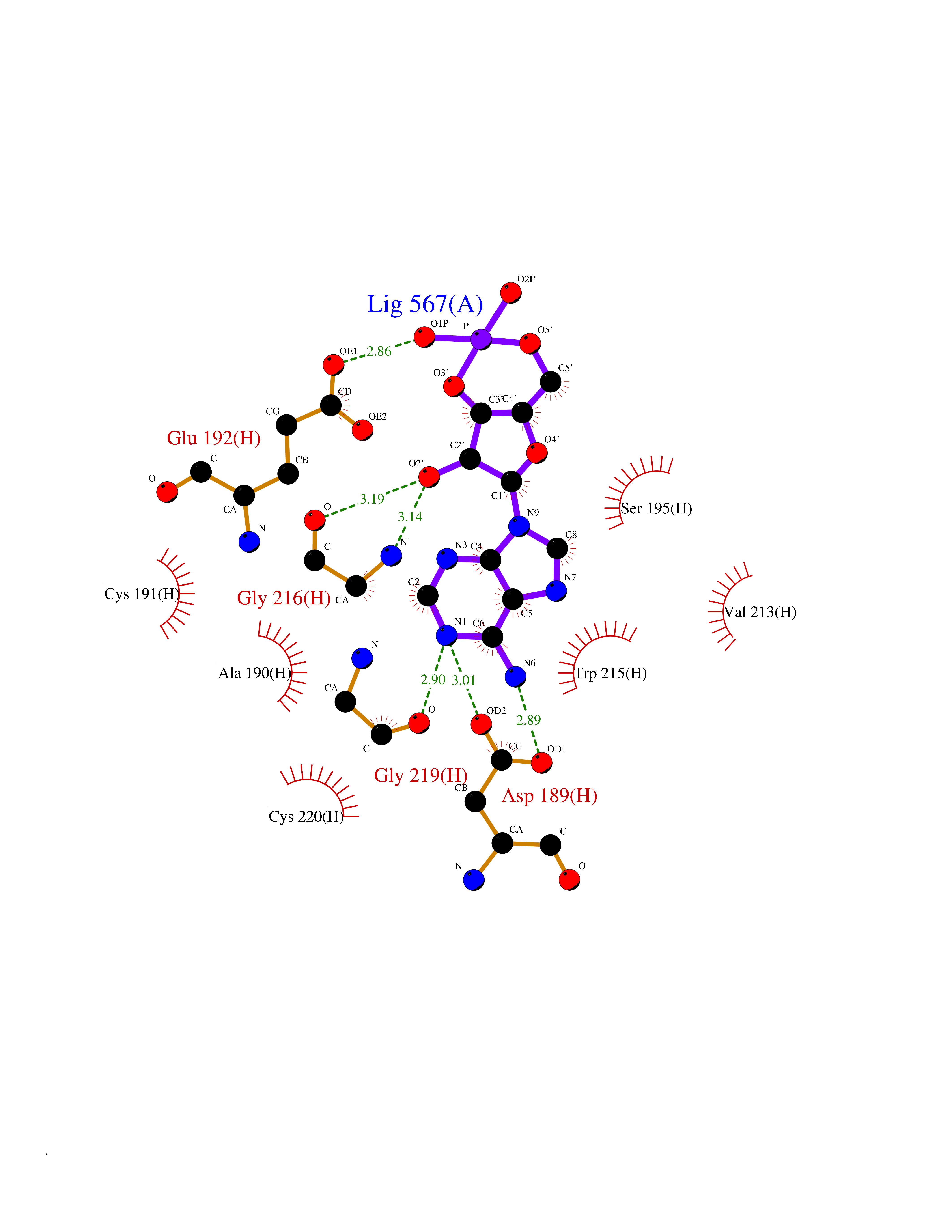 Ligplot Map 3D Binding mode Sequence IVEGSDAEIGMSPWQVMLFRSPQELLCGASLISDRWVLTAAHCLLTENDLLVRIGKHSRTRYRNIEKISMLEKIYIHPRYNWENLDRDIALMKLKKPVAFSDYIHPVCLPDRETLLQAGYKGRVTGWGNLKETWGQPSVLQVVNLPIVERPVCKDSTRIRITDNMFCAYKKRGDACEGDSGGPFVMKSNNRWYQMGIVSWGEGCRDGKYGFYTHVFRLKKWIQKVIDQFGGDFEEIPEELQCGLRPLFEKKSLE Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Non-heme chloroperoxidase | 1A8U | 6.54 | |
Target general information Gen name cpo Organism Kitasatospora aureofaciens (Streptomyces aureofaciens) Uniprot ID TTD ID NA Synonyms cpoT Protein family AB hydrolase superfamily, Bacterial non-heme haloperoxidase / perhydrolase family Biochemical class Haloperoxidase Function Chloride peroxidase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB03793 Interacts with NA EC number 1.11.1.- Uniprot keywords 3D-structure; Chloride; Oxidoreductase; Peroxidase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60428.4 Length 554 Aromaticity 0.13 Instability index 31.26 Isoelectric point 4.65 Charge (pH=7) -32.72 2D Binding mode Binding energy (Kcal/mol) -6.95  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAKPFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Oxalosuccinate decarboxylase (IDH1) | 6ADG | 6.53 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD; NADP(+)-specific ICDH; Isocitrate dehydrogenase [NADP] cytoplasmic; IDP; IDH; Cytosolic NADP-isocitrate dehydrogenase Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -7.61  Molscript Map 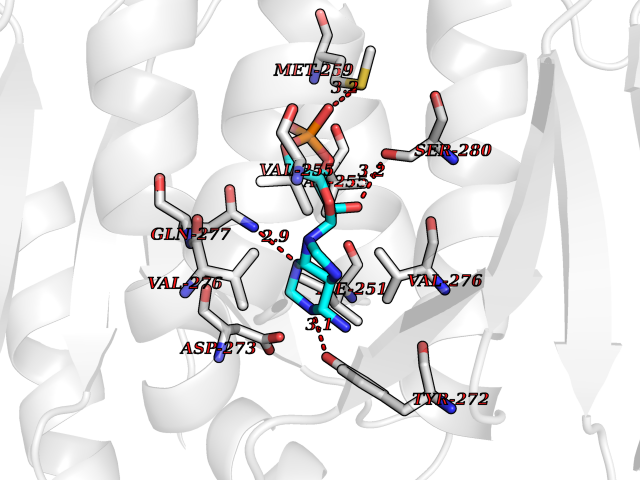 Pymol Map 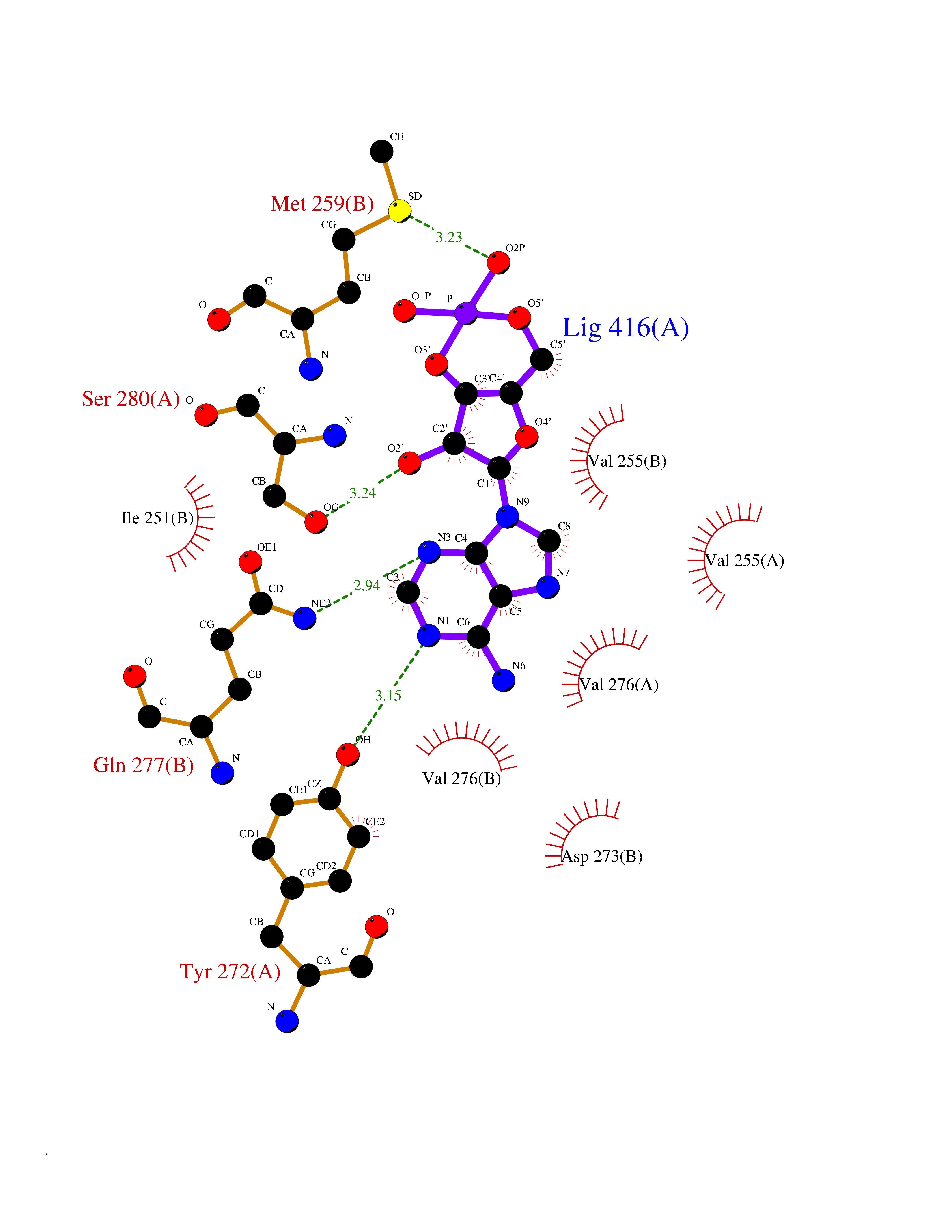 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Mutated oxalosuccinate decarboxylase (mIDH1) | 6ADG | 6.52 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD (mutated); Oxalosuccinate decarboxylase (mutated); NADP(+)-specific ICDH (mutated); Isocitrate dehydrogenase [NADP] cytoplasmic (mutated); IDP (mutated); IDH (mutated); Cytosolic NADP-isocitrate Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -7.58 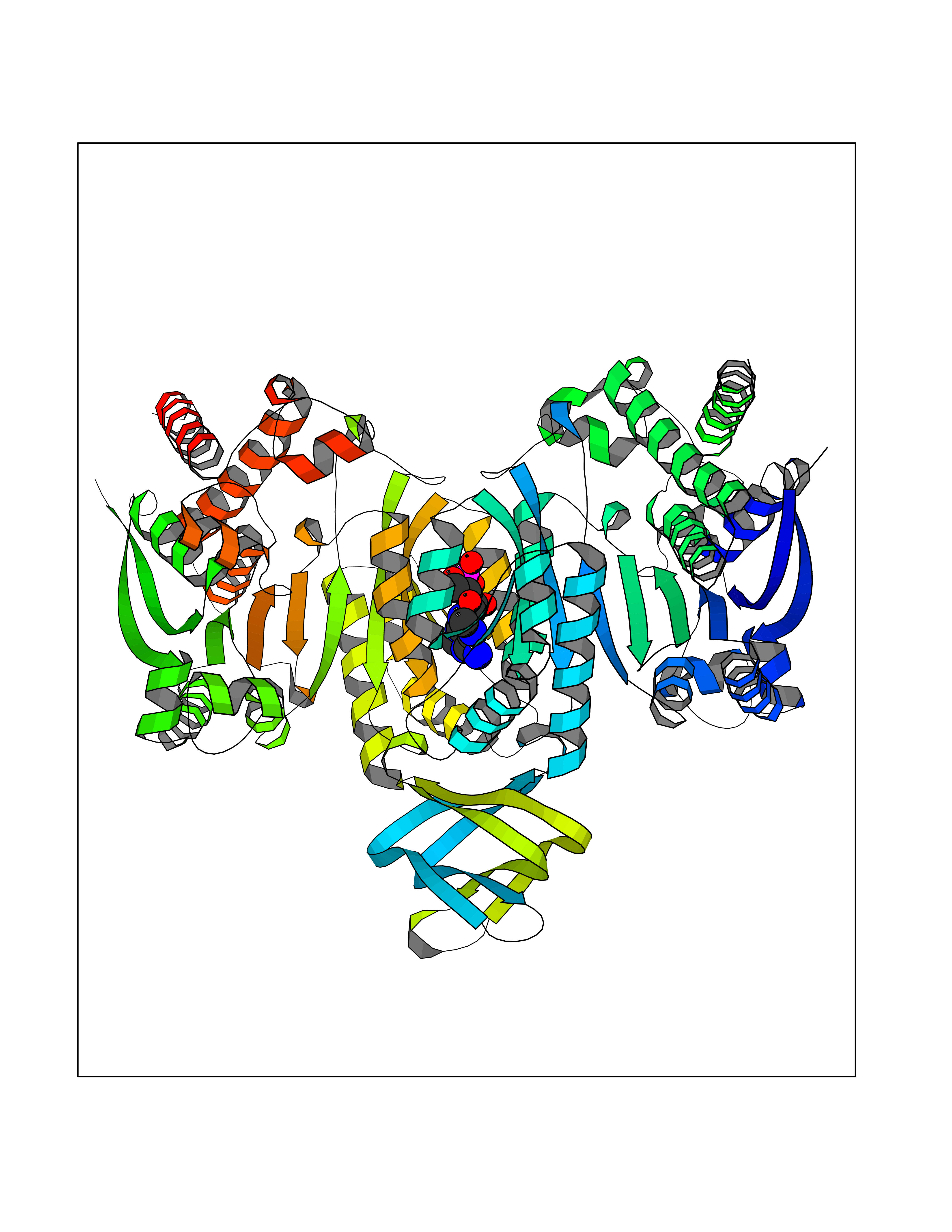 Molscript Map 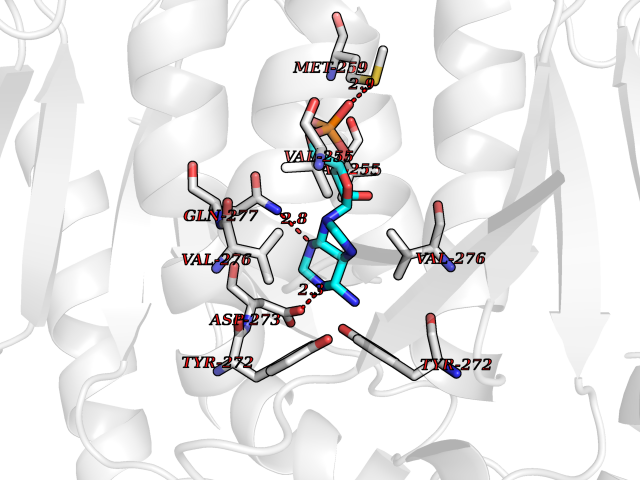 Pymol Map 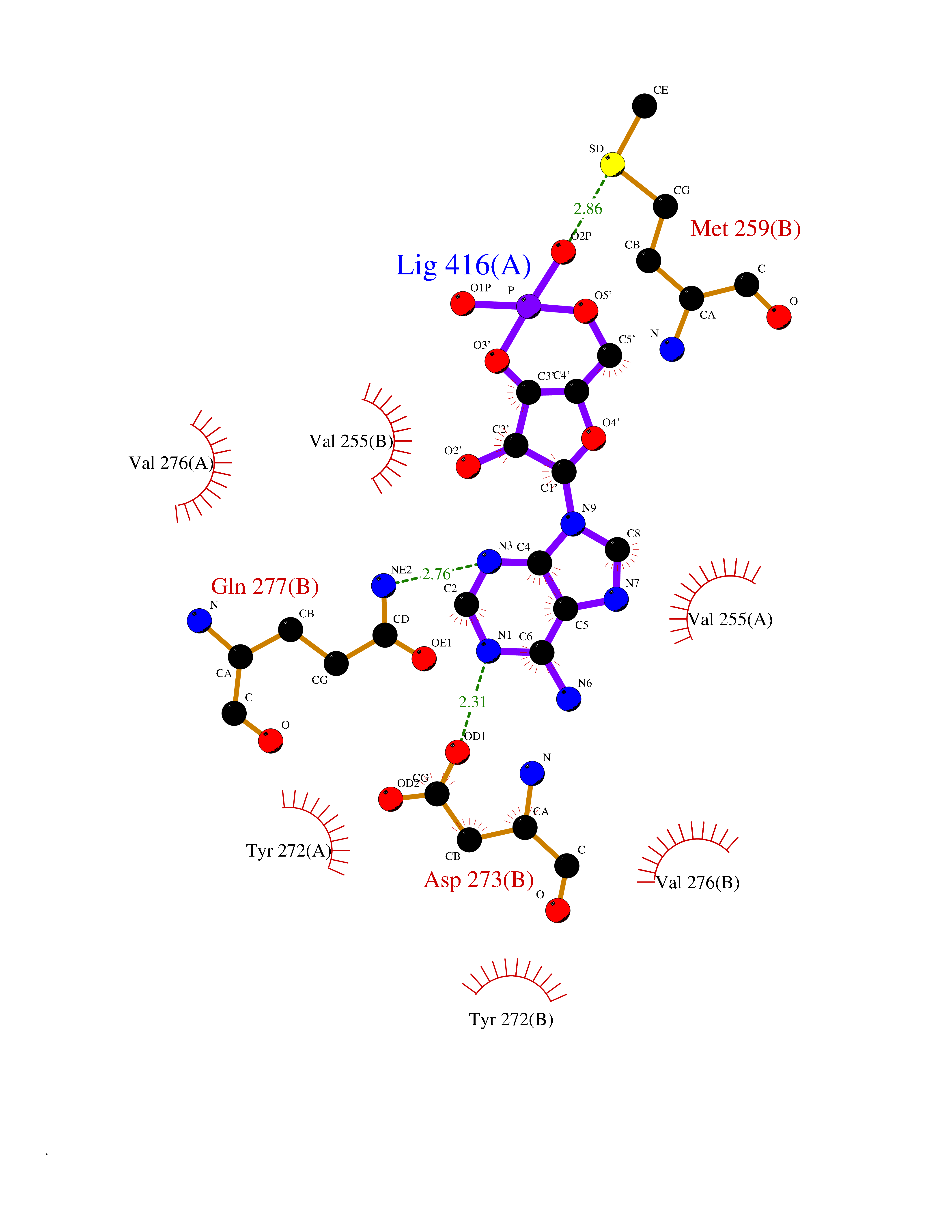 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Lanosterol 14-alpha demethylase (CYP51A1) | 4UHI | 6.52 | |
Target general information Gen name CYP51A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450LI; Cytochrome P45014DM; Cytochrome P450-14DM; Cytochrome P450 51A1 Protein family Cytochrome P450 family Biochemical class Cytochrome P450 family Function Catalyzes C14-demethylation of lanosterol; it transforms lanosterol into 4,4'-dimethyl cholesta-8,14,24-triene-3-beta-ol. Related diseases Spondyloepimetaphyseal dysplasia, short limb-hand type (SEMD-SL) [MIM:271665]: A bone disease characterized by short-limbed dwarfism, a narrow chest with pectus excavatum, brachydactyly in the hands and feet, a characteristic craniofacial appearance and premature calcifications. The radiological findings are distinctive and comprise short long bones throughout the skeleton with striking epiphyses that are stippled, flattened and fragmented and flared, irregular metaphyses. Platyspondyly in the spine with wide intervertebral spaces is observed and some vertebral bodies are pear-shaped with central humps, anterior protrusions and posterior scalloping. {ECO:0000269|PubMed:19110212, ECO:0000269|PubMed:20223752, ECO:0000269|PubMed:26463668}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Warburg-Cinotti syndrome (WRCN) [MIM:618175]: An autosomal dominant disease characterized by progressive corneal neovascularization, keloid formation, chronic skin ulcers, wasting of subcutaneous tissue, flexion contractures of the fingers, and acro-osteolysis. {ECO:0000269|PubMed:30449416}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07705; DB05667; DB01110; DB01007 Interacts with NA EC number EC 1.14.14.154 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Endoplasmic reticulum; Heme; Iron; Lipid biosynthesis; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 53013.3 Length 462 Aromaticity 0.11 Instability index 47.66 Isoelectric point 8.8 Charge (pH=7) 7 2D Binding mode Binding energy (Kcal/mol) -7.05 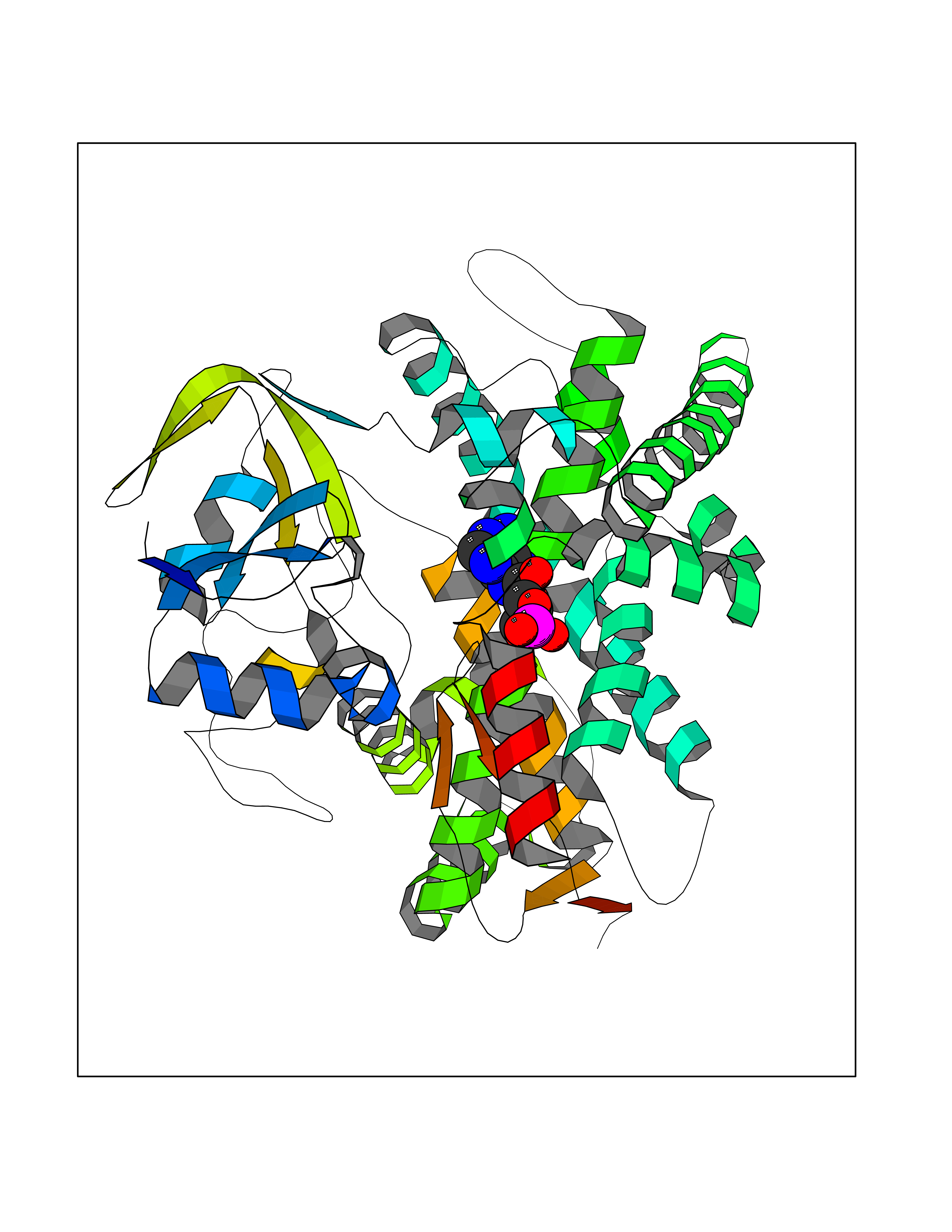 Molscript Map 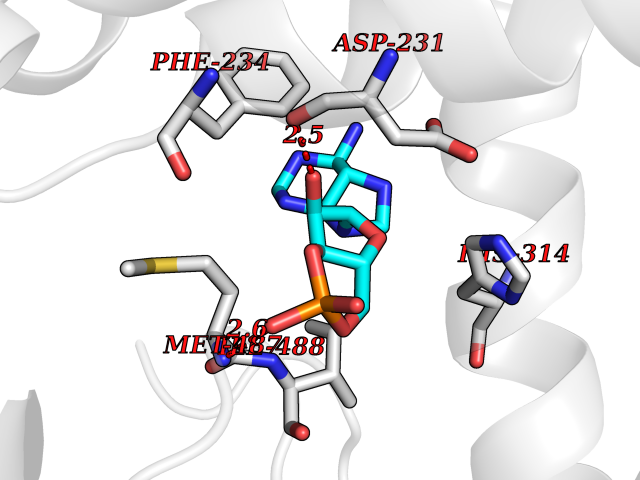 Pymol Map 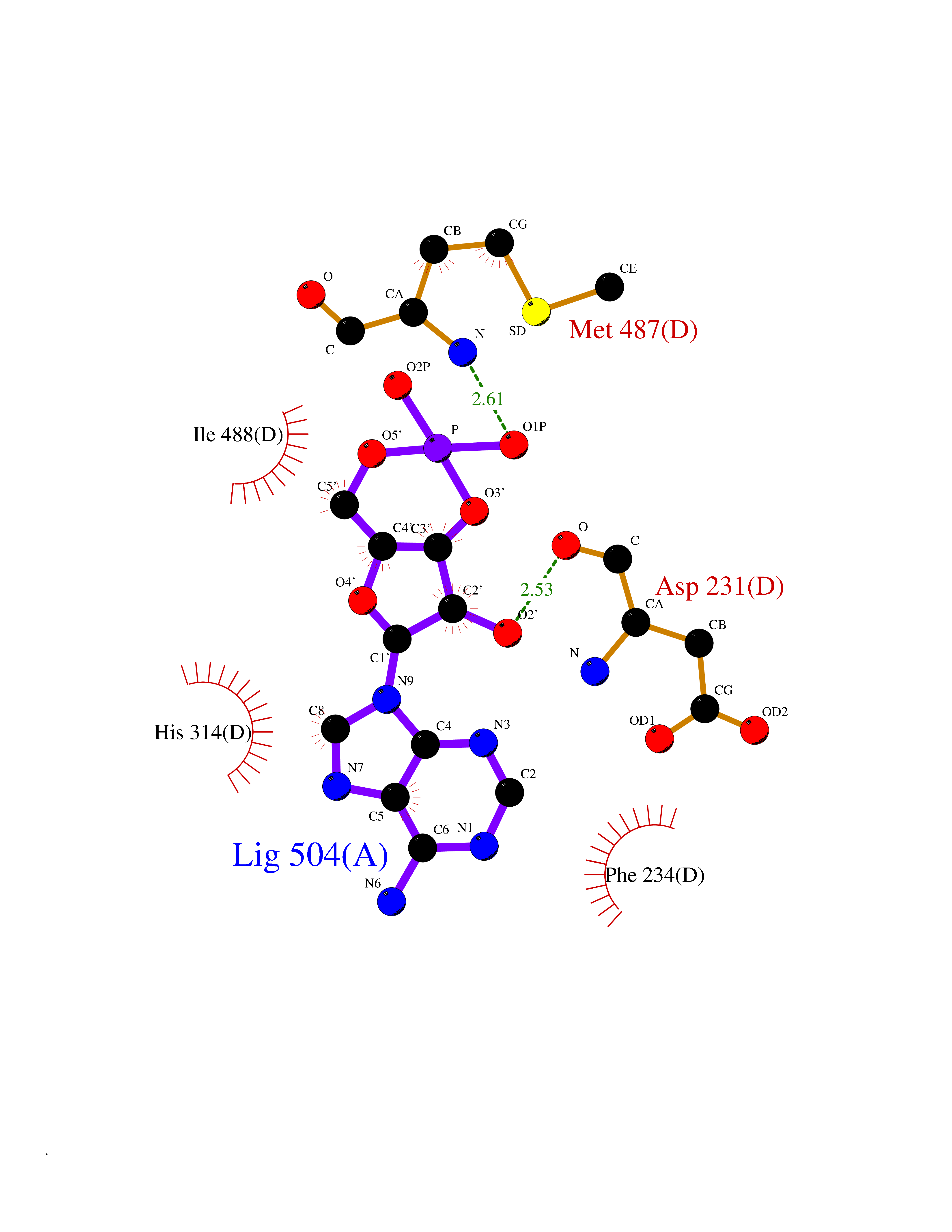 Ligplot Map 3D Binding mode Sequence PPYIFSPIPFLGHAIAFGKSPIEFLENAYEKYGPVFSFTMVGKTFTYLLGSDAAALLFNSKNEDLNAEDVYSRLTTPVFGKGVAYDVPNPVFLEQKKMLKSGLNIAHFKQHVSIIEKETKEYFESWGESGEKNVFEALSELIILTASHCLHGKEIRSQLNEKVAQLYADLDGGFSHAAWLLPGWLPLPSFRRRDRAHREIKDIFYKAIQKRRQSQEKIDDILQTLLDATYKDGRPLTDDEVAGMLIGLLLAGQHTSSTTSAWMGFFLARDKTLQKKCYLEQKTVCGENLPPLTYDQLKDLNLLDRCIKETLRLRPPIMIMMRMARTPQTVAGYTIPPGHQVCVSPTVNQRLKDSWVERLDFNPDRYLQDNPASGEKFAYVPFGAGRHRCIGENFAYVQIKTIWSTMLRLYEFDLIDGYFPTVNYTTMIHTPENPVIRYKRRSLPGWLPLPSFRRRDRAHREI Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Protein cereblon (CRBN) | 5FQD | 6.52 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -8.02 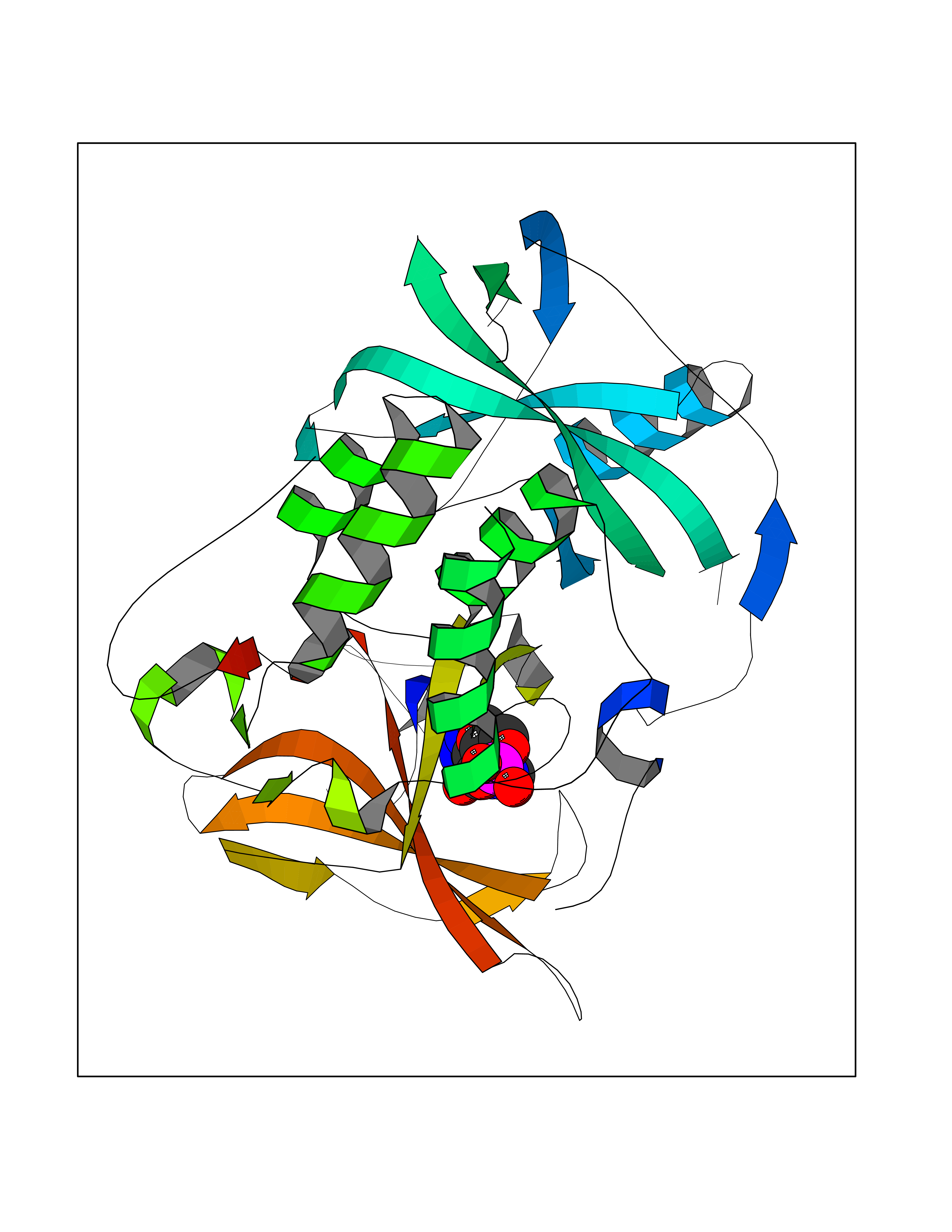 Molscript Map 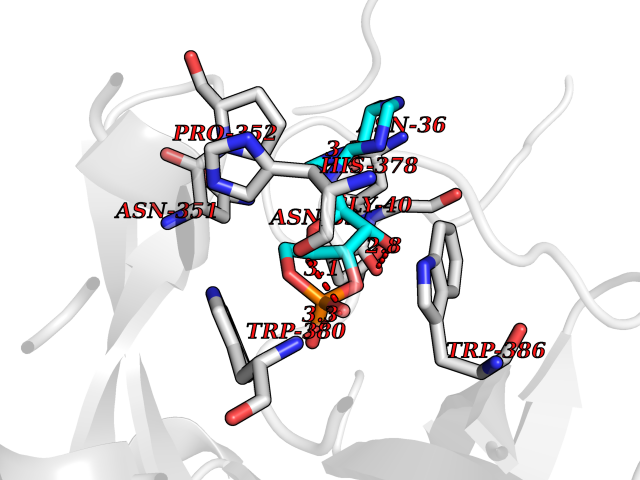 Pymol Map 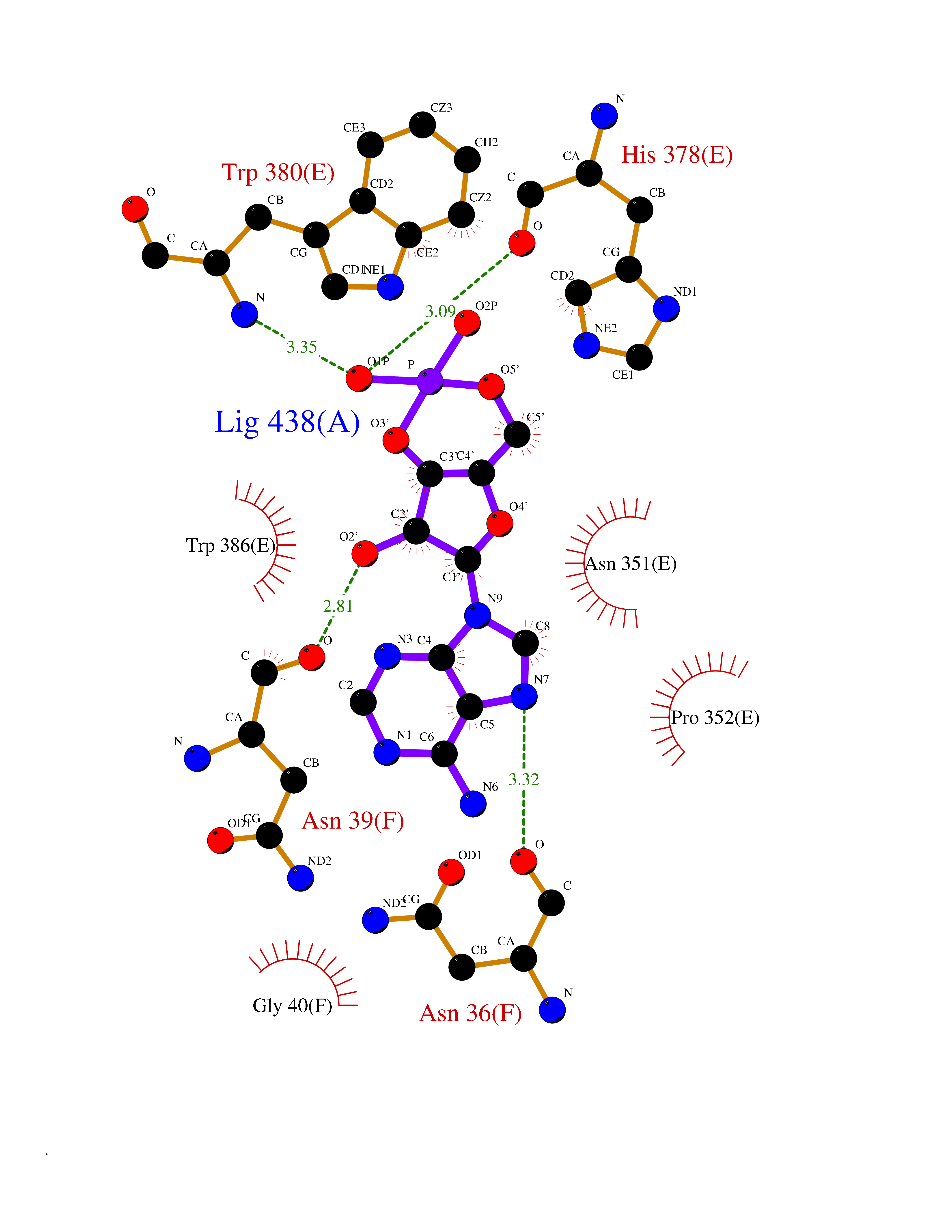 Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||