Job Results:
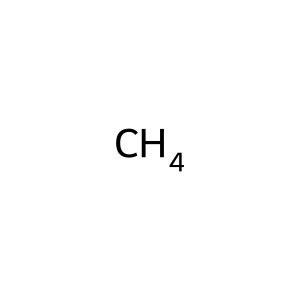
Ligand
Structure
Job ID
9d5454904e548acd822dcc3f8261b50f
Job name
NA
Time
2026-02-27 11:53:48
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Cerebroside-sulfatase (ARSA) | 1E2S | 3.95 | |
Target general information Gen name ARSA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cerebrosidesulfatase; Arylsulfatase A component C; ASA; ARSA Protein family Sulfatase family Biochemical class Sulfuric ester hydrolase Function Hydrolyzes cerebroside sulfate. Related diseases Metachromatic leukodystrophy (MLD) [MIM:250100]: An autosomal recessive disease caused by abnormal intralysosomal accumulation of cerebroside-3-sulfate in central and peripheral nervous systems, as well as other organs. MLD is clinically characterized by leukodystrophy, progressive demyelination and a variety of neurological symptoms, including gait disturbances, ataxias, optical atrophy, dementia, seizures, and spastic tetraparesis. Decreased arylsulfatase A activity is detected in urine, leukocytes, and fibroblasts of affected individuals. Several forms of the disease can be distinguished according to the age at onset and disease severity: late infantile, juvenile and adult forms, partial cerebroside sulfate deficiency, and pseudoarylsulfatase A deficiency. Individuals with pseudoarylsulfatase A deficiency have low arylsulfatase A activity but lack neurological manifestations and are apparently healthy. {ECO:0000269|PubMed:10220151, ECO:0000269|PubMed:10381328, ECO:0000269|PubMed:10477432, ECO:0000269|PubMed:10533072, ECO:0000269|PubMed:10751093, ECO:0000269|PubMed:11020646, ECO:0000269|PubMed:11061266, ECO:0000269|PubMed:11456299, ECO:0000269|PubMed:11941485, ECO:0000269|PubMed:12503099, ECO:0000269|PubMed:12788103, ECO:0000269|PubMed:1353340, ECO:0000269|PubMed:14517960, ECO:0000269|PubMed:14680985, ECO:0000269|PubMed:15026521, ECO:0000269|PubMed:15326627, ECO:0000269|PubMed:15710861, ECO:0000269|PubMed:1670590, ECO:0000269|PubMed:1673291, ECO:0000269|PubMed:1678251, ECO:0000269|PubMed:18693274, ECO:0000269|PubMed:19606494, ECO:0000269|PubMed:20339381, ECO:0000269|PubMed:21265945, ECO:0000269|PubMed:2574462, ECO:0000269|PubMed:7581401, ECO:0000269|PubMed:7825603, ECO:0000269|PubMed:7860068, ECO:0000269|PubMed:7902317, ECO:0000269|PubMed:7906588, ECO:0000269|PubMed:7909527, ECO:0000269|PubMed:8095918, ECO:0000269|PubMed:8101038, ECO:0000269|PubMed:8101083, ECO:0000269|PubMed:8104633, ECO:0000269|PubMed:8891236, ECO:0000269|PubMed:9090526, ECO:0000269|PubMed:9272717, ECO:0000269|PubMed:9452102, ECO:0000269|PubMed:9490297, ECO:0000269|PubMed:9600244, ECO:0000269|PubMed:9819708}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Multiple sulfatase deficiency (MSD) [MIM:272200]: A clinically and biochemically heterogeneous disorder caused by the simultaneous impairment of all sulfatases, due to defective post-translational modification and activation. It combines features of individual sulfatase deficiencies such as metachromatic leukodystrophy, mucopolysaccharidosis, chondrodysplasia punctata, hydrocephalus, ichthyosis, neurologic deterioration and developmental delay. {ECO:0000269|PubMed:15146462}. The protein represented in this entry is involved in disease pathogenesis. Arylsulfatase A activity is impaired in multiple sulfatase deficiency due to mutations in SUMF1 (PubMed:15146462). SUMF1 mutations result in defective post-translational modification of ARSA at residue Cys-69 that is not converted to 3-oxoalanine (PubMed:7628016). {ECO:0000269|PubMed:15146462, ECO:0000269|PubMed:7628016}. Drugs (DrugBank ID) DB03821; DB01800; DB01141; DB04786 Interacts with P50995; Q6P5X5; Q13554-3; O60826; Q96D98; Q9H0I2; Q12951-2; Q16512; P28069; O75360; Q9BQY4; Q15645; O95231 EC number EC 3.1.6.8 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Hydrolase; Ichthyosis; Leukodystrophy; Lipid metabolism; Lysosome; Metachromatic leukodystrophy; Metal-binding; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID P Molecular weight (Da) 51173.7 Length 481 Aromaticity 0.09 Instability index 47.42 Isoelectric point 5.59 Charge (pH=7) -12.84 2D Binding mode Binding energy (Kcal/mol) -5.39 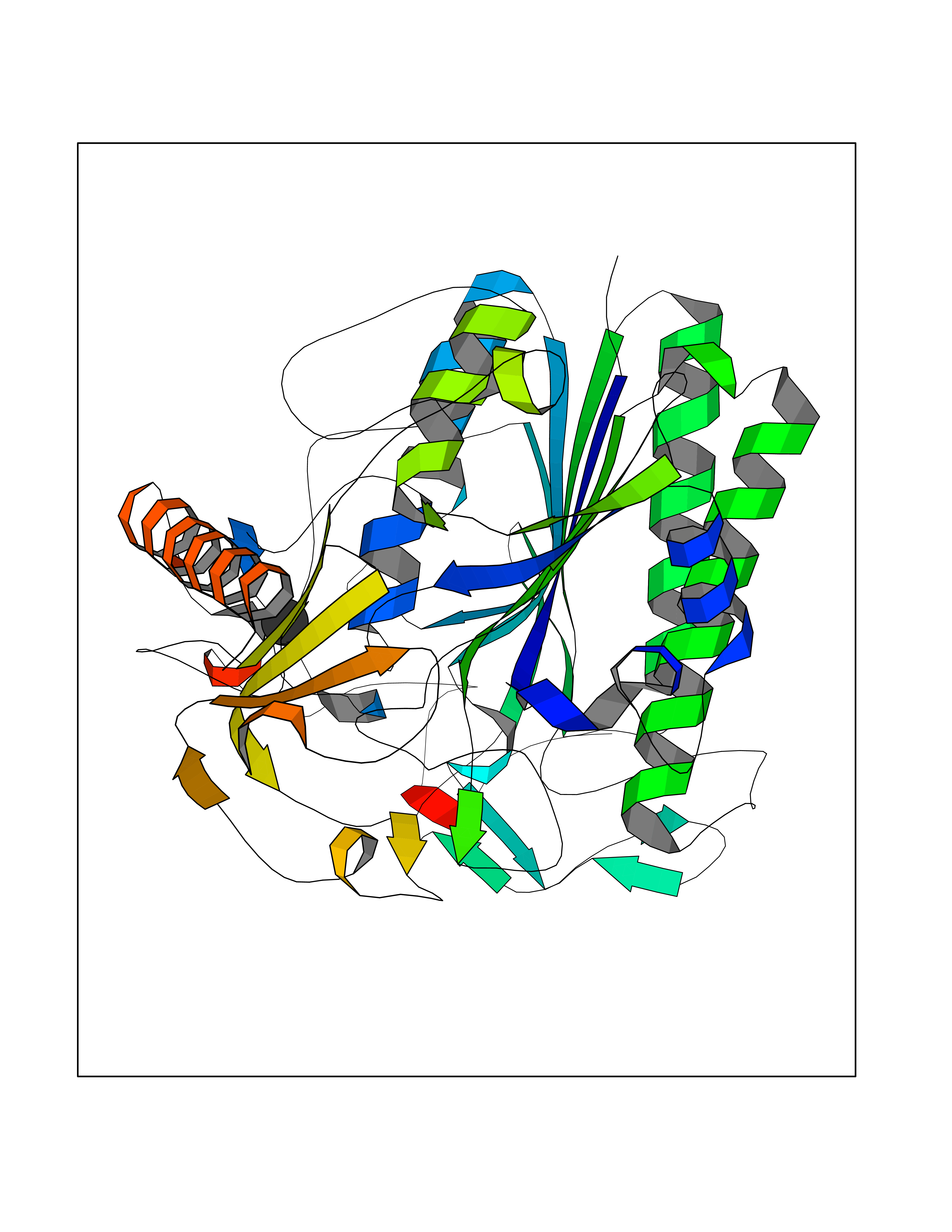 Molscript Map 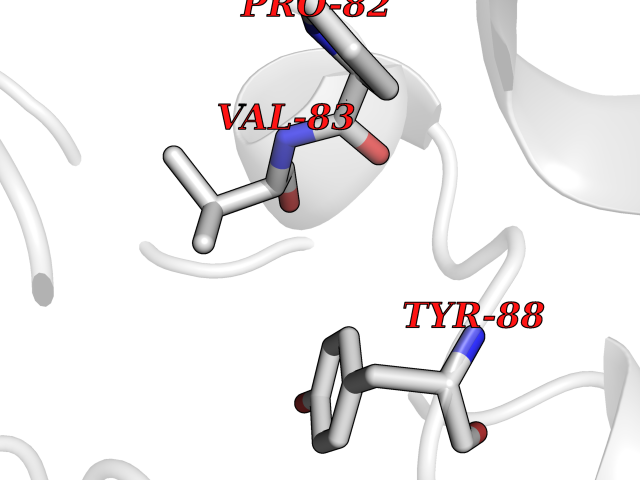 Pymol Map 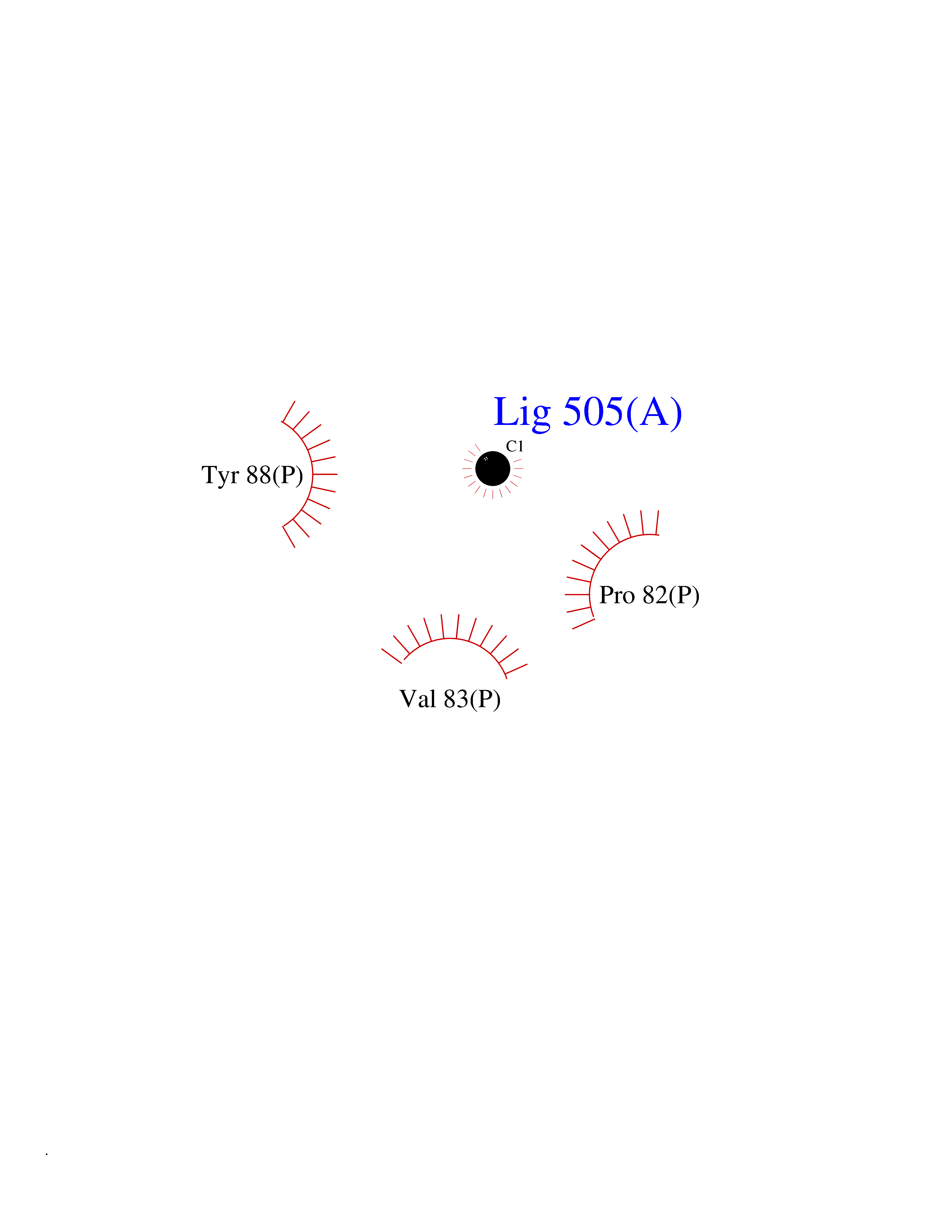 Ligplot Map 3D Binding mode Sequence RPPNIVLIFADDLGYGDLGCYGHPSSTTPNLDQLAAGGLRFTDFYVPVSLATPSRAALLTGRLPVRMGMYPGVLVPSSRGGLPLEEVTVAEVLAARGYLTGMAGKWHLGVGPEGAFLPPHQGFHRFLGIPYSHDQGPCQNLTCFPPATPCDGGCDQGLVPIPLLANLSVEAQPPWLPGLEARYMAFAHDLMADAQRQDRPFFLYYASHHTHYPQFSGQSFAERSGRGPFGDSLMELDAAVGTLMTAIGDLGLLEETLVIFTADNGPETMRMSRGGCSGLLRCGKGTTYEGGVREPALAFWPGHIAPGVTHELASSLDLLPTLAALAGAPLPNVTLDGFDLSPLLLGTGKSPRQSLFFYPSYPDEVRGVFAVRTGKYKAHFFTQGSAHSDTTADPACHASSSLTAHEPPLLYDLSKDPGENYNLLGATPEVLQALKQLQLLKAQLDAAVTFGPSQVARGEDPALQICCHPGCTPRPACCHCP Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Argininosuccinate synthase | 1K97 | 3.94 | |
Target general information Gen name argG Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3172;JW3140 Protein family Argininosuccinate synthase family, Type 2 subfamily Biochemical class Ligase Function Argininosuccinate synthase activity.ATP binding.Identical protein binding.Protein homodimerization activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P0A6E4; P24228 EC number 6.3.4.5 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Arginine biosynthesis; ATP-binding; Cytoplasm; Direct protein sequencing; Ligase; Nucleotide-binding; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 42479.6 Length 380 Aromaticity 0.09 Instability index 41.7 Isoelectric point 5.23 Charge (pH=7) -13.68 2D Binding mode Binding energy (Kcal/mol) -5.37 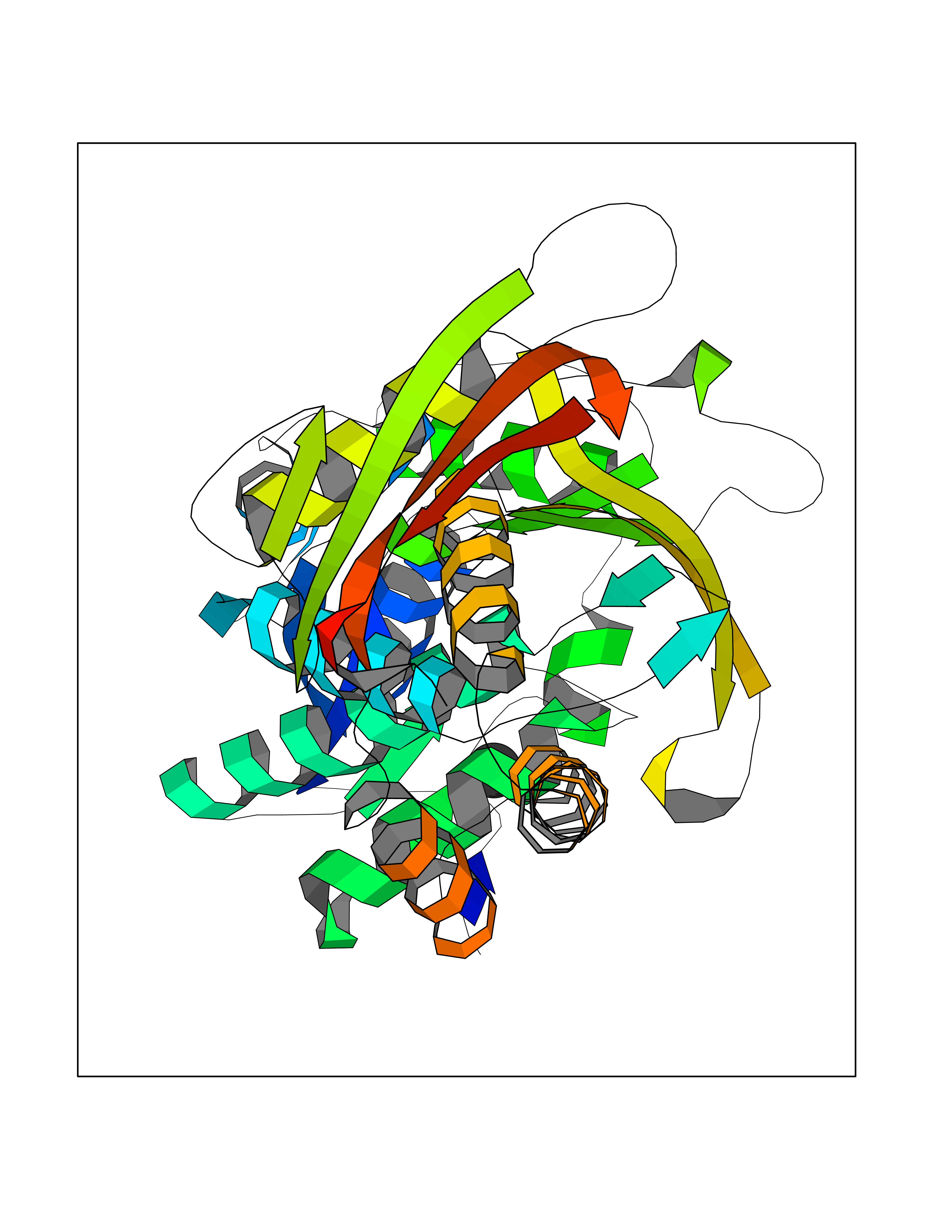 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence TTILKHLPVGQRIGIAFSGGLDTSAALLWMRQKGAVPYAYTANLGQPDEEDYDAIPRRAMEYGAENARLIDCRKQLVAEGIAAIQCGAFHNTTGGLTYFNTTPLGRAVTGTMLVAAMKEDGVNIWGDGSTYKGNDIERFYRYGLLTNAELQIYKPWLDTDFIDELGGRHEMSEFMIACGFDYKMSVEKAYSTDSNMLGATHEAKDLEYLNSSVKIVNPIMGVKFWDESVKIPAEEVTVRFEQGHPVALNGKTFSDDVEMMLEANRIGGRHGLGMSDQIENRIIEAKSRGIYEAPGMALLHIAYERLLTGIHNEDTIEQYHAHGRQLGRLLYQGRWFDSQALMLRDSLQRWVASQITGEVTLELRRGNDYSILNTVSENLT Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Tyrosine 3-monooxygenase (TH) | 2XSN | 3.94 | |
Target general information Gen name TH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine 3-hydroxylase; TH Protein family Biopterin-dependent aromatic amino acid hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Plays an important role in the physiology of adrenergic neurones. Related diseases Segawa syndrome autosomal recessive (ARSEGS) [MIM:605407]: A form of DOPA-responsive dystonia presenting in infancy or early childhood. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Some cases present with parkinsonian symptoms in infancy. Unlike all other forms of dystonia, it is an eminently treatable condition, due to a favorable response to L-DOPA. {ECO:0000269|PubMed:10585338, ECO:0000269|PubMed:11196107, ECO:0000269|PubMed:11246459, ECO:0000269|PubMed:15505183, ECO:0000269|PubMed:15747353, ECO:0000269|PubMed:16049992, ECO:0000269|PubMed:17696123, ECO:0000269|PubMed:18058633, ECO:0000269|PubMed:18554280, ECO:0000269|PubMed:19491146, ECO:0000269|PubMed:20056467, ECO:0000269|PubMed:20430833, ECO:0000269|PubMed:21940685, ECO:0000269|PubMed:22264700, ECO:0000269|PubMed:22815559, ECO:0000269|PubMed:23762320, ECO:0000269|PubMed:23939262, ECO:0000269|PubMed:24753243, ECO:0000269|PubMed:7814018, ECO:0000269|PubMed:8528210, ECO:0000269|PubMed:8817341, ECO:0000269|PubMed:9613851, ECO:0000269|PubMed:9703425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: May play a role in the pathogenesis of Parkinson disease (PD). A genome-wide copy number variation analysis has identified a 34 kilobase deletion over the TH gene in a PD patient but not in any controls. {ECO:0000269|PubMed:20809526}. Drugs (DrugBank ID) DB03552; DB04400; DB00765; DB00120; DB00360; DB00135 Interacts with P29762; P61978-2; Q99750; P08651-5; O75928-2; Q9UHX1-2; P0DJD3-2; P07101-3; Q9UJ04; C9J7I0; Q5MCW4 EC number EC 1.14.16.2 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Cell projection; Cytoplasm; Cytoplasmic vesicle; Disease variant; Dystonia; Iron; Metal-binding; Monooxygenase; Neurotransmitter biosynthesis; Nucleus; Oxidoreductase; Parkinson disease; Parkinsonism; Phosphoprotein; Proteomics identification; Reference proteome; Synapse Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 34997 Length 306 Aromaticity 0.12 Instability index 42.59 Isoelectric point 5.32 Charge (pH=7) -12.31 2D Binding mode Binding energy (Kcal/mol) -5.38 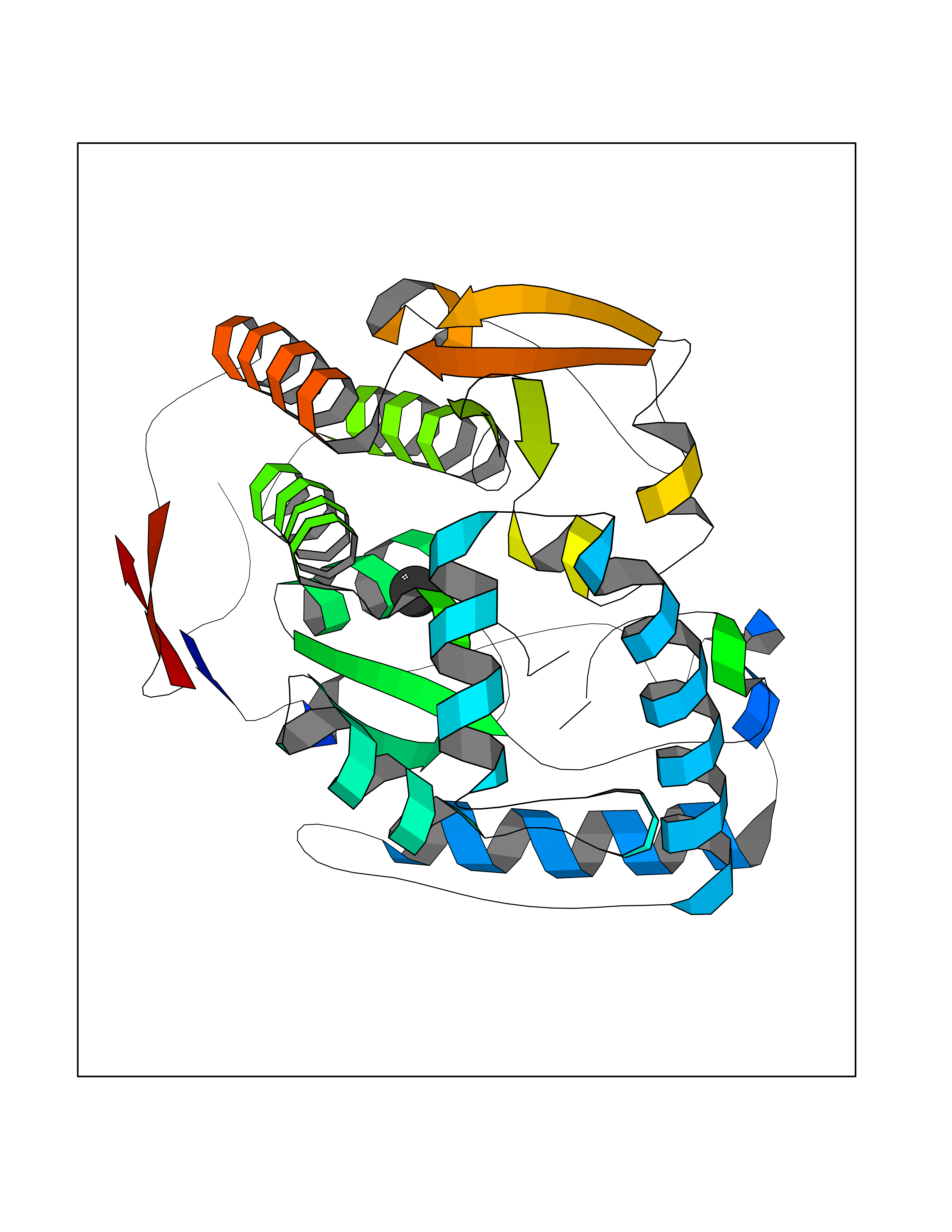 Molscript Map 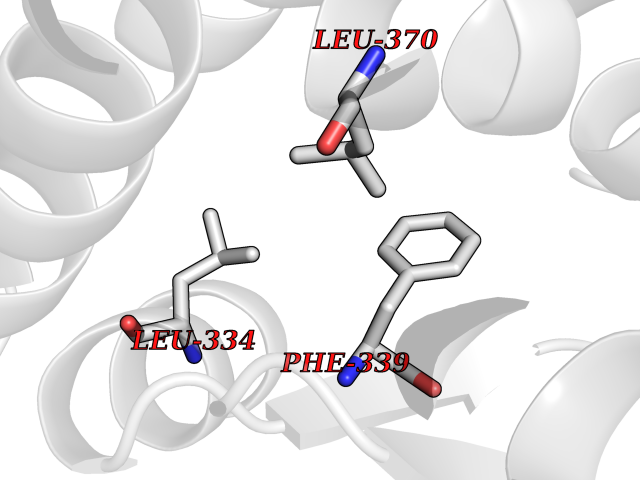 Pymol Map 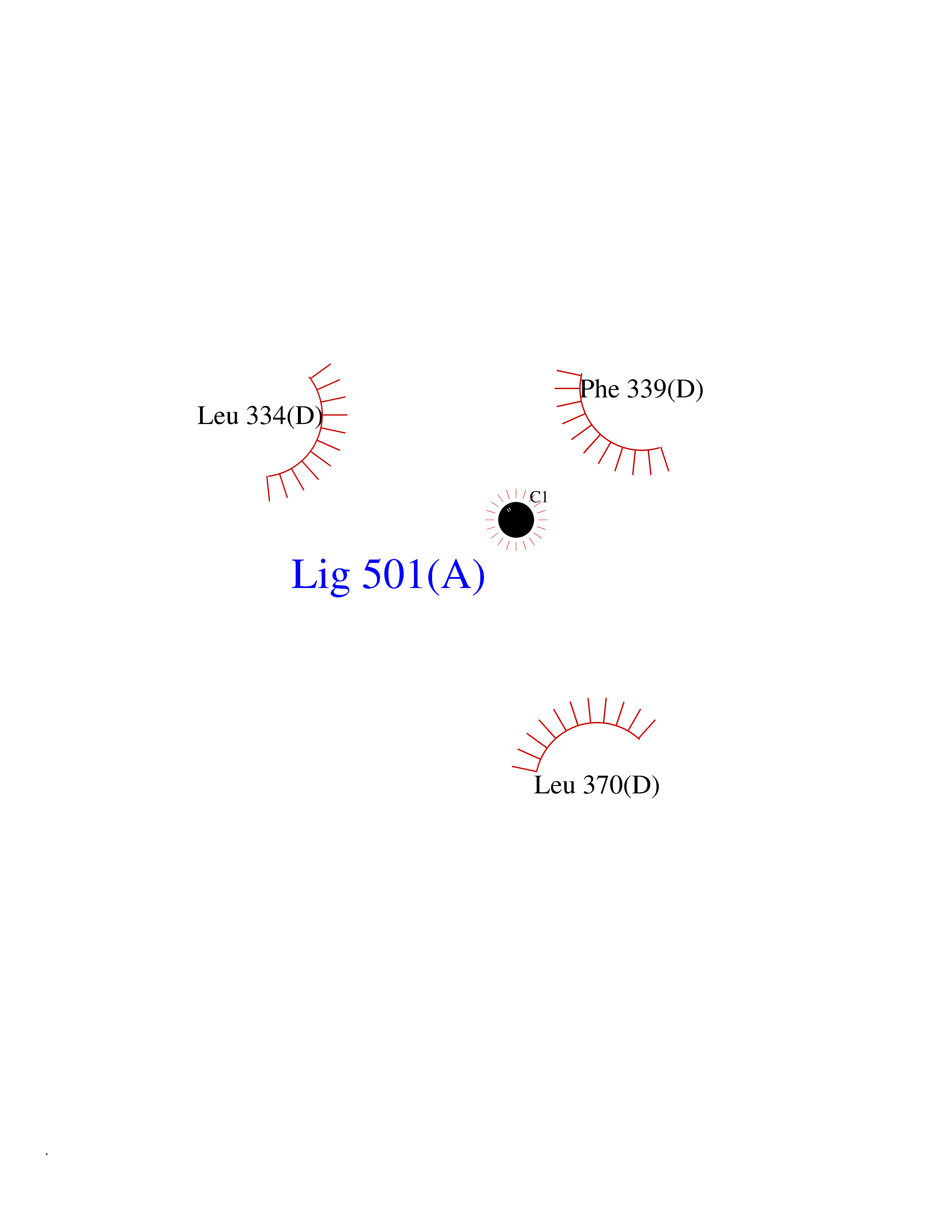 Ligplot Map 3D Binding mode Sequence VPWFPRKVSELDKCHHLVTKFDPDLDLDHPGFSDQVYRQRRKLIAEIAFQYRHGDPIPRVEYTAEEIATWKEVYTTLKGLYATHACGEHLEAFALLERFSGYREDNIPQLEDVSRFLKERTGFQLRPVAGLLSARDFLASLAFRVFQCTQYIRHASSPMHSPEPDCCHELLGHVPMLADRTFAQFSQDIGLASLGASDEEIEKLSTLYWFTVEFGLCKQNGEVKAYGAGLLSSYGELLHCLSEEPEIRAFDPEAAAVQPYQDQTYQSVYFVSESFSDAKDKLRSYASRIQRPFSVKFDPYTLAIDV Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Tyrosine 3-monooxygenase (TH) | 2XSN | 3.94 | |
Target general information Gen name TH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine 3-hydroxylase; TH Protein family Biopterin-dependent aromatic amino acid hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Plays an important role in the physiology of adrenergic neurones. Related diseases Segawa syndrome autosomal recessive (ARSEGS) [MIM:605407]: A form of DOPA-responsive dystonia presenting in infancy or early childhood. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Some cases present with parkinsonian symptoms in infancy. Unlike all other forms of dystonia, it is an eminently treatable condition, due to a favorable response to L-DOPA. {ECO:0000269|PubMed:10585338, ECO:0000269|PubMed:11196107, ECO:0000269|PubMed:11246459, ECO:0000269|PubMed:15505183, ECO:0000269|PubMed:15747353, ECO:0000269|PubMed:16049992, ECO:0000269|PubMed:17696123, ECO:0000269|PubMed:18058633, ECO:0000269|PubMed:18554280, ECO:0000269|PubMed:19491146, ECO:0000269|PubMed:20056467, ECO:0000269|PubMed:20430833, ECO:0000269|PubMed:21940685, ECO:0000269|PubMed:22264700, ECO:0000269|PubMed:22815559, ECO:0000269|PubMed:23762320, ECO:0000269|PubMed:23939262, ECO:0000269|PubMed:24753243, ECO:0000269|PubMed:7814018, ECO:0000269|PubMed:8528210, ECO:0000269|PubMed:8817341, ECO:0000269|PubMed:9613851, ECO:0000269|PubMed:9703425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: May play a role in the pathogenesis of Parkinson disease (PD). A genome-wide copy number variation analysis has identified a 34 kilobase deletion over the TH gene in a PD patient but not in any controls. {ECO:0000269|PubMed:20809526}. Drugs (DrugBank ID) DB03552; DB04400; DB00765; DB00120; DB00360; DB00135 Interacts with P29762; P61978-2; Q99750; P08651-5; O75928-2; Q9UHX1-2; P0DJD3-2; P07101-3; Q9UJ04; C9J7I0; Q5MCW4 EC number EC 1.14.16.2 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Cell projection; Cytoplasm; Cytoplasmic vesicle; Disease variant; Dystonia; Iron; Metal-binding; Monooxygenase; Neurotransmitter biosynthesis; Nucleus; Oxidoreductase; Parkinson disease; Parkinsonism; Phosphoprotein; Proteomics identification; Reference proteome; Synapse Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 34997 Length 306 Aromaticity 0.12 Instability index 42.59 Isoelectric point 5.32 Charge (pH=7) -12.31 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map 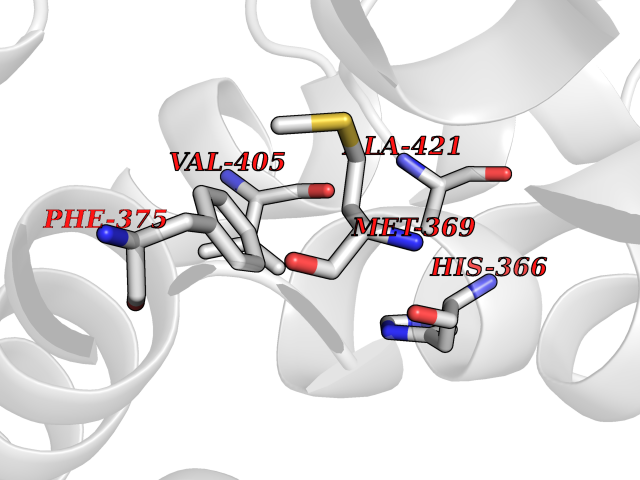 Pymol Map 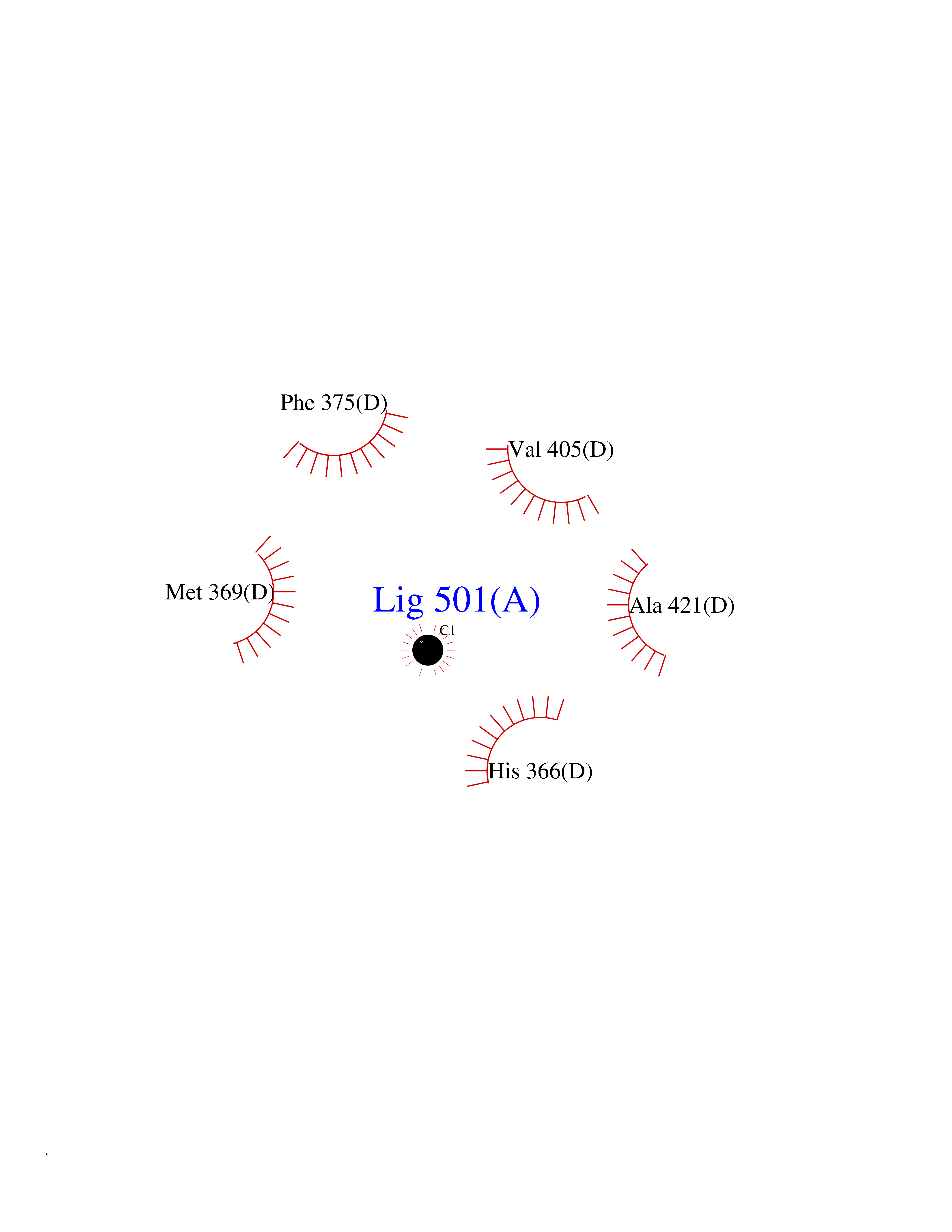 Ligplot Map 3D Binding mode Sequence VPWFPRKVSELDKCHHLVTKFDPDLDLDHPGFSDQVYRQRRKLIAEIAFQYRHGDPIPRVEYTAEEIATWKEVYTTLKGLYATHACGEHLEAFALLERFSGYREDNIPQLEDVSRFLKERTGFQLRPVAGLLSARDFLASLAFRVFQCTQYIRHASSPMHSPEPDCCHELLGHVPMLADRTFAQFSQDIGLASLGASDEEIEKLSTLYWFTVEFGLCKQNGEVKAYGAGLLSSYGELLHCLSEEPEIRAFDPEAAAVQPYQDQTYQSVYFVSESFSDAKDKLRSYASRIQRPFSVKFDPYTLAIDV Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Pyruvate dehydrogenase [ubiquinone] | 3EYA | 3.94 | |
Target general information Gen name poxB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b0871;JW0855 Protein family TPP enzyme family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Identical protein binding.Lipid binding.Magnesium ion binding.Pyruvate dehydrogenase (quinone) activity.Thiamine pyrophosphate binding. Related diseases Glycogen storage disease 6 (GSD6) [MIM:232700]: A metabolic disorder characterized by mild to moderate hypoglycemia, mild ketosis, growth retardation, and prominent hepatomegaly. Heart and skeletal muscle are not affected. {ECO:0000269|PubMed:9529348}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07003 EC number 1.2.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Lipid-binding; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Oxidoreductase; Pyruvate; Reference proteome; Thiamine pyrophosphate; Ubiquinone Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I,J,K,L Molecular weight (Da) 113027 Length 1046 Aromaticity 0.07 Instability index 35.99 Isoelectric point 5.75 Charge (pH=7) -24.38 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map 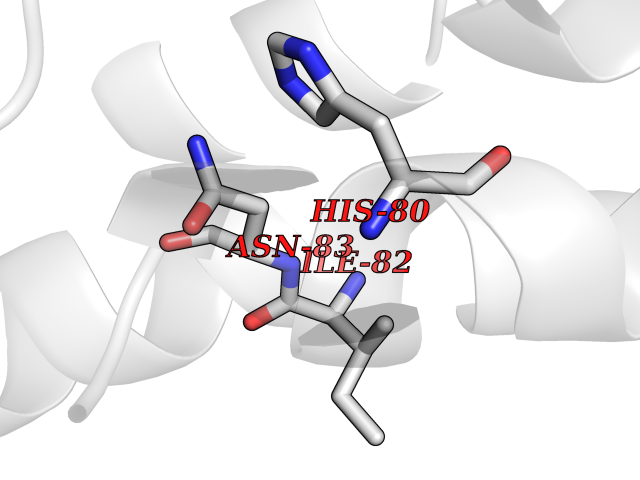 Pymol Map 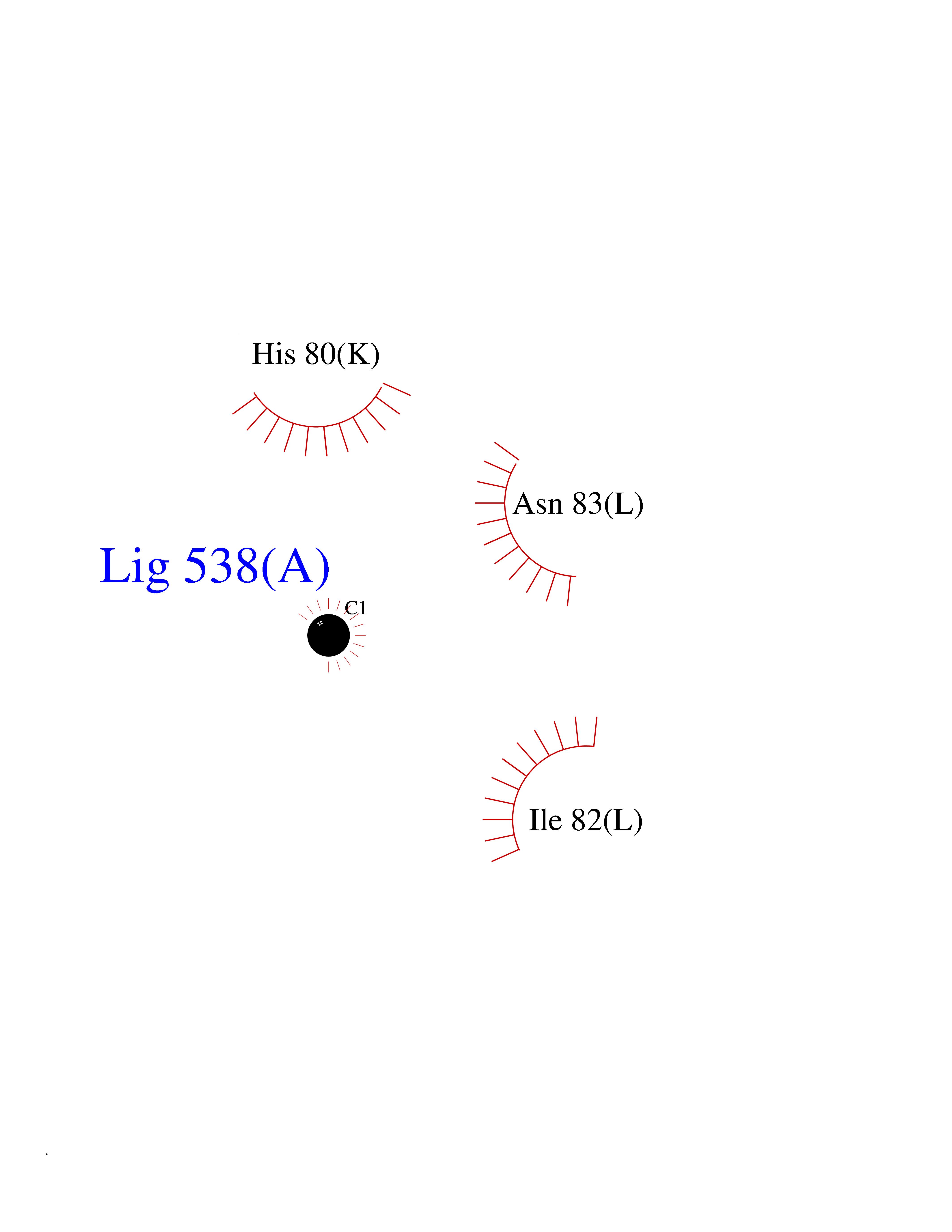 Ligplot Map 3D Binding mode Sequence MKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFDGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIPMKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFVGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIP Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Cytochrome c oxidase subunit 2 | 3VRJ | 3.94 | |
Target general information Gen name MT-CO2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MTCO2;COXII;COII;COX2 Protein family Cytochrome c oxidase subunit 2 family Biochemical class Immune system Function Copper ion binding.Cytochrome-c oxidase activity. Related diseases Mitochondrial complex IV deficiency (MT-C4D) [MIM:220110]: A disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations, ranging from isolated myopathy to severe multisystem disease affecting several tissues and organs. Features include hypertrophic cardiomyopathy, hepatomegaly and liver dysfunction, hypotonia, muscle weakness, exercise intolerance, developmental delay, delayed motor development and intellectual disability. Some affected individuals manifest a fatal hypertrophic cardiomyopathy resulting in neonatal death. A subset of patients manifest Leigh syndrome. {ECO:0000269|PubMed:10486321}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02659; DB04464; DB05412 Interacts with Q9NZ94-2; P49281-3 EC number 7.1.1.9 Uniprot keywords 3D-structure; Copper; Disease variant; Electron transport; Magnesium; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Primary mitochondrial disease; Proteomics identification; Reference proteome; Respiratory chain; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID C Molecular weight (Da) 21687.9 Length 189 Aromaticity 0.11 Instability index 38 Isoelectric point 5.68 Charge (pH=7) -3.26 2D Binding mode Binding energy (Kcal/mol) -5.37 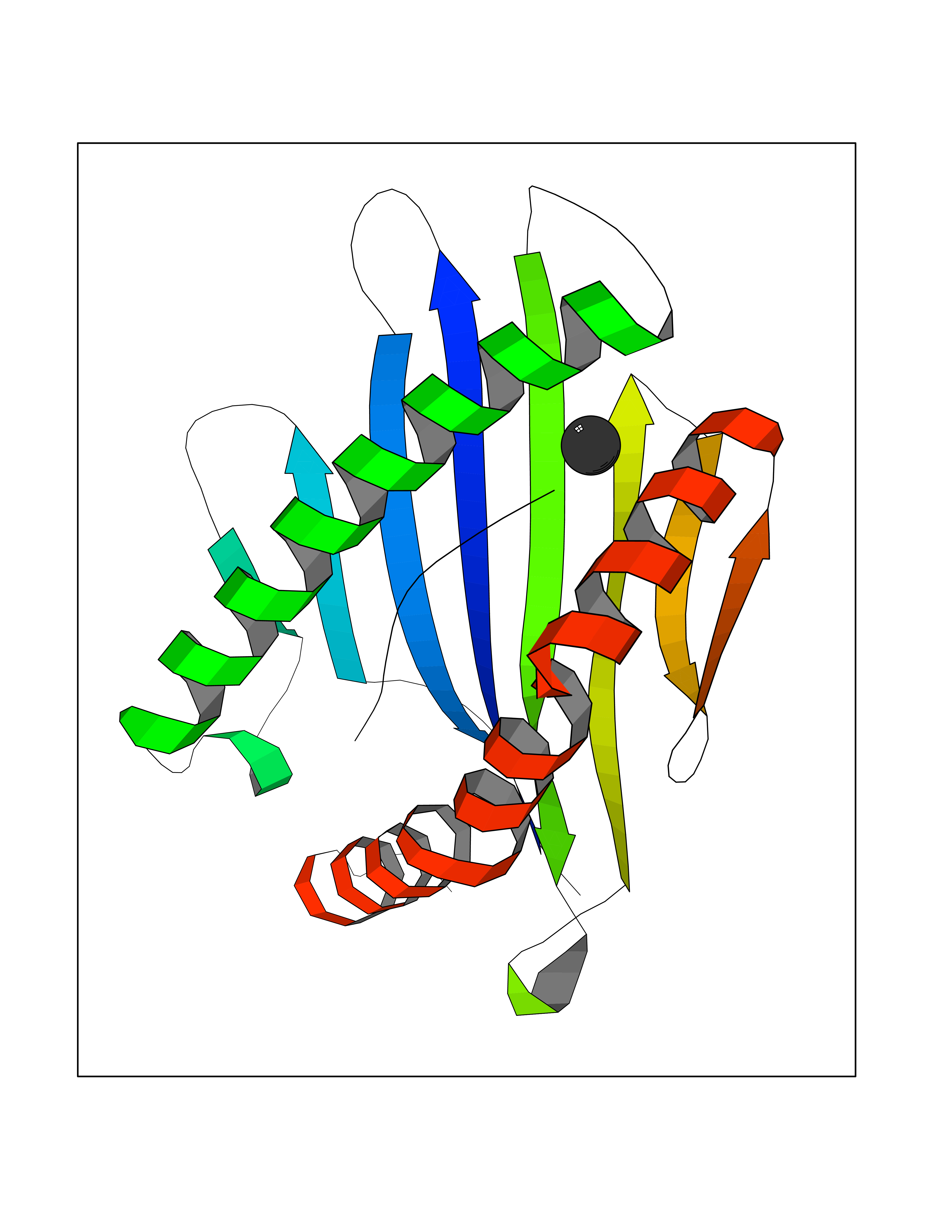 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRMAPRAPWIEQEGPEYWDGETRNMKASAQTYRENLRIALRYYNQSEAGSHIIQVMYGCDVGPDGRLLRGHDQSAYDGKDYIALNEDLSSWTAADTAAQITQRKWEAARVAEQLRAYLEGLCVEWLRRYLENGKETLQLTTKLTNTNI Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Hypoxanthine-guanine phosphoribosyltransferase | 5HIA | 3.94 | |
Target general information Gen name HPRT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HPRT Protein family Purine/pyrimidine phosphoribosyltransferase family Biochemical class Transferase Function Guanine phosphoribosyltransferase activity.Hypoxanthine phosphoribosyltransferase activity.Identical protein binding.Magnesium ion binding.Nucleotide binding.Protein homodimerization activity. Related diseases Lesch-Nyhan syndrome (LNS) [MIM:300322]: Characterized by complete lack of enzymatic activity that results in hyperuricemia, choreoathetosis, intellectual disability, and compulsive self-mutilation. {ECO:0000269|PubMed:15571223, ECO:0000269|PubMed:17027311, ECO:0000269|PubMed:20544509, ECO:0000269|PubMed:2071157, ECO:0000269|PubMed:2246854, ECO:0000269|PubMed:2347587, ECO:0000269|PubMed:2358296, ECO:0000269|PubMed:24940672, ECO:0000269|PubMed:2572141, ECO:0000269|PubMed:2910902, ECO:0000269|PubMed:3265398, ECO:0000269|PubMed:3384338, ECO:0000269|PubMed:6853716, ECO:0000269|PubMed:7627191, ECO:0000269|PubMed:9452051}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperuricemia, HPRT-related (HRH) [MIM:300323]: An X-linked metabolic disorder characterized by uric acid excess in the blood, renal stones, uric acid nephropathy, and renal obstruction. After puberty, the hyperuricemia may cause gout. {ECO:0000269|PubMed:15571223, ECO:0000269|PubMed:17027311, ECO:0000269|PubMed:20544509, ECO:0000269|PubMed:24940672, ECO:0000269|PubMed:2896620, ECO:0000269|PubMed:2909537, ECO:0000269|PubMed:3198771, ECO:0000269|PubMed:3358423, ECO:0000269|PubMed:6572373, ECO:0000269|PubMed:6706936, ECO:0000269|PubMed:6853490, ECO:0000269|PubMed:7987318}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03153; DB02309; DB01632; DB04356; DB00993; DB01033; DB00352 Interacts with Q96G04; Q8N7B9-2; Q14749; P00492; P42858; Q9H1K1; Q96HA8; Q9NRG1; O00560; Q9NZD8; Q7Z699; Q8N9Q2 EC number 2.4.2.8 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Gout; Isopeptide bond; Magnesium; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 23099.5 Length 205 Aromaticity 0.09 Instability index 18.85 Isoelectric point 6.2 Charge (pH=7) -2.09 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map  Pymol Map 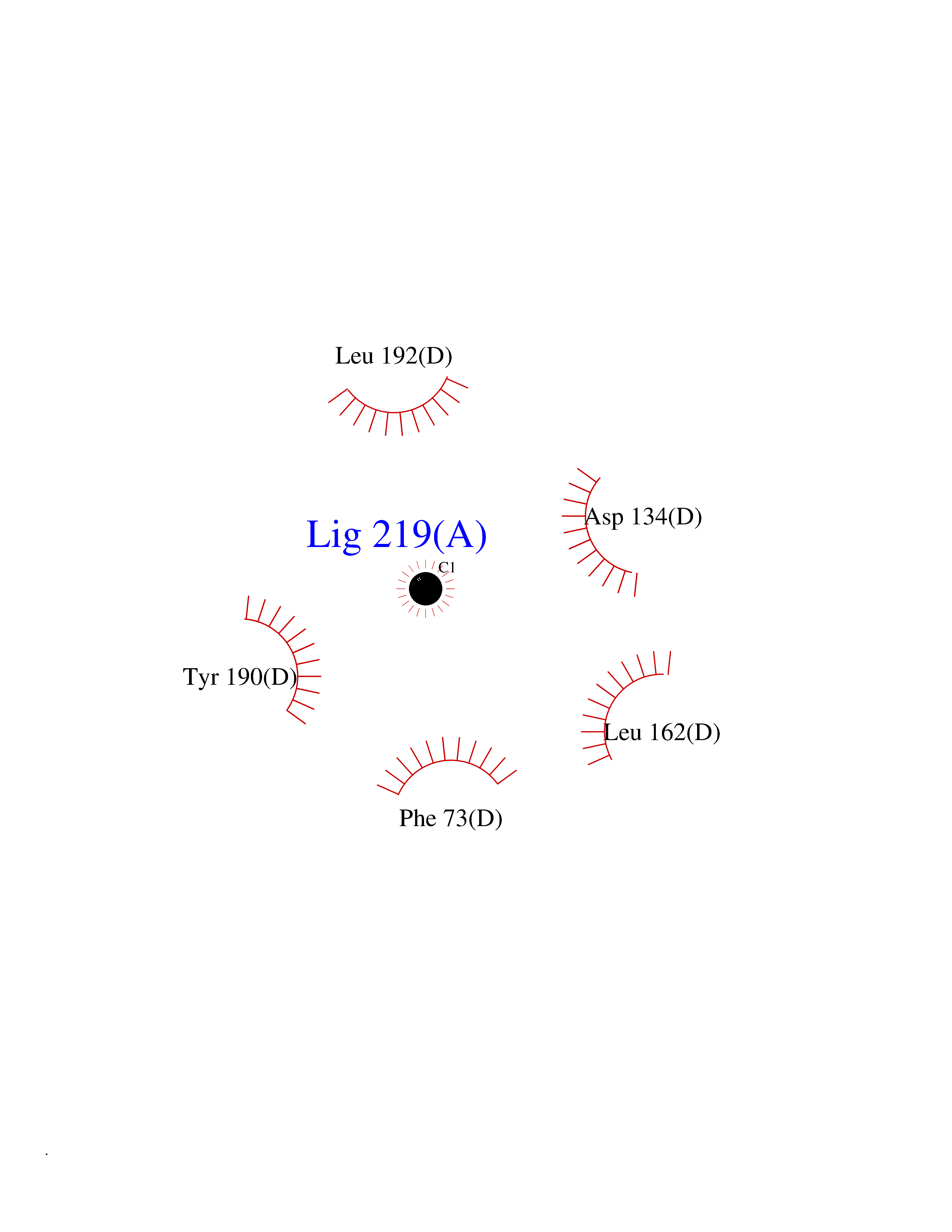 Ligplot Map 3D Binding mode Sequence SPGVVISDDEPGYDLDLFAIPNHYAEDLERVFIPHGLIMDRTERLARDVMKEMGGHHIVALCVLKGGYKFFADLLDYIKALNRNSDRSIPMTVDFIRLKDIKVIGGDDLSTLTGKNVLIVEDIIDTGKTMQTLLSLVRQYNPKMVKVASLLVKRTPRSVGYKPDFVGFEIPDKFVVGYALDYNEYFRDLNHVAVISETGKAKYKA Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Vitamin D3 receptor (VDR) | 3B0T | 3.94 | |
Target general information Gen name VDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin D(3) receptor; Nuclear vitamin D receptor; Nuclear receptor subfamily 1 group I member 1; NR1I1; 1,25-dihydroxyvitamin D3 receptor Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Enters the nucleus upon vitamin D3 binding where it forms heterodimers with the retinoid X receptor/RXR. The VDR-RXR heterodimers bind to specific response elements on DNA and activate the transcription of vitamin D3-responsive target genes. Plays a central role in calcium homeostasis. Nuclear receptor for calcitriol, the active form of vitamin D3 which mediates the action of this vitamin on cells. Related diseases Rickets vitamin D-dependent 2A (VDDR2A) [MIM:277440]: A disorder of vitamin D metabolism resulting in severe rickets, hypocalcemia and secondary hyperparathyroidism. Most patients have total alopecia in addition to rickets. {ECO:0000269|PubMed:1652893, ECO:0000269|PubMed:17970811, ECO:0000269|PubMed:2177843, ECO:0000269|PubMed:2849209, ECO:0000269|PubMed:28698609, ECO:0000269|PubMed:7828346, ECO:0000269|PubMed:8106618, ECO:0000269|PubMed:8381803, ECO:0000269|PubMed:8392085, ECO:0000269|PubMed:8675579, ECO:0000269|PubMed:8961271, ECO:0000269|PubMed:9005998}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07530; DB08742; DB01436; DB04891; DB00146; DB02300; DB00136; DB00169; DB04540; DB05024; DB11672; DB14635; DB01070; DB06410; DB05295; DB06194; DB00153; DB04796; DB03451; DB00910; DB04258; DB11094 Interacts with P35222; Q09472; Q15648; P50222; Q15788; P26045; P19793; Q13573; Q13501; P04637; Q15645; Q9JLI4; P28700; X5D778; Q96HA8; Q01804; Q96S38; P48443 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 28781 Length 254 Aromaticity 0.07 Instability index 47.69 Isoelectric point 6.15 Charge (pH=7) -3.44 2D Binding mode Binding energy (Kcal/mol) -5.37 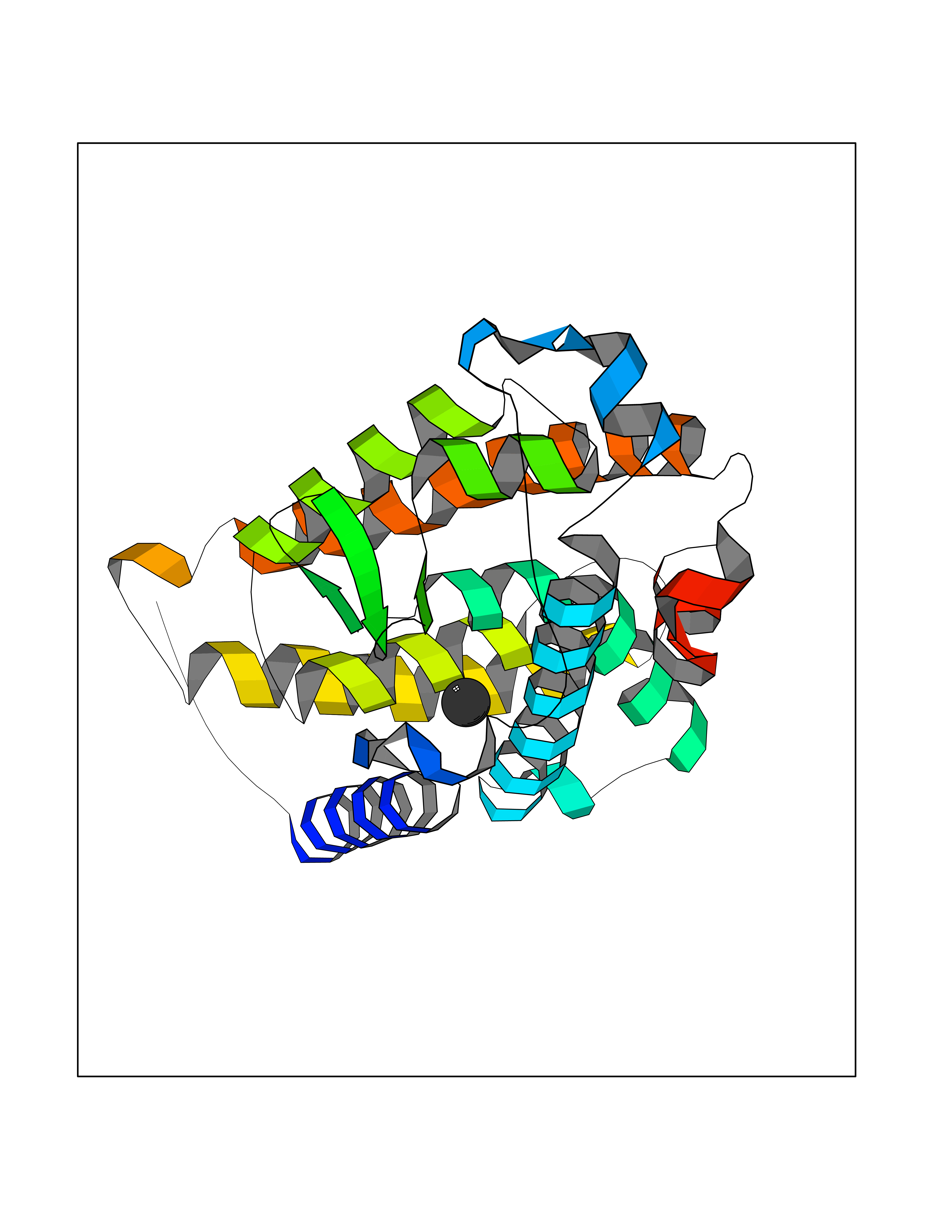 Molscript Map 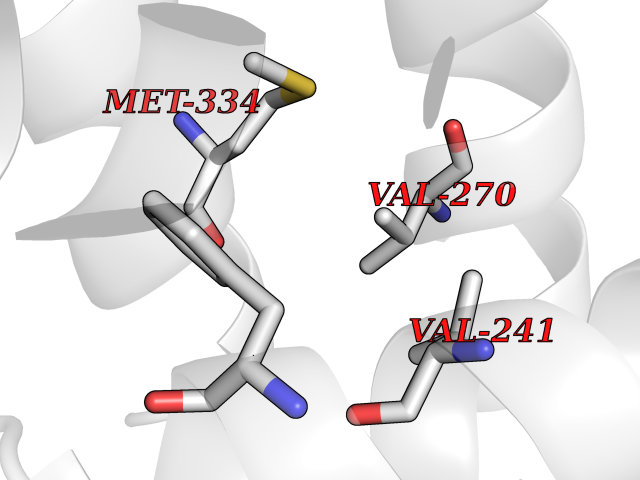 Pymol Map 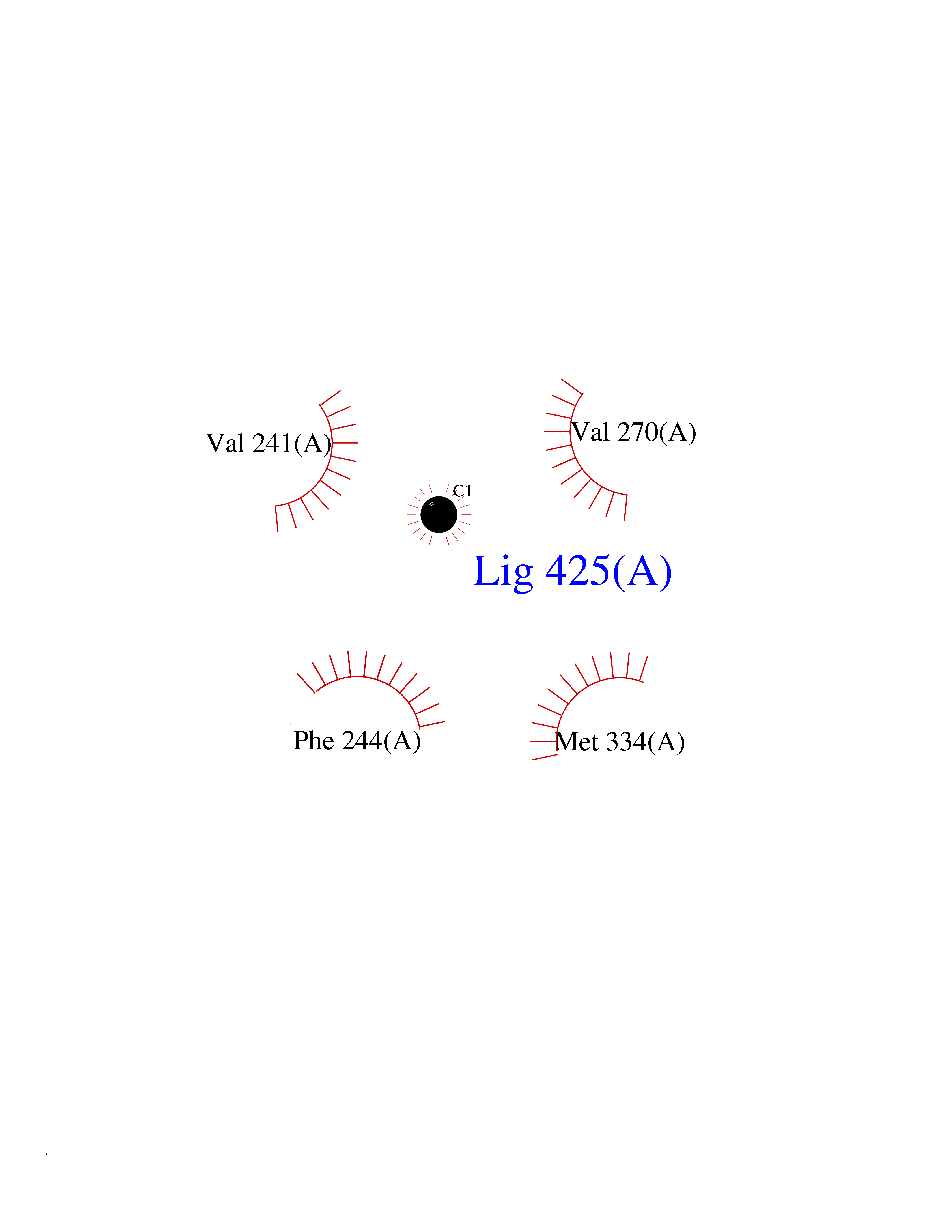 Ligplot Map 3D Binding mode Sequence ALRPKLSEEQQRIIAILLDAHHKTYDPTYSDFCQFRPPVRVNDGGGSVTLELSQLSMLPHLADLVSYSIQKVIGFAKMIPGFRDLTSEDQIVLLKSSAIEVIMLRSNESFTMDDMSWTCGNQDYKYRVSDVTKAGHSLELIEPLIKFQVGLKKLNLHEEEHVLLMAICIVSPDRPGVQDAALIEAIQDRLSNTLQTYIRCRHPPPGSHLLYAKMIQKLADLRSLNEEHSKQYRCLSFQPECSMKLTPLVLEVFG Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Dopamine beta-hydroxylase | 4ZEL | 3.94 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -5.37 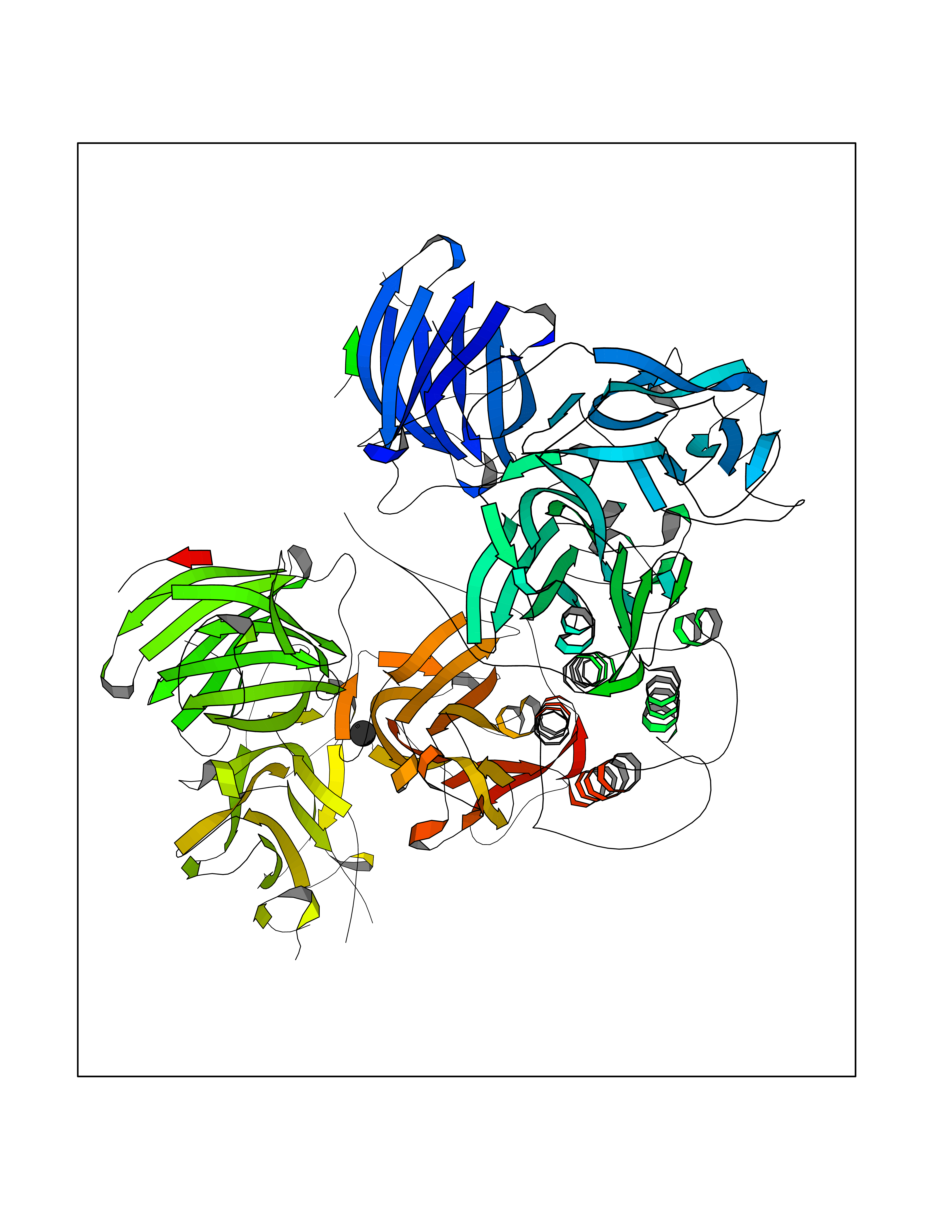 Molscript Map 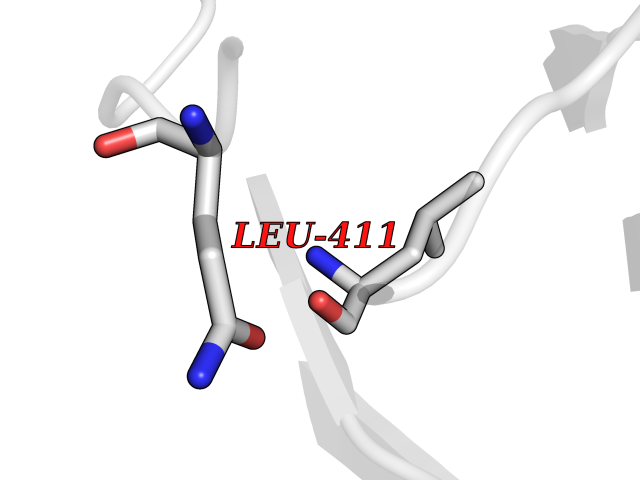 Pymol Map 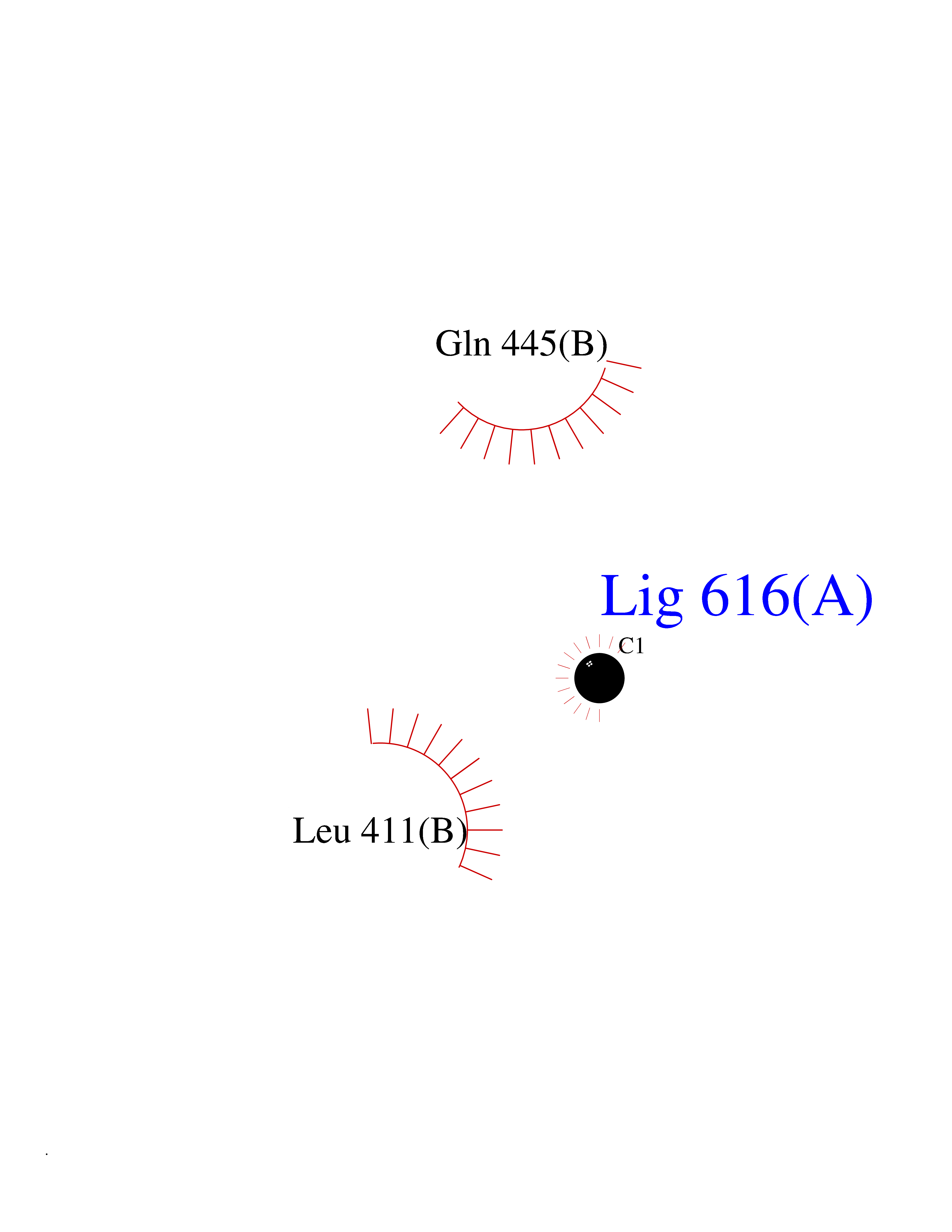 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Natriuretic peptides B | 1YK1 | 3.94 | |
Target general information Gen name NPPB Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Natriuretic peptide family Biochemical class Hormone / growth factor receptor Function Diuretic hormone activity.Hormone activity.Peptide hormone receptor binding.Receptor binding. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01136; DB06412 Interacts with A8MQ03; P57678; Q6A162; P60411; Q7Z3S9; P25788; Q9UJW9 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disulfide bond; Glycoprotein; Hormone; Pharmaceutical; Proteoglycan; Proteomics identification; Reference proteome; Secreted; Signal; Vasoactive; Vasodilator Protein physicochemical properties Chain ID E Molecular weight (Da) 46353.1 Length 415 Aromaticity 0.1 Instability index 37.91 Isoelectric point 5.51 Charge (pH=7) -12.09 2D Binding mode Binding energy (Kcal/mol) -5.38 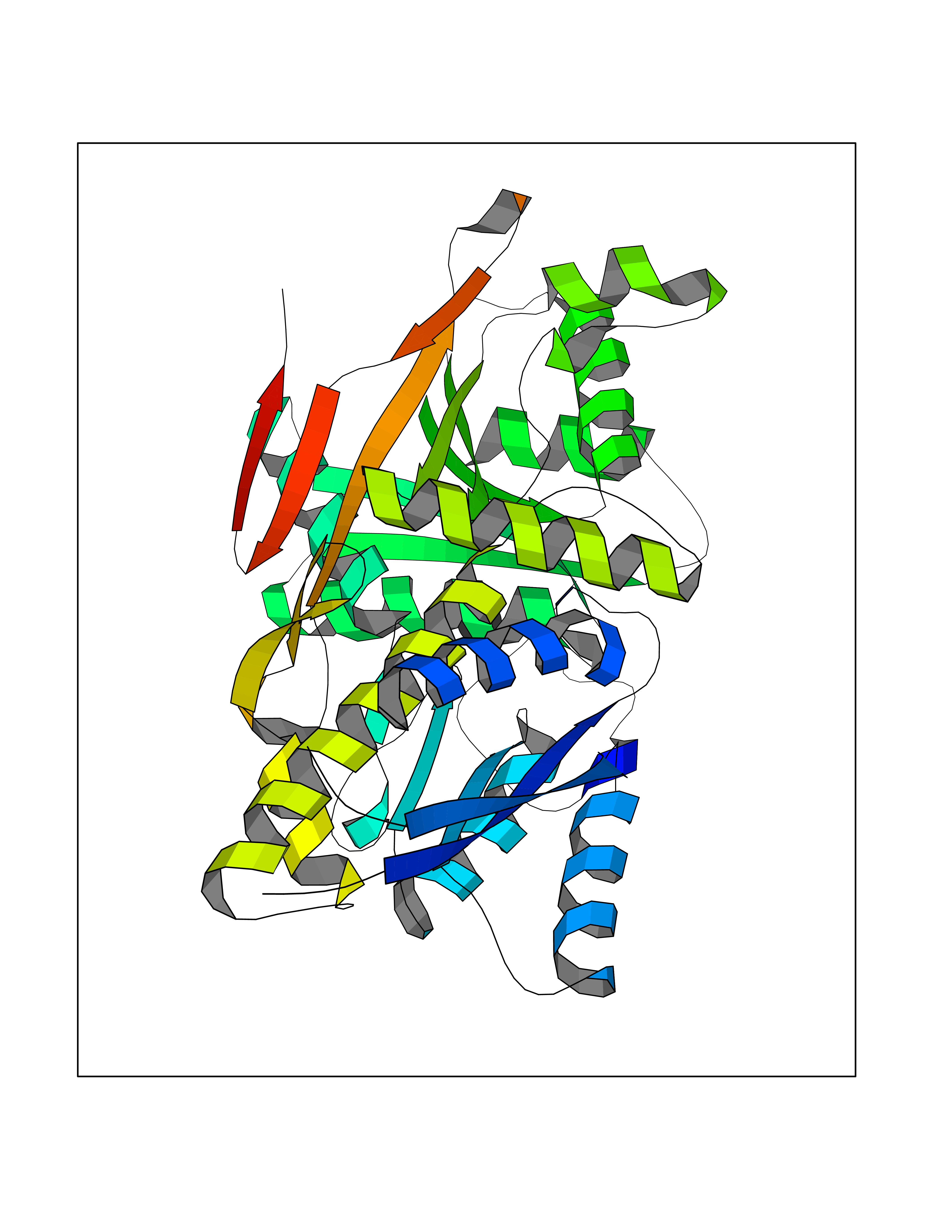 Molscript Map 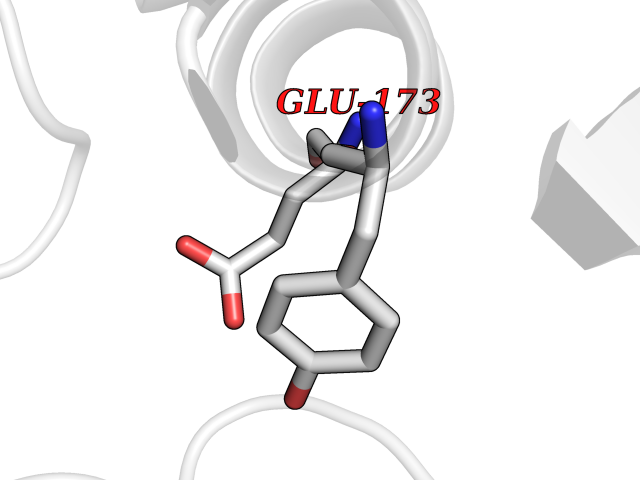 Pymol Map 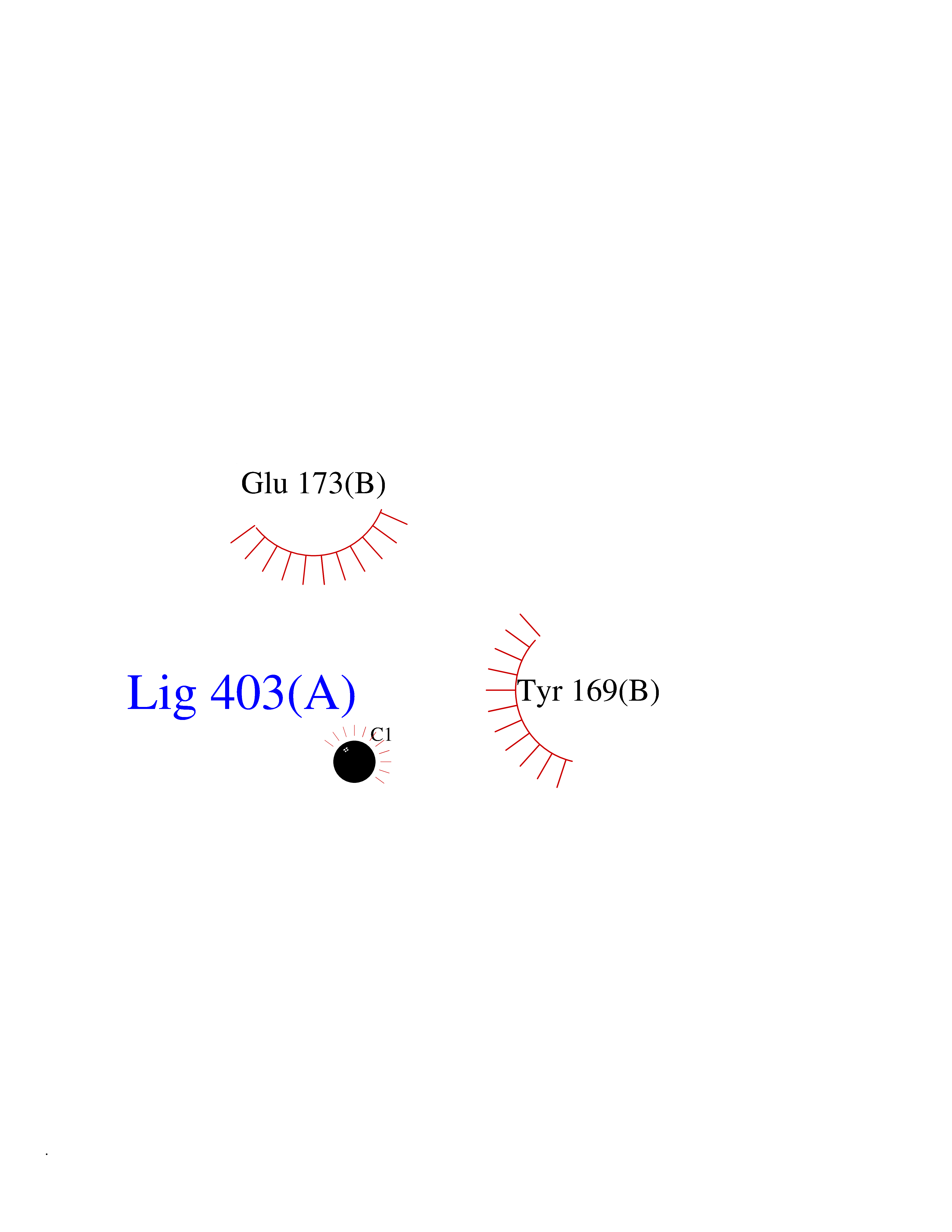 Ligplot Map 3D Binding mode Sequence GCFGRKMDRISSSSGLGCKVLALPPQKIEVLVLLPQDDSYLFSLTRVRPAIEYALRSVEGLLPPGTRFQVAYEDSDCGNRALFSLVDRVAAARGAKPDLILGPVCEYAAAPVARLASHWDLPMLSAGALAAGFQHKDSEYSHLTRVAPAYAKMGEMMLALFRHHHWSRAALVYSDDKLERNCYFTLEGVHEVFQEEGLHTSIYSFDETKDLDLEDIVRNIQASERVVIMCASSDTIRSIMLVAHRHGMTSGDYAFFNIELFNSSSYGDGSWKRGDKHDFEAKQAYSSLQTVTLLRTVKPEFEKFSMEVKSSVEKQGLNMEDYVNMFVEGFHDAILLYVLALHEVLRAGYSKKDGGKIIQQTWNRTFEGIAGQVSIDANGDRYGDFSVIAMTDVEAGTQEVIGDYFGKEGRFEMRP Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Prothrombin | 4UD9 | 3.94 | |
Target general information Gen name F2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase S1 family Biochemical class Hydrolase Function Calcium ion binding.Growth factor activity.Heparin binding.Lipopolysaccharide binding.Receptor binding.Serine-type endopeptidase activity.Thrombospondin receptor activity. Related diseases Factor II deficiency (FA2D) [MIM:613679]: A very rare blood coagulation disorder characterized by mucocutaneous bleeding symptoms. The severity of the bleeding manifestations correlates with blood factor II levels. {ECO:0000269|PubMed:1349838, ECO:0000269|PubMed:1354985, ECO:0000269|PubMed:1421398, ECO:0000269|PubMed:14962227, ECO:0000269|PubMed:2719946, ECO:0000269|PubMed:3242619, ECO:0000269|PubMed:3567158, ECO:0000269|PubMed:3771562, ECO:0000269|PubMed:3801671, ECO:0000269|PubMed:6405779, ECO:0000269|PubMed:7792730, ECO:0000269|PubMed:7865694}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Thrombophilia due to thrombin defect (THPH1) [MIM:188050]: A multifactorial disorder of hemostasis characterized by abnormal platelet aggregation in response to various agents and recurrent thrombi formation. {ECO:0000269|PubMed:2825773}. The disease is caused by variants affecting the gene represented in this entry. A common genetic variation in the 3-prime untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increased risk of venous thrombosis.; DISEASE: Pregnancy loss, recurrent, 2 (RPRGL2) [MIM:614390]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:11506076}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07211; DB07796; DB07016; DB07521; DB06850; DB07091; DB06845; DB07088; DB07131; DB07095; DB07515; DB07897; DB06878; DB06947; DB08624; DB06869; DB06929; DB07400; DB04771; DB04772; DB02287; DB07277; DB07550; DB07549; DB07548; DB07105; DB04722; DB07366; DB08254; DB01725; DB08062; DB07639; DB07461; DB07120; DB07190; DB07741; DB07353; DB07508; DB07809; DB08546; DB08061; DB07718; DB03136; DB02723; DB07440; DB07376; DB06861; DB06866; DB06865; DB03865; DB06841; DB07934; DB08422; DB07659; DB07660; DB07658; DB13151; DB00025; DB11166; DB00278; DB01766; DB07083; DB00006; DB00100; DB13152; DB09228; DB09130; DB03159; DB06911; DB06996; DB06919; DB07027; DB07133; DB07143; DB07005; DB06695; DB00055; DB01225; DB05714; DB12831; DB03847; DB07278; DB01767; DB06404; DB09332; DB00001; DB13998; DB04136; DB00170; DB06838; DB13999; DB06868; DB06942; DB06936; DB07165; DB07527; DB07522; DB07665; DB07946; DB06859; DB06853; DB06858; DB07279; DB08187; DB04591; DB07944; DB07128; DB12598; DB01123; DB04786; DB05777; DB04697; DB09109; DB14738; DB04898; DB01593; DB14487; DB08152 Interacts with P05067; P07204; Q846V4; PRO_0000032489 [P01008] EC number 3.4.21.5 Uniprot keywords 3D-structure; Acute phase; Blood coagulation; Calcium; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; Gamma-carboxyglutamic acid; Glycoprotein; Hemostasis; Hydrolase; Kringle; Pharmaceutical; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Thrombophilia; Zymogen Protein physicochemical properties Chain ID H Molecular weight (Da) 29321.6 Length 254 Aromaticity 0.1 Instability index 39.57 Isoelectric point 8.56 Charge (pH=7) 4.16 2D Binding mode Binding energy (Kcal/mol) -5.38  Molscript Map  Pymol Map 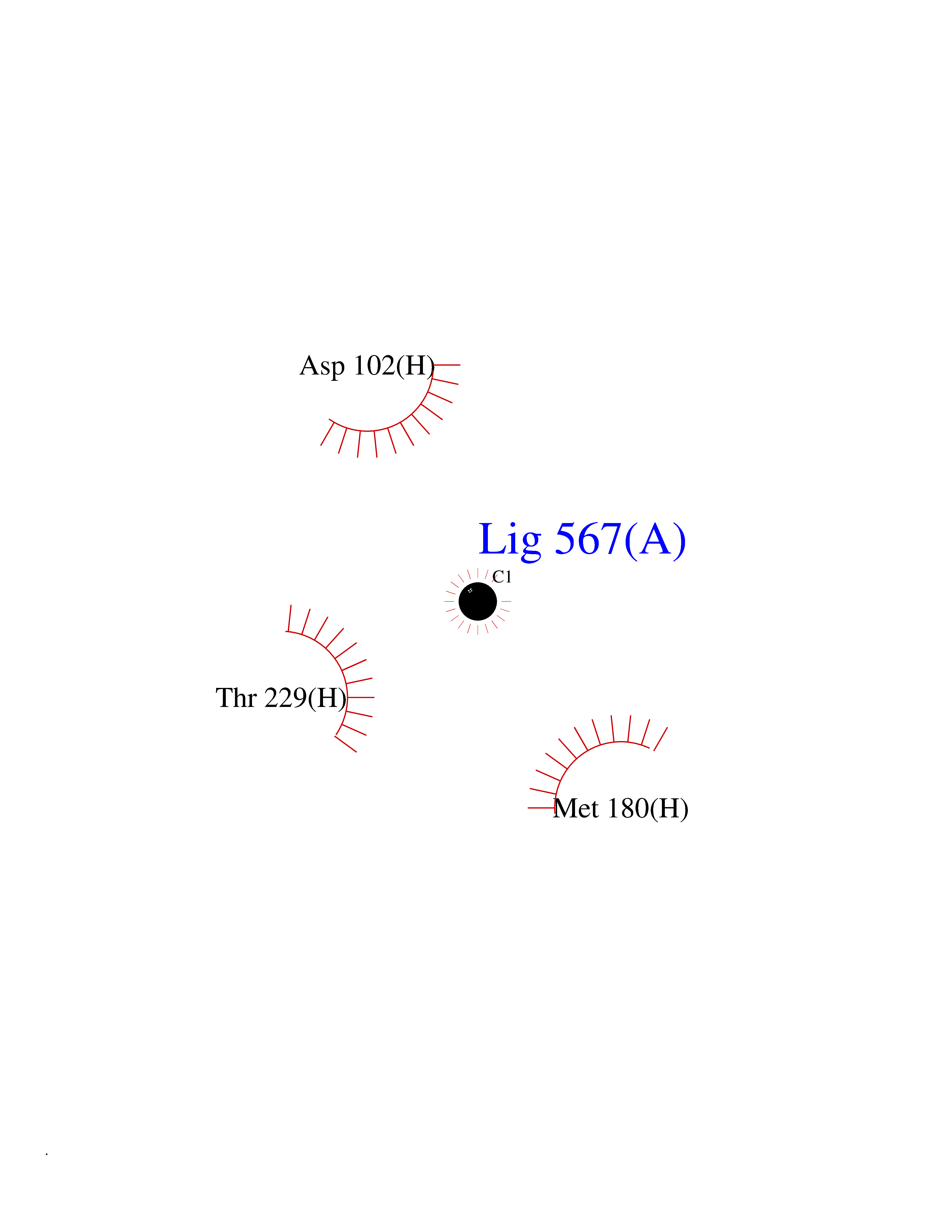 Ligplot Map 3D Binding mode Sequence IVEGSDAEIGMSPWQVMLFRSPQELLCGASLISDRWVLTAAHCLLTENDLLVRIGKHSRTRYRNIEKISMLEKIYIHPRYNWENLDRDIALMKLKKPVAFSDYIHPVCLPDRETLLQAGYKGRVTGWGNLKETWGQPSVLQVVNLPIVERPVCKDSTRIRITDNMFCAYKKRGDACEGDSGGPFVMKSNNRWYQMGIVSWGEGCRDGKYGFYTHVFRLKKWIQKVIDQFGGDFEEIPEELQCGLRPLFEKKSLE Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Non-heme chloroperoxidase | 1A8U | 3.94 | |
Target general information Gen name cpo Organism Kitasatospora aureofaciens (Streptomyces aureofaciens) Uniprot ID TTD ID NA Synonyms cpoT Protein family AB hydrolase superfamily, Bacterial non-heme haloperoxidase / perhydrolase family Biochemical class Haloperoxidase Function Chloride peroxidase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB03793 Interacts with NA EC number 1.11.1.- Uniprot keywords 3D-structure; Chloride; Oxidoreductase; Peroxidase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60428.4 Length 554 Aromaticity 0.13 Instability index 31.26 Isoelectric point 4.65 Charge (pH=7) -32.72 2D Binding mode Binding energy (Kcal/mol) -5.37 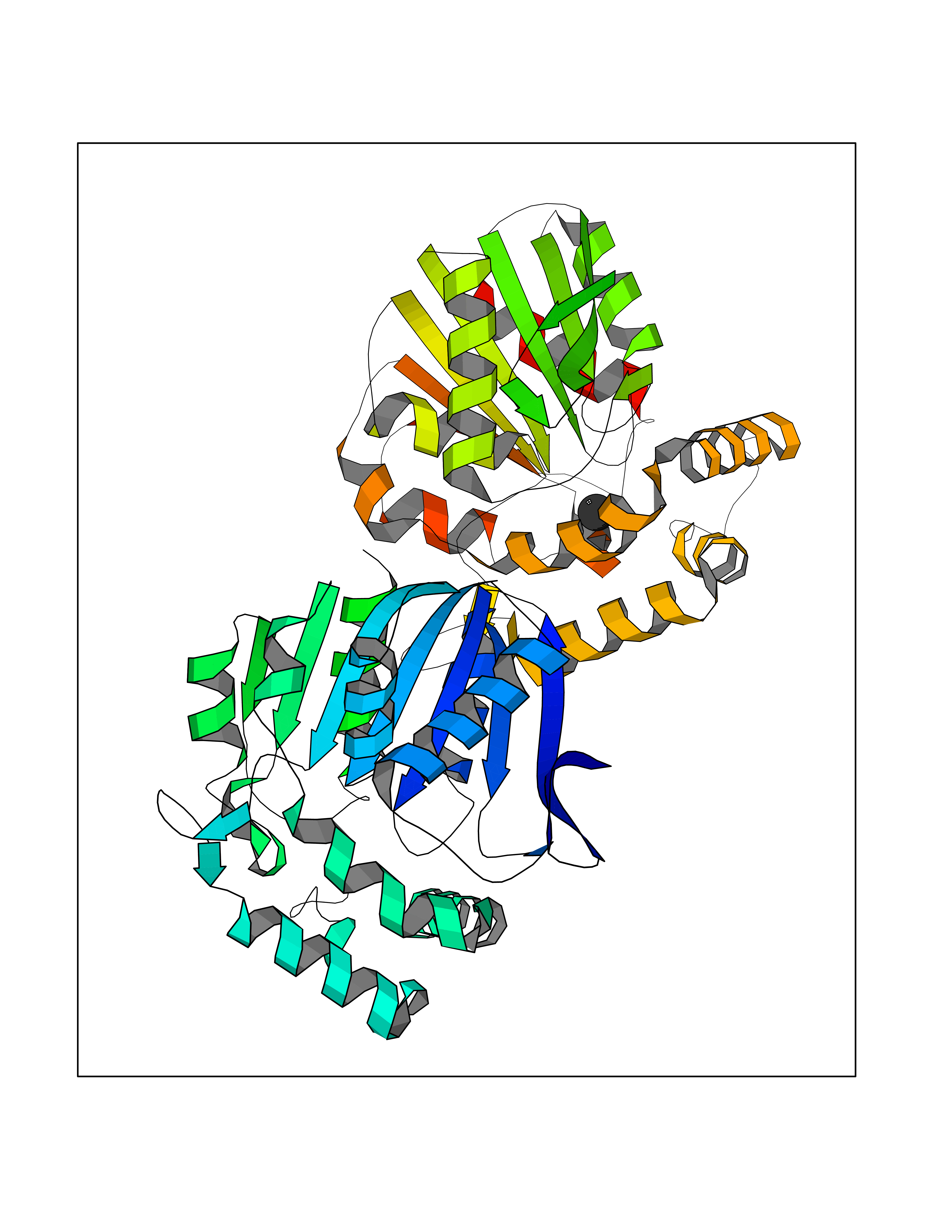 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAKPFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Protein kinase C beta (PRKCB) | 2I0E | 3.94 | |
Target general information Gen name PRKCB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein kinase C beta type; PRKCB1; PKCB; PKC-beta; PKC-B Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, PKC subfamily Biochemical class Kinase Function Plays a key role in B-cell activation by regulating BCR-induced NF-kappa-B activation. Mediates the activation of the canonical NF-kappa-B pathway (NFKB1) by direct phosphorylation of CARD11/CARMA1 at 'Ser-559', 'Ser-644' and 'Ser-652'. Phosphorylation induces CARD11/CARMA1 association with lipid rafts and recruitment of the BCL10-MALT1 complex as well as MAP3K7/TAK1, which then activates IKK complex, resulting in nuclear translocation and activation of NFKB1. Plays a direct role in the negative feedback regulation of the BCR signaling, by down-modulating BTK function via direct phosphorylation of BTK at 'Ser-180', which results in the alteration of BTK plasma membrane localization and in turn inhibition of BTK activity. Involved in apoptosis following oxidative damage: in case of oxidative conditions, specifically phosphorylates 'Ser-36' of isoform p66Shc of SHC1, leading to mitochondrial accumulation of p66Shc, where p66Shc acts as a reactive oxygen species producer. Acts as a coactivator of androgen receptor (ANDR)-dependent transcription, by being recruited to ANDR target genes and specifically mediating phosphorylation of 'Thr-6' of histone H3 (H3T6ph), a specific tag for epigenetic transcriptional activation that prevents demethylation of histone H3 'Lys-4' (H3K4me) by LSD1/KDM1A. In insulin signaling, may function downstream of IRS1 in muscle cells and mediate insulin-dependent DNA synthesis through the RAF1-MAPK/ERK signaling cascade. May participate in the regulation of glucose transport in adipocytes by negatively modulating the insulin-stimulated translocation of the glucose transporter SLC2A4/GLUT4. Under high glucose in pancreatic beta-cells, is probably involved in the inhibition of the insulin gene transcription, via regulation of MYC expression. In endothelial cells, activation of PRKCB induces increased phosphorylation of RB1, increased VEGFA-induced cell proliferation, and inhibits PI3K/AKT-dependent nitric oxide synthase (NOS3/eNOS) regulation by insulin, which causes endothelial dysfunction. Also involved in triglyceride homeostasis. Phosphorylates ATF2 which promotes cooperation between ATF2 and JUN, activating transcription. Calcium-activated, phospholipid- and diacylglycerol (DAG)-dependent serine/threonine-protein kinase involved in various cellular processes such as regulation of the B-cell receptor (BCR) signalosome, oxidative stress-induced apoptosis, androgen receptor-dependent transcription regulation, insulin signaling and endothelial cells proliferation. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14001; DB09096; DB08862; DB14002; DB04209; DB08846; DB06486; DB01738; DB02010; DB00675; DB00163 Interacts with Q8NEM2; Q9BSI4; O60341-1; Q6DHV7-2; Q9UQM7; Q6ZQX7-4; O60346; Q6ZVD8 EC number EC 2.7.11.13 Uniprot keywords 3D-structure; Acetylation; Adaptive immunity; Alternative splicing; Apoptosis; ATP-binding; Calcium; Chromatin regulator; Cytoplasm; Direct protein sequencing; Immunity; Kinase; Membrane; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Serine/threonine-protein kinase; Transcription; Transcription regulation; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34383.3 Length 302 Aromaticity 0.13 Instability index 34.04 Isoelectric point 5.22 Charge (pH=7) -10.63 2D Binding mode Binding energy (Kcal/mol) -5.37 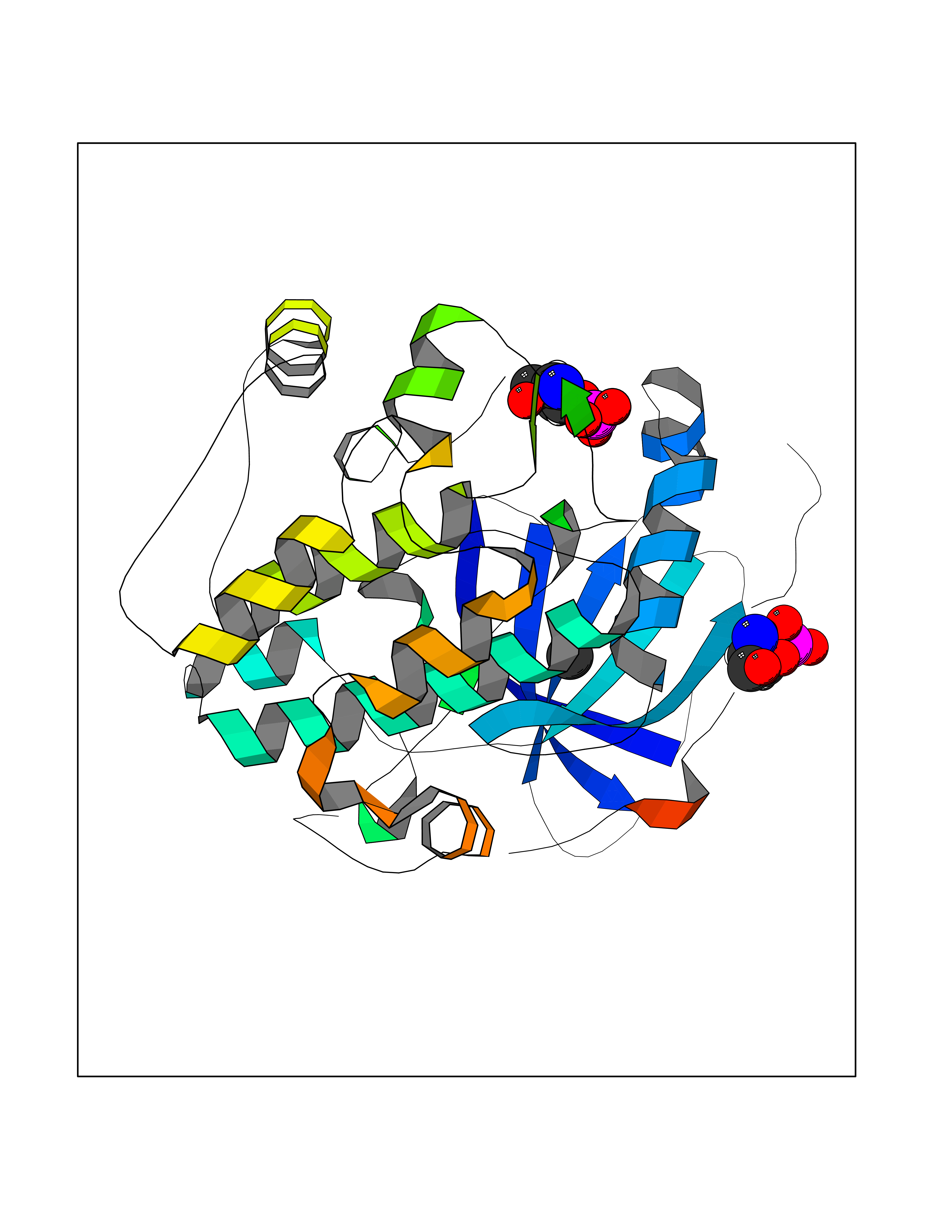 Molscript Map 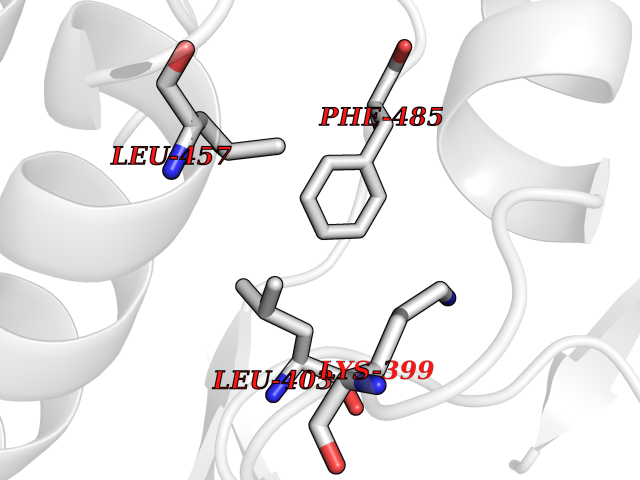 Pymol Map 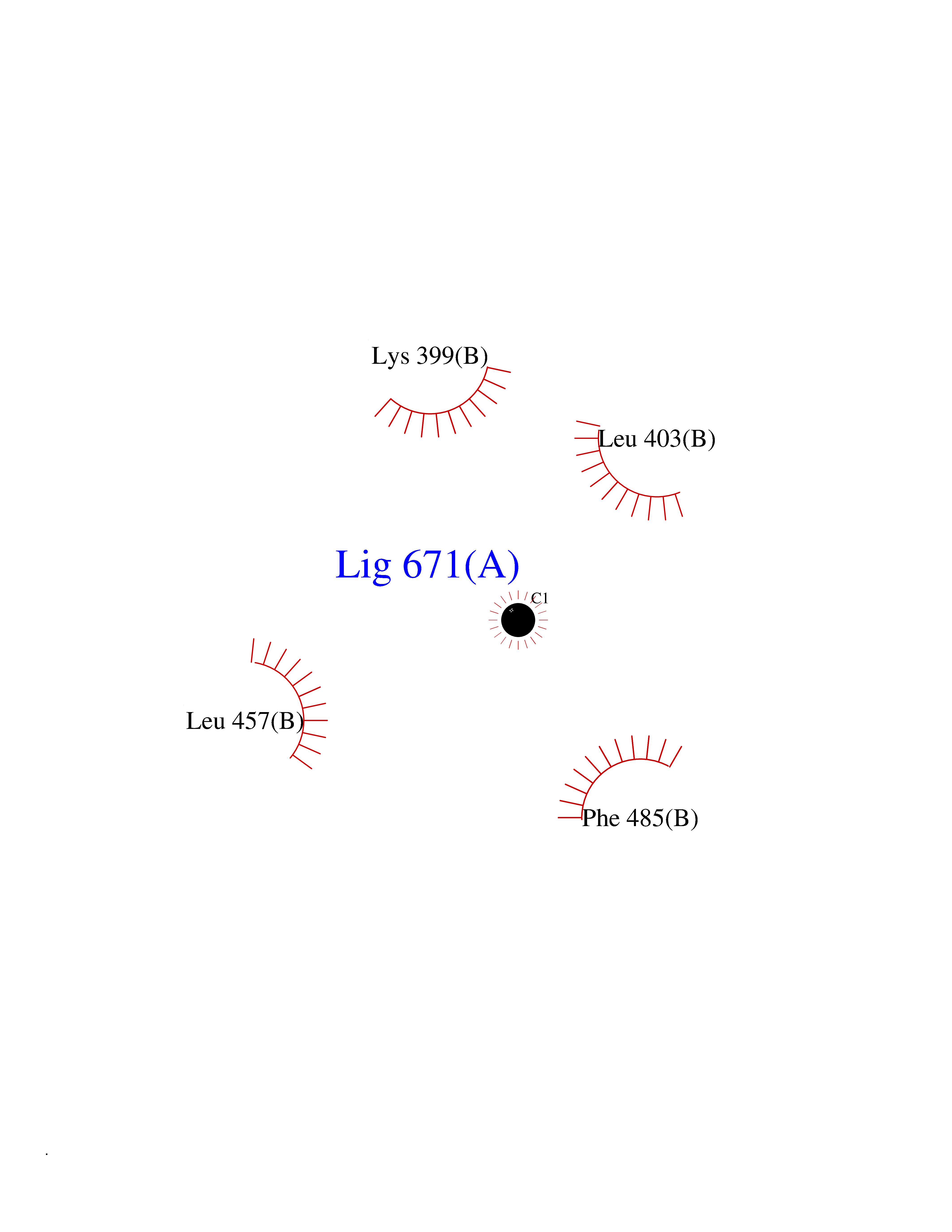 Ligplot Map 3D Binding mode Sequence LTDFNFLMVLGKGSFGKVMLSERKGTDELYAVKILKKDVVIQDDDVECTMVEKRVLALPGKPPFLTQLHSCFQTMDRLYFVMEYVNGGDLMYHIQQVGRFKEPHAVFYAAEIAIGLFFLQSKGIIYRDLKLDNVMLDSEGHIKIADFGMCKENIWDGVTTKXFCGTPDYIAPEIIAYQPYGKSVDWWAFGVLLYEMLAGQAPFEGEDEDELFQSIMEHNVAYPKSMSKEAVAICKGLMTKHPGKRLGCGPEGERDIKEHAFFRYIDWEKLERKEIQPPYKPKAIDQSEFEGFXFVNSEFLKP Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Chromodomain-helicase-DNA-binding protein 1 | 4O42 | 3.94 | |
Target general information Gen name CHD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SNF2/RAD54 helicase family Biochemical class Dna binding protein / viral protein Function ATP binding.ATP-dependent DNA helicase activity.DNA binding.Methylated histone binding. Related diseases Pilarowski-Bjornsson syndrome (PILBOS) [MIM:617682]: An autosomal dominant disorder characterized by developmental delay, speech apraxia, intellectual disability, autism, and facial dysmorphic features. Some patients may have seizures. {ECO:0000269|PubMed:28866611}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60341-1; B2BUF1; P28799; O76024 EC number 3.6.4.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromatin regulator; Cytoplasm; Disease variant; DNA-binding; Helicase; Hydrolase; Intellectual disability; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 20969.1 Length 180 Aromaticity 0.12 Instability index 46.35 Isoelectric point 5.88 Charge (pH=7) -2.83 2D Binding mode Binding energy (Kcal/mol) -5.37 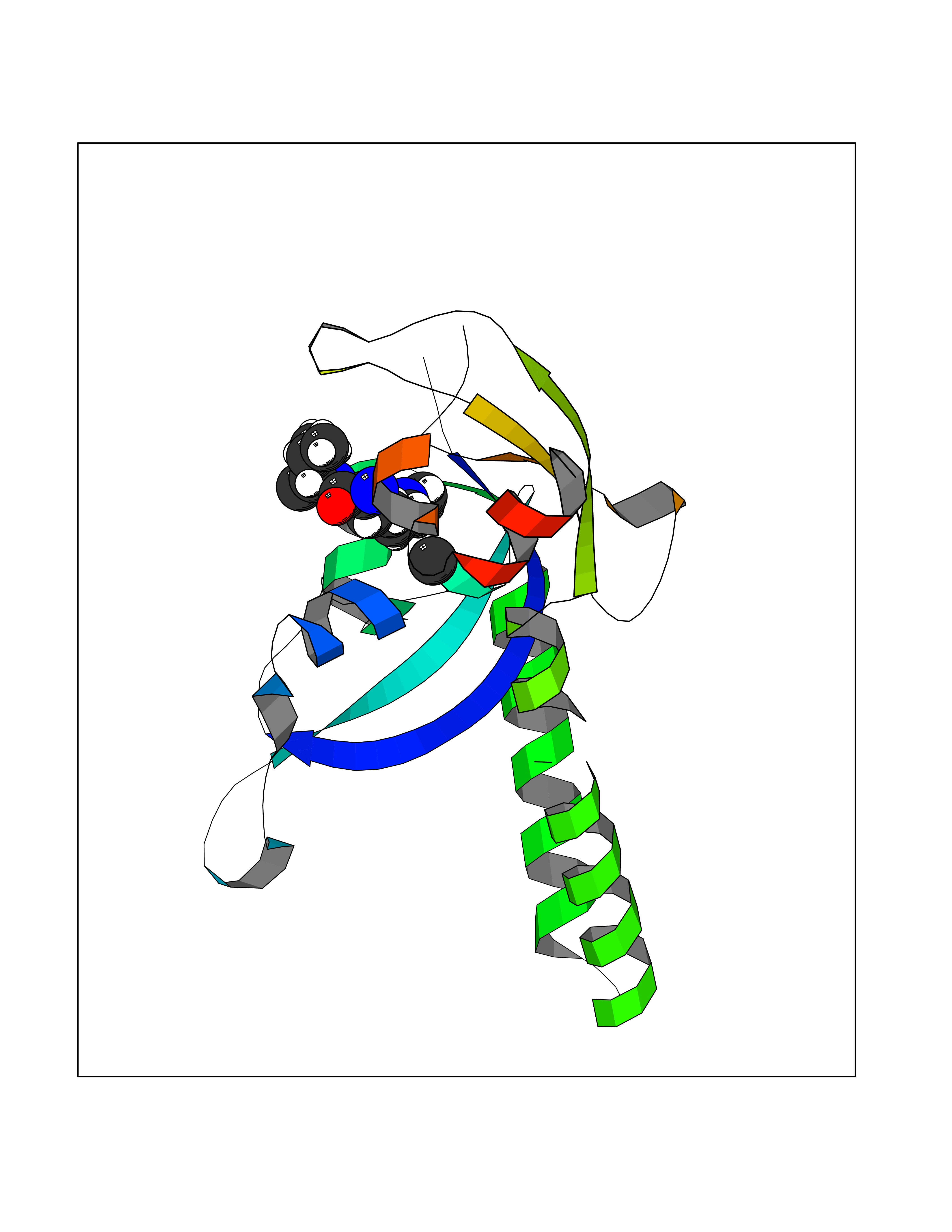 Molscript Map 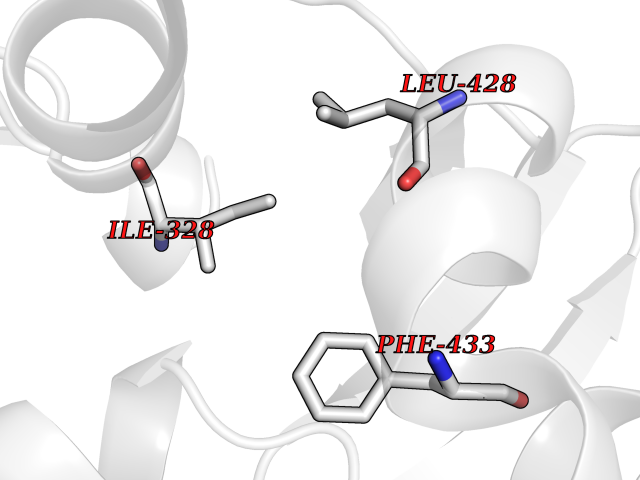 Pymol Map 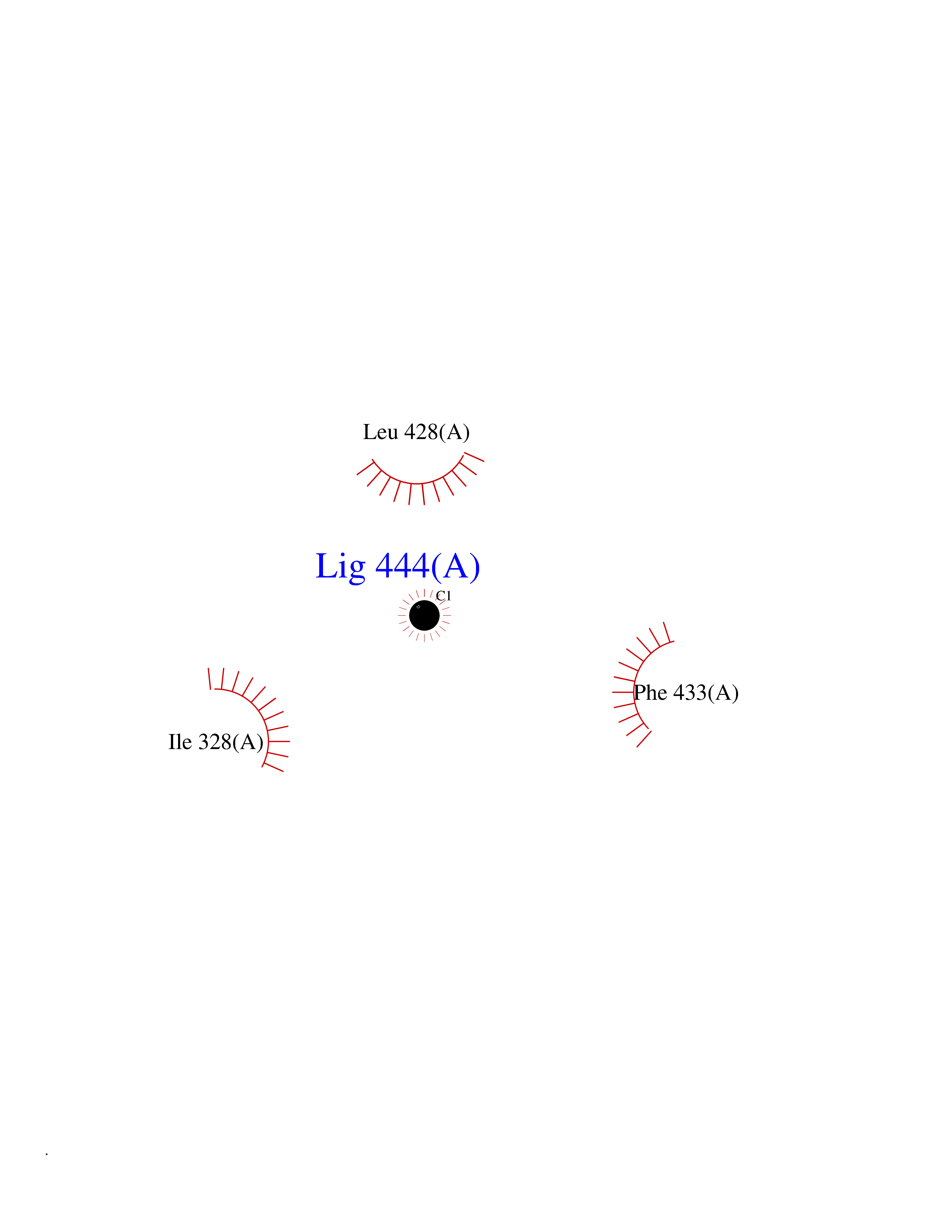 Ligplot Map 3D Binding mode Sequence EFETIERFMDCRIGRKGATGATTTIYAVEADGDPNAGFEKNKEPGEIQYLIKWKGWSHIHNTWETEETLKQQNVRGMKKLDNYKKKDQETKRWLKNASPEDVEYYNCQQELTDDLHKQYQIVERIIAHSNQKSAAGYPDYYCKWQGLPYSECSWEDGALISKKFQACIDEYFSRTARSXV Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Tyrosine aminotransferase | 3DYD | 3.94 | |
Target general information Gen name TAT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transferase Function Amino acid binding.L-phenylalanine:2-oxoglutarate aminotransferase activity.L-tyrosine:2-oxoglutarate aminotransferase activity.Pyridoxal phosphate binding. Related diseases Tyrosinemia 2 (TYRSN2) [MIM:276600]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, and oculocutaneous manifestations. Typical features include palmoplantar keratosis, painful corneal ulcers, and intellectual disability. {ECO:0000269|PubMed:1357662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00142; DB00120; DB00114; DB00135 Interacts with P15104; P28799; P28799-2; P17735; Q05086; Q05086-3 EC number 2.6.1.5 Uniprot keywords 3D-structure; Acetylation; Aminotransferase; Disease variant; Intellectual disability; Palmoplantar keratoderma; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Tyrosine catabolism Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42209.5 Length 380 Aromaticity 0.08 Instability index 51.79 Isoelectric point 5.29 Charge (pH=7) -10.66 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map 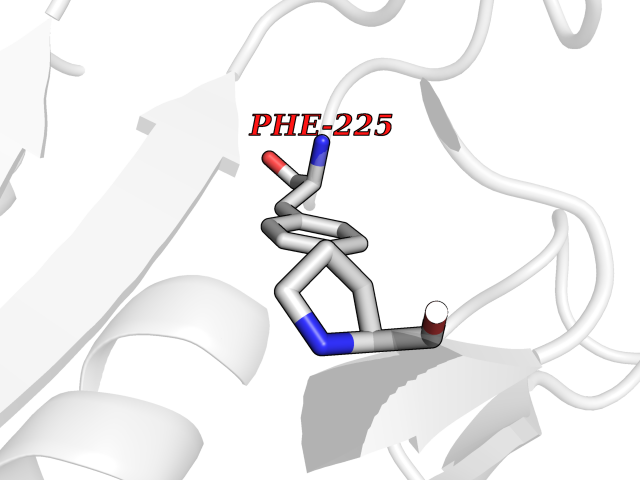 Pymol Map 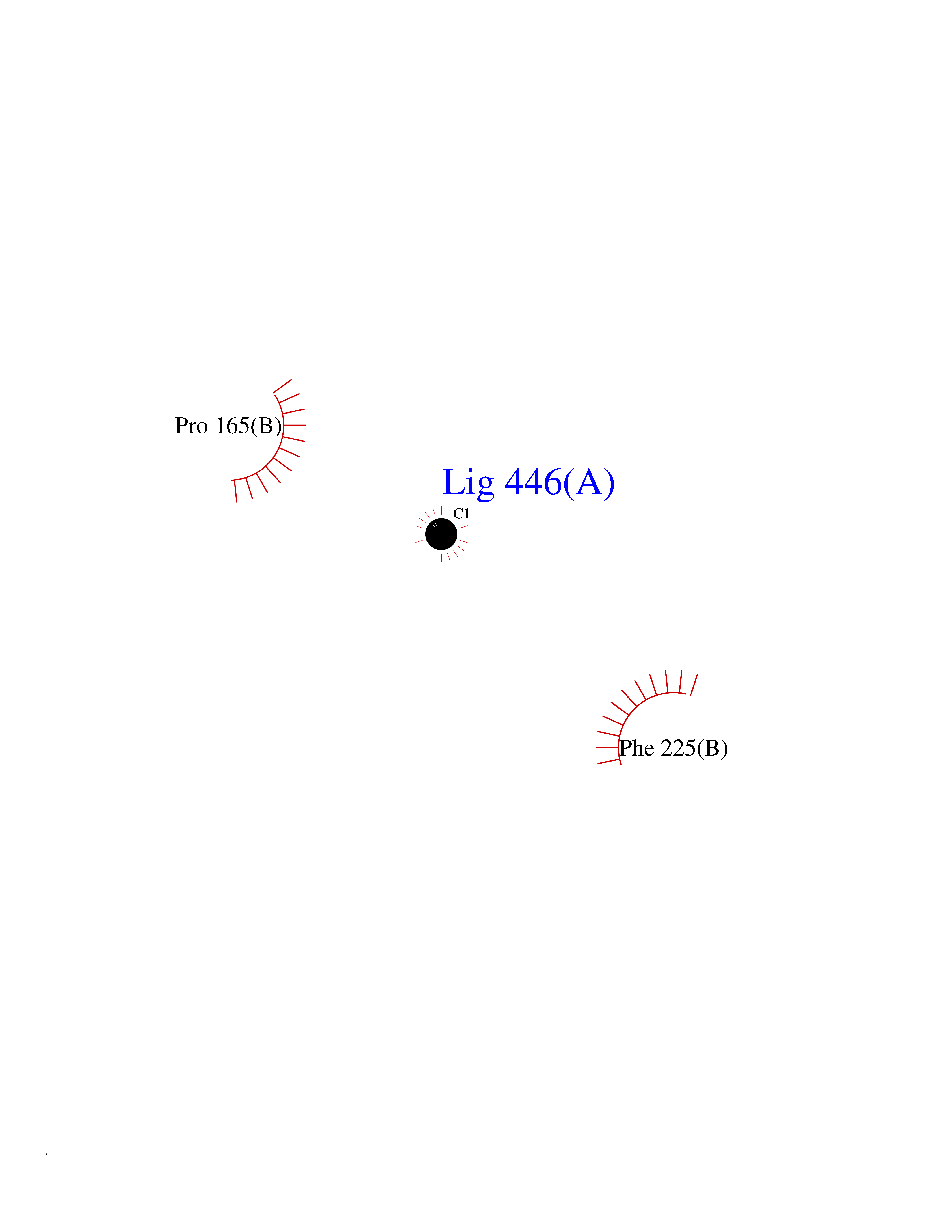 Ligplot Map 3D Binding mode Sequence VKPNPNKTMISLSIGDPTVFGNLPTDPEVTQAMKDALDSGKYNGYAPSIGFLSSREEIASYYHCPEAPLEAKDVILTSGCSQAIDLCLAVLANPGQNILVPRPGFSLYKTLAESMGIEVKLYNLLPEKSWEIDLKQLEYLIDEKTACLIVNNPSNPCGSVFSKRHLQKILAVAARQCVPILADEIYGDMVFSDCKYEPLATLSTDVPILSCGGLAKRWLVPGWRLGWILIHDRRDIFGNEIRDGLVKLSQRILGPCTIVQGALKSILCRTPGEFYHNTLSFLKSNADLCYGALAAIPGLRPVRPSGAMYLMVGIEMEHFPEFENDVEFTERLVAEQSVHCLPATCFEYPNFIRVVITVPEVMMLEACSRIQEFCEQHYHC Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Acetylcholinesterase (AChE) | 4M0E | 3.94 | |
Target general information Gen name ACHE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms YT; N-ACHE; ARACHE Protein family Type-B carboxylesterase/lipase family Biochemical class Carboxylic ester hydrolase Function Role in neuronal apoptosis. Terminates signal transduction at the neuromuscular junction by rapid hydrolysis of the acetylcholine released into the synaptic cleft. Related diseases Phosphoribosylaminoimidazole carboxylase deficiency (PAICSD) [MIM:619859]: An autosomal recessive inborn error of purine metabolism, clinically characterized by multiple congenital anomalies and early neonatal death. {ECO:0000269|PubMed:31600779}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07846; DB02673; DB04617; DB04614; DB04615; DB07756; DB07701; DB02404; DB03814; DB08615; DB02343; DB02226; DB03005; DB04114; DB03128; DB01122; DB03283; DB00411; DB00122; DB14006; DB01245; DB00944; DB08357; DB08996; DB00449; DB00843; DB01010; DB01364; DB00898; DB00674; DB00483; DB06525; DB04864; DB03348; DB07555; DB00677; DB04924; DB03359; DB00358; DB00940; DB02825; DB02845; DB08167; DB04021; DB00805; DB01805; DB03740; DB04556; DB01400; DB04892; DB00981; DB00733; DB02166; DB00545; DB00863; DB00989; DB00382; DB04616; DB12816; DB01199; DB13503; DB04859 Interacts with Q9Y215; P06733; P63244 EC number EC 3.1.1.7 Uniprot keywords 3D-structure; Alternative splicing; Blood group antigen; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; Neurotransmitter degradation; Nucleus; Proteomics identification; Reference proteome; Secreted; Serine esterase; Signal; Synapse Protein physicochemical properties Chain ID A,B Molecular weight (Da) 58804.1 Length 537 Aromaticity 0.11 Instability index 40.85 Isoelectric point 5.73 Charge (pH=7) -8.18 2D Binding mode Binding energy (Kcal/mol) -5.37 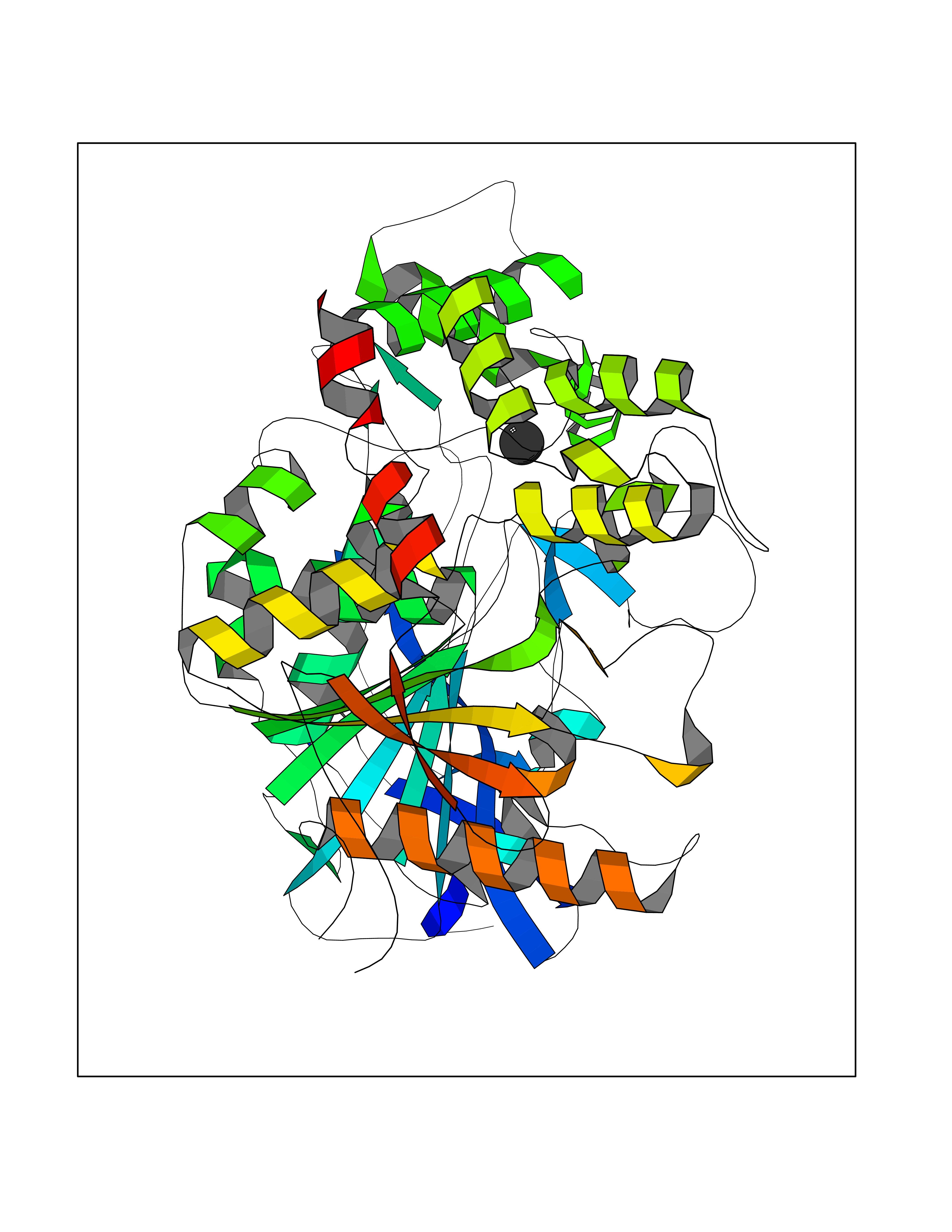 Molscript Map 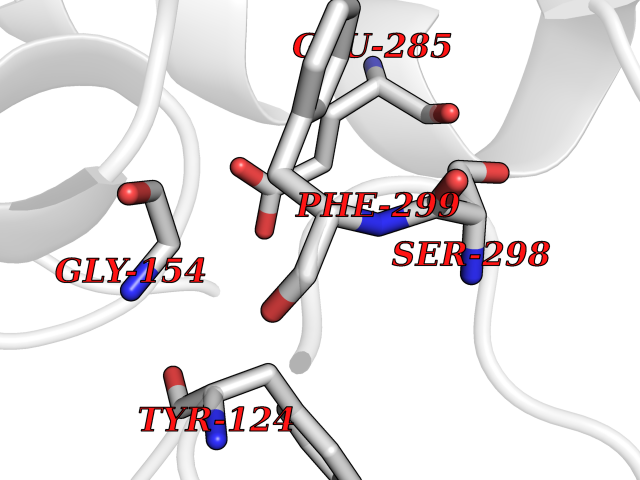 Pymol Map 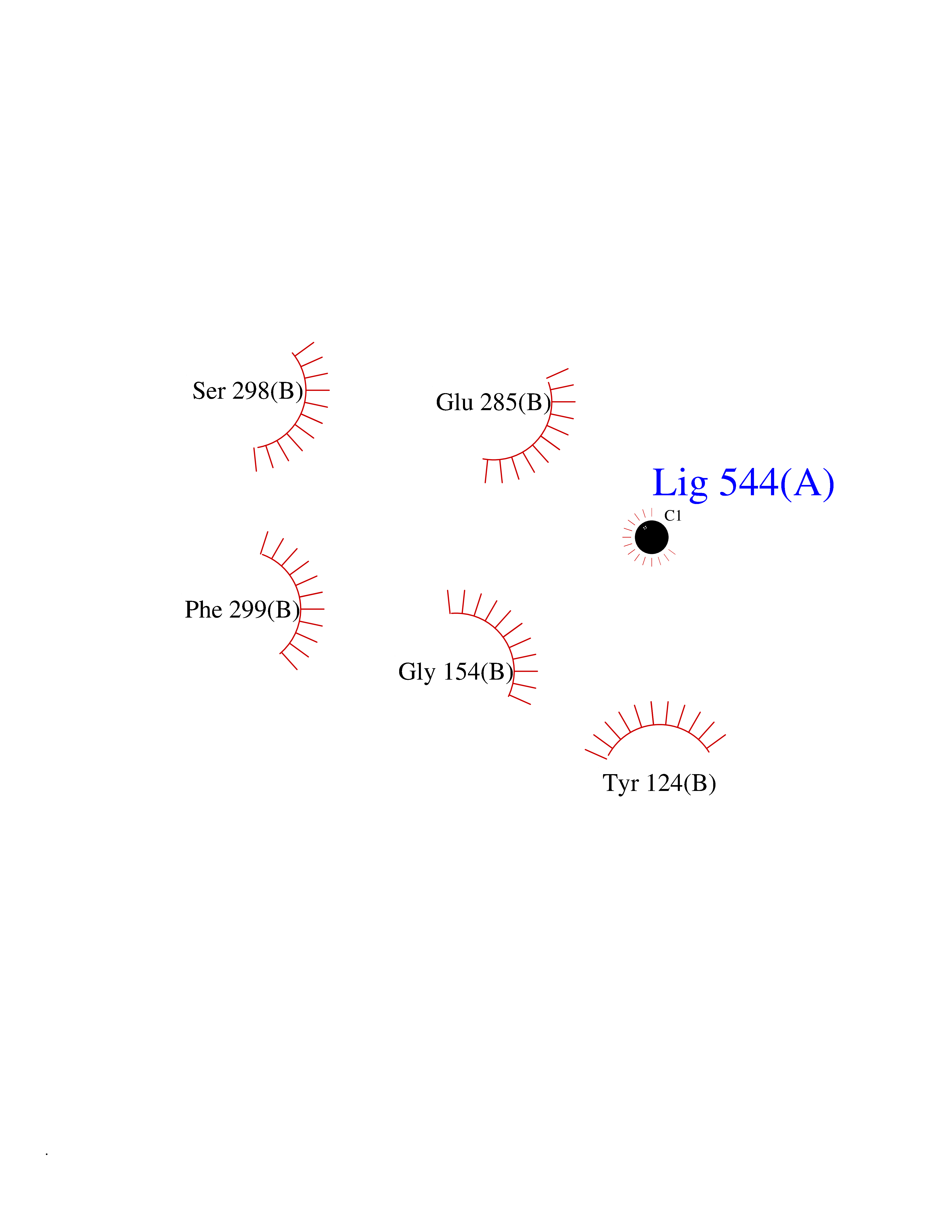 Ligplot Map 3D Binding mode Sequence EDAELLVTVRGGRLRGIRLKTPGGPVSAFLGIPFAEPPMGPRRFLPPEPKQPWSGVVDATTFQSVCYQYVDTLYPGFEGTEMWNPNRELSEDCLYLNVWTPYPRPTSPTPVLVWIYGGGFYSGASSLDVYDGRFLVQAERTVLVSMNYRVGAFGFLALPGSREAPGNVGLLDQRLALQWVQENVAAFGGDPTSVTLFGESAGAASVGMHLLSPPSRGLFHRAVLQSGAPNGPWATVGMGEARRRATQLAHLVGCPPGGTGGNDTELVACLRTRPAQVLVNHEWHVLPQESVFRFSFVPVVDGDFLSDTPEALINAGDFHGLQVLVGVVKDEGSYFLVYGAPGFSKDNESLISRAEFLAGVRVGVPQVSDLAAEAVVLHYTDWLHPEDPARLREALSDVVGDHNVVCPVAQLAGRLAAQGARVYAYVFEHRASTLSWPLWMGVPHGYEIEFIFGIPLDPSRNYTAEEKIFAQRLMRYWANFARTGDPNEPPKAPQWPPYTAGAQQYVSLDLRPLEVRRGLRAQACAFWNRFLPKLLSA Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Serum paraoxonase/arylesterase 1 | 1V04 | 3.94 | |
Target general information Gen name PON1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PON Protein family Paraoxonase family Biochemical class Hydrolase Function Acyl-L-homoserine-lactone lactonohydrolase activity.Aryldialkylphosphatase activity.Arylesterase activity.Calcium ion binding.Phospholipid binding.Protein homodimerization activity. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB01327; DB09130; DB01395; DB14598; DB14600; DB14596; DB00218; DB01085; DB01593; DB14487; DB14533; DB14548 Interacts with NA EC number 3.1.1.2; 3.1.1.81; 3.1.8.1 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Disulfide bond; Glycoprotein; HDL; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Secreted; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 37232.8 Length 332 Aromaticity 0.11 Instability index 35.09 Isoelectric point 5.06 Charge (pH=7) -17.08 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LFDRQKSSFQTRFNVHREVTPVELPNCNLVKGIDNGSEDLEILPNGLAFISSGLKYDKSGKILLMDLNEKEPAVSELEIIGNTLDISSFNPHGISTFIDDDNTVYLLVVNHPGSSSTVEVFKFQEEEKSLLHLKTIRHKLLPSVNDIVAVGPEHFYATNDHYFIDPYLKSWEMHLGLAWSFVTYYSPNDVRVVAEGFDFANGINISPDGKYVYIAELLAHKIHVYEKHANWTLTPLRVLSFDTLVDNISVDPVTGDLWVGCHPNGMRIFFYDAENPPGSEVLRIQDILSEEPKVTVVYAENGTVLQGSTVAAVYKGKLLIGTVFHKALYCDL Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Extracellular calcium-sensing receptor (CASR) | 5FBK | 3.94 | |
Target general information Gen name CASR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hCasR; Parathyroid cell calciumreceptor; Parathyroid cell calcium-sensing receptor 1; Parathyroid calcium receptor; Parathyroid Cell calcium-sensing receptor; PCaR1; GPRC2A; CaSR Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Senses fluctuations in the circulating calcium concentration and modulates the production of parathyroid hormone (PTH) in parathyroid glands. The activity of this receptor is mediated by a G-protein that activates a phosphatidylinositol-calcium second messenger system. The G-protein-coupled receptor activity is activated by a co-agonist mechanism: aromatic amino acids, such as Trp or Phe, act concertedly with divalent cations, such as calcium or magnesium, to achieve full receptor activation. G-protein-coupled receptor that senses changes in the extracellular concentration of calcium ions and plays a key role in maintaining calcium homeostasis. Related diseases Hypocalciuric hypercalcemia, familial 1 (HHC1) [MIM:145980]: A form of hypocalciuric hypercalcemia, a disorder of mineral homeostasis that is transmitted as an autosomal dominant trait with a high degree of penetrance. It is characterized biochemically by lifelong elevation of serum calcium concentrations and is associated with inappropriately low urinary calcium excretion and a normal or mildly elevated circulating parathyroid hormone level. Hypermagnesemia is typically present. Affected individuals are usually asymptomatic and the disorder is considered benign. However, chondrocalcinosis and pancreatitis occur in some adults. {ECO:0000269|PubMed:11762699, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:15579740, ECO:0000269|PubMed:15879434, ECO:0000269|PubMed:16598859, ECO:0000269|PubMed:16740594, ECO:0000269|PubMed:17473068, ECO:0000269|PubMed:17698911, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:19789209, ECO:0000269|PubMed:21566075, ECO:0000269|PubMed:21643651, ECO:0000269|PubMed:22114145, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25104082, ECO:0000269|PubMed:25292184, ECO:0000269|PubMed:26386835, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:7673400, ECO:0000269|PubMed:7726161, ECO:0000269|PubMed:7916660, ECO:0000269|PubMed:8636323, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9298824}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperparathyroidism, neonatal severe (NSHPT) [MIM:239200]: A disorder characterized by severe hypercalcemia, bone demineralization, and failure to thrive usually manifesting in the first 6 months of life. If untreated, NSHPT can be a devastating neurodevelopmental disorder, which in some cases is lethal without parathyroidectomy. {ECO:0000269|PubMed:14985373, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:17555508, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:8675635, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253359}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypocalcemia, autosomal dominant 1 (HYPOC1) [MIM:601198]: A disorder of mineral homeostasis characterized by blood calcium levels below normal, and low or normal serum parathyroid hormone concentrations. Disease manifestations include mild or asymptomatic hypocalcemia, paresthesias, carpopedal spasm, seizures, hypercalciuria with nephrocalcinosis or kidney stones, and ectopic and basal ganglia calcifications. Few patients manifest hypocalcemia and features of Bartter syndrome, including hypomagnesemia, hypokalemia, metabolic alkalosis, hyperreninemia, and hyperaldosteronemia. {ECO:0000269|PubMed:10487661, ECO:0000269|PubMed:12050233, ECO:0000269|PubMed:12107202, ECO:0000269|PubMed:12241879, ECO:0000269|PubMed:12574188, ECO:0000269|PubMed:12915654, ECO:0000269|PubMed:15551332, ECO:0000269|PubMed:16608894, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:22789683, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25766501, ECO:0000269|PubMed:7874174, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8733126, ECO:0000269|PubMed:8813042, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253358, ECO:0000269|PubMed:9661634, ECO:0000269|PubMed:9920108}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epilepsy, idiopathic generalized 8 (EIG8) [MIM:612899]: A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Seizure types are variable, but include myoclonic seizures, absence seizures, febrile seizures, complex partial seizures, and generalized tonic-clonic seizures. {ECO:0000269|PubMed:18756473}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11093; DB11348; DB14481; DB01012; DB12865; DB00994; DB05695; DB05255; DB00127 Interacts with Q15363; P41180-1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 52438.9 Length 467 Aromaticity 0.12 Instability index 39.62 Isoelectric point 5.63 Charge (pH=7) -10.18 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map 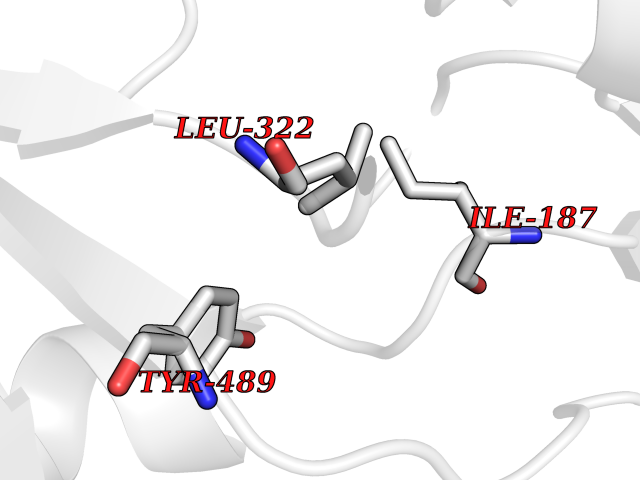 Pymol Map 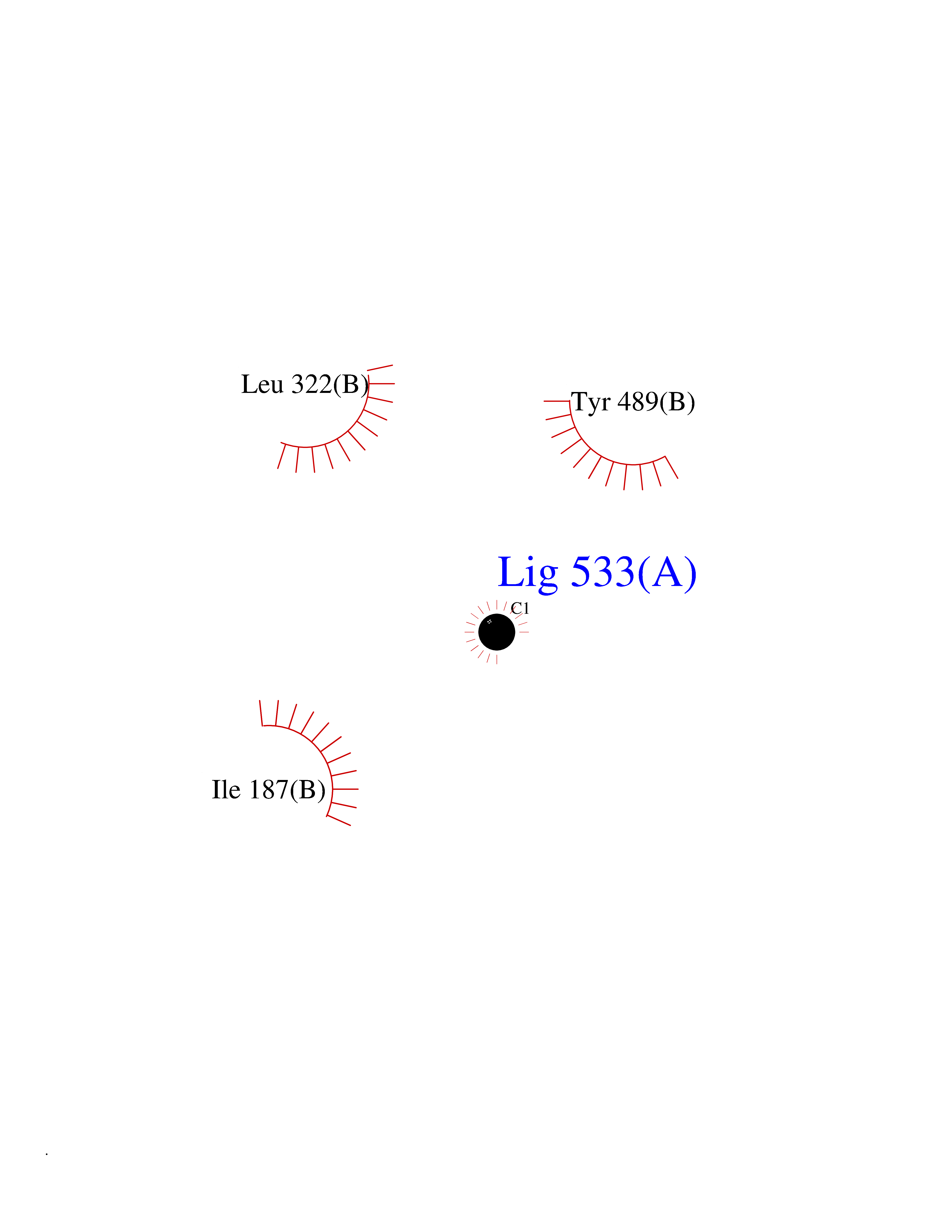 Ligplot Map 3D Binding mode Sequence GPDQRAQKKGDIILGGLFPIHFGVAAKDQDLKSRPESVECIRYNFRGFRWLQAMIFAIEEINSSPALLPNLTLGYRIFDTCNTVSKALEATLSFVAQNKIDSTIAVVGATGSGVSTAVANLLGLFYIPQVSYASSSRLLSNKNQFKSFLRTIPNDEHQATAMADIIEYFRWNWVGTIAADDDYGRPGIEKFREEAEERDIXIDFSELISQYSDEEEIQHVVEVIQNSTAKVIVVFSSGPDLEPLIKEIVRRNITGKIWLASEAWASSSLIAMPQYFHVVGGTIGFALKAGQIPGFREFLKKVHPRKSVHNGFAKEFWEETFNCHLQFRPLCTGDENISSVETPYIDYTHLRISYNVYLAVYSIAHALQDIYTCLPGRGLFTNGSCADIKKVEAWQVLKHLRHLNFTNNMGEQVTFDEXGDLVGNYSIINWHLSPEDGSIVFKEVGYYNVYAKKGERLFINEEKILWS Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Monomeric sarcosine oxidase | 2GF3 | 3.94 | |
Target general information Gen name soxA Organism Bacillus sp. (strain B-0618) Uniprot ID TTD ID NA Synonyms sox Protein family MSOX/MTOX family, MSOX subfamily Biochemical class Oxidoreductase Function Sarcosine oxidase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB03098; DB01918; DB03517; DB03147; DB03366; DB02083; DB02543 Interacts with NA EC number 1.5.3.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42606.4 Length 385 Aromaticity 0.1 Instability index 26.97 Isoelectric point 5.27 Charge (pH=7) -17.18 2D Binding mode Binding energy (Kcal/mol) -5.38  Molscript Map 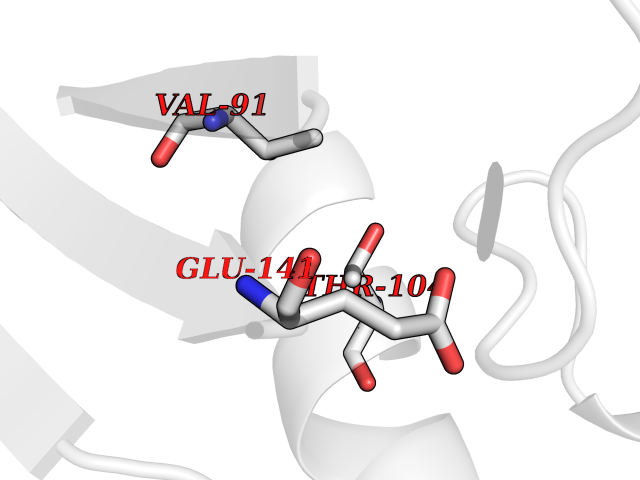 Pymol Map 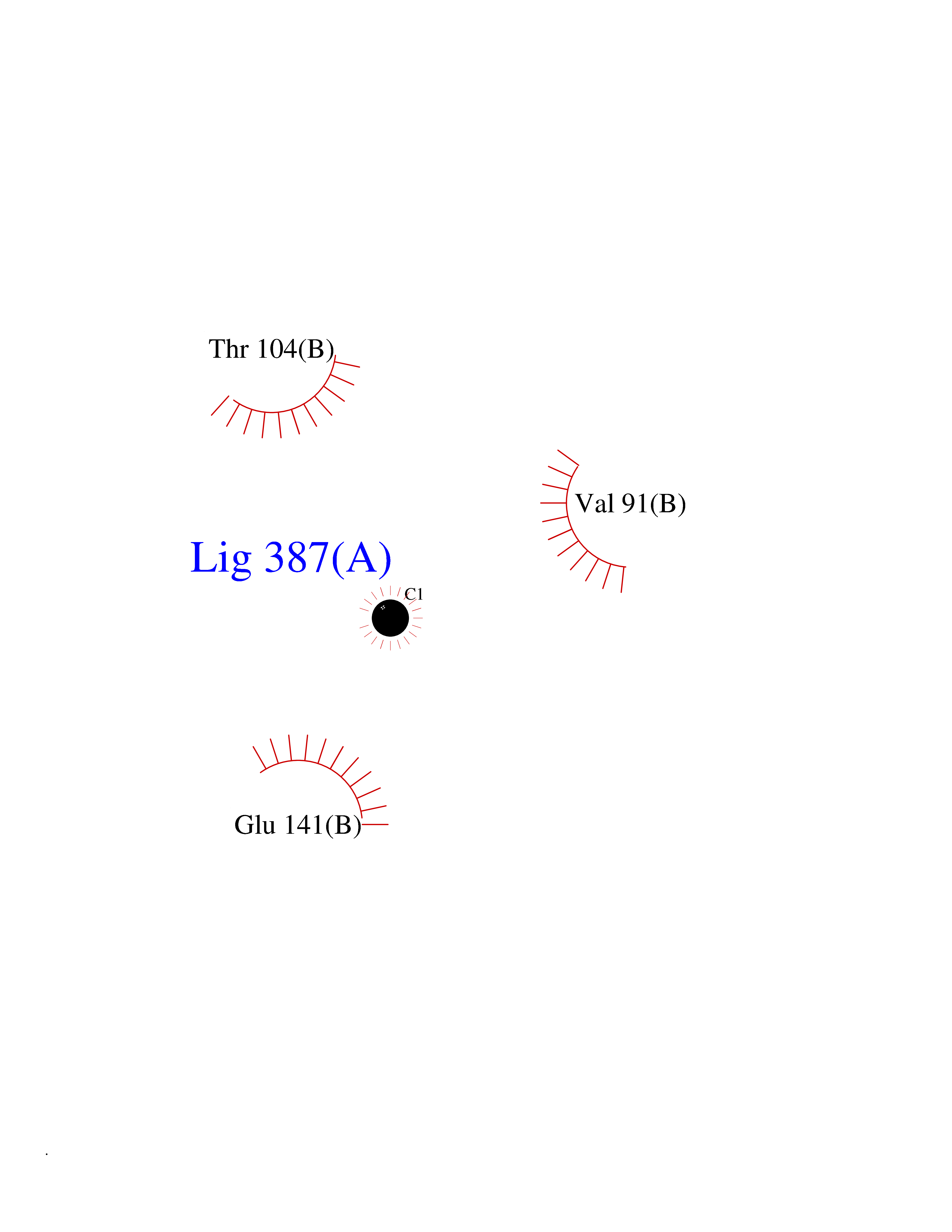 Ligplot Map 3D Binding mode Sequence STHFDVIVVGAGSMGMAAGYQLAKQGVKTLLVDAFDPPHTNGSHHGDTRIIRHAYGEGREYVPLALRSQELWYELEKETHHKIFTKTGVLVFGPKGESAFVAETMEAAKEHSLTVDLLEGDEINKRWPGITVPENYNAIFEPNSGVLFSENCIRAYRELAEARGAKVLTHTRVEDFDISPDSVKIETANGSYTADKLIVSMGAWNSKLLSKLNLDIPLQPYRQVVGFFESDESKYSNDIDFPGFMVEVPNGIYYGFPSFGGCGLKLGYHTFGQKIDPDTINREFGVYPEDESNLRAFLEEYMPGANGELKRGAVCMYTKTLDEHFIIDLHPEHSNVVIAAGFSGHGFKFSSGVGEVLSQLALTGKTEHDISIFSINRPALKESLQ Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Aldo-keto reductase family 1 member C3 | 1S1P | 3.94 | |
Target general information Gen name AKR1C3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PGFS;DDH1;HSD17B5;KIAA0119 Protein family Aldo/keto reductase family Biochemical class Oxidoreductase Function 15-hydroxyprostaglandin-D dehydrogenase (NADP+) activity.Alditol:NADP+ 1-oxidoreductase activity.Aldo-keto reductase (NADP) activity.Androsterone dehydrogenase activity.Delta4-3-oxosteroid 5beta-reductase activity.Dihydrotestosterone 17-beta-dehydrogenase activity.Geranylgeranyl reductase activity.Indanol dehydrogenase activity.Ketoreductase activity.Ketosteroid monooxygenase activity.NADP-retinol dehydrogenase activity.Oxidoreductase activity, acting on NAD(P)H, quinone or similar compound as acceptor.Phenanthrene 9,10-monooxygenase activity.Prostaglandin D2 11-ketoreductase activity.Prostaglandin-F synthase activity.Prostaglandin H2 endoperoxidase reductase activity.Retinal dehydrogenase activity.Retinol dehydrogenase activity.Testosterone 17-beta-dehydrogenase (NADP+) activity.Testosterone dehydrogenase (NAD+) activity.Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase activity. Related diseases Neurodevelopmental disorder with language impairment and behavioral abnormalities (NEDLIB) [MIM:618917]: A neurodevelopmental disorder characterized by global developmental delay, impaired intellectual development, poor or absent speech, and behavioral abnormalities, such as autism spectrum disorder, repetitive behaviors, and hyperactivity. Some patients develop seizures and manifest developmental regression. {ECO:0000269|PubMed:31300657}. The disease is caused by variants affecting the gene represented in this entry. The genetic variation producing the missense variant p.Q607E, associated with NEDLIB, is predicted to deeply affect RNA editing. In a physiological context, the adenosine (A) residue of the original glutamine (Q) codon CAG is post-transcriptionaly edited to inosine (I) by ADAR2, leading to a codon recognized by the ribosome as arginine (R). The glutamate (E) codon GAG, resulting from the genetic variation, is predicted to be edited 90% less than the normal CAG codon. If edited, the codon GIG would be translated as p.Q607G. {ECO:0000305|PubMed:31300657}. Drugs (DrugBank ID) DB07700; DB01561; DB01536; DB00997; DB01039; DB02266; DB13751; DB00328; DB06077; DB00959; DB00157; DB03461; DB09074; DB00776; DB02056; DB01698; DB02901 Interacts with P17516 EC number 1.1.1.-; 1.1.1.188; 1.1.1.210; 1.1.1.239; 1.1.1.357; 1.1.1.53; 1.1.1.62; 1.1.1.64 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Lipid metabolism; NAD; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35846.8 Length 315 Aromaticity 0.09 Instability index 47.59 Isoelectric point 8.54 Charge (pH=7) 4.51 2D Binding mode Binding energy (Kcal/mol) -5.37 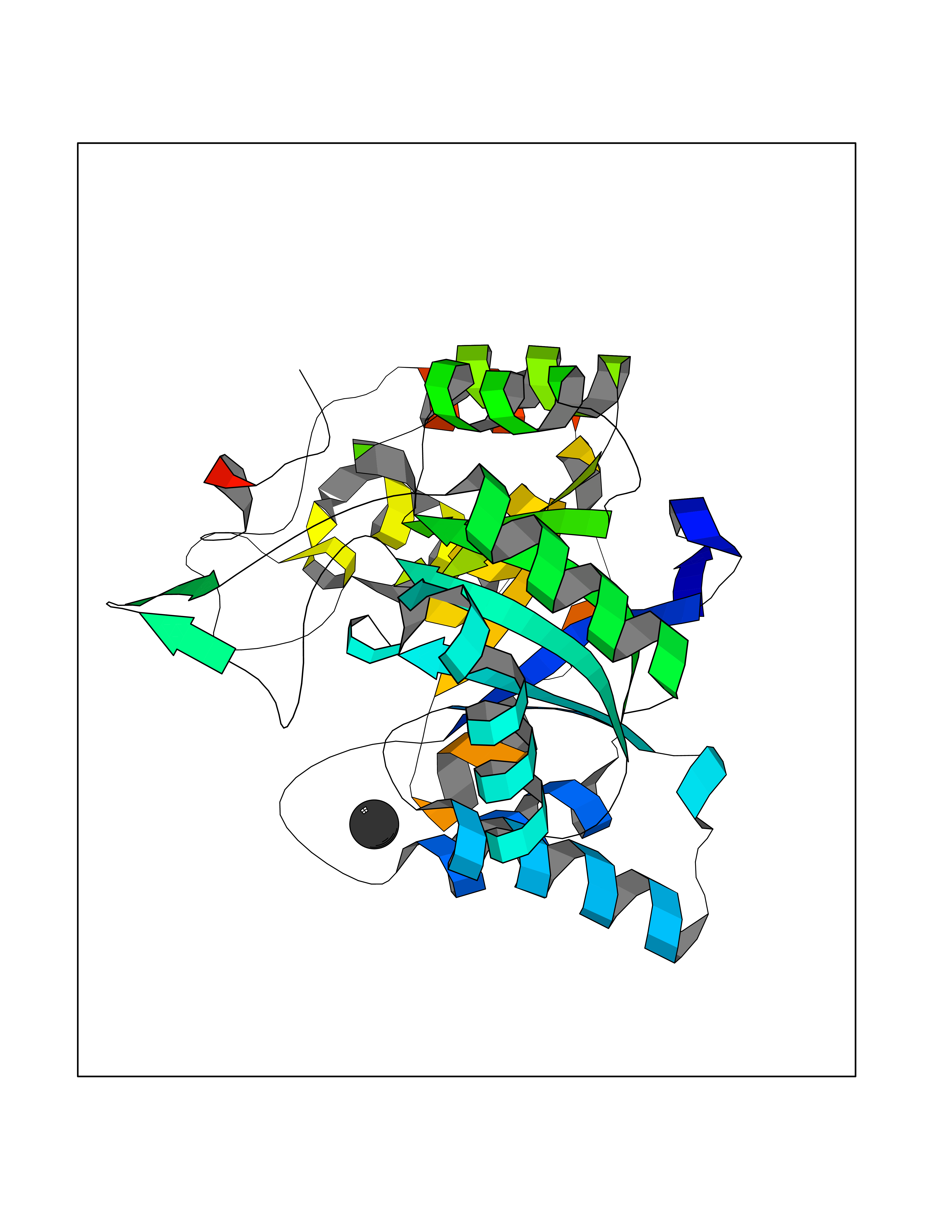 Molscript Map 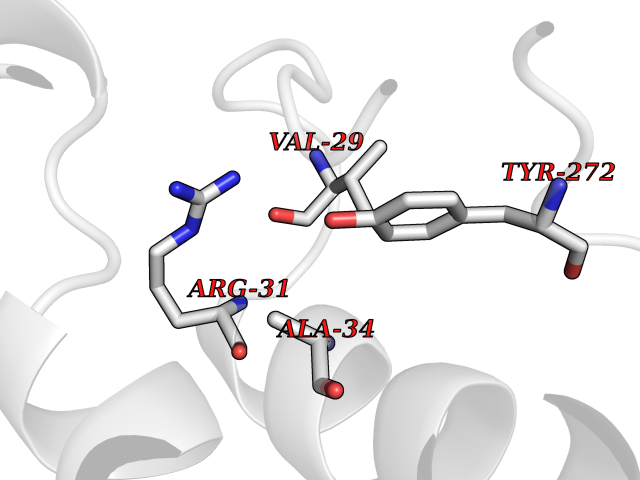 Pymol Map 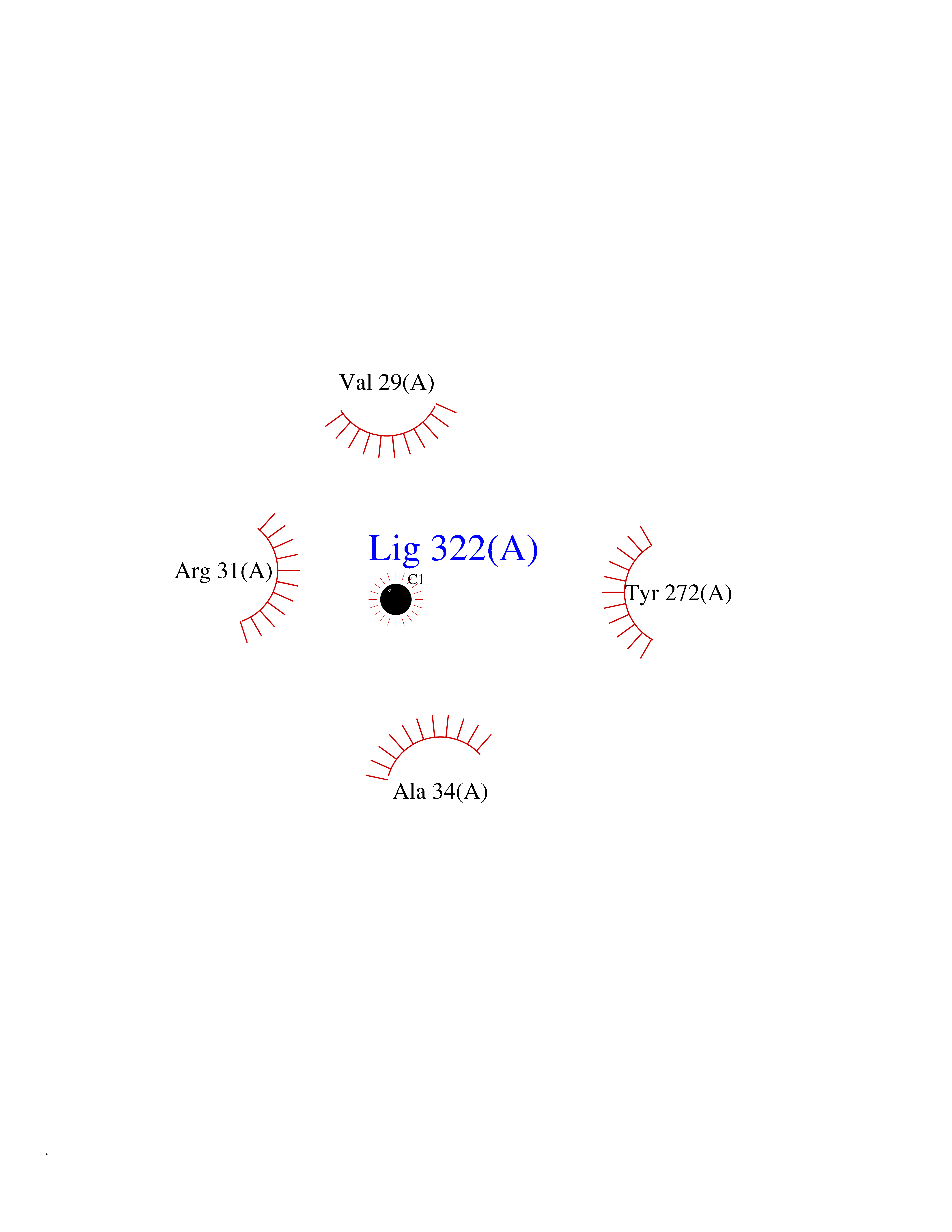 Ligplot Map 3D Binding mode Sequence QCVKLNDGHFMPVLGFGTYAPPEVPRSKALEVTKLAIEAGFRHIDSAHLYNNEEQVGLAIRSKIADGSVKREDIFYTSKLWSTFHRPELVRPALENSLKKAQLDYVDLYLIHSPMSLKPGEELSPTDENGKVIFDIVDLCTTWEAMEKCKDAGLAKSIGVSNFNRRQLEMILNKPGLKYKPVCNQVECHPYFNRSKLLDFCKSKDIVLVAYSALGSQRDKRWVDPNSPVLLEDPVLCALAKKHKRTPALIALRYQLQRGVVVLAKSYNEQRIRQNVQVFEFQLTAEDMKAIDGLDRNLHYFNSDSFASHPNYPYS Hydrogen bonds contact Hydrophobic contact | ||||