Job Results:
Ligand
Structure
Job ID
d1681d1a563fba4481a5af742fb3bb0c
Job name
NA
Time
2026-02-27 11:58:22
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Neuronal acetylcholine receptor alpha-2 (CHRNA2) | 5FJV | 4.64 | |
Target general information Gen name CHRNA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNA2 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-2/CHRNA2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Epilepsy, nocturnal frontal lobe, 4 (ENFL4) [MIM:610353]: An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking. {ECO:0000269|PubMed:16826524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Seizures, benign familial infantile, 6 (BFIS6) [MIM:610353]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS6 inheritance is autosomal dominant. {ECO:0000269|PubMed:25847220}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00732; DB00237; DB00411; DB00565; DB01245; DB00514; DB01135; DB07720; DB00898; DB00472; DB00483; DB08960; DB00657; DB01336; DB00416; DB01226; DB00184; DB01337; DB01338; DB00721; DB00728; DB05740; DB00202; DB01199; DB01339 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 48323.4 Length 413 Aromaticity 0.15 Instability index 32 Isoelectric point 5.69 Charge (pH=7) -6.58 2D Binding mode Binding energy (Kcal/mol) -6.33  Molscript Map  Pymol Map 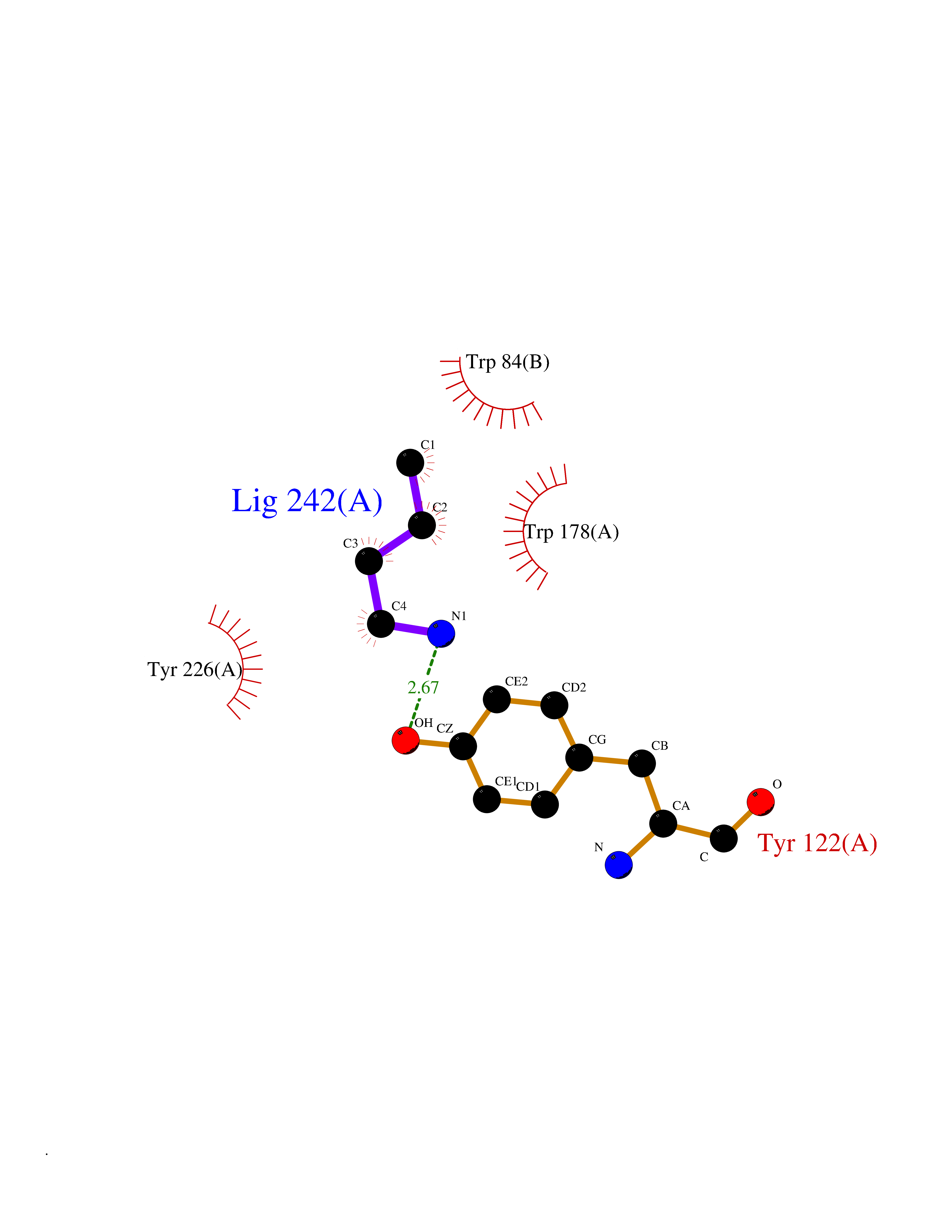 Ligplot Map 3D Binding mode Sequence DRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLP Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Lysine-specific demethylase 5B (KDM5B) | 5FY9 | 4.64 | |
Target general information Gen name KDM5B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoblastomabinding protein 2 homolog 1; Retinoblastoma-binding protein 2 homolog 1; RBP2H1; RBP2-H1; RBBP2H1; PLU1; PLU-1; Lysinespecific demethylase 5B; Jumonji/ARID domaincontaining protein 1B; J Protein family JARID1 histone demethylase family Biochemical class Paired donor oxygen oxidoreductase Function Does not demethylate histone H3 'Lys-9' or H3 'Lys-27'. Demethylates trimethylated, dimethylated and monomethylated H3 'Lys-4'. Acts as a transcriptional corepressor for FOXG1B and PAX9. Favors the proliferation of breast cancer cells by repressing tumor suppressor genes such as BRCA1 and HOXA5. In contrast, may act as a tumor suppressor for melanoma. Represses the CLOCK-ARNTL/BMAL1 heterodimer-mediated transcriptional activation of the core clock component PER2. Histone demethylase that demethylates 'Lys-4' of histone H3, thereby playing a central role in histone code. Related diseases Intellectual developmental disorder, autosomal recessive 65 (MRT65) [MIM:618109]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT65 patients have moderate to severe intellectual disability, developmental delay, and facial dysmorphism. Camptodactyly is present in some patients. {ECO:0000269|PubMed:29276005, ECO:0000269|PubMed:30409806}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P49711 EC number EC 1.14.11.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Dioxygenase; Disease variant; Intellectual disability; Iron; Isopeptide bond; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Repressor; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 53020.6 Length 460 Aromaticity 0.12 Instability index 44.23 Isoelectric point 5.28 Charge (pH=7) -18.32 2D Binding mode Binding energy (Kcal/mol) -6.32  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SMFLPPPECPVFEPSWEEFADPFAFIHKIRPIAEQTGICKVRPPPDWQPPFACDVDKLHFTPRIQRLNELEAQTRVKLGGGGARDYTLRTFGEMADAFKSDYFNMPVHMVPTELVEKEFWRLVSTIEEDVTVEYGADIASKEFGSGFPVRDIKLSPEEEEYLDSGWNLNNMPVMEQSVLAHITADICGMKLPWLYVGMCFSSFCWHIEDHWSYSINYLHWGEPKTWYGVPGYAAEQLENVMKKLAPELFVSQPDLLHQLVTIMNPNTLMTHEVPVYRTNQCAGEFVITFPRAYHSGFNQGFNFAEAVNFCTVDWLPLGRQCVEHYRLLHRYCVFSHDEMICKMASKADVLDVVVASTVQKDMAIMIEDEKALRETVRKLGVIDSERMDFELLPDDERQCVKCKTTCFMSAISCSCKPGLLVCLHHVKELCSCPPYKYKLRYRYTLDDLYPMMNALKLRAE Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Lysine N-methyltransferase 3B (NSD1) | 3OOI | 4.64 | |
Target general information Gen name NSD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor-binding SET domain-containing protein 1; NR-binding SET domain-containing protein; NR-binding SET domain containing protein; KMT3B; Hypothetical protein FLJ22263 similar to nuclear re Protein family Class V-like SAM-binding methyltransferase superfamily Biochemical class Methyltransferase Function Preferentially methylates 'Lys-36' of histone H3 and 'Lys-20' of histone H4 (in vitro). Transcriptional intermediary factor capable of both negatively or positively influencing transcription, depending on the cellular context. Histone methyltransferase. Related diseases Sotos syndrome (SOTOS) [MIM:117550]: An autosomal dominant, childhood overgrowth syndrome characterized by pre- and postnatal overgrowth, developmental delay, intellectual disability, advanced bone age, and abnormal craniofacial morphology including macrodolichocephaly with frontal bossing, frontoparietal sparseness of hair, apparent hypertelorism, downslanting palpebral fissures, and facial flushing. Common oral findings include: premature eruption of teeth; high, arched palate; pointed chin and, more rarely, prognathism. {ECO:0000269|PubMed:11896389, ECO:0000269|PubMed:12464997, ECO:0000269|PubMed:12807965, ECO:0000269|PubMed:14997421}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Beckwith-Wiedemann syndrome (BWS) [MIM:130650]: A disorder characterized by anterior abdominal wall defects including exomphalos (omphalocele), pre- and postnatal overgrowth, and macroglossia. Additional less frequent complications include specific developmental defects and a predisposition to embryonal tumors. {ECO:0000269|PubMed:14997421}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A chromosomal aberration involving NSD1 is found in childhood acute myeloid leukemia. Translocation t(5;11)(q35;p15.5) with NUP98.; DISEASE: A chromosomal aberration involving NSD1 is found in an adult form of myelodysplastic syndrome (MDS). Insertion of NUP98 into NSD1 generates a NUP98-NSD1 fusion product. {ECO:0000269|PubMed:15382262}. Drugs (DrugBank ID) NA Interacts with Q13283; Q16778; Q04206; O95994; Q86Z20; A8MQ03; Q3LI66; Q99750 EC number EC 2.1.1.43 Uniprot keywords 3D-structure; Activator; Alternative splicing; Chromatin regulator; Chromosomal rearrangement; Chromosome; Disease variant; Isopeptide bond; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; Repeat; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 26487.9 Length 232 Aromaticity 0.08 Instability index 37.29 Isoelectric point 8.1 Charge (pH=7) 2.69 2D Binding mode Binding energy (Kcal/mol) -6.32  Molscript Map 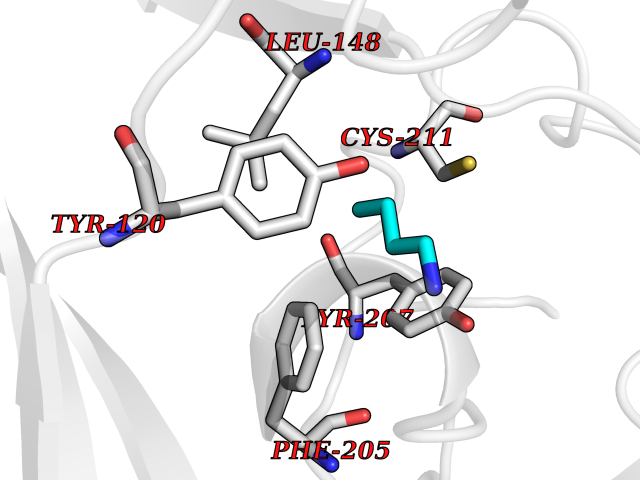 Pymol Map 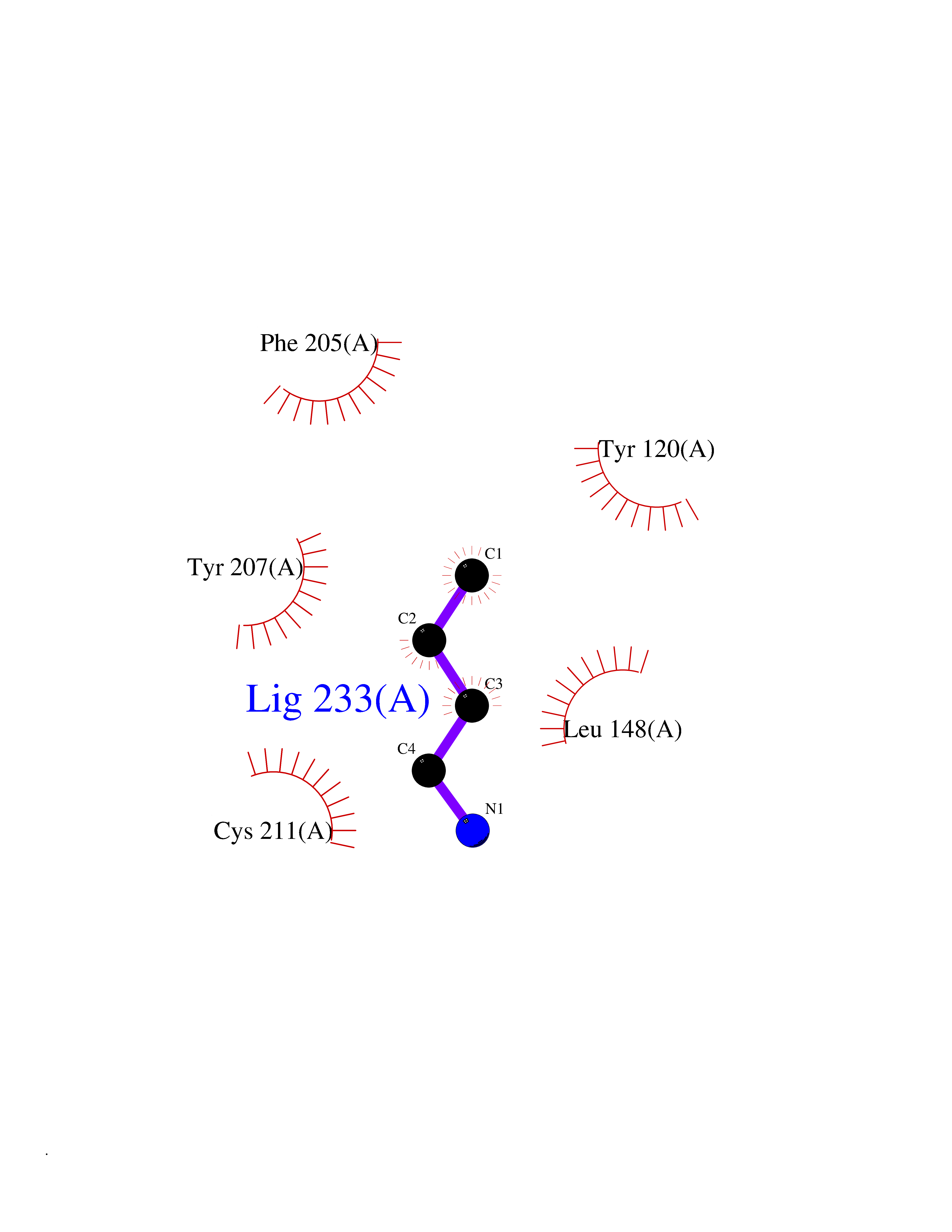 Ligplot Map 3D Binding mode Sequence SKELRQLQEDRKNDKKPPPYKHIKVNRPIGRVQIFTADLSEIPRCNCKATDENPCGIDSECINRMLLYECHPTVCPAGGRCQNQCFSKRQYPEVEIFRTLQRGWGLRTKTDIKKGEFVNEYVGELIDEEECRARIRYAQEHDITNFYMLTLDKDRIIDAGPKGNYARFMNHCCQPNCETQKWSVNGDTRVGLFALSDIKAGTELTFNYNLECLGNGKTVCKCGAPNCSGFLG Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | C-C chemokine receptor type 5 (CCR5) | 4MBS | 4.63 | |
Target general information Gen name CCR5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HIV-1 fusion coreceptor; HIV-1 fusion co-receptor; Chemokine receptor CCR5; CMKBR5; CHEMR13; CD195 antigen; CD195; CCR-5; CC-CKR-5; C-C CKR-5 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function May play a role in the control of granulocytic lineage proliferation or differentiation. Receptor for a number of inflammatory CC-chemokines including CCL3/MIP-1-alpha, CCL4/MIP-1-beta and RANTES and subsequently transduces a signal by increasing the intracellular calcium ion level. Related diseases Type 1 diabetes mellitus 22 (T1D22) [MIM:612522]: A multifactorial disorder of glucose homeostasis that is characterized by susceptibility to ketoacidosis in the absence of insulin therapy. Clinical features are polydipsia, polyphagia and polyuria which result from hyperglycemia-induced osmotic diuresis and secondary thirst. These derangements result in long-term complications that affect the eyes, kidneys, nerves, and blood vessels. {ECO:0000269|PubMed:19073967}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06497; DB05906; DB12698; DB12960; DB05941; DB04835; DB05501; DB06652 Interacts with Q16570-2; Q92583; PRO_0000005165 [P13236]; P13501; P51681; P01730; P61073; P54849; O54081 EC number NA Uniprot keywords 3D-structure; Cell membrane; Diabetes mellitus; Disulfide bond; G-protein coupled receptor; Glycoprotein; Host cell receptor for virus entry; Host-virus interaction; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Sulfation; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 33641.8 Length 291 Aromaticity 0.16 Instability index 26.12 Isoelectric point 9.4 Charge (pH=7) 11.41 2D Binding mode Binding energy (Kcal/mol) -6.31 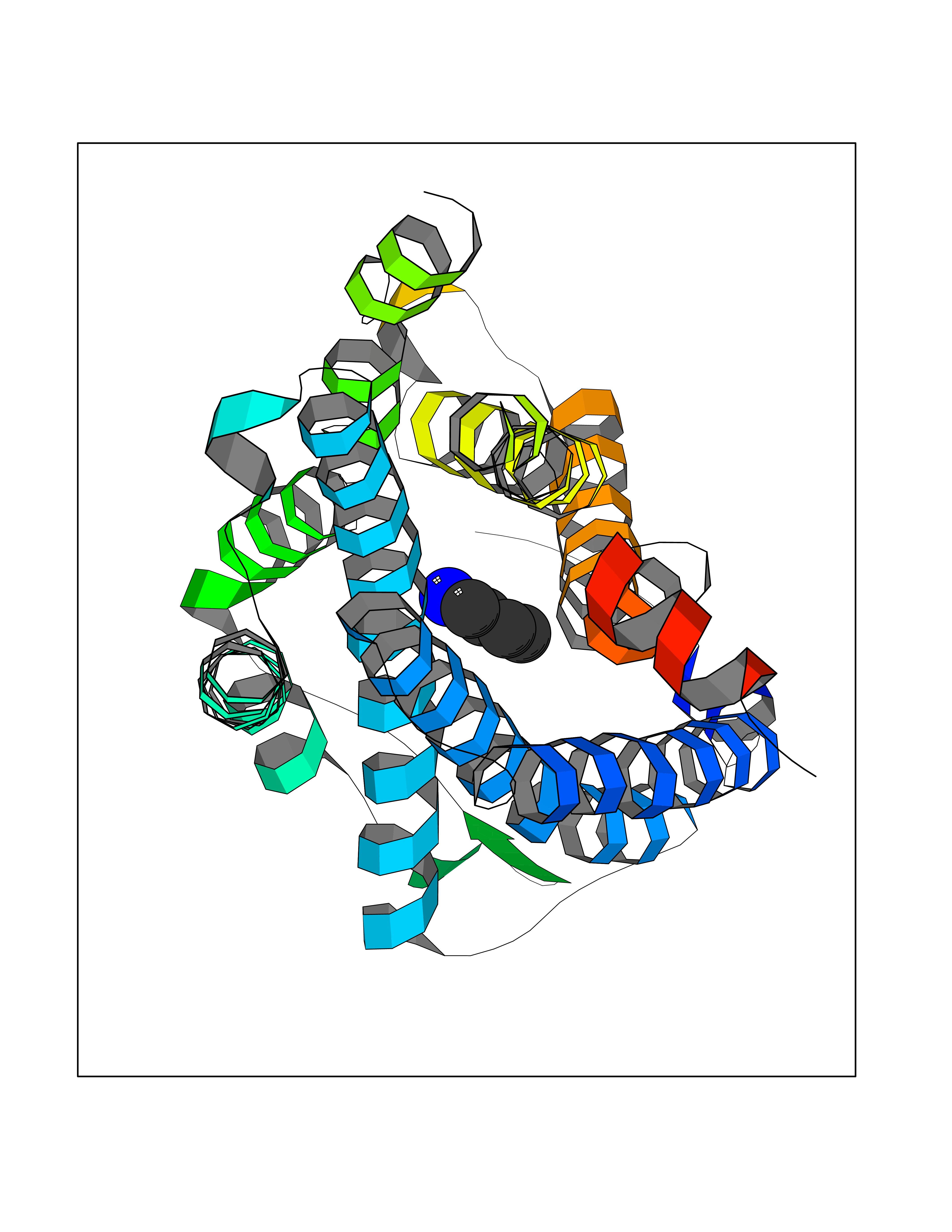 Molscript Map 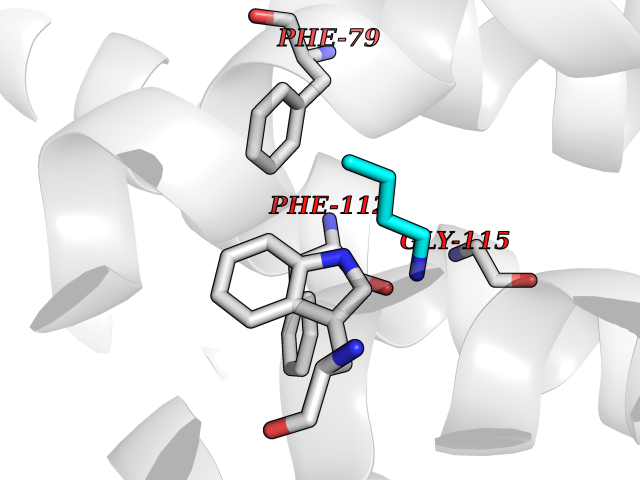 Pymol Map 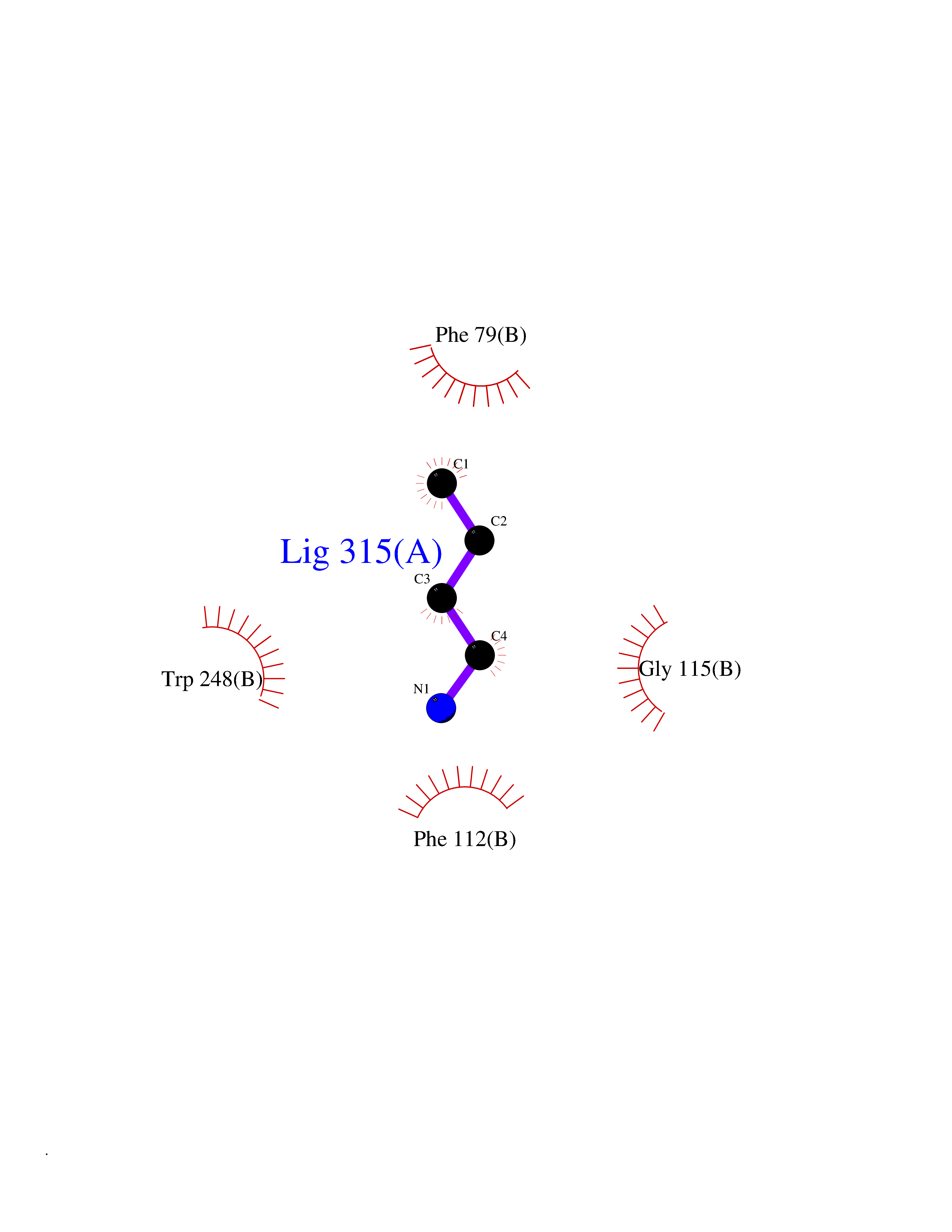 Ligplot Map 3D Binding mode Sequence PCQKINVKQIAARLLPPLYSLVFIFGFVGNMLVILILINYKRLKSMTDIYLLNLAISDLFFLLTVPFWAHYAAAQWDFGNTMCQLLTGLYFIGFFSGIFFIILLTIDRYLAVVHAVFALKARTVTFGVVTSVITWVVAVFASLPNIIFTRSQKEGLHYTCSSHFPYSQYQFWKNFQTLKIVILGLVLPLLVMVICYSGILKTLLRKKRHRDVRLIFTIMIVYFLFWAPYNIVLLLNTFQEFFGLNNCSSSNRLDQAMQVTETLGMTHCCINPIIYAFVGEEFRNYLLVFFQ Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Acetyl-CoA carboxylase 1 | 2YL2 | 4.63 | |
Target general information Gen name ACACA Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACAC;ACCA;ACC1 Protein family NA Biochemical class Ligase Function Acetyl-CoA carboxylase activity.ATP binding.Biotin carboxylase activity.Identical protein binding.Metal ion binding. Related diseases Acetyl-CoA carboxylase-alpha deficiency (ACACAD) [MIM:613933]: An autosomal recessive inborn error of de novo fatty acid synthesis associated with severe brain damage, persistent myopathy and poor growth. {ECO:0000269|PubMed:6114432}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00121 Interacts with Q13085; O60218; P38398; Q96EB6; Q9CQ20; P02654; Q92915-2; Q6NTF9-3 EC number 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative promoter usage; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Magnesium; Manganese; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 54237.7 Length 486 Aromaticity 0.09 Instability index 39.18 Isoelectric point 6.37 Charge (pH=7) -2.46 2D Binding mode Binding energy (Kcal/mol) -6.31  Molscript Map 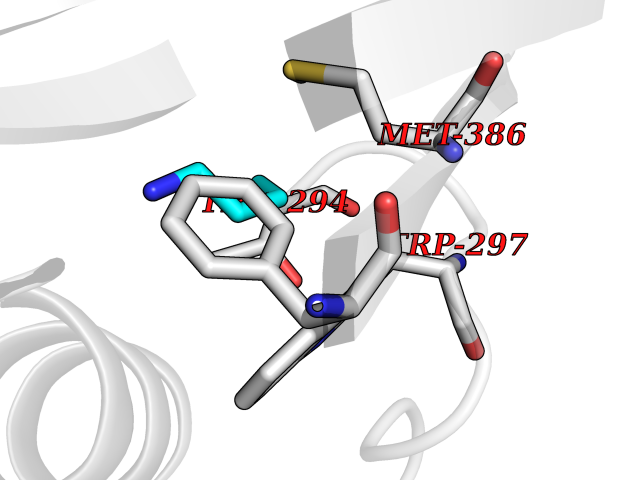 Pymol Map  Ligplot Map 3D Binding mode Sequence VASPAEFVTRFGGNKVIEKVLIANNGIAAVKCMRSIRRWSYEMFRNERAIRFVVMVTPEDLKANAEYIKMADHYVPVPGGPNNNNYANVELILDIAKRIPVQAVWAGWGHASENPKLPELLLKNGIAFMGPPSQAMWALGDKIASSIVAQTAGIPTLPWSGSGLRVDWSKRILNVPQELYEKGYVKDVDDGLQAAEEVGYPVMIKASEGGGGKGIRKVNNADDFPNLFRQVQAEVPGSPIFVMRLAKQSRHLEVQILADQYGNAISLFGRDCSVQRRHQKIIEEAPATIATPAVFEHMEQCAVKLAKMVGYVSAGTVEYLYSQDGSFYFLELNPRLQVEHPCTEMVADVNLPAAQLQIAMGIPLYRIKDIRMMYGVSPWGDSPIDFEDSAHVPCPRGHVIAARITGTVQELNFRSNKNVWGYFSVQFGHCFSWGENREEAISNMVVALKELSIRGDFRTTVEYLIKLLETESFQMNRIDTGWLDRL Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Proto-oncogene c-Met (MET) | 3DKC | 4.63 | |
Target general information Gen name MET Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase Met; Scatter factor receptor; SF receptor; Met proto-oncogene tyrosine kinase; Hepatocyte growth factor receptor; HGF/SF receptor; HGF-SF receptor; HGF receptor; C-met; C-Met r Protein family Protein kinase superfamily, Tyr protein kinase family Biochemical class Kinase Function Regulates many physiological processes including proliferation, scattering, morphogenesis and survival. Ligand binding at the cell surface induces autophosphorylation of MET on its intracellular domain that provides docking sites for downstream signaling molecules. Following activation by ligand, interacts with the PI3-kinase subunit PIK3R1, PLCG1, SRC, GRB2, STAT3 or the adapter GAB1. Recruitment of these downstream effectors by MET leads to the activation of several signaling cascades including the RAS-ERK, PI3 kinase-AKT, or PLCgamma-PKC. The RAS-ERK activation is associated with the morphogenetic effects while PI3K/AKT coordinates prosurvival effects. During embryonic development, MET signaling plays a role in gastrulation, development and migration of muscles and neuronal precursors, angiogenesis and kidney formation. In adults, participates in wound healing as well as organ regeneration and tissue remodeling. Promotes also differentiation and proliferation of hematopoietic cells. May regulate cortical bone osteogenesis. Receptor tyrosine kinase that transduces signals from the extracellular matrix into the cytoplasm by binding to hepatocyte growth factor/HGF ligand. Related diseases Activation of MET after rearrangement with the TPR gene produces an oncogenic protein.; DISEASE: Defects in MET may be associated with gastric cancer.; DISEASE: Hepatocellular carcinoma (HCC) [MIM:114550]: A primary malignant neoplasm of epithelial liver cells. The major risk factors for HCC are chronic hepatitis B virus (HBV) infection, chronic hepatitis C virus (HCV) infection, prolonged dietary aflatoxin exposure, alcoholic cirrhosis, and cirrhosis due to other causes. {ECO:0000269|PubMed:9927037}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Renal cell carcinoma papillary (RCCP) [MIM:605074]: A subtype of renal cell carcinoma tending to show a tubulo-papillary architecture formed by numerous, irregular, finger-like projections of connective tissue. Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. {ECO:0000269|PubMed:10327054, ECO:0000269|PubMed:10417759, ECO:0000269|PubMed:10433944, ECO:0000269|PubMed:9140397, ECO:0000269|PubMed:9563489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A common allele in the promoter region of the MET shows genetic association with susceptibility to autism in some families. Functional assays indicate a decrease in MET promoter activity and altered binding of specific transcription factor complexes.; DISEASE: MET activating mutations may be involved in the development of a highly malignant, metastatic syndrome known as cancer of unknown primary origin (CUP) or primary occult malignancy. Systemic neoplastic spread is generally a late event in cancer progression. However, in some instances, distant dissemination arises at a very early stage, so that metastases reach clinical relevance before primary lesions. Sometimes, the primary lesions cannot be identified in spite of the progresses in the diagnosis of malignancies.; DISEASE: Deafness, autosomal recessive, 97 (DFNB97) [MIM:616705]: A form of non-syndromic sensorineural hearing loss with prelingual onset. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:25941349}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Osteofibrous dysplasia (OSFD) [MIM:607278]: A congenital disorder of osteogenesis characterized by non-neoplastic, radiolucent lesions that affect the cortical bone immediately under the periosteum. It usually manifests as a painless swelling or anterior bowing of the long bones, most commonly the tibia and fibula. {ECO:0000269|PubMed:26637977}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Disease-associated variants identified in 4 families cause the deletion of exon 14. This results in the exclusion of an ubiquitination target site within the cytoplasmic domain, hence in protein stabilization. The persistent presence of MET at the cell surface in conditions of ligand-dependent activation retards osteoblastic differentiation. {ECO:0000269|PubMed:26637977}.; DISEASE: Arthrogryposis, distal, 11 (DA11) [MIM:620019]: A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. DA11 is an autosomal dominant form characterized mainly by camptodactyly. Other features include absent flexion creases and limited forearm supination. {ECO:0000269|PubMed:30777867}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06896; DB08791; DB06997; DB07969; DB08079; DB16695; DB12742; DB12267; DB08875; DB11791; DB08865; DB12010; DB02152; DB07369; DB06995; DB06314; DB01268; DB15133; DB12200; DB11800 Interacts with P22681; Q96EY1; Q96EY1-2; P00533; P09769; P14210; P14210-6; O15357; P35968; P06239; P07948; P08581; P41218; P15941; P16333; O43639; Q16288; P27986; O00459; Q92569; P19174; O43157; O15031; Q9ULL4; Q8TCU6; P18031; Q06124; P23467; Q12913; Q16827; P20936; Q9UQQ2; O60880; O14796; Q9NP31; Q8N5H7; Q15464; P29353; P98077; Q6S5L8; Q96IW2; Q9H6Q3; O75159; O14544; P12931; Q9ULZ2; P43405; P42680; Q9HBL0; Q63HR2; Q68CZ2; Q9UKW4; P07947; P43403; Q08048; P0DQD2; P35918; Q00944 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromosomal rearrangement; Deafness; Disease variant; Disulfide bond; Glycoprotein; Kinase; Membrane; Non-syndromic deafness; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35229.5 Length 312 Aromaticity 0.1 Instability index 37.98 Isoelectric point 7.79 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -6.32 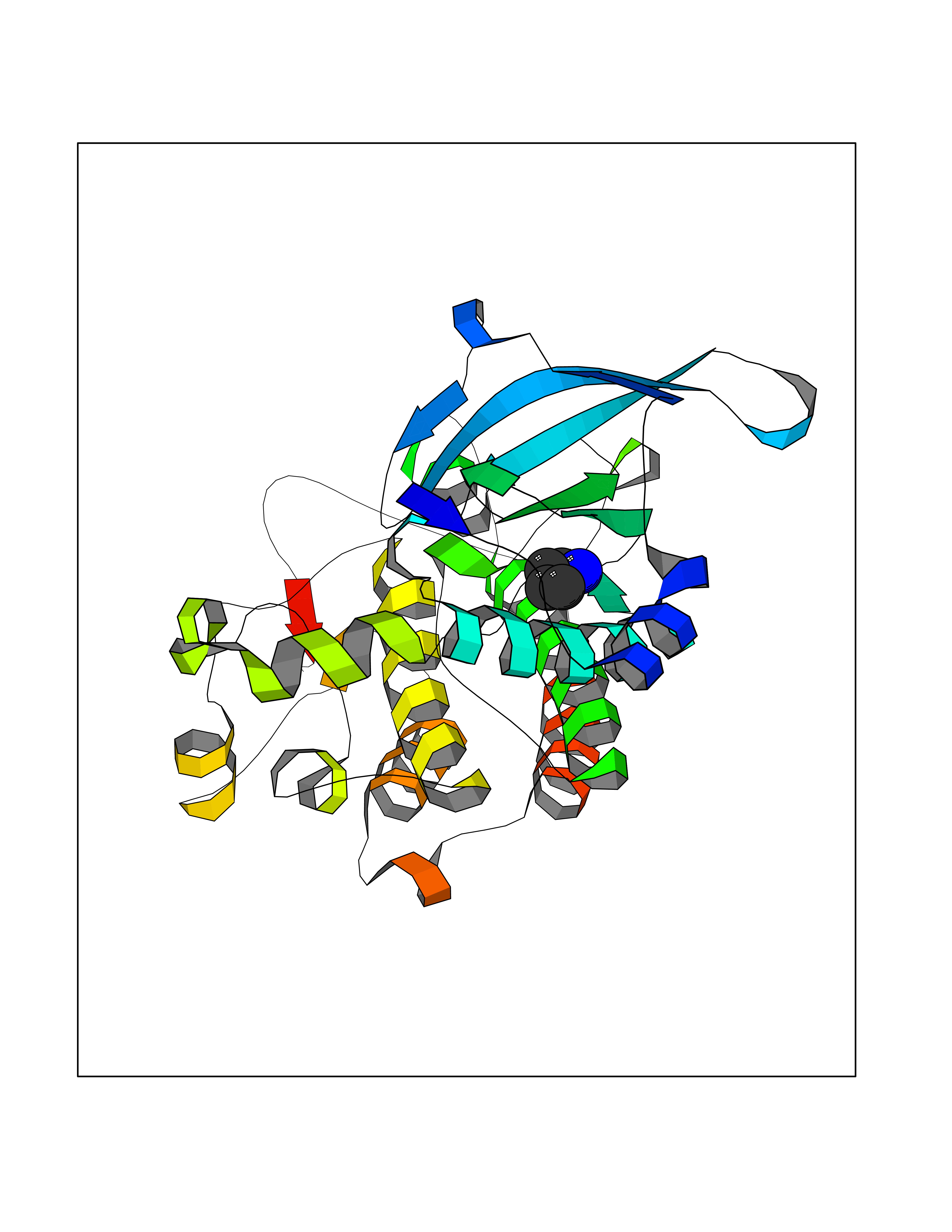 Molscript Map 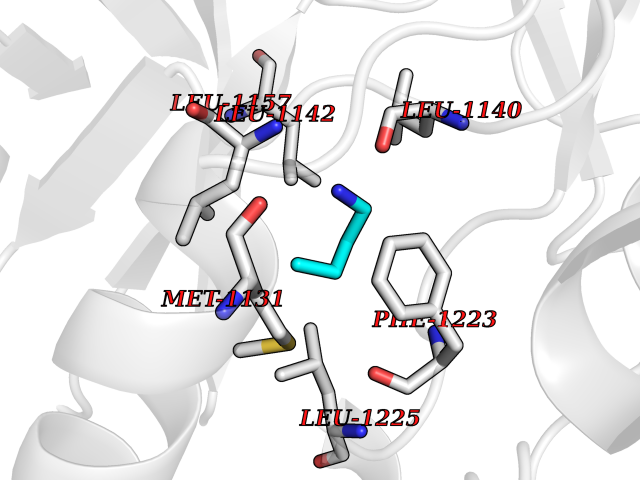 Pymol Map 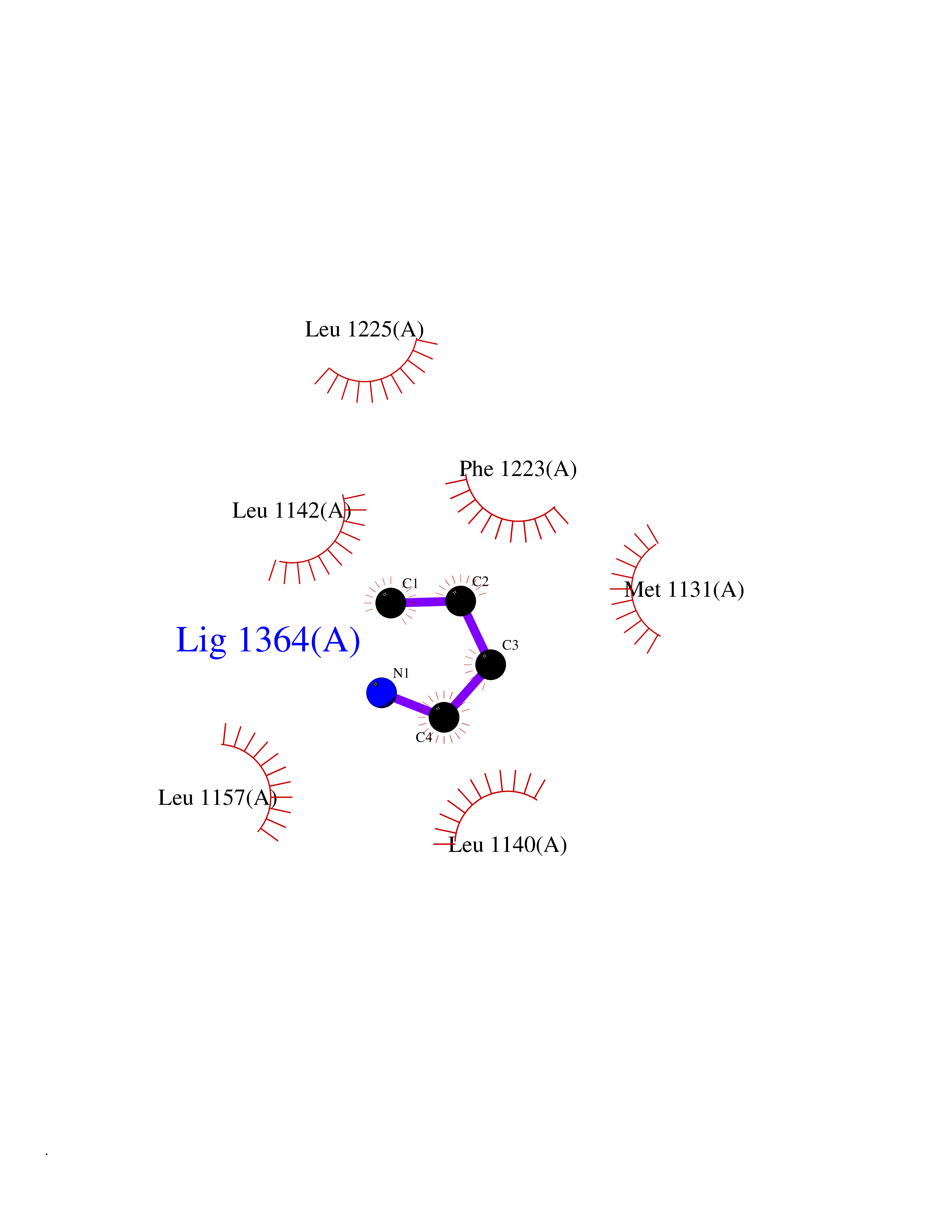 Ligplot Map 3D Binding mode Sequence VHIDLSALNPELVQAVQHVVIGPSSLIVHFNEVIGRGHFGCVYHGTLLDNDGKKIHCAVKSLNRITDIGEVSQFLTEGIIMKDFSHPNVLSLLGICLRSEGSPLVVLPYMKHGDLRNFIRNETHNPTVKDLIGFGLQVAKGMKFLASKKFVHRDLAARNCMLDEKFTVKVADFGLARDMYDKEFDSVHNKTGAKLPVKWMALESLQTQKFTTKSDVWSFGVLLWELMTRGAPPYPDVNTFDITVYLLQGRRLLQPEYCPDPLYEVMLKCWHPKAEMRPSFSELVSRISAIFSTFIGEHYVHVNATYVNVKEG Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Dipeptidyl peptidase 9 (DPP-9) | 6EOR | 4.63 | |
Target general information Gen name DPP9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dipeptidyl peptidase-like protein 9; Dipeptidyl peptidase IX; Dipeptidyl peptidase IV-related protein 2; DPRP2; DPRP-2; DPP IX; DPLP9; DP9 Protein family Peptidase S9B family, DPPIV subfamily Biochemical class Peptidase Function Dipeptidyl peptidase that cleaves off N-terminal dipeptides from proteins having a Pro or Ala residue at position 2. Related diseases Hatipoglu immunodeficiency syndrome (HATIS) [MIM:620331]: An autosomal recessive immunologic disorder manifesting in infancy or early childhood, and characterized by failure to thrive, short stature, skin pigmentation abnormalities, pancytopenia, and susceptibility to recurrent infections. {ECO:0000269|PubMed:36112693}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NXR5; Q86TI2; Q6NUP5; P46379-2; Q8WUW1; Q96A83-2; O75190-2; O14645; Q01658; P29692-2; Q06787-7; Q9Y5Q9; O14901; Q9BVL2; Q96CV9; Q06830; P14678-2; P49458; Q11203; Q13148; P14927 EC number EC 3.4.14.5 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminopeptidase; Cytoplasm; Disease variant; Hydrolase; Nucleus; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A Molecular weight (Da) 92797.4 Length 808 Aromaticity 0.12 Instability index 37.45 Isoelectric point 6.34 Charge (pH=7) -8.98 2D Binding mode Binding energy (Kcal/mol) -6.31  Molscript Map 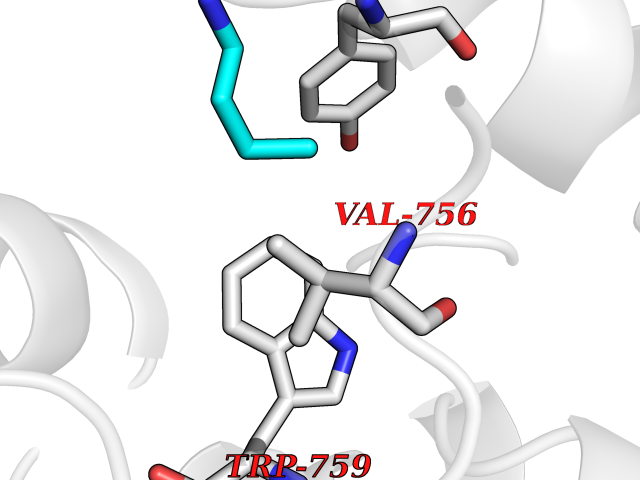 Pymol Map 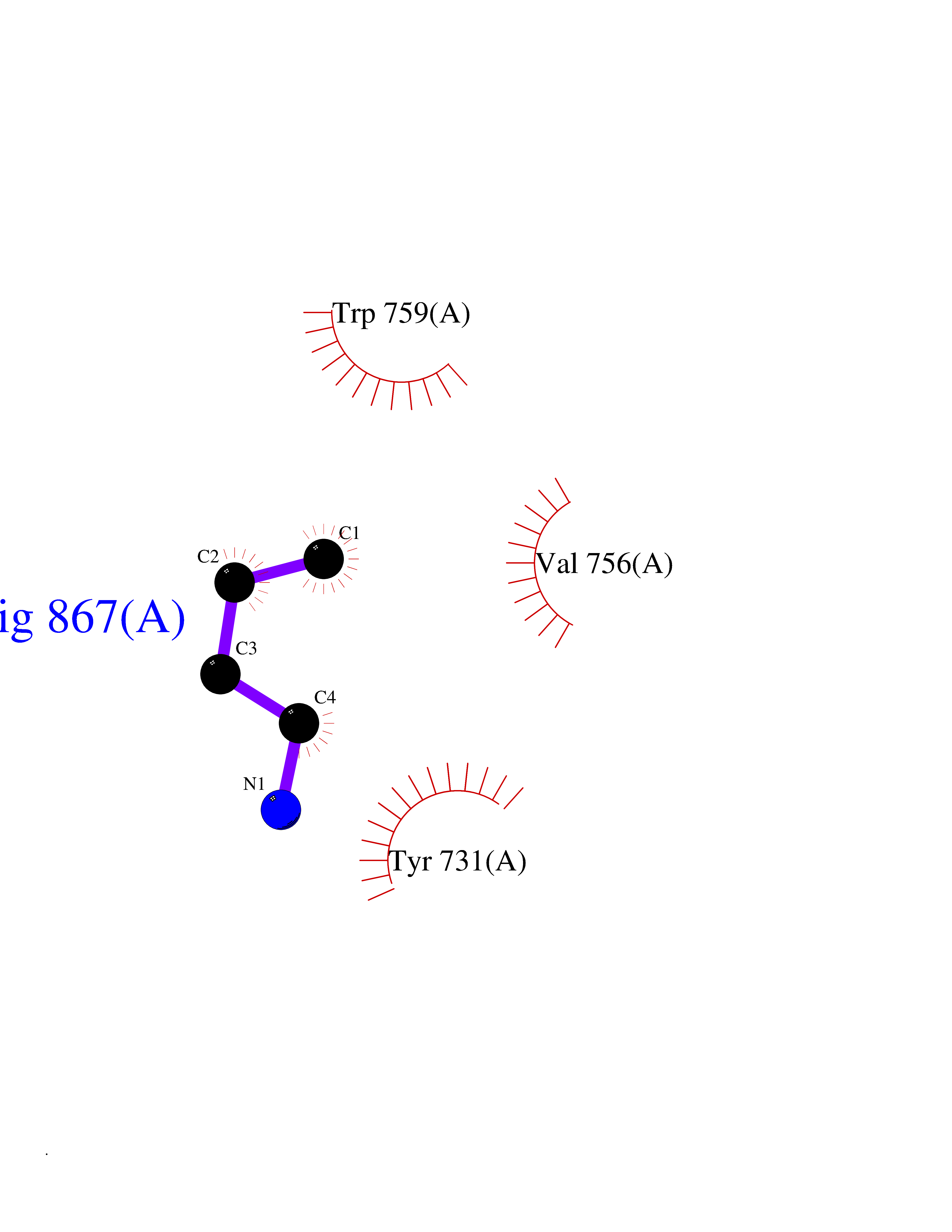 Ligplot Map 3D Binding mode Sequence AARFQVQKHSWDGLRSIIHGSRKAPHDFQFVQKSGPHSHRLYYLGMPYRENSLLYSEIPKLLLSWKQMLDHFQATPHHGVYSREEELLRERKRLGVFGITSYDFHSESGLFLFQASNSLFHCRDGGKNGFMVSPMKPLEIKTQCSGPRMDPKICPADPAFFSFINNSDLWVANIETGEERRLTFCHQNVLDDPKSAGVATFVIQEEFDRFTGYWWCPTASWEGLKTLRILYEEVDESEVEVIHVPSPALEERKTDSYRYPRTGSKNPKIALKLAEFQTDSQGKIVSTQEKELVQPFSSLFPKVEYIARAGWTRDGKYAWAMFLDRPQQWLQLVLLPPALFIPSTENEEQRLASARAVPRNVQPYVVYEEVTNVWINVHDIFYPFPQLCFLRANECKTGFCHLYKVTAVLKSQGYDWSEPFSPGEDEFKCPIKEEIALTSGEWEVLARHGSKIWVNEETKLVYFQGTKDTPLEHHLYVVSYEAAGEIVRLTTPGFSHSCSMSQNFDMFVSHYSSVSTPPCVHVYKLSGPDDDPLHKQPRFWASMMEADYVPPEIFHFHTRSDVRLYGMIYKPHALQPGKKHPTVLFVYGGPQVQLVNNSFKGIKYLRLNTLASLGYAVVVIDGRGSCQRGLRFEGALKNQMGQVEIEDQVEGLQFVAEKYGFIDLSRVAIHGWSYGGFLSLMGLIHKPQVFKVAIAGAPVTVWMAYDTGYTERYMDVPENNQHGYEAGSVALHVEKLPNEPNRLLILHGFLDENVHFFHTNFLVSQLIRAGKPYQLQIYPNERHSIRCPESGEHYEVTLLHFLQEYLHH Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Phosphatidylethanolamine-binding protein 1 (PEBP1) | 2QYQ | 4.63 | |
Target general information Gen name PEBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Raf kinase inhibitor protein; RKIP; Prostatic-binding protein; PEBP-1; PEBP; PBP; Neuropolypeptide h3; Hippocampal cholinergic neurostimulating peptide; HCNPpp; HCNP Protein family Phosphatidylethanolamine-binding protein family Biochemical class Phosphatidylethanolamine-binding protein family Function Binds ATP, opioids and phosphatidylethanolamine. Has lower affinity for phosphatidylinositol and phosphatidylcholine. Serine protease inhibitor which inhibits thrombin, neuropsin and chymotrypsin but not trypsin, tissue type plasminogen activator and elastase. Inhibits the kinase activity of RAF1 by inhibiting its activation and by dissociating the RAF1/MEK complex and acting as a competitive inhibitor of MEK phosphorylation. Related diseases Retinitis pigmentosa 49 (RP49) [MIM:613756]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:15570217, ECO:0000269|PubMed:7479749}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09130; DB09568 Interacts with P16050; Q9NRD5; P04049; Q15208; Q9NS68; Q9JLL3 EC number NA Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Direct protein sequencing; Disulfide bond; Lipid-binding; Nucleotide-binding; Phosphoprotein; Protease inhibitor; Proteomics identification; Reference proteome; Serine protease inhibitor Protein physicochemical properties Chain ID A Molecular weight (Da) 20928.3 Length 186 Aromaticity 0.1 Instability index 24.05 Isoelectric point 6.59 Charge (pH=7) -0.98 2D Binding mode Binding energy (Kcal/mol) -6.31  Molscript Map  Pymol Map 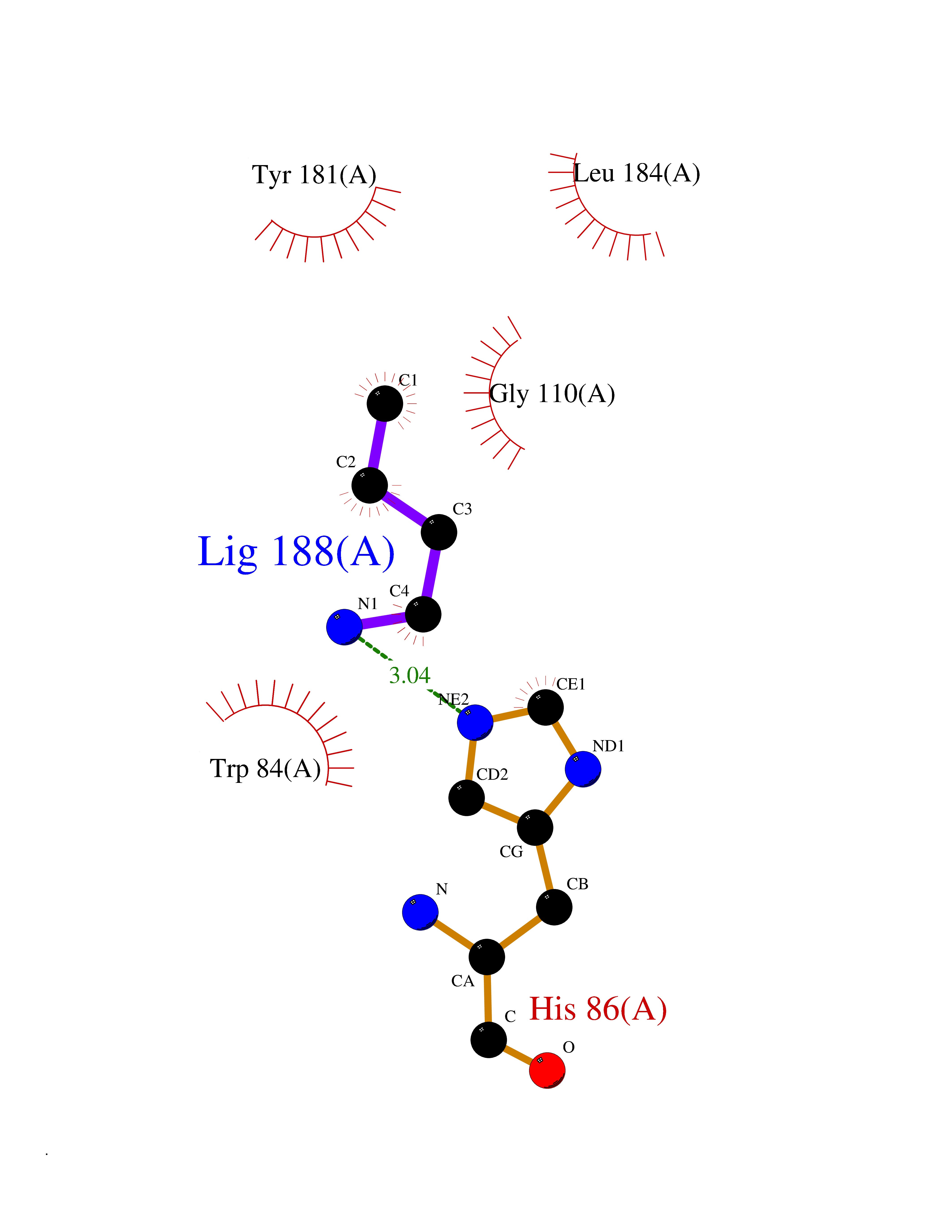 Ligplot Map 3D Binding mode Sequence MPVDLSKWSGPLSLQEVDEQPQHPLHVTYAGAAVDELGKVLTPTQVKNRPTSISWDGLDSGKLYTLVLTDPDAPSRKDPKYREWHHFLVVNMKGNDISSGTVLSDYVGSGPPKGTGLHRYVWLVYEQDRPLKCDEPILSNRSGDHRGKFKVASFRKKYELRAPVAGTCYQAEWDDYVPKLYEQLSG Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Acetylcholine receptor subunit alpha | 4ZJS | 4.62 | |
Target general information Gen name CHRNA1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACHRA;CHNRA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-1/CHRNA1 sub-subfamily Biochemical class Immune system Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ion channel activity.Ligand-gated ion channel activity. Related diseases Multiple pterygium syndrome, lethal type (LMPS) [MIM:253290]: Multiple pterygia are found infrequently in children with arthrogryposis and in fetuses with fetal akinesia syndrome. In lethal multiple pterygium syndrome there is intrauterine growth retardation, multiple pterygia, and flexion contractures causing severe arthrogryposis and fetal akinesia. Subcutaneous edema can be severe, causing fetal hydrops with cystic hygroma and lung hypoplasia. Oligohydramnios and facial anomalies are frequent. {ECO:0000269|PubMed:18252226}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: The alpha subunit is the main focus for antibody binding in myasthenia gravis. Myasthenia gravis is characterized by sporadic muscular fatigability and weakness, occurring chiefly in muscles innervated by cranial nerves, and characteristically improved by cholinesterase-inhibiting drugs.; DISEASE: Myasthenic syndrome, congenital, 1A, slow-channel (CMS1A) [MIM:601462]: A common congenital myasthenic syndrome. Congenital myasthenic syndromes are characterized by muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1A is a slow-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in prolonged AChR channel opening episodes, prolonged endplate currents, and depolarization block. This is associated with calcium overload, which may contribute to subsequent degeneration of the endplate and postsynaptic membrane. {ECO:0000269|PubMed:16685696, ECO:0000269|PubMed:7619526, ECO:0000269|PubMed:8872460, ECO:0000269|PubMed:9158151, ECO:0000269|PubMed:9221765}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 1B, fast-channel (CMS1B) [MIM:608930]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1B is a fast-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in brief opening and activity of the channel, with a rapid decay in endplate current, failure to achieve threshold depolarization of the endplate and consequent failure to fire an action potential. {ECO:0000269|PubMed:10195214, ECO:0000269|PubMed:12588888, ECO:0000269|PubMed:15079006}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08838; DB00565; DB00555 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46717.8 Length 411 Aromaticity 0.11 Instability index 38.02 Isoelectric point 4.77 Charge (pH=7) -22.31 2D Binding mode Binding energy (Kcal/mol) -6.3 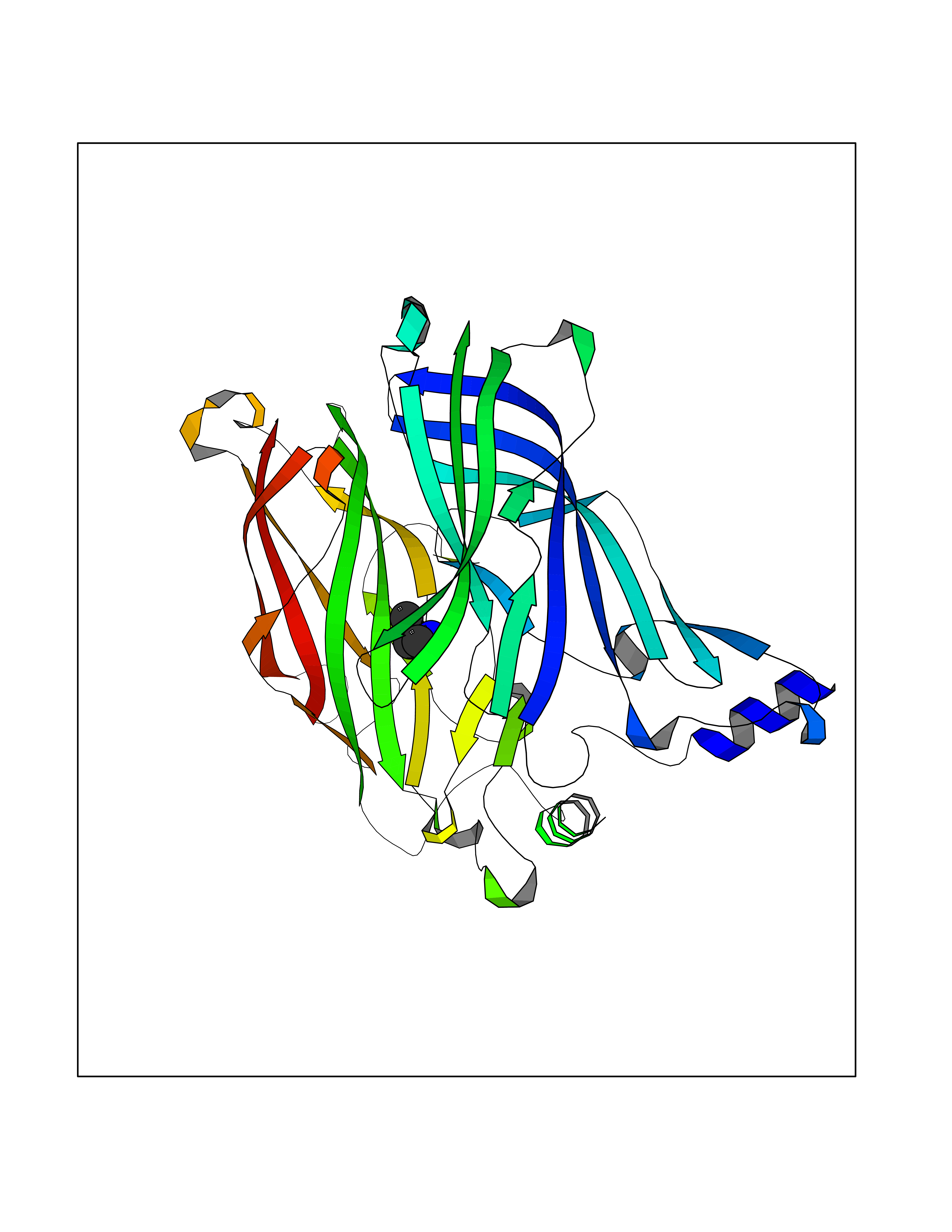 Molscript Map 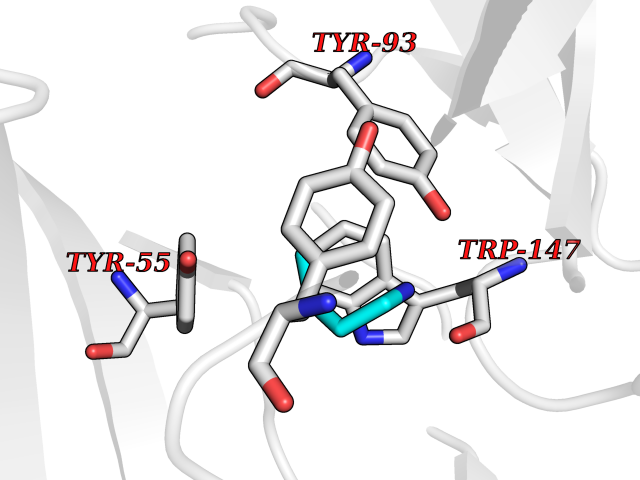 Pymol Map 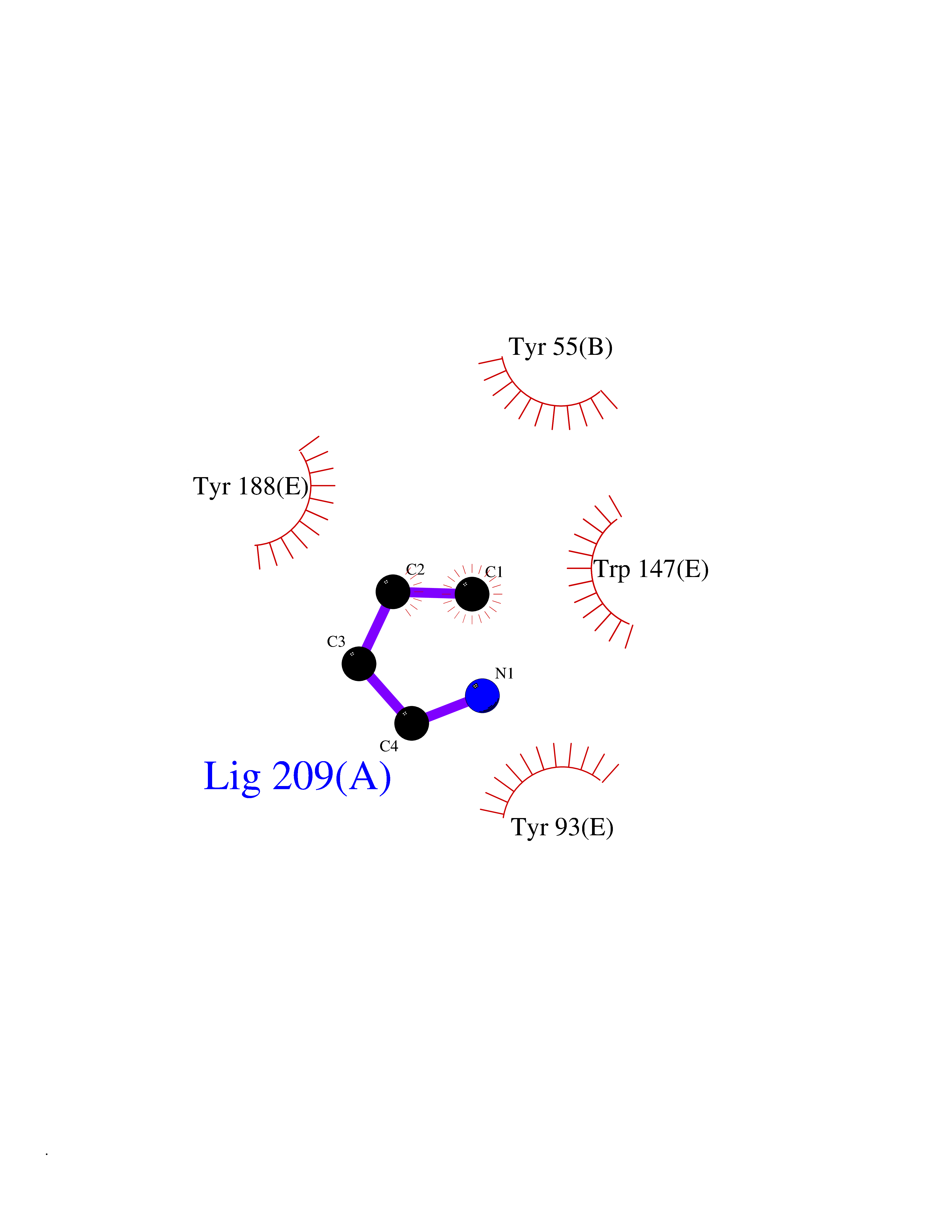 Ligplot Map 3D Binding mode Sequence EHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFREEHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFRER Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Glycogen phosphorylase, liver form | 3DDS | 4.62 | |
Target general information Gen name PYGL Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Glycogen phosphorylase family Biochemical class Transferase Function AMP binding.ATP binding.Bile acid binding.Drug binding.Glucose binding.Glycogen phosphorylase activity.Linear malto-oligosaccharide phosphorylase activity.Protein homodimerization activity.Purine nucleobase binding.Pyridoxal phosphate binding.SHG alpha-glucan phosphorylase activity.Vitamin binding. Related diseases Glycogen storage disease 6 (GSD6) [MIM:232700]: A metabolic disorder characterized by mild to moderate hypoglycemia, mild ketosis, growth retardation, and prominent hepatomegaly. Heart and skeletal muscle are not affected. {ECO:0000269|PubMed:9529348}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P11216; P11217 EC number 2.4.1.1 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Carbohydrate metabolism; Cytoplasm; Disease variant; Glycogen metabolism; Glycogen storage disease; Glycosyltransferase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89481.9 Length 778 Aromaticity 0.11 Instability index 34.96 Isoelectric point 6.49 Charge (pH=7) -3.54 2D Binding mode Binding energy (Kcal/mol) -6.3  Molscript Map  Pymol Map 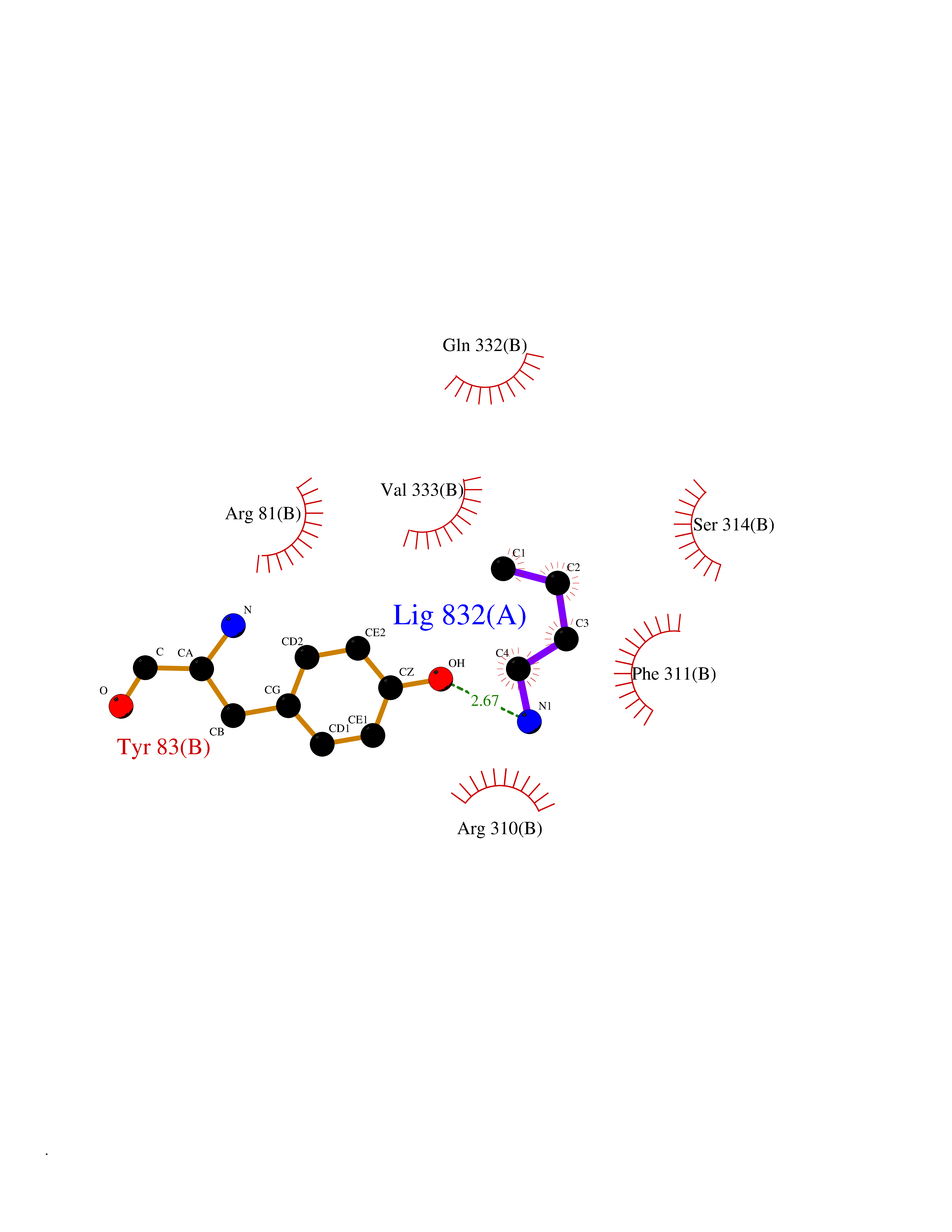 Ligplot Map 3D Binding mode Sequence NVAELKKSFNRHLHFTLVKDRNVATTRDYYFALAHTVRDHLVGRWIRTQQHYYDKCPKRVYYLSLEFYMGRTLQNTMINLGLQNACDEAIYQLGLDIEELEEIEEDAGLGNGGLGRLAACFLDSMATLGLAAYGYGIRYEYGIFNQKIRDGWQVEEADDWLRYGNPWEKSRPEFMLPVHFYGKVEHTNTGTKWIDTQVVLALPYDTPVPGYMNNTVNTMRLWSENISRVLYPNDNFFEGKELRLKQEYFVVAATLQDIIRRFKASKFGSTGTVFDAFPDQVAIQLNDTHPALAIPELMRIFVDIEKLPWSKAWELTQKTFAYTNHTVLPEALERWPVDLVEKLLPRHLEIIYEINQKHLDRIVALFPKDVDRLRRMSLIEEEGSKRINMAHLCIVGSHAVNGVAKIHSDIVKTKVFKDFSELEPDKFQNKTNGITPRRWLLLCNPGLAELIAEKIGEDYVKDLSQLTKLHSFLGDDVFLRELAKVKQENKLKFSQFLETEYKVKINPSSMFDVQVKRIHEYKRQLLNCLHVITMYNRIKKDPKKLFVPRTVIIGGKAAPGYHMAKMIIKLITSVADVVNNDPMVGSKLKVIFLENYRVSLAEKVIPATDLSEQISTAGTEASGTGNMKFMLNGALTIGTMDGANVEMAEEAGEENLFIFGMRIDDVAALDKKGYEAKEYYEALPELKLVIDQIDNGFFSPKQPDLFKDIINMLFYHDRFKVFADYEAYVKCQDKVSQLYMNPKAWNTMVLKNIAASGKFSSDRTIKEYAQNIWNVEPS Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Cytosolic 10-formyltetrahydrofolate dehydrogenase | 2CFI | 4.62 | |
Target general information Gen name ALDH1L1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FTHFD Protein family GART family; Aldehyde dehydrogenase family, ALDH1L subfamily Biochemical class Oxidoreductase Function Aldehyde dehydrogenase (NAD) activity.Catalytic activity.Formyltetrahydrofolate dehydrogenase activity.Hydroxymethyl-, formyl- and related transferase activity. Related diseases Developmental and epileptic encephalopathy 39 with leukodystrophy (DEE39) [MIM:612949]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE39 is characterized by global hypomyelination of the central nervous system, with the gray matter appearing relatively unaffected. Inheritance is autosomal recessive. {ECO:0000269|PubMed:19641205, ECO:0000269|PubMed:24515575}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00116 Interacts with Q3SY69; Q92624 EC number 1.5.1.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; NADP; One-carbon metabolism; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33869.5 Length 308 Aromaticity 0.08 Instability index 30.42 Isoelectric point 6.09 Charge (pH=7) -3.83 2D Binding mode Binding energy (Kcal/mol) -6.3 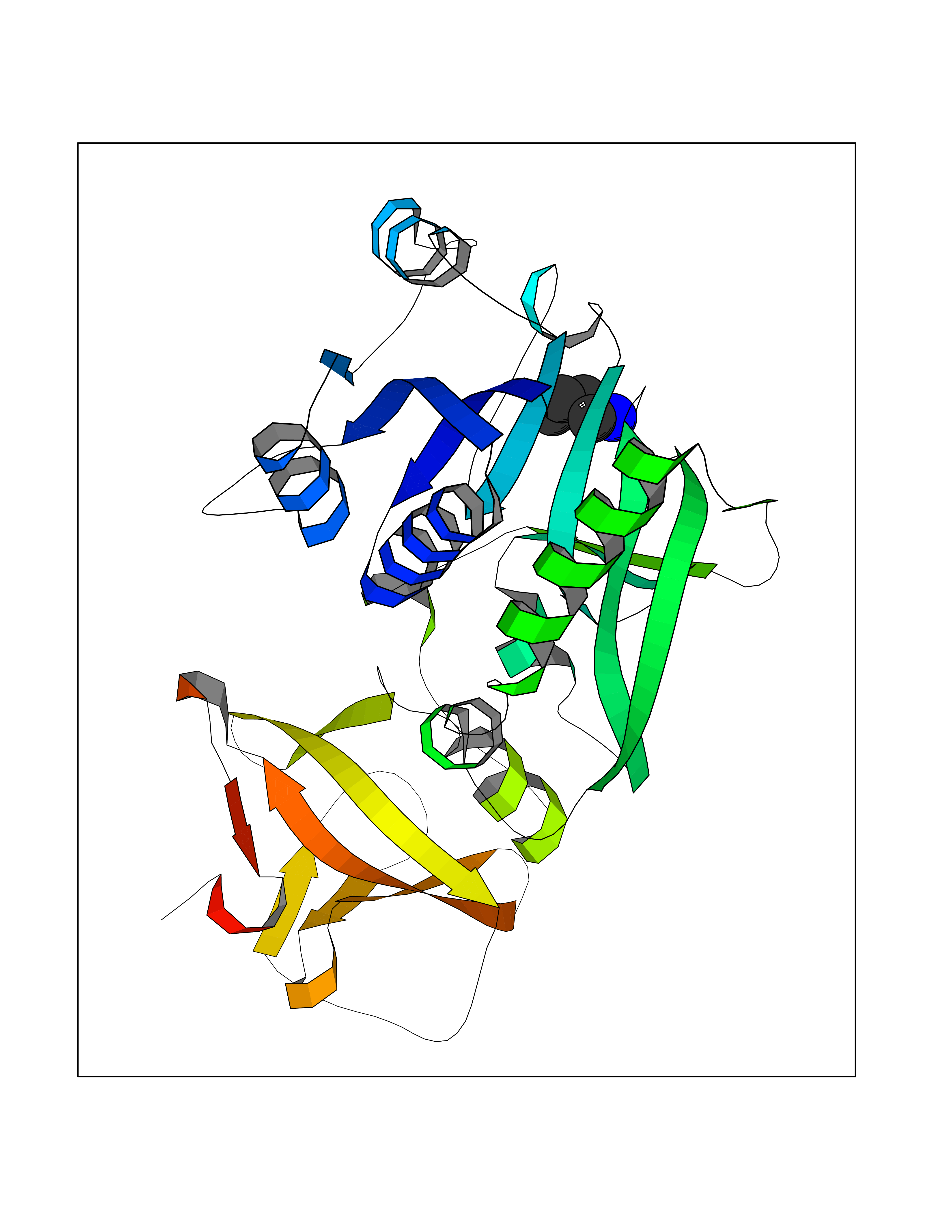 Molscript Map 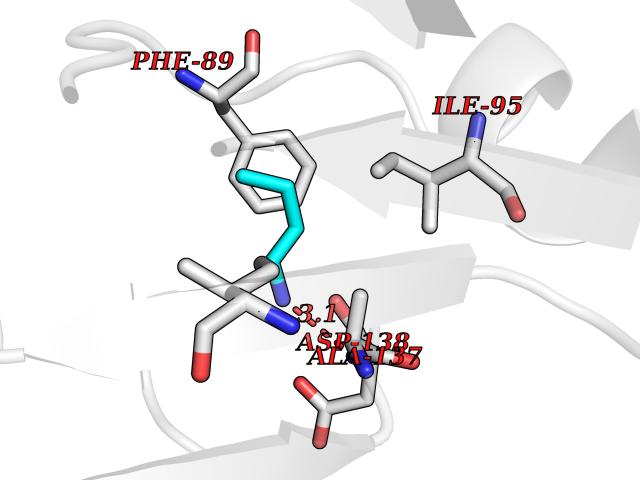 Pymol Map 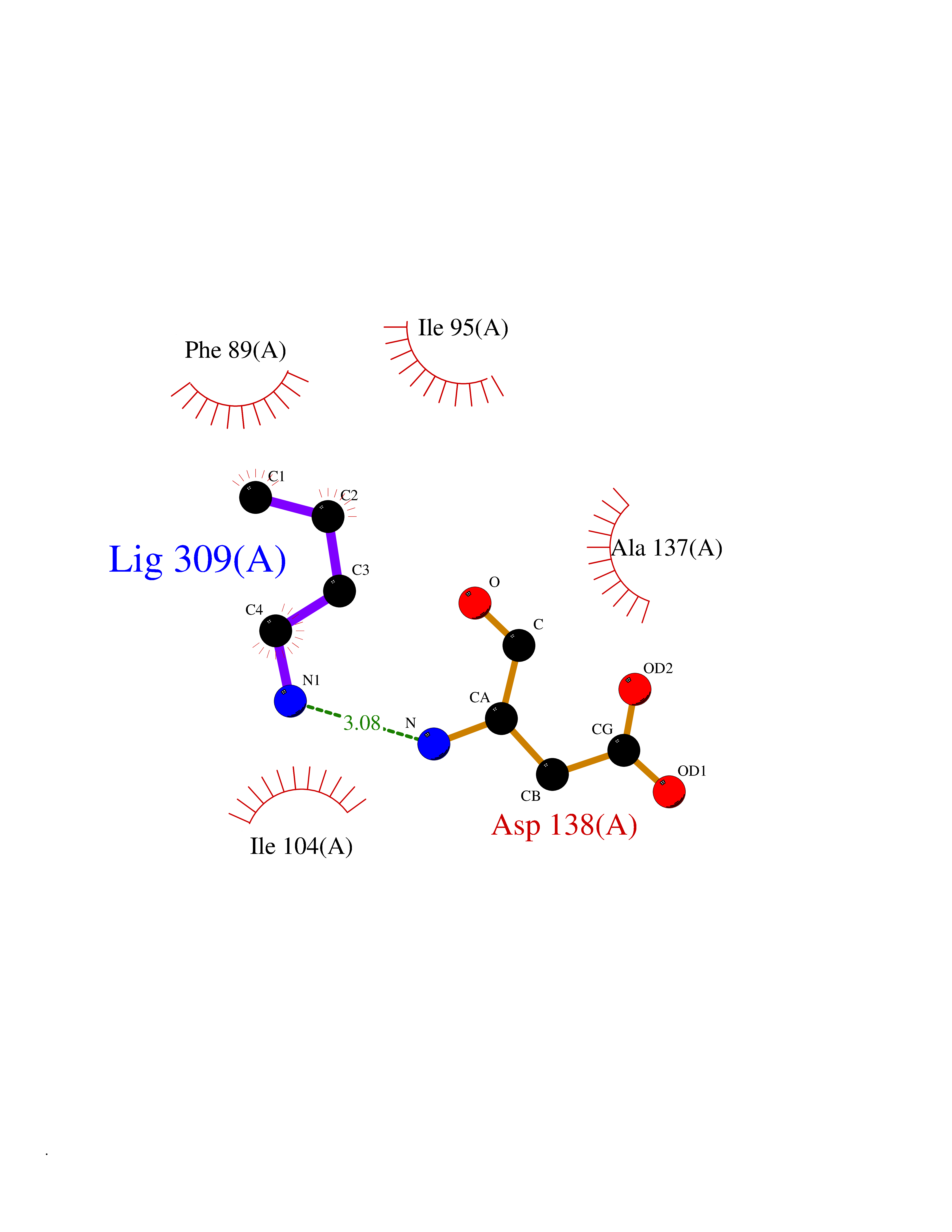 Ligplot Map 3D Binding mode Sequence SMKIAVIGQSLFGQEVYCHLRKEGHEVVGVFTVPDKDGKADPLGLEAEKDGVPVFKYSRWRAKGQALPDVVAKYQALGAELNVLPFCSQFIPMEIISAPRHGSIIYHPSLLPRHRGASAINWTLIHGDKKGGFSIFWADDGLDTGDLLLQKECEVLPDDTVSTLYNRFLFPEGIKGMVQAVRLIAEGKAPRLPQPEEGATYEGIQKKETAKINWDQPAEAIHNWIRGNDKVPGAWTEACEQKLTFFNSTLNTSGLVPEGDALPIPGAHRPGVVTKAGLILFGNDDKMLLVKNIQLEDGKMILASNFFK Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Catalase (CAT) | 1DGF | 4.62 | |
Target general information Gen name CAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Human catalase Protein family Catalase family Biochemical class Peroxide acceptor oxidoreductase Function Promotes growth of cells including T-cells, B-cells, myeloid leukemia cells, melanoma cells, mastocytoma cells and normal and transformed fibroblast cells. Occurs in almost all aerobically respiring organisms and serves to protect cells from the toxic effects of hydrogen peroxide. Related diseases Acatalasemia (ACATLAS) [MIM:614097]: A metabolic disorder characterized by a total or near total loss of catalase activity in red cells. It is often associated with ulcerating oral lesions. Acatalasemia is inherited as an autosomal recessive trait. {ECO:0000269|PubMed:2308162}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09096; DB09116; DB09061; DB01213; DB11091; DB14009; DB14011 Interacts with P04040 EC number EC 1.11.1.6 Uniprot keywords 3D-structure; Acetylation; Direct protein sequencing; Heme; Hydrogen peroxide; Iron; Metal-binding; Mitogen; NADP; Oxidoreductase; Peroxidase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 56550.9 Length 497 Aromaticity 0.12 Instability index 29.44 Isoelectric point 6.73 Charge (pH=7) -1.48 2D Binding mode Binding energy (Kcal/mol) -6.3 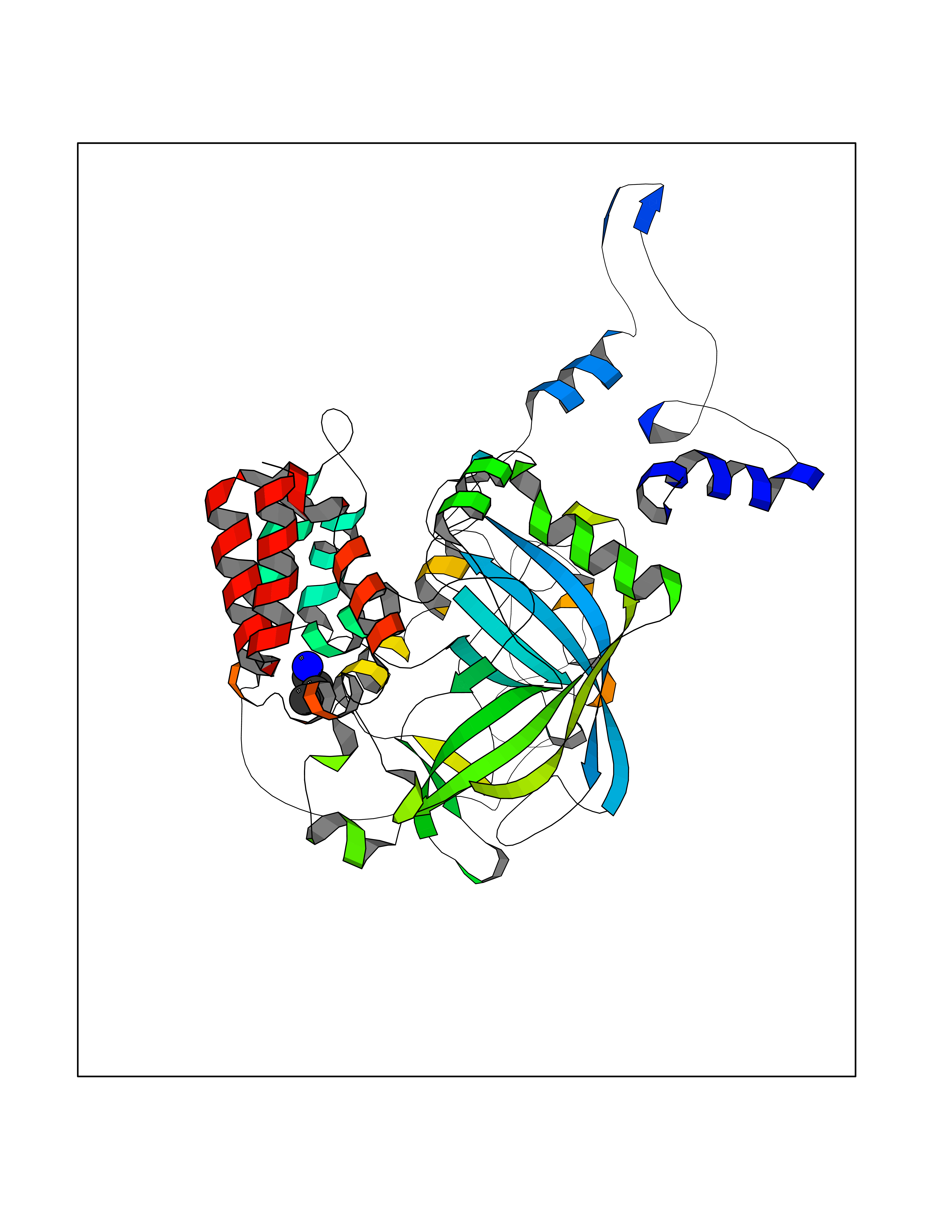 Molscript Map 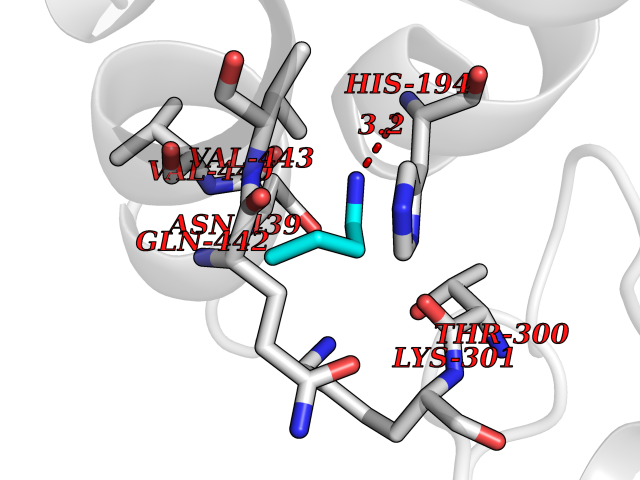 Pymol Map 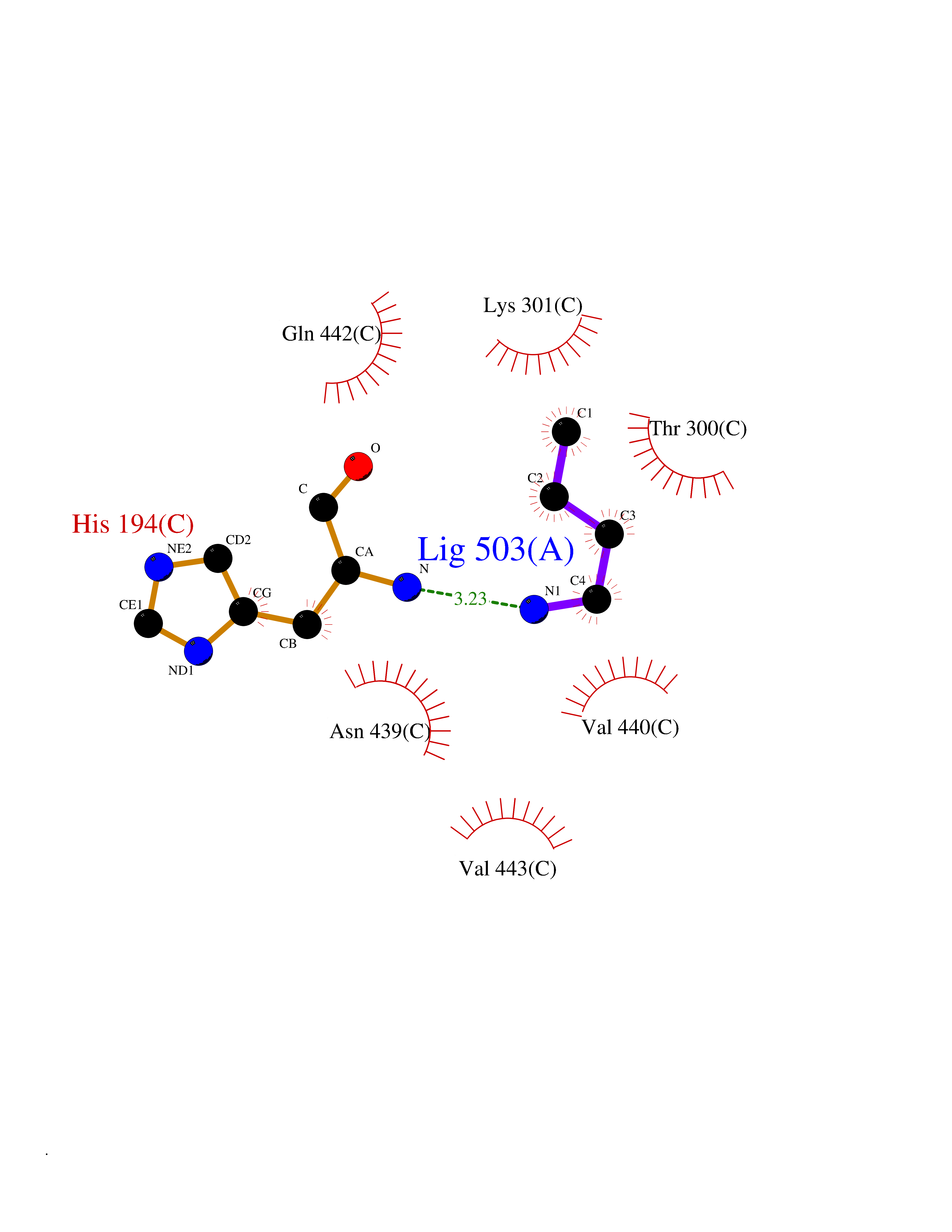 Ligplot Map 3D Binding mode Sequence RDPASDQMQHWKEQRAAQKADVLTTGAGNPVGDKLNVITVGPRGPLLVQDVVFTDEMAHFDRERIPERVVHAKGAGAFGYFEVTHDITKYSKAKVFEHIGKKTPIAVRFSTVAGESGSADTVRDPRGFAVKFYTEDGNWDLVGNNTPIFFIRDPILFPSFIHSQKRNPQTHLKDPDMVWDFWSLRPESLHQVSFLFSDRGIPDGHRHMNGYGSHTFKLVNANGEAVYCKFHYKTDQGIKNLSVEDAARLSQEDPDYGIRDLFNAIATGKYPSWTFYIQVMTFNQAETFPFNPFDLTKVWPHKDYPLIPVGKLVLNRNPVNYFAEVEQIAFDPSNMPPGIEASPDKMLQGRLFAYPDTHRHRLGPNYLHIPVNCPYRARVANYQRDGPMCMQDNQGGAPNYYPNSFGAPEQQPSALEHSIQYSGEVRRFNTANDDNVTQVRAFYVNVLNEEQRKRLCENIAGHLKDAQIFIQKKAVKNFTEVHPDYGSHIQALLDKYN Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | 5'-methylthioadenosine/S-adenosylhomocysteine nucleosidase | 4WKC | 4.62 | |
Target general information Gen name mtnN Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms pfs;b0159;yadA;JW0155;mtn Protein family PNP/UDP phosphorylase family, MtnN subfamily Biochemical class hydrolase / hydrolase inhibitor Function Adenosylhomocysteine nucleosidase activity.Identical protein binding.Methylthioadenosine nucleosidase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02158; DB08606; DB02933; DB00173; DB02281 Interacts with P0AF12 EC number 3.2.2.9 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Hydrolase; Methionine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 24353.7 Length 232 Aromaticity 0.05 Instability index 22.1 Isoelectric point 5.09 Charge (pH=7) -9.9 2D Binding mode Binding energy (Kcal/mol) -6.3  Molscript Map 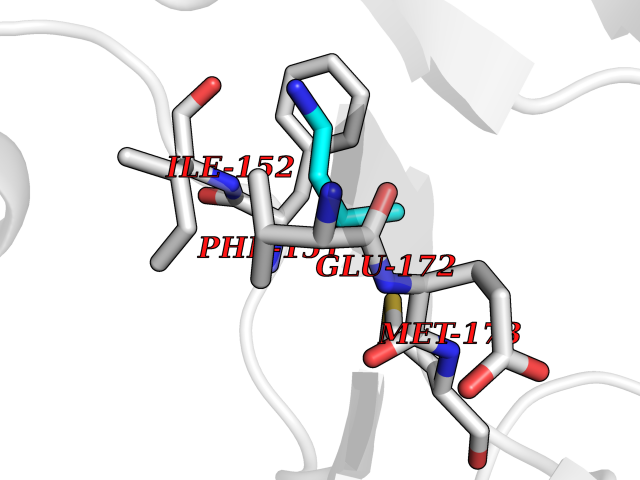 Pymol Map  Ligplot Map 3D Binding mode Sequence MKIGIIGAMEEEVTLLRDKIENRQTISLGGCEIYTGQLNGTEVALLKSGIGKVAAALGATLLLEHCKPDVIINTGSAGGLAPTLKVGDIVVSDEARYHDADVTAFGYEYGQLPGCPAGFKADDKLIAAAEACIAELNLNAVRGLIVSGDAFINGSVGLAKIRHNFPQAIAVEMEATAIAHVCHNFNVPFVVVRAISDVADQQSHLSFDEFLAVAAKQSSLMVESLVQKLAHG Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Oxygen-insensitive NADPH nitroreductase | 1F5V | 4.62 | |
Target general information Gen name nfsA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms mda18;b0851;mdaA;ybjB;JW0835 Protein family Flavin oxidoreductase frp family Biochemical class Oxidoreductase Function Chromate reductase activity.FMN binding.Oxidoreductase activity, acting on NAD(P)H, nitrogenous group as acceptor. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03247; DB00698 Interacts with P28630 EC number 1.-.-.- Uniprot keywords 3D-structure; Flavoprotein; FMN; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 53582.7 Length 480 Aromaticity 0.07 Instability index 45.43 Isoelectric point 6.47 Charge (pH=7) -3.13 2D Binding mode Binding energy (Kcal/mol) -6.3 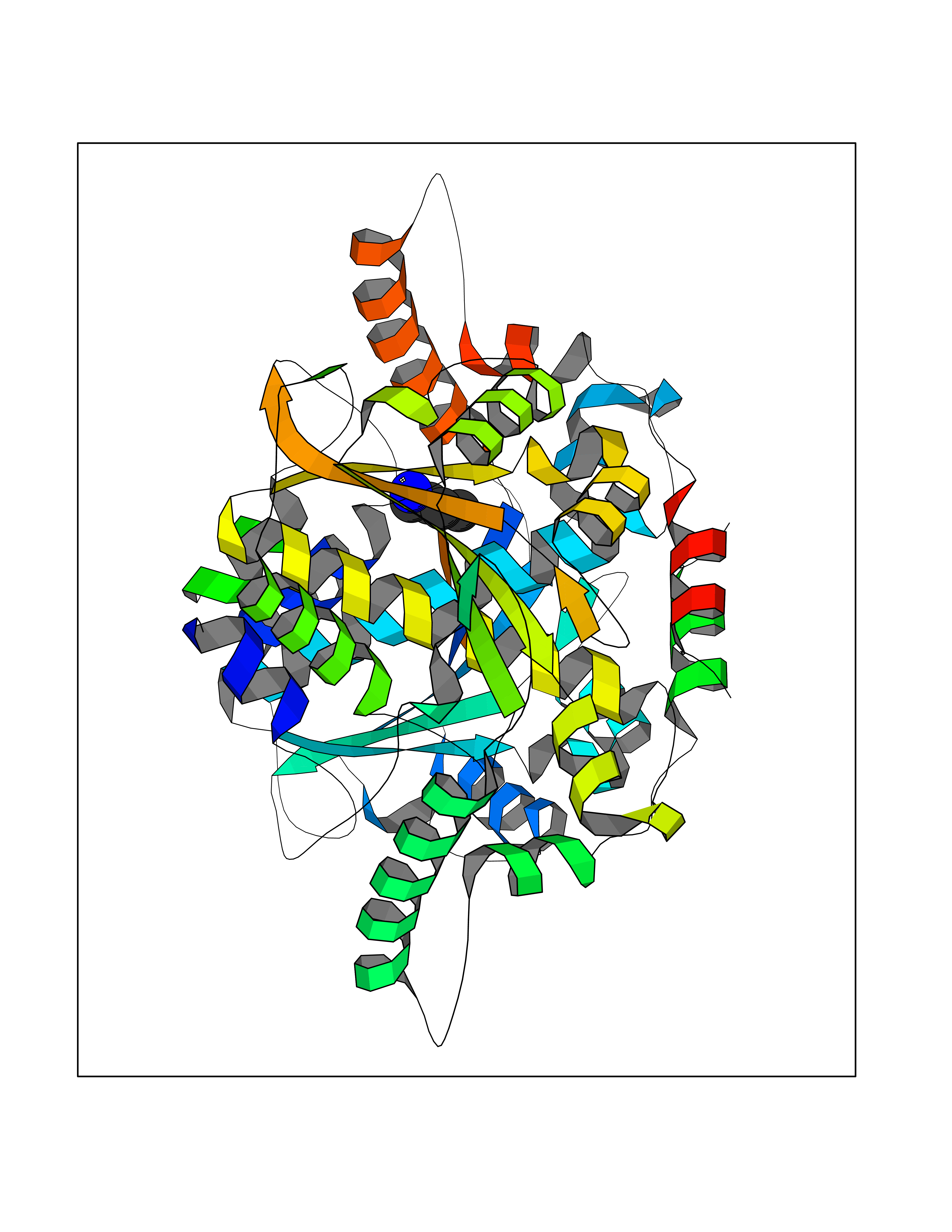 Molscript Map 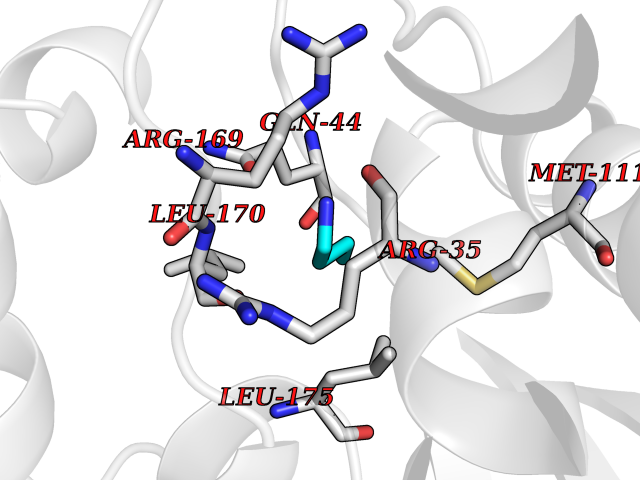 Pymol Map  Ligplot Map 3D Binding mode Sequence MTPTIELICGHRSIRHFTDEPISEAQREAIINSARATSSSSFLQCSSIIRITDKALREELVTLTGGQKHVAQAAEFWVFCADFNRHLQICPDAQLGLAEQLLLGVVDTAMMAQNALIAAESLGLGGVYIGGLRNNIEAVTKLLKLPQHVLPLFGLCLGWPADNPDLKPRLPASILVHENSYQPLDKGALAQYDEQLAEYYLTRGSNNRRDTWSDHIRRTIIKESRPFILDYLHKQGWATRMTPTIELICGHRSIRHFTDEPISEAQREAIINSARATSSSSFLQCSSIIRITDKALREELVTLTGGQKHVAQAAEFWVFCADFNRHLQICPDAQLGLAEQLLLGVVDTAMMAQNALIAAESLGLGGVYIGGLRNNIEAVTKLLKLPQHVLPLFGLCLGWPADNPDLKPRLPASILVHENSYQPLDKGALAQYDEQLAEYYLTRGSNNRRDTWSDHIRRTIIKESRPFILDYLHKQGWATR Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 4.62 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -6.3  Molscript Map 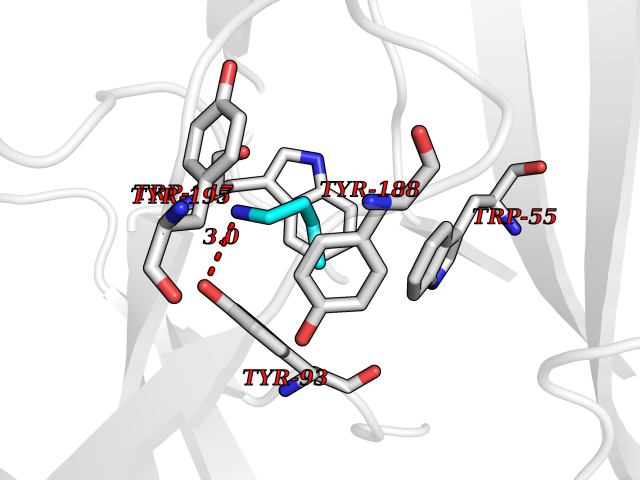 Pymol Map 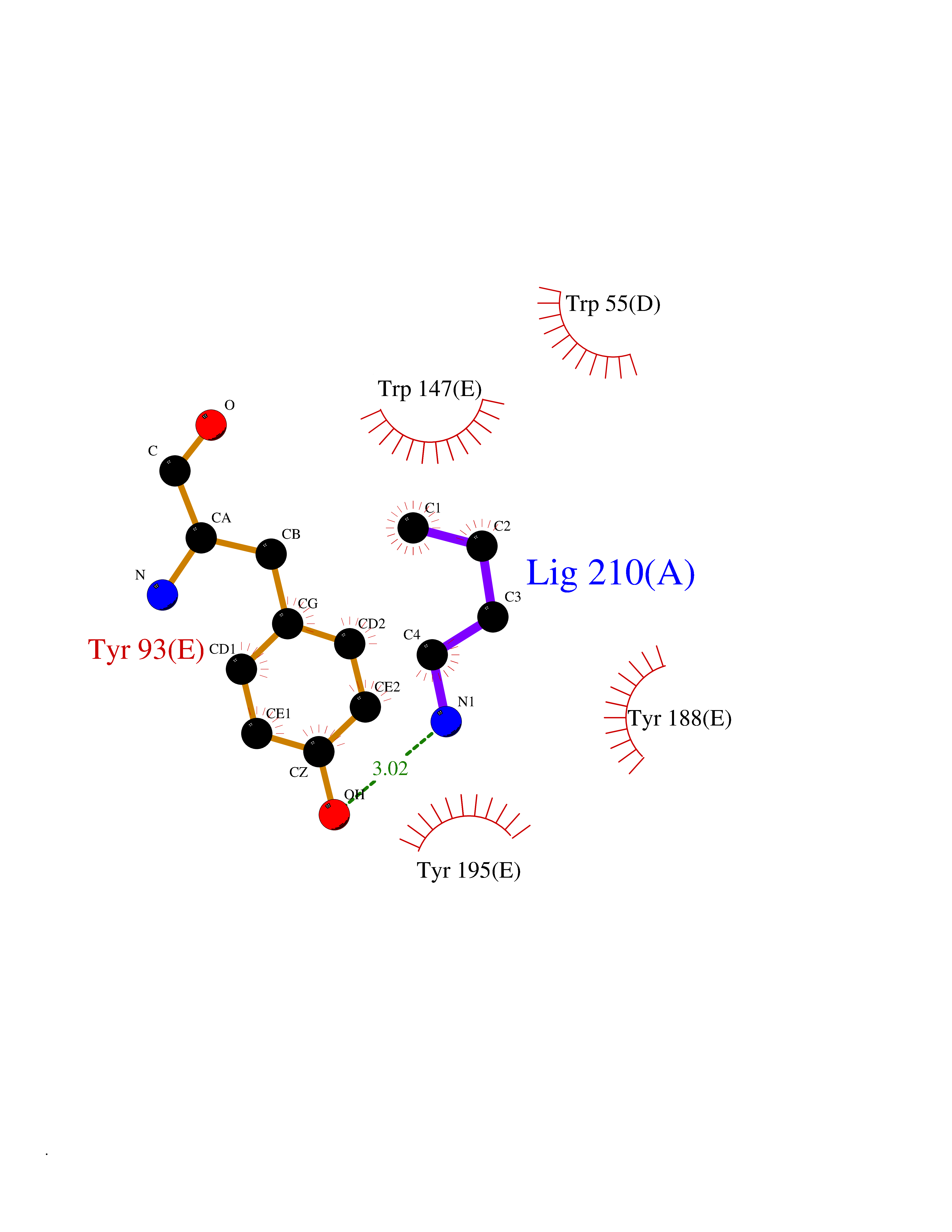 Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | 4-hydroxyphenylpyruvate dioxygenase | 3ISQ | 4.62 | |
Target general information Gen name HPD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PPD Protein family 4HPPD family Biochemical class Oxidoreductase Function 4-hydroxyphenylpyruvate dioxygenase activity.Metal ion binding. Related diseases Tyrosinemia 3 (TYRSN3) [MIM:276710]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, seizures and mild intellectual disability. {ECO:0000269|PubMed:10942115, ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hawkinsinuria (HWKS) [MIM:140350]: An inborn error of tyrosine metabolism characterized by failure to thrive, persistent metabolic acidosis, fine and sparse hair, and excretion of the unusual cyclic amino acid metabolite, hawkinsin, in the urine. {ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02850; DB00348 Interacts with NA EC number 1.13.11.27 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Dioxygenase; Disease variant; Endoplasmic reticulum; Golgi apparatus; Intellectual disability; Iron; Membrane; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 43164.8 Length 376 Aromaticity 0.11 Instability index 32.38 Isoelectric point 6.73 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -6.3 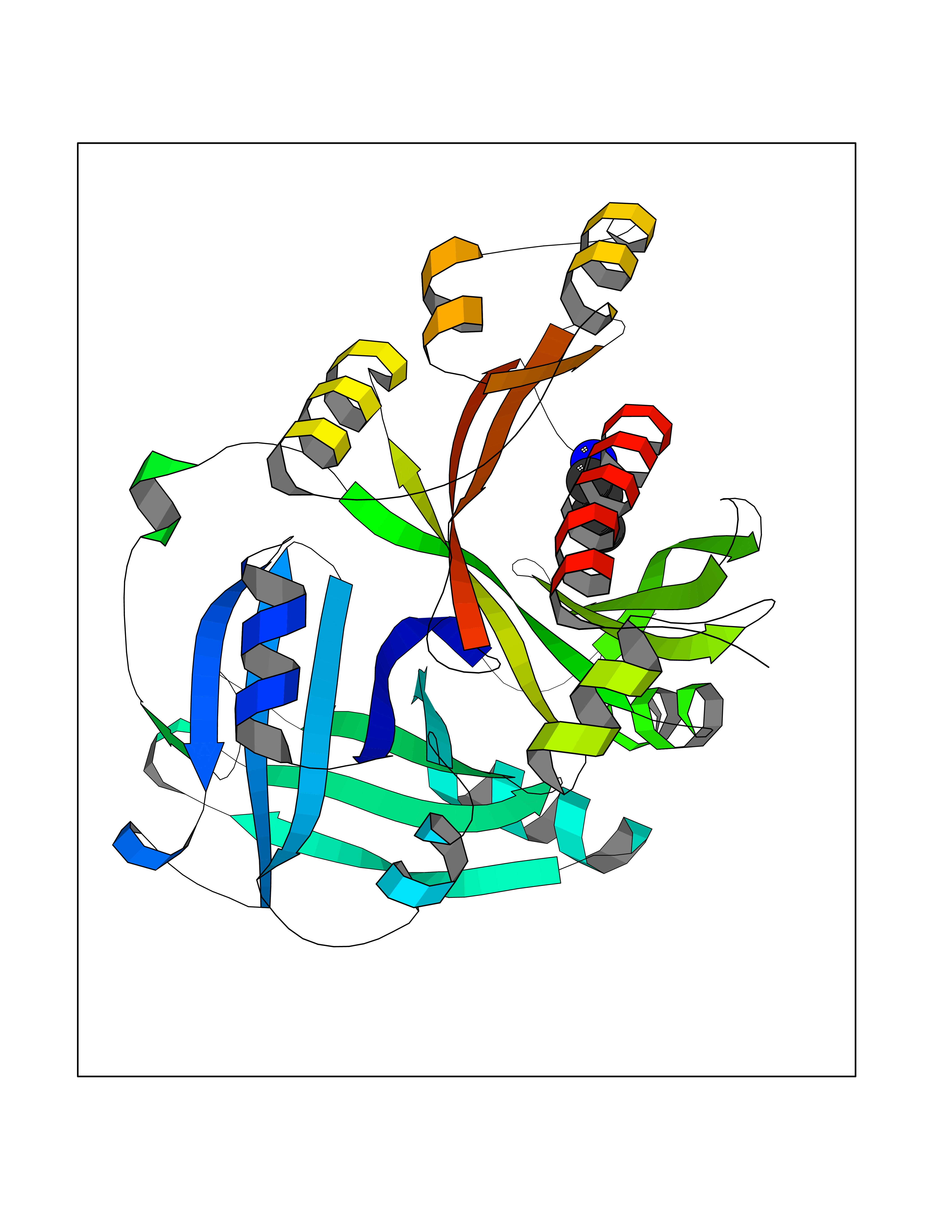 Molscript Map 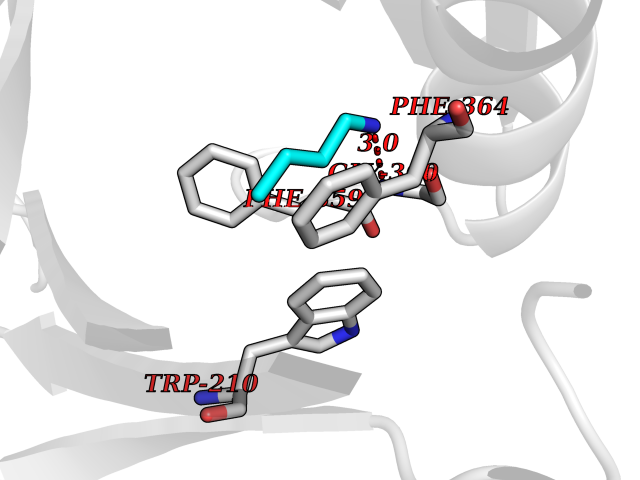 Pymol Map 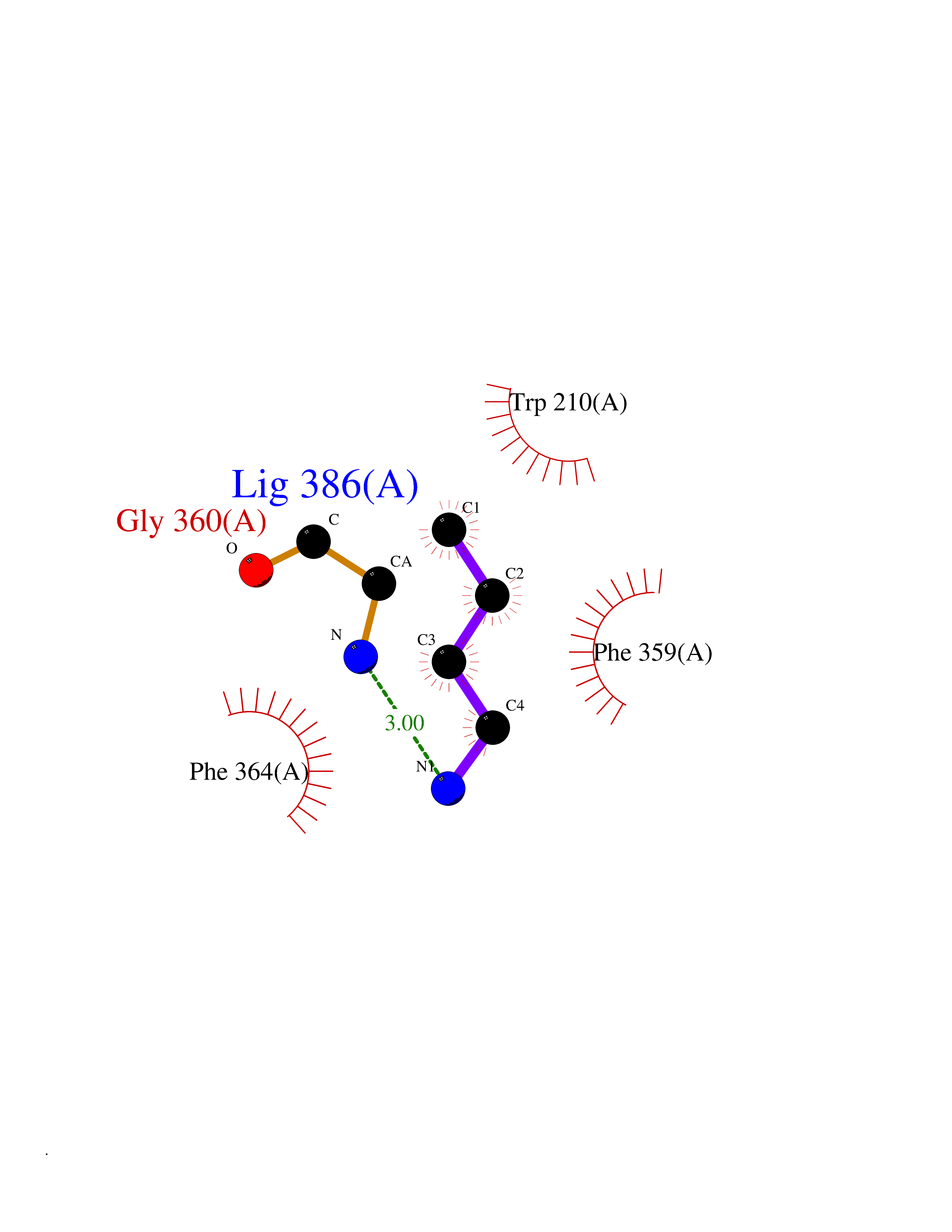 Ligplot Map 3D Binding mode Sequence AKPERGRFLHFHSVTFWVGNAKQAASFYCSKMGFEPLAYRGLETGSREVVSHVIKQGKIVFVLSSALNPWNKEMGDHLVKHGDGVKDIAFEVEDCDYIVQKARERGAKIMREPWVEQDKFGKVKFAVLQTYGDTTHTLVEKMNYIGQFLPGYEAPAFMDPLLPKLPKCSLEMIDHIVGNQPDQEMVSASEWYLKNLQFHRFWSVDDTQVHTEYSSLRSIVVANYEESIKMPINEPAPGKKKSQIQEYVDYNGGAGVQHIALKTEDIITAIRHLRERGLEFLSVPSTYYKQLREKLKTAKIKVKENIDALEELKILVDYDEKGYLLQIFTKPVQDRPTLFLEVIQRHNHQGFGAGNFNSLFKAFEEEQNLRGNLTNM Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Cocaine esterase | 3I2K | 4.62 | |
Target general information Gen name cocE Organism Rhodococcus sp. (strain MB1 Bresler) Uniprot ID TTD ID NA Synonyms NA Protein family CocE/NonD hydrolase family Biochemical class Hydrolase Function Carboxylic ester hydrolase activity.Dipeptidyl-peptidase activity. Related diseases Thiamine metabolism dysfunction syndrome 5, episodic encephalopathy type (THMD5) [MIM:614458]: An autosomal recessive metabolic disorder due to an inborn error of thiamine metabolism. The phenotype is highly variable, but in general, affected individuals have onset in early childhood of acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive neurologic dysfunction manifest as gait disturbances, ataxia, dystonia, and spasticity, which in some cases may result in loss of ability to walk. Cognitive function is usually preserved, although mildly delayed development has been reported. These episodes are usually associated with infection and metabolic decompensation. Some patients may have recovery of some neurologic deficits. {ECO:0000269|PubMed:22152682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03793; DB01795 Interacts with NA EC number 3.1.1.84 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Hydrolase; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 62127.9 Length 574 Aromaticity 0.09 Instability index 26.62 Isoelectric point 4.56 Charge (pH=7) -33.24 2D Binding mode Binding energy (Kcal/mol) -6.3  Molscript Map 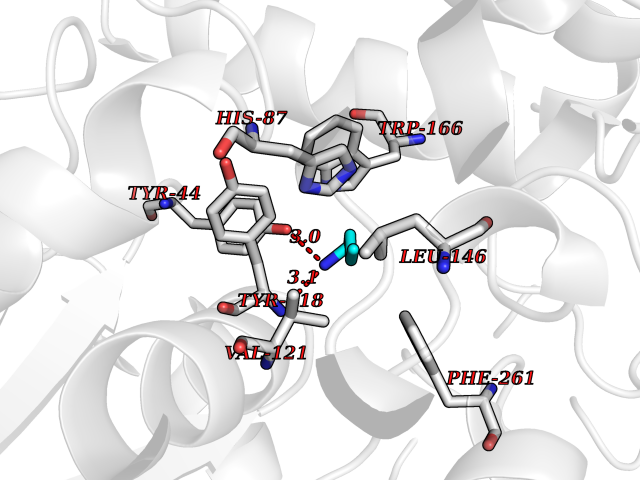 Pymol Map 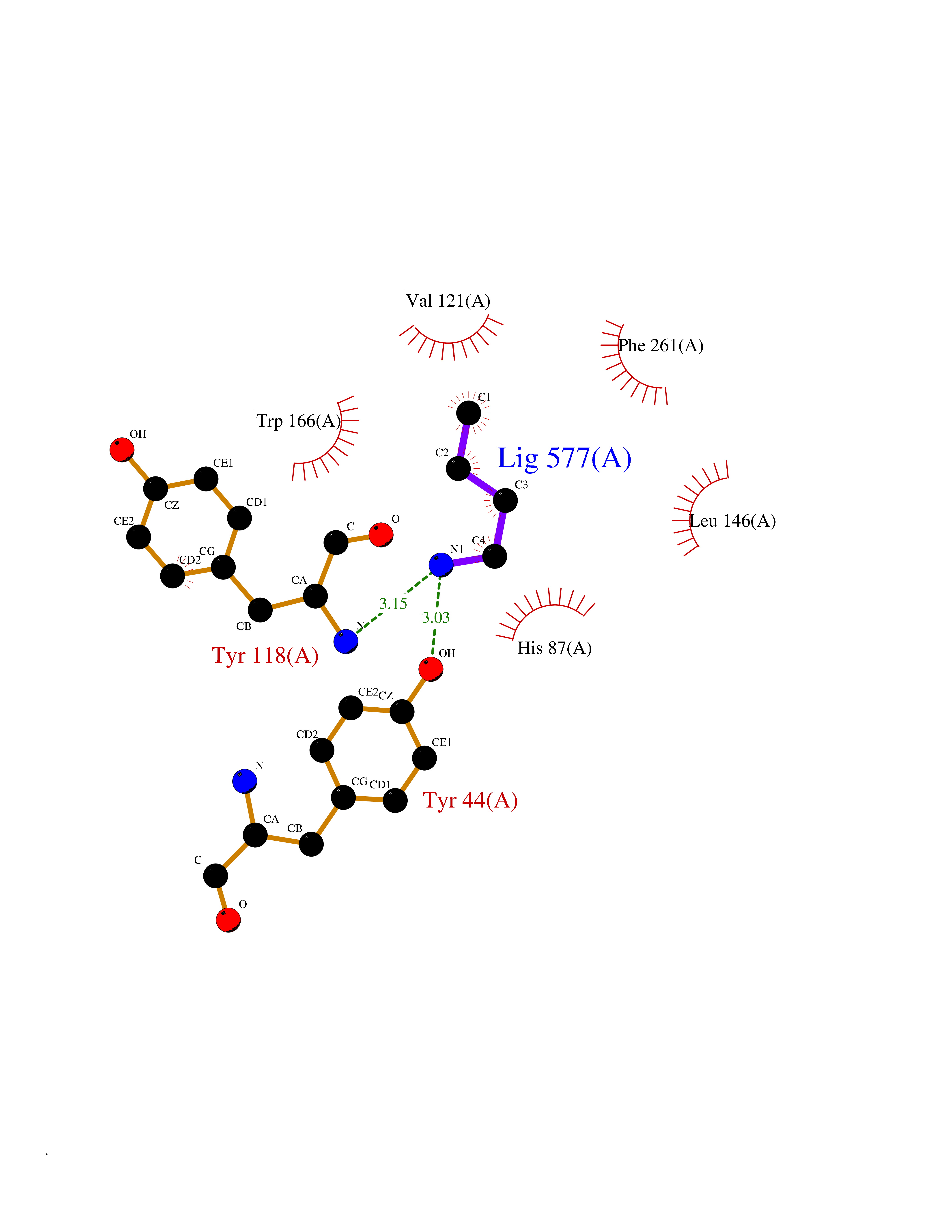 Ligplot Map 3D Binding mode Sequence VDGNYSVASNVMVPMRDGVRLAVDLYRPDADGPVPVLLVRNPYDKFDVFAWSTQSTNWLEFVRDGYAVVIQDTRGLFASEGEFVPHVDDEADAEDTLSWILEQAWCDGNVGMFGVSYLGVTQWQAAVSGVGGLKAIAPSMASADLYRAPWYGPGGALSVEALLGWSALIGTGLITSRSDARPEDAADFVQLAAILNDVAGAASVTPLAEQPLLGRLIPWVIDQVVDHPDNDESWQSISLFERLGGLATPALITAGWYDGFVGESLRTFVAVKDNADARLVVGPWSHSNLTGRNADRKFGIAATYPIQEATTMHKAFFDRHLRGETDALAGVPKVRLFVMGIDEWRDETDWPLPDTAYTPFYLGGSGAANTSTGGGTLSTSISGTESADTYLYDPADPVPSLGGTLLFHNGDNGPADQRPIHDRDDVLCYSTEVLTDPVEVTGTVSARLFVSSSAVDTDFTAKLVDVFPDGRAIALCDGIVRMRYRETLVNPTLIEAGEIYEVAIDMLATSNVFLPGHRIMVQVSSSNFPKYDRNSNTGGVIAREQLEEMCTAVNRIHRGPEHPSHIVLPIIKRK Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Vitamin K epoxide reductase complex 1 (VKORC1) | 6WV3 | 4.62 | |
Target general information Gen name VKORC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin K1 2,3-epoxide reductase subunit 1; VKORC1; VKOR; UNQ308/PRO351; MSTP576; MSTP134 Protein family VKOR family Biochemical class Short-chain dehydrogenases reductase Function Involved invitamin K metabolism. Catalytic subunit of the vitamin K epoxide reductase (VKOR) complex which reduces inactive vitamin K 2,3-epoxide to active vitamin K. Vitamin K is required for the gamma-carboxylation of various proteins, including clotting factors, and is required for normal blood coagulation, but also for normal bone development. Related diseases Combined deficiency of vitamin K-dependent clotting factors 2 (VKCFD2) [MIM:607473]: VKCFD leads to a bleeding tendency that is usually reversed by oral administration of vitamin K. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:16270630}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Coumarin resistance (CMRES) [MIM:122700]: A condition characterized by partial or complete resistance to warfarin or other 4-hydroxycoumarin derivatives. These drugs are used as anti-coagulants for the prevention of thromboembolic diseases in subjects with deep vein thrombosis, atrial fibrillation, or mechanical heart valve replacement. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:20946155}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01418; DB00266; DB09332; DB00170; DB00498; DB00946; DB01022; DB00682 Interacts with Q13323; Q7Z7G2; Q96BA8; Q9Y282; Q5JX71; Q96KR6; Q5T7V8; Q8TDT2; Q9NQG1; P15941-11; Q96TC7; Q9NR31; A0A0S2Z4U3; Q8TBB6; O15393-2; Q19QW4 EC number EC 1.17.4.4 Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Disulfide bond; Endoplasmic reticulum; Membrane; Oxidoreductase; Proteomics identification; Quinone; Redox-active center; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 42656.4 Length 381 Aromaticity 0.1 Instability index 32.12 Isoelectric point 7.73 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -6.3  Molscript Map  Pymol Map 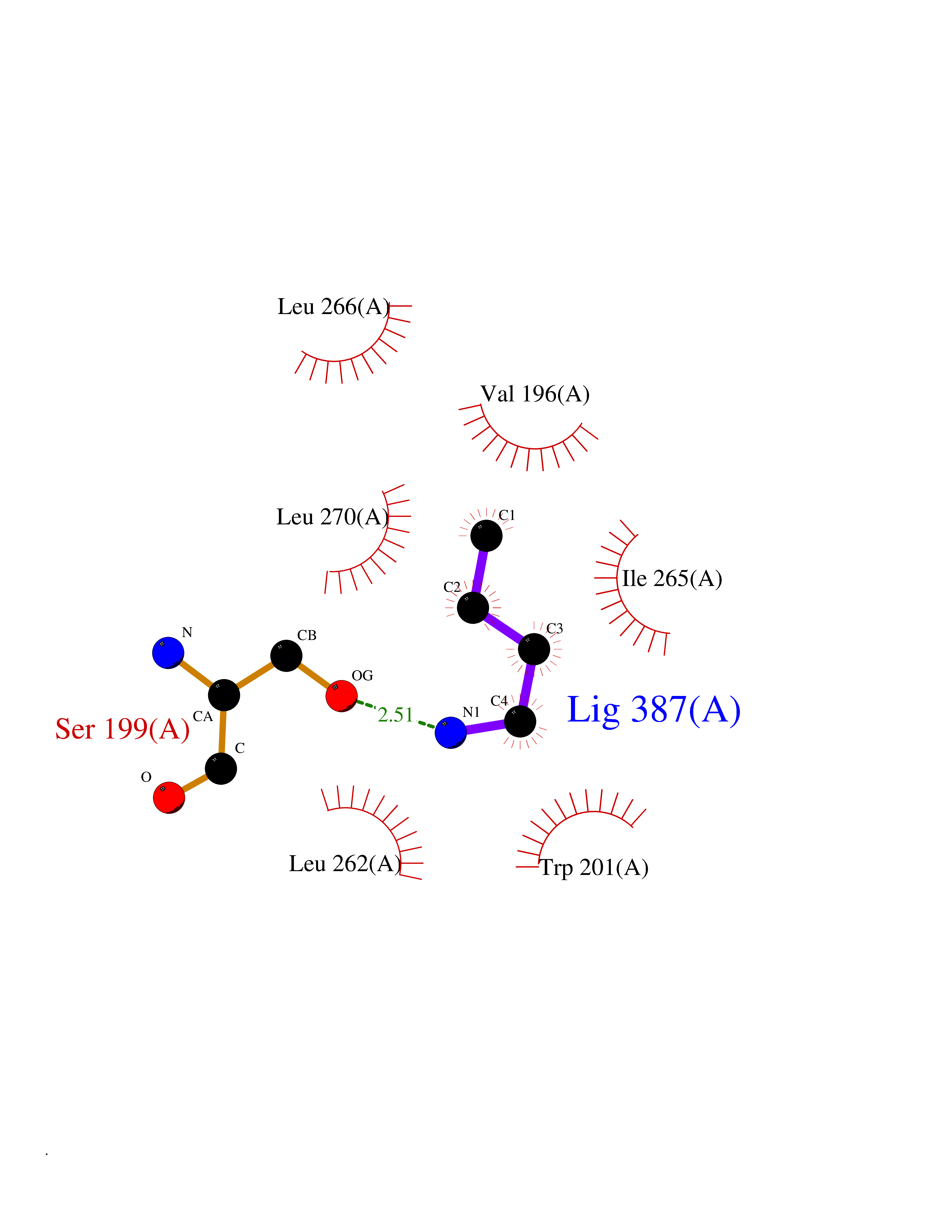 Ligplot Map 3D Binding mode Sequence KGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLXVQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNSTWGSPGWVRLALCLTGLVLSLYALHVKAARARDRDYRALCDVGTAISCSRVFSSRWGRGFGLVEHVLGQDSILNQSNSIFGCIFYTLQLLLGCLRTRWASVLMLLSSLVSLAGSVYLAWILFFVLYDFCIVCITTYAINVSLMWLSFRKVQENSHNVYITADKQKNGIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHHH Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Beta-N-acetylhexosaminidase (OGA) | 6PM9 | 4.62 | |
Target general information Gen name MGEA5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms OGA; Nuclear cytoplasmic OGlcNAcase and acetyltransferase; Meningiomaexpressed antigen 5; MGEA5; Histone acetyltransferase; Bifunctional protein NCOAT Protein family Glycosyl hydrolase 84 family Biochemical class Glycosylase Function Isoform 3: Cleaves GlcNAc but not GalNAc from O- glycosylated proteins. Can use p-nitrophenyl-beta-GlcNAc as substrate but not p-nitrophenyl-beta-GalNAc or p-nitrophenyl- alpha-GlcNAc (in vitro), but has about six times lower specific activity than isoform1. {ECO:0000269|PubMed:20673219}. Related diseases Immunodeficiency 56 (IMD56) [MIM:615207]: An autosomal recessive primary immunodeficiency characterized by B- and T-cell defects and variable dysfunction of NK cells. Patients tend to have normal numbers of lymphocytes, but show defective class-switched B-cells, low IgG, defective antibody response, and defective T-cell responses to certain antigens. {ECO:0000269|PubMed:23440042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Chromosomal aberrations involving IL21R is a cause of B-cell non-Hodgkin lymphomas (B-cell NHL). Translocation t(3;16)(q27;p11), with BCL6. Drugs (DrugBank ID) DB00428 Interacts with O60502 EC number EC 3.2.1.169 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Glycosidase; Host-virus interaction; Hydrolase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,G Molecular weight (Da) 49869.8 Length 426 Aromaticity 0.14 Instability index 46.71 Isoelectric point 5.7 Charge (pH=7) -6.64 2D Binding mode Binding energy (Kcal/mol) -6.31  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RFLCGVVEGFYGRPWVMEQRKELFRRLQKWELNTYLYAPKDDYKHRMFWREMYSVEEAEQLMTLISAAREYEIEFIYAISPGLDITFSNPKEVSTLKRKLDQVSQFGCRSFALLFDDIDHNMCAADKEVFSSFAHAQVSITNEIYQYLGEPETFLFCPTEYCGTFCYPNVSQSPYLRTVGEKLLPGIEVLWTGPKVVSKEIPVESIEEVSKIIKRAPVIWDNIHANDYDQKRLFLGPYKGRSTELIPRLKGVLTNPNCEFEANYVAIHTLATWYKSNMNGVRDVLYSPQMALKLALTEWLQEFGVPHQYTLEDLQLLADLFYLPYEHGPKGAQMLREFQWLRANKDSEKIEEWRSRAAKFEEMCGLVMGMFTRLSNCANRTILYDMYSYVWDIKSIMSMVKSFVQWQEPWAFRGGLAGEFQRLLPI Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Bacterial Botulinum toxin A (Bact botA) | 6XCF | 4.62 | |
Target general information Gen name Bact botA Organism Clostridium botulinum Uniprot ID TTD ID Synonyms botA Protein family Peptidase M27 family Biochemical class Peptidase Function Inhibits acetylcholine release. The botulinum toxin binds with high affinity to peripheral neuronal presynaptic membrane to the secretory vesicle protein SV2. It binds directly to the largest luminal loop of SV2A, SV2B and SV2C. It is then internalized by receptor-mediated endocytosis. The C-terminus of the heavy chain (H) is responsible for the adherence of the toxin to the cell surface while the N-terminus mediates transport of the light chain from the endocytic vesicle to the cytosol. After translocation, the light chain (L) hydrolyzes the 197-Gln-|-Arg- 198 bond in SNAP-25, thereby blocking neurotransmitter release. Inhibition of acetylcholine release results in flaccid paralysis, with frequent heart or respiratory failure. Related diseases Major depressive disorder (MDD) [MIM:608516]: A common psychiatric disorder. It is a complex trait characterized by one or more major depressive episodes without a history of manic, mixed, or hypomanic episodes. A major depressive episode is characterized by at least 2 weeks during which there is a new onset or clear worsening of either depressed mood or loss of interest or pleasure in nearly all activities. Four additional symptoms must also be present including changes in appetite, weight, sleep, and psychomotor activity; decreased energy; feelings of worthlessness or guilt; difficulty thinking, concentrating, or making decisions; or recurrent thoughts of death or suicidal ideation, plans, or attempts. The episode must be accompanied by distress or impairment in social, occupational, or other important areas of functioning. {ECO:0000269|PubMed:15229186}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q02563; Q496J9; Q9Z2I6 EC number NA Uniprot keywords 3D-structure; Cell wall; Direct protein sequencing; Disulfide bond; Host cell membrane; Host cytoplasm; Host cytoplasmic vesicle; Host membrane; Host synapse; Hydrolase; Lipid-binding; Membrane; Metal-binding; Metalloprotease; Neurotoxin; Pharmaceutical; Protease; Secreted; Toxin; Transmembrane; Transmembrane helix; Ubl conjugation; Virulence; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 47759.6 Length 417 Aromaticity 0.13 Instability index 21.7 Isoelectric point 6.36 Charge (pH=7) -1.96 2D Binding mode Binding energy (Kcal/mol) -6.3  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MQFVNKQFNYKDPVNGVDIAYIKIPNVGQMQPVKAFKIHNKIWVIPERDTFTNPEEGDLNPPPPVSYYDSTYLSTDNEKDNYLKGVTKLFERIYSTDLGRMLLTSIVRGIPFWGGSTIDTELKVIDTNCINVIQPDGSYRSEELNLVIIGPSADIIQFECKSFGHEVLNLTRNGYGSTQYIRFSPDFTFGFEESLEVDTNPLLGAGKFATDPAVTLAHELIHAGHRLYGIAINPNRVFKVNTNAYYEMSGLEVSFEELRTFGGHDAKFIDSLQENEFRLYYYNKFKDIASTLNKAKSIVGTTASLQYMKNVFKEKYLLSEDTSGKFSVDKLKFDKLYKMLTEIYTEDNFVKFFKVLNRKTYLNFDKAVFKINIVPKVNYTIYDGFNLRNTNLAANFNGQNTEINNMNFTKLKNFTGL Hydrogen bonds contact Hydrophobic contact | ||||