Job Results:
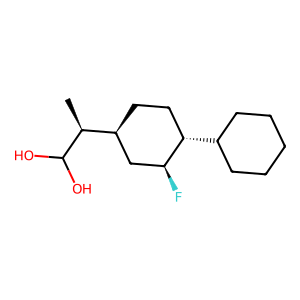
Ligand
Structure
Job ID
6a37619723fa756e073677fca12f4afa
Job name
NA
Time
2024-07-14 16:31:23
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Carbonic anhydrase II (CA-II) | 3K34 | 7.41 | |
Target general information Gen name CA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase C; Carbonic anhydrase 2; Carbonate dehydratase II; CAC Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Reversible hydration of carbon dioxide. Can hydrate cyanamide to urea. Involved in the regulation of fluid secretion into the anterior chamber of the eye. Contributes to intracellular pH regulation in the duodenal upper villous epithelium during proton-coupled peptide absorption. Stimulates the chloride-bicarbonate exchange activity of SLC26A6. Essential for bone resorption and osteoclast differentiation. Related diseases Osteopetrosis, autosomal recessive 3 (OPTB3) [MIM:259730]: A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB3 is associated with renal tubular acidosis, cerebral calcification (marble brain disease) and in some cases with intellectual disability. {ECO:0000269|PubMed:15300855, ECO:0000269|PubMed:1542674, ECO:0000269|PubMed:1928091, ECO:0000269|PubMed:8834238, ECO:0000269|PubMed:9143915}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07596; DB03333; DB08418; DB04081; DB08416; DB02479; DB07467; DB03950; DB03594; DB03294; DB04763; DB03270; DB08083; DB06954; DB08659; DB08046; DB02087; DB08156; DB04203; DB04394; DB08782; DB04549; DB02221; DB04180; DB03039; DB02861; DB02429; DB08202; DB04600; DB04601; DB01784; DB03385; DB03697; DB04002; DB07632; DB07050; DB06891; DB08645; DB08765; DB00819; DB03877; DB03262; DB03598; DB04089; DB01964; DB03526; DB04371; DB02220; DB03221; DB02602; DB02535; DB00436; DB00562; DB01194; DB00482; DB00880; DB02679; DB00606; DB02866; DB01144; DB00869; DB08846; DB01031; DB00311; DB08157; DB01942; DB00695; DB00774; DB08165; DB02292; DB03975; DB00703; DB00232; DB02610; DB07742; DB03844; DB02069; DB02986; DB08301; DB07048; DB03596; DB07476; DB01748; DB08155; DB01671; DB07710; DB01325; DB09460; DB09472; DB02894; DB00391; DB08329; DB07363; DB00273; DB01021; DB03904; DB00580; DB14533; DB14548; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Acetylation; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Lyase; Membrane; Metal-binding; Osteopetrosis; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29027.4 Length 258 Aromaticity 0.1 Instability index 20.07 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -7.7 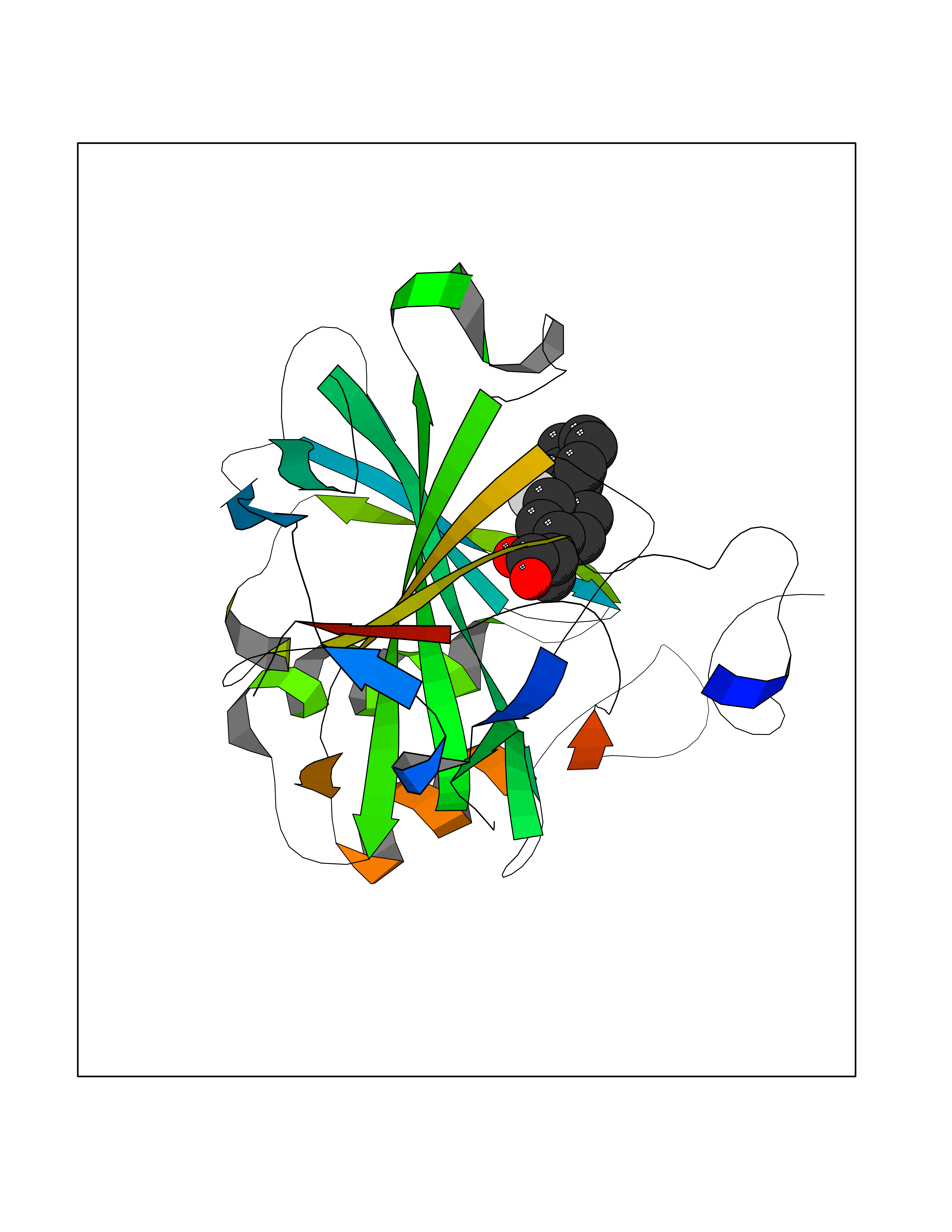 Molscript Map 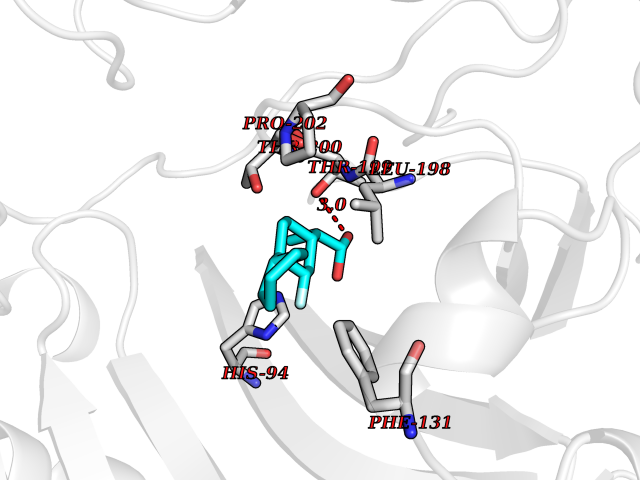 Pymol Map 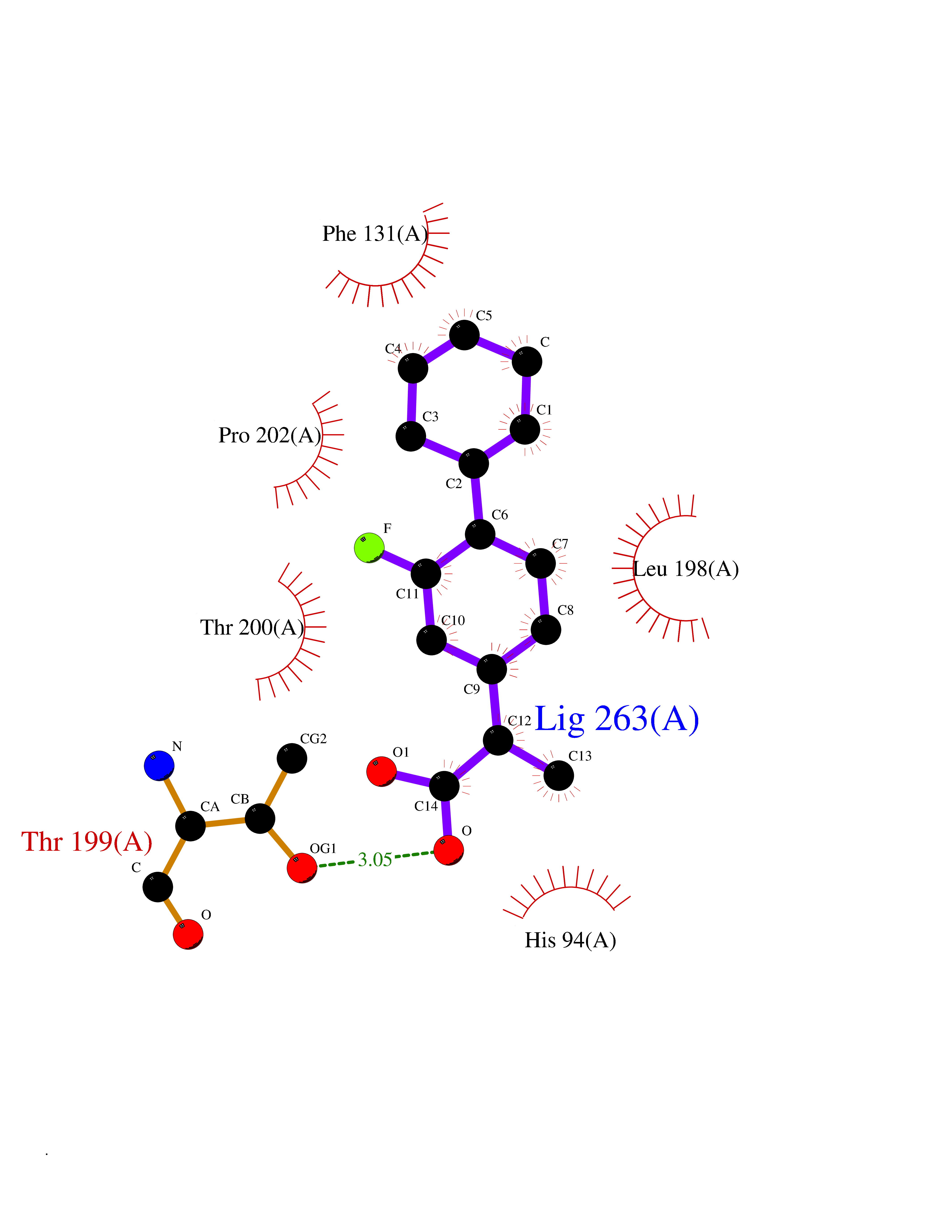 Ligplot Map 3D Binding mode Sequence HHWGYGKHNGPEHWHKDFPIAKGERQSPVDIDTHTAKYDPSLKPLSVSYDQATSLRILNNGHAFNVEFDDSQDKAVLKGGPLDGTYRLIQFHFHWGSLDGQGSEHTVDKKKYAAELHLVHWNTKYGDFGKAVQQPDGLAVLGIFLKVGSAKPGLQKVVDVLDSIKTKGKSADFTNFDPRGLLPESLDYWTYPGSLTTPPLLECVTWIVLKEPISVSSEQVLKFRKLNFNGEGEPEELMVDNWRPAQPLKNRQIKASFK Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Phosphodiesterase 4D (PDE4D) | 1Y2K | 7.41 | |
Target general information Gen name PDE4D Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-specific 3',5'-cyclic phosphodiesterase 4D; PDE43; DPDE3 Protein family Cyclic nucleotide phosphodiesterase family, PDE4 subfamily Biochemical class Phosphoric diester hydrolase Function Hydrolyzes the second messenger cAMP, which is a key regulator of many important physiological processes. Related diseases Genetic variations in PDE4D might be associated with susceptibility to stroke. PubMed:17006457 states that association with stroke has to be considered with caution. {ECO:0000269|PubMed:17006457}.; DISEASE: Acrodysostosis 2, with or without hormone resistance (ACRDYS2) [MIM:614613]: A pleiotropic disorder characterized by skeletal, endocrine, and neurological abnormalities. Skeletal features include brachycephaly, midface hypoplasia with a small upturned nose, brachydactyly, and lumbar spinal stenosis. Endocrine abnormalities include hypothyroidism and hypogonadism in males and irregular menses in females. Developmental disability is a common finding but is variable in severity and can be associated with significant behavioral problems. {ECO:0000269|PubMed:22464250, ECO:0000269|PubMed:22464252, ECO:0000269|PubMed:23033274, ECO:0000269|PubMed:23043190}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06842; DB04149; DB03606; DB03183; DB04469; DB02676; DB01959; DB07051; DB04271; DB07954; DB08299; DB00131; DB01427; DB00201; DB03849; DB05219; DB00651; DB06246; DB05266; DB01088; DB01113; DB01791; DB01656; DB01954; DB05298; DB09283; DB02918 Interacts with P32121; P38432; Q0D2H9; Q08AF8; P43360; Q07343; Q13077; P32121; P26769; P38432; Q96CV9; Q8IUH5 EC number EC 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cell membrane; Cytoplasm; Cytoskeleton; Disease variant; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37201.9 Length 322 Aromaticity 0.07 Instability index 35.83 Isoelectric point 5.02 Charge (pH=7) -21.16 2D Binding mode Binding energy (Kcal/mol) -8.62 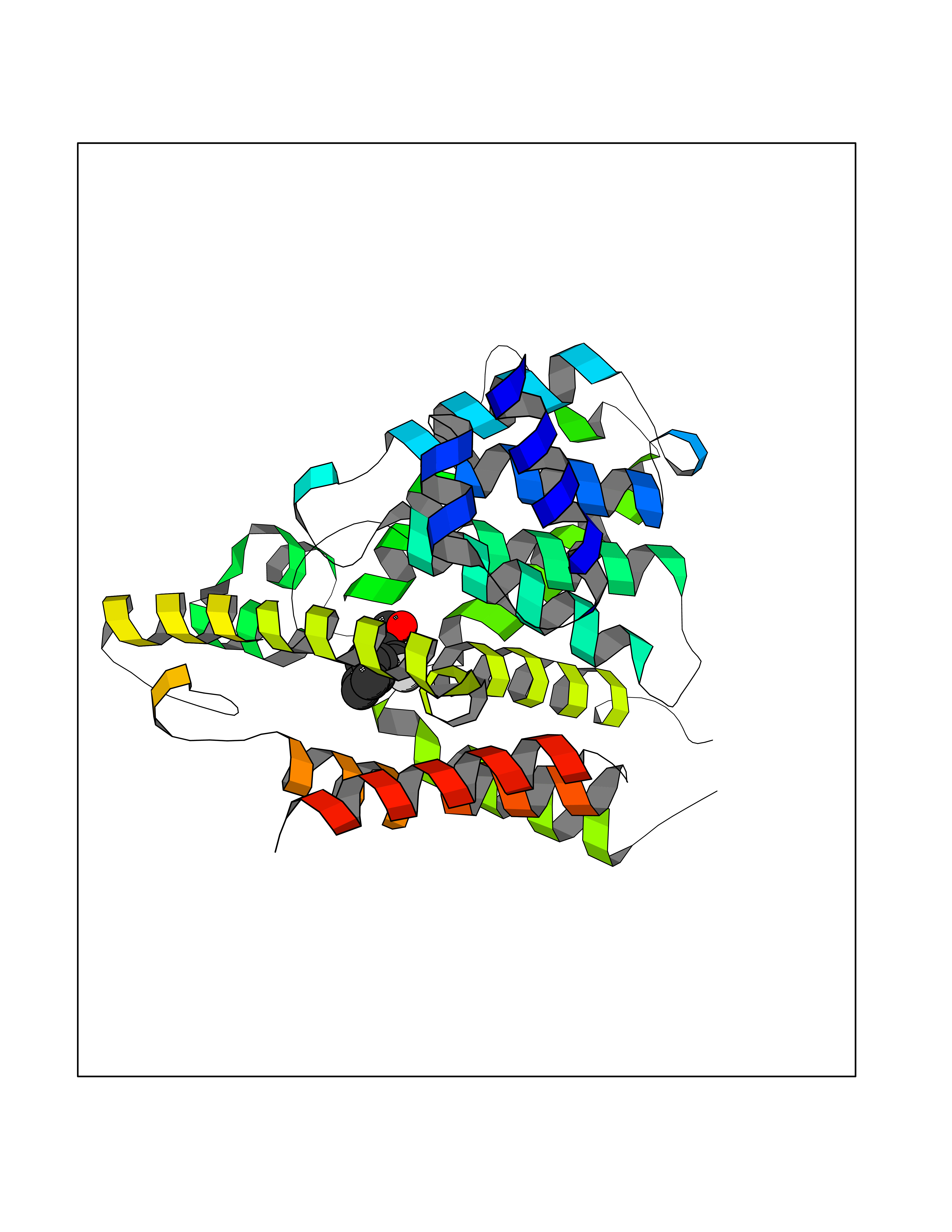 Molscript Map 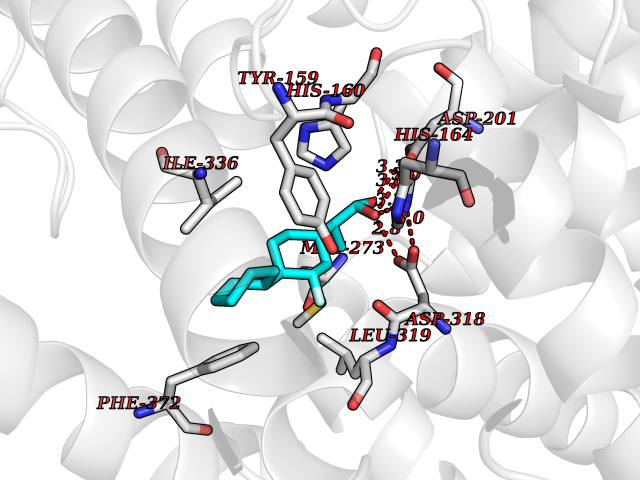 Pymol Map 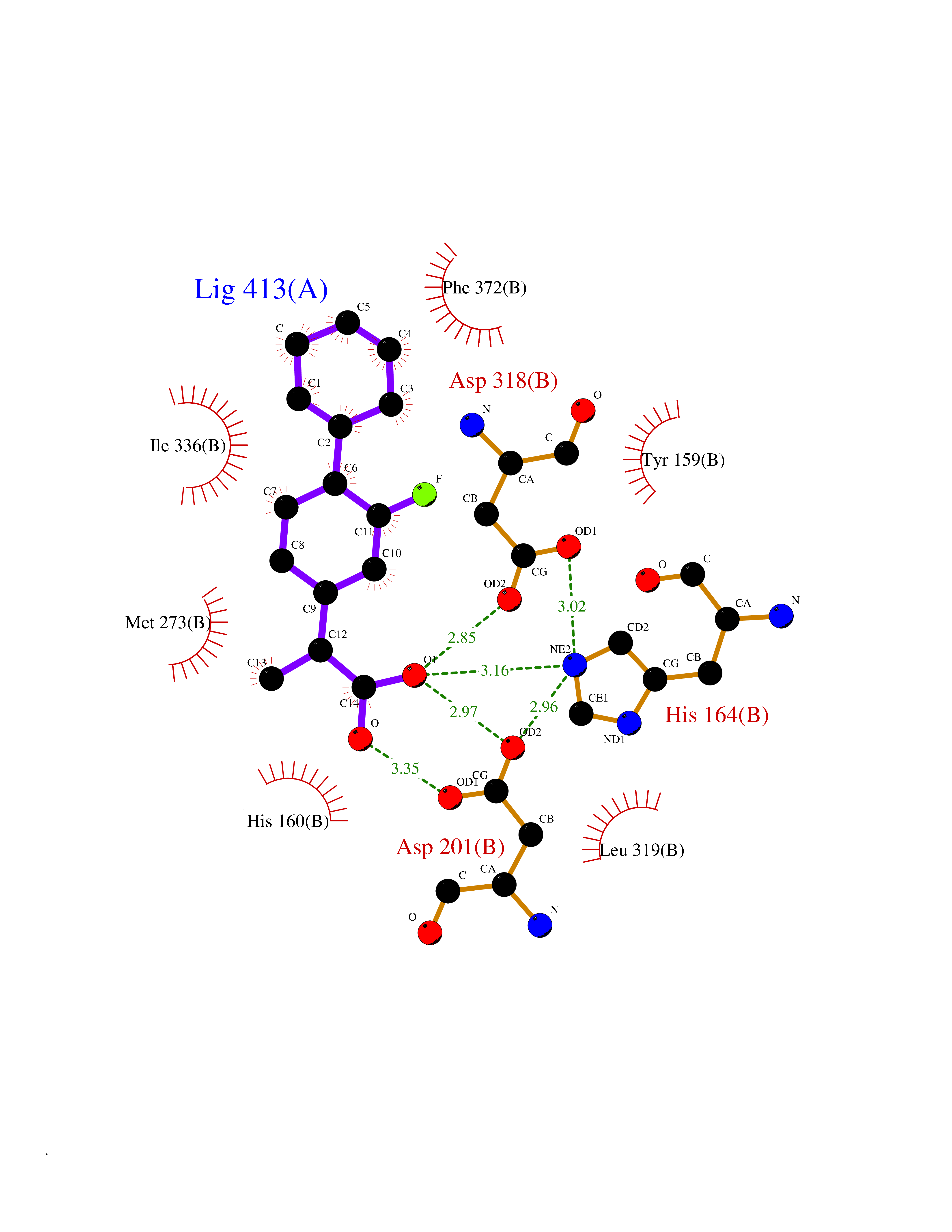 Ligplot Map 3D Binding mode Sequence TEQEDVLAKELEDVNKWGLHVFRIAELSGNRPLTVIMHTIFQERDLLKTFKIPVDTLITYLMTLEDHYHADVAYHNNIHAADVVQSTHVLLSTPALEAVFTDLEILAAIFASAIHDVDHPGVSNQFLINTNSELALMYNDSSVLENHHLAVGFKLLQEENCDIFQNLTKKQRQSLRKMVIDIVLATDMSKHMNLLADLKTMVETKKVVLLLDNYSDRIQVLQNMVHCADLSNPTKPLQLYRQWTDRIMEEFFRQGDRERERGMEISPMCDKHNASVEKSQVGFIDYIVHPLWETWADLVHPDAQDILDTLEDNREWYQSTIP Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Corynebacterium Pup-protein ligase (Cory pafA) | 4BJR | 7.41 | |
Target general information Gen name Cory pafA Organism Corynebacterium glutamicum (strain ATCC 13032 / DSM 20300 / JCM 1318 / BCRC 11384 / CCUG 27702 / LMG 3730 / NBRC 12168 / NCIMB 10025 / NRRL B-2784 / 534) Uniprot ID TTD ID Synonyms Pup-conjugating enzyme; Pup--protein ligase; Proteasome accessory factor A Protein family Pup ligase/Pup deamidase family, Pup-conjugating enzyme subfamily Biochemical class NA Function Catalyzes the covalent attachment of the prokaryotic ubiquitin-like protein modifier Pup to the proteasomal substrate proteins, thereby targeting them for proteasomal degradation. This tagging system is termed pupylation. The ligation reaction involves the side-chain carboxylate of the C-terminal glutamate of Pup and the side-chain amino group of a substrate lysine. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; ATP-binding; Ligase; Magnesium; Metal-binding; Nucleotide-binding; Reference proteome; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 110572 Length 994 Aromaticity 0.07 Instability index 42.33 Isoelectric point 5.26 Charge (pH=7) -34.19 2D Binding mode Binding energy (Kcal/mol) -9.22 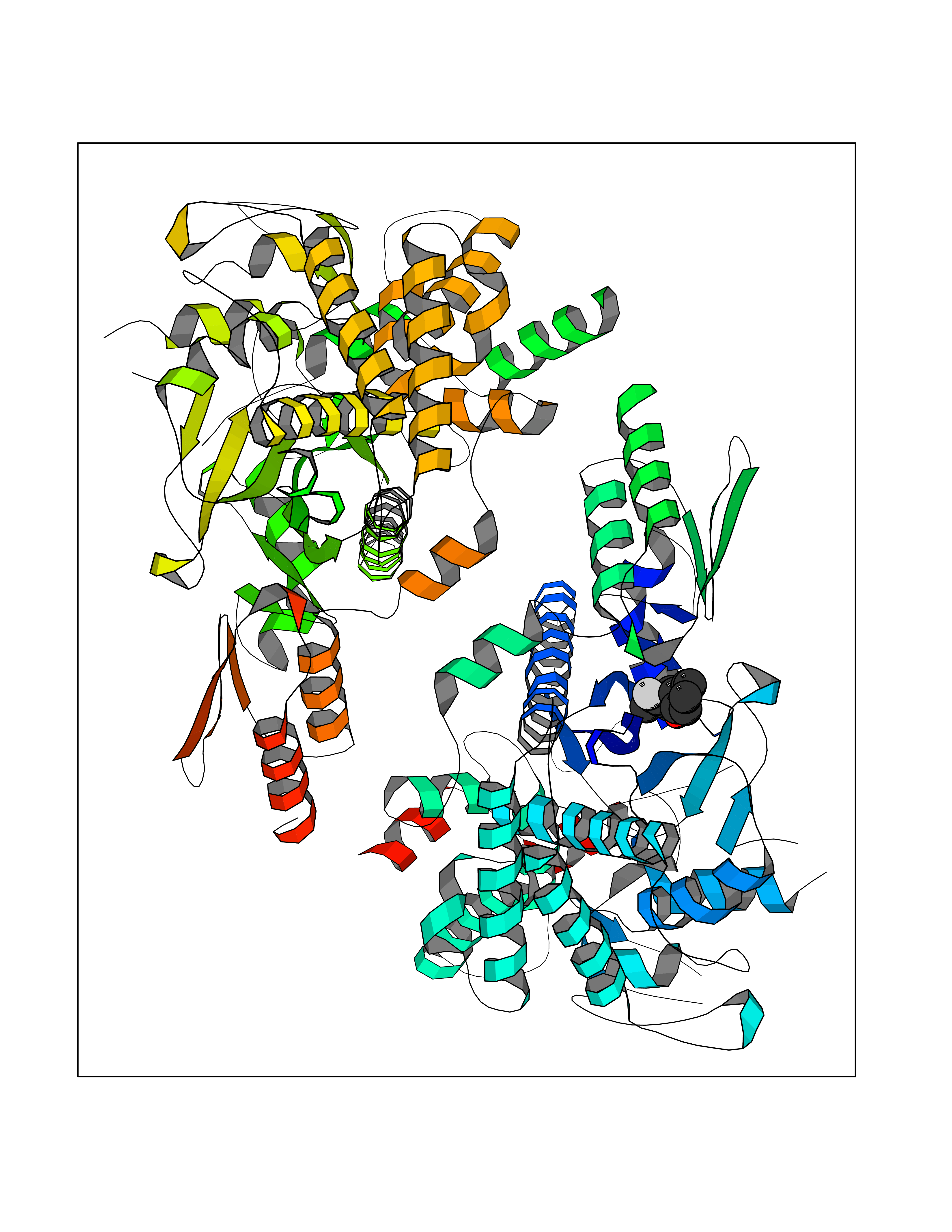 Molscript Map 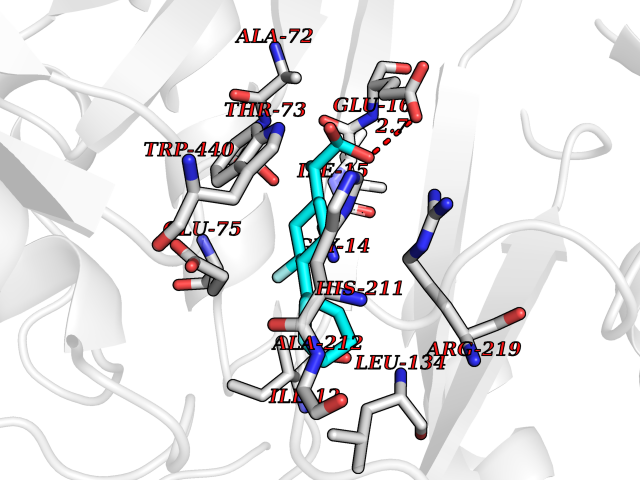 Pymol Map 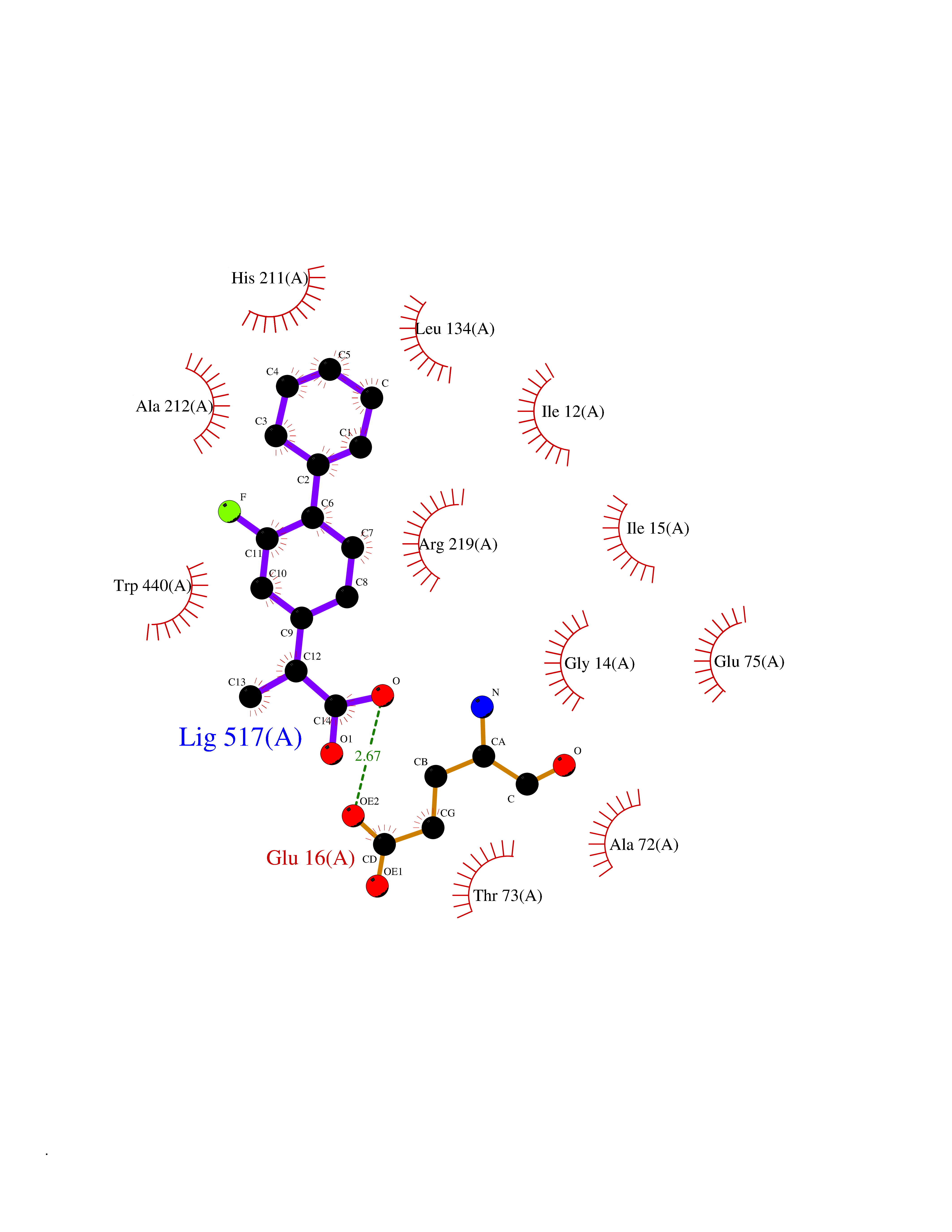 Ligplot Map 3D Binding mode Sequence TVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHAGSASGTSLLDEIDGLLENNAEEFVRSYVQKGGETVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHASLLDEIDGLLENNAEEFVRSYVQKGGE Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 7.39 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -9.37  Molscript Map 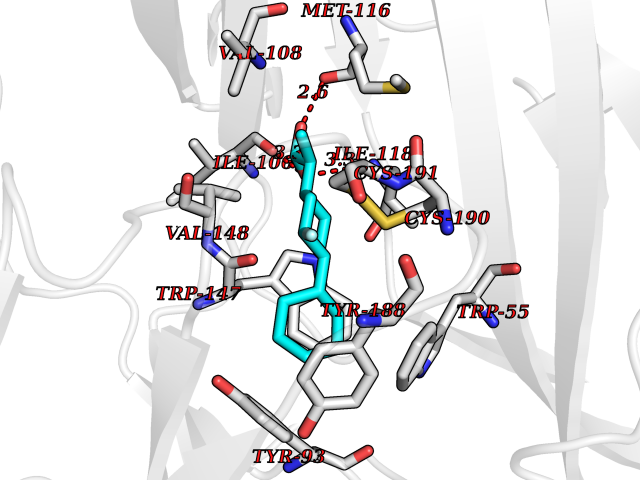 Pymol Map 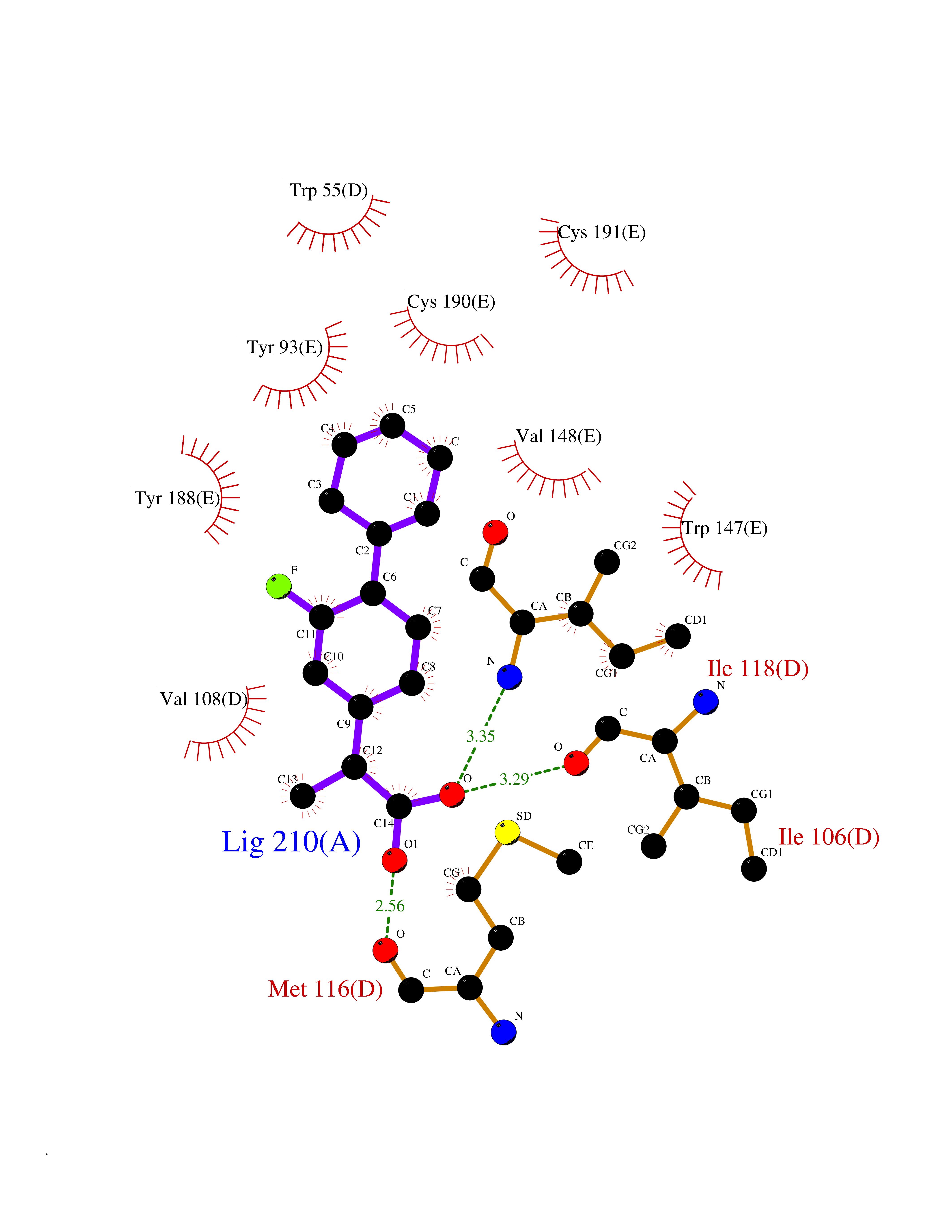 Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 7.39 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -6.86 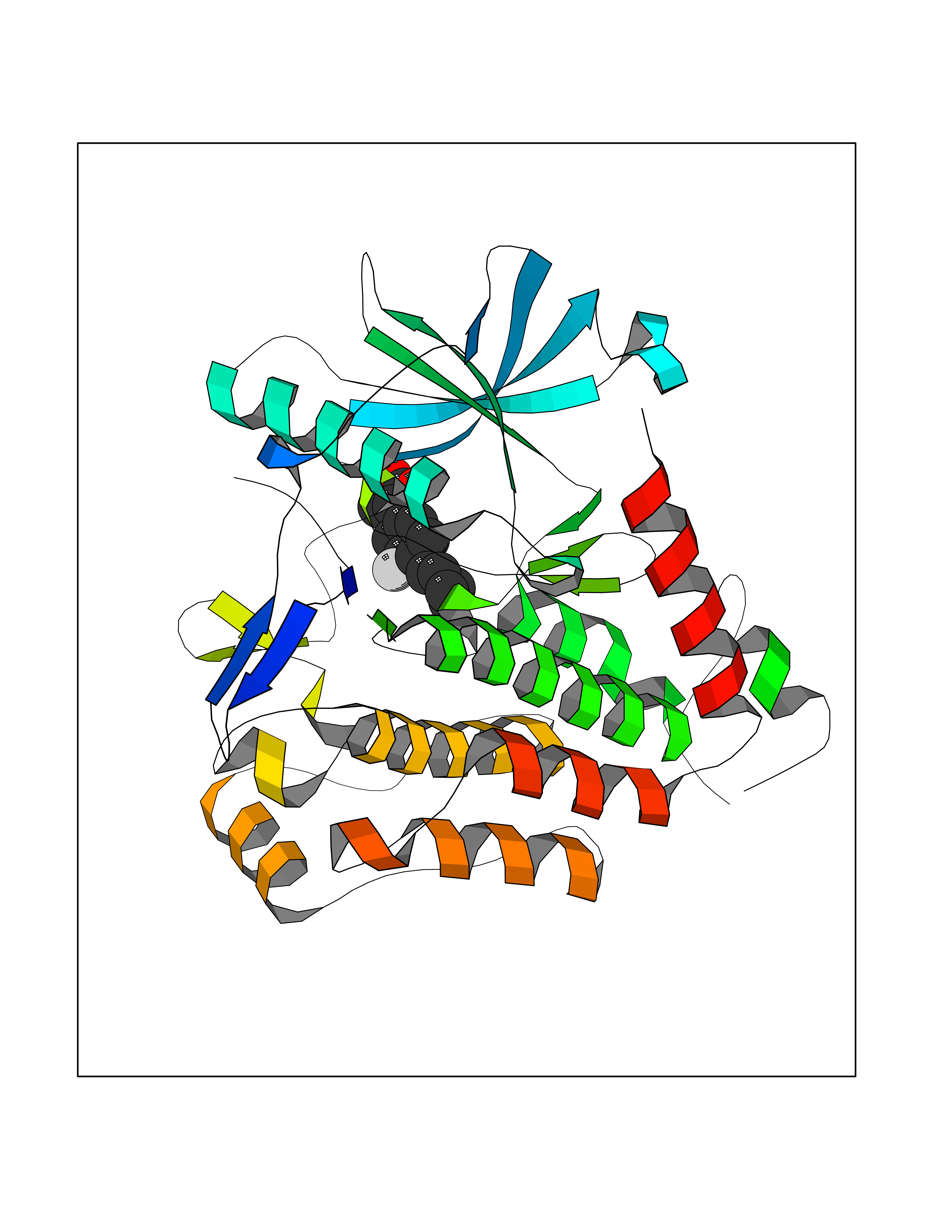 Molscript Map 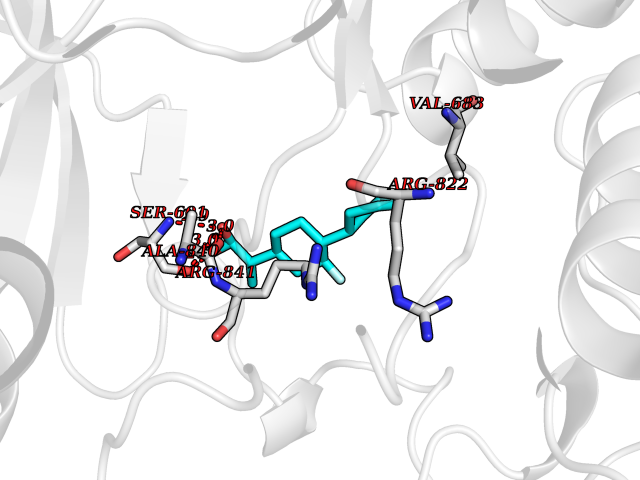 Pymol Map 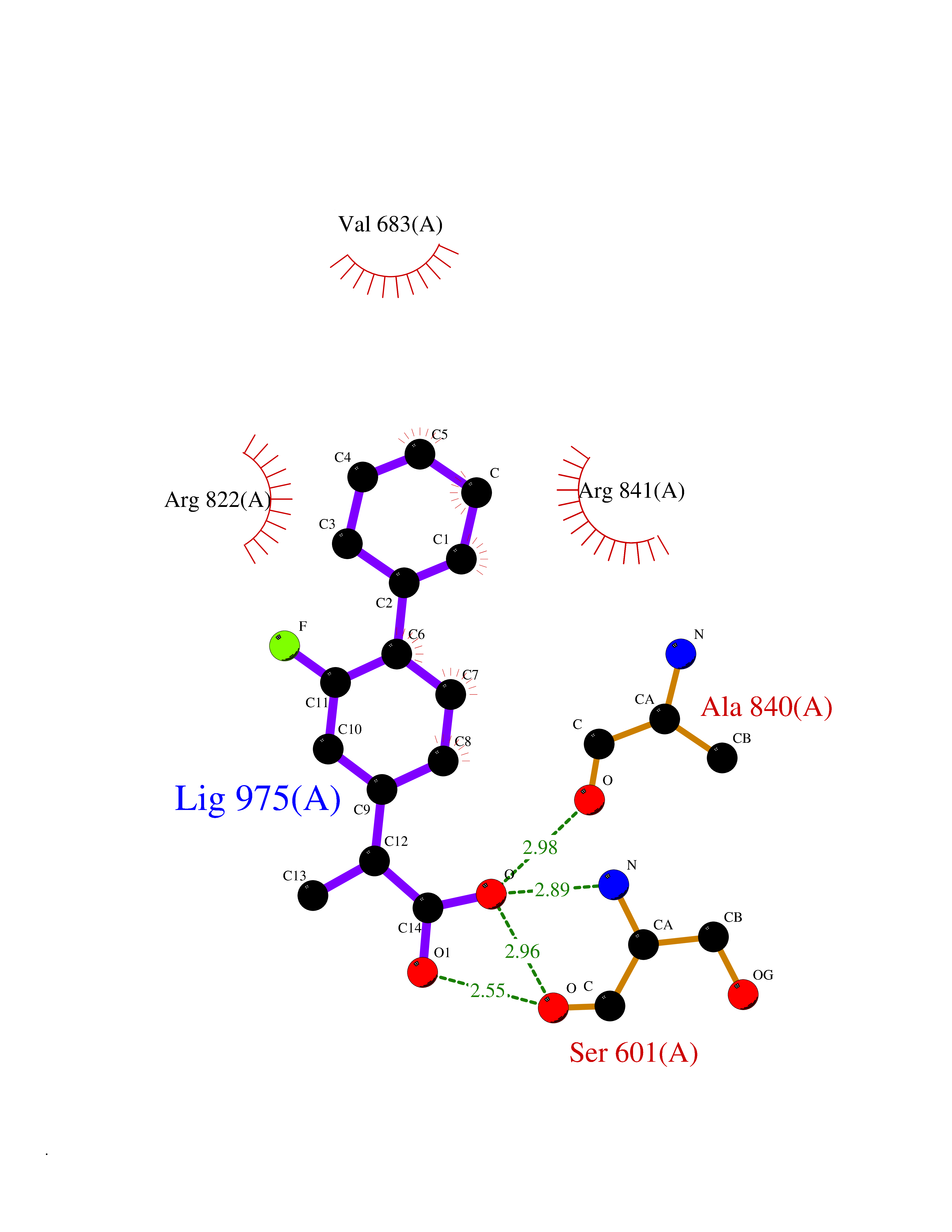 Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Proto-oncogene c-Fes (FES) | 3CBL | 7.39 | |
Target general information Gen name FES Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p93c-fes; Tyrosine-protein kinase Fes/Fps; Proto-oncogene c-Fps; Feline sarcoma/Fujinami avian sarcoma oncogene homolog; FPS Protein family Protein kinase superfamily, Tyr protein kinase family, Fes/fps subfamily Biochemical class Kinase Function Plays a role in FCER1 (high affinity immunoglobulin epsilon receptor)-mediated signaling in mast cells. Acts down-stream of the activated FCER1 receptor and the mast/stem cell growth factor receptor KIT. Plays a role in the regulation of mast cell degranulation. Plays a role in the regulation of cell differentiation and promotes neurite outgrowth in response to NGF signaling. Plays a role in cell scattering and cell migration in response to HGF-induced activation of EZR. Phosphorylates BCR and down-regulates BCR kinase activity. Phosphorylates HCLS1/HS1, PECAM1, STAT3 and TRIM28. Tyrosine-protein kinase that acts downstream of cell surface receptors and plays a role in the regulation of the actin cytoskeleton, microtubule assembly, cell attachment and cell spreading. Related diseases Has been shown to act as proto-oncogene in some types of cancer, possibly due to abnormal activation of the kinase. Has been shown to act as tumor suppressor in other types of cancer. Expressed and present as activated kinase in a subset of acute myeloid leukemia patients; promotes survival of leukemia cells (PubMed:20111072). Expression is absent in K562 leukemia cells; ectopic expression of FSP/FES restores myeloid differentiation (PubMed:2656706). May function as tumor suppressor in colorectal cancer; expression is reduced or absent in samples from some colon cancer patients (PubMed:16455651). May function as tumor suppressor in melanoma by preventing melanoma cell proliferation; expression is reduced or absent in samples from some melanoma patients (PubMed:28463229). Ectopic expression of FSP/FES suppresses anchorage-independent growth in colon cancer cell lines (PubMed:16455651). Up-regulated in prostate cancer, and might be a predictor of recurrence after radical surgery (PubMed:16455651). May promote growth of renal carcinoma cells (PubMed:19082481). {ECO:0000269|PubMed:16455651, ECO:0000269|PubMed:19082481, ECO:0000269|PubMed:20111072, ECO:0000269|PubMed:2656706, ECO:0000269|PubMed:28463229}. Drugs (DrugBank ID) DB12010 Interacts with P10275; P15924; P15311; Q13480; P10721; P54274 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell junction; Cell membrane; Coiled coil; Cytoplasm; Cytoplasmic vesicle; Cytoskeleton; Golgi apparatus; Kinase; Lipid-binding; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; SH2 domain; Transferase; Tumor suppressor; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 40460.2 Length 356 Aromaticity 0.09 Instability index 42.56 Isoelectric point 7.12 Charge (pH=7) 0.32 2D Binding mode Binding energy (Kcal/mol) -8.35 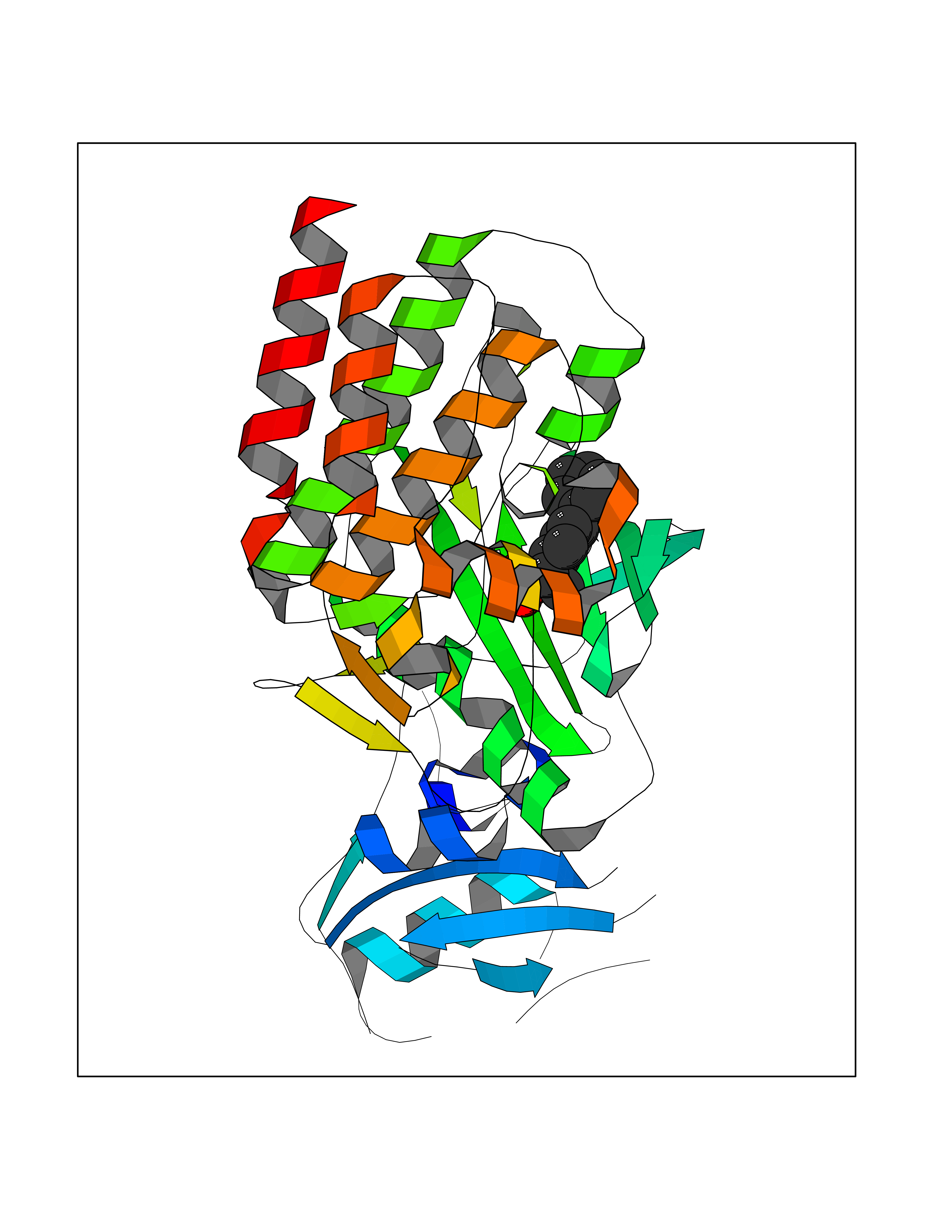 Molscript Map 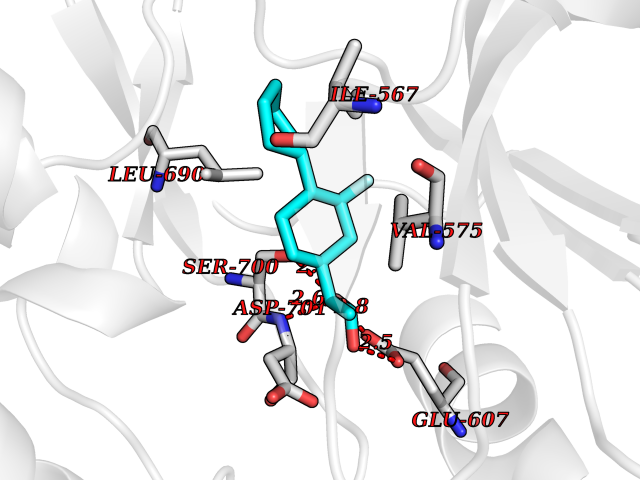 Pymol Map 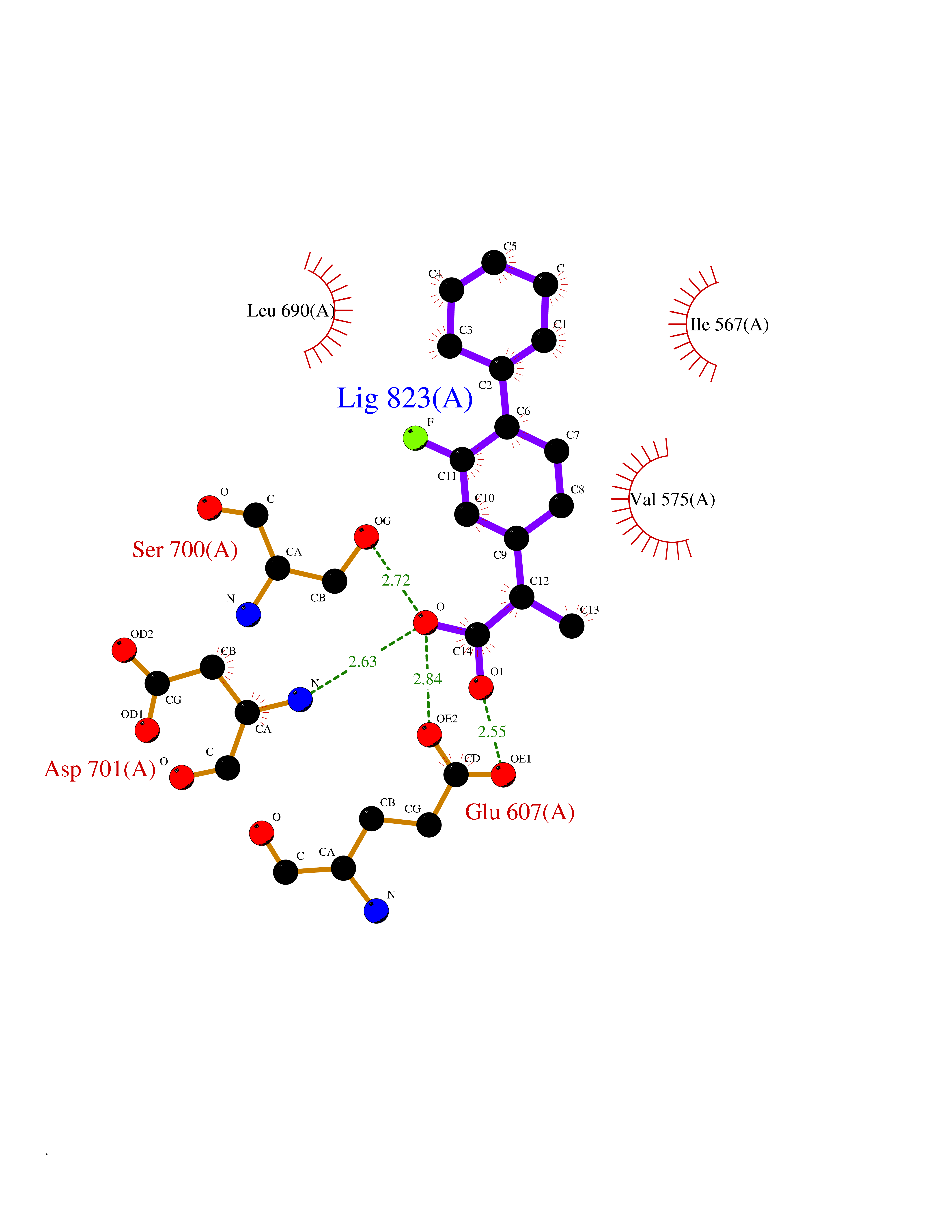 Ligplot Map 3D Binding mode Sequence SMIPEVQKPLHEQLWYHGAIPRAEVAELLVHSGDFLVRESQQEYVLSVPRHFIINLYRLFPSIPLLIDHLLSTQQPLVVLHRAVPKDKWVLNHEDLVLGEQIGRGNFGEVFSGRLRADNTLVAVKSCRETLPPDLKAKFLQEARILKQYSHPNIVRLIGVCTQKQPIYIVMELVQGGDFLTFLRTEGARLRVKTLLQMVGDAAAGMEYLESKCCIHRDLAARNCLVTEKNVLKISDFGMSREEADGVYAASGGLRQVPVKWTAPEALNYGRYSSESDVWSFGILLWETFSLGASPYPNLSNQQTREFVEKGGRLPCPELCPDAVFRLMEQCWAYEPGQRPSFSTIYQELQSIRKRH Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Aggrecanase-1 (ADAMTS4) | 2RJP | 7.39 | |
Target general information Gen name ADAMTS4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aggrecanase 1; ADMP-1; ADAMTS4; ADAM-TS4; ADAM-TS 4; A disintegrin and metalloproteinase with thrombospondin motifs 4 Protein family NA Biochemical class Peptidase Function Cleaves aggrecan, a cartilage proteoglycan, and may be involved in its turnover. May play an important role in the destruction of aggrecan in arthritic diseases. Could also be a critical factor in the exacerbation of neurodegeneration in Alzheimer disease. Cleaves aggrecan at the '392-Glu-|-Ala-393' site. Related diseases Familial male precocious puberty (FMPP) [MIM:176410]: In FMPP the receptor is constitutively activated. {ECO:0000269|PubMed:11134146, ECO:0000269|PubMed:11391350, ECO:0000269|PubMed:7629248, ECO:0000269|PubMed:7692306, ECO:0000269|PubMed:7714085, ECO:0000269|PubMed:7757065, ECO:0000269|PubMed:8281137, ECO:0000269|PubMed:8829636, ECO:0000269|PubMed:8929952, ECO:0000269|PubMed:9467560, ECO:0000269|PubMed:9661624}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Luteinizing hormone resistance (LHR) [MIM:238320]: An autosomal recessive disorder characterized by unresponsiveness to luteinizing hormone, defective sexual development in males, and defective follicular development and ovulation, amenorrhea and infertility in females. Two forms of the disorder have been defined in males. Type 1 is a severe form characterized by complete 46,XY male pseudohermaphroditism, low testosterone and high luteinizing hormone levels, total lack of responsiveness to luteinizing and chorionic gonadotropin hormones, lack of breast development, and absent development of secondary male sex characteristics. Type 2, a milder form, displays a broader range of phenotypic expression ranging from micropenis to severe hypospadias. {ECO:0000269|PubMed:12050206, ECO:0000269|PubMed:15372531, ECO:0000269|PubMed:15472221, ECO:0000269|PubMed:19551906, ECO:0000269|PubMed:7719343, ECO:0000269|PubMed:8559204, ECO:0000269|PubMed:9215288, ECO:0000269|PubMed:9514160, ECO:0000269|PubMed:9626144, ECO:0000269|PubMed:9626653}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06822 Interacts with NA EC number EC 3.4.24.82 Uniprot keywords 3D-structure; Alternative splicing; Cleavage on pair of basic residues; Direct protein sequencing; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 31309.4 Length 291 Aromaticity 0.07 Instability index 42.35 Isoelectric point 5.97 Charge (pH=7) -7.02 2D Binding mode Binding energy (Kcal/mol) -8.68 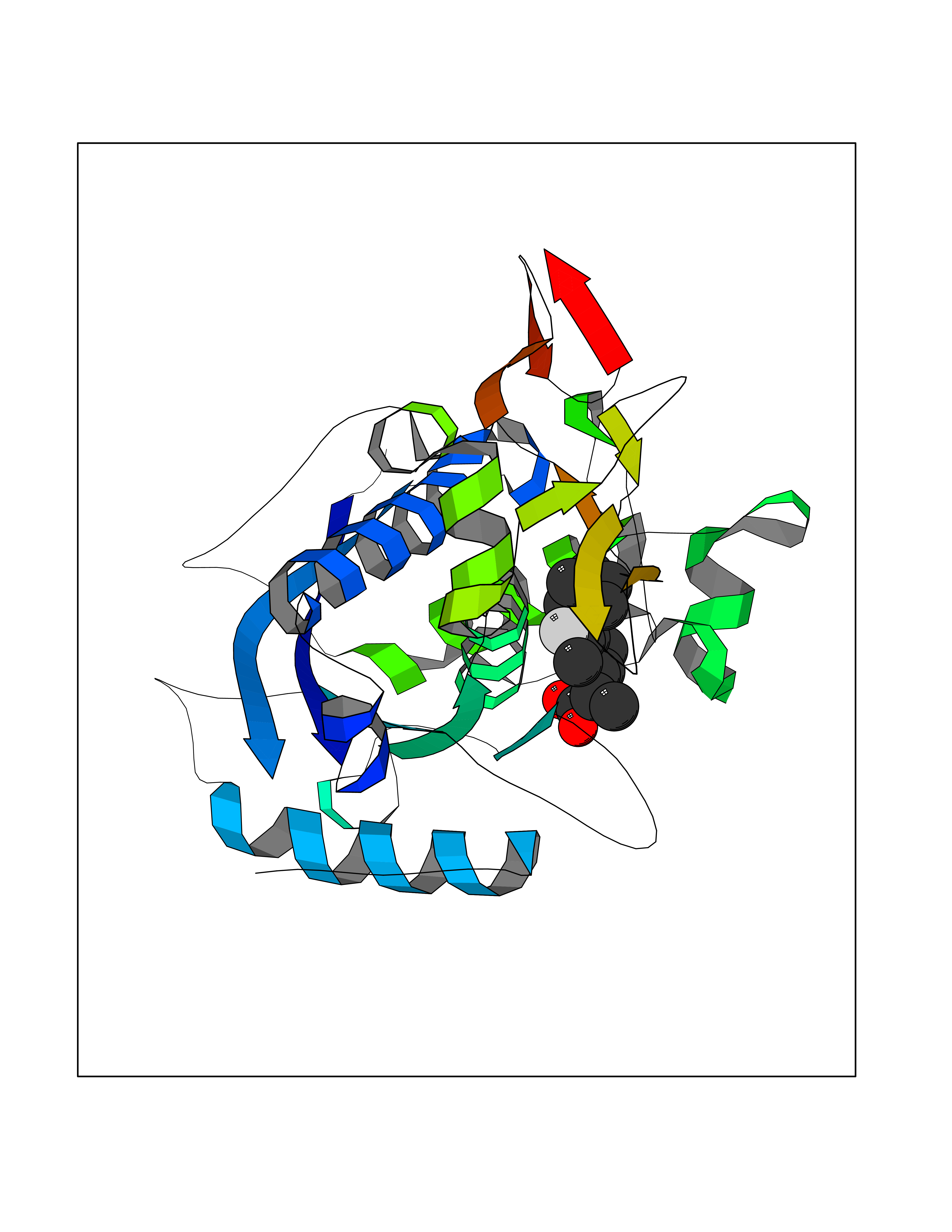 Molscript Map 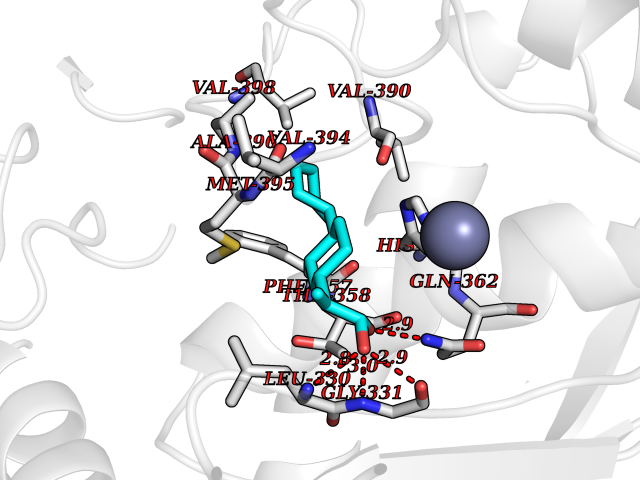 Pymol Map 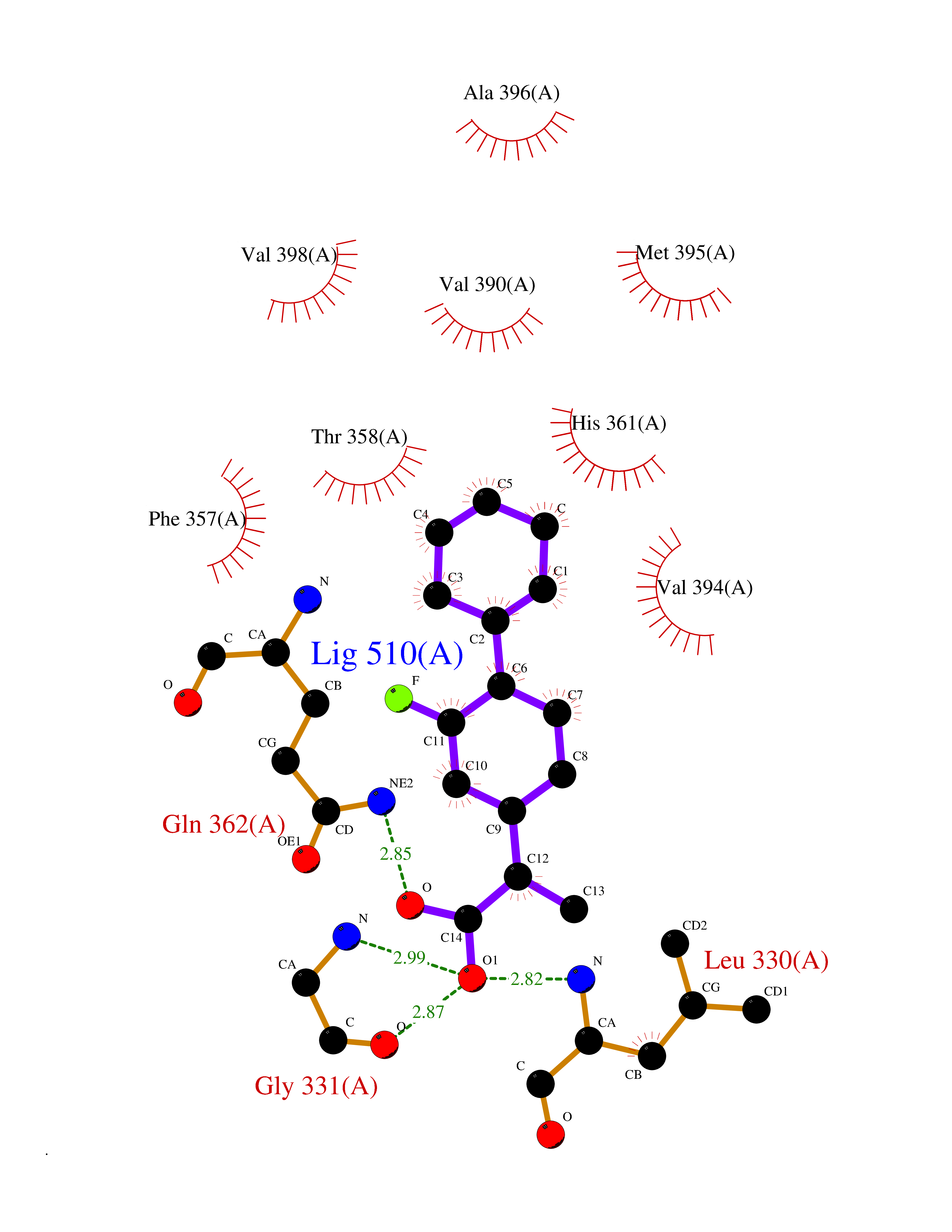 Ligplot Map 3D Binding mode Sequence ASLSRFVETLVVADDKMAAFHGAGLKRYLLTVMAAAAKAFKHPSIRNPVSLVVTRLVILEGPQVGPSAAQTLRSFCAWQRGLNTPEDSDPDHFDTAILFTRQDLCGVSTCDTLGMADVGTVCDPARSCAIVEDDGLQSAFTAAHQLGHVFNMLHDNSKPCISLNGPLSTSRHVMAPVMAHVDPEEPWSPCSARFITDFLDNGYGHCLLDKPEAPLHLPVTFPGKDYDADRQCQLTFGPDSRHCPQLPPPCAALWCSGHLNGHAMCQTKHSPWADGTPCGPAQACMGGRCLH Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Carbonic anhydrase XIV (CA-XIV) | 5CJF | 7.39 | |
Target general information Gen name CA14 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ690/PRO1335; Carbonic anhydrase 14; Carbonate dehydratase XIV Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Reversible hydration of carbon dioxide. Related diseases Isobutyryl-CoA dehydrogenase deficiency (IBDD) [MIM:611283]: An autosomal recessive metabolic disorder characterized by plasma carnitine deficiency and elevated C4-acylcarnitine. Patients manifest variable clinical features including failure to thrive, seizures, anemia, muscular hypotonia and developmental delay. Some patients may be asymptomatic. {ECO:0000269|PubMed:12359132, ECO:0000269|PubMed:15505379, ECO:0000269|PubMed:16857760}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00819; DB00562; DB00606; DB08846; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Disulfide bond; Glycoprotein; Lyase; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29397.4 Length 260 Aromaticity 0.1 Instability index 59.95 Isoelectric point 5.54 Charge (pH=7) -11 2D Binding mode Binding energy (Kcal/mol) -8.21  Molscript Map 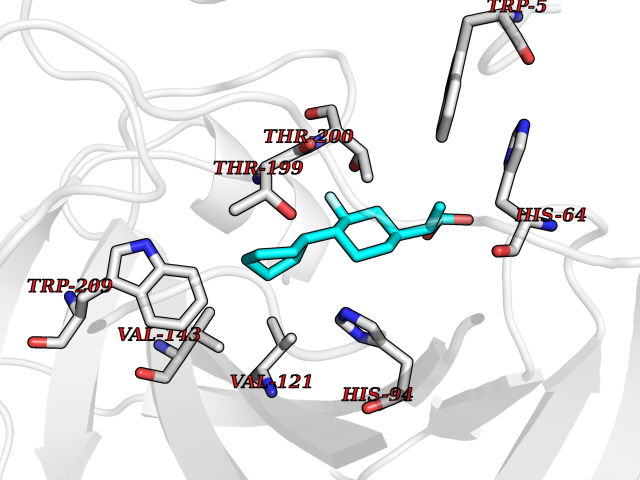 Pymol Map 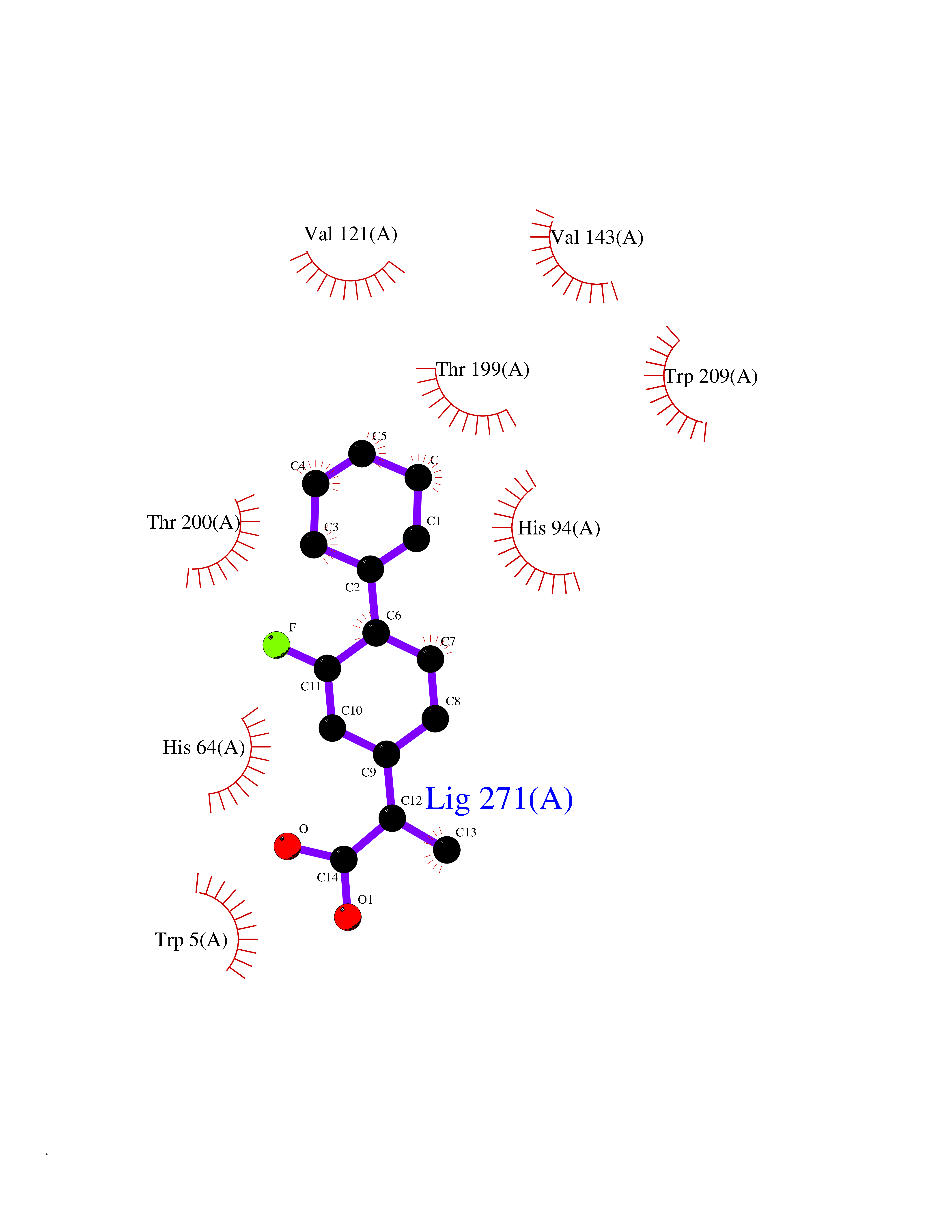 Ligplot Map 3D Binding mode Sequence HWTYEGPHGQDHWPASYPECGNNAQSPIDIQTDSVTFDPDLPALQPGYDQTEPLDLHNNGHTVQLSLPSTLYLGGLPRKYVAAQLHLHWGQKGPGGSEHQINSEATFAELHIVHYDSDSYDSLSEAAERPQGLAVLGILIEVETKNIAYEHILSHLHEVRHKDQKTSVPPFNLRELLPKQGQYFRYNGSLTTPPCYQSVLWTVFYRRSQISMEQLEKLGTLFSTEEEPSKLLVQNYRALQPLNQRMVFASFIQAGSSYTT Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Carbonic anhydrase IV (CA-IV) | 3FW3 | 7.38 | |
Target general information Gen name CA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase 4; Carbonate dehydratase IV; CAIV Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function May stimulate the sodium/bicarbonate transporter activity of SLC4A4 that acts in pH homeostasis. It is essential for acid overload removal from the retina and retina epithelium, and acid release in the choriocapillaris in the choroid. Reversible hydration of carbon dioxide. Related diseases Retinitis pigmentosa 17 (RP17) [MIM:600852]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:15563508, ECO:0000269|PubMed:17652713, ECO:0000269|PubMed:20450258}. The disease is caused by variants affecting the gene represented in this entry. Defective acid overload removal from retina and retinal epithelium, due to mutant CA4, is responsible for photoreceptor degeneration, indicating that impaired pH homeostasis is the most likely cause underlying the RP17 phenotype. Drugs (DrugBank ID) DB00819; DB00436; DB00562; DB01194; DB00606; DB01144; DB00869; DB08846; DB00311; DB00774; DB00703; DB00232; DB09460; DB00273; DB01021; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; GPI-anchor; Lipoprotein; Lyase; Membrane; Metal-binding; Proteomics identification; Reference proteome; Retinitis pigmentosa; Signal; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 27055.7 Length 235 Aromaticity 0.09 Instability index 44.3 Isoelectric point 6.87 Charge (pH=7) -0.36 2D Binding mode Binding energy (Kcal/mol) -7.55  Molscript Map 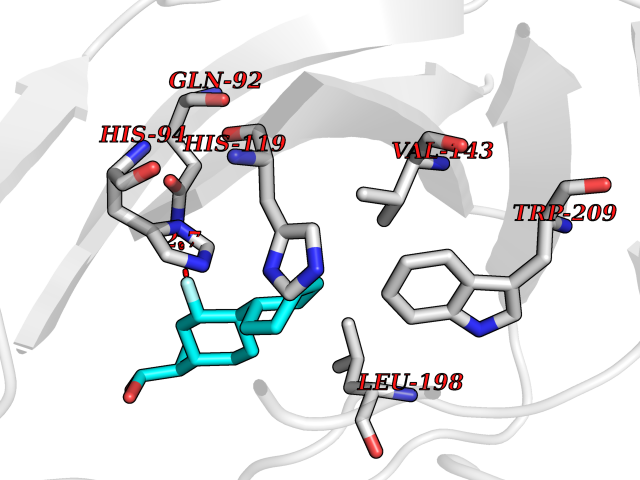 Pymol Map 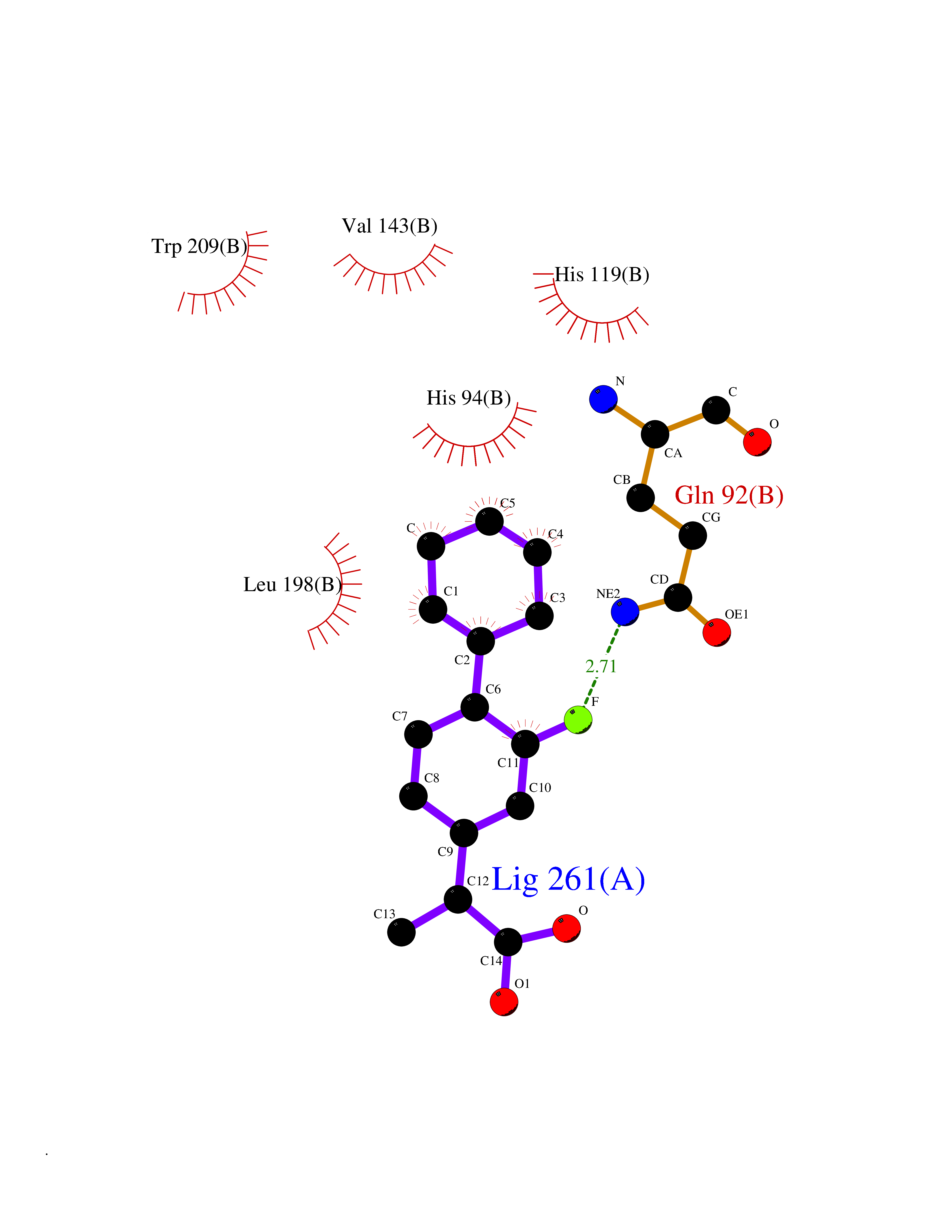 Ligplot Map 3D Binding mode Sequence HWCYEVQLVPVKWGGNCQKDRQSPINIVTTKAKVDKKLGRFFFGYDKKQTWTVQNNGHSVMMLLENKASISGGGLPAPYQAKQLHLHWSDLPYKGSEHSLDGEHFAMEMHIVHEKEEIAVLAFLVEATQVNEGFQPLVEALSNIPKPEMSTTMAESSLLDLLPEEKRHYFRYLGSLTTPTCDEKVVWTVFREPIQLHREQILAFQKLYYDKEQTVSMKDNVRPLQQLGQRTVIKS Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Carbonic anhydrase IX (CA-IX) | 5FL4 | 7.38 | |
Target general information Gen name CA9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Renal cell carcinoma-associated antigen G250; RCC-associated antigen G250; PMW1; P54/58N; Membrane antigen MN; MN; G250 antigen (MN/CA IX/G250); G250; Carbonic anhydrase 9; Carbonate dehydratase IX; C Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Participates in pH regulation. May be involved in the control of cell proliferation and transformation. Appears to be a novel specific biomarker for a cervical neoplasia. Reversible hydration of carbon dioxide. Related diseases Hydroxykynureninuria (HYXKY) [MIM:236800]: An inborn error of amino acid metabolism characterized by massive urinary excretion of large amounts of kynurenine, 3-hydroxykynurenine and xanthurenic acid. Affected individuals manifest renal tubular dysfunction, metabolic acidosis, psychomotor retardation, non-progressive encephalopathy, and muscular hypertonia. {ECO:0000269|PubMed:17334708, ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Vertebral, cardiac, renal, and limb defects syndrome 2 (VCRL2) [MIM:617661]: An autosomal recessive congenital malformation syndrome characterized by vertebral segmentation abnormalities, congenital cardiac defects, renal defects, and distal mild limb defects. {ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00562; DB00606; DB12741; DB08846; DB05304; DB00774; DB09460; DB00909 Interacts with P21291; O76003 EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Cell membrane; Cell projection; Direct protein sequencing; Disulfide bond; Glycoprotein; Lyase; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27522.8 Length 251 Aromaticity 0.08 Instability index 48.97 Isoelectric point 5.48 Charge (pH=7) -7.5 2D Binding mode Binding energy (Kcal/mol) -7.81 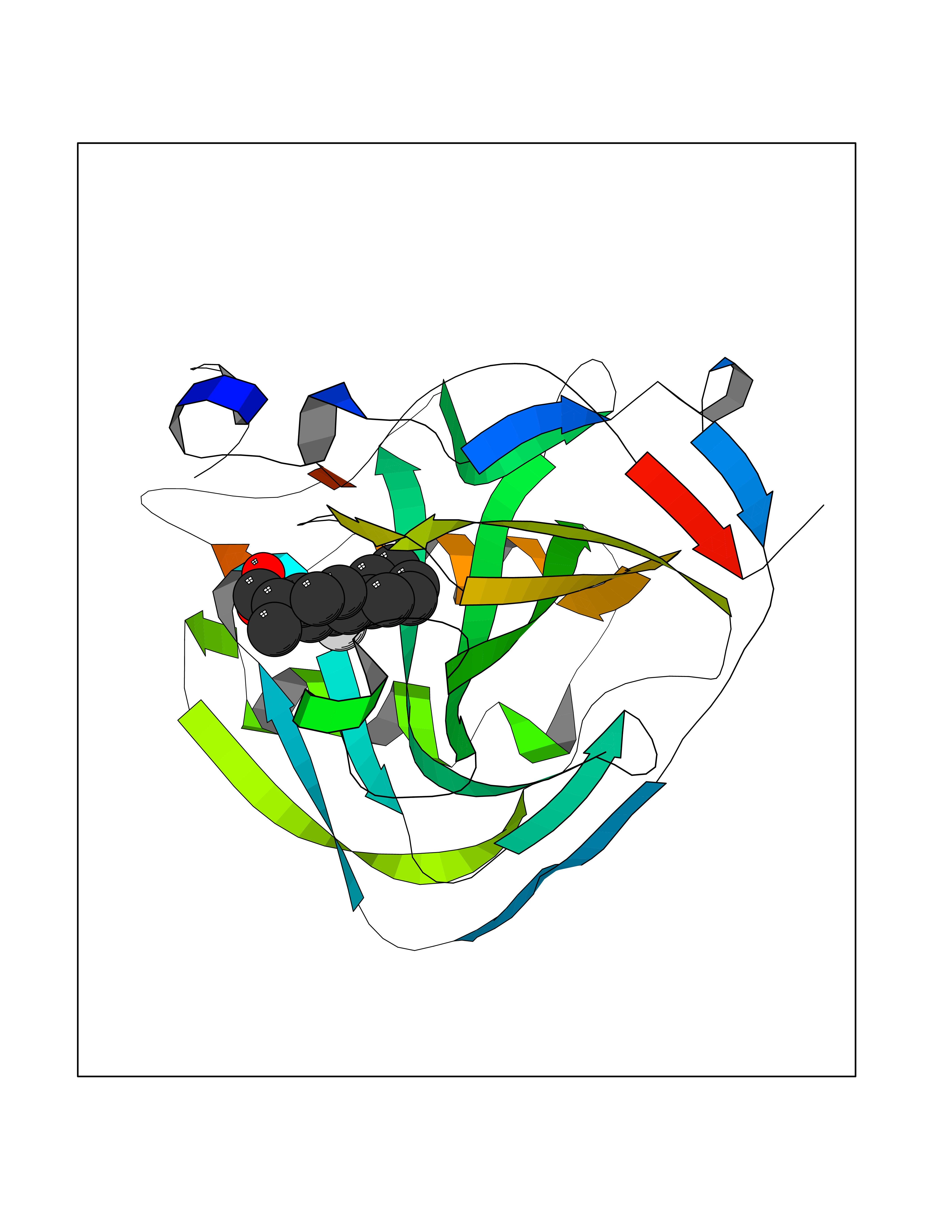 Molscript Map 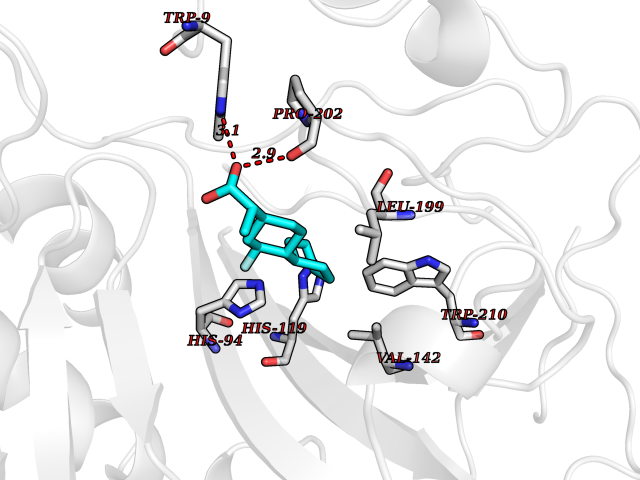 Pymol Map 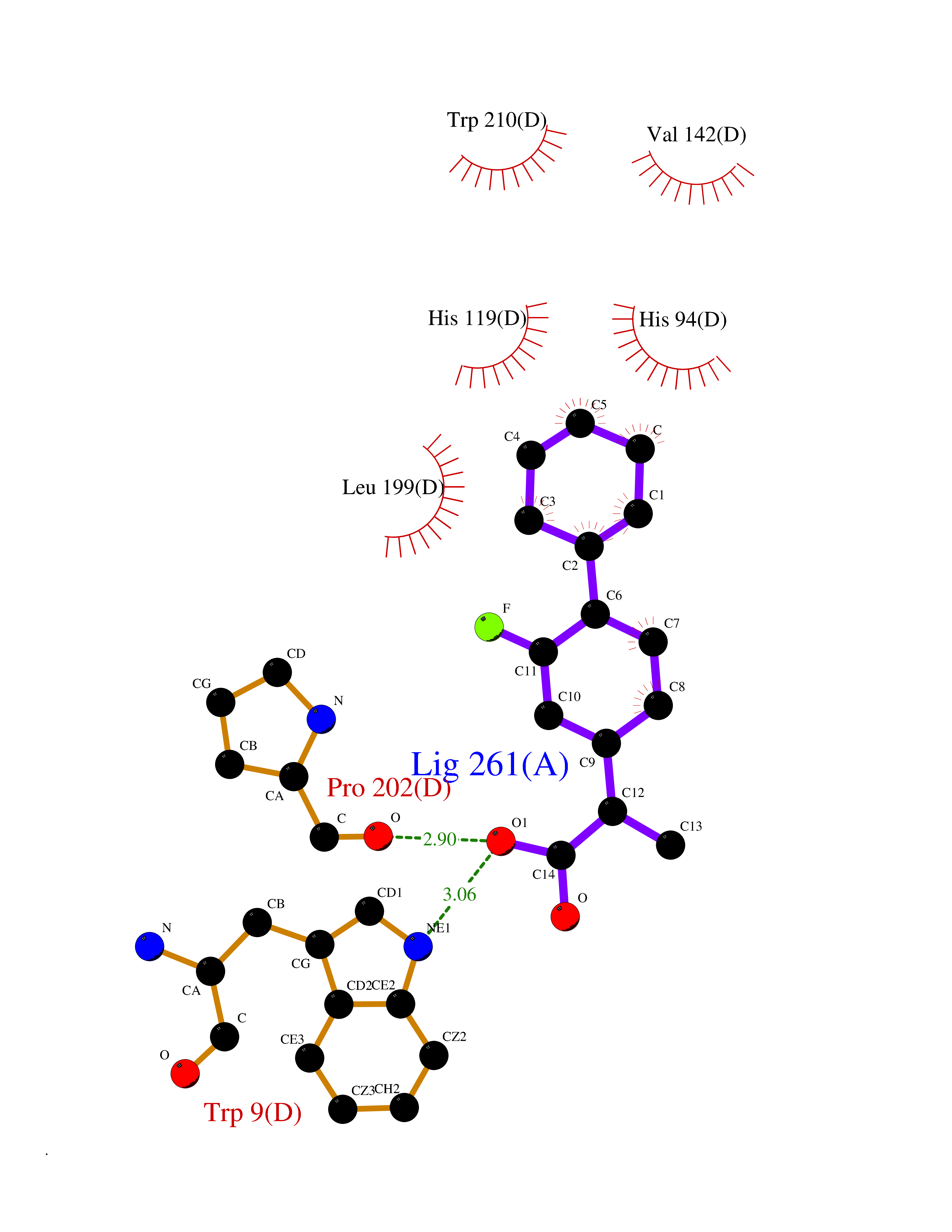 Ligplot Map 3D Binding mode Sequence WRYGGDPPWPRVSPACAGRFQSPVDIRPQLAAFSPALRPLELLGFQLPPLPELRLRNNGHSVQLTLPPGLEMALGPGREYRALQLHLHWGAAGRPGSEHTVEGHRFPAEIHVVHLSTAFARVDEALGRPGGLAVLAAFLEEGPEENSAYEQLLSRLEEIAEEGSETQVPGLDISALLPSDFSRYFQYEGSLTTPPCAQGVIWTVFNQTVMLSAKQLHTLSDTLWGPGDSRLQLNFRATQPLNGRVIEASFP Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Transferrin (TF) | 1RYO | 7.38 | |
Target general information Gen name TF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Siderophilin; Serotransferrin; PRO1400; Beta-1 metal-binding globulin Protein family Transferrin family Biochemical class Transferrin Function It is responsible for the transport of iron from sites of absorption and heme degradation to those of storage and utilization. Serum transferrin may also have a further role in stimulating cell proliferation. Transferrins are iron binding transport proteins which can bind two Fe(3+) ions in association with the binding of an anion, usually bicarbonate. Related diseases Atransferrinemia (ATRAF) [MIM:209300]: A rare autosomal recessive disorder characterized by abnormal synthesis of transferrin leading to iron overload and microcytic hypochromic anemia. {ECO:0000269|PubMed:11110675, ECO:0000269|PubMed:15466165}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01370; DB14517; DB14518; DB01294; DB14526; DB14527; DB11136; DB14528; DB14529; DB14530; DB00515; DB09130; DB11397; DB13949; DB14490; DB14491; DB14488; DB14501; DB14489; DB13257; DB06215; DB06784; DB05260; DB01592; DB00893; DB00677; DB06757; DB11182; DB14520; DB01593; DB14487; DB14533; DB14548 Interacts with O43315; O00501; Q7Z7G2; Q9GZR5; Q9Y282; Q96KR6; P01350; P08034; Q8NBJ4; O15529; Q8TED1; Q7Z5P4; A8MZ59; O15173; Q96TC7; Q3KNW5; Q9BXS9-3; Q99523; O43278-2; Q8N9I0; P02786; Q4KMG9; Q9K0U9; Q09057; Q9K0V0; P02786 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Iron; Iron transport; Metal-binding; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 35854.5 Length 324 Aromaticity 0.1 Instability index 34.17 Isoelectric point 7.58 Charge (pH=7) 1.42 2D Binding mode Binding energy (Kcal/mol) -8.08 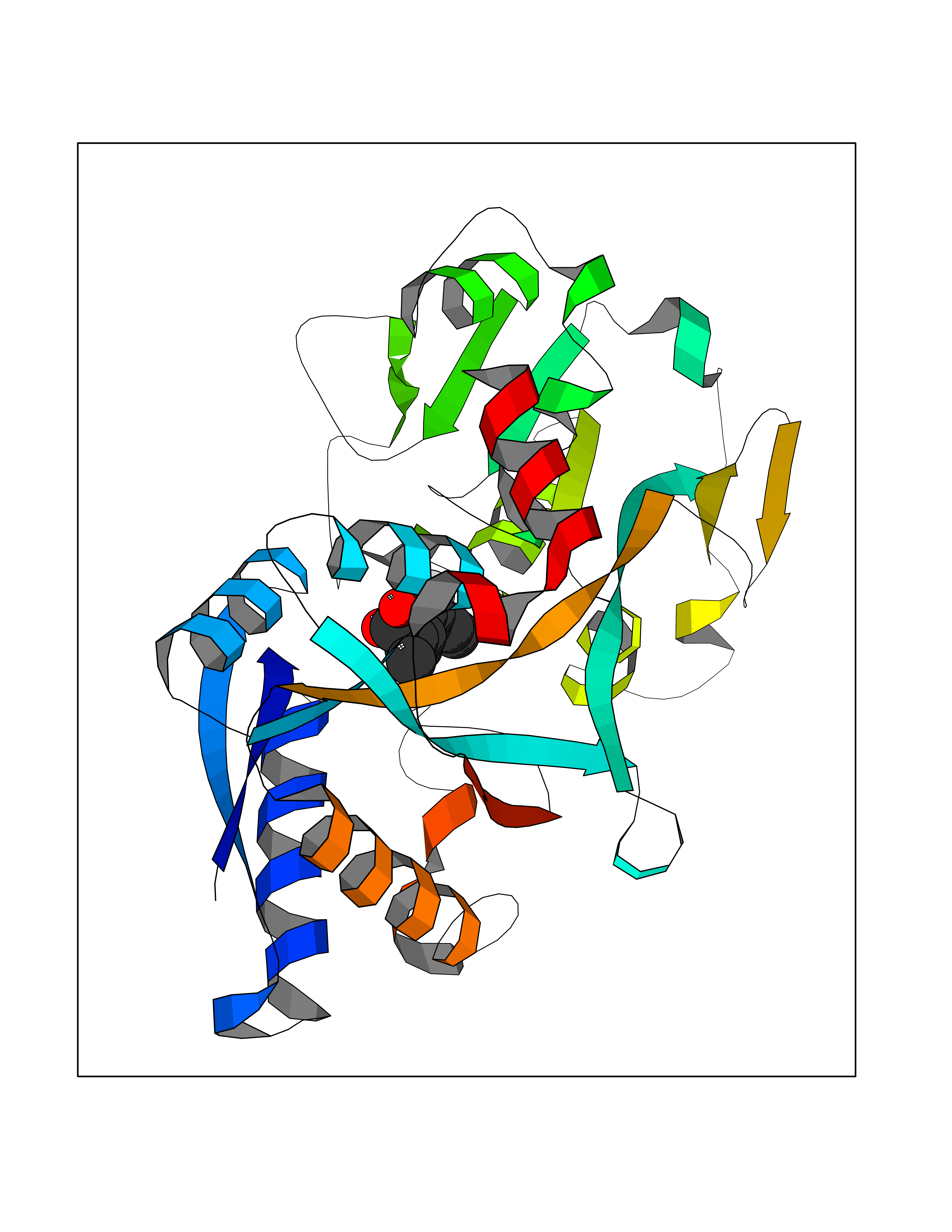 Molscript Map 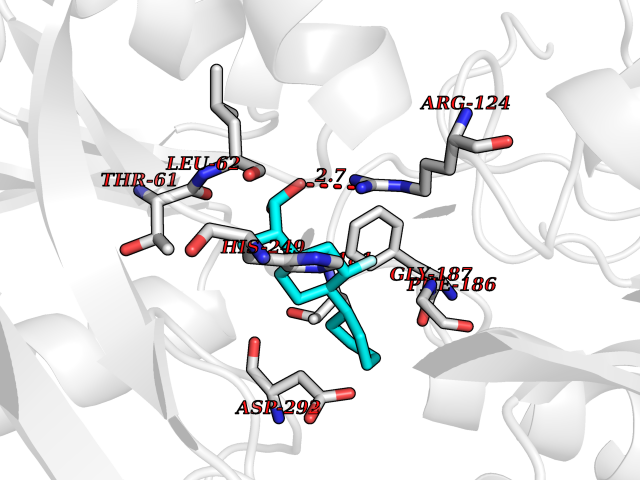 Pymol Map 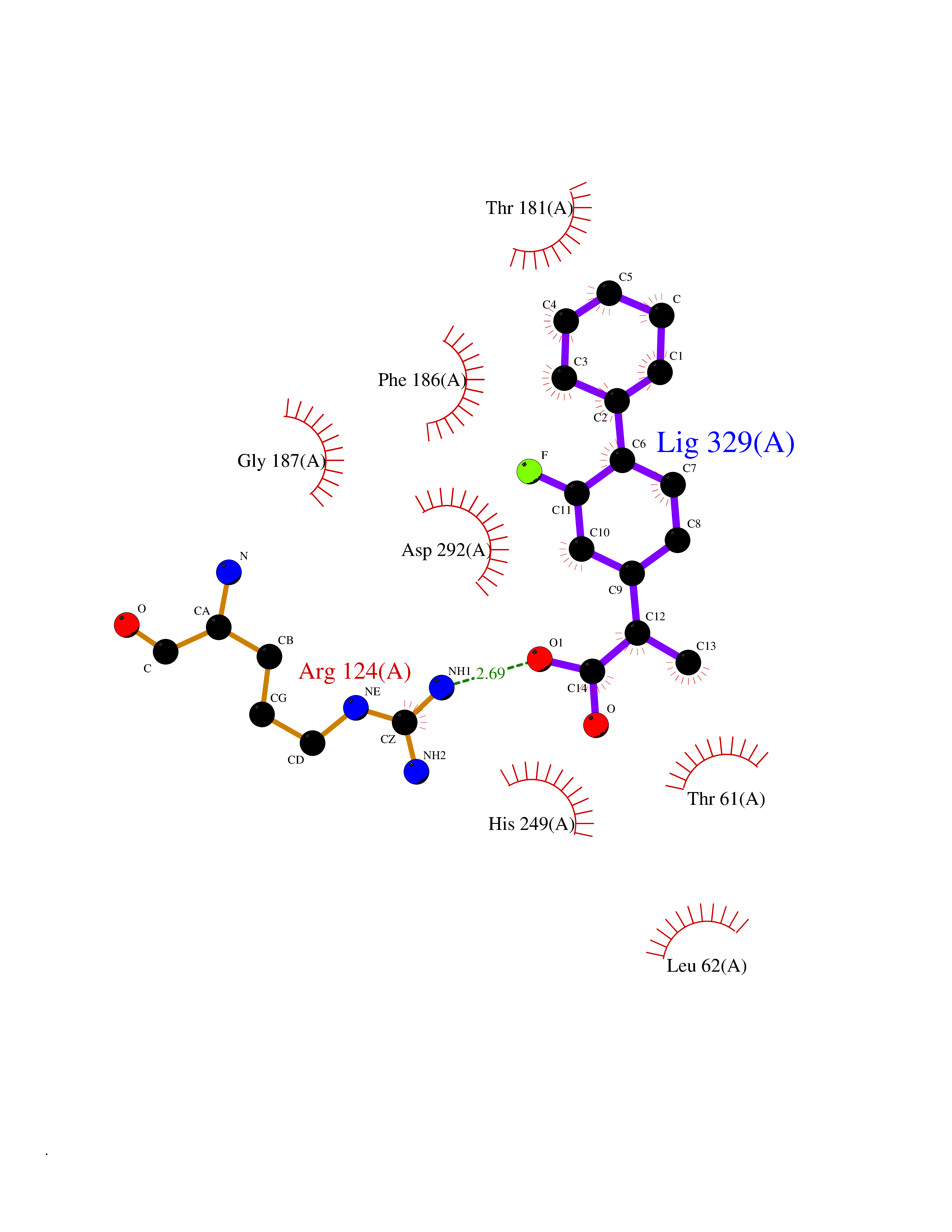 Ligplot Map 3D Binding mode Sequence KTVRWCAVSEHEATKCQSFRDHMKSVIPSDGPSVACVKKASYLDCIRAIAANEADAVTLDAGLVYDAYLAPNNLKPVVAEFYGSKEDPQTFYYAVAVVKKDSGFQMNQLRGKKSCHTGLGRSAGWNIPIGLLYCDLPEPRKPLEKAVANFFSGSCAPCADGTDFPQLCQLCPGCGCSTLNQYFGYSGAFKCLKDGAGDVAFVKHSTIFENLANKADRDQYELLCLDNTRKPVDEYKDCHLAQVPSHTVVARSMGGKEDLIWELLNQAQEHFGKDKSKEFQLFSSPHGKDLLFKDSAHGFLKVPPRMDAKMYLGYEYVTAIRNLR Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | HIF-prolyl hydroxylase 2 (HPH-2) | 6ZBO | 7.38 | |
Target general information Gen name EGLN1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SM-20; Prolyl hydroxylase domain-containing protein 2; PHD2; Hypoxia-inducible factor prolyl hydroxylase 2; HPH-2; HIF-PH2; Egl nine homolog 1; C1orf12 Protein family NA Biochemical class Paired donor oxygen oxidoreductase Function Cellular oxygen sensor that catalyzes, under normoxic conditions, the post-translational formation of 4-hydroxyproline in hypoxia-inducible factor (HIF) alpha proteins. Hydroxylates a specific proline found in each of the oxygen-dependent degradation (ODD) domains (N-terminal, NODD, and C-terminal, CODD) of HIF1A. Also hydroxylates HIF2A. Has a preference for the CODD site for both HIF1A and HIF1B. Hydroxylated HIFs are then targeted for proteasomal degradation via the von Hippel-Lindau ubiquitination complex. Under hypoxic conditions, the hydroxylation reaction is attenuated allowing HIFs to escape degradation resulting in their translocation to the nucleus, heterodimerization with HIF1B, and increased expression of hypoxy-inducible genes. EGLN1 is the most important isozyme under normoxia and, through regulating the stability of HIF1, involved in various hypoxia-influenced processes such as angiogenesis in retinal and cardiac functionality. Target proteins are preferentially recognized via a LXXLAP motif. Related diseases Erythrocytosis, familial, 3 (ECYT3) [MIM:609820]: An autosomal dominant disorder characterized by elevated serum hemoglobin and hematocrit, and normal serum erythropoietin levels. {ECO:0000269|PubMed:16407130, ECO:0000269|PubMed:17579185}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB11682; DB14490; DB14491; DB14488; DB14501; DB14489; DB08687; DB01592; DB07112; DB04847; DB12255 Interacts with Q99814; Q14318; Q16665; Q13438; PRO_0000037551 [Q9WMX2] EC number EC 1.14.11.29 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Congenital erythrocytosis; Cytoplasm; Dioxygenase; Disease variant; Iron; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; S-nitrosylation; Vitamin C; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,D Molecular weight (Da) 45717.8 Length 406 Aromaticity 0.11 Instability index 24.41 Isoelectric point 7.59 Charge (pH=7) 1.54 2D Binding mode Binding energy (Kcal/mol) -8.49 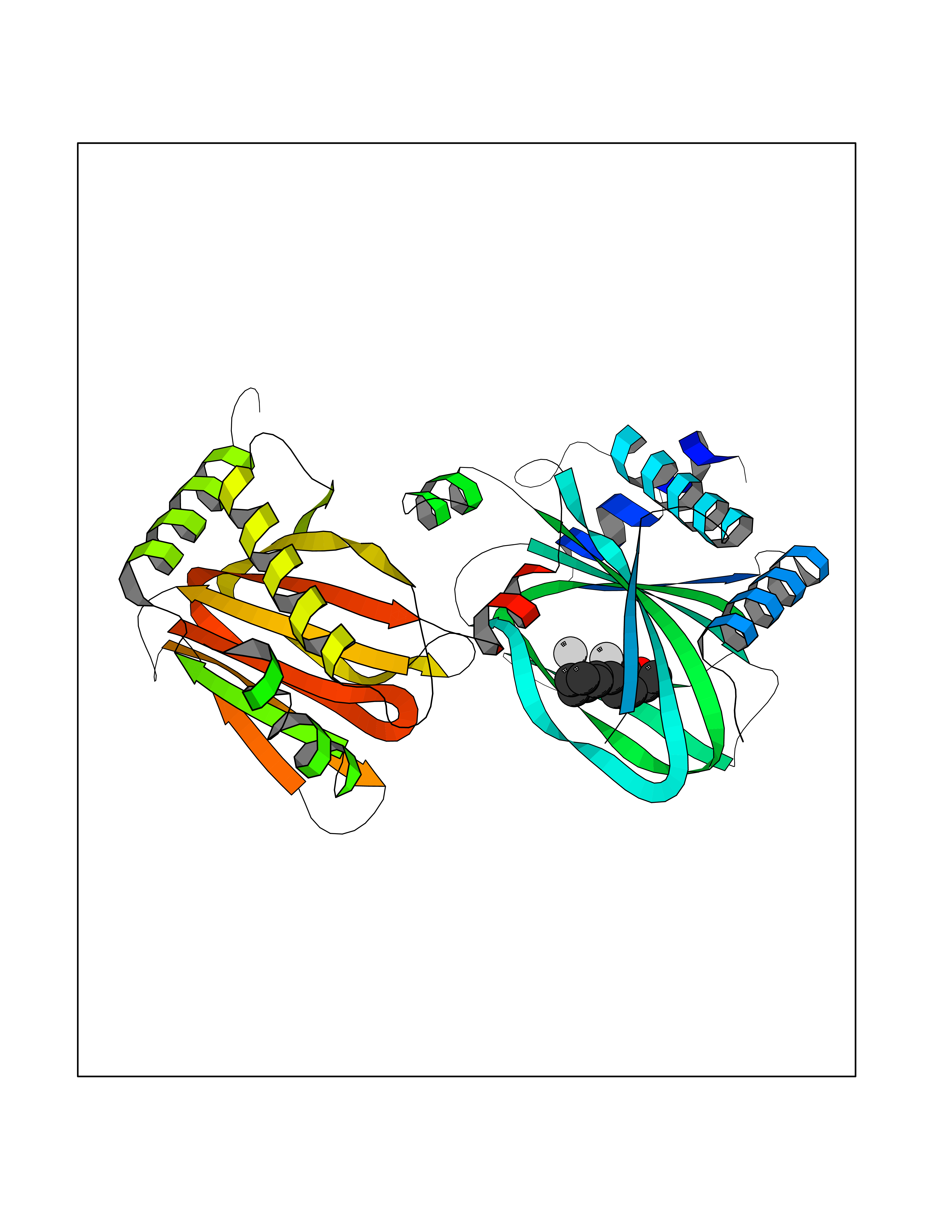 Molscript Map 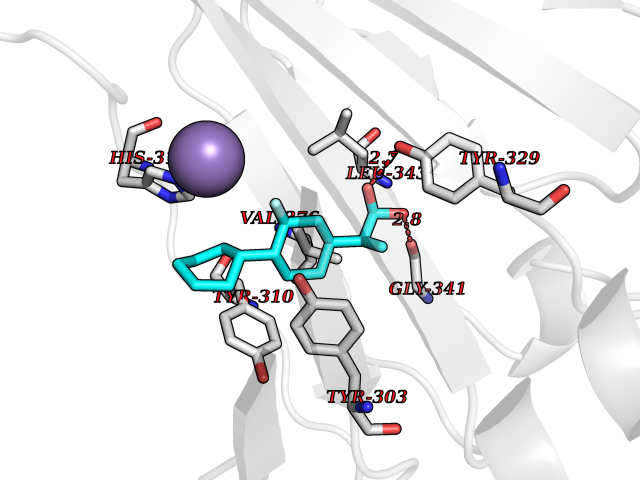 Pymol Map 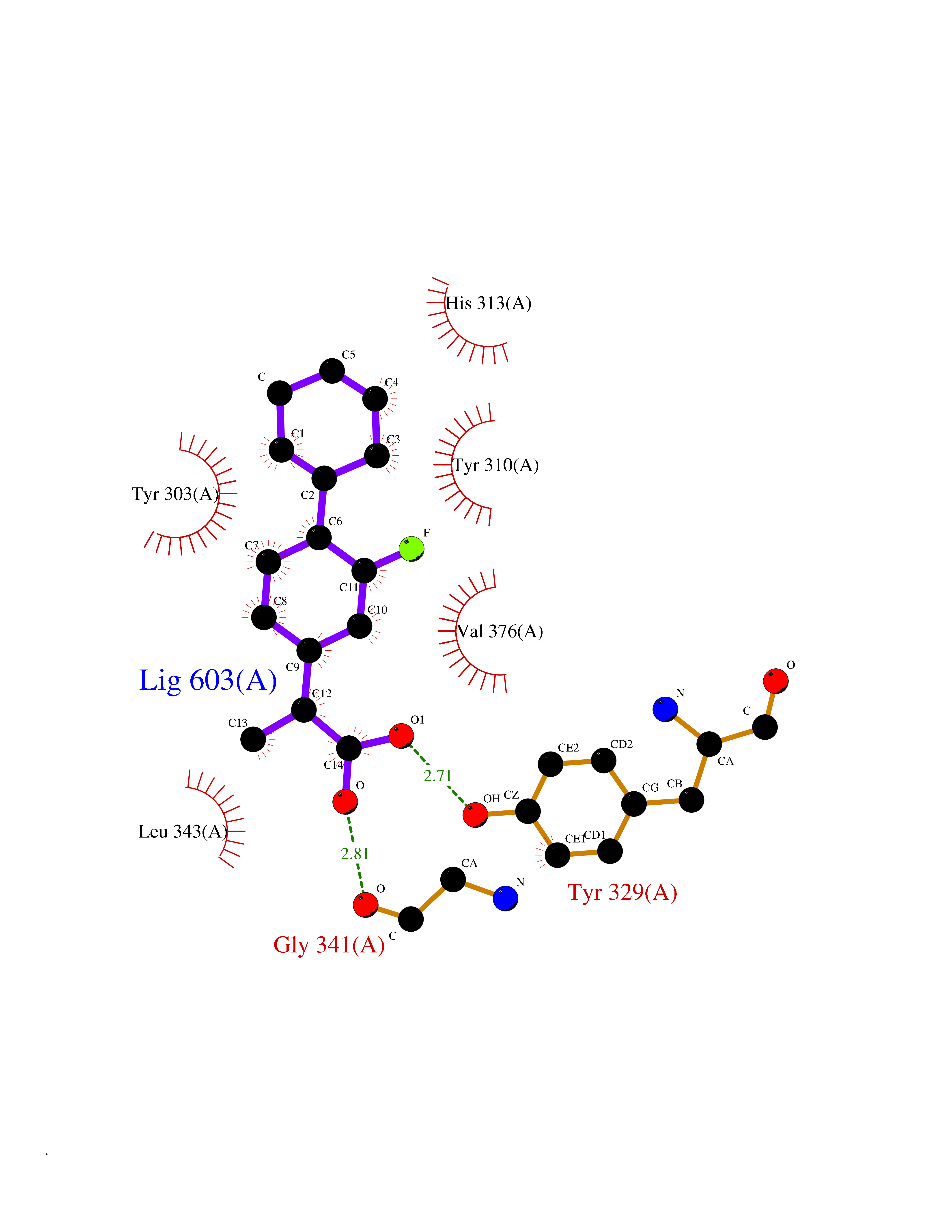 Ligplot Map 3D Binding mode Sequence LPALKLALEYIVPCMNKHGICVVDDFLGKETGQQIGDEVRALHDTGKFTGDKITWIEGKEPGCETIGLLMSSMDDLIRHCNGKLGSYKINGRTKAMVACYPGNGTGYVRHVDNPNGDGRCVTCIYYLNKDWDAKVSGGILRIFPEGKAQFADIEPKFDRLLFFWSDRRNPHEVQPAYATRYAITVWYFDADERARAKVKYLTGELPALKLALEYIVPCMNKHGICVVDDFLGKETGQQIGDEVRALHDTGKFTGDKITWIEGKEPGCETIGLLMSSMDDLIRHCNGKLGSYKINGRTKAMVACYPGNGTGYVRHVDNPNGDGRCVTCIYYLNKDWDAKVSGGILRIFPEGKAQFADIEPKFDRLLFFWSDRRNPHEVQPAYATRYAITVWYFDADERARAKVKYLT Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Phosphodiesterase 4A (PDE4A) | 2QYK | 7.35 | |
Target general information Gen name PDE4A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-specific 3',5'-cyclic phosphodiesterase 4A; Type 4A cAMP phosphodiesterase; PDE46; DPDE2 Protein family Cyclic nucleotide phosphodiesterase family, PDE4 subfamily Biochemical class Phosphoric diester hydrolase Function Hydrolyzes the second messenger cAMP, which is a key regulator of many important physiological processes. Related diseases Developmental and epileptic encephalopathy 50 (DEE50) [MIM:616457]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE50 is an autosomal recessive, progressive disease with onset in infancy and favorable response to treatment with oral uridine. {ECO:0000269|PubMed:25678555, ECO:0000269|PubMed:28087732}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06842; DB08299; DB01427; DB00201; DB05219; DB00975; DB06751; DB00651; DB00824; DB05266; DB01088; DB01303; DB01791; DB06479; DB01656; DB01954; DB00277; DB08811; DB09283 Interacts with P55212; O14569; P13473-2; Q9UJX0; P16118; O75400-2; Q9Y371 EC number EC 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cell membrane; Cell projection; Cytoplasm; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 38579.6 Length 335 Aromaticity 0.08 Instability index 37.43 Isoelectric point 5.01 Charge (pH=7) -19.92 2D Binding mode Binding energy (Kcal/mol) -8.54 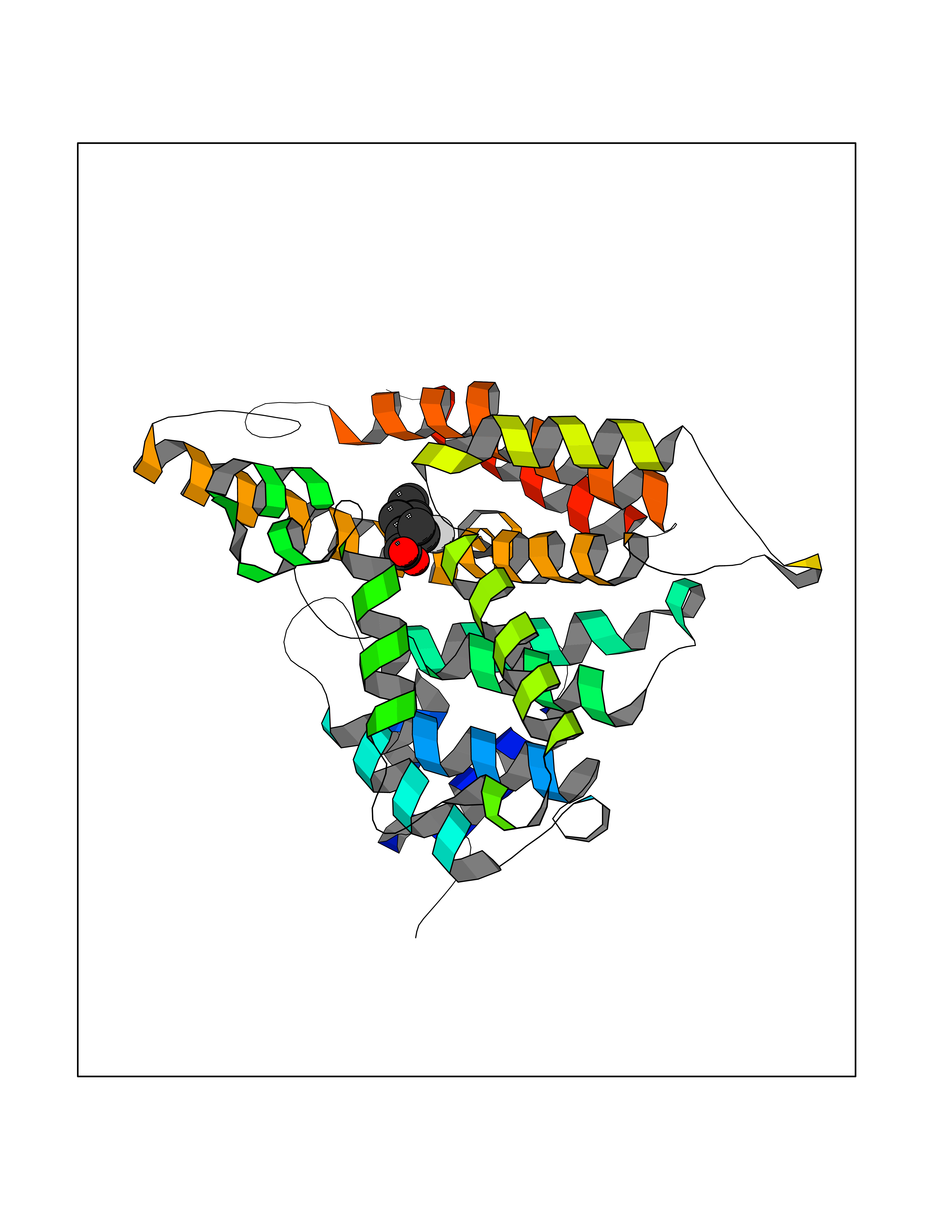 Molscript Map 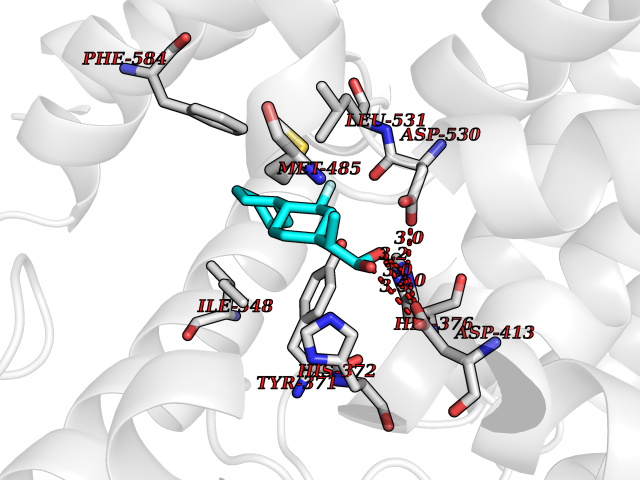 Pymol Map 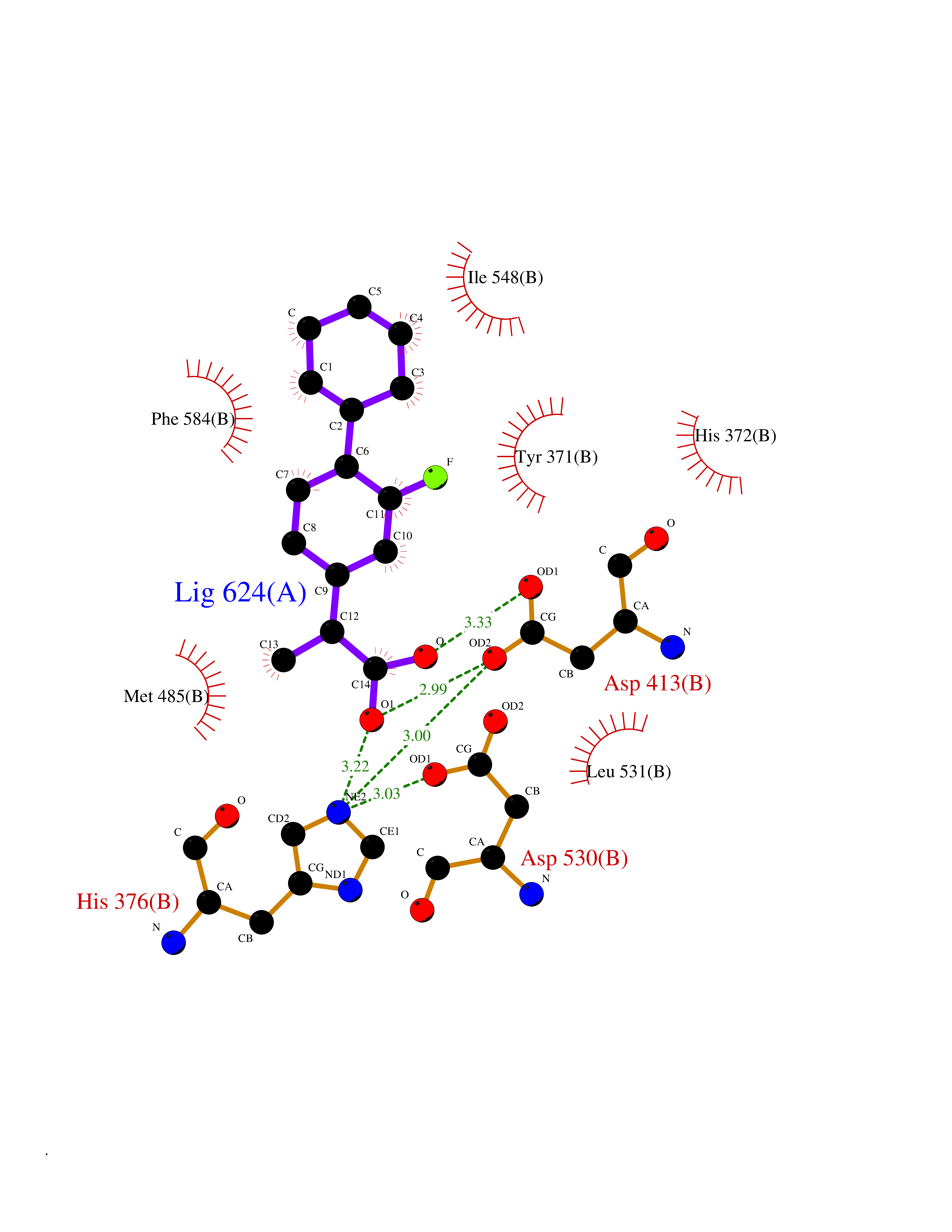 Ligplot Map 3D Binding mode Sequence HMNIPRFGVKTDQEELLAQELENLNKWGLNIFCVSDYAGGRSLTCIMYMIFQERDLLKKFRIPVDTMVTYMLTLEDHYHADVAYHNSLHAADVLQSTHVLLATPALDAVFTDLEILAALFAAAIHDVDHPGVSNQFLINTNSELALMYNDESVLENHHLAVGFKLLQEDNCDIFQNLSKRQRQSLRKMVIDMVLATDMSKHMTLLADLKTMVETKKVTSSGVLLLDNYSDRIQVLRNMVHCADLSNPTKPLELYRQWTDRIMAEFFQQGDRERERGMEISPMCDKHTASVEKSQVGFIDYIVHPLWETWADLVHPDAQEILDTLEDNRDWYYSAI Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Guanidinoacetate N-methyltransferase | 3ORH | 7.35 | |
Target general information Gen name GAMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, RMT2 methyltransferase family Biochemical class Transferase Function Guanidinoacetate N-methyltransferase activity.Methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 2 (CCDS2) [MIM:612736]: An autosomal recessive disorder characterized by developmental delay and regression, intellectual disability, severe disturbance of expressive and cognitive speech, intractable seizures, movement disturbances, severe depletion of creatine and phosphocreatine in the brain, and accumulation of guanidinoacetic acid in brain and body fluids. {ECO:0000269|PubMed:12468279, ECO:0000269|PubMed:15108290, ECO:0000269|PubMed:15651030, ECO:0000269|PubMed:16293431, ECO:0000269|PubMed:16855203, ECO:0000269|PubMed:17101918, ECO:0000269|PubMed:17466557, ECO:0000269|PubMed:19388150, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:24415674, ECO:0000269|PubMed:8651275}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00148; DB02751; DB00536; DB13191; DB01752 Interacts with O95363; Q969Q5; Q9HCM9-2 EC number 2.1.1.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Disease variant; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 24656 Length 219 Aromaticity 0.11 Instability index 46.5 Isoelectric point 5.91 Charge (pH=7) -4.34 2D Binding mode Binding energy (Kcal/mol) -8.44 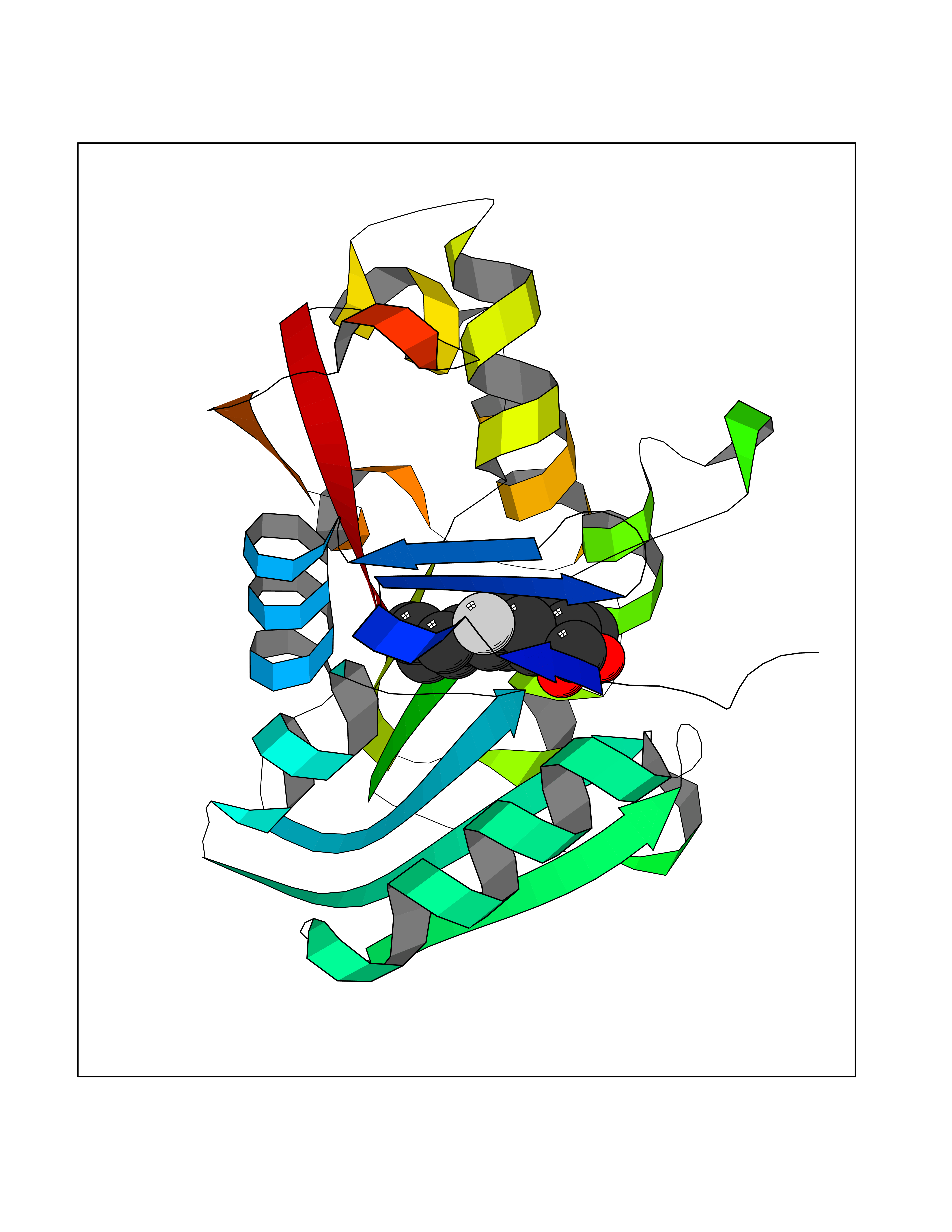 Molscript Map 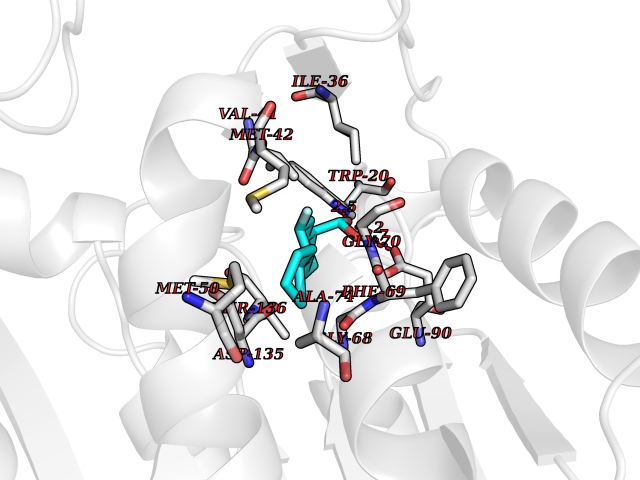 Pymol Map 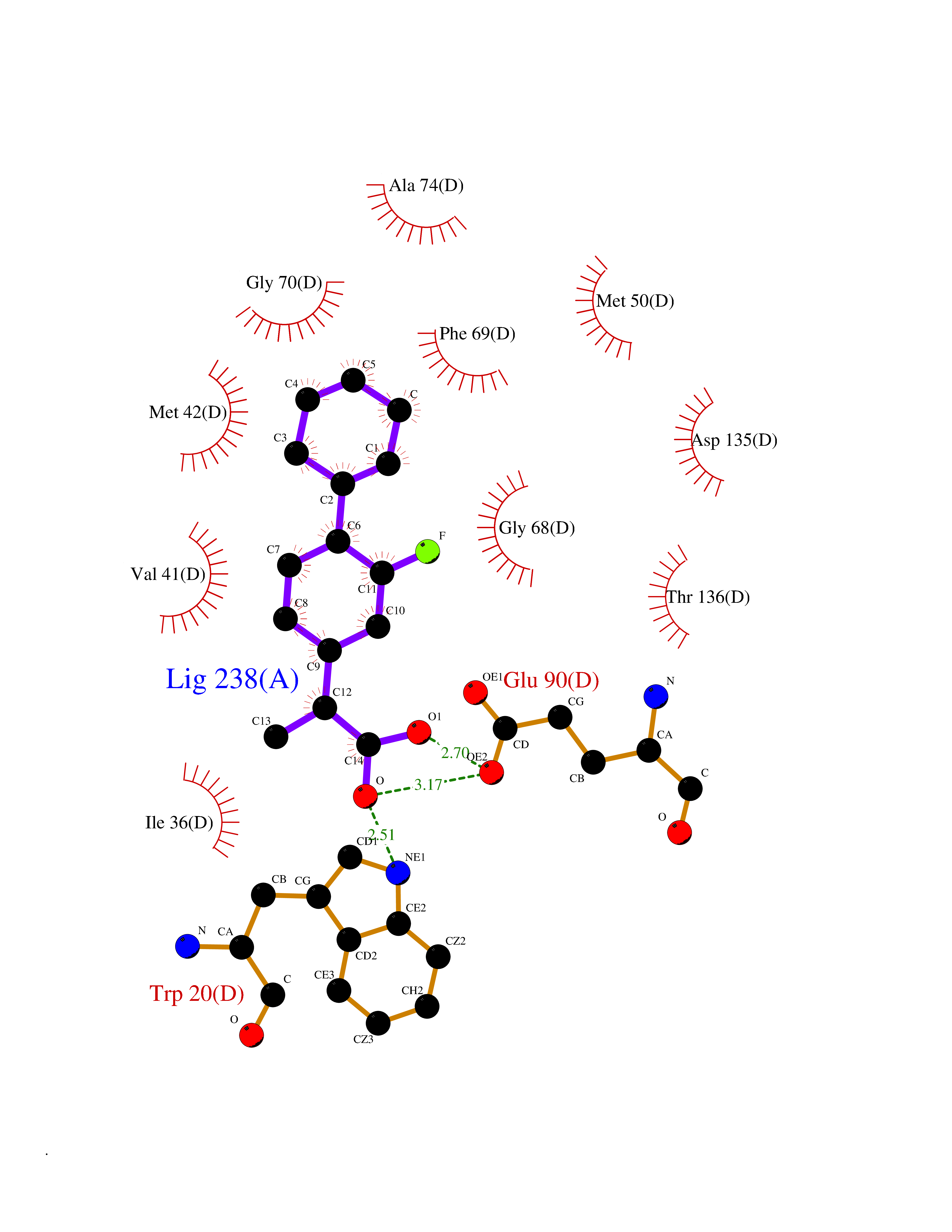 Ligplot Map 3D Binding mode Sequence PAWGAAPAAYDAADTHLRILGKPVMERWETPYMHALAAAASSKGGRVLEVGFGMAIAASKVQEAPIDEHWIIECNDGVFQRLRDWAPRQTHKVIPLKGLWEDVAPTLPDGHFDGILYDTYPLSEETWHTHQFNFIKNHAFRLLKPGGVLTYCNLTSWGELMKSKYSDITIMFEETQVPALLEAGFRRENIRTEVMALVPPADCRYYAFPQMITPLVTKG Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Aromatic-L-amino-acid decarboxylase (DDC) | 3RCH | 7.34 | |
Target general information Gen name DDC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms DOPA decarboxylase; AADC Protein family Group II decarboxylase family Biochemical class Carbon-carbon lyase Function Catalyzes the decarboxylation of L-3,4-dihydroxyphenylalanine (DOPA) to dopamine, L-5-hydroxytryptophan to serotonin and L-tryptophan to tryptamine. Related diseases Aromatic L-amino-acid decarboxylase deficiency (AADCD) [MIM:608643]: An inborn error in neurotransmitter metabolism that leads to combined serotonin and catecholamine deficiency. It causes developmental and psychomotor delay, poor feeding, lethargy, ptosis, intermittent hypothermia, gastrointestinal disturbances. The onset is early in infancy and inheritance is autosomal recessive. {ECO:0000269|PubMed:14991824, ECO:0000269|PubMed:15079002, ECO:0000269|Ref.12}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB12783; DB00190; DB00260; DB06262; DB13848; DB00875; DB01235; DB00968; DB00114; DB00150 Interacts with P10275 EC number EC 4.1.1.28 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Catecholamine biosynthesis; Decarboxylase; Disease variant; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome; Repeat Protein physicochemical properties Chain ID A,B Molecular weight (Da) 41117.4 Length 369 Aromaticity 0.09 Instability index 37.52 Isoelectric point 8.26 Charge (pH=7) 3.76 2D Binding mode Binding energy (Kcal/mol) -7.94 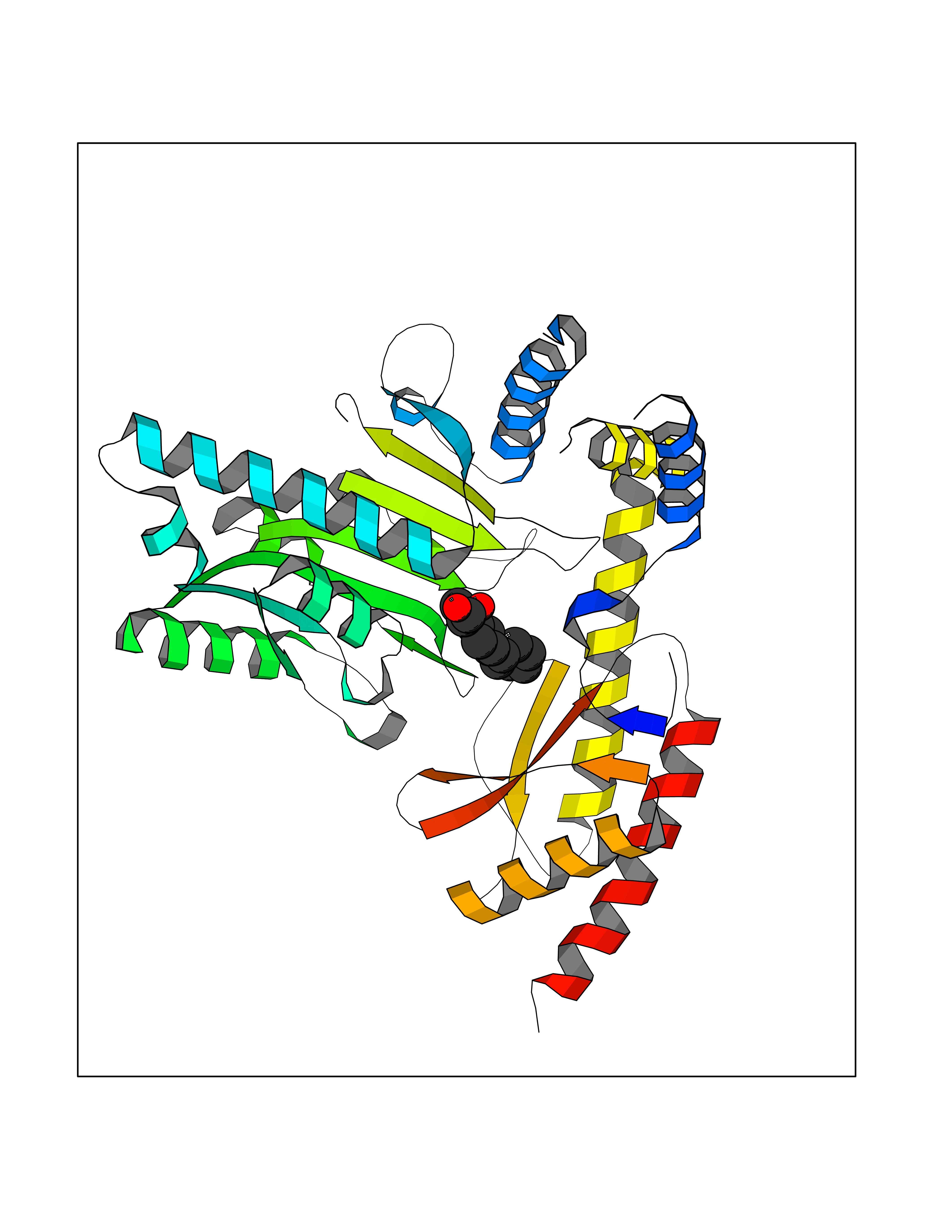 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence HSPYFFAYFPTASSYPAMLADMLCGAIGASPACTELETVMMDWLGKMLELPKAFLNEKAGEGGGVIQGSASEATLVALLAARTKVIHRLQAASPELTQAAIMEKLVAYSSDQAHSSVERAGLIGGVKLKAIPSDGNFAMRASALQEALERDKAAGLIPFFMVATLGTTTCCSFDNLLEVGPICNKEDIWLHVDAAYAGSAFICPEFRHLLNGVEFADSFNFNPHKWLLVNFDCSAMWVKKRTDLRFRSLKMWFVFRMYGVKGLQAYIRKHVQLSHEFESLVRQDPRFEICVEVILGLVCFRLKGSNKVNEALLQRINSAKKIHLVPCHLRDKFVLRFAICSRTVESAHVQRAWEHIKELAADVLRAERE Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Phosphodiesterase 3A (PDE3A) | 7EG0 | 7.33 | |
Target general information Gen name PDE3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-inhibited 3',5'-cyclic phosphodiesterase A; Phosphodiesterase 3A; Cyclic GMP-inhibited phosphodiesterase A; Cyclic GMP inhibited phosphodiesterase A; CGI-PDE A Protein family Cyclic nucleotide phosphodiesterase family, PDE3 subfamily Biochemical class Phosphoric diester hydrolase Function Cyclic nucleotide phosphodiesterase with a dual-specificity for the second messengers cAMP and cGMP, which are key regulators of many important physiological processes. Related diseases Hypertension and brachydactyly syndrome (HTNB) [MIM:112410]: A syndrome characterized by brachydactyly type E, severe salt-independent but age-dependent hypertension, an increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, and altered baroreflex blood pressure regulation. It results in death from stroke before age 50 years when untreated. Brachydactyly type E is characterized by shortening of the fingers mainly in the metacarpals and metatarsals. {ECO:0000269|PubMed:25961942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01223; DB01427; DB00261; DB00201; DB01166; DB04880; DB05266; DB00922; DB00235; DB01303; DB00277; DB08811; DB09283 Interacts with Q9Y6D6; Q9Y6D5 EC number EC 3.1.4.17 Uniprot keywords 3D-structure; cAMP; cGMP; Cytoplasm; Direct protein sequencing; Disease variant; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 108171 Length 939 Aromaticity 0.11 Instability index 39.75 Isoelectric point 6.53 Charge (pH=7) -4.15 2D Binding mode Binding energy (Kcal/mol) -8.47  Molscript Map  Pymol Map 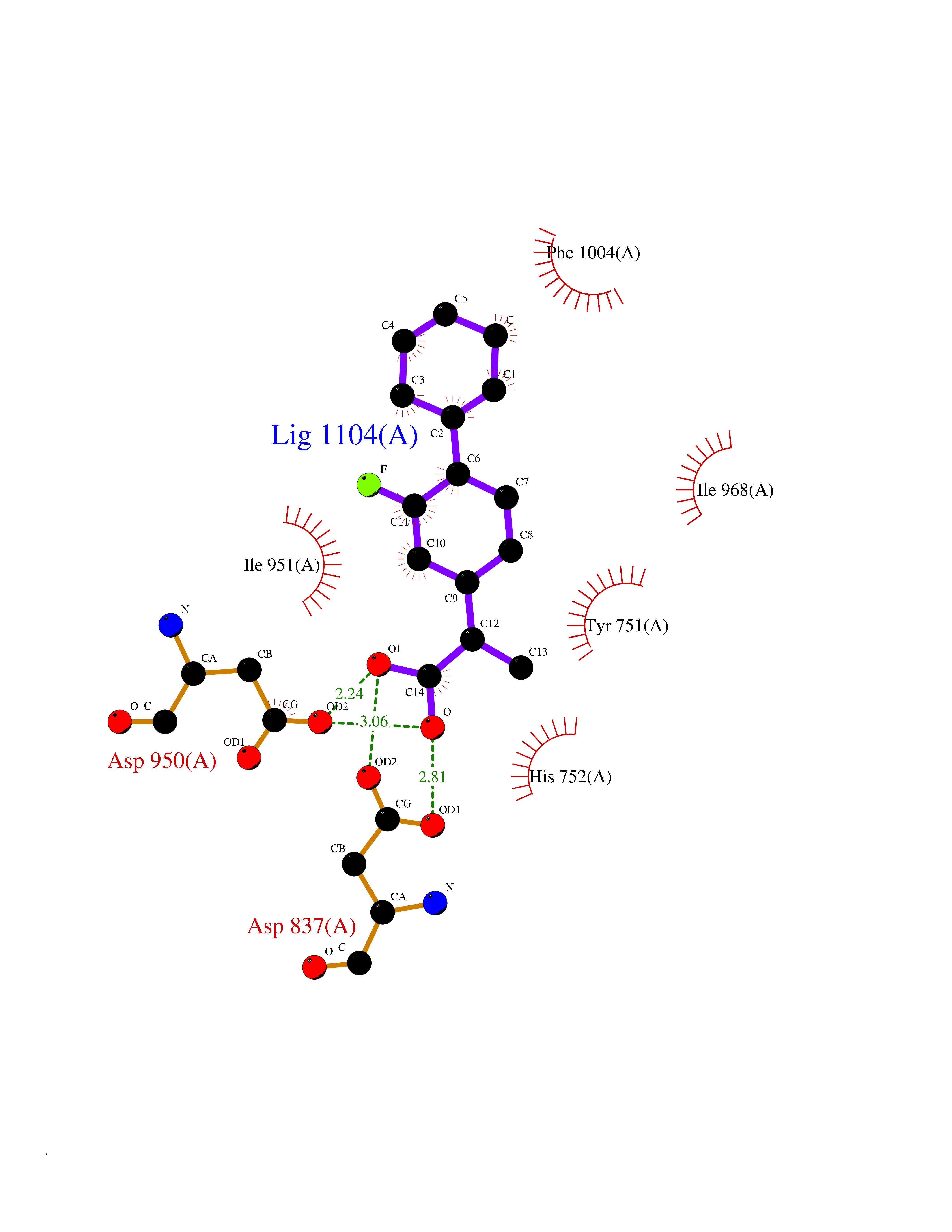 Ligplot Map 3D Binding mode Sequence KPILAPEPLVMDNLDSIMEQLNTWNFPIFDLVENIGRKCGRILSQVSYRLFEDMGLFEAFKIPIREFMNYFHALEIGYRDIPYHNRIHATDVLHAVWYLTTQPIPGLSTVGYVFSKTYNVTDDKYGCLSGNIPALELMALYVAAAMHDYDHPGRTNAFLVATSAPQAVLYNDRSVLENHHAAAAWNLFMSRPEYNFLINLDHVEFKHFRFLVIEAILATDLKKHFDFVAKFNGKVNDDVGIDWTNENDRLLVCQMCIKLADINGPAKCKELHLQWTDGIVNEFYEQGDEEASLGLPISPFMDRSAPQLANLQESFISHIVGPLCNSYDSAGLMPGKWVEKIYCQITQHLLQNHKMWKKVIEEEQRLAGIENQNISVDLETNYAELVLDVGRVTLGENSRKKMKDCKLRKKQNESVSRAMCALLNSGGGVIKAEIENEDYSYTKDGIGLDLENSFSNILLFVPEYLDFMQNGNYFLIFVKSWSLNTSGLRITTLSSNLYKRDITSAKVMNATAALEFLKDMKKTRGRLYLRPELLAKRPCVDIQEENNMKALAGVFFDRTELDRKEKLTFTESTHVEIKNFSTERLLQRIKEILPQYVSAFANTDGGYLFIGLNEDKEIIGFKAEMSDLDDLEREIEKSIRKMPVHHFCMEKKKINYSCKFLGVYDKGSLCGYVCALRVERFCCAVFAKEPDSWHVKDNRVMQLTRKEWIQFMVEAEPKFSSAYEEVISQINTSLPAPHSWPLLEWQRQRHHCPGLSGRITYTPENLCRKLFLQHEGLKQLICEEMSSVRKGSLIFSRSWSVDLGLQENHKVLCDALLISQDSPPVLYTFHMVQDEEFKGYSTQTALTLKQKLAKIGGYTKKVCVMTKIFYLSPEGMTSCQYDLRSQVIYPESYYFTRRKYLLKALFKALKRLKSLRDQFSFAENLYQIIGIDCFQKNDK Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Bifunctional aminoacyl-tRNA synthetase (EPRS) | 4HVC | 7.32 | |
Target general information Gen name EPRS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms QPRS; QARS; ProlinetRNA ligase; PIG32; GlutamatylprolyltRNA synthetase; Glutamatyl-prolyl-tRNA synthetase; GluRS; EPRS; Cell proliferationinducing gene 32 protein; Cell proliferation-inducing gene 32 Protein family Class-I aminoacyl-tRNA synthetase family, Glutamate--tRNA ligase type 2 subfamily; Class-II aminoacyl-tRNA synthetase family Biochemical class Carbon-oxygen ligase Function The phosphorylation of EPRS, induced by interferon-gamma, dissociates the protein from the aminoacyl-tRNA synthetase multienzyme complex and recruits it to the GAIT complex that binds to stem loop-containing GAIT elements in the 3'-UTR of diverse inflammatory mRNAs (such as ceruplasmin), suppressing their translation. Interferon-gamma can therefore redirect, in specific cells, the EPRS function from protein synthesis to translation inhibition. Also functions as an effector of the mTORC1 signaling pathway by promoting, through SLC27A1, the uptake of long-chain fatty acid by adipocytes. Thereby, it also plays a role in fat metabolism and more indirectly influences lifespan. Multifunctional protein which is primarily part of the aminoacyl-tRNA synthetase multienzyme complex, also know as multisynthetase complex, that catalyzes the attachment of the cognate amino acid to the corresponding tRNA in a two-step reaction: the amino acid is first activated by ATP to form a covalent intermediate with AMP and is then transferred to the acceptor end of the cognate tRNA. Related diseases Leukodystrophy, hypomyelinating, 15 (HLD15) [MIM:617951]: An autosomal recessive disorder characterized by hypomyelinating leukodystrophy with thinning of the corpus callosum. Clinical features include motor and cognitive impairment appearing in the first or second decade of life, dystonia, ataxia, spasticity, and dysphagia. Most patients develop severe optic atrophy, and some have hearing loss. {ECO:0000269|PubMed:29576217}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02684; DB02510; DB03376; DB00142; DB00172 Interacts with P07814; Q8IWL3; P41252; Q15046; P42695; P54136; O60506 EC number NA Uniprot keywords 3D-structure; Acetylation; Aminoacyl-tRNA synthetase; ATP-binding; Cytoplasm; Disease variant; Leukodystrophy; Ligase; Membrane; Metal-binding; Methylation; Multifunctional enzyme; Neurodegeneration; Nucleotide-binding; Phosphoprotein; Protein biosynthesis; Proteomics identification; Reference proteome; Repeat; RNA-binding; Translation regulation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 55039.7 Length 483 Aromaticity 0.1 Instability index 39.15 Isoelectric point 5.92 Charge (pH=7) -6.4 2D Binding mode Binding energy (Kcal/mol) -7.97  Molscript Map 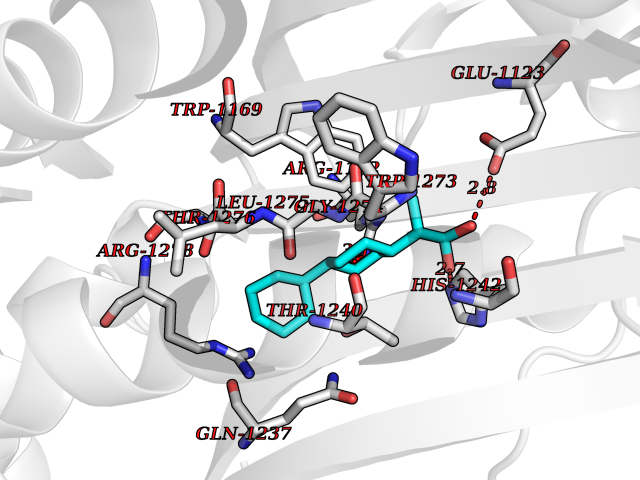 Pymol Map 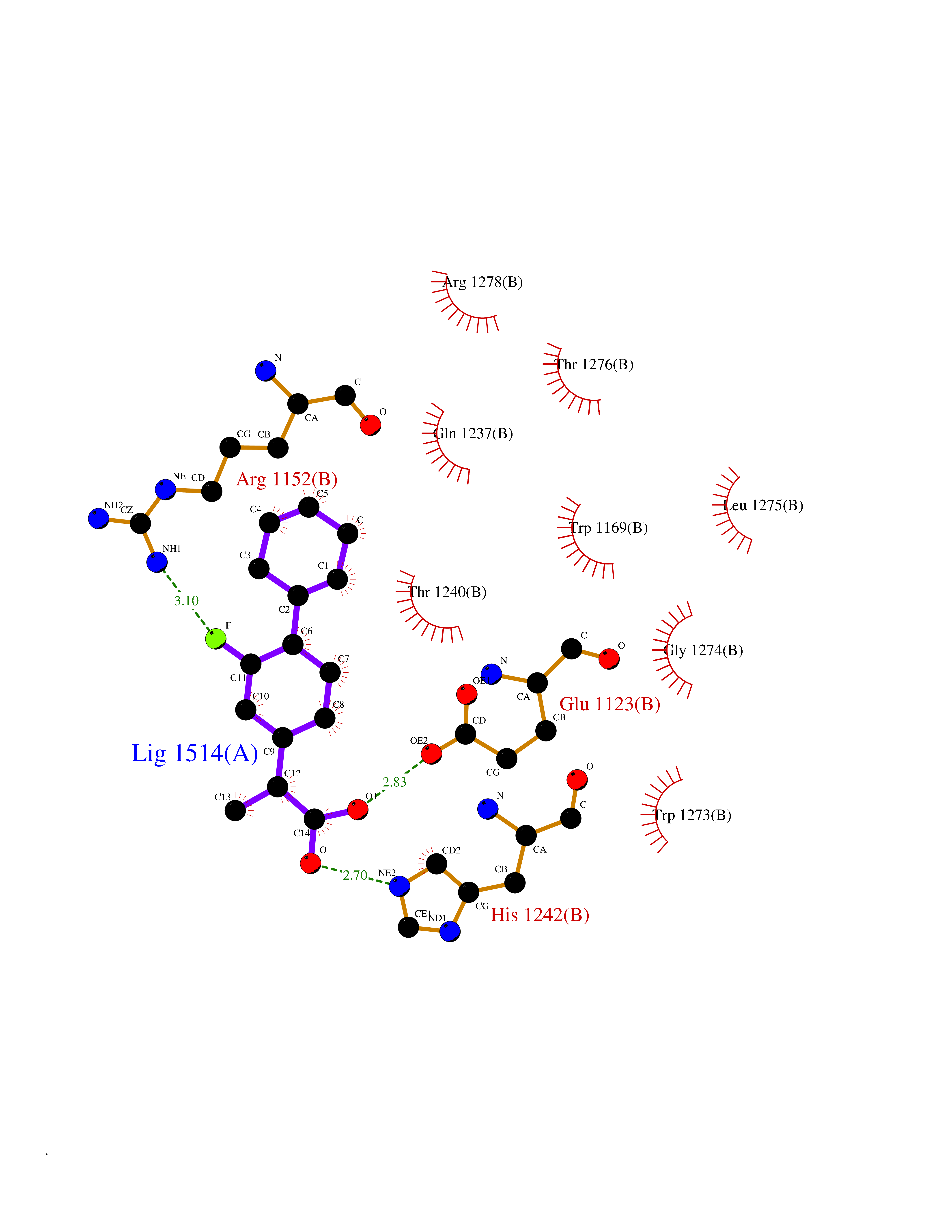 Ligplot Map 3D Binding mode Sequence GLEAKKEENLADWYSQVITKSEMIEYHDISGCYILRPWAYAIWEAIKDFFDAEIKKLGVENCYFPMFVSQSALEKEKTHVADFAPEVAWVTRSGKTELAEPIAIRPTSETVMYPAYAKWVQSHRDLPIKLNQWCNVVRWEFKHPQPFLRTREFLWQEGHSAFATMEEAAEEVLQILDLYAQVYEELLAIPVVKGRKTEKEKFAGGDYTTTIEAFISASGRAIQGGTSHHLGQNFSKMFEIVFEDPKIPGEKQFAYQNSWGLTTRTIGVMTMVHGDNMGLVLPPRVACVQVVIIPCGISEEDKEALIAKCNDYRRRLLSVNIRVRADLRDNYSPGWKFNHWELKGVPIRLEVGPRDMKSCQFVAVRRDTGEKLTVAENEAETKLQAILEDIQVTLFTRASEDLKTHMVVANTMEDFQKILDSGKIVQIPFCGEIDCEDWIKKTTAMGAKSLCIPFKPLCELQPGAKCVCGKNPAKYYTLFGRSY Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Acetylcholinesterase (AChE) (EC 3.1.1.7) | 1GPK | 7.32 | |
Target general information Gen name ache Organism Tetronarce californica (Pacific electric ray) (Torpedo californica) Uniprot ID TTD ID NA Synonyms NA Protein family Type-B carboxylesterase/lipase family Biochemical class NA Function Terminates signal transduction at the neuromuscular junction by rapid hydrolysis of the acetylcholine released into the synaptic cleft. May be involved in cell-cell interactions. Related diseases Noonan syndrome 5 (NS5) [MIM:611553]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:17603482, ECO:0000269|PubMed:17603483, ECO:0000269|PubMed:20683980}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: LEOPARD syndrome 2 (LPRD2) [MIM:611554]: A disorder characterized by lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonic stenosis, abnormalities of genitalia, retardation of growth, and sensorineural deafness. {ECO:0000269|PubMed:17603483}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cardiomyopathy, dilated, 1NN (CMD1NN) [MIM:615916]: A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. {ECO:0000269|PubMed:24777450}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.1.1.7 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; Neurotransmitter degradation; Serine esterase; Signal; Synapse Protein physicochemical properties Chain ID A Molecular weight (Da) 59779.2 Length 529 Aromaticity 0.12 Instability index 48.49 Isoelectric point 5.8 Charge (pH=7) -8.48 2D Binding mode Binding energy (Kcal/mol) -8.5  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SELLVNTKSGKVMGTRVPVLSSHISAFLGIPFAEPPVGNMRFRRPEPKKPWSGVWNASTYPNNCQQYVDEQFPGFSGSEMWNPNREMSEDCLYLNIWVPSPRPKSTTVMVWIYGGGFYSGSSTLDVYNGKYLAYTEEVVLVSLSYRVGAFGFLALHGSQEAPGNVGLLDQRMALQWVHDNIQFFGGDPKTVTIFGESAGGASVGMHILSPGSRDLFRRAILQSGSPNCPWASVSVAEGRRRAVELGRNLNCNLNSDEELIHCLREKKPQELIDVEWNVLPFDSIFRFSFVPVIDGEFFPTSLESMLNSGNFKKTQILLGVNKDEGSFFLLYGAPGFSKDSESKISREDFMSGVKLSVPHANDLGLDAVTLQYTDWMDDNNGIKNRDGLDDIVGDHNVICPLMHFVNKYTKFGNGTYLYFFNHRASNLVWPEWMGVIHGYEIEFVFGLPLVKELNYTAEEEALSRRIMHYWATFAKTGNPNEPESKWPLFTTKEQKFIDLNTEPMKVHQRLRVQMCVFWNQFLPKLLNAT Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Cyclic ADP-ribose hydrolase 1 (CD38) | 3DZG | 7.31 | |
Target general information Gen name CD38 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cADPr hydrolase 1; T10; Cyclic ADPribose hydrolase 1; ADPribosyl cyclase 1; ADPRC 1; ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1; ADP-ribosyl cyclase 1; 2'-phospho-cyclic-ADP-ribose transferase; Protein family ADP-ribosyl cyclase family Biochemical class Glycosylase Function Has cADPr hydrolase activity. Also moonlights as a receptor in cells of the immune system. Synthesizes the second messagers cyclic ADP-ribose and nicotinate-adenine dinucleotide phosphate, the former a second messenger for glucose-induced insulin secretion. Related diseases Intellectual developmental disorder with hypotonia and behavioral abnormalities (IDDHBA) [MIM:618748]: An autosomal dominant neurodevelopmental disorder with onset in infancy. IDDHBA is characterized by hypotonia, global developmental delay, learning disability, and behavioral abnormalities, such as autistic features and attention deficit-hyperactivity disorder. Additional variable features may include non-specific facial dysmorphism, congenital heart defects, ocular anomalies, and poor feeding. {ECO:0000269|PubMed:30905399}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09331; DB14811; DB16370 Interacts with NA EC number EC 3.2.2.6 Uniprot keywords 3D-structure; Alternative splicing; Diabetes mellitus; Disulfide bond; Glycoprotein; Hydrolase; Membrane; NAD; NADP; Proteomics identification; Receptor; Reference proteome; Signal-anchor; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 29222.8 Length 252 Aromaticity 0.1 Instability index 50.29 Isoelectric point 6.19 Charge (pH=7) -2.71 2D Binding mode Binding energy (Kcal/mol) -8.24  Molscript Map 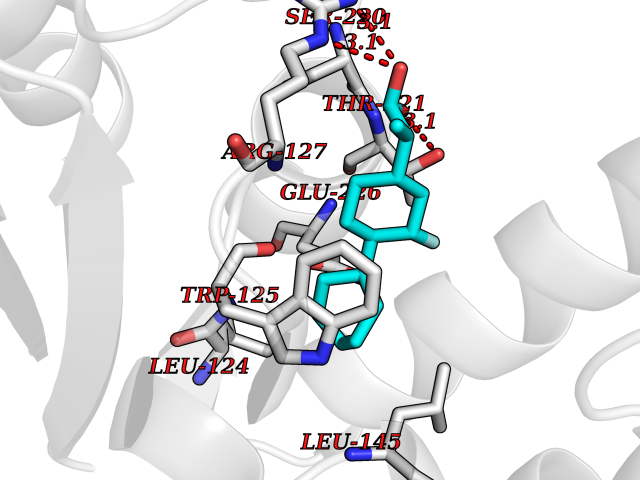 Pymol Map 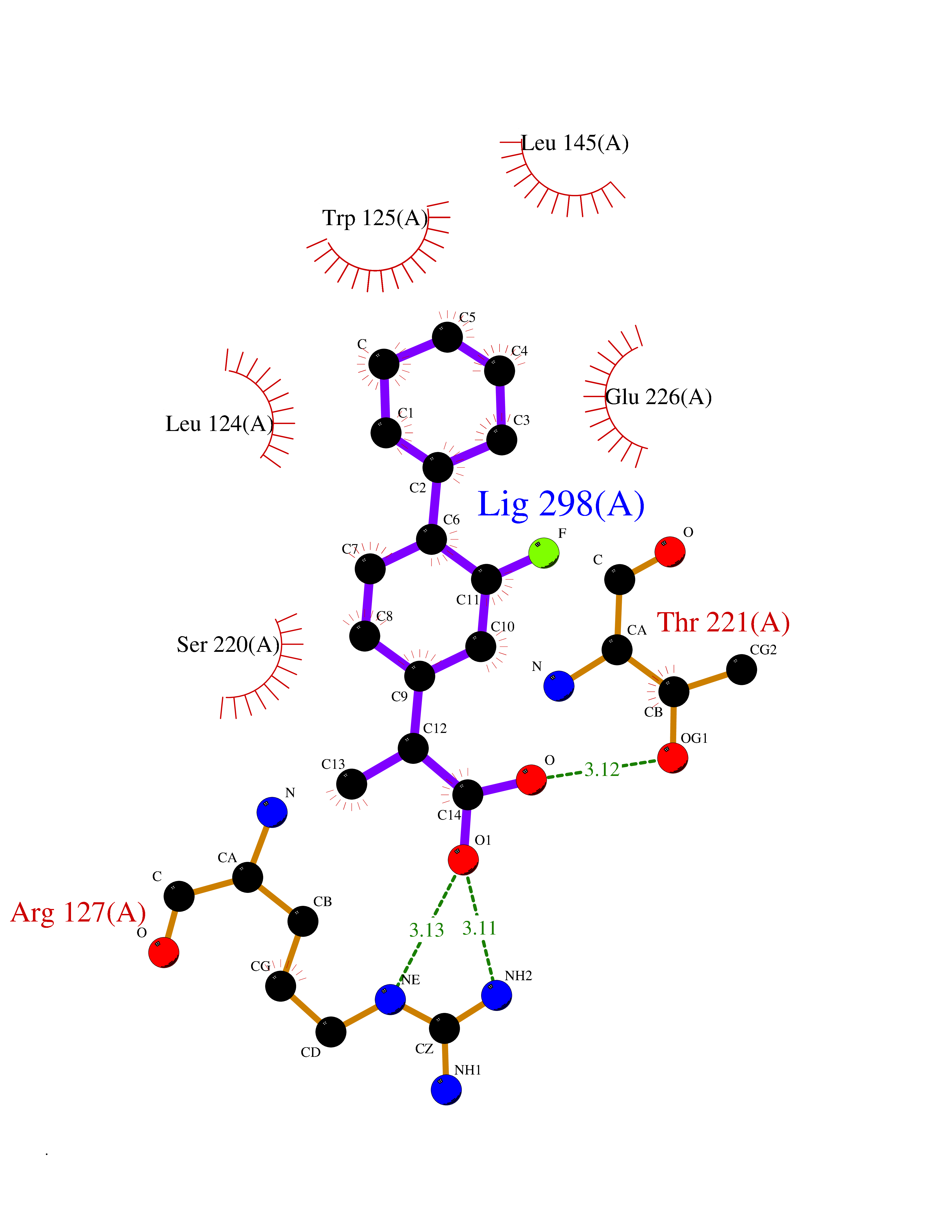 Ligplot Map 3D Binding mode Sequence RWRQTWSGPGTTKRFPETVLARCVKYTEIHPEMRHVDCQSVWDAFKGAFISKHPCDITEEDYQPLMKLGTQTVPCNKILLWSRIKDLAHQFTQVQRDMFTLEDTLLGYLADDLTWCGEFDTSKINYQSCPDWRKDCSNNPVSVFWKTVSRRFAEAACDVVHVMLDGSRSKIFDKDSTFGSVEVHNLQPEKVQTLEAWVIHGGREDSRDLCQDPTIKELESIISKRNIQFSCKNIYRPDKFLQCVKNPEDSSC Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | 1C3R | 7.30 | |
Target general information Gen name murA Organism Aquifex aeolicus (strain VF5) Uniprot ID TTD ID NA Synonyms aq_1023 Protein family Histone deacetylase family Biochemical class Lyase Function UDP-N-acetylglucosamine 1-carboxyvinyltransferase activity. Related diseases Atrial fibrillation, familial, 14 (ATFB14) [MIM:615378]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:19808477}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Genetic variations in SCN2B may be involved in Brugada syndrome (PubMed:23559163). This tachyarrhythmia is characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:23559163}. Drugs (DrugBank ID) DB04297; DB02546 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetoin catabolism; Metal-binding; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42338.9 Length 372 Aromaticity 0.12 Instability index 35.74 Isoelectric point 5.77 Charge (pH=7) -6.5 2D Binding mode Binding energy (Kcal/mol) -8.71  Molscript Map 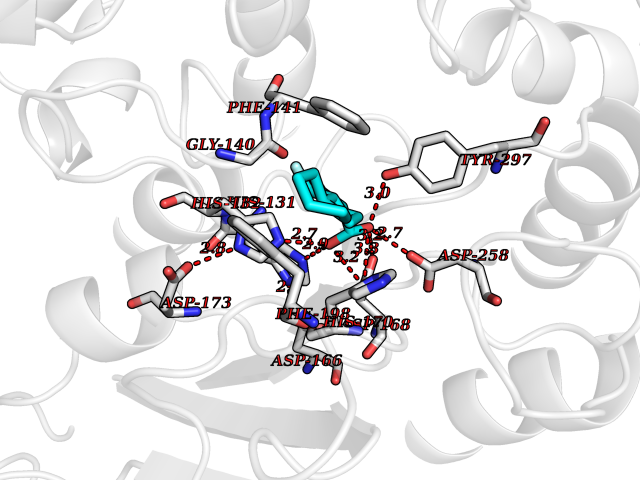 Pymol Map 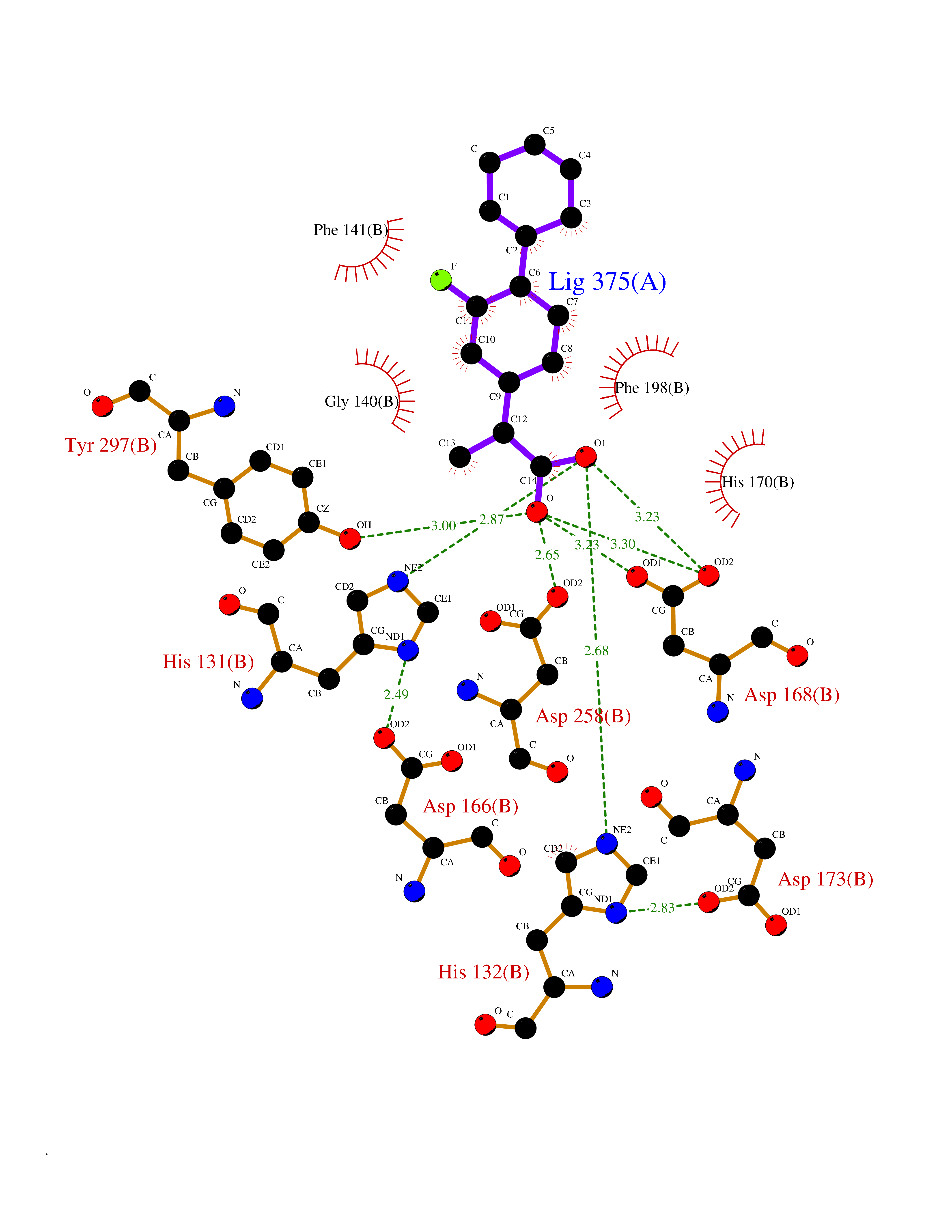 Ligplot Map 3D Binding mode Sequence KKVKLIGTLDYGKYRYPKNHPLKIPRVSLLLRFKDAMNLIDEKELIKSRPATKEELLLFHTEDYINTLMEAERSQSVPKGAREKYNIGGYENPVSYAMFTGSSLATGSTVQAIEEFLKGNVAFNPAGGMHHAFKSRANGFCYINNPAVGIEYLRKKGFKRILYIDLDAHHCDGVQEAFYDTDQVFVLSLHQSPEYAFPFEKGFLEEIGEGKGKGYNLNIPLPKGLNDNEFLFALEKSLEIVKEVFEPEVYLLQLGTDPLLEDYLSKFNLSNVAFLKAFNIVREVFGEGVYLGGGGYHPYALARAWTLIWCELSGREVPEKLNNKAKELLKSIDFEEFDDEVDRSYMLETLKDPWRGGEVRKEVKDTLEKAKA Hydrogen bonds contact Hydrophobic contact | ||||