Job Results:
Ligand
Structure
Job ID
8adf9aabe9fa12b13024fa8d07ca9b9b
Job name
NA
Time
2026-01-21 12:38:59
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Monoamine oxidase type B (MAO-B) | 2V5Z | 6.60 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -9  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Thiopurine S-methyltransferase | 2BZG | 6.60 | |
Target general information Gen name TPMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, TPMT family Biochemical class Transferase Function Thiopurine S-methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00993; DB00436; DB01327; DB01033; DB01250; DB01021 Interacts with Q8TAP4-4; Q15047-2; P61981 EC number 2.1.1.67 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 25971.5 Length 229 Aromaticity 0.12 Instability index 32.58 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -9 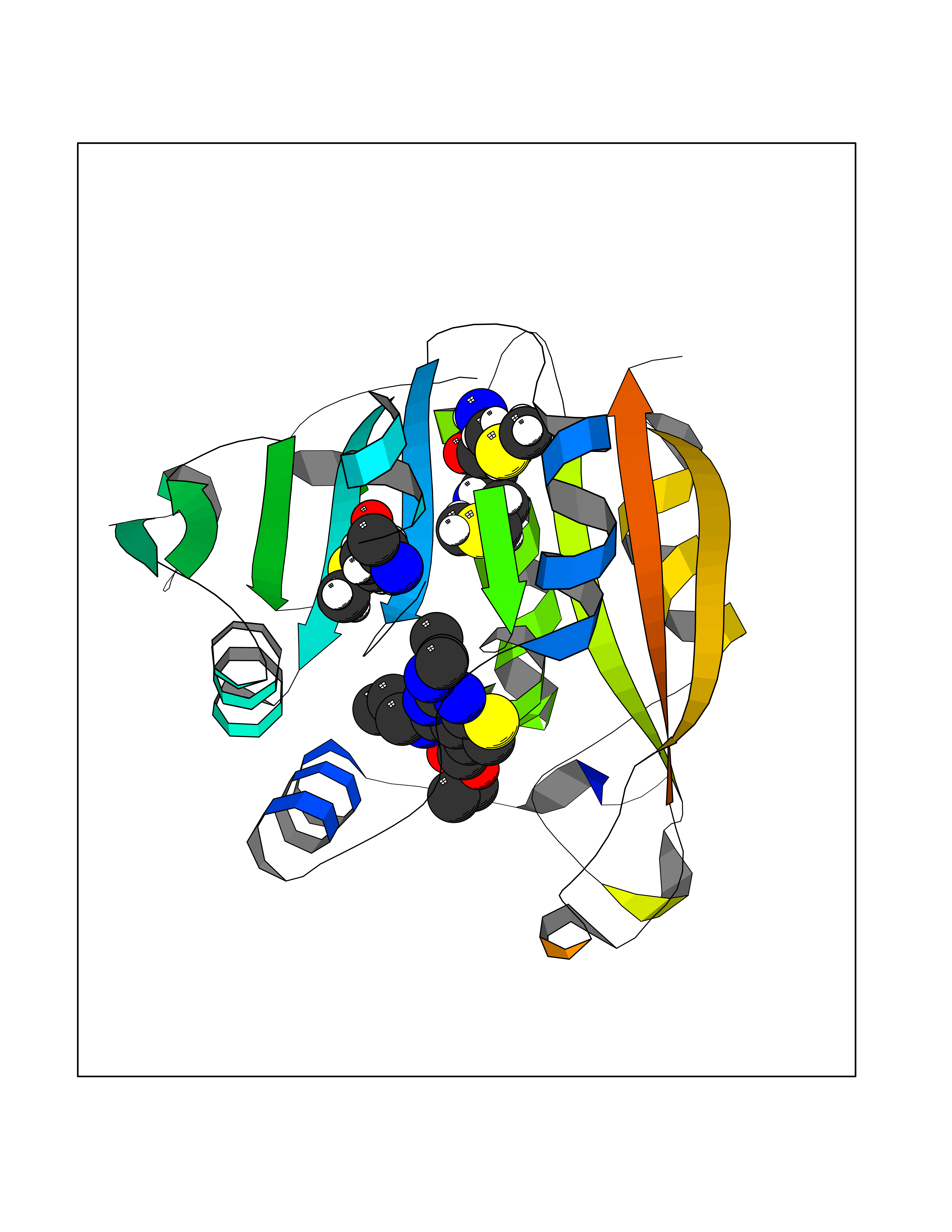 Molscript Map 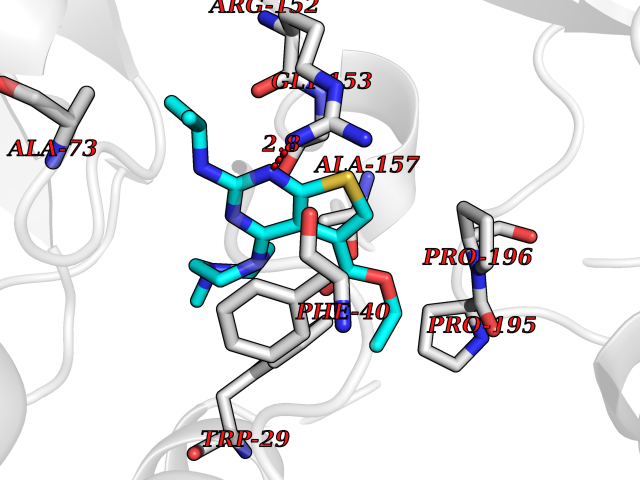 Pymol Map 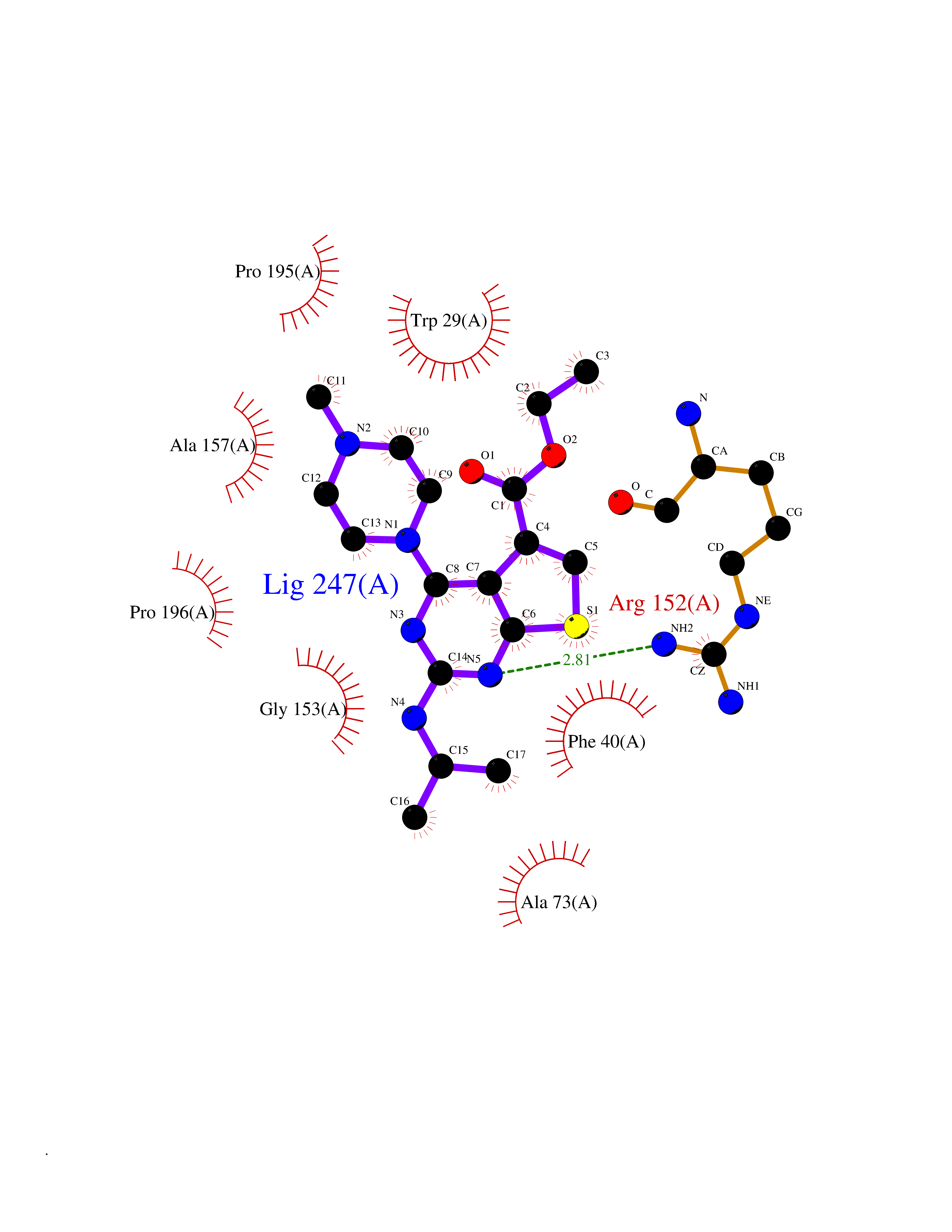 Ligplot Map 3D Binding mode Sequence EVQKNQVLTLEEWQDKWVNGKTAFHQEQGHQLLKKHLDTFLKGKSGLRVFFPLCGKAVEXKWFADRGHSVVGVEISELGIQEFFTEQNLSYSEEPITEIPGTKVFKSSSGNISLYCCSIFDLPRTNIGKFDXIWDRGALVAINPGDRKCYADTXFSLLGKKFQYLLCVLSYDPTKHPGPPFYVPHAEIERLFGKICNIRCLEKVDAFEERHKSWGIDCLFEKLYLLTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Asparagine--tRNA ligase, cytoplasmic | 4ZYA | 6.59 | |
Target general information Gen name NARS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NRS;NARS Protein family Class-II aminoacyl-tRNA synthetase family Biochemical class Ligase Function Asparagine-tRNA ligase activity.ATP binding.Nucleic acid binding. Related diseases Neurodevelopmental disorder with microcephaly, impaired language, and gait abnormalities (NEDMILG) [MIM:619091]: An autosomal recessive neurodevelopmental disorder characterized by global developmental delay apparent in infancy, moderate to profound intellectual disability, poor or absent speech and language, delayed walking with variable gait abnormalities, and progressive microcephaly. Additional variable features include hypotonia, early-onset seizures, and a peripheral demyelinating or axonal peripheral sensorimotor neuropathy. {ECO:0000269|PubMed:32738225, ECO:0000269|PubMed:32788587}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with microcephaly, impaired language, epilepsy, and gait abnormalities (NEDMILEG) [MIM:619092]: An autosomal dominant neurodevelopmental disorder characterized by global developmental delay apparent in infancy, delayed walking, ataxia, spasticity, impaired intellectual development with poor or absent speech and language, progressive microcephaly, and early-onset seizures in most patients. Facial dysmorphism and a demyelinating peripheral neuropathy may also be observed. {ECO:0000269|PubMed:32738225}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00174 Interacts with P54253 EC number 6.1.1.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminoacyl-tRNA synthetase; ATP-binding; Cytoplasm; Direct protein sequencing; Disease variant; Epilepsy; Intellectual disability; Ligase; Neurodegeneration; Nucleotide-binding; Phosphoprotein; Protein biosynthesis; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 15621.4 Length 145 Aromaticity 0.09 Instability index 35.33 Isoelectric point 9.5 Charge (pH=7) 7.08 2D Binding mode Binding energy (Kcal/mol) -8.99 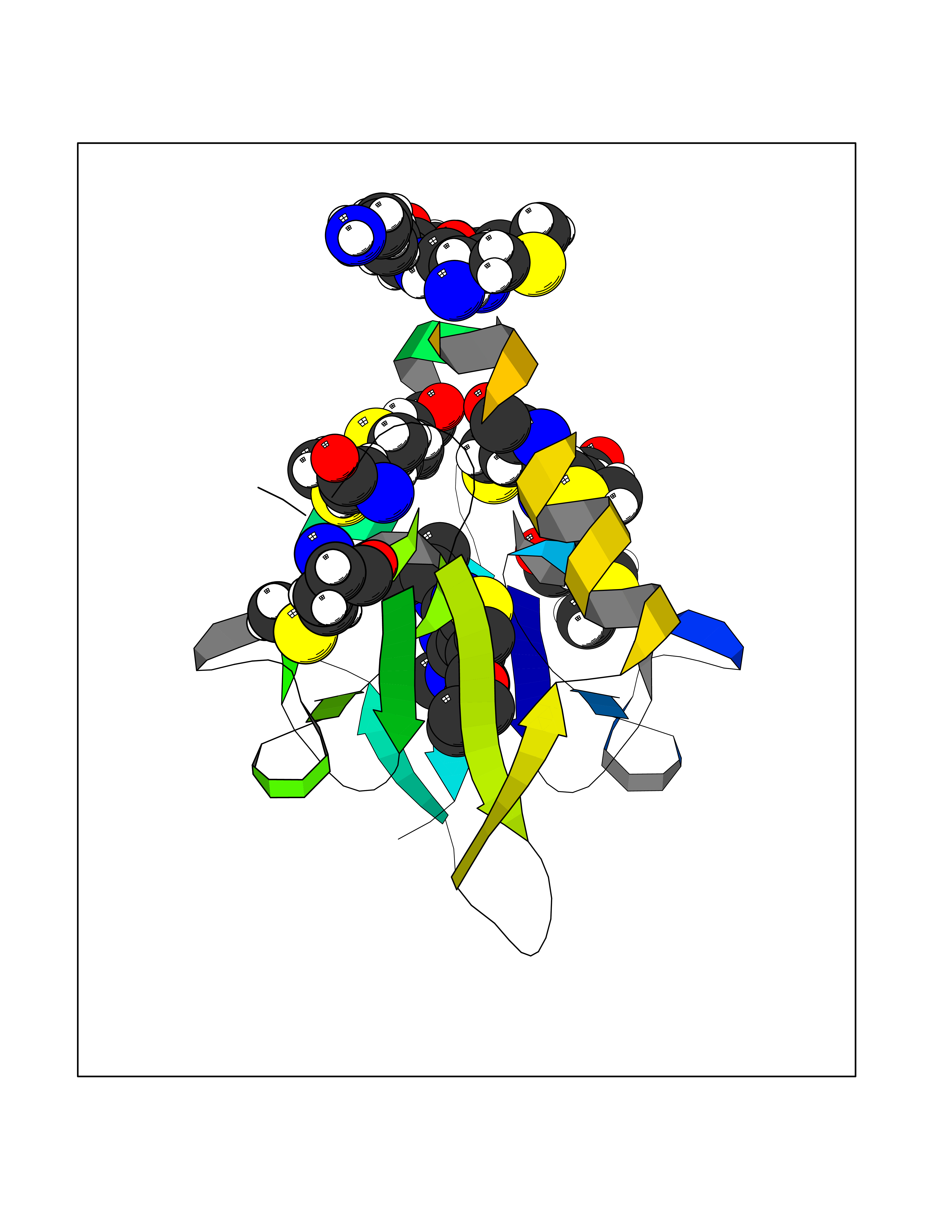 Molscript Map 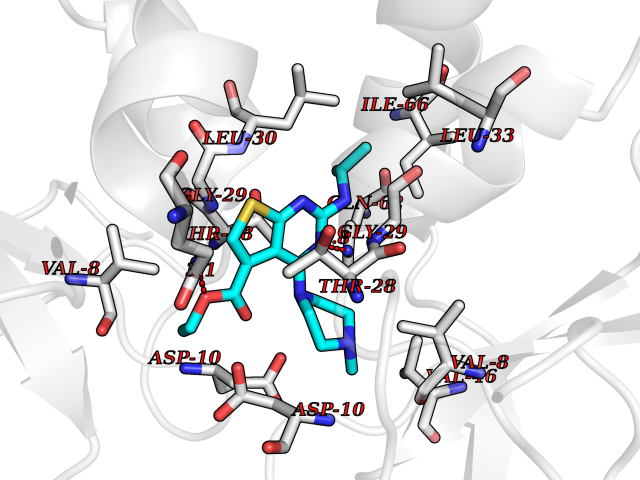 Pymol Map 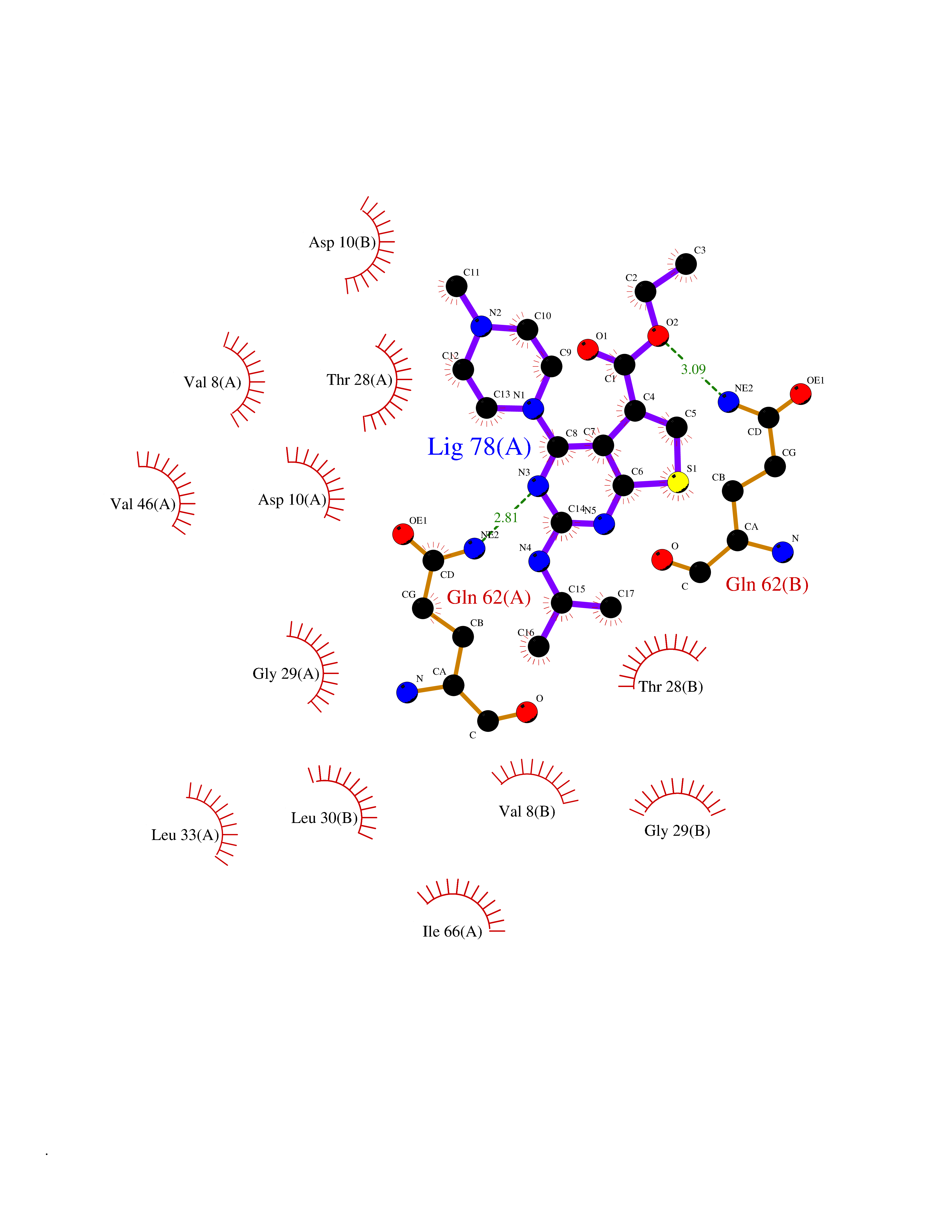 Ligplot Map 3D Binding mode Sequence XAELYVSDREGSDATGDGTKEKPFKTGLKALXTVGKEPFPTIYVDSRWNVISKSQLKNIKKXWHREQXKGHXAELYVSDREGSDATGDGTKEKPFKTGLKALXTVGKEPFPTIYVDSQKENERWNVISKSQLKNIKKXWHREQXK Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Glutamate receptor ionotropic NMDA 2A (NMDAR2A) | 5KCJ | 6.58 | |
Target general information Gen name GRIN2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NR2A; NMDA receptor NR2A; N-methyl D-aspartate receptor subtype 2A; HNR2A; Glutamate receptor ionotropic, NMDA 2A; Glutamate [NMDA] receptor subunit epsilon-1; GluN2A Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, NR1/GRIN1 subfamily Biochemical class Glutamate-gated ion channel Function Channel activation requires binding of the neurotransmitter glutamate to the epsilon subunit, glycine binding to the zeta subunit, plus membrane depolarization to eliminate channel inhibition by Mg(2+). Sensitivity to glutamate and channel kinetics depend on the subunit composition; channels containing GRIN1 and GRIN2A have higher sensitivity to glutamate and faster kinetics than channels formed by GRIN1 and GRIN2B. Contributes to the slow phase of excitatory postsynaptic current, long-term synaptic potentiation, and learning. Component of NMDA receptor complexes that function as heterotetrameric, ligand-gated ion channels with high calcium permeability and voltage-dependent sensitivity to magnesium. Related diseases Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant (NDHMSD) [MIM:614254]: An autosomal dominant neurodevelopmental disorder characterized by severe intellectual disability and developmental delay, absent speech, muscular hypotonia, dyskinesia, and hyperkinetic movements. Cortical blindness, cerebral atrophy, and seizures are present in some patients. {ECO:0000269|PubMed:21376300, ECO:0000269|PubMed:25167861, ECO:0000269|PubMed:25864721, ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:28228639, ECO:0000269|PubMed:28389307, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal recessive (NDHMSR) [MIM:617820]: An autosomal recessive neurodevelopmental disorder characterized by severe intellectual disability and psychomotor developmental delay, involuntary and stereotypic movements, spasticity, and inability to walk without support. Intractable seizures manifest in some patients. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28051072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 101 (DEE101) [MIM:619814]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE101 is an autosomal recessive, severe form characterized by onset of seizures in early infancy. Death in infancy may occur. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:34611970}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01931; DB00659; DB06151; DB08838; DB01238; DB00289; DB05824; DB04620; DB03929; DB00647; DB00843; DB00228; DB11823; DB13146; DB06741; DB00142; DB00874; DB08954; DB06738; DB09409; DB09481; DB01043; DB00454; DB00333; DB04896; DB01173; DB00312; DB01174; DB01708; DB00418; DB00193 Interacts with P05067; P35637; Q12879-1; Q13224; Q62936 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Ligand-gated ion channel; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Zinc Protein physicochemical properties Chain ID B Molecular weight (Da) 53395.6 Length 469 Aromaticity 0.11 Instability index 29.84 Isoelectric point 8.72 Charge (pH=7) 5.65 2D Binding mode Binding energy (Kcal/mol) -8.98 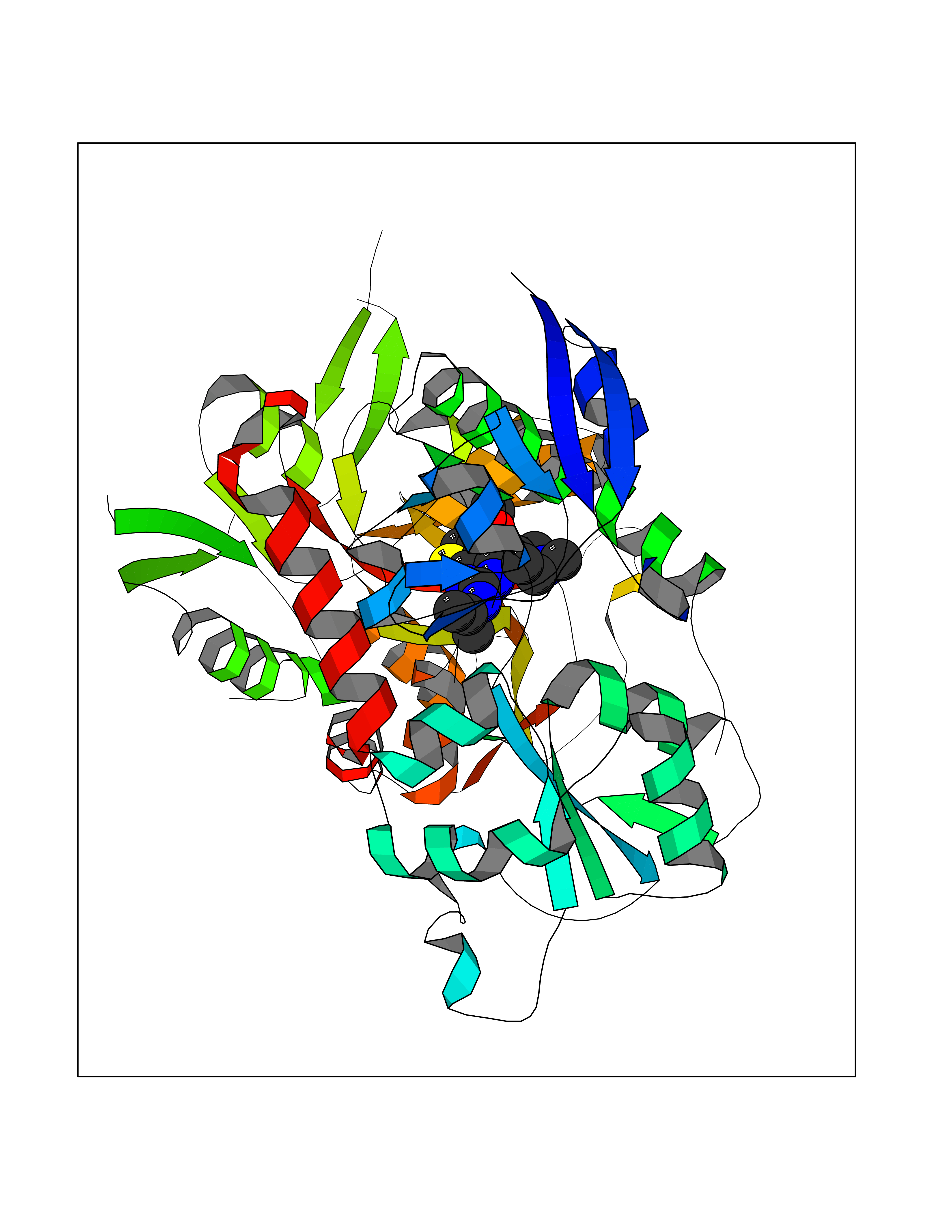 Molscript Map 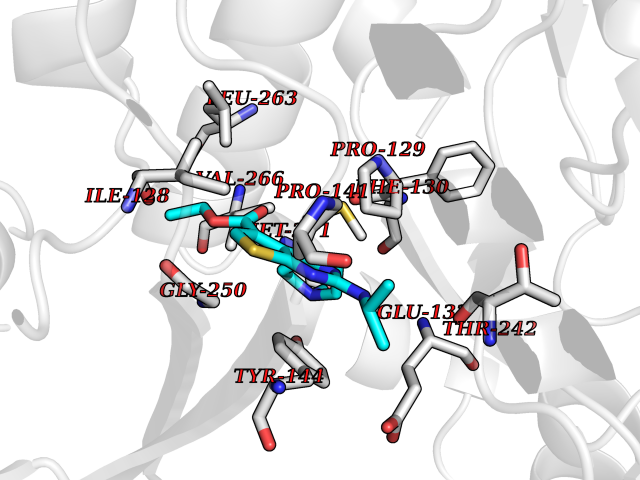 Pymol Map 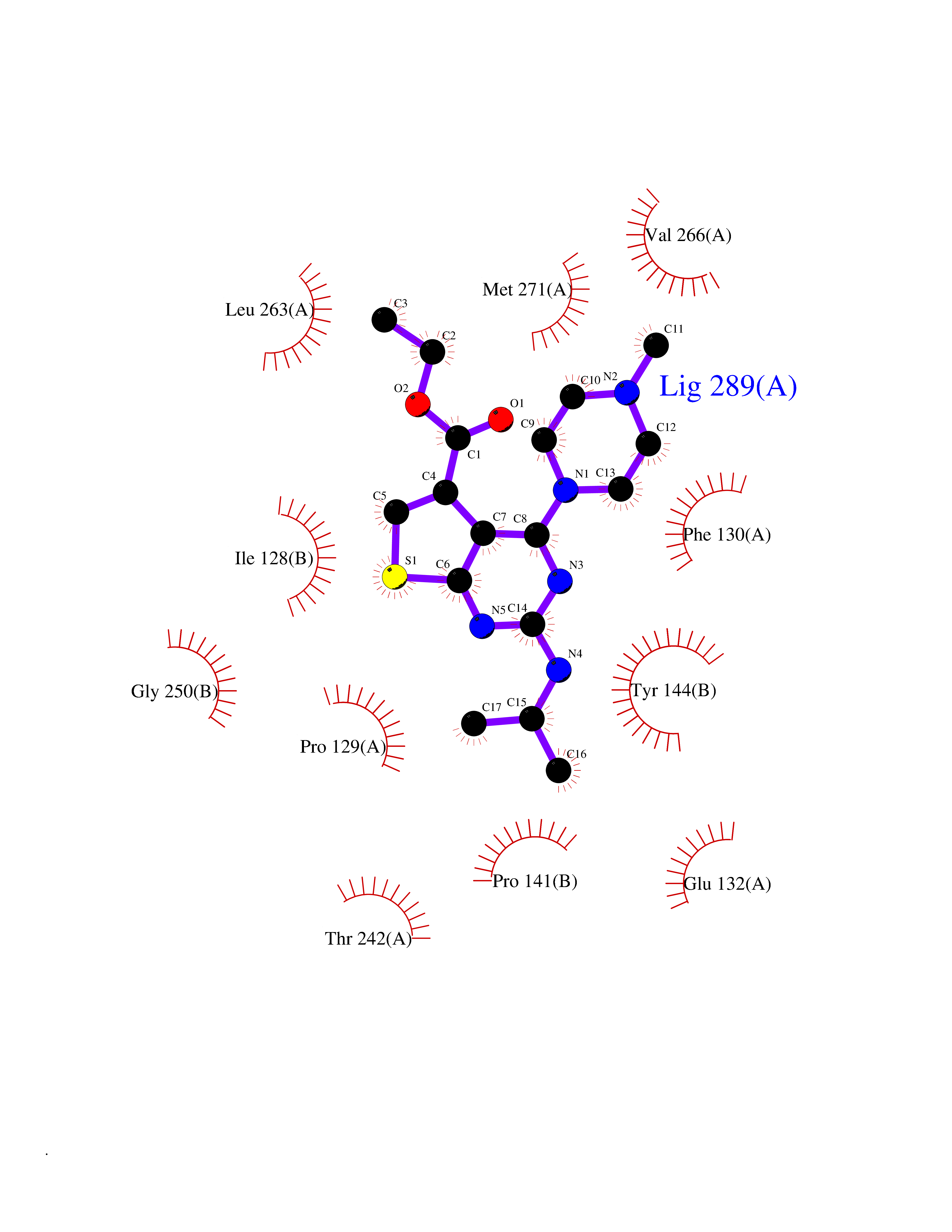 Ligplot Map 3D Binding mode Sequence DNHLSIVTLEEAPFVILKKLSRTVKFTYDLYLVTNGKHGKKVNNVWNGMIGEVVYQRAVMAVGSLTINEERSEVVDFSVPFVETGISVMVSRGTQVTGLSDKKFQRPHDYSPPFRFGTVPNGSTERNIRNNYPYMHQYMTKFNQKGVEDALVSLKTGKLDAFIYDAAVLNYKAGRDEGCKLVTIGSGYIFATTGYGIALQKGSPWKRQIDLALLQFVGDGEMEELETLWLTGICTRLKIVTIHQEPFVYYGFCIDLLIKLARTMNFTYEVHLVADGKFGTQERVNKKEWNGMMGELLSGQADMIVAPLTINNERAQYIEFSKPFKYQGLTILVKKGTRITGINDPRLRNPSDKFIYATVKQSSVDIYFRRQVELSTMYRHMEKHNYESAAEAIQAVRDNKLHAFIWDSAVLEFEASQKCDLVTTGELFFRSGFGIGMRKDSPWKQNVSLSILKSHENGFMEDLDKTWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Plasmodium Adenylosuccinate synthetase (Malaria Adss) | 1P9B | 6.58 | |
Target general information Gen name Malaria Adss Organism Plasmodium falciparum Uniprot ID TTD ID Synonyms IMP--aspartate ligase; Adenylosuccinate synthase; AdSS; AMPSase Protein family Adenylosuccinate synthetase family Biochemical class Carbon-nitrogen ligase Function Plays an important role in the salvage pathway for purine nucleotide biosynthesis. Catalyzes the first committed step in the biosynthesis of AMP from IMP. Related diseases Hypertension and brachydactyly syndrome (HTNB) [MIM:112410]: A syndrome characterized by brachydactyly type E, severe salt-independent but age-dependent hypertension, an increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, and altered baroreflex blood pressure regulation. It results in death from stroke before age 50 years when untreated. Brachydactyly type E is characterized by shortening of the fingers mainly in the metacarpals and metatarsals. {ECO:0000269|PubMed:25961942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03510; DB04315; DB02109 Interacts with NA EC number EC 6.3.4.4 Uniprot keywords 3D-structure; Cytoplasm; GTP-binding; Ligase; Magnesium; Metal-binding; Nucleotide-binding; Purine biosynthesis Protein physicochemical properties Chain ID A Molecular weight (Da) 47877.9 Length 424 Aromaticity 0.09 Instability index 31.72 Isoelectric point 7.63 Charge (pH=7) 1.58 2D Binding mode Binding energy (Kcal/mol) -8.97 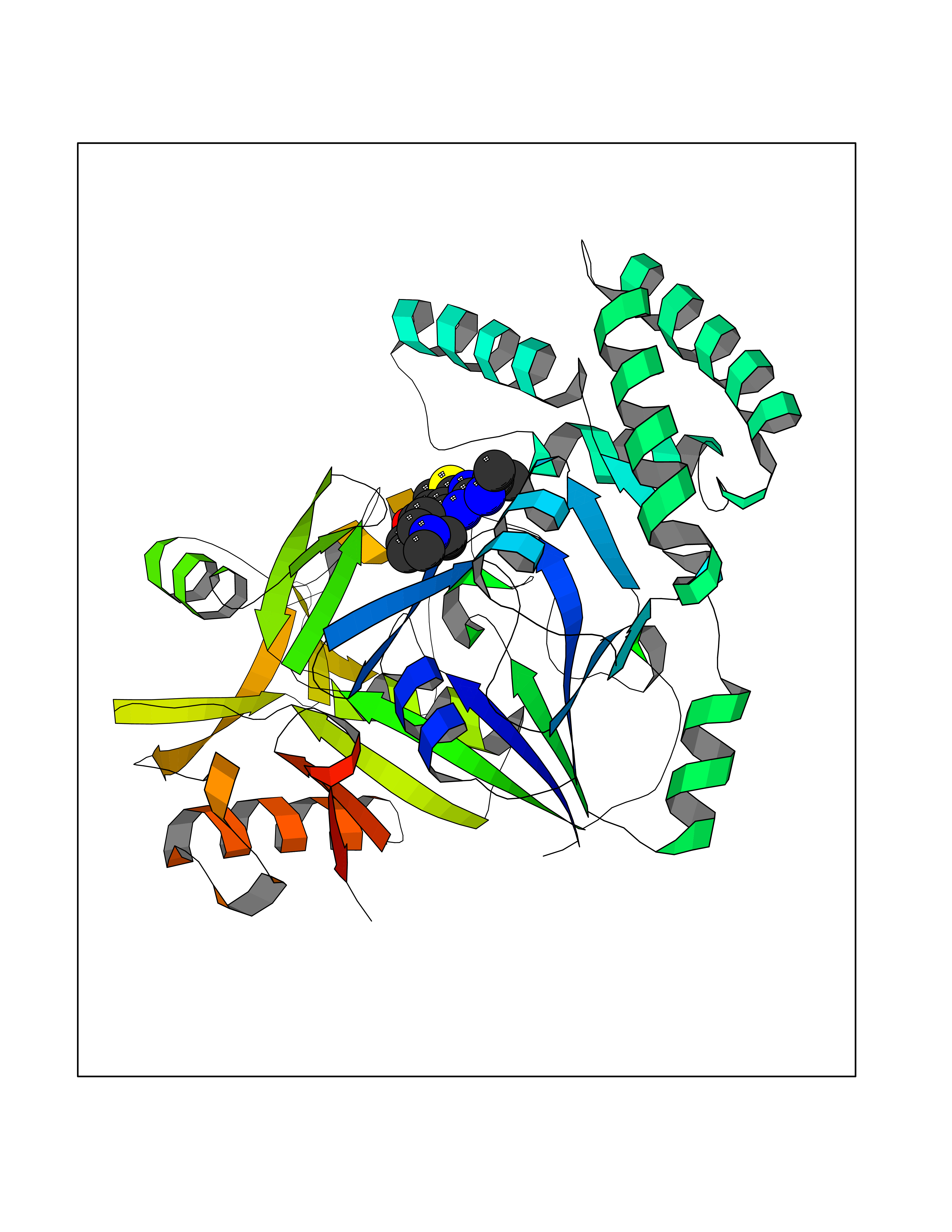 Molscript Map 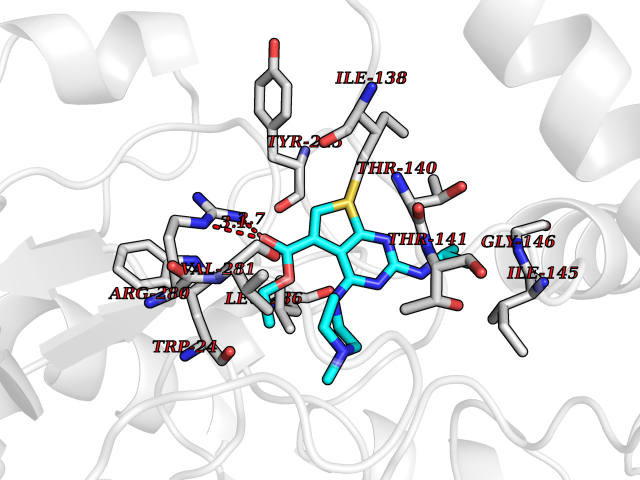 Pymol Map 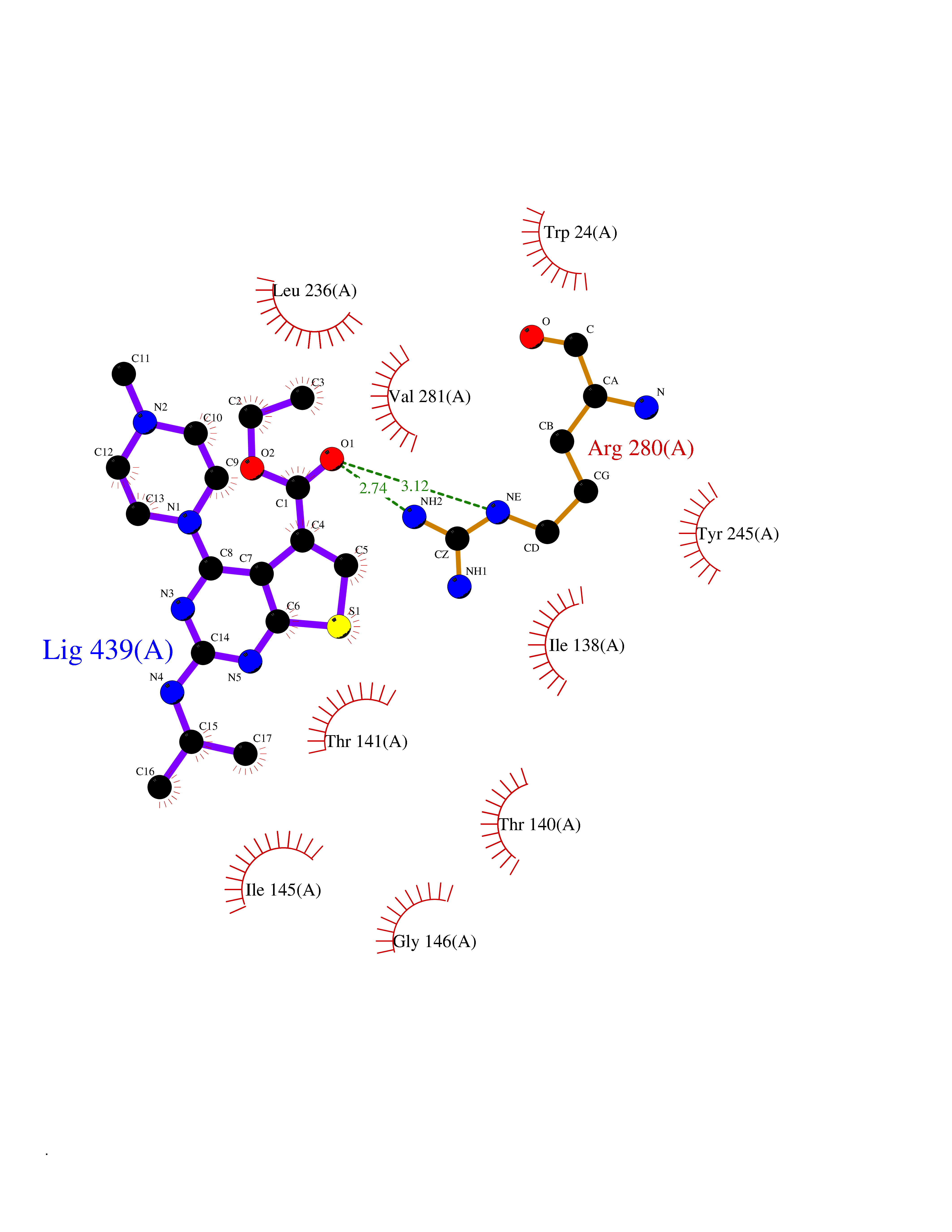 Ligplot Map 3D Binding mode Sequence GNVVAILGAQWGDEGKGKIIDMLSEYSDITCRFNGGANAGHTISVNDKKYALHLLPCGVLYDNNISVLGNGMVIHVKSLMEEIESVGGKLLDRLYLSNKAHILFDIHQIIDSIQETKKLKEGKQIGTTKRGIGPCYSTKASRIGIRLGTLKNFENFKNMYSKLIDHLMDLYNITEYDKEKELNLFYNYHIKLRDRIVDVISFMNTNLENNKKVLIEGANAAMLDIDFGTYPYVTSSCTTVGGVFSGLGIHHKKLNLVVGVVKSYLTRVGCGPFLTELNNDVGQYLREKGHEYGTTTKRPRRCGWLDIPMLLYVKCINSIDMINLTKLDVLSGLEEILLCVNFKNKKTGELLEKGCYPVEEEISEEYEPVYEKFSGWKEDISTCNEFDELPENAKKYILAIEKYLKTPIVWIGVGPNRKNMIVKK Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Apoptosis signal-regulating kinase 1 (MAP3K5) | 6OYT | 6.58 | |
Target general information Gen name MAP3K5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mitogen-activated protein kinase kinase kinase 5; MEKK5; MEKK 5; MEK kinase 5; MAPKKK5; MAPK/ERK kinase kinase 5; ASK1; ASK-1 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase kinase subfamily Biochemical class Kinase Function Plays an important role in the cascades of cellular responses evoked by changes in the environment. Mediates signaling for determination of cell fate such as differentiation and survival. Plays a crucial role in the apoptosis signal transduction pathway through mitochondria-dependent caspase activation. MAP3K5/ASK1 is required for the innate immune response, which is essential for host defense against a wide range of pathogens. Mediates signal transduction of various stressors like oxidative stress as well as by receptor-mediated inflammatory signals, such as the tumor necrosis factor (TNF) or lipopolysaccharide (LPS). Once activated, acts as an upstream activator of the MKK/JNK signal transduction cascade and the p38 MAPK signal transduction cascade through the phosphorylation and activation of several MAP kinase kinases like MAP2K4/SEK1, MAP2K3/MKK3, MAP2K6/MKK6 and MAP2K7/MKK7. These MAP2Ks in turn activate p38 MAPKs and c-jun N-terminal kinases (JNKs). Both p38 MAPK and JNKs control the transcription factors activator protein-1 (AP-1). Serine/threonine kinase which acts as an essential component of the MAP kinase signal transduction pathway. Related diseases Ataxia telangiectasia (AT) [MIM:208900]: A rare recessive disorder characterized by progressive cerebellar ataxia, dilation of the blood vessels in the conjunctiva and eyeballs, immunodeficiency, growth retardation and sexual immaturity. Patients have a strong predisposition to cancer; about 30% of patients develop tumors, particularly lymphomas and leukemias. Cells from affected individuals are highly sensitive to damage by ionizing radiation and resistant to inhibition of DNA synthesis following irradiation. {ECO:0000269|PubMed:10234507, ECO:0000269|PubMed:10425038, ECO:0000269|PubMed:10817650, ECO:0000269|PubMed:10873394, ECO:0000269|PubMed:19431188, ECO:0000269|PubMed:27664052, ECO:0000269|PubMed:7792600, ECO:0000269|PubMed:8589678, ECO:0000269|PubMed:8665503, ECO:0000269|PubMed:8698354, ECO:0000269|PubMed:8755918, ECO:0000269|PubMed:8789452, ECO:0000269|PubMed:8797579, ECO:0000269|PubMed:8808599, ECO:0000269|PubMed:8845835, ECO:0000269|PubMed:9043869, ECO:0000269|PubMed:9150358, ECO:0000269|PubMed:9443866, ECO:0000269|PubMed:9450874, ECO:0000269|PubMed:9463314, ECO:0000269|PubMed:9497252, ECO:0000269|PubMed:9521587, ECO:0000269|PubMed:9711876, ECO:0000269|PubMed:9792409, ECO:0000269|PubMed:9792410, ECO:0000269|PubMed:9872980, ECO:0000269|PubMed:9887333}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in ATM may contribute to T-cell acute lymphoblastic leukemia (TALL) and T-prolymphocytic leukemia (TPLL). TPLL is characterized by a high white blood cell count, with a predominance of prolymphocytes, marked splenomegaly, lymphadenopathy, skin lesions and serous effusion. The clinical course is highly aggressive, with poor response to chemotherapy and short survival time. TPLL occurs both in adults as a sporadic disease and in younger AT patients. {ECO:0000269|PubMed:9288106, ECO:0000269|PubMed:9334731, ECO:0000269|PubMed:9463314, ECO:0000269|PubMed:9488043, ECO:0000269|PubMed:9573030}.; DISEASE: Defects in ATM may contribute to B-cell non-Hodgkin lymphomas (BNHL), including mantle cell lymphoma (MCL). {ECO:0000269|PubMed:10397742, ECO:0000269|PubMed:10706620, ECO:0000269|PubMed:9288106}.; DISEASE: Defects in ATM may contribute to B-cell chronic lymphocytic leukemia (BCLL). BCLL is the commonest form of leukemia in the elderly. It is characterized by the accumulation of mature CD5+ B-lymphocytes, lymphadenopathy, immunodeficiency and bone marrow failure. {ECO:0000269|PubMed:10023947, ECO:0000269|PubMed:10397742, ECO:0000269|PubMed:9892178}. Drugs (DrugBank ID) NA Interacts with P31749; P49407; P32121; Q99828; P02489; Q5VWQ8; Q9UER7; P50570-2; Q14204; P20042; P41091; Q0VDC6; Q8TB36; P28799; P54652; P04792; O43464; P42858; O60333-2; Q92876; Q14114-3; P46734; Q99683; Q96EZ8; Q96HS1; P63098; P60891; Q16637; Q12933; P10599; Q9UBQ0-2; P31946; P62258; P61981; Q04917; P63104; Q969S3; Q9WTR2; Q9D1C8 EC number EC 2.7.11.25 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Coiled coil; Cytoplasm; Endoplasmic reticulum; Host-virus interaction; Immunity; Innate immunity; Kinase; Magnesium; Metal-binding; Methylation; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Stress response; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,C Molecular weight (Da) 51651.4 Length 458 Aromaticity 0.09 Instability index 36.22 Isoelectric point 5.76 Charge (pH=7) -10.26 2D Binding mode Binding energy (Kcal/mol) -8.97  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MESDLLEYDYEYDENGDRVVLGKGTYGIVYAGRDLSNQVRIAIKEIPERQPLHEEIALHKHLKHKNIVQYLGSFSENGFIKIFMEQVPGGSLSALLRSKWGPLKDNEQTIGFYTKQILEGLKYLHDNQIVHRDIKGDNVLINTYSGVLKISDFGTSKRLGKAADIWSLGCTIIEMATGKPPVHPEIPESMSAEAKAFILKCFEPDPDKRACANDLLVDEFLKLEYDYEYDENGDRVVLGKGTYGIVYAGRDLSNQVRIAIKEIPERDSRYSQPLHEEIALHKHLKHKNIVQYLGSFSENGFIKIFMEQVPGGSLSALLRSKWGPLKDNEQTIGFYTKQILEGLKYLHDNQIVHRDIKGDNVLINTYSGVLKISDFGTSKRLLQYMAPEIIDKGPRGGKAADIWSLGCTIIEMATGKPPFHPEIPESMSAEAKAFILKCFEPDPDKRACANDLLVDEFL Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 6.57 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -8.97  Molscript Map 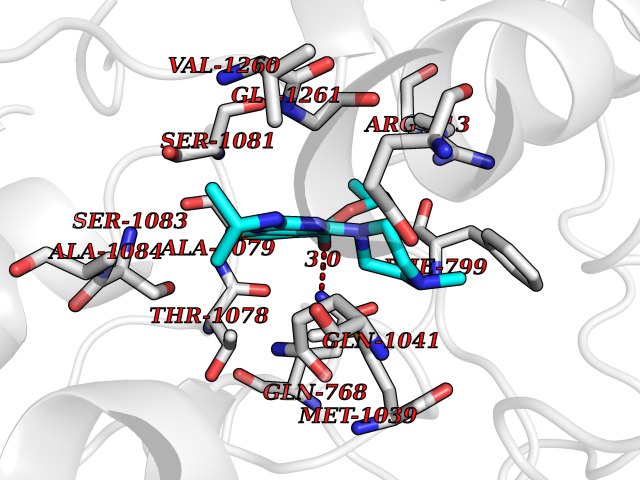 Pymol Map 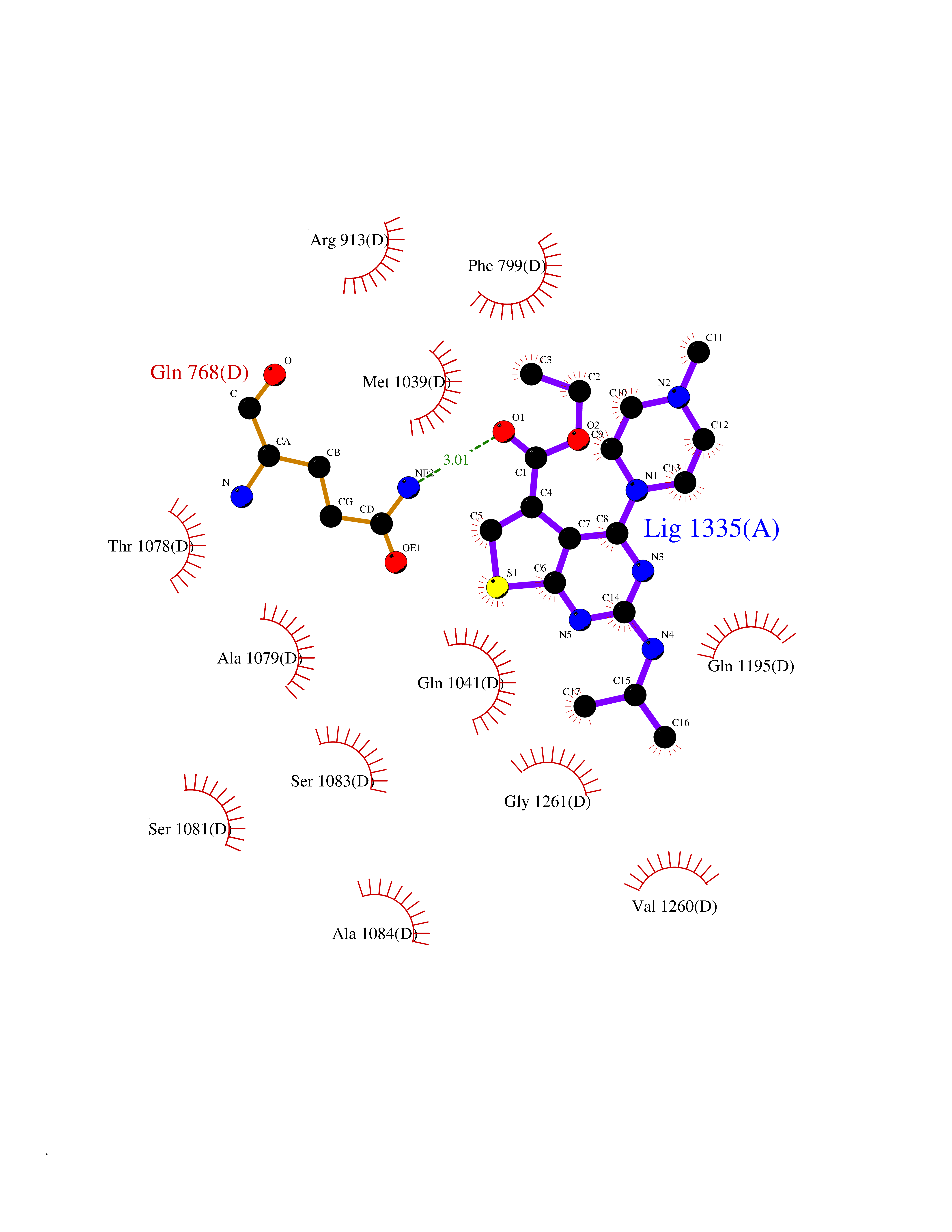 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Receptor-type protein-tyrosine phosphatase zeta (PTPRZ1) | 5H08 | 6.57 | |
Target general information Gen name PTPRZ1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Receptor protein tyrosine phosphatase zeta; R-PTP-zeta; PTPRZ1 Protein family Protein-tyrosine phosphatase family, Receptor class 5 subfamily Biochemical class Phosphoric monoester hydrolase Function Protein tyrosine phosphatase that negatively regulates oligodendrocyte precursor proliferation in the embryonic spinal cord. Required for normal differentiation of the precursor cells into mature, fully myelinating oligodendrocytes. May play a role in protecting oligondendrocytes against apoptosis. May play a role in the establishment of contextual memory, probably via the dephosphorylation of proteins that are part of important signaling cascades. Related diseases Optic atrophy 1 (OPA1) [MIM:165500]: A condition that features progressive visual loss in association with optic atrophy. Atrophy of the optic disk indicates a deficiency in the number of nerve fibers which arise in the retina and converge to form the optic disk, optic nerve, optic chiasm and optic tracts. OPA1 is characterized by an insidious onset of visual impairment in early childhood with moderate to severe loss of visual acuity, temporal optic disk pallor, color vision deficits, and centrocecal scotoma of variable density. {ECO:0000269|PubMed:11017079, ECO:0000269|PubMed:11017080, ECO:0000269|PubMed:11440988, ECO:0000269|PubMed:11440989, ECO:0000269|PubMed:11810270, ECO:0000269|PubMed:12036970, ECO:0000269|PubMed:12566046, ECO:0000269|PubMed:14961560, ECO:0000269|PubMed:15948788, ECO:0000269|PubMed:16513463, ECO:0000269|PubMed:16617242, ECO:0000269|PubMed:18204809, ECO:0000269|PubMed:18360822, ECO:0000269|PubMed:19319978, ECO:0000269|PubMed:19325939, ECO:0000269|PubMed:19969356, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:22382025, ECO:0000269|PubMed:22857269, ECO:0000269|PubMed:23401657}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Dominant optic atrophy plus syndrome (DOA+) [MIM:125250]: A neurologic disorder characterized most commonly by an insidious onset of visual loss and sensorineural hearing loss in childhood with variable presentation of other clinical manifestations including progressive external ophthalmoplegia, muscle cramps, hyperreflexia, and ataxia. There appears to be a wide range of intermediate phenotypes. {ECO:0000269|PubMed:15531309, ECO:0000269|PubMed:16240368, ECO:0000269|PubMed:18065439, ECO:0000269|PubMed:18158317, ECO:0000269|PubMed:18195150, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:21112924, ECO:0000269|PubMed:23387428}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Behr syndrome (BEHRS) [MIM:210000]: An autosomal recessive syndrome characterized by optic atrophy beginning in early childhood associated with ataxia, pyramidal signs, spasticity, intellectual disability, and posterior column sensory loss. The ataxia, spasticity, and muscle contractures, mainly of the hip adductors, hamstrings, and soleus, are progressive and become more prominent in the second decade. {ECO:0000269|PubMed:21636302, ECO:0000269|PubMed:25012220, ECO:0000269|PubMed:25146916}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mitochondrial DNA depletion syndrome 14, cardioencephalomyopathic type (MTDPS14) [MIM:616896]: An autosomal recessive mitochondrial disorder characterized by lethal infantile encephalopathy, hypertrophic cardiomyopathy and optic atrophy. Skeletal muscle biopsies show significant mtDNA depletion and abnormal mitochondria. {ECO:0000269|PubMed:26561570}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UM73; Q12860 EC number EC 3.1.3.48 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Hydrolase; Membrane; Phosphoprotein; Protein phosphatase; Proteoglycan; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32208.1 Length 282 Aromaticity 0.11 Instability index 38.98 Isoelectric point 7.35 Charge (pH=7) 0.89 2D Binding mode Binding energy (Kcal/mol) -8.97 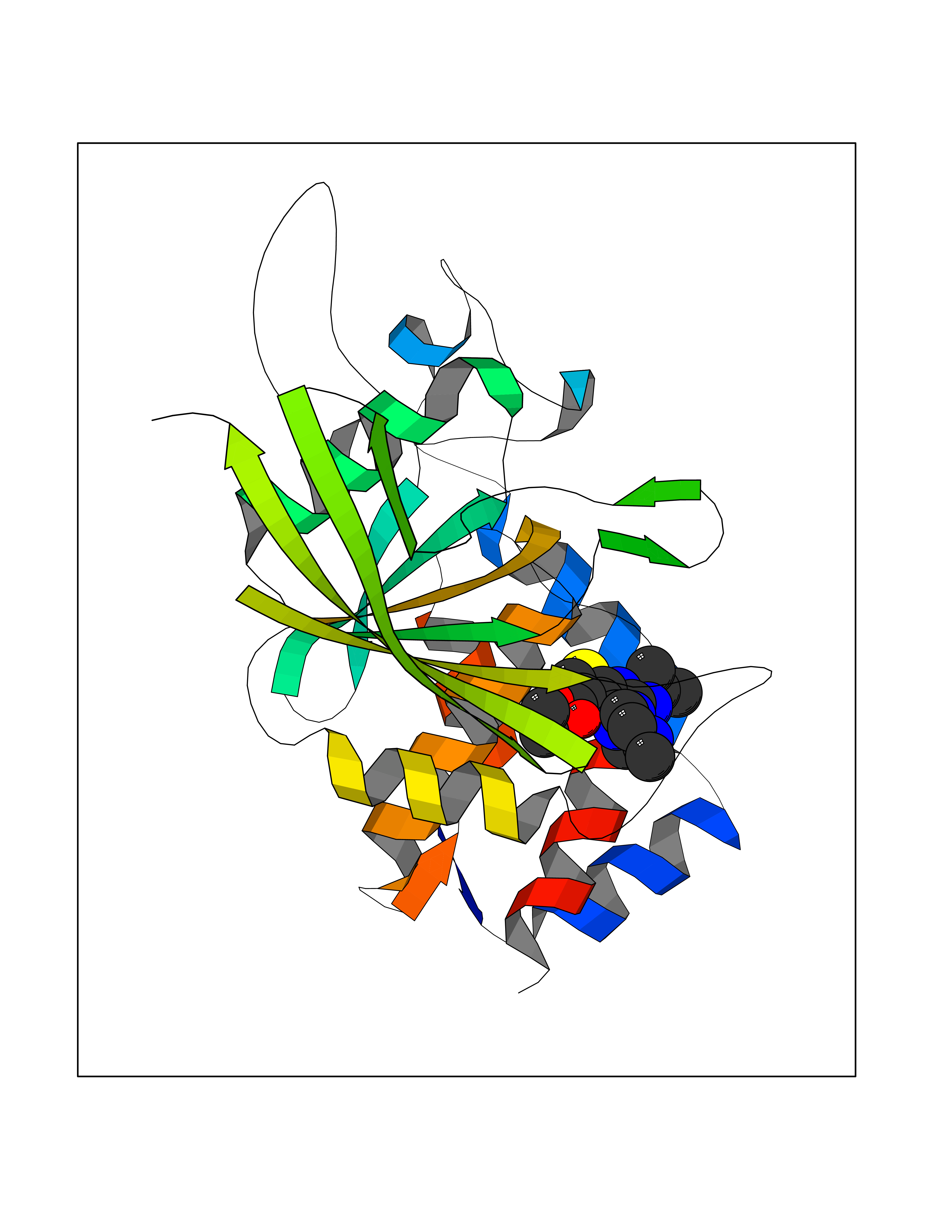 Molscript Map 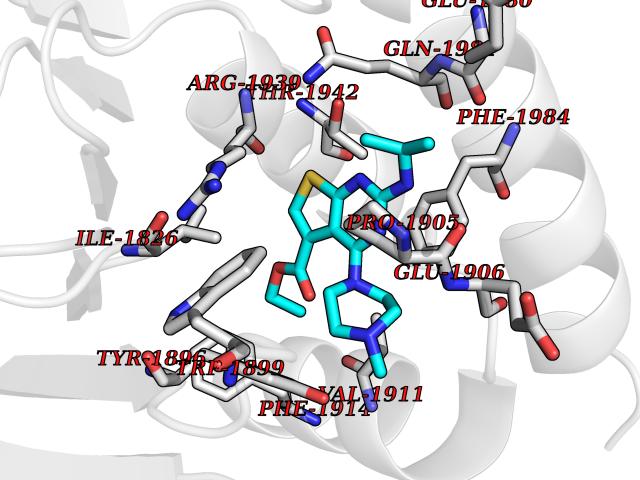 Pymol Map 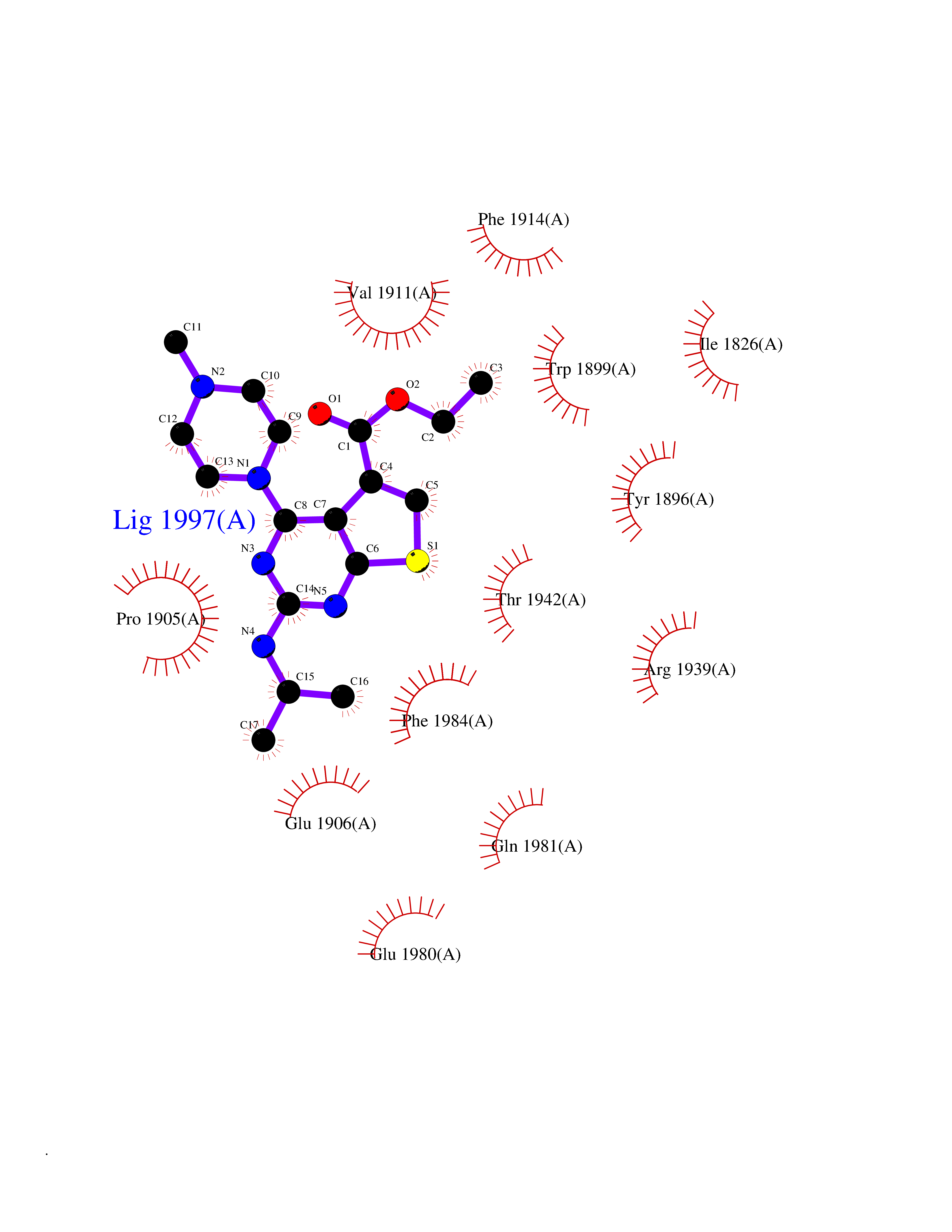 Ligplot Map 3D Binding mode Sequence GPAIPIKHFPKHVADLHASSGFTEEFEEVQSCTVDLGITADSSNHPDNKHKNRYINIVAYDHSRVKLAQLAEKDGKLTDYINANYVDGYNRPKAYIAAQGPLKSTAEDFWRMIWEHNVEVIVMITNLVEKGRRKCDQYWPADGSEEYGNFLVTQKSVQVLAYYTVRNFTLRNTKIRVVTQYHYTQWPDMGVPEYSLPVLTFVRKAAYAKRHAVGPVVVHCSAGVGRTGTYIVLDSMLQQIQHEGTVNIFGFLKHIRSQRNYLVQTEEQYVFIHDTLVEAILS Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 6.56 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -8.95  Molscript Map 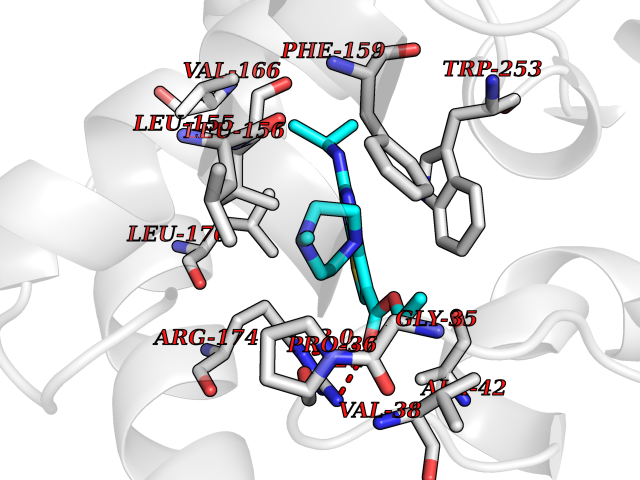 Pymol Map 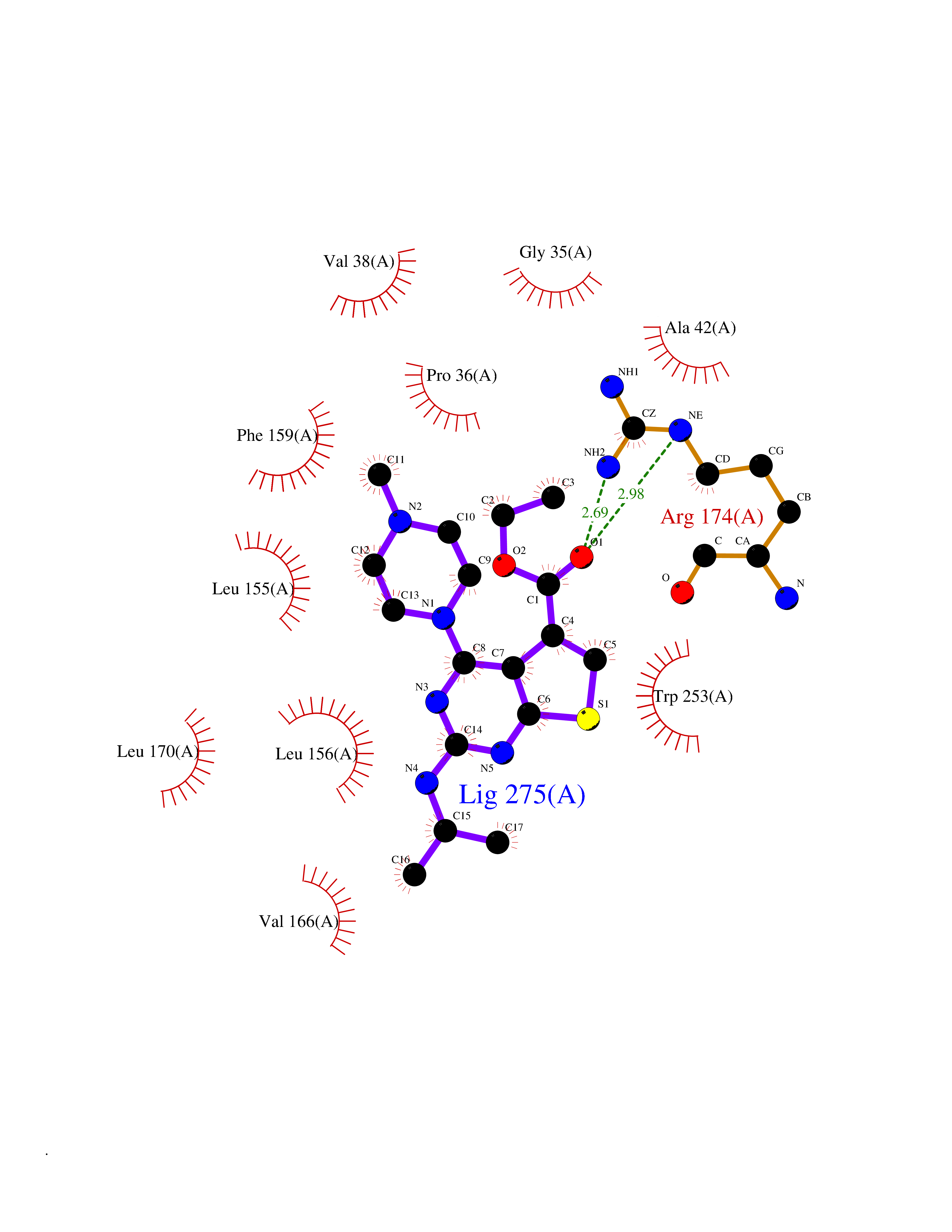 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Glutamate receptor ionotropic NMDA 2A (NMDAR2A) | 5H8Q | 6.56 | |
Target general information Gen name GRIN2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NR2A; NMDA receptor NR2A; N-methyl D-aspartate receptor subtype 2A; HNR2A; Glutamate receptor ionotropic, NMDA 2A; Glutamate [NMDA] receptor subunit epsilon-1; GluN2A Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, NR1/GRIN1 subfamily Biochemical class Glutamate-gated ion channel Function Channel activation requires binding of the neurotransmitter glutamate to the epsilon subunit, glycine binding to the zeta subunit, plus membrane depolarization to eliminate channel inhibition by Mg(2+). Sensitivity to glutamate and channel kinetics depend on the subunit composition; channels containing GRIN1 and GRIN2A have higher sensitivity to glutamate and faster kinetics than channels formed by GRIN1 and GRIN2B. Contributes to the slow phase of excitatory postsynaptic current, long-term synaptic potentiation, and learning. Component of NMDA receptor complexes that function as heterotetrameric, ligand-gated ion channels with high calcium permeability and voltage-dependent sensitivity to magnesium. Related diseases Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant (NDHMSD) [MIM:614254]: An autosomal dominant neurodevelopmental disorder characterized by severe intellectual disability and developmental delay, absent speech, muscular hypotonia, dyskinesia, and hyperkinetic movements. Cortical blindness, cerebral atrophy, and seizures are present in some patients. {ECO:0000269|PubMed:21376300, ECO:0000269|PubMed:25167861, ECO:0000269|PubMed:25864721, ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:28228639, ECO:0000269|PubMed:28389307, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal recessive (NDHMSR) [MIM:617820]: An autosomal recessive neurodevelopmental disorder characterized by severe intellectual disability and psychomotor developmental delay, involuntary and stereotypic movements, spasticity, and inability to walk without support. Intractable seizures manifest in some patients. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28051072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 101 (DEE101) [MIM:619814]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE101 is an autosomal recessive, severe form characterized by onset of seizures in early infancy. Death in infancy may occur. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:34611970}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01931; DB00659; DB06151; DB08838; DB01238; DB00289; DB05824; DB04620; DB03929; DB00647; DB00843; DB00228; DB11823; DB13146; DB06741; DB00142; DB00874; DB08954; DB06738; DB09409; DB09481; DB01043; DB00454; DB00333; DB04896; DB01173; DB00312; DB01174; DB01708; DB00418; DB00193 Interacts with P05067; P35637; Q12879-1; Q13224; Q62936 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Ligand-gated ion channel; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Zinc Protein physicochemical properties Chain ID B Molecular weight (Da) 63014.6 Length 557 Aromaticity 0.1 Instability index 28.81 Isoelectric point 8.59 Charge (pH=7) 7.66 2D Binding mode Binding energy (Kcal/mol) -8.94 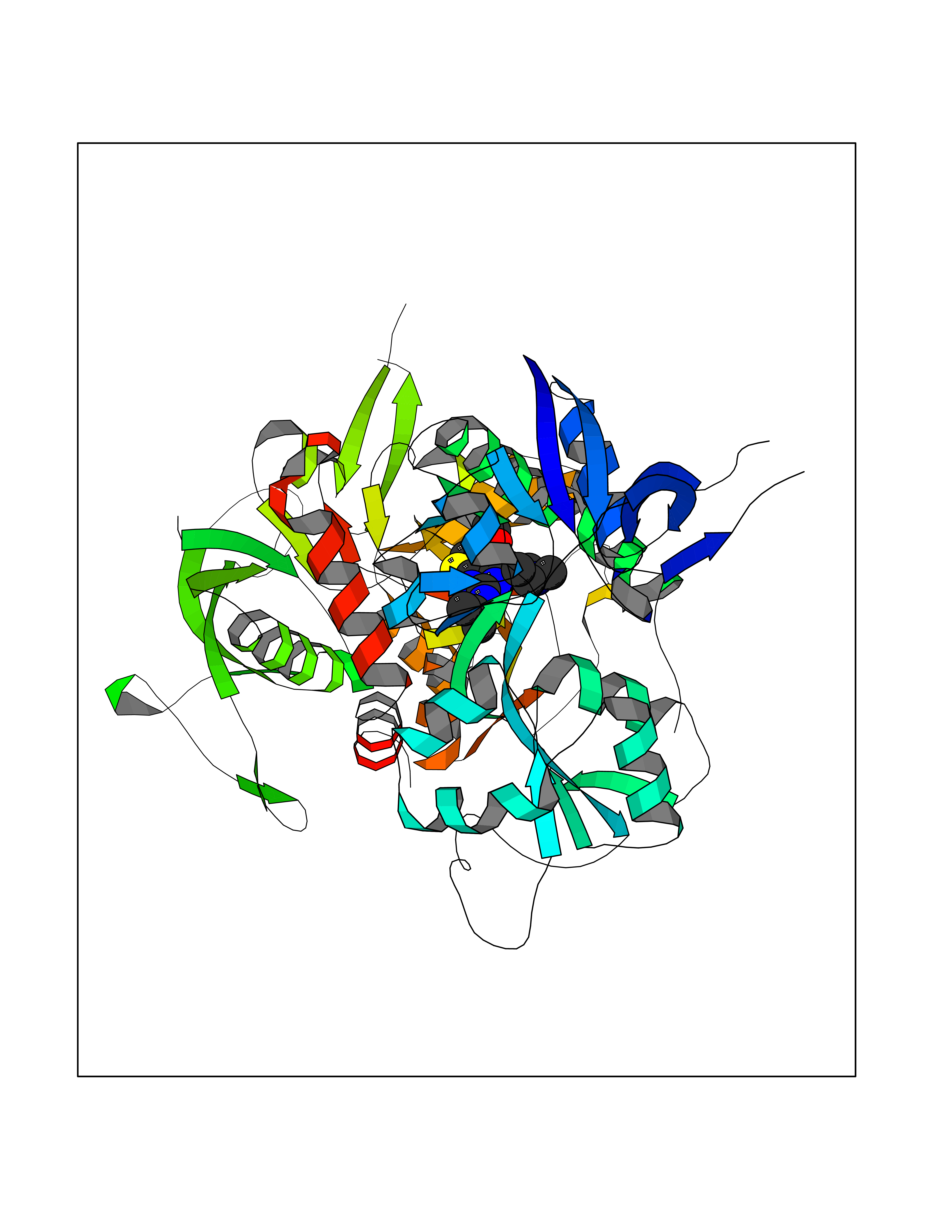 Molscript Map 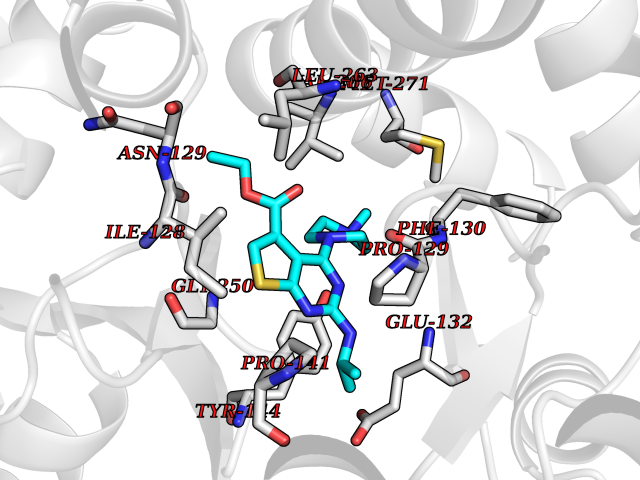 Pymol Map 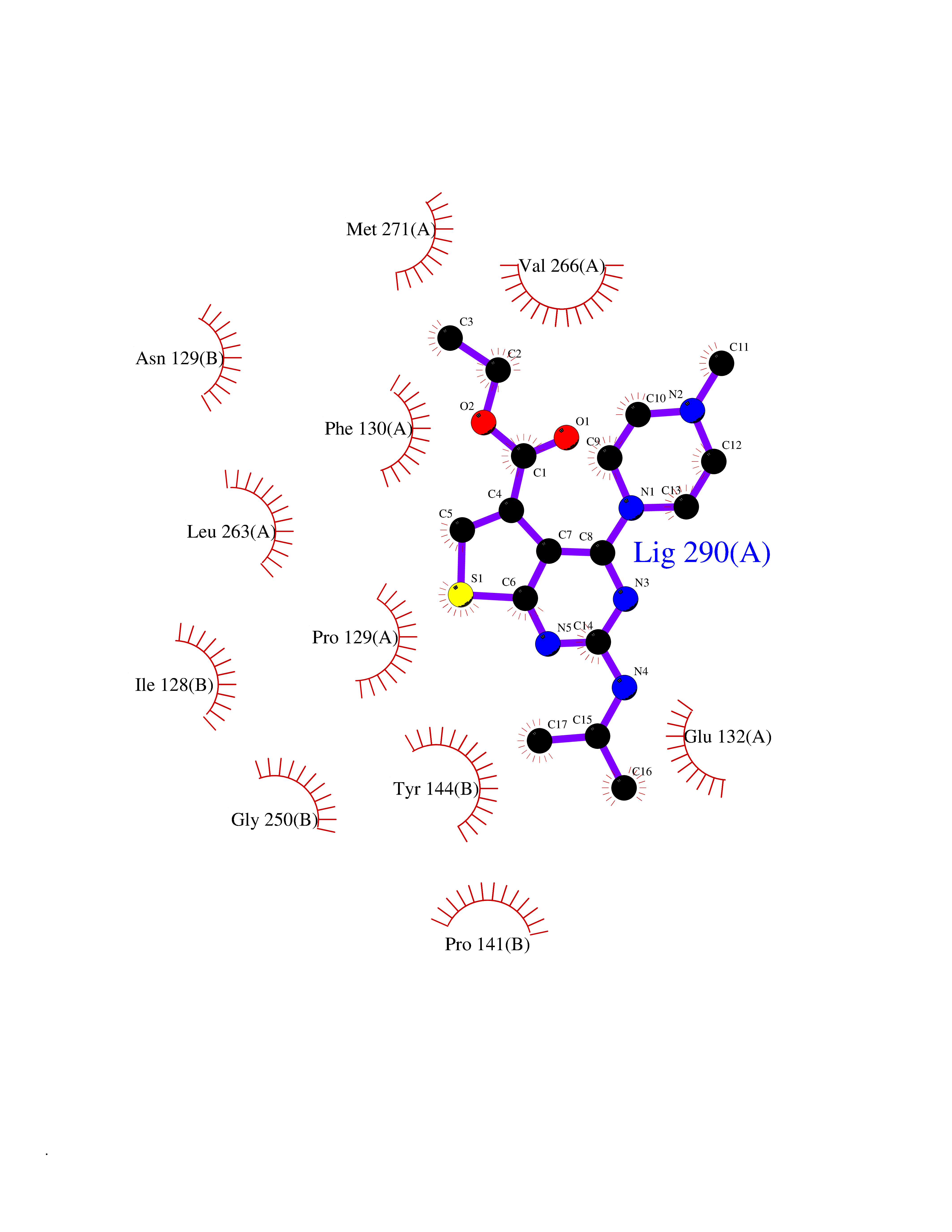 Ligplot Map 3D Binding mode Sequence NHLSIVTLEEAPFVIVEDIDPETCVRNTVPCRKFVKINNSTNEGMNVKKCCKGFCIDILKKLSRTVKFTYDLYLVTNGKHGKKVNNVWNGMIGEVVYQRAVMAVGSLTINEERSEVVDFSVPFVETGISVMVSRGTQVTGLSDKKFQRPHDYSPPFRFGTVPNGSTERNIRNNYPYMHQYMTKFNQKGVEDALVSLKTGKLDAFIYDAAVLNYKAGRDEGCKLVTIGSGYIFATTGYGIALQKGSPWKRQIDLALLQFVGDGEMEELETLWLTGICTRLKIVTIHQEPFVYVKPTLSDGTCKEEFTVNGDPVKKVICTGPNDTSPGSPRHTVPQCCYGFCIDLLIKLARTMNFTYEVHLVADGKFGTQERVNKKEWNGMMGELLSGQADMIVAPLTINNERAQYIEFSKPFKYQGLTILVKKGTRITGINDPRLRNPSDKFIYATVKQSSVDIYFRRQVELSTMYRHMEKHNYESAAEAIQAVRDNKLHAFIWDSAVLEFEASQKCDLVTTGELFFRSGFGIGMRKDSPWKQNVSLSILKSHENGFMEDLDKTWVRY Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Serine Racemase (SRR) | 3L6B | 6.56 | |
Target general information Gen name SRR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms L-serine dehydratase; L-serine ammonia-lyase; D-serine dehydratase; D-serine ammonia-lyase Protein family Serine/threonine dehydratase family Biochemical class Racemases and epimerase Function D-serine is a key coagonist with glutamate at NMDA receptors. Has dehydratase activity towards both L-serine and D-serine. Catalyzes the synthesis of D-serine from L-serine. Related diseases Cornelia de Lange syndrome 5 (CDLS5) [MIM:300882]: A form of Cornelia de Lange syndrome, a clinically heterogeneous developmental disorder associated with malformations affecting multiple systems. It is characterized by facial dysmorphisms, abnormal hands and feet, growth delay, cognitive retardation, hirsutism, gastroesophageal dysfunction and cardiac, ophthalmologic and genitourinary anomalies. {ECO:0000269|PubMed:22885700, ECO:0000269|PubMed:22889856}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00114; DB00133 Interacts with Q9H4P4; Q9GZT4 EC number EC 5.1.1.18 Uniprot keywords 3D-structure; Allosteric enzyme; ATP-binding; Direct protein sequencing; Isomerase; Lyase; Magnesium; Metal-binding; Nucleotide-binding; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation Protein physicochemical properties Chain ID A Molecular weight (Da) 34613.6 Length 322 Aromaticity 0.06 Instability index 31.62 Isoelectric point 6.35 Charge (pH=7) -1.7 2D Binding mode Binding energy (Kcal/mol) -8.95 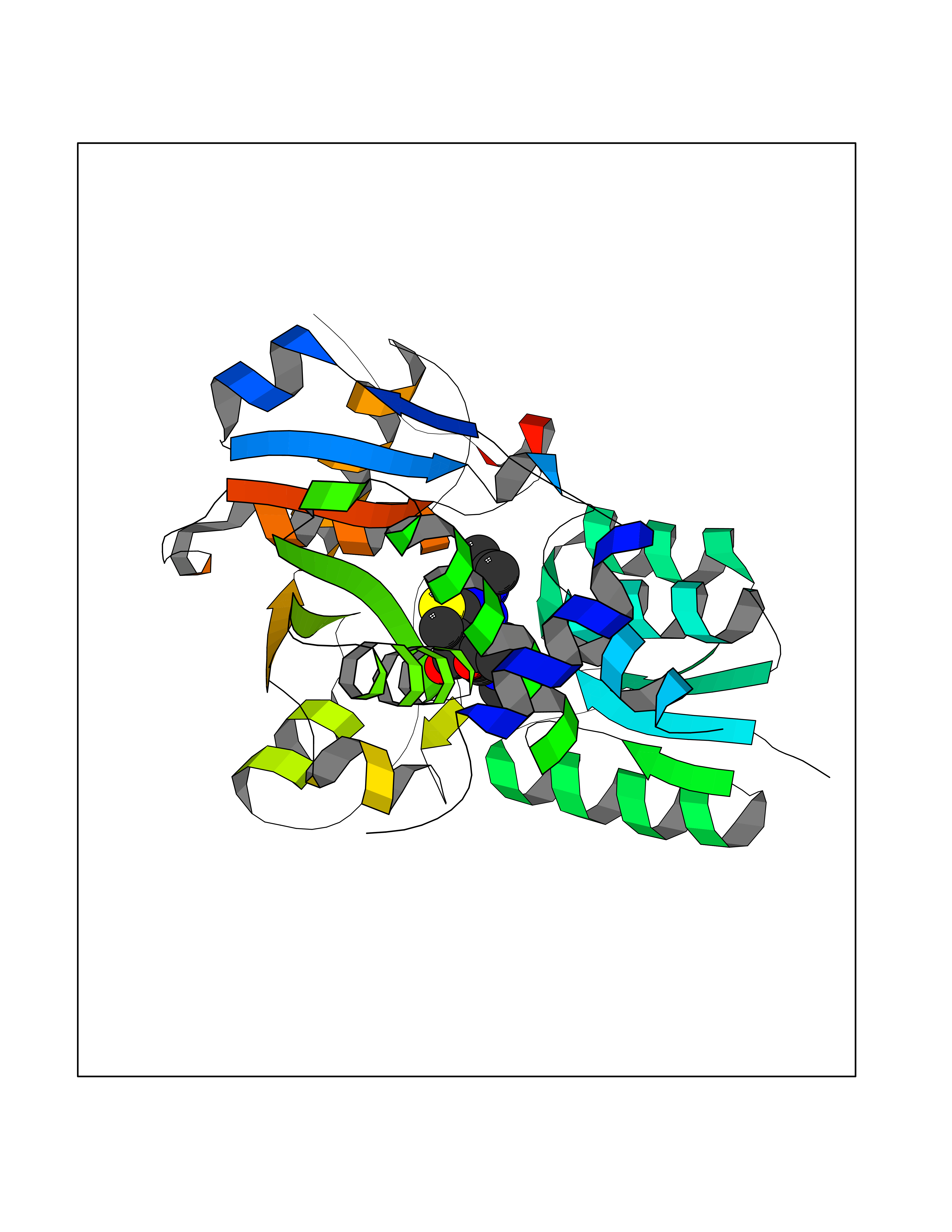 Molscript Map 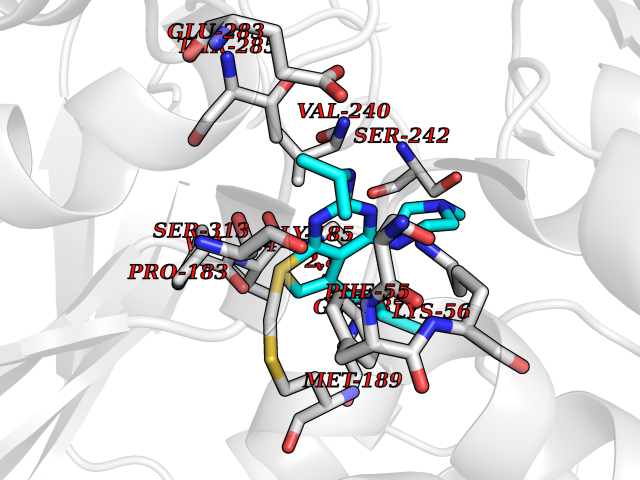 Pymol Map 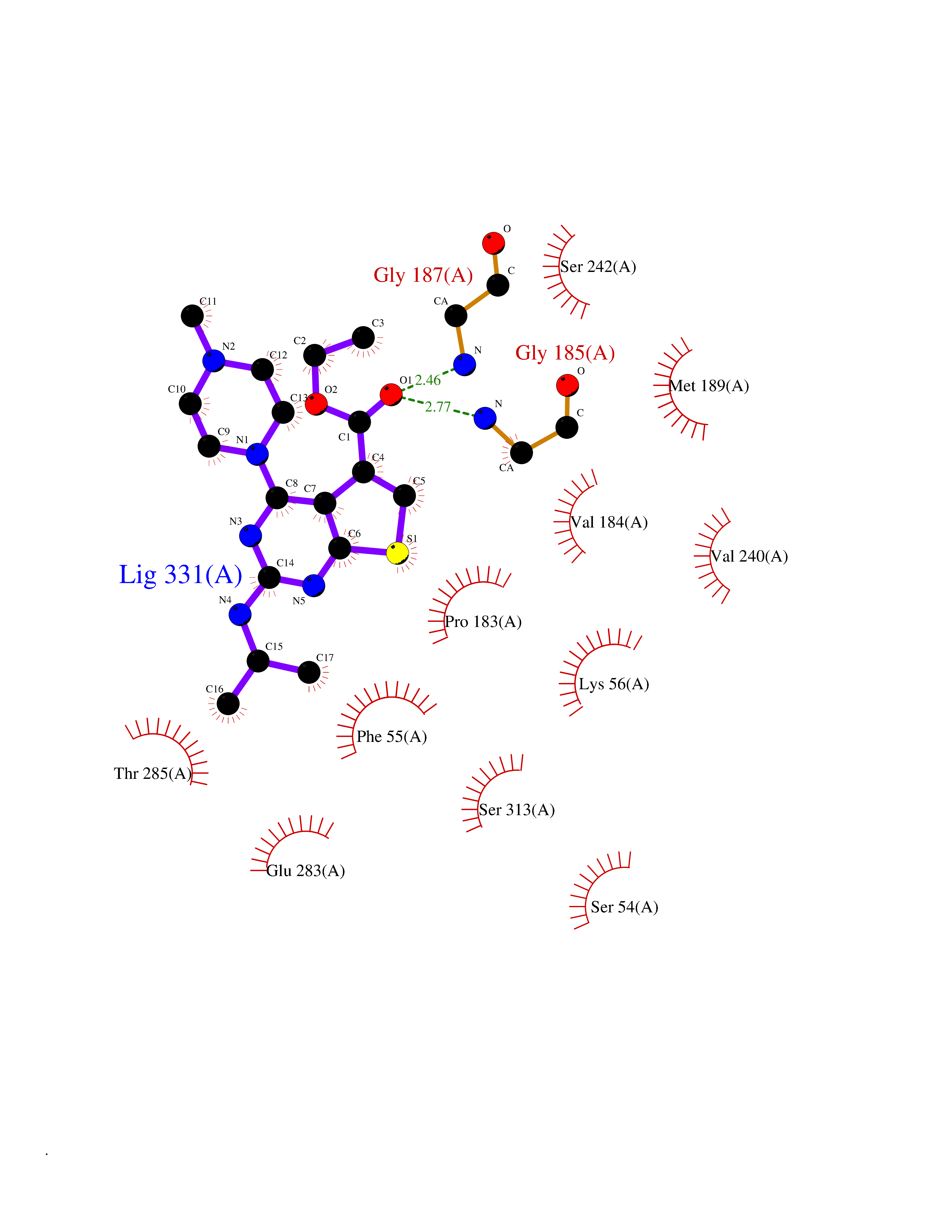 Ligplot Map 3D Binding mode Sequence AQYDISFADVEKAHINIRDSIHLTPVLTSSILNQLTGRNLFFKCELFQKTGSFKIRGALNAVRSLVRKPKAVVTHSSGNHGQALTYAAKLEGIPAYIVVPQTAPDCKKLAIQAYGASIVYCEPSDESRENVAKRVTEETEGIMVHPNQEPAVIAGQGTIALEVLNQVPLVDALVVPVGGGGMLAGIAITVKALKPSVKVYAAEPSNADDCYQSKLKGKLMPNLYPPETIADGVKSSIGLNTWPIIRDLVDDIFTVTEDEIKCATQLVWERMKLLIEPTAGVGVAAVLSQHFQTVSPEVKNICIVLSGGNVDLTSSITWVKQA Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Vitamin K epoxide reductase complex 1 (VKORC1) | 6WV3 | 6.56 | |
Target general information Gen name VKORC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin K1 2,3-epoxide reductase subunit 1; VKORC1; VKOR; UNQ308/PRO351; MSTP576; MSTP134 Protein family VKOR family Biochemical class Short-chain dehydrogenases reductase Function Involved invitamin K metabolism. Catalytic subunit of the vitamin K epoxide reductase (VKOR) complex which reduces inactive vitamin K 2,3-epoxide to active vitamin K. Vitamin K is required for the gamma-carboxylation of various proteins, including clotting factors, and is required for normal blood coagulation, but also for normal bone development. Related diseases Combined deficiency of vitamin K-dependent clotting factors 2 (VKCFD2) [MIM:607473]: VKCFD leads to a bleeding tendency that is usually reversed by oral administration of vitamin K. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:16270630}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Coumarin resistance (CMRES) [MIM:122700]: A condition characterized by partial or complete resistance to warfarin or other 4-hydroxycoumarin derivatives. These drugs are used as anti-coagulants for the prevention of thromboembolic diseases in subjects with deep vein thrombosis, atrial fibrillation, or mechanical heart valve replacement. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:20946155}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01418; DB00266; DB09332; DB00170; DB00498; DB00946; DB01022; DB00682 Interacts with Q13323; Q7Z7G2; Q96BA8; Q9Y282; Q5JX71; Q96KR6; Q5T7V8; Q8TDT2; Q9NQG1; P15941-11; Q96TC7; Q9NR31; A0A0S2Z4U3; Q8TBB6; O15393-2; Q19QW4 EC number EC 1.17.4.4 Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Disulfide bond; Endoplasmic reticulum; Membrane; Oxidoreductase; Proteomics identification; Quinone; Redox-active center; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 42656.4 Length 381 Aromaticity 0.1 Instability index 32.12 Isoelectric point 7.73 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -8.95  Molscript Map 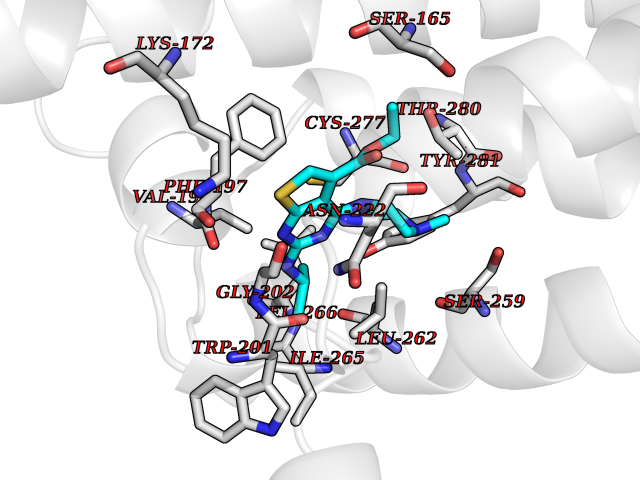 Pymol Map 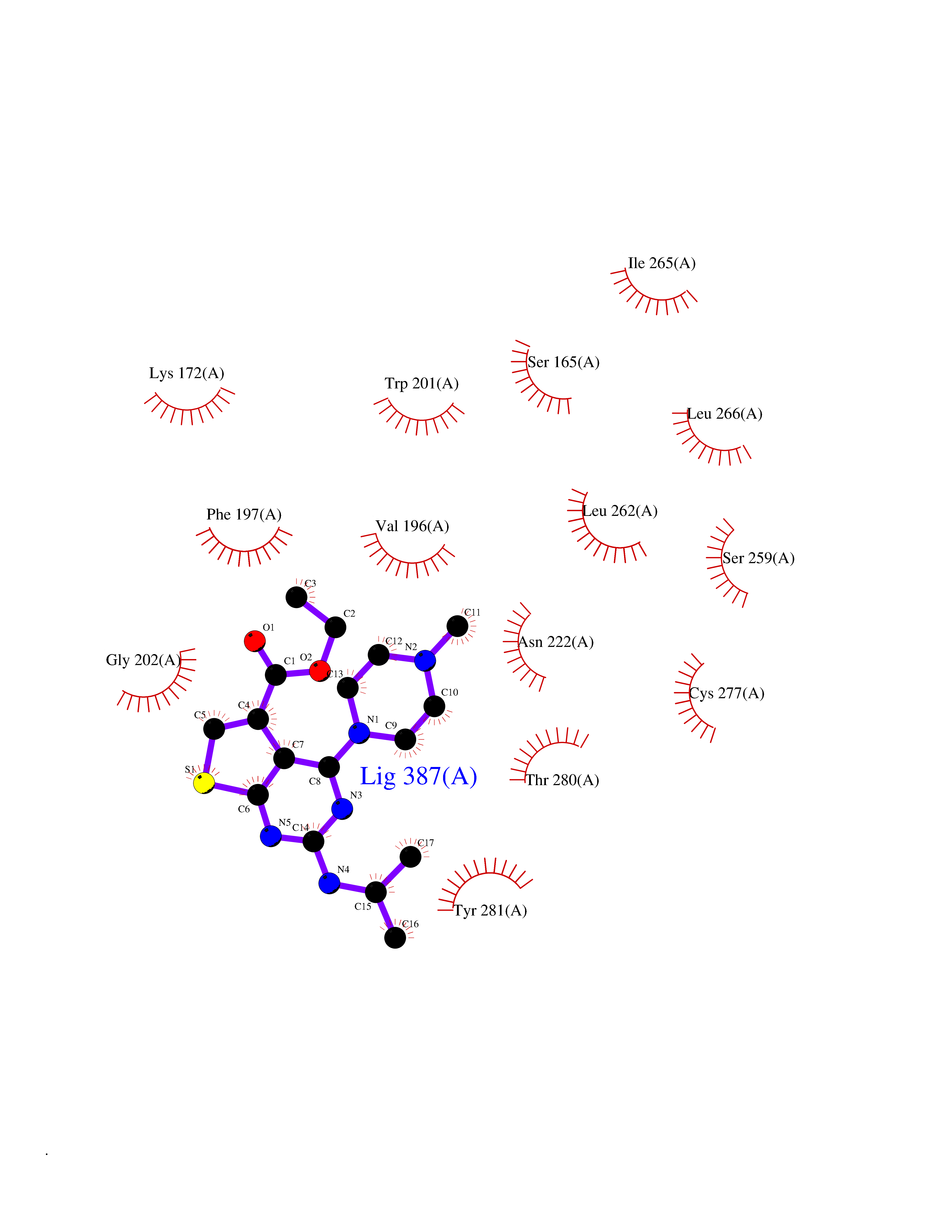 Ligplot Map 3D Binding mode Sequence KGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLXVQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNSTWGSPGWVRLALCLTGLVLSLYALHVKAARARDRDYRALCDVGTAISCSRVFSSRWGRGFGLVEHVLGQDSILNQSNSIFGCIFYTLQLLLGCLRTRWASVLMLLSSLVSLAGSVYLAWILFFVLYDFCIVCITTYAINVSLMWLSFRKVQENSHNVYITADKQKNGIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHHH Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Plasmodium Hypoxanthine-guanine phosphoribosyltransferase (Malaria LACZ) | 3OZF | 6.56 | |
Target general information Gen name Malaria LACZ Organism Plasmodium falciparum (isolate FCR-3 / Gambia) Uniprot ID TTD ID Synonyms LACZ of Plasmodium falciparum (isolate FCR-3 / Gambia); Hypoxanthine phosphoribosyltransferase; HPRT; HGPRTase; HGPRT of Plasmodium falciparum (isolate FCR-3 / Gambia); Guanine phosphoribosyltransfera Protein family Purine/pyrimidine phosphoribosyltransferase family Biochemical class Pentosyltransferase Function Converts guanine to guanosine monophosphate, and hypoxanthine to inosine monophosphate. Transfers the 5- phosphoribosyl group from 5-phosphoribosylpyrophosphate onto the purine. Plays a central role in the generation of purine nucleotides through the purine salvage pathway. Related diseases Intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) [MIM:617333]: An autosomal dominant neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, delayed language, and facial dysmorphisms, most notably ptosis. Additional features may include poor growth, hypotonia, and seizures. {ECO:0000269|PubMed:27939639, ECO:0000269|PubMed:27939640}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02075; DB11638 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cytoplasm; Glycosyltransferase; Magnesium; Metal-binding; Nucleotide-binding; Purine salvage; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 26581.3 Length 233 Aromaticity 0.12 Instability index 29.77 Isoelectric point 7.92 Charge (pH=7) 1.69 2D Binding mode Binding energy (Kcal/mol) -8.94  Molscript Map 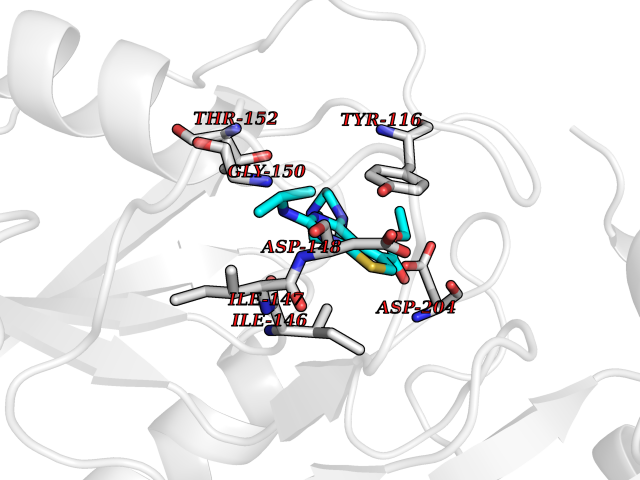 Pymol Map 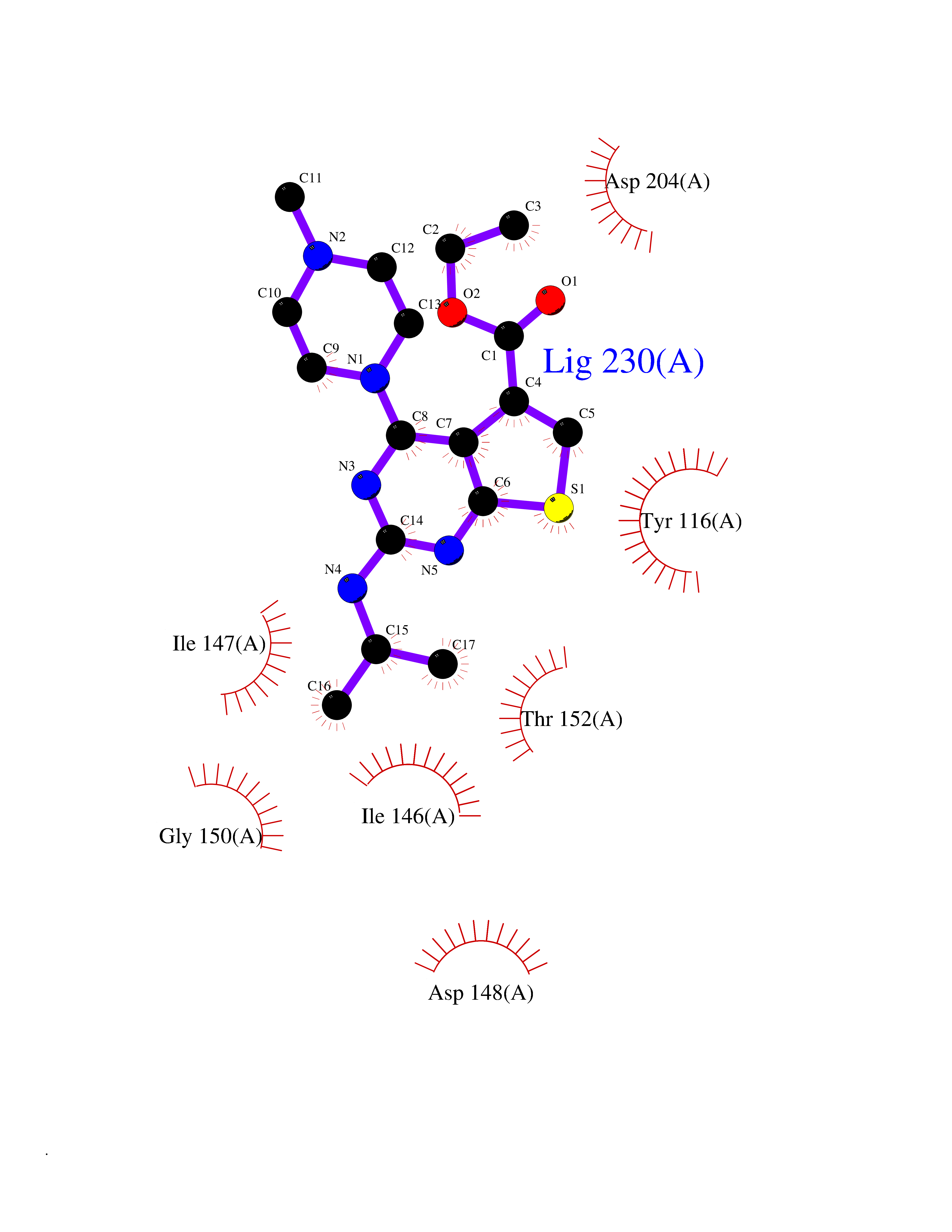 Ligplot Map 3D Binding mode Sequence PRGSHMPIPNNPGAGENAFDPVFVNDDDGYDLDSFMIPAHYKKYLTKVLVPNGVIKNRIEKLAYDIKKVYNNEEFHILCLLKGSRGFFTALLKHLSRIHNYSAVETSKPLFGEHYVRVKSYCNDQSTGTLEIVSEDLSCLKGKHVLIVEDIIDTGKTLVKFCEYLKKFEIKTVAIACLFIKRTPLWNGFKADFVGFSIPDHFVVGYSLDYNEIFRDLDHCCLVNDEGKKKYKA Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | 4-cresol dehydrogenase [hydroxylating] flavoprotein subunit | 1WVF | 6.55 | |
Target general information Gen name pchF Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Oxidoreductase Function 4-cresol dehydrogenase (hydroxylating) activity.Flavin adenine dinucleotide binding.Oxidoreductase activity, acting on CH-OH group of donors. Related diseases Dihydrolipoamide dehydrogenase deficiency (DLDD) [MIM:246900]: An autosomal recessive metabolic disorder characterized biochemically by a combined deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC), pyruvate dehydrogenase complex (PDC), and alpha-ketoglutarate dehydrogenase complex (KGDC). Clinically, affected individuals have lactic acidosis and neurologic deterioration due to sensitivity of the central nervous system to defects in oxidative metabolism. {ECO:0000269|PubMed:10448086, ECO:0000269|PubMed:11687750, ECO:0000269|PubMed:12925875, ECO:0000269|PubMed:15712224, ECO:0000269|PubMed:16442803, ECO:0000269|PubMed:16770810, ECO:0000269|PubMed:17404228, ECO:0000269|PubMed:20160912, ECO:0000269|PubMed:8506365, ECO:0000269|PubMed:8968745, ECO:0000269|PubMed:9540846, ECO:0000269|PubMed:9934985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.17.9.1 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase; Plasmid Protein physicochemical properties Chain ID A Molecular weight (Da) 57240.8 Length 515 Aromaticity 0.1 Instability index 30.94 Isoelectric point 6.06 Charge (pH=7) -4.42 2D Binding mode Binding energy (Kcal/mol) -8.94 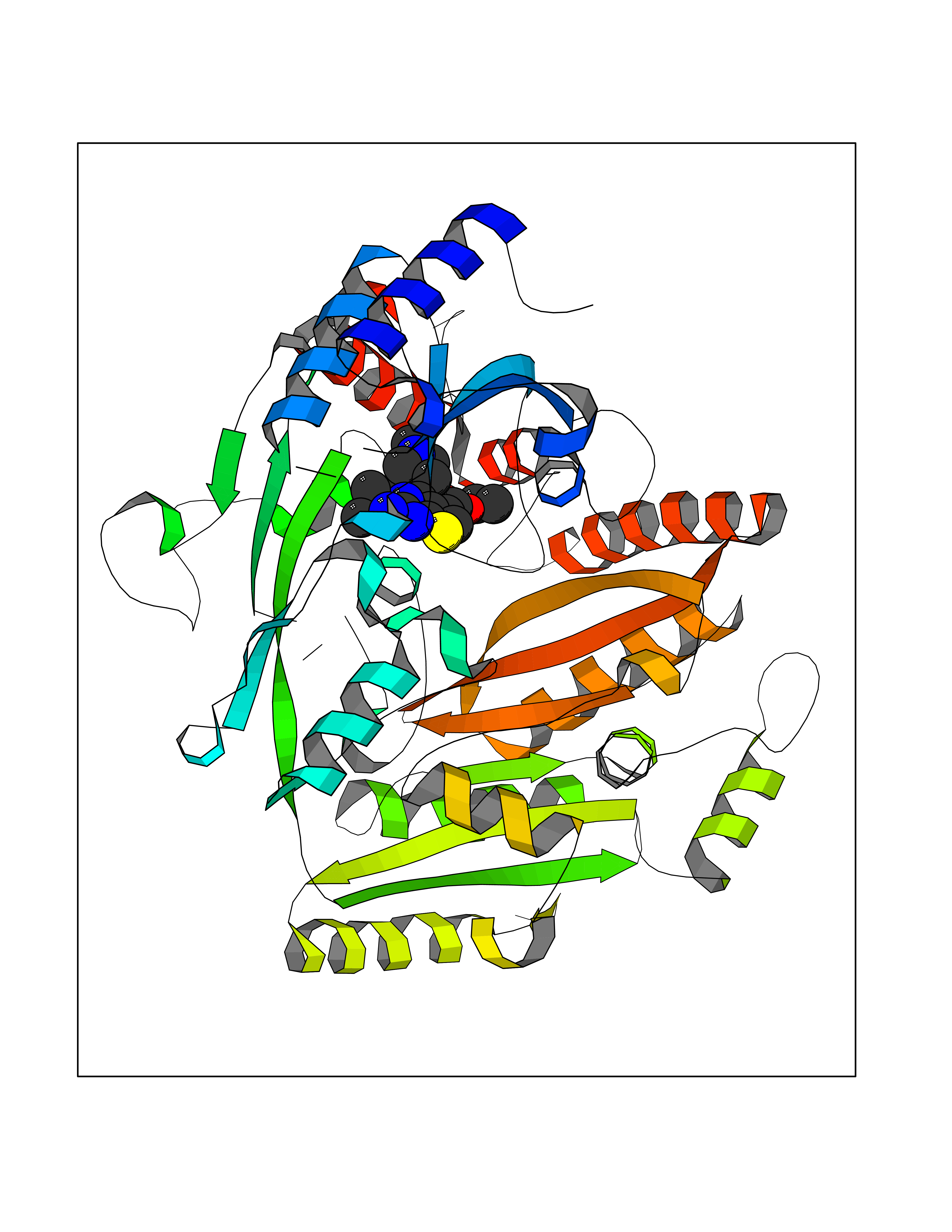 Molscript Map 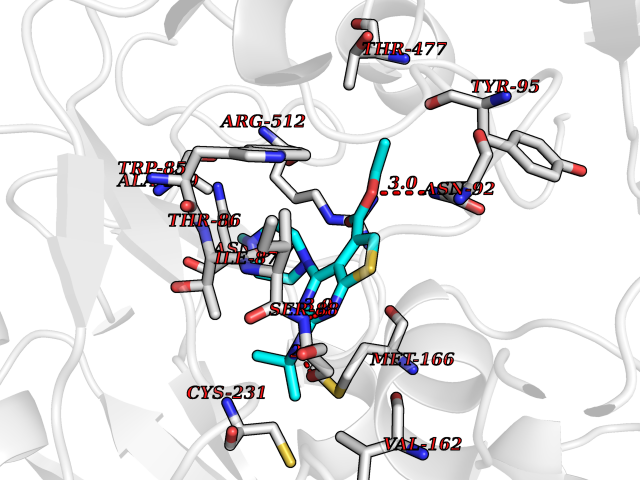 Pymol Map 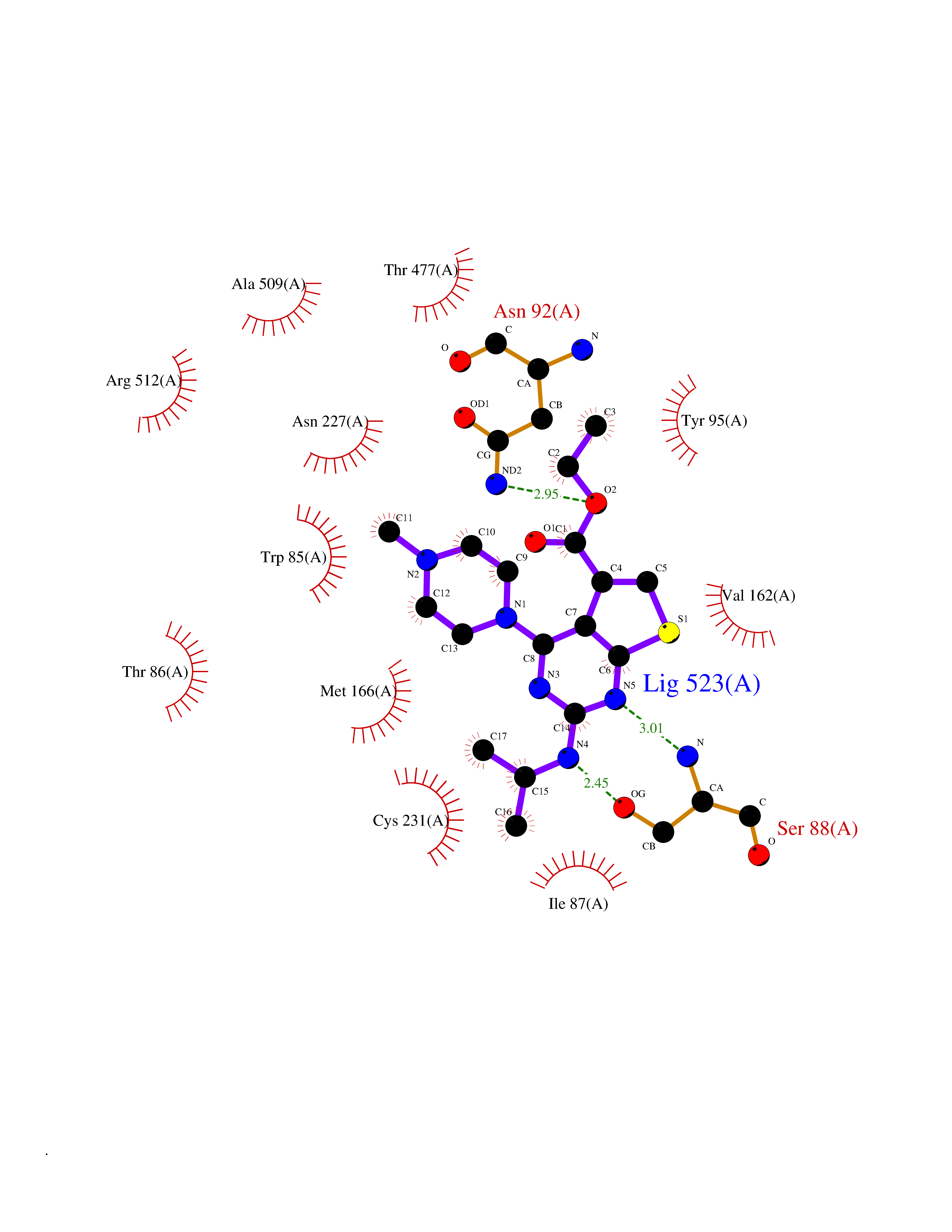 Ligplot Map 3D Binding mode Sequence AVLPKGVTQGEFNKAVQKFRALLGDDNVLVESDQLVPYNKIMMPVENAAHAPSAAVTATTVEQVQGVVKICNEHKIPIWTISTGRNFGYGSAAPVQRGQVILDLKKMNKIIKIDPEMCYALVEPGVTFGQMYDYIQENNLPVMLSFSAPSAIAGPVGNTMDRGVGYTPYGEHFMMQCGMEVVLANGDVYRTGMGGVPGSNTWQIFKWGYGPTLDGMFTQANYGICTKMGFWLMPKPPVFKPFEVIFEDEADIVEIVDALRPLRMSNTIPNSVVIASTLWEAGSAHLTRAQYTTEPGHTPDSVIKQMQKDTGMGAWNLYAALYGTQEQVDVNWKIVTDVFKKLGKGRIVTQEEAGDTQPFKYRAQLMSGVPNLQEFGLYNWRGGGGSMWFAPVSEARGSECKKQAAMAKRVLHKYGLDYVAEFIVAPRDMHHVIDVLYDRTNPEETKRADACFNELLDEFEKEGYAVYRVNTRFQDRVAQSYGPVKRKLEHAIKRAVDPNNILAPGRSGIDLNNDF Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Guanidinoacetate N-methyltransferase | 3ORH | 6.55 | |
Target general information Gen name GAMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, RMT2 methyltransferase family Biochemical class Transferase Function Guanidinoacetate N-methyltransferase activity.Methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 2 (CCDS2) [MIM:612736]: An autosomal recessive disorder characterized by developmental delay and regression, intellectual disability, severe disturbance of expressive and cognitive speech, intractable seizures, movement disturbances, severe depletion of creatine and phosphocreatine in the brain, and accumulation of guanidinoacetic acid in brain and body fluids. {ECO:0000269|PubMed:12468279, ECO:0000269|PubMed:15108290, ECO:0000269|PubMed:15651030, ECO:0000269|PubMed:16293431, ECO:0000269|PubMed:16855203, ECO:0000269|PubMed:17101918, ECO:0000269|PubMed:17466557, ECO:0000269|PubMed:19388150, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:24415674, ECO:0000269|PubMed:8651275}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00148; DB02751; DB00536; DB13191; DB01752 Interacts with O95363; Q969Q5; Q9HCM9-2 EC number 2.1.1.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Disease variant; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 24656 Length 219 Aromaticity 0.11 Instability index 46.5 Isoelectric point 5.91 Charge (pH=7) -4.34 2D Binding mode Binding energy (Kcal/mol) -8.94 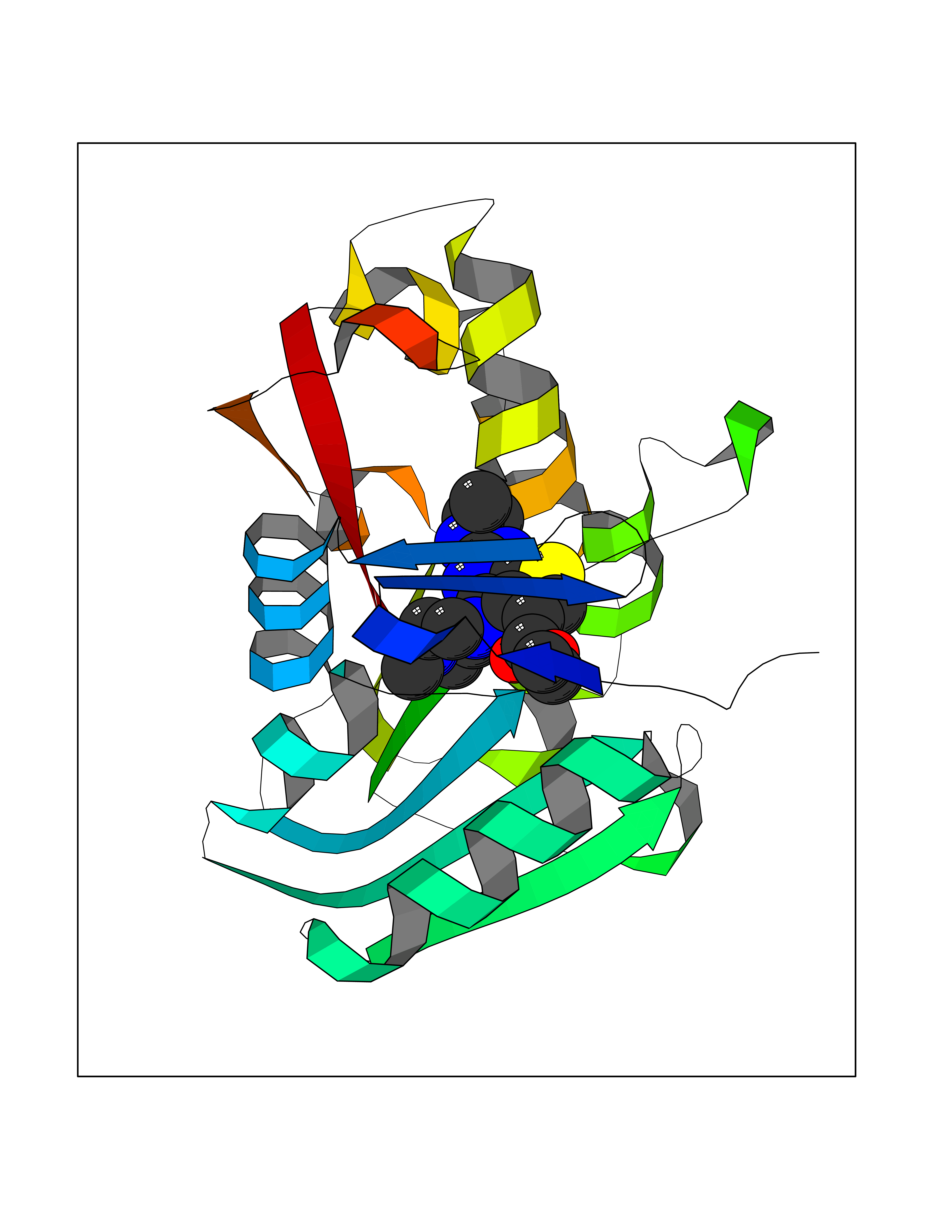 Molscript Map 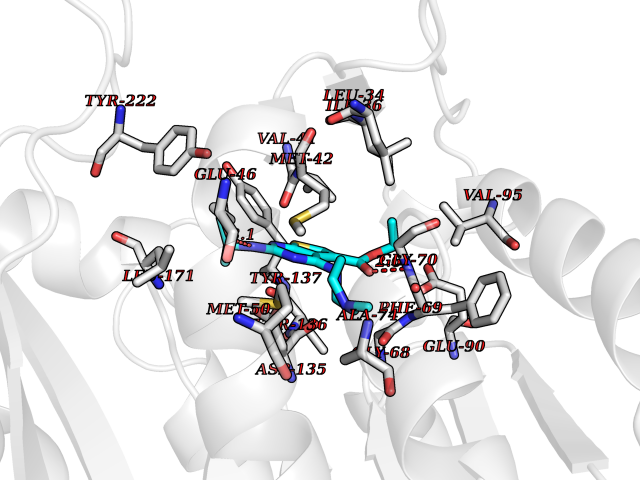 Pymol Map 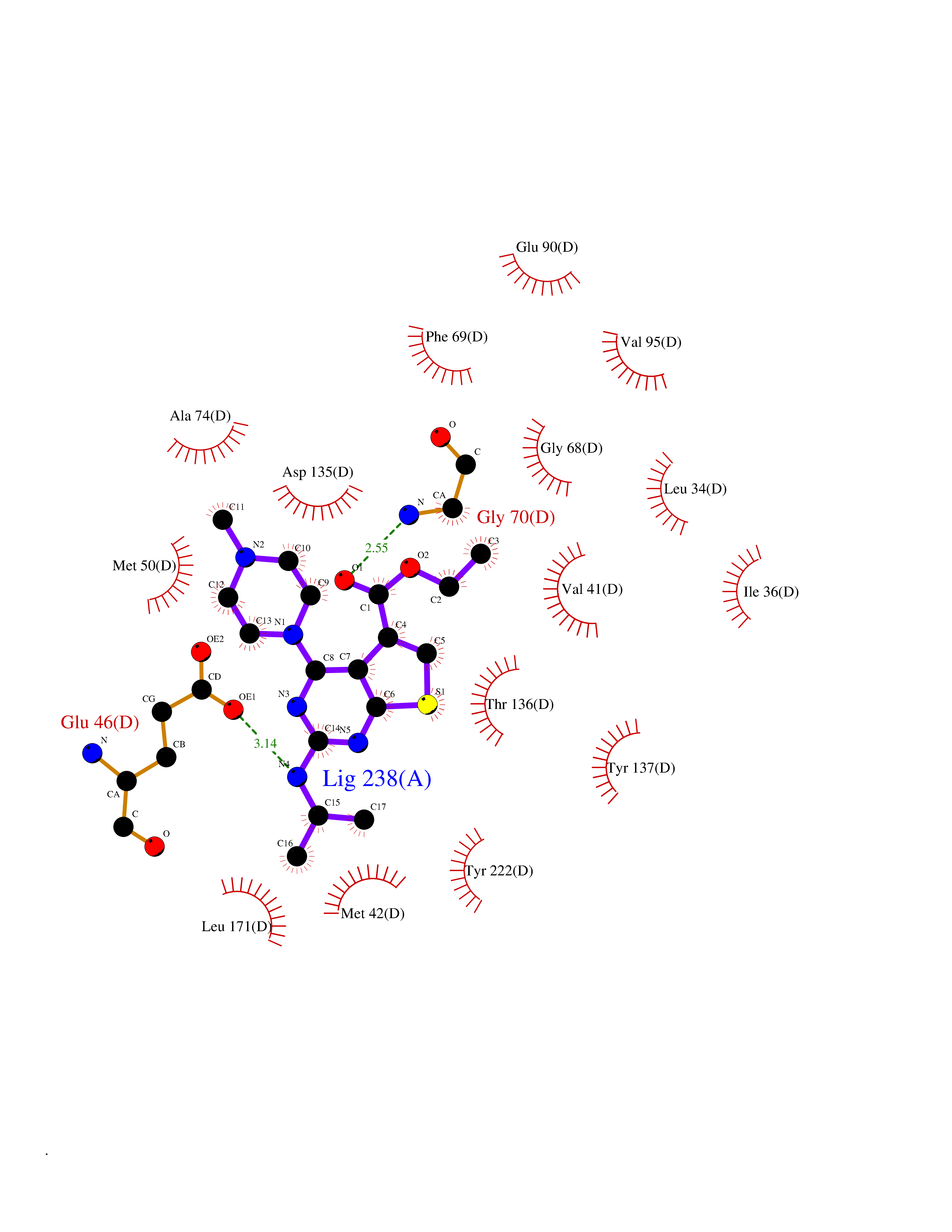 Ligplot Map 3D Binding mode Sequence PAWGAAPAAYDAADTHLRILGKPVMERWETPYMHALAAAASSKGGRVLEVGFGMAIAASKVQEAPIDEHWIIECNDGVFQRLRDWAPRQTHKVIPLKGLWEDVAPTLPDGHFDGILYDTYPLSEETWHTHQFNFIKNHAFRLLKPGGVLTYCNLTSWGELMKSKYSDITIMFEETQVPALLEAGFRRENIRTEVMALVPPADCRYYAFPQMITPLVTKG Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Aldehyde oxidoreductase | 4USA | 6.54 | |
Target general information Gen name mop Organism Megalodesulfovibrio gigas (Desulfovibrio gigas) Uniprot ID TTD ID NA Synonyms NA Protein family Xanthine dehydrogenase family Biochemical class Oxidoreductase Function 2 iron, 2 sulfur cluster binding.Aldehyde dehydrogenase (FAD-independent) activity.Electron carrier activity.Metal ion binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB02137 Interacts with NA EC number 1.2.99.7 Uniprot keywords 2Fe-2S; 3D-structure; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 96930.4 Length 907 Aromaticity 0.07 Instability index 29.17 Isoelectric point 5.69 Charge (pH=7) -17.56 2D Binding mode Binding energy (Kcal/mol) -8.92 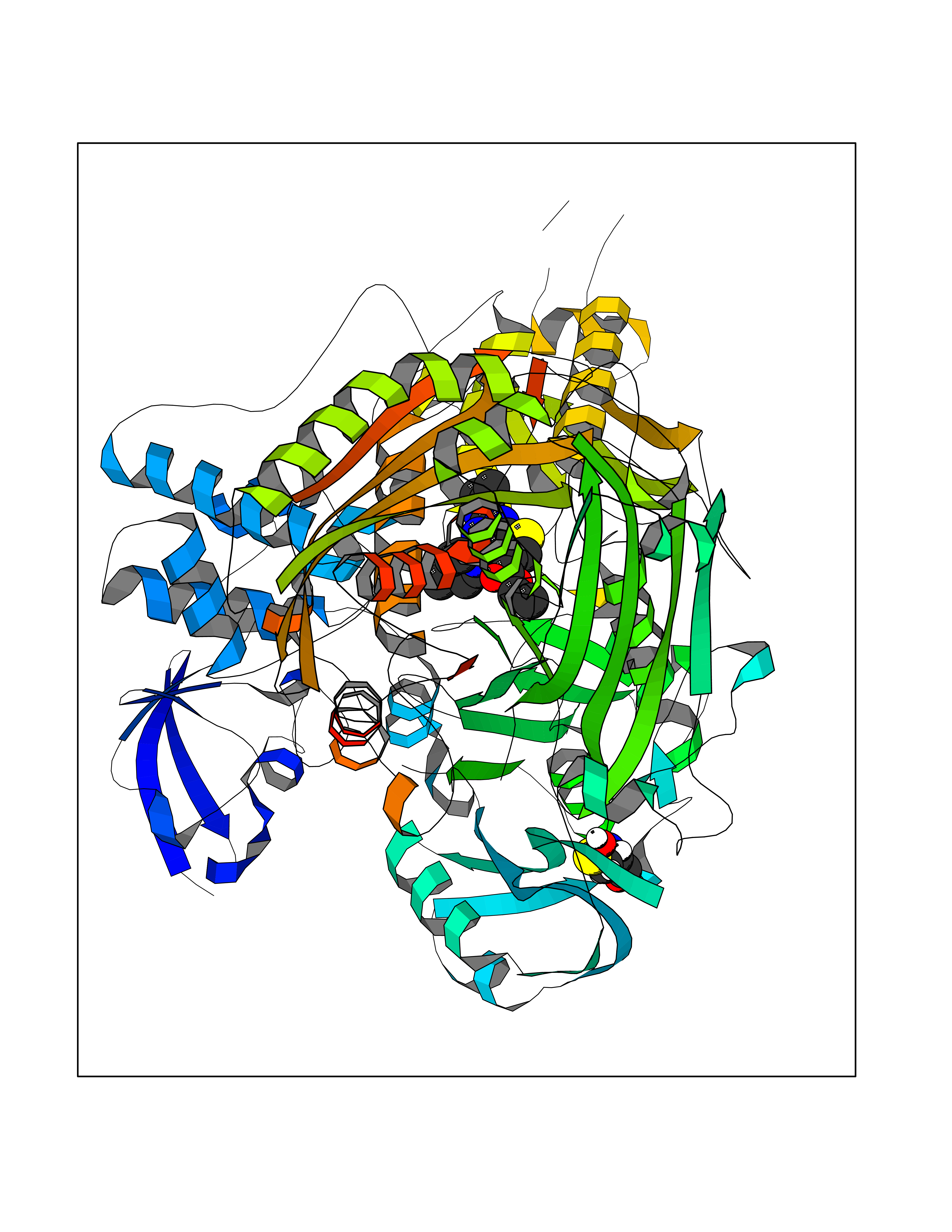 Molscript Map 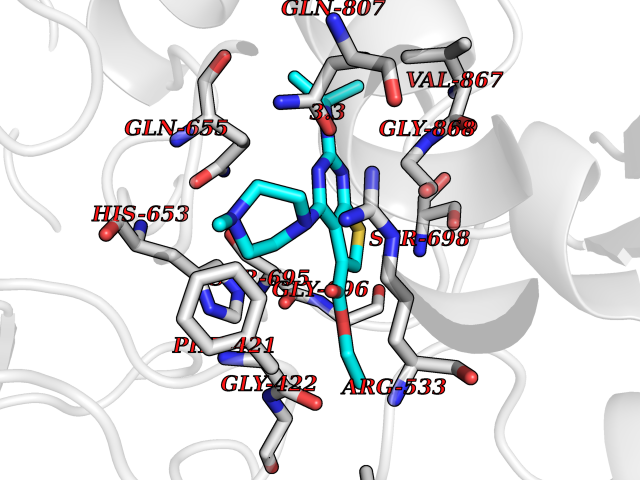 Pymol Map  Ligplot Map 3D Binding mode Sequence MIQKVITVNGIEQNLFVDAEALLSDVLRQQLGLTGVKVGCEQGQCGACSVILDGKVVRACVTKMKRVADGAQITTIEGVGQPENLHPLQKAWVLHGGAQCGFCSPGFIVSAKGLLDTNADPSREDVRDWFQKHRNACRCTGYKPLVDAVMDAAAVINGKKPETDLEFKMPADGRIWGSKYPRPTAVAKVTGTLDYGADLGLKMPAGTLHLAMVQAKVSHANIKGIDTSEALTMPGVHSVITHKDVKGKNRITGLITFPTNKGDGWDRPILXDEKVFQYGDCIALVCADSEANARAAAEKVKVDLEELPAYMSGPAAAAEDAIEIHPGTPNVYFEQPIVKGEDTGPIFASADVTVEGDFYVGRQPHMPIEPDVAFAYMGDDGKCYIHSKSIGVHLHLYMIAPGVGLEPDQLVLVANPMGGTFGYKFSPTSEALVAVAAMATGRPVHLRYNYQQQQQYTGKRSPWEMNVKFAAKKDGTLLAMESDWLVDHGPYSEFGDLLTLRGAQFIGAGYNIPNIRGLGRTVATNHVWGSAFRGYGAPQSMFASECLMDMLAEKLGMDPLELRYKNAYRPGDTNPTGQEPEVFSLPDMIDQLRPKYQAALEKAQKESTATHKKGVGISIGVYGSGLDGPDASEAWAELNADGTITVHTAWEDHGQGADIGCVGTAHEALRPMGVAPEKIKFTWPNTATTPNSGPSGGSRQQVMTGNAIRVACENLLKACEKPGGGYYTYDELKAADKPTKITGNWTASGATHCDAVTGLGKPFVVYMYGVFMAEVTVDVATGQTTVDGMTLMADLGSLCNQLATDGQIYGGLAQGIGLALSEDFEDIKKHATLVGAGFPFIKQIPDKLDIVYVNHPRPDGPFGASGVGELPLTSPHAAIINAIKSATGVRIYRLPAYPEKVLEALKA Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | cAMP-dependent protein kinase A type I (PRKAR1A) | 5KJZ | 6.54 | |
Target general information Gen name PRKAR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-dependent protein kinase type I-alpha regulatory subunit; Tissue-specific extinguisher 1; TSE1; PRKAR1; PKR1 Protein family CAMP-dependent kinase regulatory chain family Biochemical class Kinase Function Regulatory subunit of the cAMP-dependent protein kinases involved in cAMP signaling in cells. Related diseases Carney complex 1 (CNC1) [MIM:160980]: CNC is a multiple neoplasia syndrome characterized by spotty skin pigmentation, cardiac and other myxomas, endocrine tumors, and psammomatous melanotic schwannomas. {ECO:0000269|PubMed:15371594, ECO:0000269|PubMed:18241045, ECO:0000269|PubMed:22785148, ECO:0000269|PubMed:23323113, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Intracardiac myxoma (INTMYX) [MIM:255960]: Inheritance is autosomal recessive. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Primary pigmented nodular adrenocortical disease 1 (PPNAD1) [MIM:610489]: A rare bilateral adrenal defect causing ACTH-independent Cushing syndrome. Macroscopic appearance of the adrenals is characteristic with small pigmented micronodules observed in the cortex. Clinical manifestations of Cushing syndrome include facial and truncal obesity, abdominal striae, muscular weakness, osteoporosis, arterial hypertension, diabetes. PPNAD1 is most often diagnosed in patients with Carney complex, a multiple neoplasia syndrome. However it can also be observed in patients without other manifestations. {ECO:0000269|PubMed:12213893}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Acrodysostosis 1, with or without hormone resistance (ACRDYS1) [MIM:101800]: A form of skeletal dysplasia characterized by short stature, severe brachydactyly, facial dysostosis, and nasal hypoplasia. Affected individuals often have advanced bone age and obesity. Laboratory studies show resistance to multiple hormones, including parathyroid, thyrotropin, calcitonin, growth hormone-releasing hormone, and gonadotropin. However, not all patients show endocrine abnormalities. {ECO:0000269|PubMed:21651393, ECO:0000269|PubMed:22464250, ECO:0000269|PubMed:22464252, ECO:0000269|PubMed:22723333, ECO:0000269|PubMed:23043190, ECO:0000269|PubMed:23425300, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01790; DB02527; DB02315; DB05798 Interacts with Q9GZX7; P24588; O43687-2; Q9BSF0; Q9H6J7-2; Q86Y01; P0C7A2-2; Q9H0R8; Q9H8W4; P17612; P31321; P51817; P35250; Q86UC2; Q01105; Q8N0X7; O96006; P03259-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; cAMP; cAMP-binding; Cell membrane; Cushing syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat Protein physicochemical properties Chain ID A Molecular weight (Da) 17007.4 Length 149 Aromaticity 0.08 Instability index 49.36 Isoelectric point 6.36 Charge (pH=7) -0.52 2D Binding mode Binding energy (Kcal/mol) -8.93  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SILMGSTLRKRKMYEEFLSKVSILESLDKWERLTVADALEPVQFEDGQKIVVQGEPGDEFFIILEGSAAVLQRRSENEEFVEVRRLGPSDYFGEIALLMNRPRTATVVARGPLKCVKLDRPRFERVLGPCSDILKRNIQQYNSFVSLSV Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Extracellular lysophospholipase D (E-NPP2) | 4ZGA | 6.54 | |
Target general information Gen name ENPP2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LysoPLD; Ectonucleotide pyrophosphatase/phosphodiesterase family member 2; E-NPP 2; Autotaxin; ATX Protein family Nucleotide pyrophosphatase/phosphodiesterase family Biochemical class Phosphoric diester hydrolase Function Hydrolyzes lysophospholipids to produce the signaling molecule lysophosphatidic acid (LPA) in extracellular fluids. Major substrate is lysophosphatidylcholine. Also can act on sphingosylphosphorylcholine producing sphingosine-1-phosphate, a modulator of cell motility. Can hydrolyze, in vitro, bis-pNPP, to some extent pNP-TMP, and barely ATP. Involved in several motility-related processes such as angiogenesis and neurite outgrowth. Acts as an angiogenic factor by stimulating migration of smooth muscle cells and microtubule formation. Stimulates migration of melanoma cells, probably via a pertussis toxin-sensitive G protein. May have a role in induction of parturition. Possible involvement in cell proliferation and adipose tissue development (Probable). Tumor cell motility-stimulating factor. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.1.4.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Chemotaxis; Cleavage on pair of basic residues; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lipid degradation; Lipid metabolism; Metal-binding; Obesity; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 85397.4 Length 740 Aromaticity 0.12 Instability index 58.63 Isoelectric point 7.16 Charge (pH=7) 1.02 2D Binding mode Binding energy (Kcal/mol) -8.92  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GSCKGRCFELCRCDNLCKSYTSCCHDFDELCLKTARGWECTKDRCGNEENACHCEDCLARGDCCTNYQVVCKGESHWVDDDCEEIKAAECPAGFVRPPLIIFSVDGFRASYMKKGSKVMPNIEKLRSCGTHSPYMRPVYPTKTFPNLYTLATGLYPESHGIVGNSMYDPVFDATFHLRGREKFNHRWWGGQPLWITATKQGVKAGTFFWSVVIPHERRILTILQWLTLPDHERPSVYAFYSEQPDFSGHKYGPFGPEMTNPLREIDKIVGQLMDGLKQLKLHRCVNVIFVGDHGMEDVTCDRTEFLSNYLTNVDDITLVPGTLGRIRSKFDPKAIIANLTCKKPDQHFKPYLKQHLPKRLHYANNRRIEDIHLLVERRWHVARKPFFQGDHGFDNKVNSMQTVFVGYGSTFKYKTKVPPFENIELYNVMCDLLGLKPAPNNGTHGSLNHLLRTNTFRPTMPEEVTRPNYPGIMYLQSDFDLGTEERHLLYGRPAVLYRTRYDILYHTDFESGYSEIFLMPLWTSYTVSKQACVRPDVRVSPSFSQNCLAYKNDKQMSYGFLFPPYLSSSPEAKYDAFLVTNMVPMYPAFKRVWNYFQRVLVKKYASERNGVNVISGPIFDYDYDGLHDTEDKIKQYVEGSSIPVPTHYYSIITSCLDFTQPADKCDGPLSVSSFILPHRPDNEESCNSSEDESKWVEELMKMHTARVRDIEHLTSLDFFRKTSRSYPEILTLKTYLHTYE Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Trypanosoma Trypanothione reductase (Trypano TPR) | 2WBA | 6.53 | |
Target general information Gen name Trypano TPR Organism Trypanosoma brucei brucei Uniprot ID TTD ID Synonyms TRYR; TPR; Parasite-specific trypanothione reductase; N(1),N(8)-bis(glutathionyl)spermidine reductase Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Sulfur donor oxidoreductase Function Trypanothione is the parasite analog of glutathione; this enzyme is the equivalent of glutathione reductase. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.8.1.12 Uniprot keywords 3D-structure; Cytoplasm; Disulfide bond; FAD; Flavoprotein; NADP; Oxidoreductase; Redox-active center Protein physicochemical properties Chain ID A,B Molecular weight (Da) 105578 Length 978 Aromaticity 0.08 Instability index 33.76 Isoelectric point 6.25 Charge (pH=7) -6.81 2D Binding mode Binding energy (Kcal/mol) -8.9  Molscript Map 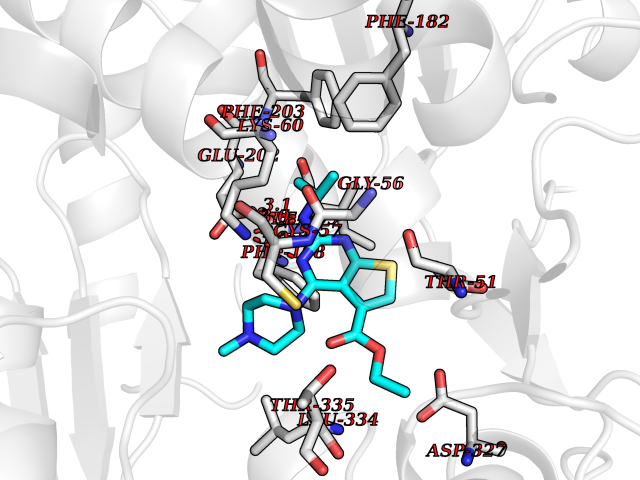 Pymol Map 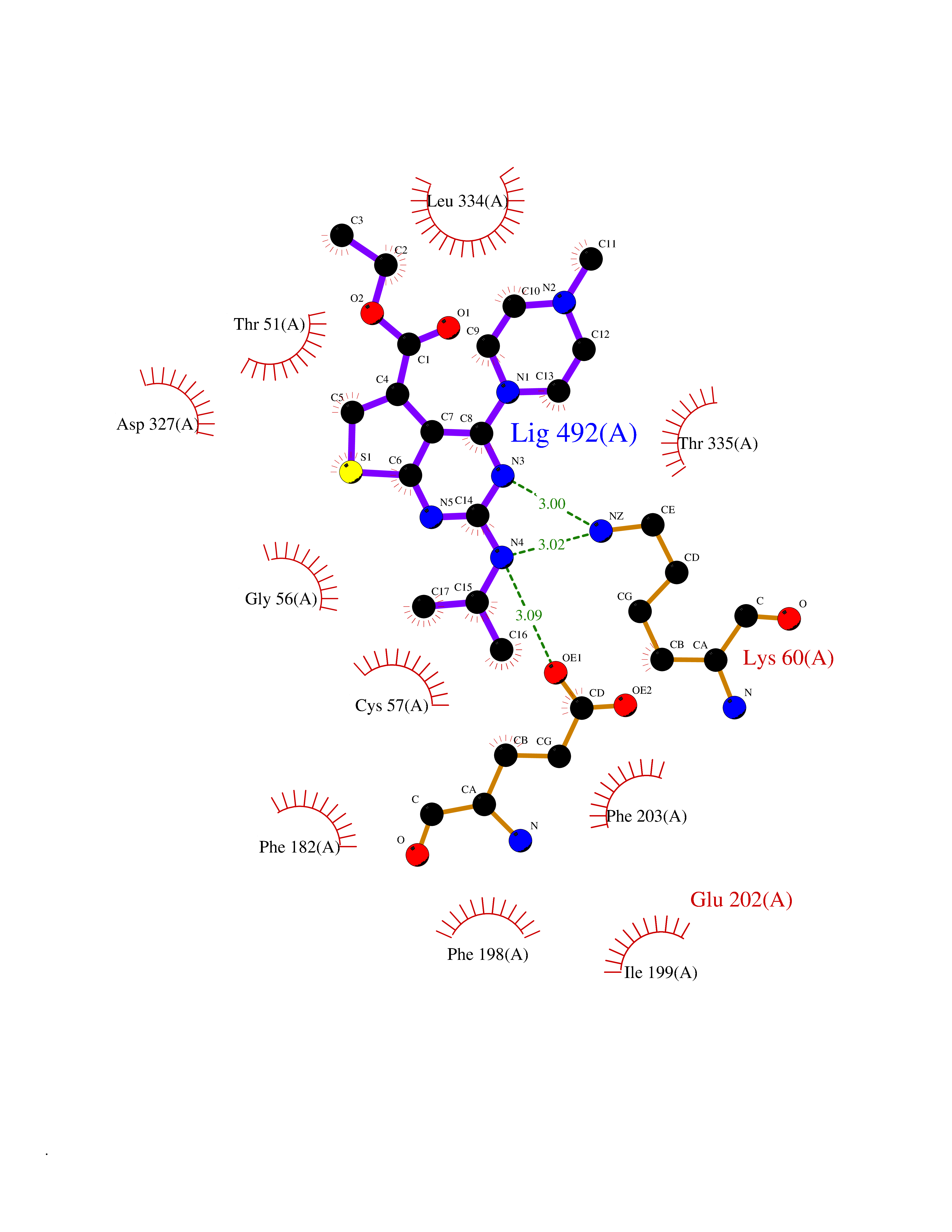 Ligplot Map 3D Binding mode Sequence SKAFDLVVIGAGSGGLEAGWNAATLYGKRVAVVDVQTSHGPPFYAALGGTCVNVGCVPKKLMVTGAQYMDHLRESAGFGWEFDGSSVKANWKKLIAAKNEAVLDINKSYEGMFNDTEGLDFFLGWGSLESKNVVVVRETADPKSAVKERLQADHILLATGSWPQMPAIPGIEHCISSNEAFYLPEPPRRVLTVGGGFISVEFAGIFNAYKPPGGKVTLCYRNNLILRGFDETIREEVTKQLTANGIEIMTNENPAKVSLNTDGSKHVTFESGKTLDVDVVMMAIGRIPRTNDLQLGNVGVKLTPKGGVQVDEFSRTNVPNIYAIGDITDRLMLTPVAINEGAALVDTVFGNKPRKTDHTRVASAVFSIPPIGTCGLIEEVAAKEFEKVAVYMSSFTPLMHNISGSKYKKFVAKIVTNHSDGTVLGVHLLGDGAPEIIQAVGVCLRLNAKISDFYNTIGVHPTSAEELCSMRTPSYYYVKGEKMEKLPDSSKAFDLVVIGAGSGGLEAGWNAATLYGKRVAVVDVQTSHGPPFYAALGGTCVNVGCVPKKLMVTGAQYMDHLRESAGFGWEFDGSSVKANWKKLIAAKNEAVLDINKSYEGMFNDTEGLDFFLGWGSLESKNVVVVRETADPKSAVKERLQADHILLATGSWPQMPAIPGIEHCISSNEAFYLPEPPRRVLTVGGGFISVEFAGIFNAYKPPGGKVTLCYRNNLILRGFDETIREEVTKQLTANGIEIMTNENPAKVSLNTDGSKHVTFESGKTLDVDVVMMAIGRIPRTNDLQLGNVGVKLTPKGGVQVDEFSRTNVPNIYAIGDITDRLMLTPVAINEGAALVDTVFGNKPRKTDHTRVASAVFSIPPIGTCGLIEEVAAKEFEKVAVYMSSFTPLMHNISGSKYKKFVAKIVTNHSDGTVLGVHLLGDGAPEIIQAVGVCLRLNAKISDFYNTIGVHPTSAEELCSMRTPSYYYVKGEKMEKLPDS Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Fumarate reductase flavoprotein subunit | 1Y0P | 6.52 | |
Target general information Gen name fccA Organism Shewanella frigidimarina Uniprot ID TTD ID NA Synonyms fcc3 Protein family FAD-dependent oxidoreductase 2 family, FRD/SDH subfamily Biochemical class Oxidoreductase Function Electron carrier activity.Fumarate reductase (menaquinone).Metal ion binding.Nucleic acid binding.Succinate dehydrogenase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04734; DB03147; DB01677; DB03343 Interacts with NA EC number 1.3.2.4 Uniprot keywords 3D-structure; Electron transport; FAD; Flavoprotein; Heme; Iron; Metal-binding; Oxidoreductase; Periplasm; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 60177.2 Length 568 Aromaticity 0.06 Instability index 27.7 Isoelectric point 6 Charge (pH=7) -8.64 2D Binding mode Binding energy (Kcal/mol) -8.9 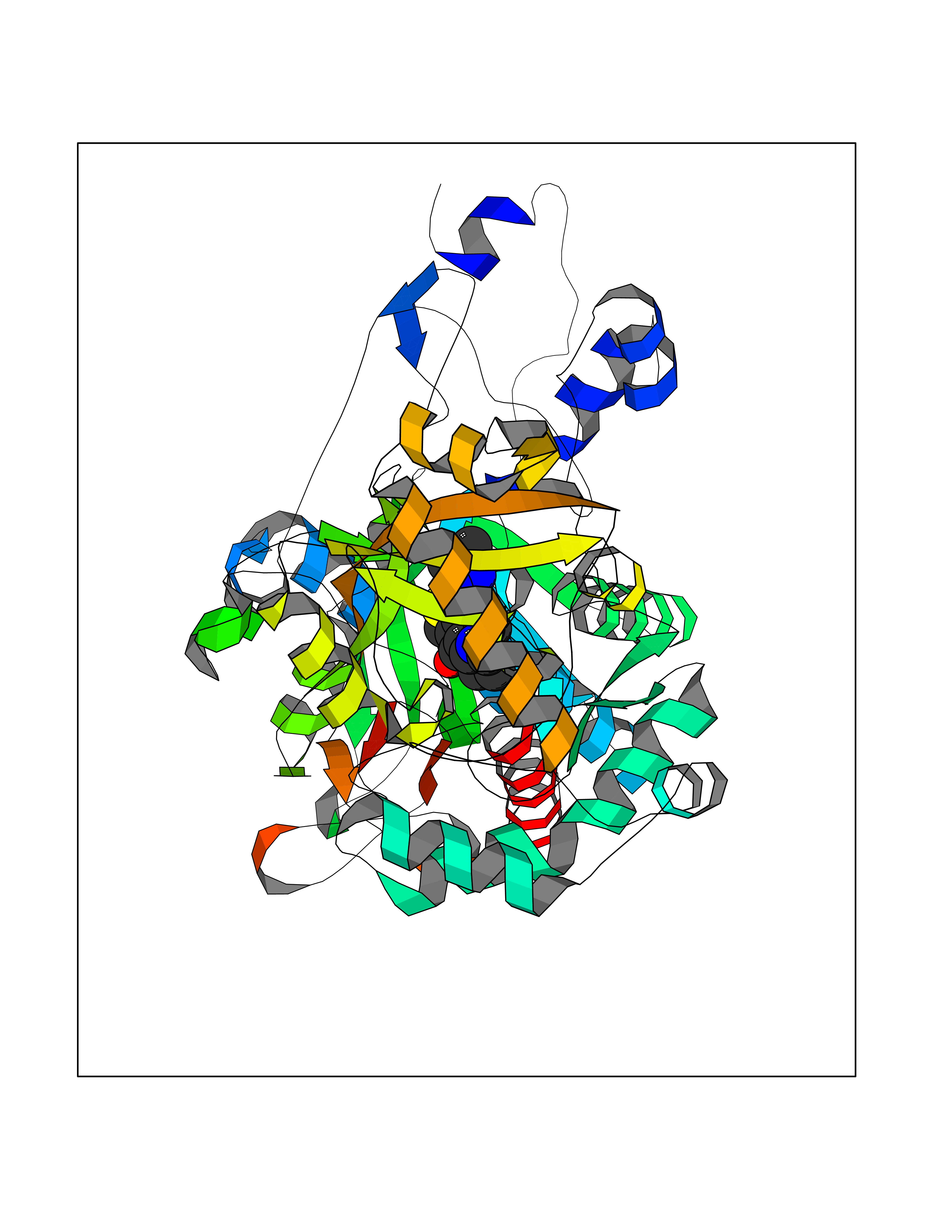 Molscript Map 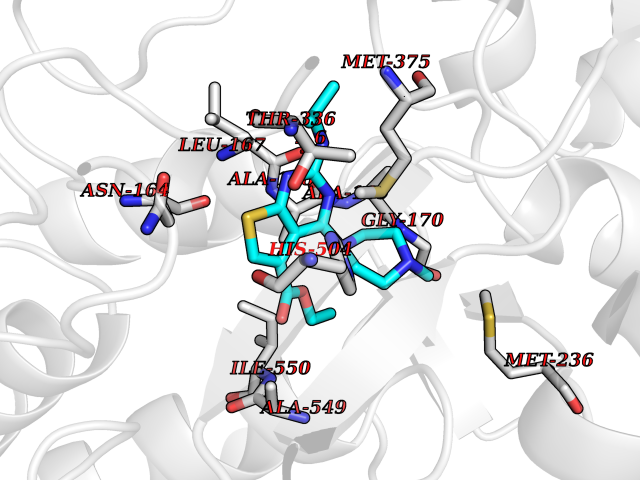 Pymol Map 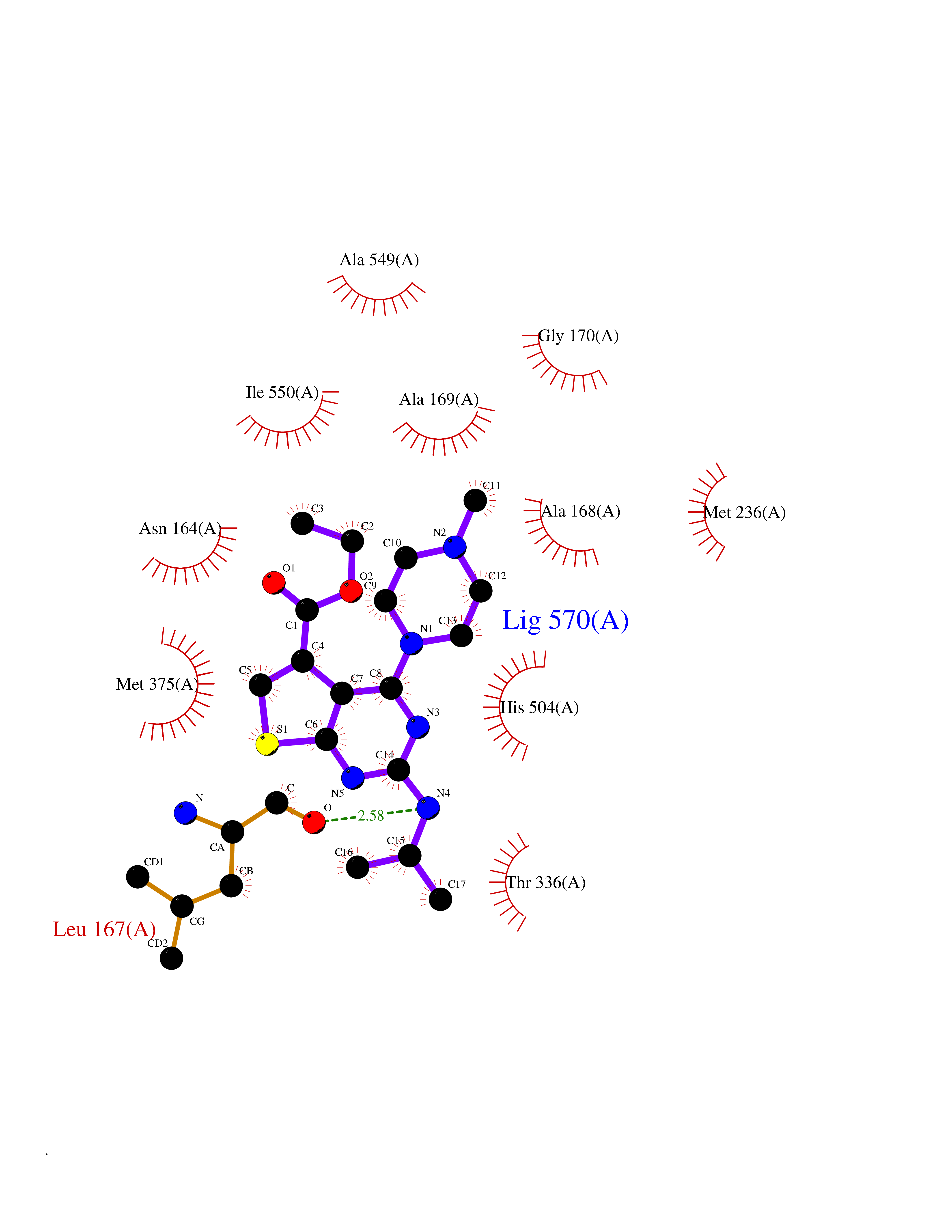 Ligplot Map 3D Binding mode Sequence ADNLAEFHVQNQECDSCHTPDGELSNDSLTYENTQCVSCHGTLAEVAETTKHEHYNAHASHFPGEVACTSCHSAHEKSMVYCDSCHSFDFNMPYAKKWLRDEPTIAELAKDKSERQAALASAPHDTVDVVVVGSGGAGFSAAISATDSGAKVILIEKEPVIGGNAKLAAGGMNAAWTDQQKAKKITDSPELMFEDTMKGGQNINDPALVKVLSSHSKDSVDWMTAMGADLTDVGMMGGASVNRAHRPTGGAGVGAHVVQVLYDNAVKRNIDLRMNTRGIEVLKDDKGTVKGILVKGMYKGYYWVKADAVILATGGFAKNNERVAKLDPSLKGFISTNQPGAVGDGLDVAENAGGALKDMQYIQAHPTLSVKGGVMVTEAVRGNGAILVNREGKRFVNEITTRDKASAAILAQTGKSAYLIFDDSVRKSLSKIDKYIGLGVAPTADSLVKLGKMEGIDGKALTETVARYNSLVSSGKDTDFERPNLPRALNEGNYYAIEVTPGVHHTMGGVMIDTKAEVMNAKKQVIPGLYGAGEVTGGVHGANRLGGNAISDIITFGRLAGEEAAKYS Hydrogen bonds contact Hydrophobic contact | ||||