Job Results:
Ligand
Structure
Job ID
5c75d0793a5c7fa3ef7fd4eba45db829
Job name
NA
Time
2026-02-27 11:58:42
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Neuronal acetylcholine receptor beta-4 (CHRNB4) | 6PV7 | 5.09 | |
Target general information Gen name CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNB4; Beta-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-4/CHRNB4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. {ECO:0000269|PubMed:20054297, ECO:0000269|PubMed:23622243, ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}. The disease may be caused by variants affecting the gene represented in this entry. Defects of SETD2 are associated with loss of DNA methylation at non-promoter regions (PubMed:23792563). SETD2 defects lead to aberrant and reduced nucleosome compaction and chromatin association of key replication proteins, such as MCM7 and DNA polymerase delta, leading to hinder replication fork progression and prevent loading of RAD51 homologous recombination repair factor at DNA breaks (PubMed:25728682). {ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}.; DISEASE: Luscan-Lumish syndrome (LLS) [MIM:616831]: An autosomal dominant syndrome with a variable phenotype. Clinical features include macrocephaly, distinctive facial appearance, postnatal overgrowth, various degrees of learning difficulties, autism spectrum disorder, and intellectual disability. {ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24852293, ECO:0000269|PubMed:26084711, ECO:0000269|PubMed:27317772}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute lymphoblastic (ALL) [MIM:613065]: A subtype of acute leukemia, a cancer of the white blood cells. ALL is a malignant disease of bone marrow and the most common malignancy diagnosed in children. The malignant cells are lymphoid precursor cells (lymphoblasts) that are arrested in an early stage of development. The lymphoblasts replace the normal marrow elements, resulting in a marked decrease in the production of normal blood cells. Consequently, anemia, thrombocytopenia, and neutropenia occur to varying degrees. The lymphoblasts also proliferate in organs other than the marrow, particularly the liver, spleen, and lymphnodes. {ECO:0000269|PubMed:24509477, ECO:0000269|PubMed:24662245}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16314571, ECO:0000269|PubMed:24509477}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Intellectual developmental disorder, autosomal dominant 70 (MRD70) [MIM:620157]: An autosomal dominant disorder characterized by mild global developmental delay, moderately impaired intellectual disability with speech difficulties, and behavioral abnormalities. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rabin-Pappas syndrome (RAPAS) [MIM:620155]: An autosomal dominant neurodevelopmental disorder characterized by severely impaired global development, intellectual disability, microcephaly, facial dysmorphism, and variable congenital anomalies affecting the skeletal, genitourinary, cardiac, and other organ systems. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00237; DB00565; DB00514; DB07720; DB00898; DB00472; DB01227; DB00184; DB01090; DB00202 Interacts with Q6FHY5 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -6.94 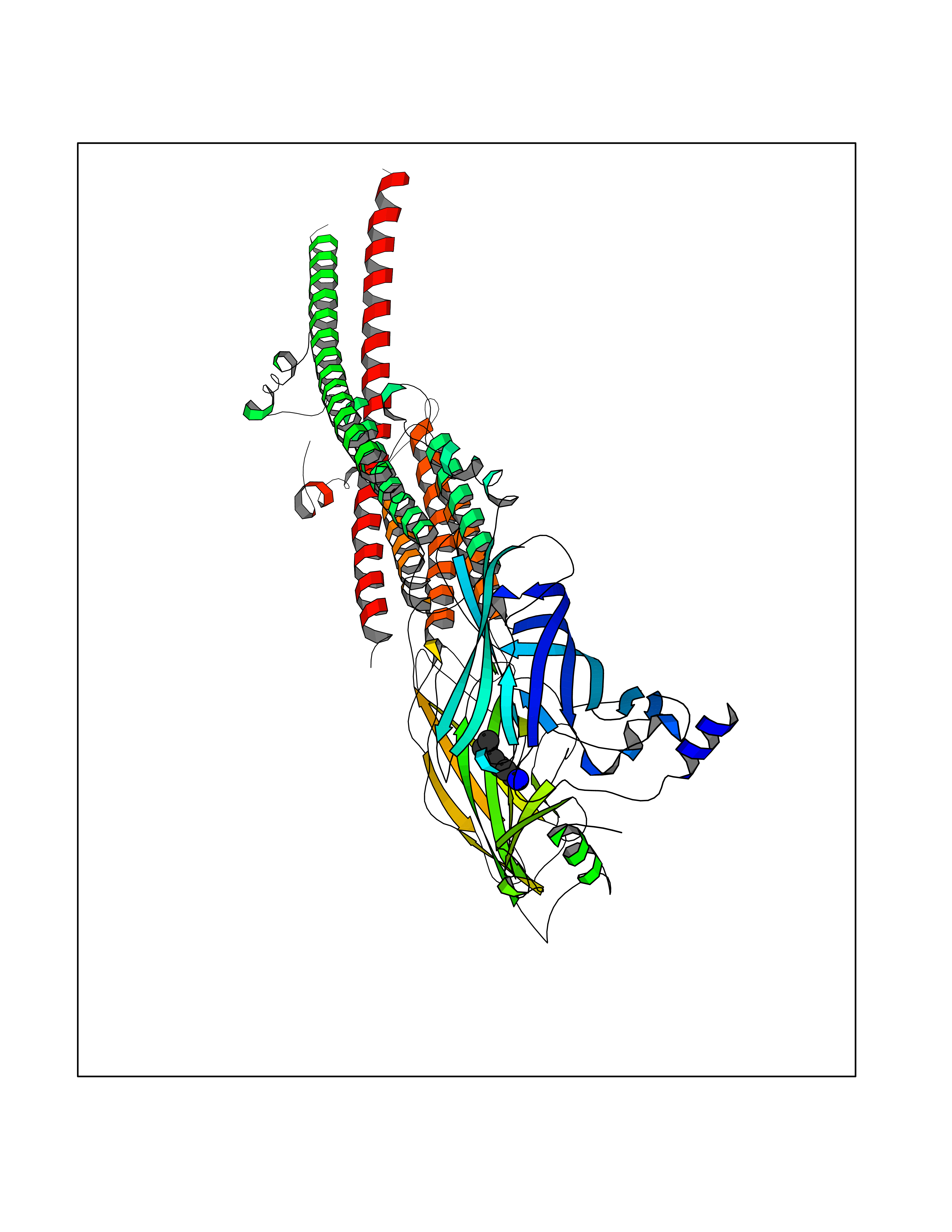 Molscript Map 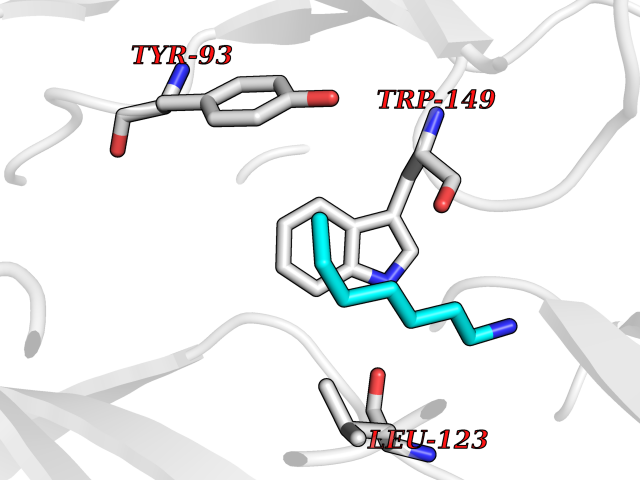 Pymol Map 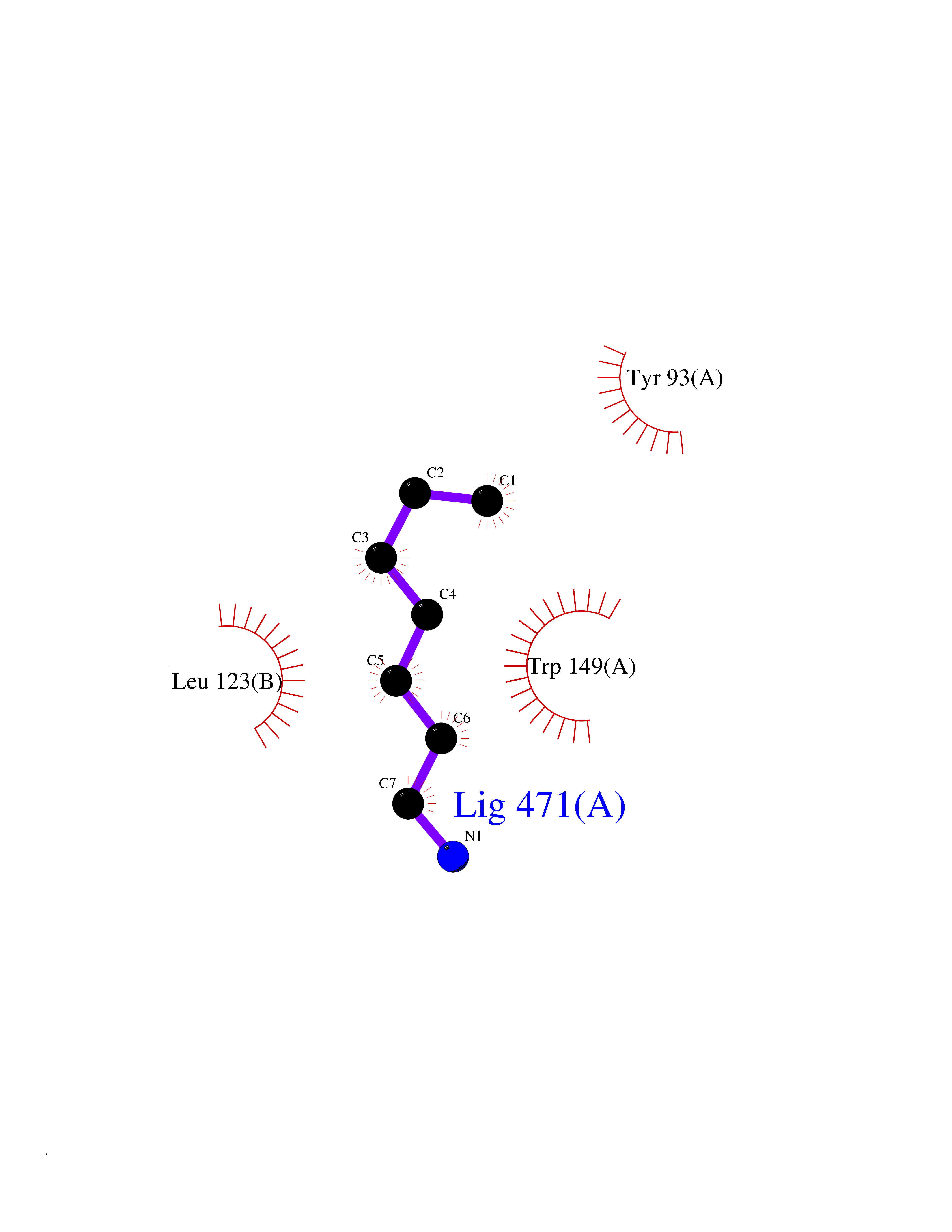 Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Cytosolic 10-formyltetrahydrofolate dehydrogenase | 2CFI | 5.08 | |
Target general information Gen name ALDH1L1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FTHFD Protein family GART family; Aldehyde dehydrogenase family, ALDH1L subfamily Biochemical class Oxidoreductase Function Aldehyde dehydrogenase (NAD) activity.Catalytic activity.Formyltetrahydrofolate dehydrogenase activity.Hydroxymethyl-, formyl- and related transferase activity. Related diseases Developmental and epileptic encephalopathy 39 with leukodystrophy (DEE39) [MIM:612949]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE39 is characterized by global hypomyelination of the central nervous system, with the gray matter appearing relatively unaffected. Inheritance is autosomal recessive. {ECO:0000269|PubMed:19641205, ECO:0000269|PubMed:24515575}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00116 Interacts with Q3SY69; Q92624 EC number 1.5.1.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; NADP; One-carbon metabolism; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33869.5 Length 308 Aromaticity 0.08 Instability index 30.42 Isoelectric point 6.09 Charge (pH=7) -3.83 2D Binding mode Binding energy (Kcal/mol) -6.93 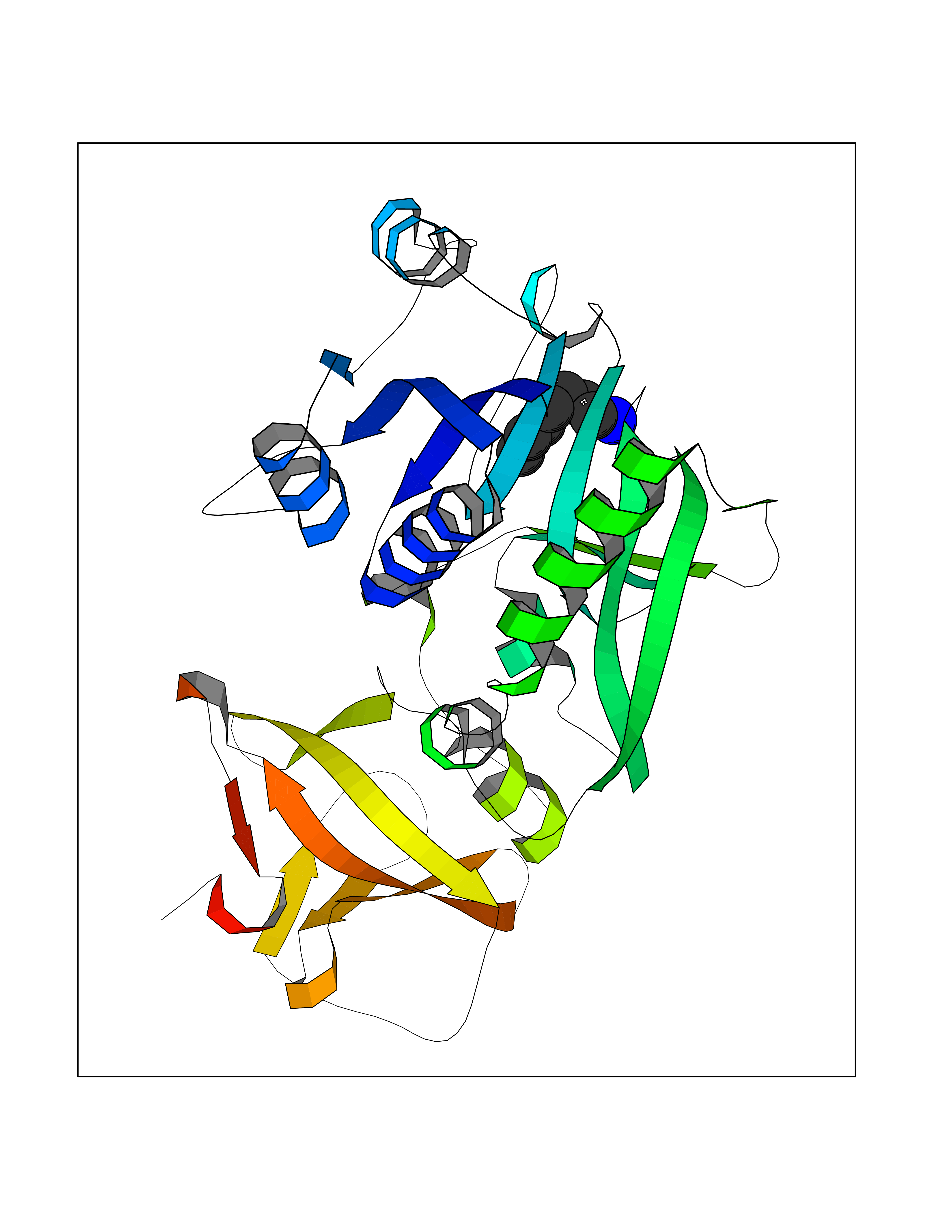 Molscript Map 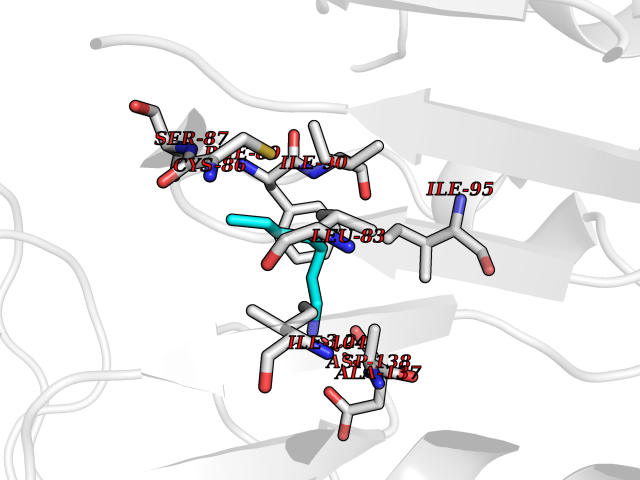 Pymol Map 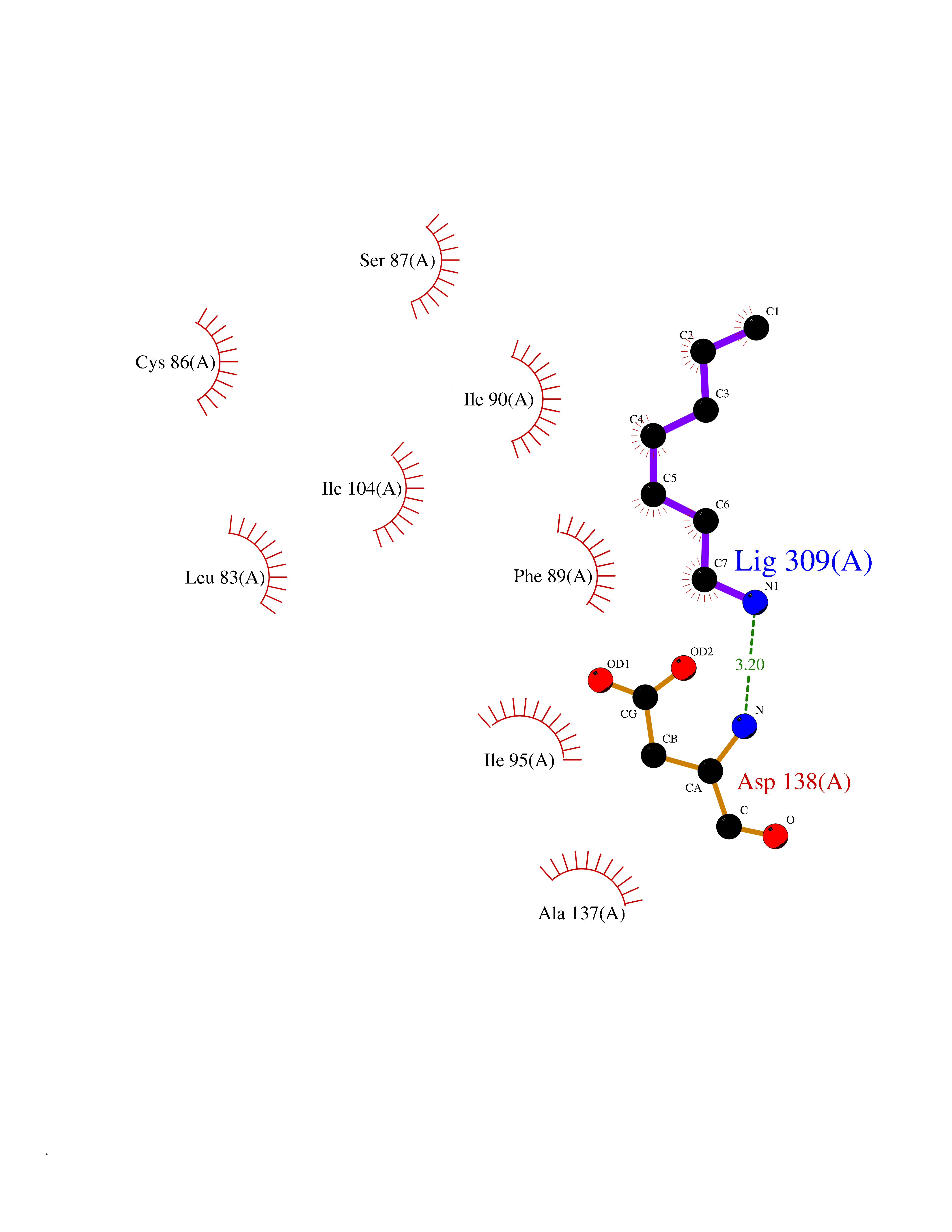 Ligplot Map 3D Binding mode Sequence SMKIAVIGQSLFGQEVYCHLRKEGHEVVGVFTVPDKDGKADPLGLEAEKDGVPVFKYSRWRAKGQALPDVVAKYQALGAELNVLPFCSQFIPMEIISAPRHGSIIYHPSLLPRHRGASAINWTLIHGDKKGGFSIFWADDGLDTGDLLLQKECEVLPDDTVSTLYNRFLFPEGIKGMVQAVRLIAEGKAPRLPQPEEGATYEGIQKKETAKINWDQPAEAIHNWIRGNDKVPGAWTEACEQKLTFFNSTLNTSGLVPEGDALPIPGAHRPGVVTKAGLILFGNDDKMLLVKNIQLEDGKMILASNFFK Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Arachidonate 5-lipoxygenase (5-LOX) | 3V99 | 5.08 | |
Target general information Gen name ALOX5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LOG5; 5-lipoxygenase; 5-LO Protein family Lipoxygenase family Biochemical class Oxygenase Function Catalyzes the first step in leukotriene biosynthesis, and thereby plays a role in inflammatory processes. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14001; DB00233; DB01014; DB09061; DB14002; DB11994; DB00586; DB00711; DB12010; DB01892; DB00159; DB04725; DB00179; DB00939; DB14009; DB00244; DB01017; DB05431; DB00471; DB09285; DB14011; DB11133; DB13168; DB02709; DB13174; DB00795; DB00163; DB00744 Interacts with Q8IYJ2-2; Q6PII3; Q6P2R3; Q8IYX8-2; Q96MT8; Q96MT8-3; Q14019; P09769; O43716; P08631; Q6UWX4; P14061; P31025; Q9Y6D9; P50221; Q6FHY5; Q86Y26; A6NGQ2; P17612; Q04864; Q7Z699; Q8N0S2; Q9P0N9; P07947 EC number EC 1.13.11.34 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cytoplasm; Dioxygenase; Direct protein sequencing; Hydrolase; Iron; Leukotriene biosynthesis; Lipid metabolism; Membrane; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60590.7 Length 523 Aromaticity 0.11 Instability index 41.73 Isoelectric point 5.69 Charge (pH=7) -10.99 2D Binding mode Binding energy (Kcal/mol) -6.93 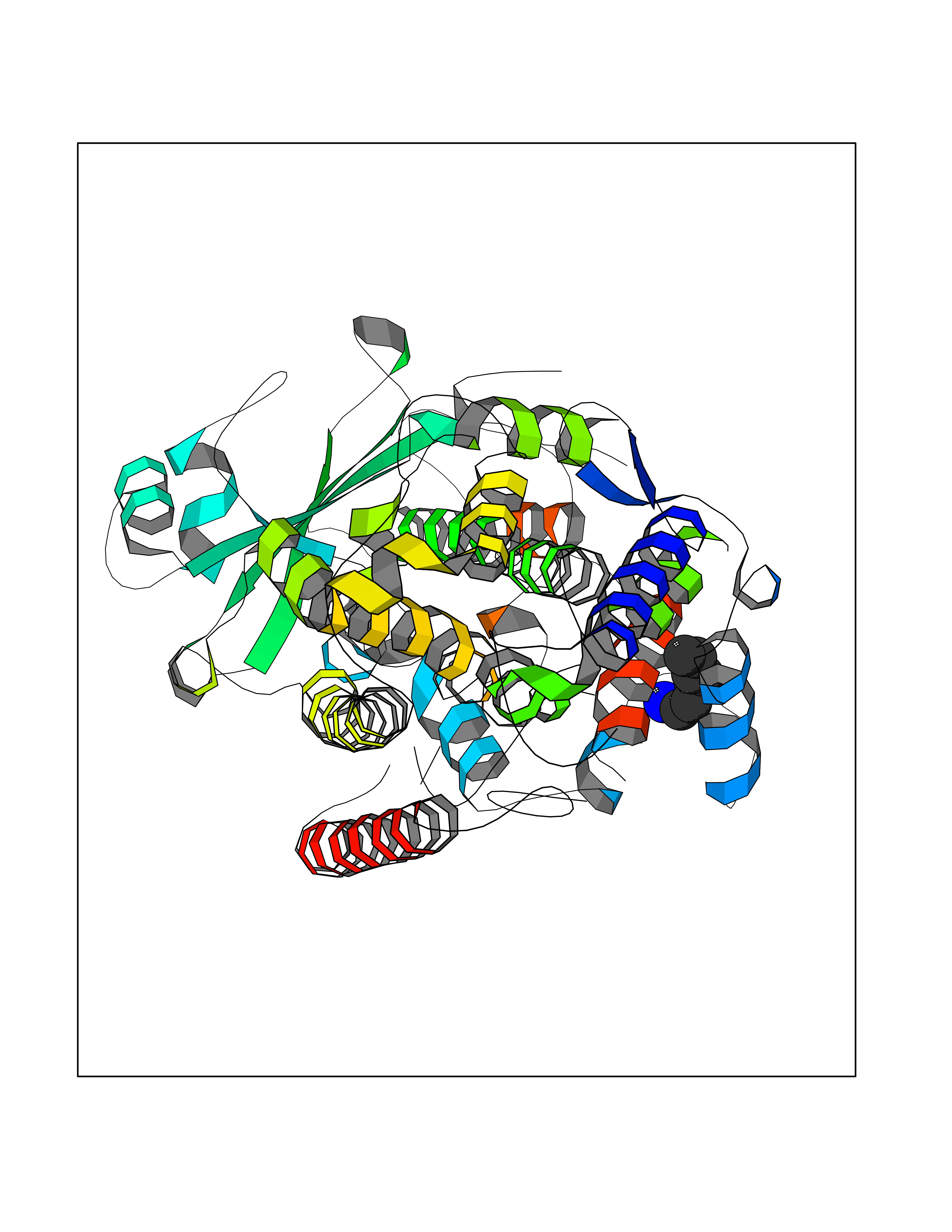 Molscript Map 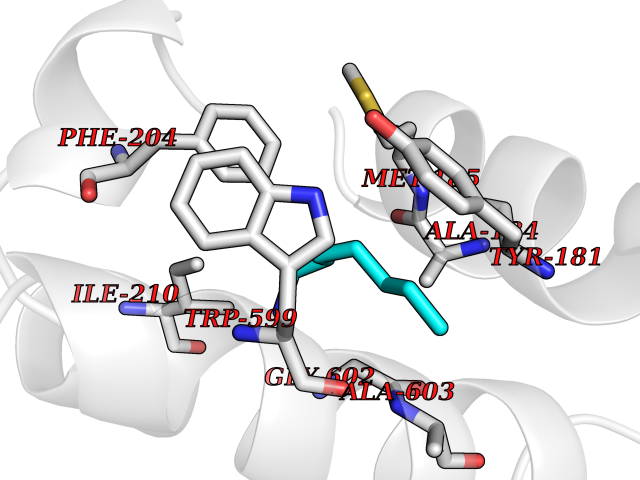 Pymol Map 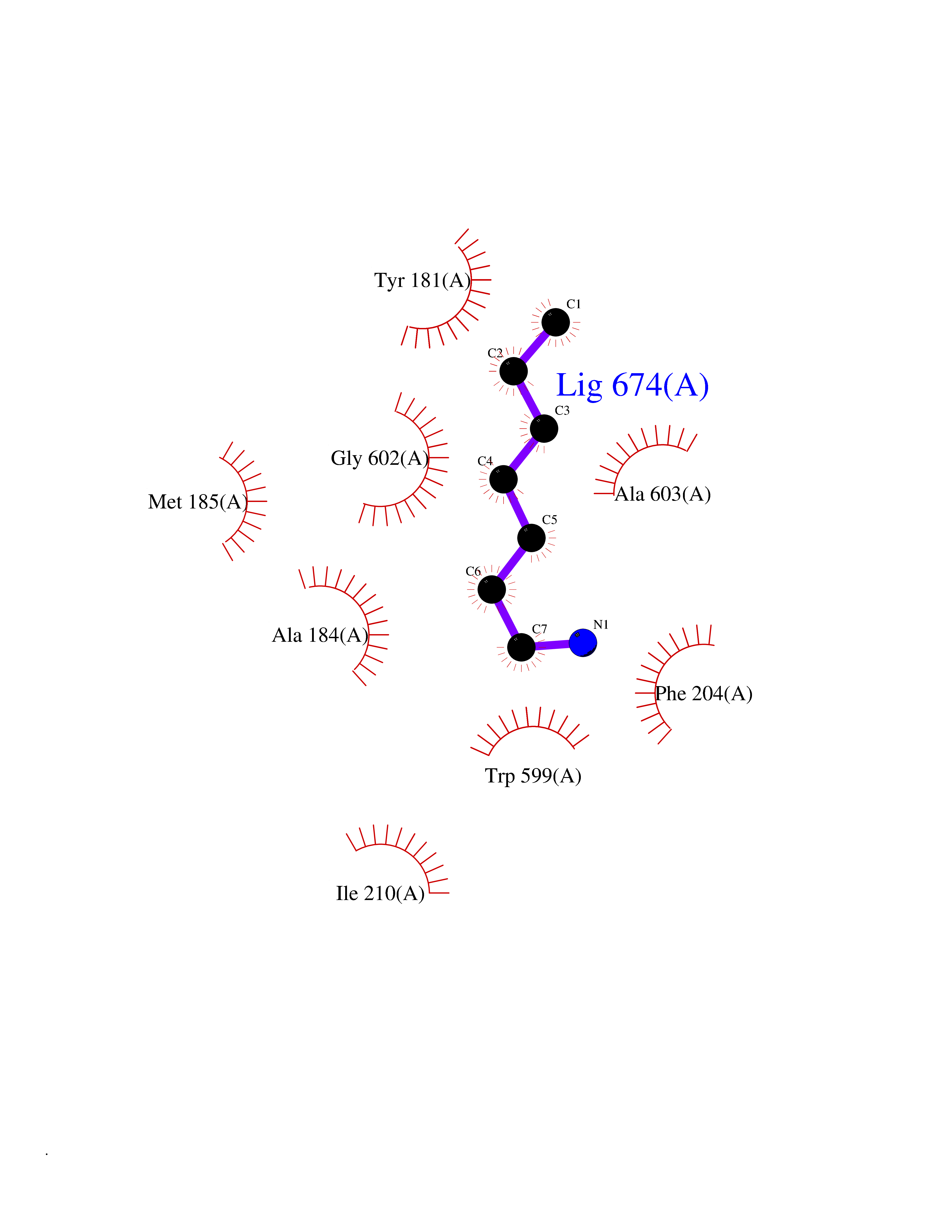 Ligplot Map 3D Binding mode Sequence RDGRAKLARDDQIHILKQHRRKELETRQKQYRWMEWNPGFPLSIDAKCHKDLPRDIQFDSFVLNYSKAMENLFQSSWNDFADFEKIFVKISNTISERVMNHWQEDLMFGYQFLNGANPVLIRRCTELPEKLPVTTEMVECSLERQLSLEQEVQQGNIFIVDFELLDGIDCTLQFLAAPICLLYKNLANKIVPIAIQLNQIPGDENPIFLPSDAKYDWLLAKIWVRSSDFHVHQTITHLLRTHLVSEVFGIAMYRQLPAVHPIFKLLVAHVRFTIAINTKAREQGGHVQMVQRAMKDLTYASLCFPEAIKARGMESKEDIPYYFYRDDGLLVWEAIRTFTAEVVDIYYEGDQVVEEDPELQDFVNDVYVYGMRGRKSSGFPKSVKSREQLSEYLTVVIFTASAQHAAVNFGQYDWASWIPNAPPTMRAPPPTAKGVVTIEQIVDTLPDRGRSCWHLGAVWALSQFELFLGMYPEEHFIEKPVKEAMARFRKNLEAIVSVIAERNENLQLPYYYLDPDRIPNSVA Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Tankyrase-2 (TNKS-2) | 3U9H | 5.08 | |
Target general information Gen name TNKS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tankyrase-related protein; Tankyrase-like protein; Tankyrase II; TRF1-interacting ankyrin-related ADP-ribose polymerase 2; TNKL; TANK2; Protein poly-ADP-ribosyltransferase tankyrase-2; Poly [ADP-ribos Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Acts as an activator of the Wnt signaling pathway by mediating poly-ADP-ribosylation of AXIN1 and AXIN2, 2 key components of the beta-catenin destruction complex: poly-ADP-ribosylated target proteins are recognized by RNF146, which mediates their ubiquitination and subsequent degradation. Also mediates poly-ADP-ribosylation of BLZF1 and CASC3, followed by recruitment of RNF146 and subsequent ubiquitination. Mediates poly-ADP-ribosylation of TERF1, thereby contributing to the regulation of telomere length. Stimulates 26S proteasome activity. Poly-ADP-ribosyltransferase involved in various processes such as Wnt signaling pathway, telomere length and vesicle trafficking. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O15084; Q7Z6K5-1; O15169; Q9NWV8; P11274; Q13698; Q9NRI5; Q6V0I7; Q9NWT6; P14652; Q9UIQ6; Q14980; Q9BZL4; Q92698; P78314; O43815; P54274; Q9C0C2; Q9UHP3; Q06649 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; ADP-ribosylation; ANK repeat; Chromosome; Cytoplasm; Glycosyltransferase; Golgi apparatus; Hydroxylation; Membrane; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Proteomics identification; Reference proteome; Repeat; Telomere; Transferase; Ubl conjugation; Wnt signaling pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 23695.5 Length 208 Aromaticity 0.11 Instability index 47.61 Isoelectric point 8.28 Charge (pH=7) 2.88 2D Binding mode Binding energy (Kcal/mol) -6.93  Molscript Map 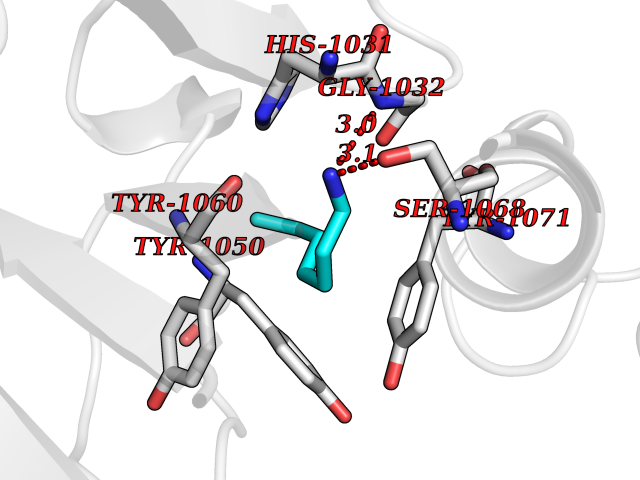 Pymol Map 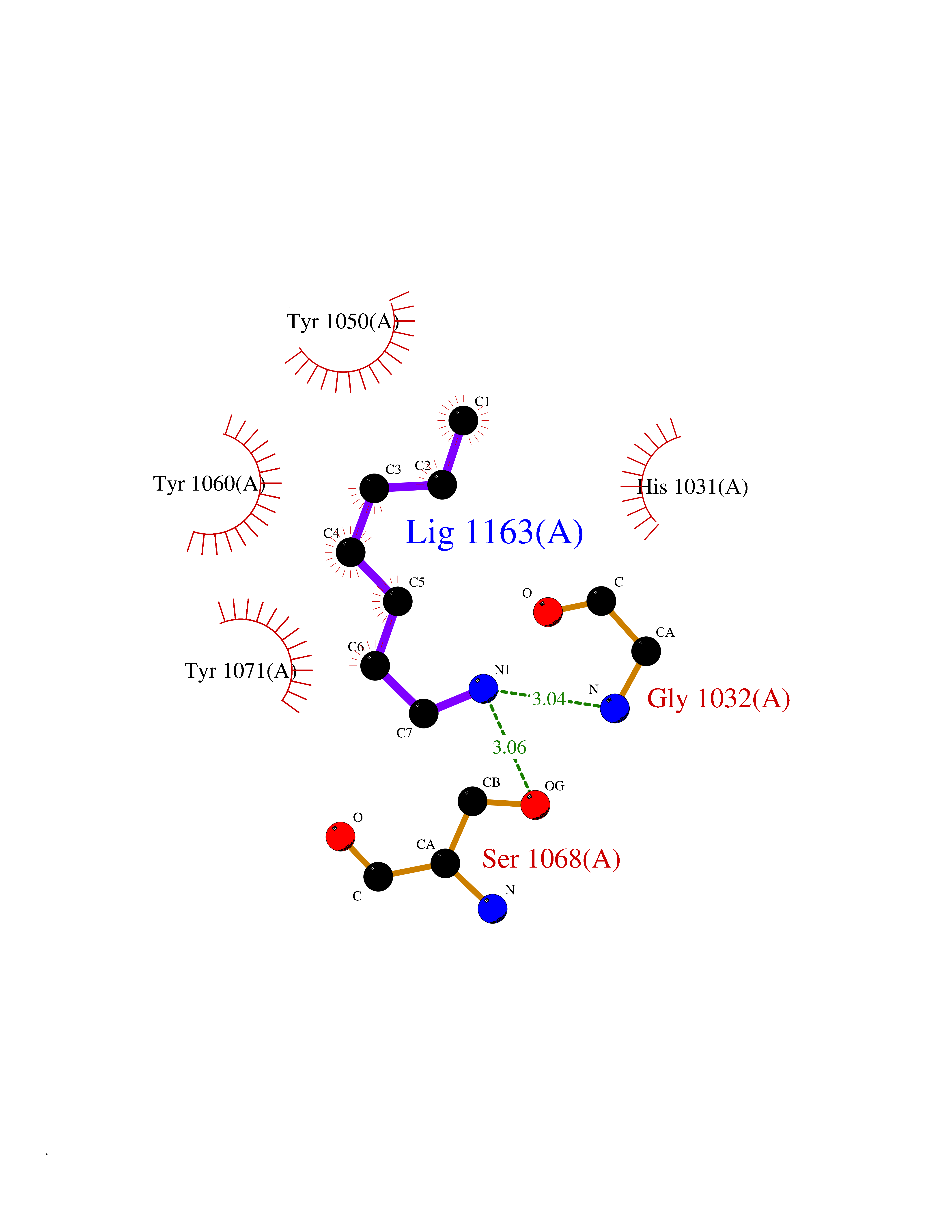 Ligplot Map 3D Binding mode Sequence GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEVSEENHNHANERMLFHGSPFVNAIIHKGFDERHAYIGGMFGAGIYFAENSSKSNQYVYGIGGGTGCPVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Lysine N-methyltransferase 3B (NSD1) | 3OOI | 5.08 | |
Target general information Gen name NSD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor-binding SET domain-containing protein 1; NR-binding SET domain-containing protein; NR-binding SET domain containing protein; KMT3B; Hypothetical protein FLJ22263 similar to nuclear re Protein family Class V-like SAM-binding methyltransferase superfamily Biochemical class Methyltransferase Function Preferentially methylates 'Lys-36' of histone H3 and 'Lys-20' of histone H4 (in vitro). Transcriptional intermediary factor capable of both negatively or positively influencing transcription, depending on the cellular context. Histone methyltransferase. Related diseases Sotos syndrome (SOTOS) [MIM:117550]: An autosomal dominant, childhood overgrowth syndrome characterized by pre- and postnatal overgrowth, developmental delay, intellectual disability, advanced bone age, and abnormal craniofacial morphology including macrodolichocephaly with frontal bossing, frontoparietal sparseness of hair, apparent hypertelorism, downslanting palpebral fissures, and facial flushing. Common oral findings include: premature eruption of teeth; high, arched palate; pointed chin and, more rarely, prognathism. {ECO:0000269|PubMed:11896389, ECO:0000269|PubMed:12464997, ECO:0000269|PubMed:12807965, ECO:0000269|PubMed:14997421}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Beckwith-Wiedemann syndrome (BWS) [MIM:130650]: A disorder characterized by anterior abdominal wall defects including exomphalos (omphalocele), pre- and postnatal overgrowth, and macroglossia. Additional less frequent complications include specific developmental defects and a predisposition to embryonal tumors. {ECO:0000269|PubMed:14997421}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A chromosomal aberration involving NSD1 is found in childhood acute myeloid leukemia. Translocation t(5;11)(q35;p15.5) with NUP98.; DISEASE: A chromosomal aberration involving NSD1 is found in an adult form of myelodysplastic syndrome (MDS). Insertion of NUP98 into NSD1 generates a NUP98-NSD1 fusion product. {ECO:0000269|PubMed:15382262}. Drugs (DrugBank ID) NA Interacts with Q13283; Q16778; Q04206; O95994; Q86Z20; A8MQ03; Q3LI66; Q99750 EC number EC 2.1.1.43 Uniprot keywords 3D-structure; Activator; Alternative splicing; Chromatin regulator; Chromosomal rearrangement; Chromosome; Disease variant; Isopeptide bond; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; Repeat; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 26487.9 Length 232 Aromaticity 0.08 Instability index 37.29 Isoelectric point 8.1 Charge (pH=7) 2.69 2D Binding mode Binding energy (Kcal/mol) -6.93 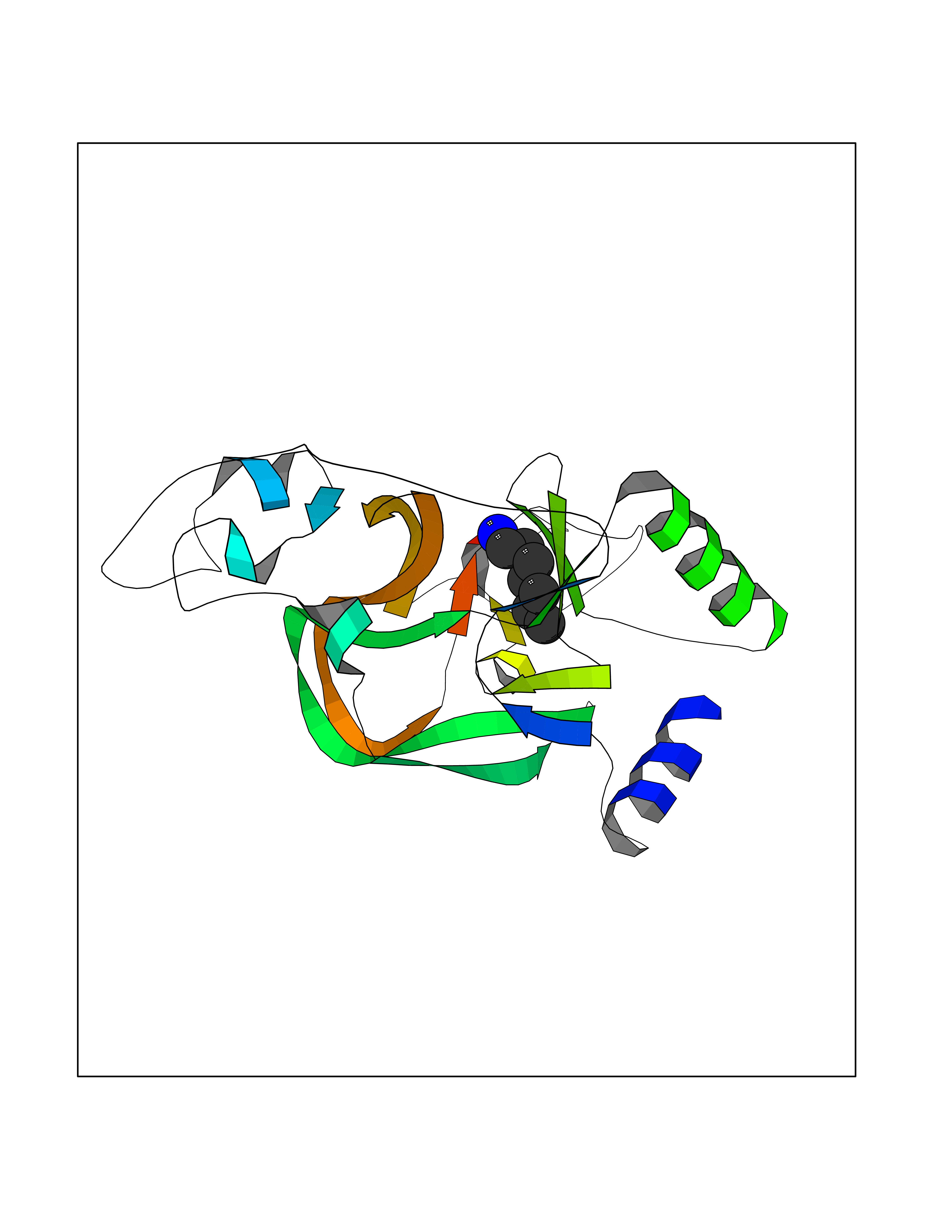 Molscript Map 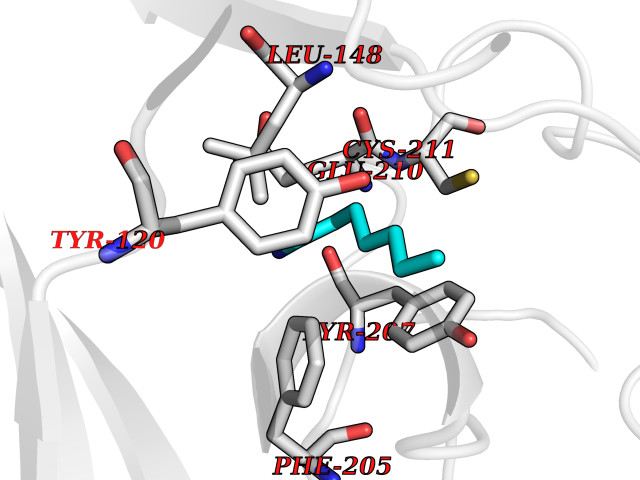 Pymol Map 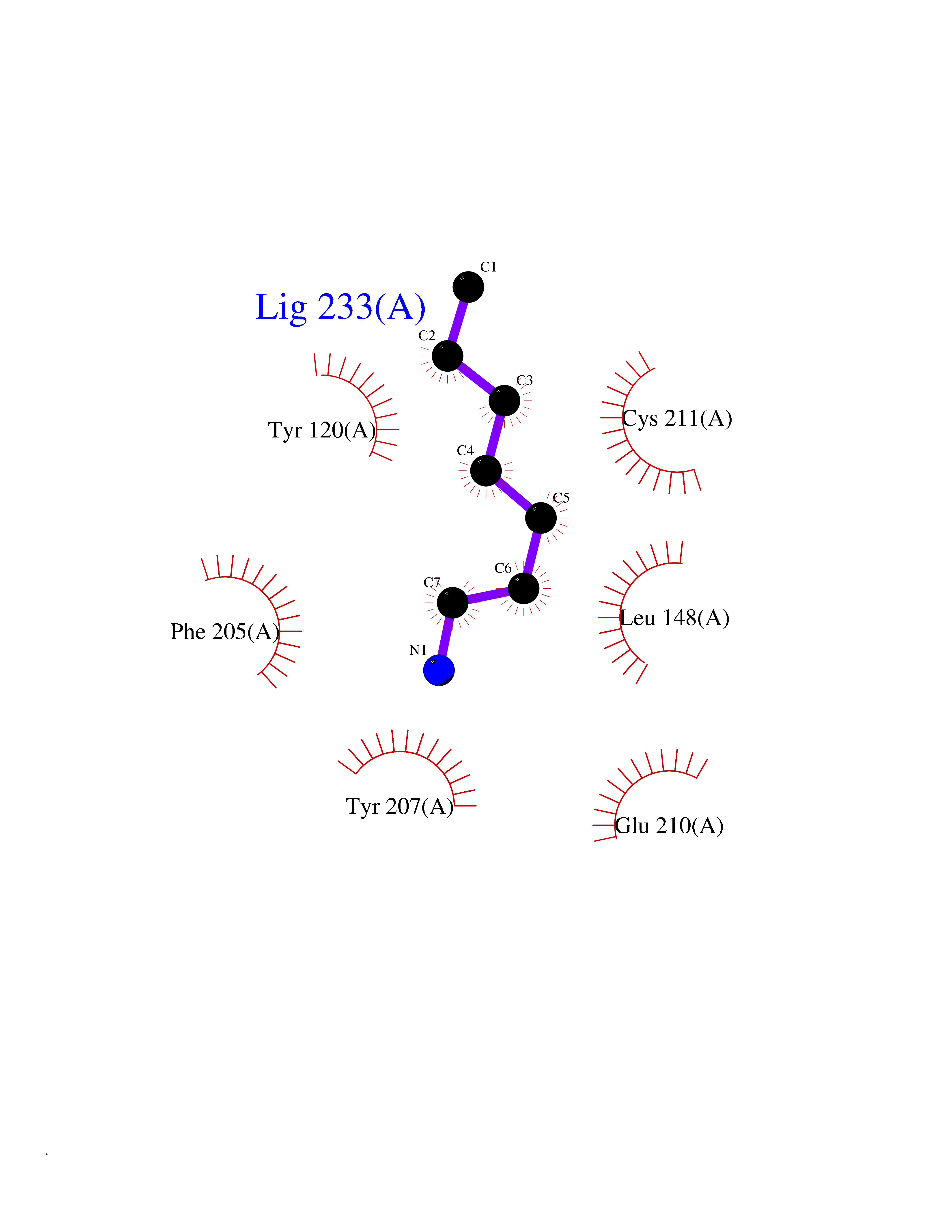 Ligplot Map 3D Binding mode Sequence SKELRQLQEDRKNDKKPPPYKHIKVNRPIGRVQIFTADLSEIPRCNCKATDENPCGIDSECINRMLLYECHPTVCPAGGRCQNQCFSKRQYPEVEIFRTLQRGWGLRTKTDIKKGEFVNEYVGELIDEEECRARIRYAQEHDITNFYMLTLDKDRIIDAGPKGNYARFMNHCCQPNCETQKWSVNGDTRVGLFALSDIKAGTELTFNYNLECLGNGKTVCKCGAPNCSGFLG Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Fibrinogen gamma chain | 1DUG | 5.07 | |
Target general information Gen name FGG Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PRO2061 Protein family NA Biochemical class transferase Function Cell adhesion molecule binding.Metal ion binding.Protein binding, bridging.Protein homodimerization activity.Receptor binding.Structural molecule activity. Related diseases Congenital afibrinogenemia (CAFBN) [MIM:202400]: Rare autosomal recessive disorder is characterized by bleeding that varies from mild to severe and by complete absence or extremely low levels of plasma and platelet fibrinogen. {ECO:0000269|PubMed:25427968}. The disease is caused by variants affecting the gene represented in this entry. Patients with congenital fibrinogen abnormalities can manifest different clinical pictures. Some cases are clinically silent, some show a tendency toward bleeding and some show a predisposition for thrombosis with or without bleeding.; DISEASE: Dysfibrinogenemia, congenital (DYSFIBRIN) [MIM:616004]: A disorder characterized by qualitative abnormalities (dysfibrinogenemia) of the circulating fibrinogen. Affected individuals are frequently asymptomatic, but some patients have bleeding diathesis, thromboembolic complications, or both. In some cases, dysfibrinogenemia is associated with low circulating fibrinogen levels (hypodysfibrinogenemia). {ECO:0000269|PubMed:15632207, ECO:0000269|PubMed:2257302, ECO:0000269|PubMed:2976995, ECO:0000269|PubMed:3708159}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00009; DB11571; DB00364; DB11300; DB11572 Interacts with P75358 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Calcium; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hemostasis; Isopeptide bond; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Sulfation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 54170 Length 468 Aromaticity 0.12 Instability index 35.94 Isoelectric point 6.08 Charge (pH=7) -7.16 2D Binding mode Binding energy (Kcal/mol) -6.92 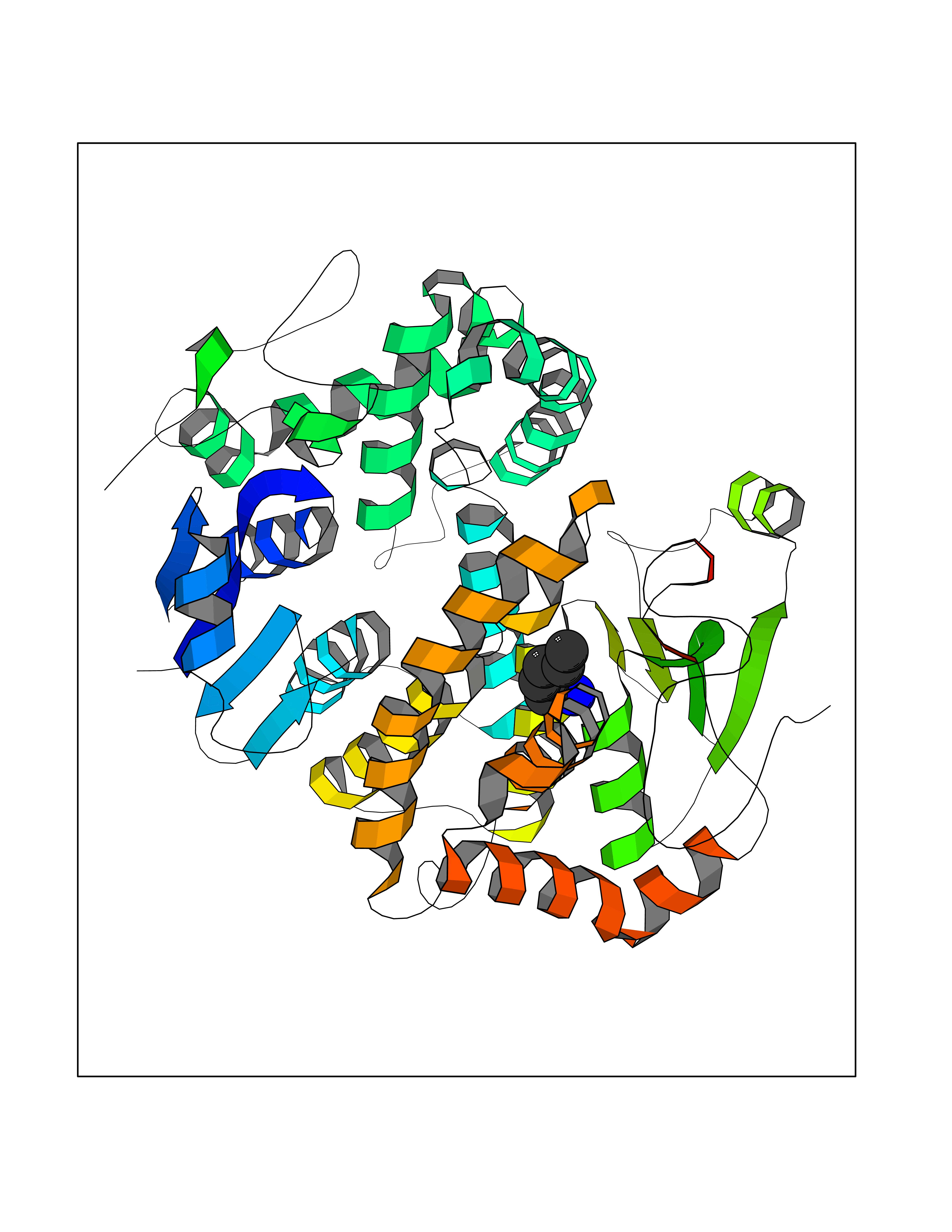 Molscript Map 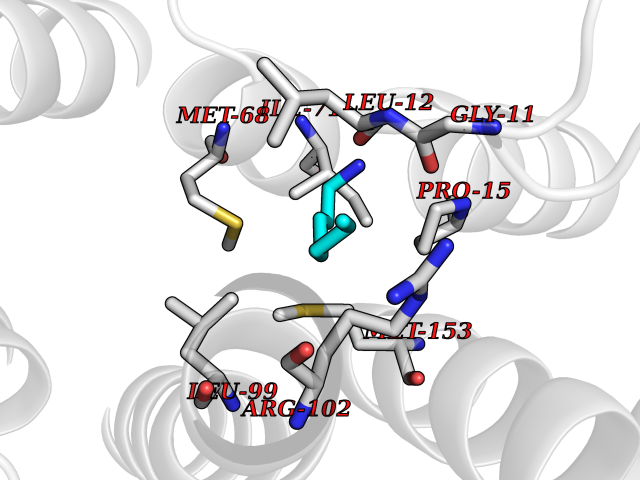 Pymol Map 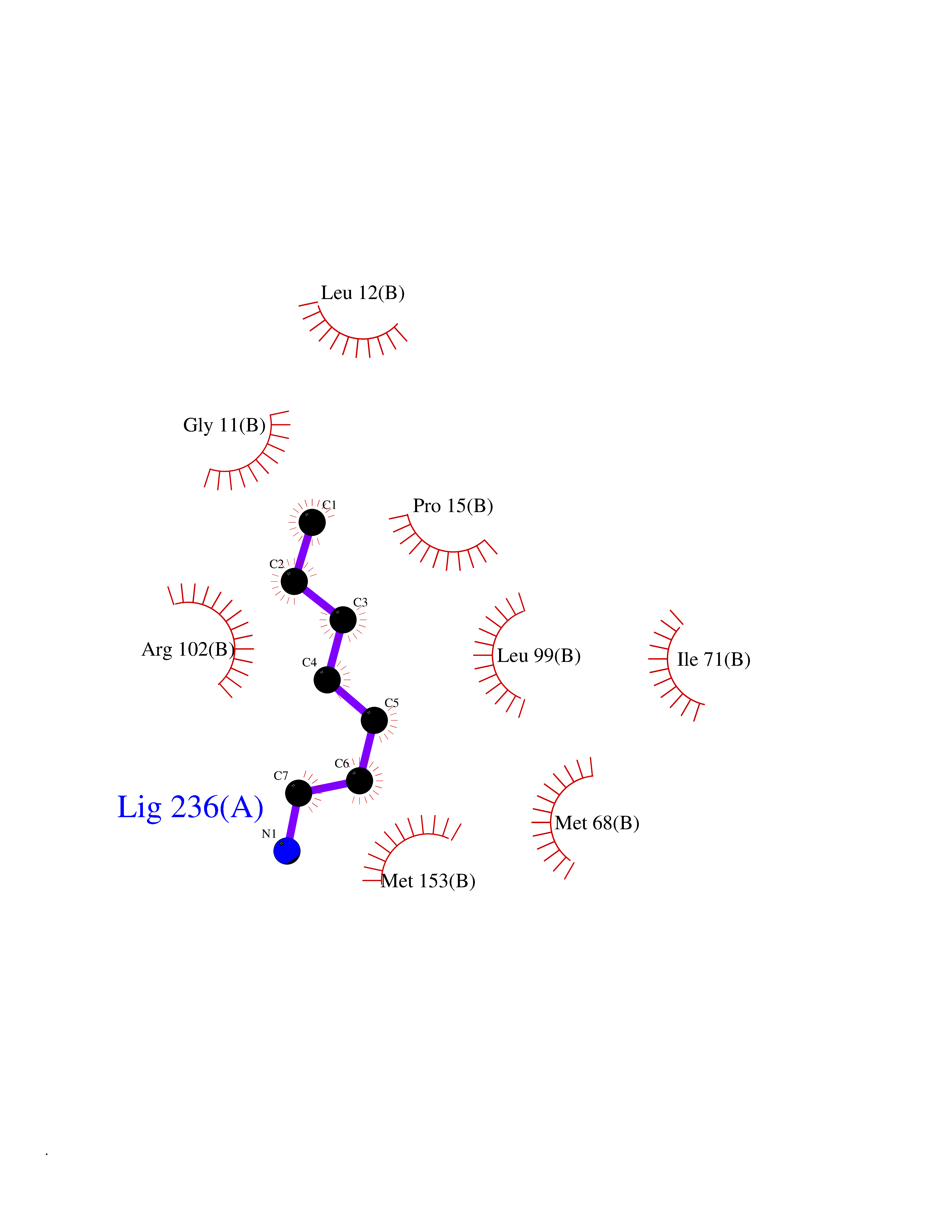 Ligplot Map 3D Binding mode Sequence SPILGYWKIKGLVQPTRLLLEYLEEKYEEHLYERDEGDKWRNKKFELGLEFPNLPYYIDGDVKLTQSMAIIRYIADKHNMLGGCPKERAEISMLEGAVLDIRYGVSRIAYSKDFETLKVDFLSKLPEMLKMFEDRLCHKTYLNGDHVTHPDFMLYDALDVVLYMDPMCLDAFPKLVCFKKRIEAIPQIDKYLKSSKYIAWPLQGWQATFGGGDHPPKSDPQQHHLGGAKQAGDVSPILGYWKIKGLVQPTRLLLEYLEEKYEEHLYERDEGDKWRNKKFELGLEFPNLPYYIDGDVKLTQSMAIIRYIADKHNMLGGCPKERAEISMLEGAVLDIRYGVSRIAYSKDFETLKVDFLSKLPEMLKMFEDRLCHKTYLNGDHVTHPDFMLYDALDVVLYMDPMCLDAFPKLVCFKKRIEAIPQIDKYLKSSKYIAWPLQGWQATFGGGDHPPKSDPQQHHLGGAKQAGDV Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Histidine decarboxylase (HDC) | 4E1O | 5.07 | |
Target general information Gen name HDC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Human histidine decarboxylase Protein family Group II decarboxylase family Biochemical class Carbon-carbon lyase Function Catalyzes the biosynthesis of histamine from histidine. Related diseases Corticosterone methyloxidase 1 deficiency (CMO-1 deficiency) [MIM:203400]: Autosomal recessive disorder of aldosterone biosynthesis. There are two biochemically different forms of selective aldosterone deficiency be termed corticosterone methyloxidase (CMO) deficiency type 1 and type 2. In CMO-1 deficiency, aldosterone is undetectable in plasma, while its immediate precursor, 18-hydroxycorticosterone, is low or normal. {ECO:0000269|PubMed:11238478, ECO:0000269|PubMed:8439335, ECO:0000269|PubMed:9177280}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Corticosterone methyloxidase 2 deficiency (CMO-2 deficiency) [MIM:610600]: Autosomal recessive disorder of aldosterone biosynthesis. In CMO-2 deficiency, aldosterone can be low or normal, but at the expense of increased secretion of 18-hydroxycorticosterone. Consequently, patients have a greatly increased ratio of 18-hydroxycorticosterone to aldosterone and a low ratio of corticosterone to 18-hydroxycorticosterone in serum. {ECO:0000269|PubMed:12788848, ECO:0000269|PubMed:1346492, ECO:0000269|PubMed:1594605, ECO:0000269|PubMed:9625333, ECO:0000269|PubMed:9814506}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00117; DB00114 Interacts with Q86UW9 EC number EC 4.1.1.22 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Decarboxylase; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 107706 Length 956 Aromaticity 0.1 Instability index 55.17 Isoelectric point 6.23 Charge (pH=7) -9.63 2D Binding mode Binding energy (Kcal/mol) -6.92 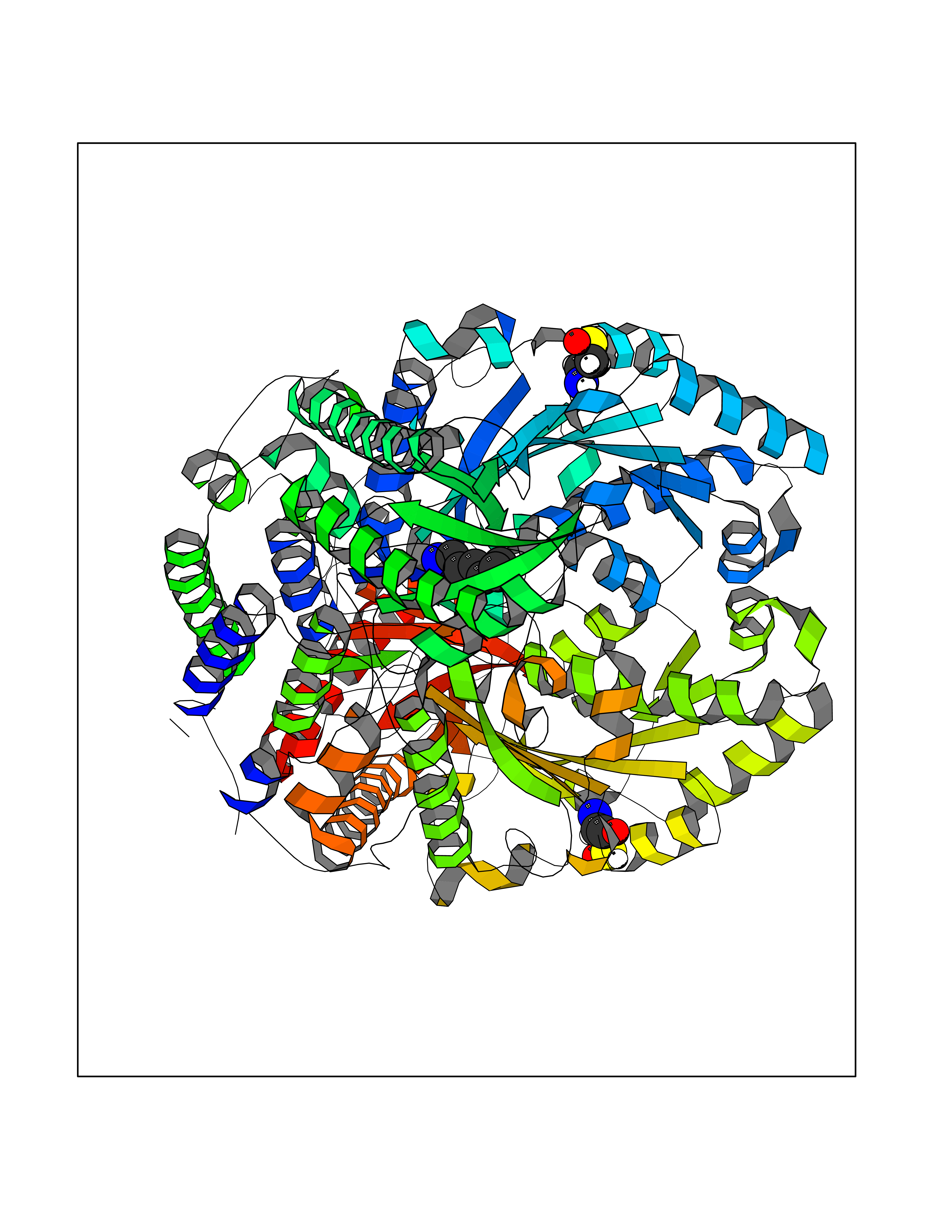 Molscript Map 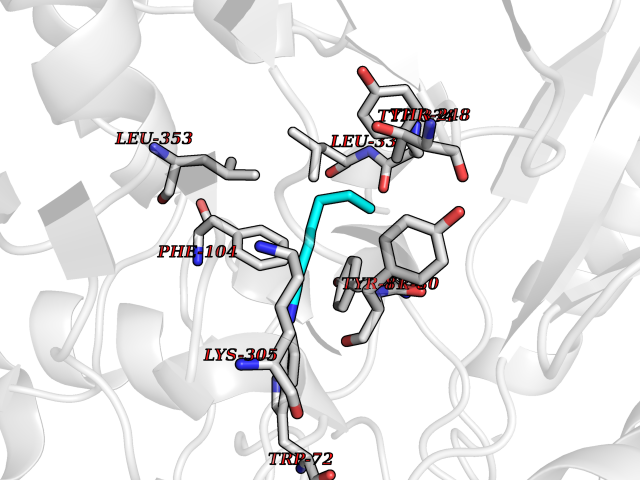 Pymol Map 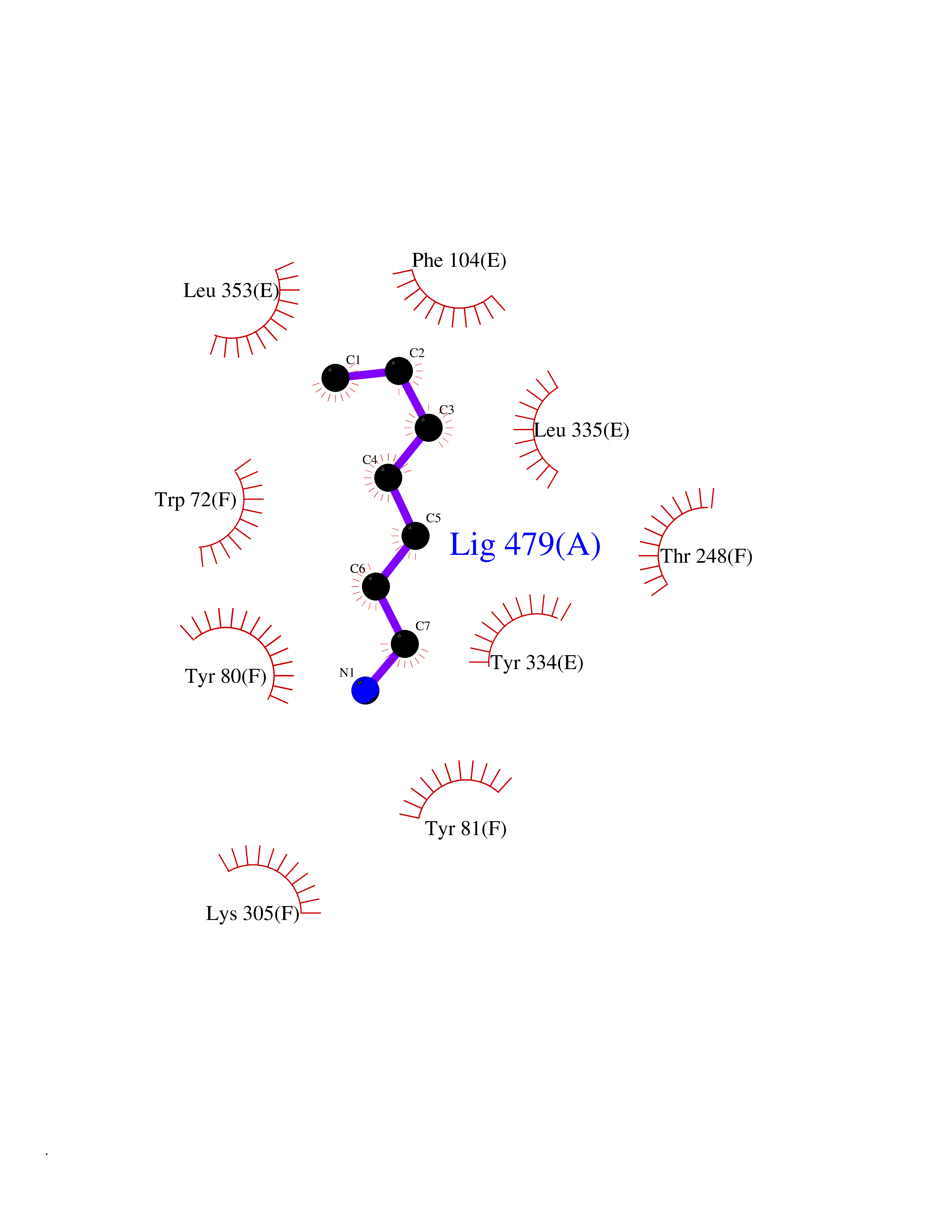 Ligplot Map 3D Binding mode Sequence GSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQGSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Nicotinamide N-methyltransferase | 2IIP | 5.07 | |
Target general information Gen name NNMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class Transferase Function Nicotinamide N-methyltransferase activity.Pyridine N-methyltransferase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB00627 Interacts with NA EC number 2.1.1.1 Uniprot keywords 3D-structure; Acetylation; Citrullination; Cytoplasm; Direct protein sequencing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27886.8 Length 251 Aromaticity 0.1 Instability index 40.66 Isoelectric point 5.23 Charge (pH=7) -5.11 2D Binding mode Binding energy (Kcal/mol) -6.92  Molscript Map 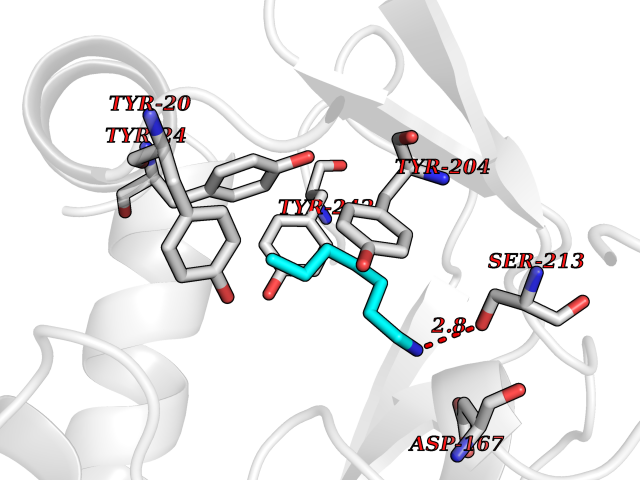 Pymol Map 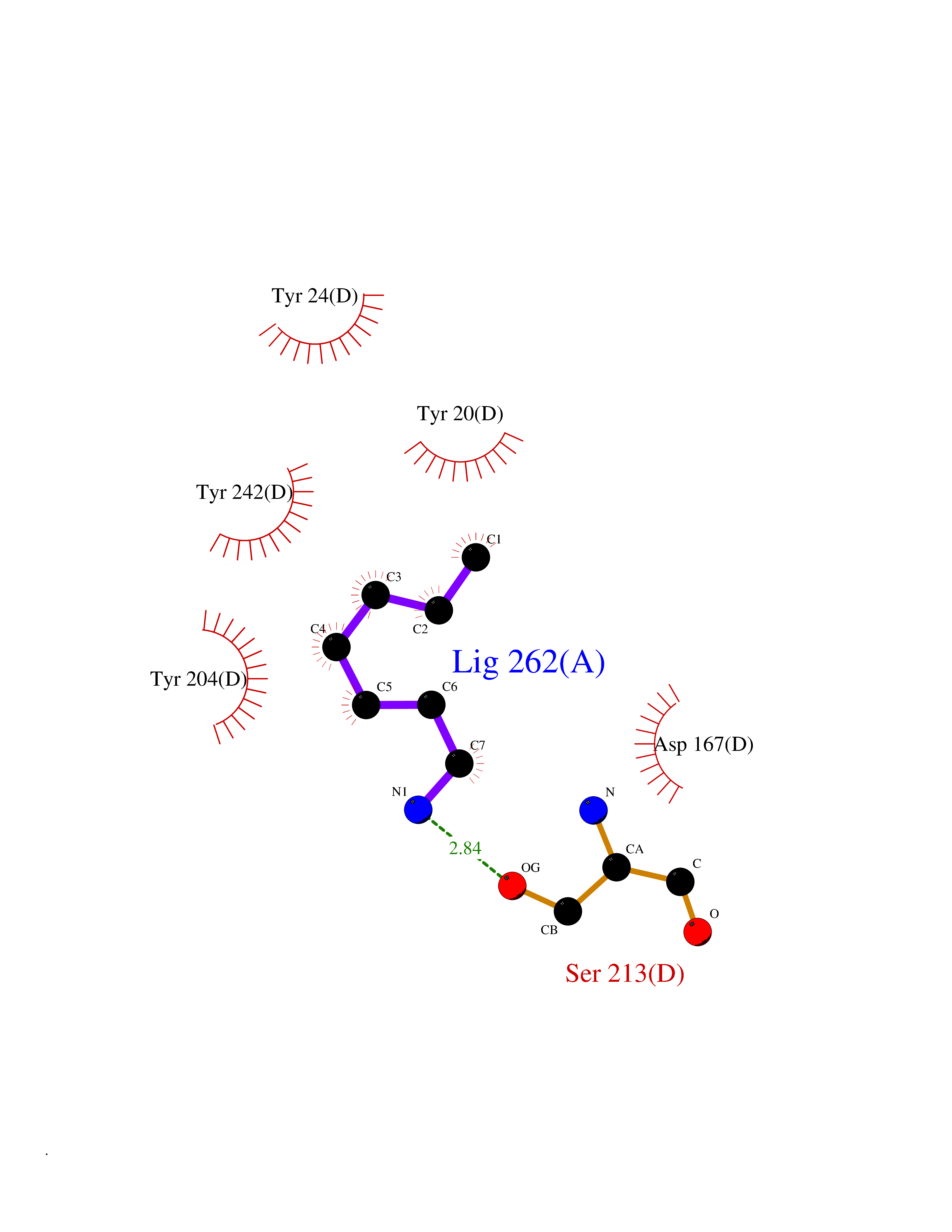 Ligplot Map 3D Binding mode Sequence GFTSKDTYLSHFNPRDYLEKYYSAESQILKHLLKNLFKIFCLDGVKGDLLIDIGSGPTIYQLLSACESFKEIVVTDYSDQNLQELEKWLKAAPAAFDWSPVVTYVCDLEGNRVKGPEKEEKLRQAVKQVLKCDVTQSQPLGAVPLPPADCVLSTLCLDAACPDLPTYCRALRNLGSLLKPGGFLVIMDALKSSYYMIGEQKFSSLPLGREAVEAAVKEAGYTIEWFEVISQSYSSTMANNEGLFSLVARKL Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Lecithin-cholesterol acyltransferase (LCAT) | 6MVD | 5.07 | |
Target general information Gen name LCAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phospholipidcholesterolacyltransferase; Phospholipid-cholesterol acyltransferase; Phosphatidylcholinesterol acyltransferase; Phosphatidylcholine-sterol acyltransferase Protein family AB hydrolase superfamily, Lipase family Biochemical class Acyltransferase Function Synthesized mainly in the liver and secreted into plasma where it converts cholesterol and phosphatidylcholines (lecithins) to cholesteryl esters and lysophosphatidylcholines on the surface of high and low density lipoproteins (HDLs and LDLs). The cholesterol ester is then transported back to the liver. Has a preference for plasma 16:0-18:2 or 18:O-18:2 phosphatidylcholines. Also produced in the brain by primary astrocytes, and esterifies free cholesterol on nascent APOE-containing lipoproteins secreted from glia and influences cerebral spinal fluid (CSF) APOE- and APOA1 levels. Together with APOE and the cholesterol transporter ABCA1, plays a key role in the maturation of glial-derived, nascent lipoproteins. Required for remodeling high-density lipoprotein particles into their spherical forms. Central enzyme in the extracellular metabolism of plasma lipoproteins. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P02647; O76024 EC number EC 2.3.1.43 Uniprot keywords 3D-structure; Acyltransferase; Cholesterol metabolism; Corneal dystrophy; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipid metabolism; Proteomics identification; Reference proteome; Secreted; Signal; Steroid metabolism; Sterol metabolism; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42715.4 Length 376 Aromaticity 0.12 Instability index 42.05 Isoelectric point 5.69 Charge (pH=7) -9.12 2D Binding mode Binding energy (Kcal/mol) -6.92  Molscript Map 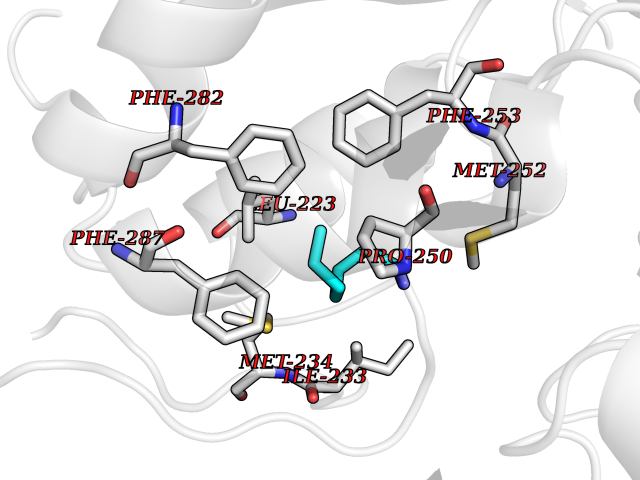 Pymol Map 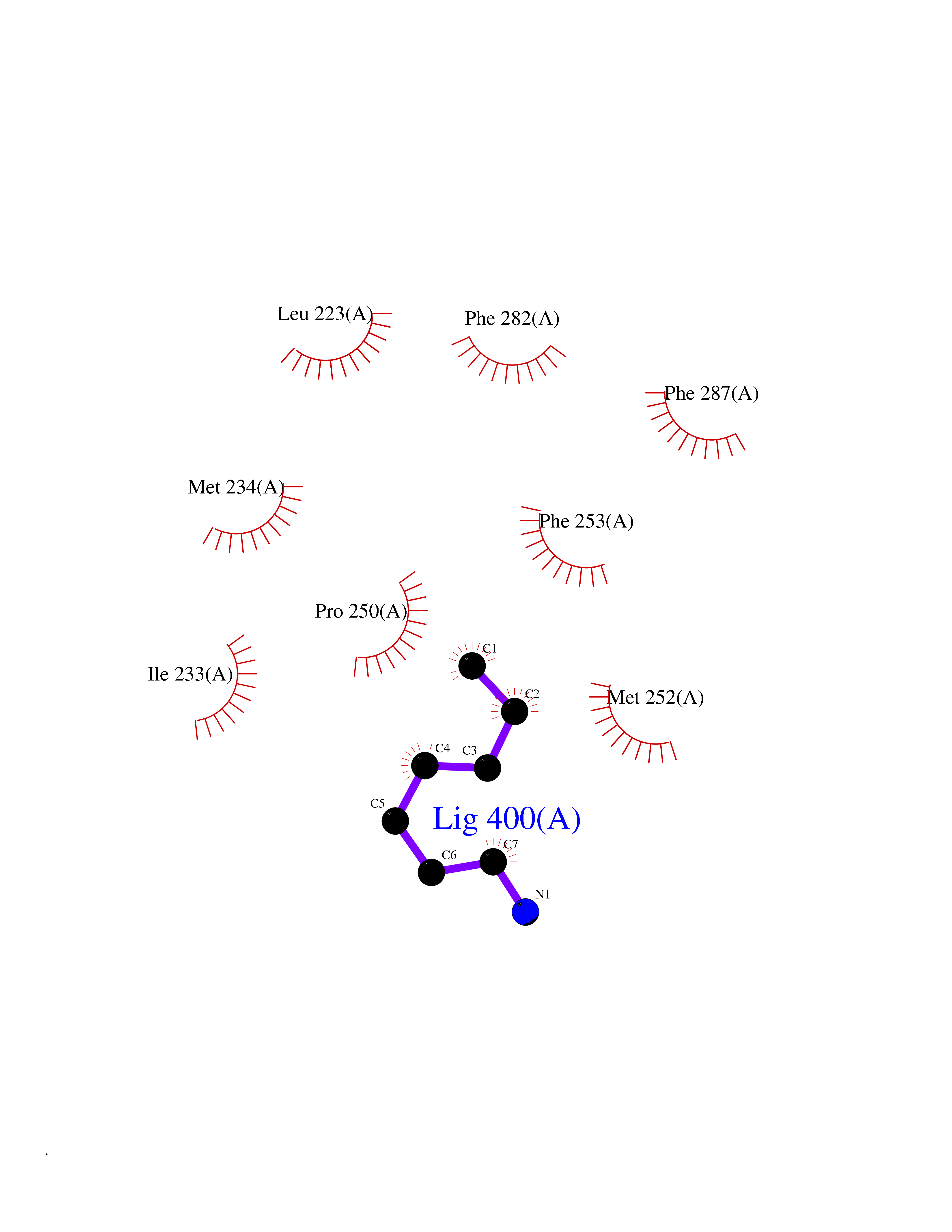 Ligplot Map 3D Binding mode Sequence HTRPVILVPGCLGNQLEAKLDKPDVVNWMCYRKTEDFFTIWLDLNMFLPLGVDCWIDNTRVVYNRSSGLVSNAPGVQIRVPGFGKTYSVEYLDSSKLAGYLHTLVQNLVNNGYVRDETVRAAPYDWRLEPGQQEEYYRKLAGLVEEMHAAYGKPVFLIGHSLGCLHLLYFLLRQPQAWKDRFIDGFISLGAPWGGSIKPMLVLASGDNQGIPIMSSIKEEQRITTTSPWMFPSRMAWPEDHVFISTPSFNYTGRDFQRFFADLHFEEGWYMWLQSRDLLAGLPAPGVEVYCLYGVGLPTPRTYIYDHGFPYTDPVGVLYEDGDDTVATRSTELCGLWQGRQPQPVHLLPLHGIQHLNMVFSNLTLEHINAILLGAH Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Acyloxyacyl hydrolase (neutrophil) | 5W7C | 5.07 | |
Target general information Gen name AOAH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Acyloxyacyl hydrolase Protein family NA Biochemical class NA Function Removes the secondary (acyloxyacyl-linked) fatty acyl chains from the lipid A region of bacterial lipopolysaccharides. By breaking down LPS, terminates the host response to bacterial infection and prevents prolonged and damaging inflammatory responses (By similarity). In peritoneal macrophages, seems to be important for recovery from a state of immune tolerance following infection by Gram-negative bacteria (By similarity). Related diseases Major depressive disorder (MDD) [MIM:608516]: A common psychiatric disorder. It is a complex trait characterized by one or more major depressive episodes without a history of manic, mixed, or hypomanic episodes. A major depressive episode is characterized by at least 2 weeks during which there is a new onset or clear worsening of either depressed mood or loss of interest or pleasure in nearly all activities. Four additional symptoms must also be present including changes in appetite, weight, sleep, and psychomotor activity; decreased energy; feelings of worthlessness or guilt; difficulty thinking, concentrating, or making decisions; or recurrent thoughts of death or suicidal ideation, plans, or attempts. The episode must be accompanied by distress or impairment in social, occupational, or other important areas of functioning. {ECO:0000269|PubMed:15229186}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q15700 EC number EC 3.1.1.77 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lipid metabolism; Metal-binding; Proteomics identification; Reference proteome; Secreted; Signal; Zymogen Protein physicochemical properties Chain ID C Molecular weight (Da) 47779.7 Length 420 Aromaticity 0.1 Instability index 43.45 Isoelectric point 7.72 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -6.91 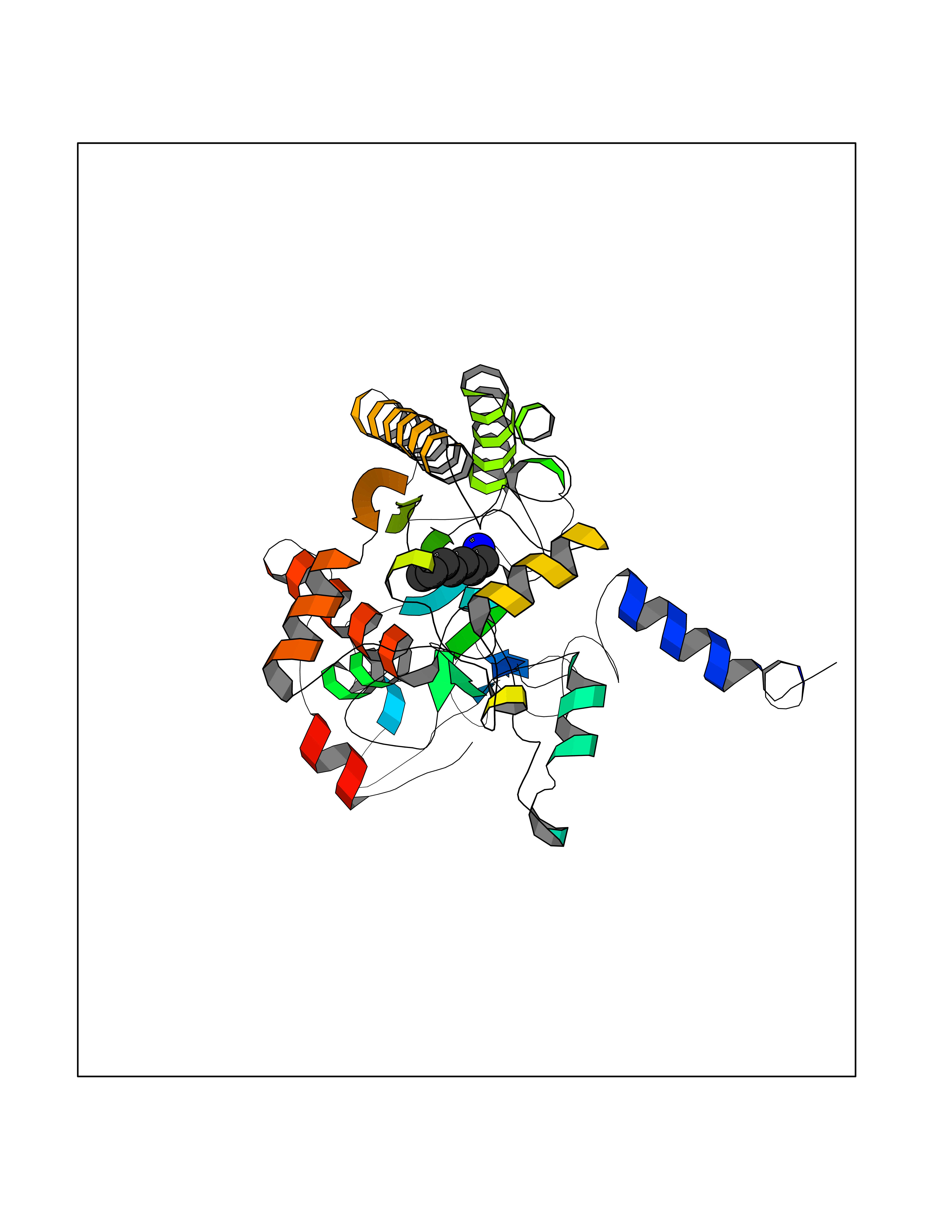 Molscript Map 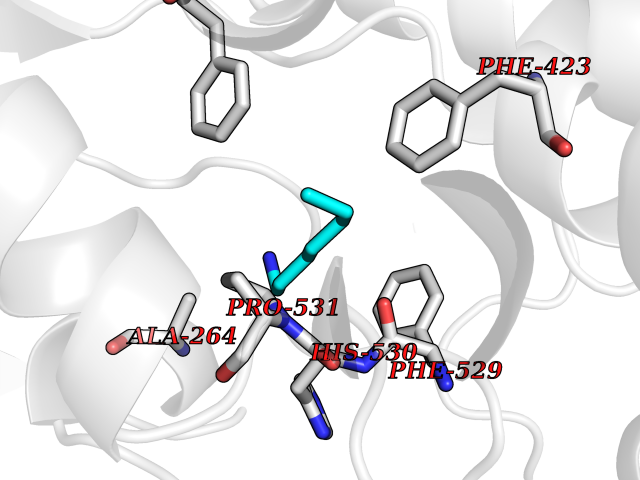 Pymol Map 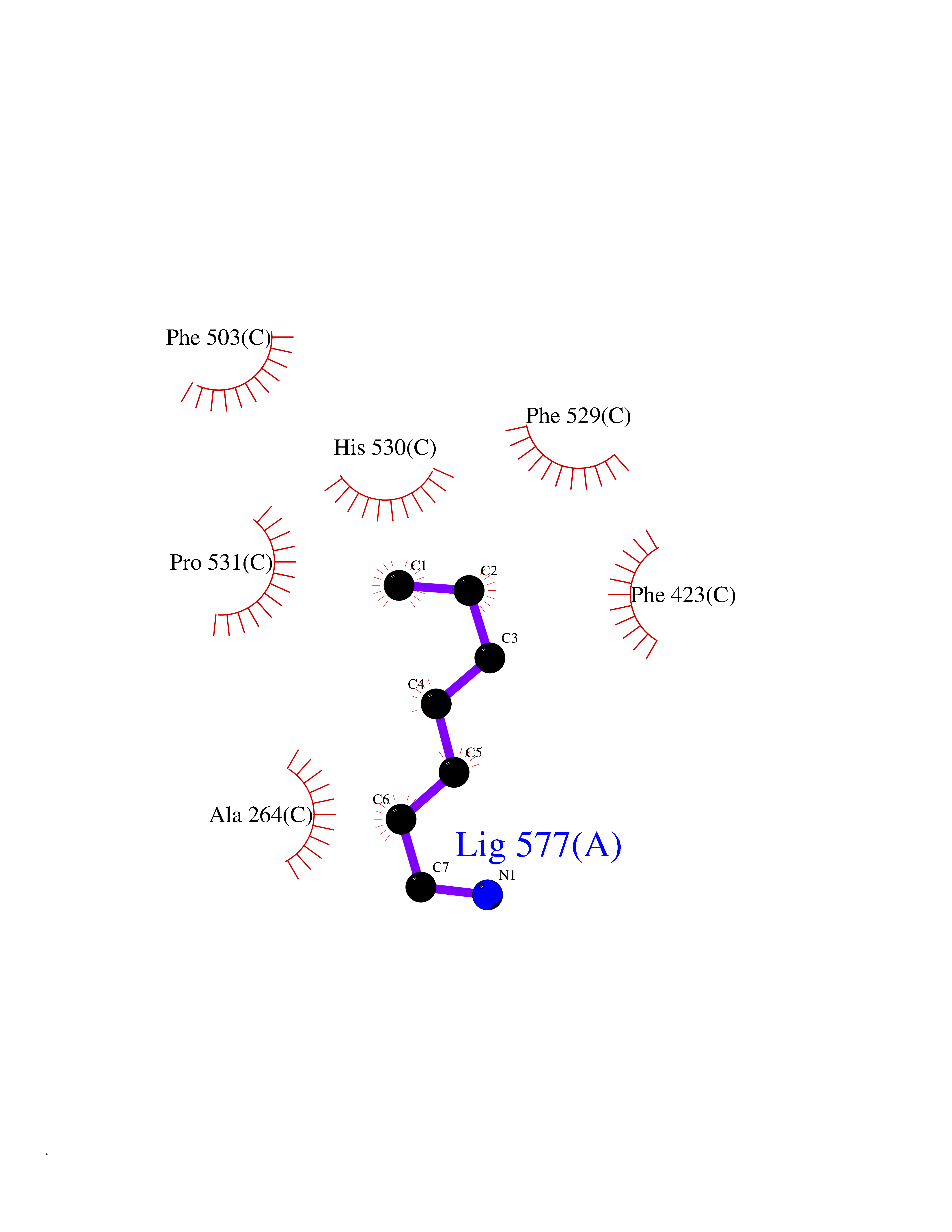 Ligplot Map 3D Binding mode Sequence GSDICSLPVLAKICQKIKLAMEQSVPFKDVDSDKYSVFPTLRGYHWRGRDCNDSDESVYPGRRPNNWDVHQDSNCNGIWGVDPKDGVPYEKKFCEGSQPRGIILLGDAAGAHFHISPEWITASQMSLNSFINLPTALTNELDWPQLSGATGFLDSTVGIKEKSIYLRLWKRNHCNHRDYQNISRNGASSRNLKKFIESLSRNKVLDYPAIVIYAMIGNDVCSGKSDPVPAMTTPEKLYSNVMQTLKHLNSHLPNGSHVILYGLPDGTFLWDNLHNRYHPLGQLNKDMTYAQLYSFLNCLQVSPCHGWMSSNKTLRTLTSERAEQLSNTLKKIAASEKFTNFNLFYMDFAFHEIIQEWQKRGGQPWQLIEPVDGFHPNEVALLLLADHFWKKVQLQWPQILGKENPFNPQIKQVFGDQGGH Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Chromodomain-helicase-DNA-binding protein 1 | 4O42 | 5.06 | |
Target general information Gen name CHD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SNF2/RAD54 helicase family Biochemical class Dna binding protein / viral protein Function ATP binding.ATP-dependent DNA helicase activity.DNA binding.Methylated histone binding. Related diseases Pilarowski-Bjornsson syndrome (PILBOS) [MIM:617682]: An autosomal dominant disorder characterized by developmental delay, speech apraxia, intellectual disability, autism, and facial dysmorphic features. Some patients may have seizures. {ECO:0000269|PubMed:28866611}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60341-1; B2BUF1; P28799; O76024 EC number 3.6.4.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromatin regulator; Cytoplasm; Disease variant; DNA-binding; Helicase; Hydrolase; Intellectual disability; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 20969.1 Length 180 Aromaticity 0.12 Instability index 46.35 Isoelectric point 5.88 Charge (pH=7) -2.83 2D Binding mode Binding energy (Kcal/mol) -6.9 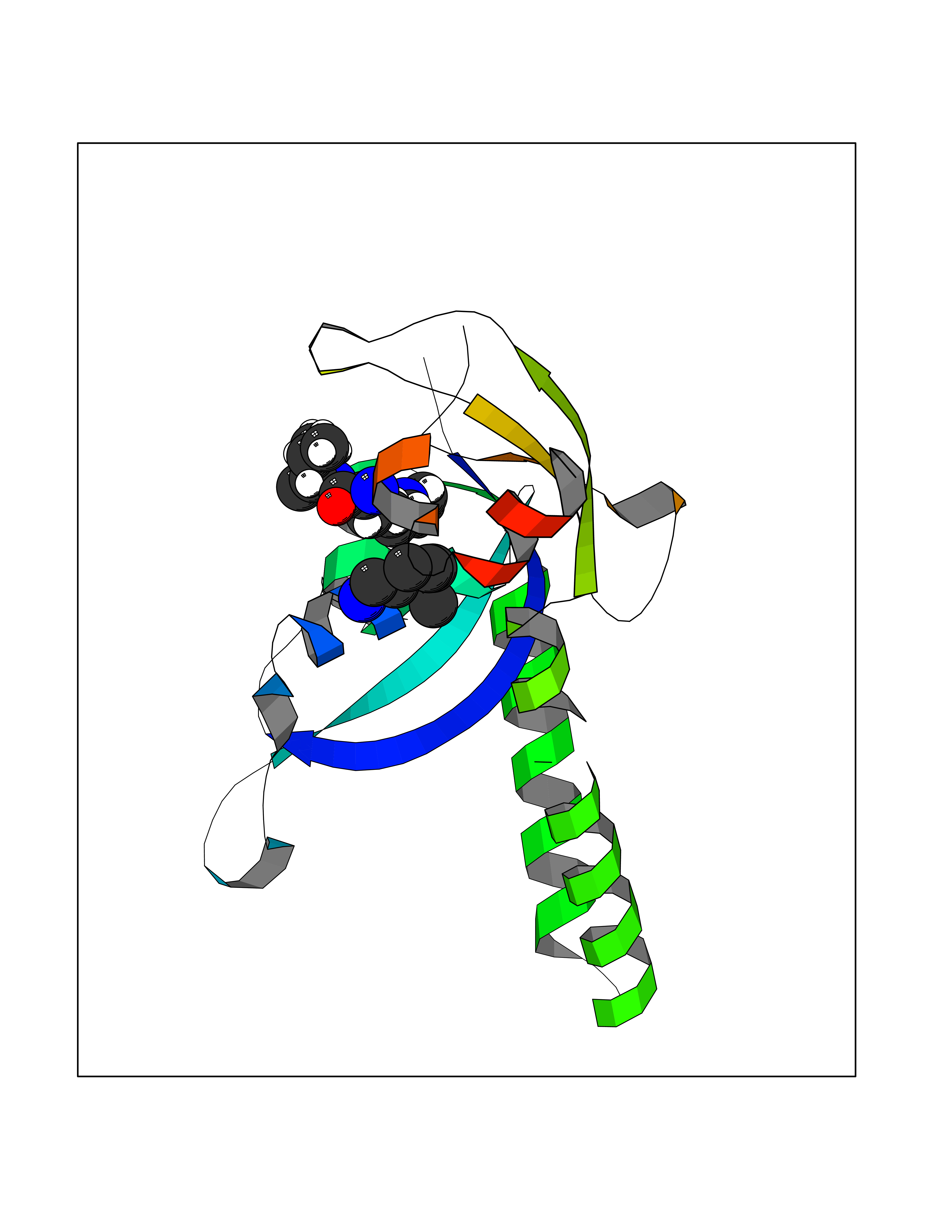 Molscript Map 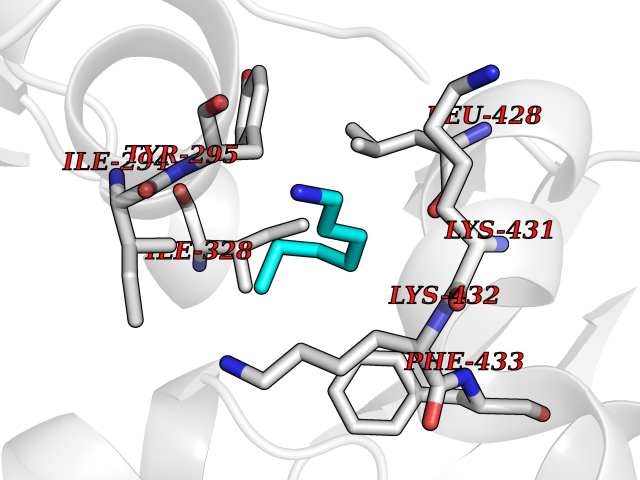 Pymol Map 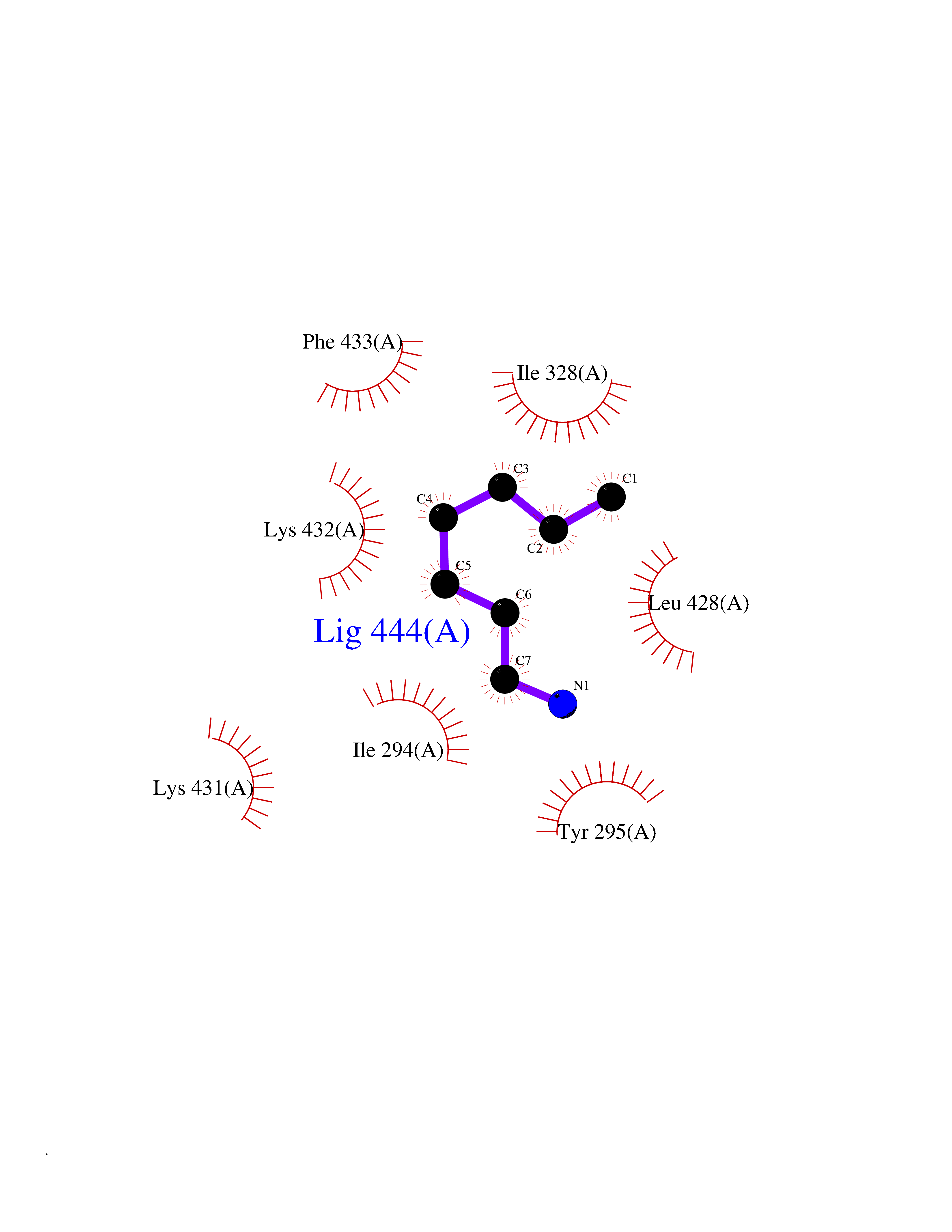 Ligplot Map 3D Binding mode Sequence EFETIERFMDCRIGRKGATGATTTIYAVEADGDPNAGFEKNKEPGEIQYLIKWKGWSHIHNTWETEETLKQQNVRGMKKLDNYKKKDQETKRWLKNASPEDVEYYNCQQELTDDLHKQYQIVERIIAHSNQKSAAGYPDYYCKWQGLPYSECSWEDGALISKKFQACIDEYFSRTARSXV Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Cytochrome b (Complex III subunit 3) (Complex III subunit III) (Cytochrome b-c1 complex subunit 3) (Ubiquinol-cytochrome-c reductase complex cytochrome b subunit) | 1SQB | 5.06 | |
Target general information Gen name MT-CYB Organism Bos taurus (Bovine) Uniprot ID TTD ID NA Synonyms CYTB;MTCYB;COB Protein family Cytochrome b family Biochemical class NA Function Component of the ubiquinol-cytochrome c reductase complex (complex III or cytochrome b-c1 complex) that is part of the mitochondrial respiratory chain. The b-c1 complex mediates electron transfer from ubiquinol to cytochrome c. Contributes to the generation of a proton gradient across the mitochondrial membrane that is then used for ATP synthesis. {ECO:0000269|PubMed:1327781, ECO:0000269|PubMed:20025846, ECO:0000269|PubMed:9485330, ECO:0000305|PubMed:189810}." Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Electron transport; Heme; Iron; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Reference proteome; Respiratory chain; Transmembrane; Transmembrane helix; Transport; Ubiquinone Protein physicochemical properties Chain ID C,D,F,G,H Molecular weight (Da) 99281.2 Length 866 Aromaticity 0.12 Instability index 43.81 Isoelectric point 8.32 Charge (pH=7) 7.12 2D Binding mode Binding energy (Kcal/mol) -6.9  Molscript Map 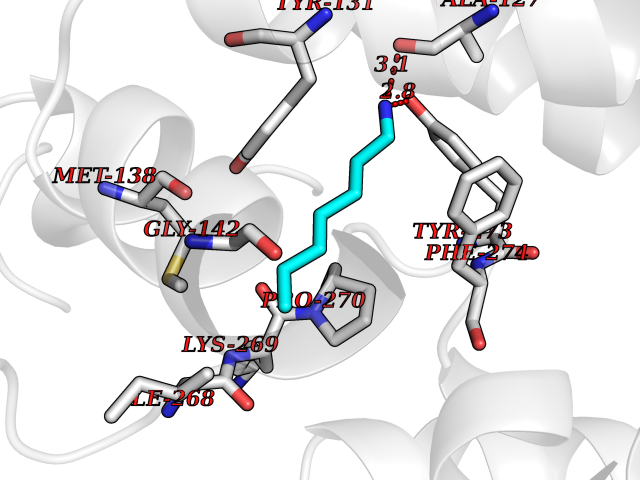 Pymol Map 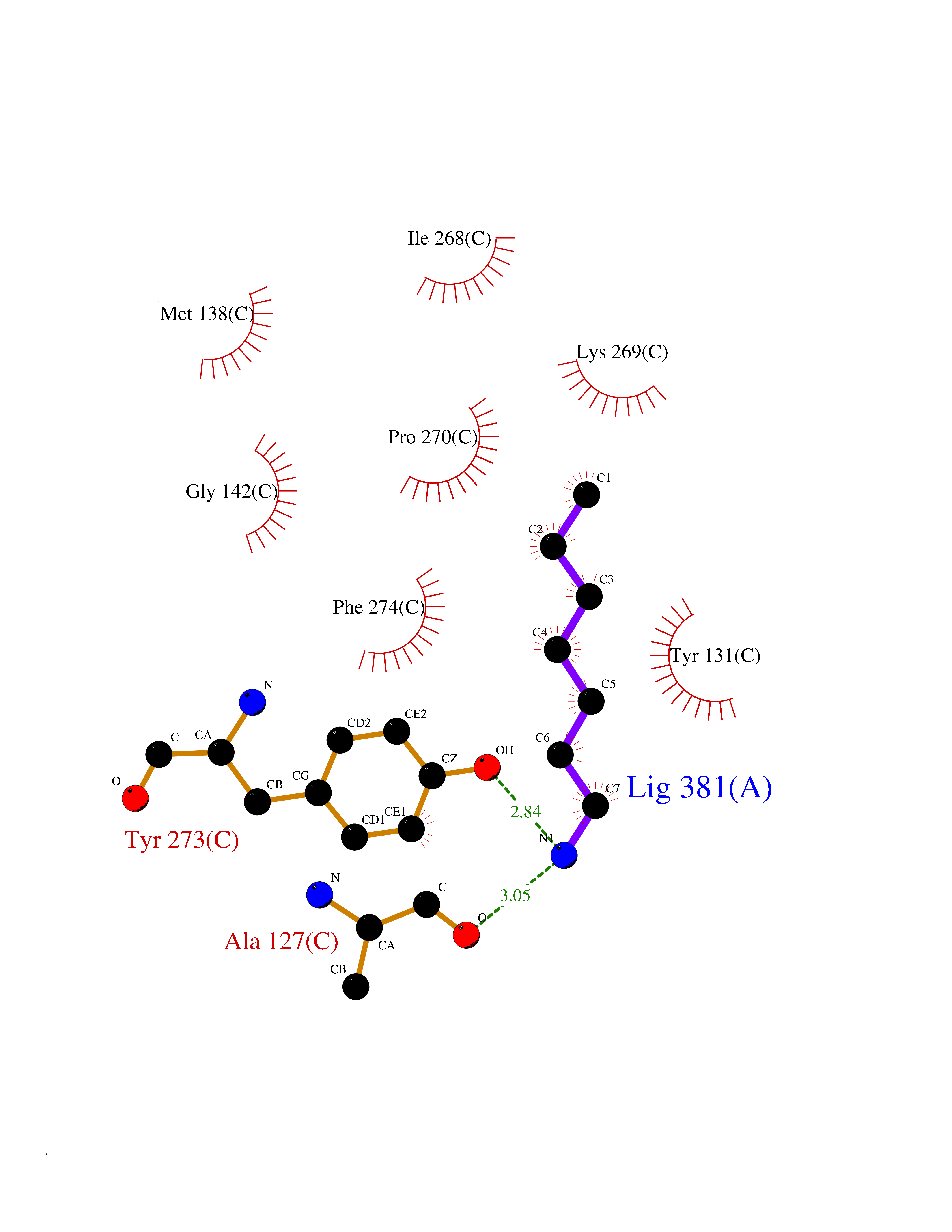 Ligplot Map 3D Binding mode Sequence VSASSRWLEGIRKWYYNAAGFNKLGLMRDDTIHENDDVKEAIRRLPENLYDDRVFRIKRALDLSMRQQILPKEQWTKYEEDKSYLEPYLKEVIRERKEREEWAKKELVDPLTTVREQCEQLEKCVKARERLELCDERVSSRSQTEEDCTEELLDFLHARDHCVAHKLFNSLKTNIRKSHPLMKIVNNAFIDLPAPSNISSWWNFGSLLGICLILQILTGLFLAMHYTSDTTTAFSSVTHICRDVNYGWIIRYMHANGASMFFICLYMHVGRGLYYGSYTFLETWNIGVILLLTVMATAFMGYVLPWGQMSFWGATVITNLLSAIPYIGTNLVEWIWGGFSVDKATLTRFFAFHFILPFIIMAIAMVHLLFLHETGSNNPTGISSDVDKIPFHPYYTIKDILGALLLILALMLLVLFAPDLLGDPDNYTPANPLNTPPHIKPEWYFLFAYAILRSIPNKLGGVLALAFSILILALIPLLHTSKQRSMMFRPLSQCLFWALVADLLTLTWIGGQPVEHPYITIGQLASVLYFLLILVLMPTAGTIENKLLKWSDLELHPPSYPWSHRGLLSSLDHTSIRRGFQVYKQVCSSCHSMDYVAYRHLVGVCYTEDEAKALAEEVEVQDGPNEDGEMFMRPGKLSDYFPKPYPNPEAARAANNGALPPDLSYIVRARHGGEDYVFSLLTGYCEPPTGVSLREGLYFNPYFPGQAIGMAPPIYNEVLEFDDGTPATMSQVAKDVCTFLRWAAEPEHDHRKRMGLKMLLMMGLLLPLVYAMKRHKWSVLKSRKLAYRPPKGRQFGHLTRVRHVITYSLSPFEQRAFPHYFSKGIPNVLRRTRACILRVAPPFVAFYLVYTWGTQEFEKSKRKNPA Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 5.06 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -6.91  Molscript Map 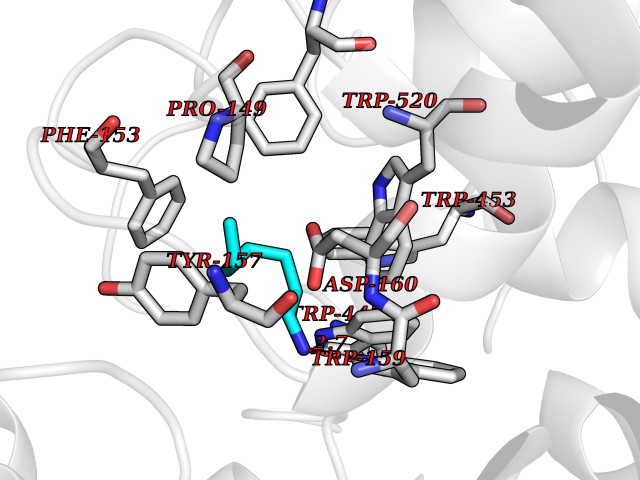 Pymol Map 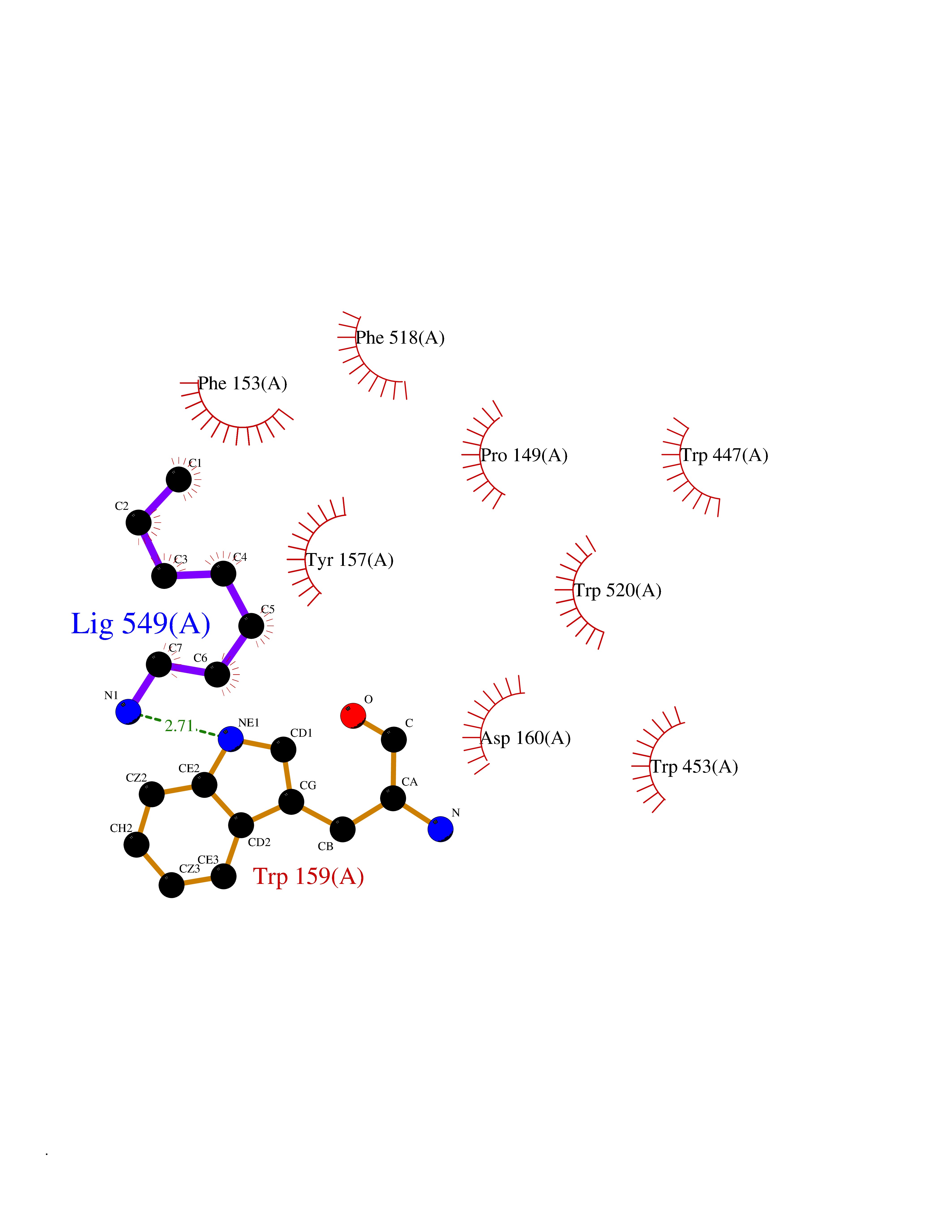 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Leukotriene A-4 hydrolase (LTA4H) | 3U9W | 5.06 | |
Target general information Gen name LTA4H Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Leukotriene A4 hydrolase; Leukotriene A(4)Leukotriene A-4 hydrolase hydrolase; Leukotriene A(4) hydrolase; LTA4; LTA-H; LTA-4hydrolase; LTA-4 hydrolase Protein family Peptidase M1 family Biochemical class Ether bond hydrolase Function Has also aminopeptidase activity. Epoxide hydrolase that catalyzes the final step in the biosynthesis of the proinflammatory mediator leukotriene B4. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07102; DB06917; DB07258; DB07094; DB07259; DB02352; DB07292; DB07104; DB06828; DB08466; DB01197; DB05177; DB03366; DB08040; DB06851; DB02062; DB07099; DB07260; DB07196; DB11781; DB03424; DB07237 Interacts with Q9BSI4 EC number EC 3.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Leukotriene biosynthesis; Lipid metabolism; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 68927 Length 608 Aromaticity 0.1 Instability index 38.84 Isoelectric point 5.87 Charge (pH=7) -9.86 2D Binding mode Binding energy (Kcal/mol) -6.91 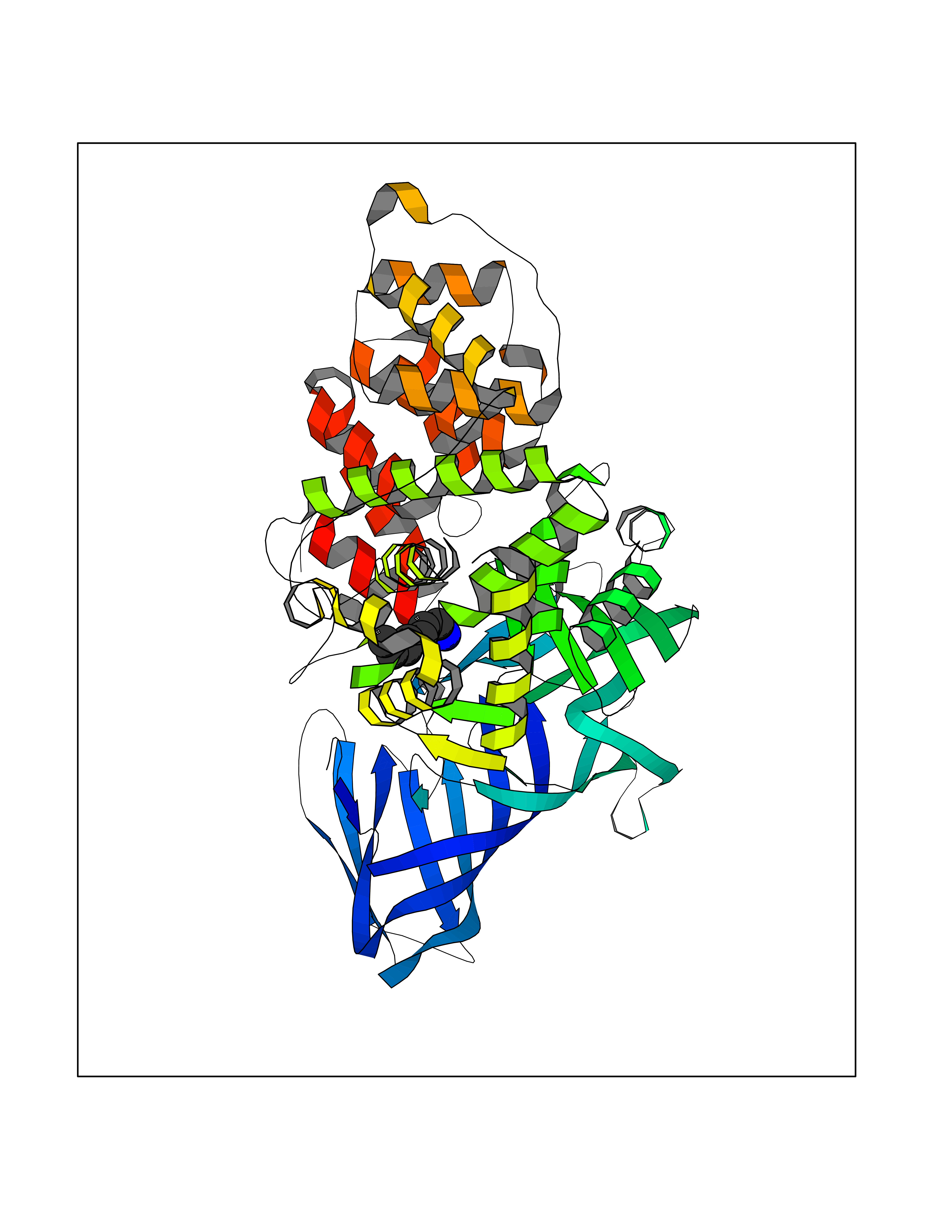 Molscript Map 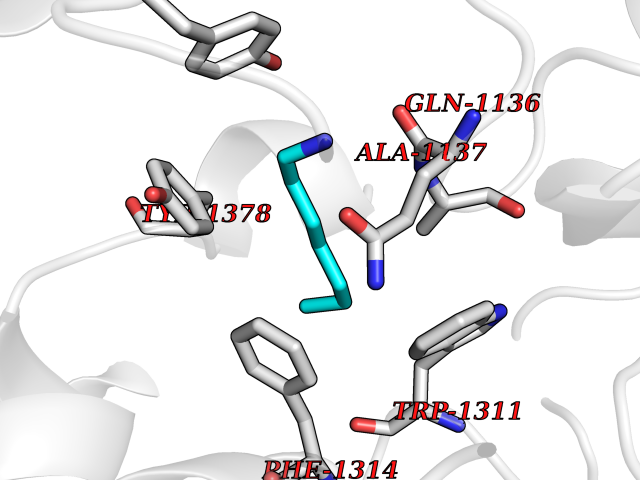 Pymol Map 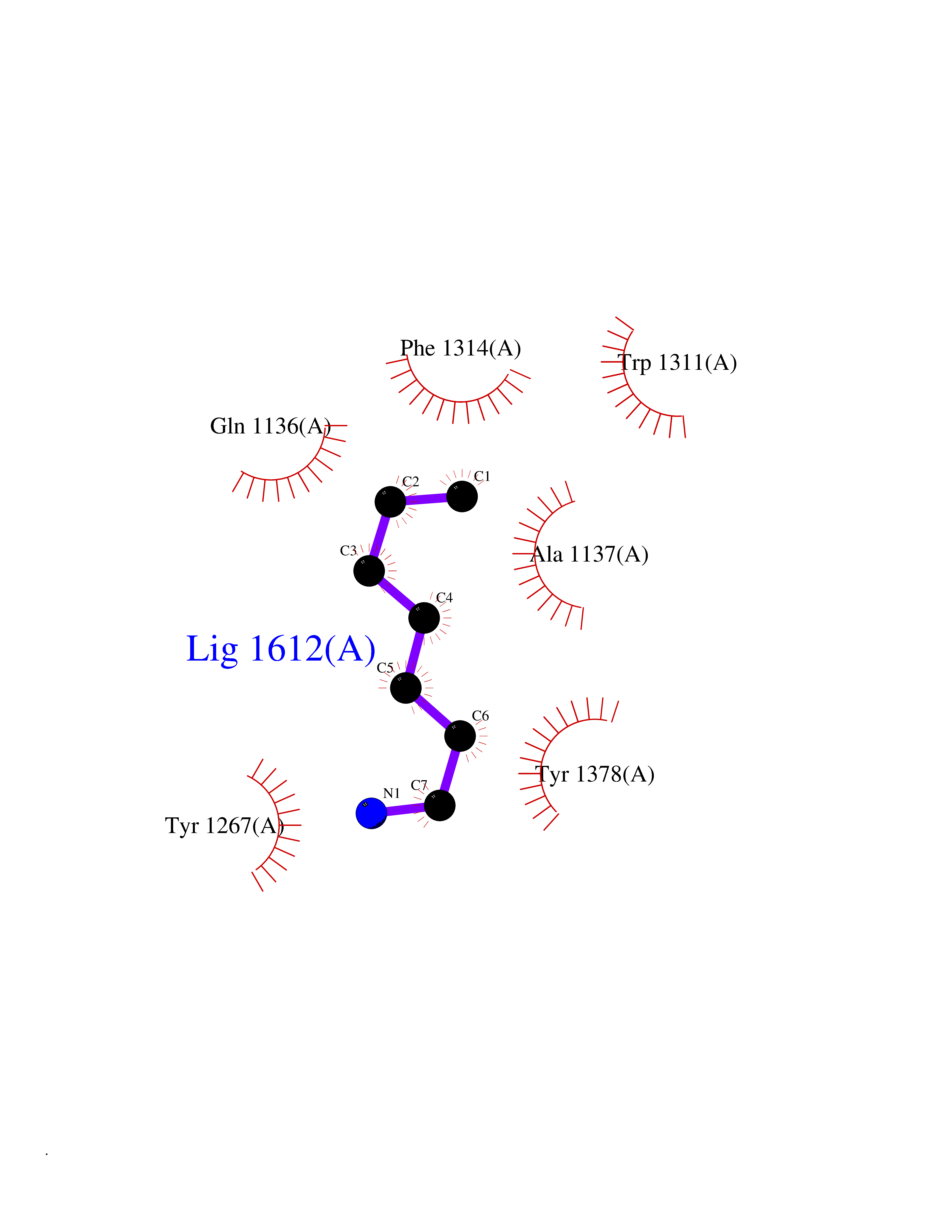 Ligplot Map 3D Binding mode Sequence IVDTCSLASPASVCRTKHLHLRCSVDFTRRTLTGTAALTVQSQEDNLRSLVLDTKDLTIEKVVINGQEVKYALGERQSYKGSPMEISLPIALSKNQEIVIEISFETSPKSSALQWLTPEQTSGKEHPYLFSQCQAIHCRAILPCQDTPSVKLTYTAEVSVPKELVALMSAIRDGETPDPEDPSRKIYKFIQKVPIPCYLIALVVGALESRQIGPRTLVWSEKEQVEKSAYEFSETESMLKIAEDLGGPYVWGQYDLLVLPPSFPYGGMENPCLTFVTPTLLAGDKSLSNVIAHEISHSWTGNLVTNKTWDHFWLNEGHTVYLERHICGRLFGEKFRHFNALGGWGELQNSVKTFGETHPFTKLVVDLTDIDPDVAYSSVPYEKGFALLFYLEQLLGGPEIFLGFLKAYVEKFSYKSITTDDWKDFLYSYFKDKVDVLNQVDWNAWLYSPGLPPIKPNYDMTLTNACIALSQRWITAKEDDLNSFNATDLKDLSSHQLNEFLAQTLQRAPLPLGHIKRMQEVYNFNAINNSEIRFRWLRLCIQSKWEDAIPLALKMATEQGRMKFTRPLFKDLAAFDKSHDQAVRTYQEHKASMHPVTAMLVGKDLKVD Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Matrix metalloproteinase-16 (MMP-16) | 1RM8 | 5.06 | |
Target general information Gen name MMP16 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Membrane-type-3 matrix metalloproteinase; Membrane-type matrix metalloproteinase 3; MTMMP3; MT3MMP; MT3-MMP; MT-MMP 3; MMPX2; MMP-X2; C8orf57 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates progelatinase A. Involved in the matrix remodeling of blood vessels. Isoform short cleaves fibronectin and also collagen type III, but at lower rate. It has no effect on type I, II, IV and V collagen. However, upon interaction with CSPG4, it may be involved in degradation and invasion of type I collagen by melanoma cells. Endopeptidase that degrades various components of the extracellular matrix, such as collagen type III and fibronectin. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03880; DB00786 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Cleavage on pair of basic residues; Collagen degradation; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18853.6 Length 169 Aromaticity 0.12 Instability index 33.65 Isoelectric point 4.88 Charge (pH=7) -12.42 2D Binding mode Binding energy (Kcal/mol) -6.91 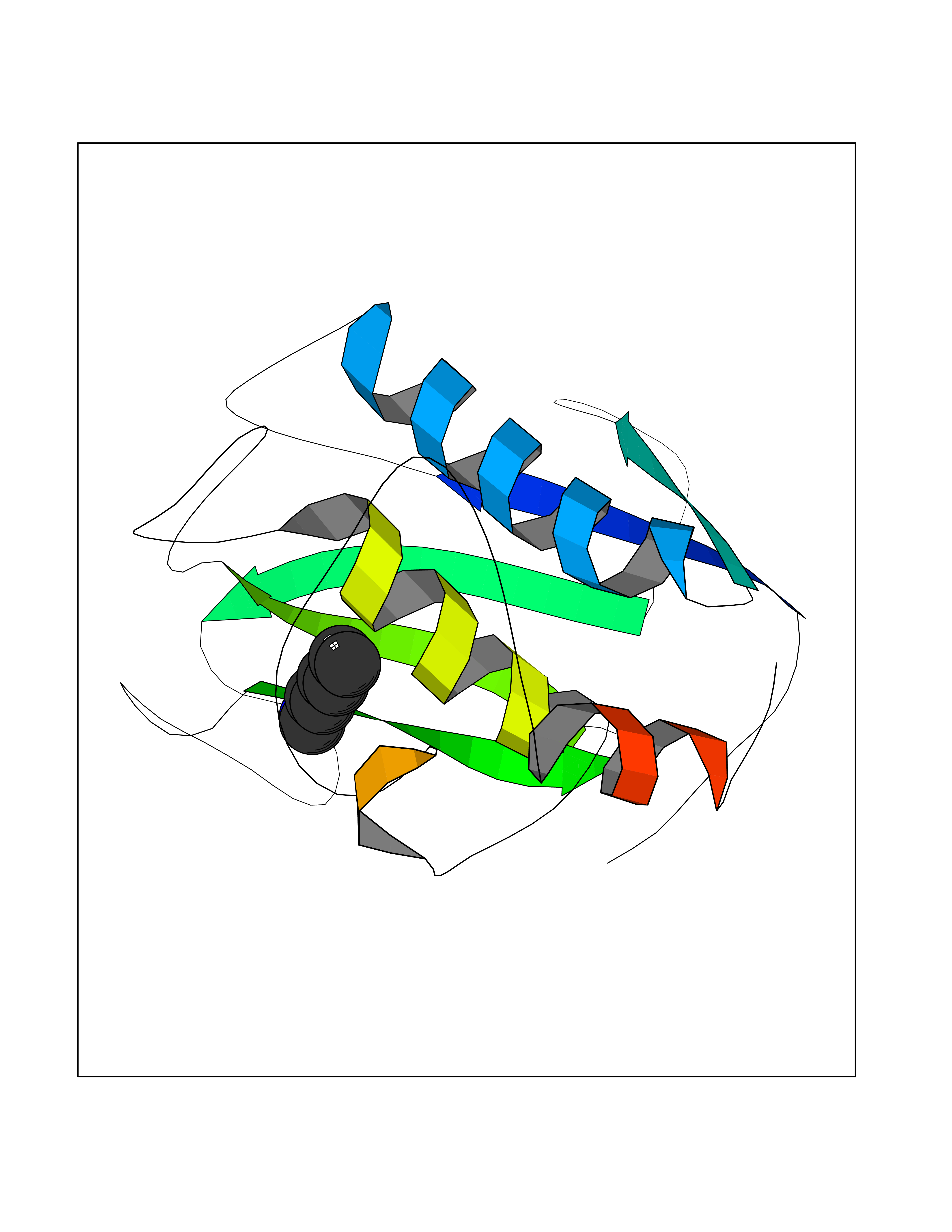 Molscript Map 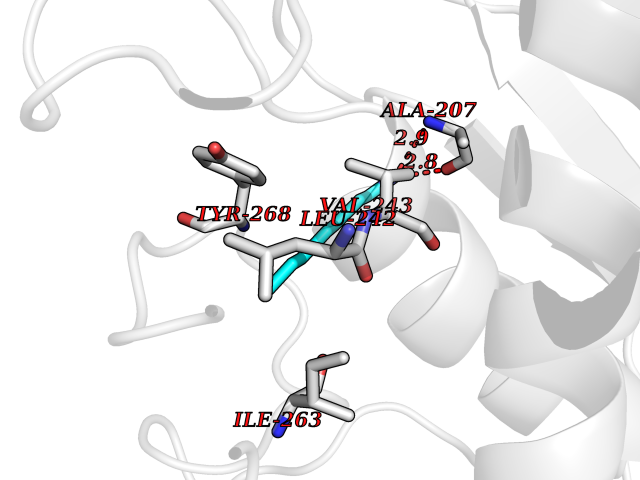 Pymol Map 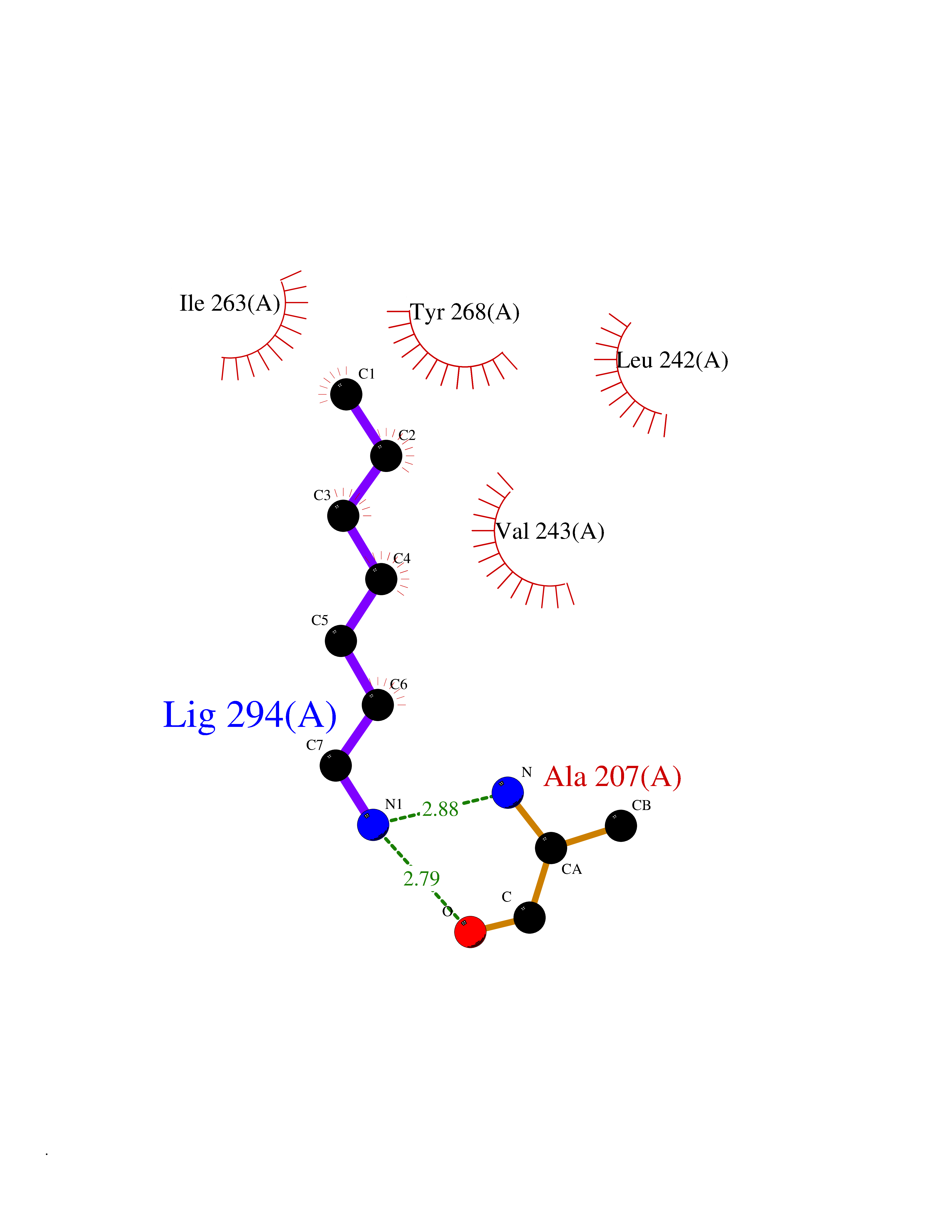 Ligplot Map 3D Binding mode Sequence GQKWQHKHITYSIKNVTPKVGDPETRKAIRRAFDVWQNVTPLTFEEVPYSELENGKRDVDITIIFASGFHGDSSPFDGEGGFLAHAYFPGPGIGGDTHFDSDEPWTLGNPNHDGNDLFLVAVHELGHALGLEHSNDPTAIMAPFYQYMETDNFKLPNDDLQGIQKIYGP Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | TetR family transcriptional regulator | 2V57 | 5.06 | |
Target general information Gen name lfrR Organism Mycolicibacterium smegmatis (Mycobacterium smegmatis) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Transcription Function DNA binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB01123 Interacts with NA EC number NA Uniprot keywords 3D-structure Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 37947.8 Length 348 Aromaticity 0.07 Instability index 36.27 Isoelectric point 5.5 Charge (pH=7) -9.3 2D Binding mode Binding energy (Kcal/mol) -6.9 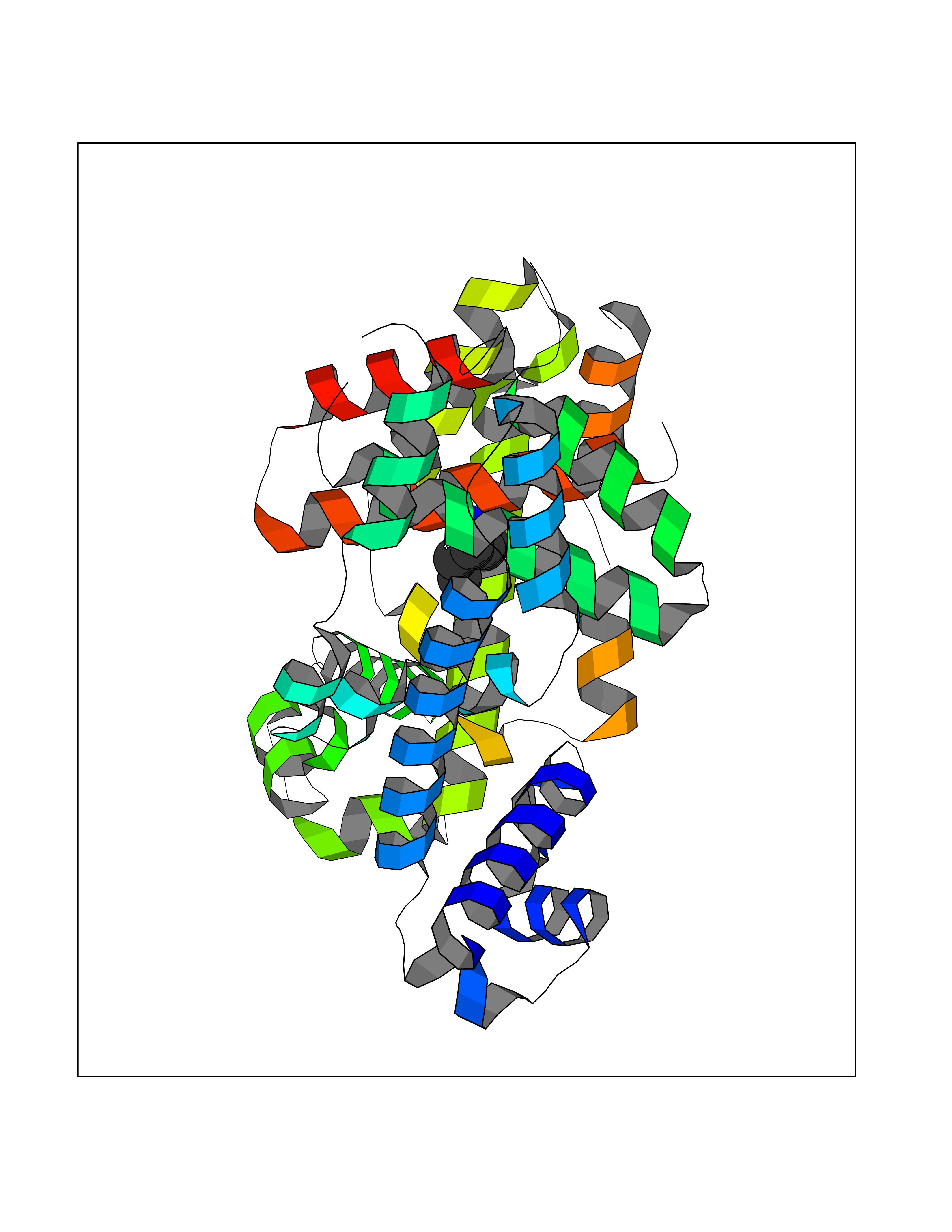 Molscript Map 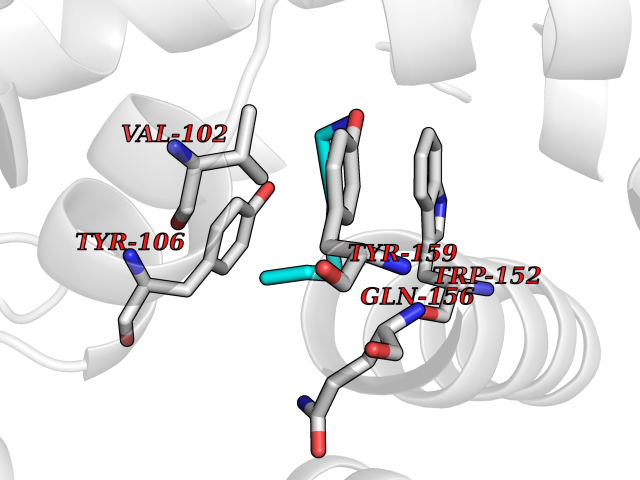 Pymol Map 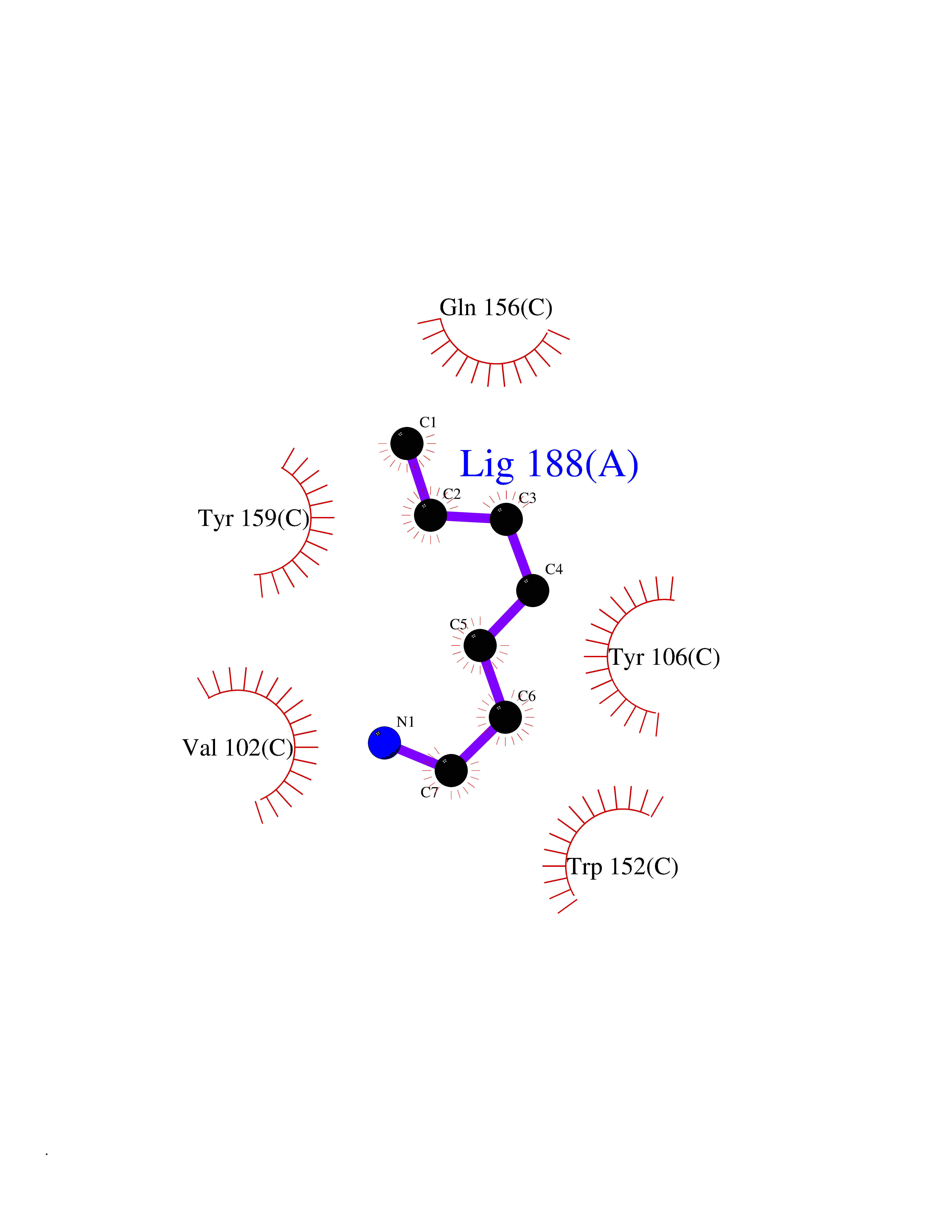 Ligplot Map 3D Binding mode Sequence GARERTRRAILDAAMLVLADHPTAALGDIAAAAGVGRSTVHRYYPERTDLLRALARHVHDLSNAAIERADPTSGPVDAALRRVVESQLDLGPIVLFVYYEPSILADPELAAYFDIGDEAIVEVLNRASTERYPPGWARRVFWALMQAGYEAAKDGMPRHQIVDAIMTSLTSGIITLARERTRRAILDAAMLVLADHPTAALGDIAAAAGVGRSTVHRYYPERTDLLRALARHVHDLSNAAIERADPTSGPVDAALRRVVESQLDLGPIVLFVYYEPSILADPELAAYFDIGDEAIVEVLNRASYPPGWARRVFWALMQAGYEAAKDGMPRHQIVDAIMTSLTSGIITL Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Ephrin type-B receptor 3 (EPHB3) | 5L6O | 5.06 | |
Target general information Gen name EPHB3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hEK2; Tyrosine-protein kinase TYRO6; TYRO6; Embryonic kinase 2; ETK2; EPH-like tyrosine kinase 2; EPH-like kinase 2; EK2 Protein family Protein kinase superfamily, Tyr protein kinase family, Ephrin receptor subfamily Biochemical class Kinase Function The signaling pathway downstream of the receptor is referred to as forward signaling while the signaling pathway downstream of the ephrin ligand is referred to as reverse signaling. Generally has an overlapping and redundant function with EPHB2. Like EPHB2, functions in axon guidance during development regulating for instance the neurons forming the corpus callosum and the anterior commissure, 2 major interhemispheric connections between the temporal lobes of the cerebral cortex. In addition to its role in axon guidance plays also an important redundant role with other ephrin-B receptors in development and maturation of dendritic spines and the formation of excitatory synapses. Controls other aspects of development through regulation of cell migration and positioning. This includes angiogenesis, palate development and thymic epithelium development for instance. Forward and reverse signaling through the EFNB2/EPHB3 complex also regulate migration and adhesion of cells that tubularize the urethra and septate the cloaca. Finally, plays an important role in intestinal epithelium differentiation segregating progenitor from differentiated cells in the crypt. Receptor tyrosine kinase which binds promiscuously transmembrane ephrin-B family ligands residing on adjacent cells, leading to contact-dependent bidirectional signaling into neighboring cells. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) NA Interacts with P37235; O75031 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Angiogenesis; ATP-binding; Cell membrane; Cell projection; Developmental protein; Disulfide bond; Glycoprotein; Kinase; Membrane; Neurogenesis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30412.9 Length 267 Aromaticity 0.09 Instability index 37.42 Isoelectric point 7.74 Charge (pH=7) 1 2D Binding mode Binding energy (Kcal/mol) -6.9 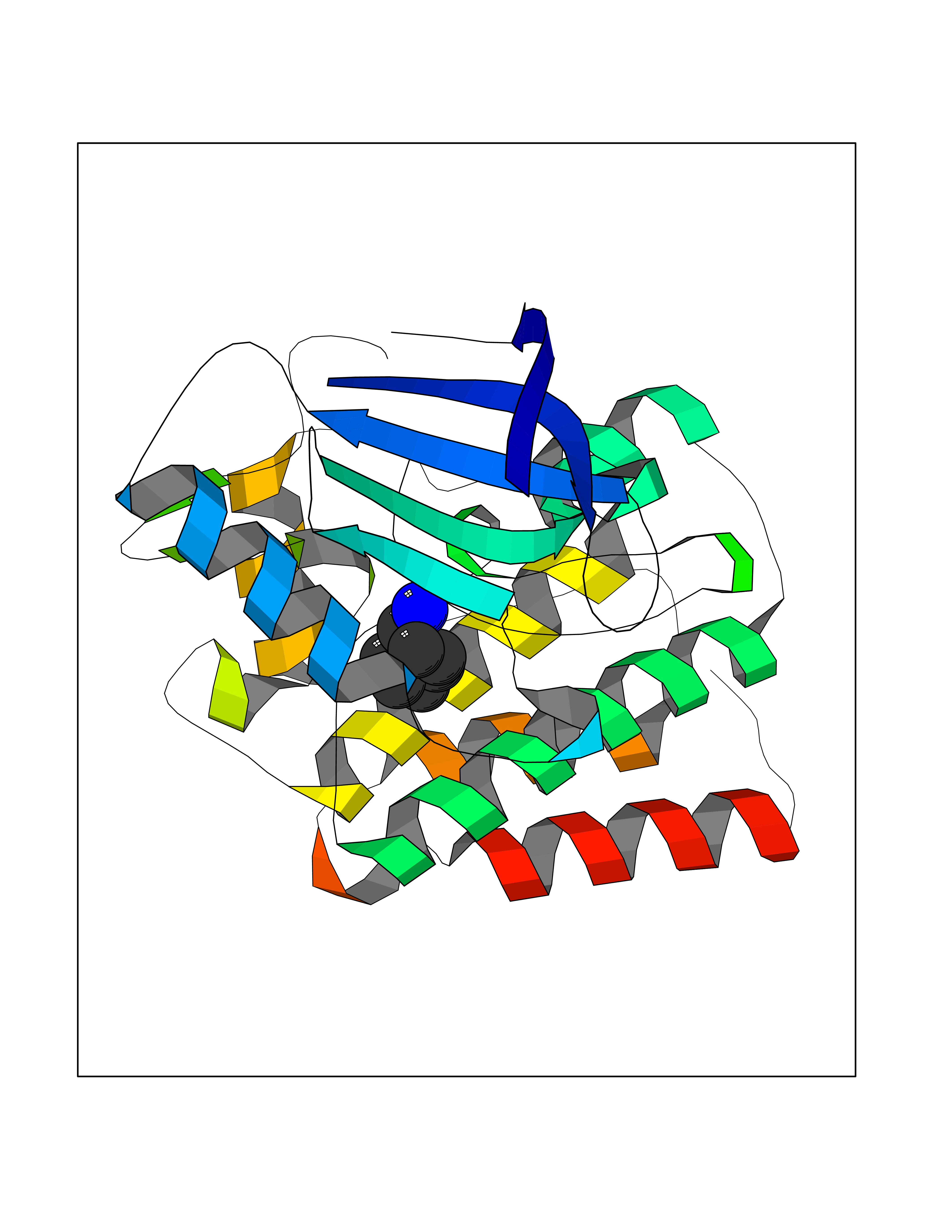 Molscript Map 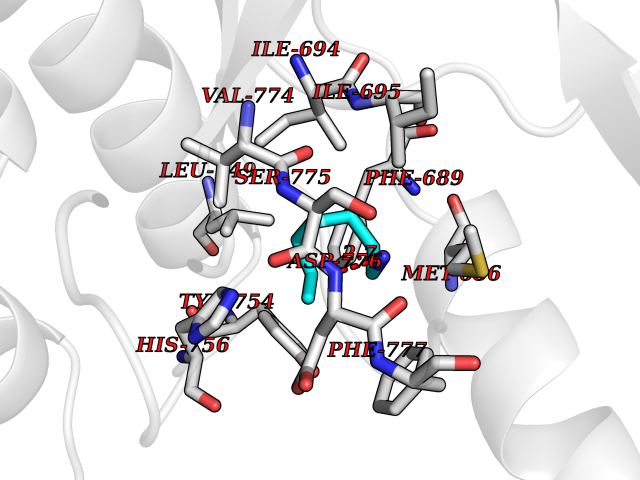 Pymol Map 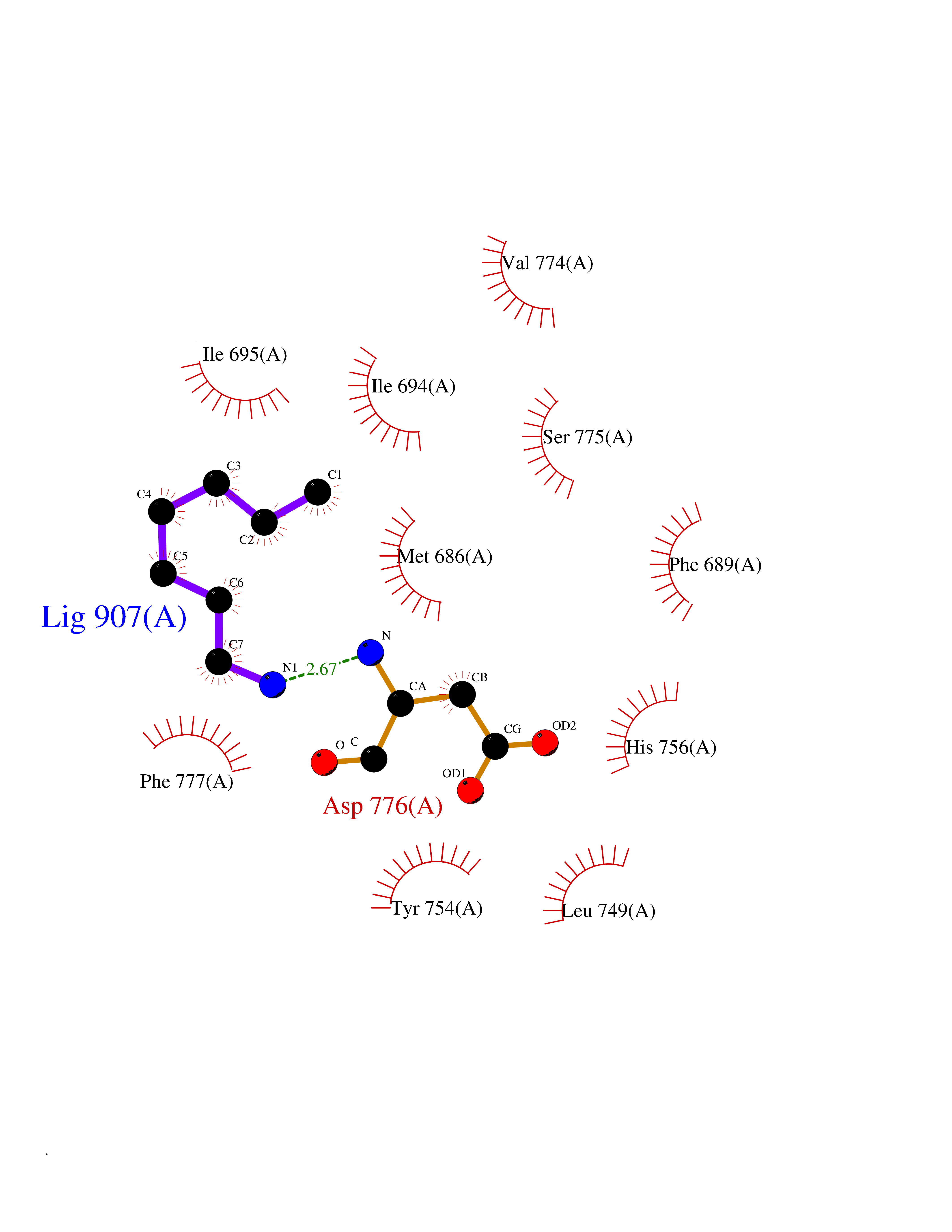 Ligplot Map 3D Binding mode Sequence CVKIEEVIGAGEVCRGRLKQPGRREVFVAIKTLKVGYTERQRRDFLSEASIMGQFDHPNIIRLEGVVTKSRPVMILTEFMENCALDSFLRLNDGQFTVIQLVGMLRGIAAGMKYLSEMNYVHRDLAARNILVNSNLVCKVSDFGLEDDPSDPTYTSSLGGKIPIRWTAPEAIAYRKFTSASDVWSYGIVMWEVMSYGERPYWDMSNQDVINAVEQDYRLPPPMDCPTALHQLMLDCWVRDRNLRPKFSQIVNTLDKLIRNPASLKVI Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Zinc finger-containing ubiquitin peptidase 1 (ZUP1) | 6EI1 | 5.06 | |
Target general information Gen name ZUP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Zinc finger with UFM1-specific peptidase domain protein; ZUFSP; Lys-63-specific deubiquitinase ZUFSP; DUB; C6orf113 Protein family Peptidase C78 family, ZUFSP subfamily Biochemical class Peptidase Function Shows only weak activity against 'Lys-11' and 'Lys-48'-linked chains. Plays an important role in genome stability pathways, functioning to prevent spontaneous DNA damage and also promote cellular survival in response to exogenous DNA damage. Modulates the ubiquitination status of replication protein A (RPA) complex proteins in response to replication stress. Deubiquitinase with endodeubiquitinase activity that specifically interacts with and cleaves 'Lys-63'-linked long polyubiquitin chains. Related diseases WHIM syndrome 2 (WHIMS2) [MIM:619407]: An autosomal recessive form of WHIM syndrome, a primary immunodeficiency disorder characterized by warts, hypogammaglobulinemia, infections, and myelokathexis. Myelokathexis is a unique form of non-cyclic severe congenital neutropenia caused by accumulation of mature and degenerating neutrophils in the bone marrow. Monocytopenia and lymphopenia, especially B lymphopenia, also commonly occur. There is significant phenotypic variation among patients, such that some individuals may have an incomplete form of the disorder in which one or more of the classic tetrad features are not present. {ECO:0000269|PubMed:24777453}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q92619; P50281; Q8WVC2 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Hydrolase; Metal-binding; Nucleus; Proteomics identification; Reference proteome; Repeat; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 46930.3 Length 410 Aromaticity 0.07 Instability index 58.67 Isoelectric point 9 Charge (pH=7) 9.7 2D Binding mode Binding energy (Kcal/mol) -6.9 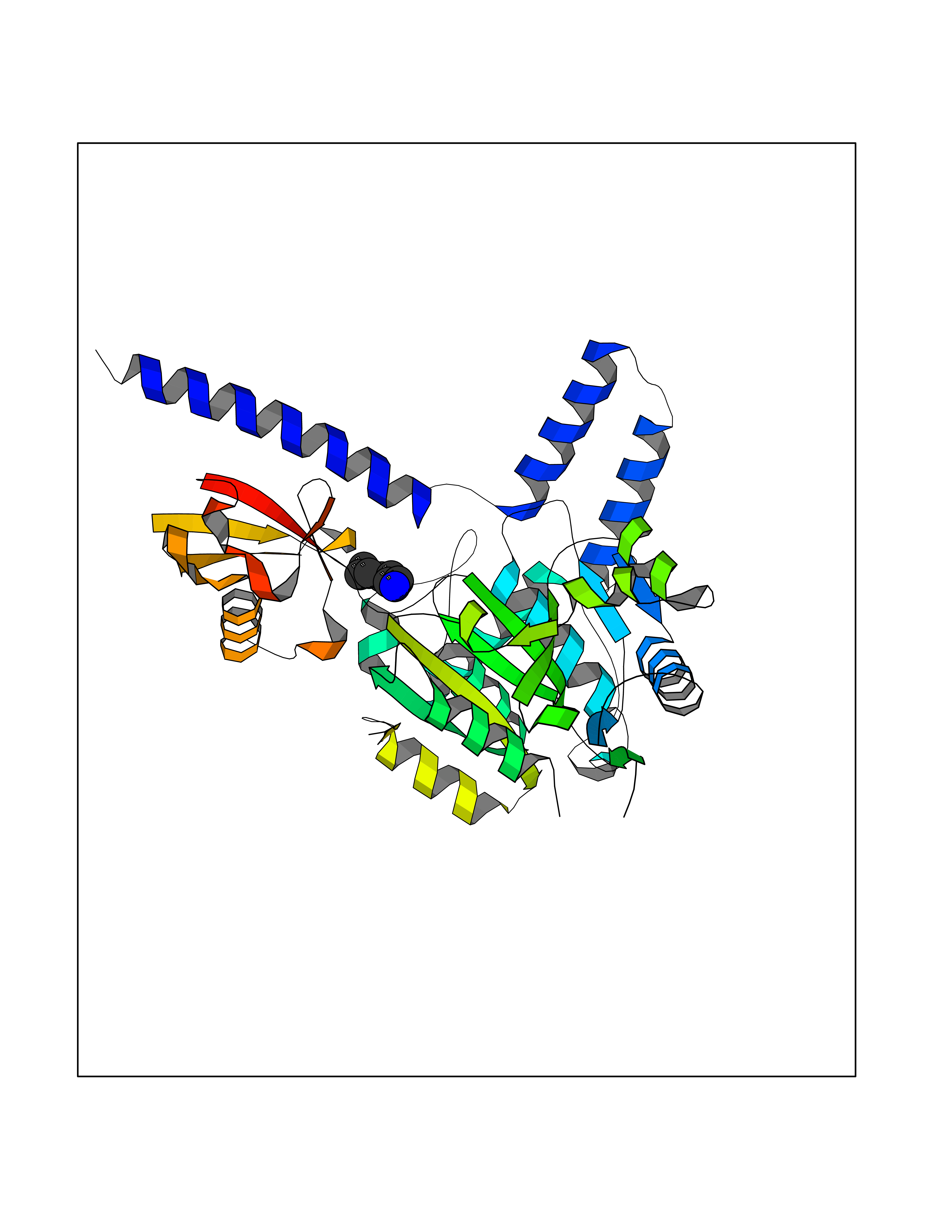 Molscript Map 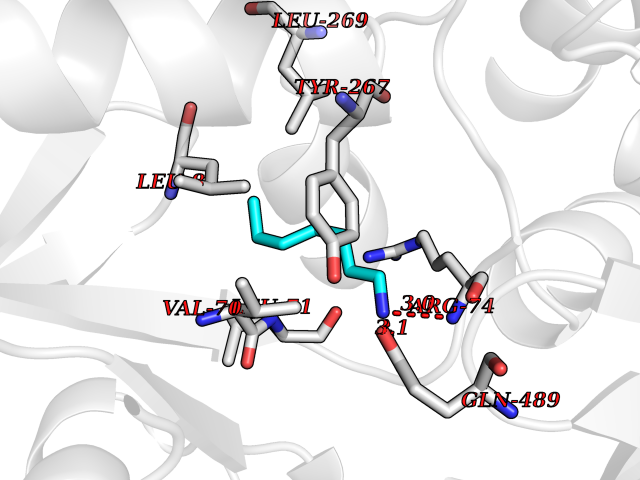 Pymol Map 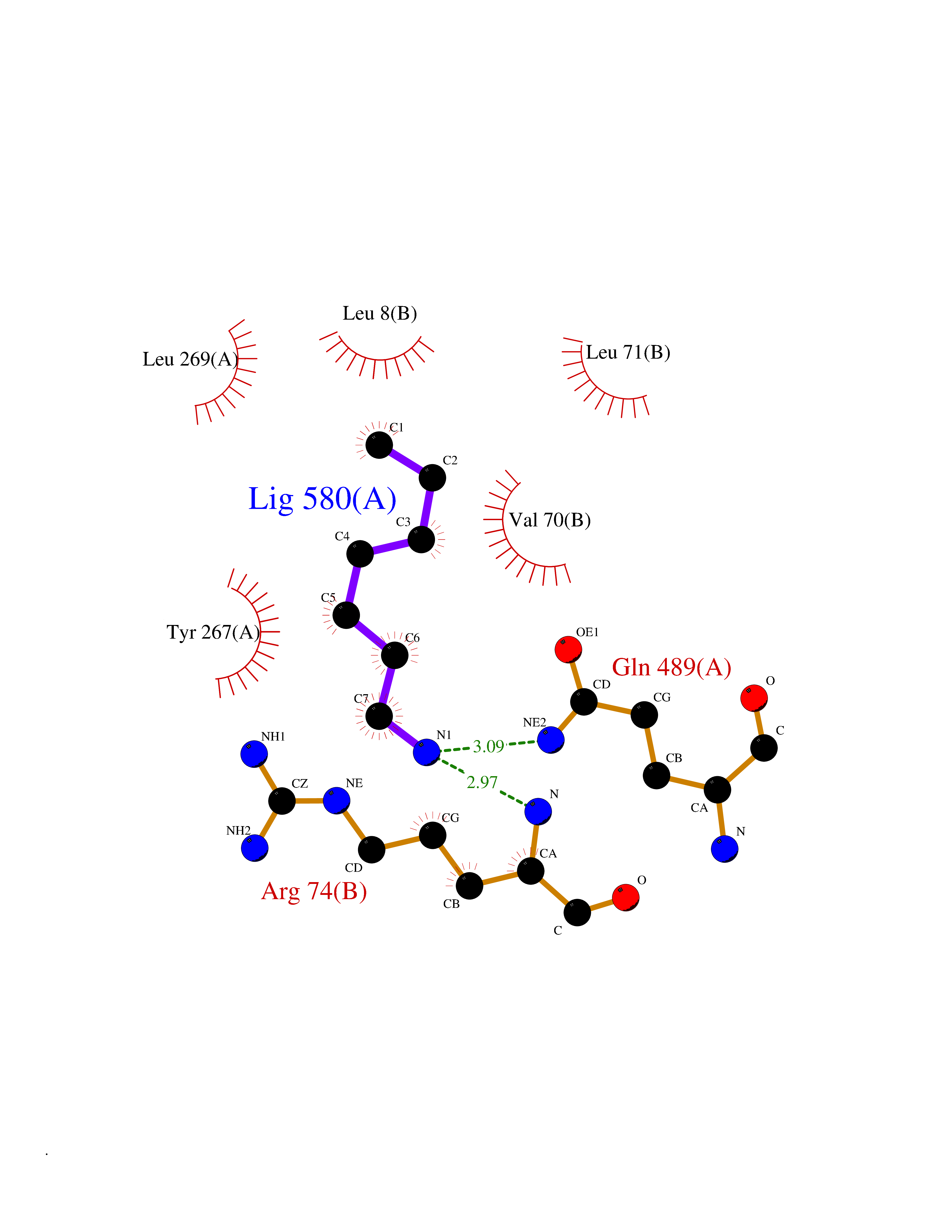 Ligplot Map 3D Binding mode Sequence LQQEEDRKRRSEESRQEIEEFQKLQRQYGLDNSGGYKQQQLRNMEIEVNRGRMPPSEFHRRKADMMESLALGFDDGKTKTSGIIEALHRYYQNAATDVRRVWLSSVVDHFHSSLGDKGWGCGYRNFQMLLSSLLQNDAYNDCLKGMLIPCIPKIQSMIEDAWKEGFDPQGASQLNNRLQGTKAWIGACEVYILLTSLRVKCHIVDFHKSTGPLGTHPRLFEWILNYYSSSPKVVCTSKPPIYLQHQGHSRTVIGIEEKKNRTLCLLILDPGCPSREMQKLLKQDIEASSLKQLRKSMGNLKHKQYQILAVEGALSLEEKLARRQASQVFTAEKIPMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRG Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Squalene synthetase (FDFT1) | 3WCM | 5.06 | |
Target general information Gen name FDFT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Squalene synthase; SS; SQS; Farnesyl-diphosphate farnesyltransferase; FPP:FPP farnesyltransferase Protein family Phytoene/squalene synthase family Biochemical class Alkyl aryl transferase Function Participates in the isoprenoid biosynthetic pathway, catalyzing a two-step reaction in which two identical molecules of farnesyl pyrophosphate (FPP) are converted into squalene, with the consumption of NADPH. Related diseases Squalene synthase deficiency (SQSD) [MIM:618156]: An autosomal recessive disorder characterized by profound developmental delay, brain abnormalities, 2/3 syndactyly of the toes, facial dysmorphisms, low total and LDL-cholesterol, and abnormal urine organic acids. {ECO:0000269|PubMed:29909962}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05317 Interacts with Q13520; Q3SXY8; P04233-2; P11912; O75503; O43889-2; Q9GZR5; Q5JX71; P48165; Q8TDT2; Q8N5M9; Q6IBW4-4; Q96RD7; Q14973; Q9NQQ7-3; Q96MV1; Q9Y320 EC number EC 2.5.1.21 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Endoplasmic reticulum; Lipid biosynthesis; Lipid metabolism; Magnesium; Membrane; Metal-binding; Multifunctional enzyme; NAD; NADP; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 37860 Length 329 Aromaticity 0.1 Instability index 40.19 Isoelectric point 5.47 Charge (pH=7) -7.65 2D Binding mode Binding energy (Kcal/mol) -6.9 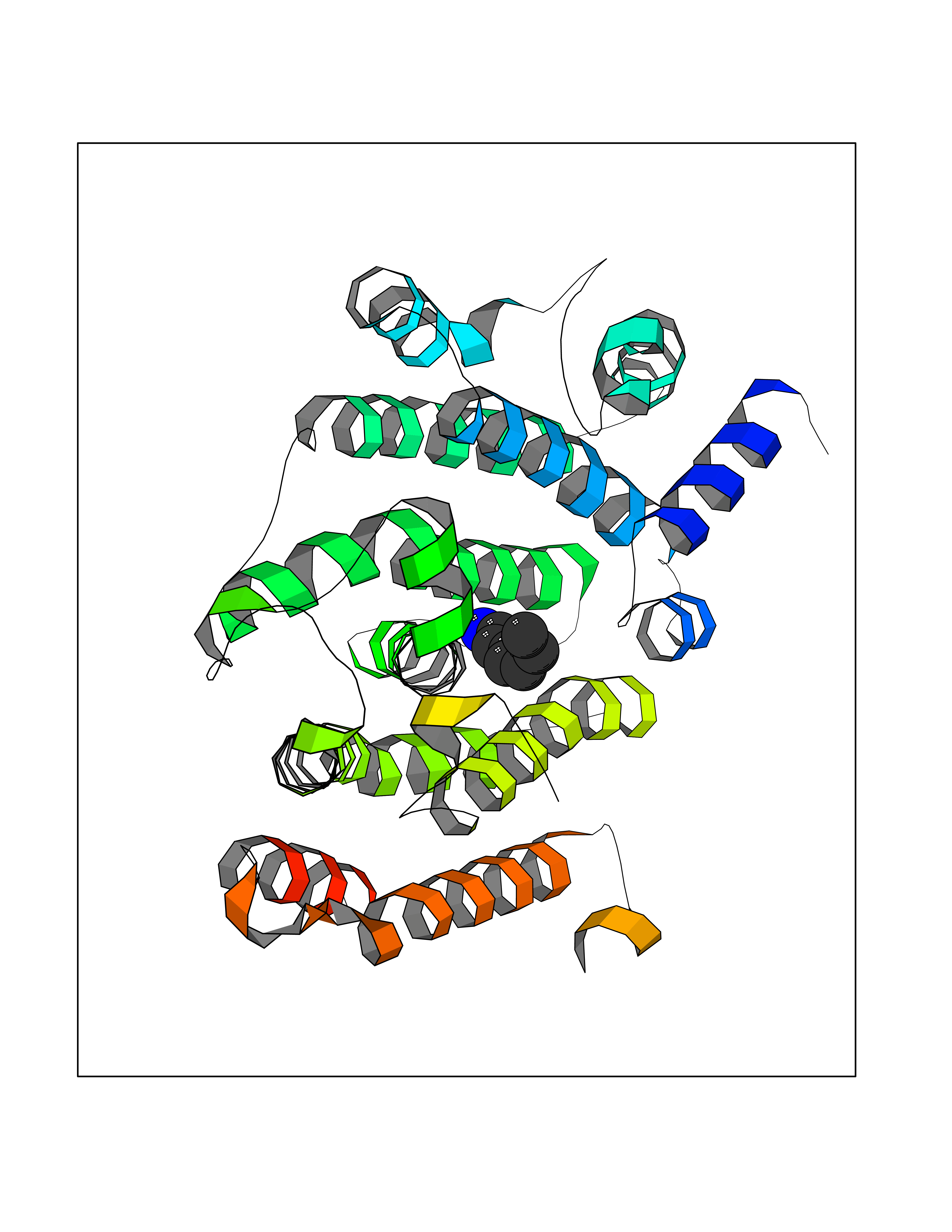 Molscript Map 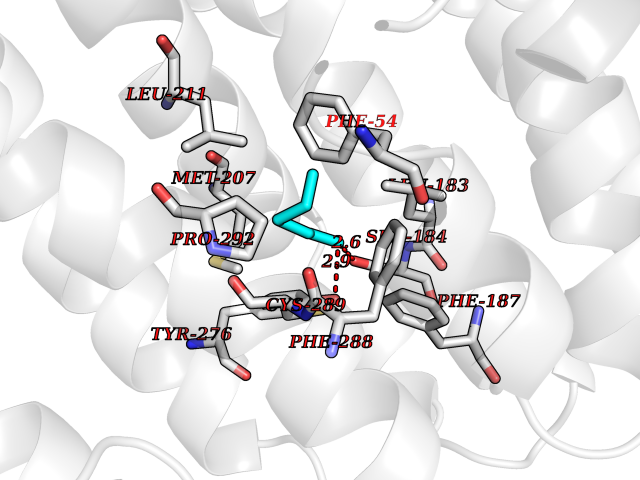 Pymol Map 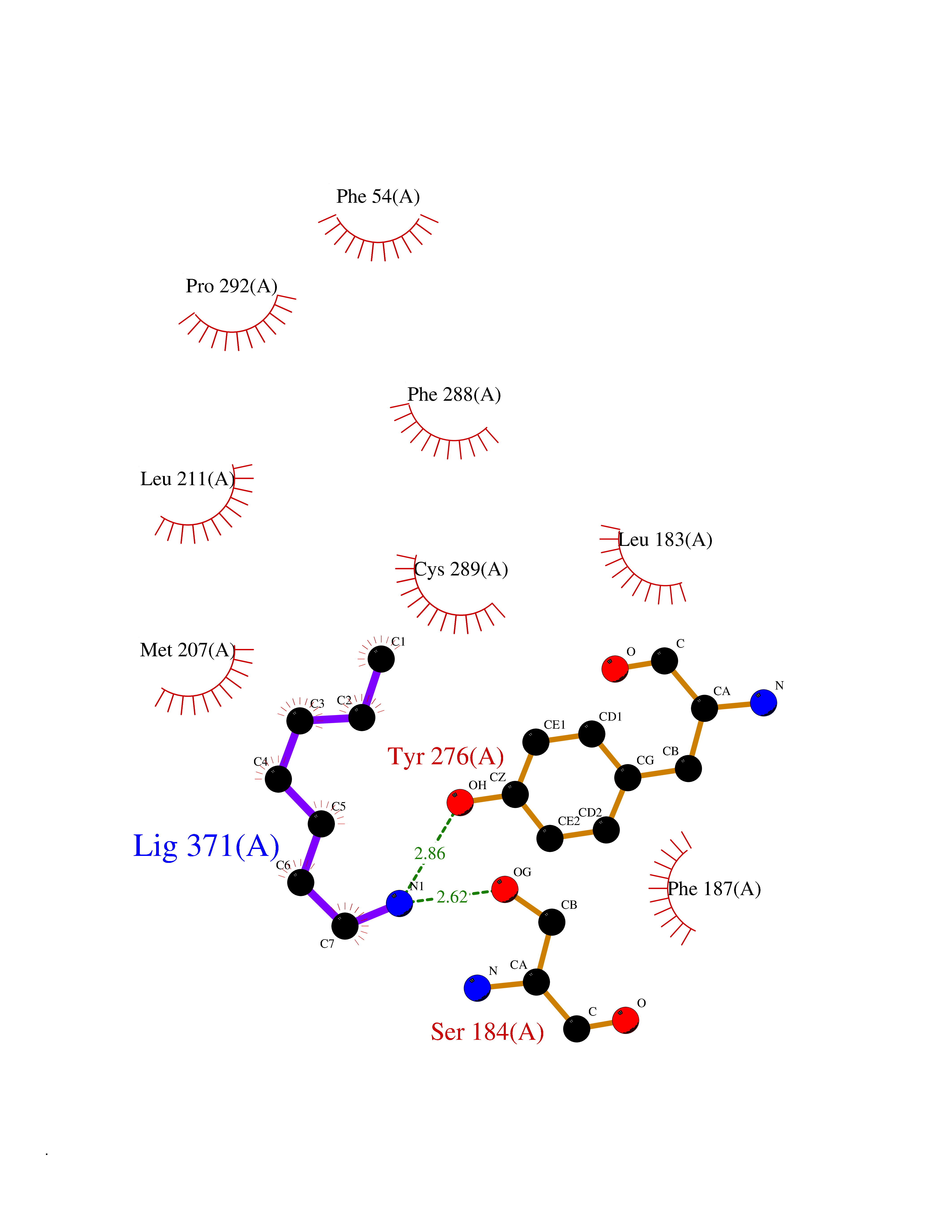 Ligplot Map 3D Binding mode Sequence LSSSLKTCYKYLNQTSRSFAAVIQALDGEMRNAVCIFYLVLRALDTLEDDMTISVEKKVPLLHNFHSFLYQPDWRFMESKEKDRQVLEDFPTISLEFRNLAEKYQTVIADICRRMGIGMAEFLDKHVTSEQEWDKYCHYVAGLVGIGLSRLFSASEFEDPLVGEDTERANSMGLFLQKTNIIRDYLEDQQGGREFWPQEVWSRYVKKLGDFALPENIDLAVQCLNELITNALHHIPDVITYLSRLRNQSVFNFCAIPQVMAIATLAACYNNQQVFKGAVLIVTLMMDATNMPAVKAIIYQYMEEIYHRIPDSNPSSSKTRQIISTIRTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Phosphatidylethanolamine-binding protein 1 (PEBP1) | 2QYQ | 5.06 | |
Target general information Gen name PEBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Raf kinase inhibitor protein; RKIP; Prostatic-binding protein; PEBP-1; PEBP; PBP; Neuropolypeptide h3; Hippocampal cholinergic neurostimulating peptide; HCNPpp; HCNP Protein family Phosphatidylethanolamine-binding protein family Biochemical class Phosphatidylethanolamine-binding protein family Function Binds ATP, opioids and phosphatidylethanolamine. Has lower affinity for phosphatidylinositol and phosphatidylcholine. Serine protease inhibitor which inhibits thrombin, neuropsin and chymotrypsin but not trypsin, tissue type plasminogen activator and elastase. Inhibits the kinase activity of RAF1 by inhibiting its activation and by dissociating the RAF1/MEK complex and acting as a competitive inhibitor of MEK phosphorylation. Related diseases Retinitis pigmentosa 49 (RP49) [MIM:613756]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:15570217, ECO:0000269|PubMed:7479749}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09130; DB09568 Interacts with P16050; Q9NRD5; P04049; Q15208; Q9NS68; Q9JLL3 EC number NA Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Direct protein sequencing; Disulfide bond; Lipid-binding; Nucleotide-binding; Phosphoprotein; Protease inhibitor; Proteomics identification; Reference proteome; Serine protease inhibitor Protein physicochemical properties Chain ID A Molecular weight (Da) 20928.3 Length 186 Aromaticity 0.1 Instability index 24.05 Isoelectric point 6.59 Charge (pH=7) -0.98 2D Binding mode Binding energy (Kcal/mol) -6.9  Molscript Map 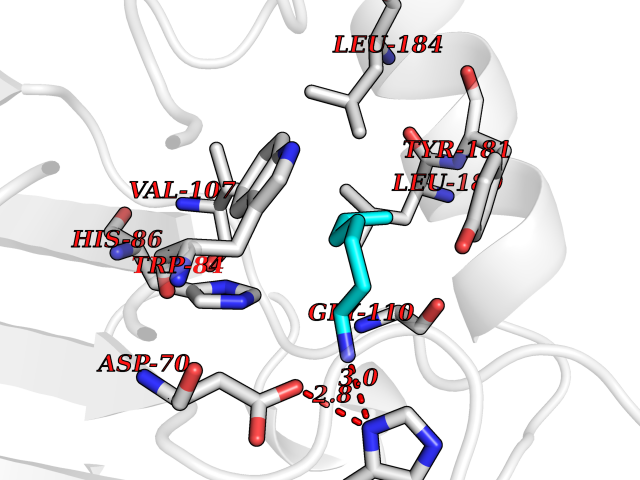 Pymol Map 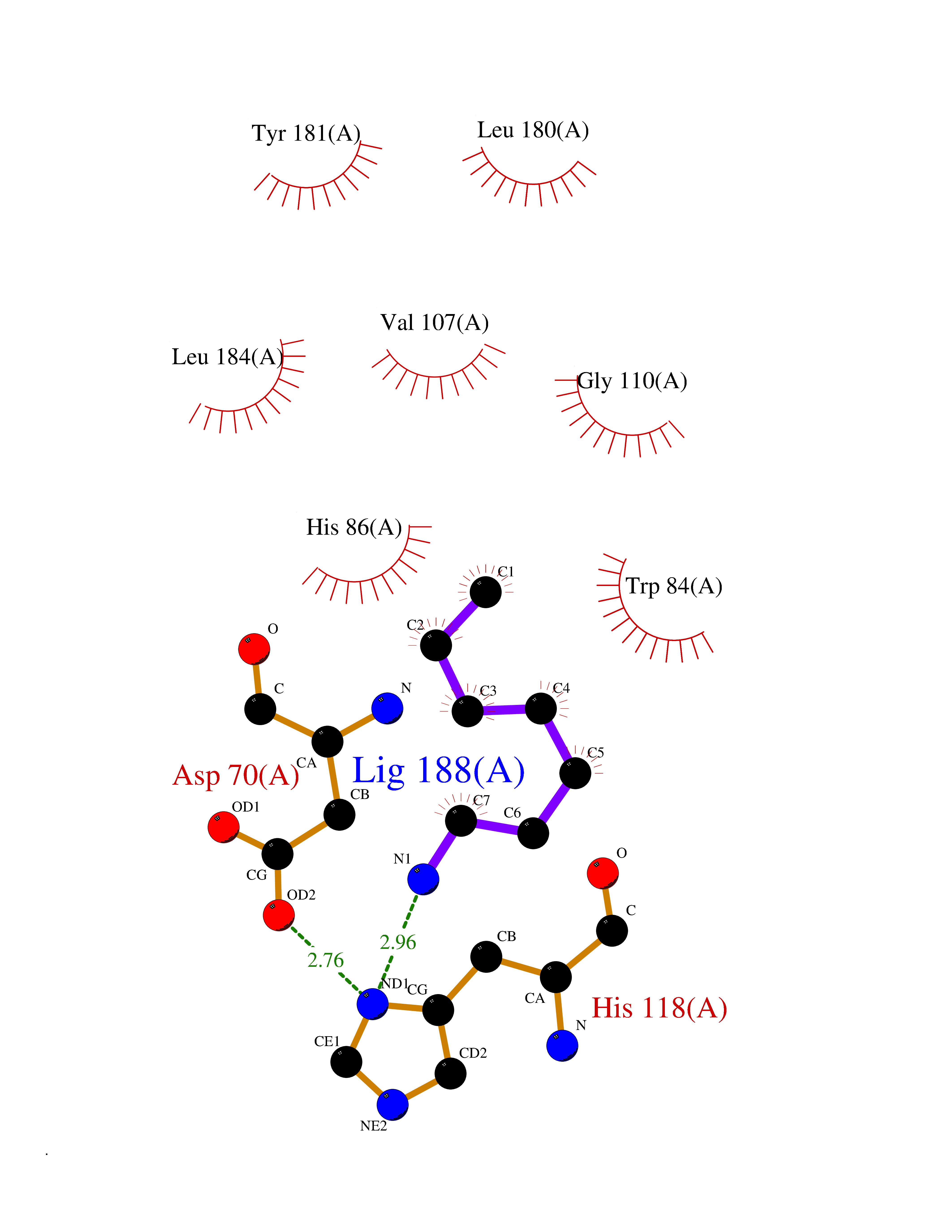 Ligplot Map 3D Binding mode Sequence MPVDLSKWSGPLSLQEVDEQPQHPLHVTYAGAAVDELGKVLTPTQVKNRPTSISWDGLDSGKLYTLVLTDPDAPSRKDPKYREWHHFLVVNMKGNDISSGTVLSDYVGSGPPKGTGLHRYVWLVYEQDRPLKCDEPILSNRSGDHRGKFKVASFRKKYELRAPVAGTCYQAEWDDYVPKLYEQLSG Hydrogen bonds contact Hydrophobic contact | ||||