Job Results:
Ligand
Structure
Job ID
0006599cba9dbe83f9144f4d36fa70dc
Job name
NA
Time
2024-05-29 06:52:44
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 41 | Cannabinoid receptor 2 (CB2) | 6PT0 | 8.13 | |
Target general information Gen name CNR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hCB2; Cannabinoid CB2 receptor; CX5; CB2B; CB2A; CB-2 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function May function in inflammatory response, nociceptive transmission and bone homeostasis. Heterotrimeric G protein-coupled receptor for endocannabinoid 2-arachidonoylglycerol mediating inhibition of adenylate cyclase. Related diseases Factor V deficiency (FA5D) [MIM:227400]: A blood coagulation disorder leading to a hemorrhagic diathesis known as parahemophilia. {ECO:0000269|PubMed:10942390, ECO:0000269|PubMed:12393490}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombophilia due to activated protein C resistance (THPH2) [MIM:188055]: A hemostatic disorder due to defective degradation of factor V by activated protein C. It is characterized by a poor anticoagulant response to activated protein C resulting in tendency to thrombosis. {ECO:0000269|PubMed:10391209, ECO:0000269|PubMed:10942390, ECO:0000269|PubMed:11435304, ECO:0000269|PubMed:11858490, ECO:0000269|PubMed:14617013, ECO:0000269|PubMed:14695241, ECO:0000269|PubMed:16710414, ECO:0000269|PubMed:8164741, ECO:0000269|PubMed:9454742}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Budd-Chiari syndrome (BDCHS) [MIM:600880]: A syndrome caused by obstruction of hepatic venous outflow involving either the hepatic veins or the terminal segment of the inferior vena cava. Obstructions are generally caused by thrombosis and lead to hepatic congestion and ischemic necrosis. Clinical manifestations observed in the majority of patients include hepatomegaly, right upper quadrant pain and abdominal ascites. Budd-Chiari syndrome is associated with a combination of disease states including primary myeloproliferative syndromes and thrombophilia due to factor V Leiden, protein C deficiency and antithrombin III deficiency. Budd-Chiari syndrome is a rare but typical complication in patients with polycythemia vera. {ECO:0000269|PubMed:9245936}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Pregnancy loss, recurrent, 1 (RPRGL1) [MIM:614389]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:11018168}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09061; DB00470; DB06202; DB14009; DB00486; DB14011; DB02955; DB16321; DB11755 Interacts with Q9UKJ8; Q15848; Q9NRZ5; P13236; P21964; Q14802-3; Q8N387; Q8IXM6; I3L0A0; Q96AA3; Q9Y6D0; Q6ICL7; Q9NP94; Q13501; Q96HH6; Q969S6; Q9NWH2; Q9H2L4; Q8N2M4; Q6ZT21; Q5TGU0; Q9Y548; Q9BSR8; Q96EC8 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; G-protein coupled receptor; Glycoprotein; Inflammatory response; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32999.2 Length 298 Aromaticity 0.11 Instability index 30.98 Isoelectric point 9.49 Charge (pH=7) 14.35 2D Binding mode Binding energy (Kcal/mol) -9.36  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MKDYMILSGPQKTAVAVLCTLLGLLSALENVAVLYLILSSHQLRRKPSYLFIGSLAGADFLASVVFACSFVNFHVFHGVDSKAVFLLKIGSVTMTFTASVGSLLLTAIDRYLCLRYPPSYKALLTRGRALVTLGIMWVLSALVSYLPLMGWTCCPRPCSELFPLIPNDYLLSWLLFIAFLFSGIIYTYGHVLWKAHQHVASLSGHQDRQVPGMARMRLDVRLAKTLGLVLAVLLICWFPVLALMAHSLATTLSDQVKKAFAFCSMLCLINSMVNPVIYALRSGEIRSSAHHCLAHWKK Hydrogen bonds contact Hydrophobic contact | ||||
| 42 | Monocarboxylate transporter 1 (SLC16A1) | 6LZ0 | 8.12 | |
Target general information Gen name SLC16A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 16 member 1; MCT1; MCT 1 Protein family Major facilitator superfamily, Monocarboxylate porter (TC 2.A.1.13) family Biochemical class Major facilitator Function Catalyzes the rapid transport across the plasma membrane of many monocarboxylates such as lactate, pyruvate, branched-chain oxo acids derived from leucine, valine and isoleucine, and the ketone bodies acetoacetate, beta-hydroxybutyrate and acetate. Depending on the tissue and on cicumstances, mediates the import or export of lactic acid and ketone bodies. Required for normal nutrient assimilation, increase of white adipose tissue and body weight gain when on a high-fat diet. Plays a role in cellular responses to a high-fat diet by modulating the cellular levels of lactate and pyruvate, small molecules that contribute to the regulation of central metabolic pathways and insulin secretion, with concomitant effects on plasma insulin levels and blood glucose homeostasis. Proton-coupled monocarboxylate transporter. Related diseases Symptomatic deficiency in lactate transport (SDLT) [MIM:245340]: Deficiency of lactate transporter may result in an acidic intracellular environment created by muscle activity with consequent degeneration of muscle and release of myoglobin and creatine kinase. This defect might compromise extreme performance in otherwise healthy individuals. {ECO:0000269|PubMed:10590411}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperinsulinemic hypoglycemia, familial, 7 (HHF7) [MIM:610021]: A form of hyperinsulinemic hypoglycemia, a clinically and genetically heterogeneous disorder characterized by inappropriate insulin secretion from the pancreatic beta-cells in the presence of low blood glucose levels. HHF7 features include exercise-induced hyperinsulinism, loss of consciousness due to hypoglycemia, and hypoglycemic seizures. HHF7 inheritance is autosomal dominant. {ECO:0000269|PubMed:17701893}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Monocarboxylate transporter 1 deficiency (MCT1D) [MIM:616095]: A metabolic disorder characterized by recurrent ketoacidosis, a pathologic state due to ketone formation exceeding ketone utilization. The clinical consequences of ketoacidosis are vomiting, osmotic diuresis, dehydration, and Kussmaul breathing. The condition may progress to decreased consciousness and, ultimately, death. {ECO:0000269|PubMed:25390740}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03166; DB01762; DB03773; DB04074; DB00345; DB00415; DB08892; DB03793; DB03066; DB07767; DB00529; DB01440; DB00142; DB04398; DB09338; DB00563; DB00731; DB00627; DB04552; DB00175; DB01032; DB00119; DB04216; DB00936; DB04348; DB00313 Interacts with Q66PJ3-4; Q92782-2; Q9UH65 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Symport; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 40405.6 Length 375 Aromaticity 0.15 Instability index 32.61 Isoelectric point 9.12 Charge (pH=7) 10.98 2D Binding mode Binding energy (Kcal/mol) -8.91 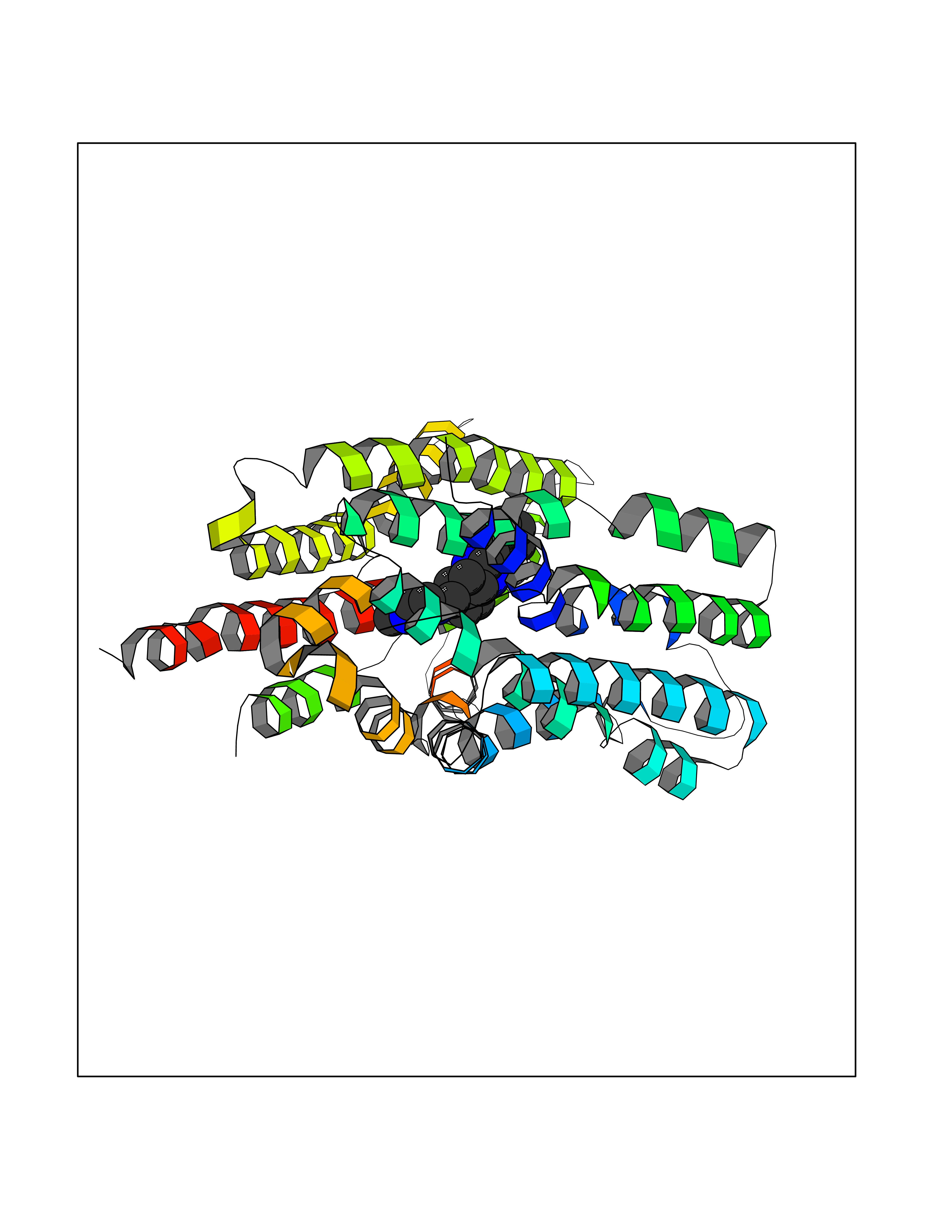 Molscript Map 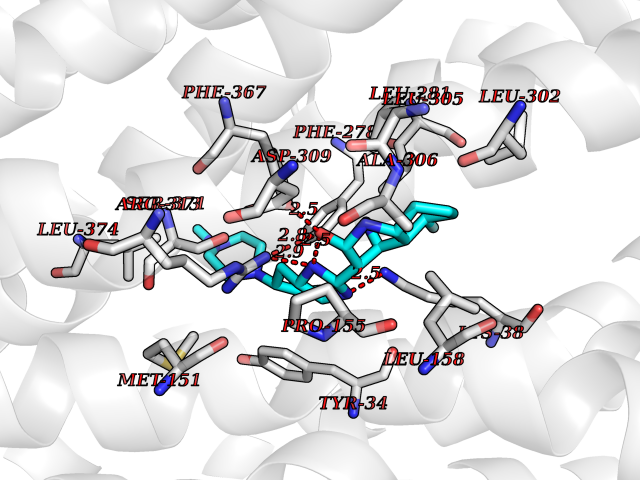 Pymol Map 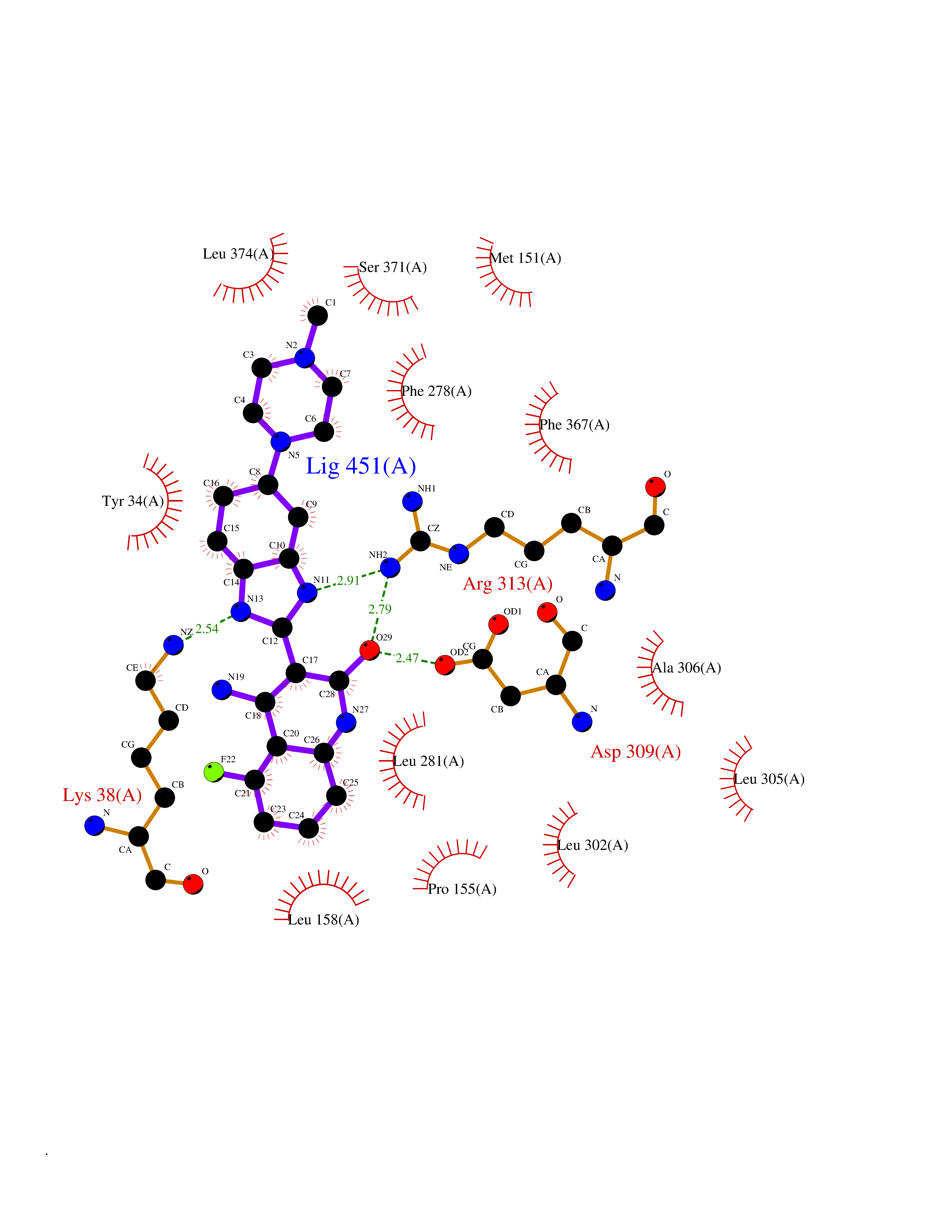 Ligplot Map 3D Binding mode Sequence GGWGWAVVIGAFISIGFSYAFPKSITVFFKEIEGIFHATTSEVSWISSIMLAVMYGGGPISSILVNKYGSRIVMIVGGCLSGCGLIAASFCNTVQQLYVCIGVIGGLGLAFNLNPALTMIGKYFYKRRPLANGLAMAGSPVFLCTLAPLNQVFFGIFGWRGSFLILGGLLLNCCVAGALMRPIGPHRGFLLYLSGNVIMFFGLFAPLVFLSSYGKSQHYSSEKSAFLLSILAFVDMVARPSMGLVANTKPIRPRIQYFFAASVVANGVCHMLAPLSTTYVGFCVYAGFFGFAFGWLSSVLFETLMDLVGPQRFSSAVGLVTIVECCPVLLGPPLLGRLNDMYGDYKYTYWACGVVLIISGIYLFIGMGINYRLLA Hydrogen bonds contact Hydrophobic contact | ||||
| 43 | Dopamine beta-hydroxylase | 4ZEL | 8.11 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -9 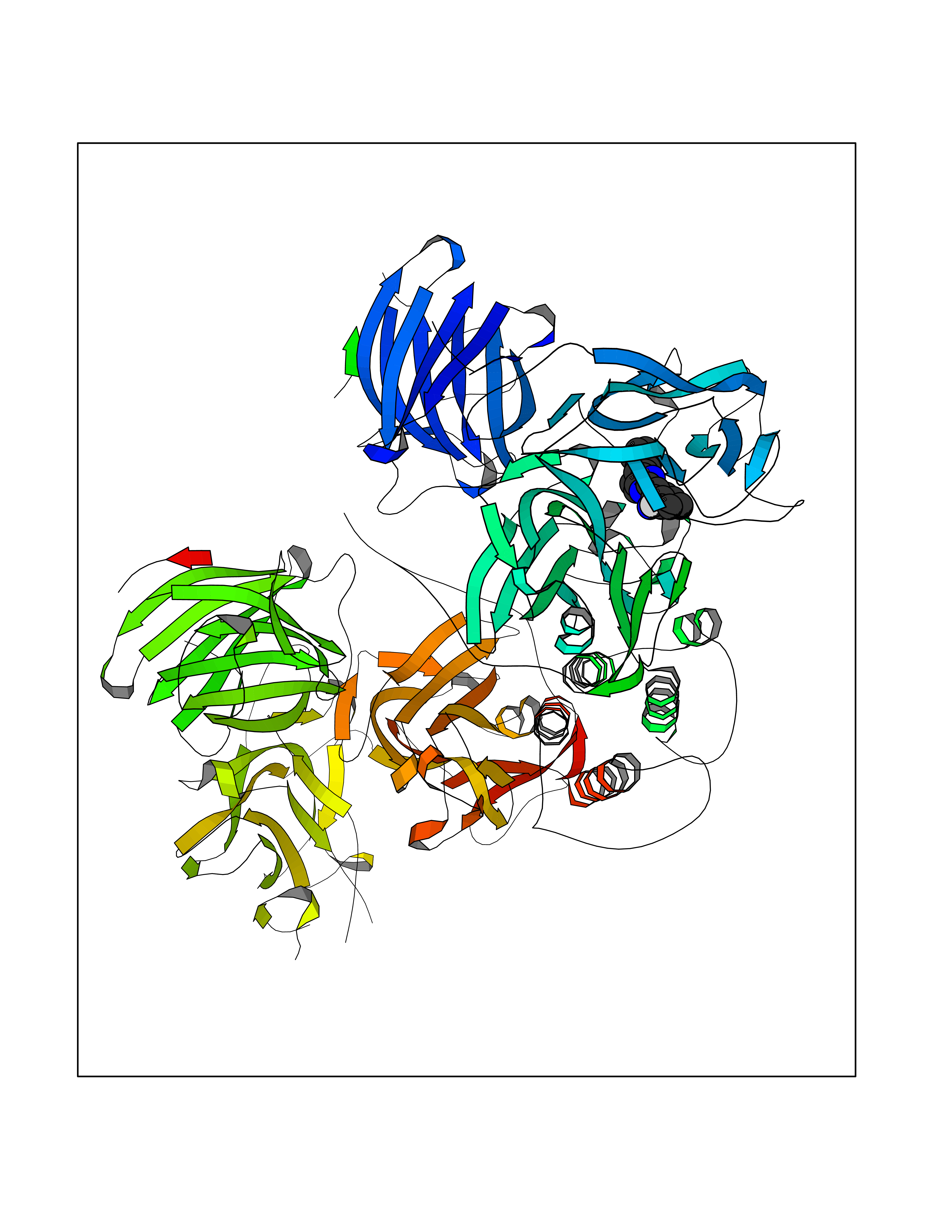 Molscript Map 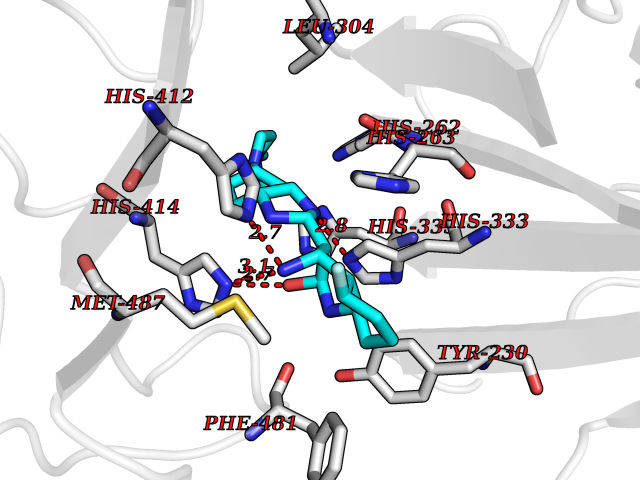 Pymol Map 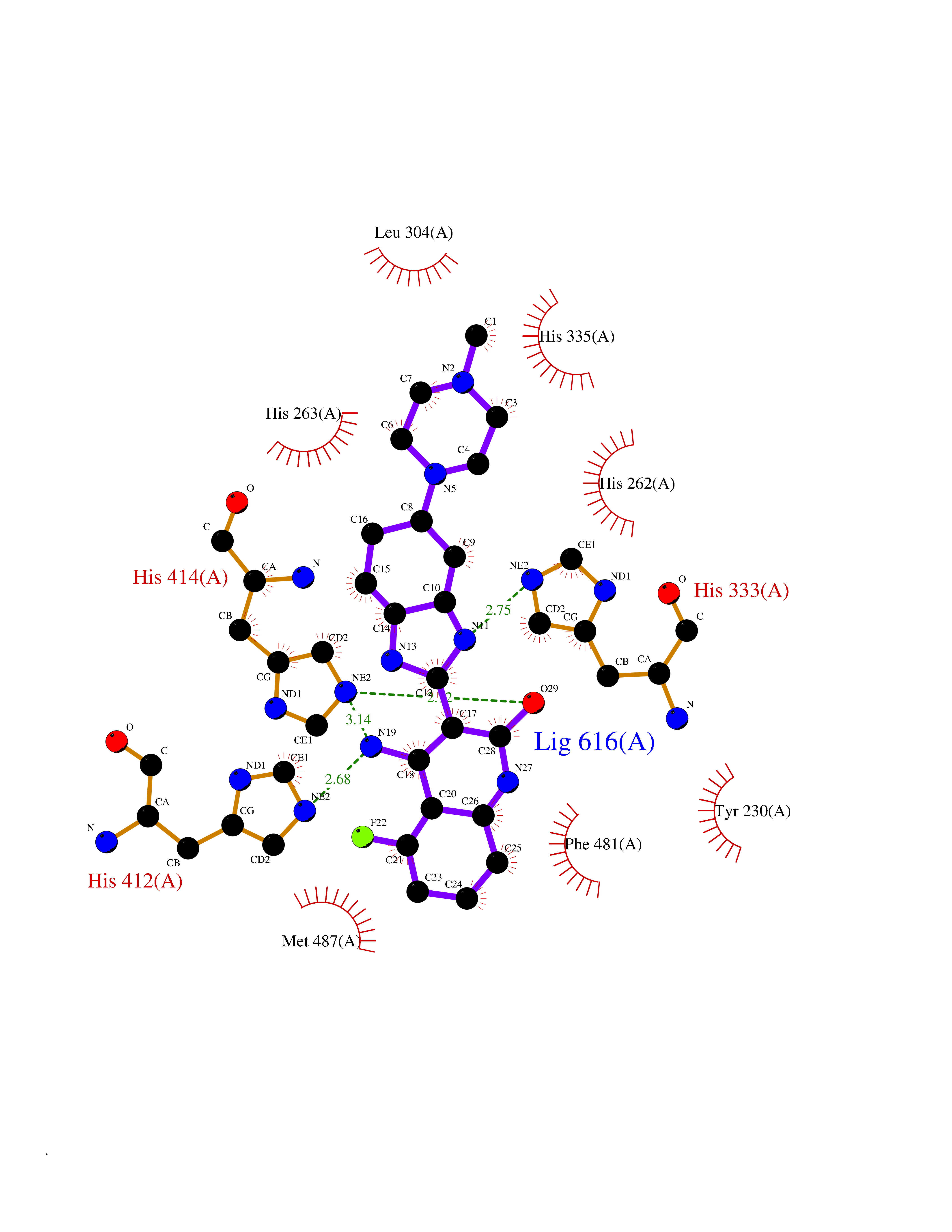 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 44 | Cholesterol oxidase | 4REK | 8.11 | |
Target general information Gen name choA Organism Streptomyces sp. (strain SA-COO) Uniprot ID TTD ID NA Synonyms NA Protein family GMC oxidoreductase family Biochemical class Oxidoreductase Function Cholesterol oxidase activity.Flavin adenine dinucleotide binding.Steroid delta-isomerase activity. Related diseases Bothnia retinal dystrophy (BRD) [MIM:607475]: A type of retinitis punctata albescens. Affected individuals show night blindness from early childhood with features consistent with retinitis punctata albescens and macular degeneration. {ECO:0000269|PubMed:10102298}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rod-cone dystrophy Newfoundland (NFRCD) [MIM:607476]: A rod-cone dystrophy reminiscent of retinitis punctata albescens but with a substantially lower age at onset and more-rapid and distinctive progression. Rod-cone dystrophies results from initial loss of rod photoreceptors, later followed by cone photoreceptors loss. {ECO:0000269|PubMed:11868161}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Retinitis punctata albescens (RPA) [MIM:136880]: A form of fleck retina disease characterized by aggregation of white flecks posteriorly in the retina, causing night blindness and delayed dark adaptation. It differs from fundus albipunctatus in being progressive and evolving to generalized atrophy of the retina. {ECO:0000269|PubMed:10102299, ECO:0000269|PubMed:11453974, ECO:0000269|PubMed:9326942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB02332 Interacts with NA EC number 1.1.3.6; 5.3.3.1 Uniprot keywords 3D-structure; Cholesterol metabolism; Direct protein sequencing; FAD; Flavoprotein; Isomerase; Lipid metabolism; Oxidoreductase; Secreted; Signal; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54367.8 Length 498 Aromaticity 0.1 Instability index 30.62 Isoelectric point 6.69 Charge (pH=7) -0.71 2D Binding mode Binding energy (Kcal/mol) -9.71  Molscript Map 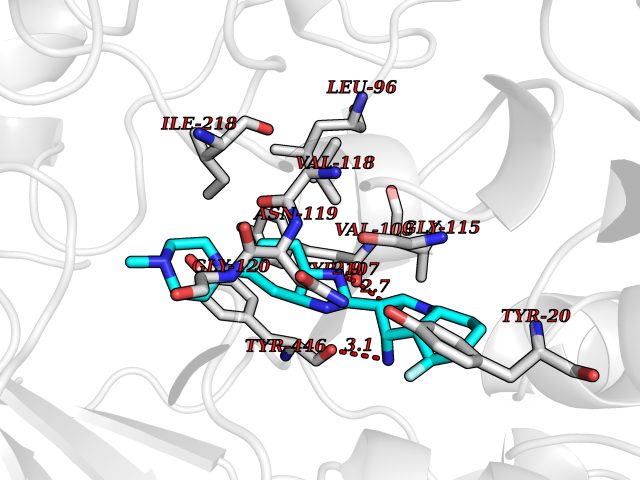 Pymol Map 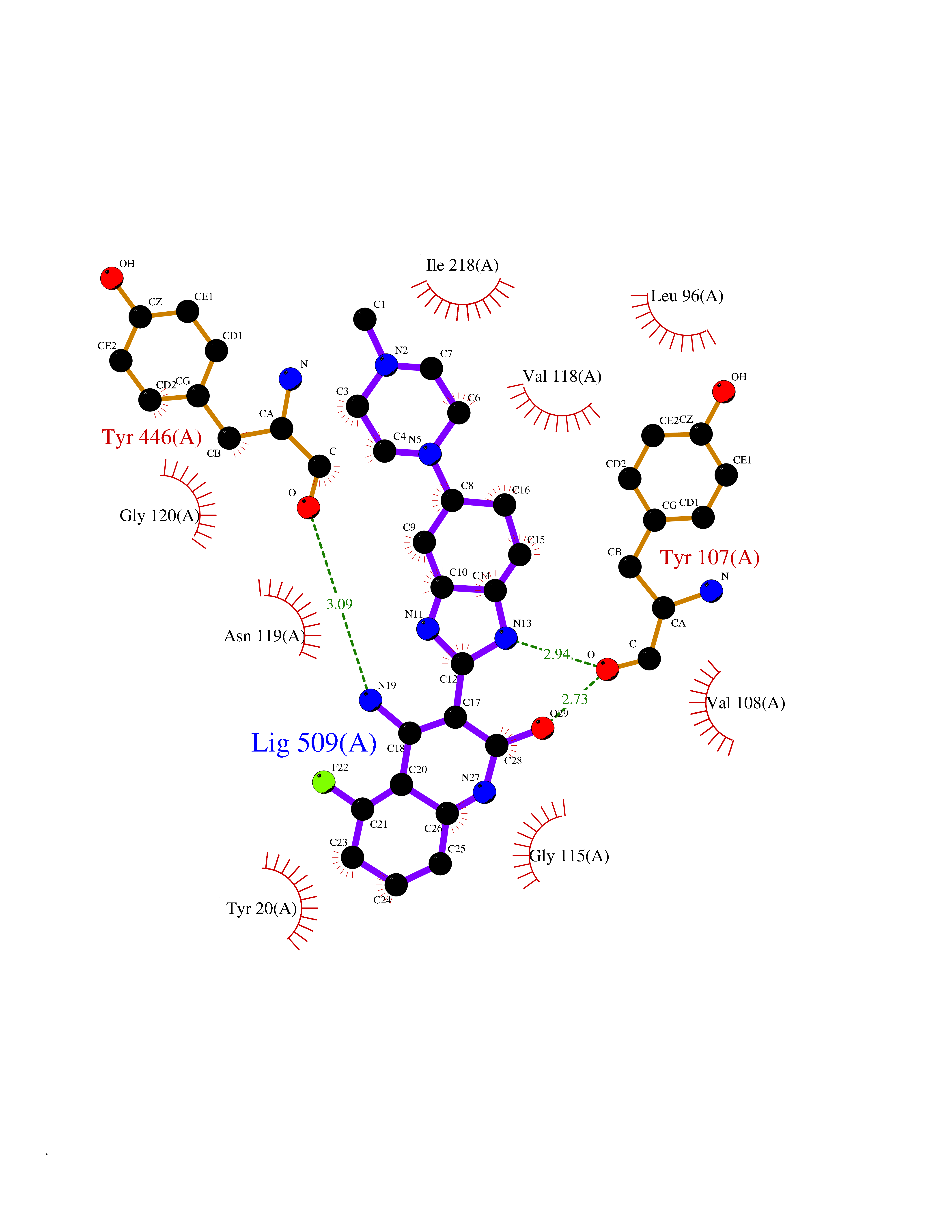 Ligplot Map 3D Binding mode Sequence GYVPAVVIGTGYGAAVSALRLGEAGVQTLMLEMGQLWNQPGPDGNIFCGMLNPDKRSSWFKNRTEAPLGSFLWLDVVNRNIDPYAGVLDRVNYDQMSVYVGRGVGGGSLVNGGMAVEPKRSYFEEILPRVDSSEMYDRYFPRANSMLRVNHIDTKWFEDTEWYKFARVSREQAGKAGLGTVFVPNVYDFGYMQREAAGEVPKSALATEVIYGNNHGKQSLDKTYLAAALGTGKVTIQTLHQVKTIRQTKDGGYALTVEQKDTDGKLLATKEISCRYLFLGAGSLGSTELLVRARDTGTLPNLNSEVGAGWGPNGNIMTARANHMWNPTGAHQSSIPALGIDAWDNSDSSVFAEIAPMPAGLETWVSLYLAITKNPQRGTFVYDAATDRAKLNWTRDQNAPAVNAAKALFDRINKANGTIYRYDLFGTQLKAFADDFCYHPLGGCVLGKATDDYGRVAGYKNLYVTDGSLIPGSVGVNPFVTITALAERNVERIIKQDV Hydrogen bonds contact Hydrophobic contact | ||||
| 45 | Folate receptor alpha (FOLR1) | 4LRH | 8.11 | |
Target general information Gen name FOLR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ovarian tumorassociated antigen MOv18; KB cells FBP; Folate receptor, adult; Folate receptor 1; FRalpha; FOLR1; Adult folatebinding protein Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pHafter receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Required for normal embryonic development and normal cell proliferation. Related diseases Neurodegeneration due to cerebral folate transport deficiency (NCFTD) [MIM:613068]: An autosomal recessive neurodegenerative disorder resulting from brain-specific folate deficiency early in life. Onset is apparent in late infancy with severe developmental regression, movement disturbances, epilepsy and leukodystrophy. {ECO:0000269|PubMed:19732866}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05595; DB00158; DB00563; DB12489; DB15413; DB05168 Interacts with Q8N357 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Endosome; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Neurodegeneration; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 24216 Length 207 Aromaticity 0.13 Instability index 49.36 Isoelectric point 8.14 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -9.58  Molscript Map 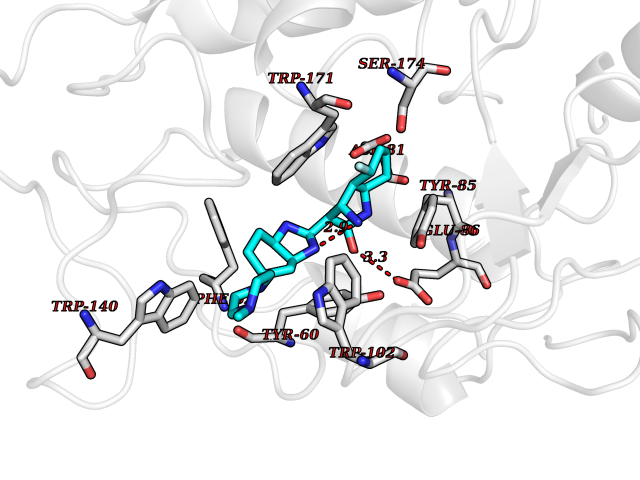 Pymol Map 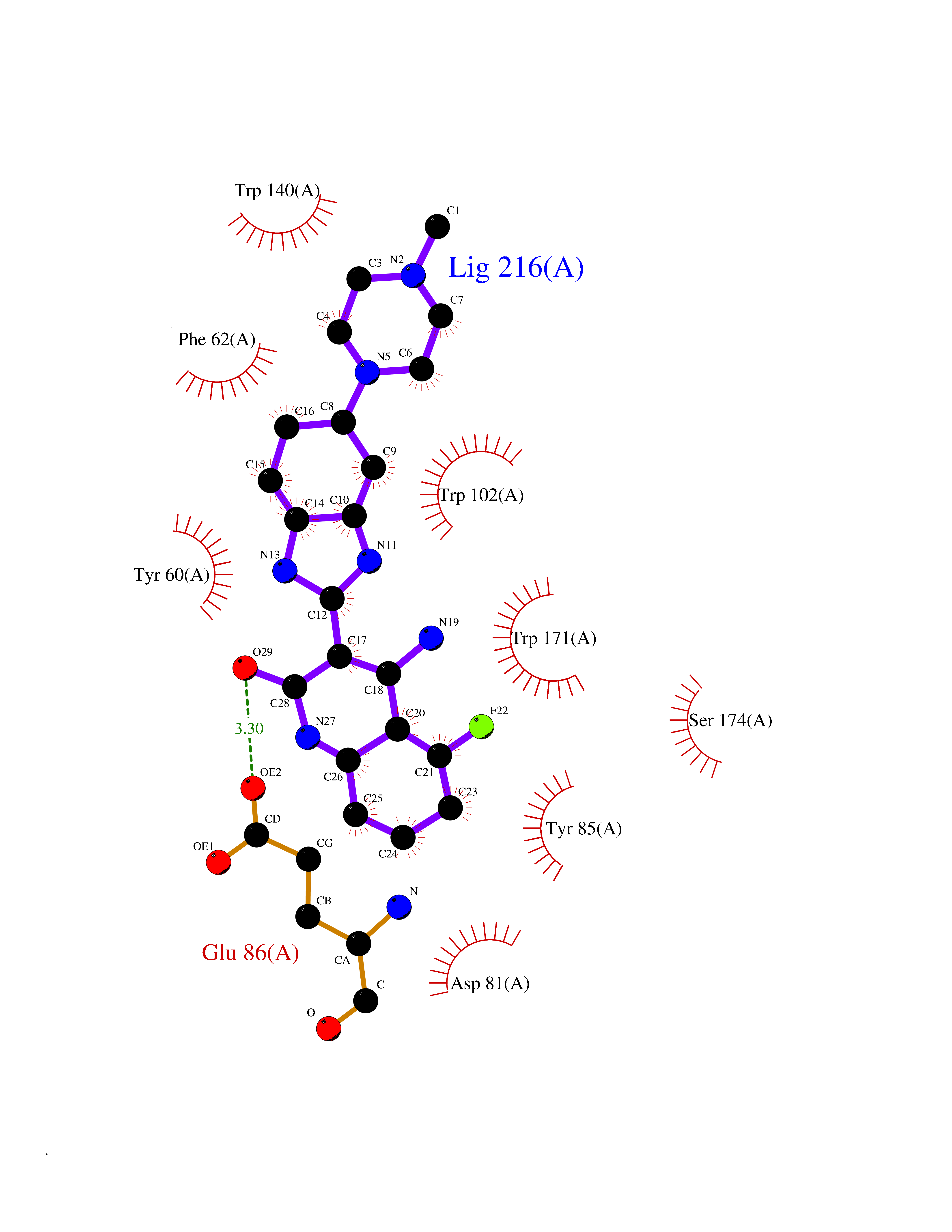 Ligplot Map 3D Binding mode Sequence RTELLNVCMNAKHHKEKPGPEDKLHEQCRPWRKNACCSTNTSQEAHKDVSYLYRFNWNHCGEMAPACKRHFIQDTCLYECSPNLGPWIQQVDQSWRKERVLNVPLCKEDCEQWWEDCRTSYTCKSNWHKGWNWTSGFNKCAVGAACQPFHFYFPTPTVLCNEIWTHSYKVSNYSRGSGRCIQMWFDPAQGNPNEEVARFYAAAMSGT Hydrogen bonds contact Hydrophobic contact | ||||
| 46 | Oxalosuccinate decarboxylase (IDH1) | 6ADG | 8.11 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD; NADP(+)-specific ICDH; Isocitrate dehydrogenase [NADP] cytoplasmic; IDP; IDH; Cytosolic NADP-isocitrate dehydrogenase Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -8.99  Molscript Map 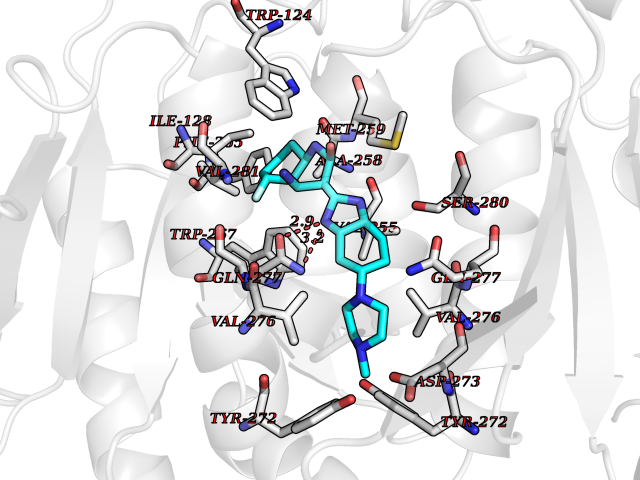 Pymol Map 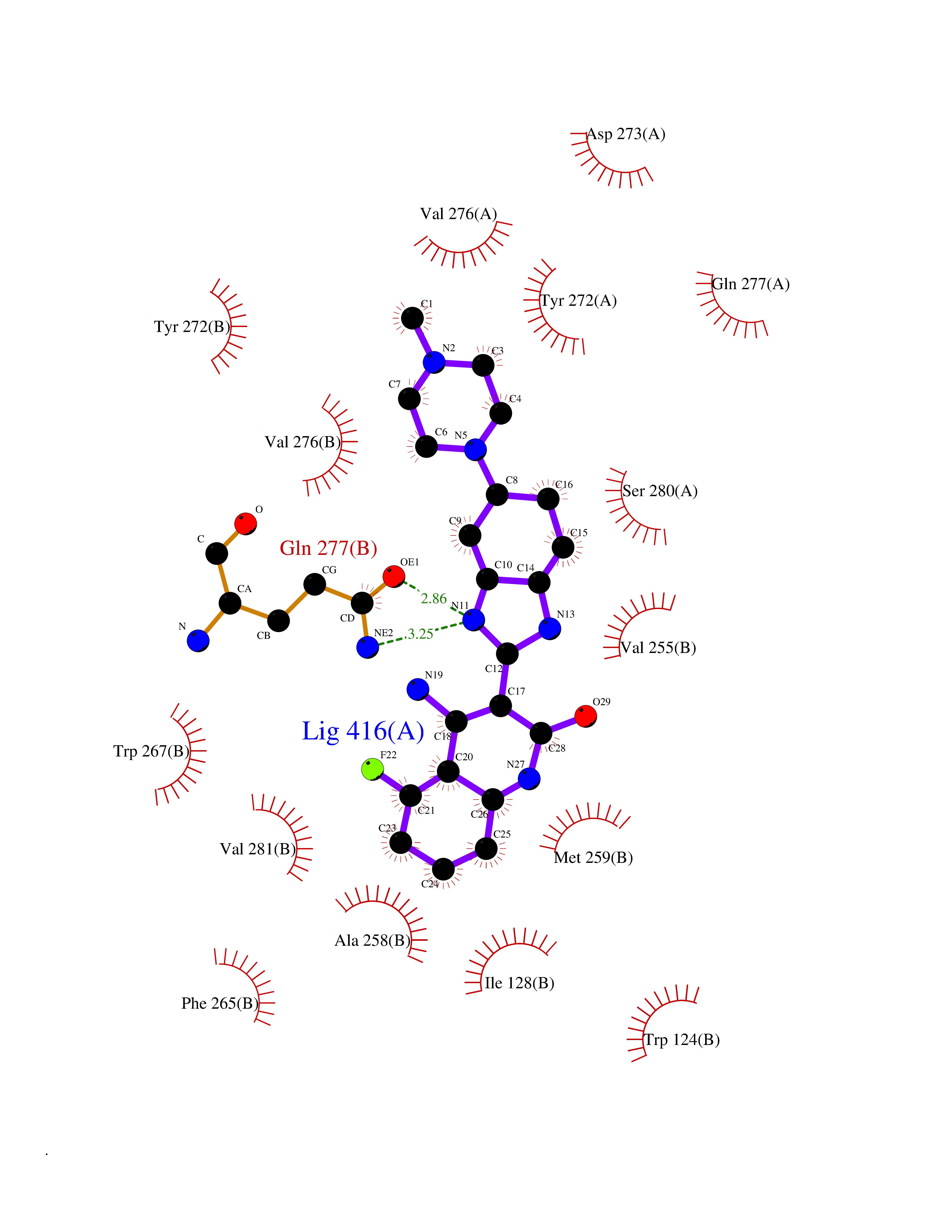 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 47 | Matrix metalloproteinase-16 (MMP-16) | 1RM8 | 8.10 | |
Target general information Gen name MMP16 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Membrane-type-3 matrix metalloproteinase; Membrane-type matrix metalloproteinase 3; MTMMP3; MT3MMP; MT3-MMP; MT-MMP 3; MMPX2; MMP-X2; C8orf57 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates progelatinase A. Involved in the matrix remodeling of blood vessels. Isoform short cleaves fibronectin and also collagen type III, but at lower rate. It has no effect on type I, II, IV and V collagen. However, upon interaction with CSPG4, it may be involved in degradation and invasion of type I collagen by melanoma cells. Endopeptidase that degrades various components of the extracellular matrix, such as collagen type III and fibronectin. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03880; DB00786 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Cleavage on pair of basic residues; Collagen degradation; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18853.6 Length 169 Aromaticity 0.12 Instability index 33.65 Isoelectric point 4.88 Charge (pH=7) -12.42 2D Binding mode Binding energy (Kcal/mol) -9.29  Molscript Map 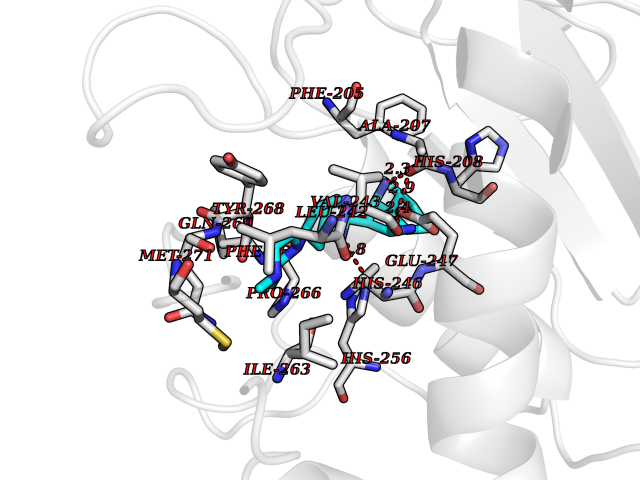 Pymol Map 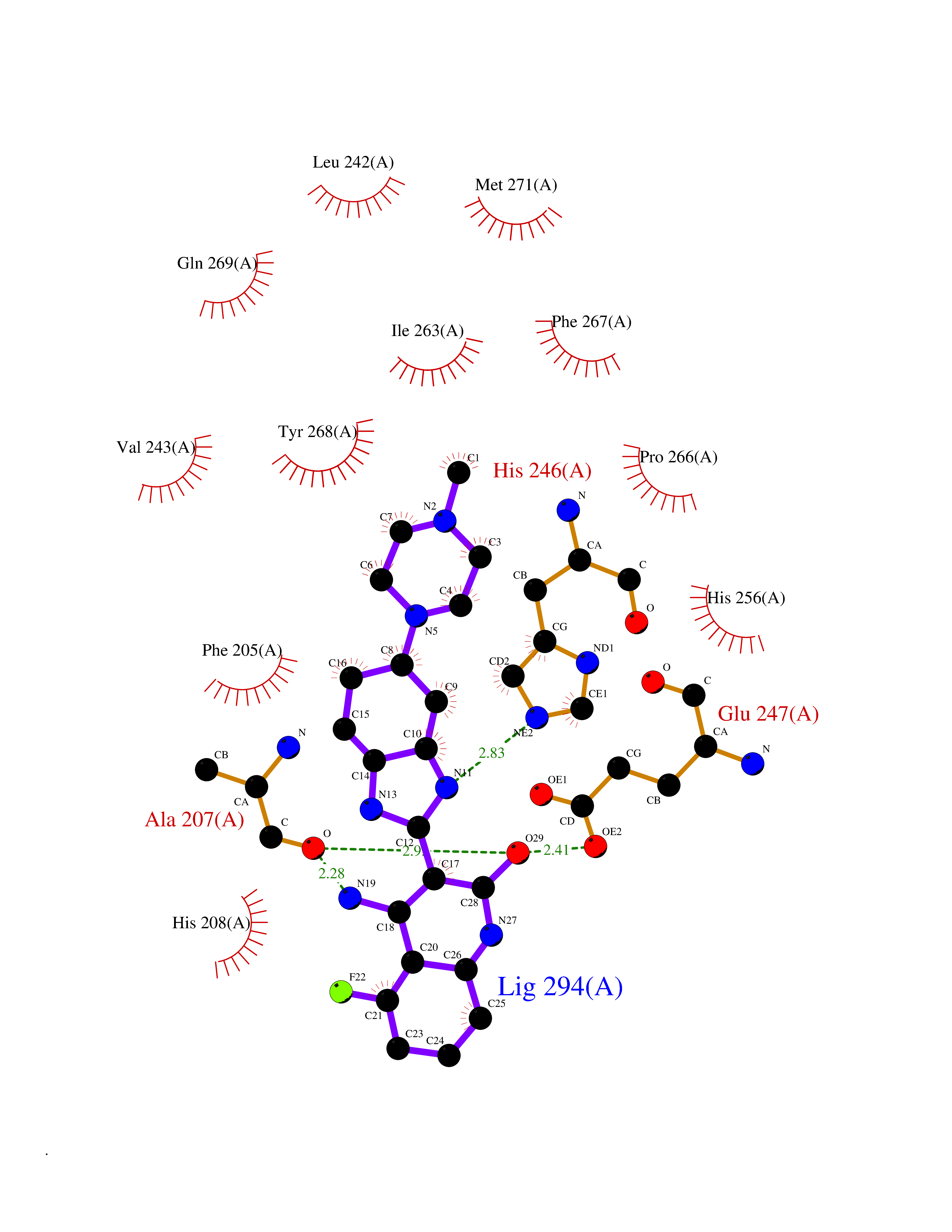 Ligplot Map 3D Binding mode Sequence GQKWQHKHITYSIKNVTPKVGDPETRKAIRRAFDVWQNVTPLTFEEVPYSELENGKRDVDITIIFASGFHGDSSPFDGEGGFLAHAYFPGPGIGGDTHFDSDEPWTLGNPNHDGNDLFLVAVHELGHALGLEHSNDPTAIMAPFYQYMETDNFKLPNDDLQGIQKIYGP Hydrogen bonds contact Hydrophobic contact | ||||
| 48 | GABA transporter GAT-1 (SLC6A1) | 7SK2 | 8.08 | |
Target general information Gen name SLC6A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 6 member 1; Sodium- and chloride-dependent GABA transporter 1; GAT1; GAT-1; GABT1; GABATR; GABA transporter 1 Protein family Sodium:neurotransmitter symporter (SNF) (TC 2.A.22) family, SLC6A1 subfamily Biochemical class Neurotransmitter:sodium symporter Function Terminates the action of GABA by its high affinity sodium-dependent reuptake into presynaptic terminals. Related diseases Myoclonic-atonic epilepsy (MAE) [MIM:616421]: A form of epilepsy characterized by myoclonic-atonic and absence seizures, appearing in early childhood. Patients have delayed development before the onset of seizures and show varying degrees of intellectual disability following seizure onset. {ECO:0000269|PubMed:25865495, ECO:0000269|PubMed:30132828}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08848; DB00906 Interacts with O75031 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Membrane; Metal-binding; Neurotransmitter transport; Phosphoprotein; Proteomics identification; Reference proteome; Sodium; Symport; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 60038.1 Length 532 Aromaticity 0.17 Instability index 32.01 Isoelectric point 8.54 Charge (pH=7) 5.89 2D Binding mode Binding energy (Kcal/mol) -9.46  Molscript Map 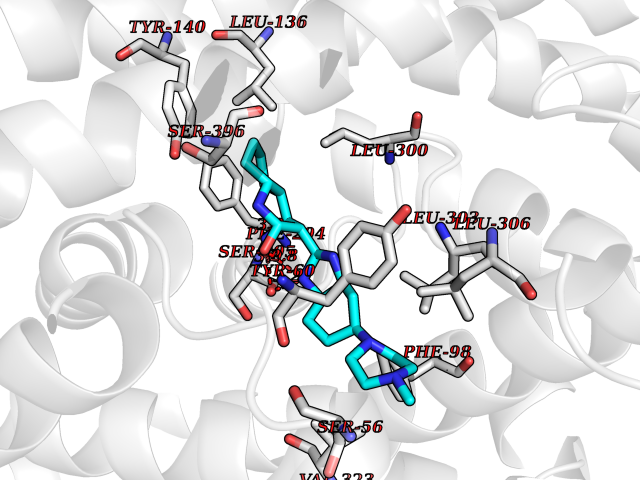 Pymol Map 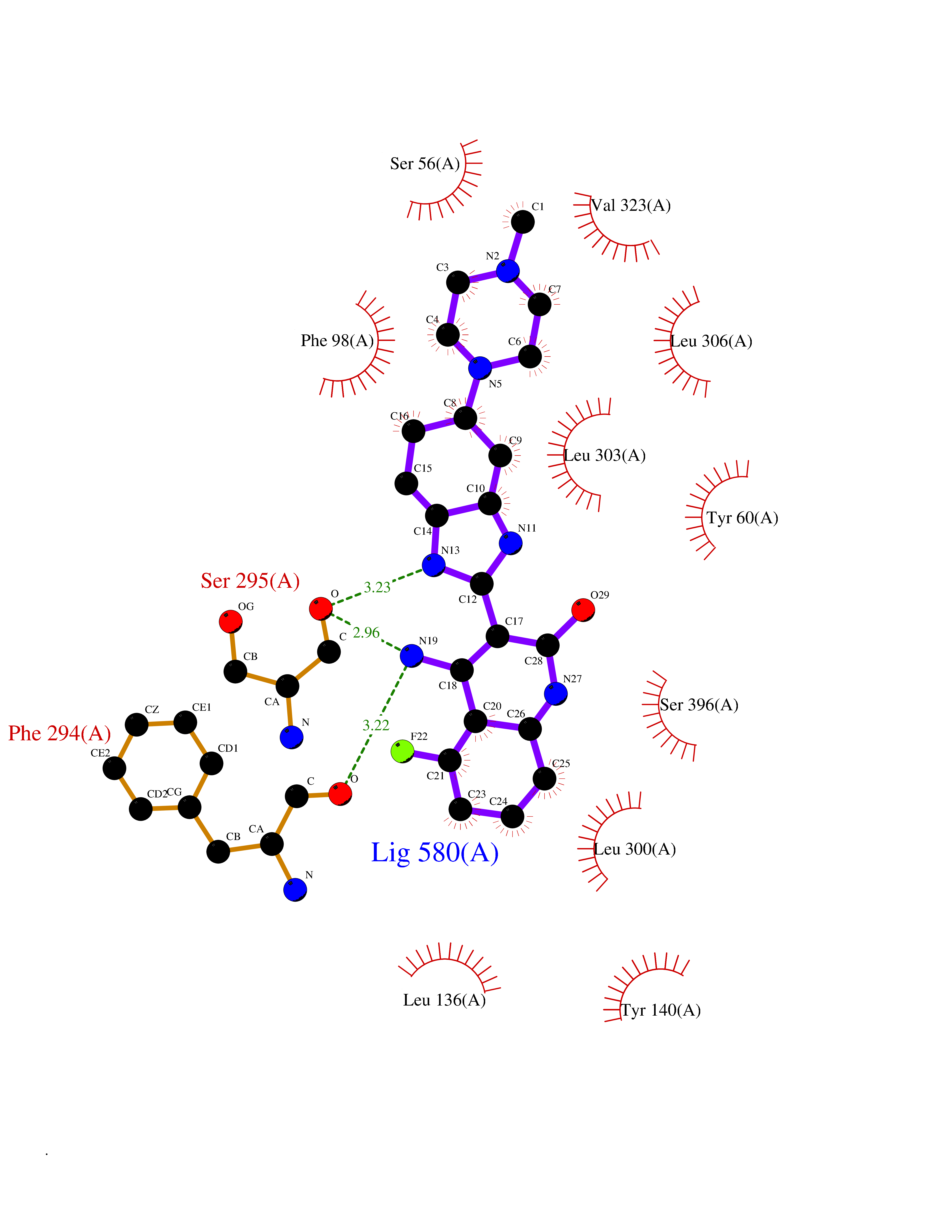 Ligplot Map 3D Binding mode Sequence WKGRFDFLMSCVGYAIGLGNVWRFPYLCGKNGGGAFLIPYFLTLIFAGVPLFLLECSLGQYTSIGGLGVWKLAPMFKGVGLAAAVLSFWLNIYYIVIISWAIYYLYNSFTTTLPWKQCDNPWNTDRCFSNYSMVNTTNMTSAVVEFWERNMHQMTDGLDKPGQIRWPLAITLAIAWILVYFCIWKGVGWTGKVVYFSATYPYIMLIILFFRGVTLPGAKEGILFYITPNFRKLSDSEVWLDAATQIFFSYGLGLGSLIALGSYNSFHNNVYRDSIIVCCINSCTSMFAGFVIFSIVGFMAHVTKRSIADVAASGPGLAFLAYPEAVTQLPISPLWAILFFSMLLMLGIDSQFCTVEGFITALVDEYPRLLRNRRELFIAAVCIISYLIGLSNITQGGIYVFKLFDYYSASGMSLLFLVFFECVSISWFYGVNRFYDNIQEMVGSRPCIWWKLCWSFFTPIIVAGVFIFSAVQMTPLTMGNYVFPKWGQGVGWLMALSSMVLIPGYMAYMFLTLKGSLKQRIQVMVQPSEDIV Hydrogen bonds contact Hydrophobic contact | ||||
| 49 | Bacterial DD-carboxypeptidase (Bact vanYB) | 5ZHW | 8.08 | |
Target general information Gen name Bact vanYB Organism Enterococcus faecalis (strain ATCC 700802 / V583) Uniprot ID TTD ID Synonyms vanYB; DD-peptidase; DD-carboxypeptidase; D-alanyl-D-alanine carboxypeptidase-transpeptidase Protein family Peptidase M15B family Biochemical class Peptidase Function Vancomycin-inducible, penicillin-resistant, DD- carboxypeptidase that hydrolyzes depsipeptide- and D-alanyl-D- alanine-containing peptidoglycan precursors. Insensitive to beta- lactams. Related diseases Brachyolmia 3 (BCYM3) [MIM:113500]: A form of brachyolmia, a clinically and genetically heterogeneous skeletal dysplasia primarily affecting the spine and characterized by a short trunk, short stature, and platyspondyly. BCYM3 is an autosomal dominant form with severe scoliosis with or without kyphosis, and flattened irregular cervical vertebrae. {ECO:0000269|PubMed:18587396}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondylometaphyseal dysplasia Kozlowski type (SMDK) [MIM:184252]: A form of spondylometaphyseal dysplasia, a group of short stature disorders distinguished by abnormalities in the vertebrae and the metaphyses of the tubular bones. It is characterized by postnatal dwarfism, significant scoliosis and mild metaphyseal abnormalities in the pelvis. The vertebrae exhibit platyspondyly and overfaced pedicles. {ECO:0000269|PubMed:19232556, ECO:0000269|PubMed:20577006, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metatropic dysplasia (MTD) [MIM:156530]: A severe spondyloepimetaphyseal dysplasia characterized by short limbs with limitation and enlargement of joints and usually severe kyphoscoliosis. Radiologic features include severe platyspondyly, severe metaphyseal enlargement and shortening of long bones. {ECO:0000269|PubMed:19232556, ECO:0000269|PubMed:20425821, ECO:0000269|PubMed:20577006, ECO:0000269|PubMed:22702953, ECO:0000269|PubMed:26249260, ECO:0000269|Ref.6}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 8 (HMND8) [MIM:600175]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:20037588, ECO:0000269|PubMed:22526352, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2C (CMT2C) [MIM:606071]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:20037586, ECO:0000269|PubMed:20037587, ECO:0000269|PubMed:20037588, ECO:0000269|PubMed:21115951, ECO:0000269|PubMed:21288981, ECO:0000269|PubMed:22702953, ECO:0000269|PubMed:25256292}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Scapuloperoneal spinal muscular atrophy (SPSMA) [MIM:181405]: A clinically variable neuromuscular disorder characterized by neurogenic scapuloperoneal amyotrophy, laryngeal palsy, congenital absence of muscles, progressive scapuloperoneal atrophy and progressive distal weakness and amyotrophy. {ECO:0000269|PubMed:20037587, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepiphyseal dysplasia Maroteaux type (SEDM) [MIM:184095]: A clinically variable spondyloepiphyseal dysplasia with manifestations limited to the musculoskeletal system. Clinical features include short stature, brachydactyly, platyspondyly, short and stubby hands and feet, epiphyseal hypoplasia of the large joints, and iliac hypoplasia. Intelligence is normal. {ECO:0000269|PubMed:20503319, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parastremmatic dwarfism (PSTD) [MIM:168400]: A bone dysplasia characterized by severe dwarfism, kyphoscoliosis, distortion and bowing of the extremities, and contractures of the large joints. Radiographically, the disease is characterized by a combination of decreased bone density, bowing of the long bones, platyspondyly and striking irregularities of endochondral ossification with areas of calcific stippling and streaking in radiolucent epiphyses, metaphyses and apophyses. {ECO:0000269|PubMed:20503319}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Digital arthropathy-brachydactyly, familial (FDAB) [MIM:606835]: A disorder characterized by irregularities in the proximal articular surfaces of the distal interphalangeal joints of the hand. Individuals appear normal at birth, with no clinical or radiographic evidence of a developmental skeletal dysplasia. The earliest changes appear during the first decade of life. By adulthood, all interphalangeal, metacarpophalangeal, and metatarsophalangeal joints are affected by a deforming, painful osteoarthritis. The remainder of the skeleton is clinically and radiographically unaffected. {ECO:0000269|PubMed:21964574}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Avascular necrosis of the femoral head, primary 2 (ANFH2) [MIM:617383]: A disease characterized by mechanical failure of the subchondral bone, and degeneration of the hip joint. It usually leads to destruction of the hip joint in the third to fifth decade of life. The clinical manifestations, such as pain on exertion, a limping gait, and a discrepancy in leg length, cause considerable disability. {ECO:0000269|PubMed:27330106}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.16.4 Uniprot keywords 3D-structure; Antibiotic resistance; Carboxypeptidase; Cell membrane; Cell shape; Cell wall biogenesis/degradation; Hydrolase; Membrane; Metal-binding; Peptidoglycan synthesis; Protease; Reference proteome; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 20547.5 Length 181 Aromaticity 0.11 Instability index 33.61 Isoelectric point 4.81 Charge (pH=7) -10.78 2D Binding mode Binding energy (Kcal/mol) -9.04  Molscript Map 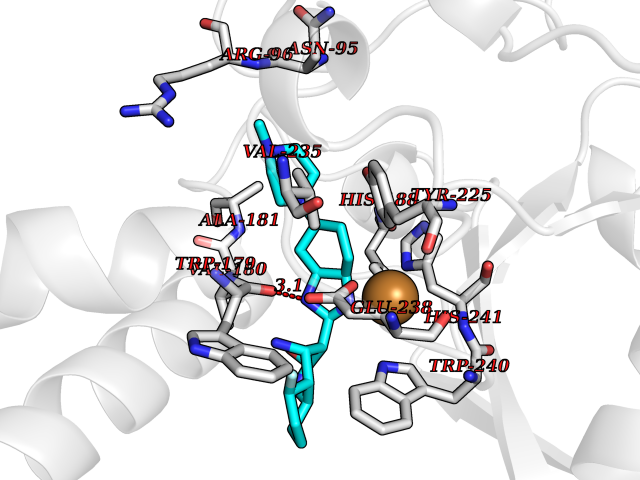 Pymol Map 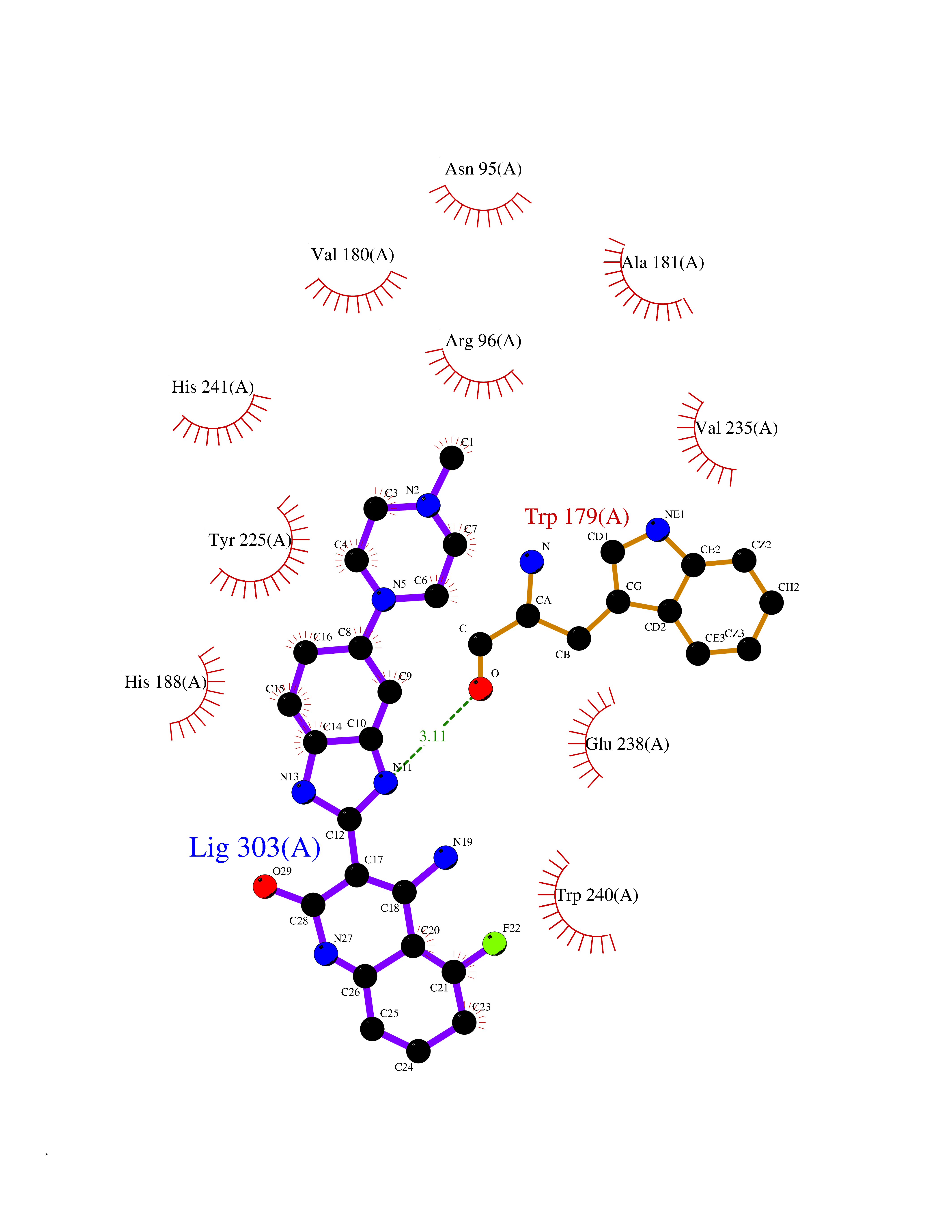 Ligplot Map 3D Binding mode Sequence EWSLILVNRQNPIPAQYDVELEQLSNGERIDIRISPYLQDLFDAARADGVYPIVASGYRTTEKQQEIXDEKVAEYKAKGYTSAQAKAEAETWVAVPGTSEHQLGLAVDINADGIHSTGNEVYRWLDENSYRFGFIRRYPPDKTEITGVSNEPWHYRYVGIEAATKIYHQGLCLEEYLNTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 50 | Clostridium histolyticum Collagenase (CH colG) | 7Z5U | 8.08 | |
Target general information Gen name CH colG Organism Hathewaya histolytica (Clostridium histolyticum) Uniprot ID TTD ID Synonyms Microbial collagenase; Gelatinase ColG; Collagenase ColG; Class I collagenase Protein family Peptidase M9B family, Collagenase subfamily Biochemical class NA Function Clostridial collagenases are among the most efficient degraders of eukaryotic collagen known; saprophytes use collagen as a carbon source while pathogens additionally digest collagen to aid in host colonization. Has both tripeptidylcarboxypeptidase on Gly-X-Y and endopeptidase activities; the endopeptidase cuts within the triple helix region of collagen while tripeptidylcarboxypeptidase successively digests the exposed ends, thus clostridial collagenases can digest large sections of collagen. Active on soluble type I collagen, insoluble collagen, azocoll, soluble PZ-peptide (all collagenase substrates) and gelatin. The full-length protein has collagenase activity, while the in vivo derived C-terminally truncated shorter versions only act on gelatin. In vitro digestion of soluble calf skin collagen fibrils requires both ColG and ColH; ColG forms missing the second collagen-binding domain are also synergistic with ColH, although their overall efficiency is decreased. The activator domain (residues 119-388) and catalytic subdomain (389-670) open and close around substrate using a Gly-rich hinge (387-397), allowing digestion when the protein is closed. Binding of collagen requires Ca(2+) and is inhibited by EGTA; the collagen-binding domain (CBD, S3a plus S3b) specifically recognizes the triple-helical conformation made by 3 collagen protein chains in the triple-helical region. Isolated CBD (S3a plus S3b) binds collagen fibrils and sheets of many tissues. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.24.3 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Hydrolase; Metal-binding; Metalloprotease; Pharmaceutical; Protease; Repeat; Secreted; Signal; Virulence; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 44751.2 Length 386 Aromaticity 0.15 Instability index 27.33 Isoelectric point 5.77 Charge (pH=7) -7.33 2D Binding mode Binding energy (Kcal/mol) -8.92 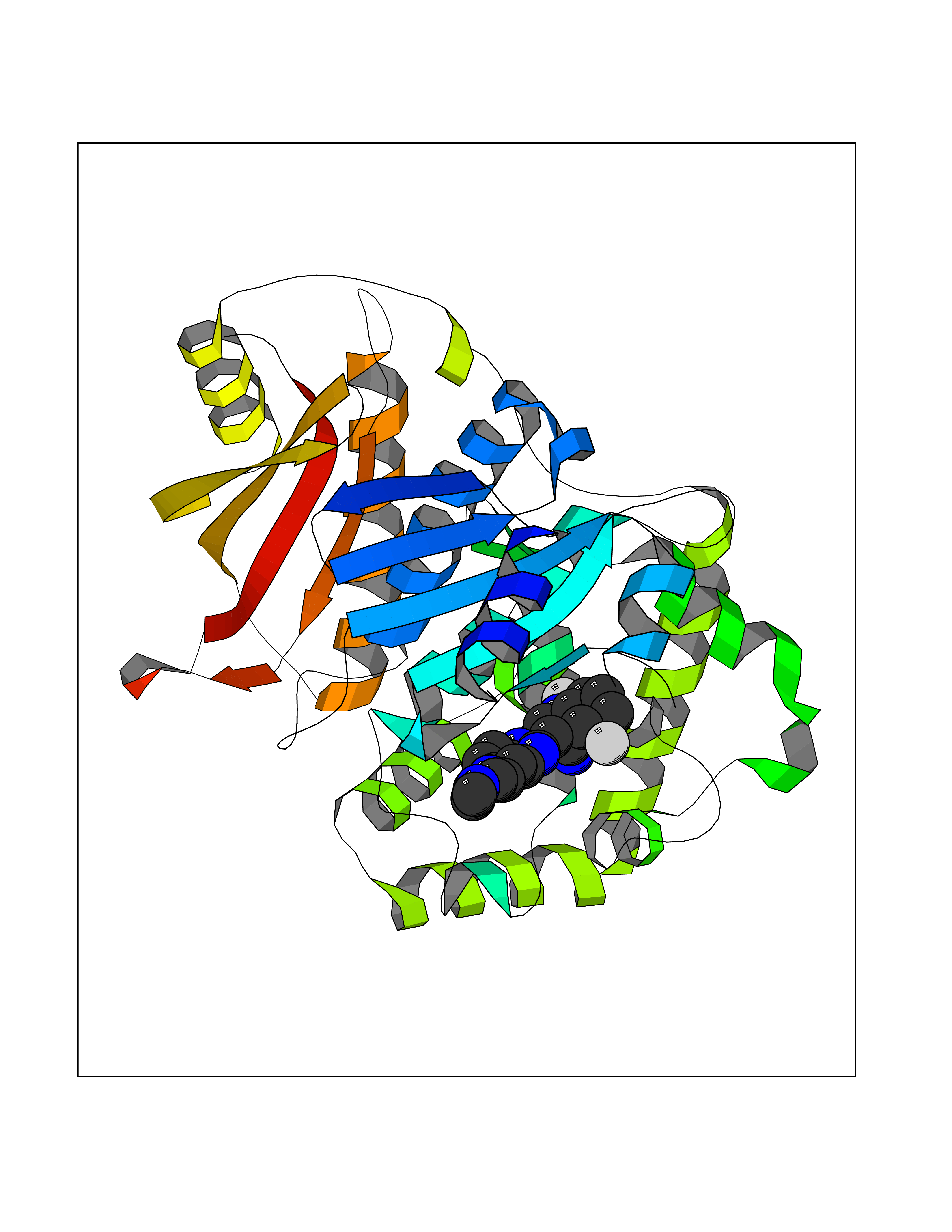 Molscript Map 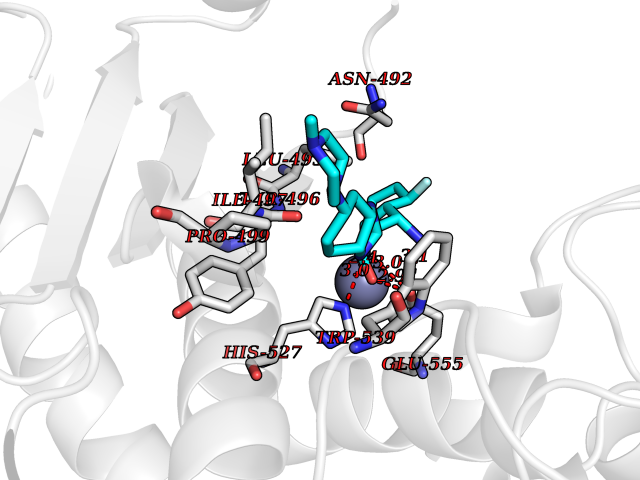 Pymol Map 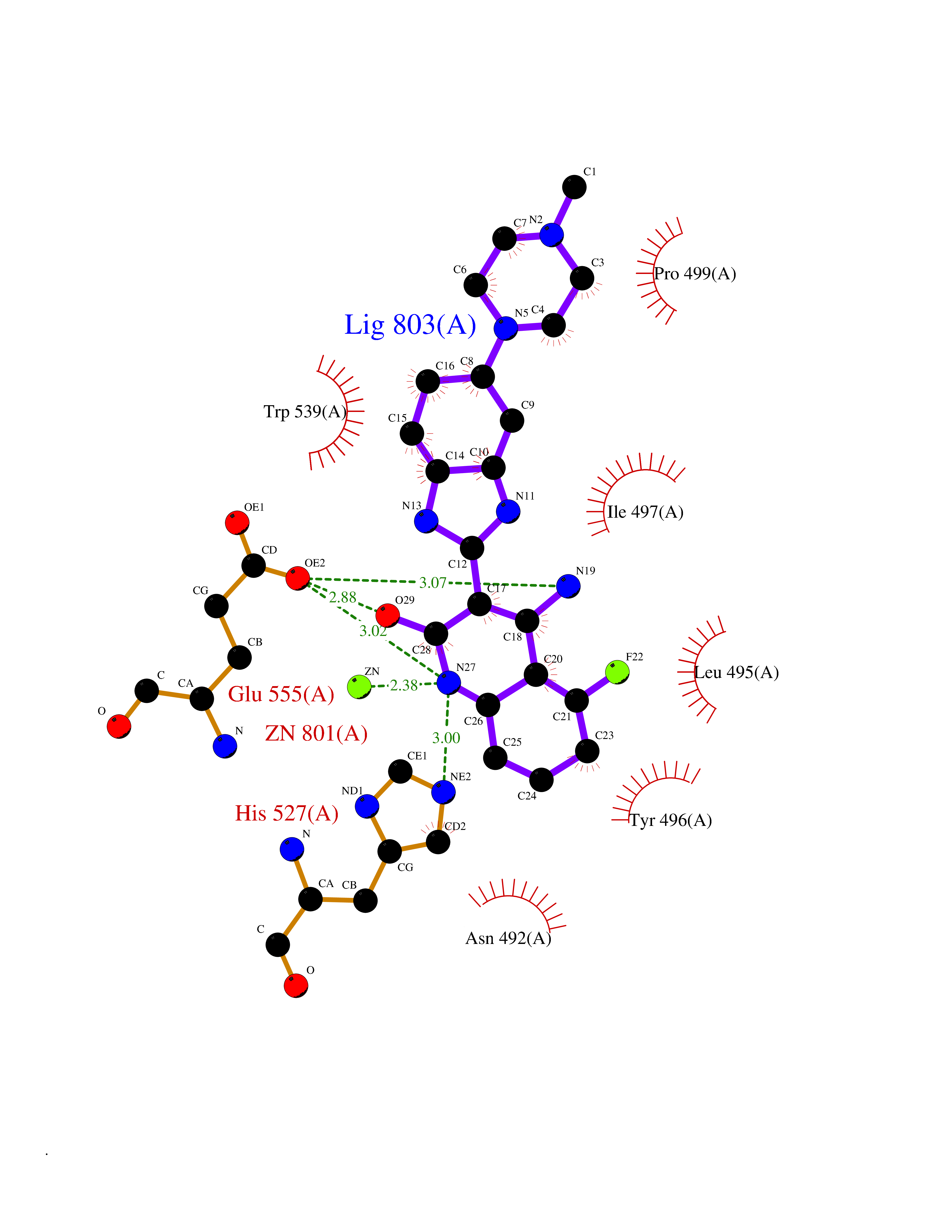 Ligplot Map 3D Binding mode Sequence DHDKFLDDAEKHYLPKTYTFDNGTFIIRAGDKVSEEKIKRLYWASREVKSQFHRVVGNDKALEVGNADDVLTMKIFNSPEEYKFNTTDNGGLYIEPRGTFYTYERTPQQSIFSLEELFRHEYTHYLQARYLVDGLWGQGPFYEKNRLTWFDEGTAEFFAGSTRTSGVLPRKLILGYLAKDKVDHRYSLKKTLNSGYDDSDWMFYNYGFAVAHYLYEKDMPTFIKMNKAILNTDVKSYDEIIKKLSDDANKNTEYQNHIQELVDKYQGAGIPLVSDDYLKDHGYKKASEVYSEISKAASLTNTSVTAEKSQYFNTFTLRGTYTGETSKGEFKDWDEMSKKLDGTLESLAKNSWSGYKTLTAYFTNYRVTSDNKVQYDVVFHGVLTDN Hydrogen bonds contact Hydrophobic contact | ||||
| 51 | Histamine H1 receptor (H1R) | 7DFL | 8.08 | |
Target general information Gen name HRH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HH1R; H1R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function In peripheral tissues, the H1 subclass of histamine receptors mediates the contraction of smooth muscles, increase in capillary permeability due to contraction of terminal venules, and catecholamine release from adrenal medulla, as well as mediating neurotransmission in the central nervous system. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01615; DB09488; DB06766; DB01246; DB00321; DB00543; DB08799; DB01238; DB14185; DB06216; DB00637; DB00719; DB00972; DB00245; DB00767; DB04890; DB06698; DB11591; DB09128; DB01237; DB00835; DB00354; DB09016; DB00748; DB06016; DB00341; DB08936; DB08800; DB01114; DB00477; DB01239; DB00568; DB00215; DB00283; DB04837; DB00363; DB01176; DB00434; DB01151; DB00967; DB00405; DB09555; DB00985; DB08801; DB01075; DB01146; DB09167; DB01142; DB00366; DB01084; DB05492; DB00751; DB01175; DB06678; DB00950; DB04841; DB00502; DB05381; DB05079; DB00557; DB04946; DB00458; DB08802; DB00920; DB00555; DB01106; DB06282; DB00455; DB09195; DB00408; DB00934; DB00737; DB06691; DB01071; DB00902; DB01403; DB06148; DB00370; DB00540; DB05080; DB06229; DB00334; DB00768; DB01173; DB01267; DB00715; DB08922; DB01619; DB01620; DB06153; DB00433; DB00420; DB01069; DB00777; DB01224; DB00912; DB00734; DB11614; DB05345; DB00342; DB04905; DB11235; DB00797; DB00656; DB00726; DB00792; DB00427; DB09185; DB00246; DB01624 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32298.5 Length 275 Aromaticity 0.15 Instability index 36.59 Isoelectric point 9.54 Charge (pH=7) 16.01 2D Binding mode Binding energy (Kcal/mol) -8.82 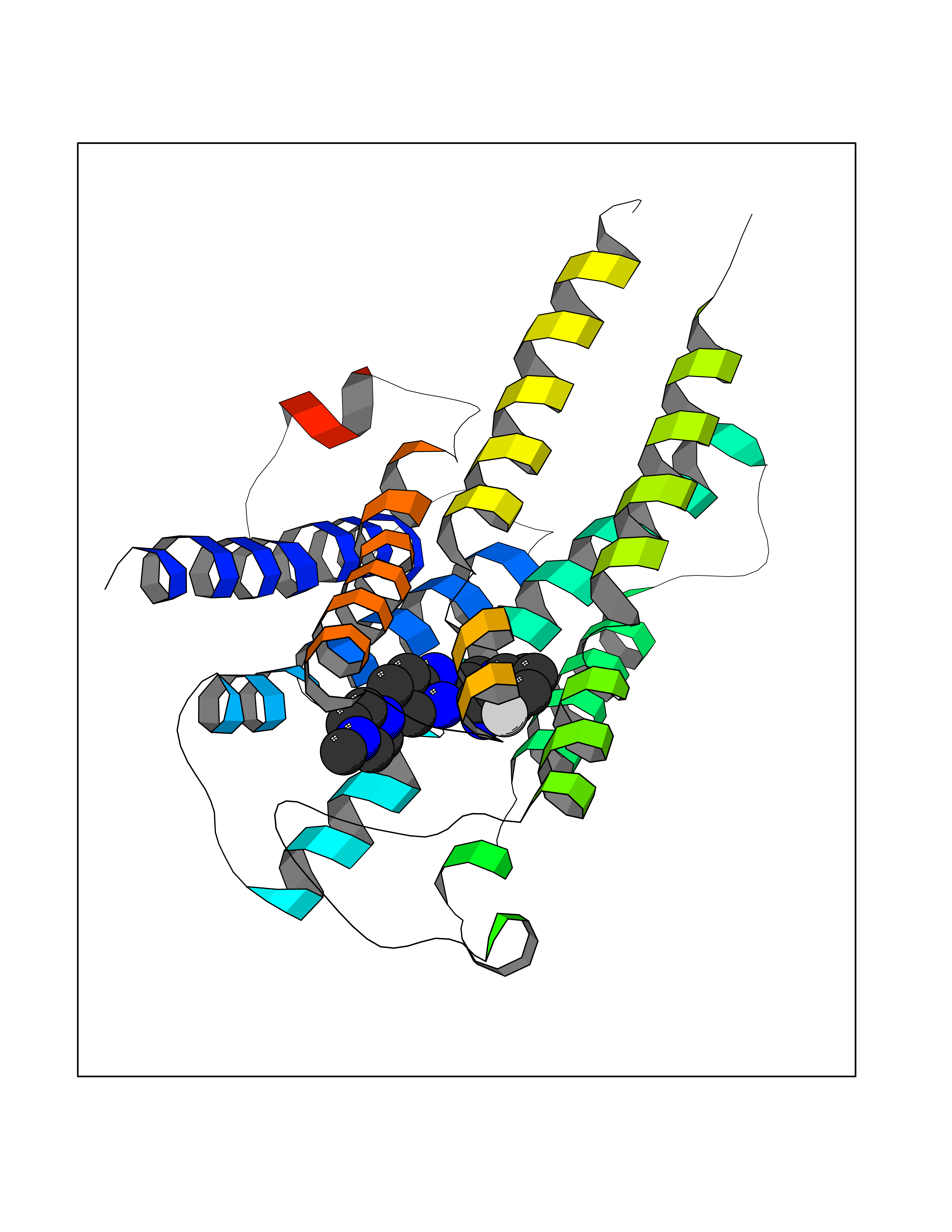 Molscript Map 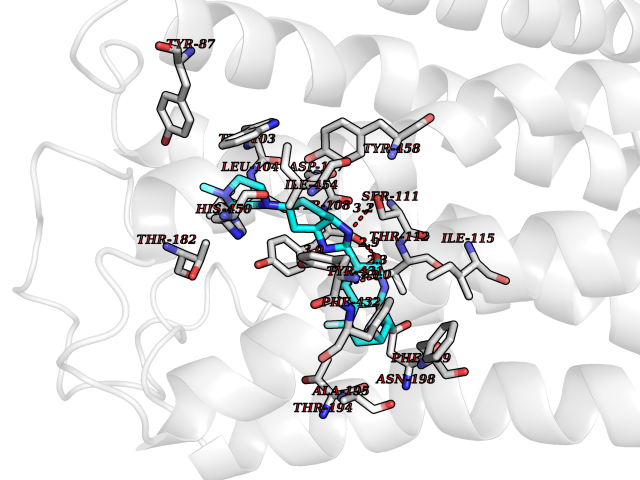 Pymol Map 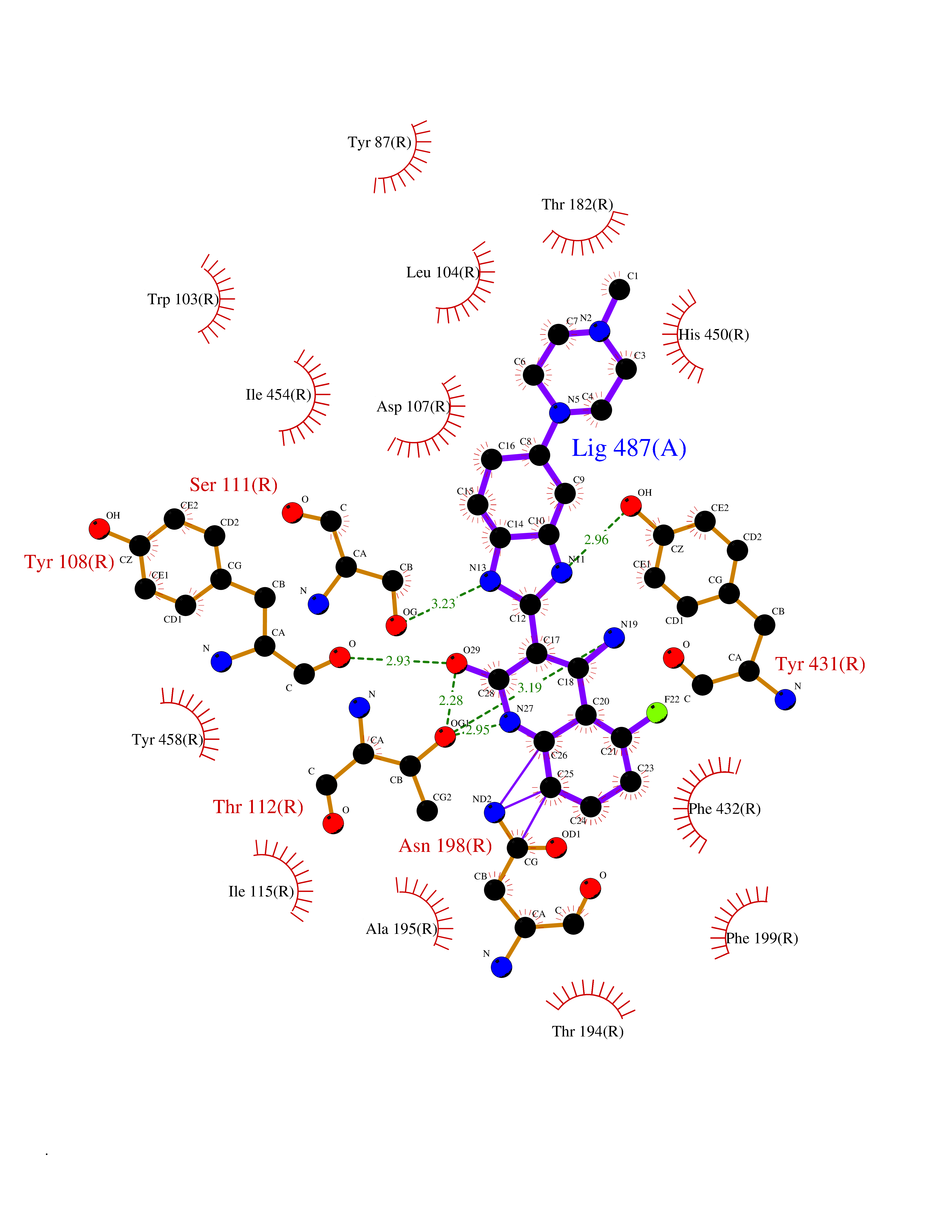 Ligplot Map 3D Binding mode Sequence MPLVVVLSTICLVTVGLNLLVLYAVRSERKLHTVGNLYIVSLSVADLIVGAVVMPMNILYLLMSKWSLGRPLCLFWLSMDYVASTASIFSVFILCIDRYRSVQQPLRYLKYRTKTRASATILGAWFLSFLWVIPILGWNHFMQQTSVRREDKCETDFYDVTWFKVMTAIINFYLPTLLMLWFYAKIYKAVRQHCLHMNRERKAAKQLGFIMAAFILCWIPYFIFFMVIAFCKNCCNEHLHMFTIWLGYINSTLNPLIYPLCNENFKKTFKRILHI Hydrogen bonds contact Hydrophobic contact | ||||
| 52 | Retinoic acid receptor RXR-beta (RXRB) | 5HJP | 8.08 | |
Target general information Gen name RXRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor beta; Nuclear receptor subfamily 2 group B member 2; NR2B2 Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE). Receptor for retinoic acid. Related diseases Noonan syndrome 13 (NS13) [MIM:619087]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. NS13 inheritance is autosomal dominant. There is considerable variability in severity. {ECO:0000269|PubMed:32721402}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08175; DB00459; DB00210; DB00523; DB00307; DB01393; DB03756; DB00926; DB01941; DB07929; DB02746; DB00412; DB00799; DB07080; DB00755 Interacts with Q00975; Q9HB07; F1D8P7; Q13133; Q13133-3; Q96RI1-1; P04150; Q9NRD5; P37231; P10276; P10276-2; P10826-2; P13631; Q6IQ16; Q13137; Q96B26; Q08379; Q6A162; Q9UJV3-2; Q13133-3; Q96RI1-1; O43586; P10276; P10826-2; Q8IUQ4-2; O75528; Q12800; Q9UBB9; Q05BL1; P14373; O94972; Q96S82 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 28845.8 Length 251 Aromaticity 0.08 Instability index 54.86 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -9.44 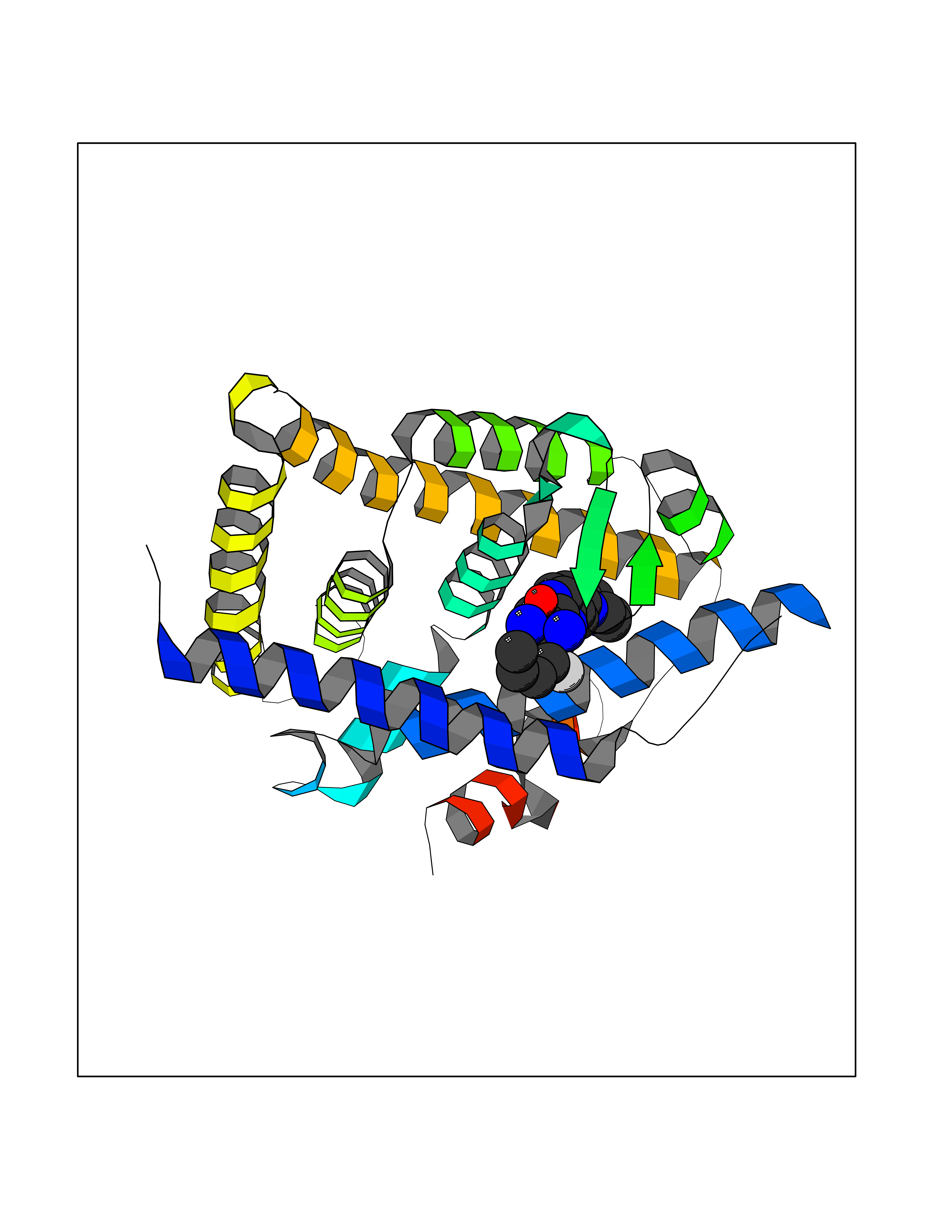 Molscript Map 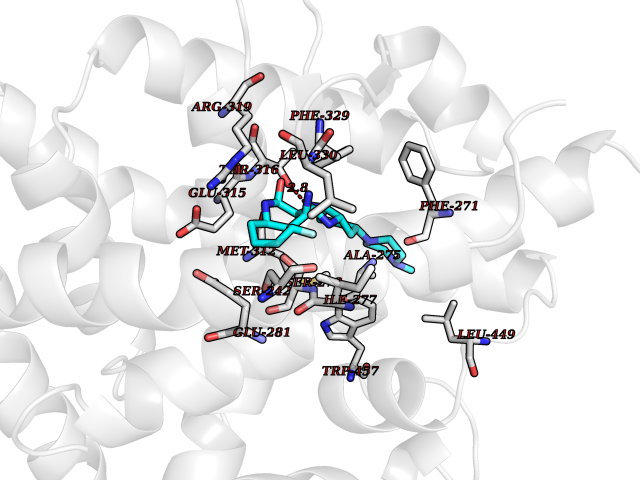 Pymol Map 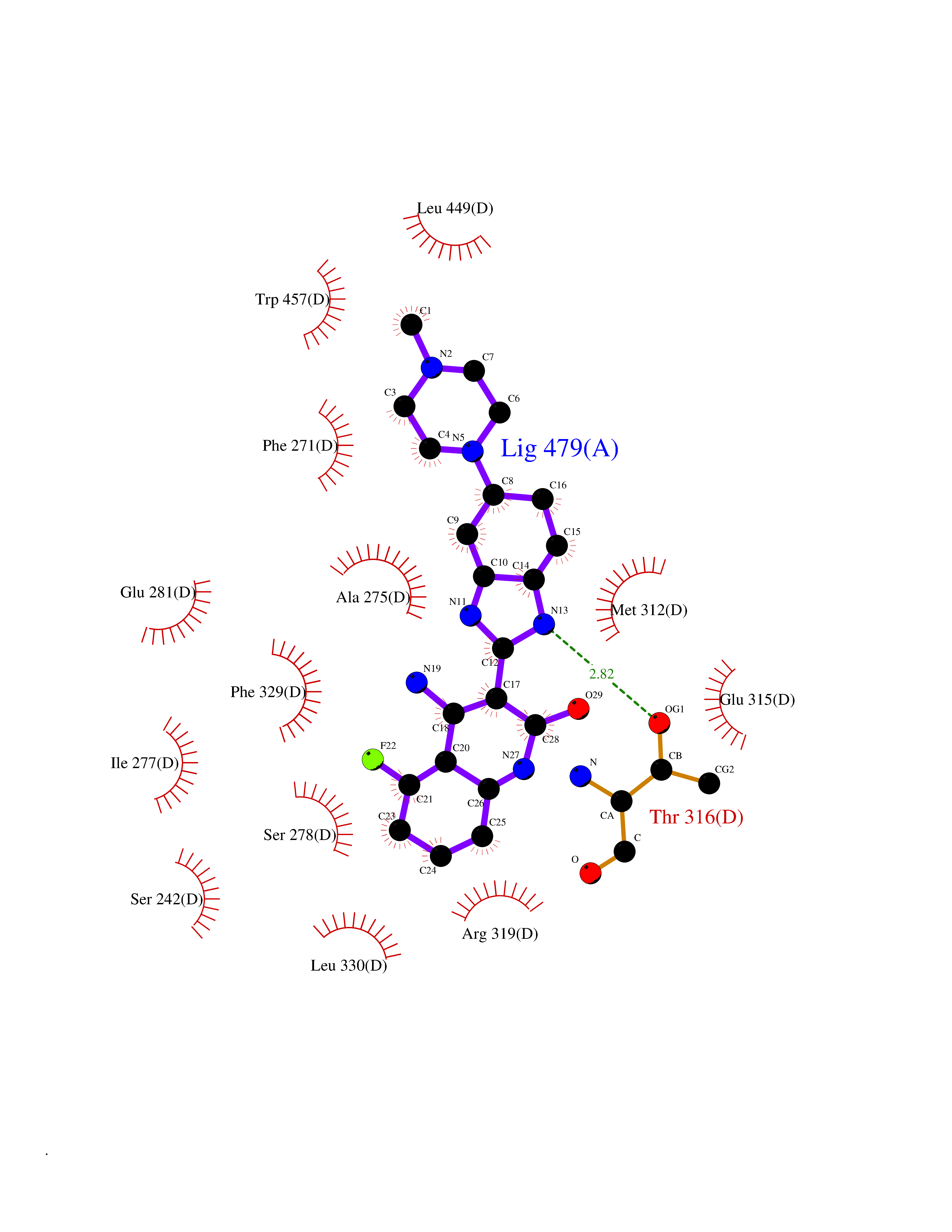 Ligplot Map 3D Binding mode Sequence QLTAAQELMIQQLVAAQLQCNKRSFSDQPKVTPWPSASQQRFAHFTELAIISVQEIVDFAKQVPGFLQLGREDQIALLKASTIEIMLLETARRYNHETECITFLKDFTYSKDDFHRAGLQVEFINPIFEFSRAMRRLGLDDAEYALLIAINIFSADRPNVQEPGRVEALQQPYVEALLSYTRIKRPQDQLRFPRMLMKLVSLRTLSSVHSEQVFALRLQDKKLPPLLSEIWDVHEGSGSGSHKILHRLLQD Hydrogen bonds contact Hydrophobic contact | ||||
| 53 | Trypanosoma Dihydrolipoamide dehydrogenase (Trypano LPD) | 2QAE | 8.06 | |
Target general information Gen name Trypano LPD Organism Trypanosoma cruzi Uniprot ID TTD ID Synonyms Lipoamide dehydrogenase; LipDH; LPD; Glycine cleavage system L protein; Dihydrolipoamide dehydrogenase Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Sulfur donor oxidoreductase Function Lipoamide dehydrogenase is a component of the glycine cleavage system as well as of the alpha-ketoacid dehydrogenase complexes. Related diseases Focal segmental glomerulosclerosis 2 (FSGS2) [MIM:603965]: A renal pathology defined by the presence of segmental sclerosis in glomeruli and resulting in proteinuria, reduced glomerular filtration rate and progressive decline in renal function. Renal insufficiency often progresses to end-stage renal disease, a highly morbid state requiring either dialysis therapy or kidney transplantation. {ECO:0000269|PubMed:15879175, ECO:0000269|PubMed:15924139, ECO:0000269|PubMed:19458060, ECO:0000269|PubMed:19936226, ECO:0000269|PubMed:20798252, ECO:0000269|PubMed:21511817, ECO:0000269|PubMed:21734084, ECO:0000269|PubMed:22732337, ECO:0000269|PubMed:23014460, ECO:0000269|PubMed:23291369, ECO:0000269|PubMed:26892346}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Disulfide bond; FAD; Flavoprotein; NAD; Oxidoreductase; Redox-active center Protein physicochemical properties Chain ID A,B Molecular weight (Da) 98183.9 Length 930 Aromaticity 0.06 Instability index 29.02 Isoelectric point 6.32 Charge (pH=7) -6.37 2D Binding mode Binding energy (Kcal/mol) -9.49  Molscript Map 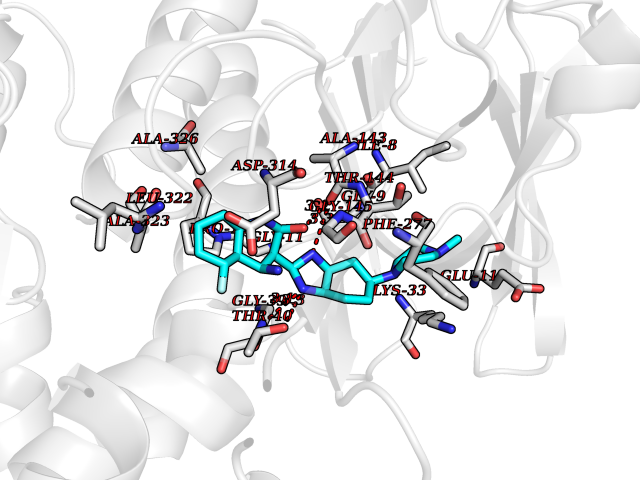 Pymol Map 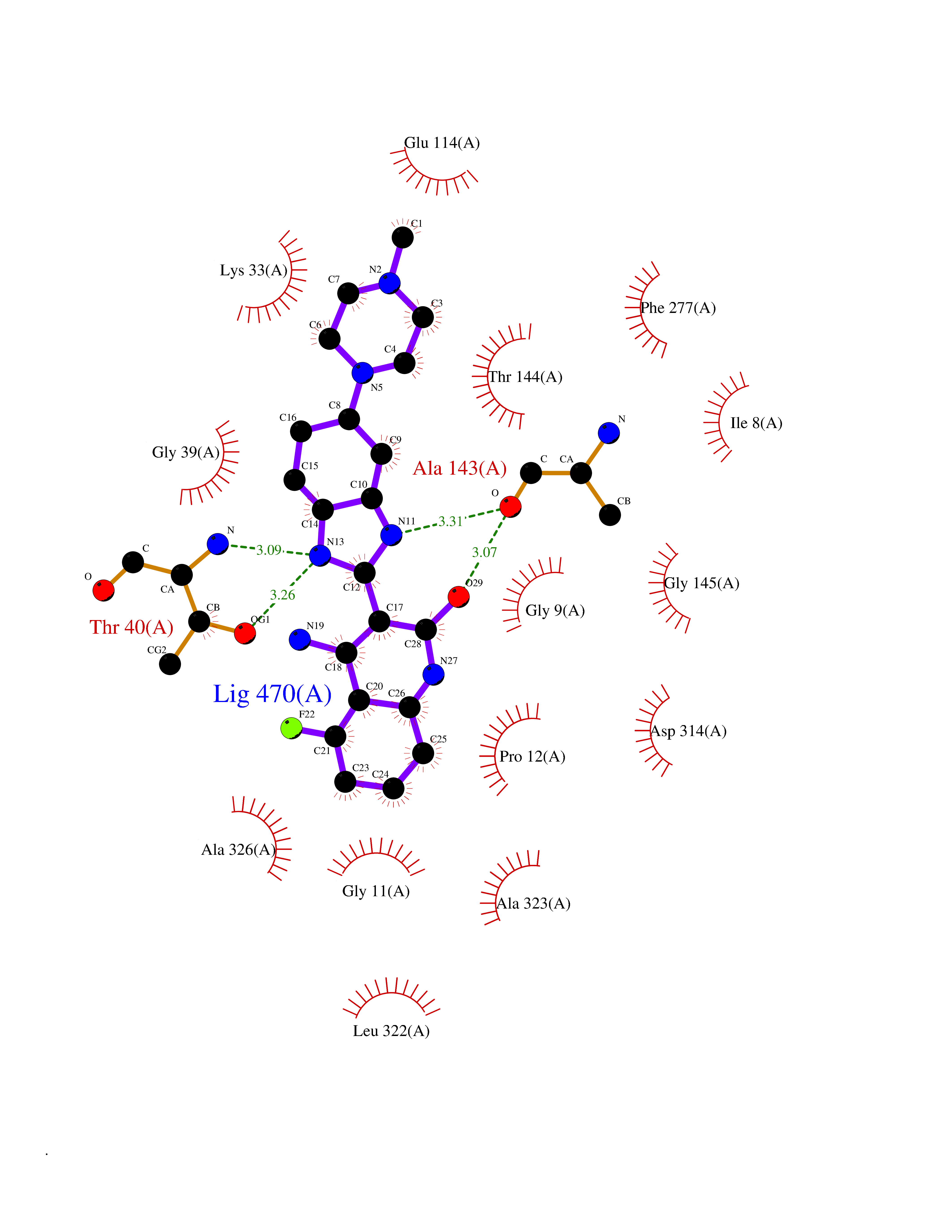 Ligplot Map 3D Binding mode Sequence NPYDVVVIGGGPGGYVASIKAAQLGMKTACVEKRGALGGTCLNVGCIPSKALLHATHLYHDAHANFARYGLMGGEGVTMDSAKMQQQKERAVKGLTGGVEYLFKKNKVTYYKGEGSFETAHSIRVNGLDGKQEMLETKKTIIATGSEPTELPFLPFDEKVVLSSTGALALPRVPKTMVVIGGGVIGLELGSVWARLGAEVTVVEFAPRCAPTLDEDVTNALVGALAKNEKMKFMTSTKVVGGTNNGDSVSLEVEGKRETVTCEALLVSVGRRPFTGGLGLDKINVAKNERGFVKIGDHFETSIPDVYAIGDVVDKGPMLAHKAEDEGVACAEILAGKPGHVNYGVIPAVIYTMPEVASVGKSEDELKKEGVAYKVGKFPFNANSRAKAVSTEDGFVKVLVDKATDRILGVHIVCTTAGELIGEACLAMEYGASSEDVGRTCHAHPTMSEALKEACMALFAKTINFNPYDVVVIGGGPGGYVASIKAAQLGMKTACVEKRGALGGTCLNVGCIPSKALLHATHLYHDAHANFARYGLMGGEGVTMDSAKMQQQKERAVKGLTGGVEYLFKKNKVTYYKGEGSFETAHSIRVNGLDGKQEMLETKKTIIATGSEPTELPFLPFDEKVVLSSTGALALPRVPKTMVVIGGGVIGLELGSVWARLGAEVTVVEFAPRCAPTLDEDVTNALVGALAKNEKMKFMTSTKVVGGTNNGDSVSLEVEGKRETVTCEALLVSVGRRPFTGGLGLDKINVAKNERGFVKIGDHFETSIPDVYAIGDVVDKGPMLAHKAEDEGVACAEILAGKPGHVNYGVIPAVIYTMPEVASVGKSEDELKKEGVAYKVGKFPFNANSRAKAVSTEDGFVKVLVDKATDRILGVHIVCTTAGELIGEACLAMEYGASSEDVGRTCHAHPTMSEALKEACMALFAKTINF Hydrogen bonds contact Hydrophobic contact | ||||
| 54 | Multidrug resistance protein 3 (ABCB4) | 6S7P | 8.05 | |
Target general information Gen name ABCB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PGY3; MDR3; ATP-binding cassette sub-family B member 4; ABCB4 Protein family ABC transporter superfamily, ABCB family, Multidrug resistance exporter (TC 3.A.1.201) subfamily Biochemical class Acid anhydrides hydrolase Function Mediates ATP-dependent export of organic anions and drugs from the cytoplasm. Hydrolyzes ATP with low efficiency. Not capable of conferring drug resistance. Mediates the translocation of phosphatidylcholine across the canalicular membrane of the hepatocyte. Related diseases Cholestasis, progressive familial intrahepatic, 3 (PFIC3) [MIM:602347]: A disorder characterized by early onset of cholestasis that progresses to hepatic fibrosis, cirrhosis, and end-stage liver disease before adulthood. PFIC3 inheritance is autosomal recessive. {ECO:0000269|PubMed:11313315, ECO:0000269|PubMed:12671900, ECO:0000269|PubMed:17726488, ECO:0000269|PubMed:21119540, ECO:0000269|PubMed:24045840, ECO:0000269|PubMed:24594635, ECO:0000269|PubMed:24806754, ECO:0000269|PubMed:28012258, ECO:0000269|PubMed:9419367}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cholestasis of pregnancy, intrahepatic 3 (ICP3) [MIM:614972]: A liver disorder of pregnancy. It presents during the second or, more commonly, the third trimester of pregnancy with intense pruritus which becomes more severe with advancing gestation and cholestasis. It causes fetal distress, spontaneous premature delivery and intrauterine death. Patients have spontaneous and progressive disappearance of cholestasis after delivery. Cholestasis results from abnormal biliary transport from the liver into the small intestine. {ECO:0000269|PubMed:10767346, ECO:0000269|PubMed:12746424, ECO:0000269|PubMed:15077010}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gallbladder disease 1 (GBD1) [MIM:600803]: One of the major digestive diseases. Gallstones composed of cholesterol (cholelithiasis) are the common manifestations in western countries. Most people with gallstones, however, remain asymptomatic through their lifetimes. {ECO:0000269|PubMed:11313316, ECO:0000269|PubMed:12891548, ECO:0000269|PubMed:22331132, ECO:0000269|PubMed:23533021, ECO:0000269|PubMed:24723470, ECO:0000269|PubMed:28012258, ECO:0000269|PubMed:28587926, ECO:0000269|Ref.2}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06414; DB06207 Interacts with NA EC number EC 7.6.2.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cytoplasm; Cytoplasmic vesicle; Disease variant; Glycoprotein; Intrahepatic cholestasis; Lipid transport; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 123919 Length 1128 Aromaticity 0.1 Instability index 29.44 Isoelectric point 8.78 Charge (pH=7) 11.08 2D Binding mode Binding energy (Kcal/mol) -8.93 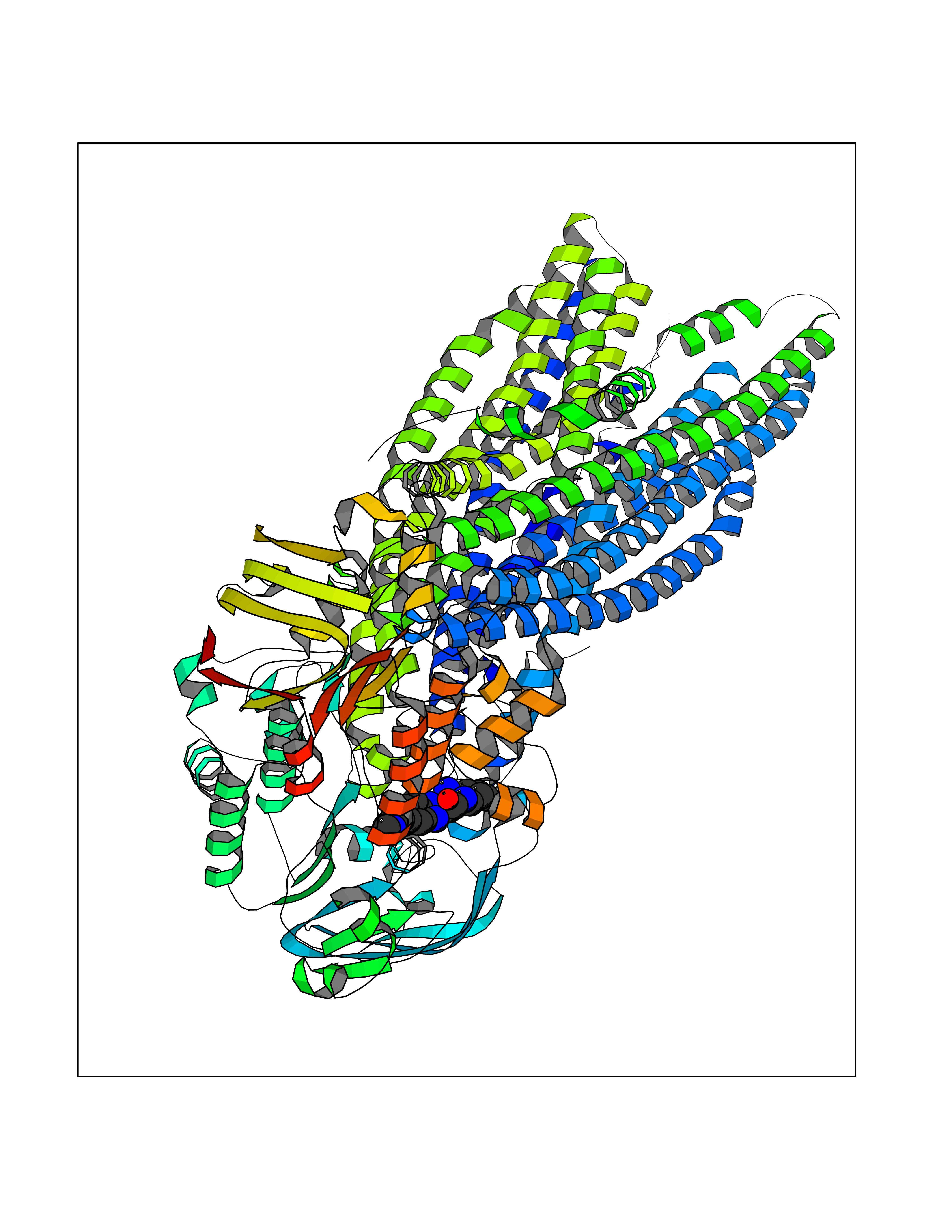 Molscript Map 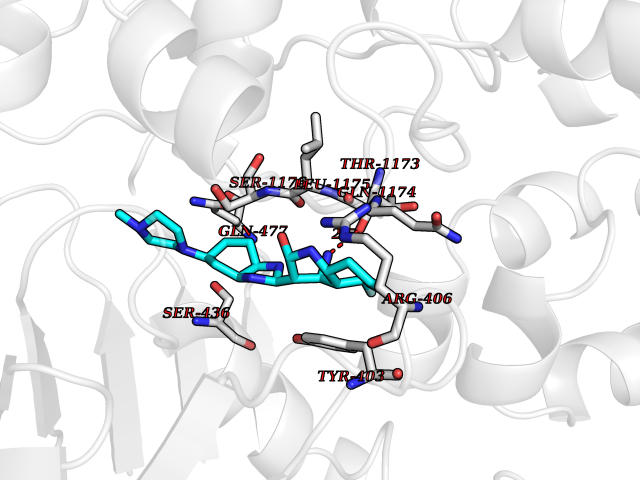 Pymol Map 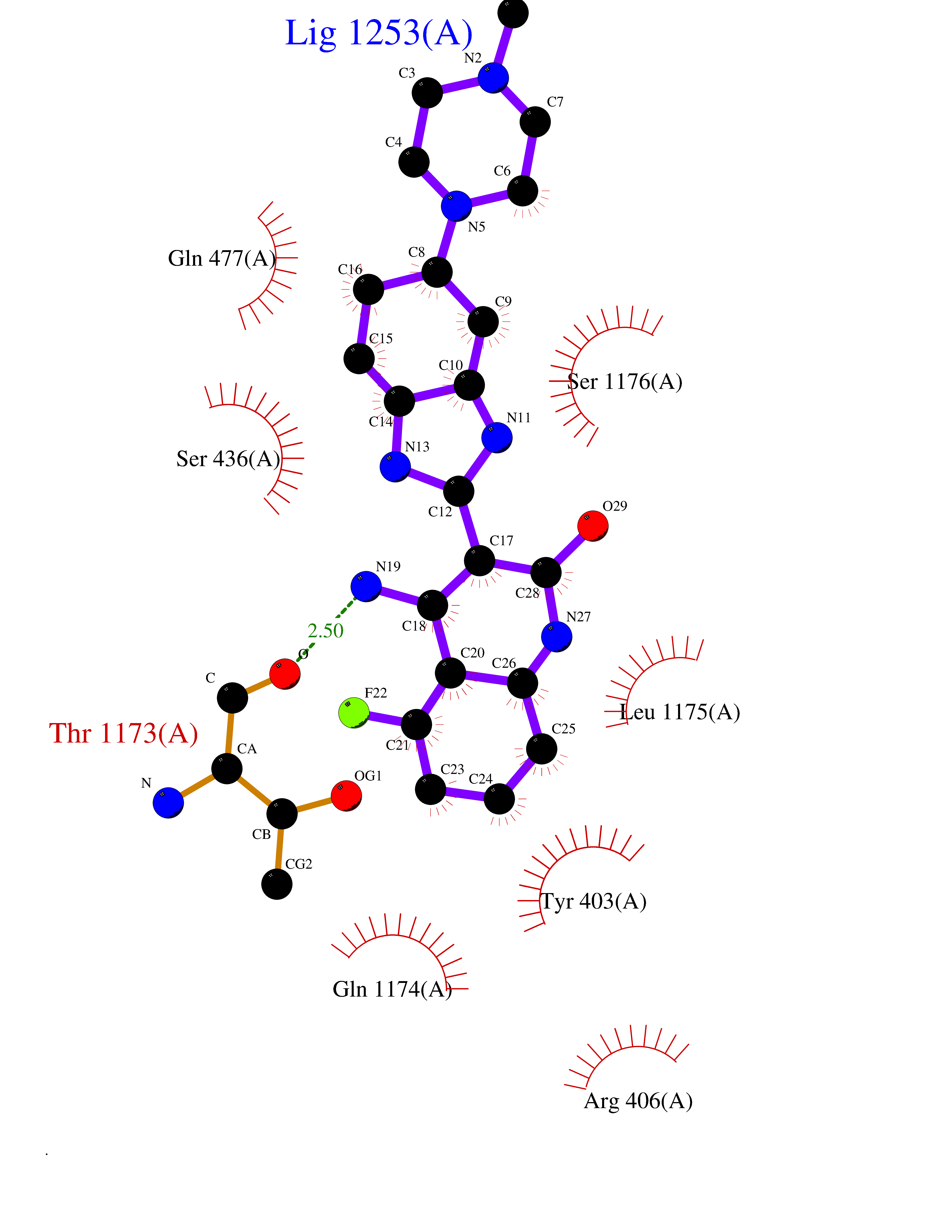 Ligplot Map 3D Binding mode Sequence VLTLFRYSDWQDKLFMSLGTIMAIAHGSGLPLMMIVFGEMTDKPGKILEEEMTRYAYYYSGLGAGVLVAAYIQVSFWTLAAGRQIRKIRQKFFHAILRQEIGWFDINDTTELNTRLTDDISKISEGIGDKVGMFFQAVATFFAGFIVGFIRGWKLTLVIMAISPILGLSAAVWAKILSAFSDKELAAYAKAGAVAEEALGAIRTVIAFGGQNKELERYQKHLENAKEIGIKKAISANISMGIAFLLIYASYALAFWYGSTLVISKEYTIGNAMTVFFSILIGAFSVGQAAPCIDAFANARGAAYVIFDIIDNNPKIDSFSERGHKPDSIKGNLEFNDVHFSYPSRANVKILKGLNLKVQSGQTVALVGSSGCGKSTTVQLIQRLYDPDEGTINIDGQDIRNFNVNYLREIIGVVSQEPVLFSTTIAENICYGRGNVTMDEIKKAVKEANAYEFIMKLPQKFDTLVGERGAQLSGGQKQRIAIARALVRNPKILLLDQATSALDTESEAEVQAALDKAREGRTTIVIAHRLSTVRNADVIAGFEDGVIVEQGSHSELMKKEGVYFKLVNVPPVSFLKVLKLNKTEWPYFVVGTVCAIANGGLQPAFSVIFSEIIAIFGPGDDAVKQQKCNIFSLIFLFLGIISFFTFFLQGFTFGKAGEILTRRLRSMAFKAMLRQDMSWFDDHKNSTGALSTRLATDAAQVQGATGTRLALIAQNIANLGTGIIISFIYGWQLTLLLLAVVPIIAVSGIVEMKLLAGNAKRDKKELEAAGKIATEAIENIRTVVSLTQERKFESMYVEKLYGPYRNSVQKAHIYGITFSISQAFMYFSYAGCFRFGAYLIVNGHMRFRDVILVFSAIVFGAVALGHASSFAPDYAKAKLSAAHLFMLFERQPLIDSYSEEGLKPDKFEGNITFNEVVFNYPTRANVPVLQGLSLEVKKGQTLALVGSSGCGKSTVVQLLERFYDPLAGTVLLDGQEAKKLNVQWLRAQLGIVSQEPILFDCSIAENIAYGDNSRVVSQDEIVSAAKAANIHPFIETLPHKYETRVGDKGTQLSGGQKQRIAIARALIRQPQILLLDQATSALDTESEKVVQEALDKAREGRTCIVIAHRLSTIQNADLIVVFQNGRVK Hydrogen bonds contact Hydrophobic contact | ||||
| 55 | Oxygen-insensitive NADPH nitroreductase | 3QDL | 8.05 | |
Target general information Gen name rdxA Organism Helicobacter pylori (strain ATCC 700392 / 26695) (Campylobacter pylori) Uniprot ID TTD ID NA Synonyms HP_0954 Protein family Nitroreductase family Biochemical class Oxidoreductase Function Oxidoreductase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB00916 Interacts with NA EC number 1.-.-.- Uniprot keywords 3D-structure; Antibiotic resistance; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 40094.3 Length 352 Aromaticity 0.08 Instability index 55.15 Isoelectric point 6.72 Charge (pH=7) -0.86 2D Binding mode Binding energy (Kcal/mol) -8.21 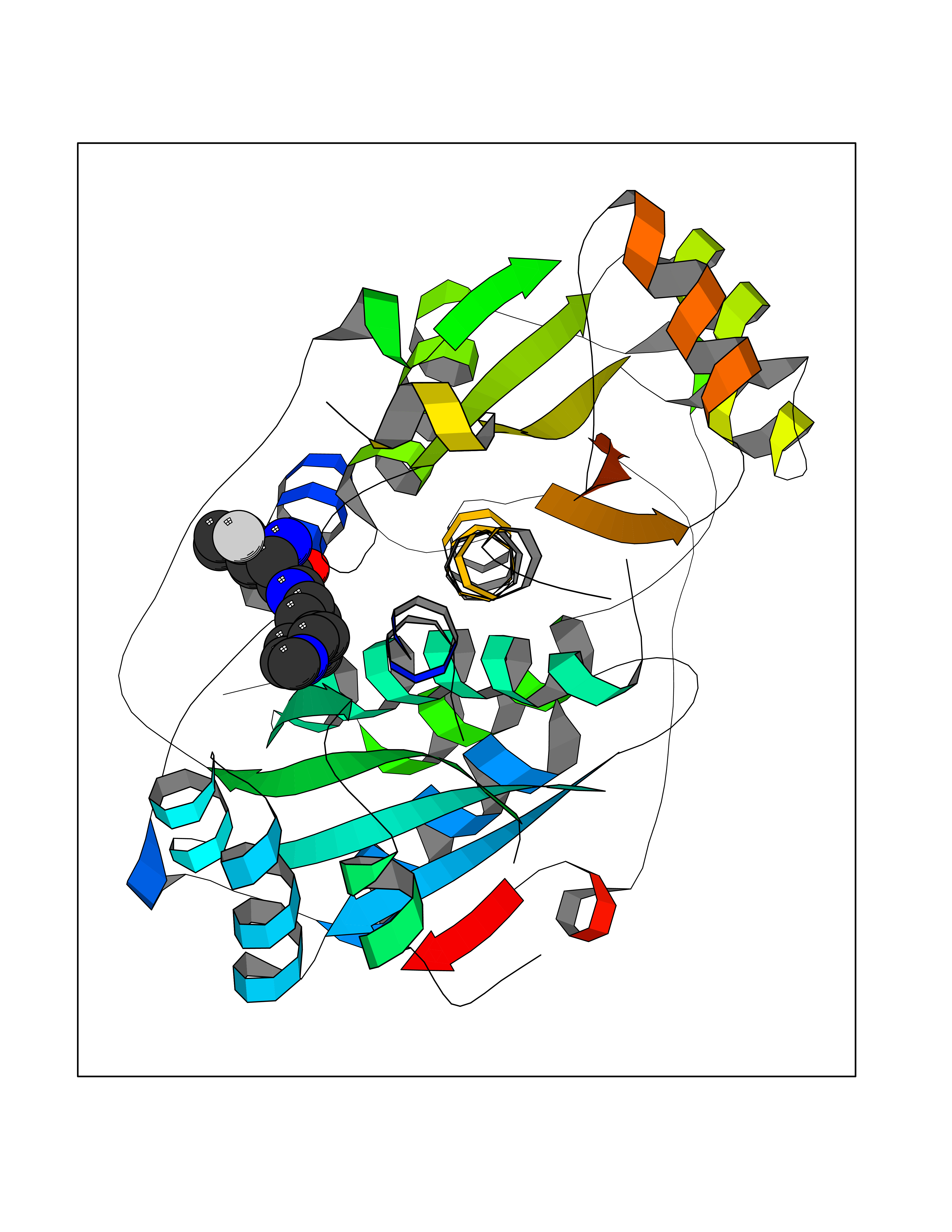 Molscript Map 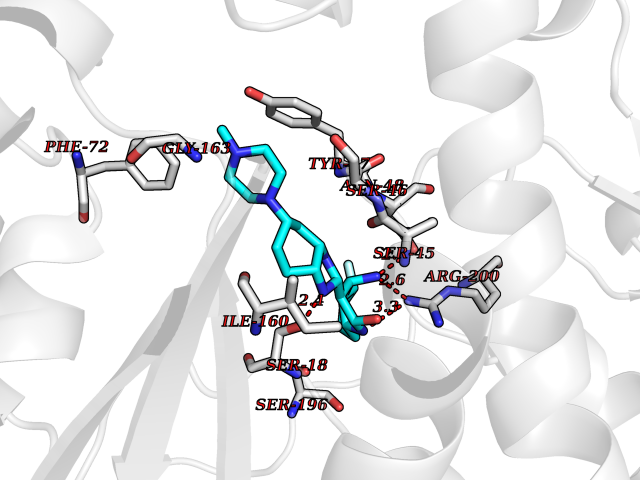 Pymol Map 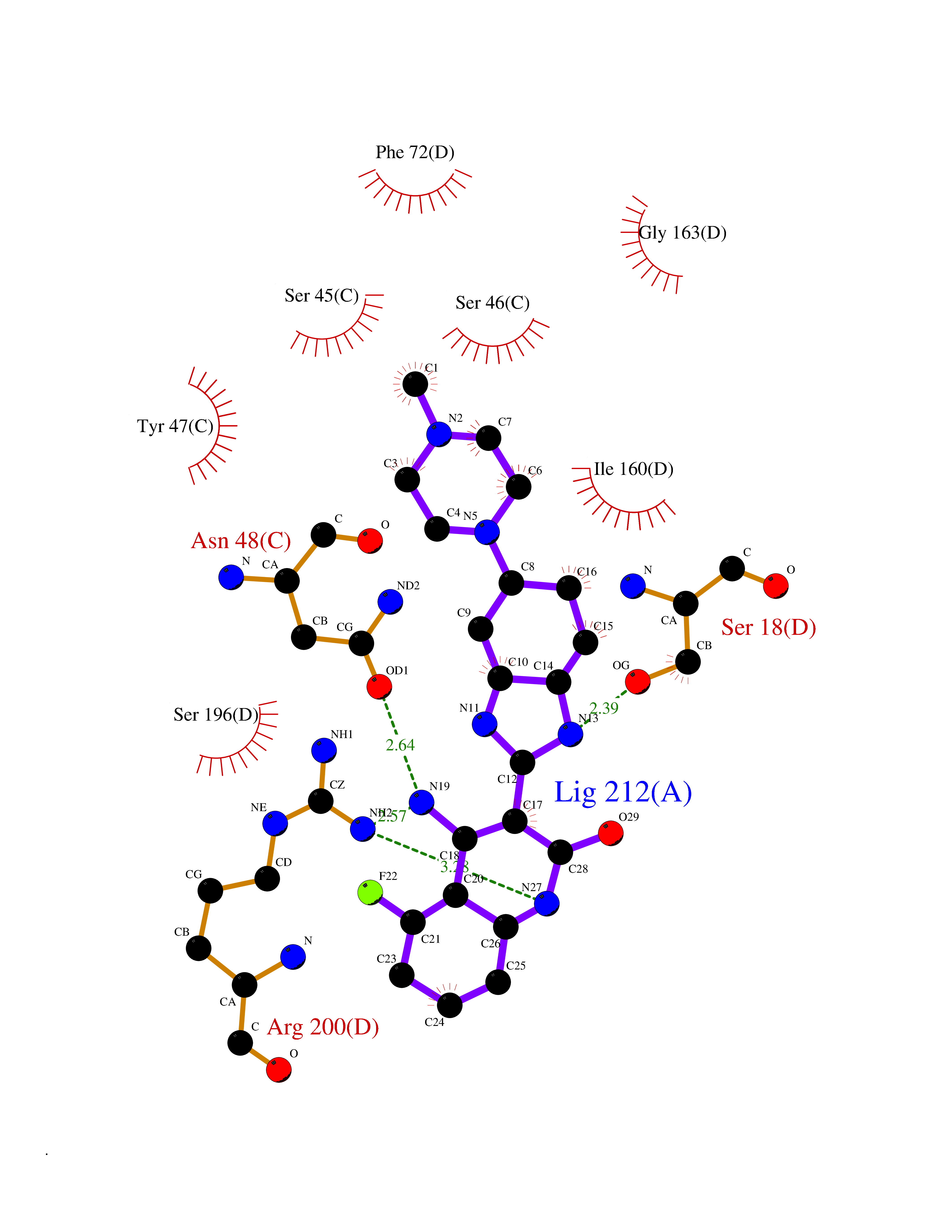 Ligplot Map 3D Binding mode Sequence MQRLESYILMKFLDQEKRRQLLNERHSCKMFDSHYEFSSTELEEIAEIARLSPSSYNTQPWHFVMVTDKDLKKQIAAHSYFNEEMIKSASALMVVCSLSYILEQCYIAVGQICMGVSLMGLDSCIIGGFDPLKVGEVLEERINPKIACLIALGKRVAEASQKSRKSKVDAITWLMKFLDQEKRRQLLNERHSCKMFDSHYEFSSTELEEIAEIARLSPSSYNTQPWHFVMVTDKDLKKQIAAHSYFNEEMIKSASALMVVCSLRPSELLPMQRLESYILEQCYIAVGQICMGVSLMGLDSCIIGGFDPLKVGEVLEERINKPKIACLIALGKRVAEASQKSRKSKVDAITWL Hydrogen bonds contact Hydrophobic contact | ||||
| 56 | Phosphodiesterase 4A (PDE4A) | 2QYK | 8.04 | |
Target general information Gen name PDE4A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-specific 3',5'-cyclic phosphodiesterase 4A; Type 4A cAMP phosphodiesterase; PDE46; DPDE2 Protein family Cyclic nucleotide phosphodiesterase family, PDE4 subfamily Biochemical class Phosphoric diester hydrolase Function Hydrolyzes the second messenger cAMP, which is a key regulator of many important physiological processes. Related diseases Developmental and epileptic encephalopathy 50 (DEE50) [MIM:616457]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE50 is an autosomal recessive, progressive disease with onset in infancy and favorable response to treatment with oral uridine. {ECO:0000269|PubMed:25678555, ECO:0000269|PubMed:28087732}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06842; DB08299; DB01427; DB00201; DB05219; DB00975; DB06751; DB00651; DB00824; DB05266; DB01088; DB01303; DB01791; DB06479; DB01656; DB01954; DB00277; DB08811; DB09283 Interacts with P55212; O14569; P13473-2; Q9UJX0; P16118; O75400-2; Q9Y371 EC number EC 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cell membrane; Cell projection; Cytoplasm; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 38579.6 Length 335 Aromaticity 0.08 Instability index 37.43 Isoelectric point 5.01 Charge (pH=7) -19.92 2D Binding mode Binding energy (Kcal/mol) -8.85 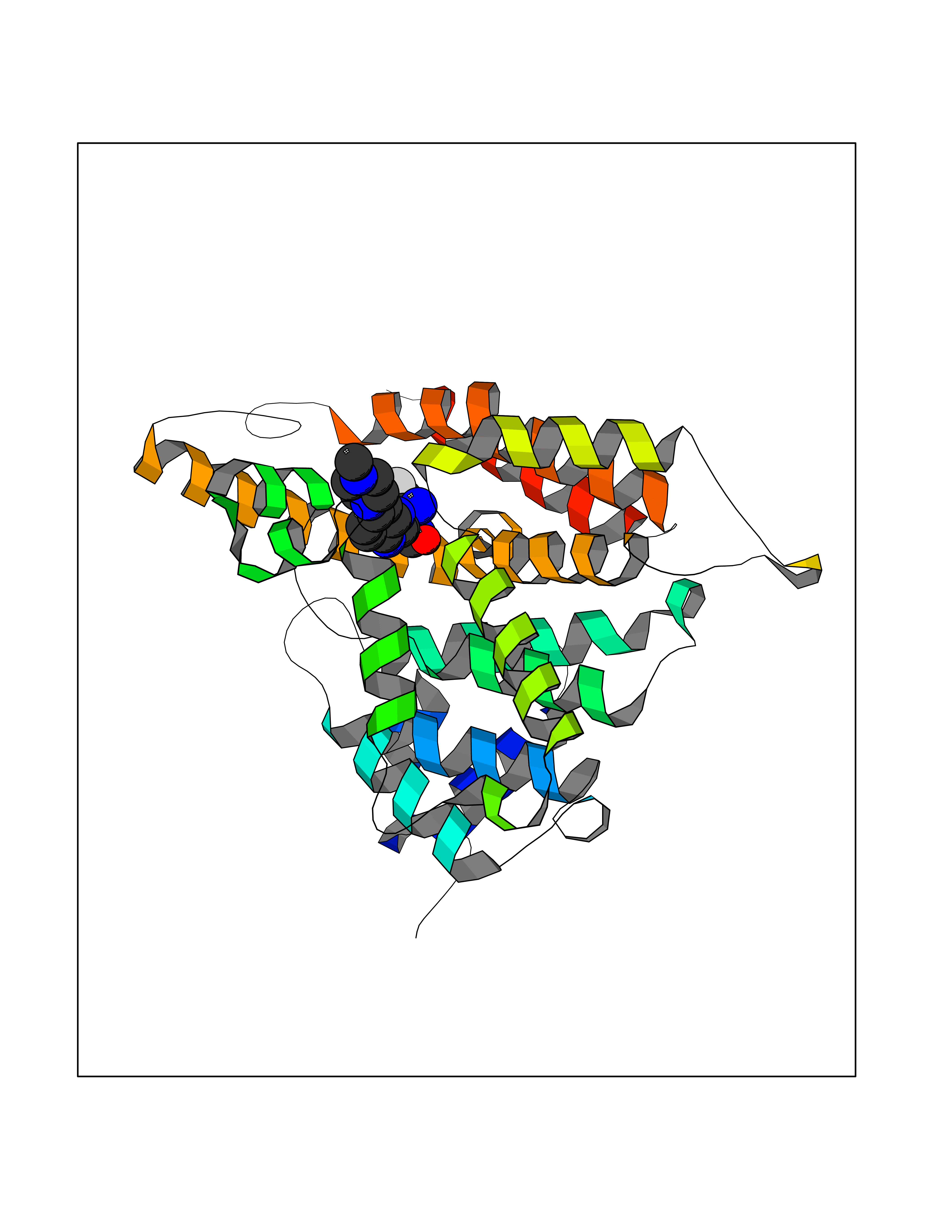 Molscript Map 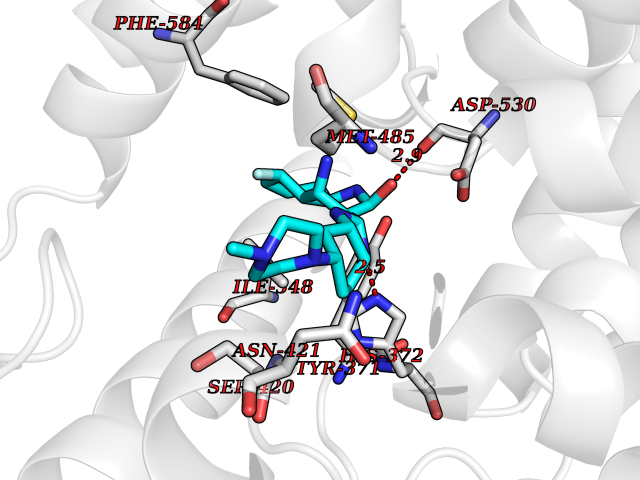 Pymol Map 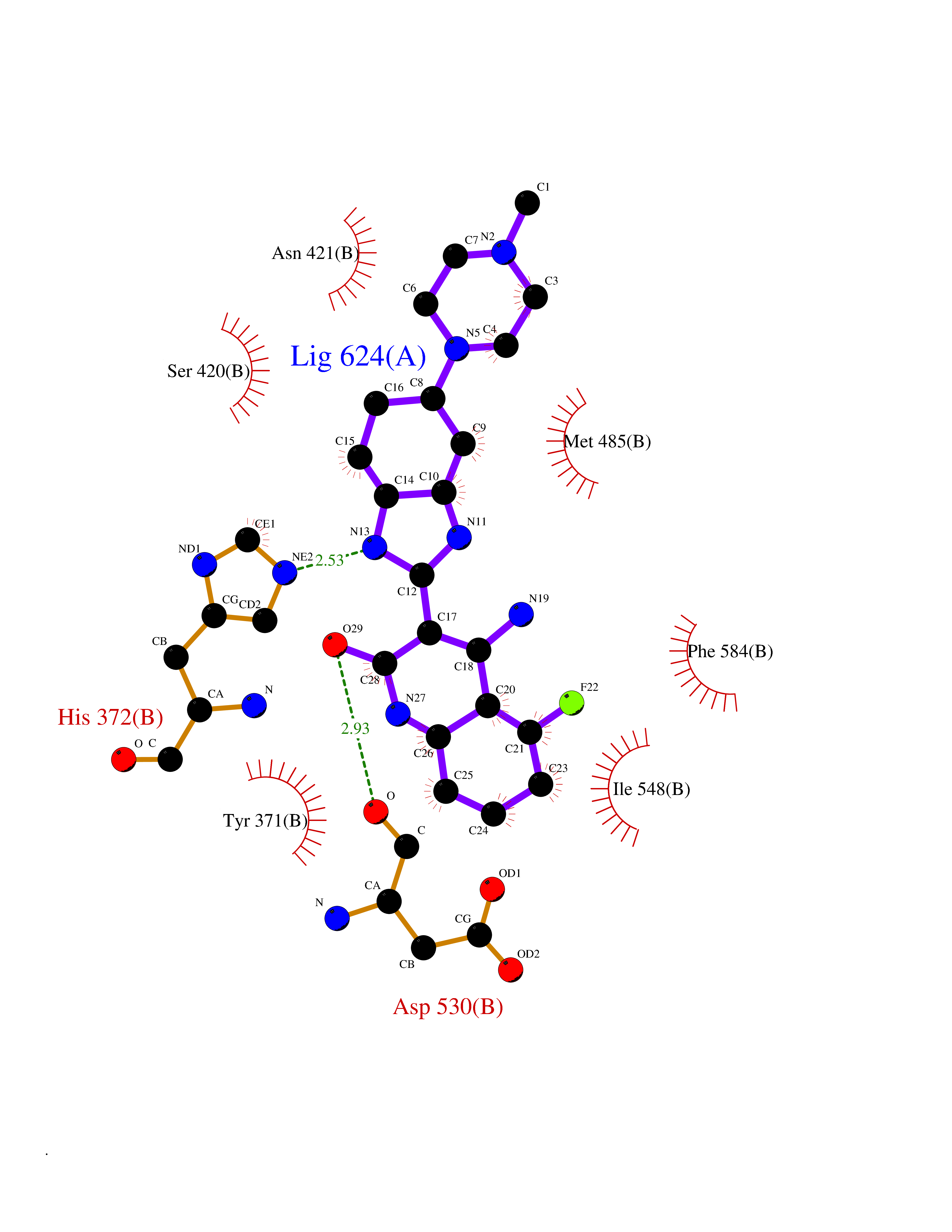 Ligplot Map 3D Binding mode Sequence HMNIPRFGVKTDQEELLAQELENLNKWGLNIFCVSDYAGGRSLTCIMYMIFQERDLLKKFRIPVDTMVTYMLTLEDHYHADVAYHNSLHAADVLQSTHVLLATPALDAVFTDLEILAALFAAAIHDVDHPGVSNQFLINTNSELALMYNDESVLENHHLAVGFKLLQEDNCDIFQNLSKRQRQSLRKMVIDMVLATDMSKHMTLLADLKTMVETKKVTSSGVLLLDNYSDRIQVLRNMVHCADLSNPTKPLELYRQWTDRIMAEFFQQGDRERERGMEISPMCDKHTASVEKSQVGFIDYIVHPLWETWADLVHPDAQEILDTLEDNRDWYYSAI Hydrogen bonds contact Hydrophobic contact | ||||
| 57 | Extracellular lysophospholipase D (E-NPP2) | 4ZGA | 8.04 | |
Target general information Gen name ENPP2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LysoPLD; Ectonucleotide pyrophosphatase/phosphodiesterase family member 2; E-NPP 2; Autotaxin; ATX Protein family Nucleotide pyrophosphatase/phosphodiesterase family Biochemical class Phosphoric diester hydrolase Function Hydrolyzes lysophospholipids to produce the signaling molecule lysophosphatidic acid (LPA) in extracellular fluids. Major substrate is lysophosphatidylcholine. Also can act on sphingosylphosphorylcholine producing sphingosine-1-phosphate, a modulator of cell motility. Can hydrolyze, in vitro, bis-pNPP, to some extent pNP-TMP, and barely ATP. Involved in several motility-related processes such as angiogenesis and neurite outgrowth. Acts as an angiogenic factor by stimulating migration of smooth muscle cells and microtubule formation. Stimulates migration of melanoma cells, probably via a pertussis toxin-sensitive G protein. May have a role in induction of parturition. Possible involvement in cell proliferation and adipose tissue development (Probable). Tumor cell motility-stimulating factor. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.1.4.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Chemotaxis; Cleavage on pair of basic residues; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lipid degradation; Lipid metabolism; Metal-binding; Obesity; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 85397.4 Length 740 Aromaticity 0.12 Instability index 58.63 Isoelectric point 7.16 Charge (pH=7) 1.02 2D Binding mode Binding energy (Kcal/mol) -8.78 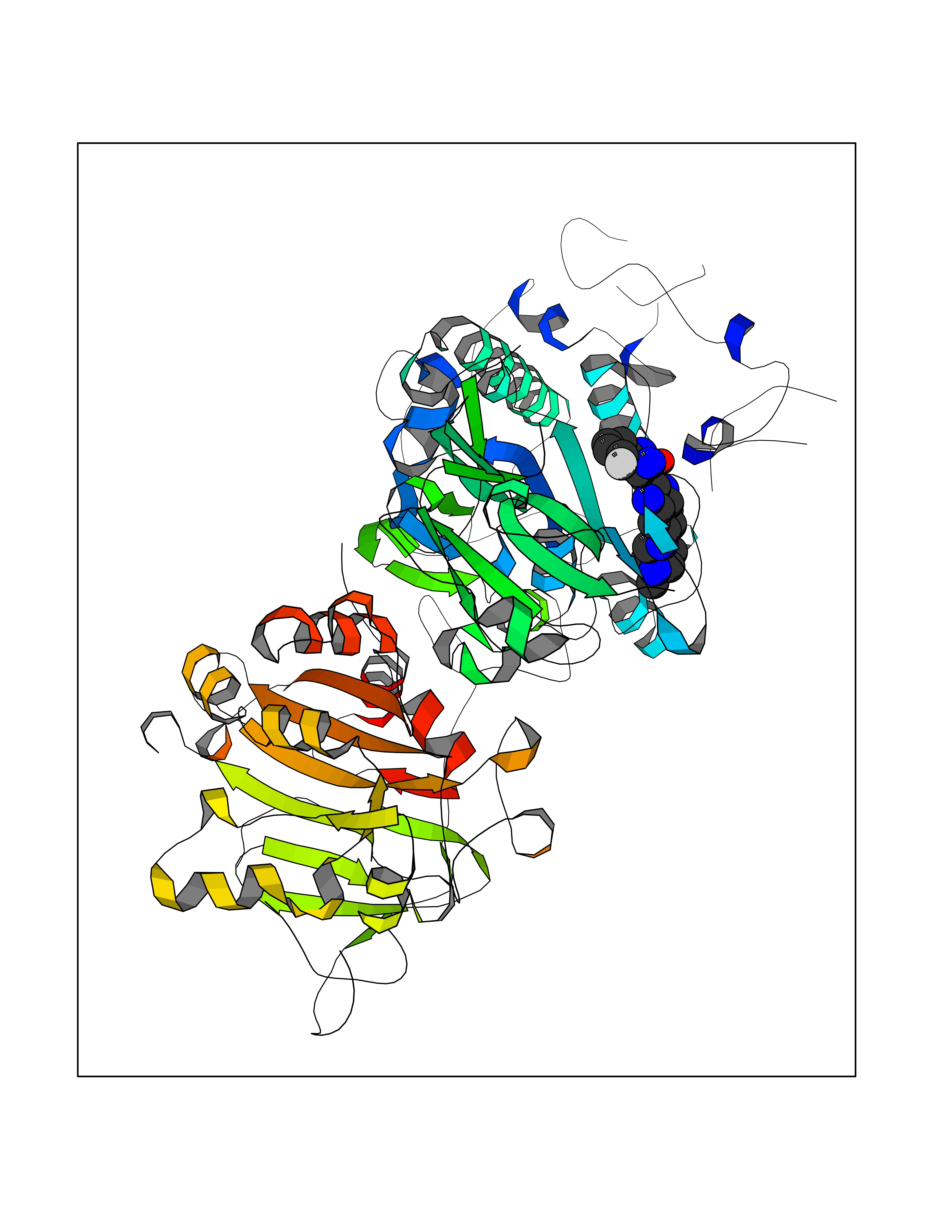 Molscript Map 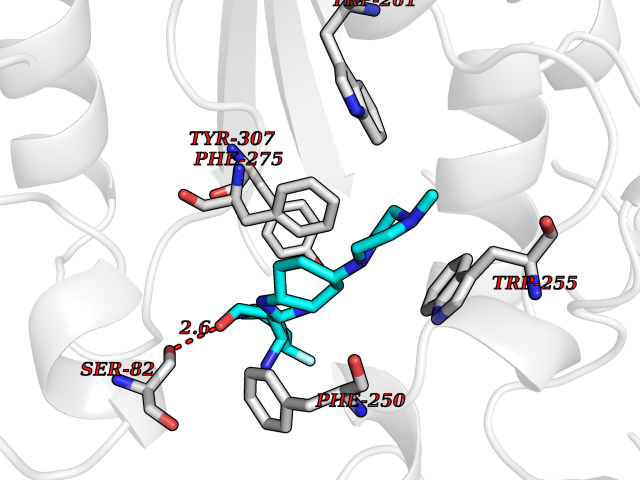 Pymol Map 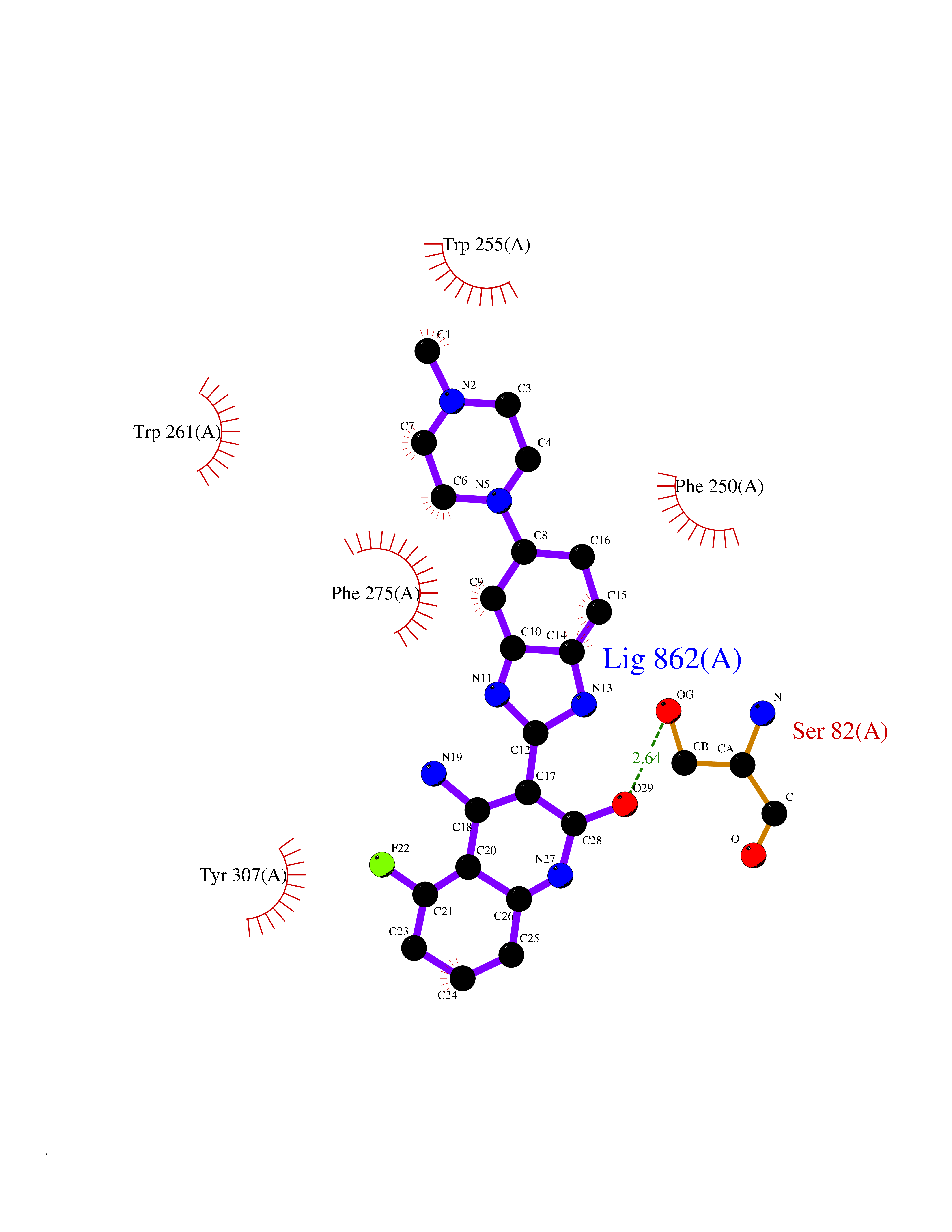 Ligplot Map 3D Binding mode Sequence GSCKGRCFELCRCDNLCKSYTSCCHDFDELCLKTARGWECTKDRCGNEENACHCEDCLARGDCCTNYQVVCKGESHWVDDDCEEIKAAECPAGFVRPPLIIFSVDGFRASYMKKGSKVMPNIEKLRSCGTHSPYMRPVYPTKTFPNLYTLATGLYPESHGIVGNSMYDPVFDATFHLRGREKFNHRWWGGQPLWITATKQGVKAGTFFWSVVIPHERRILTILQWLTLPDHERPSVYAFYSEQPDFSGHKYGPFGPEMTNPLREIDKIVGQLMDGLKQLKLHRCVNVIFVGDHGMEDVTCDRTEFLSNYLTNVDDITLVPGTLGRIRSKFDPKAIIANLTCKKPDQHFKPYLKQHLPKRLHYANNRRIEDIHLLVERRWHVARKPFFQGDHGFDNKVNSMQTVFVGYGSTFKYKTKVPPFENIELYNVMCDLLGLKPAPNNGTHGSLNHLLRTNTFRPTMPEEVTRPNYPGIMYLQSDFDLGTEERHLLYGRPAVLYRTRYDILYHTDFESGYSEIFLMPLWTSYTVSKQACVRPDVRVSPSFSQNCLAYKNDKQMSYGFLFPPYLSSSPEAKYDAFLVTNMVPMYPAFKRVWNYFQRVLVKKYASERNGVNVISGPIFDYDYDGLHDTEDKIKQYVEGSSIPVPTHYYSIITSCLDFTQPADKCDGPLSVSSFILPHRPDNEESCNSSEDESKWVEELMKMHTARVRDIEHLTSLDFFRKTSRSYPEILTLKTYLHTYE Hydrogen bonds contact Hydrophobic contact | ||||
| 58 | Somatostatin receptor type 4 (SSTR4) | 7XMT | 8.03 | |
Target general information Gen name SSTR4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SSTR4; SS4R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for somatostatin-14. The activity of this receptor is mediated by G proteins which inhibits adenylyl cyclase. It is functionally coupled not only to inhibition of adenylate cyclase, but also to activation of both arachidonate release and mitogen-activated protein (MAP) kinase cascade. Mediates antiproliferative action of somatostatin in tumor cells. Related diseases Oocyte/zygote/embryo maturation arrest 21 (OZEMA21) [MIM:620610]: An autosomal dominant, female infertility disorder characterized by zygote development arrest due to failure of pronuclei fusion. {ECO:0000269|PubMed:33948904, ECO:0000269|PubMed:33953335}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB13985; DB09099 Interacts with P35346 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 29548.3 Length 265 Aromaticity 0.12 Instability index 45.65 Isoelectric point 9.78 Charge (pH=7) 14.03 2D Binding mode Binding energy (Kcal/mol) -8.22 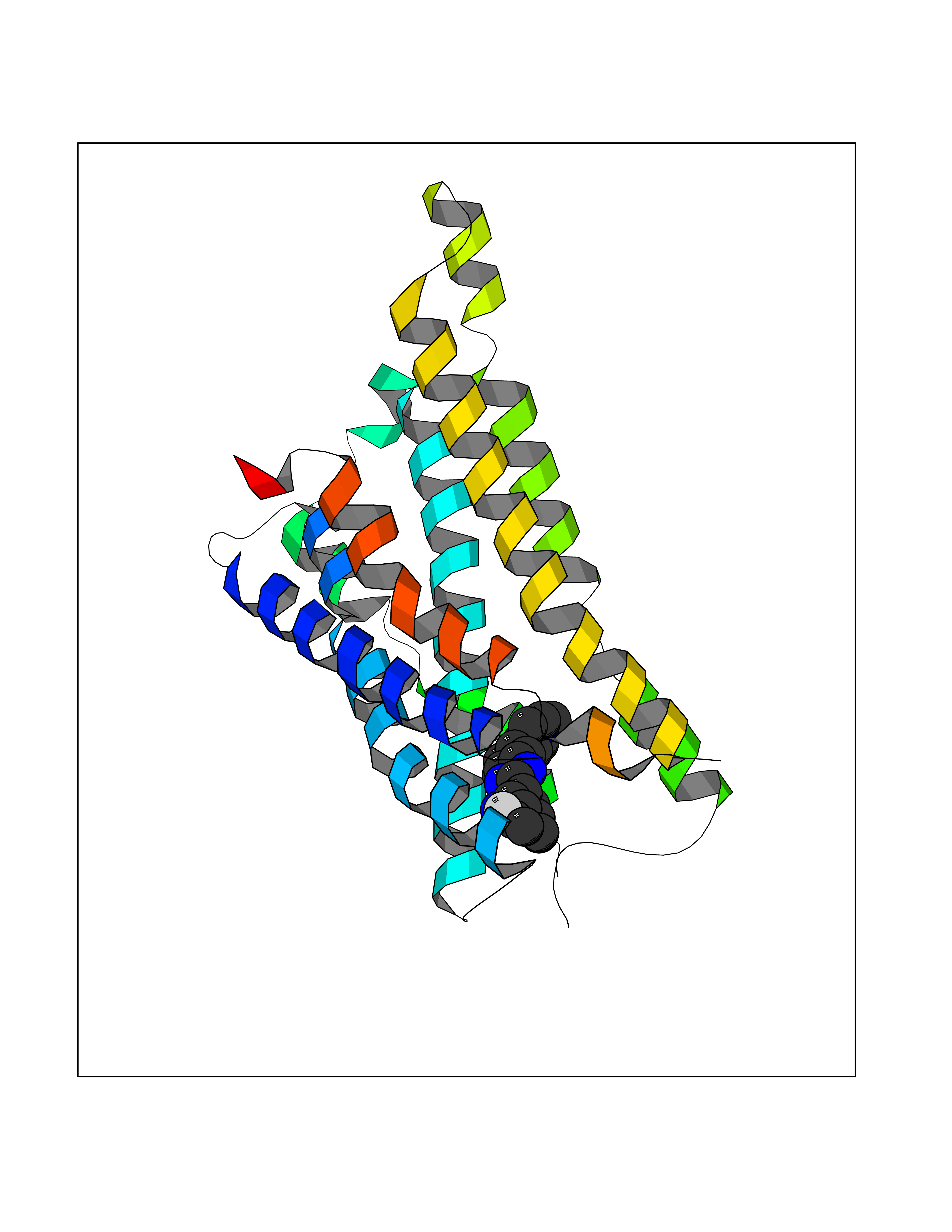 Molscript Map 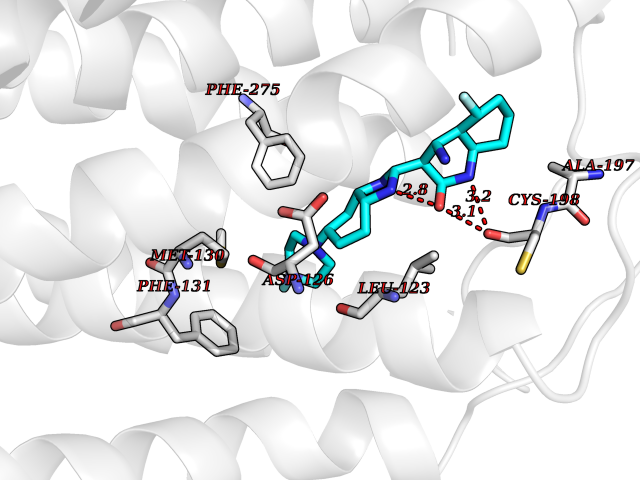 Pymol Map 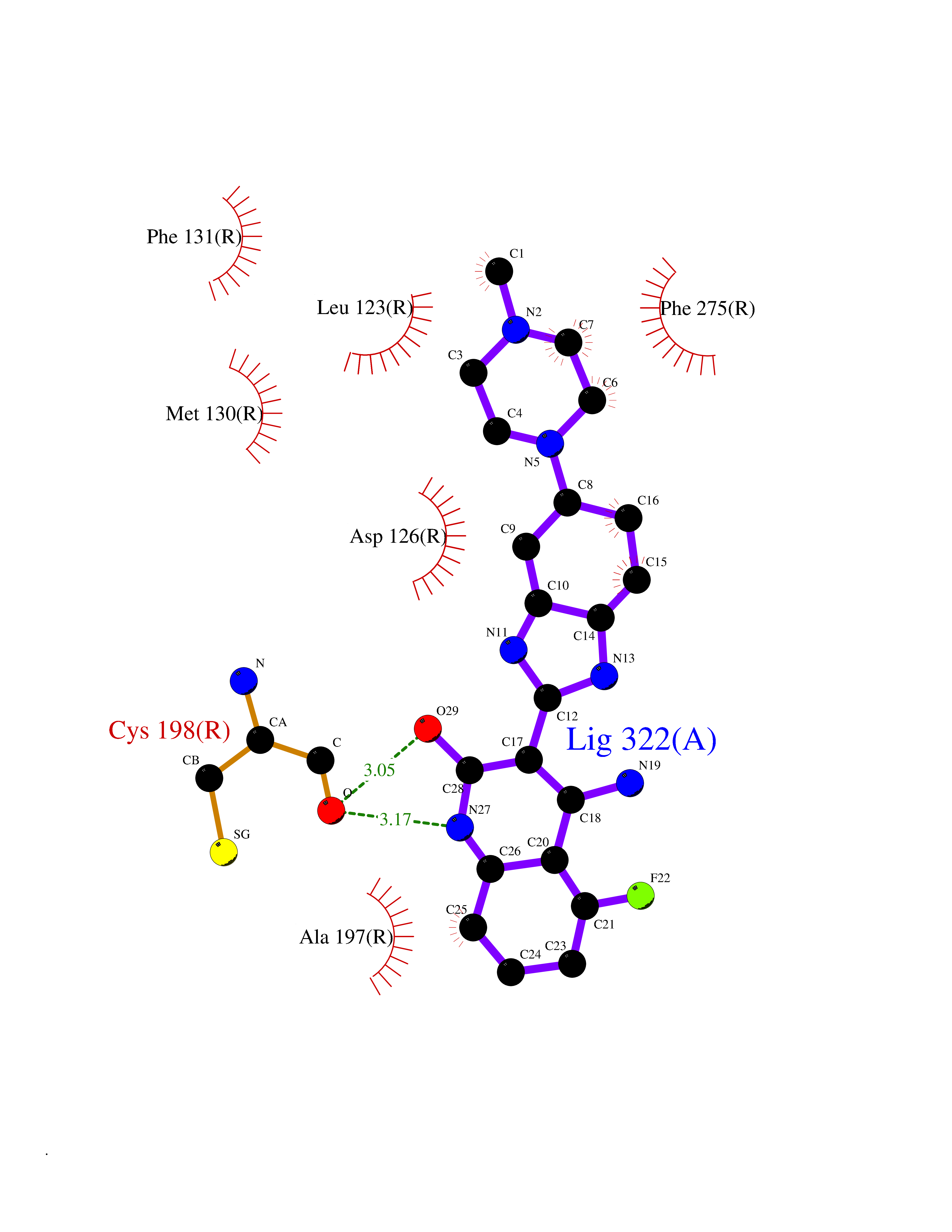 Ligplot Map 3D Binding mode Sequence GMVAIQCIYALVCLVGLVGNALVIFVILRYAKMKTATNIYLLNLAVADELFMLSVPFVASSAALRHWPFGSVLCRAVLSVDGLNMFTSVFCLTVLSVDRYVAVVHPLRAATYRRPSVAKLINLGVWLASLLVTLPIAIFADTRPACNLQWPHPAWSAVFVVYTFLLGFLLPVLAIGLCYLLIVGKMRAVALRAGWQQRRRSEKKITRLVLMFVVVFVLCWMPFYVVQLLNLFLDATVNHVSLILSYANSCANPILYGFLSDNFRR Hydrogen bonds contact Hydrophobic contact | ||||
| 59 | Protein kinase C epsilon (PRKCE) | 5LIH | 8.02 | |
Target general information Gen name PRKCE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein kinase C epsilon type; PKCE; PKC epsilon; NPKC-epsilon Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, PKC subfamily Biochemical class Kinase Function Mediates cell adhesion to the extracellular matrix via integrin-dependent signaling, by mediating angiotensin-2-induced activation of integrin beta-1 (ITGB1) in cardiac fibroblasts. Phosphorylates MARCKS, which phosphorylates and activates PTK2/FAK, leading to the spread of cardiomyocytes. Involved in the control of the directional transport of ITGB1 in mesenchymal cells by phosphorylating vimentin (VIM), an intermediate filament (IF) protein. In epithelial cells, associates with and phosphorylates keratin-8 (KRT8), which induces targeting of desmoplakin at desmosomes and regulates cell-cell contact. Phosphorylates IQGAP1, which binds to CDC42, mediating epithelial cell-cell detachment prior to migration. In HeLa cells, contributes to hepatocyte growth factor (HGF)-induced cell migration, and in human corneal epithelial cells, plays a critical role in wound healing after activation by HGF. During cytokinesis, forms a complex with YWHAB, which is crucial for daughter cell separation, and facilitates abscission by a mechanism which may implicate the regulation of RHOA. In cardiac myocytes, regulates myofilament function and excitation coupling at the Z-lines, where it is indirectly associated with F-actin via interaction with COPB1. During endothelin-induced cardiomyocyte hypertrophy, mediates activation of PTK2/FAK, which is critical for cardiomyocyte survival and regulation of sarcomere length. Plays a role in the pathogenesis of dilated cardiomyopathy via persistent phosphorylation of troponin I (TNNI3). Involved in nerve growth factor (NFG)-induced neurite outgrowth and neuron morphological change independently of its kinase activity, by inhibition of RHOA pathway, activation of CDC42 and cytoskeletal rearrangement. May be involved in presynaptic facilitation by mediating phorbol ester-induced synaptic potentiation. Phosphorylates gamma-aminobutyric acid receptor subunit gamma-2 (GABRG2), which reduces the response of GABA receptors to ethanol and benzodiazepines and may mediate acute tolerance to the intoxicating effects of ethanol. Upon PMA treatment, phosphorylates the capsaicin- and heat-activated cation channel TRPV1, which is required for bradykinin-induced sensitization of the heat response in nociceptive neurons. Is able to form a complex with PDLIM5 and N-type calcium channel, and may enhance channel activities and potentiates fast synaptic transmission by phosphorylating the pore-forming alpha subunit CACNA1B (CaV2. 2). In prostate cancer cells, interacts with and phosphorylates STAT3, which increases DNA-binding and transcriptional activity of STAT3 and seems to be essential for prostate cancer cell invasion. Downstream of TLR4, plays an important role in the lipopolysaccharide (LPS)-induced immune response by phosphorylating and activating TICAM2/TRAM, which in turn activates the transcription factor IRF3 and subsequent cytokines production. In differentiating erythroid progenitors, is regulated by EPO and controls the protection against the TNFSF10/TRAIL-mediated apoptosis, via BCL2. May be involved in the regulation of the insulin-induced phosphorylation and activation of AKT1. Calcium-independent, phospholipid- and diacylglycerol (DAG)-dependent serine/threonine-protein kinase that plays essential roles in the regulation of multiple cellular processes linked to cytoskeletal proteins, such as cell adhesion, motility, migration and cell cycle, functions in neuron growth and ion channel regulation, and is involved in immune response, cancer cell invasion and regulation of apoptosis. Related diseases Hyperinsulinemic hypoglycemia, familial, 2 (HHF2) [MIM:601820]: A form of hyperinsulinemic hypoglycemia, a clinically and genetically heterogeneous disorder characterized by inappropriate insulin secretion from the pancreatic beta-cells in the presence of low blood glucose levels. HHF2 is a common cause of persistent hypoglycemia in infancy. Unless early and aggressive intervention is undertaken, brain damage from recurrent episodes of hypoglycemia may occur. HHF2 inheritance can be autosomal dominant or autosomal recessive. {ECO:0000269|PubMed:10204114, ECO:0000269|PubMed:12364426, ECO:0000269|PubMed:15562009, ECO:0000269|PubMed:15579781, ECO:0000269|PubMed:15807877, ECO:0000269|PubMed:15998776, ECO:0000269|PubMed:16332676, ECO:0000269|PubMed:16357843, ECO:0000269|PubMed:18596924, ECO:0000269|PubMed:19357197, ECO:0000269|PubMed:7847376, ECO:0000269|PubMed:8923010}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Diabetes mellitus, permanent neonatal, 2 (PNDM2) [MIM:618856]: A form of permanent neonatal diabetes mellitus, a type of diabetes characterized by onset of persistent hyperglycemia within the first six months of life. Initial clinical manifestations include intrauterine growth retardation, hyperglycemia, glycosuria, osmotic polyuria, severe dehydration, and failure to thrive. Some PNDM2 patients may also have developmental delay, muscle weakness, epilepsy and dysmorphic features. PNDM2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:15115830, ECO:0000269|PubMed:15292329, ECO:0000269|PubMed:15448106, ECO:0000269|PubMed:15448107, ECO:0000269|PubMed:15580558, ECO:0000269|PubMed:15583126, ECO:0000269|PubMed:16609879, ECO:0000269|PubMed:16731833, ECO:0000269|PubMed:17213273, ECO:0000269|PubMed:17652641, ECO:0000269|PubMed:17855752, ECO:0000269|PubMed:20022885, ECO:0000269|PubMed:28842488}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Transient neonatal diabetes mellitus 3 (TNDM3) [MIM:610582]: Neonatal diabetes mellitus, defined as insulin-requiring hyperglycemia within the first month of life, is a rare entity. In about half of the neonates, diabetes is transient and resolves at a median age of 3 months, whereas the rest have a permanent form of diabetes. In a significant number of patients with transient neonatal diabetes mellitus, diabetes type 2 appears later in life. The onset and severity of TNDM3 is variable with childhood-onset diabetes, gestational diabetes or adult-onset diabetes described. {ECO:0000269|PubMed:15718250, ECO:0000269|PubMed:15784703}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in KCNJ11 may contribute to non-insulin-dependent diabetes mellitus (NIDDM), also known as diabetes mellitus type 2.; DISEASE: Maturity-onset diabetes of the young 13 (MODY13) [MIM:616329]: A form of diabetes that is characterized by an autosomal dominant mode of inheritance, onset in childhood or early adulthood (usually before 25 years of age), a primary defect in insulin secretion and frequent insulin-independence at the beginning of the disease. {ECO:0000269|PubMed:22701567}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09096; DB11752; DB04209; DB12010; DB06064; DB02010; DB00675 Interacts with P05067; P15056; P08238; C6GKH1; Q9BPZ7; P17252; Q15349; P31947; P37840; P62258; P63104 EC number EC 2.7.11.13 Uniprot keywords 3D-structure; ATP-binding; Cell adhesion; Cell cycle; Cell division; Cell membrane; Cytoplasm; Cytoskeleton; Immunity; Kinase; Membrane; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Serine/threonine-protein kinase; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,F Molecular weight (Da) 39129.4 Length 339 Aromaticity 0.12 Instability index 42.21 Isoelectric point 6.17 Charge (pH=7) -3.73 2D Binding mode Binding energy (Kcal/mol) -8.87 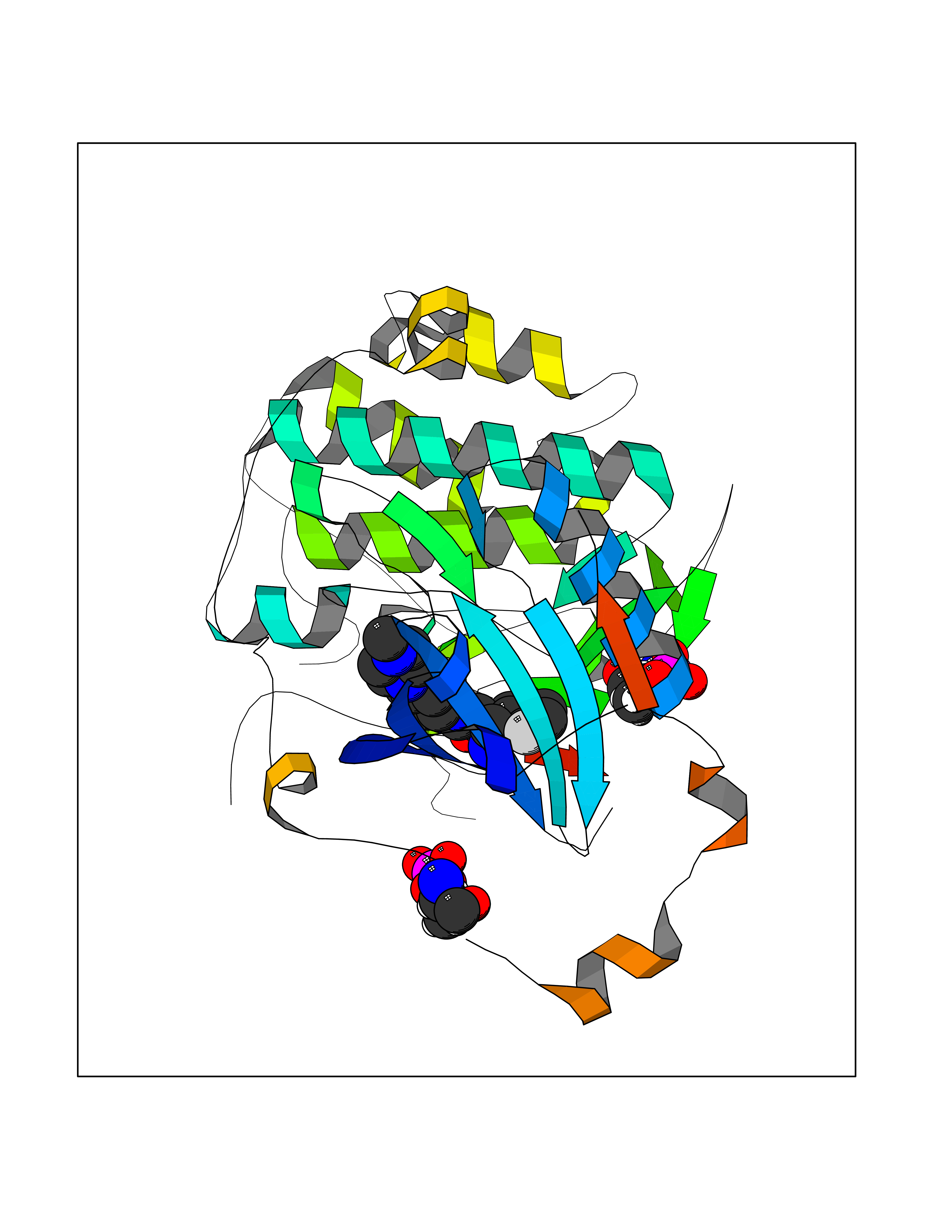 Molscript Map 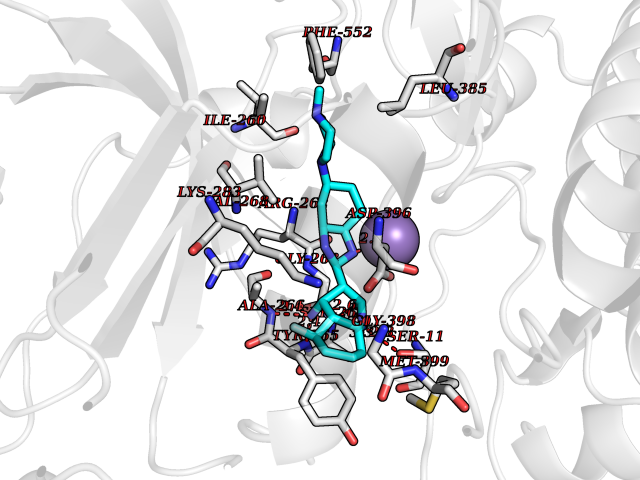 Pymol Map 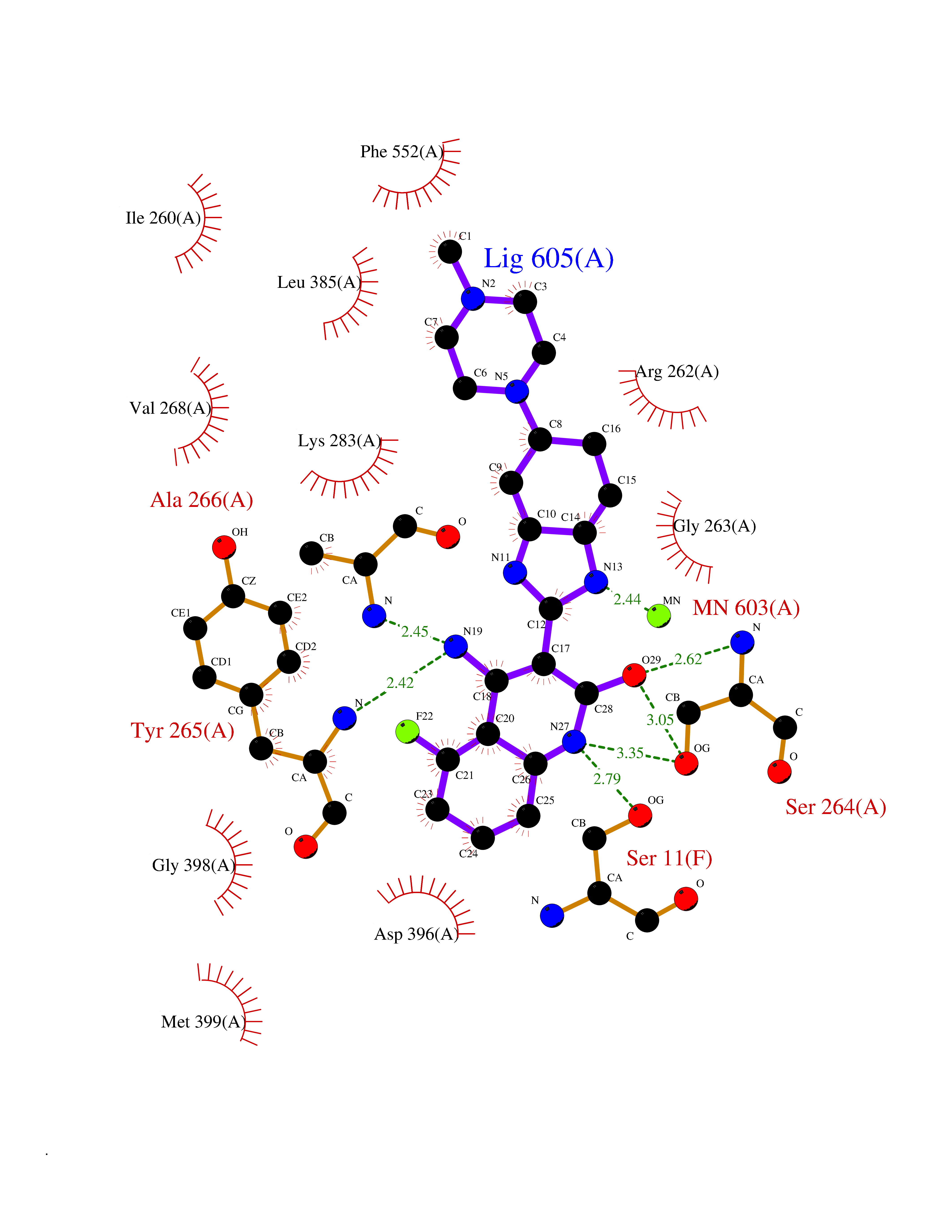 Ligplot Map 3D Binding mode Sequence SLGLQDFDLLRVIGRGSYAKVLLVRLKKTDRIYAMKVVKKELVWVQTEKHVFEQASNHPFLVGLHSCFQTESRLFFVIEYVNGGDLMFHMQRQRKLPEEHARFYSAEISLALNYLHERGIIYRDLKLDNVLLDSEGHIKLTDYGMCKEGLRPGDTTSXFCGTPNYIAPEILRGEDYGFSVDWWALGVLMFEMMAGRSPFDIQNTEDYLFQVILEKQIRIPRSLSVKAASVLKSFLNKDPKERLGCHPQTGFADIQGHPFFRNVDWDMMEQKQVVPPFKPNISGEFGLDNFDSQFTNEPVQLXPDDDDIVRKIDQSEFEGFEYINPLRMRPFKRQGSVRR Hydrogen bonds contact Hydrophobic contact | ||||
| 60 | Prothrombin | 4UD9 | 8.02 | |
Target general information Gen name F2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase S1 family Biochemical class Hydrolase Function Calcium ion binding.Growth factor activity.Heparin binding.Lipopolysaccharide binding.Receptor binding.Serine-type endopeptidase activity.Thrombospondin receptor activity. Related diseases Factor II deficiency (FA2D) [MIM:613679]: A very rare blood coagulation disorder characterized by mucocutaneous bleeding symptoms. The severity of the bleeding manifestations correlates with blood factor II levels. {ECO:0000269|PubMed:1349838, ECO:0000269|PubMed:1354985, ECO:0000269|PubMed:1421398, ECO:0000269|PubMed:14962227, ECO:0000269|PubMed:2719946, ECO:0000269|PubMed:3242619, ECO:0000269|PubMed:3567158, ECO:0000269|PubMed:3771562, ECO:0000269|PubMed:3801671, ECO:0000269|PubMed:6405779, ECO:0000269|PubMed:7792730, ECO:0000269|PubMed:7865694}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Thrombophilia due to thrombin defect (THPH1) [MIM:188050]: A multifactorial disorder of hemostasis characterized by abnormal platelet aggregation in response to various agents and recurrent thrombi formation. {ECO:0000269|PubMed:2825773}. The disease is caused by variants affecting the gene represented in this entry. A common genetic variation in the 3-prime untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increased risk of venous thrombosis.; DISEASE: Pregnancy loss, recurrent, 2 (RPRGL2) [MIM:614390]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:11506076}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07211; DB07796; DB07016; DB07521; DB06850; DB07091; DB06845; DB07088; DB07131; DB07095; DB07515; DB07897; DB06878; DB06947; DB08624; DB06869; DB06929; DB07400; DB04771; DB04772; DB02287; DB07277; DB07550; DB07549; DB07548; DB07105; DB04722; DB07366; DB08254; DB01725; DB08062; DB07639; DB07461; DB07120; DB07190; DB07741; DB07353; DB07508; DB07809; DB08546; DB08061; DB07718; DB03136; DB02723; DB07440; DB07376; DB06861; DB06866; DB06865; DB03865; DB06841; DB07934; DB08422; DB07659; DB07660; DB07658; DB13151; DB00025; DB11166; DB00278; DB01766; DB07083; DB00006; DB00100; DB13152; DB09228; DB09130; DB03159; DB06911; DB06996; DB06919; DB07027; DB07133; DB07143; DB07005; DB06695; DB00055; DB01225; DB05714; DB12831; DB03847; DB07278; DB01767; DB06404; DB09332; DB00001; DB13998; DB04136; DB00170; DB06838; DB13999; DB06868; DB06942; DB06936; DB07165; DB07527; DB07522; DB07665; DB07946; DB06859; DB06853; DB06858; DB07279; DB08187; DB04591; DB07944; DB07128; DB12598; DB01123; DB04786; DB05777; DB04697; DB09109; DB14738; DB04898; DB01593; DB14487; DB08152 Interacts with P05067; P07204; Q846V4; PRO_0000032489 [P01008] EC number 3.4.21.5 Uniprot keywords 3D-structure; Acute phase; Blood coagulation; Calcium; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; Gamma-carboxyglutamic acid; Glycoprotein; Hemostasis; Hydrolase; Kringle; Pharmaceutical; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Thrombophilia; Zymogen Protein physicochemical properties Chain ID H Molecular weight (Da) 29321.6 Length 254 Aromaticity 0.1 Instability index 39.57 Isoelectric point 8.56 Charge (pH=7) 4.16 2D Binding mode Binding energy (Kcal/mol) -8.29 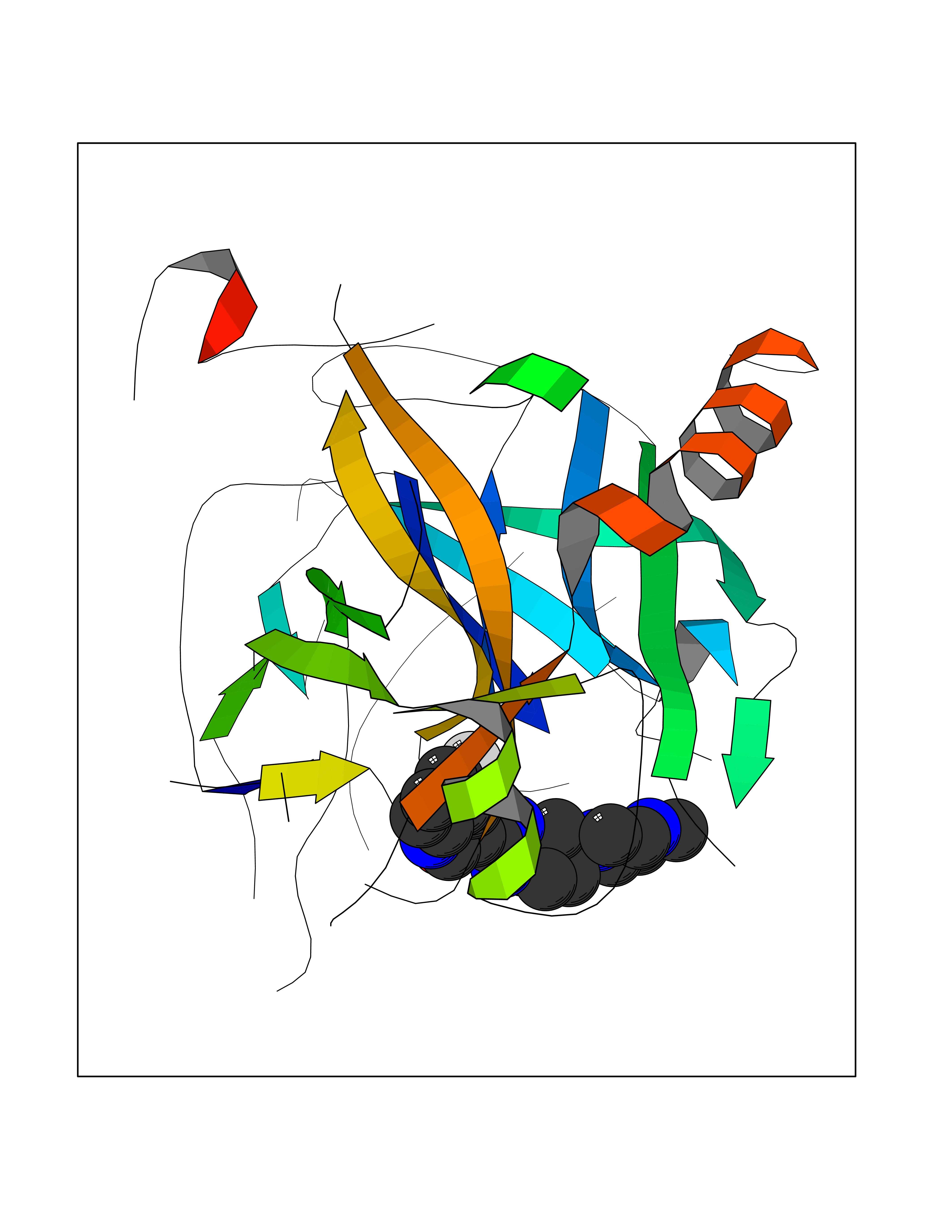 Molscript Map 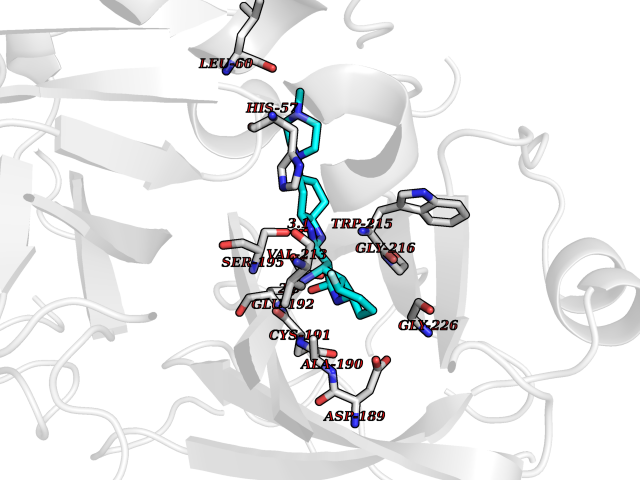 Pymol Map 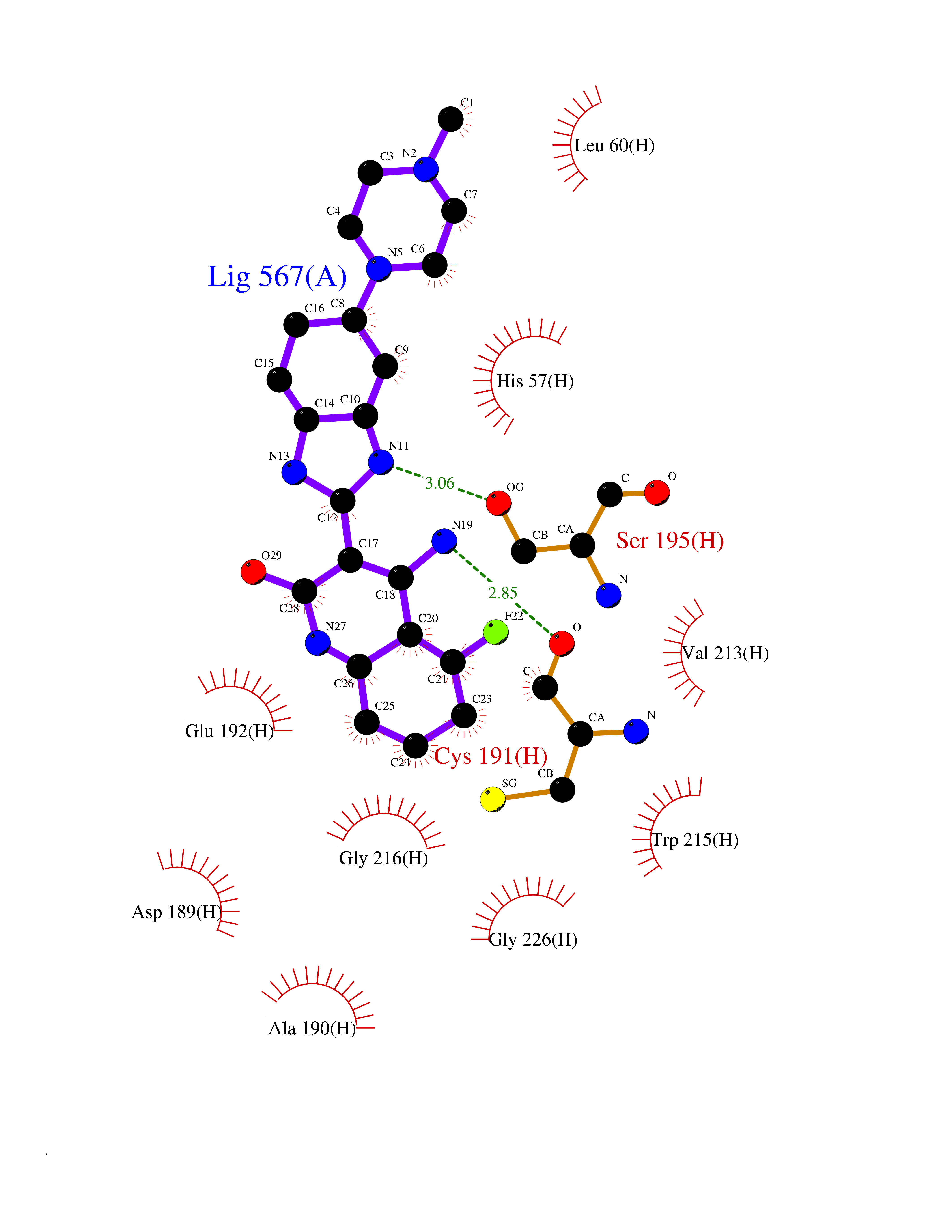 Ligplot Map 3D Binding mode Sequence IVEGSDAEIGMSPWQVMLFRSPQELLCGASLISDRWVLTAAHCLLTENDLLVRIGKHSRTRYRNIEKISMLEKIYIHPRYNWENLDRDIALMKLKKPVAFSDYIHPVCLPDRETLLQAGYKGRVTGWGNLKETWGQPSVLQVVNLPIVERPVCKDSTRIRITDNMFCAYKKRGDACEGDSGGPFVMKSNNRWYQMGIVSWGEGCRDGKYGFYTHVFRLKKWIQKVIDQFGGDFEEIPEELQCGLRPLFEKKSLE Hydrogen bonds contact Hydrophobic contact | ||||