Job Results:
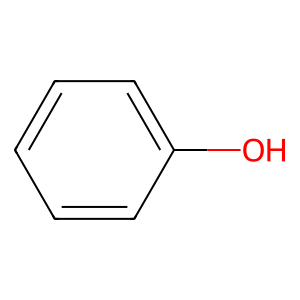
Ligand
Structure
Job ID
85eee3bcbedbd74b81bc48f797dc6af1
Job name
NA
Time
2026-02-27 13:44:11
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Chromodomain-helicase-DNA-binding protein 1 | 4O42 | 5.15 | |
Target general information Gen name CHD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SNF2/RAD54 helicase family Biochemical class Dna binding protein / viral protein Function ATP binding.ATP-dependent DNA helicase activity.DNA binding.Methylated histone binding. Related diseases Pilarowski-Bjornsson syndrome (PILBOS) [MIM:617682]: An autosomal dominant disorder characterized by developmental delay, speech apraxia, intellectual disability, autism, and facial dysmorphic features. Some patients may have seizures. {ECO:0000269|PubMed:28866611}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60341-1; B2BUF1; P28799; O76024 EC number 3.6.4.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromatin regulator; Cytoplasm; Disease variant; DNA-binding; Helicase; Hydrolase; Intellectual disability; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 20969.1 Length 180 Aromaticity 0.12 Instability index 46.35 Isoelectric point 5.88 Charge (pH=7) -2.83 2D Binding mode Binding energy (Kcal/mol) -7.02  Molscript Map 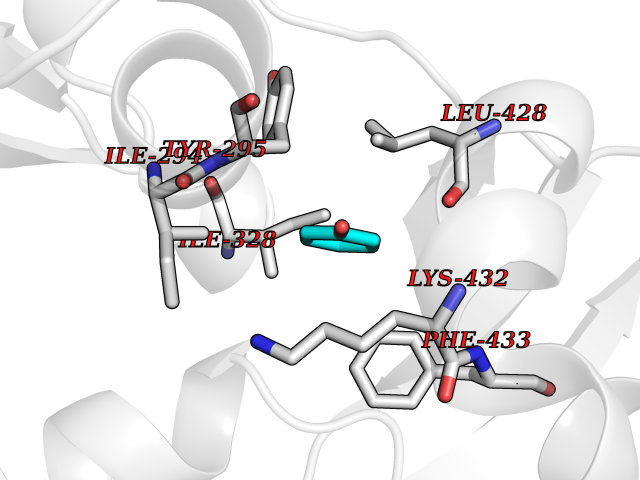 Pymol Map 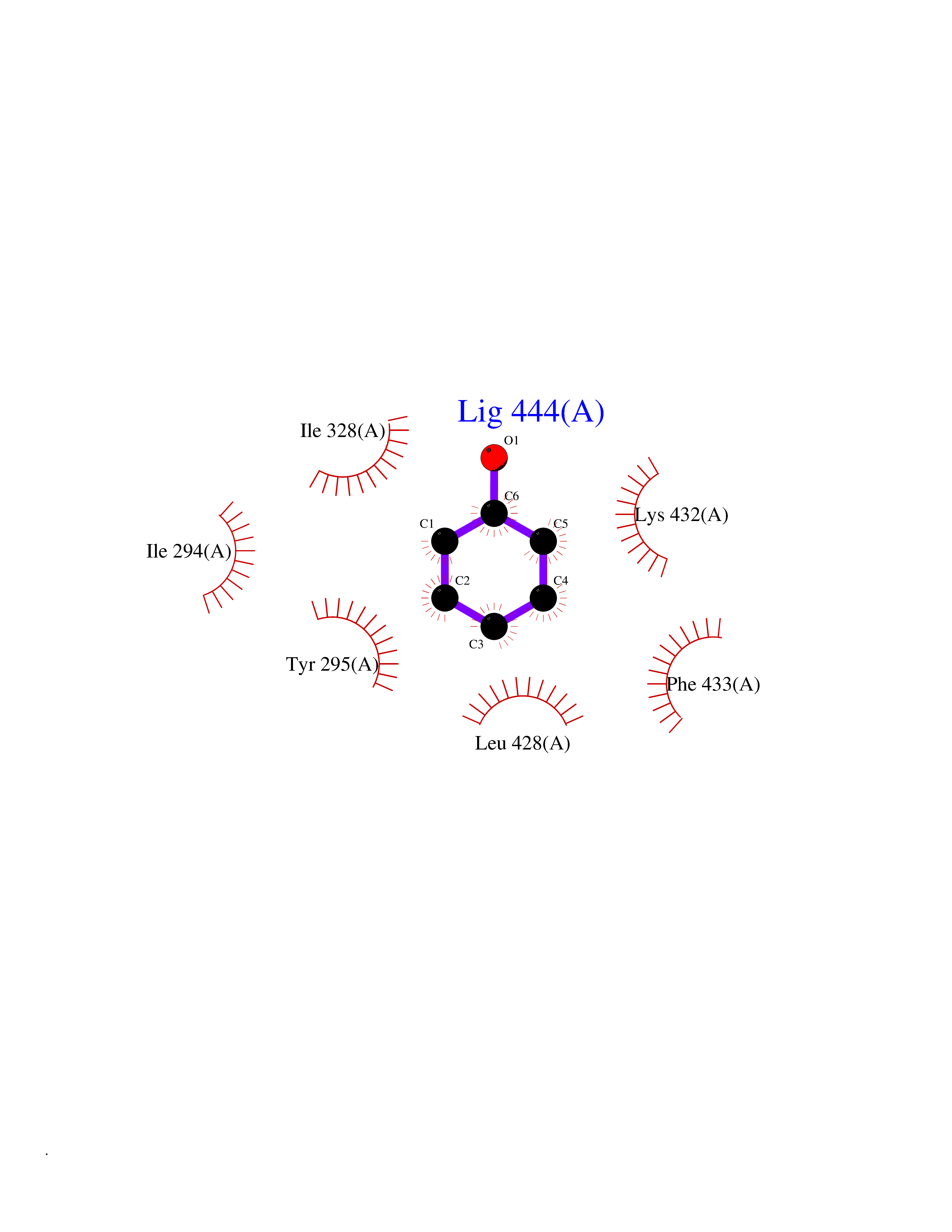 Ligplot Map 3D Binding mode Sequence EFETIERFMDCRIGRKGATGATTTIYAVEADGDPNAGFEKNKEPGEIQYLIKWKGWSHIHNTWETEETLKQQNVRGMKKLDNYKKKDQETKRWLKNASPEDVEYYNCQQELTDDLHKQYQIVERIIAHSNQKSAAGYPDYYCKWQGLPYSECSWEDGALISKKFQACIDEYFSRTARSXV Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Cerebron E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase) | 4CI1 | 5.15 | |
Target general information Gen name CUL4A/CUL4B-DDB1-CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NA Protein family Cullin family Biochemical class NA Function NA Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; Q86VP6; Q16531; Q92466; P08238; O94888; P55072 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; DNA damage; DNA repair; Host-virus interaction; Isopeptide bond; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID B Molecular weight (Da) 42669.7 Length 368 Aromaticity 0.1 Instability index 44.94 Isoelectric point 8.72 Charge (pH=7) 6.58 2D Binding mode Binding energy (Kcal/mol) -7.02  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MINFDTSLPTSHMYLGSDMEEFHGRTLHDDDSCQVIPVLPHVMVMLIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVREREAHFGTTAEIYAYREEQEYGIETVKVKAIGRQRFKVLEIRTQSDGIQQAKVQILPERVLPSTMSAVQLQSLSRRHIRAFRQWWQKYQKRKFHCASLTSWPPWLYSLYDAETLMERVKRQLHEWDENLKDESLPTNPIDFSYRVAACLPIDDALRIQLLKIGSAIQRLRELDIMNKTSLCCKQCQDTEITTKNEIFSLSLCGPMAAYVNPHGYIHETLTVYKACNLNLSGRPSTEHSWFPGYAWTIAQCRICGNHMGWKFTATKKDMSPQKFWGLTRSALLPR Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Macrophage migration inhibitory factor (MIF) | 3IJJ | 5.15 | |
Target general information Gen name MIF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phenylpyruvate tautomerase; MMIF; L-dopachrome tautomerase; L-dopachrome isomerase; Glycosylation-inhibiting factor; GLIF; GIF Protein family MIF family Biochemical class Intramolecular oxidoreductase Function Involved in the innate immune response to bacterial pathogens. The expression of MIF at sites of inflammation suggests a role as mediator in regulating the function of macrophages in host defense. Counteracts the anti-inflammatory activity of glucocorticoids. Has phenylpyruvate tautomerase and dopachrome tautomerase activity (in vitro), but the physiological substrate is not known. It is not clear whether the tautomerase activity has any physiological relevance, and whether it is important for cytokine activity. Pro-inflammatory cytokine. Related diseases Rheumatoid arthritis systemic juvenile (RASJ) [MIM:604302]: An inflammatory articular disorder with systemic onset beginning before the age of 16. It represents a subgroup of juvenile arthritis associated with severe extraarticular features and occasionally fatal complications. During active phases of the disorder, patients display a typical daily spiking fever, an evanescent macular rash, lymphadenopathy, hepatosplenomegaly, serositis, myalgia and arthritis. {ECO:0000269|PubMed:11508429}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01880; DB07888; DB08334; DB08335; DB08333; DB07718; DB08765; DB02728 Interacts with O43521-2; P00533; Q92743; P14174; Q96HA8 EC number EC 5.3.2.1 Uniprot keywords 3D-structure; Acetylation; Cytokine; Cytoplasm; Direct protein sequencing; Immunity; Inflammatory response; Innate immunity; Isomerase; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,C Molecular weight (Da) 24671.9 Length 228 Aromaticity 0.09 Instability index 31.45 Isoelectric point 8.37 Charge (pH=7) 2.26 2D Binding mode Binding energy (Kcal/mol) -7.02  Molscript Map 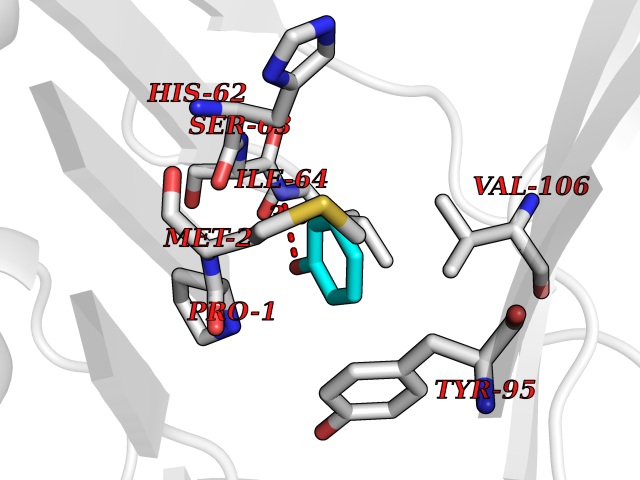 Pymol Map 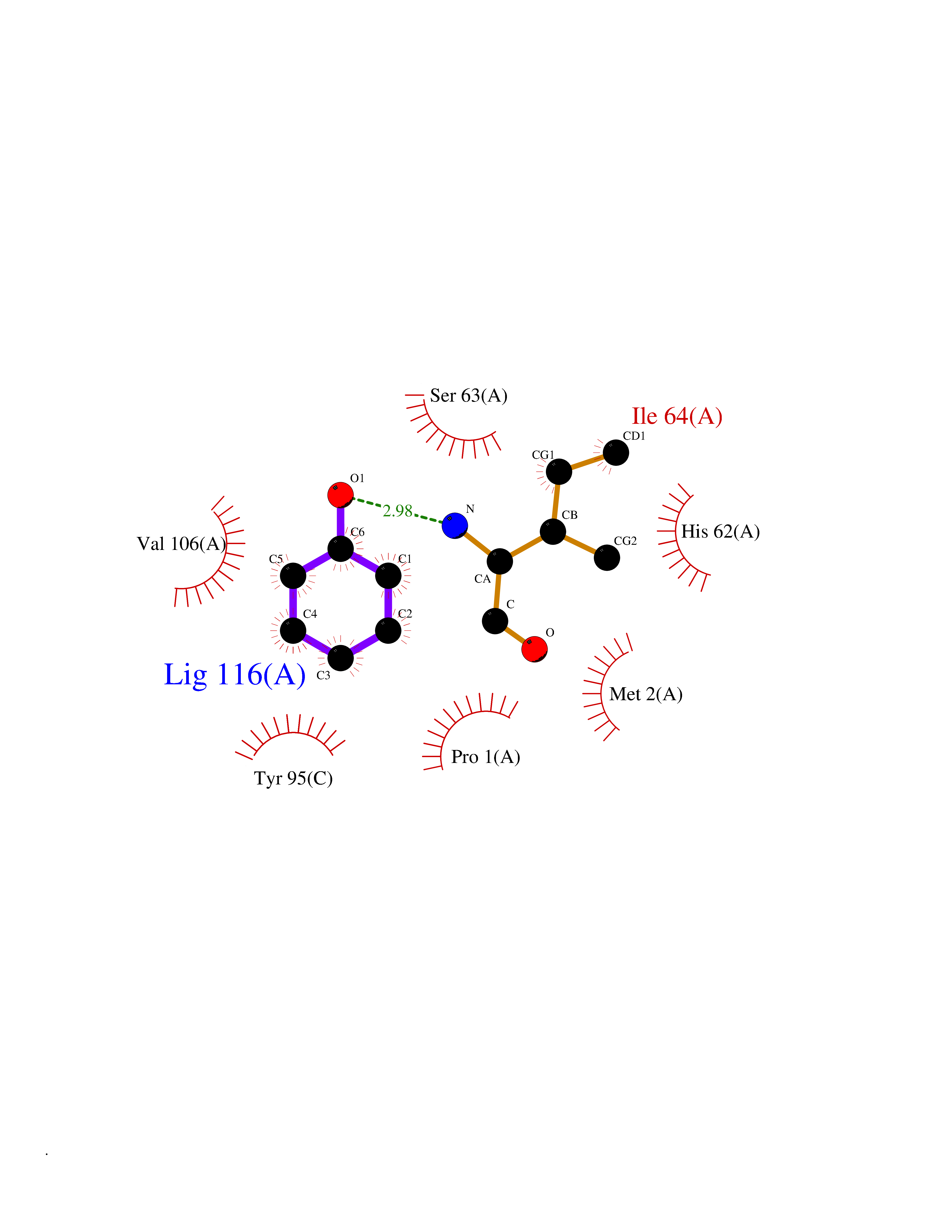 Ligplot Map 3D Binding mode Sequence PMFIVNTNVPRASVPDGFLSELTQQLAQATGKPPQYIAVHVVPDQLMAFGGSSEPCALCSLHSIGKIGGAQNRSYSKLLCGLLAERLRISPDRVYINYYDMNAANVGWNNSTFAPMFIVNTNVPRASVPDGFLSELTQQLAQATGKPPQYIAVHVVPDQLMAFGGSSEPCALCSLHSIGKIGGAQNRSYSKLLCGLLAERLRISPDRVYINYYDMNAANVGWNNSTFA Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Proto-oncogene c-Met (MET) | 3DKC | 5.15 | |
Target general information Gen name MET Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase Met; Scatter factor receptor; SF receptor; Met proto-oncogene tyrosine kinase; Hepatocyte growth factor receptor; HGF/SF receptor; HGF-SF receptor; HGF receptor; C-met; C-Met r Protein family Protein kinase superfamily, Tyr protein kinase family Biochemical class Kinase Function Regulates many physiological processes including proliferation, scattering, morphogenesis and survival. Ligand binding at the cell surface induces autophosphorylation of MET on its intracellular domain that provides docking sites for downstream signaling molecules. Following activation by ligand, interacts with the PI3-kinase subunit PIK3R1, PLCG1, SRC, GRB2, STAT3 or the adapter GAB1. Recruitment of these downstream effectors by MET leads to the activation of several signaling cascades including the RAS-ERK, PI3 kinase-AKT, or PLCgamma-PKC. The RAS-ERK activation is associated with the morphogenetic effects while PI3K/AKT coordinates prosurvival effects. During embryonic development, MET signaling plays a role in gastrulation, development and migration of muscles and neuronal precursors, angiogenesis and kidney formation. In adults, participates in wound healing as well as organ regeneration and tissue remodeling. Promotes also differentiation and proliferation of hematopoietic cells. May regulate cortical bone osteogenesis. Receptor tyrosine kinase that transduces signals from the extracellular matrix into the cytoplasm by binding to hepatocyte growth factor/HGF ligand. Related diseases Activation of MET after rearrangement with the TPR gene produces an oncogenic protein.; DISEASE: Defects in MET may be associated with gastric cancer.; DISEASE: Hepatocellular carcinoma (HCC) [MIM:114550]: A primary malignant neoplasm of epithelial liver cells. The major risk factors for HCC are chronic hepatitis B virus (HBV) infection, chronic hepatitis C virus (HCV) infection, prolonged dietary aflatoxin exposure, alcoholic cirrhosis, and cirrhosis due to other causes. {ECO:0000269|PubMed:9927037}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Renal cell carcinoma papillary (RCCP) [MIM:605074]: A subtype of renal cell carcinoma tending to show a tubulo-papillary architecture formed by numerous, irregular, finger-like projections of connective tissue. Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. {ECO:0000269|PubMed:10327054, ECO:0000269|PubMed:10417759, ECO:0000269|PubMed:10433944, ECO:0000269|PubMed:9140397, ECO:0000269|PubMed:9563489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A common allele in the promoter region of the MET shows genetic association with susceptibility to autism in some families. Functional assays indicate a decrease in MET promoter activity and altered binding of specific transcription factor complexes.; DISEASE: MET activating mutations may be involved in the development of a highly malignant, metastatic syndrome known as cancer of unknown primary origin (CUP) or primary occult malignancy. Systemic neoplastic spread is generally a late event in cancer progression. However, in some instances, distant dissemination arises at a very early stage, so that metastases reach clinical relevance before primary lesions. Sometimes, the primary lesions cannot be identified in spite of the progresses in the diagnosis of malignancies.; DISEASE: Deafness, autosomal recessive, 97 (DFNB97) [MIM:616705]: A form of non-syndromic sensorineural hearing loss with prelingual onset. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:25941349}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Osteofibrous dysplasia (OSFD) [MIM:607278]: A congenital disorder of osteogenesis characterized by non-neoplastic, radiolucent lesions that affect the cortical bone immediately under the periosteum. It usually manifests as a painless swelling or anterior bowing of the long bones, most commonly the tibia and fibula. {ECO:0000269|PubMed:26637977}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Disease-associated variants identified in 4 families cause the deletion of exon 14. This results in the exclusion of an ubiquitination target site within the cytoplasmic domain, hence in protein stabilization. The persistent presence of MET at the cell surface in conditions of ligand-dependent activation retards osteoblastic differentiation. {ECO:0000269|PubMed:26637977}.; DISEASE: Arthrogryposis, distal, 11 (DA11) [MIM:620019]: A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. DA11 is an autosomal dominant form characterized mainly by camptodactyly. Other features include absent flexion creases and limited forearm supination. {ECO:0000269|PubMed:30777867}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06896; DB08791; DB06997; DB07969; DB08079; DB16695; DB12742; DB12267; DB08875; DB11791; DB08865; DB12010; DB02152; DB07369; DB06995; DB06314; DB01268; DB15133; DB12200; DB11800 Interacts with P22681; Q96EY1; Q96EY1-2; P00533; P09769; P14210; P14210-6; O15357; P35968; P06239; P07948; P08581; P41218; P15941; P16333; O43639; Q16288; P27986; O00459; Q92569; P19174; O43157; O15031; Q9ULL4; Q8TCU6; P18031; Q06124; P23467; Q12913; Q16827; P20936; Q9UQQ2; O60880; O14796; Q9NP31; Q8N5H7; Q15464; P29353; P98077; Q6S5L8; Q96IW2; Q9H6Q3; O75159; O14544; P12931; Q9ULZ2; P43405; P42680; Q9HBL0; Q63HR2; Q68CZ2; Q9UKW4; P07947; P43403; Q08048; P0DQD2; P35918; Q00944 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromosomal rearrangement; Deafness; Disease variant; Disulfide bond; Glycoprotein; Kinase; Membrane; Non-syndromic deafness; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35229.5 Length 312 Aromaticity 0.1 Instability index 37.98 Isoelectric point 7.79 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -7.02 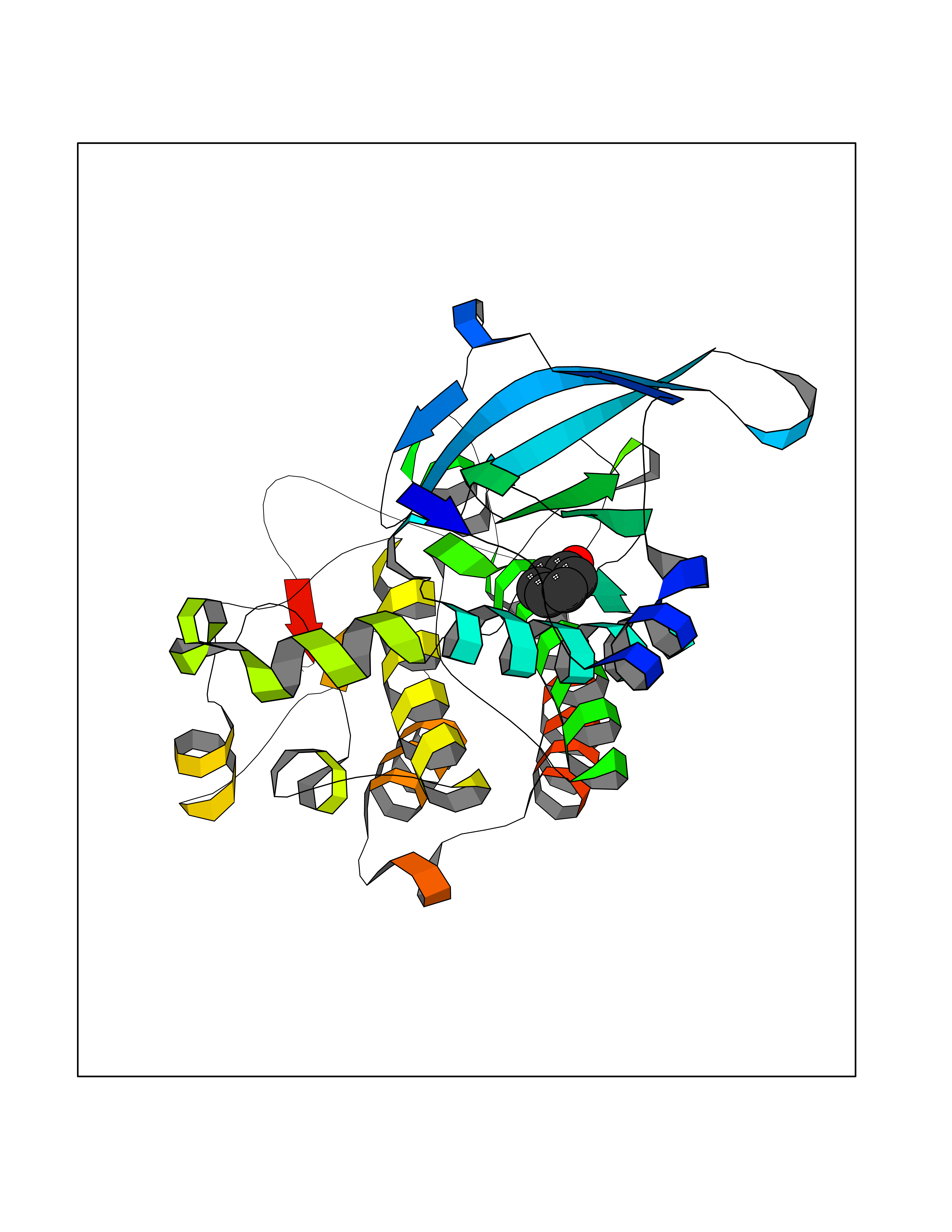 Molscript Map 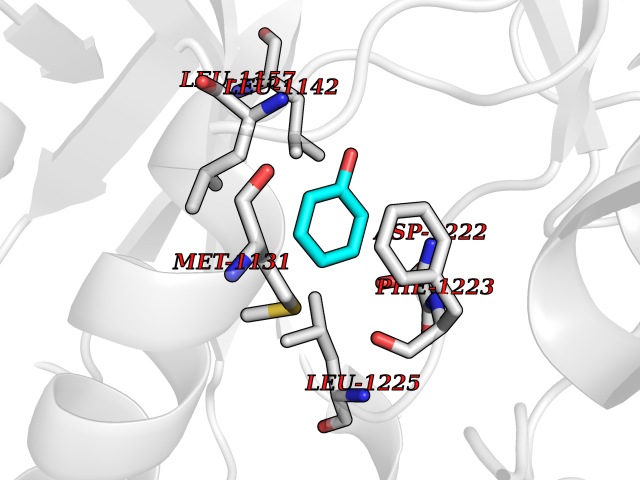 Pymol Map 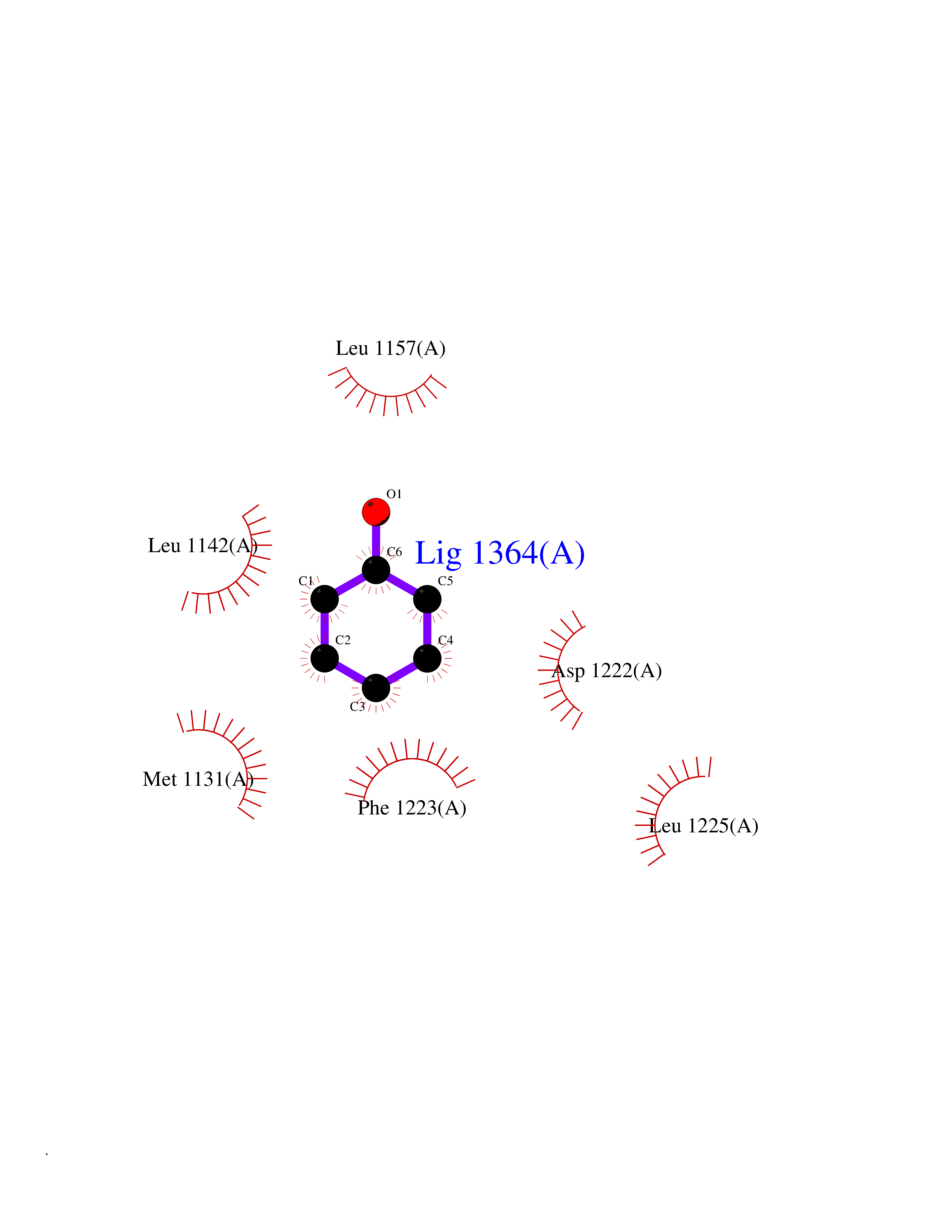 Ligplot Map 3D Binding mode Sequence VHIDLSALNPELVQAVQHVVIGPSSLIVHFNEVIGRGHFGCVYHGTLLDNDGKKIHCAVKSLNRITDIGEVSQFLTEGIIMKDFSHPNVLSLLGICLRSEGSPLVVLPYMKHGDLRNFIRNETHNPTVKDLIGFGLQVAKGMKFLASKKFVHRDLAARNCMLDEKFTVKVADFGLARDMYDKEFDSVHNKTGAKLPVKWMALESLQTQKFTTKSDVWSFGVLLWELMTRGAPPYPDVNTFDITVYLLQGRRLLQPEYCPDPLYEVMLKCWHPKAEMRPSFSELVSRISAIFSTFIGEHYVHVNATYVNVKEG Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Free fatty acid receptor 1 | 4PHU | 5.14 | |
Target general information Gen name FFAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPR40 Protein family G-protein coupled receptor 1 family Biochemical class Fatty acid binding protein / hydrolase Function Bioactive lipid receptor activity.G-protein coupled receptor activity.Guanyl-nucleotide exchange factor activity.Lipid binding. Related diseases Refsum disease (RD) [MIM:266500]: A rare autosomal recessive peroxisomal disorder characterized by the accumulation of the branched-chain fatty acid, phytanic acid, in blood and tissues. Cardinal clinical features are retinitis pigmentosa, peripheral neuropathy, cerebellar ataxia, and elevated protein levels in the cerebrospinal fluid (CSF). Half of all patients exhibit generalized, mild to moderate ichthyosis resembling ichthyosis vulgaris. Less constant features are nerve deafness, anosmia, skeletal abnormalities, cataracts and cardiac impairment. {ECO:0000269|PubMed:10709665, ECO:0000269|PubMed:10767344, ECO:0000269|PubMed:14974078, ECO:0000269|PubMed:9326939, ECO:0000269|PubMed:9326940}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00159 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 28319.1 Length 272 Aromaticity 0.11 Instability index 27.3 Isoelectric point 9.07 Charge (pH=7) 6.85 2D Binding mode Binding energy (Kcal/mol) -7.01 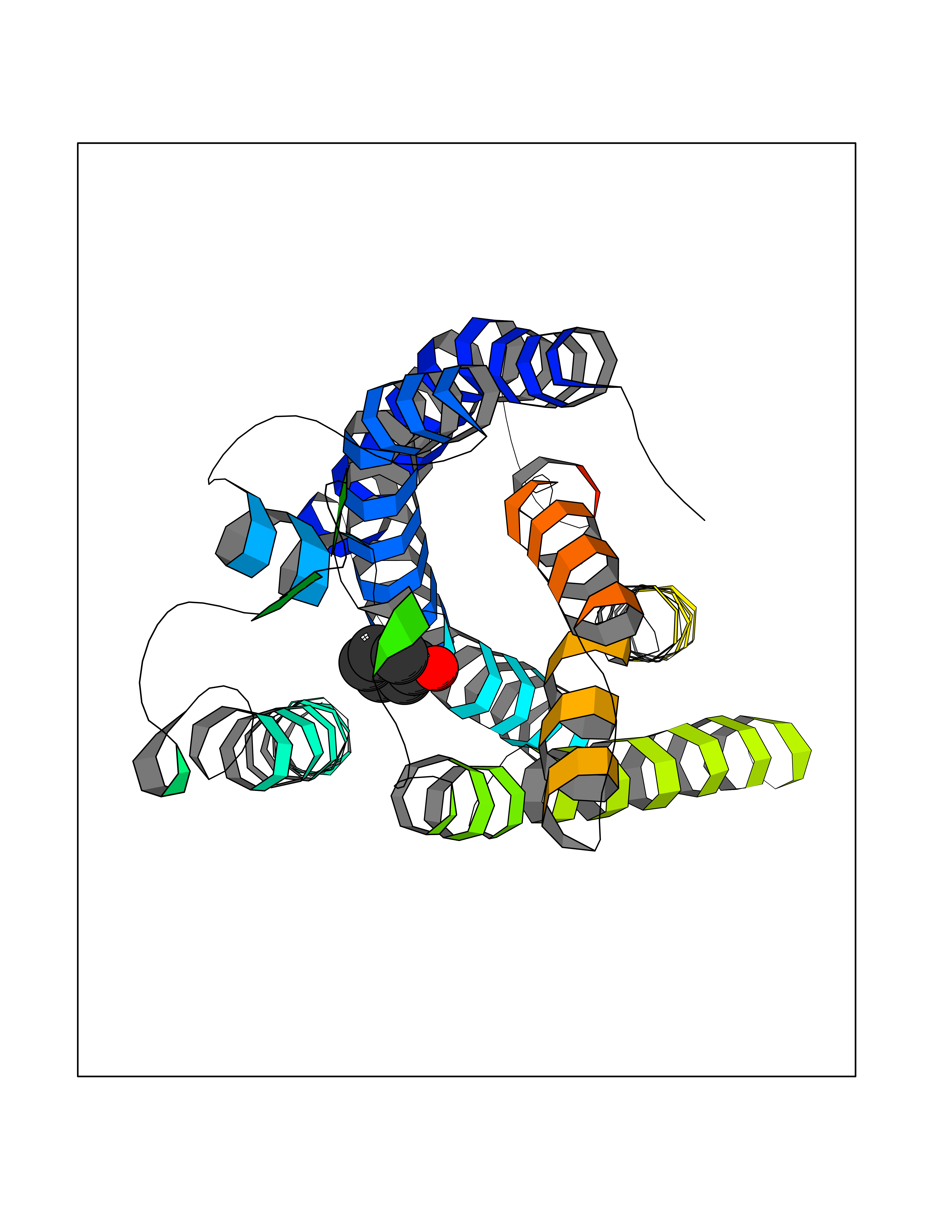 Molscript Map  Pymol Map 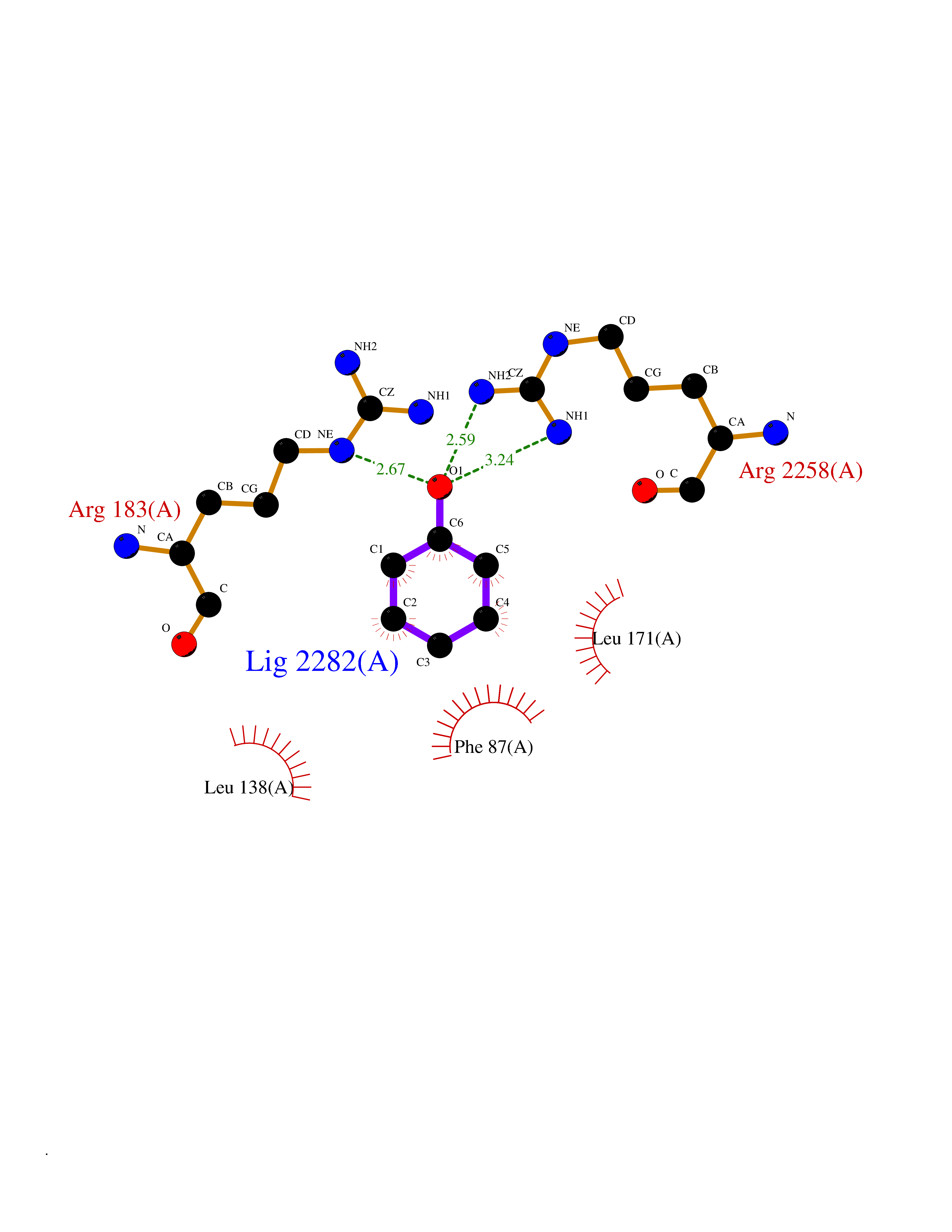 Ligplot Map 3D Binding mode Sequence MDLPPQLSFGLYVAAFALGFPLNVLAIRGATAHARLRLTPSAVYALNLGCSDLLLTVSLPLKAVEALASGAWPLPASLCPVFAVAHFAPLYAGGGFLAALSAARYLGAAFPPCYSWGVCAAIWALVLCHLGLVFGLEAPGGWLDHSNTSLGINTPVNGSPVCLEAWDPASAGPARFSLSLLLFFLPLAITAFCFVGCLRALARGSLTHRRKLRAAWVAGGALLTLLLCVGPYNASNVASFLYPNLGGSWRKLGLITGAWSVVLNPLVTGYLG Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Medium-chain specific acyl-CoA dehydrogenase, mitochondrial | 4P13 | 5.14 | |
Target general information Gen name ACADM Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Acyl-CoA dehydrogenase activity.Flavin adenine dinucleotide binding.Identical protein binding.Medium-chain-acyl-CoA dehydrogenase activity. Related diseases Acyl-CoA dehydrogenase medium-chain deficiency (ACADMD) [MIM:201450]: An inborn error of mitochondrial fatty acid beta-oxidation which causes fasting hypoglycemia, hepatic dysfunction and encephalopathy, often resulting in death in infancy. {ECO:0000269|PubMed:10767181, ECO:0000269|PubMed:11349232, ECO:0000269|PubMed:11409868, ECO:0000269|PubMed:11486912, ECO:0000269|PubMed:1363805, ECO:0000269|PubMed:1671131, ECO:0000269|PubMed:1684086, ECO:0000269|PubMed:1902818, ECO:0000269|PubMed:2251268, ECO:0000269|PubMed:2393404, ECO:0000269|PubMed:2394825, ECO:0000269|PubMed:7603790, ECO:0000269|PubMed:7929823, ECO:0000269|PubMed:8198141, ECO:0000269|PubMed:9158144, ECO:0000269|PubMed:9882619}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03415; DB03147; DB02910 Interacts with PRO_0000000502 [P11310] EC number 1.3.8.7 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; FAD; Fatty acid metabolism; Flavoprotein; Lipid metabolism; Mitochondrion; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 85080.3 Length 773 Aromaticity 0.09 Instability index 30.55 Isoelectric point 5.71 Charge (pH=7) -7.7 2D Binding mode Binding energy (Kcal/mol) -7.02 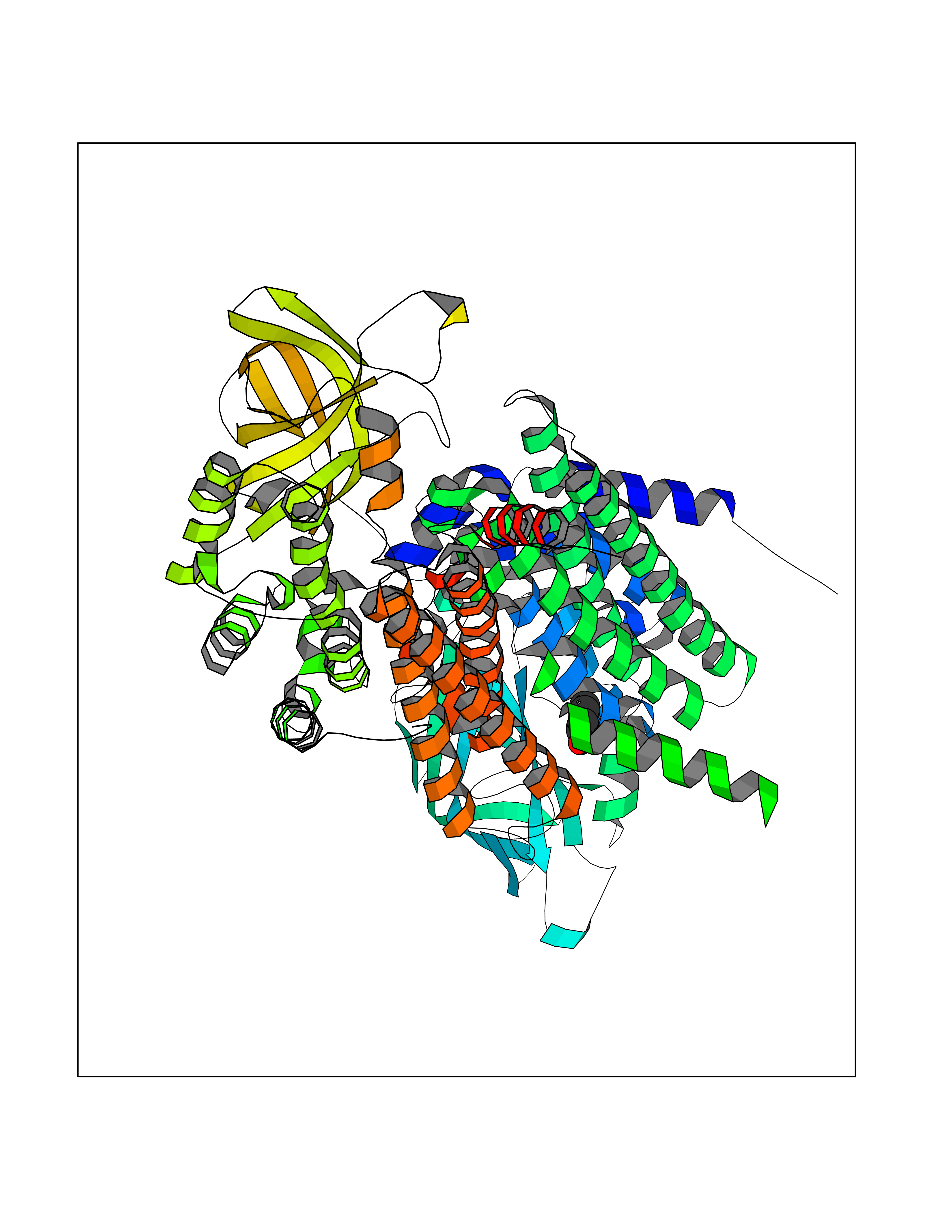 Molscript Map 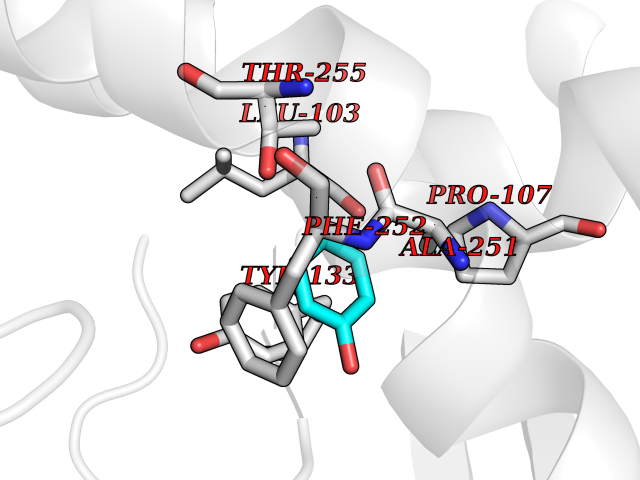 Pymol Map 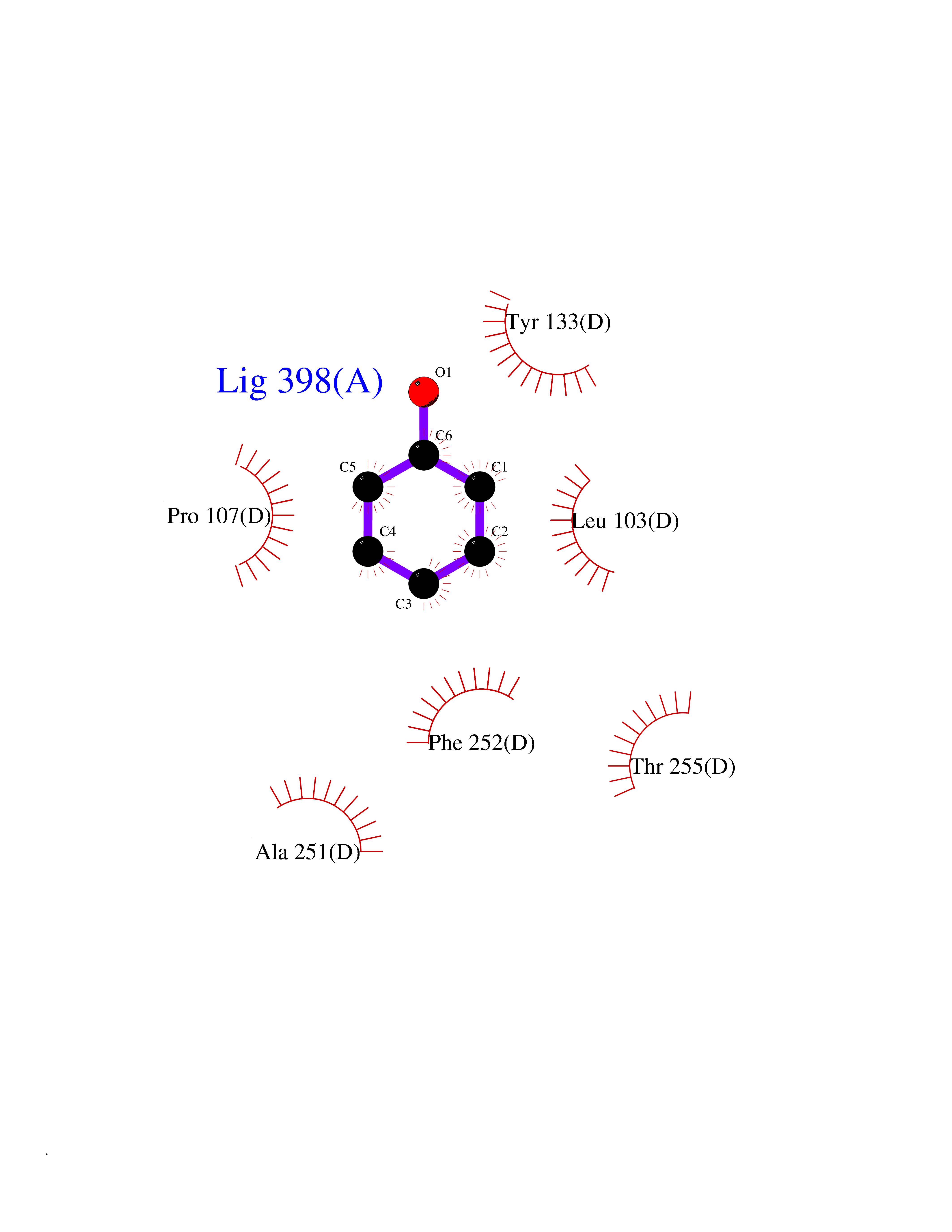 Ligplot Map 3D Binding mode Sequence LGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKLGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKN Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Lecithin-cholesterol acyltransferase (LCAT) | 6MVD | 5.14 | |
Target general information Gen name LCAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phospholipidcholesterolacyltransferase; Phospholipid-cholesterol acyltransferase; Phosphatidylcholinesterol acyltransferase; Phosphatidylcholine-sterol acyltransferase Protein family AB hydrolase superfamily, Lipase family Biochemical class Acyltransferase Function Synthesized mainly in the liver and secreted into plasma where it converts cholesterol and phosphatidylcholines (lecithins) to cholesteryl esters and lysophosphatidylcholines on the surface of high and low density lipoproteins (HDLs and LDLs). The cholesterol ester is then transported back to the liver. Has a preference for plasma 16:0-18:2 or 18:O-18:2 phosphatidylcholines. Also produced in the brain by primary astrocytes, and esterifies free cholesterol on nascent APOE-containing lipoproteins secreted from glia and influences cerebral spinal fluid (CSF) APOE- and APOA1 levels. Together with APOE and the cholesterol transporter ABCA1, plays a key role in the maturation of glial-derived, nascent lipoproteins. Required for remodeling high-density lipoprotein particles into their spherical forms. Central enzyme in the extracellular metabolism of plasma lipoproteins. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P02647; O76024 EC number EC 2.3.1.43 Uniprot keywords 3D-structure; Acyltransferase; Cholesterol metabolism; Corneal dystrophy; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipid metabolism; Proteomics identification; Reference proteome; Secreted; Signal; Steroid metabolism; Sterol metabolism; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42715.4 Length 376 Aromaticity 0.12 Instability index 42.05 Isoelectric point 5.69 Charge (pH=7) -9.12 2D Binding mode Binding energy (Kcal/mol) -7.01 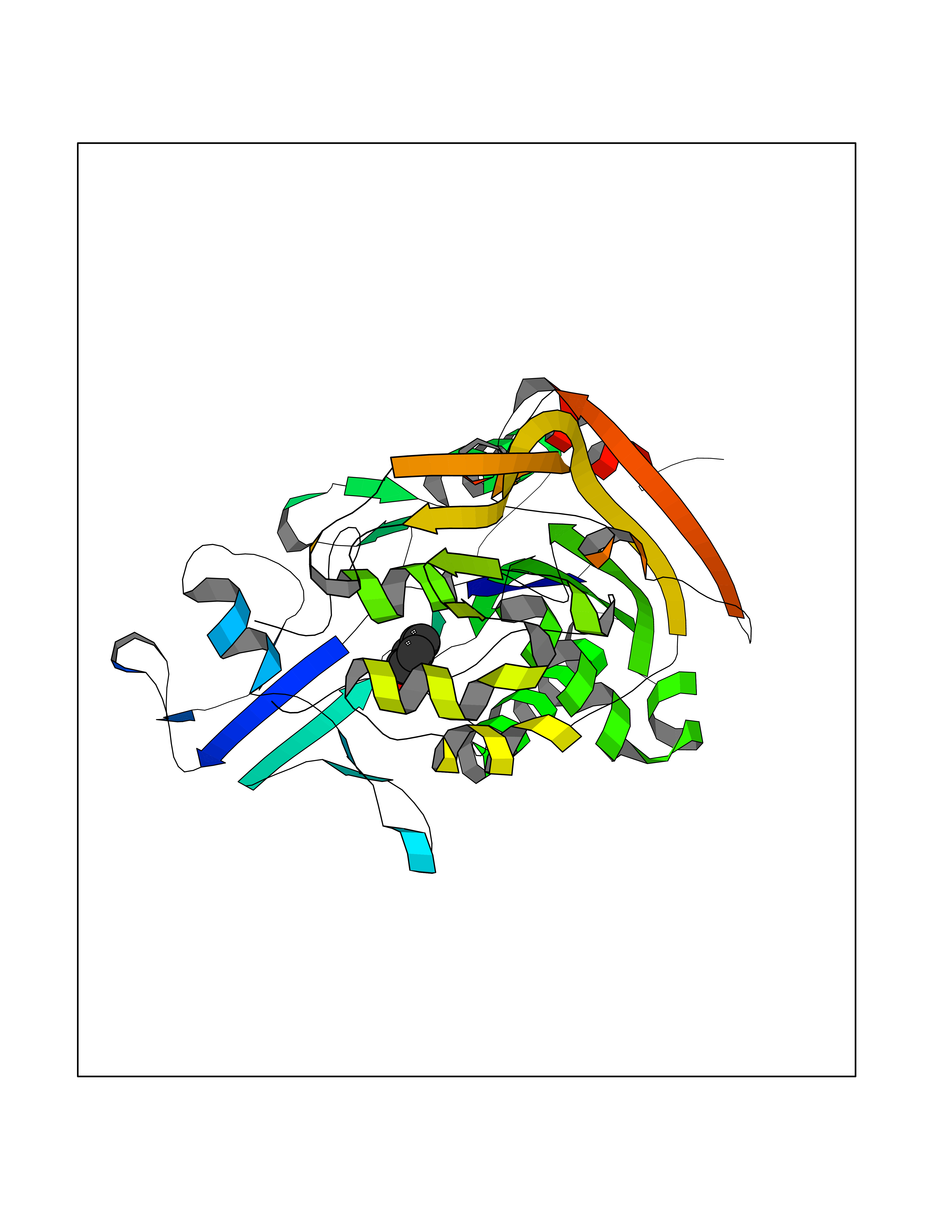 Molscript Map 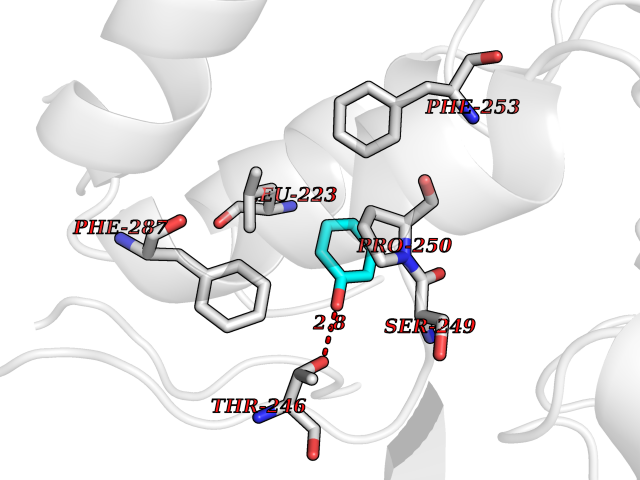 Pymol Map 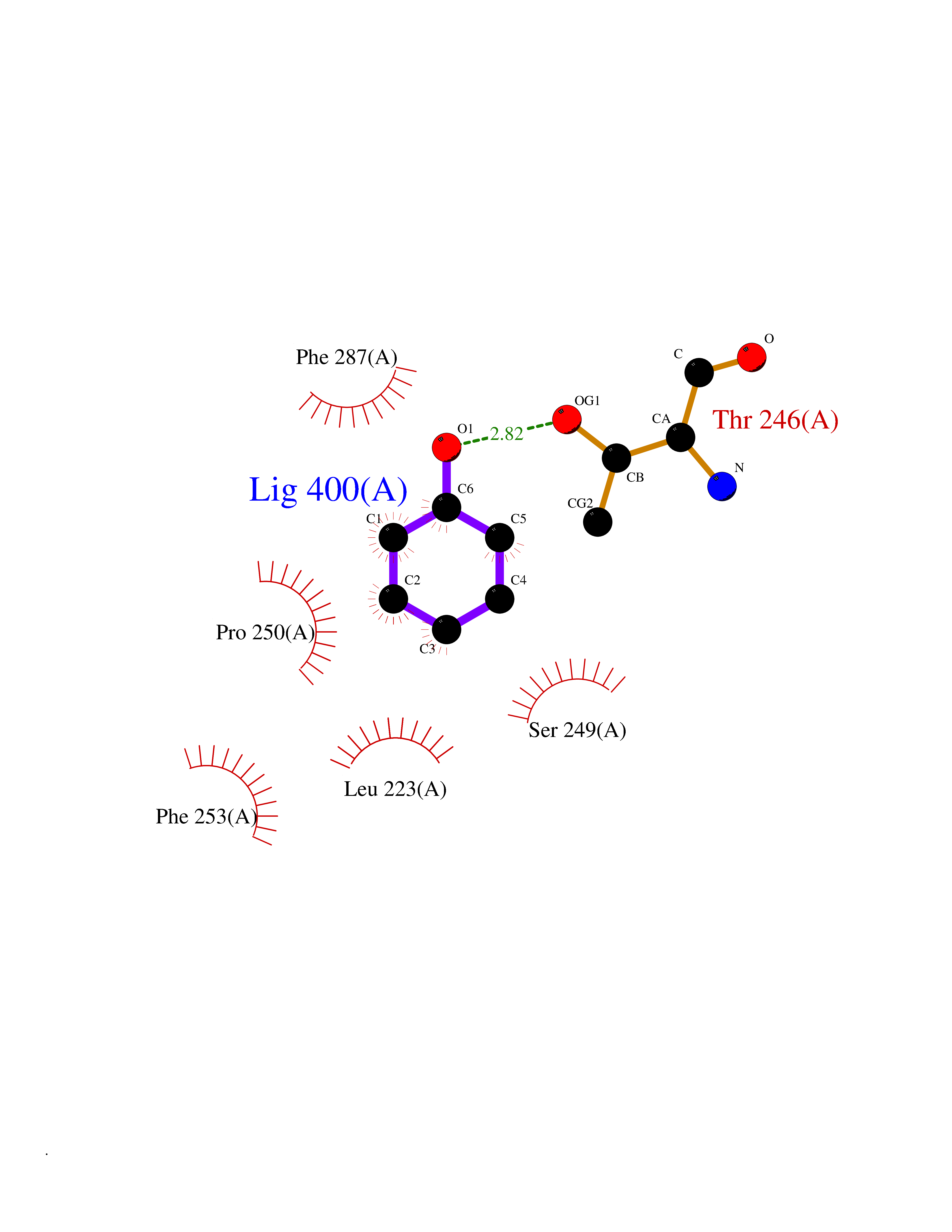 Ligplot Map 3D Binding mode Sequence HTRPVILVPGCLGNQLEAKLDKPDVVNWMCYRKTEDFFTIWLDLNMFLPLGVDCWIDNTRVVYNRSSGLVSNAPGVQIRVPGFGKTYSVEYLDSSKLAGYLHTLVQNLVNNGYVRDETVRAAPYDWRLEPGQQEEYYRKLAGLVEEMHAAYGKPVFLIGHSLGCLHLLYFLLRQPQAWKDRFIDGFISLGAPWGGSIKPMLVLASGDNQGIPIMSSIKEEQRITTTSPWMFPSRMAWPEDHVFISTPSFNYTGRDFQRFFADLHFEEGWYMWLQSRDLLAGLPAPGVEVYCLYGVGLPTPRTYIYDHGFPYTDPVGVLYEDGDDTVATRSTELCGLWQGRQPQPVHLLPLHGIQHLNMVFSNLTLEHINAILLGAH Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Dual-specificity tyrosine-phosphorylation regulated kinase 2 (DYRK2) | 6HDR | 5.14 | |
Target general information Gen name DYRK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dual specificity tyrosine-phosphorylation-regulated kinase 2 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, MNB/DYRK subfamily Biochemical class Kinase Function Functions in part via its role in ubiquitin-dependent proteasomal protein degradation. Functions downstream of ATM and phosphorylates p53/TP53 at 'Ser-46', and thereby contributes to the induction of apoptosis in response to DNA damage. Phosphorylates NFATC1, and thereby inhibits its accumulation in the nucleus and its transcription factor activity. Phosphorylates EIF2B5 at 'Ser-544', enabling its subsequent phosphorylation and inhibition by GSK3B. Likewise, phosphorylation of NFATC1, CRMP2/DPYSL2 and CRMP4/DPYSL3 promotes their subsequent phosphorylation by GSK3B. May play a general role in the priming of GSK3 substrates. Inactivates GYS1 by phosphorylation at 'Ser-641', and potentially also a second phosphorylation site, thus regulating glycogen synthesis. Mediates EDVP E3 ligase complex formation and is required for the phosphorylation and subsequent degradation of KATNA1. Phosphorylates TERT at 'Ser-457', promoting TERT ubiquitination by the EDVP complex. Phosphorylates SIAH2, and thereby increases its ubiquitin ligase activity. Promotes the proteasomal degradation of MYC and JUN, and thereby regulates progress through the mitotic cell cycle and cell proliferation. Promotes proteasomal degradation of GLI2 and GLI3, and thereby plays a role in smoothened and sonic hedgehog signaling. Plays a role in cytoskeleton organization and neurite outgrowth via its phosphorylation of DCX and DPYSL2. Phosphorylates CRMP2/DPYSL2, CRMP4/DPYSL3, DCX, EIF2B5, EIF4EBP1, GLI2, GLI3, GYS1, JUN, MDM2, MYC, NFATC1, p53/TP53, TAU/MAPT and KATNA1. Can phosphorylate histone H1, histone H3 and histone H2B (in vitro). Can phosphorylate CARHSP1 (in vitro). Serine/threonine-protein kinase involved in the regulation of the mitotic cell cycle, cell proliferation, apoptosis, organization of the cytoskeleton and neurite outgrowth. Related diseases Bone marrow failure and diabetes mellitus syndrome (BMFDMS) [MIM:620044]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFDMS is an autosomal recessive form characterized by various degrees of bone marrow failure, ranging from dyserythropoiesis to bone marrow aplasia, with onset in infancy or early childhood, and non-autoimmune insulin-dependent diabetes mellitus appearing in the first or second decades. Many patients show pigmentary skin abnormalities and short stature. {ECO:0000269|PubMed:28073829, ECO:0000269|PubMed:35611808, ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NR20; Q13422; Q9BQD3; Q9BRK4; P23497; O43379; P62258; Q96C00 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cytoplasm; Kinase; Magnesium; Manganese; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 46422.1 Length 407 Aromaticity 0.09 Instability index 44.91 Isoelectric point 9.09 Charge (pH=7) 12.37 2D Binding mode Binding energy (Kcal/mol) -7.01 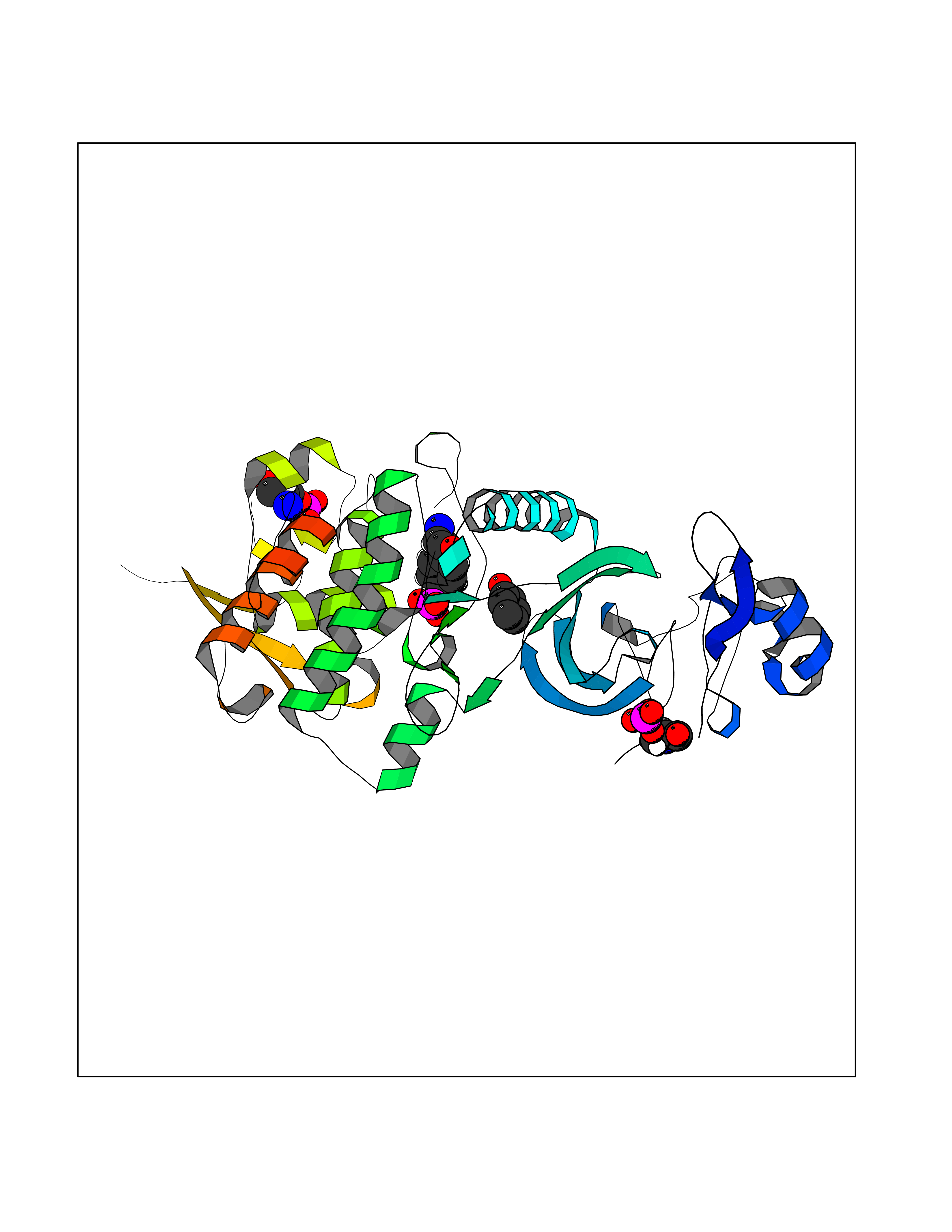 Molscript Map 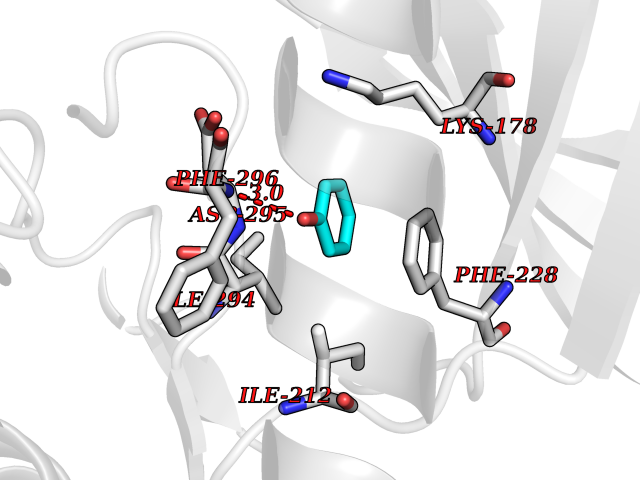 Pymol Map 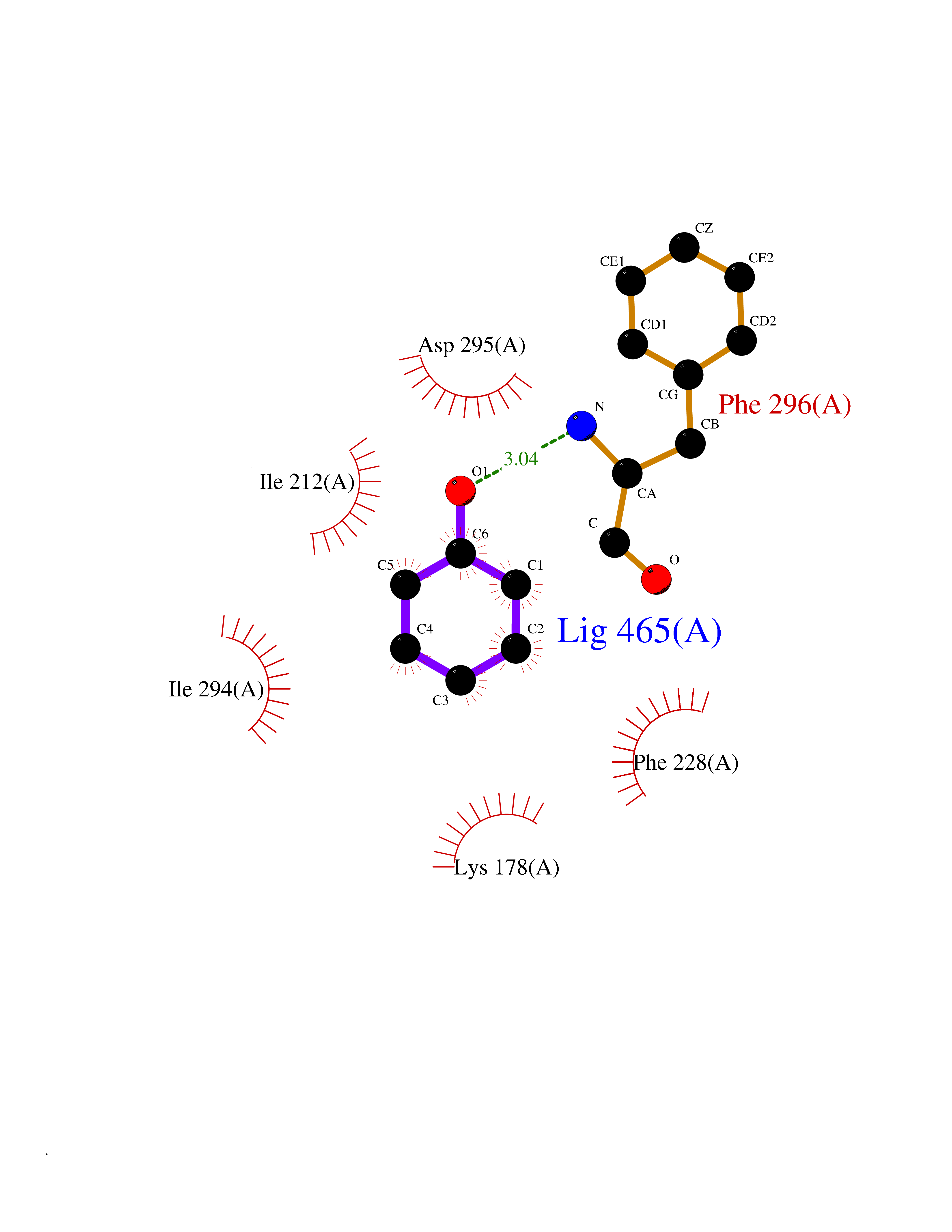 Ligplot Map 3D Binding mode Sequence HHHSXGVDLGTENLYFQSMGKVKATPMTPEQAMKQYMQKLTAFEHHEIFSYPEIYFLGLNAKKRQGMTGGPNNGGYDDDQGSYVQVPHDHVAYRYEVLKVIGKGSFGQVVKAYDHKVHQHVALKMVRNEKRFHRQAAEEIRILEHLRKQDKDNTMNVIHMLENFTFRNHICMTFELLSMNLYELIKKNKFQGFSLPLVRKFAHSILQCLDALHKNRIIHCDLKPENILLKQQGRSGIKVIDFGSSCYEHQRVYTXIQSRFYRAPEVILGARYGMPIDMWSLGCILAELLTGYPLLPGEDEGDQLACMIELLGMPSQKLLDASKRAKNFVSXKGYPRYCTVTTLSDVVLNGGRSRRGKLRGPPESREWGNALKGCDDPLFLDFLKQCLEWDPAVRMTPGQALRHPWLR Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Retinoic acid receptor alpha (RARA) | 3KMR | 5.13 | |
Target general information Gen name RARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-alpha; RAR alpha; Nuclear receptor subfamily 1 group B member 1; NR1B1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. RARA plays an essential role in the regulation of retinoic acid-induced germ cell development during spermatogenesis. Has a role in the survival of early spermatocytes at the beginning prophase of meiosis. In Sertoli cells, may promote the survival and development of early meiotic prophase spermatocytes. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Chromosomal aberrations involving RARA are commonly found in acute promyelocytic leukemia. Translocation t(11;17)(q32;q21) with ZBTB16/PLZF; translocation t(15;17)(q21;q21) with PML; translocation t(5;17)(q32;q11) with NPM. The PML-RARA oncoprotein requires both the PML ring structure and coiled-coil domain for both interaction with UBE2I, nuclear microspeckle location and sumoylation. In addition, the coiled-coil domain functions in blocking RA-mediated transactivation and cell differentiation. {ECO:0000269|PubMed:12691149, ECO:0000269|PubMed:8302850, ECO:0000269|PubMed:8562957}. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB00926; DB00982; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O43707-1; O15296; Q15699; Q96RK4; O95273; P51946; Q15910; P50148; Q9UKP3; Q96EZ8; Q15648; Q71SY5; Q15788; Q9Y6Q9; O75376; Q9Y618; Q16236; P13056-2; P48552; Q9UPP1-2; Q9H8W4; P37231; P78527; P19793; P28702; P28702-3; P48443; Q96EB6; P63165; Q8WW24; Q2M1K9; Q91XC0; P59598; Q14457; P48552; Q96CV9; P28702; P48443; Q8WW24 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Chromosomal rearrangement; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 27724.1 Length 244 Aromaticity 0.05 Instability index 50.8 Isoelectric point 5.82 Charge (pH=7) -3.61 2D Binding mode Binding energy (Kcal/mol) -7  Molscript Map 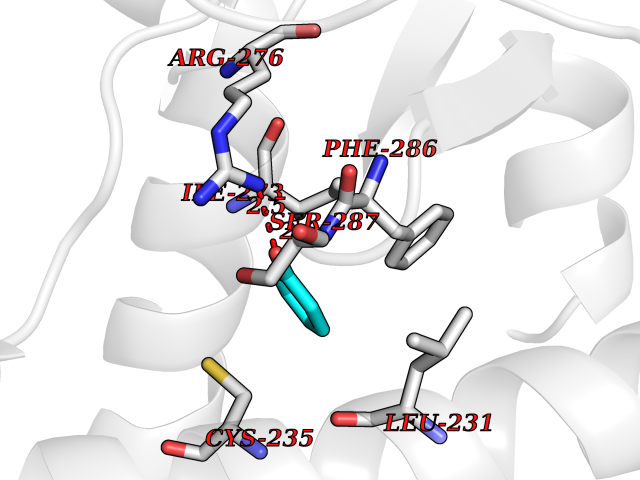 Pymol Map 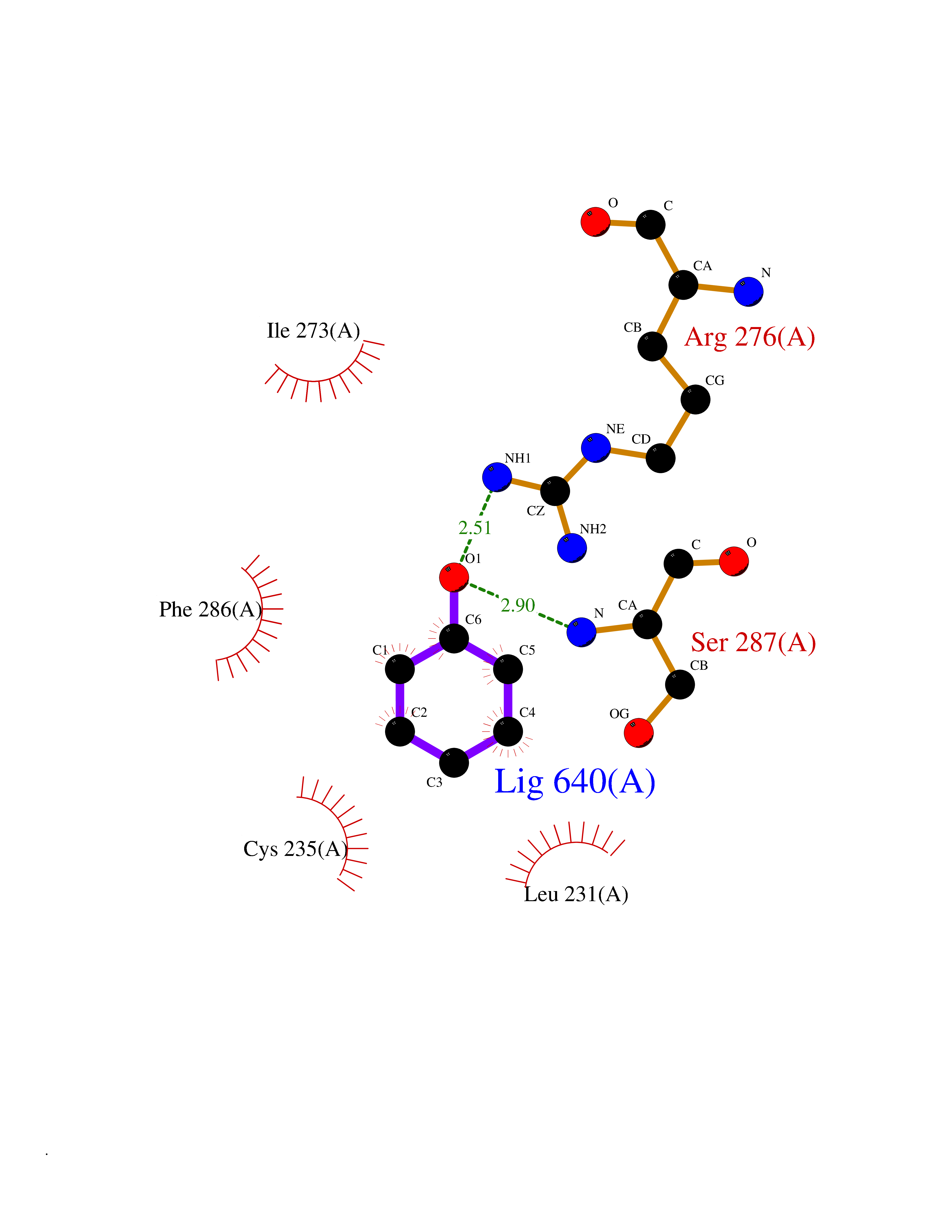 Ligplot Map 3D Binding mode Sequence PEVGELIEKVRKAHQETFPALCQLGKYTTNNSSEQRVSLDIDLWDKFSELSTKCIIKTVEFAKQLPGFTTLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFANQLLPLEMDDAETGLLSAICLICGDRQDLEQPDRVDMLQEPLLEALKVYVRKRRPSRPHMFPKMLMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEHKILHRLLQE Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Neprilysin | 1R1H | 5.13 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms EPN Protein family Peptidase M13 family Biochemical class Hydrolase Function Endopeptidase activity.Exopeptidase activity.Metalloendopeptidase activity.Metallopeptidase activity.Peptide binding.Zinc ion binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -6.99 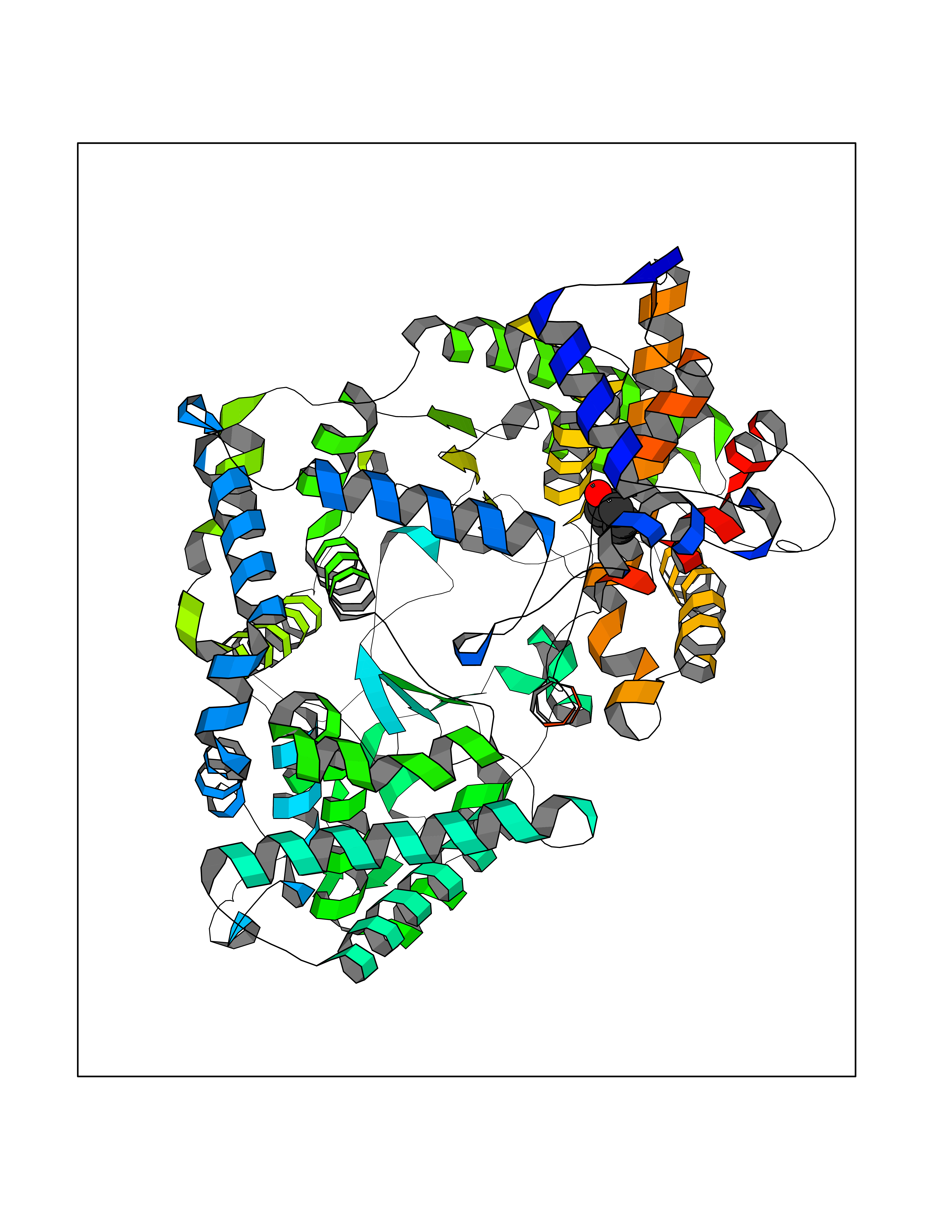 Molscript Map 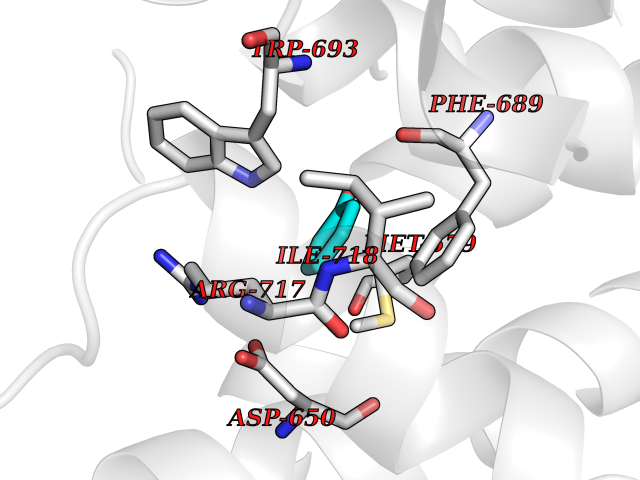 Pymol Map 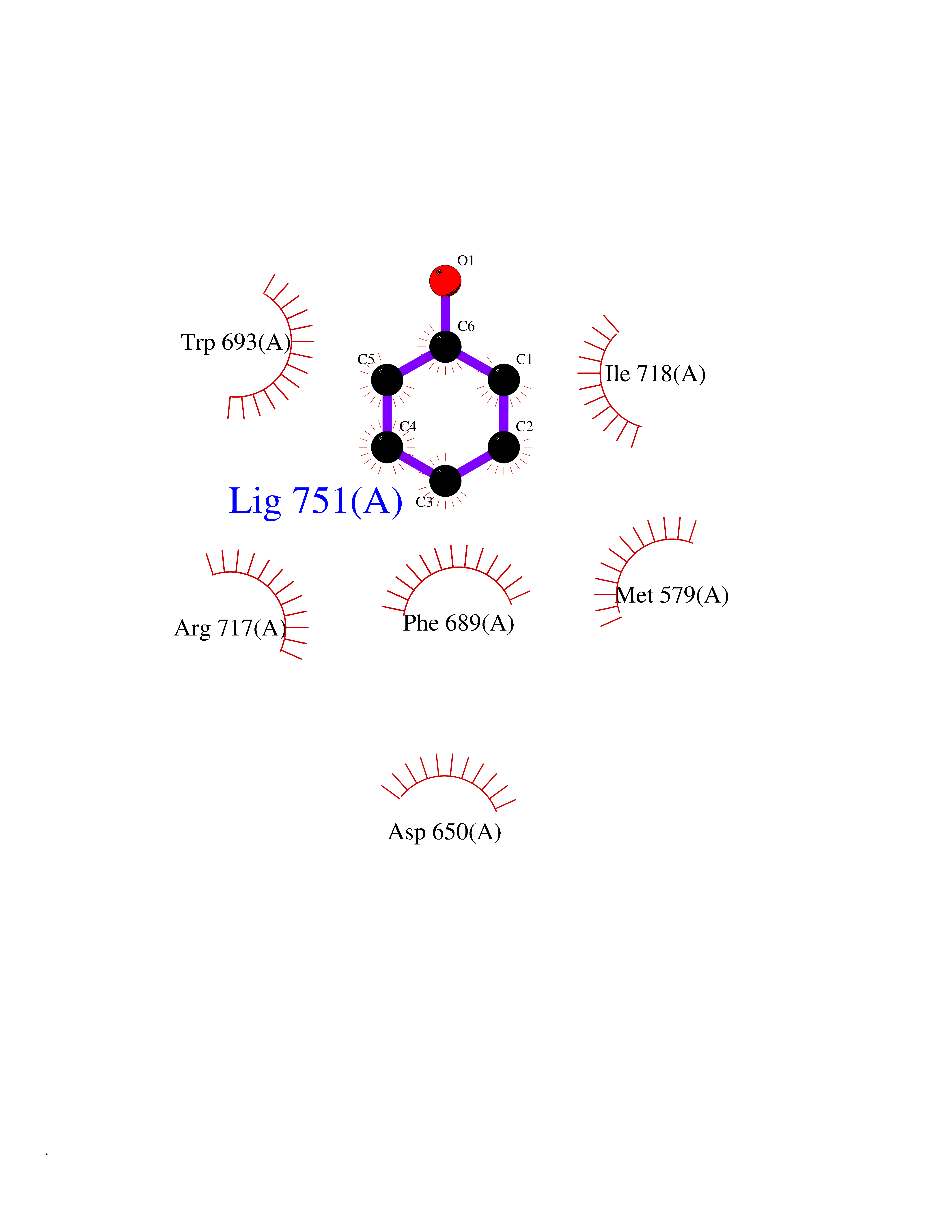 Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Retinoic acid receptor gamma (RARG) | 1FCY | 5.13 | |
Target general information Gen name RARG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-gamma; Nuclear receptor subfamily 1 group B member 3; NR1B3 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Receptor for retinoic acid. Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. Required for limb bud development. In concert with RARA or RARB, required for skeletal growth, matrix homeostasis and growth plate function (By similarity). Related diseases Cystic fibrosis (CF) [MIM:219700]: A common generalized disorder of the exocrine glands which impairs clearance of secretions in a variety of organs. It is characterized by the triad of chronic bronchopulmonary disease (with recurrent respiratory infections), pancreatic insufficiency (which leads to malabsorption and growth retardation) and elevated sweat electrolytes. It is the most common genetic disease in Caucasians, with a prevalence of about 1 in 2'000 live births. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10094564, ECO:0000269|PubMed:10869121, ECO:0000269|PubMed:10923036, ECO:0000269|PubMed:11242048, ECO:0000269|PubMed:12167682, ECO:0000269|PubMed:12394343, ECO:0000269|PubMed:12529365, ECO:0000269|PubMed:1284466, ECO:0000269|PubMed:1284468, ECO:0000269|PubMed:1284529, ECO:0000269|PubMed:1284530, ECO:0000269|PubMed:1284548, ECO:0000269|PubMed:1379210, ECO:0000269|PubMed:15528182, ECO:0000269|PubMed:15716351, ECO:0000269|PubMed:16822950, ECO:0000269|PubMed:1695717, ECO:0000269|PubMed:1699669, ECO:0000269|PubMed:17098864, ECO:0000269|PubMed:1710600, ECO:0000269|PubMed:1712898, ECO:0000269|PubMed:17182731, ECO:0000269|PubMed:20008117, ECO:0000269|PubMed:20150177, ECO:0000269|PubMed:20691141, ECO:0000269|PubMed:21884936, ECO:0000269|PubMed:2236053, ECO:0000269|PubMed:23818989, ECO:0000269|PubMed:25330774, ECO:0000269|PubMed:26846474, ECO:0000269|PubMed:27241308, ECO:0000269|PubMed:28001373, ECO:0000269|PubMed:28067262, ECO:0000269|PubMed:28087700, ECO:0000269|PubMed:32026723, ECO:0000269|PubMed:33572515, ECO:0000269|PubMed:7504969, ECO:0000269|PubMed:7505694, ECO:0000269|PubMed:7505767, ECO:0000269|PubMed:7508414, ECO:0000269|PubMed:7513296, ECO:0000269|PubMed:7517264, ECO:0000269|PubMed:7520022, ECO:0000269|PubMed:7522211, ECO:0000269|PubMed:7524909, ECO:0000269|PubMed:7524913, ECO:0000269|PubMed:7525450, ECO:0000269|PubMed:7537150, ECO:0000269|PubMed:7541273, ECO:0000269|PubMed:7541510, ECO:0000269|PubMed:7543567, ECO:0000269|PubMed:7544319, ECO:0000269|PubMed:7581407, ECO:0000269|PubMed:7606851, ECO:0000269|PubMed:7680525, ECO:0000269|PubMed:7683628, ECO:0000269|PubMed:7683954, ECO:0000269|PubMed:8081395, ECO:0000269|PubMed:8406518, ECO:0000269|PubMed:8522333, ECO:0000269|PubMed:8723693, ECO:0000269|PubMed:8723695, ECO:0000269|PubMed:8800923, ECO:0000269|PubMed:8829633, ECO:0000269|PubMed:8910473, ECO:0000269|PubMed:8956039, ECO:0000269|PubMed:9101301, ECO:0000269|PubMed:9222768, ECO:0000269|PubMed:9375855, ECO:0000269|PubMed:9401006, ECO:0000269|PubMed:9443874, ECO:0000269|PubMed:9452048, ECO:0000269|PubMed:9452054, ECO:0000269|PubMed:9452073, ECO:0000269|PubMed:9482579, ECO:0000269|PubMed:9507391, ECO:0000269|PubMed:9521595, ECO:0000269|PubMed:9554753, ECO:0000269|PubMed:9736778, ECO:0000269|PubMed:9804160, ECO:0000269|PubMed:9921909}. The disease is caused by variants affecting the gene represented in this entry. There is some evidence that the functional defect caused by the most common variant Phe-508 DEL can be corrected by the binding to the snake phospholipase A2 crotoxin basic subunit CB. This toxin both disrupts the Phe-508 DEL-cytokeratin 8 complex, allowing for the escape from degradation, and increases the chloride channel current (PubMed:27241308). {ECO:0000269|PubMed:27241308}.; DISEASE: Congenital bilateral absence of the vas deferens (CBAVD) [MIM:277180]: An autosomal recessive disease characterized by vas deferens aplasia resulting in azoospermia and male infertility. CBAVD may occur in isolation or as a manifestation of cystic fibrosis. {ECO:0000269|PubMed:10066035, ECO:0000269|PubMed:10651488, ECO:0000269|PubMed:17329263, ECO:0000269|PubMed:7529962, ECO:0000269|PubMed:7539342, ECO:0000269|PubMed:9067761, ECO:0000269|PubMed:9736778, ECO:0000269|Ref.117}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07294; DB07031; DB00459; DB00210; DB00523; DB02466; DB03466; DB02741; DB03279; DB00926; DB00982; DB05785; DB05467; DB02258; DB00799; DB00755; DB12808 Interacts with Q96RK4; P13349; P31321; P28702; P48443; O60504-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 26574.9 Length 236 Aromaticity 0.06 Instability index 49.98 Isoelectric point 5.76 Charge (pH=7) -2.95 2D Binding mode Binding energy (Kcal/mol) -6.99 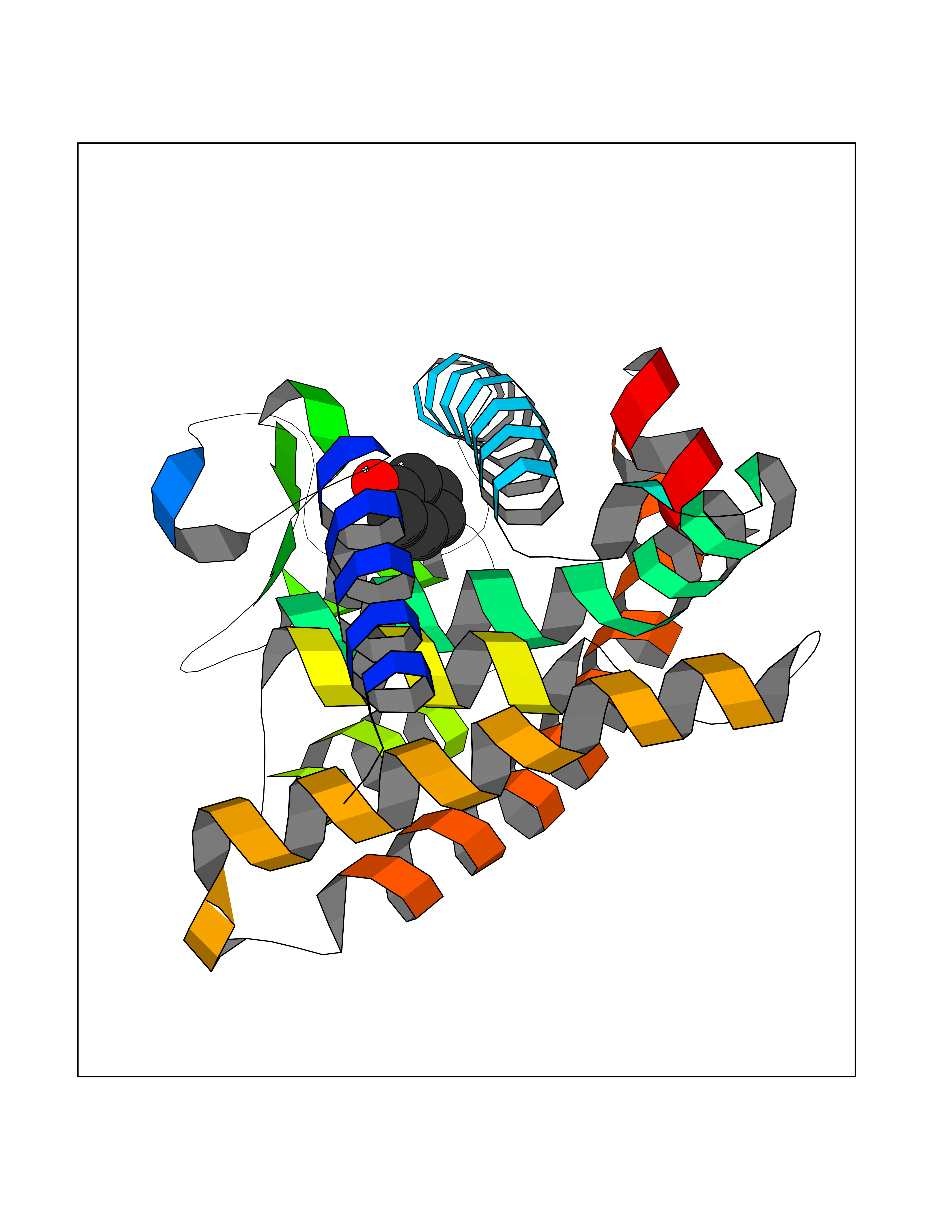 Molscript Map 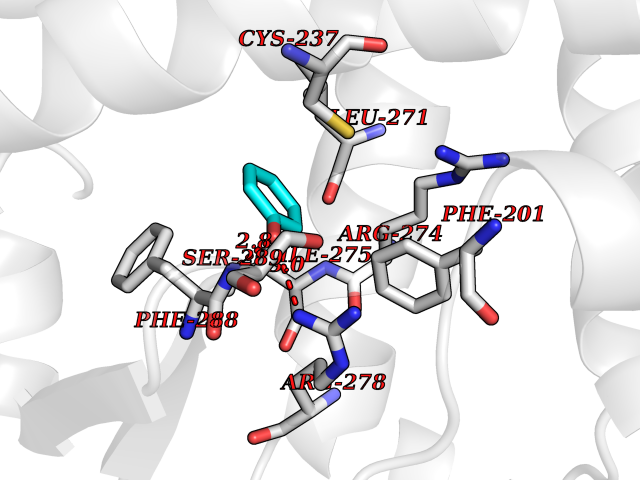 Pymol Map 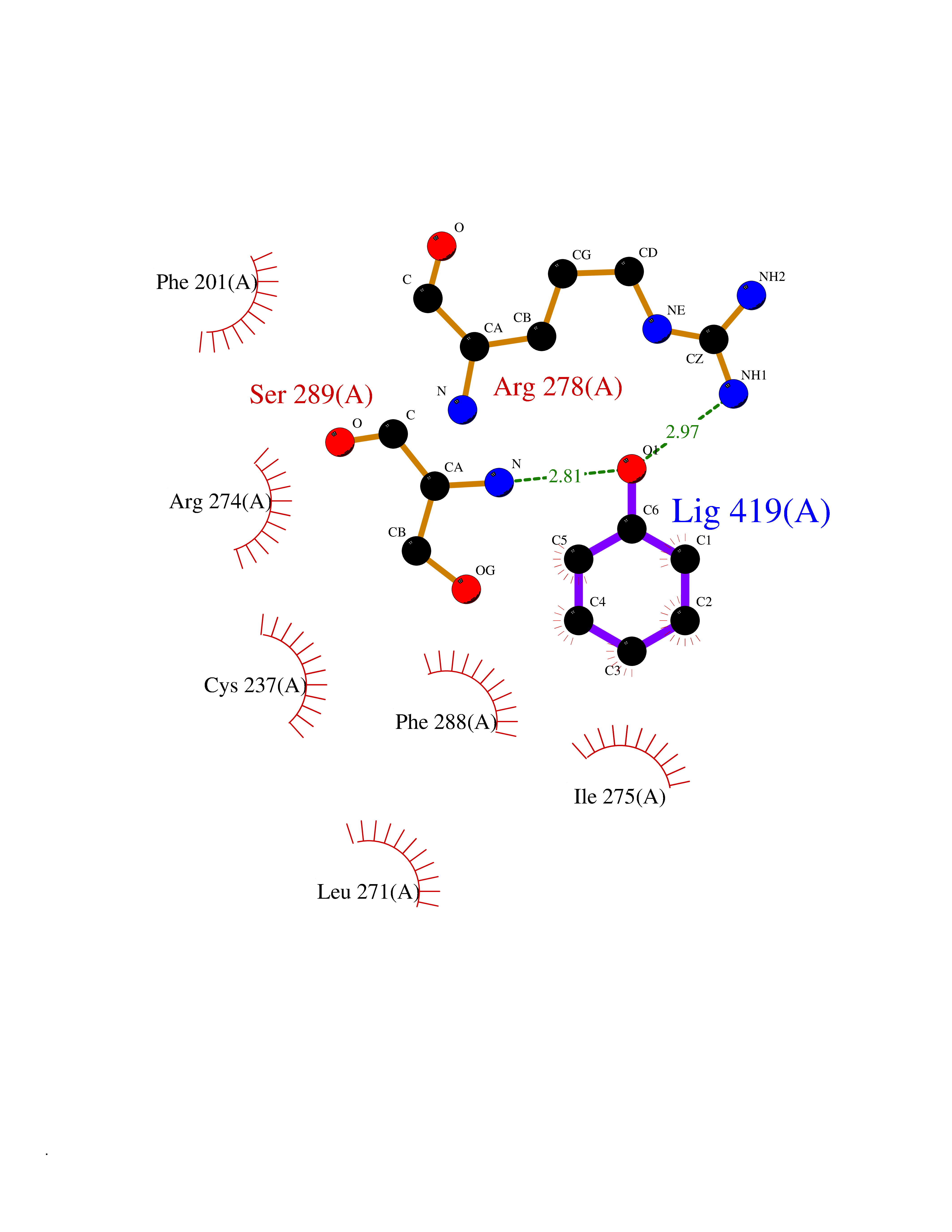 Ligplot Map 3D Binding mode Sequence ASPQLEELITKVSKAHQETFPSLCQLGKYTTNSSADHRVQLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLSIADQITLLKAACLDILMLRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFAGQLLPLEMDDTETGLLSAICLICGDRMDLEEPEKVDKLQEPLLEALRLYARRRRPSQPYMFPRMLMKITDLRGISTKGAERAITLKMEIPGPMPPLIREMLE Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Wnt-7a protein (WNT7A) | 4UZQ | 5.13 | |
Target general information Gen name WNT7A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Wnt-7a Protein family Wnt family Biochemical class NA Function Plays an important role in embryonic development, including dorsal versus ventral patterning during limb development, skeleton development and urogenital tract development. Required for central nervous system (CNS) angiogenesis and blood-brain barrier regulation. Required for normal, sexually dimorphic development of the Mullerian ducts, and for normal fertility in both sexes. Required for normal neural stem cell proliferation in the hippocampus dentate gyrus. Required for normal progress through the cell cycle in neural progenitor cells, for self-renewal of neural stem cells, and for normal neuronal differentiation and maturation. Promotes formation of synapses via its interaction with FZD5. Ligand for members of the frizzled family of seven transmembrane receptors that functions in the canonical Wnt/beta-catenin signaling pathway. Related diseases Limb pelvis hypoplasia aplasia syndrome (LPHAS) [MIM:276820]: A syndrome of severe deficiency of the extremities due to hypo- or aplasia of one or more long bones of one or more limbs. Pelvic manifestations include hip dislocation, hypoplastic iliac bone and aplastic pubic bones. Thoracic deformity, unusual facies and genitourinary anomalies can be present. {ECO:0000269|PubMed:16826533, ECO:0000269|PubMed:17431918, ECO:0000269|PubMed:20949531, ECO:0000269|PubMed:21271649, ECO:0000269|PubMed:27638328}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fuhrmann syndrome (FUHRS) [MIM:228930]: Distinct limb-malformation disorder characterized also by various degrees of limb aplasia/hypoplasia and joint dysplasia. {ECO:0000269|PubMed:16826533}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P55212; P22607; P06396; P13473-2; Q9UMX0; Q9Y5W5; Q5T9L3; Q9Z0J1 EC number NA Uniprot keywords 3D-structure; Developmental protein; Disease variant; Disulfide bond; Extracellular matrix; Glycoprotein; Lipoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Wnt signaling pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40475.5 Length 356 Aromaticity 0.1 Instability index 50.49 Isoelectric point 7.67 Charge (pH=7) 1.62 2D Binding mode Binding energy (Kcal/mol) -6.99 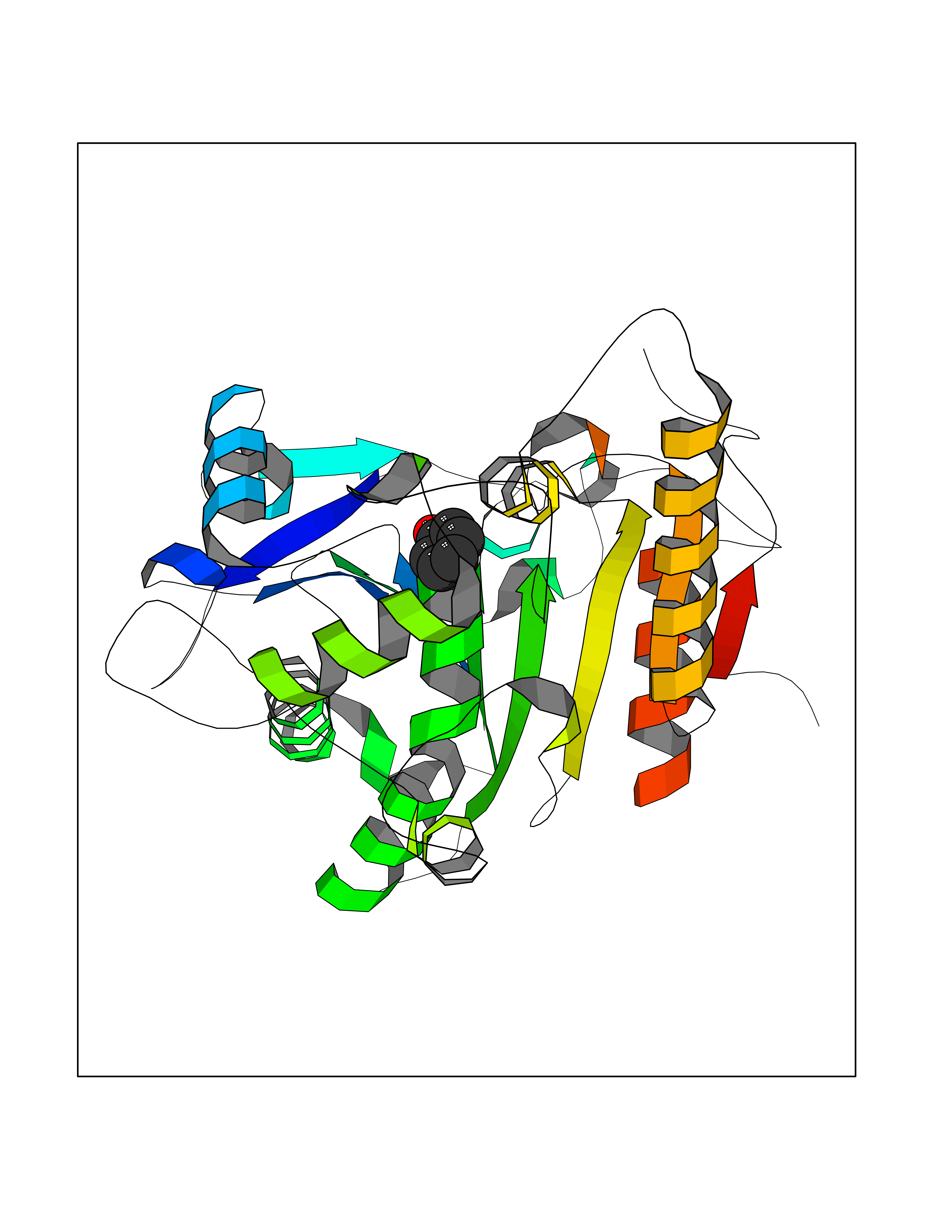 Molscript Map 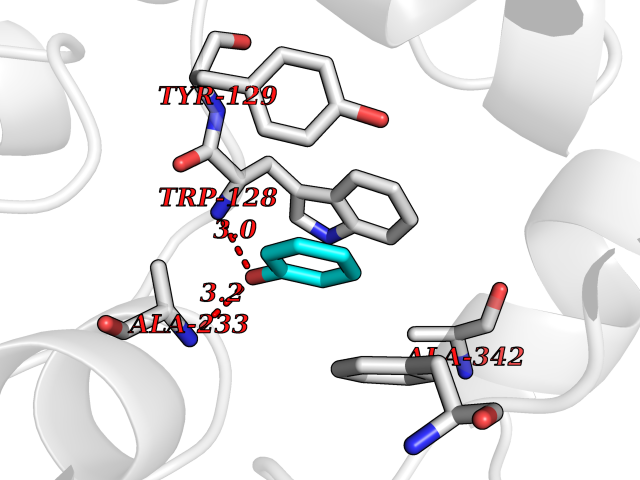 Pymol Map 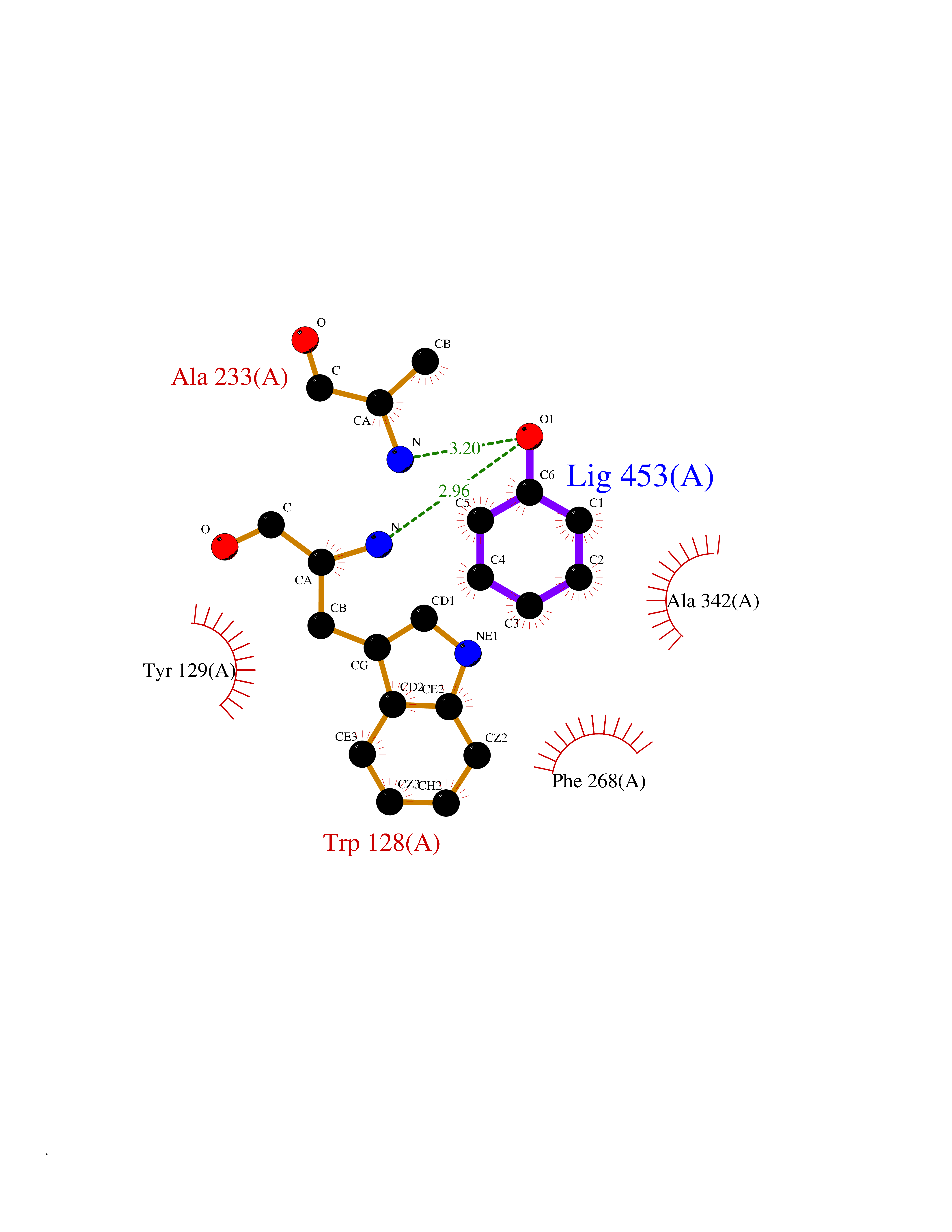 Ligplot Map 3D Binding mode Sequence EDLRLHLLLNTSVTCNDGSPAGYYLKESRGSRRWLLFLEGGWYCFNRENCDSRYDTMRRLMSSRDWPRTRTGTGILSSQPEENPYWWNANMVFIPYCSSDVWSGASSKSEKNEYAFMGALIIQEVVRELLGRGLSGAKVLLLAGSAAGGTGVLLNVDRVAEQLEKLGYPAIQVRGLADSGWFLDNKQYRHTDCVDTITCAPTEAIRRGIRYWNGVVPERCRRQFQEGEEWNCFFGYKVYPTLRSPVFVVQWLFDEAQLTVDNVHLTGQPVQEGLRLYIQNLGRELRHTLKDVPASFAPACLSHEIIIRSHWTDVQVKGTSLPRALHCWDRSLHKGCPVHLVDSCPWPHCNPSCPTS Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Natriuretic peptides B | 1YK1 | 5.12 | |
Target general information Gen name NPPB Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Natriuretic peptide family Biochemical class Hormone / growth factor receptor Function Diuretic hormone activity.Hormone activity.Peptide hormone receptor binding.Receptor binding. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01136; DB06412 Interacts with A8MQ03; P57678; Q6A162; P60411; Q7Z3S9; P25788; Q9UJW9 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disulfide bond; Glycoprotein; Hormone; Pharmaceutical; Proteoglycan; Proteomics identification; Reference proteome; Secreted; Signal; Vasoactive; Vasodilator Protein physicochemical properties Chain ID E Molecular weight (Da) 46353.1 Length 415 Aromaticity 0.1 Instability index 37.91 Isoelectric point 5.51 Charge (pH=7) -12.09 2D Binding mode Binding energy (Kcal/mol) -6.99 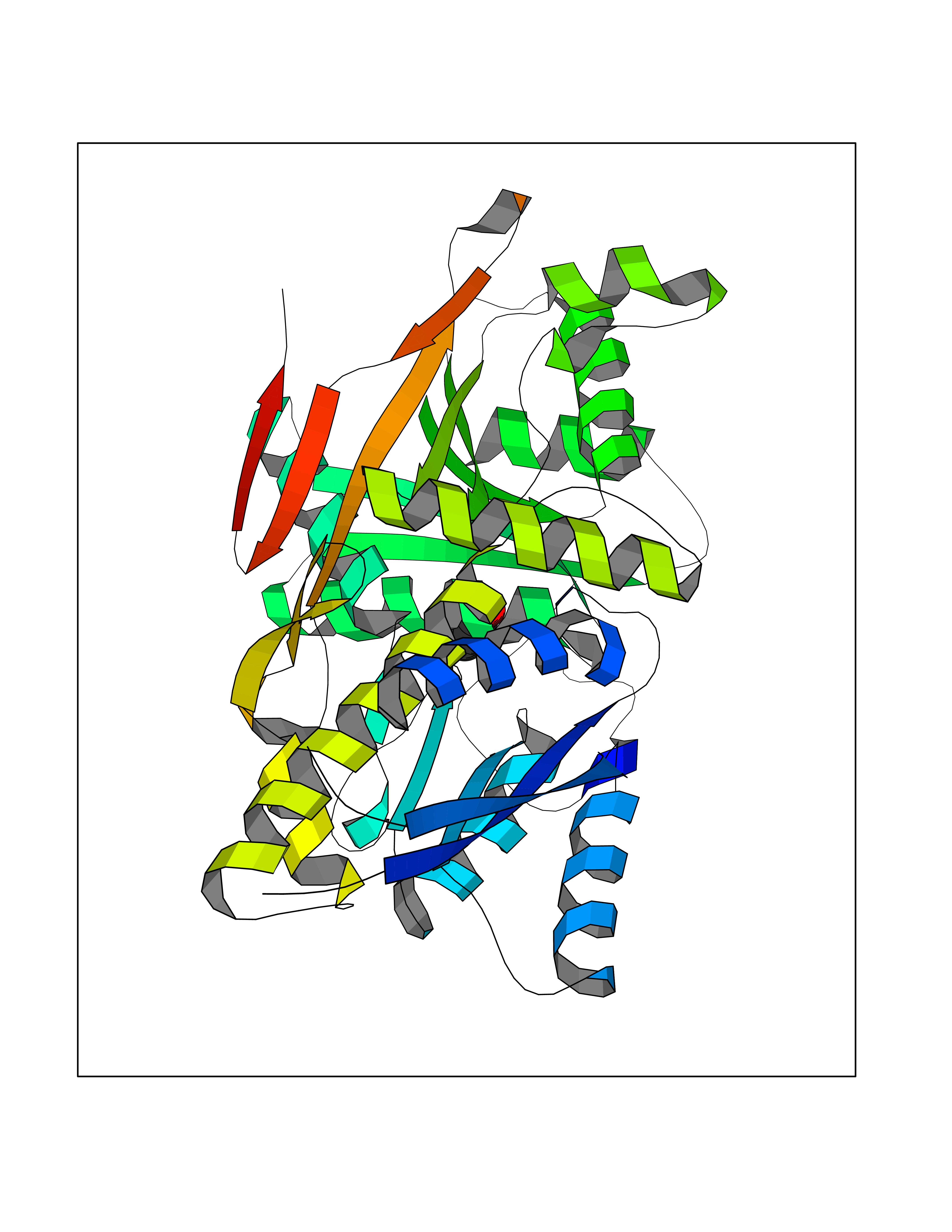 Molscript Map 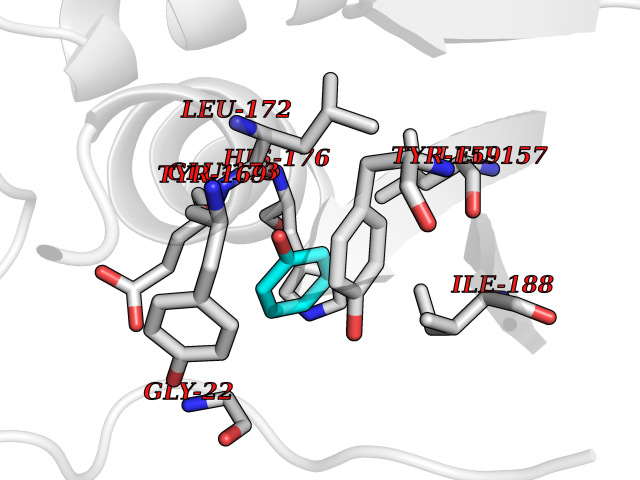 Pymol Map 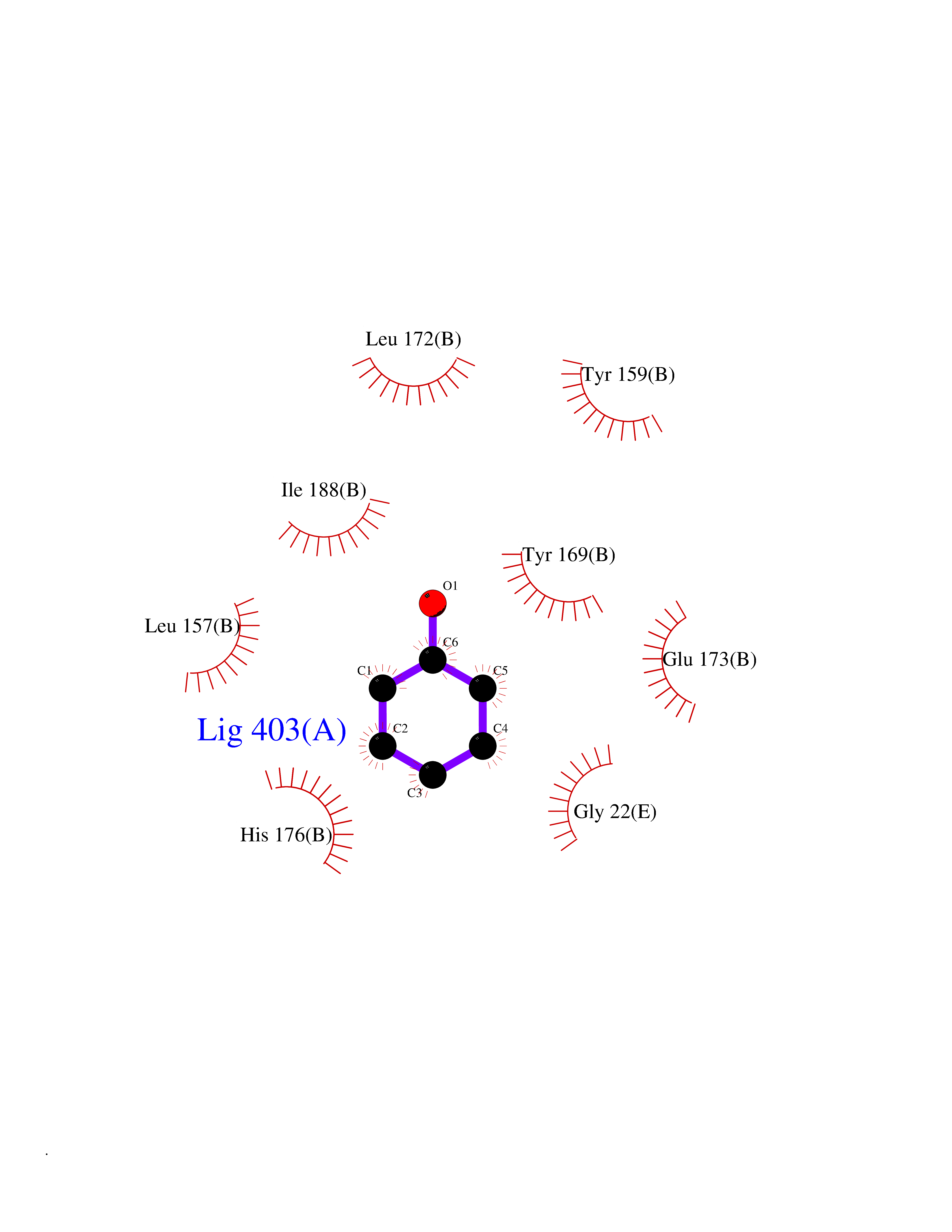 Ligplot Map 3D Binding mode Sequence GCFGRKMDRISSSSGLGCKVLALPPQKIEVLVLLPQDDSYLFSLTRVRPAIEYALRSVEGLLPPGTRFQVAYEDSDCGNRALFSLVDRVAAARGAKPDLILGPVCEYAAAPVARLASHWDLPMLSAGALAAGFQHKDSEYSHLTRVAPAYAKMGEMMLALFRHHHWSRAALVYSDDKLERNCYFTLEGVHEVFQEEGLHTSIYSFDETKDLDLEDIVRNIQASERVVIMCASSDTIRSIMLVAHRHGMTSGDYAFFNIELFNSSSYGDGSWKRGDKHDFEAKQAYSSLQTVTLLRTVKPEFEKFSMEVKSSVEKQGLNMEDYVNMFVEGFHDAILLYVLALHEVLRAGYSKKDGGKIIQQTWNRTFEGIAGQVSIDANGDRYGDFSVIAMTDVEAGTQEVIGDYFGKEGRFEMRP Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Nitric-oxide synthase inducible (NOS2) | 3E7G | 5.12 | |
Target general information Gen name NOS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms iNOS; Peptidyl-cysteine S-nitrosylase NOS2; Nitric oxide synthase, inducible; NOS2A; NOS type II; Inducible NOS; Inducible NO synthase; Hepatocyte NOS; HEP-NOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function Produces nitric oxide (NO) which is a messenger molecule with diverse functions throughout the body. In macrophages, NO mediates tumoricidal and bactericidal actions. Also has nitrosylase activity and mediates cysteine S-nitrosylation of cytoplasmic target proteins such PTGS2/COX2 (By similarity). As component of the iNOS-S100A8/9 transnitrosylase complex involved in the selective inflammatory stimulus-dependent S-nitrosylation of GAPDH on 'Cys-247' implicated in regulation of the GAIT complex activity and probably multiple targets including ANXA5, EZR, MSN and VIM. Involved in inflammation, enhances the synthesis of proinflammatory mediators such as IL6 and IL8. Related diseases Cerebellar ataxia, impaired intellectual development, and dysequilibrium syndrome 3 (CAMRQ3) [MIM:613227]: An autosomal recessive, congenital cerebellar ataxia associated with dysarthia, quadrupedal gait and intellectual disability. {ECO:0000269|PubMed:19461874}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07003; DB07007; DB07011; DB07405; DB08750; DB01997; DB07029; DB07008; DB08214; DB07002; DB01835; DB06879; DB04534; DB03100; DB02207; DB00125; DB00155; DB01234; DB14649; DB11327; DB00997; DB07306; DB07388; DB05252; DB01381; DB03366; DB05214; DB04400; DB09237; DB00244; DB01110; DB01017; DB03144; DB01686; DB03449; DB06916; DB07318; DB07389; DB02044; DB02644; DB05383; DB02234; DB03953; DB02462; DB08814 Interacts with P04406 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calmodulin-binding; Cytoplasm; FAD; Flavoprotein; FMN; Heme; Iron; Metal-binding; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 48633 Length 421 Aromaticity 0.12 Instability index 46.5 Isoelectric point 6.75 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -6.99 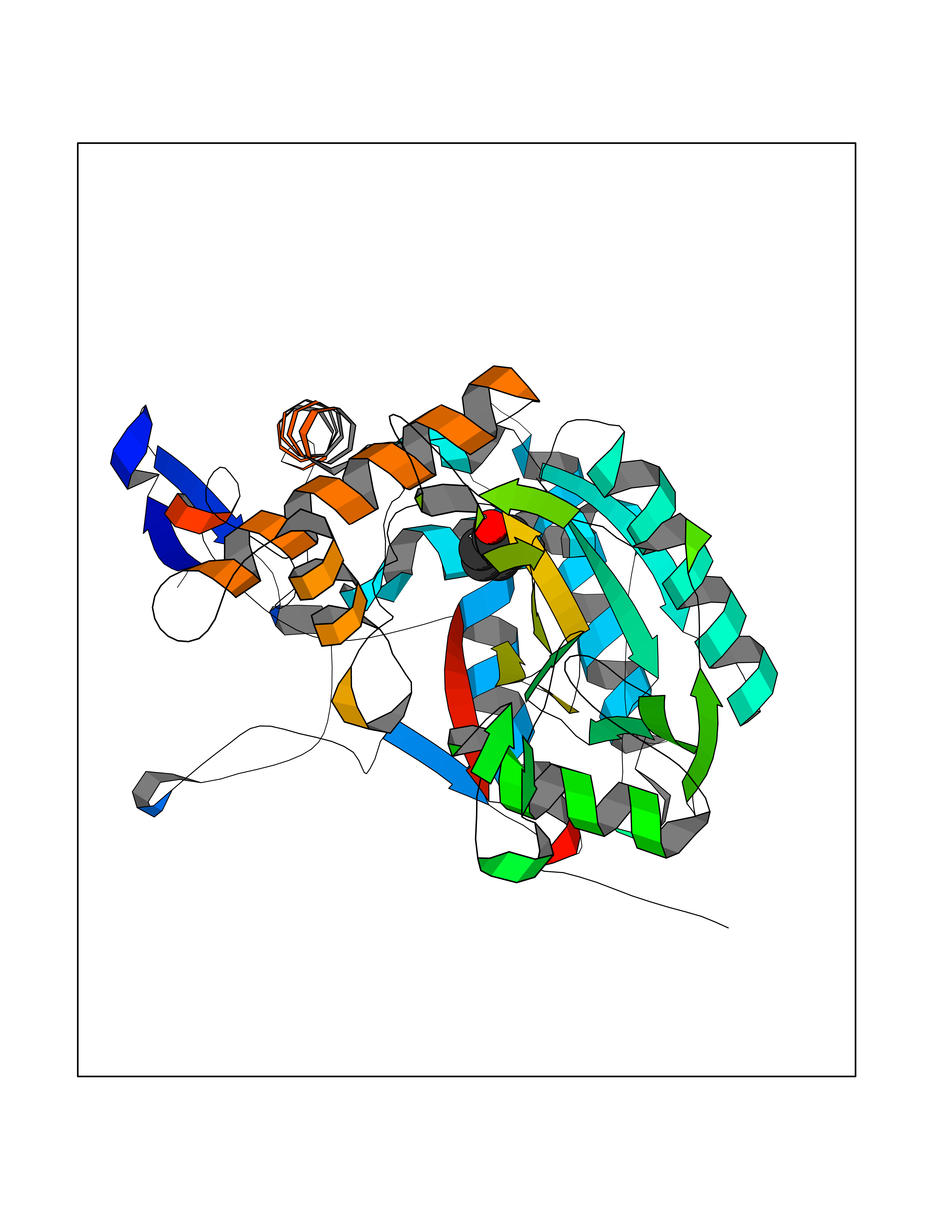 Molscript Map 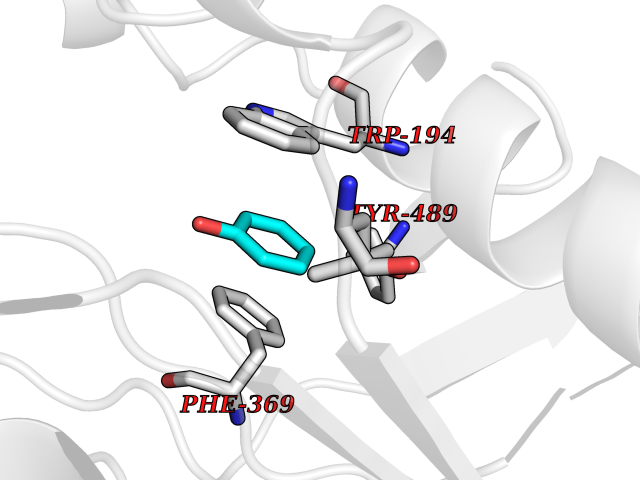 Pymol Map 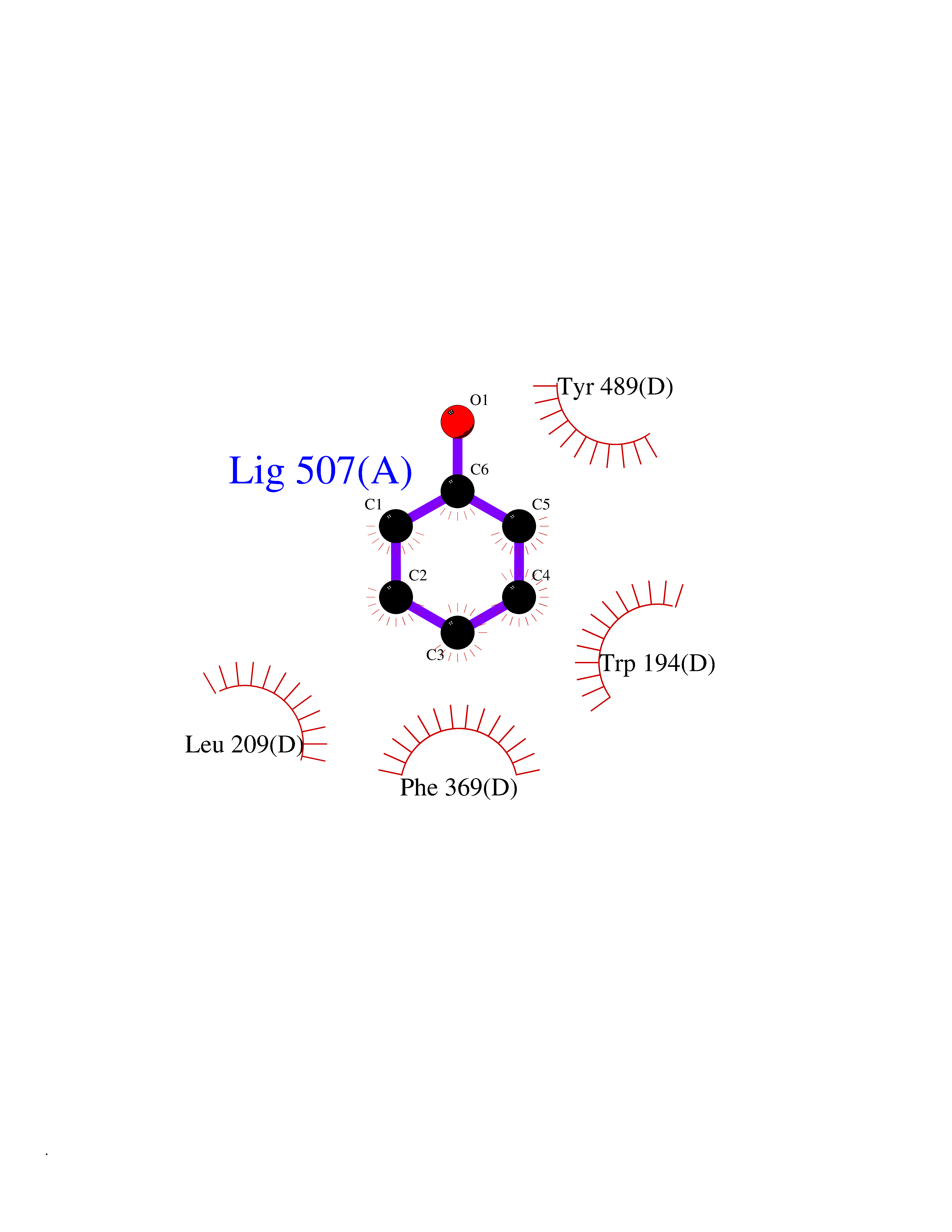 Ligplot Map 3D Binding mode Sequence RHVRIKNWGSGMTFQDTLHHKAKGILTCRSKSCLGSIMTPKSLTRGPRDKPTPPDELLPQAIEFVNQYYGSFKEAKIEEHLARVEAVTKEIETTGTYQLTGDELIFATKQAWRNAPRCIGRIQWSNLQVFDARSCSTAREMFEHICRHVRYSTNNGNIRSAITVFPQRSDGKHDFRVWNAQLIRYAGYQMPDGSIRGDPANVEFTQLCIDLGWKPKYGRFDVVPLVLQANGRDPELFEIPPDLVLEVAMEHPKYEWFRELELKWYALPAVANMLLEVGGLEFPGCPFNGWYMGTEIGVRDFCDVQRYNILEEVGRRMGLETHKLASLWKDQAVVEINIAVLHSFQKQNVTIMDHHSAAESFMKYMQNEYRSRGGCPADWIWLVPPMSGSITPVFHQEMLNYVLSPFYYYQVEAWKTHVWQD Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | SPRY domain-containing SOCS box protein 2 (SSB-2) (Gene-rich cluster protein C9) | 3EMW | 5.12 | |
Target general information Gen name SPSB2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GRCC9;SSB2 Protein family SPSB family Biochemical class NA Function Substrate recognition component of a SCF-like ECS (Elongin BC-CUL2/5-SOCS-box protein) E3 ubiquitin-protein ligase complex which mediates the ubiquitination and subsequent proteasomal degradation of target proteins (PubMed:15601820, PubMed:21199876). Negatively regulates nitric oxide (NO) production and limits cellular toxicity in activated macrophages by mediating the ubiquitination and proteasomal degradation of NOS2 (PubMed:21199876). Acts as a bridge which links NOS2 with the ECS E3 ubiquitin ligase complex components ELOC and CUL5 (PubMed:21199876). {ECO:0000269|PubMed:15601820, ECO:0000269|PubMed:21199876}." Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with G5E9A7; Q15369; Q53EP0-3; O60333-2; Q16656-4; Q96IZ0; P16284; Q92569; O60260-5; Q99873; Q6P9E2; Q9Y3C5; Q96GM5; P61086; P08670; P09052 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Host-virus interaction; Proteomics identification; Reference proteome; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 22703.1 Length 207 Aromaticity 0.09 Instability index 50.84 Isoelectric point 5.98 Charge (pH=7) -2.79 2D Binding mode Binding energy (Kcal/mol) -6.99  Molscript Map 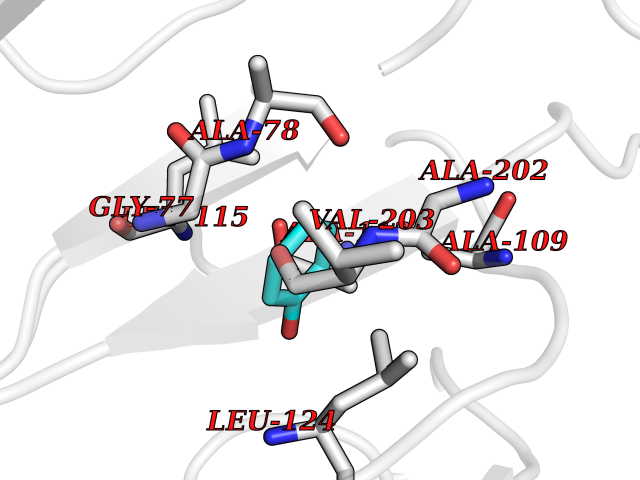 Pymol Map 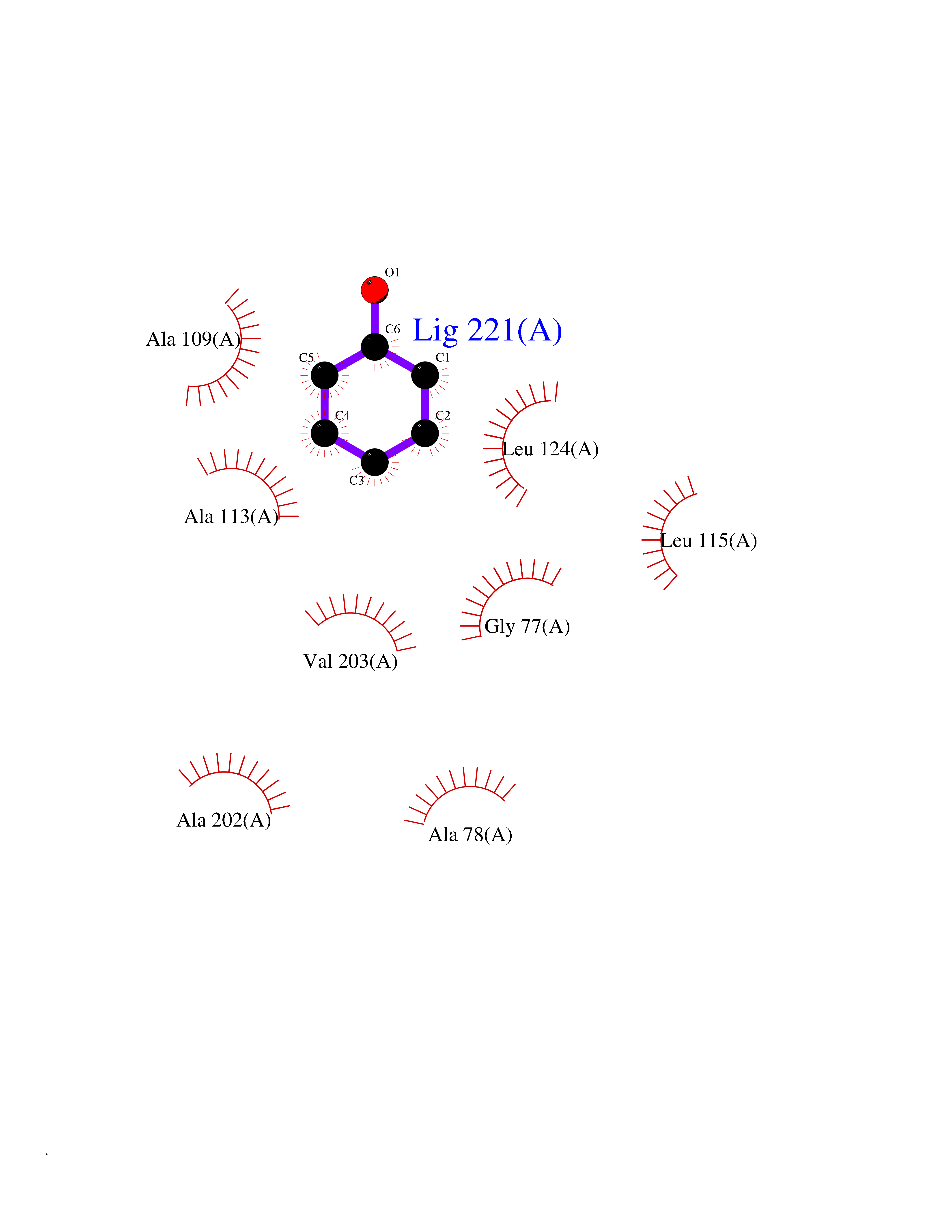 Ligplot Map 3D Binding mode Sequence LYFQSMPEGLEELLSAPPPDLGAQRRHGWNPKDCSENIEVKEGGLYFERRPVAQSTDGARGKRGYSRGLHAWEISWPLEQRGTHAVVGVATALAPLQTDHYAALLGSNSESWGWDIGRGKLYHQSKGPGAPQYPAGTQGEQLEVPERLLVVLDMEEGTLGYAIGGTYLGPAFRGLKGRTLYPAVSAVWGQCQVRIRYLGEDINNNNN Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Mitochondrial aldehyde dehydrogenase (ALDH2) | 1O04 | 5.11 | |
Target general information Gen name ALDH2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde dehydrogenase, mitochondrial; ALDM; ALDHI; ALDH-E2; ALDH class 2 Protein family Aldehyde dehydrogenase family Biochemical class Aldehyde/oxo donor oxidoreductase Function Second enzyme of the major oxidative pathway of alcohol metabolism. Catalyzes the chemical transformation from acetaldehyde to acetic acid. Additionally, functions as a protector against oxidative stress. Related diseases AMED syndrome, digenic (AMEDS) [MIM:619151]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. AMEDS is an autosomal recessive, digenic form characterized by childhood onset of bone marrow failure resulting in aplastic anemia, in association with global developmental delay, intellectual disability, and poor overall growth with short stature. {ECO:0000269|PubMed:33355142}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. AMEDS patients carry ADH5 biallelic variants and homozygous or heterozygous ALDH2 variant p.Glu504Lys, affecting protein activity. Cellular and animal studies demonstrate that the simultaneous loss of ALDH2 and ADH5 activities leads to an increase of cellular formaldehyde sensitivity and multisystem abnormalities including hematopoietic failure. {ECO:0000269|PubMed:33355142}. Drugs (DrugBank ID) DB01612; DB06770; DB04381; DB02115; DB00822; DB00536; DB00157; DB00435; DB00727; DB09117; DB06154; DB06207 Interacts with NA EC number EC 1.2.1.3 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; Dwarfism; Intellectual disability; Mitochondrion; NAD; Oxidoreductase; Proteomics identification; Reference proteome; Transit peptide; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 50223.5 Length 462 Aromaticity 0.1 Instability index 33.68 Isoelectric point 5.29 Charge (pH=7) -7.87 2D Binding mode Binding energy (Kcal/mol) -6.97 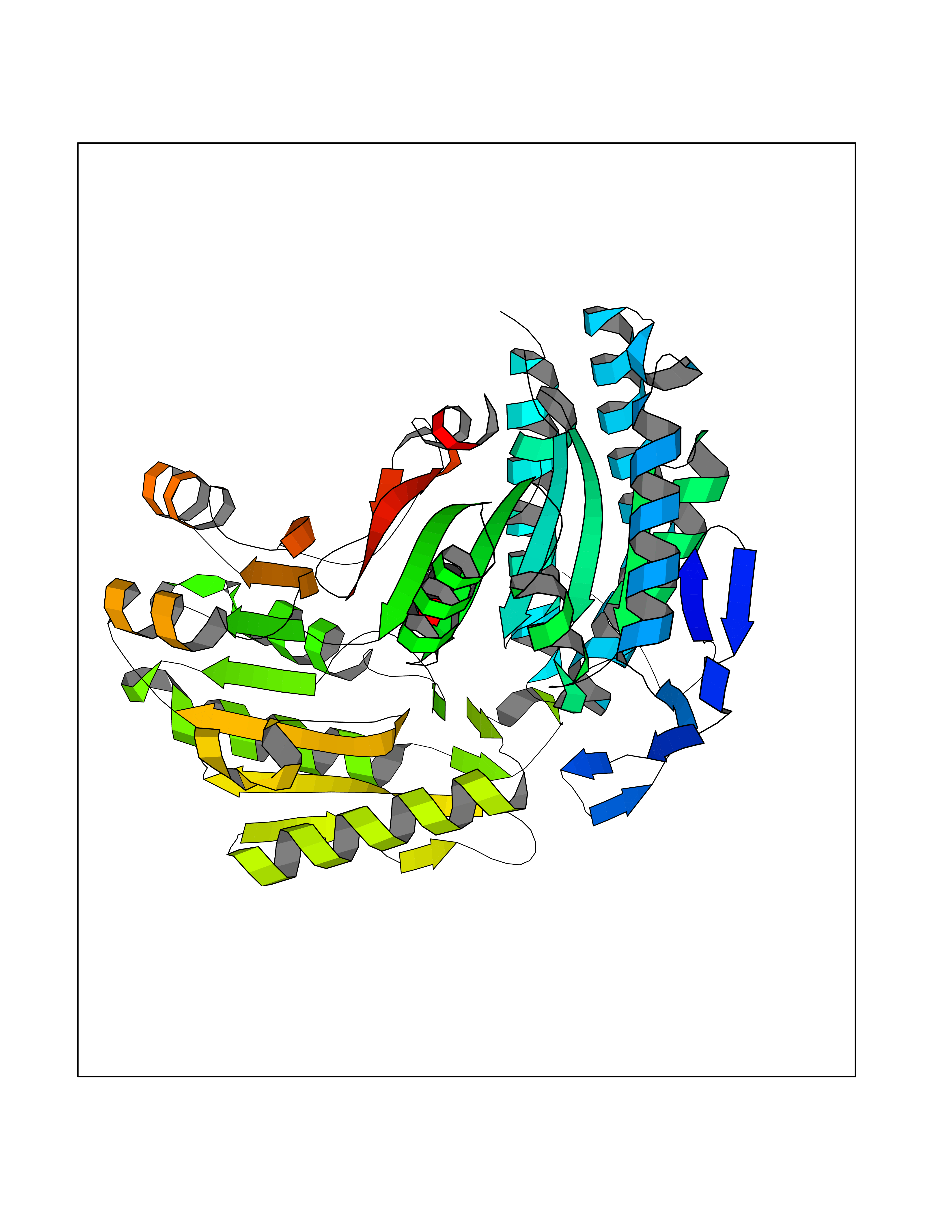 Molscript Map 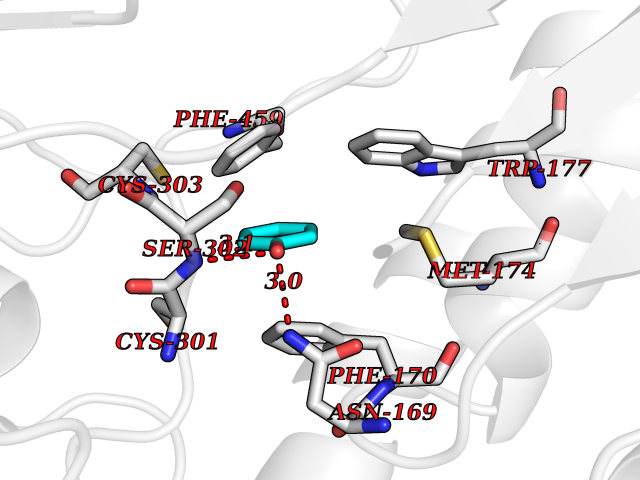 Pymol Map 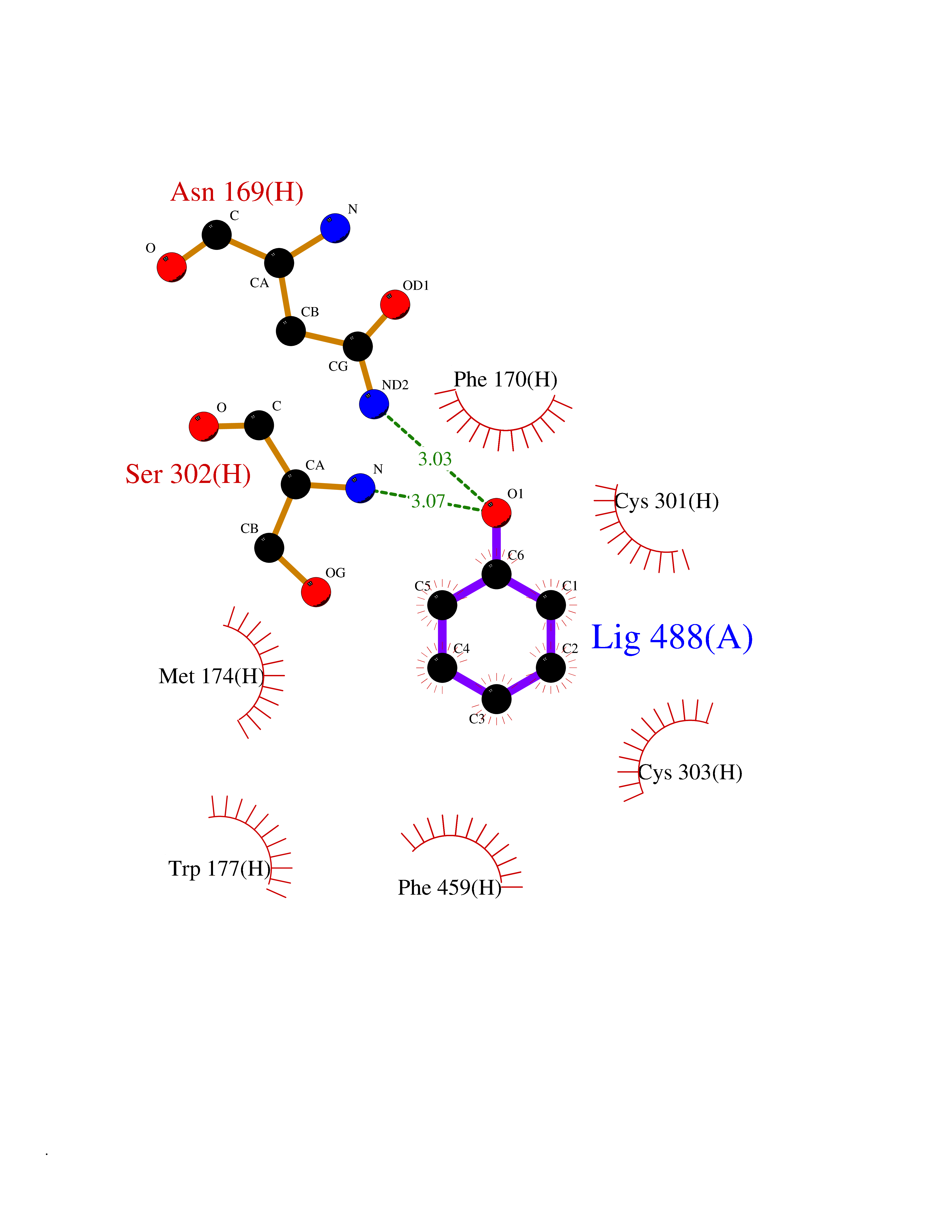 Ligplot Map 3D Binding mode Sequence AVPAPNQQPEVFCNQIFINNEWHDAVSRKTFPTVNPSTGEVICQVAEGDKEDVDKAVKAARAAFQLGSPWRRMDASHRGRLLNRLADLIERDRTYLAALETLDNGKPYVISYLVDLDMVLKCLRYYAGWADKEPVGVCGQIIPWNFPLLMQAWKLGPALATGNVVVMKVAEQTPLTALYVANLIKEAGFPPGVVNIVPGFGPTAGAAIASHEDVDKVAFTGSTEIGRVIQVAAGSSNLKRVTLELGGKSPNIIMSDADMDWAVEQAHFALFFNQGQCSCAGSRTFVQEDIYDEFVERSVARAKSRVVGNPFDSKTEQGPQVDETQFKKILGYINTGKQEGAKLLCGGGIAADRGYFIQPTVFGDVQDGMTIAKEEIFGPVMQILKFKTIEEVVGRANNSTYGLAAAVFTKDLDKANYLSQALQAGTVWVNCYDVFGAQSPFGGYKMSGSGRELGEYGLQAYT Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Dihydrofolate synthase/folylpolyglutamate synthase | 1W78 | 5.11 | |
Target general information Gen name folC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms dedC;JW2312;b2315 Protein family Folylpolyglutamate synthase family Biochemical class Synthase Function ATP binding.Dihydrofolate synthase activity.Metal ion binding.Tetrahydrofolylpolyglutamate synthase activity. Related diseases Complement hyperactivation, angiopathic thrombosis, and protein-losing enteropathy (CHAPLE) [MIM:226300]: An autosomal recessive disease characterized by abdominal pain and diarrhea, primary intestinal lymphangiectasia, edema due to hypoproteinemia, malabsorption, and less frequently, bowel inflammation, recurrent infections, and angiopathic thromboembolic disease. Patients' T lymphocytes show increased complement activation causing surface deposition of complement and the generation of soluble C5a. {ECO:0000269|PubMed:28657829, ECO:0000269|PubMed:28657861}. The disease is caused by variants affecting the gene represented in this entry. CHAPLE is caused by biallelic mutations in the CD55 gene. Drugs (DrugBank ID) DB02437; DB03830 Interacts with NA EC number 6.3.2.12; 6.3.2.17 Uniprot keywords 3D-structure; ATP-binding; Folate biosynthesis; Ligase; Magnesium; Metal-binding; Nucleotide-binding; One-carbon metabolism; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 44392.8 Length 414 Aromaticity 0.07 Instability index 29.47 Isoelectric point 5.2 Charge (pH=7) -15.5 2D Binding mode Binding energy (Kcal/mol) -6.97 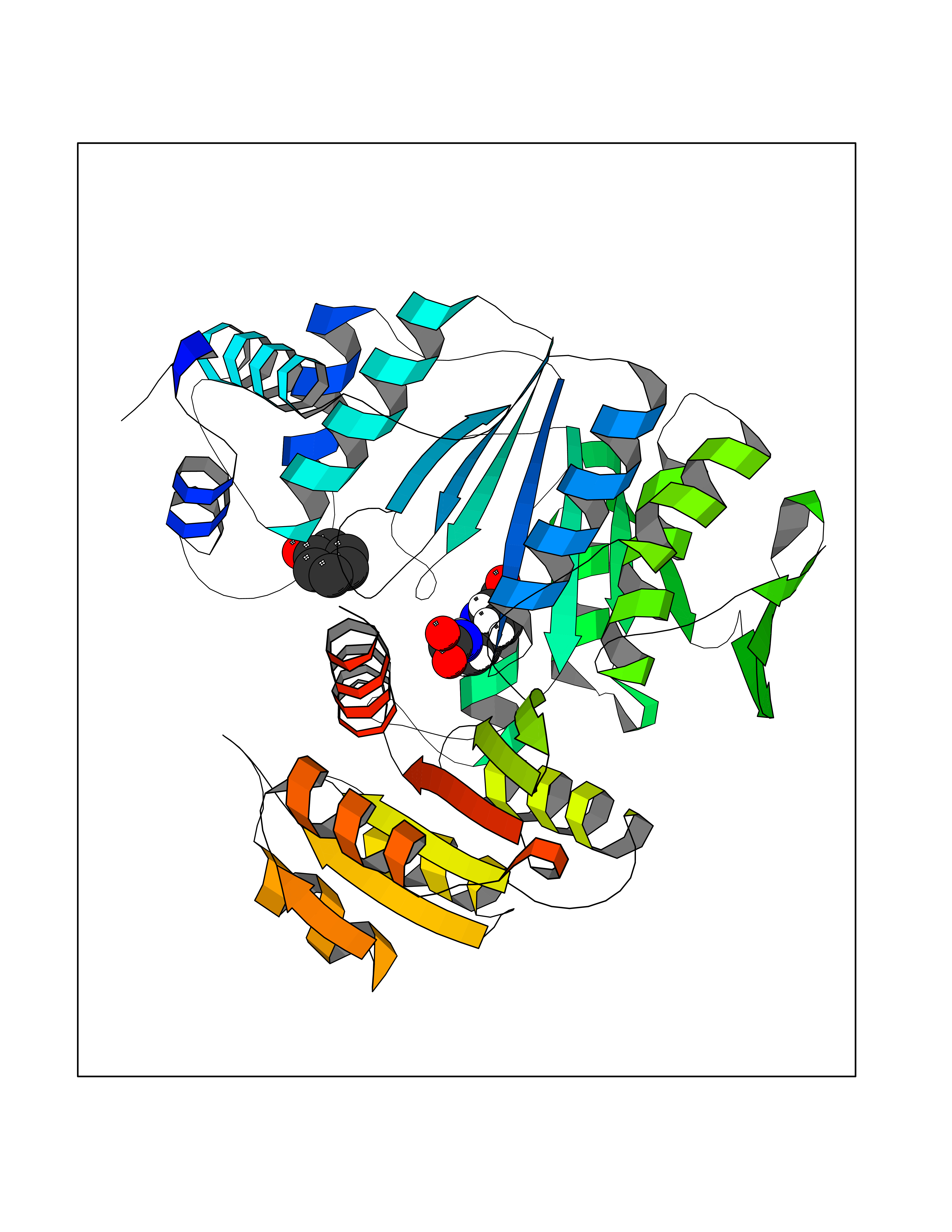 Molscript Map 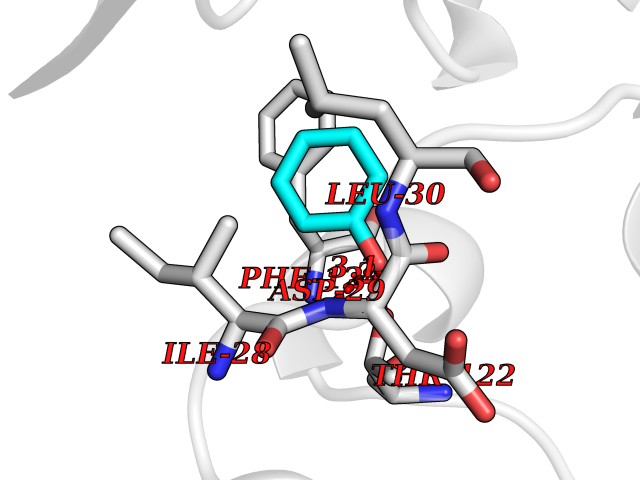 Pymol Map 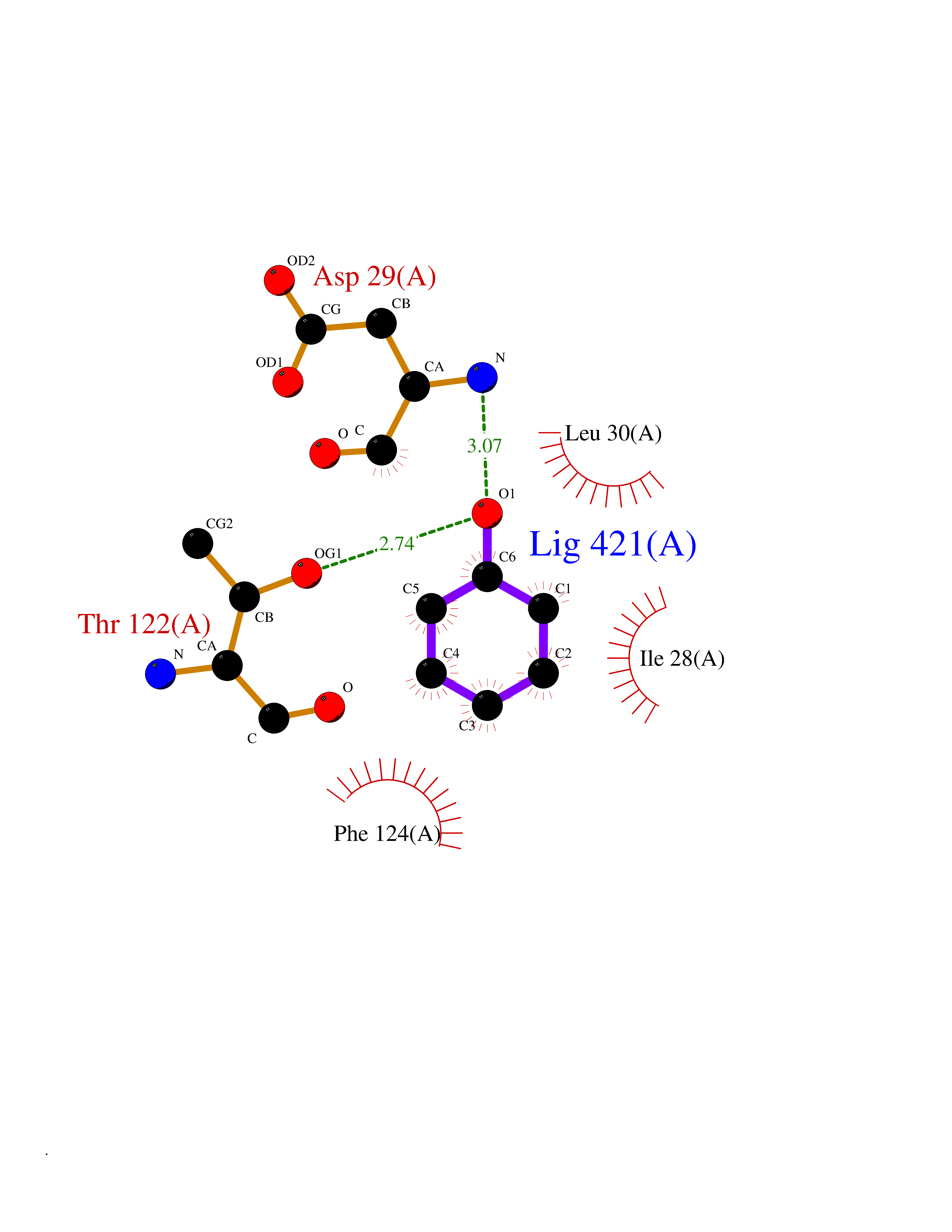 Ligplot Map 3D Binding mode Sequence TPQAASPLASWLSYLENLHSKTIDLGLERVSLVAARLGVLKPAPFVFTVAGTNGKGTTCRTLESILMAAGYKVGVYSSPHLVRYTERVRVQGQELPESAHTASFAEIESARGDISLTYFEYGTLSALWLFKQAQLDVVILEVGLGGRLDATNIVDADVAVVTSIALDHTDWLGPDRESIGREXAGIFRSEKPAIVGEPEMPSTIADVAQEKGALLQRRGVEWNYSVTDHDWAFSDAHGTLENLPLPLVPQPNAATALAALRASGLEVSENAIRDGIASAILPGRFQIVSESPRVIFDVAHNPHAAEYLTGRMKALPKNGRVLAVIGMLHDKDIAGTLAWLKSVVDDWYCAPLEGPRGATAEQLLEHLGNGKSFDSVAQAWDAAMADAKAEDTVLVCGSFHTVAHVMEVIDARRS Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Retinoic acid receptor RXR-alpha (RXRA) | 2P1T | 5.11 | |
Target general information Gen name RXRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor alpha; RXRalpha; Nuclear receptor subfamily 2 group B member 1; NR2B1 Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. The high affinity ligand for RXRs is 9-cis retinoic acid. RXRA serves as a common heterodimeric partner for a number of nuclear receptors. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. The RXRA/PPARA heterodimer is required for PPARA transcriptional activity on fatty acid oxidation genes such as ACOX1 and the P450 system genes. Receptor for retinoic acid. Related diseases Lichtenstein-Knorr syndrome (LIKNS) [MIM:616291]: An autosomal recessive neurologic disorder characterized by progressive cerebellar ataxia and severe progressive sensorineural hearing loss. {ECO:0000269|PubMed:25205112, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08063; DB08402; DB07863; DB07557; DB00459; DB00210; DB01436; DB00523; DB00132; DB04557; DB00307; DB01393; DB03756; DB00749; DB00926; DB05956; DB04224; DB02746; DB00412; DB00755; DB08601 Interacts with O14503; P35637; Q15648; Q71SY5; Q15788; Q15596; P55055; P55055-1; Q13133; P27986; P37231; P37231-1; P10276; P42224; P11473; P97792-1; Q9JLI4; P04625; PRO_0000278730 [Q03463] EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; DNA-binding; Host-virus interaction; Isopeptide bond; Metal-binding; Mitochondrion; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 24958.9 Length 221 Aromaticity 0.07 Instability index 39.47 Isoelectric point 6.16 Charge (pH=7) -3.45 2D Binding mode Binding energy (Kcal/mol) -6.97 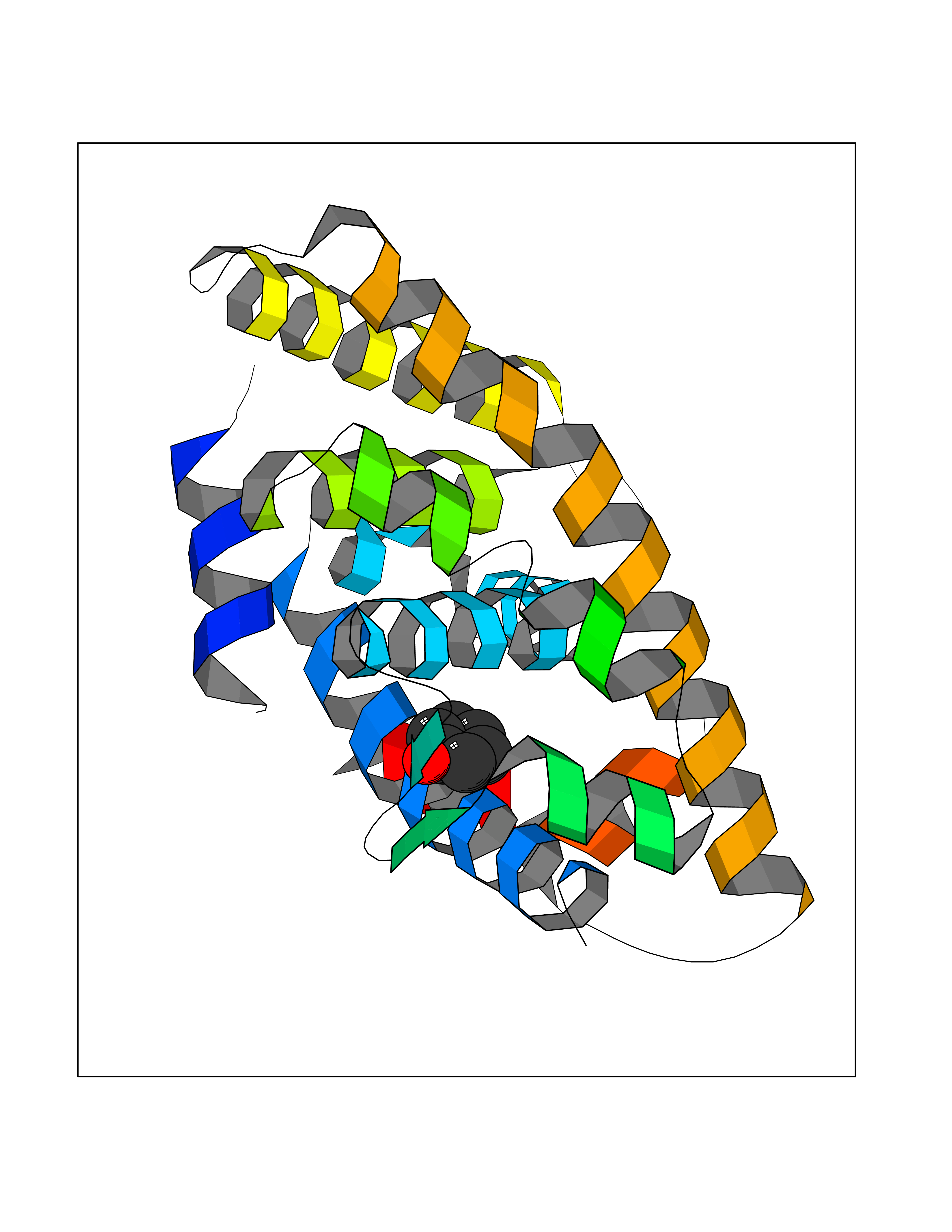 Molscript Map 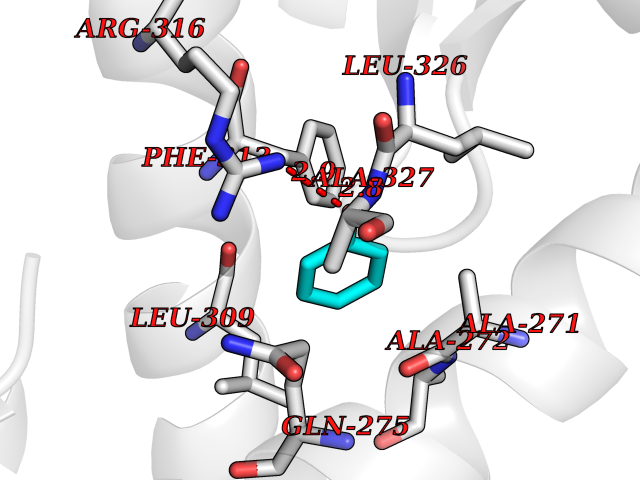 Pymol Map 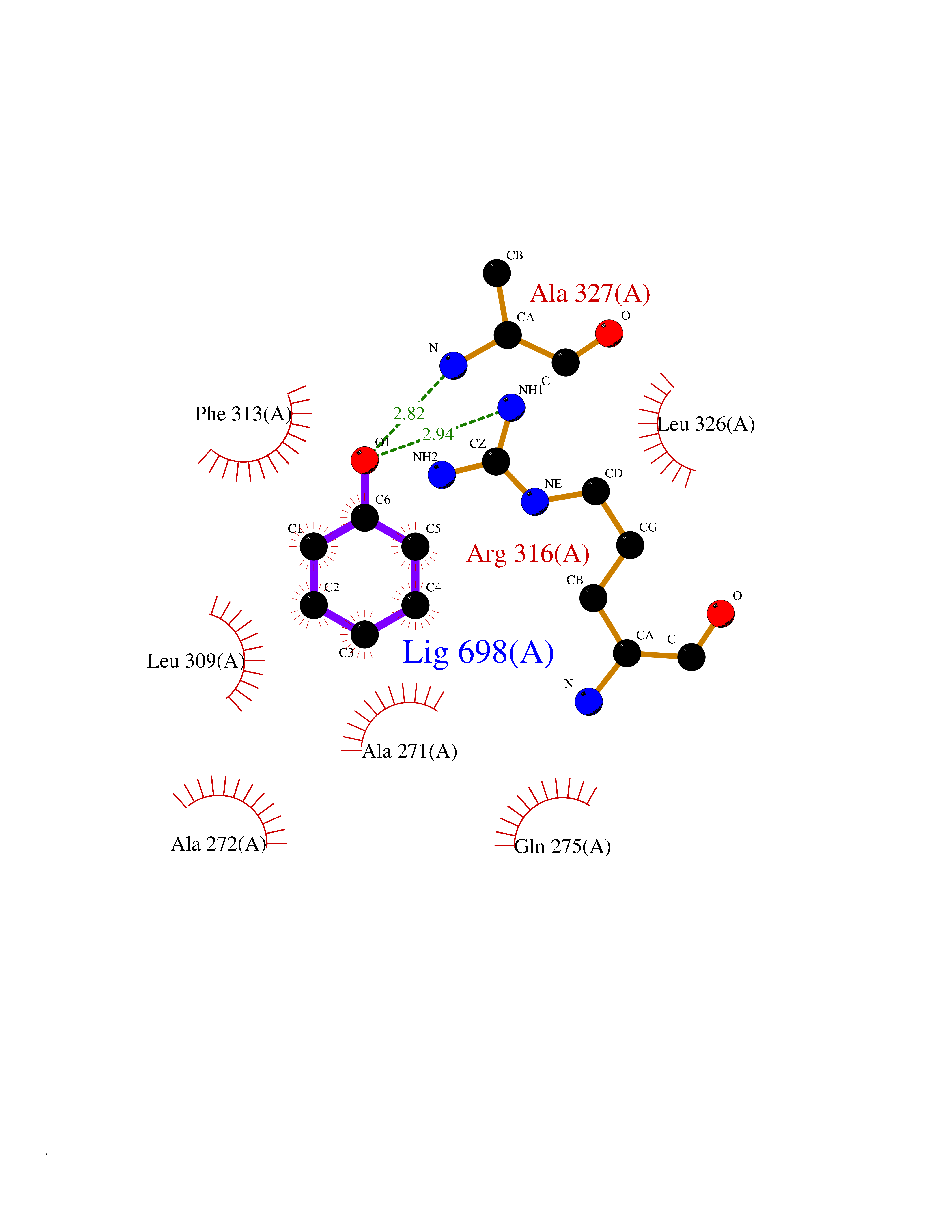 Ligplot Map 3D Binding mode Sequence DMPVERILEAELAVEDPVTNICQAADKQLFTLVEWAKRIPHFSELPLDDQVILLRAGWNELLIASFSHRSIAVKDGILLATGLHVHRNSAHSAGVGAIFDRVLTELVSKMRDMQMDKTELGCLRAIVLFNPDSKGLSNPAEVEALREKVYASLEAYCKHKYPEQPGRFAKLLLRLPALRSIGLKCLEHLFFFKLIGDTPIDTFLMEMLEAPHKILHRLLQD Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | SPRY domain-containing SOCS box protein 2 (SSB-2) (Gene-rich cluster protein C9) | 3EMW | 5.11 | |
Target general information Gen name SPSB2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GRCC9;SSB2 Protein family SPSB family Biochemical class NA Function Substrate recognition component of a SCF-like ECS (Elongin BC-CUL2/5-SOCS-box protein) E3 ubiquitin-protein ligase complex which mediates the ubiquitination and subsequent proteasomal degradation of target proteins (PubMed:15601820, PubMed:21199876). Negatively regulates nitric oxide (NO) production and limits cellular toxicity in activated macrophages by mediating the ubiquitination and proteasomal degradation of NOS2 (PubMed:21199876). Acts as a bridge which links NOS2 with the ECS E3 ubiquitin ligase complex components ELOC and CUL5 (PubMed:21199876). {ECO:0000269|PubMed:15601820, ECO:0000269|PubMed:21199876}." Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with G5E9A7; Q15369; Q53EP0-3; O60333-2; Q16656-4; Q96IZ0; P16284; Q92569; O60260-5; Q99873; Q6P9E2; Q9Y3C5; Q96GM5; P61086; P08670; P09052 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Host-virus interaction; Proteomics identification; Reference proteome; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 22703.1 Length 207 Aromaticity 0.09 Instability index 50.84 Isoelectric point 5.98 Charge (pH=7) -2.79 2D Binding mode Binding energy (Kcal/mol) -6.97  Molscript Map 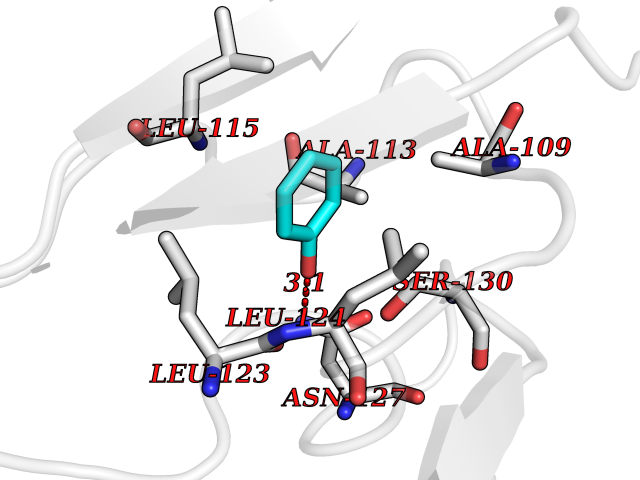 Pymol Map 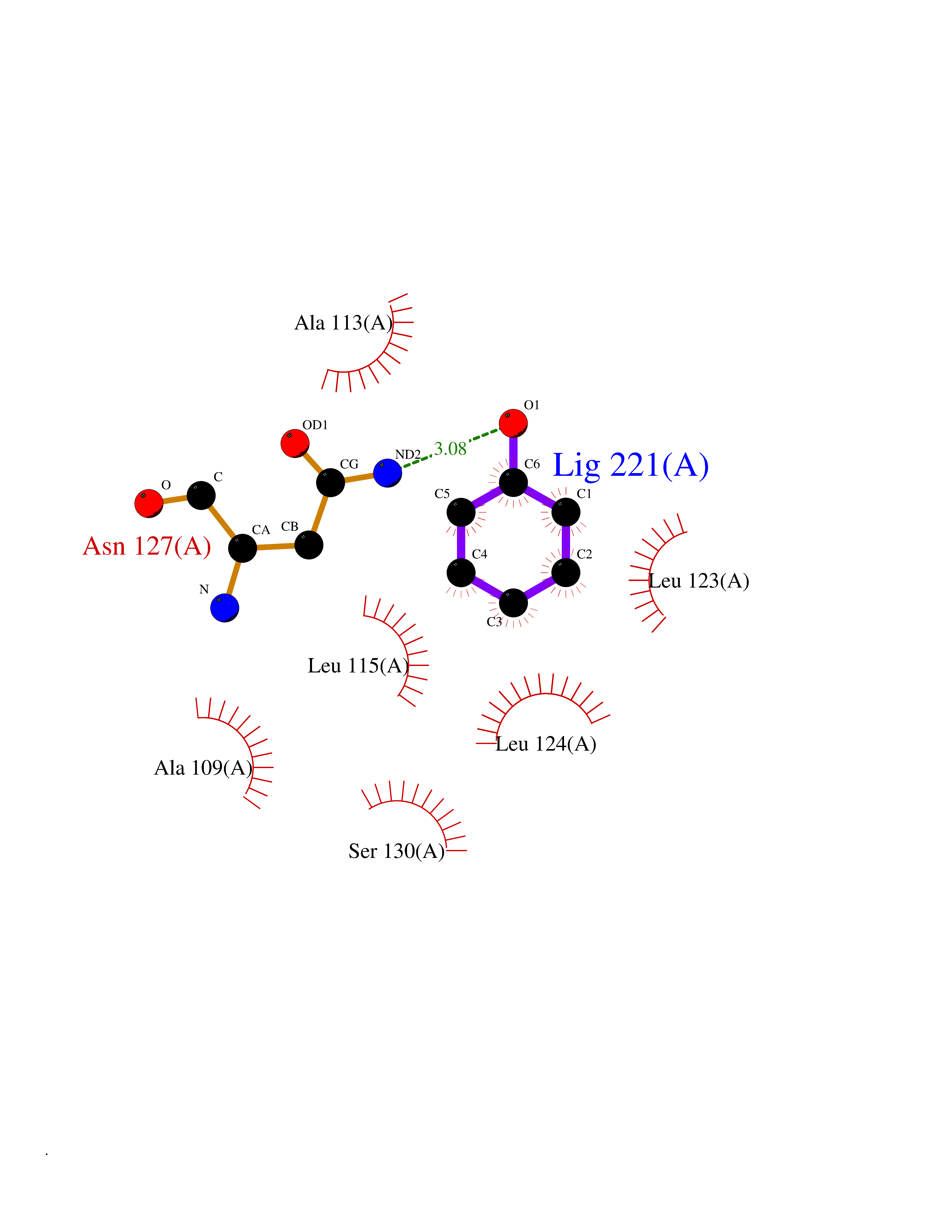 Ligplot Map 3D Binding mode Sequence LYFQSMPEGLEELLSAPPPDLGAQRRHGWNPKDCSENIEVKEGGLYFERRPVAQSTDGARGKRGYSRGLHAWEISWPLEQRGTHAVVGVATALAPLQTDHYAALLGSNSESWGWDIGRGKLYHQSKGPGAPQYPAGTQGEQLEVPERLLVVLDMEEGTLGYAIGGTYLGPAFRGLKGRTLYPAVSAVWGQCQVRIRYLGEDINNNNN Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Cytochrome P450 2A6 (CYP2A6) | 1Z11 | 5.11 | |
Target general information Gen name CYP2A6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450(I); Cytochrome P450 IIA3; Coumarin 7-hydroxylase; CYPIIA6; CYP2A6; CYP2A3 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Exhibits a high coumarin 7-hydroxylase activity. Can act in the hydroxylation of the anti-cancer drugs cyclophosphamide and ifosphamide. Competent in the metabolic activation of aflatoxin B1. Constitutes the major nicotine C-oxidase. Possesses low phenacetin O-deethylation activity. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07621; DB07623; DB00316; DB01118; DB00182; DB01435; DB05676; DB01274; DB09274; DB11586; DB00972; DB00443; DB01194; DB01222; DB09061; DB14737; DB06119; DB00356; DB00568; DB00604; DB06470; DB00257; DB00363; DB04665; DB00531; DB06292; DB01234; DB14649; DB00470; DB06374; DB00216; DB00330; DB01039; DB04841; DB01544; DB00544; DB00690; DB01213; DB00983; DB01159; DB00741; DB01181; DB00951; DB01026; DB01006; DB00281; DB06448; DB04871; DB14009; DB01043; DB00170; DB00763; DB00553; DB01028; DB00959; DB00916; DB01011; DB01110; DB00471; DB07609; DB07617; DB00238; DB00184; DB01115; DB06712; DB00717; DB00312; DB03783; DB01174; DB00252; DB01085; DB04977; DB14631; DB00635; DB00396; DB01045; DB15119; DB01037; DB06739; DB01236; DB00675; DB09256; DB09327; DB12816; DB00752; DB00755; DB12245; DB00313; DB09068; DB00495 Interacts with Q9UI14 EC number EC 1.14.13.- Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 53586.9 Length 467 Aromaticity 0.12 Instability index 40.76 Isoelectric point 9.14 Charge (pH=7) 7.77 2D Binding mode Binding energy (Kcal/mol) -6.97 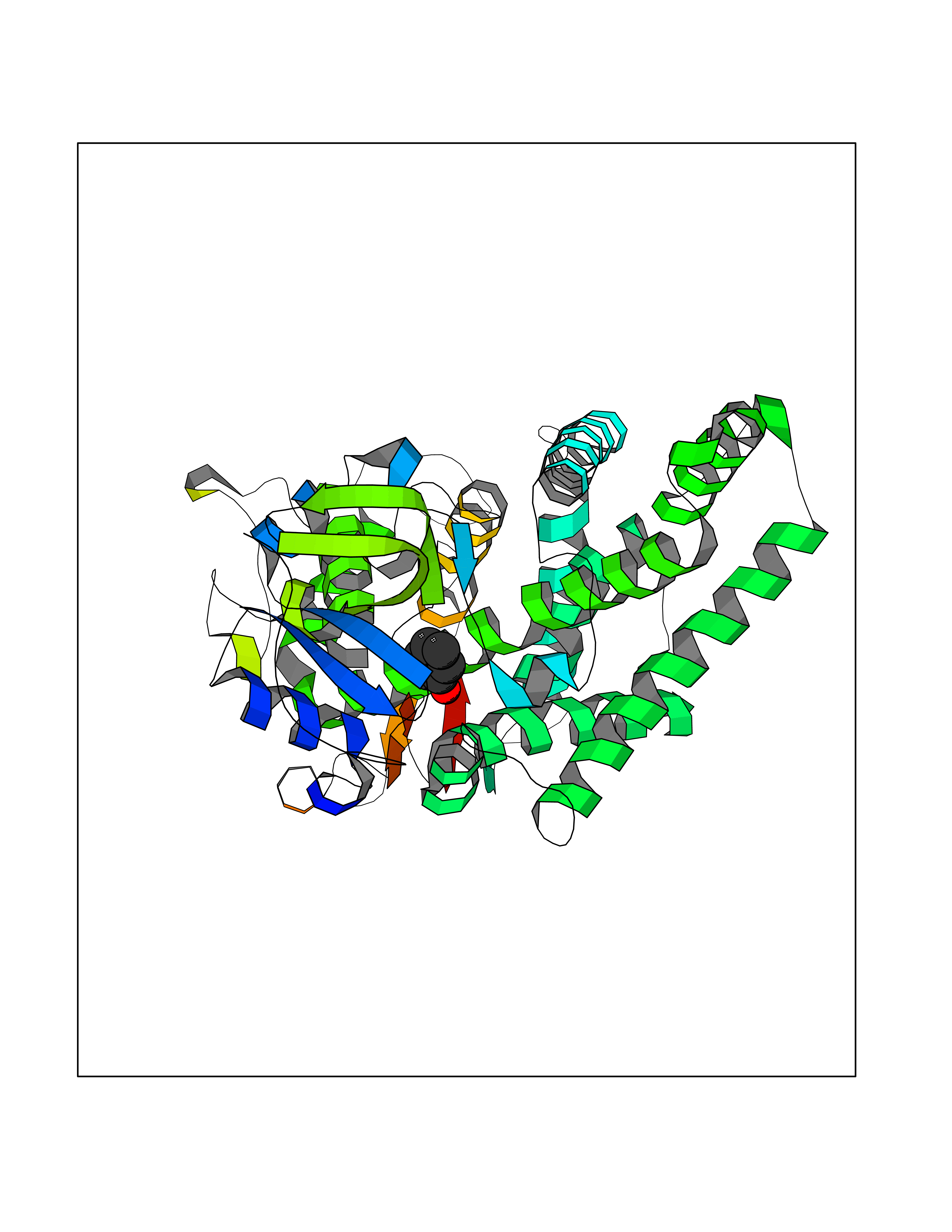 Molscript Map 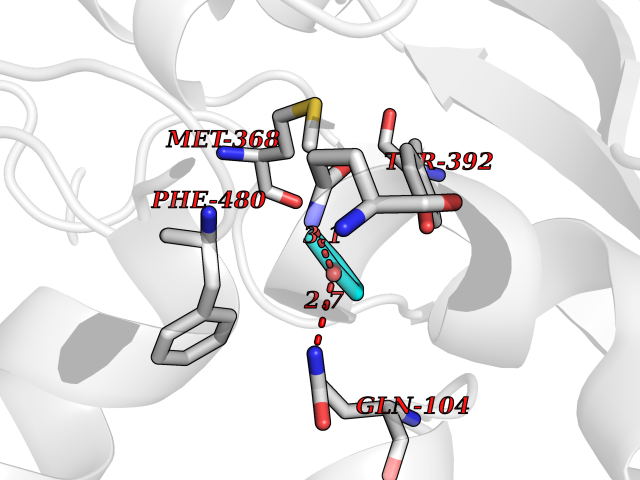 Pymol Map 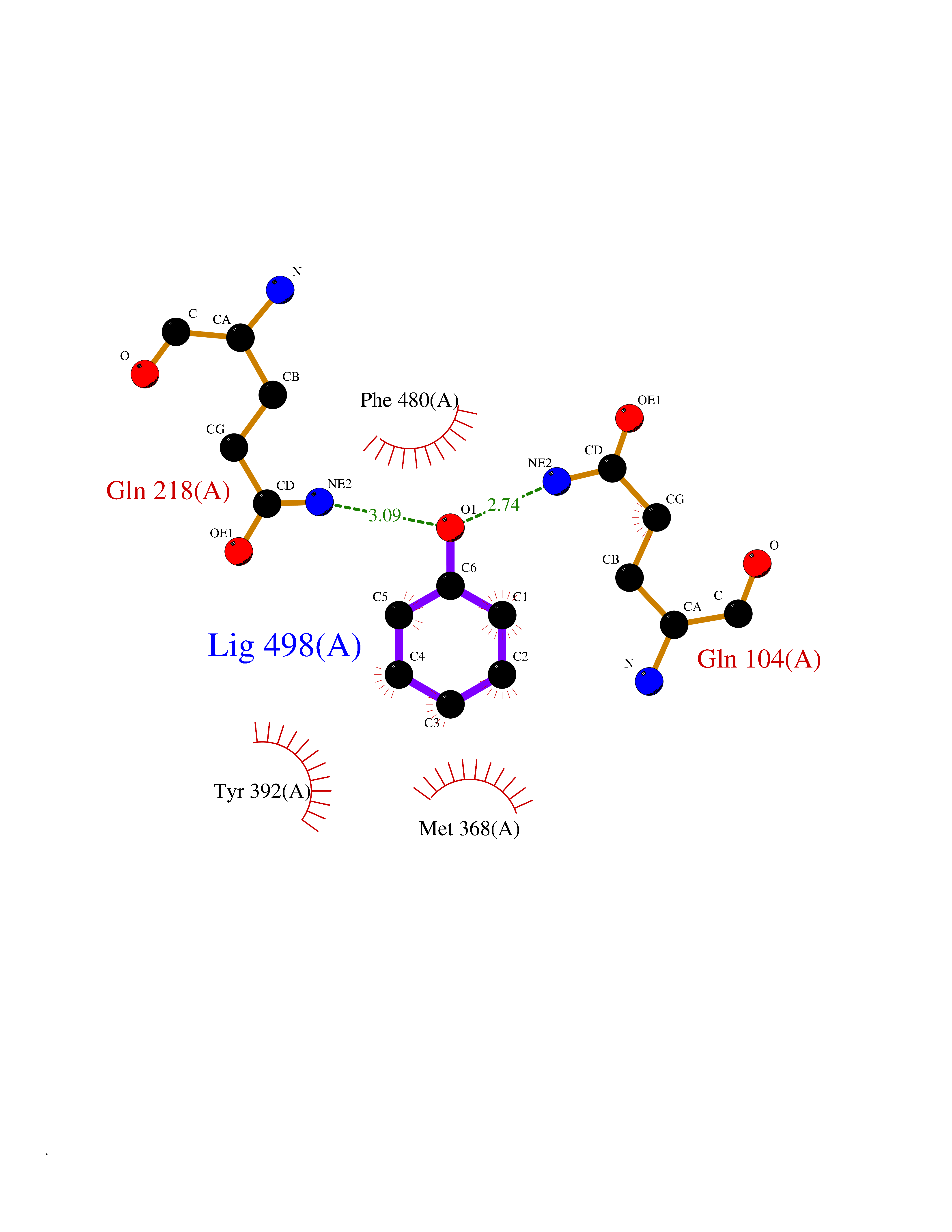 Ligplot Map 3D Binding mode Sequence KGKLPPGPTPLPFIGNYLQLNTEQMYNSLMKISERYGPVFTIHLGPRRVVVLCGHDAVREALVDQAEEFSGRGEQATFDWVFKGYGVVFSNGERAKQLRRFSIATLRDFGVGKRGIEERIQEEAGFLIDALRGTGGANIDPTFFLSRTVSNVISSIVFGDRFDYKDKEFLSLLRMMLGIFQFTSTSTGQLYEMFSSVMKHLPGPQQQAFQLLQGLEDFIAKKVEHNQRTLDPNSPRDFIDSFLIRMQEEEKNPNTEFYLKNLVMTTLNLFIGGTETVSTTLRYGFLLLMKHPEVEAKVHEEIDRVIGKNRQPKFEDRAKMPYMEAVIHEIQRFGDVIPMSLARRVKKDTKFRDFFLPKGTEVYPMLGSVLRDPSFFSNPQDFNPQHFLNEKGQFKKSDAFVPFSIGKRNCFGEGLARMELFLFFTTVMQNFRLKSSQSPKDIDVSPKHVGFATIPRNYTMSFLPRHH Hydrogen bonds contact Hydrophobic contact | ||||