Job Results:
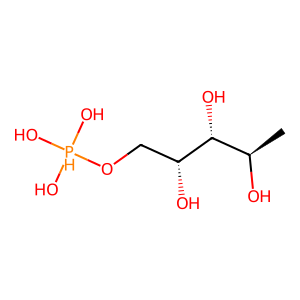
Ligand
Structure
Job ID
ea4aeeea43c0082ff76353d76181fa87
Job name
NA
Time
2024-06-15 15:52:32
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Geranylgeranyl pyrophosphate synthase | 2Q80 | 6.23 | |
Target general information Gen name GGPS1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family FPP/GGPP synthase family Biochemical class Transferase Function Dimethylallyltranstransferase activity.Farnesyltranstransferase activity.Geranyltranstransferase activity.Identical protein binding.Metal ion binding. Related diseases Muscular dystrophy, congenital hearing loss, and ovarian insufficiency syndrome (MDHLO) [MIM:619518]: An autosomal recessive disorder characterized by early-onset progressive muscle weakness, sensorineural hearing loss, and primary amenorrhea due to ovarian insufficiency. Some patients become wheelchair-bound by the second decade, whereas others have a milder phenotype and maintain independent ambulation into adulthood. Most patients have respiratory insufficiency. {ECO:0000269|PubMed:32403198}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06830; DB06931; DB07221; DB08529; DB07410; DB07780; DB04695; DB02552; DB07841; DB00710; DB04714; DB07873; DB06548; DB00282; DB00399 Interacts with O00244; O95749; O00560 EC number 2.5.1.-; 2.5.1.1; 2.5.1.10; 2.5.1.29 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Congenital muscular dystrophy; Cytoplasm; Deafness; Disease variant; Isoprene biosynthesis; Lipid metabolism; Magnesium; Metal-binding; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 32872.4 Length 284 Aromaticity 0.11 Instability index 41.09 Isoelectric point 6.17 Charge (pH=7) -3.37 2D Binding mode Binding energy (Kcal/mol) -6.26 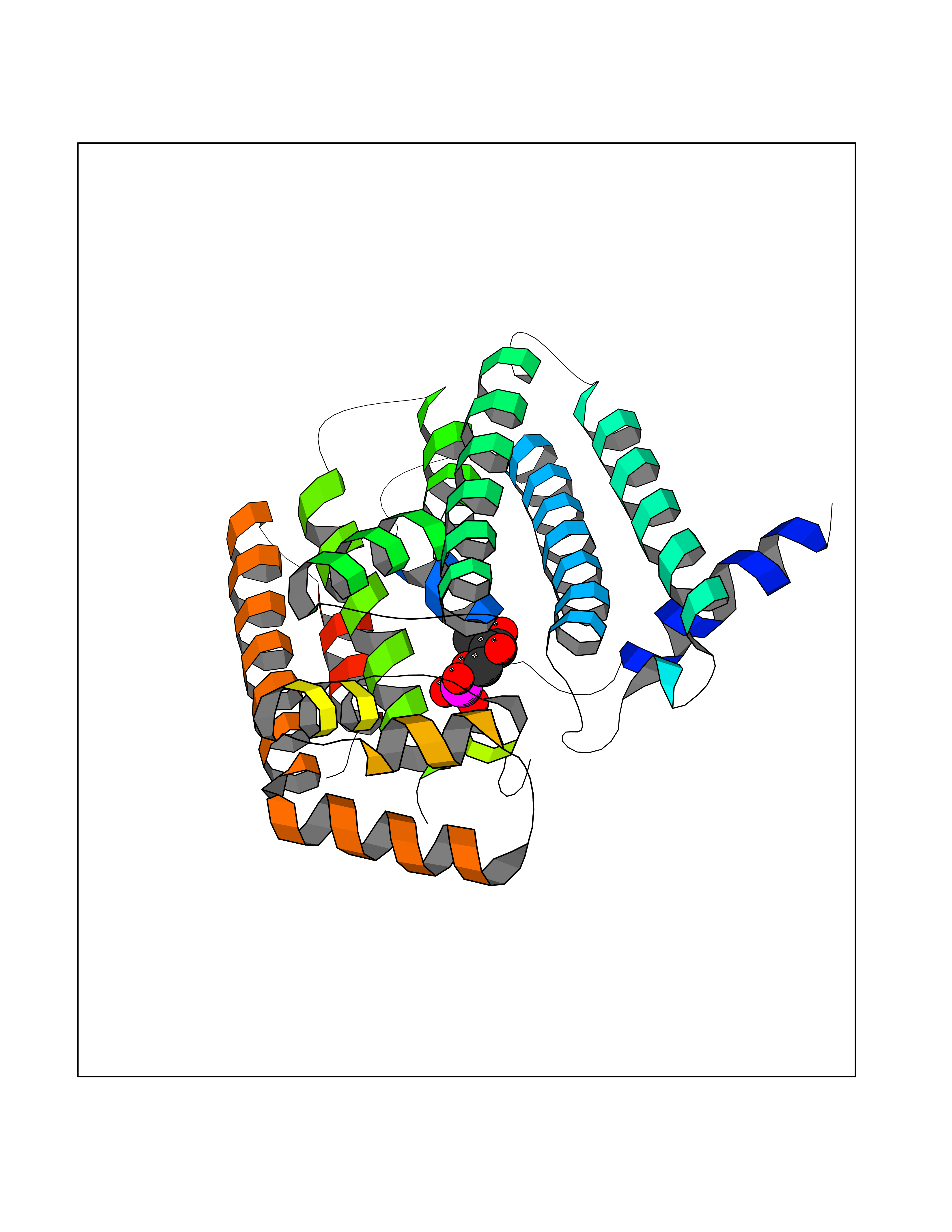 Molscript Map 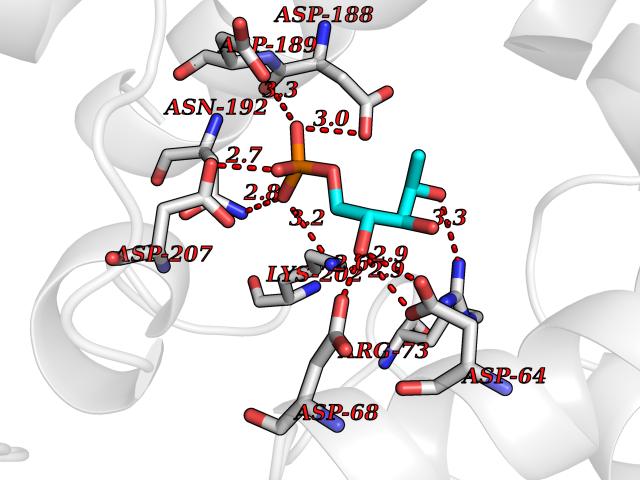 Pymol Map 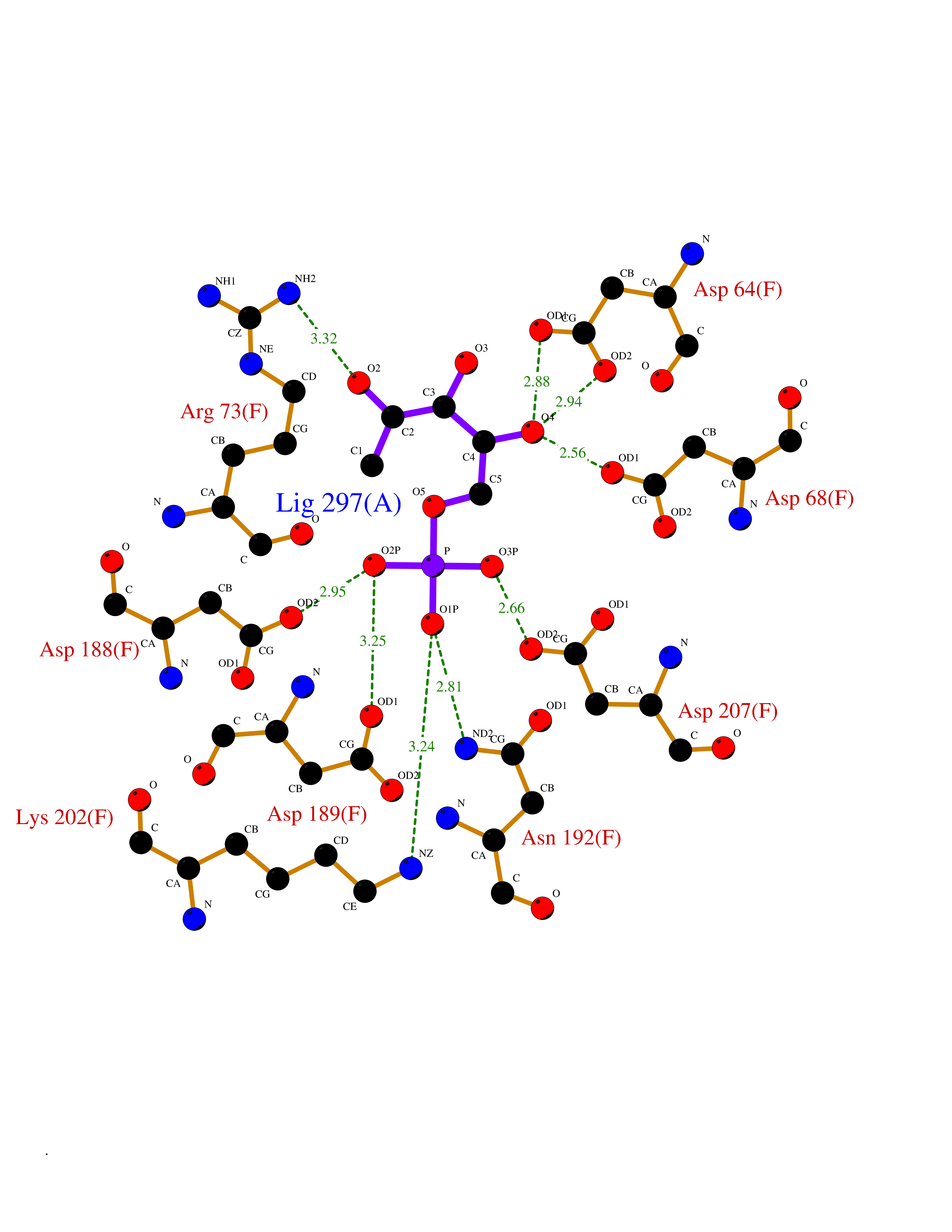 Ligplot Map 3D Binding mode Sequence ETVQRILLEPYKYLLQLPGKQVRTKLSQAFNHWLKVPEDKLQIIIEVTEMLHNASLLIDDIEDNSKLRRGFPVAHSIYGIPSVINSANYVYFLGLEKVLTLDHPDAVKLFTRQLLELHQGQGLDIYWRDNYTCPTEEEYKAMVLQKTGGLFGLAVGLMQLFSDYKEDLKPLLNTLGLFFQIRDDYANLHSKSFCEDLTEGKFSFPTIHAIWSRPESTQVQNILRQRTENIDIKKYCVHYLEDVGSFEYTRNTLKELEAKAYKQIDARGGNPELVALVKHLSKMF Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Cytomegalovirus Protease (CMV UL80) | 7TCZ | 6.23 | |
Target general information Gen name CMV UL80 Organism Human cytomegalovirus (strain AD169) (HHV-5) (Human herpesvirus 5) Uniprot ID TTD ID Synonyms UL80; Capsid protein P40; Assemblin Protein family Herpesviridae capsid scaffolding protein family Biochemical class Peptidase Function Assembly protein plays a major role in capsid assembly. Acts as a scaffold protein by binding major capsid protein UL86. Multimerizes in the nucleus such as protein UL86 forms the icosahedral T=16 capsid. Cleaved by assemblin after capsid completion. The cleavages products are evicted from the capsid before or during DNA packaging. Related diseases Hao-Fountain syndrome (HAFOUS) [MIM:616863]: An autosomal dominant neurodevelopmental disorder characterized by global developmental delay, varying degrees of intellectual disability, autism spectrum disorder, poor or absent speech, and mild facial dysmorphism. Most patients develop seizures. Additional variable features include hypotonia, hypogonadism in males, and ocular anomalies. {ECO:0000269|PubMed:26365382, ECO:0000269|PubMed:30679821}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07436; DB01973; DB03963 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative promoter usage; Host cytoplasm; Host nucleus; Hydrolase; Phosphoprotein; Protease; Reference proteome; Serine protease; Viral capsid assembly; Viral release from host cell Protein physicochemical properties Chain ID A Molecular weight (Da) 23645.4 Length 213 Aromaticity 0.08 Instability index 46.56 Isoelectric point 7.16 Charge (pH=7) 0.22 2D Binding mode Binding energy (Kcal/mol) -6.17 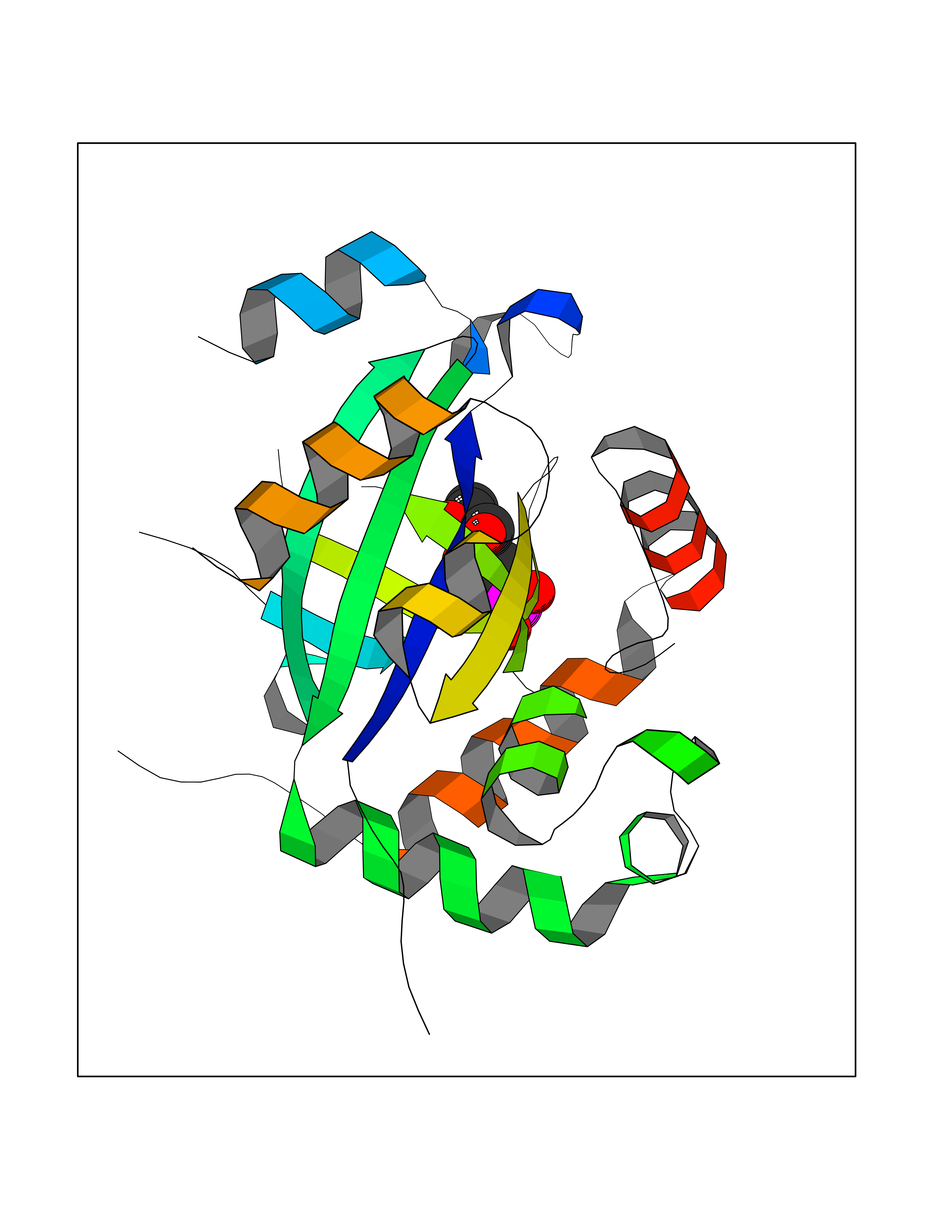 Molscript Map 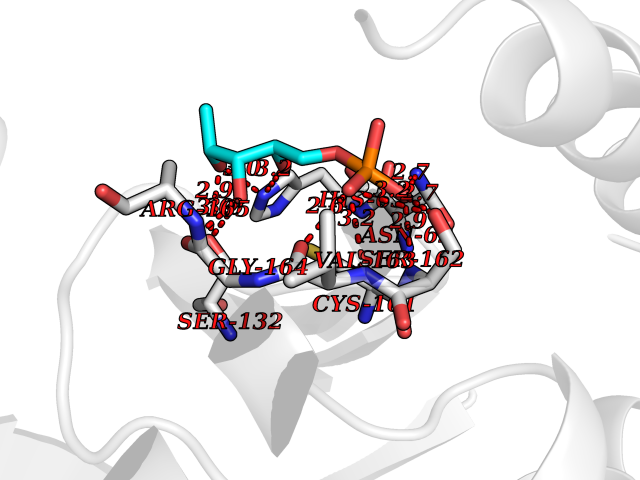 Pymol Map 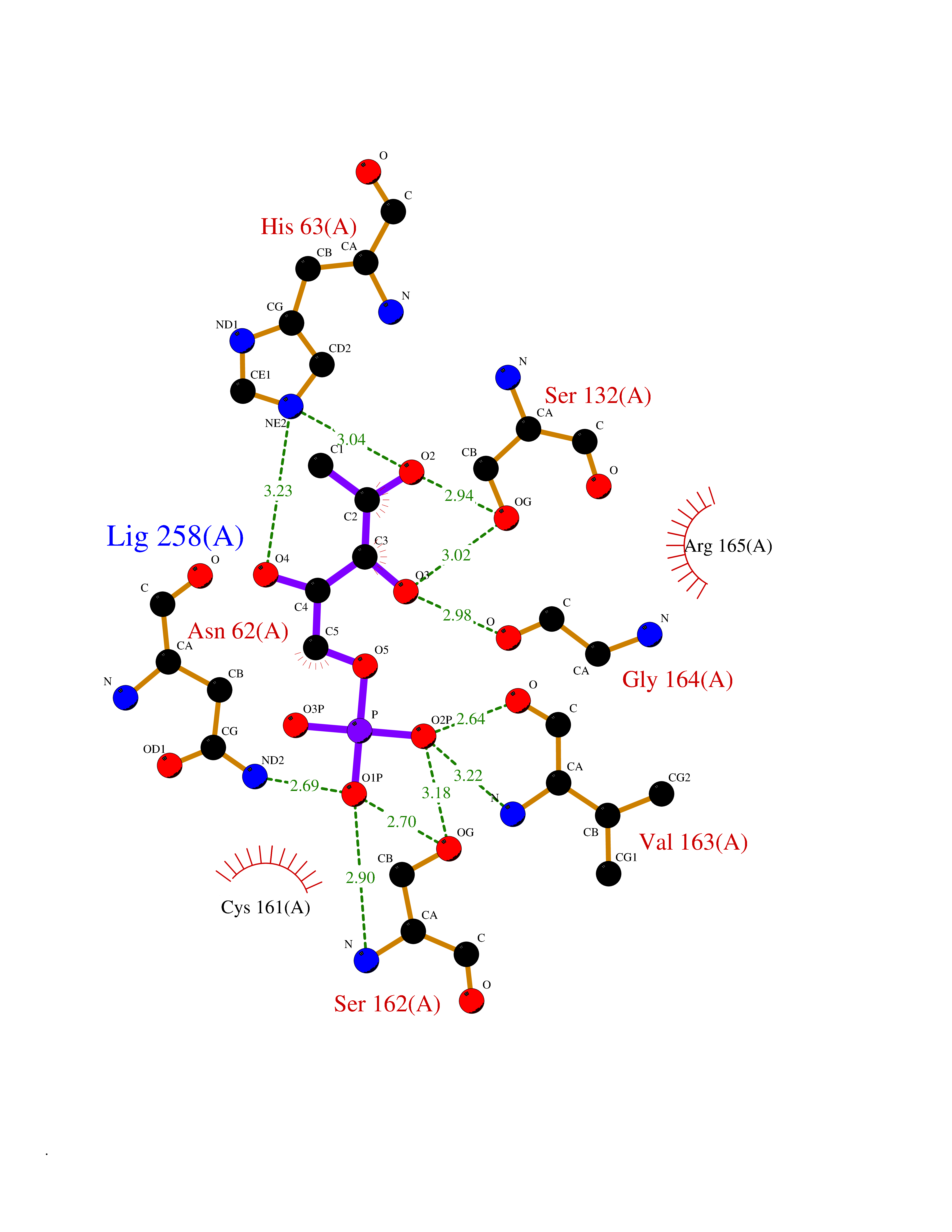 Ligplot Map 3D Binding mode Sequence QQSQAVAPVYVGGFLARYDQSPDEAELLLPRDVVEHWLHVALPLNINHDDTAVVGHVAAMQSVRDGLFALGAVTSPRFLEIVRRASEKSELVSRGPVSPLQPDKVVEFLSGSYAGLSLSFKHVALCSVGRRRGTLAVYGRDPEWVTQRFPDLTAADRDGLRAQWQRSGDPFRSDSYGLLGNSVDALYIRERLPKLRYDKQLVGVTERESYVKA Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Bacterial DD-carboxypeptidase (Bact vanYB) | 5ZHW | 6.23 | |
Target general information Gen name Bact vanYB Organism Enterococcus faecalis (strain ATCC 700802 / V583) Uniprot ID TTD ID Synonyms vanYB; DD-peptidase; DD-carboxypeptidase; D-alanyl-D-alanine carboxypeptidase-transpeptidase Protein family Peptidase M15B family Biochemical class Peptidase Function Vancomycin-inducible, penicillin-resistant, DD- carboxypeptidase that hydrolyzes depsipeptide- and D-alanyl-D- alanine-containing peptidoglycan precursors. Insensitive to beta- lactams. Related diseases Brachyolmia 3 (BCYM3) [MIM:113500]: A form of brachyolmia, a clinically and genetically heterogeneous skeletal dysplasia primarily affecting the spine and characterized by a short trunk, short stature, and platyspondyly. BCYM3 is an autosomal dominant form with severe scoliosis with or without kyphosis, and flattened irregular cervical vertebrae. {ECO:0000269|PubMed:18587396}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondylometaphyseal dysplasia Kozlowski type (SMDK) [MIM:184252]: A form of spondylometaphyseal dysplasia, a group of short stature disorders distinguished by abnormalities in the vertebrae and the metaphyses of the tubular bones. It is characterized by postnatal dwarfism, significant scoliosis and mild metaphyseal abnormalities in the pelvis. The vertebrae exhibit platyspondyly and overfaced pedicles. {ECO:0000269|PubMed:19232556, ECO:0000269|PubMed:20577006, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metatropic dysplasia (MTD) [MIM:156530]: A severe spondyloepimetaphyseal dysplasia characterized by short limbs with limitation and enlargement of joints and usually severe kyphoscoliosis. Radiologic features include severe platyspondyly, severe metaphyseal enlargement and shortening of long bones. {ECO:0000269|PubMed:19232556, ECO:0000269|PubMed:20425821, ECO:0000269|PubMed:20577006, ECO:0000269|PubMed:22702953, ECO:0000269|PubMed:26249260, ECO:0000269|Ref.6}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 8 (HMND8) [MIM:600175]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:20037588, ECO:0000269|PubMed:22526352, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2C (CMT2C) [MIM:606071]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:20037586, ECO:0000269|PubMed:20037587, ECO:0000269|PubMed:20037588, ECO:0000269|PubMed:21115951, ECO:0000269|PubMed:21288981, ECO:0000269|PubMed:22702953, ECO:0000269|PubMed:25256292}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Scapuloperoneal spinal muscular atrophy (SPSMA) [MIM:181405]: A clinically variable neuromuscular disorder characterized by neurogenic scapuloperoneal amyotrophy, laryngeal palsy, congenital absence of muscles, progressive scapuloperoneal atrophy and progressive distal weakness and amyotrophy. {ECO:0000269|PubMed:20037587, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepiphyseal dysplasia Maroteaux type (SEDM) [MIM:184095]: A clinically variable spondyloepiphyseal dysplasia with manifestations limited to the musculoskeletal system. Clinical features include short stature, brachydactyly, platyspondyly, short and stubby hands and feet, epiphyseal hypoplasia of the large joints, and iliac hypoplasia. Intelligence is normal. {ECO:0000269|PubMed:20503319, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parastremmatic dwarfism (PSTD) [MIM:168400]: A bone dysplasia characterized by severe dwarfism, kyphoscoliosis, distortion and bowing of the extremities, and contractures of the large joints. Radiographically, the disease is characterized by a combination of decreased bone density, bowing of the long bones, platyspondyly and striking irregularities of endochondral ossification with areas of calcific stippling and streaking in radiolucent epiphyses, metaphyses and apophyses. {ECO:0000269|PubMed:20503319}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Digital arthropathy-brachydactyly, familial (FDAB) [MIM:606835]: A disorder characterized by irregularities in the proximal articular surfaces of the distal interphalangeal joints of the hand. Individuals appear normal at birth, with no clinical or radiographic evidence of a developmental skeletal dysplasia. The earliest changes appear during the first decade of life. By adulthood, all interphalangeal, metacarpophalangeal, and metatarsophalangeal joints are affected by a deforming, painful osteoarthritis. The remainder of the skeleton is clinically and radiographically unaffected. {ECO:0000269|PubMed:21964574}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Avascular necrosis of the femoral head, primary 2 (ANFH2) [MIM:617383]: A disease characterized by mechanical failure of the subchondral bone, and degeneration of the hip joint. It usually leads to destruction of the hip joint in the third to fifth decade of life. The clinical manifestations, such as pain on exertion, a limping gait, and a discrepancy in leg length, cause considerable disability. {ECO:0000269|PubMed:27330106}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.16.4 Uniprot keywords 3D-structure; Antibiotic resistance; Carboxypeptidase; Cell membrane; Cell shape; Cell wall biogenesis/degradation; Hydrolase; Membrane; Metal-binding; Peptidoglycan synthesis; Protease; Reference proteome; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 20547.5 Length 181 Aromaticity 0.11 Instability index 33.61 Isoelectric point 4.81 Charge (pH=7) -10.78 2D Binding mode Binding energy (Kcal/mol) -6.87 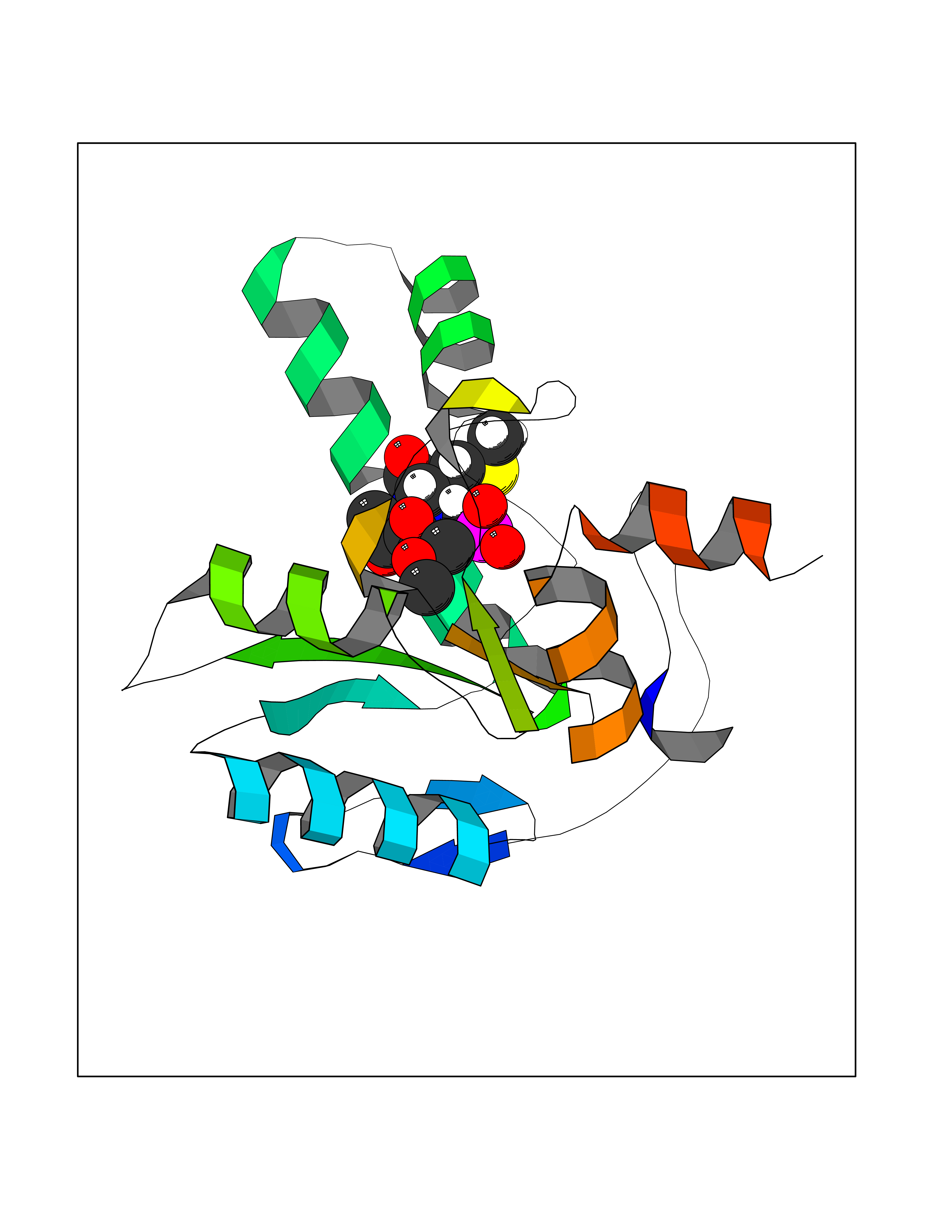 Molscript Map 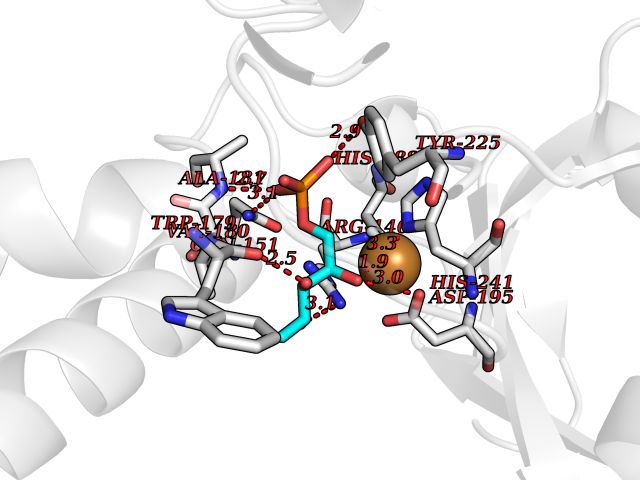 Pymol Map 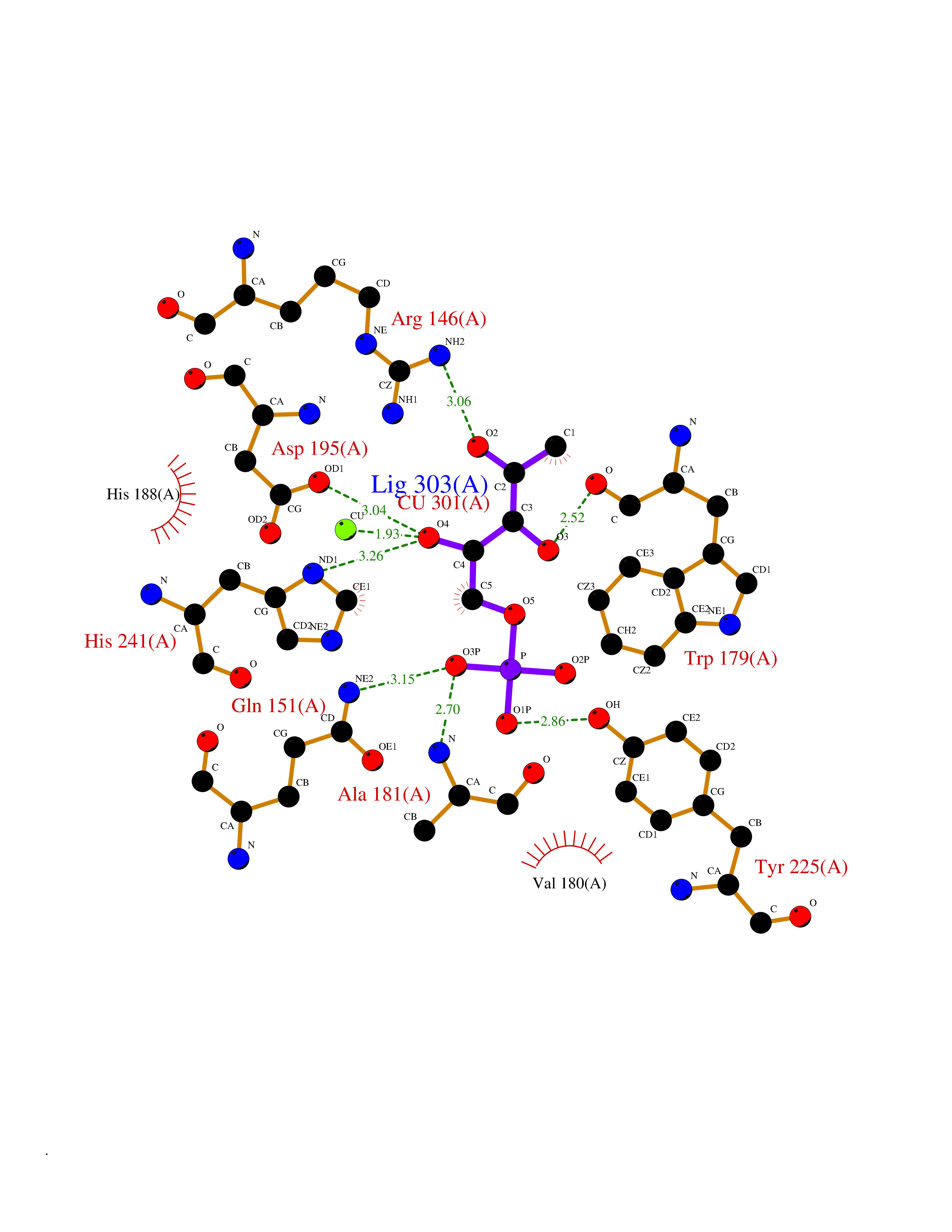 Ligplot Map 3D Binding mode Sequence EWSLILVNRQNPIPAQYDVELEQLSNGERIDIRISPYLQDLFDAARADGVYPIVASGYRTTEKQQEIXDEKVAEYKAKGYTSAQAKAEAETWVAVPGTSEHQLGLAVDINADGIHSTGNEVYRWLDENSYRFGFIRRYPPDKTEITGVSNEPWHYRYVGIEAATKIYHQGLCLEEYLNTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Endonuclease 8-like 1 | 5ITQ | 6.23 | |
Target general information Gen name NEIL1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family FPG family Biochemical class Dna binding protein / dna Function Class I DNA-(apurinic or apyrimidinic site) lyase activity.Class III/IV DNA-(apurinic or apyrimidinic site) lyase activity.Damaged DNA binding.DNA-(apurinic or apyrimidinic site) lyase activity.DNA N-glycosylase activity.Hydrolase activity, acting on glycosyl bonds.Protein C-terminus binding.Zinc ion binding. Related diseases Glutaric aciduria 1 (GA1) [MIM:231670]: An autosomal recessive metabolic disorder characterized by progressive dystonia and athetosis due to gliosis and neuronal loss in the basal ganglia. {ECO:0000269|PubMed:14707522, ECO:0000269|PubMed:18775954, ECO:0000269|PubMed:24973495, ECO:0000269|PubMed:8541831, ECO:0000269|PubMed:8900227, ECO:0000269|PubMed:8900228, ECO:0000269|PubMed:9600243, ECO:0000269|PubMed:9711871}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09130; DB14490; DB14491; DB14488; DB14501; DB14489; DB01592 Interacts with NA EC number 3.2.2.-; 4.2.99.18 Uniprot keywords 3D-structure; Chromosome; Cytoplasm; Cytoskeleton; DNA damage; DNA repair; DNA-binding; Glycosidase; Hydrolase; Lyase; Multifunctional enzyme; Nucleus; Proteomics identification; Reference proteome; RNA editing Protein physicochemical properties Chain ID A Molecular weight (Da) 32177.5 Length 286 Aromaticity 0.1 Instability index 57.82 Isoelectric point 8.89 Charge (pH=7) 5.38 2D Binding mode Binding energy (Kcal/mol) -6.2  Molscript Map 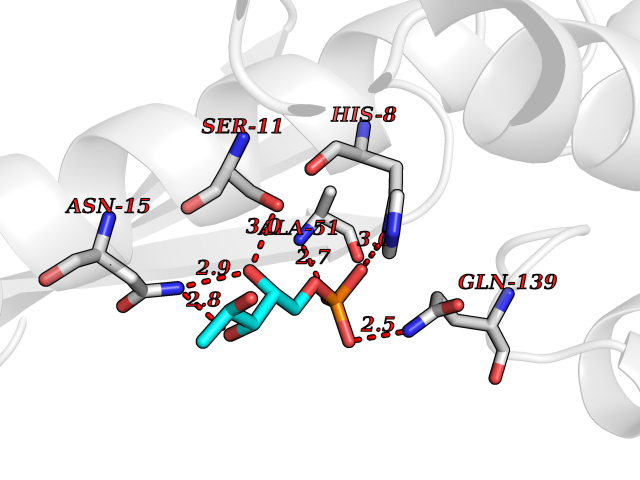 Pymol Map 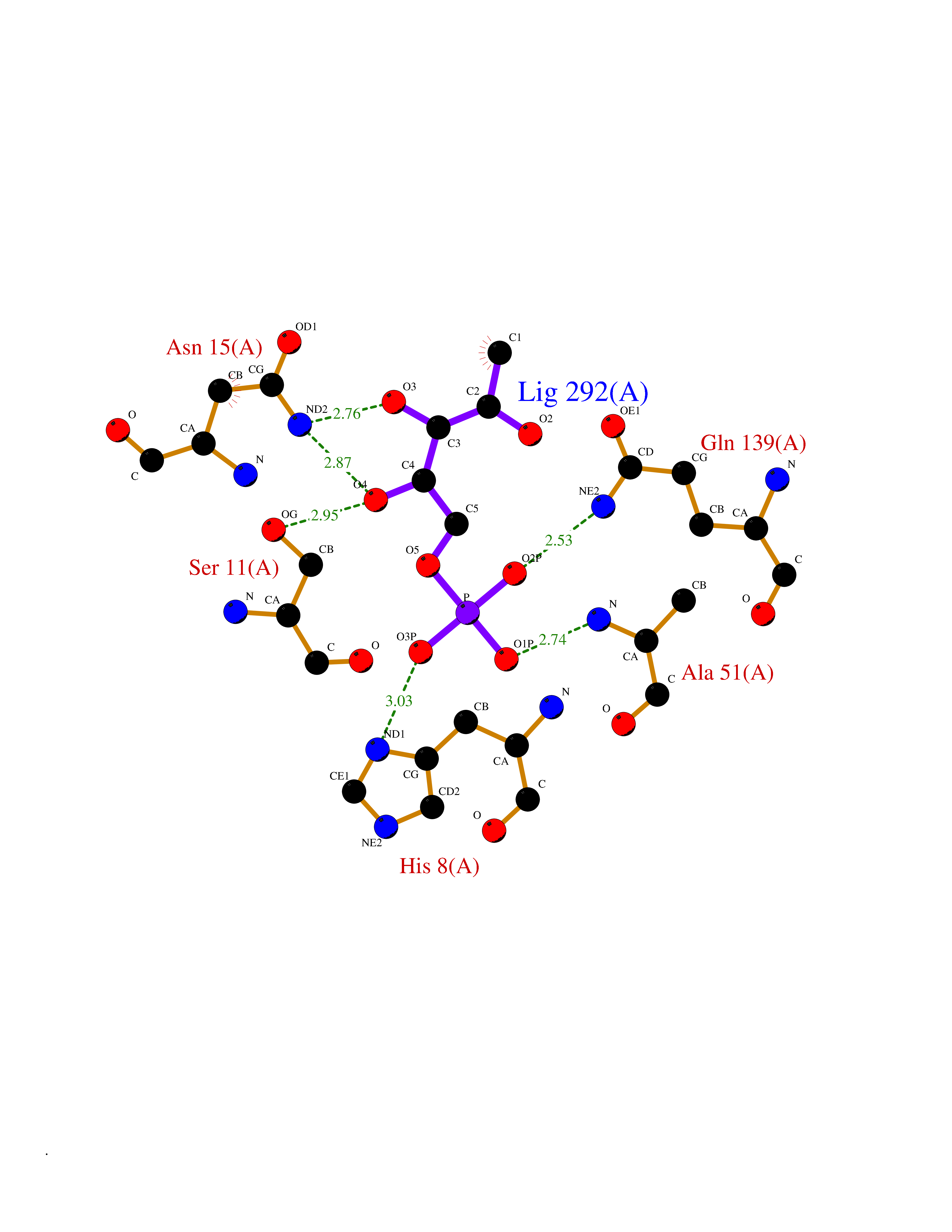 Ligplot Map 3D Binding mode Sequence PEGPELHLASQFVNEACRALVFGGCVEKSSVSRNPEVPFESSAYRISASARGKELRLILSPLPGAQPQQEPLALVFRFGMSGSFQLVPREELPRHAHLRFYTAPPGPRLALCFVDIRRFGRWDLGGKWQPGRGPCVLQEYQQFRESVLRNLADKAFDRPICEALLDQRFFNGIGNYLRAEILYRLKIPPFEKARSVLEALQQSPELTLSQKIRTKLQNPDLLELCHSVPKEVVQLGGRGYGSESGEEDFAAFRAWLRCYGMPGMSSLQDRHGRTIWFQGDPGPLAP Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Fru-2,6-P2ase 2/6PF-2-K (PFK/FBPase 2) | 5HTK | 6.22 | |
Target general information Gen name PFKFB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PFKFB2; PFK-2/FBPase-2; 6PF-2-K/Fru-2,6-P2ASE heart-typeisozyme Protein family Phosphoglycerate mutase family Biochemical class Kinase Function Synthesis and degradation of fructose 2,6-bisphosphate. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P63104 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Hydrolase; Kinase; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 49295 Length 425 Aromaticity 0.11 Instability index 37.76 Isoelectric point 6.65 Charge (pH=7) -0.78 2D Binding mode Binding energy (Kcal/mol) -6.73 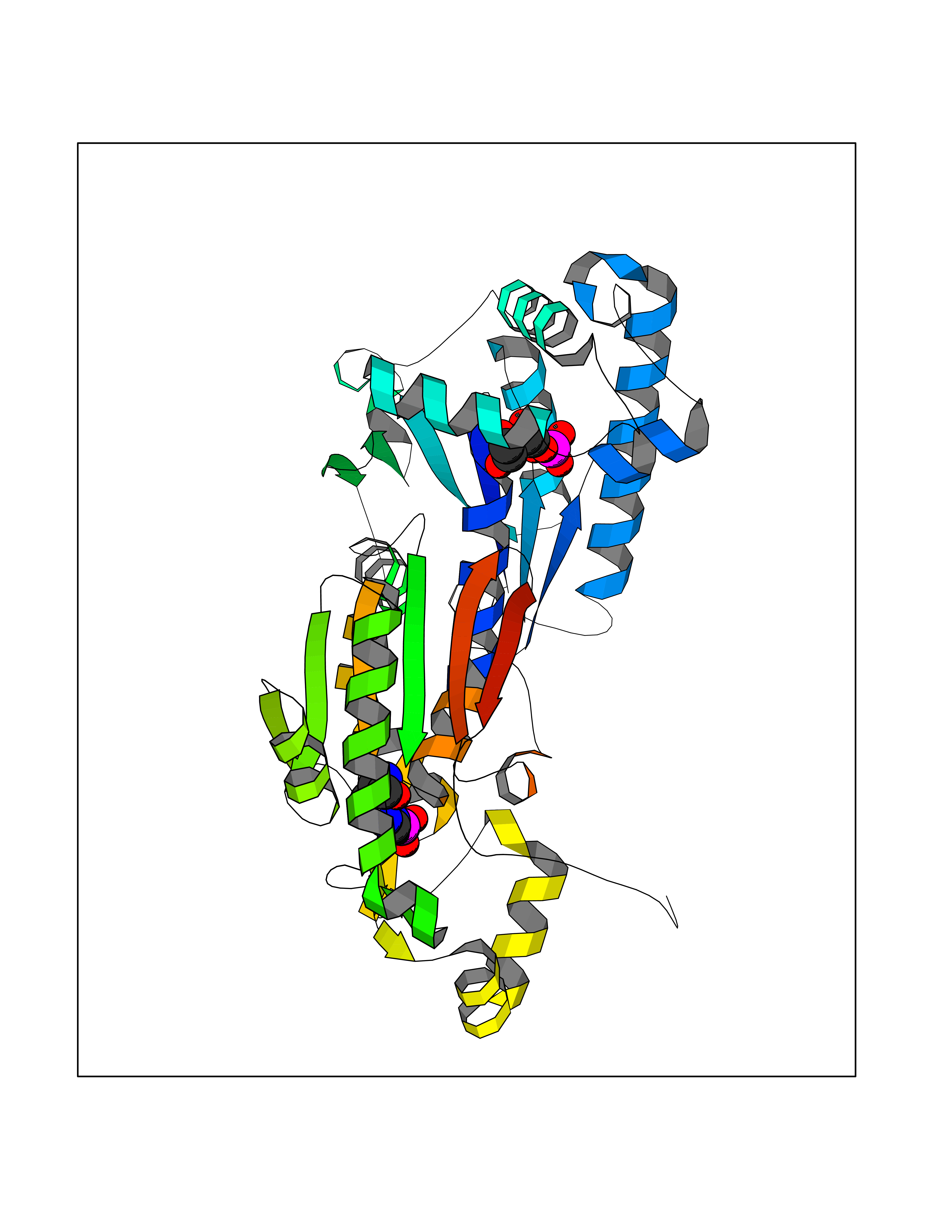 Molscript Map 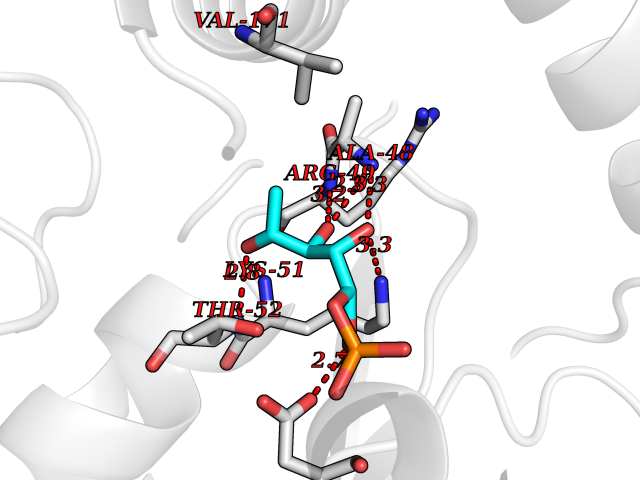 Pymol Map 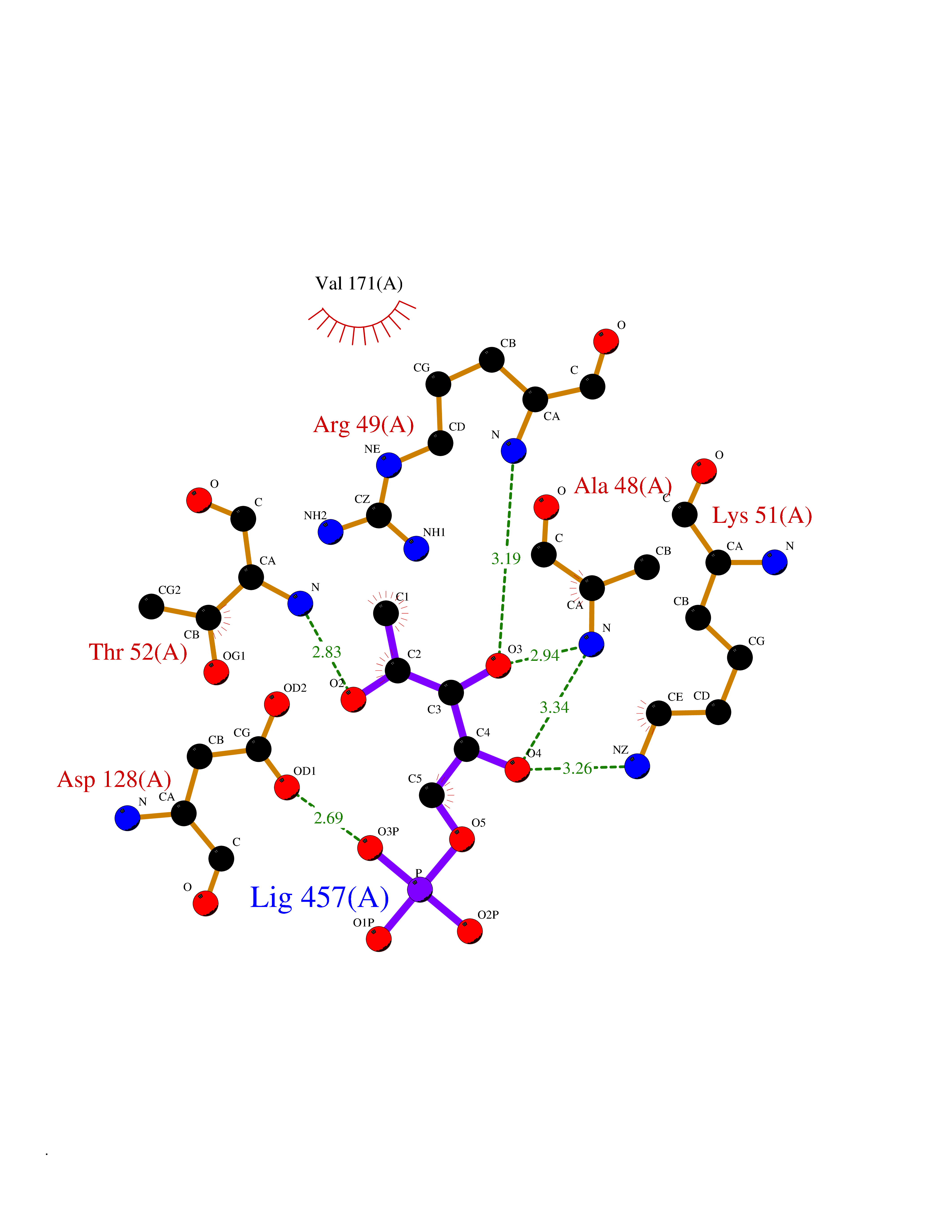 Ligplot Map 3D Binding mode Sequence ASYMTNSPTLIVMIGLPARGKTYVSKKLTRYLNWIGVPTKVFNLGVYRREAVKSYKSYDFFRHDNEEAMKIRKQCALVALEDVKAYLTEENGQIAVFDATNTTRERRDMILNFAEQNSFKVFFVESVCDDPDVIAANILEVKVSSPDYPERNRENVMEDFLKRIECYKVTYRPLDPDNYDKDLSFIKVINVGQRFLVNRVQDYIQSKIVYYLMNIHVQPRTIYLCRXGESEFNLLGKIGGDSGLSVRGKQFAQALRKFLEEQEITDLKVWTSQLKRTIQTAESLGVPYEQWKILNEIDAGVCEEMTYAEIEKRYPEEFALRDQEKYLYRYPGGESYQDLVQRLEPVIMELERQGNVLVISHQAVMRCLLAYFLDKGADELPYLRCPLHTIFKLTPVAYGCKVETIKLNVEAVNTHRDKPTNNFPK Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Mitochondrial rRNA methyltransferase 2 (MRM2) | 2NYU | 6.21 | |
Target general information Gen name MRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms rRNA methyltransferase 2, mitochondrial; Protein ftsJ homolog 2; 16S rRNA [Um1369] 2'-O-methyltransferase; 16S rRNA (uridine(1369)-2'-O)-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, RNA methyltransferase RlmE family Biochemical class Methyltransferase Function S-adenosyl-L-methionine-dependent 2'-O-ribose methyltransferase that catalyzes the formation of 2'-O-methyluridine at position 1369 (Um1369) in the 16S mitochondrial large subunit ribosomal RNA (mtLSU rRNA), a universally conserved modification in the peptidyl transferase domain of the mtLSU rRNA. Related diseases Mitochondrial DNA depletion syndrome 17 (MTDPS17) [MIM:618567]: An autosomal recessive mitochondrial disorder characterized by childhood onset of rapidly progressive encephalopathy, stroke-like episodes, lactic acidosis, hypocitrullinemia, multiple defects of oxidative phosphorylation, mitochondrial complex I and IV deficiency, and reduced mtDNA copy number. {ECO:0000269|PubMed:28973171}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.1.1.- Uniprot keywords 3D-structure; Disease variant; Methyltransferase; Mitochondrion; Primary mitochondrial disease; Proteomics identification; Reference proteome; rRNA processing; S-adenosyl-L-methionine; Transferase; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 20008.8 Length 181 Aromaticity 0.07 Instability index 53.22 Isoelectric point 8.43 Charge (pH=7) 1.79 2D Binding mode Binding energy (Kcal/mol) -6.68 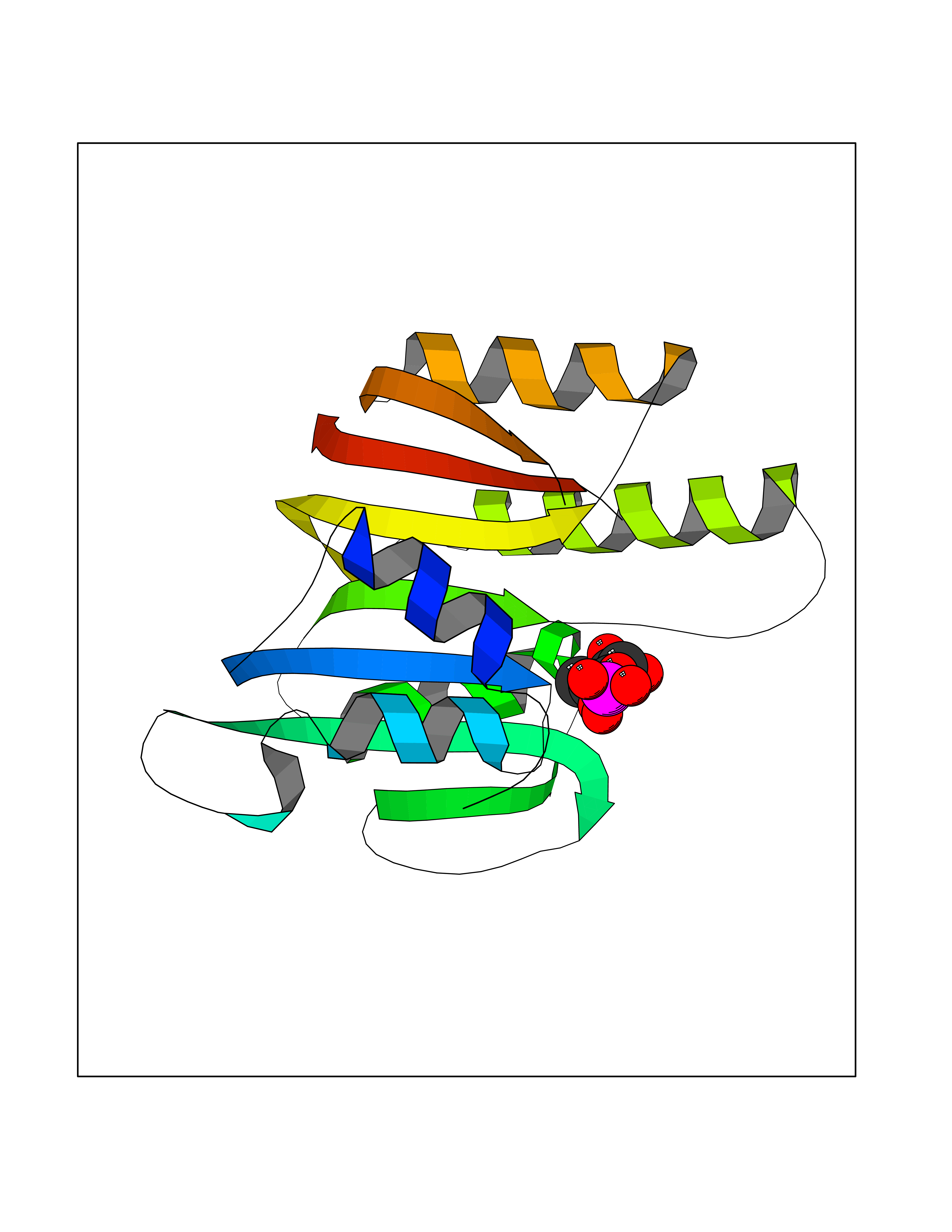 Molscript Map 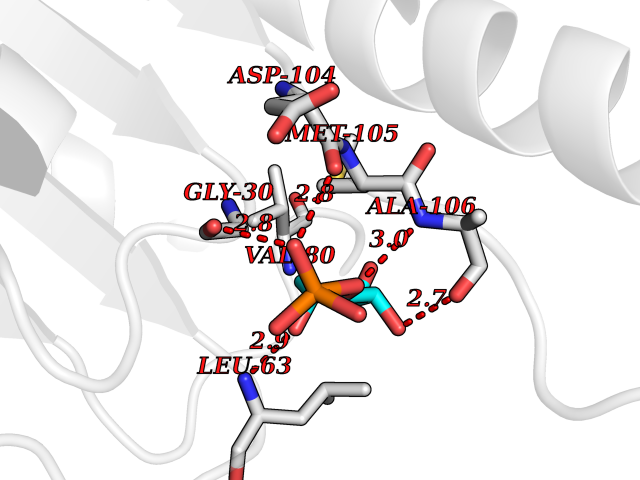 Pymol Map 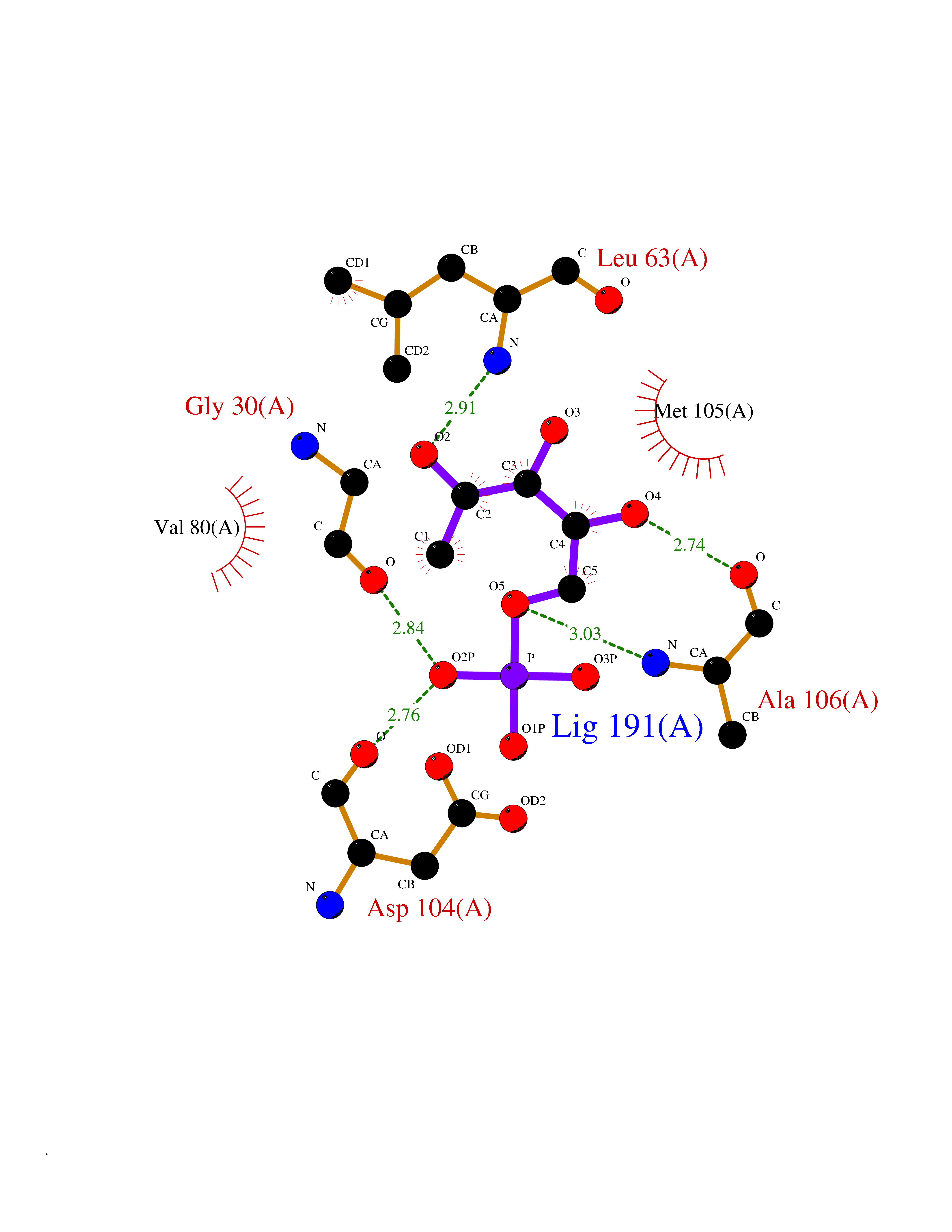 Ligplot Map 3D Binding mode Sequence SYRSRSAFKLLEVNERHQILRPGLRVLDCGAAPGAWSQVAVQKVNAAGTDPSSPVGFVLGVDLLHIFPLEGATFLCPADVTDPRTSQRILEVLPGRRADVILSDMAPNATGFRDLDHDRLISLCLTLLSVTPDILQPGGTFLCKTWAGSQSRRLQRRLTEEFQNVRIIKPEVYFLATQYHG Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Carbonic anhydrase II (CA-II) | 3K34 | 6.21 | |
Target general information Gen name CA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase C; Carbonic anhydrase 2; Carbonate dehydratase II; CAC Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Reversible hydration of carbon dioxide. Can hydrate cyanamide to urea. Involved in the regulation of fluid secretion into the anterior chamber of the eye. Contributes to intracellular pH regulation in the duodenal upper villous epithelium during proton-coupled peptide absorption. Stimulates the chloride-bicarbonate exchange activity of SLC26A6. Essential for bone resorption and osteoclast differentiation. Related diseases Osteopetrosis, autosomal recessive 3 (OPTB3) [MIM:259730]: A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB3 is associated with renal tubular acidosis, cerebral calcification (marble brain disease) and in some cases with intellectual disability. {ECO:0000269|PubMed:15300855, ECO:0000269|PubMed:1542674, ECO:0000269|PubMed:1928091, ECO:0000269|PubMed:8834238, ECO:0000269|PubMed:9143915}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07596; DB03333; DB08418; DB04081; DB08416; DB02479; DB07467; DB03950; DB03594; DB03294; DB04763; DB03270; DB08083; DB06954; DB08659; DB08046; DB02087; DB08156; DB04203; DB04394; DB08782; DB04549; DB02221; DB04180; DB03039; DB02861; DB02429; DB08202; DB04600; DB04601; DB01784; DB03385; DB03697; DB04002; DB07632; DB07050; DB06891; DB08645; DB08765; DB00819; DB03877; DB03262; DB03598; DB04089; DB01964; DB03526; DB04371; DB02220; DB03221; DB02602; DB02535; DB00436; DB00562; DB01194; DB00482; DB00880; DB02679; DB00606; DB02866; DB01144; DB00869; DB08846; DB01031; DB00311; DB08157; DB01942; DB00695; DB00774; DB08165; DB02292; DB03975; DB00703; DB00232; DB02610; DB07742; DB03844; DB02069; DB02986; DB08301; DB07048; DB03596; DB07476; DB01748; DB08155; DB01671; DB07710; DB01325; DB09460; DB09472; DB02894; DB00391; DB08329; DB07363; DB00273; DB01021; DB03904; DB00580; DB14533; DB14548; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Acetylation; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Lyase; Membrane; Metal-binding; Osteopetrosis; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29027.4 Length 258 Aromaticity 0.1 Instability index 20.07 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -6.66 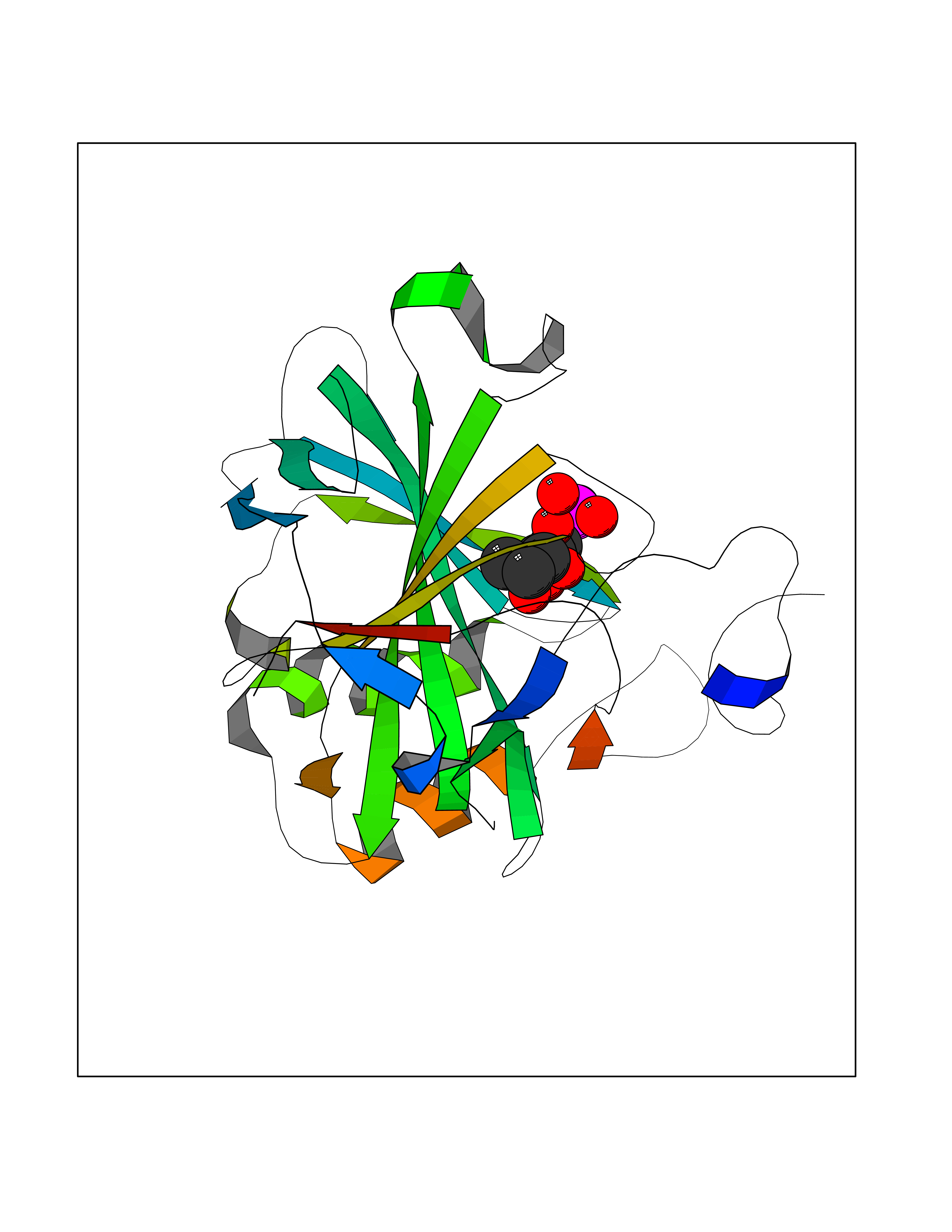 Molscript Map 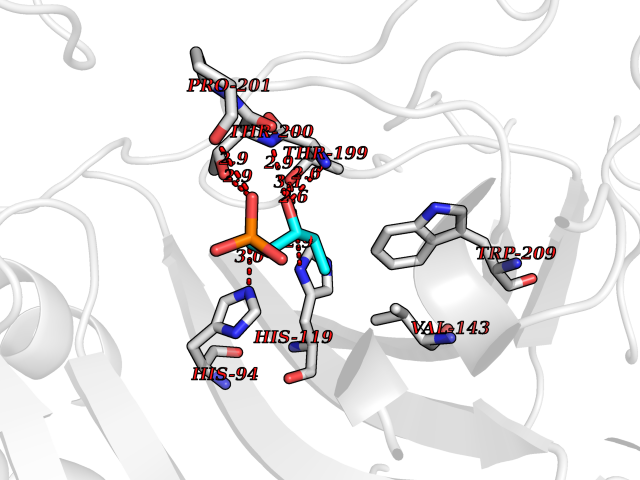 Pymol Map 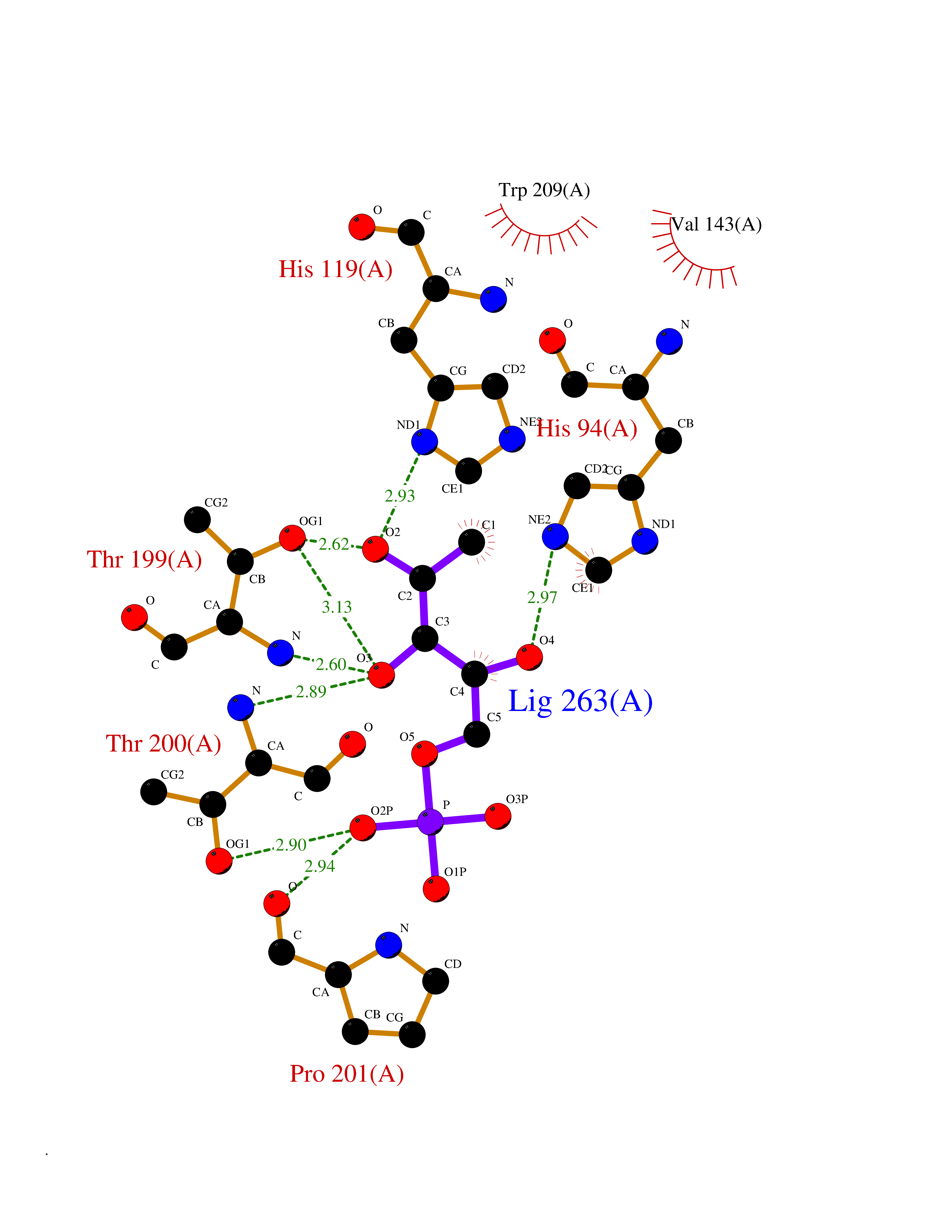 Ligplot Map 3D Binding mode Sequence HHWGYGKHNGPEHWHKDFPIAKGERQSPVDIDTHTAKYDPSLKPLSVSYDQATSLRILNNGHAFNVEFDDSQDKAVLKGGPLDGTYRLIQFHFHWGSLDGQGSEHTVDKKKYAAELHLVHWNTKYGDFGKAVQQPDGLAVLGIFLKVGSAKPGLQKVVDVLDSIKTKGKSADFTNFDPRGLLPESLDYWTYPGSLTTPPLLECVTWIVLKEPISVSSEQVLKFRKLNFNGEGEPEELMVDNWRPAQPLKNRQIKASFK Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | Caspase-3 (CASP3) | 2XYG | 6.21 | |
Target general information Gen name CASP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yama protein; SREBP cleavage activity 1; SCA-1; Protein Yama; Cysteine protease CPP32; Caspase 3; CPP32; CPP-32; CASP-3; Apopain Protein family Peptidase C14A family Biochemical class Peptidase Function At the onset of apoptosis it proteolytically cleaves poly(ADP-ribose) polymerase (PARP) at a '216-Asp-|-Gly-217' bond. Cleaves and activates sterol regulatory element binding proteins (SREBPs) between the basic helix-loop-helix leucine zipper domain and the membrane attachment domain. Cleaves and activates caspase-6, -7 and -9. Involved in the cleavage of huntingtin. Triggers cell adhesion in sympathetic neurons through RET cleavage. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Smith-Kingsmore syndrome (SKS) [MIM:616638]: An autosomal dominant syndrome characterized by intellectual disability, macrocephaly, seizures, umbilical hernia, and facial dysmorphic features. {ECO:0000269|PubMed:25851998, ECO:0000269|PubMed:26542245, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Focal cortical dysplasia 2 (FCORD2) [MIM:607341]: A form of focal cortical dysplasia, a malformation of cortical development that results in medically refractory epilepsy in the pediatric population and in adults. FCORD2 is a severe form, with onset usually in childhood, characterized by disrupted cortical lamination and specific cytological abnormalities. It is classified in 2 subtypes: type IIA characterized by dysmorphic neurons and lack of balloon cells; type IIB with dysmorphic neurons and balloon cells. {ECO:0000269|PubMed:25799227, ECO:0000269|PubMed:25878179, ECO:0000269|PubMed:26018084, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08498; DB08497; DB08213; DB06862; DB08251; DB03124; DB08229; DB00945; DB05408; DB13751; DB06255; DB07696; DB01017; DB08499; DB12843; DB13048; DB00282; DB12709 Interacts with O43823; Q9Y243; P05067; P54252; P55212; P55211; Q14203-5; P42858; Q00987; O60551; P09874; Q5JUK2; P10599; Q9BYP7; P98170 EC number EC 3.4.22.56 Uniprot keywords 3D-structure; Acetylation; Apoptosis; Cytoplasm; Direct protein sequencing; Hydrolase; Phosphoprotein; Protease; Proteomics identification; Reference proteome; S-nitrosylation; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 27483.1 Length 239 Aromaticity 0.11 Instability index 38.09 Isoelectric point 8.39 Charge (pH=7) 3.03 2D Binding mode Binding energy (Kcal/mol) -6.58 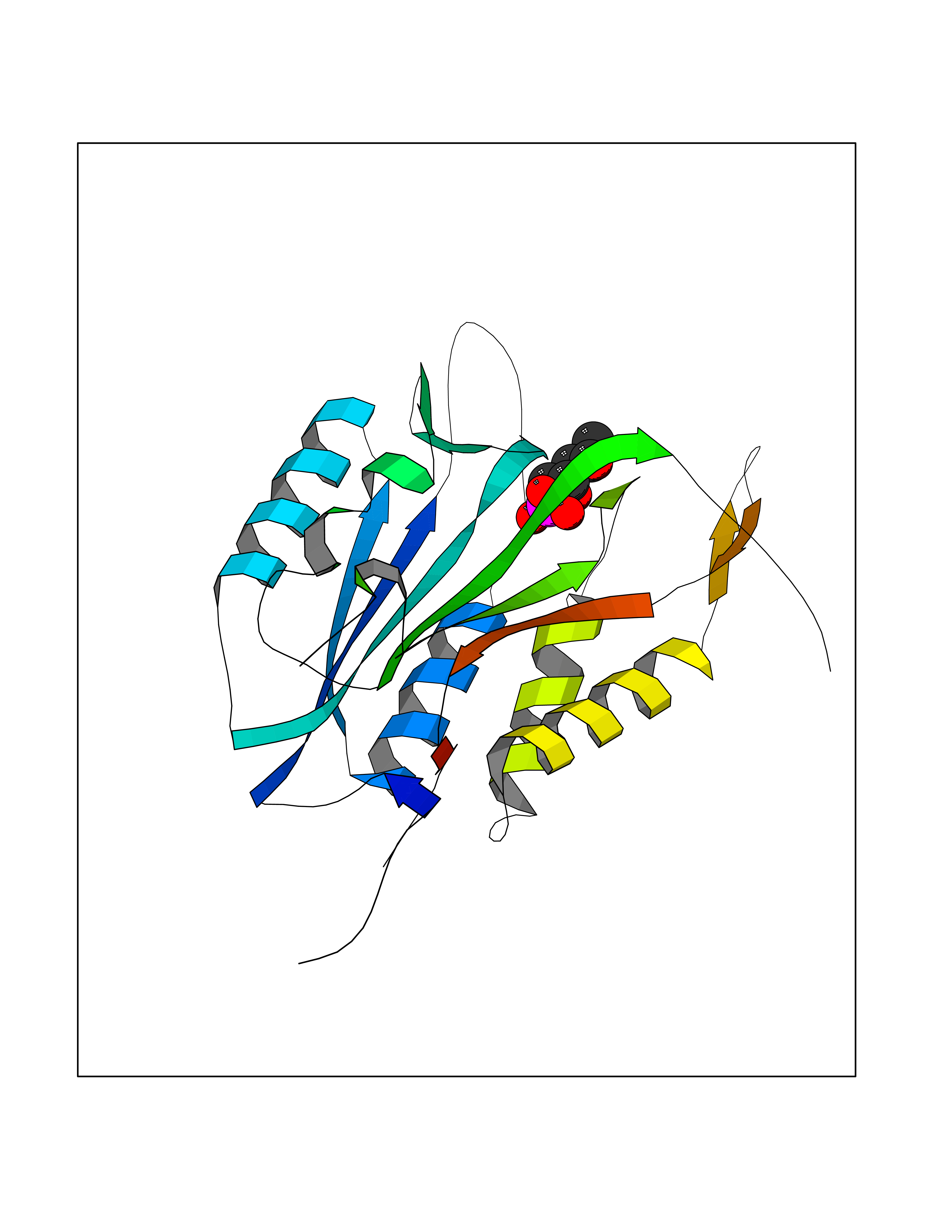 Molscript Map 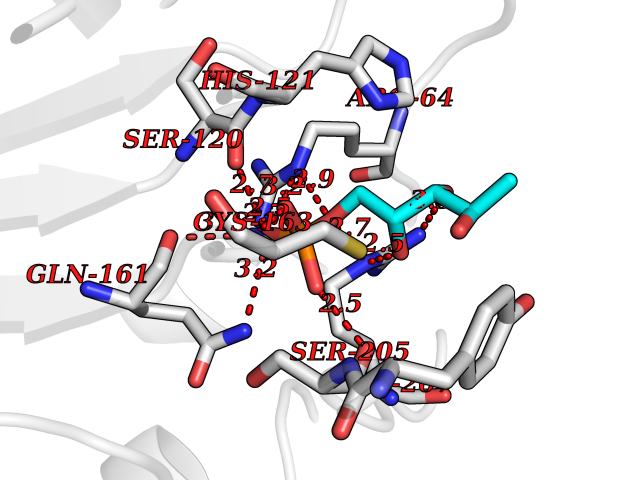 Pymol Map 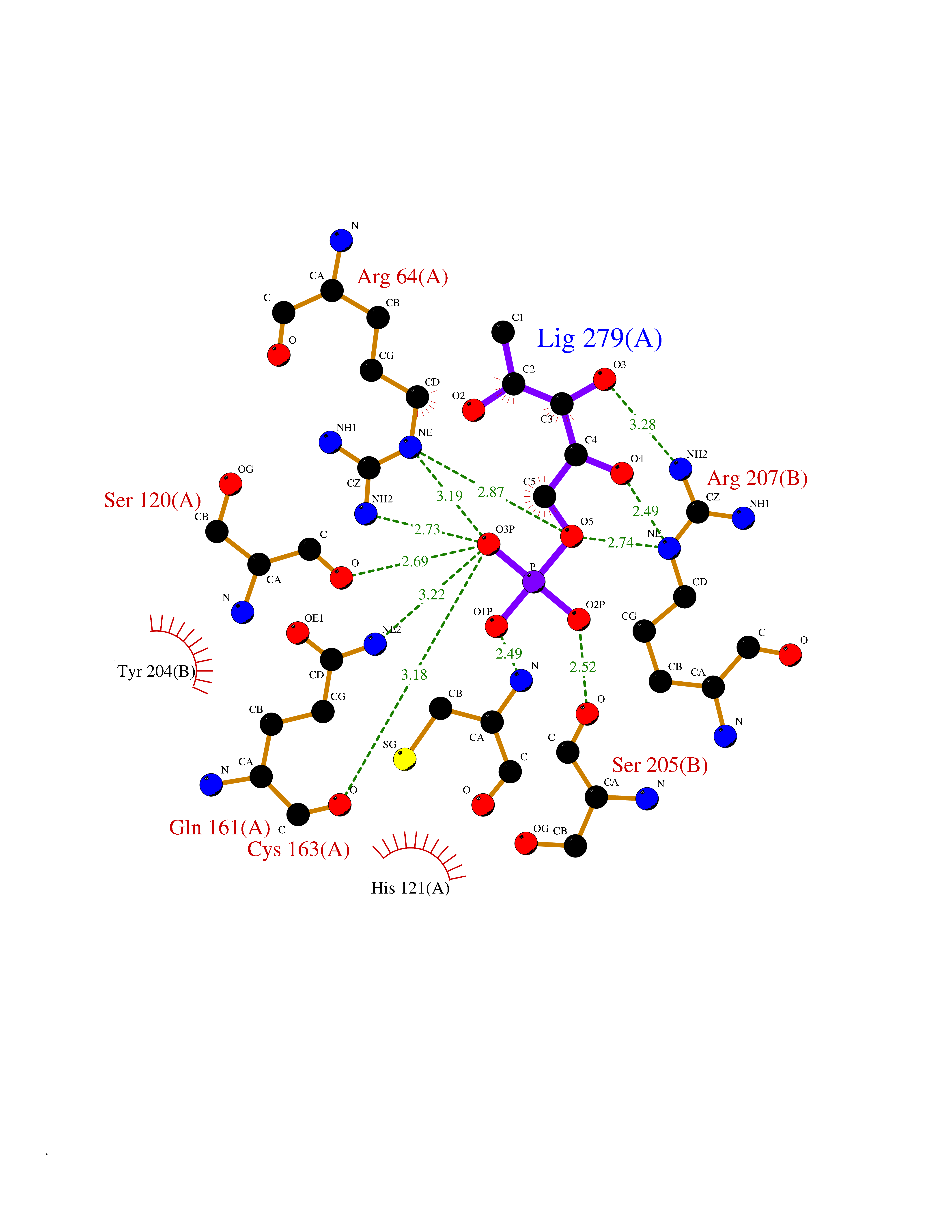 Ligplot Map 3D Binding mode Sequence SGISLDNSYKMDYPEMGLCIIINNKNFHKSTGMTSRSGTDVDAANLRETFRNLKYEVRNKNDLTREEIVELMRDVSKEDHSKRSSFVCVLLSHGEEGIIFGTNGPVDLKKITNFFRGDRCRSLTGKPKLFIIQACRGTELDCGIETHKIPVEADFLYAYSTAPGYYSWRNSKDGSWFIQSLCAMLKQYADKLEFMHILTRVNRKVATEFESFSFDATFHAKKQIPCIVSMLTKELYFYH Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Catechol-O-methyl-transferase (COMT) | 3BWY | 6.20 | |
Target general information Gen name COMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms S-COMT; MB-COMT; Catechol-O-methyltransferase; COMT Protein family Class I-like SAM-binding methyltransferase superfamily, Cation-dependent O-methyltransferase family Biochemical class Methyltransferase Function Catalyzes the O-methylation, and thereby the inactivation, of catecholamine neurotransmitters and catechol hormones. Also shortens the biological half-lives of certain neuroactive drugs, like L-DOPA, alpha-methyl DOPA and isoproterenol. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07462; DB02342; DB02105; DB08049; DB00118; DB00714; DB03336; DB00286; DB00255; DB00841; DB00988; DB15488; DB00494; DB00668; DB00783; DB00977; DB01064; DB00968; DB01141; DB03907; DB04820; DB06152; DB11632; DB00252; DB01420; DB00323 Interacts with Q6P5T0; P30518; Q8NFU1; Q8NHW4; P34972; Q96BA8; P50402; Q5JX71; O14843; O00258; P08034; O75712; Q9NTQ9; O95377; Q8TDT2; Q8N6U8; O15529; P31937; Q9H2F3; O95279; Q5SR56; A6NDP7; Q0D2K0; Q7RTS5; Q9UHJ9-5; Q8IY26; Q9H6H4; Q6NTF9-3; O75783; Q99500; Q9Y6D0; Q3KNW5; O60669; P22732; Q96G79; Q5T1Q4; Q9NY26; Q9NP94; Q6P1K1; P30825; Q9UHI5; B2RUZ4; Q9UPZ6; Q96MV1; Q9NV29; A0PK00; Q9NUH8; Q9P0S9; Q14656; Q6UW68; Q9H0R3; O95807; P34981; Q15645; Q15836; O95183; O76024; P30260; Q9H816; Q92997; P29323-3; P22607; P06396; Q15323; Q6A162; P26371; O15116; P20645; O14744; Q5T160; Q9UJD0; Q2MKA7; Q8N488; O75880; Q14141; Q9UNE7; Q15645; Q9NYH9; Q8NA23-2 EC number EC 2.1.1.6 Uniprot keywords 3D-structure; Alternative initiation; Catecholamine metabolism; Cell membrane; Cytoplasm; Direct protein sequencing; Lipid metabolism; Magnesium; Membrane; Metal-binding; Methyltransferase; Neurotransmitter degradation; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Schizophrenia; Signal-anchor; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 23851.2 Length 214 Aromaticity 0.07 Instability index 25.99 Isoelectric point 5.25 Charge (pH=7) -7.75 2D Binding mode Binding energy (Kcal/mol) -7.2 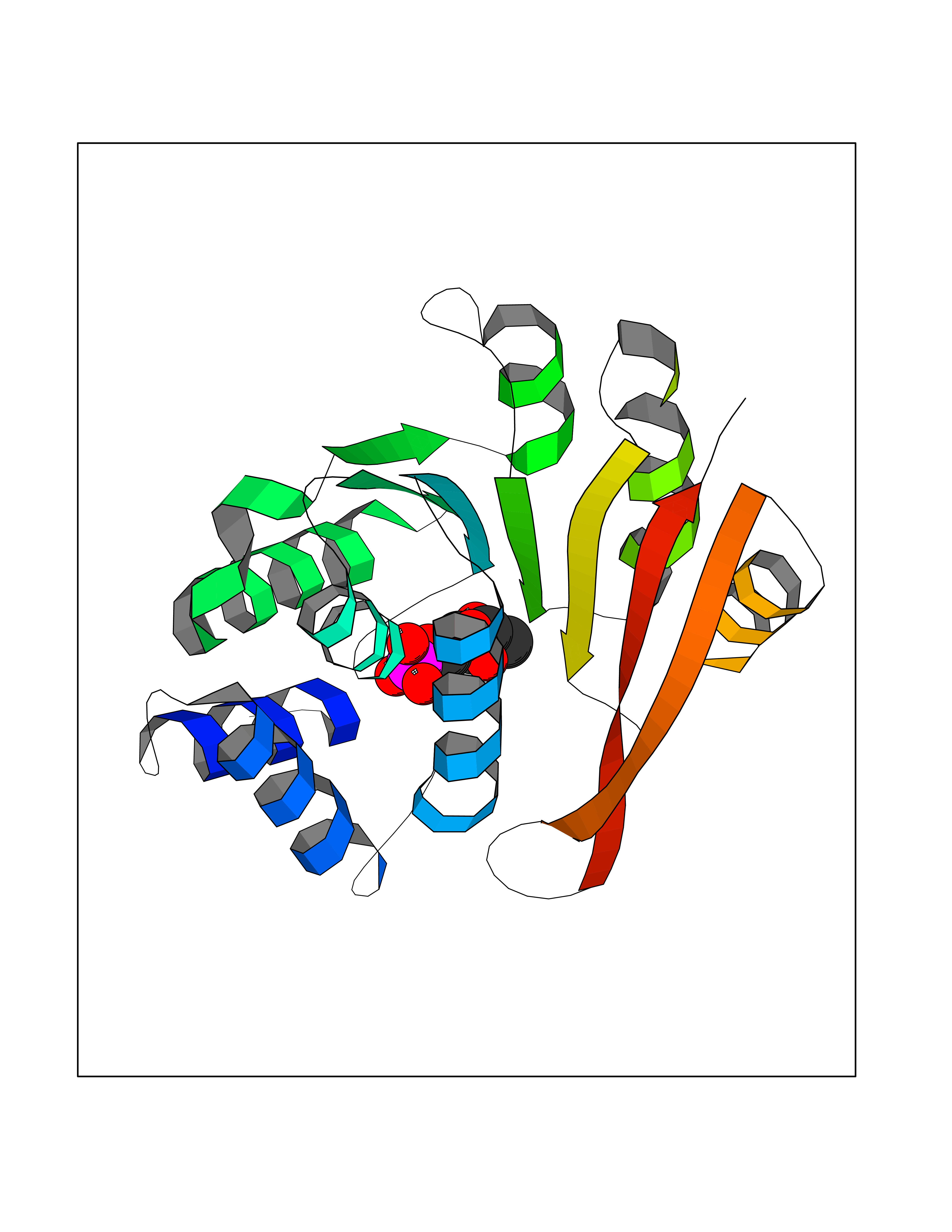 Molscript Map 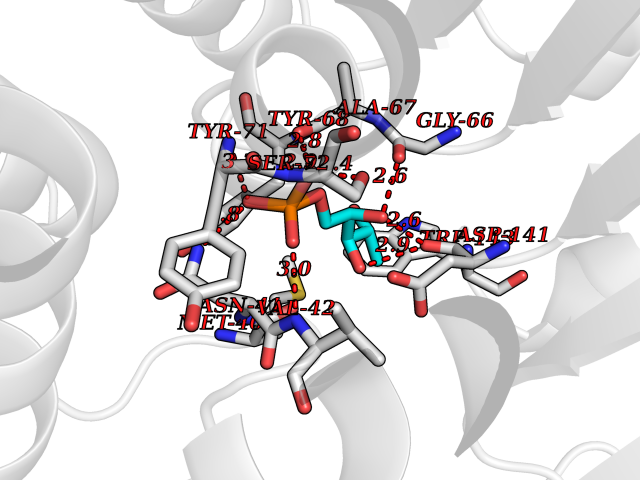 Pymol Map 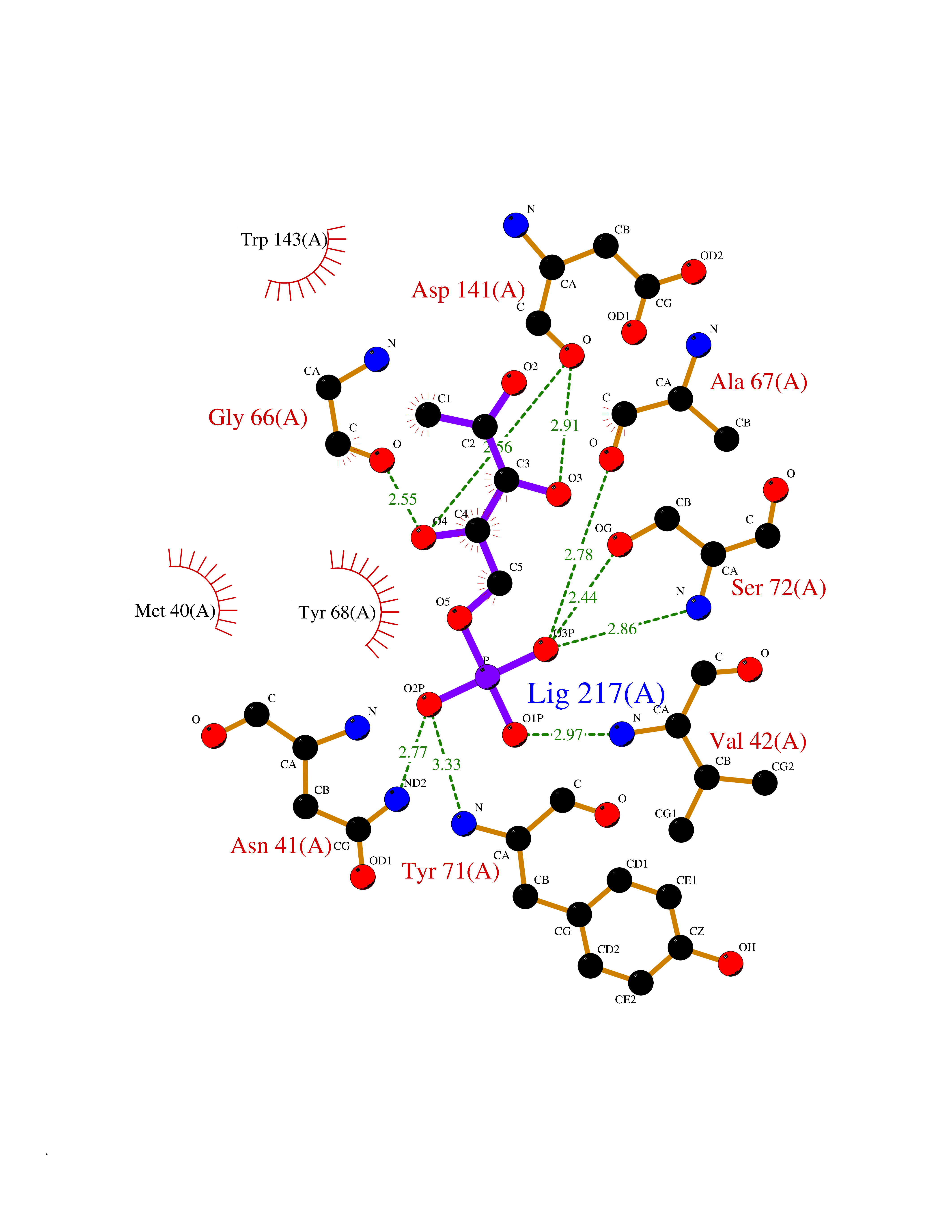 Ligplot Map 3D Binding mode Sequence GDTKEQRILNHVLQHAEPGNAQSVLEAIDTYCEQKEWAMNVGDKKGKIVDAVIQEHQPSVLLELGAYCGYSAVRMARLLSPGARLITIEINPDCAAITQRMVDFAGMKDKVTLVVGASQDIIPQLKKKYDVDTLDMVFLDHWKDRYLPDTLLLEECGLLRKGTVLLADNVICPGAPDFLAHVRGSSCFECTHYQSFLEYREVVDGLEKAIYKGP Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Opioid receptor delta (OPRD1) | 4N6H | 6.19 | |
Target general information Gen name OPRD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms OPRD; Delta-type opioid receptor; Delta opioid receptor; DOR-1; D-OR-1 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors, such as adenylate cyclase. Signaling leads to the inhibition of adenylate cyclase activity. Inhibits neurotransmitter release by reducing calcium ion currents and increasing potassium ion conductance. Plays a role in the perception of pain and in opiate-mediated analgesia. Plays a role in developing analgesic tolerance to morphine. G-protein coupled receptor that functions as receptor for endogenous enkephalins and for a subset of other opioids. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB01571; DB01439; DB05050; DB06274; DB06288; DB00321; DB01238; DB00921; DB00611; DB09173; DB09061; DB01535; DB00318; DB00514; DB00647; DB01452; DB01565; DB01444; DB01081; DB01548; DB09272; DB01497; DB00813; DB00956; DB00327; DB01221; DB06738; DB00854; DB00836; DB14146; DB14009; DB12668; DB00333; DB00295; DB06409; DB14011; DB00844; DB11691; DB06230; DB01183; DB00704; DB11130; DB00497; DB01192; DB09209; DB00899; DB12543; DB00708; DB06204; DB00193 Interacts with P16615; P27824; Q4LDR2; Q5JY77; Q9NS64; Q9Y666-2; Q9UKG4; Q0VAQ4; Q96Q45-2; P11607 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32859.3 Length 294 Aromaticity 0.11 Instability index 33.86 Isoelectric point 9.38 Charge (pH=7) 13.6 2D Binding mode Binding energy (Kcal/mol) -6.44 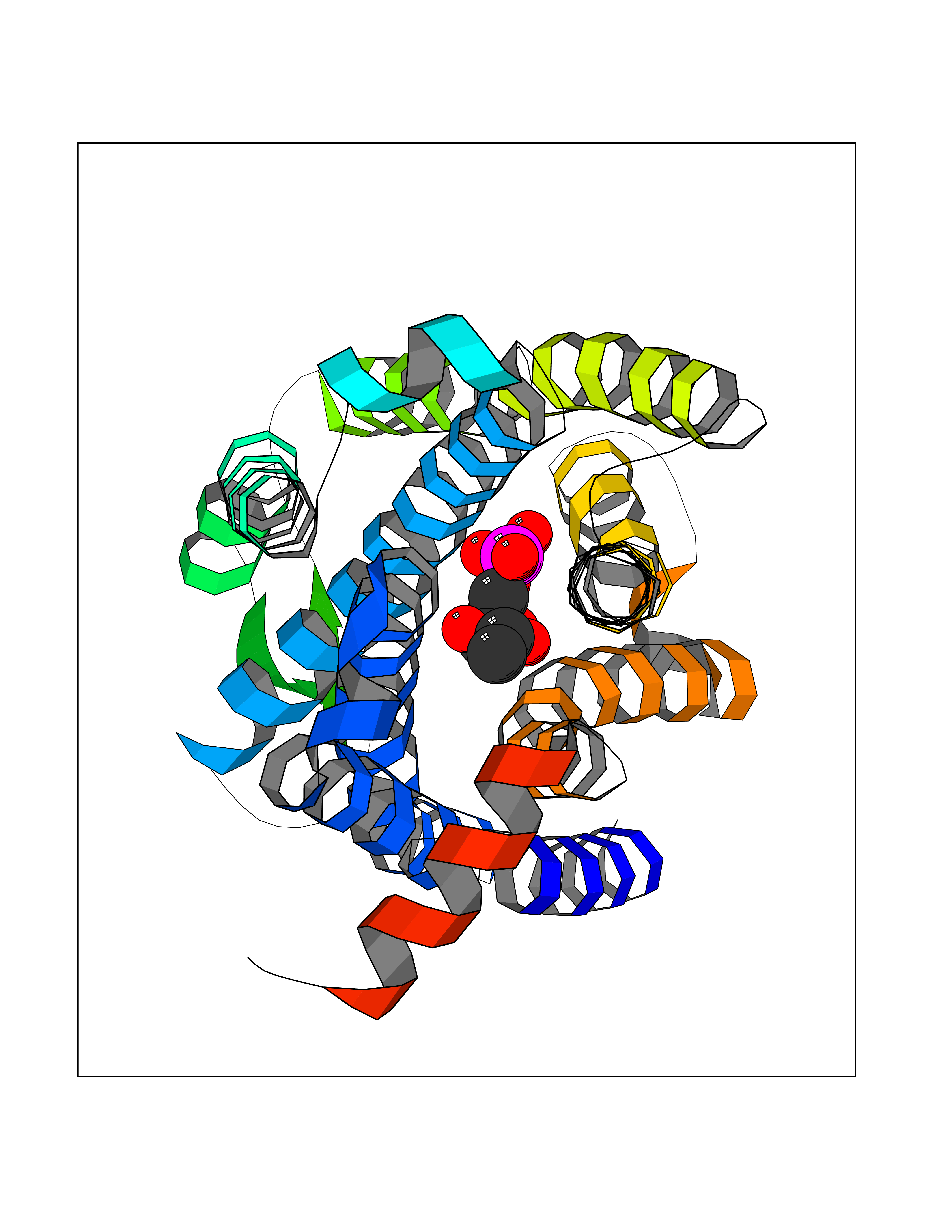 Molscript Map 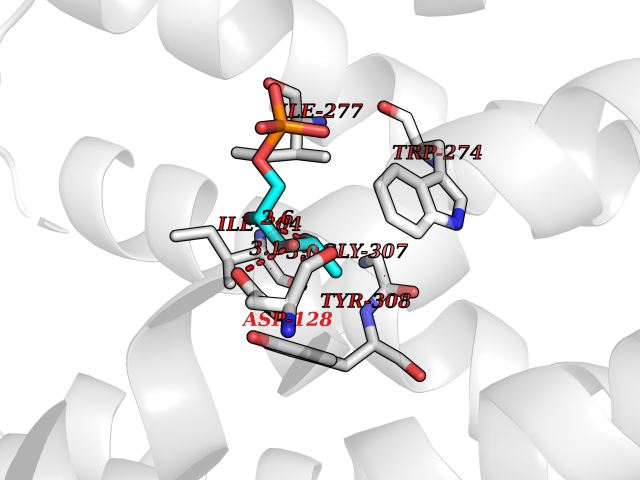 Pymol Map 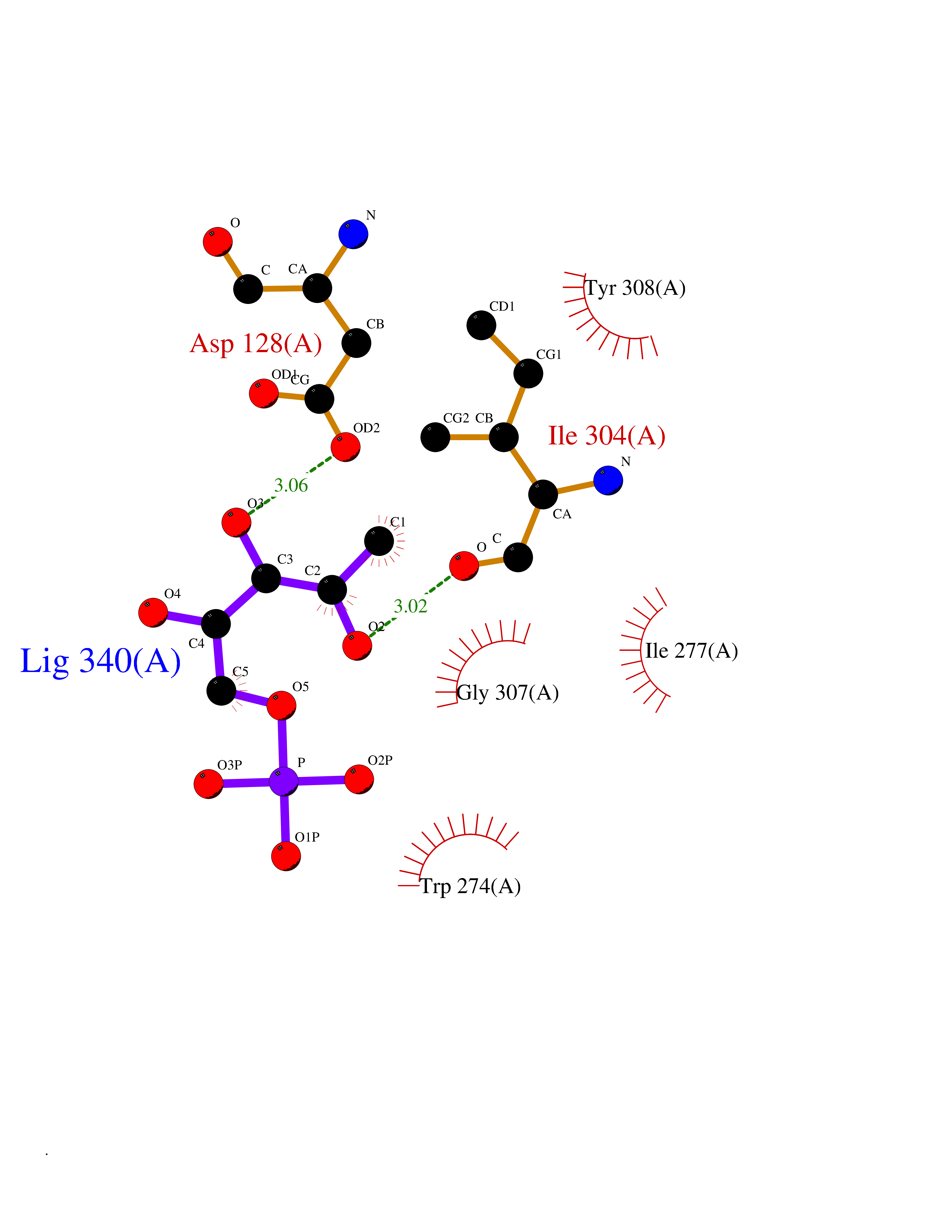 Ligplot Map 3D Binding mode Sequence SLALAIAITALYSAVCAVGLLGNVLVMFGIVRYTKMKTATNIYIFNLALADALATSTLPFQSAKYLMETWPFGELLCKAVLSIDYYNMFTSIFTLTMMSVDRYIAVCHPVKALDFRTPAKAKLINICIWVLASGVGVPIMVMAVTRPRDGAVVCMLQFPSPSWYWDTVTKICVFLFAFVVPILIITVCYGLMLLRLRSVRLLSGSKEKDRSLRRITRMVLVVVGAFVVCWAPIHIFVIVWTLVDIDRRDPLVVAALHLCIALGYANSSLNPVLYAFLDENFKRCFRQLCRKPCG Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Solute carrier family 19 member 1 (SLC19A1) | 8GOF | 6.19 | |
Target general information Gen name SLC19A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Reduced folate carrier protein; RFC1; RFC; Placental folate transporter; Intestinal folate carrier 1; IFC-1; Folate transporter 1; FOLT; FLOT1 Protein family Reduced folate carrier (RFC) transporter (TC 2.A.48) family Biochemical class NA Function Transporter for the intake of folate. Uptake of folate in human placental choriocarcinoma cells occurs by a novel mechanism called potocytosis which functionally couples three components, namely the folate receptor, the folate transporter, and a V-type H(+)-pump. Related diseases Megaloblastic anemia, folate-responsive (MEGAF) [MIM:601775]: An autosomal recessive metabolic disorder characterized by megaloblastic anemia resulting from decreased folate transport into erythrocytes. Disease manifestations include hemolytic anemia, hyperhomocysteinemia, and low vitamin B12. Serum folate levels are normal, but erythrocyte folate levels are decreased. Treatment with oral folate corrects the anemia and normalizes homocysteine. {ECO:0000269|PubMed:32276275}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency 114, folate-responsive (IMD114) [MIM:620603]: An autosomal recessive immunologic disorder manifesting in early infancy and characterized by recurrent skin and respiratory infections, mucosal bleeding, oral ulcers, chronic diarrhea, and poor overall growth. Affected individuals have lymphopenia, low serum immunoglobulins, and impaired T cell proliferation. Some patients have global developmental delay. {ECO:0000269|PubMed:36517554, ECO:0000269|PubMed:36745868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11256; DB00563; DB00642; DB06813; DB01157 Interacts with Q7Z3Y9 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Antiport; Cell membrane; Disease variant; Folate-binding; Glycoprotein; Hereditary hemolytic anemia; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 46087.7 Length 407 Aromaticity 0.15 Instability index 34.62 Isoelectric point 9.82 Charge (pH=7) 17.33 2D Binding mode Binding energy (Kcal/mol) -6.4 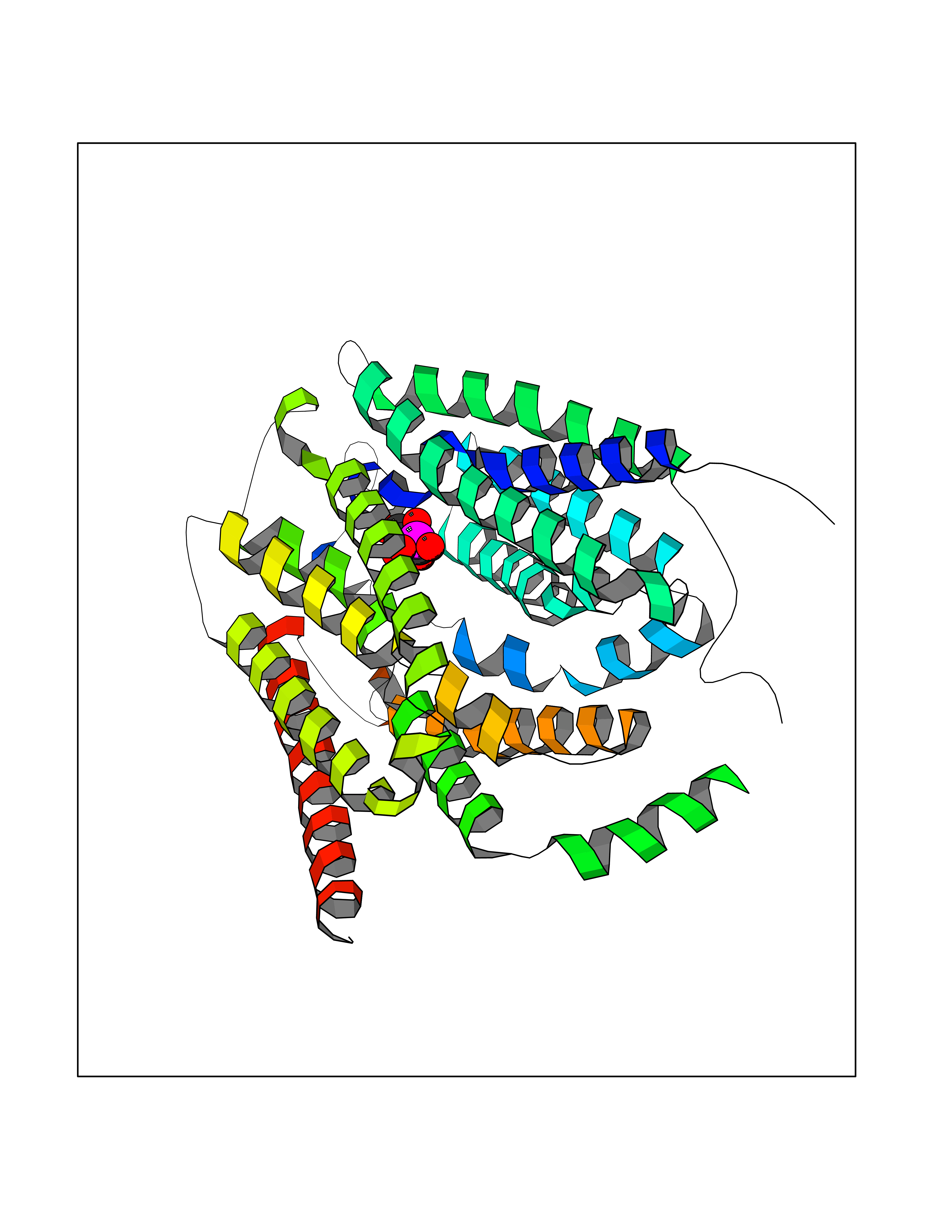 Molscript Map 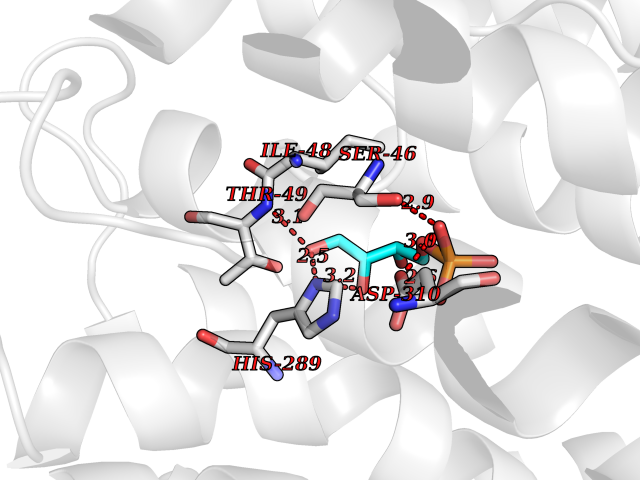 Pymol Map 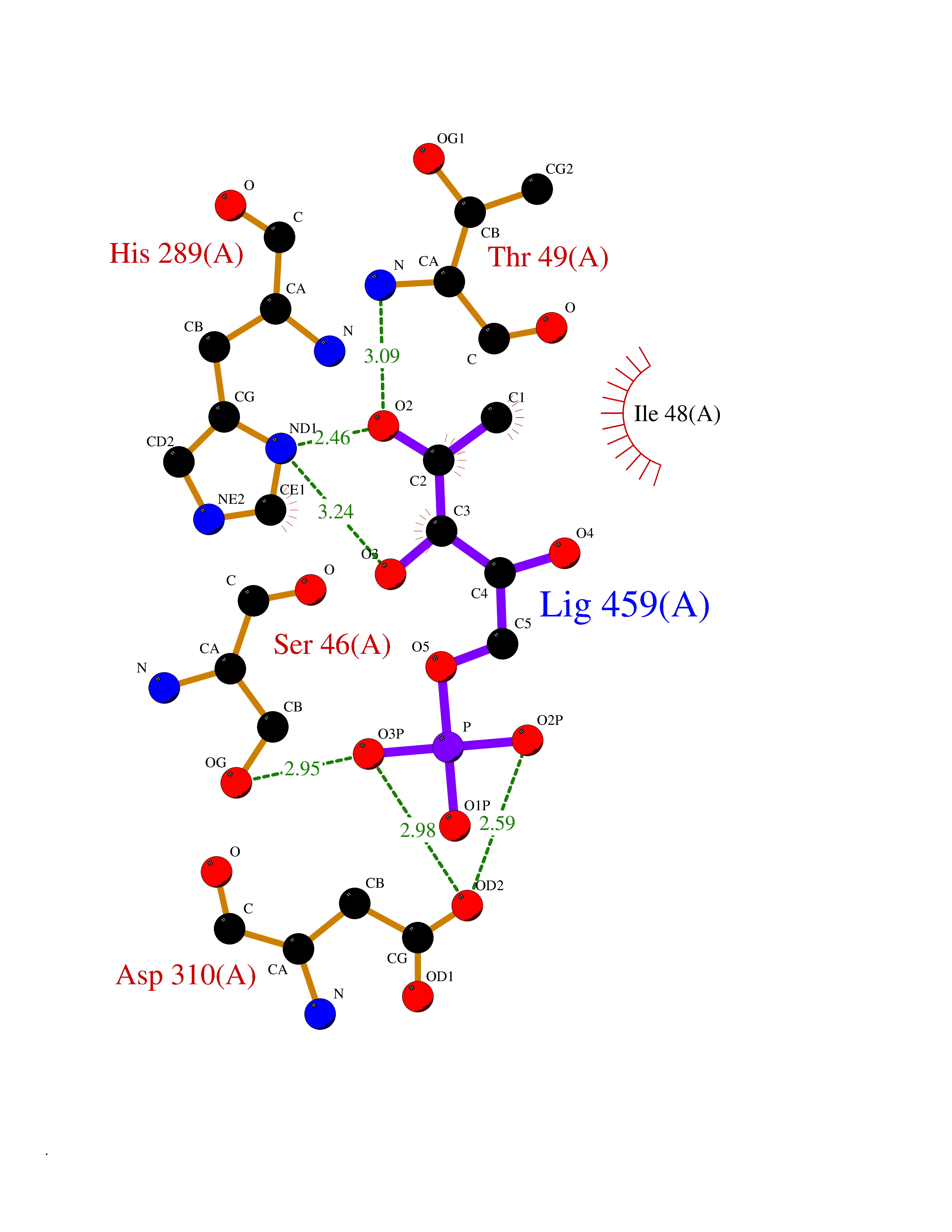 Ligplot Map 3D Binding mode Sequence DPELRSWRHLVCYLCFYGFMAQIRPGESFITPYLLGPDKNFTREQVTNEITPVLSYSYLAVLVPVFLLTDYLRYTPVLLLQGLSFVSVWLLLLLGHSVAHMQLMELFYSVTMAARIAYSSYIFSLVRPARYQRVAGYSRAAVLLGVFTSSVLGQLLVTVGRVSFSTLNYISLAFLTFSVVLALFLKRPKRSLFFNRDDSVLARMLRELGDSLRRPQLRLWSLWWVFNSAGYYLVVYYVHILWNEVDPTTNSARVYNGAADAASTLLGAITSFAAGFVKIRWARWSKLLIAGVTATQAGLVFLLAHTRHPSSIWLCYAAFVLFRGSYQFLVPIATFQIASSLSKELCALVFGVNTFFATIVKTIITFIVSDVRGLGLPVRKQFQLYSVYFLILSIIYFLGAMLDGLRH Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Probable glutathione peroxidase 8 | 3KIJ | 6.18 | |
Target general information Gen name GPX8 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms UNQ847/PRO1785 Protein family Glutathione peroxidase family Biochemical class Oxidoreductase Function Glutathione peroxidase activity.Peroxidase activity. Related diseases Neurodevelopmental disorder with spastic paraplegia and microcephaly (NEDSPM) [MIM:616281]: An autosomal recessive syndrome characterized by severe psychomotor developmental delay, dysarthria, walking difficulties, moderately to severely impaired intellectual development, poor or absent speech, and progressive microcephaly. {ECO:0000269|PubMed:25758935}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09096; DB00143; DB03310 Interacts with Q6RW13-2; Q9NVV5-2; Q9BVK2; P02656; P05090; P29972; P41181; Q92482; P07306; Q12797-6; Q92843; O15155; Q13323; O95393; Q12982; Q8WVV5; Q06432; Q8IX05; P19397; P60033; O14735; O95674; Q9BXR6; O43916; Q8N6F1-2; Q8NC01; Q6UVW9; Q96DZ9-2; O95406; Q8TBE1; P29400-2; Q4VAQ0; Q9Y5Q5; P49447; Q8NBI2; O14569; Q96LL9; Q9UPQ8; P56851; Q9UKR5; Q7L5A8; Q92520; Q96IV6; O14556; O43681; O14653; Q8TDV0; O60883; Q7Z429; P02724; Q9HCP6; P30519; P24593; Q9Y5U4; P43628; Q96E93; Q86VI4; O95214; Q8TAF8; Q9UIQ6-2; Q9UBY5; Q9Y2E5; Q9P0N8; Q9NX47; Q6N075; Q6ZSS7; Q99735; O14880; P30301; Q15546; A6NDP7; Q99519; Q92982; Q6P499; Q16617; Q8N912; Q8IXM6; Q16625; P09466; Q9NXK6; Q6TCH4; Q9UHJ9-5; Q9Y342; P26678; Q04941; Q5VZY2; Q8IY26; P54315; A5D903; Q8N0V3; Q92730; Q5QGT7; Q14108; Q14162; O00767; O75396; Q9Y6D0; Q9NRX5; Q9Y6X1; A2A2V5; P11686; Q8NHU3; Q8WWT9; Q99726; Q8N130; P78382; Q969S0; Q96JW4; Q0VAQ4; Q6UX34; Q96JF0-2; Q13277; Q9UNK0; Q9BQS2-2; P02787; P07204; Q9NPL8; P48230; P55061; Q6UX40; Q9BVC6; A0PK00; Q5BJH2-2; Q9NUH8; Q96HH6; A2RU14; Q8NBD8; Q9BU79; Q8TBM7; Q69YG0; P56557; Q9H2L4; Q8N661; Q5BJF2; Q9NRS4; Q71RG4; Q8N609; Q86UF1; Q53HI1; O75841; Q15836; O75379; O95183; Q8N0U8; Q6UX27-3; Q9UEU0; O95070; Q96EC8; Q8N966 EC number 1.11.1.9 Uniprot keywords 3D-structure; Acetylation; Membrane; Oxidoreductase; Peroxidase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 18580.2 Length 161 Aromaticity 0.13 Instability index 38.6 Isoelectric point 9.39 Charge (pH=7) 6.73 2D Binding mode Binding energy (Kcal/mol) -6.31 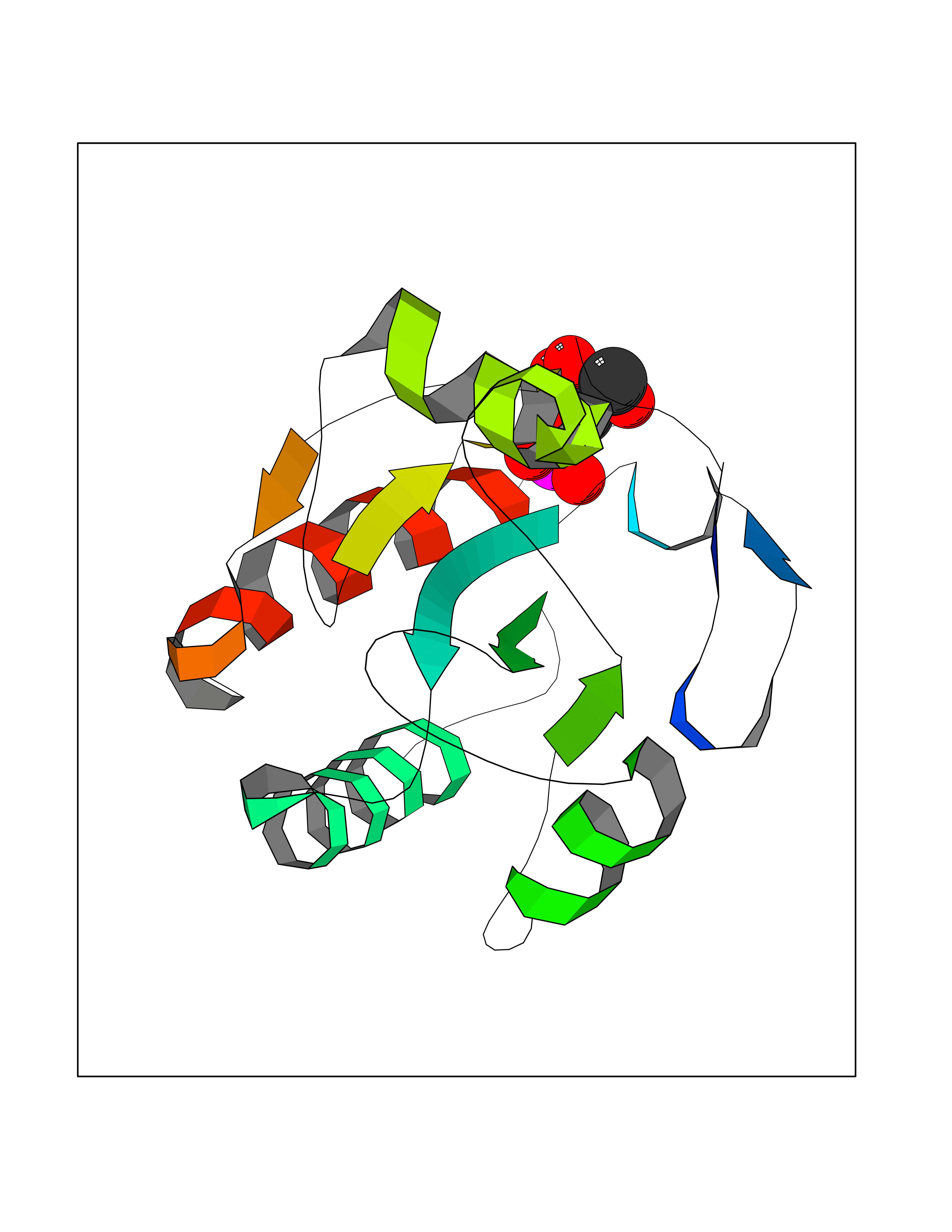 Molscript Map 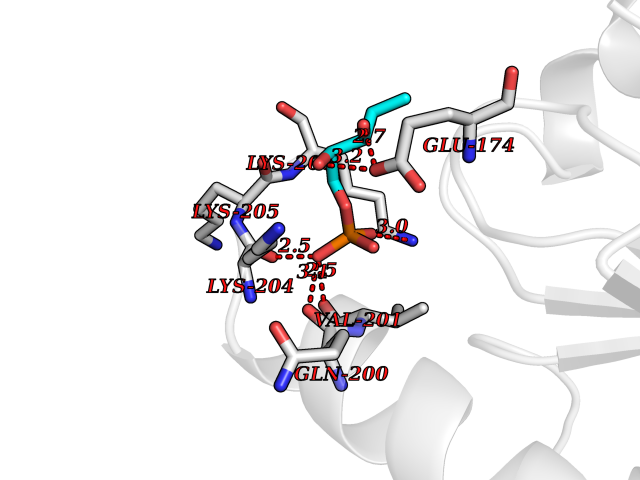 Pymol Map 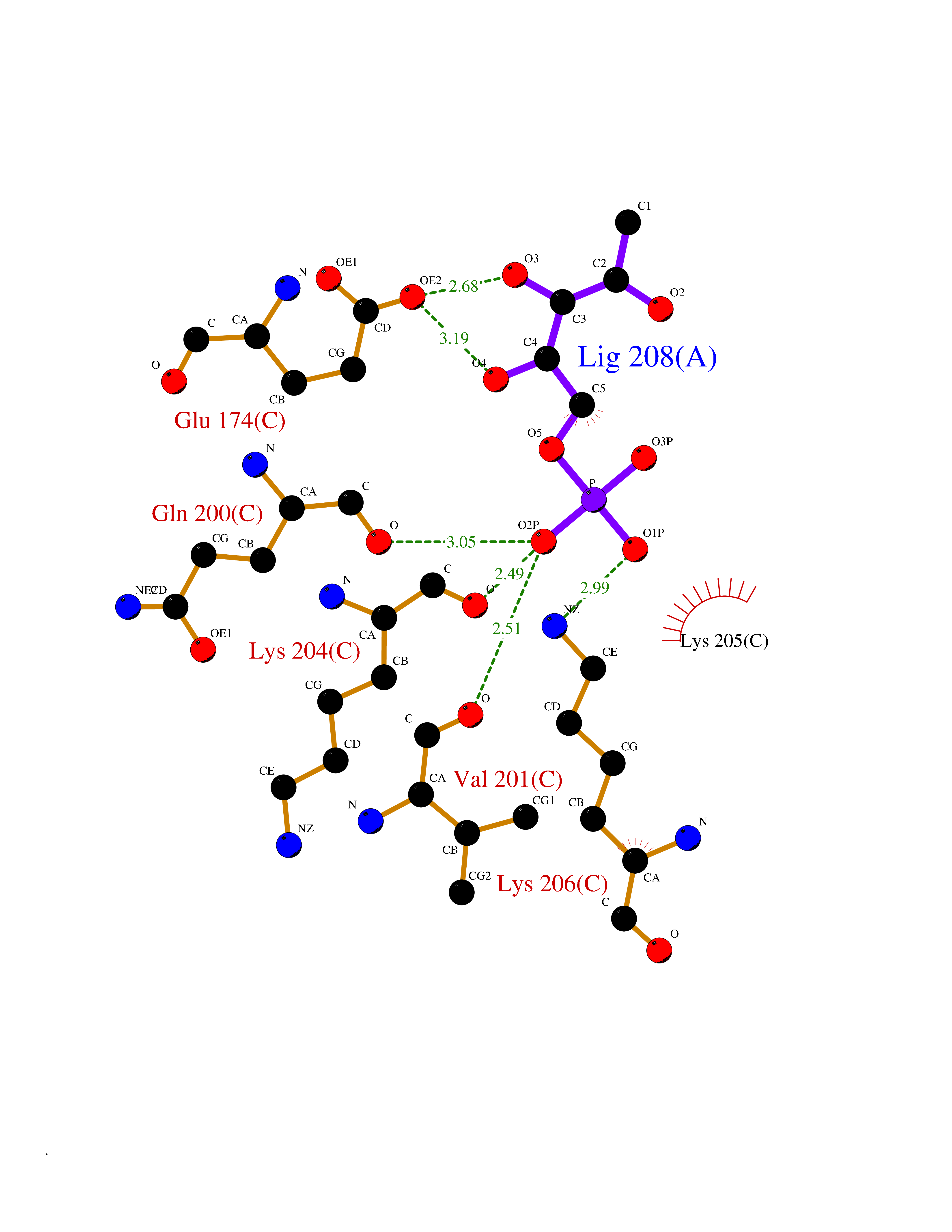 Ligplot Map 3D Binding mode Sequence SFYAFEVKDAKGRTVSLEKYKGKVSLVVNVASDCQLTDRNYLGLKELHKEFGPSHFSVLAFPCNQFGESEPRPSKEVESFARKNYGVTFPIFHKIKILGSEGEPAFRFLVDSSKKEPRWNFWKYLVNPEGQVVKFWRPEEPIEVIRPDIAALVRQVIIKKK Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Ubiquitin carboxyl-terminal hydrolase 2 (USP2) | 5XU8 | 6.18 | |
Target general information Gen name USP2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin-specific-processing protease 2; Ubiquitin thioesterase 2; UBP41; Deubiquitinating enzyme 2; 41 kDa ubiquitin-specific protease Protein family Peptidase C19 family, USP2 subfamily Biochemical class Peptidase Function Isoform 1 and isoform 4 possess both ubiquitin-specific peptidase and isopeptidase activities. Deubiquitinates MDM2 without reversing MDM2-mediated p53/TP53 ubiquitination and thus indirectly promotes p53/TP53 degradation and limits p53 activity. Has no deubiquitinase activity against p53/TP53. Prevents MDM2-mediated degradation of MDM4. Plays a role in the G1/S cell-cycle progression in normal and cancer cells. Regulates the circadian clock by modulating its intrinsic circadian rhythm and its capacity to respond to external cues. Associates with clock proteins and deubiquitinates core clock component PER1 but does not affect its overall stability. Regulates the nucleocytoplasmic shuttling and nuclear retention of PER1 and its repressive role on the clock transcription factors CLOCK and ARNTL/BMAL1. Plays a role in the regulation of myogenic differentiation of embryonic muscle cells. Hydrolase that deubiquitinates polyubiquitinated target proteins such as MDM2, MDM4 and CCND1. Related diseases Defects in AKT2 are a cause of susceptibility to breast cancer (BC). AKT2 promotes metastasis of tumor cells without affecting the latency of tumor development. May play a role in glioblastoma cell survival (PubMed:20167810). {ECO:0000269|PubMed:20167810}.; DISEASE: Type 2 diabetes mellitus (T2D) [MIM:125853]: A multifactorial disorder of glucose homeostasis caused by a lack of sensitivity to insulin. Affected individuals usually have an obese body habitus and manifestations of a metabolic syndrome characterized by diabetes, insulin resistance, hypertension and hypertriglyceridemia. The disease results in long-term complications that affect the eyes, kidneys, nerves, and blood vessels. {ECO:0000269|PubMed:15166380, ECO:0000269|PubMed:19164855}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Hypoinsulinemic hypoglycemia with hemihypertrophy (HIHGHH) [MIM:240900]: A disorder characterized by hypoglycemia, low insulin levels, low serum levels of ketone bodies and branched-chain amino acids, left-sided hemihypertrophy, neonatal macrosomia, reduced consciousness and hypoglycemic seizures. {ECO:0000269|PubMed:21979934}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NYB9-2; P12814; P35609; Q08043; Q86U10; Q86V38; P56945; Q8TD16-2; Q96CA5; A2RRN7; Q13137; Q9H257-2; Q96JN2-2; Q2TAC2; A6NC98; Q96MT8-3; Q8NHQ1; Q9BSW2; Q8N4Y2-3; Q8WTU0; O75140-2; Q9NRI5-2; Q8N9I9; Q9H596; Q8WWB3; Q5JST6; Q9NRA8; O00471; Q96B26; P57678; Q08379; Q9NYA3; A6NEM1; Q6PI77; Q14451-3; Q4V328; Q9NSC5; Q9UJC3; Q96ED9-2; Q8IYA8; Q9UKT9; Q5TA45; Q96N16; O75564-2; Q674X7-2; Q9BVG8; Q9BVG8-5; P19012; Q7Z3Y8; Q15323; Q14525; O76011; Q92764; Q6A162; Q9UBR4-2; Q969G2; Q03252; Q9BRK4; Q00987; Q9UJV3-2; Q5VZ52; Q13084; Q5JR59; Q5JR59-3; Q15742; Q9GZM8; I6L9F6; P07196; O43482; Q96CV9; Q4G0R1; Q9NRD5; Q58EX7; Q8ND90; Q16633; Q9GZV8; Q6MZQ0; Q15276; Q8HWS3; Q59EK9-3; P60903; O14492-2; O60504; Q99932-2; A6NLX3; P51692; Q86VP1; Q8WW24; Q9UBB9; Q08117-2; Q03169; Q13077; Q12933; Q9Y4K3; P36406; P14373; Q86XT4; Q15654; Q8N6Y0; Q70EL1-9; Q9UK41-2; Q8N1B4; O96006; Q9NZV7; Q9UGI0; P05067; P54253; G5E9A7; Q01658; Q00403; Q9Y5Q9; P04792; O43464; P42858; Q8WXH2; O60333-2; A0A6Q8PF08; O60260-5; P60891; Q9Y3C5; Q7Z333; P37840; P00441; Q7Z699; Q13148; O76024 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; Cell cycle; Cytoplasm; Hydrolase; Membrane; Metal-binding; Myogenesis; Nucleus; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 37785.5 Length 327 Aromaticity 0.11 Instability index 42.45 Isoelectric point 8.23 Charge (pH=7) 3.56 2D Binding mode Binding energy (Kcal/mol) -7.18  Molscript Map  Pymol Map 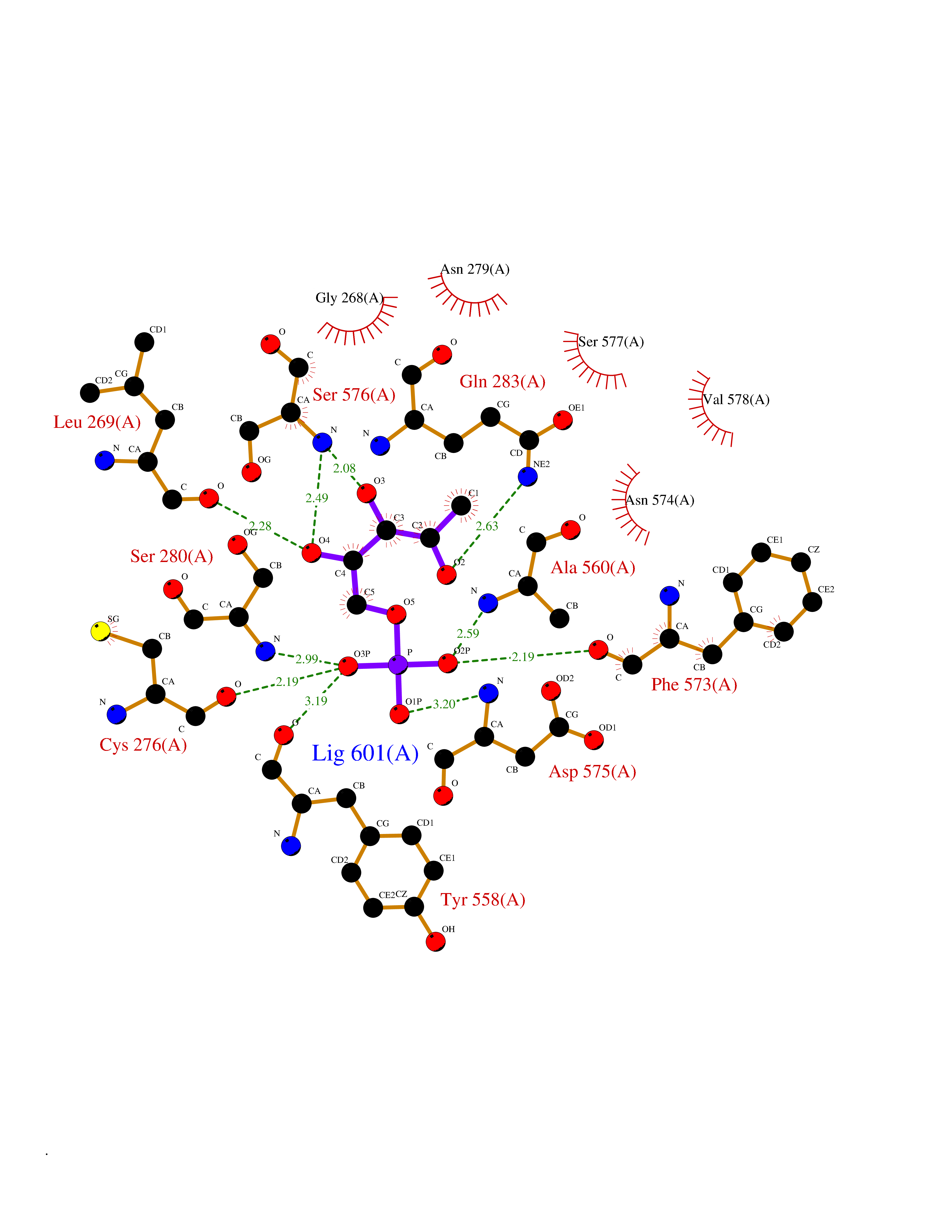 Ligplot Map 3D Binding mode Sequence QGLAGLRNLGNTCFMNSILQCLSNTRELRDYCLQRLYMRDLHHGSNAHTALVEEFAKLIQTIWTSSPNDVVSPSEFKTQIQRYAPRFVGYNQQDAQEFLRFLLDGLHNEVNRVNLDHLPDDEKGRQMWRKYLEREDSRIGDLFVGQLKSSLTCTDCGYCSTVFDPFWDLSLPIAKRGYPEVTLMDCMRLFTKEDVLDGDEKPTCCRCRGRKRCIKKFSIQRFPKILVLHLKRFSESRIRTSKLTTFVNFPLRDLDLREFASENTNHAVYNLYAVSNHSGTTMGGHYTAYCRSPGTGEWHTFNDSSVTPMSSSQVRTSDAYLLFYELA Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Lanosterol 14-alpha demethylase (CYP51A1) | 4UHI | 6.18 | |
Target general information Gen name CYP51A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450LI; Cytochrome P45014DM; Cytochrome P450-14DM; Cytochrome P450 51A1 Protein family Cytochrome P450 family Biochemical class Cytochrome P450 family Function Catalyzes C14-demethylation of lanosterol; it transforms lanosterol into 4,4'-dimethyl cholesta-8,14,24-triene-3-beta-ol. Related diseases Spondyloepimetaphyseal dysplasia, short limb-hand type (SEMD-SL) [MIM:271665]: A bone disease characterized by short-limbed dwarfism, a narrow chest with pectus excavatum, brachydactyly in the hands and feet, a characteristic craniofacial appearance and premature calcifications. The radiological findings are distinctive and comprise short long bones throughout the skeleton with striking epiphyses that are stippled, flattened and fragmented and flared, irregular metaphyses. Platyspondyly in the spine with wide intervertebral spaces is observed and some vertebral bodies are pear-shaped with central humps, anterior protrusions and posterior scalloping. {ECO:0000269|PubMed:19110212, ECO:0000269|PubMed:20223752, ECO:0000269|PubMed:26463668}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Warburg-Cinotti syndrome (WRCN) [MIM:618175]: An autosomal dominant disease characterized by progressive corneal neovascularization, keloid formation, chronic skin ulcers, wasting of subcutaneous tissue, flexion contractures of the fingers, and acro-osteolysis. {ECO:0000269|PubMed:30449416}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07705; DB05667; DB01110; DB01007 Interacts with NA EC number EC 1.14.14.154 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Endoplasmic reticulum; Heme; Iron; Lipid biosynthesis; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 53013.3 Length 462 Aromaticity 0.11 Instability index 47.66 Isoelectric point 8.8 Charge (pH=7) 7 2D Binding mode Binding energy (Kcal/mol) -6.1  Molscript Map 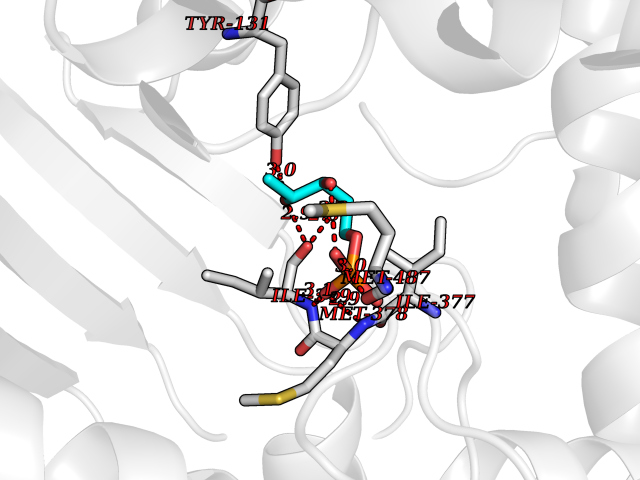 Pymol Map 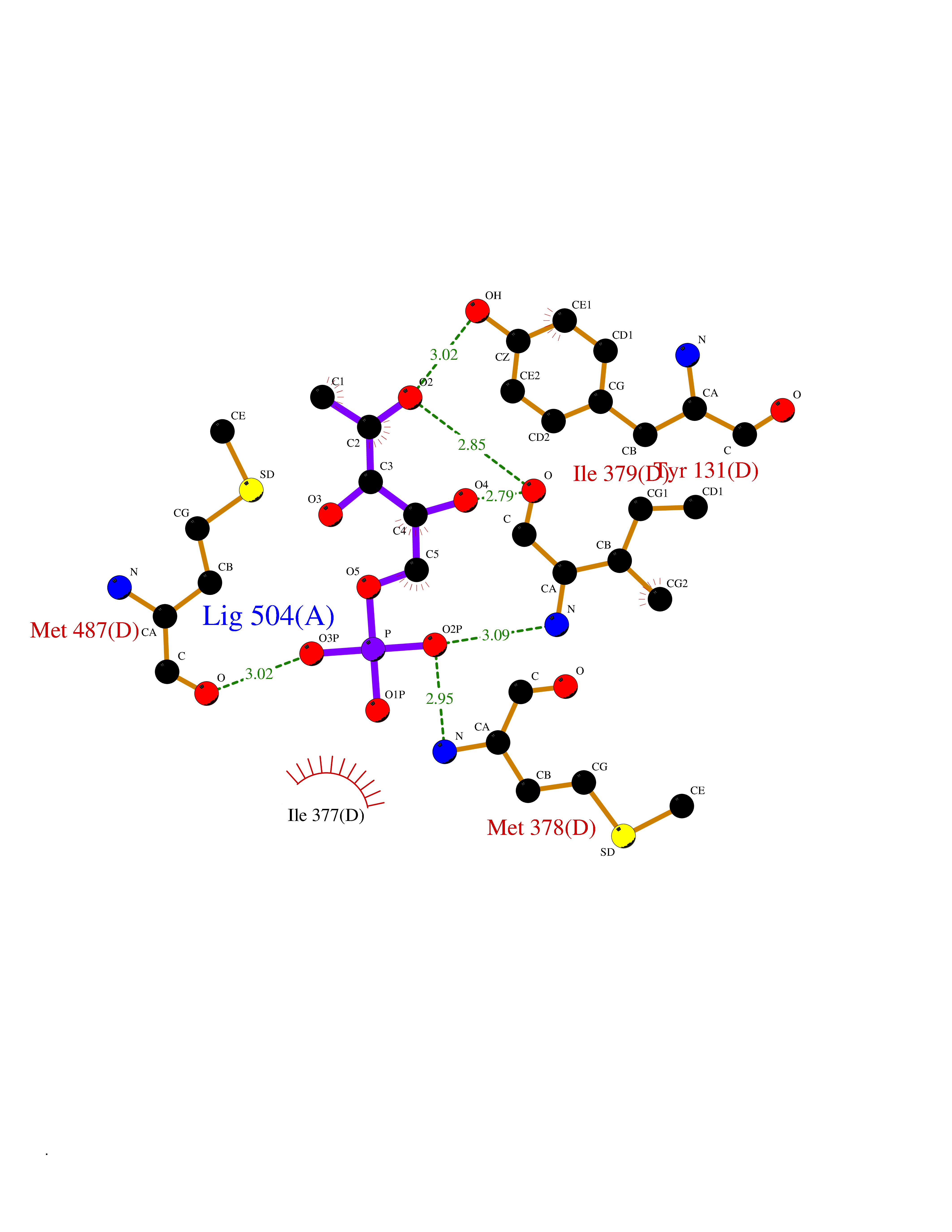 Ligplot Map 3D Binding mode Sequence PPYIFSPIPFLGHAIAFGKSPIEFLENAYEKYGPVFSFTMVGKTFTYLLGSDAAALLFNSKNEDLNAEDVYSRLTTPVFGKGVAYDVPNPVFLEQKKMLKSGLNIAHFKQHVSIIEKETKEYFESWGESGEKNVFEALSELIILTASHCLHGKEIRSQLNEKVAQLYADLDGGFSHAAWLLPGWLPLPSFRRRDRAHREIKDIFYKAIQKRRQSQEKIDDILQTLLDATYKDGRPLTDDEVAGMLIGLLLAGQHTSSTTSAWMGFFLARDKTLQKKCYLEQKTVCGENLPPLTYDQLKDLNLLDRCIKETLRLRPPIMIMMRMARTPQTVAGYTIPPGHQVCVSPTVNQRLKDSWVERLDFNPDRYLQDNPASGEKFAYVPFGAGRHRCIGENFAYVQIKTIWSTMLRLYEFDLIDGYFPTVNYTTMIHTPENPVIRYKRRSLPGWLPLPSFRRRDRAHREI Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Aspartate carbamoyltransferase (CAD) | 4C6E | 6.18 | |
Target general information Gen name CAD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CAD Protein family CarA family; CarB family; Metallo-dependent hydrolases superfamily, DHOase family, CAD subfamily; Aspartate/ornithine carbamoyltransferase superfamily, ATCase family Biochemical class Carbon-nitrogen ligase Function This protein is a "fusion" protein encoding four enzymatic activities of the pyrimidine pathway (GATase, CPSase, ATCase and DHOase). Related diseases Developmental and epileptic encephalopathy 50 (DEE50) [MIM:616457]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE50 is an autosomal recessive, progressive disease with onset in infancy and favorable response to treatment with oral uridine. {ECO:0000269|PubMed:25678555, ECO:0000269|PubMed:28087732}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00128; DB00130; DB03459 Interacts with P27708; Q8N137; P63104 EC number NA Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; ATP-binding; Congenital disorder of glycosylation; Cytoplasm; Direct protein sequencing; Disease variant; Epilepsy; Hydrolase; Ligase; Magnesium; Manganese; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Repeat; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 38268.4 Length 351 Aromaticity 0.06 Instability index 41.29 Isoelectric point 5.86 Charge (pH=7) -10.56 2D Binding mode Binding energy (Kcal/mol) -5.97 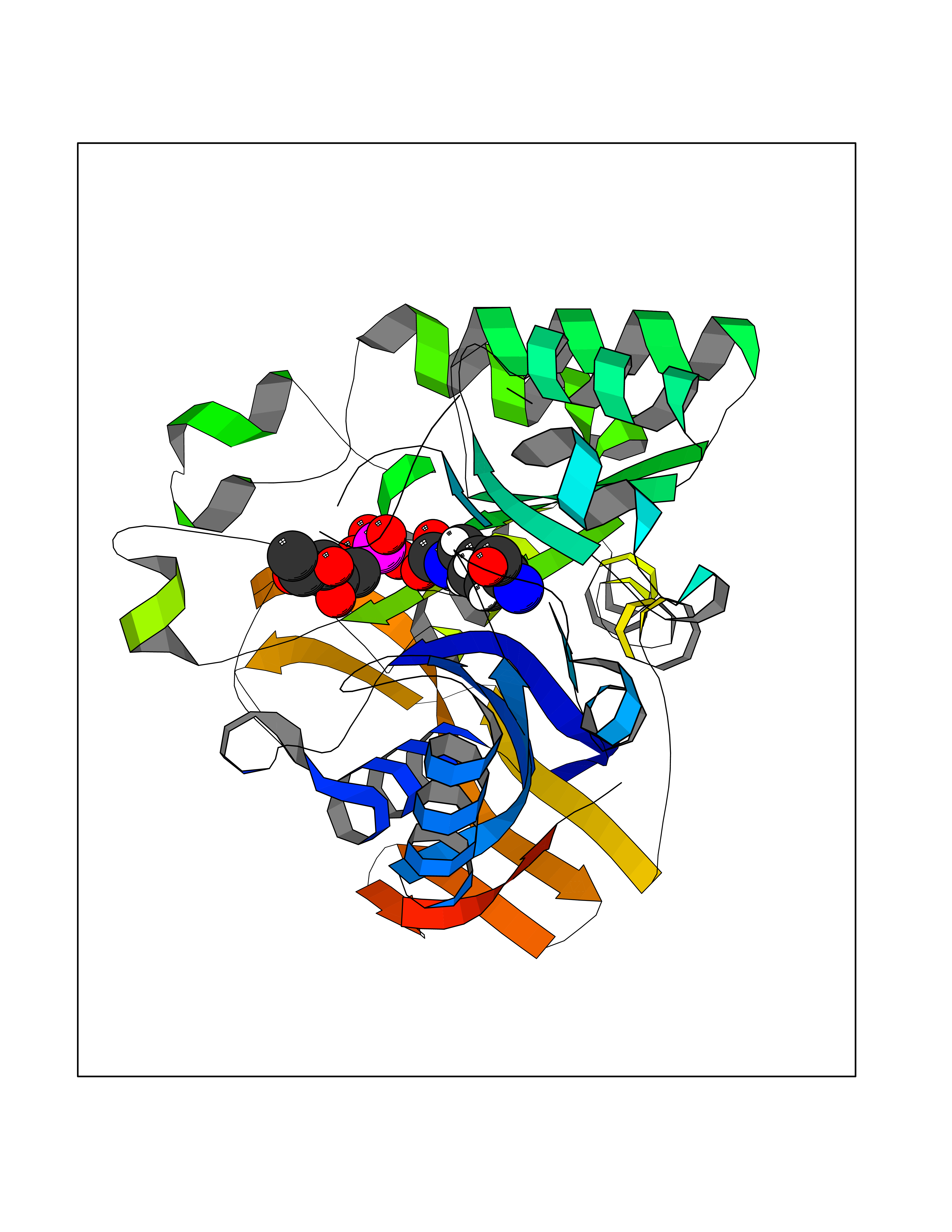 Molscript Map  Pymol Map 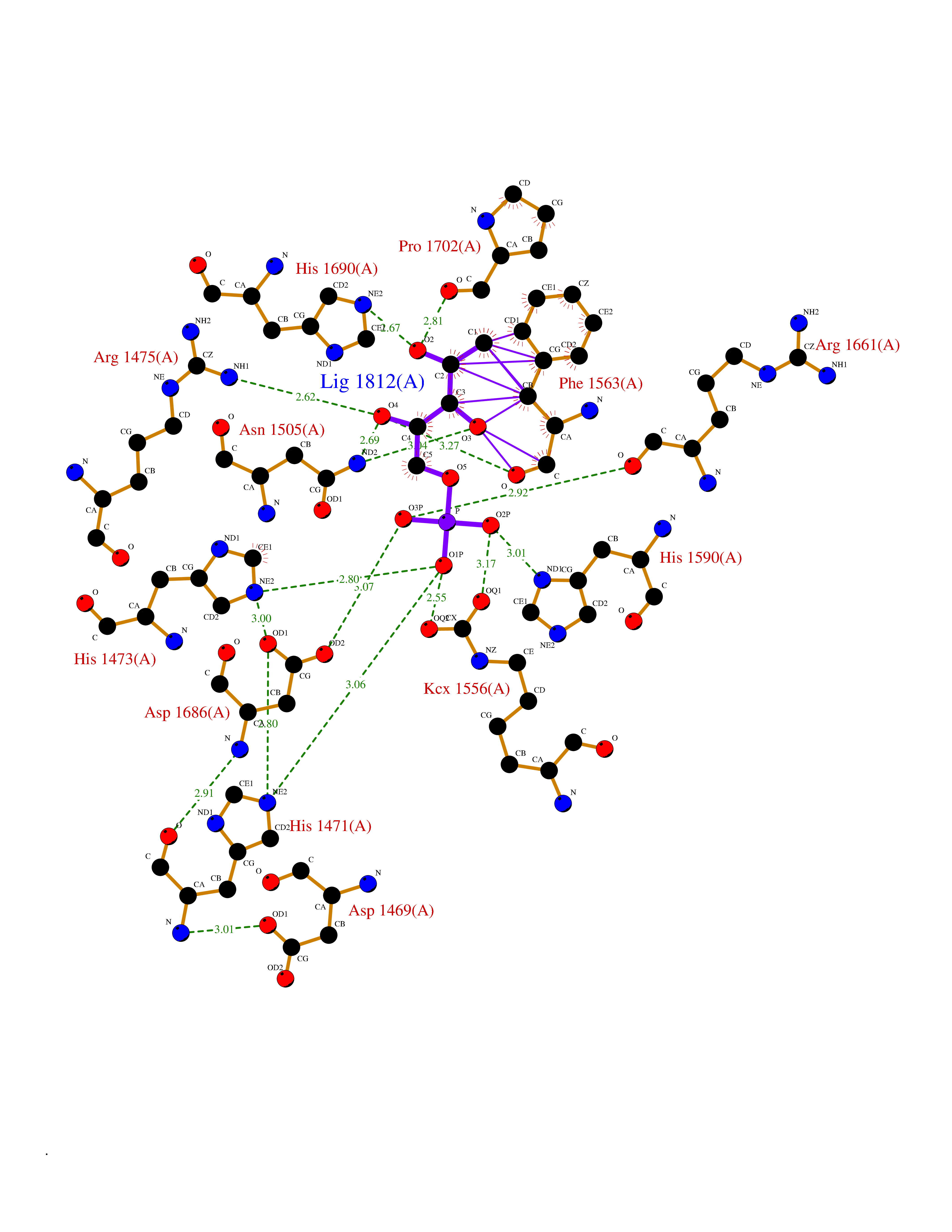 Ligplot Map 3D Binding mode Sequence KLVRLPGLIDVHVHLREPGGTHKEDFASGTAAALAGGITMVCAMPNTRPPIIDAPALALAQKLAEAGARCDFALFLGASSENAGTLGTVAGSAAGLXLYLNETFSELRLDSVVQWMEHFETWPSHLPIVAHAEQQTVAAVLMVAQLTQRSVHICHVARKEEILLIKAAKARGLPVTCEVAPHHLFLSHDDLERLGPGKGEVRPELGSRQDVEALWENMAVIDCFASDHAPHTLEEKCGSRPPPGFPGLETMLPLLLTAVSEGRLSLDDLLQRLHHNPRRIFHLPPQEDTYVEVDLEHEWTIPSHMPFSKAHWTPFEGQKVKGTVRRVVLRGEVAYIDGQVLVPPGYGQDVR Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Proteasome subunit beta type-5 | 5LF4 | 6.17 | |
Target general information Gen name PSMB5 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms X;LMPX;MB1 Protein family Peptidase T1B family Biochemical class Hydrolase Function Peptidase activity.Threonine-type endopeptidase activity. Related diseases Proteasome-associated autoinflammatory syndrome 1 (PRAAS1) [MIM:256040]: An autosomal recessive autoinflammatory disorder characterized by early childhood onset of recurrent fever, joint stiffness and severe contractures of the hands and feet, and erythematous skin lesions with subsequent development of lipodystrophy and laboratory evidence of immune dysregulation. Accompanying features may include muscle weakness and atrophy, hepatosplenomegaly, and microcytic anemia. {ECO:0000269|PubMed:21129723, ECO:0000269|PubMed:21852578, ECO:0000269|PubMed:21881205, ECO:0000269|PubMed:21953331, ECO:0000269|PubMed:26524591, ECO:0000269|PubMed:26567544}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08515; DB00188; DB08889 Interacts with O95429; Q8NCX0-3; Q92989; Q68J44; Q9Y6K9; O15116; Q96HA8; Q9Y244; P20618; P49721; P49720; P28070; Q99436; Q9BT92 EC number 3.4.25.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Direct protein sequencing; Host-virus interaction; Hydrolase; Nucleus; Protease; Proteasome; Proteomics identification; Reference proteome; Threonine protease; Zymogen Protein physicochemical properties Chain ID K,Y Molecular weight (Da) 45703.5 Length 414 Aromaticity 0.11 Instability index 27.55 Isoelectric point 8.63 Charge (pH=7) 4.98 2D Binding mode Binding energy (Kcal/mol) -7.02 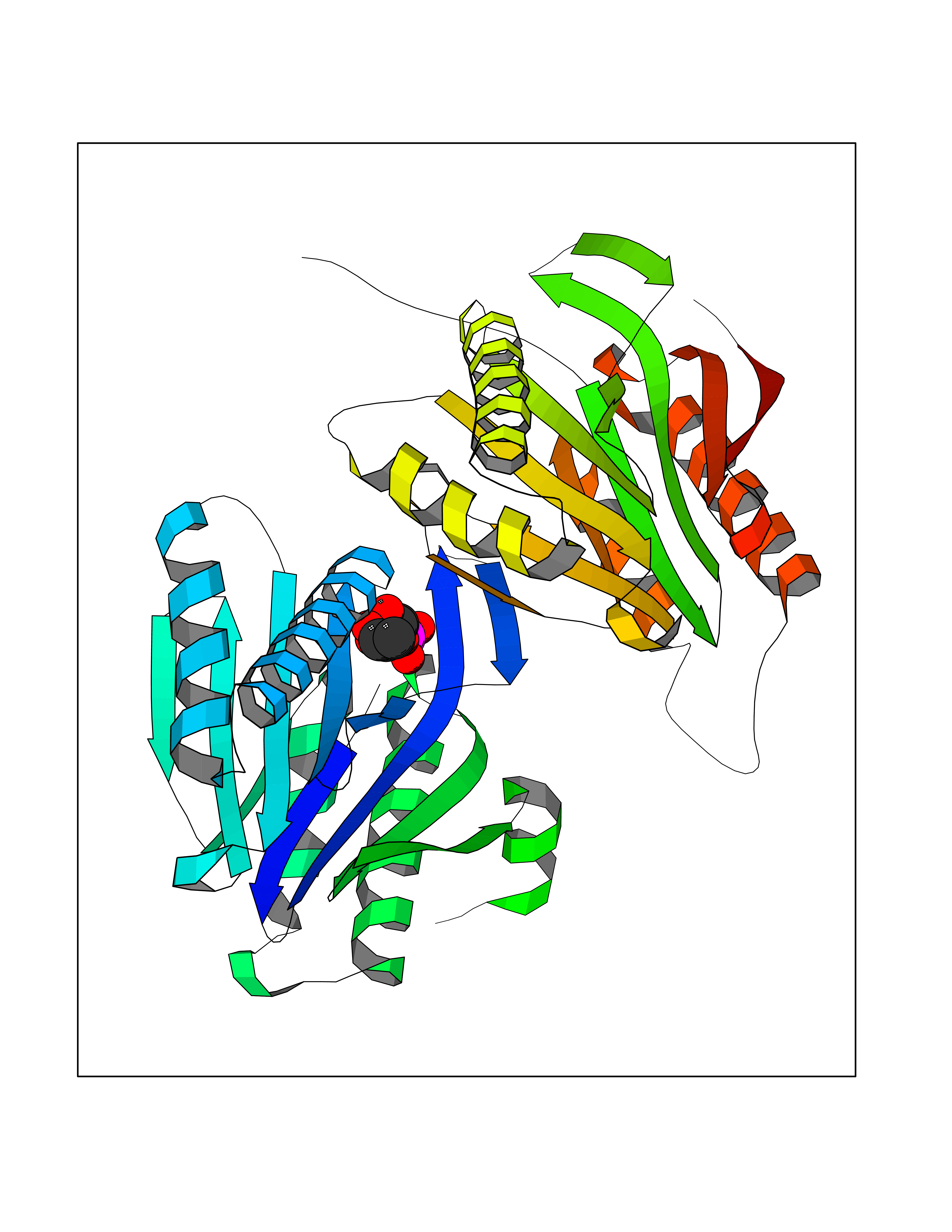 Molscript Map  Pymol Map 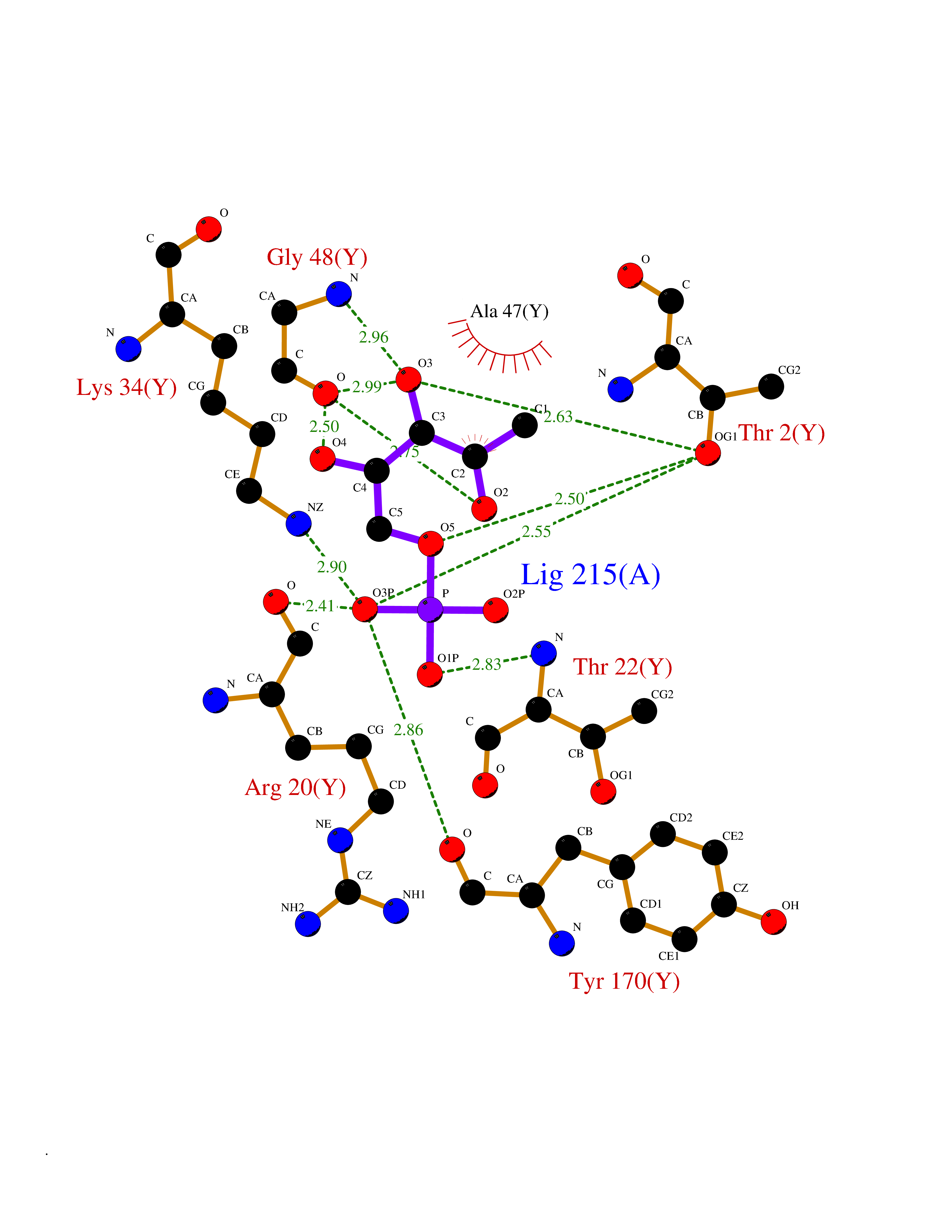 Ligplot Map 3D Binding mode Sequence TTTLAFKFRHGVIVAADSRATAGAYIASQTVKKVIEINPYLLGTMAGGAADCSFWERLLARQCRIYELRNKERISVAAASKLLANMVYQYKGMGLSMGTMICGWDKRGPGLYYVDSEGNRISGATFSVGSGSVYAYGVMDRGYSYDLEVEQAYDLARRAIYQATYRDAYSGGAVNLYHVREDGWIRVSSDNVADLHEKYSGRFSPYVFNGGTILAIAGEDFAIVASDTRLSEGFSIHTRDSPKCYKLTDKTVIGCSGFHGDCLTLTKIIEARLKMYKHSNNKAMTTGAIAAMLSTILYSRRFFPYYVYNIIGGLDEEGKGAVYSFDPVGSYQRDSFKAGGSASAMLQPLLDNQVGFKNMQNVEHVPLSLDRAMRLVKDVFISAAERDVYTGDALRICIVTKEGIREETVSLRKD Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Phosphodiesterase 3A (PDE3A) | 7EG0 | 6.17 | |
Target general information Gen name PDE3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-inhibited 3',5'-cyclic phosphodiesterase A; Phosphodiesterase 3A; Cyclic GMP-inhibited phosphodiesterase A; Cyclic GMP inhibited phosphodiesterase A; CGI-PDE A Protein family Cyclic nucleotide phosphodiesterase family, PDE3 subfamily Biochemical class Phosphoric diester hydrolase Function Cyclic nucleotide phosphodiesterase with a dual-specificity for the second messengers cAMP and cGMP, which are key regulators of many important physiological processes. Related diseases Hypertension and brachydactyly syndrome (HTNB) [MIM:112410]: A syndrome characterized by brachydactyly type E, severe salt-independent but age-dependent hypertension, an increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, and altered baroreflex blood pressure regulation. It results in death from stroke before age 50 years when untreated. Brachydactyly type E is characterized by shortening of the fingers mainly in the metacarpals and metatarsals. {ECO:0000269|PubMed:25961942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01223; DB01427; DB00261; DB00201; DB01166; DB04880; DB05266; DB00922; DB00235; DB01303; DB00277; DB08811; DB09283 Interacts with Q9Y6D6; Q9Y6D5 EC number EC 3.1.4.17 Uniprot keywords 3D-structure; cAMP; cGMP; Cytoplasm; Direct protein sequencing; Disease variant; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 108171 Length 939 Aromaticity 0.11 Instability index 39.75 Isoelectric point 6.53 Charge (pH=7) -4.15 2D Binding mode Binding energy (Kcal/mol) -6.52 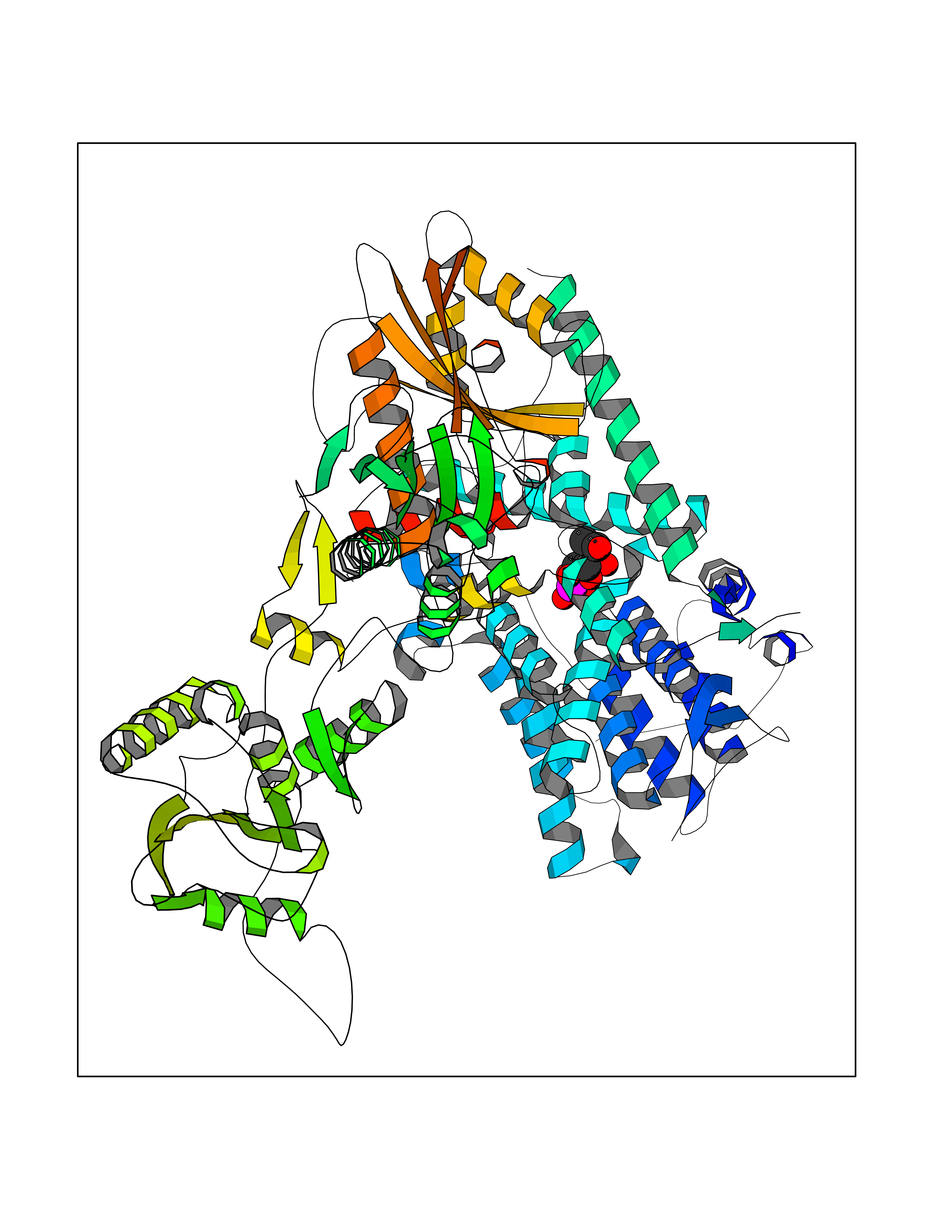 Molscript Map  Pymol Map 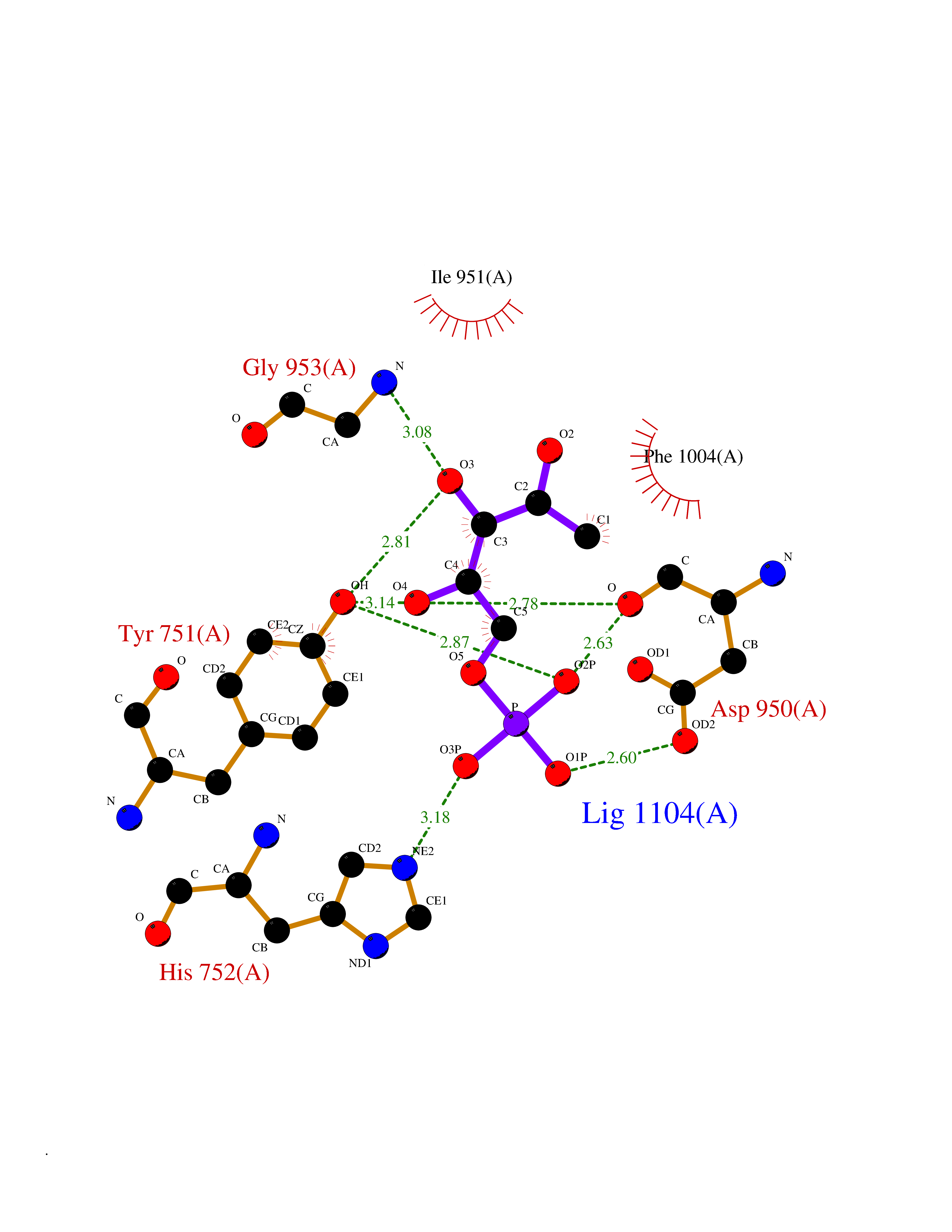 Ligplot Map 3D Binding mode Sequence KPILAPEPLVMDNLDSIMEQLNTWNFPIFDLVENIGRKCGRILSQVSYRLFEDMGLFEAFKIPIREFMNYFHALEIGYRDIPYHNRIHATDVLHAVWYLTTQPIPGLSTVGYVFSKTYNVTDDKYGCLSGNIPALELMALYVAAAMHDYDHPGRTNAFLVATSAPQAVLYNDRSVLENHHAAAAWNLFMSRPEYNFLINLDHVEFKHFRFLVIEAILATDLKKHFDFVAKFNGKVNDDVGIDWTNENDRLLVCQMCIKLADINGPAKCKELHLQWTDGIVNEFYEQGDEEASLGLPISPFMDRSAPQLANLQESFISHIVGPLCNSYDSAGLMPGKWVEKIYCQITQHLLQNHKMWKKVIEEEQRLAGIENQNISVDLETNYAELVLDVGRVTLGENSRKKMKDCKLRKKQNESVSRAMCALLNSGGGVIKAEIENEDYSYTKDGIGLDLENSFSNILLFVPEYLDFMQNGNYFLIFVKSWSLNTSGLRITTLSSNLYKRDITSAKVMNATAALEFLKDMKKTRGRLYLRPELLAKRPCVDIQEENNMKALAGVFFDRTELDRKEKLTFTESTHVEIKNFSTERLLQRIKEILPQYVSAFANTDGGYLFIGLNEDKEIIGFKAEMSDLDDLEREIEKSIRKMPVHHFCMEKKKINYSCKFLGVYDKGSLCGYVCALRVERFCCAVFAKEPDSWHVKDNRVMQLTRKEWIQFMVEAEPKFSSAYEEVISQINTSLPAPHSWPLLEWQRQRHHCPGLSGRITYTPENLCRKLFLQHEGLKQLICEEMSSVRKGSLIFSRSWSVDLGLQENHKVLCDALLISQDSPPVLYTFHMVQDEEFKGYSTQTALTLKQKLAKIGGYTKKVCVMTKIFYLSPEGMTSCQYDLRSQVIYPESYYFTRRKYLLKALFKALKRLKSLRDQFSFAENLYQIIGIDCFQKNDK Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | D-alanyl-D-alanine carboxypeptidase DacB | 2EX6 | 6.17 | |
Target general information Gen name dacB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW3149;b3182 Protein family Peptidase S13 family Biochemical class Hydrolase Function Carboxypeptidase activity.Endopeptidase activity.Penicillin binding.Serine-type D-Ala-D-Ala carboxypeptidase activity. Related diseases Ovarian dysgenesis 1 (ODG1) [MIM:233300]: An autosomal recessive disease characterized by primary amenorrhea, variable development of secondary sex characteristics, poorly developed streak ovaries, and high serum levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH). {ECO:0000269|PubMed:10551778, ECO:0000269|PubMed:11889179, ECO:0000269|PubMed:12571157, ECO:0000269|PubMed:12915623, ECO:0000269|PubMed:7553856, ECO:0000269|PubMed:9769327, ECO:0000269|PubMed:9851774}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ovarian hyperstimulation syndrome (OHSS) [MIM:608115]: Disorder which occurs either spontaneously or most often as an iatrogenic complication of ovarian stimulation treatments for in vitro fertilization. The clinical manifestations vary from abdominal distention and discomfort to potentially life-threatening, massive ovarian enlargement and capillary leak with fluid sequestration. Pathologic features of this syndrome include the presence of multiple serous and hemorrhagic follicular cysts lined by luteinized cells, a condition called hyperreactio luteinalis. {ECO:0000269|PubMed:12930927, ECO:0000269|PubMed:12930928, ECO:0000269|PubMed:15080154, ECO:0000269|PubMed:16278261, ECO:0000269|PubMed:17721928, ECO:0000269|PubMed:24058690, ECO:0000269|PubMed:25581598}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01602; DB00578; DB09319; DB14879; DB00671; DB01328; DB01329; DB01331; DB09050; DB01000; DB00303; DB04570; DB00760; DB00417 Interacts with P0A6E4 EC number 3.4.16.4; 3.4.21.- Uniprot keywords 3D-structure; Antibiotic resistance; Cell cycle; Cell division; Cell shape; Cell wall biogenesis/degradation; Direct protein sequencing; Hydrolase; Peptidoglycan synthesis; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 48811.3 Length 450 Aromaticity 0.08 Instability index 27.19 Isoelectric point 9.11 Charge (pH=7) 6.99 2D Binding mode Binding energy (Kcal/mol) -6.34  Molscript Map 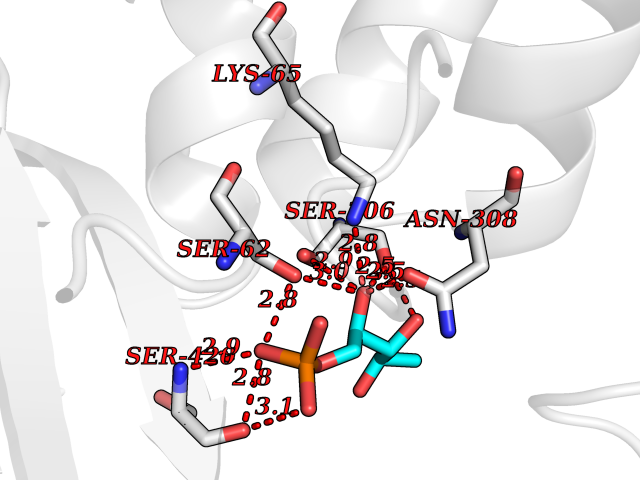 Pymol Map 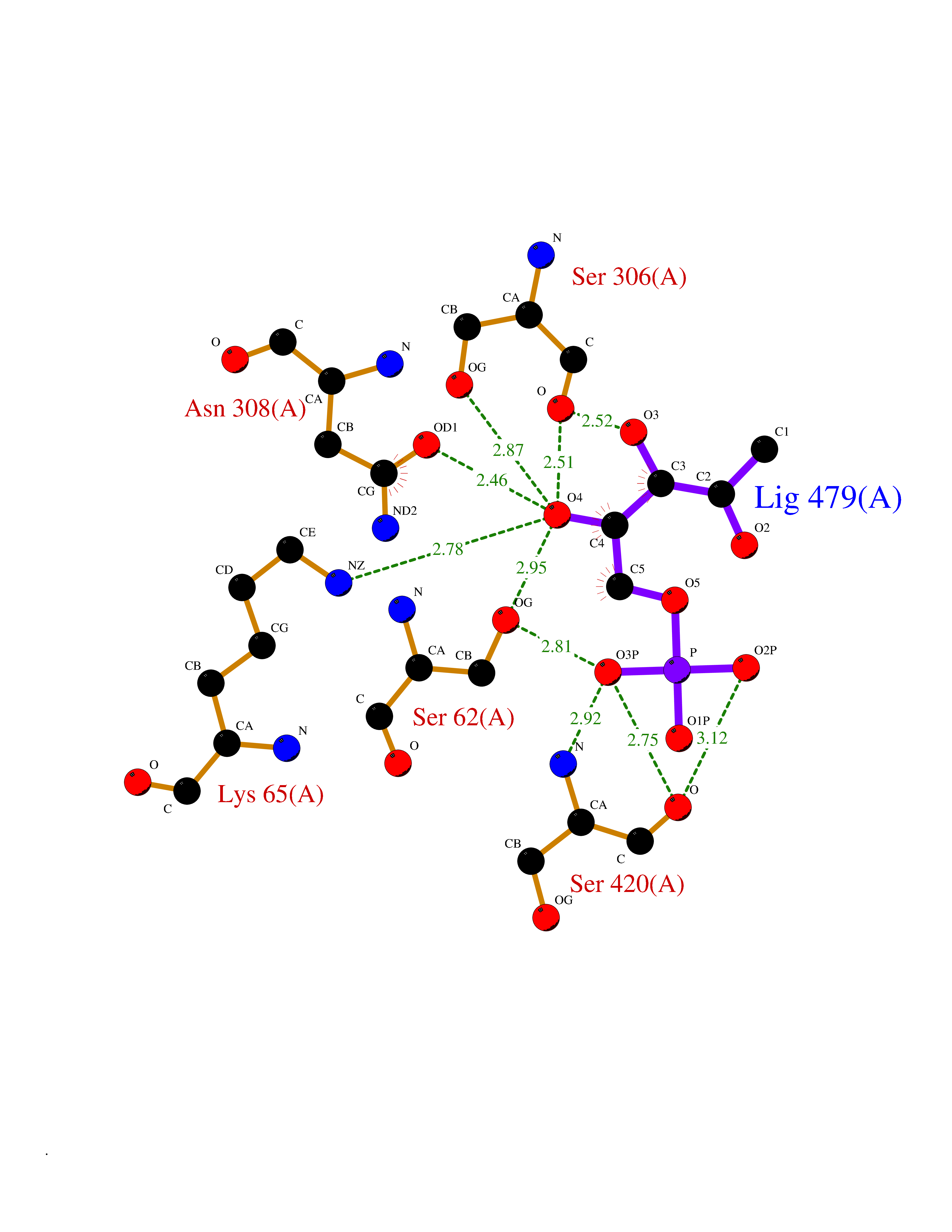 Ligplot Map 3D Binding mode Sequence TQLPAGANLALMVQKVGASAPAIDYHSQQMALPASTQKVITALAALIQLGPDFRFTTTLETKGNVENGVLKGDLVARFGADPTLKRQDIRNMVATLKKSGVNQIDGNVLIDTSIFASHDKAPGWPWNDMTQCFSAPPAAAIVDRNCFSVSLYSAPKPGDMAFIRVASYYPVTMFSQVRTLPRGSAEAQYCELDVVPGDLNRFTLTGCLPQRSEPLPLAFAVQDGASYAGAILKYELKQAGITWSGTLLRQTQVNEPGTVVASKQSAPLHDLLKIMLKKSDNMIADTVFRMIGHARFNVPGTWRAGSDAVRQILRQQAGVDIGNTIIADGSGLSRHNLIAPATMMQVLQYIAQHDNELNFISMLPLAGYDGSLQYRAGLHQAGVDGKVSAKTGSLQGVYNLAGFITTASGQRMAFVQYLSGYAVEPADQRNRRIPLVRFESRLYKDIYQNN Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Complement C1s component (C1S) | 1ELV | 6.16 | |
Target general information Gen name C1S Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complement component 1 subcomponent s; Complement C1s subcomponent; C1-esterase; C1 esterase Protein family Peptidase S1 family Biochemical class Peptidase Function C1r activates C1s so that it can, in turn, activate C2 and C4. C1s B chain is a serine protease that combines with C1q and C1r to form C1, the first component of the classical pathway of the complement system. Related diseases Complement component C1s deficiency (C1SD) [MIM:613783]: A rare defect resulting in C1 deficiency and impaired activation of the complement classical pathway. C1 deficiency generally leads to severe immune complex disease with features of systemic lupus erythematosus and glomerulonephritis. {ECO:0000269|PubMed:11390518}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ehlers-Danlos syndrome, periodontal type, 2 (EDSPD2) [MIM:617174]: A form of Ehlers-Danlos syndrome, a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDSPD2 is characterized by the association of typical features of Ehlers-Danlos syndrome with gingival recession and severe early-onset periodontal disease, leading to premature loss of permanent teeth. EDSPD2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:27745832}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02371; DB09228; DB09130; DB12831; DB06404; DB14996; DB01593; DB14487; DB14533; DB14548 Interacts with P00736; P09871; P06681; O43889-2; Q9H6H4; P05155 EC number EC 3.4.21.42 Uniprot keywords 3D-structure; Calcium; Complement pathway; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Ehlers-Danlos syndrome; Glycoprotein; Hydrolase; Hydroxylation; Immunity; Innate immunity; Metal-binding; Protease; Proteomics identification; Reference proteome; Repeat; Serine protease; Signal; Sushi Protein physicochemical properties Chain ID A Molecular weight (Da) 33278.6 Length 303 Aromaticity 0.1 Instability index 33.69 Isoelectric point 5.16 Charge (pH=7) -7.95 2D Binding mode Binding energy (Kcal/mol) -6.31  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LDCGIPESIENGKVEDPESTLFGSVIRYTCEEPYYYMEGGGEYHCAGNGSWVNEVLGPELPKCVPVCGVPREPFIIGGSDADIKNFPWQVFFDNPWAGGALINEYWVLTAAHVVEGNREPTMYVGSTSVQKMLTPEHVFIHPGWKLLAVPEGRTNFDNDIALVRLKDPVKMGPTVSPICLPGTSSDYNLMDGDLGLISGWGRTEKRDRAVRLKAARLPVAPLRKCKEVAYVFTPNMICAGGEKGMDSCKGDSGGAFAVQDPNDKTKFYAAGLVSWGPQCGTYGLYTRVKNYVDWIMKTMQENS Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Leukocyte immunoglobulin-like receptor B2 (LILRB2) | 6BCS | 6.15 | |
Target general information Gen name LILRB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LIR-2; Leukocyte immunoglobulin-like receptor 2; CD85 antigen-like family member D; Immunoglobulin-like transcript 4; ILT-4; Monocyte/macrophage immunoglobulin-like receptor 10; MIR-10; DE AltName: CD Protein family NA Biochemical class NA Function Receptor for class I MHC antigens. Recognizes a broad spectrum of HLA-A, HLA-B, HLA-C, HLA-G and HLA-F alleles. Involved in the down-regulation of the immune response and the development of tolerance. Recognizes HLA-G in complex with B2M/beta-2 microglobulin and a nonamer self-peptide (peptide-bound HLA-G-B2M) triggering differentiation of type 1 regulatory T cells and myeloid-derived suppressor cells, both of which actively maintain maternal-fetal tolerance. Competes with CD8A for binding to class I MHC antigens. Inhibits FCGR1A-mediated phosphorylation of cellular proteins and mobilization of intracellular calcium ions. Related diseases Cardiomyopathy, familial hypertrophic, 1 (CMH1) [MIM:192600]: A hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death. {ECO:0000269|PubMed:10065021, ECO:0000269|PubMed:10329202, ECO:0000269|PubMed:10521296, ECO:0000269|PubMed:10563488, ECO:0000269|PubMed:10679957, ECO:0000269|PubMed:10862102, ECO:0000269|PubMed:11113006, ECO:0000269|PubMed:11133230, ECO:0000269|PubMed:11214007, ECO:0000269|PubMed:11424919, ECO:0000269|PubMed:11733062, ECO:0000269|PubMed:11861413, ECO:0000269|PubMed:11968089, ECO:0000269|PubMed:12081993, ECO:0000269|PubMed:12566107, ECO:0000269|PubMed:12590187, ECO:0000269|PubMed:12707239, ECO:0000269|PubMed:12818575, ECO:0000269|PubMed:12820698, ECO:0000269|PubMed:12951062, ECO:0000269|PubMed:12974739, ECO:0000269|PubMed:12975413, ECO:0000269|PubMed:1417858, ECO:0000269|PubMed:15358028, ECO:0000269|PubMed:15483641, ECO:0000269|PubMed:1552912, ECO:0000269|PubMed:15563892, ECO:0000269|PubMed:15856146, ECO:0000269|PubMed:15858117, ECO:0000269|PubMed:16199542, ECO:0000269|PubMed:16267253, ECO:0000269|PubMed:1638703, ECO:0000269|PubMed:16650083, ECO:0000269|PubMed:16938236, ECO:0000269|PubMed:17095604, ECO:0000269|PubMed:17372140, ECO:0000269|PubMed:18175163, ECO:0000269|PubMed:18403758, ECO:0000269|PubMed:1975517, ECO:0000269|PubMed:25182012, ECO:0000269|PubMed:7581410, ECO:0000269|PubMed:7731997, ECO:0000269|PubMed:7848441, ECO:0000269|PubMed:7874131, ECO:0000269|PubMed:7909436, ECO:0000269|PubMed:8250038, ECO:0000269|PubMed:8254035, ECO:0000269|PubMed:8268932, ECO:0000269|PubMed:8282798, ECO:0000269|PubMed:8343162, ECO:0000269|PubMed:8435239, ECO:0000269|PubMed:8483915, ECO:0000269|PubMed:8533830, ECO:0000269|PubMed:8655135, ECO:0000269|PubMed:8899546, ECO:0000269|PubMed:9544842, ECO:0000269|PubMed:9822100, ECO:0000269|PubMed:9829907}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 7A, myosin storage, autosomal dominant (CMYO7A) [MIM:608358]: A skeletal muscle disorder characterized by prominent axial and proximal weakening, spinal stiffness, severe scoliosis, with or without respiratory and cardiac involvement. The age at symptom onset can range from early childhood to late adulthood, and disease severity ranges from asymptomatic to severe muscular weakness and respiratory insufficiency. Histopathological examination shows variable findings including subsarcolemmal hyaline bodies in type 1 fibers. {ECO:0000269|PubMed:14520662, ECO:0000269|PubMed:15136674, ECO:0000269|PubMed:16684601, ECO:0000269|PubMed:17336526}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cardiomyopathy, dilated, 1S (CMD1S) [MIM:613426]: A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. {ECO:0000269|PubMed:11106718, ECO:0000269|PubMed:12379228, ECO:0000269|PubMed:15769782, ECO:0000269|PubMed:18506004, ECO:0000269|PubMed:21127202, ECO:0000269|PubMed:21846512}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, distal, 1 (MPD1) [MIM:160500]: A muscular disorder characterized by early-onset selective weakness of the great toe and ankle dorsiflexors, followed by weakness of the finger extensors. Mild proximal weakness occasionally develops years later after the onset of the disease. {ECO:0000269|PubMed:15322983, ECO:0000269|PubMed:17548557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 7B, myosin storage, autosomal recessive (CMYO7B) [MIM:255160]: A skeletal muscle disorder characterized by the onset of scapuloperoneal muscle weakness in early childhood or young adulthood. Affected individuals have difficulty walking, steppage gait, and scapular winging due to shoulder girdle involvement. The severity and progression of the disorder is highly variable. Most patients develop respiratory insufficiency and restrictive lung disease. Some develop hypertrophic cardiomyopathy. Histopathological examination shows variable findings including subsarcolemmal hyaline bodies in type 1 fibers. {ECO:0000269|PubMed:25666907}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Left ventricular non-compaction 5 (LVNC5) [MIM:613426]: A form of left ventricular non-compaction, a cardiomyopathy due to myocardial morphogenesis arrest and characterized by a hypertrophic left ventricle, a severely thickened 2-layered myocardium, numerous prominent trabeculations, deep intertrabecular recesses, and poor systolic function. Clinical manifestations are variable. Some affected individuals experience no symptoms at all, others develop heart failure. In some cases, left ventricular non-compaction is associated with other congenital heart anomalies. LVNC5 is an autosomal dominant condition. {ECO:0000269|PubMed:18506004}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UKU9; Q86XS5; PRO_0000000092 [P05067]; P30447; Q95J06; P01889; P10321; P17693; P17693-2; P17693-5; P17693-6; P46531; P29350 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative initiation; Alternative promoter usage; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 21853.4 Length 196 Aromaticity 0.1 Instability index 50.2 Isoelectric point 8.21 Charge (pH=7) 1.8 2D Binding mode Binding energy (Kcal/mol) -6.32  Molscript Map 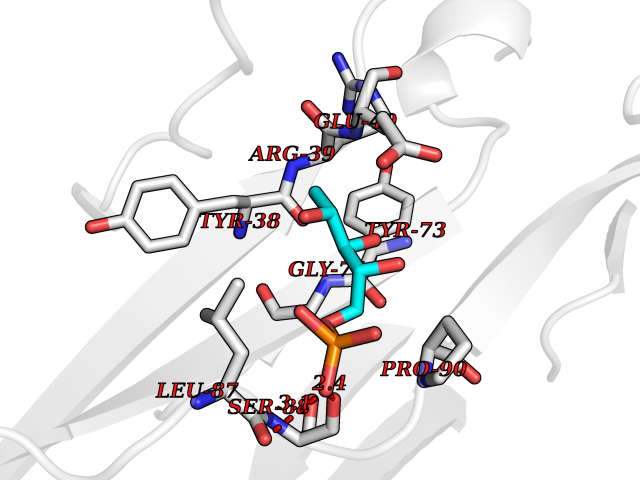 Pymol Map 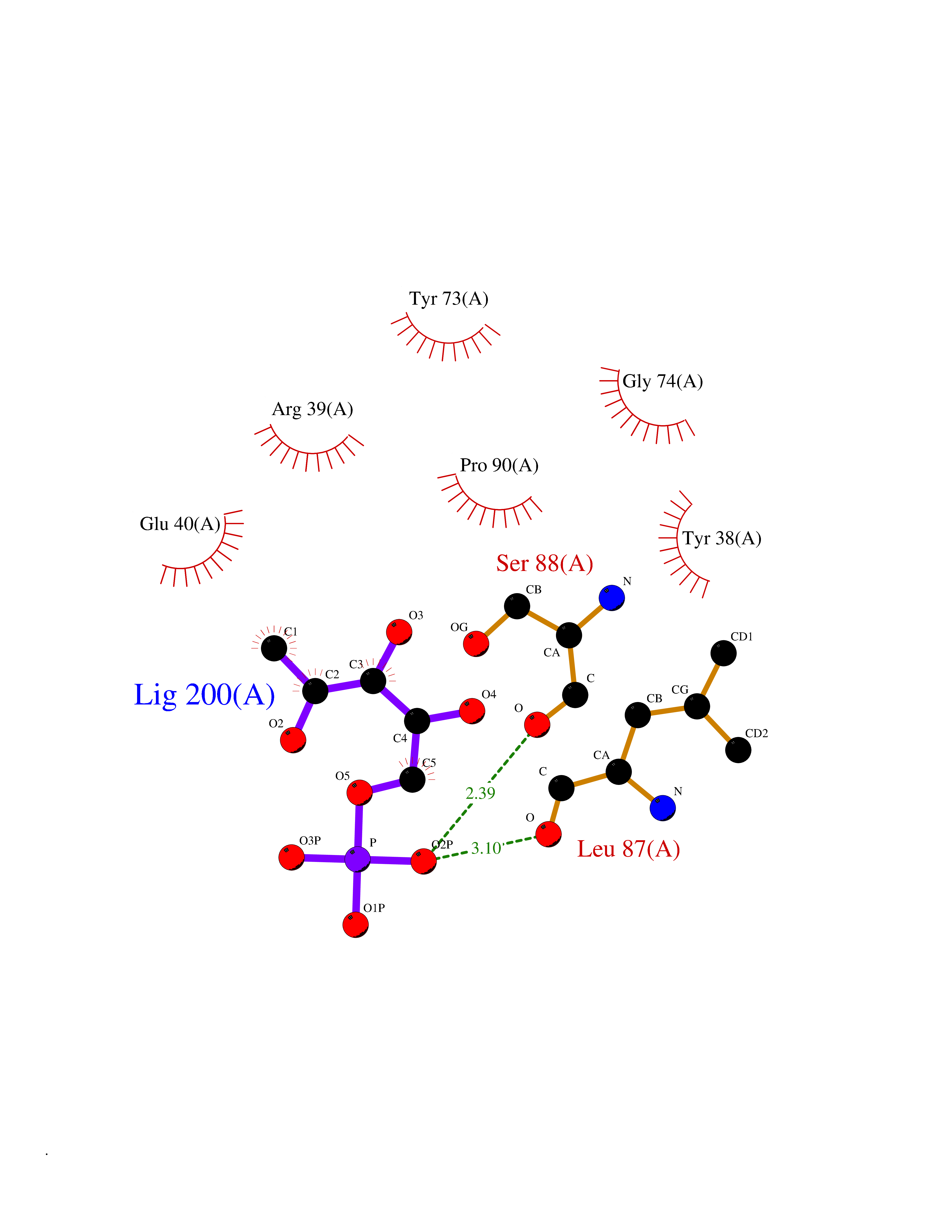 Ligplot Map 3D Binding mode Sequence TIPKPTLWAEPDSVITQGSPVTLSCQGSLEAQEYRLYREKKSASWITRIRPELVKNGQFHIPSITWEHTGRYGCQYYSRARWSELSDPLVLVMTGAYPKPTLSAQPSPVVTSGGRVTLQCESQVAFGGFILCKEGEDEHPQCLNSQPHARGSSRAIFSVGPVSPNRRWSHRCYGYDLNSPYVWSSPSDLLELLVPG Hydrogen bonds contact Hydrophobic contact | ||||