Job Results:
Ligand
Structure
Job ID
1aab99f222a82120b630ad6d44e2ad5f
Job name
NA
Time
2026-02-27 11:58:02
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Albendazole monooxygenase (CYP3A4) | 3UA1 | 4.59 | |
Target general information Gen name CYP3A4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Taurochenodeoxycholate 6-alpha-hydroxylase; Quinine 3-monooxygenase; P450-PCN1; Nifedipine oxidase; NF-25; HLp; Cytochrome P450-PCN1; Cytochrome P450 NF-25; Cytochrome P450 HLp; Cytochrome P450 3A4; C Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function In liver microsomes, this enzyme is involved in an NADPH-dependent electron transport pathway. It performs a variety of oxidation reactions (e. g. caffeine 8-oxidation, omeprazole sulphoxidation, midazolam 1'-hydroxylation and midazolam 4-hydroxylation) of structurally unrelated compounds, including steroids, fatty acids, and xenobiotics. Acts as a 1,8-cineole 2-exo-monooxygenase. The enzyme also hydroxylates etoposide. Catalyzes 4-beta-hydroxylation of cholesterol. May catalyze 25-hydroxylation of cholesterol in vitro. Catalyzes sulfoxidation of the anthelmintics albendazole and fenbendazole. Cytochromes P450 are a group of heme-thiolate monooxygenases. Related diseases Vitamin D-dependent rickets 3 (VDDR3) [MIM:619073]: An autosomal dominant disorder of vitamin D metabolism resulting in early-onset rickets, reduced serum levels of the vitamin D metabolites 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D, and deficient responsiveness to parent and activated forms of vitamin D. {ECO:0000269|PubMed:29461981}. The gene represented in this entry is involved in disease pathogenesis. Drugs (DrugBank ID) DB08496; DB14055; DB12537; DB12629; DB01456; DB04070; DB11919; DB12515; DB11932; DB12001; DB05812; DB14973; DB11703; DB01418; DB00316; DB00819; DB15568; DB00546; DB08838; DB00518; DB00240; DB00041; DB04630; DB00802; DB00346; DB09026; DB00918; DB06203; DB00969; DB12015; DB14003; DB00404; DB06403; DB06742; DB13141; DB00288; DB00357; DB01424; DB01223; DB01118; DB00321; DB00381; DB00701; DB01217; DB01536; DB01435; DB11901; DB06605; DB00714; DB05676; DB00673; DB01352; DB09229; DB00278; DB01238; DB14185; DB06413; DB01169; DB06697; DB12597; DB06216; DB00637; DB11586; DB01072; DB16098; DB01076; DB01117; DB15011; DB06237; DB15233; DB06442; DB11995; DB06318; DB06626; DB00972; DB09230; DB04957; DB00207; DB12781; DB13997; DB04975; DB01483; DB11817; DB09227; DB00394; DB08903; DB05015; DB16703; DB15463; DB13488; DB09231; DB00865; DB01244; DB15982; DB00443; DB14669; DB12236; DB00307; DB01393; DB01128; DB11799; DB04794; DB00905; DB13746; DB16536; DB00612; DB13975; DB09223; DB08873; DB00188; DB00559; DB06616; DB07348; DB08870; DB09128; DB12267; DB01194; DB05541; DB01200; DB09017; DB11752; DB01222; DB00297; DB00921; DB00490; DB01008; DB09173; DB06772; DB00248; DB08875; DB00201; DB04886; DB00136; DB08907; DB01152; DB09061; DB14737; DB12218; DB11791; DB08502; DB06774; DB00564; DB11383; DB11960; DB06016; DB13835; DB01136; DB14984; DB06634; DB00520; DB01333; DB00482; DB06119; DB09063; DB00439; DB06419; DB00185; DB06777; DB00446; DB00475; DB13528; DB00608; DB00856; DB01114; DB00477; DB00356; DB00169; DB01410; DB09201; DB09232; DB01166; DB00501; DB01012; DB00568; DB00537; DB00604; DB00215; DB01211; DB12499; DB04920; DB01190; DB00349; DB11750; DB01013; DB13158; DB14652; DB00845; DB00636; DB06470; DB01242; DB01068; DB00575; DB00758; DB13843; DB00628; DB01559; DB00257; DB00363; DB09065; DB05239; DB00907; DB00318; DB01394; DB06342; DB00872; DB00286; DB12483; DB04652; DB01285; DB14681; DB01380; DB13003; DB08865; DB11672; DB14635; DB04838; DB00924; DB00531; DB00091; DB04839; DB00987; DB08912; DB09102; DB11963; DB01764; DB01406; DB11779; DB06292; DB04884; DB11682; DB00250; DB15031; DB00496; DB09234; DB12941; DB01264; DB09183; DB01254; DB00694; DB01609; DB11921; DB11943; DB11637; DB00705; DB13857; DB01151; DB00304; DB01260; DB06780; DB01134; DB06700; DB12161; DB01234; DB14649; DB11487; DB09555; DB05351; DB04856; DB14068; DB00514; DB00647; DB14063; DB11994; DB00829; DB00586; DB00485; DB09123; DB00255; DB09095; DB06781; DB01396; DB11274; DB01551; DB11273; DB13345; DB13385; DB00320; DB00343; DB01093; DB08995; DB13347; DB00954; DB00280; DB00822; DB02520; DB01248; DB00204; DB00757; DB08930; DB01184; DB00843; DB11400; DB12301; DB06446; DB05928; DB00590; DB01142; DB00997; DB00254; DB00470; DB04855; DB01395; DB00476; DB11952; DB00378; DB11742; DB14240; DB01127; DB14598; DB14600; DB00625; DB09235; DB06374; DB11979; DB11574; DB00216; DB15444; DB09039; DB09101; DB14064; DB13874; DB11718; DB13007; DB11986; DB08899; DB08992; DB00751; DB00668; DB00700; DB12266; DB01873; DB11405; DB03515; DB02187; DB12329; DB12147; DB01049; DB01253; DB00696; DB00530; DB00199; DB01175; DB11823; DB14575; DB09119; DB00736; DB01215; DB09381; DB12235; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB01196; DB00655; DB04574; DB00402; DB00330; DB00898; DB00977; DB00593; DB08794; DB01466; DB00823; DB09166; DB00294; DB00773; DB01628; DB14766; DB06414; DB13866; DB01590; DB00990; DB00973; DB12500; DB00949; DB01023; DB08980; DB00574; DB00813; DB06702; DB12265; DB08874; DB01216; DB16165; DB13961; DB04908; DB00301; DB00196; DB00687; DB00663; DB04841; DB00180; DB01544; DB00591; DB01047; DB08971; DB00324; DB00472; DB08970; DB14634; DB09378; DB14637; DB00846; DB00690; DB13338; DB04842; DB00499; DB13867; DB08906; DB00588; DB01095; DB00176; DB12307; DB08905; DB01319; DB06717; DB14019; DB01320; DB12010; DB11796; DB11679; DB00947; DB02703; DB15149; DB00674; DB12923; DB05087; DB00317; DB01241; DB01645; DB12184; DB06730; DB11619; DB12141; DB01381; DB11978; DB13879; DB00143; DB01016; DB08909; DB00986; DB05814; DB00889; DB10534; DB11575; DB00365; DB00400; DB01018; DB06786; DB01218; DB13728; DB00502; DB01159; DB05212; DB01275; DB00956; DB00769; DB00741; DB14538; DB14539; DB14540; DB14541; DB14542; DB14543; DB14545; DB14544; DB01611; DB14570; DB06789; DB00557; DB12471; DB09053; DB01050; DB11737; DB09054; DB01181; DB04946; DB00619; DB09262; DB00458; DB00724; DB05039; DB08953; DB00808; DB00224; DB06370; DB11886; DB13293; DB01029; DB00762; DB11633; DB06636; DB00951; DB00982; DB00270; DB11757; DB01167; DB09083; DB08820; DB00602; DB14568; DB04845; DB09570; DB01221; DB01587; DB06738; DB01026; DB09309; DB05903; DB09236; DB06218; DB06791; DB00448; DB01259; DB06685; DB14723; DB12825; DB11951; DB15673; DB16217; DB09078; DB00528; DB11560; DB06469; DB12070; DB01006; DB01227; DB09237; DB01002; DB06282; DB05667; DB00825; DB08918; DB00367; DB00281; DB13766; DB08882; DB17083; DB01583; DB00589; DB09198; DB14065; DB08827; DB01206; DB06448; DB16222; DB00836; DB01601; DB00455; DB00186; DB04871; DB12130; DB09195; DB12089; DB00678; DB14596; DB00227; DB09212; DB08933; DB09280; DB06077; DB06708; DB08815; DB12674; DB12474; DB04829; DB13074; DB08932; DB09238; DB16226; DB04835; DB06234; DB14921; DB00643; DB14009; DB09124; DB00603; DB00253; DB00358; DB00351; DB11529; DB14659; DB00814; DB00170; DB00454; DB09383; DB01071; DB01357; DB04817; DB00333; DB04833; DB00763; DB00563; DB01028; DB09241; DB00353; DB00959; DB14644; DB12952; DB06710; DB00247; DB01233; DB00264; DB00916; DB01011; DB15489; DB00379; DB06148; DB01388; DB01110; DB00683; DB13456; DB06595; DB00834; DB04896; DB13287; DB08893; DB11792; DB00370; DB12489; DB16236; DB06587; DB00648; DB01204; DB16390; DB00745; DB11763; DB00764; DB14512; DB00471; DB00295; DB09205; DB00688; DB01024; DB11605; DB00486; DB14011; DB00607; DB12092; DB11691; DB06230; DB09049; DB01183; DB00731; DB04861; DB01149; DB00220; DB11828; DB09199; DB09048; DB00238; DB00627; DB00622; DB02701; DB00184; DB01115; DB09239; DB04868; DB09240; DB06712; DB04743; DB00393; DB09079; DB16691; DB12005; DB00401; DB01595; DB01054; DB00435; DB11636; DB13981; DB06713; DB14678; DB00717; DB09371; DB01059; DB00957; DB09389; DB00540; DB06174; DB06152; DB00104; DB06670; DB00334; DB09074; DB11442; DB14881; DB00768; DB16267; DB12513; DB09568; DB00338; DB00904; DB11130; DB04911; DB01083; DB01173; DB11837; DB09330; DB04938; DB13500; DB00776; DB12532; DB00239; DB01062; DB00497; DB06412; DB01192; DB12612; DB01229; DB11697; DB09073; DB01267; DB00377; DB05467; DB06603; DB00213; DB00617; DB01384; DB08439; DB00910; DB09297; DB00715; DB06663; DB03010; DB06589; DB00082; DB15102; DB13791; DB00312; DB11198; DB08883; DB01186; DB01074; DB08922; DB00850; DB12978; DB03783; DB00780; DB01174; DB00946; DB00191; DB00812; DB00252; DB13878; DB01085; DB05316; DB00337; DB01100; DB06762; DB09090; DB01132; DB13941; DB12582; DB01621; DB04951; DB17472; DB11642; DB04977; DB12240; DB08910; DB08901; DB12016; DB01263; DB05478; DB15822; DB01411; DB06209; DB01588; DB01058; DB01130; DB00860; DB15566; DB14633; DB14631; DB00635; DB14646; DB13208; DB02789; DB04825; DB05154; DB01087; DB00794; DB01032; DB00396; DB00420; DB13602; DB09288; DB01182; DB12278; DB00571; DB06480; DB00545; DB01589; DB04216; DB01224; DB01103; DB13685; DB00908; DB00468; DB01369; DB12874; DB01129; DB00481; DB00980; DB00863; DB00243; DB00234; DB08896; DB11853; DB06458; DB14761; DB00409; DB00912; DB16826; DB02709; DB01256; DB13174; DB11730; DB06233; DB00615; DB04934; DB01045; DB11753; DB01201; DB01220; DB08864; DB12457; DB00896; DB06155; DB08931; DB14840; DB15305; DB00734; DB14924; DB00503; DB06228; DB09200; DB00533; DB01656; DB13409; DB09291; DB06176; DB00296; DB00412; DB05271; DB00778; DB12332; DB06201; DB11614; DB01698; DB08877; DB06654; DB12391; DB01001; DB00938; DB12543; DB01232; DB11805; DB11767; DB06335; DB00747; DB12834; DB14583; DB11459; DB01037; DB05885; DB11362; DB11942; DB15685; DB11689; DB06731; DB06739; DB06144; DB01104; DB01236; DB01105; DB00203; DB06207; DB09036; DB06290; DB00641; DB12371; DB00877; DB01261; DB06268; DB05482; DB01591; DB09308; DB09099; DB09143; DB00398; DB12713; DB15569; DB12548; DB01323; DB09118; DB00708; DB00359; DB01015; DB01138; DB01268; DB09034; DB09317; DB09318; DB00864; DB00820; DB00675; DB00706; DB06083; DB09071; DB01349; DB08833; DB12887; DB12020; DB05521; DB00976; DB12095; DB00231; DB06287; DB11761; DB00444; DB09299; DB15133; DB00857; DB00342; DB13399; DB13725; DB04905; DB00624; DB13943; DB13944; DB01420; DB13946; DB00759; DB12093; DB14066; DB11712; DB01041; DB00277; DB01154; DB00599; DB04572; DB00906; DB09289; DB08816; DB11470; DB00911; DB01007; DB01409; DB00932; DB06137; DB16732; DB11800; DB06273; DB11635; DB11251; DB08895; DB08811; DB09216; DB01036; DB06212; DB00273; DB01685; DB00539; DB05109; DB00193; DB08911; DB07615; DB00752; DB14962; DB05773; DB00656; DB00755; DB00620; DB00897; DB12245; DB12808; DB09089; DB00347; DB00440; DB06045; DB00197; DB13179; DB11652; DB15328; DB06267; DB08867; DB14989; DB13609; DB15091; DB01586; DB12255; DB11915; DB00580; DB00313; DB15114; DB05294; DB03701; DB04894; DB00862; DB11613; DB08881; DB11581; DB00285; DB00661; DB14895; DB06652; DB09082; DB06684; DB09185; DB00570; DB00541; DB00309; DB11641; DB00361; DB12131; DB08828; DB11094; DB00163; DB11693; DB11739; DB09030; DB00582; DB09068; DB14975; DB12026; DB00682; DB13950; DB01392; DB00549; DB00962; DB15035; DB15688; DB00495; DB00744; DB04832; DB00246; DB00425; DB04828; DB00909; DB01198; DB09225; DB01624; DB15490 Interacts with O15287; Q6ZQX7-4 EC number EC 1.14.14.- Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Fatty acid metabolism; Heme; Iron; Lipid biosynthesis; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol metabolism; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 52195.6 Length 456 Aromaticity 0.11 Instability index 44.02 Isoelectric point 8.48 Charge (pH=7) 4.36 2D Binding mode Binding energy (Kcal/mol) -6.26  Molscript Map 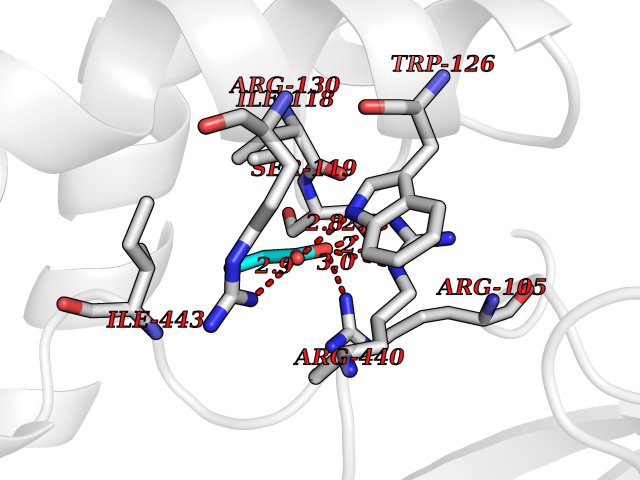 Pymol Map 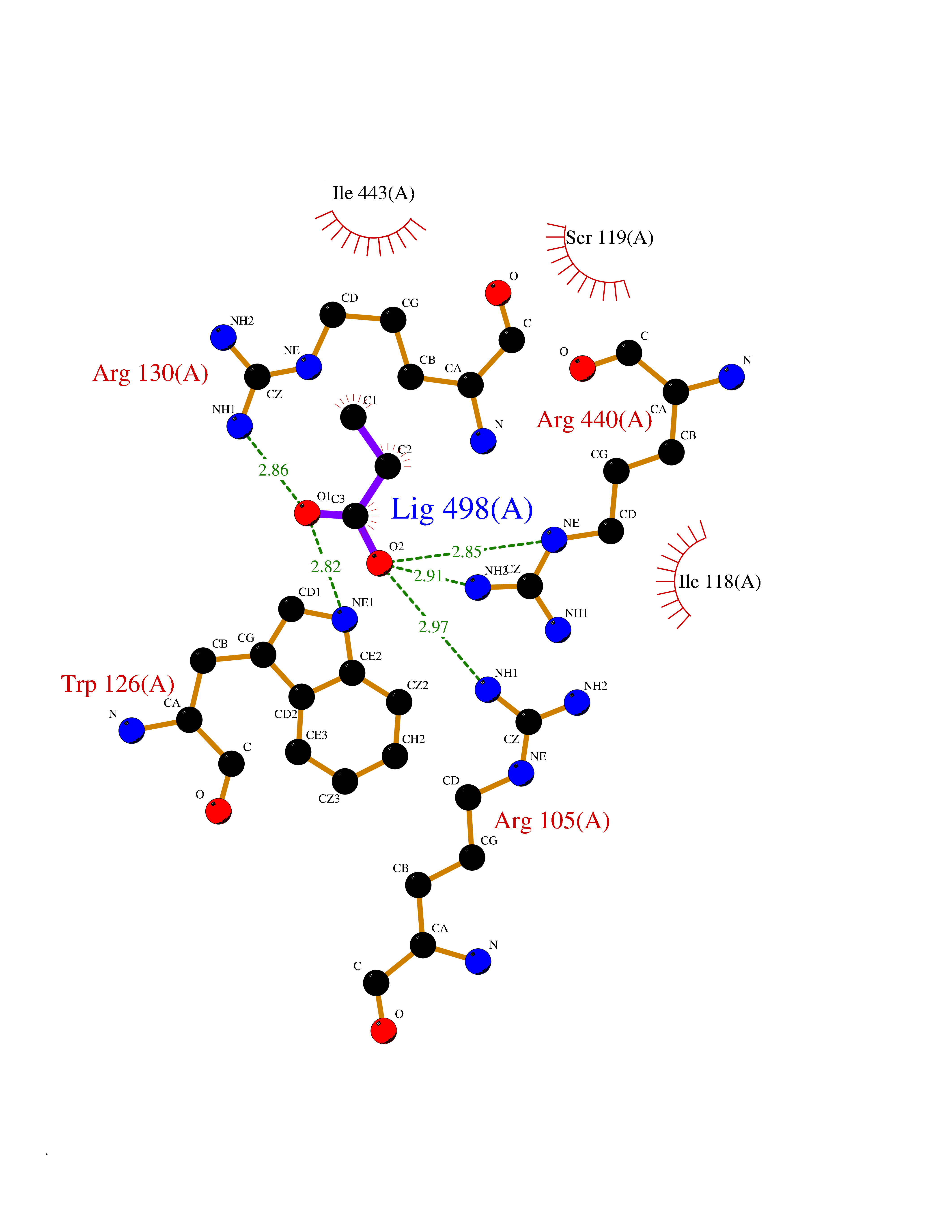 Ligplot Map 3D Binding mode Sequence HSHGLFKKLGIPGPTPLPFLGNILSYHKGFCMFDMECHKKYGKVWGFYDGQQPVLAITDPDMIKTVLVKECYSVFTNRRPFGPVGFMKSAISIAEDEEWKRLRSLLSPTFTSGKLKEMVPIIAQYGDVLVRNLRREAETGKPVTLKDVFGAYSMDVITSTSFGVNIDSLNNPQDPFVENTKKLLRFDFLDPFFLSITVFPFLIPILEVLNICVFPREVTNFLRKSVKRMKEEDTQVDFLQLMIDSQHKALSDLELVAQSIIFIFAGYETTSSVLSFIMYELATHPDVQQKLQEEIDAVLPNKAPPTYDTVLQMEYLDMVVNETLRLFPIAMRLERVCKKDVEINGMFIPKGVVVMIPSYALHRDPKYWTEPEKFLPERFSKKNKDNIDPYIYTPFGSGPRNCIGMRFALMNMKLALIRVLQNFSFKPCKETQIPLKLSLGGLLQPEKPVVLKVESR Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Deubiquitinating enzyme 1 (USP1) | 7ZH4 | 4.59 | |
Target general information Gen name USP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hUBP; Ubiquitin-specific-processing protease 1; Ubiquitin thioesterase 1; Ubiquitin carboxyl-terminal hydrolase 1 Protein family Peptidase C19 family Biochemical class Peptidase Function Involved in PCNA-mediated translesion synthesis (TLS) by deubiquitinating monoubiquitinated PCNA. Has almost no deubiquitinating activity by itself and requires the interaction with WDR48 to have a high activity. Negative regulator of DNA damage repair which specifically deubiquitinates monoubiquitinated FANCD2. Related diseases Brachydactyly A2 (BDA2) [MIM:112600]: A form of brachydactyly. Brachydactyly defines a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. In brachydactyly type A2 shortening of the middle phalanges is confined to the index finger and the second toe, all other digits being more or less normal. Because of a rhomboid or triangular shape of the affected middle phalanx, the end of the second finger usually deviates radially. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}. The gene represented in this entry is involved in disease pathogenesis. Duplications of a cis-regulatory element located approximately 110 kb downstream of BMP2 have been found in BDA2 families. They likely cause altered BMP2 expression with pathological consequences. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}.; DISEASE: Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 1 (SSFSC1) [MIM:617877]: An autosomal dominant disorder characterized by short stature, facial dysmorphism, skeletal anomalies, and variable cardiac defects. Distinctive facial features include midface retrusion, short upturned nose, long philtrum, high-arched or cleft palate, and variable degrees of micrognathia and dental crowding. Skeletal anomalies include patterning defects of the axial skeleton, characterized by 11 pairs of ribs and brachydactyly of the fifth ray. Congenital heart defects are variably observed and appear to involve primarily the cardiac outflow tract. {ECO:0000269|PubMed:29198724}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q8TAF3; Q8TAF3-1 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Autocatalytic cleavage; DNA damage; DNA repair; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID D Molecular weight (Da) 32426 Length 285 Aromaticity 0.1 Instability index 50.73 Isoelectric point 5.85 Charge (pH=7) -4.67 2D Binding mode Binding energy (Kcal/mol) -6.25 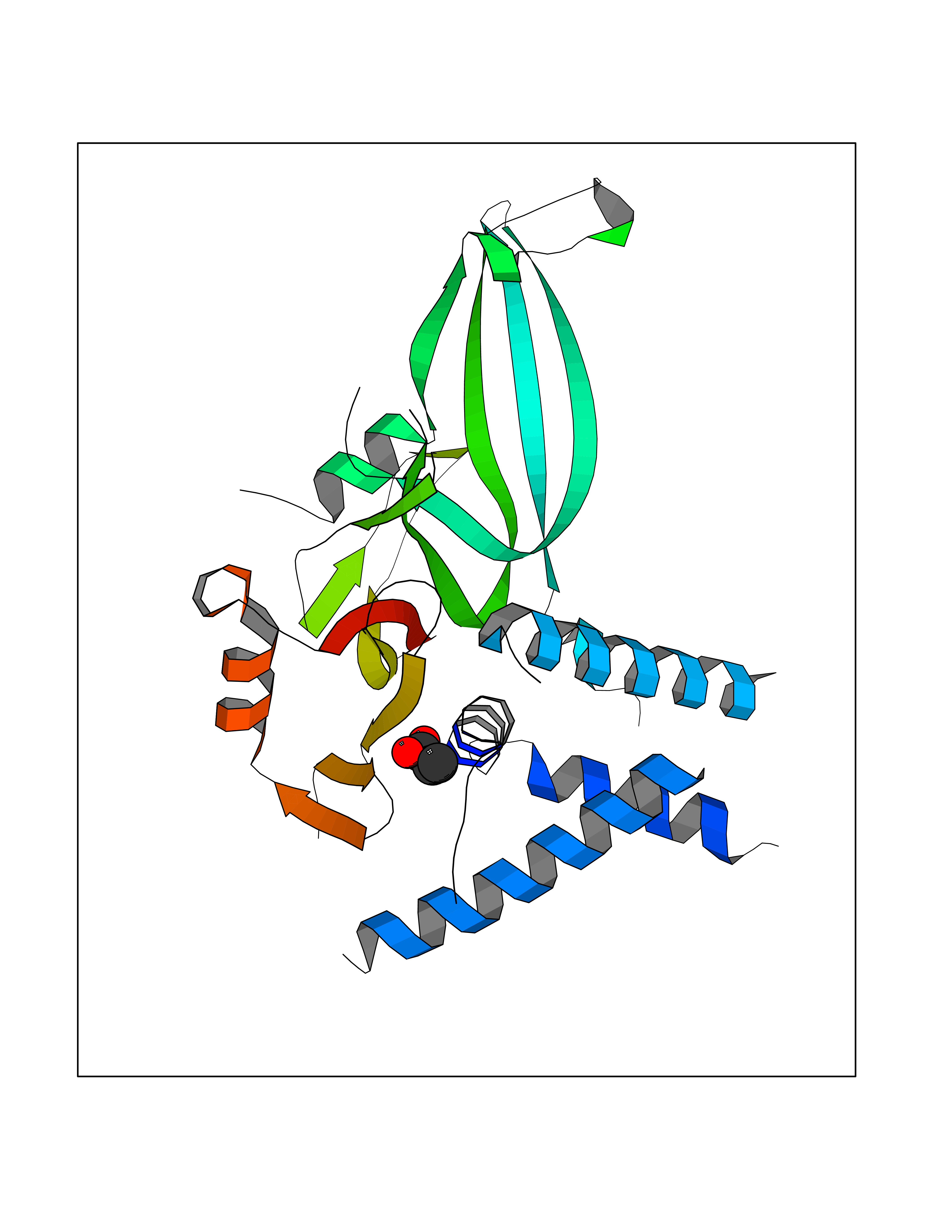 Molscript Map 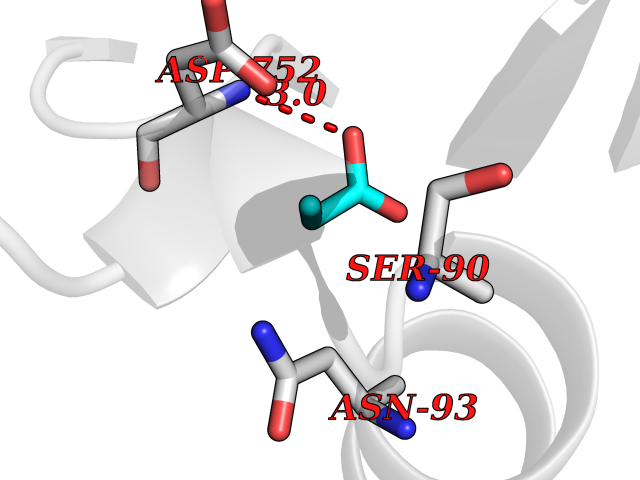 Pymol Map 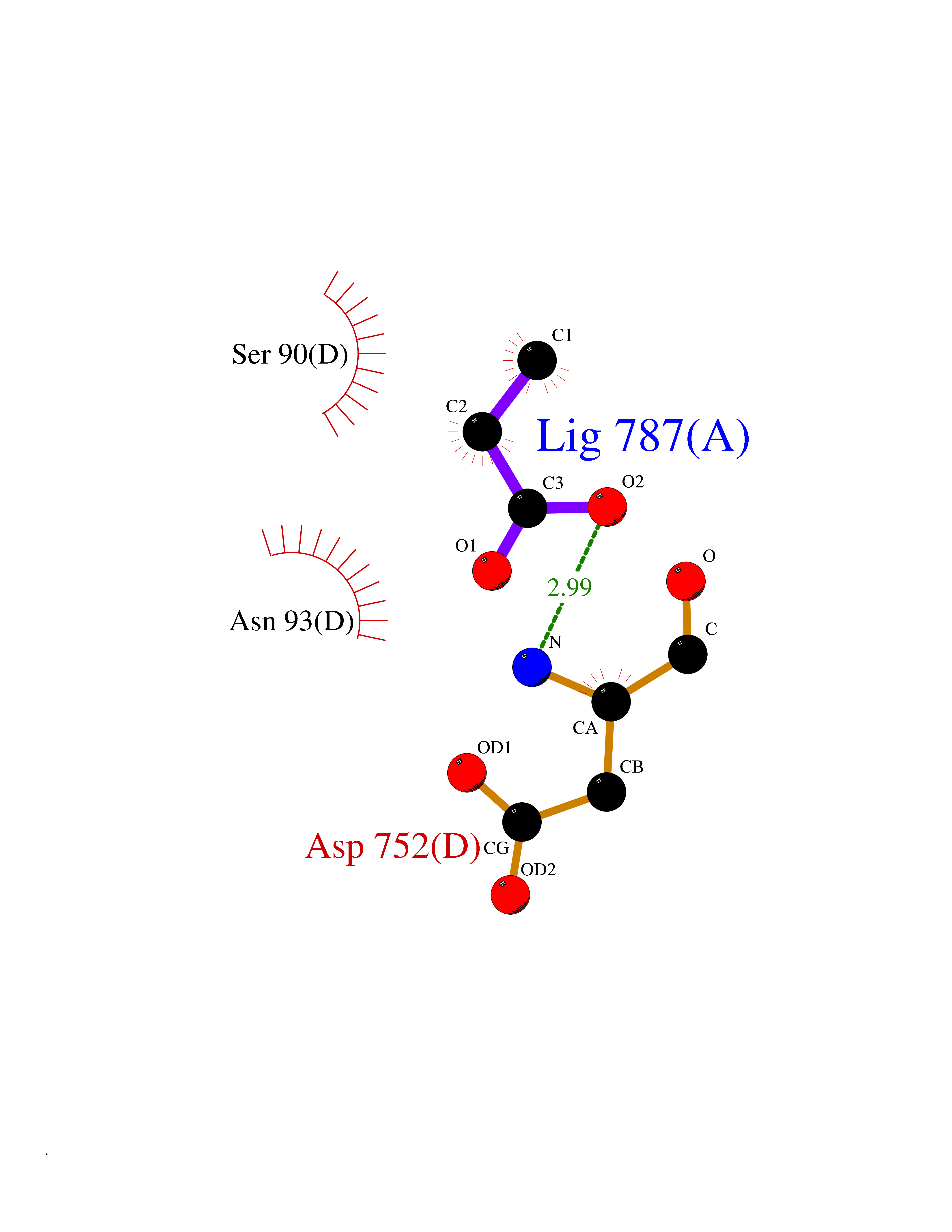 Ligplot Map 3D Binding mode Sequence GLNNLGNTSYLNSILQVLYFCPGFKSGVKHLFNIISRKKYELICSLQSLIISVEQLQASFLLNPLQHDAQEVLQCILGNIQETCQLLKKGFELVEKLFQGQLVLRTRCLECESLTERREDFQDISVPVQEDMKTLRWAISQFASVERIVGEDKYFCENCHHYTEAERSLLFDKMPEVITIHLKCFAASGLSKINTPLLTPLKLSLEEWSTKPTNDSYGLFAVVMHSGITISSGHYTASVKVTYEGKWLLFDDSEVKVTEEKDFLNSLSPSTSPTSTPYLLFYKKL Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Beta-galactosidase (GLB1) | 3THD | 4.59 | |
Target general information Gen name GLB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lactase; Elastin receptor 1; ELNR1; Acid beta-galactosidase Protein family Glycosyl hydrolase 35 family Biochemical class NA Function Isoform 1: Cleaves beta-linked terminal galactosyl residues from gangliosides, glycoproteins, and glycosaminoglycans. Related diseases GM1-gangliosidosis 1 (GM1G1) [MIM:230500]: An autosomal recessive lysosomal storage disease marked by the accumulation of GM1 gangliosides, glycoproteins and keratan sulfate primarily in neurons of the central nervous system. GM1-gangliosidosis type 1 is characterized by onset within the first three months of life, central nervous system degeneration, coarse facial features, hepatosplenomegaly, skeletal dysmorphology reminiscent of Hurler syndrome, and rapidly progressive psychomotor deterioration. Urinary oligosaccharide levels are high. It leads to death usually between the first and second year of life. {ECO:0000269|PubMed:10338095, ECO:0000269|PubMed:10737981, ECO:0000269|PubMed:10839995, ECO:0000269|PubMed:1487238, ECO:0000269|PubMed:15365997, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:15791924, ECO:0000269|PubMed:16538002, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:1928092, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8213816, ECO:0000269|Ref.28, ECO:0000269|Ref.31}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GM1-gangliosidosis 2 (GM1G2) [MIM:230600]: A gangliosidosis characterized by onset between ages 1 and 5. The main symptom is locomotor ataxia, ultimately leading to a state of decerebration with epileptic seizures. Patients do not display the skeletal changes associated with the infantile form, but they nonetheless excrete elevated amounts of beta-linked galactose-terminal oligosaccharides. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10737981, ECO:0000269|PubMed:12644936, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8213816}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GM1-gangliosidosis 3 (GM1G3) [MIM:230650]: A gangliosidosis with a variable phenotype. Patients show mild skeletal abnormalities, dysarthria, gait disturbance, dystonia and visual impairment. Visceromegaly is absent. Intellectual deficit can initially be mild or absent but progresses over time. Inheritance is autosomal recessive. {ECO:0000269|PubMed:11511921, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:15986423, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8198123, ECO:0000269|Ref.28, ECO:0000269|Ref.30}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 4B (MPS4B) [MIM:253010]: A form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life. {ECO:0000269|PubMed:11511921, ECO:0000269|PubMed:12393180, ECO:0000269|PubMed:16538002, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1928092, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:7586649}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04465 Interacts with Q8NBJ4; Q3KNW5; Q9BRI3; P30825 EC number EC 3.2.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Gangliosidosis; Glycoprotein; Glycosidase; Hydrolase; Lysosome; Mucopolysaccharidosis; Proteomics identification; Reference proteome; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 67980.6 Length 605 Aromaticity 0.13 Instability index 40.91 Isoelectric point 5.81 Charge (pH=7) -9.05 2D Binding mode Binding energy (Kcal/mol) -6.26 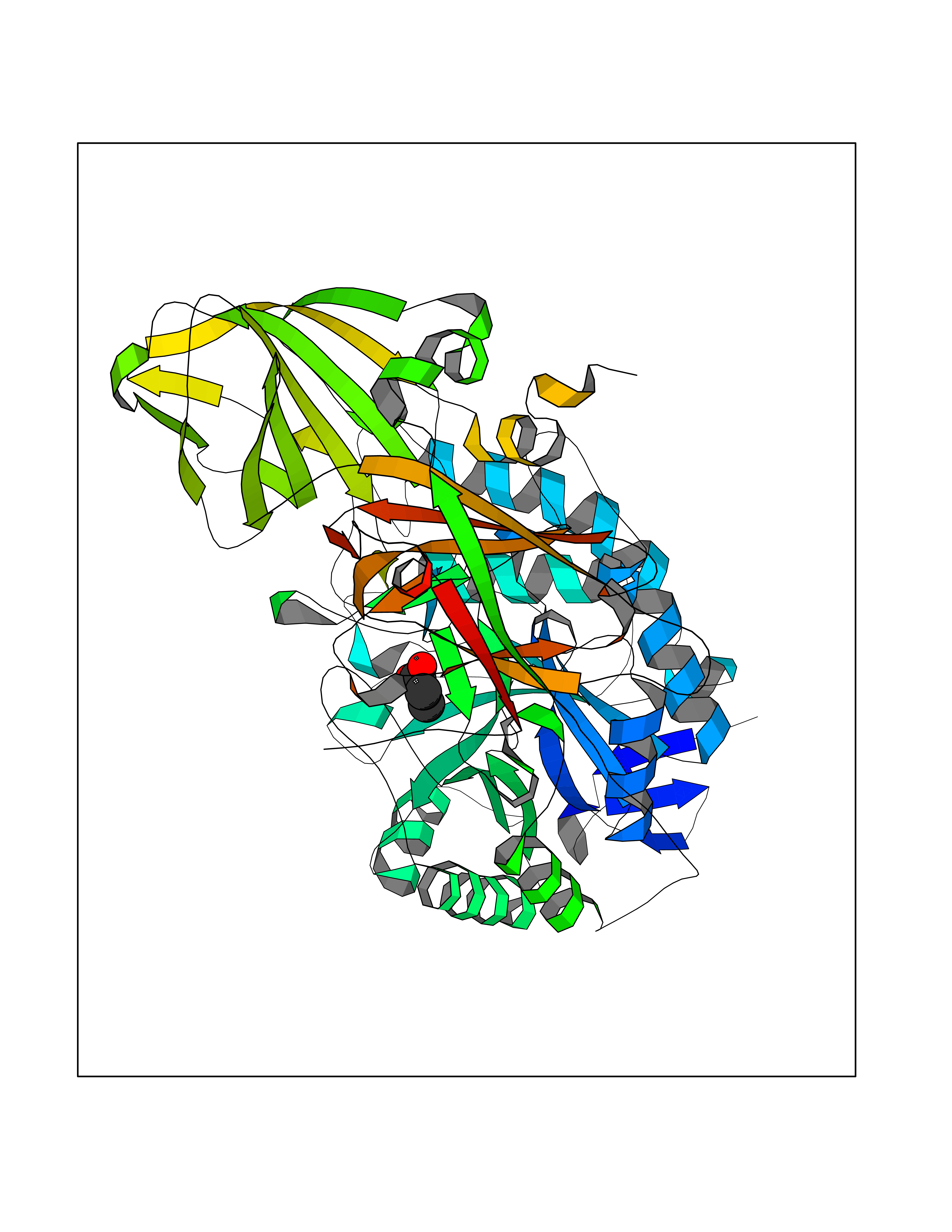 Molscript Map 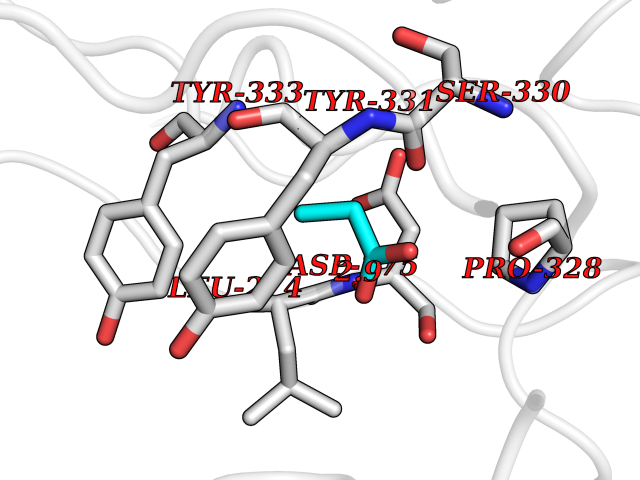 Pymol Map 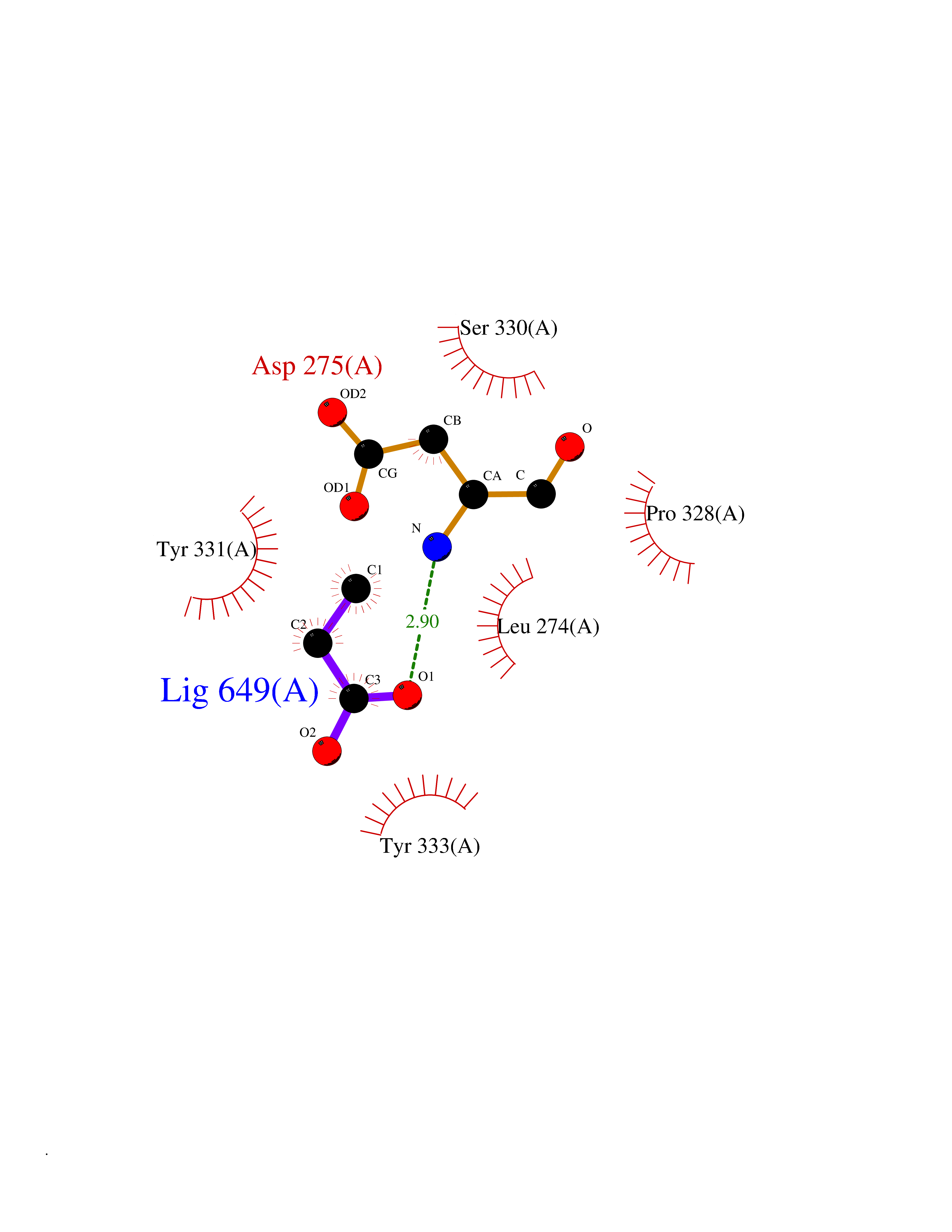 Ligplot Map 3D Binding mode Sequence QRMFEIDYSRDSFLKDGQPFRYISGSIHYSRVPRFYWKDRLLKMKMAGLNAIQTYVPWNFHEPWPGQYQFSEDHDVEYFLRLAHELGLLVILRPGPYICAEWEMGGLPAWLLEKESILLRSSDPDYLAAVDKWLGVLLPKMKPLLYQNGGPVITVQVENEYGSYFACDFDYLRFLQKRFRHHLGDDVVLFTTDGAHKTFLKCGALQGLYTTVDFGTGSNITDAFLSQRKCEPKGPLINSEFYTGWLDHWGQPHSTIKTEAVASSLYDILARGASVNLYMFIGGTNFAYWNGANSPYAAQPTSYDYDAPLSEAGDLTEKYFALRNIIQKFEKVPEGPIPPSTPKFAYGKVTLEKLKTVGAALDILCPSGPIKSLYPLTFIQVKQHYGFVLYRTTLPQDCSNPAPLSSPLNGVHDRAYVAVDGIPQGVLERNNVITLNITGKAGATLDLLVENMGRVNYGAYINDFKGLVSNLTLSSNILTDWTIFPLDTEDAVRSHLGGWGHRNYTLPAFYMGNFSIPSGIPDLPQDTFIQFPGWTKGQVWINGFNLGRYWPARGPQLTLFVPQHILMTSAPNTITVLELEWAPCSSDDPELCAVTFVDRPVIGSS Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Cyclin-dependent kinase 4 | 2W96 | 4.58 | |
Target general information Gen name CDK4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Cell cycle Function ATP binding.Cyclin binding.Cyclin-dependent protein serine/threonine kinase activity.Cyclin-dependent protein serine/threonine kinase regulator activity.Protein complex binding. Related diseases Melanoma, cutaneous malignant 3 (CMM3) [MIM:609048]: A malignant neoplasm of melanocytes, arising de novo or from a pre-existing benign nevus, which occurs most often in the skin but may also involve other sites. {ECO:0000269|PubMed:7652577, ECO:0000269|PubMed:8528263, ECO:0000269|PubMed:9311594, ECO:0000269|PubMed:9425228}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12001; DB03496; DB12010; DB09073; DB02733; DB11730; DB15442 Interacts with Q9UH17; P24385; P30279; P30281; Q16543; P50613; P38936; P46527; P49918; P42771; P42772; P42773; P55273; Q9UJC3; P08238; Q9UKT9; Q0VD86; P01106; Q9ULD0; P28749; Q08999; P09936; Q8N720 EC number 2.7.11.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Cell division; Cytoplasm; Disease variant; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 30138.4 Length 267 Aromaticity 0.09 Instability index 36.2 Isoelectric point 5.78 Charge (pH=7) -5.83 2D Binding mode Binding energy (Kcal/mol) -6.24 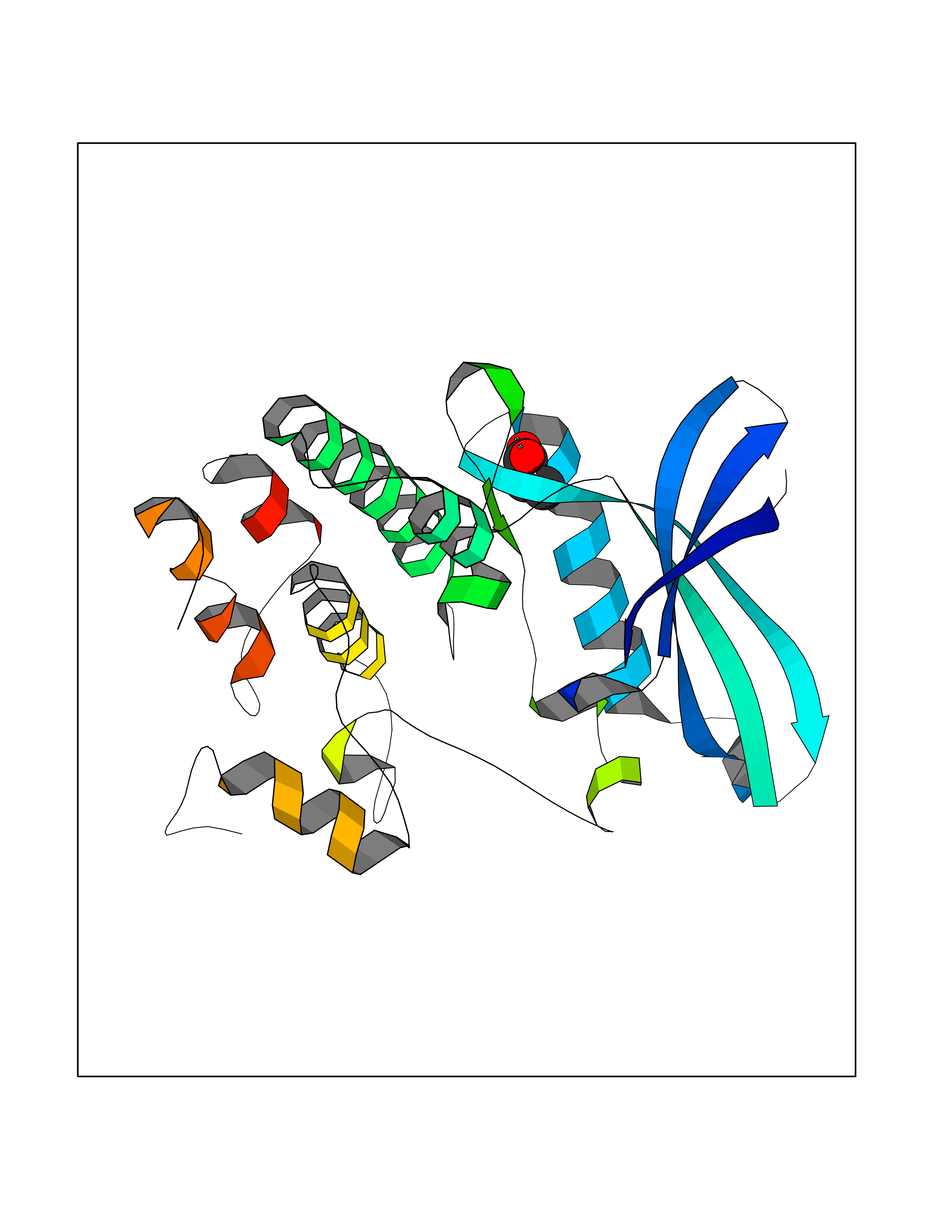 Molscript Map  Pymol Map 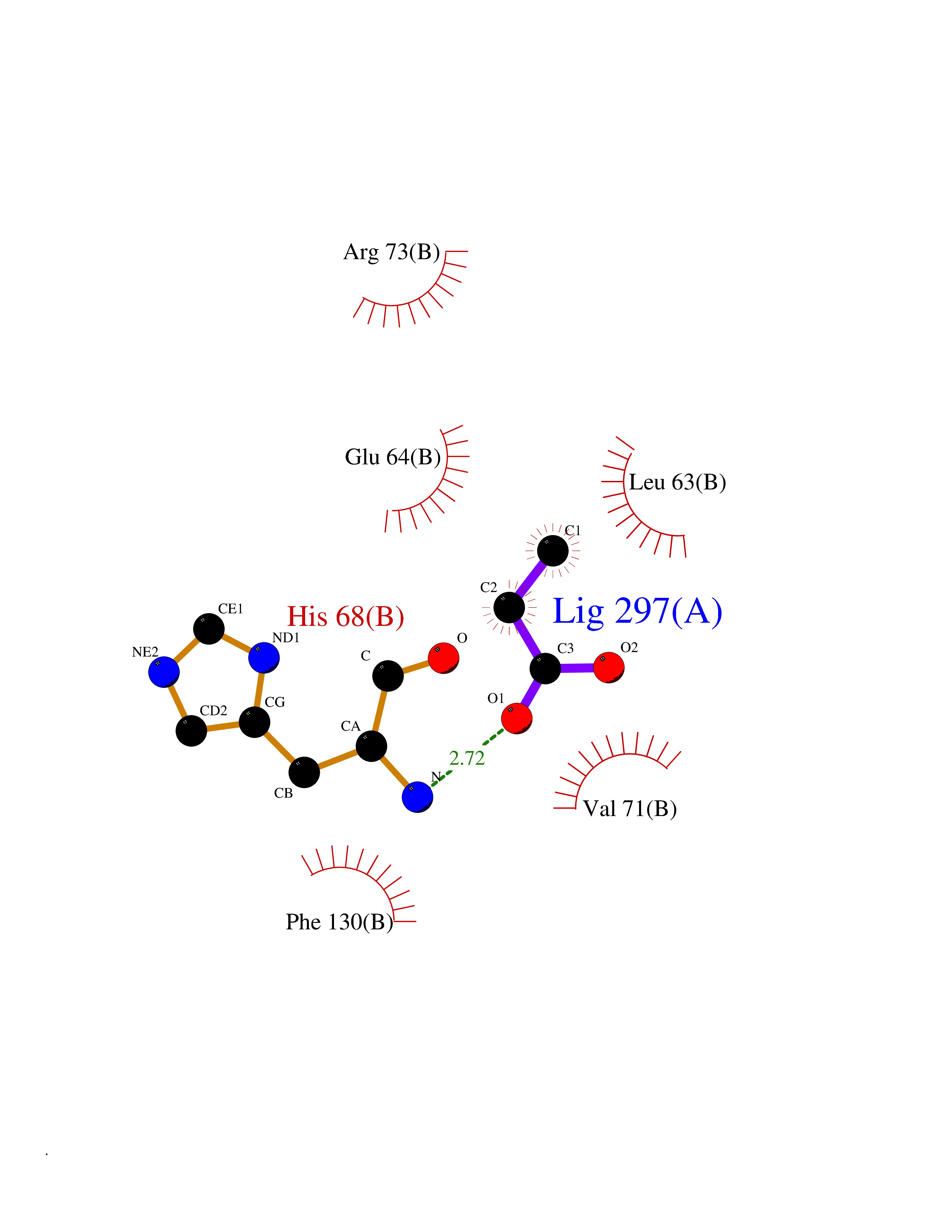 Ligplot Map 3D Binding mode Sequence SRYEPVAEIGVGAYGTVYKARDPHSGHFVALKSVRVPNGEEGLPISTVREVALLRRLEAFEHPNVVRLMDVCATSRTDREIKVTLVFEHVDQDLRTYLDKAPPPGLPAETIKDLMRQFLRGLDFLHANCIVHRDLKPENILVTSGGTVKLADFGLARIYSYQMALDPVVVTLWYRAPEVLLQSTYATPVDMWSVGCIFAEMFRRKPLFCGNSEADQLGKIFDLIGLPPEDDWVPEMEESGAQLLLEMLTFNPHKRISAFRALQHSYL Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Ornithine transcarbamylase (OTC) | 1OTH | 4.58 | |
Target general information Gen name OTC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms OTCase; Ornithine carbamoyltransferase, mitochondrial Protein family Aspartate/ornithine carbamoyltransferase superfamily, OTCase family Biochemical class NA Function Catalyzes the second step of the urea cycle, the condensation of carbamoyl phosphate with L-ornithine to form L-citrulline. The urea cycle ensures the detoxification of ammonia by converting it to urea for excretion. Related diseases Ornithine carbamoyltransferase deficiency (OTCD) [MIM:311250]: An X-linked disorder of the urea cycle which causes a form of hyperammonemia. Mutations with no residual enzyme activity are always expressed in hemizygote males by a very severe neonatal hyperammonemic coma that generally proves to be fatal. Heterozygous females are either asymptomatic or express orotic aciduria spontaneously or after protein intake. The disorder is treatable with supplemental dietary arginine and low protein diet. The arbitrary classification of patients into the 'neonatal' group (clinical hyperammonemia in the first few days of life) and 'late' onset (clinical presentation after the neonatal period) has been used to differentiate severe from mild forms. {ECO:0000269|PubMed:10070627, ECO:0000269|PubMed:10502831, ECO:0000269|PubMed:10737985, ECO:0000269|PubMed:11793483, ECO:0000269|PubMed:1480464, ECO:0000269|PubMed:1671317, ECO:0000269|PubMed:1721894, ECO:0000269|PubMed:2347583, ECO:0000269|PubMed:2474822, ECO:0000269|PubMed:2556444, ECO:0000269|PubMed:3170748, ECO:0000269|PubMed:7474905, ECO:0000269|PubMed:7951259, ECO:0000269|PubMed:8019569, ECO:0000269|PubMed:8081373, ECO:0000269|PubMed:8081398, ECO:0000269|PubMed:8099056, ECO:0000269|PubMed:8112735, ECO:0000269|PubMed:8530002, ECO:0000269|PubMed:8807340, ECO:0000269|PubMed:8830175, ECO:0000269|PubMed:8956038, ECO:0000269|PubMed:8956045, ECO:0000269|PubMed:9065786, ECO:0000269|PubMed:9143919, ECO:0000269|PubMed:9266388, ECO:0000269|PubMed:9286441, ECO:0000269|PubMed:9452024, ECO:0000269|PubMed:9452049, ECO:0000269|PubMed:9452065, ECO:0000269|Ref.32, ECO:0000269|Ref.43}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00155; DB02011; DB04185; DB00129 Interacts with NA EC number EC 2.1.3.3 Uniprot keywords 3D-structure; Acetylation; Amino-acid biosynthesis; Arginine biosynthesis; Disease variant; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Transit peptide; Urea cycle Protein physicochemical properties Chain ID A Molecular weight (Da) 36060.2 Length 321 Aromaticity 0.08 Instability index 36.44 Isoelectric point 7.87 Charge (pH=7) 1.48 2D Binding mode Binding energy (Kcal/mol) -6.24 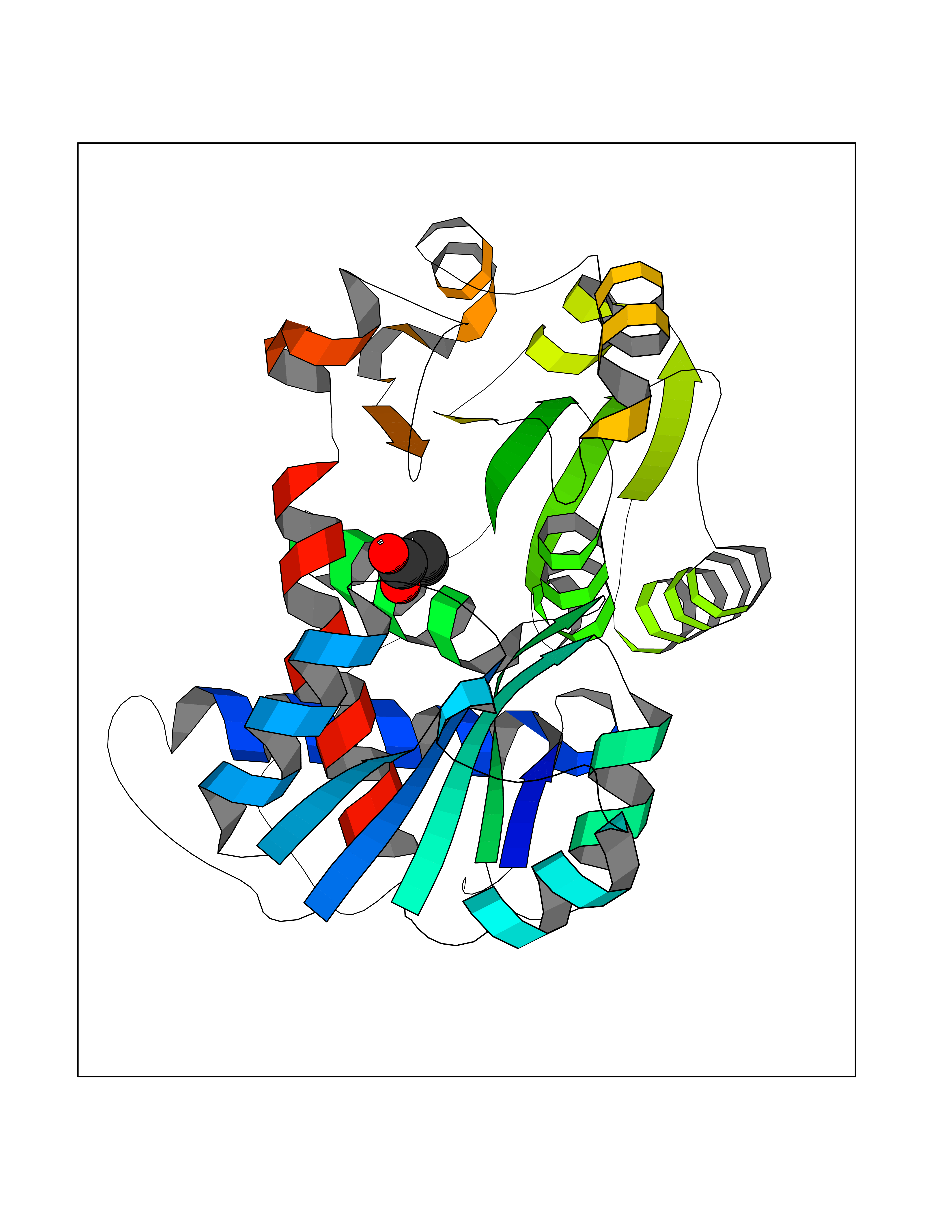 Molscript Map  Pymol Map 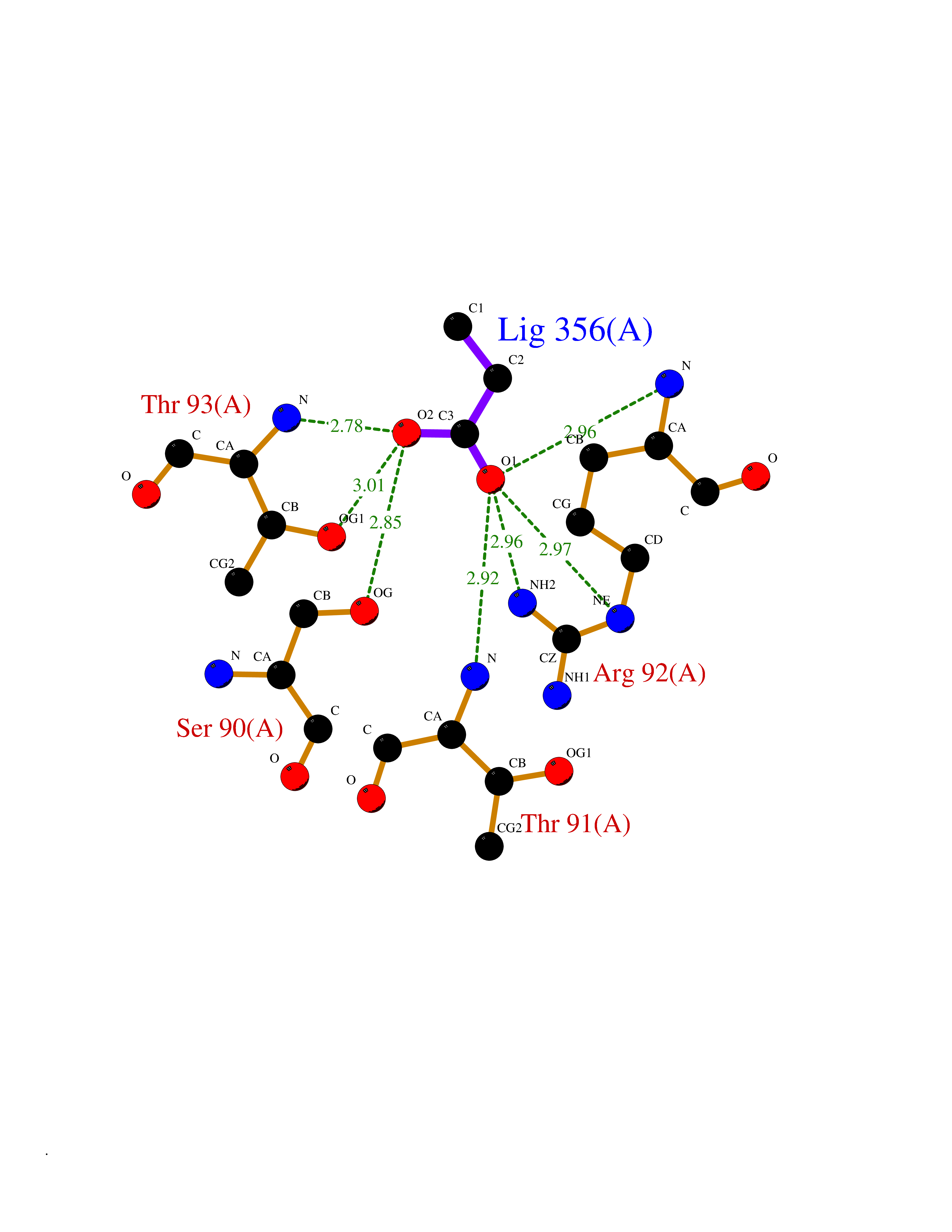 Ligplot Map 3D Binding mode Sequence KVQLKGRDLLTLKNFTGEEIKYMLWLSADLKFRIKQKGEYLPLLQGKSLGMIFEKRSTRTRLSTETGFALLGGHPCFLTTQDIHLGVNESLTDTARVLSSMADAVLARVYKQSDLDTLAKEASIPIINGLSDLYHPIQILADYLTLQEHYSSLKGLTLSWIGDGNNILHSIMMSAAKFGMHLQAATPKGYEPDASVTKLAEQYAKENGTKLLLTNDPLEAAHGGNVLITDTWISMGREEEKKKRLQAFQGYQVTMKTAKVAASDWTFLHCLPRKPEEVDDEVFYSPRSLVFPEAENRKWTIMAVMVSLLTDYSPQLQKPKF Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | "Acetolactate synthase, chloroplastic (AtALS) (EC 2.2.1.6) (Acetohydroxy-acid synthase) (Protein CHLORSULFURON RESISTANT 1)" | 5K3S | 4.58 | |
Target general information Gen name ALS Organism Arabidopsis thaliana (Mouse-ear cress) Uniprot ID TTD ID NA Synonyms At3g48560;CSR1;AHAS;T8P19.70;TZP5 Protein family TPP enzyme family Biochemical class NA Function Catalyzes the formation of acetolactate from pyruvate, the first step in valine and isoleucine biosynthesis. {ECO:0000269|PubMed:10386618, ECO:0000269|PubMed:16665813, ECO:0000269|PubMed:16667374, ECO:0000269|PubMed:16668488, ECO:0000269|PubMed:2336405, ECO:0000269|PubMed:8913312, ECO:0000269|PubMed:9355748, ECO:0000269|PubMed:9677339, ECO:0000269|Ref.9}." Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 2.2.1.6 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Branched-chain amino acid biosynthesis; Chloroplast; Coiled coil; FAD; Flavoprotein; Genetically modified food; Herbicide resistance; Magnesium; Metal-binding; Oxidation; Plastid; Reference proteome; Thiamine pyrophosphate; Transferase; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 63431 Length 583 Aromaticity 0.07 Instability index 36.62 Isoelectric point 5.4 Charge (pH=7) -15.33 2D Binding mode Binding energy (Kcal/mol) -6.24 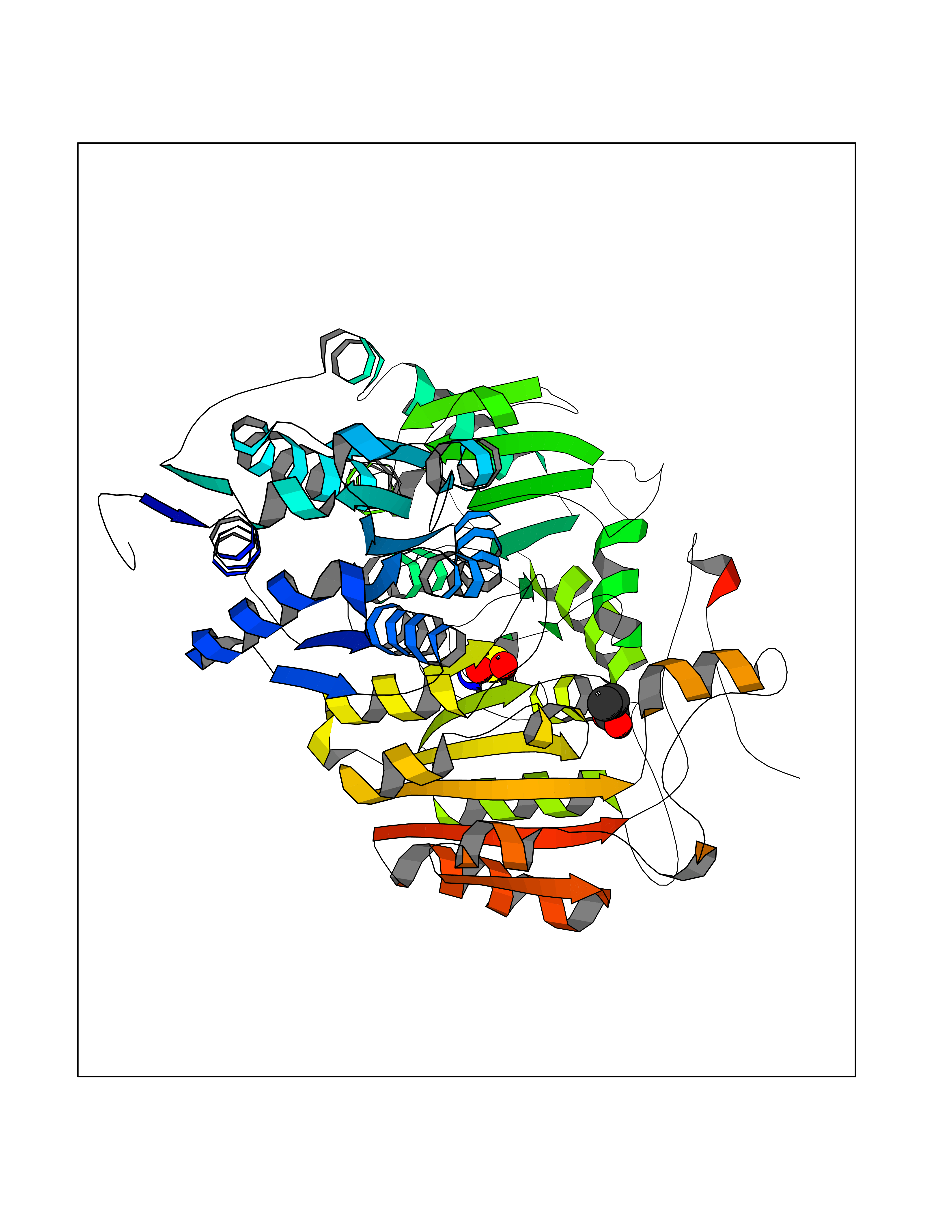 Molscript Map 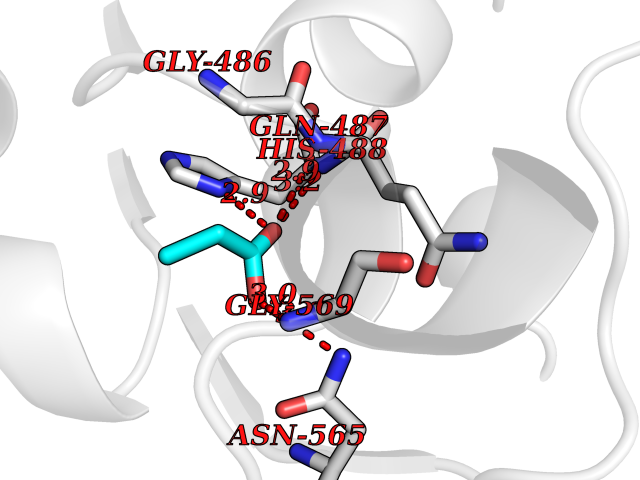 Pymol Map 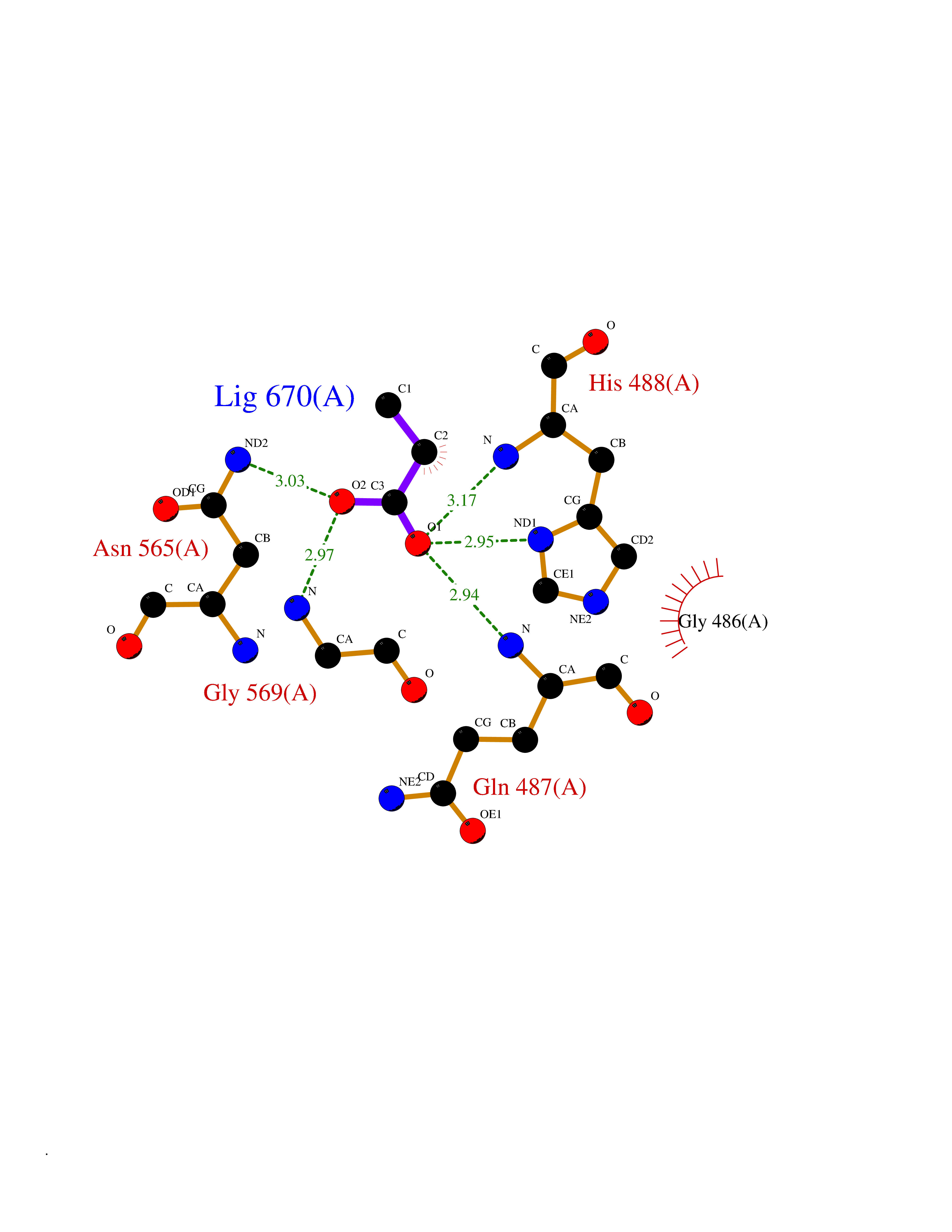 Ligplot Map 3D Binding mode Sequence TFISRFAPDQPRKGADILVEALERQGVETVFAYPGGASMEIHQALTRSSSIRNVLPRHEQGGVFAAEGYARSSGKPGICIATSGPGATNLVSGLADALLDSVPLVAITGQVPRRMIGTDAFQETPIVEVTRSITKHNYLVMDVEDIPRIIEEAFFLATSGRPGPVLVDVPKDIQQQLAIPNWEQAMRLPGYMSRMPKPPEDSHLEQIVRLISESKKPVLYVGGGCLNSSDELGRFVELTGIPVASTLMGLGSYPXDDELSLHMLGMHGTVYANYAVEHSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKTPHVSVCGDVKLALQGMNKVLENRAEELKLDFGVWRNELNVQKQKFPLSFKTFGEAIPPQYAIKVLDELTDGKAIISTGVGQHQMWAAQFYNYKKPRQWLSSGGLGAMGFGLPAAIGASVANPDAIVVDIDGDGSFIMNVQELATIRVENLPVKVLLLNNQHLGMVMQWEDRFYKANRAHTFLGDPAQEDEIFPNMLLFAAACGIPAARVTKKADLREAIQTMLDTPGPYLLDVICPHQEHVLPMIPSGGTFNDVITEGDGRL Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Beta-glucosidase A | 1E4I | 4.58 | |
Target general information Gen name bglA Organism Paenibacillus polymyxa (Bacillus polymyxa) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 1 family Biochemical class Hydrolase Function Beta-glucosidase activity.Scopolin beta-glucosidase activity. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02658; DB04282; DB04304 Interacts with NA EC number 3.2.1.21 Uniprot keywords 3D-structure; Carbohydrate metabolism; Cellulose degradation; Glycosidase; Hydrolase; Polysaccharide degradation Protein physicochemical properties Chain ID A Molecular weight (Da) 51515.2 Length 447 Aromaticity 0.14 Instability index 38.44 Isoelectric point 5.28 Charge (pH=7) -18.1 2D Binding mode Binding energy (Kcal/mol) -6.25 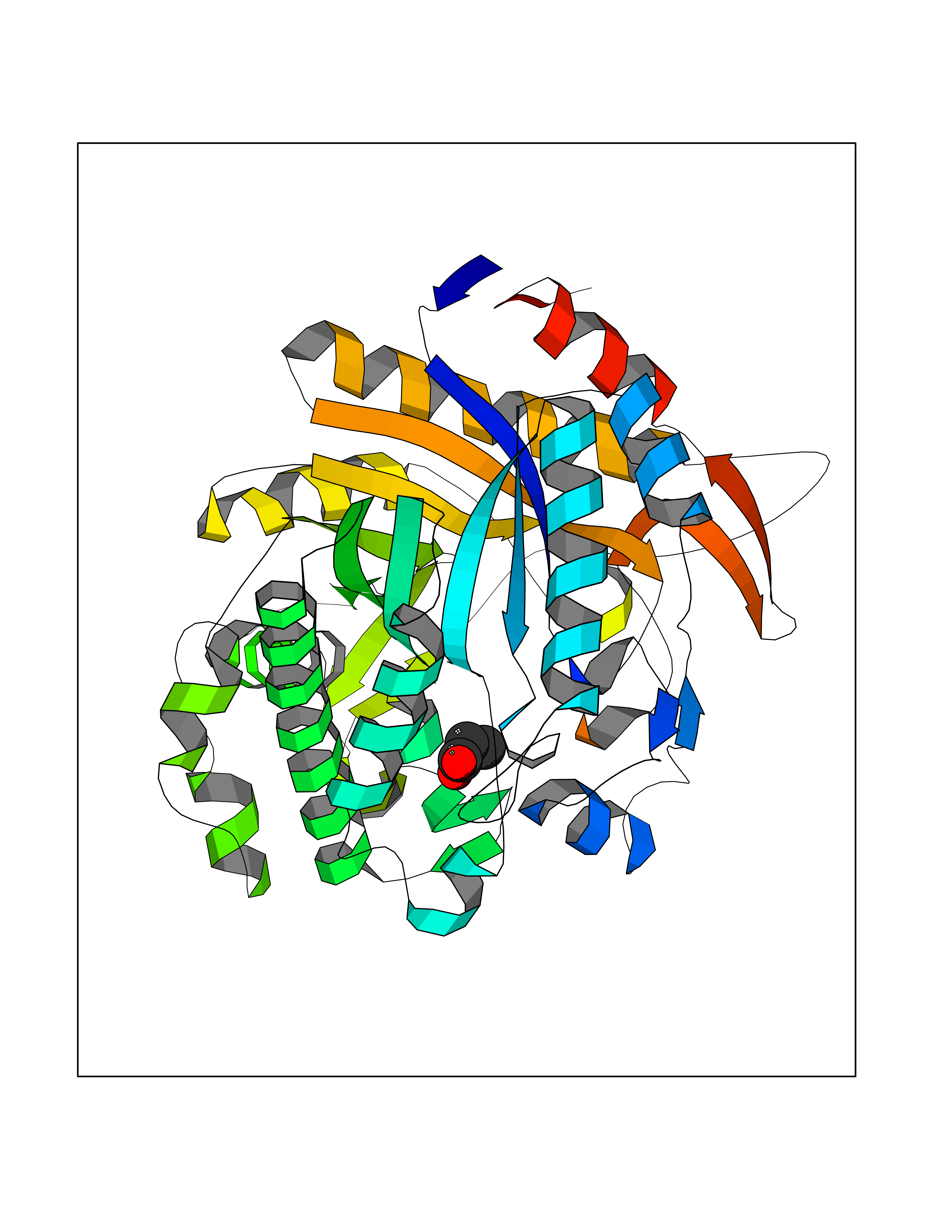 Molscript Map 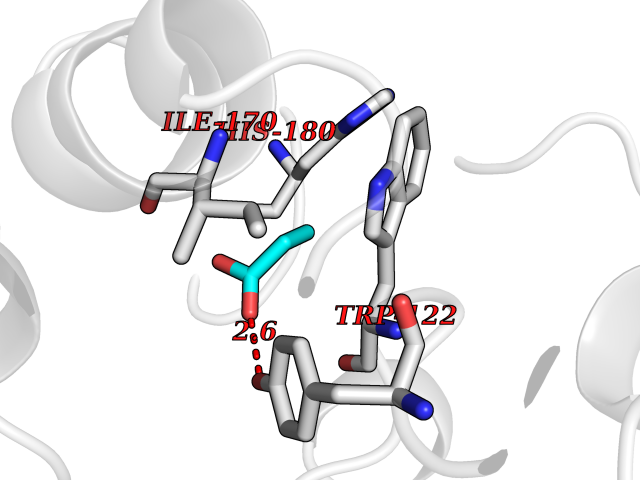 Pymol Map 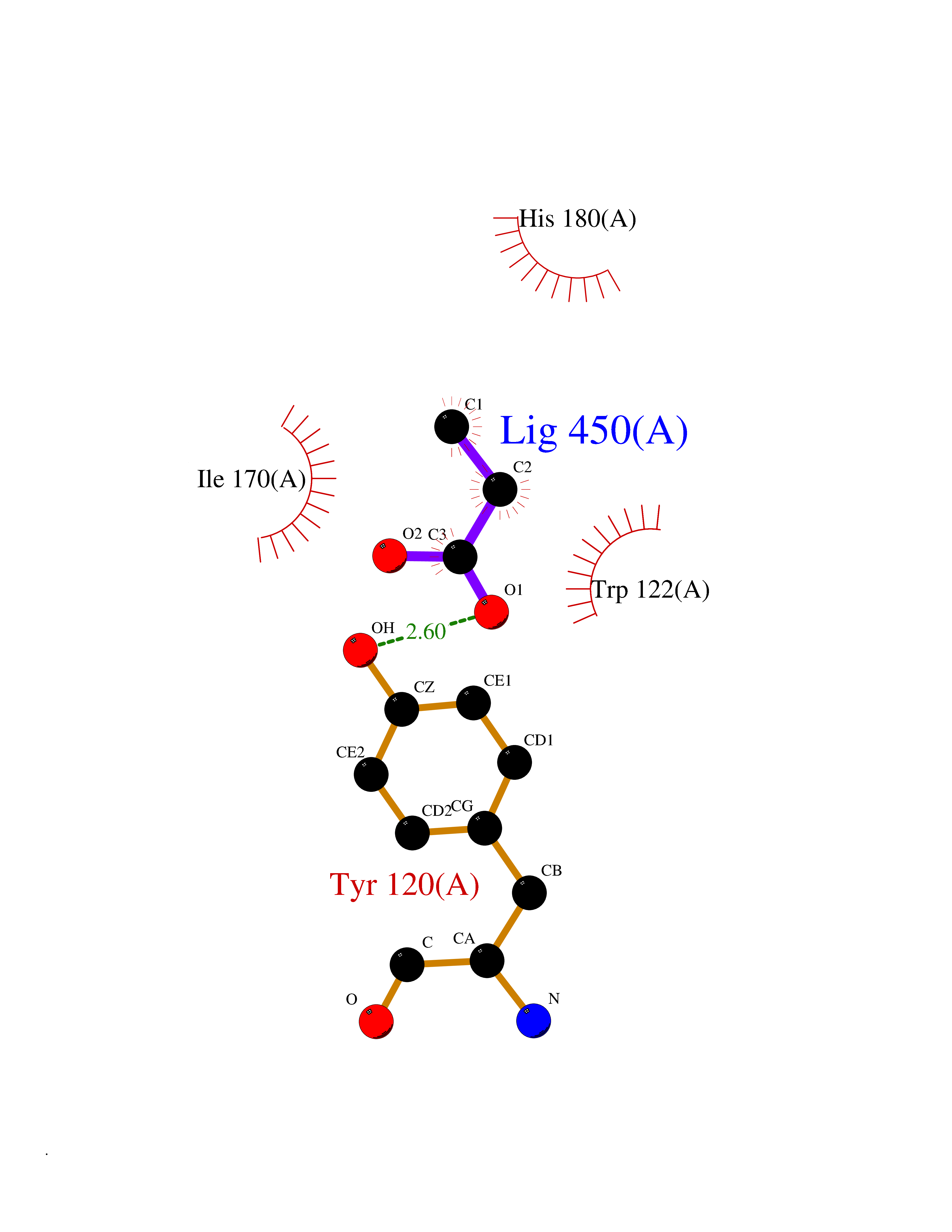 Ligplot Map 3D Binding mode Sequence TIFQFPQDFMWGTATAAYQIEGAYQEDGRGLSIWDTFAHTPGKVFNGDNGNVACDSYHRYEEDIRLMKELGIRTYRFSVSWPRIFPNGDGEVNQKGLDYYHRVVDLLNDNGIEPFCTLYHWDLPQALQDAGGWGNRRTIQAFVQFAETMFREFHGKIQHWLTFNEPWCIAFLSNMLGVHAPGLTNLQTAIDVGHHLLVAHGLSVRRFRELGTSGQIGIAPNVSWAVPYSTSEEDKAACARTISLHSDWFLQPIYQGSYPQFLVDWFAEQGATVPIQDGDMDIIGEPIDMIGINYYSMSVNRFNPEAGFLQSEEINMGLPVTDIGWPVESRGLYEVLHYLQKYGNIDIYITENGACINDEVVNGKVQDDRRISYMQQHLVQVHRTIHDGLHVKGYMAWSLLDNFEWAEGYNMRFGMIHVDFRTQVRTPKQSYYWYRNVVSNNWLETRR Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Ecto-5'-nucleotidase (CD73) | 4H2G | 4.58 | |
Target general information Gen name NT5E Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NT5; CD73 antigen; 5'-nucleotidase; 5'-NT Protein family 5'-nucleotidase family Biochemical class Phosphoric monoester hydrolase Function Exhibits AMP-, NAD-, and NMN-nucleosidase activities. Hydrolyzes extracellular nucleotides into membrane permeable nucleosides. Related diseases Calcification of joints and arteries (CALJA) [MIM:211800]: A condition characterized by adult-onset calcification of the lower extremity arteries, including the iliac, femoral and tibial arteries, and hand and foot capsule joints. Age of onset has been reported as early as the second decade of life, usually involving intense joint pain or calcification in the hands. {ECO:0000269|PubMed:21288095, ECO:0000269|PubMed:24887587}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00987; DB00806 Interacts with Q9Y225-2; Q8WWF5 EC number EC 3.1.3.5 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; Metal-binding; Nucleotide-binding; Proteomics identification; Reference proteome; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 24417.6 Length 219 Aromaticity 0.09 Instability index 40.43 Isoelectric point 5.49 Charge (pH=7) -5.75 2D Binding mode Binding energy (Kcal/mol) -6.25  Molscript Map 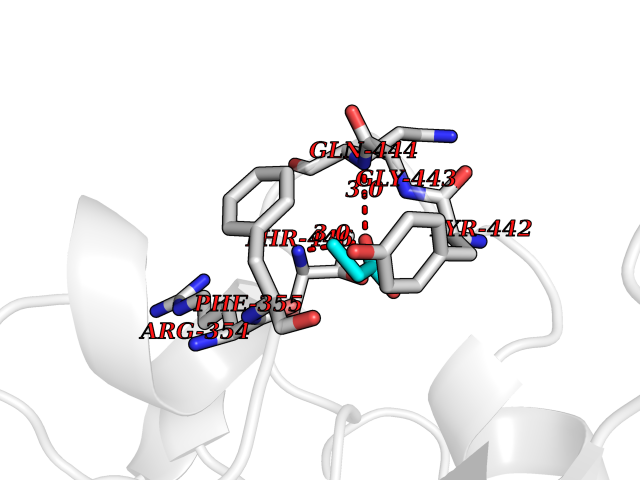 Pymol Map 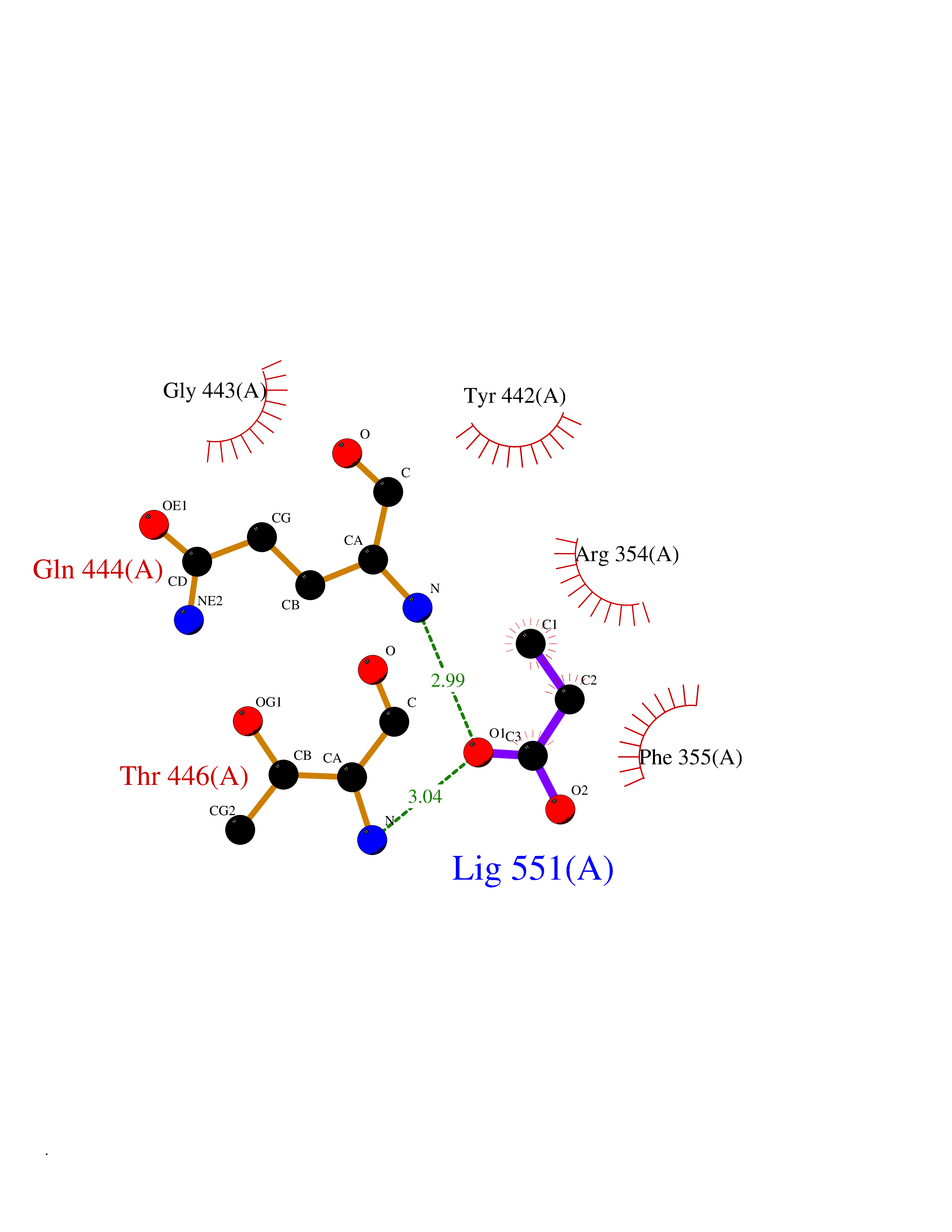 Ligplot Map 3D Binding mode Sequence LDDYSTQELGKTIVYLDGSSQSCRFRECNMGNLICDAMINNNLRHADEMFWNHVSMCILNGGGIRSPIDERNDGTITWENLAAVLPFGGTFDLVQLKGSTLKKAFEHSVHRYGQSTGEFLQVGGIHVVYDLSRKPGDRVVKLDVLCTACAVPSYDPLKMDEVYKVILPNFLANGGDGFQMIKDELLRHDSGDQDINVVSTYISKMKVIYPAVEGRIKFS Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Low molecular weight phosphotyrosine protein phosphatase | 5KQL | 4.58 | |
Target general information Gen name ACP1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Low molecular weight phosphotyrosine protein phosphatase family Biochemical class hydrolase / hydrolase inhibitor Function Acid phosphatase activity.Non-membrane spanning protein tyrosine phosphatase activity. Related diseases Waardenburg syndrome 4A (WS4A) [MIM:277580]: A disorder characterized by the association of Waardenburg features (depigmentation and deafness) with the absence of enteric ganglia in the distal part of the intestine (Hirschsprung disease). {ECO:0000269|PubMed:12189494, ECO:0000269|PubMed:8634719}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hirschsprung disease 2 (HSCR2) [MIM:600155]: A disorder of neural crest development characterized by absence of enteric ganglia along a variable length of the intestine. It is the most common cause of congenital intestinal obstruction. Early symptoms range from complete acute neonatal obstruction, characterized by vomiting, abdominal distention and failure to pass stool, to chronic constipation in the older child. {ECO:0000269|PubMed:11471546, ECO:0000269|PubMed:28236341, ECO:0000269|PubMed:8001158, ECO:0000269|PubMed:8630503, ECO:0000269|PubMed:8852660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: ABCD syndrome (ABCDS) [MIM:600501]: An autosomal recessive syndrome characterized by albinism, black lock at temporal occipital region, bilateral deafness, aganglionosis of the large intestine and total absence of neurocytes and nerve fibers in the small intestine. {ECO:0000269|PubMed:11891690}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Heterozygous mutations in EDNRB may be responsible for Waardenburg syndrome 2, an autosomal dominant disorder characterized by sensorineural deafness and pigmentary disturbances. {ECO:0000269|PubMed:28236341}. Drugs (DrugBank ID) DB04214; DB00173 Interacts with Q96CV9 EC number 3.1.3.2; 3.1.3.48 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Phosphoprotein; Protein phosphatase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 17582.8 Length 154 Aromaticity 0.09 Instability index 50.52 Isoelectric point 7 Charge (pH=7) -0 2D Binding mode Binding energy (Kcal/mol) -6.24 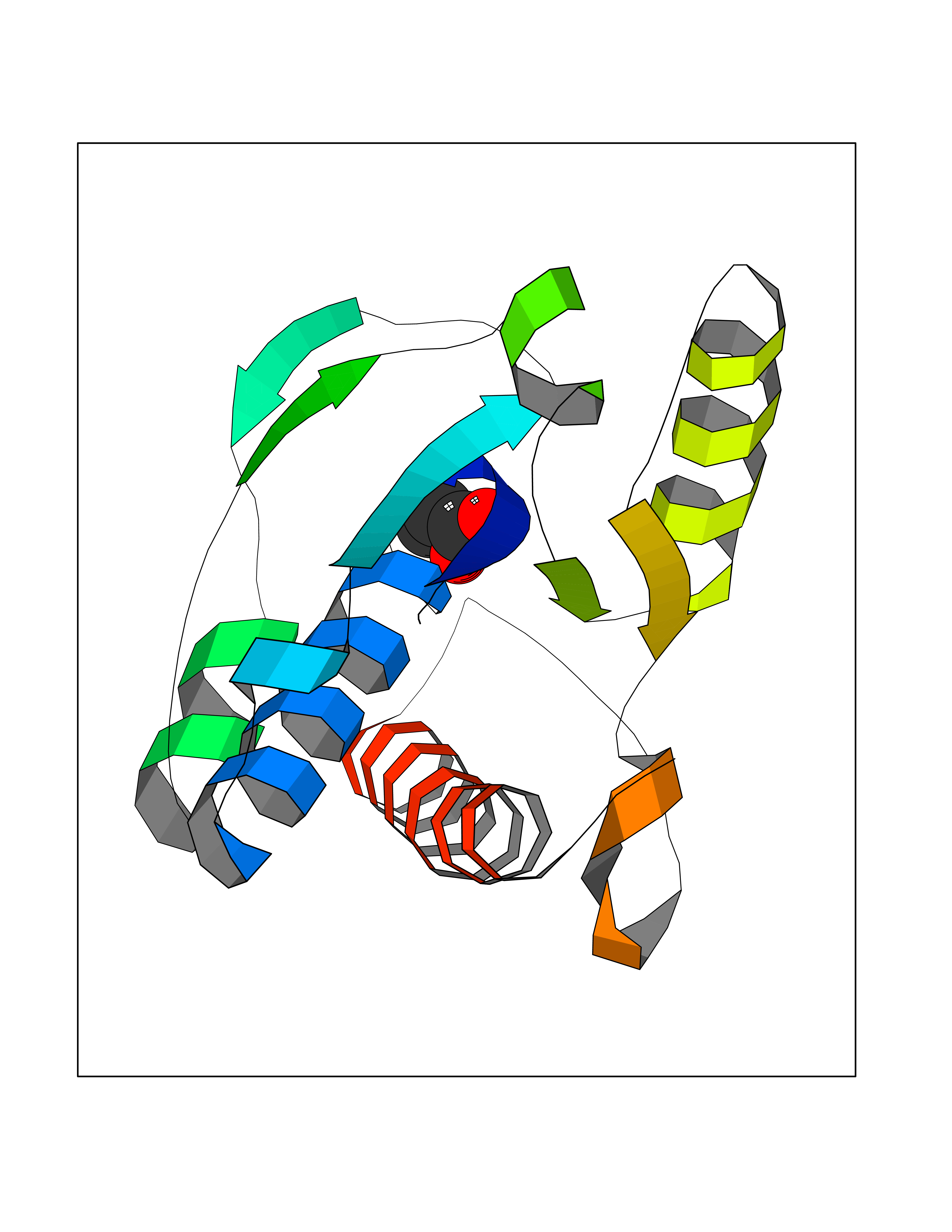 Molscript Map 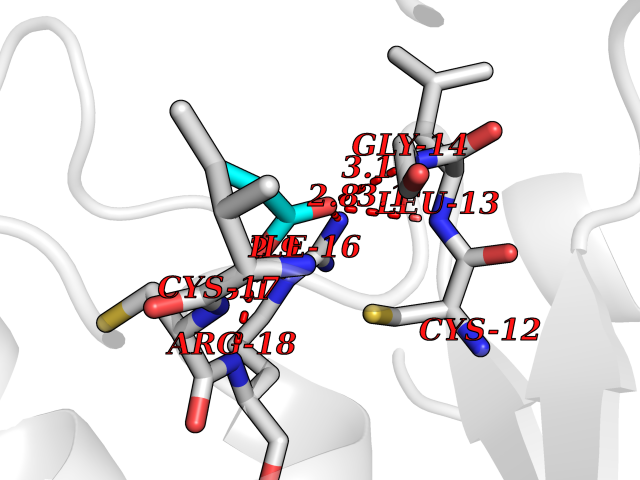 Pymol Map 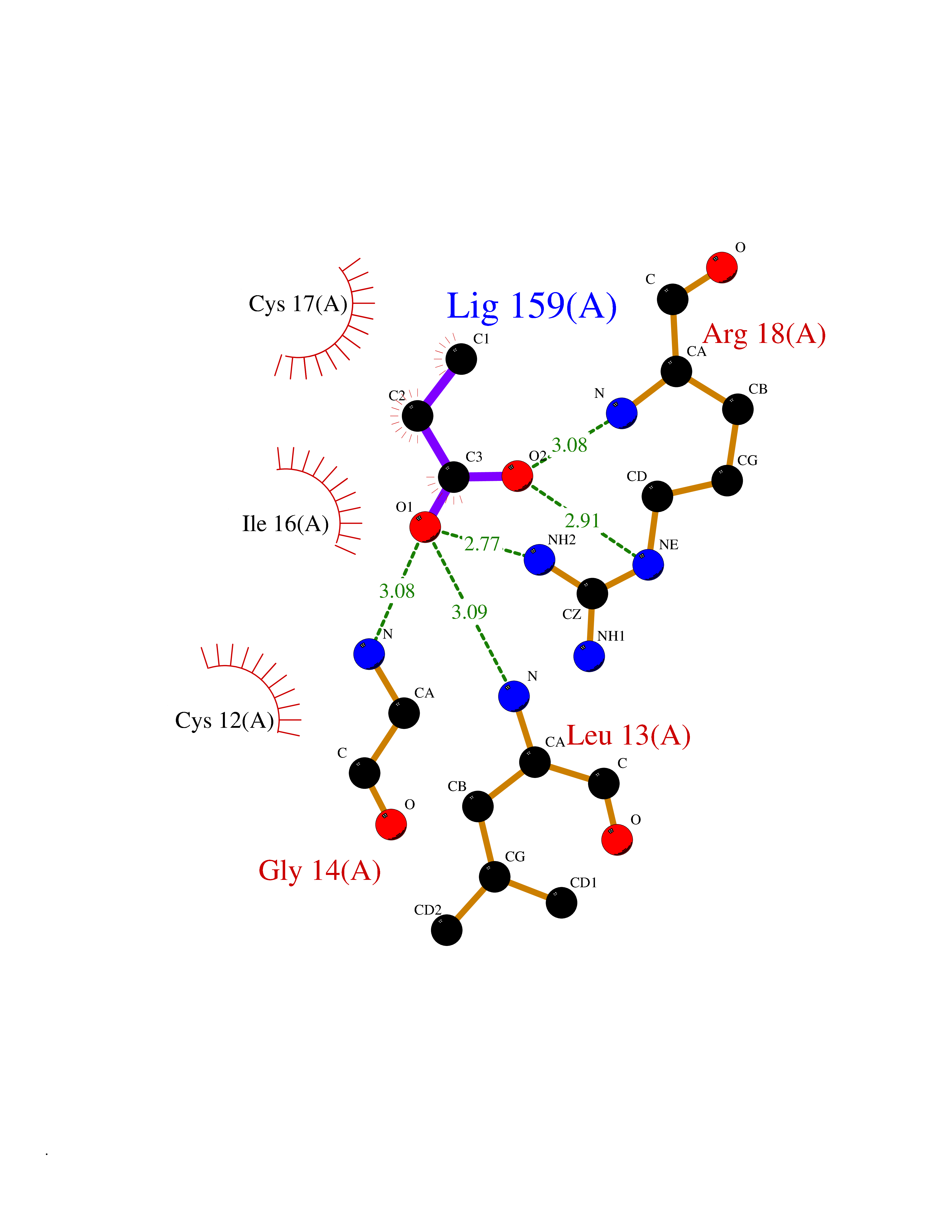 Ligplot Map 3D Binding mode Sequence ATKSVLFVCLGNICRSPIAEAVFRKLVTDQNISENWRVDSAATSGYEIGNPPDYRGQSCMKRHGIPMSHVARQITKEDFATFDYILCMDESNLRDLNRKSNQVKTCKAKIELLGSYDPQKQLIIEDPYYGNDSDFETVYQQCVRCCRAFLEKAH Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | S-adenosylmethionine synthase type-2 (MAT2A) | 5A1I | 4.58 | |
Target general information Gen name MAT2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Methionine adenosyltransferase II; Methionine adenosyltransferase 2; MAT-II; MAT 2; AdoMet synthase 2 Protein family AdoMet synthase family Biochemical class AdoMet synthase family Function Catalyzes the formation of S-adenosylmethionine from methionine and ATP. The reaction comprises two steps that are both catalyzed by the same enzyme: formation of S-adenosylmethionine (AdoMet) and triphosphate, and subsequent hydrolysis of the triphosphate. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00118; DB00134 Interacts with Q96IK1-2; Q96NX5; Q6ZP82-1; Q8IUI8; Q6P1L5; P15976-2; P80217-2; Q8WZ19; Q9UIH9; Q00266; P31153; Q9NZL9; P02795; Q9BRX2; O43663; O43741; P57052; Q8N488; P08195-4; Q13573; Q86W54-2; O95789-4 EC number EC 2.5.1.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Isopeptide bond; Magnesium; Metal-binding; Nucleotide-binding; One-carbon metabolism; Phosphoprotein; Potassium; Proteomics identification; Reference proteome; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 42071.4 Length 381 Aromaticity 0.08 Instability index 38.24 Isoelectric point 6.21 Charge (pH=7) -4.13 2D Binding mode Binding energy (Kcal/mol) -6.24 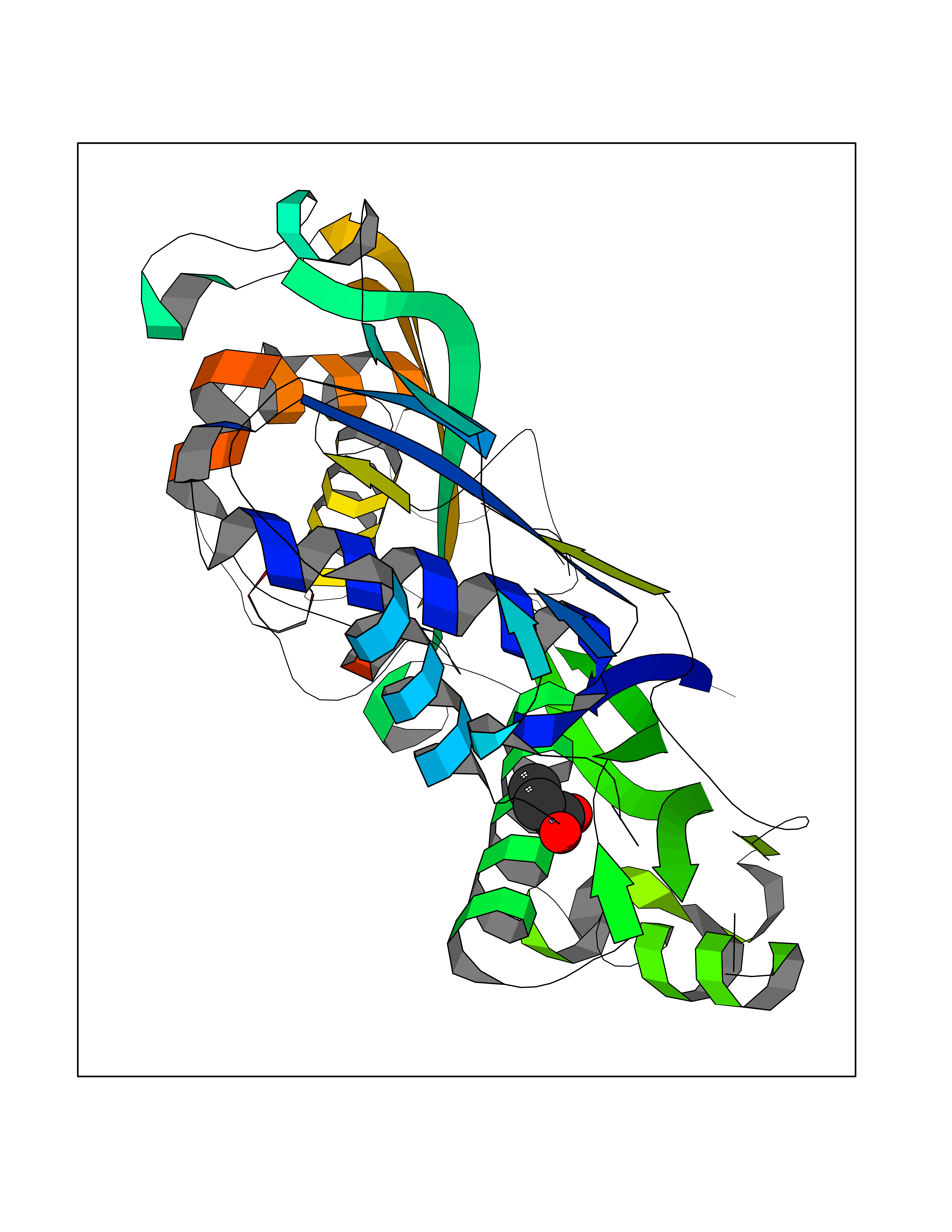 Molscript Map 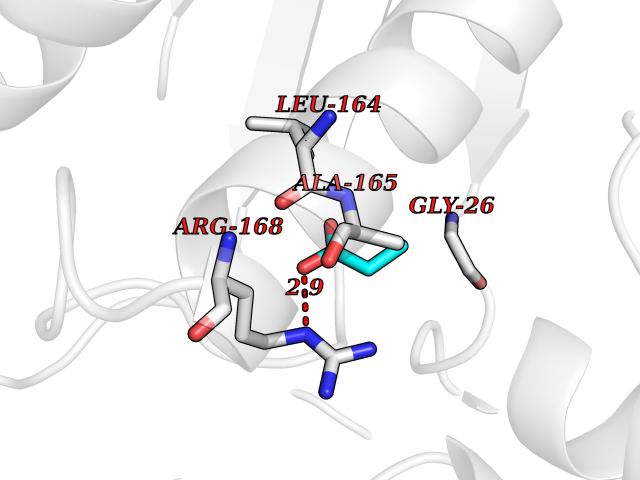 Pymol Map 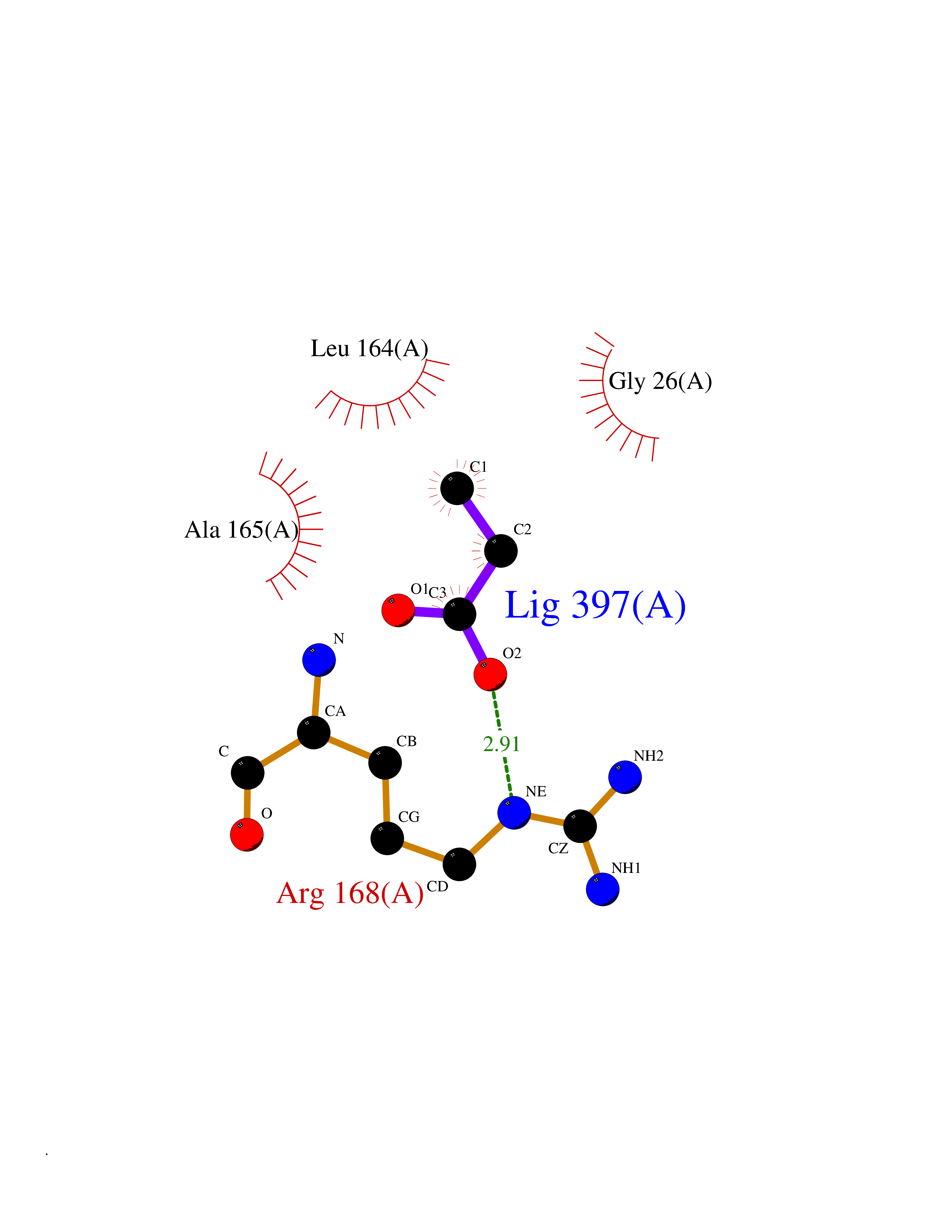 Ligplot Map 3D Binding mode Sequence EGTFLFTSESVGEGHPDKICDQISDAVLDAHLQQDPDAKVACETVAKTGMILLAGEITSRAAVDYQKVVREAVKHIGYDDSSKGFDYKTCNVLVALEQQSPDIAQGVHLDRNEEDIGAGDQGLMFGYATDETEECMPLTIVLAHKLNAKLAELRRNGTLPWLRPDSKTQVTVQYMQDRGAVLPIRVHTIVISVQHDEEVCLDEMRDALKEKVIKAVVPAKYLDEDTIYHLQPSGRFVIGGPQGDAGLTGRKIIVDTYGGWGAHGGGAFSGKDYTKVDRSAAYAARWVAKSLVKGGLCRRVLVQVSYAIGVSHPLSISIFHYGTSQKSERELLEIVKKNFDLRPGVIVRDLDLKKPIYQRTAAYGHFGRDSFPWEVPKKLKY Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Dihydroorotate dehydrogenase (quinone), mitochondrial | 4CQ8 | 4.58 | |
Target general information Gen name PFF0160c Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class Oxidoreductase Function Dihydroorotate dehydrogenase activity. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42573.5 Length 378 Aromaticity 0.1 Instability index 36.63 Isoelectric point 8.21 Charge (pH=7) 3.17 2D Binding mode Binding energy (Kcal/mol) -6.25 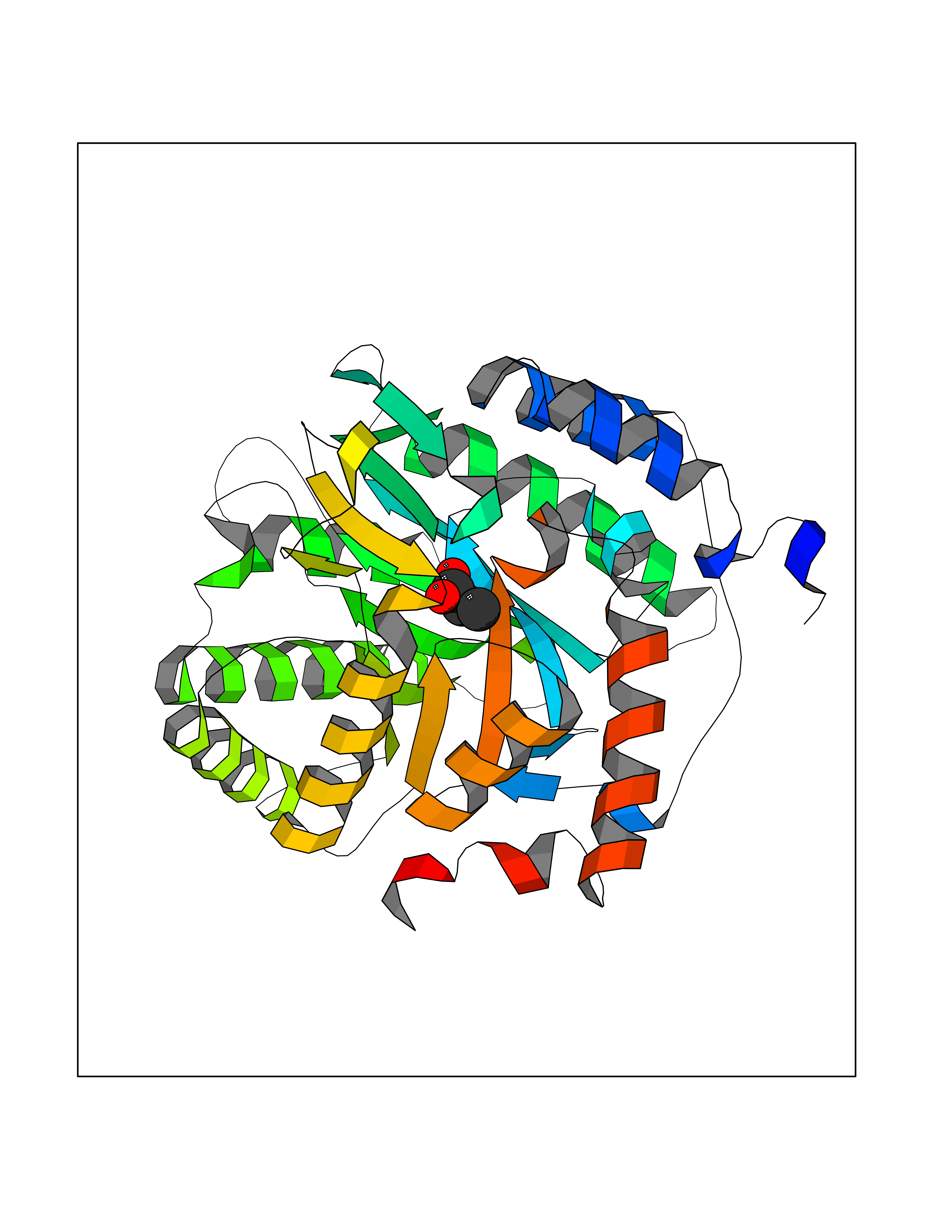 Molscript Map 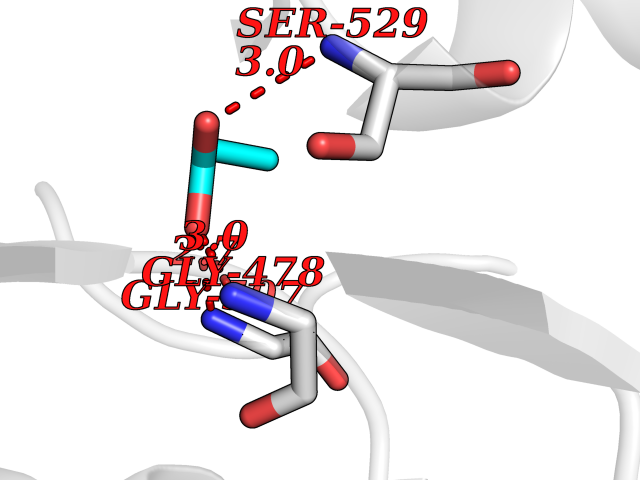 Pymol Map 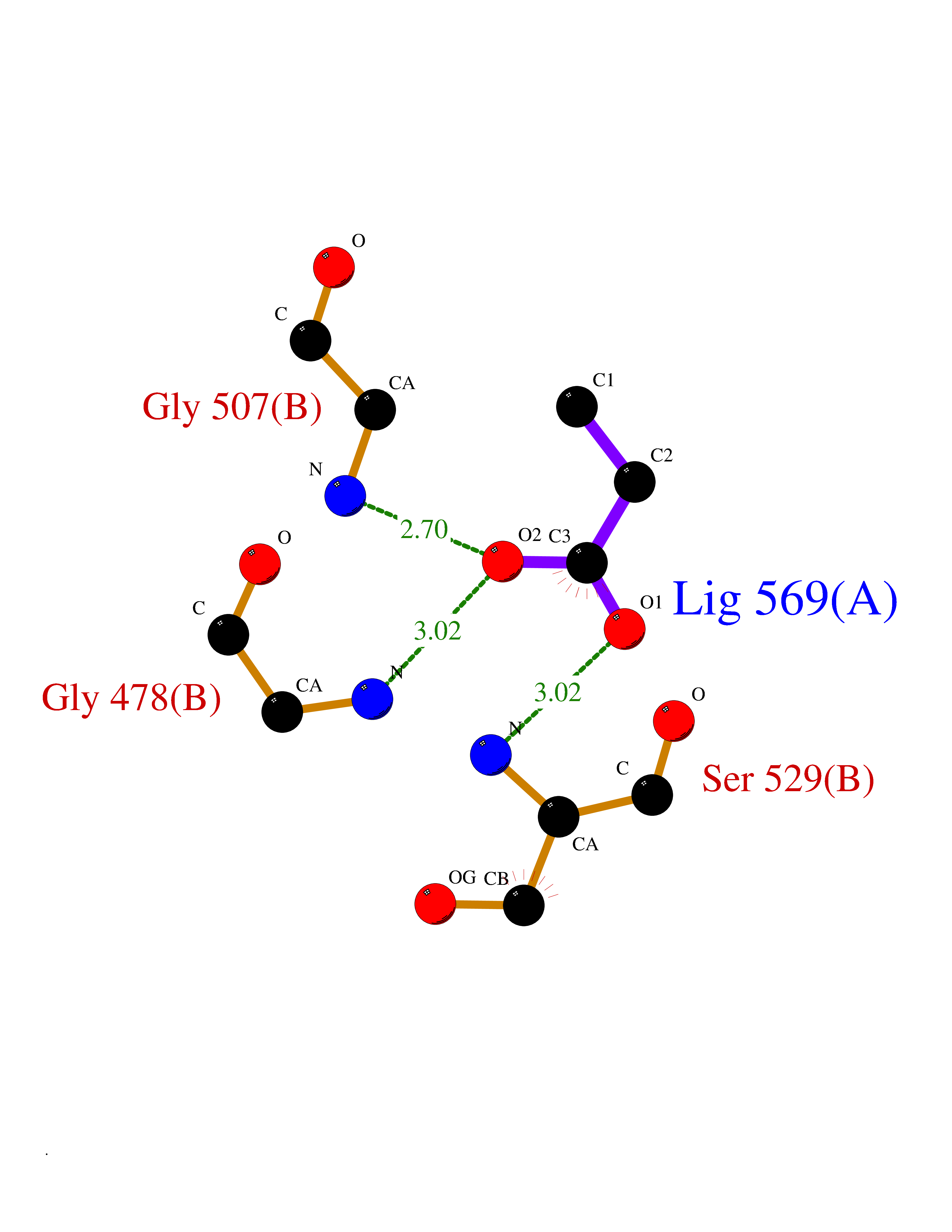 Ligplot Map 3D Binding mode Sequence ADPFESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEKNNFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Acetyl-CoA carboxylase 1 | 2YL2 | 4.58 | |
Target general information Gen name ACACA Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACAC;ACCA;ACC1 Protein family NA Biochemical class Ligase Function Acetyl-CoA carboxylase activity.ATP binding.Biotin carboxylase activity.Identical protein binding.Metal ion binding. Related diseases Acetyl-CoA carboxylase-alpha deficiency (ACACAD) [MIM:613933]: An autosomal recessive inborn error of de novo fatty acid synthesis associated with severe brain damage, persistent myopathy and poor growth. {ECO:0000269|PubMed:6114432}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00121 Interacts with Q13085; O60218; P38398; Q96EB6; Q9CQ20; P02654; Q92915-2; Q6NTF9-3 EC number 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative promoter usage; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Magnesium; Manganese; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 54237.7 Length 486 Aromaticity 0.09 Instability index 39.18 Isoelectric point 6.37 Charge (pH=7) -2.46 2D Binding mode Binding energy (Kcal/mol) -6.25 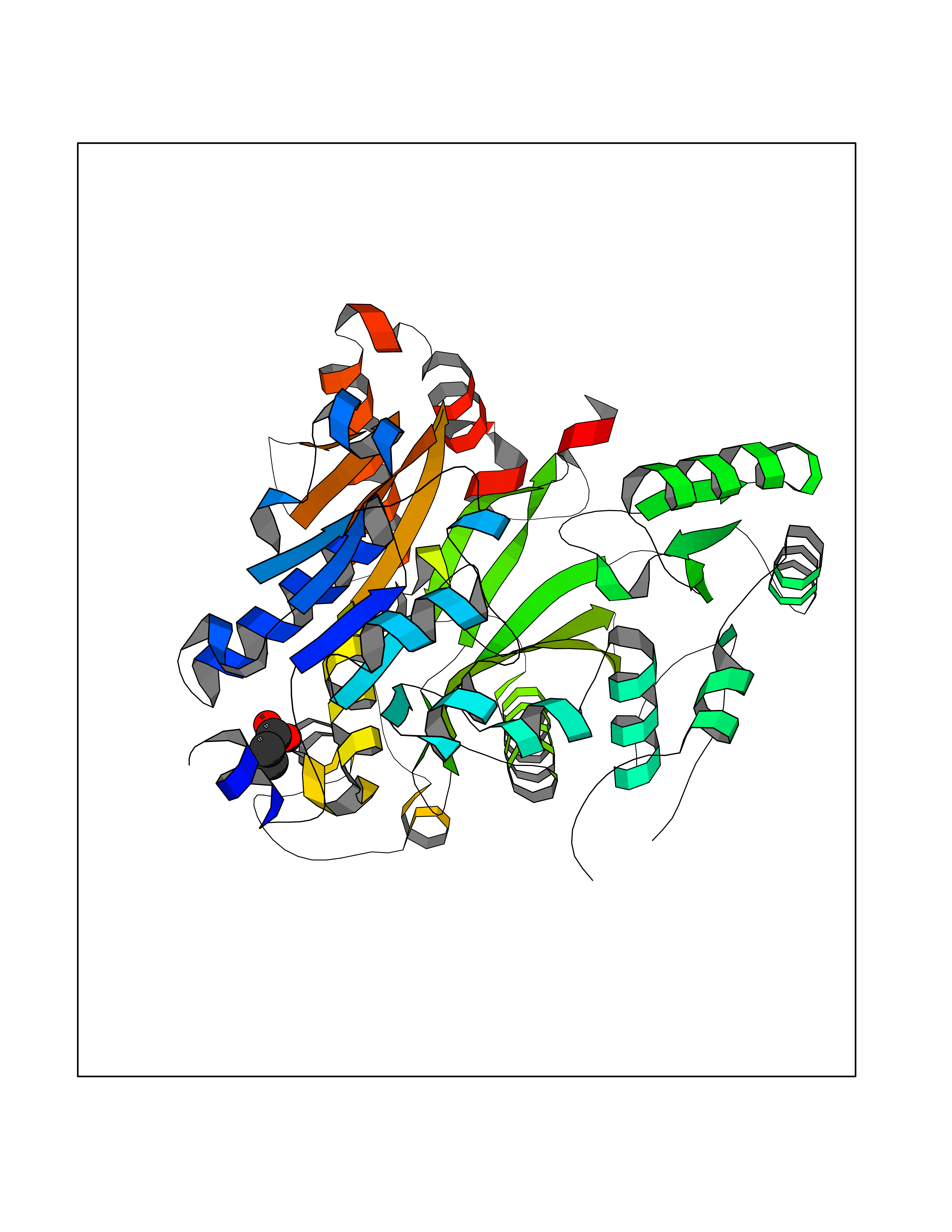 Molscript Map 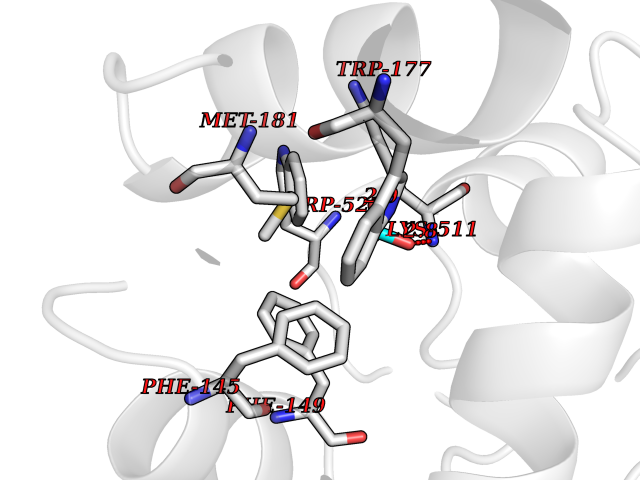 Pymol Map 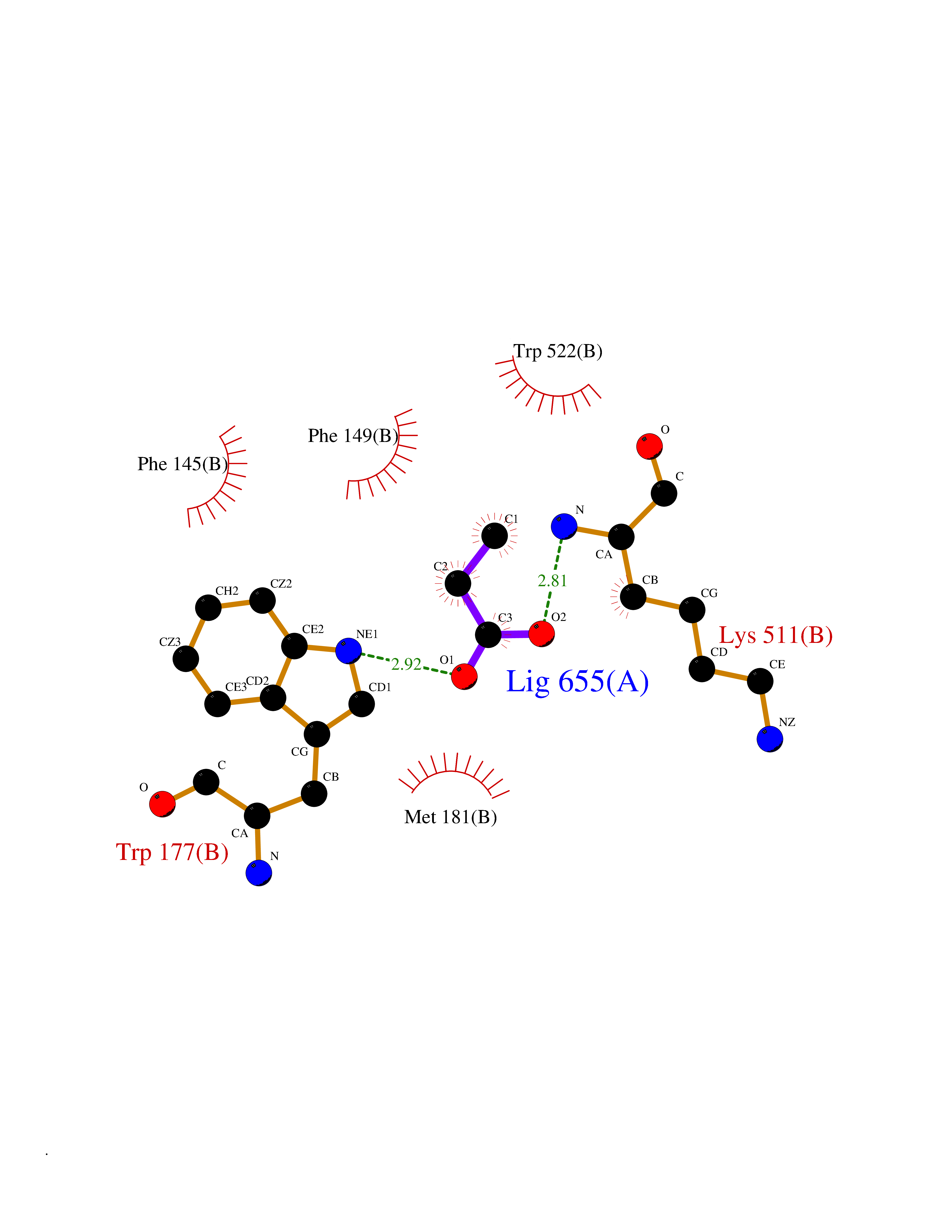 Ligplot Map 3D Binding mode Sequence VASPAEFVTRFGGNKVIEKVLIANNGIAAVKCMRSIRRWSYEMFRNERAIRFVVMVTPEDLKANAEYIKMADHYVPVPGGPNNNNYANVELILDIAKRIPVQAVWAGWGHASENPKLPELLLKNGIAFMGPPSQAMWALGDKIASSIVAQTAGIPTLPWSGSGLRVDWSKRILNVPQELYEKGYVKDVDDGLQAAEEVGYPVMIKASEGGGGKGIRKVNNADDFPNLFRQVQAEVPGSPIFVMRLAKQSRHLEVQILADQYGNAISLFGRDCSVQRRHQKIIEEAPATIATPAVFEHMEQCAVKLAKMVGYVSAGTVEYLYSQDGSFYFLELNPRLQVEHPCTEMVADVNLPAAQLQIAMGIPLYRIKDIRMMYGVSPWGDSPIDFEDSAHVPCPRGHVIAARITGTVQELNFRSNKNVWGYFSVQFGHCFSWGENREEAISNMVVALKELSIRGDFRTTVEYLIKLLETESFQMNRIDTGWLDRL Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Tyrosine-protein kinase BRK (PTK6) | 5DA3 | 4.58 | |
Target general information Gen name PTK6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein-tyrosine kinase 6; Breast tumor kinase; BRK Protein family Protein kinase superfamily, Tyr protein kinase family, BRK/PTK6/SIK subfamily Biochemical class Kinase Function Non-receptor tyrosine-protein kinase implicated in the regulation of a variety of signaling pathways that control the differentiation and maintenance of normal epithelia, as well as tumor growth. Function seems to be context dependent and differ depending on cell type, as well as its intracellular localization. A number of potential nuclear and cytoplasmic substrates have been identified. These include the RNA-binding proteins: KHDRBS1/SAM68, KHDRBS2/SLM1, KHDRBS3/SLM2 and SFPQ/PSF; transcription factors: STAT3 and STAT5A/B and a variety of signaling molecules: ARHGAP35/p190RhoGAP, PXN/paxillin, BTK/ATK, STAP2/BKS. Associates also with a variety of proteins that are likely upstream of PTK6 in various signaling pathways, or for which PTK6 may play an adapter-like role. These proteins include ADAM15, EGFR, ERBB2, ERBB3 and IRS4. In normal or non-tumorigenic tissues, PTK6 promotes cellular differentiation and apoptosis. In tumors PTK6 contributes to cancer progression by sensitizing cells to mitogenic signals and enhancing proliferation, anchorage-independent survival and migration/invasion. Association with EGFR, ERBB2, ERBB3 may contribute to mammary tumor development and growth through enhancement of EGF-induced signaling via BTK/AKT and PI3 kinase. Contributes to migration and proliferation by contributing to EGF-mediated phosphorylation of ARHGAP35/p190RhoGAP, which promotes association with RASA1/p120RasGAP, inactivating RhoA while activating RAS. EGF stimulation resulted in phosphorylation of PNX/Paxillin by PTK6 and activation of RAC1 via CRK/CrKII, thereby promoting migration and invasion. PTK6 activates STAT3 and STAT5B to promote proliferation. Nuclear PTK6 may be important for regulating growth in normal epithelia, while cytoplasmic PTK6 might activate oncogenic signaling pathways. Related diseases Periodic paralysis hypokalemic 1 (HOKPP1) [MIM:170400]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:17418573, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:7987325, ECO:0000269|PubMed:8004673}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Malignant hyperthermia 5 (MHS5) [MIM:601887]: Autosomal dominant disorder that is potentially lethal in susceptible individuals on exposure to commonly used inhalational anesthetics and depolarizing muscle relaxants. {ECO:0000269|PubMed:9199552}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Thyrotoxic periodic paralysis 1 (TTPP1) [MIM:188580]: A sporadic muscular disorder characterized by episodic weakness and hypokalemia during a thyrotoxic state. It is clinically similar to hereditary hypokalemic periodic paralysis, except for the fact that hyperthyroidism is an absolute requirement for disease manifestation. The disease presents with recurrent episodes of acute muscular weakness of the four extremities that vary in severity from paresis to complete paralysis. Attacks are triggered by ingestion of a high carbohydrate load or strenuous physical activity followed by a period of rest. Thyrotoxic periodic paralysis can occur in association with any cause of hyperthyroidism, but is most commonly associated with Graves disease. {ECO:0000269|PubMed:15001631}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 18 (CMYO18) [MIM:620246]: A congenital myopathy of variable severity, ranging from severe fetal akinesia to milder forms of muscle weakness. Most affected individuals show delayed motor development with generalized hypotonia and progressive axial and limb muscle weakness beginning soon after birth or in infancy. Additional features may include swallowing difficulties, external ophthalmoplegia, ptosis, high-arched palate, and respiratory insufficiency. Muscle biopsy shows variable morphologic abnormalities, including alveolar changes in the intermyofibrillar network, fiber size variability, focal disorganization, internal nuclei, and dilated sarcoplasmic reticulum and T-tubules. CMYO18 inheritance is autosomal dominant or recessive. {ECO:0000269|PubMed:28012042, ECO:0000269|PubMed:31227654, ECO:0000269|PubMed:33060286}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010; DB11800; DB05294; DB15035 Interacts with Q08043; Q3KP44; Q13191; Q16543; Q92841; Q8N9I9; Q5JST6; P04626; O00471; O14526; Q13480; P08238; P42858; Q9UKT9; Q5VWX1; Q5T5P2-6; P10721; O14770-4; Q13064; Q8TDC0; P78337; Q9NQX0; Q13882; Q04864; P23246; Q13239-3; O00401; Q9BYN7 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell projection; Cytoplasm; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; SH2 domain; SH3 domain; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 30240.6 Length 264 Aromaticity 0.09 Instability index 46.88 Isoelectric point 6.95 Charge (pH=7) -0.14 2D Binding mode Binding energy (Kcal/mol) -6.24  Molscript Map 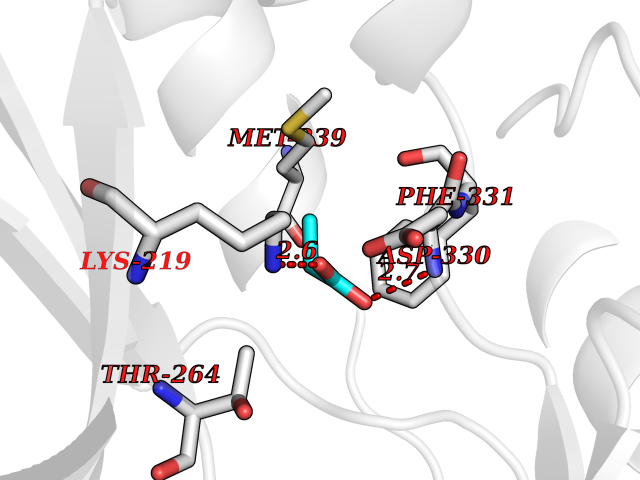 Pymol Map 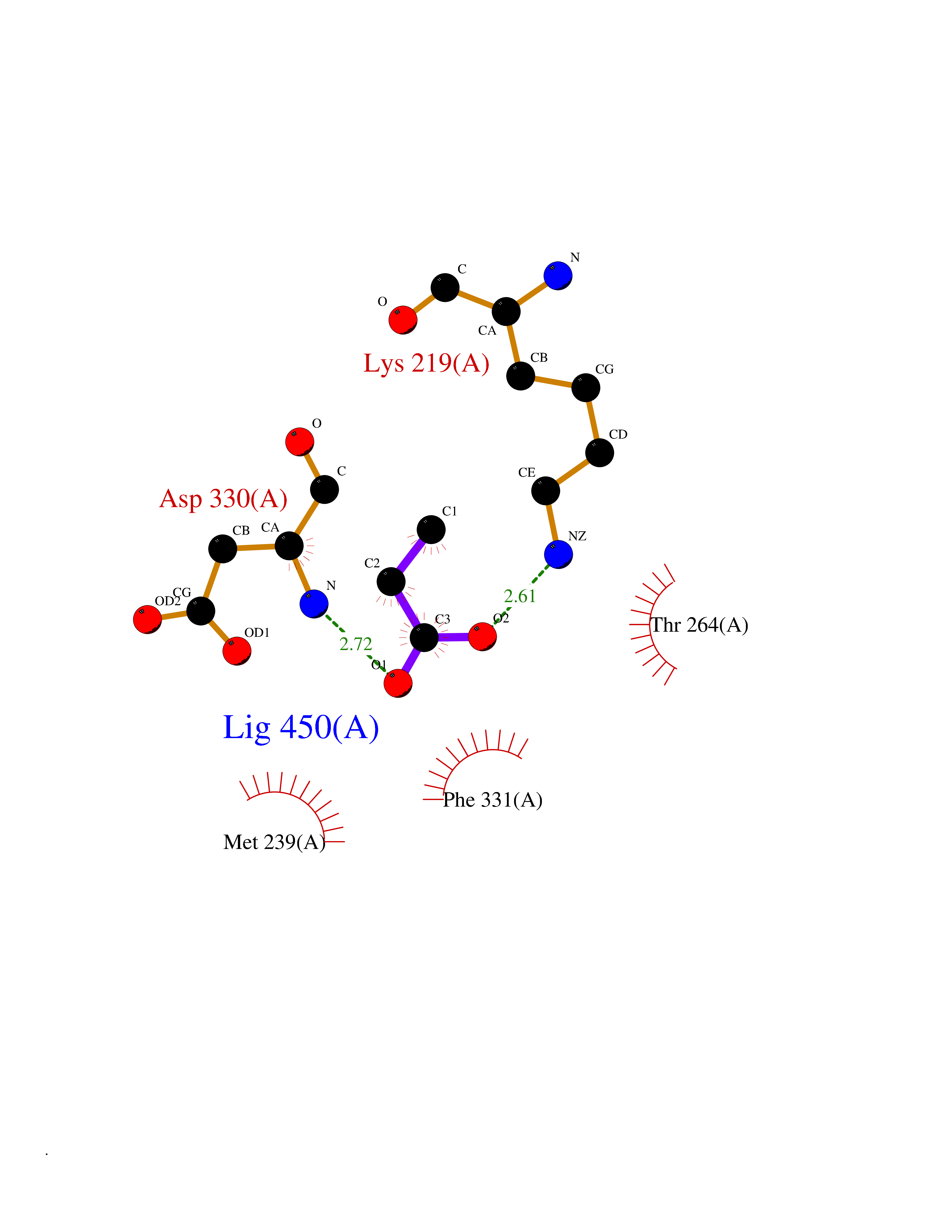 Ligplot Map 3D Binding mode Sequence XERPREEFTLCRKLGSGYFGEVFEGLWKDRVQVAIKVISRDNLLHQMLQSEIQAMKKLRHKHILALYAVVSVGDPVYIITELMAKGSLLELLRDSDEKVLPVSELLDIAWQVAEGMCYLESQNYIHRDLAARNILVGENTLCKVGDFGLARLIKEDVYLSHDHNIPYKWTAPEALSRGHYSTKSDVWSFGILLHEMFSRGQVPYPGMSNHEAFLRVDAGYRMPCPLECPPSVHKLMLTCWCRDPEQRPTFKALRERLSSFTSHH Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 4.58 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -6.25 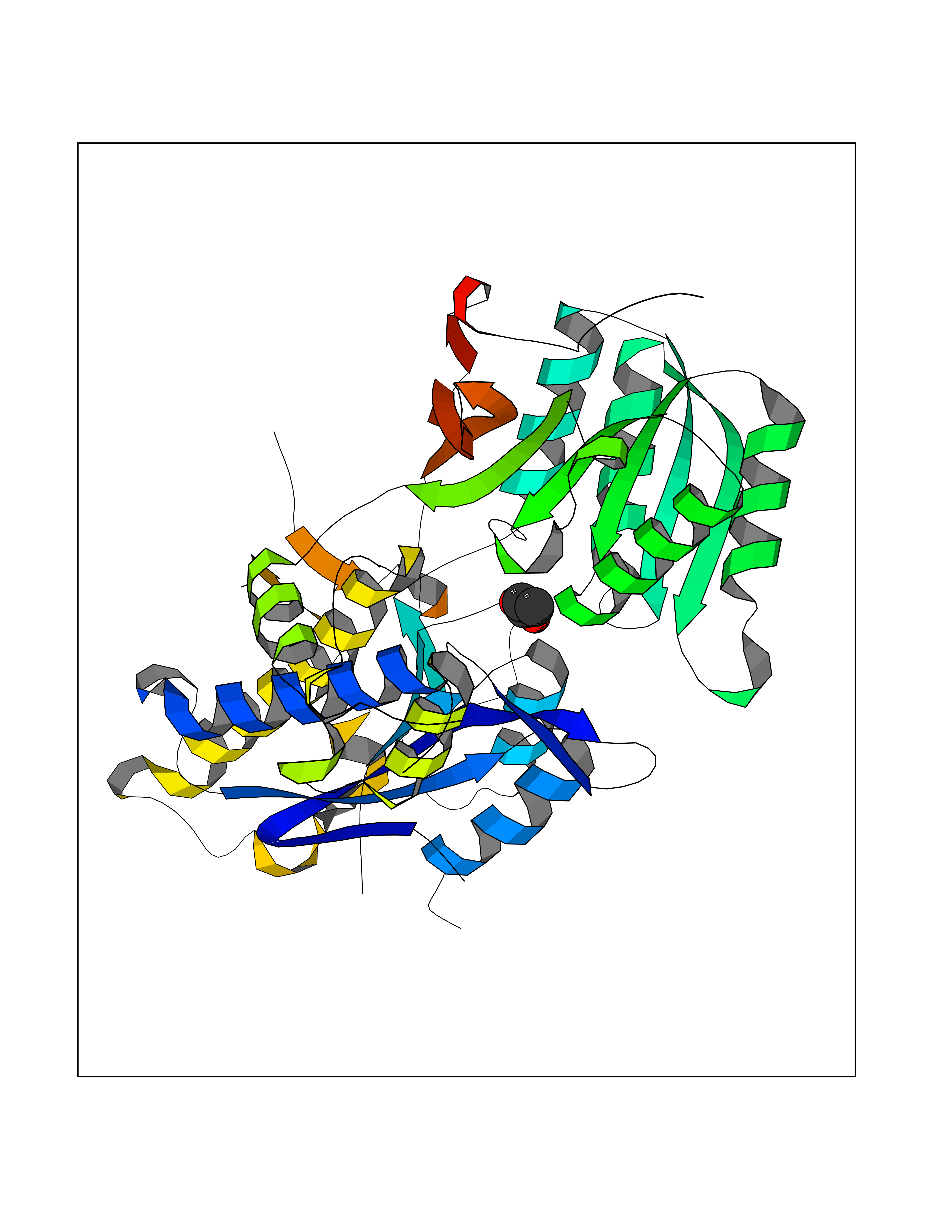 Molscript Map 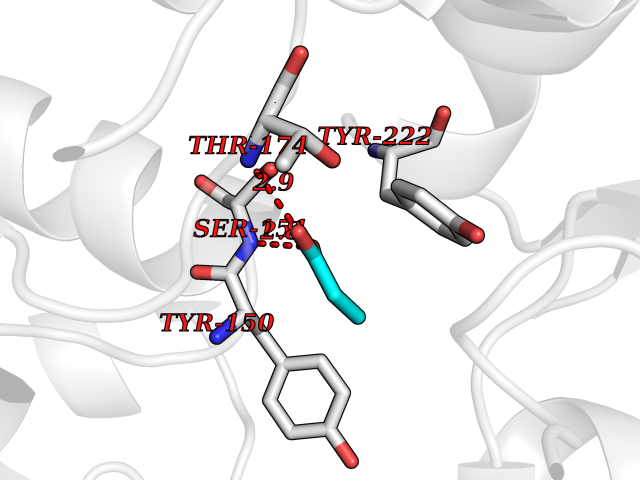 Pymol Map 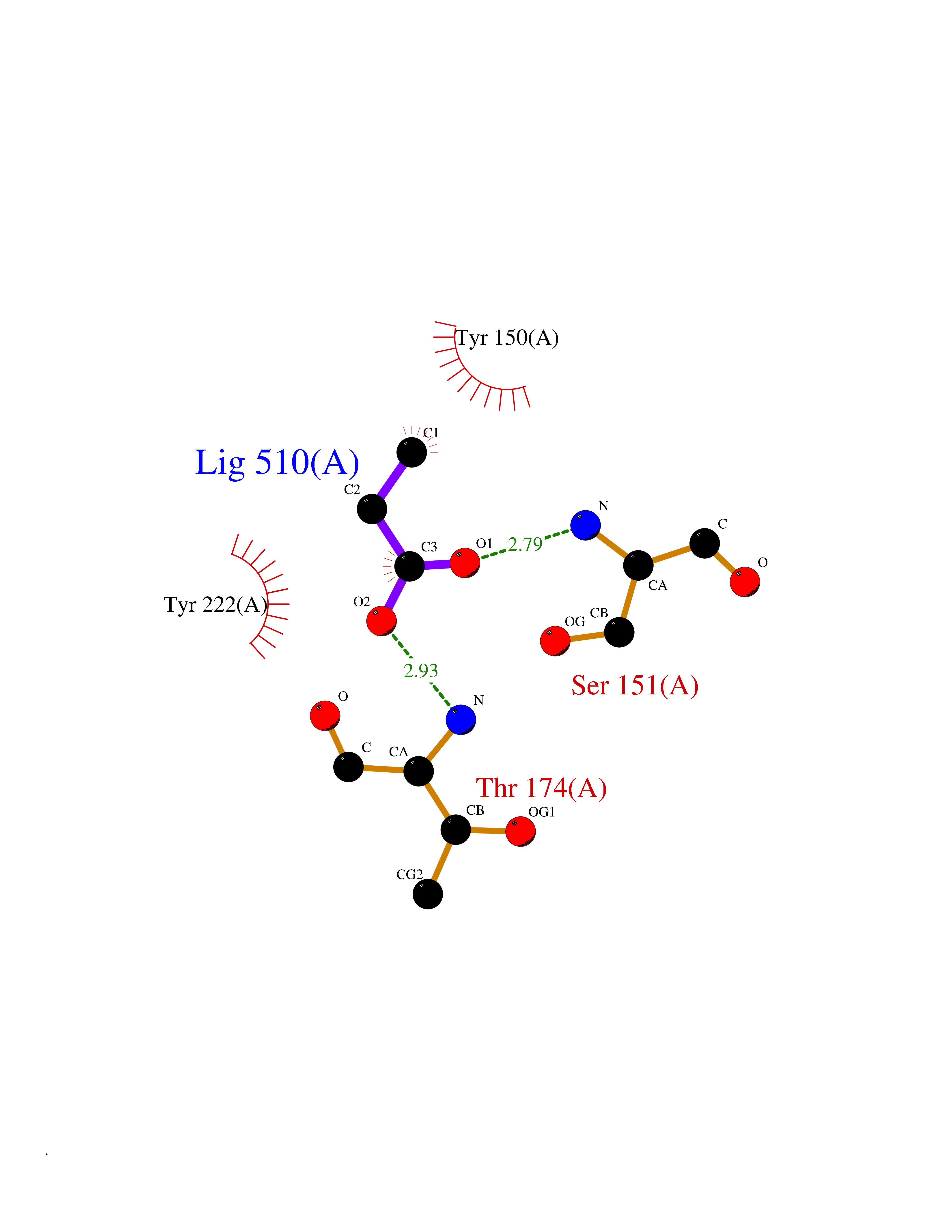 Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Orotidine 5'-monophosphate decarboxylase (UMPS) | 3MI2 | 4.58 | |
Target general information Gen name UMPS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Uridine 5'-monophosphate synthase; UMP synthase Protein family Purine/pyrimidine phosphoribosyltransferase family; OMP decarboxylase family Biochemical class Pentosyltransferase Function Catalyses the formation of uridine monophosphate (UMP), an energy-carrying molecule in many important biosynthetic pathways. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02890; DB00544 Interacts with P54764; P11172-1 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Decarboxylase; Disease variant; Glycosyltransferase; Lyase; Multifunctional enzyme; Phosphoprotein; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 55844 Length 514 Aromaticity 0.06 Instability index 22.7 Isoelectric point 6.44 Charge (pH=7) -2.99 2D Binding mode Binding energy (Kcal/mol) -6.24 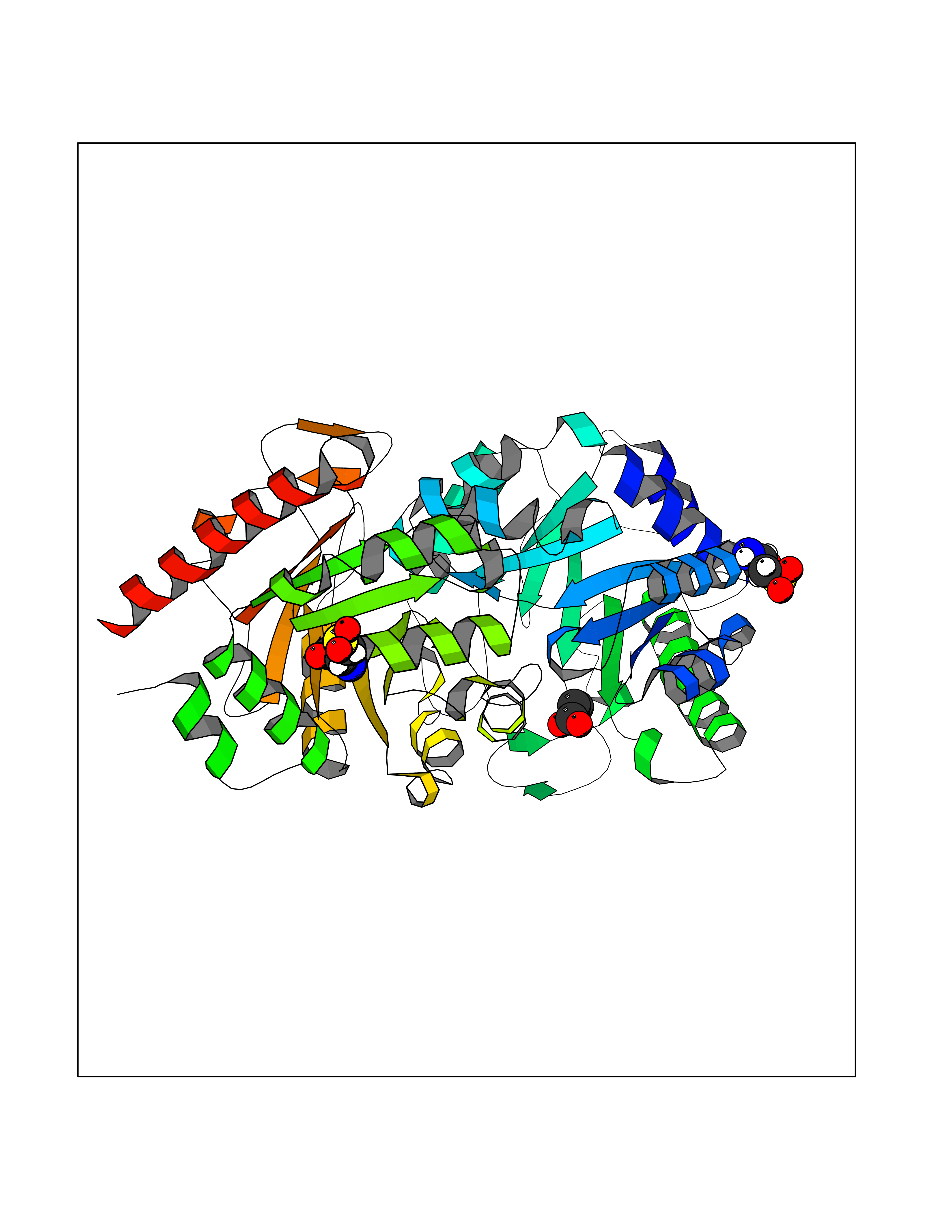 Molscript Map 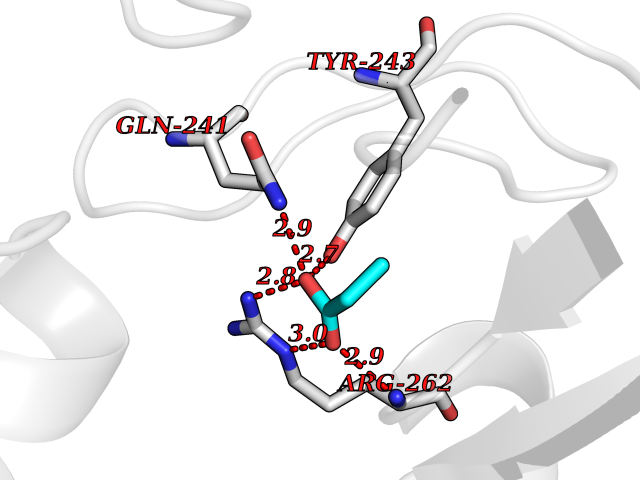 Pymol Map 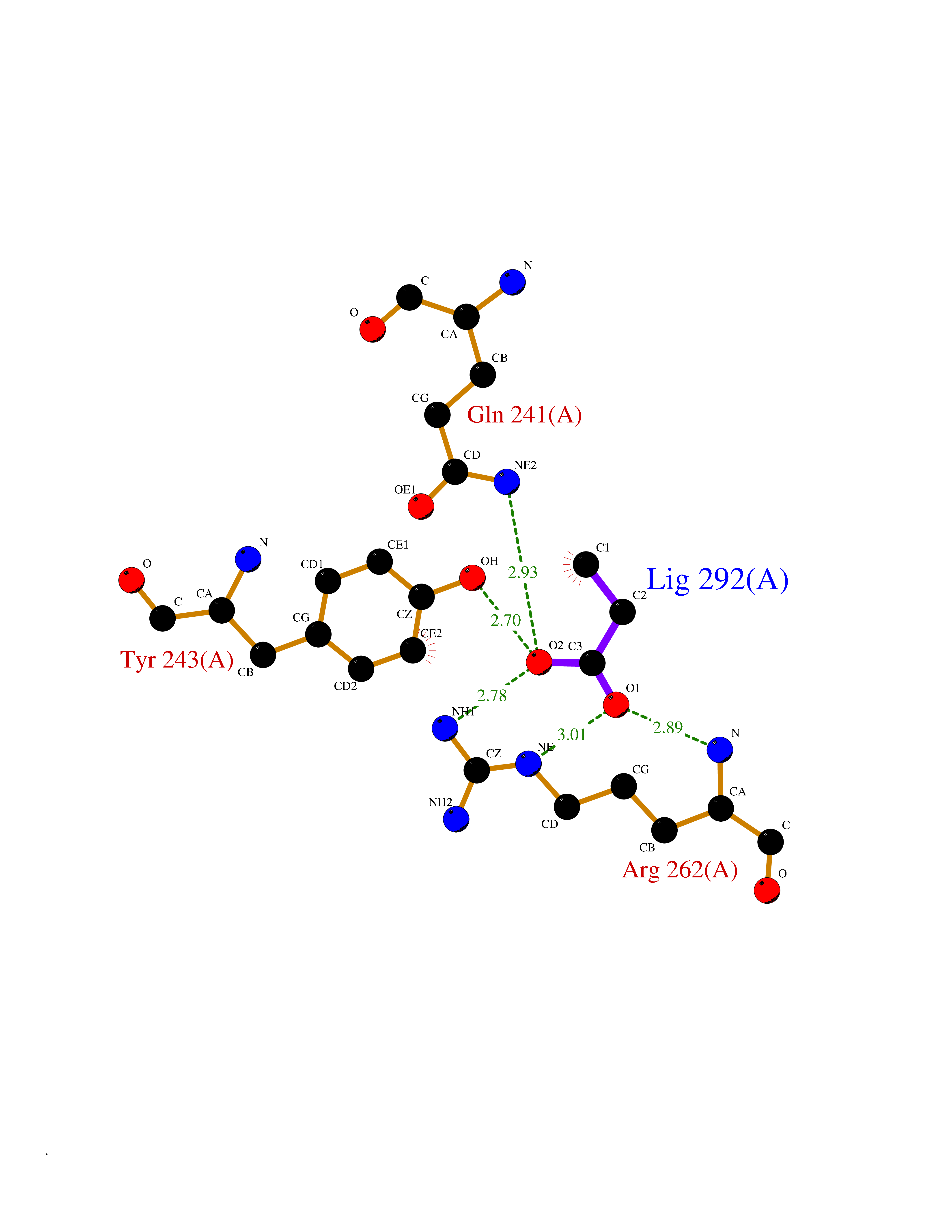 Ligplot Map 3D Binding mode Sequence KELSFGARAELPRIHPVASKLLRLMQKKETNLCLSADVSLARELLQLADALGPSICMLKTHVDILNDFTLDVMKELITLAKXHEFLIFEDRKFADIGNTVKKQYEGGIFKIASWADLVNAHVVPGSGVVKGLQEVGLPLHRGCLLIAEMSSTGSLATGDYTRAAVRMAEEHSEFVVGFISGSRVSMKPEFLHLTPGVQLEAGGDNLGQQYNSPQEVIGKRGSDIIIVGRGIISAADRLEAAEMYRKAAWEAYLSRLGKELSFGARAELPRIHPVASKLLRLMQKKETNLCLSADVSLARELLQLADALGPSICMLKTHVDILNDFTLDVMKELITLAKXHEFLIFEDRKFADIGNTVKKQYEGGIFKIASWADLVNAHVVPGSGVVKGLQEVGLPLHRGCLLIAEMSSTGSLATGDYTRAAVRMAEEHSEFVVGFISGSRVSMKPEFLHLTPGVQLEAGGDNLGQQYNSPQEVIGKRGSDIIIVGRGIISAADRLEAAEMYRKAAWEAYLSRLG Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Ubiquitin thioesterase L1 (UCHL1) | 3IFW | 4.58 | |
Target general information Gen name UCHL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin thiolesterase L1; Ubiquitin carboxyl-terminal hydrolase isozyme L1; Ubiquitin carboxy-terminal hydrolase L1; UCH-L1; PGP9.5; PGP 9.5; Neuron cytoplasmic protein 9.5 Protein family Peptidase C12 family Biochemical class Peptidase Function Ubiquitin-protein hydrolase involved both in the processing of ubiquitin precursors and of ubiquitinated proteins. This enzyme is a thiol protease that recognizes and hydrolyzes a peptide bond at the C-terminal glycine of ubiquitin. Also binds to free monoubiquitin and may prevent its degradation in lysosomes. The homodimer may have ATP-independent ubiquitin ligase activity. Related diseases Parkinson disease 5 (PARK5) [MIM:613643]: A complex neurodegenerative disorder with manifestations ranging from typical Parkinson disease to dementia with Lewy bodies. Clinical features include parkinsonian symptoms (resting tremor, rigidity, postural instability and bradykinesia), dementia, diffuse Lewy body pathology, autonomic dysfunction, hallucinations and paranoia. {ECO:0000269|PubMed:12408865, ECO:0000269|PubMed:12705903, ECO:0000269|PubMed:9774100}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Spastic paraplegia 79A, autosomal dominant, with ataxia (SPG79A) [MIM:620221]: A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG79A is a slowly progressive form characterized by late-onset spastic ataxia, neuropathy, and often optic atrophy. {ECO:0000269|PubMed:35986737}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spastic paraplegia 79B, autosomal recessive (SPG79B) [MIM:615491]: A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG79B is characterized by childhood onset blindness, cerebellar ataxia, nystagmus, dorsal column dysfunction, and spasticity with upper motor neuron dysfunction. {ECO:0000269|PubMed:23359680, ECO:0000269|PubMed:28007905}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12695 Interacts with P63010-2; P05067; P05067-2; Q8N6T3-3; P18847; Q9H1Y0; O15392; Q8WUW1; P83916; P11802; Q00535; Q9UNS2; Q92905; P00533; O60739; Q8TC29; Q9UI08-2; Q8WVV9-3; Q14164; Q6DN90-2; Q96JM7-2; P13473-2; Q9BYZ2; O95777; A4FUJ8; Q15843; O15381-5; Q9BR81; Q13113; P62826; Q8TAI7; Q9ULX5; Q15554-4; Q9NYB0; P04637; Q9Y4K3; P19474; Q9BSL1; Q7KZS0; P61086; Q9UK80; Q86WB0-2 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Glycoprotein; Hereditary spastic paraplegia; Hydrolase; Lipoprotein; Membrane; Neurodegeneration; Oxidation; Parkinson disease; Parkinsonism; Phosphoprotein; Prenylation; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 33389.8 Length 298 Aromaticity 0.07 Instability index 38.69 Isoelectric point 5.51 Charge (pH=7) -7.88 2D Binding mode Binding energy (Kcal/mol) -6.24 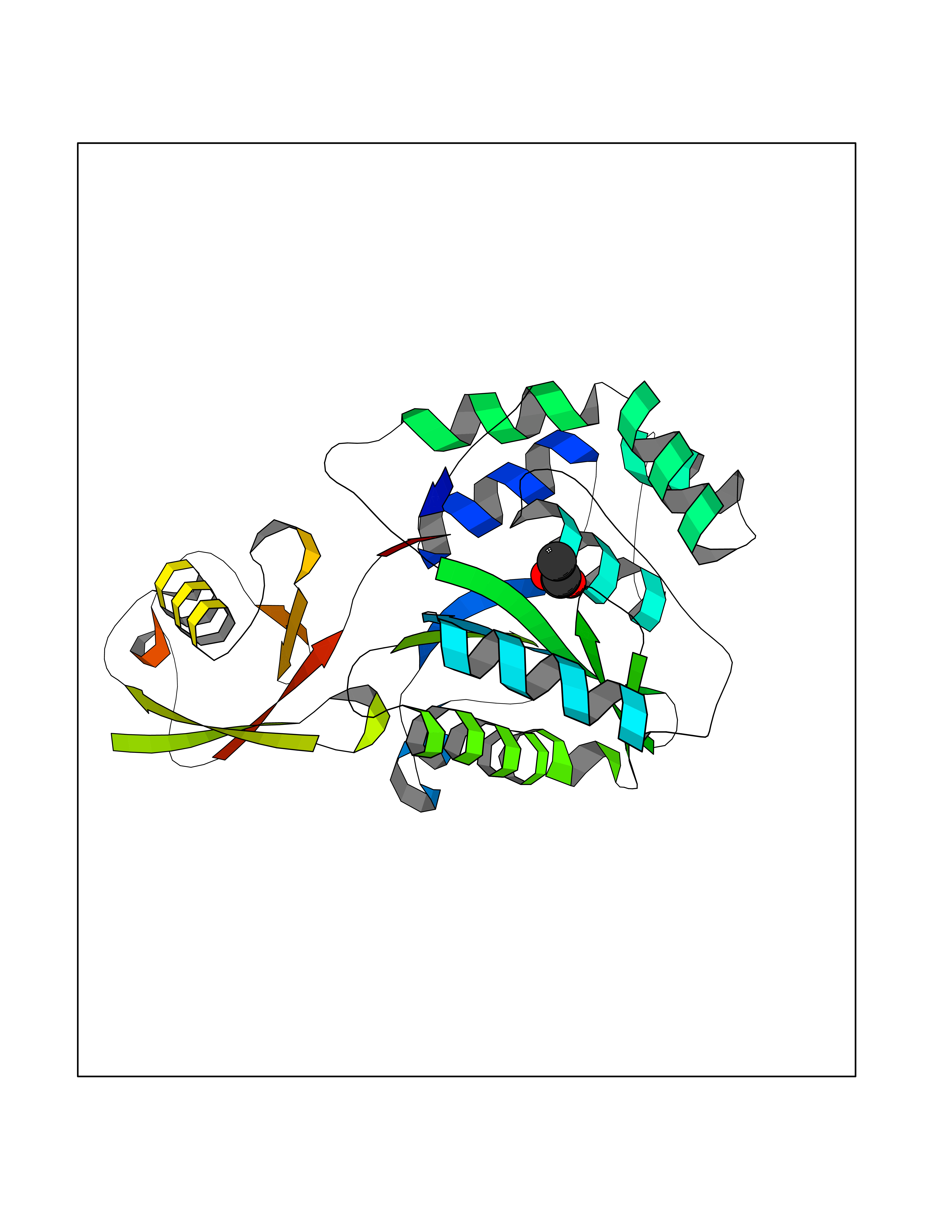 Molscript Map 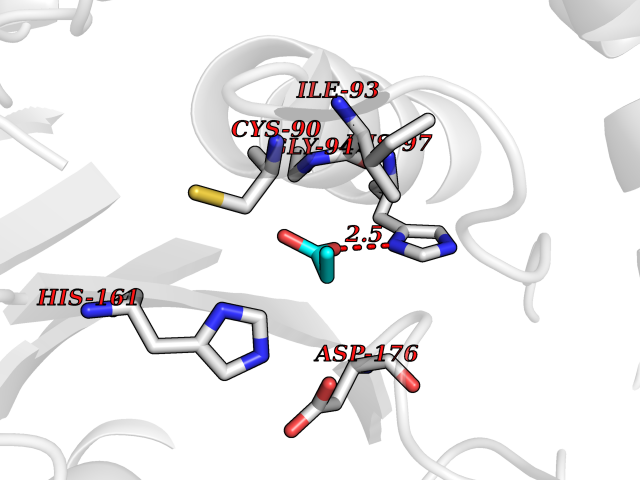 Pymol Map 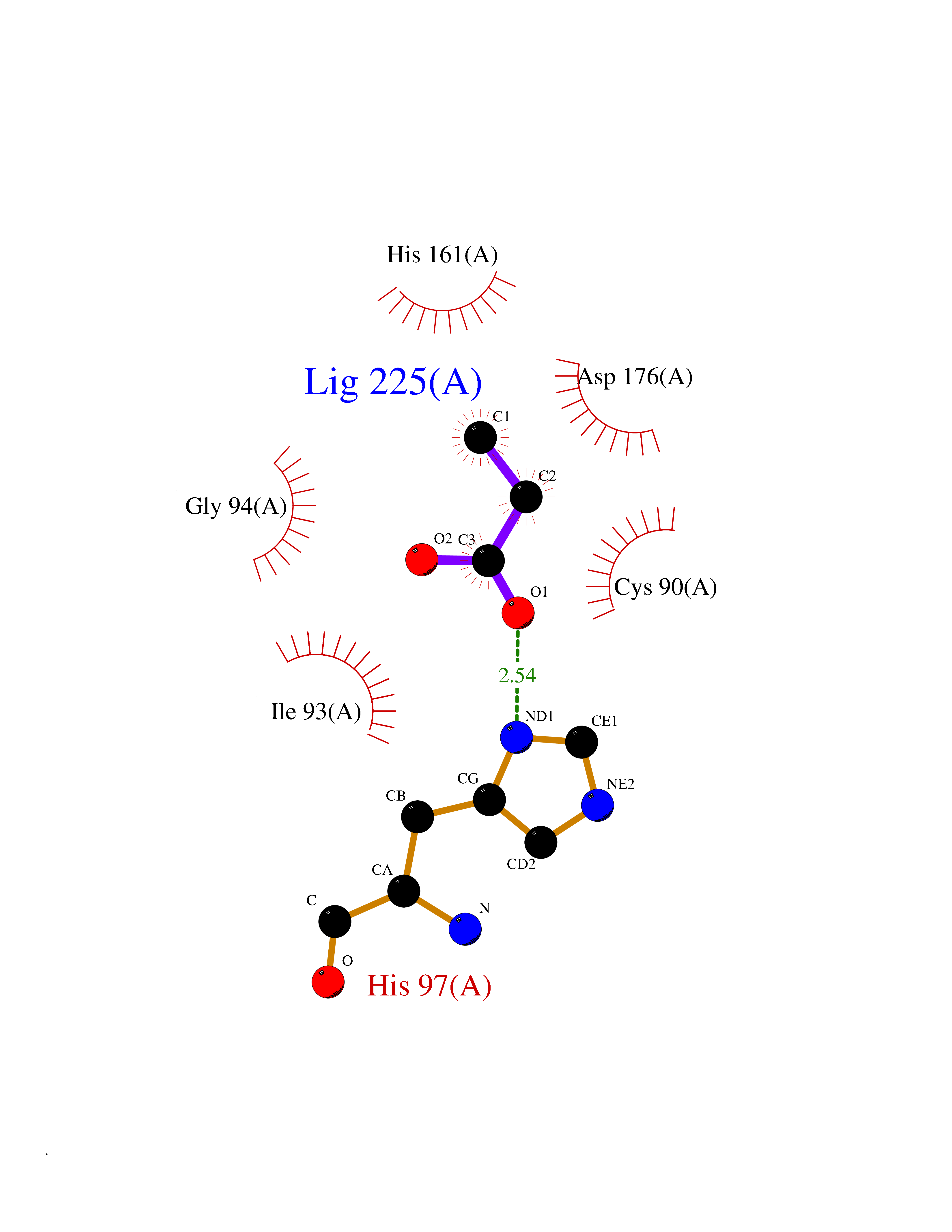 Ligplot Map 3D Binding mode Sequence MQLKPMEINPEMLNKVLYRLGVAGQWRFVDVLGLEEESLGSVPAPACALLLLFPLTAQHENFRKKQIEELKGQEVSPKVYFMKQTIGNSCGTIGLIHAVANNQDKLGFEDGSVLKQFLSETEKMSPEDRAKCFEKNEAIQAAHDAVAQEGQCRVDDKVNFHFILFNNVDGHLYELDGRMPFPVNHGASSEDTLLKDAAKVCREFTEREQGEVRFSAVALCKAAMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRG Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Gamma-aminobutyric acid type B receptor subunit 1 | 4MS4 | 4.57 | |
Target general information Gen name GABBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPRC3B;GPR51 Protein family G-protein coupled receptor 3 family, GABA-B receptor subfamily Biochemical class Signaling protein / antagonist Function G-protein coupled GABA receptor activity. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08891; DB08892; DB00181; DB00363; DB02530; DB05010; DB09072 Interacts with Q9UBS5; Q9UBS5-2; P46459; Q86UR5 EC number NA Uniprot keywords 3D-structure; Cell membrane; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Intellectual disability; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 46502.1 Length 408 Aromaticity 0.12 Instability index 50.05 Isoelectric point 5.78 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -6.23  Molscript Map 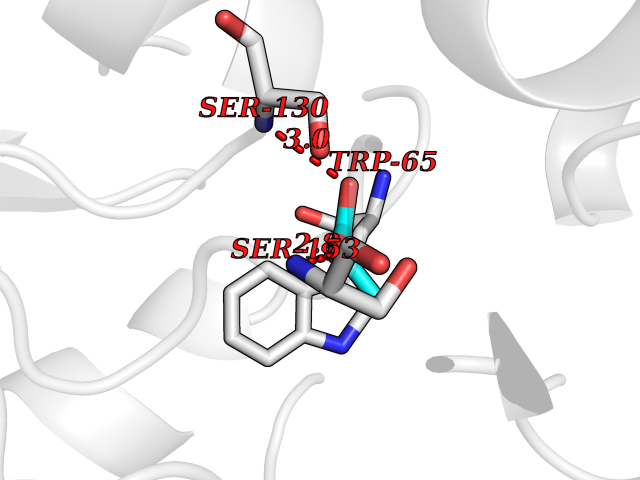 Pymol Map 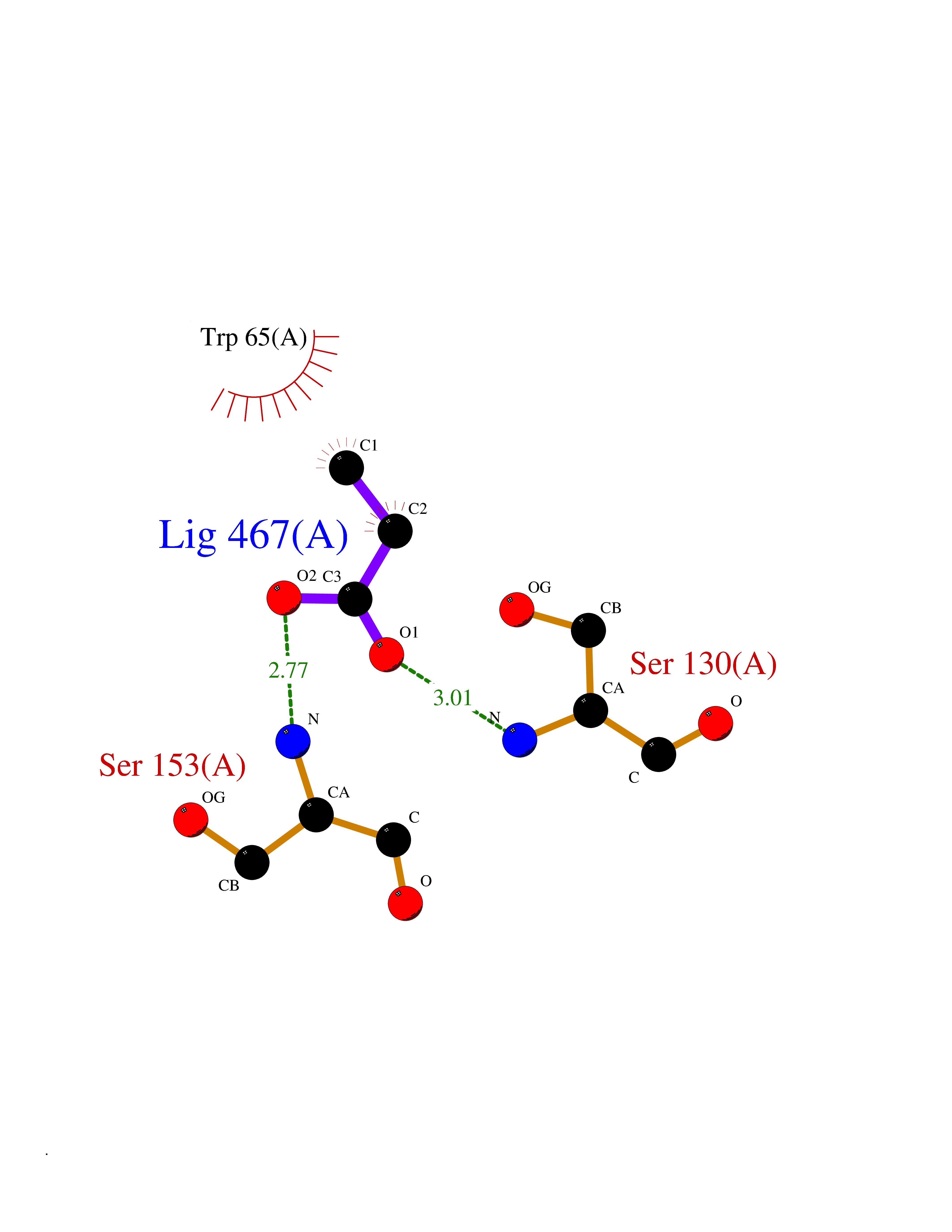 Ligplot Map 3D Binding mode Sequence RRAVYIGALFPMSGGWPGGQACQPAVEMALEDVNSRRDILPDYELKLIHHDSKCDPGQATKYLYELLYNDPIKIILMPGCSSVSTLVAEAARMWNLIVLSYGSSSPALSNRQRFPTFFRTHPSATLHNPTRVKLFEKWGWKKIATIQQTTEVFTSTLDDLEERVKEAGIEITFRQSFFSDPAVPVKNLKRQDARIIVGLFYETEARKVFCEVYKERLFGKKYVWFLIGWYADNWFKIYDPSINCTVDEMTEAVEGHITTEIVMLNPANTRSISNMTSQEFVEKLTKRLKRHPEETGGFQEAPLAYDAIWALALALNKTSRLEDFNYNNQTITDQIYRAMNSSSFEGVSGHVVFDASGSRMAWTLIEQLQGGSYKKIGYYDSTKDDLSWSKTDKWIGGSPPADDYKDDD Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | 5,10-methylenetetrahydrofolate reductase | 3FST | 4.57 | |
Target general information Gen name metF Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3941;JW3913 Protein family Methylenetetrahydrofolate reductase family Biochemical class Oxidoreductase Function FAD binding.Methylenetetrahydrofolate reductase (NAD(P)H) activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.5.1.54 Uniprot keywords 3D-structure; Amino-acid biosynthesis; FAD; Flavoprotein; Methionine biosynthesis; NAD; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,C,E Molecular weight (Da) 30855.9 Length 274 Aromaticity 0.09 Instability index 27.54 Isoelectric point 5.84 Charge (pH=7) -4.61 2D Binding mode Binding energy (Kcal/mol) -6.24 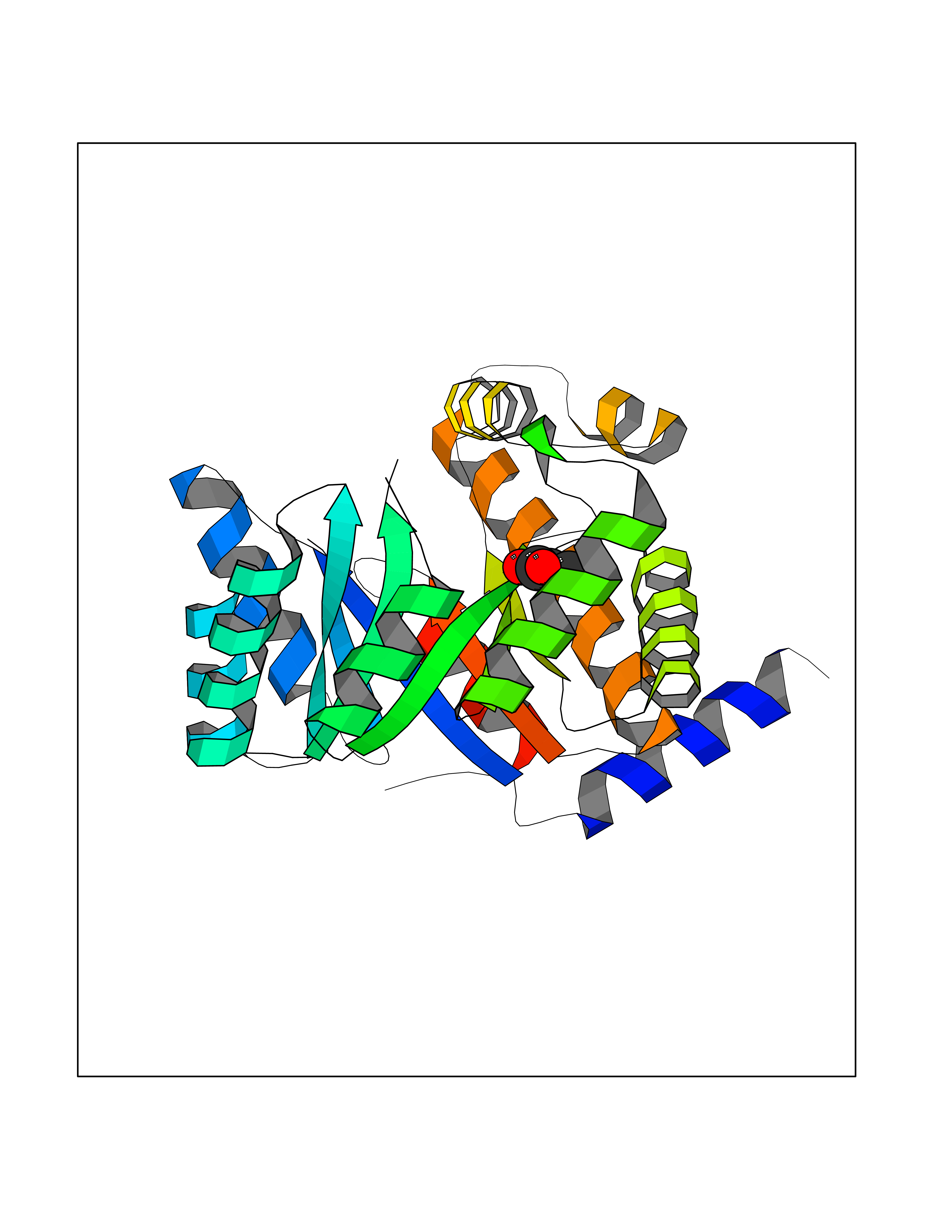 Molscript Map 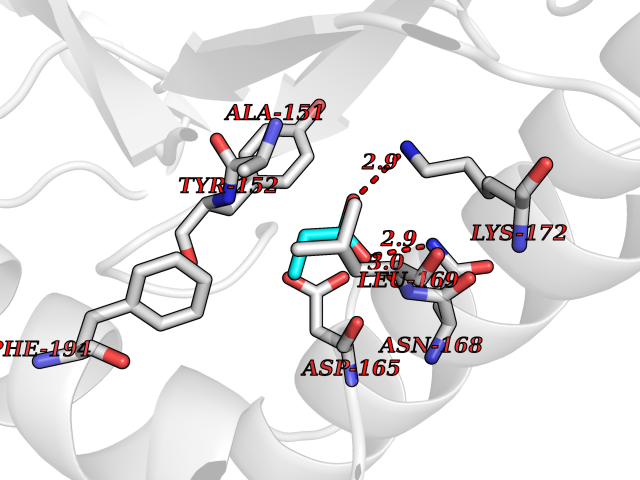 Pymol Map 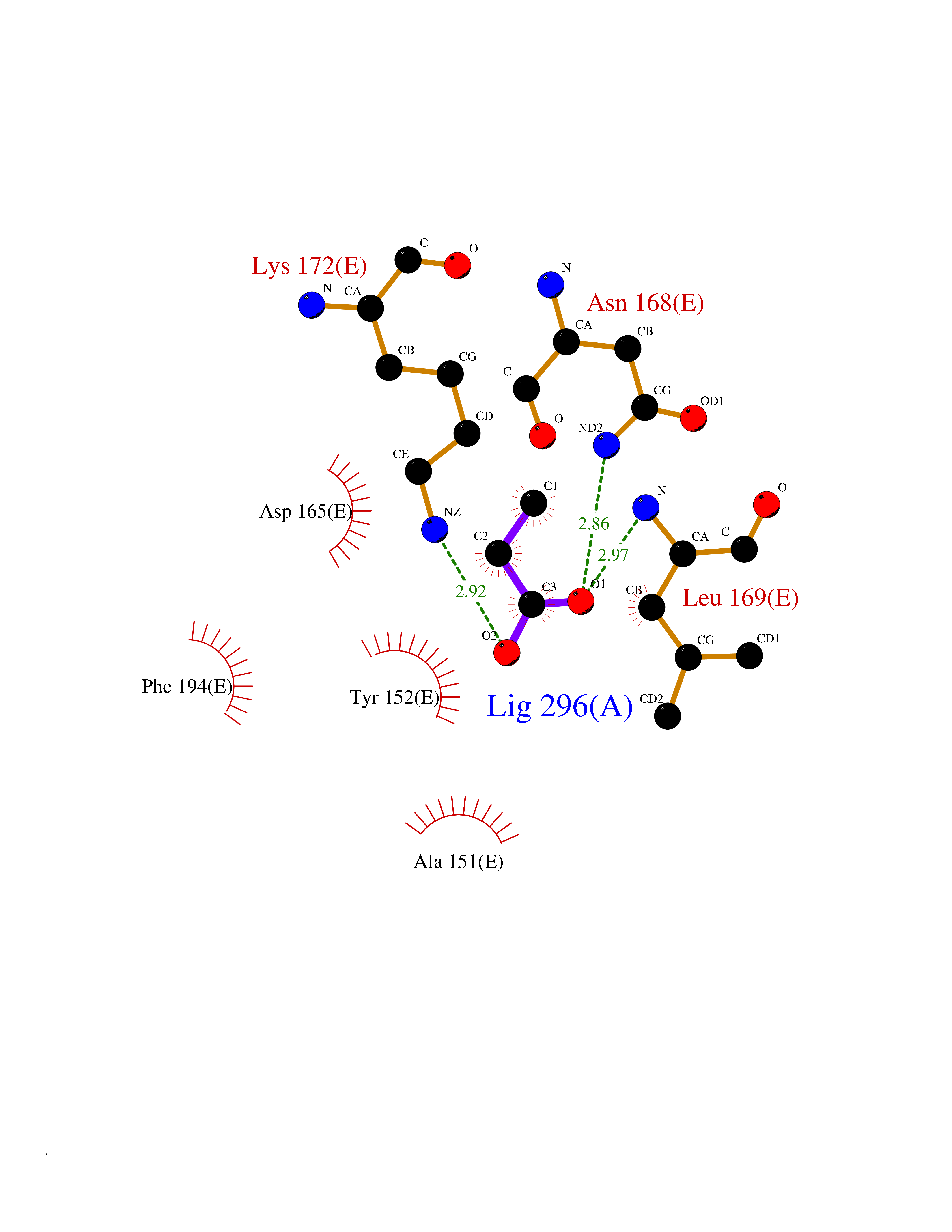 Ligplot Map 3D Binding mode Sequence FHASQRDALNQSLAEVQGQINVSFEFFPPRTSEMEQTLWNSIDRLSSLKPKFVSVTYTHSIIKGIKDRTGLEAAPHLTCIDATPDELRTIARDYWNNGIRHIVALRGDEMYASDLVTLLKEVADFDISVAAYPEVHPEAKSAQADLLNLKRKVDAGANRAITQFFFDVESYLRFRDRCVSAGIDVEIIPGILPVSNFKQAKKLADMTNVRIPAWMAQMFDGLDDDAETRKLVGANIAMDMVKILSREGVKDFHFYTLNRAEMSYAICHTLGVRP Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Dopamine D2 receptor (D2R) | 5AER | 4.57 | |
Target general information Gen name DRD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dopamine receptor 2; D(2) dopamine receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Dopamine receptor whose activity is mediated by G proteins which inhibit adenylyl cyclase. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01614; DB01063; DB01425; DB00915; DB06288; DB05964; DB00543; DB00182; DB04599; DB00714; DB01238; DB14185; DB09207; DB06216; DB04889; DB04888; DB05687; DB09223; DB04857; DB09128; DB01200; DB09018; DB00490; DB00248; DB06016; DB01038; DB00477; DB01239; DB00568; DB00363; DB01151; DB11274; DB13345; DB00320; DB01184; DB00988; DB00450; DB11275; DB01049; DB00696; DB01175; DB09194; DB00875; DB00623; DB04842; DB00502; DB04946; DB00458; DB04924; DB12579; DB01221; DB00555; DB01235; DB00589; DB00408; DB06077; DB08815; DB00934; DB09224; DB01043; DB00933; DB01403; DB01233; DB06148; DB00805; DB01618; DB08804; DB05766; DB00540; DB06229; DB00334; DB01267; DB12061; DB00715; DB01186; DB08922; DB00850; DB01100; DB09286; DB01621; DB12478; DB00413; DB00433; DB00420; DB01069; DB00777; DB01224; DB09097; DB12518; DB00409; DB00734; DB01549; DB00268; DB05271; DB06454; DB06144; DB00391; DB06477; DB04844; DB12093; DB00372; DB01622; DB00679; DB01623; DB13025; DB00831; DB00508; DB00726; DB06109; DB01392; DB00246; DB09225; DB01624 Interacts with Q9NRI5; P14416; Q01959 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Lipoprotein; Membrane; Palmitate; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID B,C Molecular weight (Da) 24300.3 Length 209 Aromaticity 0.13 Instability index 40.14 Isoelectric point 4.97 Charge (pH=7) -7.83 2D Binding mode Binding energy (Kcal/mol) -6.23 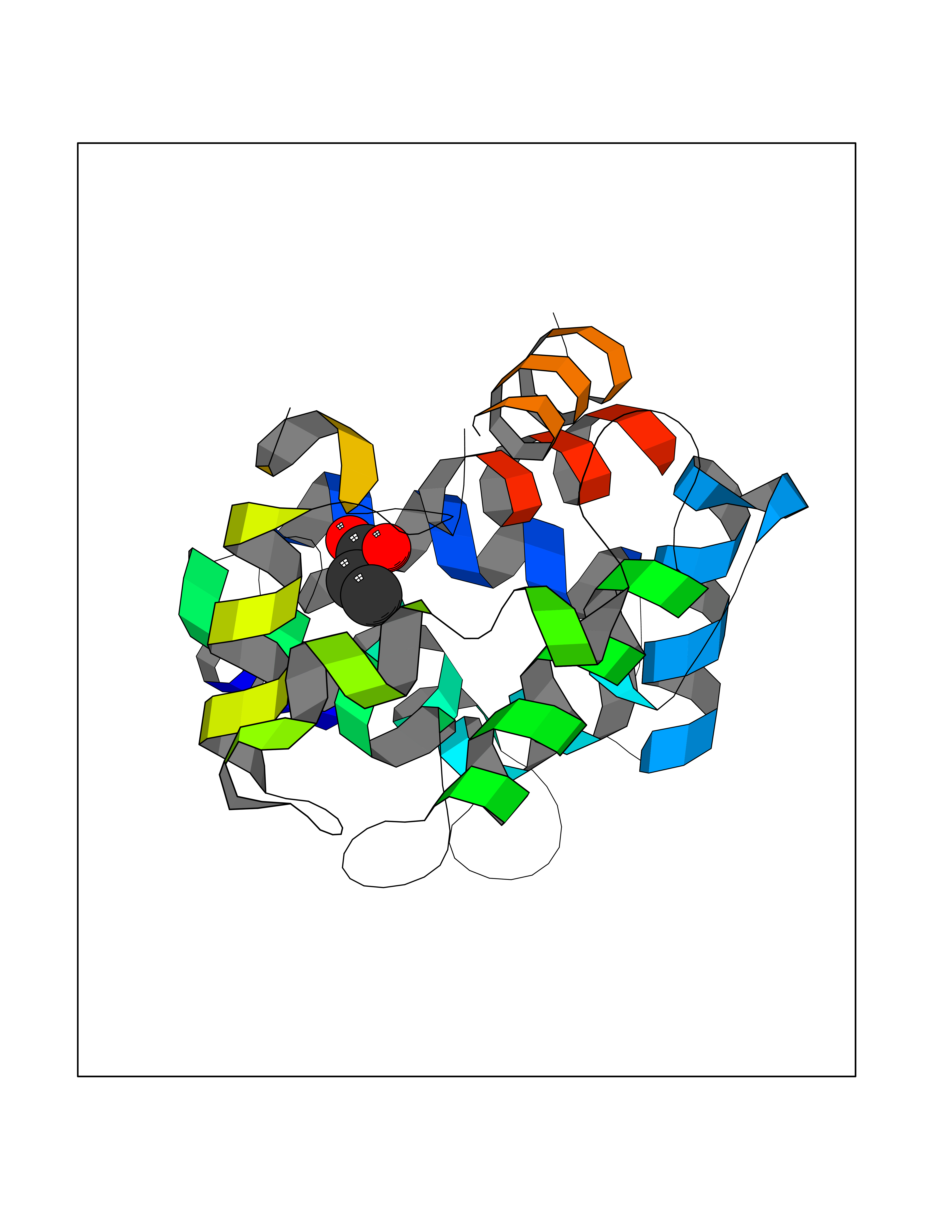 Molscript Map 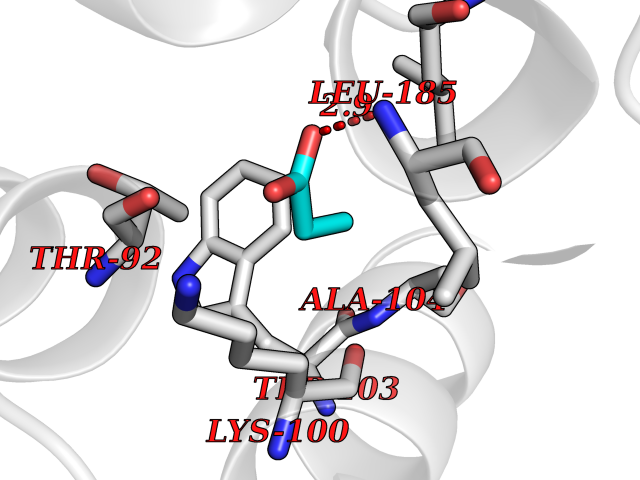 Pymol Map 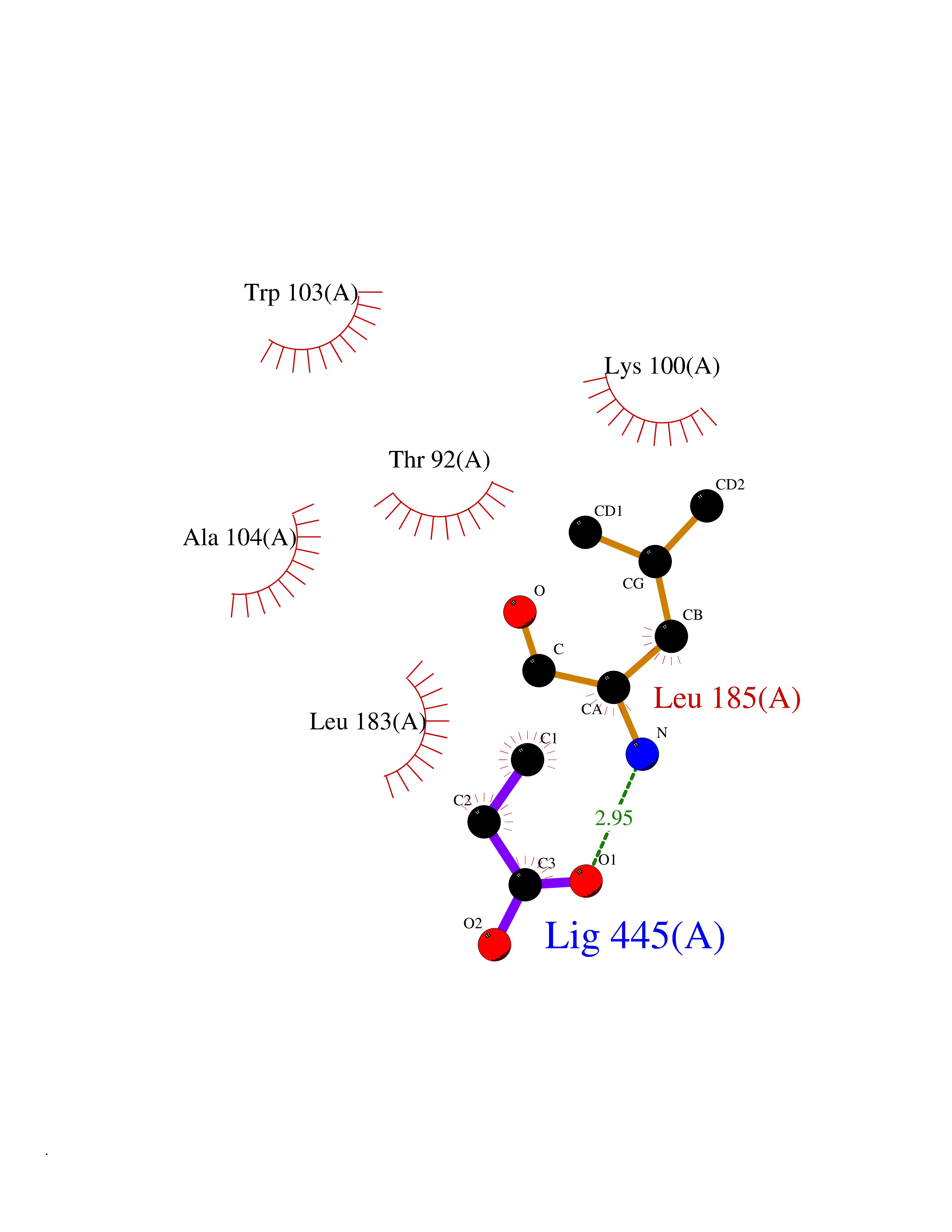 Ligplot Map 3D Binding mode Sequence PEVVEELTRKTYFTEKEVQQWYKGFIKDCPSGQLDAAGFQKIYKQFFPFGDPTKFATFVFNVFDENKDGRIEFSEFIQALSVTSRGTLDEKLRWAFKLYDLDNDGYITRNEMLDIVDAIYQMVGNTVELPEEENTPEKRVDRIFAMMDKNADGKLTLQEFQEGSKADPSIVQALSLYDGLVNIEFRKAFLKILHSNIEFRKAFLKILHS Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Vascular endothelial growth factor receptor 3 | 4BSJ | 4.57 | |
Target general information Gen name FLT4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms VEGFR3 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Transferase Function ATP binding.Growth factor binding.Protein homodimerization activity.Protein phosphatase binding.Transmembrane receptor protein tyrosine kinase activity.Vascular endothelial growth factor-activated receptor activity.VEGF-C-activated receptor activity. Related diseases Lymphatic malformation 1 (LMPHM1) [MIM:153100]: A form of primary lymphedema, a disease characterized by swelling of body parts due to developmental anomalies and functional defects of the lymphatic system. Patients with lymphedema may suffer from recurrent local infections. LMPHM1 is an autosomal dominant form with variable expression and severity. Onset is usually at birth or in early childhood but can occur later. Affected individuals manifest lymphedema, predominantly in the lower limbs, and hypoplasia of lymphatic vessels. Additional features are hemangioma and nail dysplasia or papillomatosis. {ECO:0000269|PubMed:10835628, ECO:0000269|PubMed:10856194, ECO:0000269|PubMed:12881528, ECO:0000269|PubMed:15102829, ECO:0000269|PubMed:16924388, ECO:0000269|PubMed:16965327, ECO:0000269|PubMed:17458866, ECO:0000269|PubMed:19289394, ECO:0000269|PubMed:26091405, ECO:0000269|PubMed:9817924}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hemangioma, capillary infantile (HCI) [MIM:602089]: A condition characterized by dull red, firm, dome-shaped hemangiomas, sharply demarcated from surrounding skin, usually presenting at birth or occurring within the first two or three months of life. They result from highly proliferative, localized growth of capillary endothelium and generally undergo regression and involution without scarring. {ECO:0000269|PubMed:11807987}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Plays an important role in tumor lymphangiogenesis, in cancer cell survival, migration, and formation of metastases.; DISEASE: Congenital heart defects, multiple types, 7 (CHTD7) [MIM:618780]: An autosomal dominant disorder with incomplete penetrance characterized by congenital developmental abnormalities involving structures of the heart. Common defects include tetralogy of Fallot, pulmonary stenosis or atresia, absent pulmonary valve, right aortic arch, double aortic arch, and major aortopulmonary collateral arteries. {ECO:0000269|PubMed:28991257, ECO:0000269|PubMed:30232381}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06626; DB05932; DB12010; DB11679; DB06101; DB09078; DB06080; DB09079; DB06589; DB08896; DB15685; DB00398; DB01268; DB05075; DB11800; DB04879 Interacts with P08238; P35968; P49767 EC number 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 24029.2 Length 213 Aromaticity 0.1 Instability index 47.75 Isoelectric point 8.34 Charge (pH=7) 2.35 2D Binding mode Binding energy (Kcal/mol) -6.23 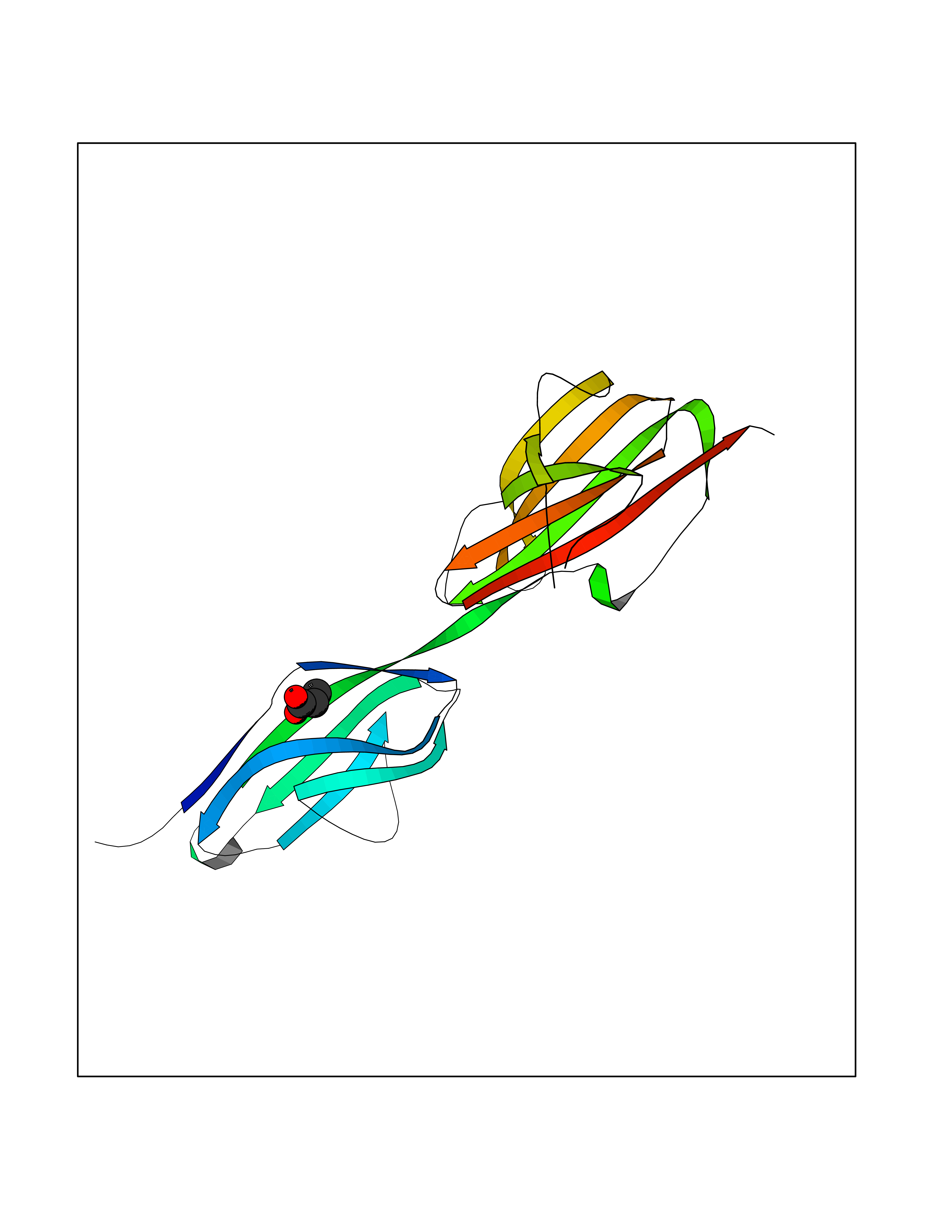 Molscript Map 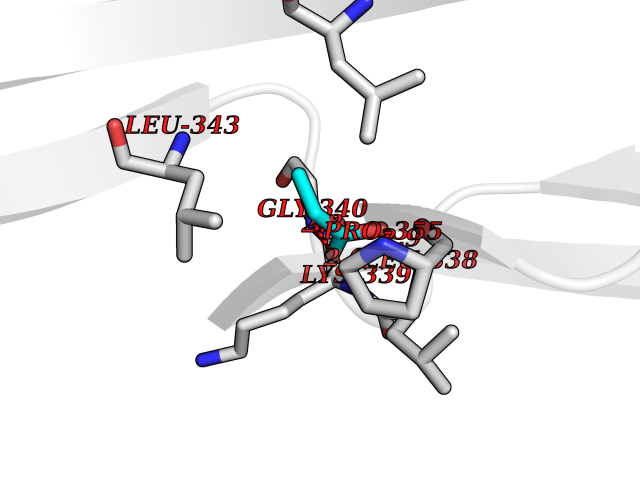 Pymol Map 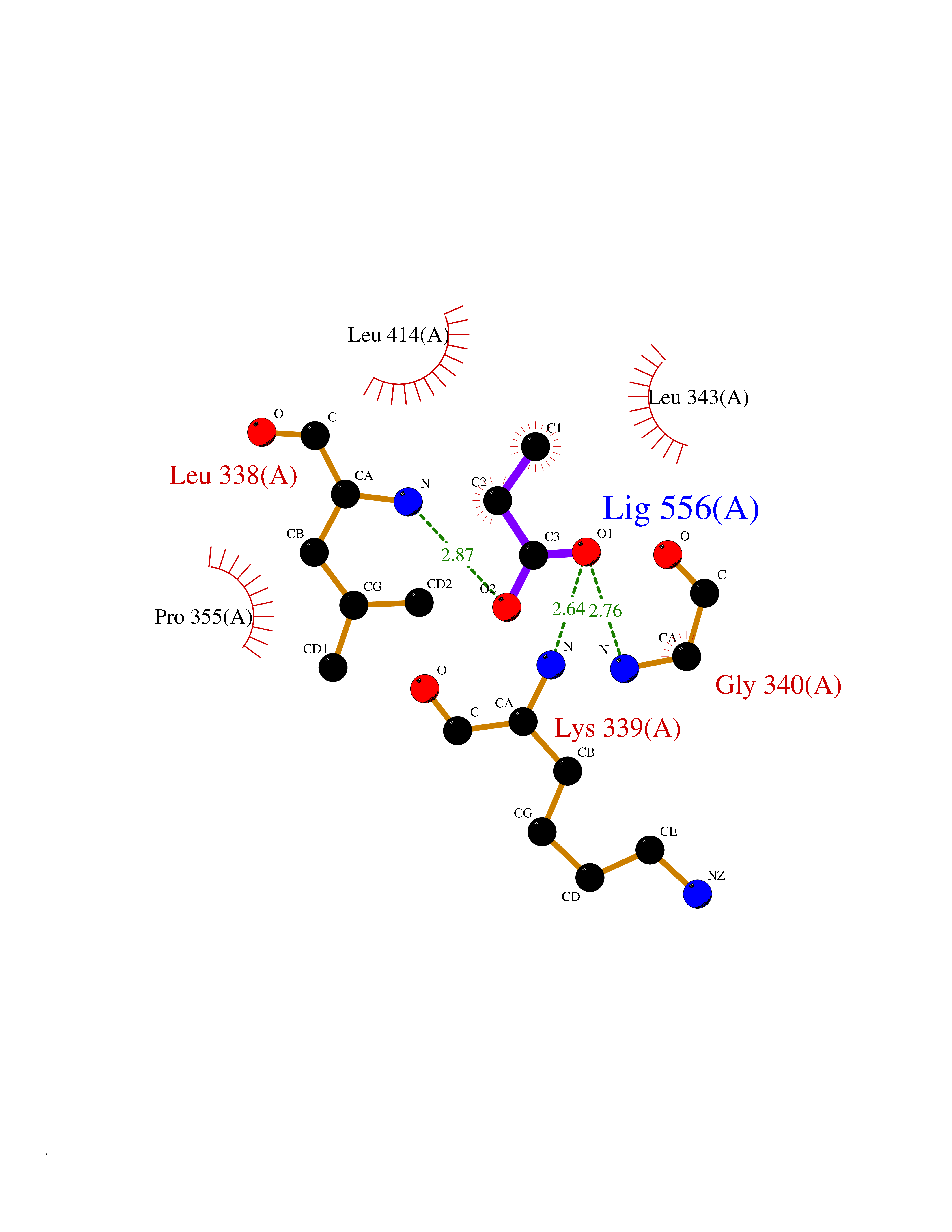 Ligplot Map 3D Binding mode Sequence DHNPFISVEWLKGPILEATAGDELVKLPVKLAAYPPPEFQWYKDGKALSGRHSPHALVLKEVTEASTGTYTLALWNSAAGLRRNISLELVVNVPPQIHEKEASSPSIYSRHSRQALTCTAYGVPLPLSIQWHWRPWTPCKMFPQCRDWRAVTTQDAVNPIESLDTWTEFVEGKNKTVSKLVIQNANVSAMYKCVVSNKVGQDERLIYFYVTTH Hydrogen bonds contact Hydrophobic contact | ||||