Job Results:
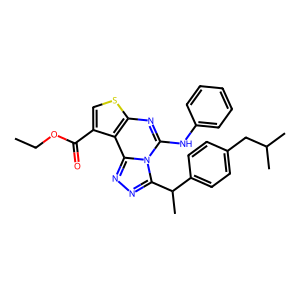
Ligand
Structure
Job ID
b5aa43c6924a21fc75a6162a600078fc
Job name
NA
Time
2026-03-04 17:25:26
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Dimethylglycine oxidase | 1PJ5 | 8.52 | |
Target general information Gen name dmg Organism Arthrobacter globiformis Uniprot ID TTD ID NA Synonyms NA Protein family GcvT family Biochemical class Oxidoreductase Function Dimethylglycine oxidase activity.Nucleotide binding. Related diseases Curry-Jones syndrome (CRJS) [MIM:601707]: A multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. The disease is caused by variants affecting the gene represented in this entry. 8 individuals have been identified with the disease-causing mutation Phe-412 and all were mosaic. The mutation could not be reliably detected in blood, greatest success rates were obtained with affected tissues obtained by invasive procedures. It is thought that the mutation has arisen postzygotically early during embryonic development (PubMed:27236920). This mutation has also been identified in ameloblastoma, medulloblastoma, meningioma, and basal cell carcinoma, and has been reported as the oncogenic driver in some of these tumors (PubMed:24859340). {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. Drugs (DrugBank ID) DB03256; DB03147 Interacts with NA EC number 1.5.3.10 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 45912.2 Length 427 Aromaticity 0.07 Instability index 43.46 Isoelectric point 4.83 Charge (pH=7) -20.69 2D Binding mode Binding energy (Kcal/mol) -11.62 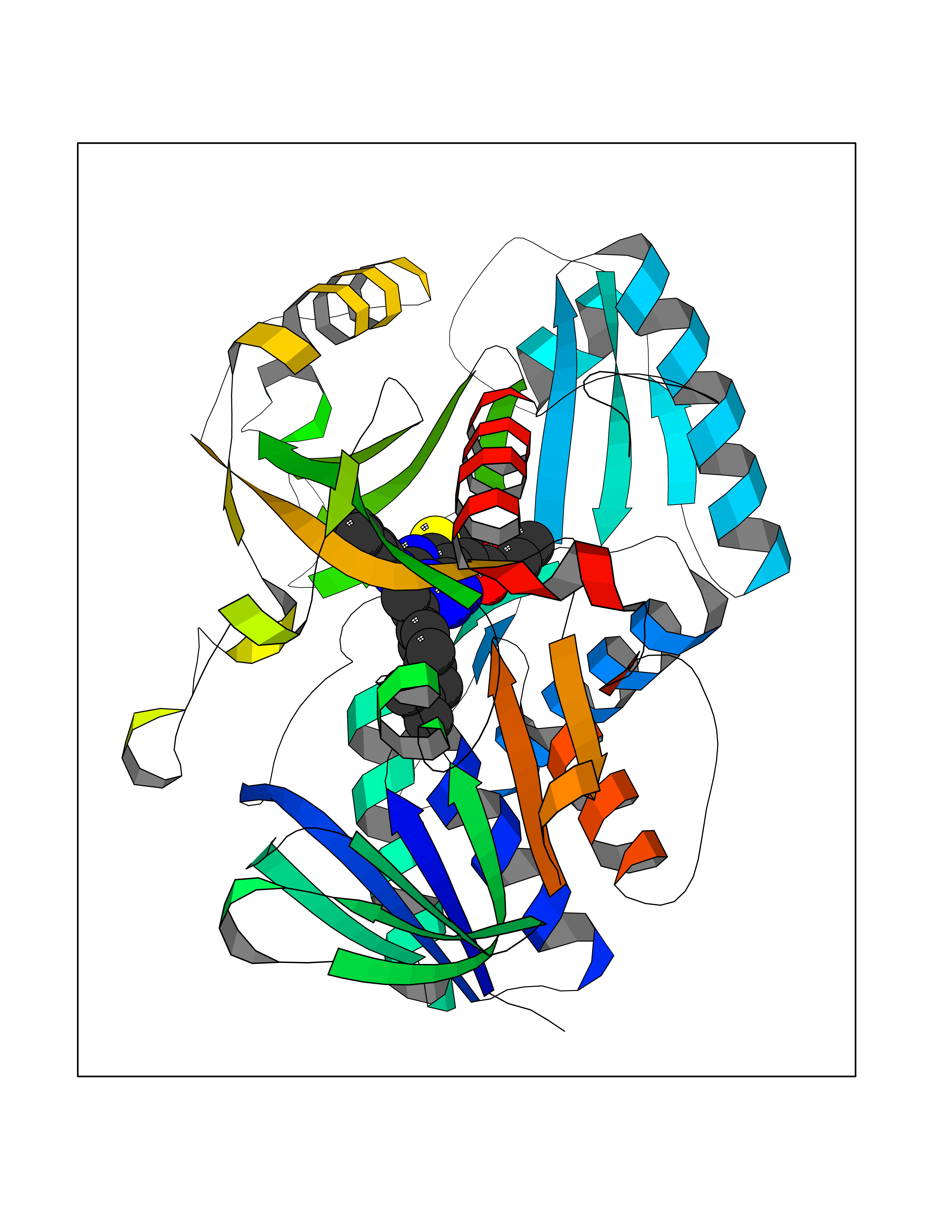 Molscript Map 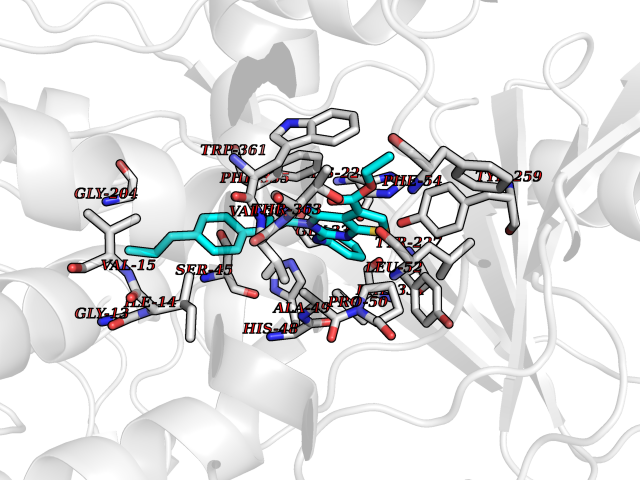 Pymol Map 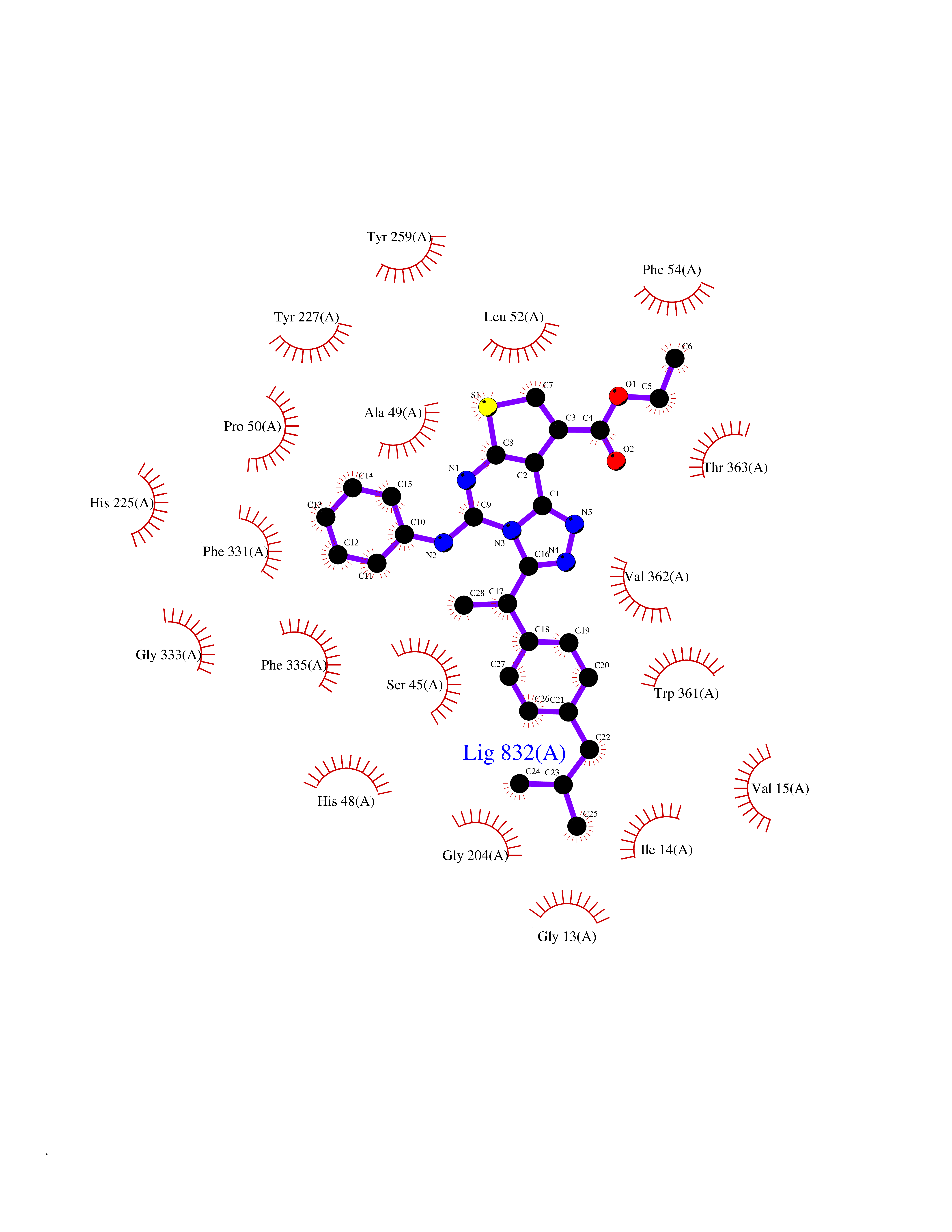 Ligplot Map 3D Binding mode Sequence TPRIVIIGAGIVGTNLADELVTRGWNNITVLDQGPLNMPGGSTSHAPGLVFQTNPSKTMASFAKYTVEKLLSLTEDGVSCFNQVGGLEVATTETRLADLKRKLGYAAAWGIEGRLLSPAECQELYPLLDGENILGGLHVPSDGLASAARAVQLLIKRTESAGVTYRGSTTVTGIEQSGGRVTGVQTADGVIPADIVVSCAGFWGAKIGAMIGMAVPLLPLAHQYVKTTPVPAQQGRNDQPNGARLPILRHQDQDLYYREHGDRYGIGSYAHRPMPVDVDTLGAYAPETVSEHHMPSRLDFTLEDFLPAWEATKQLLPALADSEIEDGFNGIFSFTPDGGPLLGESKELDGFYVAEAVWVTHSAGVAKAMAELLTTGRSETDLGECDITRFEDVQLTPEYVSETSQQNFVEIYDVLHPLQPRLSPRNL Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Dipeptidyl peptidase 8 (DPP-8) | 6EOP | 8.51 | |
Target general information Gen name DPP8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Prolyl dipeptidase DPP8; MSTP141; MSTP135; MSTP097; Dipeptidyl peptidase VIII; Dipeptidyl peptidase IV-related protein 1; DPRP1; DPRP-1; DPP VIII; DP8 Protein family Peptidase S9B family, DPPIV subfamily Biochemical class Peptidase Function Dipeptidyl peptidase that cleaves off N-terminal dipeptides from proteins having a Pro or Ala residue at position 2. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.14.5 Uniprot keywords 3D-structure; Alternative splicing; Aminopeptidase; Apoptosis; Cytoplasm; Hydrolase; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A,D Molecular weight (Da) 97764.9 Length 849 Aromaticity 0.12 Instability index 47.71 Isoelectric point 5.69 Charge (pH=7) -21.66 2D Binding mode Binding energy (Kcal/mol) -11.61 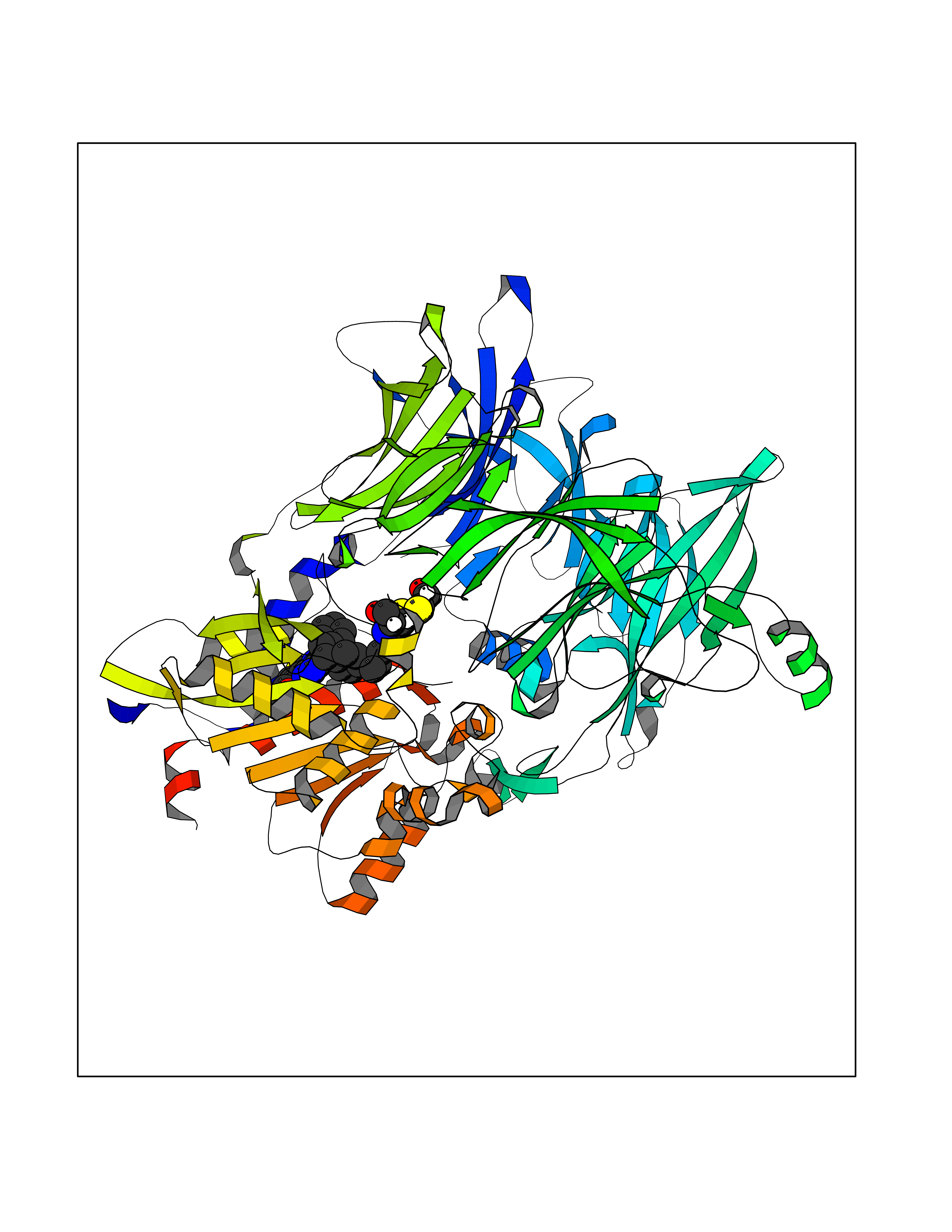 Molscript Map 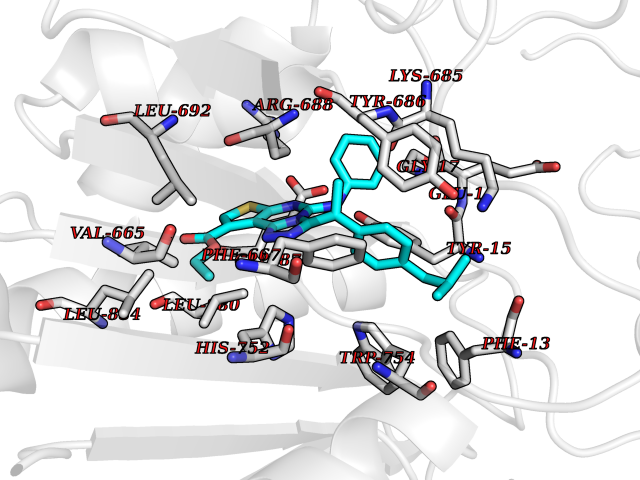 Pymol Map 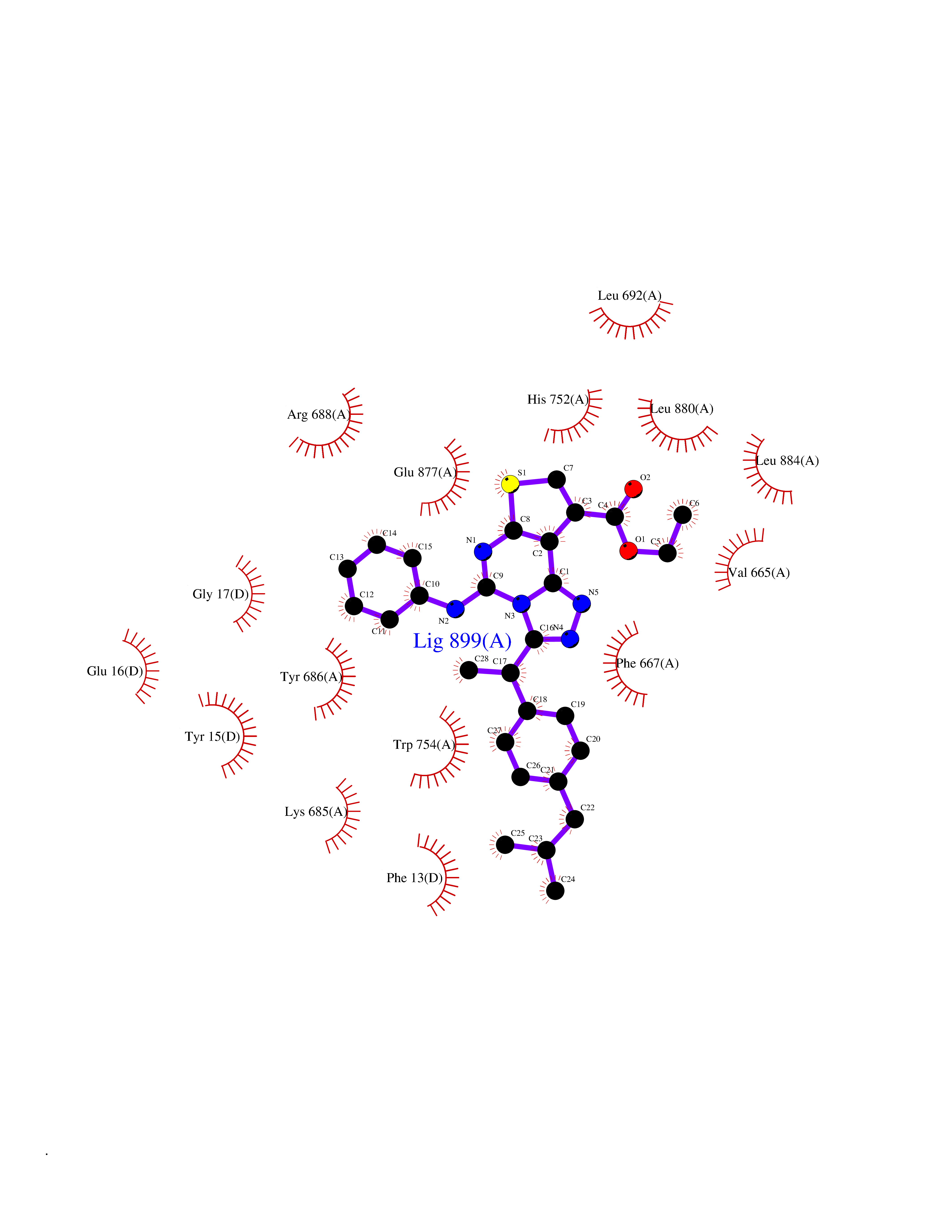 Ligplot Map 3D Binding mode Sequence LEPFYVERYSWSQLKKLLADTRKYHGYMMAKAPHDFMFVKRNDPDGPHSDRIYYLAMSNRENTLFYSEIPKTINRAAVLMLSWKPLLDLFQYSREEELLRERKRIGTVGIASYDYHQGSGTFLFQAGSGIYHVKDGGPQGFTQQPLRPNLVETSCPNIRMDPKLCPADPDWIAFIHSNDIWISNIVTREERRLTYVHNELANMEEDARSAGVATFVLQEEFDRYSGYWWCPKAETTPSGGKILRILYEENDESEVEIIHVTSPMLETRRADSFRYPKTGTANPKVTFKMSEIMIDAEGRIIDVIDKELIQPFEILFEGVEYIARAGWTPEGKYAWSILLDRSQTRLQIVLISPELFIPVEDDVMERQRLIESVPDSVTPLIIYEETTDIWINIHDIFHVFPQSHEEEIEFIFASECKTGFRHLYKITSILKESKYKRSSGGLPAPSDFKCPIKEEIAITSGEWEVLGRHGSNIQVDEVRRLVYFEGTKDSPLEHHLYVVSYVNPGEVTRLTDRGYSHSCCISQHCDFFISKYSNQKNPHCVSLYKLSSPEDDPTCKTKEFWATILDSAGPLPDYTPPEIFSFESTTGFTLYGMLYKPHDLQPGKKYPTVLFIYGGPQVQLVNNRFKGVKYFRLNTLASLGYVVVVIDNRGSXHRGLKFEGAFKYKMGQIEIDDQVEGLQYLASRYDFIDLDRVGIHGWSYGGYLSLMALMQRSDIFRVAIAGAPVTLWIFYDTGYTERYMGHPDQNEQGYYLGSVAMQAEKFPSEPNRLLLLHGFLDENVHFAHTSILLSFLVRAGKPYDLQIYPQERHSIRVPESGEHYELHLLHYLQENLGSRIAALKVSLRFLYEG Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Cannabinoid receptor 1 (CB1) | 5U09 | 8.50 | |
Target general information Gen name CNR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cannabinoid CB1 receptor; CNR; CB-R; CANN6 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Mediates many cannabinoid-induced effects, acting, among others, on food intake, memory loss, gastrointestinal motility, catalepsy, ambulatory activity, anxiety, chronic pain. Signaling typically involves reduction in cyclic AMP. In the hypothalamus, may have a dual effect on mitochondrial respiration depending upon the agonist dose and possibly upon the cell type. Increases respiration at low doses, while decreases respiration at high doses. At high doses, CNR1 signal transduction involves G-protein alpha-i protein activation and subsequent inhibition of mitochondrial soluble adenylate cyclase, decrease in cyclic AMP concentration, inhibition of protein kinase A (PKA)-dependent phosphorylation of specific subunits of the mitochondrial electron transport system, including NDUFS2. In the hypothalamus, inhibits leptin-induced reactive oxygen species (ROS) formation and mediates cannabinoid-induced increase in SREBF1 and FASN gene expression. In response to cannabinoids, drives the release of orexigenic beta-endorphin, but not that of melanocyte-stimulating hormone alpha/alpha-MSH, from hypothalamic POMC neurons, hence promoting food intake. In the hippocampus, regulates cellular respiration and energy production in response to cannabinoids. Involved in cannabinoid-dependent depolarization-induced suppression of inhibition (DSI), a process in which depolarization of CA1 postsynaptic pyramidal neurons mobilizes eCBs, which retrogradely activate presynaptic CB1 receptors, transiently decreasing GABAergic inhibitory neurotransmission. Also reduces excitatory synaptic transmission. In superior cervical ganglions and cerebral vascular smooth muscle cells, inhibits voltage-gated Ca(2+) channels in a constitutive, as well as agonist-dependent manner. In cerebral vascular smooth muscle cells, cannabinoid-induced inhibition of voltage-gated Ca(2+) channels leads to vasodilation and decreased vascular tone. Induces leptin production in adipocytes and reduces LRP2-mediated leptin clearance in the kidney, hence participating in hyperleptinemia. In adipose tissue, CNR1 signaling leads to increased expression of SREBF1, ACACA and FASN genes. In the liver, activation by endocannabinoids leads to increased de novo lipogenesis and reduced fatty acid catabolism, associated with increased expression of SREBF1/SREBP-1, GCK, ACACA, ACACB and FASN genes. May also affect de novo cholesterol synthesis and HDL-cholesteryl ether uptake. Peripherally modulates energy metabolism. In high carbohydrate diet-induced obesity, may decrease the expression of mitochondrial dihydrolipoyl dehydrogenase/DLD in striated muscles, as well as that of selected glucose/ pyruvate metabolic enzymes, hence affecting energy expenditure through mitochondrial metabolism. In response to cannabinoid anandamide, elicits a proinflammatory response in macrophages, which involves NLRP3 inflammasome activation and IL1B and IL18 secretion. In macrophages infiltrating pancreatic islets, this process may participate in the progression of type-2 diabetes and associated loss of pancreatic beta-cells. G-protein coupled receptor for endogenous cannabinoids (eCBs), including N-arachidonoylethanolamide (also called anandamide or AEA) and 2-arachidonoylglycerol (2-AG), as well as phytocannabinoids, such as delta(9)-tetrahydrocannabinol (THC). Related diseases Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:18177726}. The protein represented in this entry may be involved in disease pathogenesis. May contribute to the development of diet-induced obesity and several obesity-associated features, such as dyslipidemia and liver steatosis, regulating peripheral lipogenesis, energy expenditure and feeding behavior. CNR1 inverse agonists have been shown to reduce body weight and improve metabolic abnormalities in obese subjects, although adverse neuropsychiatric effects, including anxiety, irritability, and depressed mood, halted their therapeutic development (PubMed:18177726). In obese mice, peripherally restricted CNR1 inverse agonists have been shown to normalize metabolic abnormalities, including insulin resistance and fatty liver, and to reverse leptin resistance. {ECO:0000269|PubMed:18177726}.; DISEASE: Dysfunction of the endogenous cannabinoid system including CNR1 has been implicated in the pathogenesis of a number of central nervous system disorders, including Huntington disease, Parkinson disease, and Alzheimer disease (PubMed:32549916). In post-mortem brains from Huntington disease patients, a progressive CNR1 loss has been observed in the caudate nucleus, putamen, and substantia nigra pars reticulata, and altered expression and abnormal endocannabinoid levels precede motor symptoms in a disease mouse model (PubMed:10828533, PubMed:19524019, PubMed:8255419). In Parkinson disease, low CNR1 expression in mid-superior frontal gyrus and mid-cingulate cortex has been associated with poor mind, poor executive functioning and poor episode memory, while patients with more severe visuospatial dysfunction showed decreased receptor availability in the precuneus, mid-cingulate, supplementary motor cortex, inferior orbitofrontal gyrus and thalamus (PubMed:31342135). In an animal model for Alzheimer disease, CNR1 heterozygous deletion has been associated with decreased levels of postsynaptic density protein 95 (DLG4/PSD95) and accelerated memory impairment, suggesting synaptic dysfunction and a crucial role for CNR1 in the progression of disease symptoms (PubMed:10828533, PubMed:19524019, PubMed:30096288, PubMed:31342135, PubMed:8255419). {ECO:0000269|PubMed:10828533, ECO:0000269|PubMed:19524019, ECO:0000269|PubMed:30096288, ECO:0000269|PubMed:31342135, ECO:0000269|PubMed:32549916, ECO:0000269|PubMed:8255419}. Drugs (DrugBank ID) DB05750; DB09061; DB00470; DB14009; DB00486; DB14011; DB11745; DB09288; DB02955; DB06155; DB05077; DB11755; DB05201 Interacts with P29274; P21554 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Neurodegeneration; Obesity; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32070.3 Length 282 Aromaticity 0.13 Instability index 40.15 Isoelectric point 9.16 Charge (pH=7) 9.36 2D Binding mode Binding energy (Kcal/mol) -11.6 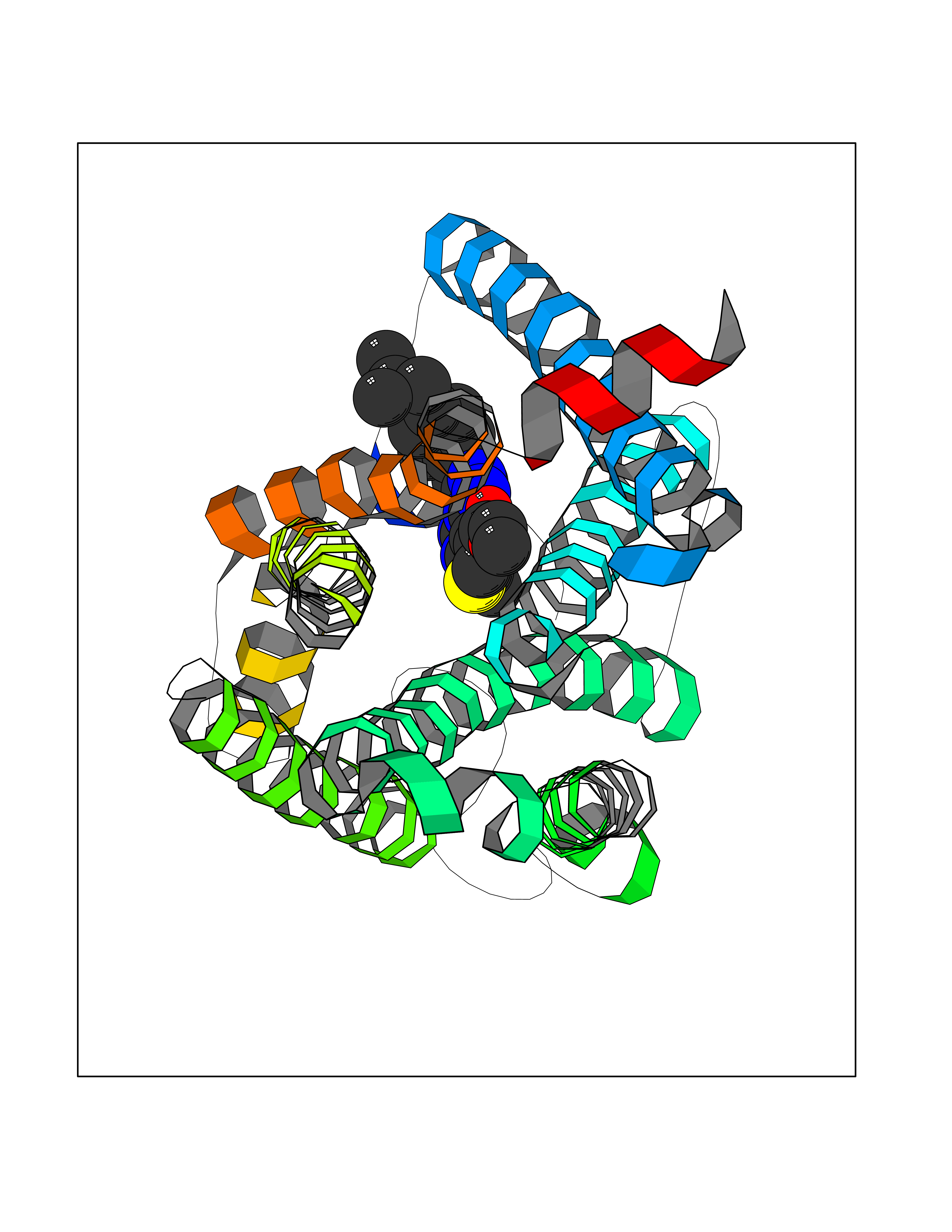 Molscript Map 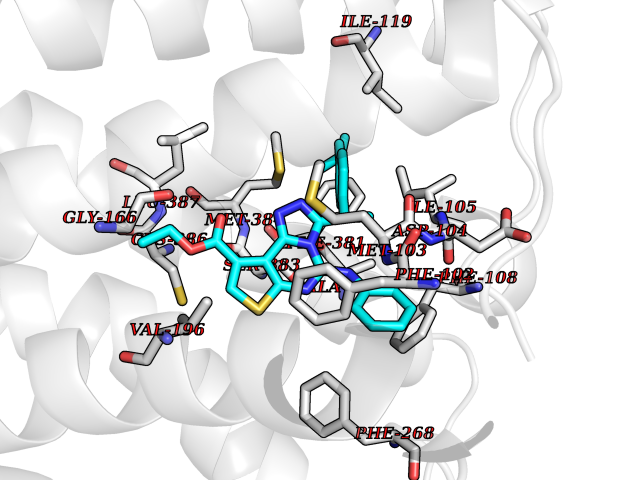 Pymol Map 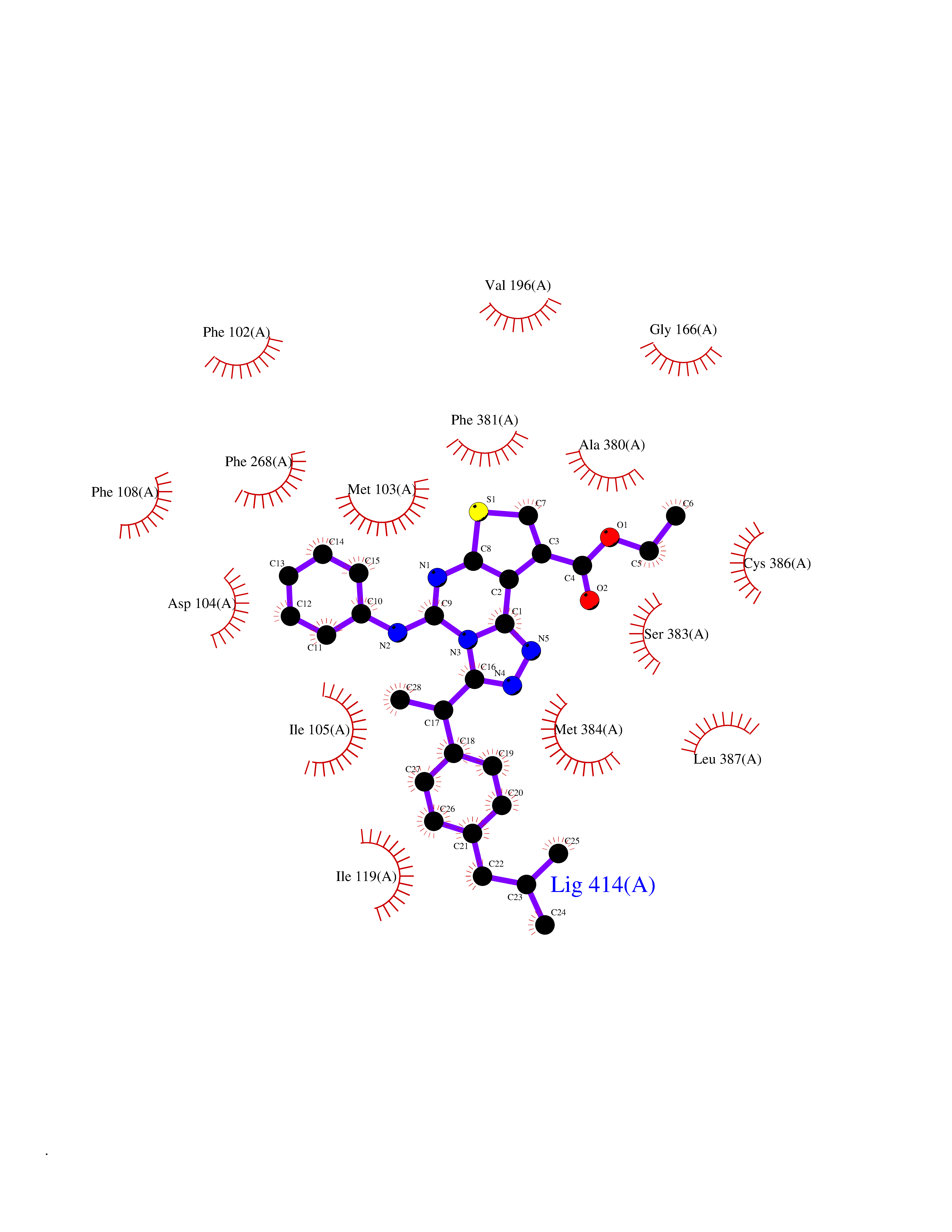 Ligplot Map 3D Binding mode Sequence ENFMDIECFMVLNPSQQLAIAVLSLTLGTFTVLENLLVLCVILHSRSLRCRPSYHFIGSLAVADLLGSVIFVYSFIDFHVFHRKDSRNVFLFKLGGVTASFTASVGSLFLAAIDRYISIHRPLAYKRIVTRPKAVVAFCLMWTIAIVIAVLPLLGWNCEKLQSVCSDIFPHIDETYLMFWIGVTSVLLLFIVYAYMYILWKADQARMDIRLAKTLVLILVVLIICWGPLLAIMVYDVFGKMNKLIKTVFAFCSMLCLLNSTVNPIIYALRSKDLRHAFRSMF Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Histidine decarboxylase (HDC) | 4E1O | 8.46 | |
Target general information Gen name HDC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Human histidine decarboxylase Protein family Group II decarboxylase family Biochemical class Carbon-carbon lyase Function Catalyzes the biosynthesis of histamine from histidine. Related diseases Corticosterone methyloxidase 1 deficiency (CMO-1 deficiency) [MIM:203400]: Autosomal recessive disorder of aldosterone biosynthesis. There are two biochemically different forms of selective aldosterone deficiency be termed corticosterone methyloxidase (CMO) deficiency type 1 and type 2. In CMO-1 deficiency, aldosterone is undetectable in plasma, while its immediate precursor, 18-hydroxycorticosterone, is low or normal. {ECO:0000269|PubMed:11238478, ECO:0000269|PubMed:8439335, ECO:0000269|PubMed:9177280}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Corticosterone methyloxidase 2 deficiency (CMO-2 deficiency) [MIM:610600]: Autosomal recessive disorder of aldosterone biosynthesis. In CMO-2 deficiency, aldosterone can be low or normal, but at the expense of increased secretion of 18-hydroxycorticosterone. Consequently, patients have a greatly increased ratio of 18-hydroxycorticosterone to aldosterone and a low ratio of corticosterone to 18-hydroxycorticosterone in serum. {ECO:0000269|PubMed:12788848, ECO:0000269|PubMed:1346492, ECO:0000269|PubMed:1594605, ECO:0000269|PubMed:9625333, ECO:0000269|PubMed:9814506}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00117; DB00114 Interacts with Q86UW9 EC number EC 4.1.1.22 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Decarboxylase; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 107706 Length 956 Aromaticity 0.1 Instability index 55.17 Isoelectric point 6.23 Charge (pH=7) -9.63 2D Binding mode Binding energy (Kcal/mol) -11.54  Molscript Map  Pymol Map 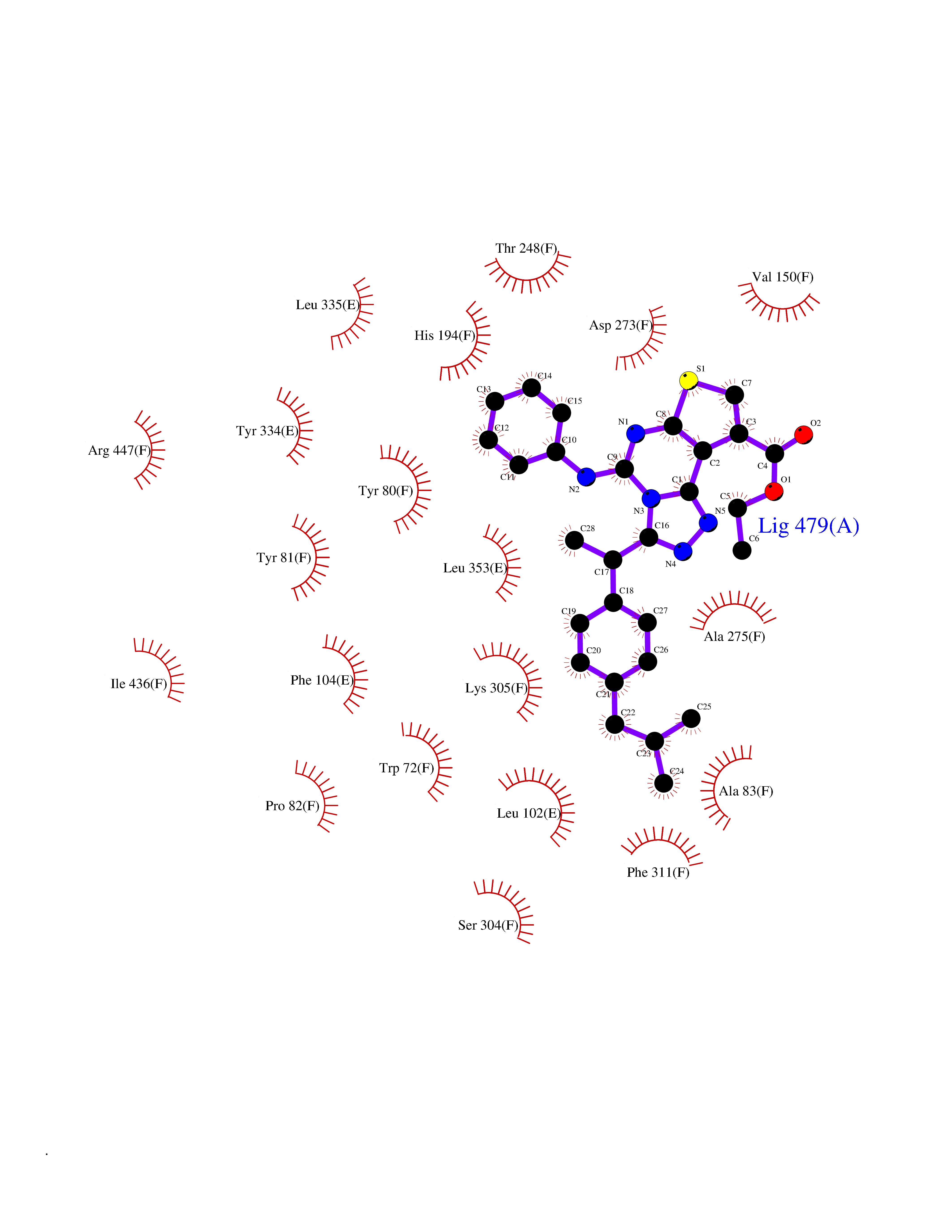 Ligplot Map 3D Binding mode Sequence GSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQGSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Metabotropic glutamate receptor 5 (mGluR5) | 4OO9 | 8.45 | |
Target general information Gen name GRM5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MGLUR5; GPRC1E Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function G-protein coupled receptor for glutamate. Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling activates a phosphatidylinositol-calcium second messenger system and generates a calcium-activated chloride current. Plays an important role in the regulation of synaptic plasticity and the modulation of the neural network activity. Related diseases Charcot-Marie-Tooth disease, axonal, 2D (CMT2D) [MIM:601472]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:17101916, ECO:0000269|PubMed:17663003, ECO:0000269|PubMed:20169446, ECO:0000269|PubMed:24604904, ECO:0000269|PubMed:25168514, ECO:0000269|PubMed:26244500, ECO:0000269|PubMed:26503042, ECO:0000269|PubMed:31173493}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 5 (HMND5) [MIM:600794]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:23279345, ECO:0000269|PubMed:24627108, ECO:0000269|PubMed:26503042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinal muscular atrophy, infantile, James type (SMAJI) [MIM:619042]: An autosomal dominant form of spinal muscular atrophy, a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. SMAJI is a severe disease characterized by hypotonia manifesting in the first weeks or months of life, delayed motor development, motor regression, and muscle weakness and atrophy primarily affecting distal muscles. Additional variable features include feeding difficulties, poor overall growth, foot deformities, kyphosis, hyperlordosis, scoliosis, vocal cord dysfunction, and respiratory insufficiency. {ECO:0000269|PubMed:32181591}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00659; DB05070; DB12733; DB06201 Interacts with P41594; Q7Z6G3 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Methylation; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 27065.4 Length 247 Aromaticity 0.13 Instability index 42.92 Isoelectric point 9.24 Charge (pH=7) 11.34 2D Binding mode Binding energy (Kcal/mol) -11.53 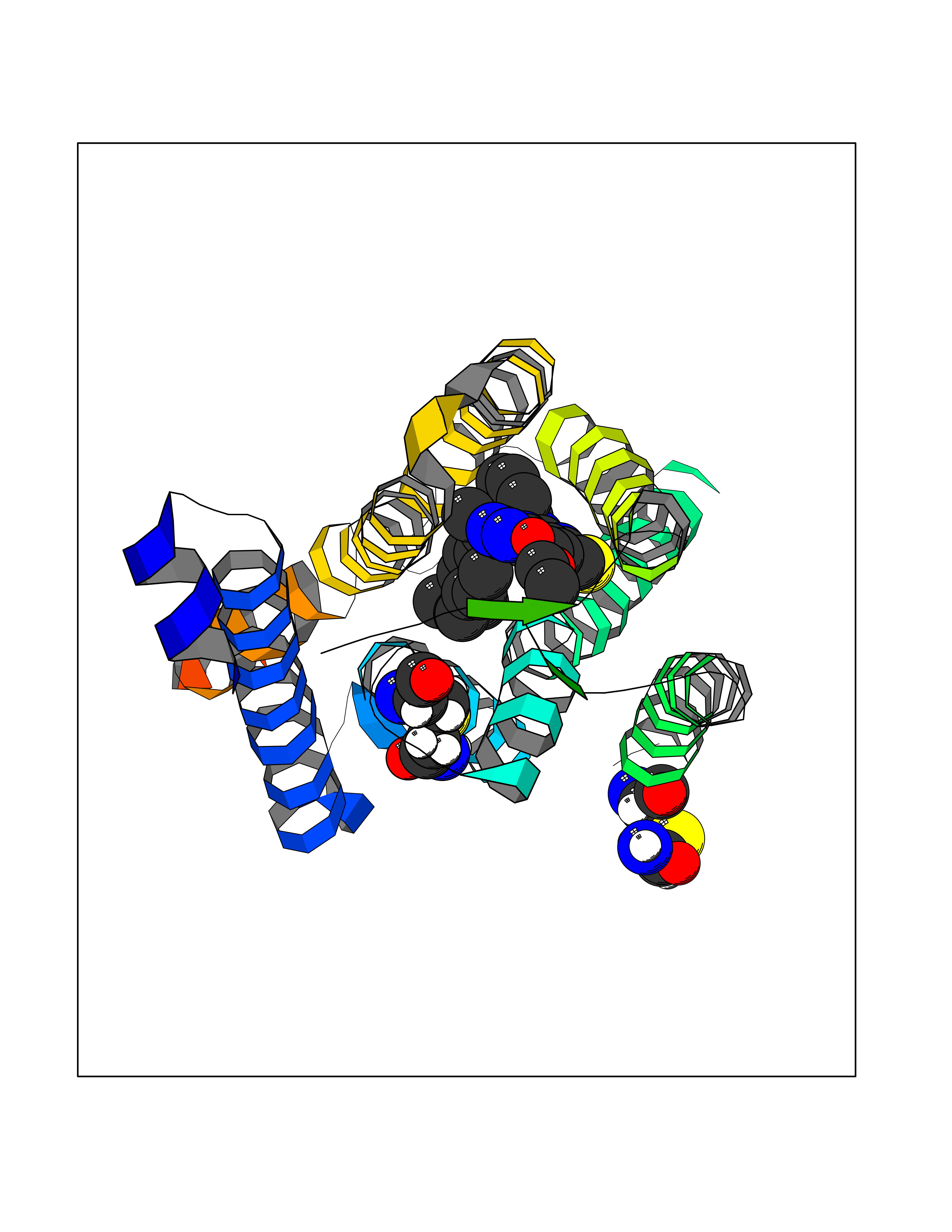 Molscript Map 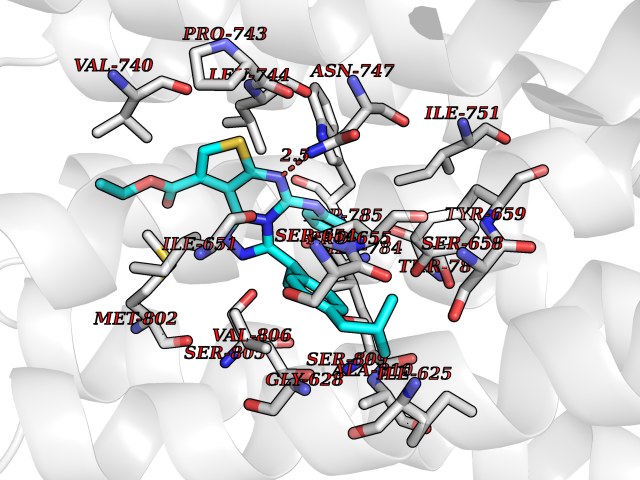 Pymol Map 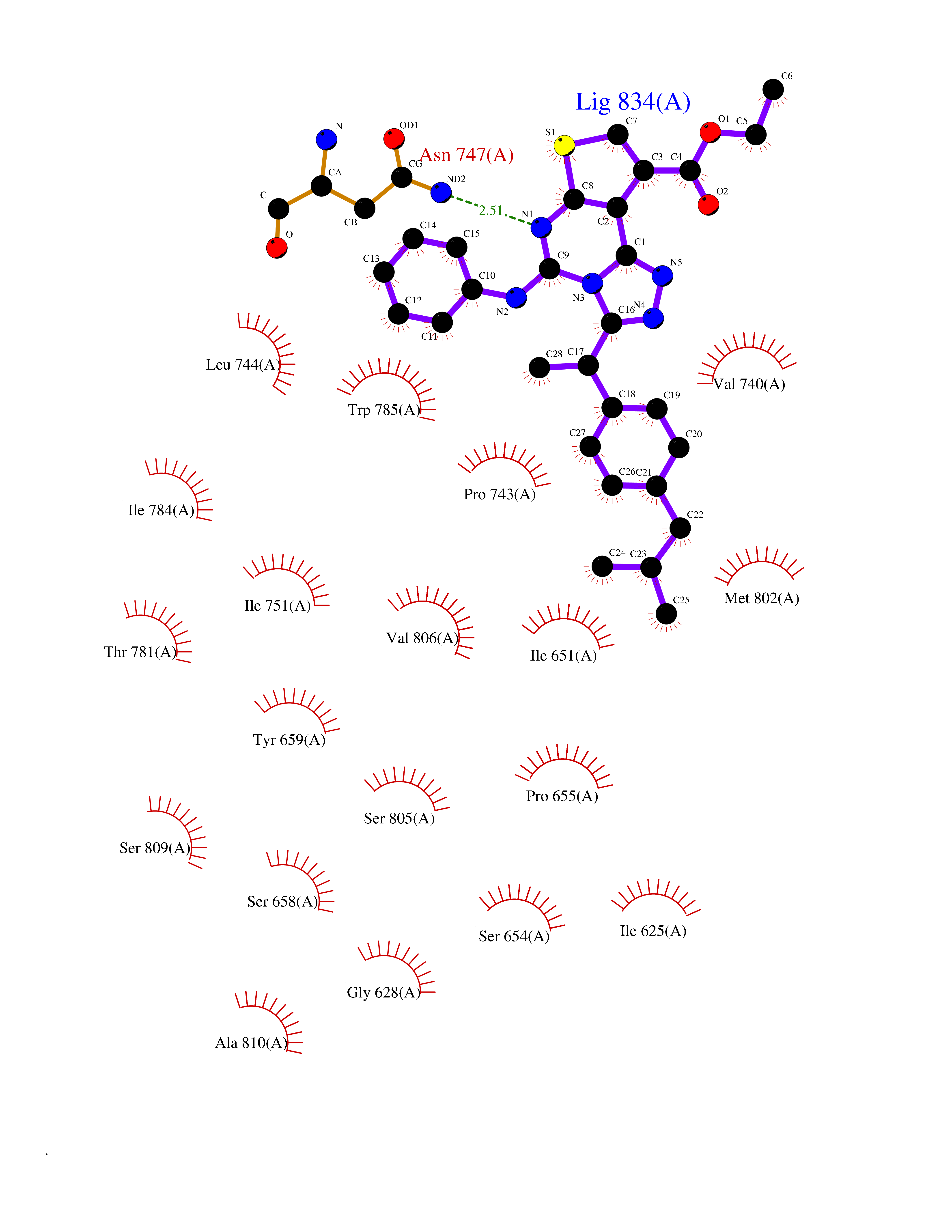 Ligplot Map 3D Binding mode Sequence SPVQYLRWGDPAPIAAVVFACLGLLATLFVTVVFIIYRDTPVVKSSSRELCYIILAGICLGYLCTFXLIAKPKQIYCYLQRIGIGLSPAMSYSALVTKTYRAARILAMSKKSAXAQLVIAFILICIQLGIIVALFIMEPPDIMVYLICNTTNLGVVAPLGYNGLLILACTFYAFKTRNVPANFNEAKYIAFTMYTTCIIWLAFVPIYFGSNYKIITMCFSVSLSATVALGCMFVPKVYIILAKPERN Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Oxalosuccinate decarboxylase (IDH1) | 6ADG | 8.45 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD; NADP(+)-specific ICDH; Isocitrate dehydrogenase [NADP] cytoplasmic; IDP; IDH; Cytosolic NADP-isocitrate dehydrogenase Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -11.52 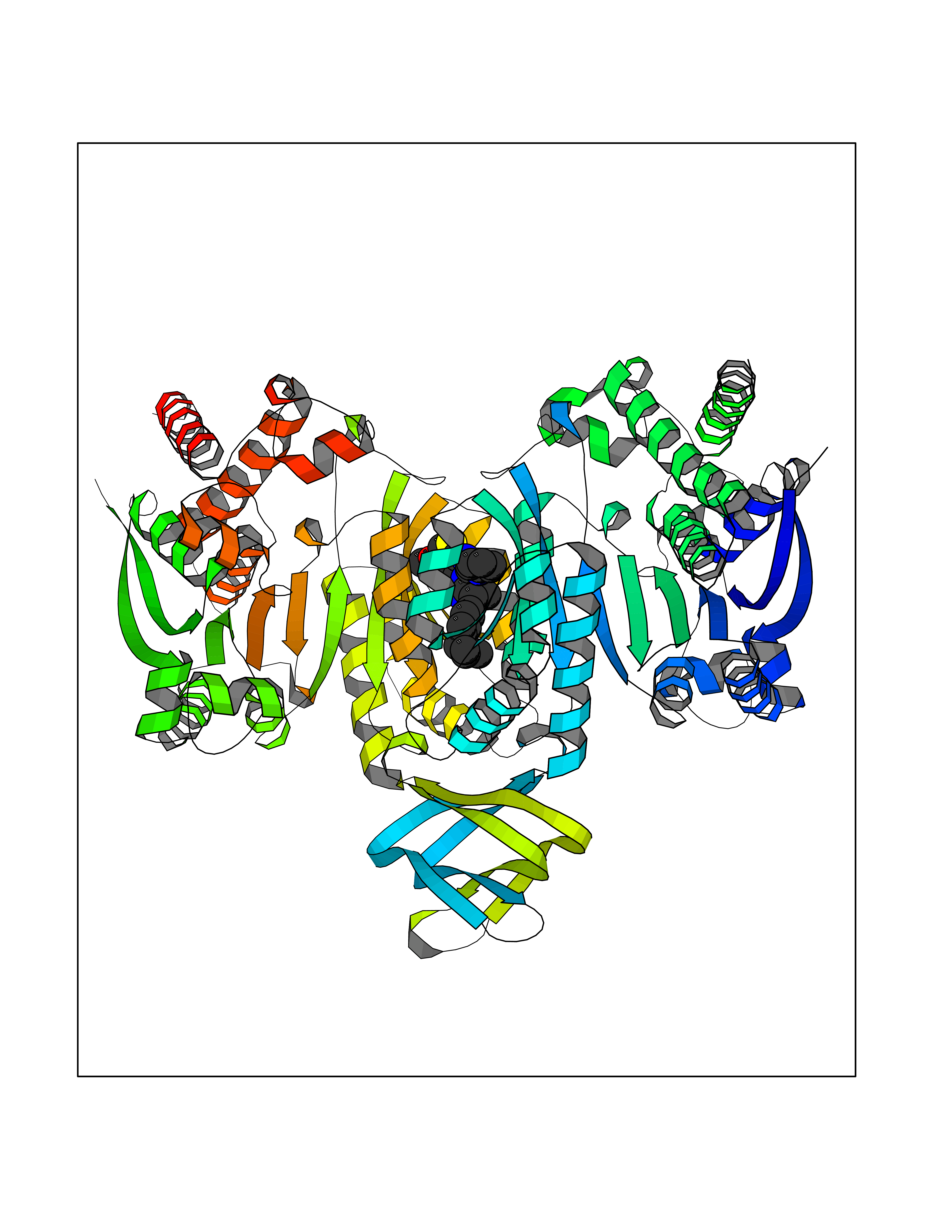 Molscript Map 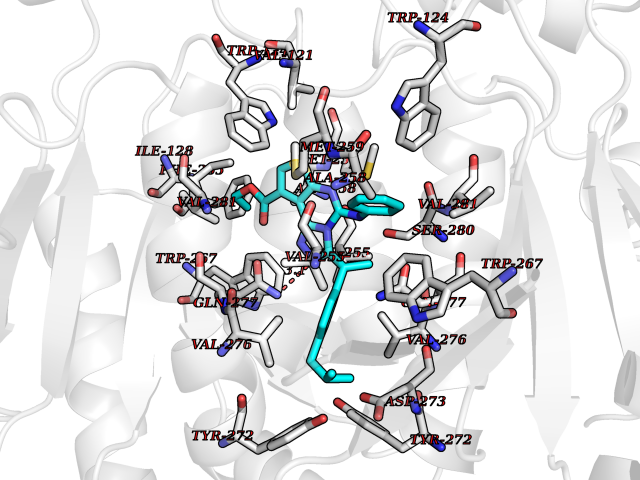 Pymol Map 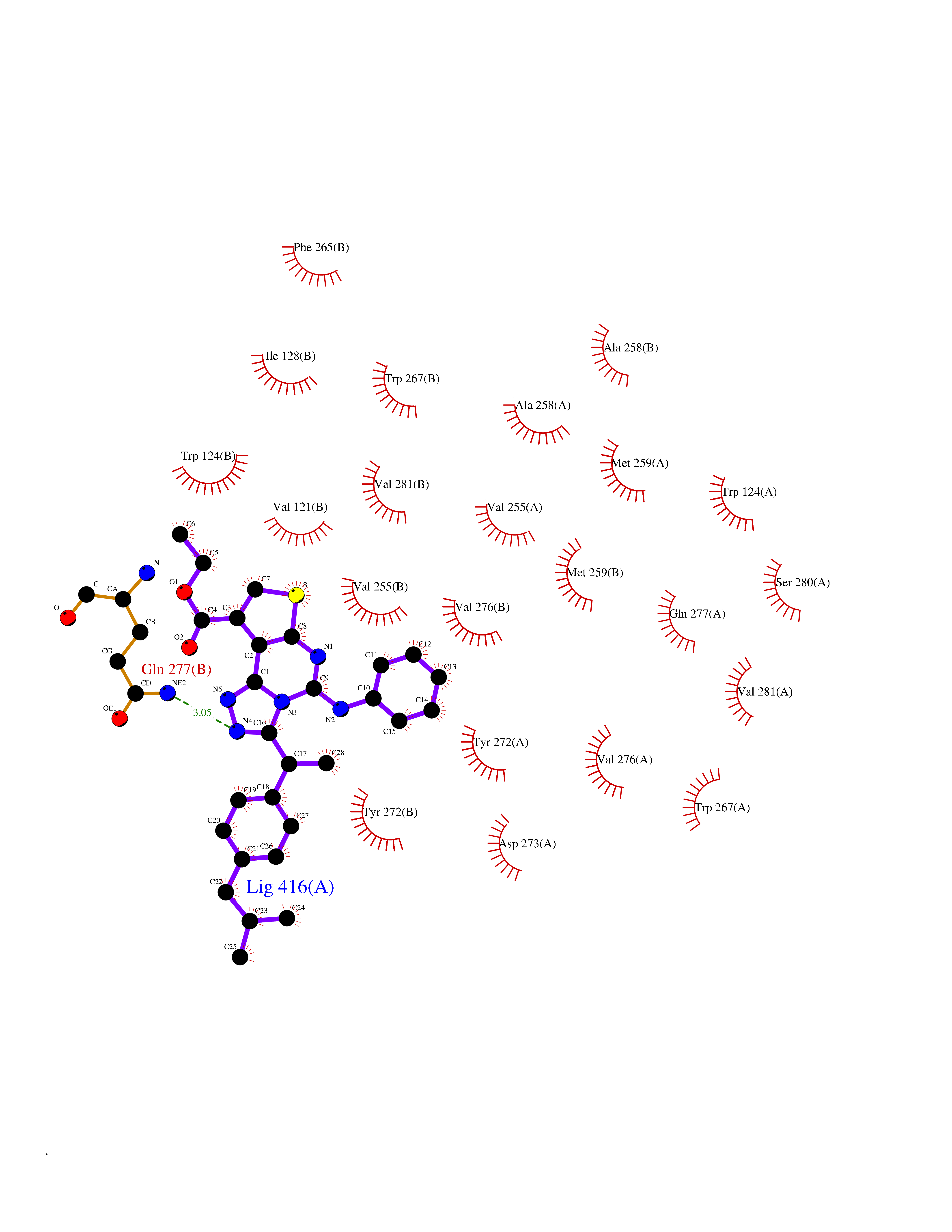 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Adrenergic receptor beta-2 (ADRB2) | 2RH1 | 8.43 | |
Target general information Gen name ADRB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Beta-2 adrenoreceptor; Beta-2 adrenoceptor; Beta-2 adrenergic receptor; B2AR; ADRB2R Protein family G-protein coupled receptor 1 family, Adrenergic receptor subfamily, ADRB2 sub-subfamily Biochemical class GPCR rhodopsin Function The beta-2-adrenergic receptor binds epinephrine with an approximately 30-fold greater affinity than it does norepinephrine. Beta-adrenergic receptors mediate the catecholamine-induced activation of adenylate cyclase through the action of G proteins. Related diseases Cortical dysplasia, complex, with other brain malformations 6 (CDCBM6) [MIM:615771]: A disorder of aberrant neuronal migration and disturbed axonal guidance. Affected individuals have microcephaly, ataxia, and severe delayed psychomotor development. Brain imaging shows variable malformations of cortical development, including white matter streaks, dysmorphic basal ganglia, corpus callosum abnormalities, brainstem and cerebellar hypoplasia, cortical dysplasia, polymicrogyria. {ECO:0000269|PubMed:23246003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Skin creases, congenital symmetric circumferential, 1 (CSCSC1) [MIM:156610]: An autosomal dominant disease characterized by multiple, symmetric, circumferential rings of folded skin, affecting primarily the limbs. Affected individuals also exhibit intellectual disability, cleft palate, and dysmorphic features. {ECO:0000269|PubMed:26637975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07543; DB01193; DB00866; DB01118; DB00182; DB01102; DB01274; DB01238; DB09204; DB06216; DB00335; DB01408; DB05590; DB09013; DB00195; DB00217; DB01295; DB00612; DB00901; DB08807; DB06726; DB08808; DB00248; DB00521; DB01136; DB04846; DB01407; DB00785; DB01151; DB11273; DB13345; DB00449; DB11278; DB00841; DB09273; DB06262; DB01363; DB01364; DB00668; DB01049; DB11587; DB01288; DB00983; DB05039; DB00221; DB01064; DB00598; DB01210; DB13139; DB01365; DB13624; DB01214; DB00264; DB01203; DB05849; DB04861; DB00368; DB00540; DB00334; DB09080; DB00816; DB01580; DB00715; DB01359; DB00925; DB00397; DB00960; DB01291; DB01366; DB01182; DB00571; DB06814; DB00852; DB01917; DB11124; DB00867; DB01001; DB00938; DB00489; DB03566; DB00127; DB00871; DB00373; DB00726; DB12248; DB09082; DB09185 Interacts with P30542; P07550; P32121; Q96B67; Q9UII2; Q9ULD4-2; Q9NSI6-4; Q5M9N0-2; A0AVK6; Q658K8; O00472; Q15910-2; Q15486; P61978; Q5TCQ9; Q99685; O14745; Q9NR21-5; Q8WVD3; Q9H0X6; Q13573; P12931; Q5T0J7-2; Q8N0U2 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Endosome; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Hydroxylation; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32266.1 Length 282 Aromaticity 0.15 Instability index 36.1 Isoelectric point 8.02 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -11.5 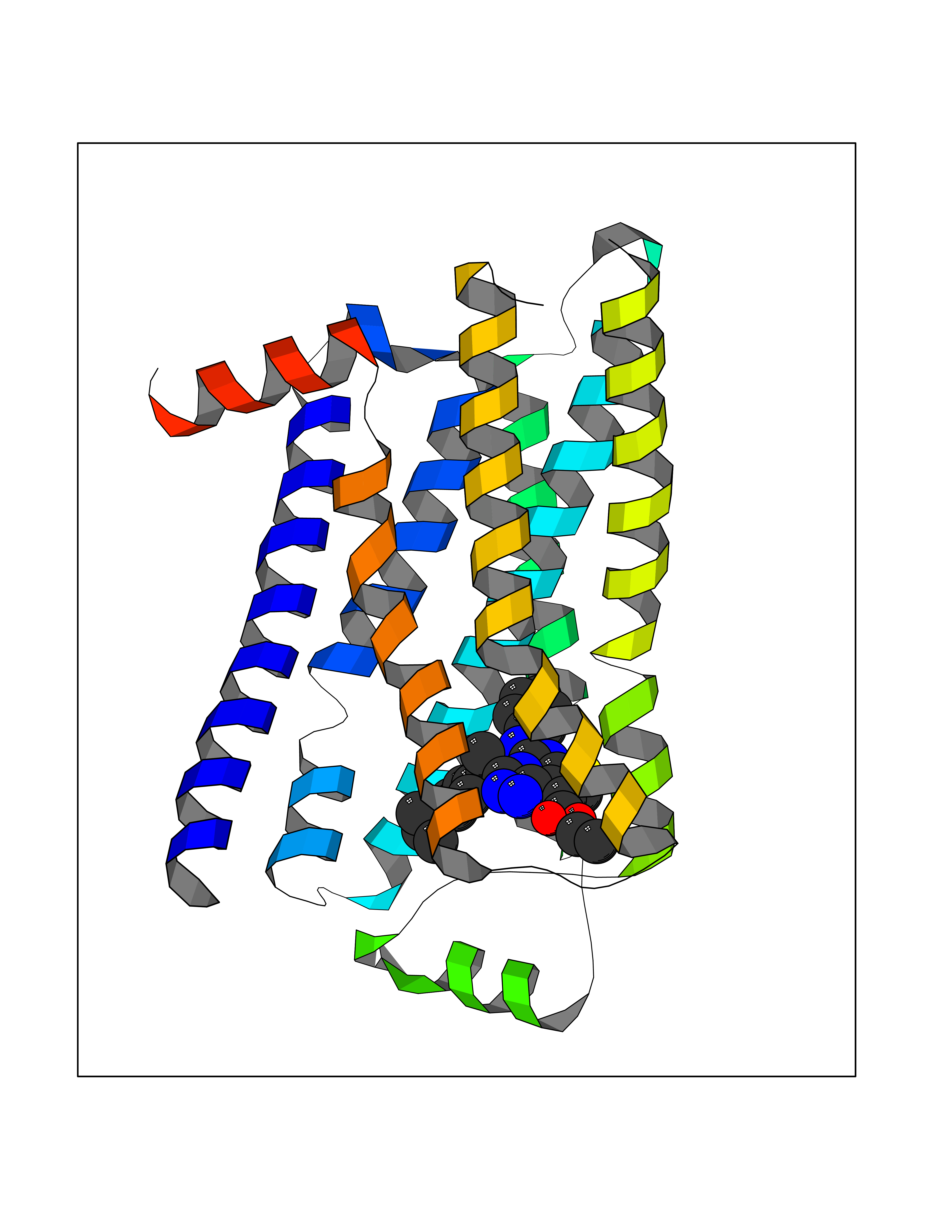 Molscript Map 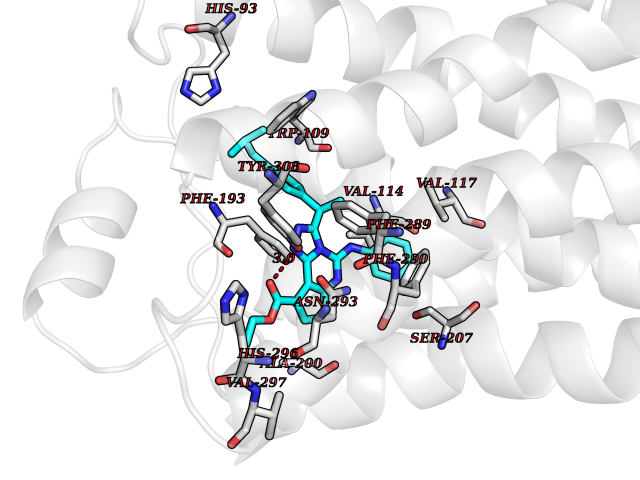 Pymol Map 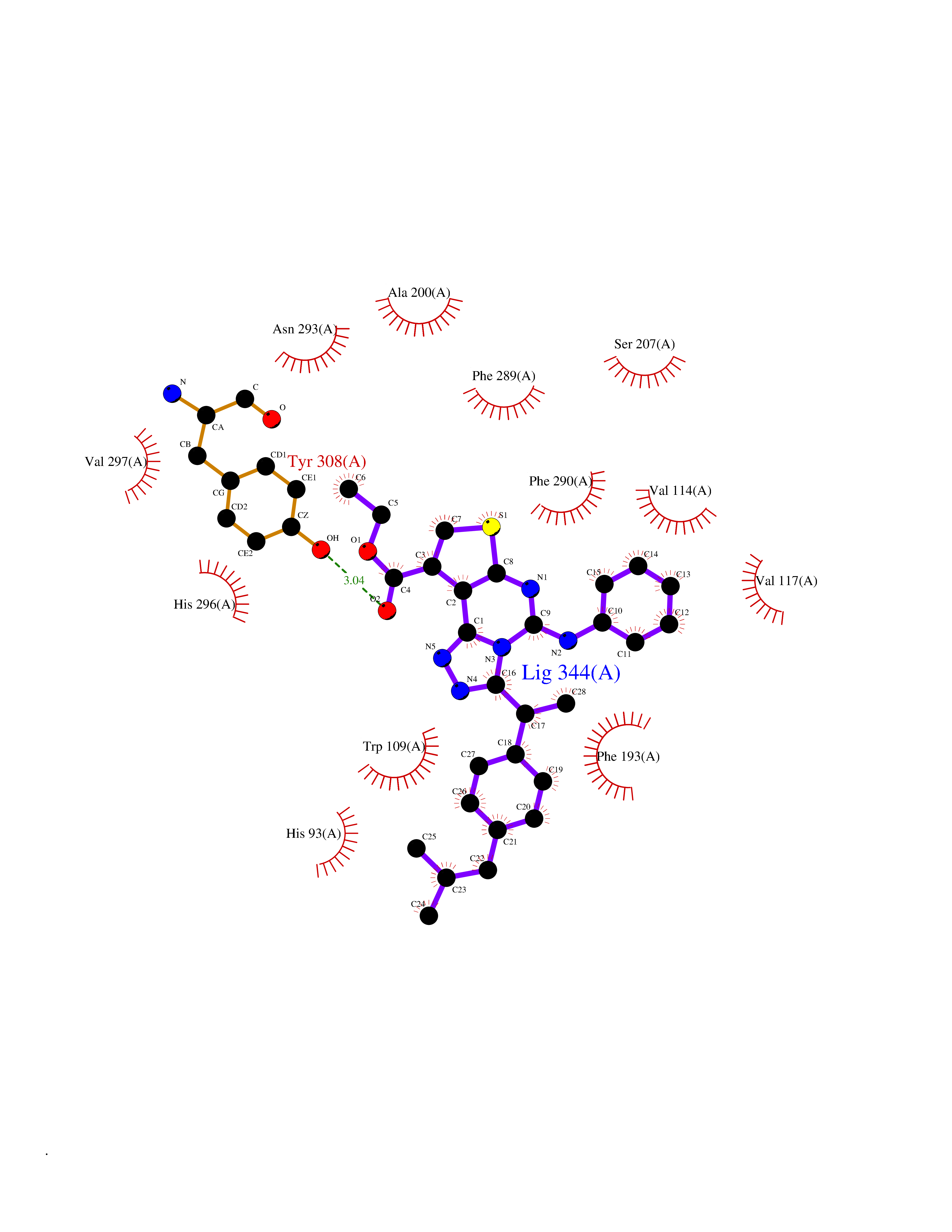 Ligplot Map 3D Binding mode Sequence DEVWVVGMGIVMSLIVLAIVFGNVLVITAIAKFERLQTVTNYFITSLACADLVMGLAVVPFGAAHILMKMWTFGNFWCEFWTSIDVLCVTASIETLCVIAVDRYFAITSPFKYQSLLTKNKARVIILMVWIVSGLTSFLPIQMHWYRATHQEAINCYAEETCCDFFTNQAYAIASSIVSFYVPLVIMVFVYSRVFQEAKRQLKFCLKEHKALKTLGIIMGTFTLCWLPFFIVNIVHVIQDNLIRKEVYILLNWIGYVNSGFNPLIYCRSPDFRIAFQELLCL Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Folate receptor beta (FOLR2) | 4KN0 | 8.43 | |
Target general information Gen name FOLR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Placental folate-binding protein; Folate receptor, fetal/placental; Folate receptor type-beta; Folate receptor 2; FR-beta; FOLR2 Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pH after receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00158; DB00563; DB05168 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disulfide bond; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23841.6 Length 205 Aromaticity 0.12 Instability index 56.78 Isoelectric point 7.92 Charge (pH=7) 2.58 2D Binding mode Binding energy (Kcal/mol) -11.5 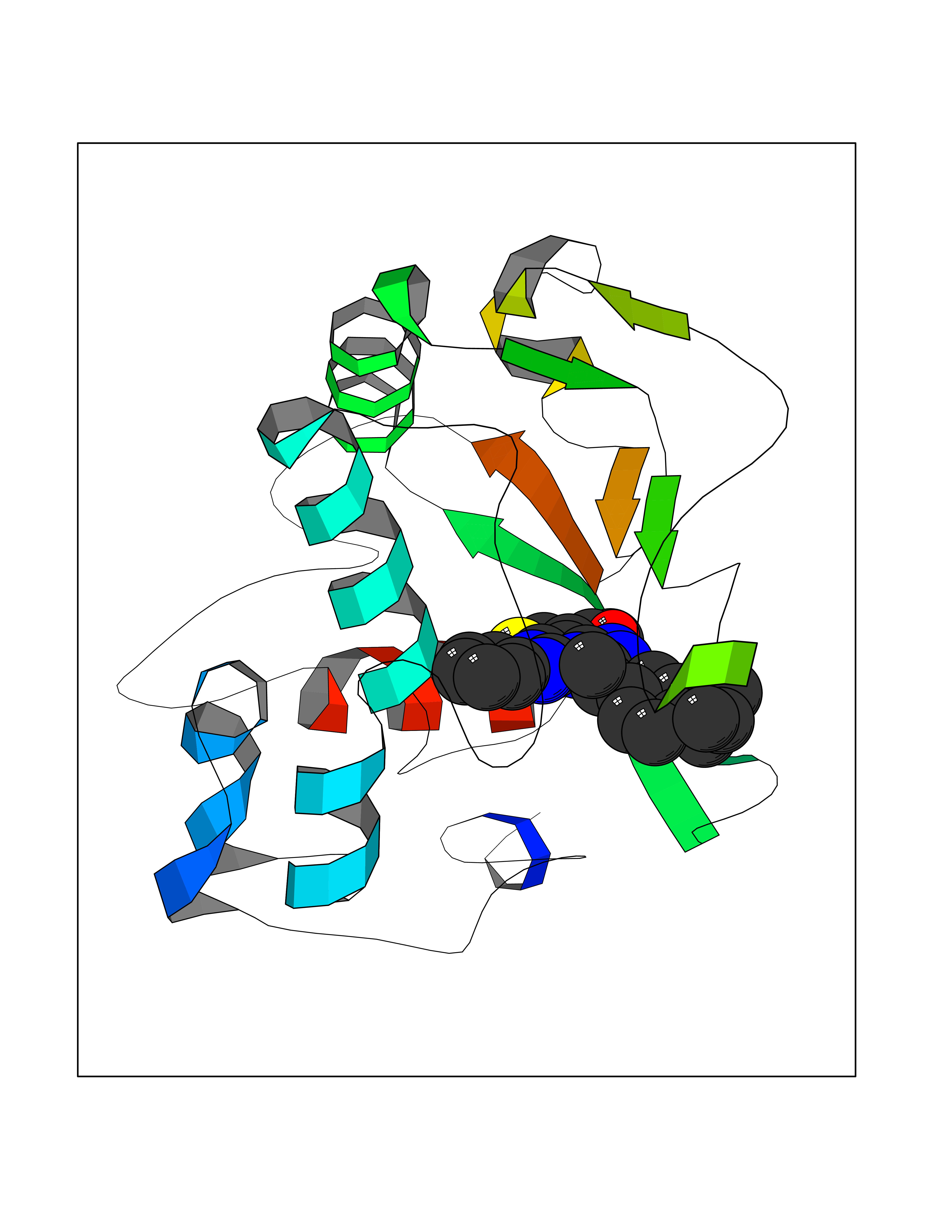 Molscript Map 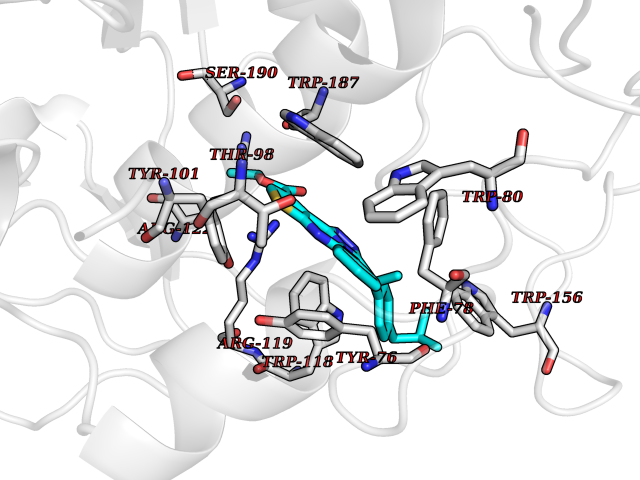 Pymol Map 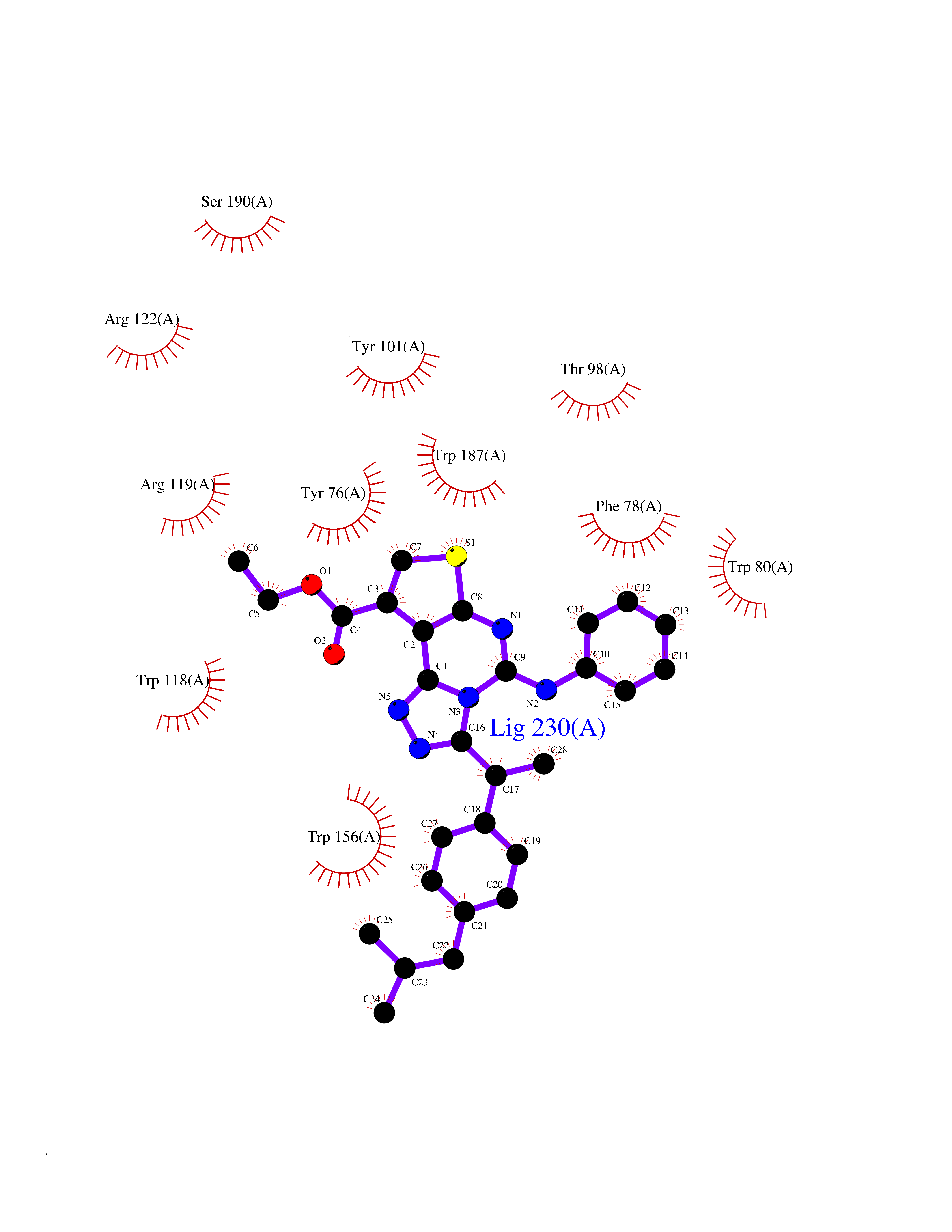 Ligplot Map 3D Binding mode Sequence RTDLLNVCMDAKHHKTKPGPEDKLHDQCSPWKKNACCTASTSQELHKDTSRLYNFNWDHCGKMEPACKRHFIQDTCLYECSPNLGPWIQQVNQSWRKERFLDVPLCKEDCQRWWEDCHTSHTCKSNWHRGWDWTSGVNKCPAGALCRTFESYFPTPAALCEGLWSHSYKVSNYSRGSGRCIQMWFDSAQGNPNEEVARFYAAAMH Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Histone-lysine N-methyltransferase EHMT2 (EHMT2) | 5VSC | 8.43 | |
Target general information Gen name EHMT2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein G9a; NG36; Lysine N-methyltransferase 1C; KMT1C; Histone H3-K9 methyltransferase 3; HLA-B-associated transcript 8; H3-K9-HMTase 3; G9A; Euchromatic histone-lysine N-methyltransferase 2; C6orf3 Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar3-9 subfamily Biochemical class Methyltransferase Function H3K9me represents a specific tag for epigenetic transcriptional repression by recruiting HP1 proteins to methylated histones. Also mediates monomethylation of 'Lys-56' of histone H3 (H3K56me1) in G1 phase, leading to promote interaction between histone H3 and PCNA and regulating DNA replication. Also weakly methylates 'Lys-27' of histone H3 (H3K27me). Also required for DNA methylation, the histone methyltransferase activity is not required for DNA methylation, suggesting that these 2 activities function independently. Probably targeted to histone H3 by different DNA-binding proteins like E2F6, MGA, MAX and/or DP1. May also methylate histone H1. In addition to the histone methyltransferase activity, also methylates non-histone proteins: mediates dimethylation of 'Lys-373' of p53/TP53. Also methylates CDYL, WIZ, ACIN1, DNMT1, HDAC1, ERCC6, KLF12 and itself. Histone methyltransferase that specifically mono- and dimethylates 'Lys-9' of histone H3 (H3K9me1 and H3K9me2, respectively) in euchromatin. Related diseases Pseudohypoaldosteronism 2C (PHA2C) [MIM:614492]: An autosomal dominant disorder characterized by severe hypertension, hyperkalemia, hyperchloremia, mild hyperchloremic metabolic acidosis in some cases, and correction of physiologic abnormalities by thiazide diuretics. {ECO:0000269|PubMed:11498583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuropathy, hereditary sensory and autonomic, 2A (HSAN2A) [MIM:201300]: A form of hereditary sensory and autonomic neuropathy, a genetically and clinically heterogeneous group of disorders characterized by degeneration of dorsal root and autonomic ganglion cells, and by sensory and/or autonomic abnormalities. HSAN2A is an autosomal recessive disorder characterized by impairment of pain, temperature and touch sensation, onset of symptoms in infancy or early childhood, occurrence of distal extremity pathologies (paronychia, whitlows, ulcers, and Charcot joints), frequent amputations, sensory loss that affects all modalities of sensation (lower and upper limbs and perhaps the trunk as well), absence or diminution of tendon reflexes (usually in all limbs), minimal autonomic dysfunction, absence of sensory nerve action potentials, and virtual absence of myelinated fibers with decreased numbers of unmyelinated fibers in sural nerves. {ECO:0000269|PubMed:15060842, ECO:0000269|PubMed:15911806, ECO:0000269|PubMed:18521183}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q6VMQ6-2; Q6P1J9; Q9UBC3; P38919; Q9UM22; P23771; Q99684; Q13547; Q96JB3; Q92831; O60341-1; Q9Y4X4; P57682; Q13330; O94776; Q9BTC8; P20592; Q9BSU3; Q99801-1; O60568; Q9NQX1; Q5JSZ5; Q7Z3Z2; Q9P2R6; Q14119; Q96GT9; O60315; Q9NWS9-2; Q96JM2; A0A0S2Z5X4; Q96BV0; Q96EG3; Q07120; O60341-1 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ANK repeat; Chromatin regulator; Chromosome; Isopeptide bond; Metal-binding; Methylation; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-adenosyl-L-methionine; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 31010.9 Length 269 Aromaticity 0.1 Instability index 47.49 Isoelectric point 5.16 Charge (pH=7) -9.31 2D Binding mode Binding energy (Kcal/mol) -11.5 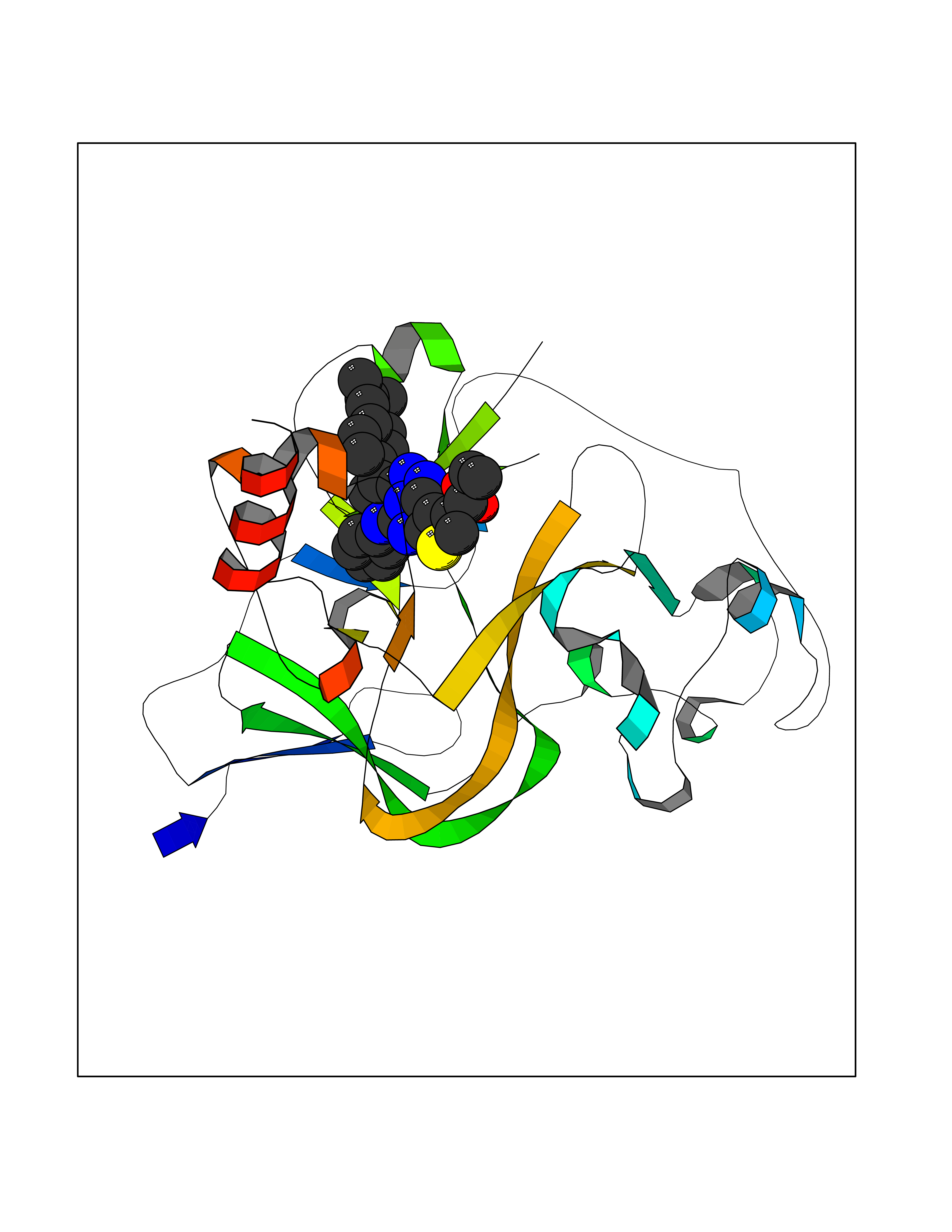 Molscript Map 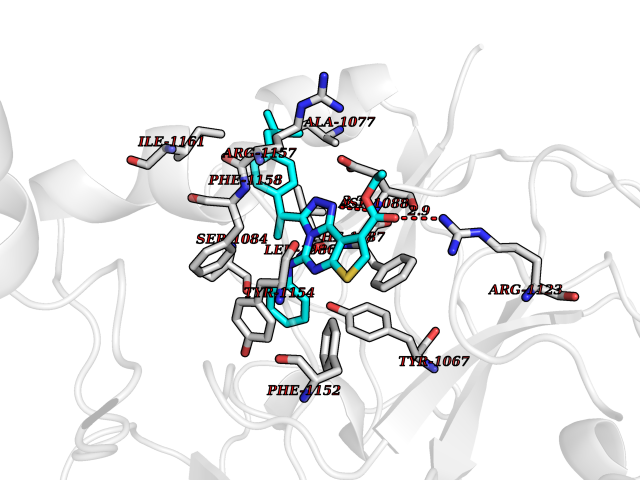 Pymol Map 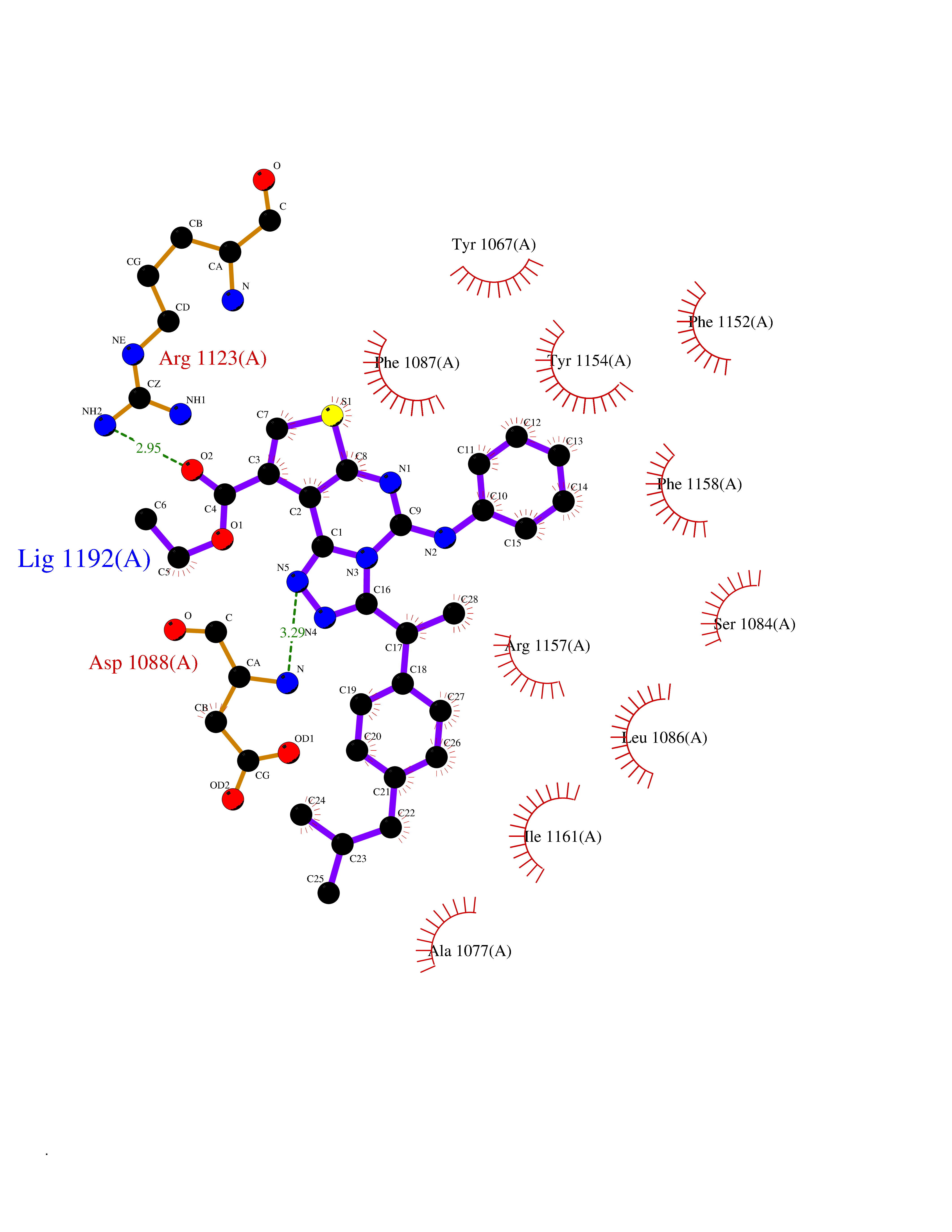 Ligplot Map 3D Binding mode Sequence TEKIICRDVARGYENVPIPCVNGVDGEPCPEDYKYISENCETSTMNIDRNITHLQHCTCVDDCSSSNCLCGQLSIRCWYDKDGRLLQEFNKIEPPLIFECNQACSCWRNCKNRVVQSGIKVRLQLYRTAKMGWGVRALQTIPQGTFICEYVGELISDAEADVREDDSYLFDLDEVYCIDARYYGNISRFINHLCDPNIIPVRVFMLHQDLRFPRIAFFSSRDIRTGEELGFDYGDRFWDIKSKYFTCQCGSEKCKHSAEAIALEQSRLA Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Pyruvate kinase M2 (PKM) | 3GR4 | 8.42 | |
Target general information Gen name PKM Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p58; Tumor M2-PK; Thyroid hormone-binding protein 1; THBP1; Pyruvate kinase muscle isozyme; Pyruvate kinase isozymes M1/M2; Pyruvate kinase PKM; Pyruvate kinase 2/3; PKM2; PK3; PK2; Opa-interacting pr Protein family Pyruvate kinase family Biochemical class Kinase Function Stimulates POU5F1-mediated transcriptional activation. Plays a general role in caspase independent cell death of tumor cells. The ratio between the highly active tetrameric form and nearly inactive dimeric form determines whether glucose carbons are channeled to biosynthetic processes or used for glycolytic ATP production. The transition between the 2 forms contributes to the control of glycolysis and is important for tumor cell proliferation and survival. Promotes in a STAT1-dependent manner, the expression of the immune checkpoint protein CD274 in ARNTL/BMAL1-deficient macrophages. Glycolytic enzyme that catalyzes the transfer of a phosphoryl group from phosphoenolpyruvate (PEP) to ADP, generating ATP. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07697; DB07692; DB02726; DB07628; DB00787; DB11638; DB09130; DB08951; DB01733; DB11263; DB00119 Interacts with P49407; P32121; Q96IK1-2; P35222; P53355; P22607; P42858; P04049; Q8N488; Q7Z699; Q9BSI4; Q9UMX0; Q9Y649; Q9WMX2; P35222; P53355; Q9H6Z9; P68431; Q16665; P27361 EC number EC 2.7.1.40 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; ATP-binding; Cytoplasm; Direct protein sequencing; Glycolysis; Hydroxylation; Isopeptide bond; Kinase; Magnesium; Metal-binding; Methylation; Nucleotide-binding; Nucleus; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; S-nitrosylation; Transferase; Translation regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 112053 Length 1024 Aromaticity 0.05 Instability index 27.06 Isoelectric point 7.34 Charge (pH=7) 1.66 2D Binding mode Binding energy (Kcal/mol) -11.49 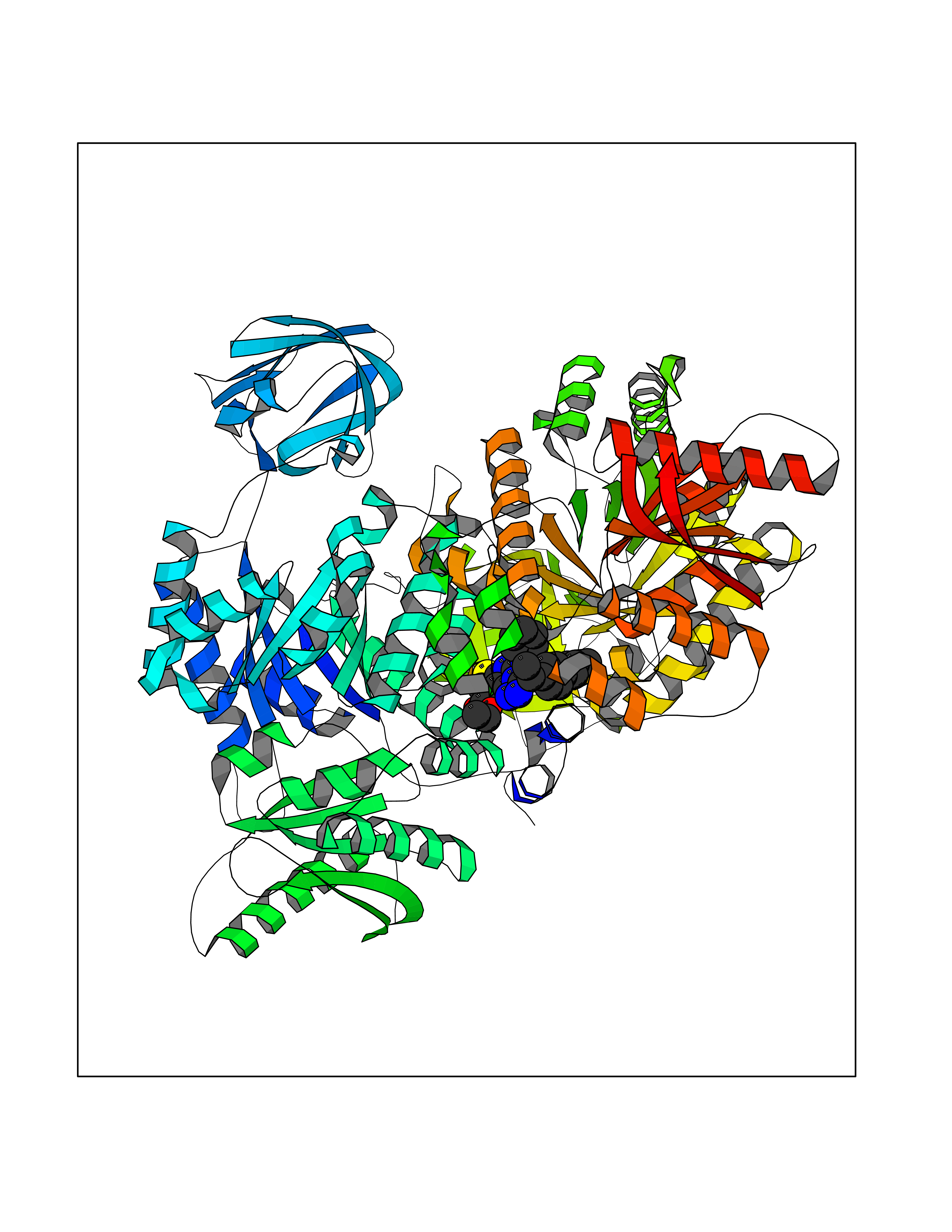 Molscript Map 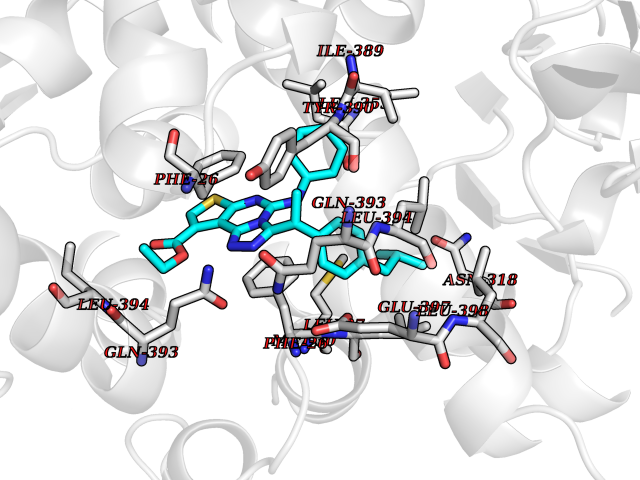 Pymol Map 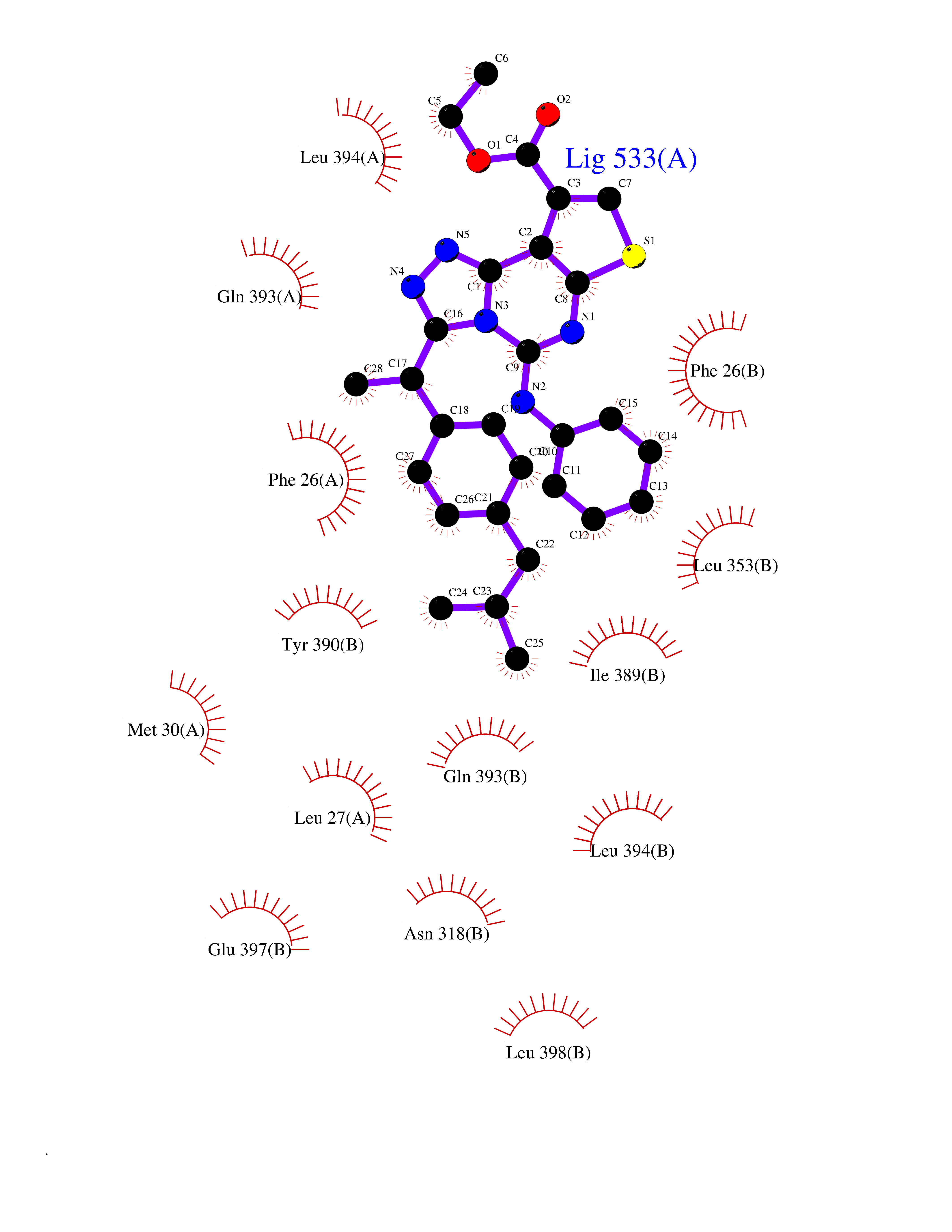 Ligplot Map 3D Binding mode Sequence IQTQQLHAAMADTFLEHMCRLDIDSPPITARNTGIICTIGPASRSVETLKEMIKSGMNVARLNFSHGTHEYHAETIKNVRTATESFASDPILYRPVAVALDTKGPEIRTGLIKGSGTAEVELKKGATLKITLDNAYMEKCDENILWLDYKNICKVVEVGSKIYVDDGLISLQVKQKGADFLVTEVENGGSLGSKKGVNLPGAAVDLPAVSEKDIQDLKFGVEQDVDMVFASFIRKASDVHEVRKVLGEKGKNIKIISKIENHEGVRRFDEILEASDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNRAGKPVICATQMLESMIKKPRPTRAEGSDVANAVLDGADCIMLSGETAKGDYPLEAVRMQHLIAREAEAAIYHLQLFEELRRLAPITSDPTEATAVGAVEASFKCCSGAIIVLTKSGRSAHQVARYRPRAPIIAVTRNPQTARQAHLYRGIFPVLCKDPVQEAWAEDVDLRVNFAMNVGKARGFFKKGDVVIVLTGWRPGSGFTNTMRVVPVPIQTQQLHAAMADTFLEHMCRLDIDSPPITARNTGIICTIGPASRSVETLKEMIKSGMNVARLNFSHGTHEYHAETIKNVRTATESFASDPILYRPVAVALDTKGPEIRTGLIKEVEATLKITLDNAYMEKCDENILWLDYKNICKVVEVGSKIYVDDGLISLQVDFLVTEVENGGSLGSKKGVNLPGAAVDLPAVSEKDIQDLKFGVEQDVDMVFASFIRKASDVHEVRKVLGEKGKNIKIISKIENHEGVRRFDEILEASDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNRAGKPVICATQMLESMIKKPRPTRAEGSDVANAVLDGADCIMLSGETAKGDYPLEAVRMQHLIAREAEAAIYHLQLFEELRRLAPITSDPTEATAVGAVEASFKCCSGAIIVLTKSGRSAHQVARYRPRAPIIAVTRNPQTARQAHLYRGIFPVLCKDPVQEAWAEDVDLRVNFAMNVGKARGFFKKGDVVIVLTGWRPGSGFTNTMRVVPVP Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | 5-HT 7 receptor (HTR7) | 7XTC | 8.42 | |
Target general information Gen name HTR7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serotonin receptor 7; 5HT7; 5-hydroxytryptamine receptor 7; 5-HT7 receptor; 5-HT7; 5-HT-X; 5-HT-7 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function The activity of this receptor is mediated by G proteins that stimulate adenylate cyclase. This is one of the several different receptors for 5-hydroxytryptamine (serotonin), a biogenic hormone that functions as a neurotransmitter, a hormone, and a mitogen. Related diseases Immunodeficiency 35 (IMD35) [MIM:611521]: A primary immunodeficiency characterized by recurrent skin abscesses, pneumonia, and highly elevated serum IgE. {ECO:0000269|PubMed:17088085}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06288; DB00321; DB00543; DB01238; DB14185; DB06216; DB09128; DB01200; DB00248; DB00477; DB01239; DB00363; DB00924; DB00434; DB11273; DB13345; DB00988; DB00751; DB01049; DB12141; DB00502; DB04946; DB00458; DB00589; DB04948; DB00408; DB08815; DB00934; DB00247; DB06148; DB01267; DB00715; DB01224; DB00734; DB00953; DB13988; DB09304; DB13025; DB09068; DB00246; DB00315; DB09225 Interacts with P43243 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 30879.7 Length 275 Aromaticity 0.14 Instability index 36.79 Isoelectric point 9.22 Charge (pH=7) 11.88 2D Binding mode Binding energy (Kcal/mol) -11.49 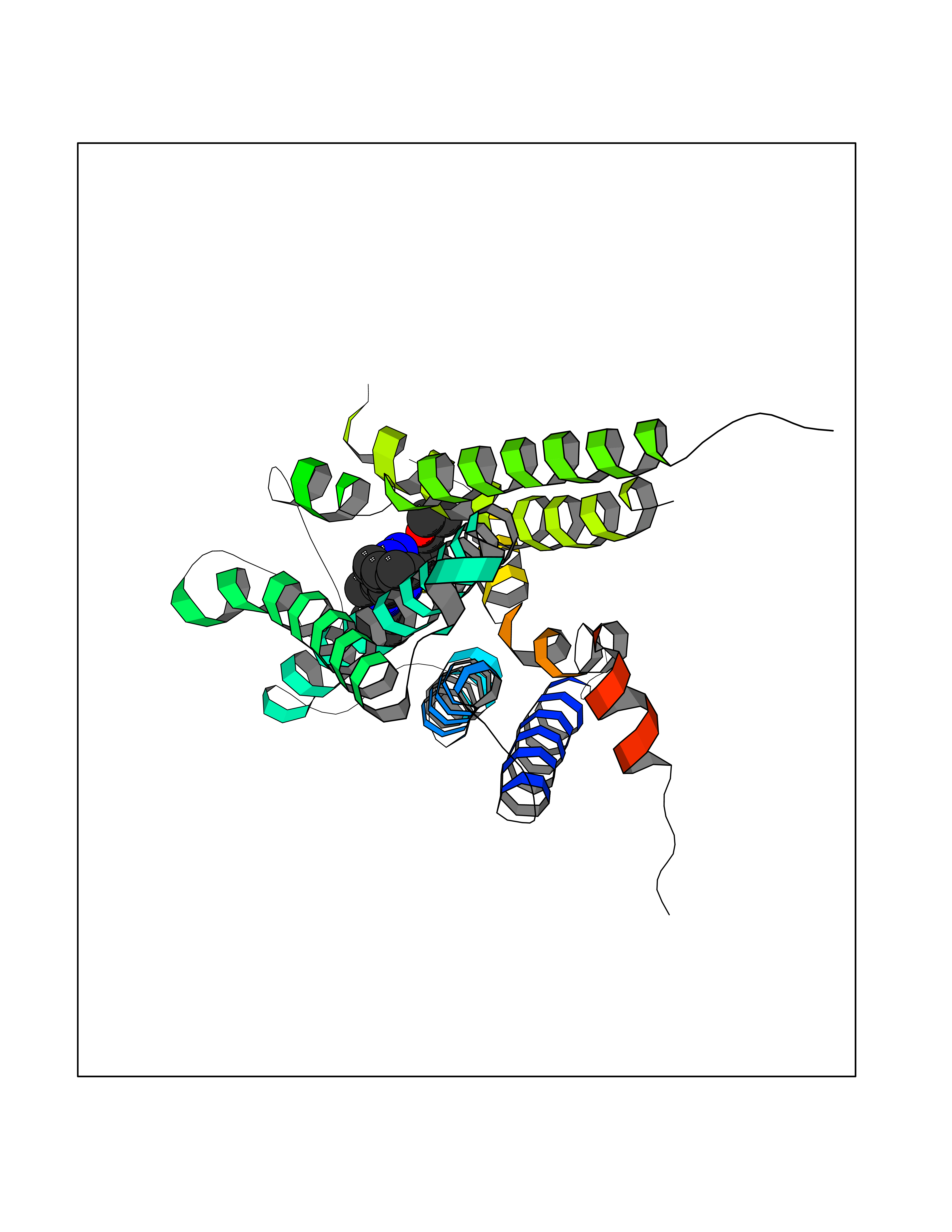 Molscript Map 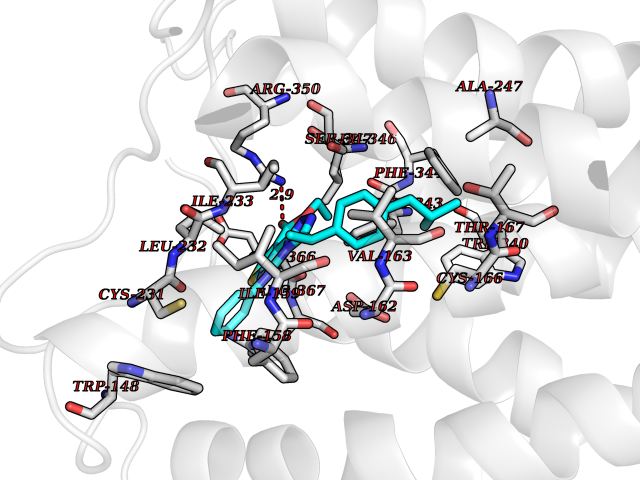 Pymol Map 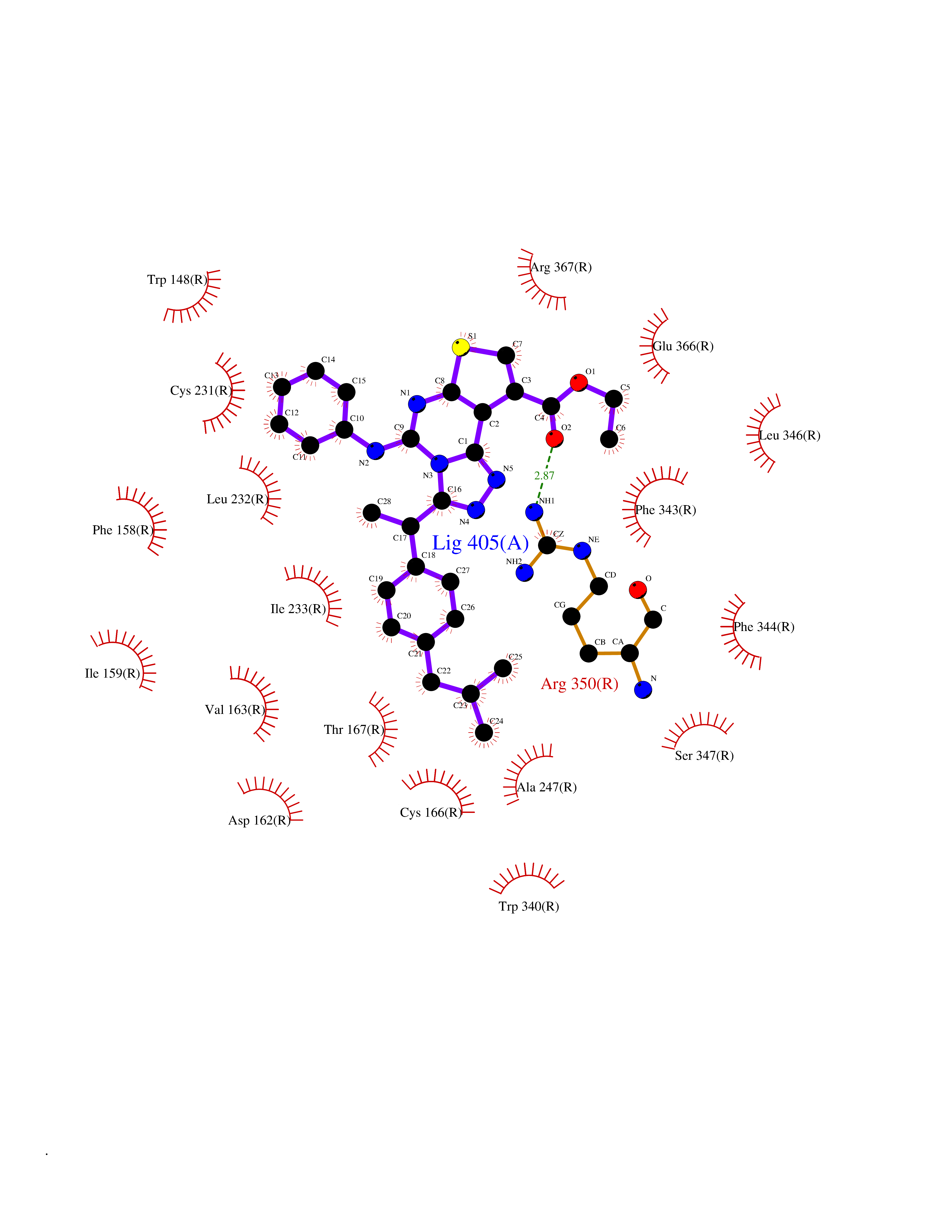 Ligplot Map 3D Binding mode Sequence EKVVIGSILTLITLLTIAGNCLVVISVCFVKKLRQPSNYLIVSLALADLSVAVAVMPFVSVTDLIGGKWIFGHFFCNVFIAMDVMCCTASIMTLCVISIDRYLGITRPLTYPVRQNGKCMAKMILSVWLLSASITLPPLFGWAQNVNDDKVCLISQDFGYTIYSTAVAFYIPMSVMLFMYYQIYKAARKSAAKHKFPGFPKAATTLGIIVGAFTVCWLPFFLLSTARPFICCIPLWVERTFLWLGYANSLINPFIYAFFNRDLRTTYRSLLQCQY Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Cytochrome c oxidase subunit 1 | 3DTU | 8.41 | |
Target general information Gen name ctaD Organism Cereibacter sphaeroides (Rhodobacter sphaeroides) Uniprot ID TTD ID NA Synonyms NA Protein family Heme-copper respiratory oxidase family Biochemical class Oxidoreductase Function Copper ion binding.Cytochrome-c oxidase activity.Heme binding.Iron ion binding. Related diseases Cystathioninuria (CSTNU) [MIM:219500]: Autosomal recessive phenotype characterized by abnormal accumulation of plasma cystathionine, leading to increased urinary excretion. {ECO:0000269|PubMed:12574942, ECO:0000269|PubMed:18476726}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03619 Interacts with Q03736 EC number 7.1.1.9 Uniprot keywords 3D-structure; Cell membrane; Copper; Electron transport; Heme; Hydrogen ion transport; Ion transport; Iron; Membrane; Metal-binding; Respiratory chain; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,C Molecular weight (Da) 88419.8 Length 794 Aromaticity 0.15 Instability index 40.59 Isoelectric point 6.09 Charge (pH=7) -10.78 2D Binding mode Binding energy (Kcal/mol) -11.47 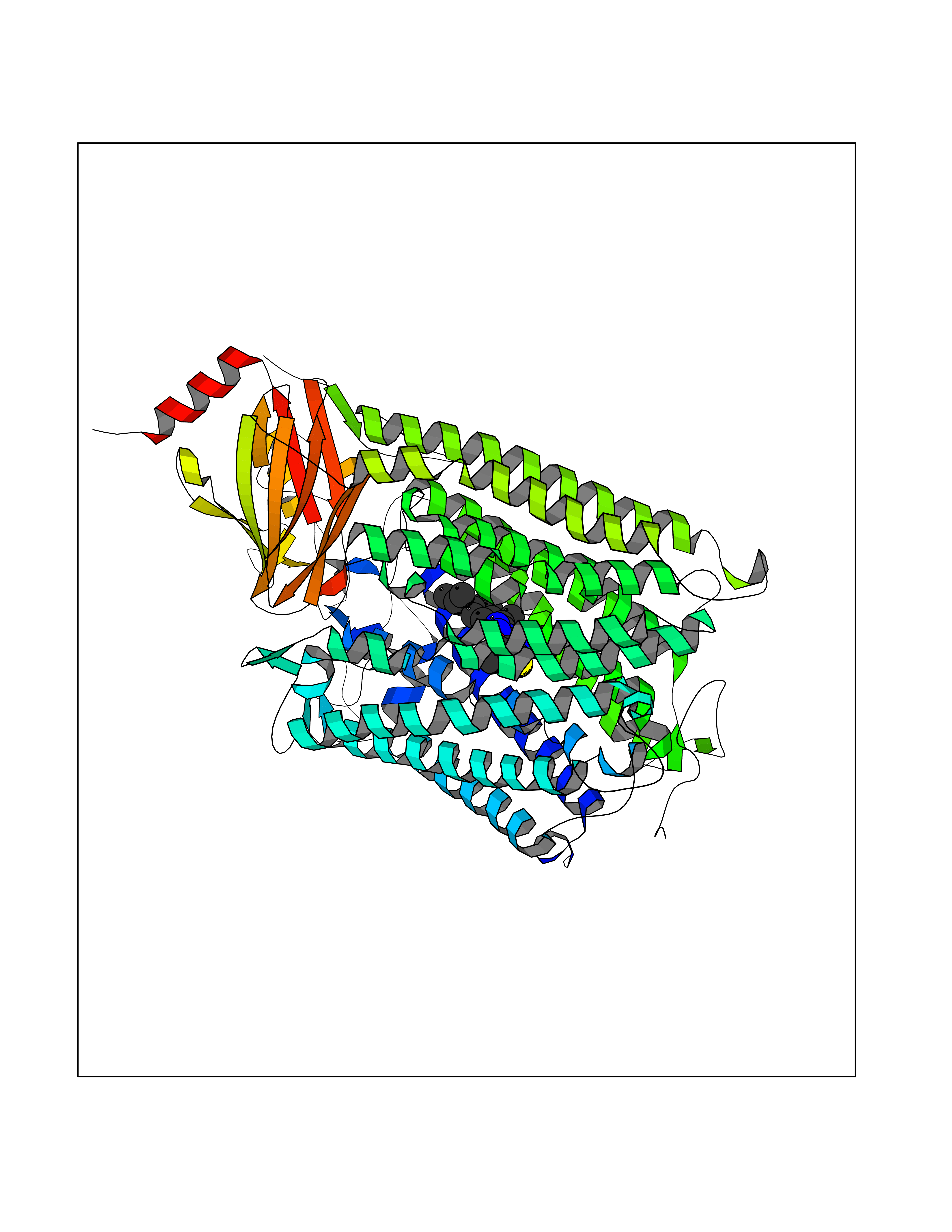 Molscript Map 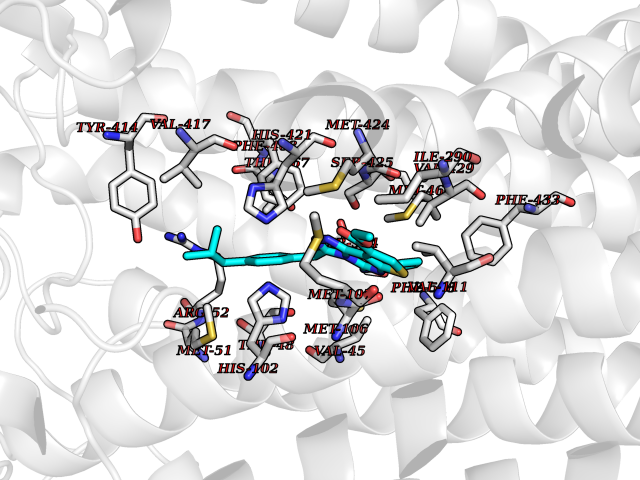 Pymol Map 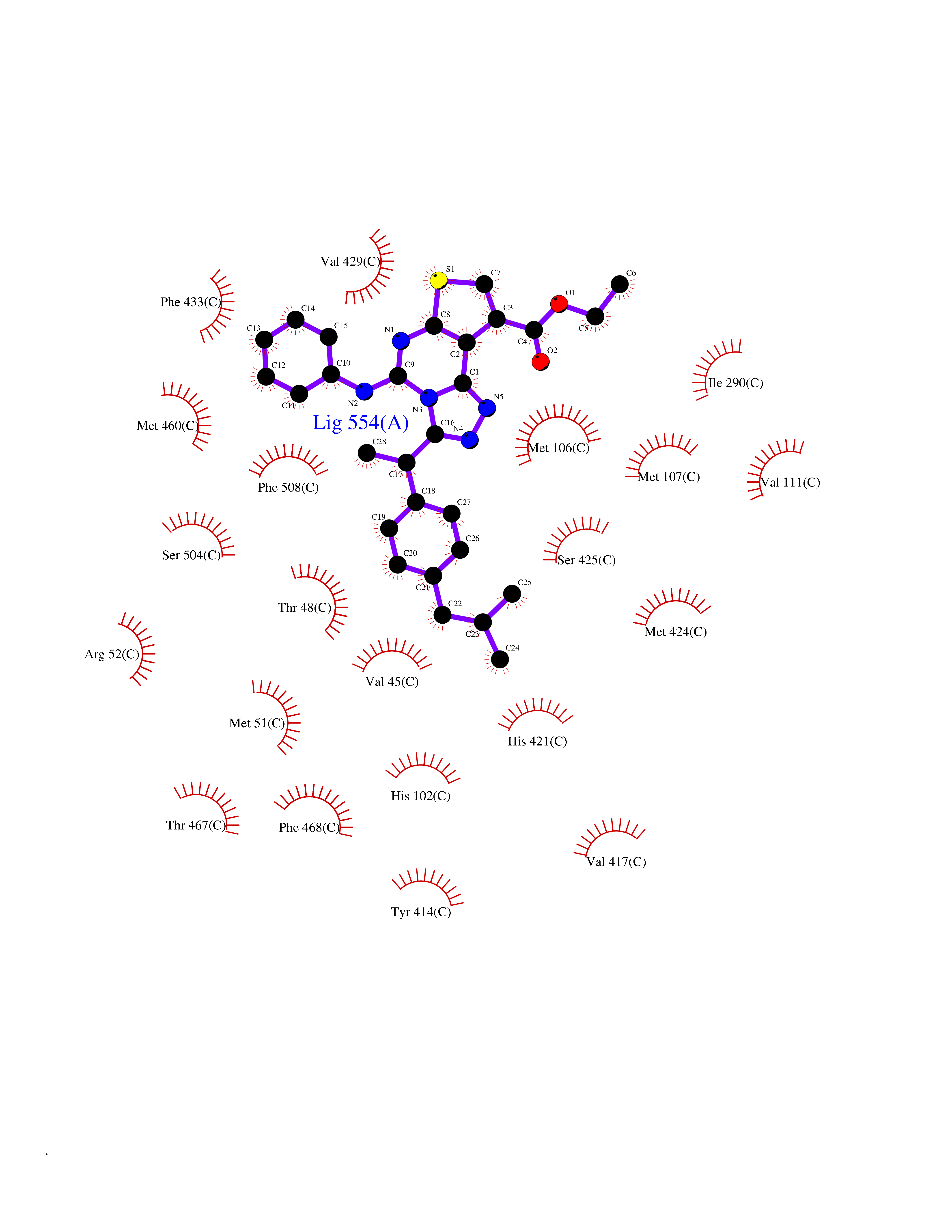 Ligplot Map 3D Binding mode Sequence FTRWFMSTNHKDIGVLYLFTGGLVGLISVAFTVYMRMELMAPGVQFMCAEHLESGLVKGFFQSLWPSAVENCTPNGHLWNVMITGHGILMMFFVVIPALFGGFGNYFMPLHIGAPDMAFPRMNNLSYWLYVAGTSLAVASLFAPGGNGQLGSGIGWVLYPPLSTSESGYSTDLAIFAVHLSGASSILGAINMITTFLNMRAPGMTMHKVPLFAWSIFVTAWLILLALPVLAGAITMLLTDRNFGTTFFQPSGGGDPVLYQHILWFFGHPEVYIIVLPAFGIVSHVIATFAKKPIFGYLPMVYAMVAIGVLGFVVWAHHMYTAGLSLTQQSYFMMATMVIAVPTGIKIFSWIATMWGGSIELKTPMLWALGFLFLFTVGGVTGIVLSQASVDRYYHDTYYVVAHFHYVMSLGAVFGIFAGIYFWIGKMSGRQYPEWAGKLHFWMMFVGANLTFFPQHFLGRQGMPRRYIDYPEAFATWNFVSSLGAFLSFASFLFFLGVIFYTLTRGARVTANNYWNEHADTLEWTLTSPPPEHTFEQSLEIIGRPQPGGTGFQPSASPVATQIHWLDGFILVIIAAITIFVTLLILYAVWRFHEKRNKVPARFTHNSPLEIAWTIVPIVILVAIGAFSLPVLFNQQEIPEADVTVKVTGYQWYWGYEYPDEEISFESYMIGSPATGGDNRMSPEVEQQLIEAGYSRDEFLLATDTAMVVPVNKTVVVQVTGADVIHSWTVPAFGVKQDAVPGRLAQLWFRAEREGIFFGQCSELCGISHAYMPITVKVVSEEAYAAWLEQHHHH Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Peroxisome proliferator-activated receptor alpha (PPARA) | 3VI8 | 8.41 | |
Target general information Gen name PPARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peroxisome proliferater-activated receptor alpha; PPARalpha; PPAR-alpha; PPAR; Nuclear receptor subfamily 1 group C member 1; NR1C1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Key regulator of lipid metabolism. Activated by the endogenous ligand 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine (16:0/18:1-GPC). Activated by oleylethanolamide, a naturally occurring lipid that regulates satiety. Receptor for peroxisome proliferators such as hypolipidemic drugs and fatty acids. Regulates the peroxisomal beta-oxidation pathway of fatty acids. Functions as transcription activator for the ACOX1 and P450 genes. Transactivation activity requires heterodimerization with RXRA and is antagonized by NR2C2. May be required for the propagation of clock information to metabolic pathways regulated by PER2. Ligand-activated transcription factor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08915; DB00132; DB01118; DB04557; DB01393; DB04519; DB05416; DB09064; DB09006; DB00636; DB09213; DB03756; DB05187; DB06521; DB01039; DB13873; DB00573; DB13961; DB02266; DB01241; DB07215; DB01050; DB00159; DB07724; DB00328; DB12007; DB03017; DB12961; DB06510; DB08231; DB11605; DB01890; DB04224; DB11133; DB03796; DB02746; DB01708; DB06533; DB04971; DB02709; DB00412; DB09422; DB03193; DB06536; DB00197; DB00313 Interacts with P02768-3; P55212; P45973; P06307; Q3L8U1-3; G5E9A7; P22607; P62993; Q14957; P06396; P42858; Q8WXH2; P13473-2; O75376; Q13133; A0A6Q8PF08; P54725; P62826; Q7Z699; P37173; P55072; P55055-1; Q13133 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Biological rhythms; DNA-binding; Lipid-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29322.1 Length 258 Aromaticity 0.09 Instability index 35.53 Isoelectric point 6.09 Charge (pH=7) -3.57 2D Binding mode Binding energy (Kcal/mol) -11.47  Molscript Map 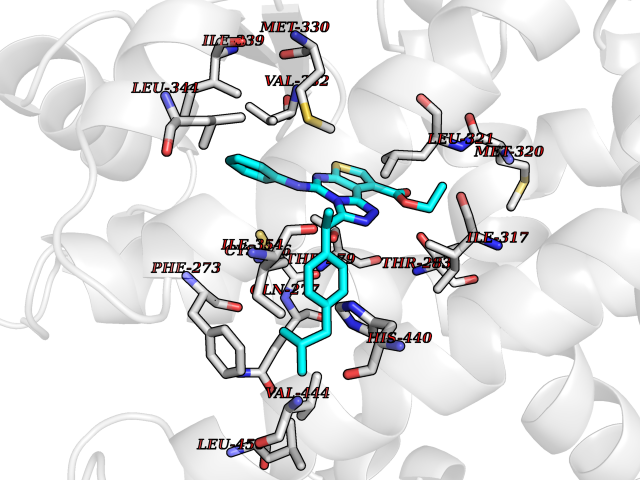 Pymol Map 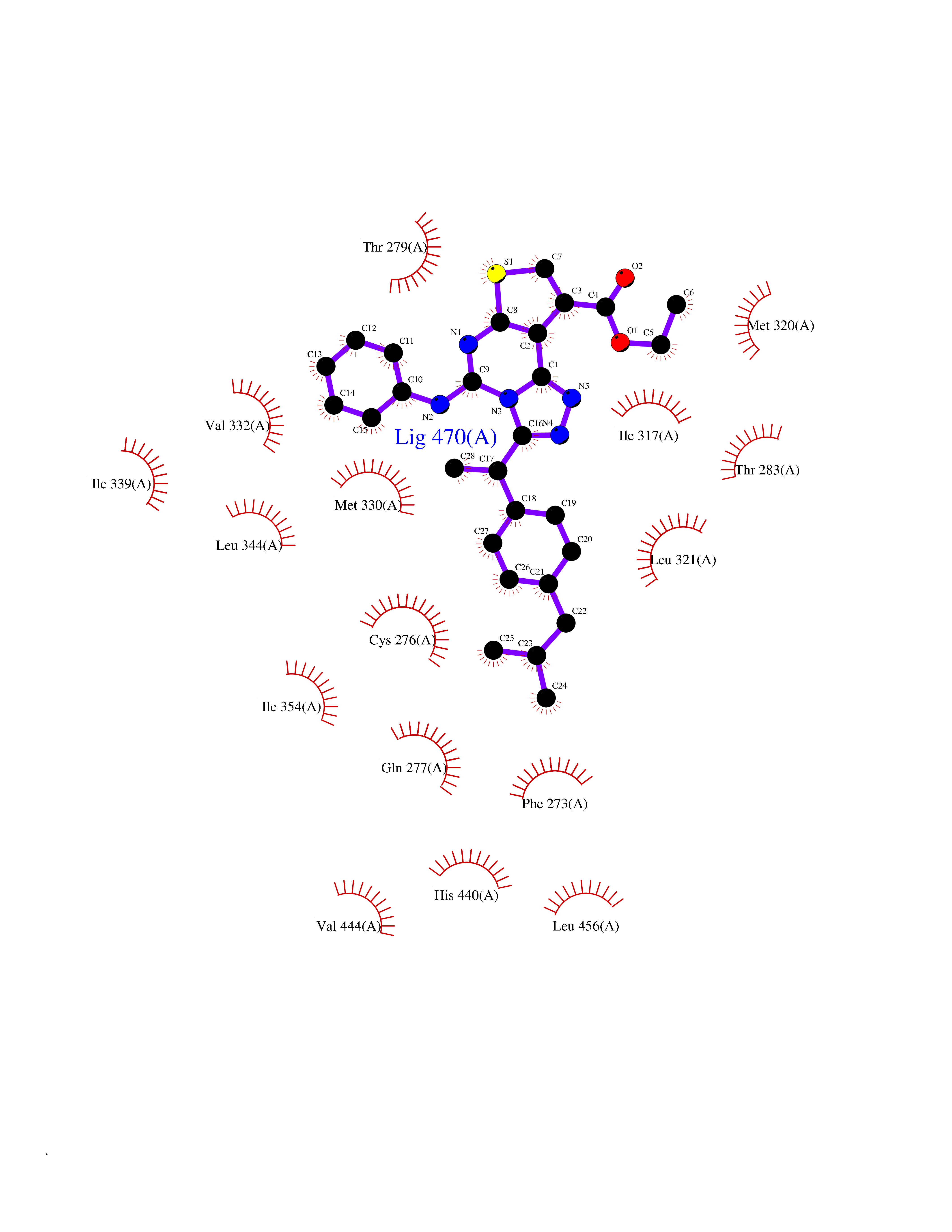 Ligplot Map 3D Binding mode Sequence DLKSLAKRIYEAYLKNFNMNKVKARVILSPFVIHDMETLCMAEKTLVAKLVANGNKEAEVRIFHCCQCTSVETVTELTEFAKAIPGFANLDLNDQVTLLKYGVYEAIFAMLSSVMNKDGMLVAYGNGFITREFLKSLRKPFCDIMEPKFDFAMKFNALELDDSDISLFVAAIICCGDRPGLLNVGHIEKMQEGIVHVLRLHLQSNHPDDIFLFPKLLQKMADLRQLVTEHAQLVQIIKKTESDAALHPLLQEIYRDMY Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Sphingosine kinase 1 (SPHK1) | 3VZB | 8.41 | |
Target general information Gen name SPHK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SPK 1; SPK; SPHK1; SK 1; Acetyltransferase SPHK1 Protein family NA Biochemical class Kinase Function Acts on D-erythro-sphingosine and to a lesser extent sphinganine, but not other lipids, such as D,L-threo-dihydrosphingosine, N,N-dimethylsphingosine, diacylglycerol, ceramide, or phosphatidylinositol. In contrast to proapoptotic SPHK2, has a negative effect on intracellular ceramide levels, enhances cell growth and inhibits apoptosis. Involved in the regulation of inflammatory response and neuroinflammation. Via the product sphingosine 1-phosphate, stimulates TRAF2 E3 ubiquitin ligase activity, and promotes activation of NF-kappa-B in response to TNF signaling leading to IL17 secretion. In response to TNF and in parallel to NF-kappa-B activation, negatively regulates RANTES inducion through p38 MAPK signaling pathway. Involved in endocytic membrane trafficking induced by sphingosine, recruited to dilate endosomes, also plays a role on later stages of endosomal maturation and membrane fusion independently of its kinase activity. In Purkinje cells, seems to be also involved in the regulation of autophagosome-lysosome fusion upon VEGFA. Catalyzes the phosphorylation of sphingosine to form sphingosine 1-phosphate (SPP), a lipid mediator with both intra- and extracellular functions. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08868 Interacts with P07858; P68104; Q14192; Q2M3C7; Q9Y4K3; P13473-2; Q9Y371 EC number EC 2.7.1.91 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Calmodulin-binding; Cell membrane; Coated pit; Cytoplasm; Endosome; Kinase; Lipid metabolism; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Synapse; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 39813 Length 360 Aromaticity 0.08 Instability index 43.79 Isoelectric point 7.34 Charge (pH=7) 0.84 2D Binding mode Binding energy (Kcal/mol) -11.47 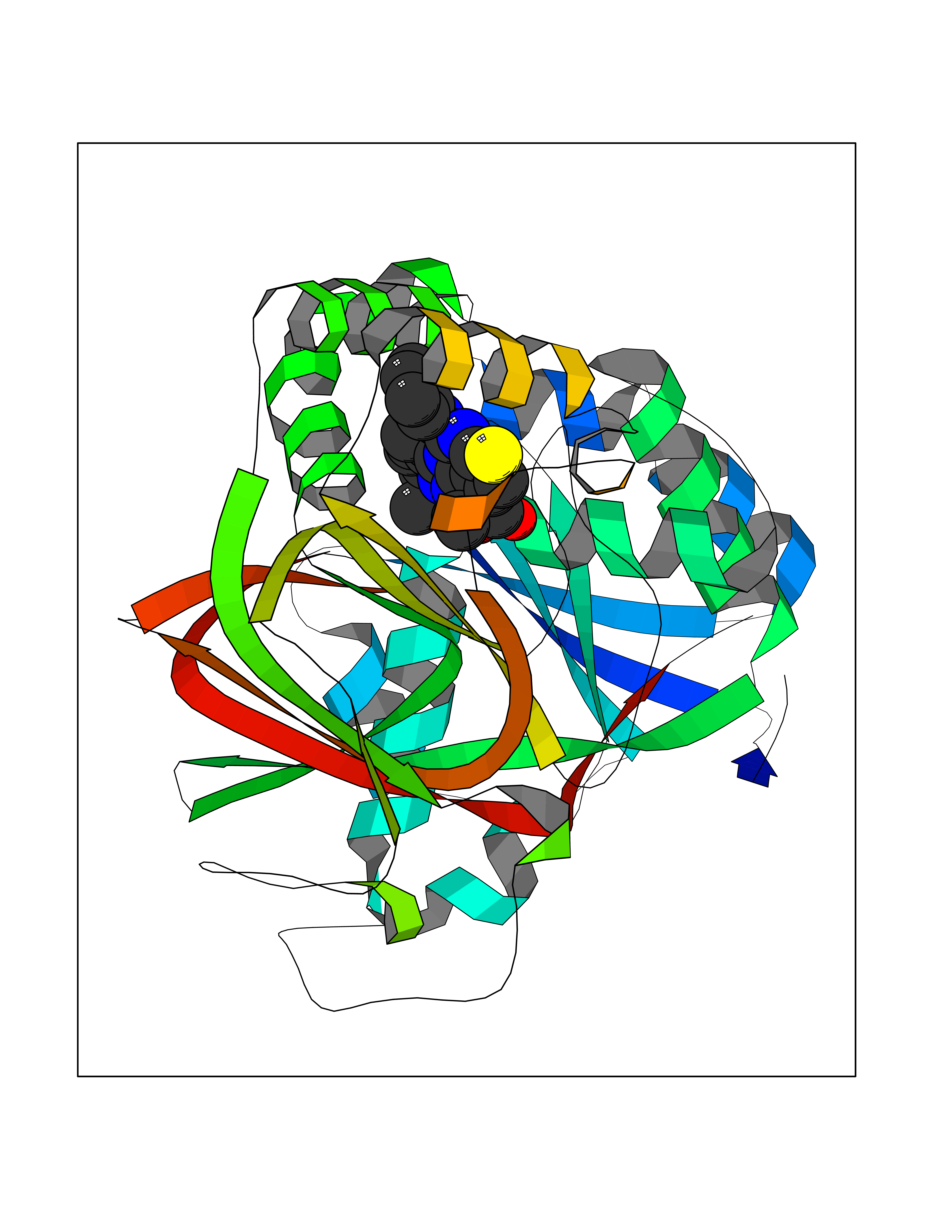 Molscript Map 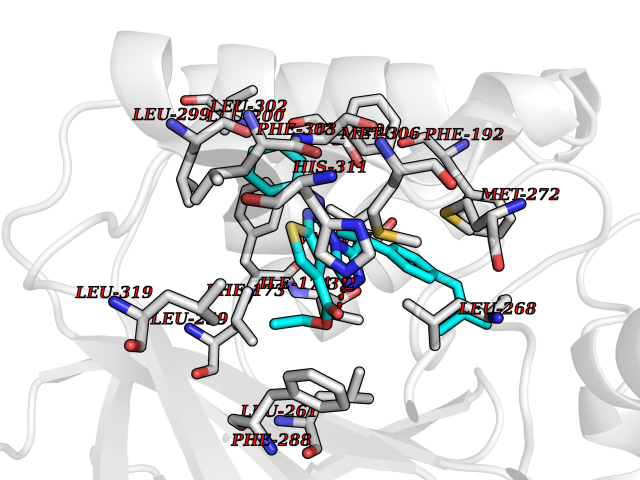 Pymol Map 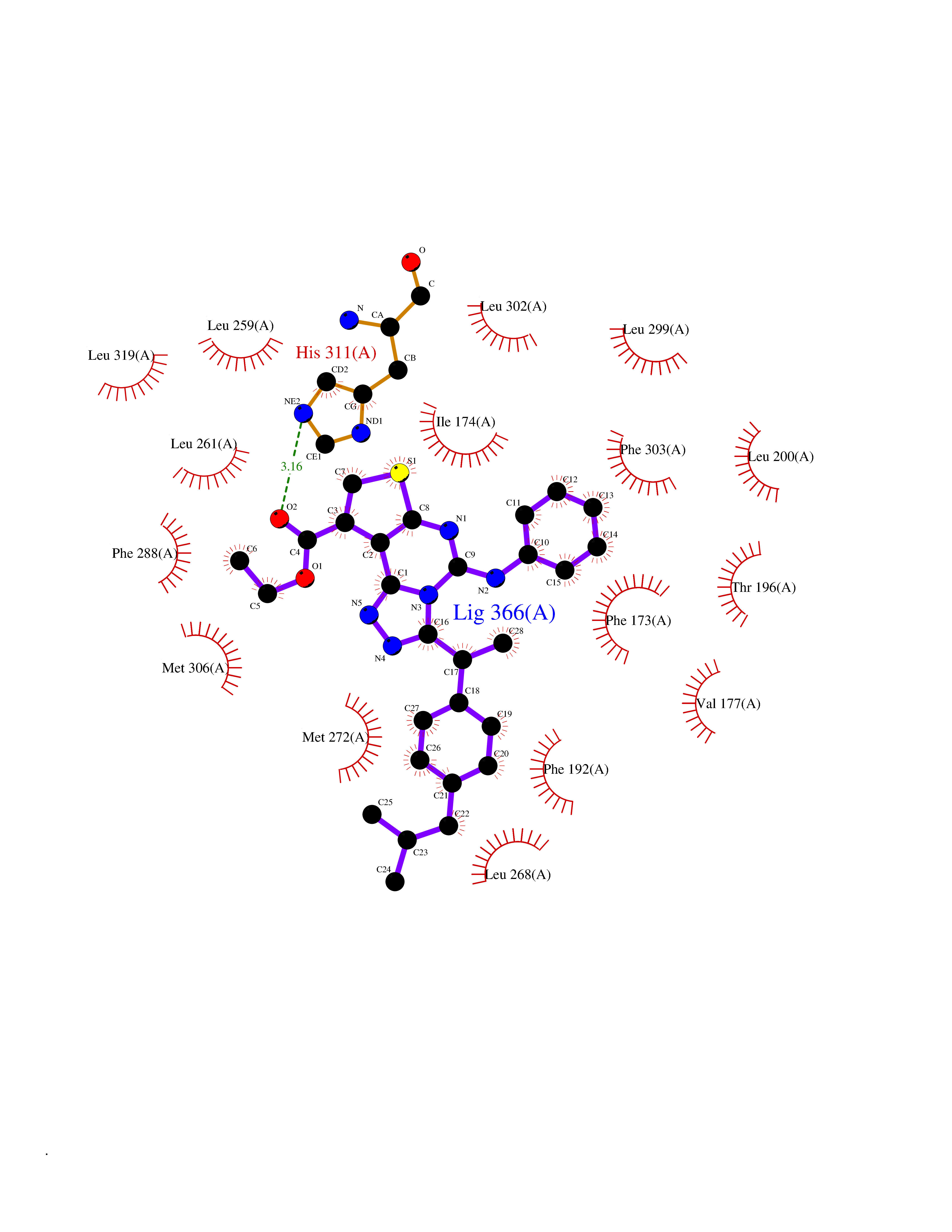 Ligplot Map 3D Binding mode Sequence AMGSGVLPRPCRVLVLLNPRGGKGKALQLFRSHVQPLLAEAEISFTLMLTERRNHARELVRSEELGRWDALVVMSGDGLMHEVVNGLMERPDWETAIQKPLCSLPAGSGNALAASLNHYAGYEQVTNEDLLTNCTLLLCRRLLSPMNLLSLHTASGLRLFSVLSLAWGFIADVDLESEKYRRLGEMRFTLGTFLRLAALRTYRGRLAYLPVGRVGSKTPASPVVVQQGPVDAHLVPLEEPVPSHWTVVPDEDFVLVLALLHSHLGSEMFAAPMGRCAAGVMHLFYVRAGVSRAMLLRLFLAMEKGRHMEYECPYLVYVPVVAFRLEPKDGKGVFAVDGELMVSEAVQGQVHPNYFWMVSG Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Urea transporter 1 (SLC14A1) | 6QD5 | 8.41 | |
Target general information Gen name SLC14A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Urea transporter, erythrocyte; UTE; UT1; Solute carrier family 14 member 1; RACH1; JK; HUT11 Protein family Urea transporter family Biochemical class Urea transporter family Function Urea channel that facilitates transmembrane urea transport down a concentration gradient. A constriction of the transmembrane channel functions as selectivity filter through which urea is expected to pass in dehydrated form. The rate of urea conduction is increased by hypotonic stress. Plays an important role in the kidney medulla collecting ducts, where it allows rapid equilibration between the lumen of the collecting ducts and the interstitium, and thereby prevents water loss driven by the high concentration of urea in the urine. Facilitates urea transport across erythrocyte membranes. May also play a role in transmembrane water transport, possibly by indirect means. Related diseases Immunodeficiency 12 (IMD12) [MIM:615468]: A primary immunodeficiency characterized by onset in infancy of recurrent bacterial and candidal infections resulting in bronchiectasis and growth delay. Manifestations include mastoiditis, aphthous ulcers, cheilitis, gingivitis, esophagitis, gastritis, duodenitis, and meningitis. Levels of absolute lymphocytes and serum immunoglobulins are normal, but specific antibody titers are low despite immunization, and T-cells show impaired proliferative responses to mitogens. {ECO:0000269|PubMed:23727036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A chromosomal aberration involving MALT1 is recurrent in low-grade mucosa-associated lymphoid tissue (MALT lymphoma). Translocation t(11;18)(q21;q21) with BIRC2. This translocation is found in approximately 50% of cytogenetically abnormal low-grade MALT lymphoma. {ECO:0000269|PubMed:10339464, ECO:0000269|PubMed:10523859, ECO:0000269|PubMed:10702396, ECO:0000269|PubMed:11090634}. Drugs (DrugBank ID) DB01005; DB03904 Interacts with Q8WVV5; Q9Y3D6; Q8WWP7; P30301; Q5QGT7; Q6UX34; P0DN84; Q9C0I4; Q5BJF2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood group antigen; Cell membrane; Glycoprotein; Membrane; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 38862.8 Length 356 Aromaticity 0.12 Instability index 40.98 Isoelectric point 7.67 Charge (pH=7) 0.95 2D Binding mode Binding energy (Kcal/mol) -11.47  Molscript Map 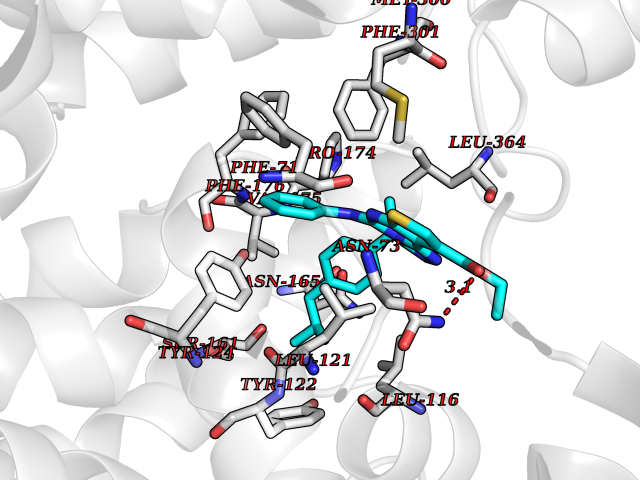 Pymol Map 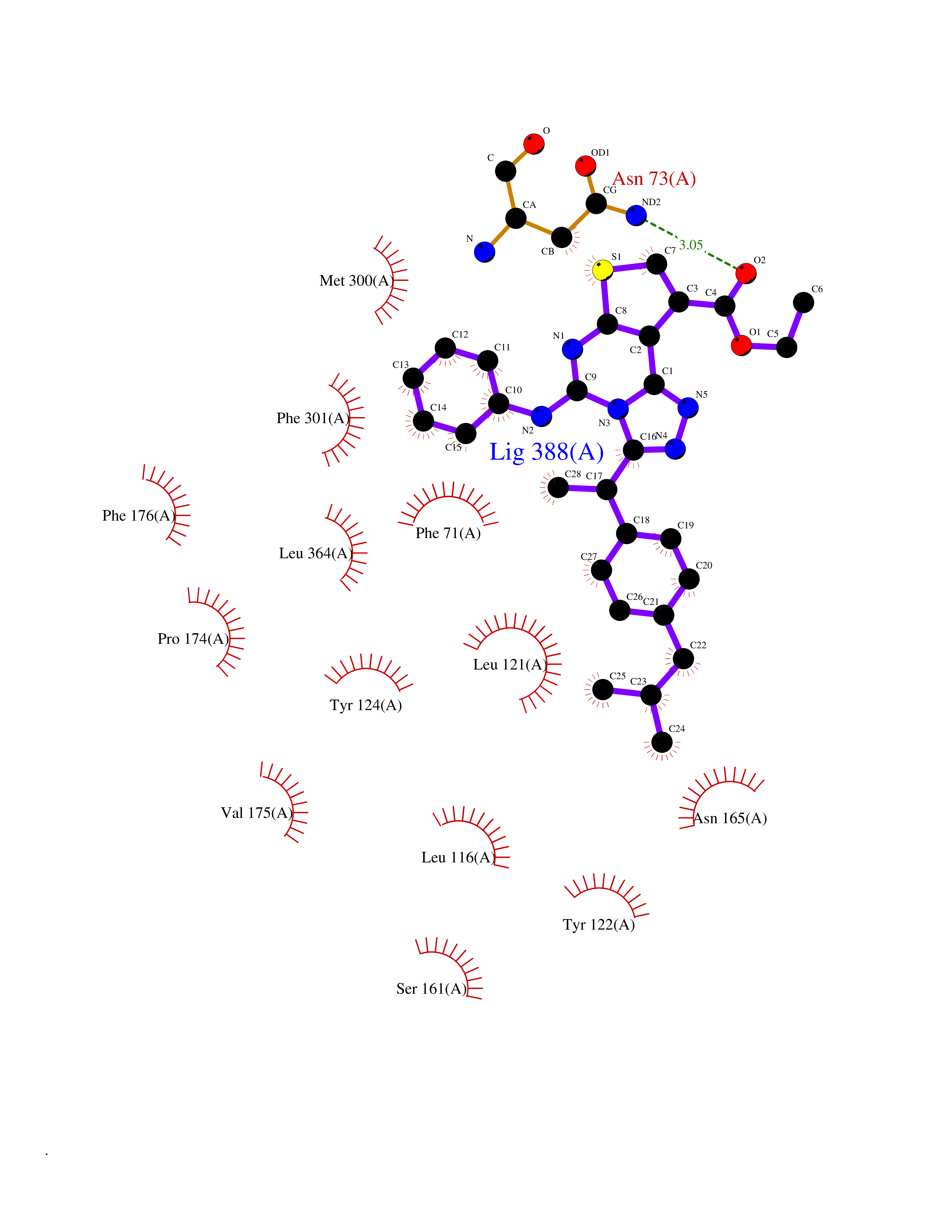 Ligplot Map 3D Binding mode Sequence FPKALGYVTGDMKELANQLKDKPVVLQFIDWILRGISQVVFVNNPVSGILILVGLLVQNPWWALTGWLGTVVSTLMALLLSQDRSLIASGLYGYNATLVGVLMAVFSDKGDYFWWLLLPVCAMSMTCPIFSSALNSVLSKWDLPVFTLPFNMALSMYLSATGHYNPFFPAKLVIPITTAPQISWSDLSALELLKSIPVGVGQIYGCDNPWTGGIFLGAILLSSPLMCLHAAIGSLLGIAAGLSLSAPFENIYFGLWGFNSSLACIAMGGMFMALTWQTHLLALGCALFTAYLGVGMANFMAEVGLPACTWPFCLATLLFLIMTTKNSNIYKMPLSKVTYPEENRIFYLQAKKRMVE Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Camphor 5-monooxygenase | 4L4E | 8.40 | |
Target general information Gen name camC Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID NA Synonyms cyp101 Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Camphor 5-monooxygenase activity.Heme binding.Iron ion binding. Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03836; DB02617; DB02817; DB03627; DB04032; DB03031; DB02125; DB04501; DB01744; DB01663; DB01011; DB01703; DB01826; DB03540; DB02851 Interacts with P00259 EC number 1.14.15.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Heme; Iron; Metal-binding; Monooxygenase; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 45446.3 Length 405 Aromaticity 0.08 Instability index 45.33 Isoelectric point 5.23 Charge (pH=7) -16.08 2D Binding mode Binding energy (Kcal/mol) -11.46 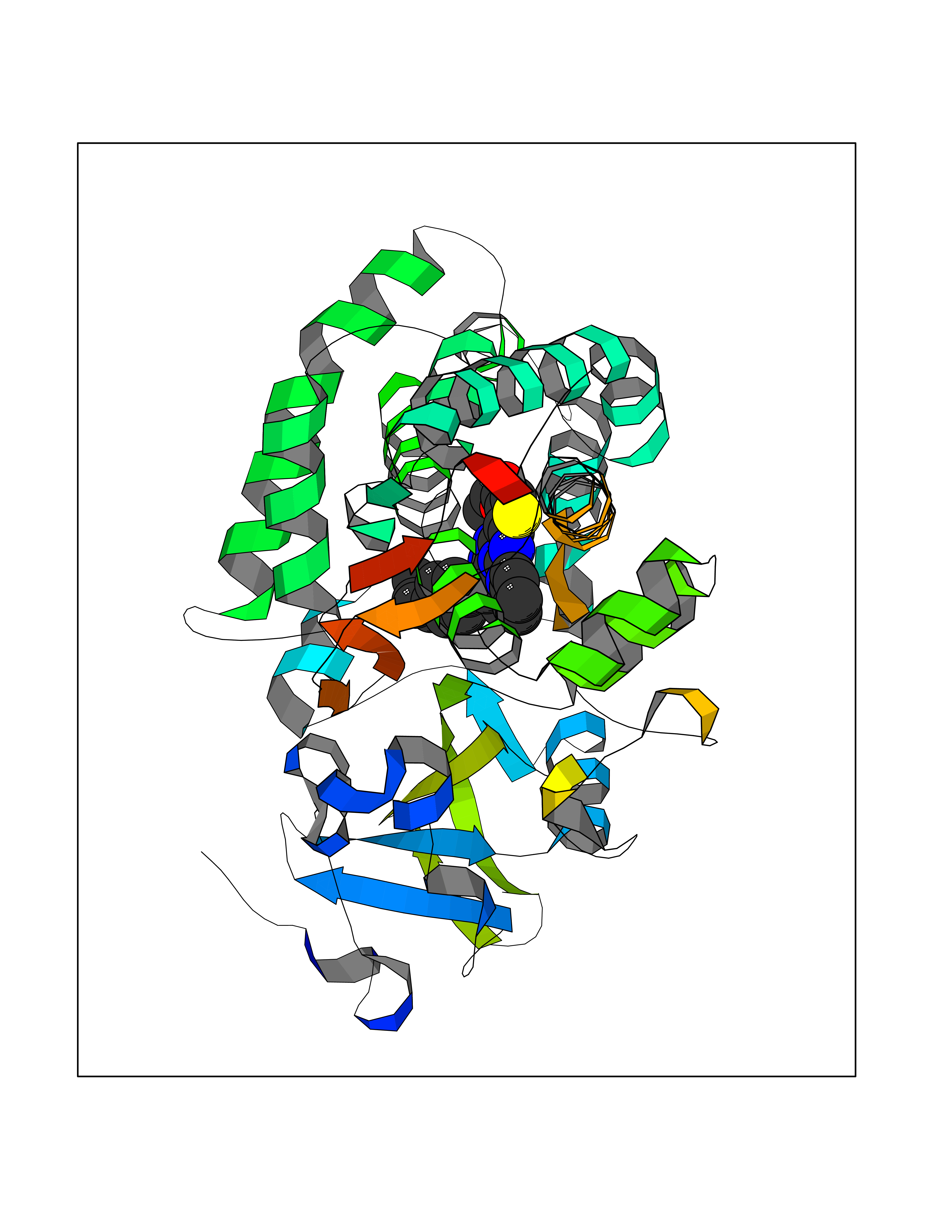 Molscript Map 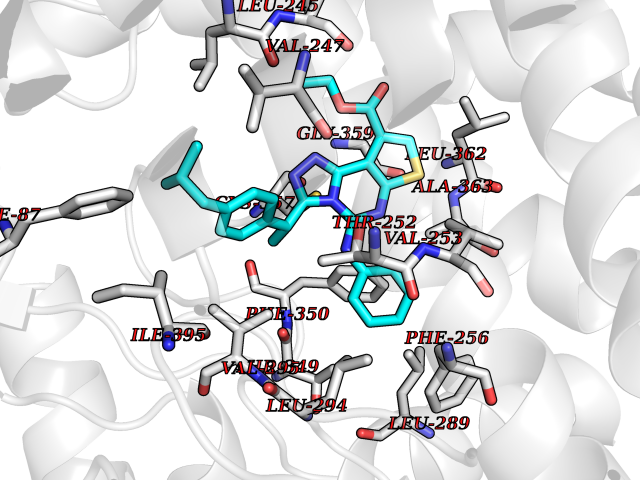 Pymol Map 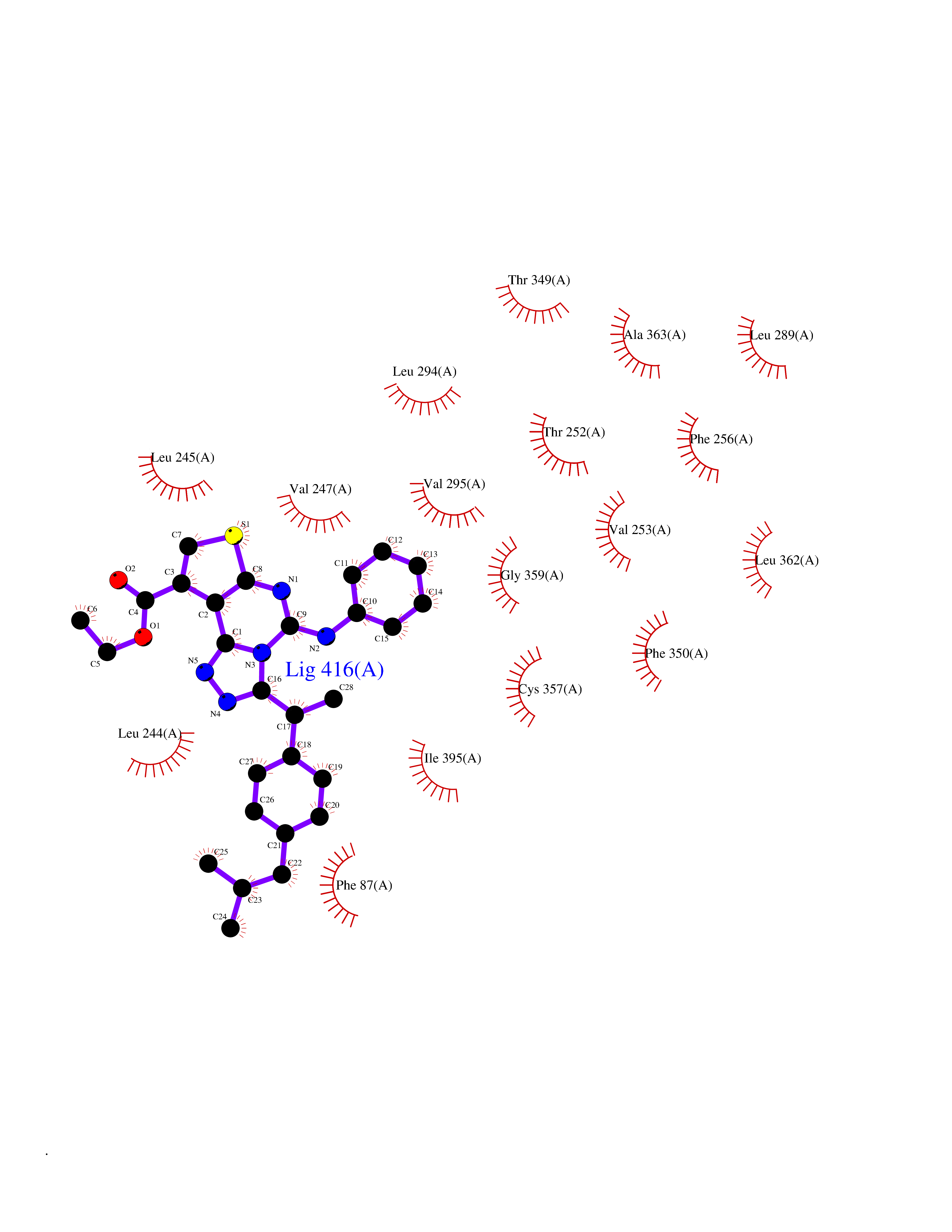 Ligplot Map 3D Binding mode Sequence NLAPLPPHVPEHLVFDFDMYNPSNLSAGVQEAWAVLQESNVPDLVWTRCNGGHWIATRGQLIREAYEDYRHFSSECPFIPREAGEAYDFIPTSMDPPEQRQFRALANQVVGMPVVDKLENRIQELACSLIESLRPQGQCNFTEDYAEPFPIRIFMLLAGLPEEDIPHLGYLTDQMTRPDGSMTFAEAKEALYDYLIPIIEQRRQKPGTDAISIVANGQVNGRPITSDEAKRMCGLLLVGGLDTVVNFLSFSMEFLAKSPEHRQELIERPERIPAACEELLRRFSLVADGRILTSDYEFHGVQLKKGDQILLPQMLSGLDERENAAPMHVDFSRQKVSHTTFGHGSHLCAGQHLARREIIVTLKEWLTRIPDFSIAPGAQIQHKSGIVSGVQALPLVWDPATTKAV Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Cannabinoid receptor 2 (CB2) | 6PT0 | 8.40 | |
Target general information Gen name CNR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hCB2; Cannabinoid CB2 receptor; CX5; CB2B; CB2A; CB-2 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function May function in inflammatory response, nociceptive transmission and bone homeostasis. Heterotrimeric G protein-coupled receptor for endocannabinoid 2-arachidonoylglycerol mediating inhibition of adenylate cyclase. Related diseases Factor V deficiency (FA5D) [MIM:227400]: A blood coagulation disorder leading to a hemorrhagic diathesis known as parahemophilia. {ECO:0000269|PubMed:10942390, ECO:0000269|PubMed:12393490}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombophilia due to activated protein C resistance (THPH2) [MIM:188055]: A hemostatic disorder due to defective degradation of factor V by activated protein C. It is characterized by a poor anticoagulant response to activated protein C resulting in tendency to thrombosis. {ECO:0000269|PubMed:10391209, ECO:0000269|PubMed:10942390, ECO:0000269|PubMed:11435304, ECO:0000269|PubMed:11858490, ECO:0000269|PubMed:14617013, ECO:0000269|PubMed:14695241, ECO:0000269|PubMed:16710414, ECO:0000269|PubMed:8164741, ECO:0000269|PubMed:9454742}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Budd-Chiari syndrome (BDCHS) [MIM:600880]: A syndrome caused by obstruction of hepatic venous outflow involving either the hepatic veins or the terminal segment of the inferior vena cava. Obstructions are generally caused by thrombosis and lead to hepatic congestion and ischemic necrosis. Clinical manifestations observed in the majority of patients include hepatomegaly, right upper quadrant pain and abdominal ascites. Budd-Chiari syndrome is associated with a combination of disease states including primary myeloproliferative syndromes and thrombophilia due to factor V Leiden, protein C deficiency and antithrombin III deficiency. Budd-Chiari syndrome is a rare but typical complication in patients with polycythemia vera. {ECO:0000269|PubMed:9245936}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Pregnancy loss, recurrent, 1 (RPRGL1) [MIM:614389]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:11018168}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09061; DB00470; DB06202; DB14009; DB00486; DB14011; DB02955; DB16321; DB11755 Interacts with Q9UKJ8; Q15848; Q9NRZ5; P13236; P21964; Q14802-3; Q8N387; Q8IXM6; I3L0A0; Q96AA3; Q9Y6D0; Q6ICL7; Q9NP94; Q13501; Q96HH6; Q969S6; Q9NWH2; Q9H2L4; Q8N2M4; Q6ZT21; Q5TGU0; Q9Y548; Q9BSR8; Q96EC8 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; G-protein coupled receptor; Glycoprotein; Inflammatory response; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32999.2 Length 298 Aromaticity 0.11 Instability index 30.98 Isoelectric point 9.49 Charge (pH=7) 14.35 2D Binding mode Binding energy (Kcal/mol) -11.45 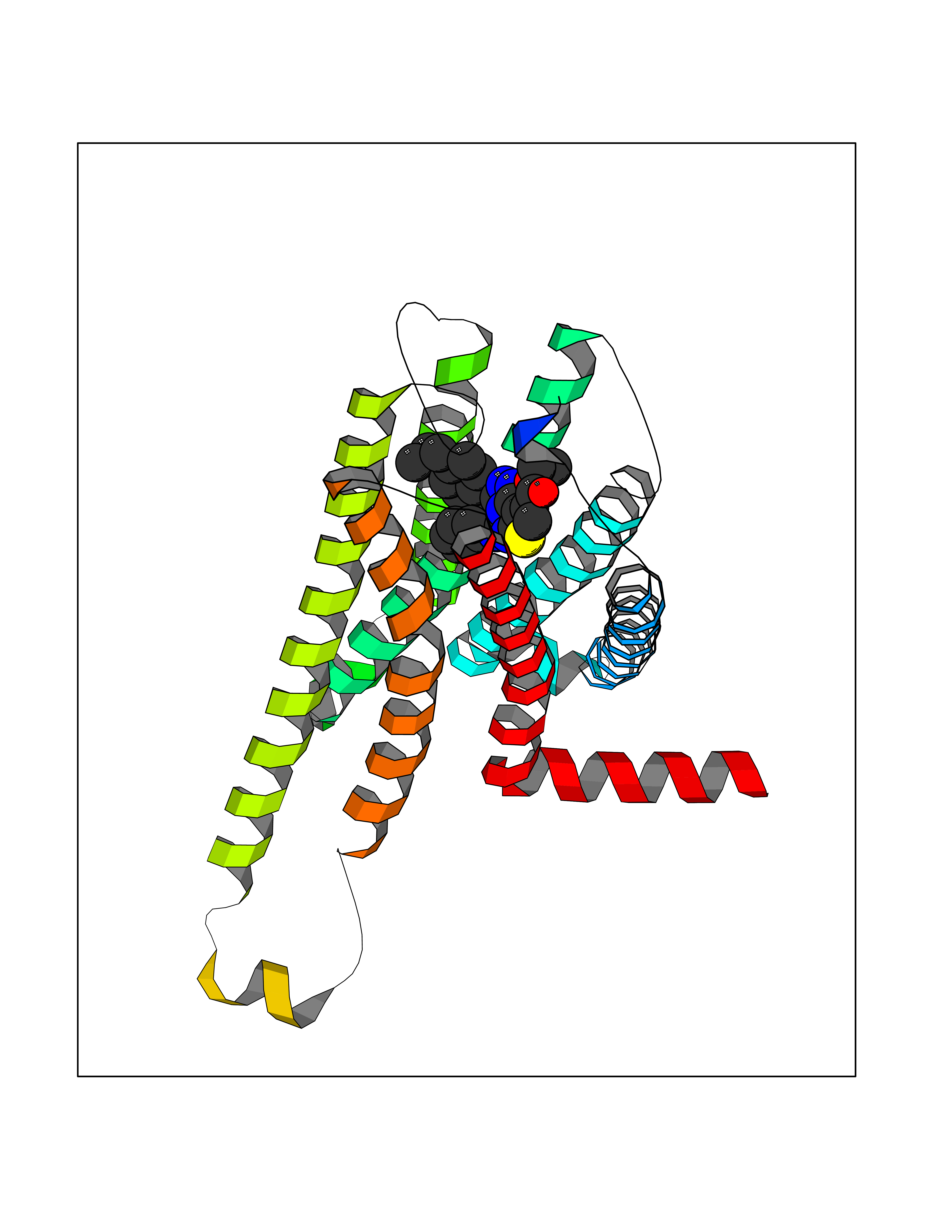 Molscript Map 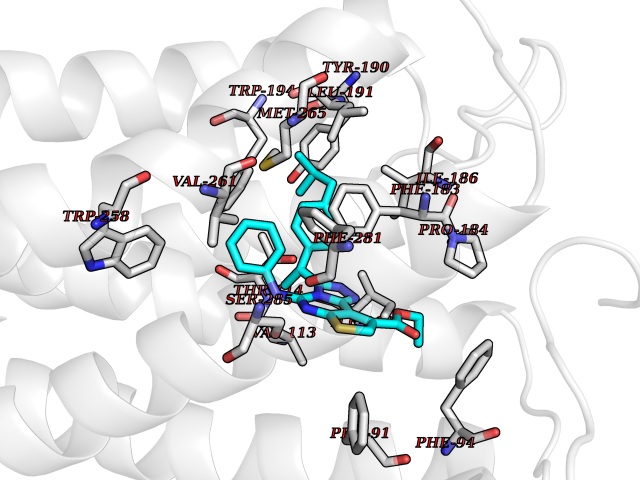 Pymol Map 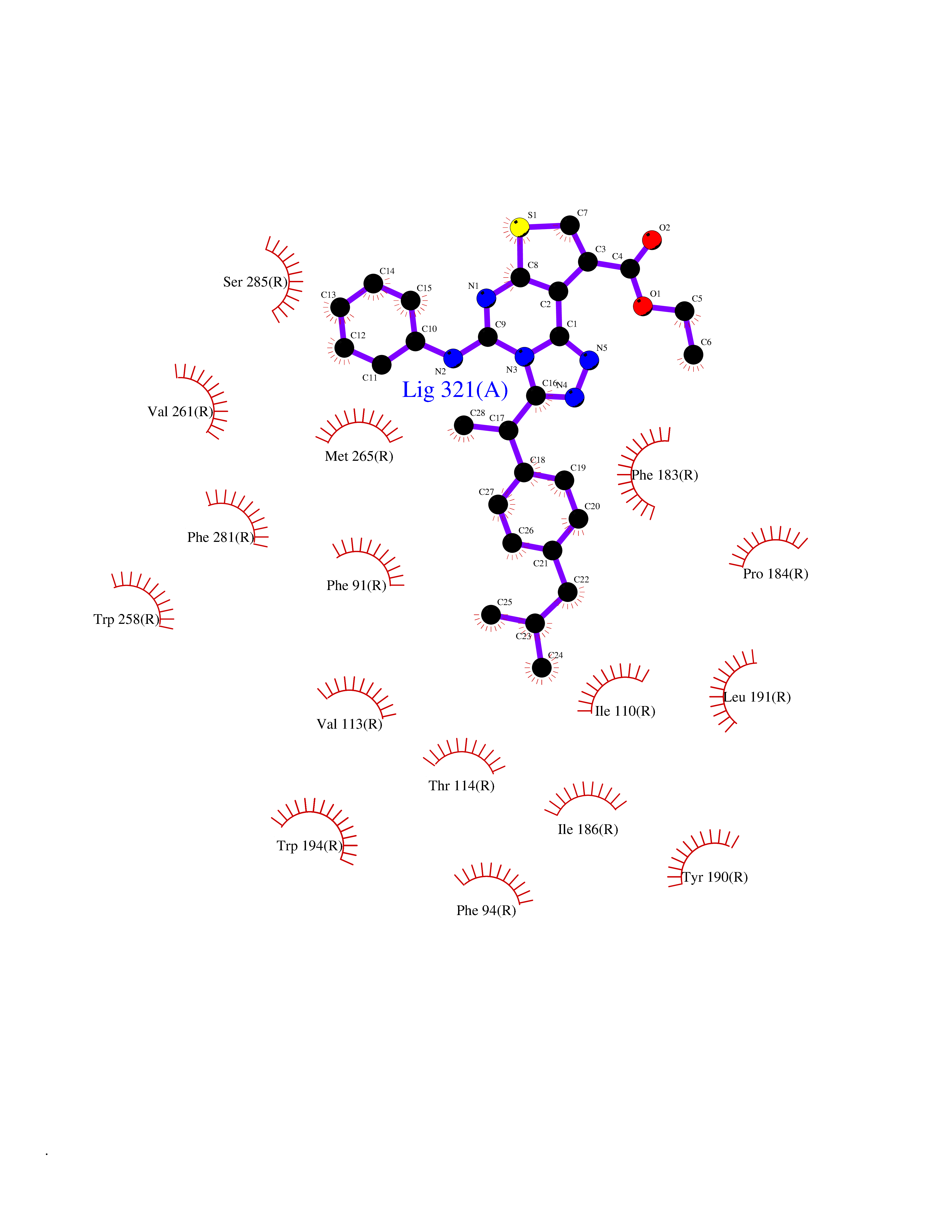 Ligplot Map 3D Binding mode Sequence MKDYMILSGPQKTAVAVLCTLLGLLSALENVAVLYLILSSHQLRRKPSYLFIGSLAGADFLASVVFACSFVNFHVFHGVDSKAVFLLKIGSVTMTFTASVGSLLLTAIDRYLCLRYPPSYKALLTRGRALVTLGIMWVLSALVSYLPLMGWTCCPRPCSELFPLIPNDYLLSWLLFIAFLFSGIIYTYGHVLWKAHQHVASLSGHQDRQVPGMARMRLDVRLAKTLGLVLAVLLICWFPVLALMAHSLATTLSDQVKKAFAFCSMLCLINSMVNPVIYALRSGEIRSSAHHCLAHWKK Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Trypanosoma Trypanothione reductase (Trypano TPR) | 2WBA | 8.40 | |
Target general information Gen name Trypano TPR Organism Trypanosoma brucei brucei Uniprot ID TTD ID Synonyms TRYR; TPR; Parasite-specific trypanothione reductase; N(1),N(8)-bis(glutathionyl)spermidine reductase Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Sulfur donor oxidoreductase Function Trypanothione is the parasite analog of glutathione; this enzyme is the equivalent of glutathione reductase. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.8.1.12 Uniprot keywords 3D-structure; Cytoplasm; Disulfide bond; FAD; Flavoprotein; NADP; Oxidoreductase; Redox-active center Protein physicochemical properties Chain ID A,B Molecular weight (Da) 105578 Length 978 Aromaticity 0.08 Instability index 33.76 Isoelectric point 6.25 Charge (pH=7) -6.81 2D Binding mode Binding energy (Kcal/mol) -11.45 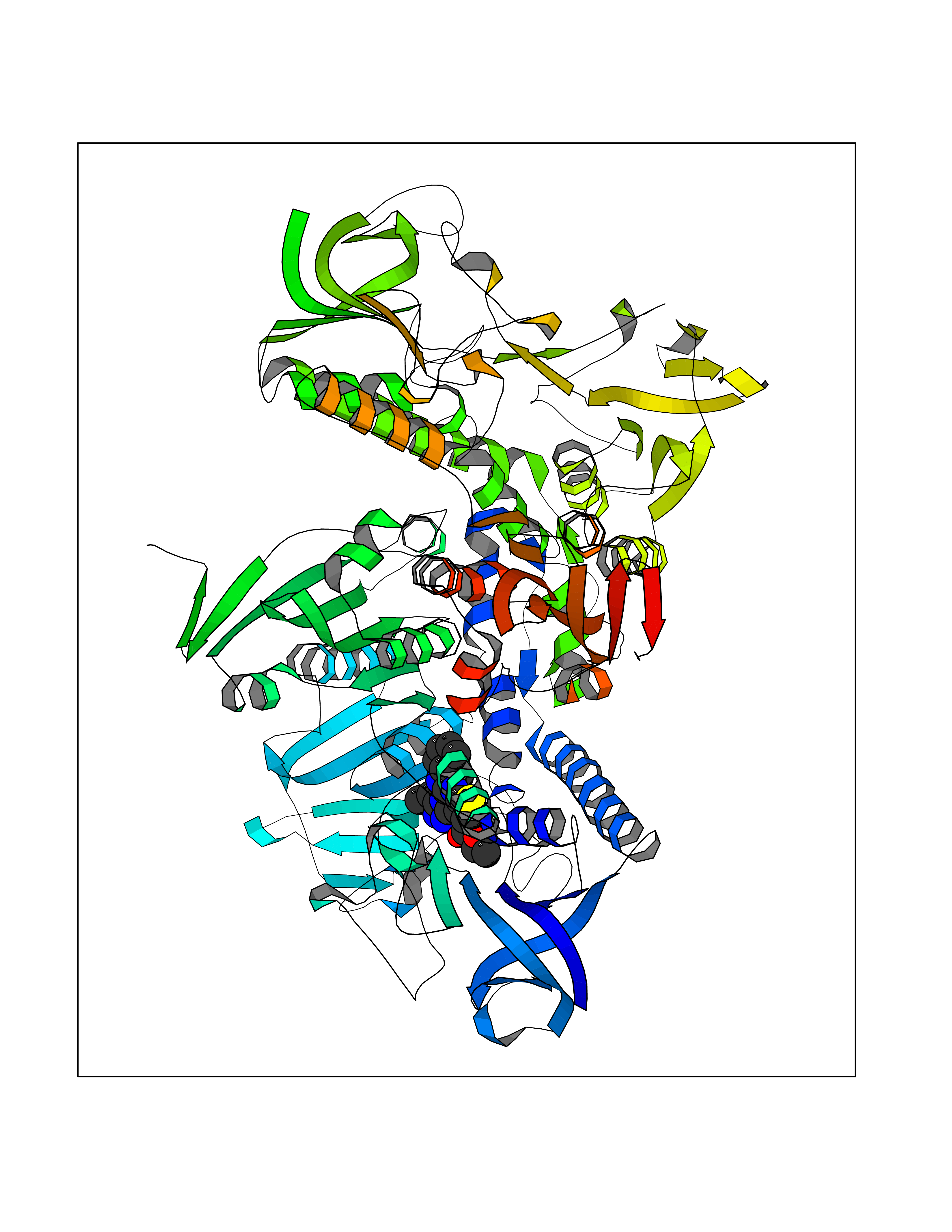 Molscript Map 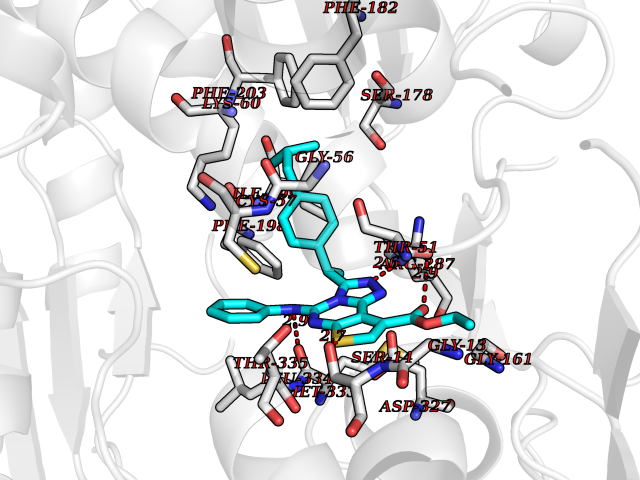 Pymol Map 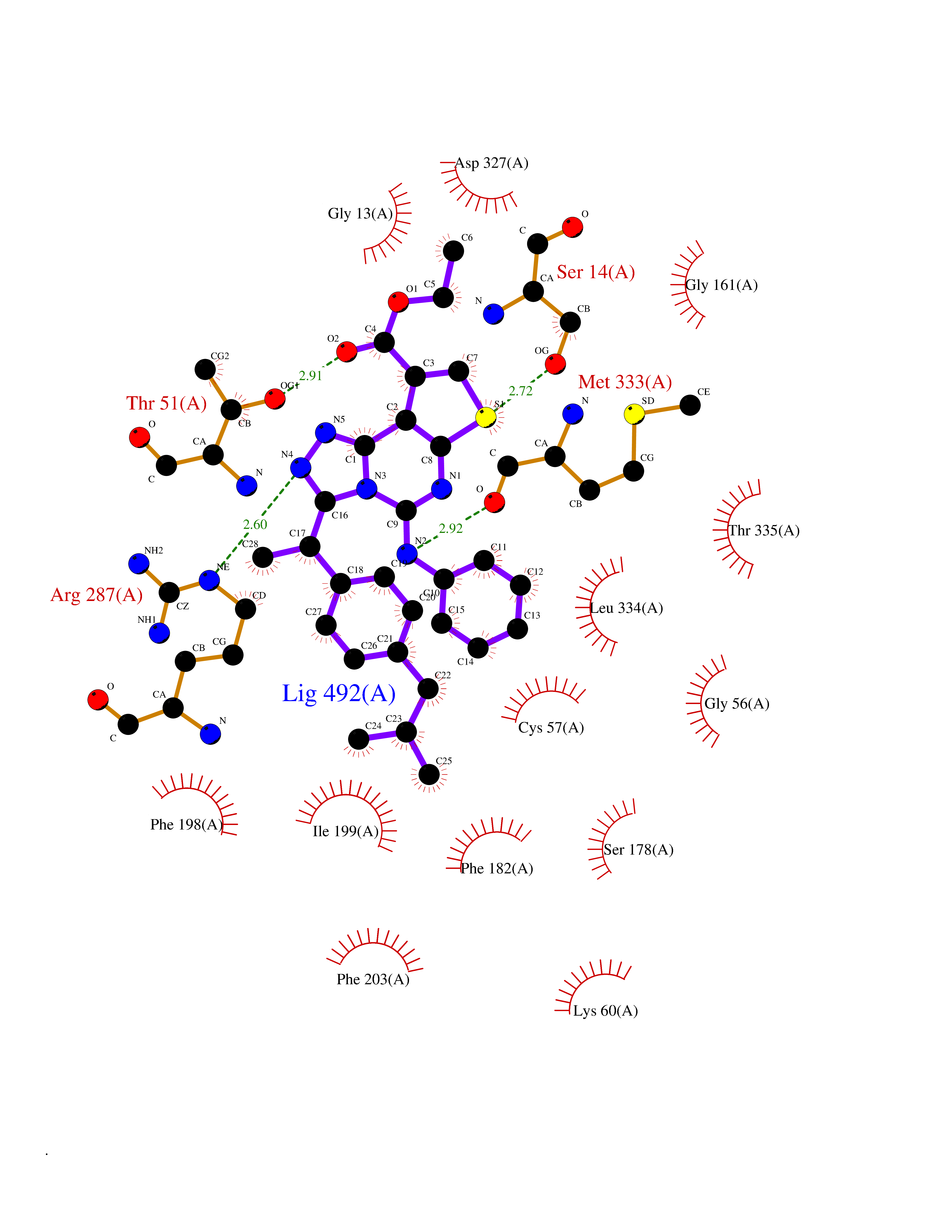 Ligplot Map 3D Binding mode Sequence SKAFDLVVIGAGSGGLEAGWNAATLYGKRVAVVDVQTSHGPPFYAALGGTCVNVGCVPKKLMVTGAQYMDHLRESAGFGWEFDGSSVKANWKKLIAAKNEAVLDINKSYEGMFNDTEGLDFFLGWGSLESKNVVVVRETADPKSAVKERLQADHILLATGSWPQMPAIPGIEHCISSNEAFYLPEPPRRVLTVGGGFISVEFAGIFNAYKPPGGKVTLCYRNNLILRGFDETIREEVTKQLTANGIEIMTNENPAKVSLNTDGSKHVTFESGKTLDVDVVMMAIGRIPRTNDLQLGNVGVKLTPKGGVQVDEFSRTNVPNIYAIGDITDRLMLTPVAINEGAALVDTVFGNKPRKTDHTRVASAVFSIPPIGTCGLIEEVAAKEFEKVAVYMSSFTPLMHNISGSKYKKFVAKIVTNHSDGTVLGVHLLGDGAPEIIQAVGVCLRLNAKISDFYNTIGVHPTSAEELCSMRTPSYYYVKGEKMEKLPDSSKAFDLVVIGAGSGGLEAGWNAATLYGKRVAVVDVQTSHGPPFYAALGGTCVNVGCVPKKLMVTGAQYMDHLRESAGFGWEFDGSSVKANWKKLIAAKNEAVLDINKSYEGMFNDTEGLDFFLGWGSLESKNVVVVRETADPKSAVKERLQADHILLATGSWPQMPAIPGIEHCISSNEAFYLPEPPRRVLTVGGGFISVEFAGIFNAYKPPGGKVTLCYRNNLILRGFDETIREEVTKQLTANGIEIMTNENPAKVSLNTDGSKHVTFESGKTLDVDVVMMAIGRIPRTNDLQLGNVGVKLTPKGGVQVDEFSRTNVPNIYAIGDITDRLMLTPVAINEGAALVDTVFGNKPRKTDHTRVASAVFSIPPIGTCGLIEEVAAKEFEKVAVYMSSFTPLMHNISGSKYKKFVAKIVTNHSDGTVLGVHLLGDGAPEIIQAVGVCLRLNAKISDFYNTIGVHPTSAEELCSMRTPSYYYVKGEKMEKLPDS Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Retinaldehyde-binding protein 1 | 3HX3 | 8.39 | |
Target general information Gen name RLBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CRALBP Protein family NA Biochemical class Transport protein Function 11-cis retinal binding.Retinol binding.Transporter activity. Related diseases Bothnia retinal dystrophy (BRD) [MIM:607475]: A type of retinitis punctata albescens. Affected individuals show night blindness from early childhood with features consistent with retinitis punctata albescens and macular degeneration. {ECO:0000269|PubMed:10102298}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rod-cone dystrophy Newfoundland (NFRCD) [MIM:607476]: A rod-cone dystrophy reminiscent of retinitis punctata albescens but with a substantially lower age at onset and more-rapid and distinctive progression. Rod-cone dystrophies results from initial loss of rod photoreceptors, later followed by cone photoreceptors loss. {ECO:0000269|PubMed:11868161}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Retinitis punctata albescens (RPA) [MIM:136880]: A form of fleck retina disease characterized by aggregation of white flecks posteriorly in the retina, causing night blindness and delayed dark adaptation. It differs from fundus albipunctatus in being progressive and evolving to generalized atrophy of the retina. {ECO:0000269|PubMed:10102299, ECO:0000269|PubMed:11453974, ECO:0000269|PubMed:9326942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00162 Interacts with Q9P2G9-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Disease variant; Proteomics identification; Reference proteome; Retinol-binding; Sensory transduction; Transport; Vision Protein physicochemical properties Chain ID A Molecular weight (Da) 28328.6 Length 250 Aromaticity 0.14 Instability index 52.64 Isoelectric point 4.96 Charge (pH=7) -9.87 2D Binding mode Binding energy (Kcal/mol) -11.44 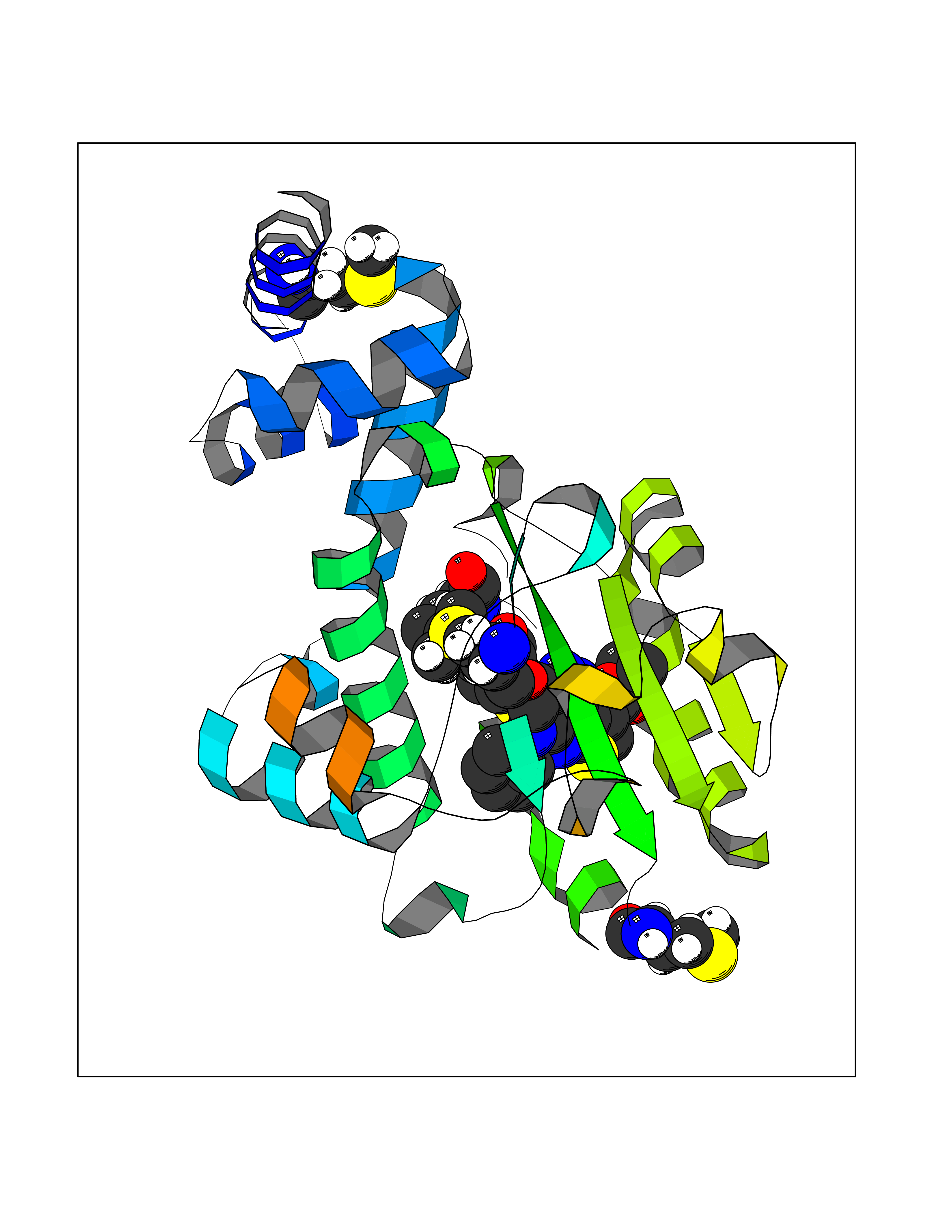 Molscript Map 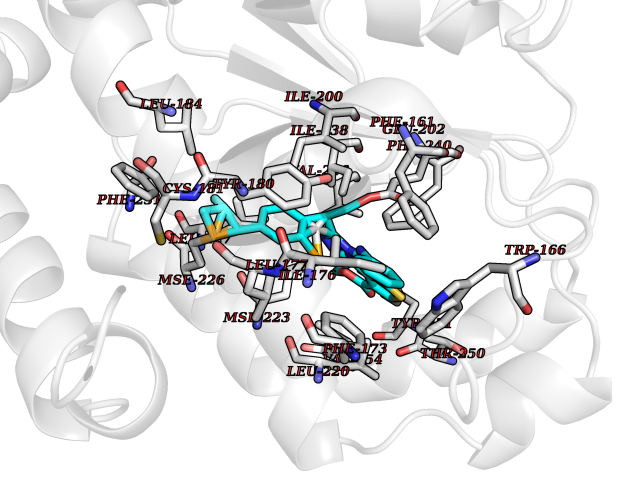 Pymol Map 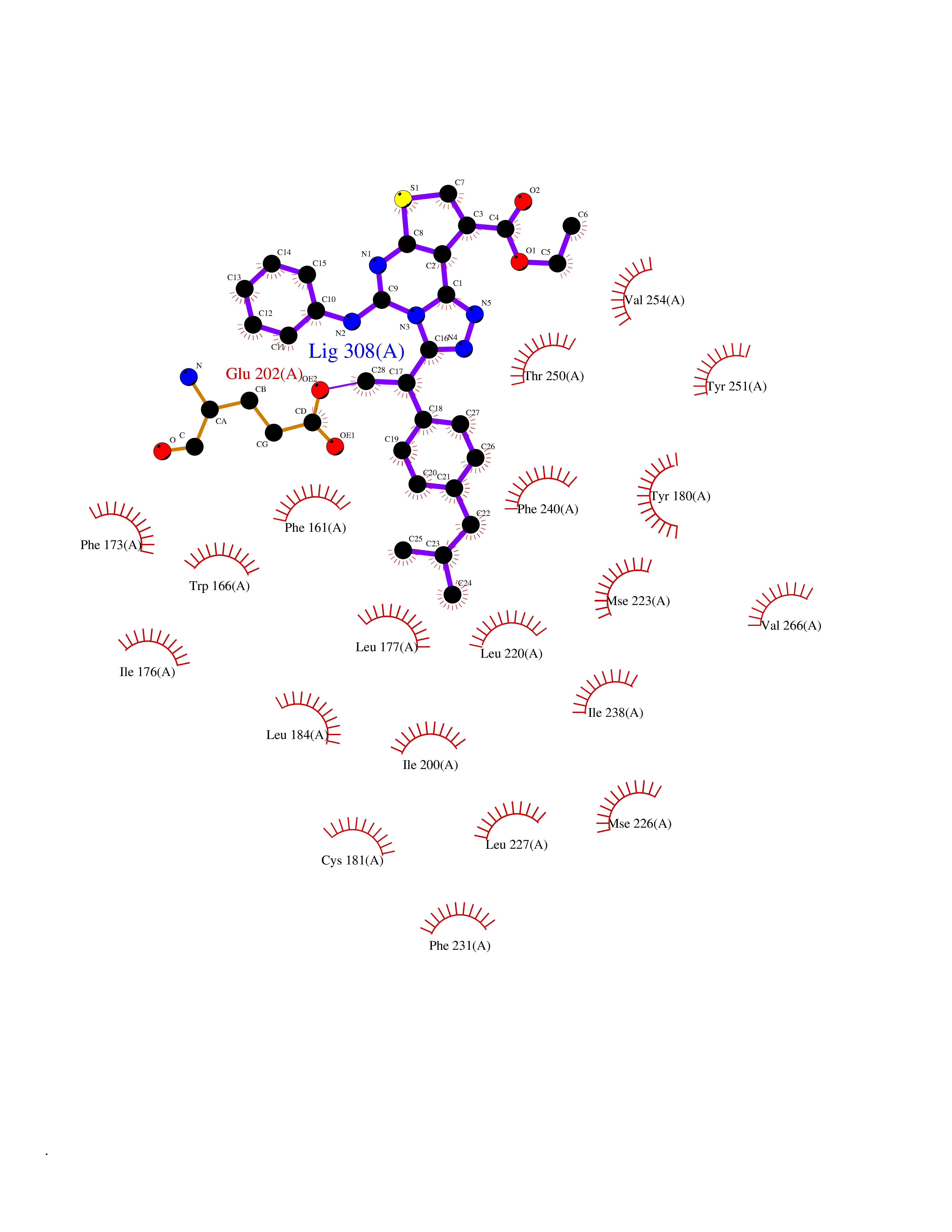 Ligplot Map 3D Binding mode Sequence ETREEAVRELQEXVQAQAASGEELAVAVAERVQEKDSGFFLRFIRARKFNVGRAYELLRGYVNFRLQYPELFDSLSPEAVRCTIEAGYPGVLSSRDKYGRVVXLFNIENWQSQEITFDEILQAYCFILEKLLENEETQINGFCIIENFKGFTXQQAASLRTSDLRKXVDXLQDSFPAWFKAIHFIHQPWYFTTTYNVVKPFLKSKLLERVFVHGDDLSGFYQEIDENILPSDFGGTLPKYDGKAVAEQLF Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Retinol-binding protein 1 | 5HBS | 8.39 | |
Target general information Gen name RBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CRBP1 Protein family Calycin superfamily, Fatty-acid binding protein (FABP) family Biochemical class Retinol-binding protein Function Retinal binding.Retinoid binding.Retinol binding.Transporter activity. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00459; DB06755; DB00162 Interacts with P49366; Q9UBN6; Q6DKK2; Q8N2K1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Lipid droplet; Methylation; Proteomics identification; Reference proteome; Retinol-binding; Transport; Vitamin A Protein physicochemical properties Chain ID A Molecular weight (Da) 16404.5 Length 139 Aromaticity 0.09 Instability index 16.54 Isoelectric point 5.62 Charge (pH=7) -5.43 2D Binding mode Binding energy (Kcal/mol) -11.44  Molscript Map 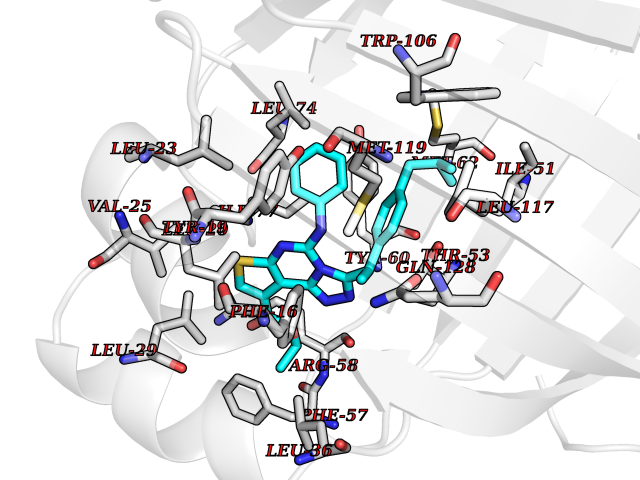 Pymol Map 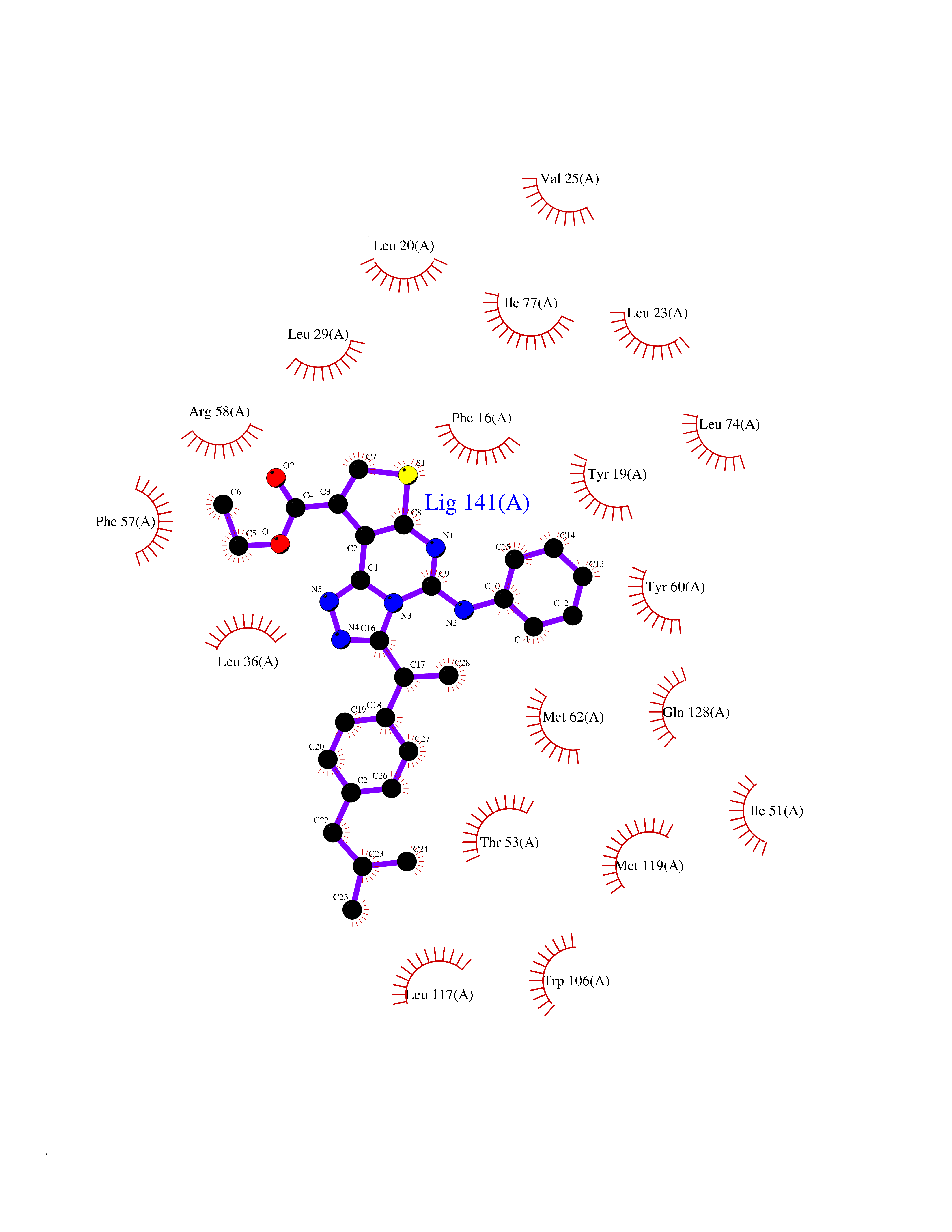 Ligplot Map 3D Binding mode Sequence PVDFTGYWKMLVNENFEEYLRALDVNVALRKIANLLKPDKEIVQDGDHMIIRTLSTFRNYIMDFQVGKEFEEDLTGIDDRKCMTTVSWDGDKLQCVQKGEKEGRGWTQWIEGDELHLEMRVEGVVCKQVFKKVQHHHHH Hydrogen bonds contact Hydrophobic contact | ||||