Job Results:
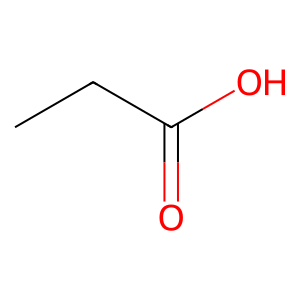
Ligand
Structure
Job ID
5217c115ac480b5185ad5781ec63f66c
Job name
NA
Time
2026-02-27 11:51:57
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 4.59 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -6.25 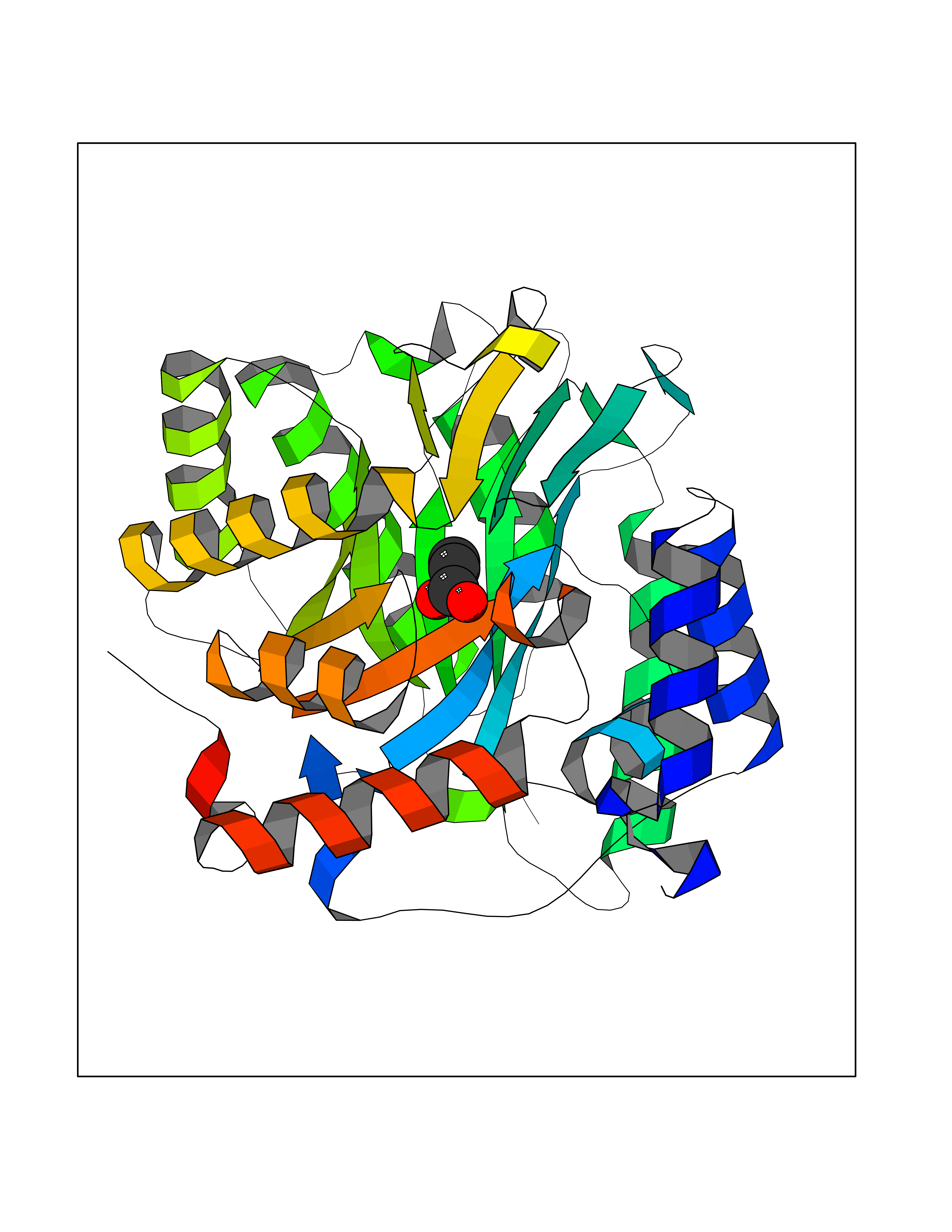 Molscript Map 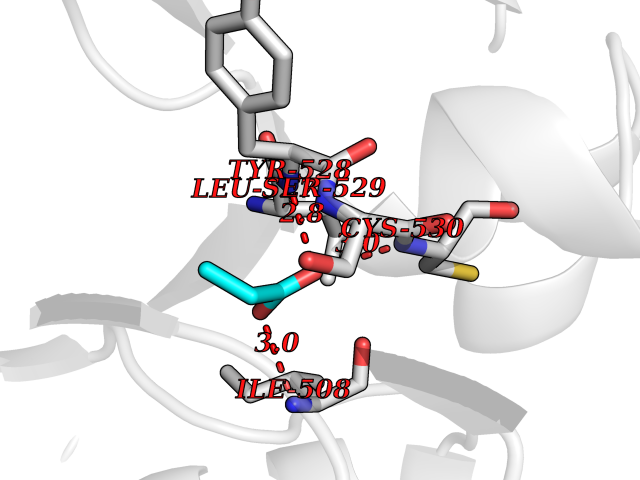 Pymol Map 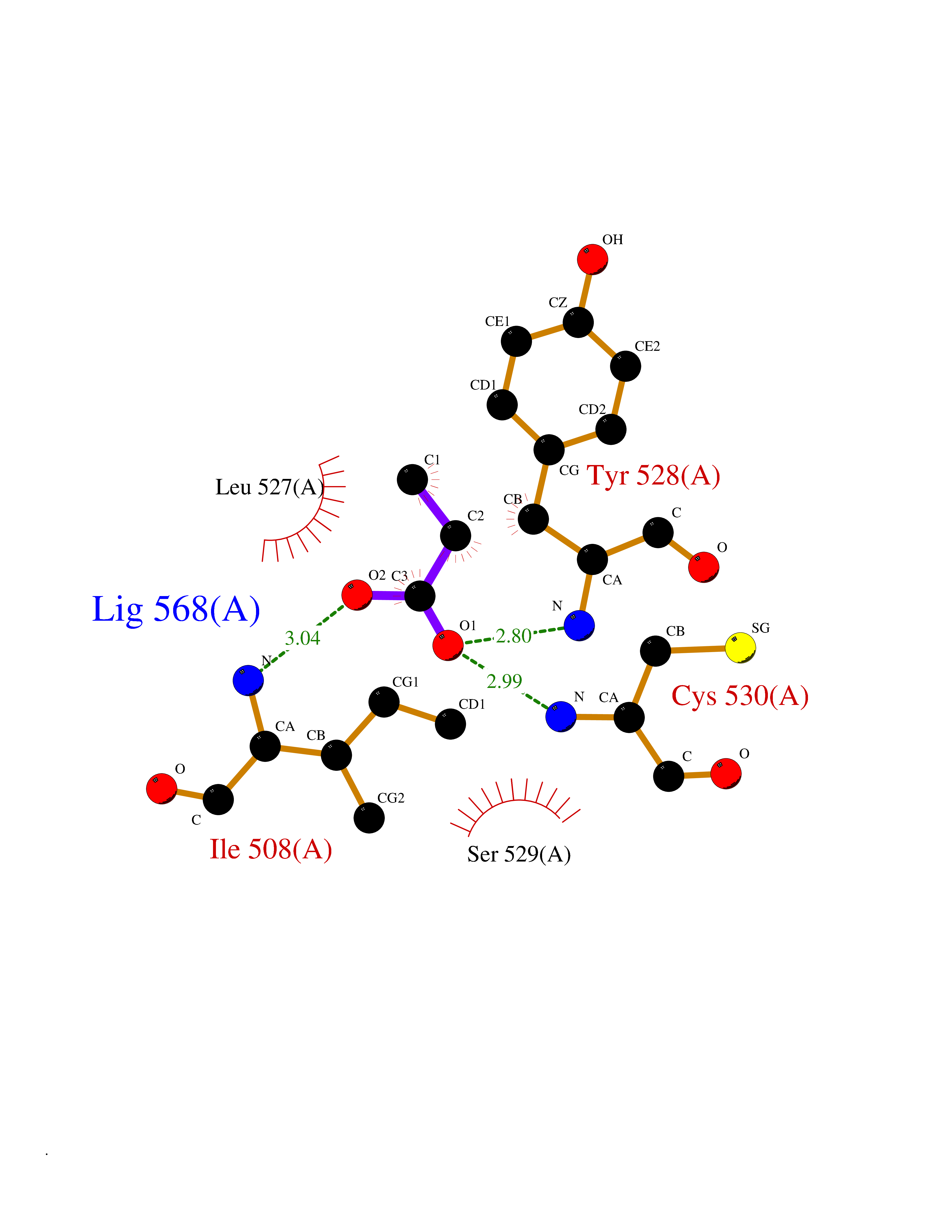 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Beta-galactosidase (GLB1) | 3THD | 4.59 | |
Target general information Gen name GLB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lactase; Elastin receptor 1; ELNR1; Acid beta-galactosidase Protein family Glycosyl hydrolase 35 family Biochemical class NA Function Isoform 1: Cleaves beta-linked terminal galactosyl residues from gangliosides, glycoproteins, and glycosaminoglycans. Related diseases GM1-gangliosidosis 1 (GM1G1) [MIM:230500]: An autosomal recessive lysosomal storage disease marked by the accumulation of GM1 gangliosides, glycoproteins and keratan sulfate primarily in neurons of the central nervous system. GM1-gangliosidosis type 1 is characterized by onset within the first three months of life, central nervous system degeneration, coarse facial features, hepatosplenomegaly, skeletal dysmorphology reminiscent of Hurler syndrome, and rapidly progressive psychomotor deterioration. Urinary oligosaccharide levels are high. It leads to death usually between the first and second year of life. {ECO:0000269|PubMed:10338095, ECO:0000269|PubMed:10737981, ECO:0000269|PubMed:10839995, ECO:0000269|PubMed:1487238, ECO:0000269|PubMed:15365997, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:15791924, ECO:0000269|PubMed:16538002, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:1928092, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8213816, ECO:0000269|Ref.28, ECO:0000269|Ref.31}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GM1-gangliosidosis 2 (GM1G2) [MIM:230600]: A gangliosidosis characterized by onset between ages 1 and 5. The main symptom is locomotor ataxia, ultimately leading to a state of decerebration with epileptic seizures. Patients do not display the skeletal changes associated with the infantile form, but they nonetheless excrete elevated amounts of beta-linked galactose-terminal oligosaccharides. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10737981, ECO:0000269|PubMed:12644936, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8213816}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GM1-gangliosidosis 3 (GM1G3) [MIM:230650]: A gangliosidosis with a variable phenotype. Patients show mild skeletal abnormalities, dysarthria, gait disturbance, dystonia and visual impairment. Visceromegaly is absent. Intellectual deficit can initially be mild or absent but progresses over time. Inheritance is autosomal recessive. {ECO:0000269|PubMed:11511921, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:15986423, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8198123, ECO:0000269|Ref.28, ECO:0000269|Ref.30}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 4B (MPS4B) [MIM:253010]: A form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life. {ECO:0000269|PubMed:11511921, ECO:0000269|PubMed:12393180, ECO:0000269|PubMed:16538002, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1928092, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:7586649}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04465 Interacts with Q8NBJ4; Q3KNW5; Q9BRI3; P30825 EC number EC 3.2.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Gangliosidosis; Glycoprotein; Glycosidase; Hydrolase; Lysosome; Mucopolysaccharidosis; Proteomics identification; Reference proteome; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 67980.6 Length 605 Aromaticity 0.13 Instability index 40.91 Isoelectric point 5.81 Charge (pH=7) -9.05 2D Binding mode Binding energy (Kcal/mol) -6.26 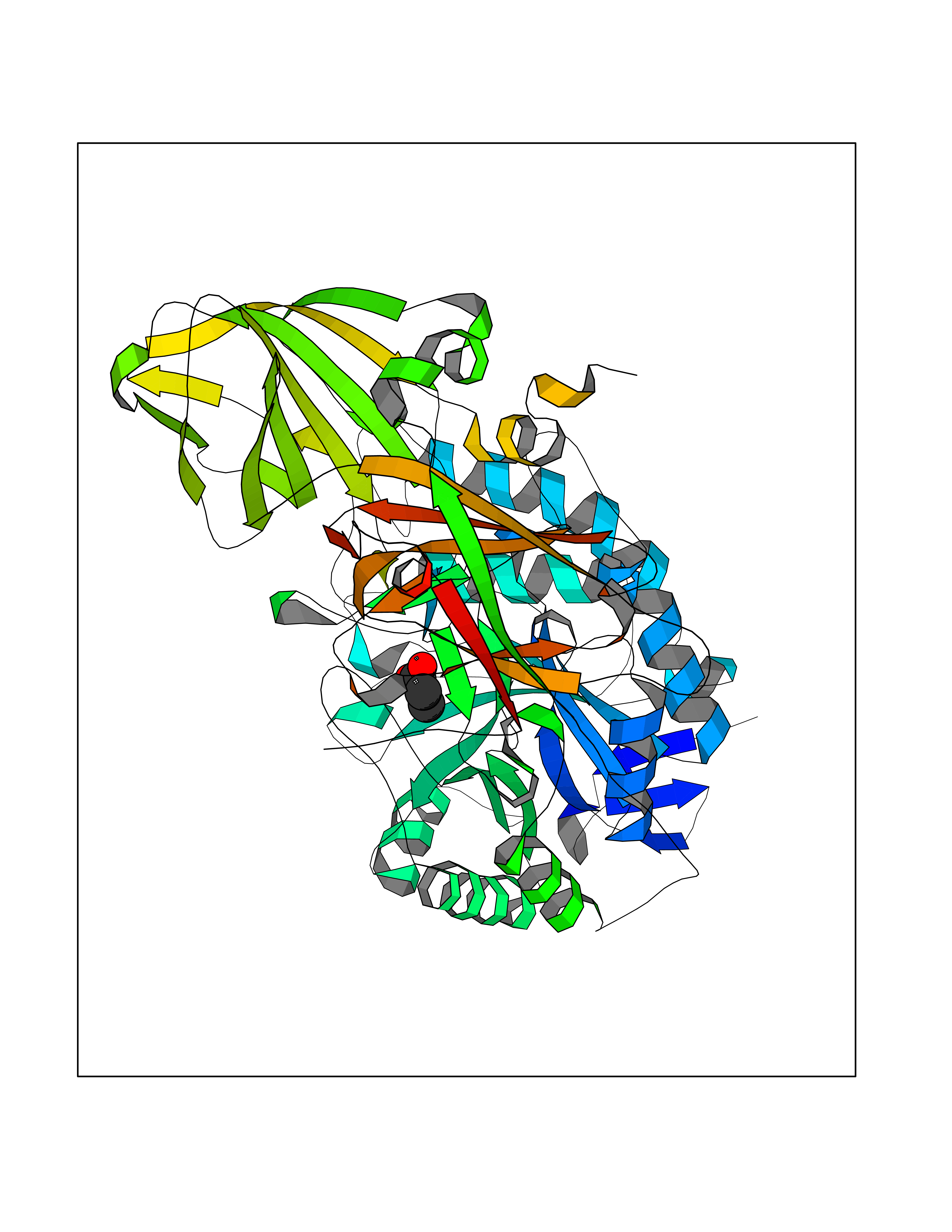 Molscript Map 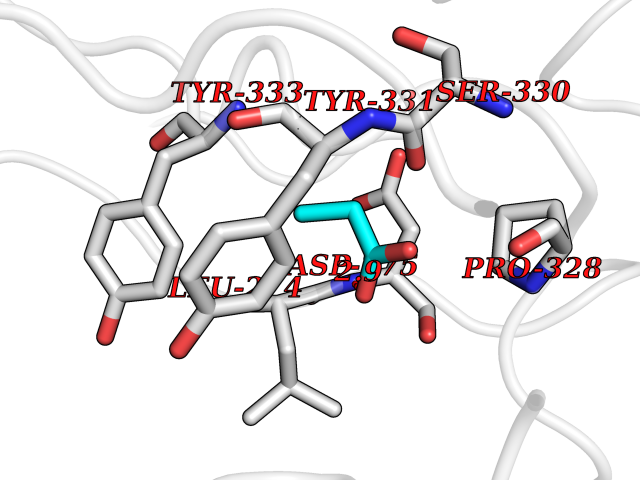 Pymol Map 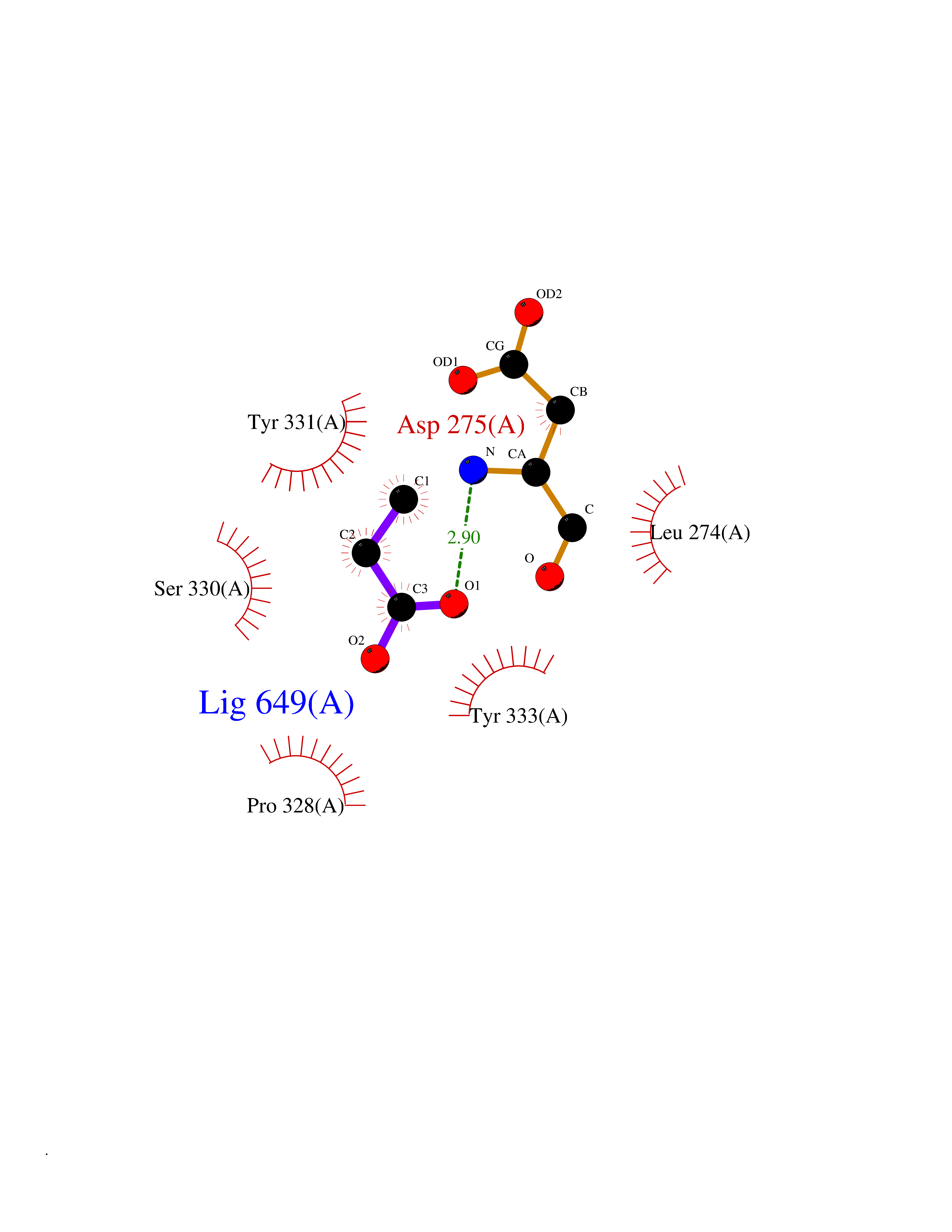 Ligplot Map 3D Binding mode Sequence QRMFEIDYSRDSFLKDGQPFRYISGSIHYSRVPRFYWKDRLLKMKMAGLNAIQTYVPWNFHEPWPGQYQFSEDHDVEYFLRLAHELGLLVILRPGPYICAEWEMGGLPAWLLEKESILLRSSDPDYLAAVDKWLGVLLPKMKPLLYQNGGPVITVQVENEYGSYFACDFDYLRFLQKRFRHHLGDDVVLFTTDGAHKTFLKCGALQGLYTTVDFGTGSNITDAFLSQRKCEPKGPLINSEFYTGWLDHWGQPHSTIKTEAVASSLYDILARGASVNLYMFIGGTNFAYWNGANSPYAAQPTSYDYDAPLSEAGDLTEKYFALRNIIQKFEKVPEGPIPPSTPKFAYGKVTLEKLKTVGAALDILCPSGPIKSLYPLTFIQVKQHYGFVLYRTTLPQDCSNPAPLSSPLNGVHDRAYVAVDGIPQGVLERNNVITLNITGKAGATLDLLVENMGRVNYGAYINDFKGLVSNLTLSSNILTDWTIFPLDTEDAVRSHLGGWGHRNYTLPAFYMGNFSIPSGIPDLPQDTFIQFPGWTKGQVWINGFNLGRYWPARGPQLTLFVPQHILMTSAPNTITVLELEWAPCSSDDPELCAVTFVDRPVIGSS Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | "Acetolactate synthase, chloroplastic (AtALS) (EC 2.2.1.6) (Acetohydroxy-acid synthase) (Protein CHLORSULFURON RESISTANT 1)" | 5K3S | 4.58 | |
Target general information Gen name ALS Organism Arabidopsis thaliana (Mouse-ear cress) Uniprot ID TTD ID NA Synonyms At3g48560;CSR1;AHAS;T8P19.70;TZP5 Protein family TPP enzyme family Biochemical class NA Function Catalyzes the formation of acetolactate from pyruvate, the first step in valine and isoleucine biosynthesis. {ECO:0000269|PubMed:10386618, ECO:0000269|PubMed:16665813, ECO:0000269|PubMed:16667374, ECO:0000269|PubMed:16668488, ECO:0000269|PubMed:2336405, ECO:0000269|PubMed:8913312, ECO:0000269|PubMed:9355748, ECO:0000269|PubMed:9677339, ECO:0000269|Ref.9}." Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 2.2.1.6 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Branched-chain amino acid biosynthesis; Chloroplast; Coiled coil; FAD; Flavoprotein; Genetically modified food; Herbicide resistance; Magnesium; Metal-binding; Oxidation; Plastid; Reference proteome; Thiamine pyrophosphate; Transferase; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 63431 Length 583 Aromaticity 0.07 Instability index 36.62 Isoelectric point 5.4 Charge (pH=7) -15.33 2D Binding mode Binding energy (Kcal/mol) -6.25 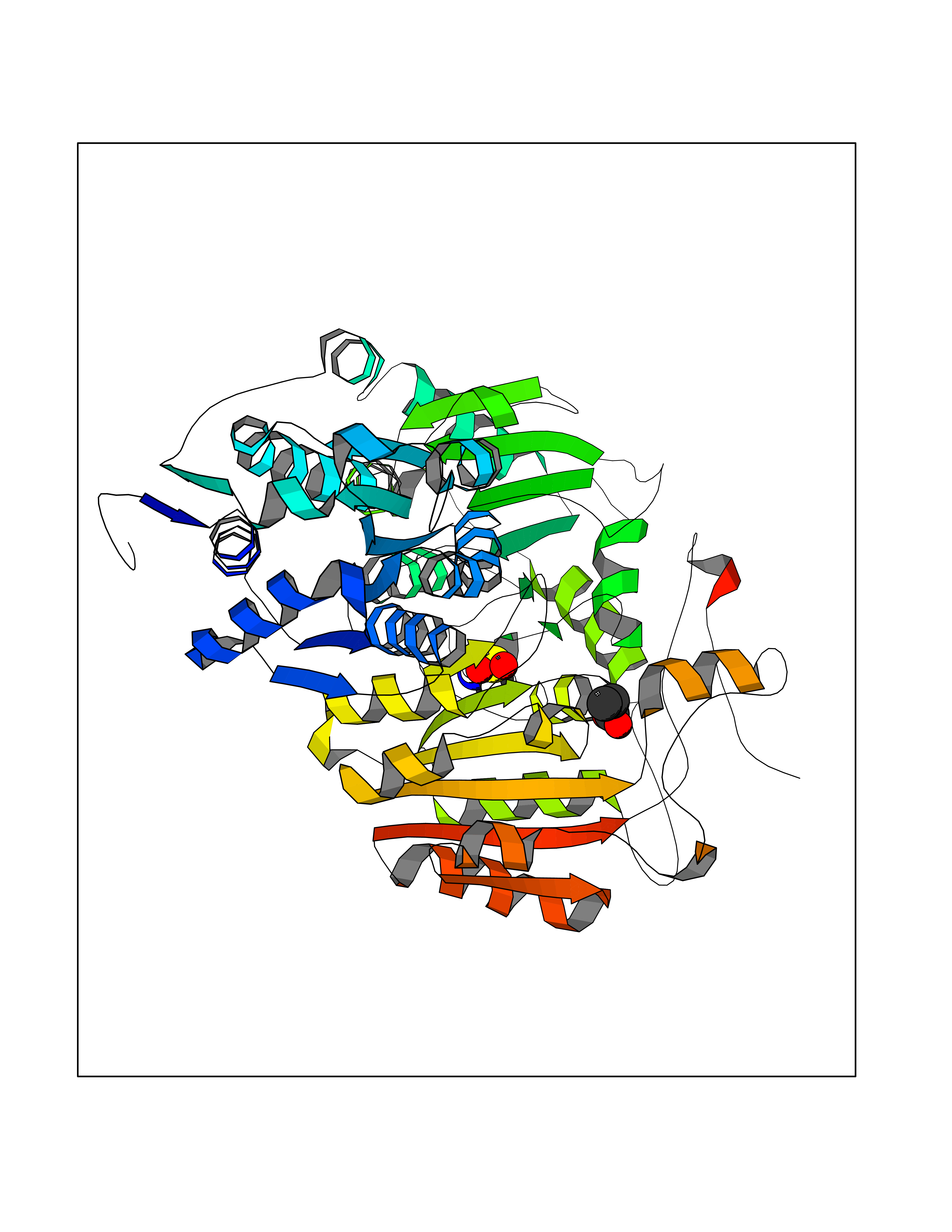 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence TFISRFAPDQPRKGADILVEALERQGVETVFAYPGGASMEIHQALTRSSSIRNVLPRHEQGGVFAAEGYARSSGKPGICIATSGPGATNLVSGLADALLDSVPLVAITGQVPRRMIGTDAFQETPIVEVTRSITKHNYLVMDVEDIPRIIEEAFFLATSGRPGPVLVDVPKDIQQQLAIPNWEQAMRLPGYMSRMPKPPEDSHLEQIVRLISESKKPVLYVGGGCLNSSDELGRFVELTGIPVASTLMGLGSYPXDDELSLHMLGMHGTVYANYAVEHSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKTPHVSVCGDVKLALQGMNKVLENRAEELKLDFGVWRNELNVQKQKFPLSFKTFGEAIPPQYAIKVLDELTDGKAIISTGVGQHQMWAAQFYNYKKPRQWLSSGGLGAMGFGLPAAIGASVANPDAIVVDIDGDGSFIMNVQELATIRVENLPVKVLLLNNQHLGMVMQWEDRFYKANRAHTFLGDPAQEDEIFPNMLLFAAACGIPAARVTKKADLREAIQTMLDTPGPYLLDVICPHQEHVLPMIPSGGTFNDVITEGDGRL Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Nitric-oxide synthase endothelial (NOS3) | 4D1P | 4.58 | |
Target general information Gen name NOS3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nitric oxide synthase, endothelial; NOSIII; NOS,type III; NOS type III; Endothelial nitric oxide synthase; Endothelial NOS; ENOS; EC-NOS; Constitutive NOS; CNOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function NO mediates vascular endothelial growth factor (VEGF)-induced angiogenesis in coronary vessels and promotes blood clotting through the activation of platelets. Produces nitric oxide (NO) which is implicated in vascular smooth muscle relaxation through a cGMP-mediated signal transduction pathway. Related diseases Variation Asp-298 in NOS3 may be associated with susceptibility to coronary spasm. {ECO:0000269|PubMed:11740345, ECO:0000269|PubMed:9737779}. Drugs (DrugBank ID) DB07001; DB02048; DB02911; DB02335; DB01997; DB03332; DB04534; DB07244; DB03100; DB03918; DB02207; DB03065; DB00125; DB02994; DB01833; DB00155; DB00997; DB07388; DB03974; DB02077; DB01821; DB09237; DB01110; DB03144; DB03305; DB01686; DB04559; DB02044; DB08019; DB08018; DB02027; DB02979; DB00435; DB04223; DB06154; DB03910; DB02141; DB03963; DB03707; DB02234; DB04018; DB00360; DB02589 Interacts with P60709; P63010-2; Q8N6T3-3; Q9Y575-3; Q96FT7-4; Q5SZD1; Q16543; Q9UNS2; Q8IUI8; P35222; Q05193; O15287; Q08379; Q71DI3; P69905; P61978; Q12891; Q9UKT9; Q9Y2M5; Q14525; Q6DKI2; P43364-2; Q8N6F8; O94851; A4FUJ8; Q8N594; Q8IVI9; Q6X4W1-6; O15381-5; Q9NV79; Q16549; Q5T2W1; O75925; Q96I34; Q6ZMI0-5; P57052; Q9GZR2; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q7Z699; Q7Z698; P50502; Q9BR01-2; Q9NVV9; Q86WT6-2; Q9H347; P58304; Q9NZC7-5; Q9UNY5; P14079 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calmodulin-binding; Cell membrane; Cytoplasm; Cytoskeleton; Direct protein sequencing; FAD; Flavoprotein; FMN; Golgi apparatus; Heme; Iron; Lipoprotein; Membrane; Metal-binding; Myristate; NADP; Oxidoreductase; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 90790.1 Length 803 Aromaticity 0.11 Instability index 50.67 Isoelectric point 6.03 Charge (pH=7) -9.56 2D Binding mode Binding energy (Kcal/mol) -6.25  Molscript Map  Pymol Map 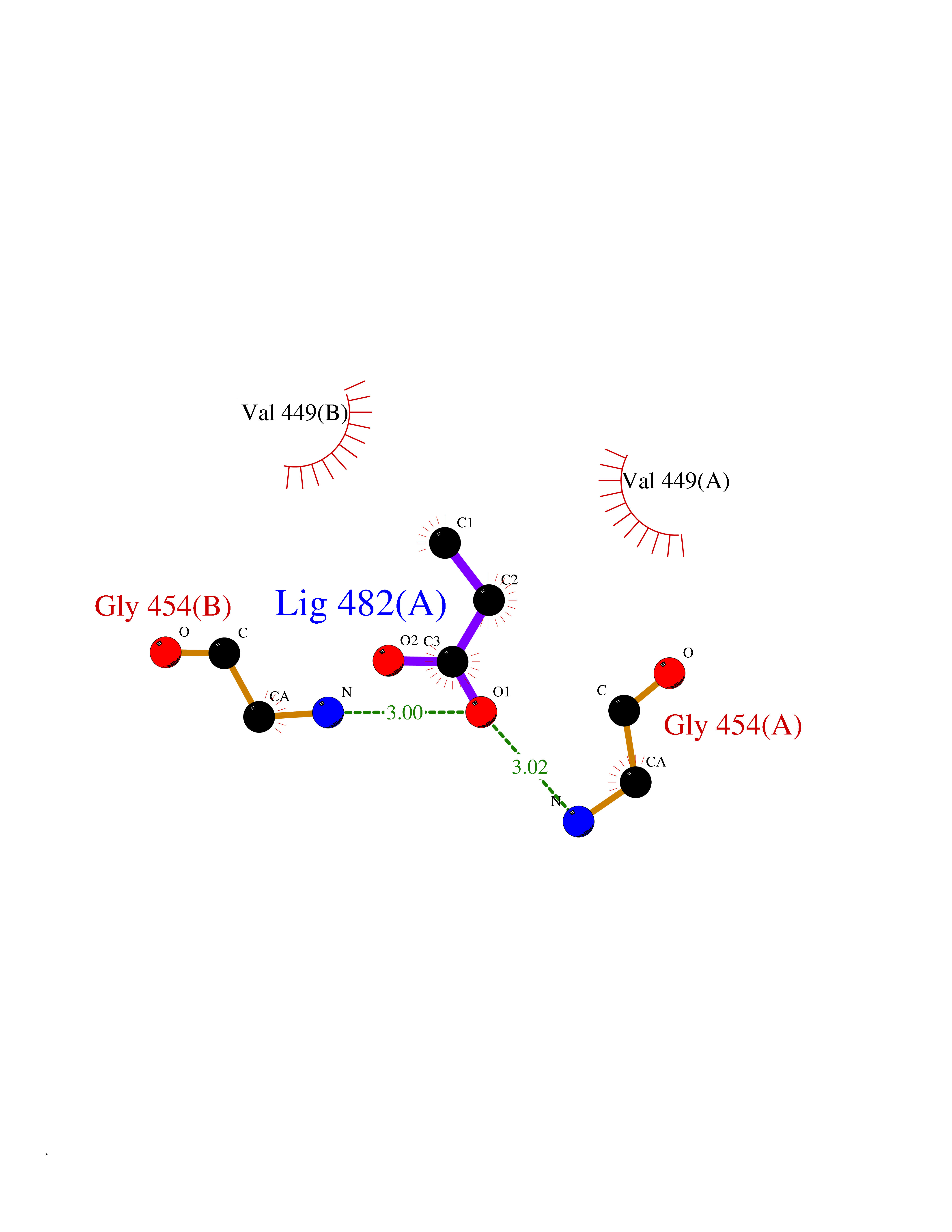 Ligplot Map 3D Binding mode Sequence FPRVKNWEVGSITYDTLSAQAQQDGPCTPRRCLGSLVFPAPEQLLSQARDFINQYYSSIKRSGSQAHEQRLQEVEAEVAATGTYQLRESELVFGAKQAWRNAPRCVGRIQWGKLQVFDARDCRSAQEMFTYICNHIKYATNRGNLRSAITVFPQRCPGRGDFRIWNSQLVRYAGYRQQDGSVRGDPANVEITELCIQHGWTPGNGRFDVLPLLLQAPDEPPELFLLPPELVLEVPLEHPTLEWFAALGLRWYALPAVSNMLLEIGGLEFPAAPFSGWYMSTEIGTRNLCDPHRYNILEDVAVCMDLDTRTTSSLWKDKAAVEINVAVLHSYQLAKVTIVDHHAATASFMKHLENEQKARGGCPADWAWIVPPISGSLTPVFHQEMVNYFLSPAFRYQPDPWKFPRVKNWEVGSITYDTLSAQAQQDGPCTPRRCLGSLVFPAPEQLLSQARDFINQYYSSIKRSGSQAHEQRLQEVEAEVAATGTYQLRESELVFGAKQAWRNAPRCVGRIQWGKLQVFDARDCRSAQEMFTYICNHIKYATNRGNLRSAITVFPQRCPGRGDFRIWNSQLVRYAGYRQQDGSVRGDPANVEITELCIQHGWTPGNGRFDVLPLLLQAPDEPPELFLLPPELVLEVPLEHPTLEWFAALGLRWYALPAVSNMLLEIGGLEFPAAPFSGWYMSTEIGTRNLCDPHRYNILEDVAVCMDLDTRTTSSLWKDKAAVEINVAVLHSYQLAKVTIVDHHAATASFMKHLENEQKARGGCPADWAWIVPPISGSLTPVFHQEMVNYFLSPAFRYQPDPW Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 4.58 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -6.24 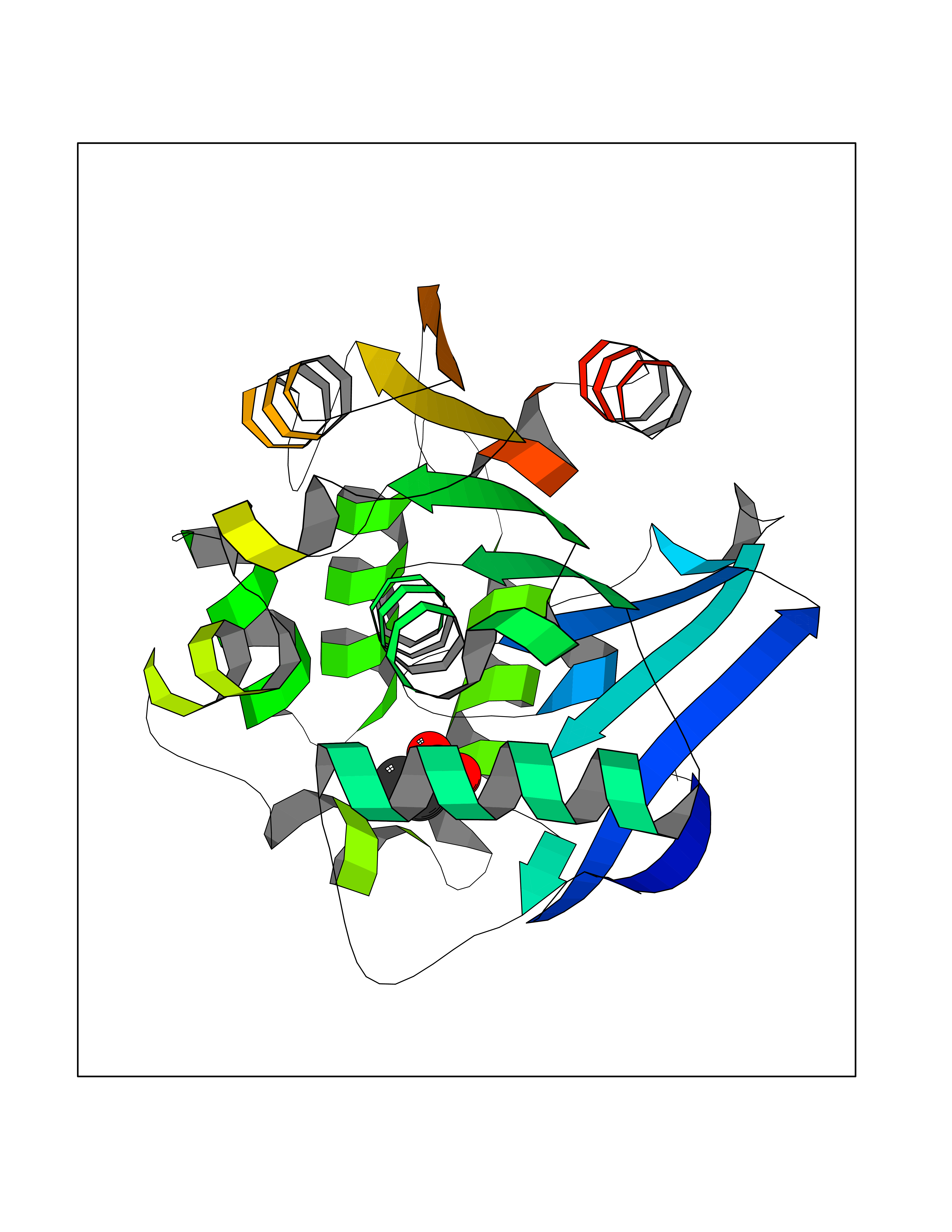 Molscript Map 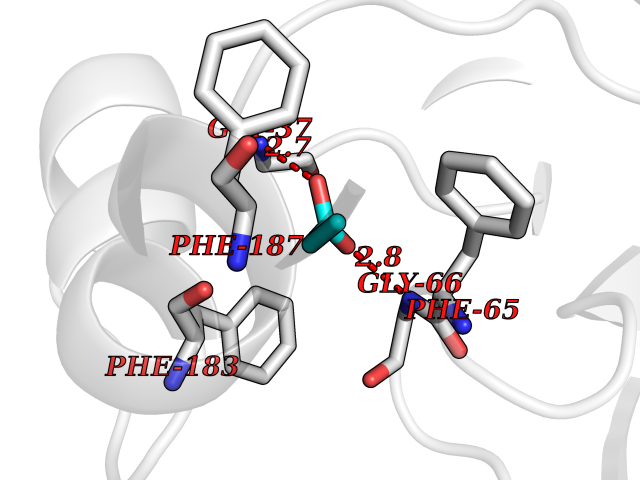 Pymol Map 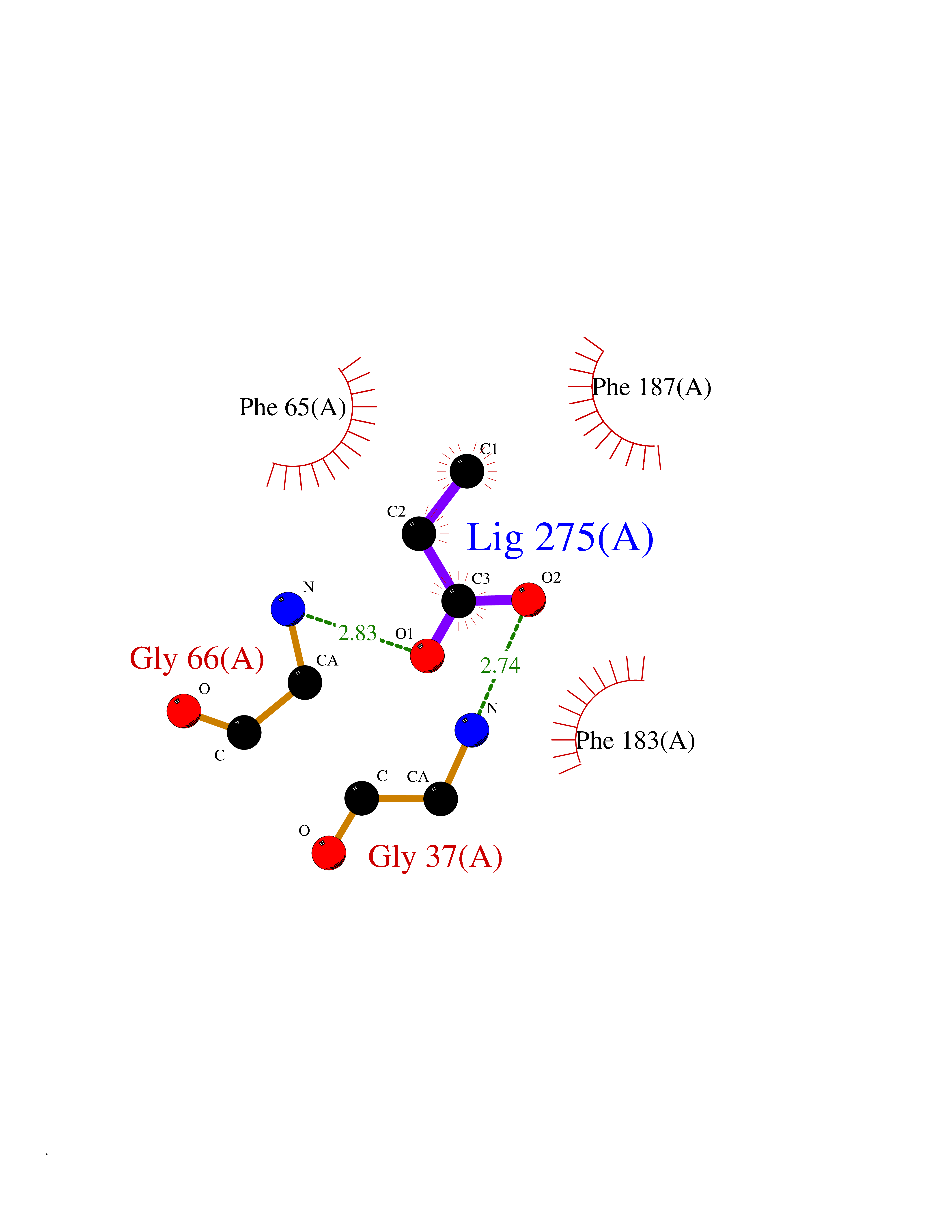 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Dihydroorotate dehydrogenase (quinone), mitochondrial | 4CQ8 | 4.58 | |
Target general information Gen name PFF0160c Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class Oxidoreductase Function Dihydroorotate dehydrogenase activity. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42573.5 Length 378 Aromaticity 0.1 Instability index 36.63 Isoelectric point 8.21 Charge (pH=7) 3.17 2D Binding mode Binding energy (Kcal/mol) -6.25 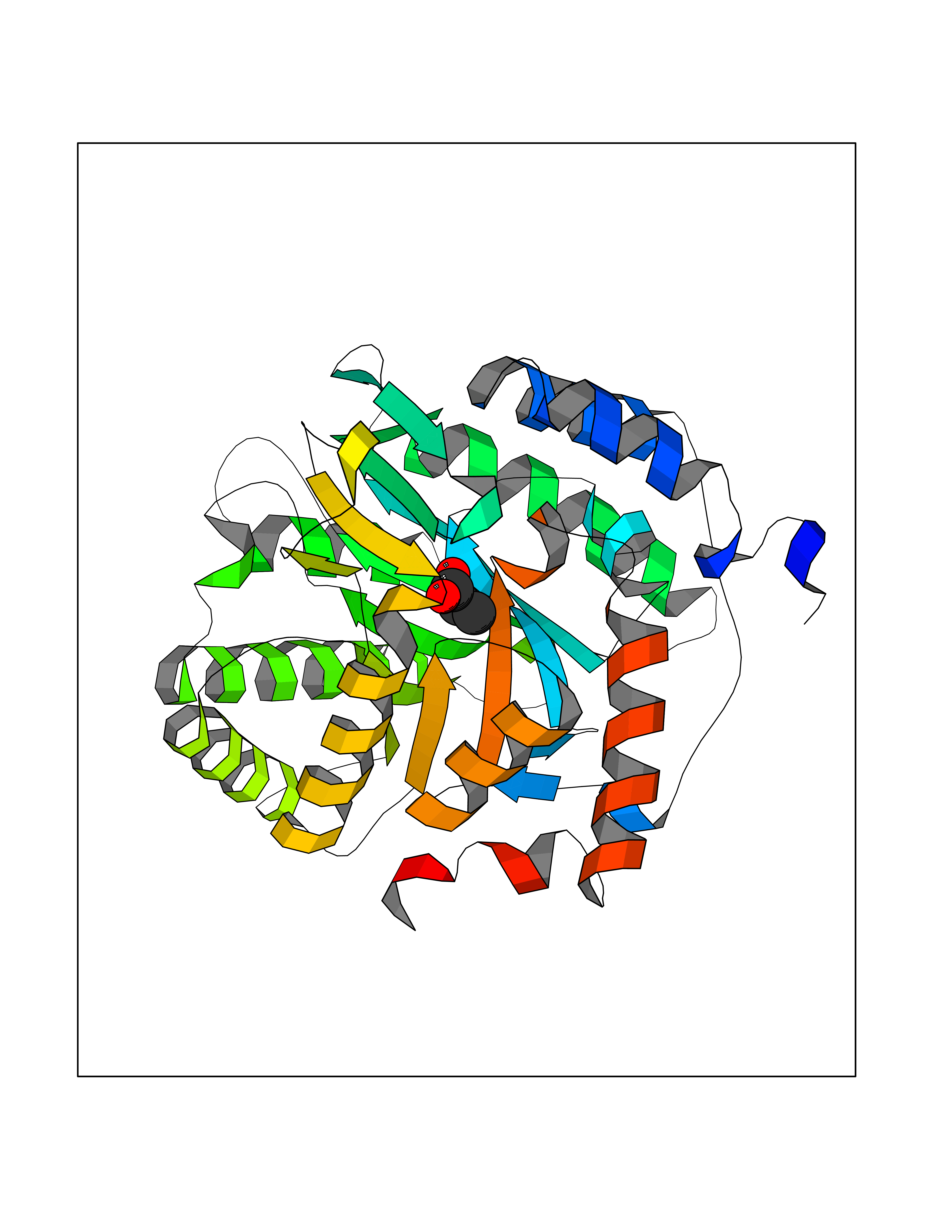 Molscript Map 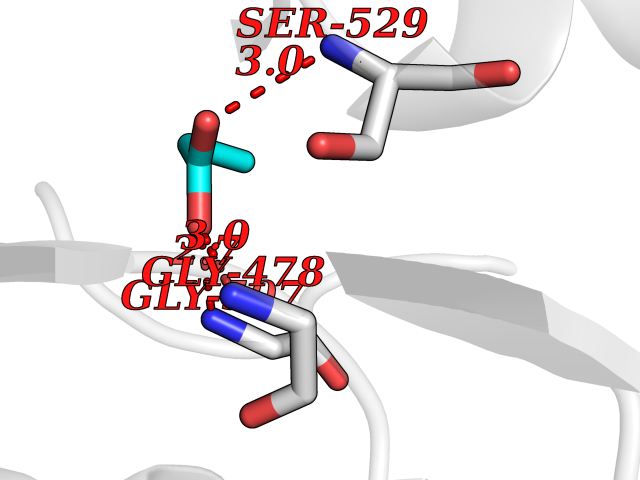 Pymol Map 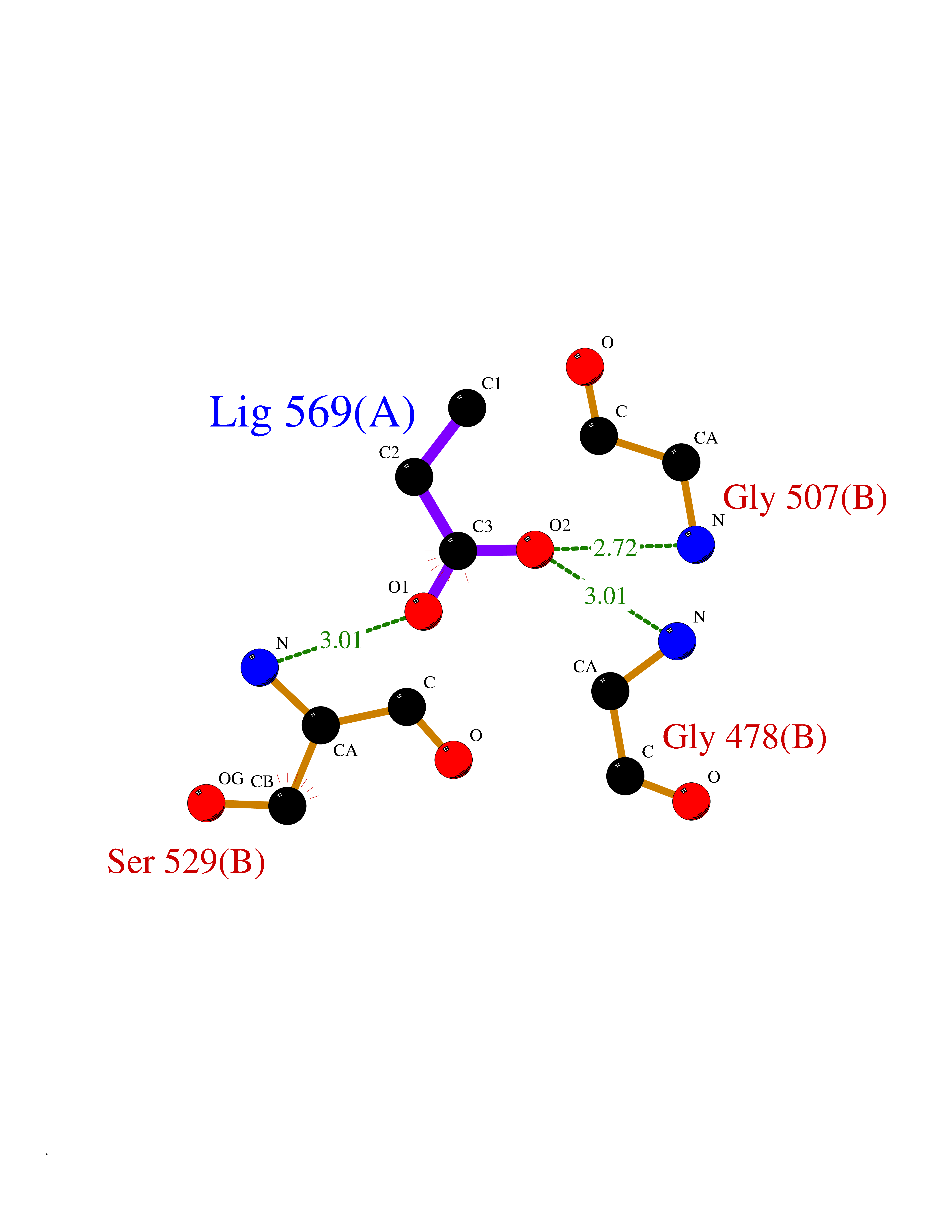 Ligplot Map 3D Binding mode Sequence ADPFESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEKNNFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 4.58 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -6.25 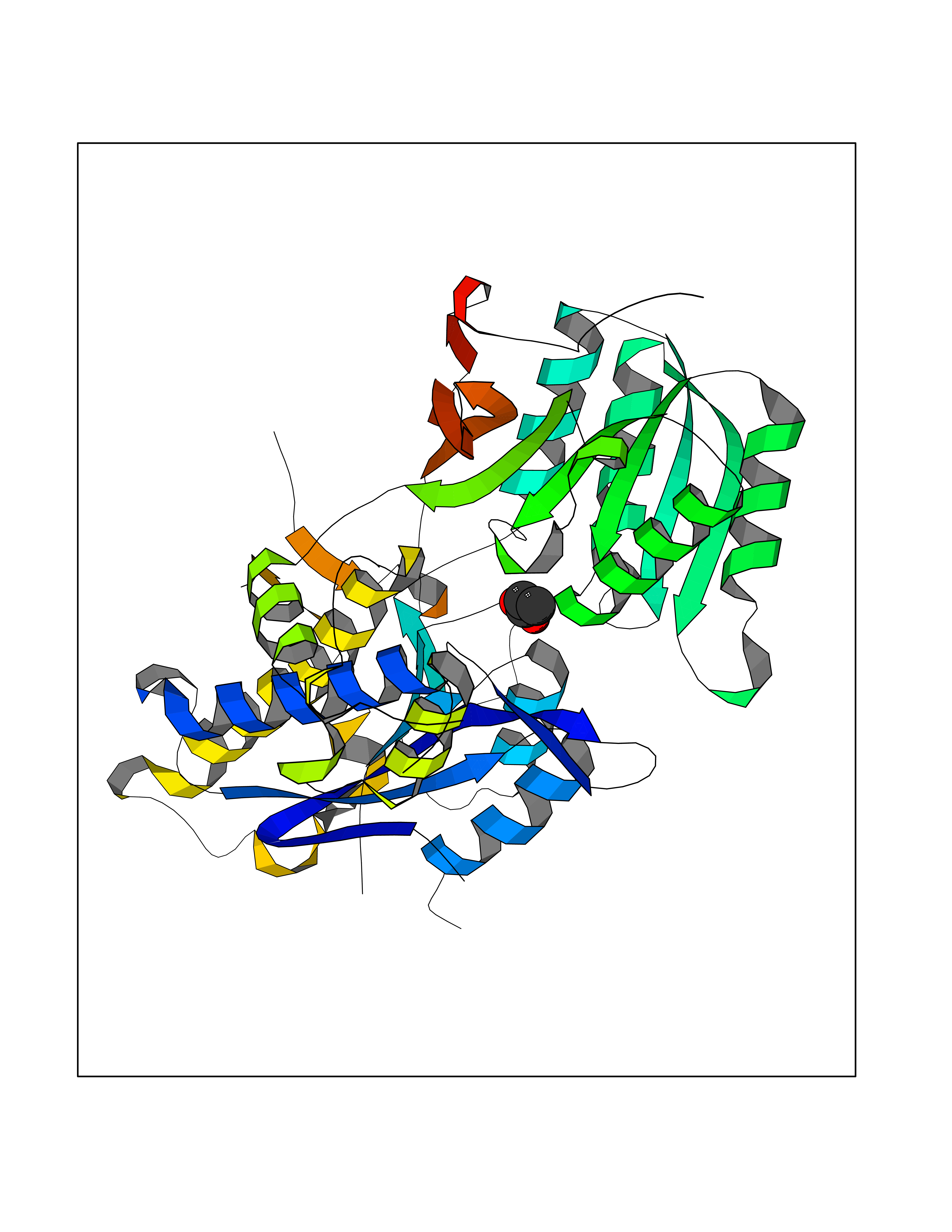 Molscript Map 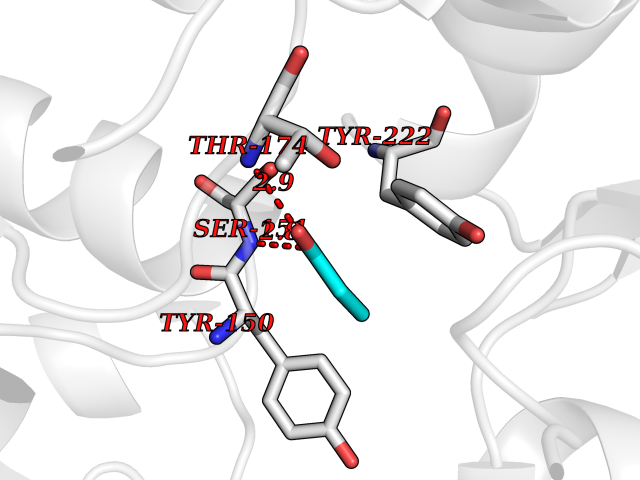 Pymol Map 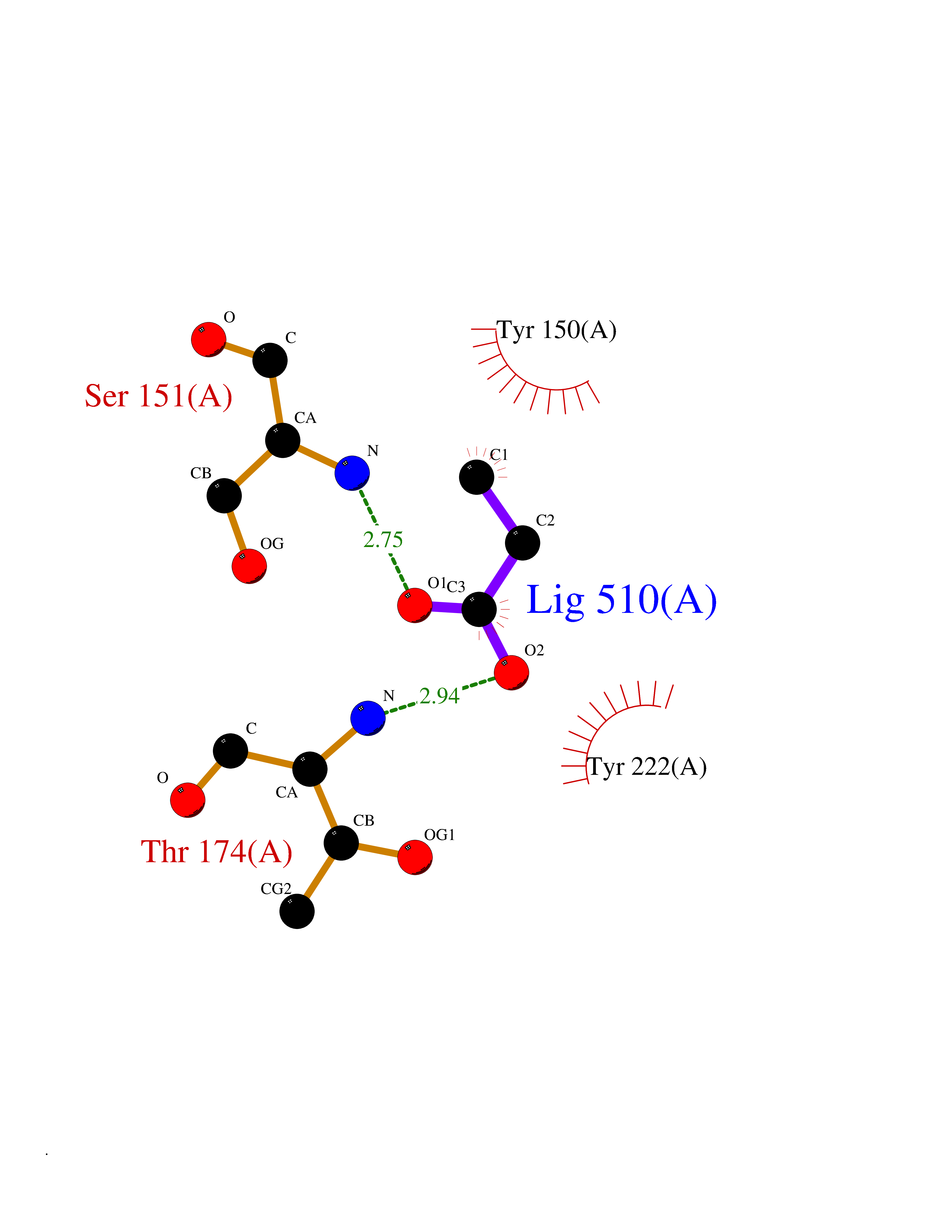 Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | C5a anaphylatoxin chemotactic receptor (C5AR1) | 5O9H | 4.58 | |
Target general information Gen name C5AR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CD88; C5aR; C5a-R; C5a anaphylatoxin chemotactic receptor 1; C5R1 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function The ligand interacts with at least two sites on the receptor: a high-affinity site on the extracellular N-terminus, and a second site in the transmembrane region which activates downstream signaling events. Receptor activation stimulates chemotaxis, granule enzyme release, intracellular calcium release and superoxide anion production. Receptor for the chemotactic and inflammatory peptide anaphylatoxin C5a. Related diseases Acrokeratosis verruciformis (AKV) [MIM:101900]: A localized disorder of keratinization, which is inherited as an autosomal dominant trait. Its onset is early in life with multiple flat-topped, flesh-colored papules on the hands and feet, punctate keratoses on the palms and soles, with varying degrees of nail involvement. The histopathology shows a distinctive pattern of epidermal features with hyperkeratosis, hypergranulosis and acanthosis together with papillomatosis. These changes are frequently associated with circumscribed elevations of the epidermis that are said to resemble church spires. There are no features of dyskeratosis or acantholysis, the typical findings in lesions of Darier disease. {ECO:0000269|PubMed:12542527}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Darier disease (DD) [MIM:124200]: A skin disorder characterized by warty papules and plaques in seborrheic areas (central trunk, flexures, scalp and forehead), palmoplantar pits and distinctive nail abnormalities. It is due to loss of adhesion between epidermal cells (acantholysis) and abnormal keratinization. Patients with mild disease may have no more than a few scattered keratotic papules or subtle nail changes, whereas those with severe disease are handicapped by widespread malodorous keratotic plaques. Some patients present with hemorrhage into acantholytic vesicles on the palms and dorsal aspects of the fingers which gives rise to black macules. In a few families affected by Darier disease, neuropsychiatric abnormalities such as mild intellectual disability, schizophrenia, bipolar disorder and epilepsy have been reported. Stress, UV exposure, heat, sweat, friction and oral contraception exacerbate disease symptoms. Clinical variants of Darier disease include hypertrophic, vesicobullous, hypopigmented, cornifying, zosteriform or linear, acute and comedonal subtypes. Comedonal Darier disease is characterized by the coexistence of acne-like comedonal lesions with typical Darier hyperkeratotic papules on light-exposed areas. At histopathologic level, comedonal Darier disease differs from classic Darier disease in the prominent follicular involvement and the presence of greatly elongated dermal villi. {ECO:0000269|PubMed:10080178, ECO:0000269|PubMed:10441323, ECO:0000269|PubMed:10441324, ECO:0000269|PubMed:10441325, ECO:0000269|PubMed:19995371, ECO:0000269|PubMed:28035777}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15011 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Chemotaxis; Cytoplasmic vesicle; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Sulfation; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 65967.4 Length 589 Aromaticity 0.12 Instability index 35.93 Isoelectric point 9.27 Charge (pH=7) 18.32 2D Binding mode Binding energy (Kcal/mol) -6.25  Molscript Map 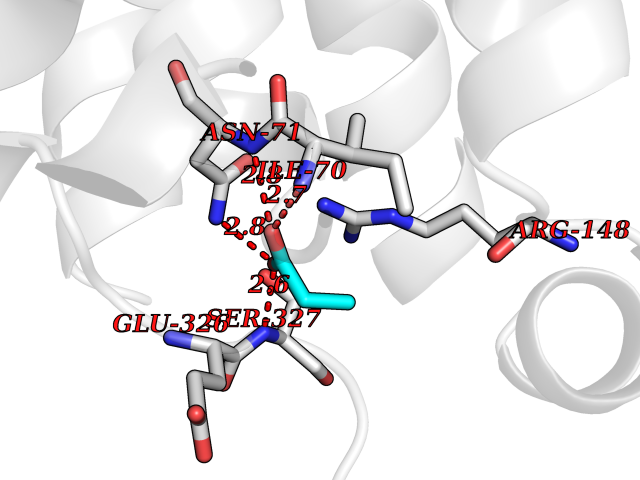 Pymol Map 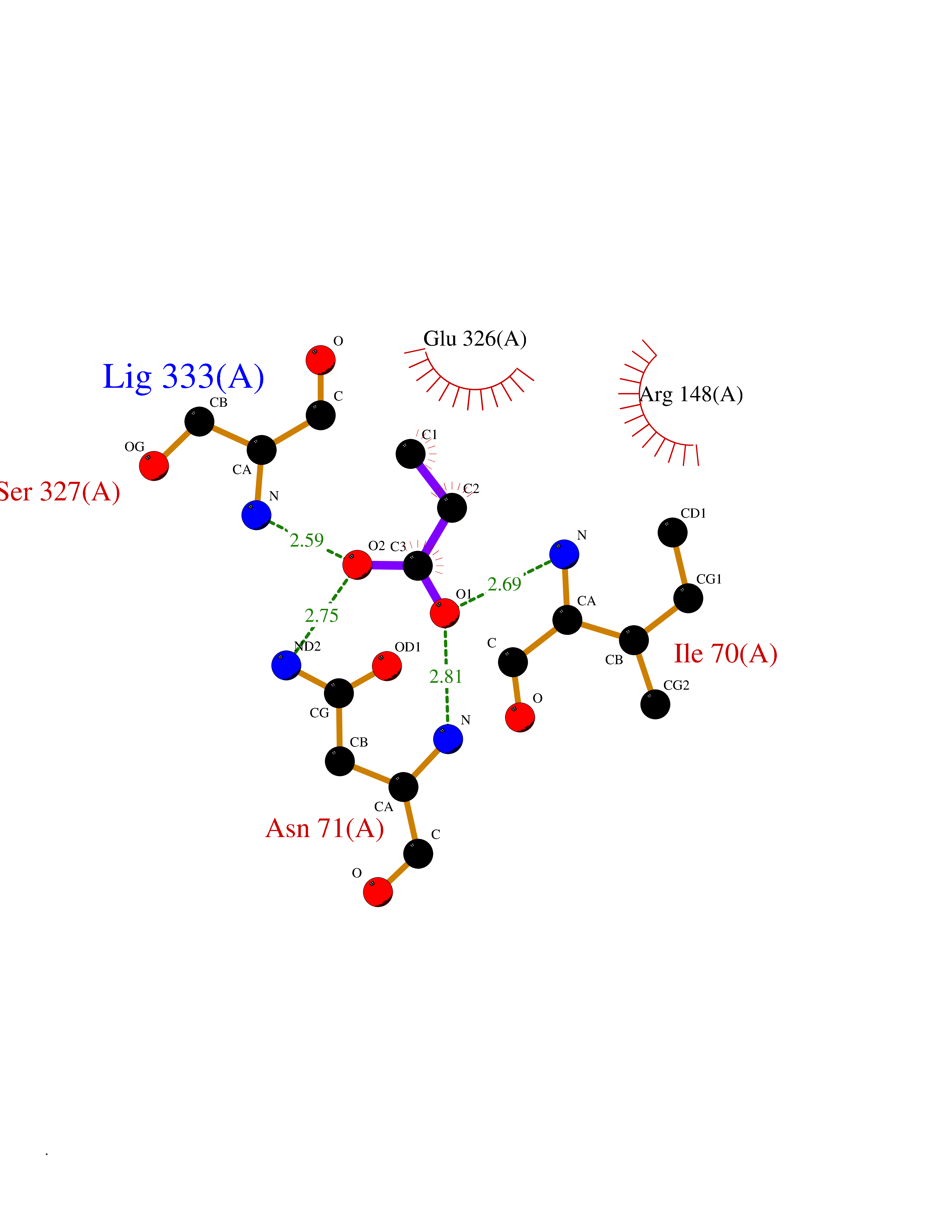 Ligplot Map 3D Binding mode Sequence NTLRVPDILALVIFAVVFLVGVLGNALVVWVTAFEAKRTINAIWFLNLAVADFLACLALPALFTSIVQHHHWPFGGAACSILPSLILLNMYASILLLATISADRFLLVFKPAWCQRFRGAGLAWILCAVAWGLALLLTIPSALYRVVREEYFPPKVLCGVDHDKRRERAVAIVRLVLGFLWPLLTLTICYTFILLRTWSARETRSTKTLKVVVAVVASFFIFWLPYQVTGIMMSFLEPSSPTFLLLKKLDSLCVSFAYINCCINPIIYVVAGQGFQKSLPELLREVLTEESVVRNTLRVPDILALVIFAVVFLVGVLGNALVVWVTAFEAKRTINAIWFLNLAVADFLACLALPALFTSIVQHHHWPFGGAACSILPSLILLNMYASILLLATISADRFLLVFKPAWCQRFRGAGLAWILCAVAWGLALLLTIPSALYRVVREEYFPPKVLCGVDYSHDKRRERAVAIVRLVLGFLWPLLTLTICYTFILLRTWSARETRSTKTLKVVVAVVASFFIFWLPYQVTGIMMSFLEPSSPTFLLLKKLDSLCVSFAYINCCINPIIYVVAGQRKSLPELLREVLTEESVVRE Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Protein kinase G2 (PRKG2) | 5BV6 | 4.58 | |
Target general information Gen name PRKG2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-dependent protein kinase II; cGMP-dependent protein kinase 2; cGKII; cGK2; cGK 2; PRKGR2 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, cGMP subfamily Biochemical class NA Function Crucial regulator of intestinal secretion and bone growth (By similarity). Phosphorylates and activates CFTR on the plasma membrane. Plays a key role in intestinal secretion by regulating cGMP-dependent translocation of CFTR in jejunum (By similarity). Acts downstream of NMDAR to activate the plasma membrane accumulation of GRIA1/GLUR1 in synapse and increase synaptic plasticity. Phosphorylates GRIA1/GLUR1 at Ser-863 (By similarity). Acts as regulator of gene expression and activator of the extracellular signal-regulated kinases MAPK3/ERK1 and MAPK1/ERK2 in mechanically stimulated osteoblasts. Under fluid shear stress, mediates ERK activation and subsequent induction of FOS, FOSL1/FRA1, FOSL2/FRA2 and FOSB that play a key role in the osteoblast anabolic response to mechanical stimulation (By similarity). Related diseases Spondylometaphyseal dysplasia, Pagnamenta type (SMDP) [MIM:619638]: A form of spondylometaphyseal dysplasia, a group of short stature disorders distinguished by abnormalities in the vertebrae and the metaphyses of the tubular bones. SMDP is an autosomal recessive form characterized by short stature and mild platyspondyly with no disproportion between the limbs. Mild metaphyseal changes are present. {ECO:0000269|PubMed:34782440}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Acromesomelic dysplasia 4 (AMD4) [MIM:619636]: A form of acromesomelic dysplasia, a skeletal disorder characterized by short stature, very short limbs and hand/foot malformations. The severity of limb abnormalities increases from proximal to distal with profoundly affected hands and feet showing brachydactyly and/or rudimentary fingers (knob-like fingers). AMD4 radiographic hallmarks include mild to moderate platyspondyly, moderate brachydactyly, iliac flaring, and metaphyseal alterations of the long bones that progressively increase with age. AMD4 inheritance is autosomal recessive. {ECO:0000269|PubMed:33106379, ECO:0000269|PubMed:34782440}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with NA EC number EC 2.7.11.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; cGMP; cGMP-binding; Coiled coil; Disease variant; Dwarfism; Kinase; Lipoprotein; Membrane; Myristate; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 17227.1 Length 150 Aromaticity 0.09 Instability index 43.7 Isoelectric point 4.88 Charge (pH=7) -8.41 2D Binding mode Binding energy (Kcal/mol) -6.24  Molscript Map 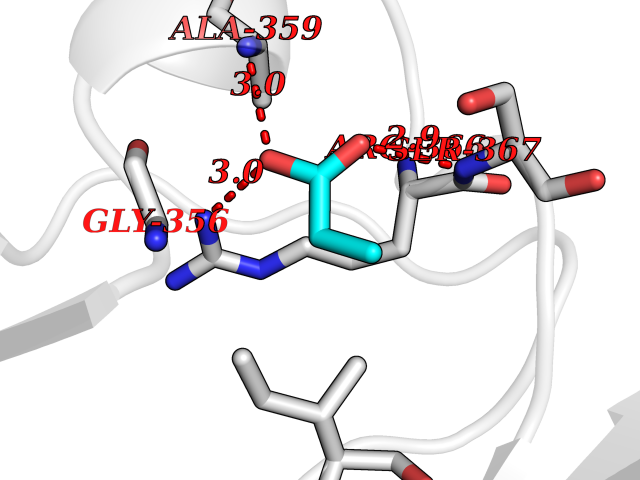 Pymol Map 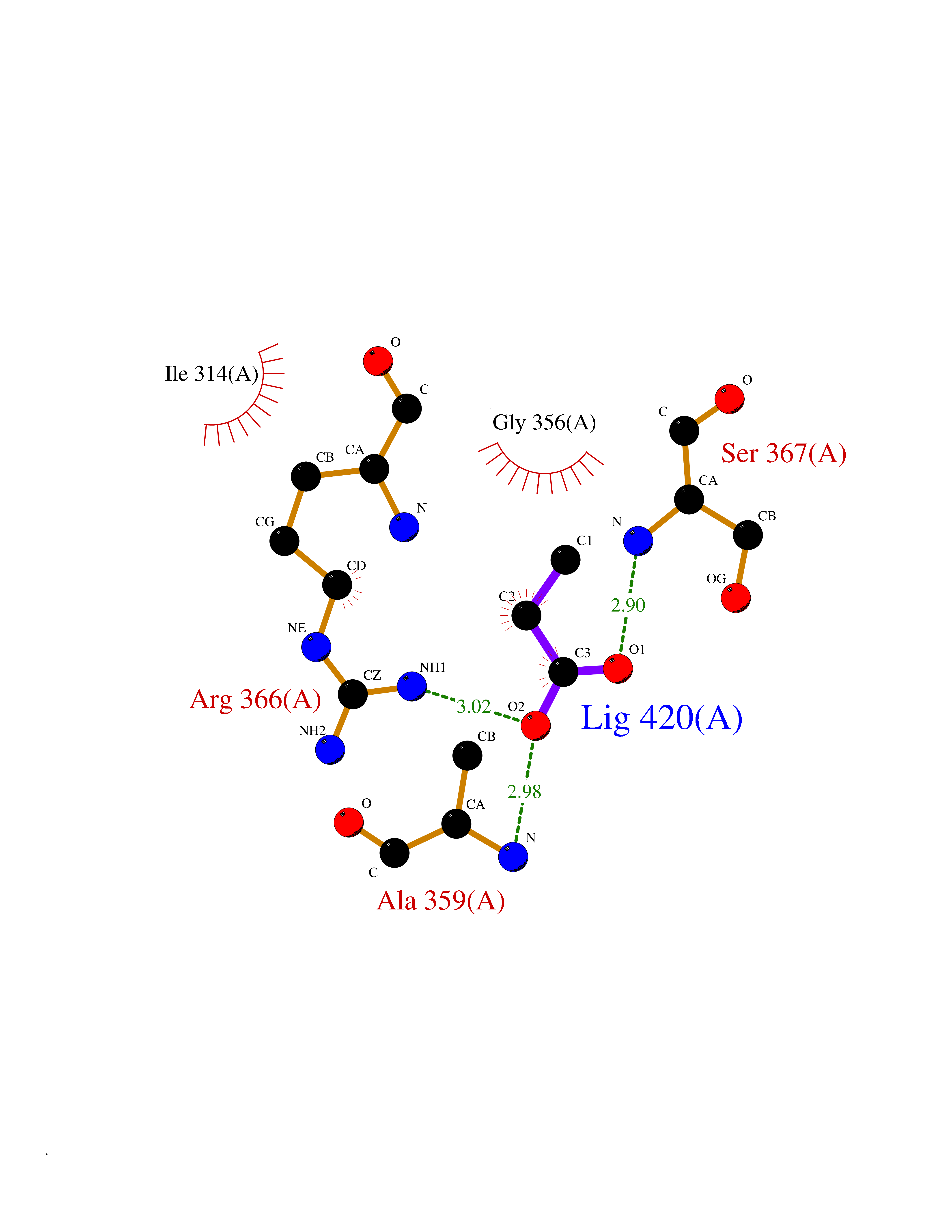 Ligplot Map 3D Binding mode Sequence TAQARDEQYRNFLRSVSLLKNLPEDKLTKIIDCLEVEYYDKGDYIIREGEEGSTFFILAKGKVKVTQSTEGHDQPQLIKTLQKGEYFGEKALISDDVRSANIIAEENDVACLVIDRETFNQTVGTFEELQKYLEGYVANLNRDDEKRHAK Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Isopeptidase T (USP5) | 3IHP | 4.58 | |
Target general information Gen name USP5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin-specific-processing protease 5; Ubiquitin thioesterase 5; Ubiquitin carboxyl-terminal hydrolase 5; ISOT; Deubiquitinating enzyme 5 Protein family Peptidase C19 family Biochemical class Peptidase Function Involved in unanchored 'Lys-48'-linked polyubiquitin disassembly. Binds linear and 'Lys-63'-linked polyubiquitin with a lower affinity. Knock-down of USP5 causes the accumulation of p53/TP53 and an increase in p53/TP53 transcriptional activity because the unanchored polyubiquitin that accumulates is able to compete with ubiquitinated p53/TP53 but not with MDM2 for proteasomal recognition. Cleaves linear and branched multiubiquitin polymers with a marked preference for branched polymers. Related diseases Defects in KAT2B has been found in a patient with isolated coloboma, a defect of the eye characterized by the absence of ocular structures due to abnormal morphogenesis of the optic cup and stalk, and the fusion of the fetal fissure (optic fissure). Isolated colobomas may be associated with an abnormally small eye (microphthalmia) or small cornea. {ECO:0000269|PubMed:28493397}. Drugs (DrugBank ID) NA Interacts with Q15038; P54727; O75528; Q8WW34; Q9NWF9; Q8N0X7; Q8WW34-2; O14773; Q9BZR9; P0CG47 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disulfide bond; Hydrolase; Metal-binding; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Thiol protease; Ubl conjugation pathway; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 84381.3 Length 748 Aromaticity 0.09 Instability index 38.12 Isoelectric point 5.22 Charge (pH=7) -25.02 2D Binding mode Binding energy (Kcal/mol) -6.25  Molscript Map 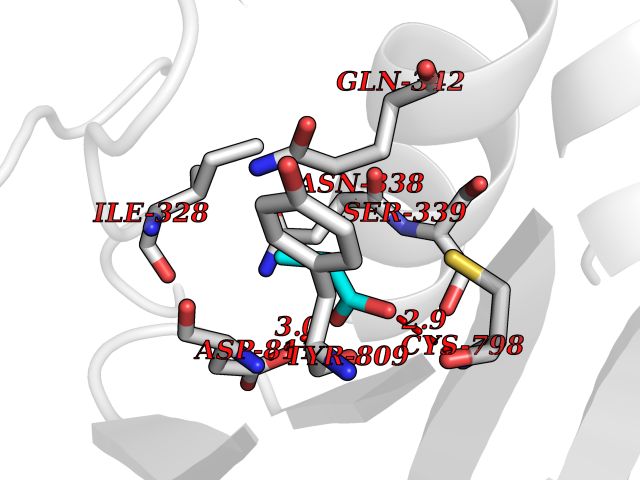 Pymol Map 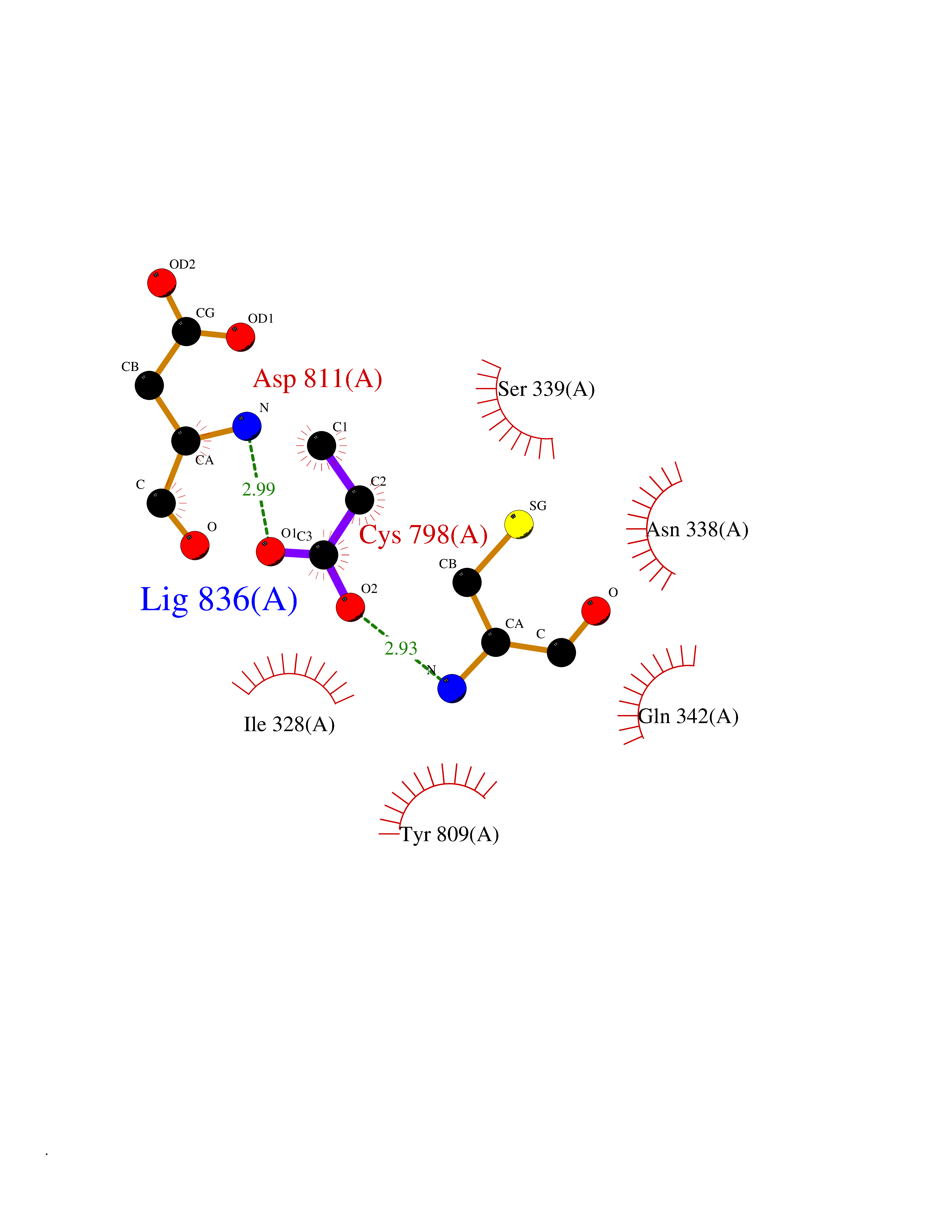 Ligplot Map 3D Binding mode Sequence MAELSEEALLSVLPTIRVPKAGDRVHKDECAFSFDTPESEGGLYICMNTFLGFGKQYVERHFNKTGQRVYLHLRRTDEDVKIVILPDYLEIARDGLGGLPDIVRDRVTSAVEALLSVRQVSKHAFSLKQLDNPARIPPCGWKCSKCDMRENLWLNLTDGSILCGRRYDGSGGNNHAVEHYRETGYPLAVKLGTITPDGADVYSYDEDDMVLDPSLAEHLSHFGIDMPLFGPGYTGIRNLGNSCYLNSVVQVLFSIPDFQRKYVDKLEKIFQNAPTDPTQDFSTQVAKLGHGLLSGEDGIAPRMFKALIGKGHPEFSTNRQQDAQEFFLHLINMVERNCRSSENPNEVFRFLVEEKIKCLATEKVKYTQRVDYIMQLPVPMDAALNKEELLEYEEKKRQAEEEKMALPELVRAQVPFSSCLEAYGAPEQVDDFWSTALQAKSVAVKTTRFASFPDYLVIQIKKFTFGLDWVPKKLDVSIEMPEELDISQLRGTGLQPGEEELPDIESVIIQLVEMGFPMDACRKAVYYTGNSGAEAAMNWVMSHMDDPDFANPLILPPPEDCVTTIVSMGFSRDQALKALRATNNSLERAVDWIFSHIDDLDAEAAMDPKVRDGPGKYQLFAFISHMGTSTMCGHYVCHIKKEGRWVIYNDQKVCASEKPPKDLGYIYFYQRVAMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRG Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Ubiquitin thioesterase L1 (UCHL1) | 3IFW | 4.58 | |
Target general information Gen name UCHL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin thiolesterase L1; Ubiquitin carboxyl-terminal hydrolase isozyme L1; Ubiquitin carboxy-terminal hydrolase L1; UCH-L1; PGP9.5; PGP 9.5; Neuron cytoplasmic protein 9.5 Protein family Peptidase C12 family Biochemical class Peptidase Function Ubiquitin-protein hydrolase involved both in the processing of ubiquitin precursors and of ubiquitinated proteins. This enzyme is a thiol protease that recognizes and hydrolyzes a peptide bond at the C-terminal glycine of ubiquitin. Also binds to free monoubiquitin and may prevent its degradation in lysosomes. The homodimer may have ATP-independent ubiquitin ligase activity. Related diseases Parkinson disease 5 (PARK5) [MIM:613643]: A complex neurodegenerative disorder with manifestations ranging from typical Parkinson disease to dementia with Lewy bodies. Clinical features include parkinsonian symptoms (resting tremor, rigidity, postural instability and bradykinesia), dementia, diffuse Lewy body pathology, autonomic dysfunction, hallucinations and paranoia. {ECO:0000269|PubMed:12408865, ECO:0000269|PubMed:12705903, ECO:0000269|PubMed:9774100}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Spastic paraplegia 79A, autosomal dominant, with ataxia (SPG79A) [MIM:620221]: A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG79A is a slowly progressive form characterized by late-onset spastic ataxia, neuropathy, and often optic atrophy. {ECO:0000269|PubMed:35986737}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spastic paraplegia 79B, autosomal recessive (SPG79B) [MIM:615491]: A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG79B is characterized by childhood onset blindness, cerebellar ataxia, nystagmus, dorsal column dysfunction, and spasticity with upper motor neuron dysfunction. {ECO:0000269|PubMed:23359680, ECO:0000269|PubMed:28007905}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12695 Interacts with P63010-2; P05067; P05067-2; Q8N6T3-3; P18847; Q9H1Y0; O15392; Q8WUW1; P83916; P11802; Q00535; Q9UNS2; Q92905; P00533; O60739; Q8TC29; Q9UI08-2; Q8WVV9-3; Q14164; Q6DN90-2; Q96JM7-2; P13473-2; Q9BYZ2; O95777; A4FUJ8; Q15843; O15381-5; Q9BR81; Q13113; P62826; Q8TAI7; Q9ULX5; Q15554-4; Q9NYB0; P04637; Q9Y4K3; P19474; Q9BSL1; Q7KZS0; P61086; Q9UK80; Q86WB0-2 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Glycoprotein; Hereditary spastic paraplegia; Hydrolase; Lipoprotein; Membrane; Neurodegeneration; Oxidation; Parkinson disease; Parkinsonism; Phosphoprotein; Prenylation; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 33389.8 Length 298 Aromaticity 0.07 Instability index 38.69 Isoelectric point 5.51 Charge (pH=7) -7.88 2D Binding mode Binding energy (Kcal/mol) -6.24  Molscript Map 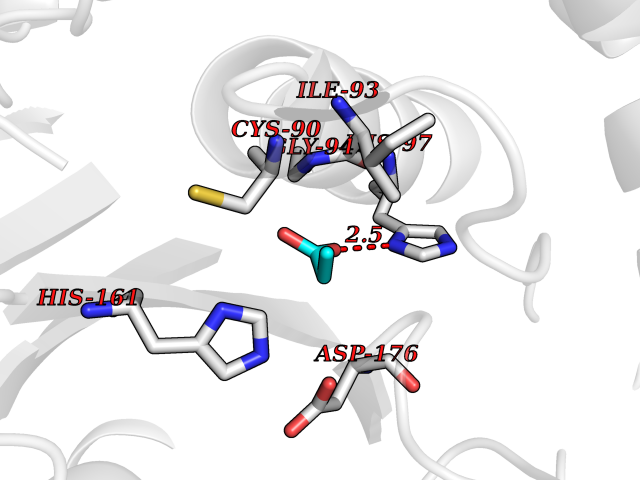 Pymol Map 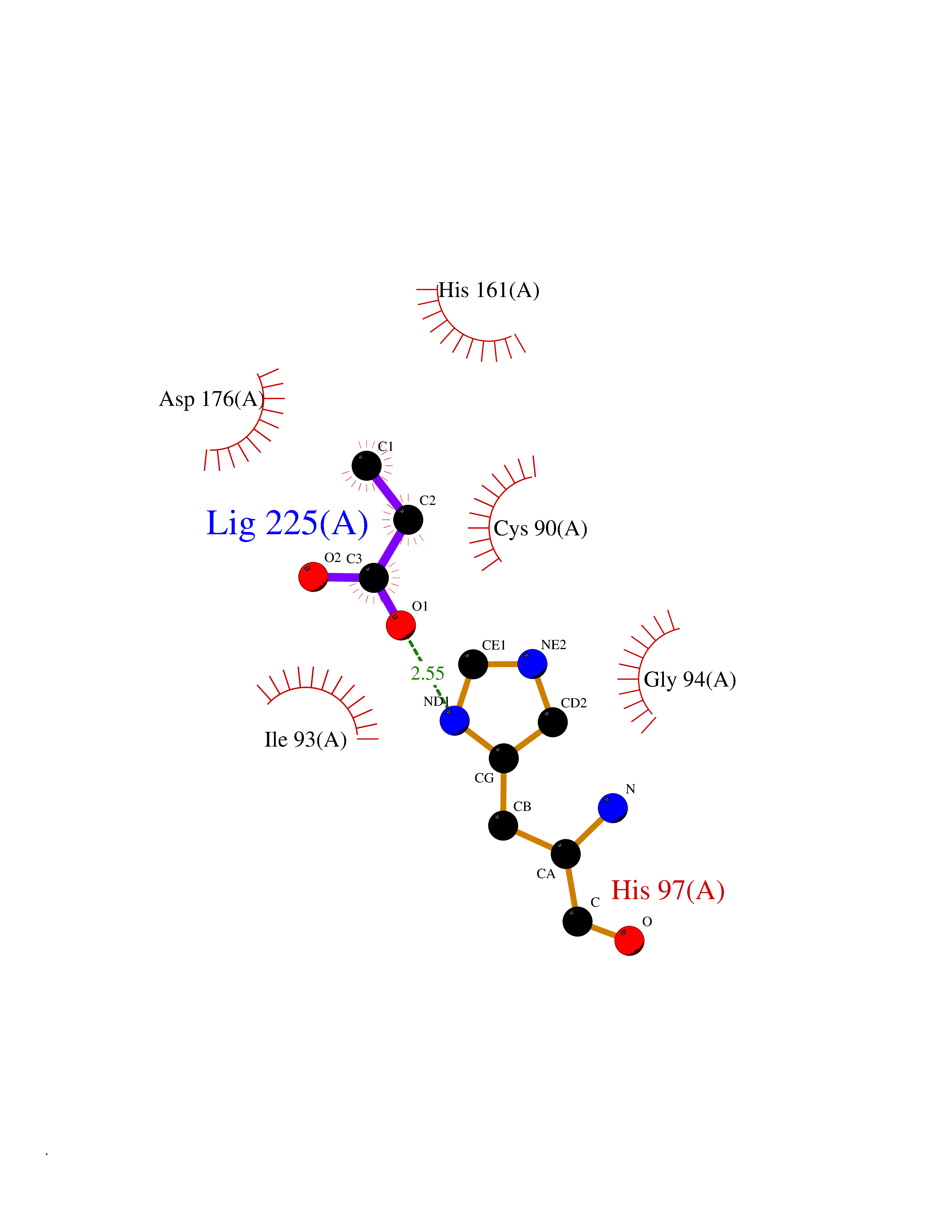 Ligplot Map 3D Binding mode Sequence MQLKPMEINPEMLNKVLYRLGVAGQWRFVDVLGLEEESLGSVPAPACALLLLFPLTAQHENFRKKQIEELKGQEVSPKVYFMKQTIGNSCGTIGLIHAVANNQDKLGFEDGSVLKQFLSETEKMSPEDRAKCFEKNEAIQAAHDAVAQEGQCRVDDKVNFHFILFNNVDGHLYELDGRMPFPVNHGASSEDTLLKDAAKVCREFTEREQGEVRFSAVALCKAAMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRG Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Plasma kallikrein (KLKB1) | 6T7P | 4.58 | |
Target general information Gen name KLKB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Plasma prekallikrein; Plasma kallikrein light chain; Plasma kallikrein heavy chain; PKK; Kininogenin; KLK3; Fletcher factor Protein family Peptidase S1 family, Plasma kallikrein subfamily Biochemical class Peptidase Function It activates, in a reciprocal reaction, factor XII after its binding to a negatively charged surface. It also releases bradykinin from HMW kininogen and may also play a role in the renin-angiotensin system by converting prorenin into renin. The enzyme cleaves Lys-Arg and Arg-Ser bonds. Related diseases Prekallikrein deficiency (PKKD) [MIM:612423]: An autosomal recessive condition characterized by a clotting defect due to prolongation of activated partial thromboplastin time. Affected individuals are clinically asymptomatic. {ECO:0000269|PubMed:14652634, ECO:0000269|PubMed:17598838, ECO:0000269|PubMed:34847617}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15982; DB09228; DB05311; DB12831; DB06404; DB14597; DB01593; DB14487; DB14533; DB14548 Interacts with Q9UI10; O00746; C9J082; O14744; Q8TAS3; O00233; Q8IYM2; Q9UMY4; O43493-5; Q8NFB2; Q8N0U8 EC number EC 3.4.21.34 Uniprot keywords 3D-structure; Blood coagulation; Direct protein sequencing; Disease variant; Disulfide bond; Fibrinolysis; Glycoprotein; Hemostasis; Hydrolase; Inflammatory response; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 26696.2 Length 237 Aromaticity 0.1 Instability index 34.33 Isoelectric point 8.07 Charge (pH=7) 2.21 2D Binding mode Binding energy (Kcal/mol) -6.25  Molscript Map 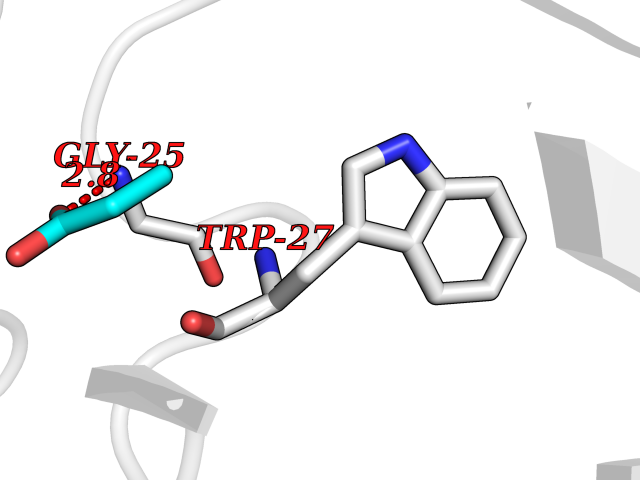 Pymol Map 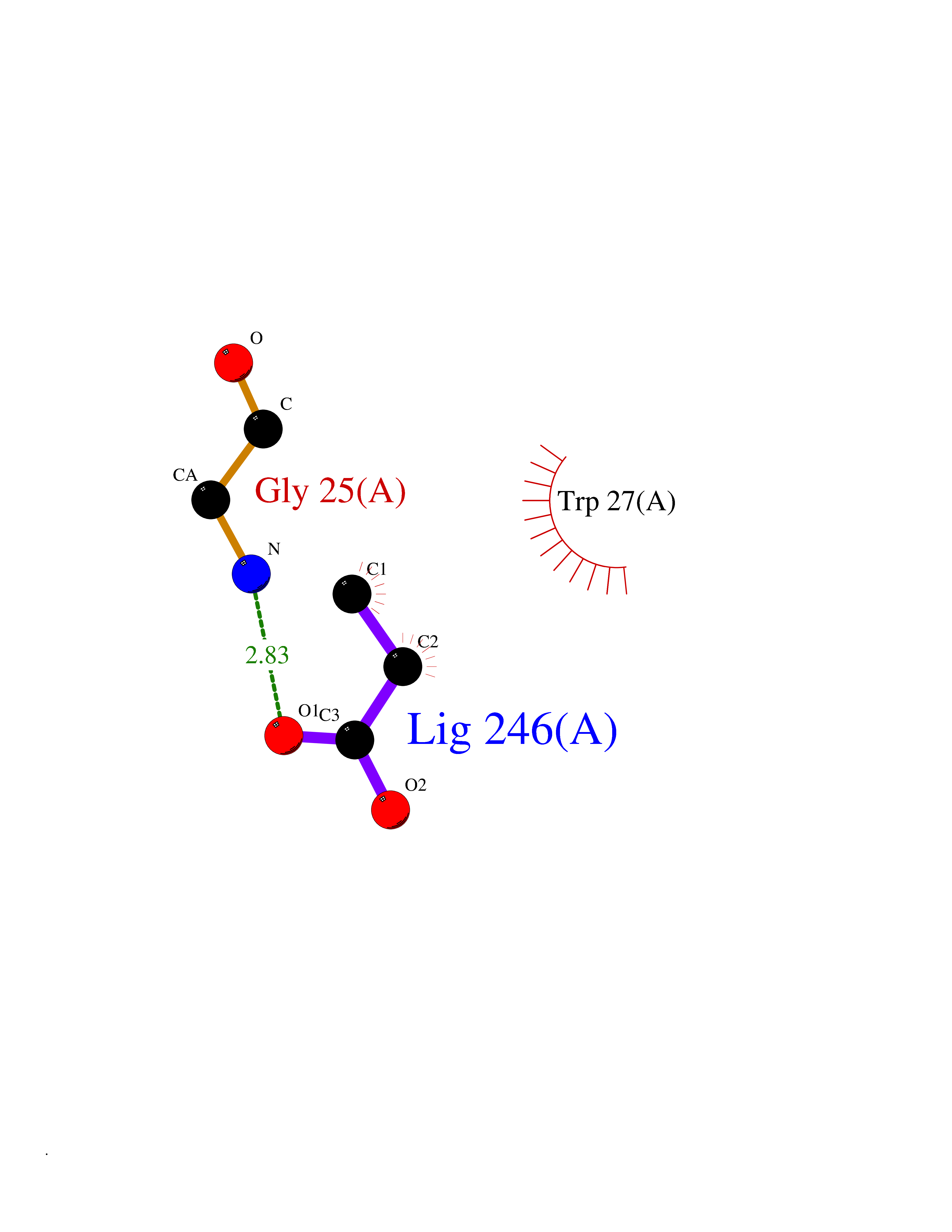 Ligplot Map 3D Binding mode Sequence IVGGTNSSWGEWPWQVSLQVKLTAQRHLCGGSLIGHQWVLTAAHCFDGLPLQDVWRIYSGILNLSDITKDTPFSQIKEIIIHQNYKVSEGNHDIALIKLQAPLNYTEFQKPICLPSKGDTSTIYTNCWVTGWGFSKEKGEIQNILQKVNIPLVTNEECQKRYQDYKITQRMVCAGYKEGGKDACKGDSGGPLVCKHNGMWRLVGITSWGEGCARREQPGVYTKVAEYMDWILEKTQS Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Dopamine D2 receptor (D2R) | 5AER | 4.57 | |
Target general information Gen name DRD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dopamine receptor 2; D(2) dopamine receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Dopamine receptor whose activity is mediated by G proteins which inhibit adenylyl cyclase. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01614; DB01063; DB01425; DB00915; DB06288; DB05964; DB00543; DB00182; DB04599; DB00714; DB01238; DB14185; DB09207; DB06216; DB04889; DB04888; DB05687; DB09223; DB04857; DB09128; DB01200; DB09018; DB00490; DB00248; DB06016; DB01038; DB00477; DB01239; DB00568; DB00363; DB01151; DB11274; DB13345; DB00320; DB01184; DB00988; DB00450; DB11275; DB01049; DB00696; DB01175; DB09194; DB00875; DB00623; DB04842; DB00502; DB04946; DB00458; DB04924; DB12579; DB01221; DB00555; DB01235; DB00589; DB00408; DB06077; DB08815; DB00934; DB09224; DB01043; DB00933; DB01403; DB01233; DB06148; DB00805; DB01618; DB08804; DB05766; DB00540; DB06229; DB00334; DB01267; DB12061; DB00715; DB01186; DB08922; DB00850; DB01100; DB09286; DB01621; DB12478; DB00413; DB00433; DB00420; DB01069; DB00777; DB01224; DB09097; DB12518; DB00409; DB00734; DB01549; DB00268; DB05271; DB06454; DB06144; DB00391; DB06477; DB04844; DB12093; DB00372; DB01622; DB00679; DB01623; DB13025; DB00831; DB00508; DB00726; DB06109; DB01392; DB00246; DB09225; DB01624 Interacts with Q9NRI5; P14416; Q01959 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Lipoprotein; Membrane; Palmitate; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID B,C Molecular weight (Da) 24300.3 Length 209 Aromaticity 0.13 Instability index 40.14 Isoelectric point 4.97 Charge (pH=7) -7.83 2D Binding mode Binding energy (Kcal/mol) -6.23  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PEVVEELTRKTYFTEKEVQQWYKGFIKDCPSGQLDAAGFQKIYKQFFPFGDPTKFATFVFNVFDENKDGRIEFSEFIQALSVTSRGTLDEKLRWAFKLYDLDNDGYITRNEMLDIVDAIYQMVGNTVELPEEENTPEKRVDRIFAMMDKNADGKLTLQEFQEGSKADPSIVQALSLYDGLVNIEFRKAFLKILHSNIEFRKAFLKILHS Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Pyridoxal kinase (PDXK) | 3KEU | 4.57 | |
Target general information Gen name PDXK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Pyridoxine kinase; PDXK Protein family Pyridoxine kinase family Biochemical class Kinase Function Required for synthesisof pyridoxal-5-phosphate from vitamin B6. Related diseases Neuropathy, hereditary motor and sensory, 6C, with optic atrophy (HMSN6C) [MIM:618511]: An autosomal recessive neurologic disorder characterized by childhood onset of axonal, sensorimotor polyneuropathy affecting mainly the lower limbs, and adult-onset optic atrophy. Clinical features include progressive distal muscle weakness and atrophy, significant standing and walking difficulties, areflexia, neurogenic pain and progressive visual impairment. {ECO:0000269|PubMed:31187503}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04776; DB03909; DB04770; DB00147; DB00165 Interacts with NA EC number EC 2.7.1.35 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Charcot-Marie-Tooth disease; Cobalt; Cytoplasm; Disease variant; Kinase; Magnesium; Manganese; Metal-binding; Neurodegeneration; Neuropathy; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Sodium; Transferase; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34370.1 Length 305 Aromaticity 0.07 Instability index 39.92 Isoelectric point 6.16 Charge (pH=7) -3.37 2D Binding mode Binding energy (Kcal/mol) -6.23  Molscript Map 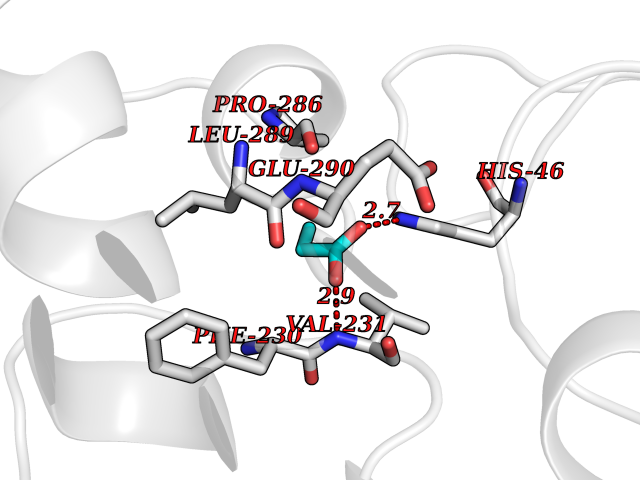 Pymol Map 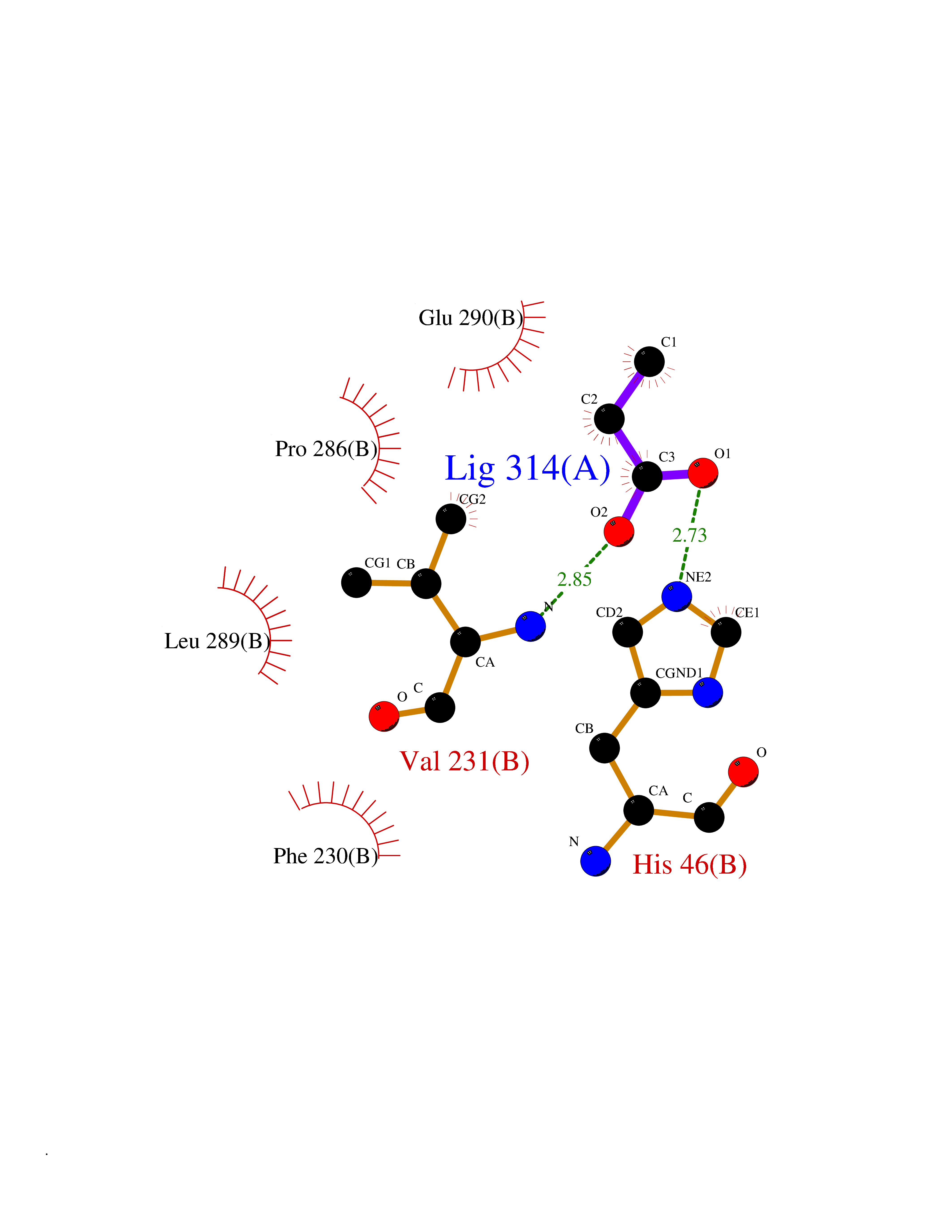 Ligplot Map 3D Binding mode Sequence ECRVLSIQSHVIRGYVGNRAATFPLQVLGFEIDAVNSVQFSNHTGYAHWKGQVLNSDELQELYEGLRLNNMNKYDYVLTGYTRDKSFLAMVVDIVQELKQQNPRLVYVCDPVLGDKWDGEGSMYVPEDLLPVYKEKVVPLADIITPNQFEAELLSGRKIHSQEEALRVMDMLHSMGPDTVVITSSDLPSPQGSNYLIVLGSQRRRNPAGSVVMERIRMDIRKVDAVFVGTGDLFAAMLLAWTHKHPNNLKVACEKTVSTLHHVLQRTIQCAKAQARPSPMQLELRMVQSKRDIEDPEIVVQATVL Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Ornithine transcarbamylase (OTC) | 1OTH | 4.57 | |
Target general information Gen name OTC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms OTCase; Ornithine carbamoyltransferase, mitochondrial Protein family Aspartate/ornithine carbamoyltransferase superfamily, OTCase family Biochemical class NA Function Catalyzes the second step of the urea cycle, the condensation of carbamoyl phosphate with L-ornithine to form L-citrulline. The urea cycle ensures the detoxification of ammonia by converting it to urea for excretion. Related diseases Ornithine carbamoyltransferase deficiency (OTCD) [MIM:311250]: An X-linked disorder of the urea cycle which causes a form of hyperammonemia. Mutations with no residual enzyme activity are always expressed in hemizygote males by a very severe neonatal hyperammonemic coma that generally proves to be fatal. Heterozygous females are either asymptomatic or express orotic aciduria spontaneously or after protein intake. The disorder is treatable with supplemental dietary arginine and low protein diet. The arbitrary classification of patients into the 'neonatal' group (clinical hyperammonemia in the first few days of life) and 'late' onset (clinical presentation after the neonatal period) has been used to differentiate severe from mild forms. {ECO:0000269|PubMed:10070627, ECO:0000269|PubMed:10502831, ECO:0000269|PubMed:10737985, ECO:0000269|PubMed:11793483, ECO:0000269|PubMed:1480464, ECO:0000269|PubMed:1671317, ECO:0000269|PubMed:1721894, ECO:0000269|PubMed:2347583, ECO:0000269|PubMed:2474822, ECO:0000269|PubMed:2556444, ECO:0000269|PubMed:3170748, ECO:0000269|PubMed:7474905, ECO:0000269|PubMed:7951259, ECO:0000269|PubMed:8019569, ECO:0000269|PubMed:8081373, ECO:0000269|PubMed:8081398, ECO:0000269|PubMed:8099056, ECO:0000269|PubMed:8112735, ECO:0000269|PubMed:8530002, ECO:0000269|PubMed:8807340, ECO:0000269|PubMed:8830175, ECO:0000269|PubMed:8956038, ECO:0000269|PubMed:8956045, ECO:0000269|PubMed:9065786, ECO:0000269|PubMed:9143919, ECO:0000269|PubMed:9266388, ECO:0000269|PubMed:9286441, ECO:0000269|PubMed:9452024, ECO:0000269|PubMed:9452049, ECO:0000269|PubMed:9452065, ECO:0000269|Ref.32, ECO:0000269|Ref.43}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00155; DB02011; DB04185; DB00129 Interacts with NA EC number EC 2.1.3.3 Uniprot keywords 3D-structure; Acetylation; Amino-acid biosynthesis; Arginine biosynthesis; Disease variant; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Transit peptide; Urea cycle Protein physicochemical properties Chain ID A Molecular weight (Da) 36060.2 Length 321 Aromaticity 0.08 Instability index 36.44 Isoelectric point 7.87 Charge (pH=7) 1.48 2D Binding mode Binding energy (Kcal/mol) -6.24  Molscript Map 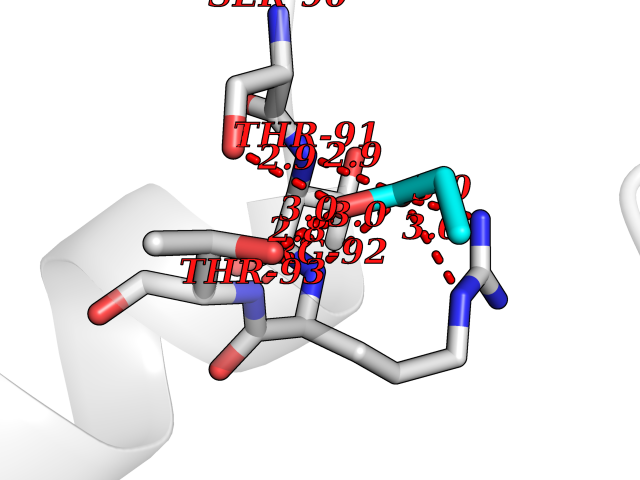 Pymol Map 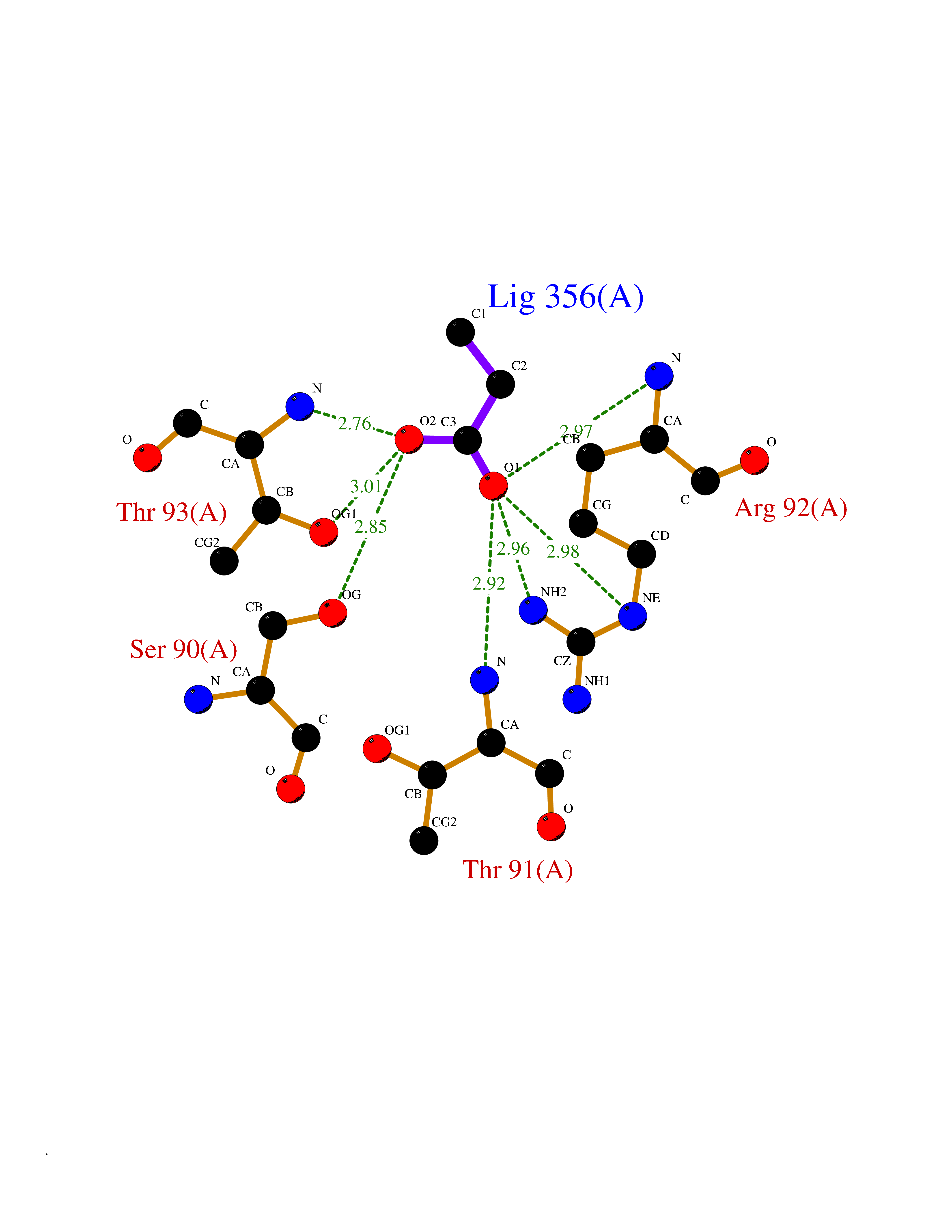 Ligplot Map 3D Binding mode Sequence KVQLKGRDLLTLKNFTGEEIKYMLWLSADLKFRIKQKGEYLPLLQGKSLGMIFEKRSTRTRLSTETGFALLGGHPCFLTTQDIHLGVNESLTDTARVLSSMADAVLARVYKQSDLDTLAKEASIPIINGLSDLYHPIQILADYLTLQEHYSSLKGLTLSWIGDGNNILHSIMMSAAKFGMHLQAATPKGYEPDASVTKLAEQYAKENGTKLLLTNDPLEAAHGGNVLITDTWISMGREEEKKKRLQAFQGYQVTMKTAKVAASDWTFLHCLPRKPEEVDDEVFYSPRSLVFPEAENRKWTIMAVMVSLLTDYSPQLQKPKF Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Cyclin-dependent kinase 4 | 2W96 | 4.57 | |
Target general information Gen name CDK4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Cell cycle Function ATP binding.Cyclin binding.Cyclin-dependent protein serine/threonine kinase activity.Cyclin-dependent protein serine/threonine kinase regulator activity.Protein complex binding. Related diseases Melanoma, cutaneous malignant 3 (CMM3) [MIM:609048]: A malignant neoplasm of melanocytes, arising de novo or from a pre-existing benign nevus, which occurs most often in the skin but may also involve other sites. {ECO:0000269|PubMed:7652577, ECO:0000269|PubMed:8528263, ECO:0000269|PubMed:9311594, ECO:0000269|PubMed:9425228}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12001; DB03496; DB12010; DB09073; DB02733; DB11730; DB15442 Interacts with Q9UH17; P24385; P30279; P30281; Q16543; P50613; P38936; P46527; P49918; P42771; P42772; P42773; P55273; Q9UJC3; P08238; Q9UKT9; Q0VD86; P01106; Q9ULD0; P28749; Q08999; P09936; Q8N720 EC number 2.7.11.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Cell division; Cytoplasm; Disease variant; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 30138.4 Length 267 Aromaticity 0.09 Instability index 36.2 Isoelectric point 5.78 Charge (pH=7) -5.83 2D Binding mode Binding energy (Kcal/mol) -6.24 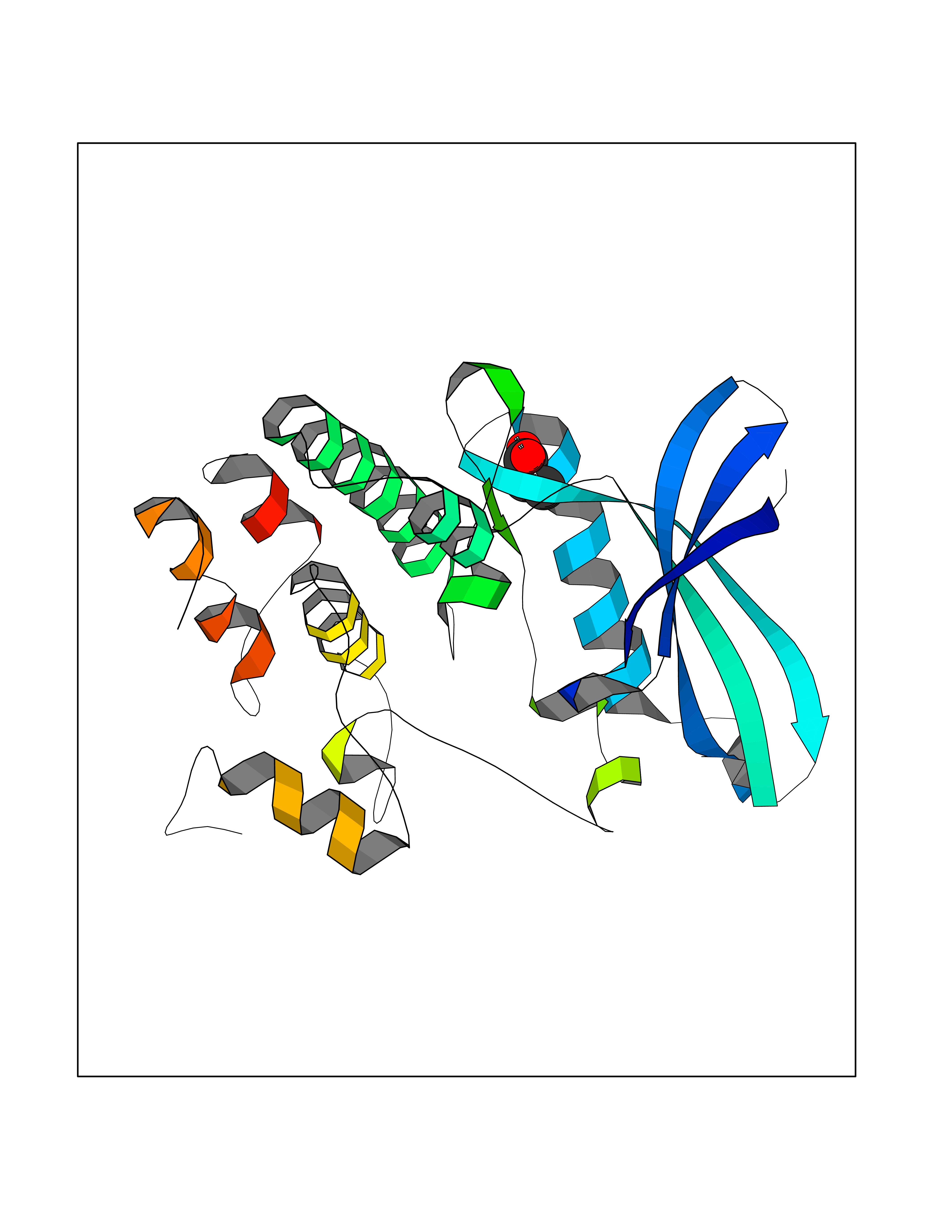 Molscript Map 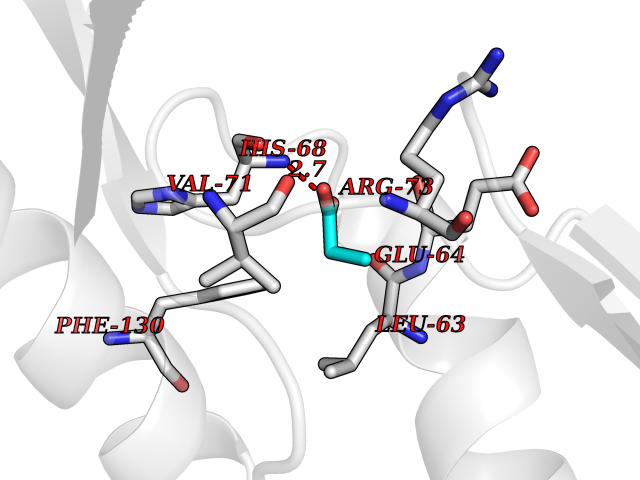 Pymol Map 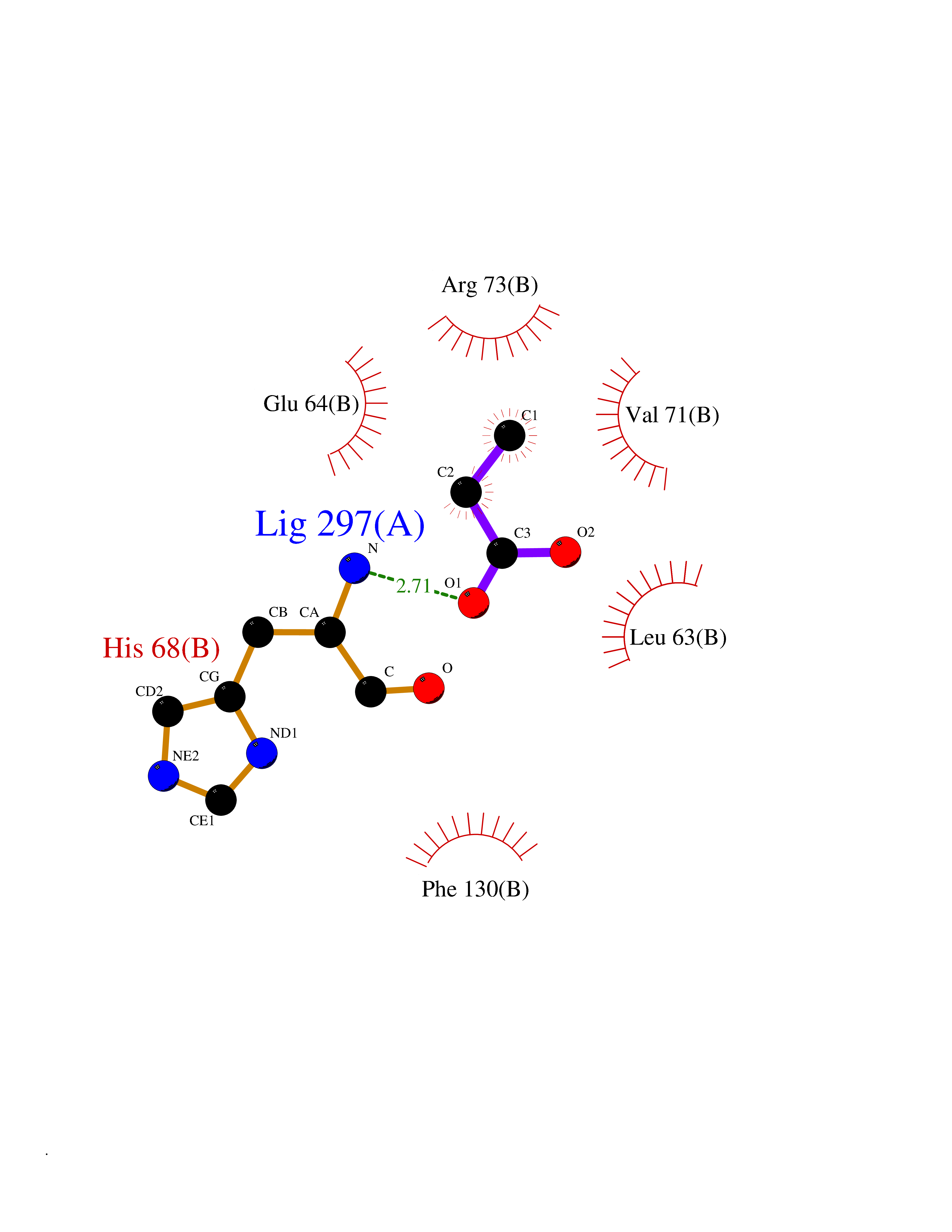 Ligplot Map 3D Binding mode Sequence SRYEPVAEIGVGAYGTVYKARDPHSGHFVALKSVRVPNGEEGLPISTVREVALLRRLEAFEHPNVVRLMDVCATSRTDREIKVTLVFEHVDQDLRTYLDKAPPPGLPAETIKDLMRQFLRGLDFLHANCIVHRDLKPENILVTSGGTVKLADFGLARIYSYQMALDPVVVTLWYRAPEVLLQSTYATPVDMWSVGCIFAEMFRRKPLFCGNSEADQLGKIFDLIGLPPEDDWVPEMEESGAQLLLEMLTFNPHKRISAFRALQHSYL Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Ornithine delta-aminotransferase (OAT) | 2OAT | 4.57 | |
Target general information Gen name OAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ornithine--oxo-acid aminotransferase; Ornithine aminotransferase, mitochondrial Protein family Class-III pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Catalyzes the transfer of the delta-amino group from L-ornithine. Related diseases Hyperornithinemia with gyrate atrophy of choroid and retina (HOGA) [MIM:258870]: A disorder clinically characterized by a triad of progressive chorioretinal degeneration, early cataract formation, and type II muscle fiber atrophy. Characteristic chorioretinal atrophy with progressive constriction of the visual fields leads to blindness at the latest during the sixth decade of life. Patients generally have normal intelligence. {ECO:0000269|PubMed:1612597, ECO:0000269|PubMed:1737786, ECO:0000269|PubMed:23076989, ECO:0000269|PubMed:2793865, ECO:0000269|PubMed:3375240, ECO:0000269|PubMed:7668253, ECO:0000269|PubMed:7887415}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02821; DB02054; DB00129; DB00114 Interacts with P05067 EC number EC 2.6.1.13 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminotransferase; Direct protein sequencing; Disease variant; Mitochondrion; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 44807.9 Length 404 Aromaticity 0.09 Instability index 26.67 Isoelectric point 5.72 Charge (pH=7) -6.54 2D Binding mode Binding energy (Kcal/mol) -6.23 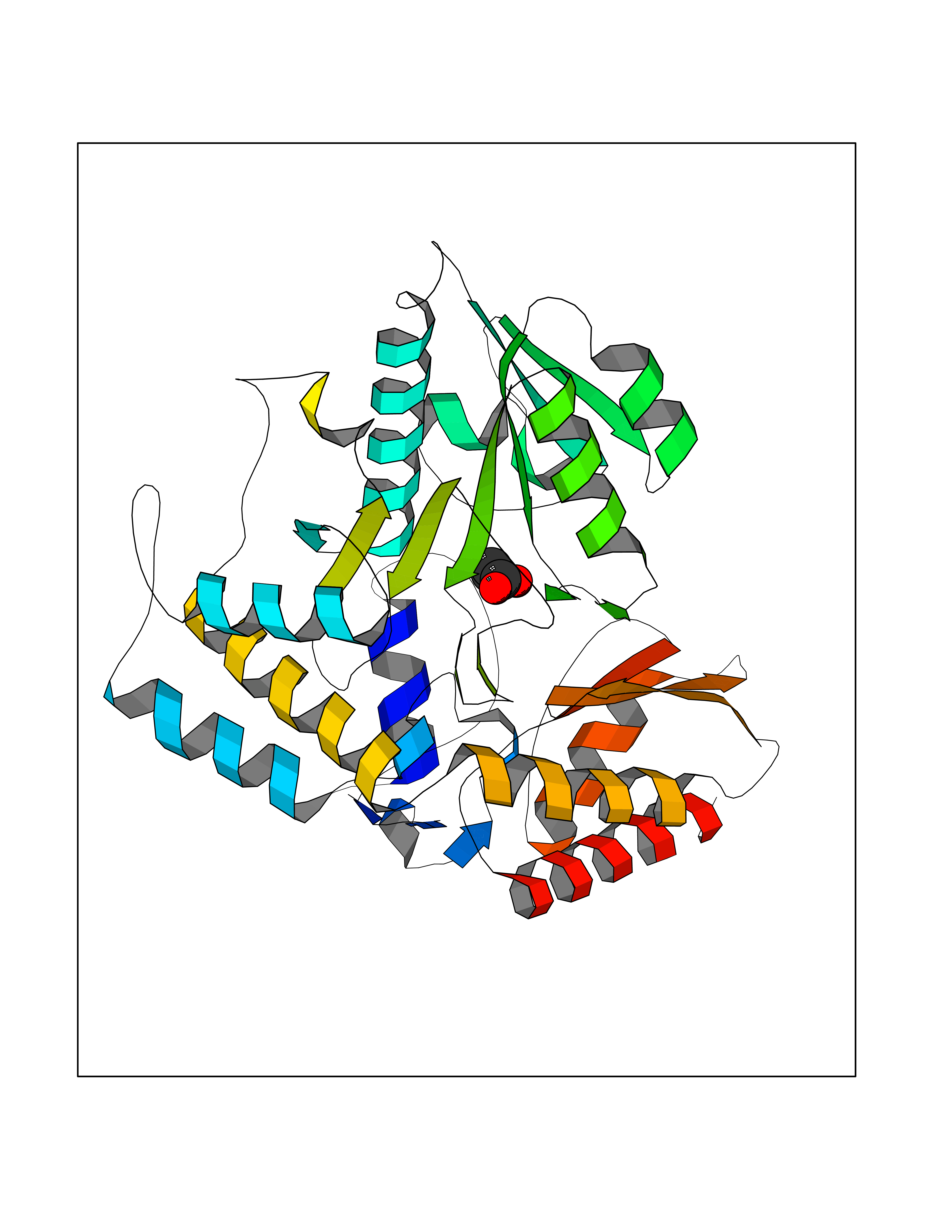 Molscript Map 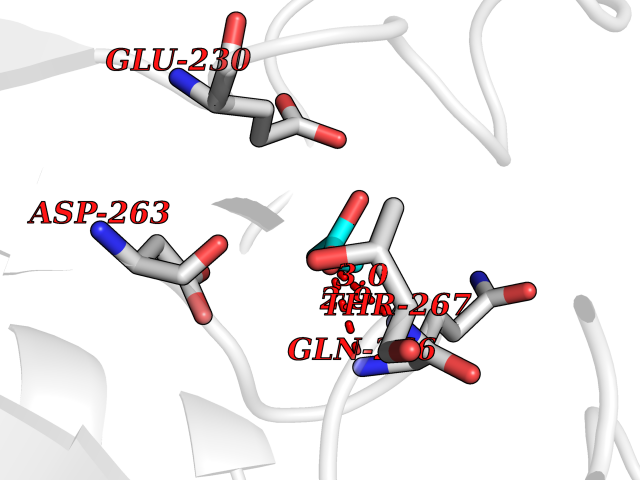 Pymol Map 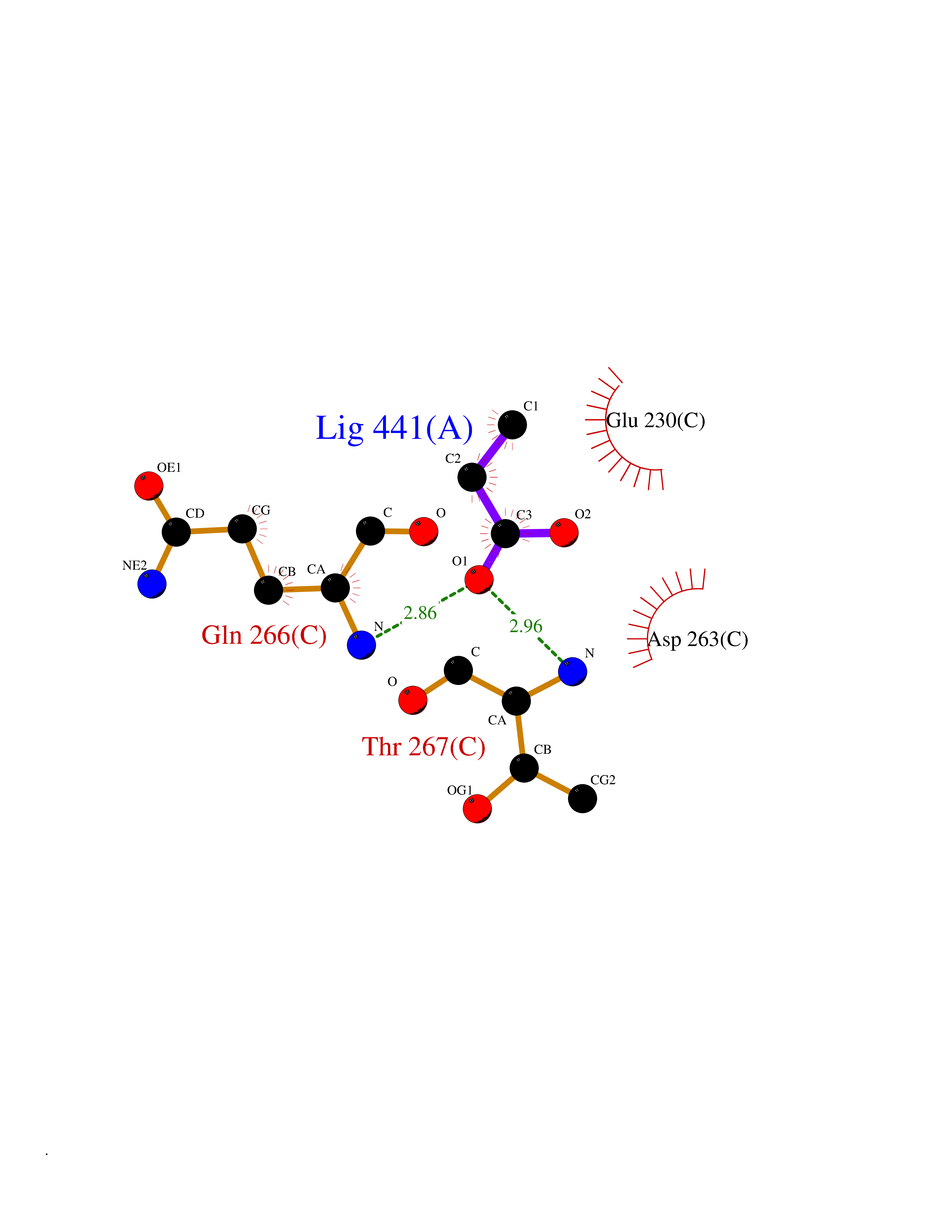 Ligplot Map 3D Binding mode Sequence GPPTSDDIFEREYKYGAHNYHPLPVALERGKGIYLWDVEGRKYFDFLSSYSAVNQGHCHPKIVNALKSQVDKLTLTSRAFYNNVLGEYEEYITKLFNYHKVLPMNTGVEAGETACKLARKWGYTVKGIQKYKAKIVFAAGNFWGRTLSAISSSTDPTSYDGFGPFMPGFDIIPYNDLPALERALQDPNVAAFMVEPIQGEAGVVVPDPGYLMGVRELCTRHQVLFIADEIQTGLARTGRWLAVDYENVRPDIVLLGKALSGGLYPVSAVLCDDDIMLTIKPGEHGSTYGGNPLGCRVAIAALEVLEEENLAENADKLGIILRNELMKLPSDVVTAVRGKGLLNAIVIKETKDWDAWKVCLRLRDNGLLAKPTHGDIIRFAPPLVIKEDELRESIEIINKTILSF Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Beta-glucosidase A | 1E4I | 4.57 | |
Target general information Gen name bglA Organism Paenibacillus polymyxa (Bacillus polymyxa) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 1 family Biochemical class Hydrolase Function Beta-glucosidase activity.Scopolin beta-glucosidase activity. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02658; DB04282; DB04304 Interacts with NA EC number 3.2.1.21 Uniprot keywords 3D-structure; Carbohydrate metabolism; Cellulose degradation; Glycosidase; Hydrolase; Polysaccharide degradation Protein physicochemical properties Chain ID A Molecular weight (Da) 51515.2 Length 447 Aromaticity 0.14 Instability index 38.44 Isoelectric point 5.28 Charge (pH=7) -18.1 2D Binding mode Binding energy (Kcal/mol) -6.24 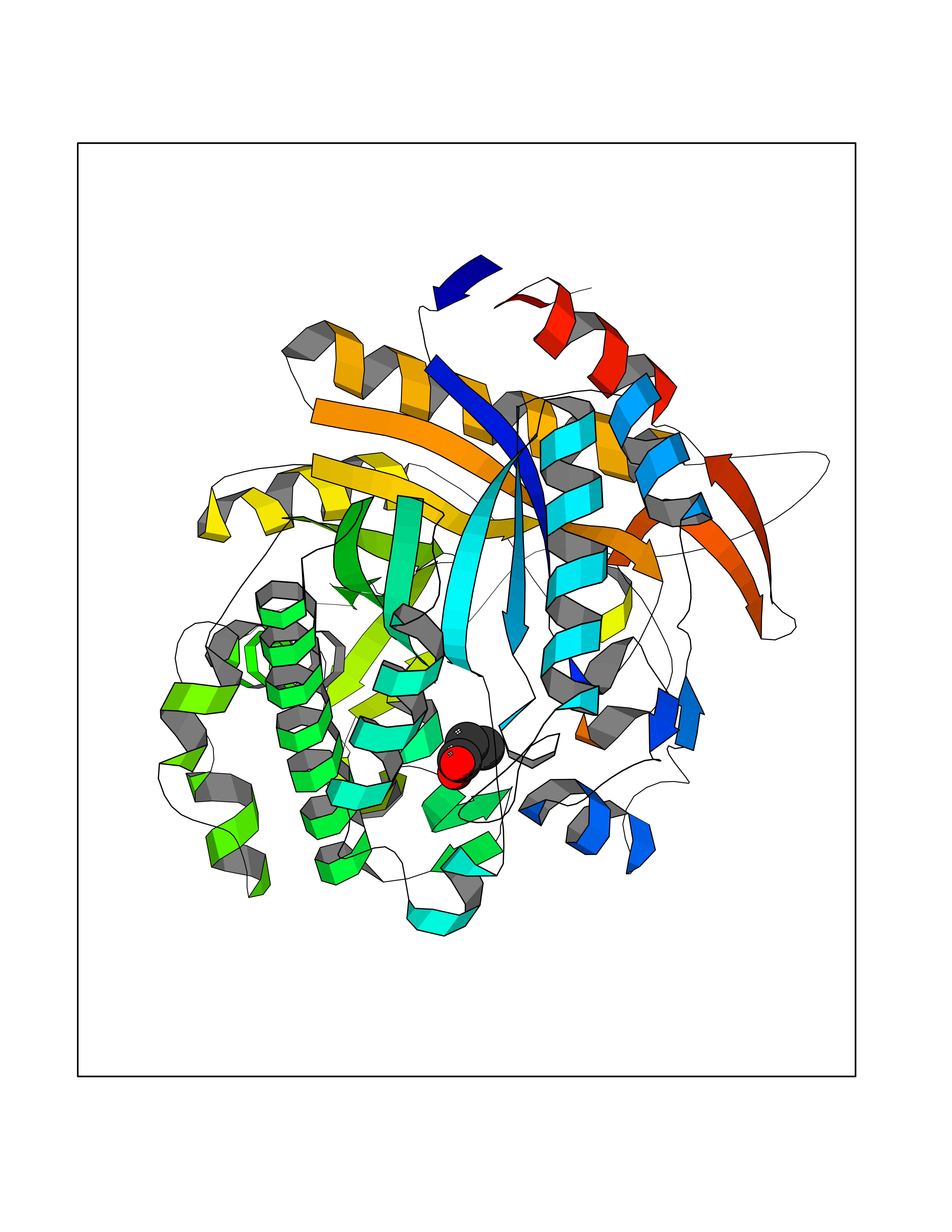 Molscript Map 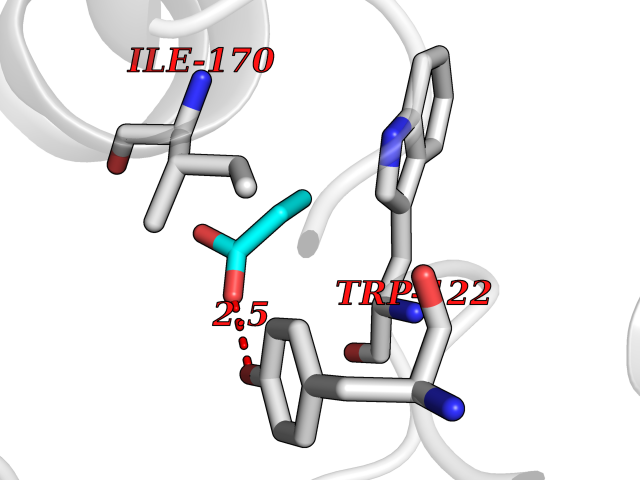 Pymol Map 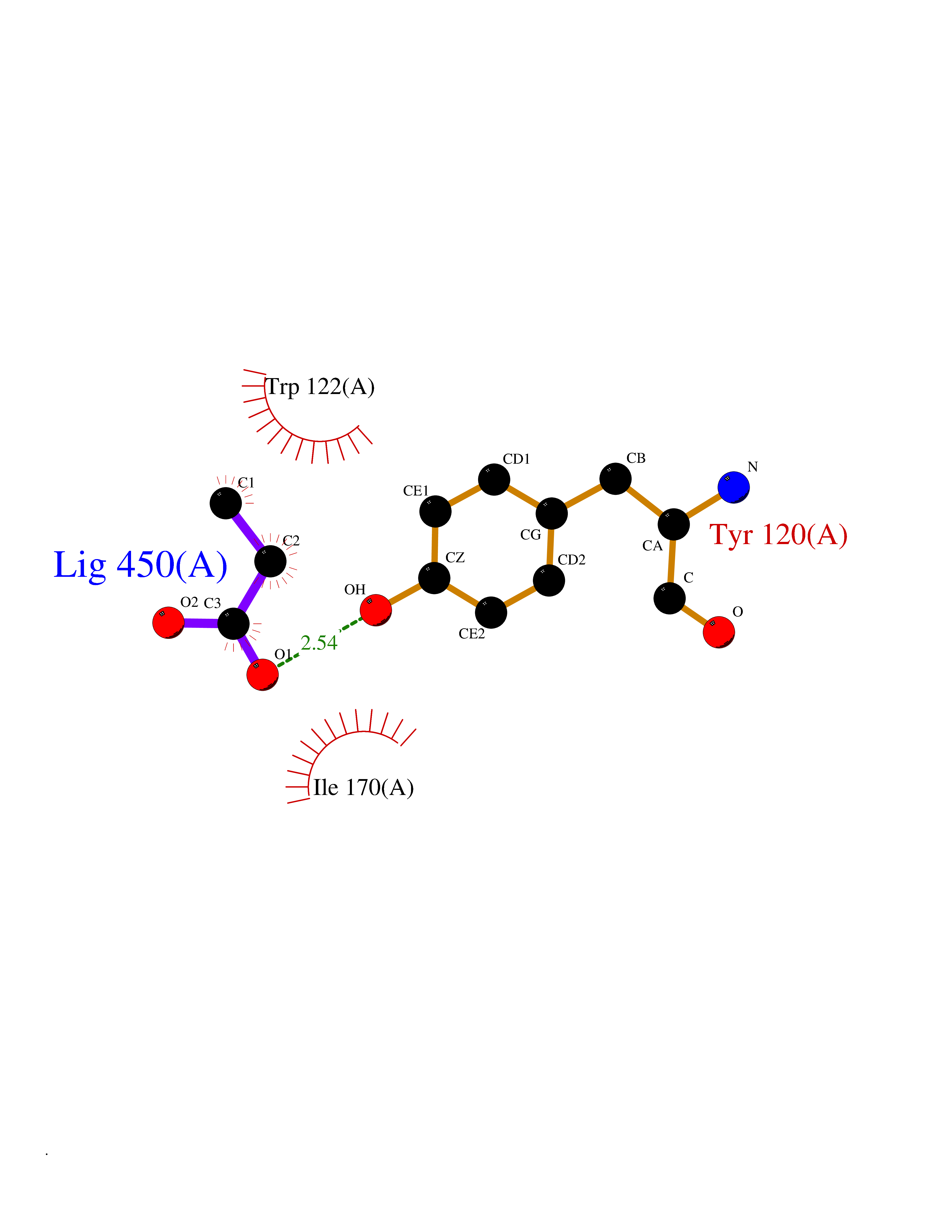 Ligplot Map 3D Binding mode Sequence TIFQFPQDFMWGTATAAYQIEGAYQEDGRGLSIWDTFAHTPGKVFNGDNGNVACDSYHRYEEDIRLMKELGIRTYRFSVSWPRIFPNGDGEVNQKGLDYYHRVVDLLNDNGIEPFCTLYHWDLPQALQDAGGWGNRRTIQAFVQFAETMFREFHGKIQHWLTFNEPWCIAFLSNMLGVHAPGLTNLQTAIDVGHHLLVAHGLSVRRFRELGTSGQIGIAPNVSWAVPYSTSEEDKAACARTISLHSDWFLQPIYQGSYPQFLVDWFAEQGATVPIQDGDMDIIGEPIDMIGINYYSMSVNRFNPEAGFLQSEEINMGLPVTDIGWPVESRGLYEVLHYLQKYGNIDIYITENGACINDEVVNGKVQDDRRISYMQQHLVQVHRTIHDGLHVKGYMAWSLLDNFEWAEGYNMRFGMIHVDFRTQVRTPKQSYYWYRNVVSNNWLETRR Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Lactaldehyde dehydrogenase | 2IMP | 4.57 | |
Target general information Gen name aldA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms ald;JW1412;b1415 Protein family Aldehyde dehydrogenase family Biochemical class Oxidoreductase Function Glycolaldehyde dehydrogenase activity.Identical protein binding.Lactaldehyde dehydrogenase activity.NAD binding.Succinate-semialdehyde dehydrogenase (NAD+) activity. Related diseases 3-ketothiolase deficiency (3KTD) [MIM:203750]: An autosomal recessive inborn error of isoleucine catabolism characterized by intermittent ketoacidotic attacks associated with unconsciousness. Some patients die during an attack or are mentally retarded. Urinary excretion of 2-methyl-3-hydroxybutyric acid, 2-methylacetoacetic acid, triglylglycine, butanone is increased. It seems likely that the severity of this disease correlates better with the environmental or acquired factors than with the ACAT1 genotype. {ECO:0000269|PubMed:1346617, ECO:0000269|PubMed:1715688, ECO:0000269|PubMed:7728148, ECO:0000269|PubMed:9744475}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03619 Interacts with NA EC number 1.2.1.21; 1.2.1.22 Uniprot keywords 3D-structure; Direct protein sequencing; NAD; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 48991.4 Length 451 Aromaticity 0.08 Instability index 26.75 Isoelectric point 5.14 Charge (pH=7) -11.76 2D Binding mode Binding energy (Kcal/mol) -6.24 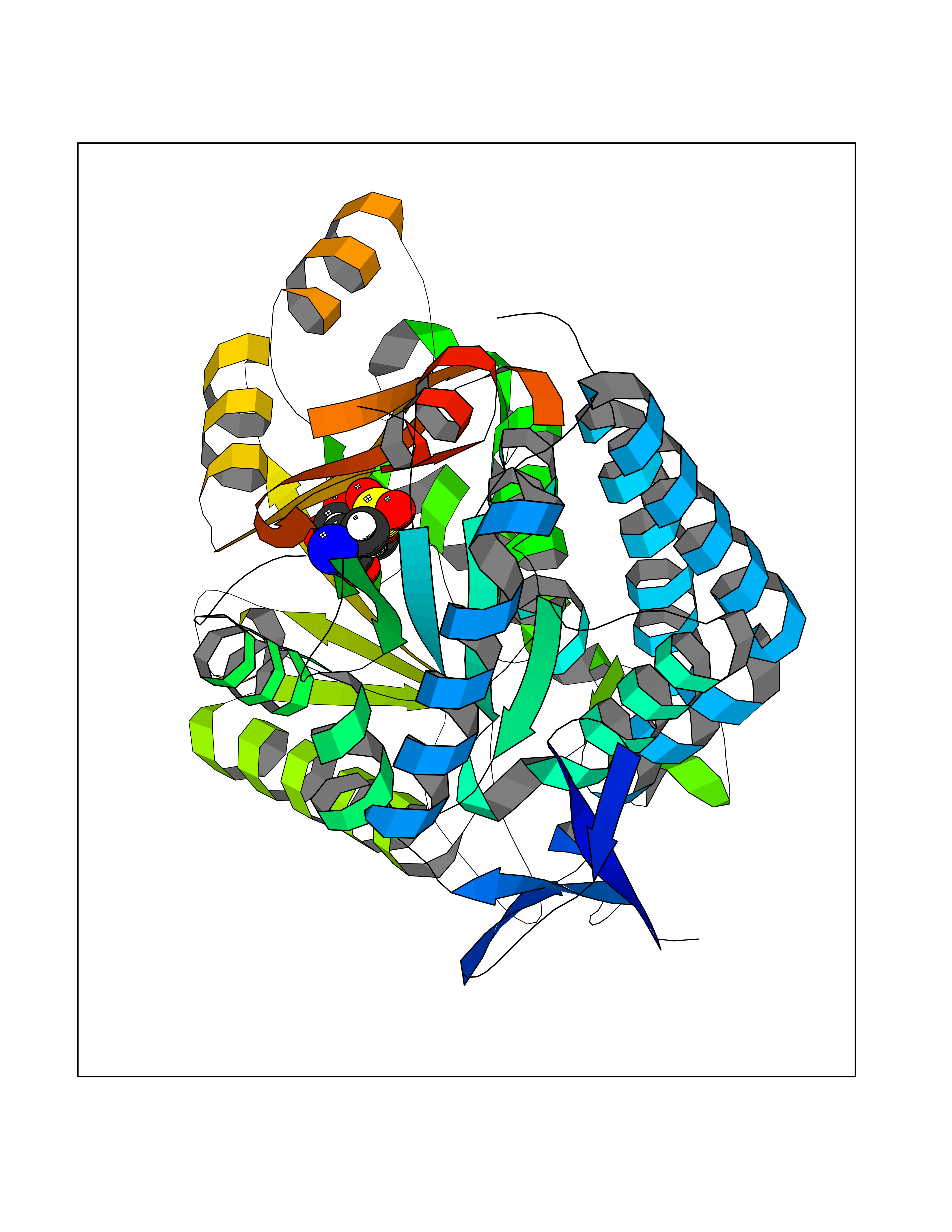 Molscript Map 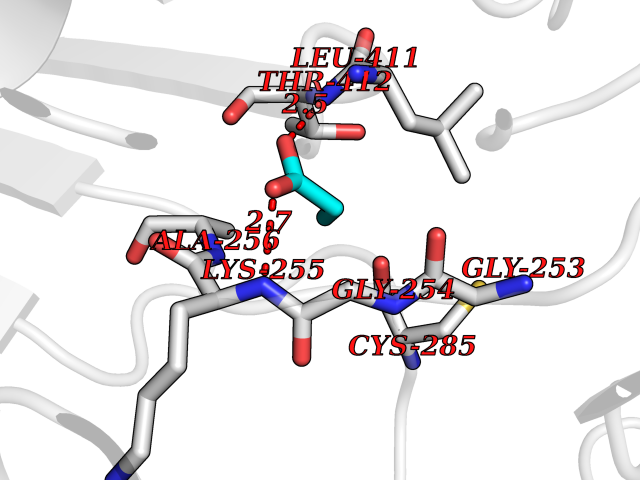 Pymol Map 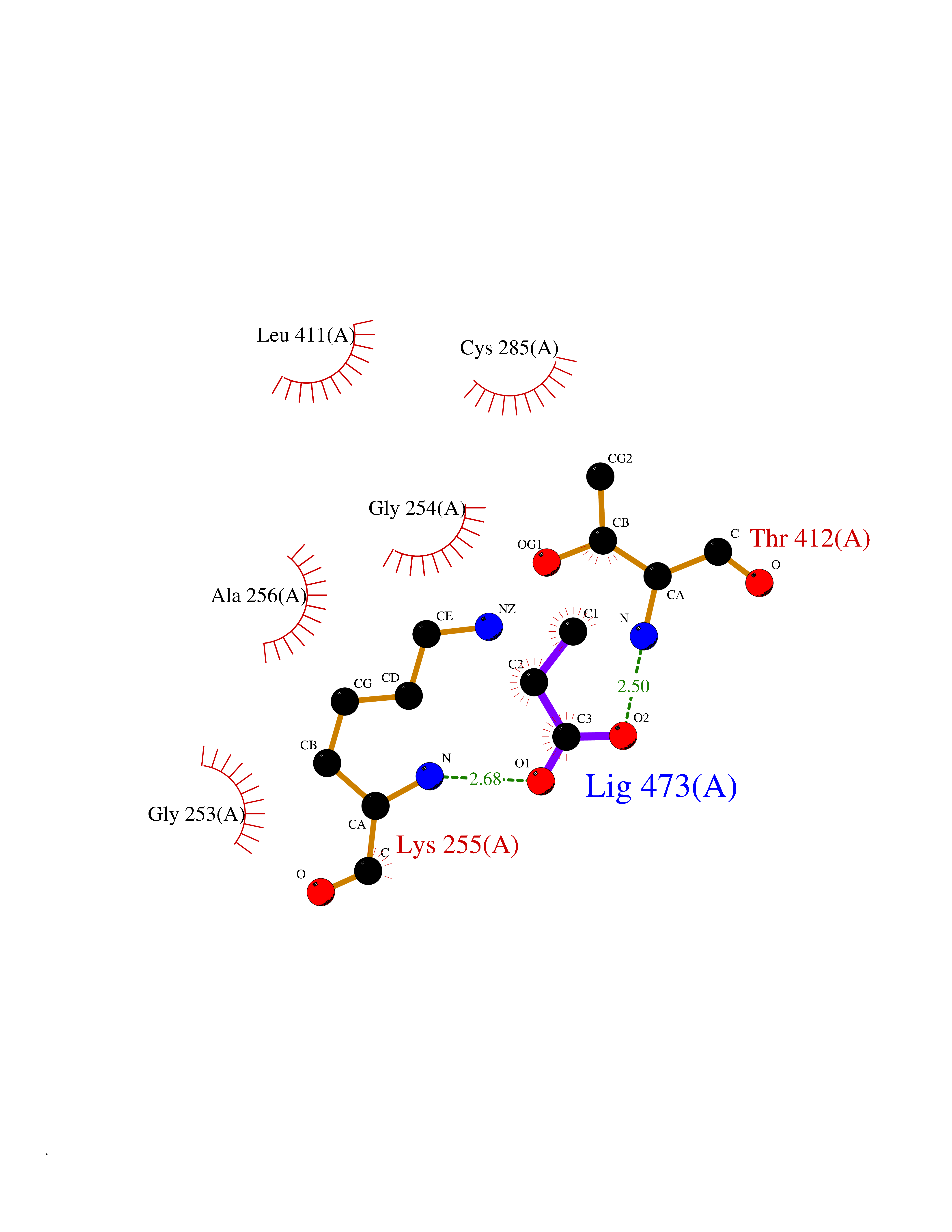 Ligplot Map 3D Binding mode Sequence VPVQHPMYIDGQFVTWRGDAWIDVVNPATEAVISRIPDGQAEDARKAIDAAERAQPEWEALPAIERASWLRKISAGIRERASEISALIVEEGGKIQQLAEVEVAFTADYIDYMAEWARRYRALGVTTGILPWNFPFFLIARKMAPALLTGNTIVIKPSEFTPNNAIAFAKIVDEIGLPRGVFNLVLGRGETVGQELAGNPKVAMVSMTGSVSAGEKIMATAAKNITKVXLELGGKAPAIVMDDADLELAVKAIVDSRVINSGQVCNCAERVYVQKGIYDQFVNRLGEAMQAVQFGNPAERNDIAMGPLINAAALERVEQKVARAVEEGARVAFGGKAVEGKGYYYPPTLLLDVRQEMSIMHEETFGPVLPVVAFDTLEDAISMANDSDYGLTSSIYTQNLNVAMKAIKGLKFGETYINRENFEAMQGFHAGWRKSGIGGADGKHGLHEYLQ Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Low molecular weight phosphotyrosine protein phosphatase | 5KQL | 4.57 | |
Target general information Gen name ACP1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Low molecular weight phosphotyrosine protein phosphatase family Biochemical class hydrolase / hydrolase inhibitor Function Acid phosphatase activity.Non-membrane spanning protein tyrosine phosphatase activity. Related diseases Waardenburg syndrome 4A (WS4A) [MIM:277580]: A disorder characterized by the association of Waardenburg features (depigmentation and deafness) with the absence of enteric ganglia in the distal part of the intestine (Hirschsprung disease). {ECO:0000269|PubMed:12189494, ECO:0000269|PubMed:8634719}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hirschsprung disease 2 (HSCR2) [MIM:600155]: A disorder of neural crest development characterized by absence of enteric ganglia along a variable length of the intestine. It is the most common cause of congenital intestinal obstruction. Early symptoms range from complete acute neonatal obstruction, characterized by vomiting, abdominal distention and failure to pass stool, to chronic constipation in the older child. {ECO:0000269|PubMed:11471546, ECO:0000269|PubMed:28236341, ECO:0000269|PubMed:8001158, ECO:0000269|PubMed:8630503, ECO:0000269|PubMed:8852660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: ABCD syndrome (ABCDS) [MIM:600501]: An autosomal recessive syndrome characterized by albinism, black lock at temporal occipital region, bilateral deafness, aganglionosis of the large intestine and total absence of neurocytes and nerve fibers in the small intestine. {ECO:0000269|PubMed:11891690}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Heterozygous mutations in EDNRB may be responsible for Waardenburg syndrome 2, an autosomal dominant disorder characterized by sensorineural deafness and pigmentary disturbances. {ECO:0000269|PubMed:28236341}. Drugs (DrugBank ID) DB04214; DB00173 Interacts with Q96CV9 EC number 3.1.3.2; 3.1.3.48 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Phosphoprotein; Protein phosphatase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 17582.8 Length 154 Aromaticity 0.09 Instability index 50.52 Isoelectric point 7 Charge (pH=7) -0 2D Binding mode Binding energy (Kcal/mol) -6.24  Molscript Map 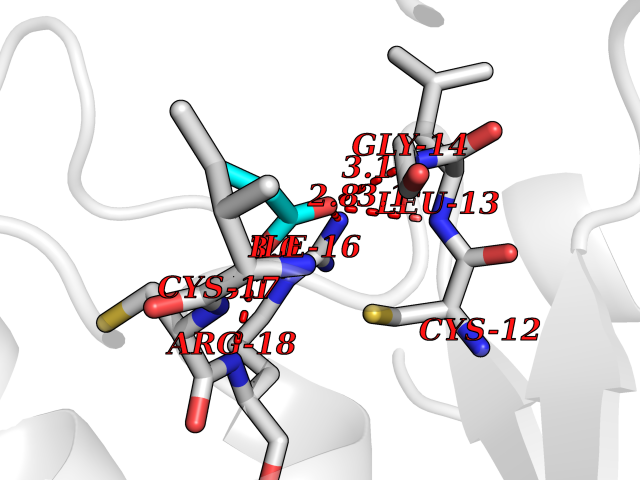 Pymol Map 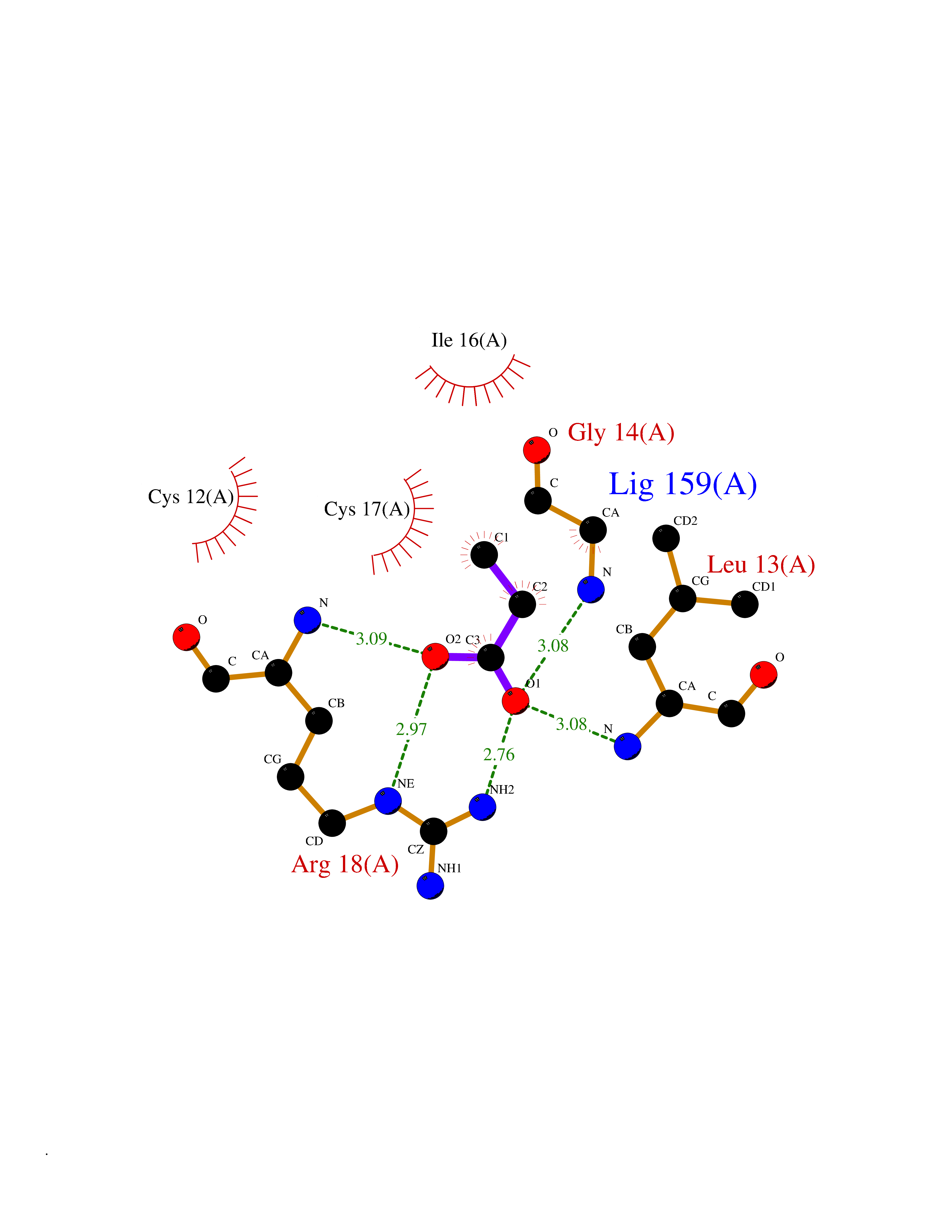 Ligplot Map 3D Binding mode Sequence ATKSVLFVCLGNICRSPIAEAVFRKLVTDQNISENWRVDSAATSGYEIGNPPDYRGQSCMKRHGIPMSHVARQITKEDFATFDYILCMDESNLRDLNRKSNQVKTCKAKIELLGSYDPQKQLIIEDPYYGNDSDFETVYQQCVRCCRAFLEKAH Hydrogen bonds contact Hydrophobic contact | ||||