Job Results:
Ligand
Structure
Job ID
6365174e5cc200e6a930804dda23a0ba
Job name
NA
Time
2026-02-27 16:33:59
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | N-acylethanolamine-hydrolyzing acidamidase (NAAA) | 6DXX | 4.42 | |
Target general information Gen name NAAA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nacylsphingosine amidohydrolaselike; Nacylethanolaminehydrolyzing acid amidase subunit beta; NAAA; Acid ceramidaselike protein; ASAHlike protein Protein family Acid ceramidase family Biochemical class Carbon-nitrogen hydrolase Function Degrades bioactive fatty acid amides to their corresponding acids, with the following preference: N- palmitoylethanolamine > N-myristoylethanolamine > N- lauroylethanolamine = N-stearoylethanolamine > N- arachidonoylethanolamine > N-oleoylethanolamine. Also exhibits weak hydrolytic activity against the ceramides N- lauroylsphingosine and N-palmitoylsphingosine. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09061; DB14009; DB14011 Interacts with NA EC number EC 3.5.1.- Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Direct protein sequencing; Disulfide bond; Fatty acid metabolism; Glycoprotein; Hydrolase; Lipid degradation; Lipid metabolism; Lysosome; Membrane; Proteomics identification; Reference proteome; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36877.8 Length 328 Aromaticity 0.11 Instability index 44.37 Isoelectric point 7.72 Charge (pH=7) 1.08 2D Binding mode Binding energy (Kcal/mol) -6.02  Molscript Map 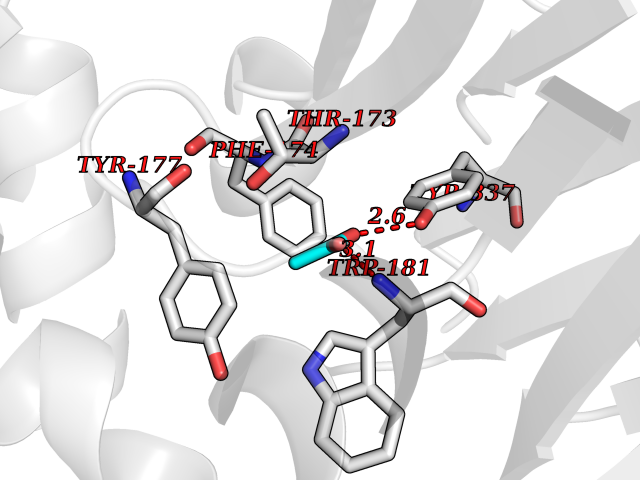 Pymol Map 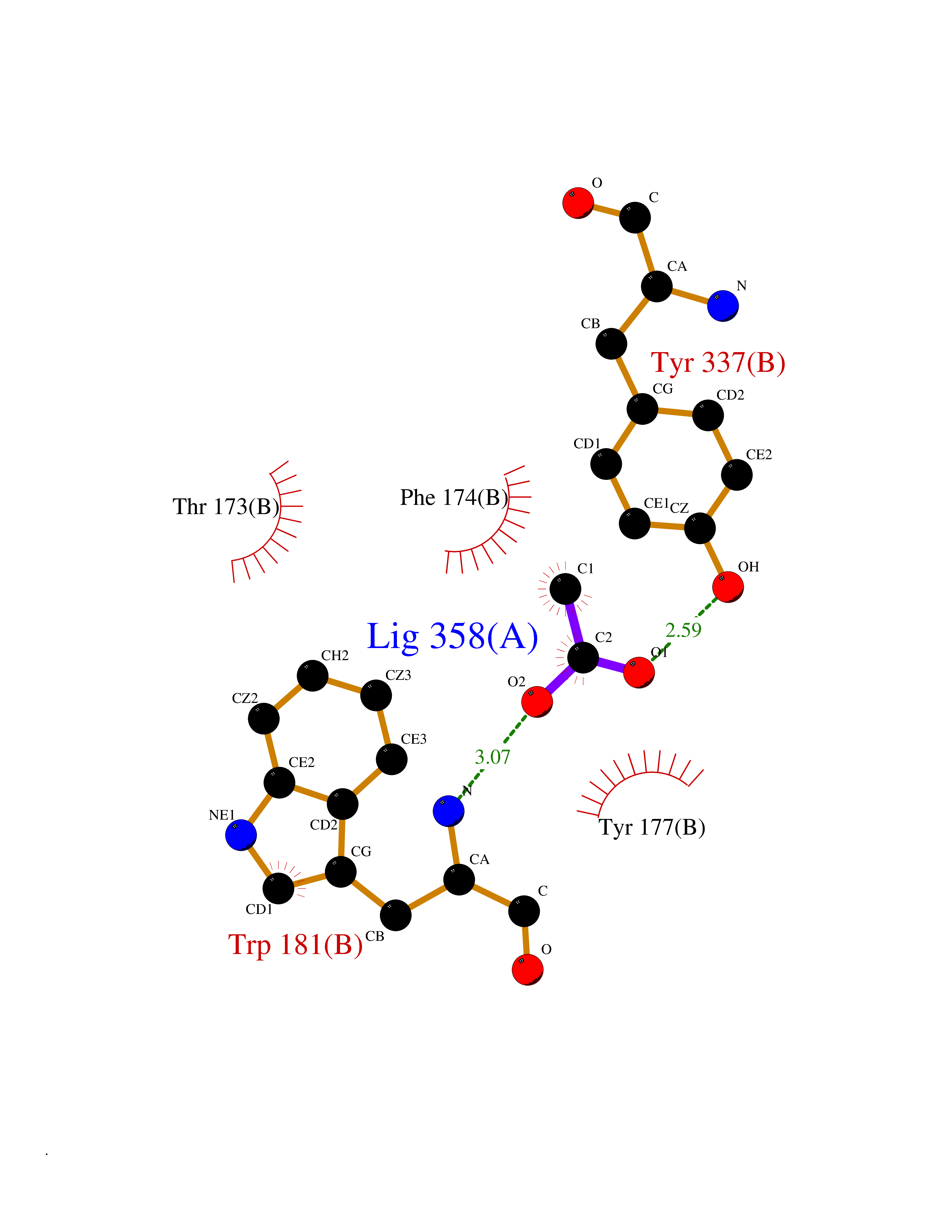 Ligplot Map 3D Binding mode Sequence SPPAAPRFNVSLDSVPELRWLPVLRHYDLDLVRAAMAQVIGDRVPKWVHVLIGKVVLELERFLPQPFTGEIRGMCDFMNLSLADCLLVNLAYESSVFCTSIVAQDSRGHIYHGRNLDYPFGNVLRKLTVDVQFLKNGQIAFTGTTFIGYVGLWTGQSPHKFTVSGDERDKGWWWENAIAALFRRHIPVSWLIRATLSESENFEAAVGKLAKTPLIADVYYIVGGTSPREGVVITRNRDGPADIWPLDPLNGAWFRVETNYDHWKPAPKEDDRRTSAIKALNATGQANLSLEALFQILSVVPVYNNFTIYTTVMSAGSPDKYMTRIRNP Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | C-C chemokine receptor type 2 (CCR2) | 5T1A | 4.42 | |
Target general information Gen name CCR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monocyte chemoattractant protein 1 receptor; MCP-1-R; Chemokine receptor CCR2B; CMKBR2; CD192; CCR-2; CC-CKR-2; C-C CKR-2 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Its binding with CCL2 on monocytes and macrophages mediates chemotaxis and migration induction through the activation of the PI3K cascade, the small G protein Rac and lamellipodium protrusion. Also acts as a receptor for the beta-defensin DEFB106A/DEFB106B. Regulates the expression of T-cell inflammatory cytokines and T-cell differentiation, promoting the differentiation of T-cells into T-helper 17 cells (Th17) during inflammation. Faciltates the export of mature thymocytes by enhancing directional movement of thymocytes to sphingosine-1-phosphate stimulation and up-regulation of S1P1R expression; signals through the JAK-STAT pathway to regulate FOXO1 activity leading to an increased expression of S1P1R. Plays an important role in mediating peripheral nerve injury-induced neuropathic pain. Increases NMDA-mediated synaptic transmission in both dopamine D1 and D2 receptor-containing neurons, which may be caused by MAPK/ERK-dependent phosphorylation of GRIN2B/NMDAR2B. Mediates the recruitment of macrophages and monocytes to the injury site following brain injury. Key functional receptor for CCL2 but can also bind CCL7 and CCL12. Related diseases Polycystic lung disease (PCLUD) [MIM:219600]: An autosomal recessive disease characterized by pulmonary alveolar proteinosis, marked peribronchovascular and parenchymal lymphocytosis, peribronchiolar pulmonary fibrosis, progressive diffuse parenchymal lung cyst formation and enlargement, progressive obstructive airflow limitation, and recurrent secondary infections. Additional features may include digital clubbing, allergies, and atopic dermatitis. {ECO:0000269|PubMed:38157855}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05159; DB11758; DB05130; DB12520 Interacts with Q6S8J3; Q9BW27 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; G-protein coupled receptor; Glycoprotein; Host-virus interaction; Inflammatory response; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Sulfation; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50270.6 Length 445 Aromaticity 0.13 Instability index 29.96 Isoelectric point 9.49 Charge (pH=7) 17.19 2D Binding mode Binding energy (Kcal/mol) -6.02  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence VKQIGAQLLPPLYSLVFIFGFVGNMLVVLILINCKKLKCLTDIYLLNLAISDLLFLITLPLWAHSAANEWVFGNAMCKLFTGLYHIGYFGGIFFIILLTIDRYLAIVHAVFALKARTVTFGVVTSVITWLVAVFASVPGIIFTKXQKEDSVYVCGPYFPRGWNNFHTIMRNILGLVLPLLIMVICYSGISRASKSRINIFEMLRIDEGLRLKIYKDTEGYYTIGIGHLLTKSPSLNAAKSELDKAIGRNTNGVITKDEAEKLFNQDVDAAVRGILRNAKLKPVYDSLDAVRRAALINMVFQMGETGVAGFTNSLRMLQQKRWDEAAVNLAKSRWYNQTPNRAKRVITTFRTGTWDAYPPPSREKKAVRVIFTIMIVYFLFWTPYNIVILLNTFQEFFGLSNCESTSQLDQATQVTETLGMTHCCINPIIYAFVGEKFRRYLSVFF Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Phosphatidylethanolamine-binding protein 1 (PEBP1) | 2QYQ | 4.42 | |
Target general information Gen name PEBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Raf kinase inhibitor protein; RKIP; Prostatic-binding protein; PEBP-1; PEBP; PBP; Neuropolypeptide h3; Hippocampal cholinergic neurostimulating peptide; HCNPpp; HCNP Protein family Phosphatidylethanolamine-binding protein family Biochemical class Phosphatidylethanolamine-binding protein family Function Binds ATP, opioids and phosphatidylethanolamine. Has lower affinity for phosphatidylinositol and phosphatidylcholine. Serine protease inhibitor which inhibits thrombin, neuropsin and chymotrypsin but not trypsin, tissue type plasminogen activator and elastase. Inhibits the kinase activity of RAF1 by inhibiting its activation and by dissociating the RAF1/MEK complex and acting as a competitive inhibitor of MEK phosphorylation. Related diseases Retinitis pigmentosa 49 (RP49) [MIM:613756]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:15570217, ECO:0000269|PubMed:7479749}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09130; DB09568 Interacts with P16050; Q9NRD5; P04049; Q15208; Q9NS68; Q9JLL3 EC number NA Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Direct protein sequencing; Disulfide bond; Lipid-binding; Nucleotide-binding; Phosphoprotein; Protease inhibitor; Proteomics identification; Reference proteome; Serine protease inhibitor Protein physicochemical properties Chain ID A Molecular weight (Da) 20928.3 Length 186 Aromaticity 0.1 Instability index 24.05 Isoelectric point 6.59 Charge (pH=7) -0.98 2D Binding mode Binding energy (Kcal/mol) -6.03  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MPVDLSKWSGPLSLQEVDEQPQHPLHVTYAGAAVDELGKVLTPTQVKNRPTSISWDGLDSGKLYTLVLTDPDAPSRKDPKYREWHHFLVVNMKGNDISSGTVLSDYVGSGPPKGTGLHRYVWLVYEQDRPLKCDEPILSNRSGDHRGKFKVASFRKKYELRAPVAGTCYQAEWDDYVPKLYEQLSG Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Cerebroside-sulfatase (ARSA) | 1E2S | 4.42 | |
Target general information Gen name ARSA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cerebrosidesulfatase; Arylsulfatase A component C; ASA; ARSA Protein family Sulfatase family Biochemical class Sulfuric ester hydrolase Function Hydrolyzes cerebroside sulfate. Related diseases Metachromatic leukodystrophy (MLD) [MIM:250100]: An autosomal recessive disease caused by abnormal intralysosomal accumulation of cerebroside-3-sulfate in central and peripheral nervous systems, as well as other organs. MLD is clinically characterized by leukodystrophy, progressive demyelination and a variety of neurological symptoms, including gait disturbances, ataxias, optical atrophy, dementia, seizures, and spastic tetraparesis. Decreased arylsulfatase A activity is detected in urine, leukocytes, and fibroblasts of affected individuals. Several forms of the disease can be distinguished according to the age at onset and disease severity: late infantile, juvenile and adult forms, partial cerebroside sulfate deficiency, and pseudoarylsulfatase A deficiency. Individuals with pseudoarylsulfatase A deficiency have low arylsulfatase A activity but lack neurological manifestations and are apparently healthy. {ECO:0000269|PubMed:10220151, ECO:0000269|PubMed:10381328, ECO:0000269|PubMed:10477432, ECO:0000269|PubMed:10533072, ECO:0000269|PubMed:10751093, ECO:0000269|PubMed:11020646, ECO:0000269|PubMed:11061266, ECO:0000269|PubMed:11456299, ECO:0000269|PubMed:11941485, ECO:0000269|PubMed:12503099, ECO:0000269|PubMed:12788103, ECO:0000269|PubMed:1353340, ECO:0000269|PubMed:14517960, ECO:0000269|PubMed:14680985, ECO:0000269|PubMed:15026521, ECO:0000269|PubMed:15326627, ECO:0000269|PubMed:15710861, ECO:0000269|PubMed:1670590, ECO:0000269|PubMed:1673291, ECO:0000269|PubMed:1678251, ECO:0000269|PubMed:18693274, ECO:0000269|PubMed:19606494, ECO:0000269|PubMed:20339381, ECO:0000269|PubMed:21265945, ECO:0000269|PubMed:2574462, ECO:0000269|PubMed:7581401, ECO:0000269|PubMed:7825603, ECO:0000269|PubMed:7860068, ECO:0000269|PubMed:7902317, ECO:0000269|PubMed:7906588, ECO:0000269|PubMed:7909527, ECO:0000269|PubMed:8095918, ECO:0000269|PubMed:8101038, ECO:0000269|PubMed:8101083, ECO:0000269|PubMed:8104633, ECO:0000269|PubMed:8891236, ECO:0000269|PubMed:9090526, ECO:0000269|PubMed:9272717, ECO:0000269|PubMed:9452102, ECO:0000269|PubMed:9490297, ECO:0000269|PubMed:9600244, ECO:0000269|PubMed:9819708}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Multiple sulfatase deficiency (MSD) [MIM:272200]: A clinically and biochemically heterogeneous disorder caused by the simultaneous impairment of all sulfatases, due to defective post-translational modification and activation. It combines features of individual sulfatase deficiencies such as metachromatic leukodystrophy, mucopolysaccharidosis, chondrodysplasia punctata, hydrocephalus, ichthyosis, neurologic deterioration and developmental delay. {ECO:0000269|PubMed:15146462}. The protein represented in this entry is involved in disease pathogenesis. Arylsulfatase A activity is impaired in multiple sulfatase deficiency due to mutations in SUMF1 (PubMed:15146462). SUMF1 mutations result in defective post-translational modification of ARSA at residue Cys-69 that is not converted to 3-oxoalanine (PubMed:7628016). {ECO:0000269|PubMed:15146462, ECO:0000269|PubMed:7628016}. Drugs (DrugBank ID) DB03821; DB01800; DB01141; DB04786 Interacts with P50995; Q6P5X5; Q13554-3; O60826; Q96D98; Q9H0I2; Q12951-2; Q16512; P28069; O75360; Q9BQY4; Q15645; O95231 EC number EC 3.1.6.8 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Hydrolase; Ichthyosis; Leukodystrophy; Lipid metabolism; Lysosome; Metachromatic leukodystrophy; Metal-binding; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID P Molecular weight (Da) 51173.7 Length 481 Aromaticity 0.09 Instability index 47.42 Isoelectric point 5.59 Charge (pH=7) -12.84 2D Binding mode Binding energy (Kcal/mol) -6.03  Molscript Map  Pymol Map 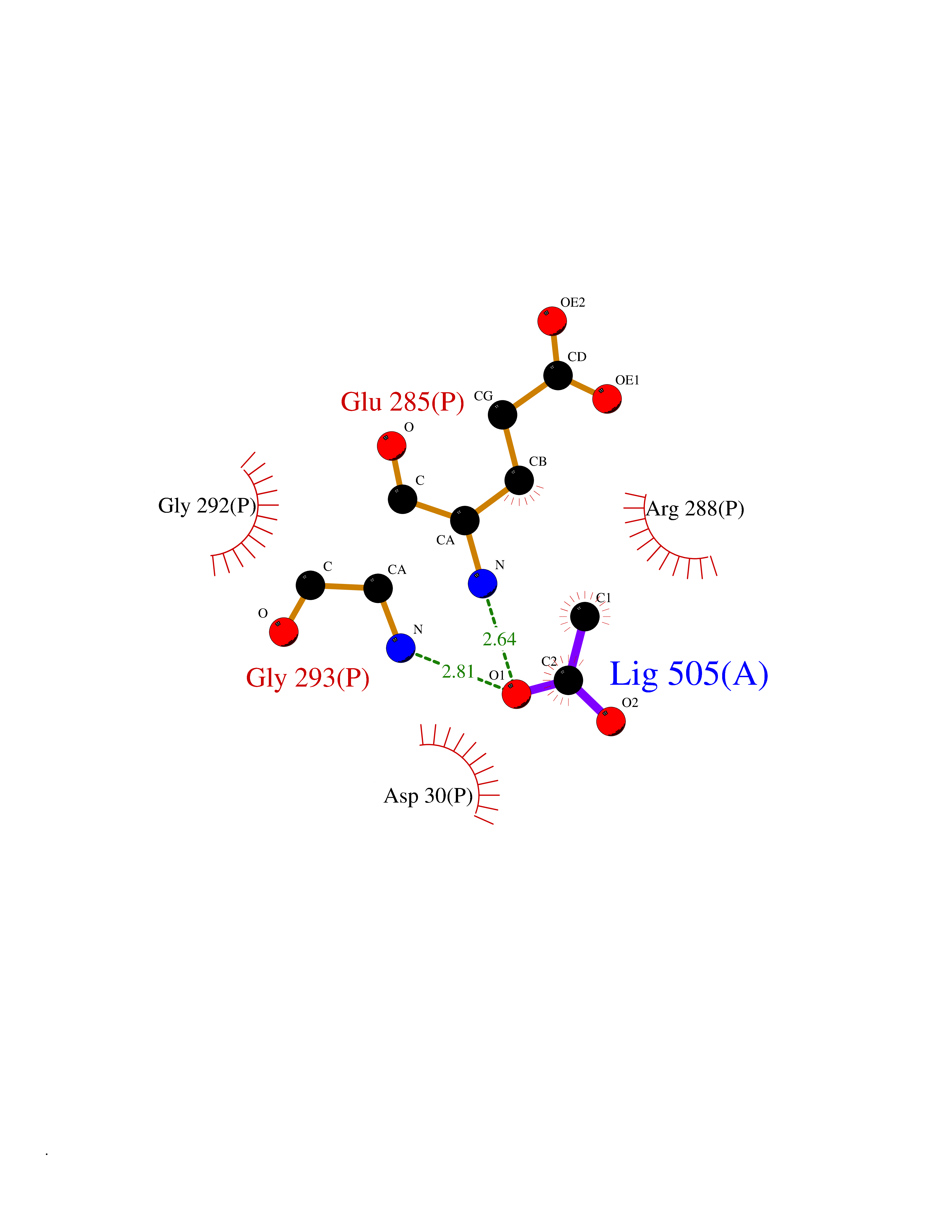 Ligplot Map 3D Binding mode Sequence RPPNIVLIFADDLGYGDLGCYGHPSSTTPNLDQLAAGGLRFTDFYVPVSLATPSRAALLTGRLPVRMGMYPGVLVPSSRGGLPLEEVTVAEVLAARGYLTGMAGKWHLGVGPEGAFLPPHQGFHRFLGIPYSHDQGPCQNLTCFPPATPCDGGCDQGLVPIPLLANLSVEAQPPWLPGLEARYMAFAHDLMADAQRQDRPFFLYYASHHTHYPQFSGQSFAERSGRGPFGDSLMELDAAVGTLMTAIGDLGLLEETLVIFTADNGPETMRMSRGGCSGLLRCGKGTTYEGGVREPALAFWPGHIAPGVTHELASSLDLLPTLAALAGAPLPNVTLDGFDLSPLLLGTGKSPRQSLFFYPSYPDEVRGVFAVRTGKYKAHFFTQGSAHSDTTADPACHASSSLTAHEPPLLYDLSKDPGENYNLLGATPEVLQALKQLQLLKAQLDAAVTFGPSQVARGEDPALQICCHPGCTPRPACCHCP Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Gamma-aminobutyric acid type B receptor subunit 1 | 4MS4 | 4.41 | |
Target general information Gen name GABBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPRC3B;GPR51 Protein family G-protein coupled receptor 3 family, GABA-B receptor subfamily Biochemical class Signaling protein / antagonist Function G-protein coupled GABA receptor activity. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08891; DB08892; DB00181; DB00363; DB02530; DB05010; DB09072 Interacts with Q9UBS5; Q9UBS5-2; P46459; Q86UR5 EC number NA Uniprot keywords 3D-structure; Cell membrane; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Intellectual disability; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 46502.1 Length 408 Aromaticity 0.12 Instability index 50.05 Isoelectric point 5.78 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -6.01  Molscript Map 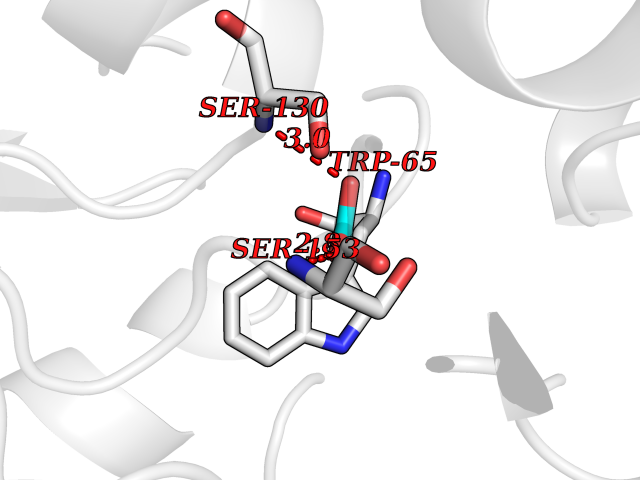 Pymol Map 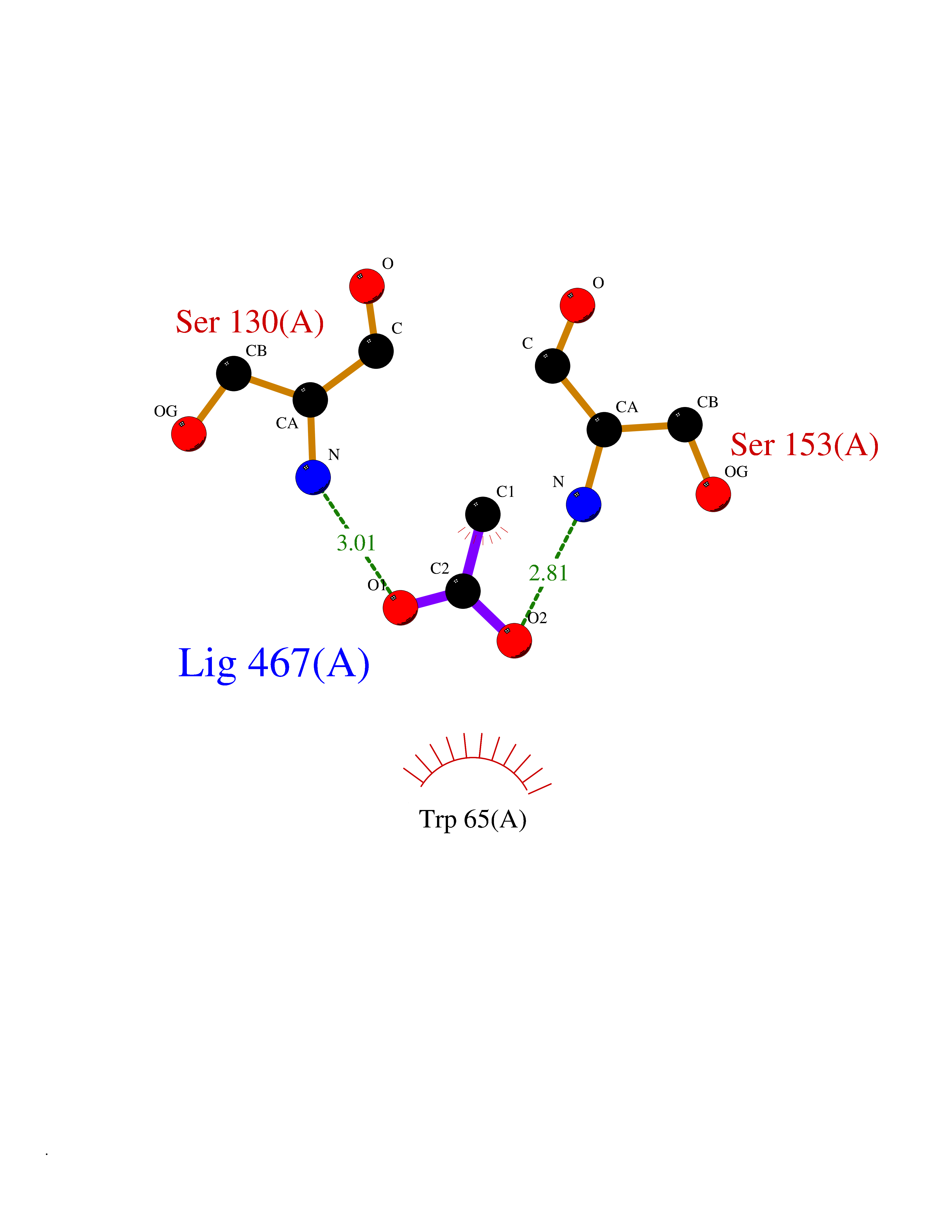 Ligplot Map 3D Binding mode Sequence RRAVYIGALFPMSGGWPGGQACQPAVEMALEDVNSRRDILPDYELKLIHHDSKCDPGQATKYLYELLYNDPIKIILMPGCSSVSTLVAEAARMWNLIVLSYGSSSPALSNRQRFPTFFRTHPSATLHNPTRVKLFEKWGWKKIATIQQTTEVFTSTLDDLEERVKEAGIEITFRQSFFSDPAVPVKNLKRQDARIIVGLFYETEARKVFCEVYKERLFGKKYVWFLIGWYADNWFKIYDPSINCTVDEMTEAVEGHITTEIVMLNPANTRSISNMTSQEFVEKLTKRLKRHPEETGGFQEAPLAYDAIWALALALNKTSRLEDFNYNNQTITDQIYRAMNSSSFEGVSGHVVFDASGSRMAWTLIEQLQGGSYKKIGYYDSTKDDLSWSKTDKWIGGSPPADDYKDDD Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | 30S ribosomal protein S3 | 4ODQ | 4.41 | |
Target general information Gen name rpsC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW3276;b3314 Protein family Universal ribosomal protein uS3 family Biochemical class Isomerase Function RRNA binding.Structural constituent of ribosome. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09093; DB12455; DB00759 Interacts with NA EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Reference proteome; Ribonucleoprotein; Ribosomal protein; RNA-binding; rRNA-binding Protein physicochemical properties Chain ID B Molecular weight (Da) 12457.7 Length 112 Aromaticity 0.05 Instability index 12.49 Isoelectric point 5.91 Charge (pH=7) -7.24 2D Binding mode Binding energy (Kcal/mol) -6.01  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MKVGQDKVVTIRYTLQVEGEVLDQGELSYLHGHRNLIPGLEEALEGREEGEAFQAHVPAEKAYGATGHPPHATLDFQVEVVKVREATPEELLHGHAHPSGHHHHHHGIVKPW Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Dopamine D2 receptor (D2R) | 5AER | 4.41 | |
Target general information Gen name DRD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dopamine receptor 2; D(2) dopamine receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Dopamine receptor whose activity is mediated by G proteins which inhibit adenylyl cyclase. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01614; DB01063; DB01425; DB00915; DB06288; DB05964; DB00543; DB00182; DB04599; DB00714; DB01238; DB14185; DB09207; DB06216; DB04889; DB04888; DB05687; DB09223; DB04857; DB09128; DB01200; DB09018; DB00490; DB00248; DB06016; DB01038; DB00477; DB01239; DB00568; DB00363; DB01151; DB11274; DB13345; DB00320; DB01184; DB00988; DB00450; DB11275; DB01049; DB00696; DB01175; DB09194; DB00875; DB00623; DB04842; DB00502; DB04946; DB00458; DB04924; DB12579; DB01221; DB00555; DB01235; DB00589; DB00408; DB06077; DB08815; DB00934; DB09224; DB01043; DB00933; DB01403; DB01233; DB06148; DB00805; DB01618; DB08804; DB05766; DB00540; DB06229; DB00334; DB01267; DB12061; DB00715; DB01186; DB08922; DB00850; DB01100; DB09286; DB01621; DB12478; DB00413; DB00433; DB00420; DB01069; DB00777; DB01224; DB09097; DB12518; DB00409; DB00734; DB01549; DB00268; DB05271; DB06454; DB06144; DB00391; DB06477; DB04844; DB12093; DB00372; DB01622; DB00679; DB01623; DB13025; DB00831; DB00508; DB00726; DB06109; DB01392; DB00246; DB09225; DB01624 Interacts with Q9NRI5; P14416; Q01959 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Lipoprotein; Membrane; Palmitate; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID B,C Molecular weight (Da) 24300.3 Length 209 Aromaticity 0.13 Instability index 40.14 Isoelectric point 4.97 Charge (pH=7) -7.83 2D Binding mode Binding energy (Kcal/mol) -6.01  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PEVVEELTRKTYFTEKEVQQWYKGFIKDCPSGQLDAAGFQKIYKQFFPFGDPTKFATFVFNVFDENKDGRIEFSEFIQALSVTSRGTLDEKLRWAFKLYDLDNDGYITRNEMLDIVDAIYQMVGNTVELPEEENTPEKRVDRIFAMMDKNADGKLTLQEFQEGSKADPSIVQALSLYDGLVNIEFRKAFLKILHSNIEFRKAFLKILHS Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Deoxycytidine kinase (DCK) | 1P5Z | 4.41 | |
Target general information Gen name DCK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms dCK Protein family DCK/DGK family Biochemical class Kinase Function Required for the phosphorylation of the deoxyribonucleosides deoxycytidine (dC), deoxyguanosine (dG) and deoxyadenosine (dA). Has broad substrate specificity, and does not display selectivity based on the chirality of the substrate. It is also an essential enzyme for the phosphorylation of numerous nucleoside analogs widely employed as antiviral and chemotherapeutic agents. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB02594; DB00242; DB00631; DB00987; DB01262; DB05494; DB00879; DB01073; DB00441; DB00709; DB01280; DB00642; DB04961; DB00943 Interacts with Q16854 EC number EC 2.7.1.74 Uniprot keywords 3D-structure; ATP-binding; Direct protein sequencing; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 27128.5 Length 229 Aromaticity 0.13 Instability index 52.5 Isoelectric point 5.26 Charge (pH=7) -7.8 2D Binding mode Binding energy (Kcal/mol) -6.02  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RIKKISIEGNIAAGKSTFVNILKQLCEDWEVVPEPVARWCNVQSTNGGNVLQMMYEKPERWSFTFQTYACLSRIRAQLASLNGKLKDAEKPVLFFERSVYSDRYIFASNLYESECMNETEWTIYQDWHDWMNNQFGQSLELDGIIYLQATPETCLHRIYLRGRNEEQGIPLEYLEKLHYKHESWLLHRTLKTNFDYLQEVPILTLDVNEDFKDKYESLVEKVKEFLSTL Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Epithelial discoidin domain receptor 1 (DDR1) | 4BKJ | 4.41 | |
Target general information Gen name DDR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase CAK; Tyrosine kinase DDR; TRKE; TRK E; RTK6; Protein-tyrosine kinase RTK-6; Protein-tyrosine kinase 3A; PTK3A; NTRK4; NEP; Mammary carcinoma kinase 10; MCK-10; HGK2; Epithelial Protein family Protein kinase superfamily, Tyr protein kinase family, Insulin receptor subfamily Biochemical class Kinase Function Collagen binding triggers a signaling pathway that involves SRC and leads to the activation of MAP kinases. Regulates remodeling of the extracellular matrix by up-regulation of the matrix metalloproteinases MMP2, MMP7 and MMP9, and thereby facilitates cell migration and wound healing. Required for normal blastocyst implantation during pregnancy, for normal mammary gland differentiation and normal lactation. Required for normal ear morphology and normal hearing. Promotes smooth muscle cell migration, and thereby contributes to arterial wound healing. Also plays a role in tumor cell invasion. Phosphorylates PTPN11. Tyrosine kinase that functions as cell surface receptor for fibrillar collagen and regulates cell attachment to the extracellular matrix, remodeling of the extracellular matrix, cell migration, differentiation, survival and cell proliferation. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010; DB00619; DB15822 Interacts with Q16832; O43639; Q06124; Q9UHD9 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Calcium; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; Kinase; Lactation; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Pregnancy; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34061.1 Length 297 Aromaticity 0.1 Instability index 42.8 Isoelectric point 6.32 Charge (pH=7) -2.01 2D Binding mode Binding energy (Kcal/mol) -6.01  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MPRVDFPRSRLRFKEKLGEGQFGEVHLCEVDSPQDLVSLDFPLNVRKGHPLLVAVKILRPDATKNARNDFLKEVKIMSRLKDPNIIRLLGVCVQDDPLCMITDYMENGDLNQFLSAHQLEDKGPTISYPMLLHVAAQIASGMRYLATLNFVHRDLATRNCLVGENFTIKIADFGMSRNLYAGDYYRAVLPIRWMAWECILMGKFTTASDVWAFGVTLWEVLMLCRAQPFGQLTDEQVIENAGEFFRDQGRQVYLSRPPACPQGLYELMLRCWSRESEQRPPFSQLHRFLAEDALNTV Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Nischarin | 3P0C | 4.41 | |
Target general information Gen name NISCH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA0975;IRAS Protein family NA Biochemical class Signaling protein Function Identical protein binding.Integrin binding.Phosphatidylinositol binding. Related diseases Dyskinesia, limb and orofacial, infantile-onset (IOLOD) [MIM:616921]: An autosomal recessive, early-onset hyperkinetic movement disorder characterized by axial hypotonia, dyskinesia of the limbs and trunk, orofacial dyskinesia, drooling, and dysarthria. The severity of the hyperkinesis is variable. {ECO:0000269|PubMed:27058446}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Striatal degeneration, autosomal dominant 2 (ADSD2) [MIM:616922]: An autosomal dominant disorder characterized by striatal degeneration and dysfunction of basal ganglia, resulting in hyperkinesis. {ECO:0000269|PubMed:27058447}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08838; DB09242; DB15133; DB00697 Interacts with Q9Y2I1; P51151; P61107; P05714 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; Cell membrane; Coiled coil; Cytoplasm; Endosome; Leucine-rich repeat; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25709.3 Length 219 Aromaticity 0.11 Instability index 13.52 Isoelectric point 8.57 Charge (pH=7) 3.01 2D Binding mode Binding energy (Kcal/mol) -6.02 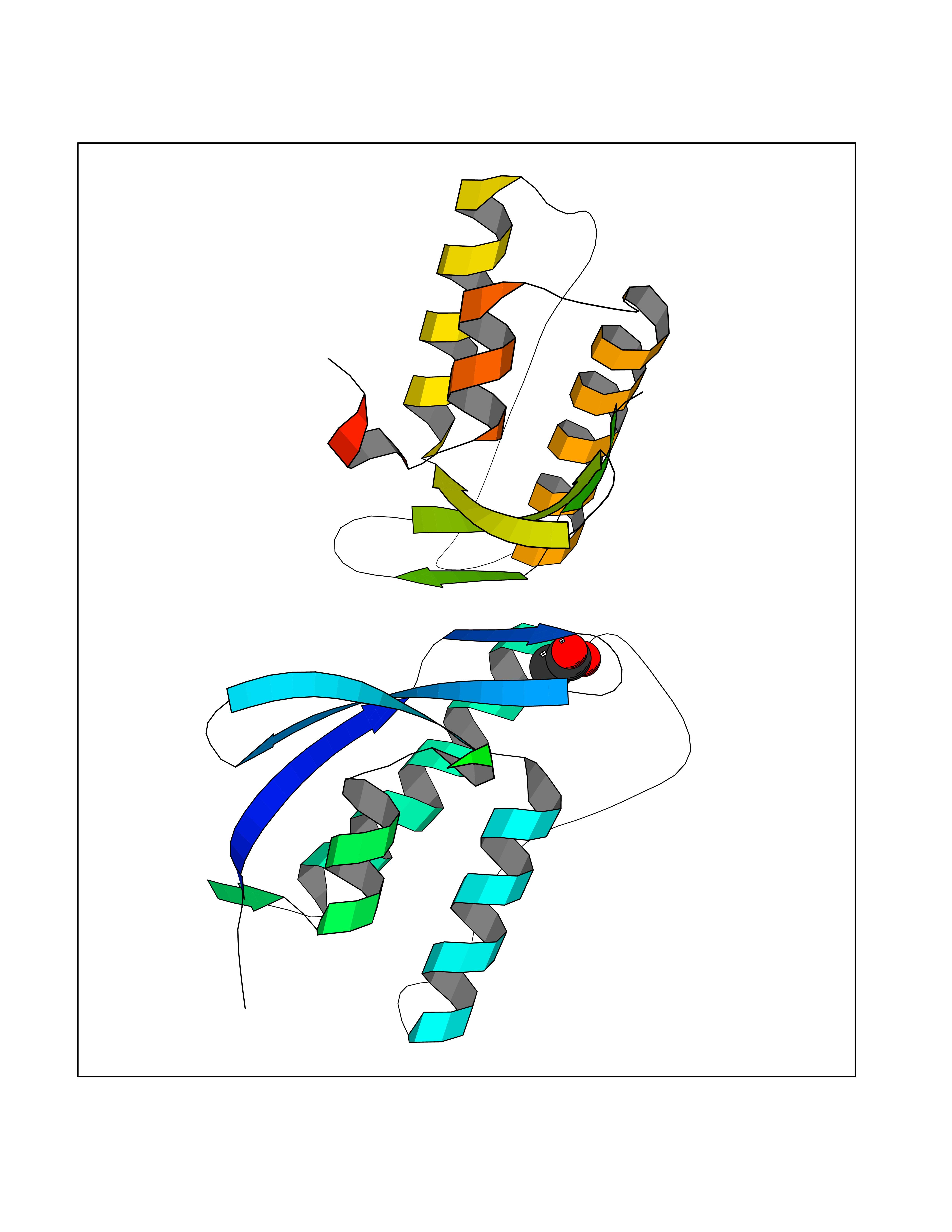 Molscript Map 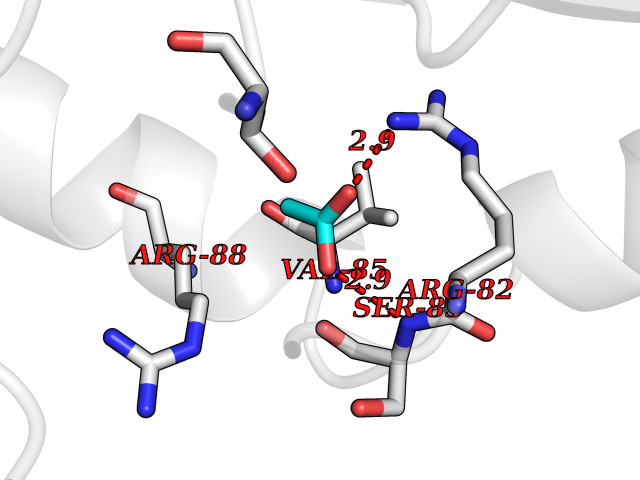 Pymol Map 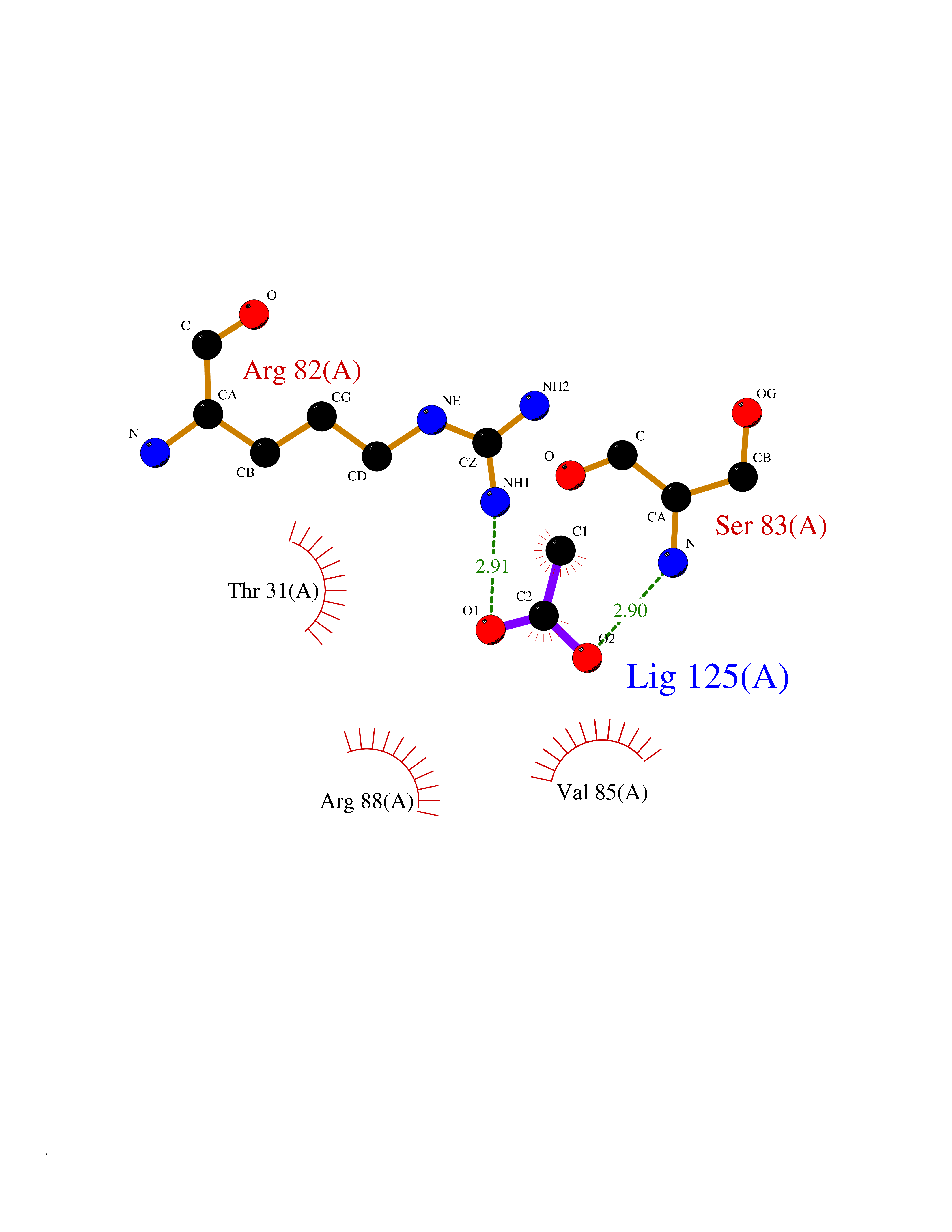 Ligplot Map 3D Binding mode Sequence NLYFQSMEARVVGSELVDTYTVYIIQVTDGSHEWTVKHRYSDFHDLHEKLVAERKIDKNLLPPKKIIGKNSRSLVEKREKDLEVYLQKLLAAFPGVTPRVLAHFLHFHFYESMEARVVGSELVDTYTVYIIQVTDGSHEWTVKHRYSDFHDLHEKLVAERKIDKNLLPPKKIIGKNSRSLVEKREKDLEVYLQKLLAAFPGVTPRVLAHFLHFHFYEIN Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Orotidine 5'-monophosphate decarboxylase (UMPS) | 3MI2 | 4.41 | |
Target general information Gen name UMPS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Uridine 5'-monophosphate synthase; UMP synthase Protein family Purine/pyrimidine phosphoribosyltransferase family; OMP decarboxylase family Biochemical class Pentosyltransferase Function Catalyses the formation of uridine monophosphate (UMP), an energy-carrying molecule in many important biosynthetic pathways. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02890; DB00544 Interacts with P54764; P11172-1 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Decarboxylase; Disease variant; Glycosyltransferase; Lyase; Multifunctional enzyme; Phosphoprotein; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 55844 Length 514 Aromaticity 0.06 Instability index 22.7 Isoelectric point 6.44 Charge (pH=7) -2.99 2D Binding mode Binding energy (Kcal/mol) -6.02 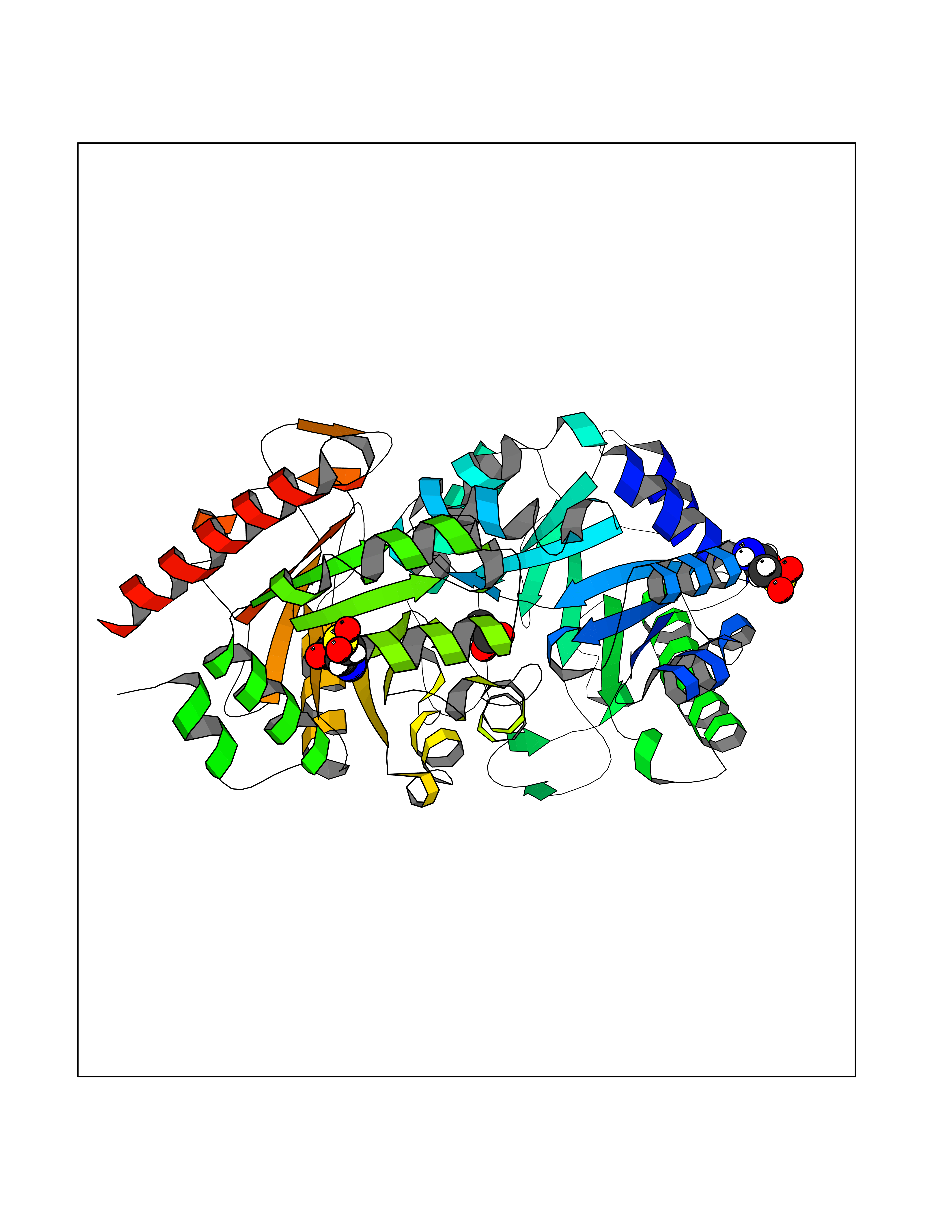 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KELSFGARAELPRIHPVASKLLRLMQKKETNLCLSADVSLARELLQLADALGPSICMLKTHVDILNDFTLDVMKELITLAKXHEFLIFEDRKFADIGNTVKKQYEGGIFKIASWADLVNAHVVPGSGVVKGLQEVGLPLHRGCLLIAEMSSTGSLATGDYTRAAVRMAEEHSEFVVGFISGSRVSMKPEFLHLTPGVQLEAGGDNLGQQYNSPQEVIGKRGSDIIIVGRGIISAADRLEAAEMYRKAAWEAYLSRLGKELSFGARAELPRIHPVASKLLRLMQKKETNLCLSADVSLARELLQLADALGPSICMLKTHVDILNDFTLDVMKELITLAKXHEFLIFEDRKFADIGNTVKKQYEGGIFKIASWADLVNAHVVPGSGVVKGLQEVGLPLHRGCLLIAEMSSTGSLATGDYTRAAVRMAEEHSEFVVGFISGSRVSMKPEFLHLTPGVQLEAGGDNLGQQYNSPQEVIGKRGSDIIIVGRGIISAADRLEAAEMYRKAAWEAYLSRLG Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Dopamine D2 receptor (D2R) | 5AER | 4.40 | |
Target general information Gen name DRD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dopamine receptor 2; D(2) dopamine receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Dopamine receptor whose activity is mediated by G proteins which inhibit adenylyl cyclase. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01614; DB01063; DB01425; DB00915; DB06288; DB05964; DB00543; DB00182; DB04599; DB00714; DB01238; DB14185; DB09207; DB06216; DB04889; DB04888; DB05687; DB09223; DB04857; DB09128; DB01200; DB09018; DB00490; DB00248; DB06016; DB01038; DB00477; DB01239; DB00568; DB00363; DB01151; DB11274; DB13345; DB00320; DB01184; DB00988; DB00450; DB11275; DB01049; DB00696; DB01175; DB09194; DB00875; DB00623; DB04842; DB00502; DB04946; DB00458; DB04924; DB12579; DB01221; DB00555; DB01235; DB00589; DB00408; DB06077; DB08815; DB00934; DB09224; DB01043; DB00933; DB01403; DB01233; DB06148; DB00805; DB01618; DB08804; DB05766; DB00540; DB06229; DB00334; DB01267; DB12061; DB00715; DB01186; DB08922; DB00850; DB01100; DB09286; DB01621; DB12478; DB00413; DB00433; DB00420; DB01069; DB00777; DB01224; DB09097; DB12518; DB00409; DB00734; DB01549; DB00268; DB05271; DB06454; DB06144; DB00391; DB06477; DB04844; DB12093; DB00372; DB01622; DB00679; DB01623; DB13025; DB00831; DB00508; DB00726; DB06109; DB01392; DB00246; DB09225; DB01624 Interacts with Q9NRI5; P14416; Q01959 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Lipoprotein; Membrane; Palmitate; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID B,C Molecular weight (Da) 24300.3 Length 209 Aromaticity 0.13 Instability index 40.14 Isoelectric point 4.97 Charge (pH=7) -7.83 2D Binding mode Binding energy (Kcal/mol) -6  Molscript Map  Pymol Map 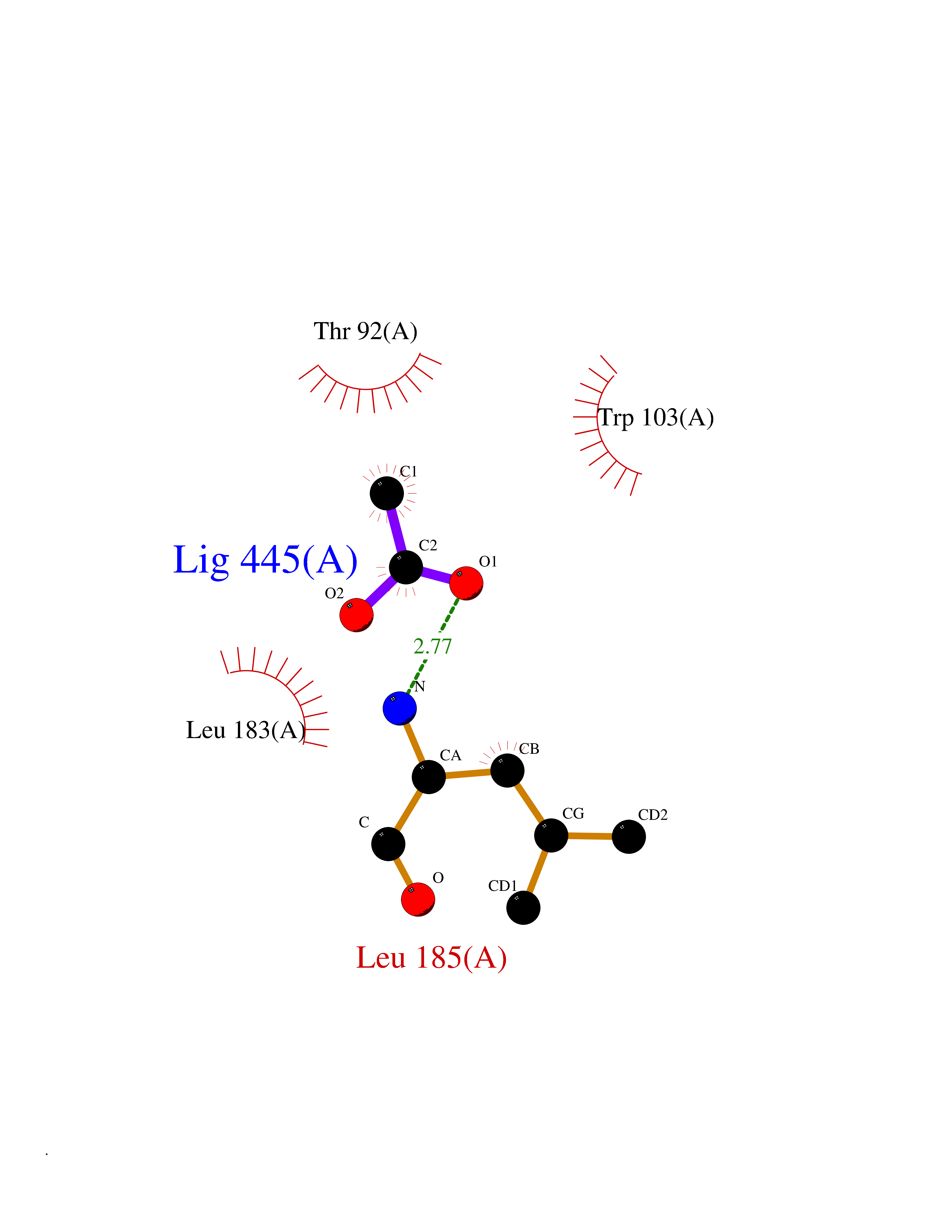 Ligplot Map 3D Binding mode Sequence PEVVEELTRKTYFTEKEVQQWYKGFIKDCPSGQLDAAGFQKIYKQFFPFGDPTKFATFVFNVFDENKDGRIEFSEFIQALSVTSRGTLDEKLRWAFKLYDLDNDGYITRNEMLDIVDAIYQMVGNTVELPEEENTPEKRVDRIFAMMDKNADGKLTLQEFQEGSKADPSIVQALSLYDGLVNIEFRKAFLKILHSNIEFRKAFLKILHS Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | DNA polymerase catalytic subunit | 1YYP | 4.40 | |
Target general information Gen name UL54 Organism Human cytomegalovirus (strain AD169) (HHV-5) (Human herpesvirus 5) Uniprot ID TTD ID NA Synonyms HFLF2 Protein family DNA polymerase type-B family Biochemical class Replication / transferase Function 3'-5' exonuclease activity.DNA binding.DNA-directed DNA polymerase activity.Nucleotide binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00369; DB00529 Interacts with NA EC number 2.7.7.7 Uniprot keywords 3D-structure; DNA replication; DNA-binding; DNA-directed DNA polymerase; Host nucleus; Nucleotidyltransferase; Reference proteome; Transferase; Viral DNA replication Protein physicochemical properties Chain ID B Molecular weight (Da) 30127.5 Length 269 Aromaticity 0.09 Instability index 36.39 Isoelectric point 8.41 Charge (pH=7) 3.48 2D Binding mode Binding energy (Kcal/mol) -6  Molscript Map  Pymol Map 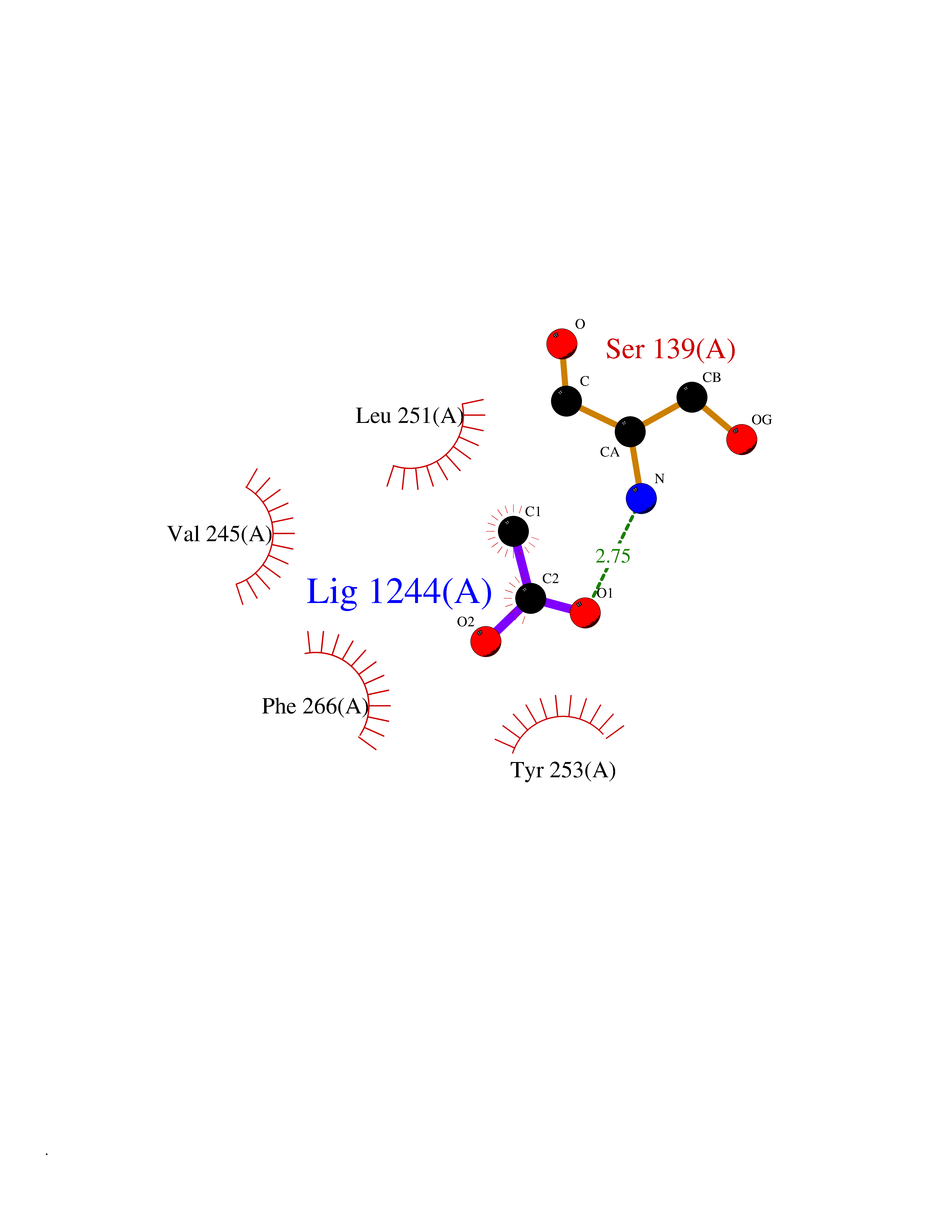 Ligplot Map 3D Binding mode Sequence EPPTLALRLKPYKTAIQQLRSVIRALKENTTVTFLPTPSLILQTVRSHCVSKITFNSSCLYITDKSFQPKTINNSTPLLGNFMYLTSSKDLTKFYVQDISDLSAKISMCAPDFNMEFSSACVHGQDIVRESENSAVHVDLDFGVVADLLKWIGPTGTVQILVHAGPPAIKFILTNGSELEFTSNNRVSFHGVKNMRINVQLKNFYQTLLNCAVTKLPCTLRIVTEHDTLLYVASRNGLFAVENFLTEEPRRLHLEPAFLPYSVKAHECC Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Ornithine decarboxylase (ODC1) | 2OO0 | 4.40 | |
Target general information Gen name ODC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ODC Protein family Orn/Lys/Arg decarboxylase class-II family Biochemical class Carbon-carbon lyase Function Polyamines are essential for cell proliferation and are implicated in cellular processes, ranging from DNA replication to apoptosis. Catalyzes the first and rate-limiting step of polyamine biosynthesis that converts ornithine into putrescine, which is the precursor for the polyamines, spermidine and spermine. Related diseases Bachmann-Bupp syndrome (BABS) [MIM:619075]: An autosomal dominant disorder characterized by global developmental delay, alopecia, absolute or relative macrocephaly, and facial dysmorphism. Neuroimaging shows white matter abnormalities, prominent Virchow-Robin spaces, periventricular cysts, and abnormalities of the corpus callosum. {ECO:0000269|PubMed:30239107, ECO:0000269|PubMed:30475435}. The disease is caused by variants affecting the gene represented in this entry. BABS is due to truncating variants that lead to a gain of function. This phenomenon apparently results from truncation proximal to or involving the C-terminal region of ODC1 protein, distal enough to allow escape from nonsense-mediated decay. A gain of function is corroborated by elevated plasma levels of N-acetylputrescine, with otherwise normal polyamine levels, in affected individuals. {ECO:0000269|PubMed:30475435}. Drugs (DrugBank ID) DB06243; DB04263; DB03856; DB04083; DB02824; DB01917; DB00114; DB02209; DB00203; DB00127; DB00313 Interacts with Q9H8Y8; Q92993; Q9UMX2; Q9UMX2-2 EC number EC 4.1.1.17 Uniprot keywords 3D-structure; Decarboxylase; Disease variant; Hypotrichosis; Lyase; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 45682.9 Length 410 Aromaticity 0.11 Instability index 40.93 Isoelectric point 5.61 Charge (pH=7) -6.68 2D Binding mode Binding energy (Kcal/mol) -6  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LMNNFGNEEFDCHFLDEGFTAKDILDQKINEVSSSDDKDAFYVADLGDILKKHLRWLKALPRVTPFYAVKCNDSKAIVKTLAATGTGFDCASKTEIQLVQSLGVPPERIIYANPCKQVSQIKYAANNGVQMMTFDSEVELMKVARAHPKAKLVLRIATDDSKAVCRLSVKFGATLRTSRLLLERAKELNIDVVGVSFHVGSGCTDPETFVQAISDARCVFDMGAEVGFSMYLLDIGGGFPGSEDVKLKFEEITGVINPALDKYFPSDSGVRIIAEPGRYYVASAFTLAVNIIAKKIVLEQTFMYYVNDGVYGSFNCILYDHAHVKPLLQKRPKPDEKYYSSSIWGPTCDGLDRIVERCDLPEMHVGDWMLFENMGAYTVAAASTFNGFQRPTIYYVMSGPAWQLMQQFQN Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Pyruvate dehydrogenase [ubiquinone] | 3EYA | 4.40 | |
Target general information Gen name poxB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b0871;JW0855 Protein family TPP enzyme family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Identical protein binding.Lipid binding.Magnesium ion binding.Pyruvate dehydrogenase (quinone) activity.Thiamine pyrophosphate binding. Related diseases Glycogen storage disease 6 (GSD6) [MIM:232700]: A metabolic disorder characterized by mild to moderate hypoglycemia, mild ketosis, growth retardation, and prominent hepatomegaly. Heart and skeletal muscle are not affected. {ECO:0000269|PubMed:9529348}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07003 EC number 1.2.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Lipid-binding; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Oxidoreductase; Pyruvate; Reference proteome; Thiamine pyrophosphate; Ubiquinone Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I,J,K,L Molecular weight (Da) 113027 Length 1046 Aromaticity 0.07 Instability index 35.99 Isoelectric point 5.75 Charge (pH=7) -24.38 2D Binding mode Binding energy (Kcal/mol) -6  Molscript Map  Pymol Map 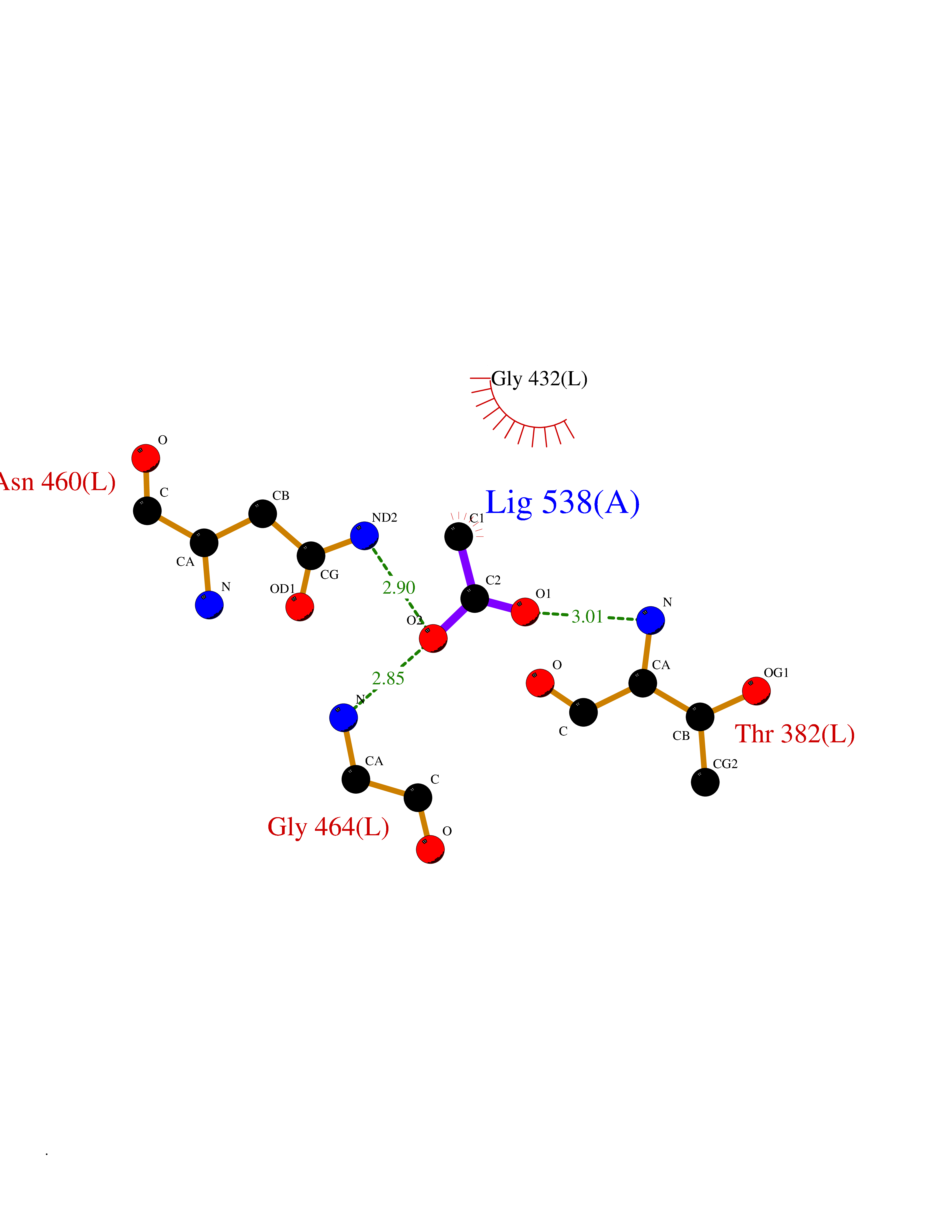 Ligplot Map 3D Binding mode Sequence MKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFDGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIPMKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFVGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIP Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Cyclin-dependent kinase 4 | 2W96 | 4.40 | |
Target general information Gen name CDK4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Cell cycle Function ATP binding.Cyclin binding.Cyclin-dependent protein serine/threonine kinase activity.Cyclin-dependent protein serine/threonine kinase regulator activity.Protein complex binding. Related diseases Melanoma, cutaneous malignant 3 (CMM3) [MIM:609048]: A malignant neoplasm of melanocytes, arising de novo or from a pre-existing benign nevus, which occurs most often in the skin but may also involve other sites. {ECO:0000269|PubMed:7652577, ECO:0000269|PubMed:8528263, ECO:0000269|PubMed:9311594, ECO:0000269|PubMed:9425228}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12001; DB03496; DB12010; DB09073; DB02733; DB11730; DB15442 Interacts with Q9UH17; P24385; P30279; P30281; Q16543; P50613; P38936; P46527; P49918; P42771; P42772; P42773; P55273; Q9UJC3; P08238; Q9UKT9; Q0VD86; P01106; Q9ULD0; P28749; Q08999; P09936; Q8N720 EC number 2.7.11.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Cell division; Cytoplasm; Disease variant; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 30138.4 Length 267 Aromaticity 0.09 Instability index 36.2 Isoelectric point 5.78 Charge (pH=7) -5.83 2D Binding mode Binding energy (Kcal/mol) -6.01 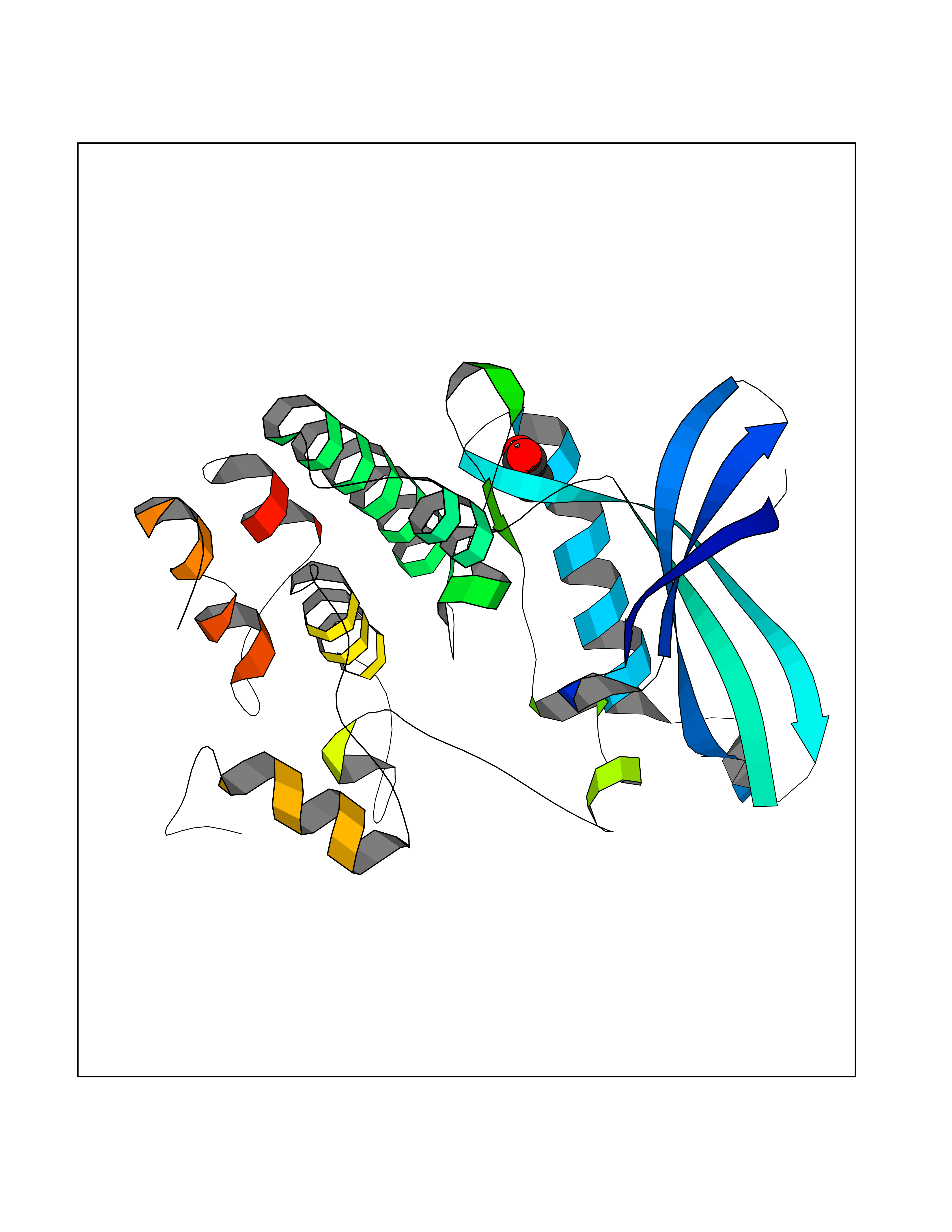 Molscript Map 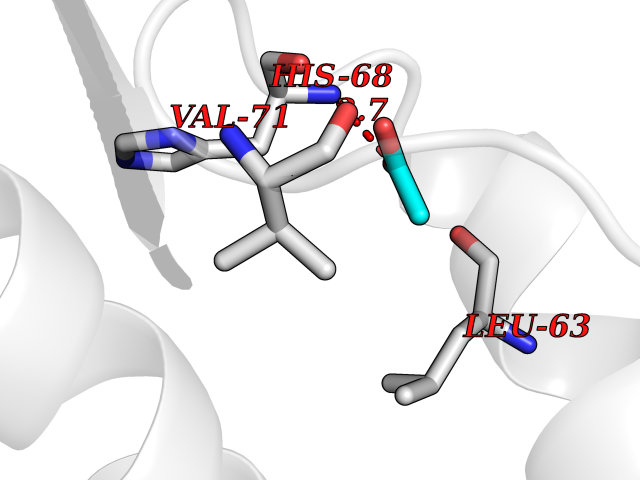 Pymol Map 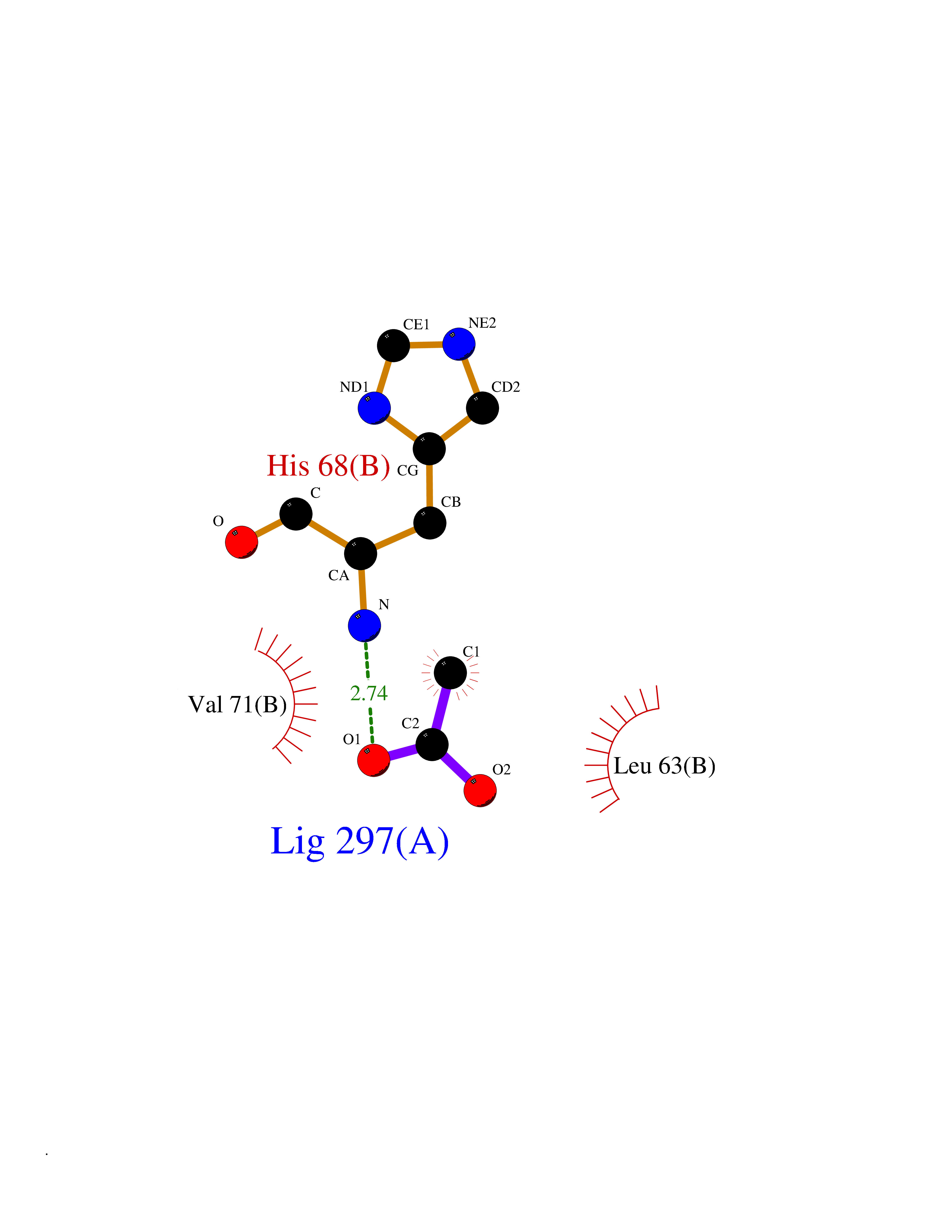 Ligplot Map 3D Binding mode Sequence SRYEPVAEIGVGAYGTVYKARDPHSGHFVALKSVRVPNGEEGLPISTVREVALLRRLEAFEHPNVVRLMDVCATSRTDREIKVTLVFEHVDQDLRTYLDKAPPPGLPAETIKDLMRQFLRGLDFLHANCIVHRDLKPENILVTSGGTVKLADFGLARIYSYQMALDPVVVTLWYRAPEVLLQSTYATPVDMWSVGCIFAEMFRRKPLFCGNSEADQLGKIFDLIGLPPEDDWVPEMEESGAQLLLEMLTFNPHKRISAFRALQHSYL Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | "Acetolactate synthase, chloroplastic (AtALS) (EC 2.2.1.6) (Acetohydroxy-acid synthase) (Protein CHLORSULFURON RESISTANT 1)" | 5K3S | 4.40 | |
Target general information Gen name ALS Organism Arabidopsis thaliana (Mouse-ear cress) Uniprot ID TTD ID NA Synonyms At3g48560;CSR1;AHAS;T8P19.70;TZP5 Protein family TPP enzyme family Biochemical class NA Function Catalyzes the formation of acetolactate from pyruvate, the first step in valine and isoleucine biosynthesis. {ECO:0000269|PubMed:10386618, ECO:0000269|PubMed:16665813, ECO:0000269|PubMed:16667374, ECO:0000269|PubMed:16668488, ECO:0000269|PubMed:2336405, ECO:0000269|PubMed:8913312, ECO:0000269|PubMed:9355748, ECO:0000269|PubMed:9677339, ECO:0000269|Ref.9}." Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 2.2.1.6 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Branched-chain amino acid biosynthesis; Chloroplast; Coiled coil; FAD; Flavoprotein; Genetically modified food; Herbicide resistance; Magnesium; Metal-binding; Oxidation; Plastid; Reference proteome; Thiamine pyrophosphate; Transferase; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 63431 Length 583 Aromaticity 0.07 Instability index 36.62 Isoelectric point 5.4 Charge (pH=7) -15.33 2D Binding mode Binding energy (Kcal/mol) -6  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence TFISRFAPDQPRKGADILVEALERQGVETVFAYPGGASMEIHQALTRSSSIRNVLPRHEQGGVFAAEGYARSSGKPGICIATSGPGATNLVSGLADALLDSVPLVAITGQVPRRMIGTDAFQETPIVEVTRSITKHNYLVMDVEDIPRIIEEAFFLATSGRPGPVLVDVPKDIQQQLAIPNWEQAMRLPGYMSRMPKPPEDSHLEQIVRLISESKKPVLYVGGGCLNSSDELGRFVELTGIPVASTLMGLGSYPXDDELSLHMLGMHGTVYANYAVEHSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKTPHVSVCGDVKLALQGMNKVLENRAEELKLDFGVWRNELNVQKQKFPLSFKTFGEAIPPQYAIKVLDELTDGKAIISTGVGQHQMWAAQFYNYKKPRQWLSSGGLGAMGFGLPAAIGASVANPDAIVVDIDGDGSFIMNVQELATIRVENLPVKVLLLNNQHLGMVMQWEDRFYKANRAHTFLGDPAQEDEIFPNMLLFAAACGIPAARVTKKADLREAIQTMLDTPGPYLLDVICPHQEHVLPMIPSGGTFNDVITEGDGRL Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Glycogen synthase kinase-3 beta (GSK-3B) | 1O6L | 4.40 | |
Target general information Gen name GSK3B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein kinase GSK3B; GSK-3 beta Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, GSK-3 subfamily Biochemical class Kinase Function Requires primed phosphorylation of the majority of its substrates. In skeletal muscle, contributes to insulin regulation of glycogen synthesis by phosphorylating and inhibiting GYS1 activity and hence glycogen synthesis. May also mediate the development of insulin resistance by regulating activation of transcription factors. Regulates protein synthesis by controlling the activity of initiation factor 2B (EIF2BE/EIF2B5) in the same manner as glycogen synthase. In Wnt signaling, GSK3B forms a multimeric complex with APC, AXIN1 and CTNNB1/beta-catenin and phosphorylates the N-terminus of CTNNB1 leading to its degradation mediated by ubiquitin/proteasomes. Phosphorylates JUN at sites proximal to its DNA-binding domain, thereby reducing its affinity for DNA. Phosphorylates NFATC1/NFATC on conserved serine residues promoting NFATC1/NFATC nuclear export, shutting off NFATC1/NFATC gene regulation, and thereby opposing the action of calcineurin. Phosphorylates MAPT/TAU on 'Thr-548', decreasing significantly MAPT/TAU ability to bind and stabilize microtubules. MAPT/TAU is the principal component of neurofibrillary tangles in Alzheimer disease. Plays an important role in ERBB2-dependent stabilization of microtubules at the cell cortex. Phosphorylates MACF1, inhibiting its binding to microtubules which is critical for its role in bulge stem cell migration and skin wound repair. Probably regulates NF-kappa-B (NFKB1) at the transcriptional level and is required for the NF-kappa-B-mediated anti-apoptotic response to TNF-alpha (TNF/TNFA). Negatively regulates replication in pancreatic beta-cells, resulting in apoptosis, loss of beta-cells and diabetes. Through phosphorylation of the anti-apoptotic protein MCL1, may control cell apoptosis in response to growth factors deprivation. Phosphorylates MUC1 in breast cancer cells, decreasing the interaction of MUC1 with CTNNB1/beta-catenin. Is necessary for the establishment of neuronal polarity and axon outgrowth. Phosphorylates MARK2, leading to inhibit its activity. Phosphorylates SIK1 at 'Thr-182', leading to sustain its activity. Phosphorylates ZC3HAV1 which enhances its antiviral activity. Phosphorylates SNAI1, leading to its BTRC-triggered ubiquitination and proteasomal degradation. Phosphorylates SFPQ at 'Thr-687' upon T-cell activation. Phosphorylates NR1D1 st 'Ser-55' and 'Ser-59' and stabilizes it by protecting it from proteasomal degradation. Regulates the circadian clock via phosphorylation of the major clock components including ARNTL/BMAL1, CLOCK and PER2. Phosphorylates CLOCK AT 'Ser-427' and targets it for proteasomal degradation. Phosphorylates ARNTL/BMAL1 at 'Ser-17' and 'Ser-21' and primes it for ubiquitination and proteasomal degradation. Phosphorylates OGT at 'Ser-3' or 'Ser-4' which positively regulates its activity. Phosphorylates MYCN in neuroblastoma cells which may promote its degradation. Regulates the circadian rhythmicity of hippocampal long-term potentiation and ARNTL/BMLA1 and PER2 expression. Acts as a regulator of autophagy by mediating phosphorylation of KAT5/TIP60 under starvation conditions, leading to activate KAT5/TIP60 acetyltransferase activity and promote acetylation of key autophagy regulators, such as ULK1 and RUBCNL/Pacer. Constitutively active protein kinase that acts as a negative regulator in the hormonal control of glucose homeostasis, Wnt signaling and regulation of transcription factors and microtubules, by phosphorylating and inactivating glycogen synthase (GYS1 or GYS2), EIF2B, CTNNB1/beta-catenin, APC, AXIN1, DPYSL2/CRMP2, JUN, NFATC1/NFATC, MAPT/TAU and MACF1. Related diseases Neuromyotonia and axonal neuropathy, autosomal recessive (NMAN) [MIM:137200]: An autosomal recessive neurologic disorder characterized by onset in the first or second decade of a peripheral axonal neuropathy predominantly affecting motor more than sensory nerves. The axonal neuropathy is reminiscent of Charcot-Marie-Tooth disease type 2 and distal hereditary motor neuropathy. Individuals with NMAN also have delayed muscle relaxation and action myotonia associated with neuromyotonic discharges on needle EMG resulting from hyperexcitability of the peripheral nerves. {ECO:0000269|PubMed:16835243, ECO:0000269|PubMed:22961002, ECO:0000269|PubMed:28691797, ECO:0000269|PubMed:29787766, ECO:0000269|PubMed:31088288}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08073; DB07149; DB07014; DB07676; DB01772; DB07859; DB07585; DB07058; DB03444; DB04014; DB01950; DB12010; DB02052; DB07947; DB14509; DB01356; DB14507; DB14508; DB07812; DB07584; DB04395; DB01793; DB02010; DB12129 Interacts with P31749; P31751; PRO_0000000093 [P05067]; O15169; Q9Y2T1; Q96G01; O75952-3; O75952-5; P35222; Q5VWQ8; Q5VWQ8-2; Q9NYF0; O75398; Q13144; Q92837; P13807; O75581; Q5S007; P10636; P10636-8; Q14596; Q8N4C6; P17612; Q01201; O95863; P37840; Q6J9G0; P04637; Q14134; O95071; P63104; Q8IX07; O35625; Q14DJ8; Q02248; Q811T9; P63085; P0DTC9; P05067; P35637; P01106; Q8BMD2-1 EC number EC 2.7.11.26 Uniprot keywords 3D-structure; ADP-ribosylation; Alternative splicing; Alzheimer disease; ATP-binding; Biological rhythms; Carbohydrate metabolism; Cell membrane; Cytoplasm; Developmental protein; Diabetes mellitus; Differentiation; Glycogen metabolism; Kinase; Lipoprotein; Membrane; Neurogenesis; Nucleotide-binding; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Signal transduction inhibitor; Transferase; Wnt signaling pathway Protein physicochemical properties Chain ID C Molecular weight (Da) 37917 Length 327 Aromaticity 0.12 Instability index 29.04 Isoelectric point 5.63 Charge (pH=7) -7.51 2D Binding mode Binding energy (Kcal/mol) -6  Molscript Map 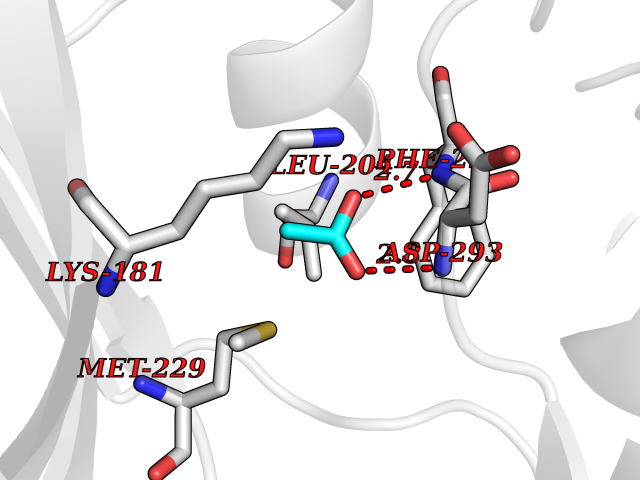 Pymol Map 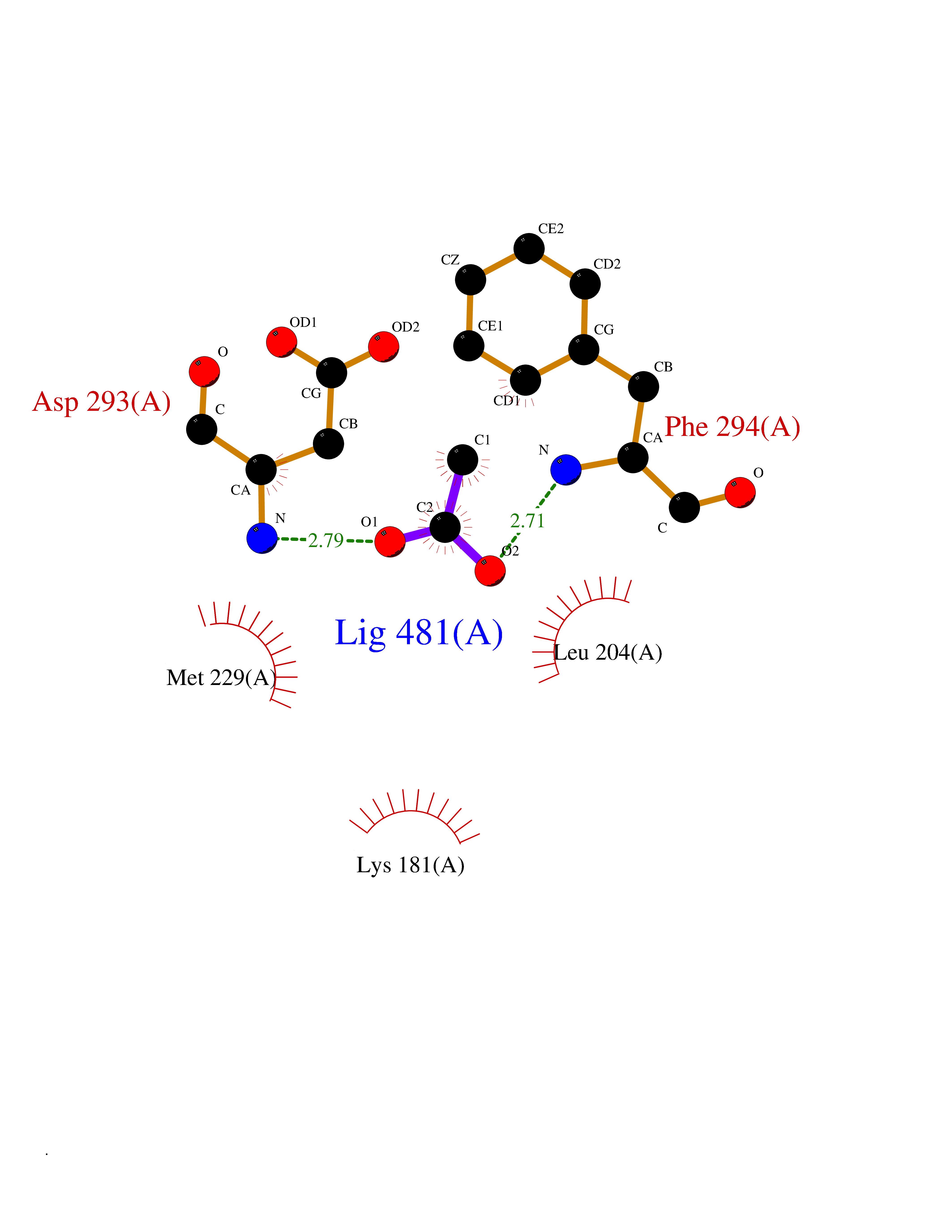 Ligplot Map 3D Binding mode Sequence KVTMNDFDYLKLLGKGTFGKVILVREKATGRYYAMKILRKEVIIAKDEVAHTVTESRVLQNTRHPFLTALKYAFQTHDRLCFVMEYANGGELFFHLSRERVFTEERARFYGAEIVSALEYLHSRDVVYRDIKLENLMLDKDGHIKITDFGLCKEGISDGATMKXFCGTPEYLAPEVLEDNDYGRAVDWWGLGVVMYEMMCGRLPFYNQDHERLFELILMEEIRFPRTLSPEAKSLLAGLLKKDPKQRLGGGPSDAKEVMEHRFFLSINWQDVVQKKLLPPFKPQVTSEVDTRYFDDEFTAQSITQEMFEDFDYIADWGRPRTTSFAE Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Oxygen-insensitive NAD(P)H nitroreductase | 1KQB | 4.40 | |
Target general information Gen name nfsB Organism Enterobacter cloacae Uniprot ID TTD ID NA Synonyms nfsI;nfnB Protein family Nitroreductase family Biochemical class Oxidoreductase Function Oxidoreductase activity. Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03793; DB03247 Interacts with NA EC number 1.-.-.- Uniprot keywords 3D-structure; Flavoprotein; FMN; NAD; NADP; Oxidoreductase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 47619.4 Length 432 Aromaticity 0.08 Instability index 38.43 Isoelectric point 5.52 Charge (pH=7) -12.98 2D Binding mode Binding energy (Kcal/mol) -6.01 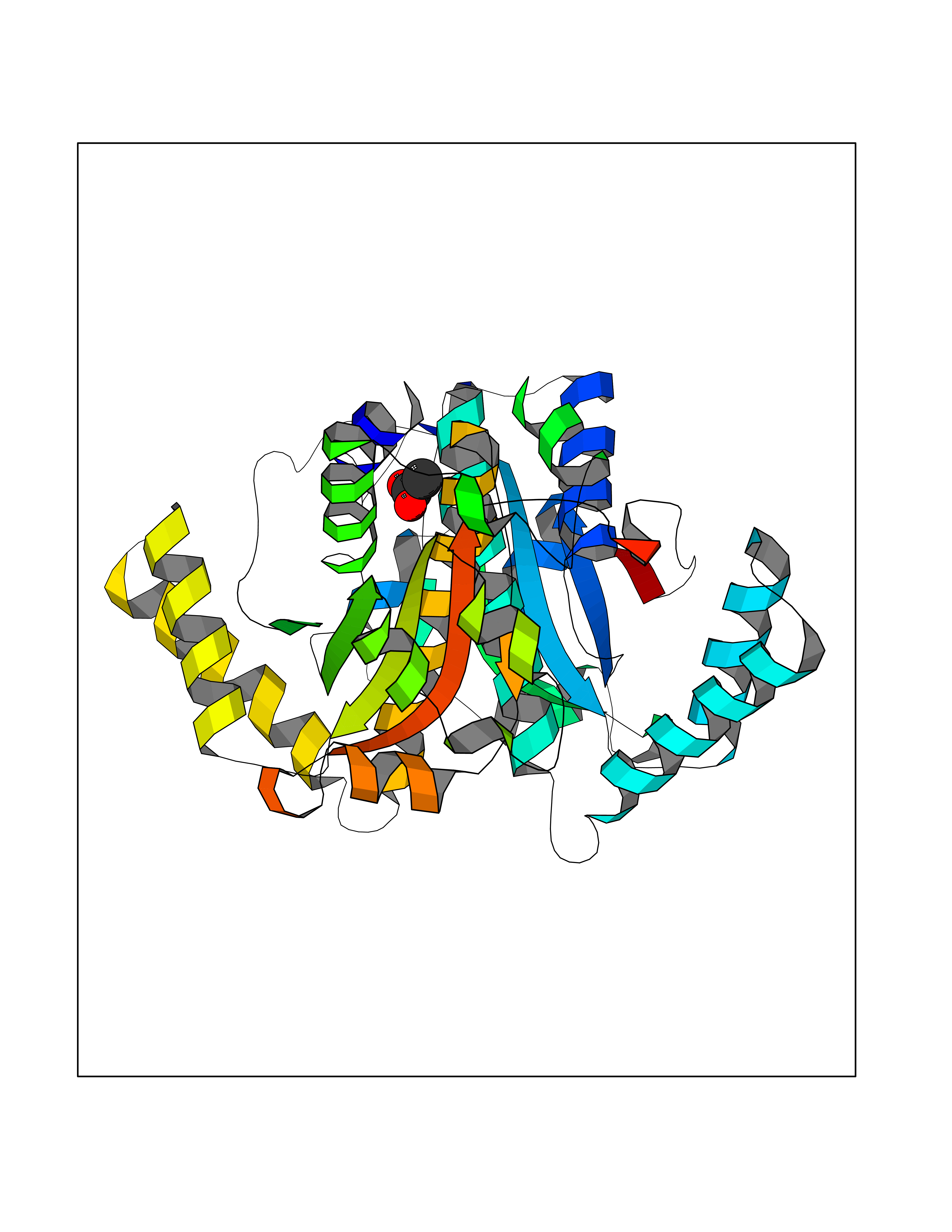 Molscript Map 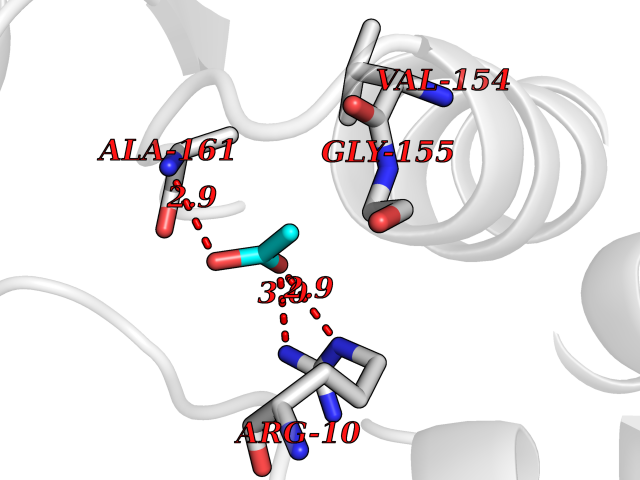 Pymol Map 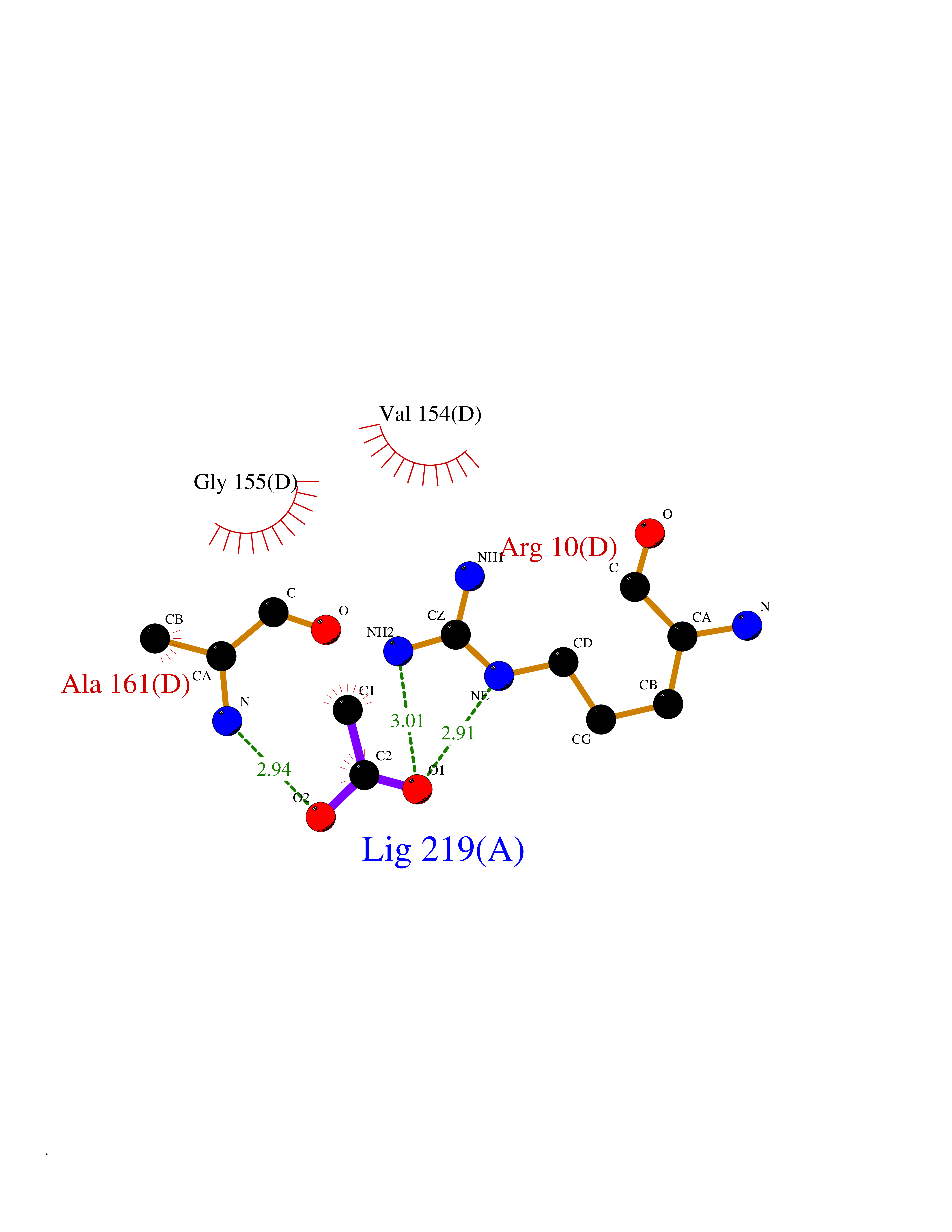 Ligplot Map 3D Binding mode Sequence DIISVALKRHSTKAFDASKKLTAEEAEKIKTLLQYSPSSTNSQPWHFIVASTEEGKARVAKSAAGTYVFNERKMLDASHVVVFCAKTAMDDAWLERVVDQEEADGRFNTPEAKAANHKGRTYFADMHRVDLKDDDQWMAKQVYLNVGNFLLGVGAMGLDAVPIEGFDAAILDEEFGLKEKGFTSLVVVPVGHHSVEDFNATLPKSRLPLSTIVTECDIISVALKRHSTKAFDASKKLTAEEAEKIKTLLQYSPSSTNSQPWHFIVASTEEGKARVAKSAAGTYVFNERKMLDASHVVVFCAKTAMDDAWLERVVDQEEADGRFNTPEAKAANHKGRTYFADMHRVDLKDDDQWMAKQVYLNVGNFLLGVGAMGLDAVPIEGFDAAILDEEFGLKEKGFTSLVVVPVGHHSVEDFNATLPKSRLPLSTIVTEC Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | NDR1 protein kinase (STK38) | 6BXI | 4.40 | |
Target general information Gen name STK38 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein kinase 38; Nuclear Dbf2-related kinase 1; NDR1 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family Biochemical class Kinase Function Converts MAP3K2 from its phosphorylated form to its non-phosphorylated form and inhibits autophosphorylation of MAP3K2. Negative regulator of MAP3K1/2 signaling. Related diseases Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:8955068}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 3 (NS3) [MIM:609942]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:16773572, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:17468812, ECO:0000269|PubMed:19396835, ECO:0000269|PubMed:20949621}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gastric cancer (GASC) [MIM:613659]: A malignant disease which starts in the stomach, can spread to the esophagus or the small intestine, and can extend through the stomach wall to nearby lymph nodes and organs. It also can metastasize to other parts of the body. The term gastric cancer or gastric carcinoma refers to adenocarcinoma of the stomach that accounts for most of all gastric malignant tumors. Two main histologic types are recognized, diffuse type and intestinal type carcinomas. Diffuse tumors are poorly differentiated infiltrating lesions, resulting in thickening of the stomach. In contrast, intestinal tumors are usually exophytic, often ulcerating, and associated with intestinal metaplasia of the stomach, most often observed in sporadic disease. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:7773929}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in KRAS are a cause of pylocytic astrocytoma (PA). Pylocytic astrocytomas are neoplasms of the brain and spinal cord derived from glial cells which vary from histologically benign forms to highly anaplastic and malignant tumors. {ECO:0000269|PubMed:16247081}.; DISEASE: Cardiofaciocutaneous syndrome 2 (CFC2) [MIM:615278]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. CFC2 patients often do not have the skin abnormalities, such as ichthyosis, hyperkeratosis, and hemangioma observed in CFC1. {ECO:0000269|PubMed:16474404, ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:20949621, ECO:0000269|PubMed:21797849}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: KRAS mutations are involved in cancer development. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:1553789, ECO:0000269|PubMed:16533793, ECO:0000269|PubMed:24623306, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:3627975, ECO:0000269|PubMed:6092920, ECO:0000269|PubMed:6695174, ECO:0000269|PubMed:7773929}.; DISEASE: Oculoectodermal syndrome (OES) [MIM:600268]: A syndrome characterized by the association of epibulbar dermoids and aplasia cutis congenita. Affected individuals show multiple, asymmetric, atrophic, non-scarring and hairless regions that may be associated with hamartomas. Ectodermal changes include linear hyperpigmentation that may follow the lines of Blaschko and rarely epidermal nevus-like lesions. Epibulbar dermoids may be uni-or bilateral. Additional ocular anomalies such as skin tags of the upper eyelid, rarely optic nerve or retinal changes, and microphthalmia can be present. The phenotypic expression is highly variable, and various other abnormalities have occasionally been reported including growth failure, lymphedema, cardiovascular defects, as well as neurodevelopmental symptoms like developmental delay, epilepsy, learning difficulties, and behavioral abnormalities. Benign tumor-like lesions such as nonossifying fibromas of the long bones and giant cell granulomas of the jaws have repeatedly been observed and appear to be age-dependent, becoming a common manifestation in individuals aged 5 years or older. {ECO:0000269|PubMed:25808193, ECO:0000269|PubMed:26970110, ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Schimmelpenning-Feuerstein-Mims syndrome (SFM) [MIM:163200]: A disease characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects. Many oral manifestations have been reported, not only including hypoplastic and malformed teeth, and mucosal papillomatosis, but also ankyloglossia, hemihyperplastic tongue, intraoral nevus, giant cell granuloma, ameloblastoma, bone cysts, follicular cysts, oligodontia, and odontodysplasia. Sebaceous nevi follow the lines of Blaschko and these can continue as linear intraoral lesions, as in mucosal papillomatosis. {ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with P49407; P32121; Q8N9N5-2; Q03135; P08238; Q9H8S9; Q70IA6; P16333; P30086; P02638 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Chromosome; Cytoplasm; Direct protein sequencing; DNA damage; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 38701.1 Length 333 Aromaticity 0.11 Instability index 38.05 Isoelectric point 5.69 Charge (pH=7) -7.8 2D Binding mode Binding energy (Kcal/mol) -6  Molscript Map 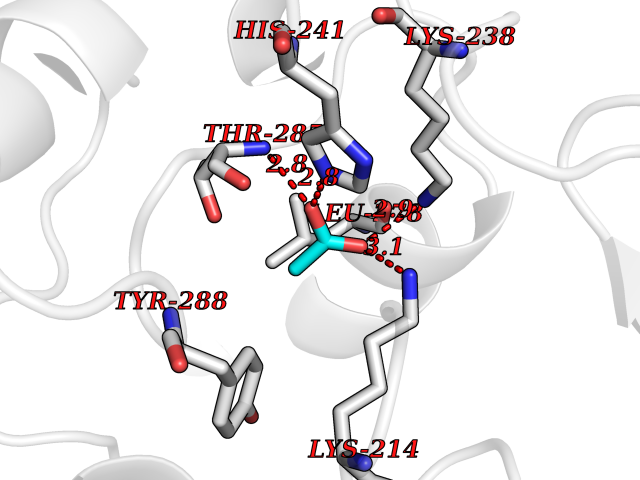 Pymol Map 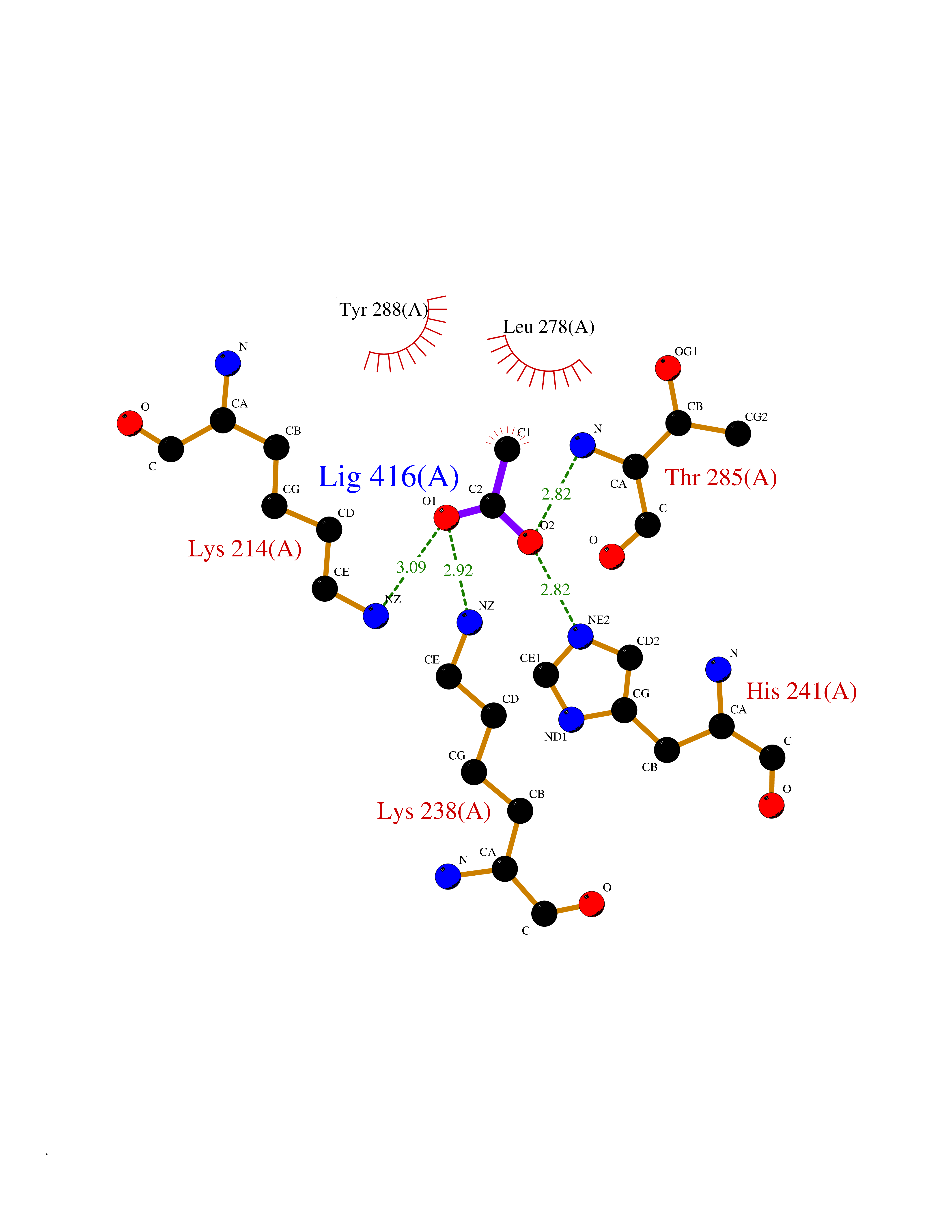 Ligplot Map 3D Binding mode Sequence TRLGLEDFESLKVIGRGAFGEVRLVQKKDTGHVYAMKILRKADMLEKEQVGHIRAERDILVEADSLWVVKMFYSFQDKLNLYLIMEFLPGGDMMTLLMKKDTLTEEETQFYIAETVLAIDSIHQLGFIHRDIKPDNLLLDSKGHVKLSDFGLCTGLKKAHRTEFYRNLNHSLPSDFTFQNMNSKRKAETWKRNRRQLAFSTVGTPDYIAPEVFMQTGYNKLCDWWSLGVIMYEMLIGYPPFCSETPQETYKKVMNWKETLTFPPEVPISEKAKDLILRFCCEWEHRIGAPGVEEIKSNSFFEGVDWEHIRERPAAISIEIKSIDDTSNFDEFP Hydrogen bonds contact Hydrophobic contact | ||||