Job Results:
Ligand
Structure
Job ID
baacf95062c374f8ce5148662fe61a2e
Job name
NA
Time
2026-02-27 15:12:55
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | S-adenosylmethionine synthase type-2 (MAT2A) | 5A1I | 4.31 | |
Target general information Gen name MAT2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Methionine adenosyltransferase II; Methionine adenosyltransferase 2; MAT-II; MAT 2; AdoMet synthase 2 Protein family AdoMet synthase family Biochemical class AdoMet synthase family Function Catalyzes the formation of S-adenosylmethionine from methionine and ATP. The reaction comprises two steps that are both catalyzed by the same enzyme: formation of S-adenosylmethionine (AdoMet) and triphosphate, and subsequent hydrolysis of the triphosphate. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00118; DB00134 Interacts with Q96IK1-2; Q96NX5; Q6ZP82-1; Q8IUI8; Q6P1L5; P15976-2; P80217-2; Q8WZ19; Q9UIH9; Q00266; P31153; Q9NZL9; P02795; Q9BRX2; O43663; O43741; P57052; Q8N488; P08195-4; Q13573; Q86W54-2; O95789-4 EC number EC 2.5.1.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Isopeptide bond; Magnesium; Metal-binding; Nucleotide-binding; One-carbon metabolism; Phosphoprotein; Potassium; Proteomics identification; Reference proteome; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 42071.4 Length 381 Aromaticity 0.08 Instability index 38.24 Isoelectric point 6.21 Charge (pH=7) -4.13 2D Binding mode Binding energy (Kcal/mol) -5.88 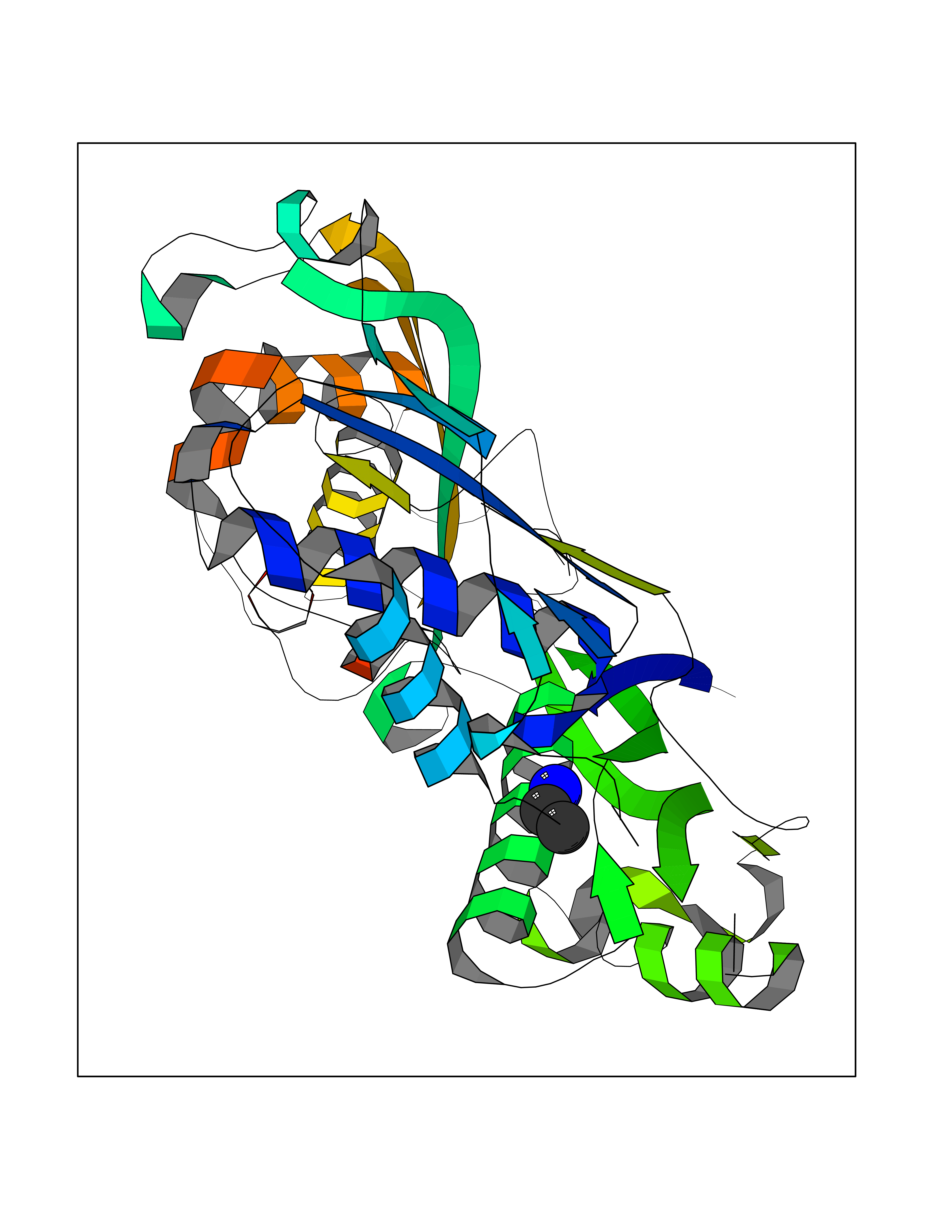 Molscript Map 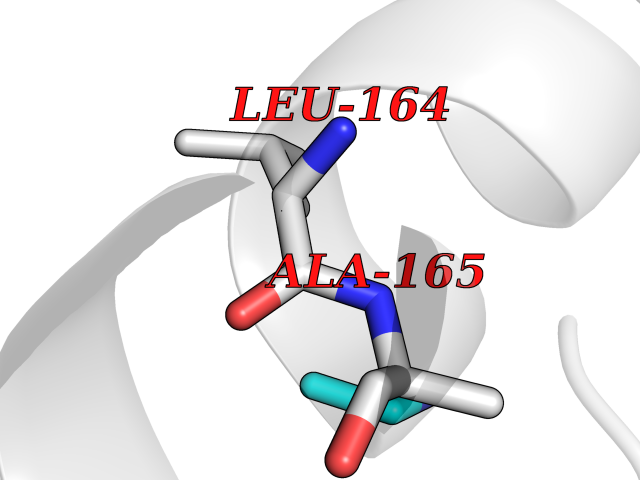 Pymol Map 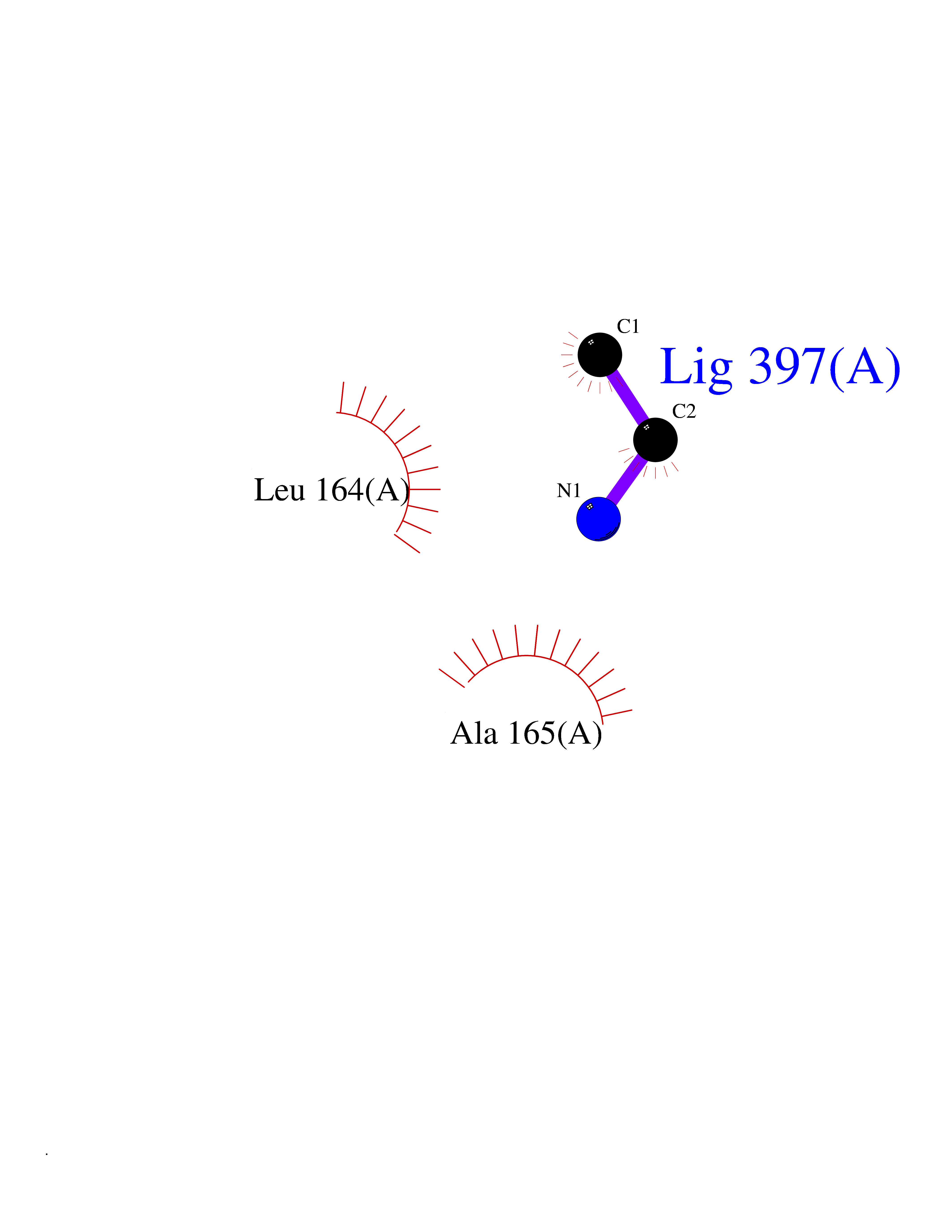 Ligplot Map 3D Binding mode Sequence EGTFLFTSESVGEGHPDKICDQISDAVLDAHLQQDPDAKVACETVAKTGMILLAGEITSRAAVDYQKVVREAVKHIGYDDSSKGFDYKTCNVLVALEQQSPDIAQGVHLDRNEEDIGAGDQGLMFGYATDETEECMPLTIVLAHKLNAKLAELRRNGTLPWLRPDSKTQVTVQYMQDRGAVLPIRVHTIVISVQHDEEVCLDEMRDALKEKVIKAVVPAKYLDEDTIYHLQPSGRFVIGGPQGDAGLTGRKIIVDTYGGWGAHGGGAFSGKDYTKVDRSAAYAARWVAKSLVKGGLCRRVLVQVSYAIGVSHPLSISIFHYGTSQKSERELLEIVKKNFDLRPGVIVRDLDLKKPIYQRTAAYGHFGRDSFPWEVPKKLKY Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Fms-like tyrosine kinase 3 (FLT-3) | 1RJB | 4.31 | |
Target general information Gen name FLT3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Stem cell tyrosine kinase 1; STK1; STK-1; Receptor-type tyrosine-protein kinase FLT3; Fetal liver kinase-2; FLT-3; FLK2; FLK-2; FL cytokine receptor; CD135 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Tyrosine-protein kinase that acts as cell-surface receptor for the cytokine FLT3LG and regulates differentiation, proliferation and survival of hematopoietic progenitor cells and of dendritic cells. Promotes phosphorylation of SHC1 and AKT1, and activation of the downstream effector MTOR. Promotes activation of RAS signaling and phosphorylation of downstream kinases, including MAPK1/ERK2 and/or MAPK3/ERK1. Promotes phosphorylation of FES, FER, PTPN6/SHP, PTPN11/SHP-2, PLCG1, and STAT5A and/or STAT5B. Activation of wild-type FLT3 causes only marginal activation of STAT5A or STAT5B. Mutations that cause constitutive kinase activity promote cell proliferation and resistance to apoptosis via the activation of multiple signaling pathways. Related diseases Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:11090077, ECO:0000269|PubMed:11290608, ECO:0000269|PubMed:11442493, ECO:0000269|PubMed:14504097, ECO:0000269|PubMed:16266983, ECO:0000269|PubMed:18305215, ECO:0000269|PubMed:8946930, ECO:0000269|PubMed:9737679}. The gene represented in this entry may be involved in disease pathogenesis. Somatic mutations that lead to constitutive activation of FLT3 are frequent in AML patients. These mutations fall into two classes, the most common being in-frame internal tandem duplications of variable length in the juxtamembrane region that disrupt the normal regulation of the kinase activity. Likewise, point mutations in the activation loop of the kinase domain can result in a constitutively activated kinase. Drugs (DrugBank ID) DB12742; DB12267; DB12500; DB12010; DB12141; DB06469; DB06080; DB06595; DB11763; DB09079; DB11697; DB12978; DB08901; DB15822; DB12874; DB00398; DB01268; DB05465; DB11800; DB05014 Interacts with P00519; P42684; P46108; P46109; P06241; Q13322; Q9Y6K9; P06239; P27986; P20936; P43405; Q8R4L0 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 34209.2 Length 298 Aromaticity 0.14 Instability index 39.68 Isoelectric point 5.57 Charge (pH=7) -4.02 2D Binding mode Binding energy (Kcal/mol) -5.88 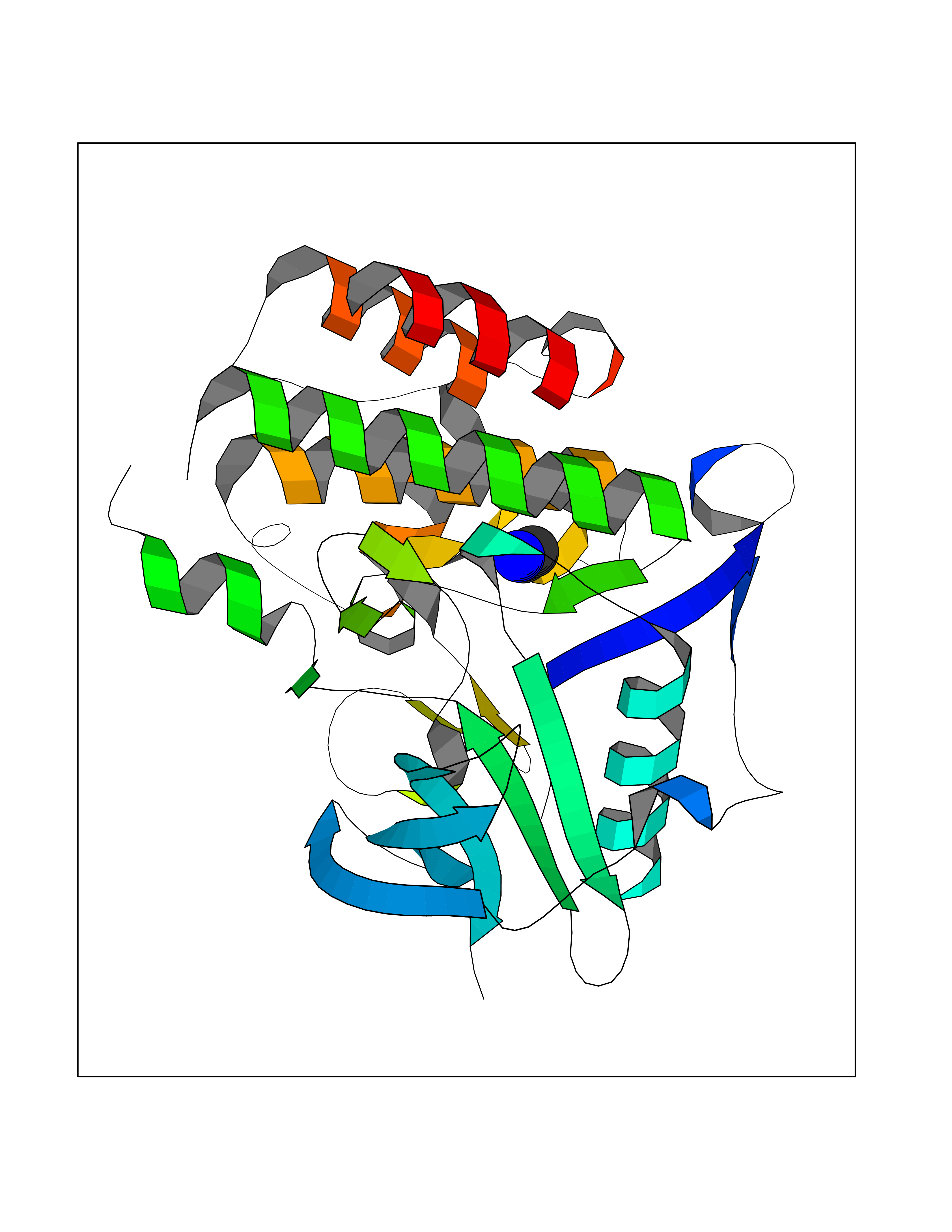 Molscript Map 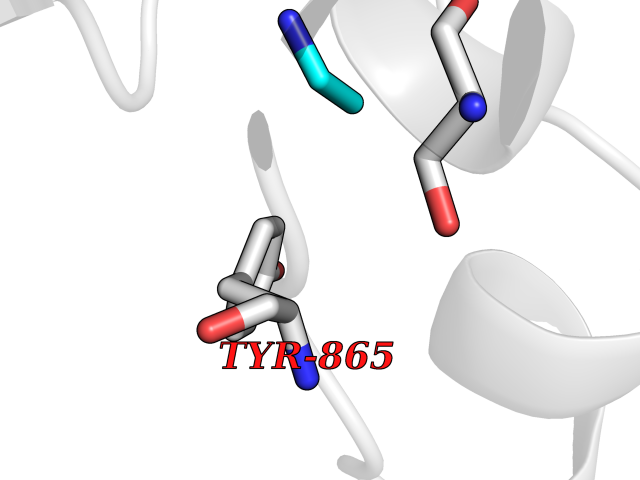 Pymol Map 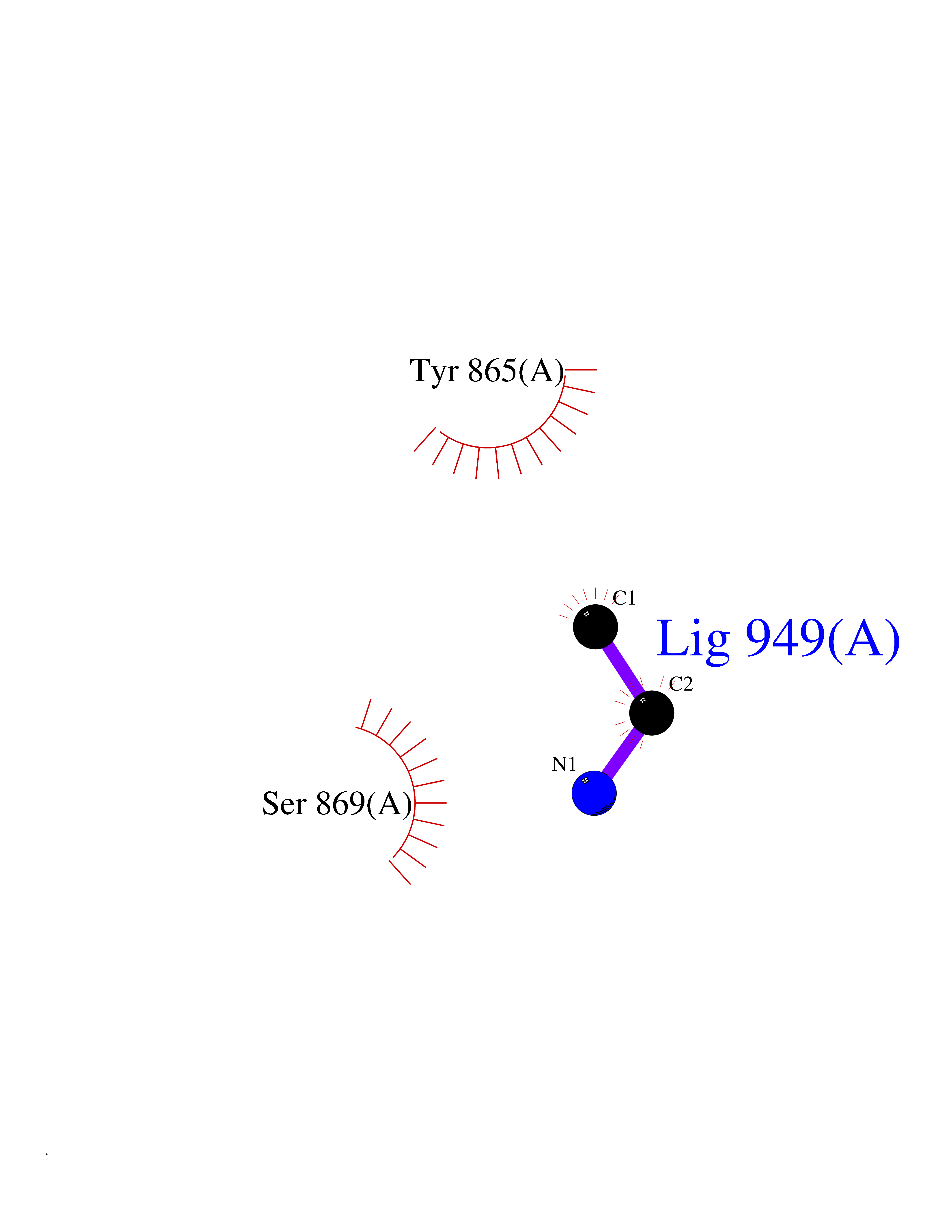 Ligplot Map 3D Binding mode Sequence YESQLQMVQVTGSSDNEYFYVDFREYEYDLKWEFPRENLEFGKVLGSGAFGKVMNATAYGISKTGVSIQVAVKMLKEREALMSELKMMTQLGSHENIVNLLGACTLSGPIYLIFEYCCYGDLLNYLRSKREKFLTFEDLLCFAYQVAKGMEFLEFKSCVHRDLAARNVLVTHGKVVKICDFGLARDIMSDSNYVVRGNARLPVKWMAPESLFEGIYTIKSDVWSYGILLWEIFSLGVNPYPGIPVDANFYKLIQNGFKMDQPFYATEEIYIIMQSCWAFDSRKRPSFPNLTSFLGCQL Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | S-adenosylmethionine synthase isoform type-1 | 2OBV | 4.31 | |
Target general information Gen name MAT1A Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MATA1;AMS1 Protein family AdoMet synthase family Biochemical class Transferase Function ATP binding.Identical protein binding.Metal ion binding.Methionine adenosyltransferase activity.Selenomethionine adenosyltransferase activity. Related diseases Methionine adenosyltransferase deficiency (MATD) [MIM:250850]: An inborn error of metabolism resulting in isolated hypermethioninemia. Most patients have no clinical abnormalities, although some neurologic symptoms may be present in rare cases with severe loss of methionine adenosyltransferase activity. {ECO:0000269|PubMed:10677294, ECO:0000269|PubMed:7560086, ECO:0000269|PubMed:8770875, ECO:0000269|PubMed:9042912}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03191; DB00118; DB03611; DB00134 Interacts with P05067; P42858; Q00266; P31153 EC number 2.5.1.6 Uniprot keywords 3D-structure; ATP-binding; Disease variant; Disulfide bond; Magnesium; Metal-binding; Nucleotide-binding; One-carbon metabolism; Potassium; Proteomics identification; Reference proteome; S-nitrosylation; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42222.9 Length 381 Aromaticity 0.08 Instability index 41.95 Isoelectric point 6.14 Charge (pH=7) -4.58 2D Binding mode Binding energy (Kcal/mol) -5.88 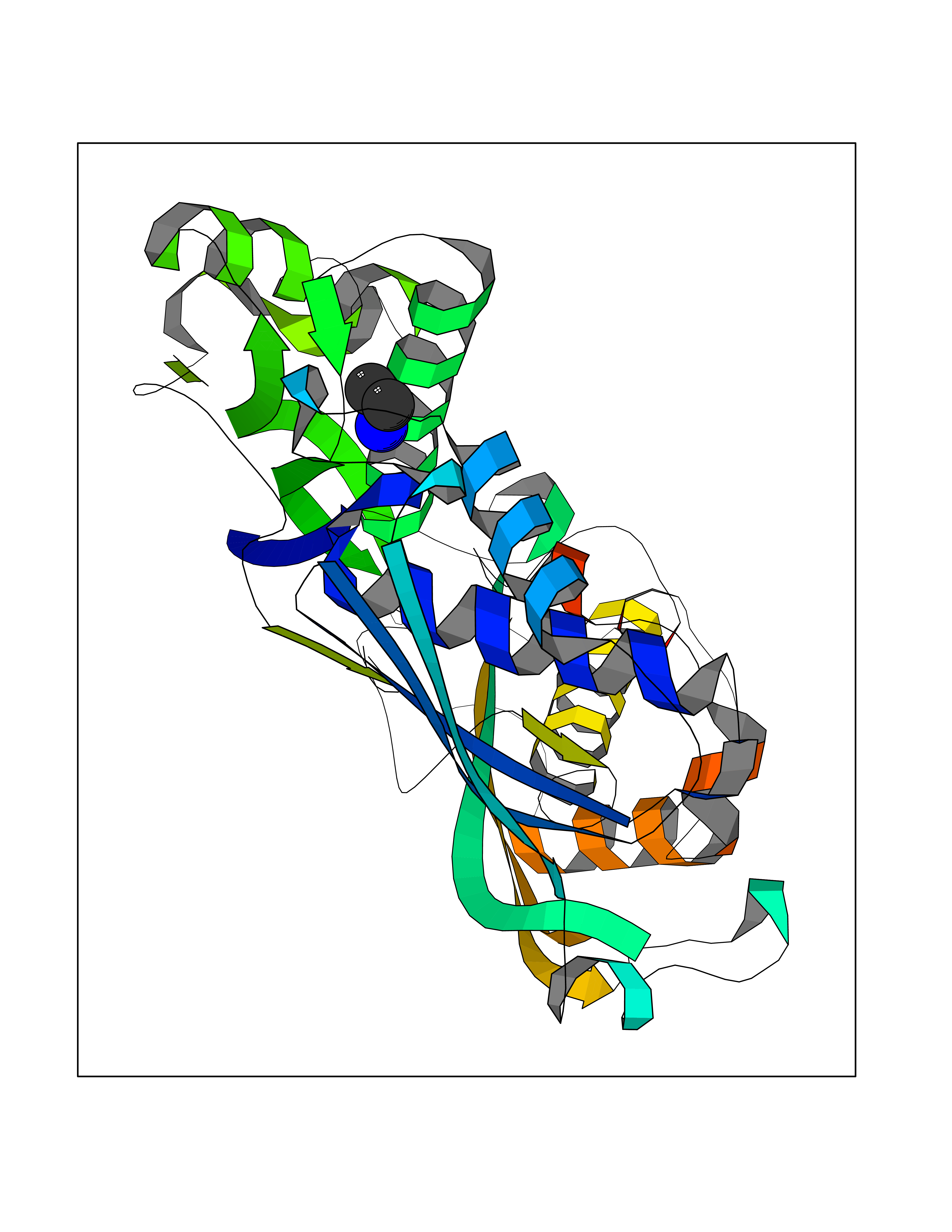 Molscript Map 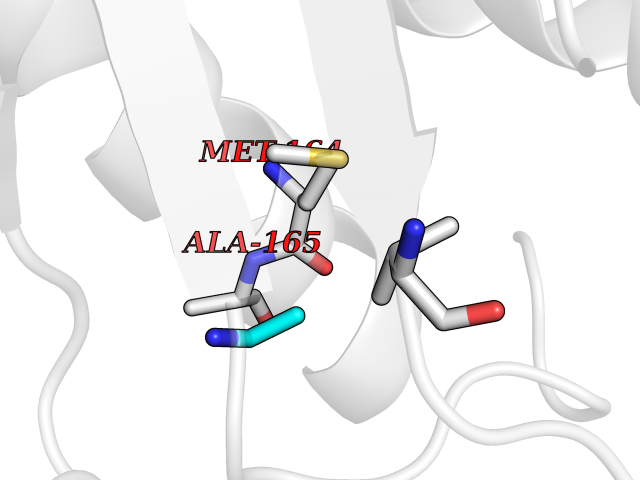 Pymol Map 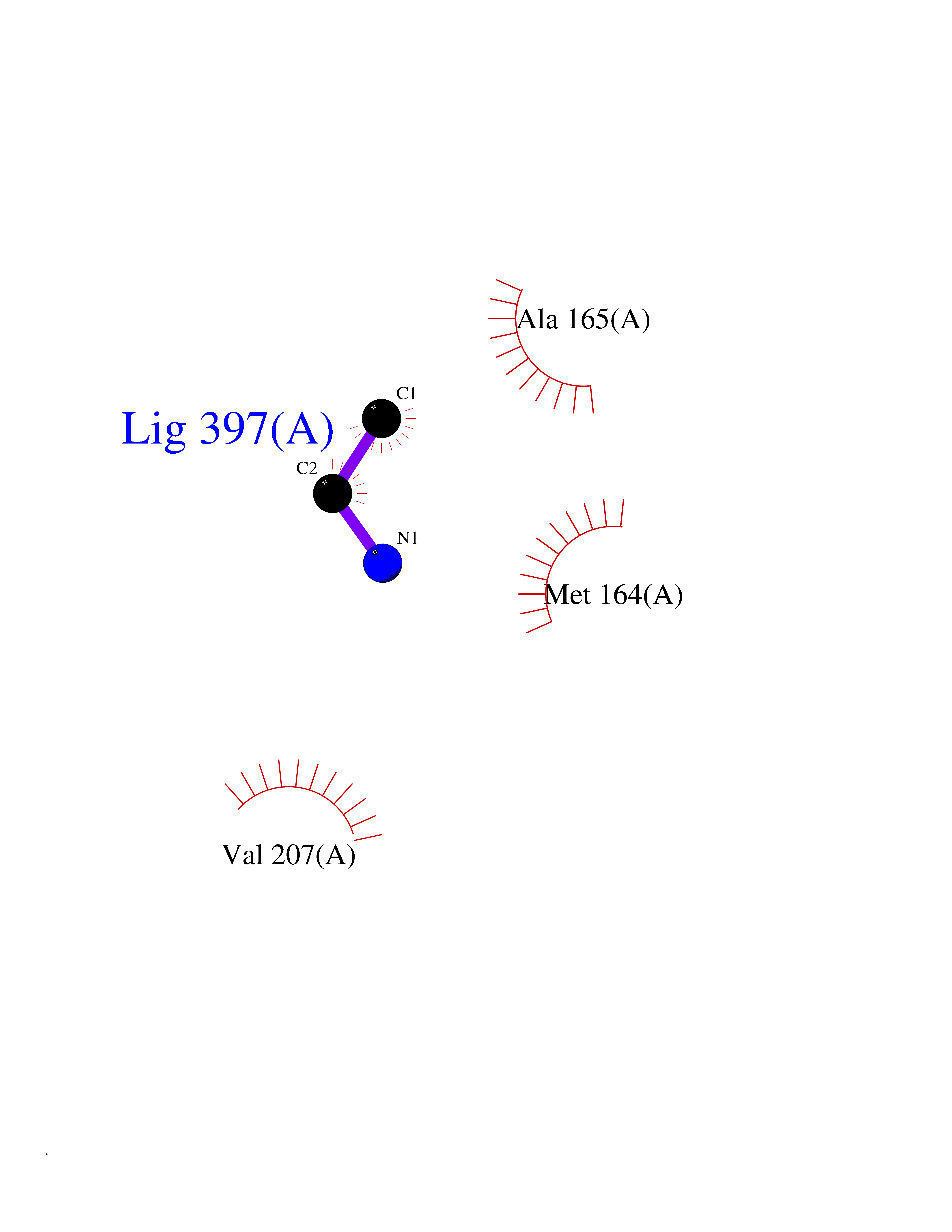 Ligplot Map 3D Binding mode Sequence MGVFMFTSESVGEGHPDKICDQISDAVLDAHLKQDPNAKVACETVCKTGMVLLCGEITSMAMVDYQRVVRDTIKHIGYDDSAKGFDFKTCNVLVALEQQSPDIAQCVHLDRNEEDVGAGDQGLMFGYATDETEECMPLTIILAHKLNARMADLRRSGLLPWLRPDSKTQVTVQYMQDNGAVIPVRIHTIVISVQHNEDITLEEMRRALKEQVIRAVVPAKYLDEDTVYHLQPSGRFVIGGPQGDAGVTGRKIIVDTYGGWGAHGGGAFSGKDYTKVDRSAAYAARWVAKSLVKAGLCRRVLVQVSYAIGVAEPLSISIFTYGTSQKTERELLDVVHKNFDLRPGVIVRDLDLKKPIYQKTACYGHFGRSEFPWEVPRKLVF Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Tissue kallikrein (KLK2) | 4NFE | 4.31 | |
Target general information Gen name KLK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hGK-1; Tissue kallikrein-2; Kallikrein-2; Glandular kallikrein-1 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function Glandular kallikreins cleave Met-Lys and Arg-Ser bonds in kininogen to release Lys-bradykinin. Related diseases Nivelon-Nivelon-Mabille syndrome (NNMS) [MIM:600092]: An autosomal recessive syndrome characterized by progressive microcephaly, cerebellar vermis hypoplasia, and skeletal dysplasia. Additional variable features include early infantile-onset seizures, intrauterine and postnatal growth retardation, generalized chondrodysplasia, and micromelia. 46,XY gonadal dysgenesis may be present. {ECO:0000269|PubMed:24784881, ECO:0000269|PubMed:30912300}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.21.35 Uniprot keywords 3D-structure; Alternative splicing; Disulfide bond; Glycoprotein; Hydrolase; Protease; Proteomics identification; Reference proteome; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 24954.3 Length 227 Aromaticity 0.08 Instability index 43.7 Isoelectric point 6.42 Charge (pH=7) -2.35 2D Binding mode Binding energy (Kcal/mol) -5.88 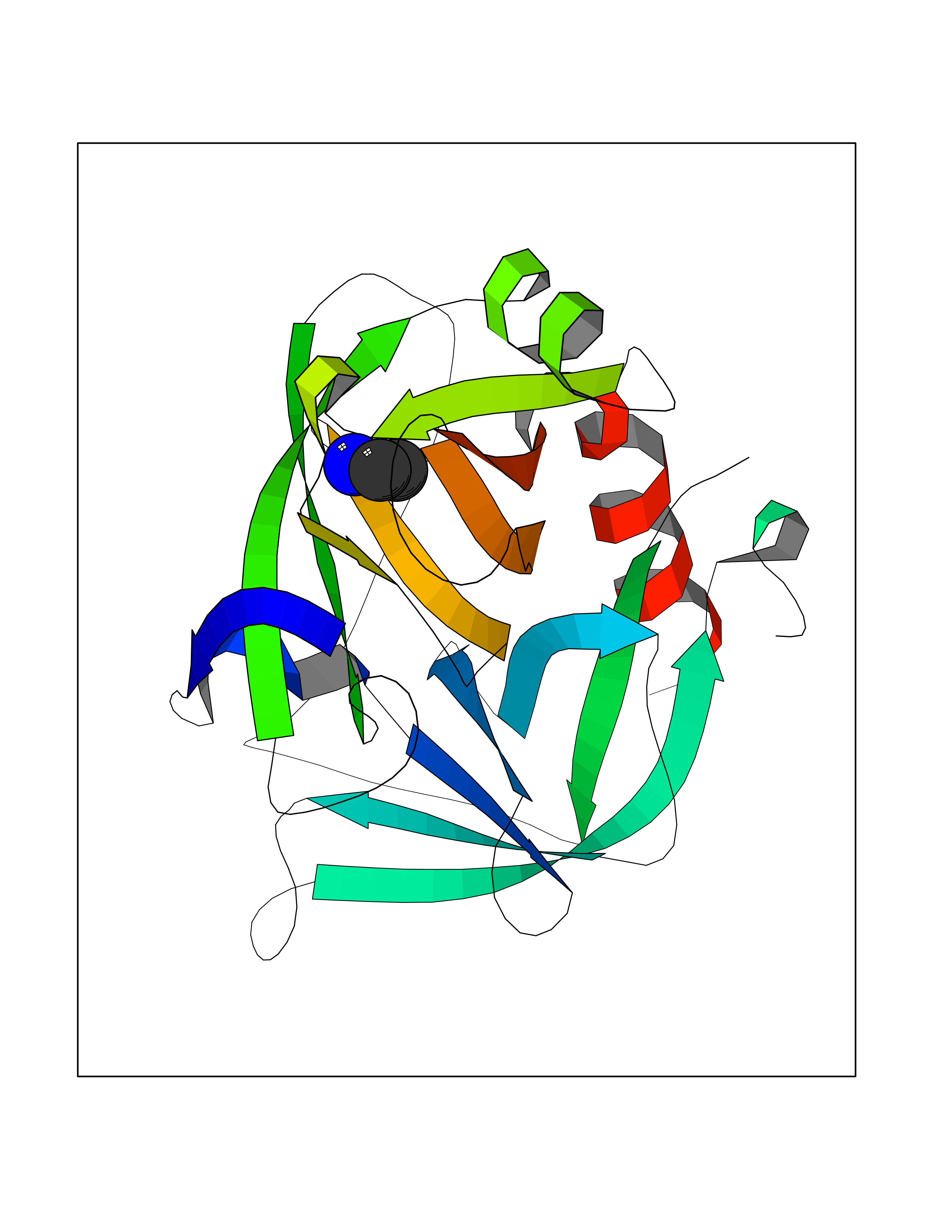 Molscript Map 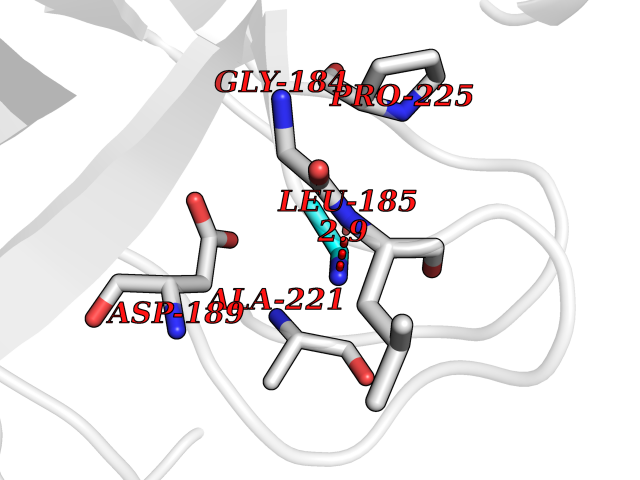 Pymol Map 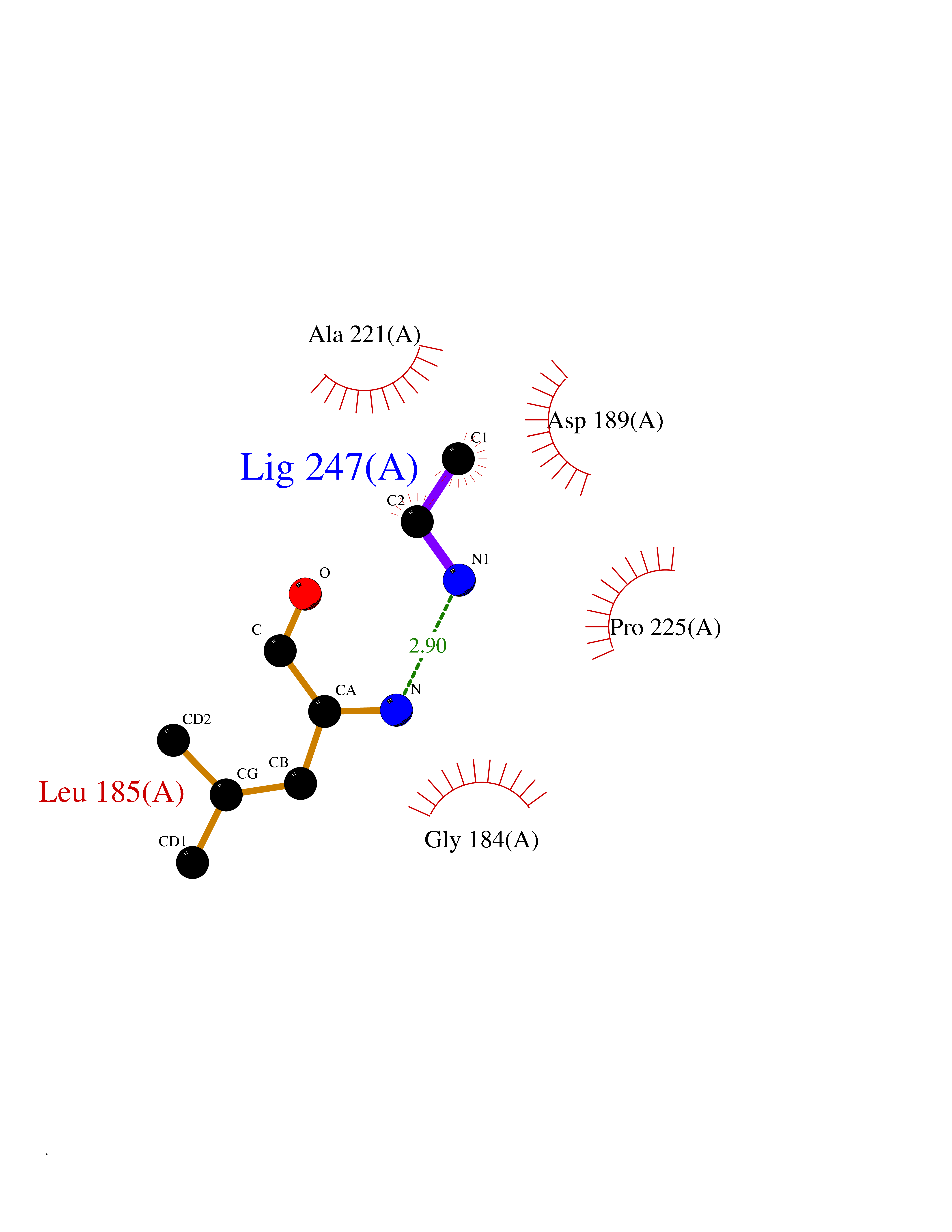 Ligplot Map 3D Binding mode Sequence IVGGWECEKHSQPWQVAVYSHGWAHCGGVLVHPQWVLTAAHCLKKNSQVWLGRHNLFEPEDTGQRVPVSHSFPHPLYNMSLDSSHDLMLLRLSEPAKITDVVKVLGLPTQEPALGTTCYASGWGSIEPEEFLRPRSLQCVSLHLLSNDMCARAYSEKVTEFMLCAGLWTGGKDTCGGDSGGPLVCNGVLQGITSWGPEPCALPEKPAVYTKVVHYRKWIKDTIAANP Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Channel-activating protease 1 (CAP1) | 3FVF | 4.31 | |
Target general information Gen name PRSS8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine protease 8; Prostasin; CAP1 Protein family Peptidase S1 family Biochemical class NA Function Possesses a trypsin-like cleavage specificity with a preference for poly-basic substrates. Stimulates epithelial sodium channel (ENaC) activity through activating cleavage of the gamma subunits (SCNN1G). Related diseases Intellectual developmental disorder with language impairment and early-onset DOPA-responsive dystonia-parkinsonism (IDLDP) [MIM:619911]: An autosomal dominant disorder characterized by global developmental delay affecting motor, cognitive, and speech domains apparent in early childhood or infancy. Most patients also show movement abnormalities, often hypotonia with later development of dopa-responsive dystonia or parkinsonism. About half of patients develop various types of seizures. {ECO:0000269|PubMed:31428396, ECO:0000269|PubMed:31922365, ECO:0000269|PubMed:32366965, ECO:0000269|PubMed:33585677}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06900 Interacts with NA EC number EC 3.4.21.- Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Membrane; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Transmembrane; Transmembrane helix; Zymogen Protein physicochemical properties Chain ID B Molecular weight (Da) 26777.8 Length 248 Aromaticity 0.08 Instability index 47.83 Isoelectric point 5.4 Charge (pH=7) -8.5 2D Binding mode Binding energy (Kcal/mol) -5.87 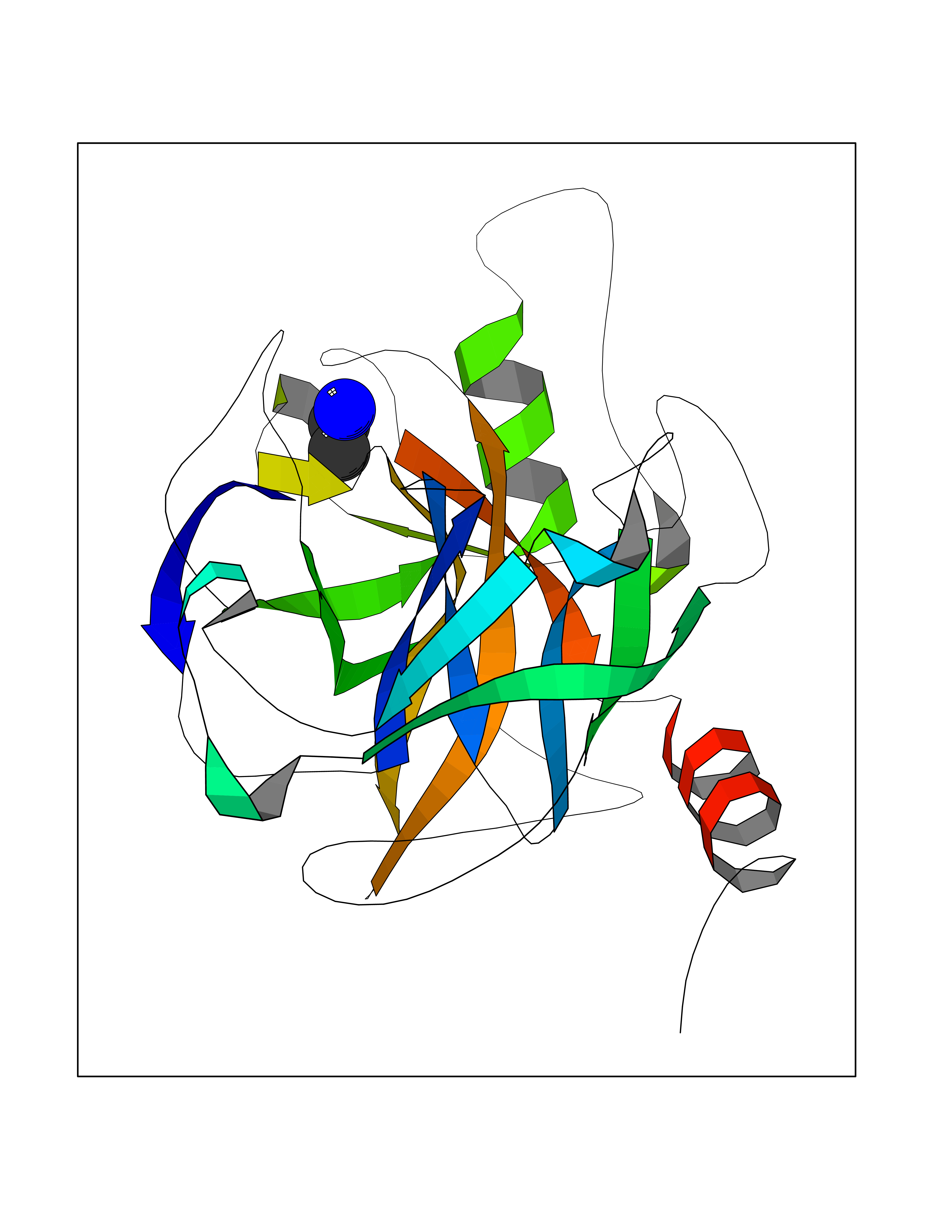 Molscript Map 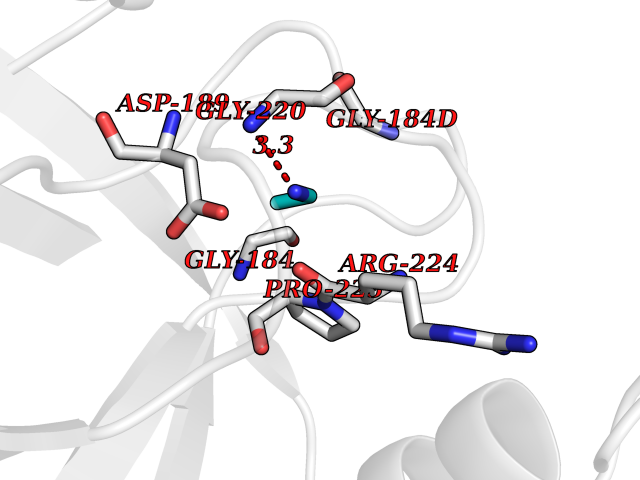 Pymol Map 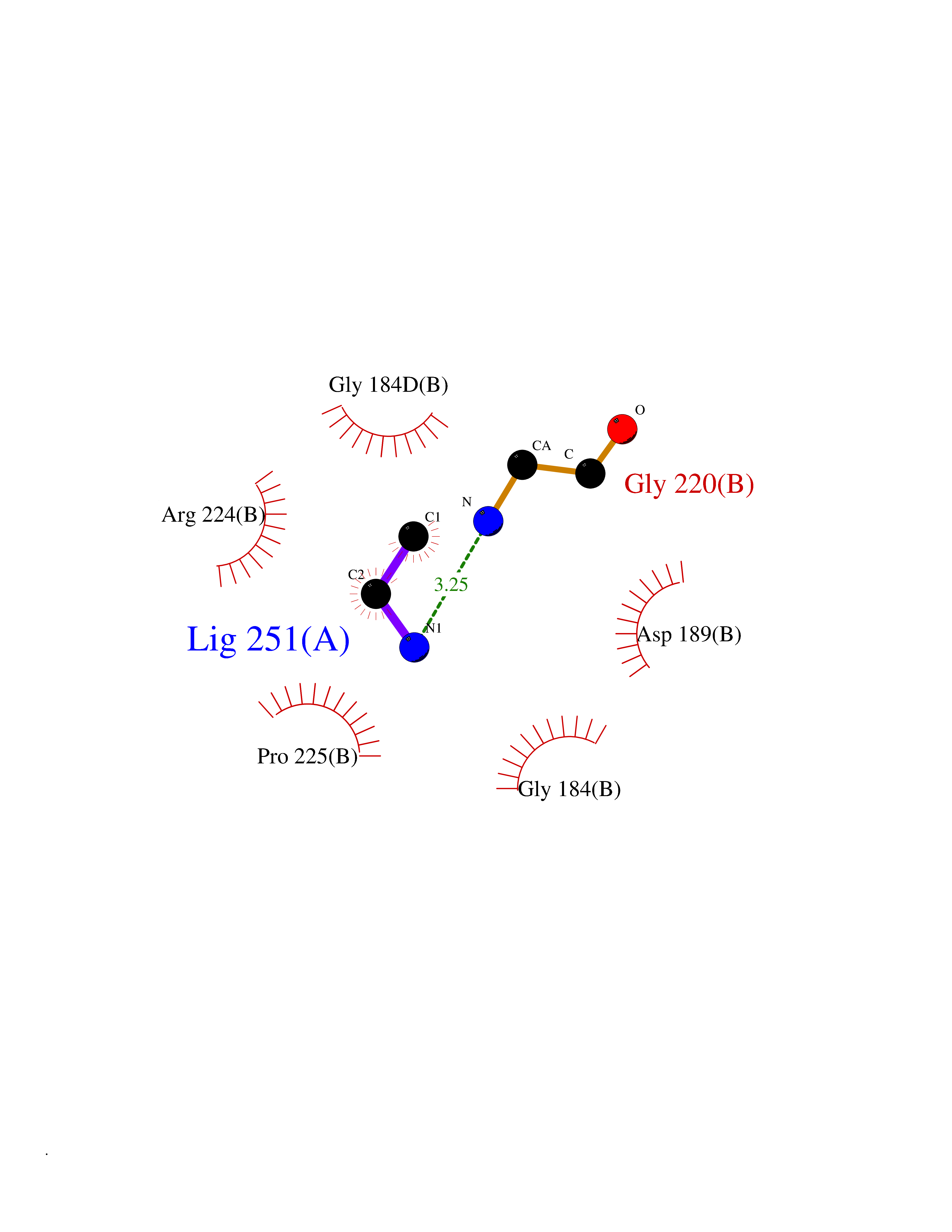 Ligplot Map 3D Binding mode Sequence ITGGSSAVAGQWPWQVSITYEGVHVCGGSLVSEQWVLSAAHCFPSEHHKEAYEVKLGAHQLDSYSEDAKVSTLKDIIPHPSYLQEGSQGDIALLQLSRPITFSRYIRPISLPAAQASFPNGLHCTVTGWGHVAPSVSLLTPKPLQQLEVPLISRETCNSLYNIDAKPEEPHFVQEDMVCAGYVEGGKDACQGDSGGPLSCPVEGLWYLTGIVSWGDACGARNRPGVYTLASSYASWIQSKVTELQPRV Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Membrane copper amine oxidase (AOC3) | 4BTX | 4.31 | |
Target general information Gen name AOC3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vascular adhesion protein-1; Vascular adhesion protein 1; VAP1; VAP-1; Semicarbazide-sensitive amine oxidase; SSAO; Membrane primary amine oxidase; HPAO; Copper amine oxidase Protein family Copper/topaquinone oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Has semicarbazide-sensitive (SSAO) monoamine oxidase activity. May play a role in adipogenesis. Cell adhesion protein that participates in lymphocyte extravasation and recirculation by mediating the binding of lymphocytes to peripheral lymph node vascular endothelial cells in an L-selectin-independent fashion. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB04334; DB01275; DB00780 Interacts with Q3SXY8; O95484; Q7Z7G2; Q96BA8; Q6PI48; Q8TBE3; Q7Z5P4; P42858; O43765; Q16623 EC number EC 1.4.3.21 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell adhesion; Cell membrane; Copper; Direct protein sequencing; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Oxidoreductase; Proteomics identification; Reference proteome; Signal-anchor; TPQ; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 157266 Length 1415 Aromaticity 0.12 Instability index 39.94 Isoelectric point 6 Charge (pH=7) -23.49 2D Binding mode Binding energy (Kcal/mol) -5.88 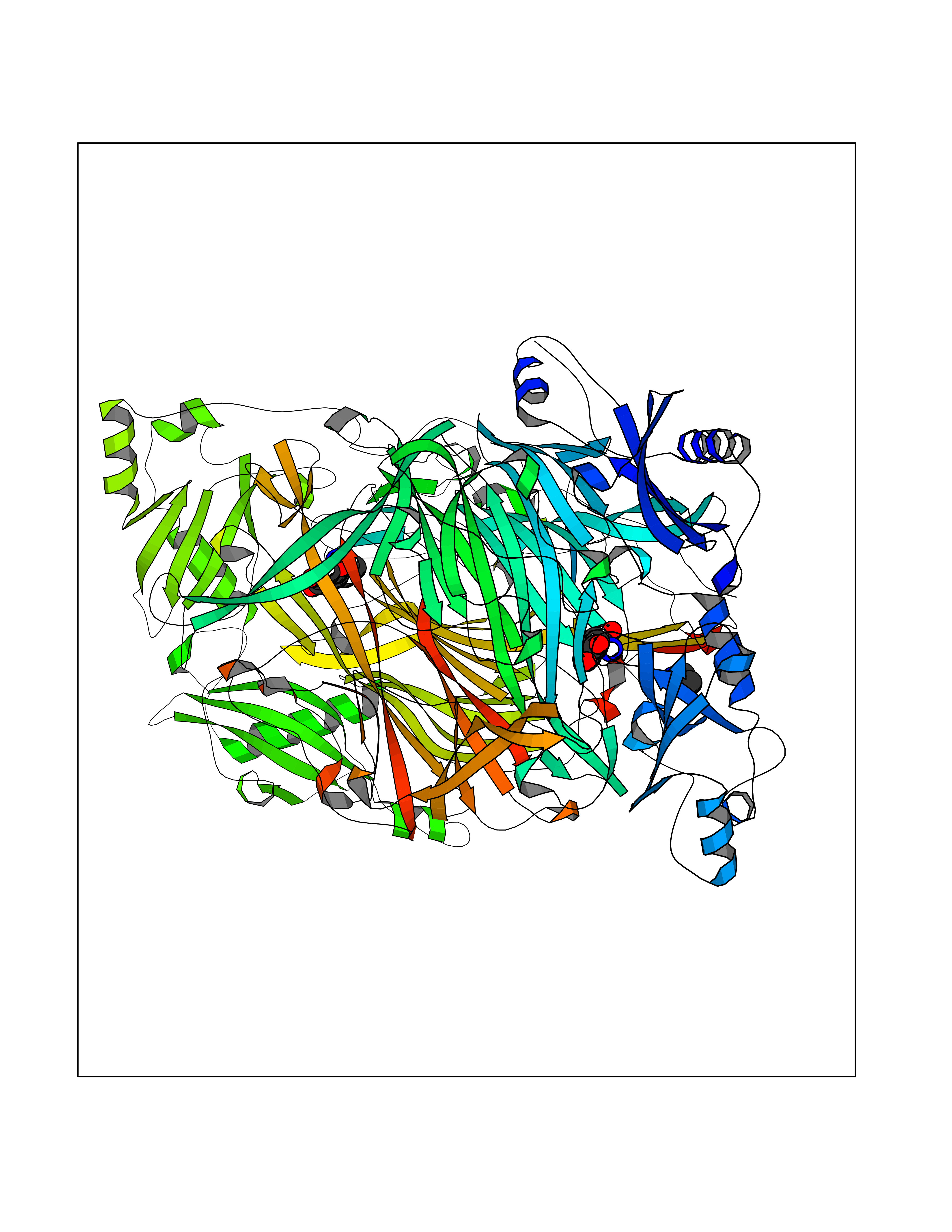 Molscript Map 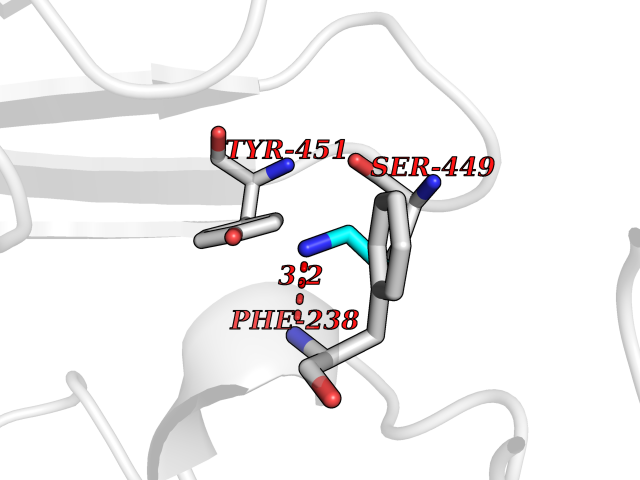 Pymol Map 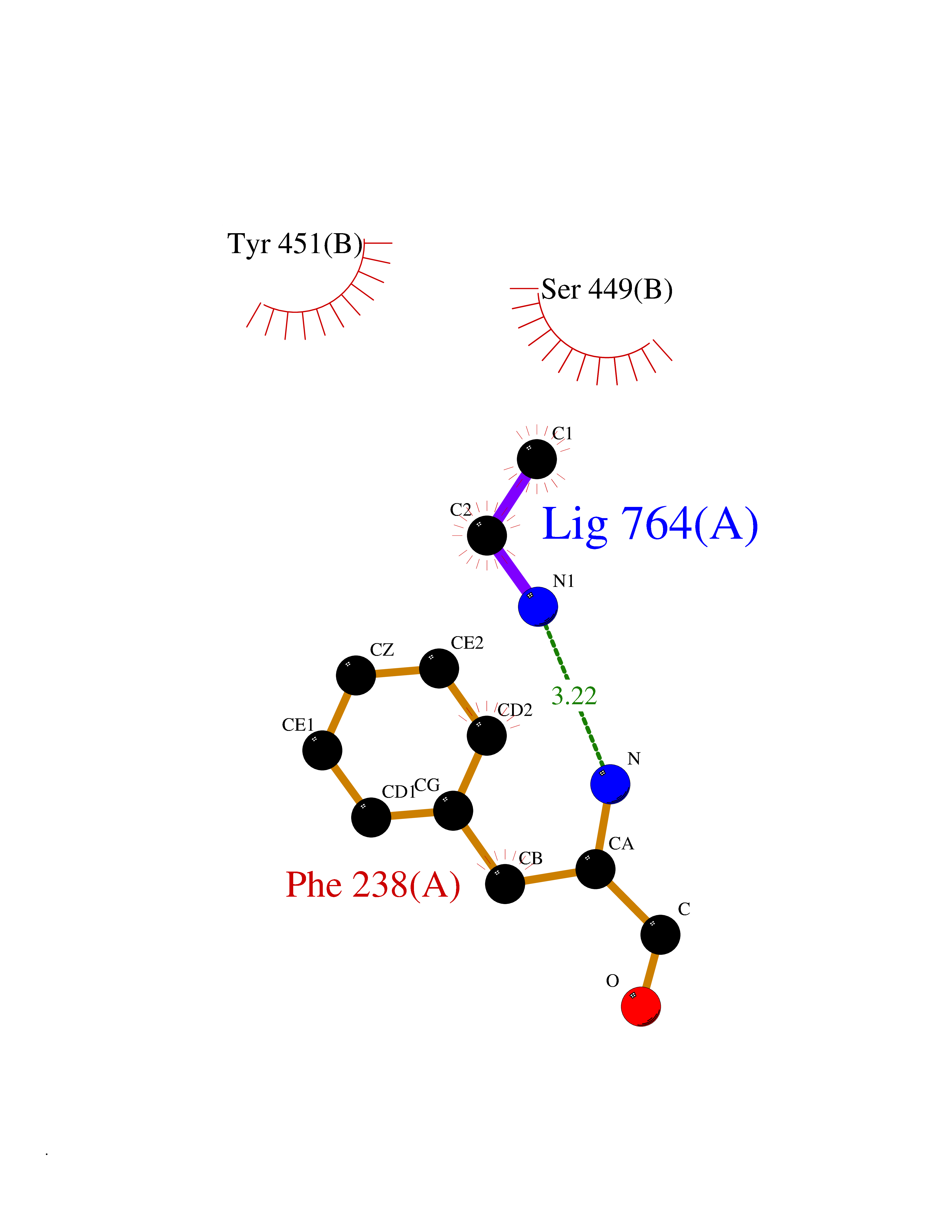 Ligplot Map 3D Binding mode Sequence PGQSQLFADLSREELTAVMRFLTQRLGPGLVDAAQARPSDNCVFSVELQLPPKAAALAHLDRGSPPPAREALAIVFFGRQPQPNVSELVVGPLPHPSYMRDVTVERHGGPLPYHRRPVLFQEYLDIDQMIFNRELPQASGLLHHCCFYKHRGRNLVTMTTAPRGLQSGDRATWFGLYYNISGAGFFLHHVGLELLVNHKALDPARWTIQKVFYQGRYYDSLAQLEAQFEAGLVNVVLIPDNGTGGSWSLKSPVPPGPAPPLQFYPQGPRFSVQGSRVASSLWTFSFGLGAFSGPRIFDVRFQGERLVYEISLQEALAIYGGNSPAAMTTRYVDGGFGMGKYTTPLTRGVDCPYLATYVDWHFLLESQAPKTIRDAFCVFEQNQGLPLRRHHSDLYSHYFGGLAETVLVVRSMSTLLNXDYVWDTVFHPSGAIEIRFYATGYISSAFLFGATGKYGNQVSEHTLGTVHTHSAHFKVDLDVAGLENWVWAEDMVFVPMAVPWSPEHQLQRLQVTRKLLEMEEQAAFLVGSATPRYLYLASNHSNKWGHPRGYRIQMLSFAGEPLPQNSSMARGFSWERYQLAVTQRKEEEPSSSSVFNQNDPWAPTVDFSDFINNETIAGKDLVAWVTAGFLHIPHAEDIPNTVTVGNGVGFFLRPYNFFDEDPSFYSADSIYFRGDQDAGACEVNPLACLPQAAACAPDLPAFSHGGFSHSQLFADLSREELTAVMRFLTQRLGPGLVDAAQARPSDNCVFSVELQLPPKAAALAHLDRGSPPPAREALAIVFFGRQPQPNVSELVVGPLPHPSYMRDVTVERHGGPLPYHRRPVLFQEYLDIDQMIFNRELPQASGLLHHCCFYKHRGRNLVTMTTAPRGLQSGDRATWFGLYYNISGAGFFLHHVGLELLVNHKALDPARWTIQKVFYQGRYYDSLAQLEAQFEAGLVNVVLIPDNGTGGSWSLKSPVPPGPAPPLQFYPQGPRFSVQGSRVASSLWTFSFGLGAFSGPRIFDVRFQGERLVYEISLQEALAIYGGNSPAAMTTRYVDGGFGMGKYTTPLTRGVDCPYLATYVDWHFLLESQAPKTIRDAFCVFEQNQGLPLRRHHSDLYSHYFGGLAETVLVVRSMSTLLNXDYVWDTVFHPSGAIEIRFYATGYISSAFLFGATGKYGNQVSEHTLGTVHTHSAHFKVDLDVAGLENWVWAEDMVFVPMAVPWSPEHQLQRLQVTRKLLEMEEQAAFLVGSATPRYLYLASNHSNKWGHPRGYRIQMLSFAGEPLPQNSSMARGFSWERYQLAVTQRKEEEPSSSSVFNQNDPWAPTVDFSDFINNETIAGKDLVAWVTAGFLHIPHAEDIPNTVTVGNGVGFFLRPYNFFDEDPSFYSADSIYFRGDQDAGACEVNPLACLPQAAACAPDLPAFSHGGFSH Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Beta-galactosidase (GLB1) | 3THD | 4.31 | |
Target general information Gen name GLB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lactase; Elastin receptor 1; ELNR1; Acid beta-galactosidase Protein family Glycosyl hydrolase 35 family Biochemical class NA Function Isoform 1: Cleaves beta-linked terminal galactosyl residues from gangliosides, glycoproteins, and glycosaminoglycans. Related diseases GM1-gangliosidosis 1 (GM1G1) [MIM:230500]: An autosomal recessive lysosomal storage disease marked by the accumulation of GM1 gangliosides, glycoproteins and keratan sulfate primarily in neurons of the central nervous system. GM1-gangliosidosis type 1 is characterized by onset within the first three months of life, central nervous system degeneration, coarse facial features, hepatosplenomegaly, skeletal dysmorphology reminiscent of Hurler syndrome, and rapidly progressive psychomotor deterioration. Urinary oligosaccharide levels are high. It leads to death usually between the first and second year of life. {ECO:0000269|PubMed:10338095, ECO:0000269|PubMed:10737981, ECO:0000269|PubMed:10839995, ECO:0000269|PubMed:1487238, ECO:0000269|PubMed:15365997, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:15791924, ECO:0000269|PubMed:16538002, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:1928092, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8213816, ECO:0000269|Ref.28, ECO:0000269|Ref.31}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GM1-gangliosidosis 2 (GM1G2) [MIM:230600]: A gangliosidosis characterized by onset between ages 1 and 5. The main symptom is locomotor ataxia, ultimately leading to a state of decerebration with epileptic seizures. Patients do not display the skeletal changes associated with the infantile form, but they nonetheless excrete elevated amounts of beta-linked galactose-terminal oligosaccharides. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10737981, ECO:0000269|PubMed:12644936, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8213816}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GM1-gangliosidosis 3 (GM1G3) [MIM:230650]: A gangliosidosis with a variable phenotype. Patients show mild skeletal abnormalities, dysarthria, gait disturbance, dystonia and visual impairment. Visceromegaly is absent. Intellectual deficit can initially be mild or absent but progresses over time. Inheritance is autosomal recessive. {ECO:0000269|PubMed:11511921, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:15986423, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8198123, ECO:0000269|Ref.28, ECO:0000269|Ref.30}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 4B (MPS4B) [MIM:253010]: A form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life. {ECO:0000269|PubMed:11511921, ECO:0000269|PubMed:12393180, ECO:0000269|PubMed:16538002, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1928092, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:7586649}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04465 Interacts with Q8NBJ4; Q3KNW5; Q9BRI3; P30825 EC number EC 3.2.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Gangliosidosis; Glycoprotein; Glycosidase; Hydrolase; Lysosome; Mucopolysaccharidosis; Proteomics identification; Reference proteome; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 67980.6 Length 605 Aromaticity 0.13 Instability index 40.91 Isoelectric point 5.81 Charge (pH=7) -9.05 2D Binding mode Binding energy (Kcal/mol) -5.88 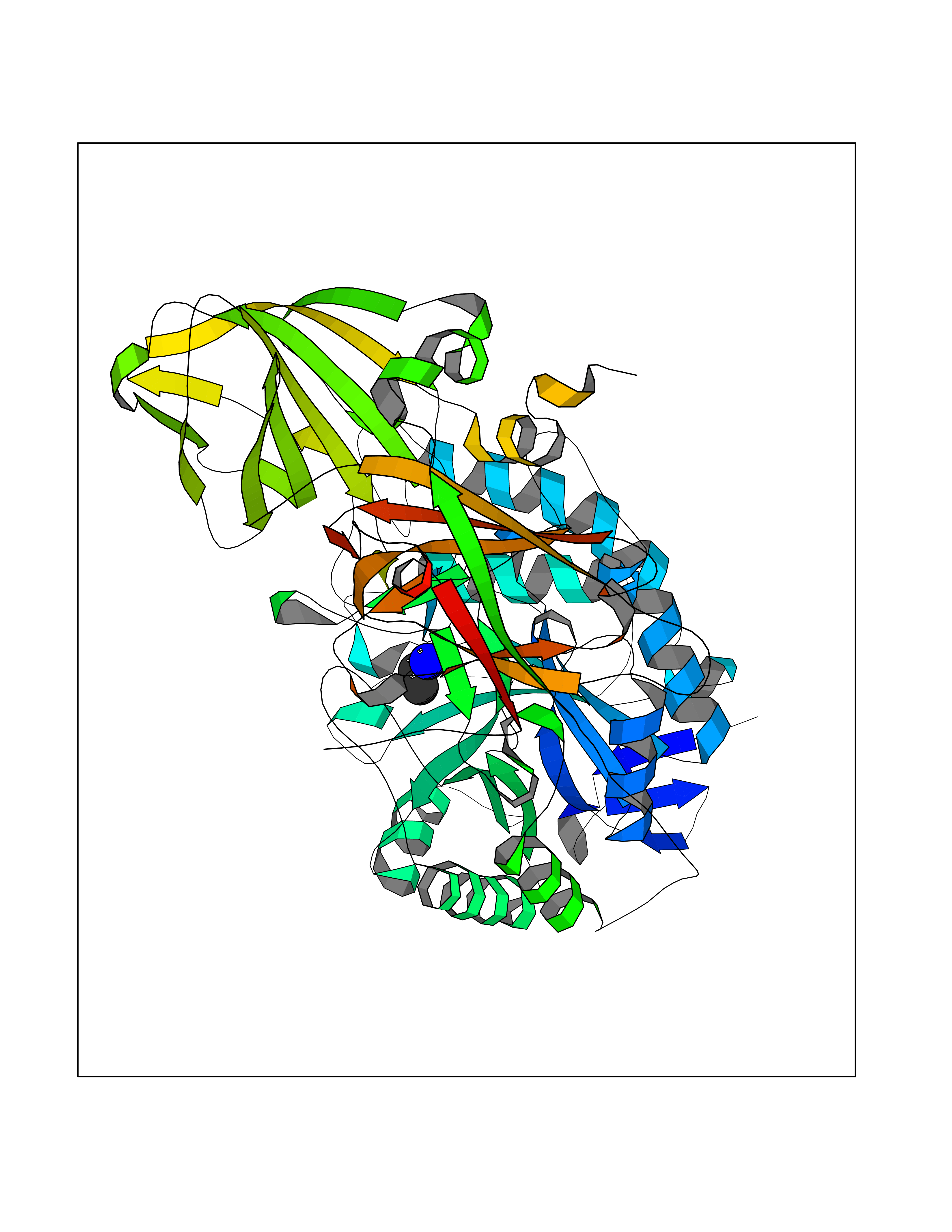 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence QRMFEIDYSRDSFLKDGQPFRYISGSIHYSRVPRFYWKDRLLKMKMAGLNAIQTYVPWNFHEPWPGQYQFSEDHDVEYFLRLAHELGLLVILRPGPYICAEWEMGGLPAWLLEKESILLRSSDPDYLAAVDKWLGVLLPKMKPLLYQNGGPVITVQVENEYGSYFACDFDYLRFLQKRFRHHLGDDVVLFTTDGAHKTFLKCGALQGLYTTVDFGTGSNITDAFLSQRKCEPKGPLINSEFYTGWLDHWGQPHSTIKTEAVASSLYDILARGASVNLYMFIGGTNFAYWNGANSPYAAQPTSYDYDAPLSEAGDLTEKYFALRNIIQKFEKVPEGPIPPSTPKFAYGKVTLEKLKTVGAALDILCPSGPIKSLYPLTFIQVKQHYGFVLYRTTLPQDCSNPAPLSSPLNGVHDRAYVAVDGIPQGVLERNNVITLNITGKAGATLDLLVENMGRVNYGAYINDFKGLVSNLTLSSNILTDWTIFPLDTEDAVRSHLGGWGHRNYTLPAFYMGNFSIPSGIPDLPQDTFIQFPGWTKGQVWINGFNLGRYWPARGPQLTLFVPQHILMTSAPNTITVLELEWAPCSSDDPELCAVTFVDRPVIGSS Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Cathepsin S (CTSS) | 2OP3 | 4.31 | |
Target general information Gen name CTSS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cysteine protease cathepsin S Protein family Peptidase C1 family Biochemical class Peptidase Function Key protease responsible for the removal of the invariant chain from MHC class II molecules. The bond-specificity of this proteinase is in part similar to the specificities of cathepsin L. Thiol protease. Related diseases Autoinflammatory disease, systemic, with vasculitis (SAIDV) [MIM:620376]: An autosomal dominant disorder characterized by systemic autoinflammation manifesting in the first hours of life with diffuse purpuric skin lesions, fever, hepatosplenomegaly, and increased C-reactive protein. Additional clinical features include periorbital edema, conjunctivitis, urticaria, atopic dermatitis, abdominal pain, and arthralgia. Laboratory studies may show leukocytosis, thrombocytopenia, and autoantibodies. {ECO:0000269|PubMed:16920712, ECO:0000269|PubMed:36122175, ECO:0000269|PubMed:36932076, ECO:0000269|Ref.67}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Constitutively phosphorylated and activated in cells from a number of chronic myelogenous leukemia (CML) and acute myeloid leukemia (AML) patients. Mediates phosphorylation of the BCR-ABL fusion protein. Abnormally elevated expression levels or activation of LYN signaling may play a role in survival and proliferation of some types of cancer cells. Drugs (DrugBank ID) DB08195; DB08611; DB12010; DB03837; DB03984; DB03767; DB07587; DB07878; DB08752; DB08755; DB07589; DB07520; DB07839 Interacts with NA EC number EC 3.4.22.27 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lysosome; Protease; Proteomics identification; Reference proteome; Secreted; Signal; Thiol protease; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 23978.7 Length 217 Aromaticity 0.12 Instability index 24.68 Isoelectric point 7.64 Charge (pH=7) 1.03 2D Binding mode Binding energy (Kcal/mol) -5.88  Molscript Map  Pymol Map 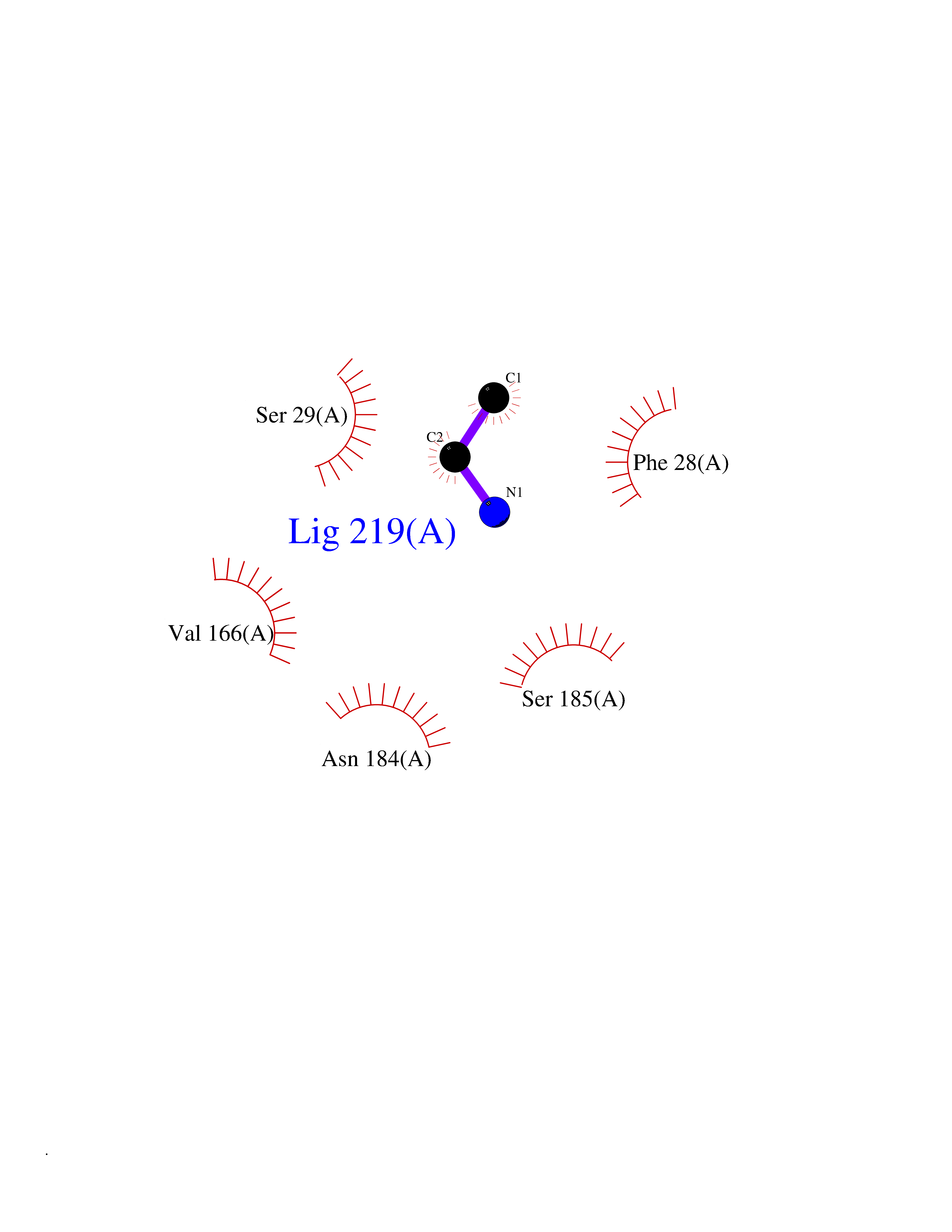 Ligplot Map 3D Binding mode Sequence LPDSVDWREKGCVTEVKYQGSCGACWAFSAVGALEAQLKLKTGKLVSLSAQNLVDCSTEKYGNKGCNGGFMTTAFQYIIDNKGIDSDASYPYKAMDQKCQYDSKYRAATCSKYTELPYGREDVLKEAVANKGPVSVGVDARHPSFFLYRSGVYYEPSCTQNVNHGVLVVGYGDLNGKEYWLVKNSWGHNFGEEGYIRMARNKGNHCGIASFPSYPEI Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Cerebroside-sulfatase (ARSA) | 1E2S | 4.31 | |
Target general information Gen name ARSA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cerebrosidesulfatase; Arylsulfatase A component C; ASA; ARSA Protein family Sulfatase family Biochemical class Sulfuric ester hydrolase Function Hydrolyzes cerebroside sulfate. Related diseases Metachromatic leukodystrophy (MLD) [MIM:250100]: An autosomal recessive disease caused by abnormal intralysosomal accumulation of cerebroside-3-sulfate in central and peripheral nervous systems, as well as other organs. MLD is clinically characterized by leukodystrophy, progressive demyelination and a variety of neurological symptoms, including gait disturbances, ataxias, optical atrophy, dementia, seizures, and spastic tetraparesis. Decreased arylsulfatase A activity is detected in urine, leukocytes, and fibroblasts of affected individuals. Several forms of the disease can be distinguished according to the age at onset and disease severity: late infantile, juvenile and adult forms, partial cerebroside sulfate deficiency, and pseudoarylsulfatase A deficiency. Individuals with pseudoarylsulfatase A deficiency have low arylsulfatase A activity but lack neurological manifestations and are apparently healthy. {ECO:0000269|PubMed:10220151, ECO:0000269|PubMed:10381328, ECO:0000269|PubMed:10477432, ECO:0000269|PubMed:10533072, ECO:0000269|PubMed:10751093, ECO:0000269|PubMed:11020646, ECO:0000269|PubMed:11061266, ECO:0000269|PubMed:11456299, ECO:0000269|PubMed:11941485, ECO:0000269|PubMed:12503099, ECO:0000269|PubMed:12788103, ECO:0000269|PubMed:1353340, ECO:0000269|PubMed:14517960, ECO:0000269|PubMed:14680985, ECO:0000269|PubMed:15026521, ECO:0000269|PubMed:15326627, ECO:0000269|PubMed:15710861, ECO:0000269|PubMed:1670590, ECO:0000269|PubMed:1673291, ECO:0000269|PubMed:1678251, ECO:0000269|PubMed:18693274, ECO:0000269|PubMed:19606494, ECO:0000269|PubMed:20339381, ECO:0000269|PubMed:21265945, ECO:0000269|PubMed:2574462, ECO:0000269|PubMed:7581401, ECO:0000269|PubMed:7825603, ECO:0000269|PubMed:7860068, ECO:0000269|PubMed:7902317, ECO:0000269|PubMed:7906588, ECO:0000269|PubMed:7909527, ECO:0000269|PubMed:8095918, ECO:0000269|PubMed:8101038, ECO:0000269|PubMed:8101083, ECO:0000269|PubMed:8104633, ECO:0000269|PubMed:8891236, ECO:0000269|PubMed:9090526, ECO:0000269|PubMed:9272717, ECO:0000269|PubMed:9452102, ECO:0000269|PubMed:9490297, ECO:0000269|PubMed:9600244, ECO:0000269|PubMed:9819708}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Multiple sulfatase deficiency (MSD) [MIM:272200]: A clinically and biochemically heterogeneous disorder caused by the simultaneous impairment of all sulfatases, due to defective post-translational modification and activation. It combines features of individual sulfatase deficiencies such as metachromatic leukodystrophy, mucopolysaccharidosis, chondrodysplasia punctata, hydrocephalus, ichthyosis, neurologic deterioration and developmental delay. {ECO:0000269|PubMed:15146462}. The protein represented in this entry is involved in disease pathogenesis. Arylsulfatase A activity is impaired in multiple sulfatase deficiency due to mutations in SUMF1 (PubMed:15146462). SUMF1 mutations result in defective post-translational modification of ARSA at residue Cys-69 that is not converted to 3-oxoalanine (PubMed:7628016). {ECO:0000269|PubMed:15146462, ECO:0000269|PubMed:7628016}. Drugs (DrugBank ID) DB03821; DB01800; DB01141; DB04786 Interacts with P50995; Q6P5X5; Q13554-3; O60826; Q96D98; Q9H0I2; Q12951-2; Q16512; P28069; O75360; Q9BQY4; Q15645; O95231 EC number EC 3.1.6.8 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Hydrolase; Ichthyosis; Leukodystrophy; Lipid metabolism; Lysosome; Metachromatic leukodystrophy; Metal-binding; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID P Molecular weight (Da) 51173.7 Length 481 Aromaticity 0.09 Instability index 47.42 Isoelectric point 5.59 Charge (pH=7) -12.84 2D Binding mode Binding energy (Kcal/mol) -5.87 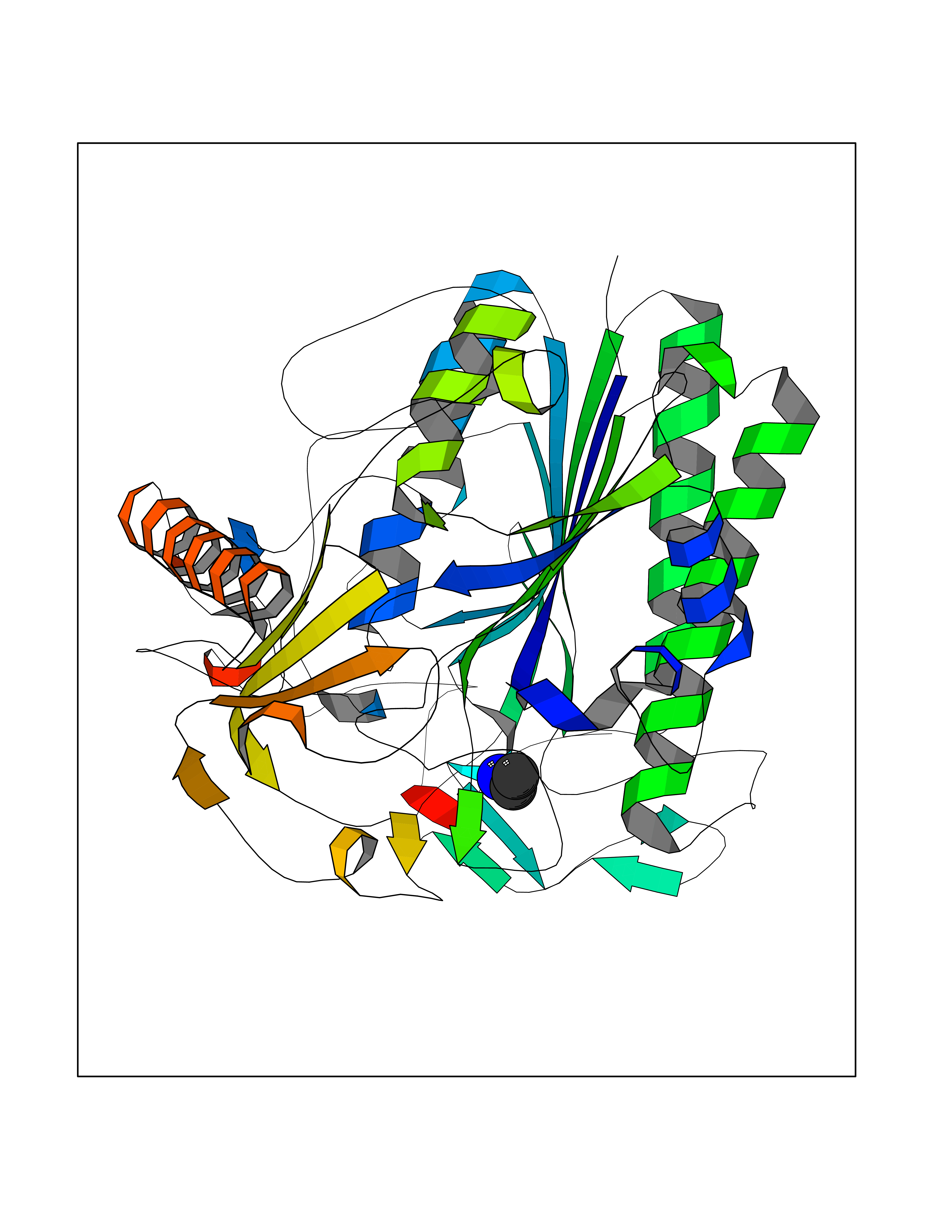 Molscript Map 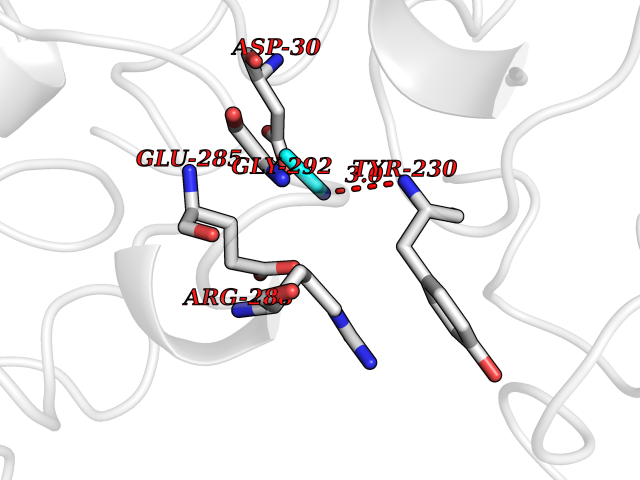 Pymol Map 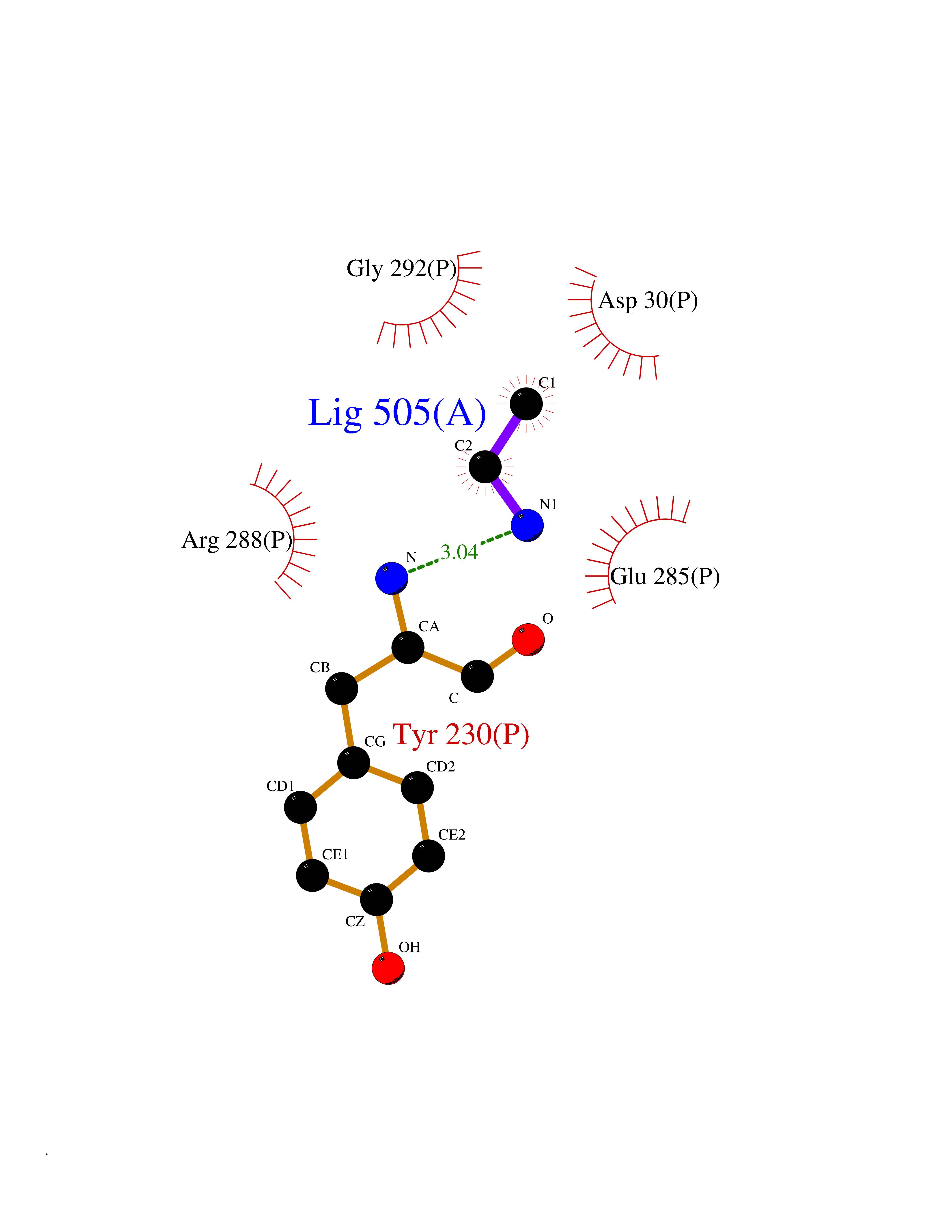 Ligplot Map 3D Binding mode Sequence RPPNIVLIFADDLGYGDLGCYGHPSSTTPNLDQLAAGGLRFTDFYVPVSLATPSRAALLTGRLPVRMGMYPGVLVPSSRGGLPLEEVTVAEVLAARGYLTGMAGKWHLGVGPEGAFLPPHQGFHRFLGIPYSHDQGPCQNLTCFPPATPCDGGCDQGLVPIPLLANLSVEAQPPWLPGLEARYMAFAHDLMADAQRQDRPFFLYYASHHTHYPQFSGQSFAERSGRGPFGDSLMELDAAVGTLMTAIGDLGLLEETLVIFTADNGPETMRMSRGGCSGLLRCGKGTTYEGGVREPALAFWPGHIAPGVTHELASSLDLLPTLAALAGAPLPNVTLDGFDLSPLLLGTGKSPRQSLFFYPSYPDEVRGVFAVRTGKYKAHFFTQGSAHSDTTADPACHASSSLTAHEPPLLYDLSKDPGENYNLLGATPEVLQALKQLQLLKAQLDAAVTFGPSQVARGEDPALQICCHPGCTPRPACCHCP Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Fumarate reductase flavoprotein subunit | 1KF6 | 4.30 | |
Target general information Gen name frdA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW4115;b4154 Protein family FAD-dependent oxidoreductase 2 family, FRD/SDH subfamily Biochemical class Oxidoreductase Function Electron carrier activity.FAD binding.Fumarate reductase (menaquinone).Succinate dehydrogenase activity. Related diseases Glycogen storage disease 11 (GSD11) [MIM:612933]: A metabolic disorder that results in exertional myoglobinuria, pain, cramps and easy fatigue. {ECO:0000269|PubMed:2334430}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07490; DB07918; DB00730 Interacts with P0AC47; P0ACB4; P76111 EC number 1.3.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; Electron transport; FAD; Flavoprotein; Membrane; Nucleotide-binding; Oxidoreductase; Reference proteome; Transport Protein physicochemical properties Chain ID A,M Molecular weight (Da) 90370.7 Length 820 Aromaticity 0.08 Instability index 28.88 Isoelectric point 5.86 Charge (pH=7) -16.21 2D Binding mode Binding energy (Kcal/mol) -5.87 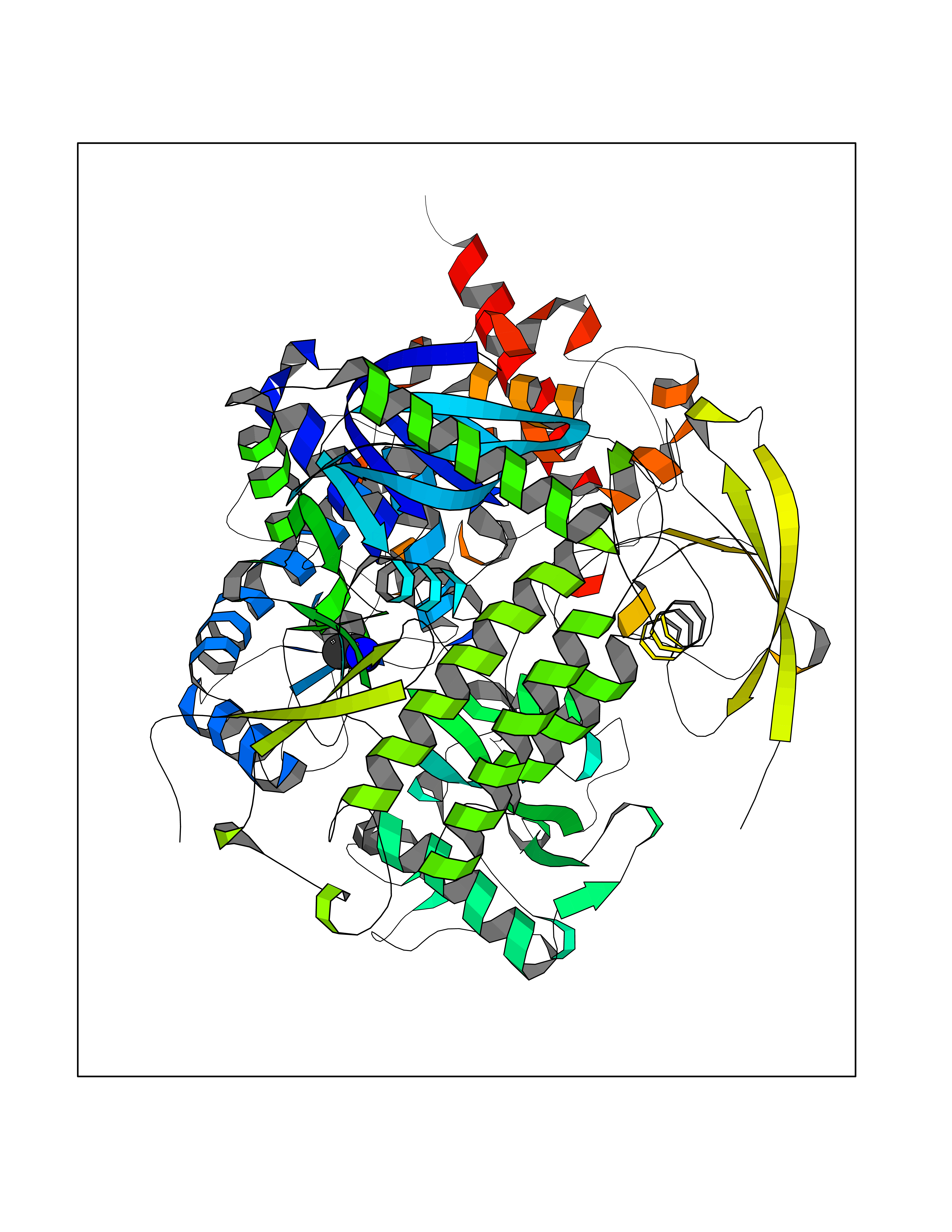 Molscript Map 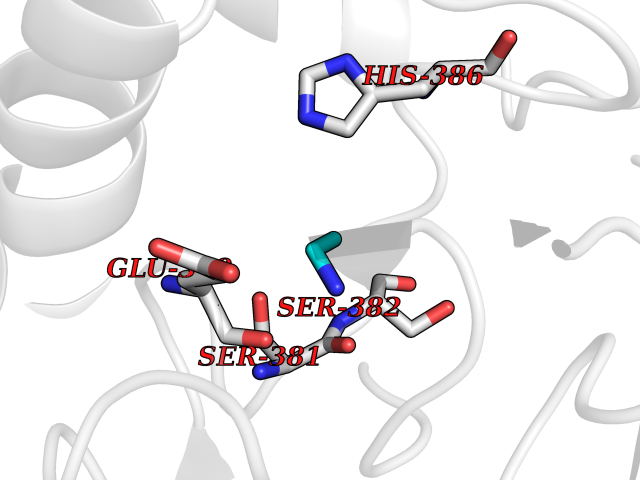 Pymol Map 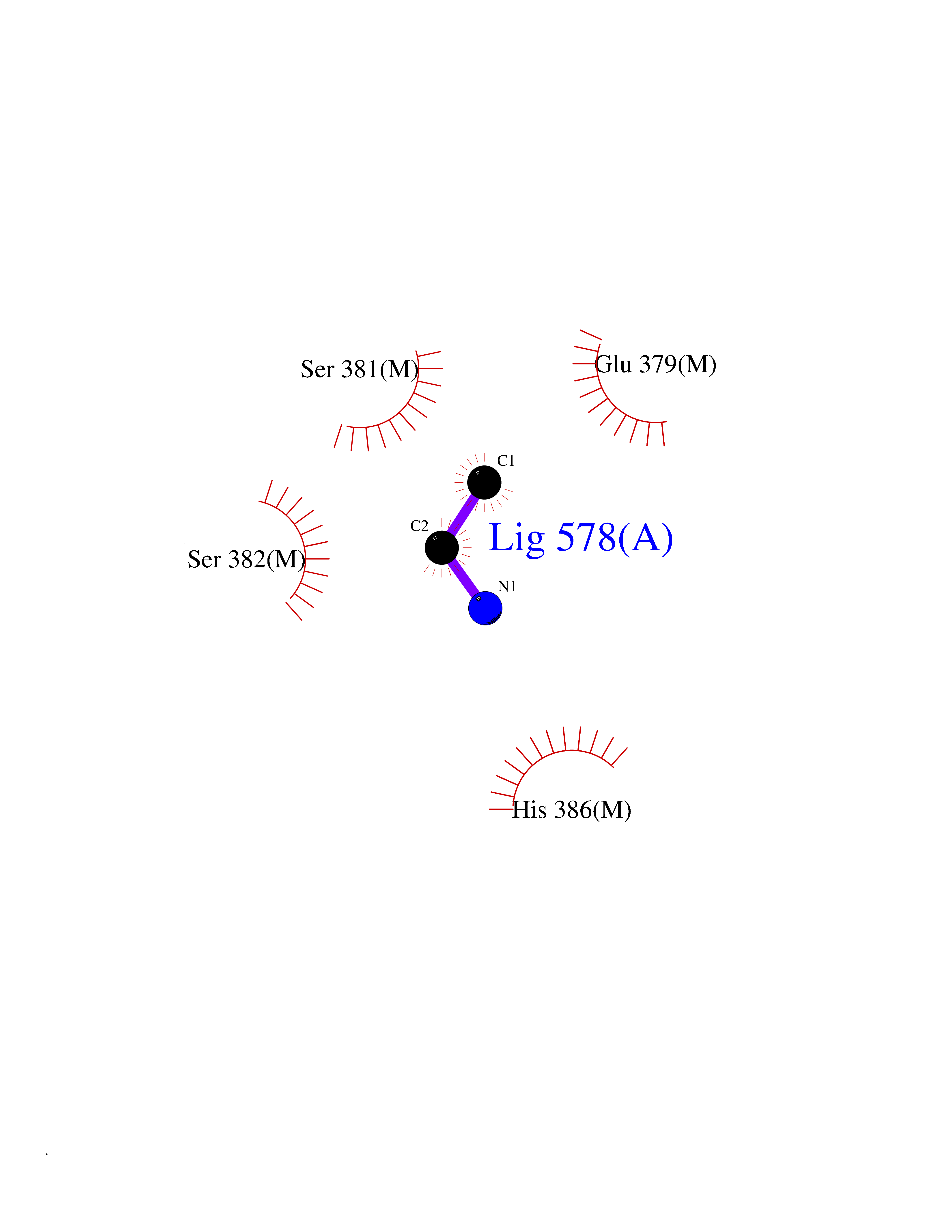 Ligplot Map 3D Binding mode Sequence MQTFQADLAIVGAGGAGLRAAIAAAQANPNAKIALISKVYPMRSHTVAAEGGSAAVAQDHDSFEYHFHDTVAGGDWLCEQDVVDYFVHHCPTEMTQLELWGCPWSRRPDGSVNVRRFGGMKIERTWFAADKTGFHMLHTLFQTSLQFPQIQRFDEHFVLDILVDDGHVRGLVAMNMMEGTLVQIRANAVVMATGGAGRVYRYNTNGGIVTGDGMGMALSHGVPLRDMEFVQYHPTGLPGSGILMTEGCRGEGGILVNKNGYRYLQDYGMGPETPLGEPKNKYMELGPRDKVSQAFWHEWRKGNTISTPRGDVVYLDLRHLGEKKLHERLPFICELAKAYVGVDPVKEPIPVRPTAHYTMGGIETDQNCETRIKGLFAVGECSSVGLHGANRLGSNSLAELVVFGRLAGEQATERAATAGNGNEAAIEAQAAGVEQRLKDLVNQDGGENWAKIRDEMGLAMEEGCGIYRTPELMQKTIDKLAELQERFKRVRITDTSSVFNTDLLYTIELGHGLNVAECMAHSAMARKESRGAHQRLDEGCTERDDVNFLKHTLAFRDADGTTRLEYSDVKITTLPPAAEMKNLKIEVVRYNPEVDTAPHSAFYEVPYDATTSLLDALGYIKDNLAPDLSYRWSCRMAICGSCGMMVNNVPKLACKTFLRDYTDGMKVEALANFPIERDLVVDMTHFIESLEAIKPYIIGNSRTADQGTNIQTPAQMAKYHQFSGCINCGLCYAACPQFGLNPEFIGPAAITLAHRYNEDSRDHGKKERMAQLNSQNGVWSCTFVGYCSEVCPKHVDPAAAIQQGKVESSKDFLIATLKPR Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Isovaleryl-CoA dehydrogenase, mitochondrial | 1IVH | 4.30 | |
Target general information Gen name IVD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Isovaleryl-CoA dehydrogenase activity. Related diseases Isovaleric acidemia (IVA) [MIM:243500]: A metabolic disorder characterized by retarded psychomotor development, a peculiar odor resembling sweaty feet, an aversion to dietary protein, and pernicious vomiting, leading to acidosis and coma. The acute neonatal form leads to massive metabolic acidosis from the first days of life and rapid death. {ECO:0000269|PubMed:2063866, ECO:0000269|PubMed:22004070, ECO:0000269|PubMed:22350545, ECO:0000269|PubMed:23587913, ECO:0000269|PubMed:28535199, ECO:0000269|PubMed:9665741}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04036; DB03147 Interacts with Q08043; Q9Y4H4 EC number 1.3.8.1; 1.3.8.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; FAD; Fatty acid metabolism; Flavoprotein; Lipid metabolism; Mitochondrion; Oxidoreductase; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 84454.2 Length 774 Aromaticity 0.08 Instability index 30.01 Isoelectric point 6.85 Charge (pH=7) -0.77 2D Binding mode Binding energy (Kcal/mol) -5.87 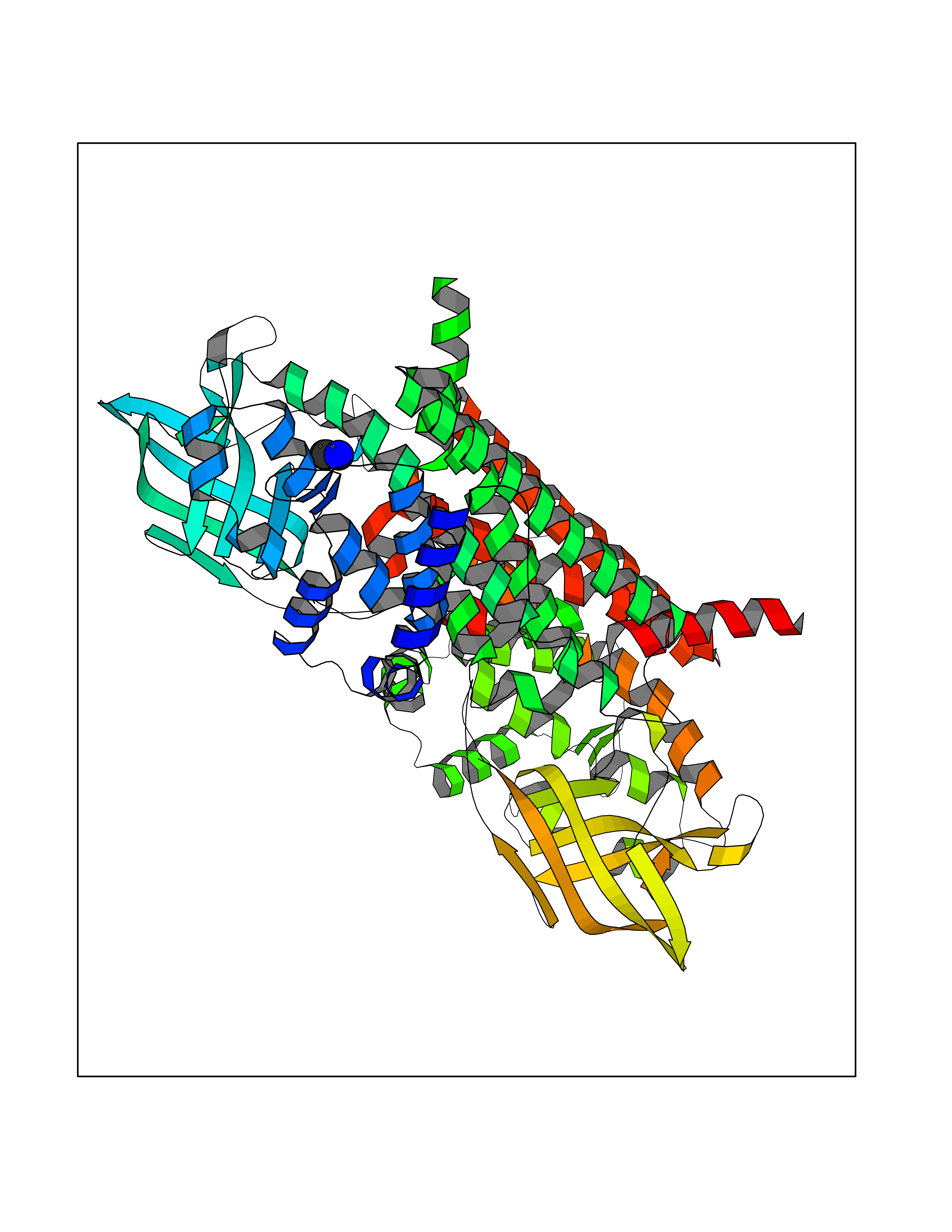 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence VDDAINGLSEEQRQLRQTMAKFLQEHLAPKAQEIDRSNEFKNLREFWKQLGNLGVLGITAPVQYGGSGLGYLEHVLVMEEISRASGAVGLSYGAHSNLCINQLVRNGNEAQKEKYLPKLISGEYIGALAMSEPNAGSDVVSMKLKAEKKGNHYILNGNKFWITNGPDADVLIVYAKTDLAAVPASRGITAFIVEKGMPGFSTSKKLDKLGMRGSNTCELIFEDCKIPAANILGHENKGVYVLMSGLDLERLVLAGGPLGLMQAVLDHTIPYLHVREAFGQKIGHFQLMQGKMADMYTRLMACRQYVYNVAKACDEGHCTAKDCAGVILYSAECATQVALDGIQCFGGNGYINDFPMGRFLRDAKLYEIGAGTSEVRRLVIGRAFNADVDDAINGLSEEQRQLRQTMAKFLQEHLAPKAQEIDRSNEFKNLREFWKQLGNLGVLGITAPVQYGGSGLGYLEHVLVMEEISRASGAVGLSYGAHSNLCINQLVRNGNEAQKEKYLPKLISGEYIGALAMSEPNAGSDVVSMKLKAEKKGNHYILNGNKFWITNGPDADVLIVYAKTDLAAVPASRGITAFIVEKGMPGFSTSKKLDKLGMRGSNTCELIFEDCKIPAANILGHENKGVYVLMSGLDLERLVLAGGPLGLMQAVLDHTIPYLHVREAFGQKIGHFQLMQGKMADMYTRLMACRQYVYNVAKACDEGHCTAKDCAGVILYSAECATQVALDGIQCFGGNGYINDFPMGRFLRDAKLYEIGAGTSEVRRLVIGRAFNAD Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Ribonucleoside-diphosphate reductase subunit M2 | 3OLJ | 4.30 | |
Target general information Gen name RRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms RR2 Protein family Ribonucleoside diphosphate reductase small chain family Biochemical class Oxidoreductase Function Metal ion binding.Ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00242; DB05260; DB05801; DB05003; DB05428 Interacts with P41002; Q9UM11; P23921; O00560 EC number 1.17.4.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Deoxyribonucleotide synthesis; Iron; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 33579.4 Length 286 Aromaticity 0.14 Instability index 43.7 Isoelectric point 5.12 Charge (pH=7) -12.86 2D Binding mode Binding energy (Kcal/mol) -5.86  Molscript Map  Pymol Map 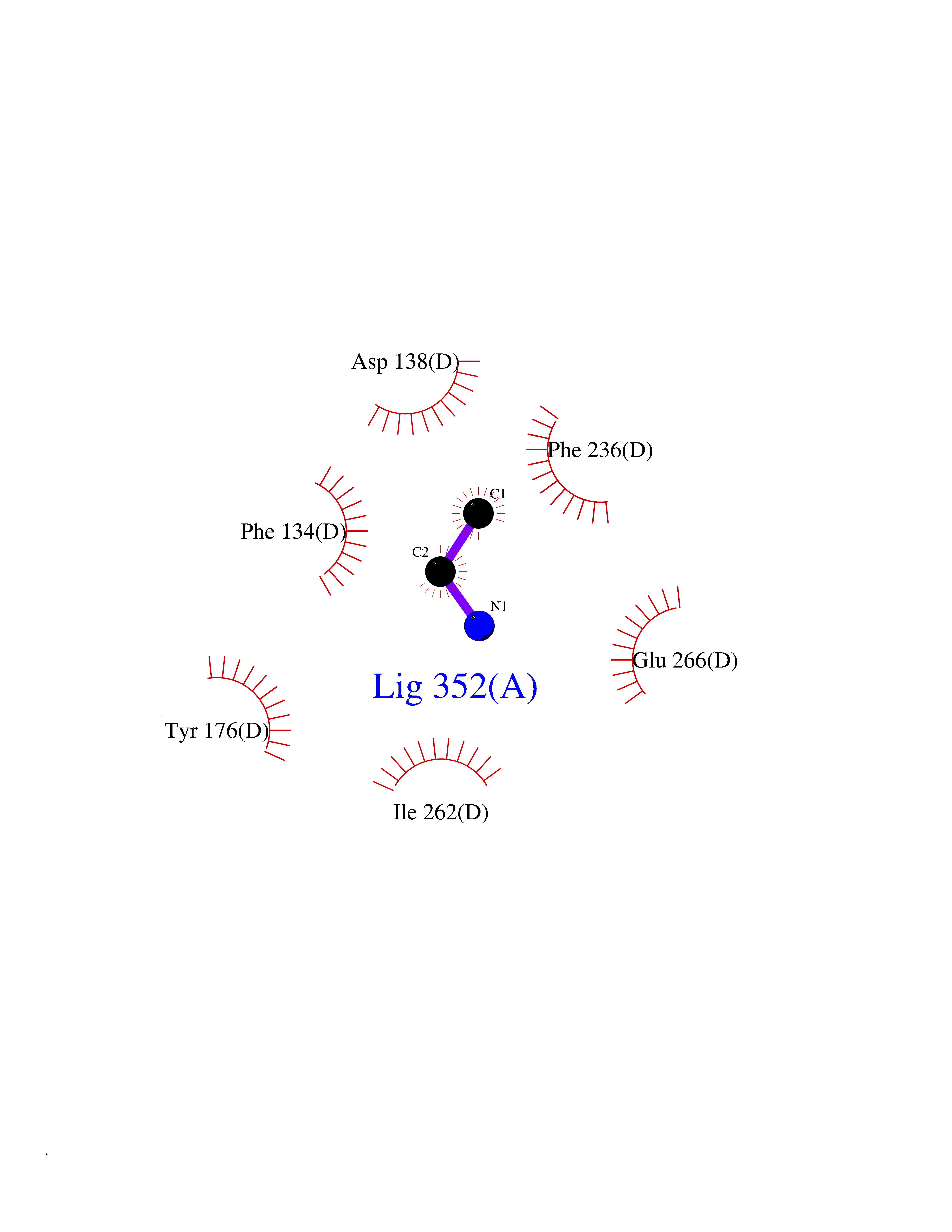 Ligplot Map 3D Binding mode Sequence MGVEDEPLLRENPRRFVIFPIEYHDIWQMYKKAEASFWTAEEVDLSKDIQHWESLKPEERYFISHVLAFFAASDGIVNENLVERFSQEVQITEARCFYGFQIAMENIHSEMYSLLIDTYIKDPKEREFLFNAIETMPCVKKKADWALRWIGDKEATYGERVVAFAAVEGIFFSGSFASIFWLKKRGLMPGLTFSNELISRDEGLHCDFACLMFKHLVHKPSEERVREIIINAVRIEQEFLTEALPVKLIGMNCTLMKQYIEFVADRLMLELGFSKVFRVENPFDFM Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Phosphoribosylaminoimidazolecarboxamide formyltransferase (ATIC) | 1PKX | 4.30 | |
Target general information Gen name ATIC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PURH; OK/SW-cl.86; Bifunctional purine biosynthesis protein PURH Protein family PurH family Biochemical class Methyltransferase Function Bifunctional enzyme that catalyzes 2 steps in purine biosynthesis. Related diseases AICA-ribosuria due to ATIC deficiency (AICAR) [MIM:608688]: A neurologically devastating inborn error of purine biosynthesis. Patients excrete massive amounts of AICA-riboside in the urine and accumulate AICA-ribotide and its derivatives in erythrocytes and fibroblasts. Clinical features include profound intellectual disability, epilepsy, dysmorphic features and congenital blindness. AICAR inheritance is autosomal recessive. {ECO:0000269|PubMed:15114530}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02309; DB03442; DB01700; DB01972; DB00563; DB04057; DB00642; DB00116 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Epilepsy; Hydrolase; Intellectual disability; Multifunctional enzyme; Proteomics identification; Purine biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 41584.9 Length 386 Aromaticity 0.06 Instability index 42 Isoelectric point 5.6 Charge (pH=7) -7.51 2D Binding mode Binding energy (Kcal/mol) -5.87 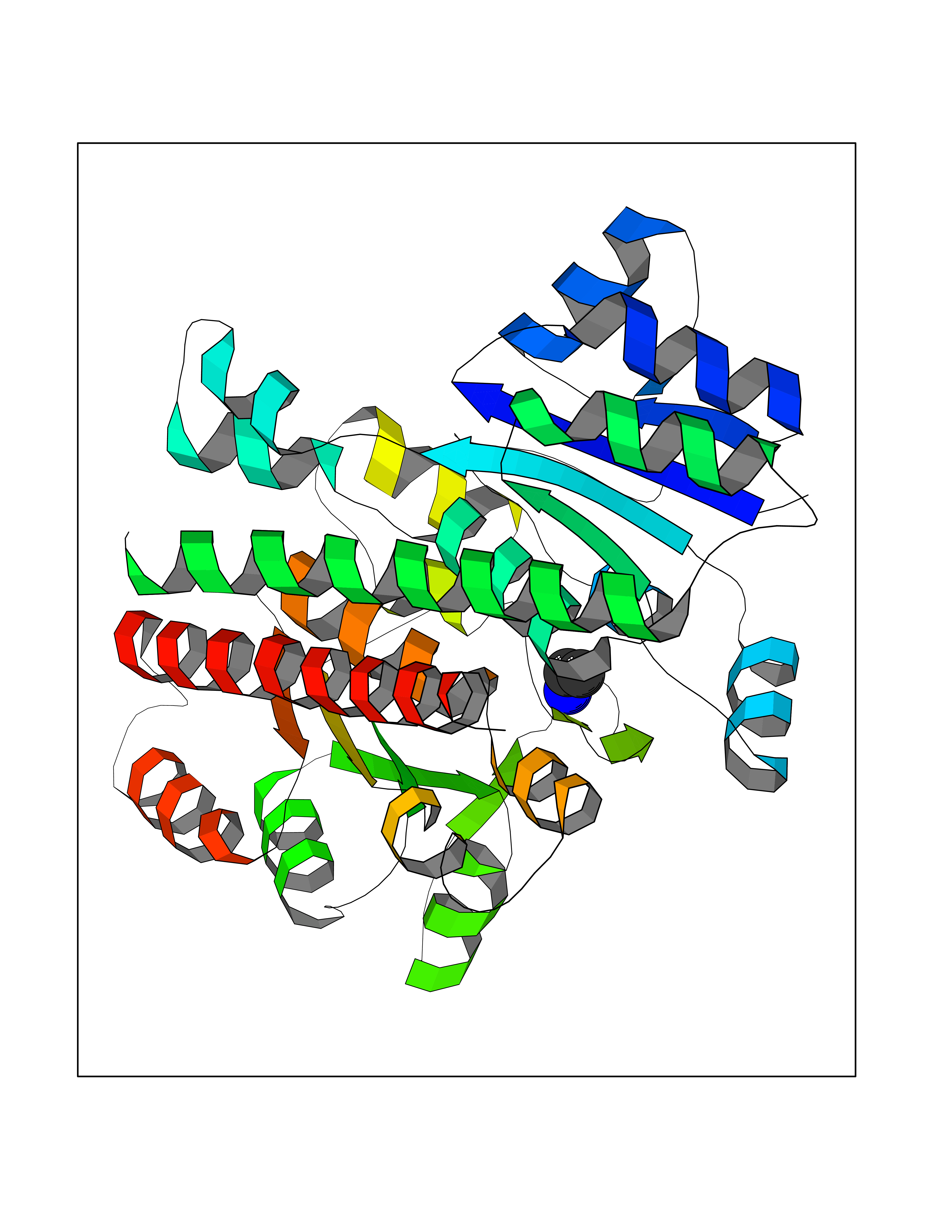 Molscript Map 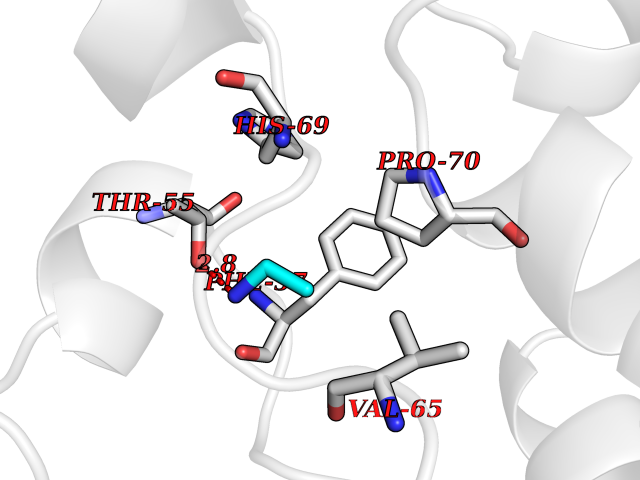 Pymol Map 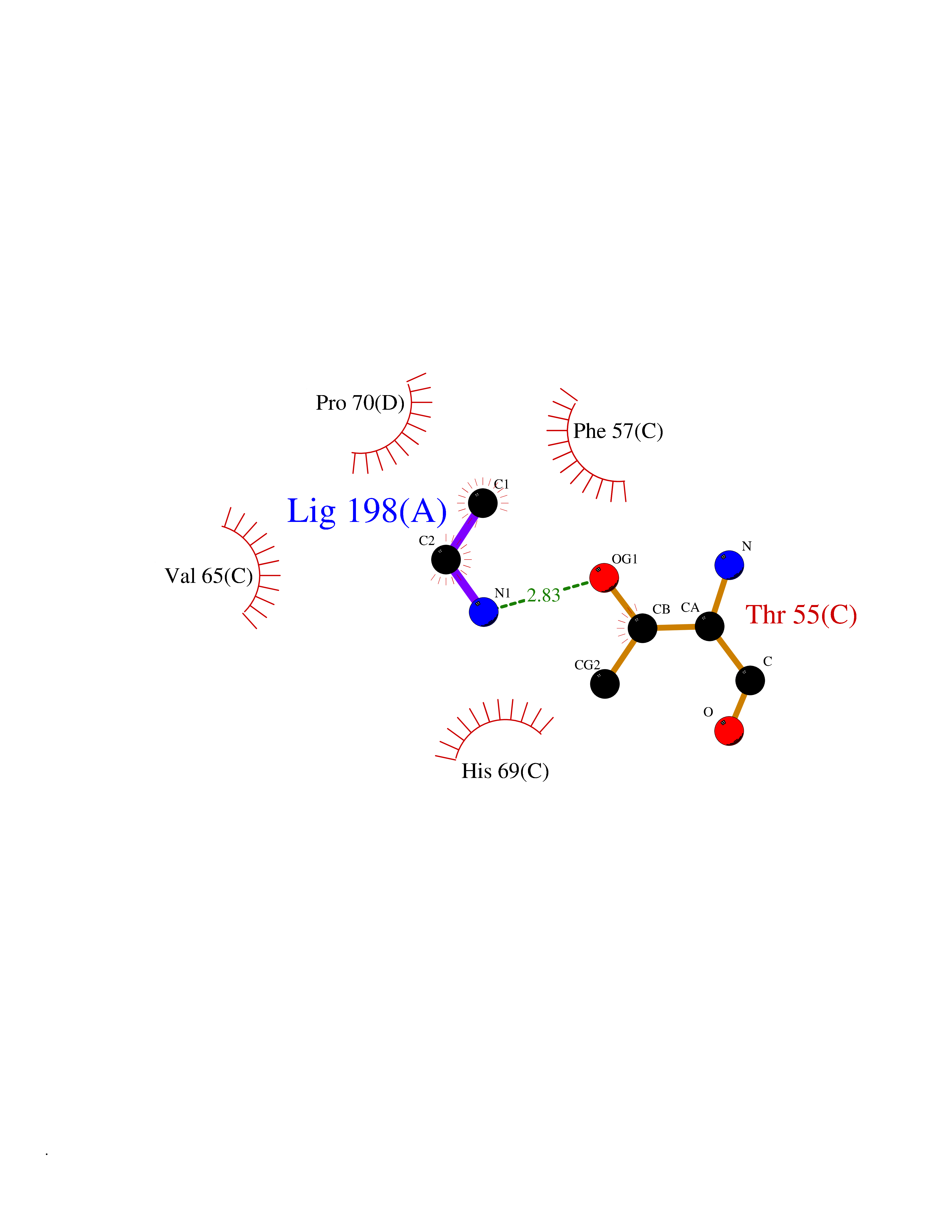 Ligplot Map 3D Binding mode Sequence GQLALFSVSDKTGLVEFARNLTALGLNLVASGGTAKALRDAGLAVRDVSELTGFPEMLGGRVKTLHPAVHAGILARNIPEDNADMARLDFNLIRVVACNLYPFVKTVASPGVTVEEAVEQIDIGGVTLLRAAAKNHARVTVVCEPEDYVVVSTEMQSSESKDTSLETRRQLALKAFTHTAQYDEAISDYFRKQGQLALFSVSDKTGLVEFARNLTALGLNLVASGGTAKALRDAGLAVRDVSELTGFPEMLGGRVKTLHPAVHAGILARNIPEDNADMARLDFNLIRVVACNLYPFVKTVASPGVTVEEAVEQIDIGGVTLLRAAAKNHARVTVVCEPEDYVVVSTEMQSSESKDTSLETRRQLALKAFTHTAQYDEAISDYFRKQ Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Cystathionine gamma-lyase (CTH) | 3COG | 4.30 | |
Target general information Gen name CTH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gamma-cystathionase; Cysteine-protein sulfhydrase Protein family Trans-sulfuration enzymes family Biochemical class NA Function Catalyzes the last step in the trans-sulfuration pathway from methionine to cysteine. Has broad substrate specificity. Converts cystathionine to cysteine, ammonia and 2-oxobutanoate. Converts two cysteine molecules to lanthionine and hydrogen sulfide. Can also accept homocysteine as substrate. Specificity depends on the levels of the endogenous substrates. Generates the endogenous signaling molecule hydrogen sulfide (H2S), and so contributes to the regulation of blood pressure. Acts as a cysteine-protein sulfhydrase by mediating sulfhydration of target proteins: sulfhydration consists of converting -SH groups into -SSH on specific cysteine residues of target proteins such as GAPDH, PTPN1 and NF-kappa-B subunit RELA, thereby regulating their function. Related diseases Cystathioninuria (CSTNU) [MIM:219500]: Autosomal recessive phenotype characterized by abnormal accumulation of plasma cystathionine, leading to increased urinary excretion. {ECO:0000269|PubMed:12574942, ECO:0000269|PubMed:18476726}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02328; DB03928; DB00151; DB04217; DB00114 Interacts with P32929; Q96NT3; Q96NT3-2; Q96HA8; Q6P9E2 EC number EC 4.4.1.1 Uniprot keywords 3D-structure; Alternative splicing; Amino-acid biosynthesis; Calmodulin-binding; Cysteine biosynthesis; Cytoplasm; Disease variant; Lipid metabolism; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 86026 Length 782 Aromaticity 0.08 Instability index 32.4 Isoelectric point 6.27 Charge (pH=7) -9.46 2D Binding mode Binding energy (Kcal/mol) -5.86  Molscript Map 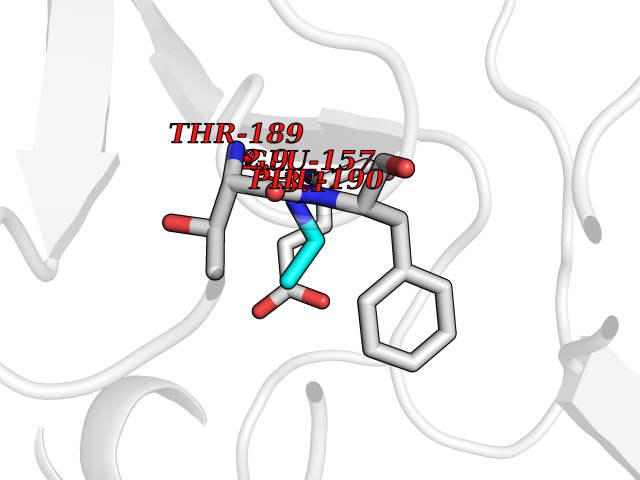 Pymol Map 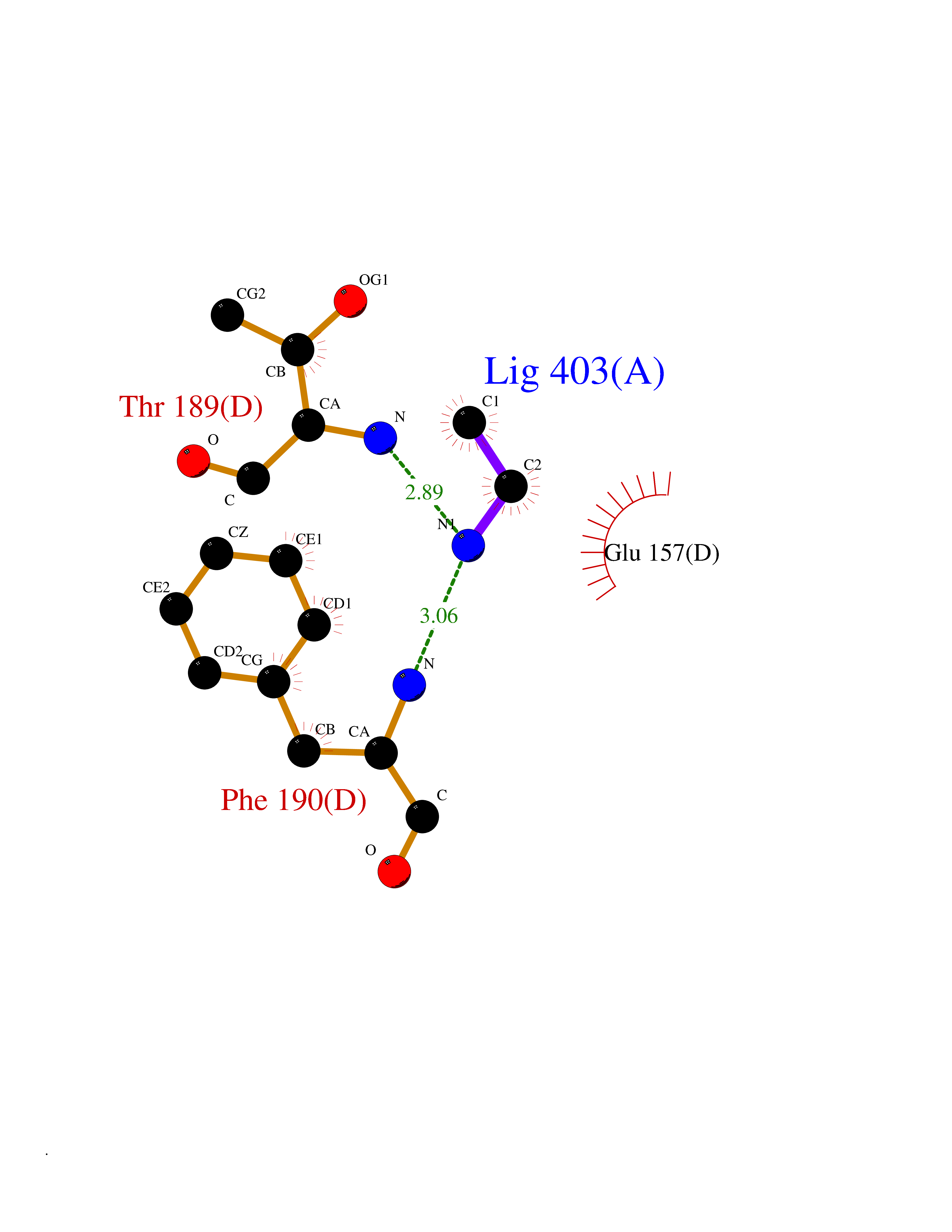 Ligplot Map 3D Binding mode Sequence GFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQHSGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPSGFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPS Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Folic acid synthesis protein FOL1 | 2BMB | 4.30 | |
Target general information Gen name FOL1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms N0848;YNL256W Protein family DHNA family; HPPK family; DHPS family Biochemical class Transferase Function 2-amino-4-hydroxy-6-hydroxymethyldihydropteridine diphosphokinase activity.7,8-dihydromonapterin aldolase activity.ATP binding.Dihydroneopterin aldolase activity.Dihydropteroate synthase activity.Kinase activity.Metal ion binding. Related diseases LIMK1 is located in the Williams-Beuren syndrome (WBS) critical region. WBS results from a hemizygous deletion of several genes on chromosome 7q11.23, thought to arise as a consequence of unequal crossing over between highly homologous low-copy repeat sequences flanking the deleted region. Drugs (DrugBank ID) DB00634 Interacts with NA EC number 2.5.1.15; 2.7.6.3; 4.1.2.25 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Folate biosynthesis; Kinase; Lyase; Magnesium; Membrane; Metal-binding; Mitochondrion; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 58205.2 Length 513 Aromaticity 0.08 Instability index 42.41 Isoelectric point 5.92 Charge (pH=7) -8.31 2D Binding mode Binding energy (Kcal/mol) -5.86  Molscript Map 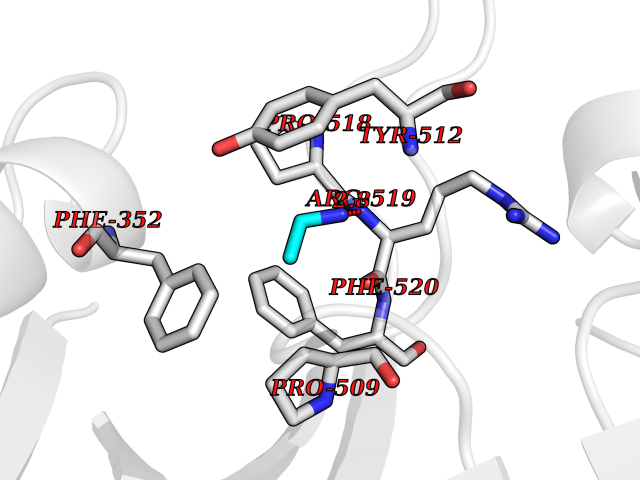 Pymol Map  Ligplot Map 3D Binding mode Sequence SWKRAFLAFGSNIGDRFKHIQMALQLLSREKTVKLRNISSIFESEPMYFKDQTPFMNGCVEVETLLTPSELLKLCKKIEYEELQRTIDLDIVMFLNSAGEDIIVNEPDLNIPHPRMLERTFVLEPLCELISPVHLHPVTAEPIVDHLKQLYDKQHDEDTLWKLVPLPYRSGVEPRFLKFKTATKTNRITVSPTYIMAIFNATPDSFSDGGEHFADIESQLNDIIKLCKDALYLHESVIIDVGGCSTRPNSIQASEEEEIRRSIPLIKAIRESTELPQDKVILSIDTYRSNVAKEAIKVGVDIINDISGGLFDSNMFAVIAENPEICYILSHTRGDISTMNRLAHYENFALGDSIQQEFVHNTDIQQLDDLKDKTVLIRNVGQEIGERYIKAIDNGVKRWQILIDPGLGFAKTWKQNLQIIRHIPILKNYSFTMNSNNSQVYVNLRNMPVLLGPSRKKFIGHITKDVDAKQRDFATGAVVASCIGFGSDMVRVHDVKNCSKSIKLADAIYKGLE Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Alpha-N-acetylglucosaminidase (NAGLU) | 4XWH | 4.30 | |
Target general information Gen name NAGLU Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UFHSD1; NAGLU; NAG; N-acetyl-alpha-glucosaminidase; Alpha-N-acetylglucosaminidase82 kDa form; Alpha-N-acetylglucosaminidase77 kDa form Protein family Glycosyl hydrolase 89 family Biochemical class Glycosylase Function Involved in the degradation of heparan sulfate. Related diseases Mucopolysaccharidosis 3B (MPS3B) [MIM:252920]: A form of mucopolysaccharidosis type 3, an autosomal recessive lysosomal storage disease due to impaired degradation of heparan sulfate. MPS3 is characterized by severe central nervous system degeneration, but only mild somatic disease. Onset of clinical features usually occurs between 2 and 6 years; severe neurologic degeneration occurs in most patients between 6 and 10 years of age, and death occurs typically during the second or third decade of life. {ECO:0000269|PubMed:10094189, ECO:0000269|PubMed:11068184, ECO:0000269|PubMed:11153910, ECO:0000269|PubMed:11286389, ECO:0000269|PubMed:11793481, ECO:0000269|PubMed:11836372, ECO:0000269|PubMed:12202988, ECO:0000269|PubMed:14984474, ECO:0000269|PubMed:15933803, ECO:0000269|PubMed:16151907, ECO:0000269|PubMed:28101780, ECO:0000269|PubMed:9443875, ECO:0000269|PubMed:9443878, ECO:0000269|PubMed:9832037, ECO:0000269|PubMed:9950362}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2V (CMT2V) [MIM:616491]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2V is an autosomal dominant sensory neuropathy with late onset. The main clinical feature is recurrent leg pain that progresses to constant painful paraesthesias in the feet and later the hands. As it evolves, some patients develop a mild sensory ataxia. {ECO:0000269|PubMed:25818867}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06773; DB00141 Interacts with NA EC number EC 3.2.1.50 Uniprot keywords 3D-structure; Charcot-Marie-Tooth disease; Direct protein sequencing; Disease variant; Glycoprotein; Glycosidase; Hydrolase; Lysosome; Mucopolysaccharidosis; Neurodegeneration; Neuropathy; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 80206.3 Length 720 Aromaticity 0.13 Instability index 41.59 Isoelectric point 6.28 Charge (pH=7) -4.09 2D Binding mode Binding energy (Kcal/mol) -5.86  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DEAREAAAVRALVARLLGPGPAADFSVSVERALAAKPGLDTYSLGGGGAARVRVRGSTGVAAAAGLHRYLRDFCGCHVAWSGSQLRLPRPLPAVPGELTEATPNRYRYYQNVCTQSYSFVWWDWARWEREIDWMALNGINLALAWSGQEAIWQRVYLALGLTQAEINEFFTGPAFLAWGRMGNLHTWDGPLPPSWHIKQLYLQHRVLDQMRSFGMTPVLPAFAGHVPEAVTRVFPQVNVTKMGSWGHFNCSYSCSFLLAPEDPIFPIIGSLFLRELIKEFGTDXIYGADTFNEMQPPSSEPSYLAAATTAVYEAMTAVDTEAVWLLQGWLFQHQPQFWGPAQIRAVLGAVPRGRLLVLDLFAESQPVYTRTASFQGQPFIWCMLHNFGGNHGLFGALEAVNGGPEAARLFPNSTMVGTGMAPEGISQNEVVYSLMAELGWRKDPVPDLAAWVTSFAARRYGVSHPDAGAAWRLLLRSVYNCSGEACRGHNRSPLVRRPSLQMNTSIWYNRSDVFEAWRLLLTSAPSLATSPAFRYDLLDLTRQAVQELVSLYYEEARSAYLSKELASLLRAGGVLAYELLPALDEVLASDSRFLLGSWLEQARAAAVSEAEADFYEQNSRYQLTLWGPEGNILDYANKQLAGLVANYYTPRWRLFLEALVDSVAQGIPFQQHQFDKNVFQLEQAFVLSKQRYPSQPRGDTVDLAKKIFLKYYPRWVAGSW Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Aminoacylase-1 | 1Q7L | 4.30 | |
Target general information Gen name ACY1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase M20A family Biochemical class Hydrolase Function Aminoacylase activity.Identical protein binding.Metal ion binding.Metallopeptidase activity. Related diseases Aminoacylase-1 deficiency (ACY1D) [MIM:609924]: An enzymatic deficiency resulting in encephalopathy, unspecific psychomotor delay, psychomotor delay with atrophy of the vermis and syringomyelia, marked muscular hypotonia or normal clinical features. Epileptic seizures are a frequent feature. All affected individuals exhibit markedly increased urinary excretion of several N-acetylated amino acids. {ECO:0000269|PubMed:16274666, ECO:0000269|PubMed:16465618, ECO:0000269|PubMed:17562838, ECO:0000269|PubMed:21414403}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06151; DB00128; DB09130 Interacts with Q03154; O75934; Q96HA8; P36639; P36639-2; Q8TCT1; P0CG20; Q96A09; P54274; O43711; Q9UPN9; Q9NZC7-5 EC number 3.5.1.14 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,C Molecular weight (Da) 31172.2 Length 275 Aromaticity 0.11 Instability index 36.46 Isoelectric point 6 Charge (pH=7) -5.26 2D Binding mode Binding energy (Kcal/mol) -5.87 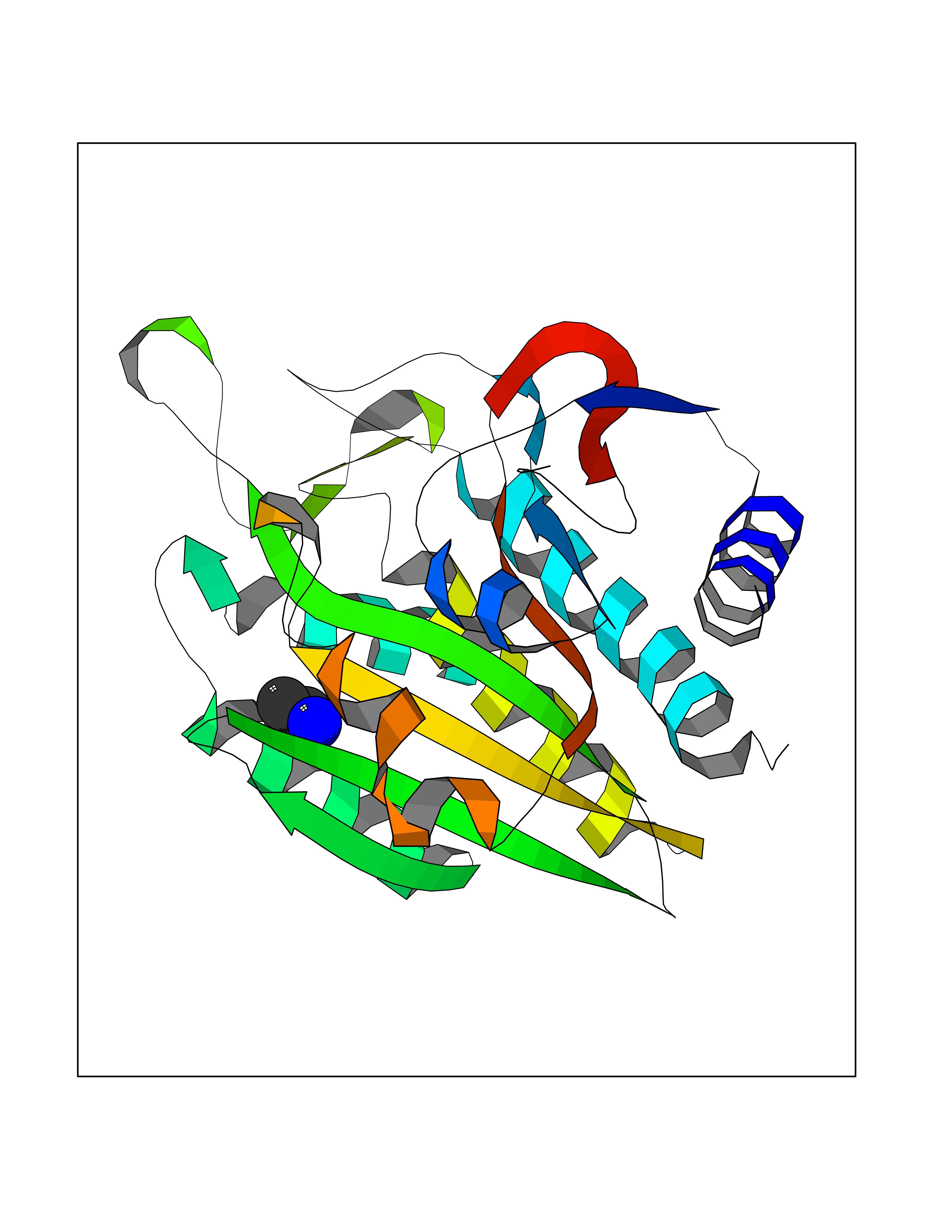 Molscript Map 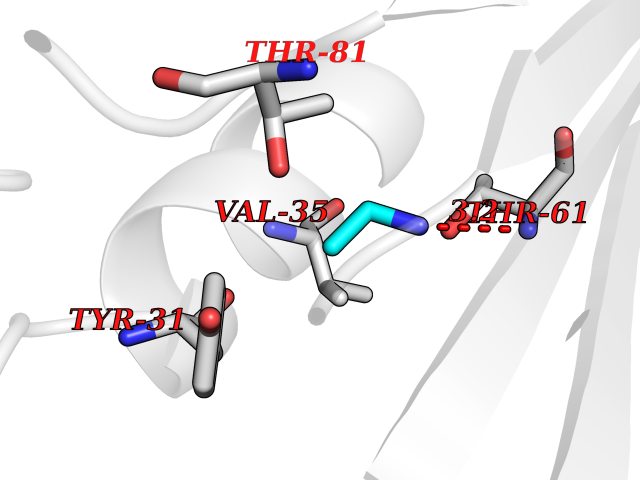 Pymol Map 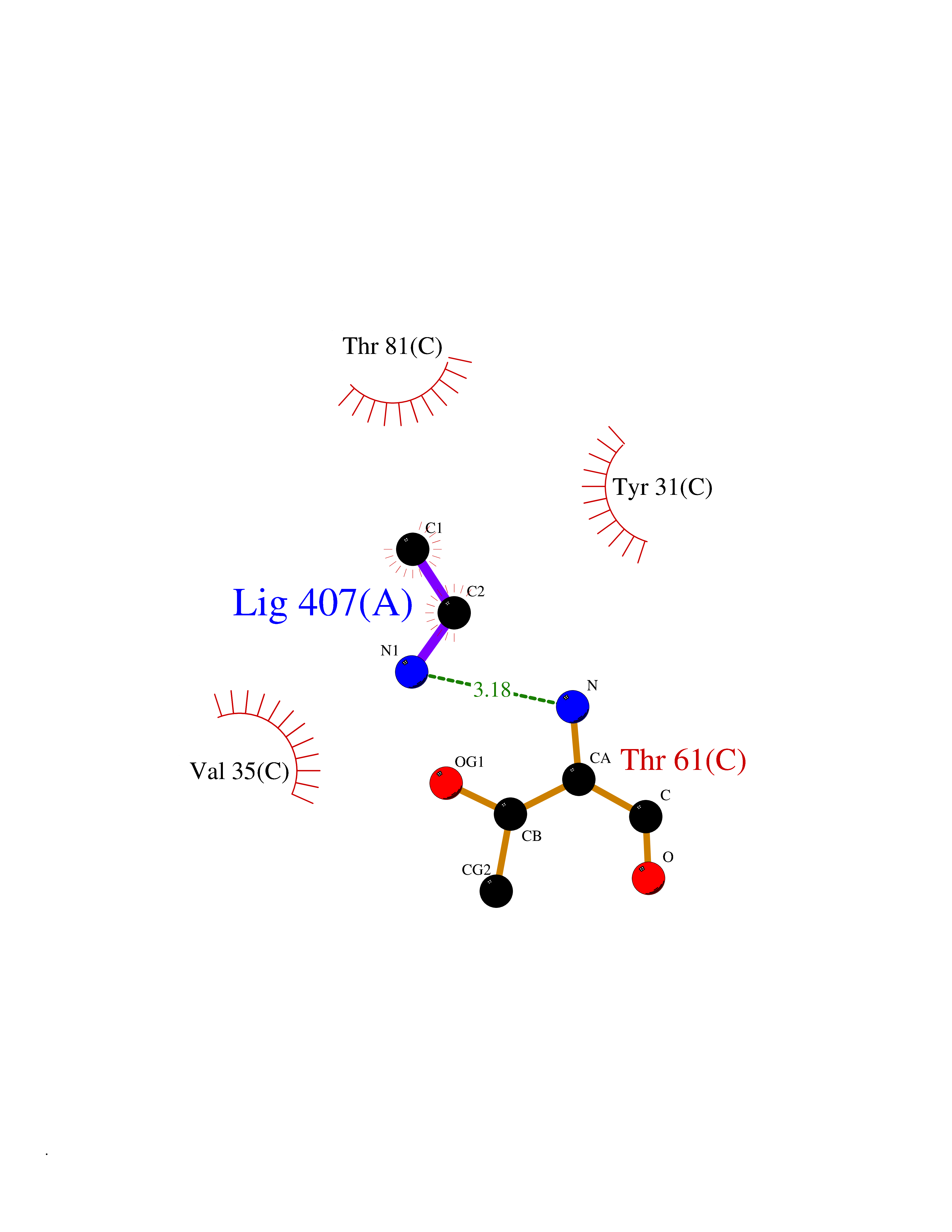 Ligplot Map 3D Binding mode Sequence NPWWAAFSRVCKDMNLTLEPEIMPAAGDNRYIRAVGVPALGFSPMNRTPVLLHDHDERLHEAVFLRGVDIYTRLLPALASVPALPEEHPSVTLFRQYLRIRTVQPKPDYGAAVAFFEETARQLGLGCQKVEVAPGYVVTVLTWPGTNPTLSSILLNSHTDVVPVFKEHWSHDPFEAFKDSEGYIYARGAQDMKCVSIQYLEAVRRLKVEGHRFPRTIHMTFVPDEEVGGHQGMELFVQRPEFHALRAGFALDEGIANPTDAFTVFYSERSPWWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Quinone oxidoreductase (CRYZ) | 1YB5 | 4.30 | |
Target general information Gen name CRYZ Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Zeta-crystallin; NADPH:quinone reductase; CRYZ Protein family Zinc-containing alcohol dehydrogenase family, Quinone oxidoreductase subfamily Biochemical class NA Function Does not have alcohol dehydrogenase activity. Binds NADP and acts through a one-electron transfer process. Orthoquinones, such as 1,2-naphthoquinone or 9,10-phenanthrenequinone, are the best substrates (in vitro). May act in the detoxification of xenobiotics. Interacts with (AU)-rich elements (ARE) in the 3'-UTR of target mRNA species. Enhances the stability of mRNA coding for BCL2. NADPH binding interferes with mRNA binding. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09061; DB00266; DB14009; DB14011; DB03461 Interacts with Q08257 EC number EC 1.6.5.5 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; RNA-binding Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34717.6 Length 324 Aromaticity 0.07 Instability index 28.36 Isoelectric point 8.57 Charge (pH=7) 3.64 2D Binding mode Binding energy (Kcal/mol) -5.86 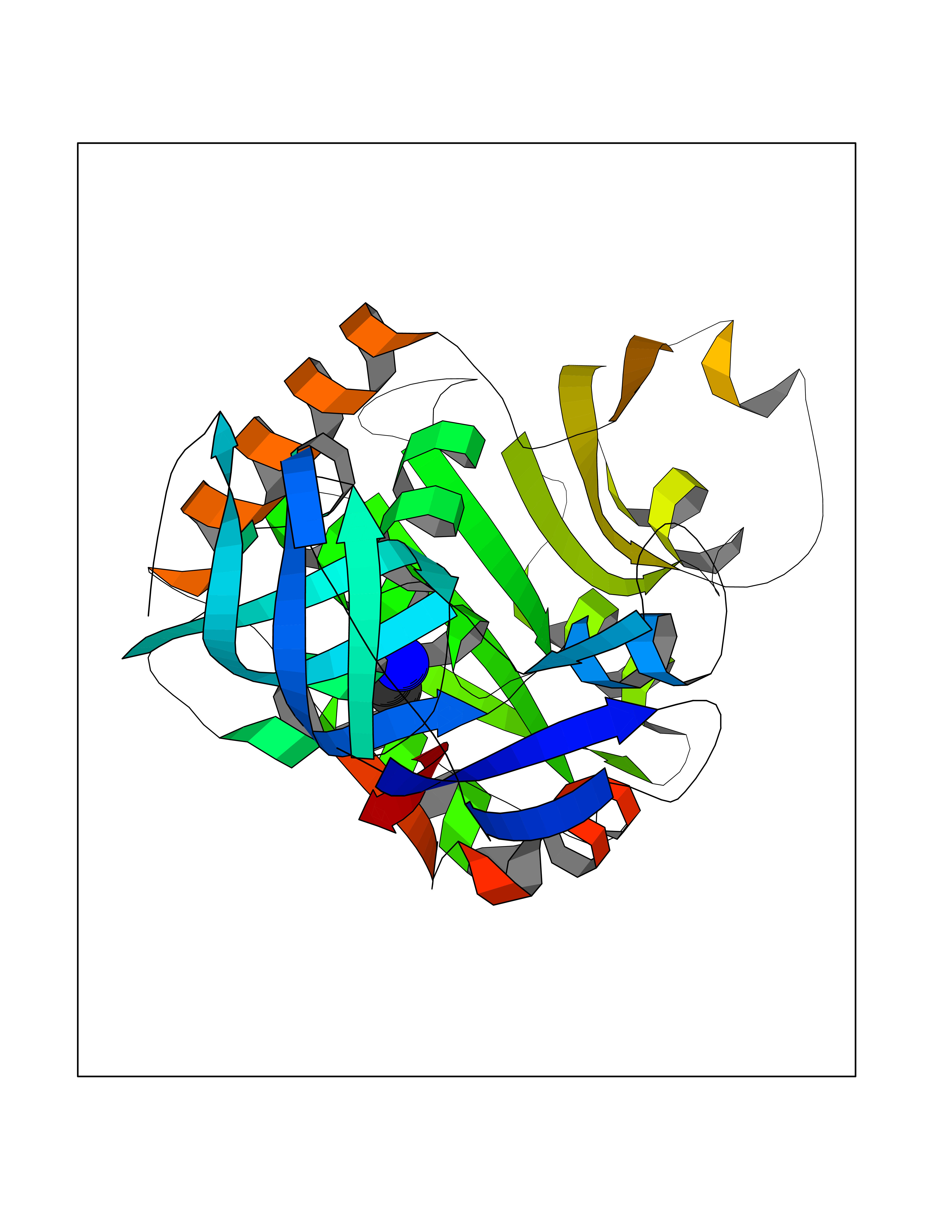 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KLMRAVRVFEFGGPEVLKLRSDIAVPIPKDHQVLIKVHACGVNPVETYIRSGTYSRKPLLPYTPGSDVAGVIEAVGDNASAFKKGDRVFTSSTISGGYAEYALAADHTVYKLPEKLDFKQGAAIGIPYFTAYRALIHSACVKAGESVLVHGASGGVGLAACQIARAYGLKILGTAGTEEGQKIVLQNGAHEVFNHREVNYIDKIKKYVGEKGIDIIIEMLANVNLSKDLSLLSHGGRVIVVGSRGTIEINPRDTMAKESSIIGVTLFSSTKEEFQQYAAALQAGMEIGWLKPVIGSQYPLEKVAEAHENIIHGSGATGKMILLL Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Peroxisomal trans-2-enoyl-CoA reductase | 1YXM | 4.30 | |
Target general information Gen name PECR Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PRO1004;SDR29C1 Protein family Short-chain dehydrogenases/reductases (SDR) family Biochemical class Oxidoreductase Function Receptor binding.Trans-2-enoyl-CoA reductase (NADPH) activity. Related diseases Cornelia de Lange syndrome 5 (CDLS5) [MIM:300882]: A form of Cornelia de Lange syndrome, a clinically heterogeneous developmental disorder associated with malformations affecting multiple systems. It is characterized by facial dysmorphisms, abnormal hands and feet, growth delay, cognitive retardation, hirsutism, gastroesophageal dysfunction and cardiac, ophthalmologic and genitourinary anomalies. {ECO:0000269|PubMed:22885700, ECO:0000269|PubMed:22889856}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00173 Interacts with NA EC number 1.3.1.38 Uniprot keywords 3D-structure; Alternative splicing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 30349.5 Length 283 Aromaticity 0.08 Instability index 38.34 Isoelectric point 8.89 Charge (pH=7) 5.26 2D Binding mode Binding energy (Kcal/mol) -5.86  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RSYLAPGLLQGQVAIVTGGATGIGKAIVKELLELGSNVVIASRKLERLKSAADELQANLPPTKQARVIPIQCNIRNEEEVNNLVKSTLDTFGKINFLVNNGGGQFLSPAEHISSKGWHAVLETNLTGTFYMCKAVYSSWMKEHGGSIVNIIVPTKAGFPLAVHSGAARAGVYNLTKSLALEWACSGIRINCVAPGVIYSQTAQSFFEGSFQKIPAKRIGVPEEVSSVVCFLLSPAASFITGQSVDVDGGRSLYTHSYEVPDHDNWPKGAGDLSVVKKMKETFK Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Valacyclovir hydrolase (BPHL) | 2OCI | 4.30 | |
Target general information Gen name BPHL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms VACVase; MCNAA; DJ40E16.6.1; Breast epithelial mucin-associated antigen; Bph-rp; Biphenyl hydrolase-like protein (Serine hydrolase) (Breast epithelial mucin-associated antigen, MCNAA, Bph-rp), variant Protein family AB hydrolase superfamily, Lipase family Biochemical class Carboxylic ester hydrolase Function Serine hydrolase that catalyzes the hydrolytic activation of amino acid ester prodrugs of nucleoside analogs such as valacyclovir and valganciclovir. Activates valacyclovir to acyclovir. May play a role in detoxification processes. It is a specific alpha-amino acid ester hydrolase that prefers small, hydrophobic, and aromatic side chains and does not have a stringent requirement for the leaving group other than preferring aprimary alcohol. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03380 Interacts with P62508-3 EC number EC 3.1.-.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Hydrolase; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 28823.7 Length 254 Aromaticity 0.11 Instability index 25.18 Isoelectric point 8.31 Charge (pH=7) 2.59 2D Binding mode Binding energy (Kcal/mol) -5.87  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SVTSAKVAVNGVQLHYQQTGEGDHAVLLLPGMLGSGETDFGPQLKNLNKKLFTVVAWDPRGYGHSRPPDRDFPADFFERDAKDAVDLMKALKFKKVSLLGWSDGGITALIAAAKYPSYIHKMVIWGANAYVTDEDSMIYEGIRDVSKWSERTRKPLEALYGYDYFARTCEKWVDGIRQFKHLPDGNICRHLLPRVQCPALIVHGEKDPLVPRFHADFIHKHVKGSRLHLMPEGKHNLHLRFADEFNKLAEDFLQ Hydrogen bonds contact Hydrophobic contact | ||||