Job Results:
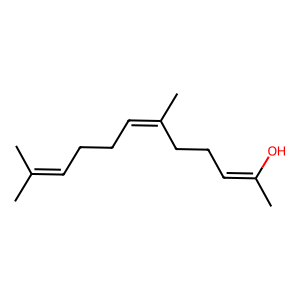
Ligand
Structure
Job ID
22a4f6254c6ea1b7fba986d94e380114
Job name
NA
Time
2026-02-27 16:46:57
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Acetylcholine receptor subunit alpha | 4ZJS | 6.19 | |
Target general information Gen name CHRNA1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACHRA;CHNRA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-1/CHRNA1 sub-subfamily Biochemical class Immune system Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ion channel activity.Ligand-gated ion channel activity. Related diseases Multiple pterygium syndrome, lethal type (LMPS) [MIM:253290]: Multiple pterygia are found infrequently in children with arthrogryposis and in fetuses with fetal akinesia syndrome. In lethal multiple pterygium syndrome there is intrauterine growth retardation, multiple pterygia, and flexion contractures causing severe arthrogryposis and fetal akinesia. Subcutaneous edema can be severe, causing fetal hydrops with cystic hygroma and lung hypoplasia. Oligohydramnios and facial anomalies are frequent. {ECO:0000269|PubMed:18252226}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: The alpha subunit is the main focus for antibody binding in myasthenia gravis. Myasthenia gravis is characterized by sporadic muscular fatigability and weakness, occurring chiefly in muscles innervated by cranial nerves, and characteristically improved by cholinesterase-inhibiting drugs.; DISEASE: Myasthenic syndrome, congenital, 1A, slow-channel (CMS1A) [MIM:601462]: A common congenital myasthenic syndrome. Congenital myasthenic syndromes are characterized by muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1A is a slow-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in prolonged AChR channel opening episodes, prolonged endplate currents, and depolarization block. This is associated with calcium overload, which may contribute to subsequent degeneration of the endplate and postsynaptic membrane. {ECO:0000269|PubMed:16685696, ECO:0000269|PubMed:7619526, ECO:0000269|PubMed:8872460, ECO:0000269|PubMed:9158151, ECO:0000269|PubMed:9221765}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 1B, fast-channel (CMS1B) [MIM:608930]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1B is a fast-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in brief opening and activity of the channel, with a rapid decay in endplate current, failure to achieve threshold depolarization of the endplate and consequent failure to fire an action potential. {ECO:0000269|PubMed:10195214, ECO:0000269|PubMed:12588888, ECO:0000269|PubMed:15079006}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08838; DB00565; DB00555 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46717.8 Length 411 Aromaticity 0.11 Instability index 38.02 Isoelectric point 4.77 Charge (pH=7) -22.31 2D Binding mode Binding energy (Kcal/mol) -8.44 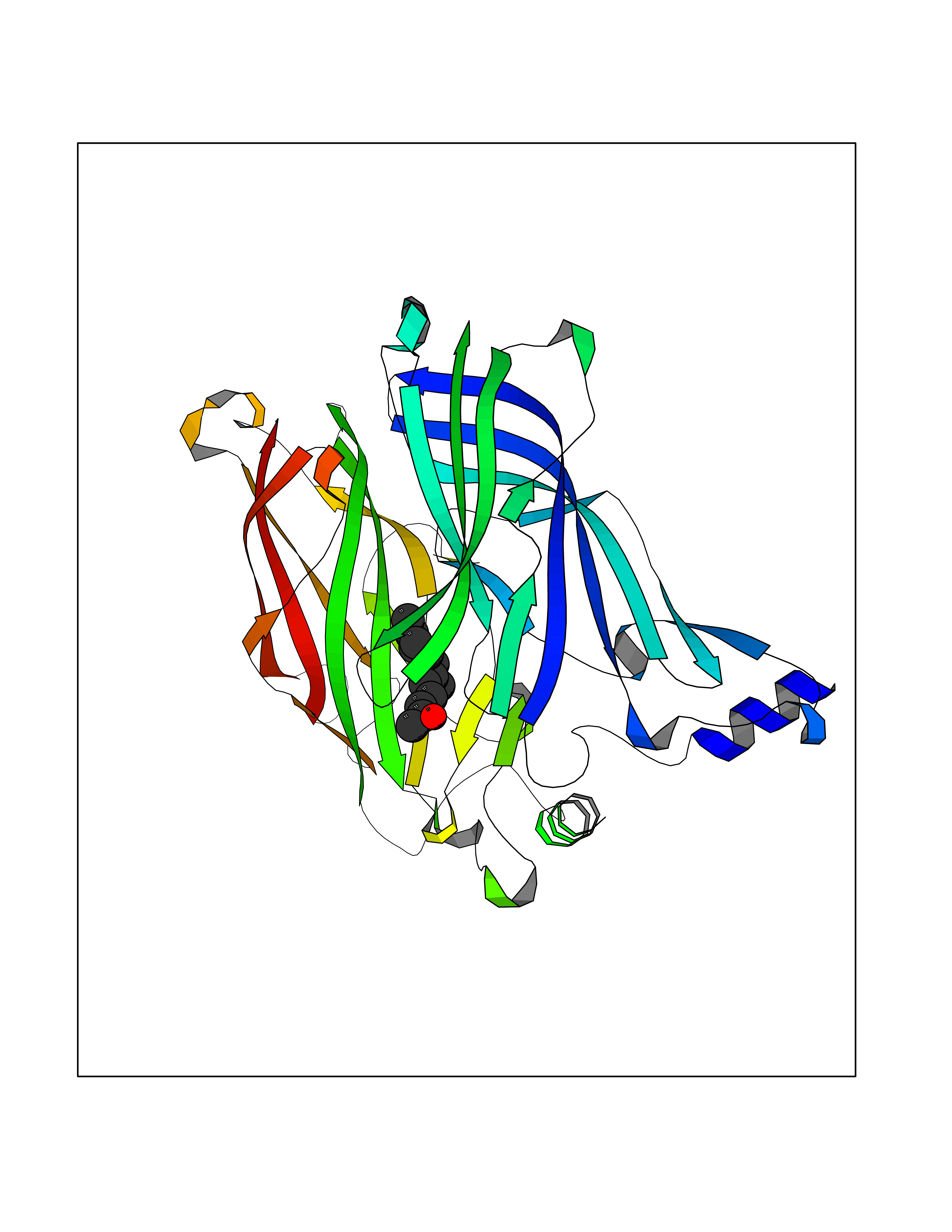 Molscript Map 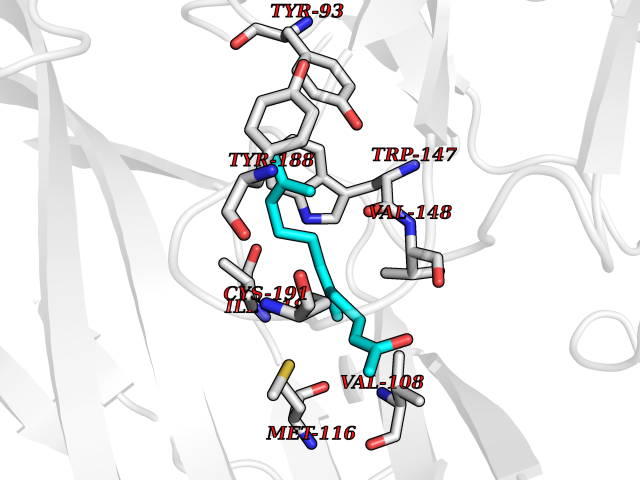 Pymol Map 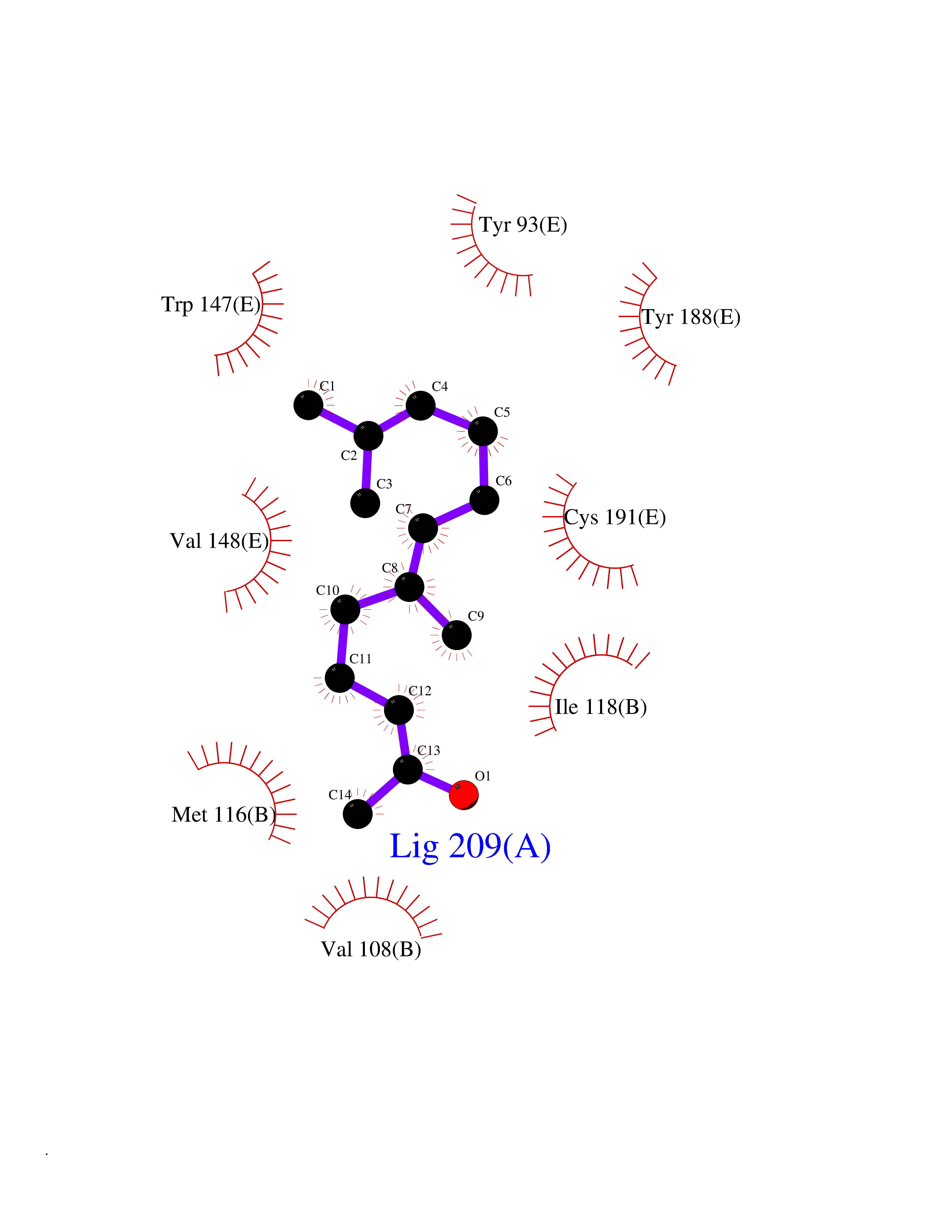 Ligplot Map 3D Binding mode Sequence EHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFREEHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFRER Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Extracellular lysophospholipase D (E-NPP2) | 4ZGA | 6.19 | |
Target general information Gen name ENPP2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LysoPLD; Ectonucleotide pyrophosphatase/phosphodiesterase family member 2; E-NPP 2; Autotaxin; ATX Protein family Nucleotide pyrophosphatase/phosphodiesterase family Biochemical class Phosphoric diester hydrolase Function Hydrolyzes lysophospholipids to produce the signaling molecule lysophosphatidic acid (LPA) in extracellular fluids. Major substrate is lysophosphatidylcholine. Also can act on sphingosylphosphorylcholine producing sphingosine-1-phosphate, a modulator of cell motility. Can hydrolyze, in vitro, bis-pNPP, to some extent pNP-TMP, and barely ATP. Involved in several motility-related processes such as angiogenesis and neurite outgrowth. Acts as an angiogenic factor by stimulating migration of smooth muscle cells and microtubule formation. Stimulates migration of melanoma cells, probably via a pertussis toxin-sensitive G protein. May have a role in induction of parturition. Possible involvement in cell proliferation and adipose tissue development (Probable). Tumor cell motility-stimulating factor. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.1.4.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Chemotaxis; Cleavage on pair of basic residues; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lipid degradation; Lipid metabolism; Metal-binding; Obesity; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 85397.4 Length 740 Aromaticity 0.12 Instability index 58.63 Isoelectric point 7.16 Charge (pH=7) 1.02 2D Binding mode Binding energy (Kcal/mol) -8.45 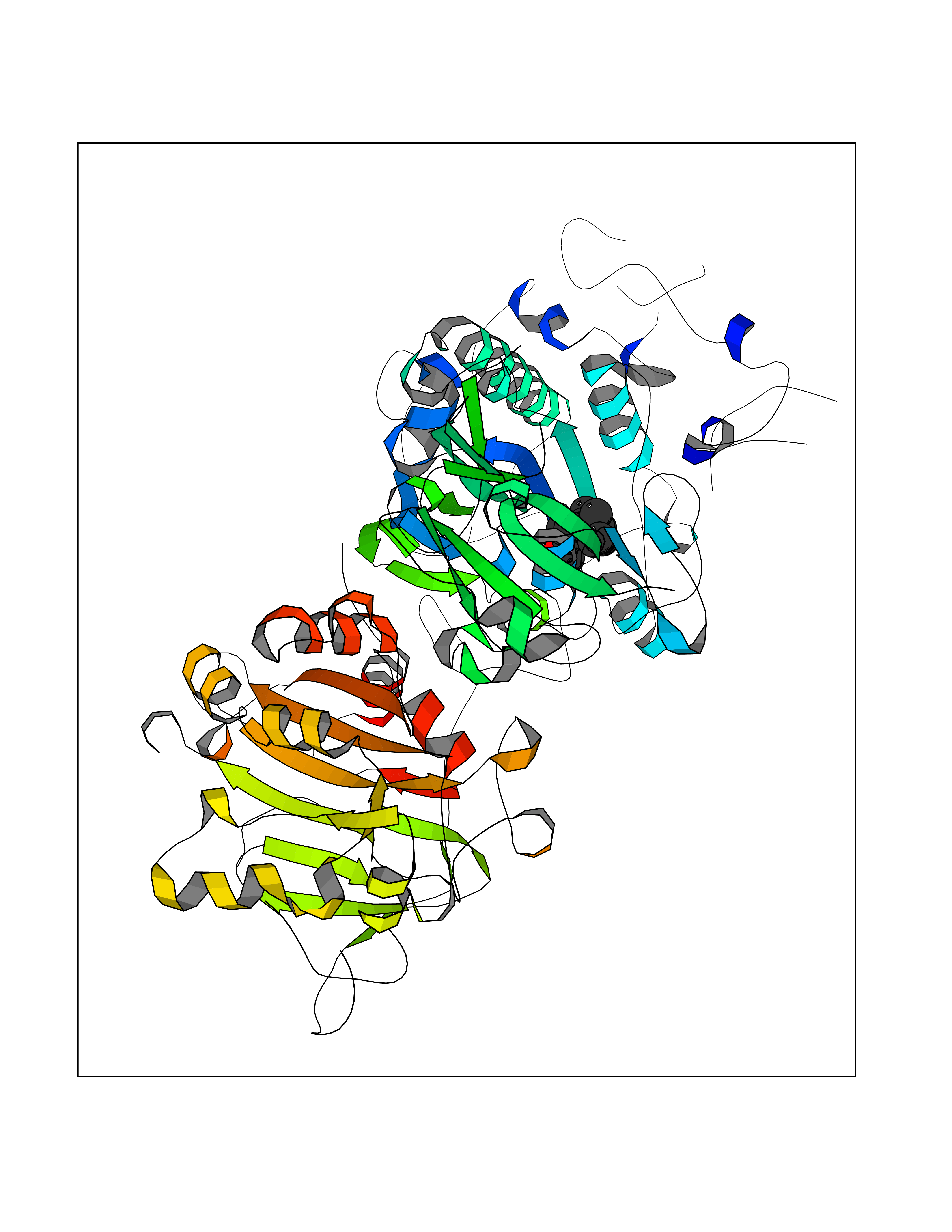 Molscript Map 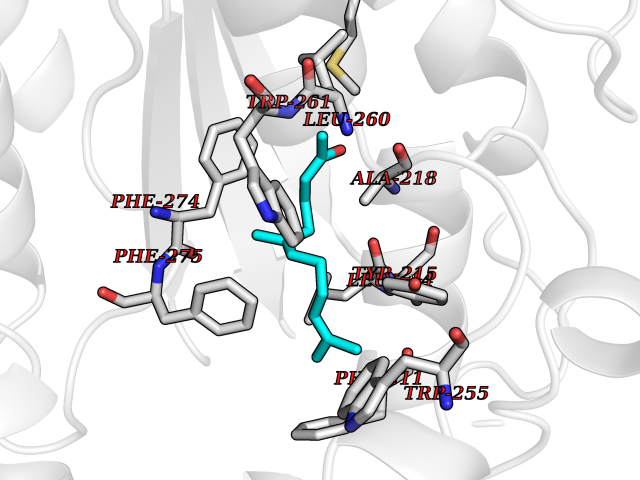 Pymol Map 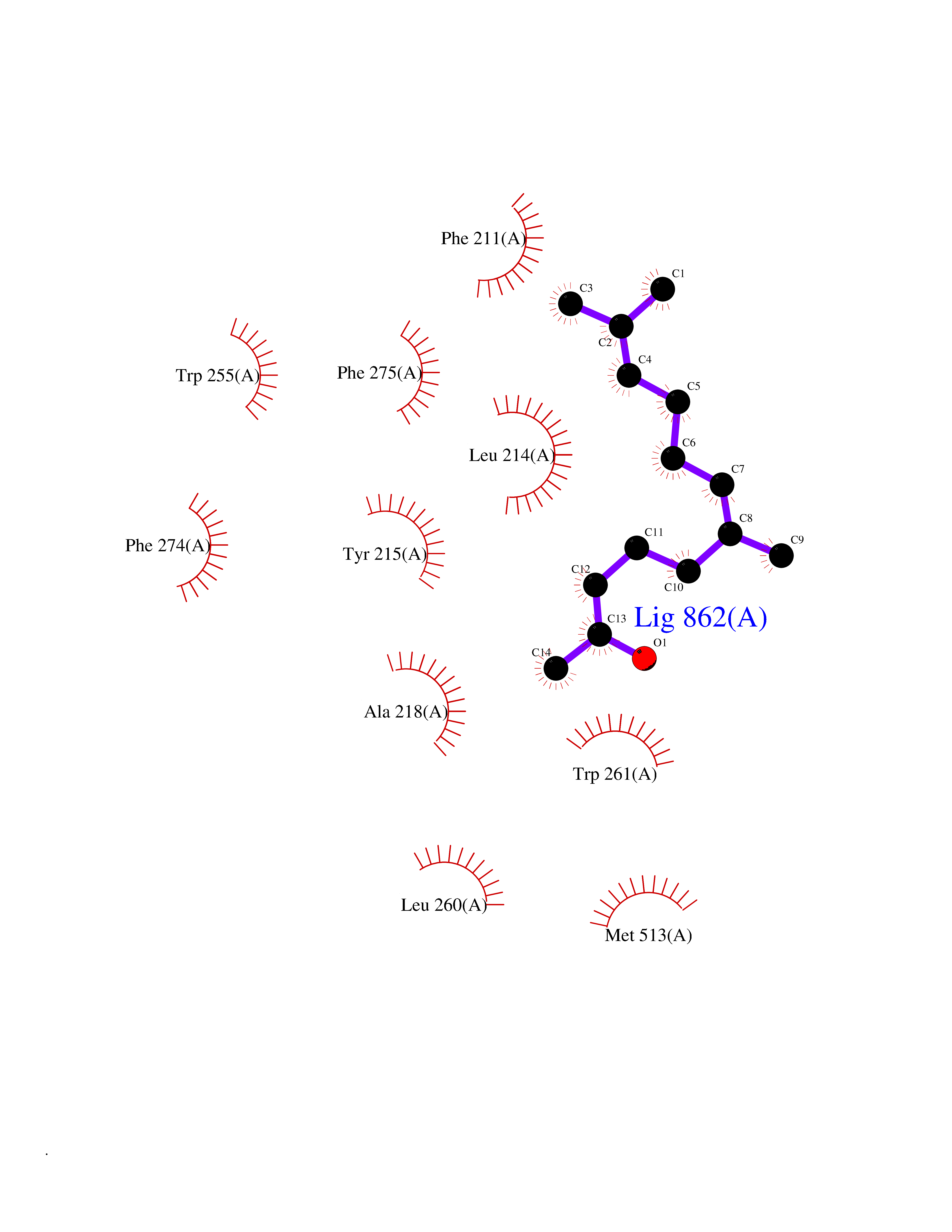 Ligplot Map 3D Binding mode Sequence GSCKGRCFELCRCDNLCKSYTSCCHDFDELCLKTARGWECTKDRCGNEENACHCEDCLARGDCCTNYQVVCKGESHWVDDDCEEIKAAECPAGFVRPPLIIFSVDGFRASYMKKGSKVMPNIEKLRSCGTHSPYMRPVYPTKTFPNLYTLATGLYPESHGIVGNSMYDPVFDATFHLRGREKFNHRWWGGQPLWITATKQGVKAGTFFWSVVIPHERRILTILQWLTLPDHERPSVYAFYSEQPDFSGHKYGPFGPEMTNPLREIDKIVGQLMDGLKQLKLHRCVNVIFVGDHGMEDVTCDRTEFLSNYLTNVDDITLVPGTLGRIRSKFDPKAIIANLTCKKPDQHFKPYLKQHLPKRLHYANNRRIEDIHLLVERRWHVARKPFFQGDHGFDNKVNSMQTVFVGYGSTFKYKTKVPPFENIELYNVMCDLLGLKPAPNNGTHGSLNHLLRTNTFRPTMPEEVTRPNYPGIMYLQSDFDLGTEERHLLYGRPAVLYRTRYDILYHTDFESGYSEIFLMPLWTSYTVSKQACVRPDVRVSPSFSQNCLAYKNDKQMSYGFLFPPYLSSSPEAKYDAFLVTNMVPMYPAFKRVWNYFQRVLVKKYASERNGVNVISGPIFDYDYDGLHDTEDKIKQYVEGSSIPVPTHYYSIITSCLDFTQPADKCDGPLSVSSFILPHRPDNEESCNSSEDESKWVEELMKMHTARVRDIEHLTSLDFFRKTSRSYPEILTLKTYLHTYE Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Neuronal acetylcholine receptor beta-4 (CHRNB4) | 6PV7 | 6.19 | |
Target general information Gen name CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNB4; Beta-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-4/CHRNB4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. {ECO:0000269|PubMed:20054297, ECO:0000269|PubMed:23622243, ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}. The disease may be caused by variants affecting the gene represented in this entry. Defects of SETD2 are associated with loss of DNA methylation at non-promoter regions (PubMed:23792563). SETD2 defects lead to aberrant and reduced nucleosome compaction and chromatin association of key replication proteins, such as MCM7 and DNA polymerase delta, leading to hinder replication fork progression and prevent loading of RAD51 homologous recombination repair factor at DNA breaks (PubMed:25728682). {ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}.; DISEASE: Luscan-Lumish syndrome (LLS) [MIM:616831]: An autosomal dominant syndrome with a variable phenotype. Clinical features include macrocephaly, distinctive facial appearance, postnatal overgrowth, various degrees of learning difficulties, autism spectrum disorder, and intellectual disability. {ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24852293, ECO:0000269|PubMed:26084711, ECO:0000269|PubMed:27317772}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute lymphoblastic (ALL) [MIM:613065]: A subtype of acute leukemia, a cancer of the white blood cells. ALL is a malignant disease of bone marrow and the most common malignancy diagnosed in children. The malignant cells are lymphoid precursor cells (lymphoblasts) that are arrested in an early stage of development. The lymphoblasts replace the normal marrow elements, resulting in a marked decrease in the production of normal blood cells. Consequently, anemia, thrombocytopenia, and neutropenia occur to varying degrees. The lymphoblasts also proliferate in organs other than the marrow, particularly the liver, spleen, and lymphnodes. {ECO:0000269|PubMed:24509477, ECO:0000269|PubMed:24662245}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16314571, ECO:0000269|PubMed:24509477}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Intellectual developmental disorder, autosomal dominant 70 (MRD70) [MIM:620157]: An autosomal dominant disorder characterized by mild global developmental delay, moderately impaired intellectual disability with speech difficulties, and behavioral abnormalities. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rabin-Pappas syndrome (RAPAS) [MIM:620155]: An autosomal dominant neurodevelopmental disorder characterized by severely impaired global development, intellectual disability, microcephaly, facial dysmorphism, and variable congenital anomalies affecting the skeletal, genitourinary, cardiac, and other organ systems. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00237; DB00565; DB00514; DB07720; DB00898; DB00472; DB01227; DB00184; DB01090; DB00202 Interacts with Q6FHY5 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -8.45 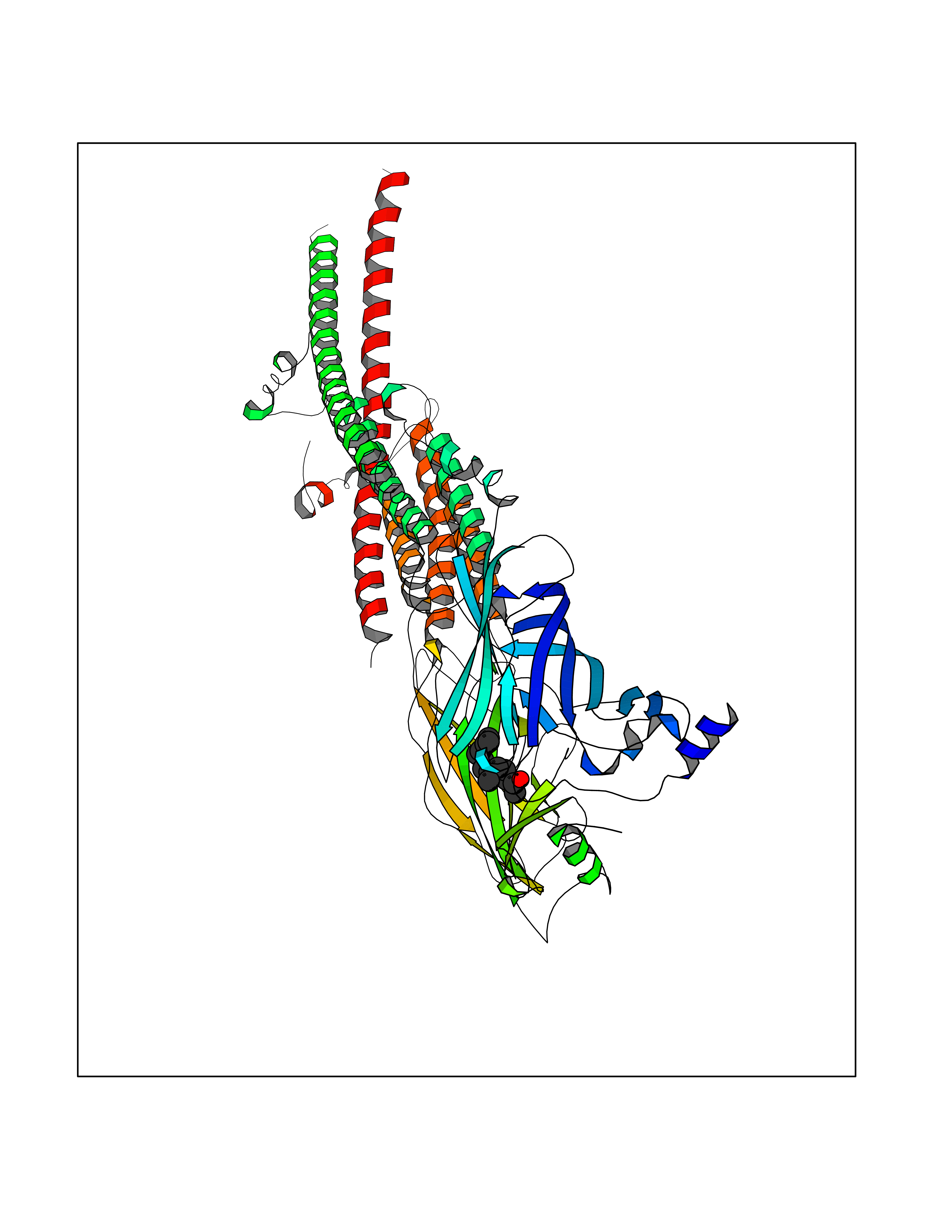 Molscript Map 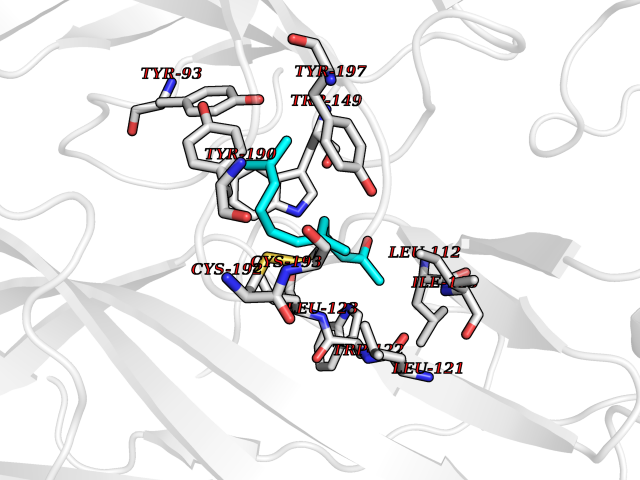 Pymol Map 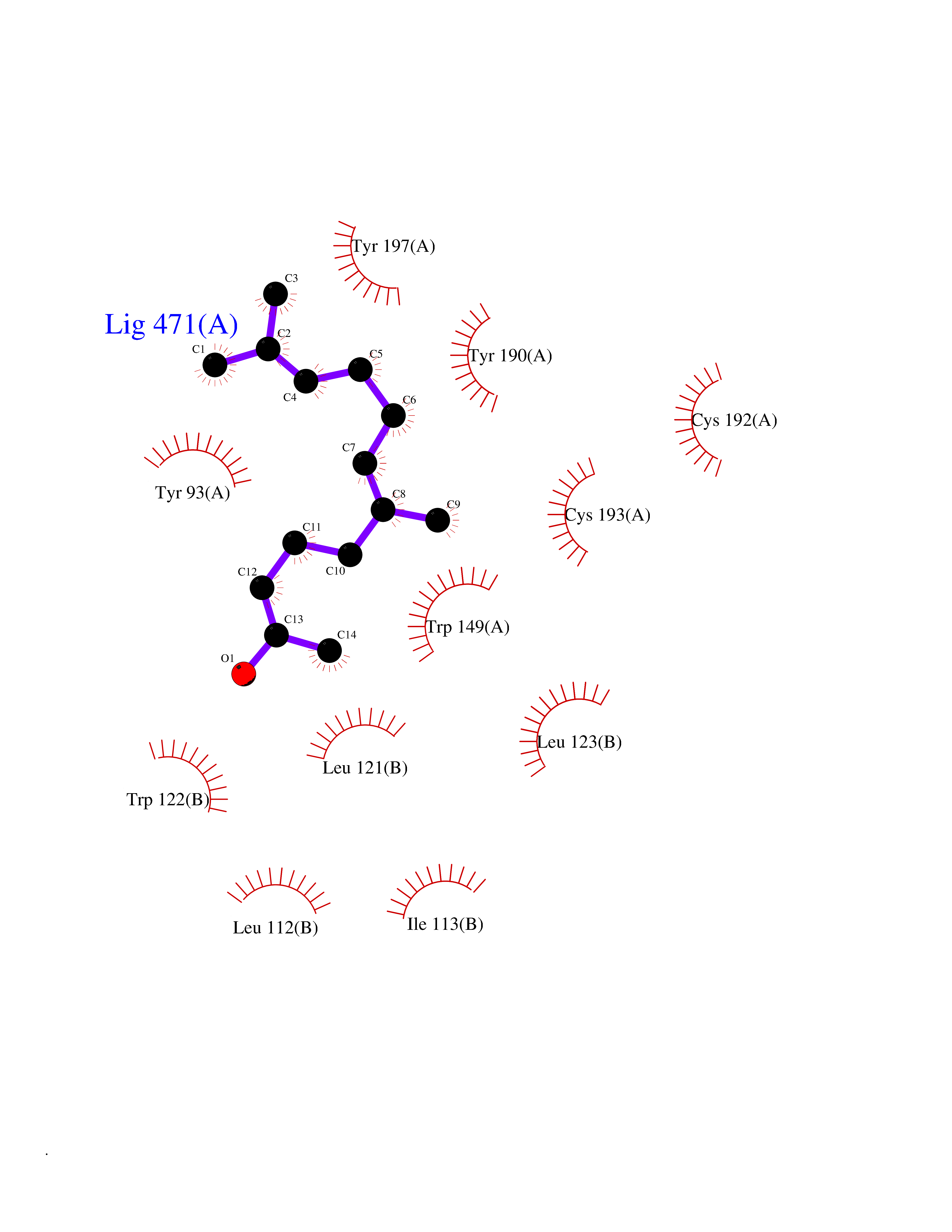 Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Aldose reductase (AKR1B1) | 1US0 | 6.18 | |
Target general information Gen name AKR1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde reductase; AKR1B1 Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies. Related diseases Glutamine deficiency, congenital (GLND) [MIM:610015]: An autosomal recessive disorder characterized by variable brain malformations, encephalopathy, severe developmental delay, seizures, and decreased glutamine levels in bodily fluids. Death in early infancy may occur. {ECO:0000269|PubMed:16267323, ECO:0000269|PubMed:26711351, ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 116 (DEE116) [MIM:620806]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE116 is autosomal dominant form characterized by severe developmental delay, seizures, and white matter abnormalities. {ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry. DEE116 is caused by variants that disrupt the canonical translation start codon in GLUL resulting in initiation of translation at Met-18 (PubMed:38579670). The resulting protein is enzymatically competent but insensitive to negative feedback regulation via glutamine-induced degradation. {ECO:0000269|PubMed:38579670}. Drugs (DrugBank ID) DB07028; DB07030; DB07450; DB02101; DB08449; DB08000; DB07139; DB07498; DB02007; DB02020; DB11859; DB02994; DB04272; DB07187; DB00694; DB00997; DB06246; DB01039; DB02021; DB16707; DB00143; DB02834; DB08084; DB01689; DB07063; DB06077; DB02518; DB00157; DB03461; DB05383; DB05533; DB05327; DB02712; DB00605; DB02383; DB02132; DB08772; DB07093; DB07999; DB08098 Interacts with Q9BUY7 EC number EC 1.1.1.300 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35447.6 Length 313 Aromaticity 0.09 Instability index 36.41 Isoelectric point 7.1 Charge (pH=7) 0.26 2D Binding mode Binding energy (Kcal/mol) -8.43 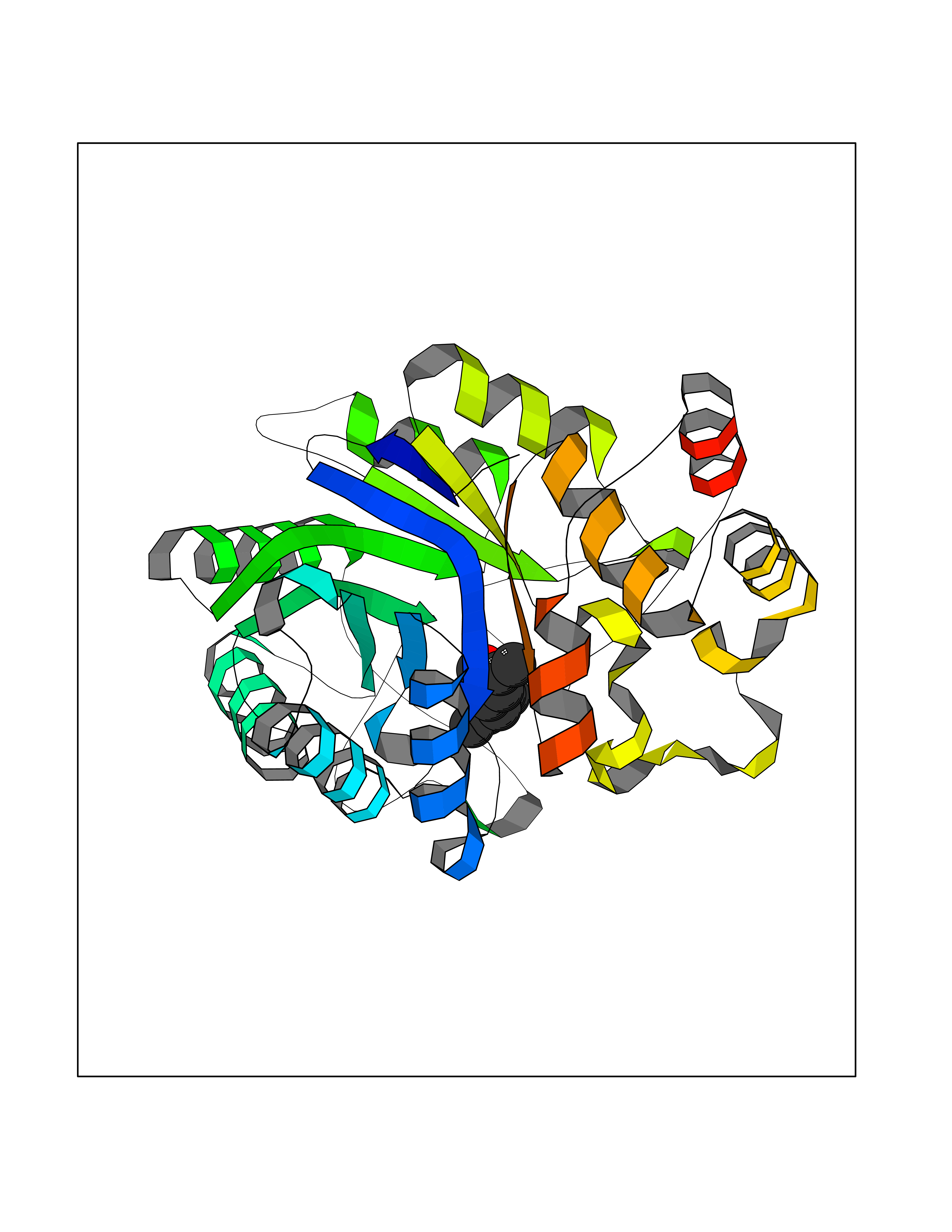 Molscript Map  Pymol Map 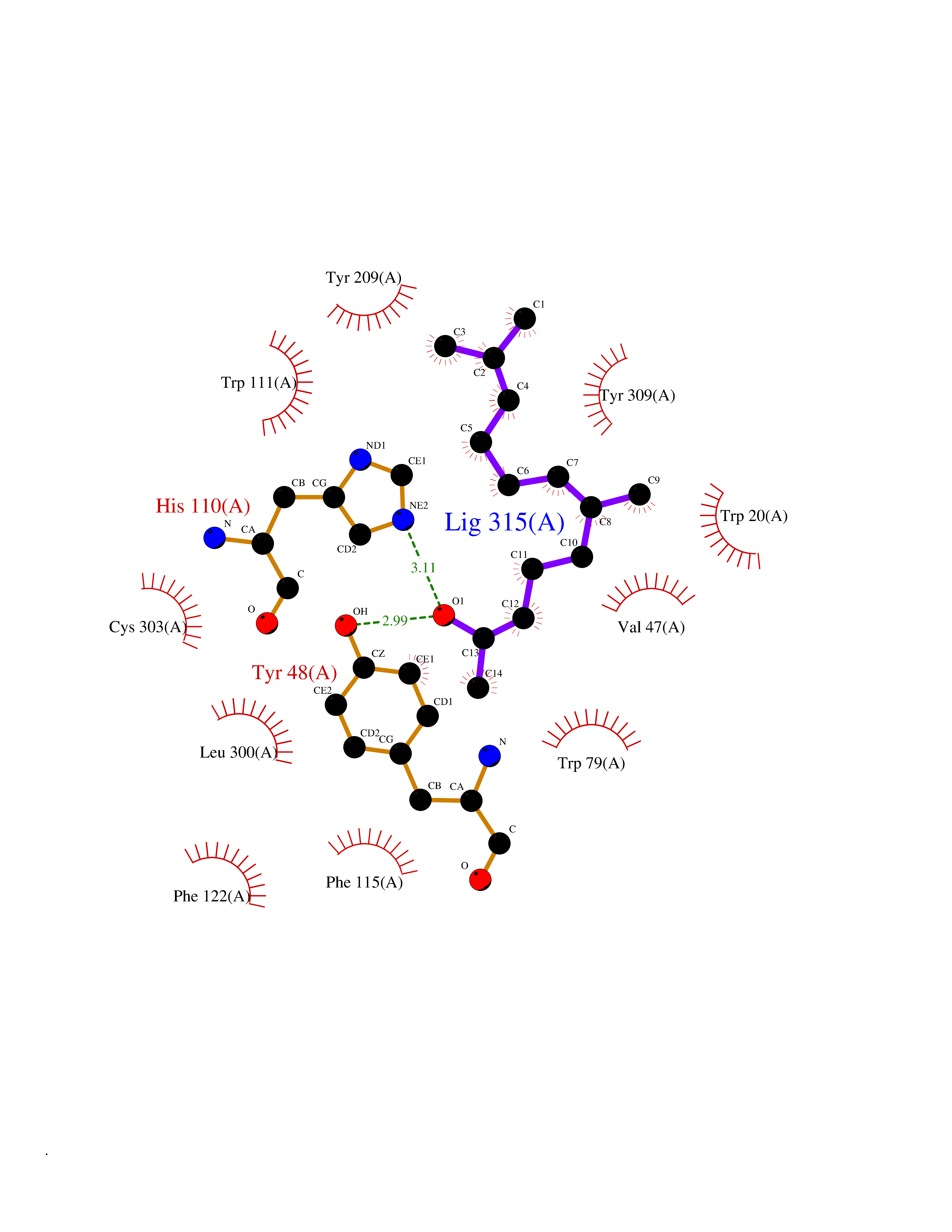 Ligplot Map 3D Binding mode Sequence MASRILLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEKLREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEFFPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAVNQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHNKTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRNWRVCALLSCTSHKDYPFH Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Protein-tyrosine phosphatase SHP-2 (PTPN11) | 2SHP | 6.18 | |
Target general information Gen name PTPN11 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein phosphatase non-receptor type 11; SHPTP2; SHP2; SHP-2; SH-PTP3; SH-PTP2; Protein-tyrosine phosphatase SHP2; Protein-tyrosine phosphatase 2C; Protein-tyrosine phosphatase 1D; PTP2C; PT Protein family Protein-tyrosine phosphatase family, Non-receptor class 2 subfamily Biochemical class Phosphoric monoester hydrolase Function Positively regulates MAPK signal transduction pathway. Dephosphorylates GAB1, ARHGAP35 and EGFR. Dephosphorylates ROCK2 at 'Tyr-722' resulting in stimulatation of its RhoA binding activity. Dephosphorylates CDC73. Acts downstream of various receptor and cytoplasmic protein tyrosine kinases to participate in the signal transduction from the cell surface to the nucleus. Related diseases LEOPARD syndrome 1 (LPRD1) [MIM:151100]: A disorder characterized by lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonic stenosis, abnormalities of genitalia, retardation of growth, and sensorineural deafness. {ECO:0000269|PubMed:12058348, ECO:0000269|PubMed:14961557, ECO:0000269|PubMed:15121796, ECO:0000269|PubMed:15389709, ECO:0000269|PubMed:15520399, ECO:0000269|PubMed:15690106, ECO:0000269|PubMed:16679933, ECO:0000269|PubMed:16733669, ECO:0000269|PubMed:24891296, ECO:0000269|PubMed:26742426}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 1 (NS1) [MIM:163950]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. Some patients with NS1 develop multiple giant cell lesions of the jaw or other bony or soft tissues, which are classified as pigmented villonodular synovitis (PVNS) when occurring in the jaw or joints. {ECO:0000269|PubMed:11704759, ECO:0000269|PubMed:11992261, ECO:0000269|PubMed:12161469, ECO:0000269|PubMed:12325025, ECO:0000269|PubMed:12529711, ECO:0000269|PubMed:12634870, ECO:0000269|PubMed:12717436, ECO:0000269|PubMed:12739139, ECO:0000269|PubMed:12960218, ECO:0000269|PubMed:15384080, ECO:0000269|PubMed:15889278, ECO:0000269|PubMed:15948193, ECO:0000269|PubMed:19020799, ECO:0000269|PubMed:24891296, ECO:0000269|PubMed:28074573}. The disease is caused by variants affecting the gene represented in this entry. Mutations in PTPN11 account for more than 50% of the cases.; DISEASE: Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:12717436, ECO:0000269|PubMed:26742426}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metachondromatosis (MC) [MIM:156250]: A skeletal disorder with radiologic features of both multiple exostoses and Ollier disease, characterized by the presence of exostoses, commonly of the bones of the hands and feet, and enchondromas of the metaphyses of long bones and iliac crest. {ECO:0000269|PubMed:20577567}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02779 Interacts with P10275; P32239; Q9BZW8; P20138; Q08345; P00533; P29317; P04626; Q8WU20; Q13480; Q9UQC2; P62993; P08069; P06213; P35568; P43628; P10721; P08581; O95297; Q15116; P09619; P16284; P49023; P49247; Q13049; P68105; Q71V39; P35570; P97710; Q6P1J9; Q13480; O75496; Q9UKI8 EC number EC 3.1.3.48 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Deafness; Disease variant; Hydrolase; Nucleus; Phosphoprotein; Protein phosphatase; Proteomics identification; Reference proteome; Repeat; SH2 domain Protein physicochemical properties Chain ID A Molecular weight (Da) 56341.2 Length 491 Aromaticity 0.09 Instability index 41.37 Isoelectric point 7.76 Charge (pH=7) 2.43 2D Binding mode Binding energy (Kcal/mol) -8.43  Molscript Map 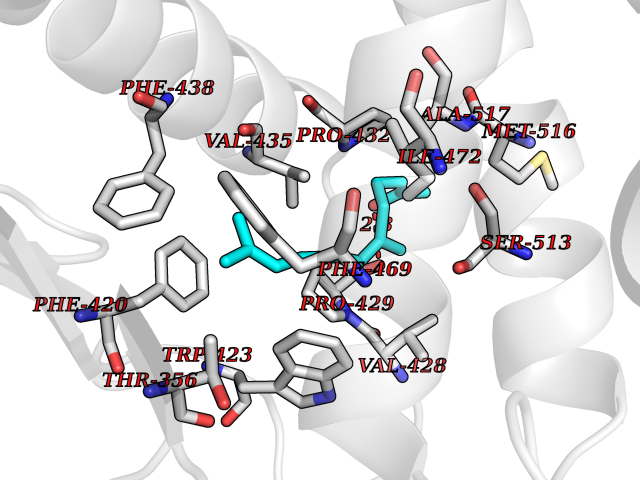 Pymol Map 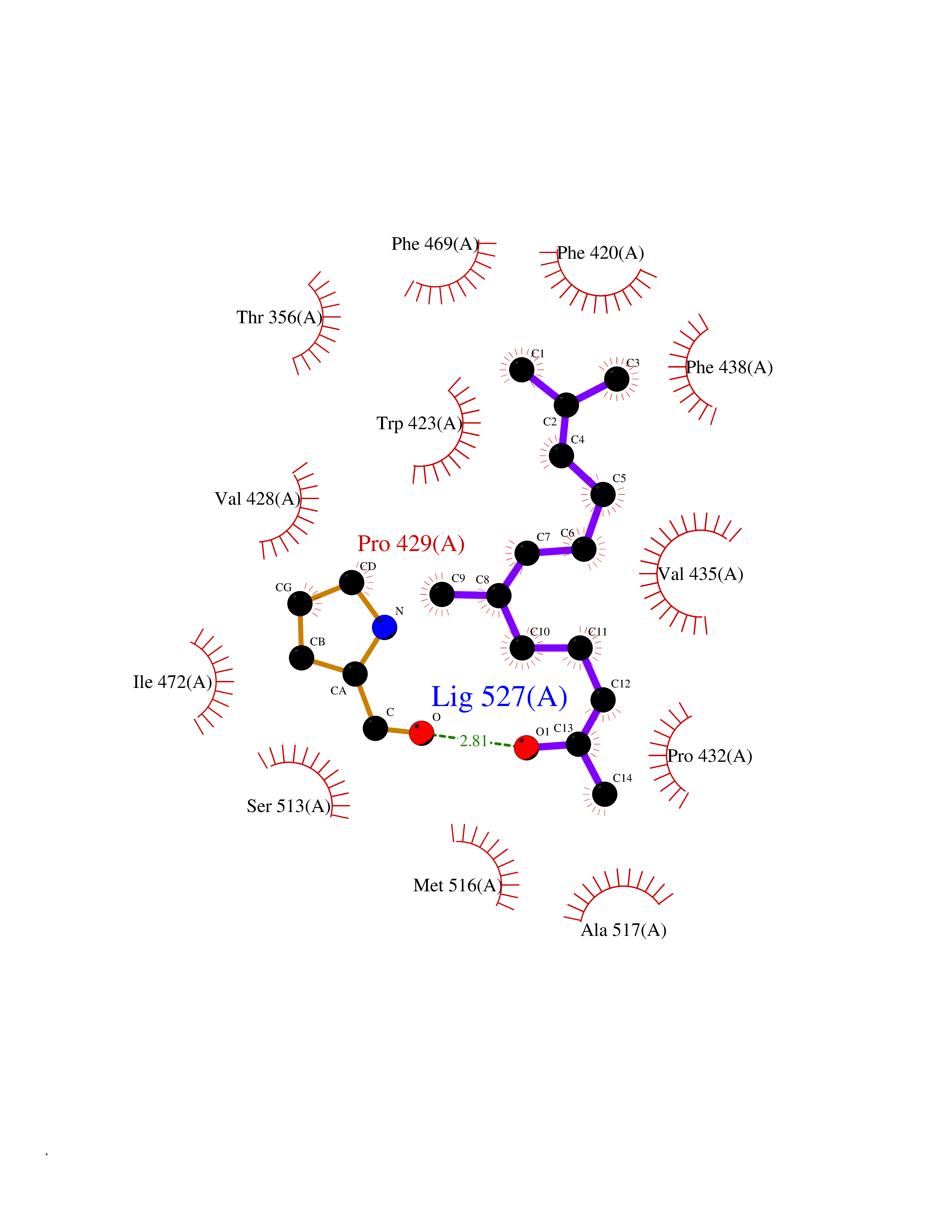 Ligplot Map 3D Binding mode Sequence KSRRWFHPNITGVEAENLLLTRGVDGSFLARPSKSNPGDLTLSVRRNGAVTHIKIQNTGDYYDLYGGEKFATLAELVQYYMEHHGQLKEKNGDVIELKYPLNCADPTSERWFHGHLSGKEAEKLLTEKGKHGSFLVRESQSHPGDFVLSVRTGDNDGKSKVTHVMIRCQELKYDVGGGERFDSLTDLVEHYKKNPMVETLGTVLQLKQPLNTTRINAAEIESRVRELSKGFWEEFETLQQQECKLLYSRKEGQRQENKNKNRYKNILPFDHTRVVLHDSDYINANIIMPKKSYIATQGCLQNTVNDFWRMVFQENSRVIVMTTKEVERGKSKCVKYWPDEYALKEYGVMRVRNVKESAAHDYTLRELKLSKVGQGNTERTVWQYHFRTWPDHGVPSDPGGVLDFLEEVHHKQESIMDAGPVVVHCSAGIGRTGTFIVIDILIDIIREKGVDCDIDVPKTIQMVRSQRSGMVQTEAQYRSIYMAVQHYIETL Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | SEC14-like protein 2 | 4OMJ | 6.17 | |
Target general information Gen name SEC14L2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA1658;C22orf6;KIAA1186 Protein family NA Biochemical class Transport protein Function Phospholipid binding.Transporter activity.Vitamin E binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Lipid-binding; Nucleus; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 31533.3 Length 274 Aromaticity 0.1 Instability index 49.26 Isoelectric point 8.34 Charge (pH=7) 2.81 2D Binding mode Binding energy (Kcal/mol) -8.42 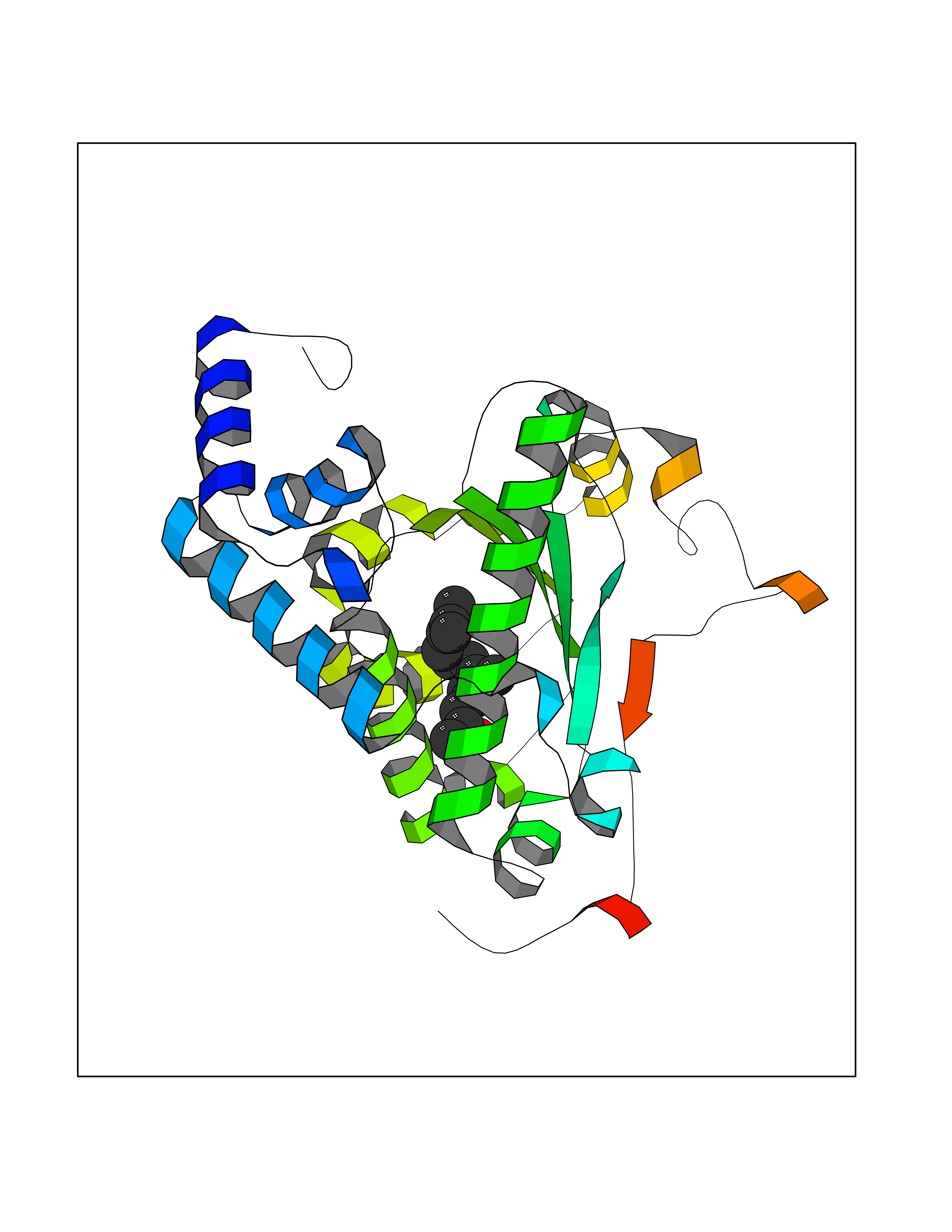 Molscript Map 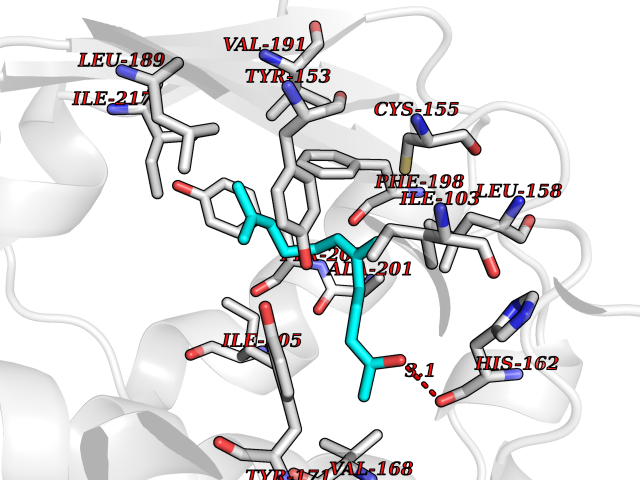 Pymol Map 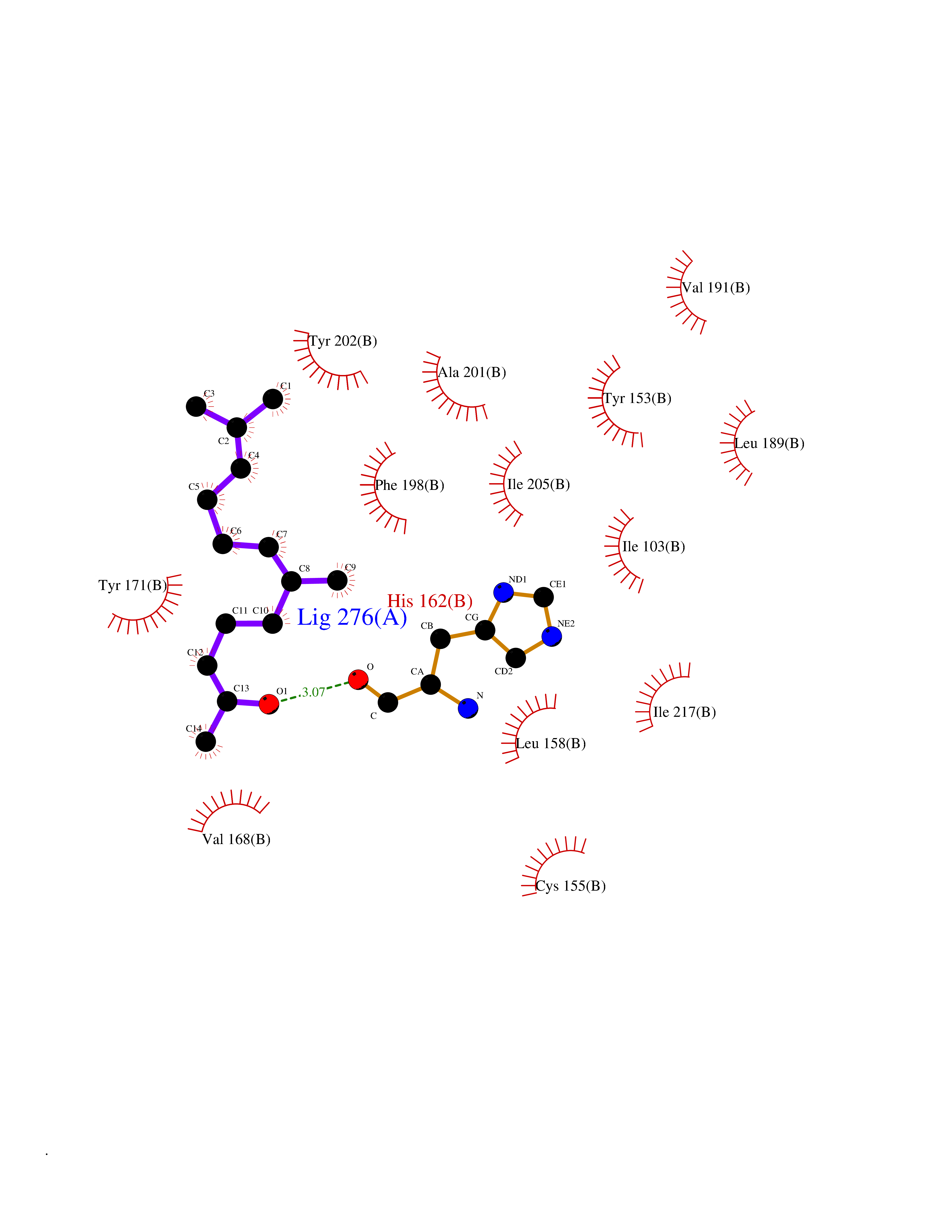 Ligplot Map 3D Binding mode Sequence MSGRVGDLSPRQKEALAKFRENVQDVLPALPNPDDYFLLRWLRARSFDLQKSEAMLRKHVEFRKQKDIDNIISWQPPEVIQQYLSGGMCGYDLDGCPVWYDIIGPLDAKGLLFSASKQDLLRTKMRECELLLQECAHQTTKLGRKVETITIIYDCEGLGLKHLWKPAVEAYGEFLCMFEENYPETLKRLFVVKAPKLFPVAYNLIKPFLSEDTRKKIMVLGANWKEVLLKHISPDQVPVEYGGTMTDPDGNPKCKSKINYGGDIPRKYYVRDQV Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 6.17 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -8.41 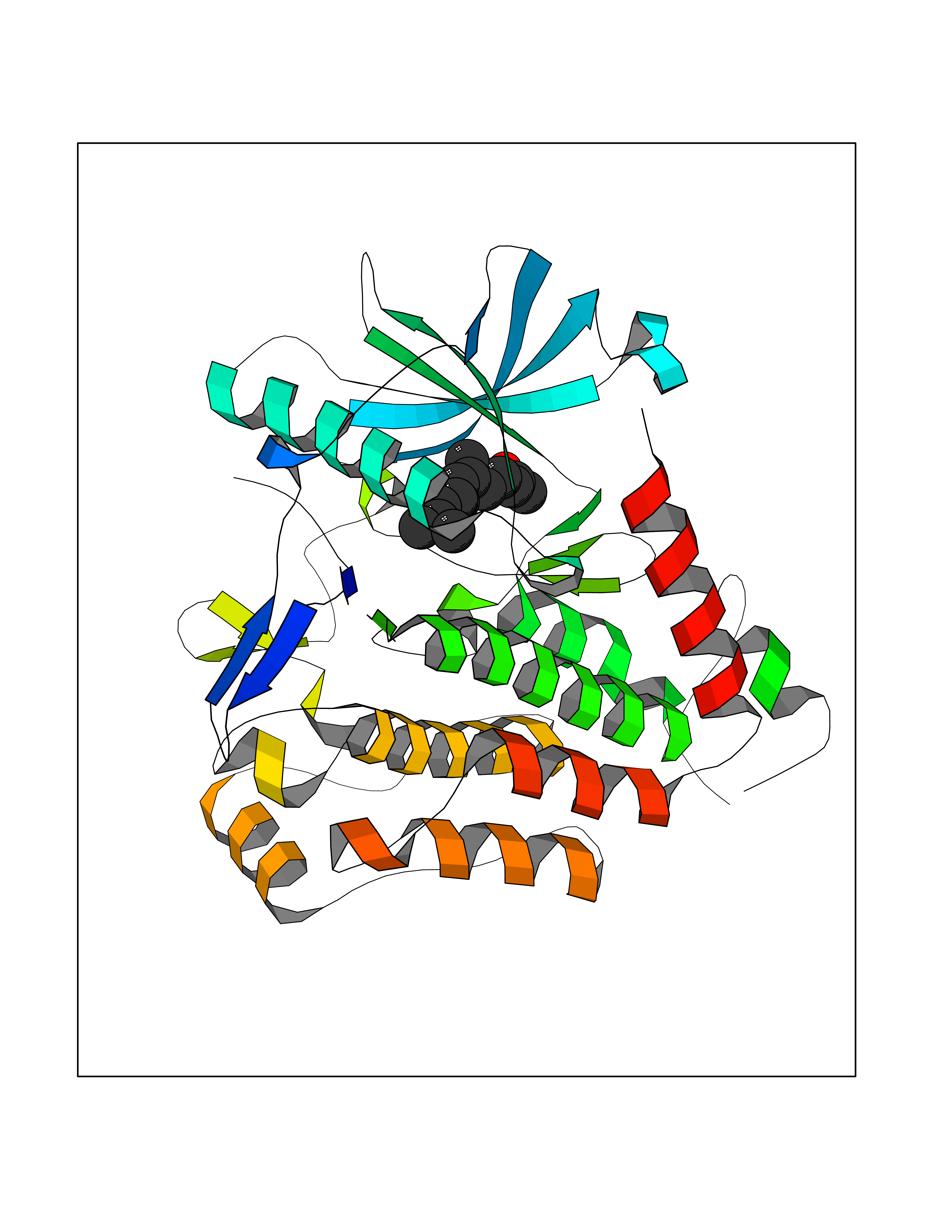 Molscript Map 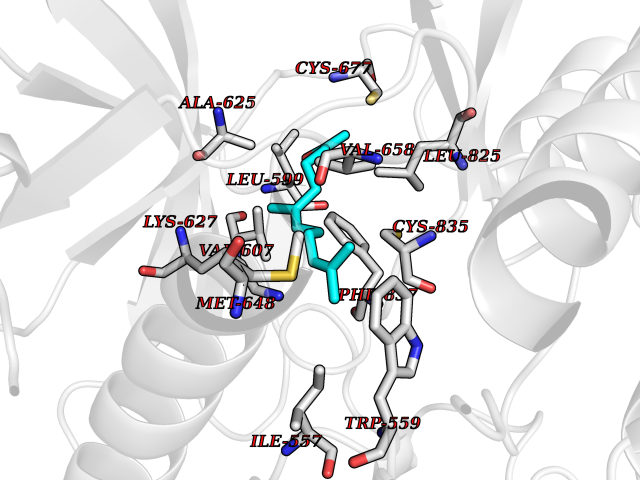 Pymol Map 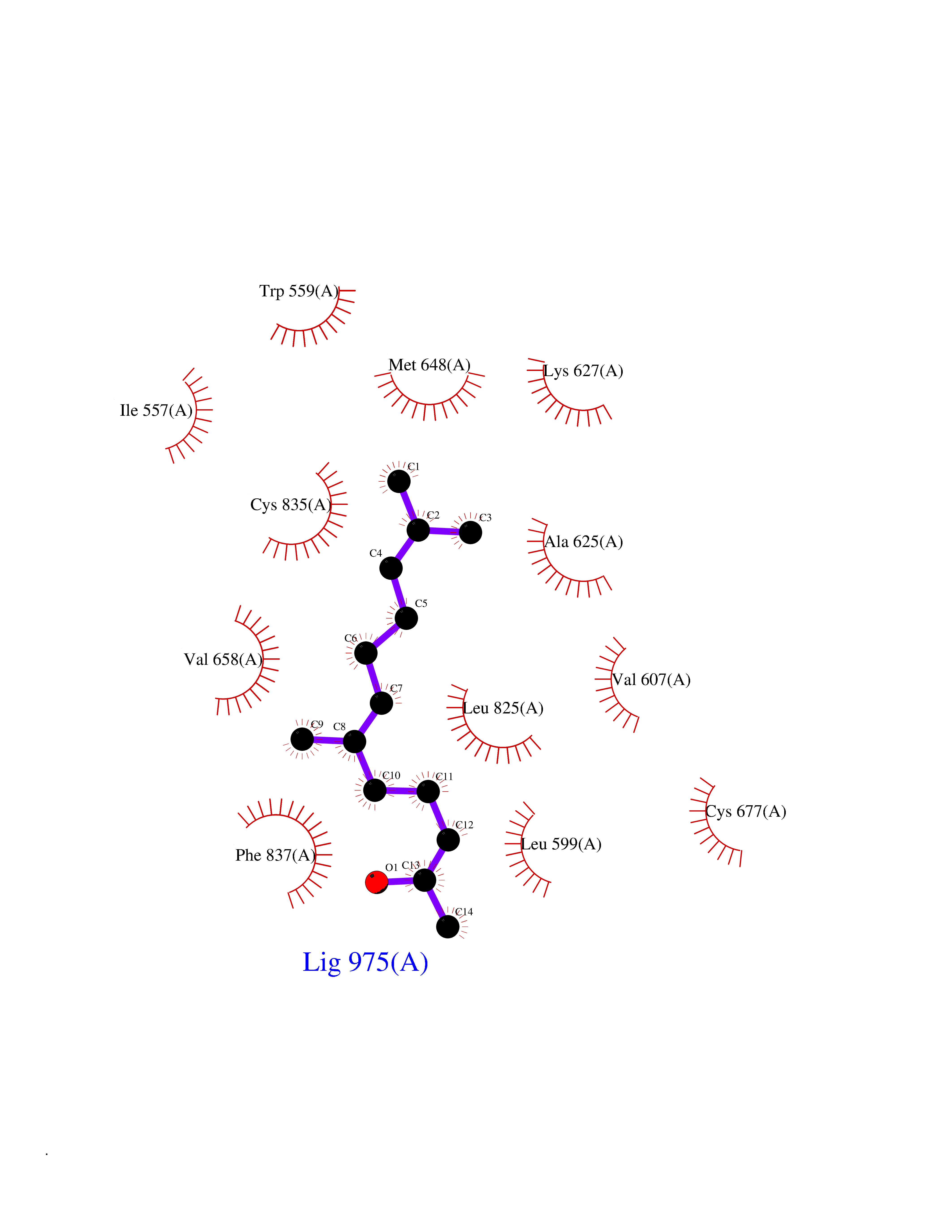 Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | 2-iminobutanoate/2-iminopropanoate deaminase | 1ONI | 6.17 | |
Target general information Gen name RIDA Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HRSP12 Protein family RutC family Biochemical class Translation Function Deaminase activity.Endoribonuclease activity, producing 3'-phosphomonoesters.Long-chain fatty acid binding.Platinum binding.Protein homodimerization activity.RNA binding.Transition metal ion binding.Xenon atom binding. Related diseases Congenital bile acid synthesis defect 2 (CBAS2) [MIM:235555]: A condition characterized by jaundice, intrahepatic cholestasis and hepatic failure. Patients with this liver disease show absence or low levels of chenodeoxycholic acid and cholic acid in plasma and urine. {ECO:0000269|PubMed:12970144, ECO:0000269|PubMed:15030995, ECO:0000269|PubMed:19175828, ECO:0000269|PubMed:20522910}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q8N9N5-2 EC number 3.5.99.10 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Hydrolase; Lipid metabolism; Mitochondrion; Nucleus; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; RNA-binding Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I Molecular weight (Da) 42624.3 Length 404 Aromaticity 0.07 Instability index 36.76 Isoelectric point 8.99 Charge (pH=7) 5.46 2D Binding mode Binding energy (Kcal/mol) -8.41 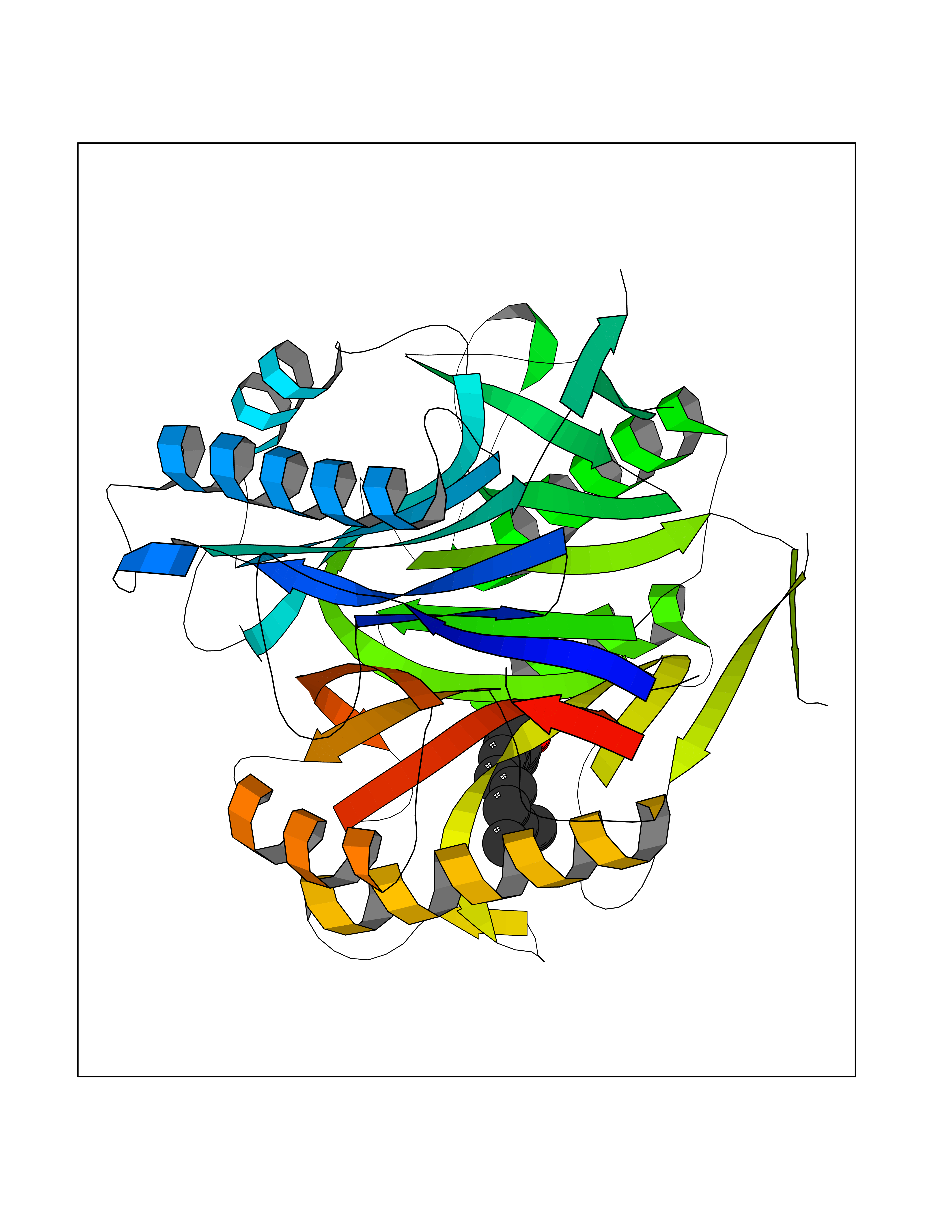 Molscript Map 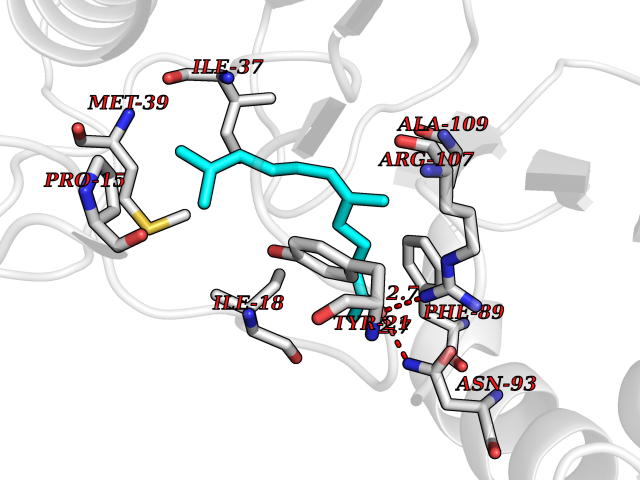 Pymol Map  Ligplot Map 3D Binding mode Sequence SSLIRRVISTAKAPGAIGPYSQAVLVDRTIYISGQIGMDPSSGQLVSGGVAEEAKQALKNMGEILKAAGCDFTNVVKTTVLLADINDFNTVNEIYKQYFKSNFPARAAYQVAALPKGSRIEIEAVAIQGPLTTASSSLIRRVISTAKAPGAIGPYSQAVLVDRTIYISGQIGMDPSSGQLVSGGVAEEAKQALKNMGEILKAAGCDFTNVVKTTVLLADINDFNTVNEIYKQYFKSNFPARAAYQVAALPKGSRIEIEAVAIQGPLTTASSSLIRRVISTAKAPGAIGPYSQAVLVDRTIYISGQIGMDPSSGQLVSGGVAEEAKQALKNMGEILKAAGCDFTNVVKTTVLLADINDFNTVNEIYKQYFKSNFPARAAYQVAALPKGSRIEIEAVAIQGPLTTA Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Neuronal acetylcholine receptor subunit alpha-2 | 5FJV | 6.17 | |
Target general information Gen name CHRNA2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-2/CHRNA2 sub-subfamily Biochemical class NA Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane." Related diseases Epilepsy, nocturnal frontal lobe, 4 (ENFL4) [MIM:610353]: An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking. {ECO:0000269|PubMed:16826524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Seizures, benign familial infantile, 6 (BFIS6) [MIM:610353]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS6 inheritance is autosomal dominant. {ECO:0000269|PubMed:25847220}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00732; DB00237; DB00411; DB00565; DB01245; DB00514; DB01135; DB07720; DB00898; DB00472; DB00483; DB08960; DB00657; DB01336; DB00416; DB01226; DB00184; DB01337; DB01338; DB00721; DB00728; DB05740; DB00202; DB01199; DB01339 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 120584 Length 1031 Aromaticity 0.14 Instability index 32.21 Isoelectric point 5.62 Charge (pH=7) -17.58 2D Binding mode Binding energy (Kcal/mol) -8.42  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLP Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Toxoplasma Calcium-dependent protein kinase 1 (Tg CDPK1) | 6BFA | 6.17 | |
Target general information Gen name Tg CDPK1 Organism Toxoplasma gondii (strain ATCC 50861 / VEG) Uniprot ID TTD ID Synonyms CAM kinase, CDPK family TgCDPK1 Protein family Protein kinase superfamily, Ser/Thr protein kinase family, CDPK subfamily Biochemical class NA Function Provides the mechanistic link between calcium signalling and motility, differentiation and invasion. Required for the microneme secretion at the apical complex and parasite proliferation. Related diseases Factor XI deficiency (FA11D) [MIM:612416]: A hemorrhagic disease characterized by reduced levels and activity of factor XI resulting in moderate bleeding symptoms, usually occurring after trauma or surgery. Patients usually do not present spontaneous bleeding but women can present with menorrhagia. Hemorrhages are usually moderate. {ECO:0000269|PubMed:10027710, ECO:0000269|PubMed:10606881, ECO:0000269|PubMed:11895778, ECO:0000269|PubMed:15026311, ECO:0000269|PubMed:15180874, ECO:0000269|PubMed:1547342, ECO:0000269|PubMed:15953011, ECO:0000269|PubMed:16607084, ECO:0000269|PubMed:18005151, ECO:0000269|PubMed:21457405, ECO:0000269|PubMed:21668437, ECO:0000269|PubMed:21999818, ECO:0000269|PubMed:22016685, ECO:0000269|PubMed:22159456, ECO:0000269|PubMed:22322133, ECO:0000269|PubMed:25158988, ECO:0000269|PubMed:2813350, ECO:0000269|PubMed:7669672, ECO:0000269|PubMed:7888672, ECO:0000269|PubMed:9401068, ECO:0000269|PubMed:9787168}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; ATP-binding; Calcium; Kinase; Metal-binding; Nucleotide-binding; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 53029.9 Length 464 Aromaticity 0.09 Instability index 29.74 Isoelectric point 5.61 Charge (pH=7) -8.87 2D Binding mode Binding energy (Kcal/mol) -8.42 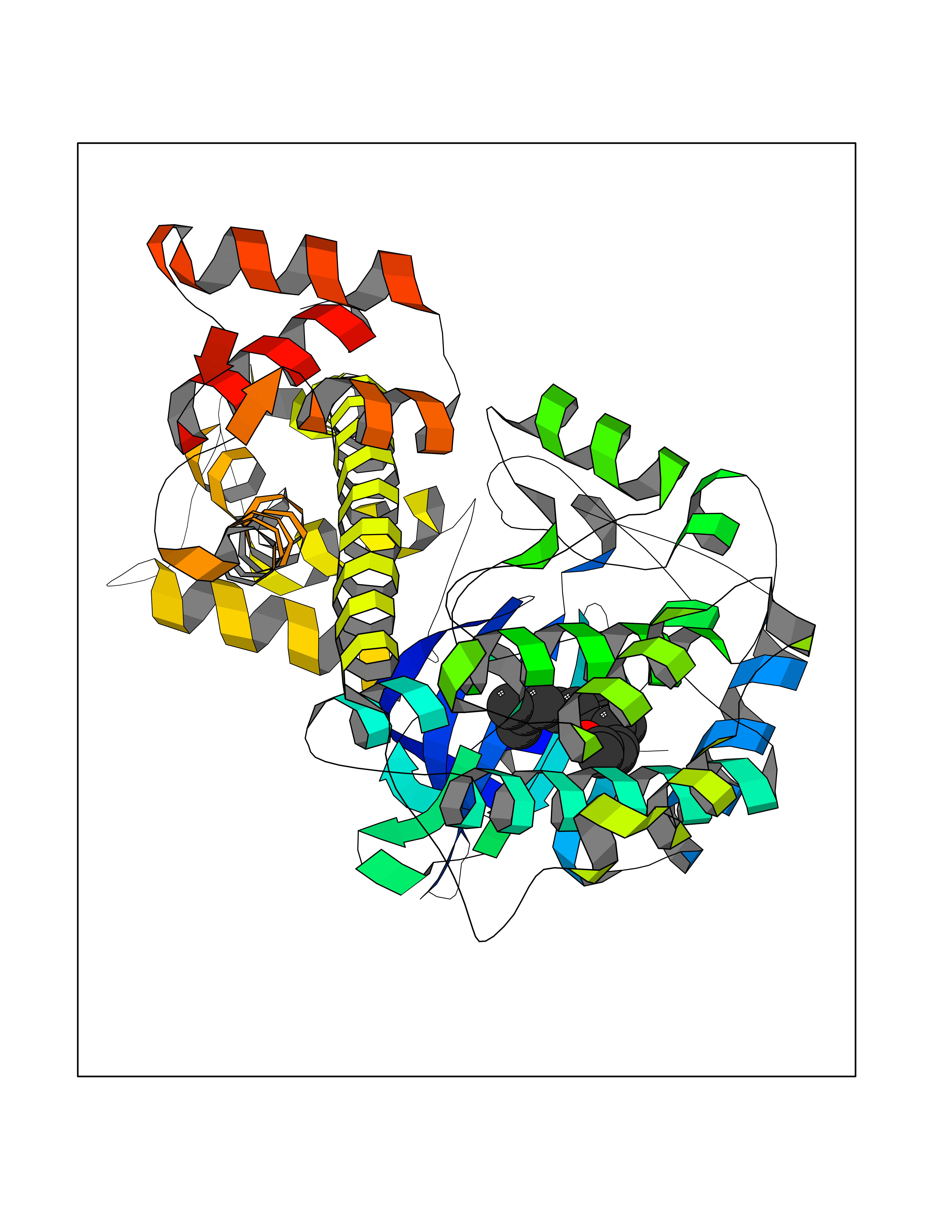 Molscript Map 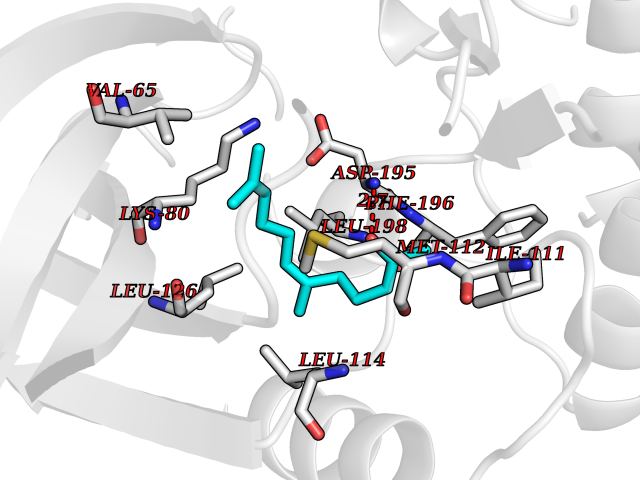 Pymol Map 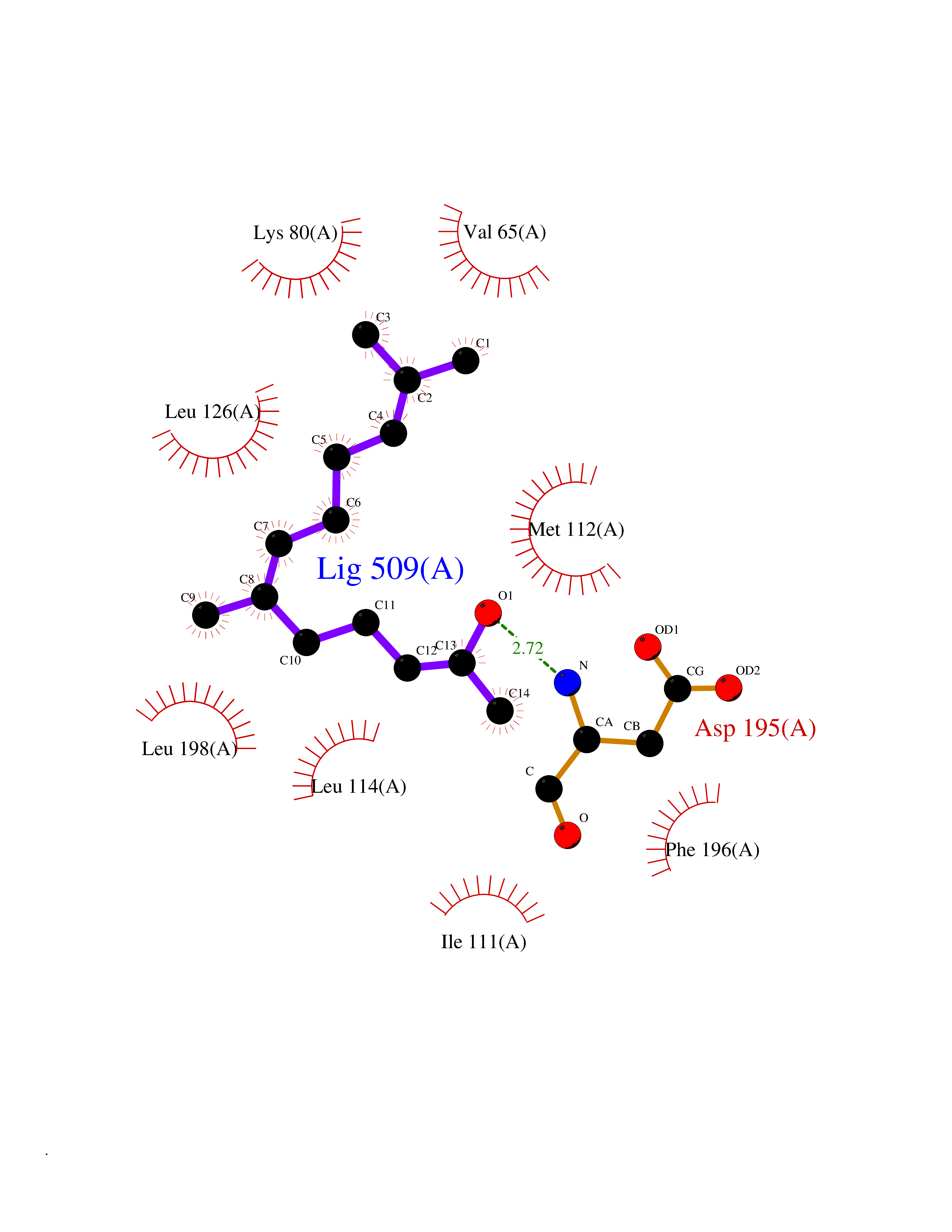 Ligplot Map 3D Binding mode Sequence TAIFSDRYKGQRVLGKGSFGEVILCKDKITGQECAVKVISKRQVKQKTDKESLLREVQLLKQLDHPNIMKLYEFFEDKGYFYLVGEVYTGGELFDEIISRKRFSEVDAARIIRQVLSGITYMHKNKIVHRDLKPENLLLESKSKDANIRIIDFGLSTHFEASKKMKDKIGTAYYIAPEVLHGTYDEKCDVWSTGVILYILLSGCPPFNGANEYDILKKVEKGKYTFELPQWKKVSESAKDLIRKMLTYVPSMRISARDALDHEWIQTYTKEQISVDVPSLDNAILNIRQFQGTQKLAQAALLYMGSKLTSQDETKELTAIFHKMDKNGDGQLDRAELIEGYKELMRMKGQDASMLDASAVEHEVDQVLDAVDFDKNGYIEYSEFVTVAMDRKTLLSRERLERAFRMFDSDNSGKISSTELATIFGVSDVDSETWKSVLSEVDKNNDGEVDFDEFQQMLLKLCGN Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Acyloxyacyl hydrolase (neutrophil) | 5W7C | 6.17 | |
Target general information Gen name AOAH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Acyloxyacyl hydrolase Protein family NA Biochemical class NA Function Removes the secondary (acyloxyacyl-linked) fatty acyl chains from the lipid A region of bacterial lipopolysaccharides. By breaking down LPS, terminates the host response to bacterial infection and prevents prolonged and damaging inflammatory responses (By similarity). In peritoneal macrophages, seems to be important for recovery from a state of immune tolerance following infection by Gram-negative bacteria (By similarity). Related diseases Major depressive disorder (MDD) [MIM:608516]: A common psychiatric disorder. It is a complex trait characterized by one or more major depressive episodes without a history of manic, mixed, or hypomanic episodes. A major depressive episode is characterized by at least 2 weeks during which there is a new onset or clear worsening of either depressed mood or loss of interest or pleasure in nearly all activities. Four additional symptoms must also be present including changes in appetite, weight, sleep, and psychomotor activity; decreased energy; feelings of worthlessness or guilt; difficulty thinking, concentrating, or making decisions; or recurrent thoughts of death or suicidal ideation, plans, or attempts. The episode must be accompanied by distress or impairment in social, occupational, or other important areas of functioning. {ECO:0000269|PubMed:15229186}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q15700 EC number EC 3.1.1.77 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lipid metabolism; Metal-binding; Proteomics identification; Reference proteome; Secreted; Signal; Zymogen Protein physicochemical properties Chain ID C Molecular weight (Da) 47779.7 Length 420 Aromaticity 0.1 Instability index 43.45 Isoelectric point 7.72 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -8.41 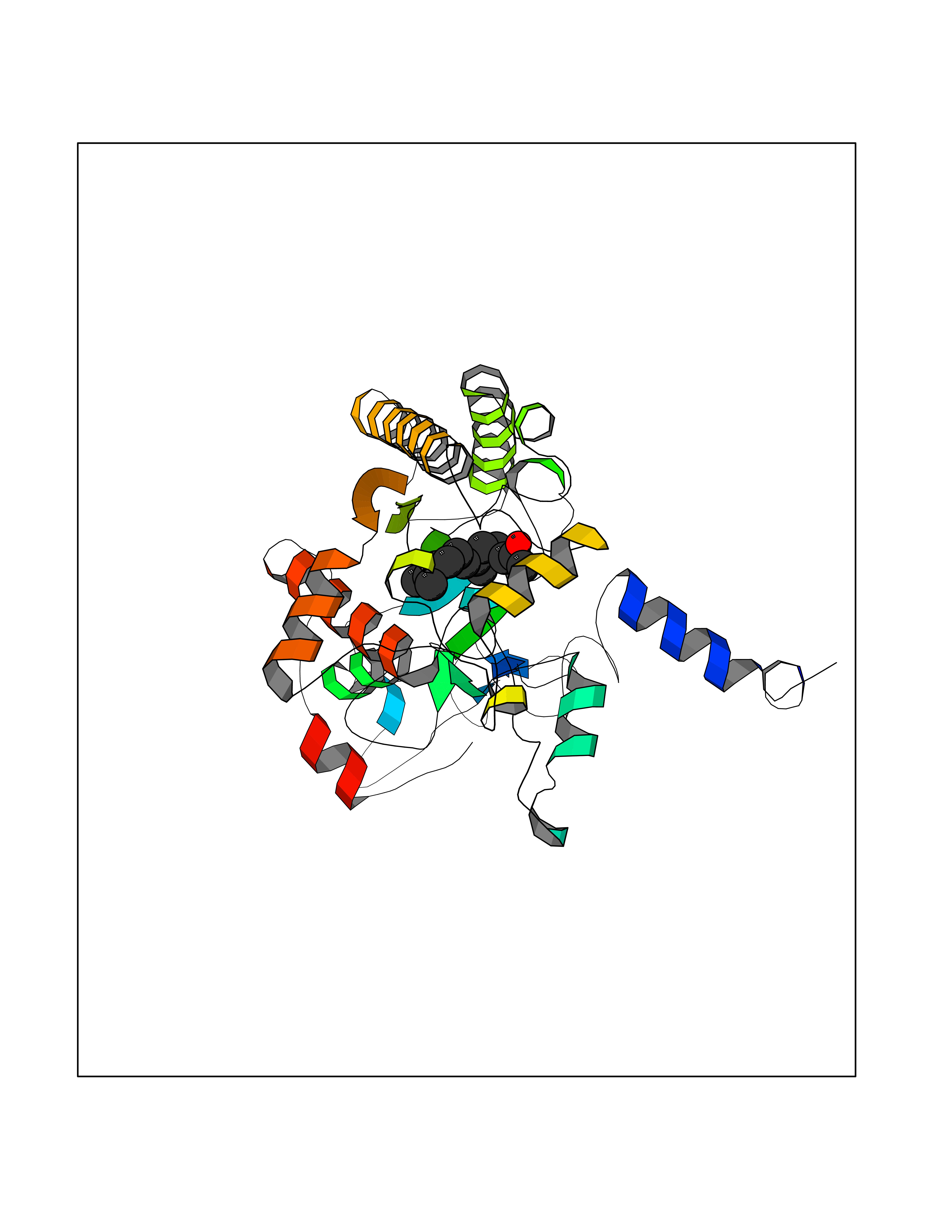 Molscript Map 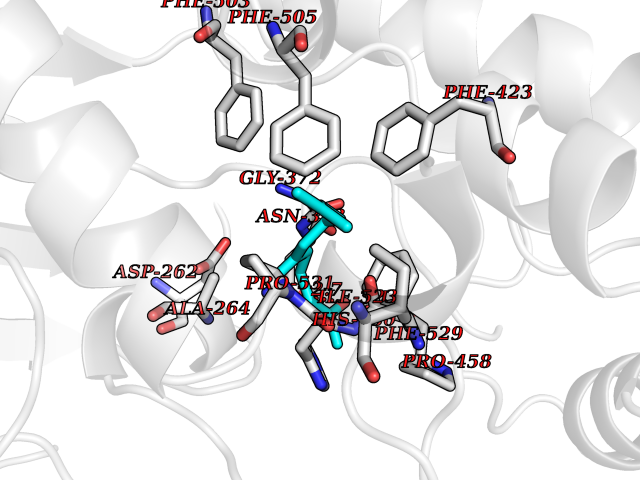 Pymol Map 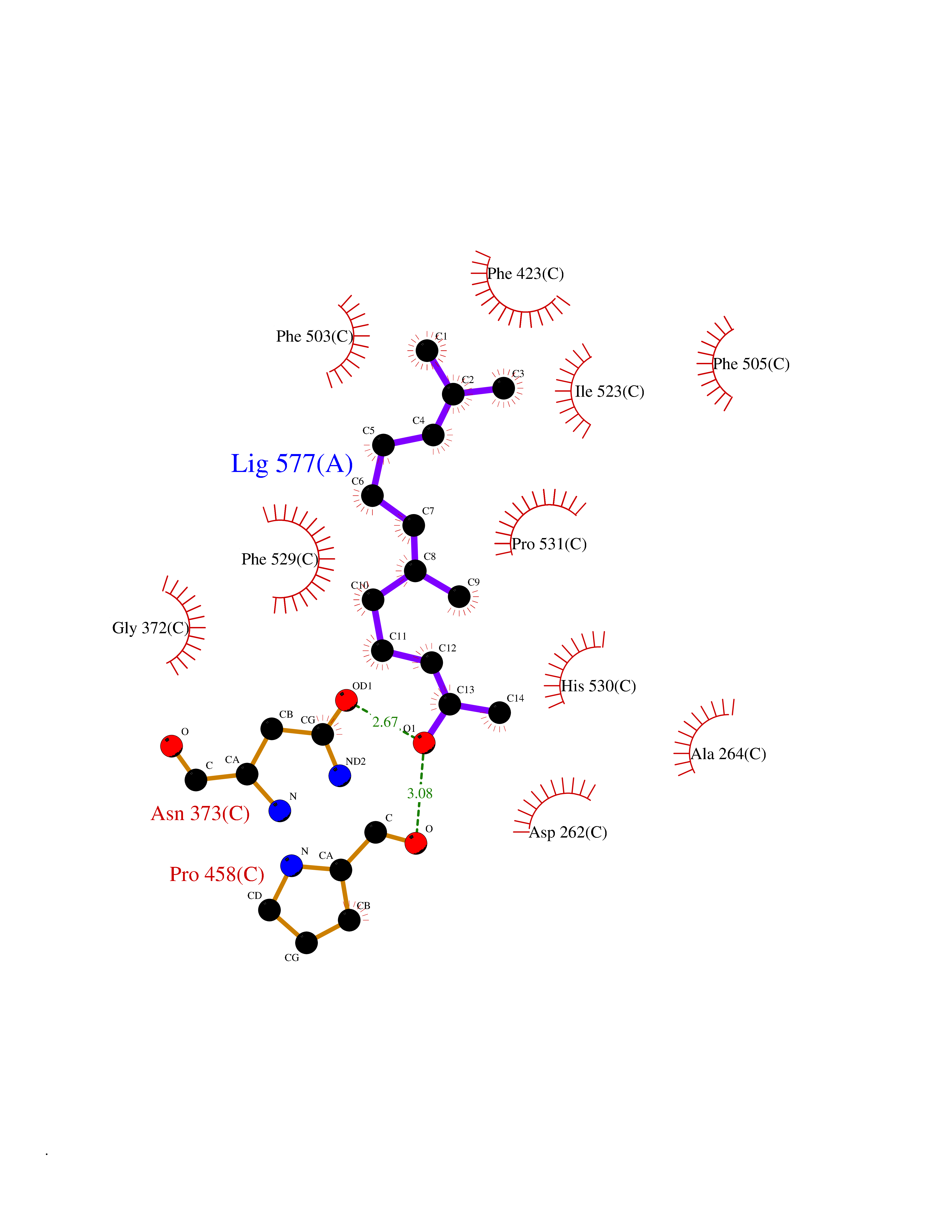 Ligplot Map 3D Binding mode Sequence GSDICSLPVLAKICQKIKLAMEQSVPFKDVDSDKYSVFPTLRGYHWRGRDCNDSDESVYPGRRPNNWDVHQDSNCNGIWGVDPKDGVPYEKKFCEGSQPRGIILLGDAAGAHFHISPEWITASQMSLNSFINLPTALTNELDWPQLSGATGFLDSTVGIKEKSIYLRLWKRNHCNHRDYQNISRNGASSRNLKKFIESLSRNKVLDYPAIVIYAMIGNDVCSGKSDPVPAMTTPEKLYSNVMQTLKHLNSHLPNGSHVILYGLPDGTFLWDNLHNRYHPLGQLNKDMTYAQLYSFLNCLQVSPCHGWMSSNKTLRTLTSERAEQLSNTLKKIAASEKFTNFNLFYMDFAFHEIIQEWQKRGGQPWQLIEPVDGFHPNEVALLLLADHFWKKVQLQWPQILGKENPFNPQIKQVFGDQGGH Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Phenylethanolamine N-methyltransferase (PNMT) | 2G72 | 6.17 | |
Target general information Gen name PNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNMTase; PENT; Noradrenaline N-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class NA Function Converts noradrenaline to adrenaline. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB08129; DB08128; DB07739; DB07798; DB07747; DB03468; DB08550; DB03824; DB04273; DB07906; DB07597; DB09571; DB00968; DB08631; DB01752; DB08654 Interacts with Q9P2G9-2; Q8TBB1 EC number EC 2.1.1.28 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29198.9 Length 264 Aromaticity 0.09 Instability index 54.33 Isoelectric point 5.91 Charge (pH=7) -3.69 2D Binding mode Binding energy (Kcal/mol) -8.41  Molscript Map 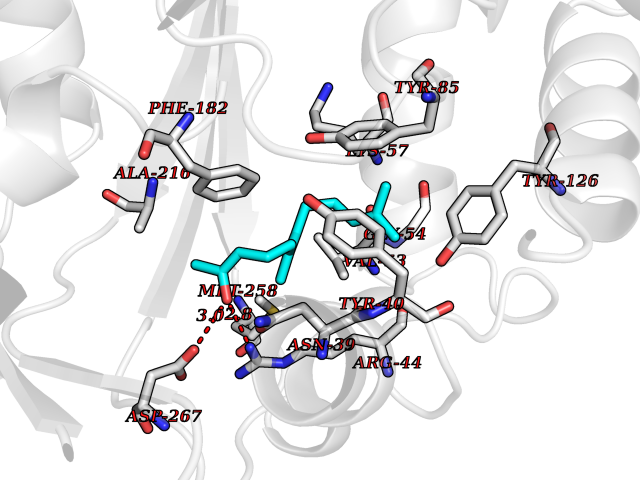 Pymol Map 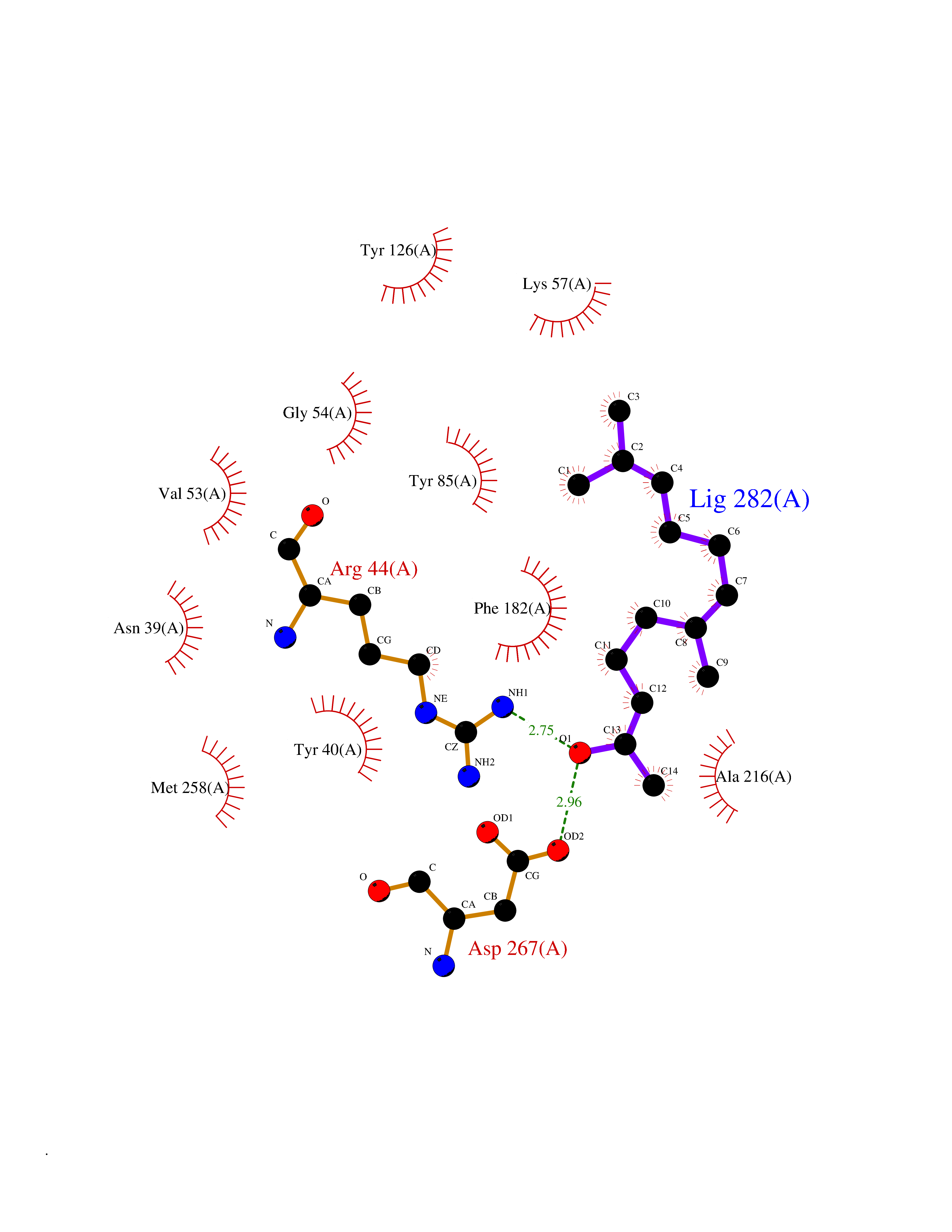 Ligplot Map 3D Binding mode Sequence APGQAAVASAYQRFEPRAYLRNNYAPPRGDLCNPNGVGPWKLRCLAQTFATGEVSGRTLIDIGSGPTVYQLLSACSHFEDITMTDFLEVNRQELGRWLQEEPGAFNWSMYSQHACLIEGKGECWQDKERQLRARVKRVLPIDVHQPQPLGAGSPAPLPADALVSAFCLEAVSPDLASFQRALDHITTLLRPGGHLLLIGALEESWYLAGEARLTVVPVSEEEVREALVRSGYKVRDLRTYIMPAHLQTGVDDVKGVFFAWAQKV Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Neuronal acetylcholine receptor alpha-3/beta-4 (CHRNA3/B4) | 6PV7 | 6.17 | |
Target general information Gen name CHRNA3-CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Neuronal acetylcholine receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Neurotransmitter receptor Function A type of nicotinic acetylcholine receptor, consisting of 3 and 4 subunits. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -8.42 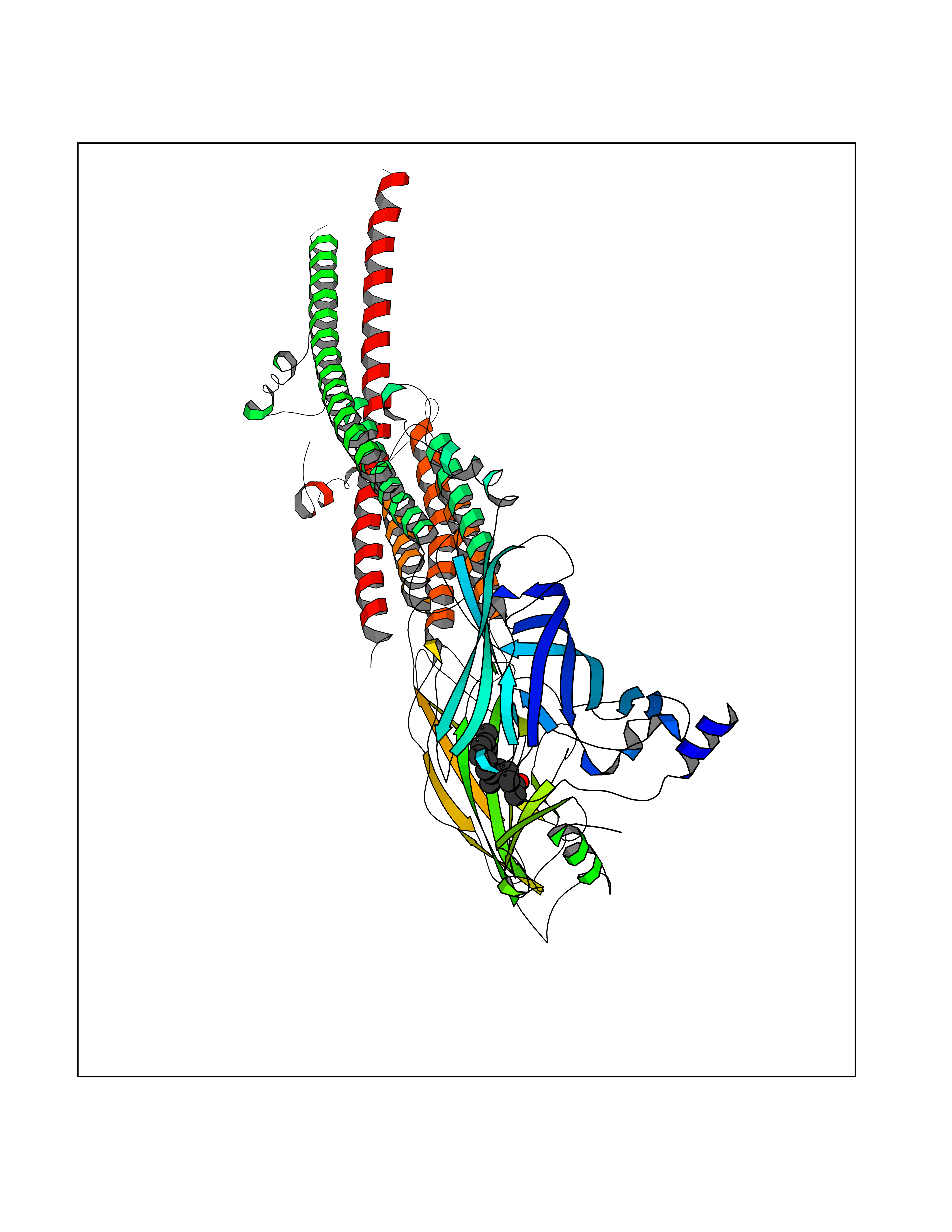 Molscript Map 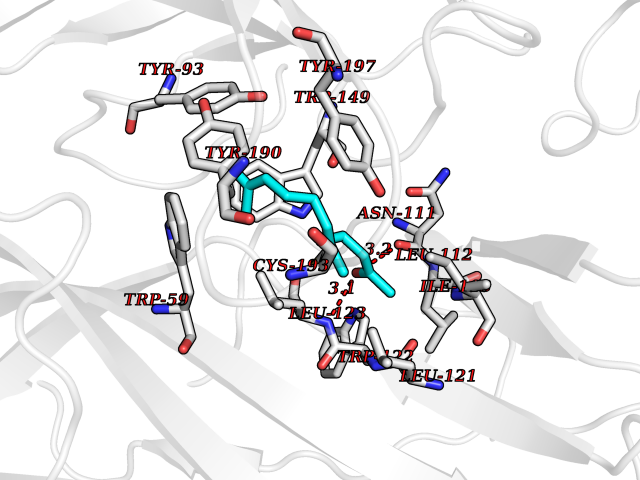 Pymol Map 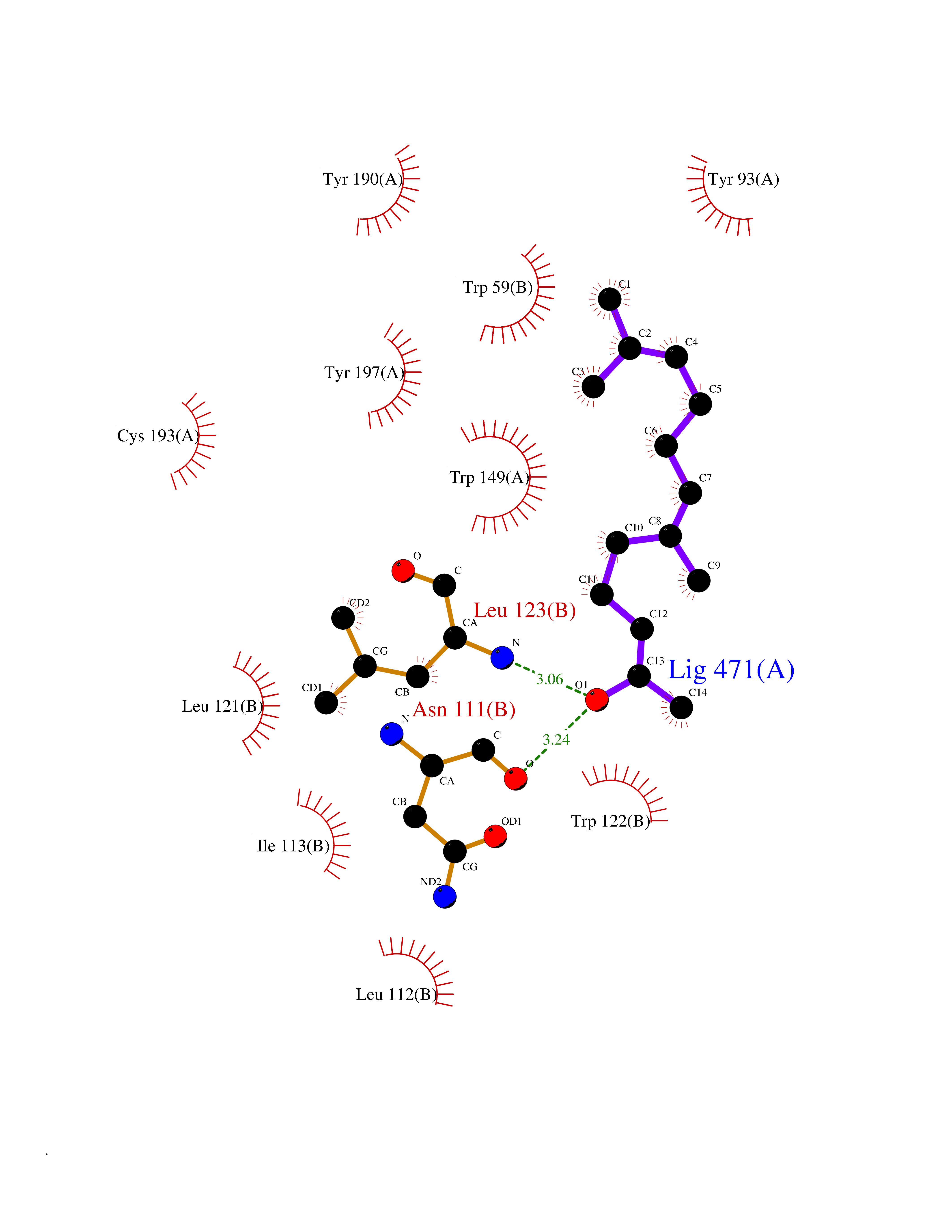 Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 6.16 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -8.41 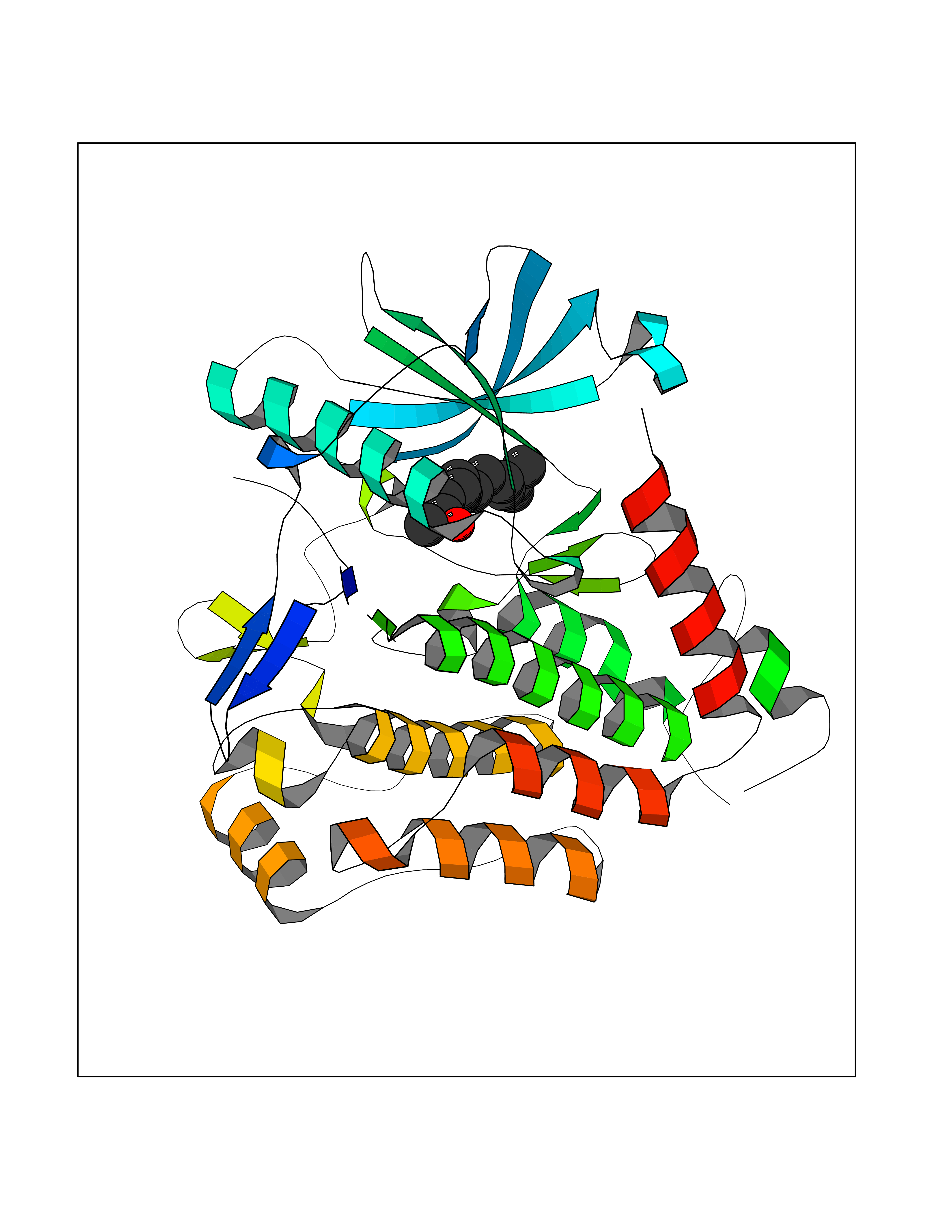 Molscript Map 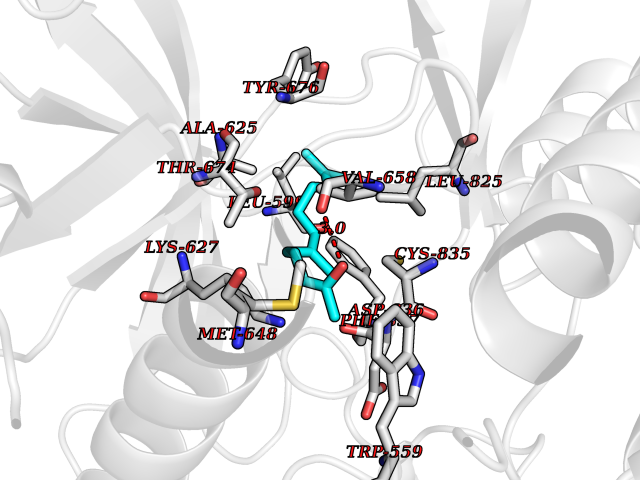 Pymol Map 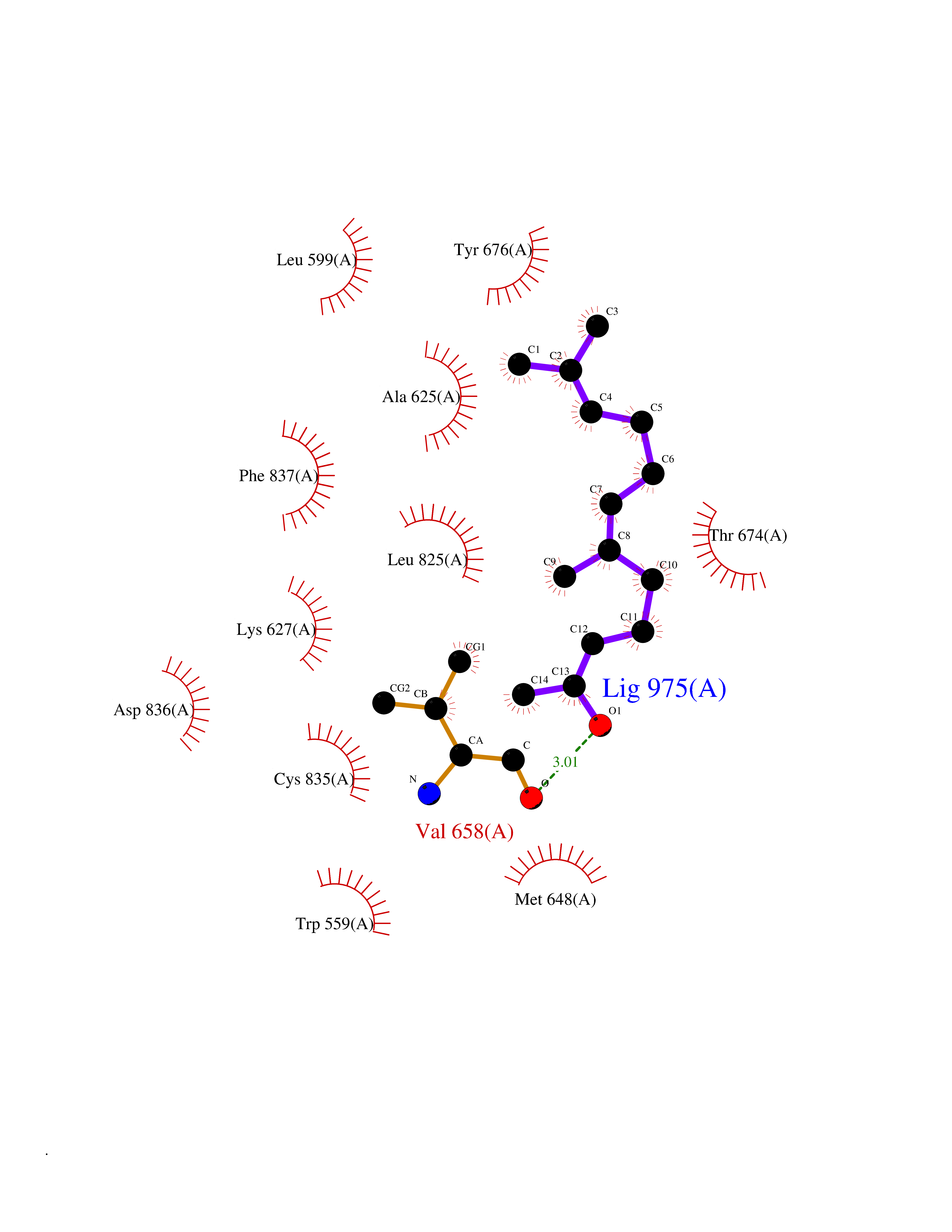 Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Dimethylglycine oxidase | 1PJ5 | 6.16 | |
Target general information Gen name dmg Organism Arthrobacter globiformis Uniprot ID TTD ID NA Synonyms NA Protein family GcvT family Biochemical class Oxidoreductase Function Dimethylglycine oxidase activity.Nucleotide binding. Related diseases Curry-Jones syndrome (CRJS) [MIM:601707]: A multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. The disease is caused by variants affecting the gene represented in this entry. 8 individuals have been identified with the disease-causing mutation Phe-412 and all were mosaic. The mutation could not be reliably detected in blood, greatest success rates were obtained with affected tissues obtained by invasive procedures. It is thought that the mutation has arisen postzygotically early during embryonic development (PubMed:27236920). This mutation has also been identified in ameloblastoma, medulloblastoma, meningioma, and basal cell carcinoma, and has been reported as the oncogenic driver in some of these tumors (PubMed:24859340). {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. Drugs (DrugBank ID) DB03256; DB03147 Interacts with NA EC number 1.5.3.10 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 45912.2 Length 427 Aromaticity 0.07 Instability index 43.46 Isoelectric point 4.83 Charge (pH=7) -20.69 2D Binding mode Binding energy (Kcal/mol) -8.4 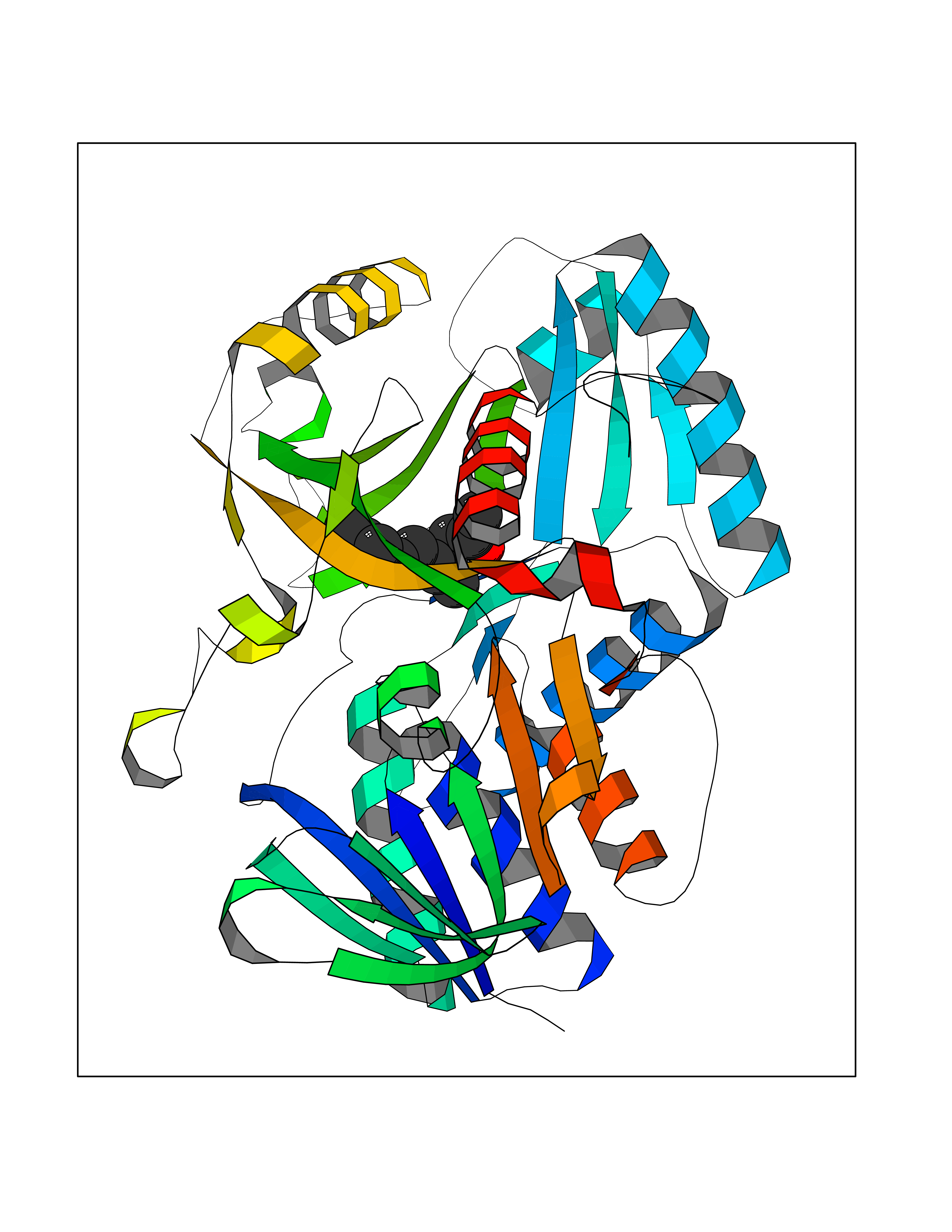 Molscript Map 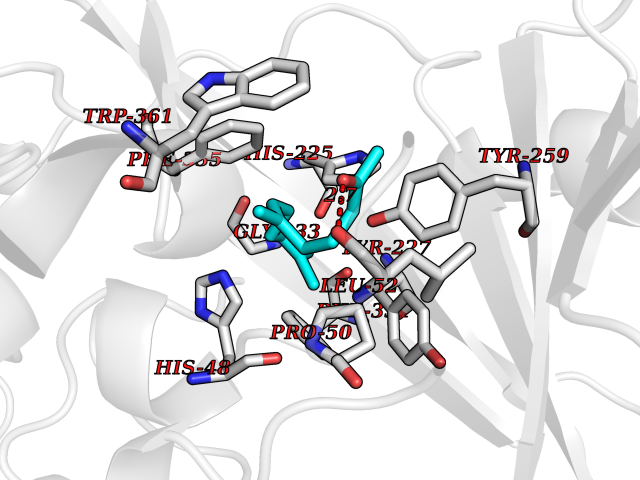 Pymol Map  Ligplot Map 3D Binding mode Sequence TPRIVIIGAGIVGTNLADELVTRGWNNITVLDQGPLNMPGGSTSHAPGLVFQTNPSKTMASFAKYTVEKLLSLTEDGVSCFNQVGGLEVATTETRLADLKRKLGYAAAWGIEGRLLSPAECQELYPLLDGENILGGLHVPSDGLASAARAVQLLIKRTESAGVTYRGSTTVTGIEQSGGRVTGVQTADGVIPADIVVSCAGFWGAKIGAMIGMAVPLLPLAHQYVKTTPVPAQQGRNDQPNGARLPILRHQDQDLYYREHGDRYGIGSYAHRPMPVDVDTLGAYAPETVSEHHMPSRLDFTLEDFLPAWEATKQLLPALADSEIEDGFNGIFSFTPDGGPLLGESKELDGFYVAEAVWVTHSAGVAKAMAELLTTGRSETDLGECDITRFEDVQLTPEYVSETSQQNFVEIYDVLHPLQPRLSPRNL Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Folate receptor beta (FOLR2) | 4KN0 | 6.15 | |
Target general information Gen name FOLR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Placental folate-binding protein; Folate receptor, fetal/placental; Folate receptor type-beta; Folate receptor 2; FR-beta; FOLR2 Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pH after receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00158; DB00563; DB05168 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disulfide bond; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23841.6 Length 205 Aromaticity 0.12 Instability index 56.78 Isoelectric point 7.92 Charge (pH=7) 2.58 2D Binding mode Binding energy (Kcal/mol) -8.38  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RTDLLNVCMDAKHHKTKPGPEDKLHDQCSPWKKNACCTASTSQELHKDTSRLYNFNWDHCGKMEPACKRHFIQDTCLYECSPNLGPWIQQVNQSWRKERFLDVPLCKEDCQRWWEDCHTSHTCKSNWHRGWDWTSGVNKCPAGALCRTFESYFPTPAALCEGLWSHSYKVSNYSRGSGRCIQMWFDSAQGNPNEEVARFYAAAMH Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Dihydroorotate dehydrogenase (quinone), mitochondrial | 4CQ8 | 6.15 | |
Target general information Gen name PFF0160c Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class Oxidoreductase Function Dihydroorotate dehydrogenase activity. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42573.5 Length 378 Aromaticity 0.1 Instability index 36.63 Isoelectric point 8.21 Charge (pH=7) 3.17 2D Binding mode Binding energy (Kcal/mol) -8.39 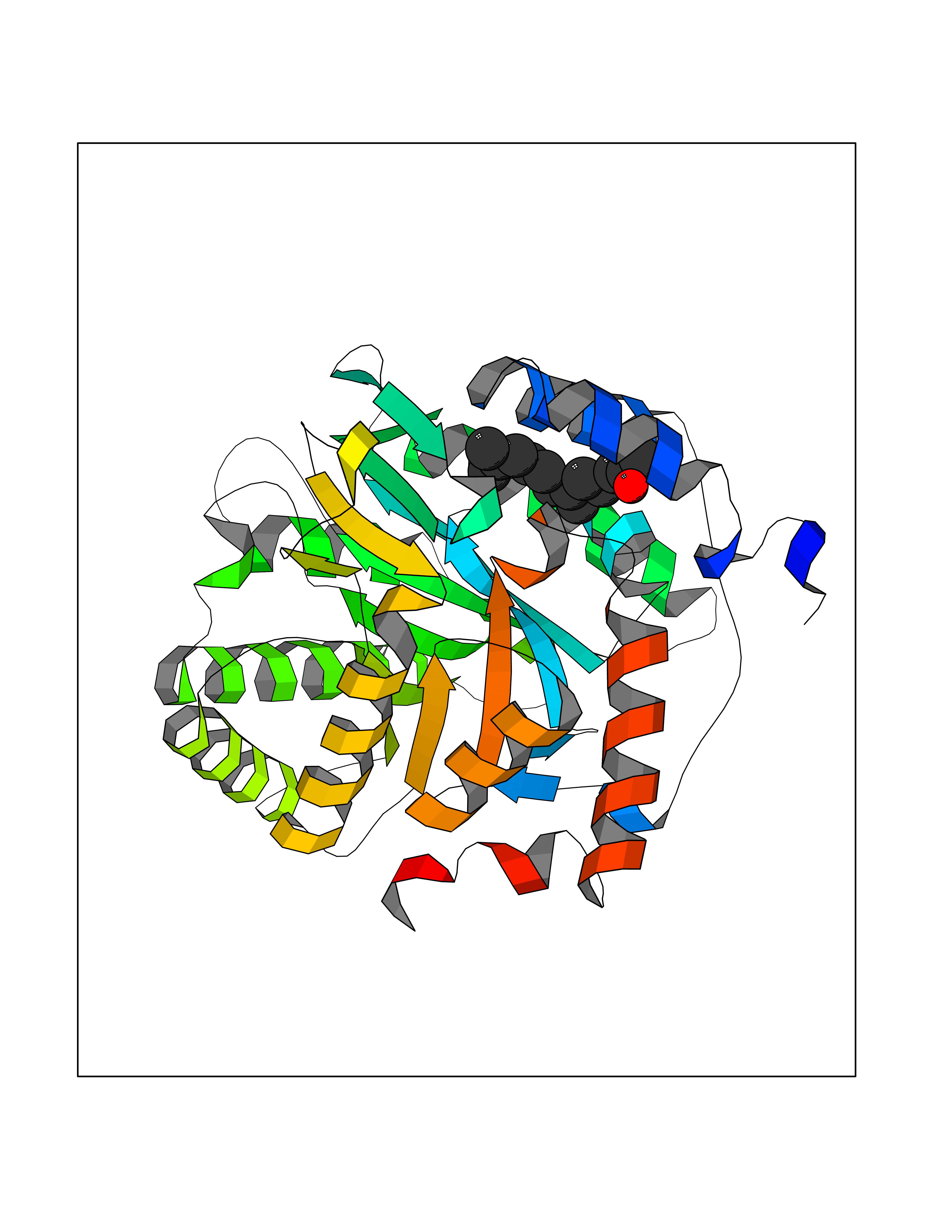 Molscript Map 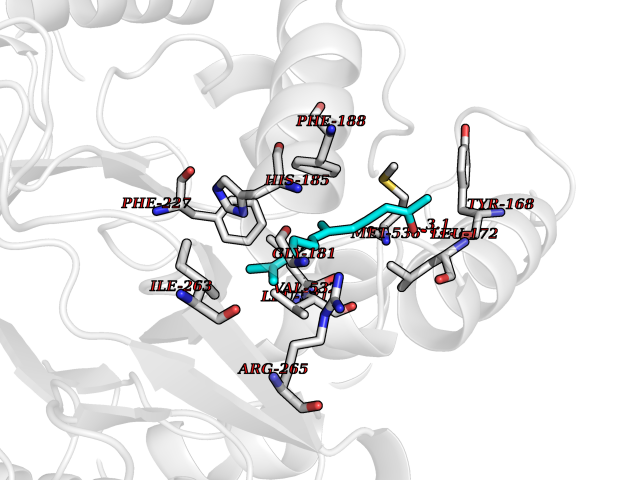 Pymol Map 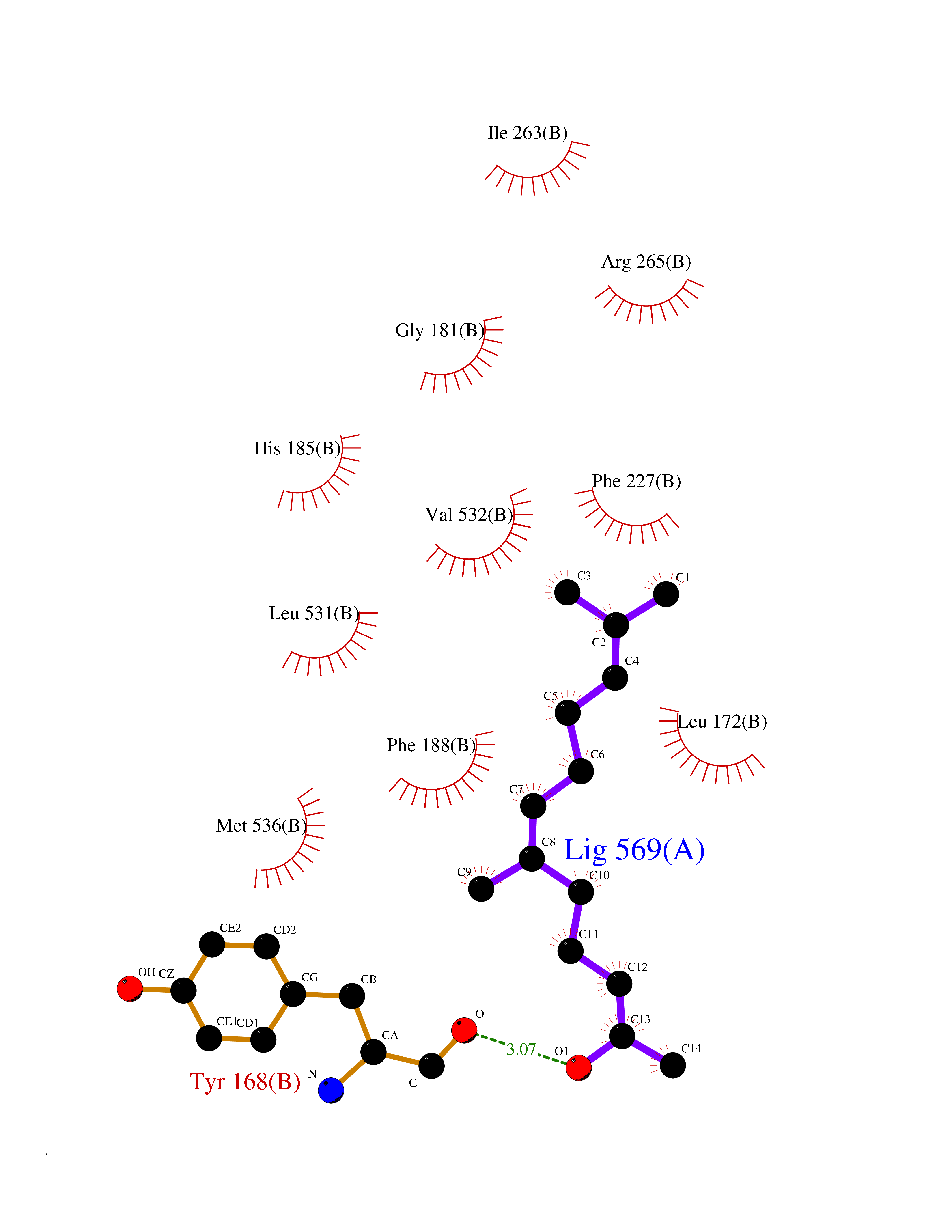 Ligplot Map 3D Binding mode Sequence ADPFESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEKNNFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Lysine-specific histone demethylase 1B (KDM1B) | 4HSU | 6.15 | |
Target general information Gen name KDM1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine-specific histone demethylase 2; LSD2; Flavin-containing amine oxidase domain-containing protein 1; C6orf193; AOF1 Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Required for de novo DNA methylation of a subset of imprinted genes during oogenesis. Acts by oxidizing the substrate by FAD to generate the corresponding imine that is subsequently hydrolyzed. Demethylates both mono- and di-methylated 'Lys-4' of histone H3. Has no effect on tri-methylated 'Lys-4', mono-, di- or tri-methylated 'Lys-9', mono-, di- or tri-methylated 'Lys-27', mono-, di- or tri-methylated 'Lys-36' of histone H3, or on mono-, di- or tri-methylated 'Lys-20' of histone H4. Histone demethylase that demethylates 'Lys-4' of histone H3, a specific tag for epigenetic transcriptional activation, thereby acting as a corepressor. Related diseases Angioedema, hereditary, 1 (HAE1) [MIM:106100]: An autosomal dominant disorder characterized by episodic local swelling involving subcutaneous or submucous tissue of the upper respiratory and gastrointestinal tracts, face, extremities, and genitalia. Hereditary angioedema due to C1 esterase inhibitor deficiency is comprised of two clinically indistinguishable forms. In hereditary angioedema type 1, serum levels of C1 esterase inhibitor are decreased, while in type 2, the levels are normal or elevated, but the protein is non-functional. {ECO:0000269|PubMed:12773530, ECO:0000269|PubMed:1363816, ECO:0000269|PubMed:1451784, ECO:0000269|PubMed:14635117, ECO:0000269|PubMed:16409206, ECO:0000269|PubMed:2118657, ECO:0000269|PubMed:2296585, ECO:0000269|PubMed:22994404, ECO:0000269|PubMed:2365061, ECO:0000269|PubMed:24456027, ECO:0000269|PubMed:3178731, ECO:0000269|PubMed:7814636, ECO:0000269|PubMed:7883978, ECO:0000269|PubMed:8172583, ECO:0000269|PubMed:8529136, ECO:0000269|PubMed:8755917, ECO:0000269|Ref.41}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q96L03 EC number EC 1.-.-.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Developmental protein; FAD; Flavoprotein; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85795.5 Length 763 Aromaticity 0.1 Instability index 37.87 Isoelectric point 8.41 Charge (pH=7) 9.16 2D Binding mode Binding energy (Kcal/mol) -8.39 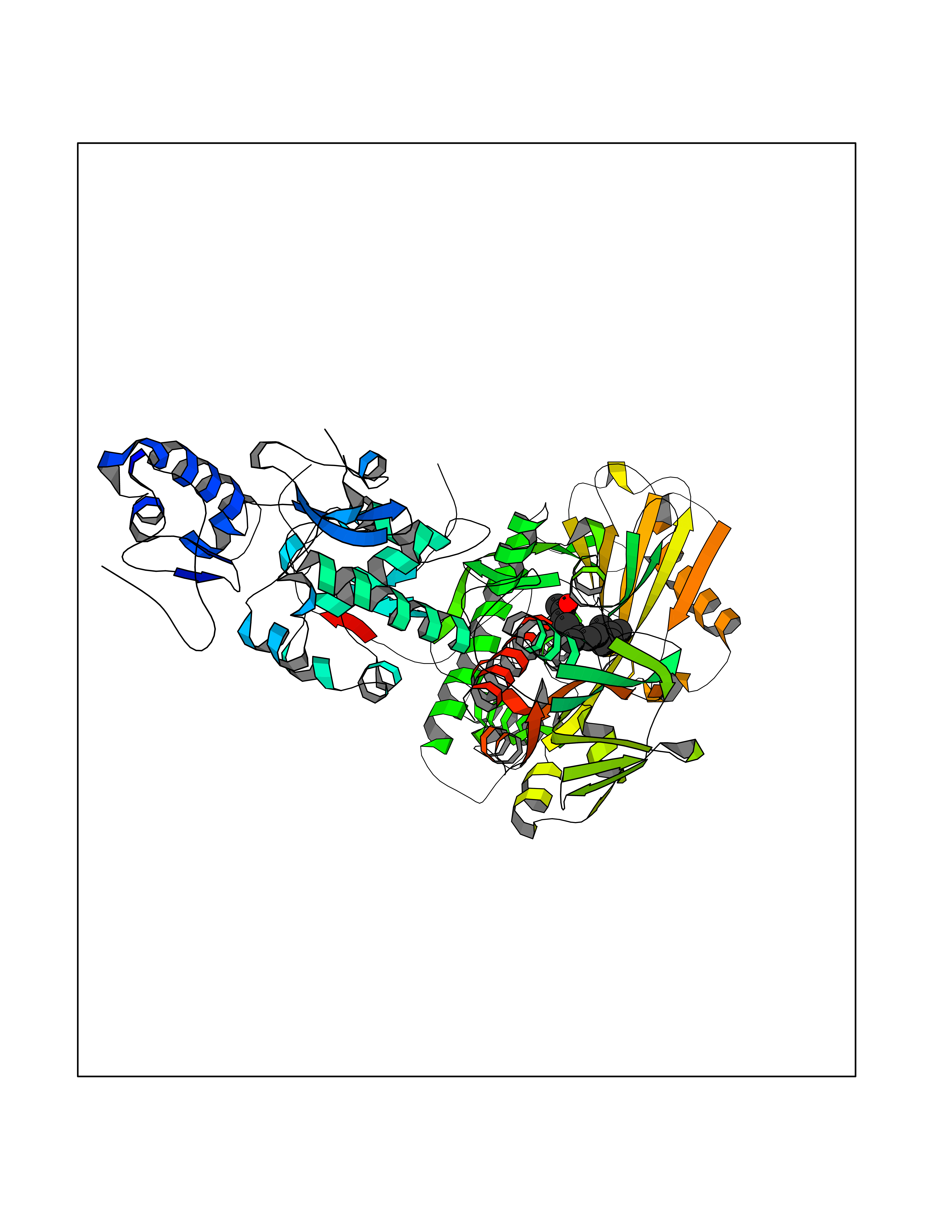 Molscript Map 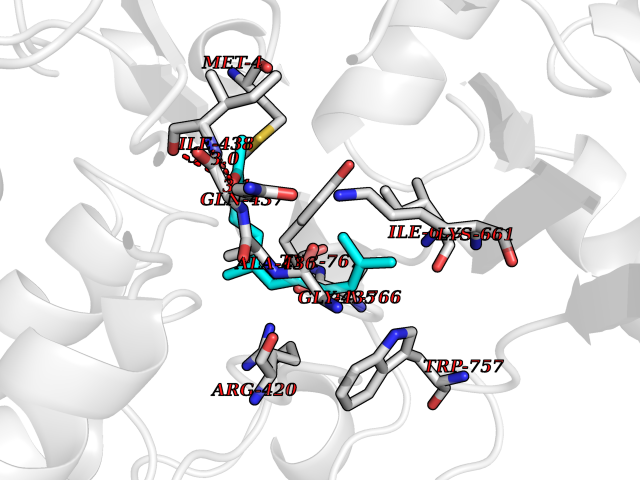 Pymol Map 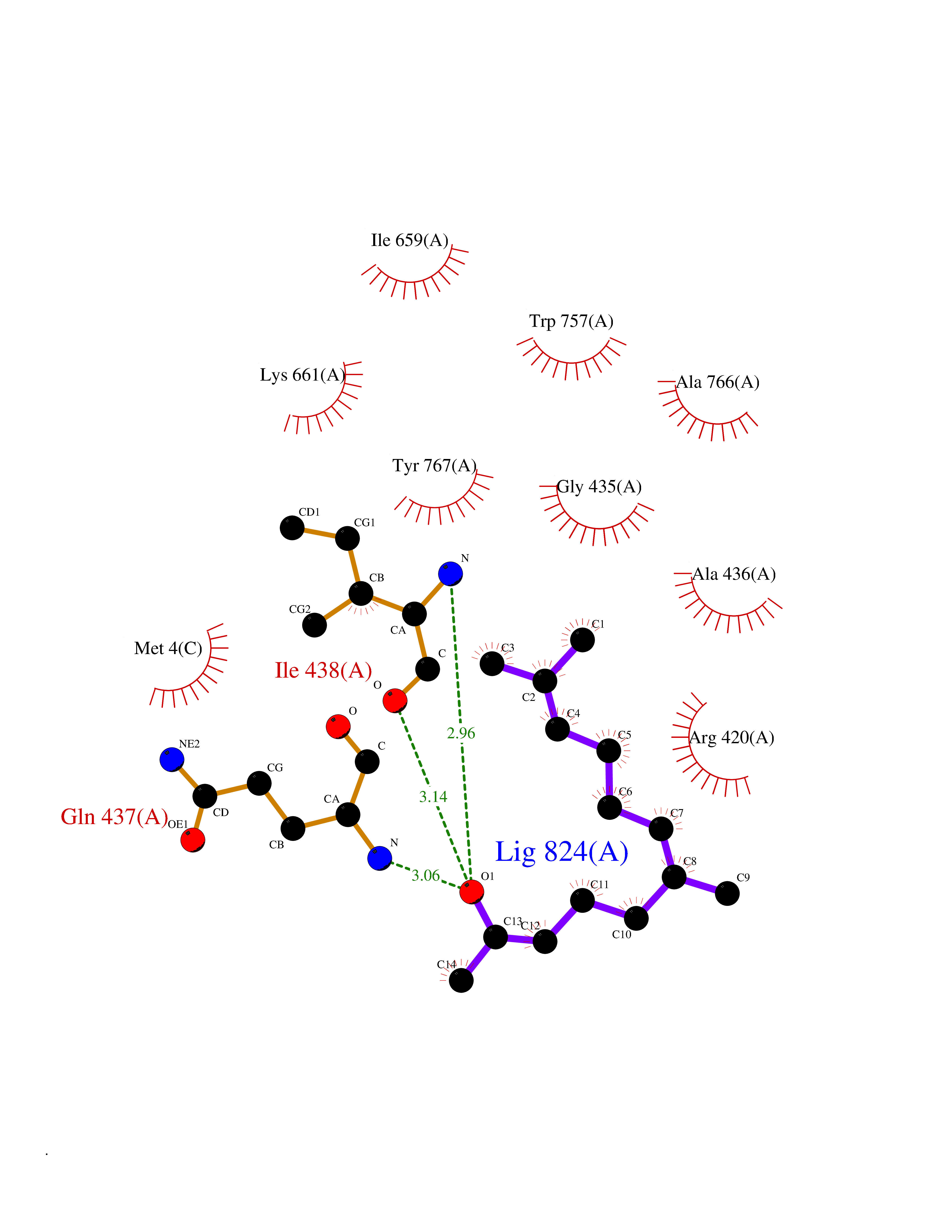 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQLATKAAR Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Acetylcholinesterase (AChE) | 4M0E | 6.14 | |
Target general information Gen name ACHE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms YT; N-ACHE; ARACHE Protein family Type-B carboxylesterase/lipase family Biochemical class Carboxylic ester hydrolase Function Role in neuronal apoptosis. Terminates signal transduction at the neuromuscular junction by rapid hydrolysis of the acetylcholine released into the synaptic cleft. Related diseases Phosphoribosylaminoimidazole carboxylase deficiency (PAICSD) [MIM:619859]: An autosomal recessive inborn error of purine metabolism, clinically characterized by multiple congenital anomalies and early neonatal death. {ECO:0000269|PubMed:31600779}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07846; DB02673; DB04617; DB04614; DB04615; DB07756; DB07701; DB02404; DB03814; DB08615; DB02343; DB02226; DB03005; DB04114; DB03128; DB01122; DB03283; DB00411; DB00122; DB14006; DB01245; DB00944; DB08357; DB08996; DB00449; DB00843; DB01010; DB01364; DB00898; DB00674; DB00483; DB06525; DB04864; DB03348; DB07555; DB00677; DB04924; DB03359; DB00358; DB00940; DB02825; DB02845; DB08167; DB04021; DB00805; DB01805; DB03740; DB04556; DB01400; DB04892; DB00981; DB00733; DB02166; DB00545; DB00863; DB00989; DB00382; DB04616; DB12816; DB01199; DB13503; DB04859 Interacts with Q9Y215; P06733; P63244 EC number EC 3.1.1.7 Uniprot keywords 3D-structure; Alternative splicing; Blood group antigen; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; Neurotransmitter degradation; Nucleus; Proteomics identification; Reference proteome; Secreted; Serine esterase; Signal; Synapse Protein physicochemical properties Chain ID A,B Molecular weight (Da) 58804.1 Length 537 Aromaticity 0.11 Instability index 40.85 Isoelectric point 5.73 Charge (pH=7) -8.18 2D Binding mode Binding energy (Kcal/mol) -8.38 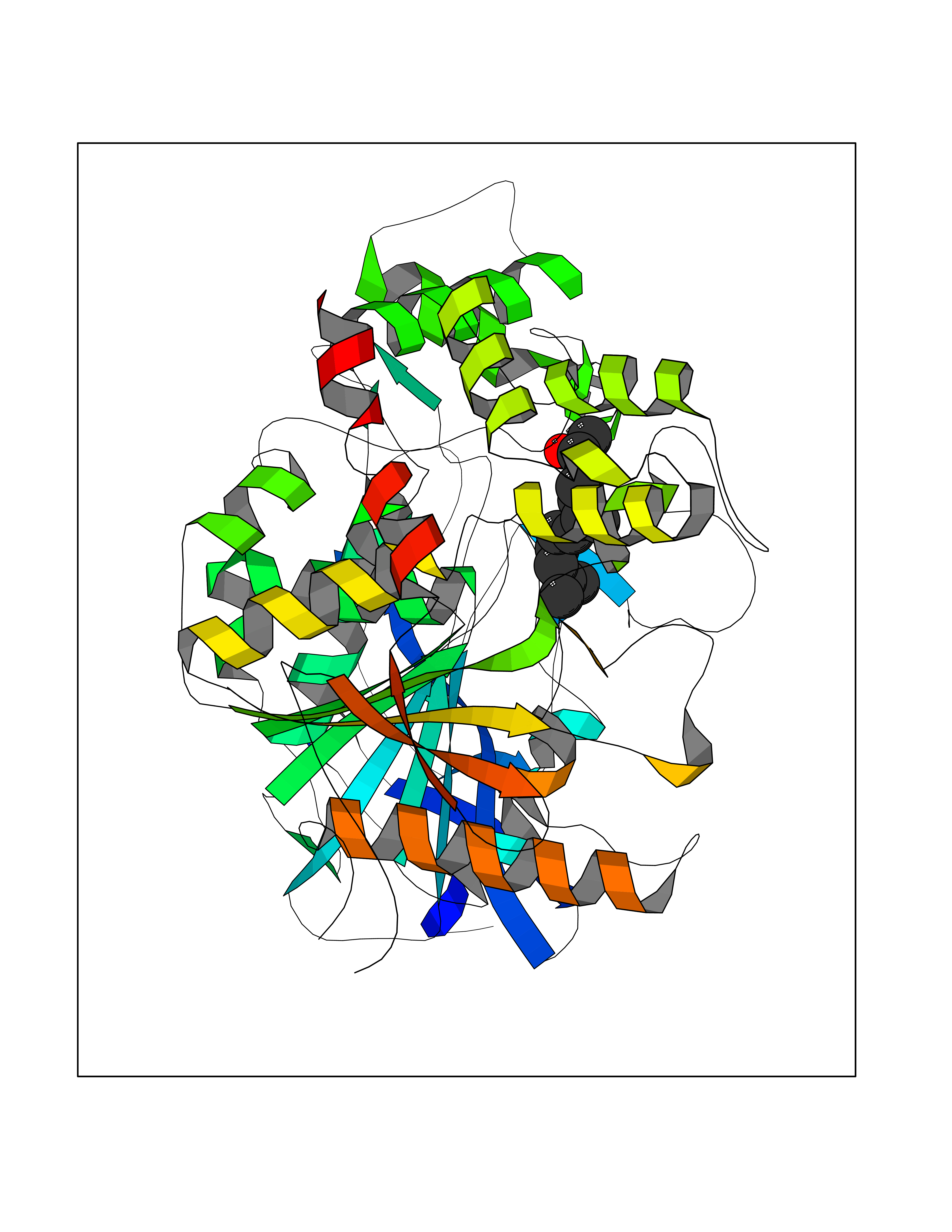 Molscript Map 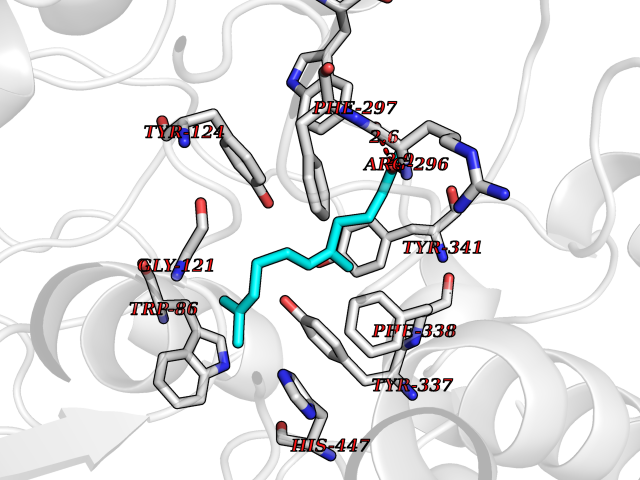 Pymol Map  Ligplot Map 3D Binding mode Sequence EDAELLVTVRGGRLRGIRLKTPGGPVSAFLGIPFAEPPMGPRRFLPPEPKQPWSGVVDATTFQSVCYQYVDTLYPGFEGTEMWNPNRELSEDCLYLNVWTPYPRPTSPTPVLVWIYGGGFYSGASSLDVYDGRFLVQAERTVLVSMNYRVGAFGFLALPGSREAPGNVGLLDQRLALQWVQENVAAFGGDPTSVTLFGESAGAASVGMHLLSPPSRGLFHRAVLQSGAPNGPWATVGMGEARRRATQLAHLVGCPPGGTGGNDTELVACLRTRPAQVLVNHEWHVLPQESVFRFSFVPVVDGDFLSDTPEALINAGDFHGLQVLVGVVKDEGSYFLVYGAPGFSKDNESLISRAEFLAGVRVGVPQVSDLAAEAVVLHYTDWLHPEDPARLREALSDVVGDHNVVCPVAQLAGRLAAQGARVYAYVFEHRASTLSWPLWMGVPHGYEIEFIFGIPLDPSRNYTAEEKIFAQRLMRYWANFARTGDPNEPPKAPQWPPYTAGAQQYVSLDLRPLEVRRGLRAQACAFWNRFLPKLLSA Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Thiopurine S-methyltransferase | 2BZG | 6.14 | |
Target general information Gen name TPMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, TPMT family Biochemical class Transferase Function Thiopurine S-methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00993; DB00436; DB01327; DB01033; DB01250; DB01021 Interacts with Q8TAP4-4; Q15047-2; P61981 EC number 2.1.1.67 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 25971.5 Length 229 Aromaticity 0.12 Instability index 32.58 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -8.37  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence EVQKNQVLTLEEWQDKWVNGKTAFHQEQGHQLLKKHLDTFLKGKSGLRVFFPLCGKAVEXKWFADRGHSVVGVEISELGIQEFFTEQNLSYSEEPITEIPGTKVFKSSSGNISLYCCSIFDLPRTNIGKFDXIWDRGALVAINPGDRKCYADTXFSLLGKKFQYLLCVLSYDPTKHPGPPFYVPHAEIERLFGKICNIRCLEKVDAFEERHKSWGIDCLFEKLYLLTEK Hydrogen bonds contact Hydrophobic contact | ||||