Job Results:
Ligand
Structure
Job ID
60cfc8edcb03d90540341cd2f0373921
Job name
NA
Time
2026-03-02 09:52:38
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | 30S ribosomal protein S3 | 4ODQ | 4.41 | |
Target general information Gen name rpsC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW3276;b3314 Protein family Universal ribosomal protein uS3 family Biochemical class Isomerase Function RRNA binding.Structural constituent of ribosome. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09093; DB12455; DB00759 Interacts with NA EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Reference proteome; Ribonucleoprotein; Ribosomal protein; RNA-binding; rRNA-binding Protein physicochemical properties Chain ID B Molecular weight (Da) 12457.7 Length 112 Aromaticity 0.05 Instability index 12.49 Isoelectric point 5.91 Charge (pH=7) -7.24 2D Binding mode Binding energy (Kcal/mol) -6.01 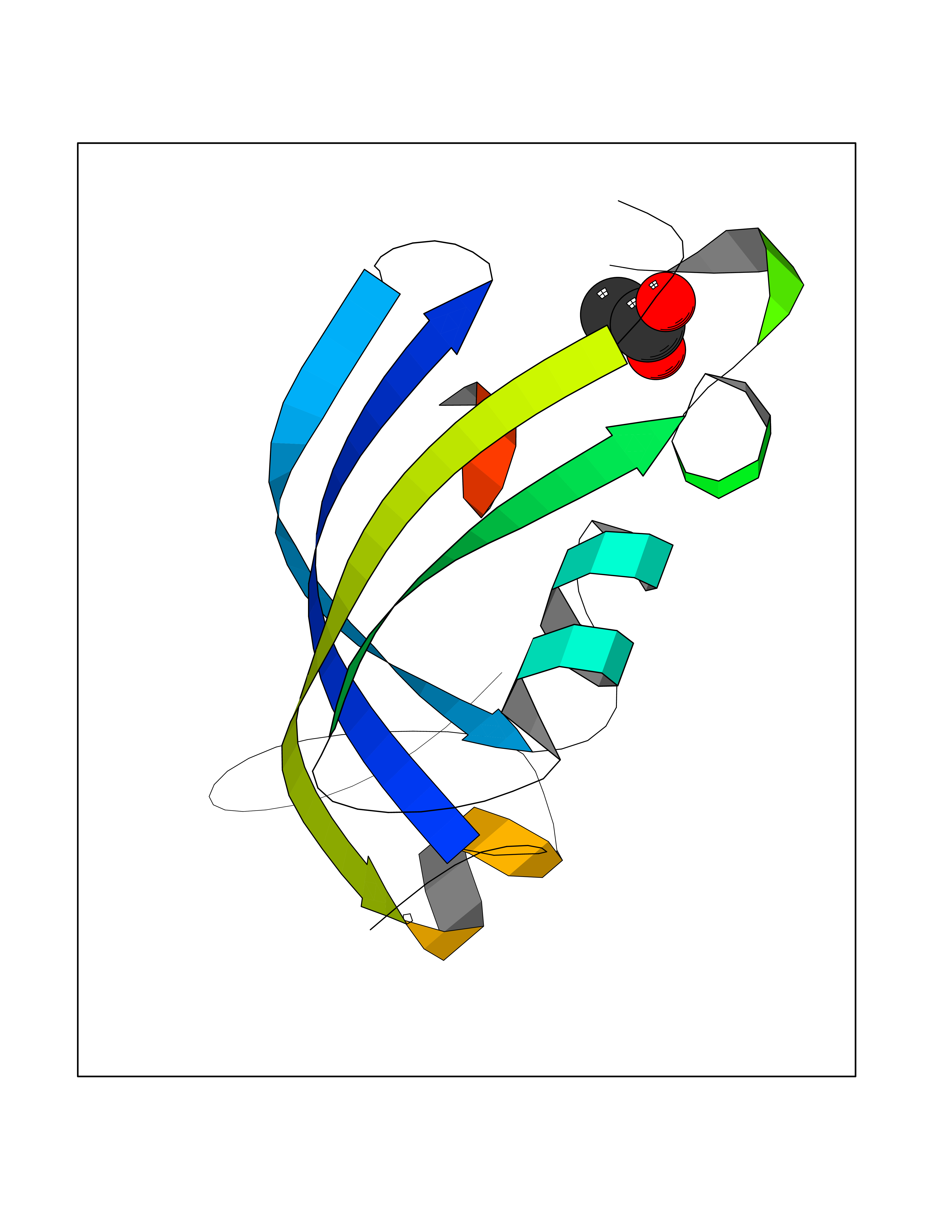 Molscript Map 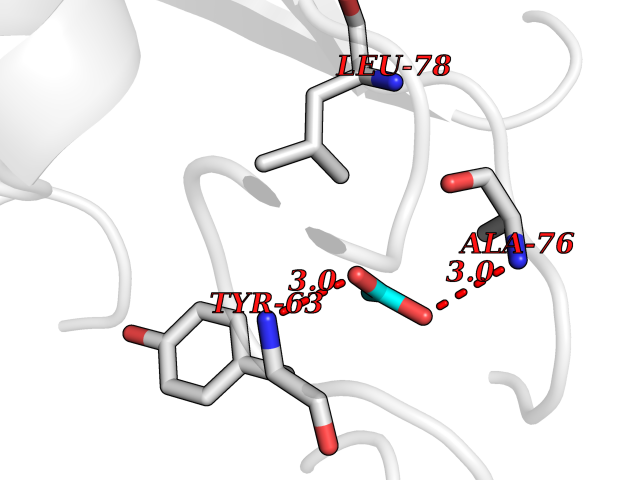 Pymol Map 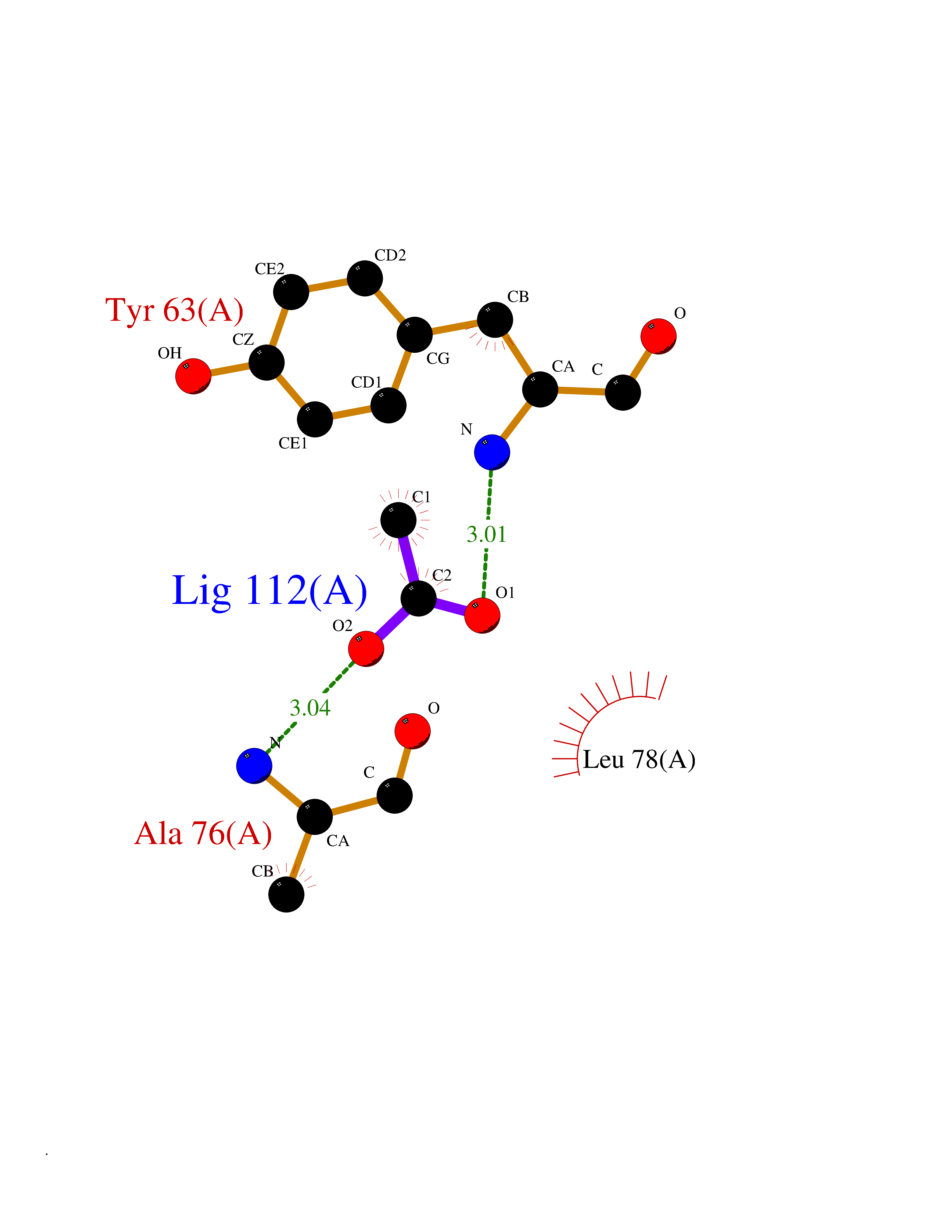 Ligplot Map 3D Binding mode Sequence MKVGQDKVVTIRYTLQVEGEVLDQGELSYLHGHRNLIPGLEEALEGREEGEAFQAHVPAEKAYGATGHPPHATLDFQVEVVKVREATPEELLHGHAHPSGHHHHHHGIVKPW Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Dopamine D2 receptor (D2R) | 5AER | 4.41 | |
Target general information Gen name DRD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dopamine receptor 2; D(2) dopamine receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Dopamine receptor whose activity is mediated by G proteins which inhibit adenylyl cyclase. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01614; DB01063; DB01425; DB00915; DB06288; DB05964; DB00543; DB00182; DB04599; DB00714; DB01238; DB14185; DB09207; DB06216; DB04889; DB04888; DB05687; DB09223; DB04857; DB09128; DB01200; DB09018; DB00490; DB00248; DB06016; DB01038; DB00477; DB01239; DB00568; DB00363; DB01151; DB11274; DB13345; DB00320; DB01184; DB00988; DB00450; DB11275; DB01049; DB00696; DB01175; DB09194; DB00875; DB00623; DB04842; DB00502; DB04946; DB00458; DB04924; DB12579; DB01221; DB00555; DB01235; DB00589; DB00408; DB06077; DB08815; DB00934; DB09224; DB01043; DB00933; DB01403; DB01233; DB06148; DB00805; DB01618; DB08804; DB05766; DB00540; DB06229; DB00334; DB01267; DB12061; DB00715; DB01186; DB08922; DB00850; DB01100; DB09286; DB01621; DB12478; DB00413; DB00433; DB00420; DB01069; DB00777; DB01224; DB09097; DB12518; DB00409; DB00734; DB01549; DB00268; DB05271; DB06454; DB06144; DB00391; DB06477; DB04844; DB12093; DB00372; DB01622; DB00679; DB01623; DB13025; DB00831; DB00508; DB00726; DB06109; DB01392; DB00246; DB09225; DB01624 Interacts with Q9NRI5; P14416; Q01959 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Lipoprotein; Membrane; Palmitate; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID B,C Molecular weight (Da) 24300.3 Length 209 Aromaticity 0.13 Instability index 40.14 Isoelectric point 4.97 Charge (pH=7) -7.83 2D Binding mode Binding energy (Kcal/mol) -6.02 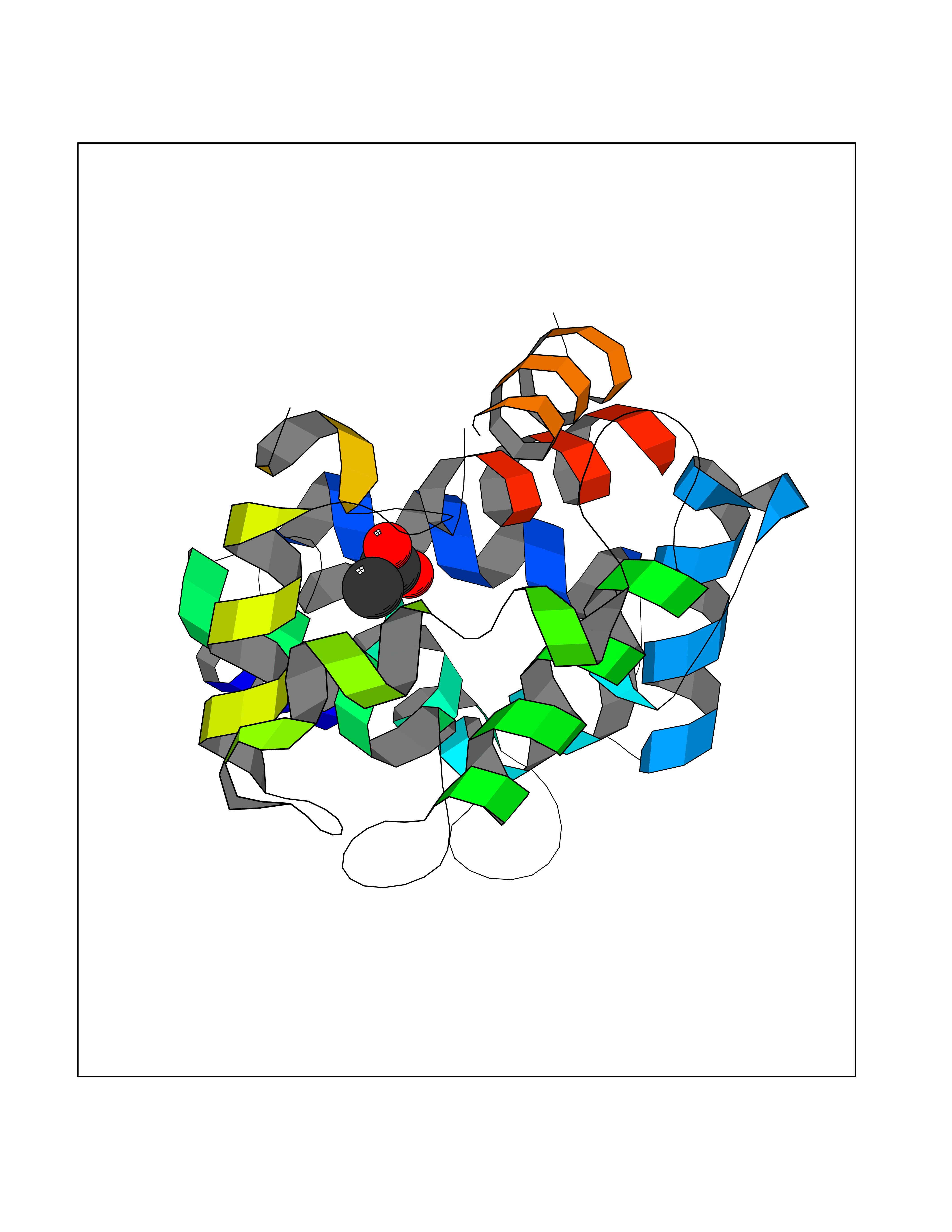 Molscript Map 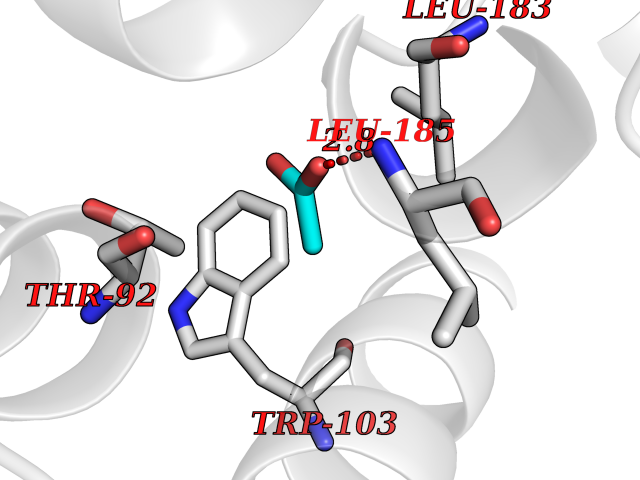 Pymol Map 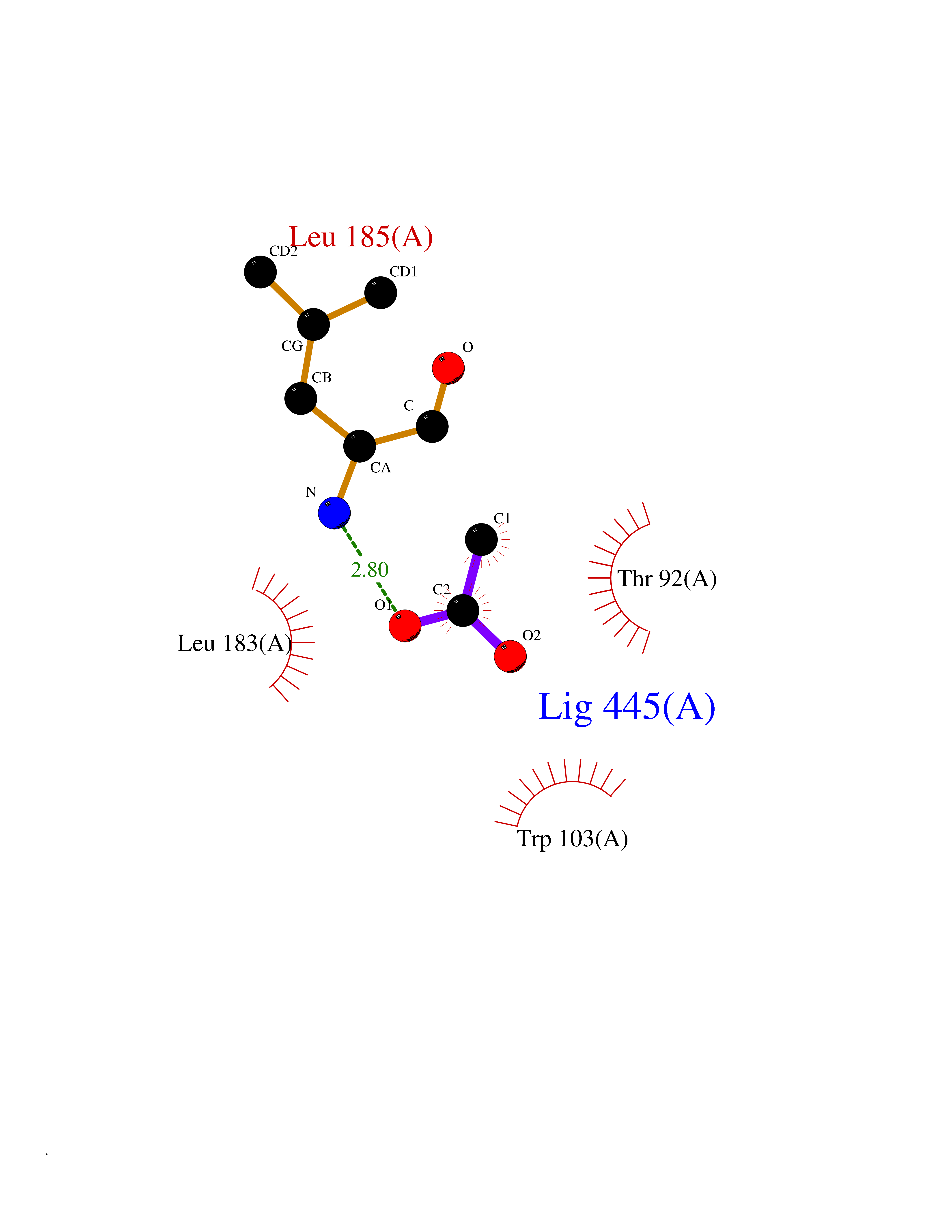 Ligplot Map 3D Binding mode Sequence PEVVEELTRKTYFTEKEVQQWYKGFIKDCPSGQLDAAGFQKIYKQFFPFGDPTKFATFVFNVFDENKDGRIEFSEFIQALSVTSRGTLDEKLRWAFKLYDLDNDGYITRNEMLDIVDAIYQMVGNTVELPEEENTPEKRVDRIFAMMDKNADGKLTLQEFQEGSKADPSIVQALSLYDGLVNIEFRKAFLKILHSNIEFRKAFLKILHS Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Deoxycytidine kinase (DCK) | 1P5Z | 4.41 | |
Target general information Gen name DCK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms dCK Protein family DCK/DGK family Biochemical class Kinase Function Required for the phosphorylation of the deoxyribonucleosides deoxycytidine (dC), deoxyguanosine (dG) and deoxyadenosine (dA). Has broad substrate specificity, and does not display selectivity based on the chirality of the substrate. It is also an essential enzyme for the phosphorylation of numerous nucleoside analogs widely employed as antiviral and chemotherapeutic agents. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB02594; DB00242; DB00631; DB00987; DB01262; DB05494; DB00879; DB01073; DB00441; DB00709; DB01280; DB00642; DB04961; DB00943 Interacts with Q16854 EC number EC 2.7.1.74 Uniprot keywords 3D-structure; ATP-binding; Direct protein sequencing; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 27128.5 Length 229 Aromaticity 0.13 Instability index 52.5 Isoelectric point 5.26 Charge (pH=7) -7.8 2D Binding mode Binding energy (Kcal/mol) -6.01 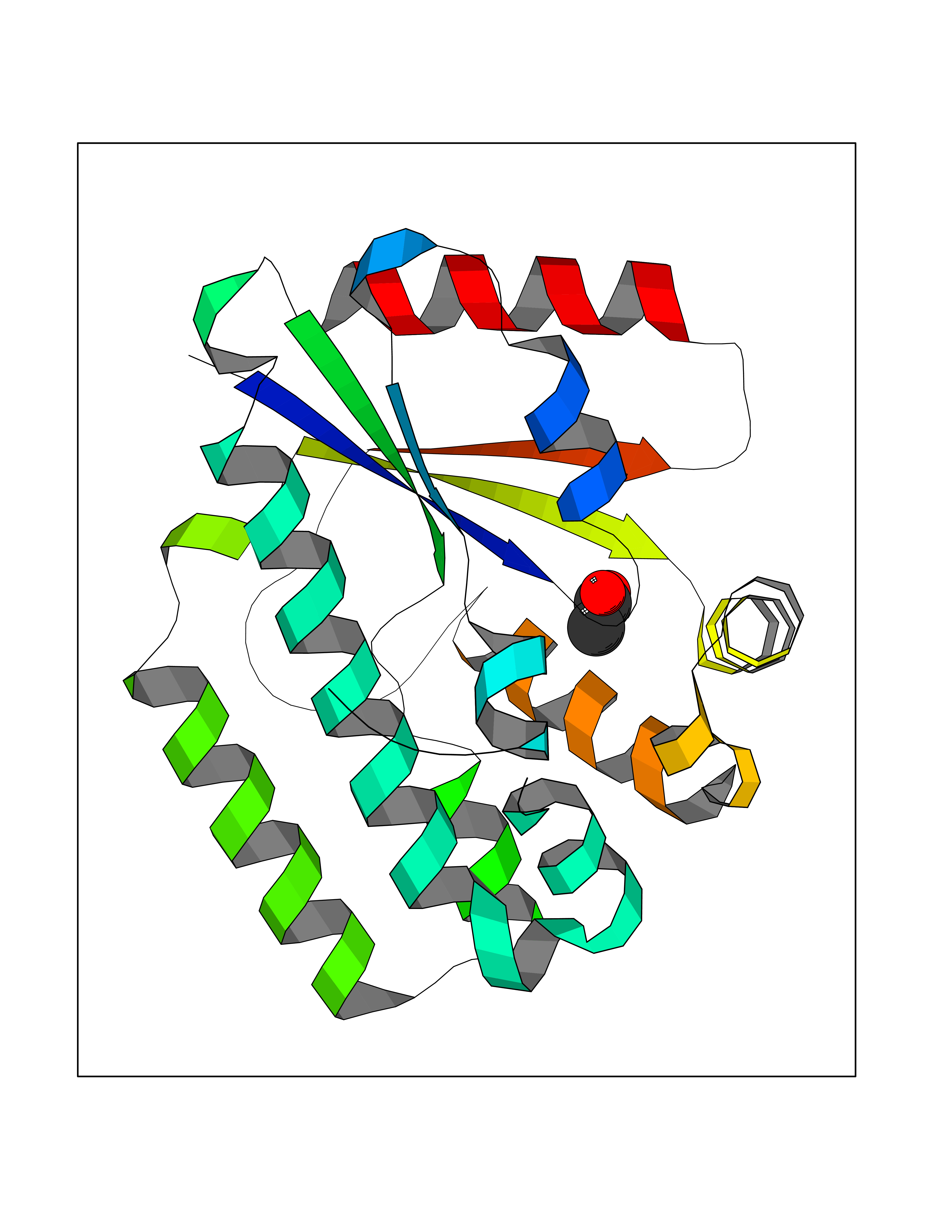 Molscript Map 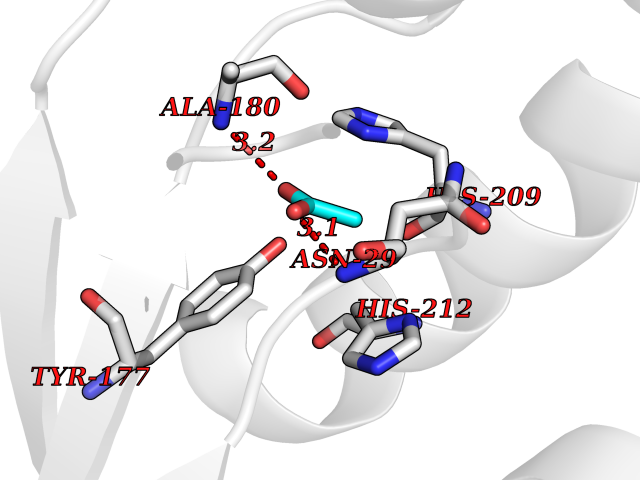 Pymol Map 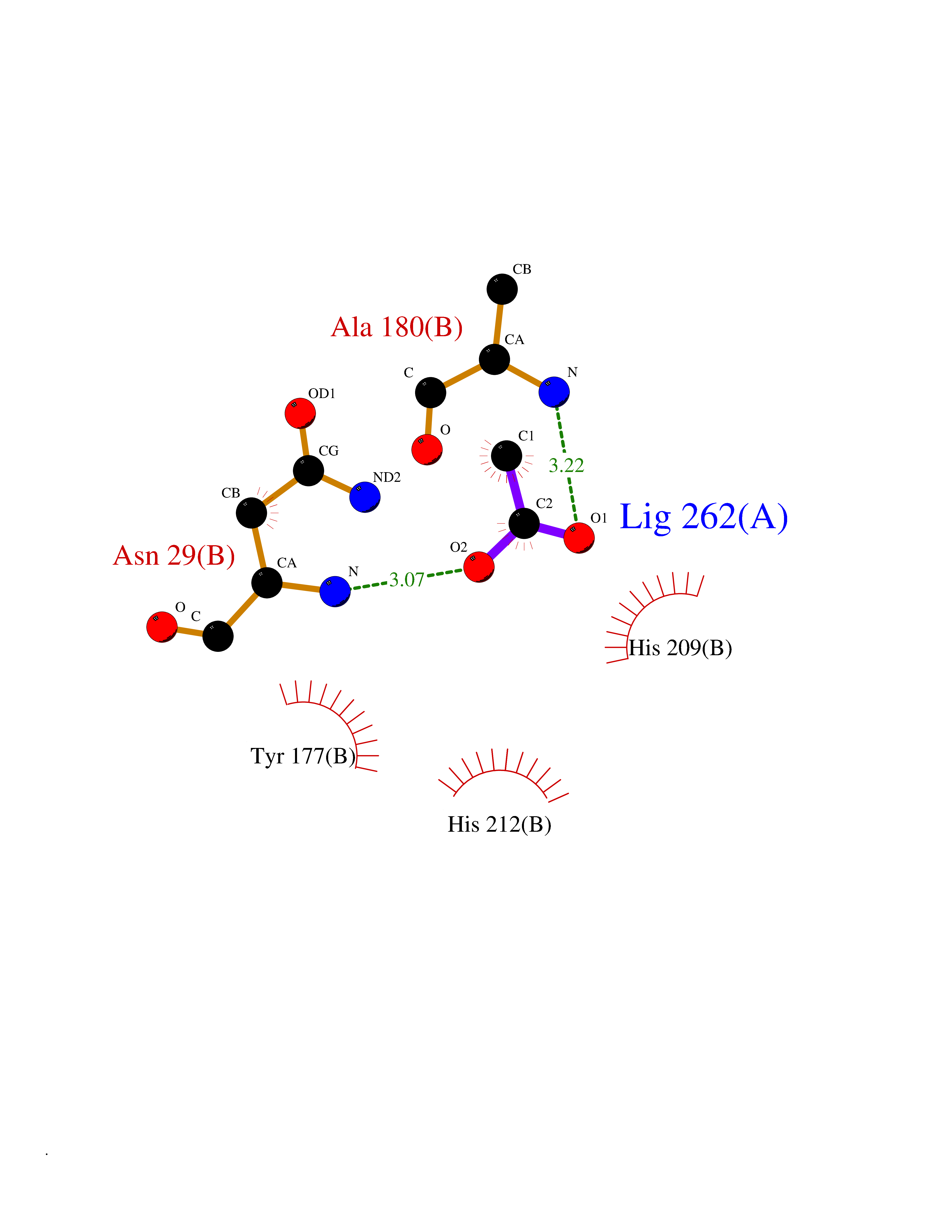 Ligplot Map 3D Binding mode Sequence RIKKISIEGNIAAGKSTFVNILKQLCEDWEVVPEPVARWCNVQSTNGGNVLQMMYEKPERWSFTFQTYACLSRIRAQLASLNGKLKDAEKPVLFFERSVYSDRYIFASNLYESECMNETEWTIYQDWHDWMNNQFGQSLELDGIIYLQATPETCLHRIYLRGRNEEQGIPLEYLEKLHYKHESWLLHRTLKTNFDYLQEVPILTLDVNEDFKDKYESLVEKVKEFLSTL Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Epithelial discoidin domain receptor 1 (DDR1) | 4BKJ | 4.41 | |
Target general information Gen name DDR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase CAK; Tyrosine kinase DDR; TRKE; TRK E; RTK6; Protein-tyrosine kinase RTK-6; Protein-tyrosine kinase 3A; PTK3A; NTRK4; NEP; Mammary carcinoma kinase 10; MCK-10; HGK2; Epithelial Protein family Protein kinase superfamily, Tyr protein kinase family, Insulin receptor subfamily Biochemical class Kinase Function Collagen binding triggers a signaling pathway that involves SRC and leads to the activation of MAP kinases. Regulates remodeling of the extracellular matrix by up-regulation of the matrix metalloproteinases MMP2, MMP7 and MMP9, and thereby facilitates cell migration and wound healing. Required for normal blastocyst implantation during pregnancy, for normal mammary gland differentiation and normal lactation. Required for normal ear morphology and normal hearing. Promotes smooth muscle cell migration, and thereby contributes to arterial wound healing. Also plays a role in tumor cell invasion. Phosphorylates PTPN11. Tyrosine kinase that functions as cell surface receptor for fibrillar collagen and regulates cell attachment to the extracellular matrix, remodeling of the extracellular matrix, cell migration, differentiation, survival and cell proliferation. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010; DB00619; DB15822 Interacts with Q16832; O43639; Q06124; Q9UHD9 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Calcium; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; Kinase; Lactation; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Pregnancy; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34061.1 Length 297 Aromaticity 0.1 Instability index 42.8 Isoelectric point 6.32 Charge (pH=7) -2.01 2D Binding mode Binding energy (Kcal/mol) -6.02 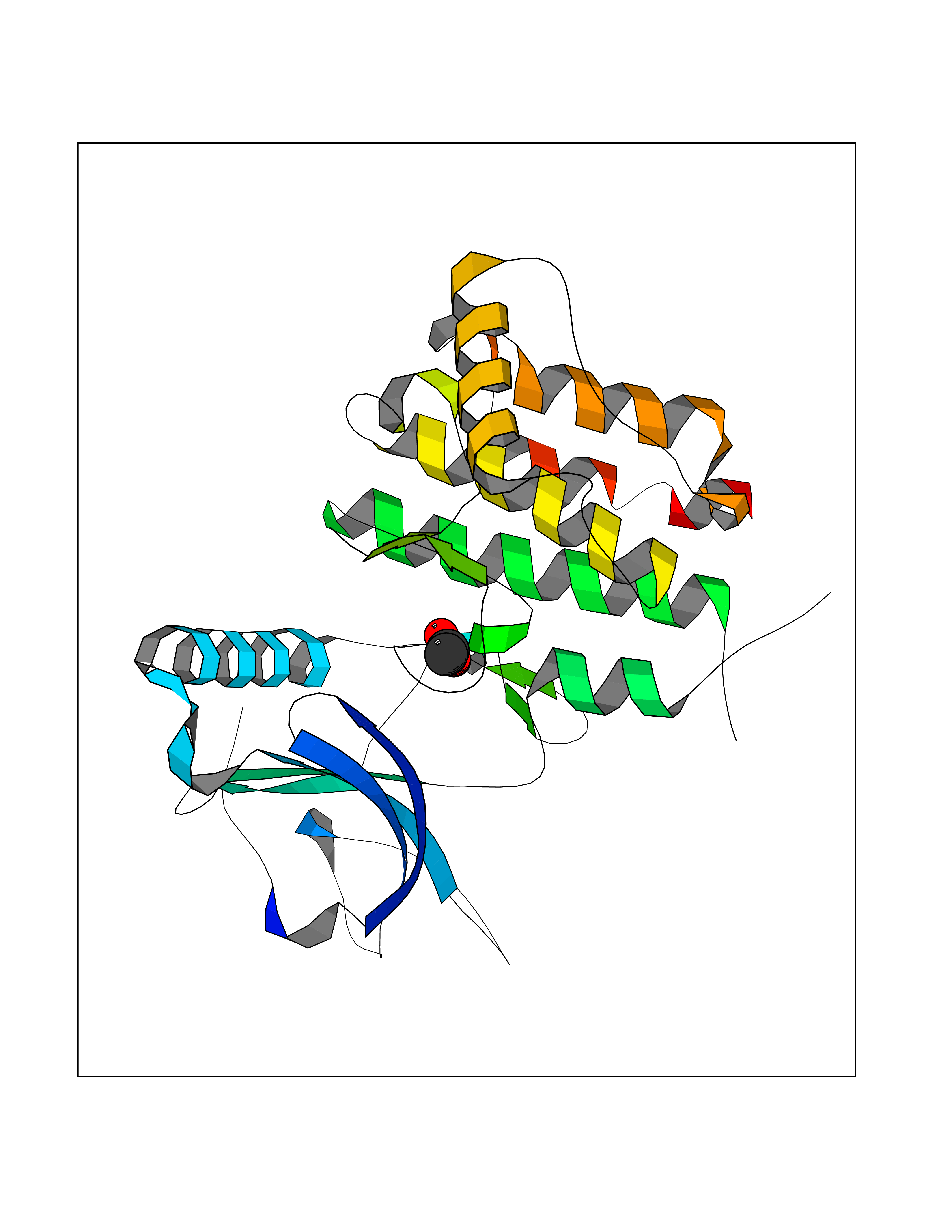 Molscript Map 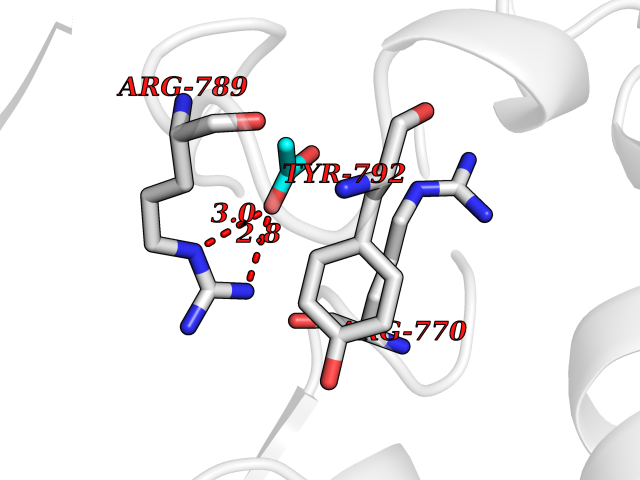 Pymol Map 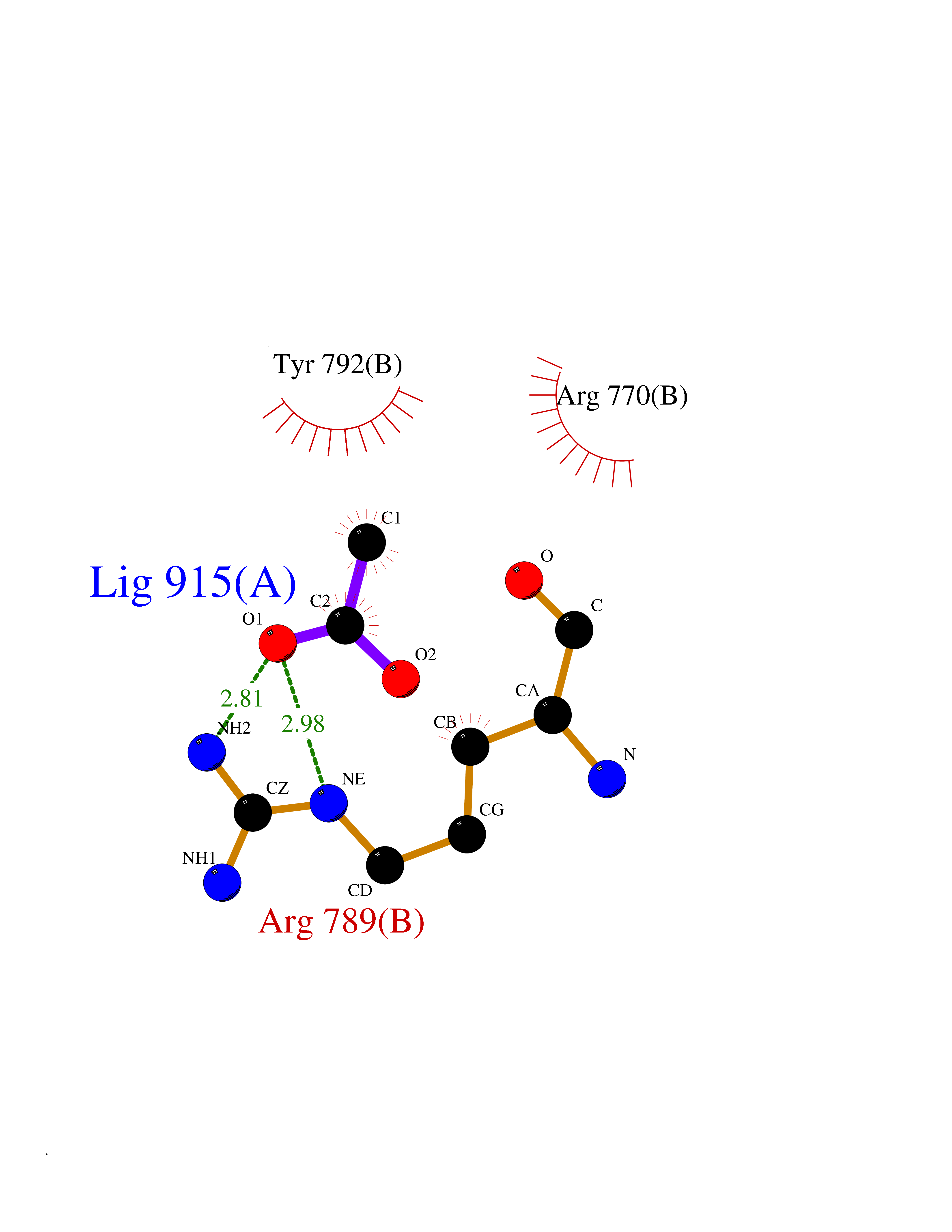 Ligplot Map 3D Binding mode Sequence MPRVDFPRSRLRFKEKLGEGQFGEVHLCEVDSPQDLVSLDFPLNVRKGHPLLVAVKILRPDATKNARNDFLKEVKIMSRLKDPNIIRLLGVCVQDDPLCMITDYMENGDLNQFLSAHQLEDKGPTISYPMLLHVAAQIASGMRYLATLNFVHRDLATRNCLVGENFTIKIADFGMSRNLYAGDYYRAVLPIRWMAWECILMGKFTTASDVWAFGVTLWEVLMLCRAQPFGQLTDEQVIENAGEFFRDQGRQVYLSRPPACPQGLYELMLRCWSRESEQRPPFSQLHRFLAEDALNTV Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Nischarin | 3P0C | 4.41 | |
Target general information Gen name NISCH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA0975;IRAS Protein family NA Biochemical class Signaling protein Function Identical protein binding.Integrin binding.Phosphatidylinositol binding. Related diseases Dyskinesia, limb and orofacial, infantile-onset (IOLOD) [MIM:616921]: An autosomal recessive, early-onset hyperkinetic movement disorder characterized by axial hypotonia, dyskinesia of the limbs and trunk, orofacial dyskinesia, drooling, and dysarthria. The severity of the hyperkinesis is variable. {ECO:0000269|PubMed:27058446}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Striatal degeneration, autosomal dominant 2 (ADSD2) [MIM:616922]: An autosomal dominant disorder characterized by striatal degeneration and dysfunction of basal ganglia, resulting in hyperkinesis. {ECO:0000269|PubMed:27058447}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08838; DB09242; DB15133; DB00697 Interacts with Q9Y2I1; P51151; P61107; P05714 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; Cell membrane; Coiled coil; Cytoplasm; Endosome; Leucine-rich repeat; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25709.3 Length 219 Aromaticity 0.11 Instability index 13.52 Isoelectric point 8.57 Charge (pH=7) 3.01 2D Binding mode Binding energy (Kcal/mol) -6.02 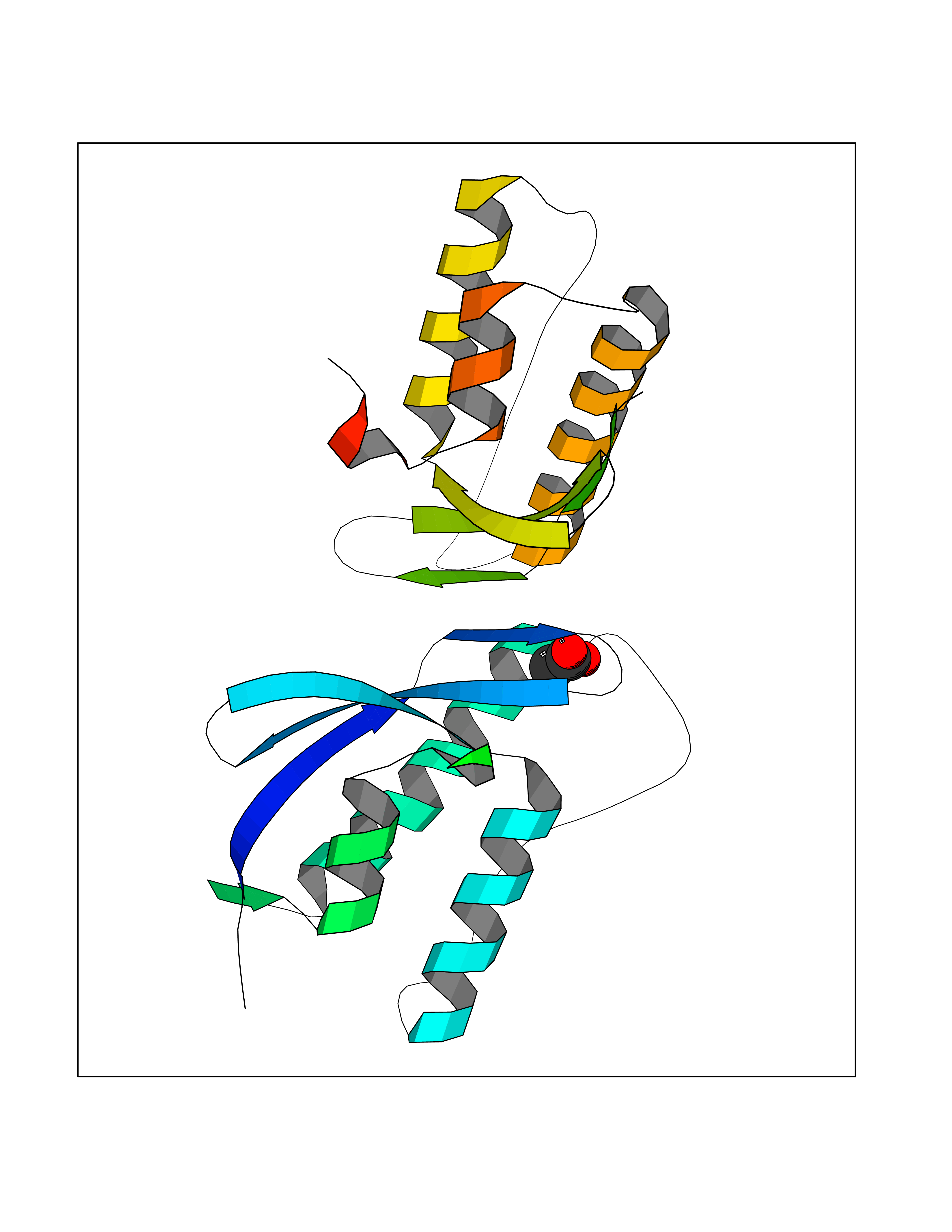 Molscript Map  Pymol Map 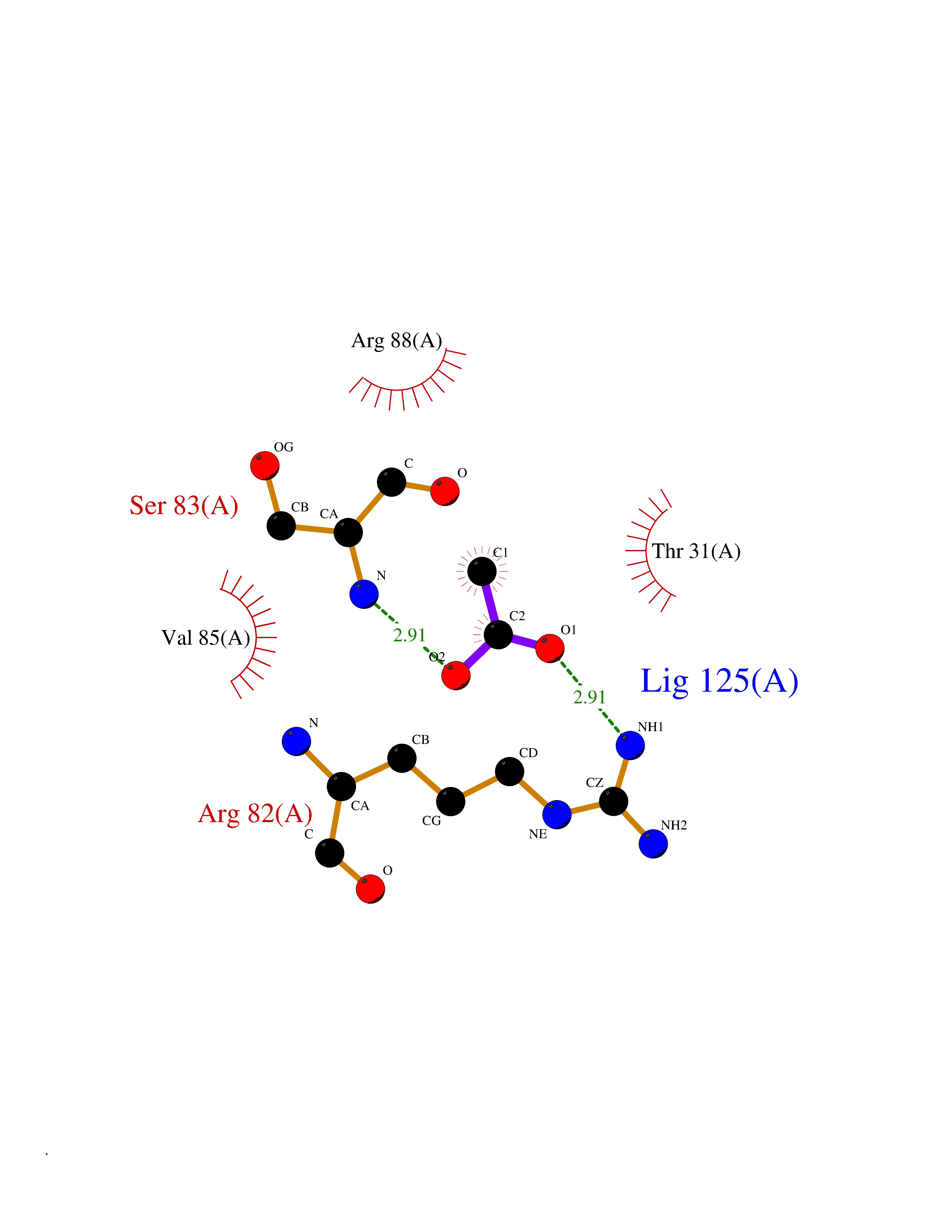 Ligplot Map 3D Binding mode Sequence NLYFQSMEARVVGSELVDTYTVYIIQVTDGSHEWTVKHRYSDFHDLHEKLVAERKIDKNLLPPKKIIGKNSRSLVEKREKDLEVYLQKLLAAFPGVTPRVLAHFLHFHFYESMEARVVGSELVDTYTVYIIQVTDGSHEWTVKHRYSDFHDLHEKLVAERKIDKNLLPPKKIIGKNSRSLVEKREKDLEVYLQKLLAAFPGVTPRVLAHFLHFHFYEIN Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | cAMP-dependent protein kinase A type I (PRKAR1A) | 5KJZ | 4.41 | |
Target general information Gen name PRKAR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-dependent protein kinase type I-alpha regulatory subunit; Tissue-specific extinguisher 1; TSE1; PRKAR1; PKR1 Protein family CAMP-dependent kinase regulatory chain family Biochemical class Kinase Function Regulatory subunit of the cAMP-dependent protein kinases involved in cAMP signaling in cells. Related diseases Carney complex 1 (CNC1) [MIM:160980]: CNC is a multiple neoplasia syndrome characterized by spotty skin pigmentation, cardiac and other myxomas, endocrine tumors, and psammomatous melanotic schwannomas. {ECO:0000269|PubMed:15371594, ECO:0000269|PubMed:18241045, ECO:0000269|PubMed:22785148, ECO:0000269|PubMed:23323113, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Intracardiac myxoma (INTMYX) [MIM:255960]: Inheritance is autosomal recessive. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Primary pigmented nodular adrenocortical disease 1 (PPNAD1) [MIM:610489]: A rare bilateral adrenal defect causing ACTH-independent Cushing syndrome. Macroscopic appearance of the adrenals is characteristic with small pigmented micronodules observed in the cortex. Clinical manifestations of Cushing syndrome include facial and truncal obesity, abdominal striae, muscular weakness, osteoporosis, arterial hypertension, diabetes. PPNAD1 is most often diagnosed in patients with Carney complex, a multiple neoplasia syndrome. However it can also be observed in patients without other manifestations. {ECO:0000269|PubMed:12213893}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Acrodysostosis 1, with or without hormone resistance (ACRDYS1) [MIM:101800]: A form of skeletal dysplasia characterized by short stature, severe brachydactyly, facial dysostosis, and nasal hypoplasia. Affected individuals often have advanced bone age and obesity. Laboratory studies show resistance to multiple hormones, including parathyroid, thyrotropin, calcitonin, growth hormone-releasing hormone, and gonadotropin. However, not all patients show endocrine abnormalities. {ECO:0000269|PubMed:21651393, ECO:0000269|PubMed:22464250, ECO:0000269|PubMed:22464252, ECO:0000269|PubMed:22723333, ECO:0000269|PubMed:23043190, ECO:0000269|PubMed:23425300, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01790; DB02527; DB02315; DB05798 Interacts with Q9GZX7; P24588; O43687-2; Q9BSF0; Q9H6J7-2; Q86Y01; P0C7A2-2; Q9H0R8; Q9H8W4; P17612; P31321; P51817; P35250; Q86UC2; Q01105; Q8N0X7; O96006; P03259-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; cAMP; cAMP-binding; Cell membrane; Cushing syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat Protein physicochemical properties Chain ID A Molecular weight (Da) 17007.4 Length 149 Aromaticity 0.08 Instability index 49.36 Isoelectric point 6.36 Charge (pH=7) -0.52 2D Binding mode Binding energy (Kcal/mol) -6.02 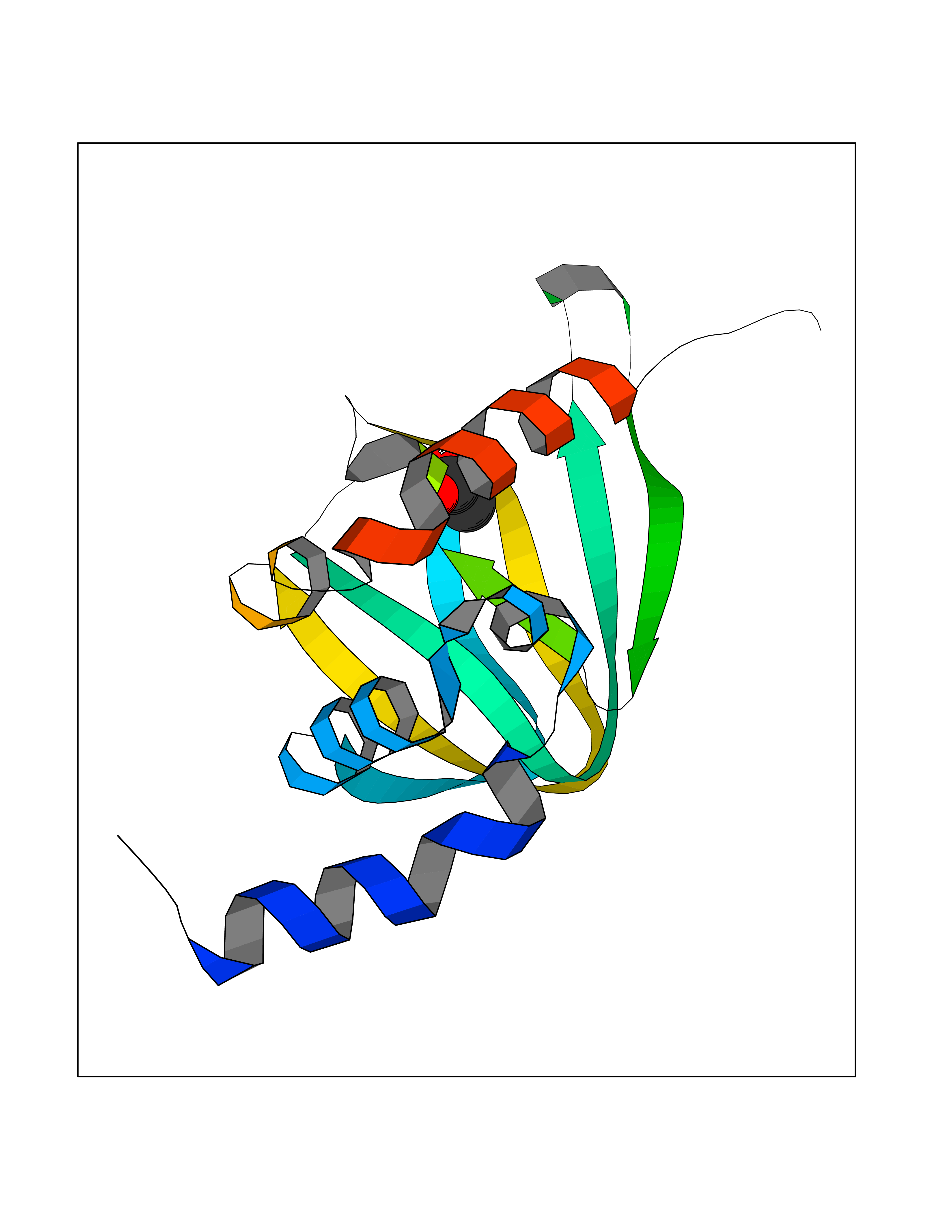 Molscript Map 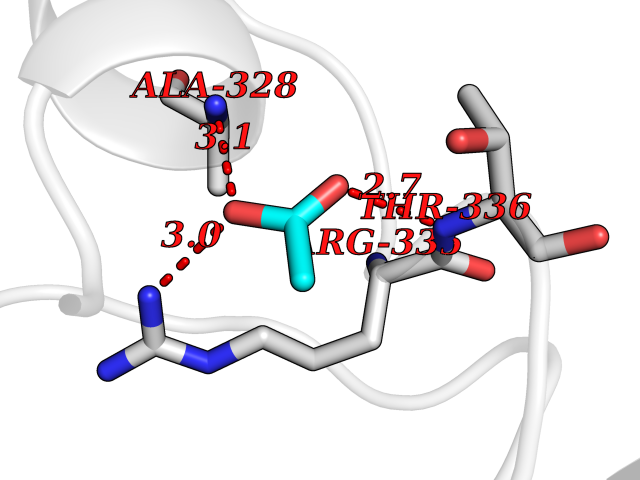 Pymol Map 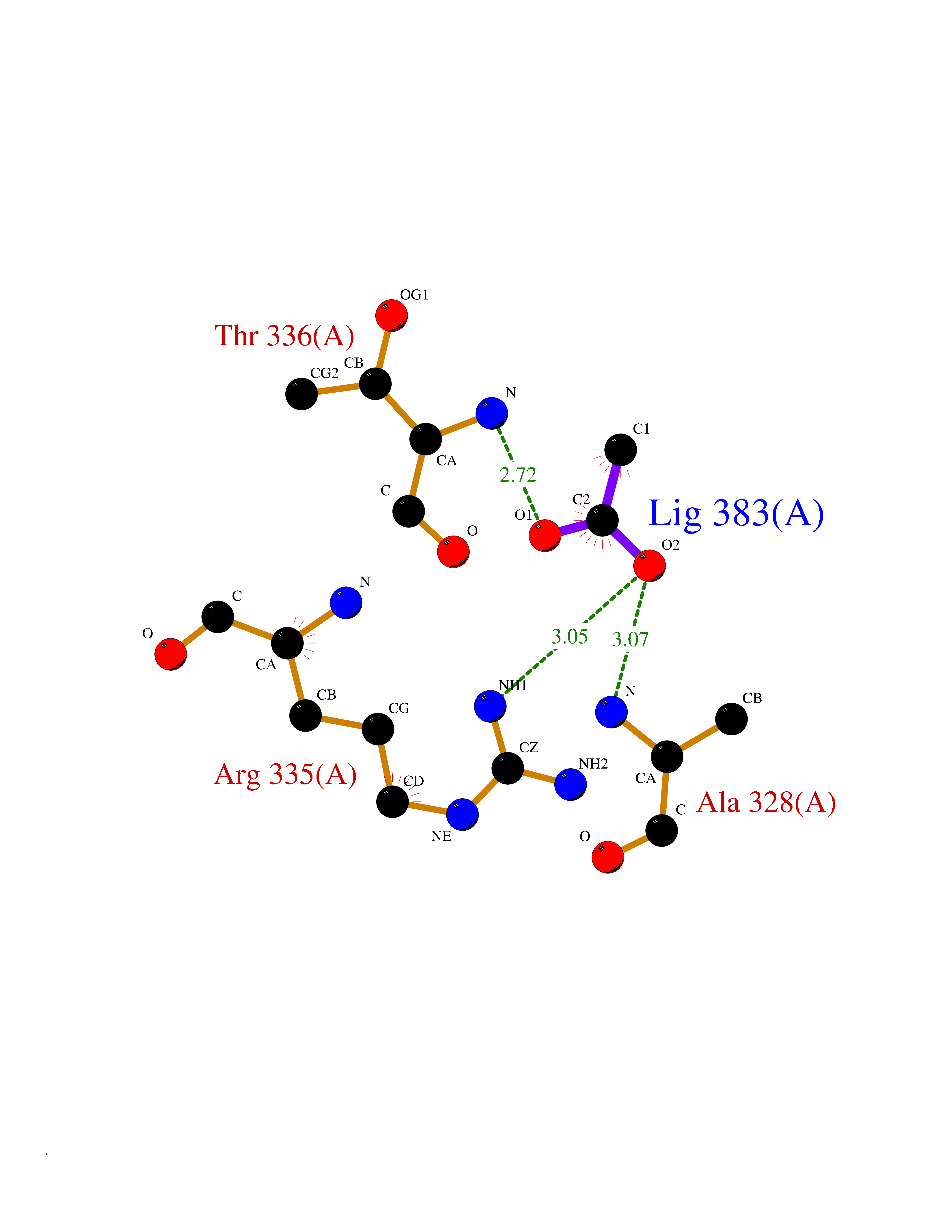 Ligplot Map 3D Binding mode Sequence SILMGSTLRKRKMYEEFLSKVSILESLDKWERLTVADALEPVQFEDGQKIVVQGEPGDEFFIILEGSAAVLQRRSENEEFVEVRRLGPSDYFGEIALLMNRPRTATVVARGPLKCVKLDRPRFERVLGPCSDILKRNIQQYNSFVSLSV Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Orotidine 5'-monophosphate decarboxylase (UMPS) | 3MI2 | 4.41 | |
Target general information Gen name UMPS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Uridine 5'-monophosphate synthase; UMP synthase Protein family Purine/pyrimidine phosphoribosyltransferase family; OMP decarboxylase family Biochemical class Pentosyltransferase Function Catalyses the formation of uridine monophosphate (UMP), an energy-carrying molecule in many important biosynthetic pathways. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02890; DB00544 Interacts with P54764; P11172-1 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Decarboxylase; Disease variant; Glycosyltransferase; Lyase; Multifunctional enzyme; Phosphoprotein; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 55844 Length 514 Aromaticity 0.06 Instability index 22.7 Isoelectric point 6.44 Charge (pH=7) -2.99 2D Binding mode Binding energy (Kcal/mol) -6.02 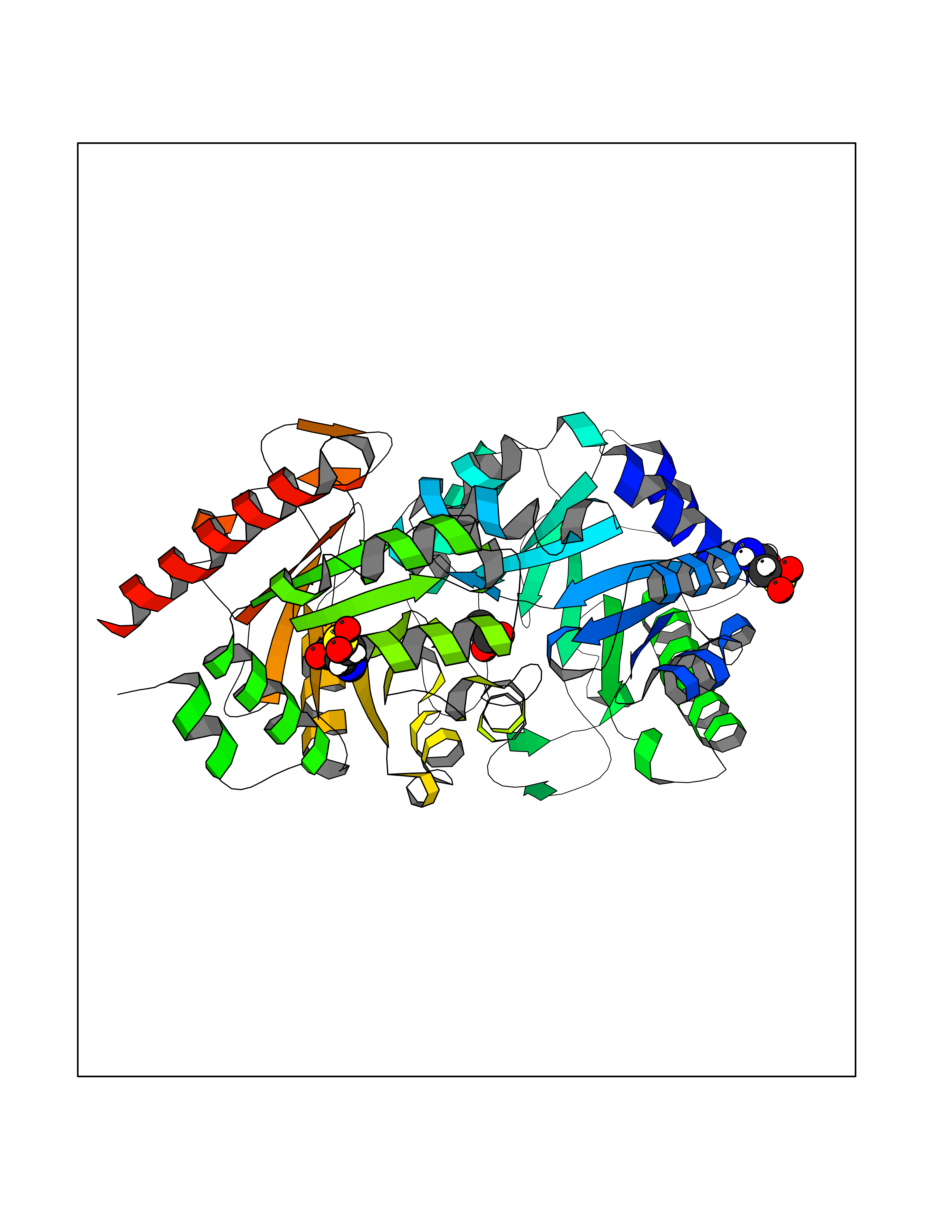 Molscript Map 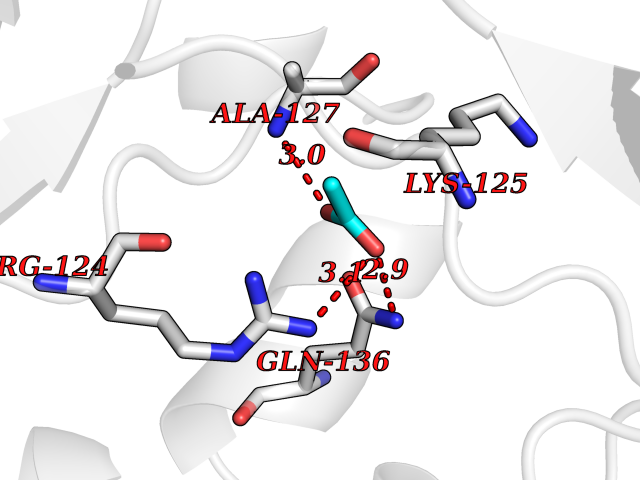 Pymol Map 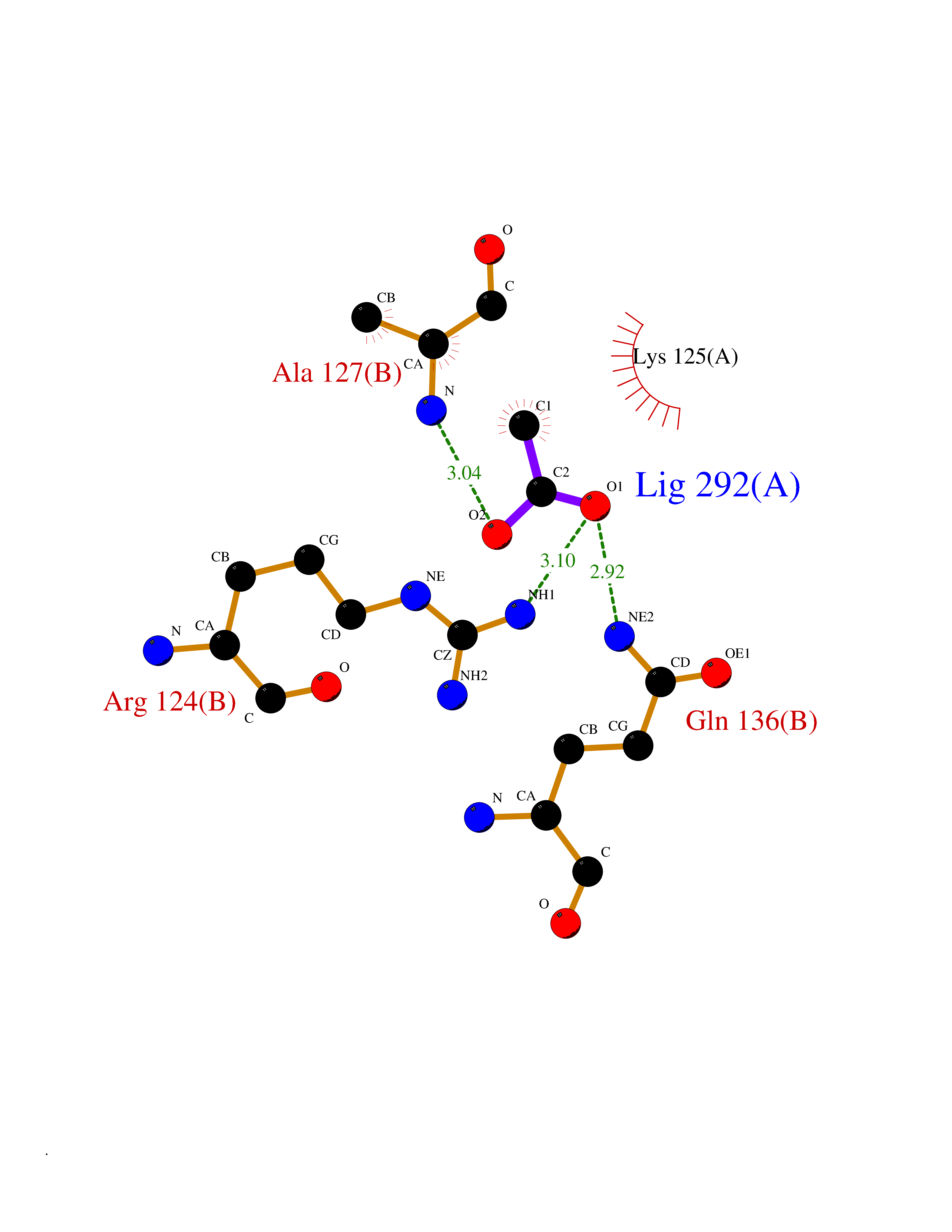 Ligplot Map 3D Binding mode Sequence KELSFGARAELPRIHPVASKLLRLMQKKETNLCLSADVSLARELLQLADALGPSICMLKTHVDILNDFTLDVMKELITLAKXHEFLIFEDRKFADIGNTVKKQYEGGIFKIASWADLVNAHVVPGSGVVKGLQEVGLPLHRGCLLIAEMSSTGSLATGDYTRAAVRMAEEHSEFVVGFISGSRVSMKPEFLHLTPGVQLEAGGDNLGQQYNSPQEVIGKRGSDIIIVGRGIISAADRLEAAEMYRKAAWEAYLSRLGKELSFGARAELPRIHPVASKLLRLMQKKETNLCLSADVSLARELLQLADALGPSICMLKTHVDILNDFTLDVMKELITLAKXHEFLIFEDRKFADIGNTVKKQYEGGIFKIASWADLVNAHVVPGSGVVKGLQEVGLPLHRGCLLIAEMSSTGSLATGDYTRAAVRMAEEHSEFVVGFISGSRVSMKPEFLHLTPGVQLEAGGDNLGQQYNSPQEVIGKRGSDIIIVGRGIISAADRLEAAEMYRKAAWEAYLSRLG Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Protein kinase G2 (PRKG2) | 5BV6 | 4.41 | |
Target general information Gen name PRKG2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-dependent protein kinase II; cGMP-dependent protein kinase 2; cGKII; cGK2; cGK 2; PRKGR2 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, cGMP subfamily Biochemical class NA Function Crucial regulator of intestinal secretion and bone growth (By similarity). Phosphorylates and activates CFTR on the plasma membrane. Plays a key role in intestinal secretion by regulating cGMP-dependent translocation of CFTR in jejunum (By similarity). Acts downstream of NMDAR to activate the plasma membrane accumulation of GRIA1/GLUR1 in synapse and increase synaptic plasticity. Phosphorylates GRIA1/GLUR1 at Ser-863 (By similarity). Acts as regulator of gene expression and activator of the extracellular signal-regulated kinases MAPK3/ERK1 and MAPK1/ERK2 in mechanically stimulated osteoblasts. Under fluid shear stress, mediates ERK activation and subsequent induction of FOS, FOSL1/FRA1, FOSL2/FRA2 and FOSB that play a key role in the osteoblast anabolic response to mechanical stimulation (By similarity). Related diseases Spondylometaphyseal dysplasia, Pagnamenta type (SMDP) [MIM:619638]: A form of spondylometaphyseal dysplasia, a group of short stature disorders distinguished by abnormalities in the vertebrae and the metaphyses of the tubular bones. SMDP is an autosomal recessive form characterized by short stature and mild platyspondyly with no disproportion between the limbs. Mild metaphyseal changes are present. {ECO:0000269|PubMed:34782440}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Acromesomelic dysplasia 4 (AMD4) [MIM:619636]: A form of acromesomelic dysplasia, a skeletal disorder characterized by short stature, very short limbs and hand/foot malformations. The severity of limb abnormalities increases from proximal to distal with profoundly affected hands and feet showing brachydactyly and/or rudimentary fingers (knob-like fingers). AMD4 radiographic hallmarks include mild to moderate platyspondyly, moderate brachydactyly, iliac flaring, and metaphyseal alterations of the long bones that progressively increase with age. AMD4 inheritance is autosomal recessive. {ECO:0000269|PubMed:33106379, ECO:0000269|PubMed:34782440}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with NA EC number EC 2.7.11.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; cGMP; cGMP-binding; Coiled coil; Disease variant; Dwarfism; Kinase; Lipoprotein; Membrane; Myristate; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 17227.1 Length 150 Aromaticity 0.09 Instability index 43.7 Isoelectric point 4.88 Charge (pH=7) -8.41 2D Binding mode Binding energy (Kcal/mol) -6.02 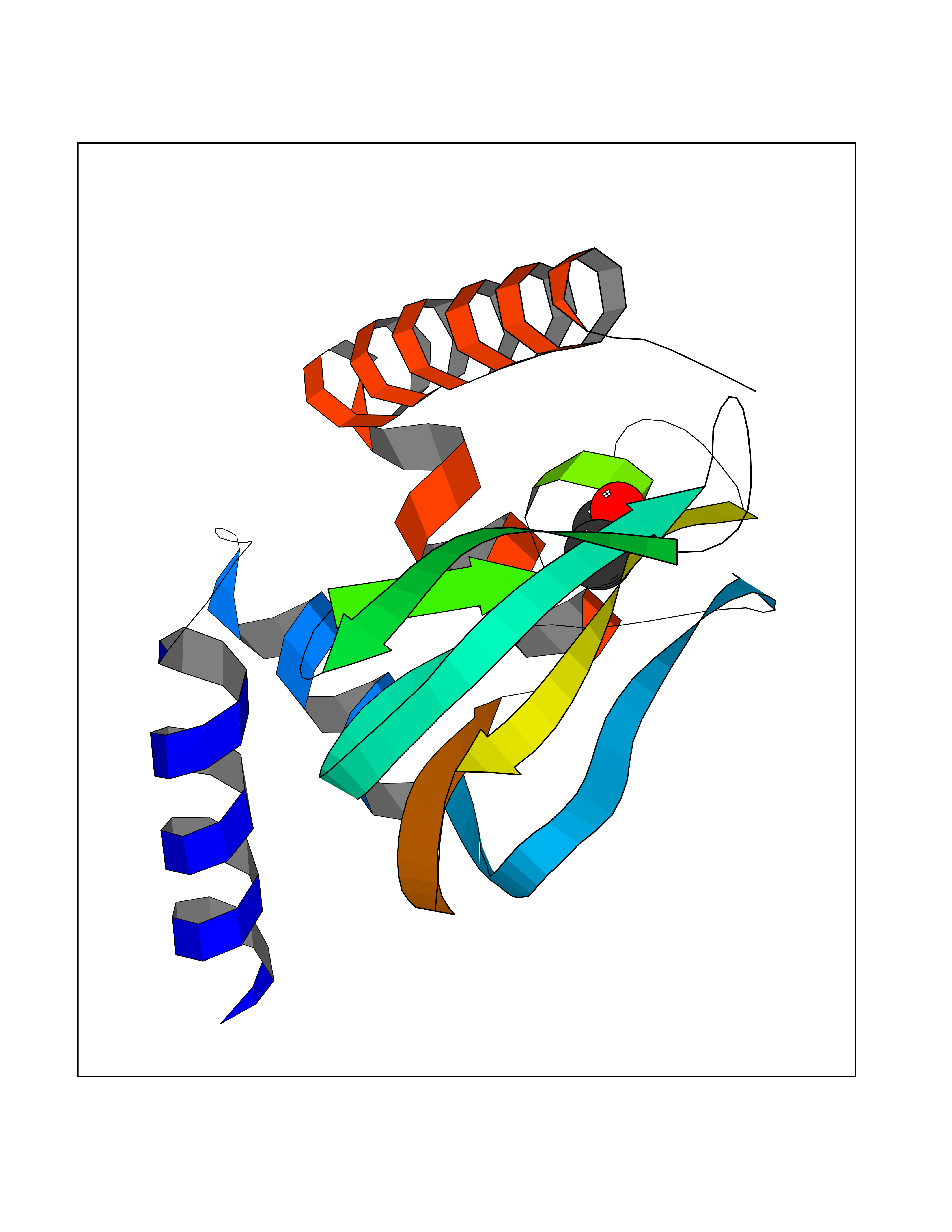 Molscript Map 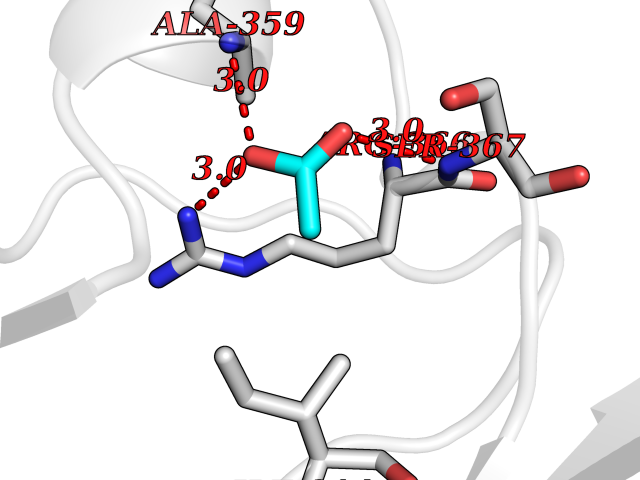 Pymol Map 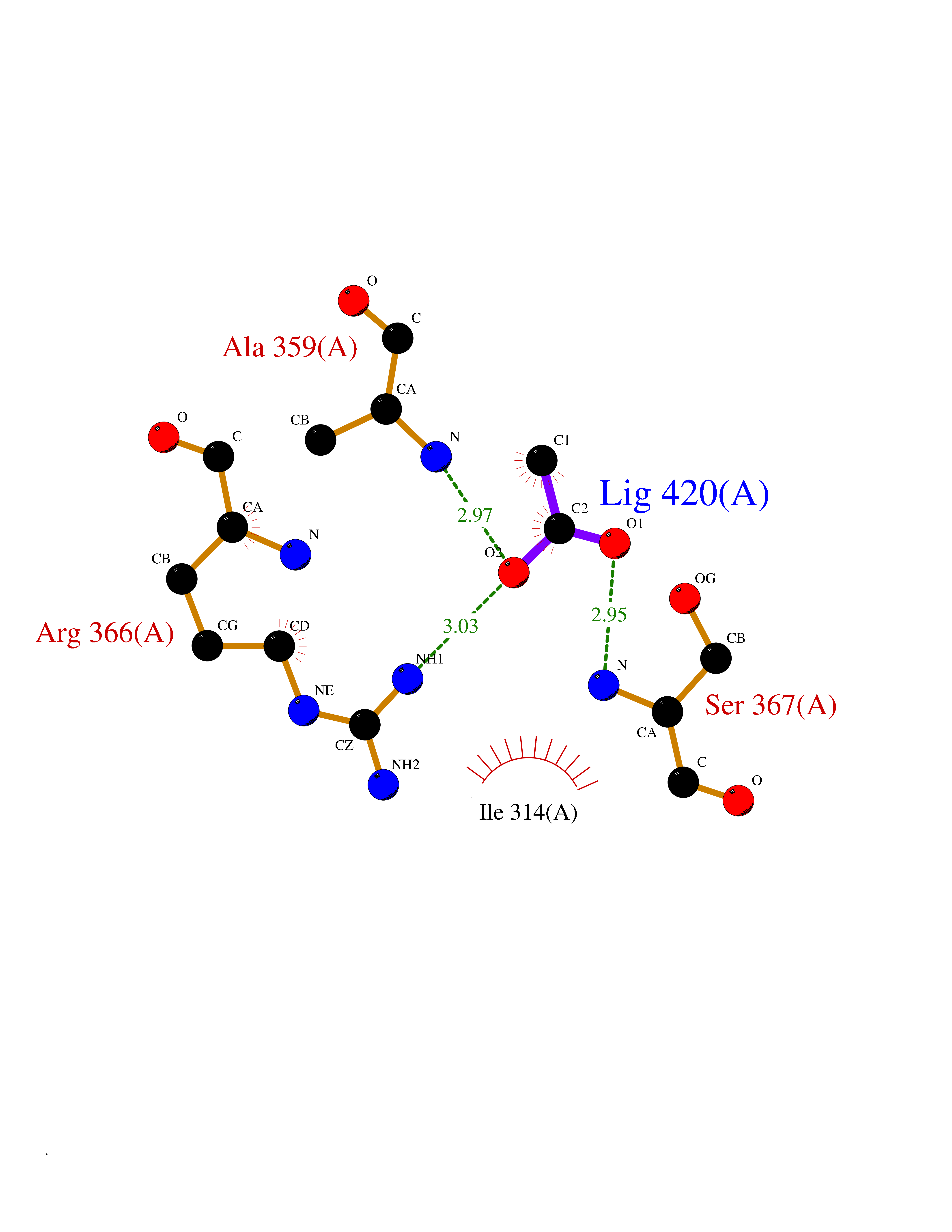 Ligplot Map 3D Binding mode Sequence TAQARDEQYRNFLRSVSLLKNLPEDKLTKIIDCLEVEYYDKGDYIIREGEEGSTFFILAKGKVKVTQSTEGHDQPQLIKTLQKGEYFGEKALISDDVRSANIIAEENDVACLVIDRETFNQTVGTFEELQKYLEGYVANLNRDDEKRHAK Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Lysine-specific histone demethylase 1B (KDM1B) | 4HSU | 4.41 | |
Target general information Gen name KDM1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine-specific histone demethylase 2; LSD2; Flavin-containing amine oxidase domain-containing protein 1; C6orf193; AOF1 Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Required for de novo DNA methylation of a subset of imprinted genes during oogenesis. Acts by oxidizing the substrate by FAD to generate the corresponding imine that is subsequently hydrolyzed. Demethylates both mono- and di-methylated 'Lys-4' of histone H3. Has no effect on tri-methylated 'Lys-4', mono-, di- or tri-methylated 'Lys-9', mono-, di- or tri-methylated 'Lys-27', mono-, di- or tri-methylated 'Lys-36' of histone H3, or on mono-, di- or tri-methylated 'Lys-20' of histone H4. Histone demethylase that demethylates 'Lys-4' of histone H3, a specific tag for epigenetic transcriptional activation, thereby acting as a corepressor. Related diseases Angioedema, hereditary, 1 (HAE1) [MIM:106100]: An autosomal dominant disorder characterized by episodic local swelling involving subcutaneous or submucous tissue of the upper respiratory and gastrointestinal tracts, face, extremities, and genitalia. Hereditary angioedema due to C1 esterase inhibitor deficiency is comprised of two clinically indistinguishable forms. In hereditary angioedema type 1, serum levels of C1 esterase inhibitor are decreased, while in type 2, the levels are normal or elevated, but the protein is non-functional. {ECO:0000269|PubMed:12773530, ECO:0000269|PubMed:1363816, ECO:0000269|PubMed:1451784, ECO:0000269|PubMed:14635117, ECO:0000269|PubMed:16409206, ECO:0000269|PubMed:2118657, ECO:0000269|PubMed:2296585, ECO:0000269|PubMed:22994404, ECO:0000269|PubMed:2365061, ECO:0000269|PubMed:24456027, ECO:0000269|PubMed:3178731, ECO:0000269|PubMed:7814636, ECO:0000269|PubMed:7883978, ECO:0000269|PubMed:8172583, ECO:0000269|PubMed:8529136, ECO:0000269|PubMed:8755917, ECO:0000269|Ref.41}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q96L03 EC number EC 1.-.-.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Developmental protein; FAD; Flavoprotein; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85795.5 Length 763 Aromaticity 0.1 Instability index 37.87 Isoelectric point 8.41 Charge (pH=7) 9.16 2D Binding mode Binding energy (Kcal/mol) -6.02 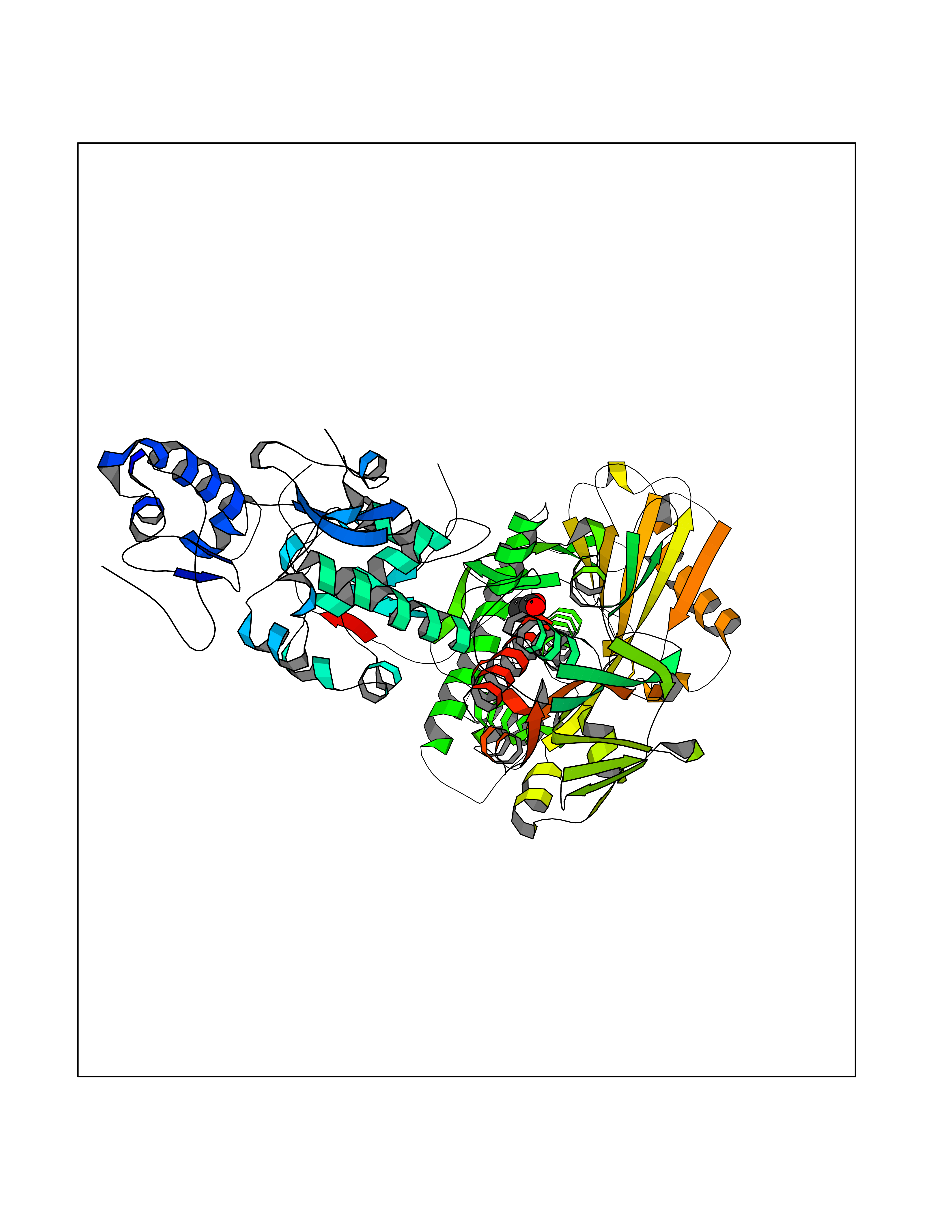 Molscript Map  Pymol Map 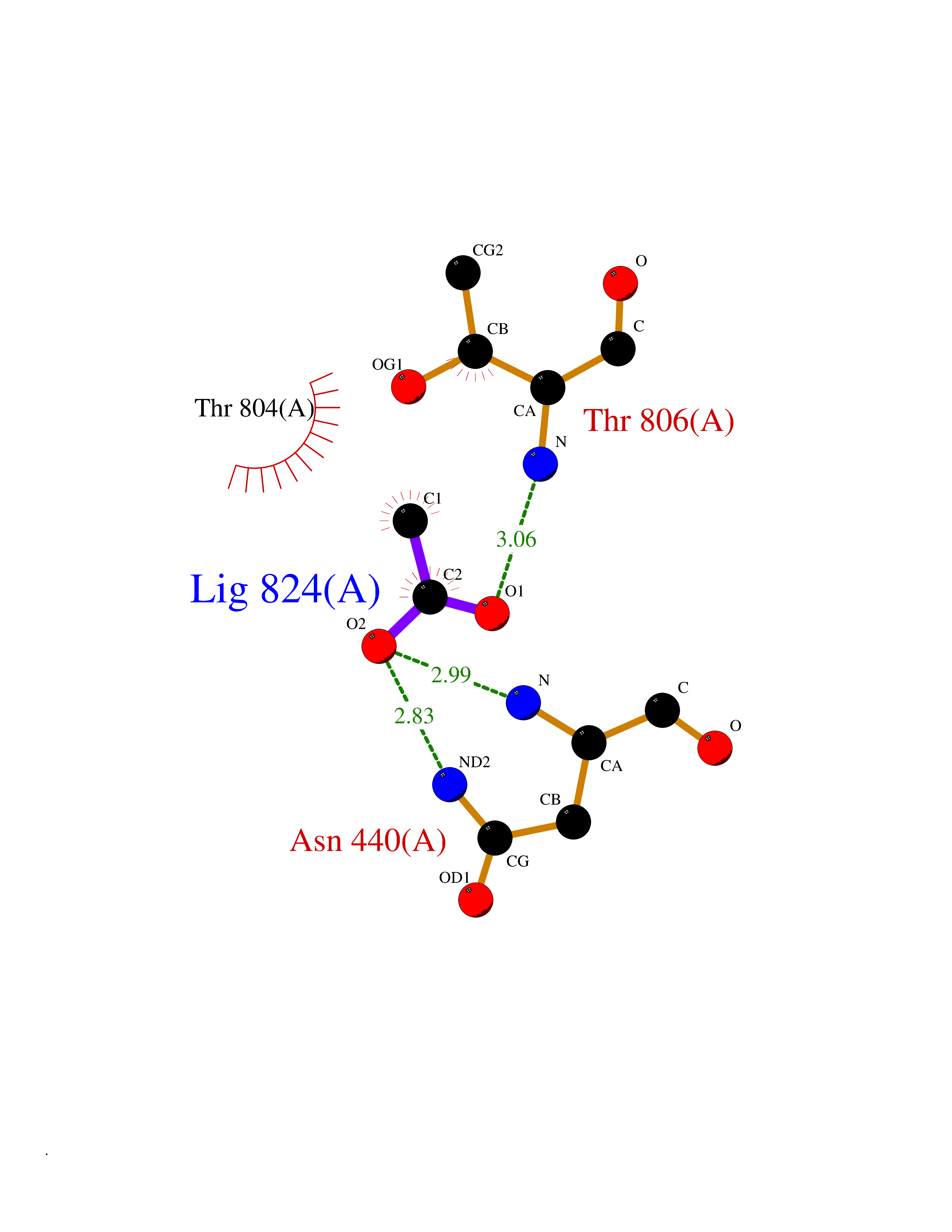 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQLATKAAR Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Thymidine kinase 1 (TK1) | 1W4R | 4.41 | |
Target general information Gen name TK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Thymidine kinase, cytosolic Protein family Thymidine kinase family Biochemical class Kinase Function cytosol, identical protein binding, thymidine kinase activity, zinc ion binding, DNA metabolic process, nucleobase-containing compound metabolic process, protein homotetramerization, pyrimidine nucleoside salvage, thymidine metabolic process Related diseases Seizures, benign familial infantile, 3 (BFIS3) [MIM:607745]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS3 inheritance is autosomal dominant. {ECO:0000269|PubMed:11371648, ECO:0000269|PubMed:12243921, ECO:0000269|PubMed:15048894, ECO:0000269|PubMed:16417554, ECO:0000269|PubMed:17021166, ECO:0000269|PubMed:17386050, ECO:0000269|PubMed:18479388, ECO:0000269|PubMed:20371507, ECO:0000269|PubMed:22612257, ECO:0000269|PubMed:23360469, ECO:0000269|PubMed:23758435, ECO:0000269|PubMed:25982755, ECO:0000269|PubMed:26291284, ECO:0000269|PubMed:29844171, ECO:0000269|PubMed:30144217}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 11 (DEE11) [MIM:613721]: An autosomal dominant seizure disorder characterized by neonatal or infantile onset of refractory seizures with resultant delayed neurologic development and persistent neurologic abnormalities. Patients may progress to West syndrome, which is characterized by tonic spasms with clustering, arrest of psychomotor development, and hypsarrhythmia on EEG. {ECO:0000269|PubMed:19783390, ECO:0000269|PubMed:19786696, ECO:0000269|PubMed:20956790, ECO:0000269|PubMed:22677033, ECO:0000269|PubMed:23033978, ECO:0000269|PubMed:23195492, ECO:0000269|PubMed:23550958, ECO:0000269|PubMed:23662938, ECO:0000269|PubMed:23708187, ECO:0000269|PubMed:23935176, ECO:0000269|PubMed:23988467, ECO:0000269|PubMed:24463883, ECO:0000269|PubMed:24579881, ECO:0000269|PubMed:24659627, ECO:0000269|PubMed:24710820, ECO:0000269|PubMed:25457084, ECO:0000269|PubMed:25459969, ECO:0000269|PubMed:25772804, ECO:0000269|PubMed:25818041, ECO:0000269|PubMed:26138355, ECO:0000269|PubMed:26291284, ECO:0000269|PubMed:26993267, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:29625812, ECO:0000269|PubMed:29844171, ECO:0000269|PubMed:30144217, ECO:0000269|PubMed:30415926}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in SCN2A are associated with genetic epilepsy with febrile seizures plus (GEFS+), a familial autosomal dominant epilepsy syndrome, a clinical subset of febrile seizures, characterized by frequent episodes after 6 years of age and various types of subsequent epilepsy. {ECO:0000269|PubMed:29635106}.; DISEASE: Defects in SCN2A are associated with autism spectrum disorders (ASD). It seems that mutations resulting in sodium channel gain of function and increased neuron excitability lead to infantile seizures, whereas variants resulting in sodium channel loss of function and decrease neuron excitability are associated with ASD. {ECO:0000269|PubMed:28256214}.; DISEASE: Episodic ataxia 9 (EA9) [MIM:618924]: An autosomal dominant neurologic disorder characterized by episodic ataxia manifesting in the first years of life, early-onset seizures, difficulty walking, dizziness, slurred speech, headache, vomiting, and pain. The duration of ataxic episodes is heterogeneous. Most patients show episodes lasting minutes to maximum several hours, but periods lasting days up to weeks have been reported. Some patients have mildly delayed development with speech delay and/or autistic features or mildly impaired intellectual development. {ECO:0000269|PubMed:26645390, ECO:0000269|PubMed:27159988, ECO:0000269|PubMed:27328862, ECO:0000269|PubMed:28065826}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01692; DB04485; DB02452; DB00432; DB00495 Interacts with P05067; A0A087WZT3; Q92993; Q1RN33; P04183 EC number EC 2.7.1.21 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cytoplasm; DNA synthesis; Kinase; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 19373.5 Length 174 Aromaticity 0.09 Instability index 36.21 Isoelectric point 8.63 Charge (pH=7) 3.88 2D Binding mode Binding energy (Kcal/mol) -6.02 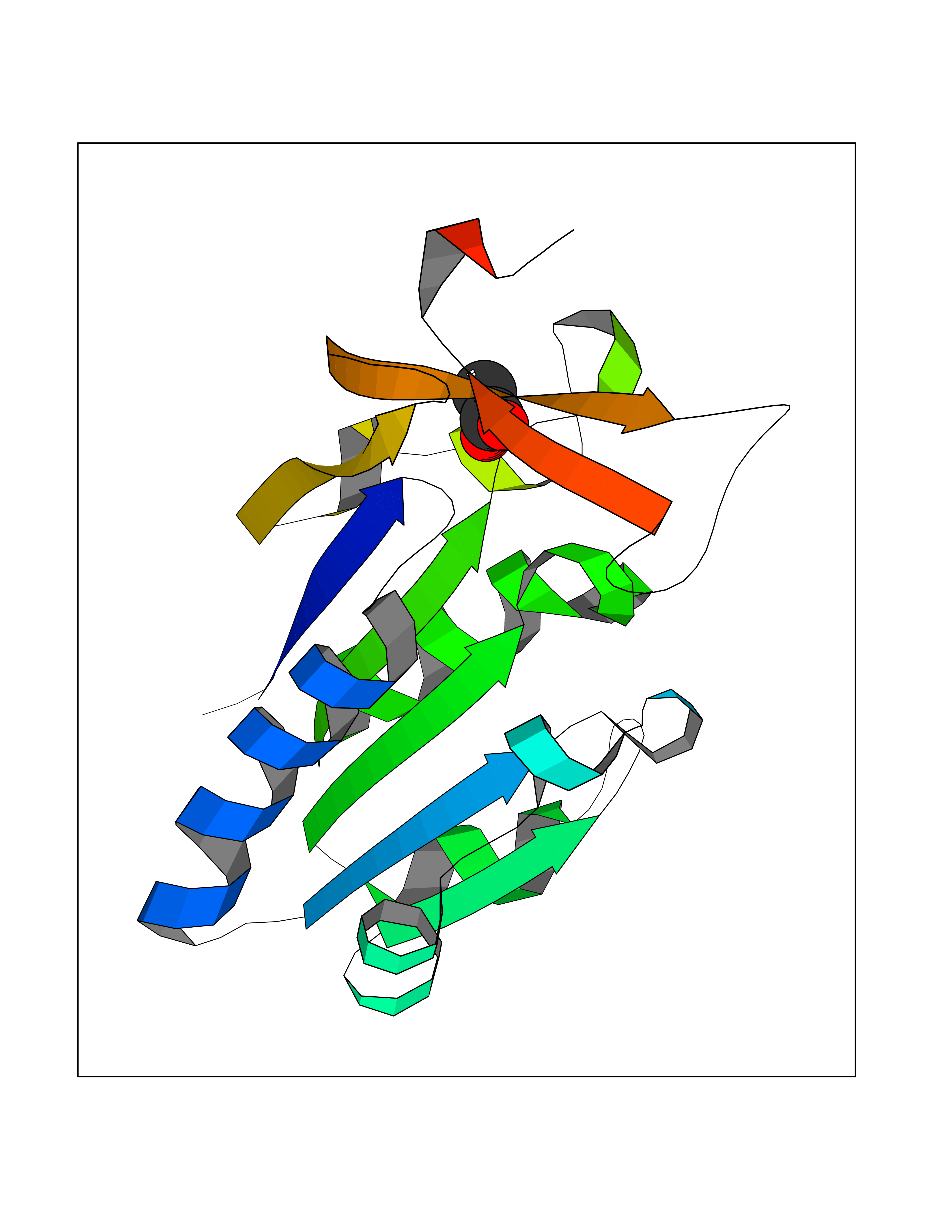 Molscript Map 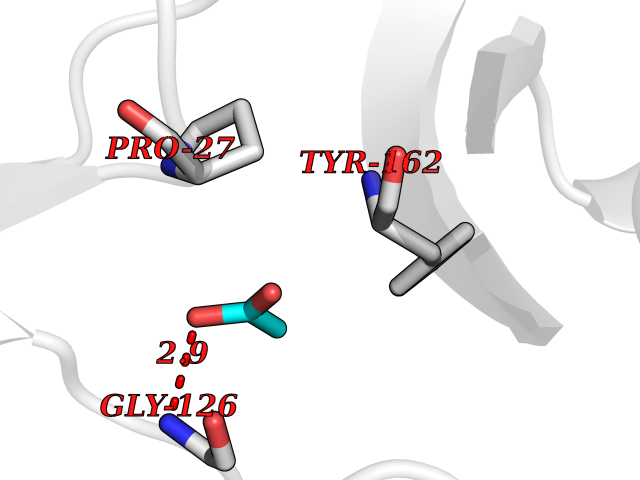 Pymol Map 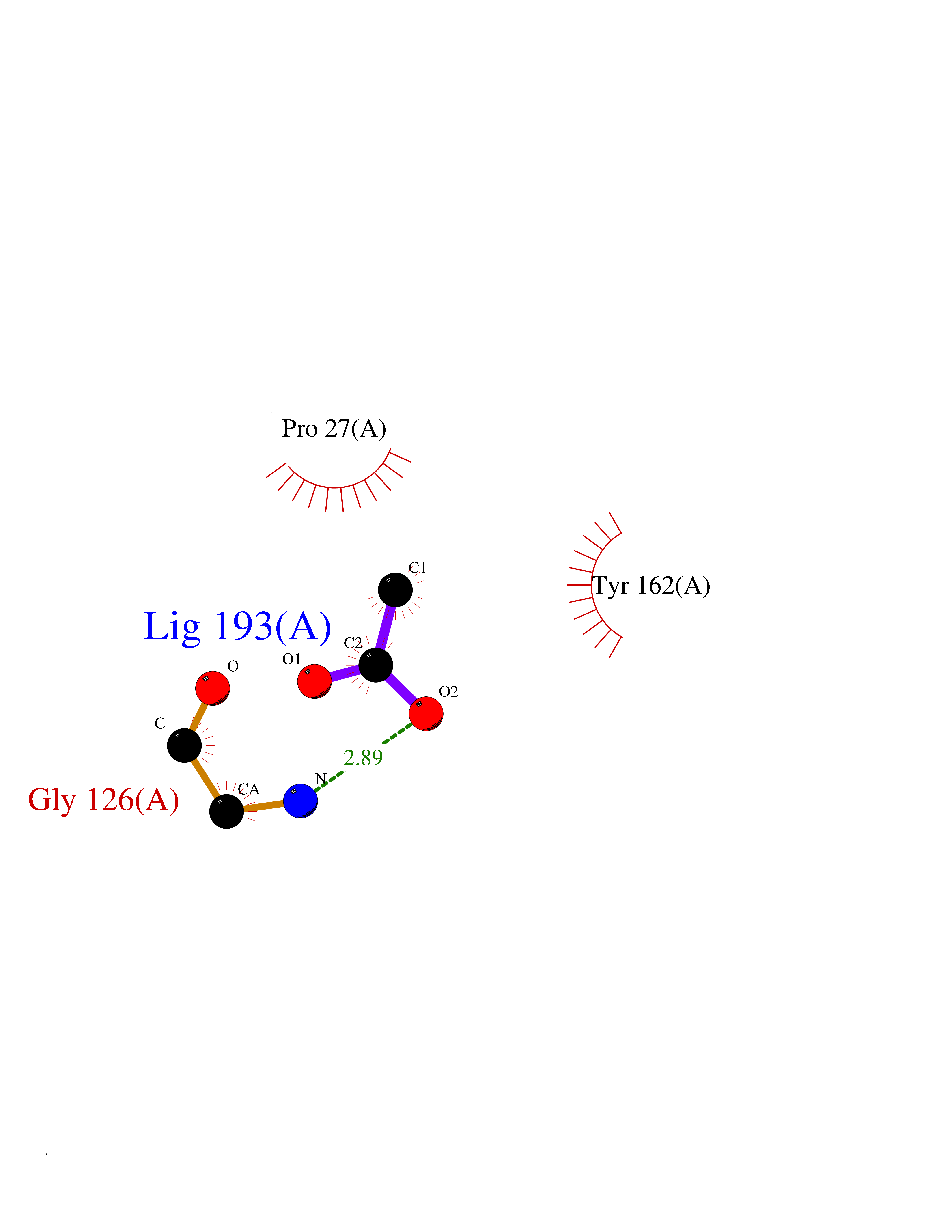 Ligplot Map 3D Binding mode Sequence RGQIQVILGPMFSGKSTELMRRVRRFQIAQYKCLVIKYAKDTRYSSSFCTHDRNTMEALPACLLRDVAQEALGVAVIGIDEGQFFPDIVEFCEAMANAGKTVIVAALDGTFQRKPFGAILNLVPLAESVVKLTAVCMECFREAAYTKRLGTEKEVEVIGGADKYHSVCRLCYFK Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Folate receptor alpha (FOLR1) | 4LRH | 4.41 | |
Target general information Gen name FOLR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ovarian tumorassociated antigen MOv18; KB cells FBP; Folate receptor, adult; Folate receptor 1; FRalpha; FOLR1; Adult folatebinding protein Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pHafter receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Required for normal embryonic development and normal cell proliferation. Related diseases Neurodegeneration due to cerebral folate transport deficiency (NCFTD) [MIM:613068]: An autosomal recessive neurodegenerative disorder resulting from brain-specific folate deficiency early in life. Onset is apparent in late infancy with severe developmental regression, movement disturbances, epilepsy and leukodystrophy. {ECO:0000269|PubMed:19732866}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05595; DB00158; DB00563; DB12489; DB15413; DB05168 Interacts with Q8N357 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Endosome; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Neurodegeneration; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 24216 Length 207 Aromaticity 0.13 Instability index 49.36 Isoelectric point 8.14 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -6.02 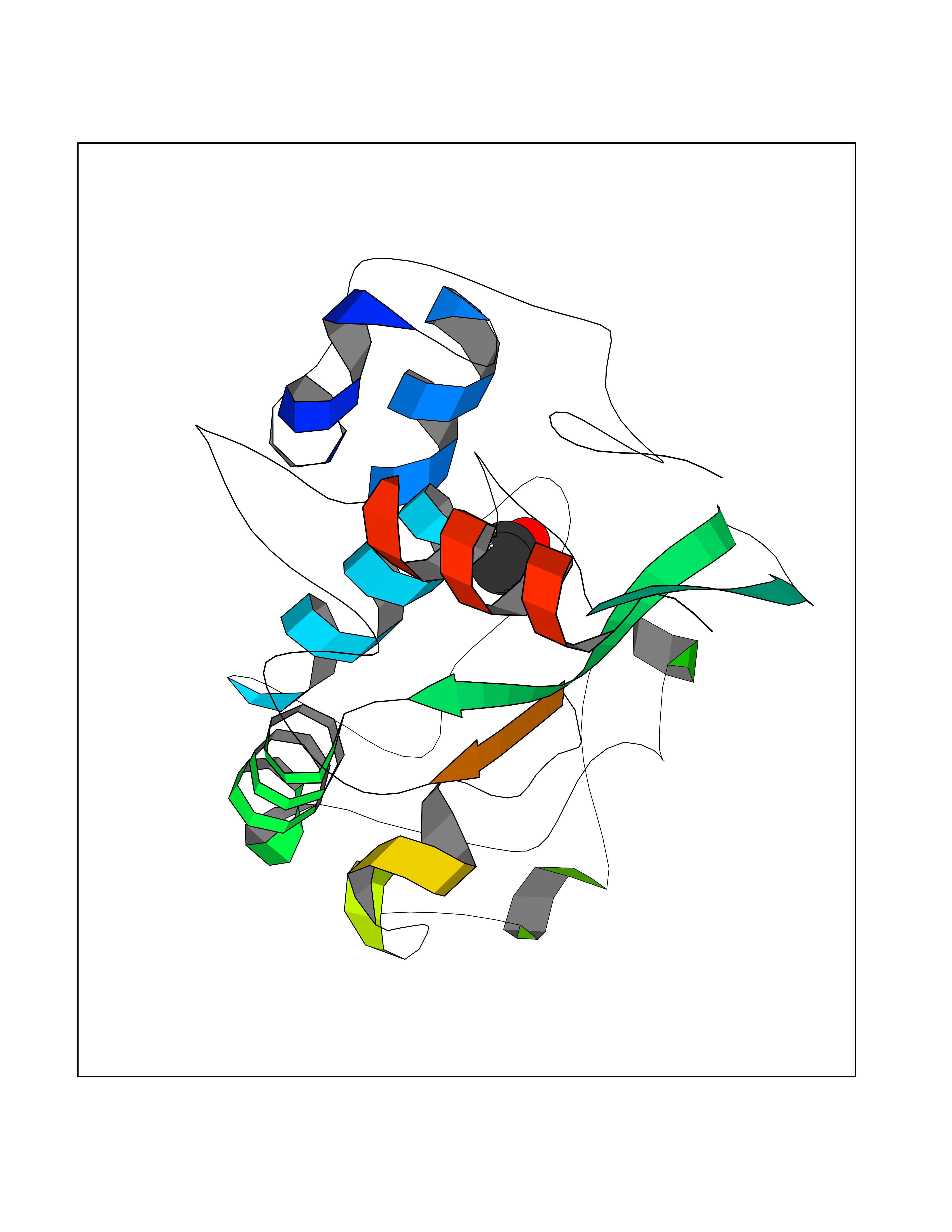 Molscript Map 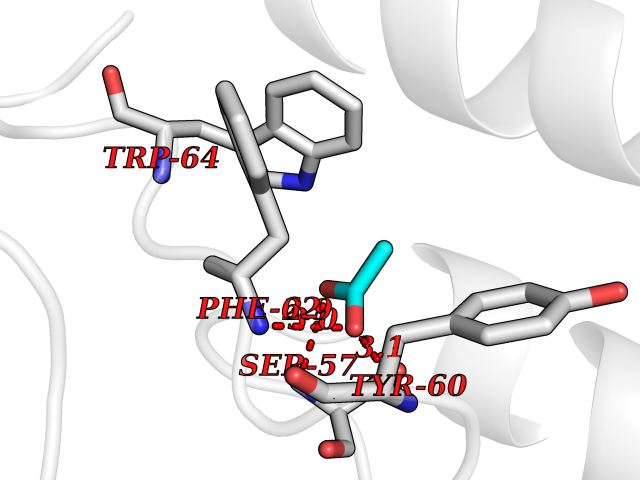 Pymol Map 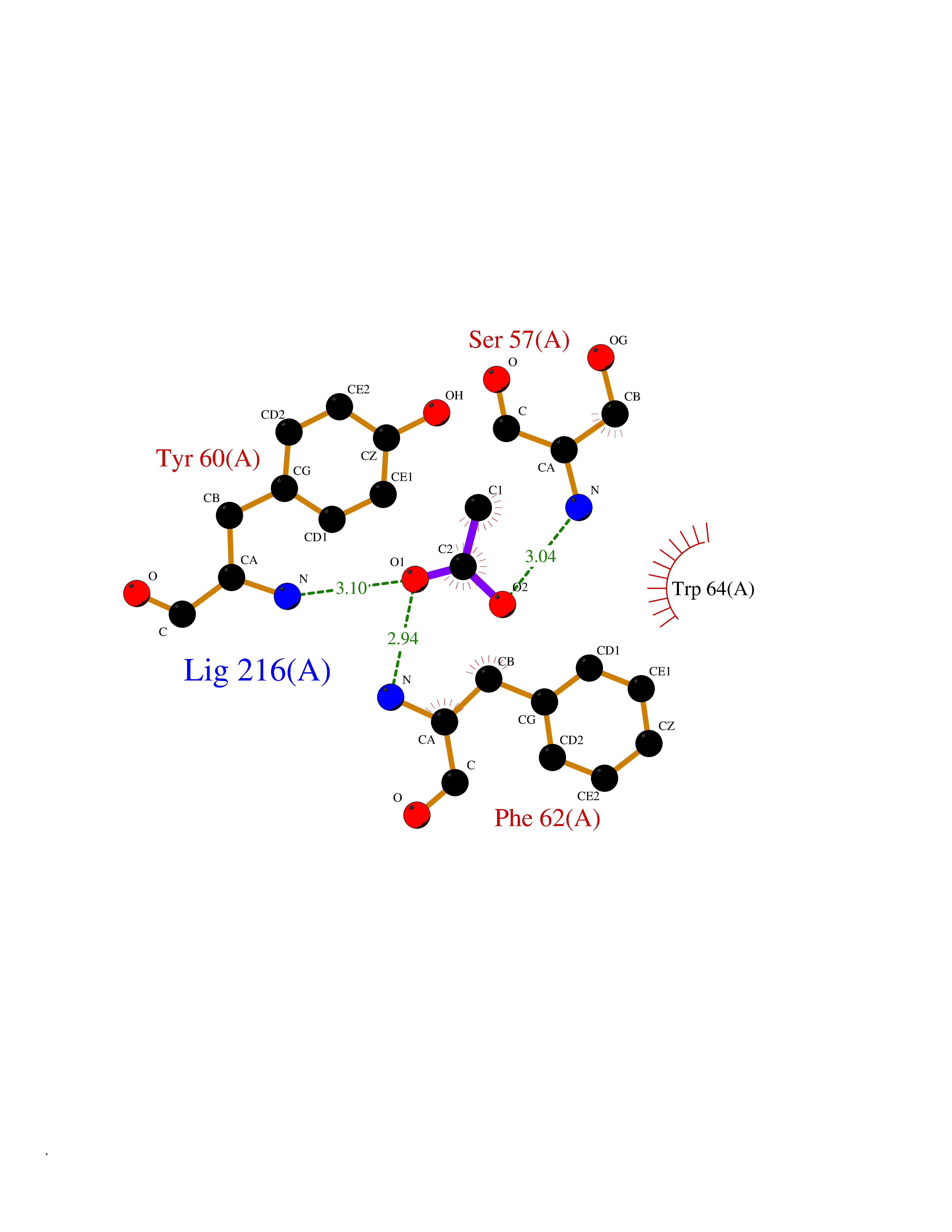 Ligplot Map 3D Binding mode Sequence RTELLNVCMNAKHHKEKPGPEDKLHEQCRPWRKNACCSTNTSQEAHKDVSYLYRFNWNHCGEMAPACKRHFIQDTCLYECSPNLGPWIQQVDQSWRKERVLNVPLCKEDCEQWWEDCRTSYTCKSNWHKGWNWTSGFNKCAVGAACQPFHFYFPTPTVLCNEIWTHSYKVSNYSRGSGRCIQMWFDPAQGNPNEEVARFYAAAMSGT Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Phosphatidylethanolamine-binding protein 1 (PEBP1) | 2QYQ | 4.41 | |
Target general information Gen name PEBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Raf kinase inhibitor protein; RKIP; Prostatic-binding protein; PEBP-1; PEBP; PBP; Neuropolypeptide h3; Hippocampal cholinergic neurostimulating peptide; HCNPpp; HCNP Protein family Phosphatidylethanolamine-binding protein family Biochemical class Phosphatidylethanolamine-binding protein family Function Binds ATP, opioids and phosphatidylethanolamine. Has lower affinity for phosphatidylinositol and phosphatidylcholine. Serine protease inhibitor which inhibits thrombin, neuropsin and chymotrypsin but not trypsin, tissue type plasminogen activator and elastase. Inhibits the kinase activity of RAF1 by inhibiting its activation and by dissociating the RAF1/MEK complex and acting as a competitive inhibitor of MEK phosphorylation. Related diseases Retinitis pigmentosa 49 (RP49) [MIM:613756]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:15570217, ECO:0000269|PubMed:7479749}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09130; DB09568 Interacts with P16050; Q9NRD5; P04049; Q15208; Q9NS68; Q9JLL3 EC number NA Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Direct protein sequencing; Disulfide bond; Lipid-binding; Nucleotide-binding; Phosphoprotein; Protease inhibitor; Proteomics identification; Reference proteome; Serine protease inhibitor Protein physicochemical properties Chain ID A Molecular weight (Da) 20928.3 Length 186 Aromaticity 0.1 Instability index 24.05 Isoelectric point 6.59 Charge (pH=7) -0.98 2D Binding mode Binding energy (Kcal/mol) -6.01 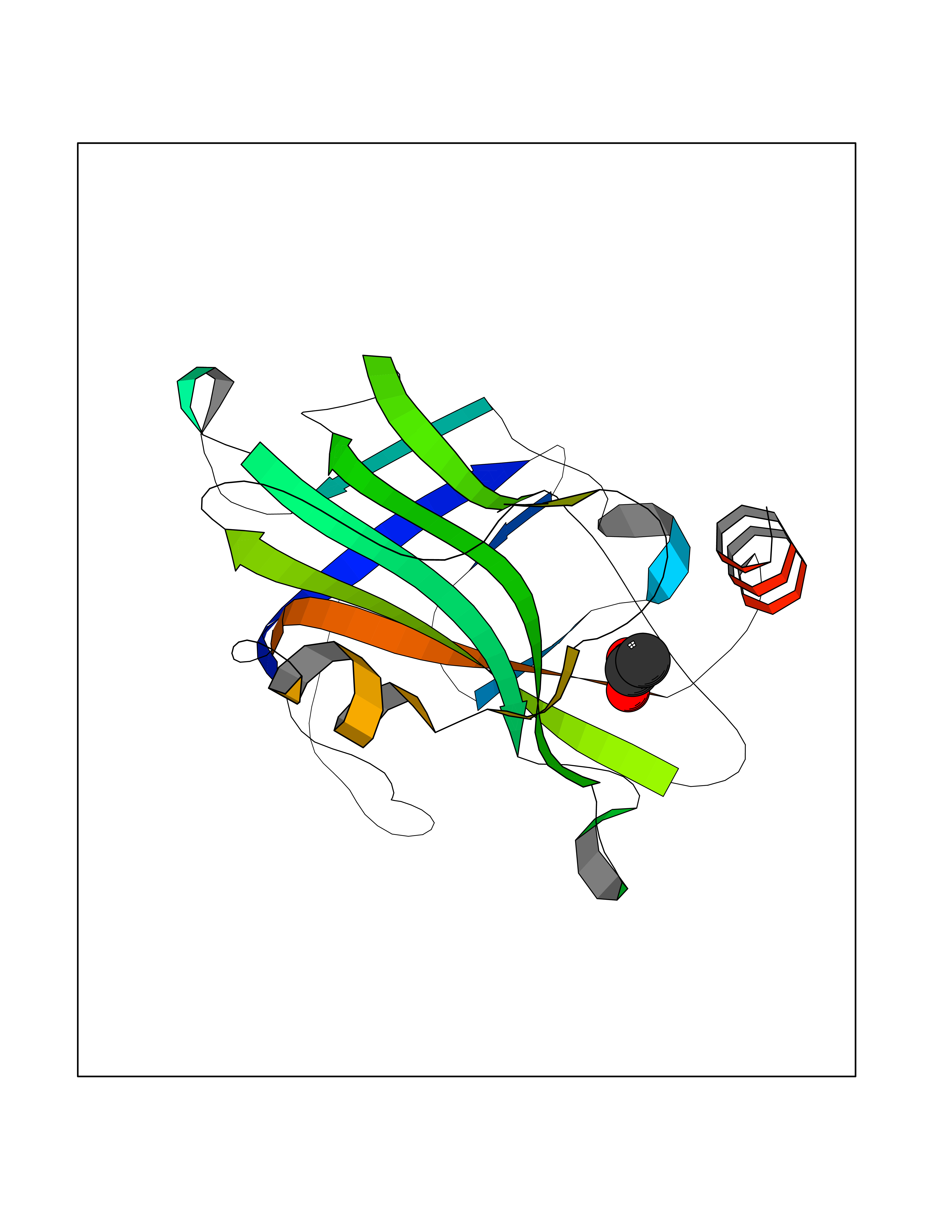 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MPVDLSKWSGPLSLQEVDEQPQHPLHVTYAGAAVDELGKVLTPTQVKNRPTSISWDGLDSGKLYTLVLTDPDAPSRKDPKYREWHHFLVVNMKGNDISSGTVLSDYVGSGPPKGTGLHRYVWLVYEQDRPLKCDEPILSNRSGDHRGKFKVASFRKKYELRAPVAGTCYQAEWDDYVPKLYEQLSG Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Pyruvate dehydrogenase [ubiquinone] | 3EYA | 4.40 | |
Target general information Gen name poxB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b0871;JW0855 Protein family TPP enzyme family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Identical protein binding.Lipid binding.Magnesium ion binding.Pyruvate dehydrogenase (quinone) activity.Thiamine pyrophosphate binding. Related diseases Glycogen storage disease 6 (GSD6) [MIM:232700]: A metabolic disorder characterized by mild to moderate hypoglycemia, mild ketosis, growth retardation, and prominent hepatomegaly. Heart and skeletal muscle are not affected. {ECO:0000269|PubMed:9529348}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07003 EC number 1.2.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Lipid-binding; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Oxidoreductase; Pyruvate; Reference proteome; Thiamine pyrophosphate; Ubiquinone Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I,J,K,L Molecular weight (Da) 113027 Length 1046 Aromaticity 0.07 Instability index 35.99 Isoelectric point 5.75 Charge (pH=7) -24.38 2D Binding mode Binding energy (Kcal/mol) -6  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFDGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIPMKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFVGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIP Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Cyclin-dependent kinase 4 | 2W96 | 4.40 | |
Target general information Gen name CDK4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Cell cycle Function ATP binding.Cyclin binding.Cyclin-dependent protein serine/threonine kinase activity.Cyclin-dependent protein serine/threonine kinase regulator activity.Protein complex binding. Related diseases Melanoma, cutaneous malignant 3 (CMM3) [MIM:609048]: A malignant neoplasm of melanocytes, arising de novo or from a pre-existing benign nevus, which occurs most often in the skin but may also involve other sites. {ECO:0000269|PubMed:7652577, ECO:0000269|PubMed:8528263, ECO:0000269|PubMed:9311594, ECO:0000269|PubMed:9425228}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12001; DB03496; DB12010; DB09073; DB02733; DB11730; DB15442 Interacts with Q9UH17; P24385; P30279; P30281; Q16543; P50613; P38936; P46527; P49918; P42771; P42772; P42773; P55273; Q9UJC3; P08238; Q9UKT9; Q0VD86; P01106; Q9ULD0; P28749; Q08999; P09936; Q8N720 EC number 2.7.11.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Cell division; Cytoplasm; Disease variant; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 30138.4 Length 267 Aromaticity 0.09 Instability index 36.2 Isoelectric point 5.78 Charge (pH=7) -5.83 2D Binding mode Binding energy (Kcal/mol) -6  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SRYEPVAEIGVGAYGTVYKARDPHSGHFVALKSVRVPNGEEGLPISTVREVALLRRLEAFEHPNVVRLMDVCATSRTDREIKVTLVFEHVDQDLRTYLDKAPPPGLPAETIKDLMRQFLRGLDFLHANCIVHRDLKPENILVTSGGTVKLADFGLARIYSYQMALDPVVVTLWYRAPEVLLQSTYATPVDMWSVGCIFAEMFRRKPLFCGNSEADQLGKIFDLIGLPPEDDWVPEMEESGAQLLLEMLTFNPHKRISAFRALQHSYL Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | DNA polymerase catalytic subunit | 1YYP | 4.40 | |
Target general information Gen name UL54 Organism Human cytomegalovirus (strain AD169) (HHV-5) (Human herpesvirus 5) Uniprot ID TTD ID NA Synonyms HFLF2 Protein family DNA polymerase type-B family Biochemical class Replication / transferase Function 3'-5' exonuclease activity.DNA binding.DNA-directed DNA polymerase activity.Nucleotide binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00369; DB00529 Interacts with NA EC number 2.7.7.7 Uniprot keywords 3D-structure; DNA replication; DNA-binding; DNA-directed DNA polymerase; Host nucleus; Nucleotidyltransferase; Reference proteome; Transferase; Viral DNA replication Protein physicochemical properties Chain ID B Molecular weight (Da) 30127.5 Length 269 Aromaticity 0.09 Instability index 36.39 Isoelectric point 8.41 Charge (pH=7) 3.48 2D Binding mode Binding energy (Kcal/mol) -6  Molscript Map  Pymol Map 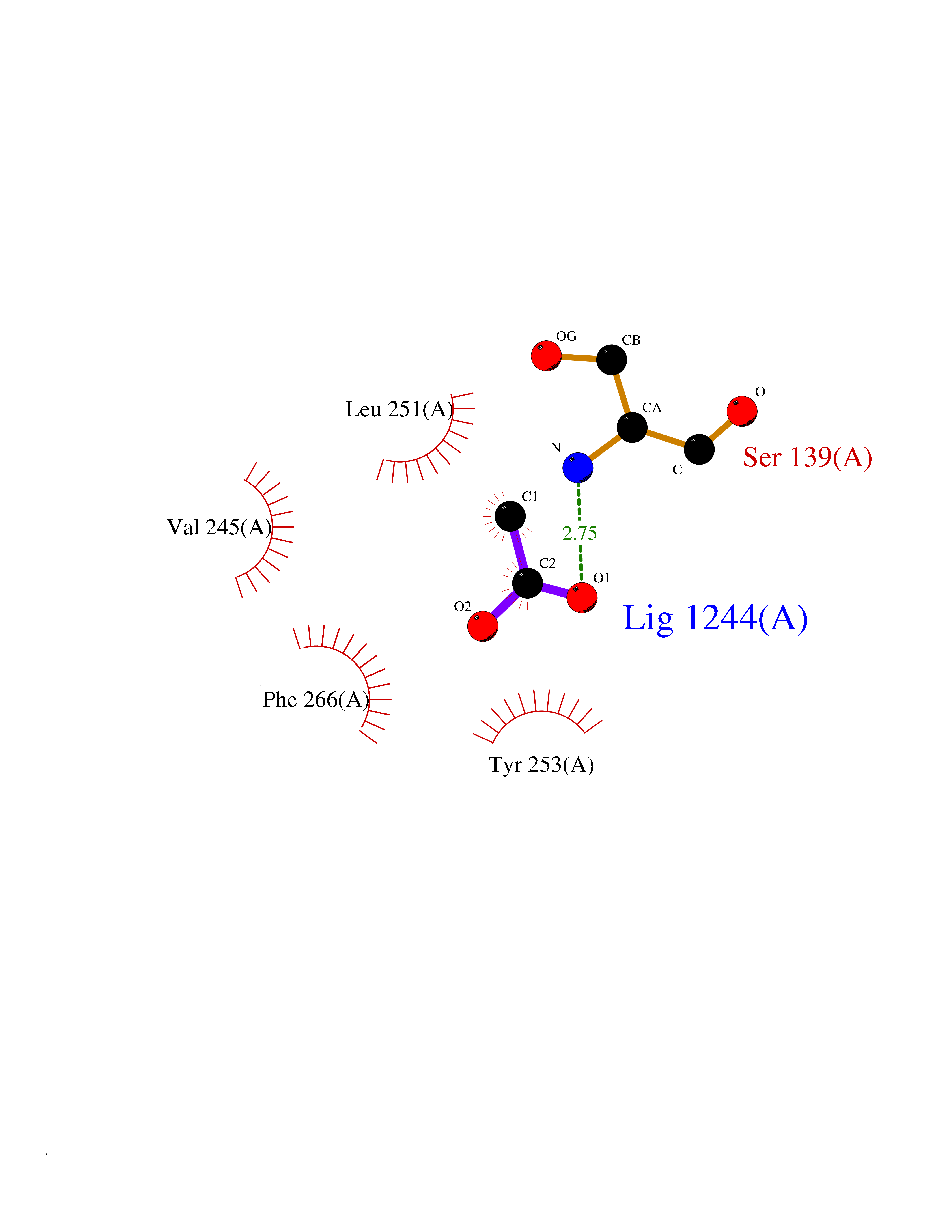 Ligplot Map 3D Binding mode Sequence EPPTLALRLKPYKTAIQQLRSVIRALKENTTVTFLPTPSLILQTVRSHCVSKITFNSSCLYITDKSFQPKTINNSTPLLGNFMYLTSSKDLTKFYVQDISDLSAKISMCAPDFNMEFSSACVHGQDIVRESENSAVHVDLDFGVVADLLKWIGPTGTVQILVHAGPPAIKFILTNGSELEFTSNNRVSFHGVKNMRINVQLKNFYQTLLNCAVTKLPCTLRIVTEHDTLLYVASRNGLFAVENFLTEEPRRLHLEPAFLPYSVKAHECC Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Leukotriene A-4 hydrolase (LTA4H) | 3U9W | 4.40 | |
Target general information Gen name LTA4H Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Leukotriene A4 hydrolase; Leukotriene A(4)Leukotriene A-4 hydrolase hydrolase; Leukotriene A(4) hydrolase; LTA4; LTA-H; LTA-4hydrolase; LTA-4 hydrolase Protein family Peptidase M1 family Biochemical class Ether bond hydrolase Function Has also aminopeptidase activity. Epoxide hydrolase that catalyzes the final step in the biosynthesis of the proinflammatory mediator leukotriene B4. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07102; DB06917; DB07258; DB07094; DB07259; DB02352; DB07292; DB07104; DB06828; DB08466; DB01197; DB05177; DB03366; DB08040; DB06851; DB02062; DB07099; DB07260; DB07196; DB11781; DB03424; DB07237 Interacts with Q9BSI4 EC number EC 3.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Leukotriene biosynthesis; Lipid metabolism; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 68927 Length 608 Aromaticity 0.1 Instability index 38.84 Isoelectric point 5.87 Charge (pH=7) -9.86 2D Binding mode Binding energy (Kcal/mol) -6  Molscript Map  Pymol Map 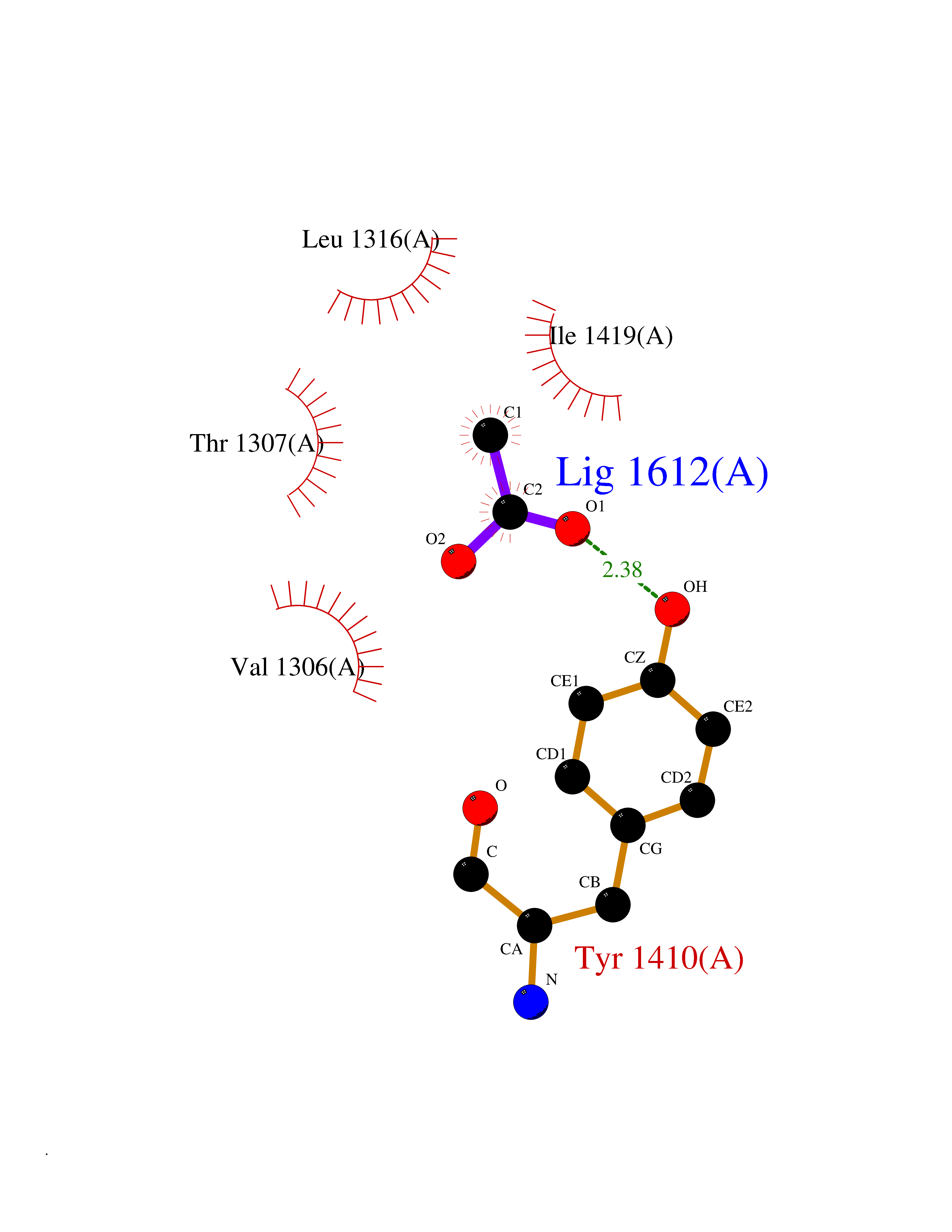 Ligplot Map 3D Binding mode Sequence IVDTCSLASPASVCRTKHLHLRCSVDFTRRTLTGTAALTVQSQEDNLRSLVLDTKDLTIEKVVINGQEVKYALGERQSYKGSPMEISLPIALSKNQEIVIEISFETSPKSSALQWLTPEQTSGKEHPYLFSQCQAIHCRAILPCQDTPSVKLTYTAEVSVPKELVALMSAIRDGETPDPEDPSRKIYKFIQKVPIPCYLIALVVGALESRQIGPRTLVWSEKEQVEKSAYEFSETESMLKIAEDLGGPYVWGQYDLLVLPPSFPYGGMENPCLTFVTPTLLAGDKSLSNVIAHEISHSWTGNLVTNKTWDHFWLNEGHTVYLERHICGRLFGEKFRHFNALGGWGELQNSVKTFGETHPFTKLVVDLTDIDPDVAYSSVPYEKGFALLFYLEQLLGGPEIFLGFLKAYVEKFSYKSITTDDWKDFLYSYFKDKVDVLNQVDWNAWLYSPGLPPIKPNYDMTLTNACIALSQRWITAKEDDLNSFNATDLKDLSSHQLNEFLAQTLQRAPLPLGHIKRMQEVYNFNAINNSEIRFRWLRLCIQSKWEDAIPLALKMATEQGRMKFTRPLFKDLAAFDKSHDQAVRTYQEHKASMHPVTAMLVGKDLKVD Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Xylose isomerase | 1XIM | 4.40 | |
Target general information Gen name xylA Organism Actinoplanes missouriensis (strain ATCC 14538 / DSM 43046 / CBS 188.64 / JCM 3121 / NBRC 102363 / NCIMB 12654 / NRRL B-3342 / UNCC 431) Uniprot ID TTD ID NA Synonyms AMIS_10350;XI Protein family Xylose isomerase family Biochemical class Isomerase(intramolecular oxidoreductase) Function Metal ion binding.Xylose isomerase activity. Related diseases Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Renal tubular dysgenesis (RTD) [MIM:267430]: Autosomal recessive severe disorder of renal tubular development characterized by persistent fetal anuria and perinatal death, probably due to pulmonary hypoplasia from early-onset oligohydramnios (the Potter phenotype). {ECO:0000269|PubMed:16116425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Microvascular complications of diabetes 3 (MVCD3) [MIM:612624]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. {ECO:0000269|PubMed:10099885}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Intracerebral hemorrhage (ICH) [MIM:614519]: A pathological condition characterized by bleeding into one or both cerebral hemispheres including the basal ganglia and the cerebral cortex. It is often associated with hypertension and craniocerebral trauma. Intracerebral bleeding is a common cause of stroke. {ECO:0000269|PubMed:15277638}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11195 Interacts with NA EC number 5.3.1.5 Uniprot keywords 3D-structure; Carbohydrate metabolism; Cytoplasm; Isomerase; Magnesium; Metal-binding; Reference proteome; Xylose metabolism Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 35820.8 Length 322 Aromaticity 0.1 Instability index 32.29 Isoelectric point 5.32 Charge (pH=7) -11.33 2D Binding mode Binding energy (Kcal/mol) -6  Molscript Map 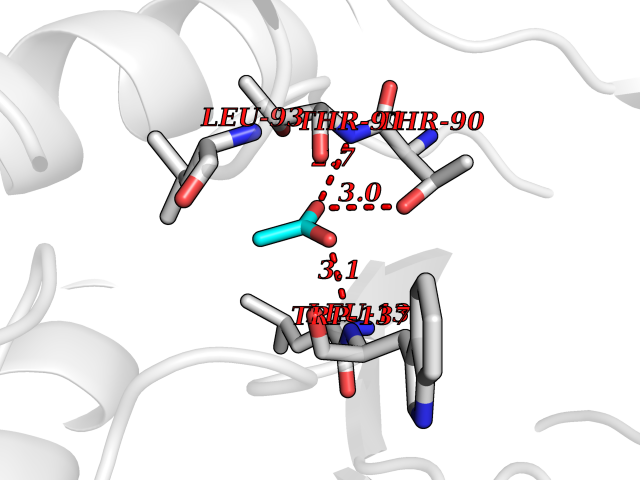 Pymol Map 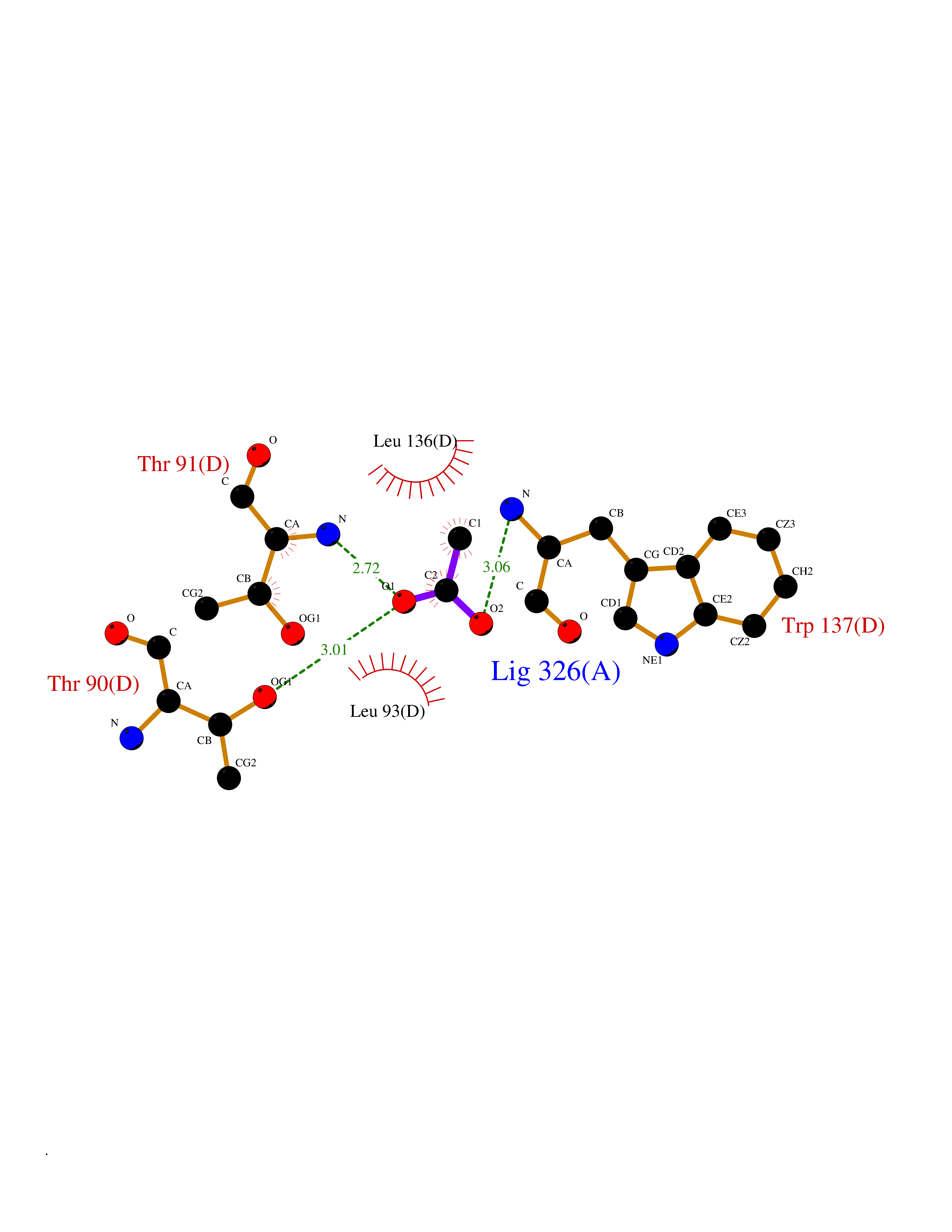 Ligplot Map 3D Binding mode Sequence VQATREDKFSFGLWTVGWQARDAFGDATRTALDPVEAVHKLAEIGAYGITFHDDDLVPFGSDAQTRDGIIAGFKKALDETGLIVPMVTTNLFTHPVFKDGGFTSNDRSVRRYAIRKVLRQMDLGAELGAKTLVLWGGREGAEYDSAKDVSAALDRYREALNLLAQYSEDRGYGLRFAIEPKPNEPRGDILLPTAGHAIAFVQELERPELFGINPETGHEQMSNLNFTQGIAQALWHKKLFHIDLNGQHGPKFDQDLVFGHGDLLNAFSLVDLLENGPDGAPAYDGPRHFDYKPSRTEDYDGVWESAKANIRMYLLLKERAKA Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Oxygen-insensitive NAD(P)H nitroreductase | 1KQB | 4.40 | |
Target general information Gen name nfsB Organism Enterobacter cloacae Uniprot ID TTD ID NA Synonyms nfsI;nfnB Protein family Nitroreductase family Biochemical class Oxidoreductase Function Oxidoreductase activity. Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03793; DB03247 Interacts with NA EC number 1.-.-.- Uniprot keywords 3D-structure; Flavoprotein; FMN; NAD; NADP; Oxidoreductase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 47619.4 Length 432 Aromaticity 0.08 Instability index 38.43 Isoelectric point 5.52 Charge (pH=7) -12.98 2D Binding mode Binding energy (Kcal/mol) -6.01  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DIISVALKRHSTKAFDASKKLTAEEAEKIKTLLQYSPSSTNSQPWHFIVASTEEGKARVAKSAAGTYVFNERKMLDASHVVVFCAKTAMDDAWLERVVDQEEADGRFNTPEAKAANHKGRTYFADMHRVDLKDDDQWMAKQVYLNVGNFLLGVGAMGLDAVPIEGFDAAILDEEFGLKEKGFTSLVVVPVGHHSVEDFNATLPKSRLPLSTIVTECDIISVALKRHSTKAFDASKKLTAEEAEKIKTLLQYSPSSTNSQPWHFIVASTEEGKARVAKSAAGTYVFNERKMLDASHVVVFCAKTAMDDAWLERVVDQEEADGRFNTPEAKAANHKGRTYFADMHRVDLKDDDQWMAKQVYLNVGNFLLGVGAMGLDAVPIEGFDAAILDEEFGLKEKGFTSLVVVPVGHHSVEDFNATLPKSRLPLSTIVTEC Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | 2-oxopropyl-CoM reductase, carboxylating | 1MO9 | 4.40 | |
Target general information Gen name xecC Organism Xanthobacter autotrophicus (strain ATCC BAA-1158 / Py2) Uniprot ID TTD ID NA Synonyms Xaut_4867 Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Oxidoreductase Function 2-oxopropyl-CoM reductase (carboxylating) activity.Flavin adenine dinucleotide binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03163; DB03147 Interacts with NA EC number 1.8.1.5 Uniprot keywords 3D-structure; Disulfide bond; FAD; Flavoprotein; NADP; Oxidoreductase; Plasmid; Redox-active center; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 114413 Length 1044 Aromaticity 0.08 Instability index 25.66 Isoelectric point 5.68 Charge (pH=7) -21.74 2D Binding mode Binding energy (Kcal/mol) -6.01  Molscript Map  Pymol Map 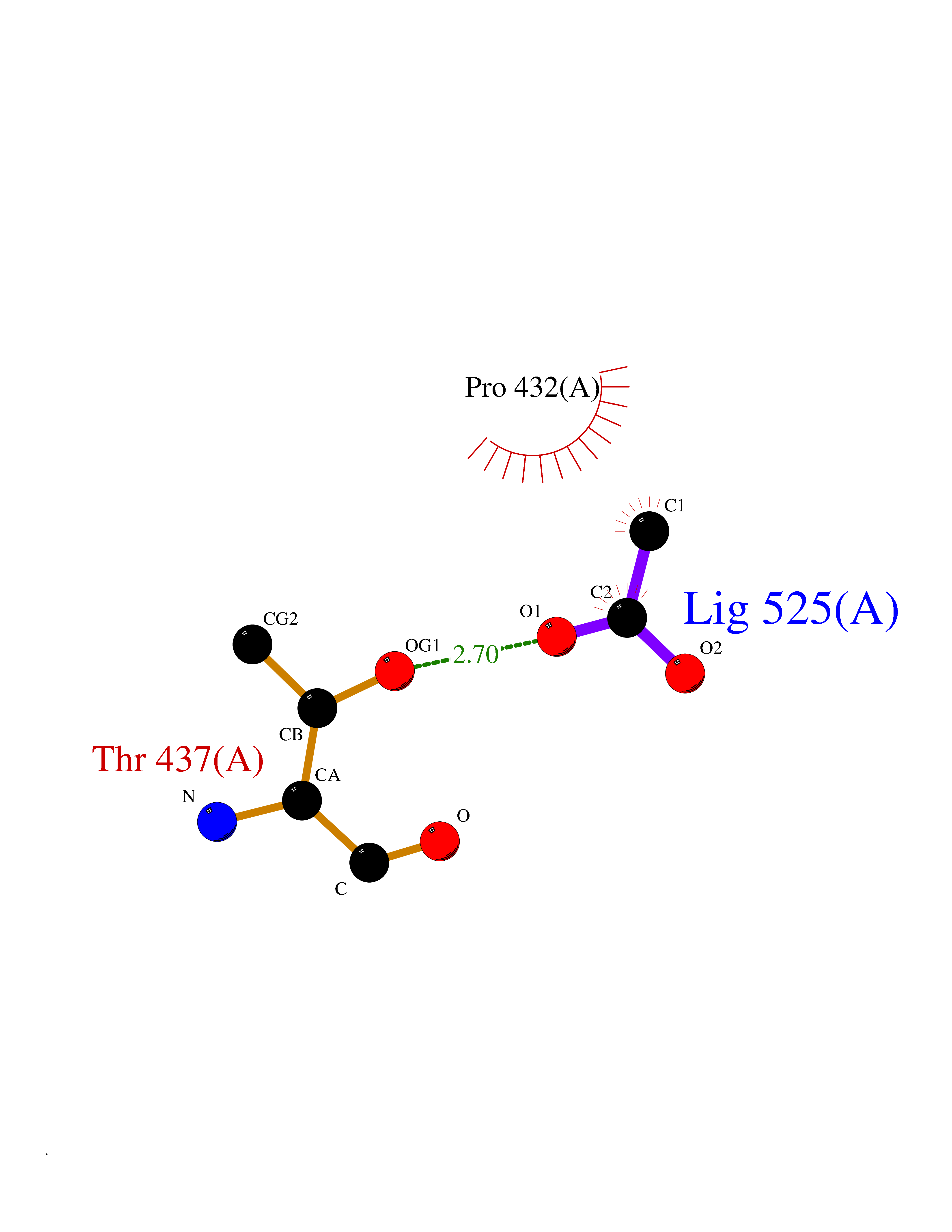 Ligplot Map 3D Binding mode Sequence KVWNARNDHLTINQWATRIDEILEAPDGGEVIYNVDENDPREYDAIFIGGGAAGRFGSAYLRAMGGRQLIVDRWPFLGGSCPHNACVPHHLFSDCAAELMLARTFSGQYWFPDMTEKVVGIKEVVDLFRAGRNGPHGIMNFQSKEQLNLEYILNCPAKVIDNHTVEAAGKVFKAKNLILAVGAGPGTLDVPGVNAKGVFDHATLVEELDYEPGSTVVVVGGSKTAVEYGCFFNATGRRTVMLVRTEPLKLIKDNETRAYVLDRMKEQGMEIISGSNVTRIEEDANGRVQAVVAMTPNGEMRIETDFVFLGLGEQPRSAELAKILGLDLGPKGEVLVNEYLQTSVPNVYAVGDLIGGPMEMFKARKSGCYAARNVMGEKISYTPKNYPDFLHTHYEVSFLGMGEEEARAAGHEIVTIKMPPDTENGLNVALPASDRTMLYAFGKGTAHMSGFQKIVIDAKTRKVLGAHHVGYGAKDAFQYLNVLIKQGLTVDELGDMDELFLNPTHFIQLSRLRAGSKNLVSLKVWNARNDHLTINQWATRIDEILEAPDGGEVIYNVDENDPREYDAIFIGGGAAGRFGSAYLRAMGGRQLIVDRWPFLGGSCPHNACVPHHLFSDCAAELMLARTFSGQYWFPDMTEKVVGIKEVVDLFRAGRNGPHGIMNFQSKEQLNLEYILNCPAKVIDNHTVEAAGKVFKAKNLILAVGAGPGTLDVPGVNAKGVFDHATLVEELDYEPGSTVVVVGGSKTAVEYGCFFNATGRRTVMLVRTEPLKLIKDNETRAYVLDRMKEQGMEIISGSNVTRIEEDANGRVQAVVAMTPNGEMRIETDFVFLGLGEQPRSAELAKILGLDLGPKGEVLVNEYLQTSVPNVYAVGDLIGGPMEMFKARKSGCYAARNVMGEKISYTPKNYPDFLHTHYEVSFLGMGEEEARAAGHEIVTIKMPPDTENGLNVALPASDRTMLYAFGKGTAHMSGFQKIVIDAKTRKVLGAHHVGYGAKDAFQYLNVLIKQGLTVDELGDMDELFLNPTHFIQLSRLRAGSKNLVSL Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Hemoglobin (HB) | 5E29 | 4.40 | |
Target general information Gen name HBA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Hemoglobin subunit alpha; Hemoglobin alpha chain; HBA1; Alpha-globin Protein family Globin family Biochemical class Pore-forming globin Function Involved in oxygen transport from the lung to the various peripheral tissues. Related diseases Heinz body anemias (HEIBAN) [MIM:140700]: Form of non-spherocytic hemolytic anemia of Dacie type 1. After splenectomy, which has little benefit, basophilic inclusions called Heinz bodies are demonstrable in the erythrocytes. Before splenectomy, diffuse or punctate basophilia may be evident. Most of these cases are probably instances of hemoglobinopathy. The hemoglobin demonstrates heat lability. Heinz bodies are observed also with the Ivemark syndrome (asplenia with cardiovascular anomalies) and with glutathione peroxidase deficiency. {ECO:0000269|PubMed:2833478}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Alpha-thalassemia (A-THAL) [MIM:604131]: A form of thalassemia. Thalassemias are common monogenic diseases occurring mostly in Mediterranean and Southeast Asian populations. The hallmark of alpha-thalassemia is an imbalance in globin-chain production in the adult HbA molecule. The level of alpha chain production can range from none to very nearly normal levels. Deletion of both copies of each of the two alpha-globin genes causes alpha(0)-thalassemia, also known as homozygous alpha thalassemia. Due to the complete absence of alpha chains, the predominant fetal hemoglobin is a tetramer of gamma-chains (Bart hemoglobin) that has essentially no oxygen carrying capacity. This causes oxygen starvation in the fetal tissues leading to prenatal lethality or early neonatal death. The loss of two alpha genes results in mild alpha-thalassemia, also known as heterozygous alpha-thalassemia. Affected individuals have small red cells and a mild anemia (microcytosis). If three of the four alpha-globin genes are functional, individuals are completely asymptomatic. Some rare forms of alpha-thalassemia are due to point mutations (non-deletional alpha-thalassemia). The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Alpha(0)-thalassemia is associated with non-immune hydrops fetalis, a generalized edema of the fetus with fluid accumulation in the body cavities due to non-immune causes. Non-immune hydrops fetalis is not a diagnosis in itself but a symptom, a feature of many genetic disorders, and the end-stage of a wide variety of disorders.; DISEASE: Hemoglobin H disease (HBH) [MIM:613978]: A form of alpha-thalassemia due to the loss of three alpha genes. This results in high levels of a tetramer of four beta chains (hemoglobin H), causing a severe and life-threatening anemia. Untreated, most patients die in childhood or early adolescence. {ECO:0000269|PubMed:10569720}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08262; DB07427; DB08077; DB07428; DB02126; DB09130; DB08486; DB15617; DB09147; DB13995; DB14490; DB14491; DB14488; DB14501; DB14489; DB13257; DB01592; DB00893; DB09112; DB09140; DB06154; DB07645; DB09517; DB08632; DB14975; DB01593; DB14487; DB14533; DB14548 Interacts with Q9NZD4; Q2TAC2; P00387; P68871; P02042; P02100; P69892; P09105; Q15323; O76011; Q6A162; P29474; P0DPK4 EC number NA Uniprot keywords 3D-structure; Acetylation; Direct protein sequencing; Disease variant; Glycation; Glycoprotein; Heme; Hereditary hemolytic anemia; Iron; Metal-binding; Oxygen transport; Phosphoprotein; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 45984.2 Length 427 Aromaticity 0.08 Instability index 7.16 Isoelectric point 8.48 Charge (pH=7) 5.25 2D Binding mode Binding energy (Kcal/mol) -6 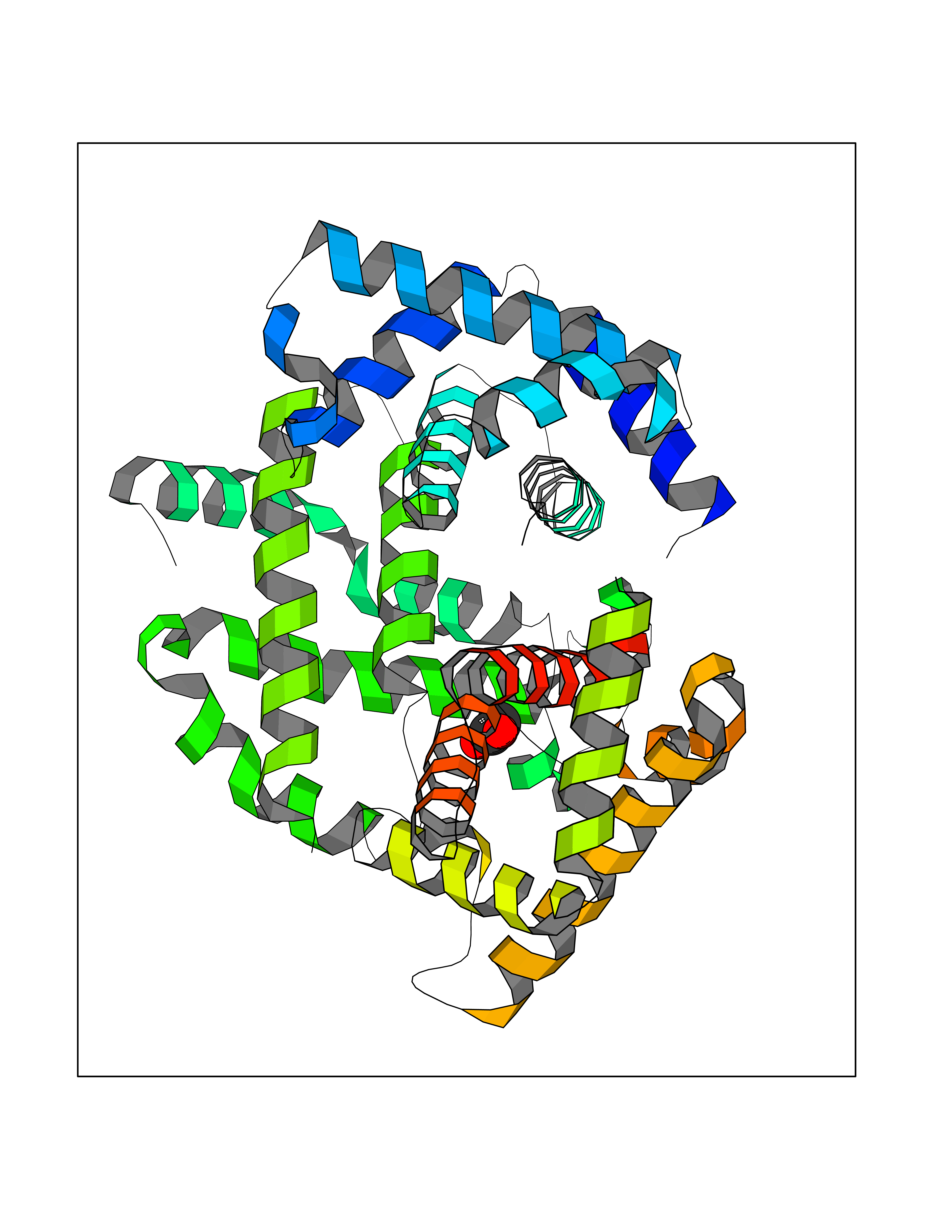 Molscript Map 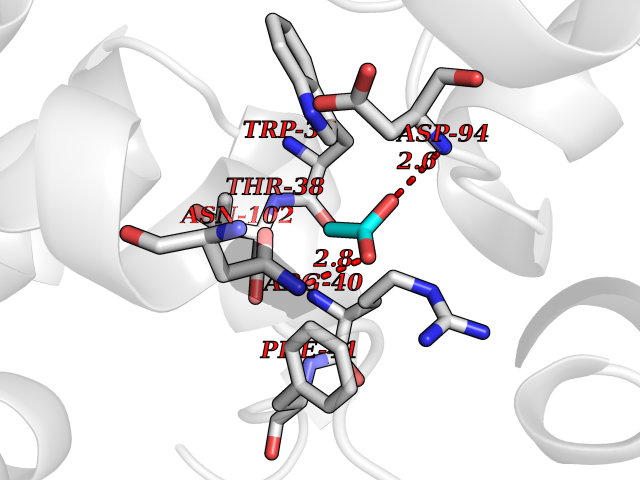 Pymol Map 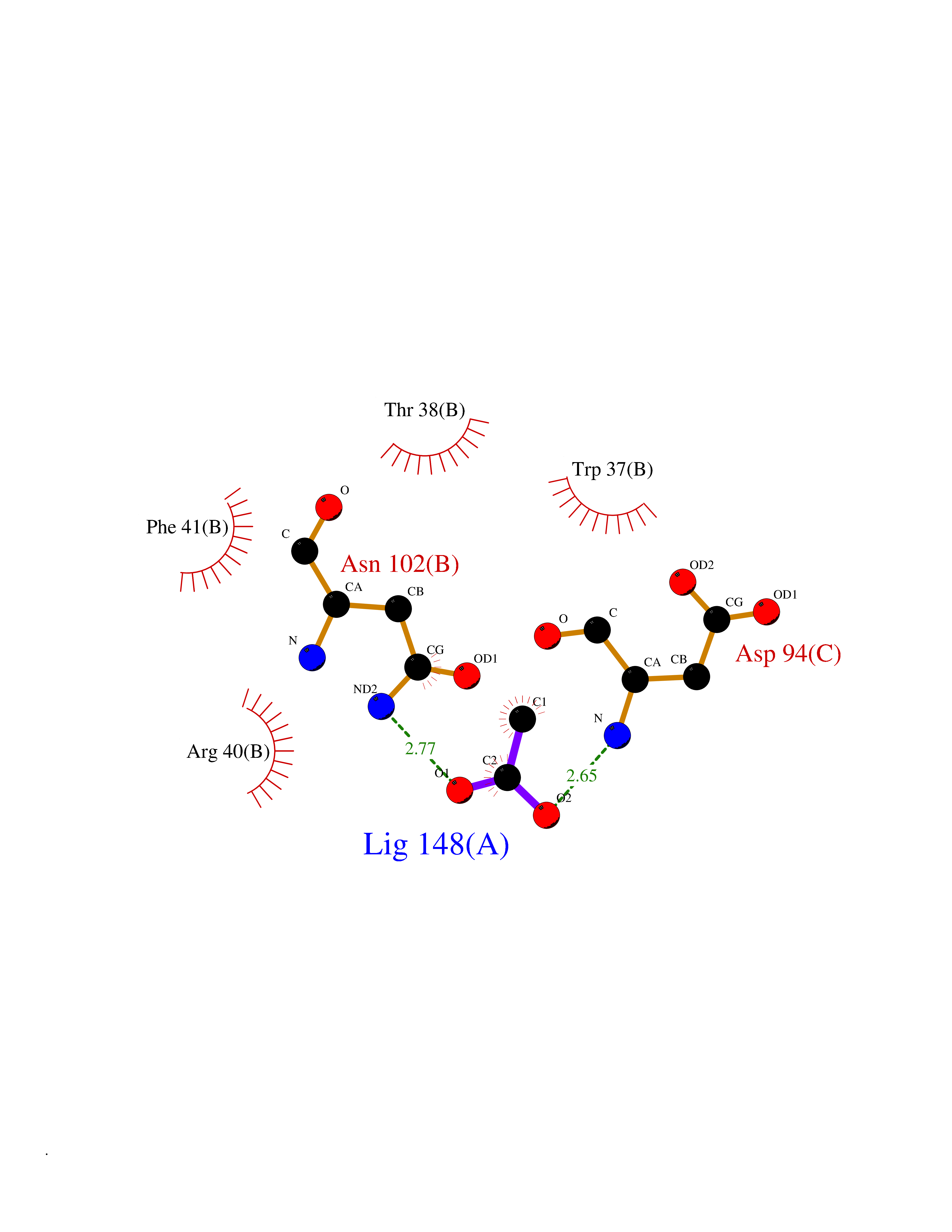 Ligplot Map 3D Binding mode Sequence VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYRHLTPEEKSAVTALWGKVNVDEVGGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAHKYHVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR Hydrogen bonds contact Hydrophobic contact | ||||