Job Results:
Ligand
Structure
Job ID
8e57bce38c3d7c3304510f6b58eb7b68
Job name
NA
Time
2026-02-27 11:59:36
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | TRANSPORT INHIBITOR RESPONSE 1 protein | 2P1Q | 4.80 | |
Target general information Gen name IAA7 Organism Arabidopsis thaliana (Mouse-ear cress) Uniprot ID TTD ID NA Synonyms AXR2;At3g23050;MXC7.8 Protein family Aux/IAA family Biochemical class Signaling protein Function DNA binding transcription factor activity. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) NA Interacts with Q9LW29; Q9C5W9; Q8RYC8; Q94AH6; Q9ZR12; P49677; Q38828; Q38829; Q38830; Q38831; O24407; O24408; O24409; P49678; O24410; Q8LAL2; Q9XFM0; Q38822; Q9M1R4; Q9C5X0; Q9C8Y3; Q39255; Q570C0 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Auxin signaling pathway; Nucleus; Reference proteome; Repressor; Transcription; Transcription regulation Protein physicochemical properties Chain ID B,C Molecular weight (Da) 65385.2 Length 581 Aromaticity 0.09 Instability index 47.83 Isoelectric point 7.46 Charge (pH=7) 1.23 2D Binding mode Binding energy (Kcal/mol) -6.54  Molscript Map 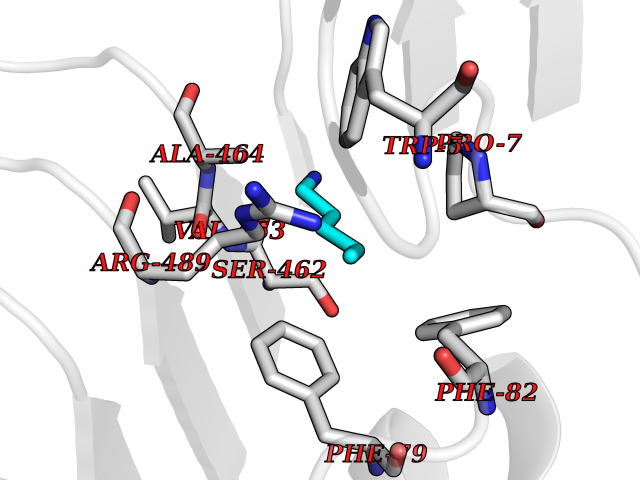 Pymol Map 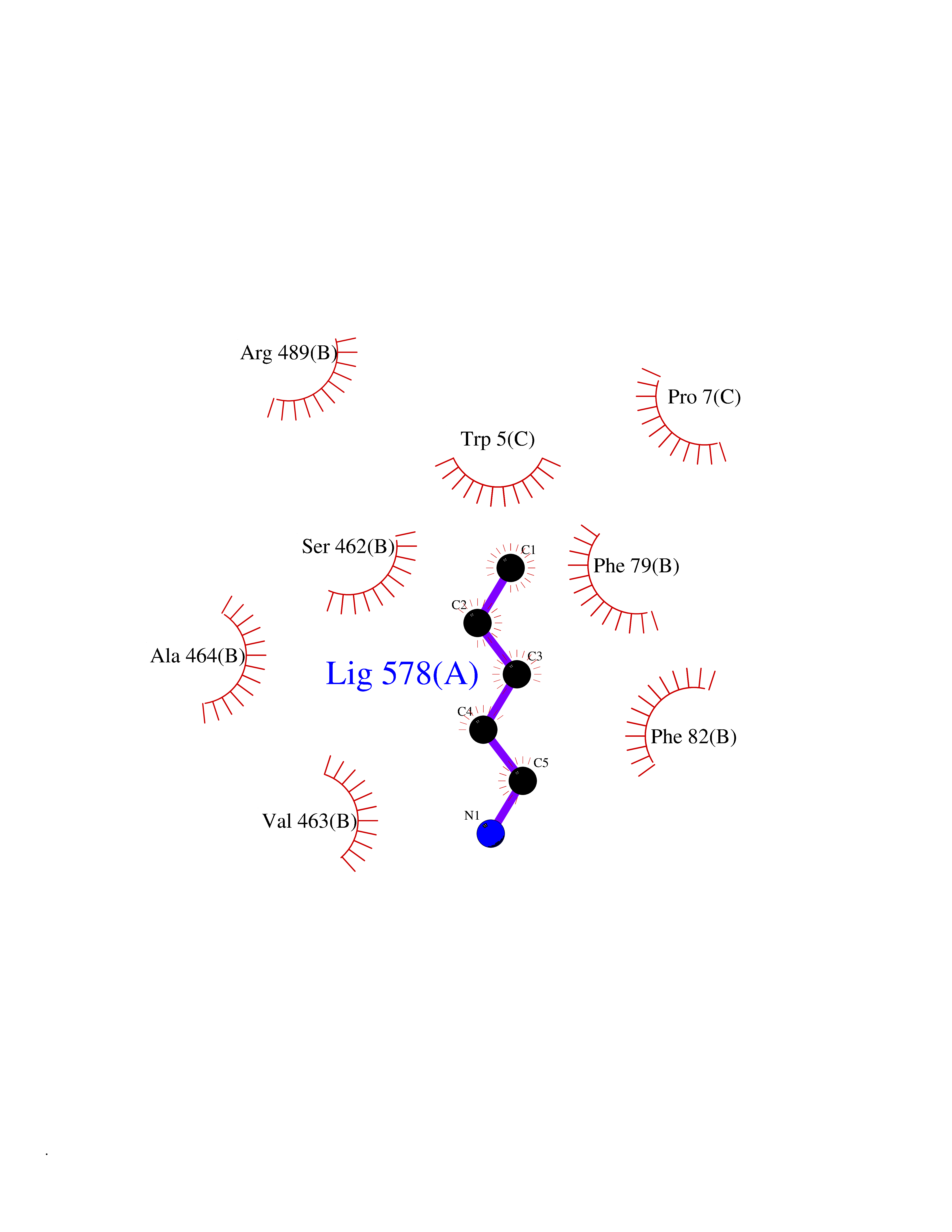 Ligplot Map 3D Binding mode Sequence QVVGWPPVRNYRKFPEEVLEHVFSFIQLDKDRNSVSLVCKSWYEIERWCRRKVFIGNCYAVSPATVIRRFPKVRSVELKGKPHFADFNLVPDGWGGYVYPWIEAMSSSYTWLEEIRLKRMVVTDDCLELIAKSFKNFKVLVLSSCEGFSTDGLAAIAATCRNLKELDLRESDVDDVSGHWLSHFPDTYTSLVSLNISCLASEVSFSALERLVTRCPNLKSLKLNRAVPLEKLATLLQRAPQLEELGTGGYTAEVRPDVYSGLSVALSGCKELRCLSGFWDAVPAYLPAVYSVCSRLTTLNLSYATVQSYDLVKLLCQCPKLQRLWVLDYIEDAGLEVLASTCKDLRELRVFPSEPFVMEPNVALTEQGLVSVSMGCPKLESVLYFCRQMTNAALITIARNRPNMTRFRLCIIEPKAPDYLTLEPLDIGFGAIVEHCKDLRRLSLSGLLTDKVFEYIGTYAKKMEMLSVAFAGDSDLGMHHVLSGCDSLRKLEIRDCPFGDKALLANASKLETMRSLWMSSCSVSFGACKLLGQKMPKLNVEVIDERGAPDSRPESCPVERVFIYRTVAGPRFDMPGFVWNM Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Lecithin-cholesterol acyltransferase (LCAT) | 6MVD | 4.80 | |
Target general information Gen name LCAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phospholipidcholesterolacyltransferase; Phospholipid-cholesterol acyltransferase; Phosphatidylcholinesterol acyltransferase; Phosphatidylcholine-sterol acyltransferase Protein family AB hydrolase superfamily, Lipase family Biochemical class Acyltransferase Function Synthesized mainly in the liver and secreted into plasma where it converts cholesterol and phosphatidylcholines (lecithins) to cholesteryl esters and lysophosphatidylcholines on the surface of high and low density lipoproteins (HDLs and LDLs). The cholesterol ester is then transported back to the liver. Has a preference for plasma 16:0-18:2 or 18:O-18:2 phosphatidylcholines. Also produced in the brain by primary astrocytes, and esterifies free cholesterol on nascent APOE-containing lipoproteins secreted from glia and influences cerebral spinal fluid (CSF) APOE- and APOA1 levels. Together with APOE and the cholesterol transporter ABCA1, plays a key role in the maturation of glial-derived, nascent lipoproteins. Required for remodeling high-density lipoprotein particles into their spherical forms. Central enzyme in the extracellular metabolism of plasma lipoproteins. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P02647; O76024 EC number EC 2.3.1.43 Uniprot keywords 3D-structure; Acyltransferase; Cholesterol metabolism; Corneal dystrophy; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipid metabolism; Proteomics identification; Reference proteome; Secreted; Signal; Steroid metabolism; Sterol metabolism; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42715.4 Length 376 Aromaticity 0.12 Instability index 42.05 Isoelectric point 5.69 Charge (pH=7) -9.12 2D Binding mode Binding energy (Kcal/mol) -6.55 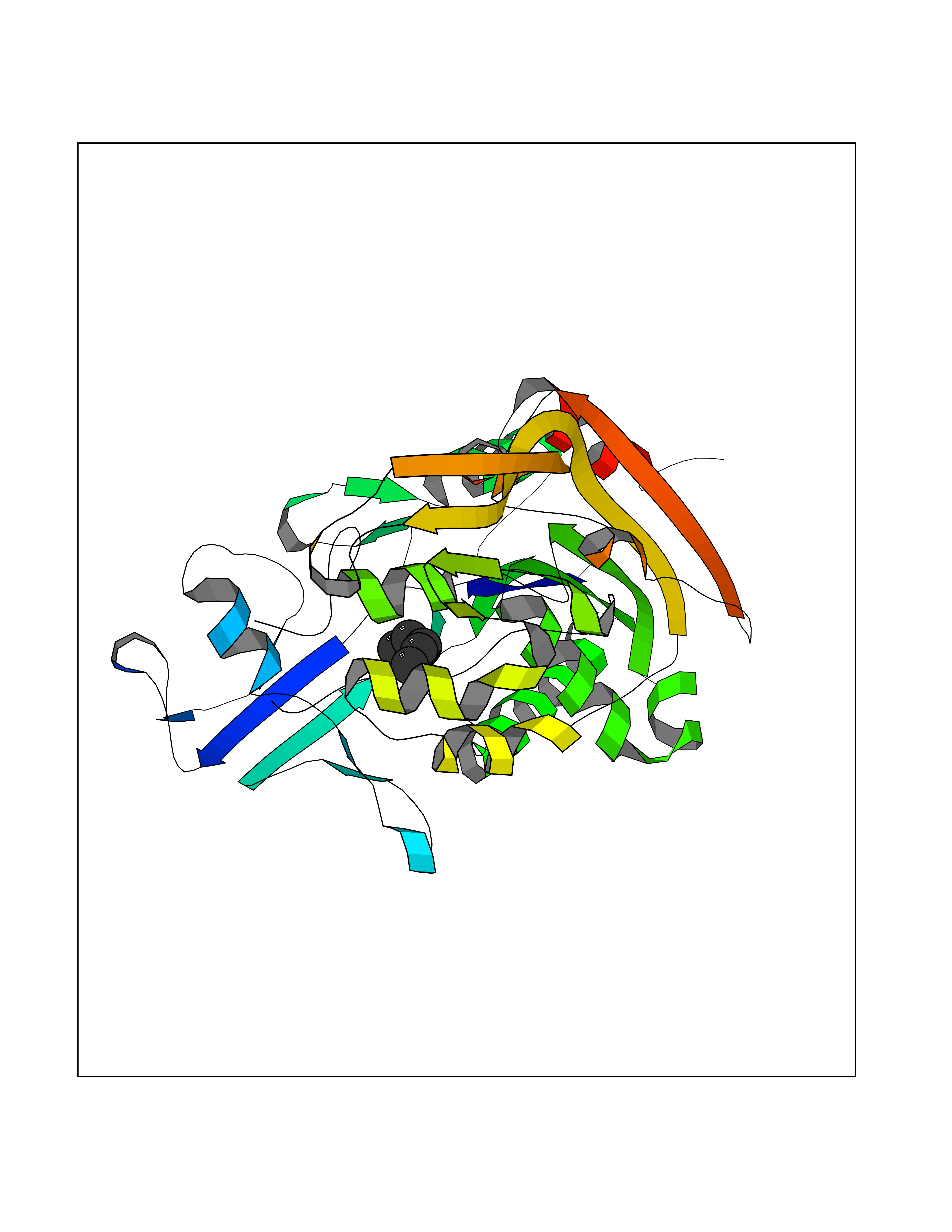 Molscript Map 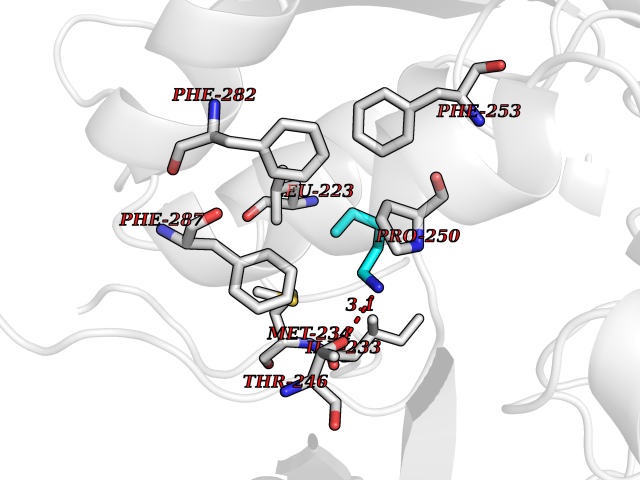 Pymol Map 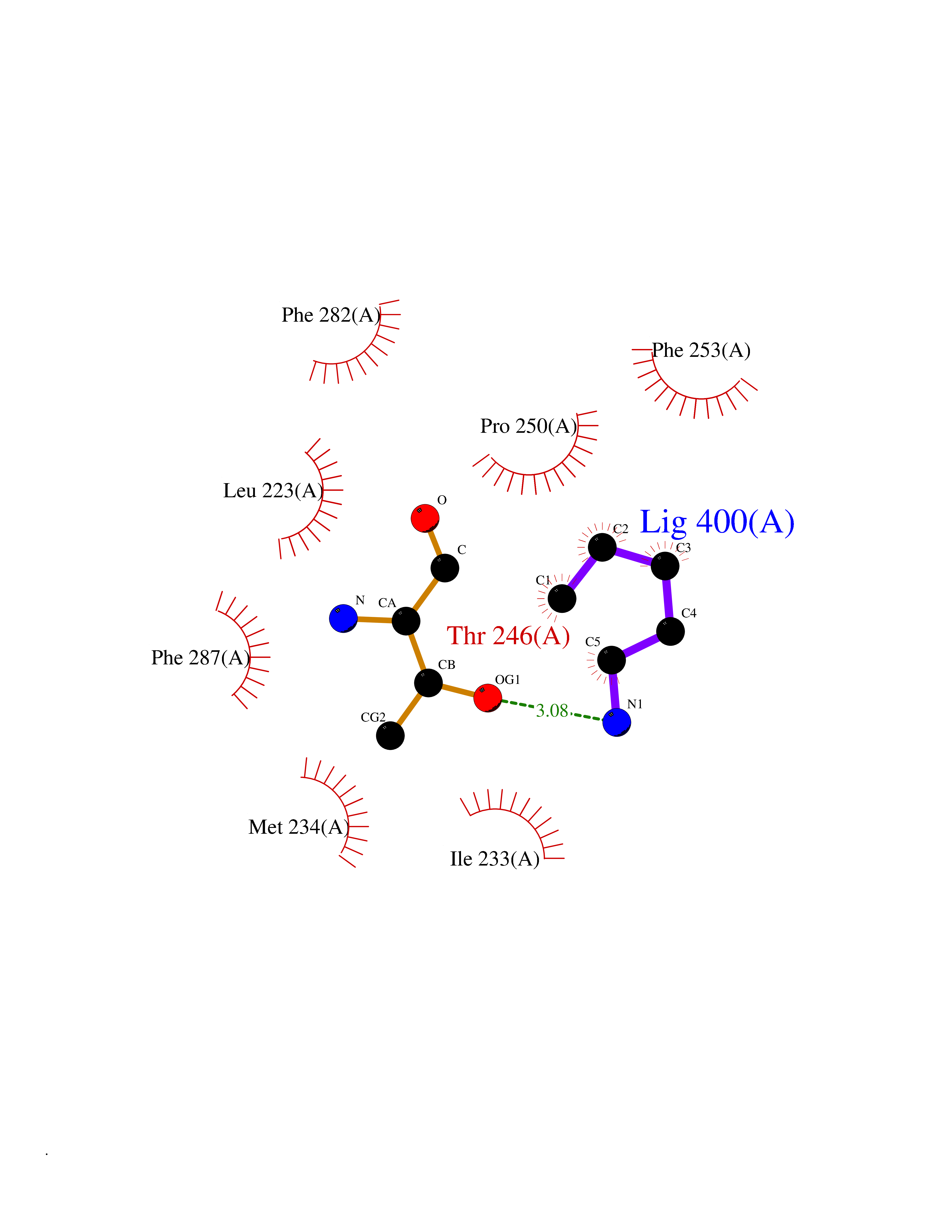 Ligplot Map 3D Binding mode Sequence HTRPVILVPGCLGNQLEAKLDKPDVVNWMCYRKTEDFFTIWLDLNMFLPLGVDCWIDNTRVVYNRSSGLVSNAPGVQIRVPGFGKTYSVEYLDSSKLAGYLHTLVQNLVNNGYVRDETVRAAPYDWRLEPGQQEEYYRKLAGLVEEMHAAYGKPVFLIGHSLGCLHLLYFLLRQPQAWKDRFIDGFISLGAPWGGSIKPMLVLASGDNQGIPIMSSIKEEQRITTTSPWMFPSRMAWPEDHVFISTPSFNYTGRDFQRFFADLHFEEGWYMWLQSRDLLAGLPAPGVEVYCLYGVGLPTPRTYIYDHGFPYTDPVGVLYEDGDDTVATRSTELCGLWQGRQPQPVHLLPLHGIQHLNMVFSNLTLEHINAILLGAH Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Deoxyribodipyrimidine photo-lyase | 1DNP | 4.79 | |
Target general information Gen name phrB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms phr;b0708;JW0698 Protein family DNA photolyase class-1 family Biochemical class Lyase (carbon-carbon) Function Deoxyribodipyrimidine photo-lyase activity.DNA binding.Nucleotide binding. Related diseases Severe combined immunodeficiency autosomal recessive T-cell-negative/B-cell-negative/NK-cell-negative due to adenosine deaminase deficiency (ADASCID) [MIM:102700]: An autosomal recessive disorder accounting for about 50% of non-X-linked SCIDs. SCID refers to a genetically and clinically heterogeneous group of rare congenital disorders characterized by impairment of both humoral and cell-mediated immunity, leukopenia, and low or absent antibody levels. Patients with SCID present in infancy with recurrent, persistent infections by opportunistic organisms. The common characteristic of all types of SCID is absence of T-cell-mediated cellular immunity due to a defect in T-cell development. ADA deficiency has been diagnosed in chronically ill teenagers and adults (late or adult onset). Population and newborn screening programs have also identified several healthy individuals with normal immunity who have partial ADA deficiency. {ECO:0000269|PubMed:10200056, ECO:0000269|PubMed:1284479, ECO:0000269|PubMed:2166947, ECO:0000269|PubMed:2783588, ECO:0000269|PubMed:3182793, ECO:0000269|PubMed:3839802, ECO:0000269|PubMed:6208479, ECO:0000269|PubMed:7599635, ECO:0000269|PubMed:8227344, ECO:0000269|PubMed:8299233, ECO:0000269|PubMed:9361033}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with P68739 EC number 4.1.99.3 Uniprot keywords 3D-structure; Chromophore; DNA damage; DNA repair; DNA-binding; FAD; Flavoprotein; Lyase; Nucleotide-binding; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 53349.8 Length 469 Aromaticity 0.1 Instability index 42.46 Isoelectric point 6.57 Charge (pH=7) -2.28 2D Binding mode Binding energy (Kcal/mol) -6.53 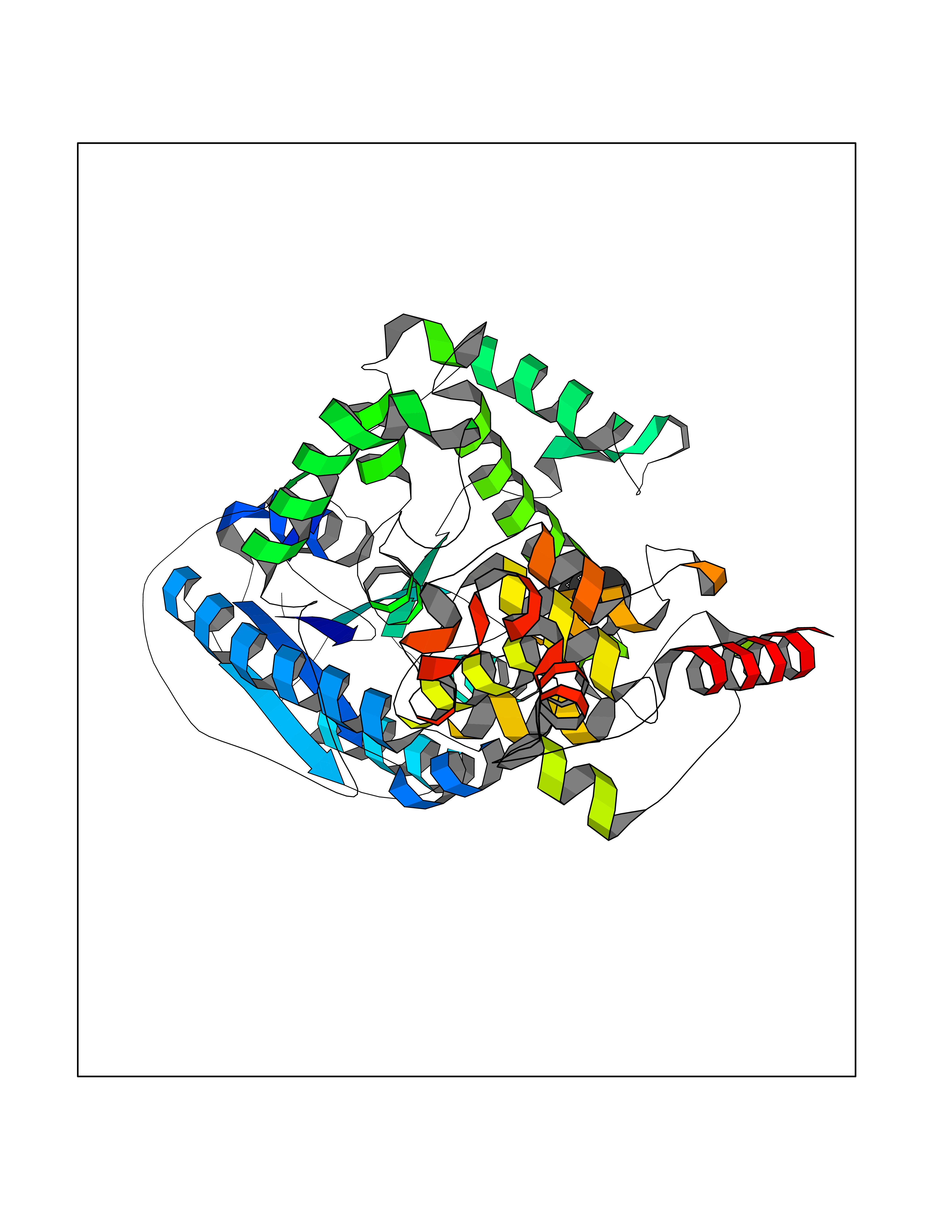 Molscript Map 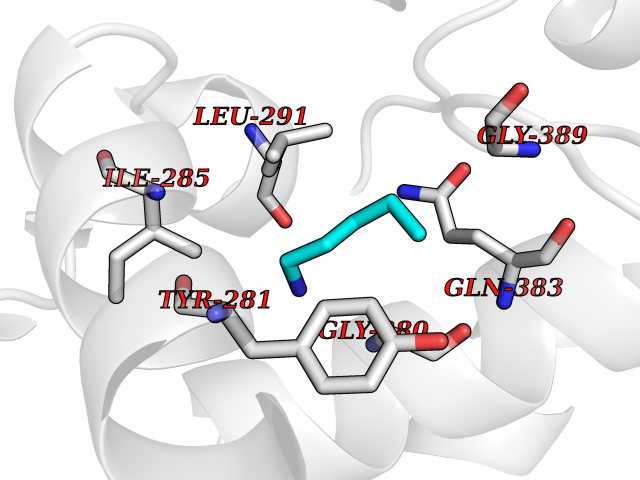 Pymol Map 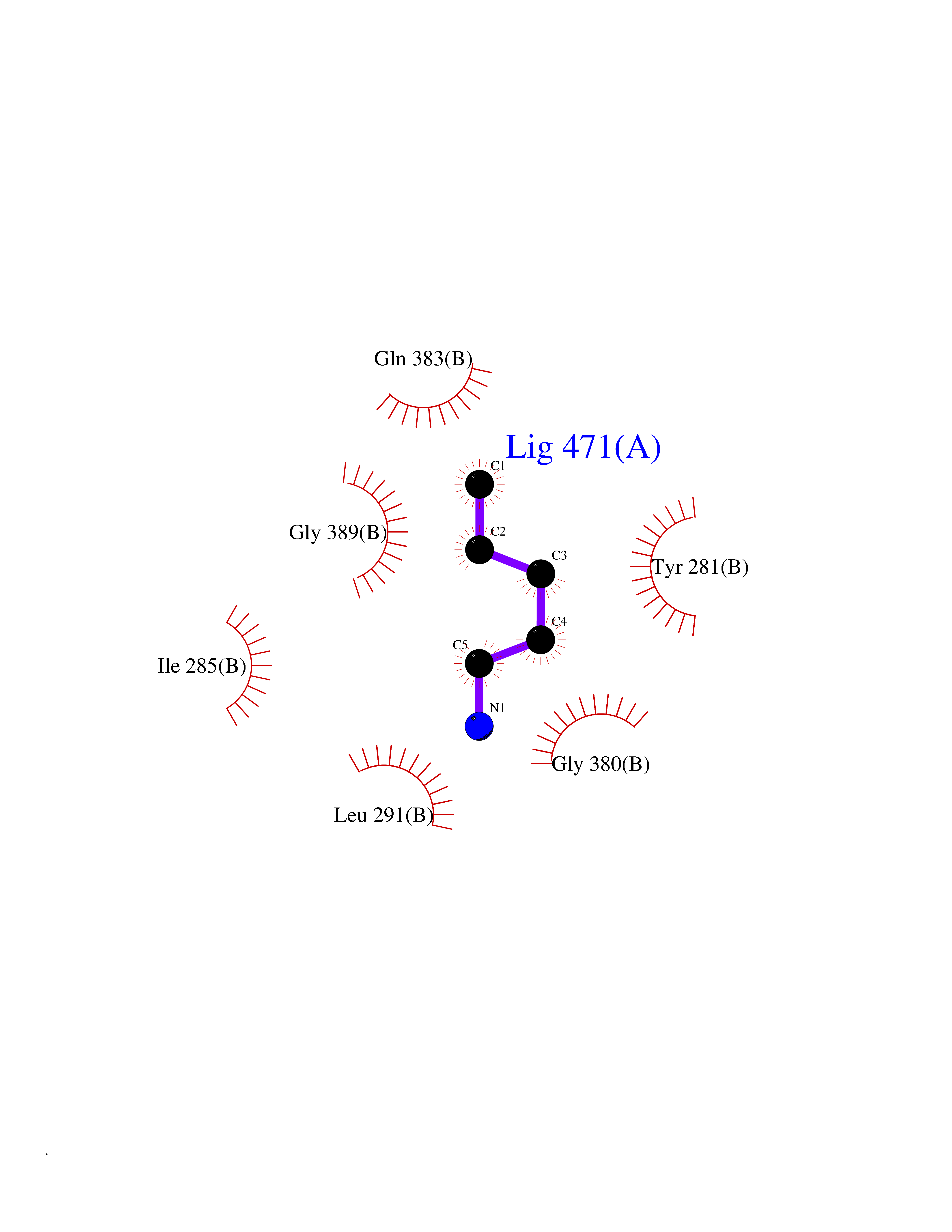 Ligplot Map 3D Binding mode Sequence TTHLVWFRQDLRLHDNLALAAACRNSSARVLALYIATPRQWATHNMSPRQAELINAQLNGLQIALAEKGIPLLFREVDDFVASVEIVKQVCAENSVTHLFYNYQYEVNERARDVEVERALRNVVCEGFDDSVILPPGAVMTGNHEMYKVFTPFKNAWLKRLREGMPECVAAPKVRSSGSIEPSPSITLNYPRQSFDTAHFPVEEKAAIAQLRQFCQNGAGEYEQQRDFPAVEGTSRLSASLATGGLSPRQCLHRLLAEQPQALDGGAGSVWLNELIWREFYRHLITYHPSLCKHRPFIAWTDRVQWQSNPAHLQAWQEGKTGYPIVDAAMRQLNSTGWMHNRLRMITASFLVKDLLIDWREGERYFMSQLIDGDLAANNGGWQWAASTGTDAAPYFRIFNPTTQGEKFDHEGEFIRQWLPELRDVPGKVVHEPWKWAQKAGVTLDYPQPIVEHKEARVQTLAAYEAARK Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Medium-chain specific acyl-CoA dehydrogenase, mitochondrial | 4P13 | 4.79 | |
Target general information Gen name ACADM Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Acyl-CoA dehydrogenase activity.Flavin adenine dinucleotide binding.Identical protein binding.Medium-chain-acyl-CoA dehydrogenase activity. Related diseases Acyl-CoA dehydrogenase medium-chain deficiency (ACADMD) [MIM:201450]: An inborn error of mitochondrial fatty acid beta-oxidation which causes fasting hypoglycemia, hepatic dysfunction and encephalopathy, often resulting in death in infancy. {ECO:0000269|PubMed:10767181, ECO:0000269|PubMed:11349232, ECO:0000269|PubMed:11409868, ECO:0000269|PubMed:11486912, ECO:0000269|PubMed:1363805, ECO:0000269|PubMed:1671131, ECO:0000269|PubMed:1684086, ECO:0000269|PubMed:1902818, ECO:0000269|PubMed:2251268, ECO:0000269|PubMed:2393404, ECO:0000269|PubMed:2394825, ECO:0000269|PubMed:7603790, ECO:0000269|PubMed:7929823, ECO:0000269|PubMed:8198141, ECO:0000269|PubMed:9158144, ECO:0000269|PubMed:9882619}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03415; DB03147; DB02910 Interacts with PRO_0000000502 [P11310] EC number 1.3.8.7 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; FAD; Fatty acid metabolism; Flavoprotein; Lipid metabolism; Mitochondrion; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 85080.3 Length 773 Aromaticity 0.09 Instability index 30.55 Isoelectric point 5.71 Charge (pH=7) -7.7 2D Binding mode Binding energy (Kcal/mol) -6.54  Molscript Map 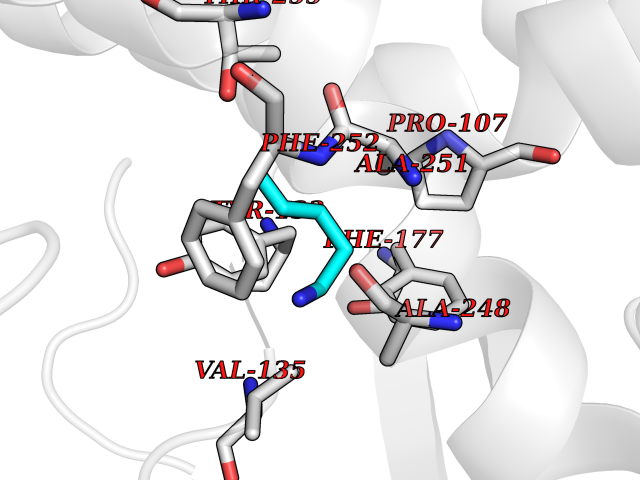 Pymol Map 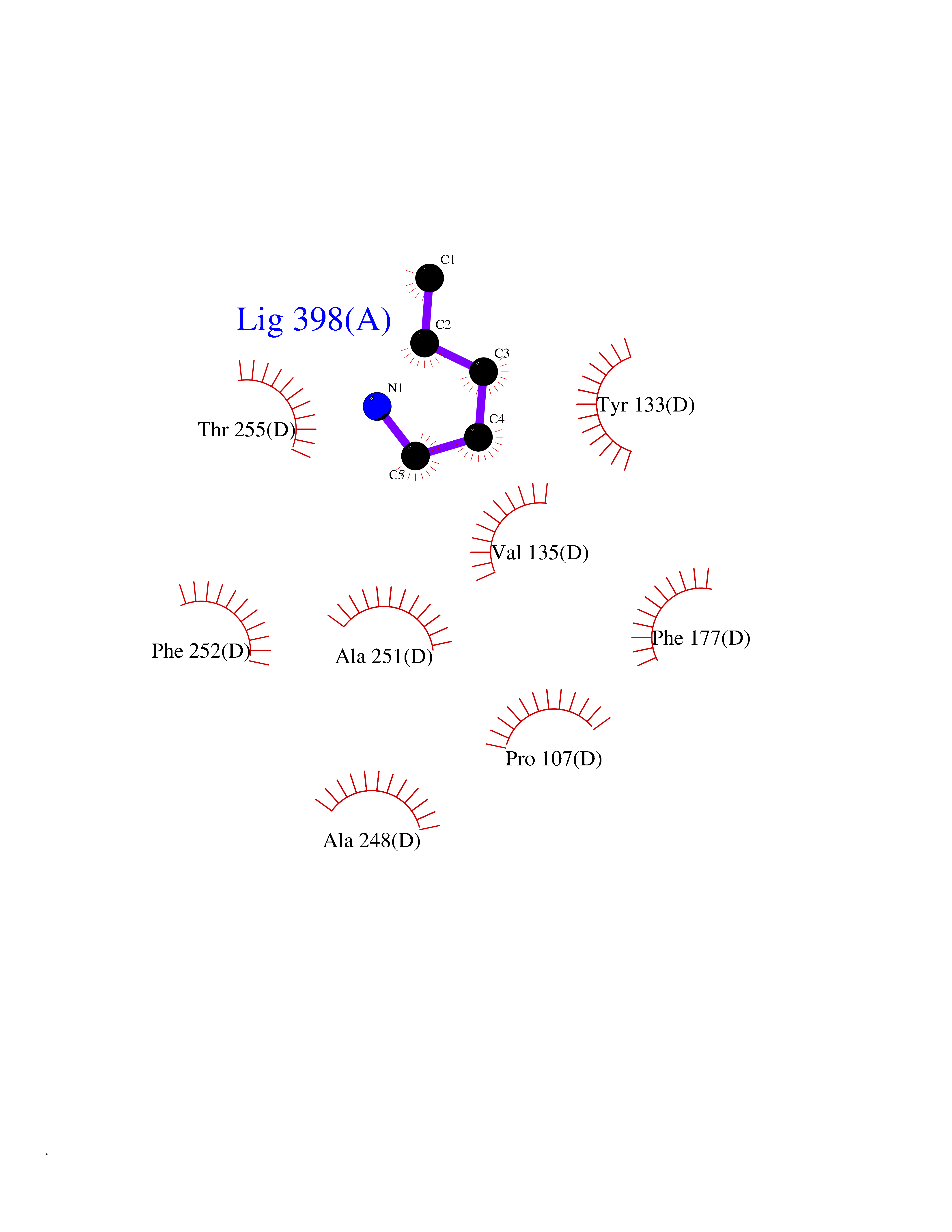 Ligplot Map 3D Binding mode Sequence LGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKLGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKN Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Cocaine esterase | 3I2K | 4.79 | |
Target general information Gen name cocE Organism Rhodococcus sp. (strain MB1 Bresler) Uniprot ID TTD ID NA Synonyms NA Protein family CocE/NonD hydrolase family Biochemical class Hydrolase Function Carboxylic ester hydrolase activity.Dipeptidyl-peptidase activity. Related diseases Thiamine metabolism dysfunction syndrome 5, episodic encephalopathy type (THMD5) [MIM:614458]: An autosomal recessive metabolic disorder due to an inborn error of thiamine metabolism. The phenotype is highly variable, but in general, affected individuals have onset in early childhood of acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive neurologic dysfunction manifest as gait disturbances, ataxia, dystonia, and spasticity, which in some cases may result in loss of ability to walk. Cognitive function is usually preserved, although mildly delayed development has been reported. These episodes are usually associated with infection and metabolic decompensation. Some patients may have recovery of some neurologic deficits. {ECO:0000269|PubMed:22152682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03793; DB01795 Interacts with NA EC number 3.1.1.84 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Hydrolase; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 62127.9 Length 574 Aromaticity 0.09 Instability index 26.62 Isoelectric point 4.56 Charge (pH=7) -33.24 2D Binding mode Binding energy (Kcal/mol) -6.53 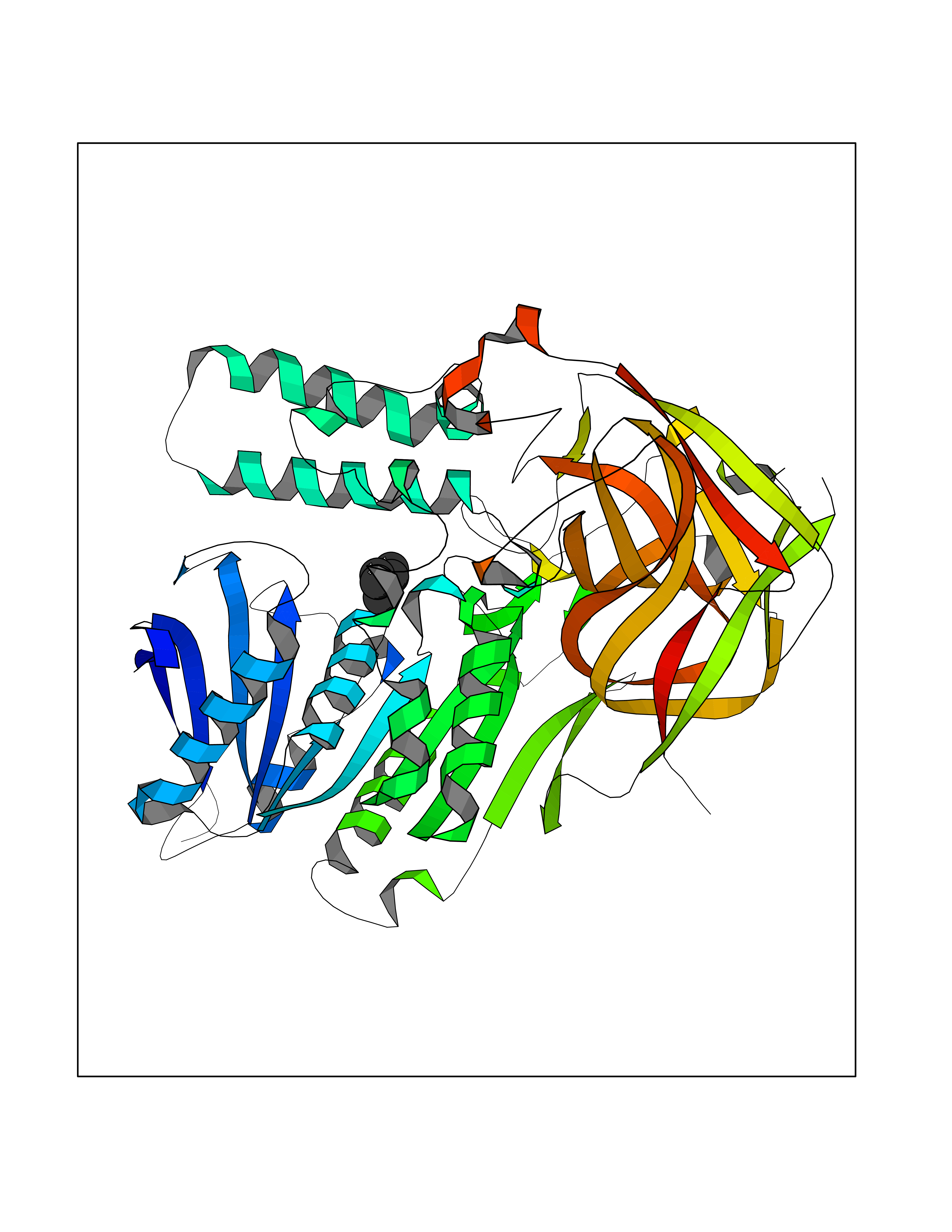 Molscript Map 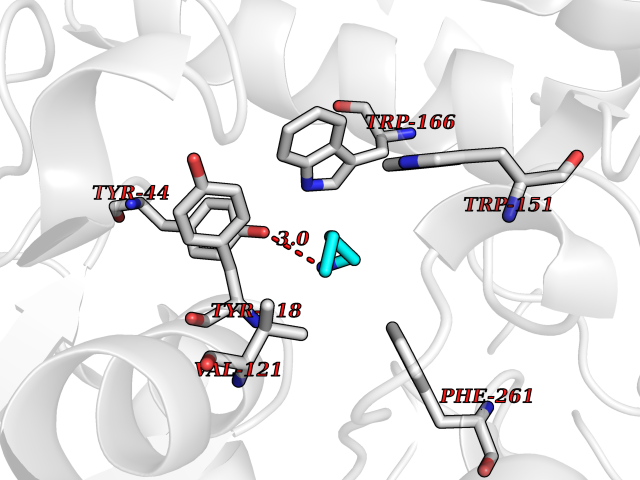 Pymol Map 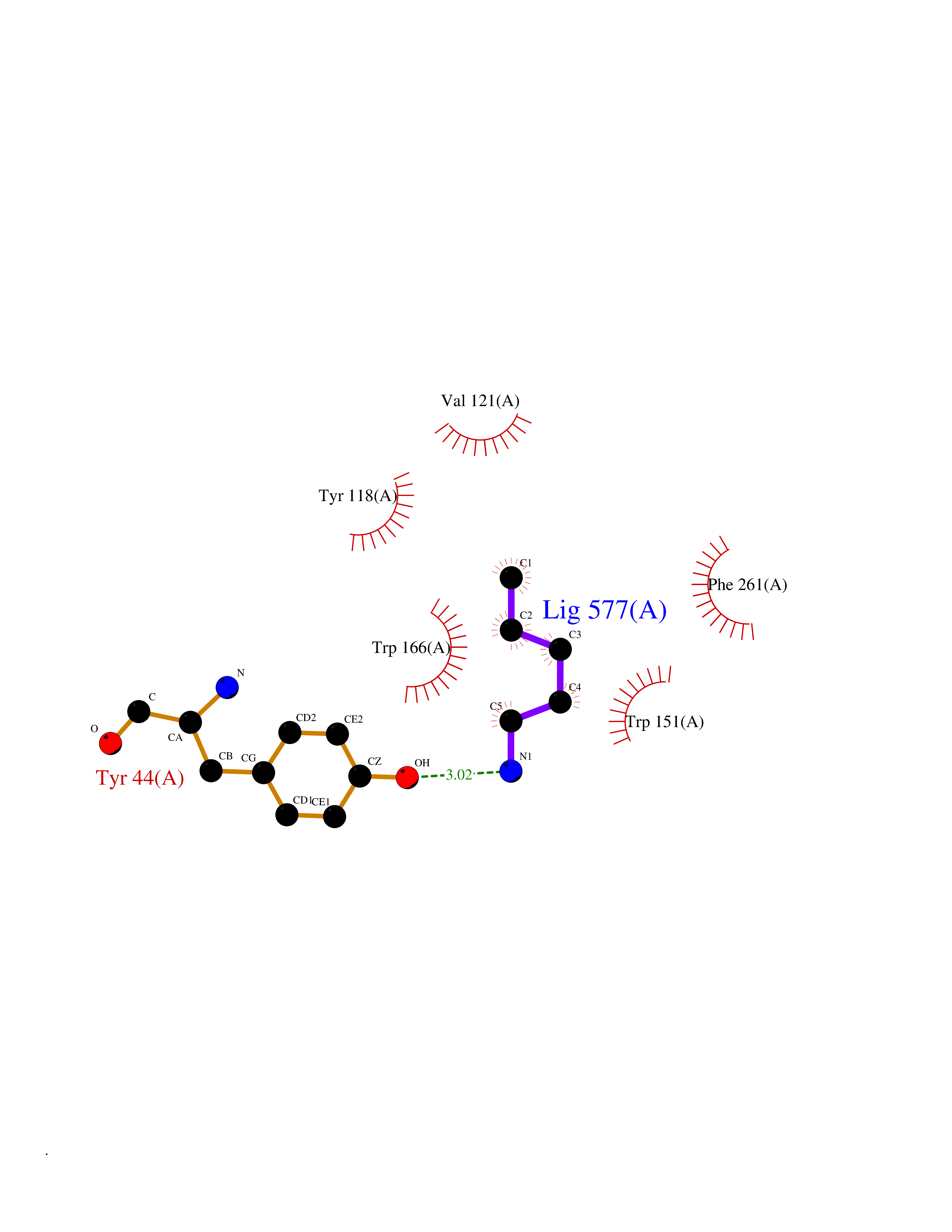 Ligplot Map 3D Binding mode Sequence VDGNYSVASNVMVPMRDGVRLAVDLYRPDADGPVPVLLVRNPYDKFDVFAWSTQSTNWLEFVRDGYAVVIQDTRGLFASEGEFVPHVDDEADAEDTLSWILEQAWCDGNVGMFGVSYLGVTQWQAAVSGVGGLKAIAPSMASADLYRAPWYGPGGALSVEALLGWSALIGTGLITSRSDARPEDAADFVQLAAILNDVAGAASVTPLAEQPLLGRLIPWVIDQVVDHPDNDESWQSISLFERLGGLATPALITAGWYDGFVGESLRTFVAVKDNADARLVVGPWSHSNLTGRNADRKFGIAATYPIQEATTMHKAFFDRHLRGETDALAGVPKVRLFVMGIDEWRDETDWPLPDTAYTPFYLGGSGAANTSTGGGTLSTSISGTESADTYLYDPADPVPSLGGTLLFHNGDNGPADQRPIHDRDDVLCYSTEVLTDPVEVTGTVSARLFVSSSAVDTDFTAKLVDVFPDGRAIALCDGIVRMRYRETLVNPTLIEAGEIYEVAIDMLATSNVFLPGHRIMVQVSSSNFPKYDRNSNTGGVIAREQLEEMCTAVNRIHRGPEHPSHIVLPIIKRK Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Beta-N-acetylhexosaminidase (OGA) | 6PM9 | 4.79 | |
Target general information Gen name MGEA5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms OGA; Nuclear cytoplasmic OGlcNAcase and acetyltransferase; Meningiomaexpressed antigen 5; MGEA5; Histone acetyltransferase; Bifunctional protein NCOAT Protein family Glycosyl hydrolase 84 family Biochemical class Glycosylase Function Isoform 3: Cleaves GlcNAc but not GalNAc from O- glycosylated proteins. Can use p-nitrophenyl-beta-GlcNAc as substrate but not p-nitrophenyl-beta-GalNAc or p-nitrophenyl- alpha-GlcNAc (in vitro), but has about six times lower specific activity than isoform1. {ECO:0000269|PubMed:20673219}. Related diseases Immunodeficiency 56 (IMD56) [MIM:615207]: An autosomal recessive primary immunodeficiency characterized by B- and T-cell defects and variable dysfunction of NK cells. Patients tend to have normal numbers of lymphocytes, but show defective class-switched B-cells, low IgG, defective antibody response, and defective T-cell responses to certain antigens. {ECO:0000269|PubMed:23440042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Chromosomal aberrations involving IL21R is a cause of B-cell non-Hodgkin lymphomas (B-cell NHL). Translocation t(3;16)(q27;p11), with BCL6. Drugs (DrugBank ID) DB00428 Interacts with O60502 EC number EC 3.2.1.169 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Glycosidase; Host-virus interaction; Hydrolase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,G Molecular weight (Da) 49869.8 Length 426 Aromaticity 0.14 Instability index 46.71 Isoelectric point 5.7 Charge (pH=7) -6.64 2D Binding mode Binding energy (Kcal/mol) -6.54 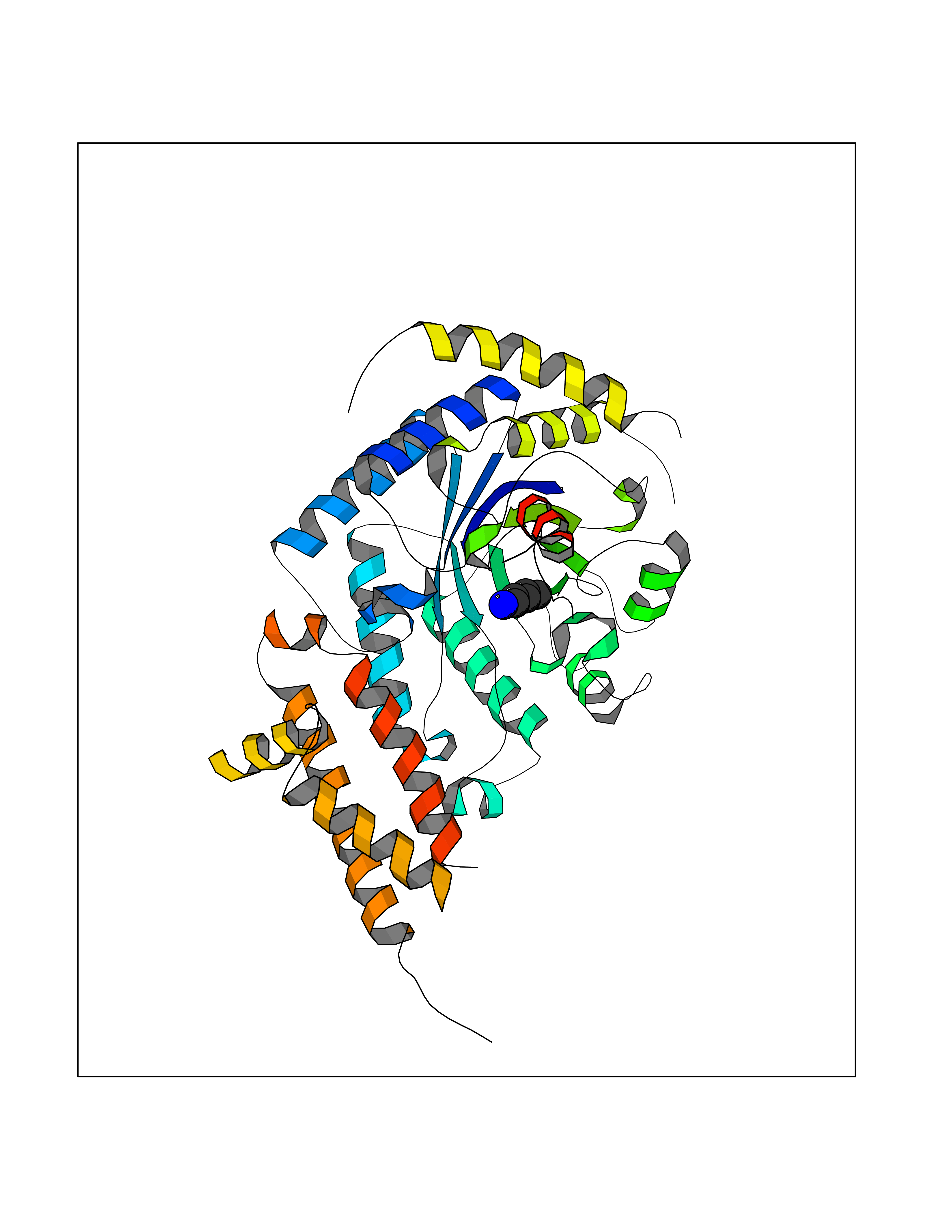 Molscript Map 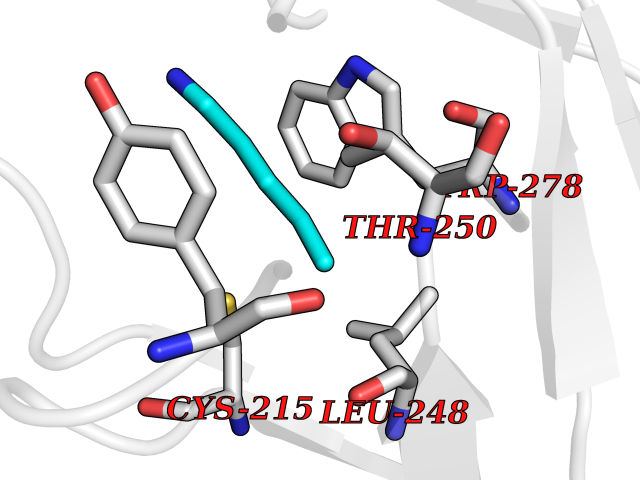 Pymol Map 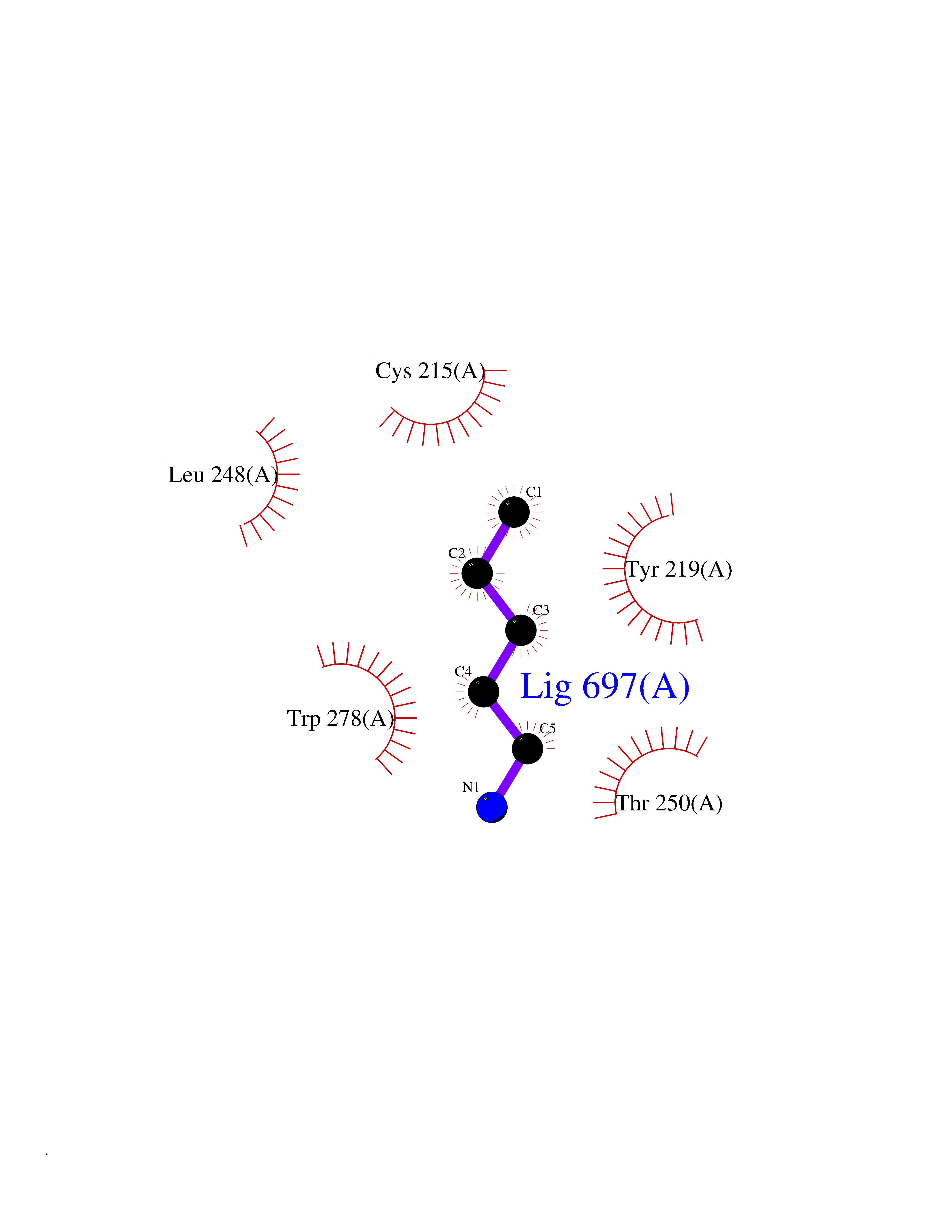 Ligplot Map 3D Binding mode Sequence RFLCGVVEGFYGRPWVMEQRKELFRRLQKWELNTYLYAPKDDYKHRMFWREMYSVEEAEQLMTLISAAREYEIEFIYAISPGLDITFSNPKEVSTLKRKLDQVSQFGCRSFALLFDDIDHNMCAADKEVFSSFAHAQVSITNEIYQYLGEPETFLFCPTEYCGTFCYPNVSQSPYLRTVGEKLLPGIEVLWTGPKVVSKEIPVESIEEVSKIIKRAPVIWDNIHANDYDQKRLFLGPYKGRSTELIPRLKGVLTNPNCEFEANYVAIHTLATWYKSNMNGVRDVLYSPQMALKLALTEWLQEFGVPHQYTLEDLQLLADLFYLPYEHGPKGAQMLREFQWLRANKDSEKIEEWRSRAAKFEEMCGLVMGMFTRLSNCANRTILYDMYSYVWDIKSIMSMVKSFVQWQEPWAFRGGLAGEFQRLLPI Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Bacterial Botulinum toxin A (Bact botA) | 6XCF | 4.79 | |
Target general information Gen name Bact botA Organism Clostridium botulinum Uniprot ID TTD ID Synonyms botA Protein family Peptidase M27 family Biochemical class Peptidase Function Inhibits acetylcholine release. The botulinum toxin binds with high affinity to peripheral neuronal presynaptic membrane to the secretory vesicle protein SV2. It binds directly to the largest luminal loop of SV2A, SV2B and SV2C. It is then internalized by receptor-mediated endocytosis. The C-terminus of the heavy chain (H) is responsible for the adherence of the toxin to the cell surface while the N-terminus mediates transport of the light chain from the endocytic vesicle to the cytosol. After translocation, the light chain (L) hydrolyzes the 197-Gln-|-Arg- 198 bond in SNAP-25, thereby blocking neurotransmitter release. Inhibition of acetylcholine release results in flaccid paralysis, with frequent heart or respiratory failure. Related diseases Major depressive disorder (MDD) [MIM:608516]: A common psychiatric disorder. It is a complex trait characterized by one or more major depressive episodes without a history of manic, mixed, or hypomanic episodes. A major depressive episode is characterized by at least 2 weeks during which there is a new onset or clear worsening of either depressed mood or loss of interest or pleasure in nearly all activities. Four additional symptoms must also be present including changes in appetite, weight, sleep, and psychomotor activity; decreased energy; feelings of worthlessness or guilt; difficulty thinking, concentrating, or making decisions; or recurrent thoughts of death or suicidal ideation, plans, or attempts. The episode must be accompanied by distress or impairment in social, occupational, or other important areas of functioning. {ECO:0000269|PubMed:15229186}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q02563; Q496J9; Q9Z2I6 EC number NA Uniprot keywords 3D-structure; Cell wall; Direct protein sequencing; Disulfide bond; Host cell membrane; Host cytoplasm; Host cytoplasmic vesicle; Host membrane; Host synapse; Hydrolase; Lipid-binding; Membrane; Metal-binding; Metalloprotease; Neurotoxin; Pharmaceutical; Protease; Secreted; Toxin; Transmembrane; Transmembrane helix; Ubl conjugation; Virulence; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 47759.6 Length 417 Aromaticity 0.13 Instability index 21.7 Isoelectric point 6.36 Charge (pH=7) -1.96 2D Binding mode Binding energy (Kcal/mol) -6.54  Molscript Map 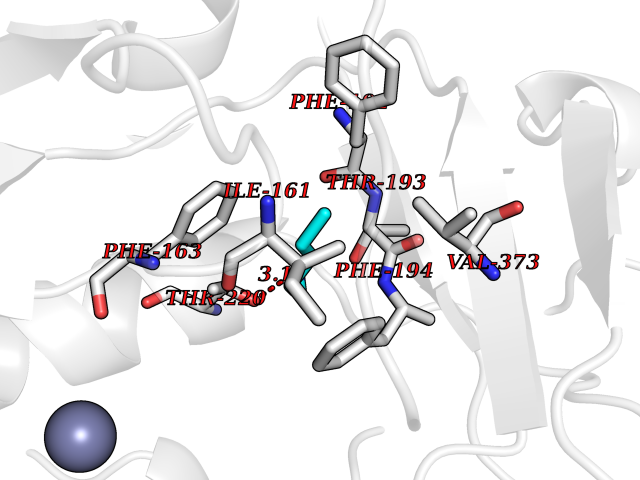 Pymol Map 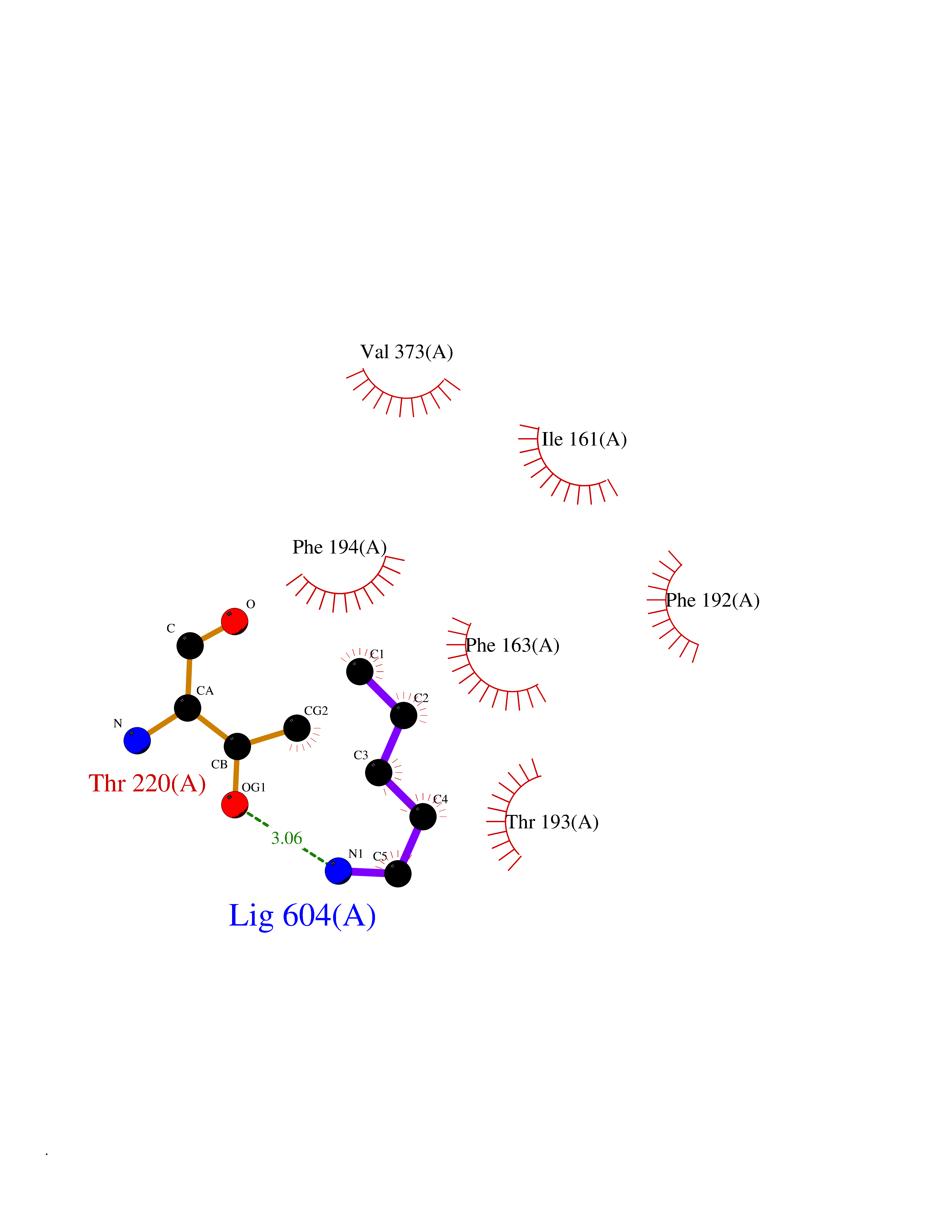 Ligplot Map 3D Binding mode Sequence MQFVNKQFNYKDPVNGVDIAYIKIPNVGQMQPVKAFKIHNKIWVIPERDTFTNPEEGDLNPPPPVSYYDSTYLSTDNEKDNYLKGVTKLFERIYSTDLGRMLLTSIVRGIPFWGGSTIDTELKVIDTNCINVIQPDGSYRSEELNLVIIGPSADIIQFECKSFGHEVLNLTRNGYGSTQYIRFSPDFTFGFEESLEVDTNPLLGAGKFATDPAVTLAHELIHAGHRLYGIAINPNRVFKVNTNAYYEMSGLEVSFEELRTFGGHDAKFIDSLQENEFRLYYYNKFKDIASTLNKAKSIVGTTASLQYMKNVFKEKYLLSEDTSGKFSVDKLKFDKLYKMLTEIYTEDNFVKFFKVLNRKTYLNFDKAVFKINIVPKVNYTIYDGFNLRNTNLAANFNGQNTEINNMNFTKLKNFTGL Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Squalene synthetase (FDFT1) | 3WCM | 4.79 | |
Target general information Gen name FDFT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Squalene synthase; SS; SQS; Farnesyl-diphosphate farnesyltransferase; FPP:FPP farnesyltransferase Protein family Phytoene/squalene synthase family Biochemical class Alkyl aryl transferase Function Participates in the isoprenoid biosynthetic pathway, catalyzing a two-step reaction in which two identical molecules of farnesyl pyrophosphate (FPP) are converted into squalene, with the consumption of NADPH. Related diseases Squalene synthase deficiency (SQSD) [MIM:618156]: An autosomal recessive disorder characterized by profound developmental delay, brain abnormalities, 2/3 syndactyly of the toes, facial dysmorphisms, low total and LDL-cholesterol, and abnormal urine organic acids. {ECO:0000269|PubMed:29909962}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05317 Interacts with Q13520; Q3SXY8; P04233-2; P11912; O75503; O43889-2; Q9GZR5; Q5JX71; P48165; Q8TDT2; Q8N5M9; Q6IBW4-4; Q96RD7; Q14973; Q9NQQ7-3; Q96MV1; Q9Y320 EC number EC 2.5.1.21 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Endoplasmic reticulum; Lipid biosynthesis; Lipid metabolism; Magnesium; Membrane; Metal-binding; Multifunctional enzyme; NAD; NADP; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 37860 Length 329 Aromaticity 0.1 Instability index 40.19 Isoelectric point 5.47 Charge (pH=7) -7.65 2D Binding mode Binding energy (Kcal/mol) -6.54 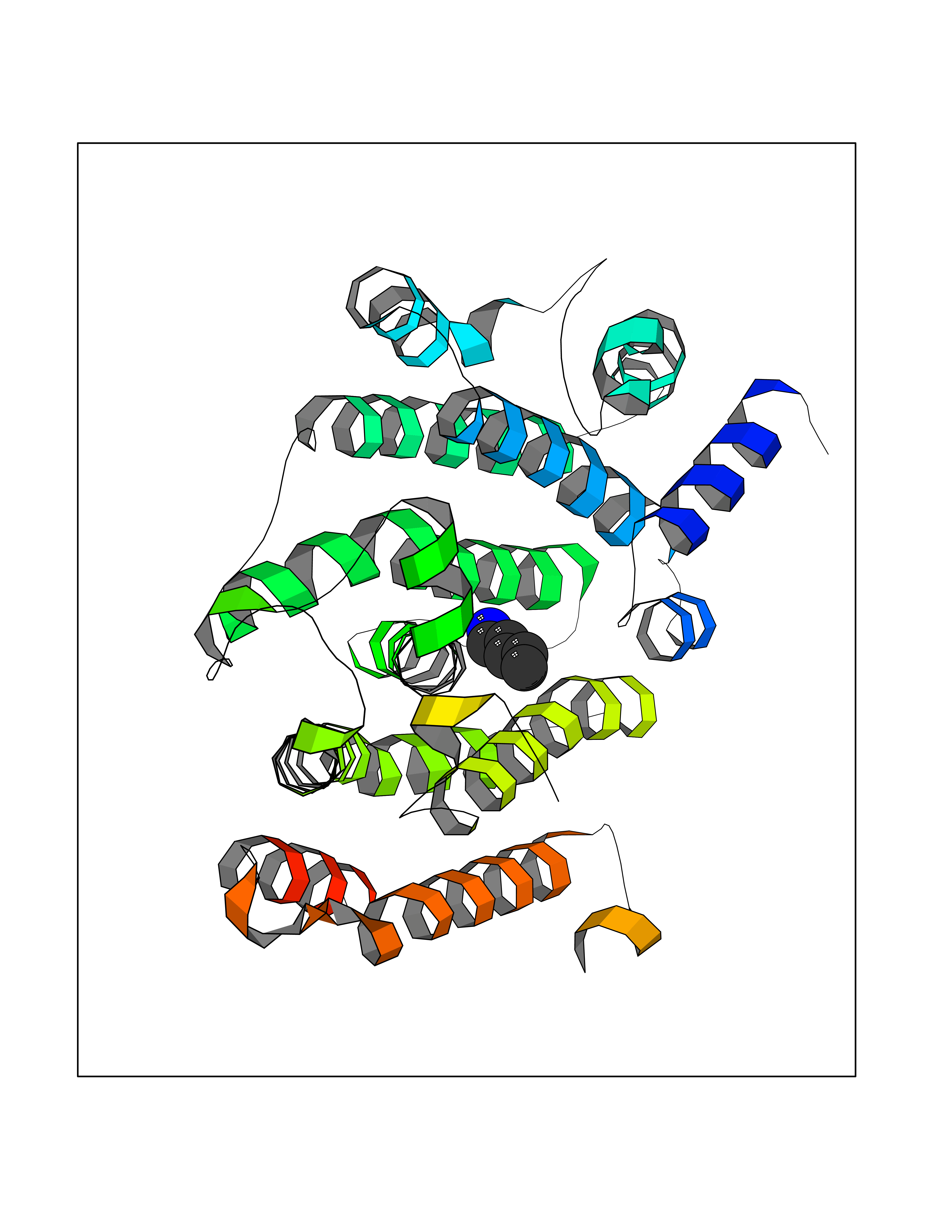 Molscript Map 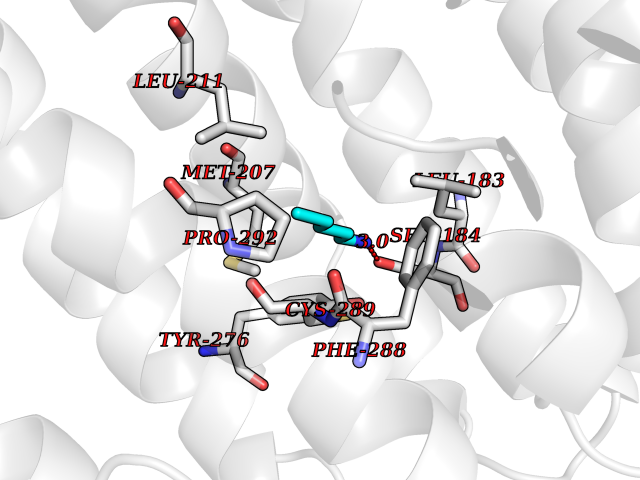 Pymol Map  Ligplot Map 3D Binding mode Sequence LSSSLKTCYKYLNQTSRSFAAVIQALDGEMRNAVCIFYLVLRALDTLEDDMTISVEKKVPLLHNFHSFLYQPDWRFMESKEKDRQVLEDFPTISLEFRNLAEKYQTVIADICRRMGIGMAEFLDKHVTSEQEWDKYCHYVAGLVGIGLSRLFSASEFEDPLVGEDTERANSMGLFLQKTNIIRDYLEDQQGGREFWPQEVWSRYVKKLGDFALPENIDLAVQCLNELITNALHHIPDVITYLSRLRNQSVFNFCAIPQVMAIATLAACYNNQQVFKGAVLIVTLMMDATNMPAVKAIIYQYMEEIYHRIPDSNPSSSKTRQIISTIRTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | 5'-methylthioadenosine/S-adenosylhomocysteine nucleosidase | 4WKC | 4.78 | |
Target general information Gen name mtnN Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms pfs;b0159;yadA;JW0155;mtn Protein family PNP/UDP phosphorylase family, MtnN subfamily Biochemical class hydrolase / hydrolase inhibitor Function Adenosylhomocysteine nucleosidase activity.Identical protein binding.Methylthioadenosine nucleosidase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02158; DB08606; DB02933; DB00173; DB02281 Interacts with P0AF12 EC number 3.2.2.9 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Hydrolase; Methionine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 24353.7 Length 232 Aromaticity 0.05 Instability index 22.1 Isoelectric point 5.09 Charge (pH=7) -9.9 2D Binding mode Binding energy (Kcal/mol) -6.52  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MKIGIIGAMEEEVTLLRDKIENRQTISLGGCEIYTGQLNGTEVALLKSGIGKVAAALGATLLLEHCKPDVIINTGSAGGLAPTLKVGDIVVSDEARYHDADVTAFGYEYGQLPGCPAGFKADDKLIAAAEACIAELNLNAVRGLIVSGDAFINGSVGLAKIRHNFPQAIAVEMEATAIAHVCHNFNVPFVVVRAISDVADQQSHLSFDEFLAVAAKQSSLMVESLVQKLAHG Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Xylose isomerase | 4XIA | 4.78 | |
Target general information Gen name xylA Organism Arthrobacter sp. (strain NRRL B3728) Uniprot ID TTD ID NA Synonyms NA Protein family Xylose isomerase family Biochemical class Isomerase(intramolecular oxidoreductase) Function Metal ion binding.Xylose isomerase activity. Related diseases Bachmann-Bupp syndrome (BABS) [MIM:619075]: An autosomal dominant disorder characterized by global developmental delay, alopecia, absolute or relative macrocephaly, and facial dysmorphism. Neuroimaging shows white matter abnormalities, prominent Virchow-Robin spaces, periventricular cysts, and abnormalities of the corpus callosum. {ECO:0000269|PubMed:30239107, ECO:0000269|PubMed:30475435}. The disease is caused by variants affecting the gene represented in this entry. BABS is due to truncating variants that lead to a gain of function. This phenomenon apparently results from truncation proximal to or involving the C-terminal region of ODC1 protein, distal enough to allow escape from nonsense-mediated decay. A gain of function is corroborated by elevated plasma levels of N-acetylputrescine, with otherwise normal polyamine levels, in affected individuals. {ECO:0000269|PubMed:30475435}. Drugs (DrugBank ID) DB02172; DB03206; DB11195 Interacts with NA EC number 5.3.1.5 Uniprot keywords 3D-structure; Carbohydrate metabolism; Cytoplasm; Direct protein sequencing; Isomerase; Magnesium; Metal-binding; Xylose metabolism Protein physicochemical properties Chain ID A,B Molecular weight (Da) 35700.7 Length 325 Aromaticity 0.11 Instability index 20.63 Isoelectric point 5.19 Charge (pH=7) -14.16 2D Binding mode Binding energy (Kcal/mol) -6.52 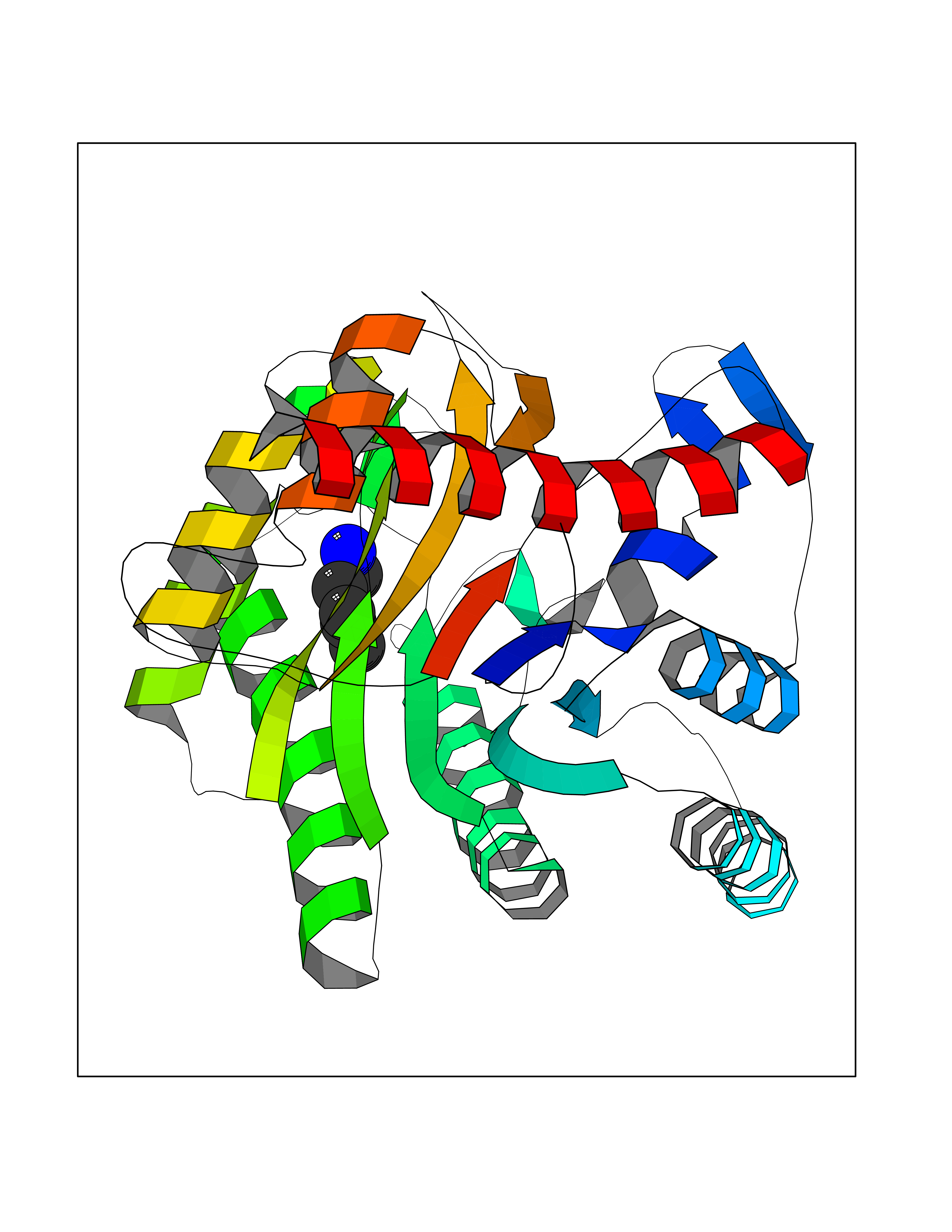 Molscript Map 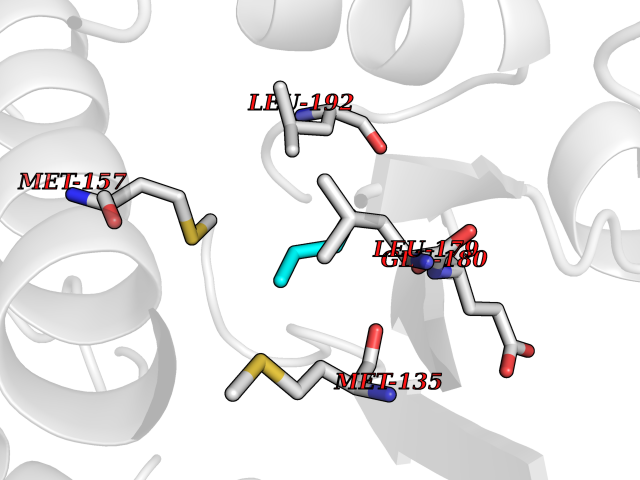 Pymol Map 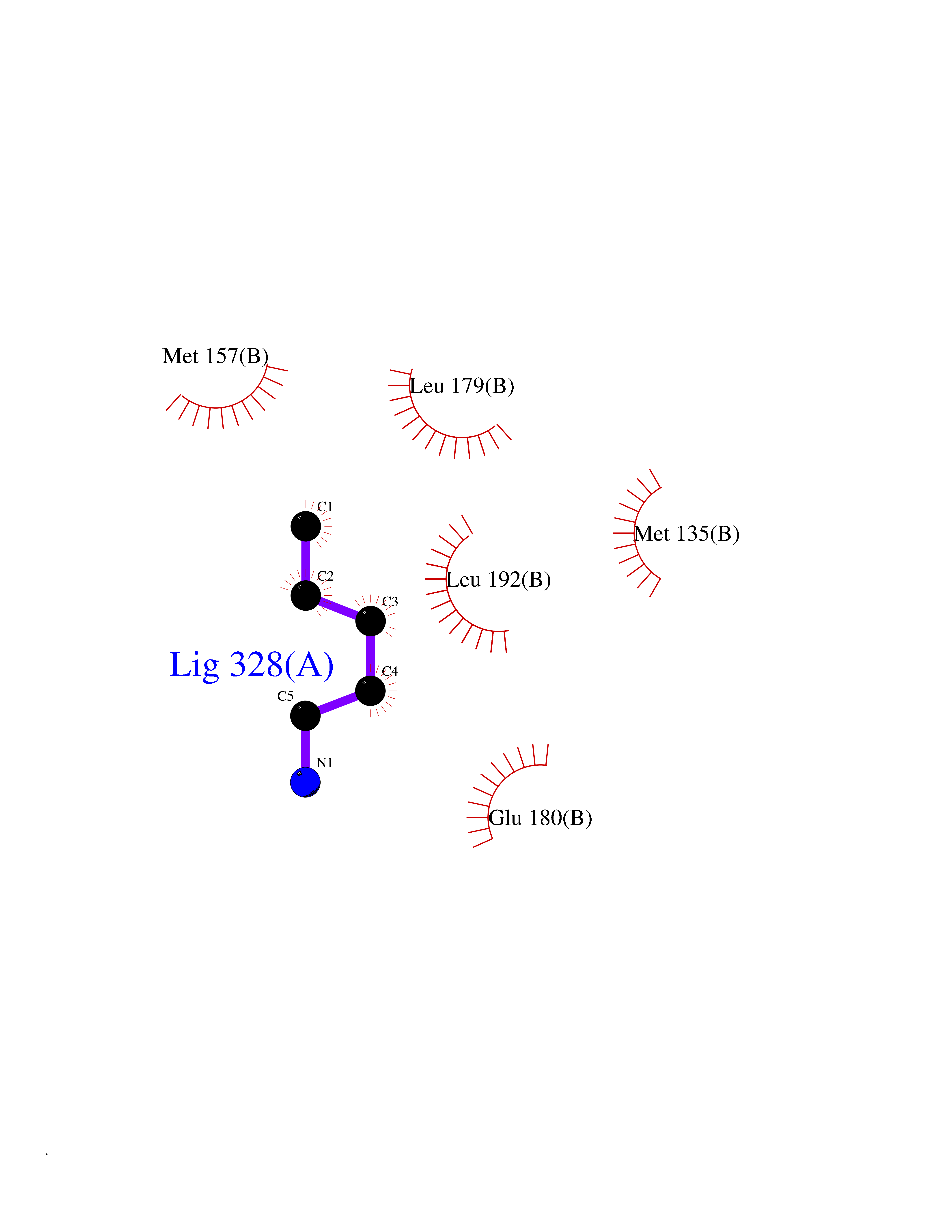 Ligplot Map 3D Binding mode Sequence VQPTPADHFTFGLWTVGWTGADPFGVATRANLDPVEAVHKLAELGAYGITFHDNDLIPFDATAAEREKILGDFNQALADTGLKVPMVTTNLFSHPVFKDGGFTSNDRSIRRFALAKVLHNIDLAAEMGAETFVMWGGREGSEYDGSKDLAAALDRMREGVDTAAGYIKDKGYNLRIALEPKPNEPRGDIFLPTVGHGLAFIEQLEHGDIVGLNPETGHEQMAGLNFTHGIAQALWAEKLFHIDLNGQRGIKYDQDLVFGHGDLTSAFFTVDLLENGFPNGGPKYTGPRHFDYKPSRTDGYDGVWDSAKANMSMYLLLKERALAFR Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Dopamine beta-hydroxylase | 4ZEL | 4.78 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -6.52 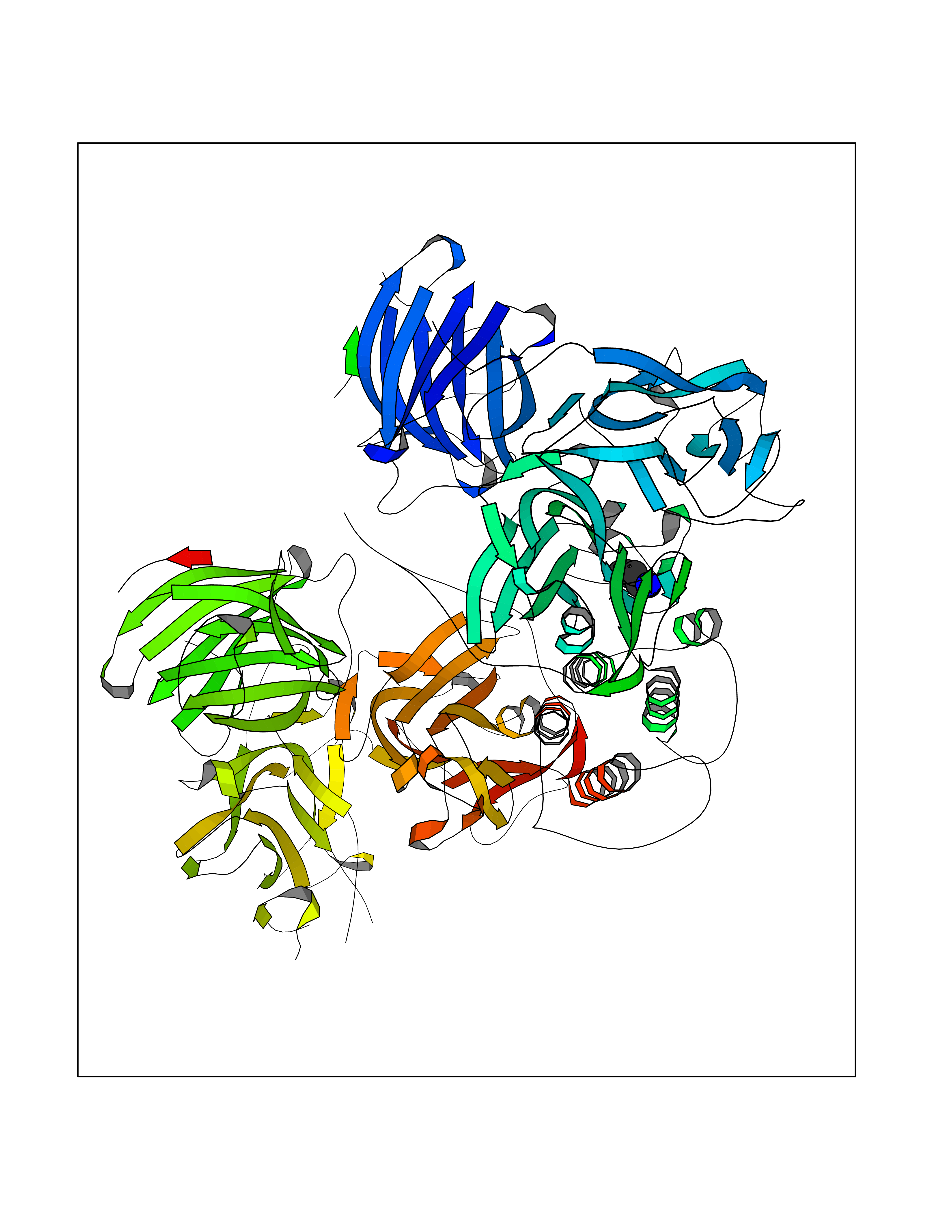 Molscript Map 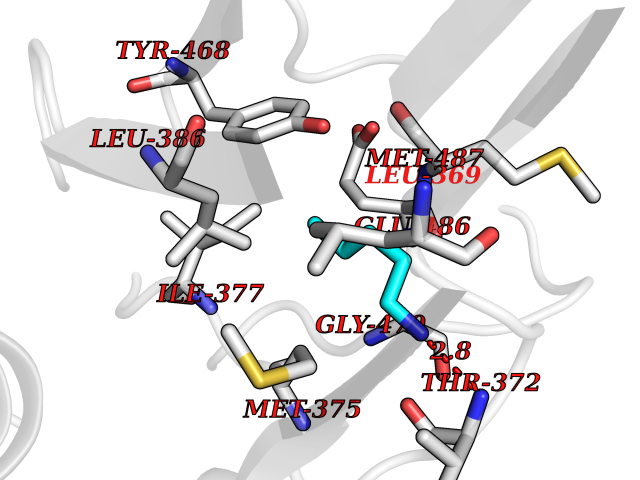 Pymol Map 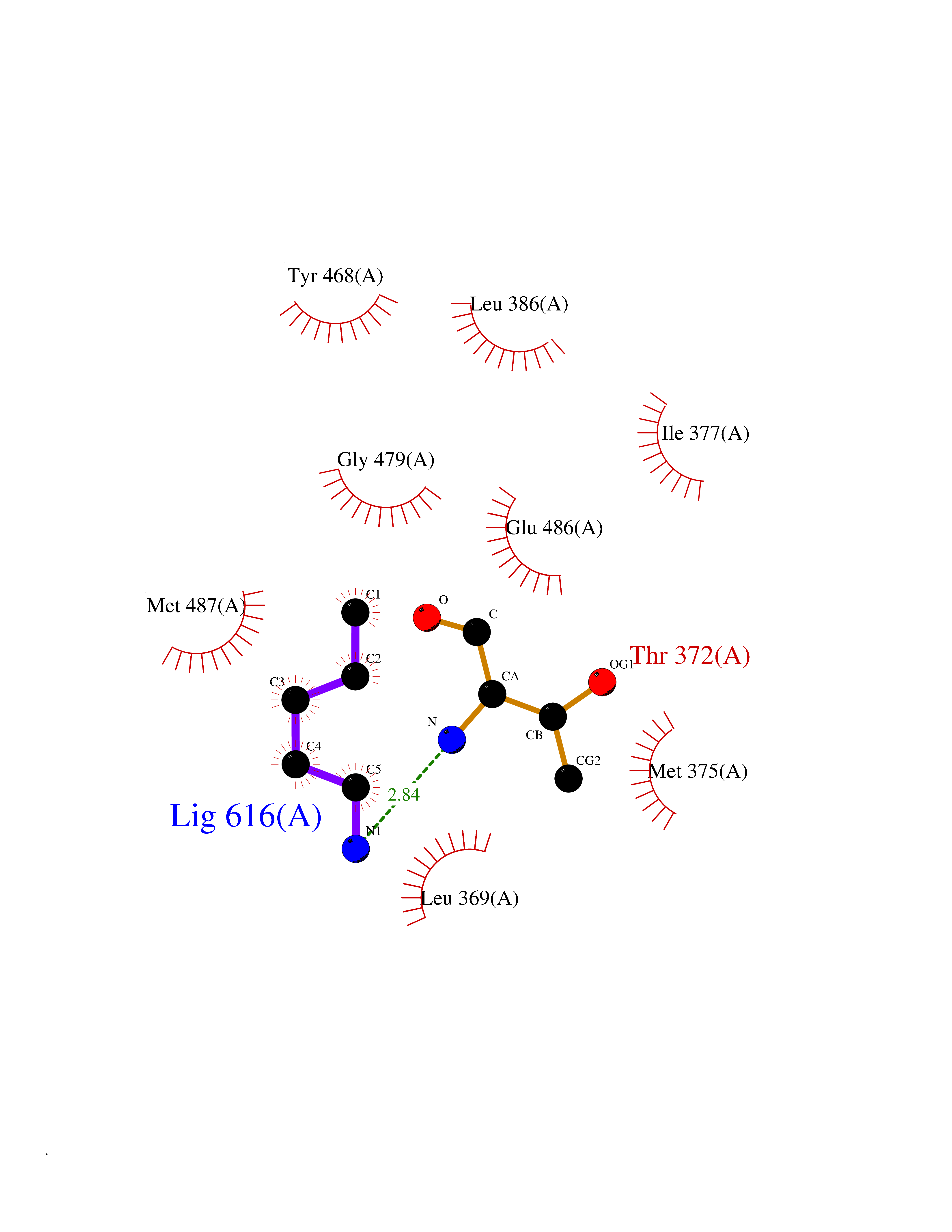 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | 4-hydroxyphenylpyruvate dioxygenase | 3ISQ | 4.78 | |
Target general information Gen name HPD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PPD Protein family 4HPPD family Biochemical class Oxidoreductase Function 4-hydroxyphenylpyruvate dioxygenase activity.Metal ion binding. Related diseases Tyrosinemia 3 (TYRSN3) [MIM:276710]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, seizures and mild intellectual disability. {ECO:0000269|PubMed:10942115, ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hawkinsinuria (HWKS) [MIM:140350]: An inborn error of tyrosine metabolism characterized by failure to thrive, persistent metabolic acidosis, fine and sparse hair, and excretion of the unusual cyclic amino acid metabolite, hawkinsin, in the urine. {ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02850; DB00348 Interacts with NA EC number 1.13.11.27 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Dioxygenase; Disease variant; Endoplasmic reticulum; Golgi apparatus; Intellectual disability; Iron; Membrane; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 43164.8 Length 376 Aromaticity 0.11 Instability index 32.38 Isoelectric point 6.73 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -6.52 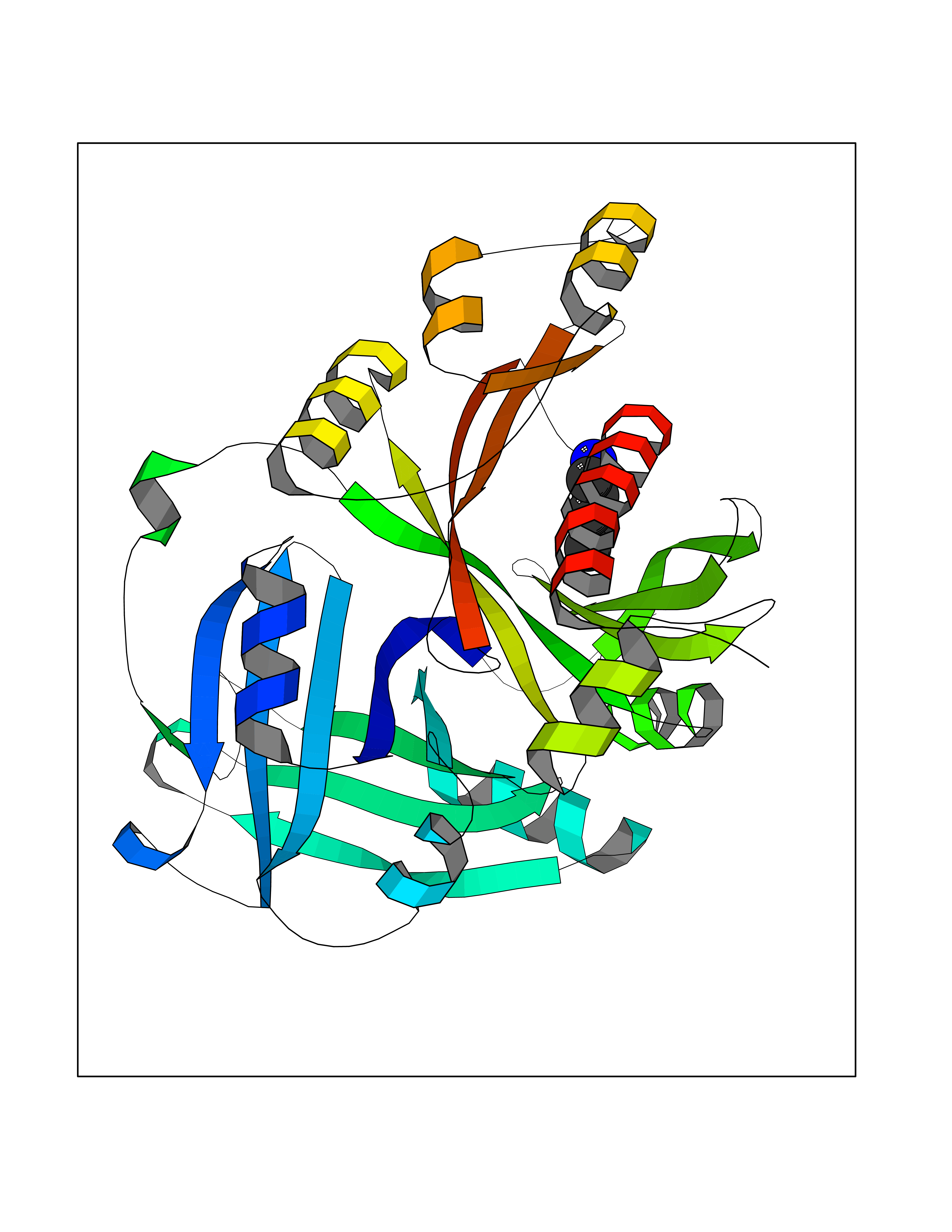 Molscript Map 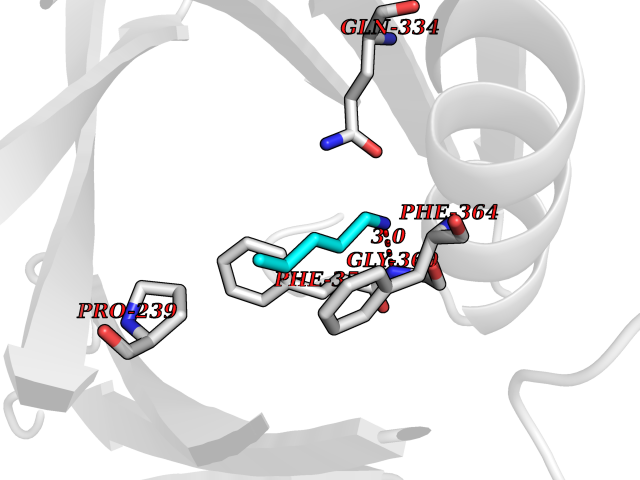 Pymol Map 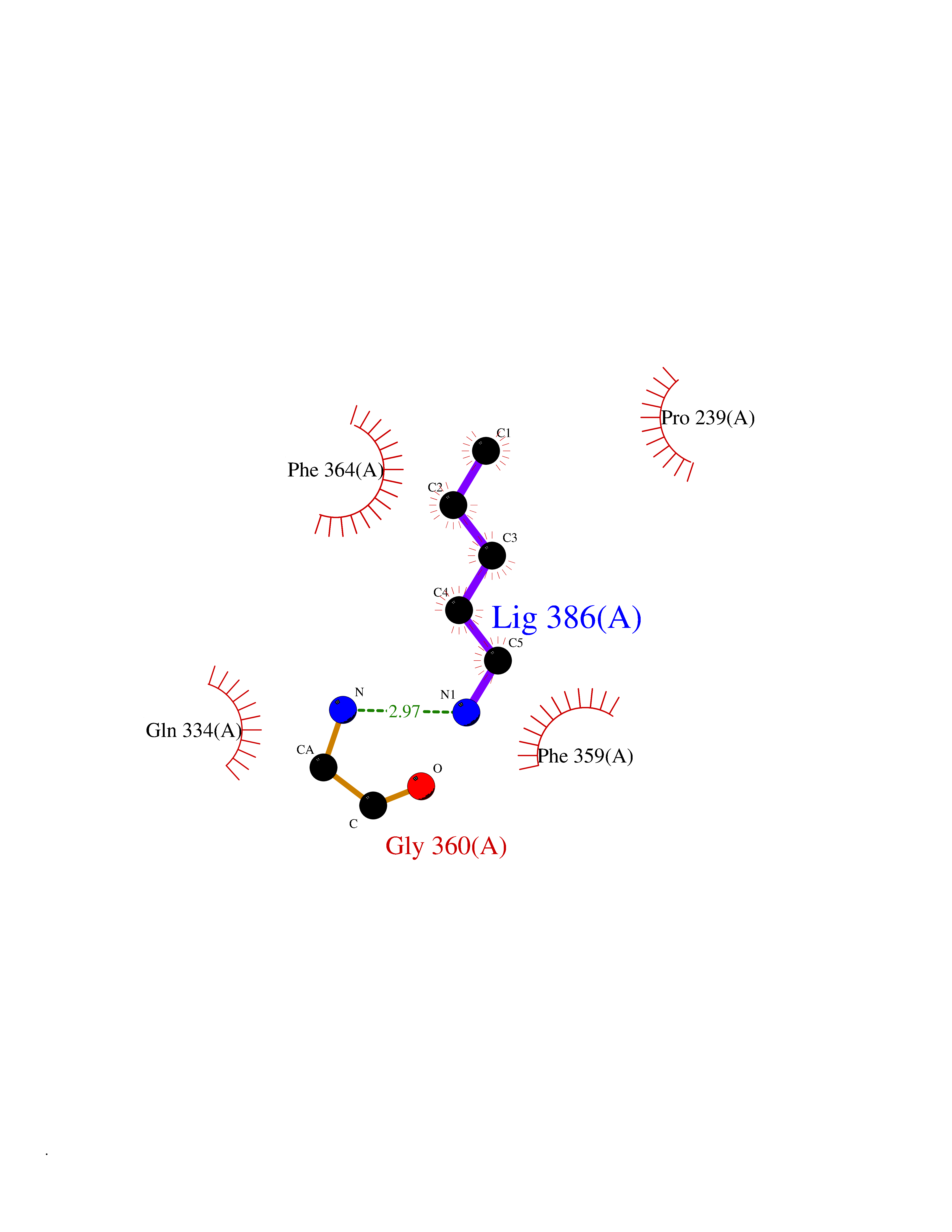 Ligplot Map 3D Binding mode Sequence AKPERGRFLHFHSVTFWVGNAKQAASFYCSKMGFEPLAYRGLETGSREVVSHVIKQGKIVFVLSSALNPWNKEMGDHLVKHGDGVKDIAFEVEDCDYIVQKARERGAKIMREPWVEQDKFGKVKFAVLQTYGDTTHTLVEKMNYIGQFLPGYEAPAFMDPLLPKLPKCSLEMIDHIVGNQPDQEMVSASEWYLKNLQFHRFWSVDDTQVHTEYSSLRSIVVANYEESIKMPINEPAPGKKKSQIQEYVDYNGGAGVQHIALKTEDIITAIRHLRERGLEFLSVPSTYYKQLREKLKTAKIKVKENIDALEELKILVDYDEKGYLLQIFTKPVQDRPTLFLEVIQRHNHQGFGAGNFNSLFKAFEEEQNLRGNLTNM Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Ribonucleoside-diphosphate reductase subunit M2 | 3OLJ | 4.78 | |
Target general information Gen name RRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms RR2 Protein family Ribonucleoside diphosphate reductase small chain family Biochemical class Oxidoreductase Function Metal ion binding.Ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00242; DB05260; DB05801; DB05003; DB05428 Interacts with P41002; Q9UM11; P23921; O00560 EC number 1.17.4.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Deoxyribonucleotide synthesis; Iron; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 33579.4 Length 286 Aromaticity 0.14 Instability index 43.7 Isoelectric point 5.12 Charge (pH=7) -12.86 2D Binding mode Binding energy (Kcal/mol) -6.52  Molscript Map 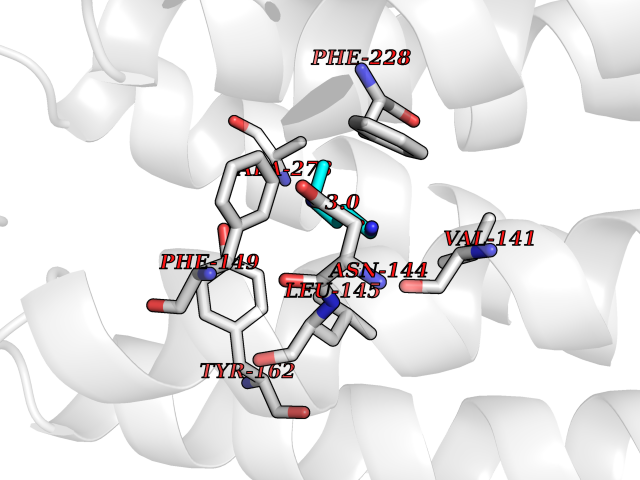 Pymol Map 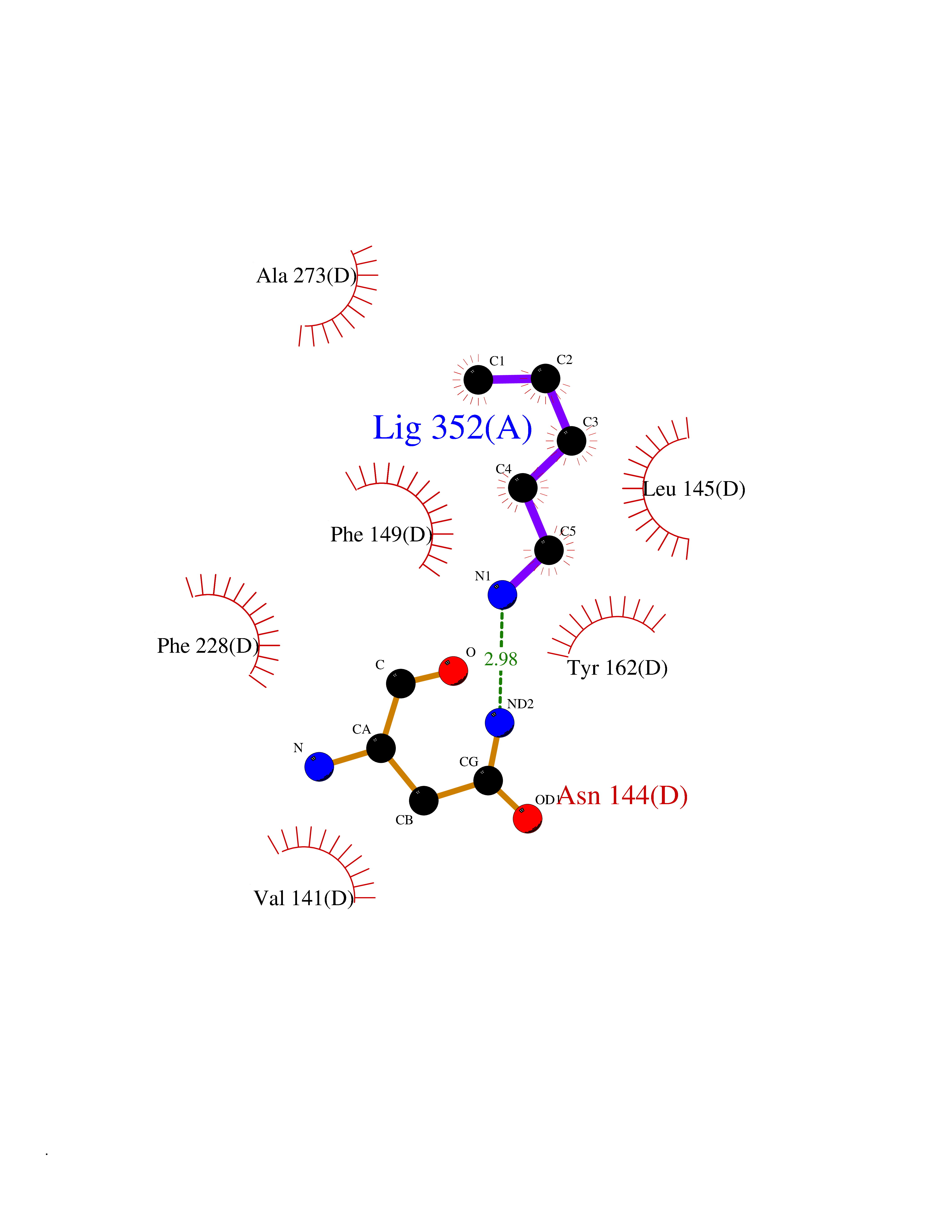 Ligplot Map 3D Binding mode Sequence MGVEDEPLLRENPRRFVIFPIEYHDIWQMYKKAEASFWTAEEVDLSKDIQHWESLKPEERYFISHVLAFFAASDGIVNENLVERFSQEVQITEARCFYGFQIAMENIHSEMYSLLIDTYIKDPKEREFLFNAIETMPCVKKKADWALRWIGDKEATYGERVVAFAAVEGIFFSGSFASIFWLKKRGLMPGLTFSNELISRDEGLHCDFACLMFKHLVHKPSEERVREIIINAVRIEQEFLTEALPVKLIGMNCTLMKQYIEFVADRLMLELGFSKVFRVENPFDFM Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Acetyl-CoA carboxylase 1 | 2YL2 | 4.78 | |
Target general information Gen name ACACA Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACAC;ACCA;ACC1 Protein family NA Biochemical class Ligase Function Acetyl-CoA carboxylase activity.ATP binding.Biotin carboxylase activity.Identical protein binding.Metal ion binding. Related diseases Acetyl-CoA carboxylase-alpha deficiency (ACACAD) [MIM:613933]: An autosomal recessive inborn error of de novo fatty acid synthesis associated with severe brain damage, persistent myopathy and poor growth. {ECO:0000269|PubMed:6114432}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00121 Interacts with Q13085; O60218; P38398; Q96EB6; Q9CQ20; P02654; Q92915-2; Q6NTF9-3 EC number 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative promoter usage; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Magnesium; Manganese; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 54237.7 Length 486 Aromaticity 0.09 Instability index 39.18 Isoelectric point 6.37 Charge (pH=7) -2.46 2D Binding mode Binding energy (Kcal/mol) -6.52 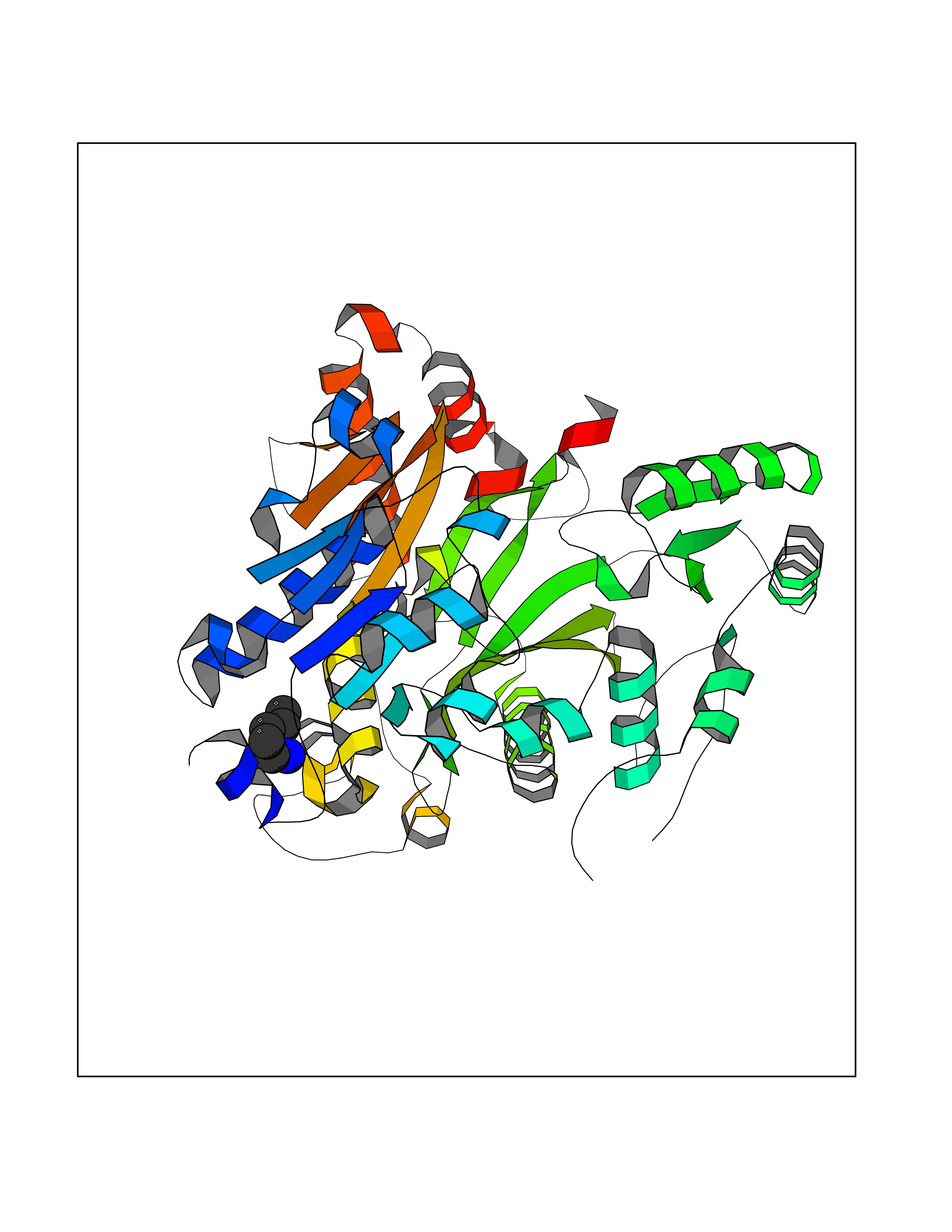 Molscript Map 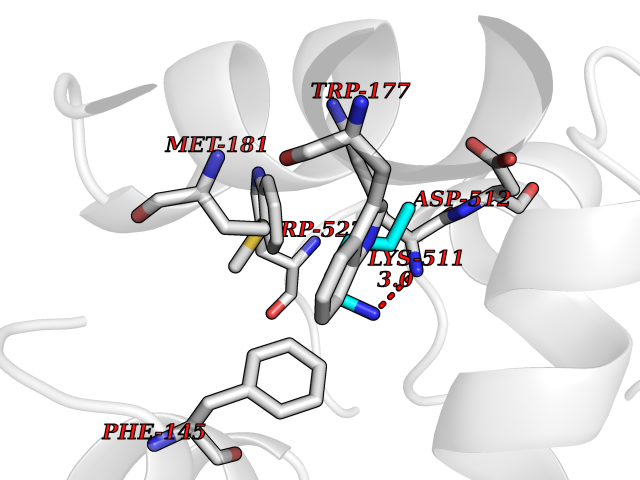 Pymol Map 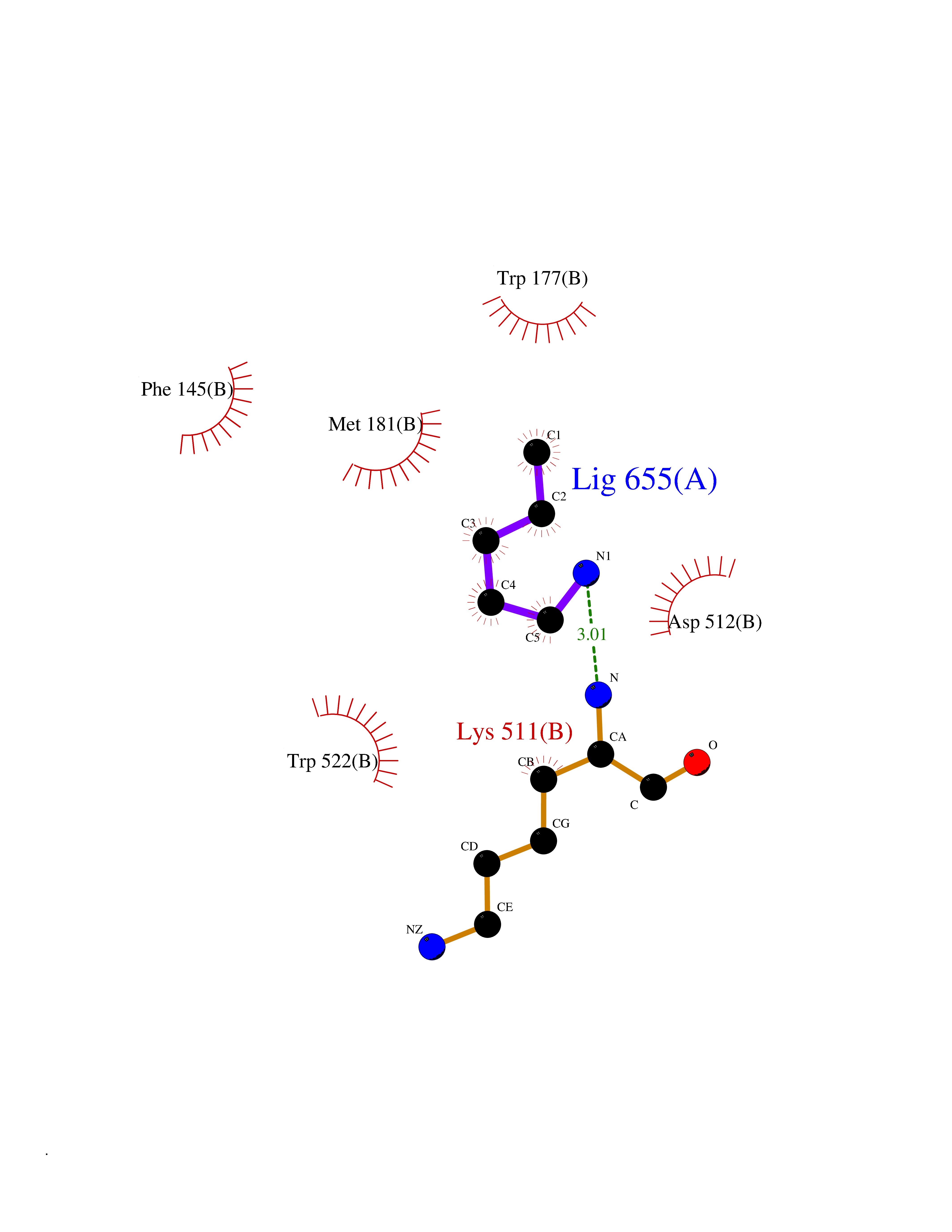 Ligplot Map 3D Binding mode Sequence VASPAEFVTRFGGNKVIEKVLIANNGIAAVKCMRSIRRWSYEMFRNERAIRFVVMVTPEDLKANAEYIKMADHYVPVPGGPNNNNYANVELILDIAKRIPVQAVWAGWGHASENPKLPELLLKNGIAFMGPPSQAMWALGDKIASSIVAQTAGIPTLPWSGSGLRVDWSKRILNVPQELYEKGYVKDVDDGLQAAEEVGYPVMIKASEGGGGKGIRKVNNADDFPNLFRQVQAEVPGSPIFVMRLAKQSRHLEVQILADQYGNAISLFGRDCSVQRRHQKIIEEAPATIATPAVFEHMEQCAVKLAKMVGYVSAGTVEYLYSQDGSFYFLELNPRLQVEHPCTEMVADVNLPAAQLQIAMGIPLYRIKDIRMMYGVSPWGDSPIDFEDSAHVPCPRGHVIAARITGTVQELNFRSNKNVWGYFSVQFGHCFSWGENREEAISNMVVALKELSIRGDFRTTVEYLIKLLETESFQMNRIDTGWLDRL Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Matrix metalloproteinase-13 (MMP-13) | 2OW9 | 4.78 | |
Target general information Gen name MMP13 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Matrix metalloproteinase 13; Collagenase-3; Collagenase 3 Protein family Peptidase M10A family Biochemical class Peptidase Function Cleaves triple helical collagens, including type I, type II and type III collagen, but has the highest activity with soluble type II collagen. Can also degrade collagen type IV, type XIV and type X. May also function by activating or degrading key regulatory proteins, such as TGFB1 and CTGF. Plays a role in wound healing, tissue remodeling, cartilage degradation, bone development, bone mineralization and ossification. Required for normal embryonic bone development and ossification. Plays a role in the healing of bone fractures via endochondral ossification. Plays a role in wound healing, probably by a mechanism that involves proteolytic activation of TGFB1 and degradation of CTGF. Plays a role in keratinocyte migration during wound healing. May play a role in cell migration and in tumor cell invasion. Plays a role in the degradation of extracellular matrix proteins including fibrillar collagen, fibronectin, TNC and ACAN. Related diseases Spondyloepimetaphyseal dysplasia, Missouri type (SEMDM) [MIM:602111]: A bone disease characterized by moderate to severe metaphyseal changes, mild epiphyseal involvement, rhizomelic shortening of the lower limbs with bowing of the femora and/or tibiae, coxa vara, genu varum and pear-shaped vertebrae in childhood. Epimetaphyseal changes improve with age. {ECO:0000269|PubMed:16167086}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metaphyseal anadysplasia 1 (MANDP1) [MIM:602111]: A bone development disorder characterized by skeletal anomalies that resolve spontaneously with age. Clinical characteristics are evident from the first months of life and include slight shortness of stature and a mild varus deformity of the legs. Patients attain a normal stature in adolescence and show improvement or complete resolution of varus deformity of the legs and rhizomelic micromelia. {ECO:0000269|PubMed:19615667}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metaphyseal dysplasia, Spahr type (MDST) [MIM:250400]: An autosomal recessive, rare disease characterized by moderate short stature, mild genua vara, and radiographic signs of metaphyseal dysplasia, but no biochemical signs of rickets. {ECO:0000269|PubMed:24648384, ECO:0000269|PubMed:24781753}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02049; DB01996; DB03033; DB07827; DB08388; DB08561; DB08490; DB06423; DB02697; DB00786; DB04759; DB04760; DB04761; DB07013; DB02071 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Calcium; Collagen degradation; Direct protein sequencing; Disease variant; Disulfide bond; Dwarfism; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18757.8 Length 167 Aromaticity 0.15 Instability index 9.97 Isoelectric point 5.47 Charge (pH=7) -7.44 2D Binding mode Binding energy (Kcal/mol) -6.52 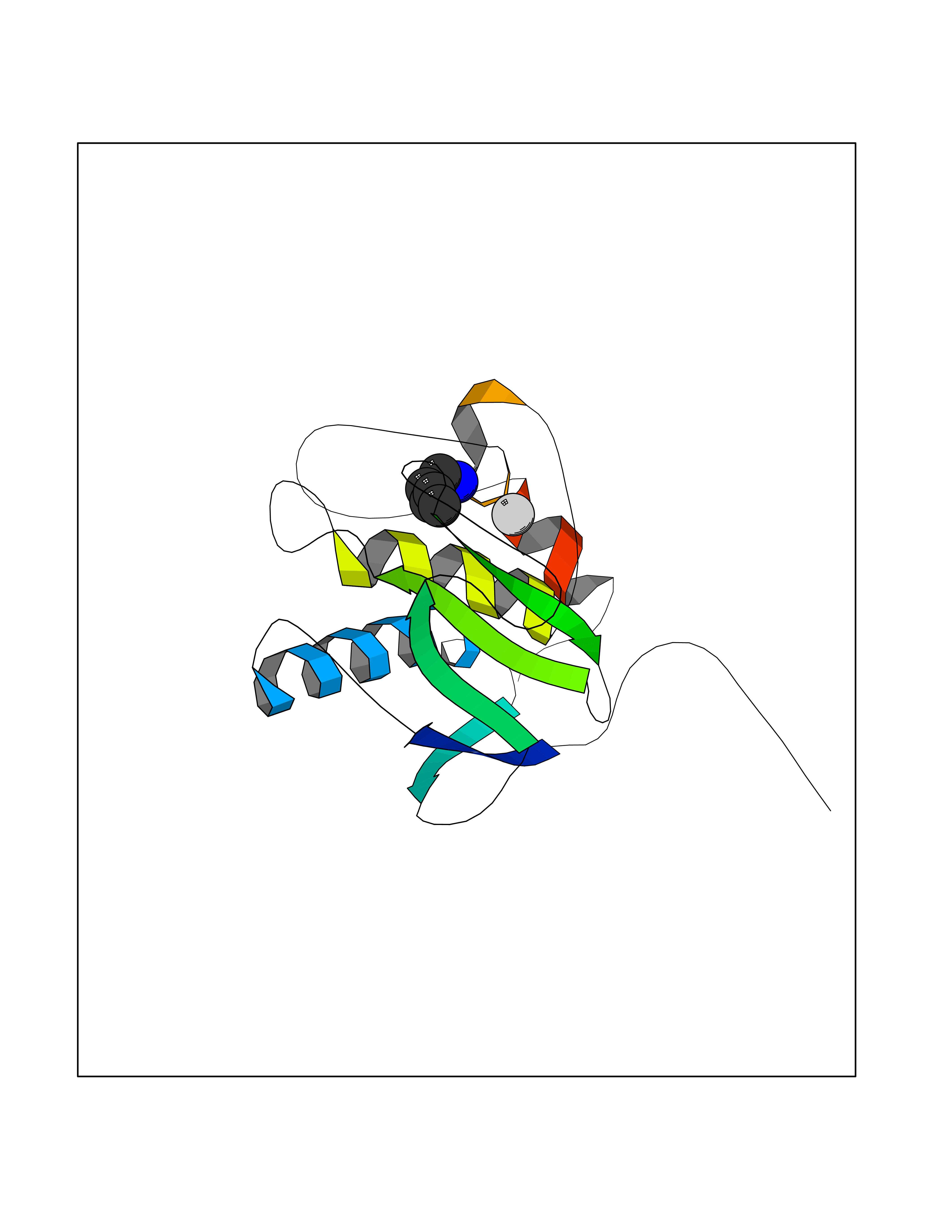 Molscript Map 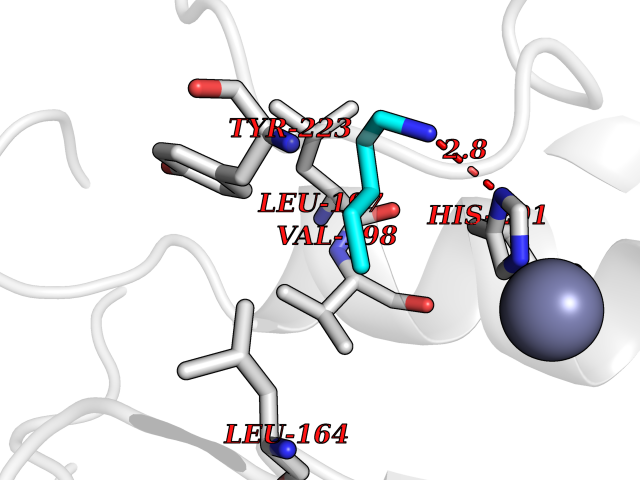 Pymol Map 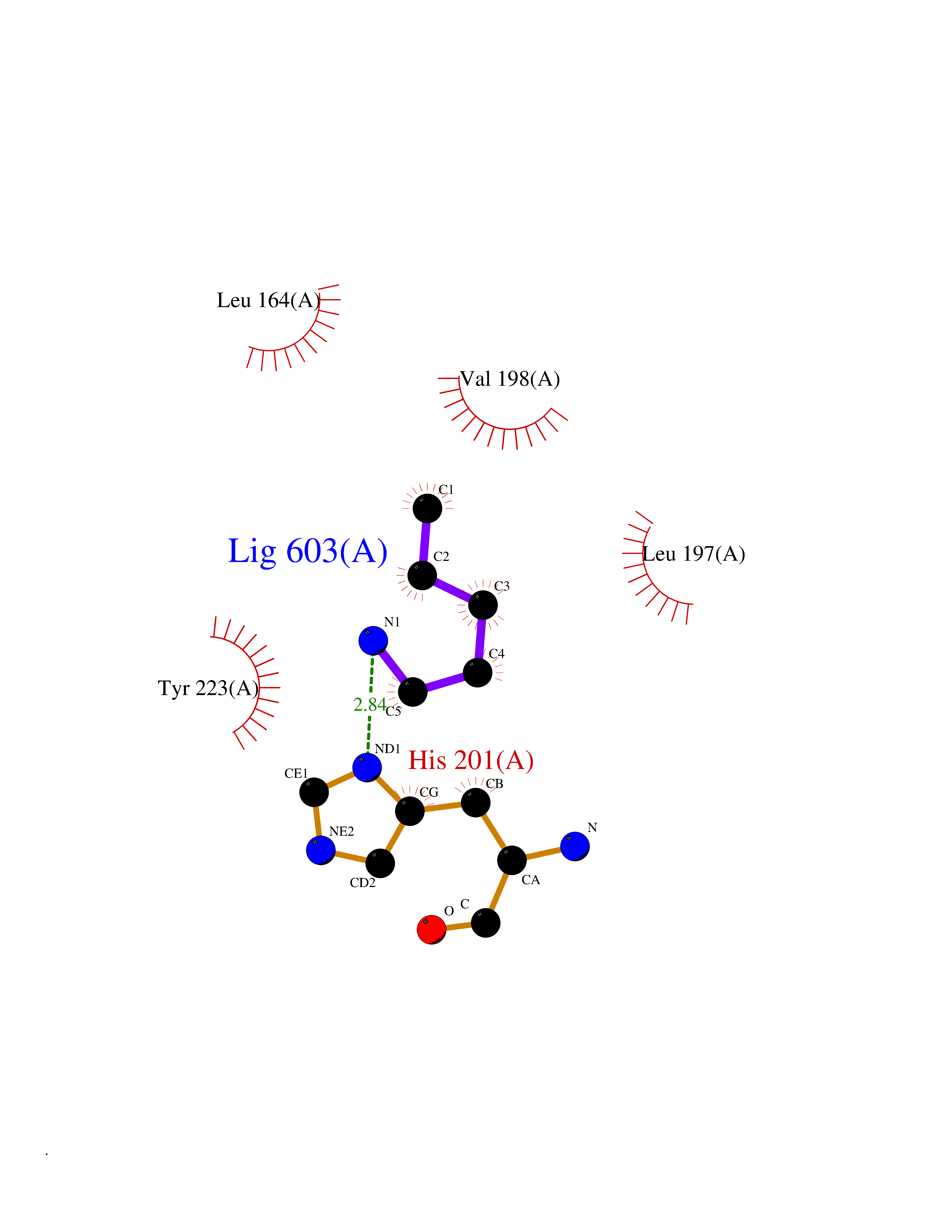 Ligplot Map 3D Binding mode Sequence YNVFPRTLKWSKMNLTYRIVNYTPDMTHSEVEKAFKKAFKVWSDVTPLNFTRLHDGIADIMISFGIKEHGDFYPFDGPSGLLAHAFPPGPNYGGDAHFDDDETWTSSSKGYNLFLVAAHEFGHSLGLDHSKDPGALMFPIYTYTGKSHFMLPDDDVQGIQSLYGPGD Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | GTPase activating protein (RASA1) | 1WQ1 | 4.78 | |
Target general information Gen name RASA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p120GAP; RasGAP; Ras p21 protein activator; Ras GTPase-activating protein 1; RASA; GTPase-activating protein; GAP Protein family NA Biochemical class NA Function Stimulates the GTPase of normal but not oncogenic Ras p21; this stimulation may be further increased in the presence of NCK1. Inhibitory regulator of the Ras-cyclic AMP pathway. Related diseases Mutations in the SH2 domain of RASA seem to be oncogenic and cause basal cell carcinomas.; DISEASE: Capillary malformation-arteriovenous malformation 1 (CMAVM1) [MIM:608354]: A disorder characterized by atypical capillary malformations that are multiple, small, round to oval in shape and pinkish red in color. These capillary malformations are associated with either arteriovenous malformation, arteriovenous fistula, or Parkes Weber syndrome. CMAVM1 inheritance is autosomal dominant. {ECO:0000269|PubMed:14639529, ECO:0000269|PubMed:24038909}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q8IZP0; P10275; P04632; Q03135; Q96QB1; P00533; P04626; P21860; P36888; Q13480; Q96IK5; P42858; P10721; P08581; P16333; P09619 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; GTPase activation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; SH2 domain; SH3 domain; Tumor suppressor Protein physicochemical properties Chain ID R,G Molecular weight (Da) 55286.9 Length 486 Aromaticity 0.07 Instability index 46.43 Isoelectric point 5.6 Charge (pH=7) -13.18 2D Binding mode Binding energy (Kcal/mol) -6.52  Molscript Map 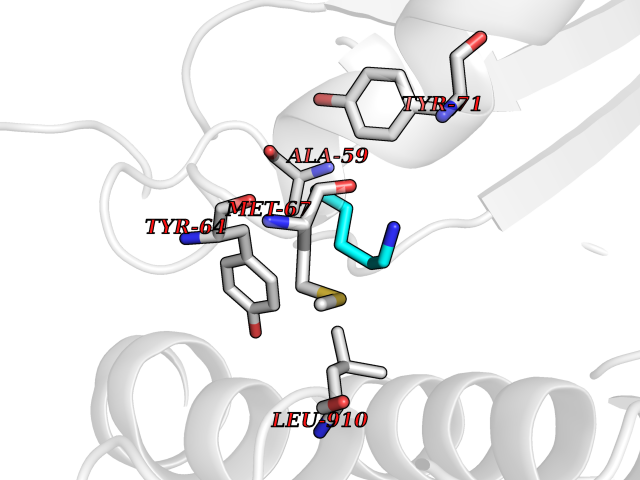 Pymol Map 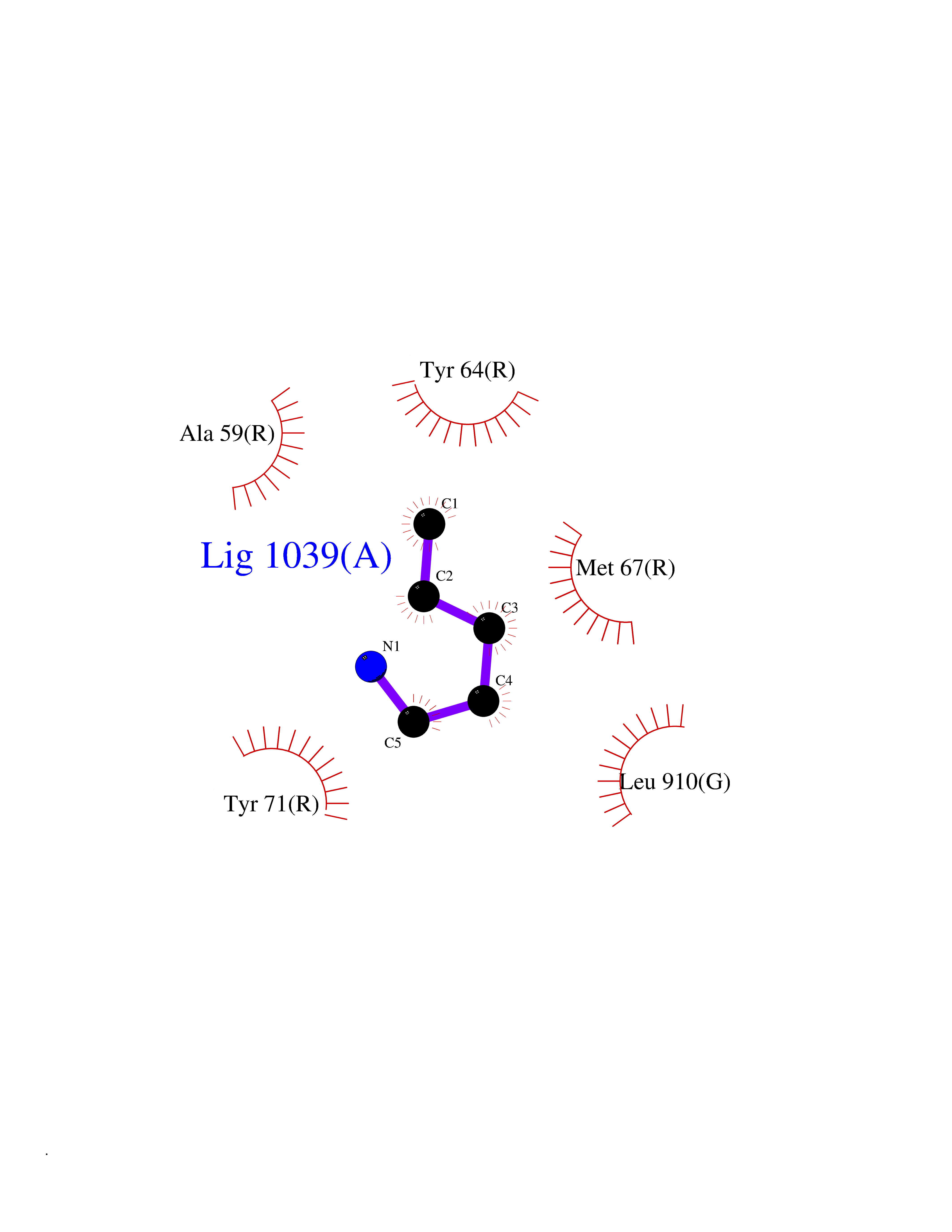 Ligplot Map 3D Binding mode Sequence MTEYKLVVVGAGGVGKSALTIQLIQNHFVDEYDPTIEDSYRKQVVIDGETCLLDILDTAGQEEYSAMRDQYMRTGEGFLCVFAINNTKSFEDIHQYREQIKRVKDSDDVPMVLVGNKCDLAARTVESRQAQDLARSYGIPYIETSAKTRQGVEDAFYTLVREIRQHMPEEEYSEFKELILQKELHVVYALSHVCGQDRTLLASILLRIFLHEKLESLLLCTLNDREISMEDEATTLFRATTLASTLMEQYMKATATQFVHHALKDSILKIMESKQSCELSPSKLEKNEDVNTNLTHLLNILSELVEKIFMASEILPPTLRYIYGCLQKSVQHKWPTNTTMRTRVVSGFVFLRLICPAILNPRMFNIISDSPSPIAARTLILVAKSVQNLANLVEFGAKEPYMEGVNPFIKSNKHRMIMFLDELGNVPELPDTTEHSRTDLSRDLAALHEICVAHSDELRTLSNERGAQQHVLKKLLAITELLQQKQ Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Dipeptidyl peptidase 9 (DPP-9) | 6EOR | 4.78 | |
Target general information Gen name DPP9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dipeptidyl peptidase-like protein 9; Dipeptidyl peptidase IX; Dipeptidyl peptidase IV-related protein 2; DPRP2; DPRP-2; DPP IX; DPLP9; DP9 Protein family Peptidase S9B family, DPPIV subfamily Biochemical class Peptidase Function Dipeptidyl peptidase that cleaves off N-terminal dipeptides from proteins having a Pro or Ala residue at position 2. Related diseases Hatipoglu immunodeficiency syndrome (HATIS) [MIM:620331]: An autosomal recessive immunologic disorder manifesting in infancy or early childhood, and characterized by failure to thrive, short stature, skin pigmentation abnormalities, pancytopenia, and susceptibility to recurrent infections. {ECO:0000269|PubMed:36112693}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NXR5; Q86TI2; Q6NUP5; P46379-2; Q8WUW1; Q96A83-2; O75190-2; O14645; Q01658; P29692-2; Q06787-7; Q9Y5Q9; O14901; Q9BVL2; Q96CV9; Q06830; P14678-2; P49458; Q11203; Q13148; P14927 EC number EC 3.4.14.5 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminopeptidase; Cytoplasm; Disease variant; Hydrolase; Nucleus; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A Molecular weight (Da) 92797.4 Length 808 Aromaticity 0.12 Instability index 37.45 Isoelectric point 6.34 Charge (pH=7) -8.98 2D Binding mode Binding energy (Kcal/mol) -6.52  Molscript Map 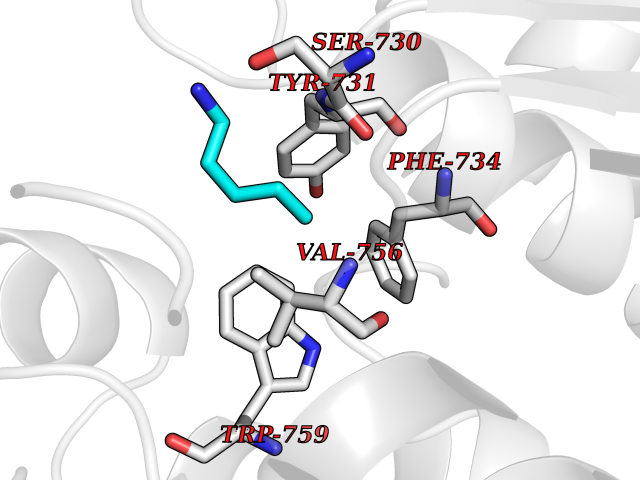 Pymol Map 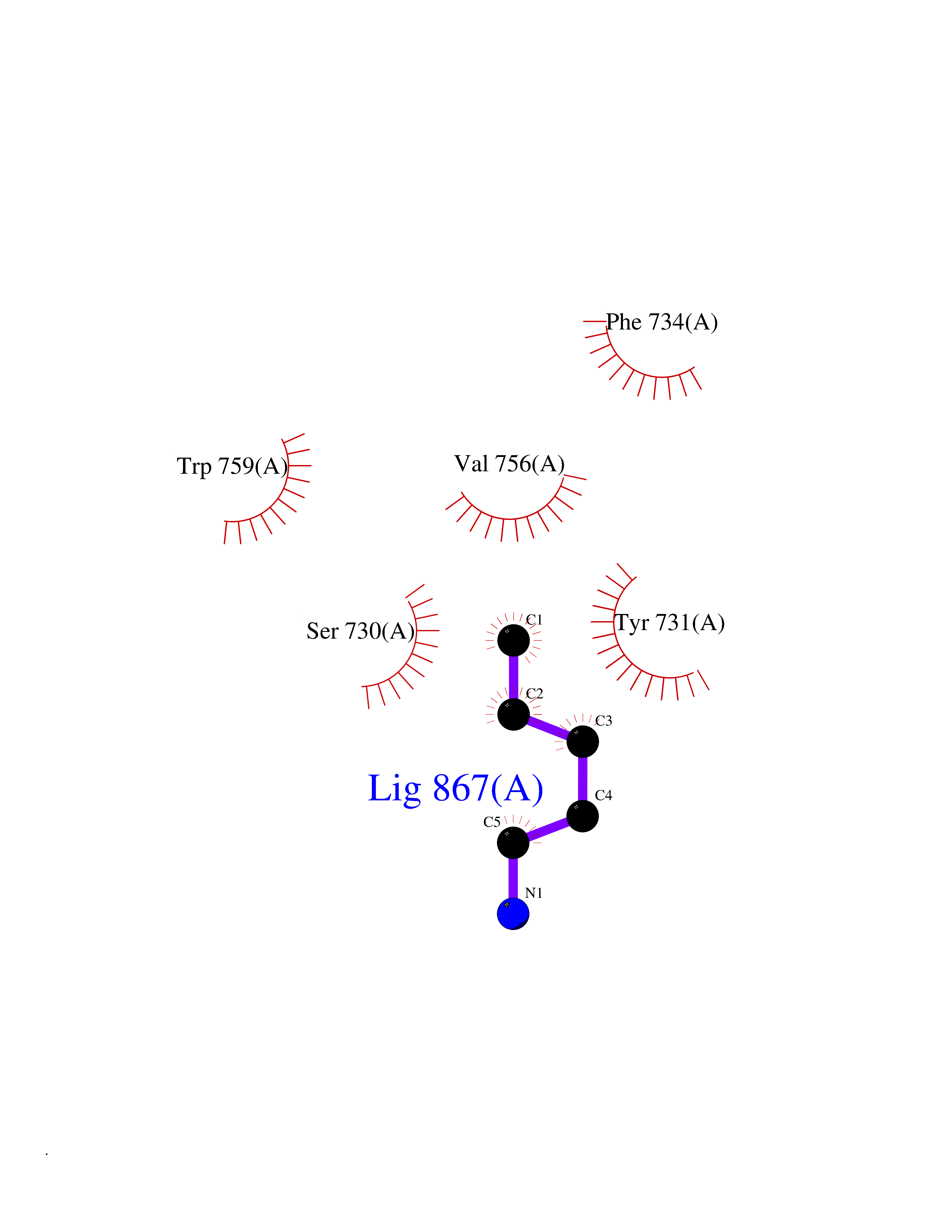 Ligplot Map 3D Binding mode Sequence AARFQVQKHSWDGLRSIIHGSRKAPHDFQFVQKSGPHSHRLYYLGMPYRENSLLYSEIPKLLLSWKQMLDHFQATPHHGVYSREEELLRERKRLGVFGITSYDFHSESGLFLFQASNSLFHCRDGGKNGFMVSPMKPLEIKTQCSGPRMDPKICPADPAFFSFINNSDLWVANIETGEERRLTFCHQNVLDDPKSAGVATFVIQEEFDRFTGYWWCPTASWEGLKTLRILYEEVDESEVEVIHVPSPALEERKTDSYRYPRTGSKNPKIALKLAEFQTDSQGKIVSTQEKELVQPFSSLFPKVEYIARAGWTRDGKYAWAMFLDRPQQWLQLVLLPPALFIPSTENEEQRLASARAVPRNVQPYVVYEEVTNVWINVHDIFYPFPQLCFLRANECKTGFCHLYKVTAVLKSQGYDWSEPFSPGEDEFKCPIKEEIALTSGEWEVLARHGSKIWVNEETKLVYFQGTKDTPLEHHLYVVSYEAAGEIVRLTTPGFSHSCSMSQNFDMFVSHYSSVSTPPCVHVYKLSGPDDDPLHKQPRFWASMMEADYVPPEIFHFHTRSDVRLYGMIYKPHALQPGKKHPTVLFVYGGPQVQLVNNSFKGIKYLRLNTLASLGYAVVVIDGRGSCQRGLRFEGALKNQMGQVEIEDQVEGLQFVAEKYGFIDLSRVAIHGWSYGGFLSLMGLIHKPQVFKVAIAGAPVTVWMAYDTGYTERYMDVPENNQHGYEAGSVALHVEKLPNEPNRLLILHGFLDENVHFFHTNFLVSQLIRAGKPYQLQIYPNERHSIRCPESGEHYEVTLLHFLQEYLHH Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Bacterial Nicotinate-nucleotide adenylyltransferase (Bact nadD) | 1K4K | 4.78 | |
Target general information Gen name Bact nadD Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms nadD of Escherichia coli (strain K12); Nicotinate mononucleotide adenylyltransferase of Escherichia coli (strain K12); NaMN adenylyltransferase of Escherichia coli (strain K12); Deamido-NAD(+)Nicotina Protein family NadD family Biochemical class Kinase Function Catalyzes the reversible adenylation of nicotinate mononucleotide (namn) to nicotinic acid adenine dinucleotide (naad). Related diseases Asthma-related traits 5 (ASRT5) [MIM:611064]: Asthma-related traits include clinical symptoms of asthma, such as coughing, wheezing, dyspnea, bronchial hyperresponsiveness as assessed by methacholine challenge test, serum IgE levels, atopy and atopic dermatitis. {ECO:0000269|PubMed:17503328}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.7.7.18 Uniprot keywords 3D-structure; ATP-binding; NAD; Nucleotide-binding; Nucleotidyltransferase; Pyridine nucleotide biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 24527.6 Length 213 Aromaticity 0.09 Instability index 48.92 Isoelectric point 5.46 Charge (pH=7) -7.78 2D Binding mode Binding energy (Kcal/mol) -6.53 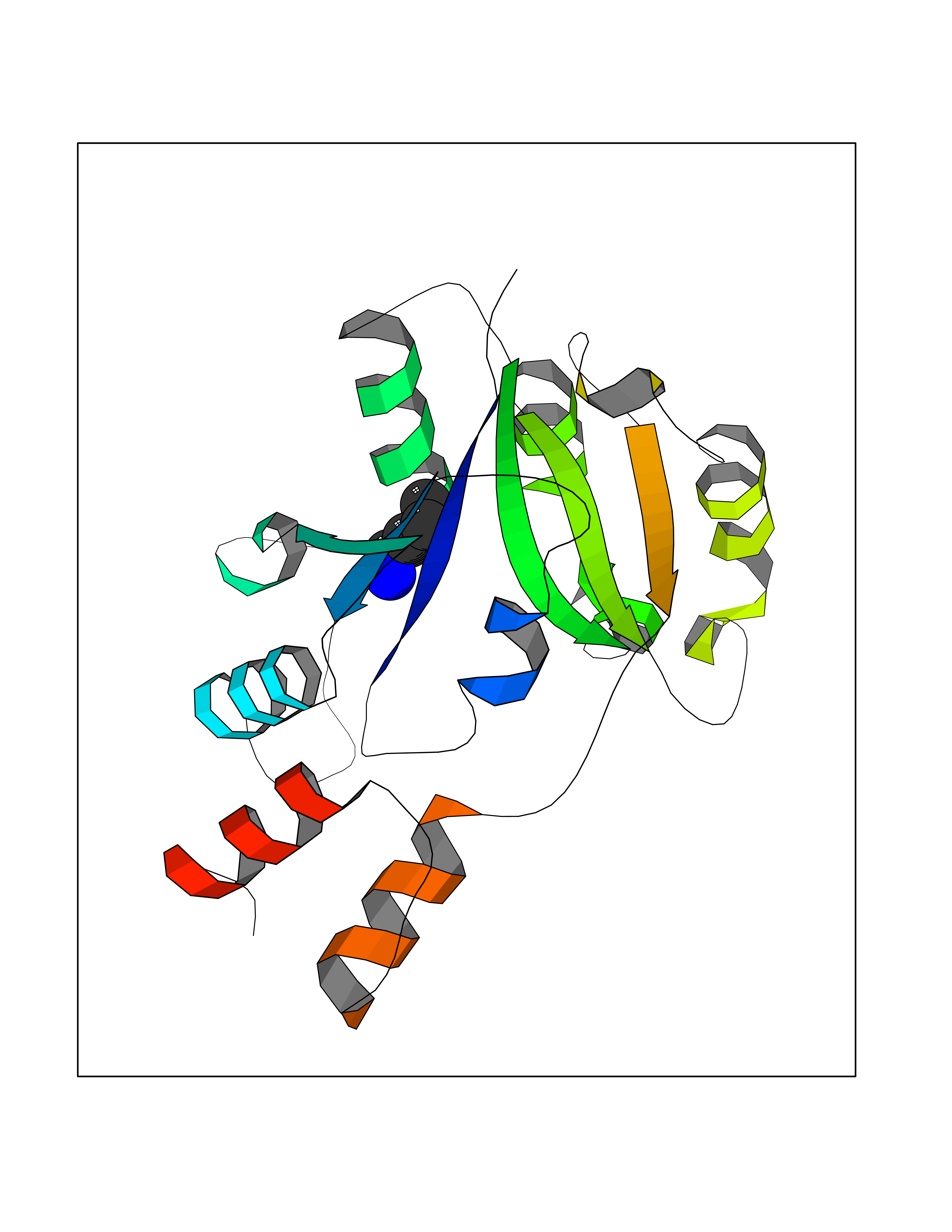 Molscript Map 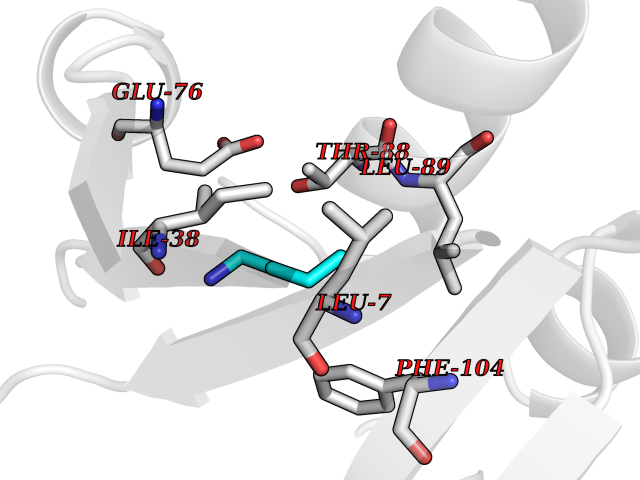 Pymol Map 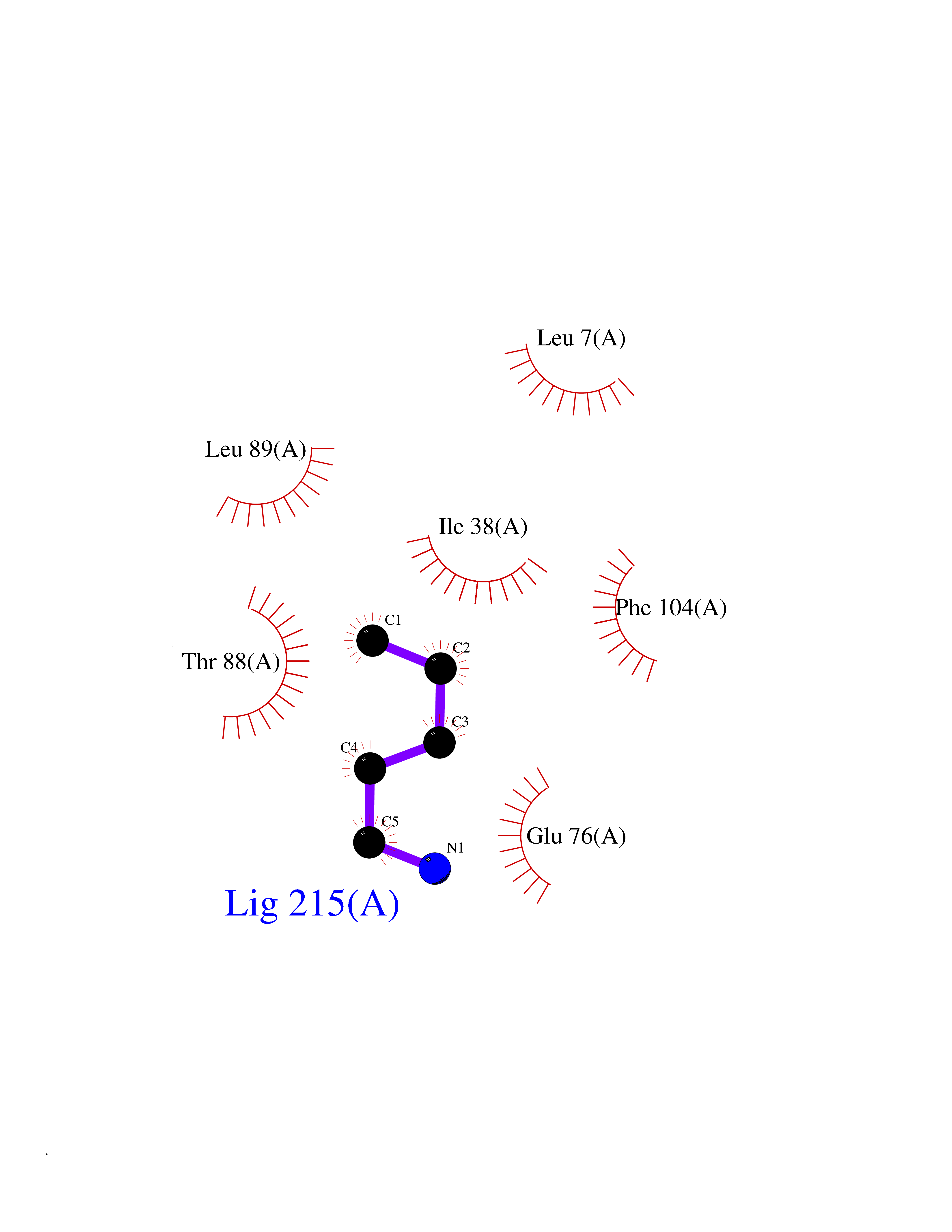 Ligplot Map 3D Binding mode Sequence MKSLQALFGGTFDPVHYGHLKPVETLANLIGLTRVTIIPNNVPPHRPQPEANSVQRKHMLELAIADKPLFTLDERELKRNAPSYTAQTLKEWRQEQGPDVPLAFIIGQDSLLTFPTWYEYETILDNAHLIVCRRPGYPLEMAQPQYQQWLEDHLTHNPEDLHLQPAGKIYLAETPWFNISATIIRERLQNGESCEDLLPEPVLTYINQQGLYR Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Vitamin D3 receptor (VDR) | 3B0T | 4.77 | |
Target general information Gen name VDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin D(3) receptor; Nuclear vitamin D receptor; Nuclear receptor subfamily 1 group I member 1; NR1I1; 1,25-dihydroxyvitamin D3 receptor Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Enters the nucleus upon vitamin D3 binding where it forms heterodimers with the retinoid X receptor/RXR. The VDR-RXR heterodimers bind to specific response elements on DNA and activate the transcription of vitamin D3-responsive target genes. Plays a central role in calcium homeostasis. Nuclear receptor for calcitriol, the active form of vitamin D3 which mediates the action of this vitamin on cells. Related diseases Rickets vitamin D-dependent 2A (VDDR2A) [MIM:277440]: A disorder of vitamin D metabolism resulting in severe rickets, hypocalcemia and secondary hyperparathyroidism. Most patients have total alopecia in addition to rickets. {ECO:0000269|PubMed:1652893, ECO:0000269|PubMed:17970811, ECO:0000269|PubMed:2177843, ECO:0000269|PubMed:2849209, ECO:0000269|PubMed:28698609, ECO:0000269|PubMed:7828346, ECO:0000269|PubMed:8106618, ECO:0000269|PubMed:8381803, ECO:0000269|PubMed:8392085, ECO:0000269|PubMed:8675579, ECO:0000269|PubMed:8961271, ECO:0000269|PubMed:9005998}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07530; DB08742; DB01436; DB04891; DB00146; DB02300; DB00136; DB00169; DB04540; DB05024; DB11672; DB14635; DB01070; DB06410; DB05295; DB06194; DB00153; DB04796; DB03451; DB00910; DB04258; DB11094 Interacts with P35222; Q09472; Q15648; P50222; Q15788; P26045; P19793; Q13573; Q13501; P04637; Q15645; Q9JLI4; P28700; X5D778; Q96HA8; Q01804; Q96S38; P48443 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 28781 Length 254 Aromaticity 0.07 Instability index 47.69 Isoelectric point 6.15 Charge (pH=7) -3.44 2D Binding mode Binding energy (Kcal/mol) -6.51 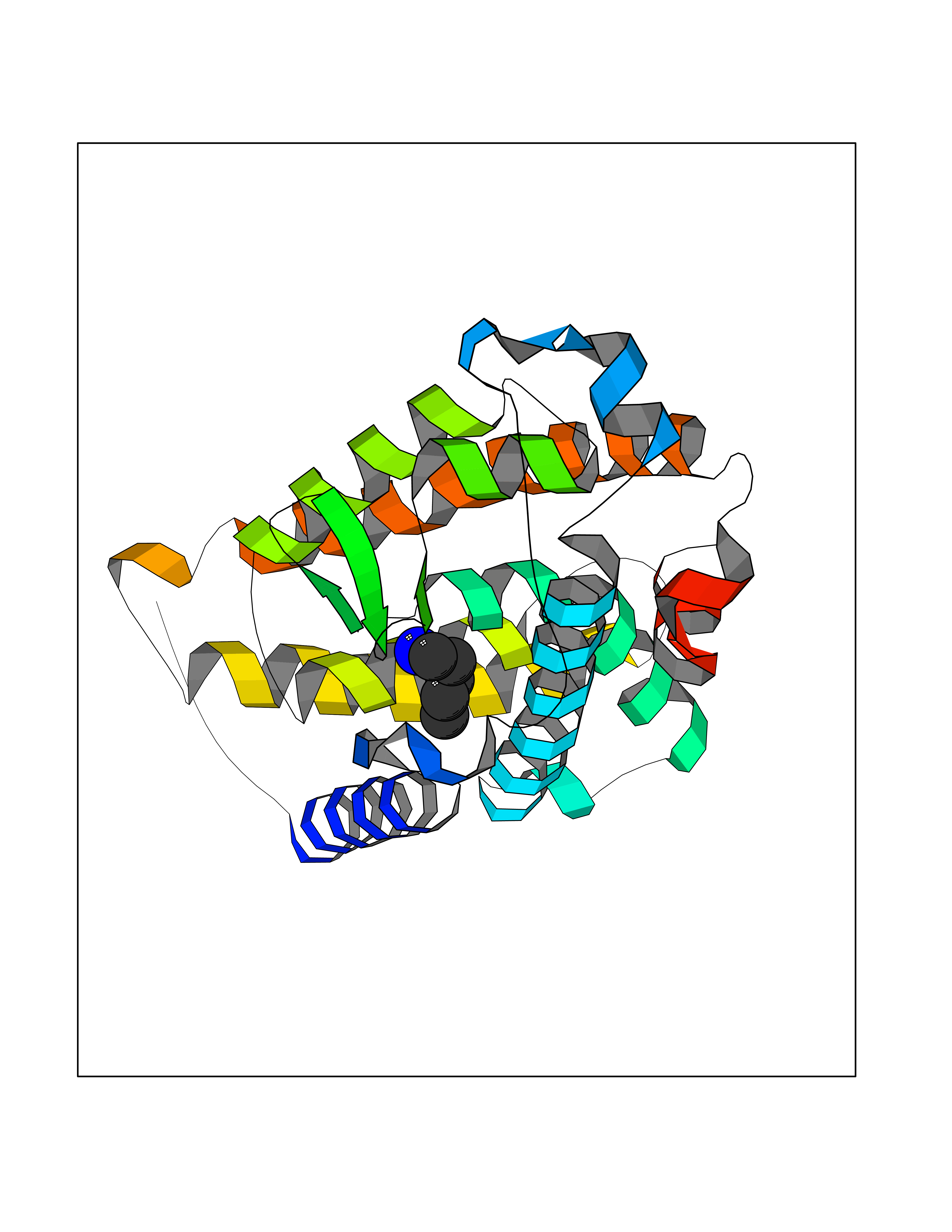 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ALRPKLSEEQQRIIAILLDAHHKTYDPTYSDFCQFRPPVRVNDGGGSVTLELSQLSMLPHLADLVSYSIQKVIGFAKMIPGFRDLTSEDQIVLLKSSAIEVIMLRSNESFTMDDMSWTCGNQDYKYRVSDVTKAGHSLELIEPLIKFQVGLKKLNLHEEEHVLLMAICIVSPDRPGVQDAALIEAIQDRLSNTLQTYIRCRHPPPGSHLLYAKMIQKLADLRSLNEEHSKQYRCLSFQPECSMKLTPLVLEVFG Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Sucrase-isomaltase, intestinal | 3LPP | 4.77 | |
Target general information Gen name SI Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 31 family Biochemical class Hydrolase Function Alpha-1,4-glucosidase activity.Carbohydrate binding.Oligo-1,6-glucosidase activity.Sucrose alpha-glucosidase activity. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00284; DB00747 Interacts with O43451 EC number 3.2.1.10; 3.2.1.48 Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Glycosidase; Hydrolase; Membrane; Multifunctional enzyme; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Signal-anchor; Sulfation; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 97633.9 Length 853 Aromaticity 0.13 Instability index 32 Isoelectric point 5.23 Charge (pH=7) -23.62 2D Binding mode Binding energy (Kcal/mol) -6.51  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LNDPVNVRINCIPEQFPTEGICAQRGCCWRPWNDSLIPWCFFVDNHGYNVQDMTTTSIGVEAKLNRIPSPTLFGNDINSVLFTTQNQTPNRFRFKITDPNNRRYEVPHQYVKEFTGPTVSDTLYDVKVAQNPFSIQVIRKSNGKTLFDTSIGPLVYSDQYLQISARLPSDYIYGIGEQVHKRFRHDLSWKTWPIFTRDQLPGDNNNNLYGHQTFFMCIEDTSGKSFGVFLMNSNAMEIFIQPTPIVTYRVTGGILDFYILLGDTPEQVVQQYQQLVGLPAMPAYWNLGFQLSRWNYKSLDVVKEVVRRNREAGIPFDTQVTDIDYMEDKKDFTYDQVAFNGLPQFVQDLHDHGQKYVIILDPAISIGRRYATYERGNTQHVWINESDGSTPIIGEVWPGLTVYPDFTNPNCIDWWANECSIFHQEVQYDGLWIDMNEVSSFIQGSTKGCNVNKLNYPPFTPDILDKLMYSKTICMDAVQNWGKQYDVHSLYGYSMAIATEQAVQKVFPNKRSFILTRSTFAGSGRHAAHWLGDNTASWEQMEWSITGMLEFSLFGIPLVGADICGFVAETTEELCRRWMQLGAFYPFSRNHNSDGYEHQDPAFFGQNSLLVKSSRQYLTIRYTLLPFLYTLFYKAHVFGETVARPVLHEFYEDTNSWIEDTEFLWGPALLITPVLKQGADTVSAYIPDAIWYDYESGAKRPWRKQRVDMYLPADKIGLHLRGGYIIPIQEPDVTTTASRKNPLGLIVALGENNTAKGDFFWDDGETKDTIQNGNYILYTFSVSNNTLDIVCTHSSYQEGTTLAFQTVKILGLTDSVTEVRVAEHSNFTYDASNQVLLIADLKLNLGRNFSVQW Hydrogen bonds contact Hydrophobic contact | ||||