Job Results:
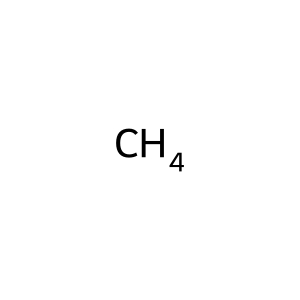
Ligand
Structure
Job ID
9069676bc0da30cae69182dac941f7de
Job name
NA
Time
2026-02-27 11:56:18
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Kallikrein-4 (KLK4) | 7JOW | 3.95 | |
Target general information Gen name KLK4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine protease 17; Prostase; PSTS; PRSS17; Kallikreinlike protein 1; Kallikrein4; Kallikrein-like protein 1; KLKL1; KLK-L1; Enamel matrix serine proteinase 1; EMSP1 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function Required during the maturation stage of tooth development for clearance of enamel proteins and normal structural patterning of the crystalline matrix. Has a major role in enamel formation. Related diseases Amelogenesis imperfecta, hypomaturation type, 2A1 (AI2A1) [MIM:204700]: A defect of enamel formation. The disorder involves both primary and secondary dentitions. The teeth have a shiny agar jelly appearance and the enamel is softer than normal. Brown pigment is present in middle layers of enamel. {ECO:0000269|PubMed:15235027}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P20155; Q06418 EC number EC 3.4.21.- Uniprot keywords 3D-structure; Alternative splicing; Amelogenesis imperfecta; Biomineralization; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Metal-binding; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID E,I Molecular weight (Da) 41763.9 Length 387 Aromaticity 0.07 Instability index 32.75 Isoelectric point 5.38 Charge (pH=7) -11.36 2D Binding mode Binding energy (Kcal/mol) -5.39 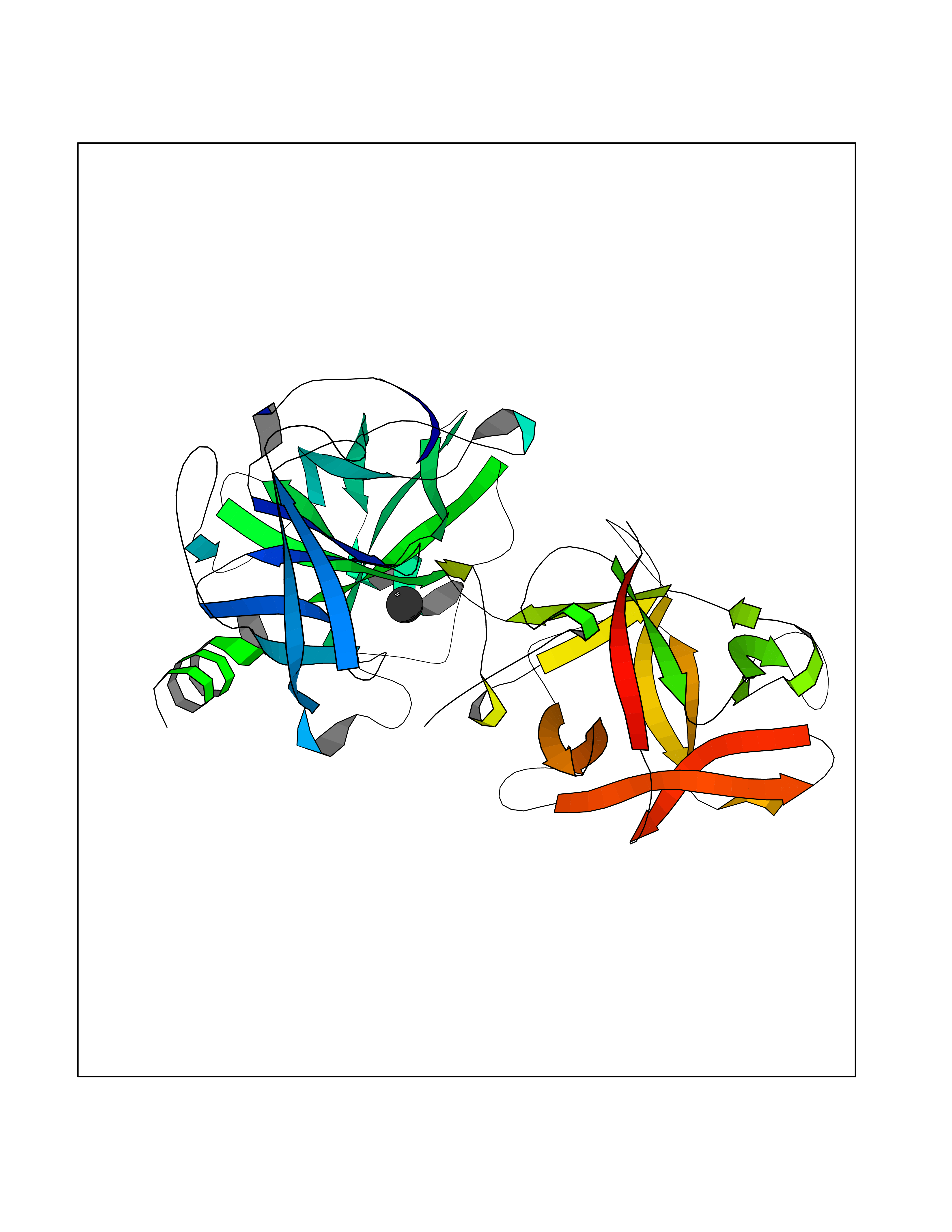 Molscript Map 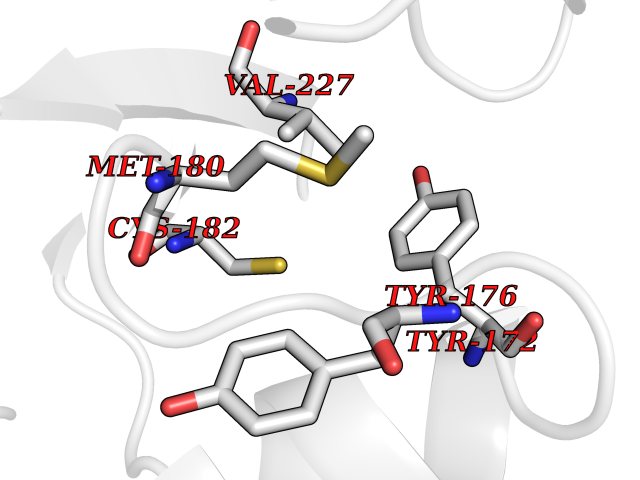 Pymol Map 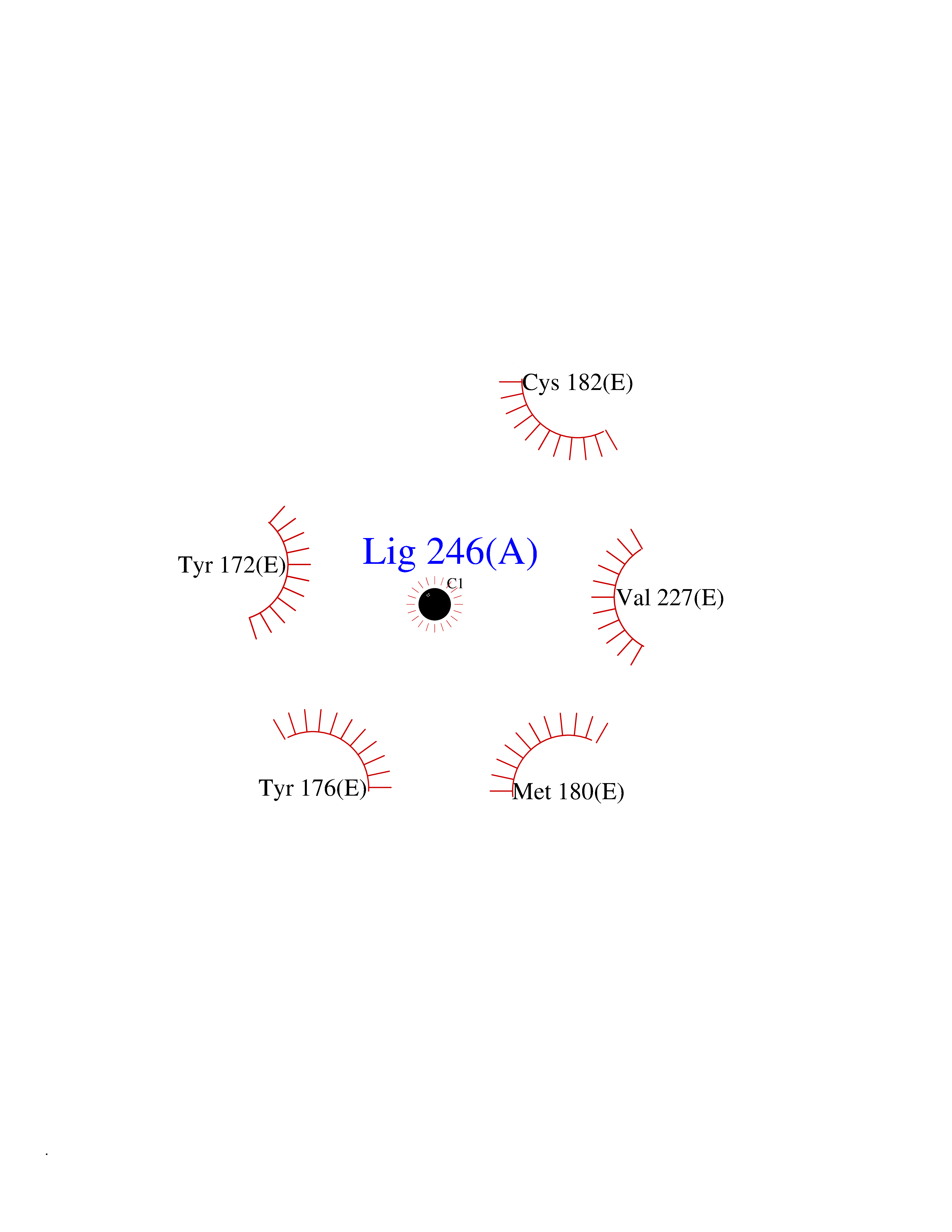 Ligplot Map 3D Binding mode Sequence IINGEDCSPHSQPWQAALVMENELFCSGVLVHPQWVLSAAHCFQNSYTIGLGLHSLEADQEPGSQMVEASLSVRHPEYNRPLLANDLMLIKLDESVSESDTIRSISIASQCPTAGNSCLVSGWGLLANGRMPTVLQCVNVSVVSEEVCSKLYDPLYHPSMFCAGGGQDQKDSCNGDSGGPLICNGYLQGLVSFGKAPCGQVGVPGVYTNLCKFTEWIEKTVQAGSSVVVDTNGQPVSNGADAYYLVPVSHGHAGLALAKIGNEAEPRAVVLDPHHRPGLPVRFESPLRINIIKESYFLNIKFGPSSSDSGVWDVIQQDPIGLAVKVTDTKSLLGPFKVEKEGEGYKIVYYPERGQTGLDIGLVHRNDKYYLAVKDGEPCVFKIRKAT Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Extracellular calcium-sensing receptor (CASR) | 5FBK | 3.94 | |
Target general information Gen name CASR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hCasR; Parathyroid cell calciumreceptor; Parathyroid cell calcium-sensing receptor 1; Parathyroid calcium receptor; Parathyroid Cell calcium-sensing receptor; PCaR1; GPRC2A; CaSR Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Senses fluctuations in the circulating calcium concentration and modulates the production of parathyroid hormone (PTH) in parathyroid glands. The activity of this receptor is mediated by a G-protein that activates a phosphatidylinositol-calcium second messenger system. The G-protein-coupled receptor activity is activated by a co-agonist mechanism: aromatic amino acids, such as Trp or Phe, act concertedly with divalent cations, such as calcium or magnesium, to achieve full receptor activation. G-protein-coupled receptor that senses changes in the extracellular concentration of calcium ions and plays a key role in maintaining calcium homeostasis. Related diseases Hypocalciuric hypercalcemia, familial 1 (HHC1) [MIM:145980]: A form of hypocalciuric hypercalcemia, a disorder of mineral homeostasis that is transmitted as an autosomal dominant trait with a high degree of penetrance. It is characterized biochemically by lifelong elevation of serum calcium concentrations and is associated with inappropriately low urinary calcium excretion and a normal or mildly elevated circulating parathyroid hormone level. Hypermagnesemia is typically present. Affected individuals are usually asymptomatic and the disorder is considered benign. However, chondrocalcinosis and pancreatitis occur in some adults. {ECO:0000269|PubMed:11762699, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:15579740, ECO:0000269|PubMed:15879434, ECO:0000269|PubMed:16598859, ECO:0000269|PubMed:16740594, ECO:0000269|PubMed:17473068, ECO:0000269|PubMed:17698911, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:19789209, ECO:0000269|PubMed:21566075, ECO:0000269|PubMed:21643651, ECO:0000269|PubMed:22114145, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25104082, ECO:0000269|PubMed:25292184, ECO:0000269|PubMed:26386835, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:7673400, ECO:0000269|PubMed:7726161, ECO:0000269|PubMed:7916660, ECO:0000269|PubMed:8636323, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9298824}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperparathyroidism, neonatal severe (NSHPT) [MIM:239200]: A disorder characterized by severe hypercalcemia, bone demineralization, and failure to thrive usually manifesting in the first 6 months of life. If untreated, NSHPT can be a devastating neurodevelopmental disorder, which in some cases is lethal without parathyroidectomy. {ECO:0000269|PubMed:14985373, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:17555508, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:8675635, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253359}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypocalcemia, autosomal dominant 1 (HYPOC1) [MIM:601198]: A disorder of mineral homeostasis characterized by blood calcium levels below normal, and low or normal serum parathyroid hormone concentrations. Disease manifestations include mild or asymptomatic hypocalcemia, paresthesias, carpopedal spasm, seizures, hypercalciuria with nephrocalcinosis or kidney stones, and ectopic and basal ganglia calcifications. Few patients manifest hypocalcemia and features of Bartter syndrome, including hypomagnesemia, hypokalemia, metabolic alkalosis, hyperreninemia, and hyperaldosteronemia. {ECO:0000269|PubMed:10487661, ECO:0000269|PubMed:12050233, ECO:0000269|PubMed:12107202, ECO:0000269|PubMed:12241879, ECO:0000269|PubMed:12574188, ECO:0000269|PubMed:12915654, ECO:0000269|PubMed:15551332, ECO:0000269|PubMed:16608894, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:22789683, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25766501, ECO:0000269|PubMed:7874174, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8733126, ECO:0000269|PubMed:8813042, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253358, ECO:0000269|PubMed:9661634, ECO:0000269|PubMed:9920108}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epilepsy, idiopathic generalized 8 (EIG8) [MIM:612899]: A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Seizure types are variable, but include myoclonic seizures, absence seizures, febrile seizures, complex partial seizures, and generalized tonic-clonic seizures. {ECO:0000269|PubMed:18756473}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11093; DB11348; DB14481; DB01012; DB12865; DB00994; DB05695; DB05255; DB00127 Interacts with Q15363; P41180-1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 52438.9 Length 467 Aromaticity 0.12 Instability index 39.62 Isoelectric point 5.63 Charge (pH=7) -10.18 2D Binding mode Binding energy (Kcal/mol) -5.37 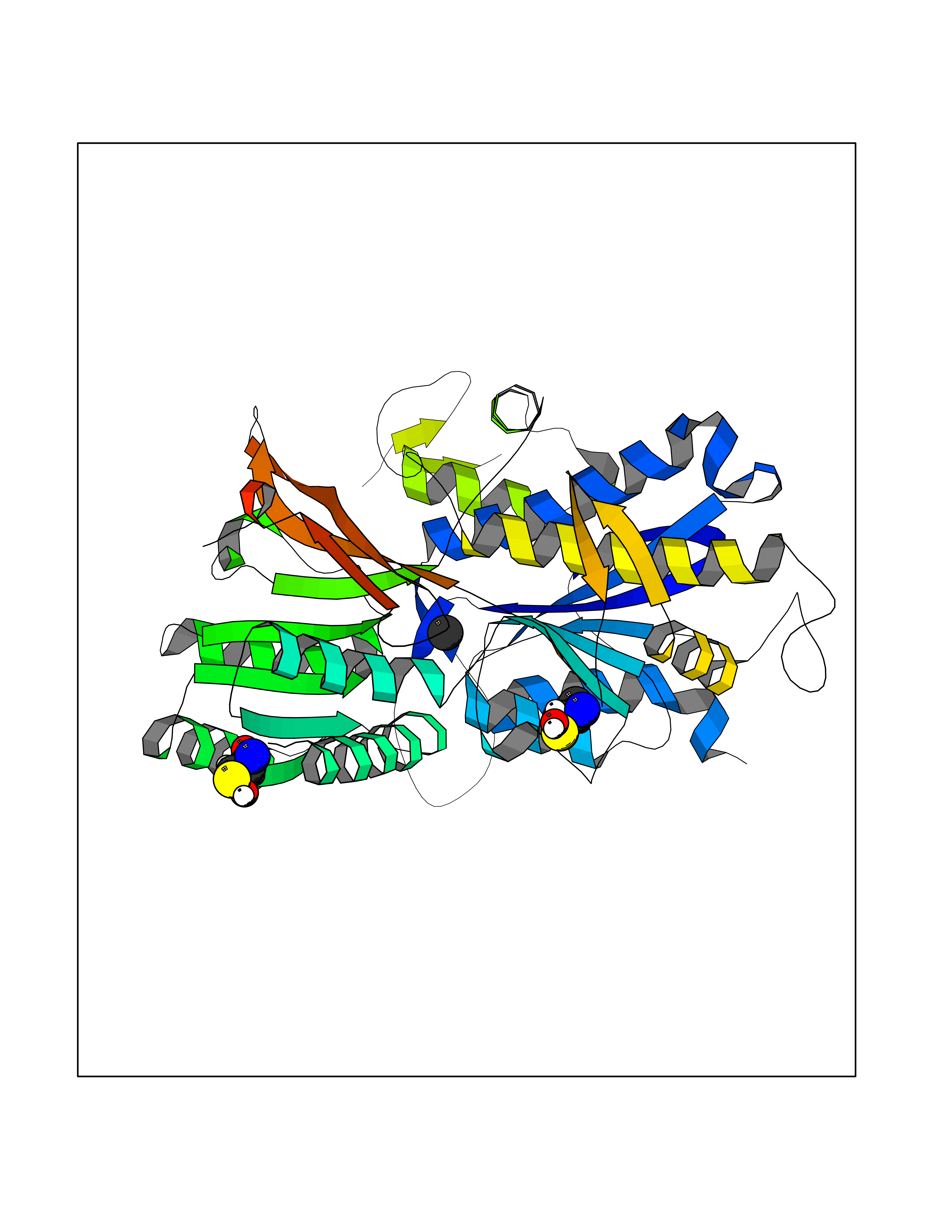 Molscript Map 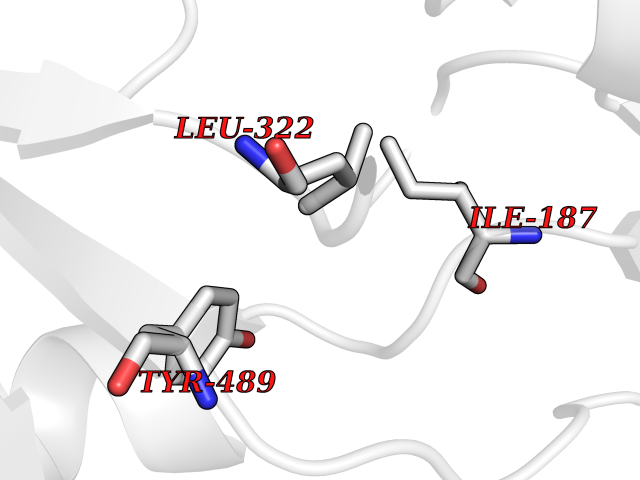 Pymol Map 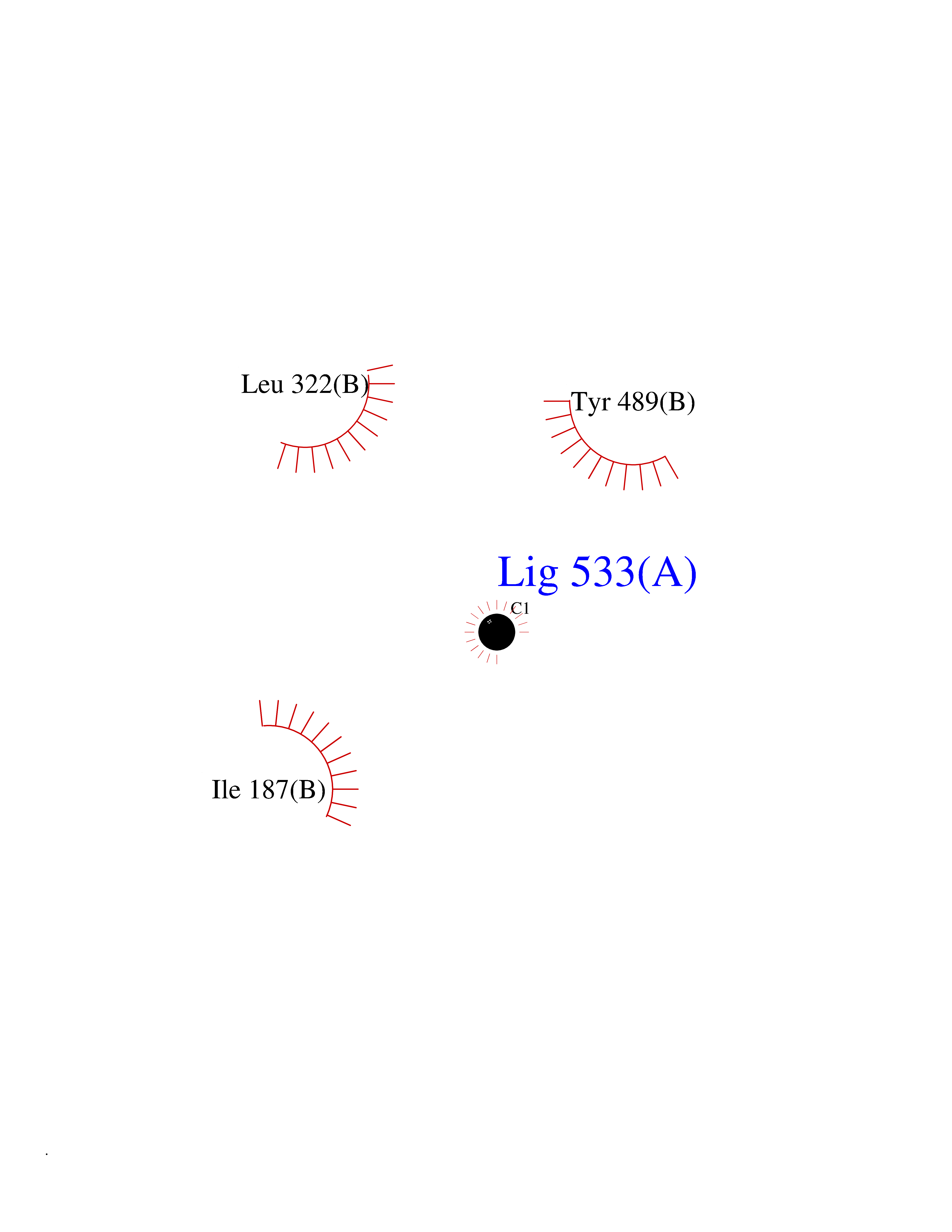 Ligplot Map 3D Binding mode Sequence GPDQRAQKKGDIILGGLFPIHFGVAAKDQDLKSRPESVECIRYNFRGFRWLQAMIFAIEEINSSPALLPNLTLGYRIFDTCNTVSKALEATLSFVAQNKIDSTIAVVGATGSGVSTAVANLLGLFYIPQVSYASSSRLLSNKNQFKSFLRTIPNDEHQATAMADIIEYFRWNWVGTIAADDDYGRPGIEKFREEAEERDIXIDFSELISQYSDEEEIQHVVEVIQNSTAKVIVVFSSGPDLEPLIKEIVRRNITGKIWLASEAWASSSLIAMPQYFHVVGGTIGFALKAGQIPGFREFLKKVHPRKSVHNGFAKEFWEETFNCHLQFRPLCTGDENISSVETPYIDYTHLRISYNVYLAVYSIAHALQDIYTCLPGRGLFTNGSCADIKKVEAWQVLKHLRHLNFTNNMGEQVTFDEXGDLVGNYSIINWHLSPEDGSIVFKEVGYYNVYAKKGERLFINEEKILWS Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Aldo-keto reductase family 1 member C3 | 1S1P | 3.94 | |
Target general information Gen name AKR1C3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PGFS;DDH1;HSD17B5;KIAA0119 Protein family Aldo/keto reductase family Biochemical class Oxidoreductase Function 15-hydroxyprostaglandin-D dehydrogenase (NADP+) activity.Alditol:NADP+ 1-oxidoreductase activity.Aldo-keto reductase (NADP) activity.Androsterone dehydrogenase activity.Delta4-3-oxosteroid 5beta-reductase activity.Dihydrotestosterone 17-beta-dehydrogenase activity.Geranylgeranyl reductase activity.Indanol dehydrogenase activity.Ketoreductase activity.Ketosteroid monooxygenase activity.NADP-retinol dehydrogenase activity.Oxidoreductase activity, acting on NAD(P)H, quinone or similar compound as acceptor.Phenanthrene 9,10-monooxygenase activity.Prostaglandin D2 11-ketoreductase activity.Prostaglandin-F synthase activity.Prostaglandin H2 endoperoxidase reductase activity.Retinal dehydrogenase activity.Retinol dehydrogenase activity.Testosterone 17-beta-dehydrogenase (NADP+) activity.Testosterone dehydrogenase (NAD+) activity.Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase activity. Related diseases Neurodevelopmental disorder with language impairment and behavioral abnormalities (NEDLIB) [MIM:618917]: A neurodevelopmental disorder characterized by global developmental delay, impaired intellectual development, poor or absent speech, and behavioral abnormalities, such as autism spectrum disorder, repetitive behaviors, and hyperactivity. Some patients develop seizures and manifest developmental regression. {ECO:0000269|PubMed:31300657}. The disease is caused by variants affecting the gene represented in this entry. The genetic variation producing the missense variant p.Q607E, associated with NEDLIB, is predicted to deeply affect RNA editing. In a physiological context, the adenosine (A) residue of the original glutamine (Q) codon CAG is post-transcriptionaly edited to inosine (I) by ADAR2, leading to a codon recognized by the ribosome as arginine (R). The glutamate (E) codon GAG, resulting from the genetic variation, is predicted to be edited 90% less than the normal CAG codon. If edited, the codon GIG would be translated as p.Q607G. {ECO:0000305|PubMed:31300657}. Drugs (DrugBank ID) DB07700; DB01561; DB01536; DB00997; DB01039; DB02266; DB13751; DB00328; DB06077; DB00959; DB00157; DB03461; DB09074; DB00776; DB02056; DB01698; DB02901 Interacts with P17516 EC number 1.1.1.-; 1.1.1.188; 1.1.1.210; 1.1.1.239; 1.1.1.357; 1.1.1.53; 1.1.1.62; 1.1.1.64 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Lipid metabolism; NAD; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35846.8 Length 315 Aromaticity 0.09 Instability index 47.59 Isoelectric point 8.54 Charge (pH=7) 4.51 2D Binding mode Binding energy (Kcal/mol) -5.37 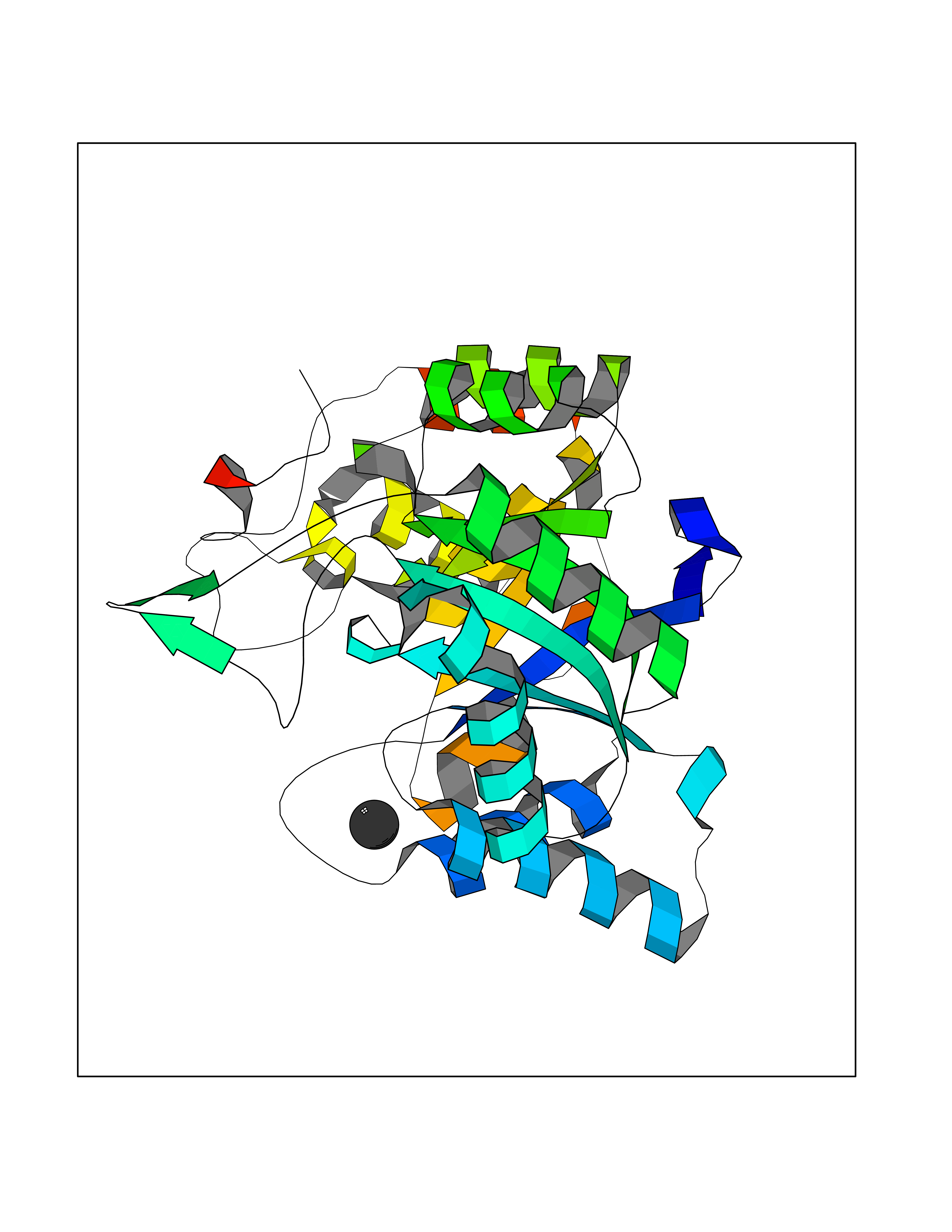 Molscript Map 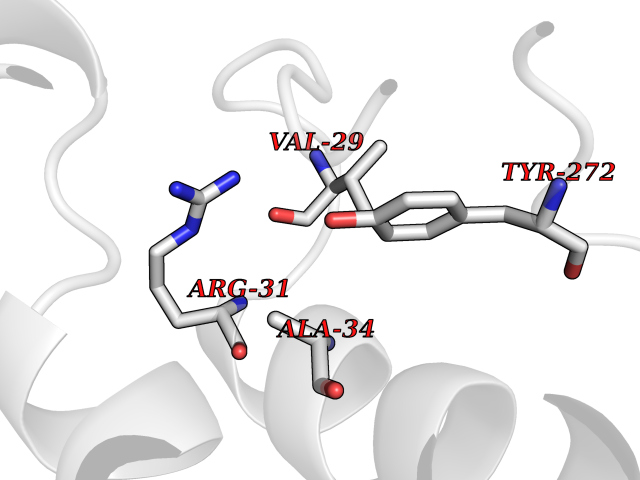 Pymol Map 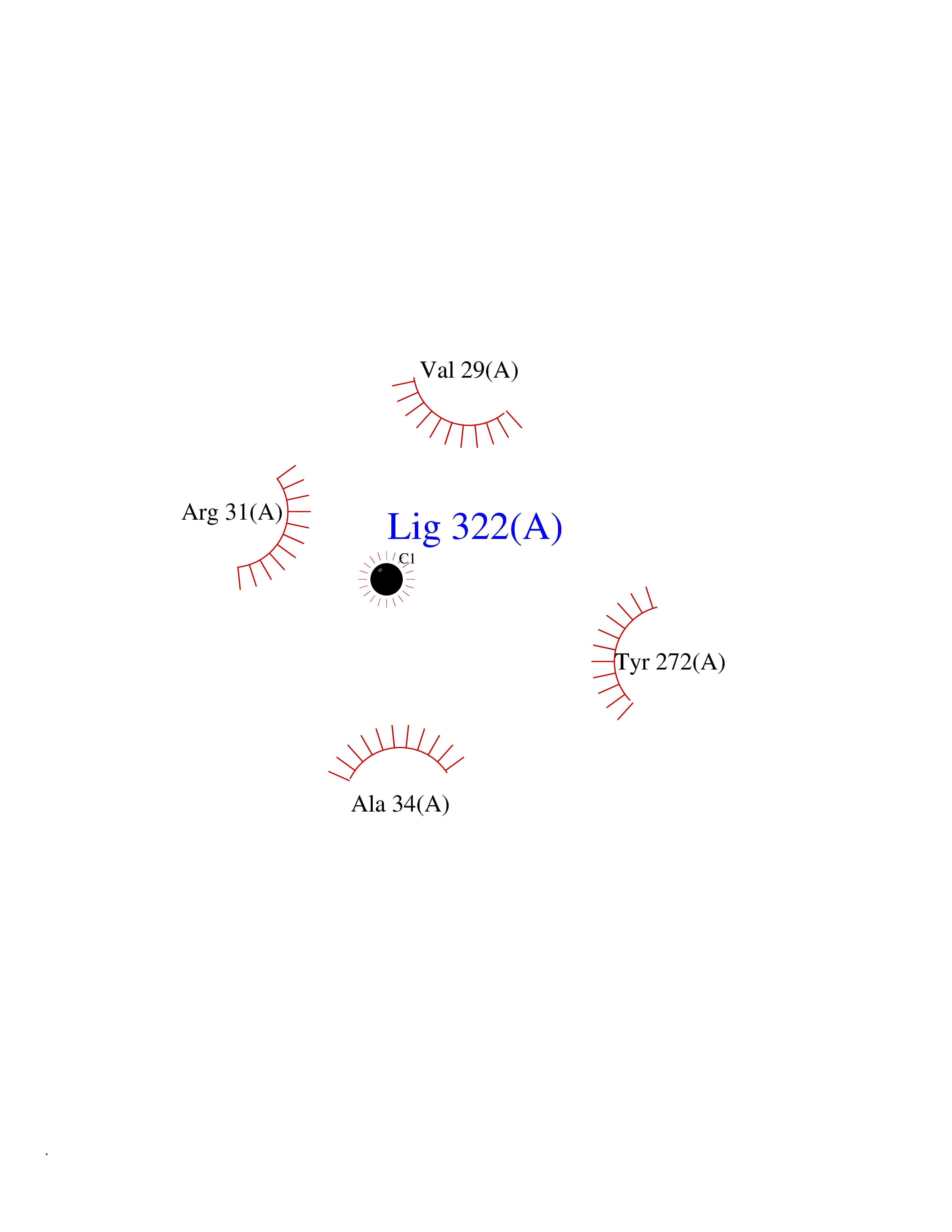 Ligplot Map 3D Binding mode Sequence QCVKLNDGHFMPVLGFGTYAPPEVPRSKALEVTKLAIEAGFRHIDSAHLYNNEEQVGLAIRSKIADGSVKREDIFYTSKLWSTFHRPELVRPALENSLKKAQLDYVDLYLIHSPMSLKPGEELSPTDENGKVIFDIVDLCTTWEAMEKCKDAGLAKSIGVSNFNRRQLEMILNKPGLKYKPVCNQVECHPYFNRSKLLDFCKSKDIVLVAYSALGSQRDKRWVDPNSPVLLEDPVLCALAKKHKRTPALIALRYQLQRGVVVLAKSYNEQRIRQNVQVFEFQLTAEDMKAIDGLDRNLHYFNSDSFASHPNYPYS Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Monomeric sarcosine oxidase | 2GF3 | 3.94 | |
Target general information Gen name soxA Organism Bacillus sp. (strain B-0618) Uniprot ID TTD ID NA Synonyms sox Protein family MSOX/MTOX family, MSOX subfamily Biochemical class Oxidoreductase Function Sarcosine oxidase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB03098; DB01918; DB03517; DB03147; DB03366; DB02083; DB02543 Interacts with NA EC number 1.5.3.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42606.4 Length 385 Aromaticity 0.1 Instability index 26.97 Isoelectric point 5.27 Charge (pH=7) -17.18 2D Binding mode Binding energy (Kcal/mol) -5.38  Molscript Map 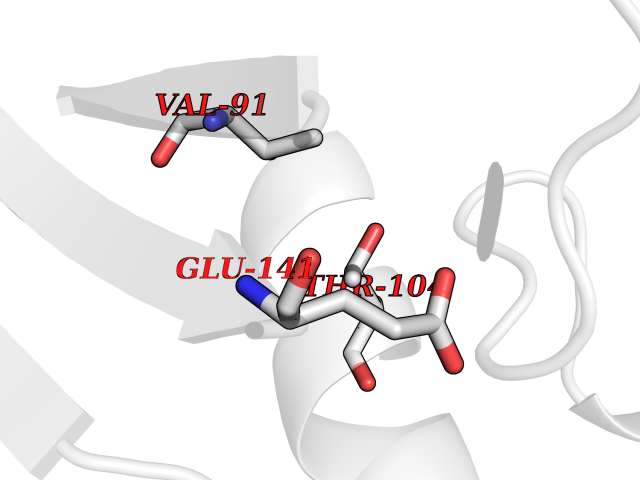 Pymol Map 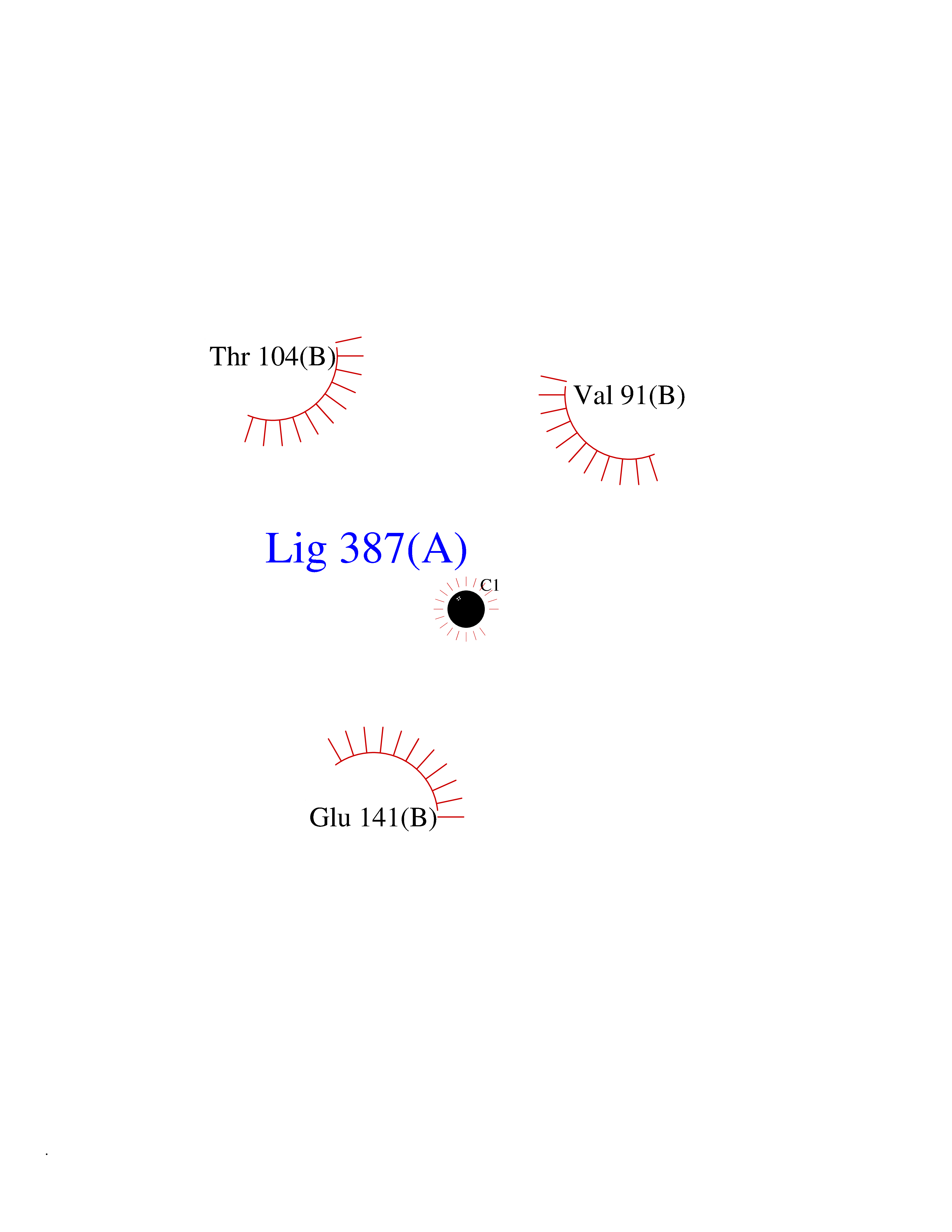 Ligplot Map 3D Binding mode Sequence STHFDVIVVGAGSMGMAAGYQLAKQGVKTLLVDAFDPPHTNGSHHGDTRIIRHAYGEGREYVPLALRSQELWYELEKETHHKIFTKTGVLVFGPKGESAFVAETMEAAKEHSLTVDLLEGDEINKRWPGITVPENYNAIFEPNSGVLFSENCIRAYRELAEARGAKVLTHTRVEDFDISPDSVKIETANGSYTADKLIVSMGAWNSKLLSKLNLDIPLQPYRQVVGFFESDESKYSNDIDFPGFMVEVPNGIYYGFPSFGGCGLKLGYHTFGQKIDPDTINREFGVYPEDESNLRAFLEEYMPGANGELKRGAVCMYTKTLDEHFIIDLHPEHSNVVIAAGFSGHGFKFSSGVGEVLSQLALTGKTEHDISIFSINRPALKESLQ Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Beta-xylanase | 2D22 | 3.94 | |
Target general information Gen name N/A Organism Streptomyces olivaceoviridis (Streptomyces corchorusii) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 10 (cellulase F) family Biochemical class Hydrolase Function Endo-1,4-beta-xylanase activity. Related diseases Mitochondrial DNA depletion syndrome 8A (MTDPS8A) [MIM:612075]: A disorder due to mitochondrial dysfunction characterized by various combinations of neonatal hypotonia, neurological deterioration, respiratory distress, lactic acidosis, and renal tubulopathy. {ECO:0000269|PubMed:17486094, ECO:0000269|PubMed:18504129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mitochondrial DNA depletion syndrome 8B (MTDPS8B) [MIM:612075]: A disease due to mitochondrial dysfunction and characterized by ophthalmoplegia, ptosis, gastrointestinal dysmotility, cachexia, peripheral neuropathy. {ECO:0000269|PubMed:19667227}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Progressive external ophthalmoplegia with mitochondrial DNA deletions, autosomal dominant, 5 (PEOA5) [MIM:613077]: A disorder characterized by progressive weakness of ocular muscles and levator muscle of the upper eyelid. In a minority of cases, it is associated with skeletal myopathy, which predominantly involves axial or proximal muscles and which causes abnormal fatigability and even permanent muscle weakness. Ragged-red fibers and atrophy are found on muscle biopsy. A large proportion of chronic ophthalmoplegias are associated with other symptoms, leading to a multisystemic pattern of this disease. Additional symptoms are variable, and may include cataracts, hearing loss, sensory axonal neuropathy, ataxia, depression, hypogonadism, and parkinsonism. {ECO:0000269|PubMed:19664747}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rod-cone dystrophy, sensorineural deafness, and Fanconi-type renal dysfunction (RCDFRD) [MIM:268315]: An autosomal recessive disease characterized by visual impairment due to rod-cone dystrophy, sensorineural hearing loss, and Fanconi-type renal dysfunction resulting in rickets-like skeletal changes. Death may occur in childhood or young adulthood due to renal failure. Disease onset is before age 5 years. {ECO:0000269|PubMed:32827185}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03389; DB02379; DB04465 Interacts with NA EC number 3.2.1.8 Uniprot keywords 3D-structure; Carbohydrate metabolism; Glycosidase; Hydrolase; Polysaccharide degradation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32170.2 Length 295 Aromaticity 0.11 Instability index 29.09 Isoelectric point 6.66 Charge (pH=7) -0.7 2D Binding mode Binding energy (Kcal/mol) -5.37 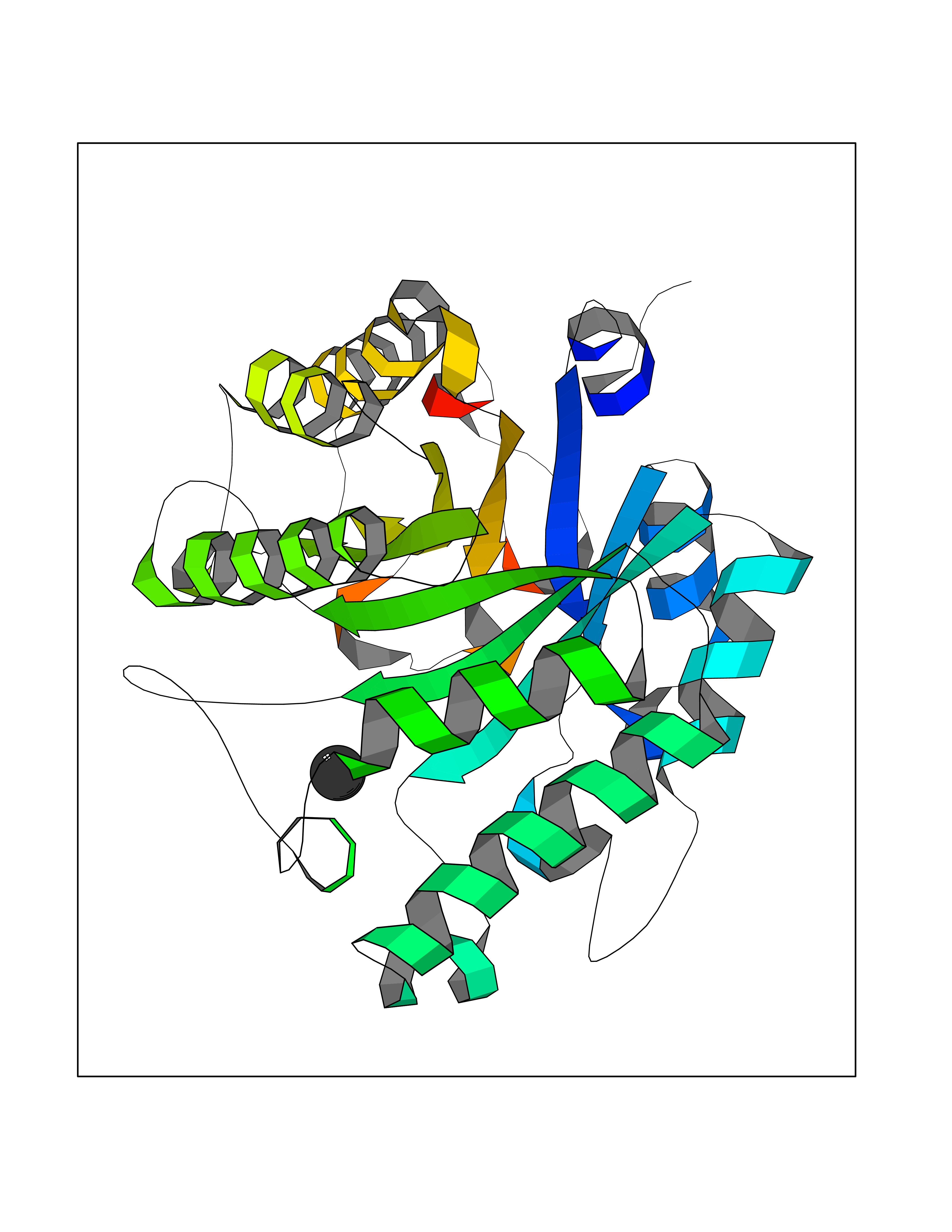 Molscript Map 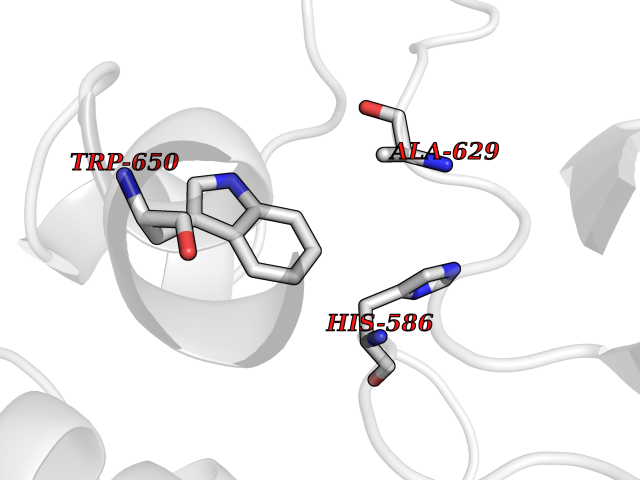 Pymol Map 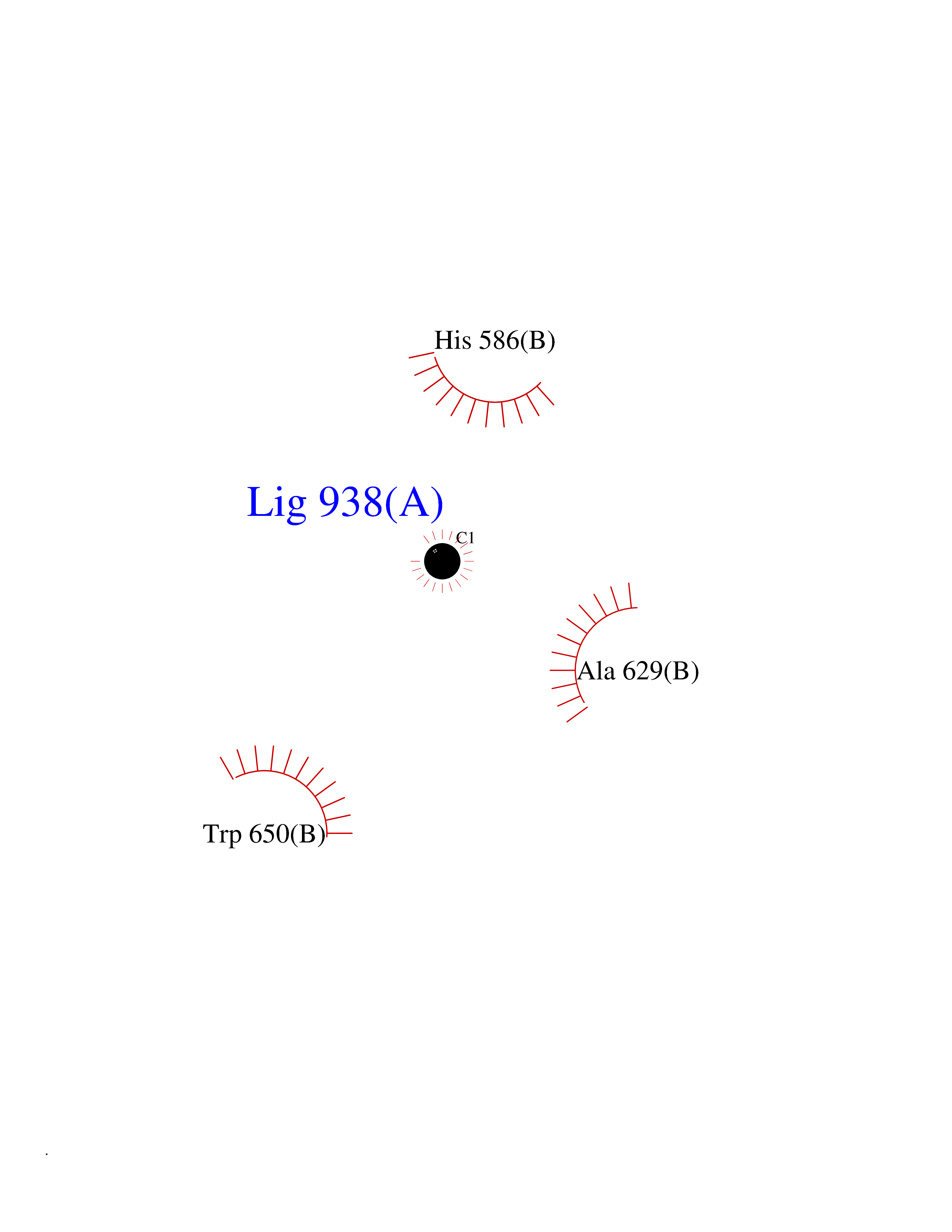 Ligplot Map 3D Binding mode Sequence AESTLGAAAAQSGRYFGTAIASGKLGDSAYTTIASREFNMVTAENEMKIDATEPQRGQFNFSAGDRVYNWAVQNGKQVRGHTLAWHSQQPGWMQSLSGSTLRQAMIDHINGVMGHYKGKIAQWDVVSHAFSDDGSGGRRDSNLQRTGNDWIEVAFRTARAADPAAKLCYNDYNIENWTWAKTQGVYNMVRDFKQRGVPIDCVGFQSHFNSGSPYNSNFRTTLQNFAALGVDVAITELDIQGASSSTYAAVTNDCLAVSRCLGITVWGVRDTDSWRSGDTPLLFNGDGSKKAAYTA Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | L-xylulose reductase | 3D3W | 3.94 | |
Target general information Gen name DCXR Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms SDR20C1 Protein family Short-chain dehydrogenases/reductases (SDR) family Biochemical class Oxidoreductase Function Identical protein binding.L-xylulose reductase (NADP+) activity.Oxidoreductase activity, acting on NAD(P)H, quinone or similar compound as acceptor. Related diseases Pentosuria (PNTSU) [MIM:260800]: An inborn error of metabolism characterized by excessive urinary excretion of L-xylulose. {ECO:0000269|PubMed:11882650, ECO:0000269|PubMed:22042873, ECO:0000269|PubMed:4392213}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02831; DB03461 Interacts with Q7Z4W1; Q9NS18 EC number 1.1.1.10 Uniprot keywords 3D-structure; Acetylation; Carbohydrate metabolism; Direct protein sequencing; Glucose metabolism; Membrane; Methylation; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Xylose metabolism Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25809.6 Length 244 Aromaticity 0.05 Instability index 27.42 Isoelectric point 8.41 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -5.37 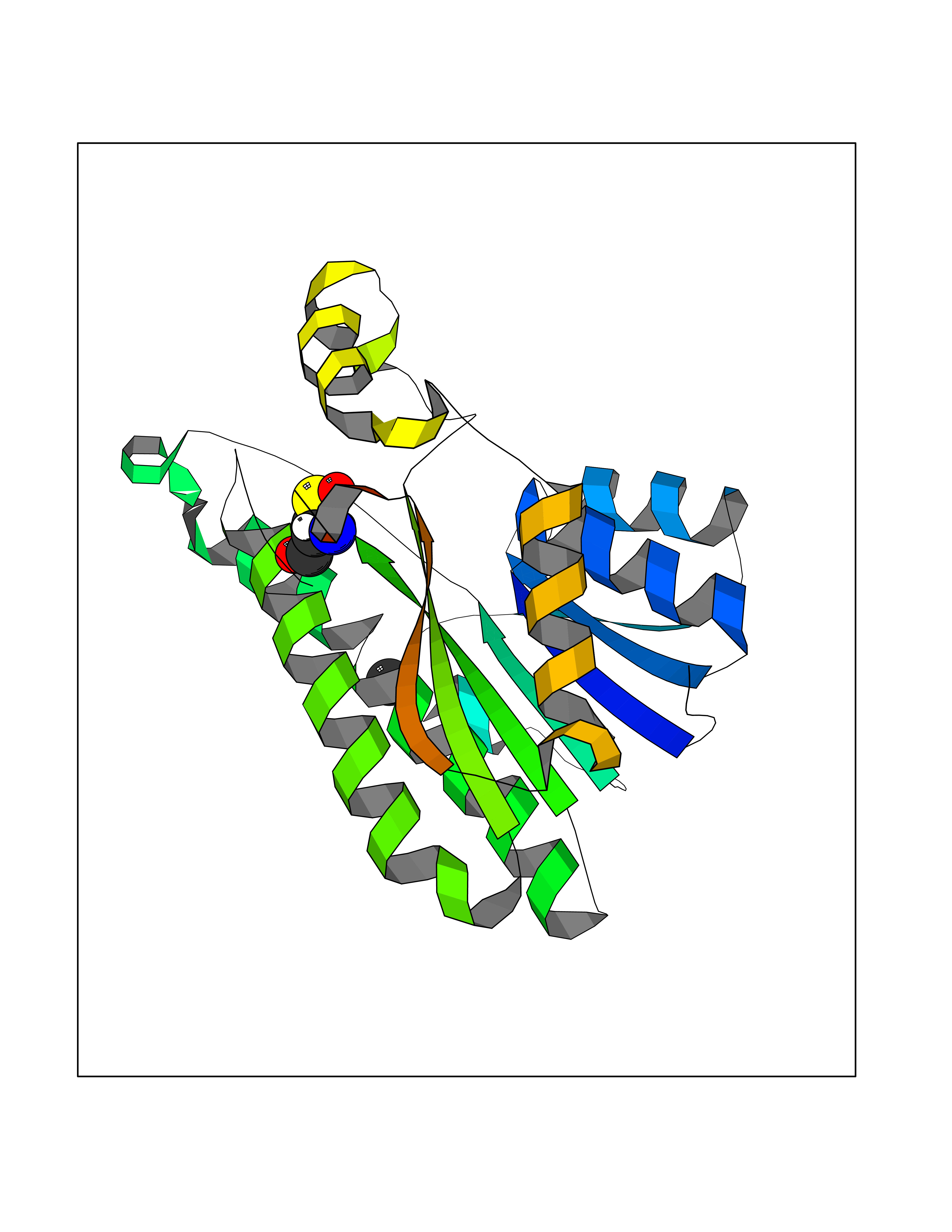 Molscript Map 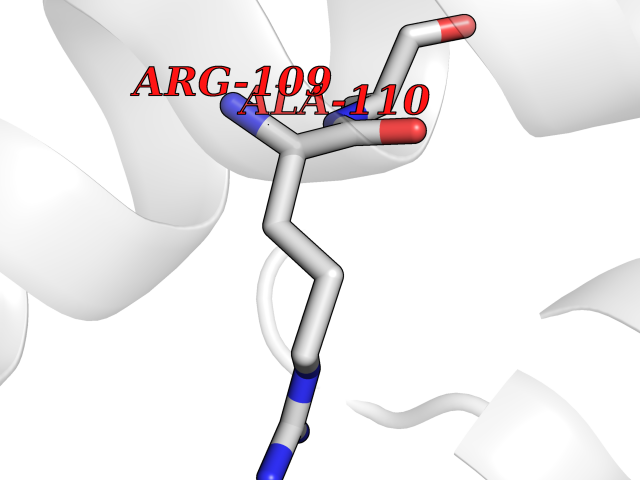 Pymol Map 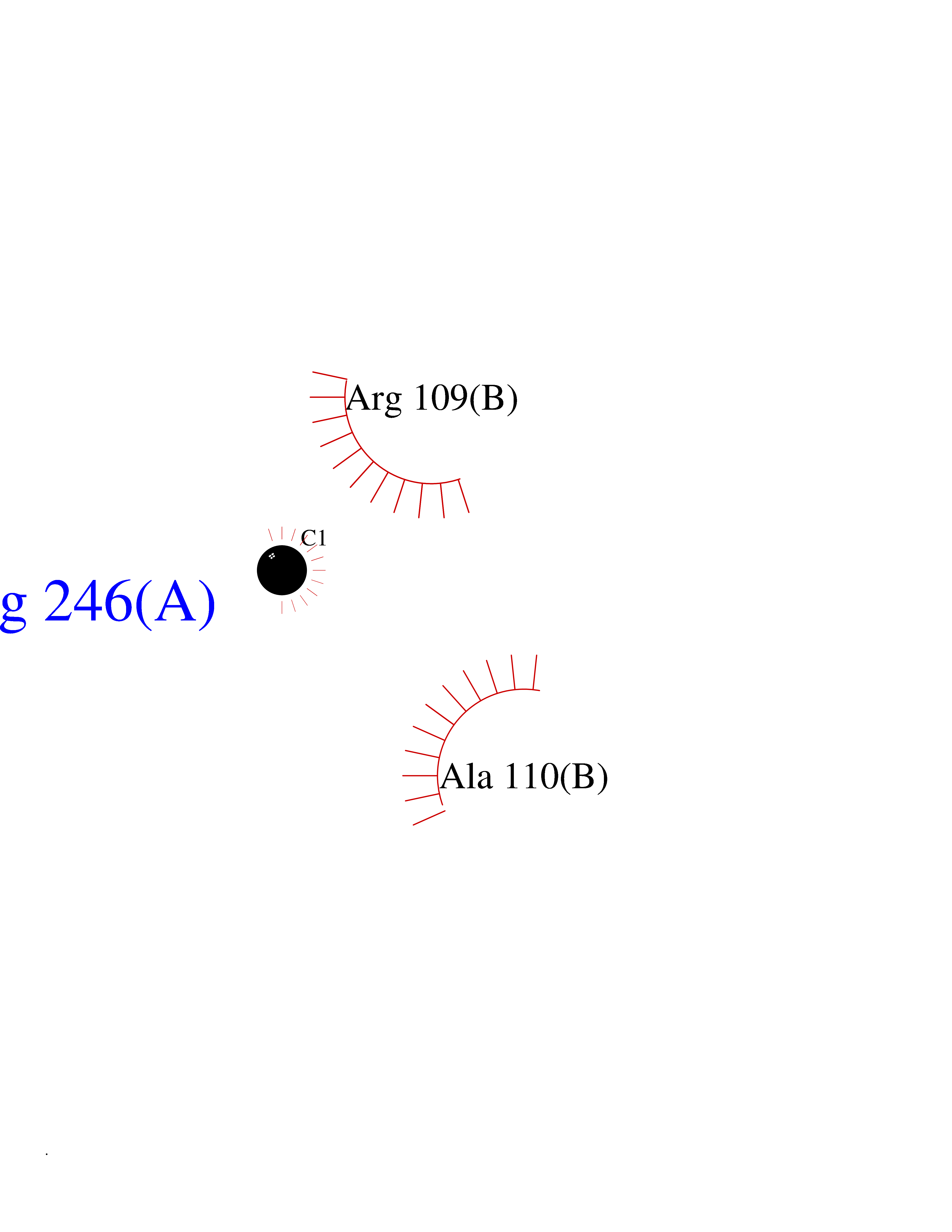 Ligplot Map 3D Binding mode Sequence MELFLAGRRVLVTGAGKGIGRGTVQALHATGARVVAVSRTQADLDSLVRECPGIEPVCVDLGDWEATERALGSVGPVDLLVNNAAVALLQPFLEVTKEAFDRSFEVNLRAVIQVSQIVARGLIARGVPGAIVNVSSQXSQRAVTNHSVYCSTKGALDMLTKVMALELGPHKIRVNAVNPTVVMTSMGQATWSDPHKAKTMLNRIPLGKFAEVEHVVNAILFLLSDRSGMTTGSTLPVEGGFWAC Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | SEC14-like protein 4 | 4TLG | 3.94 | |
Target general information Gen name SEC14L4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TAP3 Protein family NA Biochemical class Transport protein Function Lipid binding.Transporter activity. Related diseases Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia (CDP-PBHM) [MIM:300863]: A disease characterized by chondrodysplasia, severe platyspondyly, hydrocephaly, and facial features with microphthalmia. Bone abnormalities include a distinctive metaphyseal cupping of the metacarpals, metatarsals, and phalanges. Affected females show a milder phenotype with small stature, sometimes associated with body asymmetry and mild intellectual disability. {ECO:0000269|PubMed:20181727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB11635; DB11251; DB00163 Interacts with Q96LC9; O43186; P78358; Q9NYQ3; Q0VD86; Q15323; O76011; P50221; Q6FHY5; Q02548; P26367; Q9H8W4; Q04864; Q04864-2; Q9UHV2; P15884; P15884-3; Q96N21; Q9BYV2; Q8N6Y0; Q9H0C1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 23947.6 Length 210 Aromaticity 0.1 Instability index 50.84 Isoelectric point 5.55 Charge (pH=7) -3.11 2D Binding mode Binding energy (Kcal/mol) -5.38  Molscript Map  Pymol Map 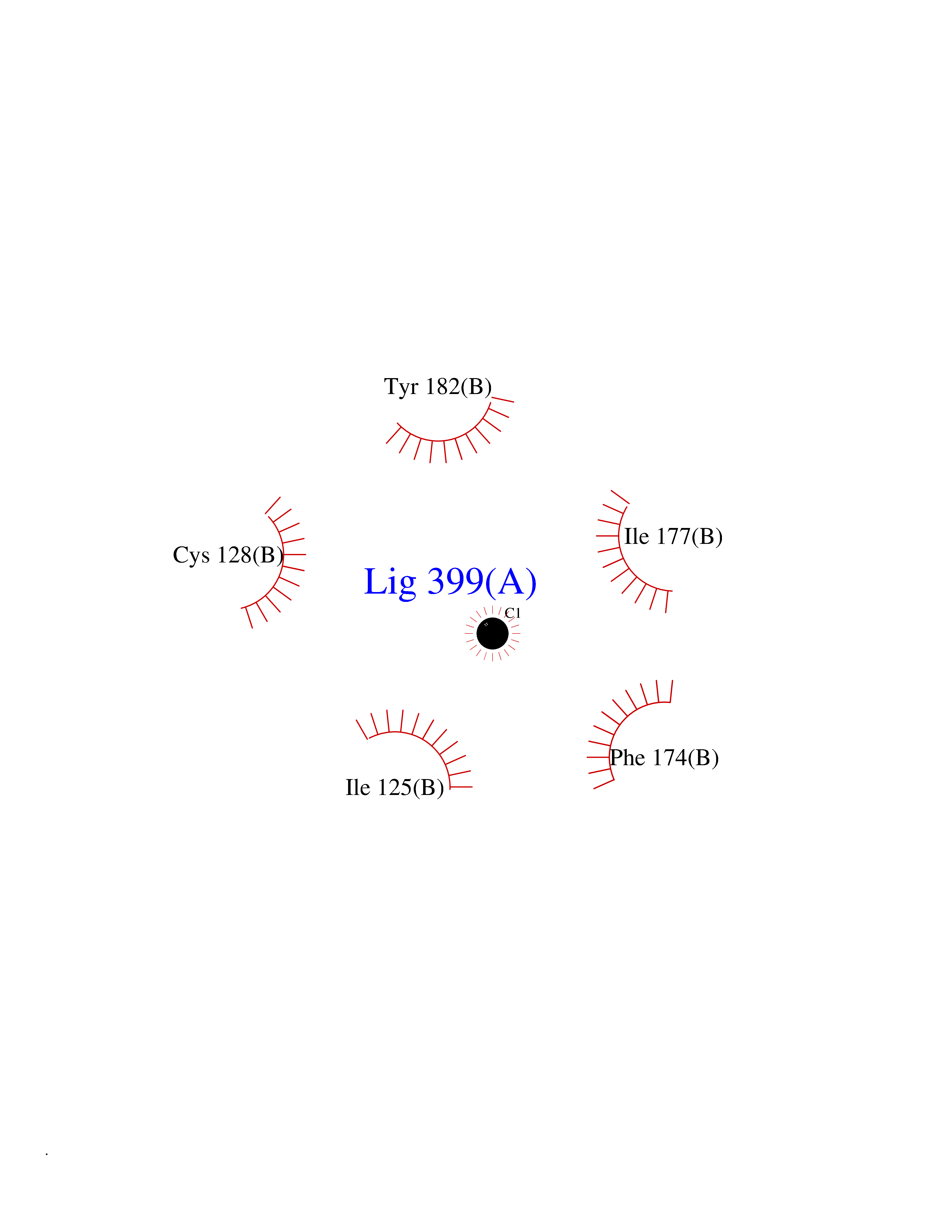 Ligplot Map 3D Binding mode Sequence VTWQPPEVIQLYDSGGLCGYDYEGCPVYFNIIGSLDPKGLLLSASKQDMIRKRIKVCELLLHECELQTQKLGRKIEMALMVFDMEGLSLKHLWKPAVEVYQQFFSILEANYPETLKNLIVIRAPKLFPVAFNLVKSFMSEETRRKIVILGDNWKQELTKFISPDQLPVEFGGTMTDPDGNPKCLTKINYGGEVPKSYYPDKASEETLQSM Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Notch-1 receptor (NOTCH1) | 5L0R | 3.94 | |
Target general information Gen name NOTCH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hN1; Translocationassociated notch protein TAN1; Translocation-associated notch protein TAN-1; TAN1; Notch 1 intracellular domain; Notch 1; Neurogenic locus notch homolog protein 1; NICD Protein family NOTCH family Biochemical class Notch protein Function Upon ligand activation through the released notch intracellular domain (NICD) it forms a transcriptional activator complex with RBPJ/RBPSUH and activates genes of the enhancer of split locus. Affects the implementation of differentiation, proliferation and apoptotic programs. Involved in angiogenesis; negatively regulates endothelial cell proliferation and migration and angiogenic sprouting. Involved in the maturation of both CD4(+) and CD8(+) cells in the thymus. Important for follicular differentiation and possibly cell fate selection within the follicle. During cerebellar development, functions as a receptor for neuronal DNER and is involved in the differentiation of Bergmann glia. Represses neuronal and myogenic differentiation. May play an essential role in postimplantation development, probably in some aspect of cell specification and/or differentiation. May be involved in mesoderm development, somite formation and neurogenesis. May enhance HIF1A function by sequestering HIF1AN away from HIF1A. Required for the THBS4 function in regulating protective astrogenesis from the subventricular zone (SVZ) niche after injury. Involved in determination of left/right symmetry by modulating the balance between motile and immotile (sensory) cilia at the left-right organiser (LRO). Functions as a receptor for membrane-bound ligands Jagged-1 (JAG1), Jagged-2 (JAG2) and Delta-1 (DLL1) to regulate cell-fate determination. Related diseases Aortic valve disease 1 (AOVD1) [MIM:109730]: A common defect in the aortic valve in which two rather than three leaflets are present. It is often associated with aortic valve calcification, stenosis and insufficiency. In extreme cases, the blood flow may be so restricted that the left ventricle fails to grow, resulting in hypoplastic left heart syndrome. {ECO:0000269|PubMed:16025100}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Adams-Oliver syndrome 5 (AOS5) [MIM:616028]: A form of Adams-Oliver syndrome, a disorder characterized by the congenital absence of skin (aplasia cutis congenita) in combination with transverse limb defects. Aplasia cutis congenita can be located anywhere on the body, but in the vast majority of the cases, it is present on the posterior parietal region where it is often associated with an underlying defect of the parietal bones. Limb abnormalities are typically limb truncation defects affecting the distal phalanges or entire digits (true ectrodactyly). Only rarely, metatarsals/metacarpals or more proximal limb structures are also affected. Apart from transverse limb defects, syndactyly, most commonly of second and third toes, can also be observed. The clinical features are highly variable and can also include cardiovascular malformations, brain abnormalities and vascular defects such as cutis marmorata and dilated scalp veins. {ECO:0000269|PubMed:25132448}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q13315; Q969H0; Q16665; P78504; O60341; Q8N423; Q92585; P19838; P46531; Q13153; Q13526; Q06330; Q06330-6; Q13573; P98170; Q8IZL2; Q96JK9 EC number NA Uniprot keywords 3D-structure; Activator; Angiogenesis; ANK repeat; Calcium; Cell membrane; Developmental protein; Differentiation; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Endosome; Glycoprotein; Hydroxylation; Isopeptide bond; Membrane; Metal-binding; Notch signaling pathway; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transcription; Transcription regulation; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 46121.2 Length 394 Aromaticity 0.13 Instability index 39.95 Isoelectric point 7.22 Charge (pH=7) 0.63 2D Binding mode Binding energy (Kcal/mol) -5.37 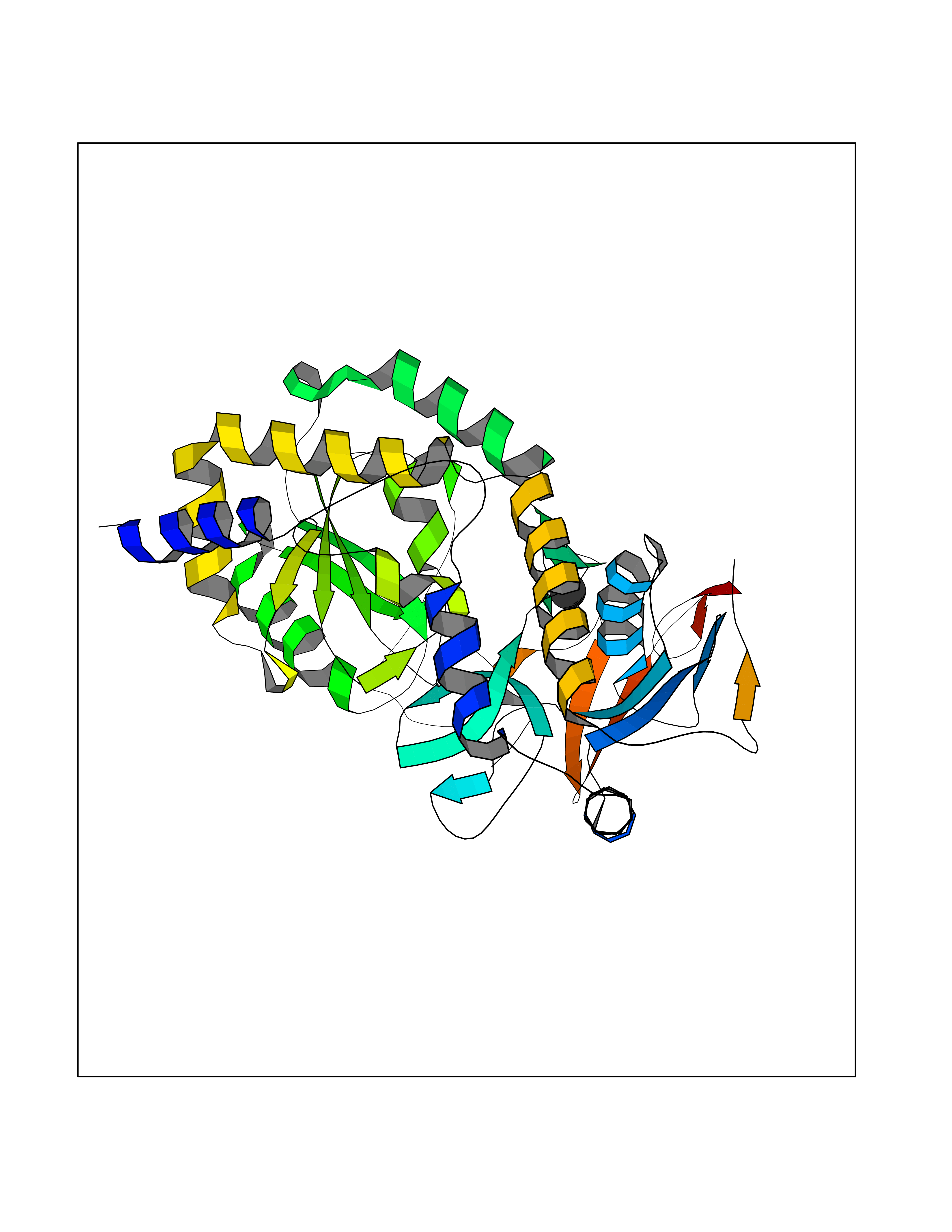 Molscript Map 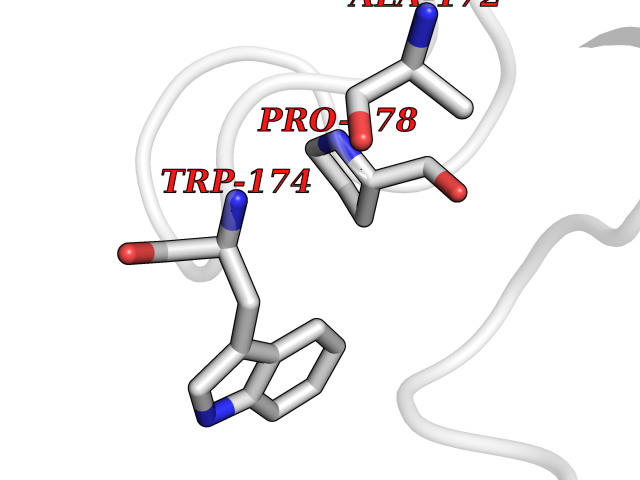 Pymol Map 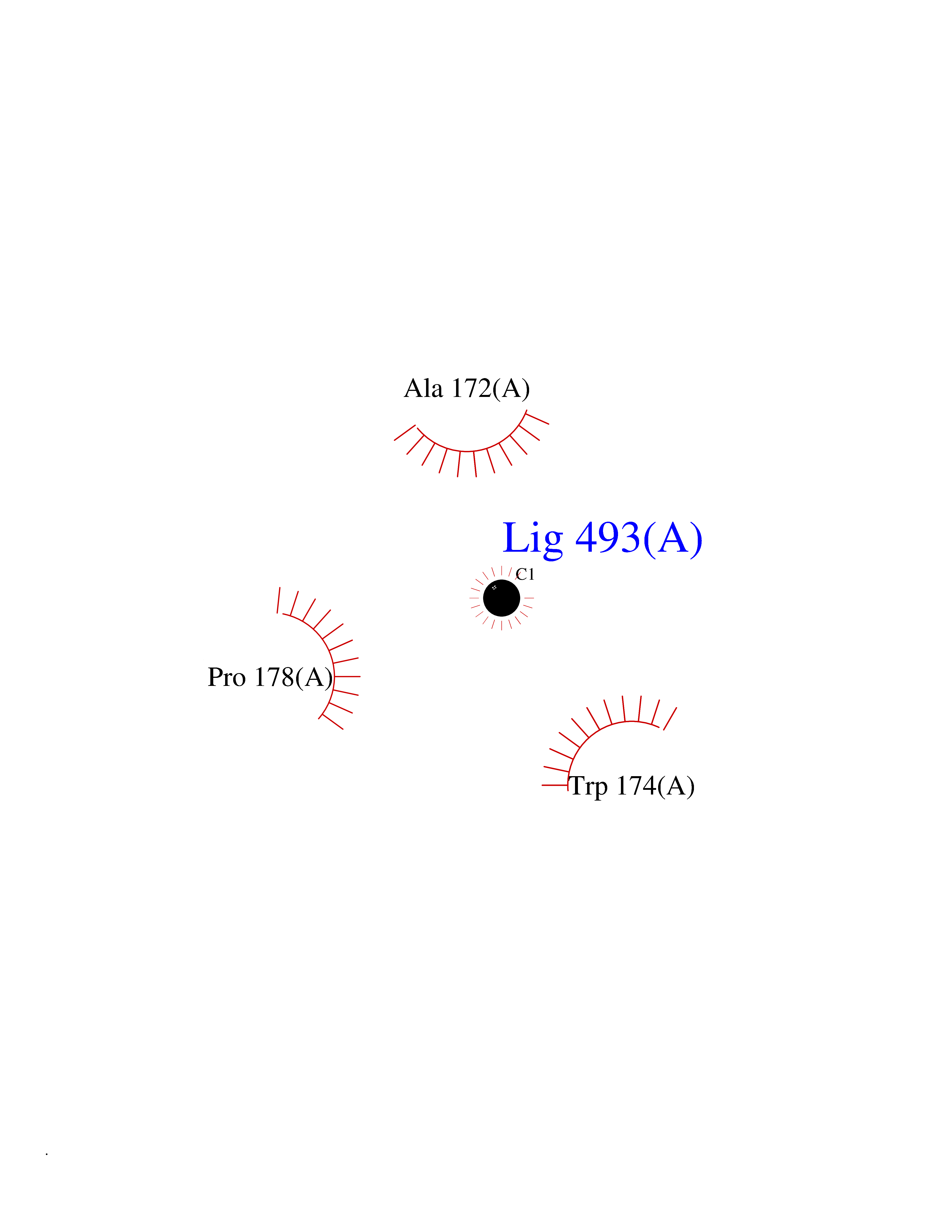 Ligplot Map 3D Binding mode Sequence KWKVFIDQINRSLENYEPCSSQNCSCYHGVIEEDLTPFRGGISRKMMAEVVRRKLGTHYQITKNRLYRENDCMFPSRCSGVEHFILEVIGRLPDMEMVINVRDYPQVPKWMEPAIPVFSFSKTSEYHDIMYPAWTFWEGGPAVWPIYPTGLGRWDLFREDLVRSAAQWPWKKKNSTAYFRGSRTSPERDPLILLSRKNPKLVDAEYTKNQAWKSMKDTLGKPAAKDVHLVDHCKYKYLFNFRGVAASFRFKHLFLCGSLVFHVGDEWLEFFYPQLKPWVHYIPVKTDLSNVQELLQFVKANDDVAQEIAERGSQFIRNHLQMDDITCYWENLLSEYSKFLSYNVTRRKGYDQIIPVNECVSNPCQNDATCLDQIGEFQCICMPGYEGVHCEVNT Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | SET and MYND domain-containing protein 2 (SMYD2) | 5ARF | 3.94 | |
Target general information Gen name SMYD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nlysine methyltransferase SMYD2; N-lysine methyltransferase SMYD2; Lysine Nmethyltransferase 3C; Lysine N-methyltransferase 3C; KMT3C; Histone methyltransferase SMYD2; HSKMB; HSKM-B Protein family Class V-like SAM-binding methyltransferase superfamily Biochemical class Methyltransferase Function Specifically methylates histone H3 'Lys-4' (H3K4me) and dimethylates histone H3 'Lys-36' (H3K36me2). Shows even higher methyltransferase activity on p53/TP53. Monomethylates 'Lys-370' of p53/TP53, leading to decreased DNA-binding activity and subsequent transcriptional regulation activity of p53/TP53. Monomethylates RB1 at 'Lys-860'. Protein-lysine N-methyltransferase that methylates both histones and non-histone proteins, including p53/TP53 and RB1. Related diseases Pulmonary hypertension, primary, 4 (PPH4) [MIM:615344]: A rare disorder characterized by plexiform lesions of proliferating endothelial cells in pulmonary arterioles. The lesions lead to elevated pulmonary arterial pression, right ventricular failure, and death. The disease can occur from infancy throughout life and it has a mean age at onset of 36 years. Penetrance is reduced. Although familial pulmonary hypertension is rare, cases secondary to known etiologies are more common and include those associated with the appetite-suppressant drugs. {ECO:0000269|PubMed:23883380}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in this gene may cause developmental delay with sleep apnea (DDSA). A disorder characterized by developmental neurologic, skeletal and respiratory anomalies including microcephaly, arthrogryposis, scoliosis, cleft palate, facial dysmorphology, bilateral talipes, feeding difficulties and central and/or obstructive sleep apnea. Malformations are detected as early as 21 weeks post gestation. Severely affected patients require ongoing treatment with nocturnal O2 or pressure-controlled ventilation. The disease is associated with recurrent de novo gain of function variants. {ECO:0000269|PubMed:36195757}. Drugs (DrugBank ID) NA Interacts with P20290-2; Q96K17; Q9UPZ9; Q9Y5W9; P04637 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Cytoplasm; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 48789.6 Length 425 Aromaticity 0.1 Instability index 51.93 Isoelectric point 6.35 Charge (pH=7) -4.02 2D Binding mode Binding energy (Kcal/mol) -5.38 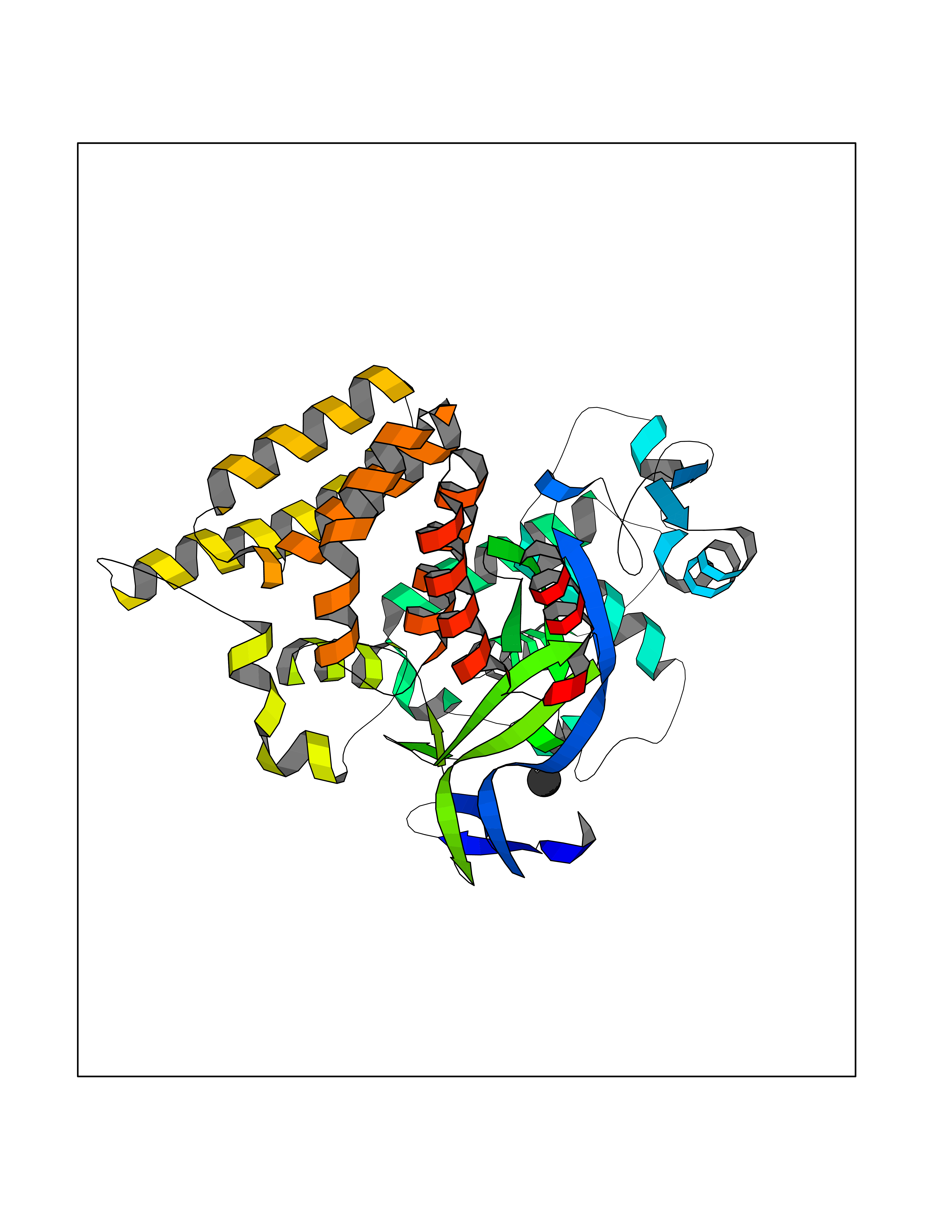 Molscript Map 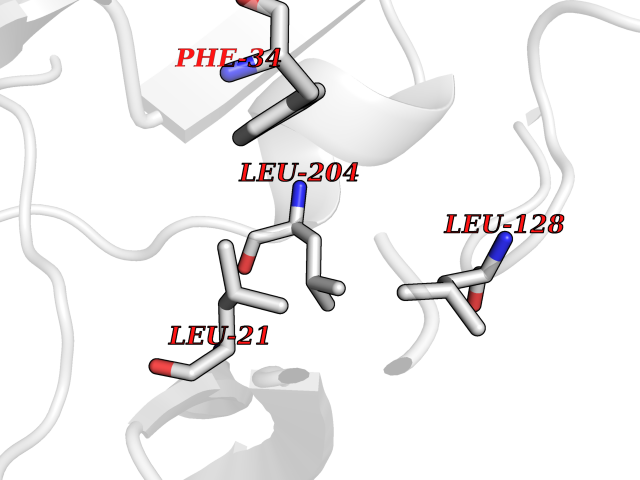 Pymol Map 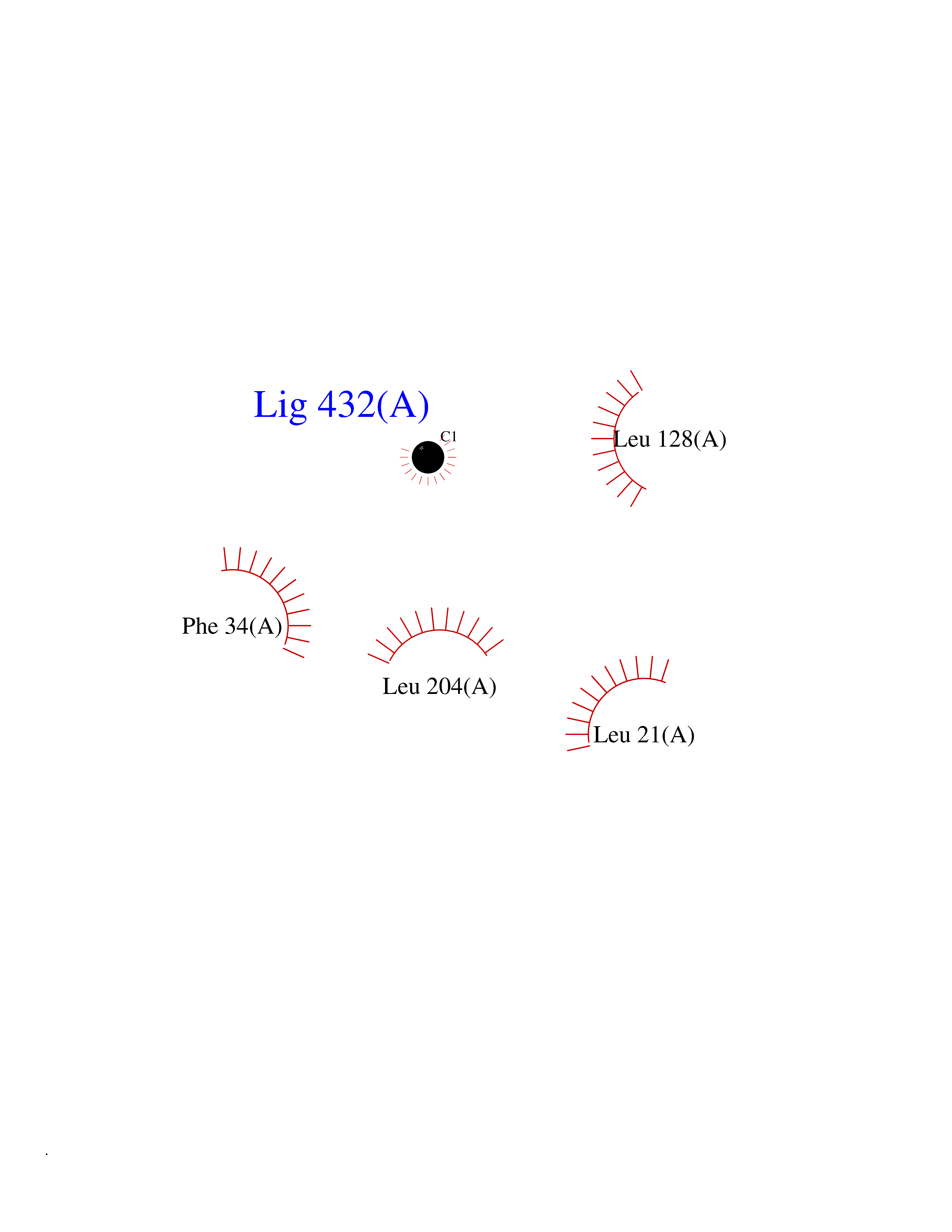 Ligplot Map 3D Binding mode Sequence LGGLERFCSPGKGRGLRALQPFQVGDLLFSCPAYAYVLTVNERGNHCEYCFTRKEGLSKCGRCKQAFYCNVECQKEDWPMHKLECSPMVVFGENWNPSETVRLTARILAKQKIHPERTPSEKLLAVKEFESHLDKLDNEKKDLIQSDIAALHHFYSKHLGFPDNDSLVVLFAQVNCNGFTIEDEELSHLGSAIFPDVALMNHSCCPNVIVTYKGTLAEVRAVQEIKPGEEVFTSYIDLLYPTEDRNDRLRDSYFFTCECQECTTKDKDKAKVEIRKLSDPPKAEAIRDMVRYARNVIEEFRRAKHYKSPSELLEICELSQEKMSSVFEDSNVYMLHMMYQAMGVCLYMQDWEGALQYGQKIIKPYSKHYPLYSLNVASMWLKLGRLYMGLEHKAAGEKALKKAIAIMEVAHGKDHPYISEIKQEI Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Caspase-6 (CASP6) | 4NBL | 3.94 | |
Target general information Gen name CASP6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH2; Caspase-6 subunit p18; Caspase-6 subunit p11; CASP-6; Apoptotic protease Mch-2 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves poly(ADP-ribose) polymerase in vitro, as well as lamins. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9Y614; Q6DHV7-2; Q6UY14-3; Q96MA6; Q5T2L2; Q96Q83-2; Q9Y303-2; Q9NU02; P09525; P06727; Q8WW27; Q66PJ3-4; Q6XD76; P18848; Q9H0Y0; Q14032; P54687-4; P06276; Q9NSI6-4; Q96Q07-2; Q9H0W9-3; Q9NQ89; Q13901; Q3SXR2; Q8N1A6; P17655; P20807-4; P42574; P55212; O00257-3; P24863; Q9NNX6-10; Q9UJX2; P42773; O95674; Q494V2-2; Q8WUX9; Q9Y3D0; Q8N365; Q3SX64; Q99966; P09496-2; Q6PJW8-3; Q96BR5; P02458-1; Q9UGL9; Q9UKG9-2; P26998; P35222; Q53TN4; P61962; O60479; Q96EY1-3; Q92782-2; Q9BPU6; A0AVK6; Q658K8; O00303; Q13347; O00472; O00423; Q6NXG1-3; Q49AJ0-4; Q8N128-2; Q8IZU1; Q6ZNL6; Q9NSA1; Q06547-3; Q49A26-4; Q9HAV0; Q6NXT2; Q9BT25; Q9NRZ9-6; Q96EW2-2; P42858; Q8N6M8-2; Q92613; P0C870; Q9UK76; Q8N5Z5; Q8TBB5-2; Q9UH77; Q8N4N3-2; Q5JUW0-3; Q8N1A0; P13473-2; Q6DKI2; Q9H2C1; Q8N0U6; Q9Y234; Q8TBB1; Q1L5Z9; Q96JB6; Q16609; Q8IYG6; P0DP58-2; Q969L2; P27338; A6NJ78-4; Q96C03-3; Q8N5J2-3; A0A0A0MR05; P34949-2; Q9BV20; Q6IN84-2; A2RUH7; P01106; Q9H7X0; Q15742-2; Q9UJ70-2; Q8NDH3-5; Q96HA8; P36639-4; Q8NFH4; Q8NFH3; Q7Z3B4; Q6N063-2; Q6GQQ9-2; Q9H8K7; Q99447; P27815-4; O15534; Q9BUL5; Q00169; P48739; P61925; Q58EX7-2; O60664; Q14181; P0DPB6; P36954; Q07869; O60927; Q6ZMI0-5; P54619; Q8NCQ7-2; P41222; P29074; Q8WUD1-2; Q5R372-9; Q9HD47-3; Q09028; Q04206; P47804-3; Q15382; Q06587; Q8N5U6; P62701; Q66K80; Q01826; O15126; P22307-3; Q9BRK5; Q9NTN9-3; P01011; Q15393; Q9NR46; Q9BZQ2; O60902-3; Q86US8; P37840; Q96H20; Q13573; Q7Z6I5; Q496A3; Q9C004; Q5W111-2; Q96BD6; Q92797-2; O60506-4; O15273; Q86WV5; Q96A09; P54274-2; P22735; O43548; Q9NQ88; Q9UIK5-2; Q53NU3; P04637; Q12888; P36406; Q86WT6-2; Q13885; P49459; Q9P1Q0-4; Q9NX94; Q8NA23-2; Q9BQA1; O00755; O95070; O43829; Q8IWT0-2; Q53FD0-2; Q05CR2; Q96JL9-2; Q96LX8; Q3KNS6-3; Q5JTY5; A0A384MDV8; B7Z3E8; Q86V28 EC number EC 3.4.22.59 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; Autocatalytic cleavage; Cytoplasm; Hydrolase; Lipoprotein; Nucleus; Palmitate; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 57170 Length 500 Aromaticity 0.12 Instability index 31.33 Isoelectric point 8.05 Charge (pH=7) 4.52 2D Binding mode Binding energy (Kcal/mol) -5.38 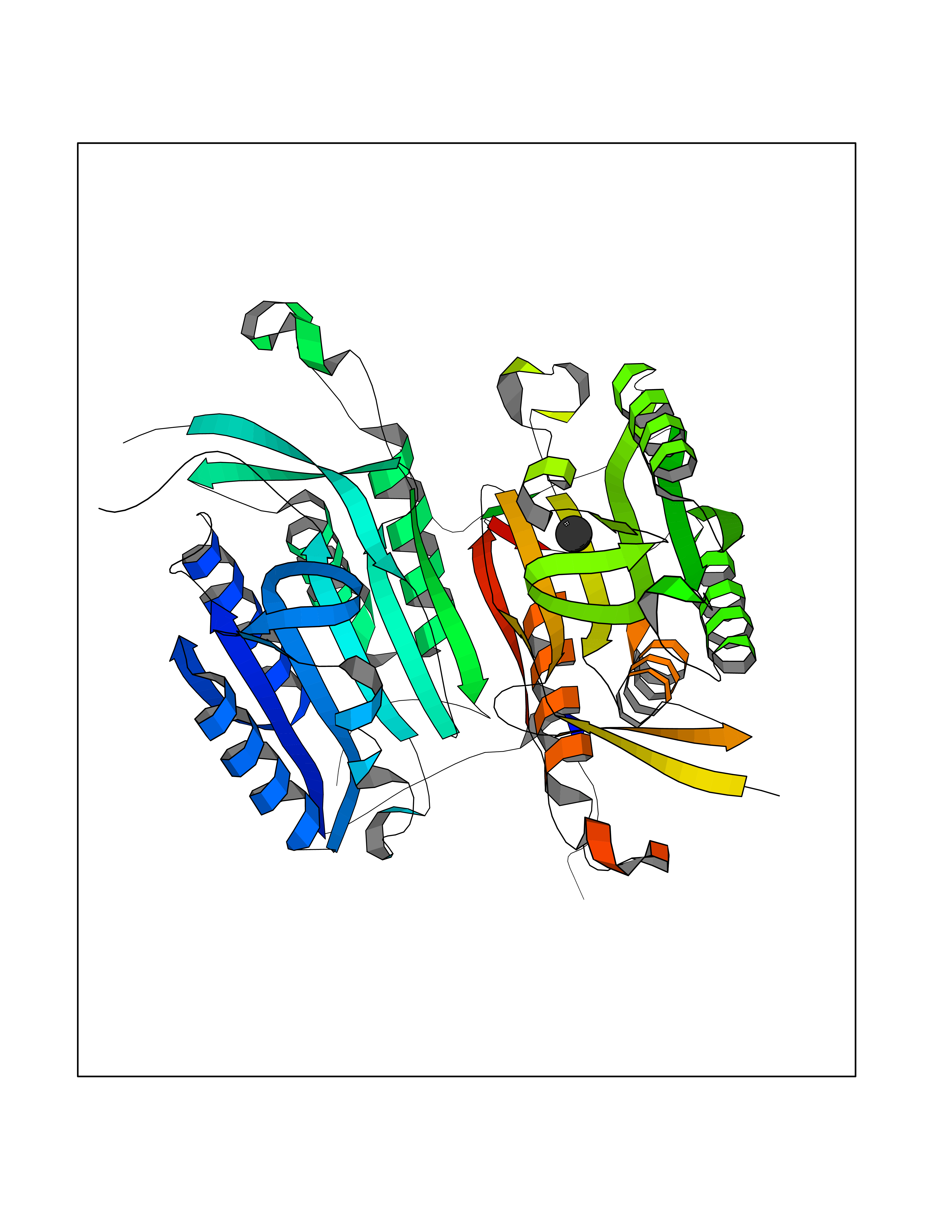 Molscript Map 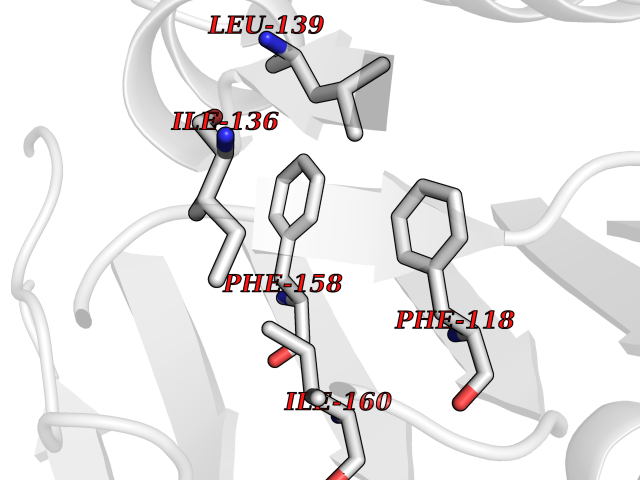 Pymol Map 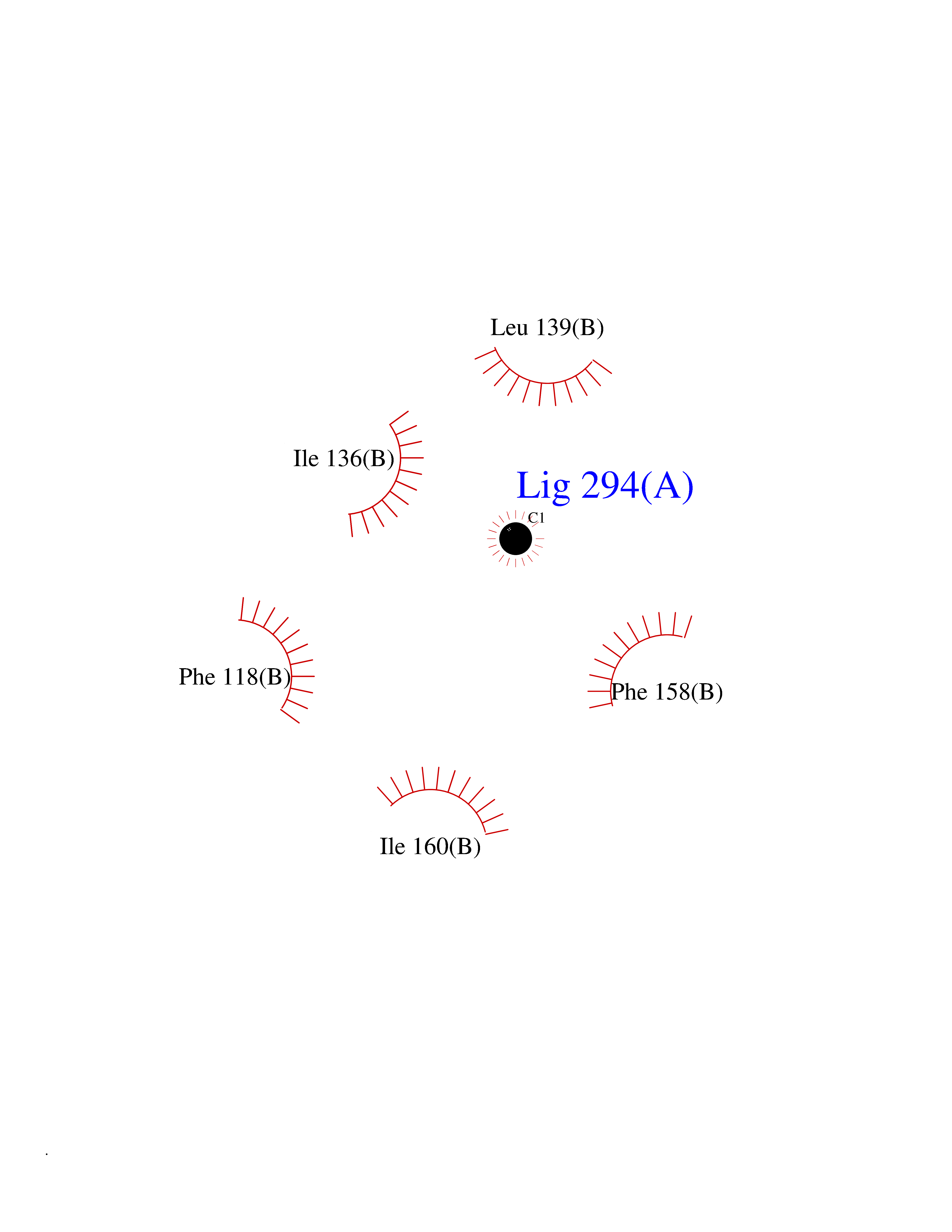 Ligplot Map 3D Binding mode Sequence AFYKREMFDPAEKYKMDHRRRGIALIFNHERFFWHLTLPERRGTCADRDNLTRRFSDLGFEVKCFNDLKAEELLLKIHEVSTVSHADADCFVCVFLSHGEGNHIYAYDAKIEIQTLTGLFKGDKCHSLVGKPKIFIIQAARGNQHDVPVIPDTNITEVDAASVYTLPAGADFLMCYSVAEGYYSHRETVNGSWYIQDLCEMLGKYGSSLEFTELLTLVNRKVSQRRVDFCKDPSAIGKKQVPCFASMLTKKLHFFPKSMFDPAEKYKMDHRRRGIALIFNHERFFWHLTLPERRGTCADRDNLTRRFSDLGFEVKCFNDLKAEELLLKIHEVSTVSHADADCFVCVFLSHGEGNHIYAYDAKIEIQTLTGLFKGDKCHSLVGKPKIFIIQAARGNTNITEVDAASVYTLPAGADFLMCYSVAEGYYSHRETVNGSWYIQDLCEMLGKYGSSLEFTELLTLVNRKVSQRRVDFCKDPSAIGKKQVPCFASMLTKKLHFFPK Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Dipeptidyl peptidase 8 (DPP-8) | 6EOP | 3.94 | |
Target general information Gen name DPP8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Prolyl dipeptidase DPP8; MSTP141; MSTP135; MSTP097; Dipeptidyl peptidase VIII; Dipeptidyl peptidase IV-related protein 1; DPRP1; DPRP-1; DPP VIII; DP8 Protein family Peptidase S9B family, DPPIV subfamily Biochemical class Peptidase Function Dipeptidyl peptidase that cleaves off N-terminal dipeptides from proteins having a Pro or Ala residue at position 2. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.14.5 Uniprot keywords 3D-structure; Alternative splicing; Aminopeptidase; Apoptosis; Cytoplasm; Hydrolase; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A,D Molecular weight (Da) 97764.9 Length 849 Aromaticity 0.12 Instability index 47.71 Isoelectric point 5.69 Charge (pH=7) -21.66 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map 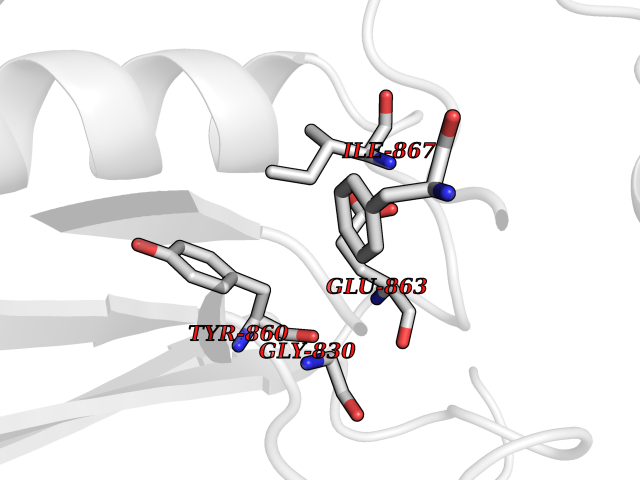 Pymol Map 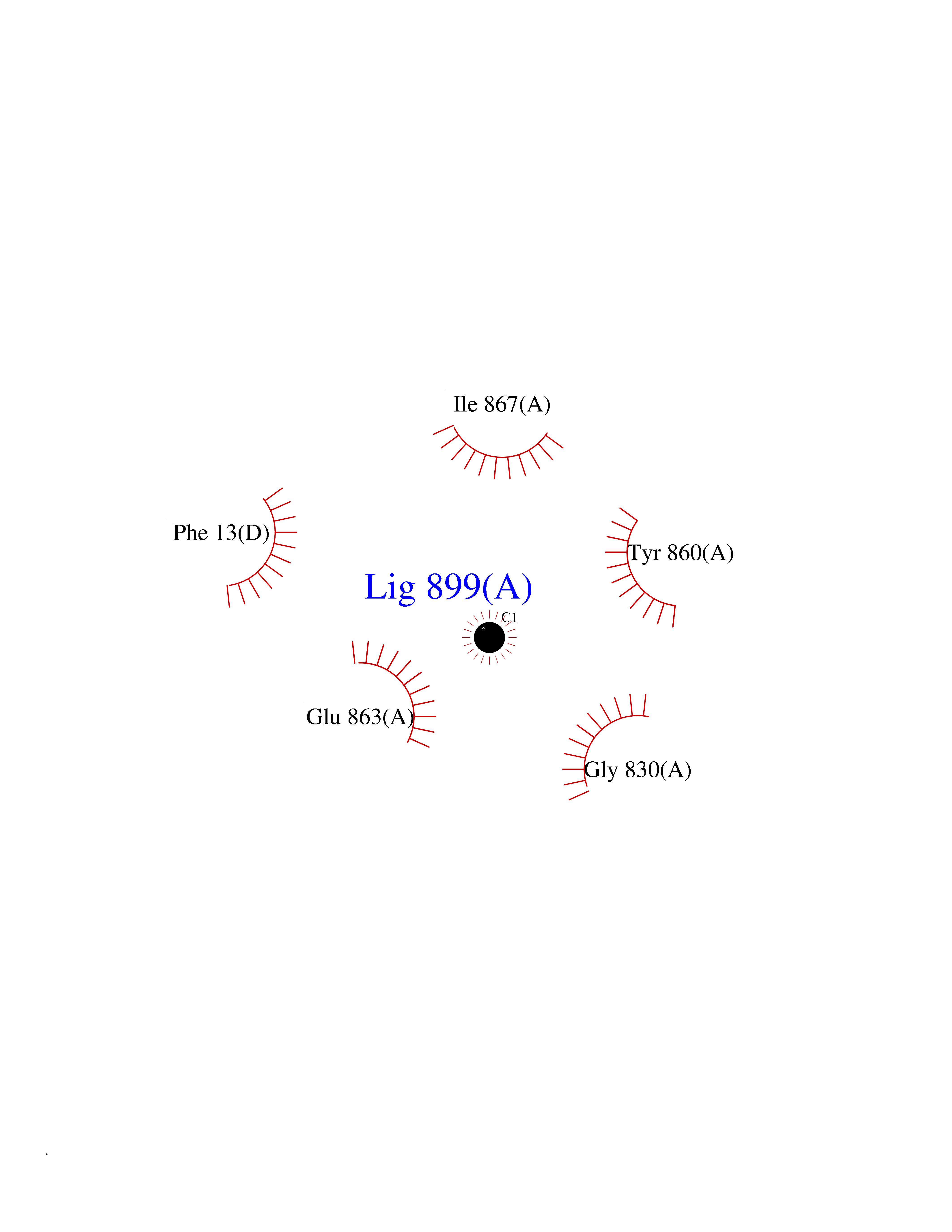 Ligplot Map 3D Binding mode Sequence LEPFYVERYSWSQLKKLLADTRKYHGYMMAKAPHDFMFVKRNDPDGPHSDRIYYLAMSNRENTLFYSEIPKTINRAAVLMLSWKPLLDLFQYSREEELLRERKRIGTVGIASYDYHQGSGTFLFQAGSGIYHVKDGGPQGFTQQPLRPNLVETSCPNIRMDPKLCPADPDWIAFIHSNDIWISNIVTREERRLTYVHNELANMEEDARSAGVATFVLQEEFDRYSGYWWCPKAETTPSGGKILRILYEENDESEVEIIHVTSPMLETRRADSFRYPKTGTANPKVTFKMSEIMIDAEGRIIDVIDKELIQPFEILFEGVEYIARAGWTPEGKYAWSILLDRSQTRLQIVLISPELFIPVEDDVMERQRLIESVPDSVTPLIIYEETTDIWINIHDIFHVFPQSHEEEIEFIFASECKTGFRHLYKITSILKESKYKRSSGGLPAPSDFKCPIKEEIAITSGEWEVLGRHGSNIQVDEVRRLVYFEGTKDSPLEHHLYVVSYVNPGEVTRLTDRGYSHSCCISQHCDFFISKYSNQKNPHCVSLYKLSSPEDDPTCKTKEFWATILDSAGPLPDYTPPEIFSFESTTGFTLYGMLYKPHDLQPGKKYPTVLFIYGGPQVQLVNNRFKGVKYFRLNTLASLGYVVVVIDNRGSXHRGLKFEGAFKYKMGQIEIDDQVEGLQYLASRYDFIDLDRVGIHGWSYGGYLSLMALMQRSDIFRVAIAGAPVTLWIFYDTGYTERYMGHPDQNEQGYYLGSVAMQAEKFPSEPNRLLLLHGFLDENVHFAHTSILLSFLVRAGKPYDLQIYPQERHSIRVPESGEHYELHLLHYLQENLGSRIAALKVSLRFLYEG Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Legumain (LGMN) | 5LU8 | 3.94 | |
Target general information Gen name LGMN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protease, cysteine 1; PRSC1; Asparaginyl endopeptidase Protein family Peptidase C13 family Biochemical class Peptidase Function Can also cleave aspartyl bonds slowly, especially under acidic conditions. Required for normal lysosomal protein degradation in renal proximal tubules. Required for normal degradation of internalized EGFR. Plays a role in the regulation of cell proliferation via its role in EGFR degradation. May be involved in the processing of proteins for MHC class II antigen presentation in the lysosomal/endosomal system. Has a strict specificity for hydrolysis of asparaginyl bonds. Related diseases Lymphatic malformation 8 (LMPHM8) [MIM:618773]: A form of primary lymphedema, a disease characterized by swelling of body parts due to developmental anomalies and functional defects of the lymphatic system. Adult patients with lymphedema may suffer from recurrent local infections. Impaired lymphatic drainage in the fetus can develop into hydrops fetalis, a severe condition characterized by excessive fluid accumulation in more than two fetal extra-vascular compartments and body cavities, placental enlargement and edema, pericardial or pleural effusion, or ascites. LMPHM8 is an autosomal recessive form characterized by onset in utero and fetal death due to non-immune hydrops fetalis. {ECO:0000269|PubMed:30115739}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with PRO_0000006639 [P01034]; PRO_0000006648 [Q15828] EC number EC 3.4.22.34 Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lysosome; Protease; Proteomics identification; Reference proteome; Signal; Thiol protease; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 29967.3 Length 263 Aromaticity 0.11 Instability index 43.86 Isoelectric point 6.04 Charge (pH=7) -6.03 2D Binding mode Binding energy (Kcal/mol) -5.37 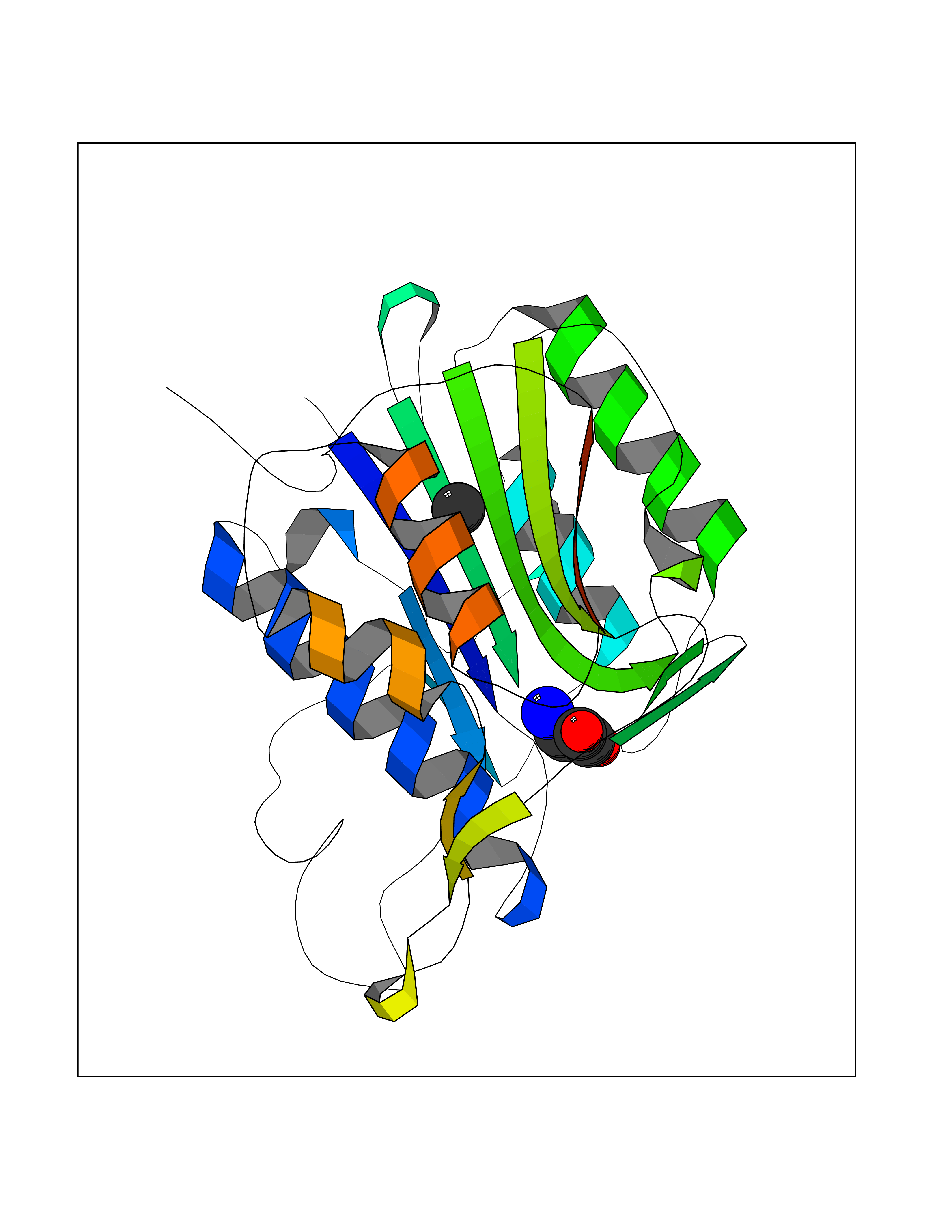 Molscript Map 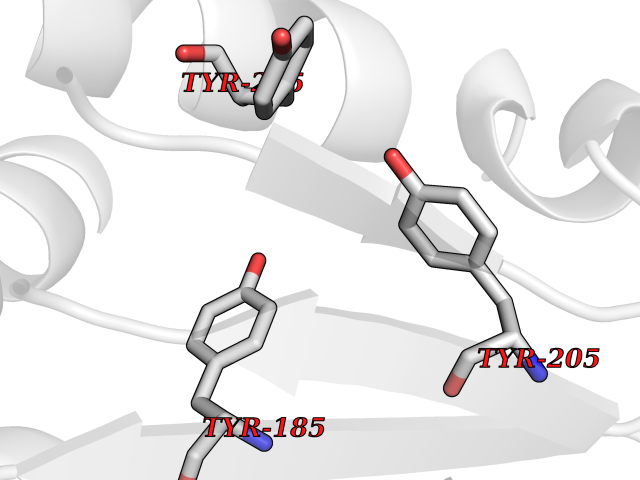 Pymol Map 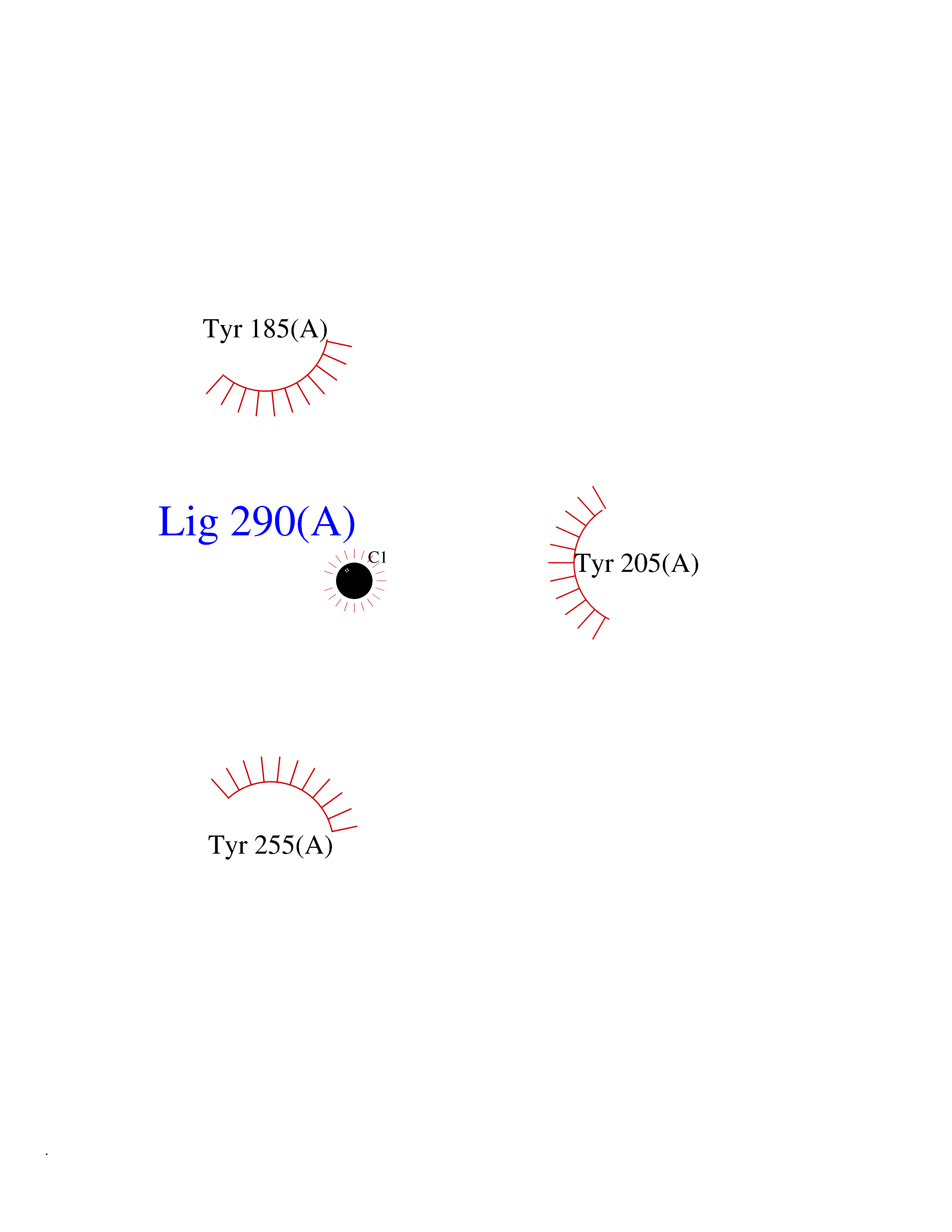 Ligplot Map 3D Binding mode Sequence GGKHWVVIVAGSNGWYNYRHQADACHAYQIIHRNGIPDEQIVVMMYDDIAYSEDNPTPGIVINRPNGTDVYQGVPKDYTGEDVTPQNFLAVLRGDAEAVKGIGSGKVLKSGPQDHVFIYFTXHGSTGILVFPNEDLHVKDLNETIHYMYKHKMYRKMVFYIEACESGSMMNHLPDNINVYATTAANPRESSYACYYDEKRSTYLGDWYSVNWMEDSDVEDLTKETLHKQYHLVKSHTQTSHVMQYGNKTISTMKVMQFQGMKR Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | PAS domain-containing serine/threonine-protein kinase (PASK) | 3DLS | 3.94 | |
Target general information Gen name PASK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hPASK; PASKIN; PAS-kinase; KIAA0135 Protein family Protein kinase superfamily, CAMK Ser/Thr protein kinase family Biochemical class NA Function Serine/threonine-protein kinase involved in energy homeostasis and protein translation. Phosphorylates EEF1A1, GYS1, PDX1 and RPS6. Probably plays a role under changing environmental conditions (oxygen, glucose, nutrition), rather than under standard conditions. Acts as a sensor involved in energy homeostasis: regulates glycogen synthase synthesis by mediating phosphorylation of GYS1, leading to GYS1 inactivation. May be involved in glucose-stimulated insulin production in pancreas and regulation of glucagon secretion by glucose in alpha cells; however such data require additional evidences. May play a role in regulation of protein translation by phosphorylating EEF1A1, leading to increase translation efficiency. May also participate to respiratory regulation. Related diseases Galactosialidosis (GSL) [MIM:256540]: A lysosomal storage disease associated with a combined deficiency of beta-galactosidase and neuraminidase, secondary to a defect in cathepsin A. All patients have clinical manifestations typical of a lysosomal disorder, such as coarse facies, cherry red spots, vertebral changes, foam cells in the bone marrow, and vacuolated lymphocytes. Three phenotypic subtypes are recognized. The early infantile form is associated with fetal hydrops, edema, ascites, visceromegaly, skeletal dysplasia, and early death. The late infantile type is characterized by hepatosplenomegaly, growth retardation, cardiac involvement, and a normal or mildly affected mental state. The juvenile/adult form is characterized by myoclonus, ataxia, angiokeratoma, intellectual disability, neurologic deterioration, absence of visceromegaly, and long survival. {ECO:0000269|PubMed:10944848, ECO:0000269|PubMed:1756715, ECO:0000269|PubMed:8514852, ECO:0000269|PubMed:8968752}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P16220; P02545; Q93100; P62753; P07293; P02542; Q28115; P02646; P20152 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cytoplasm; Kinase; Lipid-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 32470.1 Length 285 Aromaticity 0.11 Instability index 32.86 Isoelectric point 4.74 Charge (pH=7) -17.74 2D Binding mode Binding energy (Kcal/mol) -5.37 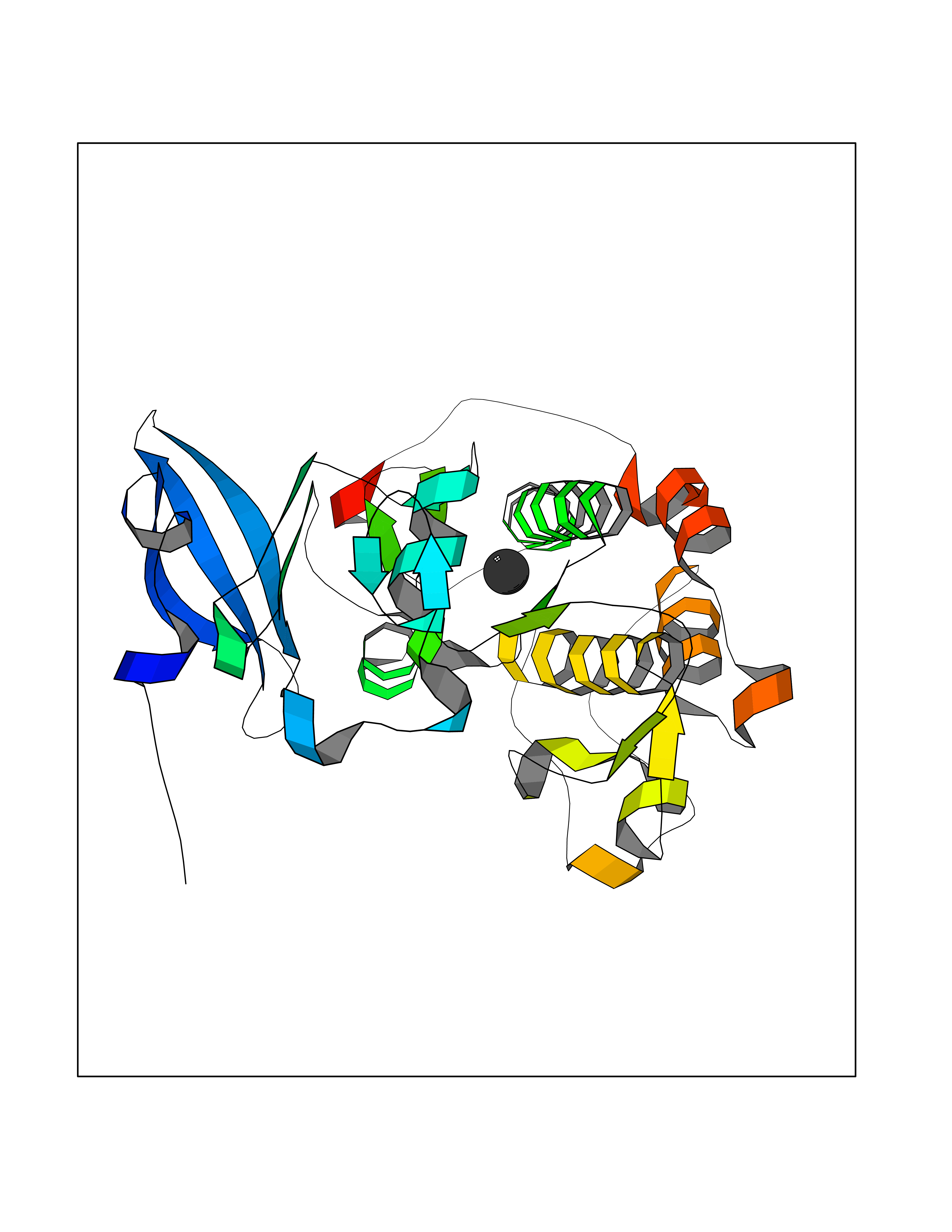 Molscript Map 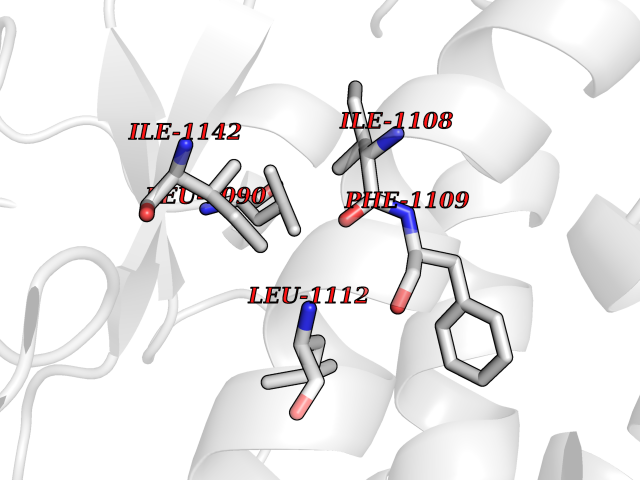 Pymol Map 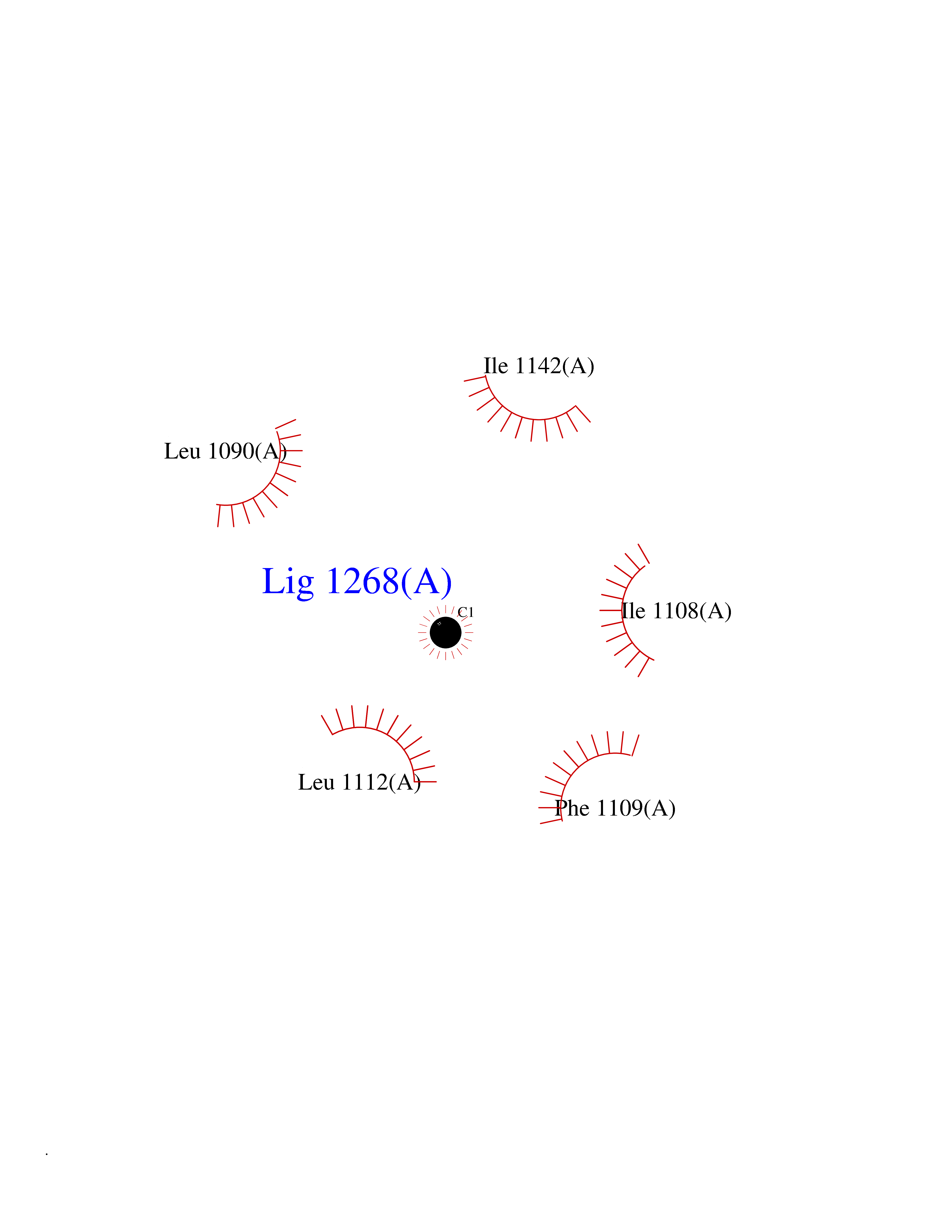 Ligplot Map 3D Binding mode Sequence AVELEGLAACEGEYSQKYSTMSPLGSGAFGFVWTAVDKEKNKEVVVKFIKKEKVLEDCWIEDPKLGKVTLEIAILSRVEHANIIKVLDIFENQGFFQLVMEKHGSGLDLFAFIDRHPRLDEPLASYIFRQLVSAVGYLRLKDIIHRDIKDENIVIAEDFTIKLIDFGSAAYLERGKLFYTFCGTIEYCAPEVLMGNPYRGPELEMWSLGVTLYTLVFEENPFCELEETVEAAIHPPYLVSKELMSLVSGLLQPVPERRTTLEKLVTDPWVTQPVNLADYTWEEVF Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Lethal(3)malignant brain tumor-like 3 (L3MBTL3) | 4FL6 | 3.94 | |
Target general information Gen name L3MBTL3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MBT1; MBT-1; Lethal(3)malignant brain tumor-like protein 3; L(3)mbt-like protein 3; KIAA1798; H-l(3)mbt-like protein 3 Protein family NA Biochemical class NA Function Putative Polycomb group (PcG) protein. PcG proteins maintain the transcriptionally repressive state of genes, probably via a modification of chromatin, rendering it heritably changed in its expressibility. Required for normal maturation of myeloid progenitor cells (By similarity). Related diseases Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 1 (SCAN1) [MIM:607250]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAN1 is an autosomal recessive cerebellar ataxia (ARCA) associated with peripheral axonal motor and sensory neuropathy, distal muscular atrophy, pes cavus and steppage gait as seen in Charcot-Marie-Tooth neuropathy. All affected individuals have normal intelligence. {ECO:0000269|PubMed:12244316, ECO:0000269|PubMed:15647511, ECO:0000269|PubMed:15920477, ECO:0000269|PubMed:16141202, ECO:0000269|PubMed:17948061}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NWX5; Q8N9N5; Q13895; Q96JK2; Q16531; P26358; Q01094; O00716; Q96JM7; Q9Y4Z0; P45984; Q9UBU8; Q15014; Q9NPG2; Q9UMX2; P18545; Q8IXK0; Q8N381; Q96S99; Q9H8W4; P62875; Q13131; P54646; P14678-2; P23497; G2XKQ0; P10827; Q6DKK2; P54253; A0A0S2Z5G4; Q13895; Q9BXJ3; Q8IUI8; Q14203-5; Q9H4E7; Q8NFF5-2; Q53EP0-3; O95995; O75031; P42858; Q14005-2; Q63ZY3; Q96JM7-2; P61968; P45984; P55081; Q9UBU8-2; Q15014; Q9NPG2; Q16656-4; Q96HA8; Q96BD5; Q92569; Q96S99; Q9H8W4; O60568; A0A6Q8PF08; P67775; P54646; P63000; O94955; Q5VUG0; P37840; P00441; Q5MJ10; Q9UMX1; Q13148; Q86TI0; Q8N8B7-2; Q9Y228; Q5T7W7; Q6DKK2; P09936; P31930; P61758; A0A0S2Z6A9; Q9H0M4-4; P36508 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 71079 Length 615 Aromaticity 0.14 Instability index 29.71 Isoelectric point 6.42 Charge (pH=7) -6.56 2D Binding mode Binding energy (Kcal/mol) -5.37 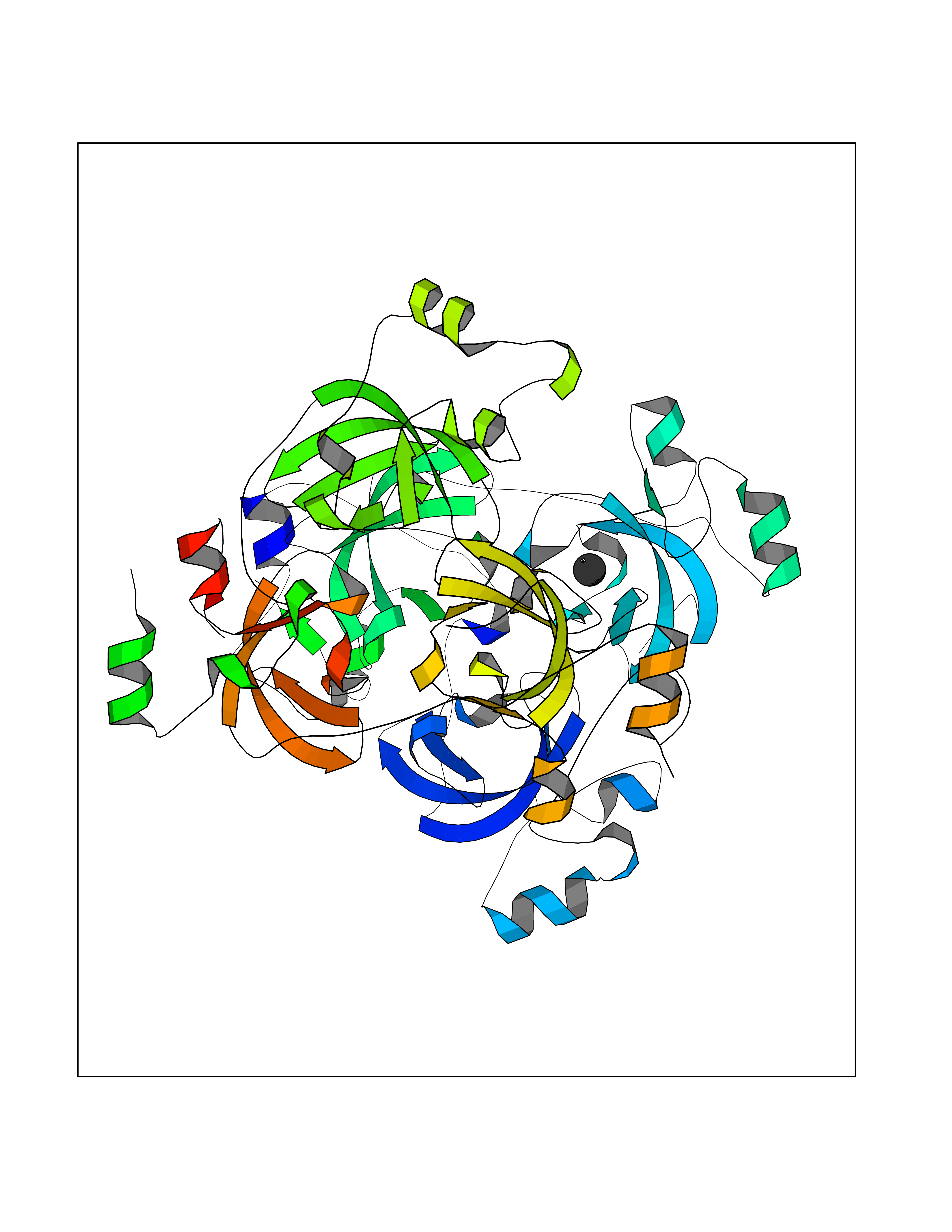 Molscript Map 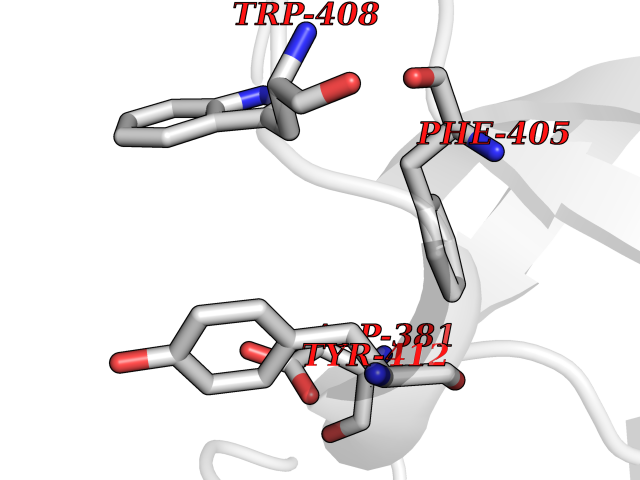 Pymol Map 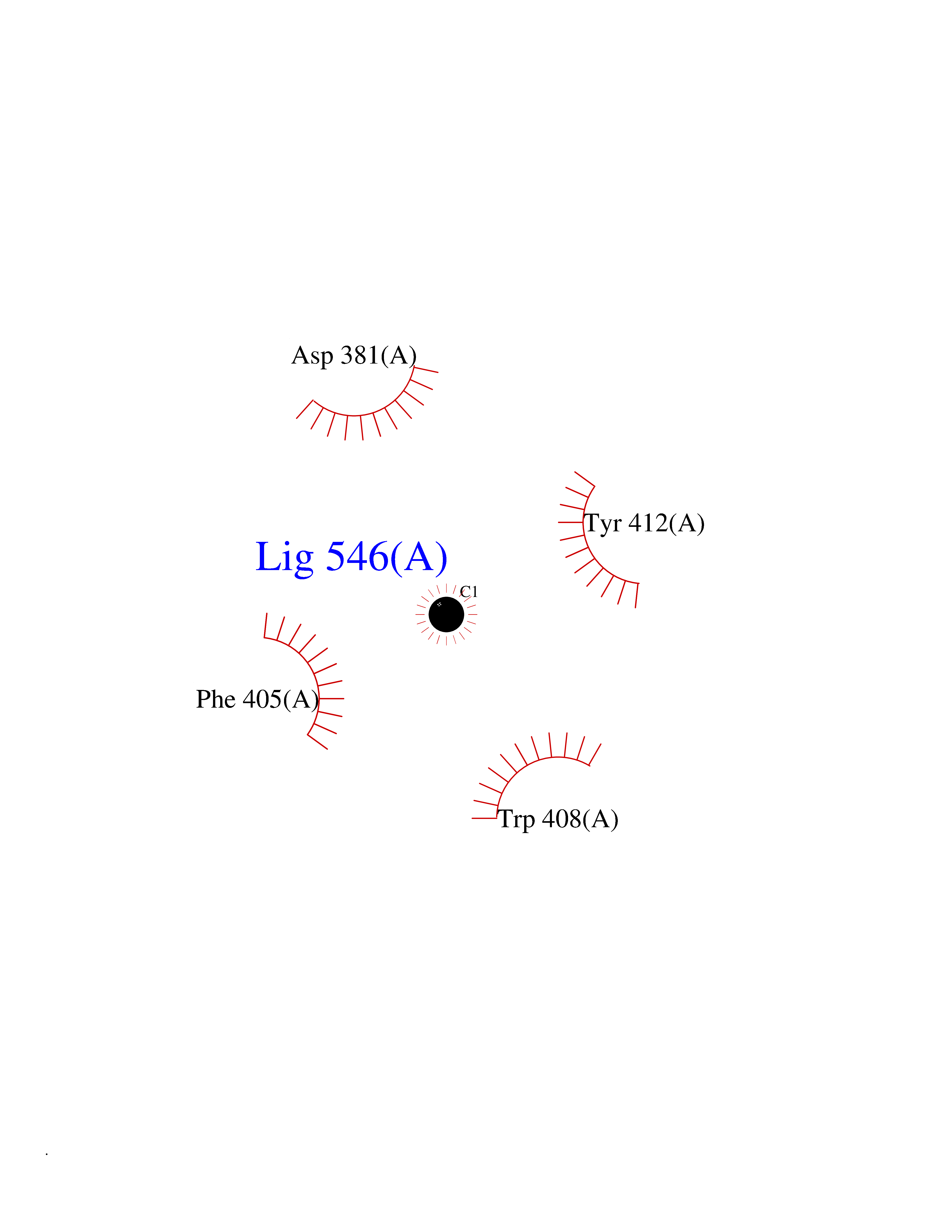 Ligplot Map 3D Binding mode Sequence AWCWASYLEEEKAVAVPAKLFKEHQSFPYNKNGFKVGMKLEGVDPEHQSVYCVLTVAEVCGYRIKLHFDGYSDCYDFWVNADALDIHPVGWCEKTGHKLHPPKGYKEEEFNWQTYLKTCKAQAAPKSLFEVIPSGFRVGMKLEAVDKKNPSFICVATVTDMVDNRFLVHFDNWDESYDYWCEASSPHIHPVGWCKEHRRTLITPPGYPNVKHFSWDKYLEETNSLPAPARAFKVKPPHGFQKKMKLEVVDKRNPMFIRVATVADTDDHRVKVHFDGWNNCYDYWIDADSPDIHPVGWCSKTGHPLQPPLAWCWASYLEEEKAVAVPAKLFKEHQSFPYNKNGFKVGMKLEGVDPEHQSVYCVLTVAEVCGYRIKLHFDGYSDCYDFWVNADALDIHPVGWCEKTGHKLHPPKGYKEEEFNWQTYLKTCKAQAAPKSLFENSGFRVGMKLEAVDKKNPSFICVATVTDMVDNRFLVHFDNWDESYDYWCEASSPHIHPVGWCKEHRRTLITPPGYPNVHFSWDKYLEETNSLPAPARAFKVKPPHGFQKKMKLEVVDKRNPMFIRVATVADTDDHRVKVHFDGWNNCYDYWIDADSPDIHPVGWCSKTGHPLQPPL Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | CDC-like kinase 1 (CLK1) | 6KHD | 3.94 | |
Target general information Gen name CLK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dual specificity protein kinase CLK1; CLK; CDClike kinase 1 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, Lammer subfamily Biochemical class Kinase Function Phosphorylates serine- and arginine-rich (SR) proteins of the spliceosomal complex and may be a constituent of a network of regulatory mechanisms that enable SR proteins to control RNA splicing. Phosphorylates: SRSF1, SRSF3 and PTPN1. Regulates the alternative splicing of tissue factor (F3) pre-mRNA in endothelial cells and adenovirus E1A pre-mRNA. Dual specificity kinase acting on both serine/threonine and tyrosine-containing substrates. Related diseases Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Genetic variations in ALOX5AP may be associated with susceptibility to myocardial infarction. Involvement in myocardial infarction is however unclear: according to some authors (PubMed:14770184), a 4-SNP haplotype in ALOX5AP confers risk of myocardial infarction, while according to other (PubMed:17304054) ALOX5AP is not implicated in this condition. {ECO:0000269|PubMed:14770184, ECO:0000269|PubMed:17304054}. Drugs (DrugBank ID) DB06376; DB04367; DB08691; DB12010 Interacts with P60409; O76083; Q8N2M8; P49760; Q92997; P60371; P60409; Q8TBB1; O76083-2; O14492-2; A7MD48; Q07955; O75494; P84103; Q16629; P62995 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 37557.7 Length 322 Aromaticity 0.1 Instability index 37.24 Isoelectric point 6.38 Charge (pH=7) -4.53 2D Binding mode Binding energy (Kcal/mol) -5.37 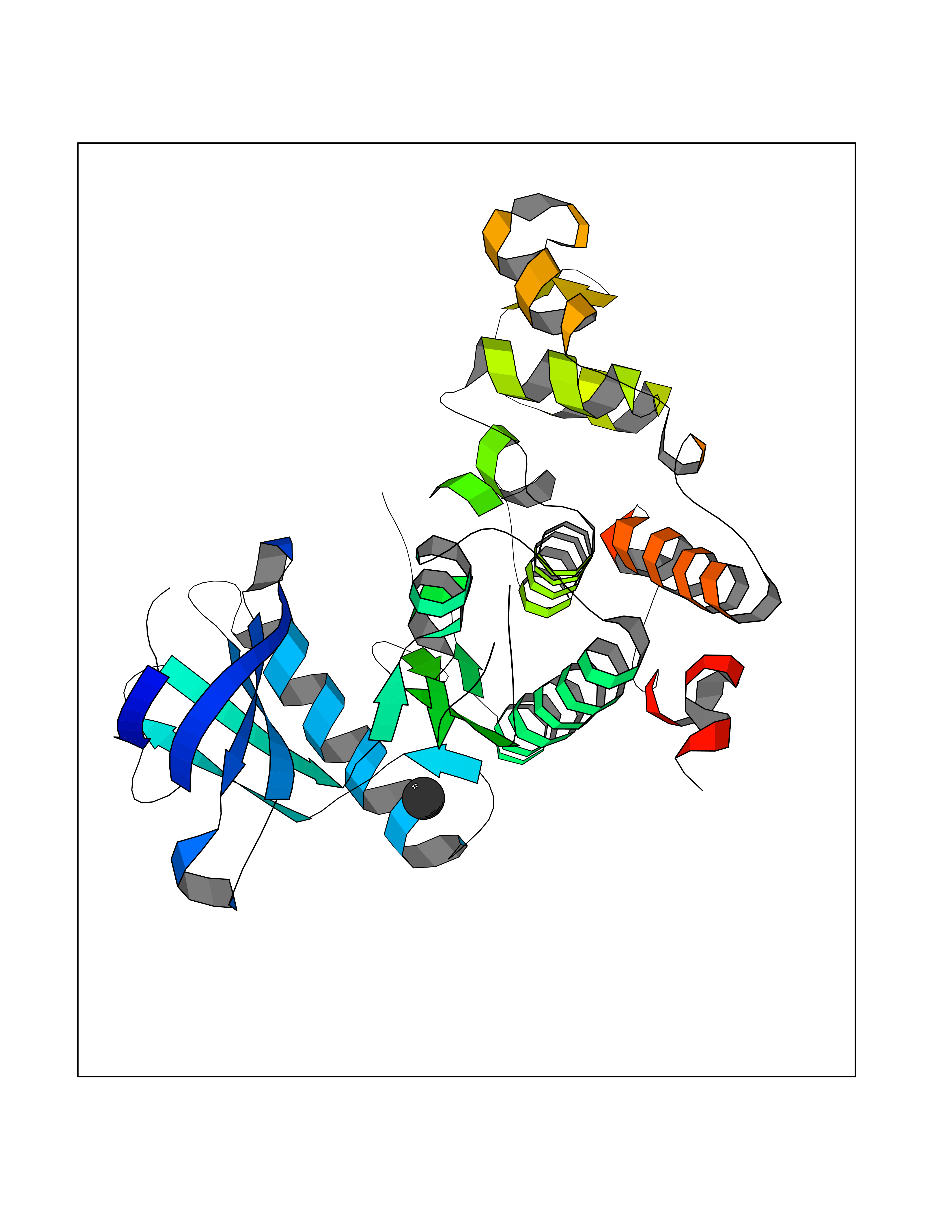 Molscript Map 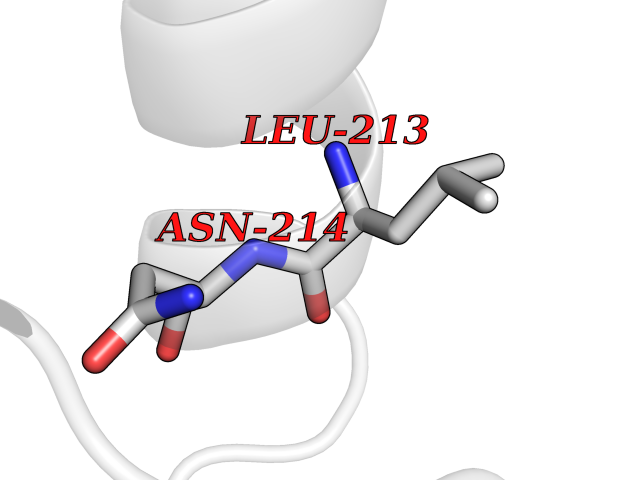 Pymol Map  Ligplot Map 3D Binding mode Sequence LICQSGDVLSARYEIVDTLGEGAFGKVVECIDHKAGGRHVAVKIVKNVDRYCEAARSEIQVLEHLNTTDPNSTFRCVQMLEWFEHHGHICIVFELLGLSTYDFIKENGFLPFRLDHIRKMAYQICKSVNFLHSNKLTHTDLKPENILFVQSDYTEERTLINPDIKVVDFGSATYDDEHHSTLVRHYRAPEVILALGWSQPCDVWSIGCILIEYYLGFTVFPTHDSKEHLAMMERILGPLPKHMIQKTRKRKYFHHDRLDWDEHSSAGRYVSRACKPLKEFMLSQDVEHERLFDLIQKMLEYDPAKRITLREALKHPFFDLLK Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Acyloxyacyl hydrolase (neutrophil) | 5W7C | 3.94 | |
Target general information Gen name AOAH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Acyloxyacyl hydrolase Protein family NA Biochemical class NA Function Removes the secondary (acyloxyacyl-linked) fatty acyl chains from the lipid A region of bacterial lipopolysaccharides. By breaking down LPS, terminates the host response to bacterial infection and prevents prolonged and damaging inflammatory responses (By similarity). In peritoneal macrophages, seems to be important for recovery from a state of immune tolerance following infection by Gram-negative bacteria (By similarity). Related diseases Major depressive disorder (MDD) [MIM:608516]: A common psychiatric disorder. It is a complex trait characterized by one or more major depressive episodes without a history of manic, mixed, or hypomanic episodes. A major depressive episode is characterized by at least 2 weeks during which there is a new onset or clear worsening of either depressed mood or loss of interest or pleasure in nearly all activities. Four additional symptoms must also be present including changes in appetite, weight, sleep, and psychomotor activity; decreased energy; feelings of worthlessness or guilt; difficulty thinking, concentrating, or making decisions; or recurrent thoughts of death or suicidal ideation, plans, or attempts. The episode must be accompanied by distress or impairment in social, occupational, or other important areas of functioning. {ECO:0000269|PubMed:15229186}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q15700 EC number EC 3.1.1.77 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lipid metabolism; Metal-binding; Proteomics identification; Reference proteome; Secreted; Signal; Zymogen Protein physicochemical properties Chain ID C Molecular weight (Da) 47779.7 Length 420 Aromaticity 0.1 Instability index 43.45 Isoelectric point 7.72 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -5.38  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GSDICSLPVLAKICQKIKLAMEQSVPFKDVDSDKYSVFPTLRGYHWRGRDCNDSDESVYPGRRPNNWDVHQDSNCNGIWGVDPKDGVPYEKKFCEGSQPRGIILLGDAAGAHFHISPEWITASQMSLNSFINLPTALTNELDWPQLSGATGFLDSTVGIKEKSIYLRLWKRNHCNHRDYQNISRNGASSRNLKKFIESLSRNKVLDYPAIVIYAMIGNDVCSGKSDPVPAMTTPEKLYSNVMQTLKHLNSHLPNGSHVILYGLPDGTFLWDNLHNRYHPLGQLNKDMTYAQLYSFLNCLQVSPCHGWMSSNKTLRTLTSERAEQLSNTLKKIAASEKFTNFNLFYMDFAFHEIIQEWQKRGGQPWQLIEPVDGFHPNEVALLLLADHFWKKVQLQWPQILGKENPFNPQIKQVFGDQGGH Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | GABA(A) receptor alpha-1 (GABRA1) | 6X3T | 3.94 | |
Target general information Gen name GABRA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gamma-aminobutyric acid receptor subunit alpha-1; GABA(A) receptor subunit alpha-1 Protein family Ligand-gated ion channel (TC 1.A.9) family, Gamma-aminobutyric acid receptor (TC 1.A.9.5) subfamily, GABRA1 sub-subfamily Biochemical class Ligand-gated ion channel Function Ligand-gated chloride channel which is a component of the heteropentameric receptor for GABA, the major inhibitory neurotransmitter in the brain. Plays an important role in the formation of functional inhibitory GABAergic synapses in addition to mediating synaptic inhibition as a GABA-gated ion channel. The gamma2 subunit is necessary but not sufficient for a rapid formation of active synaptic contacts and the synaptogenic effect of this subunit is influenced by the type of alpha and beta subunits present in the receptor pentamer (By similarity). The alpha1/beta2/gamma2 receptor and the alpha1/beta3/gamma2 receptor exhibit synaptogenic activity. GABRA1-mediated plasticity in the orbitofrontal cortex regulates context-dependent action selection (By similarity). Functions also as histamine receptor and mediates cellular responses to histamine (By similarity). Related diseases Epilepsy, childhood absence 4 (ECA4) [MIM:611136]: A subtype of idiopathic generalized epilepsy characterized by an onset at age 6-7 years, frequent absence seizures (several per day) and bilateral, synchronous, symmetric 3-Hz spike waves on EEG. Tonic-clonic seizures often develop in adolescence. Absence seizures may either remit or persist into adulthood. {ECO:0000269|PubMed:16718694}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Epilepsy, idiopathic generalized 13 (EIG13) [MIM:611136]: A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. {ECO:0000269|PubMed:21714819}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Juvenile myoclonic epilepsy 5 (EJM5) [MIM:611136]: A subtype of idiopathic generalized epilepsy. Patients have afebrile seizures only, with onset in adolescence (rather than in childhood) and myoclonic jerks which usually occur after awakening and are triggered by sleep deprivation and fatigue. {ECO:0000269|PubMed:11992121}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 19 (DEE19) [MIM:615744]: A severe neurologic disorder characterized by onset of seizures in the first months of life and usually associated with EEG abnormalities. Affected infants have convulsive seizures (hemiclonic or generalized) that are often prolonged and triggered by fever. Other seizure types include focal, myoclonic, absence seizures, and drop attacks. Development is normal in the first year of life with later slowing and intellectual disability. {ECO:0000269|PubMed:24623842, ECO:0000269|PubMed:27864847}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12537; DB00546; DB06579; DB00404; DB01351; DB00543; DB11901; DB01352; DB01483; DB14719; DB11859; DB01558; DB09017; DB00237; DB00241; DB01353; DB01489; DB00395; DB00475; DB14715; DB01594; DB00349; DB06470; DB01068; DB00628; DB01559; DB01553; DB00363; DB01511; DB01189; DB00829; DB01496; DB01341; DB13837; DB00228; DB01215; DB00402; DB00898; DB00189; DB01545; DB09166; DB00292; DB01567; DB01205; DB01544; DB00690; DB05087; DB01381; DB01437; DB00801; DB01159; DB01354; DB01355; DB00753; DB01587; DB00555; DB13643; DB00186; DB13872; DB13437; DB00603; DB01043; DB00371; DB00463; DB00474; DB01028; DB00849; DB01107; DB15489; DB00683; DB12458; DB01595; DB14028; DB00334; DB00842; DB14672; DB00312; DB01174; DB00252; DB00466; DB13335; DB01708; DB01588; DB00794; DB00837; DB00818; DB01589; DB01346; DB12404; DB00418; DB01236; DB09118; DB00306; DB01956; DB00231; DB01154; DB11582; DB00599; DB00273; DB00897; DB00962; DB00425; DB00909; DB01198; DB15490 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Chloride; Chloride channel; Cytoplasmic vesicle; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 77460 Length 672 Aromaticity 0.14 Instability index 35.95 Isoelectric point 8.75 Charge (pH=7) 7.31 2D Binding mode Binding energy (Kcal/mol) -5.37 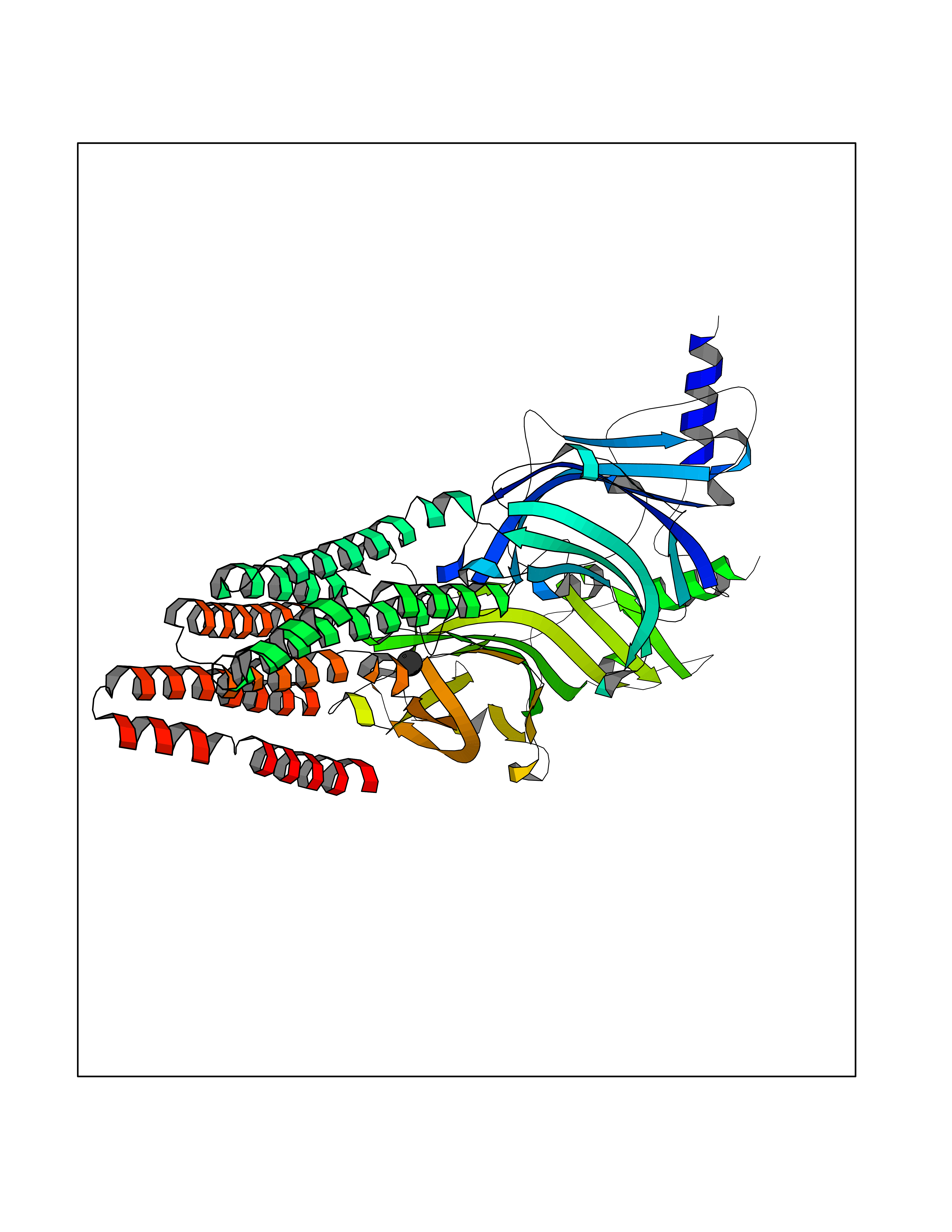 Molscript Map 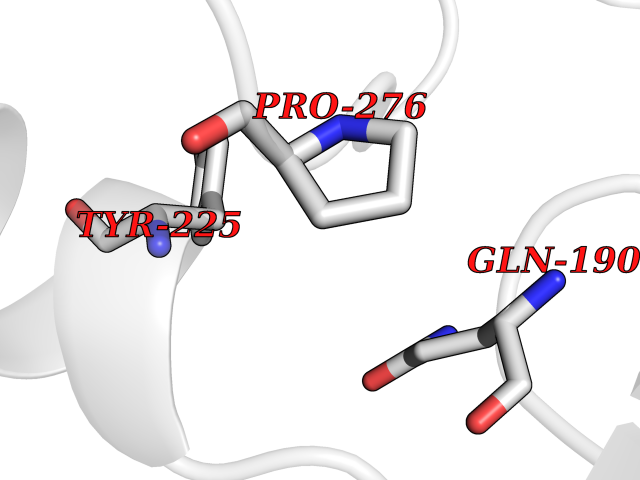 Pymol Map 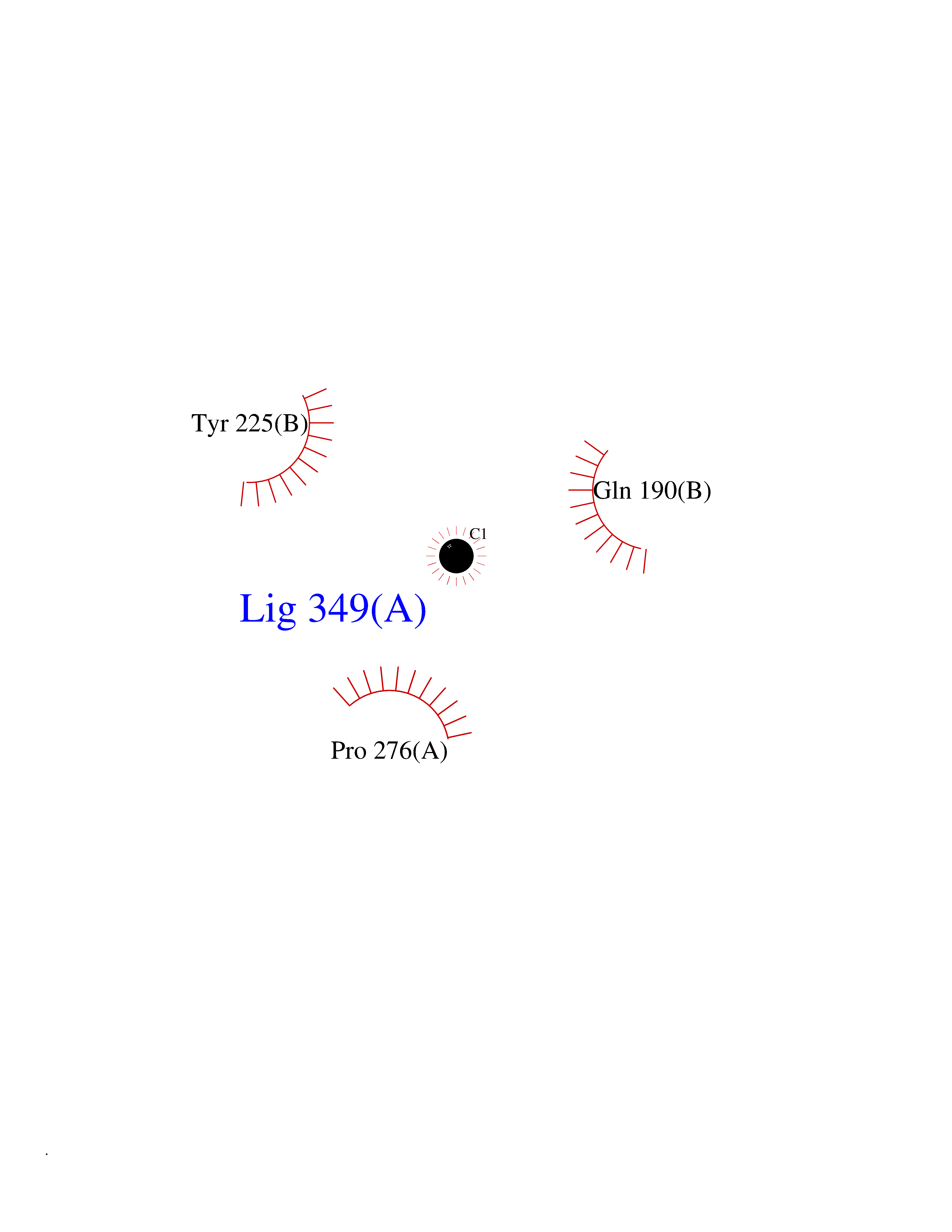 Ligplot Map 3D Binding mode Sequence SNMSLVKETVDRLLKGYDIRLRPDFGGPPVAVGMNIDIASIDMVSEVNMDYTLTMYFQQAWRDKRLSYNVIPLNLTLDNRVADQLWVPDTYFLNDKKSFVHGVTVKNRMIRLHPDGTVLYGLRITTTAACMMDLRRYPLDEQNCTLEIESYGYTTDDIEFYWRGDDNAVTGVTKIELPQFSIVDYKLITKKVVFSTGSYPRLSLSFKLKRNIGYFILQTYMPSILITILSWVSFWINYDASAARVALGITTVLTMTTINTHLRETLPKIPYVKAIDMYLMGCFVFVFMALLEYALVNYIFFSQPARAAAIDRWSRIFFPVVFSFFNIVYWLYYVDNTTVFTRILDRLLDGYDNRLRPGLGERVTEVKTDIFVTSFGPVSDHDMEYTIDVFFRQSWKDERLKFKGPMTVLRLNNLMASKIWTPDTFFHNGKKSVAHNMTMPNKLLRITEDGTLLYTMRLTVRAECPMHLEDFPMDAHACPLKFGSYAYTRAEVVYEWTREPARSVVVAEDGSRLNQYDLLGQTVDSGIVQSSTGEYVVMTTHFHLKRKIGYFVIQTYLPCIMTVILSQVSFWLNRESVPARTVFGVTTVLTMTTLSISARNSLPKVAYATAMDWFIAVCYAFVFSALIEFATVNYFTKSQPARAAKIDRLSRIAFPLLFGIFNLVYWATYLNR Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Bacterial Aspartate-semialdehyde dehydrogenase (Bact asd2) | 2R00 | 3.94 | |
Target general information Gen name Bact asd2 Organism Vibrio cholerae serotype O1 (strain ATCC 39315 / El Tor Inaba N16961) Uniprot ID TTD ID Synonyms asd2; L-Aspartate-beta-semialdehyde dehydrogenase; ASADH; ASA dehydrogenase Protein family Aspartate-semialdehyde dehydrogenase family Biochemical class Aldehyde/oxo donor oxidoreductase Function Catalyzes the NADPH-dependent formation of L-aspartate- semialdehyde (L-ASA) by the reductive dephosphorylation of L- aspartyl-4-phosphate. Related diseases A chromosomal aberration involving FHIT has been found in a lymphoblastoid cell line established from a family with renal cell carcinoma and thyroid carcinoma. Translocation t(3;8)(p14.2;q24.1) with RNF139. Although the 3p14.2 breakpoint has been shown to interrupt FHIT in its 5-prime non-coding region, it is unlikely that FHIT is causally related to renal or other malignancies. {ECO:0000269|PubMed:15007172}.; DISEASE: Associated with digestive tract cancers. Numerous tumor types are found to have aberrant forms of FHIT protein due to deletions in a coding region of chromosome 3p14.2 including the fragile site locus FRA3B. {ECO:0000269|PubMed:15007172}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Amino-acid biosynthesis; Diaminopimelate biosynthesis; Lysine biosynthesis; Methionine biosynthesis; NADP; Oxidoreductase; Reference proteome; Threonine biosynthesis Protein physicochemical properties Chain ID C Molecular weight (Da) 37140.5 Length 336 Aromaticity 0.09 Instability index 26.08 Isoelectric point 4.78 Charge (pH=7) -17.88 2D Binding mode Binding energy (Kcal/mol) -5.37 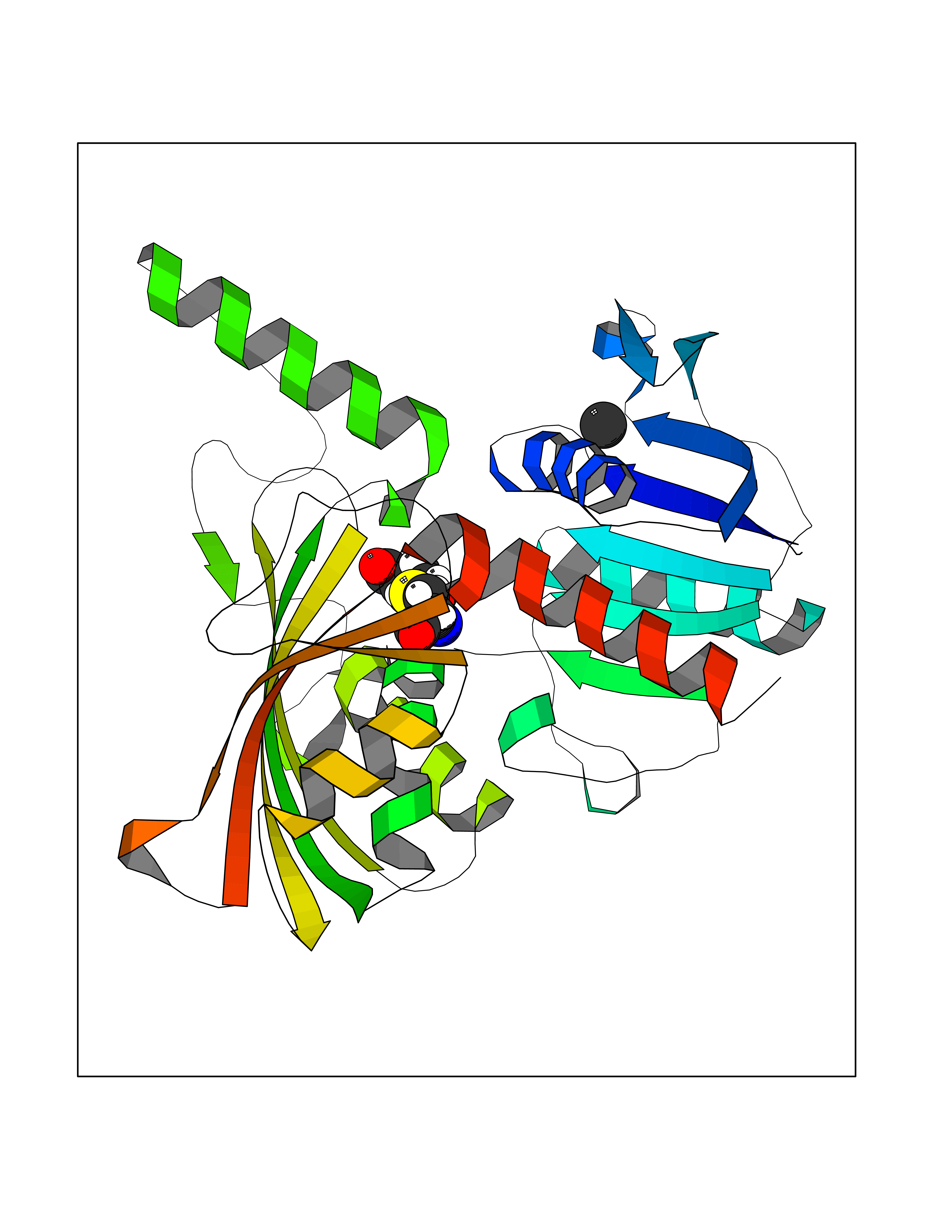 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SQQFNVAIFGATGAVGETMLEVLQEREFPVDELFLLASERSEGKTYRFNGKTVRVQNVEEFDWSQVHIALFSAGGELSAKWAPIAAEAGVVVIDNTSHFRYDYDIPLVVPEVNPEAIAEFRNRNIIANPNXSTIQMLVALKPIYDAVGIERINVTTYQSVSGAGKAGIDELAGQTAKLLNGYPAETNTFSQQIAFNCIPQIDQFMDNGYTKEEMKMVWETQKIFNDPSIMVNPTCVRVPVFYGHAEAVHVETRAPIDAEQVMDMLEQTDGIELFRGADFPTQVRDAGGKDHVLVGRVRNDISHHSGINLWVVADNVRKGAATNAVQIAELLVRDYF Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Membrane copper amine oxidase (AOC3) | 4BTX | 3.94 | |
Target general information Gen name AOC3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vascular adhesion protein-1; Vascular adhesion protein 1; VAP1; VAP-1; Semicarbazide-sensitive amine oxidase; SSAO; Membrane primary amine oxidase; HPAO; Copper amine oxidase Protein family Copper/topaquinone oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Has semicarbazide-sensitive (SSAO) monoamine oxidase activity. May play a role in adipogenesis. Cell adhesion protein that participates in lymphocyte extravasation and recirculation by mediating the binding of lymphocytes to peripheral lymph node vascular endothelial cells in an L-selectin-independent fashion. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB04334; DB01275; DB00780 Interacts with Q3SXY8; O95484; Q7Z7G2; Q96BA8; Q6PI48; Q8TBE3; Q7Z5P4; P42858; O43765; Q16623 EC number EC 1.4.3.21 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell adhesion; Cell membrane; Copper; Direct protein sequencing; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Oxidoreductase; Proteomics identification; Reference proteome; Signal-anchor; TPQ; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 157266 Length 1415 Aromaticity 0.12 Instability index 39.94 Isoelectric point 6 Charge (pH=7) -23.49 2D Binding mode Binding energy (Kcal/mol) -5.38  Molscript Map  Pymol Map 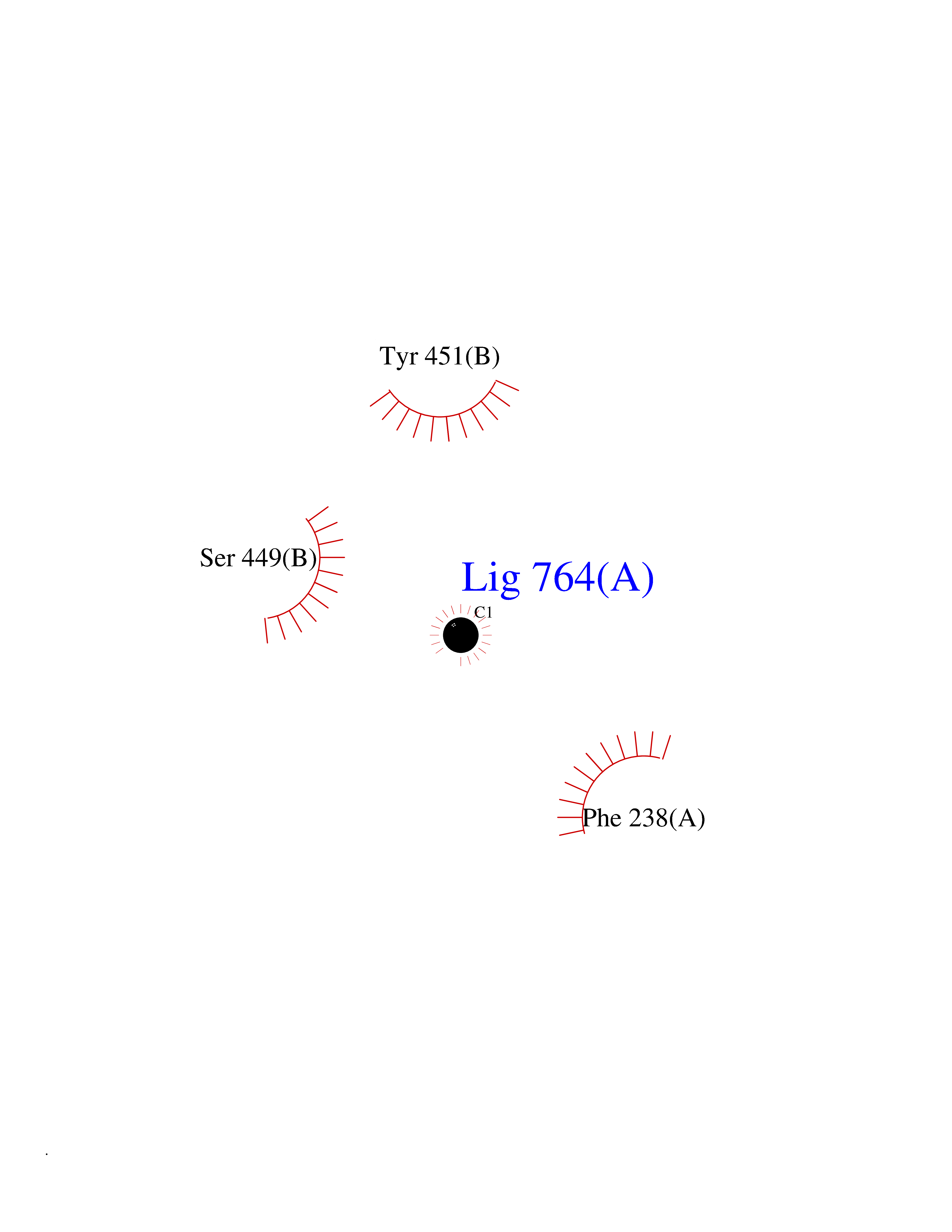 Ligplot Map 3D Binding mode Sequence PGQSQLFADLSREELTAVMRFLTQRLGPGLVDAAQARPSDNCVFSVELQLPPKAAALAHLDRGSPPPAREALAIVFFGRQPQPNVSELVVGPLPHPSYMRDVTVERHGGPLPYHRRPVLFQEYLDIDQMIFNRELPQASGLLHHCCFYKHRGRNLVTMTTAPRGLQSGDRATWFGLYYNISGAGFFLHHVGLELLVNHKALDPARWTIQKVFYQGRYYDSLAQLEAQFEAGLVNVVLIPDNGTGGSWSLKSPVPPGPAPPLQFYPQGPRFSVQGSRVASSLWTFSFGLGAFSGPRIFDVRFQGERLVYEISLQEALAIYGGNSPAAMTTRYVDGGFGMGKYTTPLTRGVDCPYLATYVDWHFLLESQAPKTIRDAFCVFEQNQGLPLRRHHSDLYSHYFGGLAETVLVVRSMSTLLNXDYVWDTVFHPSGAIEIRFYATGYISSAFLFGATGKYGNQVSEHTLGTVHTHSAHFKVDLDVAGLENWVWAEDMVFVPMAVPWSPEHQLQRLQVTRKLLEMEEQAAFLVGSATPRYLYLASNHSNKWGHPRGYRIQMLSFAGEPLPQNSSMARGFSWERYQLAVTQRKEEEPSSSSVFNQNDPWAPTVDFSDFINNETIAGKDLVAWVTAGFLHIPHAEDIPNTVTVGNGVGFFLRPYNFFDEDPSFYSADSIYFRGDQDAGACEVNPLACLPQAAACAPDLPAFSHGGFSHSQLFADLSREELTAVMRFLTQRLGPGLVDAAQARPSDNCVFSVELQLPPKAAALAHLDRGSPPPAREALAIVFFGRQPQPNVSELVVGPLPHPSYMRDVTVERHGGPLPYHRRPVLFQEYLDIDQMIFNRELPQASGLLHHCCFYKHRGRNLVTMTTAPRGLQSGDRATWFGLYYNISGAGFFLHHVGLELLVNHKALDPARWTIQKVFYQGRYYDSLAQLEAQFEAGLVNVVLIPDNGTGGSWSLKSPVPPGPAPPLQFYPQGPRFSVQGSRVASSLWTFSFGLGAFSGPRIFDVRFQGERLVYEISLQEALAIYGGNSPAAMTTRYVDGGFGMGKYTTPLTRGVDCPYLATYVDWHFLLESQAPKTIRDAFCVFEQNQGLPLRRHHSDLYSHYFGGLAETVLVVRSMSTLLNXDYVWDTVFHPSGAIEIRFYATGYISSAFLFGATGKYGNQVSEHTLGTVHTHSAHFKVDLDVAGLENWVWAEDMVFVPMAVPWSPEHQLQRLQVTRKLLEMEEQAAFLVGSATPRYLYLASNHSNKWGHPRGYRIQMLSFAGEPLPQNSSMARGFSWERYQLAVTQRKEEEPSSSSVFNQNDPWAPTVDFSDFINNETIAGKDLVAWVTAGFLHIPHAEDIPNTVTVGNGVGFFLRPYNFFDEDPSFYSADSIYFRGDQDAGACEVNPLACLPQAAACAPDLPAFSHGGFSH Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Neuronal acetylcholine receptor alpha-4 (CHRNA4) | 6CNJ | 3.94 | |
Target general information Gen name CHRNA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor alpha4; CHRNA4; Alpha-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-4/CHRNA4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasmamembrane permeable to sodium ions. Related diseases Epilepsy, nocturnal frontal lobe, 1 (ENFL1) [MIM:600513]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:10563623, ECO:0000269|PubMed:14623738, ECO:0000269|PubMed:7550350}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01351; DB01352; DB00572; DB01483; DB00237; DB00241; DB01353; DB00564; DB00565; DB09028; DB01245; DB00514; DB01496; DB07720; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00898; DB01354; DB01355; DB00753; DB00657; DB00333; DB00463; DB00849; DB00184; DB00312; DB01174; DB00981; DB05458; DB00794; DB05740; DB00747; DB00418; DB00202; DB00306; DB00599; DB01273 Interacts with Q6UY14-3; P05067; P83916; Q6UXH1-1; Q6UXH1-3; P20042; Q9NZR2; Q92673; P17787 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -5.37 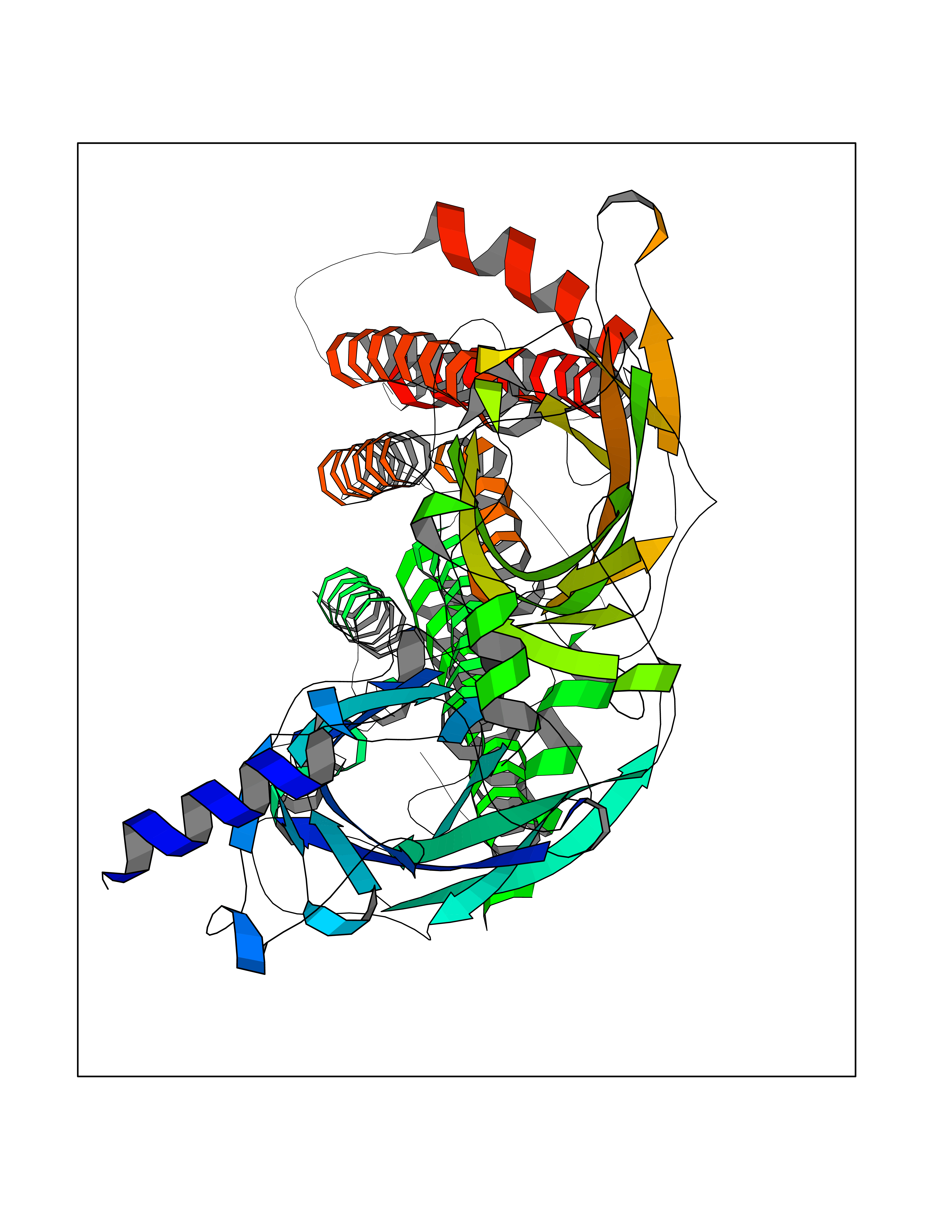 Molscript Map 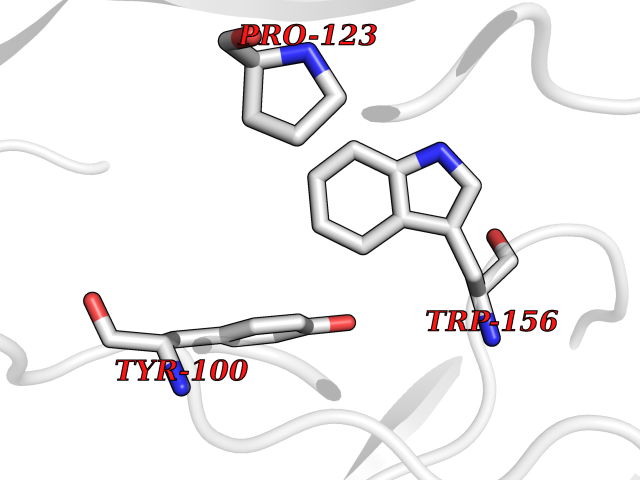 Pymol Map 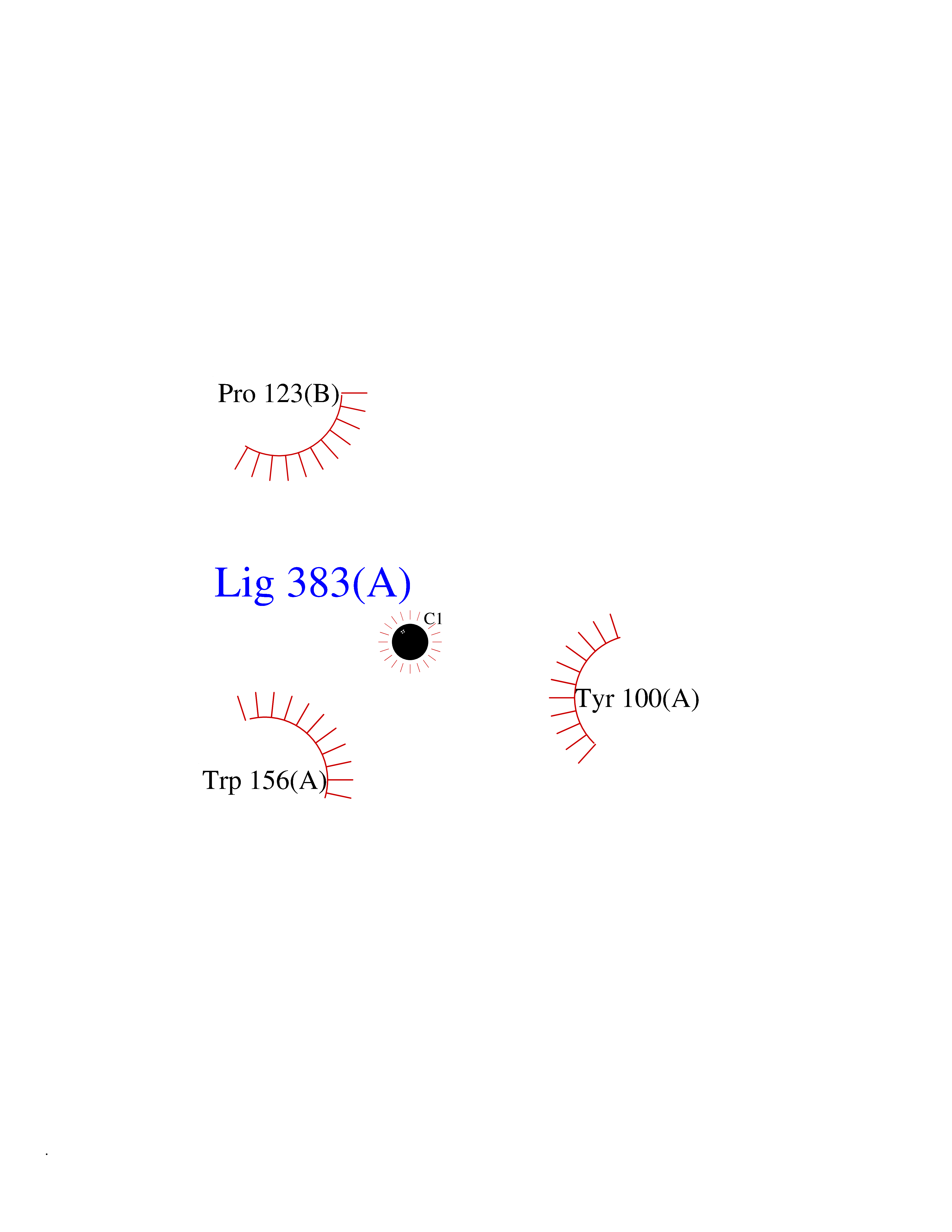 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||